
閉環状DNAを簡易に調製する方法、キット及び装置
【課題】 経費が低く、短時間で、ニックの少ない高品質のプラスミドをより少ない工数で単離する方法を提供すること。
【解決手段】 細胞から閉環状DNAを調製する方法であって、細胞を含む液体に凝集剤を添加する工程を含んでなる方法。
【解決手段】 細胞から閉環状DNAを調製する方法であって、細胞を含む液体に凝集剤を添加する工程を含んでなる方法。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は細胞から閉環状DNAを簡便に調製する方法、キット及び装置に関する。
【背景技術】
【0002】
ファージやプラスミドなどの環状二本鎖DNAは、通常、負の超らせん閉環状の構造をとっており、このような構造を持つ環状DNAはcccDNA(covalently closed circular DNA)と呼ばれている。
従来はcccDNAの分離にかかる時間が少なく、コストが低く、単離されたcccDNAの品質がよく、収量が高いという条件をすべて満たす単離法は存在しなかった。さらに、cccDNAを調製する方法としてフェノール/クロロホルム法があるが、有害な有機溶剤を使用しなければならなかった。
【0003】
公知の方法であるアルカリSDS法を主とする少検体からcccDNAを分離するキットはQiagen社のQiagen Spin Miniprep Kitが有名である。アルカリSDS法では溶液を一時的に強アルカリにする操作がある。しかし、DNAはアルカリに弱く、ニックが入る可能性が高くなり、最終調製物は環状DNAではなく、直鎖状DNAの比率が高くなる。また、作業に費やす時間が長く、さらに価格が高くなるという欠点を持つ。2004年時点で50検体用のキットは11,000円(1検体あたり220円)である。
【0004】
陰イオン交換樹脂を用いた分離法はフナコシ社のMidiprep Kit, PhoenIXがあり、2004年時点で、25検体で34000円(1検体あたり1360円)と非常に価格が高く、また作業時間も長くなるという欠点がある。
【0005】
アルカリSDS法を主とする多検体用のキットにはR.E.A.L. Prep 96Plasmid Kitがあるが、cccDNAにニックが入る可能性が高く、さらに価格も高くなるという欠点を持つ。
【0006】
また、多検体のcccDNAを一度に単離する場合、プレートの遠心分離が必須な工程であり、そのことが全自動化するにあたっての障害となっていた。すなわち、プレートの遠心分離を行って全自動化する試みは、例えば特開2001-333763号公報(特許文献1)に見られる。しかし、(1)プレート遠心機は遠心力が弱く、ペレット化したプラスミドの分離が充分に行えない(2)遠心分離機にプレートをセットする際、あるいは遠心後に遠心機からプレートを取り出す際に、ロボットアームおよび遠心機の回転を高度に制御しなければならず、実質的に製作が困難であるという問題があった。
【0007】
さらに、全自動化する試みは特開平10-155481号公報(特許文献2)にも見られる。この方法はカオトロピックイオン法を利用し、フィルター上の担体にDNAを吸着させてDNAを単離する方法である。0.6mlの大腸菌培養液から最大で4〜6μgのプラスミドDNAが得られるとされているが、実際には0.5μg程度のプラスミドDNAしか得られず、さらにゲノムDNAの夾雑も多い。
【特許文献1】特開2001-333763号公報
【特許文献2】特開平10-155481号公報
【発明の開示】
【発明が解決しようとする課題】
【0008】
本発明は、経費が低く、短時間で、ニックの少ない高品質のプラスミドをより少ない工数で無害な物質を用いて細胞から単離する方法を提供することを目的とする。
さらに、本発明は、多数のサンプルを一度に単離する全自動プラスミド調製機を提供することを目的とする。
【課題を解決するための手段】
【0009】
本発明者らは、凝集剤を使用することによって、凝集作用の違いにより、閉環状DNA(例えば、プラスミド)を他の細胞構成物、特にゲノムDNAやタンパク質から分離することに成功し、本発明を完成させるに至った。
本発明の要旨は以下の通りである。
(1) 細胞から閉環状DNAを調製する方法であって、細胞を含む液体に凝集剤を添加する工程を含んでなる方法。
(2) 以下の工程:
(a)該細胞を含む液体に少なくとも1種類のアルコール及び/又はケトンを添加する工程;
(b)該細胞を含む液体に凝集補助剤及び凝集剤を添加して、上清と沈殿に分離する工程;
(c)工程(b)で生じた上清と沈殿から上清を除く工程;
(d)工程(c)で上清を除いたあとの沈殿に細胞溶解液を添加する工程;
(e)工程(d)で生じた混合液に振動を与える工程;
(f)工程(e)で振動を与えた混合液にアルコール性溶液を添加する工程;及び
(g)工程(f)でアルコール性溶液を添加した混合液の上清を限外ろ過フィルターに通して閉環状DNAを回収する工程
を含んでなる(1)記載の方法。
(3) 遠心分離を行う工程を含まない(2)記載の方法。
(4) 工程(a)の前工程として、高張液を細胞に接触させる(2)又は(3)に記載の方法。
(5) 以下の工程:
(a’)培養した細胞を遠心分離により沈殿させ、上清を除く工程;
(b’)沈殿した細胞をバッファーもしくは培地で縣濁させ、細胞懸濁液を調製する工程;
(c’)該細胞懸濁液に少なくとも1種類のアルコール及び/又はケトンを添加する工程;
(d’)該細胞懸濁液に凝集補助剤及び凝集剤を添加する工程;
(e’)該細胞懸濁液に細胞溶解液を添加する工程;
(f’)工程(e’)で生じた混合液に振動を与える工程;
(g’)工程(f’)で振動を与えた混合液にアルコール性溶液を添加する工程;及び
(h’)工程(g’)でアルコール性溶液を添加した混合液の上清を限外ろ過フィルターに通して閉環状DNAを回収する工程
を含んでなる(1)記載の方法。
(6) 工程(b’)のバッファーもしくは培地が高張液である(5)記載の方法。
(7) 細胞が単細胞生物である(1)〜(6)のいずれかに記載の方法。
(8) 単細胞生物が細菌である(7)記載の方法。
(9) 細菌が大腸菌である(8)記載の方法。
(10) 細胞が形質転換体である(1)〜(9)のいずれかに記載の方法。
(11) 閉環状DNAが、プラスミドDNA、コスミドDNA、P1 DNA、BAC DNA、PAC DNA及びYAC DNAからなる群より選択される(1)〜(10)のいずれかに記載の方法。
(12) (1)〜(11)のいずれかに記載の方法に使用するためのキットであって、以下の成分:
(a)少なくとも1種類のアルコール及び/又はケトンを含む溶液;
(b)凝集補助剤;
(c)凝集剤;
(d)細胞溶解液;
(e)アルコール性溶液;
(f)洗浄液;及び
(g)溶出液
を含むキット。
(13) さらに、以下の要素:
(h)チップ;
(i)チップ止め装置;及び
(j)培養プレート
を含む(12)記載のキット。
(14) 閉環状DNAを調製するための装置であって、以下の装置:
(A)横移動装置;
(B)分注装置;
(C)上清吸引装置;
(D)振動装置;
(E)プレート吸引装置;及び
(F)ロボットアーム
を含む装置。
(15) 凝集剤を使用して、閉環状DNAを他の細胞構成物から分離する方法。
【0010】
本明細書で「単細胞生物」とは、大部分の生活史を単一の細胞から成る生物を言い、原核生物の大部分(細菌・藍色細菌・古細菌)及び真核生物の一部(原生生物・鞭毛藻類・珪藻類・緑藻類や紅藻類の一部・真菌類の一部)がこれに該当する。
本明細書で「閉環状DNA」とは、DNAの3’末端及び5’末端が共有結合によって自己連結された超らせん構造を持つ環状DNAを言う。DNA鎖の一部が巻き戻されるとワトソン―クリック回転が減り、その分、ねじれが生じて超らせんが形成される。ほとんどの閉環状DNAは負の超らせん構造をとっている。
本明細書で「凝集剤」とは、水の濁りや色を除去するために用いられる薬剤で、水中に懸濁する微粒子をくっつけてフロック(集塊)をつくらせ、沈降、濾過によって分離除去するのを容易にする作用があるものを言う。多くの微粒子はマイナスに帯電しており、プラスに帯電した凝集剤を入れることで微粒子同士を架橋し、そのフロックを大きくすることで沈降速度を上げる。
本明細書で「凝集補助剤」とは、凝集剤による凝集作用を高める働きのある物質のことで、水中に懸濁することで微粒子のフロックを作りやすくする作用がある。
本明細書で「細胞溶解液」とは、細胞を溶解する作用を持つ物質を含む液体を言う。
本明細書で「アルコール性溶液」とは、アルコールを含む液体を言う。
本明細書で「高張液」とは、対象とする細胞よりも高張な溶液をいう。
本明細書で「チップ」とは、ピペットマン等の先に取り付ける交換可能な器具(通常、プラスチック製)を言う。溶液の吸引や排出の際に用いる。
本明細書において、特に断りのない限り、百分率(%)は重量%を示すものとする。
【発明の効果】
【0011】
本発明により、経費が低く、短時間で、高品質の閉環状DNAをより少ない工数で無害な物質を用いて細胞から単離することができる。特に、コストが従前の約10分の1以下になることは大きな利点である。
例えば、2004年時点でQiagen社のQiagen Spin Miniprep Kitでは50検体で11,000円(1検体220円)また、R.E.A.L. Prep 96Plasmid kit(96検体同時調製)では4プレートで75,000円(1検体195円)であるのに対し、本発明では1検体18円である。
さらに、多数のサンプルから閉環状DNAを全自動で単離することができるようになった。
【発明を実施するための最良の形態】
【0012】
以下、本発明を詳細に説明する。
本発明は、細胞から閉環状DNAを調製する方法であって、細胞を含む液体に凝集剤を添加する工程を含んでなる方法を提供する。
細胞は、いかなる細胞であってもよく、細菌、古細菌などの原核細胞、酵母などの菌類細胞、原生生物、植物細胞、動物細胞などを挙げることができる。
閉環状DNAは、DNAの3’末端及び5’末端が共有結合によって自己連結された超らせん構造を持つ環状DNAであれば、いかなるものであってもよく、プラスミドDNA、コスミドDNA、P1 DNA、BAC DNA、PAC DNAまたはYAC DNAなどを挙げることができる。
細胞を含む液体は、細胞を含有するいかなる液体であってもよく、細胞の培養液、培養細胞を遠心分離で沈殿させた後にバッファーもしくは培地に懸濁させた液などを挙げることができる。
本発明の1実施態様において、本発明の方法は、以下の工程:
(a)該細胞を含む液体に少なくとも1種類のアルコール及び/又はケトンを添加する工程;
(b)該細胞を含む液体に凝集補助剤及び凝集剤を添加して、上清と沈殿に分離する工程;
(c)工程(b)で生じた上清と沈殿から上清を除く工程;
(d)工程(c)で上清を除いたあとの沈殿に細胞溶解液を添加する工程;
(e)工程(d)で生じた混合液に振動を与える工程;
(f)工程(e)で振動を与えた混合液にアルコール性溶液を添加する工程;及び
(g)工程(f)でアルコール性溶液を添加した混合液の上清を限外ろ過フィルターに通して閉環状DNAを回収する工程
を含んでなる。
本発明の方法により、遠心分離を用いることなく、実質的にゲノムDNAを含まない閉環状DNAを得ることができるが、本発明の方法は遠心分離工程を完全に排除するものではなく、本発明の方法を遠心分離と組み合わせて用いてもよい。
ここで、「実質的にゲノムDNAを含まない」とは、最終調製物中に含まれるゲノムDNAの量が、cccDNAが目視できる量を電気泳動する場合にほとんど目視できない量であることを言う。例えば、図9のレーン2は本手法で単離した最終調整物を電気泳動したもので、このレーンには2つのバンドが目視できる。上の薄いバンドは直鎖状プラスミドDNA、下の濃いバンドが閉環状プラスミドDNAであり、ゲノムDNAは目視できない。ゲノムDNAが含まれている場合、ゲノムDNAはプラスミドDNAよりもサイズが大きいので、直鎖状のプラスミドDNAのバンドの上部にバンドが現れる。
【0013】
細胞を液体培地により培養する際、0.1mL〜10Lの培地に細胞を培養し、最も細胞数が増えた状態で上記(a)〜(g)の工程を行うことが好ましい。この際、細胞が大腸菌であれば培地はLB培地(1% Trypton、0.5% Yeast Extract、1% NaCl)、2×LB培地(2% Trypton、1% Yeast Extract、1% NaCl)もしくはLB+7% Glycerol培地(1% Trypton、0.5% Yeast Extract、1% NaCl、7% Glycerol)が好ましい。特に、LB+7% Glycerol培地で培養すると、菌体数が多くなり、DNAの収量がLB培地や2×LB培地よりも多くなる。
【0014】
(a)〜(g)の工程を行う前に、高張液を細胞に接触させる(例えば、細胞よりも高張な溶液を培地に添加する)ことが好ましい。これにより細胞から水分を奪い、細胞を縮小させる。この工程は、後続のアルコール接触処理に伴う細胞破壊時の細胞質の細胞外への流出を極力抑える働きを持つ。さらに、高張液が塩の溶液であれば、(a)の工程におけるアルコールもしくはケトン添加時の細胞同士の凝集作用を高める働きがある。高張液は、安価という点で塩化ナトリウム水溶液が好ましい。終濃度0.1〜2.5Mの塩化ナトリウムが好ましく、終濃度0.83Mの塩化ナトリウム水溶液が最も好ましい。
【0015】
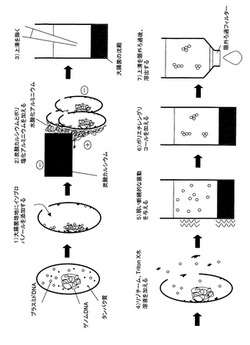
(a)の工程において、少なくとも1種類のアルコール及び/又はケトンを添加する(図13の1))。図13の1)では、「少なくとも1種類のアルコール及び/又はケトン」としてイソプロパノールを、「細胞」として大腸菌を記載している。培養した細胞に直接アルコール及び/又はケトンを添加することにより、細胞表面は脱水され、細胞同士の親水基が結びつき、大きな凝集塊を形成する(コロイドの塩析作用)。例えば、大腸菌は微弱ながらマイナスに帯電しており、それらが凝集することによって強くマイナスに帯電した凝集塊となる。また、アルコール及び/又はケトンの添加によって、細胞に穴が開き、穴からアルコール及び/又はケトンが進入する。それによって細胞内のタンパク質は変性し、凝集する。さらに、一部のタンパク質は変性時にゲノムDNAを巻き込み、固定する。閉環状DNA(例えば、プラスミドDNA)はアルコール及び/又はケトンによって脱水され、ゲノムDNAなどの細胞内物質と結合、凝集し、細胞外へ解離しない。この処理にはアルコールであればエタノールあるいはイソプロピルアルコール、ケトンであればアセトンから一種類以上選ばれることが好ましい。特にイソプロピルアルコールは少量の液量で処理できる点で最も好ましい。また、終濃度は1〜80%イソプロピルアルコールが好ましく、終濃度33.3%のイソプロピルアルコールが最も好ましい。
【0016】
(b)の工程において、凝集補助剤と凝集剤を添加する(図13の2))。凝集剤としては、ポリ塩化アルミニウム、硫酸アルミニウム、アルギン酸ナトリウム、キトサン、塩化第二鉄、ポリ硫酸第二鉄、ポリアクリルアミド系高分子凝集剤、ジメチルアミノエチルメタクリレート系高分子凝集剤、ジメチルアミノエチルアクリレート系高分子凝集剤、アミジン系高分子凝集剤、両性系高分子凝集剤を挙げることができる。凝集補助剤としては、炭酸カルシウム、粉末活性炭、活性珪酸、ベントナイトを挙げることができる。図13の2)では、「凝集補助剤」として炭酸カルシウムを、「凝集剤」としてポリ塩化アルミニウムを記載している。凝集補助剤としては粒径が大きく、水よりも比重が大きく、マイナスに帯電した不溶性物質を用いるとよい。凝集補助剤としては炭酸カルシウム(CaCO3)が好ましい。終濃度0.1〜100mg/ml炭酸カルシウムが好ましく、終濃度21.3mg/ml炭酸カルシウムが最も好ましい。炭酸カルシウムは蒸留水中で自然に沈殿になり、分離操作に扱いづらいので、炭酸カルシウムの沈降速度を遅らせるために粘度の高いグリセロール(終濃度1〜50%)を蒸留水中に加えても良い。
【0017】
さらに、(b)の工程において、マイナスに帯電した凝集補助剤と工程(a)によりマイナスに帯電した細胞(図13では、大腸菌)の凝集塊をプラスに帯電した凝集剤を用いて架橋し、凝集補助剤の重みによって共沈させる。凝集剤は終濃度3〜1000μg/mlポリ塩化アルミニウムが好ましく、終濃度83.3μg/mlポリ塩化アルミニウムが最も好ましい。沈殿が形成されるまで4〜8分待てば良いが、時間を置けば置くほど沈殿量は多くなり、数日置くことも可能である。凝集補助剤と凝集剤を添加する順序は、特に限定されることはないが、凝集補助剤が先に添付されることが好ましい。
次いで、(c)の工程において、(b)の工程で生じた上清と沈殿から上清を除く(図13の3))。
【0018】
(c)の工程で上清を除いたあとの沈殿に水分を含有する液体を加えると、水中のアルコール及び/又はケトン濃度が減少し、溶解度の高い閉環状DNAは溶解する。タンパク質は一度変性すると元に戻りにくいため、水溶液を加えた後もいくつかのタンパク質は溶解したゲノムDNAをしっかりと保持し続ける。また、水分を含有する液体として細胞溶解液を加える(工程(d)、図13の4))ことによって細胞を形成する細胞膜と細胞壁を溶かして穴を広げ、閉環状DNAを細胞外へ放出しやすくする。細胞溶解液は、細胞膜を溶解する界面活性剤、細胞溶解酵素などの成分をバッファーに溶解したものであるとよい。細胞膜を溶解する界面活性剤としてはTriton X-100、Briji、Nonidet P-40、Tween 20、Tween 80、CHAPS、SDS、NP-40、コール酸、n-octyl glycosideなどがあり、細胞溶解酵素としてはセルラーゼ、ヘミセルラーゼ、ペクチナーゼ、リゾチーム、β-1,3-グルカナーゼ、コラゲナーゼ、トリプシンなどがある。バッファーはいかなるものでもよいが、DNAの分解を補助する可能性のあるMgイオンをキレートするEDTAが入っていることが好ましい。例えばTEバッファー(10mM Tris-HCl, 1mM EDTA (pH 8))、TBEバッファー(85mM Tris, 85mM ホウ酸, 2mM EDTA)、TAEバッファー(Tris 40mM, 酢酸 40mM, 1mM EDTA)などを挙げることができる。細胞が大腸菌である場合、細胞溶解液はTriton X-100とリゾチームの混合液が好ましい。Triton X-100とリゾチームの濃度は以下の通りである。終濃度0.01〜20%のTriton X-100が好ましく、終濃度1%のTriton X-100が最も好ましい。終濃度0.01〜40mg/mlのリゾチームが好ましく、終濃度2.5mg/mlのリゾチームが最も好ましい。さらに、RNAを分解するためにRNaseを同時に加えることが望ましい。ここでRNaseはRNaseA、RNaseT、RNaseI、RNaseHを含むが、これらに限定されるものではない。RNaseがRNaseAである場合、終濃度0.1〜200μg/ml RNaseAが好ましく、終濃度34μg/ml RNaseAが最も好ましい。
【0019】
細胞膜と細胞壁が溶かされることによって、タンパク質、タンパク質―ゲノムDNA結合体、その他の細胞構成物も細胞内から分離する可能性がある。しかし、これらの不溶性細胞構成物は主としてマイナスに帯電しており、プラスに帯電した凝集剤に引き寄せられ、大きな凝集塊を作る。一方、可溶化した閉環状DNAは、凝集剤で架橋されることなく上清に溶け、沈殿と分離される。さらに、工程(e)において、振動を与える(好ましくは、弱い振動を断続的に与える(図13の5))ことで、物質同士のイオン結合による凝集作用を強化し、閉環状DNAとの分離効率を飛躍的に高める。つまり、本発明の最大の特徴は、凝集作用の違いによって閉環状DNAを他の細胞構成物、特にゲノムDNAやタンパク質から分離することである。工程(e)の振動は、500〜1700rpm程度の弱い振動を0.1〜3秒間隔で0.1〜3秒間2〜20回与える振動が好ましく、0.5秒間隔で0.5秒間10回与える振動が最も好ましい。
【0020】
(f)の工程において、上記(e)の工程で生成された閉環状DNAの溶解した上清にアルコール性溶液を添加する(図13の6))。図13の6)では、「アルコール性溶液」に含まれるアルコールとしてポリエチレングリコールを記載している。これにより、閉環状DNAをコロイド状にし、コロイドの塩析により凝集させる。アルコール性溶液の添加によって閉環状DNAは不溶化するが、遠心操作を行わないので沈殿にはならず、上清に残ったままである。アルコール性溶液に含まれるアルコールとしては、ポリエチレングリコール、エタノール、イソプロピルアルコールなどを挙げることができる。アルコール性溶液は、アルコールの種類によっても異なるが、5〜100%の濃度、好ましくは10〜100%の濃度となるように水、もしくは塩化ナトリウム、酢酸ナトリウム、塩化リチウム、酢酸アンモニウムなどの塩が混合している水溶液などと混合したものであるとよい。アルコール性溶液に含まれるアルコールはポリエチレングリコールが好ましい。終濃度は1〜20%ポリエチレングリコールが好ましく、終濃度5.7%ポリエチレングリコールが最も好ましい。またアルコール性溶液に高濃度の塩化ナトリウムを加えることにより、さらに凝集作用は促進される。終濃度0.01〜2Mの塩化ナトリウムが好ましく、終濃度0.71Mの塩化ナトリウムが最も好ましい。
【0021】
アルコール性溶液を添加後、(g)の工程において、閉環状DNA凝集物を含んだ上清だけを取り出し、フィルターを通して限外ろ過を行う(図13の7))。ここで閉環状DNAの凝集塊はフィルターの網にひっかかる。このフィルターとしては、孔径が0.01〜10μmのものが適当であり、直径0.05〜5μmの限外ろ過フィルターが好ましい。例えばMillipore社のMultiScreenFB(1μm)を用いることができる。その後、フィルターを洗浄液で洗浄した後、フィルターに残った閉環状DNAを溶出液で溶出するとよい。
洗浄液は、エタノール、イソプロピルアルコールなどを10〜100%の濃度、好ましくは40〜90%の濃度となるようにバッファー、もしくは水などに溶解したものであるとよい。バッファーはいかなるものでもよいが、例えばTEバッファー(10mM Tris-HCl, 1mM EDTA (pH 8))に塩化ナトリウム、酢酸ナトリウム、塩化リチウム、酢酸アンモニウムなどの塩を加えたものであるとよい。塩は0.01〜10Mの濃度、好ましくは0.1〜8Mの濃度となるように溶解させたものであるとよい。
溶出液は、フィルターに残った閉環状DNAを溶出することができるものであればいかなるものであってもよいが、DNAの分解を補助する可能性のあるMgイオンをキレートするEDTAが入っているバッファーが好ましい。例えばTEバッファー(10mM Tris-HCl, 1mM EDTA (pH 8))、TBEバッファー(85mM Tris, 85mM ホウ酸, 2mM EDTA)、TAEバッファー(Tris 40mM, 酢酸 40mM, 1mM EDTA)などを挙げることができる。
【0022】
上記の(a)から(d)の工程は細胞を沈殿させるための工程であるが、本発明の別の実施態様においては、遠心分離機を用いて細胞の沈殿を作らせることにより、より簡略化した工程にすることができ、さらに閉環状DNAの収量を増加することができる。
具体的には、本発明の別の実施態様において、細胞から閉環状DNAを調製する方法であって、以下の工程:
(a’)培養した細胞を遠心分離により沈殿させ、上清を除く工程;
(b’)沈殿した細胞をバッファーもしくは培地で縣濁させ、細胞懸濁液を調製する工程;
(c’)該細胞懸濁液に少なくとも1種類のアルコール及び/又はケトンを添加する工程;
(d’)該細胞懸濁液に凝集補助剤及び凝集剤を添加する工程;
(e’)該細胞懸濁液に細胞溶解液を添加する工程;
(f’)工程(e’)で生じた混合液に振動を与える工程;
(g’)工程(f’)で振動を与えた混合液にアルコール性溶液を添加する工程;及び
(h’)工程(g’)でアルコール性溶液を添加した混合液の上清を限外ろ過フィルターに通して閉環状DNAを回収する工程
を含んでなる方法が提供される。
この実施態様においては、先に説明した実施態様で(c)の工程における細胞が沈殿するまでの待機時間がなくなり、かつ上清除去の操作が必要ない。
細胞を培地により培養する際、0.1mL〜10Lの培地に細胞を培養し、最も細胞数が増えた状態で上記(a’)〜(h’)の工程を行うことが好ましい。この際、細胞が大腸菌であれば、培地は終濃度0.1〜5%のTrypton、終濃度0.05〜2.5%のYeast Extract、終濃度0〜2.5%のNaCl、終濃度0〜30%のGlycerolの組成からなる培地が好ましく、現在世界で一般的に使用されているLB培地(1% Trypton、0.5% Yeast Extract、1% NaCl)が好ましい。2×LB培地(2% Trypton、1% Yeast Extract、1% NaCl)もしくはLB+7% Glycerol培地(1% Trypton、0.5% Yeast Extract、1% NaCl、7% Glycerol)はLBよりも培養後の菌体密度が高くなり、そのためDNAの収量も多くなり、最も好ましい。
(a’)の遠心分離の工程は培養した細胞が沈殿になれば良く、いかなる遠心分離機を用いても良い。例えば、検体数が少ない場合には高速の遠心機を用いることができ、20000×gで10秒間遠心すれば良い。検体数が多く、培養プレートを用いる場合は低速のプレート遠心機を用い、1000×gで10分間遠心すれば良い。
(b’)の工程におけるバッファーもしくは培地の液量は細胞の沈殿量に依存する。細胞の沈殿量はおもに培養液の量に依存するので、培養液量の1/2〜1/40量のバッファーもしくは培地で縣濁すれば良い。また、縣濁はピペットで縣濁しても良いし、ボルテックスなどで振動を与えて縣濁してもよい。ボルテックスで振動を与える場合は、1500〜2000rpm程度の振動で5秒から10分間振動を与え続ければ良い。
さらに、(b’)の工程で細胞培養液を新たなバッファーもしくは培地に換えることで、培養中に発生したDNA調製阻害物質を取り除くことができ、閉環状DNAの収量が多くなる。縣濁用バッファーはどのようなものでも構わないが、1〜100mM Tris-HCl (pH7.5〜9)が好ましく、10mM Tris-HCl (pH8.5)が最も好ましい。また、縣濁用に無機的なバッファーを用いるよりも培地を用いた方が最終的なDNAの調整量が多い。これは培地中に含まれるタンパク質が閉環状DNAの担体として働いているからだと思われる。培地はどのようなものでも構わないが、LB培地(1% Trypton, 0.5% Yeast Extract, 1% NaCl)や2×LB培地(2% Trypton, 1% Yeast Extract, 1% NaCl)が好ましい。(b’)の工程で用いられるバッファーもしくは培地は高張液であるとよい。バッファーもしくは培地が高張液であると、細胞から水分が奪われ、細胞が縮小する。これにより、後続のアルコール接触処理に伴う細胞破壊時の細胞質の細胞外への流出が極力抑えられる。さらに、高張液が塩の溶液であれば、(c’)の工程におけるアルコール及び/又はケトン添加時の細胞同士の凝集作用を高める働きがある。高張液は、安価という点で塩化ナトリウム水溶液が好ましい。終濃度0.1〜2.5Mの塩化ナトリウムが好ましく、終濃度1Mの塩化ナトリウム水溶液が最も好ましい。
(c’)と(d’)の工程(図21の1)及び2))で用いられるアルコール及び/又はケトン、凝集補助剤、及び凝集剤は、(a)と(b)の工程(図13の1)及び2))で用いられる薬品と同様であるが、凝集補助剤として用いられる炭酸カルシウムの最適濃度は終濃度58.8mg/mlであり、凝集剤として用いられるポリ塩化アルミニウムの最適濃度は終濃度114μg/mlである。また、実験室で一般的に用いられている1.5mlチューブ、もしくは2ml培養プレートでDNAの調製を行う際、遠心機を用いない調製法では500μl以上の細胞培養液を用いることが難しかったが、遠心分離機を用いることにより、1〜1.5mlの細胞培養液からDNAを調製することが可能であり、閉環状DNAの収量が多くなる。
(e’)、(f’)、(g’)及び(h’)の工程(図21の3)、4)、5)及び6))で用いられる細胞溶解液、振動、アルコール性溶液及び限外ろ過フィルターは、(c)、(d)、(e)、(f)及び(g)の工程(図13の4)、5)、6)及び7))で用いられるものと同様である。また、フィルターを洗浄するための洗浄液、フィルターに残った閉環状DNAを溶出するための溶出液なども前述のものと同様である。
【0023】
本発明は細胞を培養した少量の培地(0.1〜1ml)に限らず、1〜10Lの大量の培養液からも閉環状DNAを単離することができる。1ml〜1Lの培養液からも単離可能であることは言うまでもない。
【0024】
本発明は閉環状DNAがコスミドDNA、P1 DNA、BAC DNA、PAC DNAまたはYAC DNAである場合も単離することができる。
【0025】
本発明は細胞が形質転換体であるときも、該形質転換体から閉環状DNAを調製することができる。細胞が大腸菌である形質転換体であるときも同様に閉環状DNAを調製することができることは言うまでもない。また、酵母、枯草菌などの形質転換体、SV40ウィルスの複製開始点を有するプラスミドで形質転換されたCOS7細胞、293T細胞などの動物細胞形質転換体であるときも、これらの細胞から閉環状DNAを調製することができる。
【0026】
本発明は下記病原性細菌から閉環状DNAを単離することができる。
炭疽菌、赤痢菌、チフス菌、サルモネラ菌、コレラ菌、腸炎ビブリオ菌、カンピロバクター、黄色ブドウ球菌、ウエルシュ菌、セレウス菌、レンサ球菌、梅毒トレポネーマ、ライム病ボレリア、ピロリ菌、髄膜炎菌、淋菌、蛍光菌、鼻疽菌、類鼻疽菌、マルタ熱菌、バング菌、ブタ流産菌、百日咳菌、野兎病菌、チフス菌、パラチフス菌、ネズミチフス菌、肺炎桿菌、エンテロバクター・クロアカ、霊菌、ペスト菌、偽結核菌、インフルエンザ菌、軟性下疳菌、フラジリス菌、メラニノゲニカス菌、ビビウス菌、ジンジバーリス菌、発疹熱リケッチア、ロッキー山紅斑熱リケッチア、恙虫病リケッチア、塹壕熱ロシャリメア、Q熱コクシエラ、肺炎マイコプラズマ、表皮ブドウ球菌、化膿レンサ球菌、肺炎レンサ球菌、セレウス菌、枯草菌、破傷風菌、ボツリヌス菌、リステリア菌、ブタ丹毒菌、ジフテリア菌、ビフィズス菌、結核菌、ウシ型菌、癩菌、ロドコッカス・エクイ、ファイトプラズマ、シゲラ菌、大腸菌
【0027】
さらにその病原性細菌が大腸菌であれば、下記大腸菌から閉環状DNAを単離することができる。
ベロ毒素産生性大腸菌(VTEC)、毒素原性大腸菌(ETEC)、組織進入性大腸菌(EIEC)、病原血清型大腸菌(EPEC)、腸管凝集付着性大腸菌(EAggEC)、K12株
【0028】
本発明は下記薬剤耐性菌から閉環状DNAを単離することができる。
分裂酵母、出芽酵母、ビブリオ属菌、バンコマイシン耐性腸球菌、黄色ブドウ球菌炭疽菌、赤痢菌、チフス菌、サルモネラ菌、コレラ菌、腸炎ビブリオ菌、カンピロバクター、黄色ブドウ球菌、ウエルシュ菌、セレウス菌、レンサ球菌、梅毒トレポネーマ、ライム病ボレリア、ピロリ菌、髄膜炎菌、淋菌、蛍光菌、鼻疽菌、類鼻疽菌、マルタ熱菌、バング菌、ブタ流産菌、百日咳菌、野兎病菌、チフス菌、パラチフス菌、ネズミチフス菌、肺炎桿菌、エンテロバクター・クロアカ、霊菌、ペスト菌、偽結核菌、インフルエンザ菌、軟性下疳菌、フラジリス菌、メラニノゲニカス菌、ビビウス菌、ジンジバーリス菌、発疹熱リケッチア、ロッキー山紅斑熱リケッチア、恙虫病リケッチア、塹壕熱ロシャリメア、Q熱コクシエラ、肺炎マイコプラズマ、表皮ブドウ球菌、化膿レンサ球菌、肺炎レンサ球菌、セレウス菌、枯草菌、破傷風菌、ボツリヌス菌、リステリア菌、ブタ丹毒菌、ジフテリア菌、ビフィズス菌、結核菌、ウシ型菌、癩菌、ロドコッカス・エクイ、ファイトプラズマ、シゲラ菌、大腸菌
薬剤耐性菌が耐性を持つ薬剤としては、ペニシリン系、セフェム系、アミノグリコシド系、テトラサイクリン系、クロラムフェニコール系、リンコマイシン系、マクロライド系、コリスチン、ホスホマイシン、ポリミキシン、バンコマイシン、トリコマイシン、サイクロセリンなどを挙げることができる。
【0029】
また、本発明は、本発明の方法に使用するためのキットであって、以下の成分:
(a)少なくとも1種類のアルコール及び/又はケトンを含む溶液;
(b)凝集補助剤;
(c)凝集剤;
(d)細胞溶解液;
(e)アルコール性溶液;
(f)洗浄液;及び
(g)溶出液
を含むキットを提供する。
少なくとも1種類のアルコール及び/又はケトンを含む溶液は、エタノール、イソプロピルアルコール及びアセトンから1種類以上を選び、それを30〜100%の濃度、好ましくは50〜100%の濃度となるように水などと混合したものであるとよい。
凝集補助剤としては、炭酸カルシウム、粉末活性炭、活性珪酸、ベントナイトを挙げることができる。本発明のキットにおいて、凝集補助剤は、水などの溶媒に0.1〜70%の濃度、好ましくは5〜50%の濃度となるように懸濁させたものであるとよい。
凝集剤としては、ポリ塩化アルミニウム、硫酸アルミニウム、アルギン酸ナトリウム、キトサン、塩化第二鉄、ポリ硫酸第二鉄、ポリアクリルアミド系高分子凝集剤、ジメチルアミノエチルメタクリレート系高分子凝集剤、ジメチルアミニエチルアクリレート系高分子凝集剤、アミジン系高分子凝集剤、両性系高分子凝集剤を挙げることができる。本発明のキットにおいて、凝集剤は、水などの溶媒に0.001〜10%の濃度、好ましくは0.01〜10%の濃度となるように溶解させたものであるとよい。
細胞溶解液中に含まれる細胞膜を溶解する界面活性剤としてはTriton X-100、Briji、Nonidet P-40、Tween 20、Tween 80、CHAPS、SDS、NP-40、コール酸、n-octyl glycosideなどがあり、細胞壁溶解酵素としてはセルラーゼ、ヘミセルラーゼ、ペクチナーゼ、リゾチーム、β-1,3-グルカナーゼ、コラゲナーゼ、トリプシンなどがある。細胞溶解液中に含まれるRNaseはRNaseA、RNaseT、RNaseI、RNaseHがある。細胞溶解液は界面活性剤、細胞壁溶解酵素、RNaseをバッファーに溶解したものであるとよい。バッファーはいかなるものでもよいが、DNAの分解を補助する可能性のあるMgイオンをキレートするEDTAが入っていることが好ましい。例えばTEバッファー(10mM Tris-HCl, 1mM EDTA (pH 8))、TBEバッファー(85mM Tris, 85mM ホウ酸, 2mM EDTA)、TAEバッファー(Tris 40mM, 酢酸 40mM, 1mM EDTA)などを挙げることができる。界面活性剤は0.01〜10%の濃度、好ましくは0.1〜10%の濃度となるように溶解させたものであると良い。細胞壁溶解酵素は0.01〜10%の濃度、好ましくは0.1〜5%の濃度となるように溶解させたものであると良い。RNaseは0.0001〜1%の濃度、好ましくは0.001〜1%の濃度となるように界面活性剤や細胞壁溶解酵素とともにバッファーに溶解させたものか、もしくは0.001〜10%の濃度、好ましくは0.01〜5%の濃度となるように水などに溶解させたものを別途小瓶で添付する方法がよい。
アルコール性溶液に含まれるアルコールとしては、ポリエチレングリコール、エタノール、イソプロピルアルコールなどを挙げることができる。アルコール性溶液は、アルコールの種類によっても異なるが、5〜100%の濃度、好ましくは10〜100%の濃度となるように水、もしくは塩化ナトリウム、酢酸ナトリウム、塩化リチウム、酢酸アンモニウムなどの塩が混合している水溶液などと混合したものであるとよい。塩は0.01〜10Mの濃度、好ましくは0.1〜8Mの濃度となるように水に溶解させたものであるとよい。
洗浄液は、エタノール、イソプロピルアルコールなどを10〜100%の濃度、好ましくは40〜90%の濃度となるようにバッファー、もしくは水などに溶解したものであるとよい。バッファーはいかなるものでもよいが、例えばTEバッファー(10mM Tris-HCl, 1mM EDTA (pH 8))に塩化ナトリウム、酢酸ナトリウム、塩化リチウム、酢酸アンモニウムなどの塩を加えたものであるとよい。塩は0.01〜10Mの濃度、好ましくは0.1〜8Mの濃度となるように溶解させたものであるとよい。
溶出液は、フィルターに残った閉環状DNAを溶出することができるものであればいかなるものであってもよいが、DNAの分解を補助する可能性のあるMgイオンをキレートするEDTAが入っているバッファーが好ましい。例えばTEバッファー(10mM Tris-HCl, 1mM EDTA (pH 8))、TBEバッファー(85mM Tris, 85mM ホウ酸, 2mM EDTA)、TAEバッファー(Tris 40mM, 酢酸 40mM, 1mM EDTA)などを挙げることができる。
【0030】
本発明のキットは、さらに、チップ、チップ止め装置、培養プレートなどを含んでもよい。これらの要素を含むキットは、多検体を処理するのに好適である。
チップは、液体を吸引・排出することができるものであればいかなるものであってもよく、例えば、バイオギルソンイエローチップ(笛吹事務所)、アイビスピペットチップ(アズワン)、Universal Fit Tip(AXY)、Beveled Pipette Tip(SSI)、Multi Fit Tip(CLP)、スタンダードピペットチップ(総研バイオ)、アシストチップ(アシスト)などを用いることができる。
多サンプルの閉環状DNAを単離する際、上清と沈殿に分かれた混合液から上清だけをプレートから抜き取るのは難しい。そこで、図8のように、一定の深さ(ここでは沈殿物のすぐ上)までチップが達した際に、チップが一定以上奥に行かないようにチップを支えて止める機能を持った装置を利用するのが便利であるが、このチップを支えて止める機能を持った装置を「チップ止め装置」と言う。チップ止め装置はチップケース内のチップを固定する装置を加工することで得ることができるが、特別に作製することもできる。図8(a)は1つのウェルの場合について示してあるが、プレートで行う際にはこれが96個並んでいる(図8(b))。また、チップ止め装置、チップ、培養プレートは図の形状に限らない。
培養プレートは、細胞を培養可能なものであればいかなるものであってもよく、例えば、Coster (Corning Incorporated)、2ml Deep Well Plate (AXY)、2.2ml 96-well Deep Well Plate (Brand)、FalconTM 96-well Deep-Well Plate (BD Biosciences)、96-Well MasterblockTM Deep Well Plate (Bellco Glass)などを用いることができる。
【0031】
本キットにはさらにフィルタープレート、シリンジ型フィルター、溶出プレート、取り扱い説明書を含めてもよい。
フィルタープレートまたはシリンジ型フィルターはフィルターの孔径が0.01〜10μm、好ましくは0.05〜5μmのものであれば、フィルターの形状及び材質はいかなるものであってもよい。フィルタープレートまたはシリンジ型フィルターは特別に作製することができるが、市販のものを使うこともできる。フィルタープレートであれば、例えばMultiScreenFB(Millipore)、コンサート96フィルタープレート(インビトロジェン)、AcroWell GHPメンブレン使用フィルタープレート(日本ジェネティクス)、ユニフィルター350(Whatman)などを用いることができる。シリンジ型フィルターはシリンジとフィルターが別になったタイプと一体になったタイプがある。シリンジとフィルターが別になったタイプであれば、例えばMILLEXTM-HV(Millipore)、25mm GD/Xシリンジフィルター(Whatman)、ミニザルトN(Sartorius)などを用いることができる。また、一体になったタイプであれば、例えばQIAfilter Midi Cartridges(Qiagen)、PhoenIXTM Filter(Qbiogene)などを用いることができる。
溶出プレートは多検体からcccDNAを調製する際に用いるが、形状はいかなるものでもよい。市販品であれば、例えばポリスチレン製プレート(Whatman)、PS-Microplate 96well V-form(Griner bio-one)、マイクロウェルプレート(アズワン)などを用いることができる。
本キットの情報、プロトコル、使用上の注意などを書いた説明書をキット内に同梱することができる。
【0032】
さらに、本発明は、閉環状DNAを調製するための装置(以下、「閉環状DNA調製装置」ということもある。)であって、以下の装置:
(A)横移動装置;
(B)分注装置;
(C)上清吸引装置;
(D)振動装置;
(E)プレート吸引装置;及び
(F)ロボットアーム
を含む装置を提供する。
【0033】
本発明の閉環状DNA調製装置の一例の概略図を図1に示す。
以下の各工程に沿って、プレート(2)は横移動装置(20及び21)に乗って移動してゆく。プレートは、上述の培養プレート、例えば、Coster (Corning Incorporated)を用いるとよい。横移動装置は、モーター(図示せず)によって駆動される。横移動装置(20及び21)のベルト構造を図2及び図3に示す。図2において、プレートはプレートを載せる枠(101)上に配置される。さらに、押さえ部材(102)は水平方向の押しばねで内側方向にプレートを押す。これにより、プレートがプレートを載せる枠(101)上に配置されると、プレートを載せる枠(101)の中心部に固定される。各プレートを載せる枠(101)は連結部材(103)で円回転するように連結されている。図2では、横移動装置の下方に振盪機(5a)が配置されている(以下の工程3)、5)、7)、9)、13)、15)の状態を示す。)。図3では、横移動装置の上にプレート(2)が乗っている(以下の工程1)〜17)の状態を示す。プレートは押さえ部材(102)によりプレートを載せる枠(101)の中心方向に押されている。
1)大腸菌が入っているプレート(2)がプレートストッカー(1)(図1)から横移動装置(20)の上に乗せられる。ここで、ストッカーの構造について図17に記す。側面Xには下方にプレート2枚分の切れ込みがあり、その切れ込みの上部にプレートストッパー1(801)を備え、下部にはロボットアーム(803)を備える。側面Yには上方に切れ込みがあり、下方にはプレートストッパー2(802)を有する。次ぎにストッカーの動作について説明する。まずロボットアーム(803)が最下部にあるプレート(16−1)をつかむ。次ぎにプレートストッパー2(802)が開く(図17(b)の(1))。次ぎにロボットアーム(803)はつかんだプレート(16−1)を横移動装置に乗せる(図17(b)の(2))。この状態ではストッカーの最下部にプレート一枚分のスペースができる。次ぎにプレートストッパー2(802)が閉じる(図17(b)の(3))。この状態でプレートストッパー1(801)が開き、ストックされているプレート(16−2、16−3及び16−4)が下方に移動する。
2)大腸菌が入っているプレート(2)に、高張液分注機(3)により塩化ナトリウム水溶液が分注される。
3)その後プレート(2)は振盪機(5a−1)により振盪される。図5に示すように、振盪機(5a)は、横移動装置の下方から上に移動し、プレートを持ち上げ、上方から降りてくる固定支持体(5b)に支持された状態で振とうされる。この工程で固定支持体は5b−1で示されている。
4)アルコール・ケトン分注機(4)により、アルコール及びケトンから選ばれる一種類以上を含有する液体がプレート(2)に分注される。
5)その後プレート(2)は振盪機(5a−2)により振とうされる。図5に示すように、振盪機(5a)は、横移動装置の下方から上に移動し、プレートを持ち上げ、上方から降りてくる固定支持体(5b)に支持された状態で振とうされる。この工程で固定支持体は5b−2で示されている。
6)凝集補助剤分注機(6)により、炭酸カルシウムを含有する液体が、プレート(2)に分注される。炭酸カルシウムを含有する液体は静置すると炭酸カルシウムが沈殿し、分注機への正確な定量吸引ができない。そこで、図14のように、後述する分注機(図6)の溶液トレー(306)に図14(a)(b)の様に、連結部(310)を介して振動装置(311)を接続する。炭酸カルシウムを含有する溶液はこの振動する溶液トレーから定量吸引される。(吸引分注方法は後述する。)溶液トレーへの溶液の供給は図14(c)に示す機構により行う。水位センサー(301)が溶液の水位の低下を感知すると信号はポンプ制御装置(309)に送られ、ポンプ(304)が作動し、溶液ビン(305)から溶液供給パイプ(303)を通して溶液トレー(306)に供給される。ここで、炭酸カルシウムを含有する液体を貯蔵する溶液ビンは振動装置(312)により常に振動攪拌されている。溶液が溶液トレーの所定の水位に達するとセンサーからポンプ制御装置に信号が送られ、ポンプは停止する。このように構成することで、炭酸カルシウムを含有する液体の正確な分注が行われる。
7)その後プレート(2)は振盪機(5a−3)により振とうされる。図5に示すように、振盪機(5a)は、横移動装置の下方から上に移動し、プレートを持ち上げ、上方から降りてくる固定支持体(5b)に支持された状態で振とうされる。この工程で固定支持体は5b−3で示されている。
8)凝集剤分注機(7)により、ポリ塩化アルミニウムを含有する液体がプレート(2)に分注される。
9)その後プレート(2)は振盪機(5a−4)により振とうされる。図5に示すように、振盪機(5a)は、横移動装置の下方から上に移動し、プレートを持ち上げ、上方から降りてくる固定支持体(5b)に支持された状態で振とうされる。この工程で固定支持体は5b−4で示されている。
10)プレート(2)は一定時間静置される。ただし、連続してプレートを処理するため、振盪機(5a−4)と上清吸引排出機(8)との間に数プレート分の間隔があり、その間は横移動のみを行う。
11)上清吸引排出機(8)により、プレート(2)中の上清が吸引廃棄される。上清吸引排出機の機構は分注装置(図16)と同様である。ただし、分注装置が溶液トレーから溶液を吸引してプレートに排出するのに対して、上清吸引排出機はプレートから上清を吸引し、排出トレーに排出する。以下に上清吸引排出機の機序を示す。上清吸引排出機は横移動装置上のプレートに向かって上清の吸引に適切な位置まで降下し、上清の吸引を行う。この時、図8のように、一定の深さ(ここでは沈殿物のすぐ上)までチップが達した際に、チップが一定以上奥に行かないようにチップを支えて止める機能を持った「チップ止め装置」を利用するのが便利である。そのあと、再び元の高さまで上昇する。上清の排出は排出トレー(図19)に行われる。排出トレーは横移動装置の側面に設置されていて、上清排出時に上清吸引排出機の真下に移動する。上清排出後、排出トレーは再び側面に移動し、元の位置に戻る。排出された上清は排出トレーに繋がった捕集ビンに集められる。上清が排出されたあと、上清吸引排出機に付属するチップはチップ洗浄装置(後述)によって洗浄される。チップ洗浄装置は横移動装置の側面に設置されていて、チップ洗浄時に上清吸引排出機の真下に移動する。その後、上清吸引排出機が降下し、チップ洗浄装置により洗浄が行われる。洗浄後、上清吸引排出機は再び元の位置まで上昇し、チップ洗浄装置は元の位置に移動する。
12)細胞溶解液分注機(9)により、Triton X―100、Tris−HCl、EDTA、NaCl、リゾチーム、RNaseを含有する液体がプレート(2)に分注される。
13)その後プレート(2)は振盪機(5a−5)により断続的に振動が与えられる。この振動は500〜1700rpm程度の振動であらかじめ0.5秒間隔で0.5秒間の振動を合計10〜20回行われるようプログラムされている。図5に示すように、振盪機(5a)は、横移動装置の下方から上に移動し、プレートを持ち上げ、上方から降りてくる固定支持体(5b)に支持された状態で振動が与えられる。この工程で固定支持体は5b−5で示されている。
14)アルコール性溶液分注機(10)により、ポリエチレングリコールとNaClを含有する液体がプレート(2)に分注される。
15)その後プレート(2)は振盪機(5a−6)により振盪される。図5に示すように、振盪機(5a)は、横移動装置の下方から上に移動し、プレートを持ち上げ、上方から降りてくる固定支持体(5b)に支持された状態で振とうされる。この工程で固定支持体は5b−6で示されている。
16)プレート(2)は一定時間静置される。ただし、連続してプレートを処理するため、振盪機(5a−6)と上清吸引移動機(11)との間に数プレート分の間隔があり、その間は横移動のみを行う。
17)上清吸引移動機(11)により、プレート(2)中の上清が吸引され、フィルタープレート(12)に移し変えられる。フィルタープレートとしては、上述のもの、例えば、MultiScreenFB (Millipore)を用いるとよい。上清吸引移動機の機構は分注装置(図16)と同様である。ただし、分注装置が溶液トレーから溶液を吸引してプレートに排出するのに対して、上清吸引移動機はプレートから上清を吸引し、フィルタープレートに排出する。以下に上清吸引移動機の機序を示す。上清の吸引移動の際は、上清吸引移動機は上清の吸引に適した高さまで降下し、上清の吸引を行う。吸引後、上清吸引移動機は再び上昇し、その後レーンBのフィルタープレートの上方まで横方向に移動する。さらに、上清のフィルタープレートへの排出に適した高さまで降下し、上清をフィルタープレートに排出する。続いて再び上昇し、元の位置に横方向に移動し、続いてチップ洗浄装置(後述)により洗浄される。チップ洗浄装置は横移動装置の側面に設置されており、洗浄時には上清吸引移動機の真下に移動して出てくる。その後、上清吸引移動機は洗浄に適した位置まで降下し、チップ洗浄装置によりチップが洗浄される。洗浄後、上清吸引移動機は元の位置まで上昇し、チップ洗浄装置は元の位置に戻る。
ここで、以上の各工程を行った第1レーン(A)に平行してなる第2レーン(B)に上清が移動するように構成してもよい。
18)フィルタープレート(12)中の溶液はプレート吸引機(13−1)で吸引され、吸引して出てきた溶液は廃棄される。
プレート吸引機とその動作の一例を図4に示す。
図4に示すように、プレート吸引容器(201)は横移動装置の下方から上方へ横移動装置に密着するように上がってくる。プレート吸引容器には吸引チューブ(202a)接続されており、吸引チューブは捕集ビン(203)に接続されている。捕集ビンにはもう1本吸引チューブ(202b)が接続されており、該吸引チューブは吸引ポンプ(204)に接続されている。このような構造をとることにより、吸引ポンプを稼動されることにより、プレート(12)から液体を吸引し、捕集ビン(203)に集めることができる。
19)フィルタープレート(12)のフィルターは洗浄液分注機(14)から供給されるエタノールで洗浄される。洗浄の際、エタノールはプレート吸引機(13−2)で吸引される。
20)上記フィルターから、溶出液分注機(15)から供給される水またはTEバッファーでプラスミドが溶出される。洗浄の際、水またはTEバッファーは吸引溶出機(13−3)で吸引される。この工程の吸引溶出機を説明する。図15に示したように、吸引溶出機(13−3)は上面および下面が開放され、上面は横移動装置(21)を介して横移動装置上のフィルタープレート(12)に接するように、タイタープレート(16)と共にロボットアーム(602−1及び602−2)で配置される(図15(b)の(2))。タイタープレートとしては、上述の溶出プレート、例えば、ポリスチレン製プレート(Whatman)を用いるとよい。ここで、タイタープレートはレーンBの横に設置されたタイタープレート供給ストッカー(17)(図1)よりロボットアーム(図示せず)により1枚ずつ供給される。吸引溶出機は中空になっており、吸引チューブ(601)により内部の空気が吸引され、内部は陰圧になる。吸引溶出機には内部(タイタープレート密着用、603)と上部(横移動装置密着用、604)に空気もれを防ぐゴムが取り付けられている。これによりフィルタープレート(12)から水またはTEバッファーと共にプラスミドをタイタープレートに回収する。その後、吸引溶出機とタイタープレートはロボットアームにより図15の(b)(1)の状態に戻り、さらにタイタープレートだけ分離され、吸引済みタイタープレートストッカー(18)(図1)に回収される。このように、溶出されたプラスミドはタイタープレート(16)に回収される。ここでタイタープレートの供給、収納機構について説明する。図18はタイタープレートの供給・収納の機序を図示したものである。タイタープレートはタイタープレート供給ストッカー(17)にストックされている。タイタープレート供給ストッカーの機構は図17と同様である。タイタープレートはロボットアーム制御機(901)により一枚ずつ取り出され、回転することにより、吸引溶出機(13−3)の下方にまで運ばれる。次ぎに、タイタープレートは押し上げられ、吸引が行われる。吸引が済むと、タイタープレートは下方に移動し、ロボットアーム制御機が回転することにより、吸引済みタイタープレートストッカー(18)の上方まで運ばれ、その中に収納される。
21)プラスミドを入れたタイタイープレート(16)はプレートストッカー(18)内にロボットアーム(602−2)(図15)によりレーンBの横に設置した吸引済みタイタープレートストッカー(18)(図1)に1枚ずつ収納される。
本発明の閉環状DNA調製装置の別の一例の概略図を図20に示す。
図20は培養液を機械装置にかけるの工程の前段階として、遠心機を用いて培養液と細胞を手作業で分離し、沈殿した細胞から閉環状DNAを調製するための装置である。
アルコール及び/又はケトンを添加する工程の前段階として、あらかじめ遠心機を用いて培養液と細胞を分離しておくことで、図1の上清吸引排出機(8)の工程を除くことができる。さらに細胞が沈殿になるまでの待機時間が必要ないので、凝集剤分注機(7)で凝集剤を添加後、振盪機(5a−4)で混合し、その直後に細胞溶解液分注機(9)で細胞溶解液を添加することができる。また、遠心分離機を用いて細胞を沈殿にすることで、細胞の保存を容易にするという利点がある。つまり、体積の大きい培養液のまま冷凍する場合には時間がかかり、解凍する際にも時間がかかるので、その間に細胞が死滅する可能性が大きい。一方、細胞を沈殿にすると、体積が小さいので、冷凍と解凍が短時間ですみ、細胞の死滅を大幅に減少することができる。細胞の冷凍保存は特に多検体から閉環状DNAを調製する際にスムーズに作業を進行させるために重要な工程である。図20の高張液分注機(3)は図1の高張液分注機(3)と同様のものであるが、図20のそれは単に高張液を分注するだけでなく、縣濁用のバッファーもしくは培地と高張液の混合液である。さらに振盪機(5a−1)で振盪するが、この際、図20の振盪機(5a−1)は図1の振盪機(5a−1)よりも強い振動が必要である。つまり、図20の振盪機(5a−1)は単に溶液を混合するだけでなく、沈殿になった細胞を縣濁するという要素が入るからである。具体的には図1の振盪機(5a−1)は1000〜1700rpmの振動、図20の振盪機(5a−1)は1500〜2000rpmの振動が良い。また、図20の装置は洗浄液分注機(14)及びプレート吸引機(13−2)が図1と比べて一台ずつ追加されている。これは2度の洗浄工程を行う方が、より夾雑物の少ない閉環状DNAを得ることができるためである。
本装置を構成する各装置はすべてインターフェイスボードを介してコンピューターに接続され、一括管理される。具体的には、動作するすべての装置のセンサーから装置毎のインターフェースを介して動作完了又は動作未完了の信号がコンピューターに送られる。コンピューターはそれらの信号を一括管理し、各装置に対して次の動作指示を出す。つまり、どの装置からもエラーが発見されなければ次の動作の信号を、エラーが一つでも発見された場合は停止の信号を各装置に送信する。
本発明の閉環状DNA調製装置における(A)横移動装置に相当するのが、上記の横移動装置(20)及び(21)(図1及び20)であり、これは、ベルト、モーターなどを備えているとよい。(B)分注装置に相当するのが、上記の高張液分注機(3)、アルコール・ケトン分注機(4)、凝集補助剤振盪分注機(6)、凝集剤分注機(7)、細胞溶解液分注機(9)、アルコール性溶液分注機(10)、洗浄液分注機(14)及び溶出液分注機(15)(図1及び20)であり、これは、水位センサー、溶液供給パイプ、ポンプ、溶液ビン、溶液トレー、チップ、ポンプ制御装置などを備えているとよい。(C)上清吸引装置に相当するのが、図1の上清吸引排出機(8)と上清吸引移動機(11)、図20の上清吸引移動機(11)であり、これは、排出トレー、捕集ビン、チップ、チップ洗浄装置などを備えているとよい。(D)振動装置に相当するのが振盪機(5a−1〜5a−6)(図1及び20)及び振動装置(311、312)(図14)であり、これは、特別に作製することができるし、市販のもの(例えば、ボルテックスなど)を利用することもできる。(E)プレート吸引装置に相当するのが、上記のプレート吸引機(図1及び20の13−1〜13−3、図4、図15)であり、これは、吸引容器、チューブ、捕集ビン、ポンプ、タイタープレートなどを備えているとよい。
【0034】
上記の工程2)、4)、6)、8)、12)及び14)で液体を分注する機構の1例を図6に示す。
分注すべき液体(302)は溶液トレー(306)にためられる。ためられた溶液はチップ(307)により、定量吸引され、プレート(2)に分注される。
溶液トレー内の溶液が減ってくると水位センサー(301)により感知され、信号がポンプ制御装置(309)に送られる。ポンプ制御装置は溶液ビン(305)から溶液を吸い上げ、溶液トレー内に溶液供給パイプ(303)を通じて溶液を供給する。一定の溶液量が溶液トレーにたまるとその水位を水位センサーが感知し、ポンプ制御装置に信号を送る。それにより、ポンプ制御装置はポンプ(304)を停止させる。
図16は分注装置の概要を透視図にしたものである。分注装置は筺体(712)を有し、その内部にモーター(702)とギアで連結され、ねじ機構(703及び704)により上下運動するピストン(706)と、筺体に固定金具(711)で固定されたシリンダ(705)を有する。コンピューターから吸引の指示が出ると、モーターに電流が流れ、上部センサー(709)に感知されるまで、ピストンが上がり、分注すべき溶液を吸引する。分注機制御装置から分注の指示が出るとモーターに逆の電流が流れ、ピストンが下部センサー(710)に感知されるまで下がり、シリンダ内の溶液が分注口(707)から排出され、分注チューブ(708)を介してチップ(307)よりプレートに溶液が分注される。
【0035】
工程内でチップを洗浄する必要が場合には、チップ洗浄装置により洗浄される。チップ洗浄装置の1例を図7に記載する。
チップ洗浄槽(403)はRO(Revers Osmosis)水で満たされている(図7(a))。洗浄すべきチップ(401)(図6のチップ(307))の先端を洗浄筒(402)内に挿入する。洗浄筒の底面部にはRO水給水口がそれぞれ設けられており、チップが挿入されると給水が行われ、チップの先端が洗浄される。洗浄筒よりあふれたRO水は排水口より排水される(図7(b))。
本発明の閉環状DNA調製装置は、全自動で、外界から閉鎖された安全な空間で、病原性の細菌から閉環状DNAを単離し、閉環状DNA由来の病原性の有無を診断することができる。さらに、病原となる遺伝子が閉環状DNA由来であるか否かを判定する研究用にも用いることができる。
さらに病原性菌が大腸菌である場合、大腸菌を外界から閉鎖された安全な空間で、大腸菌から閉環状DNAを単離し、閉環状DNA由来の病原性の有無を診断することができる。さらに、病原となる遺伝子が閉環状DNA由来であるか否かを判定する研究用にも用いることができる。
本発明の閉環状DNA調製装置により、全自動で、外界から閉鎖された安全な空間で、病原性の細菌から閉環状DNAを単離し、閉環状DNA由来の薬剤耐性の有無を診断することが可能である。
【0036】
以下、本発明を実施例により具体的に説明する。
実施例1〜3として、遠心分離を一度も行わず、その代わりに凝集剤を使用して他の細胞構成成分からcccDNAを分離する方法について述べる。
実施例4及び5として、遠心分離操作と凝集剤の使用を組み合わせて、他の細胞構成成分からcccDNAを分離する方法について述べる。
本発明の実施形態がこれらに限られるものでないことはいうまでもない。
実施例で用いた器具及び大腸菌は以下の通りである。
チップ:バイオギルソンイエローチップ(笛吹事務所)
96穴培養プレート:Costar(Corning Incorporated)
フィルタープレート:MultiScreenFB (Millipore)
溶出プレート:PS-Microplate 96 well V-form (Griner)
実施例1、3及び4の大腸菌:2002年に60770個のマウス全長cDNAのクローニングと機能解析が終了(The FANTOM Consortium and The RIKEN Genome Exploration Research Group Phase I & II Team, Analysis of the mouse transcriptome based on functional annotation of 60,770 full-length cDNA, Nature, 420, 563-573, 2002)したもののうち、その1個の遺伝子を用いて実験を行った。60770個のcDNAはC57BL/6J系統のマウスから、キャップトラップ法(Carninci, P. et al. Normalization and subtraction of cap-trapper-selected cDNAs to prepare full-length cDNA libraries for rapid discoverry of new genes. Genome Res. 10, 1617-1630, 2000)などを用いて単離された。単離された遺伝子はクローニング(ベクターにcDNAを組み込み、大腸菌に遺伝子導入)後に大腸菌が培養され、プラスミド精製が行われたあと、全塩基配列が決定され、機能解析が行われた。本実験には、pBluescript KS(+)(Stratagene)というベクターに組み込まれ、大腸菌株DH10Bに遺伝子導入された遺伝子(クローンID:9930023M19、大きさ:3255bp)を用いた。その塩基配列やアノテーションの情報はウェブページで公開されている(http://fantom2.gsc.riken.go.jp/)。
実施例2の大腸菌:2002年に60770個のマウス全長cDNAのクローニングと機能解析が終了(The FANTOM Consortium and The RIKEN Genome Exploration Research Group Phase I & II Team, Analysis of the mouse transcriptome based on functional annotation of 60,770 full-length cDNA, Nature, 420, 563-573, 2002)したもののうち、その中の5個の遺伝子を用いて実験を行った。60770個のcDNAはC57BL/6J系統のマウスから、キャップトラップ法(Carninci, P. et al. Normalization and subtraction of cap-trapper-selected cDNAs to prepare full-length cDNA libraries for rapid discoverry of new genes. Genome Res. 10, 1617-1630, 2000)などを用いて単離された。単離された遺伝子はクローニング(ベクターにcDNAを組み込み、大腸菌に遺伝子導入)後に大腸菌が培養され、プラスミド精製が行われたあと、全塩基配列が決定され、機能解析が行われた。本実験には、pBluescript KS(+)(Stratagene)というベクターに組み込まれ、大腸菌株DH10Bに遺伝子導入された遺伝子(クローンID:9930033A02、9930023M19、9930039L23、9930022D09、A130007D10)を用いた。その塩基配列やアノテーションの情報はウェブページで公開されている(http://fantom2.gsc.riken.go.jp/)。
【0037】
[実施例1]
1.5mlチューブを用いた少検体大腸菌培養液からのプラスミド調製
大腸菌DH10B株にあらかじめ組み込んでおいたpBluescript KS(+)のプラスミドDNAの単離を行った。このプラスミドDNAの中にはマウスの遺伝子DNA (クローンID:9930023M19、大きさ:3255bp)が組み込まれている。
材料:
LB培地500μl
5M 塩化ナトリウム 100μl
100% イソプロパノール 300μl
500mg/ml 炭酸カルシウム 40μl
4mg/ml ポリ塩化アルミニウム 20μl
2% Triton X-100, 4mM Tris-HCl (pH8), 0.4mM EDTA, 20mM NaCl, 5mg/ml リゾチーム, 67μg/ml RNaseA混合液 150μl
20% ポリエチレングリコール, 2.5M NaCl混合液120μl
70% エタノール 300μl
TE 100μl
MultiScreenFB (Millipore)
【0038】
手順
1)アンピシリンを含むLB培地に単一コロニーから取った大腸菌を接種し、37℃で12〜20時間インキュベートした。培養物の少なくとも4倍の体積を有する培養チューブ、もしくはフラスコを使用した。培養の際にはエアレーションが行われるようにし、約30度傾け、激しく振盪(〜300rpm)培養した。
2)約500μlの培養液を取り、1.5mlチューブに移した。
3)100μlの5M塩化ナトリウムを加え、混合した。
4)300μlの100%イソプロパノールを加え、混合した。
5)40μlの500mg/mlの炭酸カルシウムを加え、混合した。
6)20μlの4mg/mlのポリ塩化アルミニウムを加え、混合した。
7)チューブを垂直に立て、8分静置した。
8)沈殿を取らないように気をつけながら上清だけをピペットマンを使用し取り除く。わずかな上清を含む沈殿はこのとき約150μl残った。
9)この沈殿に、150μlの2%Triton X−100、4mM Tris−HCl(pH8)、0.4mM EDTA、20mM NaCl、5mg/ml リゾチーム、 67μg/ml RNaseAの混合液を加えた。
10)ただちに弱い断続的な振動、すなわち0.5秒振動→0.5秒静止、を10回繰り返した。(8)で生じた沈殿が(9)で加えた溶液と完全に混合される程度の振動(〜1700rpm)を要するが、あまり強い振動を与えると沈殿が分解して澄んだ上清が得られないので注意を要する。弱い断続的な振動を与えた後、上清が澄んでいることを確認する。
11)120μlの20%ポリエチレングリコール、2.5M NaClを加え、混合した。
12)90秒静置した。
13)限外ろ過が可能なフィルター(Millipore社のMultiScreenFB)に(12)で生じた上清を載せ、吸引ろ過を行った。
14)70%エタノールを添加し、吸引ろ過を行った。
15)100μlのTEでプラスミドDNAをフィルターから溶出させ、回収した。
【0039】
単離したプラスミドDNAの電気泳動
実施例1で単離したプラスミドDNAのアガロースゲル電気泳動を行った。
1%の濃度のAgarose-1 (和光純薬工業株式会社)を作成し、TAEバッファー(Tris 40mM, 酢酸 40mM, 1mM EDTA)を充填した電気泳動漕にセットした。実施例1記載の(15)で単離したプラスミドDNA 100μlのうち4μlを泳動した。Qiagen Mini Prep Kitで単離したプラスミドDNAも同様に溶出した100μlのうち4μlを泳動した。また、マーカーDNAとしてGeneRulerTM 1Kb DNA Ladder (MBI fermentas社)を用いた。その結果を図9に示す。マーカーは合計200ngになるように泳動した。電気泳動を行った結果、RNAは目視ではまったく観察されず、ゲノムDNAの夾雑もほとんど観察されなかった。また泳動マーカーと比較したところ、500μlの菌液から単離されるプラスミドDNAの量は約2.5μgであることが分かった。
【0040】
単離したプラスミドDNAを用いたPCR
実施例1で単離したプラスミドDNAに組み込んだマウスの遺伝子DNA (クローンID:9930023M19、大きさ:3255bp)をPCRで増幅した。増幅用のプライマーDNAとして1st-BS (TGTAAAACGACGGCCAGT)(配列番号1) と2nd-NX/X (TCCTGTGTGAAATTGTTATCCGCT)(配列番号2)を用いた。増幅用の反応バッファーは以下の通りである。
1)10×Buffer 2.5μl
2)25mM MgSO4 1μl
3)2mM dNTP 2.5μl
4)5pmol/μl プライマーDNA 各0.5μl
5)2ng/μlの単離したプラスミドDNA 1μl
6)KOD plus 0.75μl
7)蒸留水 16.25μl
また、反応温度と時間は以下の通り
1)94℃ 2分
2)94℃ 1分
3)60℃ 30秒
4)68℃ 2分
(2)〜(4)を30回繰り返す
PCRの結果を図10に示す。この図が示す通り、目的DNAの十分な量の増幅が見られ、本手法で単離したプラスミドDNAがPCRに使用できることが分かった。
【0041】
単離したプラスミドDNAのトランスフォーメーション
単離したプラスミドDNAのトランスフォーメーションを行った。大腸菌DH10B株のコンピテント細胞を用い、プラスミドDNAを2ng/μlに希釈したあと、以下の操作を行った。
1)10μlのコンピテント細胞を-80℃の冷凍庫から取り出し、氷上で溶かした
2)2ng/μlのプラスミドDNA 1μlをコンピテント細胞の中に入れ、緩やかに混合した
3)混合した液を40℃で45秒間インキュベートした
4)再び氷上に置き、抗生物質の入らないLB培地を1ml混合した
5)37℃で1時間インキュベートした
6)インキュベートした液から20μl取ってアンピシリン入りのプレート培地に塗った
7)37℃で一晩培養した
この結果、9個の大腸菌コロニーができた。このことから、本手法で単離したプラスミドDNAがトランスフォーメーションに使用できることが分かった。
【0042】
[実施例2]
96穴プレートを使った多検体の大腸菌からのプラスミド調製
大腸菌DH10B株にあらかじめ組み込んでおいたpBluescript KS(+)のプラスミドDNAの単離を行った。全ての検体はそれぞれ異なったマウスの遺伝子(クローンID:9930033A02、9930023M19、9930039L23、9930022D09、A130007D10)が入ったプラスミドを持つ。
材料:
LB培地 各500μl
5M 塩化ナトリウム 各100μl
100% イソプロパノール 各300μl
500mg/ml 炭酸カルシウム 各40μl
4mg/ml ポリ塩化アルミニウム 各20μl
2% Triton X-100, 4mM Tris-HCl (pH8), 0.4mM EDTA, 40mM NaCl, 5mg/ml リゾチーム, 67μg/ml RNaseA混合液 各150μl
20% ポリエチレングリコール, 2.5M NaCl混合液 各120μl
70% エタノール 各300μl
TE 各100μl
MultiScreenFB (Millipore)
【0043】
手順
1)それぞれのウェルにアンピシリンを含む500μlのLB培地に単一コロニーから取った大腸菌を接種し、37℃で12〜20時間インキュベートした。培養には96穴培養プレート(Costar, Corning Incorporated)を用い、エアレーションが行われるようにし、約30度傾け、激しく振盪(〜300rpm)培養した。
2)100μlの5M塩化ナトリウムを各ウェルに加え、混合した。
3)300μlの100%イソプロパノールを各ウェルに加え、混合した。
4)40μlの500mg/mlの炭酸カルシウムを各ウェルに加え、混合した。
5)20μlの4mg/mlのポリ塩化アルミニウムを各ウェルに加え、混合した。
6)8分静置した。
7)沈殿を取らないように気をつけながら上清だけを専用のチップ止め装置を用いて取り除く。わずかな上清を含む沈殿はこのとき約150μl残った。
9)この沈殿に、150μlの2%Triton X―100、4mM Tris−HCl(pH8)、0.4mM EDTA、20mM NaCl、5mg/ml リゾチーム、 67μg/ml RNaseAの混合液を各ウェルに加えた。
10)ただちに弱い断続的な振動、すなわち0.5秒振動→0.5秒静止、を10回繰り返した。8で生じた沈殿が9で加えた溶液と完全に混合される程度の振動(〜1700rpm)を要するが、あまり強い振動を与えると沈殿が分解して澄んだ上清が得られないので注意を要する。弱い断続的な振動を与えた後、上清が澄んでいることを確認する。
11)120μlの20%ポリエチレングリコール、2.5M NaClを各ウェルに加え、混合した。
12)90秒静置した。
13)限外ろ過が可能なフィルター(Millipore社のMultiScreenFB等)に(12)で生じた上清を専用のチップ止め装置を用いて載せ、吸引ろ過を行った。
14)70%エタノールを添加し、吸引ろ過を行った。
15)100μlのTEでプラスミドDNAをフィルターから溶出させ、回収した。
【0044】
単離したプラスミドDNAの電気泳動
実施例2で単離したいくつかのプラスミドDNAのアガロースゲル電気泳動を行った。
1%の濃度のAgarose-1 (和光純薬工業株式会社)を作成し、TAEバッファー(Tris 40mM, 酢酸 40mM, 1mM EDTA)を充填した電気泳動漕にセットした。実施例2記載の(15)で単離した各プラスミドDNA 100μlのうち4μlを泳動した。また、マーカーDNAとしてGeneRulerTM 1Kb DNA Ladder (MBI fermentas社)を用いた。その結果を図11に示す。マーカーは合計200ngになるように泳動した。図中の1〜5はそれぞれ別のマウス遺伝子が入ったプラスミドDNAである。図11の左から、クローンID:9930033A02、9930023M19、9930039L23、9930022D09、A130007D10。
【0045】
[実施例3]
培養チューブを用いた大腸菌培養液からのプラスミドの大量調製
大腸菌DH10B株にあらかじめ組み込んでおいたpBluescript KS(+)のプラスミドDNAの単離を行った。プラスミドDNAの中にはマウスの遺伝子DNA (クローンID:9930023M19、大きさ:3255bp)が組み込まれている。
材料:
LB培地 20ml
5M 塩化ナトリウム 4ml
100% イソプロパノール 12ml
500mg/ml 炭酸カルシウム 1.6ml
4mg/ml ポリ塩化アルミニウム 0.8ml
2% Triton X-100, 4mM Tris-HCl (pH8), 0.4mM EDTA, 20mM NaCl, 5mg/ml リゾチーム, 67μg/ml RNaseA混合液 5ml
20% ポリエチレングリコール, 2.5M NaCl混合液 4ml
70% エタノール 5ml
TE 1ml
MillexR-HV (Millipore)
【0046】
手順
1)アンピシリンを含む20mlのLB培地に単一コロニーから取った大腸菌を接種し、37℃で20時間インキュベートした。培養には50mlの培養チューブ(Falcon, Becton Dickinson)を用い、エアレーションが行われるようにし、約30度傾け、激しく振盪(〜300rpm)培養した。
2)4mlの5M塩化ナトリウムを加え、混合した。
3)12mlの100%イソプロパノールを加え、混合した。
4)1.6mlの500mg/mlの炭酸カルシウムを加え、混合した。
5)0.8mlの4mg/mlのポリ塩化アルミニウムを加え、混合した。
6)15分静置した。
7)沈殿を取らないように気をつけながら上清だけをピペットマン、もしくはピペットで除いた。ここでわずかな上清を含んだ沈殿は約5mlになった。
9)この沈殿に、5mlの2%Triton X−100、4mM Tris−HCl(pH8)、0.4mM EDTA、20mM NaCl、5mg/ml リゾチーム、 67μg/ml RNaseの混合液を加えた。
10)500μlの6M 炭酸カリウム(pH6)を加えた。
11)チューブの蓋を閉め、10回反転させた。
12)4mlの20%ポリエチレングリコール、2.5M NaClを加え、チューブを反転させた。
13)90秒静置した。ここで上清は約15ml取れた。
14)(13)で得られた上清をシリンジで3ml吸い取り、Millipore社のMillexR-HVを用いて限外ろ過を行った。
15)70%エタノールをシリンジで5ml吸い取り、フィルターを洗浄した。
16)800μlのTEでプラスミドDNAをフィルターから溶出させ、回収した。
17)上記14〜16の操作を5回繰り返し、合計4mlの溶出液を得た。
【0047】
単離したプラスミドDNAの電気泳動
実施例3で単離したプラスミドDNAのアガロースゲル電気泳動を行った。
1%の濃度のAgarose-1 (和光純薬工業株式会社)を作成し、TAEバッファー(Tris 40mM, 酢酸 40mM, 1mM EDTA)を充填した電気泳動漕にセットした。実施例1で単離したプラスミドDNA 100μlから4μl泳動したものと、実施例3記載の(17)で単離したプラスミドDNA 4mlのうち4μlを泳動したものを比較した。実施例1は0.5mlの菌液から100μl溶出し、実施例3では20mlの菌液から4ml溶出しているので、溶出の比率は同じになる。マーカーDNAとしてGeneRulerTM 1Kb DNA Ladder (MBI fermentas社)を用いた。その結果を図12に示す。マーカーは合計200ngになるように泳動した。大量に単離した場合も少量単離した場合と同様に、RNAは目視ではまったく観察されず、ゲノムDNAの夾雑もほとんど観察されなかった。また泳動マーカーと比較したところ、20mlの菌液から単離されるプラスミドDNAの量は約100μgであることが分かった。
【0048】
[実施例4]
遠心機を用いた少量検体大腸菌培養液からのプラスミド調製法(1.5mlチューブを用いた調製)
大腸菌DH10B株にあらかじめ組み込んでおいたpBluescript KS(+)のプラスミドDNAの単離を行った。このプラスミドDNAの中にはマウスの遺伝子DNA (クローンID:9930023M19、大きさ:3255bp)が組み込まれている。
材料:
培養用LB培地1ml
縣濁用LB培地80μl
5M 塩化ナトリウム 20μl
100% イソプロパノール 50μl
500mg/ml 炭酸カルシウム 20μl
4mg/ml ポリ塩化アルミニウム 5μl
2% Triton X-100, 4mM Tris-HCl (pH8), 0.4mM EDTA, 20mM NaCl, 5mg/ml リゾチーム, 67μg/ml RNaseA混合液 150μl
20% ポリエチレングリコール, 2.5M NaCl混合液120μl
70% エタノール 600μl
TE 100μl
MultiScreenFB (Millipore)
【0049】
手順
1)アンピシリンを含むLB培地に単一コロニーから取った大腸菌を接種し、37℃で約16時間インキュベートした。培養物の少なくとも4倍の体積を有する培養チューブ、もしくはフラスコを使用した。培養の際にはエアレーションが行われるようにし、約30度傾け、激しく振盪(〜300rpm)培養した。
2)1mlの培養液を取り、1.5mlチューブに移した。
3)遠心機を用い、20000×gで10秒間遠心し、菌体を沈殿にした。
4)上清をデカンテーションして除去した。残液はピペットを用いて完全に除去した。
5)80μlのLB培地と20μlの5M塩化ナトリウムよく混合し、その全量を1.5mlチューブに加え、沈殿になった菌をピペットを用いてよく縣濁した。
6)50μlの100%イソプロパノールを加え、混合した。
7)20μlの500mg/mlの炭酸カルシウムを加え、混合した。
8)5μlの4mg/mlのポリ塩化アルミニウムを加え、混合した。
9)この沈殿に、150μlの2% Triton X-100、4mM Tris-HCl (pH 8)、0.4mM EDTA、20mM NaCl、5mg/ml リゾチーム、67μg/ml RNaseAの混合液を加えた。
10)ただちに弱い断続的な振動(〜1700rpm)で、0.5秒振動→0.5秒静止、を10回繰り返した。その後、上下に5回転倒混和した。あまり強い振動を与えると沈殿が分解して澄んだ上清が得られないので注意を要する。
11)120μlの20%ポリエチレングリコール、2.5M NaClを加え、混合した。
12)90秒静置した。
13)限外ろ過が可能なフィルター(Millipore社のMultiScreenFB)に(12)で生じた上清を300ulのせ、吸引ろ過を行った。
14)70%エタノールを300μl添加し、吸引ろ過を行った。
15)再度70%エタノールを300μl添加し、吸引ろ過を行った。
16)100μlのTEでプラスミドDNAをフィルターから溶出させ、回収した。
【0050】
単離したプラスミドDNAの電気泳動
実施例4で単離したプラスミドDNAのアガロースゲル電気泳動を行った。
1%の濃度のAgarose-1 (和光純薬工業株式会社)を作成し、TAEバッファー(Tris 40mM, 酢酸 40mM, 1mM EDTA)を充填した電気泳動漕にセットした。実施例4記載の(16)で単離したプラスミドDNA 100μlのうち4μlを泳動した。比較として、実施例1記載の(16)で単離したプラスミドDNAも同時に泳動した。また、マーカーDNAとしてGeneRulerTM 1Kb DNA Ladder (MBI fermentas社)を用いた。その結果を図22に示す。マーカーDNAは合計200ngになるように泳動した。電気泳動を行った結果、RNAは目視ではまったく観察されず、ゲノムDNAの夾雑もほとんど観察されなかった。また泳動マーカーと比較したところ、1mlの菌液から単離されるプラスミドDNAの量は約8μgであることが分かった。
【産業上の利用可能性】
【0051】
本発明により、経費が低く品質の良い閉環状DNAを短時間で大量に精製でき、閉環状DNAを使用する研究ないし医療に大きく貢献し得る。
【図面の簡単な説明】
【0052】
【図1】本発明の閉環状DNA調製装置の1例の概略図である。
【図2】横移動装置の機構および振盪機の位置を表したものである。
【図3】横移動装置にプレートをセットしたものである。
【図4】プレート吸引機及びその動作を表したものである。
【図5】振盪機及び固定支持体の動きを表したものである。
【図6】溶液分注機を表したものである。(a)は各要素の構成を示す。(b)は溶液トレーから分注溶液が吸引され、プレートに分注される動作を模式的に示す。
【図7】チップ洗浄装置を表したものである。(a)は斜視図、(b)はA-Aの断面図である。
【図8】チップ止め装置を表したものである。(a)は(b)に示すA-Aの横断面図、(b)は斜視図である。
【図9】実施例1で単離したプラスミドDNAの電気泳動の結果を示す。M:マーカーDNA、1:Qiagen Mini Prep Kitで単離したプラスミドDNA、2:本発明の方法で単離したプラスミドDNA。
【図10】実施例1で単離したプラスミドDNAに組み込んだマウスの遺伝子DNAをPCRで増幅した結果を示す。M:マーカーDNA、1:Qiagen Mini Prep Kitで単離したプラスミドDNAに組み込んだマウスの遺伝子DNAをPCRで増幅したもの、2:本発明の方法で単離したプラスミドDNAに組み込んだマウスの遺伝子DNAをPCRで増幅したもの。
【図11】実施例2で単離したプラスミドDNAの電気泳動の結果を示す。M:マーカーDNA、1〜5:本発明の方法で単離したプラスミドDNA。
【図12】実施例1及び3で単離したプラスミドDNAの電気泳動の結果を示す。M:マーカーDNA、1:実施例1において本発明の方法で単離したプラスミドDNA、2:実施例3において本発明の方法で単離したプラスミドDNA。
【図13】本発明の閉環状DNA調製方法の1例のスキームを表したものである。
【図14】凝集補助剤分注機を表したものである。
【図15】吸引溶出機を表したものである。
【図16】分注装置を表したものである。
【図17】プレートストッカーの構造を表したものである。
【図18】タイタープレートの供給・収納機構を表したものである。
【図19】排出トレーを表したものである。
【図20】本発明の閉環状DNA調製装置の別の1例の概略図である。
【図21】本発明の閉環状DNA調製方法の別の1例のスキームを表したものである。
【図22】実施例1及び4で単離したプラスミドDNAの電気泳動の結果を示す。M:マーカーDNA、1:実施例1で単離したプラスミドDNA、2:実施例4で単離したプラスミドDNA。
【符号の説明】
【0053】
1:プレートストッカー
2:プレート
3:高張液分注機
4:アルコール・ケトン分注機
5a、5a−1、5a−2、5a−3、5a−4、5a−5、5a−6:振盪機
5b、5b−1、5b−2、5b−3、5b−4、5b−5、5b−6:固定支持体
6:凝集補助剤振盪分注機
7:凝集剤分注機
8:上清吸引排出機
9:細胞溶解液分注機
10:アルコール性溶液分注機
11:上清吸引移動機
12:フィルタープレート
13−1、13−2:プレート吸引機
13−3:吸引溶出機
14:洗浄液分注機
15:溶出液分注機
16:タイタープレート
16−1:プレート
16−2:プレート
16−3:プレート
16−4:プレート
17:タイタープレート供給ストッカー
18:吸引済みタイタープレートストッカー
20、21:横移動装置
A:第1レーン
B:第2レーン
101:枠
102:押さえ部材
103:連結部材
201:吸引容器
202a、202b:チューブ
203:捕集ビン
204:ポンプ
301:水位センサー
302:分注溶液
303:溶液供給パイプ
304:ポンプ
305:溶液ビン
306:溶液トレー
307:チップ
309:ポンプ制御装置
310:連結部
311:振動装置
312:振動装置
401:チップ
402:洗浄筒
403:チップ洗浄槽
501:チップ止め装置
502:チップ
503:上清
504:沈殿
601:吸引チューブ
602−1:ロボットアーム
602−2:ロボットアーム
603:密閉用ゴム
604:密閉用ゴム
701:モーター配線
702:モーター
703:ネジ機構
704:ネジ機構
705:シリンダー
706:ピストン
707:分注口
708:分注チューブ
709:センサ
710:センサ
711:固定金具
712:筺体
801:プレートストッパー1
802:プレートストッパー2
803:ロボットアーム
804:筺体
901:ロボットアーム制御機
【配列表フリーテキスト】
【0054】
<配列番号1>
配列番号1は、実施例1で増幅用のプライマーDNAとして用いた1st-BSの塩基配列を示す。
<配列番号2>
配列番号2は、実施例1で増幅用のプライマーDNAとして用いた2nd-NX/Xの塩基配列を示す。
【技術分野】
【0001】
本発明は細胞から閉環状DNAを簡便に調製する方法、キット及び装置に関する。
【背景技術】
【0002】
ファージやプラスミドなどの環状二本鎖DNAは、通常、負の超らせん閉環状の構造をとっており、このような構造を持つ環状DNAはcccDNA(covalently closed circular DNA)と呼ばれている。
従来はcccDNAの分離にかかる時間が少なく、コストが低く、単離されたcccDNAの品質がよく、収量が高いという条件をすべて満たす単離法は存在しなかった。さらに、cccDNAを調製する方法としてフェノール/クロロホルム法があるが、有害な有機溶剤を使用しなければならなかった。
【0003】
公知の方法であるアルカリSDS法を主とする少検体からcccDNAを分離するキットはQiagen社のQiagen Spin Miniprep Kitが有名である。アルカリSDS法では溶液を一時的に強アルカリにする操作がある。しかし、DNAはアルカリに弱く、ニックが入る可能性が高くなり、最終調製物は環状DNAではなく、直鎖状DNAの比率が高くなる。また、作業に費やす時間が長く、さらに価格が高くなるという欠点を持つ。2004年時点で50検体用のキットは11,000円(1検体あたり220円)である。
【0004】
陰イオン交換樹脂を用いた分離法はフナコシ社のMidiprep Kit, PhoenIXがあり、2004年時点で、25検体で34000円(1検体あたり1360円)と非常に価格が高く、また作業時間も長くなるという欠点がある。
【0005】
アルカリSDS法を主とする多検体用のキットにはR.E.A.L. Prep 96Plasmid Kitがあるが、cccDNAにニックが入る可能性が高く、さらに価格も高くなるという欠点を持つ。
【0006】
また、多検体のcccDNAを一度に単離する場合、プレートの遠心分離が必須な工程であり、そのことが全自動化するにあたっての障害となっていた。すなわち、プレートの遠心分離を行って全自動化する試みは、例えば特開2001-333763号公報(特許文献1)に見られる。しかし、(1)プレート遠心機は遠心力が弱く、ペレット化したプラスミドの分離が充分に行えない(2)遠心分離機にプレートをセットする際、あるいは遠心後に遠心機からプレートを取り出す際に、ロボットアームおよび遠心機の回転を高度に制御しなければならず、実質的に製作が困難であるという問題があった。
【0007】
さらに、全自動化する試みは特開平10-155481号公報(特許文献2)にも見られる。この方法はカオトロピックイオン法を利用し、フィルター上の担体にDNAを吸着させてDNAを単離する方法である。0.6mlの大腸菌培養液から最大で4〜6μgのプラスミドDNAが得られるとされているが、実際には0.5μg程度のプラスミドDNAしか得られず、さらにゲノムDNAの夾雑も多い。
【特許文献1】特開2001-333763号公報
【特許文献2】特開平10-155481号公報
【発明の開示】
【発明が解決しようとする課題】
【0008】
本発明は、経費が低く、短時間で、ニックの少ない高品質のプラスミドをより少ない工数で無害な物質を用いて細胞から単離する方法を提供することを目的とする。
さらに、本発明は、多数のサンプルを一度に単離する全自動プラスミド調製機を提供することを目的とする。
【課題を解決するための手段】
【0009】
本発明者らは、凝集剤を使用することによって、凝集作用の違いにより、閉環状DNA(例えば、プラスミド)を他の細胞構成物、特にゲノムDNAやタンパク質から分離することに成功し、本発明を完成させるに至った。
本発明の要旨は以下の通りである。
(1) 細胞から閉環状DNAを調製する方法であって、細胞を含む液体に凝集剤を添加する工程を含んでなる方法。
(2) 以下の工程:
(a)該細胞を含む液体に少なくとも1種類のアルコール及び/又はケトンを添加する工程;
(b)該細胞を含む液体に凝集補助剤及び凝集剤を添加して、上清と沈殿に分離する工程;
(c)工程(b)で生じた上清と沈殿から上清を除く工程;
(d)工程(c)で上清を除いたあとの沈殿に細胞溶解液を添加する工程;
(e)工程(d)で生じた混合液に振動を与える工程;
(f)工程(e)で振動を与えた混合液にアルコール性溶液を添加する工程;及び
(g)工程(f)でアルコール性溶液を添加した混合液の上清を限外ろ過フィルターに通して閉環状DNAを回収する工程
を含んでなる(1)記載の方法。
(3) 遠心分離を行う工程を含まない(2)記載の方法。
(4) 工程(a)の前工程として、高張液を細胞に接触させる(2)又は(3)に記載の方法。
(5) 以下の工程:
(a’)培養した細胞を遠心分離により沈殿させ、上清を除く工程;
(b’)沈殿した細胞をバッファーもしくは培地で縣濁させ、細胞懸濁液を調製する工程;
(c’)該細胞懸濁液に少なくとも1種類のアルコール及び/又はケトンを添加する工程;
(d’)該細胞懸濁液に凝集補助剤及び凝集剤を添加する工程;
(e’)該細胞懸濁液に細胞溶解液を添加する工程;
(f’)工程(e’)で生じた混合液に振動を与える工程;
(g’)工程(f’)で振動を与えた混合液にアルコール性溶液を添加する工程;及び
(h’)工程(g’)でアルコール性溶液を添加した混合液の上清を限外ろ過フィルターに通して閉環状DNAを回収する工程
を含んでなる(1)記載の方法。
(6) 工程(b’)のバッファーもしくは培地が高張液である(5)記載の方法。
(7) 細胞が単細胞生物である(1)〜(6)のいずれかに記載の方法。
(8) 単細胞生物が細菌である(7)記載の方法。
(9) 細菌が大腸菌である(8)記載の方法。
(10) 細胞が形質転換体である(1)〜(9)のいずれかに記載の方法。
(11) 閉環状DNAが、プラスミドDNA、コスミドDNA、P1 DNA、BAC DNA、PAC DNA及びYAC DNAからなる群より選択される(1)〜(10)のいずれかに記載の方法。
(12) (1)〜(11)のいずれかに記載の方法に使用するためのキットであって、以下の成分:
(a)少なくとも1種類のアルコール及び/又はケトンを含む溶液;
(b)凝集補助剤;
(c)凝集剤;
(d)細胞溶解液;
(e)アルコール性溶液;
(f)洗浄液;及び
(g)溶出液
を含むキット。
(13) さらに、以下の要素:
(h)チップ;
(i)チップ止め装置;及び
(j)培養プレート
を含む(12)記載のキット。
(14) 閉環状DNAを調製するための装置であって、以下の装置:
(A)横移動装置;
(B)分注装置;
(C)上清吸引装置;
(D)振動装置;
(E)プレート吸引装置;及び
(F)ロボットアーム
を含む装置。
(15) 凝集剤を使用して、閉環状DNAを他の細胞構成物から分離する方法。
【0010】
本明細書で「単細胞生物」とは、大部分の生活史を単一の細胞から成る生物を言い、原核生物の大部分(細菌・藍色細菌・古細菌)及び真核生物の一部(原生生物・鞭毛藻類・珪藻類・緑藻類や紅藻類の一部・真菌類の一部)がこれに該当する。
本明細書で「閉環状DNA」とは、DNAの3’末端及び5’末端が共有結合によって自己連結された超らせん構造を持つ環状DNAを言う。DNA鎖の一部が巻き戻されるとワトソン―クリック回転が減り、その分、ねじれが生じて超らせんが形成される。ほとんどの閉環状DNAは負の超らせん構造をとっている。
本明細書で「凝集剤」とは、水の濁りや色を除去するために用いられる薬剤で、水中に懸濁する微粒子をくっつけてフロック(集塊)をつくらせ、沈降、濾過によって分離除去するのを容易にする作用があるものを言う。多くの微粒子はマイナスに帯電しており、プラスに帯電した凝集剤を入れることで微粒子同士を架橋し、そのフロックを大きくすることで沈降速度を上げる。
本明細書で「凝集補助剤」とは、凝集剤による凝集作用を高める働きのある物質のことで、水中に懸濁することで微粒子のフロックを作りやすくする作用がある。
本明細書で「細胞溶解液」とは、細胞を溶解する作用を持つ物質を含む液体を言う。
本明細書で「アルコール性溶液」とは、アルコールを含む液体を言う。
本明細書で「高張液」とは、対象とする細胞よりも高張な溶液をいう。
本明細書で「チップ」とは、ピペットマン等の先に取り付ける交換可能な器具(通常、プラスチック製)を言う。溶液の吸引や排出の際に用いる。
本明細書において、特に断りのない限り、百分率(%)は重量%を示すものとする。
【発明の効果】
【0011】
本発明により、経費が低く、短時間で、高品質の閉環状DNAをより少ない工数で無害な物質を用いて細胞から単離することができる。特に、コストが従前の約10分の1以下になることは大きな利点である。
例えば、2004年時点でQiagen社のQiagen Spin Miniprep Kitでは50検体で11,000円(1検体220円)また、R.E.A.L. Prep 96Plasmid kit(96検体同時調製)では4プレートで75,000円(1検体195円)であるのに対し、本発明では1検体18円である。
さらに、多数のサンプルから閉環状DNAを全自動で単離することができるようになった。
【発明を実施するための最良の形態】
【0012】
以下、本発明を詳細に説明する。
本発明は、細胞から閉環状DNAを調製する方法であって、細胞を含む液体に凝集剤を添加する工程を含んでなる方法を提供する。
細胞は、いかなる細胞であってもよく、細菌、古細菌などの原核細胞、酵母などの菌類細胞、原生生物、植物細胞、動物細胞などを挙げることができる。
閉環状DNAは、DNAの3’末端及び5’末端が共有結合によって自己連結された超らせん構造を持つ環状DNAであれば、いかなるものであってもよく、プラスミドDNA、コスミドDNA、P1 DNA、BAC DNA、PAC DNAまたはYAC DNAなどを挙げることができる。
細胞を含む液体は、細胞を含有するいかなる液体であってもよく、細胞の培養液、培養細胞を遠心分離で沈殿させた後にバッファーもしくは培地に懸濁させた液などを挙げることができる。
本発明の1実施態様において、本発明の方法は、以下の工程:
(a)該細胞を含む液体に少なくとも1種類のアルコール及び/又はケトンを添加する工程;
(b)該細胞を含む液体に凝集補助剤及び凝集剤を添加して、上清と沈殿に分離する工程;
(c)工程(b)で生じた上清と沈殿から上清を除く工程;
(d)工程(c)で上清を除いたあとの沈殿に細胞溶解液を添加する工程;
(e)工程(d)で生じた混合液に振動を与える工程;
(f)工程(e)で振動を与えた混合液にアルコール性溶液を添加する工程;及び
(g)工程(f)でアルコール性溶液を添加した混合液の上清を限外ろ過フィルターに通して閉環状DNAを回収する工程
を含んでなる。
本発明の方法により、遠心分離を用いることなく、実質的にゲノムDNAを含まない閉環状DNAを得ることができるが、本発明の方法は遠心分離工程を完全に排除するものではなく、本発明の方法を遠心分離と組み合わせて用いてもよい。
ここで、「実質的にゲノムDNAを含まない」とは、最終調製物中に含まれるゲノムDNAの量が、cccDNAが目視できる量を電気泳動する場合にほとんど目視できない量であることを言う。例えば、図9のレーン2は本手法で単離した最終調整物を電気泳動したもので、このレーンには2つのバンドが目視できる。上の薄いバンドは直鎖状プラスミドDNA、下の濃いバンドが閉環状プラスミドDNAであり、ゲノムDNAは目視できない。ゲノムDNAが含まれている場合、ゲノムDNAはプラスミドDNAよりもサイズが大きいので、直鎖状のプラスミドDNAのバンドの上部にバンドが現れる。
【0013】
細胞を液体培地により培養する際、0.1mL〜10Lの培地に細胞を培養し、最も細胞数が増えた状態で上記(a)〜(g)の工程を行うことが好ましい。この際、細胞が大腸菌であれば培地はLB培地(1% Trypton、0.5% Yeast Extract、1% NaCl)、2×LB培地(2% Trypton、1% Yeast Extract、1% NaCl)もしくはLB+7% Glycerol培地(1% Trypton、0.5% Yeast Extract、1% NaCl、7% Glycerol)が好ましい。特に、LB+7% Glycerol培地で培養すると、菌体数が多くなり、DNAの収量がLB培地や2×LB培地よりも多くなる。
【0014】
(a)〜(g)の工程を行う前に、高張液を細胞に接触させる(例えば、細胞よりも高張な溶液を培地に添加する)ことが好ましい。これにより細胞から水分を奪い、細胞を縮小させる。この工程は、後続のアルコール接触処理に伴う細胞破壊時の細胞質の細胞外への流出を極力抑える働きを持つ。さらに、高張液が塩の溶液であれば、(a)の工程におけるアルコールもしくはケトン添加時の細胞同士の凝集作用を高める働きがある。高張液は、安価という点で塩化ナトリウム水溶液が好ましい。終濃度0.1〜2.5Mの塩化ナトリウムが好ましく、終濃度0.83Mの塩化ナトリウム水溶液が最も好ましい。
【0015】
(a)の工程において、少なくとも1種類のアルコール及び/又はケトンを添加する(図13の1))。図13の1)では、「少なくとも1種類のアルコール及び/又はケトン」としてイソプロパノールを、「細胞」として大腸菌を記載している。培養した細胞に直接アルコール及び/又はケトンを添加することにより、細胞表面は脱水され、細胞同士の親水基が結びつき、大きな凝集塊を形成する(コロイドの塩析作用)。例えば、大腸菌は微弱ながらマイナスに帯電しており、それらが凝集することによって強くマイナスに帯電した凝集塊となる。また、アルコール及び/又はケトンの添加によって、細胞に穴が開き、穴からアルコール及び/又はケトンが進入する。それによって細胞内のタンパク質は変性し、凝集する。さらに、一部のタンパク質は変性時にゲノムDNAを巻き込み、固定する。閉環状DNA(例えば、プラスミドDNA)はアルコール及び/又はケトンによって脱水され、ゲノムDNAなどの細胞内物質と結合、凝集し、細胞外へ解離しない。この処理にはアルコールであればエタノールあるいはイソプロピルアルコール、ケトンであればアセトンから一種類以上選ばれることが好ましい。特にイソプロピルアルコールは少量の液量で処理できる点で最も好ましい。また、終濃度は1〜80%イソプロピルアルコールが好ましく、終濃度33.3%のイソプロピルアルコールが最も好ましい。
【0016】
(b)の工程において、凝集補助剤と凝集剤を添加する(図13の2))。凝集剤としては、ポリ塩化アルミニウム、硫酸アルミニウム、アルギン酸ナトリウム、キトサン、塩化第二鉄、ポリ硫酸第二鉄、ポリアクリルアミド系高分子凝集剤、ジメチルアミノエチルメタクリレート系高分子凝集剤、ジメチルアミノエチルアクリレート系高分子凝集剤、アミジン系高分子凝集剤、両性系高分子凝集剤を挙げることができる。凝集補助剤としては、炭酸カルシウム、粉末活性炭、活性珪酸、ベントナイトを挙げることができる。図13の2)では、「凝集補助剤」として炭酸カルシウムを、「凝集剤」としてポリ塩化アルミニウムを記載している。凝集補助剤としては粒径が大きく、水よりも比重が大きく、マイナスに帯電した不溶性物質を用いるとよい。凝集補助剤としては炭酸カルシウム(CaCO3)が好ましい。終濃度0.1〜100mg/ml炭酸カルシウムが好ましく、終濃度21.3mg/ml炭酸カルシウムが最も好ましい。炭酸カルシウムは蒸留水中で自然に沈殿になり、分離操作に扱いづらいので、炭酸カルシウムの沈降速度を遅らせるために粘度の高いグリセロール(終濃度1〜50%)を蒸留水中に加えても良い。
【0017】
さらに、(b)の工程において、マイナスに帯電した凝集補助剤と工程(a)によりマイナスに帯電した細胞(図13では、大腸菌)の凝集塊をプラスに帯電した凝集剤を用いて架橋し、凝集補助剤の重みによって共沈させる。凝集剤は終濃度3〜1000μg/mlポリ塩化アルミニウムが好ましく、終濃度83.3μg/mlポリ塩化アルミニウムが最も好ましい。沈殿が形成されるまで4〜8分待てば良いが、時間を置けば置くほど沈殿量は多くなり、数日置くことも可能である。凝集補助剤と凝集剤を添加する順序は、特に限定されることはないが、凝集補助剤が先に添付されることが好ましい。
次いで、(c)の工程において、(b)の工程で生じた上清と沈殿から上清を除く(図13の3))。
【0018】
(c)の工程で上清を除いたあとの沈殿に水分を含有する液体を加えると、水中のアルコール及び/又はケトン濃度が減少し、溶解度の高い閉環状DNAは溶解する。タンパク質は一度変性すると元に戻りにくいため、水溶液を加えた後もいくつかのタンパク質は溶解したゲノムDNAをしっかりと保持し続ける。また、水分を含有する液体として細胞溶解液を加える(工程(d)、図13の4))ことによって細胞を形成する細胞膜と細胞壁を溶かして穴を広げ、閉環状DNAを細胞外へ放出しやすくする。細胞溶解液は、細胞膜を溶解する界面活性剤、細胞溶解酵素などの成分をバッファーに溶解したものであるとよい。細胞膜を溶解する界面活性剤としてはTriton X-100、Briji、Nonidet P-40、Tween 20、Tween 80、CHAPS、SDS、NP-40、コール酸、n-octyl glycosideなどがあり、細胞溶解酵素としてはセルラーゼ、ヘミセルラーゼ、ペクチナーゼ、リゾチーム、β-1,3-グルカナーゼ、コラゲナーゼ、トリプシンなどがある。バッファーはいかなるものでもよいが、DNAの分解を補助する可能性のあるMgイオンをキレートするEDTAが入っていることが好ましい。例えばTEバッファー(10mM Tris-HCl, 1mM EDTA (pH 8))、TBEバッファー(85mM Tris, 85mM ホウ酸, 2mM EDTA)、TAEバッファー(Tris 40mM, 酢酸 40mM, 1mM EDTA)などを挙げることができる。細胞が大腸菌である場合、細胞溶解液はTriton X-100とリゾチームの混合液が好ましい。Triton X-100とリゾチームの濃度は以下の通りである。終濃度0.01〜20%のTriton X-100が好ましく、終濃度1%のTriton X-100が最も好ましい。終濃度0.01〜40mg/mlのリゾチームが好ましく、終濃度2.5mg/mlのリゾチームが最も好ましい。さらに、RNAを分解するためにRNaseを同時に加えることが望ましい。ここでRNaseはRNaseA、RNaseT、RNaseI、RNaseHを含むが、これらに限定されるものではない。RNaseがRNaseAである場合、終濃度0.1〜200μg/ml RNaseAが好ましく、終濃度34μg/ml RNaseAが最も好ましい。
【0019】
細胞膜と細胞壁が溶かされることによって、タンパク質、タンパク質―ゲノムDNA結合体、その他の細胞構成物も細胞内から分離する可能性がある。しかし、これらの不溶性細胞構成物は主としてマイナスに帯電しており、プラスに帯電した凝集剤に引き寄せられ、大きな凝集塊を作る。一方、可溶化した閉環状DNAは、凝集剤で架橋されることなく上清に溶け、沈殿と分離される。さらに、工程(e)において、振動を与える(好ましくは、弱い振動を断続的に与える(図13の5))ことで、物質同士のイオン結合による凝集作用を強化し、閉環状DNAとの分離効率を飛躍的に高める。つまり、本発明の最大の特徴は、凝集作用の違いによって閉環状DNAを他の細胞構成物、特にゲノムDNAやタンパク質から分離することである。工程(e)の振動は、500〜1700rpm程度の弱い振動を0.1〜3秒間隔で0.1〜3秒間2〜20回与える振動が好ましく、0.5秒間隔で0.5秒間10回与える振動が最も好ましい。
【0020】
(f)の工程において、上記(e)の工程で生成された閉環状DNAの溶解した上清にアルコール性溶液を添加する(図13の6))。図13の6)では、「アルコール性溶液」に含まれるアルコールとしてポリエチレングリコールを記載している。これにより、閉環状DNAをコロイド状にし、コロイドの塩析により凝集させる。アルコール性溶液の添加によって閉環状DNAは不溶化するが、遠心操作を行わないので沈殿にはならず、上清に残ったままである。アルコール性溶液に含まれるアルコールとしては、ポリエチレングリコール、エタノール、イソプロピルアルコールなどを挙げることができる。アルコール性溶液は、アルコールの種類によっても異なるが、5〜100%の濃度、好ましくは10〜100%の濃度となるように水、もしくは塩化ナトリウム、酢酸ナトリウム、塩化リチウム、酢酸アンモニウムなどの塩が混合している水溶液などと混合したものであるとよい。アルコール性溶液に含まれるアルコールはポリエチレングリコールが好ましい。終濃度は1〜20%ポリエチレングリコールが好ましく、終濃度5.7%ポリエチレングリコールが最も好ましい。またアルコール性溶液に高濃度の塩化ナトリウムを加えることにより、さらに凝集作用は促進される。終濃度0.01〜2Mの塩化ナトリウムが好ましく、終濃度0.71Mの塩化ナトリウムが最も好ましい。
【0021】
アルコール性溶液を添加後、(g)の工程において、閉環状DNA凝集物を含んだ上清だけを取り出し、フィルターを通して限外ろ過を行う(図13の7))。ここで閉環状DNAの凝集塊はフィルターの網にひっかかる。このフィルターとしては、孔径が0.01〜10μmのものが適当であり、直径0.05〜5μmの限外ろ過フィルターが好ましい。例えばMillipore社のMultiScreenFB(1μm)を用いることができる。その後、フィルターを洗浄液で洗浄した後、フィルターに残った閉環状DNAを溶出液で溶出するとよい。
洗浄液は、エタノール、イソプロピルアルコールなどを10〜100%の濃度、好ましくは40〜90%の濃度となるようにバッファー、もしくは水などに溶解したものであるとよい。バッファーはいかなるものでもよいが、例えばTEバッファー(10mM Tris-HCl, 1mM EDTA (pH 8))に塩化ナトリウム、酢酸ナトリウム、塩化リチウム、酢酸アンモニウムなどの塩を加えたものであるとよい。塩は0.01〜10Mの濃度、好ましくは0.1〜8Mの濃度となるように溶解させたものであるとよい。
溶出液は、フィルターに残った閉環状DNAを溶出することができるものであればいかなるものであってもよいが、DNAの分解を補助する可能性のあるMgイオンをキレートするEDTAが入っているバッファーが好ましい。例えばTEバッファー(10mM Tris-HCl, 1mM EDTA (pH 8))、TBEバッファー(85mM Tris, 85mM ホウ酸, 2mM EDTA)、TAEバッファー(Tris 40mM, 酢酸 40mM, 1mM EDTA)などを挙げることができる。
【0022】
上記の(a)から(d)の工程は細胞を沈殿させるための工程であるが、本発明の別の実施態様においては、遠心分離機を用いて細胞の沈殿を作らせることにより、より簡略化した工程にすることができ、さらに閉環状DNAの収量を増加することができる。
具体的には、本発明の別の実施態様において、細胞から閉環状DNAを調製する方法であって、以下の工程:
(a’)培養した細胞を遠心分離により沈殿させ、上清を除く工程;
(b’)沈殿した細胞をバッファーもしくは培地で縣濁させ、細胞懸濁液を調製する工程;
(c’)該細胞懸濁液に少なくとも1種類のアルコール及び/又はケトンを添加する工程;
(d’)該細胞懸濁液に凝集補助剤及び凝集剤を添加する工程;
(e’)該細胞懸濁液に細胞溶解液を添加する工程;
(f’)工程(e’)で生じた混合液に振動を与える工程;
(g’)工程(f’)で振動を与えた混合液にアルコール性溶液を添加する工程;及び
(h’)工程(g’)でアルコール性溶液を添加した混合液の上清を限外ろ過フィルターに通して閉環状DNAを回収する工程
を含んでなる方法が提供される。
この実施態様においては、先に説明した実施態様で(c)の工程における細胞が沈殿するまでの待機時間がなくなり、かつ上清除去の操作が必要ない。
細胞を培地により培養する際、0.1mL〜10Lの培地に細胞を培養し、最も細胞数が増えた状態で上記(a’)〜(h’)の工程を行うことが好ましい。この際、細胞が大腸菌であれば、培地は終濃度0.1〜5%のTrypton、終濃度0.05〜2.5%のYeast Extract、終濃度0〜2.5%のNaCl、終濃度0〜30%のGlycerolの組成からなる培地が好ましく、現在世界で一般的に使用されているLB培地(1% Trypton、0.5% Yeast Extract、1% NaCl)が好ましい。2×LB培地(2% Trypton、1% Yeast Extract、1% NaCl)もしくはLB+7% Glycerol培地(1% Trypton、0.5% Yeast Extract、1% NaCl、7% Glycerol)はLBよりも培養後の菌体密度が高くなり、そのためDNAの収量も多くなり、最も好ましい。
(a’)の遠心分離の工程は培養した細胞が沈殿になれば良く、いかなる遠心分離機を用いても良い。例えば、検体数が少ない場合には高速の遠心機を用いることができ、20000×gで10秒間遠心すれば良い。検体数が多く、培養プレートを用いる場合は低速のプレート遠心機を用い、1000×gで10分間遠心すれば良い。
(b’)の工程におけるバッファーもしくは培地の液量は細胞の沈殿量に依存する。細胞の沈殿量はおもに培養液の量に依存するので、培養液量の1/2〜1/40量のバッファーもしくは培地で縣濁すれば良い。また、縣濁はピペットで縣濁しても良いし、ボルテックスなどで振動を与えて縣濁してもよい。ボルテックスで振動を与える場合は、1500〜2000rpm程度の振動で5秒から10分間振動を与え続ければ良い。
さらに、(b’)の工程で細胞培養液を新たなバッファーもしくは培地に換えることで、培養中に発生したDNA調製阻害物質を取り除くことができ、閉環状DNAの収量が多くなる。縣濁用バッファーはどのようなものでも構わないが、1〜100mM Tris-HCl (pH7.5〜9)が好ましく、10mM Tris-HCl (pH8.5)が最も好ましい。また、縣濁用に無機的なバッファーを用いるよりも培地を用いた方が最終的なDNAの調整量が多い。これは培地中に含まれるタンパク質が閉環状DNAの担体として働いているからだと思われる。培地はどのようなものでも構わないが、LB培地(1% Trypton, 0.5% Yeast Extract, 1% NaCl)や2×LB培地(2% Trypton, 1% Yeast Extract, 1% NaCl)が好ましい。(b’)の工程で用いられるバッファーもしくは培地は高張液であるとよい。バッファーもしくは培地が高張液であると、細胞から水分が奪われ、細胞が縮小する。これにより、後続のアルコール接触処理に伴う細胞破壊時の細胞質の細胞外への流出が極力抑えられる。さらに、高張液が塩の溶液であれば、(c’)の工程におけるアルコール及び/又はケトン添加時の細胞同士の凝集作用を高める働きがある。高張液は、安価という点で塩化ナトリウム水溶液が好ましい。終濃度0.1〜2.5Mの塩化ナトリウムが好ましく、終濃度1Mの塩化ナトリウム水溶液が最も好ましい。
(c’)と(d’)の工程(図21の1)及び2))で用いられるアルコール及び/又はケトン、凝集補助剤、及び凝集剤は、(a)と(b)の工程(図13の1)及び2))で用いられる薬品と同様であるが、凝集補助剤として用いられる炭酸カルシウムの最適濃度は終濃度58.8mg/mlであり、凝集剤として用いられるポリ塩化アルミニウムの最適濃度は終濃度114μg/mlである。また、実験室で一般的に用いられている1.5mlチューブ、もしくは2ml培養プレートでDNAの調製を行う際、遠心機を用いない調製法では500μl以上の細胞培養液を用いることが難しかったが、遠心分離機を用いることにより、1〜1.5mlの細胞培養液からDNAを調製することが可能であり、閉環状DNAの収量が多くなる。
(e’)、(f’)、(g’)及び(h’)の工程(図21の3)、4)、5)及び6))で用いられる細胞溶解液、振動、アルコール性溶液及び限外ろ過フィルターは、(c)、(d)、(e)、(f)及び(g)の工程(図13の4)、5)、6)及び7))で用いられるものと同様である。また、フィルターを洗浄するための洗浄液、フィルターに残った閉環状DNAを溶出するための溶出液なども前述のものと同様である。
【0023】
本発明は細胞を培養した少量の培地(0.1〜1ml)に限らず、1〜10Lの大量の培養液からも閉環状DNAを単離することができる。1ml〜1Lの培養液からも単離可能であることは言うまでもない。
【0024】
本発明は閉環状DNAがコスミドDNA、P1 DNA、BAC DNA、PAC DNAまたはYAC DNAである場合も単離することができる。
【0025】
本発明は細胞が形質転換体であるときも、該形質転換体から閉環状DNAを調製することができる。細胞が大腸菌である形質転換体であるときも同様に閉環状DNAを調製することができることは言うまでもない。また、酵母、枯草菌などの形質転換体、SV40ウィルスの複製開始点を有するプラスミドで形質転換されたCOS7細胞、293T細胞などの動物細胞形質転換体であるときも、これらの細胞から閉環状DNAを調製することができる。
【0026】
本発明は下記病原性細菌から閉環状DNAを単離することができる。
炭疽菌、赤痢菌、チフス菌、サルモネラ菌、コレラ菌、腸炎ビブリオ菌、カンピロバクター、黄色ブドウ球菌、ウエルシュ菌、セレウス菌、レンサ球菌、梅毒トレポネーマ、ライム病ボレリア、ピロリ菌、髄膜炎菌、淋菌、蛍光菌、鼻疽菌、類鼻疽菌、マルタ熱菌、バング菌、ブタ流産菌、百日咳菌、野兎病菌、チフス菌、パラチフス菌、ネズミチフス菌、肺炎桿菌、エンテロバクター・クロアカ、霊菌、ペスト菌、偽結核菌、インフルエンザ菌、軟性下疳菌、フラジリス菌、メラニノゲニカス菌、ビビウス菌、ジンジバーリス菌、発疹熱リケッチア、ロッキー山紅斑熱リケッチア、恙虫病リケッチア、塹壕熱ロシャリメア、Q熱コクシエラ、肺炎マイコプラズマ、表皮ブドウ球菌、化膿レンサ球菌、肺炎レンサ球菌、セレウス菌、枯草菌、破傷風菌、ボツリヌス菌、リステリア菌、ブタ丹毒菌、ジフテリア菌、ビフィズス菌、結核菌、ウシ型菌、癩菌、ロドコッカス・エクイ、ファイトプラズマ、シゲラ菌、大腸菌
【0027】
さらにその病原性細菌が大腸菌であれば、下記大腸菌から閉環状DNAを単離することができる。
ベロ毒素産生性大腸菌(VTEC)、毒素原性大腸菌(ETEC)、組織進入性大腸菌(EIEC)、病原血清型大腸菌(EPEC)、腸管凝集付着性大腸菌(EAggEC)、K12株
【0028】
本発明は下記薬剤耐性菌から閉環状DNAを単離することができる。
分裂酵母、出芽酵母、ビブリオ属菌、バンコマイシン耐性腸球菌、黄色ブドウ球菌炭疽菌、赤痢菌、チフス菌、サルモネラ菌、コレラ菌、腸炎ビブリオ菌、カンピロバクター、黄色ブドウ球菌、ウエルシュ菌、セレウス菌、レンサ球菌、梅毒トレポネーマ、ライム病ボレリア、ピロリ菌、髄膜炎菌、淋菌、蛍光菌、鼻疽菌、類鼻疽菌、マルタ熱菌、バング菌、ブタ流産菌、百日咳菌、野兎病菌、チフス菌、パラチフス菌、ネズミチフス菌、肺炎桿菌、エンテロバクター・クロアカ、霊菌、ペスト菌、偽結核菌、インフルエンザ菌、軟性下疳菌、フラジリス菌、メラニノゲニカス菌、ビビウス菌、ジンジバーリス菌、発疹熱リケッチア、ロッキー山紅斑熱リケッチア、恙虫病リケッチア、塹壕熱ロシャリメア、Q熱コクシエラ、肺炎マイコプラズマ、表皮ブドウ球菌、化膿レンサ球菌、肺炎レンサ球菌、セレウス菌、枯草菌、破傷風菌、ボツリヌス菌、リステリア菌、ブタ丹毒菌、ジフテリア菌、ビフィズス菌、結核菌、ウシ型菌、癩菌、ロドコッカス・エクイ、ファイトプラズマ、シゲラ菌、大腸菌
薬剤耐性菌が耐性を持つ薬剤としては、ペニシリン系、セフェム系、アミノグリコシド系、テトラサイクリン系、クロラムフェニコール系、リンコマイシン系、マクロライド系、コリスチン、ホスホマイシン、ポリミキシン、バンコマイシン、トリコマイシン、サイクロセリンなどを挙げることができる。
【0029】
また、本発明は、本発明の方法に使用するためのキットであって、以下の成分:
(a)少なくとも1種類のアルコール及び/又はケトンを含む溶液;
(b)凝集補助剤;
(c)凝集剤;
(d)細胞溶解液;
(e)アルコール性溶液;
(f)洗浄液;及び
(g)溶出液
を含むキットを提供する。
少なくとも1種類のアルコール及び/又はケトンを含む溶液は、エタノール、イソプロピルアルコール及びアセトンから1種類以上を選び、それを30〜100%の濃度、好ましくは50〜100%の濃度となるように水などと混合したものであるとよい。
凝集補助剤としては、炭酸カルシウム、粉末活性炭、活性珪酸、ベントナイトを挙げることができる。本発明のキットにおいて、凝集補助剤は、水などの溶媒に0.1〜70%の濃度、好ましくは5〜50%の濃度となるように懸濁させたものであるとよい。
凝集剤としては、ポリ塩化アルミニウム、硫酸アルミニウム、アルギン酸ナトリウム、キトサン、塩化第二鉄、ポリ硫酸第二鉄、ポリアクリルアミド系高分子凝集剤、ジメチルアミノエチルメタクリレート系高分子凝集剤、ジメチルアミニエチルアクリレート系高分子凝集剤、アミジン系高分子凝集剤、両性系高分子凝集剤を挙げることができる。本発明のキットにおいて、凝集剤は、水などの溶媒に0.001〜10%の濃度、好ましくは0.01〜10%の濃度となるように溶解させたものであるとよい。
細胞溶解液中に含まれる細胞膜を溶解する界面活性剤としてはTriton X-100、Briji、Nonidet P-40、Tween 20、Tween 80、CHAPS、SDS、NP-40、コール酸、n-octyl glycosideなどがあり、細胞壁溶解酵素としてはセルラーゼ、ヘミセルラーゼ、ペクチナーゼ、リゾチーム、β-1,3-グルカナーゼ、コラゲナーゼ、トリプシンなどがある。細胞溶解液中に含まれるRNaseはRNaseA、RNaseT、RNaseI、RNaseHがある。細胞溶解液は界面活性剤、細胞壁溶解酵素、RNaseをバッファーに溶解したものであるとよい。バッファーはいかなるものでもよいが、DNAの分解を補助する可能性のあるMgイオンをキレートするEDTAが入っていることが好ましい。例えばTEバッファー(10mM Tris-HCl, 1mM EDTA (pH 8))、TBEバッファー(85mM Tris, 85mM ホウ酸, 2mM EDTA)、TAEバッファー(Tris 40mM, 酢酸 40mM, 1mM EDTA)などを挙げることができる。界面活性剤は0.01〜10%の濃度、好ましくは0.1〜10%の濃度となるように溶解させたものであると良い。細胞壁溶解酵素は0.01〜10%の濃度、好ましくは0.1〜5%の濃度となるように溶解させたものであると良い。RNaseは0.0001〜1%の濃度、好ましくは0.001〜1%の濃度となるように界面活性剤や細胞壁溶解酵素とともにバッファーに溶解させたものか、もしくは0.001〜10%の濃度、好ましくは0.01〜5%の濃度となるように水などに溶解させたものを別途小瓶で添付する方法がよい。
アルコール性溶液に含まれるアルコールとしては、ポリエチレングリコール、エタノール、イソプロピルアルコールなどを挙げることができる。アルコール性溶液は、アルコールの種類によっても異なるが、5〜100%の濃度、好ましくは10〜100%の濃度となるように水、もしくは塩化ナトリウム、酢酸ナトリウム、塩化リチウム、酢酸アンモニウムなどの塩が混合している水溶液などと混合したものであるとよい。塩は0.01〜10Mの濃度、好ましくは0.1〜8Mの濃度となるように水に溶解させたものであるとよい。
洗浄液は、エタノール、イソプロピルアルコールなどを10〜100%の濃度、好ましくは40〜90%の濃度となるようにバッファー、もしくは水などに溶解したものであるとよい。バッファーはいかなるものでもよいが、例えばTEバッファー(10mM Tris-HCl, 1mM EDTA (pH 8))に塩化ナトリウム、酢酸ナトリウム、塩化リチウム、酢酸アンモニウムなどの塩を加えたものであるとよい。塩は0.01〜10Mの濃度、好ましくは0.1〜8Mの濃度となるように溶解させたものであるとよい。
溶出液は、フィルターに残った閉環状DNAを溶出することができるものであればいかなるものであってもよいが、DNAの分解を補助する可能性のあるMgイオンをキレートするEDTAが入っているバッファーが好ましい。例えばTEバッファー(10mM Tris-HCl, 1mM EDTA (pH 8))、TBEバッファー(85mM Tris, 85mM ホウ酸, 2mM EDTA)、TAEバッファー(Tris 40mM, 酢酸 40mM, 1mM EDTA)などを挙げることができる。
【0030】
本発明のキットは、さらに、チップ、チップ止め装置、培養プレートなどを含んでもよい。これらの要素を含むキットは、多検体を処理するのに好適である。
チップは、液体を吸引・排出することができるものであればいかなるものであってもよく、例えば、バイオギルソンイエローチップ(笛吹事務所)、アイビスピペットチップ(アズワン)、Universal Fit Tip(AXY)、Beveled Pipette Tip(SSI)、Multi Fit Tip(CLP)、スタンダードピペットチップ(総研バイオ)、アシストチップ(アシスト)などを用いることができる。
多サンプルの閉環状DNAを単離する際、上清と沈殿に分かれた混合液から上清だけをプレートから抜き取るのは難しい。そこで、図8のように、一定の深さ(ここでは沈殿物のすぐ上)までチップが達した際に、チップが一定以上奥に行かないようにチップを支えて止める機能を持った装置を利用するのが便利であるが、このチップを支えて止める機能を持った装置を「チップ止め装置」と言う。チップ止め装置はチップケース内のチップを固定する装置を加工することで得ることができるが、特別に作製することもできる。図8(a)は1つのウェルの場合について示してあるが、プレートで行う際にはこれが96個並んでいる(図8(b))。また、チップ止め装置、チップ、培養プレートは図の形状に限らない。
培養プレートは、細胞を培養可能なものであればいかなるものであってもよく、例えば、Coster (Corning Incorporated)、2ml Deep Well Plate (AXY)、2.2ml 96-well Deep Well Plate (Brand)、FalconTM 96-well Deep-Well Plate (BD Biosciences)、96-Well MasterblockTM Deep Well Plate (Bellco Glass)などを用いることができる。
【0031】
本キットにはさらにフィルタープレート、シリンジ型フィルター、溶出プレート、取り扱い説明書を含めてもよい。
フィルタープレートまたはシリンジ型フィルターはフィルターの孔径が0.01〜10μm、好ましくは0.05〜5μmのものであれば、フィルターの形状及び材質はいかなるものであってもよい。フィルタープレートまたはシリンジ型フィルターは特別に作製することができるが、市販のものを使うこともできる。フィルタープレートであれば、例えばMultiScreenFB(Millipore)、コンサート96フィルタープレート(インビトロジェン)、AcroWell GHPメンブレン使用フィルタープレート(日本ジェネティクス)、ユニフィルター350(Whatman)などを用いることができる。シリンジ型フィルターはシリンジとフィルターが別になったタイプと一体になったタイプがある。シリンジとフィルターが別になったタイプであれば、例えばMILLEXTM-HV(Millipore)、25mm GD/Xシリンジフィルター(Whatman)、ミニザルトN(Sartorius)などを用いることができる。また、一体になったタイプであれば、例えばQIAfilter Midi Cartridges(Qiagen)、PhoenIXTM Filter(Qbiogene)などを用いることができる。
溶出プレートは多検体からcccDNAを調製する際に用いるが、形状はいかなるものでもよい。市販品であれば、例えばポリスチレン製プレート(Whatman)、PS-Microplate 96well V-form(Griner bio-one)、マイクロウェルプレート(アズワン)などを用いることができる。
本キットの情報、プロトコル、使用上の注意などを書いた説明書をキット内に同梱することができる。
【0032】
さらに、本発明は、閉環状DNAを調製するための装置(以下、「閉環状DNA調製装置」ということもある。)であって、以下の装置:
(A)横移動装置;
(B)分注装置;
(C)上清吸引装置;
(D)振動装置;
(E)プレート吸引装置;及び
(F)ロボットアーム
を含む装置を提供する。
【0033】
本発明の閉環状DNA調製装置の一例の概略図を図1に示す。
以下の各工程に沿って、プレート(2)は横移動装置(20及び21)に乗って移動してゆく。プレートは、上述の培養プレート、例えば、Coster (Corning Incorporated)を用いるとよい。横移動装置は、モーター(図示せず)によって駆動される。横移動装置(20及び21)のベルト構造を図2及び図3に示す。図2において、プレートはプレートを載せる枠(101)上に配置される。さらに、押さえ部材(102)は水平方向の押しばねで内側方向にプレートを押す。これにより、プレートがプレートを載せる枠(101)上に配置されると、プレートを載せる枠(101)の中心部に固定される。各プレートを載せる枠(101)は連結部材(103)で円回転するように連結されている。図2では、横移動装置の下方に振盪機(5a)が配置されている(以下の工程3)、5)、7)、9)、13)、15)の状態を示す。)。図3では、横移動装置の上にプレート(2)が乗っている(以下の工程1)〜17)の状態を示す。プレートは押さえ部材(102)によりプレートを載せる枠(101)の中心方向に押されている。
1)大腸菌が入っているプレート(2)がプレートストッカー(1)(図1)から横移動装置(20)の上に乗せられる。ここで、ストッカーの構造について図17に記す。側面Xには下方にプレート2枚分の切れ込みがあり、その切れ込みの上部にプレートストッパー1(801)を備え、下部にはロボットアーム(803)を備える。側面Yには上方に切れ込みがあり、下方にはプレートストッパー2(802)を有する。次ぎにストッカーの動作について説明する。まずロボットアーム(803)が最下部にあるプレート(16−1)をつかむ。次ぎにプレートストッパー2(802)が開く(図17(b)の(1))。次ぎにロボットアーム(803)はつかんだプレート(16−1)を横移動装置に乗せる(図17(b)の(2))。この状態ではストッカーの最下部にプレート一枚分のスペースができる。次ぎにプレートストッパー2(802)が閉じる(図17(b)の(3))。この状態でプレートストッパー1(801)が開き、ストックされているプレート(16−2、16−3及び16−4)が下方に移動する。
2)大腸菌が入っているプレート(2)に、高張液分注機(3)により塩化ナトリウム水溶液が分注される。
3)その後プレート(2)は振盪機(5a−1)により振盪される。図5に示すように、振盪機(5a)は、横移動装置の下方から上に移動し、プレートを持ち上げ、上方から降りてくる固定支持体(5b)に支持された状態で振とうされる。この工程で固定支持体は5b−1で示されている。
4)アルコール・ケトン分注機(4)により、アルコール及びケトンから選ばれる一種類以上を含有する液体がプレート(2)に分注される。
5)その後プレート(2)は振盪機(5a−2)により振とうされる。図5に示すように、振盪機(5a)は、横移動装置の下方から上に移動し、プレートを持ち上げ、上方から降りてくる固定支持体(5b)に支持された状態で振とうされる。この工程で固定支持体は5b−2で示されている。
6)凝集補助剤分注機(6)により、炭酸カルシウムを含有する液体が、プレート(2)に分注される。炭酸カルシウムを含有する液体は静置すると炭酸カルシウムが沈殿し、分注機への正確な定量吸引ができない。そこで、図14のように、後述する分注機(図6)の溶液トレー(306)に図14(a)(b)の様に、連結部(310)を介して振動装置(311)を接続する。炭酸カルシウムを含有する溶液はこの振動する溶液トレーから定量吸引される。(吸引分注方法は後述する。)溶液トレーへの溶液の供給は図14(c)に示す機構により行う。水位センサー(301)が溶液の水位の低下を感知すると信号はポンプ制御装置(309)に送られ、ポンプ(304)が作動し、溶液ビン(305)から溶液供給パイプ(303)を通して溶液トレー(306)に供給される。ここで、炭酸カルシウムを含有する液体を貯蔵する溶液ビンは振動装置(312)により常に振動攪拌されている。溶液が溶液トレーの所定の水位に達するとセンサーからポンプ制御装置に信号が送られ、ポンプは停止する。このように構成することで、炭酸カルシウムを含有する液体の正確な分注が行われる。
7)その後プレート(2)は振盪機(5a−3)により振とうされる。図5に示すように、振盪機(5a)は、横移動装置の下方から上に移動し、プレートを持ち上げ、上方から降りてくる固定支持体(5b)に支持された状態で振とうされる。この工程で固定支持体は5b−3で示されている。
8)凝集剤分注機(7)により、ポリ塩化アルミニウムを含有する液体がプレート(2)に分注される。
9)その後プレート(2)は振盪機(5a−4)により振とうされる。図5に示すように、振盪機(5a)は、横移動装置の下方から上に移動し、プレートを持ち上げ、上方から降りてくる固定支持体(5b)に支持された状態で振とうされる。この工程で固定支持体は5b−4で示されている。
10)プレート(2)は一定時間静置される。ただし、連続してプレートを処理するため、振盪機(5a−4)と上清吸引排出機(8)との間に数プレート分の間隔があり、その間は横移動のみを行う。
11)上清吸引排出機(8)により、プレート(2)中の上清が吸引廃棄される。上清吸引排出機の機構は分注装置(図16)と同様である。ただし、分注装置が溶液トレーから溶液を吸引してプレートに排出するのに対して、上清吸引排出機はプレートから上清を吸引し、排出トレーに排出する。以下に上清吸引排出機の機序を示す。上清吸引排出機は横移動装置上のプレートに向かって上清の吸引に適切な位置まで降下し、上清の吸引を行う。この時、図8のように、一定の深さ(ここでは沈殿物のすぐ上)までチップが達した際に、チップが一定以上奥に行かないようにチップを支えて止める機能を持った「チップ止め装置」を利用するのが便利である。そのあと、再び元の高さまで上昇する。上清の排出は排出トレー(図19)に行われる。排出トレーは横移動装置の側面に設置されていて、上清排出時に上清吸引排出機の真下に移動する。上清排出後、排出トレーは再び側面に移動し、元の位置に戻る。排出された上清は排出トレーに繋がった捕集ビンに集められる。上清が排出されたあと、上清吸引排出機に付属するチップはチップ洗浄装置(後述)によって洗浄される。チップ洗浄装置は横移動装置の側面に設置されていて、チップ洗浄時に上清吸引排出機の真下に移動する。その後、上清吸引排出機が降下し、チップ洗浄装置により洗浄が行われる。洗浄後、上清吸引排出機は再び元の位置まで上昇し、チップ洗浄装置は元の位置に移動する。
12)細胞溶解液分注機(9)により、Triton X―100、Tris−HCl、EDTA、NaCl、リゾチーム、RNaseを含有する液体がプレート(2)に分注される。
13)その後プレート(2)は振盪機(5a−5)により断続的に振動が与えられる。この振動は500〜1700rpm程度の振動であらかじめ0.5秒間隔で0.5秒間の振動を合計10〜20回行われるようプログラムされている。図5に示すように、振盪機(5a)は、横移動装置の下方から上に移動し、プレートを持ち上げ、上方から降りてくる固定支持体(5b)に支持された状態で振動が与えられる。この工程で固定支持体は5b−5で示されている。
14)アルコール性溶液分注機(10)により、ポリエチレングリコールとNaClを含有する液体がプレート(2)に分注される。
15)その後プレート(2)は振盪機(5a−6)により振盪される。図5に示すように、振盪機(5a)は、横移動装置の下方から上に移動し、プレートを持ち上げ、上方から降りてくる固定支持体(5b)に支持された状態で振とうされる。この工程で固定支持体は5b−6で示されている。
16)プレート(2)は一定時間静置される。ただし、連続してプレートを処理するため、振盪機(5a−6)と上清吸引移動機(11)との間に数プレート分の間隔があり、その間は横移動のみを行う。
17)上清吸引移動機(11)により、プレート(2)中の上清が吸引され、フィルタープレート(12)に移し変えられる。フィルタープレートとしては、上述のもの、例えば、MultiScreenFB (Millipore)を用いるとよい。上清吸引移動機の機構は分注装置(図16)と同様である。ただし、分注装置が溶液トレーから溶液を吸引してプレートに排出するのに対して、上清吸引移動機はプレートから上清を吸引し、フィルタープレートに排出する。以下に上清吸引移動機の機序を示す。上清の吸引移動の際は、上清吸引移動機は上清の吸引に適した高さまで降下し、上清の吸引を行う。吸引後、上清吸引移動機は再び上昇し、その後レーンBのフィルタープレートの上方まで横方向に移動する。さらに、上清のフィルタープレートへの排出に適した高さまで降下し、上清をフィルタープレートに排出する。続いて再び上昇し、元の位置に横方向に移動し、続いてチップ洗浄装置(後述)により洗浄される。チップ洗浄装置は横移動装置の側面に設置されており、洗浄時には上清吸引移動機の真下に移動して出てくる。その後、上清吸引移動機は洗浄に適した位置まで降下し、チップ洗浄装置によりチップが洗浄される。洗浄後、上清吸引移動機は元の位置まで上昇し、チップ洗浄装置は元の位置に戻る。
ここで、以上の各工程を行った第1レーン(A)に平行してなる第2レーン(B)に上清が移動するように構成してもよい。
18)フィルタープレート(12)中の溶液はプレート吸引機(13−1)で吸引され、吸引して出てきた溶液は廃棄される。
プレート吸引機とその動作の一例を図4に示す。
図4に示すように、プレート吸引容器(201)は横移動装置の下方から上方へ横移動装置に密着するように上がってくる。プレート吸引容器には吸引チューブ(202a)接続されており、吸引チューブは捕集ビン(203)に接続されている。捕集ビンにはもう1本吸引チューブ(202b)が接続されており、該吸引チューブは吸引ポンプ(204)に接続されている。このような構造をとることにより、吸引ポンプを稼動されることにより、プレート(12)から液体を吸引し、捕集ビン(203)に集めることができる。
19)フィルタープレート(12)のフィルターは洗浄液分注機(14)から供給されるエタノールで洗浄される。洗浄の際、エタノールはプレート吸引機(13−2)で吸引される。
20)上記フィルターから、溶出液分注機(15)から供給される水またはTEバッファーでプラスミドが溶出される。洗浄の際、水またはTEバッファーは吸引溶出機(13−3)で吸引される。この工程の吸引溶出機を説明する。図15に示したように、吸引溶出機(13−3)は上面および下面が開放され、上面は横移動装置(21)を介して横移動装置上のフィルタープレート(12)に接するように、タイタープレート(16)と共にロボットアーム(602−1及び602−2)で配置される(図15(b)の(2))。タイタープレートとしては、上述の溶出プレート、例えば、ポリスチレン製プレート(Whatman)を用いるとよい。ここで、タイタープレートはレーンBの横に設置されたタイタープレート供給ストッカー(17)(図1)よりロボットアーム(図示せず)により1枚ずつ供給される。吸引溶出機は中空になっており、吸引チューブ(601)により内部の空気が吸引され、内部は陰圧になる。吸引溶出機には内部(タイタープレート密着用、603)と上部(横移動装置密着用、604)に空気もれを防ぐゴムが取り付けられている。これによりフィルタープレート(12)から水またはTEバッファーと共にプラスミドをタイタープレートに回収する。その後、吸引溶出機とタイタープレートはロボットアームにより図15の(b)(1)の状態に戻り、さらにタイタープレートだけ分離され、吸引済みタイタープレートストッカー(18)(図1)に回収される。このように、溶出されたプラスミドはタイタープレート(16)に回収される。ここでタイタープレートの供給、収納機構について説明する。図18はタイタープレートの供給・収納の機序を図示したものである。タイタープレートはタイタープレート供給ストッカー(17)にストックされている。タイタープレート供給ストッカーの機構は図17と同様である。タイタープレートはロボットアーム制御機(901)により一枚ずつ取り出され、回転することにより、吸引溶出機(13−3)の下方にまで運ばれる。次ぎに、タイタープレートは押し上げられ、吸引が行われる。吸引が済むと、タイタープレートは下方に移動し、ロボットアーム制御機が回転することにより、吸引済みタイタープレートストッカー(18)の上方まで運ばれ、その中に収納される。
21)プラスミドを入れたタイタイープレート(16)はプレートストッカー(18)内にロボットアーム(602−2)(図15)によりレーンBの横に設置した吸引済みタイタープレートストッカー(18)(図1)に1枚ずつ収納される。
本発明の閉環状DNA調製装置の別の一例の概略図を図20に示す。
図20は培養液を機械装置にかけるの工程の前段階として、遠心機を用いて培養液と細胞を手作業で分離し、沈殿した細胞から閉環状DNAを調製するための装置である。
アルコール及び/又はケトンを添加する工程の前段階として、あらかじめ遠心機を用いて培養液と細胞を分離しておくことで、図1の上清吸引排出機(8)の工程を除くことができる。さらに細胞が沈殿になるまでの待機時間が必要ないので、凝集剤分注機(7)で凝集剤を添加後、振盪機(5a−4)で混合し、その直後に細胞溶解液分注機(9)で細胞溶解液を添加することができる。また、遠心分離機を用いて細胞を沈殿にすることで、細胞の保存を容易にするという利点がある。つまり、体積の大きい培養液のまま冷凍する場合には時間がかかり、解凍する際にも時間がかかるので、その間に細胞が死滅する可能性が大きい。一方、細胞を沈殿にすると、体積が小さいので、冷凍と解凍が短時間ですみ、細胞の死滅を大幅に減少することができる。細胞の冷凍保存は特に多検体から閉環状DNAを調製する際にスムーズに作業を進行させるために重要な工程である。図20の高張液分注機(3)は図1の高張液分注機(3)と同様のものであるが、図20のそれは単に高張液を分注するだけでなく、縣濁用のバッファーもしくは培地と高張液の混合液である。さらに振盪機(5a−1)で振盪するが、この際、図20の振盪機(5a−1)は図1の振盪機(5a−1)よりも強い振動が必要である。つまり、図20の振盪機(5a−1)は単に溶液を混合するだけでなく、沈殿になった細胞を縣濁するという要素が入るからである。具体的には図1の振盪機(5a−1)は1000〜1700rpmの振動、図20の振盪機(5a−1)は1500〜2000rpmの振動が良い。また、図20の装置は洗浄液分注機(14)及びプレート吸引機(13−2)が図1と比べて一台ずつ追加されている。これは2度の洗浄工程を行う方が、より夾雑物の少ない閉環状DNAを得ることができるためである。
本装置を構成する各装置はすべてインターフェイスボードを介してコンピューターに接続され、一括管理される。具体的には、動作するすべての装置のセンサーから装置毎のインターフェースを介して動作完了又は動作未完了の信号がコンピューターに送られる。コンピューターはそれらの信号を一括管理し、各装置に対して次の動作指示を出す。つまり、どの装置からもエラーが発見されなければ次の動作の信号を、エラーが一つでも発見された場合は停止の信号を各装置に送信する。
本発明の閉環状DNA調製装置における(A)横移動装置に相当するのが、上記の横移動装置(20)及び(21)(図1及び20)であり、これは、ベルト、モーターなどを備えているとよい。(B)分注装置に相当するのが、上記の高張液分注機(3)、アルコール・ケトン分注機(4)、凝集補助剤振盪分注機(6)、凝集剤分注機(7)、細胞溶解液分注機(9)、アルコール性溶液分注機(10)、洗浄液分注機(14)及び溶出液分注機(15)(図1及び20)であり、これは、水位センサー、溶液供給パイプ、ポンプ、溶液ビン、溶液トレー、チップ、ポンプ制御装置などを備えているとよい。(C)上清吸引装置に相当するのが、図1の上清吸引排出機(8)と上清吸引移動機(11)、図20の上清吸引移動機(11)であり、これは、排出トレー、捕集ビン、チップ、チップ洗浄装置などを備えているとよい。(D)振動装置に相当するのが振盪機(5a−1〜5a−6)(図1及び20)及び振動装置(311、312)(図14)であり、これは、特別に作製することができるし、市販のもの(例えば、ボルテックスなど)を利用することもできる。(E)プレート吸引装置に相当するのが、上記のプレート吸引機(図1及び20の13−1〜13−3、図4、図15)であり、これは、吸引容器、チューブ、捕集ビン、ポンプ、タイタープレートなどを備えているとよい。
【0034】
上記の工程2)、4)、6)、8)、12)及び14)で液体を分注する機構の1例を図6に示す。
分注すべき液体(302)は溶液トレー(306)にためられる。ためられた溶液はチップ(307)により、定量吸引され、プレート(2)に分注される。
溶液トレー内の溶液が減ってくると水位センサー(301)により感知され、信号がポンプ制御装置(309)に送られる。ポンプ制御装置は溶液ビン(305)から溶液を吸い上げ、溶液トレー内に溶液供給パイプ(303)を通じて溶液を供給する。一定の溶液量が溶液トレーにたまるとその水位を水位センサーが感知し、ポンプ制御装置に信号を送る。それにより、ポンプ制御装置はポンプ(304)を停止させる。
図16は分注装置の概要を透視図にしたものである。分注装置は筺体(712)を有し、その内部にモーター(702)とギアで連結され、ねじ機構(703及び704)により上下運動するピストン(706)と、筺体に固定金具(711)で固定されたシリンダ(705)を有する。コンピューターから吸引の指示が出ると、モーターに電流が流れ、上部センサー(709)に感知されるまで、ピストンが上がり、分注すべき溶液を吸引する。分注機制御装置から分注の指示が出るとモーターに逆の電流が流れ、ピストンが下部センサー(710)に感知されるまで下がり、シリンダ内の溶液が分注口(707)から排出され、分注チューブ(708)を介してチップ(307)よりプレートに溶液が分注される。
【0035】
工程内でチップを洗浄する必要が場合には、チップ洗浄装置により洗浄される。チップ洗浄装置の1例を図7に記載する。
チップ洗浄槽(403)はRO(Revers Osmosis)水で満たされている(図7(a))。洗浄すべきチップ(401)(図6のチップ(307))の先端を洗浄筒(402)内に挿入する。洗浄筒の底面部にはRO水給水口がそれぞれ設けられており、チップが挿入されると給水が行われ、チップの先端が洗浄される。洗浄筒よりあふれたRO水は排水口より排水される(図7(b))。
本発明の閉環状DNA調製装置は、全自動で、外界から閉鎖された安全な空間で、病原性の細菌から閉環状DNAを単離し、閉環状DNA由来の病原性の有無を診断することができる。さらに、病原となる遺伝子が閉環状DNA由来であるか否かを判定する研究用にも用いることができる。
さらに病原性菌が大腸菌である場合、大腸菌を外界から閉鎖された安全な空間で、大腸菌から閉環状DNAを単離し、閉環状DNA由来の病原性の有無を診断することができる。さらに、病原となる遺伝子が閉環状DNA由来であるか否かを判定する研究用にも用いることができる。
本発明の閉環状DNA調製装置により、全自動で、外界から閉鎖された安全な空間で、病原性の細菌から閉環状DNAを単離し、閉環状DNA由来の薬剤耐性の有無を診断することが可能である。
【0036】
以下、本発明を実施例により具体的に説明する。
実施例1〜3として、遠心分離を一度も行わず、その代わりに凝集剤を使用して他の細胞構成成分からcccDNAを分離する方法について述べる。
実施例4及び5として、遠心分離操作と凝集剤の使用を組み合わせて、他の細胞構成成分からcccDNAを分離する方法について述べる。
本発明の実施形態がこれらに限られるものでないことはいうまでもない。
実施例で用いた器具及び大腸菌は以下の通りである。
チップ:バイオギルソンイエローチップ(笛吹事務所)
96穴培養プレート:Costar(Corning Incorporated)
フィルタープレート:MultiScreenFB (Millipore)
溶出プレート:PS-Microplate 96 well V-form (Griner)
実施例1、3及び4の大腸菌:2002年に60770個のマウス全長cDNAのクローニングと機能解析が終了(The FANTOM Consortium and The RIKEN Genome Exploration Research Group Phase I & II Team, Analysis of the mouse transcriptome based on functional annotation of 60,770 full-length cDNA, Nature, 420, 563-573, 2002)したもののうち、その1個の遺伝子を用いて実験を行った。60770個のcDNAはC57BL/6J系統のマウスから、キャップトラップ法(Carninci, P. et al. Normalization and subtraction of cap-trapper-selected cDNAs to prepare full-length cDNA libraries for rapid discoverry of new genes. Genome Res. 10, 1617-1630, 2000)などを用いて単離された。単離された遺伝子はクローニング(ベクターにcDNAを組み込み、大腸菌に遺伝子導入)後に大腸菌が培養され、プラスミド精製が行われたあと、全塩基配列が決定され、機能解析が行われた。本実験には、pBluescript KS(+)(Stratagene)というベクターに組み込まれ、大腸菌株DH10Bに遺伝子導入された遺伝子(クローンID:9930023M19、大きさ:3255bp)を用いた。その塩基配列やアノテーションの情報はウェブページで公開されている(http://fantom2.gsc.riken.go.jp/)。
実施例2の大腸菌:2002年に60770個のマウス全長cDNAのクローニングと機能解析が終了(The FANTOM Consortium and The RIKEN Genome Exploration Research Group Phase I & II Team, Analysis of the mouse transcriptome based on functional annotation of 60,770 full-length cDNA, Nature, 420, 563-573, 2002)したもののうち、その中の5個の遺伝子を用いて実験を行った。60770個のcDNAはC57BL/6J系統のマウスから、キャップトラップ法(Carninci, P. et al. Normalization and subtraction of cap-trapper-selected cDNAs to prepare full-length cDNA libraries for rapid discoverry of new genes. Genome Res. 10, 1617-1630, 2000)などを用いて単離された。単離された遺伝子はクローニング(ベクターにcDNAを組み込み、大腸菌に遺伝子導入)後に大腸菌が培養され、プラスミド精製が行われたあと、全塩基配列が決定され、機能解析が行われた。本実験には、pBluescript KS(+)(Stratagene)というベクターに組み込まれ、大腸菌株DH10Bに遺伝子導入された遺伝子(クローンID:9930033A02、9930023M19、9930039L23、9930022D09、A130007D10)を用いた。その塩基配列やアノテーションの情報はウェブページで公開されている(http://fantom2.gsc.riken.go.jp/)。
【0037】
[実施例1]
1.5mlチューブを用いた少検体大腸菌培養液からのプラスミド調製
大腸菌DH10B株にあらかじめ組み込んでおいたpBluescript KS(+)のプラスミドDNAの単離を行った。このプラスミドDNAの中にはマウスの遺伝子DNA (クローンID:9930023M19、大きさ:3255bp)が組み込まれている。
材料:
LB培地500μl
5M 塩化ナトリウム 100μl
100% イソプロパノール 300μl
500mg/ml 炭酸カルシウム 40μl
4mg/ml ポリ塩化アルミニウム 20μl
2% Triton X-100, 4mM Tris-HCl (pH8), 0.4mM EDTA, 20mM NaCl, 5mg/ml リゾチーム, 67μg/ml RNaseA混合液 150μl
20% ポリエチレングリコール, 2.5M NaCl混合液120μl
70% エタノール 300μl
TE 100μl
MultiScreenFB (Millipore)
【0038】
手順
1)アンピシリンを含むLB培地に単一コロニーから取った大腸菌を接種し、37℃で12〜20時間インキュベートした。培養物の少なくとも4倍の体積を有する培養チューブ、もしくはフラスコを使用した。培養の際にはエアレーションが行われるようにし、約30度傾け、激しく振盪(〜300rpm)培養した。
2)約500μlの培養液を取り、1.5mlチューブに移した。
3)100μlの5M塩化ナトリウムを加え、混合した。
4)300μlの100%イソプロパノールを加え、混合した。
5)40μlの500mg/mlの炭酸カルシウムを加え、混合した。
6)20μlの4mg/mlのポリ塩化アルミニウムを加え、混合した。
7)チューブを垂直に立て、8分静置した。
8)沈殿を取らないように気をつけながら上清だけをピペットマンを使用し取り除く。わずかな上清を含む沈殿はこのとき約150μl残った。
9)この沈殿に、150μlの2%Triton X−100、4mM Tris−HCl(pH8)、0.4mM EDTA、20mM NaCl、5mg/ml リゾチーム、 67μg/ml RNaseAの混合液を加えた。
10)ただちに弱い断続的な振動、すなわち0.5秒振動→0.5秒静止、を10回繰り返した。(8)で生じた沈殿が(9)で加えた溶液と完全に混合される程度の振動(〜1700rpm)を要するが、あまり強い振動を与えると沈殿が分解して澄んだ上清が得られないので注意を要する。弱い断続的な振動を与えた後、上清が澄んでいることを確認する。
11)120μlの20%ポリエチレングリコール、2.5M NaClを加え、混合した。
12)90秒静置した。
13)限外ろ過が可能なフィルター(Millipore社のMultiScreenFB)に(12)で生じた上清を載せ、吸引ろ過を行った。
14)70%エタノールを添加し、吸引ろ過を行った。
15)100μlのTEでプラスミドDNAをフィルターから溶出させ、回収した。
【0039】
単離したプラスミドDNAの電気泳動
実施例1で単離したプラスミドDNAのアガロースゲル電気泳動を行った。
1%の濃度のAgarose-1 (和光純薬工業株式会社)を作成し、TAEバッファー(Tris 40mM, 酢酸 40mM, 1mM EDTA)を充填した電気泳動漕にセットした。実施例1記載の(15)で単離したプラスミドDNA 100μlのうち4μlを泳動した。Qiagen Mini Prep Kitで単離したプラスミドDNAも同様に溶出した100μlのうち4μlを泳動した。また、マーカーDNAとしてGeneRulerTM 1Kb DNA Ladder (MBI fermentas社)を用いた。その結果を図9に示す。マーカーは合計200ngになるように泳動した。電気泳動を行った結果、RNAは目視ではまったく観察されず、ゲノムDNAの夾雑もほとんど観察されなかった。また泳動マーカーと比較したところ、500μlの菌液から単離されるプラスミドDNAの量は約2.5μgであることが分かった。
【0040】
単離したプラスミドDNAを用いたPCR
実施例1で単離したプラスミドDNAに組み込んだマウスの遺伝子DNA (クローンID:9930023M19、大きさ:3255bp)をPCRで増幅した。増幅用のプライマーDNAとして1st-BS (TGTAAAACGACGGCCAGT)(配列番号1) と2nd-NX/X (TCCTGTGTGAAATTGTTATCCGCT)(配列番号2)を用いた。増幅用の反応バッファーは以下の通りである。
1)10×Buffer 2.5μl
2)25mM MgSO4 1μl
3)2mM dNTP 2.5μl
4)5pmol/μl プライマーDNA 各0.5μl
5)2ng/μlの単離したプラスミドDNA 1μl
6)KOD plus 0.75μl
7)蒸留水 16.25μl
また、反応温度と時間は以下の通り
1)94℃ 2分
2)94℃ 1分
3)60℃ 30秒
4)68℃ 2分
(2)〜(4)を30回繰り返す
PCRの結果を図10に示す。この図が示す通り、目的DNAの十分な量の増幅が見られ、本手法で単離したプラスミドDNAがPCRに使用できることが分かった。
【0041】
単離したプラスミドDNAのトランスフォーメーション
単離したプラスミドDNAのトランスフォーメーションを行った。大腸菌DH10B株のコンピテント細胞を用い、プラスミドDNAを2ng/μlに希釈したあと、以下の操作を行った。
1)10μlのコンピテント細胞を-80℃の冷凍庫から取り出し、氷上で溶かした
2)2ng/μlのプラスミドDNA 1μlをコンピテント細胞の中に入れ、緩やかに混合した
3)混合した液を40℃で45秒間インキュベートした
4)再び氷上に置き、抗生物質の入らないLB培地を1ml混合した
5)37℃で1時間インキュベートした
6)インキュベートした液から20μl取ってアンピシリン入りのプレート培地に塗った
7)37℃で一晩培養した
この結果、9個の大腸菌コロニーができた。このことから、本手法で単離したプラスミドDNAがトランスフォーメーションに使用できることが分かった。
【0042】
[実施例2]
96穴プレートを使った多検体の大腸菌からのプラスミド調製
大腸菌DH10B株にあらかじめ組み込んでおいたpBluescript KS(+)のプラスミドDNAの単離を行った。全ての検体はそれぞれ異なったマウスの遺伝子(クローンID:9930033A02、9930023M19、9930039L23、9930022D09、A130007D10)が入ったプラスミドを持つ。
材料:
LB培地 各500μl
5M 塩化ナトリウム 各100μl
100% イソプロパノール 各300μl
500mg/ml 炭酸カルシウム 各40μl
4mg/ml ポリ塩化アルミニウム 各20μl
2% Triton X-100, 4mM Tris-HCl (pH8), 0.4mM EDTA, 40mM NaCl, 5mg/ml リゾチーム, 67μg/ml RNaseA混合液 各150μl
20% ポリエチレングリコール, 2.5M NaCl混合液 各120μl
70% エタノール 各300μl
TE 各100μl
MultiScreenFB (Millipore)
【0043】
手順
1)それぞれのウェルにアンピシリンを含む500μlのLB培地に単一コロニーから取った大腸菌を接種し、37℃で12〜20時間インキュベートした。培養には96穴培養プレート(Costar, Corning Incorporated)を用い、エアレーションが行われるようにし、約30度傾け、激しく振盪(〜300rpm)培養した。
2)100μlの5M塩化ナトリウムを各ウェルに加え、混合した。
3)300μlの100%イソプロパノールを各ウェルに加え、混合した。
4)40μlの500mg/mlの炭酸カルシウムを各ウェルに加え、混合した。
5)20μlの4mg/mlのポリ塩化アルミニウムを各ウェルに加え、混合した。
6)8分静置した。
7)沈殿を取らないように気をつけながら上清だけを専用のチップ止め装置を用いて取り除く。わずかな上清を含む沈殿はこのとき約150μl残った。
9)この沈殿に、150μlの2%Triton X―100、4mM Tris−HCl(pH8)、0.4mM EDTA、20mM NaCl、5mg/ml リゾチーム、 67μg/ml RNaseAの混合液を各ウェルに加えた。
10)ただちに弱い断続的な振動、すなわち0.5秒振動→0.5秒静止、を10回繰り返した。8で生じた沈殿が9で加えた溶液と完全に混合される程度の振動(〜1700rpm)を要するが、あまり強い振動を与えると沈殿が分解して澄んだ上清が得られないので注意を要する。弱い断続的な振動を与えた後、上清が澄んでいることを確認する。
11)120μlの20%ポリエチレングリコール、2.5M NaClを各ウェルに加え、混合した。
12)90秒静置した。
13)限外ろ過が可能なフィルター(Millipore社のMultiScreenFB等)に(12)で生じた上清を専用のチップ止め装置を用いて載せ、吸引ろ過を行った。
14)70%エタノールを添加し、吸引ろ過を行った。
15)100μlのTEでプラスミドDNAをフィルターから溶出させ、回収した。
【0044】
単離したプラスミドDNAの電気泳動
実施例2で単離したいくつかのプラスミドDNAのアガロースゲル電気泳動を行った。
1%の濃度のAgarose-1 (和光純薬工業株式会社)を作成し、TAEバッファー(Tris 40mM, 酢酸 40mM, 1mM EDTA)を充填した電気泳動漕にセットした。実施例2記載の(15)で単離した各プラスミドDNA 100μlのうち4μlを泳動した。また、マーカーDNAとしてGeneRulerTM 1Kb DNA Ladder (MBI fermentas社)を用いた。その結果を図11に示す。マーカーは合計200ngになるように泳動した。図中の1〜5はそれぞれ別のマウス遺伝子が入ったプラスミドDNAである。図11の左から、クローンID:9930033A02、9930023M19、9930039L23、9930022D09、A130007D10。
【0045】
[実施例3]
培養チューブを用いた大腸菌培養液からのプラスミドの大量調製
大腸菌DH10B株にあらかじめ組み込んでおいたpBluescript KS(+)のプラスミドDNAの単離を行った。プラスミドDNAの中にはマウスの遺伝子DNA (クローンID:9930023M19、大きさ:3255bp)が組み込まれている。
材料:
LB培地 20ml
5M 塩化ナトリウム 4ml
100% イソプロパノール 12ml
500mg/ml 炭酸カルシウム 1.6ml
4mg/ml ポリ塩化アルミニウム 0.8ml
2% Triton X-100, 4mM Tris-HCl (pH8), 0.4mM EDTA, 20mM NaCl, 5mg/ml リゾチーム, 67μg/ml RNaseA混合液 5ml
20% ポリエチレングリコール, 2.5M NaCl混合液 4ml
70% エタノール 5ml
TE 1ml
MillexR-HV (Millipore)
【0046】
手順
1)アンピシリンを含む20mlのLB培地に単一コロニーから取った大腸菌を接種し、37℃で20時間インキュベートした。培養には50mlの培養チューブ(Falcon, Becton Dickinson)を用い、エアレーションが行われるようにし、約30度傾け、激しく振盪(〜300rpm)培養した。
2)4mlの5M塩化ナトリウムを加え、混合した。
3)12mlの100%イソプロパノールを加え、混合した。
4)1.6mlの500mg/mlの炭酸カルシウムを加え、混合した。
5)0.8mlの4mg/mlのポリ塩化アルミニウムを加え、混合した。
6)15分静置した。
7)沈殿を取らないように気をつけながら上清だけをピペットマン、もしくはピペットで除いた。ここでわずかな上清を含んだ沈殿は約5mlになった。
9)この沈殿に、5mlの2%Triton X−100、4mM Tris−HCl(pH8)、0.4mM EDTA、20mM NaCl、5mg/ml リゾチーム、 67μg/ml RNaseの混合液を加えた。
10)500μlの6M 炭酸カリウム(pH6)を加えた。
11)チューブの蓋を閉め、10回反転させた。
12)4mlの20%ポリエチレングリコール、2.5M NaClを加え、チューブを反転させた。
13)90秒静置した。ここで上清は約15ml取れた。
14)(13)で得られた上清をシリンジで3ml吸い取り、Millipore社のMillexR-HVを用いて限外ろ過を行った。
15)70%エタノールをシリンジで5ml吸い取り、フィルターを洗浄した。
16)800μlのTEでプラスミドDNAをフィルターから溶出させ、回収した。
17)上記14〜16の操作を5回繰り返し、合計4mlの溶出液を得た。
【0047】
単離したプラスミドDNAの電気泳動
実施例3で単離したプラスミドDNAのアガロースゲル電気泳動を行った。
1%の濃度のAgarose-1 (和光純薬工業株式会社)を作成し、TAEバッファー(Tris 40mM, 酢酸 40mM, 1mM EDTA)を充填した電気泳動漕にセットした。実施例1で単離したプラスミドDNA 100μlから4μl泳動したものと、実施例3記載の(17)で単離したプラスミドDNA 4mlのうち4μlを泳動したものを比較した。実施例1は0.5mlの菌液から100μl溶出し、実施例3では20mlの菌液から4ml溶出しているので、溶出の比率は同じになる。マーカーDNAとしてGeneRulerTM 1Kb DNA Ladder (MBI fermentas社)を用いた。その結果を図12に示す。マーカーは合計200ngになるように泳動した。大量に単離した場合も少量単離した場合と同様に、RNAは目視ではまったく観察されず、ゲノムDNAの夾雑もほとんど観察されなかった。また泳動マーカーと比較したところ、20mlの菌液から単離されるプラスミドDNAの量は約100μgであることが分かった。
【0048】
[実施例4]
遠心機を用いた少量検体大腸菌培養液からのプラスミド調製法(1.5mlチューブを用いた調製)
大腸菌DH10B株にあらかじめ組み込んでおいたpBluescript KS(+)のプラスミドDNAの単離を行った。このプラスミドDNAの中にはマウスの遺伝子DNA (クローンID:9930023M19、大きさ:3255bp)が組み込まれている。
材料:
培養用LB培地1ml
縣濁用LB培地80μl
5M 塩化ナトリウム 20μl
100% イソプロパノール 50μl
500mg/ml 炭酸カルシウム 20μl
4mg/ml ポリ塩化アルミニウム 5μl
2% Triton X-100, 4mM Tris-HCl (pH8), 0.4mM EDTA, 20mM NaCl, 5mg/ml リゾチーム, 67μg/ml RNaseA混合液 150μl
20% ポリエチレングリコール, 2.5M NaCl混合液120μl
70% エタノール 600μl
TE 100μl
MultiScreenFB (Millipore)
【0049】
手順
1)アンピシリンを含むLB培地に単一コロニーから取った大腸菌を接種し、37℃で約16時間インキュベートした。培養物の少なくとも4倍の体積を有する培養チューブ、もしくはフラスコを使用した。培養の際にはエアレーションが行われるようにし、約30度傾け、激しく振盪(〜300rpm)培養した。
2)1mlの培養液を取り、1.5mlチューブに移した。
3)遠心機を用い、20000×gで10秒間遠心し、菌体を沈殿にした。
4)上清をデカンテーションして除去した。残液はピペットを用いて完全に除去した。
5)80μlのLB培地と20μlの5M塩化ナトリウムよく混合し、その全量を1.5mlチューブに加え、沈殿になった菌をピペットを用いてよく縣濁した。
6)50μlの100%イソプロパノールを加え、混合した。
7)20μlの500mg/mlの炭酸カルシウムを加え、混合した。
8)5μlの4mg/mlのポリ塩化アルミニウムを加え、混合した。
9)この沈殿に、150μlの2% Triton X-100、4mM Tris-HCl (pH 8)、0.4mM EDTA、20mM NaCl、5mg/ml リゾチーム、67μg/ml RNaseAの混合液を加えた。
10)ただちに弱い断続的な振動(〜1700rpm)で、0.5秒振動→0.5秒静止、を10回繰り返した。その後、上下に5回転倒混和した。あまり強い振動を与えると沈殿が分解して澄んだ上清が得られないので注意を要する。
11)120μlの20%ポリエチレングリコール、2.5M NaClを加え、混合した。
12)90秒静置した。
13)限外ろ過が可能なフィルター(Millipore社のMultiScreenFB)に(12)で生じた上清を300ulのせ、吸引ろ過を行った。
14)70%エタノールを300μl添加し、吸引ろ過を行った。
15)再度70%エタノールを300μl添加し、吸引ろ過を行った。
16)100μlのTEでプラスミドDNAをフィルターから溶出させ、回収した。
【0050】
単離したプラスミドDNAの電気泳動
実施例4で単離したプラスミドDNAのアガロースゲル電気泳動を行った。
1%の濃度のAgarose-1 (和光純薬工業株式会社)を作成し、TAEバッファー(Tris 40mM, 酢酸 40mM, 1mM EDTA)を充填した電気泳動漕にセットした。実施例4記載の(16)で単離したプラスミドDNA 100μlのうち4μlを泳動した。比較として、実施例1記載の(16)で単離したプラスミドDNAも同時に泳動した。また、マーカーDNAとしてGeneRulerTM 1Kb DNA Ladder (MBI fermentas社)を用いた。その結果を図22に示す。マーカーDNAは合計200ngになるように泳動した。電気泳動を行った結果、RNAは目視ではまったく観察されず、ゲノムDNAの夾雑もほとんど観察されなかった。また泳動マーカーと比較したところ、1mlの菌液から単離されるプラスミドDNAの量は約8μgであることが分かった。
【産業上の利用可能性】
【0051】
本発明により、経費が低く品質の良い閉環状DNAを短時間で大量に精製でき、閉環状DNAを使用する研究ないし医療に大きく貢献し得る。
【図面の簡単な説明】
【0052】
【図1】本発明の閉環状DNA調製装置の1例の概略図である。
【図2】横移動装置の機構および振盪機の位置を表したものである。
【図3】横移動装置にプレートをセットしたものである。
【図4】プレート吸引機及びその動作を表したものである。
【図5】振盪機及び固定支持体の動きを表したものである。
【図6】溶液分注機を表したものである。(a)は各要素の構成を示す。(b)は溶液トレーから分注溶液が吸引され、プレートに分注される動作を模式的に示す。
【図7】チップ洗浄装置を表したものである。(a)は斜視図、(b)はA-Aの断面図である。
【図8】チップ止め装置を表したものである。(a)は(b)に示すA-Aの横断面図、(b)は斜視図である。
【図9】実施例1で単離したプラスミドDNAの電気泳動の結果を示す。M:マーカーDNA、1:Qiagen Mini Prep Kitで単離したプラスミドDNA、2:本発明の方法で単離したプラスミドDNA。
【図10】実施例1で単離したプラスミドDNAに組み込んだマウスの遺伝子DNAをPCRで増幅した結果を示す。M:マーカーDNA、1:Qiagen Mini Prep Kitで単離したプラスミドDNAに組み込んだマウスの遺伝子DNAをPCRで増幅したもの、2:本発明の方法で単離したプラスミドDNAに組み込んだマウスの遺伝子DNAをPCRで増幅したもの。
【図11】実施例2で単離したプラスミドDNAの電気泳動の結果を示す。M:マーカーDNA、1〜5:本発明の方法で単離したプラスミドDNA。
【図12】実施例1及び3で単離したプラスミドDNAの電気泳動の結果を示す。M:マーカーDNA、1:実施例1において本発明の方法で単離したプラスミドDNA、2:実施例3において本発明の方法で単離したプラスミドDNA。
【図13】本発明の閉環状DNA調製方法の1例のスキームを表したものである。
【図14】凝集補助剤分注機を表したものである。
【図15】吸引溶出機を表したものである。
【図16】分注装置を表したものである。
【図17】プレートストッカーの構造を表したものである。
【図18】タイタープレートの供給・収納機構を表したものである。
【図19】排出トレーを表したものである。
【図20】本発明の閉環状DNA調製装置の別の1例の概略図である。
【図21】本発明の閉環状DNA調製方法の別の1例のスキームを表したものである。
【図22】実施例1及び4で単離したプラスミドDNAの電気泳動の結果を示す。M:マーカーDNA、1:実施例1で単離したプラスミドDNA、2:実施例4で単離したプラスミドDNA。
【符号の説明】
【0053】
1:プレートストッカー
2:プレート
3:高張液分注機
4:アルコール・ケトン分注機
5a、5a−1、5a−2、5a−3、5a−4、5a−5、5a−6:振盪機
5b、5b−1、5b−2、5b−3、5b−4、5b−5、5b−6:固定支持体
6:凝集補助剤振盪分注機
7:凝集剤分注機
8:上清吸引排出機
9:細胞溶解液分注機
10:アルコール性溶液分注機
11:上清吸引移動機
12:フィルタープレート
13−1、13−2:プレート吸引機
13−3:吸引溶出機
14:洗浄液分注機
15:溶出液分注機
16:タイタープレート
16−1:プレート
16−2:プレート
16−3:プレート
16−4:プレート
17:タイタープレート供給ストッカー
18:吸引済みタイタープレートストッカー
20、21:横移動装置
A:第1レーン
B:第2レーン
101:枠
102:押さえ部材
103:連結部材
201:吸引容器
202a、202b:チューブ
203:捕集ビン
204:ポンプ
301:水位センサー
302:分注溶液
303:溶液供給パイプ
304:ポンプ
305:溶液ビン
306:溶液トレー
307:チップ
309:ポンプ制御装置
310:連結部
311:振動装置
312:振動装置
401:チップ
402:洗浄筒
403:チップ洗浄槽
501:チップ止め装置
502:チップ
503:上清
504:沈殿
601:吸引チューブ
602−1:ロボットアーム
602−2:ロボットアーム
603:密閉用ゴム
604:密閉用ゴム
701:モーター配線
702:モーター
703:ネジ機構
704:ネジ機構
705:シリンダー
706:ピストン
707:分注口
708:分注チューブ
709:センサ
710:センサ
711:固定金具
712:筺体
801:プレートストッパー1
802:プレートストッパー2
803:ロボットアーム
804:筺体
901:ロボットアーム制御機
【配列表フリーテキスト】
【0054】
<配列番号1>
配列番号1は、実施例1で増幅用のプライマーDNAとして用いた1st-BSの塩基配列を示す。
<配列番号2>
配列番号2は、実施例1で増幅用のプライマーDNAとして用いた2nd-NX/Xの塩基配列を示す。
【特許請求の範囲】
【請求項1】
細胞から閉環状DNAを調製する方法であって、細胞を含む液体に凝集剤を添加する工程を含んでなる方法。
【請求項2】
以下の工程:
(a)該細胞を含む液体に少なくとも1種類のアルコール及び/又はケトンを添加する工程;
(b)該細胞を含む液体に凝集補助剤及び凝集剤を添加して、上清と沈殿に分離する工程;
(c)工程(b)で生じた上清と沈殿から上清を除く工程;
(d)工程(c)で上清を除いたあとの沈殿に細胞溶解液を添加する工程;
(e)工程(d)で生じた混合液に振動を与える工程;
(f)工程(e)で振動を与えた混合液にアルコール性溶液を添加する工程;及び
(g)工程(f)でアルコール性溶液を添加した混合液の上清を限外ろ過フィルターに通して閉環状DNAを回収する工程
を含んでなる請求項1記載の方法。
【請求項3】
遠心分離を行う工程を含まない請求項2記載の方法。
【請求項4】
工程(a)の前工程として、高張液を細胞に接触させる請求項2又は3に記載の方法。
【請求項5】
以下の工程:
(a’)培養した細胞を遠心分離により沈殿させ、上清を除く工程;
(b’)沈殿した細胞をバッファーもしくは培地で縣濁させ、細胞懸濁液を調製する工程;
(c’)該細胞懸濁液に少なくとも1種類のアルコール及び/又はケトンを添加する工程;
(d’)該細胞懸濁液に凝集補助剤及び凝集剤を添加する工程;
(e’)該細胞懸濁液に細胞溶解液を添加する工程;
(f’)工程(e’)で生じた混合液に振動を与える工程;
(g’)工程(f’)で振動を与えた混合液にアルコール性溶液を添加する工程;及び
(h’)工程(g’)でアルコール性溶液を添加した混合液の上清を限外ろ過フィルターに通して閉環状DNAを回収する工程
を含んでなる請求項1記載の方法。
【請求項6】
工程(b’)のバッファーもしくは培地が高張液である請求項5記載の方法。
【請求項7】
細胞が単細胞生物である請求項1〜6のいずれかに記載の方法。
【請求項8】
単細胞生物が細菌である請求項7記載の方法。
【請求項9】
細菌が大腸菌である請求項8記載の方法。
【請求項10】
細胞が形質転換体である請求項1〜9のいずれかに記載の方法。
【請求項11】
閉環状DNAが、プラスミドDNA、コスミドDNA、P1 DNA、BAC DNA、PAC DNA及びYAC DNAからなる群より選択される請求項1〜10のいずれかに記載の方法。
【請求項12】
請求項1〜11のいずれかに記載の方法に使用するためのキットであって、以下の成分:
(a)少なくとも1種類のアルコール及び/又はケトンを含む溶液;
(b)凝集補助剤;
(c)凝集剤;
(d)細胞溶解液;
(e)アルコール性溶液;
(f)洗浄液;及び
(g)溶出液
を含むキット。
【請求項13】
さらに、以下の要素:
(h)チップ;
(i)チップ止め装置;及び
(j)培養プレート
を含む請求項12記載のキット。
【請求項14】
閉環状DNAを調製するための装置であって、以下の装置:
(A)横移動装置;
(B)分注装置;
(C)上清吸引装置;
(D)振動装置;
(E)プレート吸引装置;及び
(F)ロボットアーム
を含む装置。
【請求項15】
凝集剤を使用して、閉環状DNAを他の細胞構成物から分離する方法。
【請求項1】
細胞から閉環状DNAを調製する方法であって、細胞を含む液体に凝集剤を添加する工程を含んでなる方法。
【請求項2】
以下の工程:
(a)該細胞を含む液体に少なくとも1種類のアルコール及び/又はケトンを添加する工程;
(b)該細胞を含む液体に凝集補助剤及び凝集剤を添加して、上清と沈殿に分離する工程;
(c)工程(b)で生じた上清と沈殿から上清を除く工程;
(d)工程(c)で上清を除いたあとの沈殿に細胞溶解液を添加する工程;
(e)工程(d)で生じた混合液に振動を与える工程;
(f)工程(e)で振動を与えた混合液にアルコール性溶液を添加する工程;及び
(g)工程(f)でアルコール性溶液を添加した混合液の上清を限外ろ過フィルターに通して閉環状DNAを回収する工程
を含んでなる請求項1記載の方法。
【請求項3】
遠心分離を行う工程を含まない請求項2記載の方法。
【請求項4】
工程(a)の前工程として、高張液を細胞に接触させる請求項2又は3に記載の方法。
【請求項5】
以下の工程:
(a’)培養した細胞を遠心分離により沈殿させ、上清を除く工程;
(b’)沈殿した細胞をバッファーもしくは培地で縣濁させ、細胞懸濁液を調製する工程;
(c’)該細胞懸濁液に少なくとも1種類のアルコール及び/又はケトンを添加する工程;
(d’)該細胞懸濁液に凝集補助剤及び凝集剤を添加する工程;
(e’)該細胞懸濁液に細胞溶解液を添加する工程;
(f’)工程(e’)で生じた混合液に振動を与える工程;
(g’)工程(f’)で振動を与えた混合液にアルコール性溶液を添加する工程;及び
(h’)工程(g’)でアルコール性溶液を添加した混合液の上清を限外ろ過フィルターに通して閉環状DNAを回収する工程
を含んでなる請求項1記載の方法。
【請求項6】
工程(b’)のバッファーもしくは培地が高張液である請求項5記載の方法。
【請求項7】
細胞が単細胞生物である請求項1〜6のいずれかに記載の方法。
【請求項8】
単細胞生物が細菌である請求項7記載の方法。
【請求項9】
細菌が大腸菌である請求項8記載の方法。
【請求項10】
細胞が形質転換体である請求項1〜9のいずれかに記載の方法。
【請求項11】
閉環状DNAが、プラスミドDNA、コスミドDNA、P1 DNA、BAC DNA、PAC DNA及びYAC DNAからなる群より選択される請求項1〜10のいずれかに記載の方法。
【請求項12】
請求項1〜11のいずれかに記載の方法に使用するためのキットであって、以下の成分:
(a)少なくとも1種類のアルコール及び/又はケトンを含む溶液;
(b)凝集補助剤;
(c)凝集剤;
(d)細胞溶解液;
(e)アルコール性溶液;
(f)洗浄液;及び
(g)溶出液
を含むキット。
【請求項13】
さらに、以下の要素:
(h)チップ;
(i)チップ止め装置;及び
(j)培養プレート
を含む請求項12記載のキット。
【請求項14】
閉環状DNAを調製するための装置であって、以下の装置:
(A)横移動装置;
(B)分注装置;
(C)上清吸引装置;
(D)振動装置;
(E)プレート吸引装置;及び
(F)ロボットアーム
を含む装置。
【請求項15】
凝集剤を使用して、閉環状DNAを他の細胞構成物から分離する方法。
【図1】


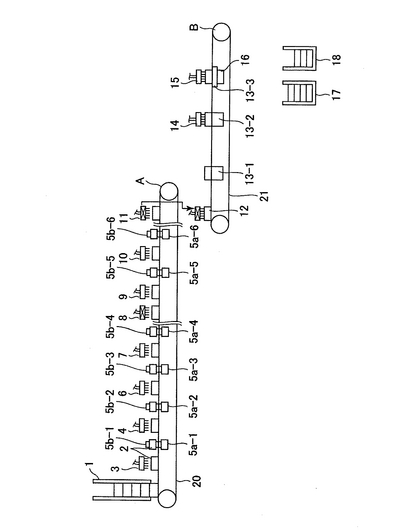
【図2】
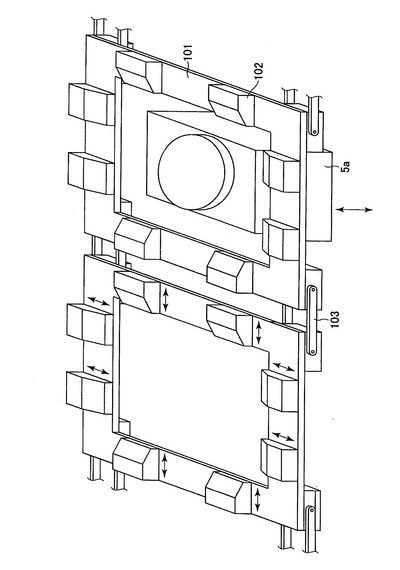
【図3】
【図4】
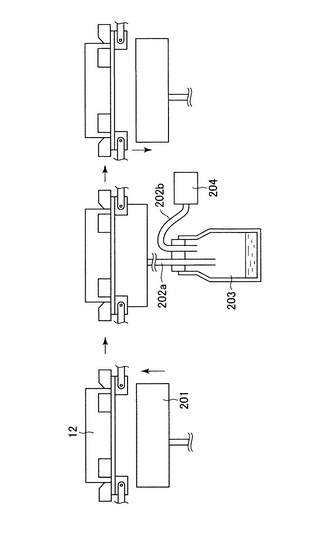
【図5】
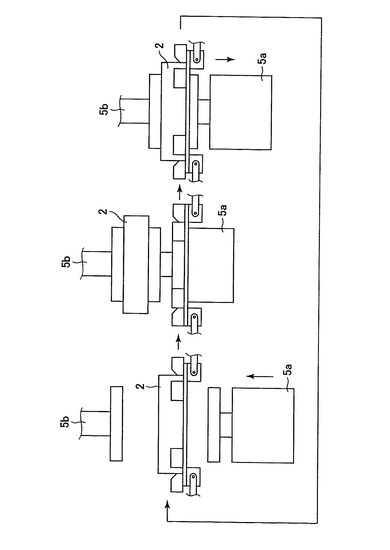
【図6】
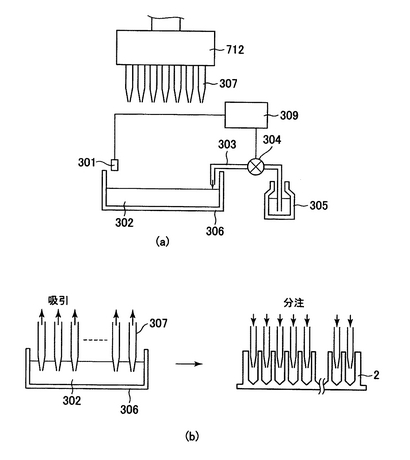
【図7】
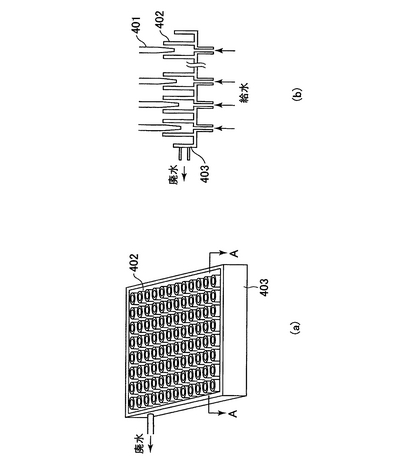
【図8】
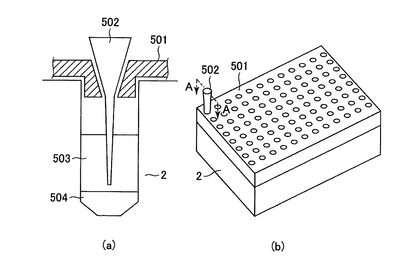
【図9】
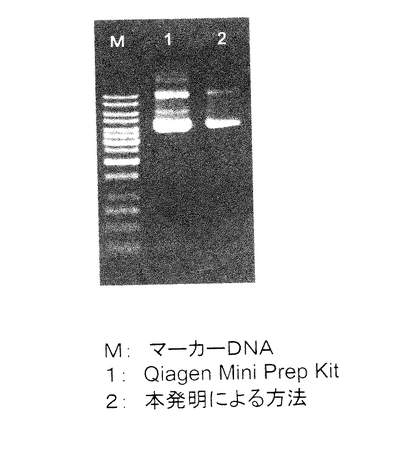
【図10】
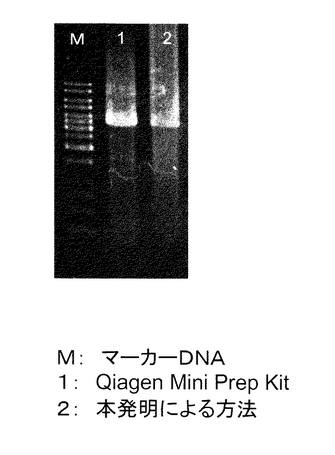
【図11】
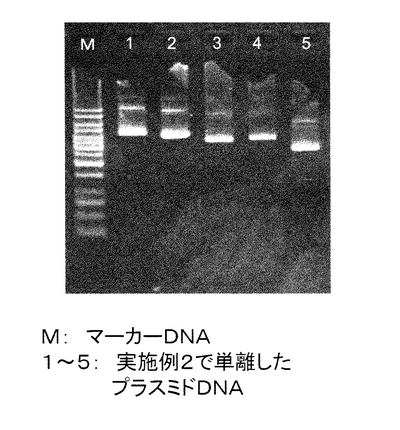
【図12】
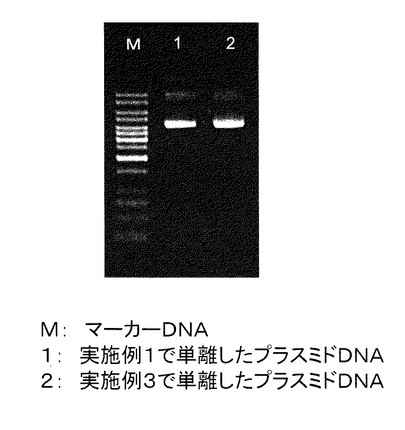
【図13】
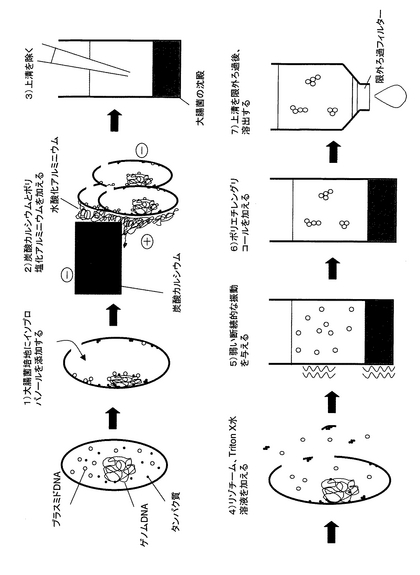
【図14】
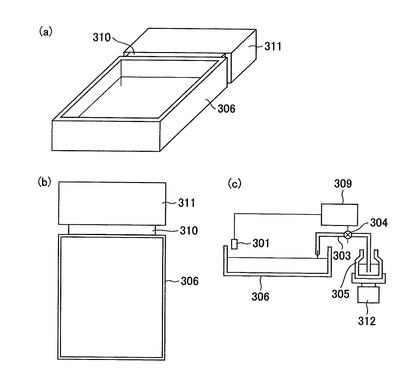
【図15】
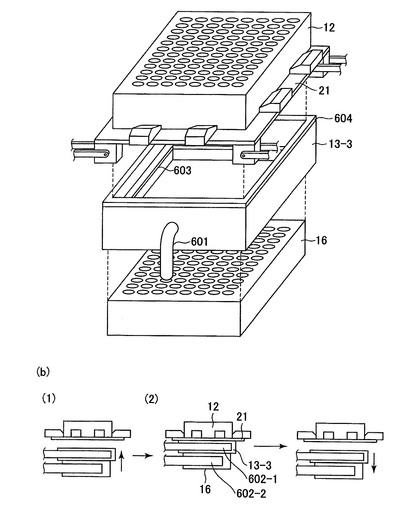
【図16】
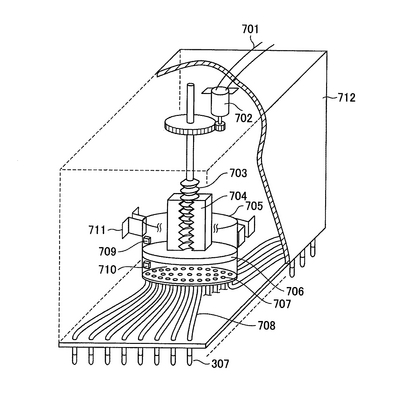
【図17】
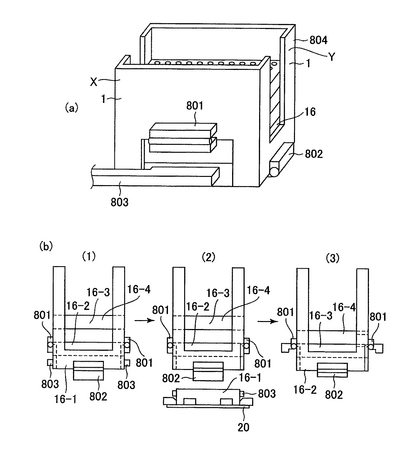
【図18】
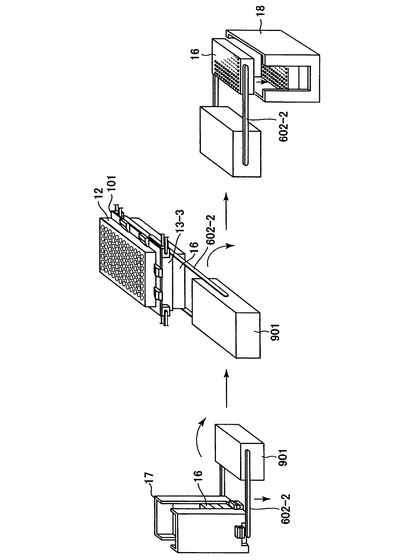
【図19】
【図20】
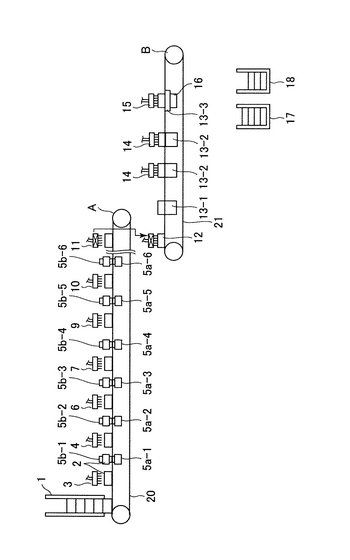
【図21】
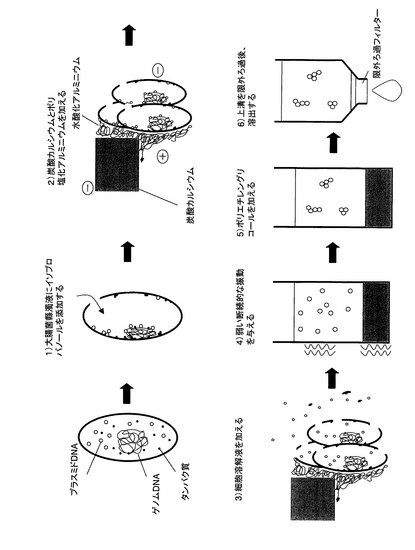
【図22】
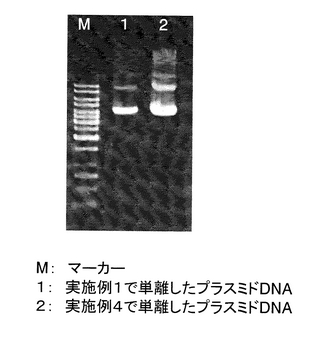
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【図11】
【図12】
【図13】
【図14】
【図15】
【図16】
【図17】
【図18】
【図19】
【図20】
【図21】
【図22】
【公開番号】特開2006−129861(P2006−129861A)
【公開日】平成18年5月25日(2006.5.25)
【国際特許分類】
【出願番号】特願2005−8565(P2005−8565)
【出願日】平成17年1月17日(2005.1.17)
【出願人】(503360115)独立行政法人科学技術振興機構 (1,734)
【出願人】(503359821)独立行政法人理化学研究所 (1,056)
【Fターム(参考)】
【公開日】平成18年5月25日(2006.5.25)
【国際特許分類】
【出願日】平成17年1月17日(2005.1.17)
【出願人】(503360115)独立行政法人科学技術振興機構 (1,734)
【出願人】(503359821)独立行政法人理化学研究所 (1,056)
【Fターム(参考)】
[ Back to top ]