
関心のある安定化された細胞を生産するための手段及び方法
本発明は、関心のある安定化された細胞を生産するための方法であって、関心のある前記細胞中に存在する場合に該関心のある細胞を安定化させることができるところの核酸配列を、該関心のある細胞の幹細胞及び/又は前駆細胞に備えること、前記幹細胞及び/又は前駆細胞を非ヒト動物に備えること、前記動物において、該関心のある細胞の生成を許すこと、及び該関心のある細胞を得ることを含む方法を提供する。前記動物は好ましくは、ヒト幹細胞及び/又はヒト前駆細胞を備えられ、ヒト細胞株の生産を許す。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、細胞生物学の分野に関する。より特に、本発明は安定化された細胞の生産に関する。
【背景技術】
【0002】
細胞のエクスビボ(ex vivo)生産は、多種多様の用途のために調査されてきた。個体から得られるサンプルの細胞は例えば、診断目的のためにエクスビボでさらに培養される。培養された細胞はまた、例えば候補薬化合物の(有用な)効果を確証することを(医学的に)試験するために使用される。その上、細胞は、前記細胞によって分泌される産物、例えば酵素、ホルモン及び/又は抗体を収穫するためにしばしば培養される。モノクロナール抗体(mAbs)の生産は、重要な用途である。モノクロナール抗体は、同じアフィニティを有する抗原に結合し且つ同じエフェクター機能を促進する単一の抗体分子の複数の同一のコピーを表わす。モノクロナール抗体は、抗原上で同じエピトープと反応する。
【0003】
mAbsの恩恵の中には、疾病原因に関与されうる特定の細胞又は化学的メディエーターをターゲットとするそれらの能力がある。この特異性は、より多くの慣用的な治療に渡るmAbsに対する或る臨床上の有利点を与え、一方、一般に低い副作用とともに、有効で、耐容性の良好な治療オプションを患者に与える。
【0004】
モノクロナール抗体は例えば、試験動物、例えばハイブリドーマ細胞を作成するために(マウス)ガン細胞に特定の抗原を用いて免疫性を与えられたマウスから得られるB細胞を融合することによって生産される。B細胞が抗体生産能力を与え、一方ガン細胞がハイブリドーマを無期限に分割し且つ細胞培養において十分に成長することを可能にする。次に、単一のリンパ球からの所望の抗体を生産する個々のハイブリドーマが、大規模培養及びmAb生産のために選択されうる。
【0005】
マウスmAbsはヒトにおける制限された治療に有用である。なぜならば、免疫応答が呼び起こされるからである。それ故に、マウスmAbsは、長期的な治療の恩恵を提供しない。この抗マウス免疫応答はまた、望まれていない副作用を引き起こす。その上、マウスmAbsは、他の重要なヒト免疫システムコンポーネントを効率的に活性化しえない。それ故に、mAbsがより多くのヒトに現れるようにするための努力がとられてきた。1つのアプローチは、抗原にまだ結合するが免疫応答を刺激しそうにないマウス/ヒト−ハイブリッド抗体をエンジニアリングすることだった。マウスCDRをもっぱら作る遺伝子領域が単離され、そして機能的な抗体分子を完成するために要求されるヒト抗体遺伝子領域に接合された。これらCDRを移植された抗体は、90%より多いヒトである。代替的に、ヒトIg遺伝子を保有するマウスが作成された。
【0006】
他のアプローチは、ファージが種々のヒト抗体可変領域(V遺伝子)を保有するように、ヒトV遺伝子についての遺伝子をバクテリオファージの遺伝子内に取り込むことによってファージディスプレイライブラリーを生産することである。変更されたファージ遺伝子は、対応するヒト抗体フラグメントを作るようにファージで感染された細菌を指図し、それはウィルス表面上に自動的に取り込まれる。
【0007】
所望の特異性を有するヒトモノクロナール抗体を直接的に得るために、そのような抗体を生産しうるB細胞を単離すること及びエクスビボでB細胞を培養することは便利であろう。しかしながら、ヒトB細を用いたハイブリドーマ技術はあまり成功していない。なぜならば、結果として生じたハイブリドーマが不安定であるからである。B細胞のエクスビボ培養をするための多くの試みが試されてきた。ヒトナイーブ及びメモリーB細胞は、IL-2、IL-4 及びIL-10を含むサイトカインの存在下でCD40の関与に続いて、制限された期間について培養されうることが十分に立証され(Arpin等、1995年;Banchereau等、1991年)、及びこのシステムは、同起源のプライムされたCD40L発現ヘルパーT細胞に向けてB細胞のイン ビボ(in vivo)応答を模倣することが信じられる。CD40ライゲーションの不存在下で、IL-10単独又はIL-2との組み合わせのIL-10が、抗体を生産する細胞に分化を誘発する(Arpin等、1997年;Malisan等、1996年)。これらの条件下で培養された成熟B細胞の生存及び増殖の調節の機構が部分的にのみ知られている。
【0008】
B細胞上のCD40の関与が、アポトーシスに対する保護、分化の(部分的な)阻害、及びB細胞によるサイトカイン応答性の誘発を含む複数の効果を有する(Foy等、1996年)。大量の細胞周期阻害剤の発現は、Rb-1及びRb-2を含むCD40関与によって減少され(Dadgostar等、2002年)、及びそのような遺伝子の下流制御は、静止から休眠B細胞をリリースしそうである。CD40トリガリング(triggering)は短い増殖応答をもたらすけれども、サイトカインは、トリガーされたB細胞の細胞周期進行を保持するのに役立つ。IL-2及びIL-4は、CD40又は表面Igを刺激されたB細胞の連続的な細胞周期進行を促進する最も効率的なサイトカインである。上記文献において記載されたB細胞培養物は、制限された期間の間に単に安定している。
【0009】
B細胞を不死化するための他のアプローチは、エプスタイン−バー(Epstein-Barr)ウィルス形質転換である。多くの試みが失敗したけれども、Traggiai等はヒトB細胞のエプスタイン−バーウイルス形質転換のための方法を報告し、該細胞は、重症急性呼吸器症候群コロナウイルス(SARS-CoV)感染から回復した患者から得られた。種々のウィルスタンパク質について特異的なモノクロナール抗体が単離された(Traggiai等、2004年)。
【0010】
B細胞を不死化するためのさらに他のアプローチが、国際公開第WO 03/052083号パンフレットに記載されている。該出願は、B細胞を安定化するための方法であって、ヒトB細胞が遺伝子導入マウスから単離され、その後該細胞が構成的に活性なSTAT(Signal Transducer of Activation and Transcription)(CA-STAT)で形質導入される方法を記載する。B細胞の長期の寿命が観察された。しかしながら、形質導入効率は低い(5〜20%の間)。これは、成功の合理的な機会で形質導入を行なうために、ある抗原に特異的に結合する抗体を生産しうる多くのB細胞が単離される必要があることを意味する。1つの特異的な抗原に対して抗体を生産するB細胞の高い量が、しばしば利用できない。
【0011】
ほとんどの先行技術方法の他の不利点は、任意の所望の抗原に対する抗体を生産しうるヒトB細胞を得ることがしばしばできないということである。例えば、自己抗原(例えばサイトカイン(例えば、インターロイキン−10及びTNF−アルファ))、及び組織(例えば、膵臓及び皮膚)又は腫瘍の非腫瘍特異的部分上で発現される抗原(例えば、悪性黒色腫上だけでなく、また正常なメラニン細胞上で発現される抗原)に対するヒトB細胞は、得ることが難しい。その上、ヒトは、所望の抗原、例えば病原体でもちろん免疫化されない。
【発明の開示】
【発明が解決しようとする課題】
【0012】
本発明の目的は、関心のある安定化された細胞を生産するための方法を提供することである。関心のある抗原に対する抗体を生産しうるヒトB細胞株を生産するための方法を提供することがさらなる目的である。
【課題を解決するための手段】
【0013】
1つの観点では、本発明は、関心のある安定化された細胞を生産するための方法であって、
−関心のある前記細胞中に存在する場合に該関心のある細胞を安定化させることができるところの核酸配列を、該関心のある細胞の幹細胞及び/又は前駆細胞に備えること;
−前記幹細胞及び/又は前駆細胞を非ヒト動物に備えること;
−前記動物において、該関心のある細胞の生成を許すこと;及び
−該関心のある細胞を得ること
を含む方法を提供する。
【0014】
好ましくは、前記幹細胞及び/又は前駆細胞が前記安定化する核酸配列を備えられた後に、前記非ヒト細胞が前記幹細胞及び/又は前駆細胞を備えられる。しかしながら、1つの実施態様では、非ヒト動物において既に存在する幹細胞及び/又は前駆細胞が、前記安定化する核酸を備えられる。
【発明を実施するための最良の形態】
【0015】
先行技術方法は、関心のある細胞が単離された後に該関心のある細胞を不死化することに注目するけれども、本発明は、前記関心のある細胞の幹細胞又は前駆細胞を遺伝子的に変更することによって関心のある安定化された細胞を生成することが可能であるという洞察を提供する。本発明に従うと、幹細胞及び/又は前駆細胞が、関心のある細胞を安定化しうる核酸配列で形質導入される場合に、該関心のある安定化された細胞が得られる。好ましくは、前記核酸の発現及び/又は活性は、スイッチオン又はオフされうる。これは例えば、誘導性プロモーターの管理下に前記核酸配列を置くことによって、及び/又は不活性化剤に前記核酸を融合させることによって行なわれる。前記形質導入された幹細胞及び/又は前駆細胞が非ヒト動物に投与された後に、前記幹細胞及び/又は前駆細胞が関心のある安定化された細胞にまだ分化しうる。不死化する核酸配列での幹細胞又は前駆細胞の形質導入は、分化することなしに複製を続ける不死化された幹細胞又は前駆細胞をもたらすだろうことが予測されうる。本発明に従うと、不滅化する核酸配列を備えられた幹細胞又は前駆細胞が、関心のある安定化された細胞にまだ分化しうる。前記関心のある安定化された細胞は好ましくは、安定化されなかった細胞の同じ種類と比較して、成長停止刺激及び/又はアポトーシス性(apoptotic)刺激にそれほど感受性でない。1つの実施態様では、前記関心のある安定化された細胞は、長期の複製寿命を有する。
【0016】
前記核酸配列は好ましくは、自然の環境下で不活性な状態にある。1つの実施態様では、不死化する核酸配列を含む関心のある幹細胞又は前駆細胞は関心のある細胞に分化し、一方前記不死化する核酸配列が活性化される場合に、関心のある結果として生じた細胞は代わりに、複製不完全な分化された細胞に迅速に分化しない。代わりに、前記関心のある結果として生じた細胞は、複製を継続しうる。前記関心のある細胞は前記動物から収穫され、及び好ましくはエクスビボでさらに培養される(例えば細胞株を生産する)。従って、本発明の不死化する核酸配列での幹細胞及び/又は前駆細胞の形質導入は、前記幹細胞及び/又は前駆細胞が分化することを妨げない。前記関心のある結果として生じた細胞は好ましくは、成長停止刺激及び/又はアポトーシス性刺激にそれほど感受性でなく、及び/又は好ましくは、誘導性不死化する核酸配列が活性化される場合に特に、複製不十分な細胞に分化しない又は有意により少ない程度まで分化しない。1つの実施態様では、前記関心のある細胞が前記非ヒト動物に存在する場合に、前記不死化する核酸配列が活性化される。これは例えば、前記非ヒト動物に活性化剤を投与することによって行われる。前記核酸配列は好ましくは、前記関心のある細胞のエクスビボ培養の間に活性化される。前記活性化剤は好ましくは、前記関心のある細胞を含む培地に投与される。
【0017】
本発明の方法は、幹細胞又は前駆細胞の形質導入効率が、関心のあるより多くの分化した細胞(例えばB細胞)の形質導入効率よりも高いという有利点を有する。それ故に、関心のあるより少ない細胞が、前記非ヒト動物から単離される必要がある。関心のある単離され細胞は、単離後に形質導入を経験する関心のある細胞と比較して、前記不死化する核酸配列を含むというより高い頻度を有する。
【0018】
関心のある安定化された細胞は、本明細書において、成長停止刺激及び/又はアポトーシス性刺激にそれほど感受性でなく、及び/又は安定化されていない同じ環境下で同じ種類の細胞と比較して長期の複製寿命を有する細胞として定義される。前記安定化された細胞はまた、本明細書において、不死化された細胞(immortalized cell)と呼ばれる。好ましくは、前記複製寿命は、少なくとも6週間を含む。より好ましくは、前記複製寿命は、少なくとも8週間、より好ましくは少なくとも3ヶ月、より好ましくは少なくとも4ヶ月、より好ましくは少なくとも6ヶ月、より好ましくは少なくとも8ヶ月、より好ましくは少なくとも10ヶ月、より好ましくは少なくとも12ヶ月を含む。
【0019】
前記関心のある細胞は好ましくは、哺乳類の細胞を含む。より好ましくは、前記関心のある細胞はヒト細胞を含む。1つの好ましい実施態様では、前記関心のある細胞は、例えば酵素、ホルモン又は抗体のような所望の製品を生産しうる細胞を含む。所望の製品を生産しうる細胞の安定化は、前記細胞のエクスビボ培養及び前記所望の製品の長期生産を許す。得られた製品は、任意に精製され、そして例えば医薬組成物及び/又は(医学的)試験キットの生産において使用される。本発明の他の好ましい実施態様では、前記細胞が免疫細胞、好ましくはB細胞、T細胞、ナチュラルキラー細胞、又は抗原提示細胞(例えば樹状細胞)を含む。免疫細胞の安定化は、医学研究及び治療を許す。1つの重要な応用は、安定化されたB細胞による(モノクロナール)抗体の生産である。ヒトB細胞株は、本発明の方法を用いて生成され、ヒト抗体生産を許す。従って、本1つの観点では、前記免疫細胞がB細胞、好ましくはヒトB細胞を含む本発明の方法が提供される。他の好ましい実施態様では、前記関心のある細胞がT細胞、より好ましくはヒトT細胞を含む。他の好ましい実施態様では、前記関心のある細胞が、肝細胞、より好ましくはヒト肝細胞を含む。従来技術では、ヒト肝細胞の拡大のための効率的な方法が利用可能でない。本発明の方法が提供された以上、ヒト幹細胞の拡大及び分化が可能になった。本発明の安定化された肝細胞は例えば、例えば急性肝不全を有する患者又は移植への懸け橋として治療するための人工肝臓システムにおける使用するために適している。前記肝細胞は、全体の臓器移植に代わりとして移植において使用するために適している。1つの実施態様では、本発明の安定化された肝細胞は、薬物の代謝を研究するために及び候補薬物化合物を試験するために使用される。
【0020】
語「所望の製品を生産しうる細胞」は、前記製品を生産するその能力を取り消しえないほどに失っていなかった細胞、及び/又は前記製品を生産しうる細胞に分化するその能力を取り消しえないほどに失っていなかった細胞を意味する。例えば、本発明の方法を用いて、複製しうるB細胞を含むB細胞株が生成される。前記B細胞が、形質細胞を生産する抗体に分化するそれらの能力を保持した。
【0021】
好ましい観点では、前記幹細胞及び/又は前駆細胞がヒト幹細胞及び/又は前駆細胞を含む本発明の方法が提供され、従って関心のある安定化されたヒト細胞が得られる。より好ましくは、前記前駆細胞が、CD34+前駆細胞を含む。本発明の方法を用いて、前記ヒト幹細胞及び/又は前駆細胞は、安定化されたヒト細胞に分化する。1つの実施態様では、前記ヒト細胞は、好ましくは活性化剤の投与によって、任意の所望の時点で安定化されうる。前記関心のあるヒト細胞は好ましくは、それが前記動物から得られた後にエクスビボでさらに培養され、ヒト細胞株を生成する。例は、ヒト(モノクロナール)抗体の生産のためのヒトB細胞株及びインスリンの生産のためのヒト膵臓ベータ−細胞株である。
【0022】
前記関心のある細胞中に存在する場合に該関心のある細胞を安定させることができるところの核酸配列がまた、本明細書において、不死化する核酸配列又は安定化する核酸配列と呼ばれる。前記核酸配列を含む幹細胞及び/又は前記細胞から得られた細胞は、成長停止刺激及び/又はアポトーシス性刺激にそれほど感受性でなく、及び/又は前記核酸配列を含まない同じ環境下で同じ種類の細胞と比較して長期の複製寿命を有する。本発明の前記核酸配列は例えば、細胞周期停止を克服し、及び/又は老化、分化及び/又はアポトーシスを妨げうる。好ましくは、前記核酸配列は、前記関心のある細胞の複製を誘発し及び/又は容易にしうる。分化が複製能力の損失(これは最終分化とよばれる)をしばしば含む故に、関心のある細胞における前記核酸配列の存在は好ましくは、例えば前記細胞の分化を妨げることによって複製寿命を延長する。例えば、T細胞依存性B細胞応答の間に、該B細胞は、広範囲の増殖を経験し、それに引き続き抗体生産形質細胞に分化し、それは細胞周期停止によって達成される。1つの実施態様では、本発明の核酸配列は、前記分化及び細胞周期停止を少なくとも部分的に妨げる。
【0023】
好ましい実施態様では、前記核酸配列は、STAT(Signal Transducer of Activation and Transcription)(STAT)タンパク質をコードする遺伝子の少なくとも機能的部分を含む。好ましくは、前記STATタンパク質は、構成的に活性なSTAT(CA-STAT)タンパク質を含む。幾つかのSTATタンパク質が知られている。IL-4がSTAT6及び間接的にSTAT5を活性化すること(Lischke等、1998年;Rolling等、1996年)、一方IL-2がSTAT3及びSTAT5を活性化すること((Leonard及びO'Shea、1998年)においてレビューされた)が従来技術において例えば記載されている。STAT3及びSTAT6分子は、B細胞展開及び分化における或る時点で関与する。STAT6は、クラススイッチ組換えの間、免疫グロブリンアイソタイプ(IgE)の選択に影響を与え(Kaplan等、1996年;Shimoda等、1996年)、一方STAT3は形質細胞分化に関与している(Reljic等、2000年)。B細胞展開及び分化におけるSTAT5の役割は、それほど十分に定義されていない。STAT5、STAT5a及びSTAT5bの2つの既知の形があり、それらは2つの異なる、タンデムに連結された遺伝子によってコードされる。それらは、種々様々の成長因子に対して細胞の応答におけるユニークな役割及び余分な役割の両方を果たす(Teglund等、1998年)。
【0024】
他の好ましい実施態様では、前記核酸配列が、BCL-6をコードする遺伝子の少なくとも機能的部分を含む。BCL-6は転写リプレッサーをコードし、それは非ホジキンリンパ腫(non-Hodgkin's lymphoma)における染色体転座によって頻繁に活性化される。BCL-6に関する染色体転座はプロモーターのみに常に影響し、及びBCL-6のオープンリーディングフレームを完全に残す。BCL-6は、正常なB細胞展開及び成熟のために要求され、及び胚中心の形成のために要求される(Ye、1997年)。BCL-6は、Blimp-1の阻害を通じて形質細胞へのB細胞の分化を妨げる。
【0025】
さらに他の好ましい実施態様では、前記核酸配列が、p53発現を少なくとも部分的に不活性化することができる。腫瘍サプレッサーp53は、照射又は遺伝毒性的メディエーター(genotoxic mediator)への暴露によって生じるDNA損傷に応答して細胞周期及びアポトーシスの調節に重要な役割を果たす。P53は、細胞型及び微環境に依存する細胞周期停止、老化、分化、及びアポトーシスを含む幾つかの細胞性応答を媒介する。変異が造血起源の腫瘍を含むヒト癌におけるp53をコードする遺伝子において生じる一方、正常なヒト造血展開におけるその機能は大部分は未調査のままである。P53は、ヒトテロメラーゼ逆転写酵素(hTERT)の抑制を通じてイン ビトロ(in vitro)で成熟ヒトT細胞の複製寿命を調節する役割を果たす。テロメアは染色体の遠心端でのDNA繰り返しであり、それは染色体末端間融合(end-to-end fusion)に対して保護する。テロメアは各細胞分裂で短くされ、そして非常に短いテロメアを有する細胞が細胞周期停止を経験しそして老いる。テロメア短縮を妨げるhTERTは、TCRを通じて刺激するとT細胞におけるトランジエントに上流調節され、そしてhTERTのドミナントネガティブ(dominant negative)変異体の発現はCD4+及びCD8+の両方の寿命を有意に減少させ、hTERTがヒトT細胞の寿命において調節的な役割を果たすことを示す。例えばRNA干渉(RNAi)によるp53の下流調節は、関心のある細胞(例えば、成熟ヒトT細胞)の寿命を延ばし、及びドミナントネガティブhTERTによる阻害を中和し、p53が初代ヒトT細胞におけるhTERT発現を調節することを示す。
【0026】
本発明に従えば、STATタンパク質及び/又はBCL-6の少なくとも機能的部分、誘導体又は類似体をコードする核酸配列を、幹細胞又は前駆細胞内に取り込むことは、関心のある安定化された細胞をもたらす。その上、p53発現を少なくとも一部不活性化しうる核酸配列(例えば、p53核酸配列をターゲットするRNA干渉誘発性DNAフラグメントのような)を、幹細胞又は前駆細胞内に取り込むことは、関心のある安定化された細胞をもたらす。好ましくは、本発明の核酸配列は、幹細胞又はB細胞前駆細胞内に取り込まれ、安定化されたB細胞及び/又は安定化されうるB細胞をもたらす。該B細胞が好ましくは、エクスビボでさらに培養される。好ましくは、国際公開第WO 03/052083号パンフレット(参照によって本明細書に取り込まれる)に記載されているように、前記STATタンパク質は構成的に活性なSTAT5タンパク質を含む。より好ましくは、前記STATタンパク質は、構成的に活性なSTAT5a、及び/又は構成的に活性なSTAT5bタンパク質を含む。
【0027】
STATタンパク質及び/又はBCL-6の少なくとも機能的部分によって、STATタンパク質及び/又はBCL-6と比較して、関心のある細胞を安定化させる種類において同じ能力を必要とし、量において同じ能力を必要としないタンパク質性分子を意味される。STATタンパク質の機能的部分は例えば、前記能力に関与していない又はほんのわずかのみ関与しているアミノ酸に欠けている。STATタンパク質及び/又はBCL-6の誘導体は、関心のある細胞を安定化する前記タンパク質の能力が種類において本質的に同じであり、量において必ずしも同じ必要はないように変更されたタンパク質として定義される。誘導体は、例えば同類のアミノ酸置換を通じて多くの様式で提供されうる。STATタンパク質及び/又はBCL-6の類似体は、関心のある細胞を安定化する同じ能力を種類において有し、必ずしも量において必要としない分子として定義される。前記類似体は、前記STATタンパク質及び/又はBCL-6から必ずしも得られない。
【0028】
p53発現を少なくとも部分的に不活性化しうる核酸配列は、本明細書において、p53をコードする核酸配列の転写及び/又は翻訳を阻害しうる核酸配列として、又はp53をコードする核酸配列の転写及び/又は翻訳を阻害しうるタンパク質性分子をコードする核酸配列として定義される。p53発現を少なくとも部分的に不活性化しうる核酸配列は例えば、p53又はp53をコードする核酸配列を特異的に結合しうる。1つの観点において、p53発現を少なくとも部分的に不活性化しうる核酸配列は、p53又はp53をコードする核酸配列を特異的に結合しうるタンパク質性分子をコードする。
【0029】
本発明の不死化する核酸の連続的な発現は、必ずしも望まれるとは限らない。例えば、関心のある化合物(例えば、抗体、ホルモン又はタンパク質)の生産は、複製する細胞の分化をしばしば要求する。一旦B細胞株が確立されると、B細胞集団の一部が抗体生産のために選択される。抗体生産は該選択されたB細胞の分化を必要とするので、複製寿命を延長しうる不死化する核酸配列の発現が好ましくは、少なくとも部分的に阻害される。それ故に、本発明の1つの観点において、本発明の前記不死化する核酸配列はスイッチオン及びオフされうる。例えば、培養された細胞の複製が調節される。
【0030】
従来技術では、核酸配列をスイッチオン又はオフする多くの様式が知られている。アンチセンス核酸が本発明の不死化する核酸配列(のmRNA)に結合しうる関心のある細胞内にアンチセンス核酸を導入し、それによってそれを不活性化することが例えば可能である。前記アンチセンス核酸は好ましくは、或る定義された環境下で生成される。一つの実施態様では、本発明の不死化する核酸配列を含む関心のある細胞は、本発明の前記不死化する核酸配列(の発現生成物)を特異的に結合しうるタンパク質性分子をコードする核酸配列を備えられる。前記タンパク質性分子をコードする前記核酸配列は好ましくは、或る定義された環境下でのみ生成され、及び/又は前記タンパク質性分子は好ましくは、或る定義された環境下でのみ本発明の前記不死化する核酸配列(の前記発現生成物)を特異的に結合しうる。当業者に知られているように、核酸配列をスイッチオン又はオフするための多くの代替方法が、従来技術において利用可能である。
【0031】
本発明の不死化する核酸配列は好ましくは、参照によって本明細書に取り込まれる国際公開第WO 03/052083号パンフレットに記載されている方法を用いてスイッチオン又はオフされる。1つの方法は、不活性化剤を、例えばSTAT5タンパク質のような前記不死化する核酸配列(の発現生成物)と関連づけるステップを含む。次に、該不活性化剤は、例えば関心のある細胞の段階に依存してスイッチオン又はオフされる。前記不活性化剤は例えば、誘導性プロモーター/切除システム、例えばcre-lox又はFLP/FRT切除システムを含む。
【0032】
一つの実施態様において、RNA干渉を誘発する核酸が使用される。例えば、二本鎖干渉RNA(RNAi)又はRNAiを生成するDNAが、本発明の不死化する核酸配列、例えばp53の発現を制御するために使用される。
【0033】
好ましい実施態様では、前記不活性化剤は、融合タンパク質として本発明の不死化する核酸配列の発現生成物(例えば、STAT5タンパク質)と関連付けられるタンパク質性分子を含む。好ましくは、前記不活性化剤をコードする核酸配列は、融合タンパク質を生産するために、前記不死化する核酸配列に融合され、そして関心のある細胞において発現される。
【0034】
国際公開第WO 03/052083号パンフレットに概略されているように、前記不活性化剤は好ましくは、前記融合タンパク質の環境を変更することによってスイッチオン又はオフされる。例えば、エストロゲン受容体及びSTAT5は、融合タンパク質(STAT5-ER)として関心のある細胞において生産される。前記エストロゲン受容体がSTAT5への不活性化剤として働く。なぜならば、前記融合タンパク質が、前記STAT5タンパク質が前記核に達することを妨げるサイトソルにおける熱ショックタンパク質と複合体を形成するために、前記融合タンパク質が不活性化されるからである。しかしながら、4-ヒドロキシタモキシフェン (4HT)とインキュベーションすると、前記融合タンパク質(STAT5-ER)が、前記熱ショックタンパク質から分離され、そしてその活性な形で核に輸送される。前記関心のある細胞の複製は、STAT5によって高められる。発現されたSTAT5及びエストロゲン受容体が熱ショックタンパク質と再び関連づけられるようになり、それ故に不活性になるので、4HTの除去は関心のある細胞の成長の停止をもたらす。従って、4HTの除去は、関心のある細胞の複製の終了をもたらす。従って、この実施態様では、前記不活性化剤ERは、4HTを使用してスイッチオン又はオフされる((国際公開第WO 03/052083号パンフレット、14〜15頁)及び(Scheeren等、(2005年) Nature Immunology, 第6巻, 第3号, 第306頁、最後のカラム、最後の段落〜第307頁、最初のカラム、最初の段落)、両方が参照によって本明細書に取り込まれる)。STAT5b-ER融合構成物の生成の非制限的な例が、Scheeren等、(2005年) Nature Immunology, 第6巻, 第3号, 311頁, 最初のカラム, 最初の全段落(3〜14行)に記載されている。この段落は、参照によって本明細書に取り込まれる。
【0035】
不活性化剤の他の例は、転写因子結合部位、好ましくは本発明の不死化する核酸配列の上流又は下流であり、それによって前記核酸配列の発現が、転写調節因子の制御下である。前記核酸の発現は、転写調節因子(トランス活性化因子)の夫々存在又は不存在によって、スイッチオン又はオフされる。前記トランス活性化因子の存在又は不存在は例えば、関心のある細胞においてその発現又は機能を操作することによって制御される。これは例えば、前記関心のある細胞に2つのプラスミド(前記転写調節因子をコードする第1のプラスミド(トランス活性化因子)及び本発明の不死化する核酸配列(例えばSTAT5)をコードする第2のプラスミド)を備えることにより慣用的に達成される。前記第2のプラスミドは好ましくはまた、前記トランス活性化因子のための核酸結合部位(好ましくは、前記不死化する核酸配列の発現が前記トランス活性化因子の制御下であるように前記不死化する核酸配列の上流)を含む。所望の場合、前記不死化する核酸配列の発現は、例えば前記細胞を、トランス活性化因子を不活性化する剤と接触させることによってスイッチオフされる。そのようなシステムの例が、例えばBujard及びその協力者(Gossen等、1995年)によって記載されているように、テトラサイクリン制御可能システムである。
【0036】
幹細胞及び/又は前駆細胞に本発明の不死化する核酸配列を備えるための多くの方法が、当業者に知られている。例えば、リン酸カルシウムトランスフェクション、DEAEデキストラン、エレクトロポレーション又はリポソームを媒介としたトランスフェクションが使用される。代替的に、該核酸の直接注入が用いられる。しかしながら、好ましくは、前記核酸がベクター、好ましくはウィルスベクターによって前記細胞内に導入される。ベクターによって細胞内へ核酸を導入することを云う様々な語が、従来技術において知られている。そのような語の例が、「形質導入」、「トランスフェクション」又は「形質転換」である。不死化する核酸配列を有するベクターを生成するための及び細胞内へ前記ベクターを導入するための技術が、従来技術において知られている。例えば抗生物質耐性のようなマーカー遺伝子又は感受性遺伝子、及び/又はマーカー(例えば、細胞表面抗原又は緑の蛍光タンパク質のような蛍光タンパク質)をコードする遺伝子が好ましくは、従来技術において周知であるように、導入された核酸を含むクローンを識別する際に使用される。
【0037】
前記ベクターは好ましくは、レトロウィルスベクターを含む。より好ましくは、前記ベクターはレンチウィルスベクターを含む。なぜならば、レンチウィルスベクターは、少なくとも2つの有利点を有するからである:それらは分裂する及び分裂しない細胞を効率的に形質導入することが可能であり、及びそれらは、展開の間にサイレンス化されず、安定な、長期の遺伝子発現を提供する。
【0038】
好ましい実施態様では、例えばSTAT若しくはBCL-6をコードする核酸配列、又はp53発現を少なくとも部分的に不活性化することができる核酸配列のような本発明の不死化する核酸配列が、誘導性プロモーターに動作可能にリンクされる。前記不死化する核酸配列の発現は、前記誘導性プロモーターによって調節される。1つの実施態様では、前記誘導性プロモーターは、関心のある細胞において活性であり及び不活性化剤の投与によって不活化される。しかしながら、好ましくは、前記誘導性プロモーターは、低いバックグラウンド発現で関心のある細胞において一般に不活性であり、従って、前記不死化する核酸配列は前記細胞において発現されない又は少しばかり発現される。誘発性剤を投与すると、前記誘導性プロモーターが活性化され、それは前記不死化する核酸配列の発現及び前記関心のある細胞の安定化をもたらす。前記誘発性剤は好ましくは、前記関心のある細胞において天然に存在しない。1つの実施態様では、誘導性プロモーターに動作可能にリンクされた前記不死化する核酸配列を含むベクターが使用される。本発明の方法において使用するために適している誘導性プロモーターが、従来技術において知られている。1つの実施態様では、参照によって本明細書に取り込まれる国際公開第WO 01/20013号パンフレットにおいて記載されているようにテトラサイクリン/又はドキシサイクリン誘発性Tetオペロンが使用される。
【0039】
本発明のベクターは好ましくは、Tet依存性転写調節因子制御システムを含む。Tet依存性転写調節因子制御システムは、イン ビトロ(in vitro)及びイン ビボ(in vivo)の両方での証明された効率を有する最も研究されたシステムのうちの1つである(Freundlich, Baron, Bonin, Gossen and Bujard, Methods Enzymology 第283巻, 第159-173頁, 1997年; Baron and Bujard, Methods Enzymology 第327巻, 第401-421頁, 2000年)。このシステムは、細菌のTn10テトラサイクリンオペレーターに基づき且つTetRが高い特異性で結合する2つのコンポーネント(テトラサイクリンリプレッサータンパク質(TetR)及びテトラサイクリンオペレーター配列(TetO))から構成される。テトラサイクリンの不存在下で、TetRは2量体になり且つTetOに結合し、そして遺伝子発現を妨げる。テトラサイクリンはTetRに結合することができ、構造的な変化を誘発し、TetOからのTetRの分離及び遺伝子発現をもたらす。
【0040】
このシステムは、テトラサイクリン応答性トランス活性化因子(tTA)を作るために単純ヘルペスウィルスのVP16トランス活性化ドメインのTetRへ融合することによって、及びTetOの7個の繰り返しを最小CMVプロモーター(Tet応答エレメント、TRE)へ融合することによってテトラサイクリン応答性プロモータエレメントを設計することによって、哺乳動物システムにおいて使用するために適合された。テトラサイクリン又はドキシサイクリン(テトラサイクリン類似体)の不存在下で、tTAは活性であり且つTREの制御下に置かれた遺伝子の転写を開始する。ドキシサイクリンの添加は、転写活性化を妨げる。
【0041】
しかしながら、好ましくは本発明のベクターは、テトラサイクリン及び/又はドキシサイクリンの不存在下で不活性であり且つテトラサイクリン及び/又はドキシサイクリンの投与によって活性化されることが可能であるTet依存性転写調節因子制御システムを含む。それ故に、本発明のベクターは好ましくは、Urlinger等(Urlinger S; Baron U; Thellmann M; Hasan M; Bujard H; Hillen W. Proc. Natl. Acad. Sci. (2000年) 第97巻, 第14号, 第7963-7968頁)に記載されているようにTetR変異体(誘導性分子が結合し且つ最終置換が二量化ドメインにおいて配置されるところの3つの置換体が配置される4個のアミノ酸置換を含む)を含む。このTetR変異体がVP16 トランス活性化因子ドメインに融合される場合、転写活性化がドキシサイクリンの存在に依存するところの逆の表現型が得られる。この変異体は、rtTAと名付けられる。
【0042】
Urlinger等(Urlinger S等; Proc. Natl. Acad. Sci. (2000年) 第97巻, 第14号, 第7963-7968頁)によって記載されたrtTAは、最大活性化のためにドキシサイクリンの高いレベル(1〜2μg/ml)を要求することを含むいくつかの制限を有し、それは、ドキシサイクリンの不存在下でTREへの残余の親和力及びVP16活性ドメインからの細胞毒性をある組織において容易に到達されえない。それ故に、本発明のベクターはより好ましくは、rtTA2-S2及びrtTA2-M2と呼ばれるrtTAの改善された変異体を含むTet依存性転写調節システムに動作可能にリンクされた不死化する核酸配列を含む(Urlinger S等;Proc. Natl. Acad. Sci. (2000年) 第97巻, 第14号, 第7963-7968頁)。これらの変異体は、TetR上のランダムな且つ定方向突然変異誘発を実行し、そして元々述べられたrtTAよりもよりよく実行した幾つかの変異体を識別することによって生成された。細胞毒性を減少するために、VP16転写活性ドメインが、細胞性転写装置(machinery)の封鎖を最小化するために、「F」型の3つの繰り返しを含む最小活性ドメインに変更される(Urlinger S等;Proc. Natl. Acad. Sci. (2000年) 第97巻, 第14巻, 第7963-7968頁)。
【0043】
ほとんどのTet調節された導入遺伝子発現システムは、2つの個々の構築物に依存し、第1はTRE(Tet応答エレメント)及び関心のある核酸(導入遺伝子)を含み、及び第2は構造性プロモーターによって駆動されるrtTAを含む。このバイナリーシステムを扱うことはイン ビトロ及びイン ビボの両方において挑戦的であることが、そこでは細胞が2つの異なるベクターによる形質導入を要求し、実験の再現性を困難にする。他の困難性は、ダブルトランスジェニック動物を生成するために、2つの個々の形質転換換え動物株及び成功した交差の生成を要求するTet制御された形質転換度物の生成にでくわす。それ故に、本発明者らは、誘導性プロモーター(PtetO7CMVm)、関心のある核酸(導入遺伝子とも呼ばれる)及びrtTA2-S2をシングルカセット内に結合し、そしてこれをレンチウィルスのベクターバックボーン内にクローニングした。2つの異なるカセットが生成され、そこではrtTA2-S2が、CMVプロモーターで構成的に発現され又は自己調節ループ内におかれ、そこではTREは、導入遺伝子及びEMCV IRESエレメントによってリンクされたrtTA2-S2の両方の発現を駆動する。本発明者らは、ヒトの初代及び確立された細胞株のパネルにおいてこれら2つのシステムを試験し、及び自己調節ループが、試験されたすべての細胞株において構成的に発現されたrtTA2-S2よりもよりよく行われたことを見つけた。それ故に、1つの観点では、本発明は、rtTAのための自己調節ループ及び導入遺伝子発現を含むベクターを提供する。そのようなシステムの有利点は、低い誘発されない発現レベルである。なぜならば、rtTA-トランス活性化因子が、ドキシサイクリンの不存在下で合成されないからである。基本的なrtTA発現の毒性が回避される。
【0044】
この概念は、複製−コンピテントHIVベースのベクターの文脈において以前に示された(Verhoef K, MarzioG, Wolfgang H, Bujard H, Berkhout B. Strict control of human immunodeficiency virus type 1 replication by a genetic switch: Tet for Tat. Journal of Virology 2001年;第75巻:第979-987頁)。本発明の好ましいベクターの例が、図15Bにおいて示される。
【0045】
このベクターシステムの潜在的な不利点は、より敏感なTet-システムが発現ループを「キック−スタート」するために要求されることである。ウィルス進化方法(Das AT, Zhou X, Vink M, Klaver B, Verhoef K, Marzio G, Berkhout B.Viral evolution as a tool to improve the tetracycline-regulated gene expression system. Journal of Biological Chemistry 2004年; 第279巻:第18776-18782頁)を介して得られたより敏感なTet-システムを使用して、本発明者らは、この遺伝子発現ループを活性化することができた。変異体rtTA(以下、本明細書でrtTA3と呼ばれる)が、定向進化(Urlinger S等; Proc. Natl. Acad. Sci. (2000年) 第97巻, 第14号, 第7963-7968頁)及びウィルス進化(Das AT等, Journal of Biological Chemistry 2004年; 第279巻:第18776-18782頁;国際公開第WO01/20013号パンフレット)から生成された自己調節カセットにおいて使用された。rtTA3は、野生型rtTAと比較してドキシサイクリンに対して13倍の増加された感受性を示す(Das AT等, Journal of Biological Chemistry 2004年; 第279巻:第18776-18782頁の表1を参照)。従って、本発明は、rtTA3を含むドキシサイクリン調節された導入遺伝子発現レンチウィルスベクターを提供し、導入遺伝子の緊密な(tight)及び段階的な(graded)発現が可能である。好ましい観点では、前記ベクターは、内部リボソームエントリー部位に動作可能にリンクされたrtTA3を含み、従って前記rtTA3は、TREが導入遺伝子及びrtTA3の両方の発現を駆動する自己調節ループ内に置かれる。本発明の方法における前記ベクターの使用がまた、本明細書で提供される。しかしながら、前記ベクターはまた、例えば関心のある細胞が得られた後に該関心のある細胞の形質導入を介して、他の様式において関心のある細胞を不死化するために適している。関心のある細胞を形質導入するためにrtTA3及び関心のある遺伝子を含むベクターの使用がまた、本明細書で提供される。好ましくは、前記rtTA3は、内部リボソームエントリー部位に動作可能にリンクされ、従って前記rtTA3は、TREが前記関心のある遺伝子及びrtTA3の両方の発現を駆動する自己調節ループ内に置かれる。より好ましくは、前記関心のある遺伝子は不死化する核酸配列を含み、従って前記関心のある細胞が安定化される。前記関心のある細胞は、好ましくは肝細胞又は免疫細胞、好ましくはB細胞を含む。
【0046】
その上、本発明のベクターは好ましくは、ヒトB型肝炎ウィルス転写後調節エレメント(HBV PRE)を含む。該HBV PREは、前記遺伝子の転写された領域に含まれる場合に、おそらく核からの接合されていないmRNAのエクスポートを刺激することによって、導入遺伝子の発現を増加させうるシス作用エレメント(cis-acting element)である(Seppen等. Journal of Hepatology第36巻 (2002年) 第459-465頁)。該PREエレメントは、開始コドン及びHBVxタンパク質のほとんどのコード領域を含む。HBVxは、ウィルスの複製のために不可欠であり、及びHBV感染に関連付けられた肝細胞癌の展開に関与している。
【0047】
関心のある不死化する核酸配列に加えて、関心のある細胞に第2の化合物を備えることが時々望まれる。例えば、関心のある細胞のテロメラーゼの短縮化が妨げられるように、該関心のある細胞は好ましくは、テロメラーゼ逆転写酵素がさらに備えられる。前記化合物は例えば、関心のある細胞に前記化合物をコードする核酸配列を備えることによって、該関心のある細胞に備えられる。好ましくは、前記不死化する核酸配列及び前記化合物をコードする核酸の両方を含むベクターが使用される。なぜならば、2つの独立のエレメントの共輸送は初代細胞において効率的でない故に、単一の形質導入工程ベクターの使用は、2つの個々のベクターの使用よりもより挑戦的でないからである。前記追加の化合物をコードする前記核酸配列は例えば、前記誘導性プロモーターと前記不死化する核酸配列との間に挿入される。もちろん、前記挿入は好ましくは、前記不死化する核酸配列を含む下流領域のフレームシフトを生じない。代替的に、前記追加の化合物をコードする前記核酸配列は、前記不死化する核酸配列の下流に挿入される。しかしながら、好ましくは、例えばUnsinger等に記載されたpBI-4からの双方向性プロモーター(Unsinger等, Retroviral vectors for the transduction of autoregulated, bidirectional expression cassettes. Molecular Therapy第4巻, 第5号, 2001年11月, 第484-489頁)のような双方向性プロモーターが使用される。前記追加の化合物をコードする前記核酸配列は好ましくは、前記双方向性プロモーターの一つの部位上に置かれ、一方前記不死化する核酸配列は、前記プロモーターの他の部位上に置かれる。好ましくは、前記双方向性プロモーターは、誘導性プロモーターである。より好ましくは、Unsinger等において記載されたpBI-4からの双方向性プロモーターのような前記プロモーターが、ドキシサイクリンによって誘発可能である。これは、前記核酸配列の(個々の)調節を許す。
【0048】
他の実施態様では、不死化する核酸配列は、不死化する核酸配列−エストロゲン受容体融合構築物としてベクター内に取り込まれる。好ましくは、実施例において記載されるER-STAT5ベクターが使用され、それはタモキシフェンによって誘発可能である(T.D.Littlewood, D.C. Hancock, P.S. Danielian, M.G. Parker and G.I. Evan, A modified oestrogen receptor ligand-binding domain as an improved switch for the regulation of heterologous proteins. Nucleic Acid Res. 第23巻 (1995年), 第1686-1690頁, 参照によって本明細書に取り込まれる)。
【0049】
従って、関心のある細胞中に存在する場合に該関心のある細胞を安定化させることができるところの核酸配列が誘導性プロモーターに動作可能にリンクされる本発明の方法が提供される。好ましくは、前記プロモーターがドキシサイクリン及び/又はタモキシフェンによって誘導可能である。他の好ましい実施態様では、関心のある細胞中に存在する場合に該関心のある細胞を安定化させることができるところの前記核酸配列を含むベクターで、好ましくはレンチウィルスベクターで、前記幹細胞及び/又は前駆細胞が形質導入される。好ましくは、前記幹細胞及び/又は前駆細胞が、ER-STAT5ベクターで形質導入される。より好ましくは、前記幹細胞及び/又は前駆細胞が、誘導性プロモーター(好ましくはPtetO7CMVm)、関心のある核酸及びrtTA2-S2を含むベクターで形質導入される。最も好ましくは、前記幹細胞及び/又は前駆細胞が、誘導性プロモーター(好ましくはPtetO7CMVm)、関心のある核酸及びrtTA3を含むベクターで形質導入される。前記rtTA2-S2及び/又はrtTA3は好ましくは、内部リボソームエントリー部位(IRES)に動作可能にリンクされ、従って前記rtTA2-S2及び/又はrtTA3が自己調節ループに置かれる。誘導性プロモーター(好ましくはPtetO7CMVm)、関心のある核酸及び(好ましくはIRESに動作可能にリンクされた)rtTA3を含む単離された又は組み換えられた核酸配列がまた、本明細書で提供される。前記関心のある核酸は好ましくは、本発明の不死化する核酸配列を含む。その上、本発明のベクターは好ましくは、ヒトB型肝炎ウィルス転写後調節エレメント(HBV PRE)を含む。最も好ましくは、本発明のベクターは図15Bにおいて記載されたベクターを含む。
【0050】
本発明の方法は特に、不死化された免疫細胞を生産するために適している。例えば、造血幹細胞(又はB細胞前駆細胞)は、不死化する核酸配列で形質導入され、そして非ヒト動物に投与される。前記動物から得られたB細胞が安定化される。1つの実施態様では、前記B細胞は、成長停止刺激及び/又はアポトーシス性刺激にそれほど感受性でない。好ましくは、前記B細胞は、長期の複製寿命を有する。好ましくは、前記非ヒト動物が、関心のある免疫細胞が得られる前に、関心のある抗原を備えられる。前記関心のある抗原が前記動物に投与される後、免疫細胞(例えばT細胞)又は前記関心のある前記抗原に対する抗体を生産することができるB細胞が得られる。これらの免疫細胞が不死化する核酸配列を既に含む故に、これらの免疫細胞はエクスビボで継続された培養のために適している。好ましくは、前記核酸は、前記培養の間に活性化される。本発明の1つの実施態様では、ヒトB細胞株が生産される。前記B細胞株が、関心のある任意の抗原に対して(モノクロナール)抗体の生産に適している。好ましくは、前記関心のある抗原は、(好ましくは例えばRSVのような(ウィルスの)病原体によって発現される)抗原、ヒト自己抗原、腫瘍関連抗原、TNF-アルファ及び/又は悪性黒色腫細胞上で発現された抗原の少なくとも免疫原部分である。非ヒト動物が使用される故に、前記動物を前記抗原で免疫化することによって所望の抗原に対するヒトB細胞を生産することが可能である。ヒト自己抗原に対する抗体を生産することができるヒトB細胞がまた生産される。なぜならば、非ヒト動物が使用されるからである。
【0051】
ヒト細胞が非ヒト動物において生産される場合、前記動物の免疫システムが好ましくは、前記ヒト細胞に対して動物免疫応答を少なくとも部分的に避けるために、少なくとも部分的に害される。これは例えば、前記動物が前記ヒト幹細胞及び/又はヒト前駆細胞を備えられる前に前記動物細胞を照射することによって達成される。しかしながら、好ましくは、ノックアウト非ヒト動物の免疫応答を担う少なくとも1つの遺伝子を欠いている該ノックアウト非ヒト動物が使用される。好ましくは、前記動物は、内在性B細胞、内在性T細胞及び/又は内在性ナチュラルキラー細胞の生産に関与する少なくとも1つの遺伝子を本質的に欠いている。ノックアウト動物は例えば、遺伝子サイレンシングによって、又は従来技術において周知の方法を使用し変異を導入することによって、生産される。遺伝子サイレンシングは例えば、前記遺伝子(の発現生成物)を特異的に結合することができる化合物を前記動物に提供することによって実行される。前記化合物は例えば、タンパク質又はアンチセンスRNAを含む。変異は例えば、部位特異的変異を使用して誘発される。ノックアウト非ヒト動物を生産するための多くの代替方法が従来技術において知られており、それは本明細書において更なる説明を必要としない。従って、前記動物が内在性造血細胞の少なくとも1つの種類を本質的に欠いているところの本発明の方法が提供される。
【0052】
好ましい実施態様では、内在性B細胞、内在性T細胞及び/又は内在性ナチュラルキラー(NK)細胞が本質的に欠けている非ヒト動物が使用される。前記非ヒト動物は好ましくは、マウスを含み、より好ましくはB、T及びNK細胞を欠くダブル変異株であるRAG2-/- γc-/-マウスを含む(Kirberg J, Berns A, von BoehmerH. Peripheral T cell survival requires continual ligation of the T cell receptor to major histocompatibility complex-encoded molecules. J Exp Med. 1997年; 第186巻:第1269-1275頁, 参照によって本明細書に取り込まれる;Weijer K, Uittenbogaart CH, Voordouw A, Couwenberg F, Seppen J, BlomB, Vyth-Dreese FA, Spits H. Intrathymic and extrathymic development of human plasmacytoid dendritic cell precursors in vivo. Blood. 2002年;第99巻:第2752-2759頁, 参照によって本明細書に取り込まれる)。前記マウス内にヒト幹細胞及び/又はヒト造血前駆細胞を移植することは、ヒト造血システムを有するマウスを生じる(Weijer等、2002年(参照によって本明細書に取り込まれる); Traggiai等、2004年)。関心のある生成され安定化されたヒト細胞はネズミ免疫応答によって攻撃されない又は小さな程度まで攻撃されない故に、前記マウスは本発明の方法において使用するために非常に適している。
【0053】
本発明の1つの観点では、前記幹細胞及び/又は前駆細胞が、誕生後1週間以内に前記動物に投与される。好ましくは、前記幹細胞及び/又は前駆細胞は、誕生後3日以内に、より好ましくは誕生後1日以内に前記動物に投与される。初期の注入は、増加されたT細胞及びB細胞移植を生じることが本発明者らによって示された。
【0054】
本発明の方法を用いて、関心のある安定化された細胞が得られ、それはさらにエクスビボで培養されることができ、例えば細胞株を生産する。それ故に、本発明は、本発明の方法によって入手可能な関心のある安定化された細胞を提供する。好ましくは、前記関心のある細胞は、免疫細胞、例えばB細胞、T細胞又は肝細胞を含む。最も好ましくは、前記関心のある細胞は、ヒトB細胞又はヒト肝細胞を含む。関心のある細胞の単離された幹細胞及び/又は前駆細胞(関心のある細胞中に存在する場合に該関心のある細胞を安定化させることができるところの核酸配列を含む前記幹細胞及び/又は前駆細胞)がまた、本明細書で提供される。ヒト複製細胞が、治療、医学的試験及び/又は関心のあるヒト化合物(例えばヒト抗体、ヒトホルモン及び/又はヒトタンパク質(例えばヒト酵素))の生産に特に適している故に、特に、関心のある安定化されたヒト細胞の生産が提供される。それ故に、本発明は好ましくは、前記先駆細胞がヒトCD34+前駆細胞を含むところの本発明の方法を提供する。好ましくは、本発明の幹細胞及び/又は前駆細胞は、STATタンパク質及び/又はBCL-6をコードする遺伝子の少なくとも機能的部分、及び/又はp53発現を少なくとも部分的に不活化することができる核酸配列を含む。STATタンパク質は好ましくは、構成的に活性なSTATタンパク質を含む。好ましい実施態様では、前記幹細胞及び/又は前駆細胞は、本発明のベクターによって形質導入される。最も好ましくは、前記ベクターは誘導性プロモーターを含み、従って前記不死化する核酸配列の発現が調節される。幹細胞及び/又は前駆細胞及び/又は本発明の関心のある細胞を含む非ヒト動物がまた、本明細書で提供される。前記動物は、関心のある細胞(例えばヒトB細胞)、好ましくは関心のある抗原に対する抗体を生産することができる細胞を生産するために適している。
【0055】
本発明の方法は特に、安定化されたB細胞の生産のために適している。従って、本発明は、B細胞株を生産するための方法であって、
−本発明の方法を用いて、B細胞を得ること、及び
−前記B細胞をエクスビボ(ex vivo)で培養すること
を含む方法を提供する。
【0056】
前記B細胞は好ましくは、ヒトB細胞を含む。前記安定化されたB細胞が特に、(モノクロナール)抗体の生産のために適している。関心のある抗原に対して特異的に向けられる抗体を生産することができるB細胞を得るために、前記関心のある抗原が、(ヒト)免疫応答を呼び出すために前記非ヒト動物に投与される。
【0057】
従って、本発明は、関心のある抗原に対して特異的に向けられた抗体を生産するための方法であって、
−核酸配列がB細胞中に存在する場合に前記B細胞を安定化させることができるところの核酸配列をB細胞の幹細胞及び/又は前駆細胞に備えること;
−前記幹細胞及び/又は前駆細胞を非ヒト動物に備えること;
−該関心のある抗原を前記非ヒト動物に備えること;
−該関心のある抗原に対して特異的に向けられた抗体を生産するB細胞の生成を許すこと;
−前記B細胞を得ること;及び
−前記B細胞によって生産された抗体を収穫すること
を含む方法を提供する。
【0058】
好ましくは、前記前駆細胞が、STATタンパク質及び/又はBCL-6をコードする遺伝子の少なくとも機能的部分、及び/又はp53発現を少なくとも部分的に不活化することができる核酸配列を備えられる。好ましくは、前記B細胞が前記動物から得られた後に、それがエクスビボでさらに培養される。1つの実施態様では、B細胞株が生成される。安定化されたヒトB細胞の生産が好ましい故に、前記前駆細胞は好ましくはヒトCD34+前駆細胞を含む。
【0059】
1つの実施態様では、前記非ヒト動物がマウスを含む。好ましくは、前記マウスはヒト造血システムの生成のために特に適している故に、前記マウスがRAG2-/-γc-/-マウスを含む。
【0060】
本発明は、下記の実施例によってさらに示される。該実施例は、本発明の範囲を何らかの方法において制限するものではない。
【0061】
図面の簡単な説明
図1
ヒト造血前駆細胞におけるp53ノック−ダウン。
(A)レンチウィルスのRNA干渉ベクターpTRIPΔU3-EF1アルファ p53の概要図。予測されるショートヘアピンRNAをターゲットとするヒトp53が示される。
(B)ヒト造血前駆細胞(CD34+)が単離され、そしてレンチウィルスベクターpTRIPΔU3-EF1アルファp53で形質導入され、そして1週間後にサイトカインでの培養において、GFPの発現に基づいて分離(ソート)された。次に、細胞がガンマ線照射され、そして6時間後に、細胞全体抽出物が調製され、10% SDS-ポリアクリルアミドゲル電気泳動上で分離され、そしてヒトp53を検出するために免疫ブロットされた。該ブロットは、ローディング対照としてベータアクチンに対する抗体でリプローブされた。
【0062】
図2
1日目、1週目又は2週目の齢における新生RAG2-/-γc-/-マウスのCD34+FL細胞を腹腔内移植。
4匹の新生RAG2-/- γc-/-マウスの3つのグループ(夫々、1日目(Δ--Δ)、1週目(O--O)又は2週目(x--x))が、0.5x106 MACS選択されたCD34+FL細胞を腹腔的に移植された。4、8及び12週で、すべてのマウスが採血され、そして該血液がCD45+ヒト細胞のパーセンテージを決定するためにFACSによる分析に付された。
【0063】
図3
胎児の肝臓からのCD34+造血幹細胞をip注射後の11週目でのRAG2-/- γc-/-マウスの胸腺の再増殖(Repopulation)。
胸腺のFACSプロファイル。胸腺細胞が、幹細胞マーカー(CD34及びCD38)、T細胞マーカー(TCRアルファ・ベータ及びTCRガンマ・デルタ、CD1、CD3、CD4、CD8、CD25、CD45RA)、NK細胞マーカー(CD16及びCD56)、形質細胞様DCマーカー(CD123及びBDCA2)、及びB細胞マーカー(CD19)に対する抗体で染色された。CD16及びCD56の分析における該細胞は、CD45+細胞上だけでなくCD3-細胞上においてもゲートされた。
【0064】
図4
胎児の肝臓からのCD34+造血幹細胞をip注射後の11週目でのRAG2-/- γc-/-マウスの骨髄、脾臓、肝臓及び肺の再増殖(repopulation )。
(A)骨髄のFACSプロファイル。骨髄細胞が、幹細胞マーカー(CD34及びCD38)、T細胞マーカー(CD3、CD4、CD25)、NK細胞マーカー(CD16及びCD56)、形質細胞様DCマーカー(CD123及びBDCA2)、B細胞マーカー(CD11、CD19、CD20、IgM及びIgD)、及び骨髄性細胞マーカー(CD11c及びCD14)に対する抗体で染色された。IgM及びIgDの分析における該細胞は、CD45+細胞上だけでなくCD19+細胞上においてもゲートされた。
(B)脾臓のFACSプロファイル。脾臓細胞は、骨髄細胞と同じ抗体で染色された。
(C)肝臓のFACSプロファイル。肝細胞が、幹細胞マーカー(CD34及びCD38)、T細胞マーカー(CD3)、形質細胞様DCマーカー(CD123及びCD4)、及びB細胞マーカー(CD19)に対する抗体で染色された。
(D)肺のFACSプロファイル。肺細胞が、T細胞マーカー(CD3)、形質細胞様DCマーカー(CD123及びCD4)、及びB細胞マーカー(CD19)に対する抗体で染色された。
【0065】
図5
レンチウィルスで形質導入されたヒト造血前駆細胞を移植されたマウスにおけるGFP+発現細胞の多分化能(multilineage)存在。
新生RAG2-/- γc-/-マウスは、胎児の肝臓から単離され且つpTRIPΔU3-EF1アルファp53 RNAiレンチウィルスで形質導入されたヒトCD34+造血幹細胞をip注射された。これらマウスの胸腺及び脾臓からの単核細胞懸濁液が、様々なヒト特異的モノクロナール抗体で染色され、そしてフローサイトメトリーによって分析された。ヒトCD45についてポジティブな細胞がゲートされ、そしてGFPポジティブ及びネガティブ集団を比較してさらに分析された。胸腺及び脾臓におけるT細胞発生の例が与えられる。細胞は、CD3、CD4、CD8及びCD56に向けられた抗体で染色され、そしてFACSによって分析された。
【0066】
図6
レンチウィルスで形質導入されたヒト造血前駆細胞を移植されたマウスにおけるp53i-GFP+を発現するT細胞からのVベータファミリー図。
【0067】
図7
レンチウィルス構築物は、形質導入されたヒトCD34+前駆細胞から得られた成熟細胞中に存在し且つ活性である。
(A)T細胞が、形質導入されたヒトCD34+細胞で再構築されたマウスの脾臓及び末梢血から単離され、イン ビトロで拡大され、そしてGFPの発現に基づいて分離(ソート)された。次に、細胞がガンマ線照射され、そして6時間後に、細胞全体の抽出物が調製され、10% SDSポリアクリルアミドゲル電気泳動上で分離され、そしてヒトp53を検出するためにイムノブロッティングされた。ベータアクチンに対する抗体でのウェスタンブロットが、対照として使用された。2匹の動物からの結果が示される。(B)p53iを発現するT細胞又は対照-GFP構築物が種々の刺激で処理され、そしてプロピジウムアイオダイド(PI)とアネキシン(Annexin) Vダブル染色の組み合わせ及びフローサイトメトリーを使用してアポトーシス誘発のために24時間後にアッセイされた。ダブルネガティブ細胞が、(パーセンテージによって示される)生存可能集団を示す。
【0068】
図8
p53の減少された含量は、ヒトCD34+前駆細胞に由来する成熟T細胞の増殖を好む。実施例1の材料及び方法において記載されているように、T細胞が、形質導入されたヒトCD34+細胞で再構築されたマウスの脾臓及び末梢血から単離され、そしてイン ビトロで拡大された。GFPポジティブ細胞のパーセンテージが、刺激後の2つの異なる時間点でのフローサイトメトリーによって確立された。
【0069】
図9
レンチウィルスで形質導入されたヒト造血前駆細胞を移植されたマウスの骨髄及び肝臓におけるGFP+発現細胞の多分化能(multilineage)存在。
新生RAG2-/- γc-/-マウスは、胎児の肝臓から単離され且つpTRIPΔU3-EF1アルファp53 RNAiレンチウィルスで形質導入されたヒトCD34+造血幹細胞をip注射された。これらマウスの肝臓及び骨髄からの単核細胞懸濁液が、様々なヒト特異的モノクロナール抗体で染色され、そしてフローサイトメトリーによって分析された。ヒトCD45についてポジティブな細胞がゲートされ、そしてGFPポジティブ及びネガティブ集団を比較してさらに分析された。形質細胞様DC、骨髄性及びB細胞の展開の例が示される。肝臓及び骨髄細胞は、CD123、BDCA2、CD11c、CD14、CD10、CD34、CD10、CD19、CD20、IgM及びIgDに向けられた抗体で染色される。
【0070】
図10
細胞パネル7TetOmCMVCMV対IRES
【0071】
図11
形質導入されたヒト繊維芽細胞MOI 1のrtTAウェスタンブロット
HeLa溶解物のウェスタンブロット
レーン1 mock形質導入された細胞、2 Lenti pgk eGFP、3、4 TRE d2eGFP CMV rtTA2-S2、5、6 TRE d2eGFP IRES rtTA2-S2。
【0072】
図12
繰り返し誘発結果(MOI0.1 HeLa)
HeLa細胞は、pgk eGFP, TRE d2eGFP CMVrtTA2-S2又はTRE d2eGFP IRES rtTA2-S2のいずれかでMOI 0.1で形質導入された。最初の誘発でGFPポジティブ細胞の数が、最初の誘発に対して正規化されたすべての他の測定で100%に設定された。
【0073】
図13
最大GFP発現のための時間
72時間でのMFIは、誘発の最大レベルとして設定され、そしてすべての他の値がこの値のパーセンテージとして基づく。
【0074】
図14
濃度(dox用量反応)7C1及び7I1ヒトFib MOI0.1
【0075】
図15
誘発可能なレンチウィルスベクターの概要図。
A:rtTAの構成的発現を有するベクター、例えばTetO7mCMV CMV rtTA3
B:自己調節発現を有するベクター、例えばTetO7mCMV IRES rtTA-S2又はTetO7mCMVIRES rtTA3。
【0076】
図16
イン ビボ及びイン ビトロにおけるヒトB細胞のSTAT5の検出
(A)ホルマリン固定され、パラフィンを埋め込まれた扁桃腺組織が、実施例3の方法の項に概説されているように、pTyr-STAT5(緑)及びCD20(赤)のためにダブル染色された。胚中心では、pTyr-STAT5及びCD20の共局在化が、暗い領域及び明るいゾーン領域においてB細胞上で観察され、パッチされ、膜様染色パターンとして観察可能である。
(B)ダブル染色されたアイソタイプ対照染色。結果は3つの実験についての代表である。元の倍率x 800。
(C)ヒトB細胞が、IL-2及びIL-4とともにCD40L-L細胞上で、示された時間について培養され、そして全体の細胞溶解物がSTAT5タンパク質のレベルについてウェスタンブロット分析によって分析された。該ブロットが取られ、そしてレーンの等しいローディングを保証するためにアクチンに対する抗体で再プローブされた。
(D)STAT5mRNAを特異的にターゲットするsiRNAプローブ(1365及び1668)のノックダウン効率。Raji B細胞株が、pSIN-対照-GFP、pSIN-siSTAT5(1365)-GFP又はpSIN-siSTAT5(1668)-GFPのいずれかで形質導入された。GFP+細胞の全体の細胞溶解物が、STAT5タンパク質発現について分析された。等しいローディングが、ブラッドフォード(Bradford)試験を使用して、タンパク質測定後に保証された。
(E)pSIN-対照-GFP又はpSIN-siSTAT5(1668)-GFPのいずれかで形質導入されたヒトB細胞の増殖応答。GFP+B細胞が分離(ソート)され、そして3日間(その最後の18時間はチミジン(1μCi/ウェル)の存在下(Chan等)で)、CD40L-L細胞、IL-2及びIL-4とともに96ウェルプレートにおいて2重に培養された。チミジンの取り込み(Chan等)は、分当たりのカウント(cpm)として表される。
【0077】
図17
CA-STAT5bの発現はB細胞の生存及び拡大をもたらし、一方WT-STAT5bの発現は生存のみをもたらす。
(A)CD19+B細胞が、扁桃腺から分離(ソート)され、CD40L、IL-2及びIL-4とともに培養され、そして対照-IRES-GFP(開いた四角)、WT-STAT5b-IRES-GFP(閉じた丸)又はCA-STAT5b-IRES-GFP(開いた丸)で形質導入され、そしてCD40L、IL-2及びIL-4でさらに培養された。示された時点でのフローサイトメトリック分析によって決定されたGFP+細胞のパーセンテージが示される。3つのうちからの代表的な実験が示される。
(B)絶対的な細胞数カウントが、(A)において決定されたGFP+細胞のパーセンテージに基づいて決定された;対照-IRES-GFP(開いた四角)、WT-STAT5b-IRES-GFP(閉じた丸)又はCA-STAT5b-IRES-GFP(開いた丸)。
(C)CD40L、IL-2及びIL-4において3ヶ月間培養されたCA-STAT5b-IRES-GFPを形質導入されたB細胞上での対照抗体染色(薄い線)と一致するアイソタイプと比較され、フローサイトメトリック分析によって決定されたCD19及びCD20(太い線)の発現。
(D)CD40L、IL-2及びIL-4において3か月間培養されたCA-STAT5b-IRES-GFPを形質導入されたB細胞におけるVH-Cμ遺伝子組み換えのRT-PCR分析。
【0078】
図18
CA-STAT5b-ERは、4HT依存様式において核に局在化する。
(A)CA-STAT5b-ER-IRES-ΔNGFRで形質導入されたB細胞が、4HTの存在しない(左パネル)又は存在する(右パネル)において、抗-NGFR-FITC(緑)及び抗-ER-TexasRed(赤)で染色され、そして共焦点レーザースキャン顕微鏡(Confocal Laser Scan Microscopy)によって分析された。
(B)STAT5(右)又はER(左)に対する抗体を使用して、CA-STAT5b-ER-IRES-ΔNGFR+ B細胞の細胞性抽出のウェスタンブロット分析。
【0079】
図19
CA-STAT5b-ERで形質導入されたB細胞の特徴
(A)扁桃腺B細胞が、CD40L、IL-2及びIL-4において培養され、そしてCA-STAT5b-ER-IRES-ΔNGFR (丸)で又は対照-IRES-ΔNGFR (四角)で形質導入された。両方の培養物が分けられ、一部は1μM 4HTなし(開いた記号)で及び他は1μM 4HTあり(閉じた記号)で培養された。形質導入後の示された時点では、形質導入された細胞のパーセンテージが、NGFRに対するAPCコンジュゲートされた抗体で染色後にフローサイトメトリック分析によって決定された。絶対的な細胞数カウントが、ΔNGFR+細胞のパーセンテージに基づいて決定された。示された実験は、行なわれた5つの独立実験についての代表である。
(B)CA-STAT5b-ER-IRES-ΔNGFRを形質導入されたB細胞が、形質導入の30日後に、4HTなしで7日間培養された。次に、等しい数の細胞が、4HTの存在(閉じた四角)又は4HTの存在なし(開いた四角)のいずれかで培養され、そして絶対的な細胞数が決定された。3つのうちからの代表的な実験が示される。
(C)4HTの存在下でCD40L、IL-2及びIL-4において培養されたCA-STAT5b-ER-IRES-ΔNGFRを形質導入されたB細胞の代表的な培養物のフローサイトメトリック分析。アイソタイプ対照染色が、薄いヒストグラムとして示される。
【0080】
図20
BCL-6-IRES-GFPで形質導入され、そしてCD40L、IL-2及びIL-4において拡大された末梢B細胞の特徴
(A)対照-IRES-GFP(開いた記号)又はBCL-6-IRES-GFP (閉じた記号)で形質導入され、そして示された期間についてCD40L、IL-2及びIL-4において培養された末梢血B細胞のGFP発現のフローサイトメトリック分析及び絶対細胞数カウント。この実験は、行なわれた8つの独立の実験についての代表である。
(B)フローサイトメトリック分析によって決定された、形質導入の77日間、CD40L、IL-2及びIL-4において培養された代表的な実験のBCL-6を形質導入されたB細胞の表現型。薄いヒストグラム線は、アイソタイプ対照染色を示す。
【0081】
図21
STAT5によるBCL-6発現の誘発
(A)4HTとともに、CD40L、IL-2及びIL-4において培養されたCA-STAT5b-ERで形質導入されたB細胞におけるBCL-6及びHPRTのRT-PCR分析。10日間、4HTなしで該細胞のインキュベーション後、4HTが、シクロヘキサミド(CH)の存在下又は不存在下で8時間添加された。この実験は3回繰り返され、同様の結果だった。
(B)抗-BCL-6抗体を使用してウェスタンブロット分析によって決定された、12日間、常に4HTの存在下で又は4HTの不存在下で、CA-STAT5b-ERを形質導入されたB細胞におけるBCL-6タンパク質の発現。アクチンに対する抗体が、タンパク質ローディング対照として使用された。同様の結果が、3つの独立した実験において得られた。
(C)デュアルルシフェラーゼリポーター分析。BCL-6遺伝子のプロモーター領域(位置-657/+471 bp)の一部が、ホタル(P. pyralis)ルシフェラーゼをコードする配列にリンクされ、そしてpGL3ベクター内にサブクローニングされた。このプロモーター領域(548/-566 bp)の1つの潜在的なSTAT5結合部位は、TTCTCTGAAからGTCTCTAAAに変異され、及び2つの異なるクローン(BCL-6*及びBCL-6**)がpGL3-ホタル・ルシフェラーゼ・バックボーン内にサブクローニングされた。これらpGL3構築物は、293T細胞内にLZRS-CA-STAT5b及びRenillaルシフェラーゼを含むベクターで共形質導入された。両方のルシフェラーゼ活性が、単一のサンプルから連続的に測定され、そして相対ルシフェラーゼ単位がRenillaルシフェラーゼ活性について計算され及び修正された。
【0082】
図22
CA-STAT5bを形質導入されたB細胞の成長に対するBCL-6ノックダウンの影響。
(A)BCL-6siRNAプローブのノックダウン効率が、BCL-6を形質導入された293T細胞内へのトランスフェクション後に、ウェスタンブロット分析によって試験された。除去後、該ブロットは、等しいタンパク質ローディングを保証するためにアクチンに対する抗体でインキュベーションされた。
(B)対照-GFP(開いた円)、siBCL-6(789)-GFP(閉じた四角)、又はsiBCL-6(970)-GFP(閉じた菱形)で形質導入され、そして示された期間の間、CD40L、IL-2及びIL-4において培養されたCA-STAT5b-ER-ΔNGFR+B細胞におけるGFP発現のフローサイトメトリック分析。この実験は4人の異なるドナーからのB細胞において繰り返され、同様の結果だった。
【0083】
実施例
【実施例1】
【0084】
材料及び方法
マウス
H-2dRAG2-/-マウス(Antonius Rolink博士及び Shunichi Takeda博士によって親切に提供された;バーゼル免疫学研究所、バーゼル、スイス国(Takeda S, Rodewald HR, Arakawa H, Bluethmann H, Shimizu T. Immunity.1996年; 第5巻:第217-228頁))が、IL-2Rγc-/-マウスとかけあわされて(Blom B, Spits H, Krimpenfort P. In: Smit Sibinga CT, Das PC, Lowenberg B編集 Cytokines and Growth Factors in Blood Transfusion. London: Kluwer Academic Publishers;1997年:第3-12頁)、H-2d RAG2-/- IL-2Rγc-/-マウスが得られた (RAG2-/-γc-/- マウスといわれる:Weijer K, Uittenbogaart CH, Voordouw A, Couwenberg F, Seppen J, Blom B, Vyth-Dreese FA, Spits H. Blood. 2002年;第99巻:第2752-2759頁; Kirberg J, Berns A, von Boehmer H. J Exp Med. 1997年; 第186巻:第1269-1275頁)。これらマウスがT、B及びNKリンパ細胞の完全な欠如を示すので、これらマウスは免疫不全である。マウスはアイソレーターにおいて飼育され且つ維持され、そしてオートクレーブされた食餌及び水を与えられた。全ての操作は、層流下で行われた。
【0085】
胎児の肝臓及びさい帯血細胞調製
ヒト胎児の肝臓が、人工妊娠中絶から得られた。妊娠期間は、頭の直径の超音波測定によって決定され、及び14〜20週に及んだ。この組織の使用は、オランダ癌研究所(Netherlands Cancer Institute)及びアカデミック医療センター(Academic Medical Center)の両方の医学倫理委員会によって承認され、及びインフォームドコンセントを条件であった。ヒト胎児の肝細胞が、機械的手段による組織の穏やかな破壊によって、引き続きFicoll-Hypaque(Lymphoprep;Nycomed Pharma、オスロ、ノルウェー)上の密度勾配遠心分離によって単離された。単一細胞懸濁液が、組織を刻み、ステンレス鋼メッシュを通じてそれらをプレスすることによって調製された。大きな集合体が除去され、そして該細胞は、CD34+細胞の単離前に培地で1回洗浄された。
【0086】
RAG2-/-γc-/- マウスへのヒトCD34+細胞の移植及びヒト細胞移植の評価
CD34+細胞(フローサイトメトリーによって評価されたときに、>98%純粋、データは示されてない)の富化が、CD34前駆細胞単離キット(Miltenyi Biotec、ベルギッシュグラートバハ、ドイツ)を使用して行なわれた。CD34+細胞(0.2〜200万個の細胞)が、半致命的に照射された(350 cGy)新生RAG2-/- γc-/- マウス(1週未満)内にip移植された。ヒト細胞移植のキネティックス(kinetics)を決定するために、末梢血が、移植後の3〜4週ごとに尾静脈から集められた。マウスが移植後の様々な時点で犠牲にされ、そして末梢血(PB)、肝臓、肺、脾臓、骨髄(BM)及び胸腺が、ヒト細胞の存在について評価された。照射されていないマウスがヒト細胞での再増殖を必ずしも示すとは限らないので、3.5Gyの全身照射(TBI)が行なわれる。単核細胞(MNC)が、Ficoll-Hypaqueを介した密度勾配遠心分離によって単離され、抗ヒトCD45(Becton and Dickinson、サンノゼ、カリフォルニア州)及び他のマーカーで染色された。これらマーカーの発現が、FACS Calibur(Becton and Dickinson)上で測定され、そしてFCS発現プログラム(Denovosoftware、オンタリオ、カナダ)で分析された。該移植されたヒト単核集団は、前方及び側方散乱光パラメーターに基づいて定義され、そして所定のマーカーについてのポジティブ細胞のパーセンテージが、白血球及びCD45+ゲート内にある細胞について決定された。移植されなかったRAG2-/-γc-/- マウスにおける抗ヒトCD45と反応した細胞存在はなかった。ヒト細胞の特異的なサブセットが、抗ヒト特異的mAbsで染色することによって定量され、それは、フルオレセインイソチオシアネート(FITC)、フィコエリトリン(PE)、ペリジニンクロロフィルタンパク質質(PerCP)又はアロフィコシアニン(APC):CD11c、CD19、CD34及びCD45RA(Coulter-Immunotech)、CD3、CD4、CD8、CD10、CD14、CD16、CD20、CD56、CD83及びCD123(Becton and Dickinson);CD38及びCD45RO(Dako、グロストラップ、デンマーク)及びBDCA-2(Miltenyi Biotec)とコンジュゲートされた。
【0087】
レンチウィルス生成
複製不完全な自己不活性化HIVベクターが、FUGENE(Roche、ナットレー、ニュージャージー州)及び3つの異なるプラスミドを使用して、293T細胞のトランジエントトランスフェクションによって生産された。使用された該プラスミドは、VSV-GエンベロープをコードするプラスミドpMD.G、ウィルス粒子を生産するためのGag、Pol、Tat及びRevタンパク質を提供するように設計されたパッケージングプラスミド pCMVDR8.91、及びトランスファーベクターpTRIPデルタU3-E1アルファ(SirvenA, Ravet E, Charneau P, Zennou V, Coulombel L, Guetard D, Pflumio F,Dubart-Kupperschmitt A. Mol Ther. 2001年;第3巻:第438-448頁)であった。pTRIPデルタU3-E1アルファは、Brummelkamp等(Brummelkamp TR, Bernards R, Agami R. Science. 2002年;第296巻:第550-553頁)によって記載されたヒトp53ターゲッティング配列5´GACTCCAGTGGTAATCTACを含むpSUPERベクターからのsiRNAカセットを含むように変更され、及びまた、伸長因子-1-アルファプロモーターによって駆動される拡張されたGFP遺伝子を運ぶ。トランスフェクションの20時間後に、培地が、Yssel培地(YsselH, De Vries JE, Koken M, van Blitterswijk W, Spits H. Methods. 1984年;第72巻:第219-227頁)によって置き換えられ、そしてウィルスの2つのサンプルが、24及び48時間で集められた。ウィルスを含む上清は、細胞を除去するために1800 rpmで10分間遠心分離され、次に0.22 mmのフィルターを通され、そして使用まで-80℃で維持された。
【0088】
形質導入プロトコル
CD34+ヒト細胞の形質導入は、サイトカインの不存在下で、レトロネクチン(宝酒造株式会社、大津、日本)で覆われた24ウェルプレート上でのウィルス上清への一晩の暴露の1サイクルによって行われた。次の日、細胞が洗浄され、そしてマウスにip注射されるか或いは夫々10 ng/mlのトロンボポイエチン、幹細胞因子及びIL-7(全てPeprotechから)の存在下でYssel培地における培養物中に維持されるかのいずれかであった。形質導入の効率が、フローサイトメトリーによって、2〜3日後に、GFPポジティブ細胞のパーセンテージを決定することによって評価された。
【0089】
ウェスタンブロット
全体の細胞抽出物が、氷上で30分間、RIPAバッファーにおいて、等しい数の細胞を溶解することによって調製された。Bio-Radタンパク質アッセイ(Bio-Rad、ミュンヘン、ドイツ)によって決定された等価な量のタンパク質が、10% SDSゲル上にロードされた。P53タンパク質がSanta Cruz Biotechnologyからのモノクロナール抗体DO-1を使用して、及びローディング対照としてベータアクチンがポリクロナール抗体I-19(またSanta Cruzから)を使用して検出された。
【0090】
ヒトT細胞培養
ヒト形質導入された幹細胞で再構築されたRAG2-/- γc-/- マウスの血液及び脾臓からのヒトT細胞の拡大は、(Spits H, Ijssel H, Terhorst C, de Vries JE. J Immunol. 1982年;第128巻:第95-99頁)に記載されているように、2人の異なるドナーからの照射された異質遺伝子型のヒト末梢血細胞(PBL)、及び照射されたEBVを形質導入されたB細胞株(JY)、PHA(Gibco、グランドアイランド、ニューヨーク州)及び組み換えヒトIL-2(rhIL-2;Roche、ナットレー、ニュージャージー州)からなるフィーダー混合物での刺激によって行われた。ヒト細胞を含む全ての培養が、Yssel培地において行われた。
【0091】
アポトーシス細胞の検出
細胞の存続可能性が、ガンマ線照射(3000rad)前若しくは24時間後、又はスタウロスポリン(1μM)又はフルダラビン(5μM)での処理後に、フローサイトメトリーによって分析された。手短に言えば、細胞が収穫され、そして氷で冷やされたHEPESバッファー(10mM HEPES、150 mM KCl、1mM MgCl2及び1.3 mM CaCl2、pH 7.4)において洗浄された。次に、細胞が、20分間、APCをラベル付けされたアネキシンV(Becton Dickinson (サンノゼ、カリフォルニア州、米国))と一緒にインキュベーションされた。フローサイトメトリーによるサンプルの分析の直前に、プロピジウムアイオダイド(PI)(Sigma、セントルイス、ミズーリ州)が添加された(最終濃度 5μg/ml)。生存可能な細胞が、アネキシンV及びPIの染色の両方についてネガティブとして定義された。
【0092】
結果
レンチウィルスを介在した遺伝子導入によるヒトCD34+細胞内へのsiRNAの導入
レンチウィルスは、他の遺伝子輸送システムに関する2つの重要な有利点を有する;それらは、ノンサイクリング細胞(non-cycling cell)を感染させることができ及び展開の間にサイレンシングされない。幾つかのグループは、哺乳動物細胞におけるRNAiの発現のために、小さい核RNAプロモーター(H1 (Brummelkamp TR, Bernards R, Agami R.Science. 2002年;第296巻:第550-553頁)及びU6(Dick JE, Lapidot T, Pflumio F. Immunol Rev. 1991年; 第124巻:第25-43頁))の使用を記載した。H1プロモーターはヒトp53タンパク質をターゲットとするように設計されたsiRNAの発現を駆動するように使用され、そしてsiRNAカセットを発現するレンチウィルスベクターが調製された(図1A)。CD34+細胞の高度に富化された集団がヒト胎児の肝臓から単離され、そして引き続きsiRNA/GFPを発現するレンチウィルスベクターで形質導入された。形質導入効率は、フローサイトメトリーによって決定されたGFP発現のレベルによって示されるように、慣用の様に20〜50%の範囲に及んだ。形質導入された細胞の細胞成長又は生存に対する明白な効果が、標題「材料及び方法」において上記されたサイトカインの存在下で3週間培養後にイン ビトロで観察されなかった。GFPマーカーを発現するCD34+由来細胞の割合は、初期集団のそれと同等のままだった(データは示されていない)。
【0093】
導入された構築物が活性であり且つこれらの細胞においてp53の発現をノックダウンしうることを示すために、我々はそれらのGFP発現に基づいて、形質導入後にCD34+細胞を分離(ソート)した。p53の発現(それは通常の状況において非常に低い)が、ガンマ線照射を含む幾つかの遺伝毒性的刺激への細胞の暴露後に、非常に迅速に上方制御される。形質導入されていない細胞及び対照を形質導入された(示されていない)細胞の両方がp53のレベルを増加させることによって放射線に応答し、一方p53 siRNA構築物を発現する細胞は応答せず(図1B)、該構築物が造血幹細胞における遺伝子発現をサイレンシングできたかったことを示す。
【0094】
T細胞展開及びホメオスタシスに対するp53のノックダウンの効果。
T細胞展開に対するp53ノックダウンの効果を検査するために、我々は、Dick及びその同僚(Dick JE, Lapidot T, Pflumio F. Immunol Rev. 1991年;第124巻:第25-43頁)によって開拓されたヒト-SCIDマウスモデルを用いた。SCIDマウスモデルは、C.B-17-Prkdcscid(SCID)マウス(Lapidot T, Pflumio F, Doedens M, Murdoch B, Williams DE, Dick JE. Science. 1992年; 第255巻:第1137-1141頁)及びより最近ではNOD/LtSz-Prkdcscid (NOD/SCID)マウス(Pflumio F等 Blood. 1996年; 第88巻:第3731-3740頁; 及びBonnet D等, Nat Med. 1997年;第3巻:第730-737頁)のBMを多分化能リンパ細胞及び骨髄細胞で再増殖するためにこれら原始細胞(SCIDを再増殖する細胞と名付けられた)の能力に基づく。しかしながら、T細胞展開の研究について、T細胞の展開が明らかにマウスにおける活性な生得の免疫システムの存在により単に時々観察された故に、これらマウスは制限を有する。最近、RAG2-/- γc-/- マウスに静脈内(iv)注射すると、CD34+細胞がB細胞及び形質細胞様DC(pDC)内に展開しうることが報告された(Weijer K等, Blood. 2002年;第99巻:第2752-2759頁)。さらに、我々は、T細胞が、CD34+細胞のiv注射に引き続きこれらの成体マウスにおいて展開することを観察した(結果は示されていない)。成功率は80%だったけれども、末梢におけるコンシステントなT細胞移植を観察するために、12〜16週平均を要した。その上、これらマウスの胸腺は、0.8×106未満の細胞で小さいままだった。6〜8週齢のRAG2-/- γc-/- マウスの胸腺の微環境がヒトT細胞展開について最適でないかもしれないことを考慮すると、我々は初期の時点でヒト前駆細胞を注射することを決定した。新生児(1週齢未満)、1週又は2週齢のマウスが、胎児の肝臓由来のCD34+細胞をip注射され、そして末梢血が注射後8及び12週間後に分析された。図2は、末梢におけるヒト細胞での移植が、年上のマウスにおけるよりも、1日齢未満のマウスにおいてより高いことを示す。1週齢マウスの注射はまた有意な移植を生じたが、2週齢マウスでは、CD34+細胞のip注入後にヒト細胞の展開がなかった(図2)。
【0095】
誕生後、1〜3日の間でip注射された78匹のマウスの分析は、64匹のマウスにおいてヒトCD45+細胞での再構築(表1)を明らかにした(83%)。32匹のマウが50%を超す再構築を示し、そして残りが10〜50%の間を示した。マウスの末梢血におけるヒトCD45+細胞を有する全てのマウスはまた、胸腺及び他の器官におけるT細胞を有した。
【0096】
【表1】

【0097】
誕生の1日後にip注射されたマウスの胸腺の構造物が、移植後10週で共焦点顕微鏡によって検査された。分化された胸腺は、外皮様の領域におけるCD1+CD4+及びCD4+CD8+細胞、並びに骨髄様領域におけるシングルポジティブCD4+及びCD8+T細胞で観察された(結果は示されていない)。
【0098】
新生マウスモデルをさらに確認にするために、我々は、形質導入されていないCD34+胎児の肝細胞を注射された大きな一連のマウスにおける胸腺及び様々な末梢器官におけるT細胞を最初に分析した。さらに、我々は、他の白血球サブセットの存在につてこれらのマウスを分析した。注射の8〜10週間後に、2〜1000万個の細胞が、胸腺から回収されることができた。広範囲のフローサイトメトリック分析(図3)は、ダブルポジティブCD4+CD8+ 細胞及びCD4+及びCD8+ シングルポジティブT細胞を含む全ての系統サブセットの存在を明らかにした。その上、CD3+high細胞のサブセットは、CD45RA+を発現した。この集団集合は、周囲への移出直前にT細胞を表わす。興味深いことに、CD25は、Stephens等(StephensLA等, Eur J Immunol. 2001年;第31巻:第1247-1254頁)によって以前に記載された制御性T(Treg)細胞でありうるCD4+T細胞のサブセット上で発現された。これらCD25+CD4+細胞がまたグルココルチコイド誘発された腫瘍壊死因子受容体ファミリー関連遺伝子(GITR)及び細胞質CTLA-4についての高度にポジティブであるという観察(結果は示されていない)は、これら細胞がTreg細胞を表わすという概念を支持する。TCRアルファ・ベータ+細胞及びCD3+CD56+に加えて、胸腺がまた、CD3-CD56+NK細胞の実質的な数を含んでいた(発現するCD16のおよそ半分を有する)。NK細胞(2%)のパーセンテージは、子どもの胸腺において通常見られるそれよりもはるかに高かった(<0.1%)。理論への結び付きなしに、マウス胸腺環境が、NK細胞の生成に好ましくすることは可能である。代替的に、NK細胞のはるかに高い割合は、正常なヒト胸腺微環境におけるよりもTCRアルファ・ベータ+細胞の低い拡大率の結果でありうる。我々はまた、TCRガンマ・デルタ+細胞(1.5%対正常な胸腺における0.1%未満)及びB細胞(2.2%対正常な胸腺における0.1%未満)の相対的に高い割合を観察し、及びこれらの高いパーセンテージはまた、TCRアルファ・ベータ+細胞の低い拡大の結果でありうる。NK細胞、B及びTCRガンマ・デルタ細胞の増加したパーセンテージと対照的に、BDCA2+CD123high pDCのそれは正常範囲内であった(0.1%)。
【0099】
胸腺において展開するT細胞は、複数の解剖位置へ移動する。T細胞の可変パーセンテージは、胸腺において見られるだけでなく、骨髄、脾臓、肝臓及び肺においても見られた(図4)。骨髄はT細胞の低い割合を有し(1%)、一方肺においてこのパーセンテージは11%であった。肝臓及び脾臓は、T細胞の同等のパーセンテージを含んでいた。CD4:CD8比(3〜4:1)は、全ての器官において同様であり及びヒトにおいて通常観察される範囲内であった。興味深いことに、脾臓及び骨髄では、Treg細胞及び最近活性化されたCD4+T細胞を推測上含むCD3+CD4+CD25+細胞の集団が検出されうる。我々の結果は、新生マウスへのCD34+細胞のip注射は、大多数のマウスにおけるヒトT細胞の相対的に迅速な再構築を生じることを示す。
【0100】
新生のRAG2-/-γc-/- マウス内に注射された場合、CD34+細胞は複数の系統に展開される。B細胞は、誕生の1日後に注射されたマウスにおける末梢血及び様々な器官において展開された最も支配的な細胞集団であった。CD19+B細胞の最高パーセンテージが、骨髄において見られた(図4A)。意外にではなく、脾臓における大多数のB細胞(図4B)が、CD10+CD20+細胞であり及びIgM及びIgDを共発現した。骨髄では、大多数のB細胞がCD20-CD10+であり、マウス骨がヒトB細胞展開のための部位であることを示唆した。骨髄の検査(図4A)は、CD34+細胞の存在を明らかにし、その小さなパーセンテージはCD38の低レベルだけを発現し、いくつかの原始的なCD34+CD38dim前駆細胞がそれらの分化していない状態にあることを示唆する。
【0101】
pDCの有意な数が、全ての器官において見られた;pDCが、骨髄(図4A)及び肝臓(図4C)において最も明らかに存在するが、また肺(図4D)においても存在した。NK細胞がまた末梢血及び様々な器官において検出されることができたが、該パーセンテージは胸腺におけるよりも一般にはるかに低かった。CD11c+及びCD14+単球が器官において観察され(図4)、ヒトリンパリンパ展開だけでなくヒト脊髄展開がこれらマウスにおいて起きることを示した。
【0102】
次に、我々は、p53siRNAで又はエンプティーベクターで形質導入されたCD34+胎児の肝細胞で一連のマウスに注射した。表2は、GFP+及びGFP-細胞における胸腺細胞のパーセンテージが、p53 siRNA又は対照を形質導入されたCD34+細胞で注射されたマウスにおける両方において同様であったことを示した。同様に、肝臓又は脾臓細胞が分析されたときに、我々は、これらの動物の末梢において有意な(決定的な)違いを検出できなかった。従って、我々は、p53の不活性化がp53 siRNAを発現する末梢のT細胞の生存の有利点をもたらさないと結論した。
【0103】
【表2】
【0104】
p53 siRNA及び対照を形質導入された細胞で注射されたマウスの胸腺の検査が、CD4CD8ダブルネガティブ、ダブルポジティブ及びシングルポジティブT細胞コンパートメントにおけるGFP+細胞の存在を明らかにした。しかしながら、p53siRNA-GFP+上でゲートする場合、我々は、形質導入されていない細胞(GFP-)又は対照-GFP+上での胸腺におけるT及び非T細胞の何らかのサブセットの表現型における差を観察しなかった(図5A)。また、我々は、何らかのp53 siRNA-GFP+におけるT細胞の存在下において、又は脾臓において形質導入されていない細胞上での差は観察されなかった(代表的なマウスのT細胞の表現型が図5において示されている)。
【0105】
DNA損傷に応答して細胞増殖の制御及びアポトーシスの誘発におけるp53の重要な役割を与え、p53の減少したレベルがTCRレパートリーの組成において効果を有しうるかを知ることは興味があった。我々は、p53i構築物で形質導入されたCD34+細胞を注射されたマウスから単離されたT細胞におけるTCR多様性を調査した。PCR分析によって、我々は、全てのVbetaファミリーがレパートリーにおいて表されることを確立できた(図6)。その上、CDR3領域の詳細な分析はこれらのマウスにおいて存在する末梢のT細胞がポリクロナールであることを明らかに示し(データは示されていない)、T細胞展開の間にp53の下流制御が幾つかのクローンの球を生じないことを示した。これらの結果を解釈するために、これらの動物の末梢から単離された細胞におけるp53の発現をノックダウンする際に該構築物の安定な発現及び効率を検証することは重要であった。形質導入されたCD34+細胞で10週早くに形質導入されたマウスの脾臓及び血液から単離されたヒト単核細胞は、2人の異なるドナーからのヒトPBMCの存在下で培養された(EBV細胞株JY, PHA及びIL-2)。これらの条件下では、T細胞は広範囲に増殖した。成熟したT細胞のみがこのプロトコルを使用して拡大されうる故に(Res P等, J Exp Med. 1997年;第185巻:第141-151頁)、我々のデータは、RAG2-/- γc-/- マウスにおいて展開されたヒトT細胞が機能的に成熟しており且つTCRライゲーションによってトリガーされたシグナルに応答しうることを示した。さらに、T細胞は、同種抗原に応答し且つイン ビトロで再刺激するとサイトカインの広いアレイを生産することができ、それらは異種抗原を認識し且つ応答することが可能であり、それらは異種抗原を認識し且つ応答することができたことを示す(データは示されていない)。イン ビボでp53 siRNAを形質導入されたCD34+細胞由来のT細胞の拡大後に、我々は、GFP+及びGFP-T細胞を分離(ソート)し、そして2つの異なる再構築された動物からのガンマ線照射後のp53のレベルを試験した。両方のマウスにおいて、p53は形質導入されていない集団において増加され、一方p53 siRNA構築物を含む細胞におけるp53レベルはほとんど検出できないままだった(図7A)。その結果、これらのp53 siRNA+細胞は、ガンマ線照射又はフルダラビンでの処置によって誘発されたアポトーシスにそれほど感受性でない。従って、これらp53 siRNA+細胞は安定される。対照的に、p53の損失は、p53独立アポトーシス誘発剤であるデキサメタゾン及びスタウロスポリンによって誘発されるアポトーシスへのT細胞の感受性に影響しなかった(図7A及びデータは示されていない)。そのアポトーシスを促進する機能(pro-apoptotic function)に加えて、p53はまた、種々の刺激に応答して成長停止の誘発に関与する。その上、p53はhTERTのネガティブ調節因子であり、それはT細胞の複製の寿命を調節する(Roth A等, Blood. 2003年;第102巻:第849-857頁)。我々は、TCR刺激後にp53 siRNAを発現する細胞の成長を研究した。我々の構築物におけるGFPの存在は、我々が、p53iで又は形質導入されていない細胞に関連する対照構築物のいずれかで形質導入されたT細胞の集団の動力学を検査することを許した。図8に示されているように、我々はp53の減少した量を有する細胞の進行的蓄積を検出し、p53 siRNA+T細胞が形質導入されていない細胞又は対照を形質導入された細胞と比較して成長有利点を有することを示した。興味深いことに、これらの差は刺激直後だけでなく、該細胞が長期間の培養において維持される場合においても検出されず、TCR刺激への即時応答がp53 siRNAを発現する細胞及び対照において同様であることを示す(図8及びデータは示されていない)。これらの結果は、CD34+細胞内に導入されたp53 siRNAがT細胞子孫における下方制御するp53タンパク質において活性のままだったことを明らかに示す。我々のデータはまた、p53の下方制御がストレスに対してT細胞を保護するがhTERTを上方制御し、それはこのシステムにおけるヒトT細胞展開又はT細胞のホメオスタシスに影響しないことを示した。
【0106】
異なる器官におけるB細胞、形質細胞様DC又は単球の展開に対する P53 RNAi の影響の欠如。
新生マウス内にCD34+細胞を注射すると複数の系統が展開されたという事実は、我々がB細胞、pDC及び単球の展開に対するp53ノックダウンの影響を分析することを同様に可能にした。表3では、GFP+ 及びGFP- 集団におけるB細胞のパーセンテージは、対照-GFP及びp53 siRNA-GFP-形質導入されたCD34+細胞から展開された細胞において類似であることが示されている。さらに、B細胞分化マーカーの発現における差が、形質導入されていない対照-GFPとp53 siRNA-GFP細胞との間で認められなかった(図9)。
【0107】
その上、GFP+集団におけるこれらの細胞のパーセンテージが形質導入されていない、対照を形質導入されたサンプルにおいて並びにp53 siRNA形質導入されたサンプルにおいて非常に類似したので、我々はまたp53をノックダウンすることがpDC及び単球の展開に対して影響しないことを示すことができた。
【0108】
【表3】
【0109】
議論
我々は、新生RAG2-/-γc-/- マウスが、関心のある安定化されたヒト細胞を生成するために適していることを本明細書に示す。ロバストなT細胞展開が、新生マウス内にCD34+細胞をip注射すると観察され、それは成体マウスで以前に観察されたものと比較して加速されたペースで進められた。メインストリームのCD4+及びCD8+T細胞に加えて、Treg細胞を表わしうるTCRガンマ・デルタ細胞、CD3+CD56+T細胞及びCD25+CD4+細胞を含むT細胞の全ての他のサブセットが観察されうる。我々はまた、サーキュレーション(circulation)及びCD34+細胞を注射されたマウスの末梢器官におけるB細胞、NK細胞、pDC及び単球の展開を観察した。CD15+CD11c+CD24+顆粒球に加えて、ヒトグリコホリンポジティブ細胞の低いがコンシステントなパーセンテージが存在した(原稿は準備中)。これらのデータはこれらのマウスにおけるヒト白血球のかなり完全なレパートリーの生成を示すとともに、これらの新生ダブルKOマウスはイン ビボで「ヒト」免疫応答を研究するために適していることを示す。
【0110】
独立プロモーターの制御下でマーカーGFPを含んだレンチウィルスベクターにおけるp53に対して向けられたsiRNAを使用することによって、我々は、イン ビボで遺伝的に組み換えられたCD34+前駆細胞の展開を追跡するためにGFPの実現可能性を示した。生化学分析は、CD34+前駆細胞において、p53 siRNA発現が、ガンマ線照射誘発されたp53発現の95%よりも多い減少をもたらしたことを明らかにした。我々は、siRNA構築物がT細胞子孫において安定的且つ機能的に発現されたままであったことを見つけた故に、siRNAが成熟白血球内でのCD34+細胞の分化の間に発現されたままであったことが示されている。
【0111】
マウスから単離されたp53 siRNA+T細胞は、対照を形質導入された又は形質導入されていないT細胞と比較して、イン ビトロで成長有利点を示した。我々は、p53の下方制御がヒトT細胞にガンマ線照射への耐性を与えることを示した。従って、p53の下方制御が、細胞の安定化をもたらす。
【実施例2】
【0112】
dox依存導入遺伝子発現を有する単一のレンチウィルスベクターが設計される。我々の設計において、我々は、IRESエレメントを使用してTRE及びGFPのrtTA下方ストリームを置くことによって、CMVプロモーターの構成的発現又は自己調節発現下でrtTAを置くことを評価する。ヒトの初代及び確立された細胞株のパネルに対する両方のベクターシステムの評価は、自己調節ループがより低い基礎導入遺伝子活性、より高い導入遺伝子誘発を有することを示す。その上、自己調節ループのための誘発動力学が、構成的に発現されたrtTAのそれよりも速く且つ長期の培養及び誘発の繰り返しサイクルに応答してより良く耐性であるように現れることが示される。
【0113】
引き続き、rtTA2-Sは、我々がrtTA3と呼ぶ最近公表されたウィルス的に発生されたrtTA(Das AT等, Journal of Biological Chemistry 2004年;279巻:第18776-18782頁)によって置き換えられた。この新しく記載されたrtTA3は、野生型rtTAと比較してdoxに対して13倍のより高い感受性及びrtTA2-M2と比較してdox濃度の広い範囲についてのより高い転写活性及び誘発の両方を有する。自己調節ループ内でのrtTA3の導入は、doxのより低い濃度でループの活性を許し、及び100 ng/mLのdox濃度に達した最大発現でGFPのdox依存段階的発現を許す。
【0114】
材料及び方法:
試薬:
Dox(Sigma)ストック溶液が、水中でdox 10mg/mlを溶解することによって作製され、そしてフィルター滅菌された。ストックは、-20で冷凍にて維持された。
【0115】
レンチウィルスベクター調製
レンチウィルスベクターは以前報告されたように調製された(Seppen,J, Rijnberg,M., Cooreman,M.P., and Oude Elferink,R.P. (2002年). Lentiviral vectors for efficient transduction of isolated primary quiescent hepatocytes. J Hepatol第36巻, 第459-65頁)。手短に言えば、HEK 293T細胞は、4つのプラスミドを含む第3世代のレンチウィルスベクターシステムで、リン酸カルシウム沈殿を使用して、トランスフェクションされた。トランスフェクションに続き24時間で、25 mM HEPES pH 7.4を補完された培地で置換された。ウィルスを含む上清がトランスフェクションに続き48時間で集められ、小分けされ、そして−80℃で冷凍された。
【0116】
細胞株及び培養
HEK293T、HeLa、HepG2、SJNB-8、HUVEC、ヒト繊維芽細胞、ヒト胎児肝細胞。すべてが、10%のウシ胎仔血清、ペニシリン/ストレプトマイシン(penn/strep)、グルタミンで補完された標準DMEM培地において培養された。
【0117】
ウィルス力価及び形質導入
ウィルス力価が、HeLa細胞の形質導入によって決定された。手短に言えば、ウィルスは、連続希釈法において、DEAEデキストランを補完されたHeLa細胞に添加され、そして4時間形質導入される。形質導入に続く72時間で細胞が収穫され、そしてGFP発現がウィルス力価を計算するためにフローサイトメトリーによって測定される。力価は、1000 ng/ml doxでの誘発によって決定された。
【0118】
SDS-PAGE及びウェスタンブロッティング
SDS-PAGE及びウェスタンブロッティングが、Seppen, J., R. R.van der, N. Looije, N. P. van Til, W. H.Lamers, and R. P. Oude Elferink. (2003年)、Long-term correction of bilirubin UDPglucuronyltransferase deficiency in rats by in utero lentiviral gene transfer. Mol. Ther. 第8巻:第593-599頁に記載されたように行われた。
【0119】
結果:
単一のTet-Onレンチウィルスベクターを構築する我々の初期の試みは、ウィルス性LTR内に配置されたTetOの2つの繰り返しを含んだ以前に記載されたtet依存条件的複製HIV-1(Verhoef等, 2001年)からの適応物だった。表4において報告されているように、我々は、これらのベクターで高いウィルスの力価を得ることができなかった。次に、我々は、セントラルポリプリン配列(cPPT)及びB型肝炎転写後制御調節エレメント(PRE)を含む第3世代レンチウィルスのベクターのバックボーンにおける最小のCMVプロモーターに融合されたTetOの7個の繰り返しからなるTREを使用することを進めた(Barry等、2001年)。これらの新しいベクターで、我々はウィルスの力価を1〜2のオーダーの大きさで増加させることができ、より特に自己調節ループベクターで、導入遺伝子発現を駆動するために構造性プロモーターを使用して得られるそれらと比較してウィルス力価を得ることができた(データは示されていない)。
【0120】
【表4】
【0121】
ヒトの初代及び確立された細胞株のパネルが、7C1又は7I1のいずれかで感染の多重度(MOI)1で形質導入され、そして1000 ng/mlのdox濃度で誘発された。誘発レベルは、doxなしにGFP発現によって分割されたdoxでのGFP発現として測定され、及び遺伝子発現全レベル及び基本的発現の両方を反映する。試験された全ての細胞株において、自己調節ループが、rtTAの構成的発現と比較してより高いレベルの誘発を与えた(表5)。
【0122】
【表5】
【0123】
我々は、我々が誘発なしにrtTAの低い基本的発現を期待したように自己調節ループを設計した。これを評価するために、我々は、Lenti TetO7mCMV CMV rtTA-S2又はLentiTetO7mCMV IRES rtTA-S2のいずれかでMOI 1で初期経過初代ヒト繊維芽細胞(early passage primary human fibroblast)を形質導入した。拡大に続き、該細胞は、72時間、1000 ng/ml で処理されず又は処理され、次に溶解物を生成するために収穫された。
【0124】
ドキシサイクリの不存在下で、我々は自己調節ループにおけるrtTAの発現を検出することができず(図11、レーン5)、我々の自己調節ループシステムの基本的発現が確かに低く、一方我々は構成的発現ウィルスにおけるrtTAの発現を明らかにみることができた(図11、レーン3)ことを確認した。
【0125】
HeLa 7C1対7I1上の繰り返し誘発
次に、我々は、7C1又は7I1のいずれかでMOI 0.1でHeLa細胞を形質導入し、doxの誘発及び撤回を変更することによって、誘発に対する繰り返しサイクルを経験するための能力のための両システムを評価し、そこでは各経過がオン状態又はオフ状態のいずれかであることを表す。対照として、我々は、MOI 0.1で、lenti pgk eGFPで形質導入されたHeLa細胞を含めた。1つのサイクルに続くGFPポジティブ細胞のパーセンテージが50%より多くだけ減少し、そして誘発の幾つかのサイクル後に、開始値の20%まで落ち、一方IRES rtTA2-S2形質導入された細胞についてのレベルが、pgk eGFP対照と比較して安定のままであり、rtTAの自己調節的制御がより良く耐性であることを示した。なぜならば、rtTAの毒性が避けられるためである。
【0126】
rtTAの自己調節発現を使用することに関する他の関心事は、最大活性が、構成的発現と比較して、相当により遅いだろうということだった。最大誘発についての時間を決定するために、我々は、TRE d2eGFP CMV rtTA2-S2又はTRE d2eGFP IRESrtTA2-S2のいずれかでMOI 1でHeLa 細胞を形質導入し、そして固定された時点でdox(1000 ng/ml)を追加した。我々は、次の72時間でGFP発現における増加がなかったことを見つけ(公表されていない観察)、従って我々はこの時間を最大発現のための任意のレベルとして設定した。図13にみられうるように、rtTAの自己調節発現は、構成的に発現されたrtTAよりも速く最大レベルに到達した(24時間、最大の70%対45%、及び48時間、100%対75%)。これらの結果は、自己調節ループを使用して誘発時間における損失がないことだけでなく、我々が構成的に発現されたrtTAと比較してより早い最大発現に到達することを示す。
【0127】
制御された遺伝子発現の重要な特徴は、誘発性分子の濃度と導入遺伝子発現のレベルとの間の相関を有することである。我々は、dox濃度(0〜1000 ng/ml)の範囲に渡る2つの異なるシステム間のGFP発現のレベルを比較した。我々は、より低いdox濃度で、我々が自己調節ループでのGFP発現を作動させることができなかったことを観察した(図14、パネルA)。基本条件下でrtTA2-S2の非常に低い発現のために、自己調節ループは、開始のためにより高い濃度のdoxを要求する。低い濃度のdoxを使用する場合に、rtTA2 S2がループを活性化するためにdoxに十分に感受性でないと我々は仮定した。この考えを試験するために、我々は、rtTA2 S2を、(Urlinger S等; Proc.Natl.Acad.Sci. (2000年) 第97巻, 第14号, 第7963-7968頁、及びDasAT等, Journal of Biological Chemistry 2004年;第279巻:第18776-18782頁)により記載された3つのアミノ酸置換を含む変異体rtTA, rtTA3で置き換え、それは野生型rtTAと比較してdoxに対して13倍のより多い感受性を有し且つ転写活性を改善した。更なる議論のために、このrtTA変異体はrtTA3と云われるだろう。我々が自己調節内にrtTA3を導入した場合、我々は、100 ng/ml doxで得られた発現の最大レベルで試験されたdox濃度の範囲にわたりGFPの段階的発現を見た(図14、パネルB)。
【実施例3】
【0128】
この実施例は、B細胞の調節及び増殖におけるSTAT5の役割を示す。
【0129】
材料及び方法
B細胞単離
B細胞が、大人の扁桃腺又は末梢血から得られた。扁桃腺摘出術が、アムステルダム自由大学の子どものための外科部門において行なわれた。T細胞が、抗-CD4及び抗-CD8コンジュゲートされたマイクロビーズ(Miltenyi Biotec, アムステルダム, オランダ)を使用して使い果たされた。次に、該細胞は、コンジュゲートされた抗-CD19 FITC (DAKO、グロストラップ、デンマーク)及びコンジュゲートされた抗-CD3 フィコエリトリン(PE)でインキュベーションされ、引き続きMoFlo (Cytomation/DAKO、グロストラップ、デンマーク)又はFacStarPlus (Becton Dickinson、サンノゼ、カリフォルニア州)を使用してCD19+CD3-集団を分離(ソート)した。該生じたB細胞は、再分析すると99%超の純度だった。
【0130】
レトロウィルス構築物及び組み換えレトロウィルスの生産
STAT5a及びbの構成的に活性な変異体が、以前に記載されていた(Ariyoshi等、2000年;大西等、1998年)。これら変異体及び野生型STAT5bをコードするDNAが、T. Kitamura(IMSUT、東京、日本)から得られた。BCL-6は、抗増殖性p19ARF-p53シグナリングの阻害剤としてネズミの繊維芽細胞における老化救出スクリーンにおいて識別された(Shvarts等、2002年)。これらDNAは、以前に記載されたLZRS-linker-IRES-GFPベクター内にライゲーションされた(Heemskerk等、1997年;Heemskerk等、1999年)。ノックダウン実験のために、Brummelkamp等によって以前に記載されたpSUPER構築物(Brummelkamp等、2002年)が適応された。形質転換された細胞の識別をフローサイトメトリーによって許すために、siRNAプローブの転写のためのpol3プロモーター及びGFP発現を駆動するPGKプロモーターが反対の位置にあるように、GFP発現カセットがpSUPER構築物に追加された。
mRNAを特異的にターゲットとするsiRNA配列が、Ambion ホームページ(http://www.ambion.com)を使用して設計された。配列が、pSUPER-GFPのBglII-HindIII部位内に挿入された。引き続き、pol3-siRNA-pgk-GFPカセットが、LZRSレトロウィルス構築物の自己不活性化誘導体内にサブクローニングされた。
【0131】
レトロウィルスのプラスミドが、製造者のプロトコルに従いFugene-6 (Roche Diagnostics Netherlands、アルメレ、オランダ)を使用して、ヘルパーウィルスのない両種性生産者細胞株Phoenix-A(ヒト杯腎臓細胞株293の誘導体)(Kinsella and Nolan、1996年)(Dr. G. Nolan, Stanford University、パロ・アルト、カルフォルニア州、の親切なギフト)内にトランスフェクションされた。2日後に、トランスフェクションされた細胞の選択が、2μg/ml ピューロマイシン(Becton Dickinson Clontech Laboratories, パロ・アルト, カルフォルニア州)の添加によって開始された。トランスフェクションの10〜14日後に、6 x 106細胞が、10 mlの完全培地(Iscove's medium (Life Technologies BV, ブレダ, オランダ)(8% ウシ胎仔血清 (FCS)における10cmペトリディッシュ (Becton Dickinson Discovery Labware、ベッドフォード、マサチューセッツ州)、ピューロマイシンなし)毎に置かれた。翌日に該培地は新たにされ、そして翌日にレトロウィルスの上清が収穫され、遠心分離され、そして-70℃で細胞フリー部分で冷凍された。このアプローチは再現可能な、迅速な、大規模の、及び3×106超(伝染性ウィルス粒子/ml)の高い力価のレトロウィルス生産を与える。
【0132】
CA及び野生型(WT)STAT5bエストロゲン受容体(ER)融合構築物が下記のように作られた:終止コドンの代わりにBglII部位を導入するために、PCRが、N604H STAT5b変異体(Ariyoshi等、2000年;Onishi等、1998年)又は野生型STAT5b cDNAsのいずれかで行なわれた。XhoI/BglII消化生成物が生成され、それはΔCA-又はΔWT-STAT5b-ERを生成するために、pBS-ER(C term)(Dr. Kurataによって親切に提供された, DNAX Institute、パロ・アルト、カルフォルニア州)(Kurata等、1999年)のBamHI/EcoRI消化、及びpBS-SK+のXhoI/EcoR1消化でライゲーションされた。次に、LZRS-CA-STAT5b-ER及びLZRS-WT-STAT5b-ERを生成するために、LZRS-CA-STAT5b-IRES-ΔNGFR又はLZRS-WT-STAT5b-IRES-ΔNGFRのXhoI/NotI消化が、pBSΔCA-STAT5b-ER又はΔWT-STAT5b-ERの部分的NotI/XhoI消化にライゲーションされた。これを使用して、我々は、IRESのCA-STAT5b-ER又はWT-STAT5b-ER下流、及びΔ神経成長因子受容体(ΔNGFR)、NGFRのシグナリング不全変異体(C. Bonini博士によって親切に提供された(Bonini等、1997年))、又は緑蛍光性タンパク質GFPを有する構築物を作った。NGFRに対するモノクロナール抗体(Chromaprobe、マウンテンビュー、カルフォルニア州)が、ΔNGFR発現細胞を視覚化するために使用された。
【0133】
レトロウィルス形質導入
組み換えヒトフィブロネクチンフラグメントCH-296形質導入手続(RetroNectin(商標); Takara, 大津, 日本)が、以前に記載されたように行われた(Heemskerk等、1997年;Heemskerk等、1999年)。非組織培養処理された24ウェルプレート(Costar、バドフーフェドルプ、オランダ)が、2時間室温で又は40℃一晩で、0.3 mlの30μg/ml 組み換えヒトフブロネクチンフラグメントCH-296で覆われた。該CH-296溶液が除去され、引き続き室温で30分間、リン酸緩衝生理食塩水(PBS)において2% ヒト血清アルブミン(HSA)とともにインキュベーションされ、引き続きPBSで一度洗われた。5x105のB細胞が、0.25mlの解凍されたレトロウィルス上清及びポリブレン(最終濃度 4 μg/ml)と混合された0.25mlの完全培地中に置かれ、そして37℃で6時間インキュベーションされた。次に、0.25mlの上清が除去され、そしてポリブレンに加えて0.25mlの新鮮なレトロウィルスの上清が追加され、そして一晩37℃でインキュベーションされた。翌朝、細胞は洗われ、そして照射された(80 Gy)CD40L(CD154)を発現するL細胞(CD40L-L)、IL-2(20U/ml)及びIL-4(50 ng/ml)を有する24ウェル組織培養処理されたプレート(Costar)に移された。
【0134】
細胞培養
B細胞は、5% CO2を含む湿った空気中、37℃で完全培地において培養された。CD40L-L細胞(80グレイで照射された)が、24ウェル組織培養処理されたプレート(Costar)においてウェル当たり5x104細胞を播かれた。5x104にソートされたB細胞が、IL-2(20U/ml)及びIL-4(50 ng/ml)とともに添加された。1週後に、該細胞はレトロウィルス形質導入のために使用された。形質導入後、B細胞が、照射されたCD40L-L細胞、IL-2及びIL-4で再び培養された。
【0135】
pTyr-STAT5及びCD20のための免疫組織学的染色
扁桃摘出術を経験する子どもから得られたヒト扁桃腺組織についてのpTyr-STAT5及びCD20の免疫組織学的分析が、以前に記載された免疫蛍光法及びCLSM分析(小さな修正を有する)を使用して行われた(Vyth-Dreese等、1995年)。手短に言えば、ホルマリン固定され、パラフィンを埋め込まれたヒト扁桃腺部分が、シトレート回収すると、5%(v/v)の正常なヤギ血清(Central Laboratory of the Netherlands Red Cross Blood Transfusion Service (CLB)、アムステルダム、オランダ)においてプレインキュベーションされた。引き続き、セクションが、一晩、4℃でウサギ抗pTyr-STAT5b抗体(Cell Signaling Technology、ベバリー、マサチューセッツ州、米国)及びマウス抗CD20 (L26, DAKO)においてインキュベーションされ、引き続きビオチン化されたヤギ抗ウサギIgG(DAKO)、ストレプタビジン/ビオチン−コンジュゲートされたホースラディッシュパーオキシダーゼ複合体(ABC-protocol, DAKO)及びTyramide-Alexa Fluor568(Molecular Probes Europe、ライデン、オランダ)においてインキュベーションされた。CD20が、Alexa Fluor 633-コンジュゲートされたヒツジ抗マウスIgG (Molecular Probes)を使用して視覚化された。インキュベーションの間に、セクションは、1%ウシ血清アルブミン(BSA, Sigma Aldrich、ズウェインドレヒト、オランダ)を含むPBS中で広範囲にすすがれた。各試験内で、アイソタイプと一致した対照抗体及び正常なウサギIgGが、ネガティブ対照として含まれていた。共焦点蛍光画像が、Kr/HeNeレーザー組み合わせを備えたLeica TCS SP (Leica Microsystems、ハイデルベルグ、ドイツ)共焦点システム上で得られた。画像は、40x 1.25 NA対物レンズを使用して得られた。カラー顕微鏡写真が、電子オーバーレイから得られた。
【0136】
STAT5発現を局地化するために、CA-STAT5b-ER-IRES-ΔNGFRを形質導入されたB細胞が、ポリシンで覆われたカバーグラス上に付けられた。15分間、室温で3.7%ホルムアルデヒドでの固定化後、細胞は、45秒間、0.1%トリトンX-100で浸透された。第1の抗体は、マウス抗NGFR及びウサギ抗ER(MC-20, Santa Cruz Biotechnology、サンタ・クルス、カリフォルニア州)であった。第2の抗体は、抗マウス-FITC(Becton Dickinson Pharmingen、ハイデルベルク、ドイツ)及び抗ウサギ-TexasRed(Molecular probes、ユージーン、オレゴン州)であった。共焦点蛍光画像が、Leica TCS SP (Leica)上で得られた。
【0137】
B細胞の表現型検査
FITC、PE又はAPCで直接ラベル付けされたヒト分子に対する抗体 IgD、IgG、CD3、CD19、CD20、CD27、CD38、CD40、CD45、CD56、CD70、CD80、CD86、HLA-DR(Becton Dickinson, Pharmingen) 、及びPE (DAKO) で直接ラベル付けされたIgM、カッパライト鎖(kappa light chain)、ラムダライト鎖(lambda lightchain)、CD138が、フローサイトメトリー分析のために使用された。染色された細胞が、FACSCalibur(商標)(Becton Dickinson Imunocytometrysystems, サンノゼ、カリフォルニア州)を使用して解析され、そしてFACSデータがCellQuest(商標)コンピュータソフトウェア(Becton Dickinson Immunocytometry systems)で処理された。
【0138】
RT-PCR
総RNAが、RNeasy(商標) mini kit (Qiagen Sciences、ジャーマンタウン、メリーランド州)を備えた解凍されたペレットから単離された。5×第1ストランドバッファー、500 μM dNTP's、25 μg/L オリゴ (dT)、200 U スーパースクリプト II RT (Life Technologies)を含むRNAが、20μLの容量で逆転写された。cDNA溶液の1マイクロリットルが、20 mM Tris-HCL、50 mM KCL、1.5 mM MgCl2、5 mM dNTP's、2.5 U Taq DNA ポリメラーゼ(Life Technologies)及び30 pmolの各プライマーを含む50μl溶液中のPCRに付された。PCR条件は下記であった:94℃で7分の変性工程、引き続き、94℃で30秒、62℃で30秒(HPRT)、52℃(LMP-1)、58℃(EBNA1/2)、58℃(BCL-6)、及び72℃で30秒の30サイクル、そして72℃で最終7分の延長。逆転写酵素(RT)PCRのために使用されるオリゴヌクレオチドは、
だった。
【0139】
VH遺伝子を検出するためのPCR条件は、(Guikema等、1999年; vonLindern等、2000年)に記載されたのと同様であり、94℃で7分の変性工程、引き続き、94℃で1分、60℃で1分及び72℃で1分の35サイクル、そして72℃で最終10分の延長を含む。RT-PCRのために使用されるオリゴヌクレオチドは次のとおりだった:
。
【0140】
ウェスタンブロッティング
細胞抽出物が、プロテアーゼ阻害剤カクテル(Boehringer Ingelheim BV, インゲルハイム, ドイツ)を補われたRIPA溶解バッファー(150 mM NaCl、1% NP-40、0.1% SDS、0.5% DOC、50 mM TRIS-HCl pH 8.0)において調製された。タンパク質は、Protranニトロセルロース転写膜(Schleicher and Schuell BioScience Inc., キーン、ニューハンプシャー州)に移された。ウェスタンブロッティングのために使用される西洋わさびパーオキシダーゼ(HRP)にコンジュゲートされた主要な抗体は、BCL-6 (C-19)、STAT5b (C-17)、ER (MC-20)及びアクチン(I-19)だった(全てSanta Cruz Biotechnology (Santa Cruz、カルフォルニア州、米国)から)。STAT5ノックダウンウェスタンの検出のために、タンパク質の等量がBradford試験(BCA protein assay reagent kit(Pierce Biotechnology Inc., Rockford、イリノイ州、米国))を使用してロードされた。他のウェスタンブロットのために、アクチンがローディング対照として使用された。タンパク質の検出が、高められた化学発光(pierce)を使用して行われた。
【0141】
ルシフェラーゼリポーター転写促進アッセイ
リポーター構築物(構成的に活性なウミシイタケ(Renilla reniformis)ルシフェラーゼ生成ベクターprL-CMV(Promega、サン・ルイ・オビスポ、カリフォルニア州、米国)及び293T細胞における発現ベクターLZRSの共トランスフェクションが、製造者の指示書に従いFugeneトランスフェクション試薬で行われた(Roche、バーゼル、スイス)。ホタル(P. pyralis)及びウミシイタケルシフェラーゼの検出が、製造者の指示書(Promega)に従いダブルルシフェラーゼアッセイキットを使用して行われた。pGL3ベーシックベクターにおけるBCL-6プロモーター領域-657/+471が、S. Hirosawa博士(東京医科歯科大、東京、日本)によって親切に提供された。BCL-6プロモーターフラグメントにおけるSTAT5結合部位の変異体が、二段階PCRアプローチによって生成された。第一工程PCRは、BCL-6(F)又はBCL-6(R)プライマー並びに対応するGLprimer2及びRVprimer4(Promega)で行なわれた。
。次に、対応するPCR製品が精製され、混合され、そしてGLprimer2及びRVprimer4プライマーを使用して再増幅された。該増幅されたPCRフラグメントは、メーカー指示書に従って、pCR2.1TOPO ベクター(Invitrogen、リーク、オランダ)内に直接的にクローニングされた。プライマーM13(F)及びM13(R)での配列決定は、染料ターミネーターサイクル配列決定キット(PerkinElmer)を使用して、ABI シーケンサー (PerkinElmer Corp、ノーウォーク、コネチカット州)で行われた。変異されたBCL-6プロモーター領域は、SacI及びBglII消化を介してpGL3においてサブクローニングされた。
【0142】
結果
チロシンリン酸化されたSTAT5は、ヒト扁桃腺における胚中心B細胞において発現される。
STAT5がイン ビトロで幾つかのB細胞成長因子によって活性化される故に、我々はチロシンリン酸化された(pTyr)-STAT5が、ヒト扁桃腺において生理学的条件下で検出されうるかどうかを調査した。我々は、pTyr-STAT5及びCD20のために共染色された扁桃腺セクションのCLSM分析を行なった。図16Aは杯中心域において、CD20dimB細胞上のpTyr-STAT5の共局在化を示し、典型的にパッチされた、膜様の染色パターンを示す。同様の染色パターンが、扁桃腺のT細胞域においてT細胞上で検出可能であった(データは示されていない)。アイソタイプ対照抗体でダブル染色された扁桃腺セクションは、何ら染色を示さなかった(図16B)。
【0143】
RNA干渉によってSTAT5をノックダウンすることが、ヒトB細胞の増殖を阻害する
ヒトB細胞が、IL-2、IL-4及びIL-10を含むサイトカインの存在におけるCD40の関与に続き、制限された期間のみについてイン ビトロで培養されうる。B細胞が数週間後に死ぬ理由は、完全に理解されていない。本明細書では、STAT5タンパク質の量が制限因子であり、それは成長因子に対して非応答性をもたらしうるかどうかを我々は扱う。ヒトCD19+B細胞が分離(ソート)され、そしてIL-2及びIL-4とともにCD40L-L細胞上で培養された。サンプルが培養の2及び17日後に取られ、そしてウェスタンブロッティングがSTAT5抗体を使用して細胞溶解物全体上で行なわれた。明らかに、タンパク質レベルが、培養の2日目と比較して、培養の17日後に、B細胞において減少された。これは、B細胞が制限されたSTAT5レベルのために結局死ぬことを示す。
【0144】
B細胞の増殖におけるSTAT5の役割を直接的に調査するために、我々は、RNA干渉によって減少するためのSTAT5を特異的にターゲッティングすることによってSTAT5レベルをノックダウンすることを選択した。STAT5 siRNAプローブ(1365及び1668)が設計され、そしてRaji B細胞を内生的に発現するSTAT5におけるそれらのノックダウン効率について試験された。STAT5 siRNAsは、PGKプロモーターによって駆動されるGFPマーカー(pSIN GFP)をまた含む我々の自己不活性化レトロウィルス構築物においてサブクローニングされた。Raji B細胞は、STAT5(1365)若しくはSTAT5(1668)siRNA発現ウィルス、又は対照ウィルスのいずれかでレトロウィルス的に形質導入され、そしてGFP発現に基づいて分離(ソート)された。図16Dにおいて示されるように、STAT5(1668)siRNAは、対照を形質導入されたRaji細胞と比較して完全に近い、STAT5タンパク質発現の量に効率的に減少され、一方STAT5(1365) siRNAはあまり有効でないがSTAT5レベルを有意に減少された。B細胞増殖に対するSTAT5の影響を分析するために、我々は、対照ウィルスと平行してCD19+分離(ソート)された末梢血B細胞内にSTAT5(1668) siRNA プローブを形質導入し、そしてIL-2及びIL-4とともにCD40L-L上でこれらを培養した。GFP+B細胞を分離(ソート)した後、3日間、IL-2及びIL-4を有するCD40L-L細胞上で2回(104細胞/ウェル)培養され、そのうち最後の18時間はチミジン(1μ Ci/ウェル)の存在下(Chan等)であった。STAT5タンパク質発現は、50%だけB細胞の増殖能力を劇的に減少した(図16E)。
【0145】
初代ヒトB細胞の拡大に対するSTAT5の影響
下記観察が、ヒトB細胞の成長の調節におけるSTAT5の役割を調べることを我々に促した:1)pTyr-STAT5は、イン ビボにおけるヒトB細胞において検出されうる;2)STAT5タンパク質レベルが、CD40L-L細胞、IL-2及びIL-4上でイン ビトロにおいて培養されたヒトB細胞において減少し、それはそれらの最終的な死に関連しうる;3)ヒトB細胞におけるSTAT5のノックダウンは、それらの増殖能力を減少する;4)pTyr-STAT5は、様々なB細胞悪性腫瘍において構成的に発現される。我々は、ヒトB細胞の成長に対する野生型(WT)並びにSTAT5a及びbの構成的に活性な(CA)変異体の影響を比較した。精製された扁桃腺CD19+B細胞が、CD40L-L細胞、IL-2及びIL-4とともに共培養された。7日目で、該細胞は、ウィルス発現CA-STAT5a-IRES-GFP、CA-STAT5b-IRES-GFP、WT-STAT5b-IRES-GFPで、又は対照-IRES-GFPで形質導入された。形質導入効率は、5%〜20%だった(データは示されていない)。CA-STAT5b-GFP(図17A)又はCA-STAT5a-GFPで形質導入された培養におけるGFP+細胞のパーセンテージ(示されない)は、時間をかけて増加した。5〜6週目で、対照-IRES-GFPを形質導入された及び形質導入されていないB細胞が死に始め、一方6週目(すなわち形質導入後の5週目)で、CA-STAT5b-GFPを形質導入された培養物の95%超がGFP+であり、且つ拡大することを続け、そして培養物中に維持された。WT-STAT5b-GFPで形質導入されたB細胞が選択性に生存したが、拡大せず(図17A及びB)、STAT5の活性はB細胞の拡大のために適していることを示す。該拡大したCA-STAT5b-GFP+B細胞は、CD19を発現したが、CD20を発現しなかった(図17C)。CA-STAT5b-GFPを形質導入された細胞の複製寿命延長がEBV形質転換によるものでなかったことを保証するために、我々は、センシティブRT-PCRによってLMP-1及びEBNA1/2mRNAを分析し、そしてこれらの遺伝子が発現されなかったことを確認した(結果は示されていない)。重要なことに、CA-STAT5b-GFPを形質導入されたB細胞は、RT-PCRによって決定されるように、Cμ(図17D)又はCガンマ(示されていない)につけられた幾つかのIgHバリアブル遺伝子セグメントを発現し、形質転換されたB細胞株がモノクロナールでないことを示した。
【0146】
同時に、これらのデータは、活性のSTAT5の構成的発現が、細胞死からのイン ビトロ培養されたヒトB細胞を救い、そしてそれらの複製寿命を拡大することを示した。
【0147】
タモキシフェンでのCA-STAT5b-ER発現の誘発に続くB細胞株の生成
STAT5bの構成的に活性だけ(及び第2の形質転換事象でない)がヒトB細胞の拡大された増殖応答をもたらすことをさらに確認するために、我々はB細胞増殖に対する調節可能なSTAT5b構築物の影響を試験した。この目的のために、我々は、CA-STAT5b又はWT-STAT5bでエストロゲン受容体(ER)の融合を調製し、及びIRES-ΔNGFRのCA-STAT5b-ER又はWT-STAT5b-ER上流を抱く組み換えウィルスの構築物を作成し、それは切り捨てられ、神経成長因子受容体の無能な変異体をシグナリングする(Bonini等、1997年)。形質導入すると、STAT5b-ER融合タンパク質は、熱ショックタンパク質によって隔離された不活性な複合体として該形質導入された細胞の細胞質において発現される。4−ヒドロキシ−タモキシフェン(4HT)とのインキュベーション後、ER融合タンパク質が熱ショックタンパク質から単離し、核へ移動する。確かに、4HTの不存在又は存在下において抗ER抗体でCA-STAT5b-ER-IRES-ΔNGFR+ B細胞の染色をすること及びCLSMを使用して分析することは、CA-STAT5bが4HT依存様式において期待されるように核に局所化する(図18A)。ウェスタンブロット分析は、形質導入された細胞における予期されたMWの融合タンパク質の存在を確認した(図18B)。
【0148】
次に、我々は、CA-STAT5b-ER-IRES-ΔNGFR構築物でヒト扁桃腺のB細胞を形質導入し、そしてこれらB細胞の増殖能力に対する4HTの影響を試験した。図19Aは、CD40L、IL-2及びIL-4におけるCA-STAT5b-ER-IRES-ΔNGFRで形質導入された扁桃腺B細胞の次の培養を示し、CA-STAT5b-ER発現細胞(ΔNGFR+)の成長選択が4HTの存在下でのみ生じた。重要なことに、CA-STAT5b-ER-IRES-ΔNGFR+を形質導入された細胞からの4HTの除去は、B細胞の成長の終了、そして最終的な死をもたらした(図19B)。4HTの不存在下で培養された、CD40L、IL-2及びIL-4で培養されたCA-STAT5b-ER-IRES-ΔNGFR+ B細胞が最初に増殖したが、それらは成長を止め、次に7〜20日後に死んだ。4HTが7日にわたり添加されたとき、該細胞は生存し、次に成長することを続けた(図19B)。これらの結果は、扁桃腺B細胞のCA-STAT5b-誘発された成長がB細胞を形質転換するCA-STAT5bによって誘発される不可逆の遺伝的変化の結果でないが、核におけるCA-STAT5bの継続的、機能的発現にのみ依存することを示す。CA-STAT5b-ER-IRES-ΔNGFRで形質導入され、そして3ヶ月の間、CD40L、IL-2、IL-4及び4HTにおいて培養された扁桃腺B細胞の一部が、細胞表面IgMを発現した(図19C)。該細胞は、細胞表面IgDについてネガティブであり、及び小さなパーセンテージ(1〜2%)がIgGを発現し、それはIgMを欠いた。すばらしいことに、CA-STAT5b-ER-IRES-ΔNGFR+ B細胞の長期の培養をすると、細胞表面Igの発現が失われた(データは示されていない)。同様の効果が、ナイーブ又はメモリーの末梢血B細胞のいずれか内にSTAT5の導入後に観察された(データは示されていない)。その上、CA-STAT5b-ER-IRES-ΔNGFR+ B細胞はCD19、CD38、CD40、CD70、CD80及びHLA-DRを発現したが、CD20及びCD27についてネガティブだった。CD25は、これらの細胞上で高度に発現されたが(図19C)、STAT5bがCD25(IL-2Ralpha)転写(John等、1996年;John等、1999年)を直接的に制御するという事実と一致する。注目すべきことに、B細胞におけるWT-STAT5b-ER融合タンパク質の発現は、B細胞におけるCA-STAT5b-ERタンパク質の発現と比較して同様の4HT依存成長効果及び表現型を生じた。思うに、これは、ERドメインがSTAT6-ER について見られたように二量化するという事実によって引き起こされ(Kurata等、1999年)、核に転位されるSTAT6-ER二量体を生成した。
【0149】
ヒト末梢血B細胞内へのBCL-6の異所性発現は、細胞の応答寿命の延長をもたらす
最近、我々は、幼い子供のヒト扁桃腺B細胞におけるBCL-6の異所性発現が、CD40L、IL-2及びIL-4において培養される場合に、B細胞の成長有利点をもたらしたことを我々は報告した(Shvarts等、2002年)。これらの細胞は、CD19を発現し且つCD3及びCD56についてネガティブだった。これらの発見を拡張するために、我々は、BCL-6がまた成人末梢血B細胞の増殖能力に影響するかどうかを調査した。それ故に、我々は、CD40L発現L細胞、IL-2及びIL-4とともに培養されたヒトB細胞内にBCL-6-IRES-GFPを導入した。図20Aは、BCL-6の発現が、BCL-6の形質導入の10〜13日後に開始するマーカーGFPを発現する末梢B細胞の成長有利点をもたらした。これらの細胞の表現型に関する情報を得るために、我々は、モノクロナール抗体のパネルで広範囲な分析を行なった。図20Bは、BCL-6-IRES-GFPを形質導入されたB細胞がCD19を発現したことを示す。加えて、BCL-6-IRES-GFPを形質導入されたB細胞はCD20を発現し、CD38及びメモリーB細胞マーカーCD27について弱いポジティブであり、しかし形質細胞マーカーCD138についてネガティブだった。その上、該細胞は、活性マーカー HLA-DR、CD40、CD70及びCD80を発現し且つCD25について弱いポジティブだった。重要なことに、BCL-6-IRES-GFP形質導入されたB細胞は、これらの細胞がそれらの分化において、細胞表面Ig-ネガティブ形質細胞内に捕捉されたという事実と一致する細胞表面Igカッパ又はラムダを発現した。
【0150】
BCL-6はSTAT5bの直接ターゲットである。
CA-STAT5及びBCL-6がヒトB細胞の成長に対して同様の効果を有するという観察は、BCL-6がSTAT5によって調節された事実にあるという可能性を高める。この概念は、BCL-6の1.5 kBプロモーター(Ohashi等、1995年)が3つの潜在的なSTAT結合部位を含むという事実によって支持される。STAT5bがBCL-6発現を規制することができるかどうかを研究するために、我々は、10日間、4HTの不存在下でSTAT5b-ER+扁桃腺B細胞を培養した。次に、4HTが添加され、そして該細胞は8時間後に収穫され、そしてRT-PCRによってBCL-6の発現について試験された。図21Aは、BCL-6転写物の発現が、このホルモンなしの培養においてよりも4HTありの培養において実質的により高いことを示す。ウェスタンブロット分析は、4HTの存在下で培養されたB細胞が4HTの不存在下で培養された細胞よりも有意に多いBCL-6タンパク質を発現したことを明らかにした。BCL-6がSTAT5bの直接又は間接のターゲットであるかどうかを判断するために、タンパク質合成阻害剤シクロヘキシミド(CH)が8時間、4HTと一緒に添加された。4HT及びCHの両方が添加された場合にBCL-6mRNAの上方制御がまた観察され(図21A)、一方CH単独の添加はBCL-6発現に影響しなかった(データは示されていない)。これらのデータは、BCL-6発現がSTAT5bによって直接的に影響されることを示す。
【0151】
この観察を拡張するために及びBCL-6が確かにSTAT5bの直接ターゲットであることを検証するために、我々はBCL-6プロモーターを使用して、ルシフェラーゼレポーター遺伝子アッセイを行なった。BCL-6プロモーターの検査は、3つの潜在的なSTAT5結合部位の存在を明らかにした:位置1863-1871 bpでのTTCTCAGAA、位置1190-1198 bpでのTTCTCTGAA及び位置548-556 bpでのTTCTCTGAA。加えて、1つの潜在的なSTAT5結合部位は、非コード第1エクソンに配置される。STAT5がBCL-6プロモーターを介して転写を活性化することができたかどうか判断するために、我々は、BCL-6プロモーターの一部(657/+471)及びルシフェラーゼコード配列にリンクされた非コード第1エクソンを含む構築物を使用した。この構築物は、開始コドンに最も近接した一つの潜在的なSTAT5結合部位(548/-556 bp)及び非コード第1エクソン内の一つを抱く。このリポーター構築物は、CA-STAT5b発現するベクター又は対照ベクターと一緒に293T細胞内にトランスフェクションされた。リポーター構築物及びCA-STAT5bの共トランスフェクションは、増加されたルシフェラーゼ活性をもたらした(図21C)。リポーター活性におけるこの増加がSTAT5活性に依存したことを示すために、変異体が(548/-556 bp)STAT5結合部位において生成された。我々は、TTCTCTGAAからGTCTCTAAAにBCL-6プロモーター領域における最初のSTAT5結合部位を変異した。CA-STAT5bが共トランスフェクションされた場合、このSTAT5結合部位の変異体は、リポーター活性を強く減じた(図21C)。これらのデータは、BCL-6プロモーターにおける(548/-556 bp)STAT5b結合部位が機能的であることを明白に示す。一緒に、我々の結果は、STAT5がBCL-6発現を直接的に制御することを示す。
【0152】
CA-STAT5 B細胞の長期の寿命は、BCL-6発現にもっぱら依存しない。
BCL-6タンパク質がSTAT5b-ER延長された寿命について重大かどうかを判断するために、我々は2つのBCL-6 siRNAプローブを設計し、それらはpSUPER内にクローニングされた。最初に、BCL-6 siRNAsのノックダウン効率を決定するために、pSUPER構築物が293T細胞におけるBCL-6を発現するベクターとともに共トランスフェクションされた。図22Aに見られるように、siBCL-6(789)及びsiBCL-6(970)の両方が、BCL-6タンパク質の量を非常に効率的に減少した。両方のBCL-6 siRNAsがpSIN-GFPレトロウィルスベクター内にサブクローニングされ、そしてウィルスがCA-STAT5b-ER-ΔNGFR+B細胞を形質導入するために生産された。対照を形質導入された細胞のGFP+細胞のパーセンテージは、時間にわたって安定していた。同じ観察が、2つBCL-6 siRNAsを形質導入されたCA-STAT5b-ER-ΔNGFR+ B細胞のいずれかについて作られ(図22B)、CA-STAT5b-ER+B細胞がそれらの生存についてBCL-6に依存しないことを示唆した。著しいことに、CA-STAT5b-ER-ΔNGFR+ B細胞におけるBCL-6ノックダウン後に、CD80発現が1.5〜2倍増加された(データは示されていない)。CD80がBCL-6によって直接的に抑制されることを記載されている故に、これはBCL-6ノックダウン構築物の効率を確認した(Niu等、2003年)。我々の発見は、BCL-6がB細胞の複製寿命に対するSTAT5bの影響の唯一の仲介人でないことを示す。
【0153】
文献
Ariyoshi, K., Nosaka, T., Yamada, K., Onishi, M., Oka, Y., Miyajima, A., and Kitamura, T. Constitutive activation of STAT5 by a point mutation in the SH2 domain. J Biol Chem 第275巻, 第24407-24413頁, (2000年)。
Barry, S.C. et al. Lentivirus vectors encoding both central polypurine tract and posttranscriptional regulatory element provide enhanced transduction and transgene expression. Hum Gene Ther 第12巻, 第1103-8頁, (2001年)。
Bonini, C., Ferrari, G., Verzeletti, S., Servida, P., Zappone, E., Ruggieri, L., Ponzoni, M., Rossini, S., Mavilio, F., Traversari, C., and Bordignon, C. HSV-TK gene transfer into donor lymphocytes for control of allogeneic graft-versus-leukemia. Science 第276巻, 第1719-1724頁, (1997年)。
Brummelkamp, T.R., Bernards, R., and Agami, R. A system for stable expression of short interfering RNAs in mammalian cells. Science 第296巻, 第550-553頁, (2002年)。
Guikema, J. E., Vellenga, E., Veeneman, J. M., Hovenga, S., Bakkus, M. H., Klip, H., and Bos, N. A. Multiple myeloma related cells in patients undergoing autologous peripheral blood stem cell transplantation. Br J Haematol 第104巻, 第748-754頁, (1999年)。
Heemskerk, M. H., Blom, B., Nolan, G., Stegmann, A. P., Bakker, A. Q., Weijer, K., Res, P. C., and Spits, H. Inhibition of T cell and promotion of natural killer cell development by the dominant negative helix loop helix factor Id3. J Exp Med 第186巻, 第1597-1602頁, (1997年)。
Heemskerk, M. H., Hooijberg, E., Ruizendaal, J. J., van der Weide, M. M., Kueter, E., Bakker, A. Q., Schumacher, T. N., and Spits, H., Enrichmentof an antigen-specific T cell response by retrovirally transduced human dendritic cells. Cell Immunol 第195巻, 第10-17頁。
John, S., Robbins, C. M., and Leonard, W. J. An IL-2 response element in the human IL-2receptor alpha chain promoter is a composite element that binds Stat5, Elf-1, HMG-I(Y) and a GATA family protein. Embo J 第15巻, 第5627-5635頁, (1996年)。
John, S., Vinkemeier, U., Soldaini, E., Darnell, J. E., and Leonard, W. J. The significance of tetramerization in promoter recruitment by Stat5. Mol Cell Biol 第19巻, 第1910-1918頁, (1999年)。
Kaplan, M. H., Schindler, U., Smiley, S. T., and Grusby, M. J. Stat6 is required formediating responses to IL-4 and for development of Th2 cells. Immunity 第4巻, 第313-319頁, (1996年)。
Kinsella, T. M., and Nolan, G. P. Episomal vectors rapidly and stably produce high-titer recombinant retrovirus. Human Gene Therapy 第7巻, 第1405-1413頁, (1996年)。
Kurata, H., Lee, H. J., O'Garra, A., and Arai, N. Ectopic expression of activated Stat6 induces the expression of Th2- specific cytokines and transcription factors in developing Th1 cells. Immunity 第11巻, 第677-688頁, (1999年)。
Leonard, W. J.,and O'Shea, J. J. Jaks and STATs: biological implications. Annu Rev Immunol 第16巻, 第293-322頁, (1998年)。
Lischke, A., Moriggl, R., Brandlein, S., Berchtold, S., Kammer, W., Sebald, W., Groner, B., Liu, X., Hennighausen, L., and Friedrich, K. The interleukin-4 receptor activates STAT5 by a mechanism that relies upon common gamma-chain. J Biol Chem 第273巻, 第31222-31229頁, (1998年)。
Niu, H., Cattoretti, G., and Dalla-Favera, R. BCL6 controls the expression of the B7-1/CD80 costimulatory receptor in germinal center B cells. J Exp Med 第198巻, 第211-221頁, (2003年)。
Onishi, M., Nosaka, T., Misawa, K., Mui, A. L., Gorman, D., McMahon, M., Miyajima, A., and Kitamura, T. Identification and characterization of a constitutively activeSTAT5 mutant that promotes cell proliferation. Mol Cell Biol 第18巻, 第3871-3879頁, (1998年)。
Reljic, R., Wagner, S. D., Peakman, L. J., and Fearon, D. T. Suppression of signal transducer and activator of transcription 3- dependent B lymphocyte terminal differentiation by BCL-6. J Exp Med 第192巻, 第1841-1848頁, (2000年)。
Rolling, C., Treton, D., Pellegrini, S., Galanaud, P., and Richard, Y. IL4 and IL13 receptors share the gamma c chain and activate STAT6, STAT3 and STAT5 proteins in normal human B cells. FEBS Lett 第393巻, 第53-56頁, (1996年)。
Scheeren, F.A., Naspetti, M., Diehl, S., Schotte, R., Nagasawa, M., Wijnands, E., Gimeno, R., Vyth-Dreese, F.A., Blom, B., and Spits, H. STAT5 regulates the self-renewal capacity and differentiation of human memory B cells and controls Bcl-6 expression. Nature Immunology. Vol 6, No. 3, 第303-313頁, (2005年)。
Seppen, J, Rijnberg, M., Cooreman, M. P., and Oude Elferink, R. P. Lentiviral vectors for efficient transduction of isolated primary quiescent hepatocytes. J Hepatol 第36巻, 第459-65頁, (2002年)。
Shimoda, K., van Deursen, J., Sangster, M. Y., Sarawar, S. R., Carson, R. T., Tripp, R. A., Chu, C., Quelle, F. W., Nosaka, T., Vignali, D. A., 等. Lack of IL-4-induced Th2 response and IgE class switching in mice with disrupted Stat6 gene. Nature 第380巻, 第630-633頁, (1996年)。
Shvarts, A., Brummelkamp, T., Scheeren, F., Koh, E., Daley, G. Q., Spits, H., and Bernards, R. A senescence rescue screen identifies BCL6 as an inhibitor of anti-proliferative p19ARF- p53 signaling. Genes Dev in press, (2002年)。
Strathdee, C.A., McLeod, M. R. and Hall, J.R. Efficient control of tetracycline-responsive gene expression from an autoregulated bi-directional expression vector. Gene 第229巻, 第21-29頁, (1999年)。
Teglund, S., McKay, C., Schuetz, E., van Deursen, J. M., Stravopodis, D., Wang, D., Brown,M., Bodner, S., Grosveld, G., and Ihle, J. N. Stat5a and Stat5b proteins have essential and nonessential, or redundant, roles in cytokine responses. Cell 第93巻, 第841-850頁, (1998年)。
Traggiai E, Chicha L, Mazzucchelli L, bronz L, Piffaretti J-C, lanzavecchia A, Manz M. Development of a human adaptive immune system in cord blood cell-transplanted mice。
Verhoef, K., Marzio, G., Hillen, W., Bujard,H. and Berkhout, B. Strict control of human immunodeficiency virus type 1 replication by a genetic switch: Tet for Tat. J Virol 第75巻, 第979-87頁, (2001年)。
Von Lindern, M., Amelsvoort, M. P., van Dijk, T., Deiner, E., van Den Akker, E., van Emst-De Vries, S., Willems, P., Beug, H., and Lowenberg, B. Protein kinase C alpha controls erythropoietin receptor signaling. J Biol Chem 第275巻, 第34719-34727頁, (2000年)。
Vyth-Dreese, F. A., Dellemijn, T. A., Majoor, D., and de Jong, D. Localizationin situ of the co-stimulatory molecules B7.1, B7.2, CD40 and their ligands in normal human lymphoid tissue. Eur J Immunol 第25巻, 第3023-3029頁, (1995年)。
Weijer K, Uittenbogaart CH, Voordouw A, Couwenberg F, Seppen J, Blom B, Vyth-Dreese FA, Spits H. Intrathymic and extrathymic development of human plasmacytoid dendritic cell precursors in vivo. Blood. 第99巻:第2752-2759頁, (2002年)。
Ye, B. H., Cattoretti, G., Shen, Q., Zhang, J., Hawe, N., de Waard, R., Leung, C., Nouri-Shirazi, M., Orazi, A., Chaganti, R. S., 等. The BCL-6 proto-oncogene controls germinal-centre formation and Th2-type inflammation.Nat Genet 第16巻, 第161-170頁, (1997年)。
【図面の簡単な説明】
【0154】
【図1】(A)レンチウィルスのRNA干渉ベクターpTRIPΔU3-EF1アルファ p53の概要図である。(B)免疫ブロットの図である。
【図2】CD45+ヒト細胞のパーセンテージのグラフである。
【図3】胸腺のFACSプロファイルの図である。
【図4A】骨髄のFACSプロファイルの図である。
【図4B】脾臓のFACSプロファイルの図である。
【図4C】肝臓のFACSプロファイルの図である。
【図4D】肺のFACSプロファイルの図である。
【図5】GFP+発現細胞の多分化能存在の図である。
【図6】Vベータファミリー図である。
【図7A】免疫ブロットの図である。
【図7B】フローサイトメトリーの図である。
【図8】GFPポジティブ細胞のパーセンテージを示す図である。
【図9】GFP+発現細胞の多分化能存在の図である。
【図10】細胞パネル7TetOmCMV CMV対IRESのグラフである。
【図11】rtTAウェスタンブロットの図である。
【図12】繰り返し誘発結果の図である。
【図13】最大GFP発現のための時間のグラフである。
【図14】濃度(dox用量反応)の図である。
【図15】誘発可能なレンチウィルスなベクターの概要図である。
【図16】(A)pTyr-STAT5及びCD20の共局在化を示す染色パターンの図である。(B)ダブル染色されたアイソタイプ対照染色の図である。(C)ウェスタンブロット分析の図である。(D)ウェスタンブロット分析の図である。(E)チミジンの取り込みのグラフである。
【図17】(A)GFP+細胞のパーセンテージのグラフである。(B)絶対的な細胞数カウントのグラフである。(C)フローサイトメトリーの図である。(D)RT-PCR分析の図である。
【図18】(A)抗-NGFR-FITC(緑)及び抗-ER-TexasRed(赤)での染色図である。(B)ウェスタンブロット分析の図である。
【図19】(A)形質導入された細胞のパーセンテージのグラフである。(B)絶対的な細胞数のグラフである。(C)フローサイトメトリーの図である。
【図20】(A)GFP発現のフローサイトメトリック分析及び絶対細胞数カウントのグラフである。(B)フローサイトメトリーの図である。
【図21】(A)RT-PCR分析の図である。(B)ウェスタンブロット分析の図である。(C)デュアルルシフェラーゼリポーター分析の図である。
【図22】(A)
【技術分野】
【0001】
本発明は、細胞生物学の分野に関する。より特に、本発明は安定化された細胞の生産に関する。
【背景技術】
【0002】
細胞のエクスビボ(ex vivo)生産は、多種多様の用途のために調査されてきた。個体から得られるサンプルの細胞は例えば、診断目的のためにエクスビボでさらに培養される。培養された細胞はまた、例えば候補薬化合物の(有用な)効果を確証することを(医学的に)試験するために使用される。その上、細胞は、前記細胞によって分泌される産物、例えば酵素、ホルモン及び/又は抗体を収穫するためにしばしば培養される。モノクロナール抗体(mAbs)の生産は、重要な用途である。モノクロナール抗体は、同じアフィニティを有する抗原に結合し且つ同じエフェクター機能を促進する単一の抗体分子の複数の同一のコピーを表わす。モノクロナール抗体は、抗原上で同じエピトープと反応する。
【0003】
mAbsの恩恵の中には、疾病原因に関与されうる特定の細胞又は化学的メディエーターをターゲットとするそれらの能力がある。この特異性は、より多くの慣用的な治療に渡るmAbsに対する或る臨床上の有利点を与え、一方、一般に低い副作用とともに、有効で、耐容性の良好な治療オプションを患者に与える。
【0004】
モノクロナール抗体は例えば、試験動物、例えばハイブリドーマ細胞を作成するために(マウス)ガン細胞に特定の抗原を用いて免疫性を与えられたマウスから得られるB細胞を融合することによって生産される。B細胞が抗体生産能力を与え、一方ガン細胞がハイブリドーマを無期限に分割し且つ細胞培養において十分に成長することを可能にする。次に、単一のリンパ球からの所望の抗体を生産する個々のハイブリドーマが、大規模培養及びmAb生産のために選択されうる。
【0005】
マウスmAbsはヒトにおける制限された治療に有用である。なぜならば、免疫応答が呼び起こされるからである。それ故に、マウスmAbsは、長期的な治療の恩恵を提供しない。この抗マウス免疫応答はまた、望まれていない副作用を引き起こす。その上、マウスmAbsは、他の重要なヒト免疫システムコンポーネントを効率的に活性化しえない。それ故に、mAbsがより多くのヒトに現れるようにするための努力がとられてきた。1つのアプローチは、抗原にまだ結合するが免疫応答を刺激しそうにないマウス/ヒト−ハイブリッド抗体をエンジニアリングすることだった。マウスCDRをもっぱら作る遺伝子領域が単離され、そして機能的な抗体分子を完成するために要求されるヒト抗体遺伝子領域に接合された。これらCDRを移植された抗体は、90%より多いヒトである。代替的に、ヒトIg遺伝子を保有するマウスが作成された。
【0006】
他のアプローチは、ファージが種々のヒト抗体可変領域(V遺伝子)を保有するように、ヒトV遺伝子についての遺伝子をバクテリオファージの遺伝子内に取り込むことによってファージディスプレイライブラリーを生産することである。変更されたファージ遺伝子は、対応するヒト抗体フラグメントを作るようにファージで感染された細菌を指図し、それはウィルス表面上に自動的に取り込まれる。
【0007】
所望の特異性を有するヒトモノクロナール抗体を直接的に得るために、そのような抗体を生産しうるB細胞を単離すること及びエクスビボでB細胞を培養することは便利であろう。しかしながら、ヒトB細を用いたハイブリドーマ技術はあまり成功していない。なぜならば、結果として生じたハイブリドーマが不安定であるからである。B細胞のエクスビボ培養をするための多くの試みが試されてきた。ヒトナイーブ及びメモリーB細胞は、IL-2、IL-4 及びIL-10を含むサイトカインの存在下でCD40の関与に続いて、制限された期間について培養されうることが十分に立証され(Arpin等、1995年;Banchereau等、1991年)、及びこのシステムは、同起源のプライムされたCD40L発現ヘルパーT細胞に向けてB細胞のイン ビボ(in vivo)応答を模倣することが信じられる。CD40ライゲーションの不存在下で、IL-10単独又はIL-2との組み合わせのIL-10が、抗体を生産する細胞に分化を誘発する(Arpin等、1997年;Malisan等、1996年)。これらの条件下で培養された成熟B細胞の生存及び増殖の調節の機構が部分的にのみ知られている。
【0008】
B細胞上のCD40の関与が、アポトーシスに対する保護、分化の(部分的な)阻害、及びB細胞によるサイトカイン応答性の誘発を含む複数の効果を有する(Foy等、1996年)。大量の細胞周期阻害剤の発現は、Rb-1及びRb-2を含むCD40関与によって減少され(Dadgostar等、2002年)、及びそのような遺伝子の下流制御は、静止から休眠B細胞をリリースしそうである。CD40トリガリング(triggering)は短い増殖応答をもたらすけれども、サイトカインは、トリガーされたB細胞の細胞周期進行を保持するのに役立つ。IL-2及びIL-4は、CD40又は表面Igを刺激されたB細胞の連続的な細胞周期進行を促進する最も効率的なサイトカインである。上記文献において記載されたB細胞培養物は、制限された期間の間に単に安定している。
【0009】
B細胞を不死化するための他のアプローチは、エプスタイン−バー(Epstein-Barr)ウィルス形質転換である。多くの試みが失敗したけれども、Traggiai等はヒトB細胞のエプスタイン−バーウイルス形質転換のための方法を報告し、該細胞は、重症急性呼吸器症候群コロナウイルス(SARS-CoV)感染から回復した患者から得られた。種々のウィルスタンパク質について特異的なモノクロナール抗体が単離された(Traggiai等、2004年)。
【0010】
B細胞を不死化するためのさらに他のアプローチが、国際公開第WO 03/052083号パンフレットに記載されている。該出願は、B細胞を安定化するための方法であって、ヒトB細胞が遺伝子導入マウスから単離され、その後該細胞が構成的に活性なSTAT(Signal Transducer of Activation and Transcription)(CA-STAT)で形質導入される方法を記載する。B細胞の長期の寿命が観察された。しかしながら、形質導入効率は低い(5〜20%の間)。これは、成功の合理的な機会で形質導入を行なうために、ある抗原に特異的に結合する抗体を生産しうる多くのB細胞が単離される必要があることを意味する。1つの特異的な抗原に対して抗体を生産するB細胞の高い量が、しばしば利用できない。
【0011】
ほとんどの先行技術方法の他の不利点は、任意の所望の抗原に対する抗体を生産しうるヒトB細胞を得ることがしばしばできないということである。例えば、自己抗原(例えばサイトカイン(例えば、インターロイキン−10及びTNF−アルファ))、及び組織(例えば、膵臓及び皮膚)又は腫瘍の非腫瘍特異的部分上で発現される抗原(例えば、悪性黒色腫上だけでなく、また正常なメラニン細胞上で発現される抗原)に対するヒトB細胞は、得ることが難しい。その上、ヒトは、所望の抗原、例えば病原体でもちろん免疫化されない。
【発明の開示】
【発明が解決しようとする課題】
【0012】
本発明の目的は、関心のある安定化された細胞を生産するための方法を提供することである。関心のある抗原に対する抗体を生産しうるヒトB細胞株を生産するための方法を提供することがさらなる目的である。
【課題を解決するための手段】
【0013】
1つの観点では、本発明は、関心のある安定化された細胞を生産するための方法であって、
−関心のある前記細胞中に存在する場合に該関心のある細胞を安定化させることができるところの核酸配列を、該関心のある細胞の幹細胞及び/又は前駆細胞に備えること;
−前記幹細胞及び/又は前駆細胞を非ヒト動物に備えること;
−前記動物において、該関心のある細胞の生成を許すこと;及び
−該関心のある細胞を得ること
を含む方法を提供する。
【0014】
好ましくは、前記幹細胞及び/又は前駆細胞が前記安定化する核酸配列を備えられた後に、前記非ヒト細胞が前記幹細胞及び/又は前駆細胞を備えられる。しかしながら、1つの実施態様では、非ヒト動物において既に存在する幹細胞及び/又は前駆細胞が、前記安定化する核酸を備えられる。
【発明を実施するための最良の形態】
【0015】
先行技術方法は、関心のある細胞が単離された後に該関心のある細胞を不死化することに注目するけれども、本発明は、前記関心のある細胞の幹細胞又は前駆細胞を遺伝子的に変更することによって関心のある安定化された細胞を生成することが可能であるという洞察を提供する。本発明に従うと、幹細胞及び/又は前駆細胞が、関心のある細胞を安定化しうる核酸配列で形質導入される場合に、該関心のある安定化された細胞が得られる。好ましくは、前記核酸の発現及び/又は活性は、スイッチオン又はオフされうる。これは例えば、誘導性プロモーターの管理下に前記核酸配列を置くことによって、及び/又は不活性化剤に前記核酸を融合させることによって行なわれる。前記形質導入された幹細胞及び/又は前駆細胞が非ヒト動物に投与された後に、前記幹細胞及び/又は前駆細胞が関心のある安定化された細胞にまだ分化しうる。不死化する核酸配列での幹細胞又は前駆細胞の形質導入は、分化することなしに複製を続ける不死化された幹細胞又は前駆細胞をもたらすだろうことが予測されうる。本発明に従うと、不滅化する核酸配列を備えられた幹細胞又は前駆細胞が、関心のある安定化された細胞にまだ分化しうる。前記関心のある安定化された細胞は好ましくは、安定化されなかった細胞の同じ種類と比較して、成長停止刺激及び/又はアポトーシス性(apoptotic)刺激にそれほど感受性でない。1つの実施態様では、前記関心のある安定化された細胞は、長期の複製寿命を有する。
【0016】
前記核酸配列は好ましくは、自然の環境下で不活性な状態にある。1つの実施態様では、不死化する核酸配列を含む関心のある幹細胞又は前駆細胞は関心のある細胞に分化し、一方前記不死化する核酸配列が活性化される場合に、関心のある結果として生じた細胞は代わりに、複製不完全な分化された細胞に迅速に分化しない。代わりに、前記関心のある結果として生じた細胞は、複製を継続しうる。前記関心のある細胞は前記動物から収穫され、及び好ましくはエクスビボでさらに培養される(例えば細胞株を生産する)。従って、本発明の不死化する核酸配列での幹細胞及び/又は前駆細胞の形質導入は、前記幹細胞及び/又は前駆細胞が分化することを妨げない。前記関心のある結果として生じた細胞は好ましくは、成長停止刺激及び/又はアポトーシス性刺激にそれほど感受性でなく、及び/又は好ましくは、誘導性不死化する核酸配列が活性化される場合に特に、複製不十分な細胞に分化しない又は有意により少ない程度まで分化しない。1つの実施態様では、前記関心のある細胞が前記非ヒト動物に存在する場合に、前記不死化する核酸配列が活性化される。これは例えば、前記非ヒト動物に活性化剤を投与することによって行われる。前記核酸配列は好ましくは、前記関心のある細胞のエクスビボ培養の間に活性化される。前記活性化剤は好ましくは、前記関心のある細胞を含む培地に投与される。
【0017】
本発明の方法は、幹細胞又は前駆細胞の形質導入効率が、関心のあるより多くの分化した細胞(例えばB細胞)の形質導入効率よりも高いという有利点を有する。それ故に、関心のあるより少ない細胞が、前記非ヒト動物から単離される必要がある。関心のある単離され細胞は、単離後に形質導入を経験する関心のある細胞と比較して、前記不死化する核酸配列を含むというより高い頻度を有する。
【0018】
関心のある安定化された細胞は、本明細書において、成長停止刺激及び/又はアポトーシス性刺激にそれほど感受性でなく、及び/又は安定化されていない同じ環境下で同じ種類の細胞と比較して長期の複製寿命を有する細胞として定義される。前記安定化された細胞はまた、本明細書において、不死化された細胞(immortalized cell)と呼ばれる。好ましくは、前記複製寿命は、少なくとも6週間を含む。より好ましくは、前記複製寿命は、少なくとも8週間、より好ましくは少なくとも3ヶ月、より好ましくは少なくとも4ヶ月、より好ましくは少なくとも6ヶ月、より好ましくは少なくとも8ヶ月、より好ましくは少なくとも10ヶ月、より好ましくは少なくとも12ヶ月を含む。
【0019】
前記関心のある細胞は好ましくは、哺乳類の細胞を含む。より好ましくは、前記関心のある細胞はヒト細胞を含む。1つの好ましい実施態様では、前記関心のある細胞は、例えば酵素、ホルモン又は抗体のような所望の製品を生産しうる細胞を含む。所望の製品を生産しうる細胞の安定化は、前記細胞のエクスビボ培養及び前記所望の製品の長期生産を許す。得られた製品は、任意に精製され、そして例えば医薬組成物及び/又は(医学的)試験キットの生産において使用される。本発明の他の好ましい実施態様では、前記細胞が免疫細胞、好ましくはB細胞、T細胞、ナチュラルキラー細胞、又は抗原提示細胞(例えば樹状細胞)を含む。免疫細胞の安定化は、医学研究及び治療を許す。1つの重要な応用は、安定化されたB細胞による(モノクロナール)抗体の生産である。ヒトB細胞株は、本発明の方法を用いて生成され、ヒト抗体生産を許す。従って、本1つの観点では、前記免疫細胞がB細胞、好ましくはヒトB細胞を含む本発明の方法が提供される。他の好ましい実施態様では、前記関心のある細胞がT細胞、より好ましくはヒトT細胞を含む。他の好ましい実施態様では、前記関心のある細胞が、肝細胞、より好ましくはヒト肝細胞を含む。従来技術では、ヒト肝細胞の拡大のための効率的な方法が利用可能でない。本発明の方法が提供された以上、ヒト幹細胞の拡大及び分化が可能になった。本発明の安定化された肝細胞は例えば、例えば急性肝不全を有する患者又は移植への懸け橋として治療するための人工肝臓システムにおける使用するために適している。前記肝細胞は、全体の臓器移植に代わりとして移植において使用するために適している。1つの実施態様では、本発明の安定化された肝細胞は、薬物の代謝を研究するために及び候補薬物化合物を試験するために使用される。
【0020】
語「所望の製品を生産しうる細胞」は、前記製品を生産するその能力を取り消しえないほどに失っていなかった細胞、及び/又は前記製品を生産しうる細胞に分化するその能力を取り消しえないほどに失っていなかった細胞を意味する。例えば、本発明の方法を用いて、複製しうるB細胞を含むB細胞株が生成される。前記B細胞が、形質細胞を生産する抗体に分化するそれらの能力を保持した。
【0021】
好ましい観点では、前記幹細胞及び/又は前駆細胞がヒト幹細胞及び/又は前駆細胞を含む本発明の方法が提供され、従って関心のある安定化されたヒト細胞が得られる。より好ましくは、前記前駆細胞が、CD34+前駆細胞を含む。本発明の方法を用いて、前記ヒト幹細胞及び/又は前駆細胞は、安定化されたヒト細胞に分化する。1つの実施態様では、前記ヒト細胞は、好ましくは活性化剤の投与によって、任意の所望の時点で安定化されうる。前記関心のあるヒト細胞は好ましくは、それが前記動物から得られた後にエクスビボでさらに培養され、ヒト細胞株を生成する。例は、ヒト(モノクロナール)抗体の生産のためのヒトB細胞株及びインスリンの生産のためのヒト膵臓ベータ−細胞株である。
【0022】
前記関心のある細胞中に存在する場合に該関心のある細胞を安定させることができるところの核酸配列がまた、本明細書において、不死化する核酸配列又は安定化する核酸配列と呼ばれる。前記核酸配列を含む幹細胞及び/又は前記細胞から得られた細胞は、成長停止刺激及び/又はアポトーシス性刺激にそれほど感受性でなく、及び/又は前記核酸配列を含まない同じ環境下で同じ種類の細胞と比較して長期の複製寿命を有する。本発明の前記核酸配列は例えば、細胞周期停止を克服し、及び/又は老化、分化及び/又はアポトーシスを妨げうる。好ましくは、前記核酸配列は、前記関心のある細胞の複製を誘発し及び/又は容易にしうる。分化が複製能力の損失(これは最終分化とよばれる)をしばしば含む故に、関心のある細胞における前記核酸配列の存在は好ましくは、例えば前記細胞の分化を妨げることによって複製寿命を延長する。例えば、T細胞依存性B細胞応答の間に、該B細胞は、広範囲の増殖を経験し、それに引き続き抗体生産形質細胞に分化し、それは細胞周期停止によって達成される。1つの実施態様では、本発明の核酸配列は、前記分化及び細胞周期停止を少なくとも部分的に妨げる。
【0023】
好ましい実施態様では、前記核酸配列は、STAT(Signal Transducer of Activation and Transcription)(STAT)タンパク質をコードする遺伝子の少なくとも機能的部分を含む。好ましくは、前記STATタンパク質は、構成的に活性なSTAT(CA-STAT)タンパク質を含む。幾つかのSTATタンパク質が知られている。IL-4がSTAT6及び間接的にSTAT5を活性化すること(Lischke等、1998年;Rolling等、1996年)、一方IL-2がSTAT3及びSTAT5を活性化すること((Leonard及びO'Shea、1998年)においてレビューされた)が従来技術において例えば記載されている。STAT3及びSTAT6分子は、B細胞展開及び分化における或る時点で関与する。STAT6は、クラススイッチ組換えの間、免疫グロブリンアイソタイプ(IgE)の選択に影響を与え(Kaplan等、1996年;Shimoda等、1996年)、一方STAT3は形質細胞分化に関与している(Reljic等、2000年)。B細胞展開及び分化におけるSTAT5の役割は、それほど十分に定義されていない。STAT5、STAT5a及びSTAT5bの2つの既知の形があり、それらは2つの異なる、タンデムに連結された遺伝子によってコードされる。それらは、種々様々の成長因子に対して細胞の応答におけるユニークな役割及び余分な役割の両方を果たす(Teglund等、1998年)。
【0024】
他の好ましい実施態様では、前記核酸配列が、BCL-6をコードする遺伝子の少なくとも機能的部分を含む。BCL-6は転写リプレッサーをコードし、それは非ホジキンリンパ腫(non-Hodgkin's lymphoma)における染色体転座によって頻繁に活性化される。BCL-6に関する染色体転座はプロモーターのみに常に影響し、及びBCL-6のオープンリーディングフレームを完全に残す。BCL-6は、正常なB細胞展開及び成熟のために要求され、及び胚中心の形成のために要求される(Ye、1997年)。BCL-6は、Blimp-1の阻害を通じて形質細胞へのB細胞の分化を妨げる。
【0025】
さらに他の好ましい実施態様では、前記核酸配列が、p53発現を少なくとも部分的に不活性化することができる。腫瘍サプレッサーp53は、照射又は遺伝毒性的メディエーター(genotoxic mediator)への暴露によって生じるDNA損傷に応答して細胞周期及びアポトーシスの調節に重要な役割を果たす。P53は、細胞型及び微環境に依存する細胞周期停止、老化、分化、及びアポトーシスを含む幾つかの細胞性応答を媒介する。変異が造血起源の腫瘍を含むヒト癌におけるp53をコードする遺伝子において生じる一方、正常なヒト造血展開におけるその機能は大部分は未調査のままである。P53は、ヒトテロメラーゼ逆転写酵素(hTERT)の抑制を通じてイン ビトロ(in vitro)で成熟ヒトT細胞の複製寿命を調節する役割を果たす。テロメアは染色体の遠心端でのDNA繰り返しであり、それは染色体末端間融合(end-to-end fusion)に対して保護する。テロメアは各細胞分裂で短くされ、そして非常に短いテロメアを有する細胞が細胞周期停止を経験しそして老いる。テロメア短縮を妨げるhTERTは、TCRを通じて刺激するとT細胞におけるトランジエントに上流調節され、そしてhTERTのドミナントネガティブ(dominant negative)変異体の発現はCD4+及びCD8+の両方の寿命を有意に減少させ、hTERTがヒトT細胞の寿命において調節的な役割を果たすことを示す。例えばRNA干渉(RNAi)によるp53の下流調節は、関心のある細胞(例えば、成熟ヒトT細胞)の寿命を延ばし、及びドミナントネガティブhTERTによる阻害を中和し、p53が初代ヒトT細胞におけるhTERT発現を調節することを示す。
【0026】
本発明に従えば、STATタンパク質及び/又はBCL-6の少なくとも機能的部分、誘導体又は類似体をコードする核酸配列を、幹細胞又は前駆細胞内に取り込むことは、関心のある安定化された細胞をもたらす。その上、p53発現を少なくとも一部不活性化しうる核酸配列(例えば、p53核酸配列をターゲットするRNA干渉誘発性DNAフラグメントのような)を、幹細胞又は前駆細胞内に取り込むことは、関心のある安定化された細胞をもたらす。好ましくは、本発明の核酸配列は、幹細胞又はB細胞前駆細胞内に取り込まれ、安定化されたB細胞及び/又は安定化されうるB細胞をもたらす。該B細胞が好ましくは、エクスビボでさらに培養される。好ましくは、国際公開第WO 03/052083号パンフレット(参照によって本明細書に取り込まれる)に記載されているように、前記STATタンパク質は構成的に活性なSTAT5タンパク質を含む。より好ましくは、前記STATタンパク質は、構成的に活性なSTAT5a、及び/又は構成的に活性なSTAT5bタンパク質を含む。
【0027】
STATタンパク質及び/又はBCL-6の少なくとも機能的部分によって、STATタンパク質及び/又はBCL-6と比較して、関心のある細胞を安定化させる種類において同じ能力を必要とし、量において同じ能力を必要としないタンパク質性分子を意味される。STATタンパク質の機能的部分は例えば、前記能力に関与していない又はほんのわずかのみ関与しているアミノ酸に欠けている。STATタンパク質及び/又はBCL-6の誘導体は、関心のある細胞を安定化する前記タンパク質の能力が種類において本質的に同じであり、量において必ずしも同じ必要はないように変更されたタンパク質として定義される。誘導体は、例えば同類のアミノ酸置換を通じて多くの様式で提供されうる。STATタンパク質及び/又はBCL-6の類似体は、関心のある細胞を安定化する同じ能力を種類において有し、必ずしも量において必要としない分子として定義される。前記類似体は、前記STATタンパク質及び/又はBCL-6から必ずしも得られない。
【0028】
p53発現を少なくとも部分的に不活性化しうる核酸配列は、本明細書において、p53をコードする核酸配列の転写及び/又は翻訳を阻害しうる核酸配列として、又はp53をコードする核酸配列の転写及び/又は翻訳を阻害しうるタンパク質性分子をコードする核酸配列として定義される。p53発現を少なくとも部分的に不活性化しうる核酸配列は例えば、p53又はp53をコードする核酸配列を特異的に結合しうる。1つの観点において、p53発現を少なくとも部分的に不活性化しうる核酸配列は、p53又はp53をコードする核酸配列を特異的に結合しうるタンパク質性分子をコードする。
【0029】
本発明の不死化する核酸の連続的な発現は、必ずしも望まれるとは限らない。例えば、関心のある化合物(例えば、抗体、ホルモン又はタンパク質)の生産は、複製する細胞の分化をしばしば要求する。一旦B細胞株が確立されると、B細胞集団の一部が抗体生産のために選択される。抗体生産は該選択されたB細胞の分化を必要とするので、複製寿命を延長しうる不死化する核酸配列の発現が好ましくは、少なくとも部分的に阻害される。それ故に、本発明の1つの観点において、本発明の前記不死化する核酸配列はスイッチオン及びオフされうる。例えば、培養された細胞の複製が調節される。
【0030】
従来技術では、核酸配列をスイッチオン又はオフする多くの様式が知られている。アンチセンス核酸が本発明の不死化する核酸配列(のmRNA)に結合しうる関心のある細胞内にアンチセンス核酸を導入し、それによってそれを不活性化することが例えば可能である。前記アンチセンス核酸は好ましくは、或る定義された環境下で生成される。一つの実施態様では、本発明の不死化する核酸配列を含む関心のある細胞は、本発明の前記不死化する核酸配列(の発現生成物)を特異的に結合しうるタンパク質性分子をコードする核酸配列を備えられる。前記タンパク質性分子をコードする前記核酸配列は好ましくは、或る定義された環境下でのみ生成され、及び/又は前記タンパク質性分子は好ましくは、或る定義された環境下でのみ本発明の前記不死化する核酸配列(の前記発現生成物)を特異的に結合しうる。当業者に知られているように、核酸配列をスイッチオン又はオフするための多くの代替方法が、従来技術において利用可能である。
【0031】
本発明の不死化する核酸配列は好ましくは、参照によって本明細書に取り込まれる国際公開第WO 03/052083号パンフレットに記載されている方法を用いてスイッチオン又はオフされる。1つの方法は、不活性化剤を、例えばSTAT5タンパク質のような前記不死化する核酸配列(の発現生成物)と関連づけるステップを含む。次に、該不活性化剤は、例えば関心のある細胞の段階に依存してスイッチオン又はオフされる。前記不活性化剤は例えば、誘導性プロモーター/切除システム、例えばcre-lox又はFLP/FRT切除システムを含む。
【0032】
一つの実施態様において、RNA干渉を誘発する核酸が使用される。例えば、二本鎖干渉RNA(RNAi)又はRNAiを生成するDNAが、本発明の不死化する核酸配列、例えばp53の発現を制御するために使用される。
【0033】
好ましい実施態様では、前記不活性化剤は、融合タンパク質として本発明の不死化する核酸配列の発現生成物(例えば、STAT5タンパク質)と関連付けられるタンパク質性分子を含む。好ましくは、前記不活性化剤をコードする核酸配列は、融合タンパク質を生産するために、前記不死化する核酸配列に融合され、そして関心のある細胞において発現される。
【0034】
国際公開第WO 03/052083号パンフレットに概略されているように、前記不活性化剤は好ましくは、前記融合タンパク質の環境を変更することによってスイッチオン又はオフされる。例えば、エストロゲン受容体及びSTAT5は、融合タンパク質(STAT5-ER)として関心のある細胞において生産される。前記エストロゲン受容体がSTAT5への不活性化剤として働く。なぜならば、前記融合タンパク質が、前記STAT5タンパク質が前記核に達することを妨げるサイトソルにおける熱ショックタンパク質と複合体を形成するために、前記融合タンパク質が不活性化されるからである。しかしながら、4-ヒドロキシタモキシフェン (4HT)とインキュベーションすると、前記融合タンパク質(STAT5-ER)が、前記熱ショックタンパク質から分離され、そしてその活性な形で核に輸送される。前記関心のある細胞の複製は、STAT5によって高められる。発現されたSTAT5及びエストロゲン受容体が熱ショックタンパク質と再び関連づけられるようになり、それ故に不活性になるので、4HTの除去は関心のある細胞の成長の停止をもたらす。従って、4HTの除去は、関心のある細胞の複製の終了をもたらす。従って、この実施態様では、前記不活性化剤ERは、4HTを使用してスイッチオン又はオフされる((国際公開第WO 03/052083号パンフレット、14〜15頁)及び(Scheeren等、(2005年) Nature Immunology, 第6巻, 第3号, 第306頁、最後のカラム、最後の段落〜第307頁、最初のカラム、最初の段落)、両方が参照によって本明細書に取り込まれる)。STAT5b-ER融合構成物の生成の非制限的な例が、Scheeren等、(2005年) Nature Immunology, 第6巻, 第3号, 311頁, 最初のカラム, 最初の全段落(3〜14行)に記載されている。この段落は、参照によって本明細書に取り込まれる。
【0035】
不活性化剤の他の例は、転写因子結合部位、好ましくは本発明の不死化する核酸配列の上流又は下流であり、それによって前記核酸配列の発現が、転写調節因子の制御下である。前記核酸の発現は、転写調節因子(トランス活性化因子)の夫々存在又は不存在によって、スイッチオン又はオフされる。前記トランス活性化因子の存在又は不存在は例えば、関心のある細胞においてその発現又は機能を操作することによって制御される。これは例えば、前記関心のある細胞に2つのプラスミド(前記転写調節因子をコードする第1のプラスミド(トランス活性化因子)及び本発明の不死化する核酸配列(例えばSTAT5)をコードする第2のプラスミド)を備えることにより慣用的に達成される。前記第2のプラスミドは好ましくはまた、前記トランス活性化因子のための核酸結合部位(好ましくは、前記不死化する核酸配列の発現が前記トランス活性化因子の制御下であるように前記不死化する核酸配列の上流)を含む。所望の場合、前記不死化する核酸配列の発現は、例えば前記細胞を、トランス活性化因子を不活性化する剤と接触させることによってスイッチオフされる。そのようなシステムの例が、例えばBujard及びその協力者(Gossen等、1995年)によって記載されているように、テトラサイクリン制御可能システムである。
【0036】
幹細胞及び/又は前駆細胞に本発明の不死化する核酸配列を備えるための多くの方法が、当業者に知られている。例えば、リン酸カルシウムトランスフェクション、DEAEデキストラン、エレクトロポレーション又はリポソームを媒介としたトランスフェクションが使用される。代替的に、該核酸の直接注入が用いられる。しかしながら、好ましくは、前記核酸がベクター、好ましくはウィルスベクターによって前記細胞内に導入される。ベクターによって細胞内へ核酸を導入することを云う様々な語が、従来技術において知られている。そのような語の例が、「形質導入」、「トランスフェクション」又は「形質転換」である。不死化する核酸配列を有するベクターを生成するための及び細胞内へ前記ベクターを導入するための技術が、従来技術において知られている。例えば抗生物質耐性のようなマーカー遺伝子又は感受性遺伝子、及び/又はマーカー(例えば、細胞表面抗原又は緑の蛍光タンパク質のような蛍光タンパク質)をコードする遺伝子が好ましくは、従来技術において周知であるように、導入された核酸を含むクローンを識別する際に使用される。
【0037】
前記ベクターは好ましくは、レトロウィルスベクターを含む。より好ましくは、前記ベクターはレンチウィルスベクターを含む。なぜならば、レンチウィルスベクターは、少なくとも2つの有利点を有するからである:それらは分裂する及び分裂しない細胞を効率的に形質導入することが可能であり、及びそれらは、展開の間にサイレンス化されず、安定な、長期の遺伝子発現を提供する。
【0038】
好ましい実施態様では、例えばSTAT若しくはBCL-6をコードする核酸配列、又はp53発現を少なくとも部分的に不活性化することができる核酸配列のような本発明の不死化する核酸配列が、誘導性プロモーターに動作可能にリンクされる。前記不死化する核酸配列の発現は、前記誘導性プロモーターによって調節される。1つの実施態様では、前記誘導性プロモーターは、関心のある細胞において活性であり及び不活性化剤の投与によって不活化される。しかしながら、好ましくは、前記誘導性プロモーターは、低いバックグラウンド発現で関心のある細胞において一般に不活性であり、従って、前記不死化する核酸配列は前記細胞において発現されない又は少しばかり発現される。誘発性剤を投与すると、前記誘導性プロモーターが活性化され、それは前記不死化する核酸配列の発現及び前記関心のある細胞の安定化をもたらす。前記誘発性剤は好ましくは、前記関心のある細胞において天然に存在しない。1つの実施態様では、誘導性プロモーターに動作可能にリンクされた前記不死化する核酸配列を含むベクターが使用される。本発明の方法において使用するために適している誘導性プロモーターが、従来技術において知られている。1つの実施態様では、参照によって本明細書に取り込まれる国際公開第WO 01/20013号パンフレットにおいて記載されているようにテトラサイクリン/又はドキシサイクリン誘発性Tetオペロンが使用される。
【0039】
本発明のベクターは好ましくは、Tet依存性転写調節因子制御システムを含む。Tet依存性転写調節因子制御システムは、イン ビトロ(in vitro)及びイン ビボ(in vivo)の両方での証明された効率を有する最も研究されたシステムのうちの1つである(Freundlich, Baron, Bonin, Gossen and Bujard, Methods Enzymology 第283巻, 第159-173頁, 1997年; Baron and Bujard, Methods Enzymology 第327巻, 第401-421頁, 2000年)。このシステムは、細菌のTn10テトラサイクリンオペレーターに基づき且つTetRが高い特異性で結合する2つのコンポーネント(テトラサイクリンリプレッサータンパク質(TetR)及びテトラサイクリンオペレーター配列(TetO))から構成される。テトラサイクリンの不存在下で、TetRは2量体になり且つTetOに結合し、そして遺伝子発現を妨げる。テトラサイクリンはTetRに結合することができ、構造的な変化を誘発し、TetOからのTetRの分離及び遺伝子発現をもたらす。
【0040】
このシステムは、テトラサイクリン応答性トランス活性化因子(tTA)を作るために単純ヘルペスウィルスのVP16トランス活性化ドメインのTetRへ融合することによって、及びTetOの7個の繰り返しを最小CMVプロモーター(Tet応答エレメント、TRE)へ融合することによってテトラサイクリン応答性プロモータエレメントを設計することによって、哺乳動物システムにおいて使用するために適合された。テトラサイクリン又はドキシサイクリン(テトラサイクリン類似体)の不存在下で、tTAは活性であり且つTREの制御下に置かれた遺伝子の転写を開始する。ドキシサイクリンの添加は、転写活性化を妨げる。
【0041】
しかしながら、好ましくは本発明のベクターは、テトラサイクリン及び/又はドキシサイクリンの不存在下で不活性であり且つテトラサイクリン及び/又はドキシサイクリンの投与によって活性化されることが可能であるTet依存性転写調節因子制御システムを含む。それ故に、本発明のベクターは好ましくは、Urlinger等(Urlinger S; Baron U; Thellmann M; Hasan M; Bujard H; Hillen W. Proc. Natl. Acad. Sci. (2000年) 第97巻, 第14号, 第7963-7968頁)に記載されているようにTetR変異体(誘導性分子が結合し且つ最終置換が二量化ドメインにおいて配置されるところの3つの置換体が配置される4個のアミノ酸置換を含む)を含む。このTetR変異体がVP16 トランス活性化因子ドメインに融合される場合、転写活性化がドキシサイクリンの存在に依存するところの逆の表現型が得られる。この変異体は、rtTAと名付けられる。
【0042】
Urlinger等(Urlinger S等; Proc. Natl. Acad. Sci. (2000年) 第97巻, 第14号, 第7963-7968頁)によって記載されたrtTAは、最大活性化のためにドキシサイクリンの高いレベル(1〜2μg/ml)を要求することを含むいくつかの制限を有し、それは、ドキシサイクリンの不存在下でTREへの残余の親和力及びVP16活性ドメインからの細胞毒性をある組織において容易に到達されえない。それ故に、本発明のベクターはより好ましくは、rtTA2-S2及びrtTA2-M2と呼ばれるrtTAの改善された変異体を含むTet依存性転写調節システムに動作可能にリンクされた不死化する核酸配列を含む(Urlinger S等;Proc. Natl. Acad. Sci. (2000年) 第97巻, 第14号, 第7963-7968頁)。これらの変異体は、TetR上のランダムな且つ定方向突然変異誘発を実行し、そして元々述べられたrtTAよりもよりよく実行した幾つかの変異体を識別することによって生成された。細胞毒性を減少するために、VP16転写活性ドメインが、細胞性転写装置(machinery)の封鎖を最小化するために、「F」型の3つの繰り返しを含む最小活性ドメインに変更される(Urlinger S等;Proc. Natl. Acad. Sci. (2000年) 第97巻, 第14巻, 第7963-7968頁)。
【0043】
ほとんどのTet調節された導入遺伝子発現システムは、2つの個々の構築物に依存し、第1はTRE(Tet応答エレメント)及び関心のある核酸(導入遺伝子)を含み、及び第2は構造性プロモーターによって駆動されるrtTAを含む。このバイナリーシステムを扱うことはイン ビトロ及びイン ビボの両方において挑戦的であることが、そこでは細胞が2つの異なるベクターによる形質導入を要求し、実験の再現性を困難にする。他の困難性は、ダブルトランスジェニック動物を生成するために、2つの個々の形質転換換え動物株及び成功した交差の生成を要求するTet制御された形質転換度物の生成にでくわす。それ故に、本発明者らは、誘導性プロモーター(PtetO7CMVm)、関心のある核酸(導入遺伝子とも呼ばれる)及びrtTA2-S2をシングルカセット内に結合し、そしてこれをレンチウィルスのベクターバックボーン内にクローニングした。2つの異なるカセットが生成され、そこではrtTA2-S2が、CMVプロモーターで構成的に発現され又は自己調節ループ内におかれ、そこではTREは、導入遺伝子及びEMCV IRESエレメントによってリンクされたrtTA2-S2の両方の発現を駆動する。本発明者らは、ヒトの初代及び確立された細胞株のパネルにおいてこれら2つのシステムを試験し、及び自己調節ループが、試験されたすべての細胞株において構成的に発現されたrtTA2-S2よりもよりよく行われたことを見つけた。それ故に、1つの観点では、本発明は、rtTAのための自己調節ループ及び導入遺伝子発現を含むベクターを提供する。そのようなシステムの有利点は、低い誘発されない発現レベルである。なぜならば、rtTA-トランス活性化因子が、ドキシサイクリンの不存在下で合成されないからである。基本的なrtTA発現の毒性が回避される。
【0044】
この概念は、複製−コンピテントHIVベースのベクターの文脈において以前に示された(Verhoef K, MarzioG, Wolfgang H, Bujard H, Berkhout B. Strict control of human immunodeficiency virus type 1 replication by a genetic switch: Tet for Tat. Journal of Virology 2001年;第75巻:第979-987頁)。本発明の好ましいベクターの例が、図15Bにおいて示される。
【0045】
このベクターシステムの潜在的な不利点は、より敏感なTet-システムが発現ループを「キック−スタート」するために要求されることである。ウィルス進化方法(Das AT, Zhou X, Vink M, Klaver B, Verhoef K, Marzio G, Berkhout B.Viral evolution as a tool to improve the tetracycline-regulated gene expression system. Journal of Biological Chemistry 2004年; 第279巻:第18776-18782頁)を介して得られたより敏感なTet-システムを使用して、本発明者らは、この遺伝子発現ループを活性化することができた。変異体rtTA(以下、本明細書でrtTA3と呼ばれる)が、定向進化(Urlinger S等; Proc. Natl. Acad. Sci. (2000年) 第97巻, 第14号, 第7963-7968頁)及びウィルス進化(Das AT等, Journal of Biological Chemistry 2004年; 第279巻:第18776-18782頁;国際公開第WO01/20013号パンフレット)から生成された自己調節カセットにおいて使用された。rtTA3は、野生型rtTAと比較してドキシサイクリンに対して13倍の増加された感受性を示す(Das AT等, Journal of Biological Chemistry 2004年; 第279巻:第18776-18782頁の表1を参照)。従って、本発明は、rtTA3を含むドキシサイクリン調節された導入遺伝子発現レンチウィルスベクターを提供し、導入遺伝子の緊密な(tight)及び段階的な(graded)発現が可能である。好ましい観点では、前記ベクターは、内部リボソームエントリー部位に動作可能にリンクされたrtTA3を含み、従って前記rtTA3は、TREが導入遺伝子及びrtTA3の両方の発現を駆動する自己調節ループ内に置かれる。本発明の方法における前記ベクターの使用がまた、本明細書で提供される。しかしながら、前記ベクターはまた、例えば関心のある細胞が得られた後に該関心のある細胞の形質導入を介して、他の様式において関心のある細胞を不死化するために適している。関心のある細胞を形質導入するためにrtTA3及び関心のある遺伝子を含むベクターの使用がまた、本明細書で提供される。好ましくは、前記rtTA3は、内部リボソームエントリー部位に動作可能にリンクされ、従って前記rtTA3は、TREが前記関心のある遺伝子及びrtTA3の両方の発現を駆動する自己調節ループ内に置かれる。より好ましくは、前記関心のある遺伝子は不死化する核酸配列を含み、従って前記関心のある細胞が安定化される。前記関心のある細胞は、好ましくは肝細胞又は免疫細胞、好ましくはB細胞を含む。
【0046】
その上、本発明のベクターは好ましくは、ヒトB型肝炎ウィルス転写後調節エレメント(HBV PRE)を含む。該HBV PREは、前記遺伝子の転写された領域に含まれる場合に、おそらく核からの接合されていないmRNAのエクスポートを刺激することによって、導入遺伝子の発現を増加させうるシス作用エレメント(cis-acting element)である(Seppen等. Journal of Hepatology第36巻 (2002年) 第459-465頁)。該PREエレメントは、開始コドン及びHBVxタンパク質のほとんどのコード領域を含む。HBVxは、ウィルスの複製のために不可欠であり、及びHBV感染に関連付けられた肝細胞癌の展開に関与している。
【0047】
関心のある不死化する核酸配列に加えて、関心のある細胞に第2の化合物を備えることが時々望まれる。例えば、関心のある細胞のテロメラーゼの短縮化が妨げられるように、該関心のある細胞は好ましくは、テロメラーゼ逆転写酵素がさらに備えられる。前記化合物は例えば、関心のある細胞に前記化合物をコードする核酸配列を備えることによって、該関心のある細胞に備えられる。好ましくは、前記不死化する核酸配列及び前記化合物をコードする核酸の両方を含むベクターが使用される。なぜならば、2つの独立のエレメントの共輸送は初代細胞において効率的でない故に、単一の形質導入工程ベクターの使用は、2つの個々のベクターの使用よりもより挑戦的でないからである。前記追加の化合物をコードする前記核酸配列は例えば、前記誘導性プロモーターと前記不死化する核酸配列との間に挿入される。もちろん、前記挿入は好ましくは、前記不死化する核酸配列を含む下流領域のフレームシフトを生じない。代替的に、前記追加の化合物をコードする前記核酸配列は、前記不死化する核酸配列の下流に挿入される。しかしながら、好ましくは、例えばUnsinger等に記載されたpBI-4からの双方向性プロモーター(Unsinger等, Retroviral vectors for the transduction of autoregulated, bidirectional expression cassettes. Molecular Therapy第4巻, 第5号, 2001年11月, 第484-489頁)のような双方向性プロモーターが使用される。前記追加の化合物をコードする前記核酸配列は好ましくは、前記双方向性プロモーターの一つの部位上に置かれ、一方前記不死化する核酸配列は、前記プロモーターの他の部位上に置かれる。好ましくは、前記双方向性プロモーターは、誘導性プロモーターである。より好ましくは、Unsinger等において記載されたpBI-4からの双方向性プロモーターのような前記プロモーターが、ドキシサイクリンによって誘発可能である。これは、前記核酸配列の(個々の)調節を許す。
【0048】
他の実施態様では、不死化する核酸配列は、不死化する核酸配列−エストロゲン受容体融合構築物としてベクター内に取り込まれる。好ましくは、実施例において記載されるER-STAT5ベクターが使用され、それはタモキシフェンによって誘発可能である(T.D.Littlewood, D.C. Hancock, P.S. Danielian, M.G. Parker and G.I. Evan, A modified oestrogen receptor ligand-binding domain as an improved switch for the regulation of heterologous proteins. Nucleic Acid Res. 第23巻 (1995年), 第1686-1690頁, 参照によって本明細書に取り込まれる)。
【0049】
従って、関心のある細胞中に存在する場合に該関心のある細胞を安定化させることができるところの核酸配列が誘導性プロモーターに動作可能にリンクされる本発明の方法が提供される。好ましくは、前記プロモーターがドキシサイクリン及び/又はタモキシフェンによって誘導可能である。他の好ましい実施態様では、関心のある細胞中に存在する場合に該関心のある細胞を安定化させることができるところの前記核酸配列を含むベクターで、好ましくはレンチウィルスベクターで、前記幹細胞及び/又は前駆細胞が形質導入される。好ましくは、前記幹細胞及び/又は前駆細胞が、ER-STAT5ベクターで形質導入される。より好ましくは、前記幹細胞及び/又は前駆細胞が、誘導性プロモーター(好ましくはPtetO7CMVm)、関心のある核酸及びrtTA2-S2を含むベクターで形質導入される。最も好ましくは、前記幹細胞及び/又は前駆細胞が、誘導性プロモーター(好ましくはPtetO7CMVm)、関心のある核酸及びrtTA3を含むベクターで形質導入される。前記rtTA2-S2及び/又はrtTA3は好ましくは、内部リボソームエントリー部位(IRES)に動作可能にリンクされ、従って前記rtTA2-S2及び/又はrtTA3が自己調節ループに置かれる。誘導性プロモーター(好ましくはPtetO7CMVm)、関心のある核酸及び(好ましくはIRESに動作可能にリンクされた)rtTA3を含む単離された又は組み換えられた核酸配列がまた、本明細書で提供される。前記関心のある核酸は好ましくは、本発明の不死化する核酸配列を含む。その上、本発明のベクターは好ましくは、ヒトB型肝炎ウィルス転写後調節エレメント(HBV PRE)を含む。最も好ましくは、本発明のベクターは図15Bにおいて記載されたベクターを含む。
【0050】
本発明の方法は特に、不死化された免疫細胞を生産するために適している。例えば、造血幹細胞(又はB細胞前駆細胞)は、不死化する核酸配列で形質導入され、そして非ヒト動物に投与される。前記動物から得られたB細胞が安定化される。1つの実施態様では、前記B細胞は、成長停止刺激及び/又はアポトーシス性刺激にそれほど感受性でない。好ましくは、前記B細胞は、長期の複製寿命を有する。好ましくは、前記非ヒト動物が、関心のある免疫細胞が得られる前に、関心のある抗原を備えられる。前記関心のある抗原が前記動物に投与される後、免疫細胞(例えばT細胞)又は前記関心のある前記抗原に対する抗体を生産することができるB細胞が得られる。これらの免疫細胞が不死化する核酸配列を既に含む故に、これらの免疫細胞はエクスビボで継続された培養のために適している。好ましくは、前記核酸は、前記培養の間に活性化される。本発明の1つの実施態様では、ヒトB細胞株が生産される。前記B細胞株が、関心のある任意の抗原に対して(モノクロナール)抗体の生産に適している。好ましくは、前記関心のある抗原は、(好ましくは例えばRSVのような(ウィルスの)病原体によって発現される)抗原、ヒト自己抗原、腫瘍関連抗原、TNF-アルファ及び/又は悪性黒色腫細胞上で発現された抗原の少なくとも免疫原部分である。非ヒト動物が使用される故に、前記動物を前記抗原で免疫化することによって所望の抗原に対するヒトB細胞を生産することが可能である。ヒト自己抗原に対する抗体を生産することができるヒトB細胞がまた生産される。なぜならば、非ヒト動物が使用されるからである。
【0051】
ヒト細胞が非ヒト動物において生産される場合、前記動物の免疫システムが好ましくは、前記ヒト細胞に対して動物免疫応答を少なくとも部分的に避けるために、少なくとも部分的に害される。これは例えば、前記動物が前記ヒト幹細胞及び/又はヒト前駆細胞を備えられる前に前記動物細胞を照射することによって達成される。しかしながら、好ましくは、ノックアウト非ヒト動物の免疫応答を担う少なくとも1つの遺伝子を欠いている該ノックアウト非ヒト動物が使用される。好ましくは、前記動物は、内在性B細胞、内在性T細胞及び/又は内在性ナチュラルキラー細胞の生産に関与する少なくとも1つの遺伝子を本質的に欠いている。ノックアウト動物は例えば、遺伝子サイレンシングによって、又は従来技術において周知の方法を使用し変異を導入することによって、生産される。遺伝子サイレンシングは例えば、前記遺伝子(の発現生成物)を特異的に結合することができる化合物を前記動物に提供することによって実行される。前記化合物は例えば、タンパク質又はアンチセンスRNAを含む。変異は例えば、部位特異的変異を使用して誘発される。ノックアウト非ヒト動物を生産するための多くの代替方法が従来技術において知られており、それは本明細書において更なる説明を必要としない。従って、前記動物が内在性造血細胞の少なくとも1つの種類を本質的に欠いているところの本発明の方法が提供される。
【0052】
好ましい実施態様では、内在性B細胞、内在性T細胞及び/又は内在性ナチュラルキラー(NK)細胞が本質的に欠けている非ヒト動物が使用される。前記非ヒト動物は好ましくは、マウスを含み、より好ましくはB、T及びNK細胞を欠くダブル変異株であるRAG2-/- γc-/-マウスを含む(Kirberg J, Berns A, von BoehmerH. Peripheral T cell survival requires continual ligation of the T cell receptor to major histocompatibility complex-encoded molecules. J Exp Med. 1997年; 第186巻:第1269-1275頁, 参照によって本明細書に取り込まれる;Weijer K, Uittenbogaart CH, Voordouw A, Couwenberg F, Seppen J, BlomB, Vyth-Dreese FA, Spits H. Intrathymic and extrathymic development of human plasmacytoid dendritic cell precursors in vivo. Blood. 2002年;第99巻:第2752-2759頁, 参照によって本明細書に取り込まれる)。前記マウス内にヒト幹細胞及び/又はヒト造血前駆細胞を移植することは、ヒト造血システムを有するマウスを生じる(Weijer等、2002年(参照によって本明細書に取り込まれる); Traggiai等、2004年)。関心のある生成され安定化されたヒト細胞はネズミ免疫応答によって攻撃されない又は小さな程度まで攻撃されない故に、前記マウスは本発明の方法において使用するために非常に適している。
【0053】
本発明の1つの観点では、前記幹細胞及び/又は前駆細胞が、誕生後1週間以内に前記動物に投与される。好ましくは、前記幹細胞及び/又は前駆細胞は、誕生後3日以内に、より好ましくは誕生後1日以内に前記動物に投与される。初期の注入は、増加されたT細胞及びB細胞移植を生じることが本発明者らによって示された。
【0054】
本発明の方法を用いて、関心のある安定化された細胞が得られ、それはさらにエクスビボで培養されることができ、例えば細胞株を生産する。それ故に、本発明は、本発明の方法によって入手可能な関心のある安定化された細胞を提供する。好ましくは、前記関心のある細胞は、免疫細胞、例えばB細胞、T細胞又は肝細胞を含む。最も好ましくは、前記関心のある細胞は、ヒトB細胞又はヒト肝細胞を含む。関心のある細胞の単離された幹細胞及び/又は前駆細胞(関心のある細胞中に存在する場合に該関心のある細胞を安定化させることができるところの核酸配列を含む前記幹細胞及び/又は前駆細胞)がまた、本明細書で提供される。ヒト複製細胞が、治療、医学的試験及び/又は関心のあるヒト化合物(例えばヒト抗体、ヒトホルモン及び/又はヒトタンパク質(例えばヒト酵素))の生産に特に適している故に、特に、関心のある安定化されたヒト細胞の生産が提供される。それ故に、本発明は好ましくは、前記先駆細胞がヒトCD34+前駆細胞を含むところの本発明の方法を提供する。好ましくは、本発明の幹細胞及び/又は前駆細胞は、STATタンパク質及び/又はBCL-6をコードする遺伝子の少なくとも機能的部分、及び/又はp53発現を少なくとも部分的に不活化することができる核酸配列を含む。STATタンパク質は好ましくは、構成的に活性なSTATタンパク質を含む。好ましい実施態様では、前記幹細胞及び/又は前駆細胞は、本発明のベクターによって形質導入される。最も好ましくは、前記ベクターは誘導性プロモーターを含み、従って前記不死化する核酸配列の発現が調節される。幹細胞及び/又は前駆細胞及び/又は本発明の関心のある細胞を含む非ヒト動物がまた、本明細書で提供される。前記動物は、関心のある細胞(例えばヒトB細胞)、好ましくは関心のある抗原に対する抗体を生産することができる細胞を生産するために適している。
【0055】
本発明の方法は特に、安定化されたB細胞の生産のために適している。従って、本発明は、B細胞株を生産するための方法であって、
−本発明の方法を用いて、B細胞を得ること、及び
−前記B細胞をエクスビボ(ex vivo)で培養すること
を含む方法を提供する。
【0056】
前記B細胞は好ましくは、ヒトB細胞を含む。前記安定化されたB細胞が特に、(モノクロナール)抗体の生産のために適している。関心のある抗原に対して特異的に向けられる抗体を生産することができるB細胞を得るために、前記関心のある抗原が、(ヒト)免疫応答を呼び出すために前記非ヒト動物に投与される。
【0057】
従って、本発明は、関心のある抗原に対して特異的に向けられた抗体を生産するための方法であって、
−核酸配列がB細胞中に存在する場合に前記B細胞を安定化させることができるところの核酸配列をB細胞の幹細胞及び/又は前駆細胞に備えること;
−前記幹細胞及び/又は前駆細胞を非ヒト動物に備えること;
−該関心のある抗原を前記非ヒト動物に備えること;
−該関心のある抗原に対して特異的に向けられた抗体を生産するB細胞の生成を許すこと;
−前記B細胞を得ること;及び
−前記B細胞によって生産された抗体を収穫すること
を含む方法を提供する。
【0058】
好ましくは、前記前駆細胞が、STATタンパク質及び/又はBCL-6をコードする遺伝子の少なくとも機能的部分、及び/又はp53発現を少なくとも部分的に不活化することができる核酸配列を備えられる。好ましくは、前記B細胞が前記動物から得られた後に、それがエクスビボでさらに培養される。1つの実施態様では、B細胞株が生成される。安定化されたヒトB細胞の生産が好ましい故に、前記前駆細胞は好ましくはヒトCD34+前駆細胞を含む。
【0059】
1つの実施態様では、前記非ヒト動物がマウスを含む。好ましくは、前記マウスはヒト造血システムの生成のために特に適している故に、前記マウスがRAG2-/-γc-/-マウスを含む。
【0060】
本発明は、下記の実施例によってさらに示される。該実施例は、本発明の範囲を何らかの方法において制限するものではない。
【0061】
図面の簡単な説明
図1
ヒト造血前駆細胞におけるp53ノック−ダウン。
(A)レンチウィルスのRNA干渉ベクターpTRIPΔU3-EF1アルファ p53の概要図。予測されるショートヘアピンRNAをターゲットとするヒトp53が示される。
(B)ヒト造血前駆細胞(CD34+)が単離され、そしてレンチウィルスベクターpTRIPΔU3-EF1アルファp53で形質導入され、そして1週間後にサイトカインでの培養において、GFPの発現に基づいて分離(ソート)された。次に、細胞がガンマ線照射され、そして6時間後に、細胞全体抽出物が調製され、10% SDS-ポリアクリルアミドゲル電気泳動上で分離され、そしてヒトp53を検出するために免疫ブロットされた。該ブロットは、ローディング対照としてベータアクチンに対する抗体でリプローブされた。
【0062】
図2
1日目、1週目又は2週目の齢における新生RAG2-/-γc-/-マウスのCD34+FL細胞を腹腔内移植。
4匹の新生RAG2-/- γc-/-マウスの3つのグループ(夫々、1日目(Δ--Δ)、1週目(O--O)又は2週目(x--x))が、0.5x106 MACS選択されたCD34+FL細胞を腹腔的に移植された。4、8及び12週で、すべてのマウスが採血され、そして該血液がCD45+ヒト細胞のパーセンテージを決定するためにFACSによる分析に付された。
【0063】
図3
胎児の肝臓からのCD34+造血幹細胞をip注射後の11週目でのRAG2-/- γc-/-マウスの胸腺の再増殖(Repopulation)。
胸腺のFACSプロファイル。胸腺細胞が、幹細胞マーカー(CD34及びCD38)、T細胞マーカー(TCRアルファ・ベータ及びTCRガンマ・デルタ、CD1、CD3、CD4、CD8、CD25、CD45RA)、NK細胞マーカー(CD16及びCD56)、形質細胞様DCマーカー(CD123及びBDCA2)、及びB細胞マーカー(CD19)に対する抗体で染色された。CD16及びCD56の分析における該細胞は、CD45+細胞上だけでなくCD3-細胞上においてもゲートされた。
【0064】
図4
胎児の肝臓からのCD34+造血幹細胞をip注射後の11週目でのRAG2-/- γc-/-マウスの骨髄、脾臓、肝臓及び肺の再増殖(repopulation )。
(A)骨髄のFACSプロファイル。骨髄細胞が、幹細胞マーカー(CD34及びCD38)、T細胞マーカー(CD3、CD4、CD25)、NK細胞マーカー(CD16及びCD56)、形質細胞様DCマーカー(CD123及びBDCA2)、B細胞マーカー(CD11、CD19、CD20、IgM及びIgD)、及び骨髄性細胞マーカー(CD11c及びCD14)に対する抗体で染色された。IgM及びIgDの分析における該細胞は、CD45+細胞上だけでなくCD19+細胞上においてもゲートされた。
(B)脾臓のFACSプロファイル。脾臓細胞は、骨髄細胞と同じ抗体で染色された。
(C)肝臓のFACSプロファイル。肝細胞が、幹細胞マーカー(CD34及びCD38)、T細胞マーカー(CD3)、形質細胞様DCマーカー(CD123及びCD4)、及びB細胞マーカー(CD19)に対する抗体で染色された。
(D)肺のFACSプロファイル。肺細胞が、T細胞マーカー(CD3)、形質細胞様DCマーカー(CD123及びCD4)、及びB細胞マーカー(CD19)に対する抗体で染色された。
【0065】
図5
レンチウィルスで形質導入されたヒト造血前駆細胞を移植されたマウスにおけるGFP+発現細胞の多分化能(multilineage)存在。
新生RAG2-/- γc-/-マウスは、胎児の肝臓から単離され且つpTRIPΔU3-EF1アルファp53 RNAiレンチウィルスで形質導入されたヒトCD34+造血幹細胞をip注射された。これらマウスの胸腺及び脾臓からの単核細胞懸濁液が、様々なヒト特異的モノクロナール抗体で染色され、そしてフローサイトメトリーによって分析された。ヒトCD45についてポジティブな細胞がゲートされ、そしてGFPポジティブ及びネガティブ集団を比較してさらに分析された。胸腺及び脾臓におけるT細胞発生の例が与えられる。細胞は、CD3、CD4、CD8及びCD56に向けられた抗体で染色され、そしてFACSによって分析された。
【0066】
図6
レンチウィルスで形質導入されたヒト造血前駆細胞を移植されたマウスにおけるp53i-GFP+を発現するT細胞からのVベータファミリー図。
【0067】
図7
レンチウィルス構築物は、形質導入されたヒトCD34+前駆細胞から得られた成熟細胞中に存在し且つ活性である。
(A)T細胞が、形質導入されたヒトCD34+細胞で再構築されたマウスの脾臓及び末梢血から単離され、イン ビトロで拡大され、そしてGFPの発現に基づいて分離(ソート)された。次に、細胞がガンマ線照射され、そして6時間後に、細胞全体の抽出物が調製され、10% SDSポリアクリルアミドゲル電気泳動上で分離され、そしてヒトp53を検出するためにイムノブロッティングされた。ベータアクチンに対する抗体でのウェスタンブロットが、対照として使用された。2匹の動物からの結果が示される。(B)p53iを発現するT細胞又は対照-GFP構築物が種々の刺激で処理され、そしてプロピジウムアイオダイド(PI)とアネキシン(Annexin) Vダブル染色の組み合わせ及びフローサイトメトリーを使用してアポトーシス誘発のために24時間後にアッセイされた。ダブルネガティブ細胞が、(パーセンテージによって示される)生存可能集団を示す。
【0068】
図8
p53の減少された含量は、ヒトCD34+前駆細胞に由来する成熟T細胞の増殖を好む。実施例1の材料及び方法において記載されているように、T細胞が、形質導入されたヒトCD34+細胞で再構築されたマウスの脾臓及び末梢血から単離され、そしてイン ビトロで拡大された。GFPポジティブ細胞のパーセンテージが、刺激後の2つの異なる時間点でのフローサイトメトリーによって確立された。
【0069】
図9
レンチウィルスで形質導入されたヒト造血前駆細胞を移植されたマウスの骨髄及び肝臓におけるGFP+発現細胞の多分化能(multilineage)存在。
新生RAG2-/- γc-/-マウスは、胎児の肝臓から単離され且つpTRIPΔU3-EF1アルファp53 RNAiレンチウィルスで形質導入されたヒトCD34+造血幹細胞をip注射された。これらマウスの肝臓及び骨髄からの単核細胞懸濁液が、様々なヒト特異的モノクロナール抗体で染色され、そしてフローサイトメトリーによって分析された。ヒトCD45についてポジティブな細胞がゲートされ、そしてGFPポジティブ及びネガティブ集団を比較してさらに分析された。形質細胞様DC、骨髄性及びB細胞の展開の例が示される。肝臓及び骨髄細胞は、CD123、BDCA2、CD11c、CD14、CD10、CD34、CD10、CD19、CD20、IgM及びIgDに向けられた抗体で染色される。
【0070】
図10
細胞パネル7TetOmCMVCMV対IRES
【0071】
図11
形質導入されたヒト繊維芽細胞MOI 1のrtTAウェスタンブロット
HeLa溶解物のウェスタンブロット
レーン1 mock形質導入された細胞、2 Lenti pgk eGFP、3、4 TRE d2eGFP CMV rtTA2-S2、5、6 TRE d2eGFP IRES rtTA2-S2。
【0072】
図12
繰り返し誘発結果(MOI0.1 HeLa)
HeLa細胞は、pgk eGFP, TRE d2eGFP CMVrtTA2-S2又はTRE d2eGFP IRES rtTA2-S2のいずれかでMOI 0.1で形質導入された。最初の誘発でGFPポジティブ細胞の数が、最初の誘発に対して正規化されたすべての他の測定で100%に設定された。
【0073】
図13
最大GFP発現のための時間
72時間でのMFIは、誘発の最大レベルとして設定され、そしてすべての他の値がこの値のパーセンテージとして基づく。
【0074】
図14
濃度(dox用量反応)7C1及び7I1ヒトFib MOI0.1
【0075】
図15
誘発可能なレンチウィルスベクターの概要図。
A:rtTAの構成的発現を有するベクター、例えばTetO7mCMV CMV rtTA3
B:自己調節発現を有するベクター、例えばTetO7mCMV IRES rtTA-S2又はTetO7mCMVIRES rtTA3。
【0076】
図16
イン ビボ及びイン ビトロにおけるヒトB細胞のSTAT5の検出
(A)ホルマリン固定され、パラフィンを埋め込まれた扁桃腺組織が、実施例3の方法の項に概説されているように、pTyr-STAT5(緑)及びCD20(赤)のためにダブル染色された。胚中心では、pTyr-STAT5及びCD20の共局在化が、暗い領域及び明るいゾーン領域においてB細胞上で観察され、パッチされ、膜様染色パターンとして観察可能である。
(B)ダブル染色されたアイソタイプ対照染色。結果は3つの実験についての代表である。元の倍率x 800。
(C)ヒトB細胞が、IL-2及びIL-4とともにCD40L-L細胞上で、示された時間について培養され、そして全体の細胞溶解物がSTAT5タンパク質のレベルについてウェスタンブロット分析によって分析された。該ブロットが取られ、そしてレーンの等しいローディングを保証するためにアクチンに対する抗体で再プローブされた。
(D)STAT5mRNAを特異的にターゲットするsiRNAプローブ(1365及び1668)のノックダウン効率。Raji B細胞株が、pSIN-対照-GFP、pSIN-siSTAT5(1365)-GFP又はpSIN-siSTAT5(1668)-GFPのいずれかで形質導入された。GFP+細胞の全体の細胞溶解物が、STAT5タンパク質発現について分析された。等しいローディングが、ブラッドフォード(Bradford)試験を使用して、タンパク質測定後に保証された。
(E)pSIN-対照-GFP又はpSIN-siSTAT5(1668)-GFPのいずれかで形質導入されたヒトB細胞の増殖応答。GFP+B細胞が分離(ソート)され、そして3日間(その最後の18時間はチミジン(1μCi/ウェル)の存在下(Chan等)で)、CD40L-L細胞、IL-2及びIL-4とともに96ウェルプレートにおいて2重に培養された。チミジンの取り込み(Chan等)は、分当たりのカウント(cpm)として表される。
【0077】
図17
CA-STAT5bの発現はB細胞の生存及び拡大をもたらし、一方WT-STAT5bの発現は生存のみをもたらす。
(A)CD19+B細胞が、扁桃腺から分離(ソート)され、CD40L、IL-2及びIL-4とともに培養され、そして対照-IRES-GFP(開いた四角)、WT-STAT5b-IRES-GFP(閉じた丸)又はCA-STAT5b-IRES-GFP(開いた丸)で形質導入され、そしてCD40L、IL-2及びIL-4でさらに培養された。示された時点でのフローサイトメトリック分析によって決定されたGFP+細胞のパーセンテージが示される。3つのうちからの代表的な実験が示される。
(B)絶対的な細胞数カウントが、(A)において決定されたGFP+細胞のパーセンテージに基づいて決定された;対照-IRES-GFP(開いた四角)、WT-STAT5b-IRES-GFP(閉じた丸)又はCA-STAT5b-IRES-GFP(開いた丸)。
(C)CD40L、IL-2及びIL-4において3ヶ月間培養されたCA-STAT5b-IRES-GFPを形質導入されたB細胞上での対照抗体染色(薄い線)と一致するアイソタイプと比較され、フローサイトメトリック分析によって決定されたCD19及びCD20(太い線)の発現。
(D)CD40L、IL-2及びIL-4において3か月間培養されたCA-STAT5b-IRES-GFPを形質導入されたB細胞におけるVH-Cμ遺伝子組み換えのRT-PCR分析。
【0078】
図18
CA-STAT5b-ERは、4HT依存様式において核に局在化する。
(A)CA-STAT5b-ER-IRES-ΔNGFRで形質導入されたB細胞が、4HTの存在しない(左パネル)又は存在する(右パネル)において、抗-NGFR-FITC(緑)及び抗-ER-TexasRed(赤)で染色され、そして共焦点レーザースキャン顕微鏡(Confocal Laser Scan Microscopy)によって分析された。
(B)STAT5(右)又はER(左)に対する抗体を使用して、CA-STAT5b-ER-IRES-ΔNGFR+ B細胞の細胞性抽出のウェスタンブロット分析。
【0079】
図19
CA-STAT5b-ERで形質導入されたB細胞の特徴
(A)扁桃腺B細胞が、CD40L、IL-2及びIL-4において培養され、そしてCA-STAT5b-ER-IRES-ΔNGFR (丸)で又は対照-IRES-ΔNGFR (四角)で形質導入された。両方の培養物が分けられ、一部は1μM 4HTなし(開いた記号)で及び他は1μM 4HTあり(閉じた記号)で培養された。形質導入後の示された時点では、形質導入された細胞のパーセンテージが、NGFRに対するAPCコンジュゲートされた抗体で染色後にフローサイトメトリック分析によって決定された。絶対的な細胞数カウントが、ΔNGFR+細胞のパーセンテージに基づいて決定された。示された実験は、行なわれた5つの独立実験についての代表である。
(B)CA-STAT5b-ER-IRES-ΔNGFRを形質導入されたB細胞が、形質導入の30日後に、4HTなしで7日間培養された。次に、等しい数の細胞が、4HTの存在(閉じた四角)又は4HTの存在なし(開いた四角)のいずれかで培養され、そして絶対的な細胞数が決定された。3つのうちからの代表的な実験が示される。
(C)4HTの存在下でCD40L、IL-2及びIL-4において培養されたCA-STAT5b-ER-IRES-ΔNGFRを形質導入されたB細胞の代表的な培養物のフローサイトメトリック分析。アイソタイプ対照染色が、薄いヒストグラムとして示される。
【0080】
図20
BCL-6-IRES-GFPで形質導入され、そしてCD40L、IL-2及びIL-4において拡大された末梢B細胞の特徴
(A)対照-IRES-GFP(開いた記号)又はBCL-6-IRES-GFP (閉じた記号)で形質導入され、そして示された期間についてCD40L、IL-2及びIL-4において培養された末梢血B細胞のGFP発現のフローサイトメトリック分析及び絶対細胞数カウント。この実験は、行なわれた8つの独立の実験についての代表である。
(B)フローサイトメトリック分析によって決定された、形質導入の77日間、CD40L、IL-2及びIL-4において培養された代表的な実験のBCL-6を形質導入されたB細胞の表現型。薄いヒストグラム線は、アイソタイプ対照染色を示す。
【0081】
図21
STAT5によるBCL-6発現の誘発
(A)4HTとともに、CD40L、IL-2及びIL-4において培養されたCA-STAT5b-ERで形質導入されたB細胞におけるBCL-6及びHPRTのRT-PCR分析。10日間、4HTなしで該細胞のインキュベーション後、4HTが、シクロヘキサミド(CH)の存在下又は不存在下で8時間添加された。この実験は3回繰り返され、同様の結果だった。
(B)抗-BCL-6抗体を使用してウェスタンブロット分析によって決定された、12日間、常に4HTの存在下で又は4HTの不存在下で、CA-STAT5b-ERを形質導入されたB細胞におけるBCL-6タンパク質の発現。アクチンに対する抗体が、タンパク質ローディング対照として使用された。同様の結果が、3つの独立した実験において得られた。
(C)デュアルルシフェラーゼリポーター分析。BCL-6遺伝子のプロモーター領域(位置-657/+471 bp)の一部が、ホタル(P. pyralis)ルシフェラーゼをコードする配列にリンクされ、そしてpGL3ベクター内にサブクローニングされた。このプロモーター領域(548/-566 bp)の1つの潜在的なSTAT5結合部位は、TTCTCTGAAからGTCTCTAAAに変異され、及び2つの異なるクローン(BCL-6*及びBCL-6**)がpGL3-ホタル・ルシフェラーゼ・バックボーン内にサブクローニングされた。これらpGL3構築物は、293T細胞内にLZRS-CA-STAT5b及びRenillaルシフェラーゼを含むベクターで共形質導入された。両方のルシフェラーゼ活性が、単一のサンプルから連続的に測定され、そして相対ルシフェラーゼ単位がRenillaルシフェラーゼ活性について計算され及び修正された。
【0082】
図22
CA-STAT5bを形質導入されたB細胞の成長に対するBCL-6ノックダウンの影響。
(A)BCL-6siRNAプローブのノックダウン効率が、BCL-6を形質導入された293T細胞内へのトランスフェクション後に、ウェスタンブロット分析によって試験された。除去後、該ブロットは、等しいタンパク質ローディングを保証するためにアクチンに対する抗体でインキュベーションされた。
(B)対照-GFP(開いた円)、siBCL-6(789)-GFP(閉じた四角)、又はsiBCL-6(970)-GFP(閉じた菱形)で形質導入され、そして示された期間の間、CD40L、IL-2及びIL-4において培養されたCA-STAT5b-ER-ΔNGFR+B細胞におけるGFP発現のフローサイトメトリック分析。この実験は4人の異なるドナーからのB細胞において繰り返され、同様の結果だった。
【0083】
実施例
【実施例1】
【0084】
材料及び方法
マウス
H-2dRAG2-/-マウス(Antonius Rolink博士及び Shunichi Takeda博士によって親切に提供された;バーゼル免疫学研究所、バーゼル、スイス国(Takeda S, Rodewald HR, Arakawa H, Bluethmann H, Shimizu T. Immunity.1996年; 第5巻:第217-228頁))が、IL-2Rγc-/-マウスとかけあわされて(Blom B, Spits H, Krimpenfort P. In: Smit Sibinga CT, Das PC, Lowenberg B編集 Cytokines and Growth Factors in Blood Transfusion. London: Kluwer Academic Publishers;1997年:第3-12頁)、H-2d RAG2-/- IL-2Rγc-/-マウスが得られた (RAG2-/-γc-/- マウスといわれる:Weijer K, Uittenbogaart CH, Voordouw A, Couwenberg F, Seppen J, Blom B, Vyth-Dreese FA, Spits H. Blood. 2002年;第99巻:第2752-2759頁; Kirberg J, Berns A, von Boehmer H. J Exp Med. 1997年; 第186巻:第1269-1275頁)。これらマウスがT、B及びNKリンパ細胞の完全な欠如を示すので、これらマウスは免疫不全である。マウスはアイソレーターにおいて飼育され且つ維持され、そしてオートクレーブされた食餌及び水を与えられた。全ての操作は、層流下で行われた。
【0085】
胎児の肝臓及びさい帯血細胞調製
ヒト胎児の肝臓が、人工妊娠中絶から得られた。妊娠期間は、頭の直径の超音波測定によって決定され、及び14〜20週に及んだ。この組織の使用は、オランダ癌研究所(Netherlands Cancer Institute)及びアカデミック医療センター(Academic Medical Center)の両方の医学倫理委員会によって承認され、及びインフォームドコンセントを条件であった。ヒト胎児の肝細胞が、機械的手段による組織の穏やかな破壊によって、引き続きFicoll-Hypaque(Lymphoprep;Nycomed Pharma、オスロ、ノルウェー)上の密度勾配遠心分離によって単離された。単一細胞懸濁液が、組織を刻み、ステンレス鋼メッシュを通じてそれらをプレスすることによって調製された。大きな集合体が除去され、そして該細胞は、CD34+細胞の単離前に培地で1回洗浄された。
【0086】
RAG2-/-γc-/- マウスへのヒトCD34+細胞の移植及びヒト細胞移植の評価
CD34+細胞(フローサイトメトリーによって評価されたときに、>98%純粋、データは示されてない)の富化が、CD34前駆細胞単離キット(Miltenyi Biotec、ベルギッシュグラートバハ、ドイツ)を使用して行なわれた。CD34+細胞(0.2〜200万個の細胞)が、半致命的に照射された(350 cGy)新生RAG2-/- γc-/- マウス(1週未満)内にip移植された。ヒト細胞移植のキネティックス(kinetics)を決定するために、末梢血が、移植後の3〜4週ごとに尾静脈から集められた。マウスが移植後の様々な時点で犠牲にされ、そして末梢血(PB)、肝臓、肺、脾臓、骨髄(BM)及び胸腺が、ヒト細胞の存在について評価された。照射されていないマウスがヒト細胞での再増殖を必ずしも示すとは限らないので、3.5Gyの全身照射(TBI)が行なわれる。単核細胞(MNC)が、Ficoll-Hypaqueを介した密度勾配遠心分離によって単離され、抗ヒトCD45(Becton and Dickinson、サンノゼ、カリフォルニア州)及び他のマーカーで染色された。これらマーカーの発現が、FACS Calibur(Becton and Dickinson)上で測定され、そしてFCS発現プログラム(Denovosoftware、オンタリオ、カナダ)で分析された。該移植されたヒト単核集団は、前方及び側方散乱光パラメーターに基づいて定義され、そして所定のマーカーについてのポジティブ細胞のパーセンテージが、白血球及びCD45+ゲート内にある細胞について決定された。移植されなかったRAG2-/-γc-/- マウスにおける抗ヒトCD45と反応した細胞存在はなかった。ヒト細胞の特異的なサブセットが、抗ヒト特異的mAbsで染色することによって定量され、それは、フルオレセインイソチオシアネート(FITC)、フィコエリトリン(PE)、ペリジニンクロロフィルタンパク質質(PerCP)又はアロフィコシアニン(APC):CD11c、CD19、CD34及びCD45RA(Coulter-Immunotech)、CD3、CD4、CD8、CD10、CD14、CD16、CD20、CD56、CD83及びCD123(Becton and Dickinson);CD38及びCD45RO(Dako、グロストラップ、デンマーク)及びBDCA-2(Miltenyi Biotec)とコンジュゲートされた。
【0087】
レンチウィルス生成
複製不完全な自己不活性化HIVベクターが、FUGENE(Roche、ナットレー、ニュージャージー州)及び3つの異なるプラスミドを使用して、293T細胞のトランジエントトランスフェクションによって生産された。使用された該プラスミドは、VSV-GエンベロープをコードするプラスミドpMD.G、ウィルス粒子を生産するためのGag、Pol、Tat及びRevタンパク質を提供するように設計されたパッケージングプラスミド pCMVDR8.91、及びトランスファーベクターpTRIPデルタU3-E1アルファ(SirvenA, Ravet E, Charneau P, Zennou V, Coulombel L, Guetard D, Pflumio F,Dubart-Kupperschmitt A. Mol Ther. 2001年;第3巻:第438-448頁)であった。pTRIPデルタU3-E1アルファは、Brummelkamp等(Brummelkamp TR, Bernards R, Agami R. Science. 2002年;第296巻:第550-553頁)によって記載されたヒトp53ターゲッティング配列5´GACTCCAGTGGTAATCTACを含むpSUPERベクターからのsiRNAカセットを含むように変更され、及びまた、伸長因子-1-アルファプロモーターによって駆動される拡張されたGFP遺伝子を運ぶ。トランスフェクションの20時間後に、培地が、Yssel培地(YsselH, De Vries JE, Koken M, van Blitterswijk W, Spits H. Methods. 1984年;第72巻:第219-227頁)によって置き換えられ、そしてウィルスの2つのサンプルが、24及び48時間で集められた。ウィルスを含む上清は、細胞を除去するために1800 rpmで10分間遠心分離され、次に0.22 mmのフィルターを通され、そして使用まで-80℃で維持された。
【0088】
形質導入プロトコル
CD34+ヒト細胞の形質導入は、サイトカインの不存在下で、レトロネクチン(宝酒造株式会社、大津、日本)で覆われた24ウェルプレート上でのウィルス上清への一晩の暴露の1サイクルによって行われた。次の日、細胞が洗浄され、そしてマウスにip注射されるか或いは夫々10 ng/mlのトロンボポイエチン、幹細胞因子及びIL-7(全てPeprotechから)の存在下でYssel培地における培養物中に維持されるかのいずれかであった。形質導入の効率が、フローサイトメトリーによって、2〜3日後に、GFPポジティブ細胞のパーセンテージを決定することによって評価された。
【0089】
ウェスタンブロット
全体の細胞抽出物が、氷上で30分間、RIPAバッファーにおいて、等しい数の細胞を溶解することによって調製された。Bio-Radタンパク質アッセイ(Bio-Rad、ミュンヘン、ドイツ)によって決定された等価な量のタンパク質が、10% SDSゲル上にロードされた。P53タンパク質がSanta Cruz Biotechnologyからのモノクロナール抗体DO-1を使用して、及びローディング対照としてベータアクチンがポリクロナール抗体I-19(またSanta Cruzから)を使用して検出された。
【0090】
ヒトT細胞培養
ヒト形質導入された幹細胞で再構築されたRAG2-/- γc-/- マウスの血液及び脾臓からのヒトT細胞の拡大は、(Spits H, Ijssel H, Terhorst C, de Vries JE. J Immunol. 1982年;第128巻:第95-99頁)に記載されているように、2人の異なるドナーからの照射された異質遺伝子型のヒト末梢血細胞(PBL)、及び照射されたEBVを形質導入されたB細胞株(JY)、PHA(Gibco、グランドアイランド、ニューヨーク州)及び組み換えヒトIL-2(rhIL-2;Roche、ナットレー、ニュージャージー州)からなるフィーダー混合物での刺激によって行われた。ヒト細胞を含む全ての培養が、Yssel培地において行われた。
【0091】
アポトーシス細胞の検出
細胞の存続可能性が、ガンマ線照射(3000rad)前若しくは24時間後、又はスタウロスポリン(1μM)又はフルダラビン(5μM)での処理後に、フローサイトメトリーによって分析された。手短に言えば、細胞が収穫され、そして氷で冷やされたHEPESバッファー(10mM HEPES、150 mM KCl、1mM MgCl2及び1.3 mM CaCl2、pH 7.4)において洗浄された。次に、細胞が、20分間、APCをラベル付けされたアネキシンV(Becton Dickinson (サンノゼ、カリフォルニア州、米国))と一緒にインキュベーションされた。フローサイトメトリーによるサンプルの分析の直前に、プロピジウムアイオダイド(PI)(Sigma、セントルイス、ミズーリ州)が添加された(最終濃度 5μg/ml)。生存可能な細胞が、アネキシンV及びPIの染色の両方についてネガティブとして定義された。
【0092】
結果
レンチウィルスを介在した遺伝子導入によるヒトCD34+細胞内へのsiRNAの導入
レンチウィルスは、他の遺伝子輸送システムに関する2つの重要な有利点を有する;それらは、ノンサイクリング細胞(non-cycling cell)を感染させることができ及び展開の間にサイレンシングされない。幾つかのグループは、哺乳動物細胞におけるRNAiの発現のために、小さい核RNAプロモーター(H1 (Brummelkamp TR, Bernards R, Agami R.Science. 2002年;第296巻:第550-553頁)及びU6(Dick JE, Lapidot T, Pflumio F. Immunol Rev. 1991年; 第124巻:第25-43頁))の使用を記載した。H1プロモーターはヒトp53タンパク質をターゲットとするように設計されたsiRNAの発現を駆動するように使用され、そしてsiRNAカセットを発現するレンチウィルスベクターが調製された(図1A)。CD34+細胞の高度に富化された集団がヒト胎児の肝臓から単離され、そして引き続きsiRNA/GFPを発現するレンチウィルスベクターで形質導入された。形質導入効率は、フローサイトメトリーによって決定されたGFP発現のレベルによって示されるように、慣用の様に20〜50%の範囲に及んだ。形質導入された細胞の細胞成長又は生存に対する明白な効果が、標題「材料及び方法」において上記されたサイトカインの存在下で3週間培養後にイン ビトロで観察されなかった。GFPマーカーを発現するCD34+由来細胞の割合は、初期集団のそれと同等のままだった(データは示されていない)。
【0093】
導入された構築物が活性であり且つこれらの細胞においてp53の発現をノックダウンしうることを示すために、我々はそれらのGFP発現に基づいて、形質導入後にCD34+細胞を分離(ソート)した。p53の発現(それは通常の状況において非常に低い)が、ガンマ線照射を含む幾つかの遺伝毒性的刺激への細胞の暴露後に、非常に迅速に上方制御される。形質導入されていない細胞及び対照を形質導入された(示されていない)細胞の両方がp53のレベルを増加させることによって放射線に応答し、一方p53 siRNA構築物を発現する細胞は応答せず(図1B)、該構築物が造血幹細胞における遺伝子発現をサイレンシングできたかったことを示す。
【0094】
T細胞展開及びホメオスタシスに対するp53のノックダウンの効果。
T細胞展開に対するp53ノックダウンの効果を検査するために、我々は、Dick及びその同僚(Dick JE, Lapidot T, Pflumio F. Immunol Rev. 1991年;第124巻:第25-43頁)によって開拓されたヒト-SCIDマウスモデルを用いた。SCIDマウスモデルは、C.B-17-Prkdcscid(SCID)マウス(Lapidot T, Pflumio F, Doedens M, Murdoch B, Williams DE, Dick JE. Science. 1992年; 第255巻:第1137-1141頁)及びより最近ではNOD/LtSz-Prkdcscid (NOD/SCID)マウス(Pflumio F等 Blood. 1996年; 第88巻:第3731-3740頁; 及びBonnet D等, Nat Med. 1997年;第3巻:第730-737頁)のBMを多分化能リンパ細胞及び骨髄細胞で再増殖するためにこれら原始細胞(SCIDを再増殖する細胞と名付けられた)の能力に基づく。しかしながら、T細胞展開の研究について、T細胞の展開が明らかにマウスにおける活性な生得の免疫システムの存在により単に時々観察された故に、これらマウスは制限を有する。最近、RAG2-/- γc-/- マウスに静脈内(iv)注射すると、CD34+細胞がB細胞及び形質細胞様DC(pDC)内に展開しうることが報告された(Weijer K等, Blood. 2002年;第99巻:第2752-2759頁)。さらに、我々は、T細胞が、CD34+細胞のiv注射に引き続きこれらの成体マウスにおいて展開することを観察した(結果は示されていない)。成功率は80%だったけれども、末梢におけるコンシステントなT細胞移植を観察するために、12〜16週平均を要した。その上、これらマウスの胸腺は、0.8×106未満の細胞で小さいままだった。6〜8週齢のRAG2-/- γc-/- マウスの胸腺の微環境がヒトT細胞展開について最適でないかもしれないことを考慮すると、我々は初期の時点でヒト前駆細胞を注射することを決定した。新生児(1週齢未満)、1週又は2週齢のマウスが、胎児の肝臓由来のCD34+細胞をip注射され、そして末梢血が注射後8及び12週間後に分析された。図2は、末梢におけるヒト細胞での移植が、年上のマウスにおけるよりも、1日齢未満のマウスにおいてより高いことを示す。1週齢マウスの注射はまた有意な移植を生じたが、2週齢マウスでは、CD34+細胞のip注入後にヒト細胞の展開がなかった(図2)。
【0095】
誕生後、1〜3日の間でip注射された78匹のマウスの分析は、64匹のマウスにおいてヒトCD45+細胞での再構築(表1)を明らかにした(83%)。32匹のマウが50%を超す再構築を示し、そして残りが10〜50%の間を示した。マウスの末梢血におけるヒトCD45+細胞を有する全てのマウスはまた、胸腺及び他の器官におけるT細胞を有した。
【0096】
【表1】
【0097】
誕生の1日後にip注射されたマウスの胸腺の構造物が、移植後10週で共焦点顕微鏡によって検査された。分化された胸腺は、外皮様の領域におけるCD1+CD4+及びCD4+CD8+細胞、並びに骨髄様領域におけるシングルポジティブCD4+及びCD8+T細胞で観察された(結果は示されていない)。
【0098】
新生マウスモデルをさらに確認にするために、我々は、形質導入されていないCD34+胎児の肝細胞を注射された大きな一連のマウスにおける胸腺及び様々な末梢器官におけるT細胞を最初に分析した。さらに、我々は、他の白血球サブセットの存在につてこれらのマウスを分析した。注射の8〜10週間後に、2〜1000万個の細胞が、胸腺から回収されることができた。広範囲のフローサイトメトリック分析(図3)は、ダブルポジティブCD4+CD8+ 細胞及びCD4+及びCD8+ シングルポジティブT細胞を含む全ての系統サブセットの存在を明らかにした。その上、CD3+high細胞のサブセットは、CD45RA+を発現した。この集団集合は、周囲への移出直前にT細胞を表わす。興味深いことに、CD25は、Stephens等(StephensLA等, Eur J Immunol. 2001年;第31巻:第1247-1254頁)によって以前に記載された制御性T(Treg)細胞でありうるCD4+T細胞のサブセット上で発現された。これらCD25+CD4+細胞がまたグルココルチコイド誘発された腫瘍壊死因子受容体ファミリー関連遺伝子(GITR)及び細胞質CTLA-4についての高度にポジティブであるという観察(結果は示されていない)は、これら細胞がTreg細胞を表わすという概念を支持する。TCRアルファ・ベータ+細胞及びCD3+CD56+に加えて、胸腺がまた、CD3-CD56+NK細胞の実質的な数を含んでいた(発現するCD16のおよそ半分を有する)。NK細胞(2%)のパーセンテージは、子どもの胸腺において通常見られるそれよりもはるかに高かった(<0.1%)。理論への結び付きなしに、マウス胸腺環境が、NK細胞の生成に好ましくすることは可能である。代替的に、NK細胞のはるかに高い割合は、正常なヒト胸腺微環境におけるよりもTCRアルファ・ベータ+細胞の低い拡大率の結果でありうる。我々はまた、TCRガンマ・デルタ+細胞(1.5%対正常な胸腺における0.1%未満)及びB細胞(2.2%対正常な胸腺における0.1%未満)の相対的に高い割合を観察し、及びこれらの高いパーセンテージはまた、TCRアルファ・ベータ+細胞の低い拡大の結果でありうる。NK細胞、B及びTCRガンマ・デルタ細胞の増加したパーセンテージと対照的に、BDCA2+CD123high pDCのそれは正常範囲内であった(0.1%)。
【0099】
胸腺において展開するT細胞は、複数の解剖位置へ移動する。T細胞の可変パーセンテージは、胸腺において見られるだけでなく、骨髄、脾臓、肝臓及び肺においても見られた(図4)。骨髄はT細胞の低い割合を有し(1%)、一方肺においてこのパーセンテージは11%であった。肝臓及び脾臓は、T細胞の同等のパーセンテージを含んでいた。CD4:CD8比(3〜4:1)は、全ての器官において同様であり及びヒトにおいて通常観察される範囲内であった。興味深いことに、脾臓及び骨髄では、Treg細胞及び最近活性化されたCD4+T細胞を推測上含むCD3+CD4+CD25+細胞の集団が検出されうる。我々の結果は、新生マウスへのCD34+細胞のip注射は、大多数のマウスにおけるヒトT細胞の相対的に迅速な再構築を生じることを示す。
【0100】
新生のRAG2-/-γc-/- マウス内に注射された場合、CD34+細胞は複数の系統に展開される。B細胞は、誕生の1日後に注射されたマウスにおける末梢血及び様々な器官において展開された最も支配的な細胞集団であった。CD19+B細胞の最高パーセンテージが、骨髄において見られた(図4A)。意外にではなく、脾臓における大多数のB細胞(図4B)が、CD10+CD20+細胞であり及びIgM及びIgDを共発現した。骨髄では、大多数のB細胞がCD20-CD10+であり、マウス骨がヒトB細胞展開のための部位であることを示唆した。骨髄の検査(図4A)は、CD34+細胞の存在を明らかにし、その小さなパーセンテージはCD38の低レベルだけを発現し、いくつかの原始的なCD34+CD38dim前駆細胞がそれらの分化していない状態にあることを示唆する。
【0101】
pDCの有意な数が、全ての器官において見られた;pDCが、骨髄(図4A)及び肝臓(図4C)において最も明らかに存在するが、また肺(図4D)においても存在した。NK細胞がまた末梢血及び様々な器官において検出されることができたが、該パーセンテージは胸腺におけるよりも一般にはるかに低かった。CD11c+及びCD14+単球が器官において観察され(図4)、ヒトリンパリンパ展開だけでなくヒト脊髄展開がこれらマウスにおいて起きることを示した。
【0102】
次に、我々は、p53siRNAで又はエンプティーベクターで形質導入されたCD34+胎児の肝細胞で一連のマウスに注射した。表2は、GFP+及びGFP-細胞における胸腺細胞のパーセンテージが、p53 siRNA又は対照を形質導入されたCD34+細胞で注射されたマウスにおける両方において同様であったことを示した。同様に、肝臓又は脾臓細胞が分析されたときに、我々は、これらの動物の末梢において有意な(決定的な)違いを検出できなかった。従って、我々は、p53の不活性化がp53 siRNAを発現する末梢のT細胞の生存の有利点をもたらさないと結論した。
【0103】
【表2】
【0104】
p53 siRNA及び対照を形質導入された細胞で注射されたマウスの胸腺の検査が、CD4CD8ダブルネガティブ、ダブルポジティブ及びシングルポジティブT細胞コンパートメントにおけるGFP+細胞の存在を明らかにした。しかしながら、p53siRNA-GFP+上でゲートする場合、我々は、形質導入されていない細胞(GFP-)又は対照-GFP+上での胸腺におけるT及び非T細胞の何らかのサブセットの表現型における差を観察しなかった(図5A)。また、我々は、何らかのp53 siRNA-GFP+におけるT細胞の存在下において、又は脾臓において形質導入されていない細胞上での差は観察されなかった(代表的なマウスのT細胞の表現型が図5において示されている)。
【0105】
DNA損傷に応答して細胞増殖の制御及びアポトーシスの誘発におけるp53の重要な役割を与え、p53の減少したレベルがTCRレパートリーの組成において効果を有しうるかを知ることは興味があった。我々は、p53i構築物で形質導入されたCD34+細胞を注射されたマウスから単離されたT細胞におけるTCR多様性を調査した。PCR分析によって、我々は、全てのVbetaファミリーがレパートリーにおいて表されることを確立できた(図6)。その上、CDR3領域の詳細な分析はこれらのマウスにおいて存在する末梢のT細胞がポリクロナールであることを明らかに示し(データは示されていない)、T細胞展開の間にp53の下流制御が幾つかのクローンの球を生じないことを示した。これらの結果を解釈するために、これらの動物の末梢から単離された細胞におけるp53の発現をノックダウンする際に該構築物の安定な発現及び効率を検証することは重要であった。形質導入されたCD34+細胞で10週早くに形質導入されたマウスの脾臓及び血液から単離されたヒト単核細胞は、2人の異なるドナーからのヒトPBMCの存在下で培養された(EBV細胞株JY, PHA及びIL-2)。これらの条件下では、T細胞は広範囲に増殖した。成熟したT細胞のみがこのプロトコルを使用して拡大されうる故に(Res P等, J Exp Med. 1997年;第185巻:第141-151頁)、我々のデータは、RAG2-/- γc-/- マウスにおいて展開されたヒトT細胞が機能的に成熟しており且つTCRライゲーションによってトリガーされたシグナルに応答しうることを示した。さらに、T細胞は、同種抗原に応答し且つイン ビトロで再刺激するとサイトカインの広いアレイを生産することができ、それらは異種抗原を認識し且つ応答することが可能であり、それらは異種抗原を認識し且つ応答することができたことを示す(データは示されていない)。イン ビボでp53 siRNAを形質導入されたCD34+細胞由来のT細胞の拡大後に、我々は、GFP+及びGFP-T細胞を分離(ソート)し、そして2つの異なる再構築された動物からのガンマ線照射後のp53のレベルを試験した。両方のマウスにおいて、p53は形質導入されていない集団において増加され、一方p53 siRNA構築物を含む細胞におけるp53レベルはほとんど検出できないままだった(図7A)。その結果、これらのp53 siRNA+細胞は、ガンマ線照射又はフルダラビンでの処置によって誘発されたアポトーシスにそれほど感受性でない。従って、これらp53 siRNA+細胞は安定される。対照的に、p53の損失は、p53独立アポトーシス誘発剤であるデキサメタゾン及びスタウロスポリンによって誘発されるアポトーシスへのT細胞の感受性に影響しなかった(図7A及びデータは示されていない)。そのアポトーシスを促進する機能(pro-apoptotic function)に加えて、p53はまた、種々の刺激に応答して成長停止の誘発に関与する。その上、p53はhTERTのネガティブ調節因子であり、それはT細胞の複製の寿命を調節する(Roth A等, Blood. 2003年;第102巻:第849-857頁)。我々は、TCR刺激後にp53 siRNAを発現する細胞の成長を研究した。我々の構築物におけるGFPの存在は、我々が、p53iで又は形質導入されていない細胞に関連する対照構築物のいずれかで形質導入されたT細胞の集団の動力学を検査することを許した。図8に示されているように、我々はp53の減少した量を有する細胞の進行的蓄積を検出し、p53 siRNA+T細胞が形質導入されていない細胞又は対照を形質導入された細胞と比較して成長有利点を有することを示した。興味深いことに、これらの差は刺激直後だけでなく、該細胞が長期間の培養において維持される場合においても検出されず、TCR刺激への即時応答がp53 siRNAを発現する細胞及び対照において同様であることを示す(図8及びデータは示されていない)。これらの結果は、CD34+細胞内に導入されたp53 siRNAがT細胞子孫における下方制御するp53タンパク質において活性のままだったことを明らかに示す。我々のデータはまた、p53の下方制御がストレスに対してT細胞を保護するがhTERTを上方制御し、それはこのシステムにおけるヒトT細胞展開又はT細胞のホメオスタシスに影響しないことを示した。
【0106】
異なる器官におけるB細胞、形質細胞様DC又は単球の展開に対する P53 RNAi の影響の欠如。
新生マウス内にCD34+細胞を注射すると複数の系統が展開されたという事実は、我々がB細胞、pDC及び単球の展開に対するp53ノックダウンの影響を分析することを同様に可能にした。表3では、GFP+ 及びGFP- 集団におけるB細胞のパーセンテージは、対照-GFP及びp53 siRNA-GFP-形質導入されたCD34+細胞から展開された細胞において類似であることが示されている。さらに、B細胞分化マーカーの発現における差が、形質導入されていない対照-GFPとp53 siRNA-GFP細胞との間で認められなかった(図9)。
【0107】
その上、GFP+集団におけるこれらの細胞のパーセンテージが形質導入されていない、対照を形質導入されたサンプルにおいて並びにp53 siRNA形質導入されたサンプルにおいて非常に類似したので、我々はまたp53をノックダウンすることがpDC及び単球の展開に対して影響しないことを示すことができた。
【0108】
【表3】
【0109】
議論
我々は、新生RAG2-/-γc-/- マウスが、関心のある安定化されたヒト細胞を生成するために適していることを本明細書に示す。ロバストなT細胞展開が、新生マウス内にCD34+細胞をip注射すると観察され、それは成体マウスで以前に観察されたものと比較して加速されたペースで進められた。メインストリームのCD4+及びCD8+T細胞に加えて、Treg細胞を表わしうるTCRガンマ・デルタ細胞、CD3+CD56+T細胞及びCD25+CD4+細胞を含むT細胞の全ての他のサブセットが観察されうる。我々はまた、サーキュレーション(circulation)及びCD34+細胞を注射されたマウスの末梢器官におけるB細胞、NK細胞、pDC及び単球の展開を観察した。CD15+CD11c+CD24+顆粒球に加えて、ヒトグリコホリンポジティブ細胞の低いがコンシステントなパーセンテージが存在した(原稿は準備中)。これらのデータはこれらのマウスにおけるヒト白血球のかなり完全なレパートリーの生成を示すとともに、これらの新生ダブルKOマウスはイン ビボで「ヒト」免疫応答を研究するために適していることを示す。
【0110】
独立プロモーターの制御下でマーカーGFPを含んだレンチウィルスベクターにおけるp53に対して向けられたsiRNAを使用することによって、我々は、イン ビボで遺伝的に組み換えられたCD34+前駆細胞の展開を追跡するためにGFPの実現可能性を示した。生化学分析は、CD34+前駆細胞において、p53 siRNA発現が、ガンマ線照射誘発されたp53発現の95%よりも多い減少をもたらしたことを明らかにした。我々は、siRNA構築物がT細胞子孫において安定的且つ機能的に発現されたままであったことを見つけた故に、siRNAが成熟白血球内でのCD34+細胞の分化の間に発現されたままであったことが示されている。
【0111】
マウスから単離されたp53 siRNA+T細胞は、対照を形質導入された又は形質導入されていないT細胞と比較して、イン ビトロで成長有利点を示した。我々は、p53の下方制御がヒトT細胞にガンマ線照射への耐性を与えることを示した。従って、p53の下方制御が、細胞の安定化をもたらす。
【実施例2】
【0112】
dox依存導入遺伝子発現を有する単一のレンチウィルスベクターが設計される。我々の設計において、我々は、IRESエレメントを使用してTRE及びGFPのrtTA下方ストリームを置くことによって、CMVプロモーターの構成的発現又は自己調節発現下でrtTAを置くことを評価する。ヒトの初代及び確立された細胞株のパネルに対する両方のベクターシステムの評価は、自己調節ループがより低い基礎導入遺伝子活性、より高い導入遺伝子誘発を有することを示す。その上、自己調節ループのための誘発動力学が、構成的に発現されたrtTAのそれよりも速く且つ長期の培養及び誘発の繰り返しサイクルに応答してより良く耐性であるように現れることが示される。
【0113】
引き続き、rtTA2-Sは、我々がrtTA3と呼ぶ最近公表されたウィルス的に発生されたrtTA(Das AT等, Journal of Biological Chemistry 2004年;279巻:第18776-18782頁)によって置き換えられた。この新しく記載されたrtTA3は、野生型rtTAと比較してdoxに対して13倍のより高い感受性及びrtTA2-M2と比較してdox濃度の広い範囲についてのより高い転写活性及び誘発の両方を有する。自己調節ループ内でのrtTA3の導入は、doxのより低い濃度でループの活性を許し、及び100 ng/mLのdox濃度に達した最大発現でGFPのdox依存段階的発現を許す。
【0114】
材料及び方法:
試薬:
Dox(Sigma)ストック溶液が、水中でdox 10mg/mlを溶解することによって作製され、そしてフィルター滅菌された。ストックは、-20で冷凍にて維持された。
【0115】
レンチウィルスベクター調製
レンチウィルスベクターは以前報告されたように調製された(Seppen,J, Rijnberg,M., Cooreman,M.P., and Oude Elferink,R.P. (2002年). Lentiviral vectors for efficient transduction of isolated primary quiescent hepatocytes. J Hepatol第36巻, 第459-65頁)。手短に言えば、HEK 293T細胞は、4つのプラスミドを含む第3世代のレンチウィルスベクターシステムで、リン酸カルシウム沈殿を使用して、トランスフェクションされた。トランスフェクションに続き24時間で、25 mM HEPES pH 7.4を補完された培地で置換された。ウィルスを含む上清がトランスフェクションに続き48時間で集められ、小分けされ、そして−80℃で冷凍された。
【0116】
細胞株及び培養
HEK293T、HeLa、HepG2、SJNB-8、HUVEC、ヒト繊維芽細胞、ヒト胎児肝細胞。すべてが、10%のウシ胎仔血清、ペニシリン/ストレプトマイシン(penn/strep)、グルタミンで補完された標準DMEM培地において培養された。
【0117】
ウィルス力価及び形質導入
ウィルス力価が、HeLa細胞の形質導入によって決定された。手短に言えば、ウィルスは、連続希釈法において、DEAEデキストランを補完されたHeLa細胞に添加され、そして4時間形質導入される。形質導入に続く72時間で細胞が収穫され、そしてGFP発現がウィルス力価を計算するためにフローサイトメトリーによって測定される。力価は、1000 ng/ml doxでの誘発によって決定された。
【0118】
SDS-PAGE及びウェスタンブロッティング
SDS-PAGE及びウェスタンブロッティングが、Seppen, J., R. R.van der, N. Looije, N. P. van Til, W. H.Lamers, and R. P. Oude Elferink. (2003年)、Long-term correction of bilirubin UDPglucuronyltransferase deficiency in rats by in utero lentiviral gene transfer. Mol. Ther. 第8巻:第593-599頁に記載されたように行われた。
【0119】
結果:
単一のTet-Onレンチウィルスベクターを構築する我々の初期の試みは、ウィルス性LTR内に配置されたTetOの2つの繰り返しを含んだ以前に記載されたtet依存条件的複製HIV-1(Verhoef等, 2001年)からの適応物だった。表4において報告されているように、我々は、これらのベクターで高いウィルスの力価を得ることができなかった。次に、我々は、セントラルポリプリン配列(cPPT)及びB型肝炎転写後制御調節エレメント(PRE)を含む第3世代レンチウィルスのベクターのバックボーンにおける最小のCMVプロモーターに融合されたTetOの7個の繰り返しからなるTREを使用することを進めた(Barry等、2001年)。これらの新しいベクターで、我々はウィルスの力価を1〜2のオーダーの大きさで増加させることができ、より特に自己調節ループベクターで、導入遺伝子発現を駆動するために構造性プロモーターを使用して得られるそれらと比較してウィルス力価を得ることができた(データは示されていない)。
【0120】
【表4】
【0121】
ヒトの初代及び確立された細胞株のパネルが、7C1又は7I1のいずれかで感染の多重度(MOI)1で形質導入され、そして1000 ng/mlのdox濃度で誘発された。誘発レベルは、doxなしにGFP発現によって分割されたdoxでのGFP発現として測定され、及び遺伝子発現全レベル及び基本的発現の両方を反映する。試験された全ての細胞株において、自己調節ループが、rtTAの構成的発現と比較してより高いレベルの誘発を与えた(表5)。
【0122】
【表5】
【0123】
我々は、我々が誘発なしにrtTAの低い基本的発現を期待したように自己調節ループを設計した。これを評価するために、我々は、Lenti TetO7mCMV CMV rtTA-S2又はLentiTetO7mCMV IRES rtTA-S2のいずれかでMOI 1で初期経過初代ヒト繊維芽細胞(early passage primary human fibroblast)を形質導入した。拡大に続き、該細胞は、72時間、1000 ng/ml で処理されず又は処理され、次に溶解物を生成するために収穫された。
【0124】
ドキシサイクリの不存在下で、我々は自己調節ループにおけるrtTAの発現を検出することができず(図11、レーン5)、我々の自己調節ループシステムの基本的発現が確かに低く、一方我々は構成的発現ウィルスにおけるrtTAの発現を明らかにみることができた(図11、レーン3)ことを確認した。
【0125】
HeLa 7C1対7I1上の繰り返し誘発
次に、我々は、7C1又は7I1のいずれかでMOI 0.1でHeLa細胞を形質導入し、doxの誘発及び撤回を変更することによって、誘発に対する繰り返しサイクルを経験するための能力のための両システムを評価し、そこでは各経過がオン状態又はオフ状態のいずれかであることを表す。対照として、我々は、MOI 0.1で、lenti pgk eGFPで形質導入されたHeLa細胞を含めた。1つのサイクルに続くGFPポジティブ細胞のパーセンテージが50%より多くだけ減少し、そして誘発の幾つかのサイクル後に、開始値の20%まで落ち、一方IRES rtTA2-S2形質導入された細胞についてのレベルが、pgk eGFP対照と比較して安定のままであり、rtTAの自己調節的制御がより良く耐性であることを示した。なぜならば、rtTAの毒性が避けられるためである。
【0126】
rtTAの自己調節発現を使用することに関する他の関心事は、最大活性が、構成的発現と比較して、相当により遅いだろうということだった。最大誘発についての時間を決定するために、我々は、TRE d2eGFP CMV rtTA2-S2又はTRE d2eGFP IRESrtTA2-S2のいずれかでMOI 1でHeLa 細胞を形質導入し、そして固定された時点でdox(1000 ng/ml)を追加した。我々は、次の72時間でGFP発現における増加がなかったことを見つけ(公表されていない観察)、従って我々はこの時間を最大発現のための任意のレベルとして設定した。図13にみられうるように、rtTAの自己調節発現は、構成的に発現されたrtTAよりも速く最大レベルに到達した(24時間、最大の70%対45%、及び48時間、100%対75%)。これらの結果は、自己調節ループを使用して誘発時間における損失がないことだけでなく、我々が構成的に発現されたrtTAと比較してより早い最大発現に到達することを示す。
【0127】
制御された遺伝子発現の重要な特徴は、誘発性分子の濃度と導入遺伝子発現のレベルとの間の相関を有することである。我々は、dox濃度(0〜1000 ng/ml)の範囲に渡る2つの異なるシステム間のGFP発現のレベルを比較した。我々は、より低いdox濃度で、我々が自己調節ループでのGFP発現を作動させることができなかったことを観察した(図14、パネルA)。基本条件下でrtTA2-S2の非常に低い発現のために、自己調節ループは、開始のためにより高い濃度のdoxを要求する。低い濃度のdoxを使用する場合に、rtTA2 S2がループを活性化するためにdoxに十分に感受性でないと我々は仮定した。この考えを試験するために、我々は、rtTA2 S2を、(Urlinger S等; Proc.Natl.Acad.Sci. (2000年) 第97巻, 第14号, 第7963-7968頁、及びDasAT等, Journal of Biological Chemistry 2004年;第279巻:第18776-18782頁)により記載された3つのアミノ酸置換を含む変異体rtTA, rtTA3で置き換え、それは野生型rtTAと比較してdoxに対して13倍のより多い感受性を有し且つ転写活性を改善した。更なる議論のために、このrtTA変異体はrtTA3と云われるだろう。我々が自己調節内にrtTA3を導入した場合、我々は、100 ng/ml doxで得られた発現の最大レベルで試験されたdox濃度の範囲にわたりGFPの段階的発現を見た(図14、パネルB)。
【実施例3】
【0128】
この実施例は、B細胞の調節及び増殖におけるSTAT5の役割を示す。
【0129】
材料及び方法
B細胞単離
B細胞が、大人の扁桃腺又は末梢血から得られた。扁桃腺摘出術が、アムステルダム自由大学の子どものための外科部門において行なわれた。T細胞が、抗-CD4及び抗-CD8コンジュゲートされたマイクロビーズ(Miltenyi Biotec, アムステルダム, オランダ)を使用して使い果たされた。次に、該細胞は、コンジュゲートされた抗-CD19 FITC (DAKO、グロストラップ、デンマーク)及びコンジュゲートされた抗-CD3 フィコエリトリン(PE)でインキュベーションされ、引き続きMoFlo (Cytomation/DAKO、グロストラップ、デンマーク)又はFacStarPlus (Becton Dickinson、サンノゼ、カリフォルニア州)を使用してCD19+CD3-集団を分離(ソート)した。該生じたB細胞は、再分析すると99%超の純度だった。
【0130】
レトロウィルス構築物及び組み換えレトロウィルスの生産
STAT5a及びbの構成的に活性な変異体が、以前に記載されていた(Ariyoshi等、2000年;大西等、1998年)。これら変異体及び野生型STAT5bをコードするDNAが、T. Kitamura(IMSUT、東京、日本)から得られた。BCL-6は、抗増殖性p19ARF-p53シグナリングの阻害剤としてネズミの繊維芽細胞における老化救出スクリーンにおいて識別された(Shvarts等、2002年)。これらDNAは、以前に記載されたLZRS-linker-IRES-GFPベクター内にライゲーションされた(Heemskerk等、1997年;Heemskerk等、1999年)。ノックダウン実験のために、Brummelkamp等によって以前に記載されたpSUPER構築物(Brummelkamp等、2002年)が適応された。形質転換された細胞の識別をフローサイトメトリーによって許すために、siRNAプローブの転写のためのpol3プロモーター及びGFP発現を駆動するPGKプロモーターが反対の位置にあるように、GFP発現カセットがpSUPER構築物に追加された。
mRNAを特異的にターゲットとするsiRNA配列が、Ambion ホームページ(http://www.ambion.com)を使用して設計された。配列が、pSUPER-GFPのBglII-HindIII部位内に挿入された。引き続き、pol3-siRNA-pgk-GFPカセットが、LZRSレトロウィルス構築物の自己不活性化誘導体内にサブクローニングされた。
【0131】
レトロウィルスのプラスミドが、製造者のプロトコルに従いFugene-6 (Roche Diagnostics Netherlands、アルメレ、オランダ)を使用して、ヘルパーウィルスのない両種性生産者細胞株Phoenix-A(ヒト杯腎臓細胞株293の誘導体)(Kinsella and Nolan、1996年)(Dr. G. Nolan, Stanford University、パロ・アルト、カルフォルニア州、の親切なギフト)内にトランスフェクションされた。2日後に、トランスフェクションされた細胞の選択が、2μg/ml ピューロマイシン(Becton Dickinson Clontech Laboratories, パロ・アルト, カルフォルニア州)の添加によって開始された。トランスフェクションの10〜14日後に、6 x 106細胞が、10 mlの完全培地(Iscove's medium (Life Technologies BV, ブレダ, オランダ)(8% ウシ胎仔血清 (FCS)における10cmペトリディッシュ (Becton Dickinson Discovery Labware、ベッドフォード、マサチューセッツ州)、ピューロマイシンなし)毎に置かれた。翌日に該培地は新たにされ、そして翌日にレトロウィルスの上清が収穫され、遠心分離され、そして-70℃で細胞フリー部分で冷凍された。このアプローチは再現可能な、迅速な、大規模の、及び3×106超(伝染性ウィルス粒子/ml)の高い力価のレトロウィルス生産を与える。
【0132】
CA及び野生型(WT)STAT5bエストロゲン受容体(ER)融合構築物が下記のように作られた:終止コドンの代わりにBglII部位を導入するために、PCRが、N604H STAT5b変異体(Ariyoshi等、2000年;Onishi等、1998年)又は野生型STAT5b cDNAsのいずれかで行なわれた。XhoI/BglII消化生成物が生成され、それはΔCA-又はΔWT-STAT5b-ERを生成するために、pBS-ER(C term)(Dr. Kurataによって親切に提供された, DNAX Institute、パロ・アルト、カルフォルニア州)(Kurata等、1999年)のBamHI/EcoRI消化、及びpBS-SK+のXhoI/EcoR1消化でライゲーションされた。次に、LZRS-CA-STAT5b-ER及びLZRS-WT-STAT5b-ERを生成するために、LZRS-CA-STAT5b-IRES-ΔNGFR又はLZRS-WT-STAT5b-IRES-ΔNGFRのXhoI/NotI消化が、pBSΔCA-STAT5b-ER又はΔWT-STAT5b-ERの部分的NotI/XhoI消化にライゲーションされた。これを使用して、我々は、IRESのCA-STAT5b-ER又はWT-STAT5b-ER下流、及びΔ神経成長因子受容体(ΔNGFR)、NGFRのシグナリング不全変異体(C. Bonini博士によって親切に提供された(Bonini等、1997年))、又は緑蛍光性タンパク質GFPを有する構築物を作った。NGFRに対するモノクロナール抗体(Chromaprobe、マウンテンビュー、カルフォルニア州)が、ΔNGFR発現細胞を視覚化するために使用された。
【0133】
レトロウィルス形質導入
組み換えヒトフィブロネクチンフラグメントCH-296形質導入手続(RetroNectin(商標); Takara, 大津, 日本)が、以前に記載されたように行われた(Heemskerk等、1997年;Heemskerk等、1999年)。非組織培養処理された24ウェルプレート(Costar、バドフーフェドルプ、オランダ)が、2時間室温で又は40℃一晩で、0.3 mlの30μg/ml 組み換えヒトフブロネクチンフラグメントCH-296で覆われた。該CH-296溶液が除去され、引き続き室温で30分間、リン酸緩衝生理食塩水(PBS)において2% ヒト血清アルブミン(HSA)とともにインキュベーションされ、引き続きPBSで一度洗われた。5x105のB細胞が、0.25mlの解凍されたレトロウィルス上清及びポリブレン(最終濃度 4 μg/ml)と混合された0.25mlの完全培地中に置かれ、そして37℃で6時間インキュベーションされた。次に、0.25mlの上清が除去され、そしてポリブレンに加えて0.25mlの新鮮なレトロウィルスの上清が追加され、そして一晩37℃でインキュベーションされた。翌朝、細胞は洗われ、そして照射された(80 Gy)CD40L(CD154)を発現するL細胞(CD40L-L)、IL-2(20U/ml)及びIL-4(50 ng/ml)を有する24ウェル組織培養処理されたプレート(Costar)に移された。
【0134】
細胞培養
B細胞は、5% CO2を含む湿った空気中、37℃で完全培地において培養された。CD40L-L細胞(80グレイで照射された)が、24ウェル組織培養処理されたプレート(Costar)においてウェル当たり5x104細胞を播かれた。5x104にソートされたB細胞が、IL-2(20U/ml)及びIL-4(50 ng/ml)とともに添加された。1週後に、該細胞はレトロウィルス形質導入のために使用された。形質導入後、B細胞が、照射されたCD40L-L細胞、IL-2及びIL-4で再び培養された。
【0135】
pTyr-STAT5及びCD20のための免疫組織学的染色
扁桃摘出術を経験する子どもから得られたヒト扁桃腺組織についてのpTyr-STAT5及びCD20の免疫組織学的分析が、以前に記載された免疫蛍光法及びCLSM分析(小さな修正を有する)を使用して行われた(Vyth-Dreese等、1995年)。手短に言えば、ホルマリン固定され、パラフィンを埋め込まれたヒト扁桃腺部分が、シトレート回収すると、5%(v/v)の正常なヤギ血清(Central Laboratory of the Netherlands Red Cross Blood Transfusion Service (CLB)、アムステルダム、オランダ)においてプレインキュベーションされた。引き続き、セクションが、一晩、4℃でウサギ抗pTyr-STAT5b抗体(Cell Signaling Technology、ベバリー、マサチューセッツ州、米国)及びマウス抗CD20 (L26, DAKO)においてインキュベーションされ、引き続きビオチン化されたヤギ抗ウサギIgG(DAKO)、ストレプタビジン/ビオチン−コンジュゲートされたホースラディッシュパーオキシダーゼ複合体(ABC-protocol, DAKO)及びTyramide-Alexa Fluor568(Molecular Probes Europe、ライデン、オランダ)においてインキュベーションされた。CD20が、Alexa Fluor 633-コンジュゲートされたヒツジ抗マウスIgG (Molecular Probes)を使用して視覚化された。インキュベーションの間に、セクションは、1%ウシ血清アルブミン(BSA, Sigma Aldrich、ズウェインドレヒト、オランダ)を含むPBS中で広範囲にすすがれた。各試験内で、アイソタイプと一致した対照抗体及び正常なウサギIgGが、ネガティブ対照として含まれていた。共焦点蛍光画像が、Kr/HeNeレーザー組み合わせを備えたLeica TCS SP (Leica Microsystems、ハイデルベルグ、ドイツ)共焦点システム上で得られた。画像は、40x 1.25 NA対物レンズを使用して得られた。カラー顕微鏡写真が、電子オーバーレイから得られた。
【0136】
STAT5発現を局地化するために、CA-STAT5b-ER-IRES-ΔNGFRを形質導入されたB細胞が、ポリシンで覆われたカバーグラス上に付けられた。15分間、室温で3.7%ホルムアルデヒドでの固定化後、細胞は、45秒間、0.1%トリトンX-100で浸透された。第1の抗体は、マウス抗NGFR及びウサギ抗ER(MC-20, Santa Cruz Biotechnology、サンタ・クルス、カリフォルニア州)であった。第2の抗体は、抗マウス-FITC(Becton Dickinson Pharmingen、ハイデルベルク、ドイツ)及び抗ウサギ-TexasRed(Molecular probes、ユージーン、オレゴン州)であった。共焦点蛍光画像が、Leica TCS SP (Leica)上で得られた。
【0137】
B細胞の表現型検査
FITC、PE又はAPCで直接ラベル付けされたヒト分子に対する抗体 IgD、IgG、CD3、CD19、CD20、CD27、CD38、CD40、CD45、CD56、CD70、CD80、CD86、HLA-DR(Becton Dickinson, Pharmingen) 、及びPE (DAKO) で直接ラベル付けされたIgM、カッパライト鎖(kappa light chain)、ラムダライト鎖(lambda lightchain)、CD138が、フローサイトメトリー分析のために使用された。染色された細胞が、FACSCalibur(商標)(Becton Dickinson Imunocytometrysystems, サンノゼ、カリフォルニア州)を使用して解析され、そしてFACSデータがCellQuest(商標)コンピュータソフトウェア(Becton Dickinson Immunocytometry systems)で処理された。
【0138】
RT-PCR
総RNAが、RNeasy(商標) mini kit (Qiagen Sciences、ジャーマンタウン、メリーランド州)を備えた解凍されたペレットから単離された。5×第1ストランドバッファー、500 μM dNTP's、25 μg/L オリゴ (dT)、200 U スーパースクリプト II RT (Life Technologies)を含むRNAが、20μLの容量で逆転写された。cDNA溶液の1マイクロリットルが、20 mM Tris-HCL、50 mM KCL、1.5 mM MgCl2、5 mM dNTP's、2.5 U Taq DNA ポリメラーゼ(Life Technologies)及び30 pmolの各プライマーを含む50μl溶液中のPCRに付された。PCR条件は下記であった:94℃で7分の変性工程、引き続き、94℃で30秒、62℃で30秒(HPRT)、52℃(LMP-1)、58℃(EBNA1/2)、58℃(BCL-6)、及び72℃で30秒の30サイクル、そして72℃で最終7分の延長。逆転写酵素(RT)PCRのために使用されるオリゴヌクレオチドは、
だった。
【0139】
VH遺伝子を検出するためのPCR条件は、(Guikema等、1999年; vonLindern等、2000年)に記載されたのと同様であり、94℃で7分の変性工程、引き続き、94℃で1分、60℃で1分及び72℃で1分の35サイクル、そして72℃で最終10分の延長を含む。RT-PCRのために使用されるオリゴヌクレオチドは次のとおりだった:
。
【0140】
ウェスタンブロッティング
細胞抽出物が、プロテアーゼ阻害剤カクテル(Boehringer Ingelheim BV, インゲルハイム, ドイツ)を補われたRIPA溶解バッファー(150 mM NaCl、1% NP-40、0.1% SDS、0.5% DOC、50 mM TRIS-HCl pH 8.0)において調製された。タンパク質は、Protranニトロセルロース転写膜(Schleicher and Schuell BioScience Inc., キーン、ニューハンプシャー州)に移された。ウェスタンブロッティングのために使用される西洋わさびパーオキシダーゼ(HRP)にコンジュゲートされた主要な抗体は、BCL-6 (C-19)、STAT5b (C-17)、ER (MC-20)及びアクチン(I-19)だった(全てSanta Cruz Biotechnology (Santa Cruz、カルフォルニア州、米国)から)。STAT5ノックダウンウェスタンの検出のために、タンパク質の等量がBradford試験(BCA protein assay reagent kit(Pierce Biotechnology Inc., Rockford、イリノイ州、米国))を使用してロードされた。他のウェスタンブロットのために、アクチンがローディング対照として使用された。タンパク質の検出が、高められた化学発光(pierce)を使用して行われた。
【0141】
ルシフェラーゼリポーター転写促進アッセイ
リポーター構築物(構成的に活性なウミシイタケ(Renilla reniformis)ルシフェラーゼ生成ベクターprL-CMV(Promega、サン・ルイ・オビスポ、カリフォルニア州、米国)及び293T細胞における発現ベクターLZRSの共トランスフェクションが、製造者の指示書に従いFugeneトランスフェクション試薬で行われた(Roche、バーゼル、スイス)。ホタル(P. pyralis)及びウミシイタケルシフェラーゼの検出が、製造者の指示書(Promega)に従いダブルルシフェラーゼアッセイキットを使用して行われた。pGL3ベーシックベクターにおけるBCL-6プロモーター領域-657/+471が、S. Hirosawa博士(東京医科歯科大、東京、日本)によって親切に提供された。BCL-6プロモーターフラグメントにおけるSTAT5結合部位の変異体が、二段階PCRアプローチによって生成された。第一工程PCRは、BCL-6(F)又はBCL-6(R)プライマー並びに対応するGLprimer2及びRVprimer4(Promega)で行なわれた。
。次に、対応するPCR製品が精製され、混合され、そしてGLprimer2及びRVprimer4プライマーを使用して再増幅された。該増幅されたPCRフラグメントは、メーカー指示書に従って、pCR2.1TOPO ベクター(Invitrogen、リーク、オランダ)内に直接的にクローニングされた。プライマーM13(F)及びM13(R)での配列決定は、染料ターミネーターサイクル配列決定キット(PerkinElmer)を使用して、ABI シーケンサー (PerkinElmer Corp、ノーウォーク、コネチカット州)で行われた。変異されたBCL-6プロモーター領域は、SacI及びBglII消化を介してpGL3においてサブクローニングされた。
【0142】
結果
チロシンリン酸化されたSTAT5は、ヒト扁桃腺における胚中心B細胞において発現される。
STAT5がイン ビトロで幾つかのB細胞成長因子によって活性化される故に、我々はチロシンリン酸化された(pTyr)-STAT5が、ヒト扁桃腺において生理学的条件下で検出されうるかどうかを調査した。我々は、pTyr-STAT5及びCD20のために共染色された扁桃腺セクションのCLSM分析を行なった。図16Aは杯中心域において、CD20dimB細胞上のpTyr-STAT5の共局在化を示し、典型的にパッチされた、膜様の染色パターンを示す。同様の染色パターンが、扁桃腺のT細胞域においてT細胞上で検出可能であった(データは示されていない)。アイソタイプ対照抗体でダブル染色された扁桃腺セクションは、何ら染色を示さなかった(図16B)。
【0143】
RNA干渉によってSTAT5をノックダウンすることが、ヒトB細胞の増殖を阻害する
ヒトB細胞が、IL-2、IL-4及びIL-10を含むサイトカインの存在におけるCD40の関与に続き、制限された期間のみについてイン ビトロで培養されうる。B細胞が数週間後に死ぬ理由は、完全に理解されていない。本明細書では、STAT5タンパク質の量が制限因子であり、それは成長因子に対して非応答性をもたらしうるかどうかを我々は扱う。ヒトCD19+B細胞が分離(ソート)され、そしてIL-2及びIL-4とともにCD40L-L細胞上で培養された。サンプルが培養の2及び17日後に取られ、そしてウェスタンブロッティングがSTAT5抗体を使用して細胞溶解物全体上で行なわれた。明らかに、タンパク質レベルが、培養の2日目と比較して、培養の17日後に、B細胞において減少された。これは、B細胞が制限されたSTAT5レベルのために結局死ぬことを示す。
【0144】
B細胞の増殖におけるSTAT5の役割を直接的に調査するために、我々は、RNA干渉によって減少するためのSTAT5を特異的にターゲッティングすることによってSTAT5レベルをノックダウンすることを選択した。STAT5 siRNAプローブ(1365及び1668)が設計され、そしてRaji B細胞を内生的に発現するSTAT5におけるそれらのノックダウン効率について試験された。STAT5 siRNAsは、PGKプロモーターによって駆動されるGFPマーカー(pSIN GFP)をまた含む我々の自己不活性化レトロウィルス構築物においてサブクローニングされた。Raji B細胞は、STAT5(1365)若しくはSTAT5(1668)siRNA発現ウィルス、又は対照ウィルスのいずれかでレトロウィルス的に形質導入され、そしてGFP発現に基づいて分離(ソート)された。図16Dにおいて示されるように、STAT5(1668)siRNAは、対照を形質導入されたRaji細胞と比較して完全に近い、STAT5タンパク質発現の量に効率的に減少され、一方STAT5(1365) siRNAはあまり有効でないがSTAT5レベルを有意に減少された。B細胞増殖に対するSTAT5の影響を分析するために、我々は、対照ウィルスと平行してCD19+分離(ソート)された末梢血B細胞内にSTAT5(1668) siRNA プローブを形質導入し、そしてIL-2及びIL-4とともにCD40L-L上でこれらを培養した。GFP+B細胞を分離(ソート)した後、3日間、IL-2及びIL-4を有するCD40L-L細胞上で2回(104細胞/ウェル)培養され、そのうち最後の18時間はチミジン(1μ Ci/ウェル)の存在下(Chan等)であった。STAT5タンパク質発現は、50%だけB細胞の増殖能力を劇的に減少した(図16E)。
【0145】
初代ヒトB細胞の拡大に対するSTAT5の影響
下記観察が、ヒトB細胞の成長の調節におけるSTAT5の役割を調べることを我々に促した:1)pTyr-STAT5は、イン ビボにおけるヒトB細胞において検出されうる;2)STAT5タンパク質レベルが、CD40L-L細胞、IL-2及びIL-4上でイン ビトロにおいて培養されたヒトB細胞において減少し、それはそれらの最終的な死に関連しうる;3)ヒトB細胞におけるSTAT5のノックダウンは、それらの増殖能力を減少する;4)pTyr-STAT5は、様々なB細胞悪性腫瘍において構成的に発現される。我々は、ヒトB細胞の成長に対する野生型(WT)並びにSTAT5a及びbの構成的に活性な(CA)変異体の影響を比較した。精製された扁桃腺CD19+B細胞が、CD40L-L細胞、IL-2及びIL-4とともに共培養された。7日目で、該細胞は、ウィルス発現CA-STAT5a-IRES-GFP、CA-STAT5b-IRES-GFP、WT-STAT5b-IRES-GFPで、又は対照-IRES-GFPで形質導入された。形質導入効率は、5%〜20%だった(データは示されていない)。CA-STAT5b-GFP(図17A)又はCA-STAT5a-GFPで形質導入された培養におけるGFP+細胞のパーセンテージ(示されない)は、時間をかけて増加した。5〜6週目で、対照-IRES-GFPを形質導入された及び形質導入されていないB細胞が死に始め、一方6週目(すなわち形質導入後の5週目)で、CA-STAT5b-GFPを形質導入された培養物の95%超がGFP+であり、且つ拡大することを続け、そして培養物中に維持された。WT-STAT5b-GFPで形質導入されたB細胞が選択性に生存したが、拡大せず(図17A及びB)、STAT5の活性はB細胞の拡大のために適していることを示す。該拡大したCA-STAT5b-GFP+B細胞は、CD19を発現したが、CD20を発現しなかった(図17C)。CA-STAT5b-GFPを形質導入された細胞の複製寿命延長がEBV形質転換によるものでなかったことを保証するために、我々は、センシティブRT-PCRによってLMP-1及びEBNA1/2mRNAを分析し、そしてこれらの遺伝子が発現されなかったことを確認した(結果は示されていない)。重要なことに、CA-STAT5b-GFPを形質導入されたB細胞は、RT-PCRによって決定されるように、Cμ(図17D)又はCガンマ(示されていない)につけられた幾つかのIgHバリアブル遺伝子セグメントを発現し、形質転換されたB細胞株がモノクロナールでないことを示した。
【0146】
同時に、これらのデータは、活性のSTAT5の構成的発現が、細胞死からのイン ビトロ培養されたヒトB細胞を救い、そしてそれらの複製寿命を拡大することを示した。
【0147】
タモキシフェンでのCA-STAT5b-ER発現の誘発に続くB細胞株の生成
STAT5bの構成的に活性だけ(及び第2の形質転換事象でない)がヒトB細胞の拡大された増殖応答をもたらすことをさらに確認するために、我々はB細胞増殖に対する調節可能なSTAT5b構築物の影響を試験した。この目的のために、我々は、CA-STAT5b又はWT-STAT5bでエストロゲン受容体(ER)の融合を調製し、及びIRES-ΔNGFRのCA-STAT5b-ER又はWT-STAT5b-ER上流を抱く組み換えウィルスの構築物を作成し、それは切り捨てられ、神経成長因子受容体の無能な変異体をシグナリングする(Bonini等、1997年)。形質導入すると、STAT5b-ER融合タンパク質は、熱ショックタンパク質によって隔離された不活性な複合体として該形質導入された細胞の細胞質において発現される。4−ヒドロキシ−タモキシフェン(4HT)とのインキュベーション後、ER融合タンパク質が熱ショックタンパク質から単離し、核へ移動する。確かに、4HTの不存在又は存在下において抗ER抗体でCA-STAT5b-ER-IRES-ΔNGFR+ B細胞の染色をすること及びCLSMを使用して分析することは、CA-STAT5bが4HT依存様式において期待されるように核に局所化する(図18A)。ウェスタンブロット分析は、形質導入された細胞における予期されたMWの融合タンパク質の存在を確認した(図18B)。
【0148】
次に、我々は、CA-STAT5b-ER-IRES-ΔNGFR構築物でヒト扁桃腺のB細胞を形質導入し、そしてこれらB細胞の増殖能力に対する4HTの影響を試験した。図19Aは、CD40L、IL-2及びIL-4におけるCA-STAT5b-ER-IRES-ΔNGFRで形質導入された扁桃腺B細胞の次の培養を示し、CA-STAT5b-ER発現細胞(ΔNGFR+)の成長選択が4HTの存在下でのみ生じた。重要なことに、CA-STAT5b-ER-IRES-ΔNGFR+を形質導入された細胞からの4HTの除去は、B細胞の成長の終了、そして最終的な死をもたらした(図19B)。4HTの不存在下で培養された、CD40L、IL-2及びIL-4で培養されたCA-STAT5b-ER-IRES-ΔNGFR+ B細胞が最初に増殖したが、それらは成長を止め、次に7〜20日後に死んだ。4HTが7日にわたり添加されたとき、該細胞は生存し、次に成長することを続けた(図19B)。これらの結果は、扁桃腺B細胞のCA-STAT5b-誘発された成長がB細胞を形質転換するCA-STAT5bによって誘発される不可逆の遺伝的変化の結果でないが、核におけるCA-STAT5bの継続的、機能的発現にのみ依存することを示す。CA-STAT5b-ER-IRES-ΔNGFRで形質導入され、そして3ヶ月の間、CD40L、IL-2、IL-4及び4HTにおいて培養された扁桃腺B細胞の一部が、細胞表面IgMを発現した(図19C)。該細胞は、細胞表面IgDについてネガティブであり、及び小さなパーセンテージ(1〜2%)がIgGを発現し、それはIgMを欠いた。すばらしいことに、CA-STAT5b-ER-IRES-ΔNGFR+ B細胞の長期の培養をすると、細胞表面Igの発現が失われた(データは示されていない)。同様の効果が、ナイーブ又はメモリーの末梢血B細胞のいずれか内にSTAT5の導入後に観察された(データは示されていない)。その上、CA-STAT5b-ER-IRES-ΔNGFR+ B細胞はCD19、CD38、CD40、CD70、CD80及びHLA-DRを発現したが、CD20及びCD27についてネガティブだった。CD25は、これらの細胞上で高度に発現されたが(図19C)、STAT5bがCD25(IL-2Ralpha)転写(John等、1996年;John等、1999年)を直接的に制御するという事実と一致する。注目すべきことに、B細胞におけるWT-STAT5b-ER融合タンパク質の発現は、B細胞におけるCA-STAT5b-ERタンパク質の発現と比較して同様の4HT依存成長効果及び表現型を生じた。思うに、これは、ERドメインがSTAT6-ER について見られたように二量化するという事実によって引き起こされ(Kurata等、1999年)、核に転位されるSTAT6-ER二量体を生成した。
【0149】
ヒト末梢血B細胞内へのBCL-6の異所性発現は、細胞の応答寿命の延長をもたらす
最近、我々は、幼い子供のヒト扁桃腺B細胞におけるBCL-6の異所性発現が、CD40L、IL-2及びIL-4において培養される場合に、B細胞の成長有利点をもたらしたことを我々は報告した(Shvarts等、2002年)。これらの細胞は、CD19を発現し且つCD3及びCD56についてネガティブだった。これらの発見を拡張するために、我々は、BCL-6がまた成人末梢血B細胞の増殖能力に影響するかどうかを調査した。それ故に、我々は、CD40L発現L細胞、IL-2及びIL-4とともに培養されたヒトB細胞内にBCL-6-IRES-GFPを導入した。図20Aは、BCL-6の発現が、BCL-6の形質導入の10〜13日後に開始するマーカーGFPを発現する末梢B細胞の成長有利点をもたらした。これらの細胞の表現型に関する情報を得るために、我々は、モノクロナール抗体のパネルで広範囲な分析を行なった。図20Bは、BCL-6-IRES-GFPを形質導入されたB細胞がCD19を発現したことを示す。加えて、BCL-6-IRES-GFPを形質導入されたB細胞はCD20を発現し、CD38及びメモリーB細胞マーカーCD27について弱いポジティブであり、しかし形質細胞マーカーCD138についてネガティブだった。その上、該細胞は、活性マーカー HLA-DR、CD40、CD70及びCD80を発現し且つCD25について弱いポジティブだった。重要なことに、BCL-6-IRES-GFP形質導入されたB細胞は、これらの細胞がそれらの分化において、細胞表面Ig-ネガティブ形質細胞内に捕捉されたという事実と一致する細胞表面Igカッパ又はラムダを発現した。
【0150】
BCL-6はSTAT5bの直接ターゲットである。
CA-STAT5及びBCL-6がヒトB細胞の成長に対して同様の効果を有するという観察は、BCL-6がSTAT5によって調節された事実にあるという可能性を高める。この概念は、BCL-6の1.5 kBプロモーター(Ohashi等、1995年)が3つの潜在的なSTAT結合部位を含むという事実によって支持される。STAT5bがBCL-6発現を規制することができるかどうかを研究するために、我々は、10日間、4HTの不存在下でSTAT5b-ER+扁桃腺B細胞を培養した。次に、4HTが添加され、そして該細胞は8時間後に収穫され、そしてRT-PCRによってBCL-6の発現について試験された。図21Aは、BCL-6転写物の発現が、このホルモンなしの培養においてよりも4HTありの培養において実質的により高いことを示す。ウェスタンブロット分析は、4HTの存在下で培養されたB細胞が4HTの不存在下で培養された細胞よりも有意に多いBCL-6タンパク質を発現したことを明らかにした。BCL-6がSTAT5bの直接又は間接のターゲットであるかどうかを判断するために、タンパク質合成阻害剤シクロヘキシミド(CH)が8時間、4HTと一緒に添加された。4HT及びCHの両方が添加された場合にBCL-6mRNAの上方制御がまた観察され(図21A)、一方CH単独の添加はBCL-6発現に影響しなかった(データは示されていない)。これらのデータは、BCL-6発現がSTAT5bによって直接的に影響されることを示す。
【0151】
この観察を拡張するために及びBCL-6が確かにSTAT5bの直接ターゲットであることを検証するために、我々はBCL-6プロモーターを使用して、ルシフェラーゼレポーター遺伝子アッセイを行なった。BCL-6プロモーターの検査は、3つの潜在的なSTAT5結合部位の存在を明らかにした:位置1863-1871 bpでのTTCTCAGAA、位置1190-1198 bpでのTTCTCTGAA及び位置548-556 bpでのTTCTCTGAA。加えて、1つの潜在的なSTAT5結合部位は、非コード第1エクソンに配置される。STAT5がBCL-6プロモーターを介して転写を活性化することができたかどうか判断するために、我々は、BCL-6プロモーターの一部(657/+471)及びルシフェラーゼコード配列にリンクされた非コード第1エクソンを含む構築物を使用した。この構築物は、開始コドンに最も近接した一つの潜在的なSTAT5結合部位(548/-556 bp)及び非コード第1エクソン内の一つを抱く。このリポーター構築物は、CA-STAT5b発現するベクター又は対照ベクターと一緒に293T細胞内にトランスフェクションされた。リポーター構築物及びCA-STAT5bの共トランスフェクションは、増加されたルシフェラーゼ活性をもたらした(図21C)。リポーター活性におけるこの増加がSTAT5活性に依存したことを示すために、変異体が(548/-556 bp)STAT5結合部位において生成された。我々は、TTCTCTGAAからGTCTCTAAAにBCL-6プロモーター領域における最初のSTAT5結合部位を変異した。CA-STAT5bが共トランスフェクションされた場合、このSTAT5結合部位の変異体は、リポーター活性を強く減じた(図21C)。これらのデータは、BCL-6プロモーターにおける(548/-556 bp)STAT5b結合部位が機能的であることを明白に示す。一緒に、我々の結果は、STAT5がBCL-6発現を直接的に制御することを示す。
【0152】
CA-STAT5 B細胞の長期の寿命は、BCL-6発現にもっぱら依存しない。
BCL-6タンパク質がSTAT5b-ER延長された寿命について重大かどうかを判断するために、我々は2つのBCL-6 siRNAプローブを設計し、それらはpSUPER内にクローニングされた。最初に、BCL-6 siRNAsのノックダウン効率を決定するために、pSUPER構築物が293T細胞におけるBCL-6を発現するベクターとともに共トランスフェクションされた。図22Aに見られるように、siBCL-6(789)及びsiBCL-6(970)の両方が、BCL-6タンパク質の量を非常に効率的に減少した。両方のBCL-6 siRNAsがpSIN-GFPレトロウィルスベクター内にサブクローニングされ、そしてウィルスがCA-STAT5b-ER-ΔNGFR+B細胞を形質導入するために生産された。対照を形質導入された細胞のGFP+細胞のパーセンテージは、時間にわたって安定していた。同じ観察が、2つBCL-6 siRNAsを形質導入されたCA-STAT5b-ER-ΔNGFR+ B細胞のいずれかについて作られ(図22B)、CA-STAT5b-ER+B細胞がそれらの生存についてBCL-6に依存しないことを示唆した。著しいことに、CA-STAT5b-ER-ΔNGFR+ B細胞におけるBCL-6ノックダウン後に、CD80発現が1.5〜2倍増加された(データは示されていない)。CD80がBCL-6によって直接的に抑制されることを記載されている故に、これはBCL-6ノックダウン構築物の効率を確認した(Niu等、2003年)。我々の発見は、BCL-6がB細胞の複製寿命に対するSTAT5bの影響の唯一の仲介人でないことを示す。
【0153】
文献
Ariyoshi, K., Nosaka, T., Yamada, K., Onishi, M., Oka, Y., Miyajima, A., and Kitamura, T. Constitutive activation of STAT5 by a point mutation in the SH2 domain. J Biol Chem 第275巻, 第24407-24413頁, (2000年)。
Barry, S.C. et al. Lentivirus vectors encoding both central polypurine tract and posttranscriptional regulatory element provide enhanced transduction and transgene expression. Hum Gene Ther 第12巻, 第1103-8頁, (2001年)。
Bonini, C., Ferrari, G., Verzeletti, S., Servida, P., Zappone, E., Ruggieri, L., Ponzoni, M., Rossini, S., Mavilio, F., Traversari, C., and Bordignon, C. HSV-TK gene transfer into donor lymphocytes for control of allogeneic graft-versus-leukemia. Science 第276巻, 第1719-1724頁, (1997年)。
Brummelkamp, T.R., Bernards, R., and Agami, R. A system for stable expression of short interfering RNAs in mammalian cells. Science 第296巻, 第550-553頁, (2002年)。
Guikema, J. E., Vellenga, E., Veeneman, J. M., Hovenga, S., Bakkus, M. H., Klip, H., and Bos, N. A. Multiple myeloma related cells in patients undergoing autologous peripheral blood stem cell transplantation. Br J Haematol 第104巻, 第748-754頁, (1999年)。
Heemskerk, M. H., Blom, B., Nolan, G., Stegmann, A. P., Bakker, A. Q., Weijer, K., Res, P. C., and Spits, H. Inhibition of T cell and promotion of natural killer cell development by the dominant negative helix loop helix factor Id3. J Exp Med 第186巻, 第1597-1602頁, (1997年)。
Heemskerk, M. H., Hooijberg, E., Ruizendaal, J. J., van der Weide, M. M., Kueter, E., Bakker, A. Q., Schumacher, T. N., and Spits, H., Enrichmentof an antigen-specific T cell response by retrovirally transduced human dendritic cells. Cell Immunol 第195巻, 第10-17頁。
John, S., Robbins, C. M., and Leonard, W. J. An IL-2 response element in the human IL-2receptor alpha chain promoter is a composite element that binds Stat5, Elf-1, HMG-I(Y) and a GATA family protein. Embo J 第15巻, 第5627-5635頁, (1996年)。
John, S., Vinkemeier, U., Soldaini, E., Darnell, J. E., and Leonard, W. J. The significance of tetramerization in promoter recruitment by Stat5. Mol Cell Biol 第19巻, 第1910-1918頁, (1999年)。
Kaplan, M. H., Schindler, U., Smiley, S. T., and Grusby, M. J. Stat6 is required formediating responses to IL-4 and for development of Th2 cells. Immunity 第4巻, 第313-319頁, (1996年)。
Kinsella, T. M., and Nolan, G. P. Episomal vectors rapidly and stably produce high-titer recombinant retrovirus. Human Gene Therapy 第7巻, 第1405-1413頁, (1996年)。
Kurata, H., Lee, H. J., O'Garra, A., and Arai, N. Ectopic expression of activated Stat6 induces the expression of Th2- specific cytokines and transcription factors in developing Th1 cells. Immunity 第11巻, 第677-688頁, (1999年)。
Leonard, W. J.,and O'Shea, J. J. Jaks and STATs: biological implications. Annu Rev Immunol 第16巻, 第293-322頁, (1998年)。
Lischke, A., Moriggl, R., Brandlein, S., Berchtold, S., Kammer, W., Sebald, W., Groner, B., Liu, X., Hennighausen, L., and Friedrich, K. The interleukin-4 receptor activates STAT5 by a mechanism that relies upon common gamma-chain. J Biol Chem 第273巻, 第31222-31229頁, (1998年)。
Niu, H., Cattoretti, G., and Dalla-Favera, R. BCL6 controls the expression of the B7-1/CD80 costimulatory receptor in germinal center B cells. J Exp Med 第198巻, 第211-221頁, (2003年)。
Onishi, M., Nosaka, T., Misawa, K., Mui, A. L., Gorman, D., McMahon, M., Miyajima, A., and Kitamura, T. Identification and characterization of a constitutively activeSTAT5 mutant that promotes cell proliferation. Mol Cell Biol 第18巻, 第3871-3879頁, (1998年)。
Reljic, R., Wagner, S. D., Peakman, L. J., and Fearon, D. T. Suppression of signal transducer and activator of transcription 3- dependent B lymphocyte terminal differentiation by BCL-6. J Exp Med 第192巻, 第1841-1848頁, (2000年)。
Rolling, C., Treton, D., Pellegrini, S., Galanaud, P., and Richard, Y. IL4 and IL13 receptors share the gamma c chain and activate STAT6, STAT3 and STAT5 proteins in normal human B cells. FEBS Lett 第393巻, 第53-56頁, (1996年)。
Scheeren, F.A., Naspetti, M., Diehl, S., Schotte, R., Nagasawa, M., Wijnands, E., Gimeno, R., Vyth-Dreese, F.A., Blom, B., and Spits, H. STAT5 regulates the self-renewal capacity and differentiation of human memory B cells and controls Bcl-6 expression. Nature Immunology. Vol 6, No. 3, 第303-313頁, (2005年)。
Seppen, J, Rijnberg, M., Cooreman, M. P., and Oude Elferink, R. P. Lentiviral vectors for efficient transduction of isolated primary quiescent hepatocytes. J Hepatol 第36巻, 第459-65頁, (2002年)。
Shimoda, K., van Deursen, J., Sangster, M. Y., Sarawar, S. R., Carson, R. T., Tripp, R. A., Chu, C., Quelle, F. W., Nosaka, T., Vignali, D. A., 等. Lack of IL-4-induced Th2 response and IgE class switching in mice with disrupted Stat6 gene. Nature 第380巻, 第630-633頁, (1996年)。
Shvarts, A., Brummelkamp, T., Scheeren, F., Koh, E., Daley, G. Q., Spits, H., and Bernards, R. A senescence rescue screen identifies BCL6 as an inhibitor of anti-proliferative p19ARF- p53 signaling. Genes Dev in press, (2002年)。
Strathdee, C.A., McLeod, M. R. and Hall, J.R. Efficient control of tetracycline-responsive gene expression from an autoregulated bi-directional expression vector. Gene 第229巻, 第21-29頁, (1999年)。
Teglund, S., McKay, C., Schuetz, E., van Deursen, J. M., Stravopodis, D., Wang, D., Brown,M., Bodner, S., Grosveld, G., and Ihle, J. N. Stat5a and Stat5b proteins have essential and nonessential, or redundant, roles in cytokine responses. Cell 第93巻, 第841-850頁, (1998年)。
Traggiai E, Chicha L, Mazzucchelli L, bronz L, Piffaretti J-C, lanzavecchia A, Manz M. Development of a human adaptive immune system in cord blood cell-transplanted mice。
Verhoef, K., Marzio, G., Hillen, W., Bujard,H. and Berkhout, B. Strict control of human immunodeficiency virus type 1 replication by a genetic switch: Tet for Tat. J Virol 第75巻, 第979-87頁, (2001年)。
Von Lindern, M., Amelsvoort, M. P., van Dijk, T., Deiner, E., van Den Akker, E., van Emst-De Vries, S., Willems, P., Beug, H., and Lowenberg, B. Protein kinase C alpha controls erythropoietin receptor signaling. J Biol Chem 第275巻, 第34719-34727頁, (2000年)。
Vyth-Dreese, F. A., Dellemijn, T. A., Majoor, D., and de Jong, D. Localizationin situ of the co-stimulatory molecules B7.1, B7.2, CD40 and their ligands in normal human lymphoid tissue. Eur J Immunol 第25巻, 第3023-3029頁, (1995年)。
Weijer K, Uittenbogaart CH, Voordouw A, Couwenberg F, Seppen J, Blom B, Vyth-Dreese FA, Spits H. Intrathymic and extrathymic development of human plasmacytoid dendritic cell precursors in vivo. Blood. 第99巻:第2752-2759頁, (2002年)。
Ye, B. H., Cattoretti, G., Shen, Q., Zhang, J., Hawe, N., de Waard, R., Leung, C., Nouri-Shirazi, M., Orazi, A., Chaganti, R. S., 等. The BCL-6 proto-oncogene controls germinal-centre formation and Th2-type inflammation.Nat Genet 第16巻, 第161-170頁, (1997年)。
【図面の簡単な説明】
【0154】
【図1】(A)レンチウィルスのRNA干渉ベクターpTRIPΔU3-EF1アルファ p53の概要図である。(B)免疫ブロットの図である。
【図2】CD45+ヒト細胞のパーセンテージのグラフである。
【図3】胸腺のFACSプロファイルの図である。
【図4A】骨髄のFACSプロファイルの図である。
【図4B】脾臓のFACSプロファイルの図である。
【図4C】肝臓のFACSプロファイルの図である。
【図4D】肺のFACSプロファイルの図である。
【図5】GFP+発現細胞の多分化能存在の図である。
【図6】Vベータファミリー図である。
【図7A】免疫ブロットの図である。
【図7B】フローサイトメトリーの図である。
【図8】GFPポジティブ細胞のパーセンテージを示す図である。
【図9】GFP+発現細胞の多分化能存在の図である。
【図10】細胞パネル7TetOmCMV CMV対IRESのグラフである。
【図11】rtTAウェスタンブロットの図である。
【図12】繰り返し誘発結果の図である。
【図13】最大GFP発現のための時間のグラフである。
【図14】濃度(dox用量反応)の図である。
【図15】誘発可能なレンチウィルスなベクターの概要図である。
【図16】(A)pTyr-STAT5及びCD20の共局在化を示す染色パターンの図である。(B)ダブル染色されたアイソタイプ対照染色の図である。(C)ウェスタンブロット分析の図である。(D)ウェスタンブロット分析の図である。(E)チミジンの取り込みのグラフである。
【図17】(A)GFP+細胞のパーセンテージのグラフである。(B)絶対的な細胞数カウントのグラフである。(C)フローサイトメトリーの図である。(D)RT-PCR分析の図である。
【図18】(A)抗-NGFR-FITC(緑)及び抗-ER-TexasRed(赤)での染色図である。(B)ウェスタンブロット分析の図である。
【図19】(A)形質導入された細胞のパーセンテージのグラフである。(B)絶対的な細胞数のグラフである。(C)フローサイトメトリーの図である。
【図20】(A)GFP発現のフローサイトメトリック分析及び絶対細胞数カウントのグラフである。(B)フローサイトメトリーの図である。
【図21】(A)RT-PCR分析の図である。(B)ウェスタンブロット分析の図である。(C)デュアルルシフェラーゼリポーター分析の図である。
【図22】(A)
【特許請求の範囲】
【請求項1】
関心のある安定化された細胞を生産するための方法であって、
−関心のある前記細胞中に存在する場合に該関心のある細胞を安定化させることができるところの核酸配列を、該関心のある細胞の幹細胞及び/又は前駆細胞に備えること;
−前記幹細胞及び/又は前駆細胞を非ヒト動物に備えること;
−前記動物において、該関心のある細胞の生成を許すこと;及び
−該関心のある細胞を得ること
を含む方法。
【請求項2】
該関心のある細胞が前記動物から得られた後に、それがエクスビボ(ex vivo)で培養される、請求項1に記載の方法。
【請求項3】
前記幹細胞及び/又は前駆細胞がヒト幹細胞及び/又は前駆細胞を含む、請求項1又は2に記載の方法。
【請求項4】
前記前駆細胞がCD34+前駆細胞を含む、請求項1〜3のいずれか一項に記載の方法。
【請求項5】
該関心のある細胞が免疫細胞を含む、請求項1〜4のいずれか一項に記載の方法。
【請求項6】
前記免疫細胞がヒトB細胞を含む、請求項5に記載の方法。
【請求項7】
該関心のある細胞が肝細胞を含む、請求項1〜4のいずれか一項に記載の方法。
【請求項8】
前記核酸配列が、該関心のある細胞の複製を誘導し及び/又は高めることができる、請求項1〜7のいずれか一項に記載の方法。
【請求項9】
前記核酸配列が、構成的に活性なSTAT(Signal Transducer of Activation and Transcription)(CA-STAT)タンパク質をコードする遺伝子の少なくとも機能的部分を含む、請求1〜8のいずれか一項に記載の方法。
【請求項10】
前記CA-STATタンパク質が、構成的に活性なSTAT5、構成的に活性なSTAT5a、及び/又は構成的に活性なSTAT5bタンパク質を含む、請求項9に記載の方法。
【請求項11】
前記核酸配列が、BCL-6をコードする遺伝子の少なくとも機能的部分を含む、請求1〜10のいずれか一項に記載の方法。
【請求項12】
前記核酸配列が、p53発現を少なくとも部分的に不活化することができる、請求項1〜11のいずれか一項に記載の方法。
【請求項13】
核酸配列が該関心のある細胞中に存在する場合に該関心のある細胞を安定させることができるところの前記核酸配列が、誘導性プロモーターに動作可能にリンクされる、請求項1〜12のいずれか一項に記載の方法。
【請求項14】
前記プロモーターが、ドキシサイクリン及び/又はタモキシフェンによって誘導可能である、請求項13に記載の方法。
【請求項15】
前記非ヒト動物が、該関心のある細胞が得られる前に、関心のある抗原を備えられる、請求項1〜14のいずれか一項に記載の方法。
【請求項16】
該関心のある抗原が、RSV抗原、ヒト自己抗原、腫瘍関連抗原、TNFアルファ及び/又は悪性黒色腫細胞上で発現された抗原の少なくとも免疫原部分である、請求項15に記載の方法。
【請求項17】
前記動物が、内在性血球細胞の少なくとも1つの種類に本質的に欠けている、請求項1〜16のいずれか一項に記載の方法。
【請求項18】
前記動物が、内在性B細胞、内在性T細胞、及び/又は内在性ナチュラルキラー(NK)細胞に本質的に欠けている、請求項〜17のいずれか一項に記載の方法。
【請求項19】
前記非ヒト動物がマウスを含む、請求項1〜18のいずれか一項に記載の方法。
【請求項20】
前記マウスがRAG2-/-γc-/-マウスを含む、請求項1〜19のいずれか一項に記載の方法。
【請求項21】
前記幹細胞及び/又は前駆細胞は、核酸配列が該関心のある細胞中に存在する場合に該関心のある細胞を安定化させることができるところの前記核酸配列を含むレンチウィルスベクターで形質導入される、請求項1〜20のいずれか一項に記載の方法。
【請求項22】
前記幹細胞及び/又は前駆細胞が、ER-STAT5ベクターで形質導入される、請求項1〜21のいずれか一項に記載の方法。
【請求項23】
前記幹細胞及び/又は前駆細胞が、誘導性プロモーターに動作可能にリンクされたrtTA3を含むベクターで形質導入される、請求項1〜22のいずれか一項に記載の方法。
【請求項24】
前記幹細胞及び/又は前駆細胞が、誕生後1週間以内に前記動物に投与される、請求項1〜23のいずれか一項に記載の方法。
【請求項25】
前記幹細胞及び/又は前駆細胞が、誕生後1日以内に前記動物に投与される、請求項1〜24のいずれか一項に記載の方法。
【請求項26】
請求項1〜24のいずれか一項に記載の方法によって入手可能な関心のある安定化された細胞。
【請求項27】
関心のある細胞の単離された幹細胞及び/又は前駆細胞であって、前記幹細胞及び/又は前駆細胞は、核酸配列が該関心のある細胞中に存在する場合に該関心のある細胞を安定化させることができるところの核酸配列を含む、前記細胞。
【請求項28】
ヒトCD34+前駆細胞を含む、請求項27に記載の細胞。
【請求項29】
前記核酸配列が、CA-STATタンパク質及び/又はBCL-6をコードする遺伝子の少なくとも機能的部分、及び/又はp53発現を少なくとも部分的に不活化することができる核酸配列を含む、請求項27又は28に記載の細胞。
【請求項30】
請求項27〜29のいずれか一項に記載の細胞を含む非ヒト動物。
【請求項31】
B細胞株を生産するための方法であって、
−請求項1〜24のいずれか一項に記載の方法でB細胞を得ること、及び
−前記B細胞をエクスビボ(ex vivo)で培養すること
を含む方法。
【請求項32】
誘導性プロモーターに動作可能にリンクされたrtTA3を含む、単離された又は組み換えベクター。
【請求項33】
関心のある抗原に対して特異的に向けられた抗体を生産するための方法であって、
−核酸配列がB細胞中に存在する場合に前記B細胞を安定化させることができるところの核酸配列をB細胞の幹細胞及び/又は前駆細胞に備えること;
−前記幹細胞及び/又は前駆細胞を非ヒト動物に備えること;
−該関心のある抗原を前記非ヒト動物に備えること;
−該関心のある抗原に対して特異的に向けられた抗体を生産するB細胞の生成を許すこと;
−前記B細胞を得ること;及び
−前記B細胞によって生産された抗体を収穫すること
を含む方法。
【請求項34】
前記B細胞が前記動物から得られた後に、それがエクスビボ(ex vivo)で培養される、請求項33に記載の方法。
【請求項35】
前記前駆細胞がヒトCD34+前駆細胞である、請求項33又は34に記載の方法。
【請求項36】
前記前駆細胞が、CA-STATタンパク質及び/又はBCL-6をコードする遺伝子の少なくとも機能的部分、及び/又はp53発現を少なくとも部分的に不活化することができる核酸配列を備えられる、請求項33〜35のいずれか一項に記載の方法。
【請求項37】
前記非ヒト動物がマウスを含む、請求項33〜36のいずれか一項に記載の方法。
【請求項38】
前記マウスがRAG2-/-γc-/-マウスを含む、請求項33〜37のいずれか一項に記載の方法。
【請求項39】
関心のある安定化された細胞を生産するために、請求項32に記載のベクターを使用する方法。
【請求項1】
関心のある安定化された細胞を生産するための方法であって、
−関心のある前記細胞中に存在する場合に該関心のある細胞を安定化させることができるところの核酸配列を、該関心のある細胞の幹細胞及び/又は前駆細胞に備えること;
−前記幹細胞及び/又は前駆細胞を非ヒト動物に備えること;
−前記動物において、該関心のある細胞の生成を許すこと;及び
−該関心のある細胞を得ること
を含む方法。
【請求項2】
該関心のある細胞が前記動物から得られた後に、それがエクスビボ(ex vivo)で培養される、請求項1に記載の方法。
【請求項3】
前記幹細胞及び/又は前駆細胞がヒト幹細胞及び/又は前駆細胞を含む、請求項1又は2に記載の方法。
【請求項4】
前記前駆細胞がCD34+前駆細胞を含む、請求項1〜3のいずれか一項に記載の方法。
【請求項5】
該関心のある細胞が免疫細胞を含む、請求項1〜4のいずれか一項に記載の方法。
【請求項6】
前記免疫細胞がヒトB細胞を含む、請求項5に記載の方法。
【請求項7】
該関心のある細胞が肝細胞を含む、請求項1〜4のいずれか一項に記載の方法。
【請求項8】
前記核酸配列が、該関心のある細胞の複製を誘導し及び/又は高めることができる、請求項1〜7のいずれか一項に記載の方法。
【請求項9】
前記核酸配列が、構成的に活性なSTAT(Signal Transducer of Activation and Transcription)(CA-STAT)タンパク質をコードする遺伝子の少なくとも機能的部分を含む、請求1〜8のいずれか一項に記載の方法。
【請求項10】
前記CA-STATタンパク質が、構成的に活性なSTAT5、構成的に活性なSTAT5a、及び/又は構成的に活性なSTAT5bタンパク質を含む、請求項9に記載の方法。
【請求項11】
前記核酸配列が、BCL-6をコードする遺伝子の少なくとも機能的部分を含む、請求1〜10のいずれか一項に記載の方法。
【請求項12】
前記核酸配列が、p53発現を少なくとも部分的に不活化することができる、請求項1〜11のいずれか一項に記載の方法。
【請求項13】
核酸配列が該関心のある細胞中に存在する場合に該関心のある細胞を安定させることができるところの前記核酸配列が、誘導性プロモーターに動作可能にリンクされる、請求項1〜12のいずれか一項に記載の方法。
【請求項14】
前記プロモーターが、ドキシサイクリン及び/又はタモキシフェンによって誘導可能である、請求項13に記載の方法。
【請求項15】
前記非ヒト動物が、該関心のある細胞が得られる前に、関心のある抗原を備えられる、請求項1〜14のいずれか一項に記載の方法。
【請求項16】
該関心のある抗原が、RSV抗原、ヒト自己抗原、腫瘍関連抗原、TNFアルファ及び/又は悪性黒色腫細胞上で発現された抗原の少なくとも免疫原部分である、請求項15に記載の方法。
【請求項17】
前記動物が、内在性血球細胞の少なくとも1つの種類に本質的に欠けている、請求項1〜16のいずれか一項に記載の方法。
【請求項18】
前記動物が、内在性B細胞、内在性T細胞、及び/又は内在性ナチュラルキラー(NK)細胞に本質的に欠けている、請求項〜17のいずれか一項に記載の方法。
【請求項19】
前記非ヒト動物がマウスを含む、請求項1〜18のいずれか一項に記載の方法。
【請求項20】
前記マウスがRAG2-/-γc-/-マウスを含む、請求項1〜19のいずれか一項に記載の方法。
【請求項21】
前記幹細胞及び/又は前駆細胞は、核酸配列が該関心のある細胞中に存在する場合に該関心のある細胞を安定化させることができるところの前記核酸配列を含むレンチウィルスベクターで形質導入される、請求項1〜20のいずれか一項に記載の方法。
【請求項22】
前記幹細胞及び/又は前駆細胞が、ER-STAT5ベクターで形質導入される、請求項1〜21のいずれか一項に記載の方法。
【請求項23】
前記幹細胞及び/又は前駆細胞が、誘導性プロモーターに動作可能にリンクされたrtTA3を含むベクターで形質導入される、請求項1〜22のいずれか一項に記載の方法。
【請求項24】
前記幹細胞及び/又は前駆細胞が、誕生後1週間以内に前記動物に投与される、請求項1〜23のいずれか一項に記載の方法。
【請求項25】
前記幹細胞及び/又は前駆細胞が、誕生後1日以内に前記動物に投与される、請求項1〜24のいずれか一項に記載の方法。
【請求項26】
請求項1〜24のいずれか一項に記載の方法によって入手可能な関心のある安定化された細胞。
【請求項27】
関心のある細胞の単離された幹細胞及び/又は前駆細胞であって、前記幹細胞及び/又は前駆細胞は、核酸配列が該関心のある細胞中に存在する場合に該関心のある細胞を安定化させることができるところの核酸配列を含む、前記細胞。
【請求項28】
ヒトCD34+前駆細胞を含む、請求項27に記載の細胞。
【請求項29】
前記核酸配列が、CA-STATタンパク質及び/又はBCL-6をコードする遺伝子の少なくとも機能的部分、及び/又はp53発現を少なくとも部分的に不活化することができる核酸配列を含む、請求項27又は28に記載の細胞。
【請求項30】
請求項27〜29のいずれか一項に記載の細胞を含む非ヒト動物。
【請求項31】
B細胞株を生産するための方法であって、
−請求項1〜24のいずれか一項に記載の方法でB細胞を得ること、及び
−前記B細胞をエクスビボ(ex vivo)で培養すること
を含む方法。
【請求項32】
誘導性プロモーターに動作可能にリンクされたrtTA3を含む、単離された又は組み換えベクター。
【請求項33】
関心のある抗原に対して特異的に向けられた抗体を生産するための方法であって、
−核酸配列がB細胞中に存在する場合に前記B細胞を安定化させることができるところの核酸配列をB細胞の幹細胞及び/又は前駆細胞に備えること;
−前記幹細胞及び/又は前駆細胞を非ヒト動物に備えること;
−該関心のある抗原を前記非ヒト動物に備えること;
−該関心のある抗原に対して特異的に向けられた抗体を生産するB細胞の生成を許すこと;
−前記B細胞を得ること;及び
−前記B細胞によって生産された抗体を収穫すること
を含む方法。
【請求項34】
前記B細胞が前記動物から得られた後に、それがエクスビボ(ex vivo)で培養される、請求項33に記載の方法。
【請求項35】
前記前駆細胞がヒトCD34+前駆細胞である、請求項33又は34に記載の方法。
【請求項36】
前記前駆細胞が、CA-STATタンパク質及び/又はBCL-6をコードする遺伝子の少なくとも機能的部分、及び/又はp53発現を少なくとも部分的に不活化することができる核酸配列を備えられる、請求項33〜35のいずれか一項に記載の方法。
【請求項37】
前記非ヒト動物がマウスを含む、請求項33〜36のいずれか一項に記載の方法。
【請求項38】
前記マウスがRAG2-/-γc-/-マウスを含む、請求項33〜37のいずれか一項に記載の方法。
【請求項39】
関心のある安定化された細胞を生産するために、請求項32に記載のベクターを使用する方法。
【図1】


【図2】
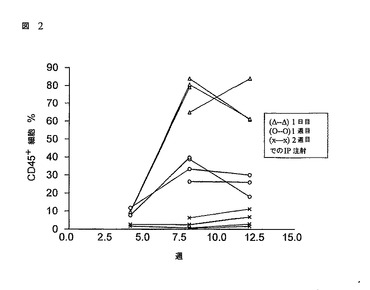
【図3】
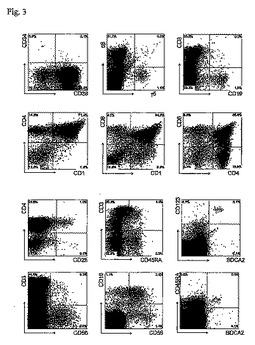
【図4A】
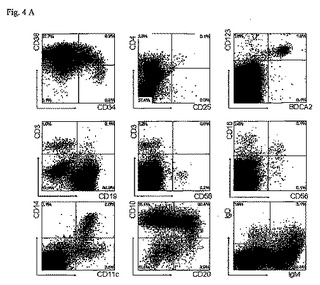
【図4B】
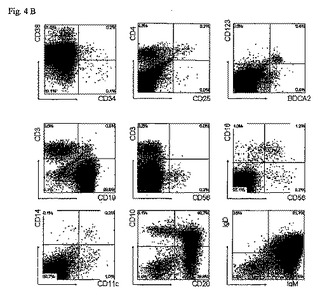
【図4C】

【図4D】

【図5】
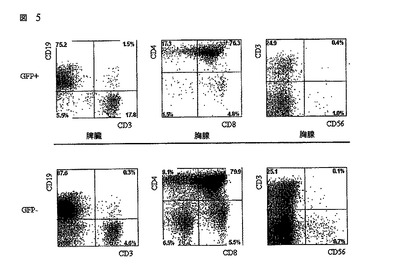
【図6】

【図7A】
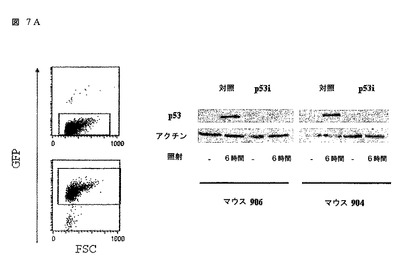
【図7B】
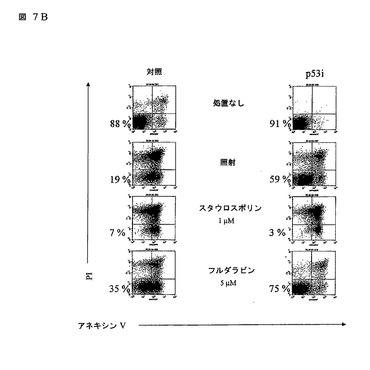
【図8】
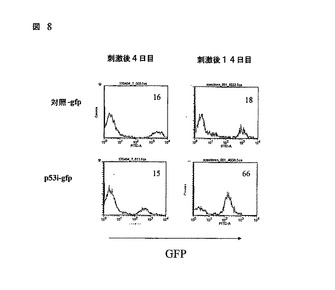
【図9】

【図10】

【図11】
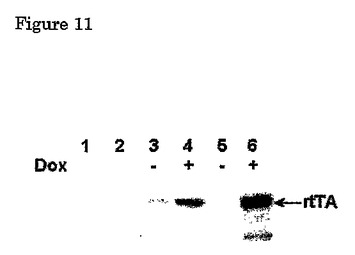
【図12】
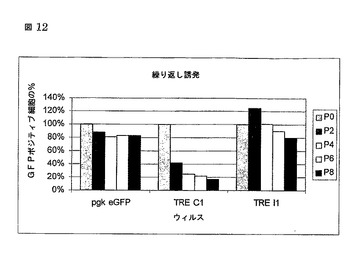
【図13】
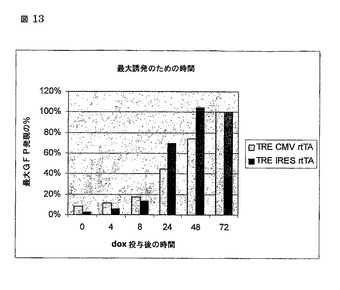
【図14】
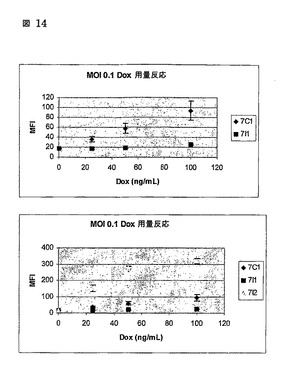
【図15】
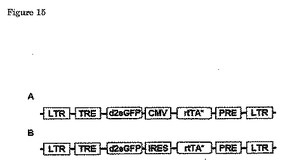
【図16】
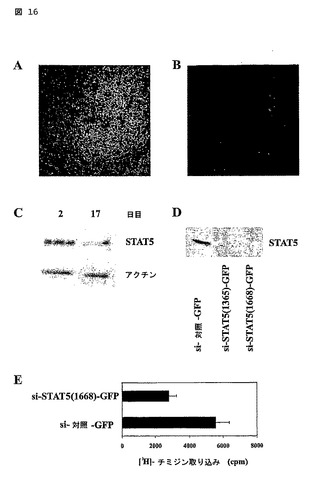
【図17】

【図18】
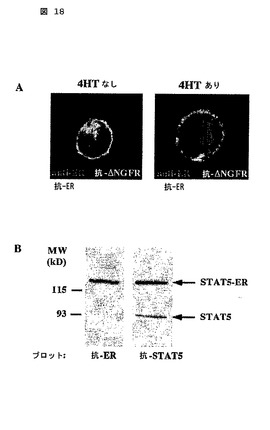
【図19】
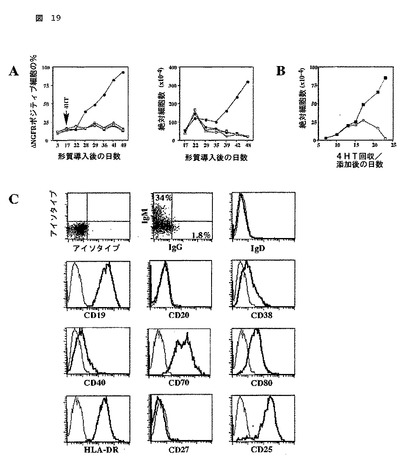
【図20】
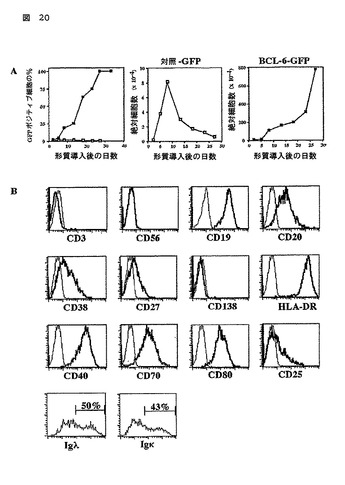
【図21】

【図22】
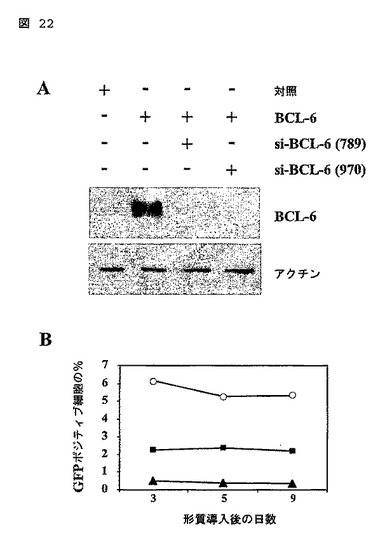
【図2】
【図3】
【図4A】
【図4B】
【図4C】
【図4D】
【図5】
【図6】
【図7A】
【図7B】
【図8】
【図9】
【図10】
【図11】
【図12】
【図13】
【図14】
【図15】
【図16】
【図17】
【図18】
【図19】
【図20】
【図21】
【図22】
【公表番号】特表2008−509663(P2008−509663A)
【公表日】平成20年4月3日(2008.4.3)
【国際特許分類】
【出願番号】特願2007−525564(P2007−525564)
【出願日】平成17年8月10日(2005.8.10)
【国際出願番号】PCT/NL2005/000581
【国際公開番号】WO2006/016808
【国際公開日】平成18年2月16日(2006.2.16)
【出願人】(507046945)アカデミシュ メディシュ セントラム ビ デ ユニベルシテイト ファン アムステルダム (1)
【Fターム(参考)】
【公表日】平成20年4月3日(2008.4.3)
【国際特許分類】
【出願日】平成17年8月10日(2005.8.10)
【国際出願番号】PCT/NL2005/000581
【国際公開番号】WO2006/016808
【国際公開日】平成18年2月16日(2006.2.16)
【出願人】(507046945)アカデミシュ メディシュ セントラム ビ デ ユニベルシテイト ファン アムステルダム (1)
【Fターム(参考)】
[ Back to top ]