
阻害剤のスクリーニングのための、蛍光的P−ループ標識キナーゼの開発
本発明は天然に存在しているか、またはP-ループに導入されているアミノ酸で標識されたキナーゼであって、前記の標識化が、前記のアミノ酸の遊離チオールまたはアミノ基で行われ、かつ、前記の標識が、(a)その環境における極性変化に感受性のある、チオールまたはアミノ反応性フルオロフォアであるか;または(b)チオール反応性スピン標識、同位体または濃縮同位体チオールまたはアミノ反応性標識であり、前記のフルオロフォア、スピン標識、同位体または濃縮同位体標識は、触媒活性を阻害せず、キナーゼの安定性を妨げない前記キナーゼに関する。本発明はさらに、本発明のキナーゼを用いた、キナーゼ阻害剤のスクリーニング方法、、リガンドの結合および/またはキナーゼ阻害剤の解離のキネティクスを決定する方法、およびキナーゼ阻害剤のスクリーニングに好適な突然変異キナーゼの産生方法に関する。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、天然に存在しているか、またはキナーゼのP-ループに導入されているアミノ酸を標識したキナーゼに関し、ここで、前記の標識は、前記のアミノ酸の遊離チオールまたはアミノ基で行われ、かつ、前記の標識は、(a)その環境における極性変化に感受性のある、チオールまたはアミノ反応性フルオロフォアであるか;または(b)チオール反応性スピン標識、同位体または濃縮同位体チオールまたはアミノ反応性標識であって、前記のフルオロフォア、スピン標識、同位体または濃縮同位体標識は、触媒活性を阻害せず、キナーゼの安定性を妨げない。さらに、本発明は、キナーゼ阻害剤のスクリーニング方法、キナーゼ阻害剤のリガンドの結合および/または解離のキネティクスの決定方法、および本発明の標識キナーゼを用いるキナーゼ阻害剤のスクリーニングに好適な変異キナーゼの製造方法に関する。
【0002】
この明細書では、特許出願書類および製造者のマニュアルを含む多数の資料を引用する。これらの資料の開示は、本発明の特許性に関係すると見なされず、参照によって全体として本書に組み込まれる。より具体的には、全ての参照した資料は、個々の資料がそれぞれ参照によって組み込まれることを特異的および個別的に示したのと同程度に、参照により組み込まれる。
【背景技術】
【0003】
プロテインキナーゼは、鍵となる細胞過程を制御する、重要な酵素の一群である。癌生物学において、異常制御されたキナーゼのシグナル伝達について理解が進んだことにより(ゲシュビント(Gschwind)およびフィッシャー(Fischer)、2004年)、望まれないキナーゼ活性の特異的標的化に使用される有機小分子が開発され、癌の標的療法の分野が開始された(ツァン(Zhang)ら、2009年)。殆どのキナーゼ阻害剤は、ダサチニブ(スプリセル(Sprycel)登録商標)等のI型阻害剤であり、キナーゼの活性「DFG-in」構造に結合し、ATPと競合してキナーゼのヒンジ領域と非常に重要な水素結合を形成する。この構造において、制御活性化ループは開環し、伸長しているため、ATPおよび基質の結合が可能である(ナイトン(Knighton)ら、1991年)。ATPのアデニンは、キナーゼのヒンジ領域と非常に重要な水素結合を形成するが、この領域はキナーゼドメインのN-およびC-末端ローブの間に位置し、短く柔軟であり、一方、ATPのβおよびγリン酸塩は、Mg2+またはMn2+イオン、保存DFGモチーフのAsp側鎖、およびATP結合間隙の上部に位置する高グリシンループのアミノ酸残基などの、いくつかの構造要素とのイオン結合性および水素結合性相互作用の複雑なネットワークによって連係する(アイムス(Aimes)ら、2000年)。しかし、これらのタイプの阻害剤の開発には、ATP結合部位内の化学的空間が使い尽くされていること、阻害剤の選択性および有効性が低いことについての問題に加え、薬物耐性の発現という問題がある。現在の医薬品化学の研究では、これらの障害を克服して、代替の(即ちアロステリック)結合部位を標的とし、および/または、酵素的能力のない不活性キナーゼ構造を安定化する阻害剤を特定および開発することによって、有効な長期的療法の開発を試みている(ツァン(Zhang)ら、2009年;アドリアン(Adrian) ら、2006年;カレジャ(Calleja) ら、2009年;フィシュマン(Fischmann) ら、2009年;キルクラント(Kirkland) およびマクイネス(McInnes) 、2009年)。これらの部位の一つは、不活性な「DFG-out」キナーゼ構造においてのみ存在し、キナーゼ研究の最前線となっている。DFG-out構造は、高度に保存されたDFGモチーフの180°反転によって引き起こされる活性化ループの構造変化によるものであり(リウ(Liu)およびグレイ(Gray)、2006年;パルゲリス(Pargellis)ら、2002年)、これは、ATP結合部位に隣接する保存性がより低いアロステリック部位にもさらされる事象である。II型およびIII型阻害剤は、この保存性がより低いアロステリック部位に結合し、優れた選択性プロファイル、向上した薬理学的特性を有すると考えられており(コープランド(Copeland)ら、2006年)、薬品開発の新たな機会を提供している(リウ(Liu)およびグレイ(Gray)、2006年)。より具体的には、ソラフェニブ(ネキサバール(Nexavar) 登録商標、ワン(Wan)ら、2004年)、イマチニブ(グリーベック(Gleevec) 登録商標、ネイガー(Nagar)ら、2002年)およびp38αの選択的阻害剤であるBIRB-796(パルゲリス(Pargellis)ら、2002年)等のII型阻害剤は、ヒンジ領域に結合し、ATP競合的であるが、このアロステリック部位中に及び、一方III型阻害剤は、専らアロステリックポケット内で結合する(パルゲリスら、2002年;シマード(Simard)ら、提出済)。最近まで、不活性DFG-out構造を安定化する阻害剤を明確に特定できるアプローチは、不十分であるか、または、新規なヒット化合物を特定する、学界および産業界で使用されるハイスループット・スクリーニング・フォーマットに適合しなかったため、このようなリガンドを検出および特性化する革新的な新規アプローチの必要性は高まっている。
【0004】
フルオロフォアのタンパク質への結合は、確立されたアプローチであり、リガンドの結合に対する反応においてタンパク質構造中の立体構造の変化を検出するために使用されている。緩衝液中の非結合脂肪酸の濃度を測定する、市販されているプローブであるアクリロダン標識脂肪酸結合タンパク質(ADIFAB;モレキュラー・プローブス(Molecular Probes))(リチェリ(Richieri)ら、1999年)に加えて、このアプローチは、アセチルコリン結合タンパク質(ヒッブス(Hibbs)ら、2004年)、インターロイキン-1β(イェム(Yem)ら、1992年)および種々の糖およびアミノ酸結合タンパク質(ドゥ・ロリミエル(de Lorimier)ら、2002年)等の、種々の他のタンパク質に適用されている。
【発明の概要】
【0005】
上記の議論から、特異的キナーゼ阻害剤のスクリーニングのための用途の広い手段および方法を有することが望ましいであろう。しかし、いくつかのキナーゼは、立体構造中のDFG-in/outスイッチに影響するか、スイッチを誘導し得るリガンドに、多少は感受性があると思われる。従って、DFG-out構造に容易に適合するキナーゼについて、および、ATP部位内部で結合し、活性化ループまたはDFG構造における変化ではない標的キナーゼにおける他の構造的変化を誘導し得るリガンドについて、DFG-outバインダーを高感度で検出する、代替のスクリーニング戦略を開発することは有用であろう。この技術的問題は、特許請求の範囲で特徴付けられる具体例を提供することによって解決される。
【図面の簡単な説明】
【0006】
【図1】p38αにおけるリガンド結合によって引き起こされる、P-ループおよび活性化ループの立体構造変化の略図である。
【図2】高グリシンループ上に標識したac-p38αを用いたリアルタイムおよび終点蛍光測定結果を示す。
【図3】P-ループac-p38αを用いたIII型およびI型リガンドのリアルタイムおよび終点蛍光測定結果を示す。
【図4−1】P-ループを標識したp38αの種々の型の阻害剤に対する蛍光特性評価および応答を示す。
【図4−2】P-ループを標識したp38αの種々の型の阻害剤に対する蛍光特性評価および応答を示す。
【図5−1】RL40、Scios-469およびCP547632の結晶構造は、P-ループの運動を裏付けることを示す図である
【図5−2】RL40、Scios-469およびCP547632の結晶構造は、P-ループの運動を裏付けることを示す図である
【図6】野生型の非標識およびアクリロダン標識p38αの反応速度論的および阻害特性評価結果を示す。
【図7】384穴フォーマットにおいて測定された、p38αによるBIRB-796(1)のKd値の時間依存性を示す。
【図8】高グリシンループを標識したMKK7を用いたII型およびI型リガンドのリアルタイムおよび終点蛍光測定結果を示す。
【発明を実施するための形態】
【0007】
従って、本発明は、天然に存在しているか、またはキナーゼのP-ループに導入されているアミノ酸で標識されたキナーゼに関し、ここで、前記の標識化は、前記のアミノ酸の遊離チオールまたはアミノ基で行われ、かつ、前記の標識は、(a)その環境における極性変化に感受性のある、チオールまたはアミノ反応性フルオロフォアであるか;または(b)チオール反応性スピン標識、同位体または濃縮同位体チオールまたはアミノ反応性標識であって、前記のフルオロフォア、スピン標識、同位体または濃縮同位体標識は、触媒活性を阻害せず、キナーゼの安定性を妨げない。
【0008】
「キナーゼ」という用語は当業界でよく知られており、リン酸基をATP等の高エネルギードナー分子からタンパク質等の特異的標的分子に移す、一種の酵素を言う。キナーゼは、酵素委員会(EC)の番号2.7に分類される。特異性によって、プロテインキナーゼは、セリン/トレオニンキナーゼ(EC 2.7.11、例えばp38α)、チロシンキナーゼ(EC 2.7.10、例えばEGFRキナーゼドメイン)、ヒスチジンキナーゼ(EC 2.7.13)、アスパラギン酸/グルタミン酸キナーゼ、および1を超える特異性を有する混合キナーゼ(EC 2.7.12、例えば、MEKは、セリン/トレオニンおよびチロシンに特異的である)に細分し得る。
【0009】
アミノ酸は、カルボン酸官能基およびアミノ官能基を有する有機分子と定義される。アミノ酸は、タンパク質の必須のビルディングブロックである。遊離チオール基を有するアミノ酸の例は、20のタンパク質構成アミノ酸に属するシステイン、および、天然アミノ酸配列には殆ど生じない、非標準的アミノ酸であるアセチルシステインである。遊離アミノ基を有するタンパク質構成アミノ酸は、リシン、ヒスチジンまたはアルギニン、および、トリプトファン等の芳香族アミンであるアミノ酸である。ピロリジン、5-ヒドロキシリジンまたはo-アミノチロシンは、遊離アミノ基を有する非標準的アミノ酸である。アミノ酸であるアスパラギンおよびグルタミンは、遊離アミノ基を有するが、標識化剤に反応性がないため、本発明においては好適ではなく、従って除外される。
【0010】
トリプトファンは、そのインドール環にアミノ基を有する芳香族アミノ酸である。芳香族アミンは弱塩基であるので、pH 7でプロトン化されない。しかし、芳香族アミンはそれでも、イソチオシアネート、塩化スルホニルまたは酸等の高反応性試薬を用いて修飾し得る。
【0011】
キナーゼは、キナーゼ中の所望の位置、即ちP-ループにおいて、アミノ酸の遊離チオールまたはアミノ基を標識する。標識化の間、従来の遊離チオールまたはアミノ基は、項目(a)および(b)により、標識したアミノ酸と標識の間の共有結合の形成に関与する。
【0012】
前記の標識されるアミノ酸は、キナーゼのP-ループに位置する。このことは、P-ループまたはそれに相当する構造を有するキナーゼのみが本発明の範囲に含まれることを意味する。P-ループ(高グリシンループとも呼ばれる)は、全てのATP/GTP結合タンパク質中で維持される、非常に柔軟な構造的特徴である(サラステ(Saraste)ら、1990年)。キナーゼにおいて、P-ループは標準的なGly-X-Gly-X-X-Glyモチーフ(Xはあらゆるアミノ酸である)を含有し、キナーゼのN-末端ローブ中に位置するが、N-末端ローブでP-ループは、ATP等のリガンドの、キナーゼのATP結合部位への侵入を導く調節ループとして働く(ウォン(Wong)ら、2005年)。
【0013】
関心のあるキナーゼ中に天然に存在し、溶媒にさらされているシステインは、P-ループの外側またはP-ループ配列の内部に位置し得る。このことは、遊離アミノ基を有するアミノ酸に同等に適用される。
【0014】
本発明の修飾キナーゼは、天然に存在しているかまたはP-ループ中に導入されているアミノ酸で標識されている。好適なアミノ酸、即ち、遊離チオールまたはアミノ基を有するアミノ酸がP-ループ中に存在しない場合、前記のアミノ酸は、当業界でよく知られた技術により導入され得、即ち、前記のアミノ酸を添加するか、存在するアミノ酸を置換することにより挿入され得る。あらゆる場合において、アミノ酸が標識試薬との反応により標識される場合、アミノ酸をP-ループ中に導入した後にのみ標識することを、誤解を避けるために理解すべきである。上記の技術は、部位特異的変異誘発、並びに他の組み換え、合成または半合成技術を含む。非標準的アミノ酸をキナーゼ中に導入する場合には、前記のアミノ酸を含有するアミノ酸ストレッチを化学的に合成した後、組み換え的または合成的に製造したものでもよいキナーゼの残りの部分に結合させてもよい。或いは、その後の標識化で所望の位置に特殊な非標準アミノ酸を組み込むために発現および設計されたキナーゼは、改変した遺伝子コードを適用することにより、組み換え技術で製造してもよい(例えば、リウ(Liu)およびシュルツ(Schltz)、2010年参照)。
【0015】
標識化方法としては、キナーゼのインキュベーションが挙げられ、例えば、本発明の変異キナーゼ(例えば、P-ループに導入されたシステインを有するキナーゼ)を、穏和な条件下でチオールまたはアミノ反応性標識とインキュベーションし、前記の変異キナーゼをP-ループ内の所望の位置で標識する。換言すれば、原理上は、チオール反応性スピン標識での標識化を除いて、キナーゼにおいて前記の所望の位置のみを標識化することが可能であり、これは好ましい態様であるが、或いは、アイソトープでの同時標識が想定される(以下参照)。穏和な条件とは、緩衝的なpH(例えば、チオール反応性プローブについてはpH 7前後)、キナーゼに対する標識の割合、インキュベーション工程の温度および長さ(チオール反応性プローブについては、例えば、4℃および暗所で一晩)を言い、これらは当業者に知られており、また、チオールおよびアミノ反応性プローブの製造者の取扱説明書により提供される。このような条件は、前記のキナーゼの標識化が所望の標識部位に特異的であることを確実にするため、選択されたチオールまたはアミノ反応性標識の反応を減速するように最適化する必要がある。フルオロフォア標識の場合、インキュベーションを暗所で実施することが必要である。露光量が増加すると、フルオロフォアの漂白が起こり、蛍光発光の強度が弱くなる。標識化の後、標識したキナーゼは、好ましくは濃縮し、ゲル濾過実験で精製するか、緩衝液で数回洗浄して、過剰な未反応標識を除去する。洗浄緩衝液は、代表的には、標識したキナーゼの保管に使用する緩衝液であり、また、所望の測定が行われる際の緩衝液であってもよい。
【0016】
「フルオロフォア」という用語は、特定の波長の光子等のエネルギーを吸収し、エネルギー、即ち光を、(生物発光の場合と同様に)化学反応の関与なく、(リン光の場合と異なり)吸収直後に、異なる(しかし同様に特異的な)波長(蛍光)で放出する、分子または分子内の官能基を示す。通常、吸収された光子の波長は紫外領域にあるが、赤外領域中にも達し得る。発光波長は、通常、可視領域内である。放出されるエネルギーの量および波長は、主にフルオロフォアの特性に依存するが、フルオロフォア周囲の化学的環境によっても影響される可能性がある。多数のフルオロフォアは、環境の変化に感受性がある。これは、極性、電荷および/またはフルオロフォアが結合している分子の立体構造の変化を含む。分子が電気的に励起された後、基底状態に緩和する時に蛍光が発生するが、UVから近赤外までのエネルギーを有する光子を放出する通常使用される蛍光化合物では、蛍光は、0.5ナノ秒および20ナノ秒の間の範囲で起こる。
【0017】
「チオールまたはアミノ反応性」という用語は、例えばフルオロフォア等の化合物が遊離チオールまたはアミノ基と特異的に反応する特性を示す。これは、チオールまたはアミノ基との特異的反応を管理する、前記の化合物中に存在する官能基のためである。これらの官能基は、遊離チオールまたはアミノ基に結合し得る特異的標識を提供するために、フルオロフォア、スピン標識または濃縮アイソトープ分子等の分子に結合していてもよい。チオール特異性化合物の例は、例えば、ヨードアセトアミド、マレイミド、Hg-LinkTMフェニル水銀化合物またはTS-linkTM試薬(両者ともインビトロゲン(Invitrogen)社)等のハロアルキル化合物である。ハロアルキル化合物は、pHに従ってチオールまたはアミノ基と反応する。
【0018】
「スピン標識」(SL)という用語は、分子、一般的には有機分子であって、通常は窒素原子上に不対電子を有し、他の分子に結合する能力を有するものを示す。スピン標識は、EPR分光法を用いるタンパク質のプローブ化のためのツールとして使用される。部位特異性スピン標識化(SDSL)技術により、タンパク質の立体構造および動力学(ダイナミクス)をモニターすることが可能になる。このような実験において、アミノ酸特異的SLを使用し得る。
【0019】
位置特異的スピン標識化は、電子スピン共鳴を使用する、タンパク質の局所力学を調査する技術である。SDSLは、アミノ酸とのスピン標識の特異的反応を基礎とする。タンパク質構造中に構築されるスピン標識は、EPR分光法によって検出し得る。SDSLにおいて、チオールまたはアミノ基等のスピン標識の結合部位は、天然に存在しない場合は、部位特異的変異誘発によって組み換え発現したタンパク質中に導入される。換言すれば、上記の技術によって、スピン標識は、キナーゼが所望の位置のみで特異的に標識されるように、キナーゼ中に導入され得る。スピン標識の内部に含有される官能基は、その特異性を決定する。中性のpHでは、タンパク質のチオール基は、メタンチオスルホネート、マレイミドおよびヨードアセトアミド等の官能基と特異的に反応し、アミノ酸のシステインと共有結合を作る。スピン標識は独特な分子リポーターであり、それらは常磁性である、即ち、不対電子を含有する。ニトロキシドスピン標識は、安定で単純なEPRシグナルを有することから、高分子の構造および動力学の研究に広く使用されている。ニトロキシルラジカル(N-O)は、通常、ピロリジン等の複素環に取り込まれ、不対電子は大部分がN-O結合に局在する。タンパク質中に取り込まれると、スピン標識の動きは、その局所環境によって決定づけられる。スピン標識は動きに極めて敏感であることから、このことは、タンパク質に結合したスピン標識のEPRスペクトルに対して大きく影響する。
【0020】
不対電子から発生するシグナルは、互いにおよび外部磁場に関し、サンプル中の不対電子の動き、距離、および配向についての情報を提供する。溶液中、自由に動く分子については、EPRはNMR(核磁気共鳴分光法)よりもかなり速いタイムスケールで作動するので、より速い分子の動きの詳細、即ちNMRでマイクロ秒であるのに対して、ナノ秒の運動の詳細を明らかにすることが可能である。電子の磁気回転比は、NMRで通常使用される核より数桁大きいため、スピン標識が必要とはいえ、この技術はより高感度である。
【0021】
「アイソトープ」という用語は、化学元素で、最も豊富な種とは異なる原子量(質量数)を有する元素の化学種を示す。元素のアイソトープは、同数のプロトンを有するが(同じ原子番号)、中性子の数が異なる核を有する。
【0022】
EPRまたはNMRに好適なアイソトープは、ゼロ以外の核スピンを有する必要がある。現在使用されている最も一般的なアイソトープは、1H、2D、15N、13C、および31Pである。
【0023】
本発明では、キナーゼのP-ループ内の特定位置に、チオール反応性スピン標識を単独で使用することも可能である一方、P-ループにおいてチオール反応性スピン標識で特異的に標識したキナーゼをアイソトープでも標識することが好ましい(さらなる詳細を以下に記載する)。しかし、アイソトープ標識のみを使用する場合、アイソトープはキナーゼのP-ループ内の特異的な所望の位置にのみ存在し、よってキナーゼの他の位置は標識されないことが好ましい。
【0024】
「濃縮アイソトープ」という用語は、例えば、アイソトープが化合物に導入されるように、チオールまたはアミノ反応性標識がアイソトープを使用して合成された化合物か、チオールまたはアミノ反応性標識がアイソトープと反応した化合物を示す。化合物は、1以上の異なる種の1以上のアイソトープを含んでいてもよい。標識の位置および数に関しては、アイソトープについても上述と同様のことが適用される。
【0025】
標識は、キナーゼの触媒活性を有意に阻止または阻害せず、その安定性を妨げないように位置しなければならない。原理上、本発明のアッセイは、本発明の標識キナーゼの触媒活性の測定には依存せず、従って、キナーゼの基質の予備知識を必要としない。しかし、本質的に触媒活性を妨げないことにより、本発明の標識キナーゼに対する潜在的阻害剤の結合活性を、本発明の標識キナーゼが由来する野生型キナーゼと、合理的に比較され得ることが好ましい。アミノ酸、例えばシステインをアイソトープで標識し、配列中に既に取り込まれたアイソトープで標識したアミノ酸を有するキナーゼを発現している宿主生物を成長させることによって製造したキナーゼの場合、キナーゼの活性阻害または安定性の妨害は起こりそうにない。一方、標識が導入されるP-ループ内の位置を選択する際には、注意もしなければならない。選択位置に好適なアミノ酸が存在しない場合、前記の位置に存在するアミノ酸は、ペプチド結合中に包含されるα-アミノ基以外の遊離チオールまたはアミノ基を含有するアミノ酸で置換しなければならない。P-ループはキナーゼにATPase活性をもたらす。従って、好適な標識位置は、キナーゼがそのATPase活性の少なくとも70%、好ましくは少なくとも80%、より好ましくは少なくとも90%、最も好ましくは100%を保持するように選択されるべきである。アミノ酸の置換前後にキナーゼの活性および安定性を評価する試験方法は、当業者によく知られており、精製タンパク質の目視検査、円偏光二色性(CD)分光法、結晶化および構造決定、酵素活性アッセイ、タンパク質融解曲線、示差走査熱量測定およびNMR分光法が挙げられる。
【0026】
上述した通り、P-ループは、ATPase/GTPase中に、それぞれATP/GTPリン酸結合モチーフとも呼ばれる、高度に保存された高グリシンモチーフG-X-G-X-X-Gを含む。保存されたグリシンは、酵素の結合基質にリン酸を効率的に運ぶためのATPまたはGTPのリン酸の最適な位置決めに、非常に重要であることが示唆される。原理上、前記の保存されたモチーフ内の全てのアミノ酸は、本発明による置換および/または標識化に選択されるかもしれないが、高グリシンモチーフの可変位置の、より保存度が低いアミノ酸(Xと示す)が選択されることが好ましい。保存されたグリシン残基の一つを選択することは、キナーゼのATPase活性を妨害する可能性があり、このことは、上記でも述べた通り、自然発生キナーゼと比較して少なくとも同等の、好ましくは本質的に変わらない触媒活性を有する標識キナーゼを得るためには、好ましくは避けるべきである。さらに好ましくは、置換されるX位置のアミノ酸は、フェニルアラニンまたはチロシン等の芳香族アミノ酸である。フェニルアラニンおよびチロシンは両者ともかさ高く、本発明による共有結合した標識、特にチオール反応性標識アクリロダンと比較した場合、結合しているリガンドとともに同様の立体構造の再配置を採ると考えられる。
【0027】
その際、好ましくは活性状態の野生型キナーゼであるキナーゼの、触媒活性の少なくとも90%が保持され、好ましくは少なくとも95%、より好ましくは少なくとも98%が保持される場合、触媒活性の阻害は存在しない。最も好ましくは、キナーゼの触媒活性は完全に保持される。従って「触媒活性を阻害しない」という用語は、触媒活性が合計100%未満であるいくつかの態様において、「本質的に触媒活性を妨げない」という意味と同じであると考えられ、この意味を有している。触媒活性は、本発明の標識キナーゼにおける阻害剤のIC50値と、それが由来する非標識キナーゼとを比較することにより、間接的に決定し得る(ATP-Kmに相当するATPの量を使用する)。IC50値が同一の範囲内である場合、即ち、IC50値が5倍を超えて異ならない場合、このことは、触媒活性が本質的に同じであること(およびキナーゼの修飾がキナーゼに対する阻害剤の親和性を変えなかったこと)を示す。本発明の標識キナーゼおよび非標識キナーゼの差が、4倍以下であることが好ましく、より好ましくは3倍以下、さらにより好ましくは2倍以下である。当業者は、5倍以下の両IC50値の差は、充分に、これらの測定に関する通常の分散の範囲内であることを承知している。このようなIC50値は、両キナーゼの触媒活性が本質的に同じであることを保証する。安定性については、導入されたアミノ酸は、タンパク質の構造的安定性を保証する必須の分子間接触を妨げないため、キナーゼは本書に記載した生物学的機能を果たし得る。
【0028】
現在存在するスクリーニング方法の欠点を克服するため、本発明は、例えば蛍光タグ付けされたキナーゼを作る、標識化戦略であって、(i)キナーゼ阻害剤の結合に感受性が高く、(ii)リガンドの結合および解離のキネティクスのリアルタイム測定に使用することが可能であり、(iii)これらのリガンドのKdの直接測定に使用することが可能であり、かつ(iv)迅速で、ロバスト性があり、再現性があり、ハイスループット・スクリーニング方法に適応可能である、標識化戦略を包含する。
【0029】
先行技術と対照的に、かつ、添付の実施例で実証した通り、本発明は、キナーゼおよびこれらのキナーゼを用いたスクリーニング方法を提供し、本発明により、削減された労力と材料で、優れた信頼性を有する阻害剤のスクリーニングが可能になる。これは、例えばキナーゼのP-ループにおける立体構造の変化が引き起こす、環境の変化に対する反応におけるキナーゼの挙動を標識が変化させるような、キナーゼの標識化戦略を提供することによって、本質的に達成される。
【0030】
キナーゼ活性のモジュレータをスクリーニングする、従来のキナーゼアッセイ以外に、最近、様々なアプローチが開発されている。しかし、これらのアプローチの多くは、深刻な問題に悩まされている。例えば、アニス(Annis)ら(2004年)は、親和性選択的質量分析(AS-MS)を用いるアプローチを記載している。この方法は、ハイスループット・スクリーニングに好適であることが記載されている。しかし、各プローブの試験の前に、サイズ排除クロマトグラフィー工程を適用しなければならず、これは時間がかかり、多量の材料が必要である。
【0031】
ドゥ・ロリミエル(De Lorimier)ら(2002年)は、小分子リガンドに結合する細菌性タンパク質に基づいた一群のバイオセンサーを記載しており、このリガンドは種々の環境感受性フルオロフォアで修飾および標識されていた。リガンドの結合に際し、フルオロフォアはそれらの発光波長および/または強度を変化させ、それによりプローブに結合した特異的リガンドの存在および/または濃度を示す。しかし、特異的阻害剤のスクリーニングにおけるキナーゼの標識および前記のキナーゼの使用については、開示も示唆もされていない。
【0032】
より最近では、p38αキナーゼ由来の、予め結合した(prebound)プローブの置換に基づく、2つのさらなる結合アッセイも報告された:1つはフルオロフォア標識阻害剤を利用し(テクル(Tecle)ら、2009年)、他方は酵素断片の相補性に基づくアプローチを使用したものである(クルター(Kluter)ら、2009年)。後者の場合、化学発光の読み出しが、予め結合した阻害剤−ペプチドプローブの置換によって発生したが、その後、β-ガラクトシダーゼを補体活性化して、アッセイの読み出しとなる化学発光を触媒する。これらのアプローチは終点測定を用いる置換リガンドの親和性の決定に好適であることが実証されたが、反応速度論的パラメータ(konおよびkoff)の分析は、p38α由来の選択されたピラゾロウレア系プローブの解離が詳しく特性化された通り緩徐であり、シグナルの検出速度が制限されることから、簡易性が劣る(パルゲリス(Pargellis)ら、2002年)。
【0033】
本発明の根底にある原理は、II型またはIII型阻害剤の結合に際し、活性化ループの立体構造変化にP-ループが反応することである。活性化ループは、活性部位の入り口付近の柔軟な断片であり、殆どのキナーゼの基質結合裂隙(cleft)を形成し、1以上のアミノ酸をリン酸化して、タンパク質キナーゼスーパーファミリー全体の重要な調節メカニズムを提供し得る(ジョンソン(Johnson)およびルイス(Lewis)、2001年;テイラー(Taylor)およびラズィオ-アンゼルム(Radzio-Andzelm)、1994年;ジョンソンら、1996年)。活性化ループは、殆どのキナーゼにおいて柔軟なループを形成するいくつかのアミノ酸より成り、ATP結合部位において高度に保存されたアスパラギン酸-フェニルアラニン-グリシン(DFG)モチーフから始まり、キナーゼのN-およびC-ローブの間まで延びる。活性化ループは、酵素的キナーゼ活性に極めて重要な構造的構成要素である。活性化ループは、基質結合裂隙の一部であり、特異的基質の認識を助けるいくつかのアミノ酸残基を含有し、また、リン酸化し得るセリン、トレオニンまたはチロシンを含む。活性化ループの立体構造は、DFG-in(活性キナーゼ)とDFG-out(不活性キナーゼ)構造の間で力学的に平衡であると考えられている。相互作用のパートナー(他のタンパク質またはDNA)のリン酸化および/または結合により、平衡はシフトする。DFG-in構造において、モチーフに含有されるアスパラギン酸は、ATP結合部位に向けられ、隣接するフェニルアラニンはATP部位から離されて、隣接するアロステリック部位に向けられる。活性化ループの一部を形成する保存されたDFGモチーフがin-構造を採用する場合、ATP-競合阻害剤(I型阻害剤)はキナーゼに結合し得る。DFG-out構造において、これらの残基の位置は、向きが180°反転する。活性化ループのout-構造は、ATPと基質の結合を妨げる。
【0034】
上述の通り、リガンドおよび基質のATP結合部位への侵入を制御することに加えて、P-ループは周囲の溶媒からのATPおよび他のリガンドの保護に役立つ。ATP結合ポケットにおけるいくつかのI型阻害剤の結合に関し、種々の立体構造を採ることが示されている(ハンクス(Hanks)およびハンター(Hunter)、1995年;マペッリ(Mapelli)ら、2005年)。
【0035】
本発明に従い、蛍光-またはスピン-標識P-ループアッセイシステムを用いて、アロステリック阻害剤(図1c参照)を検出した。結合したフルオロフォアまたはスピン標識により、キナーゼの活性化ループがDFG-out構造を採った際に起こる、P-ループにおける運動が報告される。添付の実施例に示した通り、Cys残基を部位特異的突然変異誘発によりGly-X-Gly-X-X-Glyモチーフの第三のGlyの直前の位置に導入して、環境感受性のフルオロフォアであるアクリロダンでP-ループを特異的に標識することにより、阻害剤のスクリーニングに役立つ能力を有するキナーゼが得られる。この位置の残基は、全てのヒトキナーゼのおよそ80%において、TyrまたはPheとして保存され、このことから、これらの側鎖の芳香環系について、このループと他の構造的特徴およびリガンドとのクロストークを媒介する役割を有することが示唆される。
【0036】
さらに重要なことには、この観察により、アシクロダンの平面環系の導入はキナーゼによる耐性が良好であろうことが示唆された。
【0037】
本発明者らは、最近、標的キナーゼの活性化ループにタグ付けする、ロバストなアッセイシステムを開発した(同時係属出願欧州特許EP 08 01 3340号および欧州特許EP 08 02 0341号)が、それにより、種々のリガンドの解離定数(Kd)、速度定数(kon)および解離速度定数(koff)の直接測定が可能になり、初めてcSrcおよびp38αのIII型リガンドが特定でき、強力なII型阻害剤であるゲートキーパーの変異薬物耐性cSrc-T338Mが開発された。さらに、p38αのDFG-out構造を安定化する、p38αにおけるチアゾール-尿素骨格に対する新規なIII型結合様式およびいくつかの独特なI型リガンドが特定されるかもしれない。DFG-out構造を安定化するリガンドの検出感度は、化合物ライブラリをスクリーニングするこのアプローチを使用することによって有意に上昇するが、これはリン酸化していない不活性形態のキナーゼを利用するからであり、このことは、DFG-out構造の採用を支持する。これらの以前の研究により、これらの型のリガンドのスクリーニングおよび濃縮に使用し得るアッセイが広範囲にわたって関与することが強調される。しかし、活性化ループの標識化に際し、標的キナーゼの既知の阻害剤のDFG-in/out構造的平衡の変動または親和性の有意な変化より生じる、キナーゼ活性の潜在的変化を避けるため、本発明によって提供された通りの、II型およびIII型阻害剤を特定および特性化する別の標識化戦略により、前記の活性変化が起こりにくくなる。
【0038】
添付の実施例に示した通り、本発明は、フルオロフォアの環境における変化、例えば、活性化ループのDFG-out構造への運動によるP-ループにおける立体構造変化を引き起こすII型およびIII型阻害剤等の、種々の結合モードによる阻害剤の結合を高感度に検出するP-ループ標識キナーゼの能力を実証し、また、その蛍光特性を変化させる(図2a参照)。II型およびIII型阻害剤は、HTSフォーマットにおいて蛍光シグナルまたはKdの時間依存性の変化を経時的にモニターすることによって、またはキュベットにおいてkonを測定することによって(I型バインダーは<5)、容易に識別される。また、本発明のアッセイは、独特の結合モードにより、DFG-out構造を安定化するI型リガンドを強力に検出する能力がある。このようなリガンドは、ATP-結合部位内で結合するが、阻害剤分子であるDFGモチーフの高度に保存されたPheと、P-ループ中の選択された標識位置(p38αにおけるTyr35)に典型的に見られる残基の平面環系との間に形成する、独特な環のスタッキング相互作用を利用する。最後に、DFG-in立体構造に結合する、いくつかのI型阻害剤は、記載されたP-ループのTyr/Phe側鎖と直接的に相互作用することが示された(タマヨ(Tamayo)ら、2005年)。この位置を使用してキナーゼを標識することにより、活性化ループのDFG-out構造または運動を引き起こさずに、これらの型の阻害剤を検出することも可能になる(図3B、右パネル)。活性化ループがフルオロフォアで直接標識される、最近報告されたアッセイと比較して(欧州特許出願EP 08 01 3340号およびEP 08 02 0341号)、このアッセイシステムもリン酸化されていない形態のキナーゼを利用し、II型およびIII型阻害剤によって引き起こされたもの等の、リガンド結合に相関する活性化ループの立体構造変化を検出する、強力な代替のスクリーニングツールを提供する。さらには、アロステリックポケットの薬剤可能性は、キナーゼ間で異なるようであり、キナーゼがDFG-out構造を採用する能力に感受性があり、従って、本発明は、P-ループと直接相互作用し、そこで立体構造変化を誘発する、高親和性I型化合物を検出および設計する、魅力的な代替のアプローチとなる。このようなI型リガンドを特定する利点は、それらがより保存性が低いアロステリック部位の方向に延びるII型阻害剤へのさらなる開発の出発点となり得ることから、過小評価すべきではない(リウ(Liu)およびグレイ(Gray)、2006年)。
【0039】
いくつかのI型阻害剤は、DFG-out構造も安定化する。本発明を用いてI型DFG-outバインダーを検出できる鍵は、両基質およびATPの非存在下で、非リン酸化形態のキナーゼを用いたスクリーニングを実施する能力である。上記した通り、非リン酸化形態のキナーゼは、DFG-out構造を採用する可能性が高く、この構造において、活性化ループのDFGモチーフまたはN-末端領域における残基は、リガンドと相互作用し、ATP部位中に反転してATP-競合リガンドに接触することにより、親和性を高め得る。これは、リン酸化したキナーゼを必要とする活性に基づく古典的アッセイと対照的であるが、リン酸化キナーゼはDFG-in構造中に見つかる可能性が高いため、DFG-outバインダーの親和性を低下させ、このような好ましいヒットが検出される可能性が低くなる(スィーリガー(Seeliger)ら、2007年)。スクリーニング・キャンペーンでの古典的な活性に基づくアッセイの確立された使用は、DFG-outバインダーの検出の感受性を抑制し、例えば、VEGFR2阻害剤であるCP547632の、VEGFR2以外の活性(即ち、リン酸化)キナーゼに対する結合について、文献中に情報がないことが説明されるかもしれない。CP547632は特異性が高いことが報告されており、それにより、血管新生の阻害によって腫瘍の生長および増殖を抑止する臨床試験において、VEGFR2阻害剤として適用されている。本発明のアプローチにより、非リン酸化p38αを用いて検出されるCP547632の親和性がマイクロモル以下であることを考えると、これらの知見は、この臨床的に意義のある化合物または他のキナーゼ関連疾患の治療のための類縁誘導体のさらなる研究をも刺激し得る。キナーゼは、細胞内にリン酸化および非リン酸化の両形態で存在し、これらの種の相対存在量がキナーゼ活性およびシグナル伝達経路を制御する。従って、非リン酸化キナーゼもまた、生物学的関連性を示し、魅力的な薬剤標的である。加えて、p38αと複合したCP547632について本書で示した構造的情報(即ち、新たな型であるヒンジ接触)は、この分子のアフィン部分を基礎として隣接するアロステリック部分に延びるさらなる医薬品化学への取り組みを刺激し、不活性キナーゼ立体構造に結合する、薬理学的により望ましいII型阻害剤を創出し得る。
【0040】
高グリシンループを標識することにより、適用されるキナーゼにおいてDFG-outバインダーを特定するという目標が達成されるだけでなく、キナーゼの高グリシンループにおける立体構造変化を直接誘導することでDFG-in構造に対する親和性を得るI型リガンドの検出も可能になる。この特徴は、以前のアッセイを凌ぐ、本発明のアプローチのさらなる利点である。キナーゼの例として、高グリシンループを標識したp38αを使用することにより、Scios-469等のI型阻害剤が高感度で検出されたが、この型の阻害剤はp38αのDFG-in構造に結合する。このような化合物は、リガンドの周囲の溶媒からの保護に役立つ、高グリシンループの立体構造変化を誘導することによってキナーゼに対する親和性を得る(ハンクス(Hanks)およびハンター(Hunter)、1995年;マペッリ(Mapelli)ら、2005年;パテル(Patel)ら、2009年)。常にではないが、しばしばこれらの相互作用に関与する高グリシンループ内の位置は、80%超のキナーゼにおいて芳香族TyrまたはPheとして保存されることから、本発明は、現在のスクリーニングアッセイを、容易に誘導できるDFG-in/out平衡によって制御されない多数のキナーゼ等の、さらなるキナーゼに広げる。高グリシンループと相互作用するI型阻害剤の検出は、ヒンジ領域の接触の新規な型を特定することに従来のように焦点を置くことを回避しつつ、上記の相互作用を活用する、新たな骨格の開発についての見識を提供し得る。また、高グリシンループの立体構造変化も、I型阻害剤の特異性を向上させるさらなる方法を提供し得る。
【0041】
好ましい態様において、キナーゼは、セリン/トレオニンキナーゼまたはチロシンキナーゼである。
【0042】
別の好ましい態様において、キナーゼはMEKキナーゼ、CSK、オーロラキナーゼ、GSK-3β、sSrc、EGFR、Abl、DDR1、LCK、CDK、p38αまたは別のMAPKである。
【0043】
マイトジェン活性化タンパク質(MAP)キナーゼ(EC 2.7.11.24)は、細胞外刺激(マイトジェン)に反応するセリン/トレオニン特異的タンパク質キナーゼであり、遺伝子発現、有糸分裂、分化、および細胞生存/アポトーシス等の種々の細胞活動を調節する。細胞外刺激により、MAPキナーゼ、MAPキナーゼキナーゼ(MKKまたはMAP2K)およびMAPキナーゼキナーゼキナーゼ(MKKKまたはMAP3K、EC 2.7.11.25)で構成されるシグナル伝達カスケード(「MAPKカスケード」)を介して、MAPキナーゼは活性化される。
【0044】
細胞外刺激によって活性されるMAP3Kは、そのセリンおよび/またはトレオニン残基でMAP2Kをリン酸化し、その後、このMAP2Kは、そのセリンおよび/またはチロシン残基上のリン酸化を通してMAPキナーゼを活性化する。このMAPキナーゼシグナル伝達カスケードは、酵母から哺乳類まで、進化的によく保存されている。
【0045】
これまでに、哺乳類において、明瞭な6つのグループのMAPKが特性化されている:
【0046】
1. 細胞外シグナル調節キナーゼ(ERK1、ERK2)。ERK(古典的MAPキナーゼとしても知られている)シグナル伝達経路は、好ましくは、成長因子およびホルボールエステル(発癌プロモーター)に反応して活性化され、細胞増殖および細胞分化を調節する。
2. c-Jun N-末端キナーゼ(JNK)、(MAPK8、MAPK9、MAPK10)、ストレス活性化タンパク質キナーゼ(SAPK)としても知られている。
3. p38アイソフォームは、p38α(MAPK14)、p38β(MAPK11)、p38γ(MAPK12またはERK6)およびp38δ(MAPK13またはSAPK4)である。JNKおよびp38シグナル伝達経路の両者は、サイトカイン、紫外線照射、熱ショックおよび浸透圧衝撃等のストレス刺激に反応性があり、細胞分化およびアポトーシスに関与する。p38αMAPキナーゼ(MAPK)は、RKまたはCSBPとも呼ばれるが、サイトカインおよびストレスに対する細胞反応を制御する信号伝達カスケードに関与する、酵母HOGキナーゼの哺乳類相同分子種である。SAPK/JNK経路と同様に、p38 MAPキナーゼは、浸透圧衝撃、炎症性サイトカイン、リポ多糖類(LPG)、紫外光および成長因子等の様々な細胞ストレスによって活性化される。p38 MAPキナーゼは、Thr180およびTyr182におけるリン酸化によって活性化される。
4. ERK5(MAPK7)は、最近発見されたが、成長因子およびストレス刺激のいずれでも活性化され、細胞増殖に関与する。
5. ERK3(MAPK6)およびERK4(MAPK4)は、活性化ループにSEG(セリン−グルタミン酸−グリシン)モチーフを有し、C-末端延長部においてのみ大きな差異を示す、構造的に関連した異型MAPKである。
6. ERK7/8(MAPK15)は、MAPKファミリーで最も最近発見されたメンバーであり、ERK3/4と類似の挙動を示す。
【0047】
マイトジェン活性化タンパク質キナーゼキナーゼは、マイトジェン活性化タンパク質キナーゼをリン酸化する、キナーゼのファミリーを形成する。また、これらは、MAP2K2としても知られ、EC 7.2.12.2に分類される。7つの遺伝子が存在する。これらは、MAP2K1(MEK1)、MAP2K2(MEK2)、MAP2K3(MKK3)、MAP2K4(MKK4)、MAP2K5(MKK5)、MAP2K6(aka MKK6)、MAP2K7(MKK7)をエンコードする。p38(MKK3およびMKK4)、JNK(MKK4)、およびERK(MEK1およびMEK2)の活性化剤は、独立したMAPキナーゼシグナル伝達経路を特徴付ける。
【0048】
オーロラキナーゼA(オーロラ、オーロラ-2、AIK、AIR-1、AIRK1、AYK1、BTAK、Eg2、MmIAK1、ARK1およびSTK15としても知られている)、B(オーロラ-1、AIM-1、AIK2、AIR-2、AIRK-2、ARK2、IAL-1およびSTK12としても知られている)、およびC(AIK3としても知られている)は、細胞質分裂および無調節な染色体分離等の、いくつかの生物学的プロセスに関与している。これらの重要な有糸分裂の調節因子は、多様な固形腫瘍において過剰に発現する。このセリン/トレオニンキナーゼのファミリーの一つであるヒトオーロラAは、すい臓癌における薬剤標的として提案されている。オーロラAの最近の三次元構造決定により、オーロラキナーゼは活性化ループ領域の周辺で独特な立体構造を現すことが示された。この特性は、オーロラキナーゼの阻害剤の探索および開発を促進したが、新規な発癌抑制剤としても機能する可能性がある。
【0049】
グリコーゲン合成酵素キナーゼ3(GSK-3)はセリン/トレオニンタンパク質キナーゼであり、セリン/トレオニンキナーゼ活性に加えて、チロシン残基上で自己リン酸化する独特な能力を有する。GSK-3による標的タンパク質のリン酸化は、通常、それらの活性を阻害する(グリコーゲン合成酵素およびNFATの場合と同様)。GSK-3は、キナーゼの中では他とは異なり、標的タンパク質を最初にリン酸化するのに通常「プライミングキナーゼ」が必要であり、その後にのみ、GSK-3は標的タンパク質をさらにリン酸化することが可能である。哺乳類において、GSK-3は、GSK-3αおよびβの2つの知られている遺伝子でエンコードされている。胚の発生過程でのパターン形成および細胞増殖の役割に加えて、腫瘍形成における細胞分裂およびアポトーシスの調節による役割について、最近の証拠がある。ヒトグリコーゲン合成酵素キナーゼ-3β(GSK3β)もまた、肥満、糖尿病、アルツハイマー病および双極性障害等のいくつかの病理生理学的状態と関連する。
【0050】
プロトオンコジェニックチロシンキナーゼのSrcファミリーは、細胞運動および細胞増殖の中核をなすインテグリン依存性シグナルを伝達する。Srcファミリーは、9のメンバーを含む:Src、Lck、Hck、Fyn、Blk、Lyn、Fgr、Yes、およびYrkである。これらのキナーゼは、現在の、癌を無調節な細胞成長および細胞分裂を伴う疾患として理解することに役立ってきた。CSrcプロトオンコジーンは、cSrcチロシンキナーゼをコードする。そのキナーゼドメインに加えて、cSrcはSH2ドメインおよびSH3ドメインをさらに含み、それらはSrcキナーゼドメインと多酵素複合体を形成するためのアダプタータンパク質として働く。これらのドメインもまた、cSrcキナーゼドメインの自己抑制に関与する。この遺伝子における突然変異は、癌細胞の悪性進行に関与する可能性がある。このタンパク質は、ヒト白血球特異的タンパク質チロシンキナーゼ(Lck)のTyr-504残基を特異的にリン酸化するが、この残基は、負の調節部位として作用する。またこのタンパク質は、LynおよびFynキナーゼにも作用し得る。
【0051】
白血球特異的タンパク質チロシンキナーゼ(Lck)は、T-細胞等のリンパ球の内部に見られるタンパク質である。Lckは、リンパ球の細胞内シグナル伝達経路に関与する、あるタンパク質のチロシン残基をリン酸化する、チロシンキナーゼである。LckのN-末端尾部はミリストイル化およびパルミトイル化され、タンパク質を細胞の原形質膜に繋ぐ。このタンパク質はさらにSH3ドメイン、SH2ドメインを含み、C-末端部分にはチロシンキナーゼドメインを含む。Lckで始まるチロシンリン酸化カスケードは、カルシウム(Ca2+)イオンの細胞内移動およびリンパ球内での重要なシグナル伝達カスケードの活性化に至る。これらはRas-MEK-ERK経路を含むが、これは続いて、NFAT、NFκB、およびAP-1等の転写因子を活性化し、転写因子はその後、多量の遺伝子産物の産生を調節するが、遺伝子産物は、最も顕著には、活性化リンパ球の長期の増殖および分化を促進する、インターロイキン−2等のサイトカインである。Lckの異常発現は、胸腺腫瘍、T-細胞白血病および結腸癌に関連している。
【0052】
チロシンキナーゼのSrcファミリーの触媒活性は、C末端付近に位置するチロシン残基(cSrcにおけるTyr 527)のリン酸化によって抑制されるが、これはC-末端Srcキナーゼ(Csk)によって触媒される。殆どのチロシンキナーゼの乱雑さを考えると、SrcファミリーキナーゼのC-末端尾部がCskの唯一知られている標的であることは注目に値する。おそらく代表的なSrcキナーゼである、CskとcSrcとの相互作用により、cSrcのC-末端尾部はCskの活性部位の末端に位置する。Cskは、このドッキングメカニズムが存在しない基質をリン酸化することはできないが、これは、基質を認識するために殆どのチロシンキナーゼが使用する従来の基質結合部位が、活性化ループにおける欠失により、Cskにおいて不安定化されるためである(レビンソン(Levinson)、2008年)。
【0053】
上皮成長因子受容体(EGFR;ErbB-1;ヒトにおけるHER1)は、細胞外タンパク質リガンドの上皮成長因子ファミリー(EGF-ファミリー)のメンバーに対する細胞表面受容体である。上皮成長因子受容体は、受容体のErbBファミリーの一メンバーであり、以下の4つの密接に関わる受容体チロシンキナーゼのサブファミリーである:EGFR(ErbB-1)、HER2/c-neu(ErbB-2)、Her 3(ErbB-3)およびHer 4(Erb-4)。活性EGFRは、二量体として発生する。EGFR二量化は、細胞外受容体ドメインへのリガンド結合によって誘導され、その内因性の細胞内タンパク質-チロシンキナーゼ活性を刺激する。その結果、EGFRのC-末端(細胞内)ドメインにおいて、いくつかのチロシン残基の自己リン酸化が起こる。この自己リン酸化は、自身のホスホチロシン結合SH2ドメインによってリン酸化チロシンと関係する、いくつかの他のタンパク質によって、下流の活性化およびシグナル伝達を引き起こす。また、EGFRのキナーゼドメインは、凝集した他の受容体のチロシン残基を交差リン酸化(cross-phosphorylate)することができ、そのような方法でそれ自身を活性化し得る。EGFRシグナル伝達カスケードは、いくつかの下流のシグナル伝達タンパク質を活性化するが、これらのタンパク質は次に、いくつかのシグナル形質導入カスケード、主としてMAPK、AktおよびJNK経路を開始させて、DNA合成および細胞増殖を導く。このような経路は、細胞遊走、接着、および増殖等の表現型を調節する。EGFR過剰発現(上方調節として知られている)または過剰活性を導く突然変異は、多数の癌と関係している。結果として、EGFRの突然変異は、いくつかの型の癌において特定されており、抗癌療法での拡大する分野の標的である。
【0054】
ABL1-プロトオンコジーンは、細胞質および核のタンパク質キナーゼをエンコードするが、このキナーゼは、細胞分化、細胞分裂、細胞接着およびストレス反応の過程に関係している。c-Ablタンパク質の活性は、そのSH3ドメインによって負に調節される。SH3ドメインの遺伝的欠失は、ABL1を発癌遺伝子に換える。この遺伝的欠失は、(9;22)遺伝子転座によって起こるが、慢性骨髄性白血病の多くの症例に存在するBCR(MIM:151410)およびABL1遺伝子の頭尾融合を引き起こす。遍在的に発現したABL1チロシンキナーゼのDNA結合活性は、CDC2-媒介リン酸化によって調節されるが、このことはABL1の細胞周期機能を示唆している。
【0055】
DDR1またはCD167a(分化167aのクラスター)としても知られる、ディスコイジンドメイン受容体ファミリーのメンバー1は、正常および形質転換した上皮細胞において広く発現されている受容体チロシンキナーゼ(RTK)であり、種々のタイプのコラーゲンによって活性化される。このタンパク質は、細胞外ドメインにおいて、ディクチオステリウム・ディスコイデウム(Dictyostelium discoideum)タンパク質ディスコイジンIに類似した相同領域を有する、チロシンキナーゼ受容体のサブファミリーに属する。その自己リン酸化は、これまで試験した全てのコラーゲン(I型およびVI型)により達成される。現場(In situ)研究およびノーザンブロット分析により、このエンコードしたタンパク質の発現が、特に腎、肺、胃腸管、および脳の上皮細胞に限定されることが示された。加えて、このタンパク質は、乳房、卵巣、食道、および小児脳由来のいくつかのヒト腫瘍において、有意に過剰発現している。
【0056】
上述のキナーゼは、全て、現在のところ好適な治療がないか改良した治療計画が望まれている、癌のような疾患の発症に関与するため、好ましい態様である。
【0057】
構造的情報が入手可能であったキナーゼである、p38αを使用して、本発明者らは、スクリーニングの目的での本発明の標識キナーゼの適用性を実証した。予想外に、このキナーゼは最低限の労力で標識化用に調製することができたが、また、標識したキナーゼは所望の特性を発揮し、即ち、知られている阻害剤BIRB-796およびいくつかのより小さいBIRB-796類似体の場合、導入した標識が、特異的阻害剤の結合により誘導される立体構造変化を検出するのに好適であることが判明した。
【0058】
本発明にしたがって標識した形態で、特異的阻害剤のスクリーニングに適用し得るさらなるキナーゼは、MKK7である(例えば、ワン(Wang)ら、2007年に概説)。MKK7のI型およびII型阻害剤の両者は分析されており、それらの薬理学的特性は改良されて、実施例7に詳述した通り、より強力な阻害剤を得ることができた。
【0059】
別の好ましい態様において、標識されているアミノ酸は、システイン、リシン、アルギニンまたはヒスチジンである。
【0060】
システインは、遊離チオール基を有し、一方、リシン、アルギニンまたはヒスチジンはそれぞれ、少なくとも1つの遊離アミノ基を保持する。
【0061】
別の好ましい態様において、P-ループの外部に存在する1以上の溶媒にさらされるシステインが欠失しているか置換されている。
【0062】
標識化に先立ち、興味のあるキナーゼ中に、遊離チオールまたはアミノ基を有するアミノ酸が2つ以上存在する場合、P-ループにおけるアミノ酸の特異的標識は可能ではないかもしれない。従って、上で論じた通り、キナーゼ中に存在し、遊離チオールまたはアミノ基を有するアミノ酸は、溶媒にさらされることが予測されるか示される場合、取り除かれるか、遊離チオールまたはアミノ基を有しない別のアミノ酸で置換されるべきである。興味のあるキナーゼ中に天然に存在し、溶媒にさらされるシステインは、P-ループの外部に配置し得るが、この場合、システインは取り除かれるか、遊離チオール基を有しない別のアミノ酸で置換されるべきである。このことは、遊離アミノ基を有するアミノ酸にも同様に適用され、遊離アミノ基は、反応性遊離アミノ基を有しないアミノ酸で置換されるべきである。遊離アミノ基を有する1以上のアミノ酸がP-ループ中に既に存在する場合、標識されるアミノ酸に加えて、遊離アミノ基を有し、P-ループ中に存在するアミノ酸は、置換または取り除かれるべきであるが、キナーゼに対するこれらの変異のいずれもその触媒活性を阻害しないか、その安定性を妨げない。要約すると、前記の変異により、P-ループの所望の位置で特異的に標識されるキナーゼが得られる。
【0063】
「溶媒にさらされる」という用語は、タンパク質の三次元構造の文脈において、タンパク質の一部のアミノ酸の位置に言及する。タンパク質体の内部に埋め込まれているアミノ酸は、他のアミノ酸に完全に囲まれているため、溶媒とは全く接触しない。対照的に、溶媒にさらされるアミノ酸は、部分的または完全に周囲の溶媒にさらされるため、それらアミノ酸を修飾する可能性がある化学物質に接近しやすい。このことは、例えば、遊離チオール-またはアミノ-基を有する、溶媒にさらされるアミノ酸と反応し得る、本発明に使用されるチオール-またはアミノ-反応性標識に適用する。
【0064】
「取り除く」という用語は、アミノ酸を別のアミノ酸と交換しない切除に言及し、一方「置換する」という用語は、アミノ酸の別のアミノ酸での代替に言及する。アミノ酸が別のアミノ酸で置換されるか取り除かれる場合、置換または取り除かれるアミノ酸は好ましくは、取り除かれるか置換されたアミノ酸によりキナーゼの触媒活性が阻害されず、得られるキナーゼの安定性が妨げられないように選択される。
【0065】
より好ましい態様において、キナーゼはp38αであり、標識されるシステインは配列番号1の35位に導入され、好ましくは配列番号1の119位および162位のシステインが、セリン等の遊離チオール基を有しない別のアミノ酸で置換されている。
【0066】
代わりのより好ましい態様において、キナーゼはMKK7であり、標識されるシステインは147位でP-ループ中に天然に存在し(MKK7のキナーゼドメインに対応する配列番号2における31位)、および、好ましくは218位、276位および296位のシステイン(配列番号2の102位、160位および180位)が、セリン等の遊離チオール基を有しない別のアミノ酸で置換される。218位、276位および296位にセリンに変異したシステインを有するMKK7キナーゼドメインを配列番号3に示す。
【0067】
一般に、アミノ酸置換は保護的であるべきである。システインについては、このことは、システインが好ましくはセリンで置換されることを意味する。一般に、異なるアミノ酸でのアミノ酸の置換は、PAM250 スコアリングマトリックスを使用して、置換が保護的であるかどうかの点で評価し得る。このマトリックスはしばしば、整列ペプチド配列の採点に使用され、これらの配列の類似性を決定する(ペアソン(Pearson)、1990年)。
【0068】
上述した通り、天然に存在しない場合、遊離チオールまたはアミノ基を有するアミノ酸をキナーゼのP-ループ中に導入しなければならない。p38αの場合、p38αについての入手可能な結晶構造を使用して、活性化(DFG-in)状態および不活化(DFG-out)状態の両者における構造研究を実施した。P38αはP-ループ中にシステインを保持していない。上記の構造研究により、P-ループ内に位置する35位でチロシンをシステインで置換することは、キナーゼの触媒活性または安定性に有意に影響しないことが示唆された。
【0069】
同時係属出願欧州特許EP 08 01 3340号から、配列番号1の119位と162位の2つのシステインは両者とも溶媒にさらされることが分かった。P-ループ内に位置しない2つのさらなるシステインを記録したシグナルが阻害される可能性を回避するため、これらの2つのシステインは好ましくは別のアミノ酸で置換され、好ましくはセリン等の、同等の大きさおよび構造のアミノ酸で置換される。
【0070】
p38αに相同なキナーゼを使用する場合、システインで置換されるアミノ酸の位置は、配列番号1の35位に対応し得る。キナーゼにおいていずれの位置が配列番号1の35位に対応するかを決定するため、使用したキナーゼを有する配列番号1の配列アラインメントが達成され得るが、これは例えば、CLUSTALW等の一般に入手できるプログラムを使用する。
【0071】
別の好ましい態様において、チオールまたはアミノ反応性フルオロフォアは、2つの置換基のうちの1つがチオールまたはアミノ反応性部分である、環境感受性の二置換ナフタレン化合物である。「環境感受性の」という用語は、1以上の波長でのその蛍光発光が変化するか、その全体の発光スペクトルが変化することで表される、その環境条件に対するフルオロフォアの感受性を意味する。このような変化を引き起こす条件は、例えば、活性化ループ、従ってP-ループにおける、極性変化または立体構造変化である。しかし、変化は、活性化ループに全く影響せず、P-ループで起こることもある。
【0072】
上記のタイプのフルオロフォアは、典型的には、周囲環境の極性に依存して、発光波長の強度およびシフトの両者において変化を呈する。この分類のフルオロフォアの例としては、6-アクリロイル-2-ジメチルアミノナフタレン(アクリロダン)、6-ブロモアセチル-2-ジメチルアミノ-ナフタレンバダン(バダン)、2-(4’-(ヨードアセトアミド)アニリノ)ナフタレン-6-スルホン酸ナトリウム塩(IAANS)、2-(4’-マレイミジルアニリノ)ナフタレン-6-スルホン酸ナトリウム塩(MIANS)、5-((((2-ヨードアセチル)アミノ)エチル)アミノ)ナフタレン-1-スルホン酸(1,5-IAEANS)および5-ジメチルアミノナフタレン-1-スルホニルアジリジン(ダンシルアジリジン)またはそれらの誘導体が挙げられる。
【0073】
環境の感受性によっては使用してもよい他のフルオロフォアは、クマリン系化合物、ベンゾキサジアゾール系化合物、ダポキシル系化合物、ビオシチン系化合物、フルオレスセイン;アレクサフロール(AlexaFlour)染料(モレキュラー・プローブス(Morecular Probes))、アット(Atto)フルオロフォア(アットテクノロジー(Atto Technology))またはルシファー・イエロー(Lucifer Yellow)等のスルホン化ローダミン系化合物である。クマリン系フルオロフォアは、環境に緩やかに感受性があり、例えば、7-ジエチルアミノ-3-(4’-マレイミジルフェニル)-4-メチルクマリン(CPM)が挙げられる。ベンゾキサジアゾールフルオロフォアもまた、タンパク質-フルオロフォア複合体を形成するために通常使用され、強力な環境依存性を有し、例えば7-フルオロベンズ-2-オキサ-1,3-ジアゾール-4-スルホンアミド(ABD-F)およびN-((2-(ヨードアセトキシ)エチル)-N-メチル)アミノ-7-ニトロベンズ-2-オキサ-1,3-ジアゾールエステル(IANBD)が挙げられる。PyMPOマレイミド(対チオール)またはスクシンイミドエステル(対アミン)および種々の他のダポキシル染料が良好な吸収性および極めて高い環境感受性を有する。例としては、1-(2-マレイミジルエチル)-4-(5-(4-メトキシフェニル)オキサゾール-2-イル)ピリジニウムメタンスルホネート(PyMPO-マレイミド)、臭化1-(3-(スクシンイミジルオキシカルボニル)ベンジル)-4-(5-(4-メトキシフェニル)オキサゾール-2-イル)ピリジニウム(PyMPO-スクシンイミジルエステル)およびダポキシル(2-ブロモアセトアミドエチル)スルホンアミドが挙げられる。しかし、それらの構造がより長く、より柔軟であるため、これらのプローブは、選択される標識部位によって、P-ループの運動またはP-ループと活性化ループとの間の相互作用に影響を与えるかもしれない。上記の物質の適用性は、個々のキナーゼおよび標識されるアミノ酸の位置に依存し、一部の例では本発明の方法における感受性を低下させる可能性があるとしても、原理上なお標識として適用され得る。上記の物質を好適なキナーゼと適合させることは、当業者は、本発明の教示と組み合わせた通常の手順で実施し得る。
【0074】
一般的に、キナーゼの触媒活性を阻害せず、またはキナーゼの安定性を妨げない限り、あらゆるフルオロフォアを使用し得る。このことは、フルオロフォアは好ましくは嵩高くなく、または伸長していないことを意味する。
【0075】
さらに好ましい態様において、チオール反応性スピン標識は、ニトロキシドラジカルである。
【0076】
スピン標識でタンパク質配列を部位特異的に標識する支配的な方法は、メタンチオスルホネートスピン標識とシステインとの間の反応であり、スピン標識したシステイン側鎖、CYS-SLを与える:
MeS(O)2SSR + R’SH ---> R’SSR + MeS(O)2SH
式中、Rはニトロキシド基であり、R’SHはシステインスルフヒドリルを有するタンパク質であり、R’SSRはスピン標識したタンパク質である。標識用のシステインは、固相技術または標準的組み換えDNA技術のいずれかにより、所望の配列位置に置かれる。
【0077】
さらに、本発明は、キナーゼ阻害剤のスクリーニング方法に関し、この方法は、(a) 本発明の(蛍光的またはスピン標識またはアイソトープ標識した)キナーゼを供給すること;(b) 前記の(蛍光的またはスピン標識またはアイソトープ標識した)キナーゼを候補阻害剤と接触させること;(c) 前記の工程(a)および工程(b)の蛍光標識したキナーゼの、励起の際の、1以上の波長での蛍光発光シグナルまたはスペクトルを記録すること;または(c)’ 前記の工程(a)および工程(b)のスピン標識またはアイソトープ標識したキナーゼの電子常磁性共鳴(EPR)または核磁気共鳴(NMR)スペクトルを記録すること;および(d) 工程(c)で記録される1以上の波長での蛍光発光シグナルまたはスペクトル、または工程(c)’で記録されるEPRまたはNMRスペクトルを比較することを含み;ここで、前記の工程(c)で得られる蛍光標識したキナーゼの、1以上の波長、好ましくは発光極大での蛍光強度の差、および/または、スペクトルにおける蛍光発光波長のシフト、または、前記の工程(c)’で得られるスピン標識またはアイソトープ標識したキナーゼの、EPRまたはNMRスペクトルにおける変化は、候補阻害剤がキナーゼ阻害剤であることを示す。
【0078】
キナーゼ阻害剤は、キナーゼの活性を阻害する能力のある物質である。キナーゼ阻害剤は、例えば、それらがアロステリック阻害剤(III型)であるか、またはATP結合部位に隣接するアロステリック部位に結合し、ATP結合ポケット内に到達するもの(II型)である場合、単一のキナーゼの作用をより特異的に阻害し得る。或いは、阻害剤は、多数のタンパク質キナーゼの作用を阻害し得るが、これは特に、タンパク質キナーゼ中に高度に保存されているATP結合ポケットに排他的に結合する(I型)場合である。
【0079】
候補阻害剤は、異なる化合物分類に属していてもよく、例えば、有機または無機小分子、タンパク質またはペプチド、DNAまたはRNA等の核酸が挙げられる。このような化合物は、分子ライブラリに存在するか、またはスクラッチから設計し得る。
【0080】
本発明による小分子は、分子量が2000 Da以下、好ましくは1500 Da以下、より好ましくは1000 Da以下、最も好ましくは500 Da以下の分子を含む。
【0081】
1以上の波長での蛍光発光シグナルまたはスペクトルの記録は、通常、蛍光分光計または蛍光光度計を使用して達成される。蛍光分光法または蛍光光度法または蛍光分光分析は、サンプルからの蛍光または他の放射光を分析する、電磁分光法の一種である。これらの分析は、ビーム光、通常紫外光の使用を含むが、これは特定の分子において電子を励起し、緩和の際により低いエネルギーの光を発し、必須ではないが、典型的には可視光を発する。
【0082】
本発明の方法に使用し得る、一般的な2つのタイプの装置がある:フィルター蛍光光度計は、フィルターを使用して入射光および蛍光を単離し、一方、蛍光分光計は、回折格子モノクロメーターを使用して、入射光および蛍光を単離する。両タイプは以下のスキームを利用する:励起源からの光は、フィルターまたはモノクロメーターを通過し、サンプルに当たる。一部の入射光はサンプルに吸収され、サンプル中の一部の分子は、蛍光を発する。蛍光は、すべての方向に発光する。この蛍光の一部は第二のフィルターまたはモノクロメーターを通過して検出器に到達するが、検出器は通常、入射光ビームに対し90°に設置し、透過または反射した入射光が検出器に到達するリスクを最小限に抑える。励起源として、種々の光源が使用され得るが、例えば、レーザー光線、フォトダイオード、およびランプが挙げられ;特にキセノンランプおよび水銀灯が挙げられる。検出器は、シングルチャネルまたはマルチチャネルのいずれであってもよい。シングルチャネル検出器は、一度に一つの波長の強度のみを検出し得るが、一方マルチチャネルは同時に全ての波長の強度を検出し、発光モノクロメーターまたはフィルターが不要になる。種々のタイプの検出器が利点および欠点の両方を有する。二重モノクロメーターおよび連続的励起光源を有する、最も用途の広い蛍光光度計は、励起スペクトルおよび蛍光スペクトルの両者を記録し得る。蛍光スペクトルを測定する場合、励起光の波長は、好ましくは高吸収の波長で一定に保たれ、発光モノクロメーターがスペクトルをスキャンする。励起スペクトルの測定には、発光フィルターまたはモノクロメーターを通過する波長を一定に保ち、励起モノクロメーターがスキャンする。蛍光強度が吸収に比例するため、一般に励起スペクトルは吸収スペクトルと一致する(概説は、レンデル(Rendell)、1987年;シャルマ(Sharma)およびシュルマン(Schulman)、1999年;ガウグリッツ(Gauglitz)およびボ-ディン(Vo-Dinh)、2003年;ラコビッツ(Lakowics)、1999年参照)。
【0083】
核磁気共鳴(NMR)は、原子核の量子力学的磁気特性に基づく物理現象である。奇数の陽子または中性子を含有する全ての核は、固有磁気モーメントおよび角運動量を有する。最も普通に測定される核は水素(1H)(天然存在度で最も受容的なアイソトープ)および炭素(13C)であるが、多くの他の元素のアイソトープ(例えば、113Cd、15N、14N、19F、31P、17O、29Si、10B、11B、23Na、35Cl、195Pt)由来の核も観測できる。特定の物質についてのNMR共鳴周波数は、ラーモアの歳差運動周波数の方程式に従って、印加磁場の強度に直接比例する。NMRは、一定の印加磁場でそれらを配列し、直交する交番磁界でこの配列に摂動を与えることによって、磁性核を測定する。摂動磁場に対して得られる応答は、NMR分光法および磁気共鳴映像法に利用される現象であり、高スペクトル分解能を達成するために非常に強力な印加磁場を使用するが、詳細は化学シフトおよびゼーマン効果により説明される。
【0084】
本発明において、P-ループ内の好適なアミノ酸は、アイソトープまたは濃縮アイソトープを含有するチオール/アミノ反応性小分子で標識し得る。この場合、唯一のシグナルは、P-ループ上の豊富な分子に由来するが、これは、選択された標識部位に依存する、タンパク質立体構造に感受性がある。
【0085】
好ましいアイソトープは、13C、15N等であり、これらは1Dまたは2D NMRスペクトルとして測定し得る。例えば阻害剤の結合によるタンパク質の立体構造の変化により、標識に対応するNMRの化学シフトがシフトする。
【0086】
電子常磁性共鳴(EPR)または電子スピン共鳴(ESR)分光法は、上記に簡潔に記載した通り、有機および無機フリーラジカルまたは遷移金属イオンを有する無機錯体等の、1以上の不対電子を有する化学種を研究する技術である。EPRの基本的な物理的概念は、核磁気共鳴(NMR)と類似しているが、原子核スピンの代わりに励起されるものは電子スピンである。最も安定な分子は、全ての電子を対で有するため、EPR技術はNMRより利用の幅が狭い。しかし、この常磁性種への制限もまた、普通の化学溶媒およびマトリクスがEPRスペクトルを生じないため、EPR技術が優れた特異性を有することを意味する。
【0087】
EPR技術はスピン標識を利用する。この場合、試験されるキナーゼは、13Cおよび15N等のアイソトープの存在下で細菌または他の好適な宿主細胞において発現されて、発現されるにつれて、タンパク質全体にこれらのアイソトープが取り込まれる。アイソトープを濃縮したタンパク質を精製した後、上述の通り、スピン蛍光をP-ループに結合させる。この場合、タンパク質中のアイソトープの2D NMRスペクトルが記録される。P-ループおよびスピン標識が立体構造を変化させるにつれて、スピン標識は取り込まれたアイソトープに由来するタンパク質シグナルのいくつかを変化させるが、阻害剤が結合するにつれて、この取り込まれたアイソトープはP-ループまたはスピン標識により近く接触するようになる。スピン標識が接近するにつれて、ピークは幅広くなるであろう。
【0088】
工程(c)または(c)’で得られる、種々のEPRスペクトル、または1以上の波長、好ましくは発光極大における種々の蛍光発光シグナル、または種々の蛍光発光スペクトルは、候補化合物の結合によって起こる、キナーゼにおける立体構造変化を示す。このことは、ATP-結合ポケットに隣接するアロステリック部位に、ある場合ではATP-結合ポケットそのものに化合物が結合することによって、DFGモチーフの摂動、活性化ループにおける立体構造変化を生じ、それにより、P-ループにおいて、隣接する原子の核と結合したスピン標識における極性変化および/または遊離電子の相互作用変化を招くという事実による。EPRまたはNMRスペクトルまたは蛍光発光の比較に際し、本発明の方法により、候補化合物が好適なキナーゼ阻害剤として有効であるかどうか、例えば、高親和性阻害剤であるだけではなく、あるキナーゼの活性を特異的に阻害するものであるかどうかが明らかになる。候補阻害剤を用いないキナーゼの記録データと、前記の候補阻害剤と接触させたキナーゼの記録データを比較する。蛍光発光シグナルの場合、1以上の特異的波長でいずれかのシグナルを記録および比較することができ、特定の波長でのシグナル強度の変化が検出可能になる。或いは、完全なスペクトルを記録および比較することができ、極大発光波長における変化の観察も可能になる。
【0089】
好ましくは、前記の方法は、ハイスループットフォーマットにおいて達成される。ハイスループットアッセイは、生化学分析、細胞分析または他のアッセイと独立して、一般にはマイクロタイタープレートのウェル中で実施してもよく、ここで各プレートは、96、384または1536ウェルを含有していてもよい。室温以外でのインキュベーション等のプレートの取扱、および試験化合物、この場合推定阻害剤のアッセイ混合物との接触は、好ましくは、ピペット装置を含む1以上のコンピュータ制御ロボットシステムによって達成される。試験化合物の大きなライブラリがスクリーニングされ、および/またはスクリーニングが短時間に達成される必要がある場合、例えば10、20、30、40、50または100の試験化合物の混合物を各ウェルに添加してもよい。一つのウェルが阻害活性を呈する場合、前記の試験阻害剤の混合物は、前記の活性を生じる前記の混合物中の1以上の試験阻害剤を特定するため、簡素化(de-convoluted)してもよい。
【0090】
或いは、1つの試験阻害剤のみをウェルに添加してもよく、ここで各試験阻害剤は種々の濃度で適用される。例えば、試験阻害剤は、種々の濃度で2、3または4つのウェルにおいて試験してもよい。この最初のスクリーニングにおいて、濃度は、例えば10 nMから10μMまでの広範囲をカバーしてもよい。最初のスクリーニングはヒットを見い出すためであり、即ち試験阻害剤が少なくとも1つの濃度で、好ましくは2つ、より好ましくは適用した全ての濃度で阻害活性があることを見い出し、ここで、阻害活性が検出され得る濃度がより低い範囲内である場合、ヒットはより有望である。この代案は、本発明の1つの好ましい態様となる。
【0091】
次に、ヒットと考えられる試験阻害剤は、より広い範囲の阻害剤濃度、例えば10 nM〜20μMの濃度を用いて、さらに試験され得る。これらの測定に適用される方法は以下に記載される。
【0092】
さらに、本発明は、キナーゼ阻害剤のリガンドの結合および/もしくは会合または解離のキネティクスを決定する方法に関し、(a)本発明による蛍光標識キナーゼを種々の濃度の阻害剤と接触させること、または(a)’阻害剤に結合した本発明による蛍光標識キナーゼを、種々の濃度の非標識キナーゼと接触させること;(b)前記の蛍光標識したキナーゼの、1以上の波長での蛍光発光シグナルまたはスペクトルを、各濃度の阻害剤および/または非標識キナーゼについて、励起に際して記録すること;(c)工程(b)で記録された1以上の波長での蛍光発光シグナルまたはスペクトルから、各濃度の速度定数を決定すること、または(c1)各濃度の阻害剤について、工程(b)で記録された1以上の波長での蛍光発光シグナルまたはスペクトルから、Kdを決定すること、または(c2)各濃度の非標識キナーゼについて、工程(b)で記録された1以上の波長での蛍光発光シグナルまたはスペクトルから、Kaおよび逆Kdを決定すること;(d)工程(b)で得られた種々の濃度の阻害剤についてのシグナルまたはスペクトルから工程(c)で決定した速度定数より、直接、konを決定すること、および/または、koffを外挿すること、または(d)’工程(b)で得られた種々の濃度の非標識キナーゼについてのシグナルまたはスペクトルから工程(c)で決定した速度定数より、直接、koffを決定すること、および/または、konを外挿すること;および任意に、(e)工程(d)または(d)’で得られたkonおよびkoffから、Kdおよび/またはKaを計算することを含む。
【0093】
標識キナーゼを種々の濃度の阻害剤と接触させた後、適用した各濃度について蛍光発光を決定することによって、阻害剤の結合親和性を測定し得る。各濃度について、結合および非結合阻害剤の割合は異なるであろうが、これは阻害剤の濃度の上昇を反映し、また前記のキナーゼに対する前記の阻害剤の特異的結合親和性も反映する。
【0094】
反対のアプローチは、その後、阻害剤が結合していない非標識キナーゼにより、結合した阻害剤を含有する標識キナーゼを滴定し得る。
【0095】
化学反応速度論において、速度定数kは、化学反応の速度を数値化する。物質AおよびBが反応してCを産生する場合の化学反応について、反応速度は以下の式を有する:
【数1】

【0096】
式中、k(T)は、温度に依存する反応速度定数である。
[A]および[B]は、それぞれ、物質AおよびBの濃度であり、反応が溶液の容量全体で起こっていると仮定した、溶液の容量に対するモルで表す。
指数mおよびnは次数であり、反応メカニズムに依存する。それらは実験的に決定し得る。
【0097】
また、一段階反応は次の通りにも表し得る:
【数2】
【0098】
Eaは活性化エネルギーであり、Rは気体定数である。温度Tで分子はボルツマン分布によるエネルギーを有するので、Eaより大きいエネルギーを有する衝突の割合は、e-Ea/RTと共に変化することが予測され得る。Aは前指数(pre-exponential)因子または頻度因子である。
【0099】
konおよびkoffは、非共有平衡結合を表す定数である。リガンドが受容体と相互作用する場合、または基質が酵素と相互作用する場合、結合は質量作用の法則に従う。
【数3】
【0100】
この方程式において、Rは遊離受容体の濃度であり、Lは遊離リガンドの濃度であり、RLは受容体-リガンド複合体の濃度である。酵素反応速度論の場合、Rは酵素であるか、または、この場合タンパク質キナーゼであり、Lは基質であるか、または、この場合、候補阻害剤または知られている阻害剤である。会合速度定数konは、M-1秒-1の単位で表される。RL形成速度は、R x L x konに等しい。解離速度定数koffは、秒-1の単位で表される。RL解離速度は、RL x koffに等しい。平衡において、逆(解離)反応は順(会合)反応に等しい。結合の研究では、特異的結合を測定するが、これはRLの指標である。酵素反応速度分析では、酵素の速度を評価するが、これは酵素-基質複合体濃度であるRLに比例する。
【0101】
【数4】
【0102】
平衡解離定数Kdは、モルの単位で表され、koff/konと等しく規定され、以下のようになる:
【数5】
【0103】
解離定数(Kd)は、特定のタンパク質上の結合部位の半分が塞がれた、リガンドの濃度(L)に対応し、即ち、リガンドが結合したタンパク質の濃度(RL)が、リガンドが結合していないタンパク質の濃度(R)と等しい時の、リガンドの濃度に対応する。解離定数が小さいほど、リガンドは強く結合するか、リガンドとタンパク質との親和性が高くなる。
【0104】
従って、会合定数Kaは、逆Kdとも呼ばれ、1/ Kdと規定される。特定のリガンド-タンパク質相互作用の解離定数は、溶液の条件(例えば、温度、pHおよび塩濃度)に伴い有意に変化し得る。
【0105】
本発明の上記の方法では、その後の一連の工程により、KdまたはKaは直接的または間接的に測定し得る。
【0106】
KdまたはKaをそれぞれ直接的に測定するため、工程(b)の後、このタイプの測定では最後の工程である工程(c1)または(c2)を行う。このタイプの測定は終点測定と呼ばれ、添付の実施例でも説明される。速度定数を用いる計算によるKdまたはKaの間接的決定と異なり、蛍光の経時変化よりも、平衡での最終蛍光発光が測定される。これらの測定を使用し、種々の阻害剤濃度(Kd決定のため)または非標識キナーゼの濃度(Ka決定のため)を用いて結合曲線を作成し得る。これらの曲線から、KdまたはKaを直接的に得ることができる。
【0107】
KdまたはKaを間接的に得るため、工程(b)で記録された、1以上の波長での蛍光発光シグナルまたはスペクトルからの速度定数を、工程(c)で実施した各濃度について決定しなければならない。滴定のタイプにより、即ち、標識キナーゼを阻害剤で滴定するか、または阻害剤に結合した標識キナーゼを非標識キナーゼで滴定するかにより、konまたはkoffのいずれかを、測定した速度定数から直接的に決定し得る。konの決定のため、工程(d)が適用されるが、これによりkoffの外挿も可能になる。従って、工程(d)’は、koffの直接的決定に適用され、同様にkonの外挿も可能にする。工程(d)または(d)’で得られるkonおよび/またはkoffから、上記で論じた方程式に従って、Kdおよび/またはKaを計算し得る。
【0108】
上記の方法は、ハイスループット・スクリーニングにも適用し得る。例えば、本発明のキナーゼ阻害剤のスクリーニング方法を用いて、キナーゼに対する阻害活性を有する化合物が特定された場合、本発明の方法は、前記の阻害剤をさらに特性化するために使用し得る。例えば、ハイスループットフォーマットを使用して、多数の異なる濃度の阻害剤(変数(a))または非標識キナーゼ(変数(b))について、1以上の波長での蛍光発光シグナルからKaまたはKdを決定し得る。試験される濃度範囲は、例えば、10 nMから20μMまでであり、評価する濃度の間で1、2および5の繰り返しの連続数(即ち、10、20、50、100、200、500 nM等)である。
【0109】
別の態様において、本発明は、キナーゼ阻害剤の解離または会合を決定する方法に関し、(a)本発明によるスピン標識またはアイソトープ標識したキナーゼを、種々の濃度の阻害剤と接触させること、または(a)’阻害剤に結合した本発明によるスピン標識またはアイソトープ標識したキナーゼを、種々の濃度の非標識キナーゼと接触させること;(b)前記のスピン標識またはアイソトープ標識したキナーゼのEPRまたはNMRスペクトルを、各濃度の阻害剤および/または非標識キナーゼについて記録すること;および(c)種々の濃度の阻害剤について、工程(b)で記録されたEPRまたはNMRスペクトルからKdを決定すること;または(c)’種々の濃度の非標識キナーゼについて、工程(b)で記録されたEPRまたはNMRスペクトルからKaを決定することを含む。
【0110】
蛍光標識キナーゼを用いて反応速度定数を決定することに関して上記でさらに開示した方法と同様に、本発明により、キナーゼと阻害剤との反応の会合または解離定数の直接的測定が可能になる。蛍光標識キナーゼとは異なり、機器の制限およびNMRおよびEPR測定結果の回収に要する時間は、多くの場合、高速スケールの阻害剤結合には適合せず、konまたはkoffの直接的決定はできない。また、キナーゼへの結合に数時間を要する化合物についての決定も可能かもしれない。
【0111】
反応速度論的データの決定に関する本発明の方法は、ハイスループットフォーマットにも適用し得る。例えば、上述の本発明のスクリーニング方法で特定される潜在的阻害剤は、種々の濃度の前記の阻害剤をキナーゼに適用してKdを決定して、さらに特性化し得る。阻害剤の好適な濃度範囲は10 nMと20μMとの間であるが、制限されない。
【0112】
ハイスループットフォーマットに適用されるより集中的な濃度範囲でkonおよびkoffを決定することにより、例えば、添付の実施例において実施した通り、キュベットアプローチおよびリアルタイム反応速度測定を用いて、より高感度でKd測定値が得られる。
【0113】
さらに本発明は、キナーゼ阻害剤のスクリーニングに好適な突然変異キナーゼの産生方法に関し、(a)関心のあるキナーゼにおいて存在する場合、P-ループの外部に遊離チオールまたはアミノ基を有する、溶媒にさらされているアミノ酸、または、P-ループ内部の好適でない位置に遊離チオールまたはアミノ基を有するアミノ酸を、遊離チオールまたはアミノ基を有しないアミノ酸で置換すること;(b)P-ループ内に遊離チオールまたはアミノ基を有するアミノ酸が存在しない場合、前記の関心のあるキナーゼのP-ループにおけるアミノ酸を突然変異して、遊離チオールまたはアミノ基を有するアミノ酸とすること;(c)関心のあるキナーゼを、その環境における極性変化に感受性のあるチオール-またはアミノ-反応性フルオロフォア、チオール反応性スピン標識、アイソトープまたは濃縮アイソトープチオール-またはアミノ-反応性標識で標識することであって、前記のフルオロフォア、スピン標識、アイソトープまたは高アイソトープ標識は、キナーゼの触媒活性を阻害せず、および/または、キナーゼの安定性を妨げない;(d)工程(c)で得られたキナーゼを、前記のキナーゼの知られている阻害剤と接触させること;および(e)工程(c)および(d)の前記の蛍光標識キナーゼの、励起の際の、1以上の波長での蛍光発光シグナルまたはスペクトルを記録すること、または(e)’工程(c)および(d)の前記のスピン標識キナーゼのEPRまたはNMRスペクトルを記録すること;および (f) 工程(e)で記録された1以上の波長での蛍光発光シグナルまたはスペクトル、または、工程(e)’で記録されたEPRまたはNMRスペクトルを比較することを含むが、ここで、工程(e)で得られる、少なくとも1つの波長、好ましくは発光極大での蛍光強度の差、および/または、前記の蛍光標識キナーゼのスペクトルにおける蛍光発光波長のシフト、または、工程(e)’で得られる前記のスピン標識またはアイソトープ標識キナーゼのEPRまたはNMRスペクトルの変化から、該キナーゼがキナーゼ阻害剤のスクリーニングに好適であることが示される。
【0114】
ハイスループットフォーマットに適合させて、多数のキナーゼまたは同一のキナーゼに異なる標識をした変種をスクリーニングし得る。
【0115】
本発明による「好適ではない位置」という用語は、本発明の標識したアミノ酸に好適ではないことが示された、P-ループにおける位置を示す。このことは、その環境変化に対する標識の感受性が低下しているため、または、前記の位置により標識を有するキナーゼの感受性が低下するという構造的考察に基づく予測のためであり得る。またこの用語は、潜在的に好適な位置にあるアミノ酸も包含し、ここでは、異なる位置がより適当であると考えられる。P-ループにおいて遊離チオールまたはアミノ基を有するアミノ酸の数が1を超えた場合は即座に、好適ではないとみなされるアミノ酸を変異させるべきである。
【0116】
アミノ酸を変異させることとしては、この変異により得られるキナーゼの触媒活性を阻害せず、または安定性を妨げない場合に、前記のアミノ酸を別のアミノ酸で置換することまたは取り除くことが挙げられる。工程(b)は、遊離チオールまたはアミノ基が関心のある前記のキナーゼのP-ループに存在しない場合に、実施される。挿入されるか別のアミノ酸を置換するアミノ酸は、標識化するため、遊離チオールまたはアミノ基を有していなければならない。
【0117】
本発明の方法の好ましい態様において、キナーゼ阻害剤は、キナーゼのATP結合部位に隣接するアロステリック部位に排他的に結合するか、または、アロステリック部位からATP部位中に伸長する。これらのタイプの阻害剤は、それぞれIII型またはII型阻害剤とも呼ばれる。これらの阻害剤は、キナーゼのATP-ポケットに結合するI型阻害剤と比較して、より高い特異性を有するキナーゼに結合するが、ATP-ポケットは全てのキナーゼにおいて、構造中に高度に保存されている。
【0118】
実施例において実証される通り、本発明は、ATP-競合的およびATP-非競合的阻害剤を差別化し、特異的阻害剤の迅速な選択を可能にする手段を提供する。本発明は、キナーゼのP-ループの運動を検出するために設計されるが、これは活性化ループの運動によって生じ、従って全てのII型およびIII型阻害剤に感受性がある。さらに、特異的結合形態(上記参照)を有するか、DFG-in構造においてキナーゼによりP-ループと直接的に相互作用する、あるI型阻害剤が検出される。結合速度が決定される蛍光の経時変化の測定(即ち、終点測定ではない)のみが、I型阻害剤をII型およびIII型阻害剤と区別することを可能にする。以下の実施例の一つにおいて示した通り、検出されたATP-競合的阻害剤は、瞬間的な蛍光変化(典型的には<5秒)を生じ、一方、II型およびIII型阻害剤は、はるかにゆっくり結合する(数秒から数分)。
【0119】
本発明のキナーゼまたは方法の別の好ましい態様において、キナーゼは、天然に存在するか、またはP-ループ中に導入されたシステインで標識される。
【0120】
タンパク質におけるシステインの量は通常は非常に少ないため、本発明のキナーゼは、P-ループ中のアミノ酸をシステインで置換し、かつ、任意に、溶媒にさらされているシステインを他のアミノ酸で置換することにより、直接的に製造し得る。ヒスチジン、アルギニンまたはリシンまたはそれらの誘導体等の反応性アミンを含有するアミノ酸は、はるかに豊富に存在し、周囲の溶媒と接触しているタンパク質表面で容易に見つかる。従って、導入されたシステインと特異的に反応し得る、チオール反応性標識を使用することが好ましい。
【0121】
より好ましい態様において、キナーゼ阻害剤のスクリーニング方法または変異キナーゼの産生方法は、さらに、工程(c1):工程(c)で記録された2つの波長の蛍光強度比を測定し、正規化した強度変化の平均強度変化(ΔIstd)に対する割合を得ることを含む。加えて、または、代わりに、本発明により標識したキナーゼの阻害剤が結合したものと阻害剤を有しないものとの間の、最大標準強度変化(ΔRmax)を評価してもよい。(ΔIstd)が>0.25であるか、および/または、(ΔRmax)が>0.75あり、Z因子が>0.5である場合、候補化合物はキナーゼ阻害剤と見なされるか、または、蛍光標識したキナーゼは、キナーゼ阻害剤のスクリーニングに好適であると見なされる。上述の通り、この態様は、本発明の方法のハイスループットスケールへの拡張に関する。
【0122】
ΔIstdは、蛍光発光の平均強度に対する、正規化した強度変化の割合である。ロリミエル(Lorimier)ら(2002年)によれば、ΔIstdは、高感度の蛍光分光法に好適な蛍光タンパク質複合体を特性化する、最も重要な基準の1つである。理想的には、ΔIstdの値は>0.25であるべきであり、以下によって計算される:
【数6】
ここで、λstd = (λmax, 非結合 + λmax, 飽和)/2であり、I1、I2はそれぞれ、各スペクトルのλstdでの蛍光強度である。
【0123】
ΔRmaxは、飽和および不飽和キナーゼの間の、蛍光発光の最大標準強度変化である。ドゥ・ロリミエル(De Lorimier)ら(2002年)によれば、ΔRmaxは、高感度の蛍光分析法に好適な蛍光タンパク質複合体を特性化する、別の重要な基準である。理想的には、ΔRmaxの値は>1.25であるべきであり、以下によって計算される:
【数7】
ここで、oA1、oA2はリガンドが存在しない領域であり、∞A1、∞A2は飽和しているリガンドが存在する領域である。コンピュータプログラムを使用して、2つのスペクトルにおける波長バンドの全ての可能なペアについて、ΔRを列挙して、最適な検知条件を特定し、最大値をΔRと規定し得る。
【0124】
Z因子は、ハイスループット・スクリーニング(HTS)分析の質および検出力の統計的尺度である。HTS作戦では、未知のサンプルの多数の単一測定値を確立した陽性および陰性対照と比較して、もしあるとすれば、単一測定値のいずれが陰性対照と有意に異なるかを決定する。大規模なスクリーニング作戦を開始する前に、多くの作業を行って、より小規模の分析の質を評価し、その分析がハイスループットの設定で有用であるかどうかを予測する。Z因子は、分析を何百万ものサンプルにスケールアップした場合に、有用なデータが期待できるかどうかを予測する。Z因子は以下により計算される:
【数8】
ここでは、陽性(p)および陰性(n)対照の両者の、平均(μ)および標準偏差(σ)の両者(それぞれ、μp、σp、μn、σn)が考慮される。
【0125】
ΔIstdおよびΔRmaxの測定、ならびにZ因子の決定により、選択された標識が阻害剤のスクリーニングに好適かどうかの決定に有用であることが判明するかもしれない。ドゥ・ロリミエル(De Lorimier)は、蛍光タグ付けされたタンパク質を用いて得られた、測定した反応速度およびKdが、使用したタンパク質、リガンドおよびフルオロフォアに依存することを論じている。従って、同一のキナーゼに結合する同一の阻害剤は、使用される標識によって、Kd値が異なる可能性がある。上記の値の決定により、選択された標識が好適であるかどうか、または、異なる標識を使用すべきかどうかが示唆される可能性がある。
【0126】
さらに好ましい態様において、フルオロフォアまたはスピン標識は、標識キナーゼ中に存在することが知られているか、または予測されているリン酸化部位に、またはそのリン酸化部位に隣接して、位置しない。このことは、標識化が、P-ループの動力学または通常の活性を妨げず、かつ、リン酸化および脱リン酸化によって大きく影響されるキナーゼの調節を妨げないことを確実にする。
【0127】
別の好ましい態様において、P-ループにおける前記の候補アミノ酸は、前記のキナーゼについて入手できる構造および/または配列データに基づいて特定される。
【0128】
あるキナーゼについて、例えば結晶形態の構造データまたはNMR構造は入手可能であり、ここで、キナーゼは、活性化および/または不活性化状態で捕えられる。このようなデータがキナーゼについて入手可能である場合、これにより、標識化の目的で置換される、P-ループ中のアミノ酸の位置の選択が容易になる。実際の選択は、ほとんどの場合TyrまたはPheであるG-X-G-X-X-Gモチーフの3番目のGlyの前の位置での、リガンドの結合および/または立体構造変化に応答して、どの残基がP-ループにおいて最大の変動を示す傾向があるかに基づく。非常に少ないケースであるが、この残基がTyrであり、特定のキナーゼにおいてリン酸化されることが知られている場合、この位置の標識化は、キナーゼ活性を妨害すると思われ、この位置は本発明において好適でなくなる。同様に、前記の位置でのアミノ酸の他のアミノ酸との接触も試験する。前記の接触がキナーゼの触媒活性および安定性に必須であると思われる場合、殆どの場合、この位置は置換に好適ではない。加えて、アミノ酸位置の選択は、特定のアミノ酸がタンパク質の立体構造変化に伴って変動する距離に基づいて行われ、距離が大きくなると、環境の変化が検出される機会が増えるようにする。しかし、変動距離は、特定の位置が標識化に有用かどうかの指標であるが、結合した標識によって検出される、観察される変化と直接相関する、環境における実際の変化である。
【0129】
好ましい態様において、阻害剤のスクリーニングに関する本発明の方法は、会合および解離等の反応速度論的パラメータの決定と変異したキナーゼの産生とを組み合わせて、種々のキナーゼについての特異的阻害剤を得るための直接的方法論を得る。この点で、本発明の方法の全ての好ましい態様は、本発明の他の方法の態様と組み合わせてもよい。この様相のより好ましい態様において、初期のスクリーニングは、キナーゼ阻害剤のハイスループット・スクリーニング方法を用いて実施し、その後、リガンドの結合および/もしくは会合または解離のキネティクスを決定するための本発明の方法で、上述の通り、広範な濃度範囲の阻害剤を用いてスクリーニングする。後者の工程は、とりわけ、指標であるKdおよび/またはKa値を得るために実施する。この工程は、KdまたはKaをより正確に測定するため、濃度範囲をより絞って測定を実施することにより、再度繰り返す。各阻害剤をさらに特性化するため、これらの測定は、キュベットアプローチを用いた滴定系列として、および/または、キュベットにおけるリアルタイム反応速度測定(konおよびkoff)として実施してもよい。代わりに、または、加えて、キナーゼ阻害剤として特定される化合物の結合形態は、関心のあるキナーゼとの複合体において化合物を結晶化することによってさらに特性化してもよく、これは詳細を最も明瞭に示す。任意に、この一連の方法は、他のキナーゼまたは異なる標識をした同一のキナーゼに委ねられる。この態様は、ハイスループット・スクリーニングが、多数のキナーゼまたは同一のキナーゼに異なる標識をした変異体について、大多数の阻害剤をスクリーニングおよび特性化できるように設計される。
【0130】
より具体的には、このような組み合わせ方法は、キナーゼのATP結合部位に隣接するアロステリック部位に、部分的または完全に結合するキナーゼ阻害剤の特定方法であって、(a) 本発明のキナーゼ阻害剤のスクリーニング方法に従って、阻害剤をスクリーニングすること、および(b) 工程(a)で特定された阻害剤のキナーゼに対する速度定数を決定することを含み、ここで、工程(b)で決定された<0.140秒-1の結合速度は、特定されたキナーゼ阻害剤が、キナーゼのATP結合部位に隣接するアロステリック部位に部分的または完全に結合することを示す。>0.140秒-1の速度定数は、特定されたキナーゼ阻害剤が、ATP結合部位において結合し、隣接するアロステリック部位中に伸長しないことを示す。速度定数は、反応時間(結合速度)t1/2: t1/2 = ln(2)/kobsに相関する。従って、<0.140秒-1の速度定数(kobs)は、>5秒の反応時間t1/2に対応する。
【0131】
速度定数または結合速度は、好ましくは、本発明の標識キナーゼの特性を使用して決定する。例えば、本発明のキナーゼは阻害剤と接触し、標識によっては、1以上の波長での蛍光標識キナーゼの蛍光発光シグナル、または、スピン標識またはアイソトープ標識したキナーゼの電子常磁性共鳴または核磁気共鳴スペクトルを、経時的に記録し得る。これは、本発明のキナーゼ阻害剤のリガンド結合および/もしくは会合または解離のキネティクスの決定方法の工程(a)〜(c)、または、本発明のキナーゼ阻害剤の解離または会合の決定方法の工程(a)および(b)に対応する。結合速度、即ち、蛍光、またはNMRまたはEPRスペクトルにおける測定可能な変化が、阻害剤の適用後5秒より大きい場合、このことは、阻害剤がII型またはIII型阻害剤であることを示す。
【0132】
キナーゼ阻害剤のスクリーニング方法の別の好ましい態様において、この方法は、さらに、(その後)前記のキナーゼの阻害剤として特定された候補化合物の薬理学的特性を最適化することを含む。
【0133】
一般にリード化合物と言われる、スクリーニングにおいて特定される化合物の薬理学的特性の最適化方法は、当業界において知られており、リード化合物として特定される化合物の、以下を達成するための修飾方法を含む:(a)作用部位、活性スペクトル、器官特異性を修飾する、および/または(b)効力を向上させる、および/または(c)毒性を減じる(治療指数を向上させる)、および/または(d) 副作用を減じる、および/または(e)治療効果の発現、効果持続期間を修飾する、および/または(f)薬物動態パラメータ(吸収、分布、代謝および排泄)を修飾する、および/または(g)物理化学的パラメータ(溶解性、吸湿性、色、味、臭気、安定性、状態)を修飾する、および/または(h)全般的特異性、器官/組織特異性を向上させる、および/または(i)適用形態および経路を以下によって最適化する、a. カルボキシル基のエステル化、またはb. ヒドロキシル基のカルボン酸によるエステル化、またはc. ヒドロキシル基の、例えばリン酸エステル、ピロリン酸エステルまたは硫酸エステルまたはヘミコハク酸エステルへのエステル化、またはd. 薬学的に許容される塩の形成、またはe. 薬学的に許容される複合体の形成、またはf. 薬理学的活性ポリマーの合成、またはg. 親水性部分の導入、またはh. 芳香環または側鎖上の置換基の導入/交換、置換パターンの変更、またはi. 等比体積または生物学的等価部分の導入による修飾、またはj. 同族化合物の合成、またはk. 分枝状側鎖の導入、またはl. アルキル置換基の環状アナログへの変換、またはm. ヒドロキシル基のケタール、アセタールへの誘導化、またはn. アミド、フェニルカルバメートへのN-アセチル化、またはo. マンニッヒ塩基、イミンの合成、またはp. ケトンまたはアルデヒドの、シッフ塩基、オキシム、アセタール、ケタール、エノールエステル、オキサゾリジン、チアゾリジンまたはそれらの組み合わせへの変換。候補化合物の修飾の前に、候補化合物の関心のあるキナーゼへの正確な結合形態を特性化し、キナーゼへの阻害特性に対する、ある化学修飾の候補化合物への効果を予測するために、(i) 既に知られている結晶構造中への提案分子のドッキング、(ii)相同モデル(標的タンパク質に最も近い相同体の知られている結晶構造に基づいて産生されたモデル構造)中への提案分子のドッキング、および/または(iii)キナーゼ阻害剤複合体の結晶化等の、いくつかの分析/予測ツールを適用してもよい。
【0134】
候補化合物にもたらされる修飾は、その後、上記に列記したあらゆる技術によって再度、さらに分析してもよく、その結果、多数のそのようなサイクルの後、候補化合物の特性が最適化されている。
【0135】
上記で挙げた種々の工程は、当業界で一般的に知られている。それらは、さらに、定量的構造活性相関(QSAR)分析(クビニイ(Kubinyi)、"Hausch-Analysis and Related Approaches", VCH Verlag, Weinheim, 1992年)、コンビナトリアル生化学、古典的化学等を包含するかそれらに基づく(例えば、ホルツグラーベ(Holzgrabe)およびベフトルド(Bechtold)、Deutsche Apotheker Zeitung 140(8), 813-823頁, 2000年参照)。
【0136】
図1、p38αにおけるリガンド結合によって引き起こされる、P-ループおよび活性化ループの立体構造変化の略図。a) 活性化キナーゼドメイン(DFG-in)の構造的に重要な領域(P-ループ;へリックスC;ヒンジ領域)が標識される。b) 活性化ループへの共鳴における、活性化ループの移動性。II/III型阻害剤は、DFG-outキナーゼ立体構造にのみ存在する部位をふさぐ。このアロステリックポケットの側面には、DFG-モチーフおよびヘリックスCが配置されている。アロステリック部位に結合するII/III型阻害剤は、活性化ループ(黒)の立体構造変化を引き起こし、P-ループ(黒)がより伸長した立体構造を採用し得る。c) Cys残基は、その後環境感受性フルオロフォア(大型球体)で標識するため、p38αのP-ループ中で変異して、感受性P-ループ結合アッセイを作出する。キナーゼの活性(DFG-in)および不活性(DFG-out)構造は平衡状態にあり、これらは活性化ループの構造変化により生じる。活性化ループの構造変化は、疎水性界面を通ってP-ループに移り、P-ループに結合するフルオロフォアの化学的環境を変化させる。I型阻害剤(表面が、左パネルの大型球体の背部)は、活性化キナーゼ(DFG-in)のヒンジ領域に結合する(左パネル)。この特別の場合において、P-ループは折り重なり、阻害剤と直接相互作用する。活性化キナーゼ(DFG-in)由来のリガンドの非存在下で、P-ループは、より伸長した立体構造を採る(中央パネル)。II/III型阻害剤(表面が、右パネルの大型球体の下部)はキナーゼの不活性(DFG-out)構造に結合する。
【0137】
図2、高グリシンループ上に標識したac-p38αを用いたリアルタイムおよび終点蛍光測定。475 nmでのアクリロダン発光は、BIRB-796の結合により減少して、結合状態における最大放出波長が赤方偏移(より長波長へのシフト)する(A)。終点平衡測定はKdを直接得るために実施し得る。レシオメトリック蛍光データ(R = 512 nm/475 nm)を阻害剤濃度の対数目盛りに対してプロットして、Kdを得た(B)。レシオメトリック蛍光データ(R = 445 nm / 475 nm)もまた、Kdを得るために使用し得る(データは示さず)。蛍光痕跡もまた、単一波長(475 nm)でリアルタイム測定して、様々な反応速度定数を決定し得る。種々の量のBIRB-796(大きな矢印)から生じる蛍光減衰は、一次減衰関数に適合して(灰色線)kobsを得た(C)。その後、実験的に決定したkobs値をプロットして、BIRB-796の全てのリガンドについてkonを決定し得る。過剰の非標識p38αを用いてac-p38αからBIRB-796を抽出することにより、一次関数に適合する(灰色線)koff が直接決定可能である(D)。上記に表したデータは、高グリシンループ上に標識したac-p38αを用いてBIRB-796について実施した、典型的な1セットの実験を代表する。
【0138】
図3、P-ループac-p38αを用いたIII型およびI型リガンドのリアルタイムおよび終点蛍光測定。475 nmでのアクリロダン発光は、III型リガンドRL36の結合により減少して、結合状態における最大放出波長が赤方偏移する(a)。P-ループと相互作用することによるp38αのDFG-out立体構造を安定化することが知られている、I型リガンドであるSB203580について、類似であるがより強い変化が観察された(b)。III型およびII型リガンド(図2参照)は、高グリシンループの関連する構造転位を生じる、活性化ループの立体構造変化を引き起こすことから(図1a参照)、スペクトルの形状および強度の損失の両者は、両方の型の阻害剤について同等の変化をする。蛍光痕跡を単一波長(475 nm)でリアルタイム測定して、リガンドの結合および解離速度を決定した。100 nMのRL36の添加(黒の矢印)により生じる蛍光減衰は、一次減衰関数に適合して(灰色線)kobs.onを得た((a)中央)。SB203580等のI型リガンドは、典型的には<5秒に結合し((b)中央)、正確な曲線の一致は、測定の時間分解能を増すストップフロー蛍光分光法を使用しないと不可能である。ac-p38αからの各阻害剤の抽出は、過剰の非標識p38αを同じサンプルに添加することにより達成された(白色矢印)。koffは、全ての型の阻害剤について、kobs,onより有意に遅いことが知られているので、蛍光の増加を一次関数に適合させる(灰色線)ことにより、各阻害剤についてkoffを決定することが可能であった。レシオメトリック蛍光データ(R = 512 nm / 475 nm)を阻害剤濃度の対数目盛りに対してプロットして、RL36((a)右)およびSB203580((b)右)についてのKdを得た。II型阻害剤であるイマチニブはp38αに結合しないので、RL36の陰性対照とした((a)右、黒色四角)。I型阻害剤であるダサチニブはp38αに結合するが、高グリシンループと相互作用しないので、SB203580の陰性対照とした((b)右、黒色四角)。上記に示したデータは、高グリシンループ上に標識したac-p38αを用いて実施した、典型的な1セットの実験を代表する。
【0139】
図4、P-ループを標識したp38αの阻害剤の種々の型に対する蛍光特性評価および応答。化合物スクリーニングにおいて特定された2つのヒットであるScios-469およびRL40の構造に加えて、種々の知られている阻害剤型(I、IIまたはIII型)の構造を示す。P-ループは、チオール反応性フルオロフォアであるアクリロダンでp38αのY35Cを共有結合的に修飾することによって標識し、変化する蛍光特性をp38αの知られているDFG-outおよびDFG-inバインダーの結合に際して試験した。理想的フルオロフォア-タンパク質複合体と見なされる基準(ドゥ・ロリミエル(deLorimier)ら、2003年)を満たすΔRmaxおよびΔIstdの値は全て、太字テキストで表す。古典的なDFG-outバインダー(II型およびIII型阻害剤)またはP-ループと直接的に相互作用するいくつかのI型阻害剤の場合、アクリロダンは大きな発光シフトを起こすが、〜475 nmと比べて〜512 nmにおいて発光の増加があり、最適ではないΔRmaxにもかかわらず、なお信頼性の高い結合曲線が測定可能である。しかし、これらの同一の型の阻害剤の場合、優れたΔIstd値が得られた。SB203580は、DFG-out構造を安定化することが知られている、p38αのI型阻害剤である(欧州特許EP 08 02 0341号に開示の通り)。ダサチニブはキナーゼのヒンジ領域に結合し、DFGモチーフまたはP-ループと相互作用せず(トカルスキ(Tokarski)ら、2006年)、このアッセイ系では検出されなかった(ND)。SB203580等のI型阻害剤およびDFG-out結合II型(BIRB-796)およびIII型(RL36)阻害剤は、高感度で検出され、Kdおよび反応速度測定を可能にする。反応速度測定により、非常に速く(本実施例において<2秒)結合するI型阻害剤を、p38αに対しゆっくり結合することが知られているII/III型リガンド(パルゲリス(Pargellis)ら、2003年)から、区別することが可能になる。このような2つのリガンドである、RL40およびScios-469は、スクリーニング計画において検出された。これらの2つのリガンドの検出の背後にある構造の詳細を理解するために、その後、タンパク質X線結晶学が利用された。[注:* ΔIstdは、リガンドの存在下および非存在下で445 nmおよび475 nmでの発光強度を用いて計算し(R = 445 / 475 nmはリガンド結合の検出に最適である);** ΔRmaxは、リガンドの存在下および非存在下で475 nmおよび512 nmでの発光強度を用いて計算した(R = 512 / 475 nmは結合形態の判別に最適である)。]
【0140】
図5、RL40、Scios-469およびCP547632の結晶構造は、P-ループの運動を裏付ける。p38αとの複合体(a)中のRL40の構造から、以前に報告された(欧州特許EP 08 02 0341号)SB203580の構造において観察されたものと類似の、独特で予期されなかった結合形態が明らかになるが、ここで、リガンドは、DFGモチーフのPhe側鎖で独特のπ-πスタッキングを形成することにより、P-ループと相互作用する。この相互作用の結果、DFG-out構造が安定化する。p38αにおけるSB203580およびRL40の構造の重なりにより、両阻害剤の芳香核が良好に重なり合い、P-ループおよび活性化ループと同一の型のスタッキング相互作用を形成することが明らかになる(b)。RL40のアナログは、典型的には、キナーゼのヒンジ領域に結合することが観察され、P-ループとは相互作用せず(ピエルセ(Pierce)ら、2005年)、従ってこれらの独特な結合形態を利用するリガンドを増すという、P-ループ標識キナーゼを使用する利益が強調される。加えて、P-ループ標識キナーゼアッセイは、Scios-469の結合を強力に検出した。我々は、Scios-469を野生型p38αと共結晶化し、構造を2.5Åの分解度で解明した(c)。我々は、p38αのアポ構造と比較し、P-ループにおける劇的な運動を観察した。この運動は、化合物の疎水性特徴によるP-ループTyr35(アッセイのために選択された標識位置)のスタッキング相互作用により誘導され、かつ安定化される。このことから、P-ループ標識キナーゼアッセイが、どのように、P-ループの立体構造を直接的に変化させるいくつかのI型リガンドを高感度に検出し得るかという例が提供される。(d)Scios-469のピペラジン環に結合するカルボニルは、ヒンジ領域(ピンク)への2つの水素結合を形成する(赤色破線)(Met109およびGly110の骨格NH)。高グリシンループ(緑)は折り重なって、阻害剤と直接相互作用し、インドール部分およびピペリジン環を溶媒から保護する。DFG-モチーフ(オレンジ)は、ATP結合部位に向かうAsp168を有する”in”構造である。(e)Scios-469と同様に、CP547632のハロゲン置換メトキシベンゼンはゲートキーパー(Thr106)にかぶさるように曲がり、疎水性サブポケットに向かう。チアゾール環に結合するカルボキシアミドおよび尿素は両者とも、ヒンジ領域への水素結合を形成する(His107の骨格CO、Met109のNHおよびCO)。ピロリジン-ブタン部分は90°ねじれて、溶媒からATPポケット方向に向かう。高グリシンループは、電子密度中で見えにくく、DFG-モチーフは明らかに”out”構造である。
【0141】
図6、野生型の非標識およびアクリロダン標識p38αの反応速度論的および阻害特性評価。注:反応速度論的パラメータは、シスビオ(Cisbio)のHTRF登録商標アッセイを使用して決定したが、これにより、導入された変異(p38αにおけるCys119Ser/Cys162Ser/Tyr35Cys)はキナーゼのATPに対する親和性(ATP-Km)を有意に変化させないことが実証される。各変異体のKmで実施したIC50の比較では、2、3の知られているI型およびII型p38α阻害剤のIC50に対し、変異または標識化の有意な効果は見られず、従って、本発明の標識化アプローチのために選択された高グリシンループ標識部位の正当性が立証される。報告された全ての値は、それぞれ2回実施した、独立した実験で、平均± s.d.が少なくとも3である。
【0142】
図7、384穴フォーマットにおいて測定された、p38αによるBIRB-796(1)のKd値の時間依存性。I型阻害剤であるSB203580、Scios-469およびCP547632の結合曲線、および、結合速度の遅いII型阻害剤であるBIRB-796の結合曲線を、p38αを使用して得て、阻害剤結合形態が、結合のリアルタイム反応速度の測定に加えて(図2参照)、HTSフォーマットにおいて予測され得ることが実証された。各リガンドについて、ある濃度範囲にわたり、5、30、90および300分にレシオメトリック蛍光(R = Iλ512 / Iλ475)を測定してプロットし、各時点での各リガンドのKdを決定した。SB203580、Scios-469およびCP547632のKdは、室温で高グリシンループを標識したp38αと共に5分間インキュベーションした後、有意に変化しなかった。BIRB-796のKdは、90分間に〜3倍低下した。室温での90分のインキュベーション時間は、II型阻害剤がキナーゼとの結合平衡に達するのに充分であった。384穴フォーマットにおいて決定されたKd値は、キュベットフォーマットで測定された場合(表1参照)より2〜3倍高かったが、これは、より高いDMSO濃度およびHTSプレートでのスクリーニングのための洗浄剤の添加に起因することが多い。報告された全てのKd値は、それぞれ三回実施した、独立した実験で、平均± s.d.が4である。
【0143】
図8、高グリシンループを標識したMKK7を用いたII型およびI型リガンドのリアルタイムおよび終点蛍光測定。(A) K252aの結合により、標識タンパク質の蛍光強度、および2つの波長(R = 472 nm / 510 nm)におけるレシオメトリック発光の検出可能な変化が減少する。(B) Kdを直接決定する終点方法を用い、これらの発光の割合を阻害剤濃度に対してプロットして、K252aについて38 nMのKdを得たが、これは、これらの化合物について予測した正確な範囲内である(カラマン(Karaman)ら、2008年)。陰性対照として、10 μM以下では検出されない、II型阻害剤であるソラフェニブを含有させた。これらの知見はMKK7について予測される結果と一致しており、このことは、DFG-out構造および、DFG-out構造を誘導または安定化する阻害剤に対する非感受性を示す(カラマンら、2008年)。アッセイ応答がI型阻害剤結合に際するP-ループの運動によることを実証するため、ダサニチブもまた陰性対照として含有させた。ダサニチブは、ATP競合的阻害剤であるか、cSrcおよびAblキナーゼであり、MKK1およびMKK2のみを阻害するが、報告されたKd値は> 1 μMである(カラマンら、2008年)。従って、このI型阻害剤の添加および検出は、MKK7については予測されなかったが、このことはデータで確認される(C)。K252aの結合および解離のリアルタイム反応速度論的測定および検出。p38αについて図3に示す通り、結合に伴って生じる蛍光変化は、標識キナーゼからリガンドを抽出するための過剰の非標識MKK7の添加に際して可逆的である。K252aはI型阻害剤であるため、図3Bに示すp38αのI型阻害剤SB203580のように、これらのプロセスの反応速度は速い。
【実施例】
【0144】
本発明を実施例によって説明する。
実施例1:好適なキナーゼの選択
我々は、このアッセイを展開するため、以下の理由からp38αを扱うことを選択した:i) 入手できる構造的情報が豊富であること、ii) その活性および不活性立体構造の両者の結晶構造が入手できること(図1A)およびiii) 強固に結合するII型およびIII型アロステリック阻害剤が入手できること。第一の工程において、p38αの結晶構造を綿密に調査して、アロステリックバインダーを検出する可能性のある、好適なフルオロフォア結合部位を特定した。この変異の候補残基は、ミカエル付加によるフルオロフォアの結合を可能にするため、溶媒にさらされなければならず、リガンドの結合に際して、有意な運動を呈する。また、タンパク質安定性、触媒活性の維持に非常に重要な残基、または、知られているリン酸化部位に近接する残基を選択しないように注意した。
【0145】
P-ループにおいて一つの位置(Tyr35)を選択し、その後、システイン残基に変異させた(図1B、C)。フルオロフォアとしてアクリロダンを選択したが、これは、相対的にサイズが小さいこと(トリプトファン側鎖と同等)、極性変化に対して感受性が高いこと、市販されていること、および相対的に安価であることによる。また、アクリロダンは、強い応答を生じることが知られており、アロステリック阻害剤の結合に際して、活性化ループの運動を検出するはずである(図1D)。タンパク質を標識する前に、フルオロフォアが、あらゆる他の溶媒にさらされるシステイン残基に結合する機会を減じることが必要であった。再度、構造的情報を用いて、p38α中に4つの還元システイン残基を配置した。これらのシステインのうちの2つはタンパク質中に埋め込まれ、他の2つは溶媒にさらされて保存的にセリンに変異させた。最後に、F327L変異を組み込んで、要すれば、酵素活性アッセイに使用するためアクリロダン標識したp38α(ac-p38α)を部分的に活性化した(アスカリ(Askari)ら、2007年;アヴィゾウア(Avitzour)ら、2007年)が、これはアッセイ自身の機能性には必須ではない。
【0146】
リン酸化した活性p38α変異体それぞれ(変異/非標識およびアクリロダン標識)のATP-Kmを測定し、野生型p38α(図6)と比較する、活性に基づくアッセイを用いることにより、チロシンの同等の大きさのアクリロダンで標識したシステインでの置換は、キナーゼ耐性が良好であることが示された。同様に、3つの知られているp38α阻害剤のIC50値について、高グリシンループの変異または標識化に際し有意な変化は観察されなかったが、このことは、本発明が、最終的には、検出されたリガンドについて、欧州特許EP 08 01 3340号および欧州特許EP 08 02 0341号に記載されたアッセイと類似の親和性データを提供することを実証する。
【0147】
実施例2:タンパク質標識化および蛍光特性化
タンパク質標識化
全部で4つの変異(システイン2つ→セリン、および標識化のためシステイン1つを導入)を含む、p38α構築物を、BL21(DE3)大腸菌株へ形質転換し、過剰発現させ、アフィニティクロマトグラフィー、アニオン交換クロマトグラフィーおよびサイズ排除クロマトグラフィーで精製し、その後、純粋なタンパク質を標識化に使用した。タンパク質および遊離アクリロダンを1:1.5の割合で混合し、4℃の暗所で一晩反応させた。接合したタンパク質(ac-p38α)を濃縮し、分割して、-20℃で凍結した。タンパク質の100%のモノ標識化をESI-MSによって確認した。正確に標識したシステインの確認は、現在は、非標識および標識p38αのトリプシン断片を分析し、その後HPLCとESI-MSまたはMALDIの組み合わせによって行われている。
【0148】
蛍光特性化
標識化に続いて、プローブの蛍光的性質を特性化し、ピラゾロ尿素系II型アロステリック阻害剤であるBIRB-796の様々な誘導体を用いて最初の実験を実施した(パルゲリス(Pargellis)ら、2002年;ドゥマス(Dumas)ら、2000年(aおよびb);モス(Moss)ら、2007年;レーガン(Regan)ら、2002年;レーガンら、2003年)。P-ループを標識したac-p38タンパク質は、リガンド結合に伴い、475 nmから512 nmまで、わずかな赤方偏移を示す(図2A)。2つの波長(R = 512 nm / 475 nm)の割合を測定することにより種々のサンプルの間の希釈誤差を除き得る(図2B)。これら2つの波長を用いて、平均のZ因子は0.53 ± 0.04と計算し得る。同様に、445 nmに存在する小さな極大点は、リガンドの結合に対して相対的に感度が低く、その結果、2つの波長(R = 445 nm / 475 nm)の割合もまたこの目的に使用し得る。これらの2つの波長を用いて、平均のZ因子は0.85 ± 0.05と計算し得る。475 nmでの大きな変化によっても、単一波長での反応速度測定の可能性が認められる(図2C-D)。
【0149】
512 nmおよび475 nmの波長を用いて、平均強度(ΔIstd)と比較した、正規化強度変化を決定したところ、全ての検出した阻害剤の型について0.55〜1.57の間の範囲であり、飽和および不飽和ac-p38αの間の最大標準強度変化(ΔRmax)は、全ての検出した阻害剤の型について0.23〜0.56の間の範囲であった。これらは、蛍光分光法の最も重要な基準のうちの2つである(ドゥ・ロリミエル(de Lorimier)ら、2002年)。2つの異なるレシオメトリックな測定値について決定されたZ因子とともに、ΔIstd値は、これを蛍光アッセイにおける使用に好適なプローブと特性化する。
【0150】
実施例3:終点および反応速度論的測定 − 方法
本発明の標識化戦略を特性化するため、高グリシンループをアクリロダンで標識したp38α(50 nM)を、キナーゼのDFG-inまたはDFG-out構造に特権的に結合することが一般に知られている、骨格に基づく小さなサブセット(〜400)の化合物に対してスクリーニングした。終点蛍光測定を、ポリスチレンキュベットまたは384穴プレートにおいて実施して、各化合物のKdを決定する前に、キナーゼを種々の濃度の阻害剤と予めインキュベートした。全ての実験で標準緩衝液(50 mM Hepes、200 mM NaCl、pH 7.45)を使用した。キュベット測定では、p38αについて4℃の暗所で一晩インキュベーションを実施した。HTSフォーマットでは、室温で5時間までインキュベーションを実施した。II型阻害剤のp38αへの結合の時間依存性を説明するためには、長時間のインキュベーション時間が必要である(パルゲリス(Pargellis)ら、2002年)。
【0151】
キュベットフォーマットにおいて、阻害剤のストック(DMSO中、0.01、0.1、1.0および10.0 mM)を用いて、種々の量の阻害剤を含有する一連のキュベットを調製した。キュベットの全ての測定は、JASCO FP-6500蛍光分光計(JASCO社、独国グロス・ウムシュタット(Gross-Umstadt))で行った。384穴プレートフォーマットにおける蛍光測定値の測定には、Tecan SafireII(Tecan Deutschland社、独国)を使用した。DMSOの%v/vは、キュベットでは0.2%を超えず、384穴プレートでは5%v/vであった。p38αの場合、高グリシンループの運動を引き起こすリガンドの陽性対照として飽和量のBIRB-796またはソラフェニブを使用して、平均Z’因子は、キュベットおよび384穴フォーマットでそれぞれ0.67 ± 0.05(n = 6)および0.64 ± 0.10(n = 6)と決定された。陰性対照としては担体(DMSO)を使用した。
【0152】
アクリロダン標識p38αについて、レシオメトリック蛍光値(R = Iλ512/Iλ475)により、検出化合物の信頼し得る結合曲線のプロットが可能になったが、これはリガンド結合のKdの直接決定を可能にする。検出化合物の結合形態は、種々の時点で一次および/または二次スクリーニングのプレートを測定することによって、HTSフォーマットで初期に評価し得ることに注目すべきである。最大レシオメトリックシグナルまたは経時的Kdを変化させる化合物は、II型/III型阻害剤であると思われる。あるいは、選択された化合物の会合(kon)および解離(koff)の反応速度を、キュベットを用いて決定し得る。konを決定するため、各キュベットの底に小さな攪拌棒を設置して、阻害剤がキュベット上部に位置する注入ポートを通って送達されるに伴い、確実に迅速に攪拌する。蛍光の変化は、JASCO FP-6500蛍光分光器(JASCO社、独国グロス・ウムシュタット(Gros-Umstadt))を用いて、リアルタイムでp38αについて、475 nmでモニターした。殆ど即時の結合反応速度(<5秒)がI型阻害剤の特徴であり、一方、より遅い反応速度(>10秒)は、II型またはIII型阻害剤がDFG-out構造に、よりゆっくり結合していることを示唆する。結合の後、10倍過剰の非標識キナーゼを添加して、結合平衡を標識キナーゼからシフトさせることにより、koffを決定した。過剰の非標識キナーゼの添加により、阻害剤を再分配しアクリロダン標識p38αから解離させて、蛍光シグナルを回収する。全ての結合および解離曲線は、一つの指数方程式に一致した:F(t) = F(∞) + F(0) exp(-t*kobs):ここで、tは時間であり、F(0)は初期の蛍光強度であり、F(∞)はt = ∞における蛍光である。蛍光減衰の半減期(t1/2)は、以下の方程式で計算した:t1/2 = ln 2/kobs。
【0153】
実施例4:反応速度論的発現および精製
p38α構築物は、pOPINE、pOPINFまたはpOPINMベクター中にクローン化し、Precision Protease 切断部位を有するN-末端His-タグ化構築物として、BL21(DE3)大腸菌、BL21(DE3)コドン+RIL大腸菌またはBL21(DE3)ロゼッタ大腸菌中に形質転換した。培養物は、OD600が0.6になるまで37℃で成長させ、室温まで30分間で冷却した後、160 rpmで攪拌しながら、18℃で一晩(〜20時間)1 mM IPTGを用いて発現させた。細胞を緩衝液A(50 mMトリス pH 8.0、500 mM NaCl + 5%グリセロール+ 25 mMイミダゾール)で溶解させ、30 mLのNi-カラム(自己充填)に負荷し、3 CVのNi緩衝液Aで洗浄した後、2 CVを超えるNi緩衝液B(Ni緩衝液A + 500 mMイミダゾール)を用いて0-50%の直線勾配で溶出した。12-30 mL容量の10-MWCO透析カセット(Thermo Scientific)において、透析緩衝液(50 mMトリス pH 7.5、5%グリセロール、150 mM NaCl、1 mM EDTA、1 mM DTT)中4℃で一晩、PreScission Protease(最終濃度50 μg/mL)と共にインキュベーションすることにより、タンパク質を切断した。その後、タンパク質は〜13,000 rpmで15分間遠心分離して、切断工程の間に形成された可能性がある全ての析出物を除去した。その後、上澄を取り、少なくとも4倍のアニオン緩衝液A(50 mMトリス pH 7.4、5%グリセロール、50 mM NaCl、1 mM DTT)で希釈し、1 mLセファロースQ FFカラム(GEヘルスケア)に負荷し、10 CVのアニオン緩衝液Aで洗浄した。タンパク質を、20 CVを超えるアニオン緩衝液B(アニオン緩衝液A + 600 mM NaCl)により0-100%の直線勾配で溶出した。タンパク質をプールし、2 mLまで濃縮して、サイズ排除緩衝液(20 mMトリス pH 7.4、5%グリセロール、200 mM NaCl、1 mM DTT)で平衡化したセファデックスHiLoad 26/60 Superdex 75カラムに2 mL/分の速度で通過させた。その後、溶出したタンパク質は、〜10 mg/mLまで濃縮し、分割して-80℃で凍結させた。
【0154】
実施例5:高グリシンループを標識したac-p38αを用いたリアルタイムおよび終点蛍光測定により、II/III型阻害剤およびいくつかのI型阻害剤の検出が可能になる。
本発明を、p38αの活性化ループを標識した欧州特許EP 08 01 3340号および欧州特許EP 08 02 0341号に記載された方法と比較し、高グリシンループをアクリロダンで標識することが信頼し得る代替のアプローチとなることを実証するため、II型p38α阻害剤であるBIRB-796を使用して蛍光反応を特性化した。終点測定は、BIRB-796濃度を上昇させながら、高グリシンループを標識したp38αの発光スペクトルを測定することにより実施した(図2A)。その後、レシオメトリック蛍光値(R = Iλ512/Iλ475)をリガンドの濃度に対し、対数スケールでプロットして、BIRB-796についてKd = 9.5 ± 2.7 nMと直接的に決定したが(図2B)、これは、活性化ループを標識したp38αを用いて得られた値7.5 ± 2.3 nMと同等であり、この代替のアプローチで得られたDFG-outバインダーのKd値の信頼性が実証される。
【0155】
475 nmでの発光の有意な変化によって、種々の濃度のリガンドについて、リアルタイムでの解離および会合の反応速度論の研究が可能になった。II型(例えばBIRB-796)およびIII型(例えばRL36)阻害剤両者の結合に際し、475 nmでのアクリロダン発光は減少し、結合状態における最大発光波長の赤方偏移が起こる(図2Aおよび3A左パネル)。平衡において、P-ループを標識したp38αの発光スペクトルは、512 nmおよび475 nmでの発光強度がほぼ同等であるように変化する(即ち、R = 512/475 nmは通常〜1.0の値である)。このレシオメトリック蛍光アウトプットを用い、終点平衡測定を実施して、蛍光データを阻害剤濃度の対数スケールに対してプロットすることにより、これらのリガンドのKdを直接的に得ることが可能になる(図2Bおよび3A右)。この反応は、ATP部位に隣接するアロステリック部位をふさぎ、活性化ループにおいて立体構造変化を要する、全てのII型またはIII型阻害剤の特性である。上述の通り、この立体構造変化によって、特定の方法でアクリロダンの蛍光を変化させる、P-ループにおける特徴的変化が起こる。P-ループとDFGモチーフのPheとの間のスタッキングにより、DFG-out構造を安定化することが知られているI型リガンドもまた、高感度で検出され、II/III型リガンドと比較した場合、475 nmでさらに大きく強度が損われる(図3B左)。
【0156】
各リガンドの結合反応速度を調査することにより、このようなI型リガンドはII型またはIII型リガンドから容易に識別される。反応速度の測定は、リガンドを緩衝液中の標識キナーゼ懸濁液に添加する際に、単一波長(475 nm)で発光強度の減少をモニターすることによって行われる。BIRB-796等のII型阻害剤(図2C)およびRL36等のIII型阻害剤(図3a中央)の場合、結合の反応速度はI型阻害剤よりも有意に遅いが、これはI型阻害剤がDFG-inまたはDFG-out構造を安定化するかどうかに関係しない(図4の反応速度論的値を参照)。種々のII型またはIII型阻害剤の添加による蛍光減衰はより遅く、一次減衰関数に容易に適合して、kobsを得る(図2Cおよび図3a中央)。次に、実験的に決定されたkobs値をプロットして、BIRB-796について、欧州特許EP 08 01 3340号および欧州特許EP 08 02 0341号に記載された全てのリガンドのkonを決定し得る。konは、BIRB-796について、〜4.0 x 104 M-1秒-1と決定された。結合しているI型阻害剤の蛍光減衰は非常に速く、ストップトフロー蛍光分光法を使用しないと正確には適切ではない。過剰の非標識p38αを用いてac-p38αから阻害剤を抽出することにより、475 nmでの蛍光強度が上向きに変化するが、これも、一次関数蛍光に適合し、koffの直接的決定が可能であった(図2D)。これらの測定により、蛍光反応の信頼性が実証され、かつ、DFG-inおよびDFG-out立体構造の間に存在する平衡が変化することが実証される。阻害剤の型に関わらず、タンパク質からのリガンドの解離速度は、常に結合速度よりも遅く、これはよく知られている観察結果であるが、p38αについては特に結合速度より遅い(パルゲリス(Pargellis)ら、2003年)。
【0157】
高グリシンループを標識したp38αの蛍光反応を特性化した後、入手可能な少数のサブセットの化合物におけるII型およびIII型阻害剤の高感度の検出を目的としたHTSフォーマットに、アッセイを適合させた(クルター(Kluter)ら、2009年;ゲトリク(Getlik)ら、2009年;ミカルチク(Michalczyk)ら、2008年;パワー(Pawar)ら、2010年;ソス(Sos)ら、2010年)が、これは、キナーゼのDFG-inまたはDFG-out構造に特権的に結合することが知られている種々の骨格を含む。3つの固定した阻害剤濃度(0.5、5および50μM)での最初のプレスクリーニングの後、いくつかは知られているp38α阻害剤の誘導体である、選択された化合物は、濃度依存性直接結合測定を用いてさらなる研究を行い、kon、koffおよびKdを決定し(図3および4)、さらに、妥当性を確認するため、本発明の方法と欧州特許EP 08 01 3340号および欧州特許EP 08 02 0341号に開示された方法とで検出された化合物の親和性を比較した。
【0158】
予測された通り、知られているDFG-outバインダーであるBIRB-796は、5時間にわたり、明確な時間依存性(図7)を示し、欧州特許EP 08 02 0341号の活性化ループを標識したp38αを用いて得られたものと同等のKd値を有することがわかった。加えて、BIRB-796およびRL36は、親和性の一次決定因子として、konよりも主にkoffに関して異なることがわかったが、これは、ピラゾロ尿素系化合物のp38αのDFG-out構造への結合について、非常に特徴的な観察である(パルゲリス(Pargells)ら、2002年;シマード(Simard)ら、2009年)。知られているI型p38α阻害剤であるSB203580もまた高感度で検出され、欧州特許EP 08 02 0341号に開示されたものと同等のKd値であった。欧州特許EP 08 02 0341号では、I型結合形態を採用し、キナーゼヒンジ領域に接触するにもかかわらず、SB203580がp38αに結合し、高グリシンループのDFG Phe169とTyr35の間にπ-πスタッキング相互作用を形成することによってDFG-out構造を安定化し得ることが開示されるが、これによって、欧州特許EP 08 02 0341号の方法および本発明の両者を用いて、この化合物が高感度で検出されることが説明される。SB203580のI型結合形態は、結合および解離反応速度がより迅速であるため、および、DFG-outバインダーであるBIRB-796と同等のKdの時間依存性を示さなかったため(図7)、HTSフォーマットにおいてII/III型バインダーから容易に識別された。従って、本発明の方法によっても、検出されたリガンドのタンパク質との共結晶化を要せずに、阻害剤結合形態の容易な予備的評価が可能になる。
【0159】
ダサチニブは、Kdが27 nMと報告されたp38αに結合するI型阻害剤であるが(カラマン(Karaman)ら、2008年)、高グリシンループを標識したp38αを用いて検出されず、このことから、ダサチニブは、Abl(PDBコード:2GQG)およびcSrc(PDBコード:3G5D)のDFG-in構造に見られる、予測された/公表された結合形態をとっていることが示唆される。しかし、インドール誘導体であるScios-469および2-フェニル置換キナゾリンであるRL40は両者とも、古典的なI型結合形態をとり、ヒンジ領域に接触することが知られており(ムラリ(Murali)ら、2007年;ピエルセ(Pierce)ら、2005年)、これらの化合物がこのアプローチを用いて検出されたことは、驚くべき観察であった。Scios-469の場合、我々は、Kd値を8.2 ± 2.9 nMと決定したが、これは、この化合物について報告されたIC50(〜9 nM)(ムラリ・ダール(Murali Dhar)ら、2007年)とよく一致し、本発明の方法がこのリガンドに非常に高感度であることを実証する。加えて、VEGFR2阻害剤であるCP547632(CP-547632)もまた検出され、Kd値は99 ±/- 13 nMであることが分かった。この化合物は、VEGFR2が媒介する血管新生および腫瘍成長の阻害によって作用する抗癌剤として、臨床検査中である(ベーベ(Beebe)ら、2003年)。現在まで、キナーゼとの複合体中のCP547632の結晶構造は公表されていないが、以前に報告された薬物動態的研究によって、CP547632が、ATP競合的にVEGFR2を阻害することが明らかになった(ベーべら、2003年)。RL40、Scios-469およびCP547632のリアルタイム反応速度測定により、3つの化合物は全てp38αに迅速に結合する(<2秒)ことが示されるが、これは、予測されたI型結合形態と一致する。しかし、活性化ループまたは高グリシンループのいずれかを標識したp38αを用いて、CP547632のみが特性化される可能性があるが;後者のみがPL40およびScios-469を検出した。このことから、おそらくアクリロダンで標識した高グリシンループにさらに接触し、その立体構造を変化させることによって独特な立体構造変化を起こし得る、あるI型阻害剤を検出するという追加利点を、本発明が有することが示唆される。
【0160】
p38αに結合することが予測される検出されたリガンドについて、決定されたKd値に関し、親和性は、他の方法を用いて他で決定および公表されたものと殆ど一致している(パルゲリス(Pargellis)ら、2003年、欧州特許EP 08 01 3340号および欧州特許EP 08 02 0341号)。P-ループ標識アッセイおよび他の方法を用いて報告されたKd値と反応速度論的傾向がほぼ一致することにより、このアッセイ系の正当性が立証される。加えて、p38αに結合しないことが知られているII型リガンド(イマチニブ)およびp38αを強力に阻害するが(カラマン(Karaman)ら、2008年)、DFGモチーフまたはP-ループと相互作用しないI型阻害剤(ダサチニブ)は、このアッセイでは検出されず、陰性対照として使用し得た(図3a右;図3b右)。
【0161】
実施例6:RL40、Scios-469およびCP547632の結晶構造は、P-ループの運動を裏付ける。
我々は、P-ループを標識したp38αに対していくつかの化合物をスクリーニングし、アッセイで高感度で検出し、RL40、Scios-469およびCP547632を特定した。これらのリガンドの検出を説明する構造を詳細に理解するため、我々は、RL40、Scios-469およびCP547632を野生型p38αと共結晶化した。阻害剤は非標識p38αと共結晶化した。簡単に説明すると、30μLのp38α(10 mg/mL)を0.3μLの阻害剤(DMSO中、100 mM)と混合し、この混合物を氷上で1〜2時間インキュベートすることにより、タンパク質阻害剤複合体を調製した。サンプルを13,000 rpmで5分間遠心分離して、過剰の阻害剤を除去した。懸滴蒸気拡散法を用い、1.5μLのタンパク質-阻害剤溶液を0.5μLのリザーバー(100 mM MES、pH 5.6-6.2、20-30% PEG4000および50 mM n-オクチル-β-D-グルコピラノシド)と混合することにより、24穴結晶化プレートにおいて、結晶を成長させた。p38αとの複合体中のRL40の構造(図5A)により、これまでに報告されているSB203580の構造中に見られるもの(欧州特許EP 08 01 3340号および欧州特許EP 08 02 0341号)と類似の、独特で予測しなかった結合形態が明らかになる。リガンドは、DFGモチーフのPheと独特のπ-πスタッキングを形成することにより、P-ループと相互作用し、それによりDFG-out構造を安定化し、アッセイにおけるその検出を合理的に説明する。この構造とSB203580-p38α複合体の構造との重なり(図5B)により、両阻害剤の特徴が良好に重なることが明らかになり、親和性を改善する次世代の化学修飾を理解する上での手掛かりとなる。しかし、SB203580と異なり、化合物RL40はヒンジ領域に接触せず、結合したアクリルアミドは、欧州特許EP 08 01 3340号および欧州特許EP 08 02 0341号に開示された活性化ループの標識に使用される位置の方向に、さらに伸長する。従って、この位置での活性化ループの標識化は、本書に記載したRL40の結合形態と適合しない可能性があり、これまでにRL40のp38αへの結合を検出し報告することが不可能であった理由が説明されるかもしれない。この独特の結合形態は、GSK3との複合体中のRL40のこれまでに報告された構造類似体(ピエルセ(Pierce)ら、2005年)、およびカルモジュリン依存性タンパク質キナーゼ1D(PDBコード:2JC6)とは有意に異なるが、ここで、阻害剤はATP結合部位内で完全に異なる配向を採り、阻害剤のアミノ-ピラゾール部分は3つの水素結合によってヒンジ領域と強力に相互作用し、キナーゼはDFG-in構造中に見られる。RL40の類似体は代表的にはキナーゼのヒンジ領域に結合するため、この結合形態は予測されなかった。これらの知見により、RL40およびSB203580等のこの独特の結合形態を利用するリガンドを増やすためにP-ループを標識したキナーゼを使用する利点が強調される。
【0162】
加えて、P-ループ標識キナーゼアッセイは、Scios-469の結合を強力に検出した。我々は、Scios-469を野生型p38αと共結晶化し(図5C)、阻害剤がヒンジ領域への水素結合を形成することを観察したが、これは、構造的に近い類似体(PDBコード:2QD9)についてこれまで報告されたものと類似している。Scios-469のフルオロフェニル部分はゲートキーパー残基を越えて伸長し、p38αに対する化合物の親和性を増すことが知られている相互作用である疎水性サブポケットをふさぐ(ラフォン(Lafont)ら、2007年)。DFGモチーフは活性「in」構造中に見られるが、高グリシンループの立体構造は、Scios-469と結合したp38αの場合、有意に変化することから、本発明の方法のみを用いた高感度な検出が説明される。高グリシンループは阻害剤上で折り畳まれ、Tyr35の側鎖がScios-469のメチル置換ピペラジン環およびクロロ置換インドール環を溶媒から部分的に保護し、それによって阻害剤の結合を、おそらく疎水性相互作用によって安定化する。その結果、このリガンド表面から秩序化した水分子を除去することにより、Scios-469はp38αに高親和性で結合する。検出される全てのDFG-outバインダーの他に、Scios-469によって、P-ループ標識キナーゼアッセイがP-ループの立体構造を直接的に変化させる、いくつかのI型リガンドをどのように高感度で検出し得るかの例が示される。
【0163】
p38αとの複合体中でのCP547632の結晶構造(図5d)から、キナーゼが、ヒンジ領域にも結合する阻害剤(I型)により、DFG-out構造において安定化されることが明らかになった。阻害剤のコアのイソチアゾールに結合するカルボキサミドは、ヒンジ領域への2つの平行な水素結合を形成する(His107のCOおよびMet109のNH)。我々の知る限りでは、これは新たなヒンジ領域結合モチーフを示す。加えて、尿素部分の2つのNHは、Met109の骨格COに向かい、水素結合ドナーとなる。可溶化ピロリジン-ブタン部分の脂肪族リンカーは、驚くべきことに、溶媒に向かわず、ATPポケットに向けて内側に折り畳まれる。高グリシンループは、この複合体において相対的に移動性であり、結晶構造中、部分的に観察されない。従って、活性化ループの、その不活性立体構造への運動、および、CP547632によるその安定化により、p38αのアクリロダン標識高グリシンループの上方向の運動が誘導される(図1参照)。このことから、p38αの活性化ループを標識する方法を用いた場合と同様に、本発明の方法でCP547632が高感度に検出されることが説明される。
【0164】
実施例7:高グリシンループに標識したac-MKK7を用いるリアルタイムおよび終点蛍光測定
全ての実験は、MKK7のキナーゼドメイン(配列番号2)のみを用いて、図3のp38αについて実施した。標識したシステインは、P-ループの147位(配列番号2の31位)に天然に存在する。218、276および296位のシステイン(配列番号2の102、160および180位)をセリンで置換した(配列番号3)。
【0165】
MKK7は、II型およびIII型阻害剤の結合の影響を受けやすいDFG-out構造に感受性がないかもしれないキナーゼの一例を示す。本発明は、アッセイの、DFG-out構造に結合する(より遅い結合反応速度)リガンド間を識別する能力を実証するが、P-ループと直接相互作用し、その立体構造を修飾する多数のI型阻害剤に高感度でもある。カラマン(Karaman)ら、2008年による、300を超えるキナーゼのパネルに対するいくつかの阻害剤についてのキナーゼのプロファイリングによれば、MKK7の最も近い類似体は全て(MKK1-6、またMEK1-6としても知られている)、Kd値が<10μMのII型阻害剤で阻害されない。しかし、それらは、Kd値が3.4〜70 nMの範囲のスタウロスポリンによって強力に阻害される。スタウロスポリンおよびその最も近い誘導体は、全てのキナーゼの>90%をATP競合的方法で強力に阻害する。MKK7は、その時点でこのキナーゼのパネルの一部ではなかったが、同様の阻害剤選好およびその近い類似体としてのプロファイルが予測される。従って、P-ループ標識MKK7に対するI型阻害剤(K252a)のKdを測定した。K252aは無差別的I型阻害剤であり、スタウロスポリンの構造的に近い類似体である。図8Aにより、K252aの結合が標識タンパク質の蛍光強度の低下、および2つの波長(R = 472 nm / 510 nm)でのレシオメトリック発光の検出可能な変化を引き起こすことが示される。Kdを直接的に決定する終点法を用いて、これらの発光の割合を阻害剤濃度に対してプロットし、K252aについてKdが38 nMと求めることができるが(図8B)、これは、これらの化合物について予測された正しい範囲内である。陰性対照としてソラフェニブが含まれるが、これはII型阻害剤であり、10μM以下では検出されなかった。これらの知見は、MKK7について予測された結果と一致するが、これはDFG-out構造に対する非感受性を示す。このアッセイ反応が、I型阻害剤結合に対する反応におけるP-ループの運動によることを実証するため、ダサチニブも陰性対照として含めた。ダサチニブは、ATP競合的阻害剤であるか、cSrcおよびAblキナーゼであり、報告されたKd値は> 1μMであるが(カラマン(Karaman)ら、2008年)MKK1およびMKK2のみを阻害し、代表的には、知られているあらゆる結晶構造において、キナーゼのP-ループと相互作用しない。従って、このI型阻害剤の添加および検出は、MKK7については予測されなかったが、このデータは10μM以下を裏付ける。図8Cにより、リアルタイムの反応速度測定およびK252aの結合および解離の検出が強調される。図3のとおり、p38αについて、結合により生じる蛍光変化は、標識キナーゼからリガンドを抽出するための過剰の非標識MKK7の添加に際して可逆的である。K252aはI型阻害剤であるため、図3Bに示されるp38αのI型阻害剤SB203580の様に、これらのプロセスの反応速度は速い。
【0166】
参考文献
Adrian, F. J., Ding, Q., Sin, T., Velentza, A., Sloan, C., Liu, Y., Zhang, G., Hur, W., Ding, S., Manley, P., Mestan, J., Fabbro, D., Gray, N. S.. Nat Chem Biol 2006, 2, 95-102.
Annis, D. A., Nazef, N., Chuang, C. C., Scott, M. P., and Nash, H. M. (2004) A general technique to rank protein-ligand binding affinities and determine allosteric versus direct binding site competition in compound mixtures, Journal of the American Chemical Society 126, 15495-15503.
R. T. Aimes, W. Hemmer, S. S. Taylor. Serine-53 at the tip of the glycine-rich loop of cAMP-dependent protein kinase: role in catalysis, P-site specificity, and interaction with inhibitors. Biochemistry 2000, 39, 8325.
B. Apsel, J. A. Blair, B. Gonzalez, T. M. Nazif, M. E. Feldman, B. Aizenstein, R. Hoffman, R. L. Williams, K. M. Shokat, Z. A. Knight, Nat Chem Biol 2008, 4, 691.
【0167】
A. C. Backes, B. Zech, B. Felber, B. Klebl, G. Muller, Expert Opin Drug Discovery 2008, 3, 1409.
A. C. Backes, B. Zech, B. Felber, B. Klebl, G. Muller, Expert Opin Drug Discovery 2008, 3, 1427.
Beebe, J. S., Jani, J. P., Knauth, E., Goodwin, P., Higdon, C., Rossi, A. M., Emerson, E., Finkelstein, M., Floyd, E., Harriman, S., Atherton, J., Hillerman, S., Soderstrom, C., Kou, K., Gant, T., Noe, M. C., Foster, B., Rastinejad, F., Marx, M. A., Schaeffer, T., Whalen, P. M., Roberts, W. G. (2003). Pharmacological characterization of CP-547,632, a novel vascular endothelial growth factor receptor-2 tyrosine kinase inhibitor for cancer therapy. Cancer Res 63, 7301-9.
J. A. Blair, D. Rauh, C. Kung, C. H. Yun, Q. W. Fan, H. Rode, C. Zhang, M. J. Eck, W. A. Weiss, K. M. Shokat, Nat Chem Biol 2007, 3, 229.
【0168】
Calleja, V., Laguerre, M., Parker, P. J., Larijani, B.. PLoS Biol 2009, 7, 189-200.R. A. Copeland, D. L. Pompliano, T. D. Meek, Nat Rev Drug Discov 2006, 5, 730.
Dar, A. C., Lopez, M. S., and Shokat, K. M., Small Molecule Recognition of c-Src via the Imatinib-Binding Conformation. Chem Biol 15 (10), 1015 (2008).DeLano, W.L., The PyMOL Molecular Graphics System. http://www.pymol.org (2002).
de Lorimier, R. M., Smith, J. J., Dwyer, M. A., Looger, L. L., Sali, K. M., Paavola, C. D., Rizk, S. S., Sadigov, S., Conrad, D. W., Loew, L., and Hellinga, H. W. (2002) Construction of a fluorescent biosensor family, Protein Sci 11, 2655-2675.Dumas, J., Hatoum-Mokdad, H., Sibley, R., Riedl, B., Scott, W. J., Monahan, M. K., Lowinger, T. B., Brennan, C., Natero, R., Turner, T., Johnson, J. S., Schoenleber, R., Bhargava, A., Wilhelm, S. M., Housley, T. J., Ranges, G. E., and Shrikhande, A. (2000) 1-Phenyl-5-pyrazolyl ureas: potent and selective p38 kinase inhibitors, Bioorganic & medicinal chemistry letters 10, 2051-2054.
【0169】
Emsley, P. and Cowtan, K., Coot: model‐building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr 60 (Pt 12 Pt 1), 2126 (2004).
Eyers, P. A., Churchill, M. E., Maller, J. L. (2005). The Aurora A and Aurora B protein kinases: a single amino acid difference controls intrinsic activity and activation by TPX2. Cell Cycle 4, 784-9.
Fischmann, T., Smith, C., Mayhood, T., Myers, J., Reichert, P., Mannarino, A., Carr, D., Zhu, H., Wong, J., Yang, R. S., Le, H., Madison, V., Biochemistry 2009.
M. J. Garnett, R. Marais, Cancer Cell 2004, 6, 313.Gauglitz, G. and Vo-Dinh, T. (2003). Handbook of spectroscopy. Wiley-VCH.
Getlik, M., Grutter, C., Simard, J. R., Kluter, S., Rabiller, M., Rode, H. B., Robubi, A., Rauh, D. (2009). Hybrid compound design to overcome the gatekeeper T338M mutation in cSrc. J Med Chem 52, 3915-26.
Gschwind, A., Fischer, O. M. and Ullrich, A., The discovery of receptor tyrosine kinases: targets for cancer therapy. Nat Rev Cancer 4 (5), 361 (2004).
S. K. Hanks, T. Hunter, FASEB J 1995, 9, 576.
Hibbs, R. E., Talley, T. T., and Taylor, P. (2004) Acrylodan-conjugated cysteine side chains reveal conformational state and ligand site locations of the acetylcholine-binding protein, The Journal of biological chemistry 279, 28483-28491.
【0170】
Johnson, L. N., Noble, M. E. M. and Owen, D. J. (1996) Active and inactive protein kinases : structural basis for regulation. Cell 85, 149-158.Johnson, L. N. and Lewis, R. E. (2001). Structural basis for control by phosphorylation. Chem Rev. 2001 Aug;101(8):2209-42.
Kabsch, W., Automatic processing of rotation diffraction data from crystals of initially unknown symmetry and cell constants. . J. Appl. Cryst. 26, 795 (1993).
Karaman, M. W., Herrgard, S., Treiber, D. K., Gallant, P., Atteridge, C. E., Campbell, B. T., Chan, K. W., Ciceri, P., Davis, M. I., Edeen, P. T., Faraoni, R., Floyd, M., Hunt, J. P., Lockhart, D. J., Milanov, Z. V., Morrison, M. J., Pallares, G., Patel, H. K., Pritchard, S., Wodicka, L. M., Zarrinkar P.P. (2008). A quantitative analysis of kinase inhibitor selectivity. Nat Biotechnol 26, 127-32.
Kirkland, L. O., McInnes, C.. Biochem Pharmacol 2009.
【0171】
Kluter, S., Grutter, C., Naqvi, T., Rabiller, M., Simard, J. R., Pawar, V., Getlik, M., Rauh, D. (2009). Displacement assay for the detection of stabilizers of inactive kinase conformations. J Med Chem
Knighton, D. R., Zheng, J. H., Ten Eyck, L. F., Xuong, N. H, Taylor, S. S., Sowadski, J. M. (1991). Structure of a peptide inhibitor bound to the catalytic subunit of cyclic adenosine monophosphate-dependent protein kinase. Science 253, 414-20.
Lafont, V., Armstrong, A. A., Ohtaka, H., Kiso, Y., Mario Amzel, L., Freire, E. (2007). Chem Biol Drug Des 69, 413-22.
Lakowicz, J. R. (1999). Principles of Fluorescence Spectroscopy. Kluwer Academic / Plenum Publishers
Laskowski, R. A., MacArthur, M. W., Moss, D. S., and Thornton, J. M., PROCHECK: a program to check the stereochemical quality of protein structures. J Appl Cryst 26, 283 (1993).
【0172】
Larkin, M. A. et al., Clustal W and Clustal X version 2.0. Bioinformatics 23 (21), 2947 (2007).
Levinson, N. M. et al., A Src-like inactive conformation in the abl tyrosine kinase domain. PLoS Biol 4 (5), e144 (2006).
Liu, Y. and Gray, N. S., Rational design of inhibitors that bind to inactive kinase conformations. Nat Chem Biol 2 (7), 358 (2006).
Liu, C. C. and Schultz, P. G. (2010). Adding new chemistries to the genetic code. Annu Rev Biochem 79, 15.1-15.32.
M. Mapelli, L. Massimiliano, C. Crovace, M. A. Seeliger, L. H. Tsai, L. Meijer, A. Musacchio, J Med Chem 2005, 48, 671.
Michalczyk, A. et al., Structural insights into how irreversible inhibitors can overcome drug resistance in EGFR. Bioorg Med Chem 16 (7), 3482 (2008).
【0173】
Murali Dhar, T. G., Wrobleski, S. T., Lin, S., Furch, J. A., Nirschl, D. S., Fan, Y., Todderuf, G., Pitt, S., Doweyko, A. M., Sack, J. S., Mathur, A., McKinnon, M., Barrish, J. C., Dodd, J. H., Schieven, G. L., Keftheris, K. (2007). Synthesis and SAR of p38alpha MAP kinase inhibitors based on heterobicyclic scaffolds. Bioorg Med Chem Lett 17, 5019-24.
Murshudov, G. N., Vagin, A. A., and Dodson, E. J., Refinement of macromolecular structures by the maximum‐likelihood method. Acta Crystallogr D Biol Crystallogr 53 (Pt 3), 240 (1997).
B. Nagar, W. G. Bornmann, P. Pellicena, T. Schindler, D. R. Veach, W. T. Miller, B. Clarkson, J. Kuriyan, Cancer Res 2002, 62, 4236.
Nienaber, V. L. et al., Discovering novel ligands for macromolecules using X-ray crystallographic screening. Nature biotechnology 18 (10), 1105 (2000).
【0174】
Pargellis, C., Tong, L., Churchill, L., Cirillo, P. F., Gilmore, T., Graham, A. G., Grob, P. M., Hickey, E. R., Moss, N., Pav, S., and Regan, J. (2002) Inhibition of p38 MAP kinase by utilizing a novel allosteric binding site, Nature structural biology 9, 268-272.
Patel, S. B., Cameron, P. M., O`Keefe, S. J., Frantz-Wattley, B., Thompson, J., O`Neill, E. A., Tennis, T., Liu, L., Becker, J. W., Scapin, G. Acta Crystallogr D Biol Crystallogr 2009, 65, 777-85.
Pawar, V. G., Sos, M. L., Rode, H. B., Rabiller, M., Heynck, S., van Otterlo, W. A. L., Thomas, R. K., Rauh, D. (2010), Synthesis and biological evaluation of 4-anilinoquinolines as potent inhibitors of epidermal growth factor receptor. J Med Chem 53(7), 2892-901.
【0175】
W. A Pearson, Rapid and Sensitive Sequence Comparison with FASTP and FASTA, in Methods in Enzymology, ed. R. Doolittle Academic Press, San Diego) 183(1990)63-98.
Pierce AC, ter Haar E, Binch HM, Kay DP, Patel SR, Li P. J Med Chem. 2005 Feb 24;48(4):1278-81.
Read, R. J., Pushing the boundaries of molecular replacement with maximum likelihood. Acta Crystallogr D 57 (Pt 10), 1373 (2001).
Reich, M. et al., GenePattern 2.0. Nat Genet 38 (5), 500 (2006).
Regan, J., Breitfelder, S., Cirillo, P., Gilmore, T., Graham, A. G., Hickey, E., Klaus, B., Madwed, J., Moriak, M., Moss, N., Pargellis, C., Pav, S., Proto, A., Swinamer, A., Tong, L., and Torcellini, C. (2002) Pyrazole urea-based inhibitors of p38 MAP kinase: from lead compound to clinical candidate, Journal of medicinal chemistry 45, 2994-3008.
Regan, J., Pargellis, C. A., Cirillo, P. F., Gilmore, T., Hickey, E. R., Peet, G. W., Proto, A., Swinamer, A., and Moss, N. (2003) The kinetics of binding to p38MAP kinase by analogues of BIRB 796, Bioorganic & medicinal chemistry letters 13, 3101-3104.
【0176】
Rendell, D. (1987). Fluorescence and Phosphorescence. Crown
Richieri, G. V., Ogata, R. T., and Kleinfeld, A. M. (1999) The measurement of free fatty acid concentration with the fluorescent probe ADIFAB: a practical guide for the use of the ADIFAB probe, Molecular and cellular biochemistry 192, 87-94.M. Saraste, P. R. Sibbald, A. Wittinghofer, Trends Biochem Sci 1990, 15, 430.
Schuttelkopf, A. W. and van Aalten, D. M., PRODRG: a tool for high‐throughput crystallography of protein‐ligand complexes. Acta Crystallogr D Biol Crystallogr 60 (Pt 8), 1355 (2004).
Sharma, A. and Schulman, S. G. (1999). Introduction to Fluorescence Spectroscopy. Wiley interscience.Sullivan, J. E., Holdgate, G. A., Campbell, D., Timms, D., Gerhardt, S., Breed, J., Breeze, A. L., Bermingham, A., Pauptit, R. A., Norman, R. A., Embrey, K. J., Read, J., VanScyoc, W. S., and Ward, W. H. (2005) Prevention of MKK6-dependent activation by binding to p38alpha MAP kinase, Biochemistry 44, 16475-16490.
【0177】
M. A. Seeliger, B. Nagar, F. Frank, X. Cao, M. N. Henderson, J. Kuriyan, Structure 2007, 15, 299.
Shuker, S. B., Hajduk, P. J., Meadows, R. P., and Fesik, S. W., Discovering high-affinity ligands for proteins: SAR by NMR. Science 274 (5292), 1531 (1996).
J. R. Simard, S. Kluter, C. Grutter, M. Getlik, M. Rabiller, H. B. Rode, D. Rauh, Nat Chem Biol 2009, 5, 394-396.
Simard, J. R., Getlik, M., Grutter, C., Pawar, V., Wulfert, S., Rabiller, M., Rauh, D. (2009). Development of a fluorescent-tagged kinase assay system for the detection and characterization of allosteric kinase inhibitors. J Am Chem Soc 131, 13286-13296.
Simard, J. R., Pawar, V., Aust, B., Wolf, A., Rabiller, M., Wulfert, S., Robubi, A., Kluter, S., Ottmann, C., Rauh, D. (2009). High-throughput screening to identify inhibitors which stabilize inactive kinase conformations in p38alpha. J Am Chem Soc
【0178】
Sos, M. L., Rode, H. B., Heynck, S., Peifer, M., Fischer, F., Kluter, S., Pawar, V. G., Reuter, C., Heuckmann, J., Weiss, J., Ruddigkeit, L., Rabiller, M., Koker, M., Simard, J. R., Getlik, M., Yuza, Y., Chen, T., Greulich, H., Meyerson, M., Thomas, R. K., Rauh, D. (2010). Chemogenomic profiling provides insights into the limited activity of irreversible EGFR Inhibitors in tumor cells expressing the T790M EGFR resistance mutation. Cancer Research 70(3):868-74.
Tamayo, N, Liao, L., Goldberg,M., Powers, D., Tudor, Y-Y, Yu, V., Wong, L. M., Henkle, B., Middleton, S., Syed, R., Harvey, T., Jang, G., Hungate, R. and Celia Dominguez (2005). Design and synthesis of potent pyridazine inhibitors of p38 MAP kinase, Bioorganic & Medicinal Chemistry Letters 15:2409-2413.
Tecle, H., Feru, F., Liu, H., Kuhn, C., Rennie, G., Morris, M., Shao, J., Cheng, A. C., Gikunju, D., Miret, J., Coli, R., Xi, S. H., Clugston, S. L., Low, S., Kazmirski, S., Ding, Y. H., Cao, Q., Johnson, T. L., Deshmukh, G. D., DiNitto, J. P., Wu, J. C.; English, J. M. (2009). The design, synthesis and potential utility of fluorescence probes that target DFG-out conformation of p38alpha for high throughput screening binding assay. Chem Biol Drug Des 74, 547-59.
【0179】
Taylor, S. S. and Radzio-Andzelm, E. (1994) Three protein kinase structures define a common motif. Structure 2, 345-355; 43.Vogtherr, M., Saxena, K., Hoelder, S., Grimme, S., Betz, M., Schieborr, U., Pescatore, B., Robin, M., Delarbre, L., Langer, T., Wendt, K. U., and Schwalbe, H. (2006) NMR characterization of kinase p38 dynamics in free and ligand-bound forms, Angewandte Chemie (International ed 45, 993-997.Yeatman, T. J., A renaissance for SRC. Nat Rev Cancer 4 (6), 470 (2004).
Tokarski, J.S., Newitt, J., Chang, C.Y.J., Cheng, J.D., Wittekind, M., Kiefer, S.E., Kish, K., Lee, F.Y.F., Borzilerri, R., Lombardo, L.J., Xie, D., Zhang, Y., Klei, H.E. (2006) CANCER RES. 66: 5790-5797.
Wan, P. T., Garnett, M. J., Roe, S. M., Lee, S., Niculescu-Duvaz, D., Good, V. M., Jones, C. M., Marshall, C. J., Springer, C. J., Barford, D., Marais, R., Cell 2004, 116, 855.
【0180】
Wang, X, Destrument, A, Tournier, C. (2007). Physiological roles of MKK4 and MKK7: Insights from animal models. Biochimica et Biophysica Acta 1773 (2007), 1349-1357.
Wong, L., Lieser, S. A., Miyashita, O., Miller, M., Tasken, K., Onuchic, J. N., Adams, J. A., V. L. Woods, Jr., P. A. Jennings, J Mol Biol 2005, 351, 131.
Yem, A. W., Epps, D. E., Mathews, W. R., Guido, D. M., Richard, K. A., Staite, N. D., and Deibel, M. R., Jr. (1992) Site-specific chemical modification of interleukin-1 beta by acrylodan at cysteine 8 and lysine 103, The Journal of biological chemistry 267, 3122-3128.
J. Zhang, P. L. Yang, N. S. Gray, Nat. Rev. Cancer 2009, 9, 28.
【技術分野】
【0001】
本発明は、天然に存在しているか、またはキナーゼのP-ループに導入されているアミノ酸を標識したキナーゼに関し、ここで、前記の標識は、前記のアミノ酸の遊離チオールまたはアミノ基で行われ、かつ、前記の標識は、(a)その環境における極性変化に感受性のある、チオールまたはアミノ反応性フルオロフォアであるか;または(b)チオール反応性スピン標識、同位体または濃縮同位体チオールまたはアミノ反応性標識であって、前記のフルオロフォア、スピン標識、同位体または濃縮同位体標識は、触媒活性を阻害せず、キナーゼの安定性を妨げない。さらに、本発明は、キナーゼ阻害剤のスクリーニング方法、キナーゼ阻害剤のリガンドの結合および/または解離のキネティクスの決定方法、および本発明の標識キナーゼを用いるキナーゼ阻害剤のスクリーニングに好適な変異キナーゼの製造方法に関する。
【0002】
この明細書では、特許出願書類および製造者のマニュアルを含む多数の資料を引用する。これらの資料の開示は、本発明の特許性に関係すると見なされず、参照によって全体として本書に組み込まれる。より具体的には、全ての参照した資料は、個々の資料がそれぞれ参照によって組み込まれることを特異的および個別的に示したのと同程度に、参照により組み込まれる。
【背景技術】
【0003】
プロテインキナーゼは、鍵となる細胞過程を制御する、重要な酵素の一群である。癌生物学において、異常制御されたキナーゼのシグナル伝達について理解が進んだことにより(ゲシュビント(Gschwind)およびフィッシャー(Fischer)、2004年)、望まれないキナーゼ活性の特異的標的化に使用される有機小分子が開発され、癌の標的療法の分野が開始された(ツァン(Zhang)ら、2009年)。殆どのキナーゼ阻害剤は、ダサチニブ(スプリセル(Sprycel)登録商標)等のI型阻害剤であり、キナーゼの活性「DFG-in」構造に結合し、ATPと競合してキナーゼのヒンジ領域と非常に重要な水素結合を形成する。この構造において、制御活性化ループは開環し、伸長しているため、ATPおよび基質の結合が可能である(ナイトン(Knighton)ら、1991年)。ATPのアデニンは、キナーゼのヒンジ領域と非常に重要な水素結合を形成するが、この領域はキナーゼドメインのN-およびC-末端ローブの間に位置し、短く柔軟であり、一方、ATPのβおよびγリン酸塩は、Mg2+またはMn2+イオン、保存DFGモチーフのAsp側鎖、およびATP結合間隙の上部に位置する高グリシンループのアミノ酸残基などの、いくつかの構造要素とのイオン結合性および水素結合性相互作用の複雑なネットワークによって連係する(アイムス(Aimes)ら、2000年)。しかし、これらのタイプの阻害剤の開発には、ATP結合部位内の化学的空間が使い尽くされていること、阻害剤の選択性および有効性が低いことについての問題に加え、薬物耐性の発現という問題がある。現在の医薬品化学の研究では、これらの障害を克服して、代替の(即ちアロステリック)結合部位を標的とし、および/または、酵素的能力のない不活性キナーゼ構造を安定化する阻害剤を特定および開発することによって、有効な長期的療法の開発を試みている(ツァン(Zhang)ら、2009年;アドリアン(Adrian) ら、2006年;カレジャ(Calleja) ら、2009年;フィシュマン(Fischmann) ら、2009年;キルクラント(Kirkland) およびマクイネス(McInnes) 、2009年)。これらの部位の一つは、不活性な「DFG-out」キナーゼ構造においてのみ存在し、キナーゼ研究の最前線となっている。DFG-out構造は、高度に保存されたDFGモチーフの180°反転によって引き起こされる活性化ループの構造変化によるものであり(リウ(Liu)およびグレイ(Gray)、2006年;パルゲリス(Pargellis)ら、2002年)、これは、ATP結合部位に隣接する保存性がより低いアロステリック部位にもさらされる事象である。II型およびIII型阻害剤は、この保存性がより低いアロステリック部位に結合し、優れた選択性プロファイル、向上した薬理学的特性を有すると考えられており(コープランド(Copeland)ら、2006年)、薬品開発の新たな機会を提供している(リウ(Liu)およびグレイ(Gray)、2006年)。より具体的には、ソラフェニブ(ネキサバール(Nexavar) 登録商標、ワン(Wan)ら、2004年)、イマチニブ(グリーベック(Gleevec) 登録商標、ネイガー(Nagar)ら、2002年)およびp38αの選択的阻害剤であるBIRB-796(パルゲリス(Pargellis)ら、2002年)等のII型阻害剤は、ヒンジ領域に結合し、ATP競合的であるが、このアロステリック部位中に及び、一方III型阻害剤は、専らアロステリックポケット内で結合する(パルゲリスら、2002年;シマード(Simard)ら、提出済)。最近まで、不活性DFG-out構造を安定化する阻害剤を明確に特定できるアプローチは、不十分であるか、または、新規なヒット化合物を特定する、学界および産業界で使用されるハイスループット・スクリーニング・フォーマットに適合しなかったため、このようなリガンドを検出および特性化する革新的な新規アプローチの必要性は高まっている。
【0004】
フルオロフォアのタンパク質への結合は、確立されたアプローチであり、リガンドの結合に対する反応においてタンパク質構造中の立体構造の変化を検出するために使用されている。緩衝液中の非結合脂肪酸の濃度を測定する、市販されているプローブであるアクリロダン標識脂肪酸結合タンパク質(ADIFAB;モレキュラー・プローブス(Molecular Probes))(リチェリ(Richieri)ら、1999年)に加えて、このアプローチは、アセチルコリン結合タンパク質(ヒッブス(Hibbs)ら、2004年)、インターロイキン-1β(イェム(Yem)ら、1992年)および種々の糖およびアミノ酸結合タンパク質(ドゥ・ロリミエル(de Lorimier)ら、2002年)等の、種々の他のタンパク質に適用されている。
【発明の概要】
【0005】
上記の議論から、特異的キナーゼ阻害剤のスクリーニングのための用途の広い手段および方法を有することが望ましいであろう。しかし、いくつかのキナーゼは、立体構造中のDFG-in/outスイッチに影響するか、スイッチを誘導し得るリガンドに、多少は感受性があると思われる。従って、DFG-out構造に容易に適合するキナーゼについて、および、ATP部位内部で結合し、活性化ループまたはDFG構造における変化ではない標的キナーゼにおける他の構造的変化を誘導し得るリガンドについて、DFG-outバインダーを高感度で検出する、代替のスクリーニング戦略を開発することは有用であろう。この技術的問題は、特許請求の範囲で特徴付けられる具体例を提供することによって解決される。
【図面の簡単な説明】
【0006】
【図1】p38αにおけるリガンド結合によって引き起こされる、P-ループおよび活性化ループの立体構造変化の略図である。
【図2】高グリシンループ上に標識したac-p38αを用いたリアルタイムおよび終点蛍光測定結果を示す。
【図3】P-ループac-p38αを用いたIII型およびI型リガンドのリアルタイムおよび終点蛍光測定結果を示す。
【図4−1】P-ループを標識したp38αの種々の型の阻害剤に対する蛍光特性評価および応答を示す。
【図4−2】P-ループを標識したp38αの種々の型の阻害剤に対する蛍光特性評価および応答を示す。
【図5−1】RL40、Scios-469およびCP547632の結晶構造は、P-ループの運動を裏付けることを示す図である
【図5−2】RL40、Scios-469およびCP547632の結晶構造は、P-ループの運動を裏付けることを示す図である
【図6】野生型の非標識およびアクリロダン標識p38αの反応速度論的および阻害特性評価結果を示す。
【図7】384穴フォーマットにおいて測定された、p38αによるBIRB-796(1)のKd値の時間依存性を示す。
【図8】高グリシンループを標識したMKK7を用いたII型およびI型リガンドのリアルタイムおよび終点蛍光測定結果を示す。
【発明を実施するための形態】
【0007】
従って、本発明は、天然に存在しているか、またはキナーゼのP-ループに導入されているアミノ酸で標識されたキナーゼに関し、ここで、前記の標識化は、前記のアミノ酸の遊離チオールまたはアミノ基で行われ、かつ、前記の標識は、(a)その環境における極性変化に感受性のある、チオールまたはアミノ反応性フルオロフォアであるか;または(b)チオール反応性スピン標識、同位体または濃縮同位体チオールまたはアミノ反応性標識であって、前記のフルオロフォア、スピン標識、同位体または濃縮同位体標識は、触媒活性を阻害せず、キナーゼの安定性を妨げない。
【0008】
「キナーゼ」という用語は当業界でよく知られており、リン酸基をATP等の高エネルギードナー分子からタンパク質等の特異的標的分子に移す、一種の酵素を言う。キナーゼは、酵素委員会(EC)の番号2.7に分類される。特異性によって、プロテインキナーゼは、セリン/トレオニンキナーゼ(EC 2.7.11、例えばp38α)、チロシンキナーゼ(EC 2.7.10、例えばEGFRキナーゼドメイン)、ヒスチジンキナーゼ(EC 2.7.13)、アスパラギン酸/グルタミン酸キナーゼ、および1を超える特異性を有する混合キナーゼ(EC 2.7.12、例えば、MEKは、セリン/トレオニンおよびチロシンに特異的である)に細分し得る。
【0009】
アミノ酸は、カルボン酸官能基およびアミノ官能基を有する有機分子と定義される。アミノ酸は、タンパク質の必須のビルディングブロックである。遊離チオール基を有するアミノ酸の例は、20のタンパク質構成アミノ酸に属するシステイン、および、天然アミノ酸配列には殆ど生じない、非標準的アミノ酸であるアセチルシステインである。遊離アミノ基を有するタンパク質構成アミノ酸は、リシン、ヒスチジンまたはアルギニン、および、トリプトファン等の芳香族アミンであるアミノ酸である。ピロリジン、5-ヒドロキシリジンまたはo-アミノチロシンは、遊離アミノ基を有する非標準的アミノ酸である。アミノ酸であるアスパラギンおよびグルタミンは、遊離アミノ基を有するが、標識化剤に反応性がないため、本発明においては好適ではなく、従って除外される。
【0010】
トリプトファンは、そのインドール環にアミノ基を有する芳香族アミノ酸である。芳香族アミンは弱塩基であるので、pH 7でプロトン化されない。しかし、芳香族アミンはそれでも、イソチオシアネート、塩化スルホニルまたは酸等の高反応性試薬を用いて修飾し得る。
【0011】
キナーゼは、キナーゼ中の所望の位置、即ちP-ループにおいて、アミノ酸の遊離チオールまたはアミノ基を標識する。標識化の間、従来の遊離チオールまたはアミノ基は、項目(a)および(b)により、標識したアミノ酸と標識の間の共有結合の形成に関与する。
【0012】
前記の標識されるアミノ酸は、キナーゼのP-ループに位置する。このことは、P-ループまたはそれに相当する構造を有するキナーゼのみが本発明の範囲に含まれることを意味する。P-ループ(高グリシンループとも呼ばれる)は、全てのATP/GTP結合タンパク質中で維持される、非常に柔軟な構造的特徴である(サラステ(Saraste)ら、1990年)。キナーゼにおいて、P-ループは標準的なGly-X-Gly-X-X-Glyモチーフ(Xはあらゆるアミノ酸である)を含有し、キナーゼのN-末端ローブ中に位置するが、N-末端ローブでP-ループは、ATP等のリガンドの、キナーゼのATP結合部位への侵入を導く調節ループとして働く(ウォン(Wong)ら、2005年)。
【0013】
関心のあるキナーゼ中に天然に存在し、溶媒にさらされているシステインは、P-ループの外側またはP-ループ配列の内部に位置し得る。このことは、遊離アミノ基を有するアミノ酸に同等に適用される。
【0014】
本発明の修飾キナーゼは、天然に存在しているかまたはP-ループ中に導入されているアミノ酸で標識されている。好適なアミノ酸、即ち、遊離チオールまたはアミノ基を有するアミノ酸がP-ループ中に存在しない場合、前記のアミノ酸は、当業界でよく知られた技術により導入され得、即ち、前記のアミノ酸を添加するか、存在するアミノ酸を置換することにより挿入され得る。あらゆる場合において、アミノ酸が標識試薬との反応により標識される場合、アミノ酸をP-ループ中に導入した後にのみ標識することを、誤解を避けるために理解すべきである。上記の技術は、部位特異的変異誘発、並びに他の組み換え、合成または半合成技術を含む。非標準的アミノ酸をキナーゼ中に導入する場合には、前記のアミノ酸を含有するアミノ酸ストレッチを化学的に合成した後、組み換え的または合成的に製造したものでもよいキナーゼの残りの部分に結合させてもよい。或いは、その後の標識化で所望の位置に特殊な非標準アミノ酸を組み込むために発現および設計されたキナーゼは、改変した遺伝子コードを適用することにより、組み換え技術で製造してもよい(例えば、リウ(Liu)およびシュルツ(Schltz)、2010年参照)。
【0015】
標識化方法としては、キナーゼのインキュベーションが挙げられ、例えば、本発明の変異キナーゼ(例えば、P-ループに導入されたシステインを有するキナーゼ)を、穏和な条件下でチオールまたはアミノ反応性標識とインキュベーションし、前記の変異キナーゼをP-ループ内の所望の位置で標識する。換言すれば、原理上は、チオール反応性スピン標識での標識化を除いて、キナーゼにおいて前記の所望の位置のみを標識化することが可能であり、これは好ましい態様であるが、或いは、アイソトープでの同時標識が想定される(以下参照)。穏和な条件とは、緩衝的なpH(例えば、チオール反応性プローブについてはpH 7前後)、キナーゼに対する標識の割合、インキュベーション工程の温度および長さ(チオール反応性プローブについては、例えば、4℃および暗所で一晩)を言い、これらは当業者に知られており、また、チオールおよびアミノ反応性プローブの製造者の取扱説明書により提供される。このような条件は、前記のキナーゼの標識化が所望の標識部位に特異的であることを確実にするため、選択されたチオールまたはアミノ反応性標識の反応を減速するように最適化する必要がある。フルオロフォア標識の場合、インキュベーションを暗所で実施することが必要である。露光量が増加すると、フルオロフォアの漂白が起こり、蛍光発光の強度が弱くなる。標識化の後、標識したキナーゼは、好ましくは濃縮し、ゲル濾過実験で精製するか、緩衝液で数回洗浄して、過剰な未反応標識を除去する。洗浄緩衝液は、代表的には、標識したキナーゼの保管に使用する緩衝液であり、また、所望の測定が行われる際の緩衝液であってもよい。
【0016】
「フルオロフォア」という用語は、特定の波長の光子等のエネルギーを吸収し、エネルギー、即ち光を、(生物発光の場合と同様に)化学反応の関与なく、(リン光の場合と異なり)吸収直後に、異なる(しかし同様に特異的な)波長(蛍光)で放出する、分子または分子内の官能基を示す。通常、吸収された光子の波長は紫外領域にあるが、赤外領域中にも達し得る。発光波長は、通常、可視領域内である。放出されるエネルギーの量および波長は、主にフルオロフォアの特性に依存するが、フルオロフォア周囲の化学的環境によっても影響される可能性がある。多数のフルオロフォアは、環境の変化に感受性がある。これは、極性、電荷および/またはフルオロフォアが結合している分子の立体構造の変化を含む。分子が電気的に励起された後、基底状態に緩和する時に蛍光が発生するが、UVから近赤外までのエネルギーを有する光子を放出する通常使用される蛍光化合物では、蛍光は、0.5ナノ秒および20ナノ秒の間の範囲で起こる。
【0017】
「チオールまたはアミノ反応性」という用語は、例えばフルオロフォア等の化合物が遊離チオールまたはアミノ基と特異的に反応する特性を示す。これは、チオールまたはアミノ基との特異的反応を管理する、前記の化合物中に存在する官能基のためである。これらの官能基は、遊離チオールまたはアミノ基に結合し得る特異的標識を提供するために、フルオロフォア、スピン標識または濃縮アイソトープ分子等の分子に結合していてもよい。チオール特異性化合物の例は、例えば、ヨードアセトアミド、マレイミド、Hg-LinkTMフェニル水銀化合物またはTS-linkTM試薬(両者ともインビトロゲン(Invitrogen)社)等のハロアルキル化合物である。ハロアルキル化合物は、pHに従ってチオールまたはアミノ基と反応する。
【0018】
「スピン標識」(SL)という用語は、分子、一般的には有機分子であって、通常は窒素原子上に不対電子を有し、他の分子に結合する能力を有するものを示す。スピン標識は、EPR分光法を用いるタンパク質のプローブ化のためのツールとして使用される。部位特異性スピン標識化(SDSL)技術により、タンパク質の立体構造および動力学(ダイナミクス)をモニターすることが可能になる。このような実験において、アミノ酸特異的SLを使用し得る。
【0019】
位置特異的スピン標識化は、電子スピン共鳴を使用する、タンパク質の局所力学を調査する技術である。SDSLは、アミノ酸とのスピン標識の特異的反応を基礎とする。タンパク質構造中に構築されるスピン標識は、EPR分光法によって検出し得る。SDSLにおいて、チオールまたはアミノ基等のスピン標識の結合部位は、天然に存在しない場合は、部位特異的変異誘発によって組み換え発現したタンパク質中に導入される。換言すれば、上記の技術によって、スピン標識は、キナーゼが所望の位置のみで特異的に標識されるように、キナーゼ中に導入され得る。スピン標識の内部に含有される官能基は、その特異性を決定する。中性のpHでは、タンパク質のチオール基は、メタンチオスルホネート、マレイミドおよびヨードアセトアミド等の官能基と特異的に反応し、アミノ酸のシステインと共有結合を作る。スピン標識は独特な分子リポーターであり、それらは常磁性である、即ち、不対電子を含有する。ニトロキシドスピン標識は、安定で単純なEPRシグナルを有することから、高分子の構造および動力学の研究に広く使用されている。ニトロキシルラジカル(N-O)は、通常、ピロリジン等の複素環に取り込まれ、不対電子は大部分がN-O結合に局在する。タンパク質中に取り込まれると、スピン標識の動きは、その局所環境によって決定づけられる。スピン標識は動きに極めて敏感であることから、このことは、タンパク質に結合したスピン標識のEPRスペクトルに対して大きく影響する。
【0020】
不対電子から発生するシグナルは、互いにおよび外部磁場に関し、サンプル中の不対電子の動き、距離、および配向についての情報を提供する。溶液中、自由に動く分子については、EPRはNMR(核磁気共鳴分光法)よりもかなり速いタイムスケールで作動するので、より速い分子の動きの詳細、即ちNMRでマイクロ秒であるのに対して、ナノ秒の運動の詳細を明らかにすることが可能である。電子の磁気回転比は、NMRで通常使用される核より数桁大きいため、スピン標識が必要とはいえ、この技術はより高感度である。
【0021】
「アイソトープ」という用語は、化学元素で、最も豊富な種とは異なる原子量(質量数)を有する元素の化学種を示す。元素のアイソトープは、同数のプロトンを有するが(同じ原子番号)、中性子の数が異なる核を有する。
【0022】
EPRまたはNMRに好適なアイソトープは、ゼロ以外の核スピンを有する必要がある。現在使用されている最も一般的なアイソトープは、1H、2D、15N、13C、および31Pである。
【0023】
本発明では、キナーゼのP-ループ内の特定位置に、チオール反応性スピン標識を単独で使用することも可能である一方、P-ループにおいてチオール反応性スピン標識で特異的に標識したキナーゼをアイソトープでも標識することが好ましい(さらなる詳細を以下に記載する)。しかし、アイソトープ標識のみを使用する場合、アイソトープはキナーゼのP-ループ内の特異的な所望の位置にのみ存在し、よってキナーゼの他の位置は標識されないことが好ましい。
【0024】
「濃縮アイソトープ」という用語は、例えば、アイソトープが化合物に導入されるように、チオールまたはアミノ反応性標識がアイソトープを使用して合成された化合物か、チオールまたはアミノ反応性標識がアイソトープと反応した化合物を示す。化合物は、1以上の異なる種の1以上のアイソトープを含んでいてもよい。標識の位置および数に関しては、アイソトープについても上述と同様のことが適用される。
【0025】
標識は、キナーゼの触媒活性を有意に阻止または阻害せず、その安定性を妨げないように位置しなければならない。原理上、本発明のアッセイは、本発明の標識キナーゼの触媒活性の測定には依存せず、従って、キナーゼの基質の予備知識を必要としない。しかし、本質的に触媒活性を妨げないことにより、本発明の標識キナーゼに対する潜在的阻害剤の結合活性を、本発明の標識キナーゼが由来する野生型キナーゼと、合理的に比較され得ることが好ましい。アミノ酸、例えばシステインをアイソトープで標識し、配列中に既に取り込まれたアイソトープで標識したアミノ酸を有するキナーゼを発現している宿主生物を成長させることによって製造したキナーゼの場合、キナーゼの活性阻害または安定性の妨害は起こりそうにない。一方、標識が導入されるP-ループ内の位置を選択する際には、注意もしなければならない。選択位置に好適なアミノ酸が存在しない場合、前記の位置に存在するアミノ酸は、ペプチド結合中に包含されるα-アミノ基以外の遊離チオールまたはアミノ基を含有するアミノ酸で置換しなければならない。P-ループはキナーゼにATPase活性をもたらす。従って、好適な標識位置は、キナーゼがそのATPase活性の少なくとも70%、好ましくは少なくとも80%、より好ましくは少なくとも90%、最も好ましくは100%を保持するように選択されるべきである。アミノ酸の置換前後にキナーゼの活性および安定性を評価する試験方法は、当業者によく知られており、精製タンパク質の目視検査、円偏光二色性(CD)分光法、結晶化および構造決定、酵素活性アッセイ、タンパク質融解曲線、示差走査熱量測定およびNMR分光法が挙げられる。
【0026】
上述した通り、P-ループは、ATPase/GTPase中に、それぞれATP/GTPリン酸結合モチーフとも呼ばれる、高度に保存された高グリシンモチーフG-X-G-X-X-Gを含む。保存されたグリシンは、酵素の結合基質にリン酸を効率的に運ぶためのATPまたはGTPのリン酸の最適な位置決めに、非常に重要であることが示唆される。原理上、前記の保存されたモチーフ内の全てのアミノ酸は、本発明による置換および/または標識化に選択されるかもしれないが、高グリシンモチーフの可変位置の、より保存度が低いアミノ酸(Xと示す)が選択されることが好ましい。保存されたグリシン残基の一つを選択することは、キナーゼのATPase活性を妨害する可能性があり、このことは、上記でも述べた通り、自然発生キナーゼと比較して少なくとも同等の、好ましくは本質的に変わらない触媒活性を有する標識キナーゼを得るためには、好ましくは避けるべきである。さらに好ましくは、置換されるX位置のアミノ酸は、フェニルアラニンまたはチロシン等の芳香族アミノ酸である。フェニルアラニンおよびチロシンは両者ともかさ高く、本発明による共有結合した標識、特にチオール反応性標識アクリロダンと比較した場合、結合しているリガンドとともに同様の立体構造の再配置を採ると考えられる。
【0027】
その際、好ましくは活性状態の野生型キナーゼであるキナーゼの、触媒活性の少なくとも90%が保持され、好ましくは少なくとも95%、より好ましくは少なくとも98%が保持される場合、触媒活性の阻害は存在しない。最も好ましくは、キナーゼの触媒活性は完全に保持される。従って「触媒活性を阻害しない」という用語は、触媒活性が合計100%未満であるいくつかの態様において、「本質的に触媒活性を妨げない」という意味と同じであると考えられ、この意味を有している。触媒活性は、本発明の標識キナーゼにおける阻害剤のIC50値と、それが由来する非標識キナーゼとを比較することにより、間接的に決定し得る(ATP-Kmに相当するATPの量を使用する)。IC50値が同一の範囲内である場合、即ち、IC50値が5倍を超えて異ならない場合、このことは、触媒活性が本質的に同じであること(およびキナーゼの修飾がキナーゼに対する阻害剤の親和性を変えなかったこと)を示す。本発明の標識キナーゼおよび非標識キナーゼの差が、4倍以下であることが好ましく、より好ましくは3倍以下、さらにより好ましくは2倍以下である。当業者は、5倍以下の両IC50値の差は、充分に、これらの測定に関する通常の分散の範囲内であることを承知している。このようなIC50値は、両キナーゼの触媒活性が本質的に同じであることを保証する。安定性については、導入されたアミノ酸は、タンパク質の構造的安定性を保証する必須の分子間接触を妨げないため、キナーゼは本書に記載した生物学的機能を果たし得る。
【0028】
現在存在するスクリーニング方法の欠点を克服するため、本発明は、例えば蛍光タグ付けされたキナーゼを作る、標識化戦略であって、(i)キナーゼ阻害剤の結合に感受性が高く、(ii)リガンドの結合および解離のキネティクスのリアルタイム測定に使用することが可能であり、(iii)これらのリガンドのKdの直接測定に使用することが可能であり、かつ(iv)迅速で、ロバスト性があり、再現性があり、ハイスループット・スクリーニング方法に適応可能である、標識化戦略を包含する。
【0029】
先行技術と対照的に、かつ、添付の実施例で実証した通り、本発明は、キナーゼおよびこれらのキナーゼを用いたスクリーニング方法を提供し、本発明により、削減された労力と材料で、優れた信頼性を有する阻害剤のスクリーニングが可能になる。これは、例えばキナーゼのP-ループにおける立体構造の変化が引き起こす、環境の変化に対する反応におけるキナーゼの挙動を標識が変化させるような、キナーゼの標識化戦略を提供することによって、本質的に達成される。
【0030】
キナーゼ活性のモジュレータをスクリーニングする、従来のキナーゼアッセイ以外に、最近、様々なアプローチが開発されている。しかし、これらのアプローチの多くは、深刻な問題に悩まされている。例えば、アニス(Annis)ら(2004年)は、親和性選択的質量分析(AS-MS)を用いるアプローチを記載している。この方法は、ハイスループット・スクリーニングに好適であることが記載されている。しかし、各プローブの試験の前に、サイズ排除クロマトグラフィー工程を適用しなければならず、これは時間がかかり、多量の材料が必要である。
【0031】
ドゥ・ロリミエル(De Lorimier)ら(2002年)は、小分子リガンドに結合する細菌性タンパク質に基づいた一群のバイオセンサーを記載しており、このリガンドは種々の環境感受性フルオロフォアで修飾および標識されていた。リガンドの結合に際し、フルオロフォアはそれらの発光波長および/または強度を変化させ、それによりプローブに結合した特異的リガンドの存在および/または濃度を示す。しかし、特異的阻害剤のスクリーニングにおけるキナーゼの標識および前記のキナーゼの使用については、開示も示唆もされていない。
【0032】
より最近では、p38αキナーゼ由来の、予め結合した(prebound)プローブの置換に基づく、2つのさらなる結合アッセイも報告された:1つはフルオロフォア標識阻害剤を利用し(テクル(Tecle)ら、2009年)、他方は酵素断片の相補性に基づくアプローチを使用したものである(クルター(Kluter)ら、2009年)。後者の場合、化学発光の読み出しが、予め結合した阻害剤−ペプチドプローブの置換によって発生したが、その後、β-ガラクトシダーゼを補体活性化して、アッセイの読み出しとなる化学発光を触媒する。これらのアプローチは終点測定を用いる置換リガンドの親和性の決定に好適であることが実証されたが、反応速度論的パラメータ(konおよびkoff)の分析は、p38α由来の選択されたピラゾロウレア系プローブの解離が詳しく特性化された通り緩徐であり、シグナルの検出速度が制限されることから、簡易性が劣る(パルゲリス(Pargellis)ら、2002年)。
【0033】
本発明の根底にある原理は、II型またはIII型阻害剤の結合に際し、活性化ループの立体構造変化にP-ループが反応することである。活性化ループは、活性部位の入り口付近の柔軟な断片であり、殆どのキナーゼの基質結合裂隙(cleft)を形成し、1以上のアミノ酸をリン酸化して、タンパク質キナーゼスーパーファミリー全体の重要な調節メカニズムを提供し得る(ジョンソン(Johnson)およびルイス(Lewis)、2001年;テイラー(Taylor)およびラズィオ-アンゼルム(Radzio-Andzelm)、1994年;ジョンソンら、1996年)。活性化ループは、殆どのキナーゼにおいて柔軟なループを形成するいくつかのアミノ酸より成り、ATP結合部位において高度に保存されたアスパラギン酸-フェニルアラニン-グリシン(DFG)モチーフから始まり、キナーゼのN-およびC-ローブの間まで延びる。活性化ループは、酵素的キナーゼ活性に極めて重要な構造的構成要素である。活性化ループは、基質結合裂隙の一部であり、特異的基質の認識を助けるいくつかのアミノ酸残基を含有し、また、リン酸化し得るセリン、トレオニンまたはチロシンを含む。活性化ループの立体構造は、DFG-in(活性キナーゼ)とDFG-out(不活性キナーゼ)構造の間で力学的に平衡であると考えられている。相互作用のパートナー(他のタンパク質またはDNA)のリン酸化および/または結合により、平衡はシフトする。DFG-in構造において、モチーフに含有されるアスパラギン酸は、ATP結合部位に向けられ、隣接するフェニルアラニンはATP部位から離されて、隣接するアロステリック部位に向けられる。活性化ループの一部を形成する保存されたDFGモチーフがin-構造を採用する場合、ATP-競合阻害剤(I型阻害剤)はキナーゼに結合し得る。DFG-out構造において、これらの残基の位置は、向きが180°反転する。活性化ループのout-構造は、ATPと基質の結合を妨げる。
【0034】
上述の通り、リガンドおよび基質のATP結合部位への侵入を制御することに加えて、P-ループは周囲の溶媒からのATPおよび他のリガンドの保護に役立つ。ATP結合ポケットにおけるいくつかのI型阻害剤の結合に関し、種々の立体構造を採ることが示されている(ハンクス(Hanks)およびハンター(Hunter)、1995年;マペッリ(Mapelli)ら、2005年)。
【0035】
本発明に従い、蛍光-またはスピン-標識P-ループアッセイシステムを用いて、アロステリック阻害剤(図1c参照)を検出した。結合したフルオロフォアまたはスピン標識により、キナーゼの活性化ループがDFG-out構造を採った際に起こる、P-ループにおける運動が報告される。添付の実施例に示した通り、Cys残基を部位特異的突然変異誘発によりGly-X-Gly-X-X-Glyモチーフの第三のGlyの直前の位置に導入して、環境感受性のフルオロフォアであるアクリロダンでP-ループを特異的に標識することにより、阻害剤のスクリーニングに役立つ能力を有するキナーゼが得られる。この位置の残基は、全てのヒトキナーゼのおよそ80%において、TyrまたはPheとして保存され、このことから、これらの側鎖の芳香環系について、このループと他の構造的特徴およびリガンドとのクロストークを媒介する役割を有することが示唆される。
【0036】
さらに重要なことには、この観察により、アシクロダンの平面環系の導入はキナーゼによる耐性が良好であろうことが示唆された。
【0037】
本発明者らは、最近、標的キナーゼの活性化ループにタグ付けする、ロバストなアッセイシステムを開発した(同時係属出願欧州特許EP 08 01 3340号および欧州特許EP 08 02 0341号)が、それにより、種々のリガンドの解離定数(Kd)、速度定数(kon)および解離速度定数(koff)の直接測定が可能になり、初めてcSrcおよびp38αのIII型リガンドが特定でき、強力なII型阻害剤であるゲートキーパーの変異薬物耐性cSrc-T338Mが開発された。さらに、p38αのDFG-out構造を安定化する、p38αにおけるチアゾール-尿素骨格に対する新規なIII型結合様式およびいくつかの独特なI型リガンドが特定されるかもしれない。DFG-out構造を安定化するリガンドの検出感度は、化合物ライブラリをスクリーニングするこのアプローチを使用することによって有意に上昇するが、これはリン酸化していない不活性形態のキナーゼを利用するからであり、このことは、DFG-out構造の採用を支持する。これらの以前の研究により、これらの型のリガンドのスクリーニングおよび濃縮に使用し得るアッセイが広範囲にわたって関与することが強調される。しかし、活性化ループの標識化に際し、標的キナーゼの既知の阻害剤のDFG-in/out構造的平衡の変動または親和性の有意な変化より生じる、キナーゼ活性の潜在的変化を避けるため、本発明によって提供された通りの、II型およびIII型阻害剤を特定および特性化する別の標識化戦略により、前記の活性変化が起こりにくくなる。
【0038】
添付の実施例に示した通り、本発明は、フルオロフォアの環境における変化、例えば、活性化ループのDFG-out構造への運動によるP-ループにおける立体構造変化を引き起こすII型およびIII型阻害剤等の、種々の結合モードによる阻害剤の結合を高感度に検出するP-ループ標識キナーゼの能力を実証し、また、その蛍光特性を変化させる(図2a参照)。II型およびIII型阻害剤は、HTSフォーマットにおいて蛍光シグナルまたはKdの時間依存性の変化を経時的にモニターすることによって、またはキュベットにおいてkonを測定することによって(I型バインダーは<5)、容易に識別される。また、本発明のアッセイは、独特の結合モードにより、DFG-out構造を安定化するI型リガンドを強力に検出する能力がある。このようなリガンドは、ATP-結合部位内で結合するが、阻害剤分子であるDFGモチーフの高度に保存されたPheと、P-ループ中の選択された標識位置(p38αにおけるTyr35)に典型的に見られる残基の平面環系との間に形成する、独特な環のスタッキング相互作用を利用する。最後に、DFG-in立体構造に結合する、いくつかのI型阻害剤は、記載されたP-ループのTyr/Phe側鎖と直接的に相互作用することが示された(タマヨ(Tamayo)ら、2005年)。この位置を使用してキナーゼを標識することにより、活性化ループのDFG-out構造または運動を引き起こさずに、これらの型の阻害剤を検出することも可能になる(図3B、右パネル)。活性化ループがフルオロフォアで直接標識される、最近報告されたアッセイと比較して(欧州特許出願EP 08 01 3340号およびEP 08 02 0341号)、このアッセイシステムもリン酸化されていない形態のキナーゼを利用し、II型およびIII型阻害剤によって引き起こされたもの等の、リガンド結合に相関する活性化ループの立体構造変化を検出する、強力な代替のスクリーニングツールを提供する。さらには、アロステリックポケットの薬剤可能性は、キナーゼ間で異なるようであり、キナーゼがDFG-out構造を採用する能力に感受性があり、従って、本発明は、P-ループと直接相互作用し、そこで立体構造変化を誘発する、高親和性I型化合物を検出および設計する、魅力的な代替のアプローチとなる。このようなI型リガンドを特定する利点は、それらがより保存性が低いアロステリック部位の方向に延びるII型阻害剤へのさらなる開発の出発点となり得ることから、過小評価すべきではない(リウ(Liu)およびグレイ(Gray)、2006年)。
【0039】
いくつかのI型阻害剤は、DFG-out構造も安定化する。本発明を用いてI型DFG-outバインダーを検出できる鍵は、両基質およびATPの非存在下で、非リン酸化形態のキナーゼを用いたスクリーニングを実施する能力である。上記した通り、非リン酸化形態のキナーゼは、DFG-out構造を採用する可能性が高く、この構造において、活性化ループのDFGモチーフまたはN-末端領域における残基は、リガンドと相互作用し、ATP部位中に反転してATP-競合リガンドに接触することにより、親和性を高め得る。これは、リン酸化したキナーゼを必要とする活性に基づく古典的アッセイと対照的であるが、リン酸化キナーゼはDFG-in構造中に見つかる可能性が高いため、DFG-outバインダーの親和性を低下させ、このような好ましいヒットが検出される可能性が低くなる(スィーリガー(Seeliger)ら、2007年)。スクリーニング・キャンペーンでの古典的な活性に基づくアッセイの確立された使用は、DFG-outバインダーの検出の感受性を抑制し、例えば、VEGFR2阻害剤であるCP547632の、VEGFR2以外の活性(即ち、リン酸化)キナーゼに対する結合について、文献中に情報がないことが説明されるかもしれない。CP547632は特異性が高いことが報告されており、それにより、血管新生の阻害によって腫瘍の生長および増殖を抑止する臨床試験において、VEGFR2阻害剤として適用されている。本発明のアプローチにより、非リン酸化p38αを用いて検出されるCP547632の親和性がマイクロモル以下であることを考えると、これらの知見は、この臨床的に意義のある化合物または他のキナーゼ関連疾患の治療のための類縁誘導体のさらなる研究をも刺激し得る。キナーゼは、細胞内にリン酸化および非リン酸化の両形態で存在し、これらの種の相対存在量がキナーゼ活性およびシグナル伝達経路を制御する。従って、非リン酸化キナーゼもまた、生物学的関連性を示し、魅力的な薬剤標的である。加えて、p38αと複合したCP547632について本書で示した構造的情報(即ち、新たな型であるヒンジ接触)は、この分子のアフィン部分を基礎として隣接するアロステリック部分に延びるさらなる医薬品化学への取り組みを刺激し、不活性キナーゼ立体構造に結合する、薬理学的により望ましいII型阻害剤を創出し得る。
【0040】
高グリシンループを標識することにより、適用されるキナーゼにおいてDFG-outバインダーを特定するという目標が達成されるだけでなく、キナーゼの高グリシンループにおける立体構造変化を直接誘導することでDFG-in構造に対する親和性を得るI型リガンドの検出も可能になる。この特徴は、以前のアッセイを凌ぐ、本発明のアプローチのさらなる利点である。キナーゼの例として、高グリシンループを標識したp38αを使用することにより、Scios-469等のI型阻害剤が高感度で検出されたが、この型の阻害剤はp38αのDFG-in構造に結合する。このような化合物は、リガンドの周囲の溶媒からの保護に役立つ、高グリシンループの立体構造変化を誘導することによってキナーゼに対する親和性を得る(ハンクス(Hanks)およびハンター(Hunter)、1995年;マペッリ(Mapelli)ら、2005年;パテル(Patel)ら、2009年)。常にではないが、しばしばこれらの相互作用に関与する高グリシンループ内の位置は、80%超のキナーゼにおいて芳香族TyrまたはPheとして保存されることから、本発明は、現在のスクリーニングアッセイを、容易に誘導できるDFG-in/out平衡によって制御されない多数のキナーゼ等の、さらなるキナーゼに広げる。高グリシンループと相互作用するI型阻害剤の検出は、ヒンジ領域の接触の新規な型を特定することに従来のように焦点を置くことを回避しつつ、上記の相互作用を活用する、新たな骨格の開発についての見識を提供し得る。また、高グリシンループの立体構造変化も、I型阻害剤の特異性を向上させるさらなる方法を提供し得る。
【0041】
好ましい態様において、キナーゼは、セリン/トレオニンキナーゼまたはチロシンキナーゼである。
【0042】
別の好ましい態様において、キナーゼはMEKキナーゼ、CSK、オーロラキナーゼ、GSK-3β、sSrc、EGFR、Abl、DDR1、LCK、CDK、p38αまたは別のMAPKである。
【0043】
マイトジェン活性化タンパク質(MAP)キナーゼ(EC 2.7.11.24)は、細胞外刺激(マイトジェン)に反応するセリン/トレオニン特異的タンパク質キナーゼであり、遺伝子発現、有糸分裂、分化、および細胞生存/アポトーシス等の種々の細胞活動を調節する。細胞外刺激により、MAPキナーゼ、MAPキナーゼキナーゼ(MKKまたはMAP2K)およびMAPキナーゼキナーゼキナーゼ(MKKKまたはMAP3K、EC 2.7.11.25)で構成されるシグナル伝達カスケード(「MAPKカスケード」)を介して、MAPキナーゼは活性化される。
【0044】
細胞外刺激によって活性されるMAP3Kは、そのセリンおよび/またはトレオニン残基でMAP2Kをリン酸化し、その後、このMAP2Kは、そのセリンおよび/またはチロシン残基上のリン酸化を通してMAPキナーゼを活性化する。このMAPキナーゼシグナル伝達カスケードは、酵母から哺乳類まで、進化的によく保存されている。
【0045】
これまでに、哺乳類において、明瞭な6つのグループのMAPKが特性化されている:
【0046】
1. 細胞外シグナル調節キナーゼ(ERK1、ERK2)。ERK(古典的MAPキナーゼとしても知られている)シグナル伝達経路は、好ましくは、成長因子およびホルボールエステル(発癌プロモーター)に反応して活性化され、細胞増殖および細胞分化を調節する。
2. c-Jun N-末端キナーゼ(JNK)、(MAPK8、MAPK9、MAPK10)、ストレス活性化タンパク質キナーゼ(SAPK)としても知られている。
3. p38アイソフォームは、p38α(MAPK14)、p38β(MAPK11)、p38γ(MAPK12またはERK6)およびp38δ(MAPK13またはSAPK4)である。JNKおよびp38シグナル伝達経路の両者は、サイトカイン、紫外線照射、熱ショックおよび浸透圧衝撃等のストレス刺激に反応性があり、細胞分化およびアポトーシスに関与する。p38αMAPキナーゼ(MAPK)は、RKまたはCSBPとも呼ばれるが、サイトカインおよびストレスに対する細胞反応を制御する信号伝達カスケードに関与する、酵母HOGキナーゼの哺乳類相同分子種である。SAPK/JNK経路と同様に、p38 MAPキナーゼは、浸透圧衝撃、炎症性サイトカイン、リポ多糖類(LPG)、紫外光および成長因子等の様々な細胞ストレスによって活性化される。p38 MAPキナーゼは、Thr180およびTyr182におけるリン酸化によって活性化される。
4. ERK5(MAPK7)は、最近発見されたが、成長因子およびストレス刺激のいずれでも活性化され、細胞増殖に関与する。
5. ERK3(MAPK6)およびERK4(MAPK4)は、活性化ループにSEG(セリン−グルタミン酸−グリシン)モチーフを有し、C-末端延長部においてのみ大きな差異を示す、構造的に関連した異型MAPKである。
6. ERK7/8(MAPK15)は、MAPKファミリーで最も最近発見されたメンバーであり、ERK3/4と類似の挙動を示す。
【0047】
マイトジェン活性化タンパク質キナーゼキナーゼは、マイトジェン活性化タンパク質キナーゼをリン酸化する、キナーゼのファミリーを形成する。また、これらは、MAP2K2としても知られ、EC 7.2.12.2に分類される。7つの遺伝子が存在する。これらは、MAP2K1(MEK1)、MAP2K2(MEK2)、MAP2K3(MKK3)、MAP2K4(MKK4)、MAP2K5(MKK5)、MAP2K6(aka MKK6)、MAP2K7(MKK7)をエンコードする。p38(MKK3およびMKK4)、JNK(MKK4)、およびERK(MEK1およびMEK2)の活性化剤は、独立したMAPキナーゼシグナル伝達経路を特徴付ける。
【0048】
オーロラキナーゼA(オーロラ、オーロラ-2、AIK、AIR-1、AIRK1、AYK1、BTAK、Eg2、MmIAK1、ARK1およびSTK15としても知られている)、B(オーロラ-1、AIM-1、AIK2、AIR-2、AIRK-2、ARK2、IAL-1およびSTK12としても知られている)、およびC(AIK3としても知られている)は、細胞質分裂および無調節な染色体分離等の、いくつかの生物学的プロセスに関与している。これらの重要な有糸分裂の調節因子は、多様な固形腫瘍において過剰に発現する。このセリン/トレオニンキナーゼのファミリーの一つであるヒトオーロラAは、すい臓癌における薬剤標的として提案されている。オーロラAの最近の三次元構造決定により、オーロラキナーゼは活性化ループ領域の周辺で独特な立体構造を現すことが示された。この特性は、オーロラキナーゼの阻害剤の探索および開発を促進したが、新規な発癌抑制剤としても機能する可能性がある。
【0049】
グリコーゲン合成酵素キナーゼ3(GSK-3)はセリン/トレオニンタンパク質キナーゼであり、セリン/トレオニンキナーゼ活性に加えて、チロシン残基上で自己リン酸化する独特な能力を有する。GSK-3による標的タンパク質のリン酸化は、通常、それらの活性を阻害する(グリコーゲン合成酵素およびNFATの場合と同様)。GSK-3は、キナーゼの中では他とは異なり、標的タンパク質を最初にリン酸化するのに通常「プライミングキナーゼ」が必要であり、その後にのみ、GSK-3は標的タンパク質をさらにリン酸化することが可能である。哺乳類において、GSK-3は、GSK-3αおよびβの2つの知られている遺伝子でエンコードされている。胚の発生過程でのパターン形成および細胞増殖の役割に加えて、腫瘍形成における細胞分裂およびアポトーシスの調節による役割について、最近の証拠がある。ヒトグリコーゲン合成酵素キナーゼ-3β(GSK3β)もまた、肥満、糖尿病、アルツハイマー病および双極性障害等のいくつかの病理生理学的状態と関連する。
【0050】
プロトオンコジェニックチロシンキナーゼのSrcファミリーは、細胞運動および細胞増殖の中核をなすインテグリン依存性シグナルを伝達する。Srcファミリーは、9のメンバーを含む:Src、Lck、Hck、Fyn、Blk、Lyn、Fgr、Yes、およびYrkである。これらのキナーゼは、現在の、癌を無調節な細胞成長および細胞分裂を伴う疾患として理解することに役立ってきた。CSrcプロトオンコジーンは、cSrcチロシンキナーゼをコードする。そのキナーゼドメインに加えて、cSrcはSH2ドメインおよびSH3ドメインをさらに含み、それらはSrcキナーゼドメインと多酵素複合体を形成するためのアダプタータンパク質として働く。これらのドメインもまた、cSrcキナーゼドメインの自己抑制に関与する。この遺伝子における突然変異は、癌細胞の悪性進行に関与する可能性がある。このタンパク質は、ヒト白血球特異的タンパク質チロシンキナーゼ(Lck)のTyr-504残基を特異的にリン酸化するが、この残基は、負の調節部位として作用する。またこのタンパク質は、LynおよびFynキナーゼにも作用し得る。
【0051】
白血球特異的タンパク質チロシンキナーゼ(Lck)は、T-細胞等のリンパ球の内部に見られるタンパク質である。Lckは、リンパ球の細胞内シグナル伝達経路に関与する、あるタンパク質のチロシン残基をリン酸化する、チロシンキナーゼである。LckのN-末端尾部はミリストイル化およびパルミトイル化され、タンパク質を細胞の原形質膜に繋ぐ。このタンパク質はさらにSH3ドメイン、SH2ドメインを含み、C-末端部分にはチロシンキナーゼドメインを含む。Lckで始まるチロシンリン酸化カスケードは、カルシウム(Ca2+)イオンの細胞内移動およびリンパ球内での重要なシグナル伝達カスケードの活性化に至る。これらはRas-MEK-ERK経路を含むが、これは続いて、NFAT、NFκB、およびAP-1等の転写因子を活性化し、転写因子はその後、多量の遺伝子産物の産生を調節するが、遺伝子産物は、最も顕著には、活性化リンパ球の長期の増殖および分化を促進する、インターロイキン−2等のサイトカインである。Lckの異常発現は、胸腺腫瘍、T-細胞白血病および結腸癌に関連している。
【0052】
チロシンキナーゼのSrcファミリーの触媒活性は、C末端付近に位置するチロシン残基(cSrcにおけるTyr 527)のリン酸化によって抑制されるが、これはC-末端Srcキナーゼ(Csk)によって触媒される。殆どのチロシンキナーゼの乱雑さを考えると、SrcファミリーキナーゼのC-末端尾部がCskの唯一知られている標的であることは注目に値する。おそらく代表的なSrcキナーゼである、CskとcSrcとの相互作用により、cSrcのC-末端尾部はCskの活性部位の末端に位置する。Cskは、このドッキングメカニズムが存在しない基質をリン酸化することはできないが、これは、基質を認識するために殆どのチロシンキナーゼが使用する従来の基質結合部位が、活性化ループにおける欠失により、Cskにおいて不安定化されるためである(レビンソン(Levinson)、2008年)。
【0053】
上皮成長因子受容体(EGFR;ErbB-1;ヒトにおけるHER1)は、細胞外タンパク質リガンドの上皮成長因子ファミリー(EGF-ファミリー)のメンバーに対する細胞表面受容体である。上皮成長因子受容体は、受容体のErbBファミリーの一メンバーであり、以下の4つの密接に関わる受容体チロシンキナーゼのサブファミリーである:EGFR(ErbB-1)、HER2/c-neu(ErbB-2)、Her 3(ErbB-3)およびHer 4(Erb-4)。活性EGFRは、二量体として発生する。EGFR二量化は、細胞外受容体ドメインへのリガンド結合によって誘導され、その内因性の細胞内タンパク質-チロシンキナーゼ活性を刺激する。その結果、EGFRのC-末端(細胞内)ドメインにおいて、いくつかのチロシン残基の自己リン酸化が起こる。この自己リン酸化は、自身のホスホチロシン結合SH2ドメインによってリン酸化チロシンと関係する、いくつかの他のタンパク質によって、下流の活性化およびシグナル伝達を引き起こす。また、EGFRのキナーゼドメインは、凝集した他の受容体のチロシン残基を交差リン酸化(cross-phosphorylate)することができ、そのような方法でそれ自身を活性化し得る。EGFRシグナル伝達カスケードは、いくつかの下流のシグナル伝達タンパク質を活性化するが、これらのタンパク質は次に、いくつかのシグナル形質導入カスケード、主としてMAPK、AktおよびJNK経路を開始させて、DNA合成および細胞増殖を導く。このような経路は、細胞遊走、接着、および増殖等の表現型を調節する。EGFR過剰発現(上方調節として知られている)または過剰活性を導く突然変異は、多数の癌と関係している。結果として、EGFRの突然変異は、いくつかの型の癌において特定されており、抗癌療法での拡大する分野の標的である。
【0054】
ABL1-プロトオンコジーンは、細胞質および核のタンパク質キナーゼをエンコードするが、このキナーゼは、細胞分化、細胞分裂、細胞接着およびストレス反応の過程に関係している。c-Ablタンパク質の活性は、そのSH3ドメインによって負に調節される。SH3ドメインの遺伝的欠失は、ABL1を発癌遺伝子に換える。この遺伝的欠失は、(9;22)遺伝子転座によって起こるが、慢性骨髄性白血病の多くの症例に存在するBCR(MIM:151410)およびABL1遺伝子の頭尾融合を引き起こす。遍在的に発現したABL1チロシンキナーゼのDNA結合活性は、CDC2-媒介リン酸化によって調節されるが、このことはABL1の細胞周期機能を示唆している。
【0055】
DDR1またはCD167a(分化167aのクラスター)としても知られる、ディスコイジンドメイン受容体ファミリーのメンバー1は、正常および形質転換した上皮細胞において広く発現されている受容体チロシンキナーゼ(RTK)であり、種々のタイプのコラーゲンによって活性化される。このタンパク質は、細胞外ドメインにおいて、ディクチオステリウム・ディスコイデウム(Dictyostelium discoideum)タンパク質ディスコイジンIに類似した相同領域を有する、チロシンキナーゼ受容体のサブファミリーに属する。その自己リン酸化は、これまで試験した全てのコラーゲン(I型およびVI型)により達成される。現場(In situ)研究およびノーザンブロット分析により、このエンコードしたタンパク質の発現が、特に腎、肺、胃腸管、および脳の上皮細胞に限定されることが示された。加えて、このタンパク質は、乳房、卵巣、食道、および小児脳由来のいくつかのヒト腫瘍において、有意に過剰発現している。
【0056】
上述のキナーゼは、全て、現在のところ好適な治療がないか改良した治療計画が望まれている、癌のような疾患の発症に関与するため、好ましい態様である。
【0057】
構造的情報が入手可能であったキナーゼである、p38αを使用して、本発明者らは、スクリーニングの目的での本発明の標識キナーゼの適用性を実証した。予想外に、このキナーゼは最低限の労力で標識化用に調製することができたが、また、標識したキナーゼは所望の特性を発揮し、即ち、知られている阻害剤BIRB-796およびいくつかのより小さいBIRB-796類似体の場合、導入した標識が、特異的阻害剤の結合により誘導される立体構造変化を検出するのに好適であることが判明した。
【0058】
本発明にしたがって標識した形態で、特異的阻害剤のスクリーニングに適用し得るさらなるキナーゼは、MKK7である(例えば、ワン(Wang)ら、2007年に概説)。MKK7のI型およびII型阻害剤の両者は分析されており、それらの薬理学的特性は改良されて、実施例7に詳述した通り、より強力な阻害剤を得ることができた。
【0059】
別の好ましい態様において、標識されているアミノ酸は、システイン、リシン、アルギニンまたはヒスチジンである。
【0060】
システインは、遊離チオール基を有し、一方、リシン、アルギニンまたはヒスチジンはそれぞれ、少なくとも1つの遊離アミノ基を保持する。
【0061】
別の好ましい態様において、P-ループの外部に存在する1以上の溶媒にさらされるシステインが欠失しているか置換されている。
【0062】
標識化に先立ち、興味のあるキナーゼ中に、遊離チオールまたはアミノ基を有するアミノ酸が2つ以上存在する場合、P-ループにおけるアミノ酸の特異的標識は可能ではないかもしれない。従って、上で論じた通り、キナーゼ中に存在し、遊離チオールまたはアミノ基を有するアミノ酸は、溶媒にさらされることが予測されるか示される場合、取り除かれるか、遊離チオールまたはアミノ基を有しない別のアミノ酸で置換されるべきである。興味のあるキナーゼ中に天然に存在し、溶媒にさらされるシステインは、P-ループの外部に配置し得るが、この場合、システインは取り除かれるか、遊離チオール基を有しない別のアミノ酸で置換されるべきである。このことは、遊離アミノ基を有するアミノ酸にも同様に適用され、遊離アミノ基は、反応性遊離アミノ基を有しないアミノ酸で置換されるべきである。遊離アミノ基を有する1以上のアミノ酸がP-ループ中に既に存在する場合、標識されるアミノ酸に加えて、遊離アミノ基を有し、P-ループ中に存在するアミノ酸は、置換または取り除かれるべきであるが、キナーゼに対するこれらの変異のいずれもその触媒活性を阻害しないか、その安定性を妨げない。要約すると、前記の変異により、P-ループの所望の位置で特異的に標識されるキナーゼが得られる。
【0063】
「溶媒にさらされる」という用語は、タンパク質の三次元構造の文脈において、タンパク質の一部のアミノ酸の位置に言及する。タンパク質体の内部に埋め込まれているアミノ酸は、他のアミノ酸に完全に囲まれているため、溶媒とは全く接触しない。対照的に、溶媒にさらされるアミノ酸は、部分的または完全に周囲の溶媒にさらされるため、それらアミノ酸を修飾する可能性がある化学物質に接近しやすい。このことは、例えば、遊離チオール-またはアミノ-基を有する、溶媒にさらされるアミノ酸と反応し得る、本発明に使用されるチオール-またはアミノ-反応性標識に適用する。
【0064】
「取り除く」という用語は、アミノ酸を別のアミノ酸と交換しない切除に言及し、一方「置換する」という用語は、アミノ酸の別のアミノ酸での代替に言及する。アミノ酸が別のアミノ酸で置換されるか取り除かれる場合、置換または取り除かれるアミノ酸は好ましくは、取り除かれるか置換されたアミノ酸によりキナーゼの触媒活性が阻害されず、得られるキナーゼの安定性が妨げられないように選択される。
【0065】
より好ましい態様において、キナーゼはp38αであり、標識されるシステインは配列番号1の35位に導入され、好ましくは配列番号1の119位および162位のシステインが、セリン等の遊離チオール基を有しない別のアミノ酸で置換されている。
【0066】
代わりのより好ましい態様において、キナーゼはMKK7であり、標識されるシステインは147位でP-ループ中に天然に存在し(MKK7のキナーゼドメインに対応する配列番号2における31位)、および、好ましくは218位、276位および296位のシステイン(配列番号2の102位、160位および180位)が、セリン等の遊離チオール基を有しない別のアミノ酸で置換される。218位、276位および296位にセリンに変異したシステインを有するMKK7キナーゼドメインを配列番号3に示す。
【0067】
一般に、アミノ酸置換は保護的であるべきである。システインについては、このことは、システインが好ましくはセリンで置換されることを意味する。一般に、異なるアミノ酸でのアミノ酸の置換は、PAM250 スコアリングマトリックスを使用して、置換が保護的であるかどうかの点で評価し得る。このマトリックスはしばしば、整列ペプチド配列の採点に使用され、これらの配列の類似性を決定する(ペアソン(Pearson)、1990年)。
【0068】
上述した通り、天然に存在しない場合、遊離チオールまたはアミノ基を有するアミノ酸をキナーゼのP-ループ中に導入しなければならない。p38αの場合、p38αについての入手可能な結晶構造を使用して、活性化(DFG-in)状態および不活化(DFG-out)状態の両者における構造研究を実施した。P38αはP-ループ中にシステインを保持していない。上記の構造研究により、P-ループ内に位置する35位でチロシンをシステインで置換することは、キナーゼの触媒活性または安定性に有意に影響しないことが示唆された。
【0069】
同時係属出願欧州特許EP 08 01 3340号から、配列番号1の119位と162位の2つのシステインは両者とも溶媒にさらされることが分かった。P-ループ内に位置しない2つのさらなるシステインを記録したシグナルが阻害される可能性を回避するため、これらの2つのシステインは好ましくは別のアミノ酸で置換され、好ましくはセリン等の、同等の大きさおよび構造のアミノ酸で置換される。
【0070】
p38αに相同なキナーゼを使用する場合、システインで置換されるアミノ酸の位置は、配列番号1の35位に対応し得る。キナーゼにおいていずれの位置が配列番号1の35位に対応するかを決定するため、使用したキナーゼを有する配列番号1の配列アラインメントが達成され得るが、これは例えば、CLUSTALW等の一般に入手できるプログラムを使用する。
【0071】
別の好ましい態様において、チオールまたはアミノ反応性フルオロフォアは、2つの置換基のうちの1つがチオールまたはアミノ反応性部分である、環境感受性の二置換ナフタレン化合物である。「環境感受性の」という用語は、1以上の波長でのその蛍光発光が変化するか、その全体の発光スペクトルが変化することで表される、その環境条件に対するフルオロフォアの感受性を意味する。このような変化を引き起こす条件は、例えば、活性化ループ、従ってP-ループにおける、極性変化または立体構造変化である。しかし、変化は、活性化ループに全く影響せず、P-ループで起こることもある。
【0072】
上記のタイプのフルオロフォアは、典型的には、周囲環境の極性に依存して、発光波長の強度およびシフトの両者において変化を呈する。この分類のフルオロフォアの例としては、6-アクリロイル-2-ジメチルアミノナフタレン(アクリロダン)、6-ブロモアセチル-2-ジメチルアミノ-ナフタレンバダン(バダン)、2-(4’-(ヨードアセトアミド)アニリノ)ナフタレン-6-スルホン酸ナトリウム塩(IAANS)、2-(4’-マレイミジルアニリノ)ナフタレン-6-スルホン酸ナトリウム塩(MIANS)、5-((((2-ヨードアセチル)アミノ)エチル)アミノ)ナフタレン-1-スルホン酸(1,5-IAEANS)および5-ジメチルアミノナフタレン-1-スルホニルアジリジン(ダンシルアジリジン)またはそれらの誘導体が挙げられる。
【0073】
環境の感受性によっては使用してもよい他のフルオロフォアは、クマリン系化合物、ベンゾキサジアゾール系化合物、ダポキシル系化合物、ビオシチン系化合物、フルオレスセイン;アレクサフロール(AlexaFlour)染料(モレキュラー・プローブス(Morecular Probes))、アット(Atto)フルオロフォア(アットテクノロジー(Atto Technology))またはルシファー・イエロー(Lucifer Yellow)等のスルホン化ローダミン系化合物である。クマリン系フルオロフォアは、環境に緩やかに感受性があり、例えば、7-ジエチルアミノ-3-(4’-マレイミジルフェニル)-4-メチルクマリン(CPM)が挙げられる。ベンゾキサジアゾールフルオロフォアもまた、タンパク質-フルオロフォア複合体を形成するために通常使用され、強力な環境依存性を有し、例えば7-フルオロベンズ-2-オキサ-1,3-ジアゾール-4-スルホンアミド(ABD-F)およびN-((2-(ヨードアセトキシ)エチル)-N-メチル)アミノ-7-ニトロベンズ-2-オキサ-1,3-ジアゾールエステル(IANBD)が挙げられる。PyMPOマレイミド(対チオール)またはスクシンイミドエステル(対アミン)および種々の他のダポキシル染料が良好な吸収性および極めて高い環境感受性を有する。例としては、1-(2-マレイミジルエチル)-4-(5-(4-メトキシフェニル)オキサゾール-2-イル)ピリジニウムメタンスルホネート(PyMPO-マレイミド)、臭化1-(3-(スクシンイミジルオキシカルボニル)ベンジル)-4-(5-(4-メトキシフェニル)オキサゾール-2-イル)ピリジニウム(PyMPO-スクシンイミジルエステル)およびダポキシル(2-ブロモアセトアミドエチル)スルホンアミドが挙げられる。しかし、それらの構造がより長く、より柔軟であるため、これらのプローブは、選択される標識部位によって、P-ループの運動またはP-ループと活性化ループとの間の相互作用に影響を与えるかもしれない。上記の物質の適用性は、個々のキナーゼおよび標識されるアミノ酸の位置に依存し、一部の例では本発明の方法における感受性を低下させる可能性があるとしても、原理上なお標識として適用され得る。上記の物質を好適なキナーゼと適合させることは、当業者は、本発明の教示と組み合わせた通常の手順で実施し得る。
【0074】
一般的に、キナーゼの触媒活性を阻害せず、またはキナーゼの安定性を妨げない限り、あらゆるフルオロフォアを使用し得る。このことは、フルオロフォアは好ましくは嵩高くなく、または伸長していないことを意味する。
【0075】
さらに好ましい態様において、チオール反応性スピン標識は、ニトロキシドラジカルである。
【0076】
スピン標識でタンパク質配列を部位特異的に標識する支配的な方法は、メタンチオスルホネートスピン標識とシステインとの間の反応であり、スピン標識したシステイン側鎖、CYS-SLを与える:
MeS(O)2SSR + R’SH ---> R’SSR + MeS(O)2SH
式中、Rはニトロキシド基であり、R’SHはシステインスルフヒドリルを有するタンパク質であり、R’SSRはスピン標識したタンパク質である。標識用のシステインは、固相技術または標準的組み換えDNA技術のいずれかにより、所望の配列位置に置かれる。
【0077】
さらに、本発明は、キナーゼ阻害剤のスクリーニング方法に関し、この方法は、(a) 本発明の(蛍光的またはスピン標識またはアイソトープ標識した)キナーゼを供給すること;(b) 前記の(蛍光的またはスピン標識またはアイソトープ標識した)キナーゼを候補阻害剤と接触させること;(c) 前記の工程(a)および工程(b)の蛍光標識したキナーゼの、励起の際の、1以上の波長での蛍光発光シグナルまたはスペクトルを記録すること;または(c)’ 前記の工程(a)および工程(b)のスピン標識またはアイソトープ標識したキナーゼの電子常磁性共鳴(EPR)または核磁気共鳴(NMR)スペクトルを記録すること;および(d) 工程(c)で記録される1以上の波長での蛍光発光シグナルまたはスペクトル、または工程(c)’で記録されるEPRまたはNMRスペクトルを比較することを含み;ここで、前記の工程(c)で得られる蛍光標識したキナーゼの、1以上の波長、好ましくは発光極大での蛍光強度の差、および/または、スペクトルにおける蛍光発光波長のシフト、または、前記の工程(c)’で得られるスピン標識またはアイソトープ標識したキナーゼの、EPRまたはNMRスペクトルにおける変化は、候補阻害剤がキナーゼ阻害剤であることを示す。
【0078】
キナーゼ阻害剤は、キナーゼの活性を阻害する能力のある物質である。キナーゼ阻害剤は、例えば、それらがアロステリック阻害剤(III型)であるか、またはATP結合部位に隣接するアロステリック部位に結合し、ATP結合ポケット内に到達するもの(II型)である場合、単一のキナーゼの作用をより特異的に阻害し得る。或いは、阻害剤は、多数のタンパク質キナーゼの作用を阻害し得るが、これは特に、タンパク質キナーゼ中に高度に保存されているATP結合ポケットに排他的に結合する(I型)場合である。
【0079】
候補阻害剤は、異なる化合物分類に属していてもよく、例えば、有機または無機小分子、タンパク質またはペプチド、DNAまたはRNA等の核酸が挙げられる。このような化合物は、分子ライブラリに存在するか、またはスクラッチから設計し得る。
【0080】
本発明による小分子は、分子量が2000 Da以下、好ましくは1500 Da以下、より好ましくは1000 Da以下、最も好ましくは500 Da以下の分子を含む。
【0081】
1以上の波長での蛍光発光シグナルまたはスペクトルの記録は、通常、蛍光分光計または蛍光光度計を使用して達成される。蛍光分光法または蛍光光度法または蛍光分光分析は、サンプルからの蛍光または他の放射光を分析する、電磁分光法の一種である。これらの分析は、ビーム光、通常紫外光の使用を含むが、これは特定の分子において電子を励起し、緩和の際により低いエネルギーの光を発し、必須ではないが、典型的には可視光を発する。
【0082】
本発明の方法に使用し得る、一般的な2つのタイプの装置がある:フィルター蛍光光度計は、フィルターを使用して入射光および蛍光を単離し、一方、蛍光分光計は、回折格子モノクロメーターを使用して、入射光および蛍光を単離する。両タイプは以下のスキームを利用する:励起源からの光は、フィルターまたはモノクロメーターを通過し、サンプルに当たる。一部の入射光はサンプルに吸収され、サンプル中の一部の分子は、蛍光を発する。蛍光は、すべての方向に発光する。この蛍光の一部は第二のフィルターまたはモノクロメーターを通過して検出器に到達するが、検出器は通常、入射光ビームに対し90°に設置し、透過または反射した入射光が検出器に到達するリスクを最小限に抑える。励起源として、種々の光源が使用され得るが、例えば、レーザー光線、フォトダイオード、およびランプが挙げられ;特にキセノンランプおよび水銀灯が挙げられる。検出器は、シングルチャネルまたはマルチチャネルのいずれであってもよい。シングルチャネル検出器は、一度に一つの波長の強度のみを検出し得るが、一方マルチチャネルは同時に全ての波長の強度を検出し、発光モノクロメーターまたはフィルターが不要になる。種々のタイプの検出器が利点および欠点の両方を有する。二重モノクロメーターおよび連続的励起光源を有する、最も用途の広い蛍光光度計は、励起スペクトルおよび蛍光スペクトルの両者を記録し得る。蛍光スペクトルを測定する場合、励起光の波長は、好ましくは高吸収の波長で一定に保たれ、発光モノクロメーターがスペクトルをスキャンする。励起スペクトルの測定には、発光フィルターまたはモノクロメーターを通過する波長を一定に保ち、励起モノクロメーターがスキャンする。蛍光強度が吸収に比例するため、一般に励起スペクトルは吸収スペクトルと一致する(概説は、レンデル(Rendell)、1987年;シャルマ(Sharma)およびシュルマン(Schulman)、1999年;ガウグリッツ(Gauglitz)およびボ-ディン(Vo-Dinh)、2003年;ラコビッツ(Lakowics)、1999年参照)。
【0083】
核磁気共鳴(NMR)は、原子核の量子力学的磁気特性に基づく物理現象である。奇数の陽子または中性子を含有する全ての核は、固有磁気モーメントおよび角運動量を有する。最も普通に測定される核は水素(1H)(天然存在度で最も受容的なアイソトープ)および炭素(13C)であるが、多くの他の元素のアイソトープ(例えば、113Cd、15N、14N、19F、31P、17O、29Si、10B、11B、23Na、35Cl、195Pt)由来の核も観測できる。特定の物質についてのNMR共鳴周波数は、ラーモアの歳差運動周波数の方程式に従って、印加磁場の強度に直接比例する。NMRは、一定の印加磁場でそれらを配列し、直交する交番磁界でこの配列に摂動を与えることによって、磁性核を測定する。摂動磁場に対して得られる応答は、NMR分光法および磁気共鳴映像法に利用される現象であり、高スペクトル分解能を達成するために非常に強力な印加磁場を使用するが、詳細は化学シフトおよびゼーマン効果により説明される。
【0084】
本発明において、P-ループ内の好適なアミノ酸は、アイソトープまたは濃縮アイソトープを含有するチオール/アミノ反応性小分子で標識し得る。この場合、唯一のシグナルは、P-ループ上の豊富な分子に由来するが、これは、選択された標識部位に依存する、タンパク質立体構造に感受性がある。
【0085】
好ましいアイソトープは、13C、15N等であり、これらは1Dまたは2D NMRスペクトルとして測定し得る。例えば阻害剤の結合によるタンパク質の立体構造の変化により、標識に対応するNMRの化学シフトがシフトする。
【0086】
電子常磁性共鳴(EPR)または電子スピン共鳴(ESR)分光法は、上記に簡潔に記載した通り、有機および無機フリーラジカルまたは遷移金属イオンを有する無機錯体等の、1以上の不対電子を有する化学種を研究する技術である。EPRの基本的な物理的概念は、核磁気共鳴(NMR)と類似しているが、原子核スピンの代わりに励起されるものは電子スピンである。最も安定な分子は、全ての電子を対で有するため、EPR技術はNMRより利用の幅が狭い。しかし、この常磁性種への制限もまた、普通の化学溶媒およびマトリクスがEPRスペクトルを生じないため、EPR技術が優れた特異性を有することを意味する。
【0087】
EPR技術はスピン標識を利用する。この場合、試験されるキナーゼは、13Cおよび15N等のアイソトープの存在下で細菌または他の好適な宿主細胞において発現されて、発現されるにつれて、タンパク質全体にこれらのアイソトープが取り込まれる。アイソトープを濃縮したタンパク質を精製した後、上述の通り、スピン蛍光をP-ループに結合させる。この場合、タンパク質中のアイソトープの2D NMRスペクトルが記録される。P-ループおよびスピン標識が立体構造を変化させるにつれて、スピン標識は取り込まれたアイソトープに由来するタンパク質シグナルのいくつかを変化させるが、阻害剤が結合するにつれて、この取り込まれたアイソトープはP-ループまたはスピン標識により近く接触するようになる。スピン標識が接近するにつれて、ピークは幅広くなるであろう。
【0088】
工程(c)または(c)’で得られる、種々のEPRスペクトル、または1以上の波長、好ましくは発光極大における種々の蛍光発光シグナル、または種々の蛍光発光スペクトルは、候補化合物の結合によって起こる、キナーゼにおける立体構造変化を示す。このことは、ATP-結合ポケットに隣接するアロステリック部位に、ある場合ではATP-結合ポケットそのものに化合物が結合することによって、DFGモチーフの摂動、活性化ループにおける立体構造変化を生じ、それにより、P-ループにおいて、隣接する原子の核と結合したスピン標識における極性変化および/または遊離電子の相互作用変化を招くという事実による。EPRまたはNMRスペクトルまたは蛍光発光の比較に際し、本発明の方法により、候補化合物が好適なキナーゼ阻害剤として有効であるかどうか、例えば、高親和性阻害剤であるだけではなく、あるキナーゼの活性を特異的に阻害するものであるかどうかが明らかになる。候補阻害剤を用いないキナーゼの記録データと、前記の候補阻害剤と接触させたキナーゼの記録データを比較する。蛍光発光シグナルの場合、1以上の特異的波長でいずれかのシグナルを記録および比較することができ、特定の波長でのシグナル強度の変化が検出可能になる。或いは、完全なスペクトルを記録および比較することができ、極大発光波長における変化の観察も可能になる。
【0089】
好ましくは、前記の方法は、ハイスループットフォーマットにおいて達成される。ハイスループットアッセイは、生化学分析、細胞分析または他のアッセイと独立して、一般にはマイクロタイタープレートのウェル中で実施してもよく、ここで各プレートは、96、384または1536ウェルを含有していてもよい。室温以外でのインキュベーション等のプレートの取扱、および試験化合物、この場合推定阻害剤のアッセイ混合物との接触は、好ましくは、ピペット装置を含む1以上のコンピュータ制御ロボットシステムによって達成される。試験化合物の大きなライブラリがスクリーニングされ、および/またはスクリーニングが短時間に達成される必要がある場合、例えば10、20、30、40、50または100の試験化合物の混合物を各ウェルに添加してもよい。一つのウェルが阻害活性を呈する場合、前記の試験阻害剤の混合物は、前記の活性を生じる前記の混合物中の1以上の試験阻害剤を特定するため、簡素化(de-convoluted)してもよい。
【0090】
或いは、1つの試験阻害剤のみをウェルに添加してもよく、ここで各試験阻害剤は種々の濃度で適用される。例えば、試験阻害剤は、種々の濃度で2、3または4つのウェルにおいて試験してもよい。この最初のスクリーニングにおいて、濃度は、例えば10 nMから10μMまでの広範囲をカバーしてもよい。最初のスクリーニングはヒットを見い出すためであり、即ち試験阻害剤が少なくとも1つの濃度で、好ましくは2つ、より好ましくは適用した全ての濃度で阻害活性があることを見い出し、ここで、阻害活性が検出され得る濃度がより低い範囲内である場合、ヒットはより有望である。この代案は、本発明の1つの好ましい態様となる。
【0091】
次に、ヒットと考えられる試験阻害剤は、より広い範囲の阻害剤濃度、例えば10 nM〜20μMの濃度を用いて、さらに試験され得る。これらの測定に適用される方法は以下に記載される。
【0092】
さらに、本発明は、キナーゼ阻害剤のリガンドの結合および/もしくは会合または解離のキネティクスを決定する方法に関し、(a)本発明による蛍光標識キナーゼを種々の濃度の阻害剤と接触させること、または(a)’阻害剤に結合した本発明による蛍光標識キナーゼを、種々の濃度の非標識キナーゼと接触させること;(b)前記の蛍光標識したキナーゼの、1以上の波長での蛍光発光シグナルまたはスペクトルを、各濃度の阻害剤および/または非標識キナーゼについて、励起に際して記録すること;(c)工程(b)で記録された1以上の波長での蛍光発光シグナルまたはスペクトルから、各濃度の速度定数を決定すること、または(c1)各濃度の阻害剤について、工程(b)で記録された1以上の波長での蛍光発光シグナルまたはスペクトルから、Kdを決定すること、または(c2)各濃度の非標識キナーゼについて、工程(b)で記録された1以上の波長での蛍光発光シグナルまたはスペクトルから、Kaおよび逆Kdを決定すること;(d)工程(b)で得られた種々の濃度の阻害剤についてのシグナルまたはスペクトルから工程(c)で決定した速度定数より、直接、konを決定すること、および/または、koffを外挿すること、または(d)’工程(b)で得られた種々の濃度の非標識キナーゼについてのシグナルまたはスペクトルから工程(c)で決定した速度定数より、直接、koffを決定すること、および/または、konを外挿すること;および任意に、(e)工程(d)または(d)’で得られたkonおよびkoffから、Kdおよび/またはKaを計算することを含む。
【0093】
標識キナーゼを種々の濃度の阻害剤と接触させた後、適用した各濃度について蛍光発光を決定することによって、阻害剤の結合親和性を測定し得る。各濃度について、結合および非結合阻害剤の割合は異なるであろうが、これは阻害剤の濃度の上昇を反映し、また前記のキナーゼに対する前記の阻害剤の特異的結合親和性も反映する。
【0094】
反対のアプローチは、その後、阻害剤が結合していない非標識キナーゼにより、結合した阻害剤を含有する標識キナーゼを滴定し得る。
【0095】
化学反応速度論において、速度定数kは、化学反応の速度を数値化する。物質AおよびBが反応してCを産生する場合の化学反応について、反応速度は以下の式を有する:
【数1】
【0096】
式中、k(T)は、温度に依存する反応速度定数である。
[A]および[B]は、それぞれ、物質AおよびBの濃度であり、反応が溶液の容量全体で起こっていると仮定した、溶液の容量に対するモルで表す。
指数mおよびnは次数であり、反応メカニズムに依存する。それらは実験的に決定し得る。
【0097】
また、一段階反応は次の通りにも表し得る:
【数2】
【0098】
Eaは活性化エネルギーであり、Rは気体定数である。温度Tで分子はボルツマン分布によるエネルギーを有するので、Eaより大きいエネルギーを有する衝突の割合は、e-Ea/RTと共に変化することが予測され得る。Aは前指数(pre-exponential)因子または頻度因子である。
【0099】
konおよびkoffは、非共有平衡結合を表す定数である。リガンドが受容体と相互作用する場合、または基質が酵素と相互作用する場合、結合は質量作用の法則に従う。
【数3】
【0100】
この方程式において、Rは遊離受容体の濃度であり、Lは遊離リガンドの濃度であり、RLは受容体-リガンド複合体の濃度である。酵素反応速度論の場合、Rは酵素であるか、または、この場合タンパク質キナーゼであり、Lは基質であるか、または、この場合、候補阻害剤または知られている阻害剤である。会合速度定数konは、M-1秒-1の単位で表される。RL形成速度は、R x L x konに等しい。解離速度定数koffは、秒-1の単位で表される。RL解離速度は、RL x koffに等しい。平衡において、逆(解離)反応は順(会合)反応に等しい。結合の研究では、特異的結合を測定するが、これはRLの指標である。酵素反応速度分析では、酵素の速度を評価するが、これは酵素-基質複合体濃度であるRLに比例する。
【0101】
【数4】
【0102】
平衡解離定数Kdは、モルの単位で表され、koff/konと等しく規定され、以下のようになる:
【数5】
【0103】
解離定数(Kd)は、特定のタンパク質上の結合部位の半分が塞がれた、リガンドの濃度(L)に対応し、即ち、リガンドが結合したタンパク質の濃度(RL)が、リガンドが結合していないタンパク質の濃度(R)と等しい時の、リガンドの濃度に対応する。解離定数が小さいほど、リガンドは強く結合するか、リガンドとタンパク質との親和性が高くなる。
【0104】
従って、会合定数Kaは、逆Kdとも呼ばれ、1/ Kdと規定される。特定のリガンド-タンパク質相互作用の解離定数は、溶液の条件(例えば、温度、pHおよび塩濃度)に伴い有意に変化し得る。
【0105】
本発明の上記の方法では、その後の一連の工程により、KdまたはKaは直接的または間接的に測定し得る。
【0106】
KdまたはKaをそれぞれ直接的に測定するため、工程(b)の後、このタイプの測定では最後の工程である工程(c1)または(c2)を行う。このタイプの測定は終点測定と呼ばれ、添付の実施例でも説明される。速度定数を用いる計算によるKdまたはKaの間接的決定と異なり、蛍光の経時変化よりも、平衡での最終蛍光発光が測定される。これらの測定を使用し、種々の阻害剤濃度(Kd決定のため)または非標識キナーゼの濃度(Ka決定のため)を用いて結合曲線を作成し得る。これらの曲線から、KdまたはKaを直接的に得ることができる。
【0107】
KdまたはKaを間接的に得るため、工程(b)で記録された、1以上の波長での蛍光発光シグナルまたはスペクトルからの速度定数を、工程(c)で実施した各濃度について決定しなければならない。滴定のタイプにより、即ち、標識キナーゼを阻害剤で滴定するか、または阻害剤に結合した標識キナーゼを非標識キナーゼで滴定するかにより、konまたはkoffのいずれかを、測定した速度定数から直接的に決定し得る。konの決定のため、工程(d)が適用されるが、これによりkoffの外挿も可能になる。従って、工程(d)’は、koffの直接的決定に適用され、同様にkonの外挿も可能にする。工程(d)または(d)’で得られるkonおよび/またはkoffから、上記で論じた方程式に従って、Kdおよび/またはKaを計算し得る。
【0108】
上記の方法は、ハイスループット・スクリーニングにも適用し得る。例えば、本発明のキナーゼ阻害剤のスクリーニング方法を用いて、キナーゼに対する阻害活性を有する化合物が特定された場合、本発明の方法は、前記の阻害剤をさらに特性化するために使用し得る。例えば、ハイスループットフォーマットを使用して、多数の異なる濃度の阻害剤(変数(a))または非標識キナーゼ(変数(b))について、1以上の波長での蛍光発光シグナルからKaまたはKdを決定し得る。試験される濃度範囲は、例えば、10 nMから20μMまでであり、評価する濃度の間で1、2および5の繰り返しの連続数(即ち、10、20、50、100、200、500 nM等)である。
【0109】
別の態様において、本発明は、キナーゼ阻害剤の解離または会合を決定する方法に関し、(a)本発明によるスピン標識またはアイソトープ標識したキナーゼを、種々の濃度の阻害剤と接触させること、または(a)’阻害剤に結合した本発明によるスピン標識またはアイソトープ標識したキナーゼを、種々の濃度の非標識キナーゼと接触させること;(b)前記のスピン標識またはアイソトープ標識したキナーゼのEPRまたはNMRスペクトルを、各濃度の阻害剤および/または非標識キナーゼについて記録すること;および(c)種々の濃度の阻害剤について、工程(b)で記録されたEPRまたはNMRスペクトルからKdを決定すること;または(c)’種々の濃度の非標識キナーゼについて、工程(b)で記録されたEPRまたはNMRスペクトルからKaを決定することを含む。
【0110】
蛍光標識キナーゼを用いて反応速度定数を決定することに関して上記でさらに開示した方法と同様に、本発明により、キナーゼと阻害剤との反応の会合または解離定数の直接的測定が可能になる。蛍光標識キナーゼとは異なり、機器の制限およびNMRおよびEPR測定結果の回収に要する時間は、多くの場合、高速スケールの阻害剤結合には適合せず、konまたはkoffの直接的決定はできない。また、キナーゼへの結合に数時間を要する化合物についての決定も可能かもしれない。
【0111】
反応速度論的データの決定に関する本発明の方法は、ハイスループットフォーマットにも適用し得る。例えば、上述の本発明のスクリーニング方法で特定される潜在的阻害剤は、種々の濃度の前記の阻害剤をキナーゼに適用してKdを決定して、さらに特性化し得る。阻害剤の好適な濃度範囲は10 nMと20μMとの間であるが、制限されない。
【0112】
ハイスループットフォーマットに適用されるより集中的な濃度範囲でkonおよびkoffを決定することにより、例えば、添付の実施例において実施した通り、キュベットアプローチおよびリアルタイム反応速度測定を用いて、より高感度でKd測定値が得られる。
【0113】
さらに本発明は、キナーゼ阻害剤のスクリーニングに好適な突然変異キナーゼの産生方法に関し、(a)関心のあるキナーゼにおいて存在する場合、P-ループの外部に遊離チオールまたはアミノ基を有する、溶媒にさらされているアミノ酸、または、P-ループ内部の好適でない位置に遊離チオールまたはアミノ基を有するアミノ酸を、遊離チオールまたはアミノ基を有しないアミノ酸で置換すること;(b)P-ループ内に遊離チオールまたはアミノ基を有するアミノ酸が存在しない場合、前記の関心のあるキナーゼのP-ループにおけるアミノ酸を突然変異して、遊離チオールまたはアミノ基を有するアミノ酸とすること;(c)関心のあるキナーゼを、その環境における極性変化に感受性のあるチオール-またはアミノ-反応性フルオロフォア、チオール反応性スピン標識、アイソトープまたは濃縮アイソトープチオール-またはアミノ-反応性標識で標識することであって、前記のフルオロフォア、スピン標識、アイソトープまたは高アイソトープ標識は、キナーゼの触媒活性を阻害せず、および/または、キナーゼの安定性を妨げない;(d)工程(c)で得られたキナーゼを、前記のキナーゼの知られている阻害剤と接触させること;および(e)工程(c)および(d)の前記の蛍光標識キナーゼの、励起の際の、1以上の波長での蛍光発光シグナルまたはスペクトルを記録すること、または(e)’工程(c)および(d)の前記のスピン標識キナーゼのEPRまたはNMRスペクトルを記録すること;および (f) 工程(e)で記録された1以上の波長での蛍光発光シグナルまたはスペクトル、または、工程(e)’で記録されたEPRまたはNMRスペクトルを比較することを含むが、ここで、工程(e)で得られる、少なくとも1つの波長、好ましくは発光極大での蛍光強度の差、および/または、前記の蛍光標識キナーゼのスペクトルにおける蛍光発光波長のシフト、または、工程(e)’で得られる前記のスピン標識またはアイソトープ標識キナーゼのEPRまたはNMRスペクトルの変化から、該キナーゼがキナーゼ阻害剤のスクリーニングに好適であることが示される。
【0114】
ハイスループットフォーマットに適合させて、多数のキナーゼまたは同一のキナーゼに異なる標識をした変種をスクリーニングし得る。
【0115】
本発明による「好適ではない位置」という用語は、本発明の標識したアミノ酸に好適ではないことが示された、P-ループにおける位置を示す。このことは、その環境変化に対する標識の感受性が低下しているため、または、前記の位置により標識を有するキナーゼの感受性が低下するという構造的考察に基づく予測のためであり得る。またこの用語は、潜在的に好適な位置にあるアミノ酸も包含し、ここでは、異なる位置がより適当であると考えられる。P-ループにおいて遊離チオールまたはアミノ基を有するアミノ酸の数が1を超えた場合は即座に、好適ではないとみなされるアミノ酸を変異させるべきである。
【0116】
アミノ酸を変異させることとしては、この変異により得られるキナーゼの触媒活性を阻害せず、または安定性を妨げない場合に、前記のアミノ酸を別のアミノ酸で置換することまたは取り除くことが挙げられる。工程(b)は、遊離チオールまたはアミノ基が関心のある前記のキナーゼのP-ループに存在しない場合に、実施される。挿入されるか別のアミノ酸を置換するアミノ酸は、標識化するため、遊離チオールまたはアミノ基を有していなければならない。
【0117】
本発明の方法の好ましい態様において、キナーゼ阻害剤は、キナーゼのATP結合部位に隣接するアロステリック部位に排他的に結合するか、または、アロステリック部位からATP部位中に伸長する。これらのタイプの阻害剤は、それぞれIII型またはII型阻害剤とも呼ばれる。これらの阻害剤は、キナーゼのATP-ポケットに結合するI型阻害剤と比較して、より高い特異性を有するキナーゼに結合するが、ATP-ポケットは全てのキナーゼにおいて、構造中に高度に保存されている。
【0118】
実施例において実証される通り、本発明は、ATP-競合的およびATP-非競合的阻害剤を差別化し、特異的阻害剤の迅速な選択を可能にする手段を提供する。本発明は、キナーゼのP-ループの運動を検出するために設計されるが、これは活性化ループの運動によって生じ、従って全てのII型およびIII型阻害剤に感受性がある。さらに、特異的結合形態(上記参照)を有するか、DFG-in構造においてキナーゼによりP-ループと直接的に相互作用する、あるI型阻害剤が検出される。結合速度が決定される蛍光の経時変化の測定(即ち、終点測定ではない)のみが、I型阻害剤をII型およびIII型阻害剤と区別することを可能にする。以下の実施例の一つにおいて示した通り、検出されたATP-競合的阻害剤は、瞬間的な蛍光変化(典型的には<5秒)を生じ、一方、II型およびIII型阻害剤は、はるかにゆっくり結合する(数秒から数分)。
【0119】
本発明のキナーゼまたは方法の別の好ましい態様において、キナーゼは、天然に存在するか、またはP-ループ中に導入されたシステインで標識される。
【0120】
タンパク質におけるシステインの量は通常は非常に少ないため、本発明のキナーゼは、P-ループ中のアミノ酸をシステインで置換し、かつ、任意に、溶媒にさらされているシステインを他のアミノ酸で置換することにより、直接的に製造し得る。ヒスチジン、アルギニンまたはリシンまたはそれらの誘導体等の反応性アミンを含有するアミノ酸は、はるかに豊富に存在し、周囲の溶媒と接触しているタンパク質表面で容易に見つかる。従って、導入されたシステインと特異的に反応し得る、チオール反応性標識を使用することが好ましい。
【0121】
より好ましい態様において、キナーゼ阻害剤のスクリーニング方法または変異キナーゼの産生方法は、さらに、工程(c1):工程(c)で記録された2つの波長の蛍光強度比を測定し、正規化した強度変化の平均強度変化(ΔIstd)に対する割合を得ることを含む。加えて、または、代わりに、本発明により標識したキナーゼの阻害剤が結合したものと阻害剤を有しないものとの間の、最大標準強度変化(ΔRmax)を評価してもよい。(ΔIstd)が>0.25であるか、および/または、(ΔRmax)が>0.75あり、Z因子が>0.5である場合、候補化合物はキナーゼ阻害剤と見なされるか、または、蛍光標識したキナーゼは、キナーゼ阻害剤のスクリーニングに好適であると見なされる。上述の通り、この態様は、本発明の方法のハイスループットスケールへの拡張に関する。
【0122】
ΔIstdは、蛍光発光の平均強度に対する、正規化した強度変化の割合である。ロリミエル(Lorimier)ら(2002年)によれば、ΔIstdは、高感度の蛍光分光法に好適な蛍光タンパク質複合体を特性化する、最も重要な基準の1つである。理想的には、ΔIstdの値は>0.25であるべきであり、以下によって計算される:
【数6】
ここで、λstd = (λmax, 非結合 + λmax, 飽和)/2であり、I1、I2はそれぞれ、各スペクトルのλstdでの蛍光強度である。
【0123】
ΔRmaxは、飽和および不飽和キナーゼの間の、蛍光発光の最大標準強度変化である。ドゥ・ロリミエル(De Lorimier)ら(2002年)によれば、ΔRmaxは、高感度の蛍光分析法に好適な蛍光タンパク質複合体を特性化する、別の重要な基準である。理想的には、ΔRmaxの値は>1.25であるべきであり、以下によって計算される:
【数7】
ここで、oA1、oA2はリガンドが存在しない領域であり、∞A1、∞A2は飽和しているリガンドが存在する領域である。コンピュータプログラムを使用して、2つのスペクトルにおける波長バンドの全ての可能なペアについて、ΔRを列挙して、最適な検知条件を特定し、最大値をΔRと規定し得る。
【0124】
Z因子は、ハイスループット・スクリーニング(HTS)分析の質および検出力の統計的尺度である。HTS作戦では、未知のサンプルの多数の単一測定値を確立した陽性および陰性対照と比較して、もしあるとすれば、単一測定値のいずれが陰性対照と有意に異なるかを決定する。大規模なスクリーニング作戦を開始する前に、多くの作業を行って、より小規模の分析の質を評価し、その分析がハイスループットの設定で有用であるかどうかを予測する。Z因子は、分析を何百万ものサンプルにスケールアップした場合に、有用なデータが期待できるかどうかを予測する。Z因子は以下により計算される:
【数8】
ここでは、陽性(p)および陰性(n)対照の両者の、平均(μ)および標準偏差(σ)の両者(それぞれ、μp、σp、μn、σn)が考慮される。
【0125】
ΔIstdおよびΔRmaxの測定、ならびにZ因子の決定により、選択された標識が阻害剤のスクリーニングに好適かどうかの決定に有用であることが判明するかもしれない。ドゥ・ロリミエル(De Lorimier)は、蛍光タグ付けされたタンパク質を用いて得られた、測定した反応速度およびKdが、使用したタンパク質、リガンドおよびフルオロフォアに依存することを論じている。従って、同一のキナーゼに結合する同一の阻害剤は、使用される標識によって、Kd値が異なる可能性がある。上記の値の決定により、選択された標識が好適であるかどうか、または、異なる標識を使用すべきかどうかが示唆される可能性がある。
【0126】
さらに好ましい態様において、フルオロフォアまたはスピン標識は、標識キナーゼ中に存在することが知られているか、または予測されているリン酸化部位に、またはそのリン酸化部位に隣接して、位置しない。このことは、標識化が、P-ループの動力学または通常の活性を妨げず、かつ、リン酸化および脱リン酸化によって大きく影響されるキナーゼの調節を妨げないことを確実にする。
【0127】
別の好ましい態様において、P-ループにおける前記の候補アミノ酸は、前記のキナーゼについて入手できる構造および/または配列データに基づいて特定される。
【0128】
あるキナーゼについて、例えば結晶形態の構造データまたはNMR構造は入手可能であり、ここで、キナーゼは、活性化および/または不活性化状態で捕えられる。このようなデータがキナーゼについて入手可能である場合、これにより、標識化の目的で置換される、P-ループ中のアミノ酸の位置の選択が容易になる。実際の選択は、ほとんどの場合TyrまたはPheであるG-X-G-X-X-Gモチーフの3番目のGlyの前の位置での、リガンドの結合および/または立体構造変化に応答して、どの残基がP-ループにおいて最大の変動を示す傾向があるかに基づく。非常に少ないケースであるが、この残基がTyrであり、特定のキナーゼにおいてリン酸化されることが知られている場合、この位置の標識化は、キナーゼ活性を妨害すると思われ、この位置は本発明において好適でなくなる。同様に、前記の位置でのアミノ酸の他のアミノ酸との接触も試験する。前記の接触がキナーゼの触媒活性および安定性に必須であると思われる場合、殆どの場合、この位置は置換に好適ではない。加えて、アミノ酸位置の選択は、特定のアミノ酸がタンパク質の立体構造変化に伴って変動する距離に基づいて行われ、距離が大きくなると、環境の変化が検出される機会が増えるようにする。しかし、変動距離は、特定の位置が標識化に有用かどうかの指標であるが、結合した標識によって検出される、観察される変化と直接相関する、環境における実際の変化である。
【0129】
好ましい態様において、阻害剤のスクリーニングに関する本発明の方法は、会合および解離等の反応速度論的パラメータの決定と変異したキナーゼの産生とを組み合わせて、種々のキナーゼについての特異的阻害剤を得るための直接的方法論を得る。この点で、本発明の方法の全ての好ましい態様は、本発明の他の方法の態様と組み合わせてもよい。この様相のより好ましい態様において、初期のスクリーニングは、キナーゼ阻害剤のハイスループット・スクリーニング方法を用いて実施し、その後、リガンドの結合および/もしくは会合または解離のキネティクスを決定するための本発明の方法で、上述の通り、広範な濃度範囲の阻害剤を用いてスクリーニングする。後者の工程は、とりわけ、指標であるKdおよび/またはKa値を得るために実施する。この工程は、KdまたはKaをより正確に測定するため、濃度範囲をより絞って測定を実施することにより、再度繰り返す。各阻害剤をさらに特性化するため、これらの測定は、キュベットアプローチを用いた滴定系列として、および/または、キュベットにおけるリアルタイム反応速度測定(konおよびkoff)として実施してもよい。代わりに、または、加えて、キナーゼ阻害剤として特定される化合物の結合形態は、関心のあるキナーゼとの複合体において化合物を結晶化することによってさらに特性化してもよく、これは詳細を最も明瞭に示す。任意に、この一連の方法は、他のキナーゼまたは異なる標識をした同一のキナーゼに委ねられる。この態様は、ハイスループット・スクリーニングが、多数のキナーゼまたは同一のキナーゼに異なる標識をした変異体について、大多数の阻害剤をスクリーニングおよび特性化できるように設計される。
【0130】
より具体的には、このような組み合わせ方法は、キナーゼのATP結合部位に隣接するアロステリック部位に、部分的または完全に結合するキナーゼ阻害剤の特定方法であって、(a) 本発明のキナーゼ阻害剤のスクリーニング方法に従って、阻害剤をスクリーニングすること、および(b) 工程(a)で特定された阻害剤のキナーゼに対する速度定数を決定することを含み、ここで、工程(b)で決定された<0.140秒-1の結合速度は、特定されたキナーゼ阻害剤が、キナーゼのATP結合部位に隣接するアロステリック部位に部分的または完全に結合することを示す。>0.140秒-1の速度定数は、特定されたキナーゼ阻害剤が、ATP結合部位において結合し、隣接するアロステリック部位中に伸長しないことを示す。速度定数は、反応時間(結合速度)t1/2: t1/2 = ln(2)/kobsに相関する。従って、<0.140秒-1の速度定数(kobs)は、>5秒の反応時間t1/2に対応する。
【0131】
速度定数または結合速度は、好ましくは、本発明の標識キナーゼの特性を使用して決定する。例えば、本発明のキナーゼは阻害剤と接触し、標識によっては、1以上の波長での蛍光標識キナーゼの蛍光発光シグナル、または、スピン標識またはアイソトープ標識したキナーゼの電子常磁性共鳴または核磁気共鳴スペクトルを、経時的に記録し得る。これは、本発明のキナーゼ阻害剤のリガンド結合および/もしくは会合または解離のキネティクスの決定方法の工程(a)〜(c)、または、本発明のキナーゼ阻害剤の解離または会合の決定方法の工程(a)および(b)に対応する。結合速度、即ち、蛍光、またはNMRまたはEPRスペクトルにおける測定可能な変化が、阻害剤の適用後5秒より大きい場合、このことは、阻害剤がII型またはIII型阻害剤であることを示す。
【0132】
キナーゼ阻害剤のスクリーニング方法の別の好ましい態様において、この方法は、さらに、(その後)前記のキナーゼの阻害剤として特定された候補化合物の薬理学的特性を最適化することを含む。
【0133】
一般にリード化合物と言われる、スクリーニングにおいて特定される化合物の薬理学的特性の最適化方法は、当業界において知られており、リード化合物として特定される化合物の、以下を達成するための修飾方法を含む:(a)作用部位、活性スペクトル、器官特異性を修飾する、および/または(b)効力を向上させる、および/または(c)毒性を減じる(治療指数を向上させる)、および/または(d) 副作用を減じる、および/または(e)治療効果の発現、効果持続期間を修飾する、および/または(f)薬物動態パラメータ(吸収、分布、代謝および排泄)を修飾する、および/または(g)物理化学的パラメータ(溶解性、吸湿性、色、味、臭気、安定性、状態)を修飾する、および/または(h)全般的特異性、器官/組織特異性を向上させる、および/または(i)適用形態および経路を以下によって最適化する、a. カルボキシル基のエステル化、またはb. ヒドロキシル基のカルボン酸によるエステル化、またはc. ヒドロキシル基の、例えばリン酸エステル、ピロリン酸エステルまたは硫酸エステルまたはヘミコハク酸エステルへのエステル化、またはd. 薬学的に許容される塩の形成、またはe. 薬学的に許容される複合体の形成、またはf. 薬理学的活性ポリマーの合成、またはg. 親水性部分の導入、またはh. 芳香環または側鎖上の置換基の導入/交換、置換パターンの変更、またはi. 等比体積または生物学的等価部分の導入による修飾、またはj. 同族化合物の合成、またはk. 分枝状側鎖の導入、またはl. アルキル置換基の環状アナログへの変換、またはm. ヒドロキシル基のケタール、アセタールへの誘導化、またはn. アミド、フェニルカルバメートへのN-アセチル化、またはo. マンニッヒ塩基、イミンの合成、またはp. ケトンまたはアルデヒドの、シッフ塩基、オキシム、アセタール、ケタール、エノールエステル、オキサゾリジン、チアゾリジンまたはそれらの組み合わせへの変換。候補化合物の修飾の前に、候補化合物の関心のあるキナーゼへの正確な結合形態を特性化し、キナーゼへの阻害特性に対する、ある化学修飾の候補化合物への効果を予測するために、(i) 既に知られている結晶構造中への提案分子のドッキング、(ii)相同モデル(標的タンパク質に最も近い相同体の知られている結晶構造に基づいて産生されたモデル構造)中への提案分子のドッキング、および/または(iii)キナーゼ阻害剤複合体の結晶化等の、いくつかの分析/予測ツールを適用してもよい。
【0134】
候補化合物にもたらされる修飾は、その後、上記に列記したあらゆる技術によって再度、さらに分析してもよく、その結果、多数のそのようなサイクルの後、候補化合物の特性が最適化されている。
【0135】
上記で挙げた種々の工程は、当業界で一般的に知られている。それらは、さらに、定量的構造活性相関(QSAR)分析(クビニイ(Kubinyi)、"Hausch-Analysis and Related Approaches", VCH Verlag, Weinheim, 1992年)、コンビナトリアル生化学、古典的化学等を包含するかそれらに基づく(例えば、ホルツグラーベ(Holzgrabe)およびベフトルド(Bechtold)、Deutsche Apotheker Zeitung 140(8), 813-823頁, 2000年参照)。
【0136】
図1、p38αにおけるリガンド結合によって引き起こされる、P-ループおよび活性化ループの立体構造変化の略図。a) 活性化キナーゼドメイン(DFG-in)の構造的に重要な領域(P-ループ;へリックスC;ヒンジ領域)が標識される。b) 活性化ループへの共鳴における、活性化ループの移動性。II/III型阻害剤は、DFG-outキナーゼ立体構造にのみ存在する部位をふさぐ。このアロステリックポケットの側面には、DFG-モチーフおよびヘリックスCが配置されている。アロステリック部位に結合するII/III型阻害剤は、活性化ループ(黒)の立体構造変化を引き起こし、P-ループ(黒)がより伸長した立体構造を採用し得る。c) Cys残基は、その後環境感受性フルオロフォア(大型球体)で標識するため、p38αのP-ループ中で変異して、感受性P-ループ結合アッセイを作出する。キナーゼの活性(DFG-in)および不活性(DFG-out)構造は平衡状態にあり、これらは活性化ループの構造変化により生じる。活性化ループの構造変化は、疎水性界面を通ってP-ループに移り、P-ループに結合するフルオロフォアの化学的環境を変化させる。I型阻害剤(表面が、左パネルの大型球体の背部)は、活性化キナーゼ(DFG-in)のヒンジ領域に結合する(左パネル)。この特別の場合において、P-ループは折り重なり、阻害剤と直接相互作用する。活性化キナーゼ(DFG-in)由来のリガンドの非存在下で、P-ループは、より伸長した立体構造を採る(中央パネル)。II/III型阻害剤(表面が、右パネルの大型球体の下部)はキナーゼの不活性(DFG-out)構造に結合する。
【0137】
図2、高グリシンループ上に標識したac-p38αを用いたリアルタイムおよび終点蛍光測定。475 nmでのアクリロダン発光は、BIRB-796の結合により減少して、結合状態における最大放出波長が赤方偏移(より長波長へのシフト)する(A)。終点平衡測定はKdを直接得るために実施し得る。レシオメトリック蛍光データ(R = 512 nm/475 nm)を阻害剤濃度の対数目盛りに対してプロットして、Kdを得た(B)。レシオメトリック蛍光データ(R = 445 nm / 475 nm)もまた、Kdを得るために使用し得る(データは示さず)。蛍光痕跡もまた、単一波長(475 nm)でリアルタイム測定して、様々な反応速度定数を決定し得る。種々の量のBIRB-796(大きな矢印)から生じる蛍光減衰は、一次減衰関数に適合して(灰色線)kobsを得た(C)。その後、実験的に決定したkobs値をプロットして、BIRB-796の全てのリガンドについてkonを決定し得る。過剰の非標識p38αを用いてac-p38αからBIRB-796を抽出することにより、一次関数に適合する(灰色線)koff が直接決定可能である(D)。上記に表したデータは、高グリシンループ上に標識したac-p38αを用いてBIRB-796について実施した、典型的な1セットの実験を代表する。
【0138】
図3、P-ループac-p38αを用いたIII型およびI型リガンドのリアルタイムおよび終点蛍光測定。475 nmでのアクリロダン発光は、III型リガンドRL36の結合により減少して、結合状態における最大放出波長が赤方偏移する(a)。P-ループと相互作用することによるp38αのDFG-out立体構造を安定化することが知られている、I型リガンドであるSB203580について、類似であるがより強い変化が観察された(b)。III型およびII型リガンド(図2参照)は、高グリシンループの関連する構造転位を生じる、活性化ループの立体構造変化を引き起こすことから(図1a参照)、スペクトルの形状および強度の損失の両者は、両方の型の阻害剤について同等の変化をする。蛍光痕跡を単一波長(475 nm)でリアルタイム測定して、リガンドの結合および解離速度を決定した。100 nMのRL36の添加(黒の矢印)により生じる蛍光減衰は、一次減衰関数に適合して(灰色線)kobs.onを得た((a)中央)。SB203580等のI型リガンドは、典型的には<5秒に結合し((b)中央)、正確な曲線の一致は、測定の時間分解能を増すストップフロー蛍光分光法を使用しないと不可能である。ac-p38αからの各阻害剤の抽出は、過剰の非標識p38αを同じサンプルに添加することにより達成された(白色矢印)。koffは、全ての型の阻害剤について、kobs,onより有意に遅いことが知られているので、蛍光の増加を一次関数に適合させる(灰色線)ことにより、各阻害剤についてkoffを決定することが可能であった。レシオメトリック蛍光データ(R = 512 nm / 475 nm)を阻害剤濃度の対数目盛りに対してプロットして、RL36((a)右)およびSB203580((b)右)についてのKdを得た。II型阻害剤であるイマチニブはp38αに結合しないので、RL36の陰性対照とした((a)右、黒色四角)。I型阻害剤であるダサチニブはp38αに結合するが、高グリシンループと相互作用しないので、SB203580の陰性対照とした((b)右、黒色四角)。上記に示したデータは、高グリシンループ上に標識したac-p38αを用いて実施した、典型的な1セットの実験を代表する。
【0139】
図4、P-ループを標識したp38αの阻害剤の種々の型に対する蛍光特性評価および応答。化合物スクリーニングにおいて特定された2つのヒットであるScios-469およびRL40の構造に加えて、種々の知られている阻害剤型(I、IIまたはIII型)の構造を示す。P-ループは、チオール反応性フルオロフォアであるアクリロダンでp38αのY35Cを共有結合的に修飾することによって標識し、変化する蛍光特性をp38αの知られているDFG-outおよびDFG-inバインダーの結合に際して試験した。理想的フルオロフォア-タンパク質複合体と見なされる基準(ドゥ・ロリミエル(deLorimier)ら、2003年)を満たすΔRmaxおよびΔIstdの値は全て、太字テキストで表す。古典的なDFG-outバインダー(II型およびIII型阻害剤)またはP-ループと直接的に相互作用するいくつかのI型阻害剤の場合、アクリロダンは大きな発光シフトを起こすが、〜475 nmと比べて〜512 nmにおいて発光の増加があり、最適ではないΔRmaxにもかかわらず、なお信頼性の高い結合曲線が測定可能である。しかし、これらの同一の型の阻害剤の場合、優れたΔIstd値が得られた。SB203580は、DFG-out構造を安定化することが知られている、p38αのI型阻害剤である(欧州特許EP 08 02 0341号に開示の通り)。ダサチニブはキナーゼのヒンジ領域に結合し、DFGモチーフまたはP-ループと相互作用せず(トカルスキ(Tokarski)ら、2006年)、このアッセイ系では検出されなかった(ND)。SB203580等のI型阻害剤およびDFG-out結合II型(BIRB-796)およびIII型(RL36)阻害剤は、高感度で検出され、Kdおよび反応速度測定を可能にする。反応速度測定により、非常に速く(本実施例において<2秒)結合するI型阻害剤を、p38αに対しゆっくり結合することが知られているII/III型リガンド(パルゲリス(Pargellis)ら、2003年)から、区別することが可能になる。このような2つのリガンドである、RL40およびScios-469は、スクリーニング計画において検出された。これらの2つのリガンドの検出の背後にある構造の詳細を理解するために、その後、タンパク質X線結晶学が利用された。[注:* ΔIstdは、リガンドの存在下および非存在下で445 nmおよび475 nmでの発光強度を用いて計算し(R = 445 / 475 nmはリガンド結合の検出に最適である);** ΔRmaxは、リガンドの存在下および非存在下で475 nmおよび512 nmでの発光強度を用いて計算した(R = 512 / 475 nmは結合形態の判別に最適である)。]
【0140】
図5、RL40、Scios-469およびCP547632の結晶構造は、P-ループの運動を裏付ける。p38αとの複合体(a)中のRL40の構造から、以前に報告された(欧州特許EP 08 02 0341号)SB203580の構造において観察されたものと類似の、独特で予期されなかった結合形態が明らかになるが、ここで、リガンドは、DFGモチーフのPhe側鎖で独特のπ-πスタッキングを形成することにより、P-ループと相互作用する。この相互作用の結果、DFG-out構造が安定化する。p38αにおけるSB203580およびRL40の構造の重なりにより、両阻害剤の芳香核が良好に重なり合い、P-ループおよび活性化ループと同一の型のスタッキング相互作用を形成することが明らかになる(b)。RL40のアナログは、典型的には、キナーゼのヒンジ領域に結合することが観察され、P-ループとは相互作用せず(ピエルセ(Pierce)ら、2005年)、従ってこれらの独特な結合形態を利用するリガンドを増すという、P-ループ標識キナーゼを使用する利益が強調される。加えて、P-ループ標識キナーゼアッセイは、Scios-469の結合を強力に検出した。我々は、Scios-469を野生型p38αと共結晶化し、構造を2.5Åの分解度で解明した(c)。我々は、p38αのアポ構造と比較し、P-ループにおける劇的な運動を観察した。この運動は、化合物の疎水性特徴によるP-ループTyr35(アッセイのために選択された標識位置)のスタッキング相互作用により誘導され、かつ安定化される。このことから、P-ループ標識キナーゼアッセイが、どのように、P-ループの立体構造を直接的に変化させるいくつかのI型リガンドを高感度に検出し得るかという例が提供される。(d)Scios-469のピペラジン環に結合するカルボニルは、ヒンジ領域(ピンク)への2つの水素結合を形成する(赤色破線)(Met109およびGly110の骨格NH)。高グリシンループ(緑)は折り重なって、阻害剤と直接相互作用し、インドール部分およびピペリジン環を溶媒から保護する。DFG-モチーフ(オレンジ)は、ATP結合部位に向かうAsp168を有する”in”構造である。(e)Scios-469と同様に、CP547632のハロゲン置換メトキシベンゼンはゲートキーパー(Thr106)にかぶさるように曲がり、疎水性サブポケットに向かう。チアゾール環に結合するカルボキシアミドおよび尿素は両者とも、ヒンジ領域への水素結合を形成する(His107の骨格CO、Met109のNHおよびCO)。ピロリジン-ブタン部分は90°ねじれて、溶媒からATPポケット方向に向かう。高グリシンループは、電子密度中で見えにくく、DFG-モチーフは明らかに”out”構造である。
【0141】
図6、野生型の非標識およびアクリロダン標識p38αの反応速度論的および阻害特性評価。注:反応速度論的パラメータは、シスビオ(Cisbio)のHTRF登録商標アッセイを使用して決定したが、これにより、導入された変異(p38αにおけるCys119Ser/Cys162Ser/Tyr35Cys)はキナーゼのATPに対する親和性(ATP-Km)を有意に変化させないことが実証される。各変異体のKmで実施したIC50の比較では、2、3の知られているI型およびII型p38α阻害剤のIC50に対し、変異または標識化の有意な効果は見られず、従って、本発明の標識化アプローチのために選択された高グリシンループ標識部位の正当性が立証される。報告された全ての値は、それぞれ2回実施した、独立した実験で、平均± s.d.が少なくとも3である。
【0142】
図7、384穴フォーマットにおいて測定された、p38αによるBIRB-796(1)のKd値の時間依存性。I型阻害剤であるSB203580、Scios-469およびCP547632の結合曲線、および、結合速度の遅いII型阻害剤であるBIRB-796の結合曲線を、p38αを使用して得て、阻害剤結合形態が、結合のリアルタイム反応速度の測定に加えて(図2参照)、HTSフォーマットにおいて予測され得ることが実証された。各リガンドについて、ある濃度範囲にわたり、5、30、90および300分にレシオメトリック蛍光(R = Iλ512 / Iλ475)を測定してプロットし、各時点での各リガンドのKdを決定した。SB203580、Scios-469およびCP547632のKdは、室温で高グリシンループを標識したp38αと共に5分間インキュベーションした後、有意に変化しなかった。BIRB-796のKdは、90分間に〜3倍低下した。室温での90分のインキュベーション時間は、II型阻害剤がキナーゼとの結合平衡に達するのに充分であった。384穴フォーマットにおいて決定されたKd値は、キュベットフォーマットで測定された場合(表1参照)より2〜3倍高かったが、これは、より高いDMSO濃度およびHTSプレートでのスクリーニングのための洗浄剤の添加に起因することが多い。報告された全てのKd値は、それぞれ三回実施した、独立した実験で、平均± s.d.が4である。
【0143】
図8、高グリシンループを標識したMKK7を用いたII型およびI型リガンドのリアルタイムおよび終点蛍光測定。(A) K252aの結合により、標識タンパク質の蛍光強度、および2つの波長(R = 472 nm / 510 nm)におけるレシオメトリック発光の検出可能な変化が減少する。(B) Kdを直接決定する終点方法を用い、これらの発光の割合を阻害剤濃度に対してプロットして、K252aについて38 nMのKdを得たが、これは、これらの化合物について予測した正確な範囲内である(カラマン(Karaman)ら、2008年)。陰性対照として、10 μM以下では検出されない、II型阻害剤であるソラフェニブを含有させた。これらの知見はMKK7について予測される結果と一致しており、このことは、DFG-out構造および、DFG-out構造を誘導または安定化する阻害剤に対する非感受性を示す(カラマンら、2008年)。アッセイ応答がI型阻害剤結合に際するP-ループの運動によることを実証するため、ダサニチブもまた陰性対照として含有させた。ダサニチブは、ATP競合的阻害剤であるか、cSrcおよびAblキナーゼであり、MKK1およびMKK2のみを阻害するが、報告されたKd値は> 1 μMである(カラマンら、2008年)。従って、このI型阻害剤の添加および検出は、MKK7については予測されなかったが、このことはデータで確認される(C)。K252aの結合および解離のリアルタイム反応速度論的測定および検出。p38αについて図3に示す通り、結合に伴って生じる蛍光変化は、標識キナーゼからリガンドを抽出するための過剰の非標識MKK7の添加に際して可逆的である。K252aはI型阻害剤であるため、図3Bに示すp38αのI型阻害剤SB203580のように、これらのプロセスの反応速度は速い。
【実施例】
【0144】
本発明を実施例によって説明する。
実施例1:好適なキナーゼの選択
我々は、このアッセイを展開するため、以下の理由からp38αを扱うことを選択した:i) 入手できる構造的情報が豊富であること、ii) その活性および不活性立体構造の両者の結晶構造が入手できること(図1A)およびiii) 強固に結合するII型およびIII型アロステリック阻害剤が入手できること。第一の工程において、p38αの結晶構造を綿密に調査して、アロステリックバインダーを検出する可能性のある、好適なフルオロフォア結合部位を特定した。この変異の候補残基は、ミカエル付加によるフルオロフォアの結合を可能にするため、溶媒にさらされなければならず、リガンドの結合に際して、有意な運動を呈する。また、タンパク質安定性、触媒活性の維持に非常に重要な残基、または、知られているリン酸化部位に近接する残基を選択しないように注意した。
【0145】
P-ループにおいて一つの位置(Tyr35)を選択し、その後、システイン残基に変異させた(図1B、C)。フルオロフォアとしてアクリロダンを選択したが、これは、相対的にサイズが小さいこと(トリプトファン側鎖と同等)、極性変化に対して感受性が高いこと、市販されていること、および相対的に安価であることによる。また、アクリロダンは、強い応答を生じることが知られており、アロステリック阻害剤の結合に際して、活性化ループの運動を検出するはずである(図1D)。タンパク質を標識する前に、フルオロフォアが、あらゆる他の溶媒にさらされるシステイン残基に結合する機会を減じることが必要であった。再度、構造的情報を用いて、p38α中に4つの還元システイン残基を配置した。これらのシステインのうちの2つはタンパク質中に埋め込まれ、他の2つは溶媒にさらされて保存的にセリンに変異させた。最後に、F327L変異を組み込んで、要すれば、酵素活性アッセイに使用するためアクリロダン標識したp38α(ac-p38α)を部分的に活性化した(アスカリ(Askari)ら、2007年;アヴィゾウア(Avitzour)ら、2007年)が、これはアッセイ自身の機能性には必須ではない。
【0146】
リン酸化した活性p38α変異体それぞれ(変異/非標識およびアクリロダン標識)のATP-Kmを測定し、野生型p38α(図6)と比較する、活性に基づくアッセイを用いることにより、チロシンの同等の大きさのアクリロダンで標識したシステインでの置換は、キナーゼ耐性が良好であることが示された。同様に、3つの知られているp38α阻害剤のIC50値について、高グリシンループの変異または標識化に際し有意な変化は観察されなかったが、このことは、本発明が、最終的には、検出されたリガンドについて、欧州特許EP 08 01 3340号および欧州特許EP 08 02 0341号に記載されたアッセイと類似の親和性データを提供することを実証する。
【0147】
実施例2:タンパク質標識化および蛍光特性化
タンパク質標識化
全部で4つの変異(システイン2つ→セリン、および標識化のためシステイン1つを導入)を含む、p38α構築物を、BL21(DE3)大腸菌株へ形質転換し、過剰発現させ、アフィニティクロマトグラフィー、アニオン交換クロマトグラフィーおよびサイズ排除クロマトグラフィーで精製し、その後、純粋なタンパク質を標識化に使用した。タンパク質および遊離アクリロダンを1:1.5の割合で混合し、4℃の暗所で一晩反応させた。接合したタンパク質(ac-p38α)を濃縮し、分割して、-20℃で凍結した。タンパク質の100%のモノ標識化をESI-MSによって確認した。正確に標識したシステインの確認は、現在は、非標識および標識p38αのトリプシン断片を分析し、その後HPLCとESI-MSまたはMALDIの組み合わせによって行われている。
【0148】
蛍光特性化
標識化に続いて、プローブの蛍光的性質を特性化し、ピラゾロ尿素系II型アロステリック阻害剤であるBIRB-796の様々な誘導体を用いて最初の実験を実施した(パルゲリス(Pargellis)ら、2002年;ドゥマス(Dumas)ら、2000年(aおよびb);モス(Moss)ら、2007年;レーガン(Regan)ら、2002年;レーガンら、2003年)。P-ループを標識したac-p38タンパク質は、リガンド結合に伴い、475 nmから512 nmまで、わずかな赤方偏移を示す(図2A)。2つの波長(R = 512 nm / 475 nm)の割合を測定することにより種々のサンプルの間の希釈誤差を除き得る(図2B)。これら2つの波長を用いて、平均のZ因子は0.53 ± 0.04と計算し得る。同様に、445 nmに存在する小さな極大点は、リガンドの結合に対して相対的に感度が低く、その結果、2つの波長(R = 445 nm / 475 nm)の割合もまたこの目的に使用し得る。これらの2つの波長を用いて、平均のZ因子は0.85 ± 0.05と計算し得る。475 nmでの大きな変化によっても、単一波長での反応速度測定の可能性が認められる(図2C-D)。
【0149】
512 nmおよび475 nmの波長を用いて、平均強度(ΔIstd)と比較した、正規化強度変化を決定したところ、全ての検出した阻害剤の型について0.55〜1.57の間の範囲であり、飽和および不飽和ac-p38αの間の最大標準強度変化(ΔRmax)は、全ての検出した阻害剤の型について0.23〜0.56の間の範囲であった。これらは、蛍光分光法の最も重要な基準のうちの2つである(ドゥ・ロリミエル(de Lorimier)ら、2002年)。2つの異なるレシオメトリックな測定値について決定されたZ因子とともに、ΔIstd値は、これを蛍光アッセイにおける使用に好適なプローブと特性化する。
【0150】
実施例3:終点および反応速度論的測定 − 方法
本発明の標識化戦略を特性化するため、高グリシンループをアクリロダンで標識したp38α(50 nM)を、キナーゼのDFG-inまたはDFG-out構造に特権的に結合することが一般に知られている、骨格に基づく小さなサブセット(〜400)の化合物に対してスクリーニングした。終点蛍光測定を、ポリスチレンキュベットまたは384穴プレートにおいて実施して、各化合物のKdを決定する前に、キナーゼを種々の濃度の阻害剤と予めインキュベートした。全ての実験で標準緩衝液(50 mM Hepes、200 mM NaCl、pH 7.45)を使用した。キュベット測定では、p38αについて4℃の暗所で一晩インキュベーションを実施した。HTSフォーマットでは、室温で5時間までインキュベーションを実施した。II型阻害剤のp38αへの結合の時間依存性を説明するためには、長時間のインキュベーション時間が必要である(パルゲリス(Pargellis)ら、2002年)。
【0151】
キュベットフォーマットにおいて、阻害剤のストック(DMSO中、0.01、0.1、1.0および10.0 mM)を用いて、種々の量の阻害剤を含有する一連のキュベットを調製した。キュベットの全ての測定は、JASCO FP-6500蛍光分光計(JASCO社、独国グロス・ウムシュタット(Gross-Umstadt))で行った。384穴プレートフォーマットにおける蛍光測定値の測定には、Tecan SafireII(Tecan Deutschland社、独国)を使用した。DMSOの%v/vは、キュベットでは0.2%を超えず、384穴プレートでは5%v/vであった。p38αの場合、高グリシンループの運動を引き起こすリガンドの陽性対照として飽和量のBIRB-796またはソラフェニブを使用して、平均Z’因子は、キュベットおよび384穴フォーマットでそれぞれ0.67 ± 0.05(n = 6)および0.64 ± 0.10(n = 6)と決定された。陰性対照としては担体(DMSO)を使用した。
【0152】
アクリロダン標識p38αについて、レシオメトリック蛍光値(R = Iλ512/Iλ475)により、検出化合物の信頼し得る結合曲線のプロットが可能になったが、これはリガンド結合のKdの直接決定を可能にする。検出化合物の結合形態は、種々の時点で一次および/または二次スクリーニングのプレートを測定することによって、HTSフォーマットで初期に評価し得ることに注目すべきである。最大レシオメトリックシグナルまたは経時的Kdを変化させる化合物は、II型/III型阻害剤であると思われる。あるいは、選択された化合物の会合(kon)および解離(koff)の反応速度を、キュベットを用いて決定し得る。konを決定するため、各キュベットの底に小さな攪拌棒を設置して、阻害剤がキュベット上部に位置する注入ポートを通って送達されるに伴い、確実に迅速に攪拌する。蛍光の変化は、JASCO FP-6500蛍光分光器(JASCO社、独国グロス・ウムシュタット(Gros-Umstadt))を用いて、リアルタイムでp38αについて、475 nmでモニターした。殆ど即時の結合反応速度(<5秒)がI型阻害剤の特徴であり、一方、より遅い反応速度(>10秒)は、II型またはIII型阻害剤がDFG-out構造に、よりゆっくり結合していることを示唆する。結合の後、10倍過剰の非標識キナーゼを添加して、結合平衡を標識キナーゼからシフトさせることにより、koffを決定した。過剰の非標識キナーゼの添加により、阻害剤を再分配しアクリロダン標識p38αから解離させて、蛍光シグナルを回収する。全ての結合および解離曲線は、一つの指数方程式に一致した:F(t) = F(∞) + F(0) exp(-t*kobs):ここで、tは時間であり、F(0)は初期の蛍光強度であり、F(∞)はt = ∞における蛍光である。蛍光減衰の半減期(t1/2)は、以下の方程式で計算した:t1/2 = ln 2/kobs。
【0153】
実施例4:反応速度論的発現および精製
p38α構築物は、pOPINE、pOPINFまたはpOPINMベクター中にクローン化し、Precision Protease 切断部位を有するN-末端His-タグ化構築物として、BL21(DE3)大腸菌、BL21(DE3)コドン+RIL大腸菌またはBL21(DE3)ロゼッタ大腸菌中に形質転換した。培養物は、OD600が0.6になるまで37℃で成長させ、室温まで30分間で冷却した後、160 rpmで攪拌しながら、18℃で一晩(〜20時間)1 mM IPTGを用いて発現させた。細胞を緩衝液A(50 mMトリス pH 8.0、500 mM NaCl + 5%グリセロール+ 25 mMイミダゾール)で溶解させ、30 mLのNi-カラム(自己充填)に負荷し、3 CVのNi緩衝液Aで洗浄した後、2 CVを超えるNi緩衝液B(Ni緩衝液A + 500 mMイミダゾール)を用いて0-50%の直線勾配で溶出した。12-30 mL容量の10-MWCO透析カセット(Thermo Scientific)において、透析緩衝液(50 mMトリス pH 7.5、5%グリセロール、150 mM NaCl、1 mM EDTA、1 mM DTT)中4℃で一晩、PreScission Protease(最終濃度50 μg/mL)と共にインキュベーションすることにより、タンパク質を切断した。その後、タンパク質は〜13,000 rpmで15分間遠心分離して、切断工程の間に形成された可能性がある全ての析出物を除去した。その後、上澄を取り、少なくとも4倍のアニオン緩衝液A(50 mMトリス pH 7.4、5%グリセロール、50 mM NaCl、1 mM DTT)で希釈し、1 mLセファロースQ FFカラム(GEヘルスケア)に負荷し、10 CVのアニオン緩衝液Aで洗浄した。タンパク質を、20 CVを超えるアニオン緩衝液B(アニオン緩衝液A + 600 mM NaCl)により0-100%の直線勾配で溶出した。タンパク質をプールし、2 mLまで濃縮して、サイズ排除緩衝液(20 mMトリス pH 7.4、5%グリセロール、200 mM NaCl、1 mM DTT)で平衡化したセファデックスHiLoad 26/60 Superdex 75カラムに2 mL/分の速度で通過させた。その後、溶出したタンパク質は、〜10 mg/mLまで濃縮し、分割して-80℃で凍結させた。
【0154】
実施例5:高グリシンループを標識したac-p38αを用いたリアルタイムおよび終点蛍光測定により、II/III型阻害剤およびいくつかのI型阻害剤の検出が可能になる。
本発明を、p38αの活性化ループを標識した欧州特許EP 08 01 3340号および欧州特許EP 08 02 0341号に記載された方法と比較し、高グリシンループをアクリロダンで標識することが信頼し得る代替のアプローチとなることを実証するため、II型p38α阻害剤であるBIRB-796を使用して蛍光反応を特性化した。終点測定は、BIRB-796濃度を上昇させながら、高グリシンループを標識したp38αの発光スペクトルを測定することにより実施した(図2A)。その後、レシオメトリック蛍光値(R = Iλ512/Iλ475)をリガンドの濃度に対し、対数スケールでプロットして、BIRB-796についてKd = 9.5 ± 2.7 nMと直接的に決定したが(図2B)、これは、活性化ループを標識したp38αを用いて得られた値7.5 ± 2.3 nMと同等であり、この代替のアプローチで得られたDFG-outバインダーのKd値の信頼性が実証される。
【0155】
475 nmでの発光の有意な変化によって、種々の濃度のリガンドについて、リアルタイムでの解離および会合の反応速度論の研究が可能になった。II型(例えばBIRB-796)およびIII型(例えばRL36)阻害剤両者の結合に際し、475 nmでのアクリロダン発光は減少し、結合状態における最大発光波長の赤方偏移が起こる(図2Aおよび3A左パネル)。平衡において、P-ループを標識したp38αの発光スペクトルは、512 nmおよび475 nmでの発光強度がほぼ同等であるように変化する(即ち、R = 512/475 nmは通常〜1.0の値である)。このレシオメトリック蛍光アウトプットを用い、終点平衡測定を実施して、蛍光データを阻害剤濃度の対数スケールに対してプロットすることにより、これらのリガンドのKdを直接的に得ることが可能になる(図2Bおよび3A右)。この反応は、ATP部位に隣接するアロステリック部位をふさぎ、活性化ループにおいて立体構造変化を要する、全てのII型またはIII型阻害剤の特性である。上述の通り、この立体構造変化によって、特定の方法でアクリロダンの蛍光を変化させる、P-ループにおける特徴的変化が起こる。P-ループとDFGモチーフのPheとの間のスタッキングにより、DFG-out構造を安定化することが知られているI型リガンドもまた、高感度で検出され、II/III型リガンドと比較した場合、475 nmでさらに大きく強度が損われる(図3B左)。
【0156】
各リガンドの結合反応速度を調査することにより、このようなI型リガンドはII型またはIII型リガンドから容易に識別される。反応速度の測定は、リガンドを緩衝液中の標識キナーゼ懸濁液に添加する際に、単一波長(475 nm)で発光強度の減少をモニターすることによって行われる。BIRB-796等のII型阻害剤(図2C)およびRL36等のIII型阻害剤(図3a中央)の場合、結合の反応速度はI型阻害剤よりも有意に遅いが、これはI型阻害剤がDFG-inまたはDFG-out構造を安定化するかどうかに関係しない(図4の反応速度論的値を参照)。種々のII型またはIII型阻害剤の添加による蛍光減衰はより遅く、一次減衰関数に容易に適合して、kobsを得る(図2Cおよび図3a中央)。次に、実験的に決定されたkobs値をプロットして、BIRB-796について、欧州特許EP 08 01 3340号および欧州特許EP 08 02 0341号に記載された全てのリガンドのkonを決定し得る。konは、BIRB-796について、〜4.0 x 104 M-1秒-1と決定された。結合しているI型阻害剤の蛍光減衰は非常に速く、ストップトフロー蛍光分光法を使用しないと正確には適切ではない。過剰の非標識p38αを用いてac-p38αから阻害剤を抽出することにより、475 nmでの蛍光強度が上向きに変化するが、これも、一次関数蛍光に適合し、koffの直接的決定が可能であった(図2D)。これらの測定により、蛍光反応の信頼性が実証され、かつ、DFG-inおよびDFG-out立体構造の間に存在する平衡が変化することが実証される。阻害剤の型に関わらず、タンパク質からのリガンドの解離速度は、常に結合速度よりも遅く、これはよく知られている観察結果であるが、p38αについては特に結合速度より遅い(パルゲリス(Pargellis)ら、2003年)。
【0157】
高グリシンループを標識したp38αの蛍光反応を特性化した後、入手可能な少数のサブセットの化合物におけるII型およびIII型阻害剤の高感度の検出を目的としたHTSフォーマットに、アッセイを適合させた(クルター(Kluter)ら、2009年;ゲトリク(Getlik)ら、2009年;ミカルチク(Michalczyk)ら、2008年;パワー(Pawar)ら、2010年;ソス(Sos)ら、2010年)が、これは、キナーゼのDFG-inまたはDFG-out構造に特権的に結合することが知られている種々の骨格を含む。3つの固定した阻害剤濃度(0.5、5および50μM)での最初のプレスクリーニングの後、いくつかは知られているp38α阻害剤の誘導体である、選択された化合物は、濃度依存性直接結合測定を用いてさらなる研究を行い、kon、koffおよびKdを決定し(図3および4)、さらに、妥当性を確認するため、本発明の方法と欧州特許EP 08 01 3340号および欧州特許EP 08 02 0341号に開示された方法とで検出された化合物の親和性を比較した。
【0158】
予測された通り、知られているDFG-outバインダーであるBIRB-796は、5時間にわたり、明確な時間依存性(図7)を示し、欧州特許EP 08 02 0341号の活性化ループを標識したp38αを用いて得られたものと同等のKd値を有することがわかった。加えて、BIRB-796およびRL36は、親和性の一次決定因子として、konよりも主にkoffに関して異なることがわかったが、これは、ピラゾロ尿素系化合物のp38αのDFG-out構造への結合について、非常に特徴的な観察である(パルゲリス(Pargells)ら、2002年;シマード(Simard)ら、2009年)。知られているI型p38α阻害剤であるSB203580もまた高感度で検出され、欧州特許EP 08 02 0341号に開示されたものと同等のKd値であった。欧州特許EP 08 02 0341号では、I型結合形態を採用し、キナーゼヒンジ領域に接触するにもかかわらず、SB203580がp38αに結合し、高グリシンループのDFG Phe169とTyr35の間にπ-πスタッキング相互作用を形成することによってDFG-out構造を安定化し得ることが開示されるが、これによって、欧州特許EP 08 02 0341号の方法および本発明の両者を用いて、この化合物が高感度で検出されることが説明される。SB203580のI型結合形態は、結合および解離反応速度がより迅速であるため、および、DFG-outバインダーであるBIRB-796と同等のKdの時間依存性を示さなかったため(図7)、HTSフォーマットにおいてII/III型バインダーから容易に識別された。従って、本発明の方法によっても、検出されたリガンドのタンパク質との共結晶化を要せずに、阻害剤結合形態の容易な予備的評価が可能になる。
【0159】
ダサチニブは、Kdが27 nMと報告されたp38αに結合するI型阻害剤であるが(カラマン(Karaman)ら、2008年)、高グリシンループを標識したp38αを用いて検出されず、このことから、ダサチニブは、Abl(PDBコード:2GQG)およびcSrc(PDBコード:3G5D)のDFG-in構造に見られる、予測された/公表された結合形態をとっていることが示唆される。しかし、インドール誘導体であるScios-469および2-フェニル置換キナゾリンであるRL40は両者とも、古典的なI型結合形態をとり、ヒンジ領域に接触することが知られており(ムラリ(Murali)ら、2007年;ピエルセ(Pierce)ら、2005年)、これらの化合物がこのアプローチを用いて検出されたことは、驚くべき観察であった。Scios-469の場合、我々は、Kd値を8.2 ± 2.9 nMと決定したが、これは、この化合物について報告されたIC50(〜9 nM)(ムラリ・ダール(Murali Dhar)ら、2007年)とよく一致し、本発明の方法がこのリガンドに非常に高感度であることを実証する。加えて、VEGFR2阻害剤であるCP547632(CP-547632)もまた検出され、Kd値は99 ±/- 13 nMであることが分かった。この化合物は、VEGFR2が媒介する血管新生および腫瘍成長の阻害によって作用する抗癌剤として、臨床検査中である(ベーベ(Beebe)ら、2003年)。現在まで、キナーゼとの複合体中のCP547632の結晶構造は公表されていないが、以前に報告された薬物動態的研究によって、CP547632が、ATP競合的にVEGFR2を阻害することが明らかになった(ベーべら、2003年)。RL40、Scios-469およびCP547632のリアルタイム反応速度測定により、3つの化合物は全てp38αに迅速に結合する(<2秒)ことが示されるが、これは、予測されたI型結合形態と一致する。しかし、活性化ループまたは高グリシンループのいずれかを標識したp38αを用いて、CP547632のみが特性化される可能性があるが;後者のみがPL40およびScios-469を検出した。このことから、おそらくアクリロダンで標識した高グリシンループにさらに接触し、その立体構造を変化させることによって独特な立体構造変化を起こし得る、あるI型阻害剤を検出するという追加利点を、本発明が有することが示唆される。
【0160】
p38αに結合することが予測される検出されたリガンドについて、決定されたKd値に関し、親和性は、他の方法を用いて他で決定および公表されたものと殆ど一致している(パルゲリス(Pargellis)ら、2003年、欧州特許EP 08 01 3340号および欧州特許EP 08 02 0341号)。P-ループ標識アッセイおよび他の方法を用いて報告されたKd値と反応速度論的傾向がほぼ一致することにより、このアッセイ系の正当性が立証される。加えて、p38αに結合しないことが知られているII型リガンド(イマチニブ)およびp38αを強力に阻害するが(カラマン(Karaman)ら、2008年)、DFGモチーフまたはP-ループと相互作用しないI型阻害剤(ダサチニブ)は、このアッセイでは検出されず、陰性対照として使用し得た(図3a右;図3b右)。
【0161】
実施例6:RL40、Scios-469およびCP547632の結晶構造は、P-ループの運動を裏付ける。
我々は、P-ループを標識したp38αに対していくつかの化合物をスクリーニングし、アッセイで高感度で検出し、RL40、Scios-469およびCP547632を特定した。これらのリガンドの検出を説明する構造を詳細に理解するため、我々は、RL40、Scios-469およびCP547632を野生型p38αと共結晶化した。阻害剤は非標識p38αと共結晶化した。簡単に説明すると、30μLのp38α(10 mg/mL)を0.3μLの阻害剤(DMSO中、100 mM)と混合し、この混合物を氷上で1〜2時間インキュベートすることにより、タンパク質阻害剤複合体を調製した。サンプルを13,000 rpmで5分間遠心分離して、過剰の阻害剤を除去した。懸滴蒸気拡散法を用い、1.5μLのタンパク質-阻害剤溶液を0.5μLのリザーバー(100 mM MES、pH 5.6-6.2、20-30% PEG4000および50 mM n-オクチル-β-D-グルコピラノシド)と混合することにより、24穴結晶化プレートにおいて、結晶を成長させた。p38αとの複合体中のRL40の構造(図5A)により、これまでに報告されているSB203580の構造中に見られるもの(欧州特許EP 08 01 3340号および欧州特許EP 08 02 0341号)と類似の、独特で予測しなかった結合形態が明らかになる。リガンドは、DFGモチーフのPheと独特のπ-πスタッキングを形成することにより、P-ループと相互作用し、それによりDFG-out構造を安定化し、アッセイにおけるその検出を合理的に説明する。この構造とSB203580-p38α複合体の構造との重なり(図5B)により、両阻害剤の特徴が良好に重なることが明らかになり、親和性を改善する次世代の化学修飾を理解する上での手掛かりとなる。しかし、SB203580と異なり、化合物RL40はヒンジ領域に接触せず、結合したアクリルアミドは、欧州特許EP 08 01 3340号および欧州特許EP 08 02 0341号に開示された活性化ループの標識に使用される位置の方向に、さらに伸長する。従って、この位置での活性化ループの標識化は、本書に記載したRL40の結合形態と適合しない可能性があり、これまでにRL40のp38αへの結合を検出し報告することが不可能であった理由が説明されるかもしれない。この独特の結合形態は、GSK3との複合体中のRL40のこれまでに報告された構造類似体(ピエルセ(Pierce)ら、2005年)、およびカルモジュリン依存性タンパク質キナーゼ1D(PDBコード:2JC6)とは有意に異なるが、ここで、阻害剤はATP結合部位内で完全に異なる配向を採り、阻害剤のアミノ-ピラゾール部分は3つの水素結合によってヒンジ領域と強力に相互作用し、キナーゼはDFG-in構造中に見られる。RL40の類似体は代表的にはキナーゼのヒンジ領域に結合するため、この結合形態は予測されなかった。これらの知見により、RL40およびSB203580等のこの独特の結合形態を利用するリガンドを増やすためにP-ループを標識したキナーゼを使用する利点が強調される。
【0162】
加えて、P-ループ標識キナーゼアッセイは、Scios-469の結合を強力に検出した。我々は、Scios-469を野生型p38αと共結晶化し(図5C)、阻害剤がヒンジ領域への水素結合を形成することを観察したが、これは、構造的に近い類似体(PDBコード:2QD9)についてこれまで報告されたものと類似している。Scios-469のフルオロフェニル部分はゲートキーパー残基を越えて伸長し、p38αに対する化合物の親和性を増すことが知られている相互作用である疎水性サブポケットをふさぐ(ラフォン(Lafont)ら、2007年)。DFGモチーフは活性「in」構造中に見られるが、高グリシンループの立体構造は、Scios-469と結合したp38αの場合、有意に変化することから、本発明の方法のみを用いた高感度な検出が説明される。高グリシンループは阻害剤上で折り畳まれ、Tyr35の側鎖がScios-469のメチル置換ピペラジン環およびクロロ置換インドール環を溶媒から部分的に保護し、それによって阻害剤の結合を、おそらく疎水性相互作用によって安定化する。その結果、このリガンド表面から秩序化した水分子を除去することにより、Scios-469はp38αに高親和性で結合する。検出される全てのDFG-outバインダーの他に、Scios-469によって、P-ループ標識キナーゼアッセイがP-ループの立体構造を直接的に変化させる、いくつかのI型リガンドをどのように高感度で検出し得るかの例が示される。
【0163】
p38αとの複合体中でのCP547632の結晶構造(図5d)から、キナーゼが、ヒンジ領域にも結合する阻害剤(I型)により、DFG-out構造において安定化されることが明らかになった。阻害剤のコアのイソチアゾールに結合するカルボキサミドは、ヒンジ領域への2つの平行な水素結合を形成する(His107のCOおよびMet109のNH)。我々の知る限りでは、これは新たなヒンジ領域結合モチーフを示す。加えて、尿素部分の2つのNHは、Met109の骨格COに向かい、水素結合ドナーとなる。可溶化ピロリジン-ブタン部分の脂肪族リンカーは、驚くべきことに、溶媒に向かわず、ATPポケットに向けて内側に折り畳まれる。高グリシンループは、この複合体において相対的に移動性であり、結晶構造中、部分的に観察されない。従って、活性化ループの、その不活性立体構造への運動、および、CP547632によるその安定化により、p38αのアクリロダン標識高グリシンループの上方向の運動が誘導される(図1参照)。このことから、p38αの活性化ループを標識する方法を用いた場合と同様に、本発明の方法でCP547632が高感度に検出されることが説明される。
【0164】
実施例7:高グリシンループに標識したac-MKK7を用いるリアルタイムおよび終点蛍光測定
全ての実験は、MKK7のキナーゼドメイン(配列番号2)のみを用いて、図3のp38αについて実施した。標識したシステインは、P-ループの147位(配列番号2の31位)に天然に存在する。218、276および296位のシステイン(配列番号2の102、160および180位)をセリンで置換した(配列番号3)。
【0165】
MKK7は、II型およびIII型阻害剤の結合の影響を受けやすいDFG-out構造に感受性がないかもしれないキナーゼの一例を示す。本発明は、アッセイの、DFG-out構造に結合する(より遅い結合反応速度)リガンド間を識別する能力を実証するが、P-ループと直接相互作用し、その立体構造を修飾する多数のI型阻害剤に高感度でもある。カラマン(Karaman)ら、2008年による、300を超えるキナーゼのパネルに対するいくつかの阻害剤についてのキナーゼのプロファイリングによれば、MKK7の最も近い類似体は全て(MKK1-6、またMEK1-6としても知られている)、Kd値が<10μMのII型阻害剤で阻害されない。しかし、それらは、Kd値が3.4〜70 nMの範囲のスタウロスポリンによって強力に阻害される。スタウロスポリンおよびその最も近い誘導体は、全てのキナーゼの>90%をATP競合的方法で強力に阻害する。MKK7は、その時点でこのキナーゼのパネルの一部ではなかったが、同様の阻害剤選好およびその近い類似体としてのプロファイルが予測される。従って、P-ループ標識MKK7に対するI型阻害剤(K252a)のKdを測定した。K252aは無差別的I型阻害剤であり、スタウロスポリンの構造的に近い類似体である。図8Aにより、K252aの結合が標識タンパク質の蛍光強度の低下、および2つの波長(R = 472 nm / 510 nm)でのレシオメトリック発光の検出可能な変化を引き起こすことが示される。Kdを直接的に決定する終点法を用いて、これらの発光の割合を阻害剤濃度に対してプロットし、K252aについてKdが38 nMと求めることができるが(図8B)、これは、これらの化合物について予測された正しい範囲内である。陰性対照としてソラフェニブが含まれるが、これはII型阻害剤であり、10μM以下では検出されなかった。これらの知見は、MKK7について予測された結果と一致するが、これはDFG-out構造に対する非感受性を示す。このアッセイ反応が、I型阻害剤結合に対する反応におけるP-ループの運動によることを実証するため、ダサチニブも陰性対照として含めた。ダサチニブは、ATP競合的阻害剤であるか、cSrcおよびAblキナーゼであり、報告されたKd値は> 1μMであるが(カラマン(Karaman)ら、2008年)MKK1およびMKK2のみを阻害し、代表的には、知られているあらゆる結晶構造において、キナーゼのP-ループと相互作用しない。従って、このI型阻害剤の添加および検出は、MKK7については予測されなかったが、このデータは10μM以下を裏付ける。図8Cにより、リアルタイムの反応速度測定およびK252aの結合および解離の検出が強調される。図3のとおり、p38αについて、結合により生じる蛍光変化は、標識キナーゼからリガンドを抽出するための過剰の非標識MKK7の添加に際して可逆的である。K252aはI型阻害剤であるため、図3Bに示されるp38αのI型阻害剤SB203580の様に、これらのプロセスの反応速度は速い。
【0166】
参考文献
Adrian, F. J., Ding, Q., Sin, T., Velentza, A., Sloan, C., Liu, Y., Zhang, G., Hur, W., Ding, S., Manley, P., Mestan, J., Fabbro, D., Gray, N. S.. Nat Chem Biol 2006, 2, 95-102.
Annis, D. A., Nazef, N., Chuang, C. C., Scott, M. P., and Nash, H. M. (2004) A general technique to rank protein-ligand binding affinities and determine allosteric versus direct binding site competition in compound mixtures, Journal of the American Chemical Society 126, 15495-15503.
R. T. Aimes, W. Hemmer, S. S. Taylor. Serine-53 at the tip of the glycine-rich loop of cAMP-dependent protein kinase: role in catalysis, P-site specificity, and interaction with inhibitors. Biochemistry 2000, 39, 8325.
B. Apsel, J. A. Blair, B. Gonzalez, T. M. Nazif, M. E. Feldman, B. Aizenstein, R. Hoffman, R. L. Williams, K. M. Shokat, Z. A. Knight, Nat Chem Biol 2008, 4, 691.
【0167】
A. C. Backes, B. Zech, B. Felber, B. Klebl, G. Muller, Expert Opin Drug Discovery 2008, 3, 1409.
A. C. Backes, B. Zech, B. Felber, B. Klebl, G. Muller, Expert Opin Drug Discovery 2008, 3, 1427.
Beebe, J. S., Jani, J. P., Knauth, E., Goodwin, P., Higdon, C., Rossi, A. M., Emerson, E., Finkelstein, M., Floyd, E., Harriman, S., Atherton, J., Hillerman, S., Soderstrom, C., Kou, K., Gant, T., Noe, M. C., Foster, B., Rastinejad, F., Marx, M. A., Schaeffer, T., Whalen, P. M., Roberts, W. G. (2003). Pharmacological characterization of CP-547,632, a novel vascular endothelial growth factor receptor-2 tyrosine kinase inhibitor for cancer therapy. Cancer Res 63, 7301-9.
J. A. Blair, D. Rauh, C. Kung, C. H. Yun, Q. W. Fan, H. Rode, C. Zhang, M. J. Eck, W. A. Weiss, K. M. Shokat, Nat Chem Biol 2007, 3, 229.
【0168】
Calleja, V., Laguerre, M., Parker, P. J., Larijani, B.. PLoS Biol 2009, 7, 189-200.R. A. Copeland, D. L. Pompliano, T. D. Meek, Nat Rev Drug Discov 2006, 5, 730.
Dar, A. C., Lopez, M. S., and Shokat, K. M., Small Molecule Recognition of c-Src via the Imatinib-Binding Conformation. Chem Biol 15 (10), 1015 (2008).DeLano, W.L., The PyMOL Molecular Graphics System. http://www.pymol.org (2002).
de Lorimier, R. M., Smith, J. J., Dwyer, M. A., Looger, L. L., Sali, K. M., Paavola, C. D., Rizk, S. S., Sadigov, S., Conrad, D. W., Loew, L., and Hellinga, H. W. (2002) Construction of a fluorescent biosensor family, Protein Sci 11, 2655-2675.Dumas, J., Hatoum-Mokdad, H., Sibley, R., Riedl, B., Scott, W. J., Monahan, M. K., Lowinger, T. B., Brennan, C., Natero, R., Turner, T., Johnson, J. S., Schoenleber, R., Bhargava, A., Wilhelm, S. M., Housley, T. J., Ranges, G. E., and Shrikhande, A. (2000) 1-Phenyl-5-pyrazolyl ureas: potent and selective p38 kinase inhibitors, Bioorganic & medicinal chemistry letters 10, 2051-2054.
【0169】
Emsley, P. and Cowtan, K., Coot: model‐building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr 60 (Pt 12 Pt 1), 2126 (2004).
Eyers, P. A., Churchill, M. E., Maller, J. L. (2005). The Aurora A and Aurora B protein kinases: a single amino acid difference controls intrinsic activity and activation by TPX2. Cell Cycle 4, 784-9.
Fischmann, T., Smith, C., Mayhood, T., Myers, J., Reichert, P., Mannarino, A., Carr, D., Zhu, H., Wong, J., Yang, R. S., Le, H., Madison, V., Biochemistry 2009.
M. J. Garnett, R. Marais, Cancer Cell 2004, 6, 313.Gauglitz, G. and Vo-Dinh, T. (2003). Handbook of spectroscopy. Wiley-VCH.
Getlik, M., Grutter, C., Simard, J. R., Kluter, S., Rabiller, M., Rode, H. B., Robubi, A., Rauh, D. (2009). Hybrid compound design to overcome the gatekeeper T338M mutation in cSrc. J Med Chem 52, 3915-26.
Gschwind, A., Fischer, O. M. and Ullrich, A., The discovery of receptor tyrosine kinases: targets for cancer therapy. Nat Rev Cancer 4 (5), 361 (2004).
S. K. Hanks, T. Hunter, FASEB J 1995, 9, 576.
Hibbs, R. E., Talley, T. T., and Taylor, P. (2004) Acrylodan-conjugated cysteine side chains reveal conformational state and ligand site locations of the acetylcholine-binding protein, The Journal of biological chemistry 279, 28483-28491.
【0170】
Johnson, L. N., Noble, M. E. M. and Owen, D. J. (1996) Active and inactive protein kinases : structural basis for regulation. Cell 85, 149-158.Johnson, L. N. and Lewis, R. E. (2001). Structural basis for control by phosphorylation. Chem Rev. 2001 Aug;101(8):2209-42.
Kabsch, W., Automatic processing of rotation diffraction data from crystals of initially unknown symmetry and cell constants. . J. Appl. Cryst. 26, 795 (1993).
Karaman, M. W., Herrgard, S., Treiber, D. K., Gallant, P., Atteridge, C. E., Campbell, B. T., Chan, K. W., Ciceri, P., Davis, M. I., Edeen, P. T., Faraoni, R., Floyd, M., Hunt, J. P., Lockhart, D. J., Milanov, Z. V., Morrison, M. J., Pallares, G., Patel, H. K., Pritchard, S., Wodicka, L. M., Zarrinkar P.P. (2008). A quantitative analysis of kinase inhibitor selectivity. Nat Biotechnol 26, 127-32.
Kirkland, L. O., McInnes, C.. Biochem Pharmacol 2009.
【0171】
Kluter, S., Grutter, C., Naqvi, T., Rabiller, M., Simard, J. R., Pawar, V., Getlik, M., Rauh, D. (2009). Displacement assay for the detection of stabilizers of inactive kinase conformations. J Med Chem
Knighton, D. R., Zheng, J. H., Ten Eyck, L. F., Xuong, N. H, Taylor, S. S., Sowadski, J. M. (1991). Structure of a peptide inhibitor bound to the catalytic subunit of cyclic adenosine monophosphate-dependent protein kinase. Science 253, 414-20.
Lafont, V., Armstrong, A. A., Ohtaka, H., Kiso, Y., Mario Amzel, L., Freire, E. (2007). Chem Biol Drug Des 69, 413-22.
Lakowicz, J. R. (1999). Principles of Fluorescence Spectroscopy. Kluwer Academic / Plenum Publishers
Laskowski, R. A., MacArthur, M. W., Moss, D. S., and Thornton, J. M., PROCHECK: a program to check the stereochemical quality of protein structures. J Appl Cryst 26, 283 (1993).
【0172】
Larkin, M. A. et al., Clustal W and Clustal X version 2.0. Bioinformatics 23 (21), 2947 (2007).
Levinson, N. M. et al., A Src-like inactive conformation in the abl tyrosine kinase domain. PLoS Biol 4 (5), e144 (2006).
Liu, Y. and Gray, N. S., Rational design of inhibitors that bind to inactive kinase conformations. Nat Chem Biol 2 (7), 358 (2006).
Liu, C. C. and Schultz, P. G. (2010). Adding new chemistries to the genetic code. Annu Rev Biochem 79, 15.1-15.32.
M. Mapelli, L. Massimiliano, C. Crovace, M. A. Seeliger, L. H. Tsai, L. Meijer, A. Musacchio, J Med Chem 2005, 48, 671.
Michalczyk, A. et al., Structural insights into how irreversible inhibitors can overcome drug resistance in EGFR. Bioorg Med Chem 16 (7), 3482 (2008).
【0173】
Murali Dhar, T. G., Wrobleski, S. T., Lin, S., Furch, J. A., Nirschl, D. S., Fan, Y., Todderuf, G., Pitt, S., Doweyko, A. M., Sack, J. S., Mathur, A., McKinnon, M., Barrish, J. C., Dodd, J. H., Schieven, G. L., Keftheris, K. (2007). Synthesis and SAR of p38alpha MAP kinase inhibitors based on heterobicyclic scaffolds. Bioorg Med Chem Lett 17, 5019-24.
Murshudov, G. N., Vagin, A. A., and Dodson, E. J., Refinement of macromolecular structures by the maximum‐likelihood method. Acta Crystallogr D Biol Crystallogr 53 (Pt 3), 240 (1997).
B. Nagar, W. G. Bornmann, P. Pellicena, T. Schindler, D. R. Veach, W. T. Miller, B. Clarkson, J. Kuriyan, Cancer Res 2002, 62, 4236.
Nienaber, V. L. et al., Discovering novel ligands for macromolecules using X-ray crystallographic screening. Nature biotechnology 18 (10), 1105 (2000).
【0174】
Pargellis, C., Tong, L., Churchill, L., Cirillo, P. F., Gilmore, T., Graham, A. G., Grob, P. M., Hickey, E. R., Moss, N., Pav, S., and Regan, J. (2002) Inhibition of p38 MAP kinase by utilizing a novel allosteric binding site, Nature structural biology 9, 268-272.
Patel, S. B., Cameron, P. M., O`Keefe, S. J., Frantz-Wattley, B., Thompson, J., O`Neill, E. A., Tennis, T., Liu, L., Becker, J. W., Scapin, G. Acta Crystallogr D Biol Crystallogr 2009, 65, 777-85.
Pawar, V. G., Sos, M. L., Rode, H. B., Rabiller, M., Heynck, S., van Otterlo, W. A. L., Thomas, R. K., Rauh, D. (2010), Synthesis and biological evaluation of 4-anilinoquinolines as potent inhibitors of epidermal growth factor receptor. J Med Chem 53(7), 2892-901.
【0175】
W. A Pearson, Rapid and Sensitive Sequence Comparison with FASTP and FASTA, in Methods in Enzymology, ed. R. Doolittle Academic Press, San Diego) 183(1990)63-98.
Pierce AC, ter Haar E, Binch HM, Kay DP, Patel SR, Li P. J Med Chem. 2005 Feb 24;48(4):1278-81.
Read, R. J., Pushing the boundaries of molecular replacement with maximum likelihood. Acta Crystallogr D 57 (Pt 10), 1373 (2001).
Reich, M. et al., GenePattern 2.0. Nat Genet 38 (5), 500 (2006).
Regan, J., Breitfelder, S., Cirillo, P., Gilmore, T., Graham, A. G., Hickey, E., Klaus, B., Madwed, J., Moriak, M., Moss, N., Pargellis, C., Pav, S., Proto, A., Swinamer, A., Tong, L., and Torcellini, C. (2002) Pyrazole urea-based inhibitors of p38 MAP kinase: from lead compound to clinical candidate, Journal of medicinal chemistry 45, 2994-3008.
Regan, J., Pargellis, C. A., Cirillo, P. F., Gilmore, T., Hickey, E. R., Peet, G. W., Proto, A., Swinamer, A., and Moss, N. (2003) The kinetics of binding to p38MAP kinase by analogues of BIRB 796, Bioorganic & medicinal chemistry letters 13, 3101-3104.
【0176】
Rendell, D. (1987). Fluorescence and Phosphorescence. Crown
Richieri, G. V., Ogata, R. T., and Kleinfeld, A. M. (1999) The measurement of free fatty acid concentration with the fluorescent probe ADIFAB: a practical guide for the use of the ADIFAB probe, Molecular and cellular biochemistry 192, 87-94.M. Saraste, P. R. Sibbald, A. Wittinghofer, Trends Biochem Sci 1990, 15, 430.
Schuttelkopf, A. W. and van Aalten, D. M., PRODRG: a tool for high‐throughput crystallography of protein‐ligand complexes. Acta Crystallogr D Biol Crystallogr 60 (Pt 8), 1355 (2004).
Sharma, A. and Schulman, S. G. (1999). Introduction to Fluorescence Spectroscopy. Wiley interscience.Sullivan, J. E., Holdgate, G. A., Campbell, D., Timms, D., Gerhardt, S., Breed, J., Breeze, A. L., Bermingham, A., Pauptit, R. A., Norman, R. A., Embrey, K. J., Read, J., VanScyoc, W. S., and Ward, W. H. (2005) Prevention of MKK6-dependent activation by binding to p38alpha MAP kinase, Biochemistry 44, 16475-16490.
【0177】
M. A. Seeliger, B. Nagar, F. Frank, X. Cao, M. N. Henderson, J. Kuriyan, Structure 2007, 15, 299.
Shuker, S. B., Hajduk, P. J., Meadows, R. P., and Fesik, S. W., Discovering high-affinity ligands for proteins: SAR by NMR. Science 274 (5292), 1531 (1996).
J. R. Simard, S. Kluter, C. Grutter, M. Getlik, M. Rabiller, H. B. Rode, D. Rauh, Nat Chem Biol 2009, 5, 394-396.
Simard, J. R., Getlik, M., Grutter, C., Pawar, V., Wulfert, S., Rabiller, M., Rauh, D. (2009). Development of a fluorescent-tagged kinase assay system for the detection and characterization of allosteric kinase inhibitors. J Am Chem Soc 131, 13286-13296.
Simard, J. R., Pawar, V., Aust, B., Wolf, A., Rabiller, M., Wulfert, S., Robubi, A., Kluter, S., Ottmann, C., Rauh, D. (2009). High-throughput screening to identify inhibitors which stabilize inactive kinase conformations in p38alpha. J Am Chem Soc
【0178】
Sos, M. L., Rode, H. B., Heynck, S., Peifer, M., Fischer, F., Kluter, S., Pawar, V. G., Reuter, C., Heuckmann, J., Weiss, J., Ruddigkeit, L., Rabiller, M., Koker, M., Simard, J. R., Getlik, M., Yuza, Y., Chen, T., Greulich, H., Meyerson, M., Thomas, R. K., Rauh, D. (2010). Chemogenomic profiling provides insights into the limited activity of irreversible EGFR Inhibitors in tumor cells expressing the T790M EGFR resistance mutation. Cancer Research 70(3):868-74.
Tamayo, N, Liao, L., Goldberg,M., Powers, D., Tudor, Y-Y, Yu, V., Wong, L. M., Henkle, B., Middleton, S., Syed, R., Harvey, T., Jang, G., Hungate, R. and Celia Dominguez (2005). Design and synthesis of potent pyridazine inhibitors of p38 MAP kinase, Bioorganic & Medicinal Chemistry Letters 15:2409-2413.
Tecle, H., Feru, F., Liu, H., Kuhn, C., Rennie, G., Morris, M., Shao, J., Cheng, A. C., Gikunju, D., Miret, J., Coli, R., Xi, S. H., Clugston, S. L., Low, S., Kazmirski, S., Ding, Y. H., Cao, Q., Johnson, T. L., Deshmukh, G. D., DiNitto, J. P., Wu, J. C.; English, J. M. (2009). The design, synthesis and potential utility of fluorescence probes that target DFG-out conformation of p38alpha for high throughput screening binding assay. Chem Biol Drug Des 74, 547-59.
【0179】
Taylor, S. S. and Radzio-Andzelm, E. (1994) Three protein kinase structures define a common motif. Structure 2, 345-355; 43.Vogtherr, M., Saxena, K., Hoelder, S., Grimme, S., Betz, M., Schieborr, U., Pescatore, B., Robin, M., Delarbre, L., Langer, T., Wendt, K. U., and Schwalbe, H. (2006) NMR characterization of kinase p38 dynamics in free and ligand-bound forms, Angewandte Chemie (International ed 45, 993-997.Yeatman, T. J., A renaissance for SRC. Nat Rev Cancer 4 (6), 470 (2004).
Tokarski, J.S., Newitt, J., Chang, C.Y.J., Cheng, J.D., Wittekind, M., Kiefer, S.E., Kish, K., Lee, F.Y.F., Borzilerri, R., Lombardo, L.J., Xie, D., Zhang, Y., Klei, H.E. (2006) CANCER RES. 66: 5790-5797.
Wan, P. T., Garnett, M. J., Roe, S. M., Lee, S., Niculescu-Duvaz, D., Good, V. M., Jones, C. M., Marshall, C. J., Springer, C. J., Barford, D., Marais, R., Cell 2004, 116, 855.
【0180】
Wang, X, Destrument, A, Tournier, C. (2007). Physiological roles of MKK4 and MKK7: Insights from animal models. Biochimica et Biophysica Acta 1773 (2007), 1349-1357.
Wong, L., Lieser, S. A., Miyashita, O., Miller, M., Tasken, K., Onuchic, J. N., Adams, J. A., V. L. Woods, Jr., P. A. Jennings, J Mol Biol 2005, 351, 131.
Yem, A. W., Epps, D. E., Mathews, W. R., Guido, D. M., Richard, K. A., Staite, N. D., and Deibel, M. R., Jr. (1992) Site-specific chemical modification of interleukin-1 beta by acrylodan at cysteine 8 and lysine 103, The Journal of biological chemistry 267, 3122-3128.
J. Zhang, P. L. Yang, N. S. Gray, Nat. Rev. Cancer 2009, 9, 28.
【特許請求の範囲】
【請求項1】
天然に存在しているか、またはP-ループに導入されているアミノ酸で標識されたキナーゼであって、前記の標識化は、前記のアミノ酸の遊離チオールまたはアミノ基で行われ、かつ、前記の標識が、
(a)その環境における極性変化に感受性のある、チオールまたはアミノ反応性フルオロフォアであるか;または
(b)チオール反応性スピン標識、同位体または濃縮同位体チオールまたはアミノ反応性標識であり、
前記のフルオロフォア、スピン標識、同位体または濃縮同位体標識は、触媒活性を阻害せず、キナーゼの安定性を妨げない前記キナーゼ。
【請求項2】
セリン/トレオニンキナーゼ、またはチロシンキナーゼである請求項1に記載のキナーゼ。
【請求項3】
MEKキナーゼ、CSK、オーロラキナーゼ、GSK-3β、sSrc、EGFR、Abl、DDR1、AKT、LCK、CDK、p38αまたは別のMAPKである請求項1または2に記載のキナーゼ。
【請求項4】
標識されているアミノ酸が、システイン、リシン、アルギニンまたはヒスチジンである請求項1〜3のいずれかに記載のキナーゼ。
【請求項5】
P-ループの外側に存在する、溶媒にさらされているシステインが欠失しているか置換されている請求項1〜4のいずれかに記載のキナーゼ。
【請求項6】
p38αであり、かつ、標識されるシステインは配列番号1の35位に導入され、好ましくは配列番号1の119位および162位のシステインが、別のアミノ酸で置換されている請求項3〜5のいずれかに記載のキナーゼ。
【請求項7】
チオールまたはアミノ反応性フルオロフォアが、二置換ナフタレン化合物、クマリン系化合物、ベンゾキサジアゾール系化合物、ダポキシル系化合物、ビオシチン系化合物、フルオレスセイン、スルホン化ローダミン系化合物、アット(Atto)フルオロフォア、もしくはルシファー・イエロー(Lucifer Yellow)、または環境変化に対する感受性を示す上記いずれかの誘導体である請求項1〜6のいずれかに記載のキナーゼ。
【請求項8】
チオール反応性スピン標識がニトロキシドラジカルである請求項1〜6のいずれかに記載のキナーゼ。
【請求項9】
キナーゼ阻害剤のスクリーニング方法であって、
(a)請求項1〜8のいずれかに記載のキナーゼを供給すること;
(b)前記の、蛍光的またはスピン標識またはアイソトープ標識したキナーゼを候補阻害剤と接触させること;
(c)工程(a)および工程(b)の蛍光標識したキナーゼの、励起の際の、1以上の波長での蛍光発光シグナルまたはスペクトルを記録すること;または
(c)’工程(a)および工程(b)のスピン標識またはアイソトープ標識したキナーゼの電子常磁性共鳴(EPR)または核磁気共鳴(NMR)スペクトルを記録すること;および
(d)工程(c)で記録される1以上の波長での蛍光発光シグナルまたはスペクトル、または工程(c)’で記録されるEPRまたはNMRスペクトルを比較することを含み;
ここで、前記の工程(c)で得られる前記の蛍光標識したキナーゼの、1以上の波長、好ましくは発光極大での蛍光強度の差、および/または、スペクトルにおける蛍光発光波長のシフト、または、前記の工程(c)’で得られるスピン標識またはアイソトープ標識したキナーゼの、EPRまたはNMRスペクトルにおける変化は、候補阻害剤がキナーゼ阻害剤であることを示すスクリーニング方法。
【請求項10】
キナーゼ阻害剤のリガンドの結合および/もしくは会合または解離のキネティクスを決定する方法であって、
(a)請求項1〜8のいずれかに記載の蛍光標識キナーゼを種々の濃度の阻害剤と接触させること、または、
(a)’阻害剤に結合した請求項1〜8のいずれかに記載の蛍光標識キナーゼを、種々の濃度の非標識キナーゼと接触させること;
(b)前記の蛍光標識したキナーゼの、励起の際の、1以上の波長での蛍光発光シグナルまたはスペクトルを、各濃度について記録すること;
(c)工程(b)で記録された1以上の波長での蛍光発光シグナルまたはスペクトルから、各濃度の速度定数を決定すること、または
(c1)各濃度の阻害剤について、工程(b)で記録された1以上の波長での蛍光発光シグナルまたはスペクトルから、Kdを決定すること、または
(c2)各濃度の非標識キナーゼについて、工程(b)で記録された1以上の波長での蛍光発光シグナルまたはスペクトルから、Kaを決定すること;
(d)工程(b)で得られた種々の濃度の阻害剤についてのシグナルまたはスペクトルから工程(c)で決定した速度定数より、直接、konを決定すること、および/または、koffを外挿すること、または
(d)’工程(b)で得られた種々の濃度の非標識キナーゼについてのシグナルまたはスペクトルから工程(c)で決定した速度定数より、直接、koffを決定すること、および/または、konを外挿すること;および
(e)任意に、工程(d)または(d)’で得られたkonおよびkoffから、Kdおよび/またはKaを計算することを含む方法。
【請求項11】
キナーゼ阻害剤の解離または会合を決定する方法であって、
(a)請求項1〜8のいずれかに記載のスピン標識またはアイソトープ標識したキナーゼを、種々の濃度の阻害剤と接触させること、または
(a)’阻害剤に結合した請求項1〜8のいずれかに記載のスピン標識またはアイソトープ標識したキナーゼを、種々の濃度の非標識キナーゼと接触させること;
(b)前記のスピン標識またはアイソトープ標識したキナーゼのEPRまたはNMRスペクトルを、各濃度の阻害剤および/または非標識キナーゼについて記録すること;および
(c)種々の濃度の阻害剤について、工程(b)で記録されたEPRまたはNMRスペクトルからKdを決定すること;または
(c)’種々の濃度の非標識キナーゼについて、工程(b)で記録されたEPRまたはNMRスペクトルからKaを決定することを含む方法。
【請求項12】
キナーゼ阻害剤のスクリーニングに好適な突然変異キナーゼの産生方法であって、
(a)関心のあるキナーゼにおいて存在する場合、P-ループの外部に遊離チオールまたはアミノ基を有する、溶媒にさらされているアミノ酸、および/または、P-ループ内部の好適でない位置に遊離チオールまたはアミノ基を有するアミノ酸を、遊離チオールまたはアミノ基を有しないアミノ酸で置換すること;
(b)P-ループ内に遊離チオールまたはアミノ基を有するアミノ酸が存在しない場合、前記の関心のあるキナーゼのP-ループにおけるアミノ酸を突然変異して、遊離チオールまたはアミノ基を有するアミノ酸とすること;
(c)関心のあるキナーゼを、その環境における極性変化に感受性のあるチオール-またはアミノ-反応性フルオロフォア、チオール反応性スピン標識、アイソトープまたは濃縮アイソトープチオール-またはアミノ-反応性標識で標識することであって、前記のフルオロフォア、スピン標識、アイソトープまたは高アイソトープ標識は、キナーゼの触媒活性を阻害せず、および/または、キナーゼの安定性を妨げない;
(d)工程(c)で得られたキナーゼを、前記のキナーゼの知られている阻害剤と接触させること;および
(e)工程(c)および(d)の前記の蛍光標識キナーゼの、励起の際の、1以上の波長での蛍光発光シグナルまたはスペクトルを記録すること、または
(e)’工程(c)および(d)の前記のスピン標識キナーゼのEPRまたはNMRスペクトルを記録すること;および
(f)工程(e)で記録された蛍光発光スペクトル、または、工程(e)’で記録されたEPRまたはNMRスペクトルを比較することを含み、
工程(e)で得られる、少なくとも1つの波長、好ましくは発光極大での蛍光強度の差、および/または、前記の蛍光標識キナーゼのスペクトルにおける蛍光発光波長のシフト、または、工程(e)’で得られる前記のスピン標識またはアイソトープ標識キナーゼのEPRまたはNMRスペクトルの変化から、該キナーゼがキナーゼ阻害剤のスクリーニングに好適であることが示される方法。
【請求項13】
キナーゼ阻害剤がキナーゼのATP結合部位に隣接するアロステリック部位に部分的または完全に結合する請求項9〜12のいずれかに記載の方法。
【請求項14】
キナーゼのATP結合部位に隣接するアロステリック部位に、部分的または完全に結合するキナーゼ阻害剤の特定方法であって、
(a)請求項10の方法に従って、阻害剤をスクリーニングすること、および
(b)工程(a)で特定された阻害剤の速度定数を決定することを含み、
工程(b)で決定された<0.140秒-1の結合速度は、特定されたキナーゼ阻害剤が、キナーゼのATP結合部位に隣接するアロステリック部位に部分的または完全に結合することを示す方法。
【請求項15】
キナーゼが天然に存在しているか、またはP-ループに導入されているシステインで標識されている、請求項1〜8のいずれかに記載のキナーゼまたは請求項9〜14のいずれかに記載の方法。
【請求項16】
前記のキナーゼの阻害剤として特定された化合物の薬理学的特性を最適化することをさらに含む請求項9または12〜15のいずれかに記載の方法。
【請求項17】
最適化が、前記のキナーゼの阻害剤として特定された阻害剤を修飾して以下を達成することを含む請求項16に記載の方法:
(a) 修飾された活性スペクトル、器官特異性、および/または
(b) 効力の向上、および/または
(c) 毒性の減少(治療指数の向上)、および/または
(d) 副作用の減少、および/または
(e) 修飾された治療効果の発現、効果持続期間、および/または
(f) 修飾された薬物動態パラメータ(吸収、分布、代謝および排泄)、および/または
(g) 修飾された物理化学的パラメータ(溶解性、吸湿性、色、味、臭気、安定性、状態)、および/または
(h) 全般的特異性、器官/組織特異性の向上、および/または
(i) 適用形態および経路の以下による最適化、
a. カルボキシル基のエステル化、または
b. ヒドロキシル基のカルボン酸によるエステル化、または
c. ヒドロキシル基の、例えばリン酸エステル、ピロリン酸エステルまたは硫酸エステルまたはヘミコハク酸エステルへのエステル化、または
d. 薬学的に許容される塩の形成、または
e. 薬学的に許容される複合体の形成、または
f. 薬理学的活性ポリマーの合成、または
g. 親水性部分の導入、または
h. 芳香環または側鎖上の置換基の導入/交換、置換パターンの変更、または
i. 等比体積または生物学的等価部分の導入による修飾、または
j. 同族化合物の合成、または
k. 分枝状側鎖の導入、または
l. アルキル置換基の環状アナログへの変換、または
m. ヒドロキシル基のケタール、アセタールへの誘導化、または
n. アミド、フェニルカルバメートへのN-アセチル化、または
o. マンニッヒ塩基、イミンの合成、または
p. ケトンまたはアルデヒドの、シッフ塩基、オキシム、アセタール、ケタール、エノールエステル、オキサゾリジン、チアゾリジンへの変換またはそれらの組み合わせ。
【請求項1】
天然に存在しているか、またはP-ループに導入されているアミノ酸で標識されたキナーゼであって、前記の標識化は、前記のアミノ酸の遊離チオールまたはアミノ基で行われ、かつ、前記の標識が、
(a)その環境における極性変化に感受性のある、チオールまたはアミノ反応性フルオロフォアであるか;または
(b)チオール反応性スピン標識、同位体または濃縮同位体チオールまたはアミノ反応性標識であり、
前記のフルオロフォア、スピン標識、同位体または濃縮同位体標識は、触媒活性を阻害せず、キナーゼの安定性を妨げない前記キナーゼ。
【請求項2】
セリン/トレオニンキナーゼ、またはチロシンキナーゼである請求項1に記載のキナーゼ。
【請求項3】
MEKキナーゼ、CSK、オーロラキナーゼ、GSK-3β、sSrc、EGFR、Abl、DDR1、AKT、LCK、CDK、p38αまたは別のMAPKである請求項1または2に記載のキナーゼ。
【請求項4】
標識されているアミノ酸が、システイン、リシン、アルギニンまたはヒスチジンである請求項1〜3のいずれかに記載のキナーゼ。
【請求項5】
P-ループの外側に存在する、溶媒にさらされているシステインが欠失しているか置換されている請求項1〜4のいずれかに記載のキナーゼ。
【請求項6】
p38αであり、かつ、標識されるシステインは配列番号1の35位に導入され、好ましくは配列番号1の119位および162位のシステインが、別のアミノ酸で置換されている請求項3〜5のいずれかに記載のキナーゼ。
【請求項7】
チオールまたはアミノ反応性フルオロフォアが、二置換ナフタレン化合物、クマリン系化合物、ベンゾキサジアゾール系化合物、ダポキシル系化合物、ビオシチン系化合物、フルオレスセイン、スルホン化ローダミン系化合物、アット(Atto)フルオロフォア、もしくはルシファー・イエロー(Lucifer Yellow)、または環境変化に対する感受性を示す上記いずれかの誘導体である請求項1〜6のいずれかに記載のキナーゼ。
【請求項8】
チオール反応性スピン標識がニトロキシドラジカルである請求項1〜6のいずれかに記載のキナーゼ。
【請求項9】
キナーゼ阻害剤のスクリーニング方法であって、
(a)請求項1〜8のいずれかに記載のキナーゼを供給すること;
(b)前記の、蛍光的またはスピン標識またはアイソトープ標識したキナーゼを候補阻害剤と接触させること;
(c)工程(a)および工程(b)の蛍光標識したキナーゼの、励起の際の、1以上の波長での蛍光発光シグナルまたはスペクトルを記録すること;または
(c)’工程(a)および工程(b)のスピン標識またはアイソトープ標識したキナーゼの電子常磁性共鳴(EPR)または核磁気共鳴(NMR)スペクトルを記録すること;および
(d)工程(c)で記録される1以上の波長での蛍光発光シグナルまたはスペクトル、または工程(c)’で記録されるEPRまたはNMRスペクトルを比較することを含み;
ここで、前記の工程(c)で得られる前記の蛍光標識したキナーゼの、1以上の波長、好ましくは発光極大での蛍光強度の差、および/または、スペクトルにおける蛍光発光波長のシフト、または、前記の工程(c)’で得られるスピン標識またはアイソトープ標識したキナーゼの、EPRまたはNMRスペクトルにおける変化は、候補阻害剤がキナーゼ阻害剤であることを示すスクリーニング方法。
【請求項10】
キナーゼ阻害剤のリガンドの結合および/もしくは会合または解離のキネティクスを決定する方法であって、
(a)請求項1〜8のいずれかに記載の蛍光標識キナーゼを種々の濃度の阻害剤と接触させること、または、
(a)’阻害剤に結合した請求項1〜8のいずれかに記載の蛍光標識キナーゼを、種々の濃度の非標識キナーゼと接触させること;
(b)前記の蛍光標識したキナーゼの、励起の際の、1以上の波長での蛍光発光シグナルまたはスペクトルを、各濃度について記録すること;
(c)工程(b)で記録された1以上の波長での蛍光発光シグナルまたはスペクトルから、各濃度の速度定数を決定すること、または
(c1)各濃度の阻害剤について、工程(b)で記録された1以上の波長での蛍光発光シグナルまたはスペクトルから、Kdを決定すること、または
(c2)各濃度の非標識キナーゼについて、工程(b)で記録された1以上の波長での蛍光発光シグナルまたはスペクトルから、Kaを決定すること;
(d)工程(b)で得られた種々の濃度の阻害剤についてのシグナルまたはスペクトルから工程(c)で決定した速度定数より、直接、konを決定すること、および/または、koffを外挿すること、または
(d)’工程(b)で得られた種々の濃度の非標識キナーゼについてのシグナルまたはスペクトルから工程(c)で決定した速度定数より、直接、koffを決定すること、および/または、konを外挿すること;および
(e)任意に、工程(d)または(d)’で得られたkonおよびkoffから、Kdおよび/またはKaを計算することを含む方法。
【請求項11】
キナーゼ阻害剤の解離または会合を決定する方法であって、
(a)請求項1〜8のいずれかに記載のスピン標識またはアイソトープ標識したキナーゼを、種々の濃度の阻害剤と接触させること、または
(a)’阻害剤に結合した請求項1〜8のいずれかに記載のスピン標識またはアイソトープ標識したキナーゼを、種々の濃度の非標識キナーゼと接触させること;
(b)前記のスピン標識またはアイソトープ標識したキナーゼのEPRまたはNMRスペクトルを、各濃度の阻害剤および/または非標識キナーゼについて記録すること;および
(c)種々の濃度の阻害剤について、工程(b)で記録されたEPRまたはNMRスペクトルからKdを決定すること;または
(c)’種々の濃度の非標識キナーゼについて、工程(b)で記録されたEPRまたはNMRスペクトルからKaを決定することを含む方法。
【請求項12】
キナーゼ阻害剤のスクリーニングに好適な突然変異キナーゼの産生方法であって、
(a)関心のあるキナーゼにおいて存在する場合、P-ループの外部に遊離チオールまたはアミノ基を有する、溶媒にさらされているアミノ酸、および/または、P-ループ内部の好適でない位置に遊離チオールまたはアミノ基を有するアミノ酸を、遊離チオールまたはアミノ基を有しないアミノ酸で置換すること;
(b)P-ループ内に遊離チオールまたはアミノ基を有するアミノ酸が存在しない場合、前記の関心のあるキナーゼのP-ループにおけるアミノ酸を突然変異して、遊離チオールまたはアミノ基を有するアミノ酸とすること;
(c)関心のあるキナーゼを、その環境における極性変化に感受性のあるチオール-またはアミノ-反応性フルオロフォア、チオール反応性スピン標識、アイソトープまたは濃縮アイソトープチオール-またはアミノ-反応性標識で標識することであって、前記のフルオロフォア、スピン標識、アイソトープまたは高アイソトープ標識は、キナーゼの触媒活性を阻害せず、および/または、キナーゼの安定性を妨げない;
(d)工程(c)で得られたキナーゼを、前記のキナーゼの知られている阻害剤と接触させること;および
(e)工程(c)および(d)の前記の蛍光標識キナーゼの、励起の際の、1以上の波長での蛍光発光シグナルまたはスペクトルを記録すること、または
(e)’工程(c)および(d)の前記のスピン標識キナーゼのEPRまたはNMRスペクトルを記録すること;および
(f)工程(e)で記録された蛍光発光スペクトル、または、工程(e)’で記録されたEPRまたはNMRスペクトルを比較することを含み、
工程(e)で得られる、少なくとも1つの波長、好ましくは発光極大での蛍光強度の差、および/または、前記の蛍光標識キナーゼのスペクトルにおける蛍光発光波長のシフト、または、工程(e)’で得られる前記のスピン標識またはアイソトープ標識キナーゼのEPRまたはNMRスペクトルの変化から、該キナーゼがキナーゼ阻害剤のスクリーニングに好適であることが示される方法。
【請求項13】
キナーゼ阻害剤がキナーゼのATP結合部位に隣接するアロステリック部位に部分的または完全に結合する請求項9〜12のいずれかに記載の方法。
【請求項14】
キナーゼのATP結合部位に隣接するアロステリック部位に、部分的または完全に結合するキナーゼ阻害剤の特定方法であって、
(a)請求項10の方法に従って、阻害剤をスクリーニングすること、および
(b)工程(a)で特定された阻害剤の速度定数を決定することを含み、
工程(b)で決定された<0.140秒-1の結合速度は、特定されたキナーゼ阻害剤が、キナーゼのATP結合部位に隣接するアロステリック部位に部分的または完全に結合することを示す方法。
【請求項15】
キナーゼが天然に存在しているか、またはP-ループに導入されているシステインで標識されている、請求項1〜8のいずれかに記載のキナーゼまたは請求項9〜14のいずれかに記載の方法。
【請求項16】
前記のキナーゼの阻害剤として特定された化合物の薬理学的特性を最適化することをさらに含む請求項9または12〜15のいずれかに記載の方法。
【請求項17】
最適化が、前記のキナーゼの阻害剤として特定された阻害剤を修飾して以下を達成することを含む請求項16に記載の方法:
(a) 修飾された活性スペクトル、器官特異性、および/または
(b) 効力の向上、および/または
(c) 毒性の減少(治療指数の向上)、および/または
(d) 副作用の減少、および/または
(e) 修飾された治療効果の発現、効果持続期間、および/または
(f) 修飾された薬物動態パラメータ(吸収、分布、代謝および排泄)、および/または
(g) 修飾された物理化学的パラメータ(溶解性、吸湿性、色、味、臭気、安定性、状態)、および/または
(h) 全般的特異性、器官/組織特異性の向上、および/または
(i) 適用形態および経路の以下による最適化、
a. カルボキシル基のエステル化、または
b. ヒドロキシル基のカルボン酸によるエステル化、または
c. ヒドロキシル基の、例えばリン酸エステル、ピロリン酸エステルまたは硫酸エステルまたはヘミコハク酸エステルへのエステル化、または
d. 薬学的に許容される塩の形成、または
e. 薬学的に許容される複合体の形成、または
f. 薬理学的活性ポリマーの合成、または
g. 親水性部分の導入、または
h. 芳香環または側鎖上の置換基の導入/交換、置換パターンの変更、または
i. 等比体積または生物学的等価部分の導入による修飾、または
j. 同族化合物の合成、または
k. 分枝状側鎖の導入、または
l. アルキル置換基の環状アナログへの変換、または
m. ヒドロキシル基のケタール、アセタールへの誘導化、または
n. アミド、フェニルカルバメートへのN-アセチル化、または
o. マンニッヒ塩基、イミンの合成、または
p. ケトンまたはアルデヒドの、シッフ塩基、オキシム、アセタール、ケタール、エノールエステル、オキサゾリジン、チアゾリジンへの変換またはそれらの組み合わせ。
【図1】

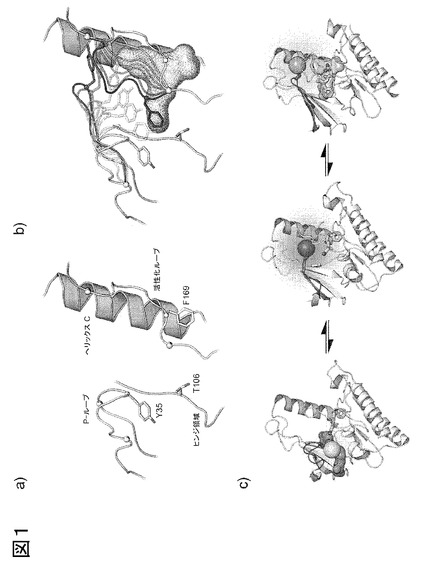
【図2】
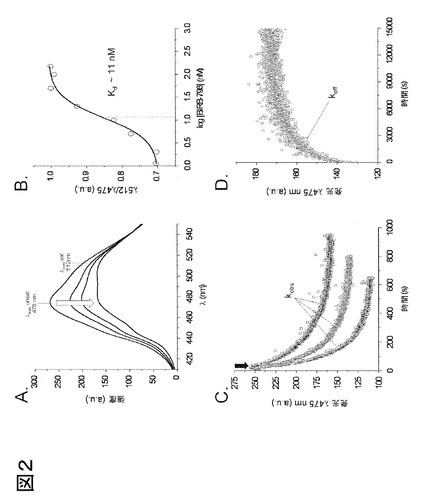
【図3】
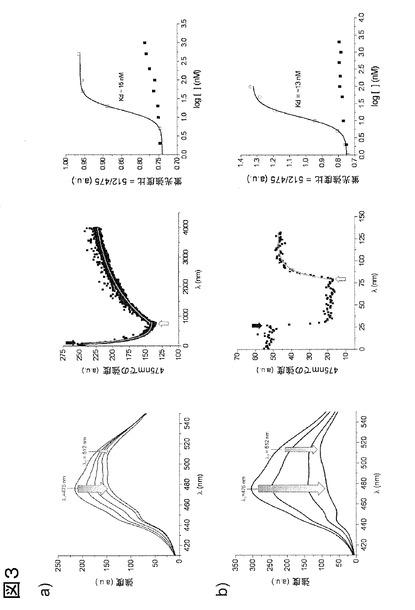
【図4−1】
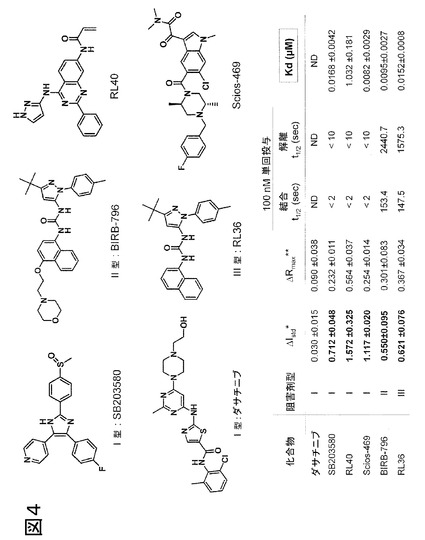
【図4−2】
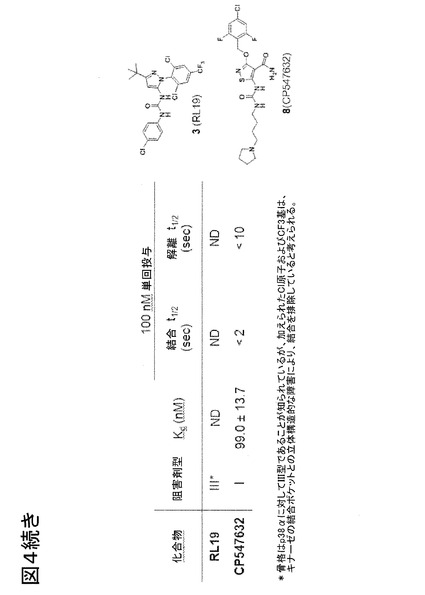
【図5−1】
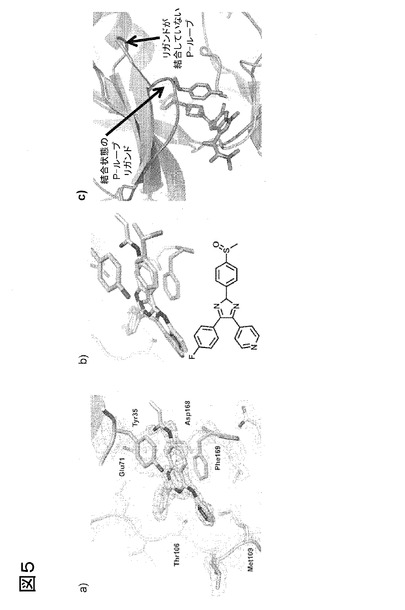
【図5−2】
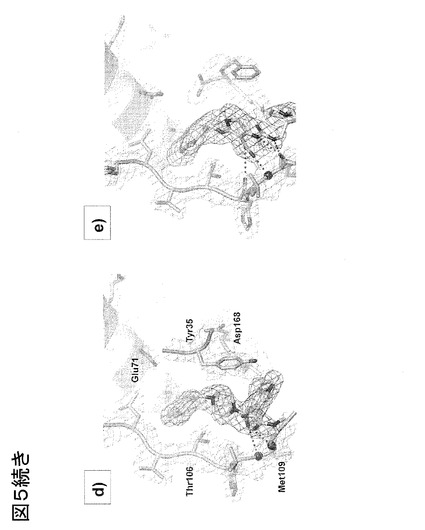
【図6】

【図7】
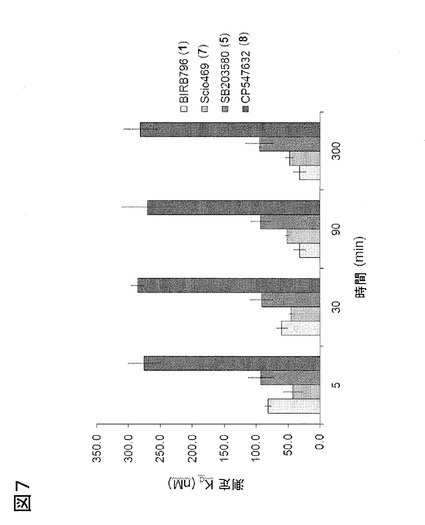
【図8】
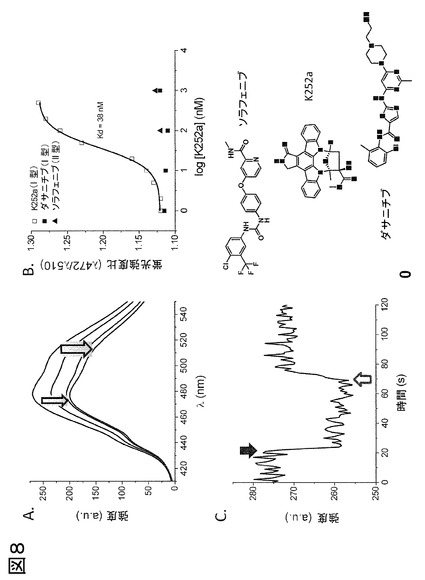
【図2】
【図3】
【図4−1】
【図4−2】
【図5−1】
【図5−2】
【図6】
【図7】
【図8】
【公表番号】特表2013−504302(P2013−504302A)
【公表日】平成25年2月7日(2013.2.7)
【国際特許分類】
【出願番号】特願2012−505187(P2012−505187)
【出願日】平成22年4月19日(2010.4.19)
【国際出願番号】PCT/EP2010/055129
【国際公開番号】WO2010/119138
【国際公開日】平成22年10月21日(2010.10.21)
【出願人】(500449363)マックス−プランク−ゲゼルシャフト ツール フェルデルンク デル ヴィッセンシャフテン エー.ファウ. (5)
【Fターム(参考)】
【公表日】平成25年2月7日(2013.2.7)
【国際特許分類】
【出願日】平成22年4月19日(2010.4.19)
【国際出願番号】PCT/EP2010/055129
【国際公開番号】WO2010/119138
【国際公開日】平成22年10月21日(2010.10.21)
【出願人】(500449363)マックス−プランク−ゲゼルシャフト ツール フェルデルンク デル ヴィッセンシャフテン エー.ファウ. (5)
【Fターム(参考)】
[ Back to top ]