
電気伝導性ナノ構造体の薄膜を成長させる汎用溶液
導電性ポリマー、ナノ構造体、特にカーボンナノ構造体、およびこれらを組み合わせたものからなるナノ構造体を堆積させる方法について記載されている。この方法は、水性相および有機相からなる非混和性の組合せを含む液体組成物中にナノ構造体を入れるステップを含む。当該混合物は、エマルジョンを形成するのに十分な時間混合され、次に相がそのまま分離するように静置される。その結果、ナノ構造物質が、形成する相の界面に位置し、当該界面に沿って均一に分散される。次に、ナノ構造物質からなる膜が界面を横切る基材上に形成し、前記基材は、相を放置して落ち着かせ、分離させる前に、当該混合物中に予め配置されている。

【発明の詳細な説明】
【技術分野】
【0001】
2010年1月14日出願の、米国特許仮出願第61/295,116号の利益を主張する。
【0002】
本発明は、全米科学財団により授与された認可番号第DMR0507294号の政府支援により実現した。米国政府は、本発明において所定の権利を有する。
【0003】
本出願は、電気伝導性のポリマー、カーボンナノ構造体、およびこれらを組み合わせたものから薄膜を形成する一般的方法に関連する。
【背景技術】
【0004】
導電性ポリマーは、様々な用途で用いられる安価で可撓性の材料として有望であり、これには非限定的に太陽電池、発光ダイオード、および化学抵抗器型検出器(例えば、非特許文献1〜3を参照)が含まれる。均質な薄膜の制御可能な堆積は、電子デバイス工学にとって必須である。数え切れない数の膜形成法が文献で報告されており、そのような方法としてin−situ堆積法(例えば、非特許文献4、5を参照)、溶液中での静電吸着法(例えば、非特許文献6を参照)、ドロップキャスティング法(drop−casting)(例えば、非特許文献7を参照)、電気化学堆積法(例えば、非特許文献8を参照)、スピンコーティング法(例えば、非特許文献1を参照)、グラフト法(例えば、非特許文献9を参照)、およびインクジェット印刷法(例えば、非特許文献10を参照)が挙げられるが、電気伝導性ポリマーもしくは導電性のナノ構造体、またはこれらを組み合わせたものを利用して、基材上に電気伝導性膜を確実に堆積させる簡便で汎用性のある方法について明らかにニーズが認められる。
【先行技術文献】
【非特許文献】
【0005】
【非特許文献1】Bravo-Grimaldo, E., Hachey, S., Cameron, C. G. & Freund, M. S. "Metastable Reaction Mixtures For The In Situ Polymerization Of Conducting Polymers". Macromolecules 40, 7166-7170 (2007)
【非特許文献2】Zhou, Y. et al. "Investigation on Polymer Anode Design For Flexible Polymer Solar Cells". Appl. Phys. Lett. 92, 233308/233301-233308/233303 (2008)
【非特許文献3】Zaumseil, J., Friend, R. H. & Sirringhaus, H. "Spatial Control Of The Recombination Zone In An Ambipolar Light-Emitting Organic Transistor". Nat. Mater. 5, 69-74 (2006)
【非特許文献4】Chiou, N.-R., Lu, C, Guan, J., Lee, L. J. & Epstein, A. J. "Growth And Alignment Of Polyaniline Nanofibres With Superhydrophobic, Superhydrophilic And Other Properties". Nature Nanotech. 2, 354-357 (2007)
【非特許文献5】Zhang, X., Goux, W. J. & Manohar, S. K. "Synthesis Of Polyaniline Nanofibers By "Nanofiber Seeding". J. Am. Chem. Soc. 126, 4502-4503 (2004)
【非特許文献6】Li, D. & Kaner, R. B. "Processable Stabilizer-Free Polyaniline Nanofiber Aqueous Colloids". Chem. Commun. 26, 3286-3288 (2005)
【非特許文献7】Huang, J., Virji, S., Weiller, B. H. & Kaner, R. B. "Nanostructured Polyaniline Sensors". Chem.-A Eur. J. 10, 1314-1319 (2004)
【非特許文献8】Valaski, R., Canestraro, C. D., Micaroni, L., Mello, R. M. Q. & Roman, L. S. "Organic Photovoltaic Devices Based On Polythiophene Films Electrodeposited On FTO Substrates". Sol. Energy Mater. Sol. Cells 91, 684-688 (2007)
【非特許文献9】Sawall, D. D., Villahermosa, R. M., Lipeles, R. A. & Hopkins, A. R. "Interfacial Polymerization of Polyaniline Nanofibers Grafted To Au Surfaces". Chem. Mater. 16, 1606-1608 (2004)
【非特許文献10】Murphy, A. R. & Frechet, J. M. J. "Organic Semiconducting Oligomers For Use In Thin Film Transistors". Chem. Rev. 107, 1066-1096 (2007)
【非特許文献11】Mayya, K. S. & Sastry, M. "A New Technique For The Spontaneous Growth Of Colloidal Nanoparticle Superlattices". Langmuir 15, 1902-1904 (1999)
【非特許文献12】Cheng, H.-L. & Velankar, S. S. "Film Climbing Of Particle- Laden Interfaces". Colloids Surf., A 315, 275-284 (2008)
【非特許文献13】Binks Bernard, P., Clint John, H., Fletcher Paul, D. I., Lees Timothy, J. G. & Taylor, P. "Particle Film Growth Driven By Foam Bubble Coalescence". Chem. Commun. 33, 3531-3533 (2006)
【非特許文献14】Binks, B. P., Clint, J. H., Fletcher, P. D. I., Lees, T. J. G. & Taylor, P. :Growth Of Gold Nanoparticle Films Driven By The Coalescence Of Particle-Stabilized Emulsion Drops". Langmuir 22, 4100-4103 (2006)
【非特許文献15】Jager, E. W. H., Smela, E. & Inganas, O. "Microfabricating Conjugated Polymer Actuators", Science 290, 1540-1546 (2000)
【非特許文献16】Goedel, W. A. "A Simple Theory Of Particle- Assisted Wetting". Europhys. Lett. 62, 607-613 (2003)
【非特許文献17】Pesach, D. & Marmur, A. "Marangoni Effects In The Spreading Of Liquid Mixtures On A Solid". Langmuir 3, 519-524 (1987)
【非特許文献18】Farahi, R. H., Passian, A., Ferrell, T. L. & Thundat, T. "Microfluidic Manipulation Via Marangoni Forces". Appl. Phys. Lett. 85, 4237-4239 (2004)
【非特許文献19】Sarma, T. K. & Chattopadhyay, A. "Visible Spectroscopic Observation Of Controlled Fluid Flow Up Along A Soap Bubble Film From A Pool Of Solution". J. Phys. Chem. B 105, 12503-12507 (2001)
【非特許文献20】Cai, Y. & Zhang Newby, B.-m. "Marangoni Flow-Induced Self- Assembly Of Hexagonal and Stripelike Nanoparticle Patterns". J. Am. Chem. Soc. 130, 6076-6077 (2008)
【非特許文献21】Melle, S., Lask, M. & Fuller, G. G. Pickering Emulsions with controllable stability. Langmuir 21, 2158-2162 (2005)
【非特許文献22】Ata, S. "coalescence Of Bubbles Covered By Particles". Langmuir 24, 6085-6091 (2008)
【非特許文献23】Lucassen, J., Lucassen-Reynders, E. H., Prins, A. & Sams, P. J. "Capillary Engineering For Zero Gravity". Critical wetting on axisymmetric solid surfaces. Langmuir 8, 3093-3098 (1992)
【非特許文献24】Rey, A. D. "Stability Analysis Of Catenoidal Shaped Liquid Crystalline Polymer Networks". Macromolecules 30, 7582-7587 (1997)
【非特許文献25】Chengara, A., Nikolov Alex, D., Wasan Darsh, T., Trokhymchuk, A. & Henderson, D. "Spreading Of Nanofluids Driven By The Structural Disjoining Pressure Gradient". J. Colloid Interface Sci. 280, 192-201 (2004)
【非特許文献26】Bestehorn, M., Pototsky, A. & Thiele, U. "3D Large Scale Marangoni Convection In Liquid Films". Eur. Phys. J. B 33, 457-467 (2003)
【非特許文献27】Pruneanu, S., Veress, E., Marian, I. & Oniciu, L. "Characterization Of Polyaniline By Cyclic Voltammetry And UV-V Is Absorption Spectroscopy". J. Mater. Sci. 34, 2733-2739 (1999)
【非特許文献28】Patil, A. O., Heeger, A. J. & Wudl, F. "Optical Properties Of Conducting Polymers". Chem. Rev. 88, 183-200 (1988)
【非特許文献29】Tran, H. D., Wang, Y., D'Arcy, J. M. & Kaner, R. B. "Toward An Understanding Of The Formation Of Conducting Polymer Nanofibers". ACS Nano 2, 1841-1848 (2008)
【非特許文献30】Sawistowski, H. "Surface Tension-Induced Interfacial Convection And Its Effect On Rates Of Mass Transfer". Chem. -Ing. -Tek. 45, 1093-1098 (1973)
【非特許文献31】Tung, V. C; Allen, M. J.; Yang, Y.; Kaner, R. B. Nat. Nanotechnol. 2009, 4, 25-29
【発明の概要】
【0006】
ナノ構造体、特に導電性のポリマー、カーボンナノ構造体、およびこれらの組合せの膜を堆積させる方法を記載する。ナノスケールで再現性のある厚み制御、および形態的均質性を可能にする簡便でスケール拡張性のある膜製造技術は、工業用途として魅力的な選択肢である。容積、ドープ、およびポリマー濃度が適切な条件の下で、導電性ポリマーナノファイバー、例えばポリアニリンやポリチオフェン、グラフェン、カーボンナノチューブ、またはこれらを組み合わせたものの単層からなる膜は、およそ数秒間のうちに製造可能である。熱力学的に推進される溶液ベースのプロセスは、界面において吸着されたナノファイバーからなる透明薄膜の成長を引き起こす。高品質の透明薄膜は、周囲条件で、事実上任意の基材上に堆積される。無傷の状態の膜を基材から取り出す方法も開示される。この安価なプロセスは、リサイクル可能な溶液を使用し、またポリマーと共に水性相および有機相を含む二相溶液を用いて、広い基材領域を導電性材料でコーティングする新しい技術を提供する。
【図面の簡単な説明】
【0007】
【図1】ポリアニリンナノファイバー膜が大きくなり、且つ広がる機構を説明し、プロセスを概略的に示す図である。
【図2】ポリアニリンナノファイバー膜が大きくなり、且つ広がる機構を説明し、プロセスを概略的に示す図である。
【図3】ポリアニリンナノファイバー膜が大きくなり、且つ広がる機構を説明し、プロセスを概略的に示す図である。
【図4】ポリアニリンナノファイバー膜が大きくなり、且つ広がる機構を説明し、時間系列による界面膜形成を示す図である。
【図5】ガラス製の顕微鏡スライド上に収集して得られた透明薄膜、3例を示す写真であり、左側のスライドは、Cl-をドープしたポリチオフェンナノファイバーからなる導電性ポリマー膜を含み(網掛け部分は赤色を表す)、中央のスライドは、ドープされたポリアニリンナノファイバー膜を示し(網掛け部分は緑色を表す)、および右側のスライドは、脱ドープされたポリアニリンナノファイバー膜を示す(網掛け部分は青色を表す)。
【図6】ガラス基材上に収集したポリアニリンナノファイバーの薄膜を倍率を上げながら示したSEM画像である(目盛りは、2μmである)。
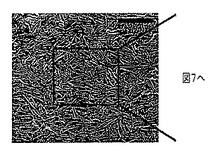
【図7】ガラス基材上に収集したポリアニリンナノファイバーの薄膜を倍率を上げながら示したSEM画像である(目盛りは、1μmである)。図6中の四角で囲繞された領域の拡大図である。
【図8】ガラス基材上に収集したポリアニリンナノファイバーの薄膜を倍率を上げながら示したSEM画像である(目盛りは、500nmである)。図7中の四角で囲繞された領域の拡大図である。
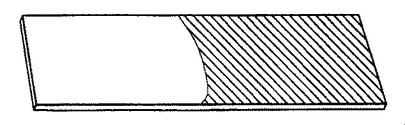
【図9】ポリアニリンナノファイバーの、酸化膜(緑色である図9の網掛け部分)から還元膜(青色である図10の網掛け部分)へのレドックススイッチングを説明するガラス製のスライドの概略図である。
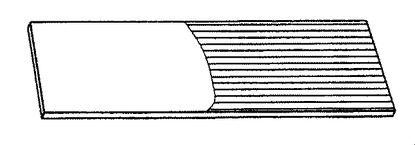
【図10】ポリアニリンナノファイバーの、酸化膜(緑色である図9の網掛け部分)から還元膜(青色である図10の網掛け部分)へのレドックススイッチングを説明するガラス製のスライドの概略図である。
【図11】図9〜10に示す変化を表す図であり、網掛け部分は図9および10の指定領域と同じ領域に対応する。
【図12】紫外可視吸光度分光法(absorbance UV−vis spectroscopy)によりモニターされた、膜形成期間中の厚さ制御を説明する図である。
【図13】紫外可視吸光度分光法によりモニターされた、膜形成期間中の厚さ制御を説明する図である。
【図14】図13にグラフで示した膜の可撓的な性質を表す図である。
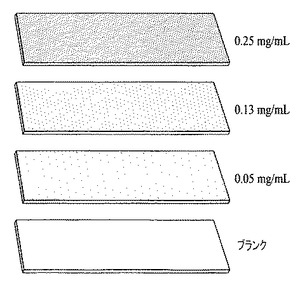
【図15】ヒドラジン中に分散した0.25mg/ml、0.13mg/ml、および0.05mg/mlのグラフェン分散物をガラス製のスライドに塗布して作製された、3つのグラフェン膜を表す図である。基材上に堆積された材料の量と、膜の成長に用いられる分散物中の固形物の濃度との間に直線相関関係が認められ、前記濃度は密度の相違(網点の相違により図中に示す)により表される。
【図16】ヒドラジン中のグラフェン分散物(2mg/ml)、0.5mlを含む、シリコン基材上で収集された高度還元グラファイト酸化物膜およびグラフェン膜からなるシートのSEM画像である。
【図17】グラフェン分散物、0.1mlを含む、シリコン基材上で収集された高度還元グラファイト酸化物膜およびグラフェン膜からなるシートのSEM画像である。
【図18】シリコン基材上で収集された高度還元グラファイト酸化物膜およびグラフェン膜からなるシートのSEM画像である。
【図19】シリコン基材上で収集された高度還元グラファイト酸化物膜およびグラフェン膜からなるシートのSEM画像である。
【図20】シリコン基材上で収集されたグラフェン膜からなる単層シートのSEM画像である。
【図21】シリコン基材上で収集されたグラフェン膜からなる単層シートのSEM画像である。
【図22】シリコン基材上で収集されたグラフェン膜からなる単層シートのSEM画像である。
【図23】シリコン基材上で収集されたグラフェン膜からなる単層シートのSEM画像である。
【図24】ガラス製のスライド上に堆積された単層カーボンナノチューブからなる3つの異なる膜を示す図であり、異なる膜密度を表すために図には網点が施されている。
【図25】シリコン基材上で水性媒体から収集した単層カーボンナノチューブ(SWCNT)からなる膜のSEM画像である。
【図26】シリコン基材上で水性媒体から収集した単層カーボンナノチューブ(SWCNT)からなる膜のSEM画像である。
【図27】シリコン基材上で水性媒体から収集した単層カーボンナノチューブ(SWCNT)からなる膜のSEM画像である。
【図28】シリコン基材上で水性媒体から収集した単層カーボンナノチューブ(SWCNT)からなる膜のSEM画像である。
【図29】シリコン基材上で水性媒体から収集した単層カーボンナノチューブ(SWCNT)膜のさらなるSEM画像である。
【図30】シリコン基材上で水性媒体から収集した単層カーボンナノチューブ(SWCNT)膜のさらなるSEM画像である。
【図31】シリコン基材上で水性媒体から収集した単層カーボンナノチューブ(SWCNT)膜のさらなるSEM画像である。
【図32】シリコン基材上で塩基性水性媒体から収集した単層カーボンナノチューブ(SWCNT)膜のさらなるSEM画像である。
【図33】シリコン基材上で塩基性水性媒体から収集した単層カーボンナノチューブ(SWCNT)膜のさらなるSEM画像である。
【図34】膜が大きくなる前に、超音波処理を延長してこれに曝露した後に生成したSWCNT−グラフェン複合物からなる膜のSEM画像であり、膜はシリコン基材上に形成されている。
【図35】膜が大きくなる前に、超音波処理を延長してこれに曝露した後に生成したSWCNT−グラフェン複合物からなる膜のSEM画像であり、膜はシリコン基材上に形成されている。
【図36】膜が大きくなる前に、超音波処理を延長してこれに曝露した後に生成したSWCNT−グラフェン複合物からなる膜のSEM画像であり、膜はシリコン基材上に形成されている。
【図37】膜が大きくなる前に、超音波処理を延長してこれに曝露した後に生成したSWCNT−グラフェン複合物からなる膜のSEM画像であり、膜はシリコン基材上に形成されている。
【図38】膜が大きくなる前に、時間間隔を変化させて超音波処理を行い、そしてシリコン基材上で収集したSWCNT−グラフェン複合物からなる膜のさらなるSEM画像である。
【図39】膜が大きくなる前に、時間間隔を変化させて超音波処理を行い、そしてシリコン基材上で収集したSWCNT−グラフェン複合物からなる膜のさらなるSEM画像である。
【図40】膜が大きくなる前に、時間間隔を変化させて超音波処理を行い、そしてシリコン基材上で収集したSWCNT−グラフェン複合物からなる膜のさらなるSEM画像である。
【図41】膜が大きくなる前に、時間間隔を変化させて超音波処理を行い、そしてシリコン基材上で収集したSWCNT−グラフェン複合物からなる膜のさらなるSEM画像である。
【図42】本明細書に記載する方法を用いて、シリコン基材上で収集したポリアニリンナノファイバーグラフェン複合物からなる膜のSEM画像である。
【図43】本明細書に記載する方法を用いて、シリコン基材上で収集したポリアニリンナノファイバーグラフェン複合物からなる膜のSEM画像である。
【図44】本明細書に記載する方法を用いて、シリコン基材上で収集したポリアニリンナノファイバーグラフェン複合物からなる膜のSEM画像である。
【図45】本明細書に記載する方法を用いて、シリコン基材上で収集したポリアニリンナノファイバーグラフェン複合物からなる膜のSEM画像である。
【図46】シリコン基材上で収集した、ポリアニリンナノファイバーSWCNT複合物からなる膜のSEM画像である。
【図47】シリコン基材上で収集した、ポリアニリンナノファイバーSWCNT複合物からなる膜のSEM画像である。
【図48】シリコン基材上で収集した、ポリアニリンナノファイバーSWCNT複合物からなる膜のSEM画像である。
【図49】シリコン基材上で収集した、ポリアニリンナノファイバーSWCNT複合物からなる膜のSEM画像である。
【図50】シリコン基材上で収集した、ポリアニリンナノファイバーSWCNT複合物からなる膜のSEM画像である。
【図51】シリコン基材上で収集したポリ(3−ヘキシルチオフェン)ナノファイバーからなる膜のSEM画像である(目盛りは、10μmである)。
【図52】シリコン基材上で収集したポリ(3−ヘキシルチオフェン)ナノファイバーからなる膜のSEM画像である(目盛りは、3μmである)。図51中の四角で囲繞された領域の拡大図である。
【図53】シリコン基材上で収集したポリ(3−ヘキシルチオフェン)ナノファイバーからなる膜のSEM画像である(目盛りは、1μmである)。図52中の四角で囲繞された領域の拡大図である。
【図54】基材上に膜を形成するために、本発明の特徴を組み込む手順の概略図である。
【図55】基材上に膜を形成するために、本発明の特徴を組み込む手順の概略図である。
【図56】基材上に膜を形成するために、本発明の特徴を組み込む手順の概略図である。
【図57】基材上に膜を形成するために、本発明の特徴を組み込む手順の概略図である。
【図58】基材上に膜を形成するために、本発明の特徴を組み込む手順の概略図である。
【図59】基材上に膜を形成するために、本発明の特徴を組み込む手順の概略図である。
【図60】図54〜59に示すプロセスを用いて形成された膜のラマンスペクトル表すグラフである。
【図61】は、膜の抵抗および透過率を、エマルジョン撹拌時間の関数として示すグラフである。
【図62】自動化された膜形成プロセスの概略図である。
【図63】自動化された膜形成プロセスの概略図である。
【図64】自動化された膜形成プロセスの概略図である。
【図65】膜の抵抗を膜層数の関数として示すグラフである。
【図66】膜の透過率を膜層数の関数として示すグラフである。
【図67】ガラス製のスライドで収集した、グラファイト酸化物シートからなる膜のSEM画像である(目盛りは、50μmである)。
【図68】ガラス製のスライドで収集した、グラファイト酸化物シートからなる膜のSEM画像である(目盛りは、30μmである)。図67中の四角で囲繞された領域の拡大図である。
【図69】ガラス製のスライドで収集した、グラファイト酸化物シートからなる膜のSEM画像である(目盛りは、10μmである)。図68中の四角で囲繞された領域の拡大図である。
【発明を実施するための形態】
【0008】
様々なナノ材料、特にポリアニリンおよびポリチオフェンナノファイバー、ならびにカーボンナノ構造体、例えばグラフェンシートおよびカーボンナノチューブ等からなる透明な薄膜を、事実上任意の基材上に、周囲条件下で成長させる溶液ベースの方法について説明する。2つ非混和性の液体およびポリマーナノファイバーを乳化することにより、界面張力勾配、粘性流、および膜の広がりが引き起こされる。表面張力の差異が、無機ナノ粒子膜を形成するためにこれまで用いられてきた(例えば、非特許文献11〜14を参照)。膜は、電気伝導性有機ポリマーを含み、またナノファイバーの単層により特徴付けられるナノスケール秩序を有する。導電性ポリマーを対象とするこの新しい膜成長技術は、容易にスケールアップ可能であり、また溶液はリサイクルすることができる。膜を形成する本方法は、形態的均質性、厚さ制御の再現性、および簡便性を有し、これらは、上記導電性ポリマーの電気特性を利用するデバイスの製造について独自の可能性をもたらす。
【0009】
一方、クロロホルムを用いた液体抽出により一次元ポリアニリンナノファイバーの水性分散物を精製中に、分液漏斗の壁にポリマーの透明な膜が形成されることを、出願者は発見した。この溶媒混合物に振動を加えるとこの膜は除去されたが、放置すると膜は迅速に再編成する。この発見に基づき、アクチュエーターおよびセンサーを非限定的に含む様々な用途で用いられる膜を調製するために、ナノ構造を有する導電性ポリマーからなる膜を成長させる溶液ベースの方法を開発したが、上記用途はこれまでに、文献(非特許文献15を参照)で開示されている。
【0010】
水および高密度油、例えばクロロベンゼン等を激しく撹拌すると、油相中に分散された水滴が形成される。液滴の水/油界面は、界面活性剤等の表面活性種および固体粒子にとって吸着部位として機能する。界面に存在する表面張力は、吸着した種の濃度に比例して減少し、また吸収された種の濃度が不均等に分布すると、界面張力勾配が発生する。次に、この勾配は流体膜が固体表面全体にわたり広がる原因となり、これはマランゴニ(Marangoni)効果として公知である。この種の指向性のある流体の流れは、生物の自己防衛機構に認められ(例えば、非特許文献16を参照)、また潤滑用途で利用可能であり(例えば、非特許文献17を参照)、マイクロフルイディクス(例えば、非特許文献18を参照)、ラブ−オン−チップ設計(例えば、非特許文献19を参照)、そして高密度データストレージで利用できる可能性がある(例えば、非特許文献20を参照)。
【0011】
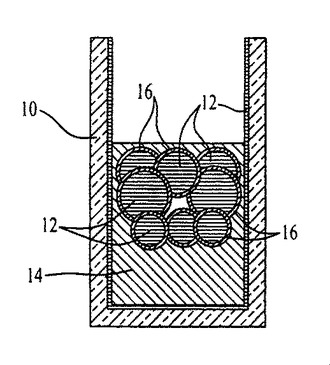
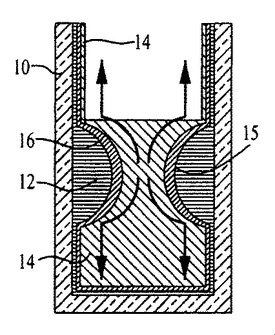
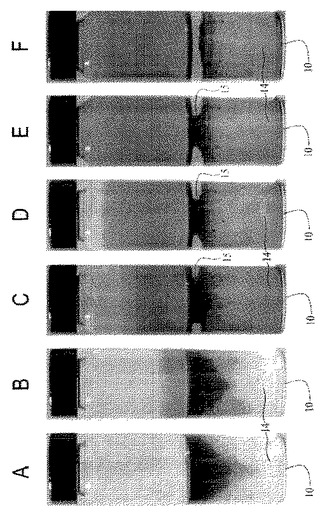
事実上任意の基材上で成長させたポリマーナノファイバー、またはその他のナノ構造体からなる極めて透明度が高く、均質な薄膜を形成するために、出願者らは、図1〜3、54〜59、および62〜64で概略的に、ならびに図4では写真を用いて示す方法を開発した。ナノファイバーまたはナノ構造体は、水および高密度油と激しく混合され、次に生成する界面をコーティング対象表面に曝露する。この乳化プロセスは、部分的に膜成長に関与する。撹拌により、容器の親水性の壁が水でコーティングされ、そして水性の液滴が油相中に分散されるようになる。図1〜3および図4の文字が施された様々な部分を参照すると、例えば水12、高密度油14、およびポリマーナノファイバー16がガラス容器10内で混合され、そして激しくかき混ぜられてエマルジョンを形成する(図1、図4A、B)。混合を止めると、油14中に分散しポリマーナノファイバー16で覆われた水滴12(図1)は、油相の上部に上昇する(図2、図4C)。液滴が合体すると、界面において吸着したナノファイバーの濃度勾配、水により形成されたカテノイド15、および指向性の流体が生み出され、その結果、単層のナノファイバー16が容器の壁を上下方向に広がる(図3、図4D、E)。このカテノイドは、水12が上部で油14が底部の2つの異なるバルク液相に分断する(図3、図4F)。ナノファイバー16は、空気に隣接した水/油の界面、およびバルク油相を覆う別の界面に堆積する。バルク液相間に生じ、膜成長が停止した後に残留するポリマーリザーバーは、追加の基材をコーティングするのに利用可能な過剰のナノファイバーを含有する。図4に示す膜成長の流れを参照すると、時間は(A)0秒、(B)0.5秒、(C)1秒、(D)10秒、(E)30秒、(F)35秒である。
【0012】
ナノファイバー等の固体粒子は、非混和性の液の間の界面張力を低下させることにより、ピッカリングエマルジョンと呼ばれる安定剤として機能することができる(例えば、非特許文献21を参照)。混合により、ポリマーナノファイバーを両方の液体と溶媒和させるのに必要とされる機械的エネルギーが提供され、こうして本質的に不可逆な吸着プロセスによってナノファイバーが水/油界面でトラップされる。理論的な試験により、吸着した粒子を任意の界面から除去するのに必要とされるエネルギーは、界面においてこれらの粒子を分離するのに必要とされるエネルギーをかなり上回ることが明らかにされた(例えば、非特許文献22を参照)。したがって、乳化されたナノファイバーは、引っ張り力を受け、そして同ナノファイバーは界面において広がる。撹拌が中止されると、機械エネルギーのインプットが弱まり、水滴が油層の上部に上昇し、合体することが可能となる。油およびナノファイバーを液滴から排除する合体期間中に界面の総表面積は低下し、不可逆的に吸着されたナノファイバーに自然発生的な濃度勾配が生成し、その結果水/油界面にマランゴニ圧を生み出す。界面張力がより高い領域に排除されたナノファイバーを引き寄せる界面張力の勾配が生じるが、一方ナノファイバーの膜が、水油間で搾り出された単層として容器の壁を上下に広がる(図2、図4C,D,E)。水/油界面が存在しないので、バルク水性相を取り巻くガラス壁上では膜は成長しないことに留意されたい(図3、図4F)。
【0013】
膜成長期間中、水層は、ナノファイバーの大部分を含有する内部油チャンネルを備えるカテノイドの形成に寄与する。水は、この形状を採ることによりその表面自由エネルギーを最低限に抑える(例えば、非特許文献23を参照)。カテノイド内部の粘性流は、チャンネルの最も薄いセクションから最も厚いセクションに向かって上下両方向に流体運動を生み出す(例えば、非特許文献24を参照)。次に、合体により内部チャンネルが狭くなり(図4C〜E)、そして最終的にカテノイドによる分断が生じ、粘性流が終結する。2つの異なるバルク相が確立され、ナノファイバーの再分布を引き起こす(図3、図4F)。ナノファイバーを含有する水/油界面が、空気に隣接した側およびバルク水層下方の両方に認められる。界面上部には、ナノファイバーの濃度勾配が存在し、この濃度勾配は、カテノイド分断後の数秒間、膜成長を上方に継続して推進する。この濃縮物は、ガラス製のスライドが溶液から引き出される際に、これをコーティングするのに利用される。底部側の界面は、ナノファイバーからなるポリマーリザーバーを備え、さらなる膜の成長に用いられる。
【0014】
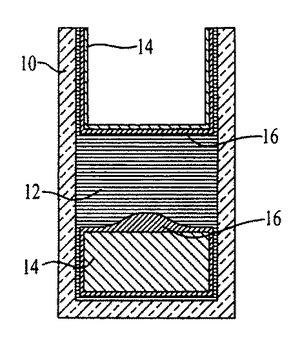
図54〜59および62〜64中のプロセスフロー図は、親水性の基材表面20、44上における膜の成長および堆積機構を表す。SiO2(図54)等の基材は、この図では金属製の電極22を有するが、ピラニア溶液中で煮沸され、酸素プラズマ中でエッチングされ、そして均質な水層24を誘発するために水に浸漬される(図55)。濡れた基材が、ナノ材料(図56)を含有するピッカリングエマルジョン26と接触すると、液滴26が合体する結果膜成長が生じる(図57〜59)。ナノ材料の透明なコーティング28は、数秒のうちに広がるが、これはコーティングされた表面全域にわたり導電的に連続している。
【0015】
膜成長の最適実験条件を調べるために、水および高密度ハロゲン化溶媒からなる異なる二元混合物を用いて、ポリアニリンナノファイバー膜を、ガラス製のスライド上で成長させた。最大到達可能な広がる長さが、各膜を成長させるのに用いられた非混和性の二元混合物の界面張力と比較された。この結果から、界面張力が大きいほど、上方に広がる膜の這い上がる高さはより大きくなることが示唆された。界面張力が大きい方が、より小さなものよりも強い力でナノファイバーを引き上げ、そして膜が重力に抗してより長時間基材上を這い上がるのを可能にし、その結果、広がる高さがより大きくなる。1つの比較では、水および四塩化炭素(界面張力は45dynes/cm)を用い、その後に水およびクロロホルム(32.8dynes/cm)、そして最終的に水および塩化メチレン(28.3dynes/cm)が続いたときに、ナノファイバー膜は最も高く這い上がった。膜の成長は、系の総界面自由エネルギーを最小化することにより推進される(例えば、非特許文献25を参照)。
【0016】
容器および基材の両方の寸法および材料について、それらの特性が膜の成長にどのように影響を及ぼすか調べるために試験された。容器の直径が大きいほど(例えば、直径約2.0〜約10.0インチ)、2液体間により広い界面表面積を提供し、したがって多数のバブル形成と高度に活発な合体、複数のカテノイド、および膜の高速成長を引き起こすことが判明した。一方、上方に這い上がる膜の被覆面積は、小さな直径を有する容器(例えば、直径約0.5〜約2.0インチ)よりも大きな直径の容器の方が小さいので、これを用いる迅速な膜製造は便利であり得る。疎水表面も、例えばアルゴン−酸素プラズマを用いて最初に表面を活性化させることにより膜成長基材として利用可能である。
【0017】
導電性ポリマーナノファイバーからなる透明な薄膜は、様々な色で製造可能である。図5(網掛け部分は色を示す)は、ガラス製のスライド上のポリアニリンおよびポリチオフェン膜を示した。左から右に向かって、塩化物をドープした赤色のポリチオフェン、過塩素酸をドープした緑色のポリアニリン、および脱ドープした青色のポリアニリンの膜である。膜、特に過塩素酸をドープしたポリアニリン膜は、優れた光透過性を有し、光透過率は60%を上回る。p−トルエンスルホン酸(p−TSA)をドープしたナノファイバーおよびクロロホルムからなる水性分散物を用いて、ポリアニリン膜を成長させた。次に、膜を脱ドープまたはさらにドープするために、塩基または酸の蒸気のいずれかに膜を曝露すると、当該膜はそれぞれ青色または緑色となった。
【0018】
界面において吸着したナノファイバーの表面自由エネルギーと基材との間の分子相互作用は、膜の形態を規定し得る(例えば、非特許文献26を参照)。過塩素酸をドープしたポリアニリンは、図6〜8に示すように、単層ナノファイバーの平均厚さを有する膜を形成する。この一連のSEM画像(52°の角度で傾いている)は、クロロホルムを用いて成長させて、HClO4に部分的をドープしたポリアニリンナノファイバー膜に特徴的である。ナノスケールの形態は、倍率を高めながら示すように、単層のナノファイバーから実質的に構成され、図6〜8の目盛りは、それぞれ(a)2μm、(b)1μm、(c)500nmを表す。図8は図7中の四角で囲繞された領域の拡大図であり、また図7は、図6中の四角で囲繞された領域の拡大図である。ナノファイバーは、油層および水層間にサンドイッチされたときに、界面において押し出されるので、この現象が生ずる。膜をゆっくり乾燥させると、次に毛管力が秩序を誘発し得る。この現象を図8に示すが、この場合部分的をドープしたナノファイバーは、互いに側面同士を接触させて配向している。個々の単層膜は、p−トルエンスルホン酸またはカンファースルホン酸等のドーパントを用いても、形成可能である。
【0019】
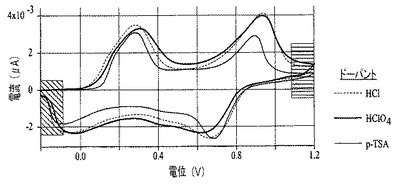
ポリアニリンナノファイバー膜の電気化学的挙動は、図11に示す通りサイクリックボルタンメトリー(CV)を用いて特徴づけられた。塩酸、過塩素酸、およびp−トルエンスルホン酸をドープしたポリアニリン膜は、すべて0.25Vと0.95Vに2つの還元ピークを示し、またこれらの対応する酸化ピークは−0.15Vと0.68Vに認められる。これらのサイクリックボルタモグラムは、ポリアニリンのエメラルジン酸化状態を示唆する(例えば、非特許文献27を参照)。エメラルジンポリアニリンの緑色のドープ形態(図9の網掛け部分)から青色のドープ形態へのスイッチングは、図10で、右手部分を異なる網掛けをして示した。これらのナノファイバー膜は、透明、堅牢で、また複数サイクルのCVを扱う能力を有する。電解質に浸漬された電極部分(図9および10ではスライドの右側部分)に限り、電位の方向が切り替ると変色する。ITO上で成長した膜の透明度は、膜を通して画像がどのくらい明瞭に見えるかにより実証可能である。エメラルジン型のポリアニリンナノファイバー膜は、図9に示す通り、電解質に途中まで浸漬され、そして電気化学的に酸化されて、ドープされた(緑色に着色)塩の状態となる。グラフ(図11)は、塩酸(HCl)、過塩素酸(HClO4)、およびp−トルエンスルホン酸(p−TSA)でドープしたポリアニリンナノファイバーのCV曲線を示す。エレクトロクロミック転移を図10に概略的に表す;図9のポリアニリンナノファイバー膜は還元され、そして溶液に浸漬された図9の透明で緑色の電極部分は青色に変化する。
【0020】
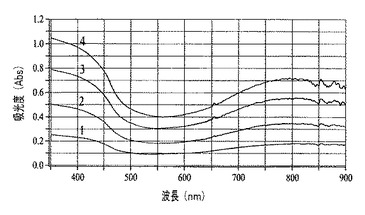
マランゴニ流により生成した膜厚は、ドープしたポリアニリンナノファイバー膜の層を連続して堆積させることにより制御可能である(図12)。図12では、ガラス上で成長させてp−TSAでドープしたポリアニリンナノファイバー膜からなる、連続して得られた4つの層それぞれが、吸収が段階的に増加(約0.2単位)することにより観察可能である。紫外可視スペクトル(図13)から、新しい層はいずれも、約0.2吸光度単位の光学濃度を生成することが明らかである。膜の各層は、スペクトルを収集する前に、周囲条件で30分間乾燥放置された。異なる高さでサンプリングされた膜について得られたポリチオフェンの紫外可視吸収(図13)は、予想された吸収ピークを示す(例えば、非特許文献28を参照)。堆積されるポリマーの質量は、膜の高さに反比例して変化するので、膜厚は、膜が成長する角度によって制御可能である。したがって、膜が基材を這い上がるにつれて光学濃度は低下する。これは、濃度勾配により膜厚を制御する能力を実証する。
【0021】
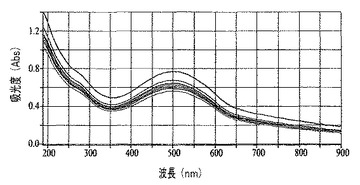
図13は、60°の角度で成長させたポリチオフェンナノファイバー膜の高さ方向の異なる位置で収集された一連のスペクトルを表し、膜が成長する角度により光学濃度は制御可能であることを実証する。図14から、ITO−ポリエチレンテレフタレートからなるプラスチック基材上を60°の角度で成長させたポリチオフェンナノファイバー膜は、軽い圧力を加えることにより実証されるように可撓性であることが明らかである(挿入物の右側および左側の端部のより暗い部分は、膜を曲げている者の手袋で覆われた指の先端である)。
【0022】
下記のいくつかの例では、導電性膜の形成手順について説明し、前記手順および得られた生成物は本発明の特徴を取り込んでいる。本明細書では、「超音波処理」と呼ぶとき、それは、容器内に収納された基材または混合物を、水で満たされた超音波バスに配置し、そして60Hzで稼動させること、あるいは混合物を収納する容器内に超音波ホーンを配置することに関係した。
【0023】
基材表面の処理:
ガラス。事前洗浄された75mm×25mm×1mmの顕微鏡用ガラス製スライド(Corning2947)を基材として用いた。膜を収集する前に、これをイソプロピルアルコールで洗浄し、そして圧縮空気で乾燥させた。a)水の中で超音波処理を30分間、b)硝酸および水の中で交互に煮沸処理、またはc)酸素プラズマ処理を5分間行う追加の表面処理を実施した。
【0024】
石英。75mm×25mm×1mmの基材(QSI Quartz Scientific)を、ガラスについて上記した方法を用いて、またはクロム酸およびDI水の中で連続して煮沸し、その後オーブン乾燥(400℃、1時間)することにより処理した。
【0025】
シリコン。Si基材をイソプロピルアルコールの中で超音波処理し(30分)、次にワイプ(Kimtech)で軽く擦り、その後酸素プラズマ処理を5分間行った。
【0026】
ITO−ガラス。Nanocs Inc.より入手したガラス製の顕微鏡スライド上にコーティングされたインジウムすず酸化物(ITO)を、イソプロピルアルコールを含有するワイプで軽く摩擦することにより洗浄し、その後水の中で30分間超音波処理を行い、および/または酸素プラズマ処理を5分間行った。
【0027】
ITO−ポリエチレンテレフタレート。PET基材(CPFilms Inc.)を、60mlのポリプロピレン製チューブの内側にぴったりと納まる大きさにした。膜成長させる前に、当該基材の表面を、酸素プラズマを用いて、3分間処理した。
【0028】
上記基材表面処理は例示であり、表面処理される材料の範囲を限定する意図はない。本明細書の教示に関係する当業者は、本明細書に記載する方法で使用するために、その他の表面処理法または好適に処理されるその他の基材材料に置き換えることができる。
【実施例】
【0029】
ナノファイバー
(実施例1)
ポリチオフェンナノファイバーの合成。ポリチオフェンナノファイバーを作製するプロセスは、文献(例えば、非特許文献29を参照)で報告されている。この手順には、2つの溶液、すなわち1)アセトニトリル、10mlに溶解したFeCl3(0.333g、2.1×10-3モル)および2)1,2−ジクロロベンゼン、10mlに溶解したチオフェン(0.133ml、1.74×10-3モル)およびテルチオフェン(0.0065g、2.61×10-5モル)を調製するステップが含まれる。これらの2溶液を一緒にし、10秒間混合し、そして7日間静置した。次に、この反応溶液を遠心分離法を用いて精製した。
【0030】
(実施例2)
ポリチオフェンナノファイバー膜の成長。実施例1から得られたポリチオフェン導電性ポリマーナノファイバーを、より少ない水性相(約0.2mlから約5.0ml、好ましくは約1.5ml)およびより多くの有機層(約5.0mlから約30.0ml、好ましくは約18ml)から構成され、水/有機比が約1/10〜1/20、好ましくは約1/12となる二元非混和性溶液を用いて界面膜に形成した。この非対称の容積分配はマランゴニ流を引き起こす。例として、75mm×25mm×1mmのガラス製スライドを、下記のようにポリチオフェンナノファイバーでコーティングした:スライドを、60mlのポリプロピレン製チューブ(BD Falcon(商標)コニカルチューブ)内に配置し、アセトニトリルに分散したナノファイバー分散物(2g/L)を1ml、DI水を0.6ml、およびクロロベンゼンを10ml、当該チューブに添加した。激しく揺動した後、ポリプロピレン製容器を水平に寝かせ(長い壁を床と平行にする)、次に容器の長い方の端部が床と平行になりスライドが直立するまで回転させた。このようなスライド方向を規定するように容器を回転させれば、広がるポリマー膜にとって這い上がる距離がより短くなって基材全体を覆うことができ、したがって、縦横比が大きい基材でも完全に被覆可能である。膜成長期間中に容器を定期的にタッピングすることにより、バブル合体速度が高まり、膜成長が促進される。膜形成後、スライドを取り出し、当該膜を有機蒸気雰囲気内でゆっくりと乾燥する。
【0031】
(実施例3)
ポリアニリンナノファイバーの合成。ドーパントとして下記の酸を用いて、ポリアニリンナノファイバーを調製した:(a)塩酸、(b)p−トルエンスルホン酸、(c)カンファースルホン酸、および(d)過塩素酸。代表的な反応として、アニリン(0.16ml、1.75×10-3モル)をペルオキシ二硫酸アンモニウム(0.1002g、4.39×10-4モル)に溶解するステップ、および1MのHCl、8mlを添加するステップが含まれた(溶液A)。ダイマー開始剤、N−フェニル−1,4−フェニルジアミン(0.0032g、1.74×10-5モル)を、MeOH、1mlに溶解し、そして5分間超音波処理した(溶液C)。次に、溶液Aおよび溶液Cを混合したが、追加の1MのHCl、8mlと一緒にして溶液Bとする前に、平衡状態で5分間放置した。次に、容器を5秒間揺動させた。オーバーナイトで静置して重合を進めた。精製は、最終生成物をDI水に対して透析することにより実施され、部分的に脱ドープした材料が得られた。
【0032】
(実施例4)
ポリアニリンナノファイバー膜の成長。高密度ポリエチレン容器(60mlのNalgene(商標)広口型)を用いて、実施例3から得られた部分的にドープされたポリアニリンナノファイバーからなる水性コロイド分散物(4g/L)、1mlを、DI水、4mlと混合した。水性分散物を30秒間混合し、次にクロロベンゼン(またはクロロホルム)、6mlを添加し、そして容器を激しく揺動した。基材、例えば洗浄した顕微鏡用ガラス製スライド(Corning2947)を容器内に配置し、そして10秒間揺動した。容器が静止状態になり次第、ポリマー膜の成長が開始した。バブルを破壊し、膜成長を促進するために、容器の壁を定期的にタッピングした。様々な試験膜を基材上で成長させた。ポリアニリンナノファイバーからなる両面透明膜を分析用に選択した。膜の肉眼的な均質性およびナノスケール形態を保つために、周囲条件下でゆっくりと膜を乾燥させる必要があった。膜と基材との接着力は乾燥プロセス期間中に高まった;55℃で48時間さらに加熱すると、安定な膜、例えばサイクリックボルタンメトリー(図11)による特徴付けに供するのに十分堅牢な膜が得られる。一方、新規に形成された濡れた状態の膜は、水により基材から移すことができる。膜は、ドープした、または部分的に脱ドープしたナノファイバーのいずれか一方から作製可能である。入念にドープされたナノファイバーが、ポリマー溶液中で用いられる場合には、揺動すると安定なバブルが生成し、合体または膜成長が生じない。親水性であることから、ドープされたポリマーは、脱ドープされたポリマーよりも速くガラス壁を這い上がる。
【0033】
(実施例5)
ポリアニリンナノファイバー膜−分析。サイクリックボルタンメトリー。ΙΤO−ガラス基材上で成長したポリアニリンナノファイバー膜について、サイクリックボルタンメトリー(CV)を実施した。実施例4に記載されている方法を用いて、単層のナノファイバーを堆積させた。電気化学測定用としてITO上に膜を調製するプロトコールには、25℃で12時間、その後55℃で48時間、膜を乾燥ステップが含まれた。Princeton Applied Research社製のポテンシオスタット263Aを用いて、−0.2Vから+1.2Vまで、次に元の−0.2Vまでサイクルして、データを収集した。用いたスキャン速度は、50mV/秒であった。1MのHCl電解質溶液をアルゴンガスで30秒間パージし、そして電位を印加する前に20秒間平衡状態で放置した。補助電極として洗浄したPtワイヤーを用い、塩化カリウムで飽和したカロメル電極が参照電極として機能し、そして単層のポリアニリンナノファイバーで被覆された25mm×75mm×1mmのITOコーティングガラス製スライドが作用電極を構成した。ポテンシオスタットのリード線と接続するために、導電性の銅テープ(3M(登録商標))を、作用電極の末端に配置した。
【0034】
走査型電子顕微鏡。基材上で収集した様々な膜のナノスケール形態を、SEM(FEI Nova600)を用いて画像化した;しかるべき導電性を確保するために、最初にサンプルをプラチナ層でプラズマスパッタリングした。サンプルおよび装置の間で電気的回路を閉じるために、導電性の銅テープを用いた。
【0035】
紫外可視分光法。紫外可視における特徴付けを行うために、ポリアニリンナノファイバー単層をガラスおよび石英スライド上で成長させた。装置内で各スライドの位置が一定となるように保証する設計がなされたホルダーに基材を入れて、紫外可視分光光度計(Hewlett−Packard HP8453 Diode−Array)内に導入した。
【0036】
上記方法は、導電性ポリマーナノファイバーからなる透明な薄膜を成長させるための簡便かつ安価な溶液を提供する。低めの表面張力を有する流体(油)は、常に、表面張力がより高い流体(水)上に広がることが公知であるが(例えば、非特許文献30を参照)、出願者らは、油膜は、ガラス表面上に存在する水性の層全体にわたり、溶媒和した有機ナノ構造体を有効に担持し得ることを今回実証した。膜は、周囲条件において数秒内で堆積し、数分内に乾燥し、そして溶媒はリサイクル可能である。上記手順を用いれば、広い基材領域を均質かつ再現性のあるやり方でコーティング可能であり、薄膜の品質も高い。
【0037】
この方法の有用性は、電気伝導性有機ポリマーに限定されることはなく、その他のナノ材料またはナノ材料を組み合わせたものからなる膜を形成するのにも利用可能である。
【0038】
グラファイト酸化物膜およびグラフェン膜
カーボンナノ構造体からなる二次元(2D)シートは、ピッカリングエマルジョンにおいて安定剤として機能し、液/液界面で界面活性剤様の吸着特性および化学特性を有する。2D液/液界面は、平らなシートと幾何学的に類似し、したがって理想的な調和環境である。2Dカーボンシートで長さスケールが極端に異なると、縦横比が大きくなり、界面で熱力学的に有利な吸着を引き起こす。グラファイト酸化物は、水および油間の界面で分子状およびコロイド状の両方の界面活性剤として働く、単原子厚の(single−atomic−thick)両親媒性物質であり、界面張力を低減する。エマルジョンを構成する液滴が合体すると、その後生じた指向性の流体は、グラファイト酸化物シートが広い領域上の界面で広がるのを促進する。改変されたHummerの方法(例えば、非特許文献31を参照)により生成されたグラファイト酸化物シートは、クロロベンゼンを併用してMilli−Q水中に分散され、そして均質な薄膜(図67〜69)に処理される。一般的に、グラファイト酸化物シートからなる0.2mg/mLの水性分散物が、クロロベンゼンを用いて1:4の比で30秒間超音波処理することにより乳化される。容器を手動により撹拌し、静置した後、数秒以内に、透明な淡黄色に着色した膜が、ガラス製のスライドをコーティングする。グラファイト酸化物膜の堆積は、膜の成長がpH依存性であるため、中性に近いpHで実施される。末端の−COOH基の脱プロトン化が生じてグラファイト酸化物がより親水性になり、またエマルジョンの合体が生じてグラファイト酸化物が水性相に排除されるので、広がることは高pHでは生じない。
【0039】
グラフェンは、ハニカム結晶格子内に稠密充填されたsp2結合型の炭素原子からなる、単原子厚の平面状シートである。グラフェンシートは、超音波処理の支援を受けてヒドラジン中に分散される。少なくとも20分間、好ましくは約2時間超音波処理すると、シートサイズは減少する可能性がある。超音波処理時間が長くなるほど、シート寸法の減少は大きくなる。高度還元グラファイト酸化物およびグラフェンの透明薄膜を生成するために、ヒドラジン分散物を、水酸化アンモニウム水溶液と混合した。部分的な酸化が生ずるが、グラフェンシートの一部は溶液中に留まる。次に、グラフェンを含有する希薄なヒドラジン分散物が用いられる場合には、単層のグラフェンシートを含有する基材上の透明薄膜が、上記プロセスを用いて得られた。基材上に堆積される材料の量を、膜を成長させるのに用いられる水性分散物中に存在するカーボン材料の濃度を変化させることにより制御した。顕微鏡用のガラス製スライドを基材として用い、そして異なる濃度の分散物からなる透明薄膜を作製した(図16〜23)。シート抵抗が23kΩの、高度還元グラファイト酸化物およびグラフェンからなる膜を、0.25mg/mlの水性分散物を用いて成長させた。
【0040】
(実施例6)
60mlの高密度ポリエチレン容器を用いた。一般的なプロセスとして、グラフェン(1〜10mg/ml)を含有するヒドラジン分散物を、数分から数時間、超音波処理した。一般的に、グラフェン分散物(1mg/ml)、0.4mlを、14wt%の水酸化アンモニウム水溶液、4〜5ml中で超音波処理した。次に、クロロベンゼン等の有機溶媒、8〜12mlを超音波処理したグラフェンに添加し、当該溶液をさらに超音波処理した。次に、ポリアニリンナノファイバー膜を作製するために上記と同じ揺動および静置プロセスにより膜を作製した。
【0041】
20mlシンチレーションバイアル、およびヒドラジン溶液(2〜3ml)中で、1〜5mg/ml(好ましくは1.0mg/ml)のグラフェン/mlを含有するグラフェン分散物、0.1〜0.5ml(好ましくは0.4ml)、14wt%の水酸化アンモニウム水溶液、および有機溶媒(クロロベンゼン、クロロホルム、四塩化炭素、トルエン、またはベンゼン)、2〜4ml(好ましくは4ml)を含む混合物を用いて、膜をシリコン基材上に堆積させた。超音波処理は、ナノ構造体の分散および均質な膜の生成、ならびにシートのより小さなサイズへの破壊において、これらを助ける。基材上に膜を堆積させる前に、基材を酸素プラズマで5分間処理した。図16は、ヒドラジン中に分散したグラフェン分散物(2mg/ml)、0.5mlから形成された膜を示し、図17は、グラフェン分散物、0.1mlから形成された膜を示す。
【0042】
シート間の結合性により、導電性のネットワークが実現する。この材料からなる透明な膜は、上記プロセスを用いて、石英およびガラス製のスライド上で得られる。高度還元グラファイト酸化物およびグラフェンシートを、超音波処理により塩基性の水性媒体中に分散した。グラフェンからなるより大きなシートを、超音波処理曝露時間を約0.5分まで短縮することにより作製した。次に、グラフェンシートのヒドラジン分散物を14wt%NH4OH水溶液と混合することにより、膜を収集した。ピッカリングエマルジョンを形成するために、有機相としてクロロベンゼンを用いた。水酸化アンモニウム溶液の代わりに脱イオン水を用いる場合には、基材領域の被覆率は、膜が這い上がる最大高さと共に低下する。図18は、シートが端部を共有している膜の平面図である。図19は、52°傾いた同一膜のSEMによる画像である。
【0043】
図20〜23を参照すると、グラフェンからなる単層シートを含有する膜を、希薄濃度のグラフェンを用いてヒドラジン(1mg/ml)中で堆積させた。最初にグラフェンをヒドラジン中で完全に還元し、その後14wt%の水酸化アンモニウム水溶液と一緒にした。クロロベンゼンと共にグラフェンを含有する塩基性のNH4OH水性分散物を混合することにより膜を得、バイアルを激しく揺動させ、次に静置して膜成長を開始した。他の溶媒、例えば四塩化炭素、クロロホルム、トルエン、およびベンゼン等は、クロロベンゼンの代用とすることができる。図20〜23の各図中に示す長方形は、単層グラフェンシートを表す。
【0044】
カーボンナノチューブ
単層カーボンナノチューブ(SWCNT)(Carbon Solutions Inc.)をカルボン酸およびヒドロキシル基により官能化した。
【0045】
(実施例7)
ガラス製スライド(75mm×25mm×1mm)上でのSWCNT膜の成長
単層カーボンナノチューブ(SWCNT(Carbon Solutions Inc.)、0.0011g)を、20mlのガラス製シンチレーションバイアル内で水、4mlと共に混合し、そして15分間超音波処理を行った。次に、クロロベンゼン、11mlを添加し、その後さらに15分間超音波処理を行った。濃HCl、3滴を添加、混合し、そして当該溶液を60mlのプロピレン製容器(BD Falcon(商標)チューブ)に移した。ガラス製スライド(Corning 29470)を、イソプロピルアルコールに浸漬したKimtech(登録商標)ワイプで洗浄し、圧縮空気で乾燥し、そして容器内に配置した。撹拌および静置を反復して実施した。約5分後に最高品質の膜を得た。
【0046】
(実施例8)
シリコン(49mm×10mm×1mm)上でのSWCNT膜の成長
SWCNT、0.1mgを、20mlのガラス製シンチレーションバイアル内で、脱イオン水、2mlと混合し、そして15分間超音波処理を行った。次に、クロロベンゼン、5mlを添加し、その後さらに15分間超音波処理を行った。次に、濃HCl、3滴を添加し、当該混合物を揺動させた。当該溶液は、約5分内に高品質の膜を生成した。酸を使用すると凝集を引き起こすのが認められた。
【0047】
(実施例9)
ガラス製スライド上のSWCNT片面膜
膜を、Falconチューブ、水、およびクロロベンゼンを用いて収集した。各成分を添加して混合物となし、その後15分間超音波処理を行った。図24は、ガラス製スライド上のカーボンナノファイバーからなる膜を概略的に表し、異なる膜密度を表すために異なる網点が施されており、
下方のスライドはコーティングされていないスライドブランクであり、
次は、SWCNTを0.0058g、水を6ml、および有機性の油を15ml用いて形成された膜で被覆されたガラス製スライドであり、
3番目の画像は、SWCNTを0.0027g、水を4ml、および有機性の油を11ml用いて形成された膜で被覆されたガラス製スライドであるが、当該混合物の撹拌後、基材上に膜を形成する前に当該混合物には、濃縮された酸が5滴添加されており、
および上方の画像は、SWCNTを0.0013g、10%エタノール水溶液を3ml、有機性の油を9ml、および濃HClを4滴用いて、ガラス製スライド上に形成された膜を表し、有機性の油はいずれの場合もクロロベンゼンであった。他のアルコールもエタノールの代用とし得る。
【0048】
【表1】
【0049】
基材に堆積される固形物の質量は、膜の透明度と反比例の関係を有する。異なる濃度のSWCNT分散物を用いることにより、ある範囲の透明度で膜が生成する。0.01mg/mLおよび0.1mg/mLの水性分散物から、95%および90%の透明度を有する膜が得られる。2%エタノールを添加すると、透過率70%の膜が得られる。エタノールは、SWCNTの表面電荷を下げ、またその界面エネルギーを低下させて、SWCNTが液/液界面で会合することを可能にする。透過率90%の膜は、1kΩのシート抵抗を有する。
【0050】
生成後に、300℃で12時間アニーリングを行うことにより、整列したSWCNTからなる膜の充填密度を制御することができ、十分に分離したカーボンロープ、および基材へのより強固な膜接着力が得られる。あるいは、水性分散物の混合プロトコールも充填密度を制御する。0.1mg/mLの水性分散物を用いて、標準的な超音波バス内で2時間の延長した超音波処理を行うと、十分に分離したカーボンロープ、および充填密度の低い整列したSWCNTからなるコーティングが得られる。
【0051】
ラマン分光法は、膜の高さ軸に沿って低から高シグナル強度勾配を示す(図60)。界面における濃度勾配の広がりは、質量の異方性分布を引き起こし、またこの広がりは、基材の領域が高くなるほど、強度勾配は強いシグナルを示す理由を説明する。
【0052】
図25〜28はSWCNT膜の顕微鏡写真である。
【0053】
図25は、水、2mlおよびクロロベンゼン、6ml中のSWCNT、0.0005gを用いて収集した、シリコン基材上の膜を示す。図25は、上記のようにシリコン基材上で成長させた単層カーボンナノチューブ膜のSEM画像である。この膜は、カーボンナノチューブロープの配列状態を示す。基材がロープの間に認められ、このような膜に典型的な多孔性の形態を示している。ロープの直径は、超音波処理の程度により制御可能である。SEM画像は、30分間超音波処理されたSWCNT分散物から形成された膜を示し、サンプルは52°傾いている。
【0054】
図26は、図25と同一の方式で形成され、15分間超音波処理した別の膜を2.5倍拡大したSEM画像である。より高い倍率では、カーボンナノチューブのロープは、超音波処理がより少ないため、図25のロープのように十分に分散されていないことが明らかである。このSEM画像は、顕微鏡に対して垂直に配置されたサンプルを用いて収集された。
【0055】
図27は、シリコン基材上で成長させた単層カーボンナノチューブ膜のSEM画像である(目盛りは1マイクロメートル)。膜の堆積で酸を用いたので、からみ合い(カーボンナノチューブロープの凝集物)が存在するが、これはより淡い色のついた外観を有する。酸はカーボンナノチューブをプロトン化し、そしてより高度の水素結合を引き起こすので、使用する酸の濃度は、凝集物の形成に直接的な影響を有する。このSEM画像は、顕微鏡に対して垂直に配置されたサンプルを用いて収集された。
【0056】
図28は、シリコン基材上で成長させた単層カーボンナノチューブ膜のSEM画像である。SWCNTの水性分散物を、45分間超音波処理することにより、十分に分散された形態が得られるが、これにより、高度に整列したカーボンナノチューブロープが認められる。このSEM画像はサンプルを52°傾けて得られた。
【0057】
図29〜31に示す膜は、脱イオン水、2mlにSWCNT、0.1〜1.0mgを添加し、20mlのガラスシンチレーションバイアル中で混合および超音波処理を10分間行った水性分散物から調製された。次に、クロロベンゼン(3〜6ml)、(好ましくは5ml)を添加し、そして当該溶液をさらに10分間超音波処理した。超音波処理プロセス全体を通じて、バイアルを反復して揺動させた。次に、当該溶液を静置した。酸素プラズマ中で5分間事前処理されたシリコン基材上で、静置した溶液から膜を形成した。次に、当該膜を、バイアル中でクロロベンゼン蒸気に曝露して10分間乾燥させ、バイアルから取り出し、次に周囲条件で2時間さらに乾燥させた。図29〜31は、濃度1mg/mlのSWCNTから形成された膜の様々な倍率における顕微鏡写真である。
【0058】
希釈および高度に精製された水性分散物からキャスティングすると、個々のSWCNTが、膜として堆積する。体積としてヘキサフルオロイソプロパノールを30%含有するSWCNTからなる5mg/mLの水性分散物を、氷浴中でホーンチップを用いて、100%出力で1時間、超音波処理した。112×gで30分間遠心分離を行い、上清上部を分離し、脱イオン水を用いて50%に希釈し、そして延長して超音波処理を行うと、精製された安定な分散物が生成する。高度に希釈された、透明なSWCNT水性分散物を得るために、この精製プロセスを4回反復した。浸漬型ホーンチップを用いながら延長して超音波処理を行うことにより、1mLのアリコートと4mLのクロロホルムが混合され、ピッカリングエマルジョンの混合物が分散するようになる。
【0059】
図62〜64を参照すると、膜の堆積は、容器40内で成分を乳化するのに超音波処理チップ38を用いることにより、自動化可能である。コーティングの対象となる濡れた基材44の端部42のみが、乳化組成物46内に配置される。超音波エネルギーが休止すると、濡れた基材44上で合体および膜48の成長が進行する。この手順により、十分に分離したSWCNTロープおよび個別のカーボンナノチューブからなる薄膜が生成する。
【0060】
複合膜
図32および33では、グラフェン膜を成長させる上記技術と同一の技術を用いて生成した膜を示す。図32は、膜中に存在する非結晶性の炭素を示し、おそらくこれは、ヒドラジンおよび超音波処理に起因する。走査型電子顕微鏡を用いて加速電圧(18.00KV)を高めることにより、膜表面のアニーリング(図32の暗くなった中央部分)を行ったが、このアニーリングにより、非結晶性の炭素が消失し、一方カーボンナノチューブが残る。図33は、図32のアニーリングされた領域を示す拡大画像であり、カーボンナノチューブのネットワークが非結晶性炭素層の下に認められる。
【0061】
図34、35、36、および37は、膜成長前に延長した超音波処理に曝露した後に生成した、SWCNT−グラフェン複合物からなる膜のSEM画像であり、当該膜はシリコン基材上に形成されている。両方の材料を、ヒドラジン中で混合、分散し、そして膜成長前に超音波処理する。画像は、超音波処理を延長して行った(少なくとも約20分間)後に認められるグラフェンシートサイズの減少を説明する。複合物からなる膜は、上記で議論したグラフェン膜を成長させるのに用いられる技術と同一の技術により得られる。図34および35は、低濃度のSWCNT(0.1mg/ml)から形成された膜を示す。図36は、高めの濃度(1.0mg/ml)の、高度還元グラファイト酸化物およびグラフェンヒドラジン分散物から形成された膜を説明し、高密度ナノ構造ネットワークを形成する。図37は、カーボンナノチューブにより相互に連結した、高度還元グラファイト酸化物およびグラフェンシートからなる高密度ネットワークを形成するために、ヒドラジン分散物中のSWCNTの濃度(2.0mg/ml)を高めて形成した膜を表す。
【0062】
図38、39、40、および41は、膜成長前に約15分間超音波処理し、そしてシリコン基材上で収集した、SWCNT−グラフェン複合物からなる膜のSEM画像である。
【0063】
図38〜41の膜は、5mg/mlのグラフェンおよびSWCNTからなるヒドラジン分散物、0.15mlと、14wt%のNH4OH溶液、2mlを混合することにより生成した。この溶液を60秒間超音波処理した。次にクロロベンゼン(3〜4ml)を添加し、そして溶液を揺動させ、バイアルを静置した後に膜を基材上に形成した。
【0064】
図38〜41は、同一の手順を用いて調製された4つの異なる膜の例であり、図39〜41は高倍率である。
【0065】
図42、43、44、および45は、本明細書に記載するプロセスを用いてシリコン基材上で収集した、ポリアニリンナノファイバーグラフェン複合物からなる膜のSEM画像である。グラフェン膜を、上記のように実施例6の方法を用いて最初に作製した。この膜を、乾燥するために1時間放置し、次をドープしたポリアニリンナノファイバー分散物を、予め成長させたグラフェン膜の上部にポリアニリンナノファイバー膜を成長させるために用いた。図42および43は、水酸化アンモニウム蒸気に曝露した後の膜を示し、ポリアニリンナノファイバーは、水酸化アンモニウム曝露に起因して脱ドープされる。図43は図42の高倍率画像である。図44および45は、高度還元グラファイト酸化物およびグラフェンシート最上部に位置するドープされたポリアニリンナノファイバーからなる膜を、2つの異なる倍率で示す(図44は図45の2倍)。
【0066】
図46〜50は、シリコン基材上で収集した、ポリアニリンナノファイバーSWCNT複合物からなる膜のSEM画像であり、これら5つの画像は異なる倍率での膜を示している(参照用に各画像の下側右隅に寸法線を示す)。過塩素酸をドープしたポリアニリンナノファイバーからなる水性分散物(4g/L)、0.4mlをSWCNT水性分散物に添加したことを除き、上記方法と同一の方法を用いてこれらの膜を成長させた。塩基は用いず、またSWCNTはヒドラジン中で事前分散せずに、固体状態から直接水と混合される。クロロベンゼンおよびSi基材を用いて膜成長を行う。ポリアニリンナノファイバーと絡み合う、束状のSWCNTからなるカーボンロープを示す。
【0067】
同様に、有機溶媒がヘキサンまたはヘプタン等のアルカンである場合を除き、ポリチオフェン膜を作製するのと同一の方法を用いて、基材上にポリ(3−ヘキシルチオフェン)ナノファイバーからなる透明な膜を成長させた。図51〜53は、本明細書に記載する方法を用いて二酸化ケイ素基材上に形成した、ポリ(3−ヘキシルチオフェン)ナノファイバー膜の3つの異なる倍率におけるSEM画像である。図53は、図52の中央部分を拡大した画像であり、図52は、図51の中央部分を拡大した画像である。
【0068】
活性化されていない疎水表面も、例えば図54〜59または62〜64の手順、および極性反対の非混和性の溶媒からなる二元混合物を用いて、透明で導電的に連続的な膜でコーティング可能である。この二元溶媒系が固体と接触すると、いわゆる選択的濡れで、固体表面の流体が別の液体に自発的に置き換わる。表面エネルギーが低い固体上に薄膜を堆積させる場合、かかる堆積では表面張力が極めて低い溶媒を必要とする。例として、水およびフルオロカーボンを混合すると、選択的濡れが生じ、そして活性化されていない疎水表面上で膜が成長する。表面張力が低い液体であるフルオロカーボンは、プラスチックを濡らし、ポリプロピレン膜キャパシタに充填され、そしてポリエステル繊維に撥水性を付与する。フルオロカーボン分子の間の凝集力は極めて低く、プラスチックの完全な濡れを実現する。フルオロカーボンの低い表面張力は、フッ素原子の低い分極率に起因し、表面張力が高い(72.8mN/m)極性液体である水と混ざらない。フルオロカーボン、例えばパーフルオロ−2−ブチルテトラヒドロフラン、パーフルオロ(メチルシクロヘキサン)、Fluorinert(登録商標)FC−40(16mN/m)、Fluorinert(登録商標)FC−70(18mN/m)、およびFluorinert(登録商標)FC−77(15mN/m)(Fluorinert(登録商標)は3Mの商標である)等を、本手順で採用した。
【0069】
水、フルオロカーボン、およびカーボンナノチューブを激しく撹拌すると液滴が生ずる。活性化されていない疎水性基材と接触すると、フルオロカーボンは、表面由来の水と置換して選択的濡れを引き起こす。液滴の合体は、溶媒の間で表面張力が極度に相違する結果、非常に活発である。両方の溶媒により部分的にコーティングされたカーボンナノチューブは、液滴の外に速やかに排除され、そして基材上に存在する水/フルオロカーボン界面に吸着する。吸着は、SWCNTおよびフルオロカーボン間の疎水的相互作用により強化される。界面間の広がりは、系の総界面表面エネルギーを最低限に抑え、そしてSWCNTの吸着および堆積を引き起こす。
【0070】
カーボンナノ構造体からなる高品質の透明な膜は、複数の液滴からなるペルフルオロ化されたエマルジョン内に浸漬された後、適する基材をコーティングする。基材が液滴と接触した状態に留まる時間は、吸着する炭素の量を決定する。時間が増加すると、吸着した固形物の濃度が高まり、また基材の濡れ性を疎水性から親水性に変化させる。カーボンナノチューブからなる高密度の膜が基材をコーティングする場合、表面エネルギーが変化し、基材は親水性表面のように振る舞い、水に濡れる。したがって、SWCNTの高密度コーティングが吸着した後に、親水性表面上で一般的に認められる垂直方向への膜の広がりが誘発されうる。
【0071】
SWCNTからなる大型で透明な導電性膜の堆積は、活性化されていない疎水性の可撓性基材、例えばポリエステル基材の22×22cm2の領域を最初にコーティングすることにより実現可能である。ホーン型超音波発生装置を用いて、Fluorinert FC−40、400mL、およびSWCNTからなる0.05mg/mLの水性分散物、200mLを混合することにより、コーティングエマルジョンを作製する。超音波発生装置を100%出力に設定し、そして氷浴中で2時間混合する。次に、基材およびカーボンエマルジョンを、ぴったり納まる容器内に収納し、そして手動により10分間激しくかき混ぜた。コーティングした後、配向したポリエステル基材(Grafix(登録商標)Plastics)を収納容器から取り出し、周囲条件で乾燥した。フルオロカーボンは表面からきれいに蒸発し、残留物はまったく残らない。透明度が90%を上回り、シート抵抗が1kΩの両面膜を、本手順を用いて作製した。
【0072】
活性化されていないプラスチック上のSWCNTからなる透明な膜は、SWCNTからなる0.1mg/mLの水性分散物、2mL、およびペルフルオロ化された炭化水素、例えばFluorinert FC−40、8mLを組み合わせることにより光学的に透明なビニル製スライド上に堆積可能である。次に、ぴったり納まる容器内で、手動により成分を1分間かき混ぜることにより、乳化を実施した。コーティングされたスライドを溶液から取り出し、過剰の吸着物を除去するために水で洗浄し、そして周囲条件で乾燥放置した。図61は、透明度が98%でシート抵抗が1ΜΩの、導電的に連続な膜の特性を示すグラフである。プラスチック基材の端部は、均質にコーティングされており、また均一なシート抵抗を有するが、中心部は2桁高い抵抗を有する。この均一性の欠如は、5分後の抵抗の安定性に改善が認められることから実証されるように、1分よりも長くかき混ぜさえすれば改善される。基材および合体した液滴間の接触は、SWCNTの吸着を引き起こし、長く接触するほど、膜のバルク均一性が増す。7分間撹拌すると、コーティングされた全表面領域にわたり、90%の透明度と1kΩのシート抵抗が得られる。
【0073】
別の例として、過塩素酸をドープしたポリアニリンナノファイバーからなる透明な膜を、配向したポリエステル基材(10.2cm×8.4cm×0.0254cm)上に堆積させたが、これには、水性ポリマー分散物[4g/L]を6mL、水を3mL、およびペルフルオロ化された液、例えばFluorinert FC−40(登録商標)を60mL用いて指向性流体をコーティングした。すべての薬品を、250mLのガラス製広口ジャー内で混合し、また激しくかき混ぜ、次に洗浄した疎水性基材をガラス製ジャーの液/液界面に導入した。次に、この構成物を激しくかき混ぜると、緑色の膜がプラスチック基材上に速やかに形成された。1分間撹拌した後、コーティングされた緑色に着色した基材を取り出し、水で洗浄し、そして周囲条件で乾燥放置すると、連続的で導電性の膜が生成した。
【0074】
界面において吸着したナノファイバーの表面自由エネルギーおよび基材間の分子相互作用は、膜形態を規定し得る。過塩素酸をドープしたポリアニリンは、単層ナノファイバーの平均厚さを有する膜を形成する。これは、例えば連続的な膜の形態で会合したナノファイバーの拡大画像を表す図8で明らかなように、油層および水層間でサンドイッチされたときに、ナノファイバーが界面において押し出されるために生ずる。また、ポリアニリンナノファイバーからなる単層の単層も、ドーパント、例えばカンファースルホン酸またはp−トルエンスルホン酸等を用いれば堆積可能である。膜は、最大3Scm-1の伝導度を有し、PDMSスタンプを用いてパターン化され得る。
【0075】
部分的に濡れた膜を、親水性表面上に存在する空気/水界面から、液体リザーバー内に存在する空気/水界面に移動することにより、基材を有さない膜を作製することができる。膜内の濡れ具合を制御することにより、空気/水界面で層間剥離が実現する。堆積後5分間、容器のフタを閉める状態に保つことにより、ガラス製スライド上のSWCNTからなる膜をゆっくりと乾燥放置した。膜を、1MのHCl水溶液を用いて層間剥離する前に、周囲条件下で1分間乾燥させた。−COOH官能基がプロトン化すると、カーボンナノチューブ間の水素結合および2Dの稠密充填が引き起こされ、層間剥離した浮遊膜は圧縮を必要としない。膜は、膜構造を含む炭素両親媒性物質が有する凝集性の分子相互作用により、完全な1片として残る。単層のSWCNT膜を製造し、また層間剥離を行うための従来法は、高分子分散剤、例えばポリ(3−ヘキシルチオフェン)、ヒドラジン処理、および3時間プロセスを必要とした。本明細書に記載する手順を用いれば、高分子分散剤を用いずに、数分のうちに自立SWCNT膜が生成する。膜全体が、空気/水界面で単層片として層間剥離し、そして均質な自立膜は、数日間浮遊した状態で残る。
【0076】
SWCNTからなる自立膜をガラスからSiO2に移動するとき、膜の形態は、マイクロメートルスケールで整列した状態に残る。自立膜は、電気的に連続的であり、また水面からこれをすくい上げることにより、任意のタイプの基材に移すことができる。酸性媒体上で層間剥離する際に、膜は、−COOH官能基のプロトン化により収縮し、また充填密度は、より強い凝集相互作用により増加する。逐次積層堆積法は、複数の層をすくい上げ、既に堆積された膜上に別の膜を堆積させる前に、それぞれを100℃で4時間アニーリングすることにより実施される。
【0077】
自立層を層間剥離し、移動することにより調製される、多層化SWCNT膜のオプトエレクトロニック特性を図65および66に示す。単層の移動層は、500kΩのシート抵抗を有し(図65)、また透明度は82%を上回る。2番目の層を堆積させると、シート抵抗は1桁下がり、また浸透閾値が規定される。3層以上重ねても、シート抵抗に対する効果はより小さい。2層以上重ねた後には、曲線の正の傾きがより小さくなることから実証されるように、膜の電気的安定性は高まる。各層は、平均10吸光度単位ずつ透明度を低下させる(図66)。この高い光学濃度は、1Mの酸で層間剥離する際に、膜の収縮および充填密度の増加が引き起こされてSWCNTが圧縮されることに起因する。層間剥離媒体を5%エタノール溶液に置き換えると、より高い透明度が得られる。
【0078】
当業者は、上記の記載内容および実施例に基づき、本発明は、前記代表的実施例により限定されず、また本発明の変形形態も本発明の範囲内であることを認識するであろう。例えば、様々な異なる材料を含む様々なナノファイバーおよびナノ構造体が開示される。しかし、本明細書に開示される方法は、その他のナノ構造体およびその他の材料、例えばデオキシリボ核酸、およびポリ(3,4−エチレンジオキシチオフェン)等のその他のチオフェンを含む様々なナノ形態のチオフェン、ならびにポリスチレンナノビーズ、およびその他のナノ形態のカーボン、例えばカーボンナノスクロールまたはカーボンブラックナノ粒子等への適用も検討対象である。そしてさらに、非常に多くの非混和性で有機性の液体、例えばニトロメタン、カーボンジスルフィド、ペルフルオロ化された炭化水素、例えばFluorinate(登録商標)FC−40、FC−75、およびFC−77等、酢酸エチル、ジメチルホルムアミド、ジエチルエーテル、様々なハロゲン化された炭化水素、例えばジクロロメタン、ジクロロエタン、およびテトラクロロエチレン等、ベンゼンおよびトルエン、ならびにハロゲン化された芳香族、例えばハロゲン化されたベンゼンまたはトルエン、例えばクロロ−、ジクロロ−、およびトリクロロ−ベンゼン等を含む、ただしこれらに限定されない様々な芳香族炭化水素が利用可能である。ある特定のナノ材料、または水性相もしくは有機相で用いられる化合物の開示が存在しないからといって、当該材料または液体の使用が除外されるとみなしてはならず、その使用はまだ評価されていないということを示唆するにすぎない。
【0079】
当業者は、非常に多くの代替基材、例えばマイカ、アルミフォイルまたは銅フォイル等の金属フォイル、ならびにビニル、ポリ塩化ビニル、ポリエチレン、およびポリエステルフィルム(例えば、Mylar(登録商標))を含む、ただしこれらに限定されない広範囲のポリマーシート材料も利用可能であることを認識するであろう。特定の基材または開示された基材のための表面処理法の開示が存在しないからといって、当該基材または表面処理法の使用が除外されるとみなしてはならず、その使用はまだ評価されていないということを示唆するにすぎない。さらに、上記記載内容は、プラズマにより活性化された疎水性基材の使用を開示するが、有機相を適切に選択すれば、活性化されていない疎水基材も利用可能である。特に、非混和性で有機性の液体がペルフルオロ化炭化水素であれば、開示された方法を用いて、疎水性基材上で膜を成長させることができる。そしてさらに、長方形の基材の使用が開示されるが、本方法の有用性は、基材の幾何学的形状に限定されるものではなく、その他の形状(四角形、三角形、円形または楕円形のディスク等)は、球等の三次元の基材を含め利用可能である。
【0080】
さらに、処理時間、液体の体積、および様々な成分の比は、代表例にすぎず、また現在のしかるべき好ましい操作条件を開示しているのであって、これらは、利用可能な様々な液体、ナノ材料、基材、および処理容器についてプロセスを最適化するために変更可能である。そしてさらに、上記手順は、水溶液の調整法を開示する。当業者は、様々な異なる酸または塩基が利用可能であることを認識するであろう。例えば、pHを調整するのに適する酸および塩基として、塩酸、過塩素酸、リン酸、ヒアルロン酸、硫酸、ポリスチレンスルホン酸、カンファースルホン酸、トルエンスルホン酸、ドデシルベンゼンスルホン酸を含むスルホン酸、その他の有機硫酸塩、樟脳酸、硝酸、酢酸、クエン酸、ヒドラジンおよび様々なヒドロキシル化合物、例えばアンモニウム、ナトリウム、カルシウム、リチウム、およびカリウムの水酸化物等が挙げられるが、ただしこれらに限定されない。pH調整に用いられる特定の酸または塩基の開示が存在しないからといって、当該材料の使用が除外されるとみなしてはならず、その使用はまだ評価されていないということを示唆するにすぎない。
【技術分野】
【0001】
2010年1月14日出願の、米国特許仮出願第61/295,116号の利益を主張する。
【0002】
本発明は、全米科学財団により授与された認可番号第DMR0507294号の政府支援により実現した。米国政府は、本発明において所定の権利を有する。
【0003】
本出願は、電気伝導性のポリマー、カーボンナノ構造体、およびこれらを組み合わせたものから薄膜を形成する一般的方法に関連する。
【背景技術】
【0004】
導電性ポリマーは、様々な用途で用いられる安価で可撓性の材料として有望であり、これには非限定的に太陽電池、発光ダイオード、および化学抵抗器型検出器(例えば、非特許文献1〜3を参照)が含まれる。均質な薄膜の制御可能な堆積は、電子デバイス工学にとって必須である。数え切れない数の膜形成法が文献で報告されており、そのような方法としてin−situ堆積法(例えば、非特許文献4、5を参照)、溶液中での静電吸着法(例えば、非特許文献6を参照)、ドロップキャスティング法(drop−casting)(例えば、非特許文献7を参照)、電気化学堆積法(例えば、非特許文献8を参照)、スピンコーティング法(例えば、非特許文献1を参照)、グラフト法(例えば、非特許文献9を参照)、およびインクジェット印刷法(例えば、非特許文献10を参照)が挙げられるが、電気伝導性ポリマーもしくは導電性のナノ構造体、またはこれらを組み合わせたものを利用して、基材上に電気伝導性膜を確実に堆積させる簡便で汎用性のある方法について明らかにニーズが認められる。
【先行技術文献】
【非特許文献】
【0005】
【非特許文献1】Bravo-Grimaldo, E., Hachey, S., Cameron, C. G. & Freund, M. S. "Metastable Reaction Mixtures For The In Situ Polymerization Of Conducting Polymers". Macromolecules 40, 7166-7170 (2007)
【非特許文献2】Zhou, Y. et al. "Investigation on Polymer Anode Design For Flexible Polymer Solar Cells". Appl. Phys. Lett. 92, 233308/233301-233308/233303 (2008)
【非特許文献3】Zaumseil, J., Friend, R. H. & Sirringhaus, H. "Spatial Control Of The Recombination Zone In An Ambipolar Light-Emitting Organic Transistor". Nat. Mater. 5, 69-74 (2006)
【非特許文献4】Chiou, N.-R., Lu, C, Guan, J., Lee, L. J. & Epstein, A. J. "Growth And Alignment Of Polyaniline Nanofibres With Superhydrophobic, Superhydrophilic And Other Properties". Nature Nanotech. 2, 354-357 (2007)
【非特許文献5】Zhang, X., Goux, W. J. & Manohar, S. K. "Synthesis Of Polyaniline Nanofibers By "Nanofiber Seeding". J. Am. Chem. Soc. 126, 4502-4503 (2004)
【非特許文献6】Li, D. & Kaner, R. B. "Processable Stabilizer-Free Polyaniline Nanofiber Aqueous Colloids". Chem. Commun. 26, 3286-3288 (2005)
【非特許文献7】Huang, J., Virji, S., Weiller, B. H. & Kaner, R. B. "Nanostructured Polyaniline Sensors". Chem.-A Eur. J. 10, 1314-1319 (2004)
【非特許文献8】Valaski, R., Canestraro, C. D., Micaroni, L., Mello, R. M. Q. & Roman, L. S. "Organic Photovoltaic Devices Based On Polythiophene Films Electrodeposited On FTO Substrates". Sol. Energy Mater. Sol. Cells 91, 684-688 (2007)
【非特許文献9】Sawall, D. D., Villahermosa, R. M., Lipeles, R. A. & Hopkins, A. R. "Interfacial Polymerization of Polyaniline Nanofibers Grafted To Au Surfaces". Chem. Mater. 16, 1606-1608 (2004)
【非特許文献10】Murphy, A. R. & Frechet, J. M. J. "Organic Semiconducting Oligomers For Use In Thin Film Transistors". Chem. Rev. 107, 1066-1096 (2007)
【非特許文献11】Mayya, K. S. & Sastry, M. "A New Technique For The Spontaneous Growth Of Colloidal Nanoparticle Superlattices". Langmuir 15, 1902-1904 (1999)
【非特許文献12】Cheng, H.-L. & Velankar, S. S. "Film Climbing Of Particle- Laden Interfaces". Colloids Surf., A 315, 275-284 (2008)
【非特許文献13】Binks Bernard, P., Clint John, H., Fletcher Paul, D. I., Lees Timothy, J. G. & Taylor, P. "Particle Film Growth Driven By Foam Bubble Coalescence". Chem. Commun. 33, 3531-3533 (2006)
【非特許文献14】Binks, B. P., Clint, J. H., Fletcher, P. D. I., Lees, T. J. G. & Taylor, P. :Growth Of Gold Nanoparticle Films Driven By The Coalescence Of Particle-Stabilized Emulsion Drops". Langmuir 22, 4100-4103 (2006)
【非特許文献15】Jager, E. W. H., Smela, E. & Inganas, O. "Microfabricating Conjugated Polymer Actuators", Science 290, 1540-1546 (2000)
【非特許文献16】Goedel, W. A. "A Simple Theory Of Particle- Assisted Wetting". Europhys. Lett. 62, 607-613 (2003)
【非特許文献17】Pesach, D. & Marmur, A. "Marangoni Effects In The Spreading Of Liquid Mixtures On A Solid". Langmuir 3, 519-524 (1987)
【非特許文献18】Farahi, R. H., Passian, A., Ferrell, T. L. & Thundat, T. "Microfluidic Manipulation Via Marangoni Forces". Appl. Phys. Lett. 85, 4237-4239 (2004)
【非特許文献19】Sarma, T. K. & Chattopadhyay, A. "Visible Spectroscopic Observation Of Controlled Fluid Flow Up Along A Soap Bubble Film From A Pool Of Solution". J. Phys. Chem. B 105, 12503-12507 (2001)
【非特許文献20】Cai, Y. & Zhang Newby, B.-m. "Marangoni Flow-Induced Self- Assembly Of Hexagonal and Stripelike Nanoparticle Patterns". J. Am. Chem. Soc. 130, 6076-6077 (2008)
【非特許文献21】Melle, S., Lask, M. & Fuller, G. G. Pickering Emulsions with controllable stability. Langmuir 21, 2158-2162 (2005)
【非特許文献22】Ata, S. "coalescence Of Bubbles Covered By Particles". Langmuir 24, 6085-6091 (2008)
【非特許文献23】Lucassen, J., Lucassen-Reynders, E. H., Prins, A. & Sams, P. J. "Capillary Engineering For Zero Gravity". Critical wetting on axisymmetric solid surfaces. Langmuir 8, 3093-3098 (1992)
【非特許文献24】Rey, A. D. "Stability Analysis Of Catenoidal Shaped Liquid Crystalline Polymer Networks". Macromolecules 30, 7582-7587 (1997)
【非特許文献25】Chengara, A., Nikolov Alex, D., Wasan Darsh, T., Trokhymchuk, A. & Henderson, D. "Spreading Of Nanofluids Driven By The Structural Disjoining Pressure Gradient". J. Colloid Interface Sci. 280, 192-201 (2004)
【非特許文献26】Bestehorn, M., Pototsky, A. & Thiele, U. "3D Large Scale Marangoni Convection In Liquid Films". Eur. Phys. J. B 33, 457-467 (2003)
【非特許文献27】Pruneanu, S., Veress, E., Marian, I. & Oniciu, L. "Characterization Of Polyaniline By Cyclic Voltammetry And UV-V Is Absorption Spectroscopy". J. Mater. Sci. 34, 2733-2739 (1999)
【非特許文献28】Patil, A. O., Heeger, A. J. & Wudl, F. "Optical Properties Of Conducting Polymers". Chem. Rev. 88, 183-200 (1988)
【非特許文献29】Tran, H. D., Wang, Y., D'Arcy, J. M. & Kaner, R. B. "Toward An Understanding Of The Formation Of Conducting Polymer Nanofibers". ACS Nano 2, 1841-1848 (2008)
【非特許文献30】Sawistowski, H. "Surface Tension-Induced Interfacial Convection And Its Effect On Rates Of Mass Transfer". Chem. -Ing. -Tek. 45, 1093-1098 (1973)
【非特許文献31】Tung, V. C; Allen, M. J.; Yang, Y.; Kaner, R. B. Nat. Nanotechnol. 2009, 4, 25-29
【発明の概要】
【0006】
ナノ構造体、特に導電性のポリマー、カーボンナノ構造体、およびこれらの組合せの膜を堆積させる方法を記載する。ナノスケールで再現性のある厚み制御、および形態的均質性を可能にする簡便でスケール拡張性のある膜製造技術は、工業用途として魅力的な選択肢である。容積、ドープ、およびポリマー濃度が適切な条件の下で、導電性ポリマーナノファイバー、例えばポリアニリンやポリチオフェン、グラフェン、カーボンナノチューブ、またはこれらを組み合わせたものの単層からなる膜は、およそ数秒間のうちに製造可能である。熱力学的に推進される溶液ベースのプロセスは、界面において吸着されたナノファイバーからなる透明薄膜の成長を引き起こす。高品質の透明薄膜は、周囲条件で、事実上任意の基材上に堆積される。無傷の状態の膜を基材から取り出す方法も開示される。この安価なプロセスは、リサイクル可能な溶液を使用し、またポリマーと共に水性相および有機相を含む二相溶液を用いて、広い基材領域を導電性材料でコーティングする新しい技術を提供する。
【図面の簡単な説明】
【0007】
【図1】ポリアニリンナノファイバー膜が大きくなり、且つ広がる機構を説明し、プロセスを概略的に示す図である。
【図2】ポリアニリンナノファイバー膜が大きくなり、且つ広がる機構を説明し、プロセスを概略的に示す図である。
【図3】ポリアニリンナノファイバー膜が大きくなり、且つ広がる機構を説明し、プロセスを概略的に示す図である。
【図4】ポリアニリンナノファイバー膜が大きくなり、且つ広がる機構を説明し、時間系列による界面膜形成を示す図である。
【図5】ガラス製の顕微鏡スライド上に収集して得られた透明薄膜、3例を示す写真であり、左側のスライドは、Cl-をドープしたポリチオフェンナノファイバーからなる導電性ポリマー膜を含み(網掛け部分は赤色を表す)、中央のスライドは、ドープされたポリアニリンナノファイバー膜を示し(網掛け部分は緑色を表す)、および右側のスライドは、脱ドープされたポリアニリンナノファイバー膜を示す(網掛け部分は青色を表す)。
【図6】ガラス基材上に収集したポリアニリンナノファイバーの薄膜を倍率を上げながら示したSEM画像である(目盛りは、2μmである)。
【図7】ガラス基材上に収集したポリアニリンナノファイバーの薄膜を倍率を上げながら示したSEM画像である(目盛りは、1μmである)。図6中の四角で囲繞された領域の拡大図である。
【図8】ガラス基材上に収集したポリアニリンナノファイバーの薄膜を倍率を上げながら示したSEM画像である(目盛りは、500nmである)。図7中の四角で囲繞された領域の拡大図である。
【図9】ポリアニリンナノファイバーの、酸化膜(緑色である図9の網掛け部分)から還元膜(青色である図10の網掛け部分)へのレドックススイッチングを説明するガラス製のスライドの概略図である。
【図10】ポリアニリンナノファイバーの、酸化膜(緑色である図9の網掛け部分)から還元膜(青色である図10の網掛け部分)へのレドックススイッチングを説明するガラス製のスライドの概略図である。
【図11】図9〜10に示す変化を表す図であり、網掛け部分は図9および10の指定領域と同じ領域に対応する。
【図12】紫外可視吸光度分光法(absorbance UV−vis spectroscopy)によりモニターされた、膜形成期間中の厚さ制御を説明する図である。
【図13】紫外可視吸光度分光法によりモニターされた、膜形成期間中の厚さ制御を説明する図である。
【図14】図13にグラフで示した膜の可撓的な性質を表す図である。
【図15】ヒドラジン中に分散した0.25mg/ml、0.13mg/ml、および0.05mg/mlのグラフェン分散物をガラス製のスライドに塗布して作製された、3つのグラフェン膜を表す図である。基材上に堆積された材料の量と、膜の成長に用いられる分散物中の固形物の濃度との間に直線相関関係が認められ、前記濃度は密度の相違(網点の相違により図中に示す)により表される。
【図16】ヒドラジン中のグラフェン分散物(2mg/ml)、0.5mlを含む、シリコン基材上で収集された高度還元グラファイト酸化物膜およびグラフェン膜からなるシートのSEM画像である。
【図17】グラフェン分散物、0.1mlを含む、シリコン基材上で収集された高度還元グラファイト酸化物膜およびグラフェン膜からなるシートのSEM画像である。
【図18】シリコン基材上で収集された高度還元グラファイト酸化物膜およびグラフェン膜からなるシートのSEM画像である。
【図19】シリコン基材上で収集された高度還元グラファイト酸化物膜およびグラフェン膜からなるシートのSEM画像である。
【図20】シリコン基材上で収集されたグラフェン膜からなる単層シートのSEM画像である。
【図21】シリコン基材上で収集されたグラフェン膜からなる単層シートのSEM画像である。
【図22】シリコン基材上で収集されたグラフェン膜からなる単層シートのSEM画像である。
【図23】シリコン基材上で収集されたグラフェン膜からなる単層シートのSEM画像である。
【図24】ガラス製のスライド上に堆積された単層カーボンナノチューブからなる3つの異なる膜を示す図であり、異なる膜密度を表すために図には網点が施されている。
【図25】シリコン基材上で水性媒体から収集した単層カーボンナノチューブ(SWCNT)からなる膜のSEM画像である。
【図26】シリコン基材上で水性媒体から収集した単層カーボンナノチューブ(SWCNT)からなる膜のSEM画像である。
【図27】シリコン基材上で水性媒体から収集した単層カーボンナノチューブ(SWCNT)からなる膜のSEM画像である。
【図28】シリコン基材上で水性媒体から収集した単層カーボンナノチューブ(SWCNT)からなる膜のSEM画像である。
【図29】シリコン基材上で水性媒体から収集した単層カーボンナノチューブ(SWCNT)膜のさらなるSEM画像である。
【図30】シリコン基材上で水性媒体から収集した単層カーボンナノチューブ(SWCNT)膜のさらなるSEM画像である。
【図31】シリコン基材上で水性媒体から収集した単層カーボンナノチューブ(SWCNT)膜のさらなるSEM画像である。
【図32】シリコン基材上で塩基性水性媒体から収集した単層カーボンナノチューブ(SWCNT)膜のさらなるSEM画像である。
【図33】シリコン基材上で塩基性水性媒体から収集した単層カーボンナノチューブ(SWCNT)膜のさらなるSEM画像である。
【図34】膜が大きくなる前に、超音波処理を延長してこれに曝露した後に生成したSWCNT−グラフェン複合物からなる膜のSEM画像であり、膜はシリコン基材上に形成されている。
【図35】膜が大きくなる前に、超音波処理を延長してこれに曝露した後に生成したSWCNT−グラフェン複合物からなる膜のSEM画像であり、膜はシリコン基材上に形成されている。
【図36】膜が大きくなる前に、超音波処理を延長してこれに曝露した後に生成したSWCNT−グラフェン複合物からなる膜のSEM画像であり、膜はシリコン基材上に形成されている。
【図37】膜が大きくなる前に、超音波処理を延長してこれに曝露した後に生成したSWCNT−グラフェン複合物からなる膜のSEM画像であり、膜はシリコン基材上に形成されている。
【図38】膜が大きくなる前に、時間間隔を変化させて超音波処理を行い、そしてシリコン基材上で収集したSWCNT−グラフェン複合物からなる膜のさらなるSEM画像である。
【図39】膜が大きくなる前に、時間間隔を変化させて超音波処理を行い、そしてシリコン基材上で収集したSWCNT−グラフェン複合物からなる膜のさらなるSEM画像である。
【図40】膜が大きくなる前に、時間間隔を変化させて超音波処理を行い、そしてシリコン基材上で収集したSWCNT−グラフェン複合物からなる膜のさらなるSEM画像である。
【図41】膜が大きくなる前に、時間間隔を変化させて超音波処理を行い、そしてシリコン基材上で収集したSWCNT−グラフェン複合物からなる膜のさらなるSEM画像である。
【図42】本明細書に記載する方法を用いて、シリコン基材上で収集したポリアニリンナノファイバーグラフェン複合物からなる膜のSEM画像である。
【図43】本明細書に記載する方法を用いて、シリコン基材上で収集したポリアニリンナノファイバーグラフェン複合物からなる膜のSEM画像である。
【図44】本明細書に記載する方法を用いて、シリコン基材上で収集したポリアニリンナノファイバーグラフェン複合物からなる膜のSEM画像である。
【図45】本明細書に記載する方法を用いて、シリコン基材上で収集したポリアニリンナノファイバーグラフェン複合物からなる膜のSEM画像である。
【図46】シリコン基材上で収集した、ポリアニリンナノファイバーSWCNT複合物からなる膜のSEM画像である。
【図47】シリコン基材上で収集した、ポリアニリンナノファイバーSWCNT複合物からなる膜のSEM画像である。
【図48】シリコン基材上で収集した、ポリアニリンナノファイバーSWCNT複合物からなる膜のSEM画像である。
【図49】シリコン基材上で収集した、ポリアニリンナノファイバーSWCNT複合物からなる膜のSEM画像である。
【図50】シリコン基材上で収集した、ポリアニリンナノファイバーSWCNT複合物からなる膜のSEM画像である。
【図51】シリコン基材上で収集したポリ(3−ヘキシルチオフェン)ナノファイバーからなる膜のSEM画像である(目盛りは、10μmである)。
【図52】シリコン基材上で収集したポリ(3−ヘキシルチオフェン)ナノファイバーからなる膜のSEM画像である(目盛りは、3μmである)。図51中の四角で囲繞された領域の拡大図である。
【図53】シリコン基材上で収集したポリ(3−ヘキシルチオフェン)ナノファイバーからなる膜のSEM画像である(目盛りは、1μmである)。図52中の四角で囲繞された領域の拡大図である。
【図54】基材上に膜を形成するために、本発明の特徴を組み込む手順の概略図である。
【図55】基材上に膜を形成するために、本発明の特徴を組み込む手順の概略図である。
【図56】基材上に膜を形成するために、本発明の特徴を組み込む手順の概略図である。
【図57】基材上に膜を形成するために、本発明の特徴を組み込む手順の概略図である。
【図58】基材上に膜を形成するために、本発明の特徴を組み込む手順の概略図である。
【図59】基材上に膜を形成するために、本発明の特徴を組み込む手順の概略図である。
【図60】図54〜59に示すプロセスを用いて形成された膜のラマンスペクトル表すグラフである。
【図61】は、膜の抵抗および透過率を、エマルジョン撹拌時間の関数として示すグラフである。
【図62】自動化された膜形成プロセスの概略図である。
【図63】自動化された膜形成プロセスの概略図である。
【図64】自動化された膜形成プロセスの概略図である。
【図65】膜の抵抗を膜層数の関数として示すグラフである。
【図66】膜の透過率を膜層数の関数として示すグラフである。
【図67】ガラス製のスライドで収集した、グラファイト酸化物シートからなる膜のSEM画像である(目盛りは、50μmである)。
【図68】ガラス製のスライドで収集した、グラファイト酸化物シートからなる膜のSEM画像である(目盛りは、30μmである)。図67中の四角で囲繞された領域の拡大図である。
【図69】ガラス製のスライドで収集した、グラファイト酸化物シートからなる膜のSEM画像である(目盛りは、10μmである)。図68中の四角で囲繞された領域の拡大図である。
【発明を実施するための形態】
【0008】
様々なナノ材料、特にポリアニリンおよびポリチオフェンナノファイバー、ならびにカーボンナノ構造体、例えばグラフェンシートおよびカーボンナノチューブ等からなる透明な薄膜を、事実上任意の基材上に、周囲条件下で成長させる溶液ベースの方法について説明する。2つ非混和性の液体およびポリマーナノファイバーを乳化することにより、界面張力勾配、粘性流、および膜の広がりが引き起こされる。表面張力の差異が、無機ナノ粒子膜を形成するためにこれまで用いられてきた(例えば、非特許文献11〜14を参照)。膜は、電気伝導性有機ポリマーを含み、またナノファイバーの単層により特徴付けられるナノスケール秩序を有する。導電性ポリマーを対象とするこの新しい膜成長技術は、容易にスケールアップ可能であり、また溶液はリサイクルすることができる。膜を形成する本方法は、形態的均質性、厚さ制御の再現性、および簡便性を有し、これらは、上記導電性ポリマーの電気特性を利用するデバイスの製造について独自の可能性をもたらす。
【0009】
一方、クロロホルムを用いた液体抽出により一次元ポリアニリンナノファイバーの水性分散物を精製中に、分液漏斗の壁にポリマーの透明な膜が形成されることを、出願者は発見した。この溶媒混合物に振動を加えるとこの膜は除去されたが、放置すると膜は迅速に再編成する。この発見に基づき、アクチュエーターおよびセンサーを非限定的に含む様々な用途で用いられる膜を調製するために、ナノ構造を有する導電性ポリマーからなる膜を成長させる溶液ベースの方法を開発したが、上記用途はこれまでに、文献(非特許文献15を参照)で開示されている。
【0010】
水および高密度油、例えばクロロベンゼン等を激しく撹拌すると、油相中に分散された水滴が形成される。液滴の水/油界面は、界面活性剤等の表面活性種および固体粒子にとって吸着部位として機能する。界面に存在する表面張力は、吸着した種の濃度に比例して減少し、また吸収された種の濃度が不均等に分布すると、界面張力勾配が発生する。次に、この勾配は流体膜が固体表面全体にわたり広がる原因となり、これはマランゴニ(Marangoni)効果として公知である。この種の指向性のある流体の流れは、生物の自己防衛機構に認められ(例えば、非特許文献16を参照)、また潤滑用途で利用可能であり(例えば、非特許文献17を参照)、マイクロフルイディクス(例えば、非特許文献18を参照)、ラブ−オン−チップ設計(例えば、非特許文献19を参照)、そして高密度データストレージで利用できる可能性がある(例えば、非特許文献20を参照)。
【0011】
事実上任意の基材上で成長させたポリマーナノファイバー、またはその他のナノ構造体からなる極めて透明度が高く、均質な薄膜を形成するために、出願者らは、図1〜3、54〜59、および62〜64で概略的に、ならびに図4では写真を用いて示す方法を開発した。ナノファイバーまたはナノ構造体は、水および高密度油と激しく混合され、次に生成する界面をコーティング対象表面に曝露する。この乳化プロセスは、部分的に膜成長に関与する。撹拌により、容器の親水性の壁が水でコーティングされ、そして水性の液滴が油相中に分散されるようになる。図1〜3および図4の文字が施された様々な部分を参照すると、例えば水12、高密度油14、およびポリマーナノファイバー16がガラス容器10内で混合され、そして激しくかき混ぜられてエマルジョンを形成する(図1、図4A、B)。混合を止めると、油14中に分散しポリマーナノファイバー16で覆われた水滴12(図1)は、油相の上部に上昇する(図2、図4C)。液滴が合体すると、界面において吸着したナノファイバーの濃度勾配、水により形成されたカテノイド15、および指向性の流体が生み出され、その結果、単層のナノファイバー16が容器の壁を上下方向に広がる(図3、図4D、E)。このカテノイドは、水12が上部で油14が底部の2つの異なるバルク液相に分断する(図3、図4F)。ナノファイバー16は、空気に隣接した水/油の界面、およびバルク油相を覆う別の界面に堆積する。バルク液相間に生じ、膜成長が停止した後に残留するポリマーリザーバーは、追加の基材をコーティングするのに利用可能な過剰のナノファイバーを含有する。図4に示す膜成長の流れを参照すると、時間は(A)0秒、(B)0.5秒、(C)1秒、(D)10秒、(E)30秒、(F)35秒である。
【0012】
ナノファイバー等の固体粒子は、非混和性の液の間の界面張力を低下させることにより、ピッカリングエマルジョンと呼ばれる安定剤として機能することができる(例えば、非特許文献21を参照)。混合により、ポリマーナノファイバーを両方の液体と溶媒和させるのに必要とされる機械的エネルギーが提供され、こうして本質的に不可逆な吸着プロセスによってナノファイバーが水/油界面でトラップされる。理論的な試験により、吸着した粒子を任意の界面から除去するのに必要とされるエネルギーは、界面においてこれらの粒子を分離するのに必要とされるエネルギーをかなり上回ることが明らかにされた(例えば、非特許文献22を参照)。したがって、乳化されたナノファイバーは、引っ張り力を受け、そして同ナノファイバーは界面において広がる。撹拌が中止されると、機械エネルギーのインプットが弱まり、水滴が油層の上部に上昇し、合体することが可能となる。油およびナノファイバーを液滴から排除する合体期間中に界面の総表面積は低下し、不可逆的に吸着されたナノファイバーに自然発生的な濃度勾配が生成し、その結果水/油界面にマランゴニ圧を生み出す。界面張力がより高い領域に排除されたナノファイバーを引き寄せる界面張力の勾配が生じるが、一方ナノファイバーの膜が、水油間で搾り出された単層として容器の壁を上下に広がる(図2、図4C,D,E)。水/油界面が存在しないので、バルク水性相を取り巻くガラス壁上では膜は成長しないことに留意されたい(図3、図4F)。
【0013】
膜成長期間中、水層は、ナノファイバーの大部分を含有する内部油チャンネルを備えるカテノイドの形成に寄与する。水は、この形状を採ることによりその表面自由エネルギーを最低限に抑える(例えば、非特許文献23を参照)。カテノイド内部の粘性流は、チャンネルの最も薄いセクションから最も厚いセクションに向かって上下両方向に流体運動を生み出す(例えば、非特許文献24を参照)。次に、合体により内部チャンネルが狭くなり(図4C〜E)、そして最終的にカテノイドによる分断が生じ、粘性流が終結する。2つの異なるバルク相が確立され、ナノファイバーの再分布を引き起こす(図3、図4F)。ナノファイバーを含有する水/油界面が、空気に隣接した側およびバルク水層下方の両方に認められる。界面上部には、ナノファイバーの濃度勾配が存在し、この濃度勾配は、カテノイド分断後の数秒間、膜成長を上方に継続して推進する。この濃縮物は、ガラス製のスライドが溶液から引き出される際に、これをコーティングするのに利用される。底部側の界面は、ナノファイバーからなるポリマーリザーバーを備え、さらなる膜の成長に用いられる。
【0014】
図54〜59および62〜64中のプロセスフロー図は、親水性の基材表面20、44上における膜の成長および堆積機構を表す。SiO2(図54)等の基材は、この図では金属製の電極22を有するが、ピラニア溶液中で煮沸され、酸素プラズマ中でエッチングされ、そして均質な水層24を誘発するために水に浸漬される(図55)。濡れた基材が、ナノ材料(図56)を含有するピッカリングエマルジョン26と接触すると、液滴26が合体する結果膜成長が生じる(図57〜59)。ナノ材料の透明なコーティング28は、数秒のうちに広がるが、これはコーティングされた表面全域にわたり導電的に連続している。
【0015】
膜成長の最適実験条件を調べるために、水および高密度ハロゲン化溶媒からなる異なる二元混合物を用いて、ポリアニリンナノファイバー膜を、ガラス製のスライド上で成長させた。最大到達可能な広がる長さが、各膜を成長させるのに用いられた非混和性の二元混合物の界面張力と比較された。この結果から、界面張力が大きいほど、上方に広がる膜の這い上がる高さはより大きくなることが示唆された。界面張力が大きい方が、より小さなものよりも強い力でナノファイバーを引き上げ、そして膜が重力に抗してより長時間基材上を這い上がるのを可能にし、その結果、広がる高さがより大きくなる。1つの比較では、水および四塩化炭素(界面張力は45dynes/cm)を用い、その後に水およびクロロホルム(32.8dynes/cm)、そして最終的に水および塩化メチレン(28.3dynes/cm)が続いたときに、ナノファイバー膜は最も高く這い上がった。膜の成長は、系の総界面自由エネルギーを最小化することにより推進される(例えば、非特許文献25を参照)。
【0016】
容器および基材の両方の寸法および材料について、それらの特性が膜の成長にどのように影響を及ぼすか調べるために試験された。容器の直径が大きいほど(例えば、直径約2.0〜約10.0インチ)、2液体間により広い界面表面積を提供し、したがって多数のバブル形成と高度に活発な合体、複数のカテノイド、および膜の高速成長を引き起こすことが判明した。一方、上方に這い上がる膜の被覆面積は、小さな直径を有する容器(例えば、直径約0.5〜約2.0インチ)よりも大きな直径の容器の方が小さいので、これを用いる迅速な膜製造は便利であり得る。疎水表面も、例えばアルゴン−酸素プラズマを用いて最初に表面を活性化させることにより膜成長基材として利用可能である。
【0017】
導電性ポリマーナノファイバーからなる透明な薄膜は、様々な色で製造可能である。図5(網掛け部分は色を示す)は、ガラス製のスライド上のポリアニリンおよびポリチオフェン膜を示した。左から右に向かって、塩化物をドープした赤色のポリチオフェン、過塩素酸をドープした緑色のポリアニリン、および脱ドープした青色のポリアニリンの膜である。膜、特に過塩素酸をドープしたポリアニリン膜は、優れた光透過性を有し、光透過率は60%を上回る。p−トルエンスルホン酸(p−TSA)をドープしたナノファイバーおよびクロロホルムからなる水性分散物を用いて、ポリアニリン膜を成長させた。次に、膜を脱ドープまたはさらにドープするために、塩基または酸の蒸気のいずれかに膜を曝露すると、当該膜はそれぞれ青色または緑色となった。
【0018】
界面において吸着したナノファイバーの表面自由エネルギーと基材との間の分子相互作用は、膜の形態を規定し得る(例えば、非特許文献26を参照)。過塩素酸をドープしたポリアニリンは、図6〜8に示すように、単層ナノファイバーの平均厚さを有する膜を形成する。この一連のSEM画像(52°の角度で傾いている)は、クロロホルムを用いて成長させて、HClO4に部分的をドープしたポリアニリンナノファイバー膜に特徴的である。ナノスケールの形態は、倍率を高めながら示すように、単層のナノファイバーから実質的に構成され、図6〜8の目盛りは、それぞれ(a)2μm、(b)1μm、(c)500nmを表す。図8は図7中の四角で囲繞された領域の拡大図であり、また図7は、図6中の四角で囲繞された領域の拡大図である。ナノファイバーは、油層および水層間にサンドイッチされたときに、界面において押し出されるので、この現象が生ずる。膜をゆっくり乾燥させると、次に毛管力が秩序を誘発し得る。この現象を図8に示すが、この場合部分的をドープしたナノファイバーは、互いに側面同士を接触させて配向している。個々の単層膜は、p−トルエンスルホン酸またはカンファースルホン酸等のドーパントを用いても、形成可能である。
【0019】
ポリアニリンナノファイバー膜の電気化学的挙動は、図11に示す通りサイクリックボルタンメトリー(CV)を用いて特徴づけられた。塩酸、過塩素酸、およびp−トルエンスルホン酸をドープしたポリアニリン膜は、すべて0.25Vと0.95Vに2つの還元ピークを示し、またこれらの対応する酸化ピークは−0.15Vと0.68Vに認められる。これらのサイクリックボルタモグラムは、ポリアニリンのエメラルジン酸化状態を示唆する(例えば、非特許文献27を参照)。エメラルジンポリアニリンの緑色のドープ形態(図9の網掛け部分)から青色のドープ形態へのスイッチングは、図10で、右手部分を異なる網掛けをして示した。これらのナノファイバー膜は、透明、堅牢で、また複数サイクルのCVを扱う能力を有する。電解質に浸漬された電極部分(図9および10ではスライドの右側部分)に限り、電位の方向が切り替ると変色する。ITO上で成長した膜の透明度は、膜を通して画像がどのくらい明瞭に見えるかにより実証可能である。エメラルジン型のポリアニリンナノファイバー膜は、図9に示す通り、電解質に途中まで浸漬され、そして電気化学的に酸化されて、ドープされた(緑色に着色)塩の状態となる。グラフ(図11)は、塩酸(HCl)、過塩素酸(HClO4)、およびp−トルエンスルホン酸(p−TSA)でドープしたポリアニリンナノファイバーのCV曲線を示す。エレクトロクロミック転移を図10に概略的に表す;図9のポリアニリンナノファイバー膜は還元され、そして溶液に浸漬された図9の透明で緑色の電極部分は青色に変化する。
【0020】
マランゴニ流により生成した膜厚は、ドープしたポリアニリンナノファイバー膜の層を連続して堆積させることにより制御可能である(図12)。図12では、ガラス上で成長させてp−TSAでドープしたポリアニリンナノファイバー膜からなる、連続して得られた4つの層それぞれが、吸収が段階的に増加(約0.2単位)することにより観察可能である。紫外可視スペクトル(図13)から、新しい層はいずれも、約0.2吸光度単位の光学濃度を生成することが明らかである。膜の各層は、スペクトルを収集する前に、周囲条件で30分間乾燥放置された。異なる高さでサンプリングされた膜について得られたポリチオフェンの紫外可視吸収(図13)は、予想された吸収ピークを示す(例えば、非特許文献28を参照)。堆積されるポリマーの質量は、膜の高さに反比例して変化するので、膜厚は、膜が成長する角度によって制御可能である。したがって、膜が基材を這い上がるにつれて光学濃度は低下する。これは、濃度勾配により膜厚を制御する能力を実証する。
【0021】
図13は、60°の角度で成長させたポリチオフェンナノファイバー膜の高さ方向の異なる位置で収集された一連のスペクトルを表し、膜が成長する角度により光学濃度は制御可能であることを実証する。図14から、ITO−ポリエチレンテレフタレートからなるプラスチック基材上を60°の角度で成長させたポリチオフェンナノファイバー膜は、軽い圧力を加えることにより実証されるように可撓性であることが明らかである(挿入物の右側および左側の端部のより暗い部分は、膜を曲げている者の手袋で覆われた指の先端である)。
【0022】
下記のいくつかの例では、導電性膜の形成手順について説明し、前記手順および得られた生成物は本発明の特徴を取り込んでいる。本明細書では、「超音波処理」と呼ぶとき、それは、容器内に収納された基材または混合物を、水で満たされた超音波バスに配置し、そして60Hzで稼動させること、あるいは混合物を収納する容器内に超音波ホーンを配置することに関係した。
【0023】
基材表面の処理:
ガラス。事前洗浄された75mm×25mm×1mmの顕微鏡用ガラス製スライド(Corning2947)を基材として用いた。膜を収集する前に、これをイソプロピルアルコールで洗浄し、そして圧縮空気で乾燥させた。a)水の中で超音波処理を30分間、b)硝酸および水の中で交互に煮沸処理、またはc)酸素プラズマ処理を5分間行う追加の表面処理を実施した。
【0024】
石英。75mm×25mm×1mmの基材(QSI Quartz Scientific)を、ガラスについて上記した方法を用いて、またはクロム酸およびDI水の中で連続して煮沸し、その後オーブン乾燥(400℃、1時間)することにより処理した。
【0025】
シリコン。Si基材をイソプロピルアルコールの中で超音波処理し(30分)、次にワイプ(Kimtech)で軽く擦り、その後酸素プラズマ処理を5分間行った。
【0026】
ITO−ガラス。Nanocs Inc.より入手したガラス製の顕微鏡スライド上にコーティングされたインジウムすず酸化物(ITO)を、イソプロピルアルコールを含有するワイプで軽く摩擦することにより洗浄し、その後水の中で30分間超音波処理を行い、および/または酸素プラズマ処理を5分間行った。
【0027】
ITO−ポリエチレンテレフタレート。PET基材(CPFilms Inc.)を、60mlのポリプロピレン製チューブの内側にぴったりと納まる大きさにした。膜成長させる前に、当該基材の表面を、酸素プラズマを用いて、3分間処理した。
【0028】
上記基材表面処理は例示であり、表面処理される材料の範囲を限定する意図はない。本明細書の教示に関係する当業者は、本明細書に記載する方法で使用するために、その他の表面処理法または好適に処理されるその他の基材材料に置き換えることができる。
【実施例】
【0029】
ナノファイバー
(実施例1)
ポリチオフェンナノファイバーの合成。ポリチオフェンナノファイバーを作製するプロセスは、文献(例えば、非特許文献29を参照)で報告されている。この手順には、2つの溶液、すなわち1)アセトニトリル、10mlに溶解したFeCl3(0.333g、2.1×10-3モル)および2)1,2−ジクロロベンゼン、10mlに溶解したチオフェン(0.133ml、1.74×10-3モル)およびテルチオフェン(0.0065g、2.61×10-5モル)を調製するステップが含まれる。これらの2溶液を一緒にし、10秒間混合し、そして7日間静置した。次に、この反応溶液を遠心分離法を用いて精製した。
【0030】
(実施例2)
ポリチオフェンナノファイバー膜の成長。実施例1から得られたポリチオフェン導電性ポリマーナノファイバーを、より少ない水性相(約0.2mlから約5.0ml、好ましくは約1.5ml)およびより多くの有機層(約5.0mlから約30.0ml、好ましくは約18ml)から構成され、水/有機比が約1/10〜1/20、好ましくは約1/12となる二元非混和性溶液を用いて界面膜に形成した。この非対称の容積分配はマランゴニ流を引き起こす。例として、75mm×25mm×1mmのガラス製スライドを、下記のようにポリチオフェンナノファイバーでコーティングした:スライドを、60mlのポリプロピレン製チューブ(BD Falcon(商標)コニカルチューブ)内に配置し、アセトニトリルに分散したナノファイバー分散物(2g/L)を1ml、DI水を0.6ml、およびクロロベンゼンを10ml、当該チューブに添加した。激しく揺動した後、ポリプロピレン製容器を水平に寝かせ(長い壁を床と平行にする)、次に容器の長い方の端部が床と平行になりスライドが直立するまで回転させた。このようなスライド方向を規定するように容器を回転させれば、広がるポリマー膜にとって這い上がる距離がより短くなって基材全体を覆うことができ、したがって、縦横比が大きい基材でも完全に被覆可能である。膜成長期間中に容器を定期的にタッピングすることにより、バブル合体速度が高まり、膜成長が促進される。膜形成後、スライドを取り出し、当該膜を有機蒸気雰囲気内でゆっくりと乾燥する。
【0031】
(実施例3)
ポリアニリンナノファイバーの合成。ドーパントとして下記の酸を用いて、ポリアニリンナノファイバーを調製した:(a)塩酸、(b)p−トルエンスルホン酸、(c)カンファースルホン酸、および(d)過塩素酸。代表的な反応として、アニリン(0.16ml、1.75×10-3モル)をペルオキシ二硫酸アンモニウム(0.1002g、4.39×10-4モル)に溶解するステップ、および1MのHCl、8mlを添加するステップが含まれた(溶液A)。ダイマー開始剤、N−フェニル−1,4−フェニルジアミン(0.0032g、1.74×10-5モル)を、MeOH、1mlに溶解し、そして5分間超音波処理した(溶液C)。次に、溶液Aおよび溶液Cを混合したが、追加の1MのHCl、8mlと一緒にして溶液Bとする前に、平衡状態で5分間放置した。次に、容器を5秒間揺動させた。オーバーナイトで静置して重合を進めた。精製は、最終生成物をDI水に対して透析することにより実施され、部分的に脱ドープした材料が得られた。
【0032】
(実施例4)
ポリアニリンナノファイバー膜の成長。高密度ポリエチレン容器(60mlのNalgene(商標)広口型)を用いて、実施例3から得られた部分的にドープされたポリアニリンナノファイバーからなる水性コロイド分散物(4g/L)、1mlを、DI水、4mlと混合した。水性分散物を30秒間混合し、次にクロロベンゼン(またはクロロホルム)、6mlを添加し、そして容器を激しく揺動した。基材、例えば洗浄した顕微鏡用ガラス製スライド(Corning2947)を容器内に配置し、そして10秒間揺動した。容器が静止状態になり次第、ポリマー膜の成長が開始した。バブルを破壊し、膜成長を促進するために、容器の壁を定期的にタッピングした。様々な試験膜を基材上で成長させた。ポリアニリンナノファイバーからなる両面透明膜を分析用に選択した。膜の肉眼的な均質性およびナノスケール形態を保つために、周囲条件下でゆっくりと膜を乾燥させる必要があった。膜と基材との接着力は乾燥プロセス期間中に高まった;55℃で48時間さらに加熱すると、安定な膜、例えばサイクリックボルタンメトリー(図11)による特徴付けに供するのに十分堅牢な膜が得られる。一方、新規に形成された濡れた状態の膜は、水により基材から移すことができる。膜は、ドープした、または部分的に脱ドープしたナノファイバーのいずれか一方から作製可能である。入念にドープされたナノファイバーが、ポリマー溶液中で用いられる場合には、揺動すると安定なバブルが生成し、合体または膜成長が生じない。親水性であることから、ドープされたポリマーは、脱ドープされたポリマーよりも速くガラス壁を這い上がる。
【0033】
(実施例5)
ポリアニリンナノファイバー膜−分析。サイクリックボルタンメトリー。ΙΤO−ガラス基材上で成長したポリアニリンナノファイバー膜について、サイクリックボルタンメトリー(CV)を実施した。実施例4に記載されている方法を用いて、単層のナノファイバーを堆積させた。電気化学測定用としてITO上に膜を調製するプロトコールには、25℃で12時間、その後55℃で48時間、膜を乾燥ステップが含まれた。Princeton Applied Research社製のポテンシオスタット263Aを用いて、−0.2Vから+1.2Vまで、次に元の−0.2Vまでサイクルして、データを収集した。用いたスキャン速度は、50mV/秒であった。1MのHCl電解質溶液をアルゴンガスで30秒間パージし、そして電位を印加する前に20秒間平衡状態で放置した。補助電極として洗浄したPtワイヤーを用い、塩化カリウムで飽和したカロメル電極が参照電極として機能し、そして単層のポリアニリンナノファイバーで被覆された25mm×75mm×1mmのITOコーティングガラス製スライドが作用電極を構成した。ポテンシオスタットのリード線と接続するために、導電性の銅テープ(3M(登録商標))を、作用電極の末端に配置した。
【0034】
走査型電子顕微鏡。基材上で収集した様々な膜のナノスケール形態を、SEM(FEI Nova600)を用いて画像化した;しかるべき導電性を確保するために、最初にサンプルをプラチナ層でプラズマスパッタリングした。サンプルおよび装置の間で電気的回路を閉じるために、導電性の銅テープを用いた。
【0035】
紫外可視分光法。紫外可視における特徴付けを行うために、ポリアニリンナノファイバー単層をガラスおよび石英スライド上で成長させた。装置内で各スライドの位置が一定となるように保証する設計がなされたホルダーに基材を入れて、紫外可視分光光度計(Hewlett−Packard HP8453 Diode−Array)内に導入した。
【0036】
上記方法は、導電性ポリマーナノファイバーからなる透明な薄膜を成長させるための簡便かつ安価な溶液を提供する。低めの表面張力を有する流体(油)は、常に、表面張力がより高い流体(水)上に広がることが公知であるが(例えば、非特許文献30を参照)、出願者らは、油膜は、ガラス表面上に存在する水性の層全体にわたり、溶媒和した有機ナノ構造体を有効に担持し得ることを今回実証した。膜は、周囲条件において数秒内で堆積し、数分内に乾燥し、そして溶媒はリサイクル可能である。上記手順を用いれば、広い基材領域を均質かつ再現性のあるやり方でコーティング可能であり、薄膜の品質も高い。
【0037】
この方法の有用性は、電気伝導性有機ポリマーに限定されることはなく、その他のナノ材料またはナノ材料を組み合わせたものからなる膜を形成するのにも利用可能である。
【0038】
グラファイト酸化物膜およびグラフェン膜
カーボンナノ構造体からなる二次元(2D)シートは、ピッカリングエマルジョンにおいて安定剤として機能し、液/液界面で界面活性剤様の吸着特性および化学特性を有する。2D液/液界面は、平らなシートと幾何学的に類似し、したがって理想的な調和環境である。2Dカーボンシートで長さスケールが極端に異なると、縦横比が大きくなり、界面で熱力学的に有利な吸着を引き起こす。グラファイト酸化物は、水および油間の界面で分子状およびコロイド状の両方の界面活性剤として働く、単原子厚の(single−atomic−thick)両親媒性物質であり、界面張力を低減する。エマルジョンを構成する液滴が合体すると、その後生じた指向性の流体は、グラファイト酸化物シートが広い領域上の界面で広がるのを促進する。改変されたHummerの方法(例えば、非特許文献31を参照)により生成されたグラファイト酸化物シートは、クロロベンゼンを併用してMilli−Q水中に分散され、そして均質な薄膜(図67〜69)に処理される。一般的に、グラファイト酸化物シートからなる0.2mg/mLの水性分散物が、クロロベンゼンを用いて1:4の比で30秒間超音波処理することにより乳化される。容器を手動により撹拌し、静置した後、数秒以内に、透明な淡黄色に着色した膜が、ガラス製のスライドをコーティングする。グラファイト酸化物膜の堆積は、膜の成長がpH依存性であるため、中性に近いpHで実施される。末端の−COOH基の脱プロトン化が生じてグラファイト酸化物がより親水性になり、またエマルジョンの合体が生じてグラファイト酸化物が水性相に排除されるので、広がることは高pHでは生じない。
【0039】
グラフェンは、ハニカム結晶格子内に稠密充填されたsp2結合型の炭素原子からなる、単原子厚の平面状シートである。グラフェンシートは、超音波処理の支援を受けてヒドラジン中に分散される。少なくとも20分間、好ましくは約2時間超音波処理すると、シートサイズは減少する可能性がある。超音波処理時間が長くなるほど、シート寸法の減少は大きくなる。高度還元グラファイト酸化物およびグラフェンの透明薄膜を生成するために、ヒドラジン分散物を、水酸化アンモニウム水溶液と混合した。部分的な酸化が生ずるが、グラフェンシートの一部は溶液中に留まる。次に、グラフェンを含有する希薄なヒドラジン分散物が用いられる場合には、単層のグラフェンシートを含有する基材上の透明薄膜が、上記プロセスを用いて得られた。基材上に堆積される材料の量を、膜を成長させるのに用いられる水性分散物中に存在するカーボン材料の濃度を変化させることにより制御した。顕微鏡用のガラス製スライドを基材として用い、そして異なる濃度の分散物からなる透明薄膜を作製した(図16〜23)。シート抵抗が23kΩの、高度還元グラファイト酸化物およびグラフェンからなる膜を、0.25mg/mlの水性分散物を用いて成長させた。
【0040】
(実施例6)
60mlの高密度ポリエチレン容器を用いた。一般的なプロセスとして、グラフェン(1〜10mg/ml)を含有するヒドラジン分散物を、数分から数時間、超音波処理した。一般的に、グラフェン分散物(1mg/ml)、0.4mlを、14wt%の水酸化アンモニウム水溶液、4〜5ml中で超音波処理した。次に、クロロベンゼン等の有機溶媒、8〜12mlを超音波処理したグラフェンに添加し、当該溶液をさらに超音波処理した。次に、ポリアニリンナノファイバー膜を作製するために上記と同じ揺動および静置プロセスにより膜を作製した。
【0041】
20mlシンチレーションバイアル、およびヒドラジン溶液(2〜3ml)中で、1〜5mg/ml(好ましくは1.0mg/ml)のグラフェン/mlを含有するグラフェン分散物、0.1〜0.5ml(好ましくは0.4ml)、14wt%の水酸化アンモニウム水溶液、および有機溶媒(クロロベンゼン、クロロホルム、四塩化炭素、トルエン、またはベンゼン)、2〜4ml(好ましくは4ml)を含む混合物を用いて、膜をシリコン基材上に堆積させた。超音波処理は、ナノ構造体の分散および均質な膜の生成、ならびにシートのより小さなサイズへの破壊において、これらを助ける。基材上に膜を堆積させる前に、基材を酸素プラズマで5分間処理した。図16は、ヒドラジン中に分散したグラフェン分散物(2mg/ml)、0.5mlから形成された膜を示し、図17は、グラフェン分散物、0.1mlから形成された膜を示す。
【0042】
シート間の結合性により、導電性のネットワークが実現する。この材料からなる透明な膜は、上記プロセスを用いて、石英およびガラス製のスライド上で得られる。高度還元グラファイト酸化物およびグラフェンシートを、超音波処理により塩基性の水性媒体中に分散した。グラフェンからなるより大きなシートを、超音波処理曝露時間を約0.5分まで短縮することにより作製した。次に、グラフェンシートのヒドラジン分散物を14wt%NH4OH水溶液と混合することにより、膜を収集した。ピッカリングエマルジョンを形成するために、有機相としてクロロベンゼンを用いた。水酸化アンモニウム溶液の代わりに脱イオン水を用いる場合には、基材領域の被覆率は、膜が這い上がる最大高さと共に低下する。図18は、シートが端部を共有している膜の平面図である。図19は、52°傾いた同一膜のSEMによる画像である。
【0043】
図20〜23を参照すると、グラフェンからなる単層シートを含有する膜を、希薄濃度のグラフェンを用いてヒドラジン(1mg/ml)中で堆積させた。最初にグラフェンをヒドラジン中で完全に還元し、その後14wt%の水酸化アンモニウム水溶液と一緒にした。クロロベンゼンと共にグラフェンを含有する塩基性のNH4OH水性分散物を混合することにより膜を得、バイアルを激しく揺動させ、次に静置して膜成長を開始した。他の溶媒、例えば四塩化炭素、クロロホルム、トルエン、およびベンゼン等は、クロロベンゼンの代用とすることができる。図20〜23の各図中に示す長方形は、単層グラフェンシートを表す。
【0044】
カーボンナノチューブ
単層カーボンナノチューブ(SWCNT)(Carbon Solutions Inc.)をカルボン酸およびヒドロキシル基により官能化した。
【0045】
(実施例7)
ガラス製スライド(75mm×25mm×1mm)上でのSWCNT膜の成長
単層カーボンナノチューブ(SWCNT(Carbon Solutions Inc.)、0.0011g)を、20mlのガラス製シンチレーションバイアル内で水、4mlと共に混合し、そして15分間超音波処理を行った。次に、クロロベンゼン、11mlを添加し、その後さらに15分間超音波処理を行った。濃HCl、3滴を添加、混合し、そして当該溶液を60mlのプロピレン製容器(BD Falcon(商標)チューブ)に移した。ガラス製スライド(Corning 29470)を、イソプロピルアルコールに浸漬したKimtech(登録商標)ワイプで洗浄し、圧縮空気で乾燥し、そして容器内に配置した。撹拌および静置を反復して実施した。約5分後に最高品質の膜を得た。
【0046】
(実施例8)
シリコン(49mm×10mm×1mm)上でのSWCNT膜の成長
SWCNT、0.1mgを、20mlのガラス製シンチレーションバイアル内で、脱イオン水、2mlと混合し、そして15分間超音波処理を行った。次に、クロロベンゼン、5mlを添加し、その後さらに15分間超音波処理を行った。次に、濃HCl、3滴を添加し、当該混合物を揺動させた。当該溶液は、約5分内に高品質の膜を生成した。酸を使用すると凝集を引き起こすのが認められた。
【0047】
(実施例9)
ガラス製スライド上のSWCNT片面膜
膜を、Falconチューブ、水、およびクロロベンゼンを用いて収集した。各成分を添加して混合物となし、その後15分間超音波処理を行った。図24は、ガラス製スライド上のカーボンナノファイバーからなる膜を概略的に表し、異なる膜密度を表すために異なる網点が施されており、
下方のスライドはコーティングされていないスライドブランクであり、
次は、SWCNTを0.0058g、水を6ml、および有機性の油を15ml用いて形成された膜で被覆されたガラス製スライドであり、
3番目の画像は、SWCNTを0.0027g、水を4ml、および有機性の油を11ml用いて形成された膜で被覆されたガラス製スライドであるが、当該混合物の撹拌後、基材上に膜を形成する前に当該混合物には、濃縮された酸が5滴添加されており、
および上方の画像は、SWCNTを0.0013g、10%エタノール水溶液を3ml、有機性の油を9ml、および濃HClを4滴用いて、ガラス製スライド上に形成された膜を表し、有機性の油はいずれの場合もクロロベンゼンであった。他のアルコールもエタノールの代用とし得る。
【0048】
【表1】
【0049】
基材に堆積される固形物の質量は、膜の透明度と反比例の関係を有する。異なる濃度のSWCNT分散物を用いることにより、ある範囲の透明度で膜が生成する。0.01mg/mLおよび0.1mg/mLの水性分散物から、95%および90%の透明度を有する膜が得られる。2%エタノールを添加すると、透過率70%の膜が得られる。エタノールは、SWCNTの表面電荷を下げ、またその界面エネルギーを低下させて、SWCNTが液/液界面で会合することを可能にする。透過率90%の膜は、1kΩのシート抵抗を有する。
【0050】
生成後に、300℃で12時間アニーリングを行うことにより、整列したSWCNTからなる膜の充填密度を制御することができ、十分に分離したカーボンロープ、および基材へのより強固な膜接着力が得られる。あるいは、水性分散物の混合プロトコールも充填密度を制御する。0.1mg/mLの水性分散物を用いて、標準的な超音波バス内で2時間の延長した超音波処理を行うと、十分に分離したカーボンロープ、および充填密度の低い整列したSWCNTからなるコーティングが得られる。
【0051】
ラマン分光法は、膜の高さ軸に沿って低から高シグナル強度勾配を示す(図60)。界面における濃度勾配の広がりは、質量の異方性分布を引き起こし、またこの広がりは、基材の領域が高くなるほど、強度勾配は強いシグナルを示す理由を説明する。
【0052】
図25〜28はSWCNT膜の顕微鏡写真である。
【0053】
図25は、水、2mlおよびクロロベンゼン、6ml中のSWCNT、0.0005gを用いて収集した、シリコン基材上の膜を示す。図25は、上記のようにシリコン基材上で成長させた単層カーボンナノチューブ膜のSEM画像である。この膜は、カーボンナノチューブロープの配列状態を示す。基材がロープの間に認められ、このような膜に典型的な多孔性の形態を示している。ロープの直径は、超音波処理の程度により制御可能である。SEM画像は、30分間超音波処理されたSWCNT分散物から形成された膜を示し、サンプルは52°傾いている。
【0054】
図26は、図25と同一の方式で形成され、15分間超音波処理した別の膜を2.5倍拡大したSEM画像である。より高い倍率では、カーボンナノチューブのロープは、超音波処理がより少ないため、図25のロープのように十分に分散されていないことが明らかである。このSEM画像は、顕微鏡に対して垂直に配置されたサンプルを用いて収集された。
【0055】
図27は、シリコン基材上で成長させた単層カーボンナノチューブ膜のSEM画像である(目盛りは1マイクロメートル)。膜の堆積で酸を用いたので、からみ合い(カーボンナノチューブロープの凝集物)が存在するが、これはより淡い色のついた外観を有する。酸はカーボンナノチューブをプロトン化し、そしてより高度の水素結合を引き起こすので、使用する酸の濃度は、凝集物の形成に直接的な影響を有する。このSEM画像は、顕微鏡に対して垂直に配置されたサンプルを用いて収集された。
【0056】
図28は、シリコン基材上で成長させた単層カーボンナノチューブ膜のSEM画像である。SWCNTの水性分散物を、45分間超音波処理することにより、十分に分散された形態が得られるが、これにより、高度に整列したカーボンナノチューブロープが認められる。このSEM画像はサンプルを52°傾けて得られた。
【0057】
図29〜31に示す膜は、脱イオン水、2mlにSWCNT、0.1〜1.0mgを添加し、20mlのガラスシンチレーションバイアル中で混合および超音波処理を10分間行った水性分散物から調製された。次に、クロロベンゼン(3〜6ml)、(好ましくは5ml)を添加し、そして当該溶液をさらに10分間超音波処理した。超音波処理プロセス全体を通じて、バイアルを反復して揺動させた。次に、当該溶液を静置した。酸素プラズマ中で5分間事前処理されたシリコン基材上で、静置した溶液から膜を形成した。次に、当該膜を、バイアル中でクロロベンゼン蒸気に曝露して10分間乾燥させ、バイアルから取り出し、次に周囲条件で2時間さらに乾燥させた。図29〜31は、濃度1mg/mlのSWCNTから形成された膜の様々な倍率における顕微鏡写真である。
【0058】
希釈および高度に精製された水性分散物からキャスティングすると、個々のSWCNTが、膜として堆積する。体積としてヘキサフルオロイソプロパノールを30%含有するSWCNTからなる5mg/mLの水性分散物を、氷浴中でホーンチップを用いて、100%出力で1時間、超音波処理した。112×gで30分間遠心分離を行い、上清上部を分離し、脱イオン水を用いて50%に希釈し、そして延長して超音波処理を行うと、精製された安定な分散物が生成する。高度に希釈された、透明なSWCNT水性分散物を得るために、この精製プロセスを4回反復した。浸漬型ホーンチップを用いながら延長して超音波処理を行うことにより、1mLのアリコートと4mLのクロロホルムが混合され、ピッカリングエマルジョンの混合物が分散するようになる。
【0059】
図62〜64を参照すると、膜の堆積は、容器40内で成分を乳化するのに超音波処理チップ38を用いることにより、自動化可能である。コーティングの対象となる濡れた基材44の端部42のみが、乳化組成物46内に配置される。超音波エネルギーが休止すると、濡れた基材44上で合体および膜48の成長が進行する。この手順により、十分に分離したSWCNTロープおよび個別のカーボンナノチューブからなる薄膜が生成する。
【0060】
複合膜
図32および33では、グラフェン膜を成長させる上記技術と同一の技術を用いて生成した膜を示す。図32は、膜中に存在する非結晶性の炭素を示し、おそらくこれは、ヒドラジンおよび超音波処理に起因する。走査型電子顕微鏡を用いて加速電圧(18.00KV)を高めることにより、膜表面のアニーリング(図32の暗くなった中央部分)を行ったが、このアニーリングにより、非結晶性の炭素が消失し、一方カーボンナノチューブが残る。図33は、図32のアニーリングされた領域を示す拡大画像であり、カーボンナノチューブのネットワークが非結晶性炭素層の下に認められる。
【0061】
図34、35、36、および37は、膜成長前に延長した超音波処理に曝露した後に生成した、SWCNT−グラフェン複合物からなる膜のSEM画像であり、当該膜はシリコン基材上に形成されている。両方の材料を、ヒドラジン中で混合、分散し、そして膜成長前に超音波処理する。画像は、超音波処理を延長して行った(少なくとも約20分間)後に認められるグラフェンシートサイズの減少を説明する。複合物からなる膜は、上記で議論したグラフェン膜を成長させるのに用いられる技術と同一の技術により得られる。図34および35は、低濃度のSWCNT(0.1mg/ml)から形成された膜を示す。図36は、高めの濃度(1.0mg/ml)の、高度還元グラファイト酸化物およびグラフェンヒドラジン分散物から形成された膜を説明し、高密度ナノ構造ネットワークを形成する。図37は、カーボンナノチューブにより相互に連結した、高度還元グラファイト酸化物およびグラフェンシートからなる高密度ネットワークを形成するために、ヒドラジン分散物中のSWCNTの濃度(2.0mg/ml)を高めて形成した膜を表す。
【0062】
図38、39、40、および41は、膜成長前に約15分間超音波処理し、そしてシリコン基材上で収集した、SWCNT−グラフェン複合物からなる膜のSEM画像である。
【0063】
図38〜41の膜は、5mg/mlのグラフェンおよびSWCNTからなるヒドラジン分散物、0.15mlと、14wt%のNH4OH溶液、2mlを混合することにより生成した。この溶液を60秒間超音波処理した。次にクロロベンゼン(3〜4ml)を添加し、そして溶液を揺動させ、バイアルを静置した後に膜を基材上に形成した。
【0064】
図38〜41は、同一の手順を用いて調製された4つの異なる膜の例であり、図39〜41は高倍率である。
【0065】
図42、43、44、および45は、本明細書に記載するプロセスを用いてシリコン基材上で収集した、ポリアニリンナノファイバーグラフェン複合物からなる膜のSEM画像である。グラフェン膜を、上記のように実施例6の方法を用いて最初に作製した。この膜を、乾燥するために1時間放置し、次をドープしたポリアニリンナノファイバー分散物を、予め成長させたグラフェン膜の上部にポリアニリンナノファイバー膜を成長させるために用いた。図42および43は、水酸化アンモニウム蒸気に曝露した後の膜を示し、ポリアニリンナノファイバーは、水酸化アンモニウム曝露に起因して脱ドープされる。図43は図42の高倍率画像である。図44および45は、高度還元グラファイト酸化物およびグラフェンシート最上部に位置するドープされたポリアニリンナノファイバーからなる膜を、2つの異なる倍率で示す(図44は図45の2倍)。
【0066】
図46〜50は、シリコン基材上で収集した、ポリアニリンナノファイバーSWCNT複合物からなる膜のSEM画像であり、これら5つの画像は異なる倍率での膜を示している(参照用に各画像の下側右隅に寸法線を示す)。過塩素酸をドープしたポリアニリンナノファイバーからなる水性分散物(4g/L)、0.4mlをSWCNT水性分散物に添加したことを除き、上記方法と同一の方法を用いてこれらの膜を成長させた。塩基は用いず、またSWCNTはヒドラジン中で事前分散せずに、固体状態から直接水と混合される。クロロベンゼンおよびSi基材を用いて膜成長を行う。ポリアニリンナノファイバーと絡み合う、束状のSWCNTからなるカーボンロープを示す。
【0067】
同様に、有機溶媒がヘキサンまたはヘプタン等のアルカンである場合を除き、ポリチオフェン膜を作製するのと同一の方法を用いて、基材上にポリ(3−ヘキシルチオフェン)ナノファイバーからなる透明な膜を成長させた。図51〜53は、本明細書に記載する方法を用いて二酸化ケイ素基材上に形成した、ポリ(3−ヘキシルチオフェン)ナノファイバー膜の3つの異なる倍率におけるSEM画像である。図53は、図52の中央部分を拡大した画像であり、図52は、図51の中央部分を拡大した画像である。
【0068】
活性化されていない疎水表面も、例えば図54〜59または62〜64の手順、および極性反対の非混和性の溶媒からなる二元混合物を用いて、透明で導電的に連続的な膜でコーティング可能である。この二元溶媒系が固体と接触すると、いわゆる選択的濡れで、固体表面の流体が別の液体に自発的に置き換わる。表面エネルギーが低い固体上に薄膜を堆積させる場合、かかる堆積では表面張力が極めて低い溶媒を必要とする。例として、水およびフルオロカーボンを混合すると、選択的濡れが生じ、そして活性化されていない疎水表面上で膜が成長する。表面張力が低い液体であるフルオロカーボンは、プラスチックを濡らし、ポリプロピレン膜キャパシタに充填され、そしてポリエステル繊維に撥水性を付与する。フルオロカーボン分子の間の凝集力は極めて低く、プラスチックの完全な濡れを実現する。フルオロカーボンの低い表面張力は、フッ素原子の低い分極率に起因し、表面張力が高い(72.8mN/m)極性液体である水と混ざらない。フルオロカーボン、例えばパーフルオロ−2−ブチルテトラヒドロフラン、パーフルオロ(メチルシクロヘキサン)、Fluorinert(登録商標)FC−40(16mN/m)、Fluorinert(登録商標)FC−70(18mN/m)、およびFluorinert(登録商標)FC−77(15mN/m)(Fluorinert(登録商標)は3Mの商標である)等を、本手順で採用した。
【0069】
水、フルオロカーボン、およびカーボンナノチューブを激しく撹拌すると液滴が生ずる。活性化されていない疎水性基材と接触すると、フルオロカーボンは、表面由来の水と置換して選択的濡れを引き起こす。液滴の合体は、溶媒の間で表面張力が極度に相違する結果、非常に活発である。両方の溶媒により部分的にコーティングされたカーボンナノチューブは、液滴の外に速やかに排除され、そして基材上に存在する水/フルオロカーボン界面に吸着する。吸着は、SWCNTおよびフルオロカーボン間の疎水的相互作用により強化される。界面間の広がりは、系の総界面表面エネルギーを最低限に抑え、そしてSWCNTの吸着および堆積を引き起こす。
【0070】
カーボンナノ構造体からなる高品質の透明な膜は、複数の液滴からなるペルフルオロ化されたエマルジョン内に浸漬された後、適する基材をコーティングする。基材が液滴と接触した状態に留まる時間は、吸着する炭素の量を決定する。時間が増加すると、吸着した固形物の濃度が高まり、また基材の濡れ性を疎水性から親水性に変化させる。カーボンナノチューブからなる高密度の膜が基材をコーティングする場合、表面エネルギーが変化し、基材は親水性表面のように振る舞い、水に濡れる。したがって、SWCNTの高密度コーティングが吸着した後に、親水性表面上で一般的に認められる垂直方向への膜の広がりが誘発されうる。
【0071】
SWCNTからなる大型で透明な導電性膜の堆積は、活性化されていない疎水性の可撓性基材、例えばポリエステル基材の22×22cm2の領域を最初にコーティングすることにより実現可能である。ホーン型超音波発生装置を用いて、Fluorinert FC−40、400mL、およびSWCNTからなる0.05mg/mLの水性分散物、200mLを混合することにより、コーティングエマルジョンを作製する。超音波発生装置を100%出力に設定し、そして氷浴中で2時間混合する。次に、基材およびカーボンエマルジョンを、ぴったり納まる容器内に収納し、そして手動により10分間激しくかき混ぜた。コーティングした後、配向したポリエステル基材(Grafix(登録商標)Plastics)を収納容器から取り出し、周囲条件で乾燥した。フルオロカーボンは表面からきれいに蒸発し、残留物はまったく残らない。透明度が90%を上回り、シート抵抗が1kΩの両面膜を、本手順を用いて作製した。
【0072】
活性化されていないプラスチック上のSWCNTからなる透明な膜は、SWCNTからなる0.1mg/mLの水性分散物、2mL、およびペルフルオロ化された炭化水素、例えばFluorinert FC−40、8mLを組み合わせることにより光学的に透明なビニル製スライド上に堆積可能である。次に、ぴったり納まる容器内で、手動により成分を1分間かき混ぜることにより、乳化を実施した。コーティングされたスライドを溶液から取り出し、過剰の吸着物を除去するために水で洗浄し、そして周囲条件で乾燥放置した。図61は、透明度が98%でシート抵抗が1ΜΩの、導電的に連続な膜の特性を示すグラフである。プラスチック基材の端部は、均質にコーティングされており、また均一なシート抵抗を有するが、中心部は2桁高い抵抗を有する。この均一性の欠如は、5分後の抵抗の安定性に改善が認められることから実証されるように、1分よりも長くかき混ぜさえすれば改善される。基材および合体した液滴間の接触は、SWCNTの吸着を引き起こし、長く接触するほど、膜のバルク均一性が増す。7分間撹拌すると、コーティングされた全表面領域にわたり、90%の透明度と1kΩのシート抵抗が得られる。
【0073】
別の例として、過塩素酸をドープしたポリアニリンナノファイバーからなる透明な膜を、配向したポリエステル基材(10.2cm×8.4cm×0.0254cm)上に堆積させたが、これには、水性ポリマー分散物[4g/L]を6mL、水を3mL、およびペルフルオロ化された液、例えばFluorinert FC−40(登録商標)を60mL用いて指向性流体をコーティングした。すべての薬品を、250mLのガラス製広口ジャー内で混合し、また激しくかき混ぜ、次に洗浄した疎水性基材をガラス製ジャーの液/液界面に導入した。次に、この構成物を激しくかき混ぜると、緑色の膜がプラスチック基材上に速やかに形成された。1分間撹拌した後、コーティングされた緑色に着色した基材を取り出し、水で洗浄し、そして周囲条件で乾燥放置すると、連続的で導電性の膜が生成した。
【0074】
界面において吸着したナノファイバーの表面自由エネルギーおよび基材間の分子相互作用は、膜形態を規定し得る。過塩素酸をドープしたポリアニリンは、単層ナノファイバーの平均厚さを有する膜を形成する。これは、例えば連続的な膜の形態で会合したナノファイバーの拡大画像を表す図8で明らかなように、油層および水層間でサンドイッチされたときに、ナノファイバーが界面において押し出されるために生ずる。また、ポリアニリンナノファイバーからなる単層の単層も、ドーパント、例えばカンファースルホン酸またはp−トルエンスルホン酸等を用いれば堆積可能である。膜は、最大3Scm-1の伝導度を有し、PDMSスタンプを用いてパターン化され得る。
【0075】
部分的に濡れた膜を、親水性表面上に存在する空気/水界面から、液体リザーバー内に存在する空気/水界面に移動することにより、基材を有さない膜を作製することができる。膜内の濡れ具合を制御することにより、空気/水界面で層間剥離が実現する。堆積後5分間、容器のフタを閉める状態に保つことにより、ガラス製スライド上のSWCNTからなる膜をゆっくりと乾燥放置した。膜を、1MのHCl水溶液を用いて層間剥離する前に、周囲条件下で1分間乾燥させた。−COOH官能基がプロトン化すると、カーボンナノチューブ間の水素結合および2Dの稠密充填が引き起こされ、層間剥離した浮遊膜は圧縮を必要としない。膜は、膜構造を含む炭素両親媒性物質が有する凝集性の分子相互作用により、完全な1片として残る。単層のSWCNT膜を製造し、また層間剥離を行うための従来法は、高分子分散剤、例えばポリ(3−ヘキシルチオフェン)、ヒドラジン処理、および3時間プロセスを必要とした。本明細書に記載する手順を用いれば、高分子分散剤を用いずに、数分のうちに自立SWCNT膜が生成する。膜全体が、空気/水界面で単層片として層間剥離し、そして均質な自立膜は、数日間浮遊した状態で残る。
【0076】
SWCNTからなる自立膜をガラスからSiO2に移動するとき、膜の形態は、マイクロメートルスケールで整列した状態に残る。自立膜は、電気的に連続的であり、また水面からこれをすくい上げることにより、任意のタイプの基材に移すことができる。酸性媒体上で層間剥離する際に、膜は、−COOH官能基のプロトン化により収縮し、また充填密度は、より強い凝集相互作用により増加する。逐次積層堆積法は、複数の層をすくい上げ、既に堆積された膜上に別の膜を堆積させる前に、それぞれを100℃で4時間アニーリングすることにより実施される。
【0077】
自立層を層間剥離し、移動することにより調製される、多層化SWCNT膜のオプトエレクトロニック特性を図65および66に示す。単層の移動層は、500kΩのシート抵抗を有し(図65)、また透明度は82%を上回る。2番目の層を堆積させると、シート抵抗は1桁下がり、また浸透閾値が規定される。3層以上重ねても、シート抵抗に対する効果はより小さい。2層以上重ねた後には、曲線の正の傾きがより小さくなることから実証されるように、膜の電気的安定性は高まる。各層は、平均10吸光度単位ずつ透明度を低下させる(図66)。この高い光学濃度は、1Mの酸で層間剥離する際に、膜の収縮および充填密度の増加が引き起こされてSWCNTが圧縮されることに起因する。層間剥離媒体を5%エタノール溶液に置き換えると、より高い透明度が得られる。
【0078】
当業者は、上記の記載内容および実施例に基づき、本発明は、前記代表的実施例により限定されず、また本発明の変形形態も本発明の範囲内であることを認識するであろう。例えば、様々な異なる材料を含む様々なナノファイバーおよびナノ構造体が開示される。しかし、本明細書に開示される方法は、その他のナノ構造体およびその他の材料、例えばデオキシリボ核酸、およびポリ(3,4−エチレンジオキシチオフェン)等のその他のチオフェンを含む様々なナノ形態のチオフェン、ならびにポリスチレンナノビーズ、およびその他のナノ形態のカーボン、例えばカーボンナノスクロールまたはカーボンブラックナノ粒子等への適用も検討対象である。そしてさらに、非常に多くの非混和性で有機性の液体、例えばニトロメタン、カーボンジスルフィド、ペルフルオロ化された炭化水素、例えばFluorinate(登録商標)FC−40、FC−75、およびFC−77等、酢酸エチル、ジメチルホルムアミド、ジエチルエーテル、様々なハロゲン化された炭化水素、例えばジクロロメタン、ジクロロエタン、およびテトラクロロエチレン等、ベンゼンおよびトルエン、ならびにハロゲン化された芳香族、例えばハロゲン化されたベンゼンまたはトルエン、例えばクロロ−、ジクロロ−、およびトリクロロ−ベンゼン等を含む、ただしこれらに限定されない様々な芳香族炭化水素が利用可能である。ある特定のナノ材料、または水性相もしくは有機相で用いられる化合物の開示が存在しないからといって、当該材料または液体の使用が除外されるとみなしてはならず、その使用はまだ評価されていないということを示唆するにすぎない。
【0079】
当業者は、非常に多くの代替基材、例えばマイカ、アルミフォイルまたは銅フォイル等の金属フォイル、ならびにビニル、ポリ塩化ビニル、ポリエチレン、およびポリエステルフィルム(例えば、Mylar(登録商標))を含む、ただしこれらに限定されない広範囲のポリマーシート材料も利用可能であることを認識するであろう。特定の基材または開示された基材のための表面処理法の開示が存在しないからといって、当該基材または表面処理法の使用が除外されるとみなしてはならず、その使用はまだ評価されていないということを示唆するにすぎない。さらに、上記記載内容は、プラズマにより活性化された疎水性基材の使用を開示するが、有機相を適切に選択すれば、活性化されていない疎水基材も利用可能である。特に、非混和性で有機性の液体がペルフルオロ化炭化水素であれば、開示された方法を用いて、疎水性基材上で膜を成長させることができる。そしてさらに、長方形の基材の使用が開示されるが、本方法の有用性は、基材の幾何学的形状に限定されるものではなく、その他の形状(四角形、三角形、円形または楕円形のディスク等)は、球等の三次元の基材を含め利用可能である。
【0080】
さらに、処理時間、液体の体積、および様々な成分の比は、代表例にすぎず、また現在のしかるべき好ましい操作条件を開示しているのであって、これらは、利用可能な様々な液体、ナノ材料、基材、および処理容器についてプロセスを最適化するために変更可能である。そしてさらに、上記手順は、水溶液の調整法を開示する。当業者は、様々な異なる酸または塩基が利用可能であることを認識するであろう。例えば、pHを調整するのに適する酸および塩基として、塩酸、過塩素酸、リン酸、ヒアルロン酸、硫酸、ポリスチレンスルホン酸、カンファースルホン酸、トルエンスルホン酸、ドデシルベンゼンスルホン酸を含むスルホン酸、その他の有機硫酸塩、樟脳酸、硝酸、酢酸、クエン酸、ヒドラジンおよび様々なヒドロキシル化合物、例えばアンモニウム、ナトリウム、カルシウム、リチウム、およびカリウムの水酸化物等が挙げられるが、ただしこれらに限定されない。pH調整に用いられる特定の酸または塩基の開示が存在しないからといって、当該材料の使用が除外されるとみなしてはならず、その使用はまだ評価されていないということを示唆するにすぎない。
【特許請求の範囲】
【請求項1】
ナノ材料の膜を形成する方法であって、
容器のなかで、水性の液体、非混和性で有機性の液体、および前記ナノ材料からなる混合物を調製し、前記混合物のエマルジョンを形成するするステップと、
基材を前記エマルジョン内に入れるステップと、
前記エマルジョンを分離させて、水性の液相および有機性の液相の間に界面を形成させ、前記基材を前記エマルジョン内に配置し、形成する界面と交差させるステップであって、
前記エマルジョンが分離する際に、前記ナノ材料は基材表面上に堆積し、その表面に沿って広がることにより、前記基材表面上に膜を形成する前記ステップと、
濡れた膜を水性の液体に浸漬して、前記基材から分離した連続性のナノ材料膜を得るステップ、または
前記基材表面上で前記濡れた膜を乾燥して、前記基材上にナノ材料膜コーティングを得るステップと
を含むことを特徴とする方法。
【請求項2】
前記エマルジョンは、前記混合物を激しく混合することにより形成され、前記混合は、前記混合物の揺動、前記混合物の超音波エネルギーへの曝露、または揺動および超音波エネルギーの併用を含むことを特徴とする請求項1に記載の方法。
【請求項3】
前記混合または超音波エネルギーへの曝露は、少なくとも約30秒間であることを特徴とする請求項2に記載の方法。
【請求項4】
前記ナノ材料は、ポリアニリン、ドープしたポリアニリンまたはポリチオフェン、ポリ(3−ヘキシルチオフェン)、ポリ(3,4−エチレンジオキシチオフェン)ナノファイバー、グラフェンまたはグラファイト酸化物シート、カーボンナノチューブ、カーボンナノスクロール、カーボンブラックナノ粒子、ポリスチレンナノスフェア、デオキシリボ核酸、またはこれらの混合物を含むことを特徴とする請求項1に記載の方法。
【請求項5】
前記非混和性で有機性の液体は、四塩化炭素、クロロホルム、塩化メチレン、ベンゼン、ハロゲン化されたベンゼン、ペルフルオロ化された炭化水素、ニトロメタン、カーボンジスルフィド、トルエン、テトラクロロエチレン、酢酸エチル、ジメチルホルムアミド、ジエチルエーテル、1つもしくは複数のアルカンもしくはハロゲン化アルカン、またはこれらの混合物であることを特徴とする請求項1に記載の方法。
【請求項6】
前記基材は、ガラス、ITOでコーティングされたガラス、シリコン、二酸化ケイ素、石英、マイカ、金属フォイル、またはプラスチック製基材であることを特徴とする請求項1に記載の方法。
【請求項7】
前記プラスチック製基材は、ITO−ポリエチレンテレフタレート、ビニル、ポリ塩化ビニル、ポリエステル、ポリエチレンを含むことを特徴とする請求項6に記載の方法。
【請求項8】
前記基材表面は、親水性であることを特徴とする請求項1に記載の方法。
【請求項9】
前記基材表面は、親水性となるように活性化されることを特徴とする請求項1に記載の方法。
【請求項10】
前記基材表面は、疎水性であり、アルゴン−酸素プラズマに曝露することにより活性化されることを特徴とする請求項9に記載の方法。
【請求項11】
前記基材表面は、疎水性であり、前記ナノ材料は、極性反対の非混和性溶媒からなる二元混合物から堆積されることを特徴とする請求項1に記載の方法。
【請求項12】
前記基材上に形成される前記膜は、膜を着色する着色した添加物または反応物質の添加により着色されることを特徴とする請求項1に記載の方法。
【請求項13】
前記ナノ材料は、塩酸をドープした、トルエンスルホン酸をドープした、ポリスチレンスルホン酸をドープした、過塩素酸をドープした、カンファースルホン酸をドープした、若しくは脱ドープされたポリアニリンナノファイバー、または塩化物をドープしたポリチオフェンであることを特徴とする請求項1に記載の方法。
【請求項14】
前記基材上の前記膜は、周囲環境条件下で少なくとも約5分間乾燥されることを特徴とする請求項1に記載の方法。
【請求項15】
前記基材は、2つの長い端部および2つの短い端部を有する長方形の形状であり、前記長い端部が、前記形成する界面と平行であり、前記形成する界面の両側にあることを特徴とする請求項1に記載の方法。
【請求項16】
前記水性の液体は、水、pHが調整された水溶液、アセトニトリル水溶液、ヒドラジン、またはアルコール溶液であることを特徴とする請求項1に記載の方法。
【請求項17】
前記pHは、塩酸、過塩素酸、硫酸、ポリスチレンスルホン酸、樟脳酸、カンファースルホン酸、トルエンスルホン酸、ドデシルベンゼンスルホン酸、硝酸、酢酸、クエン酸、リン酸、ヒアルロン酸、水酸化アンモニウム、ヒドラジン、ナトリウム、カルシウム、カリウム、またはリチウムの水酸化物、または重炭酸ナトリウムを用いて調整されることを特徴とする請求項16に記載の方法。
【請求項18】
前記膜は、周囲環境条件下で乾燥される前に、前記容器内の前記非混和性で有機性の液体上の気相中で最初に乾燥されることを特徴とする請求項42に記載の方法。
【請求項19】
前記有機相は、前記水性相より大きい体積を有することを特徴とする請求項1に記載の方法。
【請求項20】
前記水性相は、約0.2から約5mlであり、前記有機相は、約5mlから約30mlであることを特徴とする請求項19に記載の方法。
【請求項21】
前記有機相は、前記水性相の体積の約3から約20倍の体積を有することを特徴とする請求項119に記載の方法。
【請求項22】
前記有機相は、前記水性相の体積の10倍から約20倍の体積を有することを特徴とする請求項19に記載の方法。
【請求項23】
前記基材表面は、疎水性であり、それおよび前記有機性の液体は、パーフルオロカーボンであることを特徴とする請求項6または11に記載の方法。
【請求項24】
前記基材上の前記膜は、周囲環境条件下で最長約2時間乾燥されることを特徴とする請求項114に記載の方法。
【請求項1】
ナノ材料の膜を形成する方法であって、
容器のなかで、水性の液体、非混和性で有機性の液体、および前記ナノ材料からなる混合物を調製し、前記混合物のエマルジョンを形成するするステップと、
基材を前記エマルジョン内に入れるステップと、
前記エマルジョンを分離させて、水性の液相および有機性の液相の間に界面を形成させ、前記基材を前記エマルジョン内に配置し、形成する界面と交差させるステップであって、
前記エマルジョンが分離する際に、前記ナノ材料は基材表面上に堆積し、その表面に沿って広がることにより、前記基材表面上に膜を形成する前記ステップと、
濡れた膜を水性の液体に浸漬して、前記基材から分離した連続性のナノ材料膜を得るステップ、または
前記基材表面上で前記濡れた膜を乾燥して、前記基材上にナノ材料膜コーティングを得るステップと
を含むことを特徴とする方法。
【請求項2】
前記エマルジョンは、前記混合物を激しく混合することにより形成され、前記混合は、前記混合物の揺動、前記混合物の超音波エネルギーへの曝露、または揺動および超音波エネルギーの併用を含むことを特徴とする請求項1に記載の方法。
【請求項3】
前記混合または超音波エネルギーへの曝露は、少なくとも約30秒間であることを特徴とする請求項2に記載の方法。
【請求項4】
前記ナノ材料は、ポリアニリン、ドープしたポリアニリンまたはポリチオフェン、ポリ(3−ヘキシルチオフェン)、ポリ(3,4−エチレンジオキシチオフェン)ナノファイバー、グラフェンまたはグラファイト酸化物シート、カーボンナノチューブ、カーボンナノスクロール、カーボンブラックナノ粒子、ポリスチレンナノスフェア、デオキシリボ核酸、またはこれらの混合物を含むことを特徴とする請求項1に記載の方法。
【請求項5】
前記非混和性で有機性の液体は、四塩化炭素、クロロホルム、塩化メチレン、ベンゼン、ハロゲン化されたベンゼン、ペルフルオロ化された炭化水素、ニトロメタン、カーボンジスルフィド、トルエン、テトラクロロエチレン、酢酸エチル、ジメチルホルムアミド、ジエチルエーテル、1つもしくは複数のアルカンもしくはハロゲン化アルカン、またはこれらの混合物であることを特徴とする請求項1に記載の方法。
【請求項6】
前記基材は、ガラス、ITOでコーティングされたガラス、シリコン、二酸化ケイ素、石英、マイカ、金属フォイル、またはプラスチック製基材であることを特徴とする請求項1に記載の方法。
【請求項7】
前記プラスチック製基材は、ITO−ポリエチレンテレフタレート、ビニル、ポリ塩化ビニル、ポリエステル、ポリエチレンを含むことを特徴とする請求項6に記載の方法。
【請求項8】
前記基材表面は、親水性であることを特徴とする請求項1に記載の方法。
【請求項9】
前記基材表面は、親水性となるように活性化されることを特徴とする請求項1に記載の方法。
【請求項10】
前記基材表面は、疎水性であり、アルゴン−酸素プラズマに曝露することにより活性化されることを特徴とする請求項9に記載の方法。
【請求項11】
前記基材表面は、疎水性であり、前記ナノ材料は、極性反対の非混和性溶媒からなる二元混合物から堆積されることを特徴とする請求項1に記載の方法。
【請求項12】
前記基材上に形成される前記膜は、膜を着色する着色した添加物または反応物質の添加により着色されることを特徴とする請求項1に記載の方法。
【請求項13】
前記ナノ材料は、塩酸をドープした、トルエンスルホン酸をドープした、ポリスチレンスルホン酸をドープした、過塩素酸をドープした、カンファースルホン酸をドープした、若しくは脱ドープされたポリアニリンナノファイバー、または塩化物をドープしたポリチオフェンであることを特徴とする請求項1に記載の方法。
【請求項14】
前記基材上の前記膜は、周囲環境条件下で少なくとも約5分間乾燥されることを特徴とする請求項1に記載の方法。
【請求項15】
前記基材は、2つの長い端部および2つの短い端部を有する長方形の形状であり、前記長い端部が、前記形成する界面と平行であり、前記形成する界面の両側にあることを特徴とする請求項1に記載の方法。
【請求項16】
前記水性の液体は、水、pHが調整された水溶液、アセトニトリル水溶液、ヒドラジン、またはアルコール溶液であることを特徴とする請求項1に記載の方法。
【請求項17】
前記pHは、塩酸、過塩素酸、硫酸、ポリスチレンスルホン酸、樟脳酸、カンファースルホン酸、トルエンスルホン酸、ドデシルベンゼンスルホン酸、硝酸、酢酸、クエン酸、リン酸、ヒアルロン酸、水酸化アンモニウム、ヒドラジン、ナトリウム、カルシウム、カリウム、またはリチウムの水酸化物、または重炭酸ナトリウムを用いて調整されることを特徴とする請求項16に記載の方法。
【請求項18】
前記膜は、周囲環境条件下で乾燥される前に、前記容器内の前記非混和性で有機性の液体上の気相中で最初に乾燥されることを特徴とする請求項42に記載の方法。
【請求項19】
前記有機相は、前記水性相より大きい体積を有することを特徴とする請求項1に記載の方法。
【請求項20】
前記水性相は、約0.2から約5mlであり、前記有機相は、約5mlから約30mlであることを特徴とする請求項19に記載の方法。
【請求項21】
前記有機相は、前記水性相の体積の約3から約20倍の体積を有することを特徴とする請求項119に記載の方法。
【請求項22】
前記有機相は、前記水性相の体積の10倍から約20倍の体積を有することを特徴とする請求項19に記載の方法。
【請求項23】
前記基材表面は、疎水性であり、それおよび前記有機性の液体は、パーフルオロカーボンであることを特徴とする請求項6または11に記載の方法。
【請求項24】
前記基材上の前記膜は、周囲環境条件下で最長約2時間乾燥されることを特徴とする請求項114に記載の方法。
【図1】

【図2】
【図3】
【図4】
【図5】

【図6】
【図7】

【図8】

【図9】
【図10】
【図11】
【図12】
【図13】
【図14】
【図15】
【図16】

【図17】

【図18】

【図19】

【図20】

【図21】

【図22】

【図23】

【図24】
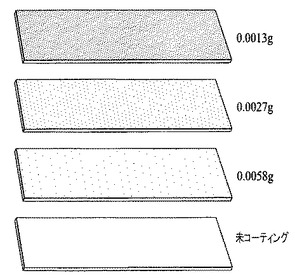
【図25】

【図26】

【図27】
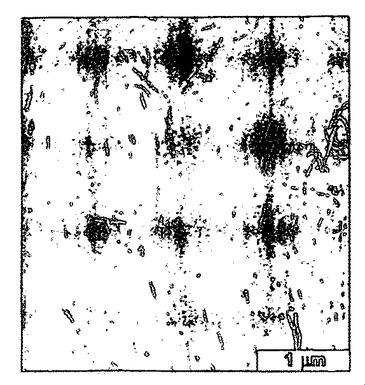
【図28】
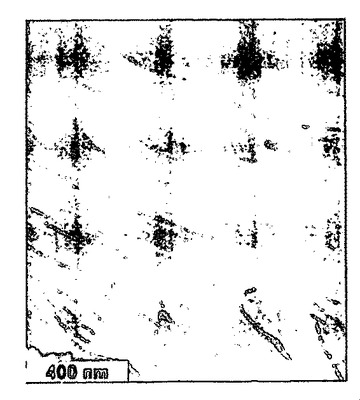
【図29】

【図30】

【図31】

【図32】

【図33】

【図34】
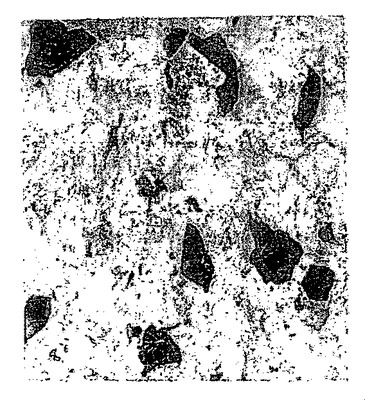
【図35】

【図36】

【図37】

【図38】
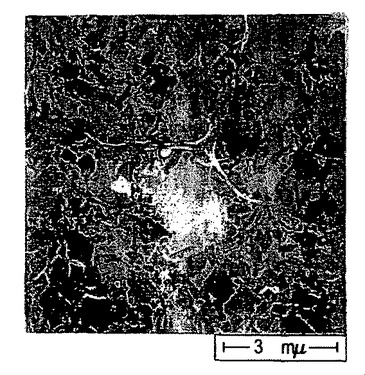
【図39】

【図40】

【図41】

【図42】
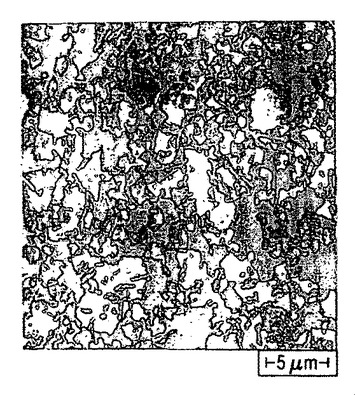
【図43】

【図44】

【図45】

【図46】

【図47】

【図48】

【図49】

【図50】

【図51】
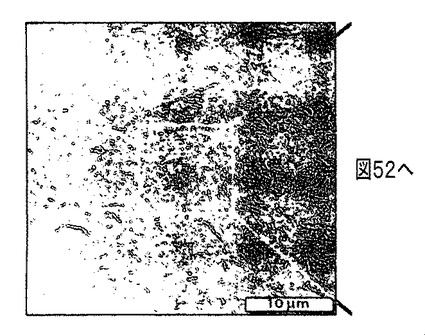
【図52】

【図53】

【図54】
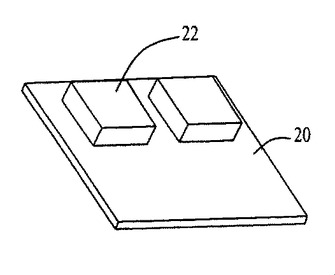
【図55】
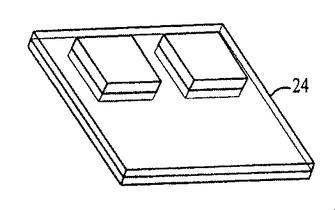
【図56】
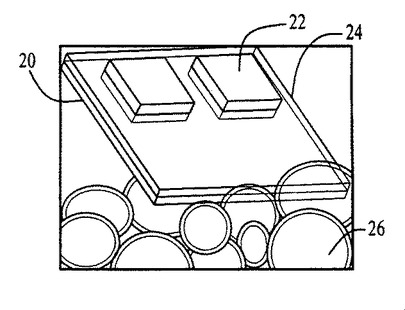
【図57】
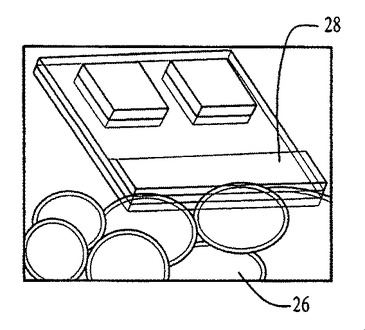
【図58】
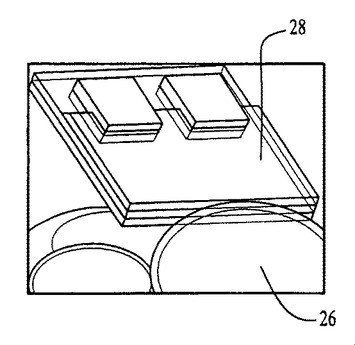
【図59】
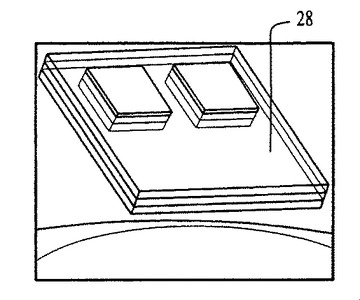
【図60】
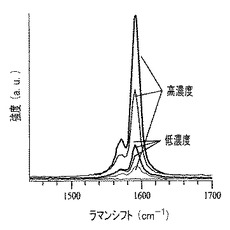
【図61】
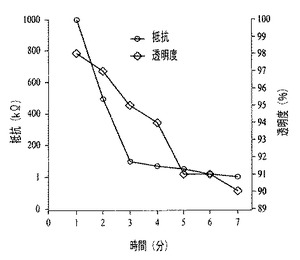
【図62】
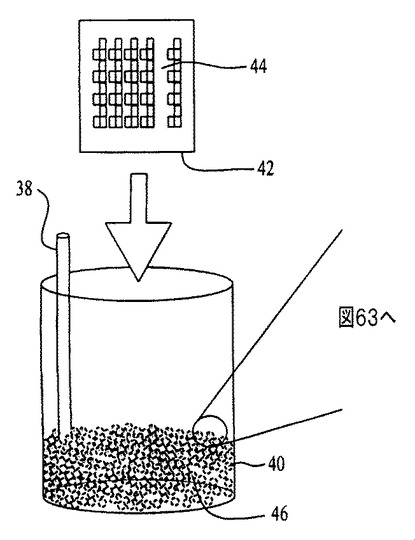
【図63】
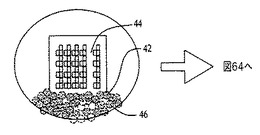
【図64】
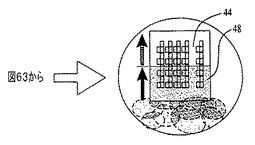
【図65】
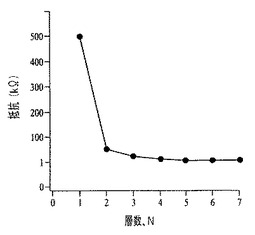
【図66】
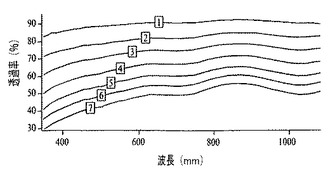
【図67】

【図68】

【図69】

【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【図11】
【図12】
【図13】
【図14】
【図15】
【図16】
【図17】
【図18】
【図19】
【図20】
【図21】
【図22】
【図23】
【図24】
【図25】
【図26】
【図27】
【図28】
【図29】
【図30】
【図31】
【図32】
【図33】
【図34】
【図35】
【図36】
【図37】
【図38】
【図39】
【図40】
【図41】
【図42】
【図43】
【図44】
【図45】
【図46】
【図47】
【図48】
【図49】
【図50】
【図51】
【図52】
【図53】
【図54】
【図55】
【図56】
【図57】
【図58】
【図59】
【図60】
【図61】
【図62】
【図63】
【図64】
【図65】
【図66】
【図67】
【図68】
【図69】
【公表番号】特表2013−517123(P2013−517123A)
【公表日】平成25年5月16日(2013.5.16)
【国際特許分類】
【出願番号】特願2012−548961(P2012−548961)
【出願日】平成23年1月13日(2011.1.13)
【国際出願番号】PCT/US2011/000071
【国際公開番号】WO2011/087913
【国際公開日】平成23年7月21日(2011.7.21)
【出願人】(506115514)ザ リージェンツ オブ ザ ユニバーシティ オブ カリフォルニア (87)
【Fターム(参考)】
【公表日】平成25年5月16日(2013.5.16)
【国際特許分類】
【出願日】平成23年1月13日(2011.1.13)
【国際出願番号】PCT/US2011/000071
【国際公開番号】WO2011/087913
【国際公開日】平成23年7月21日(2011.7.21)
【出願人】(506115514)ザ リージェンツ オブ ザ ユニバーシティ オブ カリフォルニア (87)
【Fターム(参考)】
[ Back to top ]