
電気分解による塩素製造のための電極
【課題】ゾル−ゲル法に基づいた、特に塩素製造用陽極のための、電極用触媒層の簡単かつ汎用的な製造方法を、先行技術の欠点を解消しつつ開発する。
【解決手段】導電性基材および触媒活性層を含んでなる電極であって、触媒活性層が2種の触媒活性成分に基づき、金属酸化物または混合酸化物または酸化物混合物としてイリジウム、ルテニウムまたはチタンを含んでなり、元素イリジウム、ルテニウムおよびチタンの和に基づいたルテニウムおよび/またはイリジウムの総含量が少なくとも10mol%であり、電極が導電性基材に適用された、NaClおよび/またはNaOHおよび/またはHCl含有水性電解液を透過しない少なくとも1つの酸化物基層を含んでなる、電極。
【解決手段】導電性基材および触媒活性層を含んでなる電極であって、触媒活性層が2種の触媒活性成分に基づき、金属酸化物または混合酸化物または酸化物混合物としてイリジウム、ルテニウムまたはチタンを含んでなり、元素イリジウム、ルテニウムおよびチタンの和に基づいたルテニウムおよび/またはイリジウムの総含量が少なくとも10mol%であり、電極が導電性基材に適用された、NaClおよび/またはNaOHおよび/またはHCl含有水性電解液を透過しない少なくとも1つの酸化物基層を含んでなる、電極。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、電気分解による塩素製造のための既知の貴金属触媒含有電極から出発している。本発明の分野は、向上した塩素回収のための電極用触媒被膜の製造方法(被膜は、導電性支持体材料に適用される)、およびこの方法により得ることができる新規な電極に関する。被膜は特に、緻密な割れのない基層および高度に多孔性のカバー層からなる。基層およびカバー層は例えば、Ti、RuまたはIrと遷移金属系からのドーピング元素の1種以上とに基づく混合酸化物からなる。いずれの層も、続く熱処理を伴ったゾル−ゲル変法によって、導電性支持体に適用される。
【背景技術】
【0002】
工業的な塩素の製造は、塩化ナトリウム溶液または塩酸溶液の電気分解によって実施されている。早期に使用されていたアマルガム法に代わるものとして、隔膜法およびイオン交換膜法が今日もっぱら使用されている。塩素の製造における最大のコスト要因は、電力量である。1トンの塩素を製造するために消費される電力は、塩化アルカリの電気分解(膜法)において、典型的には約2500kWhである。アマルガム法は、更に大量のエネルギーを消費する。塩素製造におけるコストを削減するためには、電解槽の設計によって、膜、電解液および電極で生じるオーム抵抗を著しく低下させる。エネルギーの消費を更に削減するため、電気分解における分解電位を低下する触媒を電極に適用する。塩素発生における陽極での電荷移動のための過電圧は、材料、表面形態、および触媒製造方法に大きく依存する。近年の陽極は通常、支持体としてのチタン、ニッケル、タンタルまたはジルコニウムで作られたエキスパンデッドメタル板に基づく。白金族元素の酸化物または酸化物混合物が通常、支持体表面に適用されている。ここでは、二酸化ルテニウム(RuO2)が塩素発生に適した触媒であることが分かっている。電気触媒活性材料としてのRuO2の重大な欠点は、電気分解中に触媒被膜が溶解するので、製造された既知の電極被膜の長期安定性が乏しいことである。そのため、使用中の陽極の触媒活性は連続的に低下する。活性ルテニウム成分を安定化させるために、例えばRuO2と二酸化チタンの混合物を塩化アルカリの電気分解において使用する。RuO2およびTiO2は、同じ結晶構造を有するので、薬品攻撃およびRuO2脱離を防ぐ固溶体を生成する。この効果は、US 3 632 498に記載されている。US 3 562 008の発明者らは、50mol%のルテニウム含量を有するRuO2−TiO2で被覆された陽極が、未被覆Pt陽極と比べて、塩素発生のための低下された槽電圧と改善された安定性を示すことを見出した。
【0003】
様々な材料および被膜形態を使用することによる、塩素発生における電気触媒効果への影響は、様々な文献に記載されているが、その効果は常には互いに完全に独立してはいない。
【0004】
DE 40 32 417 A1は、傾斜構造を有するRuO2−TiO2被膜を記載している。同文献では、層内のルテニウム含量が、陽極表面方向に40mol%から20mol%に低下している。従って、陽極酸化は、電解液との界面で実質的にもっぱら起こり、このようにして、寿命を短縮する体積侵食を回避する。
【0005】
EP 0 867 527 A1は、表面方向に適用された第一層から適用された最外層に向かって13mol%から100mol%に増加する、バルブ金属酸化物に対する貴金属酸化物の比の傾斜を有する、様々な電極用途のためのTiO2、RuO2およびIrO2からなる三成分酸化物混合物の製造方法を記載している。最外層は貴金属酸化物のみからなるので、少なくとも塩素製造の場合は、先に記載した純粋な貴金属酸化物の欠点、特に不十分な長期安定性が予想される。
【0006】
US 4 517 068では、塩素発生における改善された性能を、TiO2−RuO2と酸化パラジウムを組み合わせることによって達成している。
【0007】
T.A.F. Lassaliら(Electrochimica Acta 39, 1545 (1994))は、TiO2をPtOxで置換すると、電気化学的活性領域が劇的に増加し、従って、電気触媒活性が増加することを見出した。T.A.F. Lassaliらは、Ru0.3PtxTi0.7−xO2で示される層組成物を記載している。触媒層は、金属塩(例えば金属ハロゲン化物)の熱分解によって製造されている。欠点は、貴金属である白金を入手するコストが高いことである。更に、Pt含有被膜の化学的不安定性(ヘキサクロロプラチナート溶液としての白金の放出)は被膜の寿命を短縮する場合があり、従って、工業的使用を困難する。
【0008】
WO 2006/028443 A1もまた、Ruおよび酸化イリジウムに加えてドーピング元素としてのアンチモン、錫およびタンタルも含有できるパラジウム含有触媒混合酸化物層の、種々の金属塩の熱分解による製造方法を記載している。同文献では、非ドープ層と比べて100mV低下した、塩素発生のための過電圧が得られた。
【0009】
更に、金属基材に単一層として適用されたドープ触媒層および非ドープ触媒層の、熱分解による製造方法は、以下の文献から知られている:J. Electrochem. Soc. 129 1689 (1982)、Electrochimica Acta 42, 3525 (1997)、J. Alloys and Compounds 261, 176 (1997)。
【0010】
S. Trasatti, Electrochim. Acta, 36, 225 (1991)およびG.R.P. Malpassら、Electrochim. Acta, 52, 936 (2006)は、常套の熱分解法によって製造した電気触媒活性酸化物被膜の典型的な形態を記載している。酸化物被膜における欠陥、例えば、割れ、細孔、孔および粒界は、電解液が内部触媒活性部位を高度に利用できるようにし、従って、塩素生成反応に対する見掛けの電気触媒活性を高める。
【0011】
一方で、電解液は、電気触媒活性酸化物層における欠陥を通ってチタン基材まで浸透し、チタン基材を攻撃することができる。これは、電気触媒活性酸化物層の剥離を生じさせ、金属支持体材料と活性酸化物層の間の電気絶縁酸化チタン中間層の成長も促進し、その後の抵抗損の増加および陽極の不活性化をもたらす。このことは、L. M. Da Silvaら、J. Electroanal. Chem. 532, 141 (2002)に記載されている。
【0012】
電気分解において使用される電解液(例えば塩素の製造における塩化ナトリウムまたは塩化水素の水溶液)との接触に起因する支持体材料の不動態化を回避するために、様々な対策が開発されてきた。
【0013】
EP 0046449 A1に記載されているように、層厚さを増しかつ被覆剤の寿命を延長するため、何回もの被覆剤適用/焼成サイクルを通常採用する。適用被覆層における割れおよび孔は、その次の被覆剤適用において被覆液で少なくとも部分的に埋められる。内部欠陥の数は、更なる適用サイクル毎に低減される。
【0014】
前記方法の欠点は、多数の適用サイクルを必要とし、材料を大量に消費することである。別の欠点は、欠陥の数が少ないので、電気化学的活性領域、すなわち電気触媒活性が低減されることである。このことは、電気分解において電力量の消費が増加することにつながる。更に、被覆層の導電率は支持体材料と比べて小さいので、過度に大きい層厚さでは、電気分解における抵抗損が増加し、槽電圧が上昇することになる。
【0015】
導電率を有さないかまたは非常に小さい導電率しか有さない酸化チタン中間層の形成を回避するため、バルブ金属支持体の上側および電気触媒活性外層の下側に位置する中間層の概念が生まれてきた。そのような中間層は、用語「下層」、「バリア層」、「保護層」または「基層」によって表すこともでき、外層は「カバー層」と称することもできる。
【0016】
基層を得る1つの可能な方法は、例えばHayfield, Precious Metal Review 27(1) 1983_2-8に、チタンの陰極腐蝕防止のために記載されているような、金属白金の適用である。同方法を塩素製造において使用することについての欠点は特に、化学的不安定性(ヘキサクロロプラチナート溶液の生成)、および貴金属白金を得ることに起因する高コストである。
【0017】
別の文献には、熱分解、プラズマビームまたはプラズマ溶射による、より安定な貴金属不含有中間層の電気化学的製造方法が記載されている。US 3 882 002では、アンチモンドープ酸化錫を保護層としてバルブ金属基材に適用しており、貴金属外層も伴っている。US 3 950 240は、中間層としてのニオブドープ酸化錫と酸化ルテニウムからなるカバー層を有する陽極を記載している。US 7 211 177 B2は、塩酸の電気分解のための、炭化チタンまたはホウ化チタンからなる中間層を記載している。
【0018】
前記中間層全ての欠点は、保護層と支持体材料の間および中間層と電気触媒活性カバー層の間の接着が最適とは言えないことである。
【0019】
酸化ルテニウム系電極層の表面積を増大させることを、Y. Takasu(Electrochemica Acta 45, 4135 (2000))は、以下の5つの異なった方法を用いて調べている。
(1)浸漬被覆RuO2−Ti電極のためのアルコール性浸漬液への炭酸ナトリウムの添加、
(2)浸漬被覆によるRuO2−MOx(M:ドーピング金属)の製造、
(3)浸漬被覆およびその後の酸を用いた希土類酸化物の化学溶解による、チタン電極へのRuO2−ROx(R:希土類元素)層の製造、
(4)超微細RuO2粒子を製造するためのゾル−ゲル法における、触媒としての炭酸水素アンモニウム塩の添加、および
(5)RuO2のHxRuOy層構造への転化。
【0020】
表面構造は、製造した電極のサイクリックボルタンメトリー電荷を測定することによって電気化学的に特徴付けられた。RuO2−TiO2タイプの混合酸化物電極の場合、サイクリックボルタンメトリー電荷は12mC/cm2であり、RuO2−SnO2の場合は13mC/cm2である。最大サイクリックボルタンメトリー電荷は、RuO2−VOx系(162mC/cm2)、RuO2−MoO3系(120mC/cm2)、RuO2−CaO系(130mC/cm2)に対して得られた。しかしながら、このような方法では一般に、均一な混合酸化物ではなく、異なった酸化物の不均一な混合物が得られ、塩素製造用触媒としての純RuO2に対する先に記載した欠点を伴う。前記方法(1)、(3)、(4)および(5)によって得られた被覆組成物も、これらの欠点を示す。塩素製造用被膜の使用についての使用例は記載されていない。
【0021】
これまで開示されてきた方法により、20〜50mol%の範囲の典型的な貴金属含量を有する触媒層を製造することはできる。貴金属塩および貴金属自体は、多数の工程段階において複雑な方法で得るしかないので、大規模な工業プロセスに使用するための技術的支出は非常に高く、利用は制限される。
【0022】
RuO2を遷移金属(Ce、Nb、Sn、V、Cr、Mn、Co、Sb、Zr、Mo、W、Ta)酸化物により部分的に置換することによって、単純RuO2−TiO2系においてルテニウム含量を低下できることは示されてきた。加えて、活性、選択性および安定性の改善は、種々の遷移金属酸化物を組み合わせることによる相乗効果によって達成することができる。実験データは、S. V. Evdokimov らによって、Russian Journal of Electrochemistry 38, 657 (2002)に記載されている。10mol%のCrNbO4の添加によって、標準系RuO2−TiO2中のRuO2含量を、70mol%から30mol%に低下させることができる。このとき、電気触媒活性は、標準系と同程度である。
【0023】
US 3 776 834は、RuO2をSnO2により部分的に置換したRuO2(19mol%)、SnO2(13mol%)、TiO2(68mol%)の三成分系を使用した結果、系RuO2(33.3mol%)−TiO2(66.7mol%)を用いた塩素発生の過電圧を40mV低下できることを記載している。ドーピング元素の使用により、耐化学腐蝕性を高めることもできる。US 4 039 409およびUS 3 948 751は、ドーピング元素の塩と熱的に不安定なRu塩とを混合し、得られた混合物を適用法でチタンに直接適用することによる、ドープ触媒層の製造方法を記載している。最終的な熱処理により、ドープ触媒層が形成される。層の組成は、ドーピング元素の塩の量によって制御することができる。この方法は包括的にY. E. Roginskayaら、Electrochimica Acta 40, 817 (1995)に記載されている。同方法の欠点は、複数の成分からなる被膜が非常に不均一な微細構造を有することである。これらの知見はUS 4 668 531にも記載されている。500℃を超える温度で、触媒層と支持体との間に、緻密な電気的に絶縁されたTiO2層が生じる。そのような中間層は、電極の性能を低下させる。この関係は、WO 2008/046784 A1に記載されている。
【0024】
ゾル−ゲル法が、貴金属塩の熱分解に対する非常に良好な代替法であることが見出された。ゾル−ゲル法では、前駆体化合物の加水分解および縮合を制御することによって、混合酸化物を目的に応じて調製することができる。
【0025】
CN 1900368 A1によれば、ゾル−ゲル法によって、酸化錫、酸化イリジウム、酸化マンガンおよび酸化コバルトであるドープ種を、RuO2および高含量のCeO2からなる陽極被膜に導入する。発明者らは、ドープ三成分被膜またはドープ四成分被膜(45mol%のRuO2を含有する)についての塩素発生活性が、二成分RuO2/CeO2系と比べて増加したことを見出した。V. V. Panicら、Colloids and Surfaces A 157, 269 (1999)に記載されているように、ゾル−ゲル法によって製造した触媒被膜は、常套法(塩の熱分解)で製造した層と比べて増加した安定性および長い寿命を示す。Y. Zengら、Ceramics International 33, 1087 (2007)は、ゾル−ゲル法によって製造したRuO2−IrO2−TiO2触媒層が、減小した粒度の故に増加した活性を示すことも報告している。
【0026】
しかしながら、既知のゾル−ゲル法は、有機溶媒中での無機塩およびアルコキシドの溶解性に関して、特有の欠点を示す。十分な溶解性を得るために、強酸での変性と超音波処理が必要とされる。これらの処理は、製造工程を著しく長くする。このようにして調製した被覆液の幾つかは、溶解度の小さい成分が(特に高い濃度で)尚早に再沈澱するので、低い安定性しか有さない。そのような液は、貯蔵することができないし、極端な場合には、電極上に不均一な被膜を形成する。
【0027】
被膜の先に記載した欠点(すなわち高い貴金属含量、不十分な選択性および安定性、並びに導電性の不足)および被膜の製造方法の先に記載した欠点を示さない、新規な被膜およびその製造方法が要求されている。
【先行技術文献】
【特許文献】
【0028】
【特許文献1】US 3 632 498
【特許文献2】US 3 562 008
【特許文献3】DE 40 32 417 A1
【特許文献4】EP 0 867 527 A1
【特許文献5】US 4 517 068
【特許文献6】WO 2006/028443 A1
【特許文献7】EP 0046449 A1
【特許文献8】US 3 882 002
【特許文献9】US 3 950 240
【特許文献10】US 7 211 177 B2
【特許文献11】US 3 776 834
【特許文献12】US 4 039 409
【特許文献13】US 3 948 751
【特許文献14】US 4 668 531
【特許文献15】WO 2008/046784 A1
【特許文献16】CN 1900368 A1
【非特許文献】
【0029】
【非特許文献1】T.A.F. Lassaliら、Electrochimica Acta 39, 1545 (1994)
【非特許文献2】J. Electrochem. Soc. 129 1689 (1982)
【非特許文献3】Electrochimica Acta 42, 3525 (1997)
【非特許文献4】J. Alloys and Compounds 261, 176 (1997)
【非特許文献5】S. Trasatti, Electrochim. Acta, 36, 225 (1991)
【非特許文献6】G.R.P. Malpassら、Electrochim. Acta, 52, 936 (2006)
【非特許文献7】L. M. Da Silvaら、J. Electroanal. Chem. 532, 141 (2002)
【非特許文献8】Hayfield, Precious Metal Review 27(1) 1983_2-8
【非特許文献9】Y. Takasu(Electrochemica Acta 45, 4135 (2000)
【非特許文献10】S. V. Evdokimov ら、Russian Journal of Electrochemistry 38, 657 (2002)
【非特許文献11】Y. E. Roginskayaら、Electrochimica Acta 40, 817 (1995)
【非特許文献12】V. V. Panicら、Colloids and Surfaces A 157, 269 (1999)
【非特許文献13】Y. Zengら、Ceramics International 33, 1087 (2007)
【発明の概要】
【発明が解決しようとする課題】
【0030】
従って、本発明の目的は、ゾル−ゲル法に基づいた、特に塩素製造用陽極のための、電極用触媒層の簡単かつ汎用的な製造方法を、先に記載した欠点を解消しつつ開発することである。この層は、電気触媒活性成分、および長期安定性を確実にする安定剤からなる。更に、陽極は、先行技術に比べて、塩素過電圧および貴金属含量の低下を示さなければならない。
【課題を解決するための手段】
【0031】
本発明の態様は、導電性基材および触媒活性層を含んでなる電極であって、
触媒活性層が、2種の触媒活性成分に基づき、金属酸化物または混合酸化物または酸化物混合物としてイリジウム、ルテニウムまたはチタンを含んでなり、
元素イリジウム、ルテニウムおよびチタンの和に基づいたルテニウムおよび/またはイリジウムの総含量が、少なくとも10mol%であり、
電極が、導電性基材に適用された、NaClおよび/またはNaOHおよび/またはHCl含有水性電解液を透過しない少なくとも1つの酸化物基層を含んでなる、
電極である。
【0032】
本発明の別の態様は、少なくとも導電性基材および触媒活性層を含んでなる電極であって、
触媒活性層が、2種の触媒活性成分に基づき、金属酸化物または混合酸化物または酸化物混合物としてイリジウム、ルテニウムまたはチタンを含んでなり、
元素イリジウム、ルテニウムおよびチタンの和に基づいたルテニウムおよび/またはイリジウムの総含量が、少なくとも10mol%であり、
ルテニウムおよび/またはイリジウムの半分までが、バナジウム、ジルコニウムまたはモリブデンによって置換されている、
電極である。
【0033】
本発明の更に別の態様は、ルテニウム、イリジウム、チタンおよびそれらの混合物からなる群から選択される金属を含んでなる金属化合物の溶液または分散体を含んでなるゾル−ゲル被覆液を、導電性支持体に適用する工程、
溶媒を除去するために乾燥する工程、
酸素含有気体の存在下、少なくとも350℃の温度で焼成する工程、および
任意に、ゾル−ゲル被覆液の適用、乾燥および焼成を1回以上繰り返す工程
を含む、電極の製造方法である。
【0034】
前記した本発明の態様、および以下に記載する発明の詳細な説明は、添付の図面と併せて読むと、より良好に理解することができる。本発明の説明を補う目的で、例と考えられる代表的な態様を図面にそれぞれ示す。しかしながら、本発明が、どのような形でも、示されたまさにその態様および手段に限定されないことは理解すべきである。
【図面の簡単な説明】
【0035】
【図1】実施例6の被膜の表面の走査型電子顕微鏡写真を示す(スケール:a)10μm)。
【図2】実施例7の被膜の表面の走査型電子顕微鏡写真を示す(スケール:10μm)。
【図3】実施例8の被膜の表面の走査型電子顕微鏡写真を示す(スケール:10μm)。
【図4】実施例9bの被膜の表面の走査型電子顕微鏡写真を示す(スケール:10μm)。
【図5】実施例9の緻密基層(実線)および割れ構造物(点線)についての、電位走査速度(v)の関数としてプロットしたボルタンメトリー電荷(qa)を示す。
【図6】実施例10の被膜の表面の走査型電子顕微鏡写真を示す(スケール:10μm)。
【図7】実施例11のRu0.4Ti0.6O2被膜(実線)およびRu0.4Ti0.45La0.15O2被膜(点線)についての、サイクリックボルタモグラム数の関数としてプロットしたボルタンメトリー電荷(qa)を示す。
【図8】実施例12の被膜についての、電位走査速度(v)の関数としてプロットしたボルタンメトリー電荷(qa)を示す。
【発明を実施するための形態】
【0036】
本発明の特定の態様は、少なくとも導電性基材および触媒活性被膜を含んでなる新規な電極であって、触媒活性層が、2種の触媒活性成分に基づき、金属酸化物または混合酸化物または酸化物混合物として少なくともイリジウム、ルテニウムまたはチタンを含んでなり、元素イリジウム、ルテニウムおよびチタンの和に基づいたルテニウムおよび/またはイリジウムの総含量が、少なくとも10mol%、好ましくは10〜28mol%、好適には10〜20mol%であり、導電性基材に適用された、NaClおよび/またはNaOHまたはHCl含有水性電解液を透過しない少なくとも1つの酸化物基層が供給されていることを特徴とする電極を提供する。
【0037】
導電性基材は、好ましくはバルブ金属、特に好ましくはチタン、タンタル、ニオブおよびニッケル、或いはこれら金属の、主成分としてチタン、タンタルまたはニオブを含有する合金からなる群の金属に基づく。
【0038】
塩素発生のための新規な触媒被膜は例えば、金属酸化物の総量に基づいて10〜20mol%の含量を有する新規な活性金属成分(好ましくはRuO2またはRuO2/IrO2)、安定化成分(好ましくはTiO2)および遷移金属(好ましくは錫、ランタン、バナジウム、ジルコニウム、クロム、モリブデン)酸化物形態のドープ種からなる。ドープ種の濃度は、特に5〜15mol%である。
【0039】
基層が塩化水素水溶液、塩化ナトリウム水溶液または水酸化ナトリウム水溶液を透過しないことを特徴とする電極が好ましい。これは例えば、サイクリックボルタンメトリー実験(pH=3の3.5mol塩化ナトリウム水溶液中または0.5mol塩酸中、室温、Ag/AgCl参照電極、(Ag/AgClに対して)0.2〜1.0Vの走査範囲)において5mV/s〜200mV/sの電位走査速度範囲でサイクリックボルタモグラムの陽極ブランチを積分することによって測定されるサイクリックボルタンメトリー容量電荷(qa)が、常に10mC/cm2未満、好ましくは5mC/cm2未満、特に好ましくは2mC/cm2未満の場合である。
【0040】
基層より大きいサイクリックボルタンメトリー容量電荷(qa)を有するカバー層が付加的に供給されている電極の態様が特に好ましい。カバー層のサイクリックボルタンメトリー容量電荷(qa)は、好ましくは少なくとも10mC/cm2、特に好ましくは20mC/cm2である。
【0041】
基層は特に、0.1〜20g/m2、好ましくは0.5〜10g/m2の(酸化物としての)単位面積当たりの適用量を有し、カバー層は特に、少なくとも2g/m2、好ましくは少なくとも5g/m2の(酸化物としての)単位面積当たりの適用量を有する。
【0042】
カバー層が少なくとも2g/m2、好ましくは少なくとも5g/m2の適用量(単位面積当たりの重量)を有する電極が好ましい。新規な電極の特定の態様は、カバー層が、層厚さ方向の断面で観察して、チタンに対するイリジウムのおよび/またはチタン成分に対するルテニウムの変化する比を有することを特徴とする。
【0043】
電極の1つの態様では、カバー層におけるチタンに対するイリジウムのおよび/またはチタンに対するルテニウムの比は、層厚さ方向の断面で観察して、外側から導電性支持体に向かって低下している。
【0044】
陽極被膜の貴金属酸化物および安定化成分(例えばTiO2)を製造するための出発化合物として、塩化物、硝酸塩、アルコキシド、アセチルアセトネートを使用することが好ましい。ルテニウムアセチルアセトネート、イリジウムアセチルアセトネートまたは酢酸イリジウムが、貴金属成分を調製するための前駆体塩として好ましく使用される。TiO2は、例えばチタンイソプロポキシドまたはチタンブトキシドから得ることができる。ドープ種は特に好ましくは、前駆体塩であるバナジウムアセチルアセトネート、バナジウムテトラブトキシド、ジルコニウムn−プロポキシド、硝酸ジルコニウム、酢酸モリブデン、酢酸錫、錫イソプロポキシド、ランタンアセチルアセトネート、硝酸ランタンによって導入される。
【0045】
電極の別の態様は、カバー層が触媒活性層成分を含んでなり、孔形成組成物(特に、ランタン(とりわけ酸化ランタン)またはポリマー(とりわけポリビニルピロリドン)の組成物)を付加的に含有することを特徴とする。
【0046】
電極の別の態様では、基層は、導電性であり、少なくとも10S/m、好ましくは少なくとも1,000S/cm、特に好ましくは少なくとも10,000S/mの導電率を有する。
【0047】
先に記載した電極構造物の他に、本発明の別の態様である、少なくとも導電性基材および触媒活性被膜を含んでなる電極の別の態様も見出された。この態様は、触媒活性層が、2種の触媒活性成分に基づき、金属酸化物または混合酸化物または酸化物混合物として少なくともイリジウム、ルテニウムまたはチタンを含有し、元素イリジウム、ルテニウムおよびチタンの和に基づいたルテニウムおよび/またはイリジウムの総含量が、少なくとも10mol%、好ましくは10〜28mol%、好適には10〜20mol%であり、ルテニウムおよび/またはイリジウムの半分までが、バナジウム、ジルコニウムまたはモリブデン(好ましくはバナジウム)によって置換されていることを特徴とする。
【0048】
この選択された構造物は好ましいことに、導電性支持体に適用された、NaClおよび/またはNaOHまたはHCl含有水性電解液を透過しない酸化物基層も供給するために、最初に記載した電極の態様と組み合わせることができる。
【0049】
本発明の態様は更に、電極、特に先に記載した新規な電極の製造方法であって、第一の工程において、金属であるルテニウム、イリジウムおよびチタンの1種以上の金属化合物の溶液または分散体を含有するゾル−ゲル被覆液を、特に浸漬によって、導電性支持体に1回以上適用し;次いで、溶媒を除去するために乾燥し;続いて、乾燥金属化合物層を酸素含有気体の存在下、高温で、特に少なくとも350℃で、好ましくは少なくとも400℃で焼成し;任意に、溶液または分散体の適用、乾燥および焼成を1回以上繰り返すことを特徴とする方法を提供する。
【0050】
カバー層を製造するためには、ルテニウム、イリジウムおよびチタンからなる群からの金属の金属塩の溶液または分散体を基層に1回以上適用し、溶媒を除去し、酸素含有気体の存在下、高温で、特に少なくとも350℃で、好ましくは少なくとも450℃で焼成する方法が好ましい。
【0051】
製造方法の一態様では、基層を製造するために、金属塩液を適用した後、乾燥を高温で、特に少なくとも200℃で、好ましくは少なくとも240℃で実施する。
【0052】
1つの変法では、低級カルボン酸、特にプロピオン酸、C1〜C5アルコールまたはケトン或いはそれらの混合物を、基層および/またはカバー層を製造するための金属化合物液に添加する。
【0053】
均一かつ安定な被覆液を調製するため、プロピオン酸ゾル−ゲル法を使用することが特に好ましい。同方法では、プロピオン酸とアルコール(メタノール、エタノール、n−プロパノール、イソプロパノール、ブタノール)の様々な濃度の混合物が、先に記載した化合物の溶媒として使用される。前駆体塩の溶解は、各前駆体塩について独立して、撹拌しながら130℃を超える温度で起こる。溶解工程の時間は約1時間である。これにより、澄明かつ安定なゾル液を得る。プロピオン酸を使用することにより、使用した金属カチオンが錯体を形成するので、制御された加水分解が可能となる。
【0054】
冷却後、各前駆体塩の溶液を混合し、次いで撹拌する。この混合物から、電気触媒活性塩素発生陽極を製造するための被覆液を調製する。この被覆液は、チタンシートまたはチタン網のような基材(例えばエキスパンデッドチタンメタル)に適用することができる。適用前に、基材を物理的、化学的または電気化学的に、清浄し、磨き、粗化しなければならない。これにより、被膜の接着性を向上させる。
【0055】
常套法である塩の熱分解と比べて、ゾル−ゲル法は以下の利点を有する:
・低いプロセス温度、
・制御された化学量論量を有する混合酸化物が、異なった化合物のゾルを混合することによって容易に調製できること、
・出発物質の混合が分子レベルで起こるので、生成物の均一性が高いこと、
・最終熱処理により単純な有機基が完全に除去されるので、生成物の純度が高いこと、
・最終熱処理により、得られる層の微細構造(多孔性)に影響を与え得ること、
・浸漬被覆法、噴霧被覆法または回転被覆法により複雑な形態を被覆できること。
【0056】
ゾル−ゲル法により、溶媒(メタノール、エタノール、イソプロパノール、ブタノール)に溶解されている1種以上の易加水分解性アルコキシド、アセチルアセトネートまたは無機塩を含んでなる被覆液を調製することができる。安定なコロイド(ゾル)を調製するために、被覆液を酸触媒(塩酸、硝酸または酢酸)或いは塩基触媒(アンモニア、水酸化ナトリウム溶液)を用いて加水分解する。加水分解により、ヒドロキシ架橋およびオキソ架橋金属原子の網状構造が形成される。
【0057】
次いで、この被覆液で基材を被覆することができる。続く乾燥工程では、ゾルが基材に固定される。最終の焼成工程では、有機成分は除去され、混合酸化物はナノ構造粒子に結晶化する。ゾル−ゲル法によって、ドープ種を導入することもできる。これらのドープ種は、触媒の活性を高めることができるので、貴金属の含量を低下させることができる。
【0058】
被覆液の適用は、浸漬法、塗布法、滴下法、噴霧法または回転法によって実施することができる。次いで、特に室温で、層を乾燥し、その後、例えば250℃で少なくとも10分間、それに続いて450℃で少なくとも5分間焼成する。層を安定化するための最終の焼成工程は、例えば450℃で12〜30分間実施する。成分の酸化を向上させるため、純酸素または酸素リッチ雰囲気を使用することができる。記載した方法の逐次的繰り返し数を変えることによって、層厚さを変えることができる。この手順によって多層構造を得ることもできる。
【0059】
本発明の態様は更に、塩化ナトリウムまたは塩化水素含有液を電気分解するための電解槽であって、先に記載した新規な電極が電解槽の陽極として供給されている電解槽を提供する。
【0060】
本発明の別の態様は、塩素を電気化学的に製造するために塩化ナトリウムまたは塩化水素を電気分解するための電解槽における陽極としての、先に記載した新規な電極の使用も提供する。
【0061】
先に記載した引用文献の全ては、有用な目的全てのために、それらの全内容を引用してここに組み込む。
【0062】
本明細書では、用語および/または文脈が別途明確に記載していない限り、単数形のための用語「a」および「the」は、「1以上」および「少なくとも1」と同義であり、互いに置き換えて使用することができる。従って、例えば、明細書または特許請求の範囲における「a catalytically active component」は、「1種の触媒活性成分」または「1種以上の触媒活性成分」に言及し得る。また、数値の全ては、特に記載のない限り、用語「約」によって修飾されていると理解すべきである。
【0063】
本発明を具体的に表す特定の構造物を示し、記載しているが、本発明の概念の意図および範囲から逸脱することなく、その一部を様々に変更および修正できること、並びにそれらが本明細書に示され、記載されている特定の形態に限定されないことは当業者には明らかであろう。
【0064】
本発明の態様を、後記実施例および図面によって説明するが、それらは本発明を何ら限定するものではない。
【0065】
図1は、実施例6の被膜の表面の走査型電子顕微鏡写真を示す(スケール:a)10μm)。
図2は、実施例7の被膜の表面の走査型電子顕微鏡写真を示す(スケール:10μm)。
図3は、実施例8の被膜の表面の走査型電子顕微鏡写真を示す(スケール:10μm)。
図4は、実施例9bの被膜の表面の走査型電子顕微鏡写真を示す(スケール:10μm)。
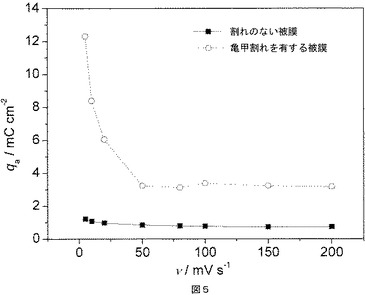
図5は、実施例9の緻密基層(実線)および割れ構造物(点線)についての、電位走査速度(v)の関数としてプロットしたボルタンメトリー電荷(qa)を示す。
図6は、実施例10の被膜の表面の走査型電子顕微鏡写真を示す(スケール:10μm)。
図7は、実施例11のRu0.4Ti0.6O2被膜(実線)およびRu0.4Ti0.45La0.15O2被膜(点線)についての、サイクリックボルタモグラム数の関数としてプロットしたボルタンメトリー電荷(qa)を示す。サイクリックボルタンメトリーは、Ag/AgCl参照電極を用い、室温で、0.5mol塩酸溶液中で実施した。電位は、v=50mV/sの電位走査速度で、(Ag/AgClに対して)0.2〜1.0Vの範囲で変化させた。
図8は、実施例12の被膜についての、電位走査速度(v)の関数としてプロットしたボルタンメトリー電荷(qa)を示す。サイクリックボルタンメトリーは、Ag/AgCl参照電極を用い、室温で、3.5mol塩酸溶液中で実施した。電位は、(Ag/AgClに対して)0.2〜1.0Vの範囲で変化させた。
【実施例】
【0066】
実施例1
15mmの直径を有するチタン板(厚さ2mm)にサンドブラストを掛け、表面を清浄および粗化し、次いで、10%濃度シュウ酸中、80℃で2時間酸洗いし、続いて、イソプロパノールで清浄して窒素流中で乾燥した。
被覆液を調製するため、99.6mgのルテニウムアセチルアセトネート(Ru(acac)3)、207.2μlのチタンイソプロポキシド(Ti(i−OPr)4)および13.3mgのバナジルアセチルアセトネート(VO(acac)2)をそれぞれ、1.45mlのイソプロパノールおよび1.45mlのプロピオン酸に溶解し、還流しながら30分間加熱した。室温に冷却後、3つの溶液を混合し、ワインレッド色の均一かつ澄明な溶液を得た。この被覆液50μlを、マイクロピペットによってチタン基材に適用し、次いで、空気中で乾燥した。層を空気中で、まず250℃で10分間、次いで450℃で10分間焼成した。これらの手順(被覆液の適用、乾燥、焼成)を8回繰り返した。9回目の被覆工程後、被覆チタン基材を450℃で1時間焼成した。これにより、金属元素に基づいてRu25mol%/Ti70mol%/V5mol%の組成を有する試料を得た。金属含量に基づいて計算すると、これは、6.4g/m2のルテニウム適用量に相当する。これは、(酸化物RuO2、TiO2、V2O5の和として)23.7g/m2の総被膜適用量に相当する。
塩素発生のための電気触媒活性は、クロノアンペロメトリー(参照電極:Ag/AgCl、電解液:3.5MのNaCl、pH:3、室温)によって測定した。実験では、定常電流密度を1kA/m2に設定した。得られた電位は1.17Vであった。
【0067】
実施例1b(比較例)
実施例1と同じ方法で、チタン基材を前処理した。
熱分解によって被膜を製造するために、2.00gの塩化ルテニウム(III)水和物(Ru含量=40.5重量%)、21.56gのn−ブタノール、0.94gの濃塩酸および5.93gのチタン酸テトラブチルTi−(O−Bu)4を含有する被覆液を調製した。被覆液の一部を、小さいチタン板にハケ刷りした。これを空気中、80℃で10分間乾燥し、次いで、空気中、470℃で10分間処理した。この手順(被覆液の適用、乾燥、熱処理)を合計8回実施した。続いて、板を空気中、520℃で1時間処理した。単位面積当たりのルテニウムの適用量は、被覆液の消費量から16g/m2と算出された。これは、31mol%のRuO2および69mol%のTiO2の組成で(酸化物として)49.2g/m2の総被膜適用量に相当する。
(実施例1と同じ方法で測定した)塩素発生のための電位は、この試料では1.25Vであった。
【0068】
実施例2
実施例1と同じ方法で、チタン基材を前処理した。
60mgのルテニウムアセチルアセトネート(Ru(acac)3)、236.8μlのチタンイソプロポキシド(Ti(i−OPr)4)および13.3mgのバナジルアセチルアセトネート(VO(acac)2)をそれぞれ、1.45mlのイソプロパノールおよび1.45mlのプロピオン酸に溶解し、次いで、(撹拌しつつ)還流しながら150℃で30分間加熱した。室温に冷却後、3つの溶液を混合し、ワインレッド色の均一かつ澄明な溶液を得た。実施例1に記載したように、被覆工程および焼成工程を実施した。
これにより、金属元素に基づいてRu15mol%/Ti80mol%/V5mol%の組成を有する試料を得た。金属含量に基づいて計算すると、これは、3.9g/m2のルテニウム適用量に相当する。これは、(酸化物RuO2、TiO2、V2O5の和として)22.7g/m2の総被膜適用量に相当する。
(実施例1と同じ方法で測定した)塩素発生のための電位は、この試料では1.17Vであった。
【0069】
実施例3
実施例1と同じ方法で、チタン基材を前処理した。
99.6mgのルテニウムアセチルアセトネート(Ru(acac)3)、207.2μlのチタンイソプロポキシド(Ti(i−OPr)4)および22.4μlのジルコニウムn−プロポキシド(n−プロパノール中70重量%)をそれぞれ、1.44mlのイソプロパノールおよび1.44mlのプロピオン酸に溶解し、次いで、(撹拌しつつ)還流しながら150℃で30分間加熱した。室温に冷却後、3つの溶液を混合し、ワインレッド色の均一かつ澄明な溶液を得た。実施例1に記載したように、被覆工程および焼成工程を実施した。
これにより、金属元素に基づいてRu25mol%/Ti70mol%/Zr5mol%の組成を有する試料を得た。金属含量に基づいて計算すると、これは、6.4g/m2のルテニウム適用量に相当する。これは、(酸化物RuO2、TiO2、ZrO2の和として)24.2g/m2の総被膜適用量に相当する。
(実施例1と同じ方法で測定した)塩素発生のための電位は、この試料では1.25Vであった。
【0070】
実施例4
実施例1と同じ方法で、チタン基材を前処理した。
99.6mgのルテニウムアセチルアセトネート(Ru(acac)3)、192.4μlのチタンイソプロポキシド(Ti(i−OPr)4)および42.8μlの酢酸モリブデン(Mo2(OCOCH3)4)をそれぞれ、1.45mlのイソプロパノールおよび1.45mlのプロピオン酸に溶解し、次いで、(撹拌しつつ)還流しながら150℃で30分間加熱した。室温に冷却後、3つの溶液を混合し、ワインレッド色の均一かつ澄明な溶液を得た。実施例1に記載したように、被覆工程および焼成工程を実施した。
これにより、金属元素に基づいてRu25mol%/Ti65mol%/Mo10mol%の組成を有する試料を得た。金属含量に基づいて計算すると、これは、6.4g/m2のルテニウム適用量に相当する。これは、(酸化物RuO2、TiO2、MoO3の和として)25.2g/m2の総被膜適用量に相当する。
(実施例1と同じ方法で測定した)塩素発生のための電位は、この試料では1.18Vであった。
【0071】
実施例5
実施例1と同じ方法で、チタン基材を前処理した。
49.8mgのルテニウムアセチルアセトネート(Ru(acac)3)、62.3mgのイリジウムアセチルアセトネート(Ir(acac)3)、16.6mgのバナジウムアセチルアセトネート(VO(acac)2)、207.6mgの錫イソプロポキシドプロパノール(Sn(i−OPr)4・C3H7OH)および129.5μlのチタンイソプロポキシド(Ti(i−OPr)4)をそれぞれ、1.11mlのイソプロパノールおよび1.11mlのプロピオン酸に溶解し、次いで、(撹拌しつつ)還流しながら150℃で30分間加熱した。室温に冷却後、5つの溶液を混合し、ワインレッド色の均一かつ澄明な溶液を得た。実施例1に記載したように、被覆工程および焼成工程を実施した。
これにより、金属元素に基づいてRu10mol%/Ir10mol%/V5mol%/Sn40mol%/Ti35mol%の組成を有する試料を得た。金属含量に基づいて計算すると、これは、3.2g/m2のルテニウム適用量に相当する。これは、(酸化物RuO2、IrO2、V2O5、SnO2、TiO2の和として)33.6g/m2の総被膜適用量に相当する。
(実施例1と同じ方法で測定した)塩素発生のための電位は、この試料では1.22Vであった。
【0072】
実施例6
実施例1と同じ方法で、チタン基材を前処理した。
149.4mgのルテニウムアセチルアセトネート(Ru(acac)3)および333.1μlのチタンイソプロポキシド(Ti(i−OPr)4)をそれぞれ、3.25mlのイソプロパノールおよび3.25mlのプロピオン酸に溶解し、次いで、(撹拌しつつ)還流しながら150℃で30分間加熱した。室温に冷却後、2つの溶液を混合し、ワインレッド色の均一かつ澄明な溶液を得た。
浸漬被覆によって、割れのない緻密な被膜を製造した。この目的のため、チタン基材を被覆液に20秒間浸漬し、次いで、167mm/分の速度で被覆液から垂直に取り出し、続いて、空気中で乾燥した。層を空気中で、まず250℃で10分間、次いで450℃で5分間焼成した。浸漬被覆−乾燥−焼成の手順を5回繰り返した。
これにより、金属元素に基づいてRu25mol%/Ti75mol%の組成を有する試料を得た。金属含量に基づいて計算すると、これは、0.16g/m2のルテニウム適用量に相当する。これは、(酸化物RuO2、TiO2の和として)0.59g/m2の総被膜適用量に相当する。
製造した被膜の表面形態を、走査型電子顕微鏡によって特徴付けた(図1参照)。
【0073】
実施例7
実施例1と同じ方法で、チタン基材を前処理した。
被覆液は、実施例6と同じである。
実施例6と同じ方法で、割れのない緻密な被膜を得た。浸漬被覆−乾燥−焼成の手順を15回繰り返した。これにより、金属元素に基づいてRu25mol%/Ti75mol%の組成を有する試料を得た。金属含量に基づいて計算すると、これは、0.50g/m2のルテニウム適用量に相当する。これは、(酸化物RuO2、TiO2の和として)1.84g/m2の総被膜適用量に相当する。
製造した被膜の表面形態を、走査型電子顕微鏡によって特徴付けた(図2参照)。
【0074】
実施例8
実施例1と同じ方法で、チタン基材を前処理した。
4.62mlのイソプロパノールに421.1mgの塩化ルテニウム(III)水和物(RuCl3・xH2O、36%Ru)を溶解し、一晩撹拌することによって、被覆液(液A)を得た。1332mlのチタンイソプロポキシド(Ti(i−OPr)4)を、2.246mlの4−ヒドロキシ−4−メチル−2−ペンタノンおよび5mlのイソプロパノールの予備混合液に添加し、30分間撹拌した(液B)。液Aおよび液Bを超音波処理によって混合し、澄明な液を得た。続いて、この液に、25.8μlの酢酸および108μlの脱イオン水を添加した。このように調製した液に蓋をし、室温で一晩撹拌した。
浸漬被覆によって、割れのない緻密な被膜を製造した。この目的のため、チタン基材を被覆液に20秒間浸漬し、次いで、193mm/分の速度で被覆液から垂直に取り出し、続いて、空気中で乾燥した。層を空気中で、まず90℃で30分間、次いで450℃で10分間焼成した。浸漬被覆−乾燥−焼成の手順を6回繰り返した。
これにより、金属元素に基づいてRu25mol%/Ti75mol%の組成を有する試料を得た。金属含量に基づいて計算すると、これは、1.0g/m2のルテニウム適用量に相当する。これは、(酸化物RuO2、TiO2の和として)3.69g/m2の総被膜適用量に相当する。
製造した被膜の表面形態を、走査型電子顕微鏡によって特徴付けた(図3参照)。
【0075】
実施例9
実施例1と同じ方法で、チタン基材を前処理した。
4.81mlのイソプロパノールに210.6mgの塩化ルテニウム(III)水和物(RuCl3・xH2O、36%Ru)を溶解し、一晩撹拌することによって、被覆液(液A)を得た。666.1mlのチタンイソプロポキシド(Ti(i−OPr)4)を、1.123mlの4−ヒドロキシ−4−メチル−2−ペンタノンおよび20mlのイソプロパノールの予備混合液に添加し、30分間撹拌した(液B)。液Aおよび液Bを超音波処理によって混合し、澄明な液を得た。続いて、この液に、12.9μlの酢酸および54μlの脱イオン水を添加した。このように調製した液に蓋をし、室温で一晩撹拌した。
浸漬被覆によって、割れのない緻密な被膜を製造した。この目的のため、チタン基材を被覆液に20秒間浸漬し、次いで、193mm/分の速度で被覆液から垂直に取り出した。湿潤被膜を空気中で乾燥し、空気中、90℃で30分間、次いで450℃で10分間焼成した。浸漬被覆−乾燥−焼成の手順を50回繰り返し、最後の焼成は450℃で1時間実施した。
これにより、金属元素に基づいてRu25mol%/Ti75mol%の組成を有する試料を得た。金属含量に基づいて計算すると、これは、2.0g/m2のルテニウム適用量に相当する。これは、(酸化物RuO2、TiO2の和として)7.38g/m2の総被膜適用量に相当する。
【0076】
実施例9b(比較例)
比較のため、実施例9と同じ被覆液を用いて、亀甲割れ構造を有する被膜を製造した。
チタン基材を実施例1と同じ方法で前処理し、マイクロピペットを用いた滴下法によって50μlの被覆液を適用した。
湿潤被膜を空気中で乾燥し、空気中、90℃で30分間、次いで450℃で10分間焼成した。滴下被覆−乾燥−焼成の手順を4回繰り返し、最後の焼成は450℃で1時間実施した。
これにより、金属元素に基づいてRu25mol%/Ti75mol%の組成を有する試料を得た。金属含量に基づいて計算すると、これは、3.4g/m2のルテニウム適用量に相当する。これは、(酸化物RuO2、TiO2の和として)12.54g/m2の総被膜適用量に相当する。
製造した被膜の表面形態を、走査型電子顕微鏡によって特徴付けた(図4参照)。
【0077】
割れのない構造を有する電極および亀甲割れ構造を有する電極を、サイクリックボルタンメトリーによって試験した。測定は、銀/塩化銀参照電極を用い、室温、pH=3、3.5mol塩化ナトリウム溶液中で実施した。電位は、5〜200mV/sの範囲の様々な電位走査速度(v)で、0.2〜1.0Vの範囲で変化した。ボルタンメトリー容量電荷(qa)は、サイクリックボルタモグラムの陽極ブランチを積分することによって、EC−Labソフトウェアを用いて得た。割れのない被膜および亀甲割れ構造を有する被膜について、ボルタンメトリー電荷(qa)を電位走査速度(v)の関数としてプロットする(図5)。亀甲割れ構造を有する被膜(図5の点線)については、電位走査速度(v)が5mV/sから50mV/sに上昇すると、ボルタンメトリー電荷は急激に低下し、その後、ほぼ一定の値をとった。このことは、低い電位走査速度(v)では、割れを通して内部の割れおよびボイドに電解液が浸透し、容量電荷が高い値となるが、より高い電位走査速度では、最外表面しか接近可能でないという事実と一致している。それとは対照的に、割れのない被膜の容量電荷は、電位走査速度に実質的には依存しない(図5の実線)。これにより、被膜が詰まっていて緻密であることが明らかである。
【0078】
実施例10
割れのない緻密な基層と割れのある電気触媒活性カバー層を有する被膜を製造するために、割れのない基層を実施例7と同じ方法で製造した。これにより、金属元素に基づいてRu25mol%/Ti75mol%の基層組成を有する試料を得た。金属含量に基づいて計算すると、これは、0.50g/m2のルテニウム適用量に相当する。これは、1.84g/m2の(酸化物RuO2、TiO2の和としての)総基層適用量に相当する。
被覆−焼成サイクルを合わせて4回実施した以外は、実施例1と同じ被覆液を用い、実施例1と同じ被覆および焼成手順によって、割れのない基層に適用された割れのあるカバー層を製造した。
これにより、金属元素に基づいてRu25mol%/Ti70mol%/V5mol%のカバー層組成を有する試料を得た。金属含量に基づいて計算すると、これは、2.6g/m2のルテニウム適用量に相当する。これは、9.64g/m2の(酸化物RuO2、TiO2、V2O5の和としての)総カバー層適用量に相当する。
製造した被膜の表面形態を、走査型電子顕微鏡によって特徴付けた(図6参照)。
塩素発生のための電極電位を、実施例1と同じ条件下で測定した。測定された電極電位は1.21Vであった。
【0079】
実施例11
割れのある多孔性電気触媒活性被膜を製造するために、実施例1と同じ方法でチタン基材を前処理した。
2mlのイソプロパノールに67.4mgの塩化ルテニウム(III)水和物(RuCl3・xH2O、36%Ru)を溶解し、一晩撹拌することによって、被覆液(液A)を得た。39mgの硝酸ランタン(III)六水和物(La(NO3)3・6H2O)を1mlのイソプロパノールに添加し、30分間撹拌した(液B)。80μlのチタンイソプロポキシド(Ti(i−OPr)4)を、224.6μlの4−ヒドロキシ−4−メチル−2−ペンタノンおよび0.66mlのイソプロパノールの予備混合液に添加し、10分間撹拌した(液C)。
液A、B、Cを超音波処理によって混合し、澄明な液を得た。続いて、この液に、5.15μlの酢酸および10.8μlの脱イオン水を添加した。このように調製した液に蓋をし、室温で一晩撹拌した。
50μlの被覆液を、マイクロピペットを用いて滴下法によって、チタン基材に適用した。
湿潤被膜を空気中で乾燥し、空気中、250℃で10分間、次いで450℃で10分間焼成した。滴下被覆−乾燥−焼成の手順を5回繰り返し、最後の焼成は450℃で1時間実施した。
これにより、金属元素に基づいてRu40mol%/Ti45mol%/La15mol%の組成を有する試料を得た。金属含量に基づいて計算すると、これは、8.5g/m2のルテニウム適用量に相当する。これは、(酸化物RuO2、TiO2、La2O3の和として)29.0g/m2の総被膜適用量に相当する。
【0080】
比較のために、Ru0.4Ti0.6O2の組成を有する被膜を製造した。この目的のために、2mlのイソプロパノールに67.4mgの塩化ルテニウム(III)水和物(RuCl3・xH2O、36%Ru)を添加し、一晩撹拌した(液A)。106.6μlのチタンイソプロポキシド(Ti(i−OPr)4)を、224.6μlの4−ヒドロキシ−4−メチル−2−ペンタノンおよび1.66mlのイソプロパノールの予備混合液に添加し、10分間撹拌した(液B)。液AおよびBを超音波処理によって混合し、澄明な液を得た。続いて、この液に、5.15μlの酢酸および10.8μlの脱イオン水を添加した。このように調製した液に蓋をし、室温で一晩撹拌した。
50μlの被覆液を、マイクロピペットを用いて滴下法によって、チタン基材に適用した。
湿潤被膜を空気中で乾燥し、空気中、250℃で10分間、次いで450℃で10分間焼成した。滴下被覆−乾燥−焼成の手順を5回繰り返し、最後の焼成は450℃で1時間実施した。
これにより、金属元素に基づいてRu40mol%/Ti60mol%の組成を有する試料を得た。金属含量に基づいて計算すると、これは、8.5g/m2のルテニウム適用量に相当する。これは、(酸化物RuO2、TiO2の和として)21.3g/m2の総被膜適用量に相当する。
【0081】
参照電極として銀/塩化銀を用い、室温で、0.5mol塩酸中でサイクリックボルタンメトリーを実施した。電位は、v=50mV/sの電位走査速度(v)で、0.2〜1.0Vの範囲で変化した。ただし、電解液に暴露された面積は1cm2であった。ボルタンメトリー容量電荷(qa)は、サイクリックボルタモグラムの陽極ブランチを積分することによって、EC−Labソフトウェアを用いて得た。Ru0.4Ti0.45La0.15O2被膜(点線)およびRu0.4Ti0.6O2被膜(実線)について、ボルタンメトリー電荷(qa)をサイクリックボルタンメトリーサイクル数の関数としてプロットする(図7)。
Ru0.4Ti0.6O2被膜のボルタンメトリー容量電荷qaは、サイクリックボルタンメトリーサイクル数に依存しない。このことは、被膜の特性が変わらないことを示している。一方、Ru0.4Ti0.45La0.15O2被膜の場合は、第2電位サイクルから第79電位サイクルまで、ボルタンメトリー電荷の連続的な上昇が見られた。このことは、被膜の多孔性の増大を同時に伴うサイクリックボルタンメトリーサイクルの間に、酸化物マトリックスから酸化ランタンが連続的に溶解していることに起因する。
【0082】
実施例12
割れのある多孔性電気触媒活性被膜を製造するために、実施例1と同じ方法でチタン基材を前処理した。
1.33mlのイソプロパノールに37.9mgの塩化ルテニウム(III)水和物(RuCl3・xH2O、36%Ru)を溶解し、一晩撹拌することによって、4つの被覆液(液A)をそれぞれ得た。130.8mgの錫イソプロポキシドプロパノレート(Sn(i−OPr)4・C3H7OH)を、1.34mlのイソプロパノールおよび1.33mlのプロピオン酸の混合物に添加し、次いで、激しく撹拌しつつ還流しながら150℃で30分間煮沸することによって、4つの液(液B)をそれぞれ得た。続いて、異なった量(すなわち39mg、29.2mg、9.7mgおよび0mg)の硝酸ランタン(III)六水和物(La(NO3)3・6H2O)を4つの熱い液Bに添加し、次いで、室温まで冷える前に、これらの液を更に20分間撹拌した。その後、これらの液を、撹拌しながら液Aに滴加した。
それぞれの場合において、50μlの被覆液を、マイクロピペットを用いて滴下法によって、チタン基材に適用した。
湿潤被膜を空気中で乾燥し、空気中、250℃で10分間、次いで450℃で10分間焼成した。続いて、被膜の酸化ランタン成分を溶解するために、被覆チタン板を5%濃度塩酸に、穏やかに撹拌しながら、60℃の温度で15分間浸漬した。
それぞれの場合において、滴下被覆−乾燥−焼成−溶解の手順を8回繰り返し、最後の焼成は450℃で1時間実施した。
これにより、金属元素に基づいてRu30mol%/Sn70mol%の組成を有する試料を4つ得た。金属含量に基づいて計算すると、それぞれの場合において、これは、7.7g/m2のルテニウム適用量に相当する。それぞれの場合において、これは、(酸化物RuO2、SnO2の和として)36.9g/m2の総被膜適用量に相当する。
【0083】
得られた被膜を、被覆液中に存在する異なった量(39mg、29.2mg、9.7mgまたは0mg)の硝酸ランタン(III)六水和物に対応して、La39、La29、La9、La0と表す。
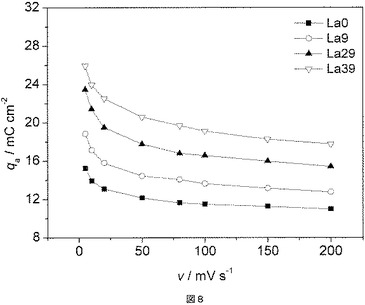
実施例9に記載したようにサイクリックボルタンメトリーによって電極を試験し、実施例9に記載したようにボルタンメトリー容量電荷(qa)を測定した。図8に、電極の電位走査速度(v)の関数としてのボルタンメトリー電荷(qa)を示す。電位走査速度が5mV/sから200mV/sに上昇すると、ボルタンメトリー電荷(qa)はかなり低下する。このことは、被膜が割れを有する多孔性構造であることを示している。硝酸ランタン(III)六水和物の量が上昇するにつれて、容量電荷も上昇した。酸化ランタンの溶解の結果、より多孔性の被膜が得られる。
【0084】
実施例13
割れのある多孔性電気触媒活性被膜を製造するために、実施例1と同じ方法でチタン基材を前処理した。
被覆液を調製するために、99.6mgのルテニウムアセチルアセトネート(Ru(acac)3)、192.4μlのチタンイソプロポキシド(Ti(i−OPr)4)および26.6mgのバナジルアセチルアセトネート(VO(acac)2)をそれぞれ、1.45mlのイソプロパノールおよび1.45mlのプロピオン酸に溶解し、各々の場合に、激しく撹拌しつつ還流しながら150℃で30分間加熱した。室温に冷却後、3つの溶液を混合し、ワインレッド色の均一かつ澄明な溶液を得た。溶液に72.2mgのポリビニルピロリドンK30(PVP)(平均分子量Mw=40,000)を添加し、溶液を超音波で30分間処理した。この被覆液50μlを、マイクロピペットを用いてチタン基材に滴下し、次いで、空気中で乾燥した。層を空気中、まず250℃で15分間、次いで450℃で20分間焼成した。これらの手順(被覆液の適用、乾燥、焼成)を8回繰り返した。その後、被覆チタン基材を450℃で1時間焼成した。
これにより、金属元素に基づいてRu25mol%/Ti65mol%/V10mol%の組成を有する試料を得た。金属含量に基づいて計算すると、これは、6.4g/m2のルテニウム適用量に相当する。これは、(酸化物RuO2、TiO2、V2O5の和として)23.9g/m2の総被膜適用量に相当する。
【0085】
Ag/AgCl参照電極を用い、室温で、3.5mol塩化ナトリウム溶液中でサイクリックボルタンメトリーを実施した。電位は、v=50mV/sの電位走査速度で、(Ag/AgClに対して)0.2〜1.0Vの範囲で変化した。ボルタンメトリー容量電荷(qa)は、実施例9に記載した様に測定した。割れを有する多孔性層について、39.2mC/cm2の値を得た。
【0086】
比較のために、ポリビニルピロリドン(PVP)を添加しなかったこと以外は同じ被覆液を用い、同じ製造方法で電極を製造し、同じ方法でボルタンメトリー容量電荷(qa)を測定した。PVPを添加しなかった被膜のボルタンメトリー容量電荷(qa)について、20.8mC/cm2の値を得た。
【技術分野】
【0001】
本発明は、電気分解による塩素製造のための既知の貴金属触媒含有電極から出発している。本発明の分野は、向上した塩素回収のための電極用触媒被膜の製造方法(被膜は、導電性支持体材料に適用される)、およびこの方法により得ることができる新規な電極に関する。被膜は特に、緻密な割れのない基層および高度に多孔性のカバー層からなる。基層およびカバー層は例えば、Ti、RuまたはIrと遷移金属系からのドーピング元素の1種以上とに基づく混合酸化物からなる。いずれの層も、続く熱処理を伴ったゾル−ゲル変法によって、導電性支持体に適用される。
【背景技術】
【0002】
工業的な塩素の製造は、塩化ナトリウム溶液または塩酸溶液の電気分解によって実施されている。早期に使用されていたアマルガム法に代わるものとして、隔膜法およびイオン交換膜法が今日もっぱら使用されている。塩素の製造における最大のコスト要因は、電力量である。1トンの塩素を製造するために消費される電力は、塩化アルカリの電気分解(膜法)において、典型的には約2500kWhである。アマルガム法は、更に大量のエネルギーを消費する。塩素製造におけるコストを削減するためには、電解槽の設計によって、膜、電解液および電極で生じるオーム抵抗を著しく低下させる。エネルギーの消費を更に削減するため、電気分解における分解電位を低下する触媒を電極に適用する。塩素発生における陽極での電荷移動のための過電圧は、材料、表面形態、および触媒製造方法に大きく依存する。近年の陽極は通常、支持体としてのチタン、ニッケル、タンタルまたはジルコニウムで作られたエキスパンデッドメタル板に基づく。白金族元素の酸化物または酸化物混合物が通常、支持体表面に適用されている。ここでは、二酸化ルテニウム(RuO2)が塩素発生に適した触媒であることが分かっている。電気触媒活性材料としてのRuO2の重大な欠点は、電気分解中に触媒被膜が溶解するので、製造された既知の電極被膜の長期安定性が乏しいことである。そのため、使用中の陽極の触媒活性は連続的に低下する。活性ルテニウム成分を安定化させるために、例えばRuO2と二酸化チタンの混合物を塩化アルカリの電気分解において使用する。RuO2およびTiO2は、同じ結晶構造を有するので、薬品攻撃およびRuO2脱離を防ぐ固溶体を生成する。この効果は、US 3 632 498に記載されている。US 3 562 008の発明者らは、50mol%のルテニウム含量を有するRuO2−TiO2で被覆された陽極が、未被覆Pt陽極と比べて、塩素発生のための低下された槽電圧と改善された安定性を示すことを見出した。
【0003】
様々な材料および被膜形態を使用することによる、塩素発生における電気触媒効果への影響は、様々な文献に記載されているが、その効果は常には互いに完全に独立してはいない。
【0004】
DE 40 32 417 A1は、傾斜構造を有するRuO2−TiO2被膜を記載している。同文献では、層内のルテニウム含量が、陽極表面方向に40mol%から20mol%に低下している。従って、陽極酸化は、電解液との界面で実質的にもっぱら起こり、このようにして、寿命を短縮する体積侵食を回避する。
【0005】
EP 0 867 527 A1は、表面方向に適用された第一層から適用された最外層に向かって13mol%から100mol%に増加する、バルブ金属酸化物に対する貴金属酸化物の比の傾斜を有する、様々な電極用途のためのTiO2、RuO2およびIrO2からなる三成分酸化物混合物の製造方法を記載している。最外層は貴金属酸化物のみからなるので、少なくとも塩素製造の場合は、先に記載した純粋な貴金属酸化物の欠点、特に不十分な長期安定性が予想される。
【0006】
US 4 517 068では、塩素発生における改善された性能を、TiO2−RuO2と酸化パラジウムを組み合わせることによって達成している。
【0007】
T.A.F. Lassaliら(Electrochimica Acta 39, 1545 (1994))は、TiO2をPtOxで置換すると、電気化学的活性領域が劇的に増加し、従って、電気触媒活性が増加することを見出した。T.A.F. Lassaliらは、Ru0.3PtxTi0.7−xO2で示される層組成物を記載している。触媒層は、金属塩(例えば金属ハロゲン化物)の熱分解によって製造されている。欠点は、貴金属である白金を入手するコストが高いことである。更に、Pt含有被膜の化学的不安定性(ヘキサクロロプラチナート溶液としての白金の放出)は被膜の寿命を短縮する場合があり、従って、工業的使用を困難する。
【0008】
WO 2006/028443 A1もまた、Ruおよび酸化イリジウムに加えてドーピング元素としてのアンチモン、錫およびタンタルも含有できるパラジウム含有触媒混合酸化物層の、種々の金属塩の熱分解による製造方法を記載している。同文献では、非ドープ層と比べて100mV低下した、塩素発生のための過電圧が得られた。
【0009】
更に、金属基材に単一層として適用されたドープ触媒層および非ドープ触媒層の、熱分解による製造方法は、以下の文献から知られている:J. Electrochem. Soc. 129 1689 (1982)、Electrochimica Acta 42, 3525 (1997)、J. Alloys and Compounds 261, 176 (1997)。
【0010】
S. Trasatti, Electrochim. Acta, 36, 225 (1991)およびG.R.P. Malpassら、Electrochim. Acta, 52, 936 (2006)は、常套の熱分解法によって製造した電気触媒活性酸化物被膜の典型的な形態を記載している。酸化物被膜における欠陥、例えば、割れ、細孔、孔および粒界は、電解液が内部触媒活性部位を高度に利用できるようにし、従って、塩素生成反応に対する見掛けの電気触媒活性を高める。
【0011】
一方で、電解液は、電気触媒活性酸化物層における欠陥を通ってチタン基材まで浸透し、チタン基材を攻撃することができる。これは、電気触媒活性酸化物層の剥離を生じさせ、金属支持体材料と活性酸化物層の間の電気絶縁酸化チタン中間層の成長も促進し、その後の抵抗損の増加および陽極の不活性化をもたらす。このことは、L. M. Da Silvaら、J. Electroanal. Chem. 532, 141 (2002)に記載されている。
【0012】
電気分解において使用される電解液(例えば塩素の製造における塩化ナトリウムまたは塩化水素の水溶液)との接触に起因する支持体材料の不動態化を回避するために、様々な対策が開発されてきた。
【0013】
EP 0046449 A1に記載されているように、層厚さを増しかつ被覆剤の寿命を延長するため、何回もの被覆剤適用/焼成サイクルを通常採用する。適用被覆層における割れおよび孔は、その次の被覆剤適用において被覆液で少なくとも部分的に埋められる。内部欠陥の数は、更なる適用サイクル毎に低減される。
【0014】
前記方法の欠点は、多数の適用サイクルを必要とし、材料を大量に消費することである。別の欠点は、欠陥の数が少ないので、電気化学的活性領域、すなわち電気触媒活性が低減されることである。このことは、電気分解において電力量の消費が増加することにつながる。更に、被覆層の導電率は支持体材料と比べて小さいので、過度に大きい層厚さでは、電気分解における抵抗損が増加し、槽電圧が上昇することになる。
【0015】
導電率を有さないかまたは非常に小さい導電率しか有さない酸化チタン中間層の形成を回避するため、バルブ金属支持体の上側および電気触媒活性外層の下側に位置する中間層の概念が生まれてきた。そのような中間層は、用語「下層」、「バリア層」、「保護層」または「基層」によって表すこともでき、外層は「カバー層」と称することもできる。
【0016】
基層を得る1つの可能な方法は、例えばHayfield, Precious Metal Review 27(1) 1983_2-8に、チタンの陰極腐蝕防止のために記載されているような、金属白金の適用である。同方法を塩素製造において使用することについての欠点は特に、化学的不安定性(ヘキサクロロプラチナート溶液の生成)、および貴金属白金を得ることに起因する高コストである。
【0017】
別の文献には、熱分解、プラズマビームまたはプラズマ溶射による、より安定な貴金属不含有中間層の電気化学的製造方法が記載されている。US 3 882 002では、アンチモンドープ酸化錫を保護層としてバルブ金属基材に適用しており、貴金属外層も伴っている。US 3 950 240は、中間層としてのニオブドープ酸化錫と酸化ルテニウムからなるカバー層を有する陽極を記載している。US 7 211 177 B2は、塩酸の電気分解のための、炭化チタンまたはホウ化チタンからなる中間層を記載している。
【0018】
前記中間層全ての欠点は、保護層と支持体材料の間および中間層と電気触媒活性カバー層の間の接着が最適とは言えないことである。
【0019】
酸化ルテニウム系電極層の表面積を増大させることを、Y. Takasu(Electrochemica Acta 45, 4135 (2000))は、以下の5つの異なった方法を用いて調べている。
(1)浸漬被覆RuO2−Ti電極のためのアルコール性浸漬液への炭酸ナトリウムの添加、
(2)浸漬被覆によるRuO2−MOx(M:ドーピング金属)の製造、
(3)浸漬被覆およびその後の酸を用いた希土類酸化物の化学溶解による、チタン電極へのRuO2−ROx(R:希土類元素)層の製造、
(4)超微細RuO2粒子を製造するためのゾル−ゲル法における、触媒としての炭酸水素アンモニウム塩の添加、および
(5)RuO2のHxRuOy層構造への転化。
【0020】
表面構造は、製造した電極のサイクリックボルタンメトリー電荷を測定することによって電気化学的に特徴付けられた。RuO2−TiO2タイプの混合酸化物電極の場合、サイクリックボルタンメトリー電荷は12mC/cm2であり、RuO2−SnO2の場合は13mC/cm2である。最大サイクリックボルタンメトリー電荷は、RuO2−VOx系(162mC/cm2)、RuO2−MoO3系(120mC/cm2)、RuO2−CaO系(130mC/cm2)に対して得られた。しかしながら、このような方法では一般に、均一な混合酸化物ではなく、異なった酸化物の不均一な混合物が得られ、塩素製造用触媒としての純RuO2に対する先に記載した欠点を伴う。前記方法(1)、(3)、(4)および(5)によって得られた被覆組成物も、これらの欠点を示す。塩素製造用被膜の使用についての使用例は記載されていない。
【0021】
これまで開示されてきた方法により、20〜50mol%の範囲の典型的な貴金属含量を有する触媒層を製造することはできる。貴金属塩および貴金属自体は、多数の工程段階において複雑な方法で得るしかないので、大規模な工業プロセスに使用するための技術的支出は非常に高く、利用は制限される。
【0022】
RuO2を遷移金属(Ce、Nb、Sn、V、Cr、Mn、Co、Sb、Zr、Mo、W、Ta)酸化物により部分的に置換することによって、単純RuO2−TiO2系においてルテニウム含量を低下できることは示されてきた。加えて、活性、選択性および安定性の改善は、種々の遷移金属酸化物を組み合わせることによる相乗効果によって達成することができる。実験データは、S. V. Evdokimov らによって、Russian Journal of Electrochemistry 38, 657 (2002)に記載されている。10mol%のCrNbO4の添加によって、標準系RuO2−TiO2中のRuO2含量を、70mol%から30mol%に低下させることができる。このとき、電気触媒活性は、標準系と同程度である。
【0023】
US 3 776 834は、RuO2をSnO2により部分的に置換したRuO2(19mol%)、SnO2(13mol%)、TiO2(68mol%)の三成分系を使用した結果、系RuO2(33.3mol%)−TiO2(66.7mol%)を用いた塩素発生の過電圧を40mV低下できることを記載している。ドーピング元素の使用により、耐化学腐蝕性を高めることもできる。US 4 039 409およびUS 3 948 751は、ドーピング元素の塩と熱的に不安定なRu塩とを混合し、得られた混合物を適用法でチタンに直接適用することによる、ドープ触媒層の製造方法を記載している。最終的な熱処理により、ドープ触媒層が形成される。層の組成は、ドーピング元素の塩の量によって制御することができる。この方法は包括的にY. E. Roginskayaら、Electrochimica Acta 40, 817 (1995)に記載されている。同方法の欠点は、複数の成分からなる被膜が非常に不均一な微細構造を有することである。これらの知見はUS 4 668 531にも記載されている。500℃を超える温度で、触媒層と支持体との間に、緻密な電気的に絶縁されたTiO2層が生じる。そのような中間層は、電極の性能を低下させる。この関係は、WO 2008/046784 A1に記載されている。
【0024】
ゾル−ゲル法が、貴金属塩の熱分解に対する非常に良好な代替法であることが見出された。ゾル−ゲル法では、前駆体化合物の加水分解および縮合を制御することによって、混合酸化物を目的に応じて調製することができる。
【0025】
CN 1900368 A1によれば、ゾル−ゲル法によって、酸化錫、酸化イリジウム、酸化マンガンおよび酸化コバルトであるドープ種を、RuO2および高含量のCeO2からなる陽極被膜に導入する。発明者らは、ドープ三成分被膜またはドープ四成分被膜(45mol%のRuO2を含有する)についての塩素発生活性が、二成分RuO2/CeO2系と比べて増加したことを見出した。V. V. Panicら、Colloids and Surfaces A 157, 269 (1999)に記載されているように、ゾル−ゲル法によって製造した触媒被膜は、常套法(塩の熱分解)で製造した層と比べて増加した安定性および長い寿命を示す。Y. Zengら、Ceramics International 33, 1087 (2007)は、ゾル−ゲル法によって製造したRuO2−IrO2−TiO2触媒層が、減小した粒度の故に増加した活性を示すことも報告している。
【0026】
しかしながら、既知のゾル−ゲル法は、有機溶媒中での無機塩およびアルコキシドの溶解性に関して、特有の欠点を示す。十分な溶解性を得るために、強酸での変性と超音波処理が必要とされる。これらの処理は、製造工程を著しく長くする。このようにして調製した被覆液の幾つかは、溶解度の小さい成分が(特に高い濃度で)尚早に再沈澱するので、低い安定性しか有さない。そのような液は、貯蔵することができないし、極端な場合には、電極上に不均一な被膜を形成する。
【0027】
被膜の先に記載した欠点(すなわち高い貴金属含量、不十分な選択性および安定性、並びに導電性の不足)および被膜の製造方法の先に記載した欠点を示さない、新規な被膜およびその製造方法が要求されている。
【先行技術文献】
【特許文献】
【0028】
【特許文献1】US 3 632 498
【特許文献2】US 3 562 008
【特許文献3】DE 40 32 417 A1
【特許文献4】EP 0 867 527 A1
【特許文献5】US 4 517 068
【特許文献6】WO 2006/028443 A1
【特許文献7】EP 0046449 A1
【特許文献8】US 3 882 002
【特許文献9】US 3 950 240
【特許文献10】US 7 211 177 B2
【特許文献11】US 3 776 834
【特許文献12】US 4 039 409
【特許文献13】US 3 948 751
【特許文献14】US 4 668 531
【特許文献15】WO 2008/046784 A1
【特許文献16】CN 1900368 A1
【非特許文献】
【0029】
【非特許文献1】T.A.F. Lassaliら、Electrochimica Acta 39, 1545 (1994)
【非特許文献2】J. Electrochem. Soc. 129 1689 (1982)
【非特許文献3】Electrochimica Acta 42, 3525 (1997)
【非特許文献4】J. Alloys and Compounds 261, 176 (1997)
【非特許文献5】S. Trasatti, Electrochim. Acta, 36, 225 (1991)
【非特許文献6】G.R.P. Malpassら、Electrochim. Acta, 52, 936 (2006)
【非特許文献7】L. M. Da Silvaら、J. Electroanal. Chem. 532, 141 (2002)
【非特許文献8】Hayfield, Precious Metal Review 27(1) 1983_2-8
【非特許文献9】Y. Takasu(Electrochemica Acta 45, 4135 (2000)
【非特許文献10】S. V. Evdokimov ら、Russian Journal of Electrochemistry 38, 657 (2002)
【非特許文献11】Y. E. Roginskayaら、Electrochimica Acta 40, 817 (1995)
【非特許文献12】V. V. Panicら、Colloids and Surfaces A 157, 269 (1999)
【非特許文献13】Y. Zengら、Ceramics International 33, 1087 (2007)
【発明の概要】
【発明が解決しようとする課題】
【0030】
従って、本発明の目的は、ゾル−ゲル法に基づいた、特に塩素製造用陽極のための、電極用触媒層の簡単かつ汎用的な製造方法を、先に記載した欠点を解消しつつ開発することである。この層は、電気触媒活性成分、および長期安定性を確実にする安定剤からなる。更に、陽極は、先行技術に比べて、塩素過電圧および貴金属含量の低下を示さなければならない。
【課題を解決するための手段】
【0031】
本発明の態様は、導電性基材および触媒活性層を含んでなる電極であって、
触媒活性層が、2種の触媒活性成分に基づき、金属酸化物または混合酸化物または酸化物混合物としてイリジウム、ルテニウムまたはチタンを含んでなり、
元素イリジウム、ルテニウムおよびチタンの和に基づいたルテニウムおよび/またはイリジウムの総含量が、少なくとも10mol%であり、
電極が、導電性基材に適用された、NaClおよび/またはNaOHおよび/またはHCl含有水性電解液を透過しない少なくとも1つの酸化物基層を含んでなる、
電極である。
【0032】
本発明の別の態様は、少なくとも導電性基材および触媒活性層を含んでなる電極であって、
触媒活性層が、2種の触媒活性成分に基づき、金属酸化物または混合酸化物または酸化物混合物としてイリジウム、ルテニウムまたはチタンを含んでなり、
元素イリジウム、ルテニウムおよびチタンの和に基づいたルテニウムおよび/またはイリジウムの総含量が、少なくとも10mol%であり、
ルテニウムおよび/またはイリジウムの半分までが、バナジウム、ジルコニウムまたはモリブデンによって置換されている、
電極である。
【0033】
本発明の更に別の態様は、ルテニウム、イリジウム、チタンおよびそれらの混合物からなる群から選択される金属を含んでなる金属化合物の溶液または分散体を含んでなるゾル−ゲル被覆液を、導電性支持体に適用する工程、
溶媒を除去するために乾燥する工程、
酸素含有気体の存在下、少なくとも350℃の温度で焼成する工程、および
任意に、ゾル−ゲル被覆液の適用、乾燥および焼成を1回以上繰り返す工程
を含む、電極の製造方法である。
【0034】
前記した本発明の態様、および以下に記載する発明の詳細な説明は、添付の図面と併せて読むと、より良好に理解することができる。本発明の説明を補う目的で、例と考えられる代表的な態様を図面にそれぞれ示す。しかしながら、本発明が、どのような形でも、示されたまさにその態様および手段に限定されないことは理解すべきである。
【図面の簡単な説明】
【0035】
【図1】実施例6の被膜の表面の走査型電子顕微鏡写真を示す(スケール:a)10μm)。
【図2】実施例7の被膜の表面の走査型電子顕微鏡写真を示す(スケール:10μm)。
【図3】実施例8の被膜の表面の走査型電子顕微鏡写真を示す(スケール:10μm)。
【図4】実施例9bの被膜の表面の走査型電子顕微鏡写真を示す(スケール:10μm)。
【図5】実施例9の緻密基層(実線)および割れ構造物(点線)についての、電位走査速度(v)の関数としてプロットしたボルタンメトリー電荷(qa)を示す。
【図6】実施例10の被膜の表面の走査型電子顕微鏡写真を示す(スケール:10μm)。
【図7】実施例11のRu0.4Ti0.6O2被膜(実線)およびRu0.4Ti0.45La0.15O2被膜(点線)についての、サイクリックボルタモグラム数の関数としてプロットしたボルタンメトリー電荷(qa)を示す。
【図8】実施例12の被膜についての、電位走査速度(v)の関数としてプロットしたボルタンメトリー電荷(qa)を示す。
【発明を実施するための形態】
【0036】
本発明の特定の態様は、少なくとも導電性基材および触媒活性被膜を含んでなる新規な電極であって、触媒活性層が、2種の触媒活性成分に基づき、金属酸化物または混合酸化物または酸化物混合物として少なくともイリジウム、ルテニウムまたはチタンを含んでなり、元素イリジウム、ルテニウムおよびチタンの和に基づいたルテニウムおよび/またはイリジウムの総含量が、少なくとも10mol%、好ましくは10〜28mol%、好適には10〜20mol%であり、導電性基材に適用された、NaClおよび/またはNaOHまたはHCl含有水性電解液を透過しない少なくとも1つの酸化物基層が供給されていることを特徴とする電極を提供する。
【0037】
導電性基材は、好ましくはバルブ金属、特に好ましくはチタン、タンタル、ニオブおよびニッケル、或いはこれら金属の、主成分としてチタン、タンタルまたはニオブを含有する合金からなる群の金属に基づく。
【0038】
塩素発生のための新規な触媒被膜は例えば、金属酸化物の総量に基づいて10〜20mol%の含量を有する新規な活性金属成分(好ましくはRuO2またはRuO2/IrO2)、安定化成分(好ましくはTiO2)および遷移金属(好ましくは錫、ランタン、バナジウム、ジルコニウム、クロム、モリブデン)酸化物形態のドープ種からなる。ドープ種の濃度は、特に5〜15mol%である。
【0039】
基層が塩化水素水溶液、塩化ナトリウム水溶液または水酸化ナトリウム水溶液を透過しないことを特徴とする電極が好ましい。これは例えば、サイクリックボルタンメトリー実験(pH=3の3.5mol塩化ナトリウム水溶液中または0.5mol塩酸中、室温、Ag/AgCl参照電極、(Ag/AgClに対して)0.2〜1.0Vの走査範囲)において5mV/s〜200mV/sの電位走査速度範囲でサイクリックボルタモグラムの陽極ブランチを積分することによって測定されるサイクリックボルタンメトリー容量電荷(qa)が、常に10mC/cm2未満、好ましくは5mC/cm2未満、特に好ましくは2mC/cm2未満の場合である。
【0040】
基層より大きいサイクリックボルタンメトリー容量電荷(qa)を有するカバー層が付加的に供給されている電極の態様が特に好ましい。カバー層のサイクリックボルタンメトリー容量電荷(qa)は、好ましくは少なくとも10mC/cm2、特に好ましくは20mC/cm2である。
【0041】
基層は特に、0.1〜20g/m2、好ましくは0.5〜10g/m2の(酸化物としての)単位面積当たりの適用量を有し、カバー層は特に、少なくとも2g/m2、好ましくは少なくとも5g/m2の(酸化物としての)単位面積当たりの適用量を有する。
【0042】
カバー層が少なくとも2g/m2、好ましくは少なくとも5g/m2の適用量(単位面積当たりの重量)を有する電極が好ましい。新規な電極の特定の態様は、カバー層が、層厚さ方向の断面で観察して、チタンに対するイリジウムのおよび/またはチタン成分に対するルテニウムの変化する比を有することを特徴とする。
【0043】
電極の1つの態様では、カバー層におけるチタンに対するイリジウムのおよび/またはチタンに対するルテニウムの比は、層厚さ方向の断面で観察して、外側から導電性支持体に向かって低下している。
【0044】
陽極被膜の貴金属酸化物および安定化成分(例えばTiO2)を製造するための出発化合物として、塩化物、硝酸塩、アルコキシド、アセチルアセトネートを使用することが好ましい。ルテニウムアセチルアセトネート、イリジウムアセチルアセトネートまたは酢酸イリジウムが、貴金属成分を調製するための前駆体塩として好ましく使用される。TiO2は、例えばチタンイソプロポキシドまたはチタンブトキシドから得ることができる。ドープ種は特に好ましくは、前駆体塩であるバナジウムアセチルアセトネート、バナジウムテトラブトキシド、ジルコニウムn−プロポキシド、硝酸ジルコニウム、酢酸モリブデン、酢酸錫、錫イソプロポキシド、ランタンアセチルアセトネート、硝酸ランタンによって導入される。
【0045】
電極の別の態様は、カバー層が触媒活性層成分を含んでなり、孔形成組成物(特に、ランタン(とりわけ酸化ランタン)またはポリマー(とりわけポリビニルピロリドン)の組成物)を付加的に含有することを特徴とする。
【0046】
電極の別の態様では、基層は、導電性であり、少なくとも10S/m、好ましくは少なくとも1,000S/cm、特に好ましくは少なくとも10,000S/mの導電率を有する。
【0047】
先に記載した電極構造物の他に、本発明の別の態様である、少なくとも導電性基材および触媒活性被膜を含んでなる電極の別の態様も見出された。この態様は、触媒活性層が、2種の触媒活性成分に基づき、金属酸化物または混合酸化物または酸化物混合物として少なくともイリジウム、ルテニウムまたはチタンを含有し、元素イリジウム、ルテニウムおよびチタンの和に基づいたルテニウムおよび/またはイリジウムの総含量が、少なくとも10mol%、好ましくは10〜28mol%、好適には10〜20mol%であり、ルテニウムおよび/またはイリジウムの半分までが、バナジウム、ジルコニウムまたはモリブデン(好ましくはバナジウム)によって置換されていることを特徴とする。
【0048】
この選択された構造物は好ましいことに、導電性支持体に適用された、NaClおよび/またはNaOHまたはHCl含有水性電解液を透過しない酸化物基層も供給するために、最初に記載した電極の態様と組み合わせることができる。
【0049】
本発明の態様は更に、電極、特に先に記載した新規な電極の製造方法であって、第一の工程において、金属であるルテニウム、イリジウムおよびチタンの1種以上の金属化合物の溶液または分散体を含有するゾル−ゲル被覆液を、特に浸漬によって、導電性支持体に1回以上適用し;次いで、溶媒を除去するために乾燥し;続いて、乾燥金属化合物層を酸素含有気体の存在下、高温で、特に少なくとも350℃で、好ましくは少なくとも400℃で焼成し;任意に、溶液または分散体の適用、乾燥および焼成を1回以上繰り返すことを特徴とする方法を提供する。
【0050】
カバー層を製造するためには、ルテニウム、イリジウムおよびチタンからなる群からの金属の金属塩の溶液または分散体を基層に1回以上適用し、溶媒を除去し、酸素含有気体の存在下、高温で、特に少なくとも350℃で、好ましくは少なくとも450℃で焼成する方法が好ましい。
【0051】
製造方法の一態様では、基層を製造するために、金属塩液を適用した後、乾燥を高温で、特に少なくとも200℃で、好ましくは少なくとも240℃で実施する。
【0052】
1つの変法では、低級カルボン酸、特にプロピオン酸、C1〜C5アルコールまたはケトン或いはそれらの混合物を、基層および/またはカバー層を製造するための金属化合物液に添加する。
【0053】
均一かつ安定な被覆液を調製するため、プロピオン酸ゾル−ゲル法を使用することが特に好ましい。同方法では、プロピオン酸とアルコール(メタノール、エタノール、n−プロパノール、イソプロパノール、ブタノール)の様々な濃度の混合物が、先に記載した化合物の溶媒として使用される。前駆体塩の溶解は、各前駆体塩について独立して、撹拌しながら130℃を超える温度で起こる。溶解工程の時間は約1時間である。これにより、澄明かつ安定なゾル液を得る。プロピオン酸を使用することにより、使用した金属カチオンが錯体を形成するので、制御された加水分解が可能となる。
【0054】
冷却後、各前駆体塩の溶液を混合し、次いで撹拌する。この混合物から、電気触媒活性塩素発生陽極を製造するための被覆液を調製する。この被覆液は、チタンシートまたはチタン網のような基材(例えばエキスパンデッドチタンメタル)に適用することができる。適用前に、基材を物理的、化学的または電気化学的に、清浄し、磨き、粗化しなければならない。これにより、被膜の接着性を向上させる。
【0055】
常套法である塩の熱分解と比べて、ゾル−ゲル法は以下の利点を有する:
・低いプロセス温度、
・制御された化学量論量を有する混合酸化物が、異なった化合物のゾルを混合することによって容易に調製できること、
・出発物質の混合が分子レベルで起こるので、生成物の均一性が高いこと、
・最終熱処理により単純な有機基が完全に除去されるので、生成物の純度が高いこと、
・最終熱処理により、得られる層の微細構造(多孔性)に影響を与え得ること、
・浸漬被覆法、噴霧被覆法または回転被覆法により複雑な形態を被覆できること。
【0056】
ゾル−ゲル法により、溶媒(メタノール、エタノール、イソプロパノール、ブタノール)に溶解されている1種以上の易加水分解性アルコキシド、アセチルアセトネートまたは無機塩を含んでなる被覆液を調製することができる。安定なコロイド(ゾル)を調製するために、被覆液を酸触媒(塩酸、硝酸または酢酸)或いは塩基触媒(アンモニア、水酸化ナトリウム溶液)を用いて加水分解する。加水分解により、ヒドロキシ架橋およびオキソ架橋金属原子の網状構造が形成される。
【0057】
次いで、この被覆液で基材を被覆することができる。続く乾燥工程では、ゾルが基材に固定される。最終の焼成工程では、有機成分は除去され、混合酸化物はナノ構造粒子に結晶化する。ゾル−ゲル法によって、ドープ種を導入することもできる。これらのドープ種は、触媒の活性を高めることができるので、貴金属の含量を低下させることができる。
【0058】
被覆液の適用は、浸漬法、塗布法、滴下法、噴霧法または回転法によって実施することができる。次いで、特に室温で、層を乾燥し、その後、例えば250℃で少なくとも10分間、それに続いて450℃で少なくとも5分間焼成する。層を安定化するための最終の焼成工程は、例えば450℃で12〜30分間実施する。成分の酸化を向上させるため、純酸素または酸素リッチ雰囲気を使用することができる。記載した方法の逐次的繰り返し数を変えることによって、層厚さを変えることができる。この手順によって多層構造を得ることもできる。
【0059】
本発明の態様は更に、塩化ナトリウムまたは塩化水素含有液を電気分解するための電解槽であって、先に記載した新規な電極が電解槽の陽極として供給されている電解槽を提供する。
【0060】
本発明の別の態様は、塩素を電気化学的に製造するために塩化ナトリウムまたは塩化水素を電気分解するための電解槽における陽極としての、先に記載した新規な電極の使用も提供する。
【0061】
先に記載した引用文献の全ては、有用な目的全てのために、それらの全内容を引用してここに組み込む。
【0062】
本明細書では、用語および/または文脈が別途明確に記載していない限り、単数形のための用語「a」および「the」は、「1以上」および「少なくとも1」と同義であり、互いに置き換えて使用することができる。従って、例えば、明細書または特許請求の範囲における「a catalytically active component」は、「1種の触媒活性成分」または「1種以上の触媒活性成分」に言及し得る。また、数値の全ては、特に記載のない限り、用語「約」によって修飾されていると理解すべきである。
【0063】
本発明を具体的に表す特定の構造物を示し、記載しているが、本発明の概念の意図および範囲から逸脱することなく、その一部を様々に変更および修正できること、並びにそれらが本明細書に示され、記載されている特定の形態に限定されないことは当業者には明らかであろう。
【0064】
本発明の態様を、後記実施例および図面によって説明するが、それらは本発明を何ら限定するものではない。
【0065】
図1は、実施例6の被膜の表面の走査型電子顕微鏡写真を示す(スケール:a)10μm)。
図2は、実施例7の被膜の表面の走査型電子顕微鏡写真を示す(スケール:10μm)。
図3は、実施例8の被膜の表面の走査型電子顕微鏡写真を示す(スケール:10μm)。
図4は、実施例9bの被膜の表面の走査型電子顕微鏡写真を示す(スケール:10μm)。
図5は、実施例9の緻密基層(実線)および割れ構造物(点線)についての、電位走査速度(v)の関数としてプロットしたボルタンメトリー電荷(qa)を示す。
図6は、実施例10の被膜の表面の走査型電子顕微鏡写真を示す(スケール:10μm)。
図7は、実施例11のRu0.4Ti0.6O2被膜(実線)およびRu0.4Ti0.45La0.15O2被膜(点線)についての、サイクリックボルタモグラム数の関数としてプロットしたボルタンメトリー電荷(qa)を示す。サイクリックボルタンメトリーは、Ag/AgCl参照電極を用い、室温で、0.5mol塩酸溶液中で実施した。電位は、v=50mV/sの電位走査速度で、(Ag/AgClに対して)0.2〜1.0Vの範囲で変化させた。
図8は、実施例12の被膜についての、電位走査速度(v)の関数としてプロットしたボルタンメトリー電荷(qa)を示す。サイクリックボルタンメトリーは、Ag/AgCl参照電極を用い、室温で、3.5mol塩酸溶液中で実施した。電位は、(Ag/AgClに対して)0.2〜1.0Vの範囲で変化させた。
【実施例】
【0066】
実施例1
15mmの直径を有するチタン板(厚さ2mm)にサンドブラストを掛け、表面を清浄および粗化し、次いで、10%濃度シュウ酸中、80℃で2時間酸洗いし、続いて、イソプロパノールで清浄して窒素流中で乾燥した。
被覆液を調製するため、99.6mgのルテニウムアセチルアセトネート(Ru(acac)3)、207.2μlのチタンイソプロポキシド(Ti(i−OPr)4)および13.3mgのバナジルアセチルアセトネート(VO(acac)2)をそれぞれ、1.45mlのイソプロパノールおよび1.45mlのプロピオン酸に溶解し、還流しながら30分間加熱した。室温に冷却後、3つの溶液を混合し、ワインレッド色の均一かつ澄明な溶液を得た。この被覆液50μlを、マイクロピペットによってチタン基材に適用し、次いで、空気中で乾燥した。層を空気中で、まず250℃で10分間、次いで450℃で10分間焼成した。これらの手順(被覆液の適用、乾燥、焼成)を8回繰り返した。9回目の被覆工程後、被覆チタン基材を450℃で1時間焼成した。これにより、金属元素に基づいてRu25mol%/Ti70mol%/V5mol%の組成を有する試料を得た。金属含量に基づいて計算すると、これは、6.4g/m2のルテニウム適用量に相当する。これは、(酸化物RuO2、TiO2、V2O5の和として)23.7g/m2の総被膜適用量に相当する。
塩素発生のための電気触媒活性は、クロノアンペロメトリー(参照電極:Ag/AgCl、電解液:3.5MのNaCl、pH:3、室温)によって測定した。実験では、定常電流密度を1kA/m2に設定した。得られた電位は1.17Vであった。
【0067】
実施例1b(比較例)
実施例1と同じ方法で、チタン基材を前処理した。
熱分解によって被膜を製造するために、2.00gの塩化ルテニウム(III)水和物(Ru含量=40.5重量%)、21.56gのn−ブタノール、0.94gの濃塩酸および5.93gのチタン酸テトラブチルTi−(O−Bu)4を含有する被覆液を調製した。被覆液の一部を、小さいチタン板にハケ刷りした。これを空気中、80℃で10分間乾燥し、次いで、空気中、470℃で10分間処理した。この手順(被覆液の適用、乾燥、熱処理)を合計8回実施した。続いて、板を空気中、520℃で1時間処理した。単位面積当たりのルテニウムの適用量は、被覆液の消費量から16g/m2と算出された。これは、31mol%のRuO2および69mol%のTiO2の組成で(酸化物として)49.2g/m2の総被膜適用量に相当する。
(実施例1と同じ方法で測定した)塩素発生のための電位は、この試料では1.25Vであった。
【0068】
実施例2
実施例1と同じ方法で、チタン基材を前処理した。
60mgのルテニウムアセチルアセトネート(Ru(acac)3)、236.8μlのチタンイソプロポキシド(Ti(i−OPr)4)および13.3mgのバナジルアセチルアセトネート(VO(acac)2)をそれぞれ、1.45mlのイソプロパノールおよび1.45mlのプロピオン酸に溶解し、次いで、(撹拌しつつ)還流しながら150℃で30分間加熱した。室温に冷却後、3つの溶液を混合し、ワインレッド色の均一かつ澄明な溶液を得た。実施例1に記載したように、被覆工程および焼成工程を実施した。
これにより、金属元素に基づいてRu15mol%/Ti80mol%/V5mol%の組成を有する試料を得た。金属含量に基づいて計算すると、これは、3.9g/m2のルテニウム適用量に相当する。これは、(酸化物RuO2、TiO2、V2O5の和として)22.7g/m2の総被膜適用量に相当する。
(実施例1と同じ方法で測定した)塩素発生のための電位は、この試料では1.17Vであった。
【0069】
実施例3
実施例1と同じ方法で、チタン基材を前処理した。
99.6mgのルテニウムアセチルアセトネート(Ru(acac)3)、207.2μlのチタンイソプロポキシド(Ti(i−OPr)4)および22.4μlのジルコニウムn−プロポキシド(n−プロパノール中70重量%)をそれぞれ、1.44mlのイソプロパノールおよび1.44mlのプロピオン酸に溶解し、次いで、(撹拌しつつ)還流しながら150℃で30分間加熱した。室温に冷却後、3つの溶液を混合し、ワインレッド色の均一かつ澄明な溶液を得た。実施例1に記載したように、被覆工程および焼成工程を実施した。
これにより、金属元素に基づいてRu25mol%/Ti70mol%/Zr5mol%の組成を有する試料を得た。金属含量に基づいて計算すると、これは、6.4g/m2のルテニウム適用量に相当する。これは、(酸化物RuO2、TiO2、ZrO2の和として)24.2g/m2の総被膜適用量に相当する。
(実施例1と同じ方法で測定した)塩素発生のための電位は、この試料では1.25Vであった。
【0070】
実施例4
実施例1と同じ方法で、チタン基材を前処理した。
99.6mgのルテニウムアセチルアセトネート(Ru(acac)3)、192.4μlのチタンイソプロポキシド(Ti(i−OPr)4)および42.8μlの酢酸モリブデン(Mo2(OCOCH3)4)をそれぞれ、1.45mlのイソプロパノールおよび1.45mlのプロピオン酸に溶解し、次いで、(撹拌しつつ)還流しながら150℃で30分間加熱した。室温に冷却後、3つの溶液を混合し、ワインレッド色の均一かつ澄明な溶液を得た。実施例1に記載したように、被覆工程および焼成工程を実施した。
これにより、金属元素に基づいてRu25mol%/Ti65mol%/Mo10mol%の組成を有する試料を得た。金属含量に基づいて計算すると、これは、6.4g/m2のルテニウム適用量に相当する。これは、(酸化物RuO2、TiO2、MoO3の和として)25.2g/m2の総被膜適用量に相当する。
(実施例1と同じ方法で測定した)塩素発生のための電位は、この試料では1.18Vであった。
【0071】
実施例5
実施例1と同じ方法で、チタン基材を前処理した。
49.8mgのルテニウムアセチルアセトネート(Ru(acac)3)、62.3mgのイリジウムアセチルアセトネート(Ir(acac)3)、16.6mgのバナジウムアセチルアセトネート(VO(acac)2)、207.6mgの錫イソプロポキシドプロパノール(Sn(i−OPr)4・C3H7OH)および129.5μlのチタンイソプロポキシド(Ti(i−OPr)4)をそれぞれ、1.11mlのイソプロパノールおよび1.11mlのプロピオン酸に溶解し、次いで、(撹拌しつつ)還流しながら150℃で30分間加熱した。室温に冷却後、5つの溶液を混合し、ワインレッド色の均一かつ澄明な溶液を得た。実施例1に記載したように、被覆工程および焼成工程を実施した。
これにより、金属元素に基づいてRu10mol%/Ir10mol%/V5mol%/Sn40mol%/Ti35mol%の組成を有する試料を得た。金属含量に基づいて計算すると、これは、3.2g/m2のルテニウム適用量に相当する。これは、(酸化物RuO2、IrO2、V2O5、SnO2、TiO2の和として)33.6g/m2の総被膜適用量に相当する。
(実施例1と同じ方法で測定した)塩素発生のための電位は、この試料では1.22Vであった。
【0072】
実施例6
実施例1と同じ方法で、チタン基材を前処理した。
149.4mgのルテニウムアセチルアセトネート(Ru(acac)3)および333.1μlのチタンイソプロポキシド(Ti(i−OPr)4)をそれぞれ、3.25mlのイソプロパノールおよび3.25mlのプロピオン酸に溶解し、次いで、(撹拌しつつ)還流しながら150℃で30分間加熱した。室温に冷却後、2つの溶液を混合し、ワインレッド色の均一かつ澄明な溶液を得た。
浸漬被覆によって、割れのない緻密な被膜を製造した。この目的のため、チタン基材を被覆液に20秒間浸漬し、次いで、167mm/分の速度で被覆液から垂直に取り出し、続いて、空気中で乾燥した。層を空気中で、まず250℃で10分間、次いで450℃で5分間焼成した。浸漬被覆−乾燥−焼成の手順を5回繰り返した。
これにより、金属元素に基づいてRu25mol%/Ti75mol%の組成を有する試料を得た。金属含量に基づいて計算すると、これは、0.16g/m2のルテニウム適用量に相当する。これは、(酸化物RuO2、TiO2の和として)0.59g/m2の総被膜適用量に相当する。
製造した被膜の表面形態を、走査型電子顕微鏡によって特徴付けた(図1参照)。
【0073】
実施例7
実施例1と同じ方法で、チタン基材を前処理した。
被覆液は、実施例6と同じである。
実施例6と同じ方法で、割れのない緻密な被膜を得た。浸漬被覆−乾燥−焼成の手順を15回繰り返した。これにより、金属元素に基づいてRu25mol%/Ti75mol%の組成を有する試料を得た。金属含量に基づいて計算すると、これは、0.50g/m2のルテニウム適用量に相当する。これは、(酸化物RuO2、TiO2の和として)1.84g/m2の総被膜適用量に相当する。
製造した被膜の表面形態を、走査型電子顕微鏡によって特徴付けた(図2参照)。
【0074】
実施例8
実施例1と同じ方法で、チタン基材を前処理した。
4.62mlのイソプロパノールに421.1mgの塩化ルテニウム(III)水和物(RuCl3・xH2O、36%Ru)を溶解し、一晩撹拌することによって、被覆液(液A)を得た。1332mlのチタンイソプロポキシド(Ti(i−OPr)4)を、2.246mlの4−ヒドロキシ−4−メチル−2−ペンタノンおよび5mlのイソプロパノールの予備混合液に添加し、30分間撹拌した(液B)。液Aおよび液Bを超音波処理によって混合し、澄明な液を得た。続いて、この液に、25.8μlの酢酸および108μlの脱イオン水を添加した。このように調製した液に蓋をし、室温で一晩撹拌した。
浸漬被覆によって、割れのない緻密な被膜を製造した。この目的のため、チタン基材を被覆液に20秒間浸漬し、次いで、193mm/分の速度で被覆液から垂直に取り出し、続いて、空気中で乾燥した。層を空気中で、まず90℃で30分間、次いで450℃で10分間焼成した。浸漬被覆−乾燥−焼成の手順を6回繰り返した。
これにより、金属元素に基づいてRu25mol%/Ti75mol%の組成を有する試料を得た。金属含量に基づいて計算すると、これは、1.0g/m2のルテニウム適用量に相当する。これは、(酸化物RuO2、TiO2の和として)3.69g/m2の総被膜適用量に相当する。
製造した被膜の表面形態を、走査型電子顕微鏡によって特徴付けた(図3参照)。
【0075】
実施例9
実施例1と同じ方法で、チタン基材を前処理した。
4.81mlのイソプロパノールに210.6mgの塩化ルテニウム(III)水和物(RuCl3・xH2O、36%Ru)を溶解し、一晩撹拌することによって、被覆液(液A)を得た。666.1mlのチタンイソプロポキシド(Ti(i−OPr)4)を、1.123mlの4−ヒドロキシ−4−メチル−2−ペンタノンおよび20mlのイソプロパノールの予備混合液に添加し、30分間撹拌した(液B)。液Aおよび液Bを超音波処理によって混合し、澄明な液を得た。続いて、この液に、12.9μlの酢酸および54μlの脱イオン水を添加した。このように調製した液に蓋をし、室温で一晩撹拌した。
浸漬被覆によって、割れのない緻密な被膜を製造した。この目的のため、チタン基材を被覆液に20秒間浸漬し、次いで、193mm/分の速度で被覆液から垂直に取り出した。湿潤被膜を空気中で乾燥し、空気中、90℃で30分間、次いで450℃で10分間焼成した。浸漬被覆−乾燥−焼成の手順を50回繰り返し、最後の焼成は450℃で1時間実施した。
これにより、金属元素に基づいてRu25mol%/Ti75mol%の組成を有する試料を得た。金属含量に基づいて計算すると、これは、2.0g/m2のルテニウム適用量に相当する。これは、(酸化物RuO2、TiO2の和として)7.38g/m2の総被膜適用量に相当する。
【0076】
実施例9b(比較例)
比較のため、実施例9と同じ被覆液を用いて、亀甲割れ構造を有する被膜を製造した。
チタン基材を実施例1と同じ方法で前処理し、マイクロピペットを用いた滴下法によって50μlの被覆液を適用した。
湿潤被膜を空気中で乾燥し、空気中、90℃で30分間、次いで450℃で10分間焼成した。滴下被覆−乾燥−焼成の手順を4回繰り返し、最後の焼成は450℃で1時間実施した。
これにより、金属元素に基づいてRu25mol%/Ti75mol%の組成を有する試料を得た。金属含量に基づいて計算すると、これは、3.4g/m2のルテニウム適用量に相当する。これは、(酸化物RuO2、TiO2の和として)12.54g/m2の総被膜適用量に相当する。
製造した被膜の表面形態を、走査型電子顕微鏡によって特徴付けた(図4参照)。
【0077】
割れのない構造を有する電極および亀甲割れ構造を有する電極を、サイクリックボルタンメトリーによって試験した。測定は、銀/塩化銀参照電極を用い、室温、pH=3、3.5mol塩化ナトリウム溶液中で実施した。電位は、5〜200mV/sの範囲の様々な電位走査速度(v)で、0.2〜1.0Vの範囲で変化した。ボルタンメトリー容量電荷(qa)は、サイクリックボルタモグラムの陽極ブランチを積分することによって、EC−Labソフトウェアを用いて得た。割れのない被膜および亀甲割れ構造を有する被膜について、ボルタンメトリー電荷(qa)を電位走査速度(v)の関数としてプロットする(図5)。亀甲割れ構造を有する被膜(図5の点線)については、電位走査速度(v)が5mV/sから50mV/sに上昇すると、ボルタンメトリー電荷は急激に低下し、その後、ほぼ一定の値をとった。このことは、低い電位走査速度(v)では、割れを通して内部の割れおよびボイドに電解液が浸透し、容量電荷が高い値となるが、より高い電位走査速度では、最外表面しか接近可能でないという事実と一致している。それとは対照的に、割れのない被膜の容量電荷は、電位走査速度に実質的には依存しない(図5の実線)。これにより、被膜が詰まっていて緻密であることが明らかである。
【0078】
実施例10
割れのない緻密な基層と割れのある電気触媒活性カバー層を有する被膜を製造するために、割れのない基層を実施例7と同じ方法で製造した。これにより、金属元素に基づいてRu25mol%/Ti75mol%の基層組成を有する試料を得た。金属含量に基づいて計算すると、これは、0.50g/m2のルテニウム適用量に相当する。これは、1.84g/m2の(酸化物RuO2、TiO2の和としての)総基層適用量に相当する。
被覆−焼成サイクルを合わせて4回実施した以外は、実施例1と同じ被覆液を用い、実施例1と同じ被覆および焼成手順によって、割れのない基層に適用された割れのあるカバー層を製造した。
これにより、金属元素に基づいてRu25mol%/Ti70mol%/V5mol%のカバー層組成を有する試料を得た。金属含量に基づいて計算すると、これは、2.6g/m2のルテニウム適用量に相当する。これは、9.64g/m2の(酸化物RuO2、TiO2、V2O5の和としての)総カバー層適用量に相当する。
製造した被膜の表面形態を、走査型電子顕微鏡によって特徴付けた(図6参照)。
塩素発生のための電極電位を、実施例1と同じ条件下で測定した。測定された電極電位は1.21Vであった。
【0079】
実施例11
割れのある多孔性電気触媒活性被膜を製造するために、実施例1と同じ方法でチタン基材を前処理した。
2mlのイソプロパノールに67.4mgの塩化ルテニウム(III)水和物(RuCl3・xH2O、36%Ru)を溶解し、一晩撹拌することによって、被覆液(液A)を得た。39mgの硝酸ランタン(III)六水和物(La(NO3)3・6H2O)を1mlのイソプロパノールに添加し、30分間撹拌した(液B)。80μlのチタンイソプロポキシド(Ti(i−OPr)4)を、224.6μlの4−ヒドロキシ−4−メチル−2−ペンタノンおよび0.66mlのイソプロパノールの予備混合液に添加し、10分間撹拌した(液C)。
液A、B、Cを超音波処理によって混合し、澄明な液を得た。続いて、この液に、5.15μlの酢酸および10.8μlの脱イオン水を添加した。このように調製した液に蓋をし、室温で一晩撹拌した。
50μlの被覆液を、マイクロピペットを用いて滴下法によって、チタン基材に適用した。
湿潤被膜を空気中で乾燥し、空気中、250℃で10分間、次いで450℃で10分間焼成した。滴下被覆−乾燥−焼成の手順を5回繰り返し、最後の焼成は450℃で1時間実施した。
これにより、金属元素に基づいてRu40mol%/Ti45mol%/La15mol%の組成を有する試料を得た。金属含量に基づいて計算すると、これは、8.5g/m2のルテニウム適用量に相当する。これは、(酸化物RuO2、TiO2、La2O3の和として)29.0g/m2の総被膜適用量に相当する。
【0080】
比較のために、Ru0.4Ti0.6O2の組成を有する被膜を製造した。この目的のために、2mlのイソプロパノールに67.4mgの塩化ルテニウム(III)水和物(RuCl3・xH2O、36%Ru)を添加し、一晩撹拌した(液A)。106.6μlのチタンイソプロポキシド(Ti(i−OPr)4)を、224.6μlの4−ヒドロキシ−4−メチル−2−ペンタノンおよび1.66mlのイソプロパノールの予備混合液に添加し、10分間撹拌した(液B)。液AおよびBを超音波処理によって混合し、澄明な液を得た。続いて、この液に、5.15μlの酢酸および10.8μlの脱イオン水を添加した。このように調製した液に蓋をし、室温で一晩撹拌した。
50μlの被覆液を、マイクロピペットを用いて滴下法によって、チタン基材に適用した。
湿潤被膜を空気中で乾燥し、空気中、250℃で10分間、次いで450℃で10分間焼成した。滴下被覆−乾燥−焼成の手順を5回繰り返し、最後の焼成は450℃で1時間実施した。
これにより、金属元素に基づいてRu40mol%/Ti60mol%の組成を有する試料を得た。金属含量に基づいて計算すると、これは、8.5g/m2のルテニウム適用量に相当する。これは、(酸化物RuO2、TiO2の和として)21.3g/m2の総被膜適用量に相当する。
【0081】
参照電極として銀/塩化銀を用い、室温で、0.5mol塩酸中でサイクリックボルタンメトリーを実施した。電位は、v=50mV/sの電位走査速度(v)で、0.2〜1.0Vの範囲で変化した。ただし、電解液に暴露された面積は1cm2であった。ボルタンメトリー容量電荷(qa)は、サイクリックボルタモグラムの陽極ブランチを積分することによって、EC−Labソフトウェアを用いて得た。Ru0.4Ti0.45La0.15O2被膜(点線)およびRu0.4Ti0.6O2被膜(実線)について、ボルタンメトリー電荷(qa)をサイクリックボルタンメトリーサイクル数の関数としてプロットする(図7)。
Ru0.4Ti0.6O2被膜のボルタンメトリー容量電荷qaは、サイクリックボルタンメトリーサイクル数に依存しない。このことは、被膜の特性が変わらないことを示している。一方、Ru0.4Ti0.45La0.15O2被膜の場合は、第2電位サイクルから第79電位サイクルまで、ボルタンメトリー電荷の連続的な上昇が見られた。このことは、被膜の多孔性の増大を同時に伴うサイクリックボルタンメトリーサイクルの間に、酸化物マトリックスから酸化ランタンが連続的に溶解していることに起因する。
【0082】
実施例12
割れのある多孔性電気触媒活性被膜を製造するために、実施例1と同じ方法でチタン基材を前処理した。
1.33mlのイソプロパノールに37.9mgの塩化ルテニウム(III)水和物(RuCl3・xH2O、36%Ru)を溶解し、一晩撹拌することによって、4つの被覆液(液A)をそれぞれ得た。130.8mgの錫イソプロポキシドプロパノレート(Sn(i−OPr)4・C3H7OH)を、1.34mlのイソプロパノールおよび1.33mlのプロピオン酸の混合物に添加し、次いで、激しく撹拌しつつ還流しながら150℃で30分間煮沸することによって、4つの液(液B)をそれぞれ得た。続いて、異なった量(すなわち39mg、29.2mg、9.7mgおよび0mg)の硝酸ランタン(III)六水和物(La(NO3)3・6H2O)を4つの熱い液Bに添加し、次いで、室温まで冷える前に、これらの液を更に20分間撹拌した。その後、これらの液を、撹拌しながら液Aに滴加した。
それぞれの場合において、50μlの被覆液を、マイクロピペットを用いて滴下法によって、チタン基材に適用した。
湿潤被膜を空気中で乾燥し、空気中、250℃で10分間、次いで450℃で10分間焼成した。続いて、被膜の酸化ランタン成分を溶解するために、被覆チタン板を5%濃度塩酸に、穏やかに撹拌しながら、60℃の温度で15分間浸漬した。
それぞれの場合において、滴下被覆−乾燥−焼成−溶解の手順を8回繰り返し、最後の焼成は450℃で1時間実施した。
これにより、金属元素に基づいてRu30mol%/Sn70mol%の組成を有する試料を4つ得た。金属含量に基づいて計算すると、それぞれの場合において、これは、7.7g/m2のルテニウム適用量に相当する。それぞれの場合において、これは、(酸化物RuO2、SnO2の和として)36.9g/m2の総被膜適用量に相当する。
【0083】
得られた被膜を、被覆液中に存在する異なった量(39mg、29.2mg、9.7mgまたは0mg)の硝酸ランタン(III)六水和物に対応して、La39、La29、La9、La0と表す。
実施例9に記載したようにサイクリックボルタンメトリーによって電極を試験し、実施例9に記載したようにボルタンメトリー容量電荷(qa)を測定した。図8に、電極の電位走査速度(v)の関数としてのボルタンメトリー電荷(qa)を示す。電位走査速度が5mV/sから200mV/sに上昇すると、ボルタンメトリー電荷(qa)はかなり低下する。このことは、被膜が割れを有する多孔性構造であることを示している。硝酸ランタン(III)六水和物の量が上昇するにつれて、容量電荷も上昇した。酸化ランタンの溶解の結果、より多孔性の被膜が得られる。
【0084】
実施例13
割れのある多孔性電気触媒活性被膜を製造するために、実施例1と同じ方法でチタン基材を前処理した。
被覆液を調製するために、99.6mgのルテニウムアセチルアセトネート(Ru(acac)3)、192.4μlのチタンイソプロポキシド(Ti(i−OPr)4)および26.6mgのバナジルアセチルアセトネート(VO(acac)2)をそれぞれ、1.45mlのイソプロパノールおよび1.45mlのプロピオン酸に溶解し、各々の場合に、激しく撹拌しつつ還流しながら150℃で30分間加熱した。室温に冷却後、3つの溶液を混合し、ワインレッド色の均一かつ澄明な溶液を得た。溶液に72.2mgのポリビニルピロリドンK30(PVP)(平均分子量Mw=40,000)を添加し、溶液を超音波で30分間処理した。この被覆液50μlを、マイクロピペットを用いてチタン基材に滴下し、次いで、空気中で乾燥した。層を空気中、まず250℃で15分間、次いで450℃で20分間焼成した。これらの手順(被覆液の適用、乾燥、焼成)を8回繰り返した。その後、被覆チタン基材を450℃で1時間焼成した。
これにより、金属元素に基づいてRu25mol%/Ti65mol%/V10mol%の組成を有する試料を得た。金属含量に基づいて計算すると、これは、6.4g/m2のルテニウム適用量に相当する。これは、(酸化物RuO2、TiO2、V2O5の和として)23.9g/m2の総被膜適用量に相当する。
【0085】
Ag/AgCl参照電極を用い、室温で、3.5mol塩化ナトリウム溶液中でサイクリックボルタンメトリーを実施した。電位は、v=50mV/sの電位走査速度で、(Ag/AgClに対して)0.2〜1.0Vの範囲で変化した。ボルタンメトリー容量電荷(qa)は、実施例9に記載した様に測定した。割れを有する多孔性層について、39.2mC/cm2の値を得た。
【0086】
比較のために、ポリビニルピロリドン(PVP)を添加しなかったこと以外は同じ被覆液を用い、同じ製造方法で電極を製造し、同じ方法でボルタンメトリー容量電荷(qa)を測定した。PVPを添加しなかった被膜のボルタンメトリー容量電荷(qa)について、20.8mC/cm2の値を得た。
【特許請求の範囲】
【請求項1】
導電性基材および触媒活性層を含んでなる電極であって、
触媒活性層が、2種の触媒活性成分に基づき、金属酸化物または混合酸化物または酸化物混合物としてイリジウム、ルテニウムまたはチタンを含んでなり、
元素イリジウム、ルテニウムおよびチタンの和に基づいたルテニウムおよび/またはイリジウムの総含量が、少なくとも10mol%であり、
電極が、導電性基材に適用された、NaClおよび/またはNaOHおよび/またはHCl含有水性電解液を透過しない少なくとも1つの酸化物基層を含んでなる、
電極。
【請求項2】
導電性基材がバルブ金属に基づく、請求項1に記載の電極。
【請求項3】
バルブ金属が、チタン、タンタル、ニオブ、ニッケル、主成分としてチタン、タンタルまたはニオブを含有するこれら金属の合金、およびそれらの混合物からなる群から選択される、請求項2に記載の電極。
【請求項4】
少なくとも1つの酸化物基層が、塩化水素水溶液、塩化ナトリウム水溶液および水酸化ナトリウム水溶液を透過しない、請求項1に記載の電極。
【請求項5】
サイクリックボルタンメトリー電荷が基層より大きいカバー層を、電極が更に含んでなる、請求項1に記載の電極。
【請求項6】
カバー層が、触媒活性層成分を含んでなり、孔形成組成物を付加的に含んでなる、請求項5に記載の電極。
【請求項7】
孔形成組成物が、酸化ランタン、ポリマーまたはそれらの混合物を含んでなる、請求項6に記載の電極。
【請求項8】
少なくとも1つの酸化物基層の厚さ(酸化物としての単位面積当たりの適用量)が0.1〜20g/m2である、請求項1に記載の電極。
【請求項9】
カバー層厚さ(酸化物としての単位面積当たりの適用量)が少なくとも2g/m2である、請求項1に記載の電極。
【請求項10】
カバー層が、層厚さ方向の断面で観察して、チタンに対するイリジウムのおよび/またはチタン成分に対するルテニウムの変化する比を有する、請求項1に記載の電極。
【請求項11】
カバー層におけるチタンに対するイリジウムのおよび/またはチタンに対するルテニウムの比が、層厚さ方向の断面で観察して、外側から導電性支持体に向かって低下している、請求項10に記載の電極。
【請求項12】
少なくとも1つの酸化物基層が導電性であり、少なくとも10S/mの導電率を有する、請求項1に記載の電極。
【請求項13】
少なくとも導電性基材および触媒活性層を含んでなる電極であって、
触媒活性層が、2種の触媒活性成分に基づき、金属酸化物または混合酸化物または酸化物混合物としてイリジウム、ルテニウムまたはチタンを含んでなり、
元素イリジウム、ルテニウムおよびチタンの和に基づいたルテニウムおよび/またはイリジウムの総含量が、少なくとも10mol%であり、
ルテニウムおよび/またはイリジウムの半分までが、バナジウム、ジルコニウムまたはモリブデンによって置換されている、
電極。
【請求項14】
電極が、導電性基材に適用された、NaClおよび/またはNaOHおよび/またはHCl含有水性電解液を透過しない少なくとも1つの酸化物基層を含んでなる、請求項13に記載の電極。
【請求項15】
ルテニウム、イリジウム、チタンおよびそれらの混合物からなる群から選択される金属を含んでなる金属化合物の溶液または分散体を含んでなるゾル−ゲル被覆液を、導電性支持体に適用する工程、
溶媒を除去するために乾燥する工程、
酸素含有気体の存在下、少なくとも350℃の温度で焼成する工程、および
任意に、ゾル−ゲル被覆液の適用、乾燥および焼成を1回以上繰り返す工程
を含む、電極の製造方法。
【請求項16】
ルテニウム、イリジウム、チタンおよびそれらの混合物からなる群から選択される金属の金属塩の溶液または分散体を、基層に1回以上適用することによって得られたカバー層を適用する工程、
溶媒を除去するために乾燥する工程、および
酸素含有気体の存在下、少なくとも350℃の温度で焼成する工程
を更に含む、請求項15に記載の方法。
【請求項17】
基層を形成するために、金属塩液の適用後、乾燥を高温で、特に少なくとも200℃で、好ましくは少なくとも240℃で実施する、請求項15に記載の方法。
【請求項18】
低級カルボン酸、C1〜C5アルコール、ケトンまたはそれらの混合物を、基層および/またはカバー層を形成するための金属化合物溶液または分散体に添加する、請求項16に記載の方法。
【請求項19】
請求項1に記載の電極を陽極として含んでなる、塩化ナトリウムまたは塩化水素を含有する溶液を電気分解するための電解槽。
【請求項20】
請求項1に記載の電極を含んでなる電解槽において、塩化ナトリウムまたは塩化水素を電気分解する工程を含む、塩素の電気化学的製造方法。
【請求項1】
導電性基材および触媒活性層を含んでなる電極であって、
触媒活性層が、2種の触媒活性成分に基づき、金属酸化物または混合酸化物または酸化物混合物としてイリジウム、ルテニウムまたはチタンを含んでなり、
元素イリジウム、ルテニウムおよびチタンの和に基づいたルテニウムおよび/またはイリジウムの総含量が、少なくとも10mol%であり、
電極が、導電性基材に適用された、NaClおよび/またはNaOHおよび/またはHCl含有水性電解液を透過しない少なくとも1つの酸化物基層を含んでなる、
電極。
【請求項2】
導電性基材がバルブ金属に基づく、請求項1に記載の電極。
【請求項3】
バルブ金属が、チタン、タンタル、ニオブ、ニッケル、主成分としてチタン、タンタルまたはニオブを含有するこれら金属の合金、およびそれらの混合物からなる群から選択される、請求項2に記載の電極。
【請求項4】
少なくとも1つの酸化物基層が、塩化水素水溶液、塩化ナトリウム水溶液および水酸化ナトリウム水溶液を透過しない、請求項1に記載の電極。
【請求項5】
サイクリックボルタンメトリー電荷が基層より大きいカバー層を、電極が更に含んでなる、請求項1に記載の電極。
【請求項6】
カバー層が、触媒活性層成分を含んでなり、孔形成組成物を付加的に含んでなる、請求項5に記載の電極。
【請求項7】
孔形成組成物が、酸化ランタン、ポリマーまたはそれらの混合物を含んでなる、請求項6に記載の電極。
【請求項8】
少なくとも1つの酸化物基層の厚さ(酸化物としての単位面積当たりの適用量)が0.1〜20g/m2である、請求項1に記載の電極。
【請求項9】
カバー層厚さ(酸化物としての単位面積当たりの適用量)が少なくとも2g/m2である、請求項1に記載の電極。
【請求項10】
カバー層が、層厚さ方向の断面で観察して、チタンに対するイリジウムのおよび/またはチタン成分に対するルテニウムの変化する比を有する、請求項1に記載の電極。
【請求項11】
カバー層におけるチタンに対するイリジウムのおよび/またはチタンに対するルテニウムの比が、層厚さ方向の断面で観察して、外側から導電性支持体に向かって低下している、請求項10に記載の電極。
【請求項12】
少なくとも1つの酸化物基層が導電性であり、少なくとも10S/mの導電率を有する、請求項1に記載の電極。
【請求項13】
少なくとも導電性基材および触媒活性層を含んでなる電極であって、
触媒活性層が、2種の触媒活性成分に基づき、金属酸化物または混合酸化物または酸化物混合物としてイリジウム、ルテニウムまたはチタンを含んでなり、
元素イリジウム、ルテニウムおよびチタンの和に基づいたルテニウムおよび/またはイリジウムの総含量が、少なくとも10mol%であり、
ルテニウムおよび/またはイリジウムの半分までが、バナジウム、ジルコニウムまたはモリブデンによって置換されている、
電極。
【請求項14】
電極が、導電性基材に適用された、NaClおよび/またはNaOHおよび/またはHCl含有水性電解液を透過しない少なくとも1つの酸化物基層を含んでなる、請求項13に記載の電極。
【請求項15】
ルテニウム、イリジウム、チタンおよびそれらの混合物からなる群から選択される金属を含んでなる金属化合物の溶液または分散体を含んでなるゾル−ゲル被覆液を、導電性支持体に適用する工程、
溶媒を除去するために乾燥する工程、
酸素含有気体の存在下、少なくとも350℃の温度で焼成する工程、および
任意に、ゾル−ゲル被覆液の適用、乾燥および焼成を1回以上繰り返す工程
を含む、電極の製造方法。
【請求項16】
ルテニウム、イリジウム、チタンおよびそれらの混合物からなる群から選択される金属の金属塩の溶液または分散体を、基層に1回以上適用することによって得られたカバー層を適用する工程、
溶媒を除去するために乾燥する工程、および
酸素含有気体の存在下、少なくとも350℃の温度で焼成する工程
を更に含む、請求項15に記載の方法。
【請求項17】
基層を形成するために、金属塩液の適用後、乾燥を高温で、特に少なくとも200℃で、好ましくは少なくとも240℃で実施する、請求項15に記載の方法。
【請求項18】
低級カルボン酸、C1〜C5アルコール、ケトンまたはそれらの混合物を、基層および/またはカバー層を形成するための金属化合物溶液または分散体に添加する、請求項16に記載の方法。
【請求項19】
請求項1に記載の電極を陽極として含んでなる、塩化ナトリウムまたは塩化水素を含有する溶液を電気分解するための電解槽。
【請求項20】
請求項1に記載の電極を含んでなる電解槽において、塩化ナトリウムまたは塩化水素を電気分解する工程を含む、塩素の電気化学的製造方法。
【図1】



【図2】

【図3】

【図4】

【図5】
【図6】

【図7】

【図8】
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【公開番号】特開2012−92449(P2012−92449A)
【公開日】平成24年5月17日(2012.5.17)
【国際特許分類】
【外国語出願】
【出願番号】特願2011−235871(P2011−235871)
【出願日】平成23年10月27日(2011.10.27)
【出願人】(504037346)バイエル・マテリアルサイエンス・アクチェンゲゼルシャフト (728)
【氏名又は名称原語表記】Bayer MaterialScience AG
【Fターム(参考)】
【公開日】平成24年5月17日(2012.5.17)
【国際特許分類】
【出願番号】特願2011−235871(P2011−235871)
【出願日】平成23年10月27日(2011.10.27)
【出願人】(504037346)バイエル・マテリアルサイエンス・アクチェンゲゼルシャフト (728)
【氏名又は名称原語表記】Bayer MaterialScience AG
【Fターム(参考)】
[ Back to top ]