
電解質
【課題】高いプロトン伝導性をもつポリベンゾイミダゾールからなる電解質膜を提供する。
【解決手段】1種以上の低分子量ホスホン酸の混合物と、随意にリン酸類とによりドープされたポリベンゾイミダゾールからなる膜。
【解決手段】1種以上の低分子量ホスホン酸の混合物と、随意にリン酸類とによりドープされたポリベンゾイミダゾールからなる膜。
【発明の詳細な説明】
【技術分野】
【0001】
ポリベンゾイミダゾール(PBI)からなる膜は、リン酸(PA)を高分子電解質膜(PEM)の電解質として含んでいる。このようにして、PAはPBI膜に固定化されている。本発明は、これら及び他の用途のための電解質を提供するものである。
【背景技術】
【0002】
後述するような驚くべきかつ新規な効果が発見された。アミノトリ(メチレンホスホン酸)(ATMP)は、低分子量のアミノホスホン酸である。ATMPおよびPAを含むPBIからなる膜は、ATMPのみからなる膜よりプロトン伝導性が高い。後者は、130℃より高い温度に対して特に適用されるものである。
【0003】
PBI膜にATMPを固定化(実施例1)し、溶液を130℃よりも高くすると、プロトン伝導性は全くなくなるか、またはわずかに存在するのみとなる。これに対して、PAを含むBPI膜(実施例2)は、同じ温度下で、明らかにプロトン伝導性が大きい。
【0004】
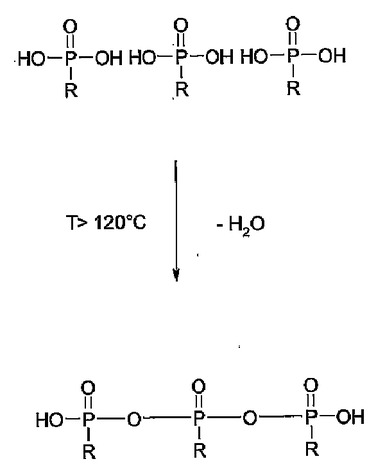
ATMPは、130℃より高い温度で縮合され、水が分離する(図1)。脱水により、ホスホン酸は酸としての機能を失い、電解質としては利用できなくなる。実施例4に従って作られるPBI膜はATMPとPAの両方を含んでいる。この膜は、実施例1および2の膜より高いプロトン伝導性をもつ。このことは全く驚くべきことであり、予想だにされていなかった。中でも、120℃より高い温度での高いプロトン伝導性は特に驚くべきことである。PBIとPAのみを含む比較例の膜に対して、200℃までの温度領域において、プロトン伝導性が明らかにある。
【0005】
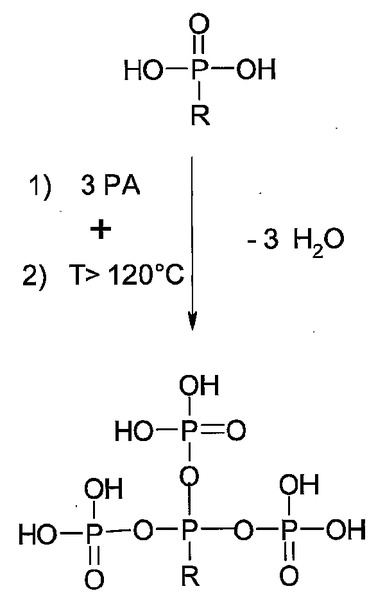
2つの機構が同定されており、これらは推定ではあるがその確度が高い。第1の機構は、ATMPとPAの間の混合縮合反応(図2)であり、第2の機構は、ATMP分子内のプロトン化された窒素(図3)による強化効果である。第2の効果により、やがて分離するプロトンの酸強度が増す。窒素をプロトン化するためのブレンステッド酸は、リン酸等の類似の分子から、または他の分子に由来する。両方とも可能であり、いずれのプロトン化源が用いられるかにより、用途が異なる。
【0006】
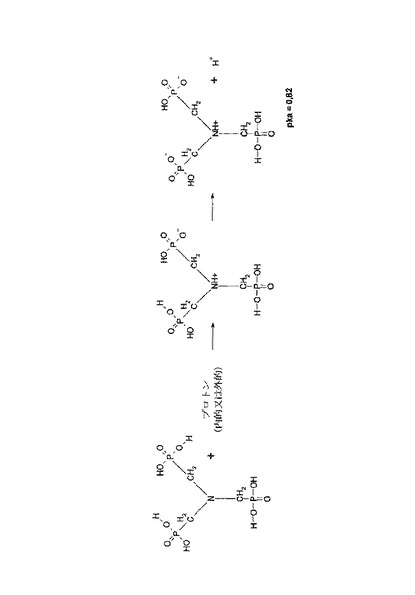
酸強度の増加を決定付けるものは、N−C−P結合における原子の配列である。Cは、一般式R2NCH2−PO3H2のCH2基に属する。ここで、Rは、互いに独立したアルキル基、アリル基、ヘテロアリル基、任意に置換されたC原子または水素である。Rは任意の官能基を有することもできる。例示的であるが非限定的に、リン酸、スルホン酸、カルボン酸、ヒドロキシル基、ニトロ基またはアミノ基を挙げることができる。酸強度または安定性を増大させるために、Rがフッ素を有することができる。ATMPの場合、残留Rは2つとも同じであり、―CH2―PO3H2である。窒素をプロトン化してR2NH+−CH2−PO3H2が得られ、ホスホン酸残基の酸強度は強くなる。その結果、高いプロトン伝導性が得られる。リン酸でドープした場合と比較して、縮合点より低い温度では、ドープされたフィルムのプロトン伝導性が高くなることがわかっている。図3はATMPのプロトン化を示している。
【図面の簡単な説明】
【0007】
【図1】ホスホン酸の縮合を示す。
【図2】PAによるホスホン酸の混合縮合の一般的な流れを示しており、式中、Rは任意の有機残基を示す。
【図3】ここでは、ATMPの酸強度の強化効果を、内部のプロトン化および2位のホスホン酸基の脱プロトン化に対する酸解離定数(pKa)値で表す。
【発明を実施するための形態】
【0008】
実施例においては、化学物質販売店アルドリッヒ(Aldrich)から購入したポリベンゾイミダゾール(PBI)を用いた。製品膜の製造には、DMAcにPBIを溶解した、10%溶液が用いられた。その溶液をガラス板にキャストし、乾燥室内で溶媒を蒸発させた。これにより、PBIからなるフィルムを製造した。
【実施例】
【0009】
1)PBIフィルムへのATMPの固定化
50重量%のATMPの水溶液1リットルに、厚さ60μのPBIフィルム(10×10cm)を浸漬した。この溶液を、24時間、60〜80℃のオーブンに入れた。その後、フィルムを取り出し、表面のパルプを乾燥した状態で拭き取り、コントロールとして重量を量った。
【0010】
80〜110℃の乾燥室内でフィルムを乾燥させ、再度、重量を量った。これは、20重量%のATMPを含んでいた。
【0011】
固定化されるATMPの量は、処理時間と、ATMP溶液の濃度および温度に依存する。溶液への浸漬と乾燥を繰り返すことで、PBIフィルム中でのATMPの濃度を40%超とすることができる。フィルムを乾燥することにより、水が取り除かれる。
【0012】
アミノホスホン酸の水溶液を非プロトン性溶媒に加えると、ATMPの吸収またはさらに他のアミノホスホン酸が増加し続けた。非プロトン性溶媒または非プロトン性溶媒の任意の混合物は、PBIフィルムの膨張に寄与する。そのような溶媒の例は、NMP、DMAc、スルホランまたはDMSOである。これらの例示は、限定的なものではない。DMSOは塩基窒素を含まないため、好適である。溶媒に対する唯一の前提条件は、PBIの膨張を促進することである。例えばアセトンはPBIを僅かにしか膨張させないため、あまり好ましくない。アミノホスホン酸は縮合された非プロトン性溶媒中では全く分解されないため、100%NMP溶液もまた不適である。したがって、水と非プロトン溶媒の間で選択される混合割合は、意図されるドープの程度により決まる。
【0013】
比較的分子量の大きいアミノホスホン酸を用いる際は、水に更なる溶媒を用いることが特に好ましい。一例としては、ジエチレントリアミノペンタメチレンホスホン酸(DTPMP)が挙げられる。PBI水溶液から2〜4%のDTPMPしか得られない。溶媒として50〜70%のNMPまたはDMSOの水溶液を用いた場合には、6%超のDTPMPをPBIフィルム中に導入することができる。
【0014】
2)PBIフィルムへのPAの固定化
50重量%のPAの水溶液1リットルに、厚さ60μのPBIフィルム(10×10cm)を浸漬した。この溶液を、24時間、80℃のオーブンに入れた。実施例1と同様にして、フィルムを乾燥した。
【0015】
3)PBI膜へのPAおよびATMPの固定化
ATMPおよびPAの水溶液1リットルに、厚さ60μのPBIフィルム(10×10cm)を浸漬した。この溶液は、25重量%のATMPおよび、25重量%のPAを含有していた。この溶液を、24時間、80℃のオーブン内に置いた。フィルムを実施例1と同様にして乾燥した。
【0016】
ここでは、非プロトン性溶媒をPAに置換する。これらの処理は、アミノホスホン酸と、ホスホン酸(PA)を同時にフィルムに取り込むことができる点で優れている。
【0017】
4)PBI膜へのPAおよびATMPの固定化
ATMPおよびPAの水溶液1リットルに、厚さ60μのPBIフィルム(10×10cm)を浸漬した。この溶液は、25重量%のATMPおよび、25重量%のPAを含有していた。この溶液を、24時間、80℃のオーブン内に置いた。膜を130℃で乾燥し、その後、再度、ATMPおよびPAからなる溶液に浸漬した。フィルムが繰り返し処理されることで、水が取り除かれ、ATMPとPAのドープの程度が上昇した。
【技術分野】
【0001】
ポリベンゾイミダゾール(PBI)からなる膜は、リン酸(PA)を高分子電解質膜(PEM)の電解質として含んでいる。このようにして、PAはPBI膜に固定化されている。本発明は、これら及び他の用途のための電解質を提供するものである。
【背景技術】
【0002】
後述するような驚くべきかつ新規な効果が発見された。アミノトリ(メチレンホスホン酸)(ATMP)は、低分子量のアミノホスホン酸である。ATMPおよびPAを含むPBIからなる膜は、ATMPのみからなる膜よりプロトン伝導性が高い。後者は、130℃より高い温度に対して特に適用されるものである。
【0003】
PBI膜にATMPを固定化(実施例1)し、溶液を130℃よりも高くすると、プロトン伝導性は全くなくなるか、またはわずかに存在するのみとなる。これに対して、PAを含むBPI膜(実施例2)は、同じ温度下で、明らかにプロトン伝導性が大きい。
【0004】
ATMPは、130℃より高い温度で縮合され、水が分離する(図1)。脱水により、ホスホン酸は酸としての機能を失い、電解質としては利用できなくなる。実施例4に従って作られるPBI膜はATMPとPAの両方を含んでいる。この膜は、実施例1および2の膜より高いプロトン伝導性をもつ。このことは全く驚くべきことであり、予想だにされていなかった。中でも、120℃より高い温度での高いプロトン伝導性は特に驚くべきことである。PBIとPAのみを含む比較例の膜に対して、200℃までの温度領域において、プロトン伝導性が明らかにある。
【0005】
2つの機構が同定されており、これらは推定ではあるがその確度が高い。第1の機構は、ATMPとPAの間の混合縮合反応(図2)であり、第2の機構は、ATMP分子内のプロトン化された窒素(図3)による強化効果である。第2の効果により、やがて分離するプロトンの酸強度が増す。窒素をプロトン化するためのブレンステッド酸は、リン酸等の類似の分子から、または他の分子に由来する。両方とも可能であり、いずれのプロトン化源が用いられるかにより、用途が異なる。
【0006】
酸強度の増加を決定付けるものは、N−C−P結合における原子の配列である。Cは、一般式R2NCH2−PO3H2のCH2基に属する。ここで、Rは、互いに独立したアルキル基、アリル基、ヘテロアリル基、任意に置換されたC原子または水素である。Rは任意の官能基を有することもできる。例示的であるが非限定的に、リン酸、スルホン酸、カルボン酸、ヒドロキシル基、ニトロ基またはアミノ基を挙げることができる。酸強度または安定性を増大させるために、Rがフッ素を有することができる。ATMPの場合、残留Rは2つとも同じであり、―CH2―PO3H2である。窒素をプロトン化してR2NH+−CH2−PO3H2が得られ、ホスホン酸残基の酸強度は強くなる。その結果、高いプロトン伝導性が得られる。リン酸でドープした場合と比較して、縮合点より低い温度では、ドープされたフィルムのプロトン伝導性が高くなることがわかっている。図3はATMPのプロトン化を示している。
【図面の簡単な説明】
【0007】
【図1】ホスホン酸の縮合を示す。
【図2】PAによるホスホン酸の混合縮合の一般的な流れを示しており、式中、Rは任意の有機残基を示す。
【図3】ここでは、ATMPの酸強度の強化効果を、内部のプロトン化および2位のホスホン酸基の脱プロトン化に対する酸解離定数(pKa)値で表す。
【発明を実施するための形態】
【0008】
実施例においては、化学物質販売店アルドリッヒ(Aldrich)から購入したポリベンゾイミダゾール(PBI)を用いた。製品膜の製造には、DMAcにPBIを溶解した、10%溶液が用いられた。その溶液をガラス板にキャストし、乾燥室内で溶媒を蒸発させた。これにより、PBIからなるフィルムを製造した。
【実施例】
【0009】
1)PBIフィルムへのATMPの固定化
50重量%のATMPの水溶液1リットルに、厚さ60μのPBIフィルム(10×10cm)を浸漬した。この溶液を、24時間、60〜80℃のオーブンに入れた。その後、フィルムを取り出し、表面のパルプを乾燥した状態で拭き取り、コントロールとして重量を量った。
【0010】
80〜110℃の乾燥室内でフィルムを乾燥させ、再度、重量を量った。これは、20重量%のATMPを含んでいた。
【0011】
固定化されるATMPの量は、処理時間と、ATMP溶液の濃度および温度に依存する。溶液への浸漬と乾燥を繰り返すことで、PBIフィルム中でのATMPの濃度を40%超とすることができる。フィルムを乾燥することにより、水が取り除かれる。
【0012】
アミノホスホン酸の水溶液を非プロトン性溶媒に加えると、ATMPの吸収またはさらに他のアミノホスホン酸が増加し続けた。非プロトン性溶媒または非プロトン性溶媒の任意の混合物は、PBIフィルムの膨張に寄与する。そのような溶媒の例は、NMP、DMAc、スルホランまたはDMSOである。これらの例示は、限定的なものではない。DMSOは塩基窒素を含まないため、好適である。溶媒に対する唯一の前提条件は、PBIの膨張を促進することである。例えばアセトンはPBIを僅かにしか膨張させないため、あまり好ましくない。アミノホスホン酸は縮合された非プロトン性溶媒中では全く分解されないため、100%NMP溶液もまた不適である。したがって、水と非プロトン溶媒の間で選択される混合割合は、意図されるドープの程度により決まる。
【0013】
比較的分子量の大きいアミノホスホン酸を用いる際は、水に更なる溶媒を用いることが特に好ましい。一例としては、ジエチレントリアミノペンタメチレンホスホン酸(DTPMP)が挙げられる。PBI水溶液から2〜4%のDTPMPしか得られない。溶媒として50〜70%のNMPまたはDMSOの水溶液を用いた場合には、6%超のDTPMPをPBIフィルム中に導入することができる。
【0014】
2)PBIフィルムへのPAの固定化
50重量%のPAの水溶液1リットルに、厚さ60μのPBIフィルム(10×10cm)を浸漬した。この溶液を、24時間、80℃のオーブンに入れた。実施例1と同様にして、フィルムを乾燥した。
【0015】
3)PBI膜へのPAおよびATMPの固定化
ATMPおよびPAの水溶液1リットルに、厚さ60μのPBIフィルム(10×10cm)を浸漬した。この溶液は、25重量%のATMPおよび、25重量%のPAを含有していた。この溶液を、24時間、80℃のオーブン内に置いた。フィルムを実施例1と同様にして乾燥した。
【0016】
ここでは、非プロトン性溶媒をPAに置換する。これらの処理は、アミノホスホン酸と、ホスホン酸(PA)を同時にフィルムに取り込むことができる点で優れている。
【0017】
4)PBI膜へのPAおよびATMPの固定化
ATMPおよびPAの水溶液1リットルに、厚さ60μのPBIフィルム(10×10cm)を浸漬した。この溶液は、25重量%のATMPおよび、25重量%のPAを含有していた。この溶液を、24時間、80℃のオーブン内に置いた。膜を130℃で乾燥し、その後、再度、ATMPおよびPAからなる溶液に浸漬した。フィルムが繰り返し処理されることで、水が取り除かれ、ATMPとPAのドープの程度が上昇した。
【特許請求の範囲】
【請求項1】
1種以上の低分子量ホスホン酸の混合物を含むことを特徴とするPBI膜。
【請求項2】
ATMPおよびリン酸を含むことを特徴とするPBI膜。
【請求項3】
PBIをドープする方法であって、連続した工程または互いに独立して、PBIからなるフィルムを
A)希釈もしくは濃縮されているリン酸溶液および/または
B)水に溶解したアミノホスホン酸もしくはリン酸の溶液
に浸漬することを特徴とする方法。
【請求項4】
固定化されたアミノホスホン酸、ホスホン酸および随意に少なくとも1種の官能基ポリマーを含有することを特徴とするPBI膜。
【請求項5】
付加的なポリマー官能基を有する、請求項4に記載のPBI膜。
【請求項6】
付加的なポリマーはスルホン化ポリマーであり、スルホン化ポリマーの割合を最大で90%とすることができる、請求項5に記載のPBI膜。
【請求項7】
アミノホスホン酸とのドーピングの間に、混合物中でスルホン化ポリマーが塩の形で存在するので、ナトリウムフォームが好ましい、請求項6に記載のPBI膜。
【請求項8】
塩基混合物が、ホスホン酸でドープされていないが、1種以上のアミノホスホン酸のみでドープされている、請求項7に記載のPBI膜。
【請求項9】
アミノホスホン酸の割合を最大で80重量%とすることのできる、請求項1〜8のいずれか一項に記載のPBI膜。
【請求項10】
請求項1〜9のいずれか一項に記載の膜の、膜処理の膜としての使用。
【請求項11】
請求項1〜10のいずれか一項に記載の膜の、特に膜気体電池、浸透気化膜、透析膜、逆浸透膜、ナノろ過膜および限外ろ過膜への使用。
【請求項1】
1種以上の低分子量ホスホン酸の混合物を含むことを特徴とするPBI膜。
【請求項2】
ATMPおよびリン酸を含むことを特徴とするPBI膜。
【請求項3】
PBIをドープする方法であって、連続した工程または互いに独立して、PBIからなるフィルムを
A)希釈もしくは濃縮されているリン酸溶液および/または
B)水に溶解したアミノホスホン酸もしくはリン酸の溶液
に浸漬することを特徴とする方法。
【請求項4】
固定化されたアミノホスホン酸、ホスホン酸および随意に少なくとも1種の官能基ポリマーを含有することを特徴とするPBI膜。
【請求項5】
付加的なポリマー官能基を有する、請求項4に記載のPBI膜。
【請求項6】
付加的なポリマーはスルホン化ポリマーであり、スルホン化ポリマーの割合を最大で90%とすることができる、請求項5に記載のPBI膜。
【請求項7】
アミノホスホン酸とのドーピングの間に、混合物中でスルホン化ポリマーが塩の形で存在するので、ナトリウムフォームが好ましい、請求項6に記載のPBI膜。
【請求項8】
塩基混合物が、ホスホン酸でドープされていないが、1種以上のアミノホスホン酸のみでドープされている、請求項7に記載のPBI膜。
【請求項9】
アミノホスホン酸の割合を最大で80重量%とすることのできる、請求項1〜8のいずれか一項に記載のPBI膜。
【請求項10】
請求項1〜9のいずれか一項に記載の膜の、膜処理の膜としての使用。
【請求項11】
請求項1〜10のいずれか一項に記載の膜の、特に膜気体電池、浸透気化膜、透析膜、逆浸透膜、ナノろ過膜および限外ろ過膜への使用。
【図1】


【図2】
【図3】
【図2】
【図3】
【公開番号】特開2013−64142(P2013−64142A)
【公開日】平成25年4月11日(2013.4.11)
【国際特許分類】
【出願番号】特願2012−248695(P2012−248695)
【出願日】平成24年11月12日(2012.11.12)
【分割の表示】特願2008−530327(P2008−530327)の分割
【原出願日】平成18年9月14日(2006.9.14)
【出願人】(507055877)
【Fターム(参考)】
【公開日】平成25年4月11日(2013.4.11)
【国際特許分類】
【出願日】平成24年11月12日(2012.11.12)
【分割の表示】特願2008−530327(P2008−530327)の分割
【原出願日】平成18年9月14日(2006.9.14)
【出願人】(507055877)
【Fターム(参考)】
[ Back to top ]