
青色素合成遺伝子
【課題】D−サイクロセリン生産性放線菌に特異的に存在する遺伝子の探索及びその機能を解明し、有用な遺伝子及びその用途を提供する。
【解決手段】以下の(a)又は(b)のタンパク質、これをコードする遺伝子。(a)特定な配列のアミノ酸配列からなるタンパク質、(b)(a)のアミノ酸配列において、1若しくは数個のアミノ酸が欠失、置換若しくは付加されたアミノ酸配列からなり、且つインジゴイジン合成能を有するタンパク質。
【解決手段】以下の(a)又は(b)のタンパク質、これをコードする遺伝子。(a)特定な配列のアミノ酸配列からなるタンパク質、(b)(a)のアミノ酸配列において、1若しくは数個のアミノ酸が欠失、置換若しくは付加されたアミノ酸配列からなり、且つインジゴイジン合成能を有するタンパク質。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、D−サイクロセリン(DCS)生産性放線菌(Streptomyces lavendulae)に特異的に存在する青色素合成遺伝子及びそれを用いた青色素の製造方法に関する。
【背景技術】
【0002】
植物の自然な発色については天然植物色素が貢献しているが、その主な物質としてクロロフィル類、カロチノイド系色素、アントシアニン系色素があげられる。青色の発色はアントシアニン系色素によるものであるが、その発色機構は複雑であり、現在も研究が進められている。
【0003】
また、生花店で販売されている植物において、マリンブルーに近い青色の花の品種は意外に少ない。バラでは古来より青いバラの作製が育種家の夢であるが、従来の育種技術では困難といわれており、近年、遺伝子工学を利用して青いバラの作成が研究されているが、未だ愛好家を満足させるものはない。
【0004】
一方、ストレプトマイセス(Streptomyces)に属する放線菌は、実用抗生物質を始めとする二次代謝産物の多くを作ることから、工業的にも重要な微生物である。近年、ペプチド系抗生物質の多くが非リボソーム型ペプチド合成酵素(non-ribosomal peptide synthetase:NRPS)により生合成されることが知られるようになってきたが(非特許文献1)、放線菌に特異的に存在するNRPSには、未だその機能が見出されていないものが数多く存在すると考えられている。
【非特許文献1】Finking, R. et al.,(2004) Annu. Rev. Microbiol. 58, 453-488.
【発明の開示】
【発明が解決しようとする課題】
【0005】
本発明は、放線菌に特異的に存在する遺伝子の探索及びその機能を解明し、有用な遺伝子及びその用途を提供することを目的とする。
【課題を解決するための手段】
【0006】
本発明者は、DCS生産性放線菌(S. lavendulae)に特異的に存在する遺伝子を探索したところ、非リボソーム型ペプチド合成酵素(NRPS)ファミリーには珍しい、単一モジュール型の非リボソーム型ペプチド合成酵素をコードする遺伝子が存在すること、さらに当該遺伝子は青色素インジゴイジン(indigoidine)合成遺伝子として機能し、青色素の製造に有用であることを見出した。
【0007】
すなわち、本発明は、以下の(1)〜(9)に係るものである。
(1)以下の(a)又は(b)のタンパク質:
(a)配列番号1に示すアミノ酸配列からなるタンパク質、
(b)(a)のアミノ酸配列において、1個若しくは数個のアミノ酸が欠失、置換若しくは付加されたアミノ酸配列からなり、且つインジゴイジン合成能を有するタンパク質。
(2)(1)に記載のタンパク質をコードする遺伝子。
(3)以下の(a)又は(b)のDNAからなる遺伝子:
(a)配列番号2に示す塩基配列からなるDNA、
(b)(a)の塩基配列からなるDNAと相補的な塩基配列からなるDNAとストリンジエントな条件下でハイブリダイズし、且つインジゴイジン合成能を有するタンパク質をコードするDNA。
(4)(2)又は(3)に記載のいずれかの遺伝子を含有する組換えベクター。
(5)(4)記載の組換えベクターを含む形質転換体。
(6)(5)記載の形質転換体を培養し、培養物から(1)記載のタンパク質を採取することを特徴とする(1)記載のタンパク質の製造方法。
(7)(2)又は(3)記載の遺伝子を導入した放線菌を培養し、培養物からインジゴイジンを採取することを特徴とするインジゴイジンの製造方法。
(8)(2)又は(3)記載の遺伝子及び4'−ホスホパンテテイニル(4'-phosphopantetheinyl)基転移酵素遺伝子を導入した宿主を培養し、培養物からインジゴイジンを採取することを特徴とするインジゴイジンの製造方法。
(9)L-グルタミンに、(1)記載のタンパク質及び4'−ホスホパンテテイニル基転移酵素を作用させることを特徴とするインジゴイジンの製造方法。
【発明の効果】
【0008】
本発明の遺伝子を用いることにより、大腸菌等を宿主として、青色素インジゴイジンを産生させることができる。また、本発明の遺伝子を、当該色素産生能を持たない植物、蚕等に導入することにより、インジゴイジンを産生するような形質転換体を得ることができ、これにより例えば青いバラやブルーシルクの創出が可能となる。
また、本発明のタンパク質を用いることにより、インビトロで、マリンブルーに近い色を呈するインジゴイジンを製造することができる。
【発明を実施するための最良の形態】
【0009】
本発明の配列番号2に示す塩基配列からなる遺伝子は、後記実施例に示すように、DCS生産性S. lavendulae ATCC11924のゲノムからDCS非産生株であるS. lavendulae JCM4055のゲノムを差し引く、いわゆるゲノムサブトラクション (genome subtraction)法を用いることにより取得されたものである(実施例1)。そして、当該遺伝子は、DCS産生菌に特異的に存在するDNA断片中に存在する非リボソーム型ペプチド合成酵素(non-ribosomal peptide synthetase: NRPS)をコードするものであった。既知のNRPSは、ほとんど多重モジュール構造を取っているが、ここで得られた遺伝子産物(配列番号1に示すアミノ酸配列からなるタンパク質)は、NRPSタンパク質ファミリーには珍しい単一モジュール型であり、OX(オキシデーション)-ドメインが存在する(実施例1)。また、当該タンパク質は、Erwinia chrysanthemiにおいて、青色素インジゴイジンの合成に必須であると同定されているIndCとアミノ酸配列において57%の同一性を示していた。
【0010】
そして、このNRPS遺伝子をS. lividans細胞に導入すると、その形質転換株は新たに青色素を産生するようになった。当該青色素は、可視部吸収スペクトル、IRスペクトル、1H-NMRスペクトル、EI-massスペクトル等により、その化学的性質と構造を解析した結果、その構造は以下に示すとおりであり、IndCにより産生されるインジゴイジン(1. Kuhn, R., et al., (1965) Arch. Mikrobiol. 51, 71-84、2. Mortimer, P. S., et al., (1966) Appl. Microbiol. 14, 870-872)と同一であった(実施例5)。
【0011】
【化1】

【0012】
このことから、当該NRPS遺伝子は青色素インジゴイジン合成遺伝子として機能するものといえ、本NRPS遺伝子をbpsA (blue-pigment synthetase A)、本遺伝子によってコードされるタンパク質をBPSAと名付けた。
【0013】
上記のS. lavendulae ATCC11924株は、インジゴイジン非生産性であるが、放線菌ホルモンに類似した物質であり、食品添加物として使用される香気成分であるγ-またはδ-ノナラクトンの添加により、インジゴイジン生産が誘導される。後記実施例に示すように、bpsA遺伝子と、さらに、その下流に存在する二つの遺伝子の転写がγ-ノナラクトンの添加により誘導される(実施例4)。
【0014】
また、放線菌由来のこの青色素インジゴイジンを大腸菌で生産させる場合には、bpsA遺伝子のほか、BPSAを4'-ホスホパンテテイニル(4'-phosphopantetheinyl)化させる、 例えば、S. verticillus ATCC15003由来 4'-ホスホパンテテイニル基転移酵素 (PPTase) 遺伝子 (svpと命名)のような、4'-ホスホパンテテイニル基転移酵素遺伝子の共発現が不可欠であった(実施例6)。すなわち、大腸菌の中で生成したBPSAが翻訳後修飾により4'-ホスホパンテテイニル化されて初めて、インジゴイジンをつくる酵素として機能する。
【0015】
さらに、BPSAの基質はL-グルタミン(L-Gln)であり、および、試験管内(in vitro)において、L-GlnにBPSAおよびsvp遺伝子がつくるタンパク質(Svpと命名)を添加すると、試験管内でインジゴイジンが合成された。
【0016】
以下に、本発明のタンパク質及び遺伝子について説明する。
尚、本明細書において遺伝子とは、2本鎖DNAのみならず、それを構成するセンス鎖およびアンチセンス鎖という各1本鎖DNAを包含する趣旨である。従って、本発明の遺伝子には、特に言及しない限り、2本鎖DNAおよびcDNAを含む1本鎖DNA(センス鎖)並びに該センス鎖と相補的な配列を有する1本鎖DNA(アンチセンス鎖)が含まれる。
【0017】
本発明のタンパク質は、具体的には、配列番号1に示すアミノ酸配列からなるタンパク質(BPSA)として示されるが、特にこれらに限定されず、当該タンパク質の変異体、例えばアレル体、ホモログ、天然の変異体であって、配列番号1に示すアミノ酸配列と90%以上、好ましくは95%以上、より好ましくは98%以上、さらにより好ましくは99%以上の同一性を有するタンパク質や、配列番号1に示すアミノ酸配列において、1若しくは数個のアミノ酸が欠失、置換若しくは付加されたアミノ酸配列からなり、且つBPSAと同様のインジゴイジン合成能を有するタンパク質が包含される。
【0018】
ここで、配列番号1に示すアミノ酸配列において、1若しくは数個のアミノ酸が欠失、置換若しくは付加されたアミノ酸配列とは、配列番号1のアミノ酸配列と等価のアミノ酸配列を意味し、1若しくは数個、好ましくは1〜10個のアミノ酸が欠失、置換若しくは付加されたアミノ酸配列を意味し、付加には、両末端への1〜数個のアミノ酸の付加が含まれる。
【0019】
本発明の遺伝子は、具体的には配列番号1に示すアミノ酸配列からなるタンパク質をコードする配列番号2に示す塩基配列からなるDNAとして示されるが、特にこれらに限定されず、上記した一定の改変を有するタンパク質をコードする塩基配列からなるDNAが包含される。また、配列番号2に示す塩基配列と、少なくとも90%以上、好ましくは95%以上、より好ましくは98%以上、更に好ましくは99%以上の同一性を有するDNAであって、且つBPSAと同様のインジゴイジン合成能を有するタンパク質をコードするDNAや、配列番号2に示す塩基配列と相補的な塩基配列からなるDNAにストリンジエントな条件下でハイブリダイズし、且つBPSAと同様のインジゴイジン合成能をするタンパク質をコードするDNAが挙げられる。
【0020】
ここで「ストリンジエントな条件下」とは、例えば、Amersham Biosciences社製 AlkPhos Direct Labeling and detection system のハイブリダイゼーション溶液にプローブとともに65℃で8〜16時間恒温し、ハイブリダイズさせる条件が挙げられる。
【0021】
上記改変のための人為的手段としては、部位特異的突然変異誘発法等の公知の手法を利用して調製することができる。例えば、部位特異的突然変異誘発法を利用した変異導入用キット(BD Bioscience社製Transformer Site-Directed Mutagenesis Kit)等を用いて変異を導入し調製することができる。
【0022】
尚、アミノ酸配列又は塩基配列の同一性は、既知の配列分析ソフトウェア、例えば大学共同利用機関法人 情報・システム研究機構 国立遺伝学研究所 日本DNAデータバンクのBLASTプログラムを使用した測定(データベースに登録されているアミノ酸配列またはDNA塩基配列との比較)によって解析できる。
【0023】
本発明遺伝子は、本明細書により開示された本発明の遺伝子の配列情報に基づいて、一般的遺伝子工学的手法により容易に製造、取得することができる(続生化学実験講座「遺伝子研究法I、II、III」、日本生化学会編(1986)など)。
具体的には、本発明遺伝子が発現される微生物、例えばDCS生産性S. lavendulae ATCC11924より、常法に従ってゲノムDNAライブラリーを調製し、該ライブラリーから、本発明遺伝子に特有の適当なプローブ等を用いて所望クローンを選択することにより製造することができる。上記において、DCS生産性S. lavendulaeからの全RNAの分離、mRNAの分離および精製、ゲノムDNAの取得およびそのクローニングなどは、いずれも常法に従って行うことができる。
【0024】
本発明遺伝子をゲノムDNAライブラリーからスクリーニングする方法も、特に制限されず、通常の各種方法に従うことができる。具体的方法としては、例えば、目的の核酸配列に選択的に結合するプローブを用いたプラークハイブリダイゼーション法、コロニーハイブリダイゼーション法など、およびこれらの組合せを例示することができる。
【0025】
上記方法において用いられるプローブとしては、本発明遺伝子の塩基配列に関する情報をもとにして化学合成されたDNAなどが一般的に使用できる。また、本発明DNAの塩基配列情報に基づき設定したセンス・プライマーおよびアンチセンス・プライマーを、スクリーニング用プローブとして用いることができる。
本発明遺伝子の取得に際しては、PCR法〔Science, 230, 1350 (1985)〕によるDNA/RNA増幅法が好適に利用できる。増幅させたDNA/RNA断片の単離精製は、前記の通り、常法に従うことができる。例えばゲル電気泳動法などによることができる。
上記方法に従い得られる本発明遺伝子は、常法、例えばジデオキシ法〔Proc. Natl. Acad. Sci., USA., 74, 5463 (1977)〕、マキサム−ギルバート法〔Methods in Enzymology, 65, 499 (1980)〕などに従って、その核酸配列を決定することができる。また、簡便には、市販のシークエンスキットなどを用いて、その核酸配列を決定することができる。
【0026】
本発明のタンパク質は、本発明により提供される遺伝子の配列情報に基づいて、通常の遺伝子組換え技術に従って調製することができる。すなわち、所望のタンパク質をコードする遺伝子が宿主細胞中で発現できる組換えDNA(発現ベクター)を作成し、これを宿主細胞に導入して形質転換し、該形質転換体を培養し、次いで得られる培養物から回収することができる。
【0027】
上記において宿主としては、例えば大腸菌(Esherichia coli BL21(DE3))などの一般的に用いられるものが広く挙げられ、好適には原核生物細胞、特に大腸菌を用いるのが好ましい。原核生物細胞を宿主とする場合は、該宿主細胞中で複製可能なベクターを用いて、このベクター中に本発明遺伝子が発現できるように該遺伝子の上流にプロモーターおよびSD(シャイン・アンド・ダルガーノ)核酸配列、更に蛋白合成開始に必要な開始コドン(例えばATG)を付与した発現プラスミドを好適に利用できる。上記ベクターとしては、一般に大腸菌由来のプラスミド、例えばpBR322、pBR325、pUC12、pUC13などがよく用いられるが、これらに限定されず既知の各種のベクターを利用することができる。大腸菌を利用した発現系に利用される上記ベクターの市販品としては、例えばpET21、pET28(Novagen社)、pGEX−4T(Amersham Pharmacia Biotech社)、pQE-30, pQE-60(QIAGEN社製)などを例示できる。
【0028】
かくして得られたベクターを用いて宿主を形質転換するにはプロトプラスト法、コンピテントセル法、エレクトロポレーション法等を用いて行うことができる。得られた形質転換体は、資化しうる炭素源、窒素源、金属塩、ビタミン等を含む培地を用いて適当な条件下で培養すればよい。得られた培養液から一般的な方法によって、タンパク質の採取、精製を行うことができる。
【0029】
次に、本発明の遺伝子を用いたインジゴイジンの製造方法について説明する。
(1)形質転換体を用いたインジゴイジンの製造
一般に、NRPSは4'−ホスホパンテテイニル基転移酵素によってホスホパンテテイニル化され、holo-formに変換されなければ活性を生じないことが知られている。本発明のタンパク質においてもインジゴイジンの産生には4'−ホスホパンテテイニル基転移酵素により、当該タンパク質がホスホパンテテイニル化される必要がある。
従って、宿主として、大腸菌(E. coli BL21(DE3))等の一般細菌を用いる場合には、本発明の遺伝子及び4'−ホスホパンテテイニル基転移酵素遺伝子を導入した宿主を培養し、培養物からインジゴイジンを採取することによりインジゴイジンを製造することができる。
【0030】
ここで、4'−ホスホパンテテイニル基転移酵素遺伝子としては、特に限定されるものではないが、例えば、生化学的機能が明らかなS. verticillus由来4'−ホスホパンテテイニル基転移酵素をコードするsvp遺伝子(配列番号4)、枯草菌Bacillus subtilis由来sfp遺伝子等が挙げられる。
共発現させるための手法としては、前記と同様公知の遺伝子工学的手法を用いることができる。例えば、本発明の遺伝子を挿入した発現ベクター(例えば、pET/bpsA)と、4'−ホスホパンテテイニル基転移酵素遺伝子を挿入した発現ベクター(例えば、pSTV/svp)を同時に、宿主に導入し、共発現している細胞を選抜する方法が挙げられる。
【0031】
ここで、培養物とは、培養上清、あるいは培養細胞若しくは培養菌体又は培養細胞若しくは培養菌体の破砕物のいずれをも意味するものである。
また、培養方法は、上記のとおり、宿主細胞培養に用いられる通常の方法に従って行えばよい。
【0032】
インジゴイジンの採取及び精製は、例えば以下のようにして行うことができる。
すなわち、5時間程度培養したS. lavendulaeの培養液に、γ-ノナラクトンを最終濃度約30μg/mlになるよう添加した後、さらに2〜3時間培養する。培地中に分泌された青色素と菌体を低速遠心により分離し、得られた遠心上清を更に高速遠心することで、青色素沈殿物を得る。この沈殿物を水およびメタノールで順次洗浄後、乾燥させることにより、粗精製青色素が得られる。これをジメチルスルホキシド等の有機溶媒に溶解後、0.2μm孔のメンブランフィルターに通すことで残存する菌体を除き、得られた濾液に対し、約5倍量の水を加えて青色素を析出させる。この溶液を高速遠心分離し、青色素沈殿物を得て、上記に示したように洗浄後、乾燥させることで精製インジゴイジンが得られる。
【0033】
また、インジゴイジン合成能のない放線菌に、本発明の遺伝子を導入することでインジゴイジン合成能もつ形質転換放線菌を得ることができる。当該放線菌を液体培養し、その培養液からインジゴイジンを採取することによってもインジゴイジンを得ることができる。この場合、チオストレプトン(50 μg/ml)を含むYEME培地で、2-5日間、約28℃にて培養することで、培地中に青色素が生産される。
【0034】
(2)インビトロにおけるインジゴイジンの製造
L-グルタミンに、本発明のタンパク質及び4'−ホスホパンテテイニル基転移酵素を作用させることにより、インジゴイジンを製造することができる。
ここで、4'−ホスホパンテテイニル基転移酵素としては、例えば、前記S. verticillus由来の4'−ホスホパンテテイニル基転移酵素(配列番号3)等が挙げられる。
本反応は、本発明のタンパク質及び4'−ホスホパンテテイニル基転移酵素に見合ったpHにおいて実施すればよく、このpHは概ね6.0〜8.0、望ましくは7.0〜7.8の範囲である。
反応溶媒は、一般に水または水−有機溶媒混合物を使用することができる。
反応は、一般に25〜30℃、望ましくは28〜30℃で、基質としてのL-グルタミンを加えた後、2〜5分、好ましくは、2〜3分である。
インジゴイジンの採取及び精製は、例えば、反応液から、約15,000gで10分間程度の遠心操作を行い、その上清を除去して得られる沈殿物を必要に応じて水およびメタノールで洗浄し、且つ上記の遠心操作を繰り返すことにより行うことができる。
【実施例】
【0035】
実施のための材料と方法
(1)材料(使用した菌株およびプラスミド、ならびに培養培地)
DCS生産菌として、S. lavendulae ATCC11924、 S. lavendulae ATCC25233、 S. lavendulae FRI-5を用いた。DCS非生産菌株として、S. coelicolor A3(2)、 S. lividans 66、 S. avermitilis K139、 およびS. lavendulae JCM4055を用いた。
Streptomyces属放線菌のプロトプラストを調製する場合には、0.5%グリシンを含むYEME培地を用いて28℃で培養した。青色素を作らせる場合や、その菌から全RNAを調製する場合にはB培地を用いた。
放線菌におけるクローニングベクターとしてはpIJ702(選択マーカーとしてメラニン色素産生遺伝子とチオストレプトン耐性遺伝子を有し、通常、目的遺伝子はメラニン色素合成遺伝子の中にある制限酵素部位に挿入させる。その結果として、メラニン色素を作らなくさせることで、目的遺伝子の挿入により選択マーカーが発黒色ではないコロニーを容易に選択できる)。
ゲノムDNAやプラスミドDNAは常法に従って精製した。目的タンパク質の発現にはベクターとして、pET-21a(+)およびpET-28a(+)が用いられ、その際の宿主として大腸菌BL21(DE3)を用いた。pACYC184の複製開始点を有するpSTV28ベクターは共発現用に用いた。タンパク質を高発現させることを目的とした場合、大腸菌はLB培地で18℃あるいは23℃で培養し、クローニングを目的とした場合、37℃で培養した。必要に応じてLB培地にはカナマイシン 30 μg/ml、アンピシリン100 μg/ml、クロラムフェニコール 34 μg/mlが添加された。
【0036】
(2)サザンハイブリダイゼーション
ハイボンドN+メンブランが用いられ、遺伝子の標識はアルフォス・ダイレクト・ラベリングおよびその検出システムを採用した。
【0037】
実施例1 DCS生産株特異的遺伝子のクローニング
DCS生産株S. lavendulae ATCC11924に特徴的な二次代謝物質の生合成遺伝子をクローニングするため、まず、DCS非生産株S. lavendulae JCM4055を対照株として用いたgenome subtraction法を用いてDCS生産株でのみ特異的に存在する遺伝子を含むDNA断片を取得した。
すなわち、PCR-select bacterial genome subtraction kit (クローンテック:Clontech社製)を用い、そのプロトコールに従って実施した。ただし、放線菌遺伝子のGC含量が高いことからハイブリダイゼーション温度は75℃とした。テスター株特異的DNA断片はpGEM-Tベクター(Promega社製)にサブクローニングし、大腸菌DH5α細胞に導入した。得られたコロニーのうち約120個の候補コロニーがランダムに選択された。各コロニーを液体培養し、それぞれの菌体からプラスミドを抽出した。テスターおよびドライバー株由来のゲノムDNAを制限酵素AluIで消化したものをプローブとし、テスター特異的DNA断片を含むプラスミドを選択した。得られたDNA断片の塩基配列を決定し、その情報をBLASTおよびFrame Plot解析した。
【0038】
S. lavendulae ATCC11924に特異的に存在する遺伝子を含む6-kb DNA断片をクローニングするため、4つのクローンのうちの一つが選ばれ、これをプローブとしたサザン解析により、BamHIで消化したS. lavendulae由来の6-kb DNA断片を得た。それをアガロース電気泳動に掛け、抽出後、BamHIで消化したpUC19に挿入し、そのキメラプラスミドを大腸菌DH5αに導入した。コロニーハイブリダイゼーションにより得られたクローンは確かにS. lavendulae ATCC11924由来の遺伝子を保有することをサザン解析にて確かめた後、A49と命名し、その塩基配列を決定した。DNA塩基配列はABI Prism310および377の自動シークエンサーにて解読され、GENETYXにて遺伝情報の解析を行った。フレーム解析はFrame Plot、ホモロジーはDDBJのプログラムのひとつFASTX、ドメイン構造はDomain Databaseなどによって解析された。
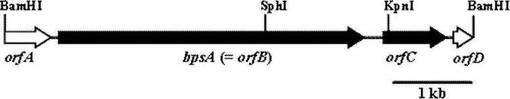
この結果、4つのOpen Reading Flame (ORF)が存在した(図1)。相同性検索より、orfBの遺伝子産物はNRPSと予想され、また、Erwinia chrysanthemi において、青色素インジゴイジンの合成に必須であると同定されているIndCと57%の同一性を示した。さらに、Domain Database検索の結果、orfBによってコードされるNRPSはCondensation domainを持たず、また、Oxidation domainをもつsingle module type-NRPSであると予想された。このorfBを含むDNA断片を放線菌vector pIJ702に連結し、S. lividans 66に導入した。この形質転換体は青色素を生産したことから、この遺伝子をbpsA (blue pigment synthetase-encoding gene A)と命名した。
【0039】
実施例2 S. lividans 66でのbspAの発現
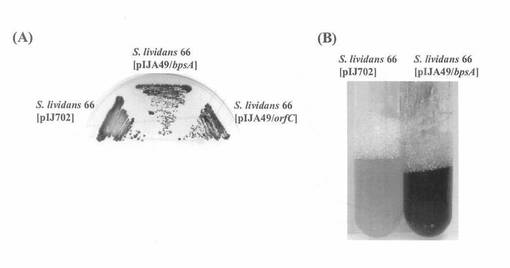
A49クローンをBamHIおよびKpnIで二重消化することによって、bspAを含む4.8-kb断片を得て、それをBglIIおよびKpnIで二重消化したpIJ702に挿入し、できたキメラプラスミドをpIJA49/bspAと命名した。一方、A49をBamHIおよびSphIで二重消化したものはorfCを含む2.6-kb断片であり、これをBglIIおよびSphIで二重消化したpIJ702に連結し、キメラプラスミドpIJA49/orfCと名付けた。それぞれのキメラプラスミドはS. lividanns 66のプロトプラストに導入し、R5培地にて再生させた。得られたコロニーはYEMEあるいはFB培地に接種し、28℃にて培養した。結果を図2に示す。
【0040】
pIJA49/bpsAを導入したS. lividans 66は、FB寒天培地上(A)およびYEME液体培地中(B)で青色素を産生した。pIJA49/bpsAを構築する際、bpsAはpIJ702に含まれるチロシナーゼ遺伝子のプロモーターとは逆向きに挿入されていることから、bpsA遺伝子の上流には自身のプロモーターが存在すると考えられる。一方、pIJ49/orfCを導入したS. lividans 66は、FB寒天培地上(A)でアクチノロジンを産生した。S. lividans 66において、アクチノロジン生合成遺伝子は通常休眠状態にあることから判断すると、orfCは転写活性化因子として機能すると考えられる。
【0041】
実施例3 他の放線菌におけるbpsAホモログの存在有無
他のDCS生産菌においてbpsAホモログが存在するかどうかを、PCR法にて確認するため、二つのオリゴヌクレオチドプライマー
bpsA-Ox-F1 (5'-GAGGGCCAGTACCGCTACGAGAAC-3') (配列番号5)と
bpsA-Ox-R1 (5'-ACGTCGATCTTGCCGTTGGCGGACA-3') (配列番号6)を設計した。
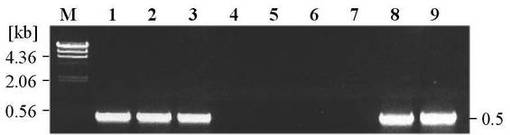
これらのプライマーを用いることによって、BPSAのOxドメインをコードする505 bpの遺伝子断片を特異的に増幅することができる。このため、他のDCS生産菌におけるbpsAホモログの存在有無を簡便に調べることができる。PCR実験は5 ngの染色体DNAを含む50 μlの反応液中にて行い、1サイクル目に熱変性 (98℃) 1分、 続いて、 熱変性 (96℃) 15秒、アニーリング (68℃) 15秒、伸長 (72℃) 30秒のサイクルを28サイクル繰り返し、増幅反応をおこなった。増幅したDNA断片は0.8%アガロースゲル中で電気泳動することによって確認した。結果を図3に示す。各レーンにおいては、以下の微生物の染色体DNAを鋳型として用いた。
【0042】
レーン1:S. lavendulae ATCC11924(サイクロセリン生産菌)
レーン2:S. lavendulae ATCC25233(サイクロセリン生産菌)
レーン3:S. garyphalus (CSH5-12)(サイクロセリン生産菌)
レーン4:S. coelicolor A3(2)(サイクロセリン非生産菌)
レーン5:S. lividans 66(サイクロセリン非生産菌)
レーン6:S. avermitilis K139(サイクロセリン非生産菌)
レーン7:S. lavendulae JCM4055(サイクロセリン非生産菌)
レーン8:S. lavendulae FRI-5(サイクロセリン生産菌)
レーン9:陽性コントロール
【0043】
遺伝子の増幅は、他のサイクロセリン生産菌由来の染色体DNAを鋳型としてPCRを行なった場合に認められ、サイクロセリン非生産菌由来の染色体DNAを鋳型とした場合では認められなかった。このことから、他のサイクロセリン生産菌にはbpsAホモログが存在すると考えられる。
【0044】
実施例4 RNaseプロテクションアッセイ
S. lavendulae ATCC11924を培地B中にて、28℃で培養した。播種後、5時間後にγ-ノナラクトン(γ−NL)を添加し、その後1時間ごとに全RNAを抽出した。全RNAの抽出はマニュアルにしたがって行った(図4)。
【0045】
orfAとbpsAの間の領域を含むリボプローブAは、以下のように合成した。まず、ふたつのオリゴヌクレオチドプライマーorfA-sense (5'-TCGAGAACGGATCTGTCACCCGGCTCCTGC-3')(配列番号7)とbpsA-antisense (5'-GTAGGCGACCGCGATCGCCTCGGGGTGTTC-3')(配列番号8)を用いて257 bpのDNA断片をPCR法にて増幅し、 pGEM-Tベクターに連結した。その後、 Ambion 社製MAXIscript in vitro transcription kitと[α-32P]UTPを用いて試験管内転写を行った。bpsAとorfCの間の領域を含むリボプローブB (279 bp) 、および、 orfCとorfDの間の領域を含むリボプローブC (305 bp) もリボプローブAと同様に合成した。ただし、リボプローブB合成のためには、 オリゴヌクレオチドプライマー bpsA-sense (5'-AGATTGACCGCGTTCTGACATACCTC-3')(配列番号9)とorfC-antisense (5'-CTTCTCGCCCCACAGCTCCCGGATGAGG-3')(配列番号10)を用い、リボプローブC合成のためには、 オリゴヌクレオチドプライマー orfC-sense (5'-AGCGGTTCGTGGTGGGCGCGGCCGTCTGAG-3')(配列番号11)と orfD-antisense (5'-CGTGCACCAGACCGGTGGTGATCAGGGTCTC-3')(配列番号12)を用いた。
【0046】
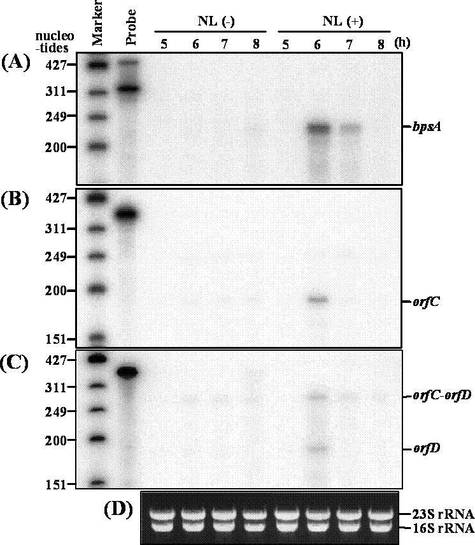
抽出したRNAサンプル (5 μg) はAmbion社製RPA III RNase protection assay kitを用い、42℃にて一晩、 放射性同位体ラベルしたリボプローブ (4 × 104 cpm) とハイブリダイズさせた。その後、 RNase AとRNase T1の混合液で処理し、沈殿させた後、8 M尿素を含む5%ポリアクリルアミド中で電気泳動した。サイズマーカーは、φX174 DNAをHinfIで切断後脱リン酸化し、 T4ポリヌクレオリドキナーゼ (東洋紡) と[γ-32P]ATPを用いて5'末端をラベル化したものを用いた。ゲル中の放射活性は、 富士フィルム社製イメージングプレートおよびBAS-2000にて検出した。結果を図4に示す。
(A):プローブAによりプロテクションされたRNA
(B):プローブBによりプロテクションされたRNA
(C):プローブCによりプロテクションされたRNA
(D):RNaseプロテクションアッセイに用いた全RNAを1%ホルムアミドアガロースゲル電気泳動で検出したもの。これにより、用いた全RNA量は等しいことが分かる。
【0047】
RNaseプロテクションアッセイの結果、bpsA、orfCおよびorfDはg-NL添加による誘導がない場合、発現しないことが分かった。一方、これらの遺伝子はg-NLによる誘導後一時間後に発現し、その後、減衰した。したがって、bpsA、orfCおよびorfDは、g-NLによりその発現が一過性に誘導されることが分かった。
【0048】
実施例5 青色素の検出、精製、およびキャラクタリゼーション
S. lavendulae ATCC11924による所定時間後における青色素の生産は、培養液を遠心して得られた上清に、 等量の水を加えた後、590 nmの吸光度を測定することで検出した。青色素の精製は以下のように行った。S. lavendulae ATCC11924をメディウムB (2 L)にて培養し、 培養開始5時間後に誘導剤としてγ-NLを添加した。その2時間後に培養液を4,400 × gで10 分間遠心し、 菌体を除いた。得られた上清をさらに、 30,000 × gにて30 分間遠心し青色素の沈殿を得た。この沈殿を水、および メタノールで2回ずつ洗浄し、真空中で乾燥させた。この沈殿にジメチルスルホキシドを加え、 ソニケーションで溶解させた後、 アドバンテック社製DISMIC-13JPフィルターを用いて微小沈殿物を除いた。その後、 5倍量の水を加え、 再び 30,000 × gにて30 分間遠心し青色素の沈殿を得た。この沈殿を水で10回、 メタノールで2回ずつ洗浄し、 真空中で乾燥させることで 精製青色素 (11 mg) を得た。
【0049】
吸光度分析のためのサンプル調整は、 精製した青色素を、 ジメチルスルホキシド (DMSO) 、 N-メチルピロリドン (NMP) 、 テトラヒドロフラン (THF) 、およびピリジンなどの各種溶媒に懸濁し、 ソニケーションで最大限溶解させた。その後、 15,000 × gにて5 分間遠心することで溶解していない粒子を取り除いた。可視部吸収スペクトルは、 日本分光社製V-550分光光度計で解析した。
EIマススペクトルは、 THFに溶解させた青色素を用い、 JEOL社製JMS-SX102Aスペクトロメーターで解析した。MALDI-TOF解析は、 ジメチルスルホキシドに溶解させた青色素を用い、 アプライドバイオシステムズ社製VoyagerTM RP-3で解析した。
【0050】
1H-NMR解析はJEOL社製JNM-LA500スペクトロメーターで解析した。内部標準としてテトラメチルシラン、 溶媒として重水素化されたDMSOを用いた。
IRスペクトルは、ヨウ化カリウム法で測定し、波数として3443, 3328, 3288, 3196, 3064, 2959, 2924, 2869, 2839, 2364, 1688, 1639, 1596, 1579, 1454, 1371, 1323, 1250, 1118, 1060, 1037, 956, 885, 824, 779, 752, 718, 689, 661, 634の周波数のところに吸収が観測された。
【0051】
結果を図5にまとめて示す。
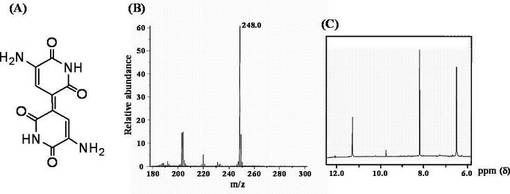
EI-mass分析においては、m/z = 248.0にピークが存在し、これは推定される青色素(インジゴイジン)の構造の分子式から計算した値と一致した。
1H-NMRスペクトル解析においては、11.30 ppm (s; NH)、8.18 ppm (s; CH)、および6.46 (s; NH2)のシグナルが得られた。
以上より、青色素は、インジゴイジンであると判断された。
【0052】
実施例6 S. verticillus由来ホスホパンテテイニル基転移酵素をコードするsvp遺伝子とbpsA遺伝子の大腸菌における共発現
bpsA遺伝子は、プライマーとして5'リン酸化した5'-CATATGACTCTTCAGGAGACCAGCGTGCTC-3'(配列番号13)(下線部はNdeIサイトを示す)、および 5'-AAGCTTCTCGCCGAGCAGGTAG CGGATGTG-3'(配列番号14)(下線部はHindIIIサイトを示す) を用い、東洋紡社製KOD-plusポリメラーゼを用いてPCR法にて増幅させた。増幅遺伝子をアガロースゲルより抽出・精製し、 pUC19のSmaIサイトに連結し、キメラプラスミドpUC/bpsAを得た。挿入されたbpsA遺伝子の塩基配列を確認した後、pUC/bpsAをNdeIおよびHindIIIにて二重消化し、 pET-28a(+)の同サイトに組み込みキメラプラスミドpET/bpsAを得た。このベクターから発現したBPSAはNおよびC末端にポリヒスチジンタグを持つ。
【0053】
S. verticillus由来ホスホパンテテイニル基転移酵素 (PPTase) をコードするsvp遺伝子は プライマーとして5'リン酸化した5'-CATATGATCGCCGCCCTCCTGCCCTCCTG-3'(配列番号15)(下線部はNdeIサイトを示す) および5'-CTCGAGCGGGACGGCGGTCCGGTCGTCCGC-3'(配列番号16)(下線部はXhoIサイトを示す) を鋳型DNAとしてS. verticillus染色体を用いて増幅した。この増幅断片をpUC19のSmaIサイトに連結し、キメラプラスミドpUC/svpを得た。挿入されたsvp遺伝子の塩基配列を確認した後、pUC/svpをNdeIおよびXhoIにて二重消化し、pET-21a(+)の同サイトに組み込み、キメラプラスミドpET/svpを得た。このベクターから発現したSvpはC末端にポリヒスチジンタグを持つ。pET/svpのうち、 ベクター由来のT7プロモーターおよびその下流に連結されたsvp遺伝子を含む2.2 kbのDNA断片を、SphIおよびPvuIで二重消化することにより切り出し、pSTV28の同サイトに連結することで、キメラプラスミドpSTV/svpを得た。
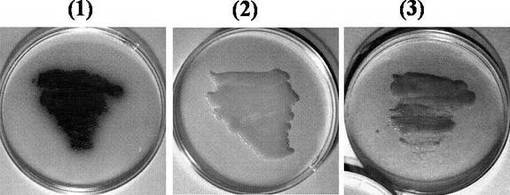
pET/bpsAおよびpSTV/svpを同時に大腸菌BL21(DE3) 株に導入し、BPSAおよびSvpの共発現株を得た。大量発現させたときの両タンパク質の溶解性を考慮し、発現実験は18℃にて行った。18℃で48時間培養した結果を、図6に示す。
【0054】
pET/bpsAおよびpSTV/svpを同時に導入した大腸菌は青色素を産生したが、pET/bpsAのみを導入した大腸菌は青色素を産生しなかった。このことから、大腸菌が保有する自身のPPTaseはBPSAをホロ型に変換できないと考えられ、また、SvpはBPSAをホロ型に変換し、ホロ型BPSAが青色素合成を触媒すると考えられた。
【0055】
実施例7 大腸菌におけるBPSAおよびBPSAΔTEの大量発現と精製
TE-ドメインを欠損したBPSAΔTE (アミノ酸番号1-1014) を生産させるため、 全長型bpsAの場合と同様に、pET-28a(+)を用いて bpsAΔTE発現用ベクターpET/bpsAΔTEを構築した。PCRプライマーは、bpsA増幅に用いた5'リン酸化センスプライマーと、5'リン酸化アンチセンスプライマー 5'-AAGCTTCTGGGCGACCTCGCGCTCCAG-3' (配列番号17)を使用した (下線部はHindIIIサイトを示す) 。BPSAΔTEタンパク質は、N末端およびC末端ヒスチジンタグ付きタンパク質として生産された。
【0056】
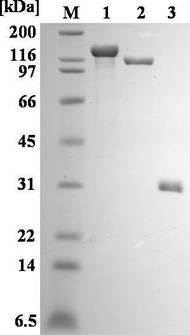
pET/bpsAあるいはpET/bpsAΔTEを保有する大腸菌BL21(DE3)株を、カナマイシンを含むLB培地2.5リットルにて18℃で培養した。発現したタンパク質の溶解性を考慮して、培養はイソプロピル-β-D-ガラクトピラノシド(IPTG)による誘導なしで30時間行った。細胞は遠心により集め、結合緩衝液 [20 mMトリス-塩酸 (pH 7.9)、500 mM塩化ナトリウム、5 mMイミダゾール] に懸濁した。懸濁以降、全ての過程は4℃にて行った。超音波により細胞を破砕し、27, 000 × g、20分間の遠心により細胞残骸物を除いた。その結果得られた上清を、予め結合緩衝液にて平衡化したHis-Bind resinカラム (1 × 10 cm、Novagen) に添加し、カラムを洗浄緩衝液 I [20 mMトリス-塩酸 (pH 7.9)、 500 mM塩化ナトリウム、30 mMイミダゾール] を用いて洗浄した。溶出は、30 mMから500 mMのイミダゾール直線濃度勾配により行った。BPSAあるいはBPSAΔTEを含む画分を集め、1 mMのエチレンジアミン四酢酸を含む結合緩衝液に対して透析し (2回) 、続いて結合緩衝液に対して透析を行った (2回)。透析した溶液を再びHis-Bind resinカラムに添加し、カラムを洗浄緩衝液II [20 mMトリス-塩酸 (pH 7.9)、500 mM塩化ナトリウム、60 mMイミダゾール] を用いて洗浄した。溶出は、60 mMから1000 mMのイミダゾール直線濃度勾配により行った。この段階で、ドデシル硫酸ナトリウム-ポリアクリルアミドゲル電気泳動から判断すると、 BPSAおよびBPSAΔTEはほぼ単一タンパク質として精製された(図7)。Ox-ドメインを保有するEpoB(Chen, H.W., O'Conner, S., Cane, D.E., and Walsh, C.T. (2001). Epothilone biosynthesis: assembly of the methylthiazolylcarboxy starter unit on the EpoB subunit. Chem. Biol. 8, 899-912.)と同様に、精製したBPSAおよびBPSAΔTEは、フラビンモノヌクレオチドを含むため黄色を呈していた。使用するまで、精製したタンパク質は50%グリセロールの存在下-20℃で保存した。
【0057】
実施例8 大腸菌におけるSvpの大量発現と精製
pET/svpを保有する大腸菌BL21(DE3)株を、アンピシリンを含むLB培地3リットルにて23℃で培養した。Svpの溶解性を考慮して、培養はIPTGによる誘導なしで24時間行った。細胞は遠心により集めた。細胞を結合緩衝液に懸濁した後は 全ての過程を4℃にて行った。超音波により細胞を破砕し、27, 000 × g、20分間の遠心により細胞残骸物を除いた。その結果得られた上清を、予め結合緩衝液にて平衡化したHis-Bind resinカラム (1 × 10 cm) に添加した。洗浄緩衝液IIによりカラムを洗浄した後、溶出は60 mMから500 mMのイミダゾール直線濃度勾配により行った。これらの過程を通して、Svpはほぼ単一タンパク質として精製された(図7)。使用するまで、精製したSvpタンパク質は50%グリセロールの存在下、-20℃で保存した。
【0058】
実施例9 試験管内おける青色素の合成
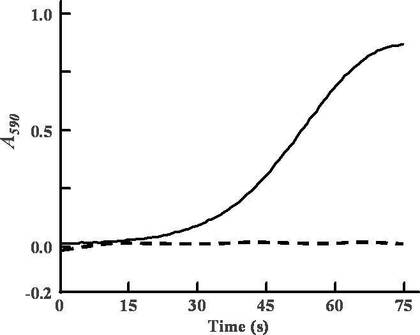
Svpを用いてBPSAを4'-ホスホパンテテイニル化するため、50 mMリン酸ナトリウム緩衝液 (pH 7.8) を用いて調製した660 nM BPSA、810 nM Svp、0.1 mM補酵素Aおよび1 mM塩化マグネシウムを含む溶液 (1.4 ml) を、30℃で10分間インキュベートした。青色素の合成は200 μlの10 mMアデノシン三リン酸溶液 (最終濃度1 mM) および400 μlの5 mM L-アミノ酸溶液 (最終濃度1 mM) を加えることにより開始した。試験管内における青色素の合成は、590 nmの吸収を測定することによりモニターした(図8)。合成された青色素の分子量は、上記で述べたように, MALDI-TOFにより分析された。
【0059】
Svpで4'-ホスホパンテテイニル化したBPSAを用いたとき、L-グルタミンを基質とした場合にのみ青色素の合成が認められた。また、Svpで4'-ホスホパンテテイニル化していないBPSAを用いたときでは、L-グルタミンを基質とした場合でも青色素の合成が認められなかった。このことから、4'-ホスホパンテテイニル化されたホロ型BPSAが、L-グルタミンを唯一の基質として青色素を合成すると考えられる。
【0060】
実施例10 アデノシン三リン酸/ピロリン酸-交換アッセイ
アデノシン三リン酸/ピロリン酸交換反応を用いて、BPSAのアデニル化ドメインの活性を測定した。
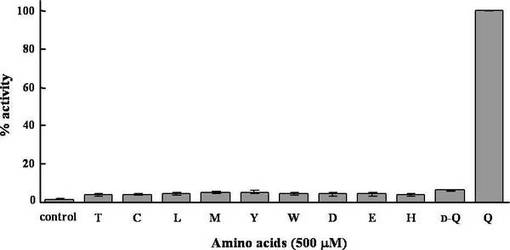
アミノ酸に依存したアデノシン三リン酸/ピロリン酸-交換アッセイは、基本的に過去に述べられた方法 (Du, L., Chen, M., Zhang, Y., and Shen, B. (2003). BimIII and BlmIV nonribosomal peptide synthetase-catalyzed biosynthesis of the bleomycin bithiazole moiety involving both in cis and in trans aminoacylation. Biochemistry 42, 9731-9740.) により行った。反応溶液 (100 μl) は, 300 nMヒスチジンタグ付きタンパク質、5 mMアデノシン三リン酸、1.72 μM [32P]ピロリン酸 (1 μCi, 60 Ci/mmol;PerkinElmer Life Siences) 、1 mMピロリン酸、1 mM塩化マグネシウム、0.1 mMエチレンジアミン四酢酸、0.5 mMアミノ酸および20 mMトリス-塩酸 (pH 7.8) を含んでいる。この溶液を30℃で20分間インキュベートした後、0.25 M過塩素酸、1.6%活性炭および0.1 Mピロリン酸四ナトリウムを含む溶液0.9 mlを加えることで、反応を停止させた。遠心により活性炭を集め、1 mlの水で2回洗浄し、0.5 mlの水に懸濁した。液体シンチレーション (ACSII、Amersham Biosciences) を加えた後、活性炭に結合した放射活性を液体シンチレーションカウンターにより測定した。なお、アミノ酸を含まない反応溶液をコントロールとして使用した。BPSAおよびBPSAΔTEの速度論的パラメーターを決定する場合、様々な濃度のL-グルタミンを用いて、反応は6分間行った。[32P]ピロリン酸の比活性を用いて、活性炭に結合した放射活性を反応速度に変換した。結果を図9に示す。図は、L-グルタミンに対する活性を100%とし、他のアミノ酸に対する活性はその相対活性として示している。
図9から、BPSAの基質はL-グルタミンであると考えられる。
【0061】
実施例11 ピロリン酸放出アッセイ
BPSAアデニル化ドメインのL-グルタミンに対する活性を、ピロリン酸放出アッセイにより測定した。
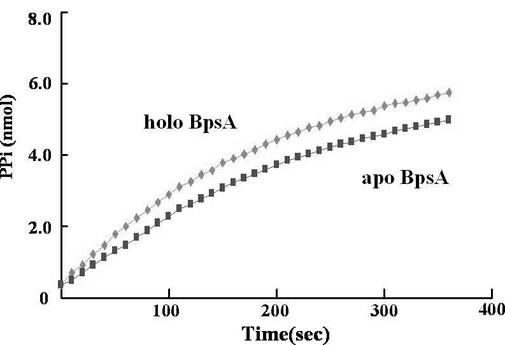
A-ドメインによるピロリン酸放出速度は、EnzCheck Pyrophosphatase Assay Kit (Molecular Probe) を用いて、グアノシンアナログであるMesGを利用した連続分光学的手法により測定した。反応溶液は、20 mMトリス-塩酸 (pH 7.8) 、1 mM塩化マグネシウム、0.1 mMエチレンジアミン四酢酸、0.2 mM MesG、0.2 Uプリンヌクレオシドホスホリラーゼ、0.2 U無機ピロホスファターゼ、5 mMアデノシン三リン酸、3 mM L-グルタミン、100 nM Svp、0.5 mM補酵素Aおよび100 nMのBPSAを含んでいる。BPSAをアポ体からホロ体へ変換する場合、予め、L-グルタミンとアデノシン三リン酸を含まない溶液にて、100 nMのBPSAを100 nMのSvpとともに30℃で30分間インキュベートした。この場合、 反応は3 mMのL-グルタミンおよび5 mMのアデノシン三リン酸を加えることにより開始し、その後360 nmの吸収を10秒ごとに6分間測定した。得られたスペクトルにおける0秒から100秒までの勾配は、標準ピロリン酸を用いて作成した標準曲線と相関した(図10)。
図10において、ひし形はホロ酵素、四角はアポ酵素を用いて得られたデータを示す。0秒から100秒における勾配を利用することにより、ホロBPSAの回転速度は107/分、アポBPSAの回転速度は76/分と算出された。
【0062】
回転速度は、アポ型をホロ型へ変換することで若干上昇した。これは、ホロ型BPSAにおいては、Aドメインにより活性化されたL-グルタミンが、速やかにTドメインへ移行するためであると考えられる。
【0063】
実施例12 BPSAΔTE T-ドメインへの[14C] L-グルタミンのアミノアシル化アッセイ
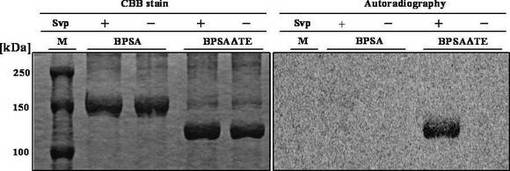
精製SvpによりT-ドメインをホスホパンテテイニル化したホロBPSAおよびホロBPSAΔTE、あるいは、Svpにより活性化しないそれぞれのアポ酵素を用いて、14C標識L-グルタミンのT-ドメインへの結合を調べた。
BPSAΔTEのT-ドメインへの[14C] L-グルタミンの結合は、オートラジオグラフィーにより調べた。100 μlの反応溶液は、20 mMトリス-塩酸 (pH 7.8) 、1 mM塩化マグネシウム、 0.1 mMエチレンジアミン四酢酸、3 mMアデノシン三リン酸、 10 mM [14C] L-グルタミン (0.2 μCi, 210 mCi/mmol; Moravek Biochemicals)、 300 nM Svp、0.5 mM補酵素Aおよび300 nMのBPSAあるいはBPSAΔTEを含んでいるが、予め[14C] L-グルタミンを含まない溶液を30℃にて30分間インキュベートすることにより、[14C] L-グルタミンを加えて反応を開始する前にそれらタンパク質のT-ドメインをホスホパンテテイニル化した。[14C] L-グルタミンを加えた後、様々な時間経過後、2%ウシ血清アルブミンを含む10%氷冷トリクロロ酢酸を0.8 ml加えることにより、反応を停止させた。沈殿したタンパク質を集め、10%氷冷トリクロロ酢酸およびアセトンで洗浄した後、その放射活性を測定した。オートラジオグラフィーを行う場合、[14C] L-グルタミンを加えてから15分後に反応を停止させ、タンパク質の沈殿を10%ドデシル硫酸ナトリウム-ポリアクリルアミドゲル電気泳動に供した。電気泳動後、乾燥させたゲルをイメージングプレートに曝露し、BAS-2000装置を用いて視覚化した(図11)。
【0064】
完全長のBPSAを用いた場合、Svpによる4'-ホスホパンテテイニル化の有無に関わらず、L-グルタミンはBPSA上に結合していなかった。一方、BPSAΔTEを用いた場合では、Svpで4'-ホスホパンテテイニル化されたBPSAΔTEにのみL-グルタミンの結合が認められた。このことから、完全長BPSAのC末端に存在するTEドメインは、Tドメインに結合したL-グルタミン(あるいはその誘導体)の切り離しを触媒すると考えられ、また、Aドメインにより活性化されたL-グルタミンは、4'-ホスホパンテテイニル化されたTドメインに結合すると考えられる。
【図面の簡単な説明】
【0065】
【図1】クローン化した約6-kbのDNA断片に存在するorfの構成とBPSAのドメイン構造を示す模式図である。白抜き矢印:不完全な遺伝子、黒矢印:完全な遺伝子
【図2】S. lividans 66でのbspAの発現を示した写真である。(A)チオストレプトン(50 μg/ml)を含むFB寒天培地上で生育する各形質転換体、(B)チオストレプトン(50 μg/ml)を含むYEME液体培地上で生育する各形質転換体
【図3】PCRで増幅した各遺伝子のアガロース電気泳動パターンを示した図である。 レーン1:S. lavendulae ATCC11924、レーン2:S. lavendulae ATCC25233、レーン3:S. garyphalus (CSH5-12)、レーン4:S. coelicolor A3(2)、レーン5:S. lividans 66、レーン6:S. avermitilis K139、レーン7:S. lavendulae JCM4055、レーン8:S. lavendulae FRI-5、レーン9:陽性コントロール
【図4】RNaseプロテクションアッセイの結果を示す図である。(A):プローブAによりプロテクションされたRNA、(B):プローブBによりプロテクションされたRNA、(C):プローブCによりプロテクションされたRNA、(D):RNaseプロテクションアッセイに用いた全RNAを1%ホルムアミドアガロースゲル電気泳動で検出したもの。NL(-):γ−NL非添加、NL(+):γ−NL添加
【図5】S. lavendulae より合成された青色素の化学的性質と構造を示した図である。 (A):構造。(B)EI-mass分析、(C)1H-NMRスペクトル
【図6】E. coliにおいて青色素の産生を示した写真である。(1)bpsAとsvpを共発現させた大腸菌BL21(DE3)株、(2)bpsAのみを発現する大腸菌BL21(DE3)株、(3)発現ベクターpET-28a(+)を保有する大腸菌BL21(DE3)株
【図7】発現および精製タンパク質のドデシル硫酸ナトリウムーポリアクリルアミドゲル電気泳動パターンを示した図である。レーン1:精製BPSA、レーン2:精製BPSAΔTE、レーン3:Svp、M:標準タンパク質、左数字:分子量。
【図8】In vitroにおける青色素の生成を示したグラフである。実線:BPSAおよびSvpを用いた場合、破線:BPSAのみを用いた場合
【図9】BPSAのアデニル化ドメインの活性を示した図である。
【図10】BPSAアデニル化ドメインのL-グルタミンに対する活性を示した図である。 ひし形:ホロ酵素、四角:アポ酵素
【図11】14C標識L-グルタミンのT-ドメインへの結合能を示した図である。左:ドデシル硫酸ナトリウムーポリアクリルアミドゲル電気泳動後、クマシー染色したもの、右:泳動後、14C標識L-グルタミンをオートラジオグラフィーで検出したもの
【技術分野】
【0001】
本発明は、D−サイクロセリン(DCS)生産性放線菌(Streptomyces lavendulae)に特異的に存在する青色素合成遺伝子及びそれを用いた青色素の製造方法に関する。
【背景技術】
【0002】
植物の自然な発色については天然植物色素が貢献しているが、その主な物質としてクロロフィル類、カロチノイド系色素、アントシアニン系色素があげられる。青色の発色はアントシアニン系色素によるものであるが、その発色機構は複雑であり、現在も研究が進められている。
【0003】
また、生花店で販売されている植物において、マリンブルーに近い青色の花の品種は意外に少ない。バラでは古来より青いバラの作製が育種家の夢であるが、従来の育種技術では困難といわれており、近年、遺伝子工学を利用して青いバラの作成が研究されているが、未だ愛好家を満足させるものはない。
【0004】
一方、ストレプトマイセス(Streptomyces)に属する放線菌は、実用抗生物質を始めとする二次代謝産物の多くを作ることから、工業的にも重要な微生物である。近年、ペプチド系抗生物質の多くが非リボソーム型ペプチド合成酵素(non-ribosomal peptide synthetase:NRPS)により生合成されることが知られるようになってきたが(非特許文献1)、放線菌に特異的に存在するNRPSには、未だその機能が見出されていないものが数多く存在すると考えられている。
【非特許文献1】Finking, R. et al.,(2004) Annu. Rev. Microbiol. 58, 453-488.
【発明の開示】
【発明が解決しようとする課題】
【0005】
本発明は、放線菌に特異的に存在する遺伝子の探索及びその機能を解明し、有用な遺伝子及びその用途を提供することを目的とする。
【課題を解決するための手段】
【0006】
本発明者は、DCS生産性放線菌(S. lavendulae)に特異的に存在する遺伝子を探索したところ、非リボソーム型ペプチド合成酵素(NRPS)ファミリーには珍しい、単一モジュール型の非リボソーム型ペプチド合成酵素をコードする遺伝子が存在すること、さらに当該遺伝子は青色素インジゴイジン(indigoidine)合成遺伝子として機能し、青色素の製造に有用であることを見出した。
【0007】
すなわち、本発明は、以下の(1)〜(9)に係るものである。
(1)以下の(a)又は(b)のタンパク質:
(a)配列番号1に示すアミノ酸配列からなるタンパク質、
(b)(a)のアミノ酸配列において、1個若しくは数個のアミノ酸が欠失、置換若しくは付加されたアミノ酸配列からなり、且つインジゴイジン合成能を有するタンパク質。
(2)(1)に記載のタンパク質をコードする遺伝子。
(3)以下の(a)又は(b)のDNAからなる遺伝子:
(a)配列番号2に示す塩基配列からなるDNA、
(b)(a)の塩基配列からなるDNAと相補的な塩基配列からなるDNAとストリンジエントな条件下でハイブリダイズし、且つインジゴイジン合成能を有するタンパク質をコードするDNA。
(4)(2)又は(3)に記載のいずれかの遺伝子を含有する組換えベクター。
(5)(4)記載の組換えベクターを含む形質転換体。
(6)(5)記載の形質転換体を培養し、培養物から(1)記載のタンパク質を採取することを特徴とする(1)記載のタンパク質の製造方法。
(7)(2)又は(3)記載の遺伝子を導入した放線菌を培養し、培養物からインジゴイジンを採取することを特徴とするインジゴイジンの製造方法。
(8)(2)又は(3)記載の遺伝子及び4'−ホスホパンテテイニル(4'-phosphopantetheinyl)基転移酵素遺伝子を導入した宿主を培養し、培養物からインジゴイジンを採取することを特徴とするインジゴイジンの製造方法。
(9)L-グルタミンに、(1)記載のタンパク質及び4'−ホスホパンテテイニル基転移酵素を作用させることを特徴とするインジゴイジンの製造方法。
【発明の効果】
【0008】
本発明の遺伝子を用いることにより、大腸菌等を宿主として、青色素インジゴイジンを産生させることができる。また、本発明の遺伝子を、当該色素産生能を持たない植物、蚕等に導入することにより、インジゴイジンを産生するような形質転換体を得ることができ、これにより例えば青いバラやブルーシルクの創出が可能となる。
また、本発明のタンパク質を用いることにより、インビトロで、マリンブルーに近い色を呈するインジゴイジンを製造することができる。
【発明を実施するための最良の形態】
【0009】
本発明の配列番号2に示す塩基配列からなる遺伝子は、後記実施例に示すように、DCS生産性S. lavendulae ATCC11924のゲノムからDCS非産生株であるS. lavendulae JCM4055のゲノムを差し引く、いわゆるゲノムサブトラクション (genome subtraction)法を用いることにより取得されたものである(実施例1)。そして、当該遺伝子は、DCS産生菌に特異的に存在するDNA断片中に存在する非リボソーム型ペプチド合成酵素(non-ribosomal peptide synthetase: NRPS)をコードするものであった。既知のNRPSは、ほとんど多重モジュール構造を取っているが、ここで得られた遺伝子産物(配列番号1に示すアミノ酸配列からなるタンパク質)は、NRPSタンパク質ファミリーには珍しい単一モジュール型であり、OX(オキシデーション)-ドメインが存在する(実施例1)。また、当該タンパク質は、Erwinia chrysanthemiにおいて、青色素インジゴイジンの合成に必須であると同定されているIndCとアミノ酸配列において57%の同一性を示していた。
【0010】
そして、このNRPS遺伝子をS. lividans細胞に導入すると、その形質転換株は新たに青色素を産生するようになった。当該青色素は、可視部吸収スペクトル、IRスペクトル、1H-NMRスペクトル、EI-massスペクトル等により、その化学的性質と構造を解析した結果、その構造は以下に示すとおりであり、IndCにより産生されるインジゴイジン(1. Kuhn, R., et al., (1965) Arch. Mikrobiol. 51, 71-84、2. Mortimer, P. S., et al., (1966) Appl. Microbiol. 14, 870-872)と同一であった(実施例5)。
【0011】
【化1】
【0012】
このことから、当該NRPS遺伝子は青色素インジゴイジン合成遺伝子として機能するものといえ、本NRPS遺伝子をbpsA (blue-pigment synthetase A)、本遺伝子によってコードされるタンパク質をBPSAと名付けた。
【0013】
上記のS. lavendulae ATCC11924株は、インジゴイジン非生産性であるが、放線菌ホルモンに類似した物質であり、食品添加物として使用される香気成分であるγ-またはδ-ノナラクトンの添加により、インジゴイジン生産が誘導される。後記実施例に示すように、bpsA遺伝子と、さらに、その下流に存在する二つの遺伝子の転写がγ-ノナラクトンの添加により誘導される(実施例4)。
【0014】
また、放線菌由来のこの青色素インジゴイジンを大腸菌で生産させる場合には、bpsA遺伝子のほか、BPSAを4'-ホスホパンテテイニル(4'-phosphopantetheinyl)化させる、 例えば、S. verticillus ATCC15003由来 4'-ホスホパンテテイニル基転移酵素 (PPTase) 遺伝子 (svpと命名)のような、4'-ホスホパンテテイニル基転移酵素遺伝子の共発現が不可欠であった(実施例6)。すなわち、大腸菌の中で生成したBPSAが翻訳後修飾により4'-ホスホパンテテイニル化されて初めて、インジゴイジンをつくる酵素として機能する。
【0015】
さらに、BPSAの基質はL-グルタミン(L-Gln)であり、および、試験管内(in vitro)において、L-GlnにBPSAおよびsvp遺伝子がつくるタンパク質(Svpと命名)を添加すると、試験管内でインジゴイジンが合成された。
【0016】
以下に、本発明のタンパク質及び遺伝子について説明する。
尚、本明細書において遺伝子とは、2本鎖DNAのみならず、それを構成するセンス鎖およびアンチセンス鎖という各1本鎖DNAを包含する趣旨である。従って、本発明の遺伝子には、特に言及しない限り、2本鎖DNAおよびcDNAを含む1本鎖DNA(センス鎖)並びに該センス鎖と相補的な配列を有する1本鎖DNA(アンチセンス鎖)が含まれる。
【0017】
本発明のタンパク質は、具体的には、配列番号1に示すアミノ酸配列からなるタンパク質(BPSA)として示されるが、特にこれらに限定されず、当該タンパク質の変異体、例えばアレル体、ホモログ、天然の変異体であって、配列番号1に示すアミノ酸配列と90%以上、好ましくは95%以上、より好ましくは98%以上、さらにより好ましくは99%以上の同一性を有するタンパク質や、配列番号1に示すアミノ酸配列において、1若しくは数個のアミノ酸が欠失、置換若しくは付加されたアミノ酸配列からなり、且つBPSAと同様のインジゴイジン合成能を有するタンパク質が包含される。
【0018】
ここで、配列番号1に示すアミノ酸配列において、1若しくは数個のアミノ酸が欠失、置換若しくは付加されたアミノ酸配列とは、配列番号1のアミノ酸配列と等価のアミノ酸配列を意味し、1若しくは数個、好ましくは1〜10個のアミノ酸が欠失、置換若しくは付加されたアミノ酸配列を意味し、付加には、両末端への1〜数個のアミノ酸の付加が含まれる。
【0019】
本発明の遺伝子は、具体的には配列番号1に示すアミノ酸配列からなるタンパク質をコードする配列番号2に示す塩基配列からなるDNAとして示されるが、特にこれらに限定されず、上記した一定の改変を有するタンパク質をコードする塩基配列からなるDNAが包含される。また、配列番号2に示す塩基配列と、少なくとも90%以上、好ましくは95%以上、より好ましくは98%以上、更に好ましくは99%以上の同一性を有するDNAであって、且つBPSAと同様のインジゴイジン合成能を有するタンパク質をコードするDNAや、配列番号2に示す塩基配列と相補的な塩基配列からなるDNAにストリンジエントな条件下でハイブリダイズし、且つBPSAと同様のインジゴイジン合成能をするタンパク質をコードするDNAが挙げられる。
【0020】
ここで「ストリンジエントな条件下」とは、例えば、Amersham Biosciences社製 AlkPhos Direct Labeling and detection system のハイブリダイゼーション溶液にプローブとともに65℃で8〜16時間恒温し、ハイブリダイズさせる条件が挙げられる。
【0021】
上記改変のための人為的手段としては、部位特異的突然変異誘発法等の公知の手法を利用して調製することができる。例えば、部位特異的突然変異誘発法を利用した変異導入用キット(BD Bioscience社製Transformer Site-Directed Mutagenesis Kit)等を用いて変異を導入し調製することができる。
【0022】
尚、アミノ酸配列又は塩基配列の同一性は、既知の配列分析ソフトウェア、例えば大学共同利用機関法人 情報・システム研究機構 国立遺伝学研究所 日本DNAデータバンクのBLASTプログラムを使用した測定(データベースに登録されているアミノ酸配列またはDNA塩基配列との比較)によって解析できる。
【0023】
本発明遺伝子は、本明細書により開示された本発明の遺伝子の配列情報に基づいて、一般的遺伝子工学的手法により容易に製造、取得することができる(続生化学実験講座「遺伝子研究法I、II、III」、日本生化学会編(1986)など)。
具体的には、本発明遺伝子が発現される微生物、例えばDCS生産性S. lavendulae ATCC11924より、常法に従ってゲノムDNAライブラリーを調製し、該ライブラリーから、本発明遺伝子に特有の適当なプローブ等を用いて所望クローンを選択することにより製造することができる。上記において、DCS生産性S. lavendulaeからの全RNAの分離、mRNAの分離および精製、ゲノムDNAの取得およびそのクローニングなどは、いずれも常法に従って行うことができる。
【0024】
本発明遺伝子をゲノムDNAライブラリーからスクリーニングする方法も、特に制限されず、通常の各種方法に従うことができる。具体的方法としては、例えば、目的の核酸配列に選択的に結合するプローブを用いたプラークハイブリダイゼーション法、コロニーハイブリダイゼーション法など、およびこれらの組合せを例示することができる。
【0025】
上記方法において用いられるプローブとしては、本発明遺伝子の塩基配列に関する情報をもとにして化学合成されたDNAなどが一般的に使用できる。また、本発明DNAの塩基配列情報に基づき設定したセンス・プライマーおよびアンチセンス・プライマーを、スクリーニング用プローブとして用いることができる。
本発明遺伝子の取得に際しては、PCR法〔Science, 230, 1350 (1985)〕によるDNA/RNA増幅法が好適に利用できる。増幅させたDNA/RNA断片の単離精製は、前記の通り、常法に従うことができる。例えばゲル電気泳動法などによることができる。
上記方法に従い得られる本発明遺伝子は、常法、例えばジデオキシ法〔Proc. Natl. Acad. Sci., USA., 74, 5463 (1977)〕、マキサム−ギルバート法〔Methods in Enzymology, 65, 499 (1980)〕などに従って、その核酸配列を決定することができる。また、簡便には、市販のシークエンスキットなどを用いて、その核酸配列を決定することができる。
【0026】
本発明のタンパク質は、本発明により提供される遺伝子の配列情報に基づいて、通常の遺伝子組換え技術に従って調製することができる。すなわち、所望のタンパク質をコードする遺伝子が宿主細胞中で発現できる組換えDNA(発現ベクター)を作成し、これを宿主細胞に導入して形質転換し、該形質転換体を培養し、次いで得られる培養物から回収することができる。
【0027】
上記において宿主としては、例えば大腸菌(Esherichia coli BL21(DE3))などの一般的に用いられるものが広く挙げられ、好適には原核生物細胞、特に大腸菌を用いるのが好ましい。原核生物細胞を宿主とする場合は、該宿主細胞中で複製可能なベクターを用いて、このベクター中に本発明遺伝子が発現できるように該遺伝子の上流にプロモーターおよびSD(シャイン・アンド・ダルガーノ)核酸配列、更に蛋白合成開始に必要な開始コドン(例えばATG)を付与した発現プラスミドを好適に利用できる。上記ベクターとしては、一般に大腸菌由来のプラスミド、例えばpBR322、pBR325、pUC12、pUC13などがよく用いられるが、これらに限定されず既知の各種のベクターを利用することができる。大腸菌を利用した発現系に利用される上記ベクターの市販品としては、例えばpET21、pET28(Novagen社)、pGEX−4T(Amersham Pharmacia Biotech社)、pQE-30, pQE-60(QIAGEN社製)などを例示できる。
【0028】
かくして得られたベクターを用いて宿主を形質転換するにはプロトプラスト法、コンピテントセル法、エレクトロポレーション法等を用いて行うことができる。得られた形質転換体は、資化しうる炭素源、窒素源、金属塩、ビタミン等を含む培地を用いて適当な条件下で培養すればよい。得られた培養液から一般的な方法によって、タンパク質の採取、精製を行うことができる。
【0029】
次に、本発明の遺伝子を用いたインジゴイジンの製造方法について説明する。
(1)形質転換体を用いたインジゴイジンの製造
一般に、NRPSは4'−ホスホパンテテイニル基転移酵素によってホスホパンテテイニル化され、holo-formに変換されなければ活性を生じないことが知られている。本発明のタンパク質においてもインジゴイジンの産生には4'−ホスホパンテテイニル基転移酵素により、当該タンパク質がホスホパンテテイニル化される必要がある。
従って、宿主として、大腸菌(E. coli BL21(DE3))等の一般細菌を用いる場合には、本発明の遺伝子及び4'−ホスホパンテテイニル基転移酵素遺伝子を導入した宿主を培養し、培養物からインジゴイジンを採取することによりインジゴイジンを製造することができる。
【0030】
ここで、4'−ホスホパンテテイニル基転移酵素遺伝子としては、特に限定されるものではないが、例えば、生化学的機能が明らかなS. verticillus由来4'−ホスホパンテテイニル基転移酵素をコードするsvp遺伝子(配列番号4)、枯草菌Bacillus subtilis由来sfp遺伝子等が挙げられる。
共発現させるための手法としては、前記と同様公知の遺伝子工学的手法を用いることができる。例えば、本発明の遺伝子を挿入した発現ベクター(例えば、pET/bpsA)と、4'−ホスホパンテテイニル基転移酵素遺伝子を挿入した発現ベクター(例えば、pSTV/svp)を同時に、宿主に導入し、共発現している細胞を選抜する方法が挙げられる。
【0031】
ここで、培養物とは、培養上清、あるいは培養細胞若しくは培養菌体又は培養細胞若しくは培養菌体の破砕物のいずれをも意味するものである。
また、培養方法は、上記のとおり、宿主細胞培養に用いられる通常の方法に従って行えばよい。
【0032】
インジゴイジンの採取及び精製は、例えば以下のようにして行うことができる。
すなわち、5時間程度培養したS. lavendulaeの培養液に、γ-ノナラクトンを最終濃度約30μg/mlになるよう添加した後、さらに2〜3時間培養する。培地中に分泌された青色素と菌体を低速遠心により分離し、得られた遠心上清を更に高速遠心することで、青色素沈殿物を得る。この沈殿物を水およびメタノールで順次洗浄後、乾燥させることにより、粗精製青色素が得られる。これをジメチルスルホキシド等の有機溶媒に溶解後、0.2μm孔のメンブランフィルターに通すことで残存する菌体を除き、得られた濾液に対し、約5倍量の水を加えて青色素を析出させる。この溶液を高速遠心分離し、青色素沈殿物を得て、上記に示したように洗浄後、乾燥させることで精製インジゴイジンが得られる。
【0033】
また、インジゴイジン合成能のない放線菌に、本発明の遺伝子を導入することでインジゴイジン合成能もつ形質転換放線菌を得ることができる。当該放線菌を液体培養し、その培養液からインジゴイジンを採取することによってもインジゴイジンを得ることができる。この場合、チオストレプトン(50 μg/ml)を含むYEME培地で、2-5日間、約28℃にて培養することで、培地中に青色素が生産される。
【0034】
(2)インビトロにおけるインジゴイジンの製造
L-グルタミンに、本発明のタンパク質及び4'−ホスホパンテテイニル基転移酵素を作用させることにより、インジゴイジンを製造することができる。
ここで、4'−ホスホパンテテイニル基転移酵素としては、例えば、前記S. verticillus由来の4'−ホスホパンテテイニル基転移酵素(配列番号3)等が挙げられる。
本反応は、本発明のタンパク質及び4'−ホスホパンテテイニル基転移酵素に見合ったpHにおいて実施すればよく、このpHは概ね6.0〜8.0、望ましくは7.0〜7.8の範囲である。
反応溶媒は、一般に水または水−有機溶媒混合物を使用することができる。
反応は、一般に25〜30℃、望ましくは28〜30℃で、基質としてのL-グルタミンを加えた後、2〜5分、好ましくは、2〜3分である。
インジゴイジンの採取及び精製は、例えば、反応液から、約15,000gで10分間程度の遠心操作を行い、その上清を除去して得られる沈殿物を必要に応じて水およびメタノールで洗浄し、且つ上記の遠心操作を繰り返すことにより行うことができる。
【実施例】
【0035】
実施のための材料と方法
(1)材料(使用した菌株およびプラスミド、ならびに培養培地)
DCS生産菌として、S. lavendulae ATCC11924、 S. lavendulae ATCC25233、 S. lavendulae FRI-5を用いた。DCS非生産菌株として、S. coelicolor A3(2)、 S. lividans 66、 S. avermitilis K139、 およびS. lavendulae JCM4055を用いた。
Streptomyces属放線菌のプロトプラストを調製する場合には、0.5%グリシンを含むYEME培地を用いて28℃で培養した。青色素を作らせる場合や、その菌から全RNAを調製する場合にはB培地を用いた。
放線菌におけるクローニングベクターとしてはpIJ702(選択マーカーとしてメラニン色素産生遺伝子とチオストレプトン耐性遺伝子を有し、通常、目的遺伝子はメラニン色素合成遺伝子の中にある制限酵素部位に挿入させる。その結果として、メラニン色素を作らなくさせることで、目的遺伝子の挿入により選択マーカーが発黒色ではないコロニーを容易に選択できる)。
ゲノムDNAやプラスミドDNAは常法に従って精製した。目的タンパク質の発現にはベクターとして、pET-21a(+)およびpET-28a(+)が用いられ、その際の宿主として大腸菌BL21(DE3)を用いた。pACYC184の複製開始点を有するpSTV28ベクターは共発現用に用いた。タンパク質を高発現させることを目的とした場合、大腸菌はLB培地で18℃あるいは23℃で培養し、クローニングを目的とした場合、37℃で培養した。必要に応じてLB培地にはカナマイシン 30 μg/ml、アンピシリン100 μg/ml、クロラムフェニコール 34 μg/mlが添加された。
【0036】
(2)サザンハイブリダイゼーション
ハイボンドN+メンブランが用いられ、遺伝子の標識はアルフォス・ダイレクト・ラベリングおよびその検出システムを採用した。
【0037】
実施例1 DCS生産株特異的遺伝子のクローニング
DCS生産株S. lavendulae ATCC11924に特徴的な二次代謝物質の生合成遺伝子をクローニングするため、まず、DCS非生産株S. lavendulae JCM4055を対照株として用いたgenome subtraction法を用いてDCS生産株でのみ特異的に存在する遺伝子を含むDNA断片を取得した。
すなわち、PCR-select bacterial genome subtraction kit (クローンテック:Clontech社製)を用い、そのプロトコールに従って実施した。ただし、放線菌遺伝子のGC含量が高いことからハイブリダイゼーション温度は75℃とした。テスター株特異的DNA断片はpGEM-Tベクター(Promega社製)にサブクローニングし、大腸菌DH5α細胞に導入した。得られたコロニーのうち約120個の候補コロニーがランダムに選択された。各コロニーを液体培養し、それぞれの菌体からプラスミドを抽出した。テスターおよびドライバー株由来のゲノムDNAを制限酵素AluIで消化したものをプローブとし、テスター特異的DNA断片を含むプラスミドを選択した。得られたDNA断片の塩基配列を決定し、その情報をBLASTおよびFrame Plot解析した。
【0038】
S. lavendulae ATCC11924に特異的に存在する遺伝子を含む6-kb DNA断片をクローニングするため、4つのクローンのうちの一つが選ばれ、これをプローブとしたサザン解析により、BamHIで消化したS. lavendulae由来の6-kb DNA断片を得た。それをアガロース電気泳動に掛け、抽出後、BamHIで消化したpUC19に挿入し、そのキメラプラスミドを大腸菌DH5αに導入した。コロニーハイブリダイゼーションにより得られたクローンは確かにS. lavendulae ATCC11924由来の遺伝子を保有することをサザン解析にて確かめた後、A49と命名し、その塩基配列を決定した。DNA塩基配列はABI Prism310および377の自動シークエンサーにて解読され、GENETYXにて遺伝情報の解析を行った。フレーム解析はFrame Plot、ホモロジーはDDBJのプログラムのひとつFASTX、ドメイン構造はDomain Databaseなどによって解析された。
この結果、4つのOpen Reading Flame (ORF)が存在した(図1)。相同性検索より、orfBの遺伝子産物はNRPSと予想され、また、Erwinia chrysanthemi において、青色素インジゴイジンの合成に必須であると同定されているIndCと57%の同一性を示した。さらに、Domain Database検索の結果、orfBによってコードされるNRPSはCondensation domainを持たず、また、Oxidation domainをもつsingle module type-NRPSであると予想された。このorfBを含むDNA断片を放線菌vector pIJ702に連結し、S. lividans 66に導入した。この形質転換体は青色素を生産したことから、この遺伝子をbpsA (blue pigment synthetase-encoding gene A)と命名した。
【0039】
実施例2 S. lividans 66でのbspAの発現
A49クローンをBamHIおよびKpnIで二重消化することによって、bspAを含む4.8-kb断片を得て、それをBglIIおよびKpnIで二重消化したpIJ702に挿入し、できたキメラプラスミドをpIJA49/bspAと命名した。一方、A49をBamHIおよびSphIで二重消化したものはorfCを含む2.6-kb断片であり、これをBglIIおよびSphIで二重消化したpIJ702に連結し、キメラプラスミドpIJA49/orfCと名付けた。それぞれのキメラプラスミドはS. lividanns 66のプロトプラストに導入し、R5培地にて再生させた。得られたコロニーはYEMEあるいはFB培地に接種し、28℃にて培養した。結果を図2に示す。
【0040】
pIJA49/bpsAを導入したS. lividans 66は、FB寒天培地上(A)およびYEME液体培地中(B)で青色素を産生した。pIJA49/bpsAを構築する際、bpsAはpIJ702に含まれるチロシナーゼ遺伝子のプロモーターとは逆向きに挿入されていることから、bpsA遺伝子の上流には自身のプロモーターが存在すると考えられる。一方、pIJ49/orfCを導入したS. lividans 66は、FB寒天培地上(A)でアクチノロジンを産生した。S. lividans 66において、アクチノロジン生合成遺伝子は通常休眠状態にあることから判断すると、orfCは転写活性化因子として機能すると考えられる。
【0041】
実施例3 他の放線菌におけるbpsAホモログの存在有無
他のDCS生産菌においてbpsAホモログが存在するかどうかを、PCR法にて確認するため、二つのオリゴヌクレオチドプライマー
bpsA-Ox-F1 (5'-GAGGGCCAGTACCGCTACGAGAAC-3') (配列番号5)と
bpsA-Ox-R1 (5'-ACGTCGATCTTGCCGTTGGCGGACA-3') (配列番号6)を設計した。
これらのプライマーを用いることによって、BPSAのOxドメインをコードする505 bpの遺伝子断片を特異的に増幅することができる。このため、他のDCS生産菌におけるbpsAホモログの存在有無を簡便に調べることができる。PCR実験は5 ngの染色体DNAを含む50 μlの反応液中にて行い、1サイクル目に熱変性 (98℃) 1分、 続いて、 熱変性 (96℃) 15秒、アニーリング (68℃) 15秒、伸長 (72℃) 30秒のサイクルを28サイクル繰り返し、増幅反応をおこなった。増幅したDNA断片は0.8%アガロースゲル中で電気泳動することによって確認した。結果を図3に示す。各レーンにおいては、以下の微生物の染色体DNAを鋳型として用いた。
【0042】
レーン1:S. lavendulae ATCC11924(サイクロセリン生産菌)
レーン2:S. lavendulae ATCC25233(サイクロセリン生産菌)
レーン3:S. garyphalus (CSH5-12)(サイクロセリン生産菌)
レーン4:S. coelicolor A3(2)(サイクロセリン非生産菌)
レーン5:S. lividans 66(サイクロセリン非生産菌)
レーン6:S. avermitilis K139(サイクロセリン非生産菌)
レーン7:S. lavendulae JCM4055(サイクロセリン非生産菌)
レーン8:S. lavendulae FRI-5(サイクロセリン生産菌)
レーン9:陽性コントロール
【0043】
遺伝子の増幅は、他のサイクロセリン生産菌由来の染色体DNAを鋳型としてPCRを行なった場合に認められ、サイクロセリン非生産菌由来の染色体DNAを鋳型とした場合では認められなかった。このことから、他のサイクロセリン生産菌にはbpsAホモログが存在すると考えられる。
【0044】
実施例4 RNaseプロテクションアッセイ
S. lavendulae ATCC11924を培地B中にて、28℃で培養した。播種後、5時間後にγ-ノナラクトン(γ−NL)を添加し、その後1時間ごとに全RNAを抽出した。全RNAの抽出はマニュアルにしたがって行った(図4)。
【0045】
orfAとbpsAの間の領域を含むリボプローブAは、以下のように合成した。まず、ふたつのオリゴヌクレオチドプライマーorfA-sense (5'-TCGAGAACGGATCTGTCACCCGGCTCCTGC-3')(配列番号7)とbpsA-antisense (5'-GTAGGCGACCGCGATCGCCTCGGGGTGTTC-3')(配列番号8)を用いて257 bpのDNA断片をPCR法にて増幅し、 pGEM-Tベクターに連結した。その後、 Ambion 社製MAXIscript in vitro transcription kitと[α-32P]UTPを用いて試験管内転写を行った。bpsAとorfCの間の領域を含むリボプローブB (279 bp) 、および、 orfCとorfDの間の領域を含むリボプローブC (305 bp) もリボプローブAと同様に合成した。ただし、リボプローブB合成のためには、 オリゴヌクレオチドプライマー bpsA-sense (5'-AGATTGACCGCGTTCTGACATACCTC-3')(配列番号9)とorfC-antisense (5'-CTTCTCGCCCCACAGCTCCCGGATGAGG-3')(配列番号10)を用い、リボプローブC合成のためには、 オリゴヌクレオチドプライマー orfC-sense (5'-AGCGGTTCGTGGTGGGCGCGGCCGTCTGAG-3')(配列番号11)と orfD-antisense (5'-CGTGCACCAGACCGGTGGTGATCAGGGTCTC-3')(配列番号12)を用いた。
【0046】
抽出したRNAサンプル (5 μg) はAmbion社製RPA III RNase protection assay kitを用い、42℃にて一晩、 放射性同位体ラベルしたリボプローブ (4 × 104 cpm) とハイブリダイズさせた。その後、 RNase AとRNase T1の混合液で処理し、沈殿させた後、8 M尿素を含む5%ポリアクリルアミド中で電気泳動した。サイズマーカーは、φX174 DNAをHinfIで切断後脱リン酸化し、 T4ポリヌクレオリドキナーゼ (東洋紡) と[γ-32P]ATPを用いて5'末端をラベル化したものを用いた。ゲル中の放射活性は、 富士フィルム社製イメージングプレートおよびBAS-2000にて検出した。結果を図4に示す。
(A):プローブAによりプロテクションされたRNA
(B):プローブBによりプロテクションされたRNA
(C):プローブCによりプロテクションされたRNA
(D):RNaseプロテクションアッセイに用いた全RNAを1%ホルムアミドアガロースゲル電気泳動で検出したもの。これにより、用いた全RNA量は等しいことが分かる。
【0047】
RNaseプロテクションアッセイの結果、bpsA、orfCおよびorfDはg-NL添加による誘導がない場合、発現しないことが分かった。一方、これらの遺伝子はg-NLによる誘導後一時間後に発現し、その後、減衰した。したがって、bpsA、orfCおよびorfDは、g-NLによりその発現が一過性に誘導されることが分かった。
【0048】
実施例5 青色素の検出、精製、およびキャラクタリゼーション
S. lavendulae ATCC11924による所定時間後における青色素の生産は、培養液を遠心して得られた上清に、 等量の水を加えた後、590 nmの吸光度を測定することで検出した。青色素の精製は以下のように行った。S. lavendulae ATCC11924をメディウムB (2 L)にて培養し、 培養開始5時間後に誘導剤としてγ-NLを添加した。その2時間後に培養液を4,400 × gで10 分間遠心し、 菌体を除いた。得られた上清をさらに、 30,000 × gにて30 分間遠心し青色素の沈殿を得た。この沈殿を水、および メタノールで2回ずつ洗浄し、真空中で乾燥させた。この沈殿にジメチルスルホキシドを加え、 ソニケーションで溶解させた後、 アドバンテック社製DISMIC-13JPフィルターを用いて微小沈殿物を除いた。その後、 5倍量の水を加え、 再び 30,000 × gにて30 分間遠心し青色素の沈殿を得た。この沈殿を水で10回、 メタノールで2回ずつ洗浄し、 真空中で乾燥させることで 精製青色素 (11 mg) を得た。
【0049】
吸光度分析のためのサンプル調整は、 精製した青色素を、 ジメチルスルホキシド (DMSO) 、 N-メチルピロリドン (NMP) 、 テトラヒドロフラン (THF) 、およびピリジンなどの各種溶媒に懸濁し、 ソニケーションで最大限溶解させた。その後、 15,000 × gにて5 分間遠心することで溶解していない粒子を取り除いた。可視部吸収スペクトルは、 日本分光社製V-550分光光度計で解析した。
EIマススペクトルは、 THFに溶解させた青色素を用い、 JEOL社製JMS-SX102Aスペクトロメーターで解析した。MALDI-TOF解析は、 ジメチルスルホキシドに溶解させた青色素を用い、 アプライドバイオシステムズ社製VoyagerTM RP-3で解析した。
【0050】
1H-NMR解析はJEOL社製JNM-LA500スペクトロメーターで解析した。内部標準としてテトラメチルシラン、 溶媒として重水素化されたDMSOを用いた。
IRスペクトルは、ヨウ化カリウム法で測定し、波数として3443, 3328, 3288, 3196, 3064, 2959, 2924, 2869, 2839, 2364, 1688, 1639, 1596, 1579, 1454, 1371, 1323, 1250, 1118, 1060, 1037, 956, 885, 824, 779, 752, 718, 689, 661, 634の周波数のところに吸収が観測された。
【0051】
結果を図5にまとめて示す。
EI-mass分析においては、m/z = 248.0にピークが存在し、これは推定される青色素(インジゴイジン)の構造の分子式から計算した値と一致した。
1H-NMRスペクトル解析においては、11.30 ppm (s; NH)、8.18 ppm (s; CH)、および6.46 (s; NH2)のシグナルが得られた。
以上より、青色素は、インジゴイジンであると判断された。
【0052】
実施例6 S. verticillus由来ホスホパンテテイニル基転移酵素をコードするsvp遺伝子とbpsA遺伝子の大腸菌における共発現
bpsA遺伝子は、プライマーとして5'リン酸化した5'-CATATGACTCTTCAGGAGACCAGCGTGCTC-3'(配列番号13)(下線部はNdeIサイトを示す)、および 5'-AAGCTTCTCGCCGAGCAGGTAG CGGATGTG-3'(配列番号14)(下線部はHindIIIサイトを示す) を用い、東洋紡社製KOD-plusポリメラーゼを用いてPCR法にて増幅させた。増幅遺伝子をアガロースゲルより抽出・精製し、 pUC19のSmaIサイトに連結し、キメラプラスミドpUC/bpsAを得た。挿入されたbpsA遺伝子の塩基配列を確認した後、pUC/bpsAをNdeIおよびHindIIIにて二重消化し、 pET-28a(+)の同サイトに組み込みキメラプラスミドpET/bpsAを得た。このベクターから発現したBPSAはNおよびC末端にポリヒスチジンタグを持つ。
【0053】
S. verticillus由来ホスホパンテテイニル基転移酵素 (PPTase) をコードするsvp遺伝子は プライマーとして5'リン酸化した5'-CATATGATCGCCGCCCTCCTGCCCTCCTG-3'(配列番号15)(下線部はNdeIサイトを示す) および5'-CTCGAGCGGGACGGCGGTCCGGTCGTCCGC-3'(配列番号16)(下線部はXhoIサイトを示す) を鋳型DNAとしてS. verticillus染色体を用いて増幅した。この増幅断片をpUC19のSmaIサイトに連結し、キメラプラスミドpUC/svpを得た。挿入されたsvp遺伝子の塩基配列を確認した後、pUC/svpをNdeIおよびXhoIにて二重消化し、pET-21a(+)の同サイトに組み込み、キメラプラスミドpET/svpを得た。このベクターから発現したSvpはC末端にポリヒスチジンタグを持つ。pET/svpのうち、 ベクター由来のT7プロモーターおよびその下流に連結されたsvp遺伝子を含む2.2 kbのDNA断片を、SphIおよびPvuIで二重消化することにより切り出し、pSTV28の同サイトに連結することで、キメラプラスミドpSTV/svpを得た。
pET/bpsAおよびpSTV/svpを同時に大腸菌BL21(DE3) 株に導入し、BPSAおよびSvpの共発現株を得た。大量発現させたときの両タンパク質の溶解性を考慮し、発現実験は18℃にて行った。18℃で48時間培養した結果を、図6に示す。
【0054】
pET/bpsAおよびpSTV/svpを同時に導入した大腸菌は青色素を産生したが、pET/bpsAのみを導入した大腸菌は青色素を産生しなかった。このことから、大腸菌が保有する自身のPPTaseはBPSAをホロ型に変換できないと考えられ、また、SvpはBPSAをホロ型に変換し、ホロ型BPSAが青色素合成を触媒すると考えられた。
【0055】
実施例7 大腸菌におけるBPSAおよびBPSAΔTEの大量発現と精製
TE-ドメインを欠損したBPSAΔTE (アミノ酸番号1-1014) を生産させるため、 全長型bpsAの場合と同様に、pET-28a(+)を用いて bpsAΔTE発現用ベクターpET/bpsAΔTEを構築した。PCRプライマーは、bpsA増幅に用いた5'リン酸化センスプライマーと、5'リン酸化アンチセンスプライマー 5'-AAGCTTCTGGGCGACCTCGCGCTCCAG-3' (配列番号17)を使用した (下線部はHindIIIサイトを示す) 。BPSAΔTEタンパク質は、N末端およびC末端ヒスチジンタグ付きタンパク質として生産された。
【0056】
pET/bpsAあるいはpET/bpsAΔTEを保有する大腸菌BL21(DE3)株を、カナマイシンを含むLB培地2.5リットルにて18℃で培養した。発現したタンパク質の溶解性を考慮して、培養はイソプロピル-β-D-ガラクトピラノシド(IPTG)による誘導なしで30時間行った。細胞は遠心により集め、結合緩衝液 [20 mMトリス-塩酸 (pH 7.9)、500 mM塩化ナトリウム、5 mMイミダゾール] に懸濁した。懸濁以降、全ての過程は4℃にて行った。超音波により細胞を破砕し、27, 000 × g、20分間の遠心により細胞残骸物を除いた。その結果得られた上清を、予め結合緩衝液にて平衡化したHis-Bind resinカラム (1 × 10 cm、Novagen) に添加し、カラムを洗浄緩衝液 I [20 mMトリス-塩酸 (pH 7.9)、 500 mM塩化ナトリウム、30 mMイミダゾール] を用いて洗浄した。溶出は、30 mMから500 mMのイミダゾール直線濃度勾配により行った。BPSAあるいはBPSAΔTEを含む画分を集め、1 mMのエチレンジアミン四酢酸を含む結合緩衝液に対して透析し (2回) 、続いて結合緩衝液に対して透析を行った (2回)。透析した溶液を再びHis-Bind resinカラムに添加し、カラムを洗浄緩衝液II [20 mMトリス-塩酸 (pH 7.9)、500 mM塩化ナトリウム、60 mMイミダゾール] を用いて洗浄した。溶出は、60 mMから1000 mMのイミダゾール直線濃度勾配により行った。この段階で、ドデシル硫酸ナトリウム-ポリアクリルアミドゲル電気泳動から判断すると、 BPSAおよびBPSAΔTEはほぼ単一タンパク質として精製された(図7)。Ox-ドメインを保有するEpoB(Chen, H.W., O'Conner, S., Cane, D.E., and Walsh, C.T. (2001). Epothilone biosynthesis: assembly of the methylthiazolylcarboxy starter unit on the EpoB subunit. Chem. Biol. 8, 899-912.)と同様に、精製したBPSAおよびBPSAΔTEは、フラビンモノヌクレオチドを含むため黄色を呈していた。使用するまで、精製したタンパク質は50%グリセロールの存在下-20℃で保存した。
【0057】
実施例8 大腸菌におけるSvpの大量発現と精製
pET/svpを保有する大腸菌BL21(DE3)株を、アンピシリンを含むLB培地3リットルにて23℃で培養した。Svpの溶解性を考慮して、培養はIPTGによる誘導なしで24時間行った。細胞は遠心により集めた。細胞を結合緩衝液に懸濁した後は 全ての過程を4℃にて行った。超音波により細胞を破砕し、27, 000 × g、20分間の遠心により細胞残骸物を除いた。その結果得られた上清を、予め結合緩衝液にて平衡化したHis-Bind resinカラム (1 × 10 cm) に添加した。洗浄緩衝液IIによりカラムを洗浄した後、溶出は60 mMから500 mMのイミダゾール直線濃度勾配により行った。これらの過程を通して、Svpはほぼ単一タンパク質として精製された(図7)。使用するまで、精製したSvpタンパク質は50%グリセロールの存在下、-20℃で保存した。
【0058】
実施例9 試験管内おける青色素の合成
Svpを用いてBPSAを4'-ホスホパンテテイニル化するため、50 mMリン酸ナトリウム緩衝液 (pH 7.8) を用いて調製した660 nM BPSA、810 nM Svp、0.1 mM補酵素Aおよび1 mM塩化マグネシウムを含む溶液 (1.4 ml) を、30℃で10分間インキュベートした。青色素の合成は200 μlの10 mMアデノシン三リン酸溶液 (最終濃度1 mM) および400 μlの5 mM L-アミノ酸溶液 (最終濃度1 mM) を加えることにより開始した。試験管内における青色素の合成は、590 nmの吸収を測定することによりモニターした(図8)。合成された青色素の分子量は、上記で述べたように, MALDI-TOFにより分析された。
【0059】
Svpで4'-ホスホパンテテイニル化したBPSAを用いたとき、L-グルタミンを基質とした場合にのみ青色素の合成が認められた。また、Svpで4'-ホスホパンテテイニル化していないBPSAを用いたときでは、L-グルタミンを基質とした場合でも青色素の合成が認められなかった。このことから、4'-ホスホパンテテイニル化されたホロ型BPSAが、L-グルタミンを唯一の基質として青色素を合成すると考えられる。
【0060】
実施例10 アデノシン三リン酸/ピロリン酸-交換アッセイ
アデノシン三リン酸/ピロリン酸交換反応を用いて、BPSAのアデニル化ドメインの活性を測定した。
アミノ酸に依存したアデノシン三リン酸/ピロリン酸-交換アッセイは、基本的に過去に述べられた方法 (Du, L., Chen, M., Zhang, Y., and Shen, B. (2003). BimIII and BlmIV nonribosomal peptide synthetase-catalyzed biosynthesis of the bleomycin bithiazole moiety involving both in cis and in trans aminoacylation. Biochemistry 42, 9731-9740.) により行った。反応溶液 (100 μl) は, 300 nMヒスチジンタグ付きタンパク質、5 mMアデノシン三リン酸、1.72 μM [32P]ピロリン酸 (1 μCi, 60 Ci/mmol;PerkinElmer Life Siences) 、1 mMピロリン酸、1 mM塩化マグネシウム、0.1 mMエチレンジアミン四酢酸、0.5 mMアミノ酸および20 mMトリス-塩酸 (pH 7.8) を含んでいる。この溶液を30℃で20分間インキュベートした後、0.25 M過塩素酸、1.6%活性炭および0.1 Mピロリン酸四ナトリウムを含む溶液0.9 mlを加えることで、反応を停止させた。遠心により活性炭を集め、1 mlの水で2回洗浄し、0.5 mlの水に懸濁した。液体シンチレーション (ACSII、Amersham Biosciences) を加えた後、活性炭に結合した放射活性を液体シンチレーションカウンターにより測定した。なお、アミノ酸を含まない反応溶液をコントロールとして使用した。BPSAおよびBPSAΔTEの速度論的パラメーターを決定する場合、様々な濃度のL-グルタミンを用いて、反応は6分間行った。[32P]ピロリン酸の比活性を用いて、活性炭に結合した放射活性を反応速度に変換した。結果を図9に示す。図は、L-グルタミンに対する活性を100%とし、他のアミノ酸に対する活性はその相対活性として示している。
図9から、BPSAの基質はL-グルタミンであると考えられる。
【0061】
実施例11 ピロリン酸放出アッセイ
BPSAアデニル化ドメインのL-グルタミンに対する活性を、ピロリン酸放出アッセイにより測定した。
A-ドメインによるピロリン酸放出速度は、EnzCheck Pyrophosphatase Assay Kit (Molecular Probe) を用いて、グアノシンアナログであるMesGを利用した連続分光学的手法により測定した。反応溶液は、20 mMトリス-塩酸 (pH 7.8) 、1 mM塩化マグネシウム、0.1 mMエチレンジアミン四酢酸、0.2 mM MesG、0.2 Uプリンヌクレオシドホスホリラーゼ、0.2 U無機ピロホスファターゼ、5 mMアデノシン三リン酸、3 mM L-グルタミン、100 nM Svp、0.5 mM補酵素Aおよび100 nMのBPSAを含んでいる。BPSAをアポ体からホロ体へ変換する場合、予め、L-グルタミンとアデノシン三リン酸を含まない溶液にて、100 nMのBPSAを100 nMのSvpとともに30℃で30分間インキュベートした。この場合、 反応は3 mMのL-グルタミンおよび5 mMのアデノシン三リン酸を加えることにより開始し、その後360 nmの吸収を10秒ごとに6分間測定した。得られたスペクトルにおける0秒から100秒までの勾配は、標準ピロリン酸を用いて作成した標準曲線と相関した(図10)。
図10において、ひし形はホロ酵素、四角はアポ酵素を用いて得られたデータを示す。0秒から100秒における勾配を利用することにより、ホロBPSAの回転速度は107/分、アポBPSAの回転速度は76/分と算出された。
【0062】
回転速度は、アポ型をホロ型へ変換することで若干上昇した。これは、ホロ型BPSAにおいては、Aドメインにより活性化されたL-グルタミンが、速やかにTドメインへ移行するためであると考えられる。
【0063】
実施例12 BPSAΔTE T-ドメインへの[14C] L-グルタミンのアミノアシル化アッセイ
精製SvpによりT-ドメインをホスホパンテテイニル化したホロBPSAおよびホロBPSAΔTE、あるいは、Svpにより活性化しないそれぞれのアポ酵素を用いて、14C標識L-グルタミンのT-ドメインへの結合を調べた。
BPSAΔTEのT-ドメインへの[14C] L-グルタミンの結合は、オートラジオグラフィーにより調べた。100 μlの反応溶液は、20 mMトリス-塩酸 (pH 7.8) 、1 mM塩化マグネシウム、 0.1 mMエチレンジアミン四酢酸、3 mMアデノシン三リン酸、 10 mM [14C] L-グルタミン (0.2 μCi, 210 mCi/mmol; Moravek Biochemicals)、 300 nM Svp、0.5 mM補酵素Aおよび300 nMのBPSAあるいはBPSAΔTEを含んでいるが、予め[14C] L-グルタミンを含まない溶液を30℃にて30分間インキュベートすることにより、[14C] L-グルタミンを加えて反応を開始する前にそれらタンパク質のT-ドメインをホスホパンテテイニル化した。[14C] L-グルタミンを加えた後、様々な時間経過後、2%ウシ血清アルブミンを含む10%氷冷トリクロロ酢酸を0.8 ml加えることにより、反応を停止させた。沈殿したタンパク質を集め、10%氷冷トリクロロ酢酸およびアセトンで洗浄した後、その放射活性を測定した。オートラジオグラフィーを行う場合、[14C] L-グルタミンを加えてから15分後に反応を停止させ、タンパク質の沈殿を10%ドデシル硫酸ナトリウム-ポリアクリルアミドゲル電気泳動に供した。電気泳動後、乾燥させたゲルをイメージングプレートに曝露し、BAS-2000装置を用いて視覚化した(図11)。
【0064】
完全長のBPSAを用いた場合、Svpによる4'-ホスホパンテテイニル化の有無に関わらず、L-グルタミンはBPSA上に結合していなかった。一方、BPSAΔTEを用いた場合では、Svpで4'-ホスホパンテテイニル化されたBPSAΔTEにのみL-グルタミンの結合が認められた。このことから、完全長BPSAのC末端に存在するTEドメインは、Tドメインに結合したL-グルタミン(あるいはその誘導体)の切り離しを触媒すると考えられ、また、Aドメインにより活性化されたL-グルタミンは、4'-ホスホパンテテイニル化されたTドメインに結合すると考えられる。
【図面の簡単な説明】
【0065】
【図1】クローン化した約6-kbのDNA断片に存在するorfの構成とBPSAのドメイン構造を示す模式図である。白抜き矢印:不完全な遺伝子、黒矢印:完全な遺伝子
【図2】S. lividans 66でのbspAの発現を示した写真である。(A)チオストレプトン(50 μg/ml)を含むFB寒天培地上で生育する各形質転換体、(B)チオストレプトン(50 μg/ml)を含むYEME液体培地上で生育する各形質転換体
【図3】PCRで増幅した各遺伝子のアガロース電気泳動パターンを示した図である。 レーン1:S. lavendulae ATCC11924、レーン2:S. lavendulae ATCC25233、レーン3:S. garyphalus (CSH5-12)、レーン4:S. coelicolor A3(2)、レーン5:S. lividans 66、レーン6:S. avermitilis K139、レーン7:S. lavendulae JCM4055、レーン8:S. lavendulae FRI-5、レーン9:陽性コントロール
【図4】RNaseプロテクションアッセイの結果を示す図である。(A):プローブAによりプロテクションされたRNA、(B):プローブBによりプロテクションされたRNA、(C):プローブCによりプロテクションされたRNA、(D):RNaseプロテクションアッセイに用いた全RNAを1%ホルムアミドアガロースゲル電気泳動で検出したもの。NL(-):γ−NL非添加、NL(+):γ−NL添加
【図5】S. lavendulae より合成された青色素の化学的性質と構造を示した図である。 (A):構造。(B)EI-mass分析、(C)1H-NMRスペクトル
【図6】E. coliにおいて青色素の産生を示した写真である。(1)bpsAとsvpを共発現させた大腸菌BL21(DE3)株、(2)bpsAのみを発現する大腸菌BL21(DE3)株、(3)発現ベクターpET-28a(+)を保有する大腸菌BL21(DE3)株
【図7】発現および精製タンパク質のドデシル硫酸ナトリウムーポリアクリルアミドゲル電気泳動パターンを示した図である。レーン1:精製BPSA、レーン2:精製BPSAΔTE、レーン3:Svp、M:標準タンパク質、左数字:分子量。
【図8】In vitroにおける青色素の生成を示したグラフである。実線:BPSAおよびSvpを用いた場合、破線:BPSAのみを用いた場合
【図9】BPSAのアデニル化ドメインの活性を示した図である。
【図10】BPSAアデニル化ドメインのL-グルタミンに対する活性を示した図である。 ひし形:ホロ酵素、四角:アポ酵素
【図11】14C標識L-グルタミンのT-ドメインへの結合能を示した図である。左:ドデシル硫酸ナトリウムーポリアクリルアミドゲル電気泳動後、クマシー染色したもの、右:泳動後、14C標識L-グルタミンをオートラジオグラフィーで検出したもの
【特許請求の範囲】
【請求項1】
以下の(a)又は(b)のタンパク質:
(a)配列番号1に示すアミノ酸配列からなるタンパク質、
(b)(a)のアミノ酸配列において、1個若しくは数個のアミノ酸が欠失、置換若しくは付加されたアミノ酸配列からなり、且つ、インジゴイジン合成能を有するタンパク質。
【請求項2】
請求項1に記載のタンパク質をコードする遺伝子。
【請求項3】
以下の(a)又は(b)のDNAからなる遺伝子:
(a)配列番号2に示す塩基配列からなるDNA、
(b)(a)の塩基配列からなるDNAと相補的な塩基配列からなるDNAとストリンジエントな条件下でハイブリダイズし、且つインジゴイジン合成能を有するタンパク質をコードするDNA。
【請求項4】
単一モジュール型の非リボソーム型ペプチド合成酵素遺伝子である請求項2又は3記載の遺伝子。
【請求項5】
請求項2又は3記載の遺伝子を含有する組換えベクター。
【請求項6】
請求項5記載の組換えベクターを含む形質転換体。
【請求項7】
請求項6記載の形質転換体を培養し、培養物から請求項1記載のタンパク質を採取することを特徴とする請求項1記載のタンパク質の製造方法。
【請求項8】
請求項2又は3記載の遺伝子を導入した放線菌を培養し、培養物からインジゴイジンを採取することを特徴とするインジゴイジンの製造方法。
【請求項9】
請求項2又は3記載の遺伝子及び4'−ホスホパンテテイニル基転移酵素遺伝子を導入した宿主を培養し、培養物からインジゴイジンを採取することを特徴とするインジゴイジンの製造方法。
【請求項10】
L-グルタミンに、請求項1記載のタンパク質及び4'−ホスホパンテテイニル基転移酵素を作用させることを特徴とするインジゴイジンの製造方法。
【請求項1】
以下の(a)又は(b)のタンパク質:
(a)配列番号1に示すアミノ酸配列からなるタンパク質、
(b)(a)のアミノ酸配列において、1個若しくは数個のアミノ酸が欠失、置換若しくは付加されたアミノ酸配列からなり、且つ、インジゴイジン合成能を有するタンパク質。
【請求項2】
請求項1に記載のタンパク質をコードする遺伝子。
【請求項3】
以下の(a)又は(b)のDNAからなる遺伝子:
(a)配列番号2に示す塩基配列からなるDNA、
(b)(a)の塩基配列からなるDNAと相補的な塩基配列からなるDNAとストリンジエントな条件下でハイブリダイズし、且つインジゴイジン合成能を有するタンパク質をコードするDNA。
【請求項4】
単一モジュール型の非リボソーム型ペプチド合成酵素遺伝子である請求項2又は3記載の遺伝子。
【請求項5】
請求項2又は3記載の遺伝子を含有する組換えベクター。
【請求項6】
請求項5記載の組換えベクターを含む形質転換体。
【請求項7】
請求項6記載の形質転換体を培養し、培養物から請求項1記載のタンパク質を採取することを特徴とする請求項1記載のタンパク質の製造方法。
【請求項8】
請求項2又は3記載の遺伝子を導入した放線菌を培養し、培養物からインジゴイジンを採取することを特徴とするインジゴイジンの製造方法。
【請求項9】
請求項2又は3記載の遺伝子及び4'−ホスホパンテテイニル基転移酵素遺伝子を導入した宿主を培養し、培養物からインジゴイジンを採取することを特徴とするインジゴイジンの製造方法。
【請求項10】
L-グルタミンに、請求項1記載のタンパク質及び4'−ホスホパンテテイニル基転移酵素を作用させることを特徴とするインジゴイジンの製造方法。
【図1】

【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【図11】
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【図11】
【公開番号】特開2007−189969(P2007−189969A)
【公開日】平成19年8月2日(2007.8.2)
【国際特許分類】
【出願番号】特願2006−12459(P2006−12459)
【出願日】平成18年1月20日(2006.1.20)
【新規性喪失の例外の表示】特許法第30条第1項適用申請有り 平成17年8月25日 社団法人日本生化学会発行の「生化学第77巻第8号」に発表
【新規性喪失の例外の表示】特許法第30条第1項適用申請有り 平成17年12月16日 社団法人日本化学会主催の「2005環太平洋国際化学会議」において文書をもって発表
【出願人】(504136568)国立大学法人広島大学 (924)
【Fターム(参考)】
【公開日】平成19年8月2日(2007.8.2)
【国際特許分類】
【出願日】平成18年1月20日(2006.1.20)
【新規性喪失の例外の表示】特許法第30条第1項適用申請有り 平成17年8月25日 社団法人日本生化学会発行の「生化学第77巻第8号」に発表
【新規性喪失の例外の表示】特許法第30条第1項適用申請有り 平成17年12月16日 社団法人日本化学会主催の「2005環太平洋国際化学会議」において文書をもって発表
【出願人】(504136568)国立大学法人広島大学 (924)
【Fターム(参考)】
[ Back to top ]