
静電噴霧法を用いる生体適合性シリカ繊維の製造方法
【課題】完全無機質、かつ、柔軟性を有する生体適合性シリカ繊維を、実験室レベルの小型で簡便な装置でも製造しうる製造方法の提供を目的とする。
【解決手段】アルコキシシランを、リン酸トリエチルを含む水と有機溶媒の混合溶媒に溶解させ、大気と接触させながらゾル状の紡績液とし、粘度を調整した紡績液を、静電噴霧法により紡糸化してシリカ不織布とする。その後、カルシウムイオンを含む水溶液中でイオン交換し、400℃以上1000℃以下の温度で焼成する。または、前記混合溶媒にリン酸トリエチルを添加せずに、静電噴霧法により紡糸化してシリカ不織布とした後、リン酸中で加熱し、その後、カルシウムイオンを含む水溶液中でイオン交換し、400℃以上1000℃以下の温度で焼成してもよい。
【解決手段】アルコキシシランを、リン酸トリエチルを含む水と有機溶媒の混合溶媒に溶解させ、大気と接触させながらゾル状の紡績液とし、粘度を調整した紡績液を、静電噴霧法により紡糸化してシリカ不織布とする。その後、カルシウムイオンを含む水溶液中でイオン交換し、400℃以上1000℃以下の温度で焼成する。または、前記混合溶媒にリン酸トリエチルを添加せずに、静電噴霧法により紡糸化してシリカ不織布とした後、リン酸中で加熱し、その後、カルシウムイオンを含む水溶液中でイオン交換し、400℃以上1000℃以下の温度で焼成してもよい。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、表面にリン酸カルシウム系物質が形成されている生体適合性シリカ不織布の製造方法に関するものである。また、本発明は、そのような生体適合性シリカ不織布を構成する生体適合性シリカ繊維に関するものである。
【背景技術】
【0002】
無機ケイ酸化合物は、ガラス、セメント、陶磁器等の原材料として古くから使用されている。一般に、無機ケイ酸化合物は化学的に安定しており、耐熱性及び耐薬品性にも優れている。また、多孔質であるため化学物質等を吸着することができるもの、触媒機能を有するために化学物質等を分解できるもの、無害で生体適合性が高いもの等、様々な機能性を備えた無機ケイ酸化合物も存在する。
【0003】
しかし、無機ケイ酸化合物の多くは粉末状であるため、そのままでは取り扱いが難しく、用途も限定されるため、商品として展開しにくい。一方、無機ケイ酸化合物を不織布として繊維状に形成すれば、容易に切断したり、曲げたり、容器に詰めたりできるため、広範囲の用途へ応用することが可能になる。
【0004】
無機ケイ酸化合物を繊維状に形成する場合、一般的には2000℃以上の高温で熔融し、その熔融液を流出させることによって繊維化するが、この製造方法では繊維径を小さくすることが極めて困難であり、また、原料である無機ケイ酸化合物を熔融するために多大なエネルギーを必要とする。さらに、高温の熔融液を取り扱うため、製造装置が大がかりなものとならざるを得ず、作業の安全性に関しても問題がある。
【0005】
アルコキシシランには、有機溶媒に溶かした状態で重合化させることによりゾル状となるものがあり、微細孔から押し出して繊維状に形成することも可能である。例えば、特許文献1には、変性ポリカルボシランと低分子量の有機金属化合物をトルエンに溶解させて紡糸原液とした後、合成繊維紡糸用装置を用いて繊維状に形成する方法が開示されている。
【0006】
一方、生体適合性を有する無機材料として、ヒドロキシアパタイトやβ-3リン酸カルシウム等のリン酸カルシウム系材料が注目されているが、これらリン酸カルシウム系材料は、すべて粉末として得られるため、合成後に成型及び焼成して使用せざるを得ない。例えば、特許文献2には、骨細胞活性を薄膜上で支えるためのリン酸カルシウム材の薄膜の改良製造方法として、リン酸アンモニウムの溶液を硝酸カルシウムの溶液と合わせてヒドロキシアパタイト含有ゾル−ゲルを形成させることによって調製し、このゾル−ゲルの膜を基層の少なくとも片側に当てがい、基層上にコーティングされた膜を焼結してリン酸カルシウム材の固体膜を形成させる方法が開示されている。
【0007】
ここで、ヒドロキシアパタイト等を繊維状に形成する方法として、熔融紡糸法やゲル紡糸後に焼成する方法(非特許文献1,2)が知られているが、いずれもヒドロキシアパタイト等を溶融する必要があるため、上述した無機ケイ酸化合物と同様の問題がある。
【0008】
また、ポリマー繊維にヒドロキシアパタイト等を分散させたコンポジット繊維も知られているが(非特許文献3)、得られる繊維は脆く、応用範囲が制限されるという問題がある。
【特許文献1】特開2003−328236号公報
【特許文献2】特開2004−209261号公報
【非特許文献1】“Preparation of Porous Ceramics with Calcium Metaphosphate Fiber Skelecton for Biomedical Use”, Journal of American ceramic society, Vol. 78, Page. 245-247, 1995.
【非特許文献2】“A study of hydroxyapatite fibers prepared via sol-gel route”, Material Letters, Vol. 58, Page. 3320-3323, 2004.
【非特許文献3】“Effect of Spinning Conditions on the Structure and Properties of PAN Fibers Containing Nano-Hydroxyatatite”, Journal of Polymer Science, Vol. 100, Page. 2881-2888, 2006.
【発明の開示】
【発明が解決しようとする課題】
【0009】
上記特許文献に開示されているシリカ繊維の製造方法は、非常に微細な細孔から高圧で紡糸原液を押し出す必要があり、装置を小型化することは困難である。また、平均繊維径を小さくするためには、細孔をさらに小さくしなければならず、より高圧で紡糸原液を押し出す必要がある。さらに、細孔を小さくすればするほど、目詰まりも生じやすくなり、装置のメインテナンス頻度も高くなる。
【0010】
一方で、ヒドロキシアパタイト等の生体適合性無機材料を簡便な方法で繊維化できれば、従来応用できなかった用途への応用も可能となる。
【0011】
本発明は、従来は不可能であった完全無機質、かつ、柔軟性を有する生体適合性シリカ繊維を、実験室レベルの小型で簡便な装置でも製造しうる製造方法、及び当該生体適合性シリカ不織布を構成する生体適合性シリカ繊維の提供を目的とする。
【課題を解決するための手段】
【0012】
アルコキシシランは、有機溶媒に溶解した状態で水と反応すると、加水分解反応と縮合反応を起こして粘性ゾルとなり、さらに縮合反応が進むと湿潤ゲル体となる。こうした現象は、ゾル−ゲル反応と呼ばれている。
【0013】
一方、試料溶液を供給するキャピラリー先端に数kVの高電圧を印加することにより 試料溶液を微細な液滴として霧化させることが可能である。この技術は静電噴霧(electrospray)と呼ばれ、薄膜や微粒子の製造に応用されてきた。
【0014】
本発明者は、鋭意研究した結果、アルコキシシランを水と有機溶媒の混合溶媒に溶解させて紡績液とし、この紡績液を静電噴霧することにより、シリカを容易に繊維化し、シリカ不織布を製造しうることを既に見出している。今回、本発明者は、この紡績液にリン分(リン成分)供給源としてリン酸トリエチル((C2H5O)3PO)を添加して静電噴霧を行い、得られたシリカ繊維を、カルシウムイオンを含む水溶液中でイオン交換し、さらに焼成することにより、シリカ繊維表面にリン酸カルシウム系無機物が形成され、タンパク質吸着性等の生体適合性を発揮することを新たに見出した。
【0015】
また、本発明者は、アルコキシシランを水と有機溶媒の混合溶媒に溶解させた紡績液を、静電噴霧することにより得られるシリカ不織布をリン酸中で加熱した後、上記と同様、カルシウムイオンを含む水溶液中でイオン交換し、さらに焼成することにより、シリカ繊維表面にリン酸カルシウム系無機物が形成され、タンパク質吸着性等の生体適合性を発揮することを新たに見出した。
【0016】
具体的に、本発明は、
アルコキシシランを、リン酸トリエチルを含む水と有機溶媒の混合溶媒に溶解させ、大気と接触させながらゾル状の紡績液とするリン分添加ゾル化工程と、
粘度調整後の前記紡績液を静電噴霧法により紡糸化するリン分添加紡糸化工程と、
前記リン分添加紡糸化工程で得られた不織布を、カルシウムイオンを含む水溶液中に浸漬することによりイオン交換させるイオン交換工程と、
前記イオン交換工程後の不織布を400℃以上1000℃以下の温度で焼成する焼成工程と、
を含む生体適合性シリカ不織布の製造方法に関する(請求項1)。
【0017】
アルコキシシランを水と有機溶媒の混合溶媒に溶解させた紡績液を、静電噴霧することにより得られるシリカ不織布(シリカ繊維)表面には、未反応アルコキシル基が残存していると考えられる。ここで、紡績液にリン分(リン成分)供給源としてリン酸トリエチルを添加すれば、静電噴霧によって得られるシリカ不織布表面にリン原子が導入され、さらにカルシウムイオンと反応させて焼成することにより、リン酸カルシウム系物質が形成されて生体適合性シリカ繊維を製造することができると推定される。
【0018】
また、本発明は、
アルコキシシランを水と有機溶媒の混合溶媒に溶解させ、大気と接触させながらゾル状の紡績液とするゾル化工程と、
粘度調整後の前記紡績液を、静電噴霧法により紡糸化する紡糸化工程と、
前記紡糸化工程で得られた不織布を、リン酸に浸漬して加熱するリン酸化工程と、
前記リン酸化工程後の不織布を、カルシウムイオンを含む水溶液中に浸漬することによりイオン交換させるイオン交換工程と、
前記イオン交換工程後の不織布を400℃以上1000℃以下の温度で焼成する焼成工程と、
を含む生体適合性シリカ不織布の製造方法に関する(請求項8)。
【0019】
シリカ不織布(シリカ繊維)表面の未反応アルコキシル基は、シリカ不織布をリン酸中で加熱することによってもリン酸化されると考えられ、リン酸化したシリカ不織布を、カルシウムイオンと反応させて焼成することにより、リン酸カルシウム系物質が形成されて生体適合性シリカ繊維を製造することができると推定される。
【0020】
なお、本発明の生体適合性シリカ不織布の平均繊維径は、10μm以下であることが好ましい。また、前記ゾル化工程においては、前記紡績液の粘度を200mPa・s以上800mPa・s以下の範囲内に調整することが好ましい。
【0021】
前記アルコキシシランはテトラエトキシシランであり、前記有機溶媒はエタノールであることが好ましい(請求項2,9)。
【0022】
本発明のシリカ不織布の製造方法において、前記混合溶媒における水の量は、アルコキシシランのモル数に対して1倍以上2倍以下であることが好ましい(請求項3,10)。
【0023】
前記混合溶媒にリン酸トリエチルを添加する製造方法においては、リン酸トリエチルの量が、アルコキシシランのモル数の2%以上10%以下であることが好ましい(請求項4)。
【0024】
前記リン分添加ゾル化工程又は前記ゾル化工程においては、前記混合溶媒に塩酸を加えることが好ましい(請求項5,11)。
【0025】
前記ゾル化工程において、加湿した大気と接触させながら撹拌することが好ましい(請求項6,12)。
【0026】
また、本発明は、請求項1乃至6、又は請求項8乃至13のいずれか1項に記載の生体適合性シリカ不織布を構成する生体適合性シリカ繊維に関する(請求項7,13)。
【発明の効果】
【0027】
本発明で製造されるシリカ不織布は、微細な繊維径のシリカ不織布の表面に、リン酸カルシウム系物質が形成された完全無機質な生体適合性シリカ不織布であり、従来にない柔軟性、強度を有する。このため、粉末又は固体のリン酸カルシウム系物質と比較して、広範囲な応用が可能である。
【0028】
また、本発明の生体適合性シリカ不織布の製造方法は、合成繊維紡糸用装置のような大がかりな装置を用いることなく、実験室レベルの小型、かつ、簡易な装置で実施することが可能である。しかも、常温付近の温度で実施することが可能であり、製造コストも非常に低い。
【発明を実施するための最良の形態】
【0029】
以下に、本発明の実施の形態について、適宜図面を参照しながら説明する。なお、本発明は、これらに限定されない。
【0030】
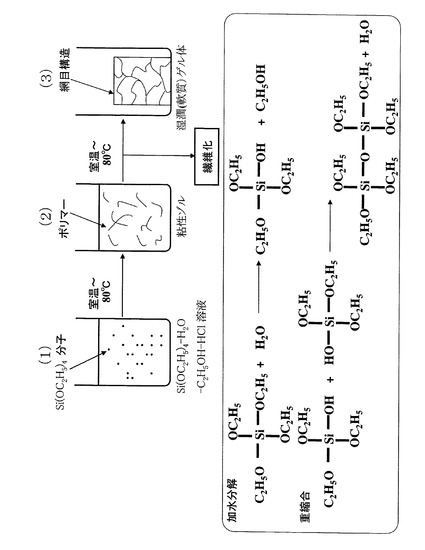
はじめに、アルコキシシランの一例として、テトラエトキシシランを水とエタノールの混合溶媒に溶解させた場合に起こる加水分解反応及び重縮合反応を、図1を参照しながら説明する。なお、加水分解反応及び重縮合反応を促進するために、水とエタノールの混合溶媒に塩酸を添加することが好ましい。塩酸の添加量は、テトラエトキシシランのモル数の1%程度が適量である。
【0031】
テトラエトキシシランを水とエタノールの混合溶媒に溶解させ(図1(1))、室温以上80℃以下で撹拌すると、テトラエトキシシランの4個の-OC2H5基のうちの1個がH+を触媒として加水分解され-OH基となり、トリエトキシシランに変化する(加水分解反応)。
【0032】
次に、トリエトキシシラン2分子がH+を触媒として脱水縮合する(重縮合反応)。さらに、脱水縮合分子の1個の-OC2H5基も加水分解されて-OH基となり、他の脱水縮合分子の-OH基との間で脱水縮合が繰り返される。この加水分解反応及び重縮合反応によって、分子同士が直線的に重合し、ポリマー化する。それに伴い、混合溶媒の粘度が上昇し、ゾル状となる(図1(2))。
【0033】
このとき、混合溶媒中の水の量が少なすぎると、テトラエトキシシランのポリマー化が途中で止まってしまう。逆に多すぎると、テトラエトキシシランの複数の-OC2H5基が同時に加水分解されため、複数の-OH基が他の脱水縮合分子の-OH基とランダムに脱水縮合する。そうすると、網目状にポリマー化が起こり、混合溶媒中でポリマーが湿潤ゲル体(図1(3)と同じ状態)となって紡績液を調製することができなくなる。
【0034】
混合溶媒中でテトラエトキシシランが直線的に重合してポリマー化すると、上述したように混合溶媒の粘度が上昇する。後述するように、静電噴霧法によって混合溶媒(紡績液)を不織布化するためには、粘度を200mPa・s以上800mPa・s以下の範囲内に調整することが好ましい。それ以上重合化が進むと、直線的に重合したポリマー分子の側鎖の-OC2H5基も加水分解されて-OH基となり、他のポリマー分子等の-OH基との間で脱水縮合を生じ、湿潤ゲル体(図1(3))となって繊維化することが不可能となる。
【0035】
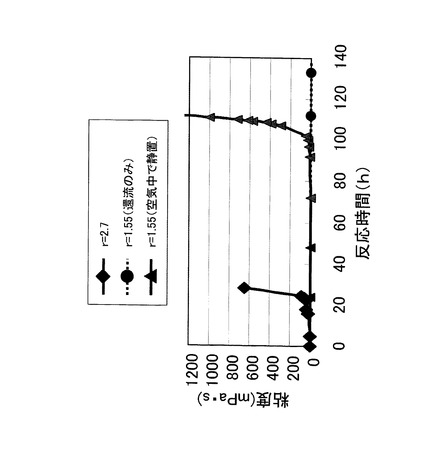
<実験例1>
テトラエトキシシランを、水とエタノールの混合溶媒に溶解させ、65℃の水浴中でエタノールを環流させた。混合溶媒は、水とテトラエトキシシランのモル比(r=[H2O]/[TEOS])が2.7及び1.55の2種類作成した。なお、テトラエトキシシランのモル数に対して1%量の塩酸を触媒として、両方の混合溶媒に添加した。混合溶媒の粘度を経時的に測定した結果を、図2に示す。
【0036】
まず、r=1.55の混合溶媒の場合、65℃で200時間還流させても粘度の上昇、すなわちテトラエトキシシランの直線的ポリマー化が認められなかった(丸印のプロット)。これは、r=1.55ではテトラエトキシシランの加水分解反応及び重合化に必要な水が不足していることが原因と思われる。
【0037】
次に、同じr=1.55の混合溶媒を、65℃で70時間還流させた後、ビーカーに移して大気と接触させながら室温で静置したところ、ビーカーに移してから約30時間経過した後(反応時間全体としては約100時間経過後)に粘度の上昇が認められ、混合溶媒がゾル状となった。これは、ビーカーに移して空気と接触させたことにより、混合溶媒中に空気中の水分が溶け込み、その水分によってテトラエトキシシランの直線的ポリマー化が起こったものと推測された。
【0038】
次に、r=2.7の混合溶媒の場合、65℃で約20時間還流させると粘度の上昇が認められた。しかし、この混合溶媒は、水の量が多すぎるためにテトラエトキシシランの網目状ポリマー化が起こり、ゾル状ではなくゲル状となって後述する静電噴霧法により不織布を製造することができなかった。
【0039】
このように、テトラエトキシシランを溶解させる水とエタノールの混合溶媒において、水の量を適正範囲に調整することにより、直線的ポリマー化を起こすことが重要である。具体的には、混合溶媒における水とテトラエトキシシランのモル比は、1以上2以下にすることが好ましい。1未満では大気と接触させても、テトラエトキシシランの直線的ポリマー化に非常に長時間を要し、2を超えるとテトラエトキシシランの網目状ポリマー化が起こりやすくなるため、静電噴霧法を用いて混合溶媒(紡績液)を紡糸化できないからである。
【0040】
<実験例2>
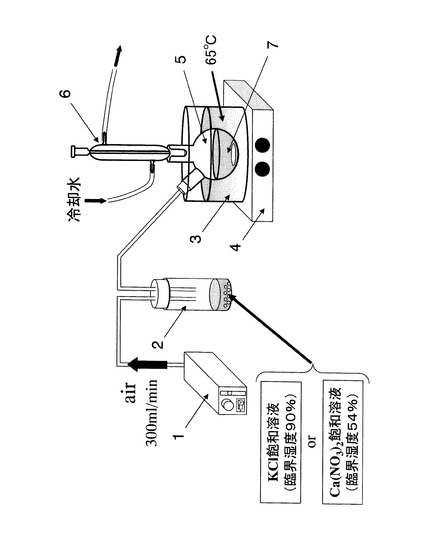
実験例1では、テトラエトキシシランを混合溶媒中で直線的にポリマー化するために、100時間以上かかった。そこで、テトラエトキシシランを溶解させた混合溶媒を、加湿した大気と接触させることにより、テトラエトキシシランの直線的重合化に必要な水を混合溶媒へと供給し、直線的重合化にかかる時間の短縮を試みた。
【0041】
本実験例で使用した装置の概略を、図3に示す。エアポンプ1からバイアル2へと300mL/minの流速で空気を送り込んだ。バイアル2からフラスコ5内のテトラエトキシシランを溶解させた混合溶媒7へ相対湿度90%の加湿空気を導入する場合には、バイアル2にKCl飽和溶液(臨界湿度90%)を入れ、相対湿度54%の加湿空気を導入する場合には、バイアル2内にCa(NO3)2飽和溶液(臨界湿度54%)を入れた。
【0042】
フラスコ5は、油浴3中で65℃に加熱され、フラスコ5内のテトラエトキシシランを溶解させた混合溶媒7は、スターラ4によって撹拌した。フラスコ5上部には冷却管6を取り付け、実験例1と同様にエタノールを環流させた。
【0043】
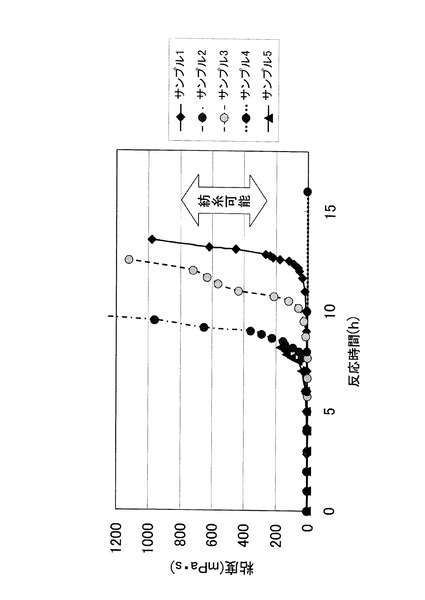
本実験例では、表1に示す条件で混合溶液を調製及び処理し、各サンプルの粘度を経時的に測定した。その結果を、図4に示す。なお、テトラエトキシシランのモル数に対して1%量の塩酸を触媒として、全ての混合溶液に添加した。また、表1のr値は、水とテトラエトキシシランのモル比(r=[H2O]/[TEOS])を意味する。
【0044】
【表1】

【0045】
サンプル1は、約12時間で粘度の上昇が認められた。r=1.7であるサンプル2は、サンプル1の約半分の反応時間で粘度の上昇が認められ、ゾル状の紡績液となった。実験例1もr=1.55であったが、65℃に加熱しながら相対湿度90%に加湿した空気と接触させることにより、テトラエトキシシランの直線的重合化に要する時間を1/10〜1/20にまで短縮することが可能であった。
【0046】
サンプル3は、r値がサンプル2と同じであるが、接触させた空気の相対湿度が54%であったため、サンプル2よりも粘度の上昇が認められるまでの反応時間が長くなった。また、r値が同じで加湿した空気と接触させなかったサンプル4は、20時間経過後も全く粘度の上昇が認められなかった。このことから、実験例1における、混合溶媒中に空気中の水分が溶け込み、その水分によってテトラエトキシシランの直線的ポリマー化が起こるという推測が正しいことが証明された。
【0047】
一方、r=4.0のサンプル5は、サンプル2とほぼ同じ反応時間で粘度の上昇が認められたが、すぐにゲル化してしまい、静電噴霧法により紡糸化することができなかった。
【0048】
なお、油浴の温度を50℃、70℃及び85℃として上記と同じ実験を行ったが、65℃の場合と同様の結果が得られたが、加熱温度が高いほど粘度の上昇が認められるまでの反応時間が、若干短くなる傾向が認められた。なお、混合溶媒7のゾル化に要する時間を短縮させるためには、50℃以上85℃以下の温度で加熱することが好ましい。
【0049】
このように、テトラエトキシシランを水とエタノールの混合溶媒に溶解させ、静電噴霧法によりシリカ不織布として紡糸化させるためには、混合溶媒における水とテトラエトキシシランのモル比を1以上2以下に調整した上で、混合溶媒を加熱しながら加湿した大気と接触させることが好ましいことが判明した。このような方法によれば、テトラエトキシシランの直線的ポリマー化を短時間で行うことができ、かつ、粘度の容易に調整することができるからである。
【0050】
<実験例3>
ここで、実験例2のサンプル1について、その一部を経時的に抜き取り、粘度を測定すると共に、図5(a)に示すような装置を用いて静電噴霧法によって紡糸化し、シリカ不織布を製造した。
【0051】
この装置は、高圧電源8と、シリンジ9と、針状電極11と、導電性捕集板12とから構成される。そして、高圧電源8及び導電性捕集板12には、アース13が取り付けられている。
【0052】
シリンジ9として、今回は、市販されているディスポタイプのプラスチック製注射用シリンジ(10mL)本体を用いた。シリンジ9は、通常注射針を取り付ける先端部分を下に向けた状態で、スタンドを用いて垂直に固定され、内部に直径1.0mmの針状電極11がセットされている。
【0053】
針状電極11は、高圧電源8に接続されており、紡糸化する際には20kVの高電圧をかけた。シリンジ9の下部には、アースを取り付けた導電性捕集板12(ここでは、ステンレス製の網)をセットし、シリンジ9下端部と導電性捕集板12の距離を、200mmに調整した。
【0054】
そして、サンプル1の一部(約4mL)を紡績液10としてシリンジ9の上部開口部から投入し、直ちに高圧電源8のスイッチをオンにして、シリンジ9から導電性捕集板12に向けて紡績液10を静電噴霧した。この静電噴霧処理により、紡績液10中の水、エタノール及び塩酸が揮散し、直線的にポリマー化していたテトラエトキシシランが、微細なシリカ繊維14として紡糸化され、図5(b)に示すように、導電性捕集板12の上にシリカ不織布15として集積する。
【0055】
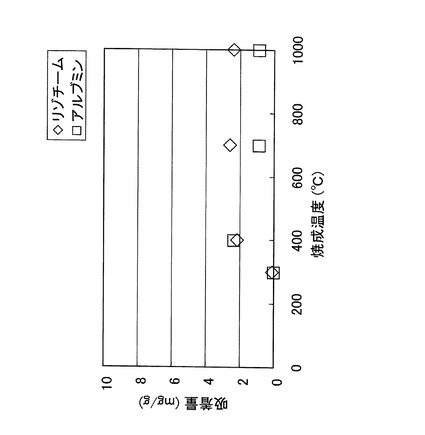
図6(a)は、紡績液10の粘度が500mPa・sの場合に製造されたシリカ不織布15の外観写真である。このシリカ不織布15の外観は、通常のグラスウールとほとんど同じであるが、触感はより滑らかで、折り曲げたりしても繊維が切断されにくい。
【0056】
図6(b)は、このシリカ不織布6の電子顕微鏡写真であり、平均繊維径は4.5μmであることが確認された。特許文献1には、合成繊維紡糸用装置を用いて変性ポリカルボシランを繊維化し、1200℃の空気中で1時間焼成することにより得られたシリカ繊維の平均直径が13μmであったこと記載されているが、本発明のシリカ不織布の製造方法は、より繊維径を小さくできることが確認された。また、本発明のシリカ不織布の製造方法は、紡績液を静電噴霧して紡糸化する際に、溶媒の留去も同時に行われるため、紡糸化操作が非常に容易である。
【0057】
シリンジから静電噴霧された紡績液(混合溶媒)は、同じ電荷に帯電しているため、導電性捕集板に到達するまでにシリカ繊維同士がくっつくことがない。そのため、できあがったシリカ繊維の繊維径は、非常に平均化されていた。さらに、高温での焼成も行う必要がなく、実験室レベルの小型、かつ、簡便な装置で製造可能であった。
【0058】
また、特許文献1の製造方法では、シリカ繊維の繊維径と同程度の微細孔から高圧で紡糸液を押し出す必要があるが、本発明のシリカ不織布の製造方法では、ディスポタイプのプラスチック製注射用シリンジから圧力をかけることなく紡糸液を静電噴霧させるため、微細孔の目詰まりという問題が発生せず、分解洗浄等、製造装置のメインテナンスも非常に容易である。
【0059】
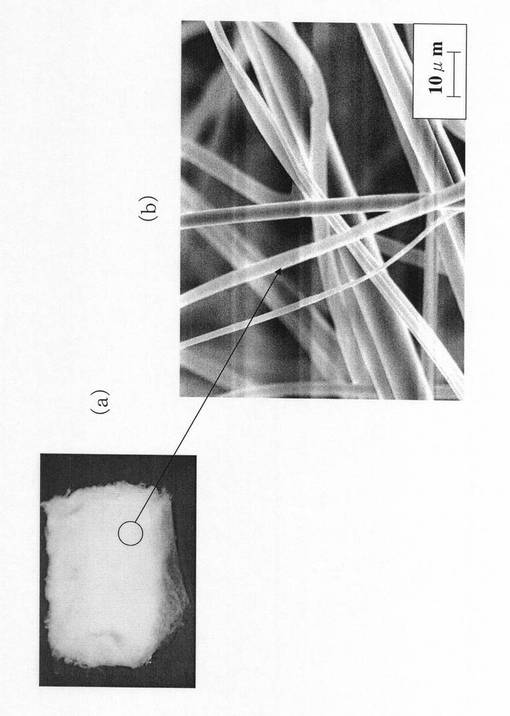
次に、静電噴霧する紡績液10の粘度が150mPa・s、500mPa・s及び920mPa・sの場合に製造されたシリカ不織布15の外観写真を、図7(a)、図7(b)及び図7(c)にそれぞれ示す。
【0060】
まず、紡績液10の粘度が150mPa・sの場合には、平均繊維径3μmの非常に細いシリカ不織布(図7(a))となったが、繊維間に空隙が少なく膜状に近い状態であり、シリカ不織布としては不適当であった。適切な空隙を有するシリカ不織布とするためには、紡績液10の粘度を200mPa・s以上とすることが好ましかった。
【0061】
紡績液10の粘度が500mPa・sの場合には、平均繊維径4.5μmのシリカ不織布が製造された(図7(b))。この不織布は、図6(a)及び図6(b)に示したシリカ不織布と同様、均質で強度も十分であった。なお、経験上、紡績液10の粘度は、600mPa・s以上800mPa・s以下とするのが最適であった。また、紡績液10の粘度が低いほど、シリカ不織布の平均繊維径が小さく、紡績液10の粘度が高いほど、シリカ不織布の平均繊維径が大きくなる傾向が認められた。
【0062】
一方、紡績液10の粘度が920mPa・sの場合には、シリカ不織布がもろくなって、繊維が折れやすいという問題が生じた(図7(c))。平均繊維径は13μmであった。紡績液3の粘度が800mPa・sを超えると、不織布が脆性的であるという問題が生じるため、適切なフレキシビリティーを有するシリカ不織布とするためには、紡績液3の粘度を800mPa・s以下とすることが好ましかった。
【0063】
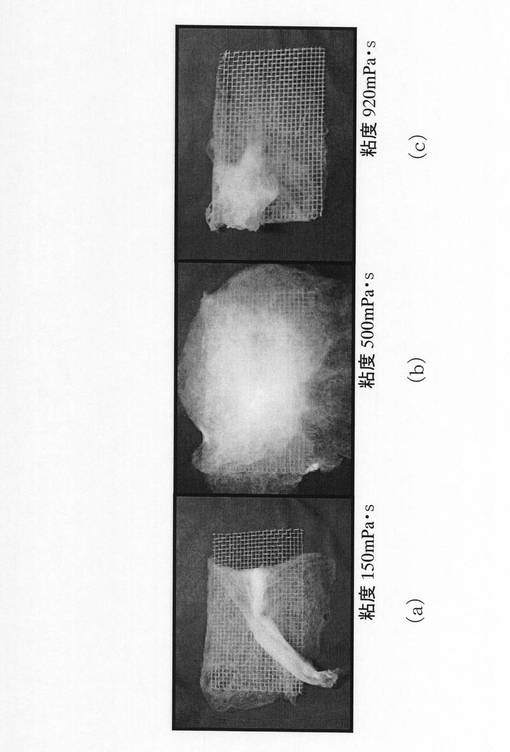
なお、ゲル化した実験例2のサンプル5を紡績液10として、図5(a)及び図5(b)に示した装置を用いて静電噴霧したところ、高圧電源8のスイッチがオフの状態では、シリンジ2の下端部からゲル状の紡績液が滴下するものの(図8(a)を参照)、高圧電源8のスイッチをオンにしても、紡績液3を静電噴霧することはできす、ゲル状の紡績液のままであった(図8(b)を参照)。
【0064】
ここで、後述する実施例1のように、混合溶媒にリン酸トリエチルをアルコキシシランのモル数の2%以上10%以下の範囲で添加した場合にも、実験例1〜実験例3と同様の実験結果が得られた。すなわち、混合溶媒にリン酸トリエチルをアルコキシシランのモル数の2%以上10%以下の範囲で添加しても、(1) 混合溶媒における好ましい水の量、(2) 紡績液の粘度として好ましい範囲、(3) 紡績液をゾル化する工程において、加湿した大気と接触させながら撹拌することが好ましいことは同じであった。
【0065】
(実施例1)
表2に示す組成の紡績液を調整し、実験例2と同様にして、粘度を500mPa・sに調整した(60℃、臨界湿度90%、空気流量300mL/min/リン分添加ゾル化工程)。そして、実験例3と同様にして静電噴霧を行い、リン分添加シリカ不織布(実施例1)を製造した(リン分添加紡糸化工程)。また、リン酸トリエチル(TPE)を添加しない紡績液についても同様の操作を行い、シリカ不織布(比較例)を製造した。
【0066】
【表2】
【0067】
次に、各シリカ不織布約1gを、酢酸カルシウム0.075mol及び塩化カルシウム0.05molを含む水溶液(pH 6.5)80mLに浸漬し、60℃で24時間加熱した(イオン交換工程)。なお、リン原子が導入された実施例1のシリカ不織布をイオン交換し、表面にカルシウムイオン(Ca2+)を導入するためには、後述するように、酢酸カルシウム0.075mol及び塩化カルシウム0.05molを含む水溶液(pH 6.5)80mLに、室温の場合には48時間以上、60℃の場合には24時間以上浸漬することが好ましい。
【0068】
次に、前記水溶液から各シリカ不織布を取り出し、精製水を用いて洗浄し、余剰のカルシウムイオンを除去した。洗浄後、各シリカ不織布を蒸発皿に投入し、昇温速度1.5℃/min、700℃に到達後2時間加熱することにより焼成した(焼成工程)。
【0069】
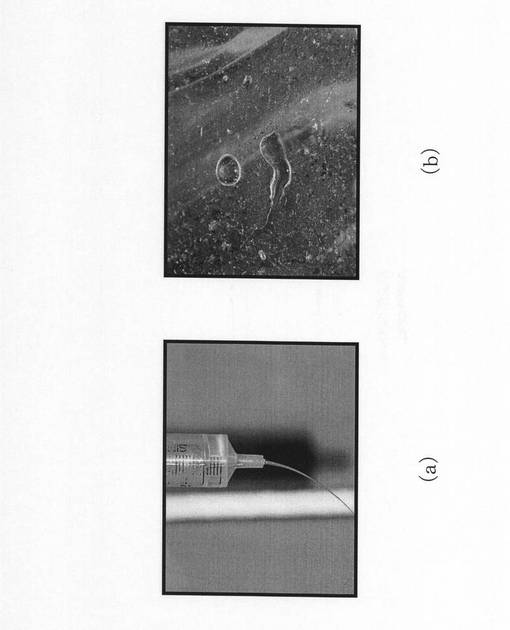
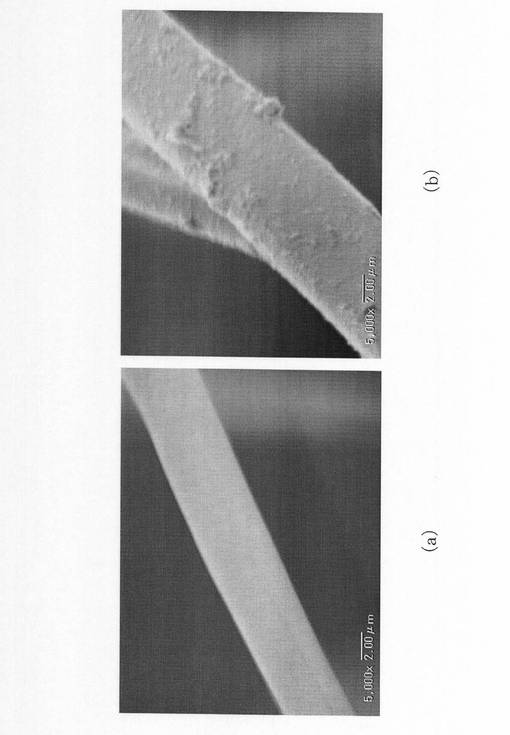
実施例1のシリカ不織布について、シリカ繊維のイオン交換工程及び焼成工程前後のSEM画像を、図9(a)及び図9(b)にそれぞれ示す。実施例1のシリカ繊維は、焼成後に繊維表面にリン酸カルシウム系物質が形成されていることが確認できた。比較例のシリカ不織布についても、シリカ繊維のイオン交換工程及び焼成工程前後のSEM画像を撮影したが、比較例のシリカ繊維には、外見上の変化が全く認められなかった。
【0070】
なお、焼成工程後の実施例1及び比較例のシリカ繊維の平均繊維径は、それぞれ6.1μm及び4.3μmであった。
【0071】
[生体適合性の確認1]
焼成後の実施例1及び比較例のシリカ不織布について、生体適合性の指標として、タンパク質吸着量を測定した。測定方法として、まず、アルブミン(ウシ血清アルブミン)又はリゾチームの100ppm水溶液10mL中に各シリカ不織布0.2gを浸漬し、25℃で2時間静置した。その後、不織布を取り出し、アルブミン又はリゾチーム水溶液の280nmにおける吸光度を、UV分光光度計を用いて測定し、タンパク質濃度の変化を求め、シリカ繊維1g当たりの吸着量を算出した。
【0072】
その結果、比較例のシリカ不織布は、アルブミン及びリゾチームを全く吸着しなかった。一方、実施例1のシリカ不織布は、アルブミン2.5mg/g又はリゾチーム8mg/gを吸着し、酸性タンパク質であるアルブミン及び塩基性タンパク質であるリゾチームの両方に対する吸着能が認められた。このため、実施例1のシリカ不織布は、通常のシリカ不織布にはないタンパク質吸着性、すなわち生体適合性を有していることが確認された。
【0073】
<イオン交換時間の影響>
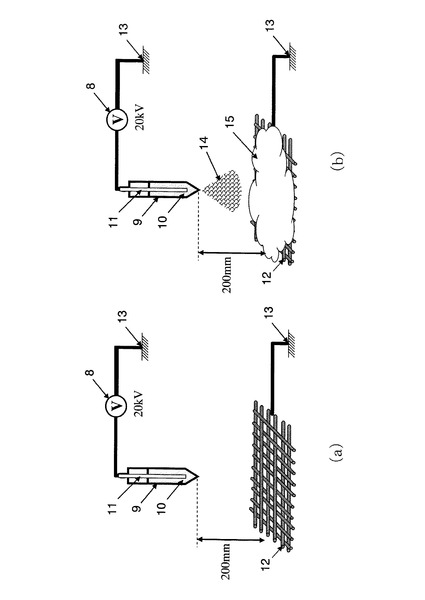
ここで、上述した実施例1の生体適合性繊維(シリカ繊維)のイオン交換工程において、60℃でカルシウムイオンを含む水溶液(酢酸カルシウム0.075mol及び塩化カルシウム0.05molを含む水溶液)に浸漬する時間を変化させ、焼成工程後のアルブミン吸着量がどのように変化するか実験した。その結果を、図10に示す。なお、焼成工程は、昇温速度1.5℃/min、900℃に到達後2時間であり、アルブミン吸着量の測定方法は、上述した通りである。
【0074】
カルシウムイオン交換時間(イオン交換工程)を24時間以上とすれば、アルブミン吸着量がほぼ平衡となったことから、イオン交換工程は、60℃の場合には24時間以上とすることが好ましいことが確認された。なお、25℃の場合には48時間以上とすればアルブミン吸着量がほぼ平衡となった。
【0075】
後述する実施例2の生体適合性繊維についても同様の実験を行ったところ、同様の結果が得られた。
【0076】
<焼成温度の影響>
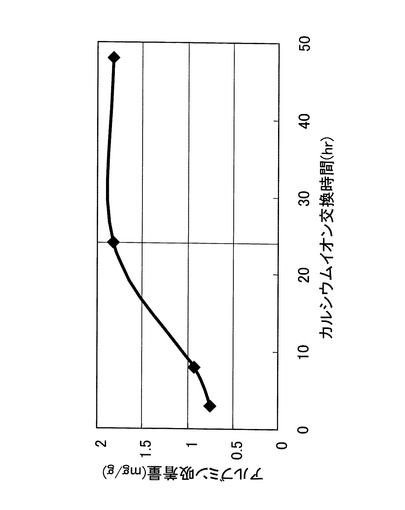
次に、上述した実施例1の生体適合性繊維のイオン交換工程において、イオン交換工程を60℃で24時間、昇温速度を1.5℃/minに固定した上で、焼成工程における焼成温度を変化させ、焼成工程後のアルブミン及びリゾチーム吸着量がどのように変化するか実験した。その結果を、図11に示す。
【0077】
アルブミン吸着量は、焼成温度400℃〜900℃の範囲でほぼ一定であり、1000℃では若干減少した。一方、リゾチーム吸着量も、焼成温度400℃以上の範囲でほぼ一定であり、1000℃では若干増加した。そして、焼成温度が300℃ではアルブミン吸着及びリゾチーム吸着が全く認められなかった。このため、焼成工程の焼成温度は、400℃以上1000℃以下が好ましく、500℃以上800℃以下がより好ましいことが確認された。
【0078】
<昇温速度の影響>
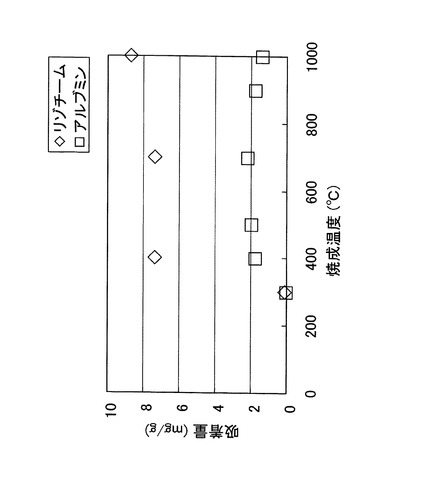
次に、上述した実施例1の生体適合性繊維のイオン交換工程において、イオン交換工程を60℃、24時間に固定し、焼成工程における焼成温度を900℃に固定した上で昇温速度を変化させ、焼成工程後のアルブミン吸着量がどのように変化するか実験した。その結果を、図12に示す。
【0079】
昇温速度が0.5℃/min及び1.5℃/minのときには、アルブミン吸着量はほぼ同じであったが、それ以上の昇温速度とすれば、焼成温度が同一であってもアルブミン吸着量が急激に低下した。このように、焼成温度が同一であっても、昇温速度が高ければシリカ繊維のタンパク質吸着量は低くなるため、昇温速度は0.5℃/min以上3℃/min以下とすることが好ましく、0.5℃/min以上1.5℃/min以下とすることがより好ましいことが確認された。
【0080】
(実施例2)
表2の比較例と同じ組成の紡績液を調整し、実験例2と同様にして、粘度を500mPa・sに調整した(60℃、臨界湿度90%、空気流量300mL/min/ゾル化工程)。そして、実験例3と同様にして静電噴霧を行い、シリカ不織布を製造した(紡糸化工程)。
【0081】
次に、このシリカ不織布をリン酸(JIS特級)中に浸漬し、120℃で1時間加熱した(リン酸化工程)。
【0082】
リン酸から取り出した不織布は、精製水を用いて洗浄し、風乾させた後、実施例1と同様、酢酸カルシウム0.075mol及び塩化カルシウム0.05molを含む水溶液(pH 6.5)80mLに浸漬し、60℃で24時間加熱した(イオン交換工程)。
【0083】
次に、前記水溶液からシリカ不織布を取り出し、精製水を用いて洗浄し、余剰のカルシウムイオンを除去した。洗浄後、各シリカ不織布を蒸発皿に投入し、昇温速度1.5℃/min、400℃に到達後2時間加熱することにより焼成した(焼成工程)。焼成工程後のシリカ不織布を、実施例2のシリカ不織布とした。
【0084】
実施例2のシリカ不織布について、シリカ繊維の焼成前後のSEM画像を、図13(a)及び図13(b)にそれぞれ示す。実施例2のシリカ繊維は、焼成後に繊維表面にリン酸カルシウム系物質が形成されていることが確認できた。
【0085】
なお、焼成工程後の実施例2のシリカ繊維の平均繊維径は、5.0μmであった。
【0086】
[生体適合性の確認2]
焼成後の実施例2のシリカ不織布について、実施例1のシリカ不織布と同様、生体適合性の指標として、タンパク質吸着量を測定した。その結果、実施例2のシリカ不織布は、アルブミン3.5mg/g又はリゾチーム3.0mg/gを吸着し、酸性タンパク質及び塩基性タンパク質の両方に対する吸着能が認められた。このため、実施例2のシリカ不織布も、通常のシリカ不織布にはないタンパク質吸着性、すなわち生体適合性を有していることが確認された。
【0087】
<焼成温度の影響>
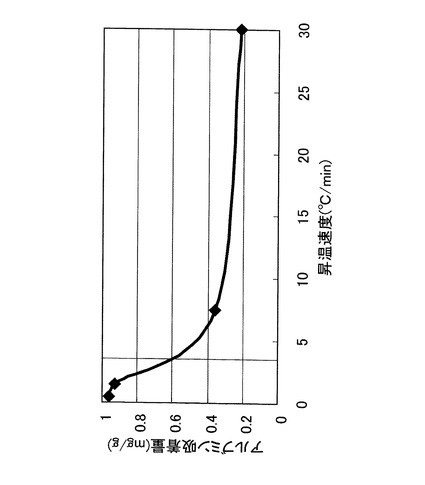
ここで、実施例1の場合と同様に、焼成工程における焼成温度を変化させ、焼成工程後のアルブミン吸着量がどのように変化するか実験した。その結果を、図14に示す。
【0088】
アルブミン吸着量は、焼成温度400℃付近が最も高く、700℃〜1000℃の範囲では減少した。一方、リゾチーム吸着量は、焼成温度400℃〜1000℃の範囲でほぼ一定であった。そして、焼成温度が300℃ではアルブミン吸着及びリゾチーム吸着が全く認められなかった。このため、焼成工程の焼成温度は、400℃以上1000℃以下が好ましく、400℃以上600℃以下がより好ましいことが確認された。
【0089】
<昇温速度の影響>
次に、焼成工程における焼成温度を400℃に固定した上で、実施例1の場合と同様に昇温速度を変化させ、焼成工程後のアルブミン吸着量がどのように変化するか実験した。その結果、実施例1と同様、焼成温度が同一であっても、昇温速度が高ければシリカ繊維のタンパク質吸着量は低くなったため、昇温速度は0.5℃/min以上3℃/min以下とすることが好ましく、0.5℃/min以上1.5℃/min以下とすることがより好ましいことが確認された。
【0090】
なお、実施例1についても焼成温度400℃に固定して、昇温速度を変化させたところ、実施例2と同様の実験結果が得られた。
【0091】
このように、本発明の製造方法によって得られる生体適合性シリカ不織布は、平均繊維径が小さく、変形容易で加工性にも富む。また、表面にリン酸カルシウム系物質が形成されているため、従来のシリカ繊維にはない生体適合性を有している。
【0092】
ヒドロキシアパタイト等のリン酸カルシウム系物質は、通常は微粒子として製造されるため、取扱が難しく、用途も限定されるが、本発明の製造方法によって得られる生体適合性シリカ不織布は、繊維状であるため、例えば、ウイルス防止用マスクや空気清浄機用フィルター等、広範囲への応用が期待できる。さらに、タンパク質吸着能を有するため、ウイルスや花粉等に対する捕集効果も高いことが期待される。
【0093】
また、本発明の生体適合性シリカ不織布の製造方法は、実験室レベルの小型、かつ、簡便な装置で、容易な操作によって実行することができる。エネルギーコストも低く、装置のメインテナンスも容易である。また、高温、高圧とならないために、安全性も高く、紡績液の粘度を調整することにより、シリカ繊維の繊維径を容易に調製することも可能であり、多成分系の紡績液についても、分子レベルで均質な生体適合性シリカ不織布を製造することが可能である。
【産業上の利用可能性】
【0094】
本発明の生体適合性シリカ不織布の製造方法、及び生体適合性シリカ繊維を構成する生体適合性シリカ繊維は、フィルター、各種衛生材料、吸着剤等、広範囲の技術分野の用途に応用することが可能である。
【図面の簡単な説明】
【0095】
【図1】ゾル−ゲル法における加水分解反応及び重合反応の概念を表す図である。
【図2】実験例1において、混合溶媒の粘度を経時的に測定した結果を示す図である。
【図3】実験例2において使用した紡糸液の製造装置の概略図である。
【図4】実験例2において、混合溶媒の粘度を経時的に測定した結果を示す図である。
【図5】本発明のシリカ不織布の製造に用いた静電噴霧装置の概略構成図であり、図5(a)は静電噴射前、図5(b)は静電噴射中の状態を示す図である。
【図6】実施例1のシリカ不織布の写真であり、図6(a)はシリカ不織布全体の外観写真、図6(b)はシリカ不織布の電子顕微鏡写真である。
【図7】実施例1のシリカ不織布の外観写真であり、図7(a)、図7(b)及び図7(c)は、混合溶媒の粘度が、それぞれ150mPa・s、500mPa・s及び920mPa・sの場合のシリカ不織布の外観を示す。
【図8】実験例2のサンプル5を紡績液として、図5に示す静電噴霧装置に用いた状態を示す写真であり、図8(a)は高圧電源のスイッチがオフの状態、図8(b)は高圧電源のスイッチがオンの状態を示す。
【図9】図9(a)は実施例1のシリカ繊維の焼成前のSEM画像であり、図9(b)は焼成後のSEM画像である。
【図10】実施例1のイオン交換工程でカルシウムイオンを含む水溶液に浸漬する時間と、焼成工程後のアルブミン吸着量との関係を表すグラフである。
【図11】実施例1の焼成工程における焼成温度と、焼成工程後のアルブミン及びリゾチーム吸着量との関係を表すグラフである。
【図12】実施例1の焼成工程における昇温速度と、焼成工程後のアルブミン吸着量との関係を表すグラフである。
【図13】図13(a)は実施例2のシリカ繊維の焼成前のSEM画像であり、図13(b)は焼成後のSEM画像である。
【図14】実施例2の焼成工程における焼成温度と、焼成工程後のアルブミン及びリゾチーム吸着量との関係を表すグラフである。
【符号の説明】
【0096】
1:エアポンプ
2:バイアル
3:油浴
4:スターラ
5:フラスコ
6:冷却管
7:テトラエトキシシランを溶解させた混合溶媒
8:高圧電源
9:シリンジ
10:紡績液
11:針状電極
12:導電性捕集板
13:アース
14:シリカ繊維
15:シリカ不織布
【技術分野】
【0001】
本発明は、表面にリン酸カルシウム系物質が形成されている生体適合性シリカ不織布の製造方法に関するものである。また、本発明は、そのような生体適合性シリカ不織布を構成する生体適合性シリカ繊維に関するものである。
【背景技術】
【0002】
無機ケイ酸化合物は、ガラス、セメント、陶磁器等の原材料として古くから使用されている。一般に、無機ケイ酸化合物は化学的に安定しており、耐熱性及び耐薬品性にも優れている。また、多孔質であるため化学物質等を吸着することができるもの、触媒機能を有するために化学物質等を分解できるもの、無害で生体適合性が高いもの等、様々な機能性を備えた無機ケイ酸化合物も存在する。
【0003】
しかし、無機ケイ酸化合物の多くは粉末状であるため、そのままでは取り扱いが難しく、用途も限定されるため、商品として展開しにくい。一方、無機ケイ酸化合物を不織布として繊維状に形成すれば、容易に切断したり、曲げたり、容器に詰めたりできるため、広範囲の用途へ応用することが可能になる。
【0004】
無機ケイ酸化合物を繊維状に形成する場合、一般的には2000℃以上の高温で熔融し、その熔融液を流出させることによって繊維化するが、この製造方法では繊維径を小さくすることが極めて困難であり、また、原料である無機ケイ酸化合物を熔融するために多大なエネルギーを必要とする。さらに、高温の熔融液を取り扱うため、製造装置が大がかりなものとならざるを得ず、作業の安全性に関しても問題がある。
【0005】
アルコキシシランには、有機溶媒に溶かした状態で重合化させることによりゾル状となるものがあり、微細孔から押し出して繊維状に形成することも可能である。例えば、特許文献1には、変性ポリカルボシランと低分子量の有機金属化合物をトルエンに溶解させて紡糸原液とした後、合成繊維紡糸用装置を用いて繊維状に形成する方法が開示されている。
【0006】
一方、生体適合性を有する無機材料として、ヒドロキシアパタイトやβ-3リン酸カルシウム等のリン酸カルシウム系材料が注目されているが、これらリン酸カルシウム系材料は、すべて粉末として得られるため、合成後に成型及び焼成して使用せざるを得ない。例えば、特許文献2には、骨細胞活性を薄膜上で支えるためのリン酸カルシウム材の薄膜の改良製造方法として、リン酸アンモニウムの溶液を硝酸カルシウムの溶液と合わせてヒドロキシアパタイト含有ゾル−ゲルを形成させることによって調製し、このゾル−ゲルの膜を基層の少なくとも片側に当てがい、基層上にコーティングされた膜を焼結してリン酸カルシウム材の固体膜を形成させる方法が開示されている。
【0007】
ここで、ヒドロキシアパタイト等を繊維状に形成する方法として、熔融紡糸法やゲル紡糸後に焼成する方法(非特許文献1,2)が知られているが、いずれもヒドロキシアパタイト等を溶融する必要があるため、上述した無機ケイ酸化合物と同様の問題がある。
【0008】
また、ポリマー繊維にヒドロキシアパタイト等を分散させたコンポジット繊維も知られているが(非特許文献3)、得られる繊維は脆く、応用範囲が制限されるという問題がある。
【特許文献1】特開2003−328236号公報
【特許文献2】特開2004−209261号公報
【非特許文献1】“Preparation of Porous Ceramics with Calcium Metaphosphate Fiber Skelecton for Biomedical Use”, Journal of American ceramic society, Vol. 78, Page. 245-247, 1995.
【非特許文献2】“A study of hydroxyapatite fibers prepared via sol-gel route”, Material Letters, Vol. 58, Page. 3320-3323, 2004.
【非特許文献3】“Effect of Spinning Conditions on the Structure and Properties of PAN Fibers Containing Nano-Hydroxyatatite”, Journal of Polymer Science, Vol. 100, Page. 2881-2888, 2006.
【発明の開示】
【発明が解決しようとする課題】
【0009】
上記特許文献に開示されているシリカ繊維の製造方法は、非常に微細な細孔から高圧で紡糸原液を押し出す必要があり、装置を小型化することは困難である。また、平均繊維径を小さくするためには、細孔をさらに小さくしなければならず、より高圧で紡糸原液を押し出す必要がある。さらに、細孔を小さくすればするほど、目詰まりも生じやすくなり、装置のメインテナンス頻度も高くなる。
【0010】
一方で、ヒドロキシアパタイト等の生体適合性無機材料を簡便な方法で繊維化できれば、従来応用できなかった用途への応用も可能となる。
【0011】
本発明は、従来は不可能であった完全無機質、かつ、柔軟性を有する生体適合性シリカ繊維を、実験室レベルの小型で簡便な装置でも製造しうる製造方法、及び当該生体適合性シリカ不織布を構成する生体適合性シリカ繊維の提供を目的とする。
【課題を解決するための手段】
【0012】
アルコキシシランは、有機溶媒に溶解した状態で水と反応すると、加水分解反応と縮合反応を起こして粘性ゾルとなり、さらに縮合反応が進むと湿潤ゲル体となる。こうした現象は、ゾル−ゲル反応と呼ばれている。
【0013】
一方、試料溶液を供給するキャピラリー先端に数kVの高電圧を印加することにより 試料溶液を微細な液滴として霧化させることが可能である。この技術は静電噴霧(electrospray)と呼ばれ、薄膜や微粒子の製造に応用されてきた。
【0014】
本発明者は、鋭意研究した結果、アルコキシシランを水と有機溶媒の混合溶媒に溶解させて紡績液とし、この紡績液を静電噴霧することにより、シリカを容易に繊維化し、シリカ不織布を製造しうることを既に見出している。今回、本発明者は、この紡績液にリン分(リン成分)供給源としてリン酸トリエチル((C2H5O)3PO)を添加して静電噴霧を行い、得られたシリカ繊維を、カルシウムイオンを含む水溶液中でイオン交換し、さらに焼成することにより、シリカ繊維表面にリン酸カルシウム系無機物が形成され、タンパク質吸着性等の生体適合性を発揮することを新たに見出した。
【0015】
また、本発明者は、アルコキシシランを水と有機溶媒の混合溶媒に溶解させた紡績液を、静電噴霧することにより得られるシリカ不織布をリン酸中で加熱した後、上記と同様、カルシウムイオンを含む水溶液中でイオン交換し、さらに焼成することにより、シリカ繊維表面にリン酸カルシウム系無機物が形成され、タンパク質吸着性等の生体適合性を発揮することを新たに見出した。
【0016】
具体的に、本発明は、
アルコキシシランを、リン酸トリエチルを含む水と有機溶媒の混合溶媒に溶解させ、大気と接触させながらゾル状の紡績液とするリン分添加ゾル化工程と、
粘度調整後の前記紡績液を静電噴霧法により紡糸化するリン分添加紡糸化工程と、
前記リン分添加紡糸化工程で得られた不織布を、カルシウムイオンを含む水溶液中に浸漬することによりイオン交換させるイオン交換工程と、
前記イオン交換工程後の不織布を400℃以上1000℃以下の温度で焼成する焼成工程と、
を含む生体適合性シリカ不織布の製造方法に関する(請求項1)。
【0017】
アルコキシシランを水と有機溶媒の混合溶媒に溶解させた紡績液を、静電噴霧することにより得られるシリカ不織布(シリカ繊維)表面には、未反応アルコキシル基が残存していると考えられる。ここで、紡績液にリン分(リン成分)供給源としてリン酸トリエチルを添加すれば、静電噴霧によって得られるシリカ不織布表面にリン原子が導入され、さらにカルシウムイオンと反応させて焼成することにより、リン酸カルシウム系物質が形成されて生体適合性シリカ繊維を製造することができると推定される。
【0018】
また、本発明は、
アルコキシシランを水と有機溶媒の混合溶媒に溶解させ、大気と接触させながらゾル状の紡績液とするゾル化工程と、
粘度調整後の前記紡績液を、静電噴霧法により紡糸化する紡糸化工程と、
前記紡糸化工程で得られた不織布を、リン酸に浸漬して加熱するリン酸化工程と、
前記リン酸化工程後の不織布を、カルシウムイオンを含む水溶液中に浸漬することによりイオン交換させるイオン交換工程と、
前記イオン交換工程後の不織布を400℃以上1000℃以下の温度で焼成する焼成工程と、
を含む生体適合性シリカ不織布の製造方法に関する(請求項8)。
【0019】
シリカ不織布(シリカ繊維)表面の未反応アルコキシル基は、シリカ不織布をリン酸中で加熱することによってもリン酸化されると考えられ、リン酸化したシリカ不織布を、カルシウムイオンと反応させて焼成することにより、リン酸カルシウム系物質が形成されて生体適合性シリカ繊維を製造することができると推定される。
【0020】
なお、本発明の生体適合性シリカ不織布の平均繊維径は、10μm以下であることが好ましい。また、前記ゾル化工程においては、前記紡績液の粘度を200mPa・s以上800mPa・s以下の範囲内に調整することが好ましい。
【0021】
前記アルコキシシランはテトラエトキシシランであり、前記有機溶媒はエタノールであることが好ましい(請求項2,9)。
【0022】
本発明のシリカ不織布の製造方法において、前記混合溶媒における水の量は、アルコキシシランのモル数に対して1倍以上2倍以下であることが好ましい(請求項3,10)。
【0023】
前記混合溶媒にリン酸トリエチルを添加する製造方法においては、リン酸トリエチルの量が、アルコキシシランのモル数の2%以上10%以下であることが好ましい(請求項4)。
【0024】
前記リン分添加ゾル化工程又は前記ゾル化工程においては、前記混合溶媒に塩酸を加えることが好ましい(請求項5,11)。
【0025】
前記ゾル化工程において、加湿した大気と接触させながら撹拌することが好ましい(請求項6,12)。
【0026】
また、本発明は、請求項1乃至6、又は請求項8乃至13のいずれか1項に記載の生体適合性シリカ不織布を構成する生体適合性シリカ繊維に関する(請求項7,13)。
【発明の効果】
【0027】
本発明で製造されるシリカ不織布は、微細な繊維径のシリカ不織布の表面に、リン酸カルシウム系物質が形成された完全無機質な生体適合性シリカ不織布であり、従来にない柔軟性、強度を有する。このため、粉末又は固体のリン酸カルシウム系物質と比較して、広範囲な応用が可能である。
【0028】
また、本発明の生体適合性シリカ不織布の製造方法は、合成繊維紡糸用装置のような大がかりな装置を用いることなく、実験室レベルの小型、かつ、簡易な装置で実施することが可能である。しかも、常温付近の温度で実施することが可能であり、製造コストも非常に低い。
【発明を実施するための最良の形態】
【0029】
以下に、本発明の実施の形態について、適宜図面を参照しながら説明する。なお、本発明は、これらに限定されない。
【0030】
はじめに、アルコキシシランの一例として、テトラエトキシシランを水とエタノールの混合溶媒に溶解させた場合に起こる加水分解反応及び重縮合反応を、図1を参照しながら説明する。なお、加水分解反応及び重縮合反応を促進するために、水とエタノールの混合溶媒に塩酸を添加することが好ましい。塩酸の添加量は、テトラエトキシシランのモル数の1%程度が適量である。
【0031】
テトラエトキシシランを水とエタノールの混合溶媒に溶解させ(図1(1))、室温以上80℃以下で撹拌すると、テトラエトキシシランの4個の-OC2H5基のうちの1個がH+を触媒として加水分解され-OH基となり、トリエトキシシランに変化する(加水分解反応)。
【0032】
次に、トリエトキシシラン2分子がH+を触媒として脱水縮合する(重縮合反応)。さらに、脱水縮合分子の1個の-OC2H5基も加水分解されて-OH基となり、他の脱水縮合分子の-OH基との間で脱水縮合が繰り返される。この加水分解反応及び重縮合反応によって、分子同士が直線的に重合し、ポリマー化する。それに伴い、混合溶媒の粘度が上昇し、ゾル状となる(図1(2))。
【0033】
このとき、混合溶媒中の水の量が少なすぎると、テトラエトキシシランのポリマー化が途中で止まってしまう。逆に多すぎると、テトラエトキシシランの複数の-OC2H5基が同時に加水分解されため、複数の-OH基が他の脱水縮合分子の-OH基とランダムに脱水縮合する。そうすると、網目状にポリマー化が起こり、混合溶媒中でポリマーが湿潤ゲル体(図1(3)と同じ状態)となって紡績液を調製することができなくなる。
【0034】
混合溶媒中でテトラエトキシシランが直線的に重合してポリマー化すると、上述したように混合溶媒の粘度が上昇する。後述するように、静電噴霧法によって混合溶媒(紡績液)を不織布化するためには、粘度を200mPa・s以上800mPa・s以下の範囲内に調整することが好ましい。それ以上重合化が進むと、直線的に重合したポリマー分子の側鎖の-OC2H5基も加水分解されて-OH基となり、他のポリマー分子等の-OH基との間で脱水縮合を生じ、湿潤ゲル体(図1(3))となって繊維化することが不可能となる。
【0035】
<実験例1>
テトラエトキシシランを、水とエタノールの混合溶媒に溶解させ、65℃の水浴中でエタノールを環流させた。混合溶媒は、水とテトラエトキシシランのモル比(r=[H2O]/[TEOS])が2.7及び1.55の2種類作成した。なお、テトラエトキシシランのモル数に対して1%量の塩酸を触媒として、両方の混合溶媒に添加した。混合溶媒の粘度を経時的に測定した結果を、図2に示す。
【0036】
まず、r=1.55の混合溶媒の場合、65℃で200時間還流させても粘度の上昇、すなわちテトラエトキシシランの直線的ポリマー化が認められなかった(丸印のプロット)。これは、r=1.55ではテトラエトキシシランの加水分解反応及び重合化に必要な水が不足していることが原因と思われる。
【0037】
次に、同じr=1.55の混合溶媒を、65℃で70時間還流させた後、ビーカーに移して大気と接触させながら室温で静置したところ、ビーカーに移してから約30時間経過した後(反応時間全体としては約100時間経過後)に粘度の上昇が認められ、混合溶媒がゾル状となった。これは、ビーカーに移して空気と接触させたことにより、混合溶媒中に空気中の水分が溶け込み、その水分によってテトラエトキシシランの直線的ポリマー化が起こったものと推測された。
【0038】
次に、r=2.7の混合溶媒の場合、65℃で約20時間還流させると粘度の上昇が認められた。しかし、この混合溶媒は、水の量が多すぎるためにテトラエトキシシランの網目状ポリマー化が起こり、ゾル状ではなくゲル状となって後述する静電噴霧法により不織布を製造することができなかった。
【0039】
このように、テトラエトキシシランを溶解させる水とエタノールの混合溶媒において、水の量を適正範囲に調整することにより、直線的ポリマー化を起こすことが重要である。具体的には、混合溶媒における水とテトラエトキシシランのモル比は、1以上2以下にすることが好ましい。1未満では大気と接触させても、テトラエトキシシランの直線的ポリマー化に非常に長時間を要し、2を超えるとテトラエトキシシランの網目状ポリマー化が起こりやすくなるため、静電噴霧法を用いて混合溶媒(紡績液)を紡糸化できないからである。
【0040】
<実験例2>
実験例1では、テトラエトキシシランを混合溶媒中で直線的にポリマー化するために、100時間以上かかった。そこで、テトラエトキシシランを溶解させた混合溶媒を、加湿した大気と接触させることにより、テトラエトキシシランの直線的重合化に必要な水を混合溶媒へと供給し、直線的重合化にかかる時間の短縮を試みた。
【0041】
本実験例で使用した装置の概略を、図3に示す。エアポンプ1からバイアル2へと300mL/minの流速で空気を送り込んだ。バイアル2からフラスコ5内のテトラエトキシシランを溶解させた混合溶媒7へ相対湿度90%の加湿空気を導入する場合には、バイアル2にKCl飽和溶液(臨界湿度90%)を入れ、相対湿度54%の加湿空気を導入する場合には、バイアル2内にCa(NO3)2飽和溶液(臨界湿度54%)を入れた。
【0042】
フラスコ5は、油浴3中で65℃に加熱され、フラスコ5内のテトラエトキシシランを溶解させた混合溶媒7は、スターラ4によって撹拌した。フラスコ5上部には冷却管6を取り付け、実験例1と同様にエタノールを環流させた。
【0043】
本実験例では、表1に示す条件で混合溶液を調製及び処理し、各サンプルの粘度を経時的に測定した。その結果を、図4に示す。なお、テトラエトキシシランのモル数に対して1%量の塩酸を触媒として、全ての混合溶液に添加した。また、表1のr値は、水とテトラエトキシシランのモル比(r=[H2O]/[TEOS])を意味する。
【0044】
【表1】
【0045】
サンプル1は、約12時間で粘度の上昇が認められた。r=1.7であるサンプル2は、サンプル1の約半分の反応時間で粘度の上昇が認められ、ゾル状の紡績液となった。実験例1もr=1.55であったが、65℃に加熱しながら相対湿度90%に加湿した空気と接触させることにより、テトラエトキシシランの直線的重合化に要する時間を1/10〜1/20にまで短縮することが可能であった。
【0046】
サンプル3は、r値がサンプル2と同じであるが、接触させた空気の相対湿度が54%であったため、サンプル2よりも粘度の上昇が認められるまでの反応時間が長くなった。また、r値が同じで加湿した空気と接触させなかったサンプル4は、20時間経過後も全く粘度の上昇が認められなかった。このことから、実験例1における、混合溶媒中に空気中の水分が溶け込み、その水分によってテトラエトキシシランの直線的ポリマー化が起こるという推測が正しいことが証明された。
【0047】
一方、r=4.0のサンプル5は、サンプル2とほぼ同じ反応時間で粘度の上昇が認められたが、すぐにゲル化してしまい、静電噴霧法により紡糸化することができなかった。
【0048】
なお、油浴の温度を50℃、70℃及び85℃として上記と同じ実験を行ったが、65℃の場合と同様の結果が得られたが、加熱温度が高いほど粘度の上昇が認められるまでの反応時間が、若干短くなる傾向が認められた。なお、混合溶媒7のゾル化に要する時間を短縮させるためには、50℃以上85℃以下の温度で加熱することが好ましい。
【0049】
このように、テトラエトキシシランを水とエタノールの混合溶媒に溶解させ、静電噴霧法によりシリカ不織布として紡糸化させるためには、混合溶媒における水とテトラエトキシシランのモル比を1以上2以下に調整した上で、混合溶媒を加熱しながら加湿した大気と接触させることが好ましいことが判明した。このような方法によれば、テトラエトキシシランの直線的ポリマー化を短時間で行うことができ、かつ、粘度の容易に調整することができるからである。
【0050】
<実験例3>
ここで、実験例2のサンプル1について、その一部を経時的に抜き取り、粘度を測定すると共に、図5(a)に示すような装置を用いて静電噴霧法によって紡糸化し、シリカ不織布を製造した。
【0051】
この装置は、高圧電源8と、シリンジ9と、針状電極11と、導電性捕集板12とから構成される。そして、高圧電源8及び導電性捕集板12には、アース13が取り付けられている。
【0052】
シリンジ9として、今回は、市販されているディスポタイプのプラスチック製注射用シリンジ(10mL)本体を用いた。シリンジ9は、通常注射針を取り付ける先端部分を下に向けた状態で、スタンドを用いて垂直に固定され、内部に直径1.0mmの針状電極11がセットされている。
【0053】
針状電極11は、高圧電源8に接続されており、紡糸化する際には20kVの高電圧をかけた。シリンジ9の下部には、アースを取り付けた導電性捕集板12(ここでは、ステンレス製の網)をセットし、シリンジ9下端部と導電性捕集板12の距離を、200mmに調整した。
【0054】
そして、サンプル1の一部(約4mL)を紡績液10としてシリンジ9の上部開口部から投入し、直ちに高圧電源8のスイッチをオンにして、シリンジ9から導電性捕集板12に向けて紡績液10を静電噴霧した。この静電噴霧処理により、紡績液10中の水、エタノール及び塩酸が揮散し、直線的にポリマー化していたテトラエトキシシランが、微細なシリカ繊維14として紡糸化され、図5(b)に示すように、導電性捕集板12の上にシリカ不織布15として集積する。
【0055】
図6(a)は、紡績液10の粘度が500mPa・sの場合に製造されたシリカ不織布15の外観写真である。このシリカ不織布15の外観は、通常のグラスウールとほとんど同じであるが、触感はより滑らかで、折り曲げたりしても繊維が切断されにくい。
【0056】
図6(b)は、このシリカ不織布6の電子顕微鏡写真であり、平均繊維径は4.5μmであることが確認された。特許文献1には、合成繊維紡糸用装置を用いて変性ポリカルボシランを繊維化し、1200℃の空気中で1時間焼成することにより得られたシリカ繊維の平均直径が13μmであったこと記載されているが、本発明のシリカ不織布の製造方法は、より繊維径を小さくできることが確認された。また、本発明のシリカ不織布の製造方法は、紡績液を静電噴霧して紡糸化する際に、溶媒の留去も同時に行われるため、紡糸化操作が非常に容易である。
【0057】
シリンジから静電噴霧された紡績液(混合溶媒)は、同じ電荷に帯電しているため、導電性捕集板に到達するまでにシリカ繊維同士がくっつくことがない。そのため、できあがったシリカ繊維の繊維径は、非常に平均化されていた。さらに、高温での焼成も行う必要がなく、実験室レベルの小型、かつ、簡便な装置で製造可能であった。
【0058】
また、特許文献1の製造方法では、シリカ繊維の繊維径と同程度の微細孔から高圧で紡糸液を押し出す必要があるが、本発明のシリカ不織布の製造方法では、ディスポタイプのプラスチック製注射用シリンジから圧力をかけることなく紡糸液を静電噴霧させるため、微細孔の目詰まりという問題が発生せず、分解洗浄等、製造装置のメインテナンスも非常に容易である。
【0059】
次に、静電噴霧する紡績液10の粘度が150mPa・s、500mPa・s及び920mPa・sの場合に製造されたシリカ不織布15の外観写真を、図7(a)、図7(b)及び図7(c)にそれぞれ示す。
【0060】
まず、紡績液10の粘度が150mPa・sの場合には、平均繊維径3μmの非常に細いシリカ不織布(図7(a))となったが、繊維間に空隙が少なく膜状に近い状態であり、シリカ不織布としては不適当であった。適切な空隙を有するシリカ不織布とするためには、紡績液10の粘度を200mPa・s以上とすることが好ましかった。
【0061】
紡績液10の粘度が500mPa・sの場合には、平均繊維径4.5μmのシリカ不織布が製造された(図7(b))。この不織布は、図6(a)及び図6(b)に示したシリカ不織布と同様、均質で強度も十分であった。なお、経験上、紡績液10の粘度は、600mPa・s以上800mPa・s以下とするのが最適であった。また、紡績液10の粘度が低いほど、シリカ不織布の平均繊維径が小さく、紡績液10の粘度が高いほど、シリカ不織布の平均繊維径が大きくなる傾向が認められた。
【0062】
一方、紡績液10の粘度が920mPa・sの場合には、シリカ不織布がもろくなって、繊維が折れやすいという問題が生じた(図7(c))。平均繊維径は13μmであった。紡績液3の粘度が800mPa・sを超えると、不織布が脆性的であるという問題が生じるため、適切なフレキシビリティーを有するシリカ不織布とするためには、紡績液3の粘度を800mPa・s以下とすることが好ましかった。
【0063】
なお、ゲル化した実験例2のサンプル5を紡績液10として、図5(a)及び図5(b)に示した装置を用いて静電噴霧したところ、高圧電源8のスイッチがオフの状態では、シリンジ2の下端部からゲル状の紡績液が滴下するものの(図8(a)を参照)、高圧電源8のスイッチをオンにしても、紡績液3を静電噴霧することはできす、ゲル状の紡績液のままであった(図8(b)を参照)。
【0064】
ここで、後述する実施例1のように、混合溶媒にリン酸トリエチルをアルコキシシランのモル数の2%以上10%以下の範囲で添加した場合にも、実験例1〜実験例3と同様の実験結果が得られた。すなわち、混合溶媒にリン酸トリエチルをアルコキシシランのモル数の2%以上10%以下の範囲で添加しても、(1) 混合溶媒における好ましい水の量、(2) 紡績液の粘度として好ましい範囲、(3) 紡績液をゾル化する工程において、加湿した大気と接触させながら撹拌することが好ましいことは同じであった。
【0065】
(実施例1)
表2に示す組成の紡績液を調整し、実験例2と同様にして、粘度を500mPa・sに調整した(60℃、臨界湿度90%、空気流量300mL/min/リン分添加ゾル化工程)。そして、実験例3と同様にして静電噴霧を行い、リン分添加シリカ不織布(実施例1)を製造した(リン分添加紡糸化工程)。また、リン酸トリエチル(TPE)を添加しない紡績液についても同様の操作を行い、シリカ不織布(比較例)を製造した。
【0066】
【表2】
【0067】
次に、各シリカ不織布約1gを、酢酸カルシウム0.075mol及び塩化カルシウム0.05molを含む水溶液(pH 6.5)80mLに浸漬し、60℃で24時間加熱した(イオン交換工程)。なお、リン原子が導入された実施例1のシリカ不織布をイオン交換し、表面にカルシウムイオン(Ca2+)を導入するためには、後述するように、酢酸カルシウム0.075mol及び塩化カルシウム0.05molを含む水溶液(pH 6.5)80mLに、室温の場合には48時間以上、60℃の場合には24時間以上浸漬することが好ましい。
【0068】
次に、前記水溶液から各シリカ不織布を取り出し、精製水を用いて洗浄し、余剰のカルシウムイオンを除去した。洗浄後、各シリカ不織布を蒸発皿に投入し、昇温速度1.5℃/min、700℃に到達後2時間加熱することにより焼成した(焼成工程)。
【0069】
実施例1のシリカ不織布について、シリカ繊維のイオン交換工程及び焼成工程前後のSEM画像を、図9(a)及び図9(b)にそれぞれ示す。実施例1のシリカ繊維は、焼成後に繊維表面にリン酸カルシウム系物質が形成されていることが確認できた。比較例のシリカ不織布についても、シリカ繊維のイオン交換工程及び焼成工程前後のSEM画像を撮影したが、比較例のシリカ繊維には、外見上の変化が全く認められなかった。
【0070】
なお、焼成工程後の実施例1及び比較例のシリカ繊維の平均繊維径は、それぞれ6.1μm及び4.3μmであった。
【0071】
[生体適合性の確認1]
焼成後の実施例1及び比較例のシリカ不織布について、生体適合性の指標として、タンパク質吸着量を測定した。測定方法として、まず、アルブミン(ウシ血清アルブミン)又はリゾチームの100ppm水溶液10mL中に各シリカ不織布0.2gを浸漬し、25℃で2時間静置した。その後、不織布を取り出し、アルブミン又はリゾチーム水溶液の280nmにおける吸光度を、UV分光光度計を用いて測定し、タンパク質濃度の変化を求め、シリカ繊維1g当たりの吸着量を算出した。
【0072】
その結果、比較例のシリカ不織布は、アルブミン及びリゾチームを全く吸着しなかった。一方、実施例1のシリカ不織布は、アルブミン2.5mg/g又はリゾチーム8mg/gを吸着し、酸性タンパク質であるアルブミン及び塩基性タンパク質であるリゾチームの両方に対する吸着能が認められた。このため、実施例1のシリカ不織布は、通常のシリカ不織布にはないタンパク質吸着性、すなわち生体適合性を有していることが確認された。
【0073】
<イオン交換時間の影響>
ここで、上述した実施例1の生体適合性繊維(シリカ繊維)のイオン交換工程において、60℃でカルシウムイオンを含む水溶液(酢酸カルシウム0.075mol及び塩化カルシウム0.05molを含む水溶液)に浸漬する時間を変化させ、焼成工程後のアルブミン吸着量がどのように変化するか実験した。その結果を、図10に示す。なお、焼成工程は、昇温速度1.5℃/min、900℃に到達後2時間であり、アルブミン吸着量の測定方法は、上述した通りである。
【0074】
カルシウムイオン交換時間(イオン交換工程)を24時間以上とすれば、アルブミン吸着量がほぼ平衡となったことから、イオン交換工程は、60℃の場合には24時間以上とすることが好ましいことが確認された。なお、25℃の場合には48時間以上とすればアルブミン吸着量がほぼ平衡となった。
【0075】
後述する実施例2の生体適合性繊維についても同様の実験を行ったところ、同様の結果が得られた。
【0076】
<焼成温度の影響>
次に、上述した実施例1の生体適合性繊維のイオン交換工程において、イオン交換工程を60℃で24時間、昇温速度を1.5℃/minに固定した上で、焼成工程における焼成温度を変化させ、焼成工程後のアルブミン及びリゾチーム吸着量がどのように変化するか実験した。その結果を、図11に示す。
【0077】
アルブミン吸着量は、焼成温度400℃〜900℃の範囲でほぼ一定であり、1000℃では若干減少した。一方、リゾチーム吸着量も、焼成温度400℃以上の範囲でほぼ一定であり、1000℃では若干増加した。そして、焼成温度が300℃ではアルブミン吸着及びリゾチーム吸着が全く認められなかった。このため、焼成工程の焼成温度は、400℃以上1000℃以下が好ましく、500℃以上800℃以下がより好ましいことが確認された。
【0078】
<昇温速度の影響>
次に、上述した実施例1の生体適合性繊維のイオン交換工程において、イオン交換工程を60℃、24時間に固定し、焼成工程における焼成温度を900℃に固定した上で昇温速度を変化させ、焼成工程後のアルブミン吸着量がどのように変化するか実験した。その結果を、図12に示す。
【0079】
昇温速度が0.5℃/min及び1.5℃/minのときには、アルブミン吸着量はほぼ同じであったが、それ以上の昇温速度とすれば、焼成温度が同一であってもアルブミン吸着量が急激に低下した。このように、焼成温度が同一であっても、昇温速度が高ければシリカ繊維のタンパク質吸着量は低くなるため、昇温速度は0.5℃/min以上3℃/min以下とすることが好ましく、0.5℃/min以上1.5℃/min以下とすることがより好ましいことが確認された。
【0080】
(実施例2)
表2の比較例と同じ組成の紡績液を調整し、実験例2と同様にして、粘度を500mPa・sに調整した(60℃、臨界湿度90%、空気流量300mL/min/ゾル化工程)。そして、実験例3と同様にして静電噴霧を行い、シリカ不織布を製造した(紡糸化工程)。
【0081】
次に、このシリカ不織布をリン酸(JIS特級)中に浸漬し、120℃で1時間加熱した(リン酸化工程)。
【0082】
リン酸から取り出した不織布は、精製水を用いて洗浄し、風乾させた後、実施例1と同様、酢酸カルシウム0.075mol及び塩化カルシウム0.05molを含む水溶液(pH 6.5)80mLに浸漬し、60℃で24時間加熱した(イオン交換工程)。
【0083】
次に、前記水溶液からシリカ不織布を取り出し、精製水を用いて洗浄し、余剰のカルシウムイオンを除去した。洗浄後、各シリカ不織布を蒸発皿に投入し、昇温速度1.5℃/min、400℃に到達後2時間加熱することにより焼成した(焼成工程)。焼成工程後のシリカ不織布を、実施例2のシリカ不織布とした。
【0084】
実施例2のシリカ不織布について、シリカ繊維の焼成前後のSEM画像を、図13(a)及び図13(b)にそれぞれ示す。実施例2のシリカ繊維は、焼成後に繊維表面にリン酸カルシウム系物質が形成されていることが確認できた。
【0085】
なお、焼成工程後の実施例2のシリカ繊維の平均繊維径は、5.0μmであった。
【0086】
[生体適合性の確認2]
焼成後の実施例2のシリカ不織布について、実施例1のシリカ不織布と同様、生体適合性の指標として、タンパク質吸着量を測定した。その結果、実施例2のシリカ不織布は、アルブミン3.5mg/g又はリゾチーム3.0mg/gを吸着し、酸性タンパク質及び塩基性タンパク質の両方に対する吸着能が認められた。このため、実施例2のシリカ不織布も、通常のシリカ不織布にはないタンパク質吸着性、すなわち生体適合性を有していることが確認された。
【0087】
<焼成温度の影響>
ここで、実施例1の場合と同様に、焼成工程における焼成温度を変化させ、焼成工程後のアルブミン吸着量がどのように変化するか実験した。その結果を、図14に示す。
【0088】
アルブミン吸着量は、焼成温度400℃付近が最も高く、700℃〜1000℃の範囲では減少した。一方、リゾチーム吸着量は、焼成温度400℃〜1000℃の範囲でほぼ一定であった。そして、焼成温度が300℃ではアルブミン吸着及びリゾチーム吸着が全く認められなかった。このため、焼成工程の焼成温度は、400℃以上1000℃以下が好ましく、400℃以上600℃以下がより好ましいことが確認された。
【0089】
<昇温速度の影響>
次に、焼成工程における焼成温度を400℃に固定した上で、実施例1の場合と同様に昇温速度を変化させ、焼成工程後のアルブミン吸着量がどのように変化するか実験した。その結果、実施例1と同様、焼成温度が同一であっても、昇温速度が高ければシリカ繊維のタンパク質吸着量は低くなったため、昇温速度は0.5℃/min以上3℃/min以下とすることが好ましく、0.5℃/min以上1.5℃/min以下とすることがより好ましいことが確認された。
【0090】
なお、実施例1についても焼成温度400℃に固定して、昇温速度を変化させたところ、実施例2と同様の実験結果が得られた。
【0091】
このように、本発明の製造方法によって得られる生体適合性シリカ不織布は、平均繊維径が小さく、変形容易で加工性にも富む。また、表面にリン酸カルシウム系物質が形成されているため、従来のシリカ繊維にはない生体適合性を有している。
【0092】
ヒドロキシアパタイト等のリン酸カルシウム系物質は、通常は微粒子として製造されるため、取扱が難しく、用途も限定されるが、本発明の製造方法によって得られる生体適合性シリカ不織布は、繊維状であるため、例えば、ウイルス防止用マスクや空気清浄機用フィルター等、広範囲への応用が期待できる。さらに、タンパク質吸着能を有するため、ウイルスや花粉等に対する捕集効果も高いことが期待される。
【0093】
また、本発明の生体適合性シリカ不織布の製造方法は、実験室レベルの小型、かつ、簡便な装置で、容易な操作によって実行することができる。エネルギーコストも低く、装置のメインテナンスも容易である。また、高温、高圧とならないために、安全性も高く、紡績液の粘度を調整することにより、シリカ繊維の繊維径を容易に調製することも可能であり、多成分系の紡績液についても、分子レベルで均質な生体適合性シリカ不織布を製造することが可能である。
【産業上の利用可能性】
【0094】
本発明の生体適合性シリカ不織布の製造方法、及び生体適合性シリカ繊維を構成する生体適合性シリカ繊維は、フィルター、各種衛生材料、吸着剤等、広範囲の技術分野の用途に応用することが可能である。
【図面の簡単な説明】
【0095】
【図1】ゾル−ゲル法における加水分解反応及び重合反応の概念を表す図である。
【図2】実験例1において、混合溶媒の粘度を経時的に測定した結果を示す図である。
【図3】実験例2において使用した紡糸液の製造装置の概略図である。
【図4】実験例2において、混合溶媒の粘度を経時的に測定した結果を示す図である。
【図5】本発明のシリカ不織布の製造に用いた静電噴霧装置の概略構成図であり、図5(a)は静電噴射前、図5(b)は静電噴射中の状態を示す図である。
【図6】実施例1のシリカ不織布の写真であり、図6(a)はシリカ不織布全体の外観写真、図6(b)はシリカ不織布の電子顕微鏡写真である。
【図7】実施例1のシリカ不織布の外観写真であり、図7(a)、図7(b)及び図7(c)は、混合溶媒の粘度が、それぞれ150mPa・s、500mPa・s及び920mPa・sの場合のシリカ不織布の外観を示す。
【図8】実験例2のサンプル5を紡績液として、図5に示す静電噴霧装置に用いた状態を示す写真であり、図8(a)は高圧電源のスイッチがオフの状態、図8(b)は高圧電源のスイッチがオンの状態を示す。
【図9】図9(a)は実施例1のシリカ繊維の焼成前のSEM画像であり、図9(b)は焼成後のSEM画像である。
【図10】実施例1のイオン交換工程でカルシウムイオンを含む水溶液に浸漬する時間と、焼成工程後のアルブミン吸着量との関係を表すグラフである。
【図11】実施例1の焼成工程における焼成温度と、焼成工程後のアルブミン及びリゾチーム吸着量との関係を表すグラフである。
【図12】実施例1の焼成工程における昇温速度と、焼成工程後のアルブミン吸着量との関係を表すグラフである。
【図13】図13(a)は実施例2のシリカ繊維の焼成前のSEM画像であり、図13(b)は焼成後のSEM画像である。
【図14】実施例2の焼成工程における焼成温度と、焼成工程後のアルブミン及びリゾチーム吸着量との関係を表すグラフである。
【符号の説明】
【0096】
1:エアポンプ
2:バイアル
3:油浴
4:スターラ
5:フラスコ
6:冷却管
7:テトラエトキシシランを溶解させた混合溶媒
8:高圧電源
9:シリンジ
10:紡績液
11:針状電極
12:導電性捕集板
13:アース
14:シリカ繊維
15:シリカ不織布
【特許請求の範囲】
【請求項1】
アルコキシシランを、リン酸トリエチルを含む水と有機溶媒の混合溶媒に溶解させ、大気と接触させながらゾル状の紡績液とするリン分添加ゾル化工程と、
粘度調整後の前記紡績液を静電噴霧法により紡糸化するリン分添加紡糸化工程と、
前記リン分添加紡糸化工程で得られた不織布を、カルシウムイオンを含む水溶液中に浸漬することによりイオン交換させるイオン交換工程と、
前記イオン交換工程後の不織布を400℃以上1000℃以下の温度で焼成する焼成工程と、
を含む生体適合性シリカ不織布の製造方法。
【請求項2】
アルコキシシランがテトラエトキシシランであり、有機溶媒がエタノールである請求項1に記載の生体適合性シリカ不織布の製造方法。
【請求項3】
前記混合溶媒における水の量が、アルコキシシランのモル数に対して1倍以上2倍以下である請求項1又は2に記載の生体適合性シリカ不織布の製造方法。
【請求項4】
前記混合溶媒におけるリン酸トリエチルの量が、アルコキシシランのモル数の2%以上10%以下である請求項1乃至3のいずれか1項に記載の生体適合性シリカ不織布の製造方法。
【請求項5】
前記リン分添加ゾル化工程において、前記混合溶媒に塩酸を加えることを特徴とする請求項1乃至4のいずれか1項に記載の生体適合性シリカ不織布の製造方法。
【請求項6】
前記リン分添加ゾル化工程において、加湿した大気と接触させながら撹拌することを特徴とする請求項1乃至5のいずれか1項に記載の生体適合性シリカ不織布の製造方法。
【請求項7】
請求項1乃至6のいずれか1項に記載の生体適合性シリカ不織布を構成する生体適合性シリカ繊維。
【請求項8】
アルコキシシランを水と有機溶媒の混合溶媒に溶解させ、大気と接触させながらゾル状の紡績液とするゾル化工程と、
粘度調整後の前記紡績液を、静電噴霧法により紡糸化する紡糸化工程と、
前記紡糸化工程で得られた不織布を、リン酸に浸漬して加熱するリン酸化工程と、
前記リン酸化工程後の不織布を、カルシウムイオンを含む水溶液中に浸漬することによりイオン交換させるイオン交換工程と、
前記イオン交換工程後の不織布を400℃以上1000℃以下の温度で焼成する焼成工程と、
を含む生体適合性シリカ不織布の製造方法。
【請求項9】
前記アルコキシシランがテトラエトキシシランであり、前記有機溶媒がエタノールであることを特徴とする請求項8に記載の生体適合性シリカ不織布の製造方法。
【請求項10】
前記混合溶媒における水の量が、アルコキシシランのモル数に対して1倍以上2倍以下であることを特徴とする請求項8又は9に記載の生体適合性シリカ不織布の製造方法。
【請求項11】
前記ゾル化工程において、前記混合溶媒に塩酸を加えることを特徴とする請求項8乃至10のいずれか1項に記載の生体適合性シリカ不織布の製造方法。
【請求項12】
前記ゾル化工程において、加湿した大気と接触させながら撹拌することを特徴とする請求項8乃至11のいずれか1項に記載の生体適合性シリカ不織布の製造方法。
【請求項13】
請求項8乃至12のいずれか1項に記載の生体適合性シリカ不織布を構成する生体適合性シリカ繊維。
【請求項1】
アルコキシシランを、リン酸トリエチルを含む水と有機溶媒の混合溶媒に溶解させ、大気と接触させながらゾル状の紡績液とするリン分添加ゾル化工程と、
粘度調整後の前記紡績液を静電噴霧法により紡糸化するリン分添加紡糸化工程と、
前記リン分添加紡糸化工程で得られた不織布を、カルシウムイオンを含む水溶液中に浸漬することによりイオン交換させるイオン交換工程と、
前記イオン交換工程後の不織布を400℃以上1000℃以下の温度で焼成する焼成工程と、
を含む生体適合性シリカ不織布の製造方法。
【請求項2】
アルコキシシランがテトラエトキシシランであり、有機溶媒がエタノールである請求項1に記載の生体適合性シリカ不織布の製造方法。
【請求項3】
前記混合溶媒における水の量が、アルコキシシランのモル数に対して1倍以上2倍以下である請求項1又は2に記載の生体適合性シリカ不織布の製造方法。
【請求項4】
前記混合溶媒におけるリン酸トリエチルの量が、アルコキシシランのモル数の2%以上10%以下である請求項1乃至3のいずれか1項に記載の生体適合性シリカ不織布の製造方法。
【請求項5】
前記リン分添加ゾル化工程において、前記混合溶媒に塩酸を加えることを特徴とする請求項1乃至4のいずれか1項に記載の生体適合性シリカ不織布の製造方法。
【請求項6】
前記リン分添加ゾル化工程において、加湿した大気と接触させながら撹拌することを特徴とする請求項1乃至5のいずれか1項に記載の生体適合性シリカ不織布の製造方法。
【請求項7】
請求項1乃至6のいずれか1項に記載の生体適合性シリカ不織布を構成する生体適合性シリカ繊維。
【請求項8】
アルコキシシランを水と有機溶媒の混合溶媒に溶解させ、大気と接触させながらゾル状の紡績液とするゾル化工程と、
粘度調整後の前記紡績液を、静電噴霧法により紡糸化する紡糸化工程と、
前記紡糸化工程で得られた不織布を、リン酸に浸漬して加熱するリン酸化工程と、
前記リン酸化工程後の不織布を、カルシウムイオンを含む水溶液中に浸漬することによりイオン交換させるイオン交換工程と、
前記イオン交換工程後の不織布を400℃以上1000℃以下の温度で焼成する焼成工程と、
を含む生体適合性シリカ不織布の製造方法。
【請求項9】
前記アルコキシシランがテトラエトキシシランであり、前記有機溶媒がエタノールであることを特徴とする請求項8に記載の生体適合性シリカ不織布の製造方法。
【請求項10】
前記混合溶媒における水の量が、アルコキシシランのモル数に対して1倍以上2倍以下であることを特徴とする請求項8又は9に記載の生体適合性シリカ不織布の製造方法。
【請求項11】
前記ゾル化工程において、前記混合溶媒に塩酸を加えることを特徴とする請求項8乃至10のいずれか1項に記載の生体適合性シリカ不織布の製造方法。
【請求項12】
前記ゾル化工程において、加湿した大気と接触させながら撹拌することを特徴とする請求項8乃至11のいずれか1項に記載の生体適合性シリカ不織布の製造方法。
【請求項13】
請求項8乃至12のいずれか1項に記載の生体適合性シリカ不織布を構成する生体適合性シリカ繊維。
【図1】

【図2】
【図3】
【図4】
【図5】
【図10】
【図11】
【図12】
【図14】
【図6】
【図7】
【図8】
【図9】

【図13】
【図2】
【図3】
【図4】
【図5】
【図10】
【図11】
【図12】
【図14】
【図6】
【図7】
【図8】
【図9】
【図13】
【公開番号】特開2008−174872(P2008−174872A)
【公開日】平成20年7月31日(2008.7.31)
【国際特許分類】
【出願番号】特願2007−10058(P2007−10058)
【出願日】平成19年1月19日(2007.1.19)
【出願人】(592216384)兵庫県 (258)
【Fターム(参考)】
【公開日】平成20年7月31日(2008.7.31)
【国際特許分類】
【出願日】平成19年1月19日(2007.1.19)
【出願人】(592216384)兵庫県 (258)
【Fターム(参考)】
[ Back to top ]