
非内在性の構成的に活性化されるヒトGタンパク質共役型受容体
【課題】受容体は、内在性リガンドもしくは薬物のような化合物により活性状態で安定化させることができ、リガンドに依存しない手段による構成的受容体活性化した受容体を提供する。
【解決手段】hARE−3(F313K)等の各種ヒトオーファン受容体を含む、非内在性で、構成的に活性化される変種ヒトGタンパク質共役型受容体(GPCR)をコードするcDNAとベクター、該GPCR蛋白質、およびそれらを含む宿主細胞。
【解決手段】hARE−3(F313K)等の各種ヒトオーファン受容体を含む、非内在性で、構成的に活性化される変種ヒトGタンパク質共役型受容体(GPCR)をコードするcDNAとベクター、該GPCR蛋白質、およびそれらを含む宿主細胞。
【発明の詳細な説明】
【技術分野】
【0001】
本特許出願は、1998年10月13日に米国特許・商標庁に出願された米国
第09/170,496号の一部継続出願であり、かつそれからの優先権を主張
する。本出願は、以下の仮出願(全部は示された日付に米国特許・商標庁に米国
エクスプレスメール(U.S.Express Mail)を介して出願された
)、すなわち1998年11月27日に出願された米国仮出願第60/110,
060号;1999年2月16日に出願された米国仮出願第60/120,41
6号;1998年11月20日に出願された米国仮出願第60/109,213
号の利益を主張する1999年2月26日に出願された米国仮出願第60/12
1,852号;1999年3月12日に出願された米国仮出願第60/123,
944号;1999年3月12日に出願された米国仮出願第60/123,94
5号;1999年3月12日に出願された米国仮出願第60/123,948号
;1999年3月12日に出願された米国仮出願第60/123,951号;1
999年3月12日に出願された米国仮出願第60/123,946号;199
9年3月12日に出願された米国仮出願第60/123,949号;1999年
8月27日に出願された米国仮出願第60/151,114号および1998年
11月12日に出願された米国仮出願第60/108,029号の利益を主張す
る1999年9月3日に出願された米国仮出願第60/152,524号;19
99年5月28日に出願された米国仮出願第60/136,436号;1999
年5月28日に出願された米国仮出願第60/136,439号;1999年5
月28日に出願された米国仮出願第60/136,567号;1999年5月2
8日に出願された米国仮出願第60/137,127号;1999年5月28日
に出願された米国仮出願第60/137,131号;1999年5月28日に出
願された米国仮出願第60/136,437号の利益を主張する1999年6月
29日に出願された米国仮出願第60/141,448号;1999年9月29
日に出願された米国仮出願第60/156,633号;1999年9月29日に
出願された米国仮出願第60/156,555号;1999年9月29日に出願
された米国仮出願第60/156,634号;1999年9月29日に出願され
た米国仮出願第____号(アリーナ ファーマシューティカルズ インク(A
rena Pharmaceuticals,Inc.)処理予定表番号:CH
N10−1);1999年10月1日に出願された米国仮出願第____号(ア
リーナ ファーマシューティカルズ インク(Arena Pharmaceu
ticals,Inc.)処理予定表番号:RUP6−1);1999年10月
1日に出願された米国仮出願第____号(アリーナ ファーマシューティカル
ズ インク(Arena Pharmaceuticals,Inc.)処理予
定表番号:RUP7−1);1999年10月1日に出願された米国仮出願第_
___号(アリーナ ファーマシューティカルズ インク(Arena Pha
rmaceuticals,Inc.)処理予定表番号:CHN6−1);19
99年10月1日に出願された米国仮出願第____号(アリーナ ファーマシ
ューティカルズ インク(Arena Pharmaceuticals,In
c.)処理予定表番号:RUP5−1);ならびに1999年10月1日に出願
された米国仮出願第____号(アリーナ ファーマシューティカルズ インク
(Arena Pharmaceuticals,Inc.)処理予定表番号:
CHN9−1)からの優先権の利益もまた主張する。本出願は、1999年10
月12日に(米国エクスプレスメール(U.S.Express Mail)を
介して)出願された同時係属中の米国第____号(ウッドコック(Woodc
ock)、ウォッシュバーン(Washburn)、クルツ(Kurtz)、マ
キエヴィッツ(Makiewicz)とノリス(Norris))、LLP処理
予定表番号AREN−0050)および1999年7月30日に出願された米国
第09/364,425号(双方は引用により本明細書に組み込まれる)にもま
た関する。本出願は、1999年10月12日に(米国エクスプレスメール(U
.S.Express Mail)を介して)出願された米国第____号(ウ
ッドコック(Woodcock)、ウォッシュバーン(Washburn)、ク
ルツ(Kurtz)、マキエヴィッツ(Makiewicz)とノリス(Nor
ris))、LLP処理予定表番号AREN−0054)(そっくりそのまま引
用により本明細書に組み込まれる)に対する優先権もまた主張する。前述の出願
のそれぞれはそっくりそのまま引用により本明細書に組み込まれる。
【0002】
(発明の分野)
本特許明細書に開示される発明は、膜貫通受容体、およびより具体的にはヒト
Gタンパク質共役型受容体、そしてとりわけ該受容体の構成的活性を確立するも
しくは高めるよう改変されているGPCRに関する。好ましくは、改変されたG
PCRは、治療薬として潜在的応用性を有する受容体のアゴニスト、反作用薬も
しくは部分的アゴニストとしての候補化合物の直接の同定に使用される。
【背景技術】
【0003】
(発明の背景)
ヒトでは多数の受容体のクラスが存在するが、はるかに最も豊富かつ治療に関
係するものは、Gタンパク質共役型受容体(GPCRもしくは複数GPCR)の
クラスにより代表される。ヒトゲノム内には数十万個の遺伝子が存在することが
推定されており、そしてこれらのなかでおよそ2%もしくは2,000個の遺伝
子がGPCRをコードすると推定されている。内在性リガンドが同定されている
、GPCRを包含する受容体は「既知」受容体と称される一方、内在性リガンド
が同定されていない受容体は「オーファン」受容体と称される。GPCRは製薬
学的製品の開発に重要な一領域を代表する。すなわち、100種の既知のGPC
Rのおよそ20種から全処方薬の60%が開発されている。
【0004】
GPCRは1つの共通な構造モチーフを共有する。全部のこれらの受容体は、
7個のαヘリックスを形成する22ないし24個の間の疎水性アミノ酸の7個の
連なりを有し、そのそれぞれは膜にまたがる(各広がり(span)は数字により同定
されている。すなわち膜貫通−1(TM−1)、膜貫通−2(TM−2)など)
。膜貫通ヘリックスは、細胞膜の外もしくは「細胞外」側で、膜貫通−2と膜貫
通−3、膜貫通−4と膜貫通−5、および膜貫通−6と膜貫通−7の間でアミノ
酸の鎖により結合されている(これらはそれぞれ「細胞外」領域1、2および3
(EC−1、EC−2およびEC−3)と称される)。膜貫通ヘリックスは、細
胞膜の内もしくは「細胞内」側で、膜貫通−1と膜貫通−2、膜貫通−3と膜貫
通−4、および膜貫通−5と膜貫通−6の間でもまたアミノ酸の鎖により結合さ
れている(これらはそれぞれ「細胞内」領域1、2および3(IC−1、IC−
2およびIC−3)と称される)。受容体の「カルボキシ」(「C」)末端は細
胞内の細胞内空隙中に存し、また、受容体の「アミノ」(「N」)末端は細胞の
外側の細胞外空隙中に存する。
【0005】
一般に、内在性リガンドが受容体と結合する場合(しばしば受容体の「活性化
」と称される)、細胞内領域のコンホメーションの変化が存在し、それは細胞内
領域と細胞内の「Gタンパク質」との間の共役(coupling)を見込む。GPCRは
Gタンパク質に関して「混雑して」いる、すなわちGPCRは1種以上のGタン
パク質と相互作用する可能性があることが報告されている。ケナキン(Kena
kin,T.)、43 Life Sciences 1095(1988)を
参照されたい。他のGタンパク質が存在するが、現在のところGq、Gs、Gi
、GzおよびGoが同定されたGタンパク質である。Gタンパク質との内在性リ
ガンドで活性化されたGPCRの共役がシグナル伝達カスケード過程(「シグナ
ル伝達」と称される)を開始する。正常な条件下では、シグナル伝達は最終的に
細胞の活性化もしくは細胞の阻害をもたらす。受容体のIC−3ループならびに
カルボキシ末端がGタンパク質と相互作用すると考えられている。
【0006】
GPCRは、生理学的条件下では2種の異なるコンホメーション、すなわち「
不活性」状態と「活性状態」との間の平衡で細胞膜中に存在する。不活性状態の
受容体は、細胞内のシグナル伝達経路に連結して生物学的応答を生じさせること
が不可能である。受容体のコンホメーションの活性状態への変化は(Gタンパク
質を介する)伝達経路への連鎖を可能にし、そして生物学的応答を生じさせる。
【0007】
受容体は、内在性リガンドもしくは薬物のような化合物により活性状態で安定
化させることができる。独占的に限定されるものでないが受容体のアミノ酸配列
に対する改変を挙げることができる最近の発見は、活性状態のコンホメーション
にある受容体を助長かつ安定化するための内在性リガンドもしくは薬物以外の手
段を提供する。これらの手段は、受容体への内在性リガンドの結合の効果を刺激
することにより、受容体を活性状態で効果的に安定化する。こうしたリガンドに
依存しない手段による安定化を「構成的受容体活性化」と命名する。
【発明の概要】
【課題を解決するための手段】
【0008】
(発明の要約)
内在性ヒトGPCRの非内在性変種およびそれらの用途を本明細書で開示する
。
本発明は、例えば、以下を提供する:
(項目1) hARE−3(F313K)を含んで成る、非内在性で、構
成的に活性化される変種ヒトGタンパク質共役型受容体をコードするcDNA。
(項目2) 項目1記載のcDNAによりコードされる、非内在性の変
種ヒトGタンパク質共役型受容体。
(項目3) ベクターおよび項目1記載のcDNAを含んで成るプラス
ミド。
(項目4) 項目3記載のプラスミドを含んで成る宿主細胞。
(項目5) hARE−4(V233K)を含んで成る、非内在性で、構
成的に活性化される変種ヒトGタンパク質共役型受容体をコードするcDNA。
(項目6) 項目5記載のcDNAによりコードされる、非内在性の変
種ヒトGタンパク質共役型受容体。
(項目7) ベクターおよび項目5記載のcDNAを含んで成るプラス
ミド。
(項目8) 項目7記載のプラスミドを含んで成る宿主細胞。
(項目9) hARE−5(A240K)を含んで成る、非内在性で、構
成的に活性化される変種ヒトGタンパク質共役型受容体をコードするcDNA。
(項目10) 項目9記載のcDNAによりコードされる、非内在性の
変種ヒトGタンパク質共役型受容体。
(項目11) ベクターおよび項目5記載のcDNAを含んで成るプラ
スミド。
(項目12) 項目11記載のプラスミドを含んで成る宿主細胞。
(項目13) hGPCR14(L257K)を含んで成る、非内在性で
、構成的に活性化される変種ヒトGタンパク質共役型受容体をコードするcDN
A。
(項目14) 項目13記載のcDNAによりコードされる、非内在性
の変種ヒトGタンパク質共役型受容体。
(項目15) ベクターおよび項目13記載のcDNAを含んで成るプ
ラスミド。
(項目16) 項目15記載のプラスミドを含んで成る宿主細胞。
(項目17) hGPCR27(C283K)を含んで成る、非内在性で
、構成的に活性化される変種ヒトGタンパク質共役型受容体をコードするcDN
A。
(項目18) 項目17記載のcDNAによりコードされる、非内在性
の変種ヒトGタンパク質共役型受容体。
(項目19) ベクターおよび項目17記載のcDNAを含んで成るプ
ラスミド。
(項目20) 項目19記載のプラスミドを含んで成る宿主細胞。
(項目21) hARE−1(E232K)を含んで成る、非内在性で、
構成的に活性化される変種ヒトGタンパク質共役型受容体をコードするcDNA
。
(項目22) 項目21記載のcDNAによりコードされる、非内在性
の変種ヒトGタンパク質共役型受容体。
(項目23) ベクターおよび項目21記載のcDNAを含んで成るプ
ラスミド。
(項目24) 項目23記載のプラスミドを含んで成る宿主細胞。
(項目25) hARE−2(G285K)を含んで成る、非内在性で、
構成的に活性化される変種ヒトGタンパク質共役型受容体をコードするcDNA
。
(項目26) 項目25記載のcDNAによりコードされる、非内在性
の変種ヒトGタンパク質共役型受容体。
(項目27) ベクターおよび項目25記載のcDNAを含んで成るプ
ラスミド。
(項目28) 項目27記載のプラスミドを含んで成る宿主細胞。
(項目29) hPPR1(L239K)を含んで成る、非内在性で、構
成的に活性化される変種ヒトGタンパク質共役型受容体をコードするcDNA。
(項目30) 項目29記載のcDNAによりコードされる、非内在性
の変種ヒトGタンパク質共役型受容体。
(項目31) ベクターおよび項目29記載のcDNAを含んで成るプ
ラスミド。
(項目32) 項目31記載のプラスミドを含んで成る宿主細胞。
(項目33) hG2A(K232A)を含んで成る、非内在性で、構成
的に活性化される変種ヒトGタンパク質共役型受容体をコードするcDNA。
(項目34) 項目33記載のcDNAによりコードされる、非内在性
の変種ヒトGタンパク質共役型受容体。
(項目35) ベクターおよび項目33記載のcDNAを含んで成るプ
ラスミド。
(項目36) 項目35記載のプラスミドを含んで成る宿主細胞。
(項目37) hRUP3(L224K)を含んで成る、非内在性で、構
成的に活性化される変種ヒトGタンパク質共役型受容体をコードするcDNA。
(項目38) 項目37記載のcDNAによりコードされる、非内在性
の変種ヒトGタンパク質共役型受容体。
(項目39) ベクターおよび項目37記載のcDNAを含んで成るプ
ラスミド。
(項目40) 項目39記載のプラスミドを含んで成る宿主細胞。
(項目41) hRUP5(A236K)を含んで成る、非内在性で、構
成的に活性化される変種ヒトGタンパク質共役型受容体をコードするcDNA。
(項目42) 項目41記載のcDNAによりコードされる、非内在性
の変種ヒトGタンパク質共役型受容体。
(項目43) ベクターおよび項目41記載のcDNAを含んで成るプ
ラスミド。
(項目44) 項目42記載のプラスミドを含んで成る宿主細胞。
(項目45) hRUP6(N267K)を含んで成る、非内在性で、構
成的に活性化される変種ヒトGタンパク質共役型受容体をコードするcDNA。
(項目46) 項目45記載のcDNAによりコードされる、非内在性
の変種ヒトGタンパク質共役型受容体。
(項目47) ベクターおよび項目45記載のcDNAを含んで成るプ
ラスミド。
(項目48) 項目47記載のプラスミドを含んで成る宿主細胞。
(項目49) hRUP7(A302K)を含んで成る、非内在性で、構
成的に活性化される変種ヒトGタンパク質共役型受容体をコードするcDNA。
(項目50) 項目49記載のcDNAによりコードされる、非内在性
の変種ヒトGタンパク質共役型受容体。
(項目51) ベクターおよび項目49記載のcDNAを含んで成るプ
ラスミド。
(項目52) 項目51記載のプラスミドを含んで成る宿主細胞。
(項目53) hCHN4(V236K)を含んで成る、非内在性で、構
成的に活性化される変種ヒトGタンパク質共役型受容体をコードするcDNA。
(項目54) 項目53記載のcDNAによりコードされる、非内在性
の変種ヒトGタンパク質共役型受容体。
(項目55) ベクターおよび項目53記載のcDNAを含んで成るプ
ラスミド。
(項目56) 項目55記載のプラスミドを含んで成る宿主細胞。
(項目57) hMC4(A244K)を含んで成る、非内在性で、構成
的に活性化される変種ヒトGタンパク質共役型受容体をコードするcDNA。
(項目58) 項目57記載のcDNAによりコードされる、非内在性
の変種ヒトGタンパク質共役型受容体。
(項目59) ベクターおよび項目57記載のcDNAを含んで成るプ
ラスミド。
(項目60) 項目60記載のプラスミドを含んで成る宿主細胞。
(項目61) hCHN3(S284K)を含んで成る、非内在性で、構
成的に活性化される変種ヒトGタンパク質共役型受容体をコードするcDNA。
(項目62) 項目61記載のcDNAによりコードされる、非内在性
の変種ヒトGタンパク質共役型受容体。
(項目63) ベクターおよび項目61記載のcDNAを含んで成るプ
ラスミド。
(項目64) 項目63記載のプラスミドを含んで成る宿主細胞。
(項目65) hCHN6(L352K)を含んで成る、非内在性で、構
成的に活性化される変種ヒトGタンパク質共役型受容体をコードするcDNA。
(項目66) 項目65記載のcDNAによりコードされる、非内在性
の変種ヒトGタンパク質共役型受容体。
(項目67) ベクターおよび項目65記載のcDNAを含んで成るプ
ラスミド。
(項目68) 項目67記載のプラスミドを含んで成る宿主細胞。
(項目69) hCHN8(N235K)を含んで成る、非内在性で、構
成的に活性化される変種ヒトGタンパク質共役型受容体をコードするcDNA。
(項目70) 項目69記載のcDNAによりコードされる、非内在性
の変種ヒトGタンパク質共役型受容体。
(項目71) ベクターおよび項目69記載のcDNAを含んで成るプ
ラスミド。
(項目72) 項目71記載のプラスミドを含んで成る宿主細胞。
(項目73) hH9(F236K)を含んで成る、非内在性で、構成的
に活性化される変種ヒトGタンパク質共役型受容体をコードするcDNA。
(項目74) 項目73記載のcDNAによりコードされる、非内在性
の変種ヒトGタンパク質共役型受容体。
(項目75) ベクターおよび項目73記載のcDNAを含んで成るプ
ラスミド。
(項目76) 項目74記載のプラスミドを含んで成る宿主細胞。
(項目77) hAT1(F239K);hAT1(N111A);hA
T1(AT2K255IC3);およびhAT1(A243+)より成る群から
選択される、非内在性で、構成的に活性化される変種ヒトGタンパク質共役型A
T1受容体をコードするcDNA。
(項目78) 項目77記載のcDNAによりコードされる、非内在性
の変種ヒトGタンパク質共役型受容体。
(項目79) ベクターおよび項目77記載のcDNAを含んで成るプ
ラスミド。
(項目80) 項目79記載のプラスミドを含んで成る宿主細胞。
【図面の簡単な説明】
【0009】
【図1】発明にかかる、8XCRE−Lucレポータープラスミドの表示である(実施例4(c)3を参照されたい)。
【図2】発明にかかる、内在性TDAG8へのATPおよびADPの結合の結果のグラフ表示(2A)ならびに血清および血清を含まない培地中での比較(2B)である。
【図3】発明にかかる、GPCR融合タンパク質H9(F236K):Gsαに対するCMVの比較的なシグナル伝達の結果のグラフ表示である。
【発明を実施するための形態】
【0010】
(詳細な記述)
受容体を中核として発展してきた学術文献は、受容体に対する多様な影響を有
するリガンドを指す多数の用語を採用している。明瞭化および一貫性のため、本
特許明細書を通じて以下の定義を使用するであろう。これらの定義がこれらの用
語の他の定義と矛盾する限り、以下の定義が支配する:
アゴニストは、それらが受容体に結合する場合に細胞内応答を活性化する、も
しくは膜へのGTP結合を高める物質(例えばリガンド、候補化合物)を意味す
る。
【0011】
本明細書で使用されるアミノ酸の略語を表Aに示す:
【0012】
【表1】

【0013】
部分的アゴニストは、それらが受容体に結合する場合にアゴニストが活性化す
るより小さい度合(degree)/程度(extent)まで細胞内応答を活性化する、もしく
はアゴニストが高めるより小さい度合(degree)/程度(extent)まで膜へのGTP
結合を高める物質(例えばリガンド、候補化合物)を意味する。
【0014】
アンタゴニストは、アゴニストと同一の部位で受容体に競争的に結合するがし
かし活性型の受容体により開始される細胞内応答を活性化せず、そしてそれによ
りアゴニストもしくは部分的アゴニストによる細胞内応答を阻害する可能性のあ
る物質(例えばリガンド、候補化合物)を意味する。アンタゴニストは、アゴニ
ストもしくは部分的アゴニストの非存在下で基線の細胞内応答を減少させない。
【0015】
候補化合物は、スクリーニング技術に基づいて分析できる分子(例えば、そし
て制限でなく化合物)を意味する。好ましくは、「候補化合物」という句は、間
接的同定方法により既に決定されたような、受容体に対する反作用薬、アゴニス
トもしくはアンタゴニストより成る群から選択される化合物であることが公知で
あった化合物(「間接的に同定された化合物」)を包含せず;より好ましくは、
最低1種の哺乳動物で治療上の効能を有することが既に決定されている間接的に
同定された化合物を包含せず;そして最も好ましくは、ヒトで治療上の有用性を
有することが既に決定されている間接的に同定された化合物を包含しない。
【0016】
組成物は最低1種の成分を含んで成る物質を意味し;「製薬学的組成物」は組
成物の一例である。
【0017】
化合物の効能は、受容体結合親和性と対照的に、受容体の機能性を阻害もしく
は刺激する化合物の能力の大きさを意味する。化合物の効能の検出の例示的手段
を本特許明細書の実施例のセクションに開示する。
【0018】
コドンは、リン酸基に結合されたヌクレオシド(アデノシン(A)、グアノシ
ン(G)、シチジン(C)、ウリジン(U)およびチミジン(T))を一般に含
んで成りかつ翻訳される場合に1個のアミノ酸をコードするひとそろいの3個の
ヌクレオチド(もしくはヌクレオチドに対する同等物)を意味する。
【0019】
構成的に活性化される受容体は構成的受容体活性化にさらされる受容体を意味
する。構成的に活性化される受容体は内在性もしくは非内在性であることができ
る。
【0020】
構成的な受容体の活性化は、その内在性リガンドもしくはその化学的同等物と
の受容体の結合以外の手段による、活性状態での受容体の安定化を意味する。
【0021】
接触させるもしくは接触することは、インビトロの系であろうとインビボの系
であろうと最低2つの部分を一緒にすることを意味する。
【0022】
直接同定するもしくは直接同定されるは、「候補化合物」という句に関係して
、構成的に活性化される受容体、好ましくは構成的に活性化されるオーファン受
容体、そして最も好ましくは構成的に活性化されるGタンパク質共役型の細胞表
面のオーファン受容体に対する候補化合物のスクリーニング、およびこうした化
合物の化合物の効能の評価を意味する。この句は、どの状況下でも、「間接的に
同定する」もしくは「間接的に同定される」という句により包含されるもしくは
それを包含すると解釈もしくは理解されるべきでない。
【0023】
内在性は哺乳動物が天然に産生する物質を意味する。例えば、そして制限でな
く「受容体」という用語に関しての内在性は、哺乳動物(例えば、そして制限で
なくヒト)もしくはウイルスにより天然に産生されるものを意味する。対照的に
、これに関して非内在性という用語は、哺乳動物(例えば、そして制限でなくヒ
ト)もしくはウイルスにより天然に産生されないものを意味する。例えば、そし
て制限でなく、その内在性の形態で構成的に活性でないがしかし操作される場合
に構成的に活性になる受容体は、本明細書で「非内在性の構成的に活性化される
受容体」と最も好ましく称される。双方の用語は「インビボ」および「インビト
ロ」双方の系を記述するのに利用することができる。例えば、そして制限でなく
、スクリーニングのアプローチにおいて、内在性もしくは非内在性の受容体はイ
ンビトロのスクリーニング系に関してであってよい。さらなる一例として、そし
て制限としてでなく、非内在性の構成的に活性化される受容体を包含するように
哺乳動物のゲノムが操作されている場合には、インビボ系による候補化合物のス
クリーニングが実現可能である。
【0024】
Gタンパク質共役型受容体融合タンパク質およびGPCR融合タンパク質は、
本明細書に開示される本発明に関して、それぞれ、最低1種のGタンパク質、最
も好ましくはこうしたGタンパク質のαサブユニット(これはGTPを結合する
サブユニットである)に融合された、内在性の構成的に活性化されるGPCRも
しくは非内在性の構成的に活性化されるGPCRを含んで成る非内在性タンパク
質を意味し、Gタンパク質は好ましくは内在性のオーファンGPCRと天然に共
役するGタンパク質と同一の型のものである。例えば、そして制限でなく、内在
性の状態において、Gタンパク質「Gsα」がGPCRと共役する主なGタンパ
ク質である場合、特定のGPCRに基づくGPCR融合タンパク質はGsαに融
合されたGPCRを含んで成る非内在性タンパク質であることができ;下に示さ
れるであろうようないくつかの状況では、主なものでないGタンパク質をGPC
Rに融合することができる。Gタンパク質は構成的に活性なGPCRのC末端に
直接融合することができるか、もしくは2者の間にスペーサーが存在してよい。
【0025】
宿主細胞は、その中に組み込まれたプラスミドおよび/もしくはベクターを有
することが可能な細胞を意味する。原核生物宿主細胞の場合には、プラスミドは
宿主細胞の複製物のような自律的分子として典型的に複製され(一般に、プラス
ミドはその後真核生物宿主細胞への導入のため単離される);真核生物宿主細胞
の場合には、真核生物宿主細胞が複製する場合にプラスミドが複製するように、
プラスミドが宿主細胞の細胞DNAに組み込まれる。好ましくは、本明細書に開
示される本発明の目的上、宿主細胞は真核生物、より好ましくは哺乳動物であり
、そして最も好ましくは293、293TおよびCOS−7細胞より成る群から
選択される。
【0026】
間接的に同定するもしくは間接的に同定されるは、内在性受容体に特異的な内
在性リガンドの同定、リガンド−受容体の相互作用を妨害および/もしくはそれ
と競争するものの決定のための受容体に対する候補化合物のスクリーニング、な
らびに活性化された受容体に関連する最低1種のセカンドメッセンジャー経路に
影響を及ぼす化合物の効能の評価を必要とする、薬物発見過程への伝統的アプロ
ーチを意味する。
【0027】
「応答」という用語に関係した阻害するもしくは阻害は、化合物の非存在下と
対照的に、化合物の存在下で応答が低下もしくは予防されることを意味する。
【0028】
反作用薬は、内在性の形態の受容体もしくは構成的に活性化された形態の受容
体のいずれかに結合し、かつ、アゴニストもしくは部分的アゴニストの非存在下
で観察される正常の基準レベルの活性より低い、活性の形態の受容体により開始
される基線の細胞内応答を阻害するか、もしくは膜へのGTP結合を減少させる
物質(例えばリガンド、候補化合物)を意味する。好ましくは、基線の細胞内応
答は、反作用薬の非存在下の基礎の応答に比較して、反作用薬の存在下で最低3
0%、より好ましくは最低50%、そして最も好ましくは最低75%阻害される
。
【0029】
既知の受容体は、その受容体に特異的な内在性リガンドが同定されている内在
性受容体を意味する。
【0030】
リガンドは、内在性の天然に存在する受容体に特異的な内在性の天然に存在す
る分子を意味する。
【0031】
内在性受容体の核酸および/もしくはアミノ酸の配列に関しての突然変異体も
しくは突然変異は、突然変異された形態の内在性の構成的に活性化されない受容
体が該受容体の構成的活性化を明示するような、こうした内在性の配列に対する
指定された1個の変化もしくは複数の変化を意味する。特定の配列に対する同等
物に関して、(a)その後の突然変異された形態のヒト受容体の構成的活性化の
レベルが該受容体の第一の突然変異により明示されるものと実質的に同一であり
;そして(b)その後の突然変異された形態の受容体と該受容体の第一の突然変
異との間の配列(アミノ酸および/もしくは核酸)の相同性のパーセントが最低
約80%、より好ましくは最低約90%、そして最も好ましくは最低95%であ
る場合、その後の突然変異された形態のヒト受容体は該ヒト受容体の第一の突然
変異に同等であるとみなされる。理想的には、また、構成的活性化を達成するた
めの本明細書に開示される最も好ましいカセットはGPCRの内在性の形態と非
内在性の形態との間の単一のアミノ酸および/もしくはコドンの変化を包含する
という事実のために、配列の相同性のパーセントは最低98%であるべきである
。
【0032】
非オーファン受容体は、内在性の天然に存在するリガンドに特異的な内在性の
天然に存在する分子を意味し、ここで、受容体へのリガンドの結合が細胞内シグ
ナル伝達経路を活性化する。
【0033】
オーファン受容体は、その受容体に特異的な内在性リガンドが同定されていな
いかもしくは未知である内在性受容体を意味する。
【0034】
製薬学的組成物は最低1種の有効成分を含んで成る組成物を意味し、それによ
り該組成物は哺乳動物(例えば、そして制限でなくヒト)における特定の効能の
ある結果についての検討に基づいて分析できる。当業者は、ある有効成分が当業
者のニーズに基づき所望の効能のある結果を有するかどうかを決定するのに適切
な技術を理解かつ認識するであろう。
【0035】
プラスミドはベクターおよびcDNAの組み合わせを意味する。一般に、プラ
スミドは、cDNAの複製および/もしくはそのタンパク質としての発現の目的
上、宿主細胞に導入される。
【0036】
「応答」という用語に関係した刺激するもしくは刺激することは、化合物の非
存在下と対照的に化合物の存在下で応答が増大されることを意味する。
【0037】
cDNAに関してのベクターは、最低1個のcDNAを組み込むことが可能か
つ宿主細胞への組み込みが可能な環状DNAを意味する。
【0038】
以下のセクションの階層は表象的な効能について示され、また、後に続く開示
もしくは請求の範囲に対する制限として意図されず、またそのように解釈される
べきでもない。
A.緒言
受容体の伝統的な研究は、発見が受容体に影響を及ぼす可能性のあるアンタゴ
ニストおよび他の分子を見出すよう進めることができる前に、内在性リガンドを
最初に同定しなければならないという(歴史的に基づく)演繹的仮定から常に進
められてきた。アンタゴニストを最初に知ることができた場合であっても、内在
性リガンドを探すように研究が直ちに拡大されてきた。この思考様式は、構成的
に活性化される受容体の発見の後でさえ、受容体の研究で持続してきた。これま
で認識されていなかったことは、受容体のアゴニスト、部分的アゴニストおよび
反作用薬の発見に最も有用であるのは活性状態の受容体であるということである
。過度に活性の受容体もしくは過小活性の受容体から生じる疾患に対して、治療
薬で望まれるものは、必ずしも内在性リガンドに対するアンタゴニストである薬
物でなく、それぞれ、受容体の活性状態を低下させるもしくは受容体の活性を高
めるよう作用する化合物である。これは、活性の受容体状態の活性を低下させる
もしくは高める化合物は内在性リガンドと同一の部位で結合する必要がないため
である。従って、本発明の方法により教示されるとおり、治療的化合物について
のいかなる研究も、リガンドに依存しない活性状態に対して化合物をスクリーニ
ングすることにより開始すべきである。
B.ヒトGPCRの同定
ヒトゲノムプロジェクトの努力の成果は、ヒトゲノム内に配置されている核酸
配列に関する夥しい量の情報の同定につながり;それは、いずれかの特定のゲノ
ム配列がヒトタンパク質を翻訳する読取り枠情報を含有するもしくは含有するか
も知れないかどうかに関する理解もしくは認識を伴わずに、遺伝子配列情報が利
用可能にされているというこの努力において真実である。ヒトゲノム内の核酸配
列のいくつかの同定方法は当業者の範囲内にある。例えば、そして制限でなく、
本明細書で開示される多様なヒトGPCRは、ジェンバンク[GenBank]
(商標)データベースを再検討することにより発見された一方、他のGPCRは
、既に配列決定されたGPCRの核酸配列を利用してESTデータベースのBL
AST(商標)検索を実施することにより発見された。下の表Bは、GPCRの
それぞれの相同受容体と一緒に、われわれが発見した数種の内在性GPCRを列
挙する。
【0039】
【表2】
【0040】
受容体の相同性は、人体内での受容体の役割の正しい認識を得ることに関して
有用である。本特許明細書が進めるように、われわれは、これらの受容体の非内
在性で構成的に活性化される変種を確立するようにこれらの受容体を突然変異す
るための技術を開示するであろう。
【0041】
本明細書に開示される技術は、本特許明細書が進行するに従って明らかになる
であろうように、技術既知の他のヒトのオーファンGPCRにもまた適用されて
いる。
C.受容体スクリーニング
本明細書に開示される、非内在性で、構成的に活性化される変種ヒトGPCR
に対する候補化合物のスクリーニングは、受容体の内在性リガンドの使用を必要
とすることなく、この細胞表面受容体で作用する候補化合物の直接の同定を許す
。本明細書に開示される内在性の変種ヒトGPCRが発現および/もしくは過剰
発現されている身体内の領域を決定することにより、受容体の発現および/もし
くは過剰発現を伴う関連疾患/障害状態を決定することが可能であり;こうした
一アプローチを本特許明細書で開示する。
【0042】
本明細書に開示されるヒトGPCRの構成的活性化を明示することができる突
然変異の創製に関して、GPCRのTM6内に配置されると想定されているプロ
リン残基からの距離に基づき;このアルゴリズム技術は、引用により本明細書に
組み込まれる同時係属中かつ共通に譲渡される特許明細書米国第09/170,
496号に開示されている。該アルゴリズム技術は、伝統的な配列の「整列」で
はなく、しかしむしろ前述のTM6のプロリン残基からの指定された距離に基づ
く。(思うに受容体のIC3領域中に配置される)この残基から16アミノ酸残
基に配置されるアミノ酸残基を最も好ましくはリシン残基に突然変異することに
より、こうした活性化を得てよい。この目的を達成するためにはこの位置の突然
変異で他のアミノ酸残基が有用であるかも知れない。
D.疾患/障害の同定および/もしくは選択
下により詳細に示されるであろうとおり、最も好ましくは非内在性で、構成的
に活性化されるGPCRに対する反作用薬を本発明の方法論により同定すること
ができる。こうした反作用薬は、本受容体に関連する疾患治療のための薬物発見
プログラムでのリード化合物として理想的な候補である。GPCRに対する反作
用薬を直接同定してそれにより製薬学的組成物の開発を可能にする能力のため、
GPCRに関連する疾患および障害の研究が適切である。例えば、GPCRの存
在について疾患に罹った組織サンプルおよび正常な組織サンプルの双方を走査す
ることは、今や、学究的修練、もしくは特定のGPCRに対する内在性リガンド
を同定する途に沿って追跡することができるもの以上となっている。組織の走査
は広範な健康なおよび疾患に罹った組織にわたって実施することができる。こう
した組織の走査は、特定の受容体のある疾患および/もしくは障害との関連付け
の好ましい第一段階を提供する。例えば、本明細書に開示されるGPCRのいく
つかの例示的ドットブロットおよびRT−PCRの結果については、同時係属中
の出願(処理予定表番号ARE−0050)を参照されたい。
【0043】
好ましくは、(a)組織mRNAに対するドットブロット分析、および/もし
くは(b)組織サンプルでの受容体の発現のRT−PCR同定のためのプローブ
を作成するのにヒトGPCRのDNA配列を使用する。組織供給源、もしくは疾
患に罹った組織中での受容体の存在、または正常組織に比較して疾患に罹った組
織中での上昇された濃度での受容体の存在は、限定されるものでないがその疾患
に関連する疾患を挙げることができる治療レジメンとの相関を同定するのに好ま
しく利用することができる。受容体はこの技術により器官の領域に等しく十分に
局在化する可能性がある。受容体が局在化されている特定の組織の既知の機能に
基づき、受容体の推定の機能上の役割を推定することができる。
E.候補化合物のスクリーニング
1.包括的GPCRスクリーニングアッセイの技術
Gタンパク質受容体が構成的に活性になった場合、それはGタンパク質(例え
ばGq、Gs、Gi、Gz、Go)に結合しかつGタンパク質へのGTPの結合
を刺激する。その後、Gタンパク質がGTPアーゼとして作用し、そしてGTP
をGDPにゆっくりと加水分解し、それにより受容体は正常条件下で失活したよ
うになる。しかしながら、構成的に活性化された受容体はGDPをGTPに交換
し続ける。構成的に活性化される受容体を発現する膜への高められた結合をモニ
ターするのに、GTPの加水分解不可能な類似物[35S]GTPγSを使用する
ことができる。[35S]GTPγSはリガンドの非存在下および存在下での膜へ
のGタンパク質の共役をモニターするのに使用することができることが報告され
ている。このモニタリングの一例、なかんずく当業者に公知かつ利用可能な例は
、1995年にトレイノル(Traynor)とナホルスキ(Nahorski
)により報告された。本アッセイ系の好ましい使用は候補化合物の初期スクリー
ニングのためである。なぜなら、該系は、受容体の細胞内ドメインと相互作用す
る特定のGタンパク質に関係なく、全部のGタンパク質共役型受容体に包括的に
応用可能であるからである。
2.特異的GPCRスクリーニングアッセイの技術
「包括的」Gタンパク質共役型受容体アッセイ(すなわちアゴニスト、部分的
アゴニストもしくは反作用薬である化合物を選択するためのアッセイ)を使用し
て候補化合物が同定されれば、該化合物が受容体部位で相互作用していることを
確認するためのさらなるスクリーニングが好ましい。例えば、「包括的」アッセ
イにより同定された化合物は受容体に結合しないかも知れないが、しかし代わり
に細胞内ドメインからGタンパク質を単に「分離する」かも知れない。
a.Gs、GzおよびGi
Gsは酵素アデニリルシクラーゼを刺激する。他方、Gi(ならびにGzおよ
びGo)はこの酵素を阻害する。アデニリルシクラーゼはATPのcAMPへの
転化を触媒し;従って、Gsタンパク質を共役する構成的に活性化されたGPC
Rは、cAMPの増大された細胞レベルと関連する。他方、Gi(もしくはGz
、Go)タンパク質を共役する構成的に活性化されたGPCRは、cAMPの低
下された細胞レベルと関連する。一般に、“Indirect Mechani
sms of Synaptic Transmission,”第8章、Fr
om Neuron To Brain(第3版)ニコルス(Nichols,
J.G.)ら編 サイナウア アソシエーツ インク(Sinauer Ass
ociates,Inc.)(1992)を参照されたい。従って、cAMPを
検出するアッセイを利用して、ある候補化合物が例えば受容体に対する反作用薬
(すなわちこうした化合物はcAMPのレベルを低下させることができる)であ
るかどうかを決定することができる。cAMPを測定するための当該技術分野で
既知の多様なアプローチを利用することができ;最も好ましいアプローチはEL
ISAに基づく形式での抗cAMP抗体の使用に頼る。利用することができる別
の型のアッセイは全細胞セカンドメッセンジャーレポーター系アッセイである。
遺伝子のプロモーターは特定の遺伝子がコードするタンパク質の発現を司る。環
状AMPは、cAMP応答性のDNA結合タンパク質もしくは転写因子(CRE
B)(その後cAMP応答要素と呼ばれる特定の部位でプロモーターに結合しそ
して遺伝子の発現を司る)の結合を促進することにより遺伝子発現を司る。レポ
ーター遺伝子(例えばβ−ガラクトシダーゼもしくはルシフェラーゼ)の前に複
数のcAMP応答要素を含有するプロモーターを有するレポーター系を構築する
ことができる。従って、構成的に活性化されたGsに結合された受容体は、cA
MPの蓄積を引き起こし、これがその後遺伝子およびレポータータンパク質の発
現を活性化する。その後、標準的な生化学的アッセイを使用して、β−ガラクト
シダーゼもしくはルシフェラーゼのようなレポータータンパク質を検出すること
ができる(チェン(Chen)ら 1995)。
b.GoおよびGq
GqおよびGoは酵素ホスホリパーゼCの活性化に関連し、この酵素は順にリ
ン脂質PIP2を加水分解して、2種の細胞内メッセンジャー、すなわちジアシ
ルグリセロール(DAG)およびイノシトール1,4,5−三リン酸(IP3)
を放出する。IP3の増大された蓄積はGqおよびGo会合型受容体の活性化と
関連する。一般に、“Indirect Mechanisms of Syn
aptic Transmission,”第8章、From Neuron
To Brain(第3版)ニコルス(Nichols,J.G.)ら編 サイ
ナウア アソシエーツ インク(Sinauer Associates,In
c.)(1992)を参照されたい。IP3の蓄積を検出するアッセイは、候補
化合物が例えばGqもしくはGo会合型受容体に対する反作用薬である(すなわ
ちこうした化合物はIP3のレベルを低下させることができる)かどうかを決定
するのに利用することができる。Gq会合型受容体はAP1レポーターアッセイ
(ここでGq依存性のホスホリパーゼCがAP1要素を含有する遺伝子の活性化
を引き起こす)を使用してもまた検査することができ;従って、活性化されたG
q会合型受容体は、こうした遺伝子の発現の増大を明示することができ、それに
より、それに対する反作用薬はこうした発現の減少を明示することができ、また
、アゴニストはこうした発現の増大を明示することができる。こうした検出のた
めの商業的に入手可能なアッセイが利用可能である。
3.GPCR融合タンパク質
反作用薬、アゴニストおよび部分的アゴニストの直接の同定のための候補化合
物のスクリーニングでの使用のための内在性で、構成的に活性化されるオーファ
ンGPCRもしくは非内在性で、構成的に活性化されるオーファンGPCRの使
用は興味深いスクリーニングの挑戦を提供し、ここでは当然、それに結合される
内在性リガンドの非存在下でさえ該受容体は活性である。従って、こうした化合
物が反作用薬、アゴニスト、部分的アゴニストであることができるかどうか、も
しくはこうした受容体に対する影響を有することができるかどうかに関しての理
解を許すこうした識別の目的をもって、例えば候補化合物の存在下の非内在性受
容体とその化合物の非存在下の非内在性受容体とを識別するために、こうした識
別を高める可能性のあるアプローチを利用することが好ましい。好ましい一アプ
ローチはGPCR融合タンパク質の使用である。
【0044】
一般に、非内在性のオーファンGPCRが上で示されたアッセイ技術(ならび
に他者)を使用して構成的に活性化されていることを決定すれば、内在性GPC
Rと共役する優勢なGタンパク質を決定することが可能である。GPCRへのG
タンパク質の共役はシグナル伝達経路を提供し、これは評価することが可能であ
る。哺乳動物発現系の使用によりスクリーニングが行われることが最も好ましい
ため、こうした系はその中に内在性Gタンパク質を有することが期待されよう。
従って、当然、こうした系においては、非内在性の構成的に活性化されたオーフ
ァンGPCRが連続的にシグナルを発するであろう。この点に関して、例えば受
容体に対する反作用薬の存在下で、とりわけスクリーニングに関してそれが反作
用薬と接触される場合にそれが受容体をより容易に識別することが可能であろう
ことがよりありそうであるように、このシグナルを高めることが好ましい。
【0045】
GPCR融合タンパク質は、非内在性GPCRと共役するGタンパク質の効能
を高めることを意図している。GPCR融合タンパク質は非内在性の構成的に活
性化されたGPCRを用いるスクリーニングに好ましい。なぜなら、こうしたア
プローチは、こうしたスクリーニング技術で最も好ましく利用されるシグナルを
増大させるからである。これは大きな「S/N」比の助長で重要であり;こうし
た大きな比は本明細書で開示されるような候補化合物のスクリーニングに重要か
つ好ましい。
【0046】
GPCR融合タンパク質の発現に有用な構築物の構築は当業者の範囲内にある
。商業的に入手可能な発現ベクターおよび系が、研究者の特定のニーズに合う可
能性のある多様なアプローチを提供する。こうしたGPCR融合タンパク質構築
物に対する重要な基準は、内在性のGPCR配列およびGタンパク質配列の双方
が同じ読み枠にある(好ましくは内在性GPCRの配列はGタンパク質配列の上
流である)こと、および、GPCRの発現に際してGタンパク質もまた発現する
ことが可能であるようにGPCRの「終止」コドンを欠失もしくは置き換えなく
てはならないことである。GPCRはGタンパク質に直接連結することができる
か、もしくは2者の間にスペーサー残基(好ましくは約12を越えないが、この
数字は当業者により容易に確かめることが可能である)が存在することができる
。われわれは、使用されないいくつかの制限部位が発現に際して効果的にスペー
サーとなるであろうことに、スペーサーの使用が(便宜性に基づくと)好ましい
。最も好ましくは、GPCR融合タンパク質構築物の創製に先立ち非内在性GP
CRに共役するGタンパク質を同定することができる。同定された数種のGタン
パク質が存在するにすぎないため、その中の内在性のGPCR配列の挿入にGタ
ンパク質の配列を含んで成る構築物(すなわち普遍的Gタンパク質構築物)が利
用可能であることが好ましく;これは、異なる配列を有する多様な異なった内在
性GPCRの大スケールのスクリーニングにおいて効率を提供する。
【0047】
上に示されたとおり、Gi、GzおよびGoに共役する構成的に活性化される
GPCRは、これらの型のGPCRに基づくアッセイを挑戦的にするcAMPの
形成を阻害すると期待される(すなわち、cAMPシグナルは活性化に際して減
少し、従って例えば(このシグナルをさらに減少させることができる)反作用薬
の直接の同定を興味深いものとする)。本明細書に開示されるであろうとおり、
われわれは、実現可能なシクラーゼに基づくアッセイを確立する努力において、
これらの型の受容体について内在性のGPCRの内在性のGタンパク質に基づか
ないGPCR融合タンパク質を創製することが可能であることを確かめた。従っ
て、例えば、H9のようなGz共役型受容体、Gs融合タンパク質を利用するG
PCR融合タンパク質を確立することが可能である。われわれは、こうした融合
構築物は、発現に際して非内在性のGPCRが例えば「天然の」Gzタンパク質
よりもむしろGsと共役することを「誘導」もしくは「強要」し、その結果シク
ラーゼに基づくアッセイを確立することが可能であると考える。従って、Gi、
GzおよびGo共役型受容体について、われわれは、GPCR融合タンパク質を
使用しかつアッセイがアデニルシクラーゼ活性の検出に基づく場合は、Gs(も
しくは酵素アデニリルシクラーゼの形成を刺激する同等のGタンパク質)を用い
て融合構築物を確立することを好む。
F.医薬品化学
一般に(しかし常にでなく)、候補化合物の直接の同定はコンビナトリアル化
学の技術を介して生成される化合物に関連して好ましく実施され、それにより何
千もの化合物がこうした分析のため無作為に調製される。一般に、こうしたスク
リーニングの結果は独特のコア構造を有する化合物であることができ;その後、
これらの化合物は、その医薬特性をさらに高めるため、好ましいコア構造(1種
もしくは複数)を取り巻く付加的な化学的改変に好ましくかけられる。こうした
技術は当業者に既知でありかつ本特許明細書で詳細に取り扱わないであろう。
G.製薬学的組成物
さらなる開発に選択された候補化合物は、当業者に公知の技術を使用して製薬
学的組成物に処方することができる。適する製薬学的に許容できる担体は当業者
に利用可能であり;例えば、Remingtons’s Pharmaceut
ical Sciences、第16版、1980、マック パブリッシング
カンパニー(Mack Publishing Co.)(オスロ(Oslo)
ら編)を参照されたい。
H.他の利用性
本明細書に開示される非内在性の変種ヒトGPCRの好ましい使用は、(好ま
しくは製薬学的作用物質としての使用のための)反作用薬、アゴニストもしくは
部分的アゴニストとしての候補化合物の直接の同定のためのものであることがで
きるが、これらの変種ヒトGPCRはまた研究の設定でも利用することができる
。例えば、GPCRを組み込むインビトロおよびインビボの系は、正常のおよび
疾患に罹った双方のヒトの状態でこれらの受容体が演じる役割をさらに解明かつ
理解するため、ならびに構成的活性化の役割の理解(それがシグナル伝達カスケ
ードの理解に適用されるため)に利用することができる。非内在性のヒトGPC
Rの価値は、そのための内在性リガンドが同定される前に人体でのこれらの受容
体の役割を理解するのに、それらの独特の特徴のため非内在性のヒトGPCRを
使用することができることにおいて、研究ツールとしてのそれらの利用性が高め
られることである。開示される受容体の他の用途は、とりわけ本特許明細書の検
討に基づき当業者に明らかとなるであろう。
【実施例】
【0048】
以下の実施例は本発明の解明の目的上(そして制限でなく)提示する。本明細
書で特定の核酸およびアミノ酸の配列が開示される一方、当業者は、下に報告さ
れる同一のもしくは実質的に類似の結果を達成しつつこれらの配列に対する小さ
な改変を行う能力があると信じられる。1配列から別のものまで(例えばラット
受容体からヒト受容体まで、もしくはヒト受容体Aからヒト受容体Bまで)の配
列カセットの応用もしくは理解への伝統的アプローチは、配列整列の技術(それ
により配列は共通の領域を決定する活動で整列される)に一般に基づく。本明細
書に開示される突然変異のアプローチはこのアプローチに頼らないが、しかし、
代わりにアルゴリズムのアプローチ、およびヒトGPCRのTM6領域内に配置
される保存されたプロリン残基からの位置上の距離に基づく。このアプローチが
確実にされれば、当業者は、それに対して小さな変更を行って本明細書に開示さ
れる実質的に同一の結果(すなわち構成的活性化)を達成する能力があると信じ
られる。こうした改変されたアプローチは本開示の範囲内と考えられる。
実施例1
内在性のヒトGPCR
1.ヒトGPCRの同定
ジェンバンク[GenBank](商標)データベース情報の再検討に基づき
、開示された内在性ヒトGPCRのあるものを同定した。データベースを検索す
る間に以下のcDNAクローンを下に明示されるとおり同定した(表C)。
【0049】
【表3】
【0050】
他の開示された内在性ヒトGPCRは、以下のESTクローンをクエリ配列と
して使用するESTデータベース(dbest)のBLAST(商標)検索を実
施することにより同定した。その後、同定された以下のESTクローンをプロー
ブとして使用して、ヒトゲノムライブラリーをスクリーニングした(表D)。
【0051】
【表4】
【0052】
2.完全長のクローニング
A.ヒトG2A
マウスESTクローン1179426を使用して、3種のアミノ酸G2Aコー
ディング配列を除く全部を含有するヒトゲノムクローンを得た。このコーディン
グ配列の5’は5’RACEを使用することにより得、また、PCRのための鋳
型はクロンテック(Clontech)のヒト脾マラソン−レディ[Marat
hon−Ready](商標)cDNAであった。開示されるヒトG2Aは、以
下:
5’−CTGTGTACAGCAGTTCGCAGAGTG−3’(配列番号4
1;1回目のPCR)
5’−GAGTGCCAGGCAGAGCAGGTAGAC−3’(配列番号4
2;2回目のPCR)
のような配列番号41および配列番号42に示されるような第一回および第二回
のPCRのためのG2AのcDNAに特異的なプライマーを使用するPCRによ
り増幅した。PCRは、94℃30秒間、次いで94℃5秒間および72℃4分
間の5周期;ならびに94°5秒間および70°4分間の30周期で、アドバン
テージ(Advantage)GCポリメラーゼキット(クロンテック(Clo
ntech);製造説明書に従うことができる)を使用して実施した。およそ1
.3kbのPCRフラグメントをアガロースゲルから精製し、HindIIIお
よびXbaIで消化し、そして発現ベクターpRC/CMV2(インヴィトロジ
ェン(Invitrogen))にクローン化した。T7シークェナーゼ[Se
quenase](商標)キット(USB アマーシャム(USB Amers
ham);製造元の説明書が従われた)を使用してクローン化された挿入物を配
列決定し、そして提示された配列と配列を比較した。P32標識されたフラグメン
トを用いてRNAドットブロット(クロンテック(Clontech);製造元
の説明書が従われた)をプロービングすることによりヒトG2Aの発現を検出し
た。
b.CHN9
ESTクローン1541536の配列決定は、CHN9が1個の開始コドンの
みを有する(すなわち終止コドンは欠けていた)部分的cDNAクローンである
ことを示した。CHN9をデータベース(nr)に対してBLAST検索する(b
last)のに使用した場合、CHN9の3’の配列はロイコトリエンB4受容体の
cDNAの5’非翻訳領域(CHN9のコーディング配列と同じ読み枠に終止コ
ドンを含有した)に100%相同であった。LTB4RのcDNAの5’非翻訳
領域がCHN9の3’配列であったかどうかを決定するために、CHN9に見出
される開始コドンに隣接する5’配列およびLTB4Rの5’非翻訳領域に見出
される終止コドンを取り巻く3’配列に基づくプライマーを使用してPCRを実
施した。利用された5’プライマー配列は以下のとおりであった:
5’−CCCGAATTCCTGCTTGCTCCCAGCTTGGCCC−3
’(配列番号43;センス)および
5’−TGTGGATCCTGCTGTCAAAGGTCCCATTCCGG−
3’(配列番号44;アンチセンス)
製造元により供給される緩衝液系、0.25μMの各プライマーおよび0.2m
Mの各4種のヌクレオチドとともに鋳型としての胸腺cDNAおよびrTthポ
リメラーゼ(パーキン エルマー(Perkin Elmer))を使用してP
CRを実施した。周期条件は、94℃1分間、65℃1分間ならびに72℃1分
および10秒間の30周期であった。予測された大きさと一致する1.1kbの
フラグメントをPCRから得た。本PCRフラグメントをpCMVにサブクロー
ニングし(下を参照されたい)そして配列決定した(配列番号35を参照された
い)。
c.RUP4
鋳型としてヒト脳cDNA(クロンテック(Clontech))を用いるR
T−PCRにより完全長のRUP4をクローン化した:
5’−TCACAATGCTAGGTGTGGTC−3’(配列番号45;セン
ス)および
5’−TGCATAGACAATGGGATTACAG−3’(配列番号46;
アンチセンス)。
以下の周期、すなわち94℃2分間;94℃30秒間;55℃30秒間、72℃
45秒間および72℃10分間により、タックプラス プレシジョン[TaqP
las Precision](商標)ポリメラーゼ(ストラタジーン(Str
atagene);製造説明書が従われた)を使用してPCRを実施した。周期
2から4を30回反復した。
【0053】
PCR産物を1%アガロースゲルで分離し、そして500bpのPCRフラグ
メントを単離しかつpCRII−TOPO(商標)ベクター(インヴィトロジェ
ン(Invitrogen))にクローン化し、そしてT7 DNAシークェナ
ーゼ[Sequenase](商標)キット(アマーシャム(Amersham
))およびSP6/T7プライマー(ストラタジーン(Stratagene)
)を使用して配列決定した。PCRフラグメントから示される配列分析は、実際
に、他のGPCRとの類似性をもつ1個の連続的読取り枠を有する、代替スプラ
イシングされた形態のAI307658であった。このPCRフラグメントの完
了された配列は以下のとおりであった:
【0054】
【表5】
【0055】
上の配列に基づき、2種のセンスオリゴヌクレオチドプライマーの組:
5’−CTGCTTAGAAGAGTGGACCAG−3’(配列番号48;オ
リゴ1)、
5’−CTGTGCACCAGAAGATCTACAC−3’(配列番号49;
オリゴ2)および
2種のアンチセンスオリゴヌクレオチドプライマーの組:
5’−CAAGGATGAAGGTGGTGTAGA−3’(配列番号50;オ
リゴ3)
5’−GTGTAGATCTTCTGGTGCACAGG−3’(配列番号51
;オリゴ4)
を、製造元の説明書に従って鋳型としてヒト脳マラソン−レディ[Marath
on−Ready](商標)cDNA(クロンテック(Clontech)、カ
タログ番号7400−1)を用いる3’−および5’−RACE PCRに使用
した。RACE PCRにより生成されたDNAフラグメントをpCRII−T
OPO(商標)ベクター(インヴィトロジェン(Invitrogen))にク
ローン化し、そしてSP6/T7プライマー(ストラタジーン(Stratag
ene))および数種の内的プライマーを使用して配列決定した。3’RACE
産物はポリ(A)テール(tail)およびTAA終止コドンで終了する1個の完了さ
れる読取り枠を含有した。5’RACE産物は不完全な5’末端を含有した(す
なわちATG開始コドンが存在しなかった)。
【0056】
新たな5’配列に基づき、オリゴ3および以下のプライマー:
5’−GCAATGCAGGTCATAGTGAGC−3’(配列番号52;オ
リゴ5)
を第2回の5’race PCRに使用し、そしてPCR産物を上のとおり分析
した。第3回の5’race PCRは、アンチセンスプライマー:
5’−TGGAGCATGGTGACGGGAATGCAGAAG−3’(配列
番号53;オリゴ6)および
5’−GTGATGAGCAGGTCACTGAGCGCCAAG−3’(配列
番号54;オリゴ7)
を利用して実施した。5’RACE PCRの産物の配列は開始コドンATGの
存在を示し、また、さらなる回の5’race PCRはいかなるそれ以上の5
’配列も生成しなかった。プライマーとして、センスプライマー
5’−GCAATGCAGGCGCTTAACATTAC−3’(配列番号55
;オリゴ8)
およびオリゴ4を使用するRT−PCR、ならびにヒト脳および心のcDNA鋳
型(クロンテック(Clontech)、カタログ番号7404−1)から生成
された650bpのPCR産物の配列分析により、完了された5’配列を確認し
た。オリゴ2および以下のアンチセンスプライマー:
5’−TTGGGTTACAATCTGAAGGGCA−3’(配列番号56;
オリゴ9)
を使用するRT−PCR、ならびにヒト脳および心のcDNA鋳型(クロンテッ
ク(Clontech)、カタログ番号7404−1)から生成された670b
pのPCR産物の配列分析により、完了された3’配列を確認した。
d.RUP5
以下の配列:
5’−ACTCCGTGTCCAGCAGGACTCTG−3’(配列番号57
)
5’−TGCGTGTTCCTGGACCCTCACGTG−3’(配列番号5
8)
を有した、ATG開始コドンから上流のセンスプライマー(配列番号57)、お
よび終止コドンとしてTCAを含有するアンチセンスプライマー(配列番号58
)、ならびに鋳型としてヒト末梢白血球cDNA(クロンテック(Clonte
ch))を使用するRT−PCRにより完全長のRUP5をクローン化した。段
階2から段階4が30回反復された以下の周期、すなわち94℃30秒間;94
℃15秒間;69℃40秒間;72℃3分間;および72℃6分間による50μ
lの反応中での増幅にアドバンテージ[Advantage](商標)cDNA
ポリメラーゼ(クロンテック(Clontech))を使用した。1.4kbの
PCRフラグメントを単離し、そしてpCRII−TOPO(商標)ベクター(
インヴィトロジェン(Invitrogen))を用いてクローン化し、そして
T7 DNAシークェナーゼ[Sequenase](商標)キット(アマーシ
ャム(Amersham))を使用して完全に配列決定した。配列番号9を参照
されたい。
e.RUP6
プライマー:
5’−CAGGCCTTGGATTTTAATGTCAGGGATGG−3’(
配列番号59)および
5’−GGAGAGTCAGCTCTGAAAGAATTCAGG−3’(配列
番号60)
ならびに鋳型としてヒト胸腺マラソン−レディ[Marathon−Ready
](商標)cDNA(クロンテック(Clontech))を使用するRT−P
CRにより完全長のRUP6をクローン化した。以下の周期、すなわち94℃3
0秒間;94℃5秒間;66℃40秒間;72℃2.5秒間および72℃7分間
による50μl反応中での増幅にアドバンテージ(Advantage)cDN
Aポリメラーゼ(クロンテック(Clontech)、製造元の説明書に従う)
を使用した。周期2から4を30回反復した。1.3kbのPCRフラグメント
を単離し、そしてpCRII−TOPO[商標]ベクター(インヴィトロジェン
(Invitrogen))にクローン化し、そしてABI ビッグ ダイ タ
ーミネーター[Big Dye Terminator](商標)キット(P.
E.バイオシステム(P.E.Biosystem))を使用して完全に配列決
定した(配列番号11を参照されたい)。
f.RUP7
プライマー:
5’−TGATGTGATGCCAGATACTAATAGCAC−3’(配列
番号61;センス)および
5’−CCTGATTCATTTAGGTGAGATTGAGAC−3’(配列
番号62;アンチセンス)
ならびに鋳型としてヒト末梢白血球cDNA(クロンテック(Clontech
))を使用するRT−PCRにより完全長のRUP7をクローン化した。段階2
ないし段階4が30回反復された以下の周期、すなわち94℃2分間;94℃1
5秒間;60℃20秒間;72℃2分間;72℃10分間による50μl反応中
での増幅にアドバンテージ[Advantage](商標)cDNAポリメラー
ゼ(クロンテック(Clontech))を使用した。1.25kbのPCRフ
ラグメントを単離し、そしてpCRII−TOPO(商標)ベクター(インヴィ
トロジェン(Invitrogen))にクローン化し、そしてABI ビッグ
ダイ ターミネーター[Big Dye Terminator](商標)キ
ット(P.E.バイオシステム(P.E.Biosystem))を使用して完
全に配列決定した。配列番号13を参照されたい。
3.アンジオテンシンIIタイプ1受容体(「AT1」)
製造元により供給される緩衝液系、0.25μMの各プライマーおよび0.2
mMの各4種のヌクレオチドとともに鋳型としてのゲノムDNAおよびrTth
ポリメラーゼ(パーキン エルマー(Perkin Elmer))を使用する
PCRにより、内在性のヒトアンジオテンシンIIタイプ1受容体(「AT1」
)を得た。周期条件は、94℃1分間、55℃1分間および72℃1.5分間の
30周期であった。5’PCRプライマーは、配列:
5’−CCCAAGCTTCCCCAGGTGTATTTGAT−3’(配列番
号63)
とともにHindIII部位を含有し、そして3’プライマーは以下の配列:
5’−GTTGGATCCACATAATGCATTTTCTC−3’(配列番
号64)
とともにBamHI部位を含有する。生じる1.3kbのPCRフラグメントを
HindIIIおよびBamHIで消化し、そしてpCMV発現ベクターのHi
ndIII−BamHI部位にクローン化した。cDNAクローンを完全に配列
決定した。その後、ヒトAT1の核酸(配列番号65)およびアミノ酸(配列番
号66)の配列を決定しかつ確かめた。
4.GPR38
GPR38を得るため、製造元により供給される緩衝液系、0.25μMの各
プライマーおよび0.2mMの各4種のヌクレオチドとともに鋳型としてのヒト
ゲノムcDNAおよびrTthポリメラーゼ(パーキン エルマー(Perki
n Elmer))を使用して、2種のPCRフラグメントを組み合わせること
によりPCRを実施した。各PCR反応の周期条件は94℃1分間、62℃1分
間および72℃2分間の30周期であった。
【0057】
第一のフラグメントは、以下の配列:
5’−ACCATGGGCAGCCCCTGGAACGGCAGC−3’(配列
番号67)
を伴う端部位を含有した5’PCRプライマー、および以下の配列:
5’−AGAACCACCACCAGCAGGACGCGGACGGTCTGC
CGGTGG−3’(配列番号68)
を有する3’プライマーを用いて増幅した。第二のPCRフラグメントは、以下
の配列:
5’−GTCCGCGTCCTGCTGGTGGTGGTTCTGGCATTT
ATAATT−3’(配列番号69)
を有する5’プライマー、ならびにBamHI部位を含有しかつ以下の配列:
5’−CCTGGATCCTTATCCCATCGTCTTCACGTTAGC
−3’(配列番号70)
を有する3’プライマーを用いて増幅した。配列番号67および配列番号70を
プライマーとして使用して(上に示された周期条件を使用する)、2種のフラグ
メントを鋳型として使用してGPR38を増幅した。生じる1.44kbのPC
RフラグメントをBamHIで消化し、そしてpCMV発現ベクターの平滑Ba
mHI部位にクローン化した。
5.MC4
MC4を得るため、製造元により供給される緩衝液系、0.25μMの各プラ
イマーおよび0.2mMの各4種のヌクレオチドとともに鋳型としてのヒトゲノ
ムcDNAおよびrTthポリメラーゼ(パーキン エルマー(Perkin
Elmer))を使用してPCRを実施した。各PCR反応の周期条件は94℃
1分間、54℃1分間および72℃1.5分間の30周期であった。
【0058】
5’PCRは配列:
5’−CTGGAATTCTCCTGCCAGCATGGTGA−3’(配列番
号71)
とともにEcoRI部位を含有し、そして3’プライマーは配列:
5’−GCAGGATCCTATATTGCGTGCTCTGTCCCC−3’
(配列番号72)
とともにBamHI部位を含有した。1.0kbのPCRフラグメントをEco
RIおよびBamHIで消化し、そしてpCMV発現ベクターのEcoRI−B
amHI部位にクローン化した。その後、ヒトMC4の核酸(配列番号73)お
よびアミノ酸(配列番号74)の配列を決定した。
6.CCKB
CCKBを得るため、製造元により供給される緩衝液系、0.25μMの各プ
ライマーおよび0.2mMの各4種のヌクレオチドとともに鋳型としてのヒト胃
cDNAおよびrTthポリメラーゼ(パーキン エルマー(Perkin E
lmer))を使用してPCRを実施した。各PCR反応の周期条件は94℃1
分間、65℃1分間ならびに72℃1分および30秒間の30周期であった。
【0059】
5’PCRは配列:
5’−CCGAAGCTTCGAGCTGAGTAAGGCGGCGGGCT−
3’(配列番号75)
とともにHindIII部位を含有し、また、3’プライマーは配列:
5’−GTGGAATTCATTTGCCCTGCCTCAACCCCCA−3
’(配列番号76)
とともにEcoRI部位を含有した。生じる1.44kbのPCRフラグメント
をHindIIIおよびEcoRIで消化し、そしてpCMV発現ベクターのH
indIII−EcoRI部位にクローン化した。その後、ヒトCCKBの核酸
(配列番号77)およびアミノ酸(配列番号78)の配列を決定した。
7.TDAG8
TDAG8を得るため、製造元により供給される緩衝液系、0.25μMの各
プライマーおよび0.2mMの各4種のヌクレオチドとともに鋳型としてのゲノ
ムDNAおよびrTthポリメラーゼ(パーキン エルマー(Perkin E
lmer))を使用してPCRを実施した。周期条件は94℃1分間、56℃1
分間ならびに72℃1分および20秒間の30周期であった。5’PCRプライ
マーは以下の配列:
5’−TGCAAGCTTAAAAAGGAAAAAATGAACAGC−3’
(配列番号79)
とともにHindIII部位を含有し、また、3’プライマーは以下の配列:
5’−TAAGGATCCCTTCCCTTCAAAACATCCTTG−3’
(配列番号80)
とともにBamHI部位を含有した。生じる1.1kbのPCRフラグメントを
HindIIIおよびBamHIで消化し、そしてpCMV発現ベクターのHi
ndIII−BamHI部位にクローン化した。配列決定された3種の生じるク
ローンは、ProからAlaへのアミノ酸43、LysからAsnへのアミノ酸
97、およびIleからPheへのアミノ酸130の変化を伴う3種の潜在的多
形を含有した。その後、ヒトTDAG8の核酸(配列番号81)およびアミノ酸
(配列番号82)の配列を決定した。
8.H9
H9を得るため、製造元により供給される緩衝液系、0.25μMの各プライ
マーおよび0.2mMの各4種のヌクレオチドとともに鋳型としての下垂体cD
NAおよびrTthポリメラーゼ(パーキン エルマー(Perkin Elm
er))を使用してPCRを実施した。周期条件は94℃1分間、62℃1分間
および72℃2分間の30周期であった。5’PCRプライマーは以下の配列:
5’−GGAAAGCTTAACGATCCCCAGGAGCAACAT−3’
(配列番号15)
とともにHindIII部位を含有し、また、3’プライマーは以下の配列:
5’−CTGGGATCCTACGAGAGCATTTTTCACACAG−3
’(配列番号16)
とともにBamHI部位を含有した。生じる1.9kbのPCRフラグメントを
HindIIIおよびBamHIで消化し、そしてpCMV発現ベクターのHi
ndIII−BamHI部位にクローン化した。H9は、アミノ酸P320S、
S493Nおよびアミノ酸G448Aの変化を伴う3種の潜在的多形を含有した
。その後、ヒトH9の核酸(配列番号139)およびアミノ酸(配列番号140
)の配列を決定しかつ確かめた。
実施例2
非内在性の構成的に活性化されるGPCRの調製
当業者は、核酸配列の突然変異のための技術を選択する能力があると信じられ
る。上に開示された非内在性の変種数種のヒトGPCRを創製するのに利用され
たアプローチを下に提示する。下に開示される突然変異はアルゴリズムのアプロ
ーチに基づき、それにより(TM6/IC3の界面近くのGPCRのTM6領域
に配置される)保存されるプロリン残基からの(GPCRのIC3領域に配置さ
れる)16番目のアミノ酸が、最も好ましくはリシンアミノ酸残基に突然変異さ
れる。
1.トランスフォーマー部位特異的[Transformer Site−Di
rected](商標)突然変異誘発
非内在性のヒトGPCRの調製は、製造元の説明書に従い、トランスフォーマ
ー部位特異的[Transformer Site−Directed](商標
)突然変異誘発キット(クロンテック(Clontech))を使用してヒトG
PCRで達成することができる。2種の突然変異誘発プライマー、最も好ましく
はリシン突然変異を創製するリシン突然変異誘発オリゴヌクレオチド、および選
択マーカーオリゴヌクレオチドを利用する。便宜上、ヒトGPCRに組み込まれ
るべきコドン突然変異を標準的形態でもまた示す(表E):
【0060】
【表6】
【0061】
指定される配列プライマーを使用して、上の方法に従い、以下のGPCRを突
然変異した(表F)。
【0062】
【表7】
【0063】
その後、非内在性のヒトGPCRを配列決定し、また、形成されかつ確かめら
れた核酸およびアミノ酸の配列を、下に表Gに要約されるとおり、本特許明細書
への付随する「配列表」付録に列挙する:
【0064】
【表8】
【0065】
2.非内在性ヒトGPCRの創製のための代替アプローチ
a.AT1
1.F239K突然変異
F239突然変異(核酸配列について配列番号89、およびアミノ酸配列につ
いて配列番号90を参照されたい)を創製することにより、非内在性の構成的に
活性化されるヒトAT1受容体の調製を達成した。突然変異誘発は、製造元の説
明書に従い、トランスフォーマー部位特異的突然変異誘発[Transform
er Site−Directed Mutagenesis](商標)キット
(クロンテック(Clontech))を使用して実施した。2種の突然変異誘
発プライマー、リシン突然変異誘発オリゴヌクレオチド(配列番号91)および
選択マーカーオリゴヌクレオチド(配列番号92)を使用した。これらはそれぞ
れ以下の配列を有した:
5’−CCAAGAAATGATGATATTAAAAAGATAATTATG
GC−3’(配列番号91)
5’−CTCCTTCGGTCCTCCTATCGTTGTCAGAAGT−3
’(配列番号92)
2.N111A突然変異
非内在性のヒトAT1受容体の調製もまた、N111A突然変異(核酸配列に
ついて配列番号93、およびアミノ酸配列について配列番号94を参照されたい
)を創製することにより達成した。10%DMSO、0.25μMの各プライマ
ー、および0.5mMの各4種のヌクレオチドを補充された、製造元により提供
される緩衝液系を用いてpfuポリメラーゼ(ストラタジーン(Stratag
ene))を使用して、2回のPCR反応を実施した。使用された5’PCRの
センスプライマーは以下の配列:
5’−CCCAAGCTTCCCCAGGTGTATTTGAT−3’(配列番
号95)
を有し、また、アンチセンスプライマーは以下の配列:
5’−CCTGCAGGCGAAACTGACTCTGGCTGAAG−3’(
配列番号96)
を有した。生じる400bpのPCRフラグメントをHindIII部位で消化
し、そしてpCMVベクターのHindIII−SmaI部位にサブクローニン
グした(5’構築物)。使用された3’PCRのセンスプライマーは以下の配列
:
5’−CTGTACGCTAGTGTGTTTCTACTCACGTGTCTC
AGCATTGAT−3’(配列番号97)
を有し、また、アンチセンスプライマーは以下の配列:
5’−GTTGGATCCACATAATGCATTTTCTC−3’(配列番
号98)
を有した。生じる880bpのPCRフラグメントをBamHIで消化し、そし
て5’構築物のPst(T4ポリメラーゼにより平滑化された)およびBamH
I部位に挿入して完全長のN111A構築物を生成した。周期条件は、94℃1
分間、60℃1分間および72℃1分間(5’PCR)もしくは1.5分間(3
’PCR)の25周期であった。
3.AT2K255IC3突然変異
AT2K255IC3の「ドメインスワップ(domain swap)」突然変異(核酸
配列について配列番号99を、およびアミノ酸配列について配列番号100を参
照されたい)を創製することにより、非内在性の構成的に活性化されるヒトAT
1の調製を達成した。AT1のIC3に隣接する制限部位を生成させて、アンジ
オテンシンIIタイプ2受容体(AT2)からの対応するIC3でのIC3の置
き換えを助長した。これは2回のPCR反応を実施することにより達成した。セ
ンスプライマーとして配列番号63を、およびアンチセンスプライマーとして以
下の配列:
5’−TCCGAATTCCAAAATAACTTGTAAGAATGATCA
GAAA−3’(配列番号101)
を利用することにより、5’非翻訳領域からIC3の開始までをコードする5’
PCRフラグメント(フラグメントA)を生成させた。センスプライマーとして
以下の配列:
5’−AGATCTTAAGAAGATAATTATGGCAATTGTGCT
−3’(配列番号102)
およびアンチセンスプライマーとして配列番号64を使用することにより、IC
3の終了から3’非翻訳領域までをコードする3’PCRフラグメント(フラグ
メントB)を生成させた。PCR条件は、10%DMSO、0.25μMの各プ
ライマーおよび0.5mMの各4種のヌクレオチドを補充された、製造元により
供給される緩衝液系を用いて、鋳型としての内在性のAT1 cDNAクローン
およびpfuポリメラーゼ(ストラタジーン(Stratagene))を使用
して、94℃1分間、55℃1分間および72℃1.5分間の30周期であった
。フラグメントA(720bp)をHindIIIおよびEcoRIで消化しそ
してサブクローニングした。フラグメントBはBamHIで消化し、そしてクロ
ーン化されたPCRフラグメントの5’にEcoRI部位をもつpCMVベクタ
ーにサブクローニングした。
【0066】
以下の配列:
5’AATTCGAAAACACTTACTGAAGACGAATAGCTAT
GGGAAGAACAGGATAACCCGTGACCAAG−3’(センス;
配列番号103)
5’TTAACTTGGTCACGGGTTATCCTGTTCTTCCCAT
AGCTATTCGTCTTCAGTAAGTGTTTTCG−3’(アンチセ
ンス;配列番号104)
を有する2種の合成オリゴヌクレオチドをアニーリングすることにより、L25
5K突然変異を伴いAT2のIC3をコードしそして5’にEcoRI付着端お
よび3’にAfIII付着端を含有するDNAフラグメント(フラグメントC)
を生成させた。
【0067】
フラグメントCを、EcoRIおよびAfIII部位によりフラグメントBの
前に挿入した。その後、生じるクローンをEcoRI部位によりフラグメントA
と連結して、AT2K255IC3をもつAT1を生成させた。
4.A243+突然変異
A243+突然変異(核酸配列について配列番号105、およびアミノ酸配列
について配列番号106を参照されたい)を創製することにより、非内在性のヒ
トAT1受容体の調製もまた達成した。A243+突然変異は以下のPCRに基
づく戦略を使用して構築した。すなわち、10%DMSO、0.25μMの各プ
ライマーおよび0.5mMの各4種のヌクレオチドを補充された、製造元により
提供される緩衝液系とともにpfuポリメラーゼ(ストラタジーン(Strat
agene))を使用して2回のPCR反応を実施した。利用された5’PCR
のセンスプライマーは以下の配列:
5’−CCCAAGCTTCCCCAGGTGTATTTGAT−3’(配列番
号107)
を有し、また、アンチセンスプライマーは以下の配列:
5’−AAGCACAATTGCTGCATAATTATCTTAAAAATA
TCATC−3’(配列番号108)
を有した。利用された3’PCRのセンスプライマーは、Ala挿入を含有する
以下の配列:
5’−AAGATAATTATGGCAGCAATTGTGCTTTTCTTT
TTCTTT−3’(配列番号109)
そしてアンチセンスプライマー:
5’−GTTGGATCCACATAATGCATTTTCTC−3’(配列番
号110)
を有した。周期条件は、94℃1分間、54℃1分間および72℃1.5分間の
25周期であった。その後、5’および3’のPCRのアリコートを共鋳型(co-
template)として使用して、5’PCRのセンスプライマーおよび3’PCRの
アンチセンスプライマーを使用する二次的PCRを実施した。PCR条件は、伸
長時間が2.5分であったことを除き一次PCRと同一であった。生じるPCR
フラグメントをHindIIIおよびBamHIで消化し、そしてpCMVベク
ターにサブクローニングした(配列番号105を参照されたい)。
4.CCKB
V322K突然変異(核酸配列について配列番号111、およびアミノ酸配列
について配列番号112を参照されたい)を創製することにより、非内在性の構
成的に活性化されるヒトCCKB受容体の調製を達成した。突然変異誘発は、実
施例1からの野性型CCKBを使用する増幅を介してPCRにより実施した。
【0068】
配列番号75、およびV322K突然変異を含んで成るアンチセンスプライマ
ー:
5’−CAGCAGCATGCGCTTCACGCGCTTCTTAGCCCA
G−3’(配列番号113)
を使用することにより第一のPCRフラグメント(1kb)を増幅した。V32
2K突然変異を含んで成るセンスプライマー:
5’−AGAAGCGCGTGAAGCGCATGCTGCTGGTGATCG
TT−3’(配列番号114)および配列番号76を使用することにより第二の
PCRフラグメント(0.44kb)を増幅した。その後、配列番号75および
配列番号76、ならびに上で示された系および条件を使用して、V332Kを含
んで成るCCKBを増幅するための鋳型として2種の生じるPCRフラグメント
を使用した。V332K突然変異を含有する生じる1.44kbのPCRフラグ
メントをHindIIIおよびEcoRIで消化し、そしてpCMV発現ベクタ
ーのHindIII−EcoRI部位にクローン化した(配列番号111を参照
されたい)。
3.クイックチェンジ[QuikChange](商標)部位特異的[Site
−Directed](商標)突然変異誘発
クイックチェンジ[QuikChange](商標)部位特異的[Site−
Directed](商標)突然変異誘発キット(ストラタジーン(Strat
agene)、製造元の説明書に従う)を使用することにより、非内在性のヒト
GPCRの調製もまた達成することができる。内在性GPCRを鋳型として好ま
しく使用し、また、2種の突然変異誘発プライマー、ならびに最も好ましくはリ
シン突然変異誘発オリゴヌクレオチドおよび選択マーカーオリゴヌクレオチド(
キットに包含される)を利用する。便宜上、ヒトGPCRに組み込まれたコドン
の突然変異およびそれぞれのオリゴヌクレオチドを標準的形態で示す(表H):
【0069】
【表9】
【0070】
実施例3
受容体の発現
タンパク質の発現のために多様な細胞が当該技術に使用可能であるが、哺乳動
物細胞を利用することが最も好ましい。これの主要な理由は実地的なことに基づ
く。すなわち、例えばGPCRの発現のための酵母細胞の利用が可能な一方で、
受容体共役型の遺伝子的機構を包含しなくてもよい(事実、酵母の場合は包含し
ない)非哺乳動物細胞および哺乳動物の系について発展した分泌経路をプロトコ
ルに導入し、従って、非哺乳動物細胞で得られる結果は、潜在的に有用な一方で
哺乳動物細胞から得られるものと同じくらい好ましくはない。哺乳動物細胞のう
ち、COS−7、293および293T細胞がとりわけ好ましいが、利用される
特定の哺乳動物細胞は当業者の特定のニーズに基づくことが可能である。
【0071】
第1日に、150mmプレートあたり1×107個の293T細胞をプレート
培養した。第2日に2本の反応チューブを準備した(各チューブについて後に続
く比率はプレート1枚あたりである)。すなわち、チューブAは、1.2mlの
血清を含まないDMEM(アーヴィン サイエンティフィック(Irvine
Scientific)、カリフォルニア州アーヴィン)中に20μgのDNA
(例えばpCMVベクター;受容体cDNAを含むpCMVベクター、など)を
混合することにより準備し;チューブBは1.2mlの血清を含まないDMEM
中に120μlのリポフェクタミン(ギブコ(Gibco)BRL)を混合する
ことにより準備した。チューブAおよびBは反転(数回)により混合し、次いで
室温で30〜45分間インキュベートした。混合状態を「トランスフェクション
混合物」と称する。プレート培養された293T細胞を1×PBSで洗浄し、次
いで10mlの血清を含まないDMEMを添加した。細胞に2.4mlのトラン
スフェクション混合物を添加し、次いで37℃/5%CO2で4時間インキュベ
ートした。トランスフェクション混合物を吸引により除去し、次いで25mlの
DMEM/10%ウシ胎児血清を添加した。細胞を37℃/5%CO2でインキ
ュベートした。72時間のインキュベーション後に細胞を収穫し、そして分析に
利用した。
実施例4
非内在性GPCRの構成的活性の測定のためのアッセイ
非内在性のヒトGPCRの構成的活性の評価には多様なアプローチが利用可能
である。以下は具体的説明であり;当業者は、当業者のニーズに優先的に利益を
もたらす技術を決定する能力があると信じられる。
1.膜結合アッセイ:[35S]GTPγSアッセイ
Gタンパク質共役型受容体がリガンド結合もしくは構成的活性化のいずれかの
結果としてその活性状態にある場合、受容体はGタンパク質に共役しそしてGD
Pの放出およびGTPのGタンパク質へのその後の結合を刺激する。Gタンパク
質−受容体複合体のαサブユニットがGTPアーゼとして作用し、そしてGTP
をGDPにゆっくりと加水分解し、この点で受容体が通常非活性化される。構成
的に活性化された受容体はGDPをGTPと交換し続ける。構成的に活性化され
る受容体を発現する膜への[35S]GTPγSの高められた結合を立証するのに
、加水分解不可能なGTP類似物[35S]GTPγSを利用することができる。
構成的活性化を測定するための[35S]GTPγS結合の使用の利点は:(a)
それが全部のGタンパク質共役型受容体に包括的に応用可能である;(b)それ
は膜表面の近接であり、細胞内カスケードに影響を及ぼす分子を拾い上げること
をより少なくありそうにすることである。
【0072】
該アッセイは、直接的に関連する受容体を発現する膜への[35S]GTPγS
の結合を刺激するGタンパク質共役型受容体の能力を利用する。従って、該アッ
セイは、既知の、オーファンのおよび構成的に活性化されるGタンパク質共役型
受容体に対する候補化合物をスクリーニングするための直接同定法で使用するこ
とができる。該アッセイは包括的であり、また、全部のGタンパク質共役型受容
体での薬物発見に対する応用性を有する。
【0073】
[35S]GTPγSアッセイは、20mM HEPES、および1mMと約2
0mMとの間のMgCl2(この量は結果の至適化のため調節することができる
が、20mMが好ましい)pH7.4、約0.3nMと約1.2nMとの間の[
35S]GTPγS(この量は結果の至適化のため調節することができるが、1.
2が好ましい)および12.5ないし75μgの膜タンパク質(例えば受容体を
発現するCOS−7細胞;この量は至適化のため調節することができるが、75
μgが好ましい)および1μM GDP(この量は至適化のため変更することが
できる)を含む結合緩衝液中で1時間インキュベートすることが可能である。そ
の後、コムギ胚芽アグルチニンビーズ(25μl;アマーシャム(Amersh
am))を添加し、そして混合物を室温で別の30分間インキュベートする。そ
の後、チューブを1500×gで室温で5分間遠心分離し、そしてその後シンチ
レーション計数器で計数する。
【0074】
より少なく高価なしかし同等に応用可能な代替物が同定されており、これはま
た大スケールのスクリーニングのニーズにも合致する。フラッシュプレート[F
lash plate](商標)およびワラック[Wallac](商標)シン
チストリップを利用して、高スループットの[35S]GTPγS結合アッセイの
全体構成を定めることができる。さらに、この技術を使用すれば、[35S]GT
PγS結合を介して効力をモニターするのと同一時点で受容体へのトリチウム化
されたリガンドの結合を同時にモニターするために、該アッセイを既知のGPC
Rに利用することができる。トリチウム標識プローブおよび35S標識プローブの
双方を見るようにワラック(Wallac)ベータ計数器がエネルギーウィンド
ウを切り替えることができるために、これが可能である。このアッセイはまた、
受容体の活性化をもたらす他の型の膜活性化事象を検出するのにも使用してよい
。例えば、該アッセイは多様な受容体(Gタンパク質共役型受容体およびチロシ
ンキナーゼ受容体の双方)の32Pホスホリル化をモニターするのに使用してよい
。膜がウェルの底部に遠心分離されている場合、結合された[35S]GTPγS
もしくは32P−ホスホリル化された受容体が、ウェルの被覆されているシンチラ
ントを活性化することができる。この原理を立証するのにシンチ[Scinti
](商標)ストリップ(ワラック(Wallac))が使用されている。加えて
、該アッセイはまた、放射活性に標識されたリガンドを使用して受容体へのリガ
ンド結合を測定するための有用性も有する。類似の様式で、放射標識された結合
されたリガンドがウェルの底部に遠心分離されている場合、シンチストリップの
標識は放射標識されたリガンドに接近して活性化および検出をもたらす。
2.アデニリルシクラーゼ
細胞に基づくアッセイのため設計されたフラッシュプレート[Flash P
late](商標)アデニリルシクラーゼキット(ニュー イングランド ニュ
ークリア(New England Nuclear);カタログ番号SMP0
04A)を、粗原形質膜との使用のため改変することができる。フラッシュプレ
ート(Flash Plate)のウェルはシンチラントのコーティングを含有
し、このコーティングはcAMPを認識する特異的抗体もまた含有する。cAM
P抗体への放射活性のcAMPトレーサーの結合についての直接の競争により、
ウェル中で生成されたcAMPを定量した。以下は、受容体を発現する膜中のc
AMPレベルの変化の測定のための簡潔なプロトコルとしてはたらく。
【0075】
トランスフェクションされた細胞をトランスフェクションのおよそ3日後に収
穫する。20mM HEPES、pH7.4および10mM MgCl2を含有
する緩衝液に懸濁された細胞の均質化により膜を調製した。均質化は、ブリンク
マン(Brinkman)ポリトロン[Polytron](商標)を使用し氷
上でおよそ10秒間実施する。生じるホモジェネートを4℃で49,000×g
で15分間遠心分離する。その後、生じるペレットを20mM HEPES、p
H7.4および0.1mM EDTAを含有する緩衝液に再懸濁し、10秒間均
質化し、次いで49,000×gで4℃で15分間遠心分離する。生じるペレッ
トは利用されるまで−80℃で保存することができる。測定の日に膜ペレットを
室温でゆっくりと融解させ、20mM HEPES、pH7.4および10mM
MgCl2(これらの量は至適化することができるが、本明細書に列挙される
値が好ましい)を含有する緩衝液に再懸濁して0.60mg/mlの最終タンパ
ク質濃度を生じる(再懸濁された膜は使用まで氷上に置いた)。
【0076】
cAMP標準および検出緩衝液(11mlの検出緩衝液に対し2μCiのトレ
ーサー[125I]cAMP(100μl)を含んで成る)を調製し、そして製造
元の説明書に従って維持する。アッセイ緩衝液はスクリーニングのため新たに調
製し、そして20mM HEPES、pH7.4、10mM MgCl2、20
mM(シグマ(Sigma))、0.1単位/mlのクレアチンホスホキナーゼ
(シグマ(Sigma))、50μM GTP(シグマ(Sigma))および
0.2mM ATP(シグマ(Sigma))を含有し;アッセイ緩衝液は利用
されるまで氷上で保存することができる。NEN フラッシュプレート(Fla
sh Plate)への50μlのアッセイ緩衝液の添加、次いで50μlの膜
懸濁液の添加によりアッセイを開始する。結果として生じるアッセイ混合物を室
温で60分間インキュベートし、次いで100μlの検出緩衝液を添加する。そ
の後、プレートを追加の2〜4時間インキュベートし、次いでワラック(Wal
lac)マイクロベータ[MicroBeta](商標)シンチレーション計数
器で計数する。各アッセイプレート内に含有される標準cAMP曲線からウェル
あたりのcAMPの値を外挿する。
C.レポーターに基づくアッセイ
1.CREBレポーターアッセイ(Gs会合型受容体)
Gs刺激を検出する方法はcAMP依存性の様式で活性化される転写因子CR
EBの既知の特性に依存する。293もしくは293T細胞中でのGs共役型の
活性についてアッセイするのに、パスディテクト[PathDetect](商
標)CREBトランスレポーティング(CREB trans−Reporti
ng)系(ストラタジーン(Stratagene)、カタログ番号21901
0)を利用することができる。細胞は、哺乳動物トランスフェクションキット(
ストラタジーン(Stratagene)、カタログ番号200285)を製造
元の説明書に従って使用して、この上の系のプラスミド成分、および内在性もし
くは突然変異体の受容体をコードする指定された発現プラスミドでトランスフェ
クションする。簡潔には、400ngのpFR−Luc(Gal4認識配列を含
有するルシフェラーゼレポータープラスミド)、40ngのFA2−CREB(
Gal4のDNA結合ドメインを含有するGal4−CREB融合タンパク質)
、80ngのpCMV−受容体発現プラスミド(受容体を含んで成る)および2
0ngのCMV−SEAP(分泌型アルカリホスファターゼ発現プラスミド;ト
ランスフェクションされた細胞の培地中でアルカリホスファターゼ活性を測定し
て、サンプル間のトランスフェクション効率の変動について制御する)を、キッ
トの説明書に従ってリン酸カルシウム沈殿中に組み合わせる。沈殿物の半分を9
6穴プレートの3個のウェルに同等に配分し、細胞上で一夜保ち、そして翌朝に
新鮮培地で置き換える。トランスフェクションの開始48時間後に細胞を処理し
、そして例えばルシフェラーゼ活性についてアッセイする。
2.AP1レポーターアッセイ(Gq会合型受容体)
Gq刺激を検出する方法は、それらのプロモーター中にAP1要素を含有する
遺伝子の活性化を引き起こすGq依存性ホスホリパーゼCの既知の特性に依存す
る。リン酸カルシウム沈殿物の成分が410ngのpAP1−Luc、80ng
のpCMV−受容体発現プラスミドおよび20ngのCMV−SEAPであった
ことを除いて、CREBレポーターアッセイに関して上に示されたプロトコルに
従い、パスディテクト[Pathdetect](商標)AP−1シスレポーテ
ィング(AP−1 cis−Reporting)系(ストラタジーン(Str
atagene)、カタログ番号219073)を利用することができる。
3.CRE−Lucレポーターアッセイ
293および293T細胞をウェルあたり細胞2×104個の密度で96穴プ
レート上でプレート培養し、そして翌日、製造元の説明書に従ってリポフェクタ
ミン試薬(BRL)を使用してトランスフェクションした。DNA/脂質混合物
を以下のとおり各6個のウェルのトランスフェクションのため調製する。すなわ
ち、100μlのDMEM中の260ngのプラスミドDNAを、100μlの
DMEM中2μlの脂質と穏やかに混合した(260ngのプラスミドDNAは
、200ngの8xCRE−Lucレポータープラスミド(プラスミドの一部分
の表示については下および図1を参照されたい)、50ngの内在性受容体もし
くは非内在性受容体を含んで成るpCMVまたはpCMV単独、ならびに10n
gのGPRS発現プラスミド(pcDNA3(インヴィトロジェン(Invit
rogen)中のGPRS)より成った)。8XCRE−Lucレポータープラ
スミドは以下のとおり調製した。すなわち、pβgal−基本ベクター(Bas
ic Vector)(クロンテック(Clontech))のBglV−Hi
ndIII部位にラットソマトスタチンプロモーター(−71/+51)をクロ
ーン化することにより、ベクターSRIF−β−galを得た。アデノウイルス
鋳型AdpCF126CCRE8(7 Human Gene Therapy
1883(1996)を参照されたい)からのPCRにより、8コピーのcA
MP応答要素を得、そしてKpn−BglV部位でSRIF−β−galベクタ
ーにクローン化して8xCRE−β−galレポーターベクターをもたらした。
8xCRE−Lucレポータープラスミドは、8xCRE−β−galレポータ
ーベクター中のβ−ガラクトシダーゼ遺伝子を、HindIII−BamHI部
位でpGL3−基本ベクター(プロメガ(Promega))から得られたルシ
フェラーゼ遺伝子で置き換えることにより生成した。室温で30分のインキュベ
ーション後に、400μlのDMEMでDNA/脂質混合物を希釈し、そして1
00μlの希釈された混合物を各ウェルに添加した。細胞培養インキュベーター
中での4時間のインキュベーション後に、10%FCSを含む100μlのDM
EMを各ウェルに添加した。翌日、トランスフェクションされた細胞を、10%
FCSを含むウェルあたり200μlのDMEMで変更した。8時間後、PBS
での1回の洗浄後に、ウェルをウェルあたり100μlのフェノールレッドを含
まないDMEMに変更した。翌日、製造元の説明書に従って、ルックライト[L
ucLite](商標)レポーター遺伝子アッセイキット(パッカード(Pac
kard))を使用してルシフェラーゼ活性を測定し、そして1450マイクロ
ベータ[MicroBeta](商標)シンチレーションおよび発光計数器(ワ
ラック(Wallac))で読み取った。
4.SRF−Lucレポーターアッセイ
Gq刺激を検出するための一方法は、それらのプロモーター中に血清応答因子
を含有する遺伝子の活性化を引き起こすGq依存性のホスホリパーゼCの既知の
特性に依存する。例えばCOS7細胞でのGq共役型の活性についてアッセイす
るのに、パスディテクト[Pathdetect](商標)SRF−Lucレポ
ーティング系(ストラタジーン(Stratagene))を利用することがで
きる。製造元の説明書に従って哺乳動物トランスフェクション[Mammali
an Transfection](商標)キット(ストラタジーン(Stra
tagene)、カタログ番号200285)を使用して、系のプラスミド成分
および内在性もしくは非内在性のGPCRをコードする指定された発現プラスミ
ドで細胞をトランスフェクションする。簡潔には、410ngのSRF−Luc
、80ngのpCMV受容体発現プラスミドおよび20ngのCMV−SEAP
(分泌型アルカリホスファターゼ発現プラスミド;トランスフェクションされた
細胞の培地中でアルカリホスファターゼ活性を測定して、サンプル間のトランス
フェクションの効率の変動について制御する)を、製造元の説明書に従ってリン
酸カルシウム沈殿物中で組み合わせる。沈殿物の半分を96穴プレートの3個の
ウェルに同等に配分し、血清を含まない培地中で細胞上で24時間保つ。指定さ
れた場合は、最後の5時間、細胞を1μMのアンジオテンシンとともにインキュ
ベートする。その後細胞を溶解し、そして、製造元の説明書に従ってルックライ
ト[Luclite](商標)キット(パッカード(Packard)、カタロ
グ番号6016911)および「トライルックス(Trilux)1450マイ
クロベータ(MicroBeta)」液体シンチレーションおよび発光計数器(
ワラック(Wallac))を使用してルシフェラーゼ活性についてアッセイす
る。データはグラフパッド プリズム[GraphPad Prism](商標
)2.0a(グラフパッド ソフトウェア インク(GraphPad Sof
tware Inc.))を使用して解析することができる。
5.細胞内IP3蓄積アッセイ
第1日に、受容体(内在性および/もしくは非内在性)を含んで成る細胞を2
4穴プレート上でプレート培養することができる(通常、ウェルあたり細胞1×
105個、とは言えこの数字は至適化することができる)。第2日に、ウェルあ
たり50μlの血清を含まないDMEM中の0.25μgのDNAおよびウェル
あたり50μlの血清を含まないDMEM中の2μlのリポフェクタミンを最初
に混合することにより、細胞をトランスフェクションすることができる。溶液は
穏やかに混合し、そして室温で15〜30分間インキュベートする。0.5ml
のPBSで細胞を洗浄し、そして400μlの血清を含まない培地をトランスフ
ェクション培地と混合しかつ細胞に添加する。その後、細胞を37℃/5%CO
2で3〜4時間インキュベートし、そしてその後、トランスフェクション培地を
除去しかつウェルあたり1mlの通常の成長培地で置き換える。第3日に、細胞
を3H−ミオイノシトールで標識する。簡潔には、培地を除去し、そして細胞を
0.5mlのPBSで洗浄する。その後、0.5mlのイノシトールを含まない
/血清を含まない培地(ギブコ(GIBCO)BRL)を、ウェルあたり0.2
5μCiの3H−ミオイノシトールとともにウェルあたりに添加し、そして細胞
を37℃/5%CO2で16〜18時間一夜インキュベートする。第4日に、細
胞を0.5mlのPBSで洗浄し、そして、イノシトールを含まない/血清を含
まない培地、10μMパージリン、10mM塩化リチウムを含有する0.45m
lのアッセイ培地、もしくは0.4mlのアッセイ培地および10μMの最終濃
度までの50μlの10×ケタンセリン(ket)を添加する。その後細胞を3
7℃で30分間インキュベートする。その後細胞を0.5mlのPBSで洗浄し
、そしてウェルあたり200μlの新鮮/氷冷停止溶液(1M KOH;18m
Mホウ酸ナトリウム;3.8mM EDTA)を添加する。溶液を5〜10分間
もしくは細胞が溶解されるまで氷上で保ち、そしてその後、200μlの新鮮/
氷冷中和溶液(7.5%HCl)により中和する。その後、ライセートを1.5
mlのエッペンドルフチューブに移し、そしてチューブあたり1mlのクロロホ
ルム/メタノール(1:2)を添加する。溶液を15秒間ボルテックス攪拌し、
そして上層をバイオラッド(BioRad)AG1−X8(商標)陰イオン交換
樹脂(100〜200メッシュ)に適用する。最初に樹脂を1:1.25W/V
の水で洗浄し、そして0.9mlの上層をカラムに負荷する。カラムを10ml
の5mMミオイノシトールおよび10mlの5mMホウ酸ナトリウム/60mM
ギ酸ナトリウムで洗浄する。2mlの0.1Mギ酸/1Mギ酸アンモニウムを用
い、10mlのシンチレーションカクテルを含有するシンチレーションバイアル
中にイノシトールトリスリン酸を溶出する。10mlの0.1Mギ酸/3Mギ酸
アンモニウムで洗浄することによりカラムを再生し、そしてddH2Oで2回す
すぎ、そして水中で4℃で保存する。
【0077】
例示的結果を下の表Iに提示する:
【0078】
【表10】
【0079】
C.細胞に基づく検出アッセイ(例−TDAG8)
293細胞をプレートあたり細胞1.3×107個の密度で150mmプレー
ト上でプレート培養し、そしてプレートあたり12μgのそれぞれのDNAおよ
び60μlのリポフェクタミン試薬(BRL)を使用してトランスフェクション
した。トランスフェクションされた細胞は、トランスフェクション後24時間に
実施されるアッセイのため血清を含有する培地中で成長させた。トランスフェク
ション後48時間に実施される検出アッセイ(血清および血清を含まない培地を
比較するアッセイ;図3を参照されたい)のため、初期培地を血清もしくは血清
を含まない培地のいずれかに変更した。血清を含まない培地は、単にダルベッコ
の改変イーグル(DME)高グルコース培地(アーヴィン サイエンティフィッ
ク(Irvine Scientific)#9024)から構成された。上の
DME培地に加えて、血清を含む培地は以下、すなわち10%ウシ胎児血清(ハ
イクロン(Hyclone)#SH30071.03)、1%の100mMピル
ビン酸ナトリウム(アーヴィン サイエンティフィック(Irvine Sci
entific)#9334)、1%の20mM L−グルタミン(アーヴィン
サイエンティフィック(Irvine Scientific)#9317)
および1%のペニシリン−ストレプトマイシン溶液(アーヴィン サイエンティ
フィック(Irvine Scientific)#9366)を含有した。
【0080】
96穴のアデニリルシクラーゼ活性化フラッシュプレート[Flashpla
te](商標)を使用した(NEN:#SMP004A)。最初に、アッセイの
ための標準50μlを、ウェルあたり50pmolから0pmolまでのcAM
P濃度の範囲にわたるプレート(二重(in duplicate))に添加した。標準cAM
P(NEN:#SMP004A)を水で再構成し、そして1×PBS(アーヴィ
ン サイエンティフィック(Irvine Scientific)#9240
)を使用して連続的希釈物を作成した。次に、50μlの刺激緩衝液(NEN:
#SMP004A)を全部のウェルに添加した。cAMPの活性化もしくは不活
性化を測定するのに化合物を使用する場合は、水で希釈された各化合物10μl
を三重でそのそれぞれのウェルに添加した。使用された多様な最終濃度は1μM
から1mMまでの範囲にわたる。アデノシン5’−三リン酸、ATP(リサーチ
バイオケミカルズ インターナショナル(Research Biochem
icals International:#A−141)およびアデノシン5
’−二リン酸、ADP(シグマ(Sigma):#A2754)をアッセイで使
用した。次に、それぞれのcDNA(CMVもしくはTDAG8)でトランスフ
ェクションされた293細胞を、トランスフェクション後24(血清培地中での
アッセイの検出)もしくは48(血清および血清を含まない培地を比較するアッ
セイの検出)時間で収穫した。培地を吸引し、そして細胞を1×PBSで1回洗
浄した。その後、5mlの1×PBSを3mlの細胞解離緩衝液(シグマ(Si
gma):#C−1544)とともに細胞に添加した。解離された細胞を遠心分
離チューブに移し、そして室温で5分間遠心分離した。上清を除去し、そして、
1ミリリットルあたり細胞2×106個の最終濃度を得るように、細胞ペレット
を適切な量の1×PBSに再懸濁した。化合物を含有するウェルに、1×PBS
中の細胞50μl(ウェルあたり細胞1×105個)を添加した。プレートを室
温で15分間振とう機でインキュベートした。トレーサーcAMPを含有する検
出緩衝液を調製した。11mlの検出緩衝液(NEN:#SMP004A)中で
50μl(1μCiに等しい)の[125I]cAMP(NEN:#SMP004
A)を添加した。インキュベーション後に、トレーサーcAMPを含有するこの
検出緩衝液50μlを各ウェルに添加した。プレートを振とう機に設置し、そし
て室温で2時間インキュベートした。最後に、プレートのウェルからの溶液を吸
引し、そしてワラック(Wallac)マイクロベータ[MicroBeta]
(商標)シンチレーション計数器を使用してフラッシュプレートを計数した。
【0081】
図2Aにおいて、ATPおよびADPは内在性のTDAG8に結合し、それぞ
れ約59%および約55%のcAMPの増大をもたらす。図2Bは、内在性のT
DAG8へのATPおよびADPの結合を明示し、ここでは内在性TDAG8が
トランスフェクションされ、そして血清および血清を含まない培地中で成長され
た。血清培地中で成長された内在性のTDAG8へのATPの結合は、化合物を
伴わない内在性のTDAG8に比較して、約65%のcAMPの増大を明示し;
血清を含まない培地中では約68%の増大が存在した。血清中の内在性TDAG
8へのADPの結合は約61%の増大を明示する一方、血清を含まない培地中で
はADP結合が約62%増大の増大を明示する。ATPおよびADPは、それぞ
れ139.8μMおよび120.5μMのEC50値で内在性のTDAG8に結
合する(データは示されない)。
【0082】
図2Bに提示される結果は、血清および血清を含まない培地を比較した場合に
実質的に同一の結果を示すが、われわれの選択は血清に基づく培地を使用するこ
とである。とは言え血清を含まない培地もまた利用することが可能である。
実施例6
GPCR融合タンパク質の調製
構成的に活性化されるGPCR−Gタンパク質融合構築物の設計を以下のとお
り達成した。すなわち、ラットGタンパク質Gsα(長形態;伊藤(Itoh,
H.)ら、83 PNAS 3776(1986))の5’および3’双方の端
をそれにHindIII(5’−AAGCTT−3’)配列を包含するように工
作した。正しい配列(隣接するHindIII配列を包含する)の確認後に、そ
のベクターのHindIII制限部位を使用してサブクローニングすることによ
り、配列全体をpcDNA3.1(−)(インヴィトロジェン(Invitro
gen)、カタログ番号V795−20)にsnuttleした。pcDNA3
.1(−)へのサブクローニング後にGsαの配列について正しい向きを決定し
た。HindIII配列でラットGsα遺伝子を含有する改変されたpcDNA
3.1(−)をその後確認し;このベクターは今や「普遍的」Gsαタンパク質
ベクターとして利用可能となった。pcDNA3.1(−)ベクターは、Hin
dIII部位の上流に多様な公知の制限部位を含有し、従ってGsタンパク質の
上流に内在性の構成的に活性なGPCRのコーディング配列を挿入する能力を有
益に提供する。他の「普遍的」Gタンパク質ベクターを創製するのにこの同一の
アプローチを利用することができ、そして、もちろん、当業者に既知の他の商業
的に入手可能なもしくは占有のベクターを利用することができる。重要な基準は
、GPCRの配列がGタンパク質の配列の上流にかつ同じ読み枠で存在すること
である。
【0083】
TDAG8はGsを介して共役する一方、H9はGzを介して共役する。以下
の例示的GPCR融合タンパク質についてGsαへの融合を達成した。
【0084】
TDAG8(I225K)−Gsα融合タンパク質構築物は以下のとおり作成
した。すなわち、プライマーを以下のとおり設計した:
5’−gatcTCTAGAATGAACAGCACATGTATTGAAG−
3’(配列番号125;センス)
5’−ctagGGTACCCGCTCAAGGACCTCTAATTCCAT
AG−3’(配列番号126;アンチセンス)。
【0085】
小文字のヌクレオチドはGタンパク質とTDAG8との間の制限部位中のスペ
ーサーとして包含される。該センスおよびアンチセンスプライマーはそれぞれX
baIおよびKpnIの制限部位を包含した。
【0086】
その後、それぞれについて以下のプロトコルを使用し、PCRを利用して、上
に開示されたGsα普遍的ベクター内の融合についてそれぞれの受容体配列を保
証した。すなわち、2μlの各プライマー(センスおよびアンチセンス)、3μ
Lの10mM dNTP、10μLの10×タックプラス[TaqPlus](
商標)プレシジョン(Precision)緩衝液、1μLのタックプラス[T
aqPlus](商標)プレシジョン(Precision)ポリメラーゼ(ス
トラタジーン(Stratagene):#600211)ならびに80μLの
水を含有する別個のチューブに、100ngのTDAG8のcDNAを添加した
。TDAG8についての反応温度および周期時間は以下のとおりであった。すな
わち、最初の変性段階はそれを94℃で5分間、そして、94℃30秒間;55
℃30秒間;72℃2分間の周期を行った。最後の伸長時間は72℃で10分間
行った。順向きのPCR産物(PCR product for)を1%アガロースゲル上で泳動
し、そしてその後精製した(データは示されない)。精製された産物をXbaI
およびKpnI(ニュー イングランド バイオラブス(New Englan
d Biolabs))で消化し、そして所望の挿入物を精製しかつそれぞれの
制限部位でGs普遍的ベクターに連結した。形質転換後に陽性のクローンを単離
し、そして制限酵素消化により決定し;293細胞を使用する発現は下に示され
るプロトコルに従って達成した。TDAG8:Gs融合タンパク質についてのそ
れぞれの陽性のクローンを配列決定して正しさを確かめた。
【0087】
非内在性の構成的に活性化されるTDAG8(I225K)を含んで成るGP
CR融合タンパク質を上のとおり分析し、そして構成的活性化について確かめた
。
【0088】
H9(F236K)−Gsα融合タンパク質構築物は以下のとおり作成した。
すなわち、プライマーを以下のとおり設計した:
5’−TTAgatatcGGGGCCCACCCTAGCGGT−3’(配列
番号145;センス)
5’−ggtaccCCCACAGCCATTTCATCAGGATC−3’(
配列番号146;アンチセンス)。
【0089】
小文字のヌクレオチドはGタンパク質とH9との間の制限部位中のスペーサー
として包含される。センスおよびアンチセンスプライマーはそれぞれEcoRV
およびKpnIの制限部位を包含し、その結果、スペーサー(制限部位に帰され
る)がGタンパク質とH9との間に存在する。
【0090】
その後、それぞれについて以下のプロトコルを使用し、PCRを利用して、上
に開示されたGsα普遍的ベクター内の融合についてそれぞれの受容体配列を保
証した。すなわち、100ngの各プライマー(センスおよびアンチセンス)な
らびに45μLのPCRスーパーミックス[Supermix](商標)(ギブ
コ(Gibco)−Brl、ライフテック(LifeTech))を含有する別
個のチューブに、80ngのH9のcDNAを添加した(50μLの総反応体積
)。H9についての反応温度および周期時間は以下のとおりであった。すなわち
、最初の変性段階はそれを94℃で1、そして、94℃30秒間;55℃30秒
間、72℃2分間の周期を行った。最後の伸長時間は72℃で7分間行った。順
向きのPCR産物を1%アガロースゲル上で泳動し、そしてその後精製した(デ
ータは示されない)。精製された産物をpCRII−TOPO(商標)系にクロ
ーン化し、次いで陽性のクローンを同定した。陽性のクローンを単離し、Eco
RVおよびKpnI(ニュー イングランド バイオラブス(New Engl
and Biolabs))で消化し、そして所望の挿入物を単離し、精製しか
つそれぞれの制限部位でGs普遍的ベクターに連結した。形質転換後に陽性のク
ローンを単離し、そして制限酵素消化により決定し;293細胞を使用する発現
は下に示されたプロトコルに従って達成した。H9(F236K):Gs融合タ
ンパク質についての各陽性のクローンを配列決定して正しさを確かめた。膜は利
用されるまで凍結(−80℃)した。
【0091】
(H9はGzと共役するのであるが)Gsタンパク質により媒介されるcAM
P応答を測定する能力を確かめるため、NENアデニルシクラーゼ活性化フラッ
シュプレート[Flashplate](商標)アッセイキット(96穴の形式
)に基づき、以下のcAMP膜アッセイを利用した。「結合緩衝液」は10mM
HEPES、100mM NaClmおよび10mM MgCl(pH7.4
)より成った。「再生緩衝液」は結合緩衝液中で調製し、そして20mMホスホ
クレアチン、20Uのクレアチンホスホキナーゼ、20μM GTP、0.2m
M ATPおよび0.6mM IBMXより成った。「cAMP標準」は以下の
とおり結合緩衝液中で調製した:
【0092】
【表11】
【0093】
凍結された膜(対照としてのpCMVおよび非内在性のH(−Gs融合タンパ
ク質の双方)を(溶液中まで室温で氷上で)融解した。膜を懸濁液中までポリト
ロンで均質化した(2×15秒)。ブラッドフォード(Bradford)アッ
セイプロトコル(下を参照されたい)を使用して膜タンパク質濃度を測定した。
再生緩衝液中で膜の濃度を0.5mg/mlに希釈した(最終アッセイ濃度−ウ
ェルあたり25μg)。その後、50μlの結合緩衝液を各ウェルに添加した。
対照には、ウェルあたり50μlのcAMP標準をウェル11および12A−G
に、結合緩衝液単独を12Hに添加した(96穴の形式で)。その後、ウェルあ
たり50μlのタンパク質をウェルに添加し、そして室温で(振とう機上で)6
0分間インキュベートした。検出緩衝液(下を参照されたい)中100μlの[
125I]cAMPを各ウェルに添加した(最終−11mlの検出緩衝液中に50
μlの[125I]cAMP)。これらを室温で2時間インキュベートした。プレ
ートを8チャンネルのマニホールドで吸引し、そしてプレートの蓋で封止した。
結果(結合されたcAMPのpmol)を「プロット#15」でワラック[Wa
llac](商標)1450で読み取った。結果を図3に提示する。
【0094】
図3に提示される結果は、Gs共役型融合がシクラーゼ反応を「駆動する」こ
とが可能であり、その結果H9(F236K)の構成的活性化の測定が実現可能
であったことを示す。これらの結果に基づけば、反作用薬、アゴニストおよび部
分的アゴニストである候補化合物の直接同定がシクラーゼに基づくアッセイを使
用して可能である。
実施例6
プロトコル:[35S]GTPγSを使用する反作用薬およびアゴニストの直接同
定
われわれは、全く理解されていない理由上、例えば反作用薬のような候補化合
物の直接同定に内在性の構成的に活性のGPCRを利用したが、アッセイ間の変
動が悪化されたようになる可能性がある。その場合、好ましくは、上に開示され
たようなGPCR融合タンパク質を非内在性の構成的に活性化されるGPCRと
ともにもまた利用する。われわれは、こうしたタンパク質を使用する場合にアッ
セイ間の変動が実質的に安定化されるようであり、それにより有効なS/N比が
得られることを決定した。これは候補化合物のより確固たる同定を見込むという
有益な結果を有する。従って、直接同定のためにはGPCR融合タンパク質を使
用すること、また、利用される場合は以下のアッセイプロトコルを利用すること
が好ましい。
膜の調製
目的の、および反作用薬、アゴニストもしくは部分的アゴニストとしての候補
化合物の直接同定での使用のための非内在性の構成的に活性のオーファンGPC
R融合タンパク質を含んで成る膜は、以下のように好ましく調製する:
a.材料
「膜掻き取り緩衝液」は20mM HEPESおよび10mM EDTA、p
H7.4より構成され;「膜洗浄緩衝液」は20mM HEPESおよび0.1
mM EDTA、pH7.4から構成され;「結合緩衝液」は20mM HEP
ES、100mM NaClおよび10mM MgCl2、pH7.4から構成
される。
b.手順
全部の材料は処置の間中氷上に保つ。最初に、細胞のコンフルエントな単層か
ら培地を吸引し、次いで10mlの冷PBSですすぎ、次いで吸引する。その後
、5mlの膜掻き取り緩衝液を添加して細胞を掻き取り;これに次いで細胞抽出
物を50ml遠心管に移す(20,000rpmで4℃で17分間遠心分離され
る)。その後、上清を吸引し、そしてペレットを30mlの膜洗浄緩衝液に再懸
濁し、次いで20,000rpmで4℃で17分間遠心分離する。上清をその後
吸引し、そしてペレットを結合緩衝液に再懸濁する。その後、ブリンクマン(B
rinkman)ポリトロン[polytron](商標)ホモジェナイザーを
使用してこれを均質化する(全部の物質が懸濁液中にあるまで15〜20秒破裂
させる)。これを本明細書で「膜タンパク質」と称する。
ブラッドフォード(Bradford)タンパク質アッセイ
均質化の後に、ブラッドフォード(Bradford)タンパク質アッセイを
使用して膜のタンパク質濃度を測定する(タンパク質を約1.5mg/mlに希
釈し、等分し、そして後の使用のため凍結する(−80℃)ことができ;凍結さ
れる場合、使用のためのプロトコルは以下のとおりである。すなわち、アッセイ
の日に、凍結された膜タンパク質を室温で融解し、次いでボルテックス攪拌し、
そしてその後、約5〜10秒間約12×1,000rpmでポリトロンで均質化
し;複数の調製のためには、異なる調製物の均質化の間にホモジェナイザーを徹
底的に洗浄すべきであることが言及される)。
a.材料
結合緩衝液(上に従う);ブラッドフォード(Bradford)色素試薬;
ブラッドフォード(Bradford)タンパク質標準を製造元の説明書に従っ
て(バイオラッド(BioRad)カタログ番号500−0006)利用する。
b.手順
二重のチューブを準備する(1本は膜を包含し、そして1本は対照の「ブラン
ク」として)。それぞれは800μlの結合緩衝液を含有した。その後、10μ
lのブラッドフォード(Bradford)タンパク質標準(1mg/ml)を
各チューブに添加し、そしてその後10μlの膜タンパク質を1本のチューブに
だけ添加する(ブランクでない)。その後、200μlのブラッドフォード(B
radford)色素試薬を各チューブに添加し、次いでそれぞれをボルテック
ス攪拌する。5分後にチューブを再度ボルテックス攪拌し、そして、その中の物
質をキュベットに移す。その後、波長595でCECIL 3041分光光度計
を使用してキュベットを読み取る。
直接同定アッセイ
a.材料
GDP緩衝液は37.5mlの結合緩衝液および2mgのGDP(シグマ(S
igma)、カタログ番号G−7127)より成り、次いで0.2μM GDP
を得るため結合緩衝液で一連の希釈を行い(各ウェル中でのGDPの最終濃度は
0.1μM GDPであった);候補化合物を含んで成る各ウェルは、100μ
lのGDP緩衝液(最終濃度、0.1μM GDP)、50μlの結合緩衝液中
の膜タンパク質、および50μlの結合緩衝液中の[35S]GTPγS(0.6
nM)(10mlの結合緩衝液あたり2.5μlの[35S]GTPγS)より成
る200μlの最終体積を有する。
b.手順
候補化合物は96穴プレートの形式(これらは−80℃で凍結させることがで
きる)を使用して好ましくスクリーニングする。膜タンパク質(もしくは、対照
として、GPCR融合タンパク質を除外する発現ベクターを含む膜)を懸濁液中
まで短く均質化する。その後、上に示されたブラッドフォード(Bradfor
d)タンパク質アッセイを使用してタンパク質濃度を測定する。その後、膜タン
パク質(および対照)を結合緩衝液で0.25mg/mlに希釈する(最終アッ
セイ濃度、ウェルあたり12.5μg)。その後、100μlのGDP緩衝液を
ワラック(Wallac)シンチストリップ[Scintistrip](商標
)(ワラック(Wallac))の各ウェルに添加する。その後、5μlのピン
ツール(pin-tool)を使用して、こうしたウェルに5μlの候補化合物を移す(す
なわち、200μlの総アッセイ体積中の5μlは1:40の比であり、その結
果候補化合物の最終スクリーニング濃度は10μMである)。再度、汚染を回避
するため、各移送段階の後にはピンツールを水(1×)、エタノール(1×)お
よび水(2×)を含んで成る3個の水槽ですすぐべきであり、過剰の液体は各す
すぎの後にツールから振りそして紙およびキムワイプでぬぐって乾かすべきであ
る。その後、50μlの膜タンパク質を各ウェルに添加し(GPCR融合タンパ
ク質を含まない膜を含んで成る対照ウェルもまた利用する)、そして室温で5〜
10分間前インキュベートする。その後、50μlの結合緩衝液中の[35S]G
TPγS(0.6nM)を各ウェルに添加し、次いで室温で振とう機で60分間
インキュベートする(再度、この実施例において、プレートをホイルで覆った)
。その後、22℃で4000RPMで15分間のプレートの回転によりアッセイ
を停止する。その後、プレートを8チャンネルのマニホールドで吸引し、そして
プレートの蓋で封止する。その後、(製造元の説明書に従って)「Prot.#
37」の設定を使用してワラック(Wallacc)1450でプレートを読み
取る。
実施例7
プロトコル:確認アッセイ
上に示されたような直接同定された候補化合物の確認を提供するための独立の
アッセイのアプローチを使用して、その後確認アッセイを利用することが好まし
い。この場合、好ましい確認アッセイはシクラーゼに基づくアッセイである。
【0095】
改変されたフラッシュプレート[Flash Plate](商標)アデニリ
ルシクラーゼキット(ニュー イングランド ニュークリア(New Engl
and Nuclear);カタログ番号SMP004A)を、以下のプロトコ
ルに従って、非内在性の構成的に活性化されるオーファンGPCRに対する反作
用薬およびアゴニストとして直接同定された候補化合物の確認に、好ましく利用
する。
【0096】
トランスフェクションされた細胞をトランスフェクション後およそ3日に収穫
する。20mM HEPES、pH7.4および10mM MgCl2を含有す
る緩衝液中に懸濁された細胞の均質化により膜を調製する。均質化は、ブリンク
マン(Brinkman)ポリトロン[Polytron](商標)を使用して
氷上でおよそ10秒間実施する。生じるホモジェネートは49,000×gで4
℃で15分間遠心分離する。その後、生じるペレットを、20mM HEPES
、pH7.4および0.1mM EDTAを含有する緩衝液に再懸濁し、10秒
間均質化し、次いで49,000×gで4℃で15分間遠心分離する。生じるペ
レットは利用されるまで−80℃で保存することができる。直接同定スクリーニ
ングの日に、膜ペレットを室温でゆっくりと融解し、20mM HEPES、p
H7.4および10mM MgCl2を含有する緩衝液に再懸濁して、0.60
mg/mlの最終タンパク質濃度を生じる(再懸濁された膜は使用まで氷上に置
く)。
【0097】
cAMP標準および検出緩衝液(11mlの検出緩衝液に対して2μCiのト
レーサー[125I]cAMP(100μl)を含んで成る)は、製造元の説明書
に従って調製しかつ維持する。アッセイ緩衝液はスクリーニングのために新たに
調製し、そして20mM HEPES、pH7.4、10mM MgCl2、2
0mMホスホクレアチン(シグマ(Sigma))、0.1単位/mlのクレア
チンホスホキナーゼ(シグマ(Sigma))、50μM GTP(シグマ(S
igma))および0.2mM ATP(シグマ(Sigma))を含有し;ア
ッセイ緩衝液は利用されるまで氷上で保存することができる。
【0098】
上に従って同定された候補化合物(凍結されている場合は室温で融解させる)
を、40μMの膜タンパク質(ウェルあたり30μg)および50μlのアッセ
イ緩衝液と一緒に、好ましくは96穴プレートのウェルに添加する(ウェルあた
り3μl;12μMの最終アッセイ濃度)。その後、この混合状態を穏やかに振
とうしながら室温で30分間インキュベートする。
【0099】
インキュベーション後に、各ウェルに100μlの検出緩衝液を添加し、次い
で2〜24時間インキュベートする。その後、プレートを、(製造元の説明書に
従い)「Prot.#31」を使用してワラック(Wallac)マイクロベー
タ[MicroBeta](商標)プレートリーダーで計数する。
【0100】
本特許文書で挙げられる特許、出願および印刷された刊行物のそれぞれはこれ
によりそっくりそのまま引用により組み込まれることが意図される。
【0101】
当業者は、多数の変更および改変を、本発明の技術思想から離れることなく本
発明の好ましい態様に対し行ってよいことを認識するであろう。全部のこうした
変形物は本発明の範囲内にあることが意図される。
【0102】
多様な発現ベクターが当業者に利用可能であるが、内在性および非内在性双方
のヒトGPCRに対する利用の目的上、利用されるベクターはpCMVであるこ
とが最も好ましい。このベクターは、特許手続上の微生物の寄託の国際的承認に
関するブダペスト条約(the Budapest Treaty for t
he International Recognition of the
Deposit of Microorganisms for the Pu
rpose of Patent Procedure)の規定の下に、199
8年10月13日にアメリカン タイプ カルチャー コレクション(Amer
ican Type Culture Collection)(ATCC)(
10801 University Blvd.,米国バージニア州マナサス
20110−2209)に寄託された。該DNAはATCCにより試験され、そ
してそうであることが決定された。ATCCはpCMVに対し以下の寄託番号:
ATCC#203351を割り当てている。
(配列表)
【技術分野】
【0001】
本特許出願は、1998年10月13日に米国特許・商標庁に出願された米国
第09/170,496号の一部継続出願であり、かつそれからの優先権を主張
する。本出願は、以下の仮出願(全部は示された日付に米国特許・商標庁に米国
エクスプレスメール(U.S.Express Mail)を介して出願された
)、すなわち1998年11月27日に出願された米国仮出願第60/110,
060号;1999年2月16日に出願された米国仮出願第60/120,41
6号;1998年11月20日に出願された米国仮出願第60/109,213
号の利益を主張する1999年2月26日に出願された米国仮出願第60/12
1,852号;1999年3月12日に出願された米国仮出願第60/123,
944号;1999年3月12日に出願された米国仮出願第60/123,94
5号;1999年3月12日に出願された米国仮出願第60/123,948号
;1999年3月12日に出願された米国仮出願第60/123,951号;1
999年3月12日に出願された米国仮出願第60/123,946号;199
9年3月12日に出願された米国仮出願第60/123,949号;1999年
8月27日に出願された米国仮出願第60/151,114号および1998年
11月12日に出願された米国仮出願第60/108,029号の利益を主張す
る1999年9月3日に出願された米国仮出願第60/152,524号;19
99年5月28日に出願された米国仮出願第60/136,436号;1999
年5月28日に出願された米国仮出願第60/136,439号;1999年5
月28日に出願された米国仮出願第60/136,567号;1999年5月2
8日に出願された米国仮出願第60/137,127号;1999年5月28日
に出願された米国仮出願第60/137,131号;1999年5月28日に出
願された米国仮出願第60/136,437号の利益を主張する1999年6月
29日に出願された米国仮出願第60/141,448号;1999年9月29
日に出願された米国仮出願第60/156,633号;1999年9月29日に
出願された米国仮出願第60/156,555号;1999年9月29日に出願
された米国仮出願第60/156,634号;1999年9月29日に出願され
た米国仮出願第____号(アリーナ ファーマシューティカルズ インク(A
rena Pharmaceuticals,Inc.)処理予定表番号:CH
N10−1);1999年10月1日に出願された米国仮出願第____号(ア
リーナ ファーマシューティカルズ インク(Arena Pharmaceu
ticals,Inc.)処理予定表番号:RUP6−1);1999年10月
1日に出願された米国仮出願第____号(アリーナ ファーマシューティカル
ズ インク(Arena Pharmaceuticals,Inc.)処理予
定表番号:RUP7−1);1999年10月1日に出願された米国仮出願第_
___号(アリーナ ファーマシューティカルズ インク(Arena Pha
rmaceuticals,Inc.)処理予定表番号:CHN6−1);19
99年10月1日に出願された米国仮出願第____号(アリーナ ファーマシ
ューティカルズ インク(Arena Pharmaceuticals,In
c.)処理予定表番号:RUP5−1);ならびに1999年10月1日に出願
された米国仮出願第____号(アリーナ ファーマシューティカルズ インク
(Arena Pharmaceuticals,Inc.)処理予定表番号:
CHN9−1)からの優先権の利益もまた主張する。本出願は、1999年10
月12日に(米国エクスプレスメール(U.S.Express Mail)を
介して)出願された同時係属中の米国第____号(ウッドコック(Woodc
ock)、ウォッシュバーン(Washburn)、クルツ(Kurtz)、マ
キエヴィッツ(Makiewicz)とノリス(Norris))、LLP処理
予定表番号AREN−0050)および1999年7月30日に出願された米国
第09/364,425号(双方は引用により本明細書に組み込まれる)にもま
た関する。本出願は、1999年10月12日に(米国エクスプレスメール(U
.S.Express Mail)を介して)出願された米国第____号(ウ
ッドコック(Woodcock)、ウォッシュバーン(Washburn)、ク
ルツ(Kurtz)、マキエヴィッツ(Makiewicz)とノリス(Nor
ris))、LLP処理予定表番号AREN−0054)(そっくりそのまま引
用により本明細書に組み込まれる)に対する優先権もまた主張する。前述の出願
のそれぞれはそっくりそのまま引用により本明細書に組み込まれる。
【0002】
(発明の分野)
本特許明細書に開示される発明は、膜貫通受容体、およびより具体的にはヒト
Gタンパク質共役型受容体、そしてとりわけ該受容体の構成的活性を確立するも
しくは高めるよう改変されているGPCRに関する。好ましくは、改変されたG
PCRは、治療薬として潜在的応用性を有する受容体のアゴニスト、反作用薬も
しくは部分的アゴニストとしての候補化合物の直接の同定に使用される。
【背景技術】
【0003】
(発明の背景)
ヒトでは多数の受容体のクラスが存在するが、はるかに最も豊富かつ治療に関
係するものは、Gタンパク質共役型受容体(GPCRもしくは複数GPCR)の
クラスにより代表される。ヒトゲノム内には数十万個の遺伝子が存在することが
推定されており、そしてこれらのなかでおよそ2%もしくは2,000個の遺伝
子がGPCRをコードすると推定されている。内在性リガンドが同定されている
、GPCRを包含する受容体は「既知」受容体と称される一方、内在性リガンド
が同定されていない受容体は「オーファン」受容体と称される。GPCRは製薬
学的製品の開発に重要な一領域を代表する。すなわち、100種の既知のGPC
Rのおよそ20種から全処方薬の60%が開発されている。
【0004】
GPCRは1つの共通な構造モチーフを共有する。全部のこれらの受容体は、
7個のαヘリックスを形成する22ないし24個の間の疎水性アミノ酸の7個の
連なりを有し、そのそれぞれは膜にまたがる(各広がり(span)は数字により同定
されている。すなわち膜貫通−1(TM−1)、膜貫通−2(TM−2)など)
。膜貫通ヘリックスは、細胞膜の外もしくは「細胞外」側で、膜貫通−2と膜貫
通−3、膜貫通−4と膜貫通−5、および膜貫通−6と膜貫通−7の間でアミノ
酸の鎖により結合されている(これらはそれぞれ「細胞外」領域1、2および3
(EC−1、EC−2およびEC−3)と称される)。膜貫通ヘリックスは、細
胞膜の内もしくは「細胞内」側で、膜貫通−1と膜貫通−2、膜貫通−3と膜貫
通−4、および膜貫通−5と膜貫通−6の間でもまたアミノ酸の鎖により結合さ
れている(これらはそれぞれ「細胞内」領域1、2および3(IC−1、IC−
2およびIC−3)と称される)。受容体の「カルボキシ」(「C」)末端は細
胞内の細胞内空隙中に存し、また、受容体の「アミノ」(「N」)末端は細胞の
外側の細胞外空隙中に存する。
【0005】
一般に、内在性リガンドが受容体と結合する場合(しばしば受容体の「活性化
」と称される)、細胞内領域のコンホメーションの変化が存在し、それは細胞内
領域と細胞内の「Gタンパク質」との間の共役(coupling)を見込む。GPCRは
Gタンパク質に関して「混雑して」いる、すなわちGPCRは1種以上のGタン
パク質と相互作用する可能性があることが報告されている。ケナキン(Kena
kin,T.)、43 Life Sciences 1095(1988)を
参照されたい。他のGタンパク質が存在するが、現在のところGq、Gs、Gi
、GzおよびGoが同定されたGタンパク質である。Gタンパク質との内在性リ
ガンドで活性化されたGPCRの共役がシグナル伝達カスケード過程(「シグナ
ル伝達」と称される)を開始する。正常な条件下では、シグナル伝達は最終的に
細胞の活性化もしくは細胞の阻害をもたらす。受容体のIC−3ループならびに
カルボキシ末端がGタンパク質と相互作用すると考えられている。
【0006】
GPCRは、生理学的条件下では2種の異なるコンホメーション、すなわち「
不活性」状態と「活性状態」との間の平衡で細胞膜中に存在する。不活性状態の
受容体は、細胞内のシグナル伝達経路に連結して生物学的応答を生じさせること
が不可能である。受容体のコンホメーションの活性状態への変化は(Gタンパク
質を介する)伝達経路への連鎖を可能にし、そして生物学的応答を生じさせる。
【0007】
受容体は、内在性リガンドもしくは薬物のような化合物により活性状態で安定
化させることができる。独占的に限定されるものでないが受容体のアミノ酸配列
に対する改変を挙げることができる最近の発見は、活性状態のコンホメーション
にある受容体を助長かつ安定化するための内在性リガンドもしくは薬物以外の手
段を提供する。これらの手段は、受容体への内在性リガンドの結合の効果を刺激
することにより、受容体を活性状態で効果的に安定化する。こうしたリガンドに
依存しない手段による安定化を「構成的受容体活性化」と命名する。
【発明の概要】
【課題を解決するための手段】
【0008】
(発明の要約)
内在性ヒトGPCRの非内在性変種およびそれらの用途を本明細書で開示する
。
本発明は、例えば、以下を提供する:
(項目1) hARE−3(F313K)を含んで成る、非内在性で、構
成的に活性化される変種ヒトGタンパク質共役型受容体をコードするcDNA。
(項目2) 項目1記載のcDNAによりコードされる、非内在性の変
種ヒトGタンパク質共役型受容体。
(項目3) ベクターおよび項目1記載のcDNAを含んで成るプラス
ミド。
(項目4) 項目3記載のプラスミドを含んで成る宿主細胞。
(項目5) hARE−4(V233K)を含んで成る、非内在性で、構
成的に活性化される変種ヒトGタンパク質共役型受容体をコードするcDNA。
(項目6) 項目5記載のcDNAによりコードされる、非内在性の変
種ヒトGタンパク質共役型受容体。
(項目7) ベクターおよび項目5記載のcDNAを含んで成るプラス
ミド。
(項目8) 項目7記載のプラスミドを含んで成る宿主細胞。
(項目9) hARE−5(A240K)を含んで成る、非内在性で、構
成的に活性化される変種ヒトGタンパク質共役型受容体をコードするcDNA。
(項目10) 項目9記載のcDNAによりコードされる、非内在性の
変種ヒトGタンパク質共役型受容体。
(項目11) ベクターおよび項目5記載のcDNAを含んで成るプラ
スミド。
(項目12) 項目11記載のプラスミドを含んで成る宿主細胞。
(項目13) hGPCR14(L257K)を含んで成る、非内在性で
、構成的に活性化される変種ヒトGタンパク質共役型受容体をコードするcDN
A。
(項目14) 項目13記載のcDNAによりコードされる、非内在性
の変種ヒトGタンパク質共役型受容体。
(項目15) ベクターおよび項目13記載のcDNAを含んで成るプ
ラスミド。
(項目16) 項目15記載のプラスミドを含んで成る宿主細胞。
(項目17) hGPCR27(C283K)を含んで成る、非内在性で
、構成的に活性化される変種ヒトGタンパク質共役型受容体をコードするcDN
A。
(項目18) 項目17記載のcDNAによりコードされる、非内在性
の変種ヒトGタンパク質共役型受容体。
(項目19) ベクターおよび項目17記載のcDNAを含んで成るプ
ラスミド。
(項目20) 項目19記載のプラスミドを含んで成る宿主細胞。
(項目21) hARE−1(E232K)を含んで成る、非内在性で、
構成的に活性化される変種ヒトGタンパク質共役型受容体をコードするcDNA
。
(項目22) 項目21記載のcDNAによりコードされる、非内在性
の変種ヒトGタンパク質共役型受容体。
(項目23) ベクターおよび項目21記載のcDNAを含んで成るプ
ラスミド。
(項目24) 項目23記載のプラスミドを含んで成る宿主細胞。
(項目25) hARE−2(G285K)を含んで成る、非内在性で、
構成的に活性化される変種ヒトGタンパク質共役型受容体をコードするcDNA
。
(項目26) 項目25記載のcDNAによりコードされる、非内在性
の変種ヒトGタンパク質共役型受容体。
(項目27) ベクターおよび項目25記載のcDNAを含んで成るプ
ラスミド。
(項目28) 項目27記載のプラスミドを含んで成る宿主細胞。
(項目29) hPPR1(L239K)を含んで成る、非内在性で、構
成的に活性化される変種ヒトGタンパク質共役型受容体をコードするcDNA。
(項目30) 項目29記載のcDNAによりコードされる、非内在性
の変種ヒトGタンパク質共役型受容体。
(項目31) ベクターおよび項目29記載のcDNAを含んで成るプ
ラスミド。
(項目32) 項目31記載のプラスミドを含んで成る宿主細胞。
(項目33) hG2A(K232A)を含んで成る、非内在性で、構成
的に活性化される変種ヒトGタンパク質共役型受容体をコードするcDNA。
(項目34) 項目33記載のcDNAによりコードされる、非内在性
の変種ヒトGタンパク質共役型受容体。
(項目35) ベクターおよび項目33記載のcDNAを含んで成るプ
ラスミド。
(項目36) 項目35記載のプラスミドを含んで成る宿主細胞。
(項目37) hRUP3(L224K)を含んで成る、非内在性で、構
成的に活性化される変種ヒトGタンパク質共役型受容体をコードするcDNA。
(項目38) 項目37記載のcDNAによりコードされる、非内在性
の変種ヒトGタンパク質共役型受容体。
(項目39) ベクターおよび項目37記載のcDNAを含んで成るプ
ラスミド。
(項目40) 項目39記載のプラスミドを含んで成る宿主細胞。
(項目41) hRUP5(A236K)を含んで成る、非内在性で、構
成的に活性化される変種ヒトGタンパク質共役型受容体をコードするcDNA。
(項目42) 項目41記載のcDNAによりコードされる、非内在性
の変種ヒトGタンパク質共役型受容体。
(項目43) ベクターおよび項目41記載のcDNAを含んで成るプ
ラスミド。
(項目44) 項目42記載のプラスミドを含んで成る宿主細胞。
(項目45) hRUP6(N267K)を含んで成る、非内在性で、構
成的に活性化される変種ヒトGタンパク質共役型受容体をコードするcDNA。
(項目46) 項目45記載のcDNAによりコードされる、非内在性
の変種ヒトGタンパク質共役型受容体。
(項目47) ベクターおよび項目45記載のcDNAを含んで成るプ
ラスミド。
(項目48) 項目47記載のプラスミドを含んで成る宿主細胞。
(項目49) hRUP7(A302K)を含んで成る、非内在性で、構
成的に活性化される変種ヒトGタンパク質共役型受容体をコードするcDNA。
(項目50) 項目49記載のcDNAによりコードされる、非内在性
の変種ヒトGタンパク質共役型受容体。
(項目51) ベクターおよび項目49記載のcDNAを含んで成るプ
ラスミド。
(項目52) 項目51記載のプラスミドを含んで成る宿主細胞。
(項目53) hCHN4(V236K)を含んで成る、非内在性で、構
成的に活性化される変種ヒトGタンパク質共役型受容体をコードするcDNA。
(項目54) 項目53記載のcDNAによりコードされる、非内在性
の変種ヒトGタンパク質共役型受容体。
(項目55) ベクターおよび項目53記載のcDNAを含んで成るプ
ラスミド。
(項目56) 項目55記載のプラスミドを含んで成る宿主細胞。
(項目57) hMC4(A244K)を含んで成る、非内在性で、構成
的に活性化される変種ヒトGタンパク質共役型受容体をコードするcDNA。
(項目58) 項目57記載のcDNAによりコードされる、非内在性
の変種ヒトGタンパク質共役型受容体。
(項目59) ベクターおよび項目57記載のcDNAを含んで成るプ
ラスミド。
(項目60) 項目60記載のプラスミドを含んで成る宿主細胞。
(項目61) hCHN3(S284K)を含んで成る、非内在性で、構
成的に活性化される変種ヒトGタンパク質共役型受容体をコードするcDNA。
(項目62) 項目61記載のcDNAによりコードされる、非内在性
の変種ヒトGタンパク質共役型受容体。
(項目63) ベクターおよび項目61記載のcDNAを含んで成るプ
ラスミド。
(項目64) 項目63記載のプラスミドを含んで成る宿主細胞。
(項目65) hCHN6(L352K)を含んで成る、非内在性で、構
成的に活性化される変種ヒトGタンパク質共役型受容体をコードするcDNA。
(項目66) 項目65記載のcDNAによりコードされる、非内在性
の変種ヒトGタンパク質共役型受容体。
(項目67) ベクターおよび項目65記載のcDNAを含んで成るプ
ラスミド。
(項目68) 項目67記載のプラスミドを含んで成る宿主細胞。
(項目69) hCHN8(N235K)を含んで成る、非内在性で、構
成的に活性化される変種ヒトGタンパク質共役型受容体をコードするcDNA。
(項目70) 項目69記載のcDNAによりコードされる、非内在性
の変種ヒトGタンパク質共役型受容体。
(項目71) ベクターおよび項目69記載のcDNAを含んで成るプ
ラスミド。
(項目72) 項目71記載のプラスミドを含んで成る宿主細胞。
(項目73) hH9(F236K)を含んで成る、非内在性で、構成的
に活性化される変種ヒトGタンパク質共役型受容体をコードするcDNA。
(項目74) 項目73記載のcDNAによりコードされる、非内在性
の変種ヒトGタンパク質共役型受容体。
(項目75) ベクターおよび項目73記載のcDNAを含んで成るプ
ラスミド。
(項目76) 項目74記載のプラスミドを含んで成る宿主細胞。
(項目77) hAT1(F239K);hAT1(N111A);hA
T1(AT2K255IC3);およびhAT1(A243+)より成る群から
選択される、非内在性で、構成的に活性化される変種ヒトGタンパク質共役型A
T1受容体をコードするcDNA。
(項目78) 項目77記載のcDNAによりコードされる、非内在性
の変種ヒトGタンパク質共役型受容体。
(項目79) ベクターおよび項目77記載のcDNAを含んで成るプ
ラスミド。
(項目80) 項目79記載のプラスミドを含んで成る宿主細胞。
【図面の簡単な説明】
【0009】
【図1】発明にかかる、8XCRE−Lucレポータープラスミドの表示である(実施例4(c)3を参照されたい)。
【図2】発明にかかる、内在性TDAG8へのATPおよびADPの結合の結果のグラフ表示(2A)ならびに血清および血清を含まない培地中での比較(2B)である。
【図3】発明にかかる、GPCR融合タンパク質H9(F236K):Gsαに対するCMVの比較的なシグナル伝達の結果のグラフ表示である。
【発明を実施するための形態】
【0010】
(詳細な記述)
受容体を中核として発展してきた学術文献は、受容体に対する多様な影響を有
するリガンドを指す多数の用語を採用している。明瞭化および一貫性のため、本
特許明細書を通じて以下の定義を使用するであろう。これらの定義がこれらの用
語の他の定義と矛盾する限り、以下の定義が支配する:
アゴニストは、それらが受容体に結合する場合に細胞内応答を活性化する、も
しくは膜へのGTP結合を高める物質(例えばリガンド、候補化合物)を意味す
る。
【0011】
本明細書で使用されるアミノ酸の略語を表Aに示す:
【0012】
【表1】
【0013】
部分的アゴニストは、それらが受容体に結合する場合にアゴニストが活性化す
るより小さい度合(degree)/程度(extent)まで細胞内応答を活性化する、もしく
はアゴニストが高めるより小さい度合(degree)/程度(extent)まで膜へのGTP
結合を高める物質(例えばリガンド、候補化合物)を意味する。
【0014】
アンタゴニストは、アゴニストと同一の部位で受容体に競争的に結合するがし
かし活性型の受容体により開始される細胞内応答を活性化せず、そしてそれによ
りアゴニストもしくは部分的アゴニストによる細胞内応答を阻害する可能性のあ
る物質(例えばリガンド、候補化合物)を意味する。アンタゴニストは、アゴニ
ストもしくは部分的アゴニストの非存在下で基線の細胞内応答を減少させない。
【0015】
候補化合物は、スクリーニング技術に基づいて分析できる分子(例えば、そし
て制限でなく化合物)を意味する。好ましくは、「候補化合物」という句は、間
接的同定方法により既に決定されたような、受容体に対する反作用薬、アゴニス
トもしくはアンタゴニストより成る群から選択される化合物であることが公知で
あった化合物(「間接的に同定された化合物」)を包含せず;より好ましくは、
最低1種の哺乳動物で治療上の効能を有することが既に決定されている間接的に
同定された化合物を包含せず;そして最も好ましくは、ヒトで治療上の有用性を
有することが既に決定されている間接的に同定された化合物を包含しない。
【0016】
組成物は最低1種の成分を含んで成る物質を意味し;「製薬学的組成物」は組
成物の一例である。
【0017】
化合物の効能は、受容体結合親和性と対照的に、受容体の機能性を阻害もしく
は刺激する化合物の能力の大きさを意味する。化合物の効能の検出の例示的手段
を本特許明細書の実施例のセクションに開示する。
【0018】
コドンは、リン酸基に結合されたヌクレオシド(アデノシン(A)、グアノシ
ン(G)、シチジン(C)、ウリジン(U)およびチミジン(T))を一般に含
んで成りかつ翻訳される場合に1個のアミノ酸をコードするひとそろいの3個の
ヌクレオチド(もしくはヌクレオチドに対する同等物)を意味する。
【0019】
構成的に活性化される受容体は構成的受容体活性化にさらされる受容体を意味
する。構成的に活性化される受容体は内在性もしくは非内在性であることができ
る。
【0020】
構成的な受容体の活性化は、その内在性リガンドもしくはその化学的同等物と
の受容体の結合以外の手段による、活性状態での受容体の安定化を意味する。
【0021】
接触させるもしくは接触することは、インビトロの系であろうとインビボの系
であろうと最低2つの部分を一緒にすることを意味する。
【0022】
直接同定するもしくは直接同定されるは、「候補化合物」という句に関係して
、構成的に活性化される受容体、好ましくは構成的に活性化されるオーファン受
容体、そして最も好ましくは構成的に活性化されるGタンパク質共役型の細胞表
面のオーファン受容体に対する候補化合物のスクリーニング、およびこうした化
合物の化合物の効能の評価を意味する。この句は、どの状況下でも、「間接的に
同定する」もしくは「間接的に同定される」という句により包含されるもしくは
それを包含すると解釈もしくは理解されるべきでない。
【0023】
内在性は哺乳動物が天然に産生する物質を意味する。例えば、そして制限でな
く「受容体」という用語に関しての内在性は、哺乳動物(例えば、そして制限で
なくヒト)もしくはウイルスにより天然に産生されるものを意味する。対照的に
、これに関して非内在性という用語は、哺乳動物(例えば、そして制限でなくヒ
ト)もしくはウイルスにより天然に産生されないものを意味する。例えば、そし
て制限でなく、その内在性の形態で構成的に活性でないがしかし操作される場合
に構成的に活性になる受容体は、本明細書で「非内在性の構成的に活性化される
受容体」と最も好ましく称される。双方の用語は「インビボ」および「インビト
ロ」双方の系を記述するのに利用することができる。例えば、そして制限でなく
、スクリーニングのアプローチにおいて、内在性もしくは非内在性の受容体はイ
ンビトロのスクリーニング系に関してであってよい。さらなる一例として、そし
て制限としてでなく、非内在性の構成的に活性化される受容体を包含するように
哺乳動物のゲノムが操作されている場合には、インビボ系による候補化合物のス
クリーニングが実現可能である。
【0024】
Gタンパク質共役型受容体融合タンパク質およびGPCR融合タンパク質は、
本明細書に開示される本発明に関して、それぞれ、最低1種のGタンパク質、最
も好ましくはこうしたGタンパク質のαサブユニット(これはGTPを結合する
サブユニットである)に融合された、内在性の構成的に活性化されるGPCRも
しくは非内在性の構成的に活性化されるGPCRを含んで成る非内在性タンパク
質を意味し、Gタンパク質は好ましくは内在性のオーファンGPCRと天然に共
役するGタンパク質と同一の型のものである。例えば、そして制限でなく、内在
性の状態において、Gタンパク質「Gsα」がGPCRと共役する主なGタンパ
ク質である場合、特定のGPCRに基づくGPCR融合タンパク質はGsαに融
合されたGPCRを含んで成る非内在性タンパク質であることができ;下に示さ
れるであろうようないくつかの状況では、主なものでないGタンパク質をGPC
Rに融合することができる。Gタンパク質は構成的に活性なGPCRのC末端に
直接融合することができるか、もしくは2者の間にスペーサーが存在してよい。
【0025】
宿主細胞は、その中に組み込まれたプラスミドおよび/もしくはベクターを有
することが可能な細胞を意味する。原核生物宿主細胞の場合には、プラスミドは
宿主細胞の複製物のような自律的分子として典型的に複製され(一般に、プラス
ミドはその後真核生物宿主細胞への導入のため単離される);真核生物宿主細胞
の場合には、真核生物宿主細胞が複製する場合にプラスミドが複製するように、
プラスミドが宿主細胞の細胞DNAに組み込まれる。好ましくは、本明細書に開
示される本発明の目的上、宿主細胞は真核生物、より好ましくは哺乳動物であり
、そして最も好ましくは293、293TおよびCOS−7細胞より成る群から
選択される。
【0026】
間接的に同定するもしくは間接的に同定されるは、内在性受容体に特異的な内
在性リガンドの同定、リガンド−受容体の相互作用を妨害および/もしくはそれ
と競争するものの決定のための受容体に対する候補化合物のスクリーニング、な
らびに活性化された受容体に関連する最低1種のセカンドメッセンジャー経路に
影響を及ぼす化合物の効能の評価を必要とする、薬物発見過程への伝統的アプロ
ーチを意味する。
【0027】
「応答」という用語に関係した阻害するもしくは阻害は、化合物の非存在下と
対照的に、化合物の存在下で応答が低下もしくは予防されることを意味する。
【0028】
反作用薬は、内在性の形態の受容体もしくは構成的に活性化された形態の受容
体のいずれかに結合し、かつ、アゴニストもしくは部分的アゴニストの非存在下
で観察される正常の基準レベルの活性より低い、活性の形態の受容体により開始
される基線の細胞内応答を阻害するか、もしくは膜へのGTP結合を減少させる
物質(例えばリガンド、候補化合物)を意味する。好ましくは、基線の細胞内応
答は、反作用薬の非存在下の基礎の応答に比較して、反作用薬の存在下で最低3
0%、より好ましくは最低50%、そして最も好ましくは最低75%阻害される
。
【0029】
既知の受容体は、その受容体に特異的な内在性リガンドが同定されている内在
性受容体を意味する。
【0030】
リガンドは、内在性の天然に存在する受容体に特異的な内在性の天然に存在す
る分子を意味する。
【0031】
内在性受容体の核酸および/もしくはアミノ酸の配列に関しての突然変異体も
しくは突然変異は、突然変異された形態の内在性の構成的に活性化されない受容
体が該受容体の構成的活性化を明示するような、こうした内在性の配列に対する
指定された1個の変化もしくは複数の変化を意味する。特定の配列に対する同等
物に関して、(a)その後の突然変異された形態のヒト受容体の構成的活性化の
レベルが該受容体の第一の突然変異により明示されるものと実質的に同一であり
;そして(b)その後の突然変異された形態の受容体と該受容体の第一の突然変
異との間の配列(アミノ酸および/もしくは核酸)の相同性のパーセントが最低
約80%、より好ましくは最低約90%、そして最も好ましくは最低95%であ
る場合、その後の突然変異された形態のヒト受容体は該ヒト受容体の第一の突然
変異に同等であるとみなされる。理想的には、また、構成的活性化を達成するた
めの本明細書に開示される最も好ましいカセットはGPCRの内在性の形態と非
内在性の形態との間の単一のアミノ酸および/もしくはコドンの変化を包含する
という事実のために、配列の相同性のパーセントは最低98%であるべきである
。
【0032】
非オーファン受容体は、内在性の天然に存在するリガンドに特異的な内在性の
天然に存在する分子を意味し、ここで、受容体へのリガンドの結合が細胞内シグ
ナル伝達経路を活性化する。
【0033】
オーファン受容体は、その受容体に特異的な内在性リガンドが同定されていな
いかもしくは未知である内在性受容体を意味する。
【0034】
製薬学的組成物は最低1種の有効成分を含んで成る組成物を意味し、それによ
り該組成物は哺乳動物(例えば、そして制限でなくヒト)における特定の効能の
ある結果についての検討に基づいて分析できる。当業者は、ある有効成分が当業
者のニーズに基づき所望の効能のある結果を有するかどうかを決定するのに適切
な技術を理解かつ認識するであろう。
【0035】
プラスミドはベクターおよびcDNAの組み合わせを意味する。一般に、プラ
スミドは、cDNAの複製および/もしくはそのタンパク質としての発現の目的
上、宿主細胞に導入される。
【0036】
「応答」という用語に関係した刺激するもしくは刺激することは、化合物の非
存在下と対照的に化合物の存在下で応答が増大されることを意味する。
【0037】
cDNAに関してのベクターは、最低1個のcDNAを組み込むことが可能か
つ宿主細胞への組み込みが可能な環状DNAを意味する。
【0038】
以下のセクションの階層は表象的な効能について示され、また、後に続く開示
もしくは請求の範囲に対する制限として意図されず、またそのように解釈される
べきでもない。
A.緒言
受容体の伝統的な研究は、発見が受容体に影響を及ぼす可能性のあるアンタゴ
ニストおよび他の分子を見出すよう進めることができる前に、内在性リガンドを
最初に同定しなければならないという(歴史的に基づく)演繹的仮定から常に進
められてきた。アンタゴニストを最初に知ることができた場合であっても、内在
性リガンドを探すように研究が直ちに拡大されてきた。この思考様式は、構成的
に活性化される受容体の発見の後でさえ、受容体の研究で持続してきた。これま
で認識されていなかったことは、受容体のアゴニスト、部分的アゴニストおよび
反作用薬の発見に最も有用であるのは活性状態の受容体であるということである
。過度に活性の受容体もしくは過小活性の受容体から生じる疾患に対して、治療
薬で望まれるものは、必ずしも内在性リガンドに対するアンタゴニストである薬
物でなく、それぞれ、受容体の活性状態を低下させるもしくは受容体の活性を高
めるよう作用する化合物である。これは、活性の受容体状態の活性を低下させる
もしくは高める化合物は内在性リガンドと同一の部位で結合する必要がないため
である。従って、本発明の方法により教示されるとおり、治療的化合物について
のいかなる研究も、リガンドに依存しない活性状態に対して化合物をスクリーニ
ングすることにより開始すべきである。
B.ヒトGPCRの同定
ヒトゲノムプロジェクトの努力の成果は、ヒトゲノム内に配置されている核酸
配列に関する夥しい量の情報の同定につながり;それは、いずれかの特定のゲノ
ム配列がヒトタンパク質を翻訳する読取り枠情報を含有するもしくは含有するか
も知れないかどうかに関する理解もしくは認識を伴わずに、遺伝子配列情報が利
用可能にされているというこの努力において真実である。ヒトゲノム内の核酸配
列のいくつかの同定方法は当業者の範囲内にある。例えば、そして制限でなく、
本明細書で開示される多様なヒトGPCRは、ジェンバンク[GenBank]
(商標)データベースを再検討することにより発見された一方、他のGPCRは
、既に配列決定されたGPCRの核酸配列を利用してESTデータベースのBL
AST(商標)検索を実施することにより発見された。下の表Bは、GPCRの
それぞれの相同受容体と一緒に、われわれが発見した数種の内在性GPCRを列
挙する。
【0039】
【表2】
【0040】
受容体の相同性は、人体内での受容体の役割の正しい認識を得ることに関して
有用である。本特許明細書が進めるように、われわれは、これらの受容体の非内
在性で構成的に活性化される変種を確立するようにこれらの受容体を突然変異す
るための技術を開示するであろう。
【0041】
本明細書に開示される技術は、本特許明細書が進行するに従って明らかになる
であろうように、技術既知の他のヒトのオーファンGPCRにもまた適用されて
いる。
C.受容体スクリーニング
本明細書に開示される、非内在性で、構成的に活性化される変種ヒトGPCR
に対する候補化合物のスクリーニングは、受容体の内在性リガンドの使用を必要
とすることなく、この細胞表面受容体で作用する候補化合物の直接の同定を許す
。本明細書に開示される内在性の変種ヒトGPCRが発現および/もしくは過剰
発現されている身体内の領域を決定することにより、受容体の発現および/もし
くは過剰発現を伴う関連疾患/障害状態を決定することが可能であり;こうした
一アプローチを本特許明細書で開示する。
【0042】
本明細書に開示されるヒトGPCRの構成的活性化を明示することができる突
然変異の創製に関して、GPCRのTM6内に配置されると想定されているプロ
リン残基からの距離に基づき;このアルゴリズム技術は、引用により本明細書に
組み込まれる同時係属中かつ共通に譲渡される特許明細書米国第09/170,
496号に開示されている。該アルゴリズム技術は、伝統的な配列の「整列」で
はなく、しかしむしろ前述のTM6のプロリン残基からの指定された距離に基づ
く。(思うに受容体のIC3領域中に配置される)この残基から16アミノ酸残
基に配置されるアミノ酸残基を最も好ましくはリシン残基に突然変異することに
より、こうした活性化を得てよい。この目的を達成するためにはこの位置の突然
変異で他のアミノ酸残基が有用であるかも知れない。
D.疾患/障害の同定および/もしくは選択
下により詳細に示されるであろうとおり、最も好ましくは非内在性で、構成的
に活性化されるGPCRに対する反作用薬を本発明の方法論により同定すること
ができる。こうした反作用薬は、本受容体に関連する疾患治療のための薬物発見
プログラムでのリード化合物として理想的な候補である。GPCRに対する反作
用薬を直接同定してそれにより製薬学的組成物の開発を可能にする能力のため、
GPCRに関連する疾患および障害の研究が適切である。例えば、GPCRの存
在について疾患に罹った組織サンプルおよび正常な組織サンプルの双方を走査す
ることは、今や、学究的修練、もしくは特定のGPCRに対する内在性リガンド
を同定する途に沿って追跡することができるもの以上となっている。組織の走査
は広範な健康なおよび疾患に罹った組織にわたって実施することができる。こう
した組織の走査は、特定の受容体のある疾患および/もしくは障害との関連付け
の好ましい第一段階を提供する。例えば、本明細書に開示されるGPCRのいく
つかの例示的ドットブロットおよびRT−PCRの結果については、同時係属中
の出願(処理予定表番号ARE−0050)を参照されたい。
【0043】
好ましくは、(a)組織mRNAに対するドットブロット分析、および/もし
くは(b)組織サンプルでの受容体の発現のRT−PCR同定のためのプローブ
を作成するのにヒトGPCRのDNA配列を使用する。組織供給源、もしくは疾
患に罹った組織中での受容体の存在、または正常組織に比較して疾患に罹った組
織中での上昇された濃度での受容体の存在は、限定されるものでないがその疾患
に関連する疾患を挙げることができる治療レジメンとの相関を同定するのに好ま
しく利用することができる。受容体はこの技術により器官の領域に等しく十分に
局在化する可能性がある。受容体が局在化されている特定の組織の既知の機能に
基づき、受容体の推定の機能上の役割を推定することができる。
E.候補化合物のスクリーニング
1.包括的GPCRスクリーニングアッセイの技術
Gタンパク質受容体が構成的に活性になった場合、それはGタンパク質(例え
ばGq、Gs、Gi、Gz、Go)に結合しかつGタンパク質へのGTPの結合
を刺激する。その後、Gタンパク質がGTPアーゼとして作用し、そしてGTP
をGDPにゆっくりと加水分解し、それにより受容体は正常条件下で失活したよ
うになる。しかしながら、構成的に活性化された受容体はGDPをGTPに交換
し続ける。構成的に活性化される受容体を発現する膜への高められた結合をモニ
ターするのに、GTPの加水分解不可能な類似物[35S]GTPγSを使用する
ことができる。[35S]GTPγSはリガンドの非存在下および存在下での膜へ
のGタンパク質の共役をモニターするのに使用することができることが報告され
ている。このモニタリングの一例、なかんずく当業者に公知かつ利用可能な例は
、1995年にトレイノル(Traynor)とナホルスキ(Nahorski
)により報告された。本アッセイ系の好ましい使用は候補化合物の初期スクリー
ニングのためである。なぜなら、該系は、受容体の細胞内ドメインと相互作用す
る特定のGタンパク質に関係なく、全部のGタンパク質共役型受容体に包括的に
応用可能であるからである。
2.特異的GPCRスクリーニングアッセイの技術
「包括的」Gタンパク質共役型受容体アッセイ(すなわちアゴニスト、部分的
アゴニストもしくは反作用薬である化合物を選択するためのアッセイ)を使用し
て候補化合物が同定されれば、該化合物が受容体部位で相互作用していることを
確認するためのさらなるスクリーニングが好ましい。例えば、「包括的」アッセ
イにより同定された化合物は受容体に結合しないかも知れないが、しかし代わり
に細胞内ドメインからGタンパク質を単に「分離する」かも知れない。
a.Gs、GzおよびGi
Gsは酵素アデニリルシクラーゼを刺激する。他方、Gi(ならびにGzおよ
びGo)はこの酵素を阻害する。アデニリルシクラーゼはATPのcAMPへの
転化を触媒し;従って、Gsタンパク質を共役する構成的に活性化されたGPC
Rは、cAMPの増大された細胞レベルと関連する。他方、Gi(もしくはGz
、Go)タンパク質を共役する構成的に活性化されたGPCRは、cAMPの低
下された細胞レベルと関連する。一般に、“Indirect Mechani
sms of Synaptic Transmission,”第8章、Fr
om Neuron To Brain(第3版)ニコルス(Nichols,
J.G.)ら編 サイナウア アソシエーツ インク(Sinauer Ass
ociates,Inc.)(1992)を参照されたい。従って、cAMPを
検出するアッセイを利用して、ある候補化合物が例えば受容体に対する反作用薬
(すなわちこうした化合物はcAMPのレベルを低下させることができる)であ
るかどうかを決定することができる。cAMPを測定するための当該技術分野で
既知の多様なアプローチを利用することができ;最も好ましいアプローチはEL
ISAに基づく形式での抗cAMP抗体の使用に頼る。利用することができる別
の型のアッセイは全細胞セカンドメッセンジャーレポーター系アッセイである。
遺伝子のプロモーターは特定の遺伝子がコードするタンパク質の発現を司る。環
状AMPは、cAMP応答性のDNA結合タンパク質もしくは転写因子(CRE
B)(その後cAMP応答要素と呼ばれる特定の部位でプロモーターに結合しそ
して遺伝子の発現を司る)の結合を促進することにより遺伝子発現を司る。レポ
ーター遺伝子(例えばβ−ガラクトシダーゼもしくはルシフェラーゼ)の前に複
数のcAMP応答要素を含有するプロモーターを有するレポーター系を構築する
ことができる。従って、構成的に活性化されたGsに結合された受容体は、cA
MPの蓄積を引き起こし、これがその後遺伝子およびレポータータンパク質の発
現を活性化する。その後、標準的な生化学的アッセイを使用して、β−ガラクト
シダーゼもしくはルシフェラーゼのようなレポータータンパク質を検出すること
ができる(チェン(Chen)ら 1995)。
b.GoおよびGq
GqおよびGoは酵素ホスホリパーゼCの活性化に関連し、この酵素は順にリ
ン脂質PIP2を加水分解して、2種の細胞内メッセンジャー、すなわちジアシ
ルグリセロール(DAG)およびイノシトール1,4,5−三リン酸(IP3)
を放出する。IP3の増大された蓄積はGqおよびGo会合型受容体の活性化と
関連する。一般に、“Indirect Mechanisms of Syn
aptic Transmission,”第8章、From Neuron
To Brain(第3版)ニコルス(Nichols,J.G.)ら編 サイ
ナウア アソシエーツ インク(Sinauer Associates,In
c.)(1992)を参照されたい。IP3の蓄積を検出するアッセイは、候補
化合物が例えばGqもしくはGo会合型受容体に対する反作用薬である(すなわ
ちこうした化合物はIP3のレベルを低下させることができる)かどうかを決定
するのに利用することができる。Gq会合型受容体はAP1レポーターアッセイ
(ここでGq依存性のホスホリパーゼCがAP1要素を含有する遺伝子の活性化
を引き起こす)を使用してもまた検査することができ;従って、活性化されたG
q会合型受容体は、こうした遺伝子の発現の増大を明示することができ、それに
より、それに対する反作用薬はこうした発現の減少を明示することができ、また
、アゴニストはこうした発現の増大を明示することができる。こうした検出のた
めの商業的に入手可能なアッセイが利用可能である。
3.GPCR融合タンパク質
反作用薬、アゴニストおよび部分的アゴニストの直接の同定のための候補化合
物のスクリーニングでの使用のための内在性で、構成的に活性化されるオーファ
ンGPCRもしくは非内在性で、構成的に活性化されるオーファンGPCRの使
用は興味深いスクリーニングの挑戦を提供し、ここでは当然、それに結合される
内在性リガンドの非存在下でさえ該受容体は活性である。従って、こうした化合
物が反作用薬、アゴニスト、部分的アゴニストであることができるかどうか、も
しくはこうした受容体に対する影響を有することができるかどうかに関しての理
解を許すこうした識別の目的をもって、例えば候補化合物の存在下の非内在性受
容体とその化合物の非存在下の非内在性受容体とを識別するために、こうした識
別を高める可能性のあるアプローチを利用することが好ましい。好ましい一アプ
ローチはGPCR融合タンパク質の使用である。
【0044】
一般に、非内在性のオーファンGPCRが上で示されたアッセイ技術(ならび
に他者)を使用して構成的に活性化されていることを決定すれば、内在性GPC
Rと共役する優勢なGタンパク質を決定することが可能である。GPCRへのG
タンパク質の共役はシグナル伝達経路を提供し、これは評価することが可能であ
る。哺乳動物発現系の使用によりスクリーニングが行われることが最も好ましい
ため、こうした系はその中に内在性Gタンパク質を有することが期待されよう。
従って、当然、こうした系においては、非内在性の構成的に活性化されたオーフ
ァンGPCRが連続的にシグナルを発するであろう。この点に関して、例えば受
容体に対する反作用薬の存在下で、とりわけスクリーニングに関してそれが反作
用薬と接触される場合にそれが受容体をより容易に識別することが可能であろう
ことがよりありそうであるように、このシグナルを高めることが好ましい。
【0045】
GPCR融合タンパク質は、非内在性GPCRと共役するGタンパク質の効能
を高めることを意図している。GPCR融合タンパク質は非内在性の構成的に活
性化されたGPCRを用いるスクリーニングに好ましい。なぜなら、こうしたア
プローチは、こうしたスクリーニング技術で最も好ましく利用されるシグナルを
増大させるからである。これは大きな「S/N」比の助長で重要であり;こうし
た大きな比は本明細書で開示されるような候補化合物のスクリーニングに重要か
つ好ましい。
【0046】
GPCR融合タンパク質の発現に有用な構築物の構築は当業者の範囲内にある
。商業的に入手可能な発現ベクターおよび系が、研究者の特定のニーズに合う可
能性のある多様なアプローチを提供する。こうしたGPCR融合タンパク質構築
物に対する重要な基準は、内在性のGPCR配列およびGタンパク質配列の双方
が同じ読み枠にある(好ましくは内在性GPCRの配列はGタンパク質配列の上
流である)こと、および、GPCRの発現に際してGタンパク質もまた発現する
ことが可能であるようにGPCRの「終止」コドンを欠失もしくは置き換えなく
てはならないことである。GPCRはGタンパク質に直接連結することができる
か、もしくは2者の間にスペーサー残基(好ましくは約12を越えないが、この
数字は当業者により容易に確かめることが可能である)が存在することができる
。われわれは、使用されないいくつかの制限部位が発現に際して効果的にスペー
サーとなるであろうことに、スペーサーの使用が(便宜性に基づくと)好ましい
。最も好ましくは、GPCR融合タンパク質構築物の創製に先立ち非内在性GP
CRに共役するGタンパク質を同定することができる。同定された数種のGタン
パク質が存在するにすぎないため、その中の内在性のGPCR配列の挿入にGタ
ンパク質の配列を含んで成る構築物(すなわち普遍的Gタンパク質構築物)が利
用可能であることが好ましく;これは、異なる配列を有する多様な異なった内在
性GPCRの大スケールのスクリーニングにおいて効率を提供する。
【0047】
上に示されたとおり、Gi、GzおよびGoに共役する構成的に活性化される
GPCRは、これらの型のGPCRに基づくアッセイを挑戦的にするcAMPの
形成を阻害すると期待される(すなわち、cAMPシグナルは活性化に際して減
少し、従って例えば(このシグナルをさらに減少させることができる)反作用薬
の直接の同定を興味深いものとする)。本明細書に開示されるであろうとおり、
われわれは、実現可能なシクラーゼに基づくアッセイを確立する努力において、
これらの型の受容体について内在性のGPCRの内在性のGタンパク質に基づか
ないGPCR融合タンパク質を創製することが可能であることを確かめた。従っ
て、例えば、H9のようなGz共役型受容体、Gs融合タンパク質を利用するG
PCR融合タンパク質を確立することが可能である。われわれは、こうした融合
構築物は、発現に際して非内在性のGPCRが例えば「天然の」Gzタンパク質
よりもむしろGsと共役することを「誘導」もしくは「強要」し、その結果シク
ラーゼに基づくアッセイを確立することが可能であると考える。従って、Gi、
GzおよびGo共役型受容体について、われわれは、GPCR融合タンパク質を
使用しかつアッセイがアデニルシクラーゼ活性の検出に基づく場合は、Gs(も
しくは酵素アデニリルシクラーゼの形成を刺激する同等のGタンパク質)を用い
て融合構築物を確立することを好む。
F.医薬品化学
一般に(しかし常にでなく)、候補化合物の直接の同定はコンビナトリアル化
学の技術を介して生成される化合物に関連して好ましく実施され、それにより何
千もの化合物がこうした分析のため無作為に調製される。一般に、こうしたスク
リーニングの結果は独特のコア構造を有する化合物であることができ;その後、
これらの化合物は、その医薬特性をさらに高めるため、好ましいコア構造(1種
もしくは複数)を取り巻く付加的な化学的改変に好ましくかけられる。こうした
技術は当業者に既知でありかつ本特許明細書で詳細に取り扱わないであろう。
G.製薬学的組成物
さらなる開発に選択された候補化合物は、当業者に公知の技術を使用して製薬
学的組成物に処方することができる。適する製薬学的に許容できる担体は当業者
に利用可能であり;例えば、Remingtons’s Pharmaceut
ical Sciences、第16版、1980、マック パブリッシング
カンパニー(Mack Publishing Co.)(オスロ(Oslo)
ら編)を参照されたい。
H.他の利用性
本明細書に開示される非内在性の変種ヒトGPCRの好ましい使用は、(好ま
しくは製薬学的作用物質としての使用のための)反作用薬、アゴニストもしくは
部分的アゴニストとしての候補化合物の直接の同定のためのものであることがで
きるが、これらの変種ヒトGPCRはまた研究の設定でも利用することができる
。例えば、GPCRを組み込むインビトロおよびインビボの系は、正常のおよび
疾患に罹った双方のヒトの状態でこれらの受容体が演じる役割をさらに解明かつ
理解するため、ならびに構成的活性化の役割の理解(それがシグナル伝達カスケ
ードの理解に適用されるため)に利用することができる。非内在性のヒトGPC
Rの価値は、そのための内在性リガンドが同定される前に人体でのこれらの受容
体の役割を理解するのに、それらの独特の特徴のため非内在性のヒトGPCRを
使用することができることにおいて、研究ツールとしてのそれらの利用性が高め
られることである。開示される受容体の他の用途は、とりわけ本特許明細書の検
討に基づき当業者に明らかとなるであろう。
【実施例】
【0048】
以下の実施例は本発明の解明の目的上(そして制限でなく)提示する。本明細
書で特定の核酸およびアミノ酸の配列が開示される一方、当業者は、下に報告さ
れる同一のもしくは実質的に類似の結果を達成しつつこれらの配列に対する小さ
な改変を行う能力があると信じられる。1配列から別のものまで(例えばラット
受容体からヒト受容体まで、もしくはヒト受容体Aからヒト受容体Bまで)の配
列カセットの応用もしくは理解への伝統的アプローチは、配列整列の技術(それ
により配列は共通の領域を決定する活動で整列される)に一般に基づく。本明細
書に開示される突然変異のアプローチはこのアプローチに頼らないが、しかし、
代わりにアルゴリズムのアプローチ、およびヒトGPCRのTM6領域内に配置
される保存されたプロリン残基からの位置上の距離に基づく。このアプローチが
確実にされれば、当業者は、それに対して小さな変更を行って本明細書に開示さ
れる実質的に同一の結果(すなわち構成的活性化)を達成する能力があると信じ
られる。こうした改変されたアプローチは本開示の範囲内と考えられる。
実施例1
内在性のヒトGPCR
1.ヒトGPCRの同定
ジェンバンク[GenBank](商標)データベース情報の再検討に基づき
、開示された内在性ヒトGPCRのあるものを同定した。データベースを検索す
る間に以下のcDNAクローンを下に明示されるとおり同定した(表C)。
【0049】
【表3】
【0050】
他の開示された内在性ヒトGPCRは、以下のESTクローンをクエリ配列と
して使用するESTデータベース(dbest)のBLAST(商標)検索を実
施することにより同定した。その後、同定された以下のESTクローンをプロー
ブとして使用して、ヒトゲノムライブラリーをスクリーニングした(表D)。
【0051】
【表4】
【0052】
2.完全長のクローニング
A.ヒトG2A
マウスESTクローン1179426を使用して、3種のアミノ酸G2Aコー
ディング配列を除く全部を含有するヒトゲノムクローンを得た。このコーディン
グ配列の5’は5’RACEを使用することにより得、また、PCRのための鋳
型はクロンテック(Clontech)のヒト脾マラソン−レディ[Marat
hon−Ready](商標)cDNAであった。開示されるヒトG2Aは、以
下:
5’−CTGTGTACAGCAGTTCGCAGAGTG−3’(配列番号4
1;1回目のPCR)
5’−GAGTGCCAGGCAGAGCAGGTAGAC−3’(配列番号4
2;2回目のPCR)
のような配列番号41および配列番号42に示されるような第一回および第二回
のPCRのためのG2AのcDNAに特異的なプライマーを使用するPCRによ
り増幅した。PCRは、94℃30秒間、次いで94℃5秒間および72℃4分
間の5周期;ならびに94°5秒間および70°4分間の30周期で、アドバン
テージ(Advantage)GCポリメラーゼキット(クロンテック(Clo
ntech);製造説明書に従うことができる)を使用して実施した。およそ1
.3kbのPCRフラグメントをアガロースゲルから精製し、HindIIIお
よびXbaIで消化し、そして発現ベクターpRC/CMV2(インヴィトロジ
ェン(Invitrogen))にクローン化した。T7シークェナーゼ[Se
quenase](商標)キット(USB アマーシャム(USB Amers
ham);製造元の説明書が従われた)を使用してクローン化された挿入物を配
列決定し、そして提示された配列と配列を比較した。P32標識されたフラグメン
トを用いてRNAドットブロット(クロンテック(Clontech);製造元
の説明書が従われた)をプロービングすることによりヒトG2Aの発現を検出し
た。
b.CHN9
ESTクローン1541536の配列決定は、CHN9が1個の開始コドンの
みを有する(すなわち終止コドンは欠けていた)部分的cDNAクローンである
ことを示した。CHN9をデータベース(nr)に対してBLAST検索する(b
last)のに使用した場合、CHN9の3’の配列はロイコトリエンB4受容体の
cDNAの5’非翻訳領域(CHN9のコーディング配列と同じ読み枠に終止コ
ドンを含有した)に100%相同であった。LTB4RのcDNAの5’非翻訳
領域がCHN9の3’配列であったかどうかを決定するために、CHN9に見出
される開始コドンに隣接する5’配列およびLTB4Rの5’非翻訳領域に見出
される終止コドンを取り巻く3’配列に基づくプライマーを使用してPCRを実
施した。利用された5’プライマー配列は以下のとおりであった:
5’−CCCGAATTCCTGCTTGCTCCCAGCTTGGCCC−3
’(配列番号43;センス)および
5’−TGTGGATCCTGCTGTCAAAGGTCCCATTCCGG−
3’(配列番号44;アンチセンス)
製造元により供給される緩衝液系、0.25μMの各プライマーおよび0.2m
Mの各4種のヌクレオチドとともに鋳型としての胸腺cDNAおよびrTthポ
リメラーゼ(パーキン エルマー(Perkin Elmer))を使用してP
CRを実施した。周期条件は、94℃1分間、65℃1分間ならびに72℃1分
および10秒間の30周期であった。予測された大きさと一致する1.1kbの
フラグメントをPCRから得た。本PCRフラグメントをpCMVにサブクロー
ニングし(下を参照されたい)そして配列決定した(配列番号35を参照された
い)。
c.RUP4
鋳型としてヒト脳cDNA(クロンテック(Clontech))を用いるR
T−PCRにより完全長のRUP4をクローン化した:
5’−TCACAATGCTAGGTGTGGTC−3’(配列番号45;セン
ス)および
5’−TGCATAGACAATGGGATTACAG−3’(配列番号46;
アンチセンス)。
以下の周期、すなわち94℃2分間;94℃30秒間;55℃30秒間、72℃
45秒間および72℃10分間により、タックプラス プレシジョン[TaqP
las Precision](商標)ポリメラーゼ(ストラタジーン(Str
atagene);製造説明書が従われた)を使用してPCRを実施した。周期
2から4を30回反復した。
【0053】
PCR産物を1%アガロースゲルで分離し、そして500bpのPCRフラグ
メントを単離しかつpCRII−TOPO(商標)ベクター(インヴィトロジェ
ン(Invitrogen))にクローン化し、そしてT7 DNAシークェナ
ーゼ[Sequenase](商標)キット(アマーシャム(Amersham
))およびSP6/T7プライマー(ストラタジーン(Stratagene)
)を使用して配列決定した。PCRフラグメントから示される配列分析は、実際
に、他のGPCRとの類似性をもつ1個の連続的読取り枠を有する、代替スプラ
イシングされた形態のAI307658であった。このPCRフラグメントの完
了された配列は以下のとおりであった:
【0054】
【表5】
【0055】
上の配列に基づき、2種のセンスオリゴヌクレオチドプライマーの組:
5’−CTGCTTAGAAGAGTGGACCAG−3’(配列番号48;オ
リゴ1)、
5’−CTGTGCACCAGAAGATCTACAC−3’(配列番号49;
オリゴ2)および
2種のアンチセンスオリゴヌクレオチドプライマーの組:
5’−CAAGGATGAAGGTGGTGTAGA−3’(配列番号50;オ
リゴ3)
5’−GTGTAGATCTTCTGGTGCACAGG−3’(配列番号51
;オリゴ4)
を、製造元の説明書に従って鋳型としてヒト脳マラソン−レディ[Marath
on−Ready](商標)cDNA(クロンテック(Clontech)、カ
タログ番号7400−1)を用いる3’−および5’−RACE PCRに使用
した。RACE PCRにより生成されたDNAフラグメントをpCRII−T
OPO(商標)ベクター(インヴィトロジェン(Invitrogen))にク
ローン化し、そしてSP6/T7プライマー(ストラタジーン(Stratag
ene))および数種の内的プライマーを使用して配列決定した。3’RACE
産物はポリ(A)テール(tail)およびTAA終止コドンで終了する1個の完了さ
れる読取り枠を含有した。5’RACE産物は不完全な5’末端を含有した(す
なわちATG開始コドンが存在しなかった)。
【0056】
新たな5’配列に基づき、オリゴ3および以下のプライマー:
5’−GCAATGCAGGTCATAGTGAGC−3’(配列番号52;オ
リゴ5)
を第2回の5’race PCRに使用し、そしてPCR産物を上のとおり分析
した。第3回の5’race PCRは、アンチセンスプライマー:
5’−TGGAGCATGGTGACGGGAATGCAGAAG−3’(配列
番号53;オリゴ6)および
5’−GTGATGAGCAGGTCACTGAGCGCCAAG−3’(配列
番号54;オリゴ7)
を利用して実施した。5’RACE PCRの産物の配列は開始コドンATGの
存在を示し、また、さらなる回の5’race PCRはいかなるそれ以上の5
’配列も生成しなかった。プライマーとして、センスプライマー
5’−GCAATGCAGGCGCTTAACATTAC−3’(配列番号55
;オリゴ8)
およびオリゴ4を使用するRT−PCR、ならびにヒト脳および心のcDNA鋳
型(クロンテック(Clontech)、カタログ番号7404−1)から生成
された650bpのPCR産物の配列分析により、完了された5’配列を確認し
た。オリゴ2および以下のアンチセンスプライマー:
5’−TTGGGTTACAATCTGAAGGGCA−3’(配列番号56;
オリゴ9)
を使用するRT−PCR、ならびにヒト脳および心のcDNA鋳型(クロンテッ
ク(Clontech)、カタログ番号7404−1)から生成された670b
pのPCR産物の配列分析により、完了された3’配列を確認した。
d.RUP5
以下の配列:
5’−ACTCCGTGTCCAGCAGGACTCTG−3’(配列番号57
)
5’−TGCGTGTTCCTGGACCCTCACGTG−3’(配列番号5
8)
を有した、ATG開始コドンから上流のセンスプライマー(配列番号57)、お
よび終止コドンとしてTCAを含有するアンチセンスプライマー(配列番号58
)、ならびに鋳型としてヒト末梢白血球cDNA(クロンテック(Clonte
ch))を使用するRT−PCRにより完全長のRUP5をクローン化した。段
階2から段階4が30回反復された以下の周期、すなわち94℃30秒間;94
℃15秒間;69℃40秒間;72℃3分間;および72℃6分間による50μ
lの反応中での増幅にアドバンテージ[Advantage](商標)cDNA
ポリメラーゼ(クロンテック(Clontech))を使用した。1.4kbの
PCRフラグメントを単離し、そしてpCRII−TOPO(商標)ベクター(
インヴィトロジェン(Invitrogen))を用いてクローン化し、そして
T7 DNAシークェナーゼ[Sequenase](商標)キット(アマーシ
ャム(Amersham))を使用して完全に配列決定した。配列番号9を参照
されたい。
e.RUP6
プライマー:
5’−CAGGCCTTGGATTTTAATGTCAGGGATGG−3’(
配列番号59)および
5’−GGAGAGTCAGCTCTGAAAGAATTCAGG−3’(配列
番号60)
ならびに鋳型としてヒト胸腺マラソン−レディ[Marathon−Ready
](商標)cDNA(クロンテック(Clontech))を使用するRT−P
CRにより完全長のRUP6をクローン化した。以下の周期、すなわち94℃3
0秒間;94℃5秒間;66℃40秒間;72℃2.5秒間および72℃7分間
による50μl反応中での増幅にアドバンテージ(Advantage)cDN
Aポリメラーゼ(クロンテック(Clontech)、製造元の説明書に従う)
を使用した。周期2から4を30回反復した。1.3kbのPCRフラグメント
を単離し、そしてpCRII−TOPO[商標]ベクター(インヴィトロジェン
(Invitrogen))にクローン化し、そしてABI ビッグ ダイ タ
ーミネーター[Big Dye Terminator](商標)キット(P.
E.バイオシステム(P.E.Biosystem))を使用して完全に配列決
定した(配列番号11を参照されたい)。
f.RUP7
プライマー:
5’−TGATGTGATGCCAGATACTAATAGCAC−3’(配列
番号61;センス)および
5’−CCTGATTCATTTAGGTGAGATTGAGAC−3’(配列
番号62;アンチセンス)
ならびに鋳型としてヒト末梢白血球cDNA(クロンテック(Clontech
))を使用するRT−PCRにより完全長のRUP7をクローン化した。段階2
ないし段階4が30回反復された以下の周期、すなわち94℃2分間;94℃1
5秒間;60℃20秒間;72℃2分間;72℃10分間による50μl反応中
での増幅にアドバンテージ[Advantage](商標)cDNAポリメラー
ゼ(クロンテック(Clontech))を使用した。1.25kbのPCRフ
ラグメントを単離し、そしてpCRII−TOPO(商標)ベクター(インヴィ
トロジェン(Invitrogen))にクローン化し、そしてABI ビッグ
ダイ ターミネーター[Big Dye Terminator](商標)キ
ット(P.E.バイオシステム(P.E.Biosystem))を使用して完
全に配列決定した。配列番号13を参照されたい。
3.アンジオテンシンIIタイプ1受容体(「AT1」)
製造元により供給される緩衝液系、0.25μMの各プライマーおよび0.2
mMの各4種のヌクレオチドとともに鋳型としてのゲノムDNAおよびrTth
ポリメラーゼ(パーキン エルマー(Perkin Elmer))を使用する
PCRにより、内在性のヒトアンジオテンシンIIタイプ1受容体(「AT1」
)を得た。周期条件は、94℃1分間、55℃1分間および72℃1.5分間の
30周期であった。5’PCRプライマーは、配列:
5’−CCCAAGCTTCCCCAGGTGTATTTGAT−3’(配列番
号63)
とともにHindIII部位を含有し、そして3’プライマーは以下の配列:
5’−GTTGGATCCACATAATGCATTTTCTC−3’(配列番
号64)
とともにBamHI部位を含有する。生じる1.3kbのPCRフラグメントを
HindIIIおよびBamHIで消化し、そしてpCMV発現ベクターのHi
ndIII−BamHI部位にクローン化した。cDNAクローンを完全に配列
決定した。その後、ヒトAT1の核酸(配列番号65)およびアミノ酸(配列番
号66)の配列を決定しかつ確かめた。
4.GPR38
GPR38を得るため、製造元により供給される緩衝液系、0.25μMの各
プライマーおよび0.2mMの各4種のヌクレオチドとともに鋳型としてのヒト
ゲノムcDNAおよびrTthポリメラーゼ(パーキン エルマー(Perki
n Elmer))を使用して、2種のPCRフラグメントを組み合わせること
によりPCRを実施した。各PCR反応の周期条件は94℃1分間、62℃1分
間および72℃2分間の30周期であった。
【0057】
第一のフラグメントは、以下の配列:
5’−ACCATGGGCAGCCCCTGGAACGGCAGC−3’(配列
番号67)
を伴う端部位を含有した5’PCRプライマー、および以下の配列:
5’−AGAACCACCACCAGCAGGACGCGGACGGTCTGC
CGGTGG−3’(配列番号68)
を有する3’プライマーを用いて増幅した。第二のPCRフラグメントは、以下
の配列:
5’−GTCCGCGTCCTGCTGGTGGTGGTTCTGGCATTT
ATAATT−3’(配列番号69)
を有する5’プライマー、ならびにBamHI部位を含有しかつ以下の配列:
5’−CCTGGATCCTTATCCCATCGTCTTCACGTTAGC
−3’(配列番号70)
を有する3’プライマーを用いて増幅した。配列番号67および配列番号70を
プライマーとして使用して(上に示された周期条件を使用する)、2種のフラグ
メントを鋳型として使用してGPR38を増幅した。生じる1.44kbのPC
RフラグメントをBamHIで消化し、そしてpCMV発現ベクターの平滑Ba
mHI部位にクローン化した。
5.MC4
MC4を得るため、製造元により供給される緩衝液系、0.25μMの各プラ
イマーおよび0.2mMの各4種のヌクレオチドとともに鋳型としてのヒトゲノ
ムcDNAおよびrTthポリメラーゼ(パーキン エルマー(Perkin
Elmer))を使用してPCRを実施した。各PCR反応の周期条件は94℃
1分間、54℃1分間および72℃1.5分間の30周期であった。
【0058】
5’PCRは配列:
5’−CTGGAATTCTCCTGCCAGCATGGTGA−3’(配列番
号71)
とともにEcoRI部位を含有し、そして3’プライマーは配列:
5’−GCAGGATCCTATATTGCGTGCTCTGTCCCC−3’
(配列番号72)
とともにBamHI部位を含有した。1.0kbのPCRフラグメントをEco
RIおよびBamHIで消化し、そしてpCMV発現ベクターのEcoRI−B
amHI部位にクローン化した。その後、ヒトMC4の核酸(配列番号73)お
よびアミノ酸(配列番号74)の配列を決定した。
6.CCKB
CCKBを得るため、製造元により供給される緩衝液系、0.25μMの各プ
ライマーおよび0.2mMの各4種のヌクレオチドとともに鋳型としてのヒト胃
cDNAおよびrTthポリメラーゼ(パーキン エルマー(Perkin E
lmer))を使用してPCRを実施した。各PCR反応の周期条件は94℃1
分間、65℃1分間ならびに72℃1分および30秒間の30周期であった。
【0059】
5’PCRは配列:
5’−CCGAAGCTTCGAGCTGAGTAAGGCGGCGGGCT−
3’(配列番号75)
とともにHindIII部位を含有し、また、3’プライマーは配列:
5’−GTGGAATTCATTTGCCCTGCCTCAACCCCCA−3
’(配列番号76)
とともにEcoRI部位を含有した。生じる1.44kbのPCRフラグメント
をHindIIIおよびEcoRIで消化し、そしてpCMV発現ベクターのH
indIII−EcoRI部位にクローン化した。その後、ヒトCCKBの核酸
(配列番号77)およびアミノ酸(配列番号78)の配列を決定した。
7.TDAG8
TDAG8を得るため、製造元により供給される緩衝液系、0.25μMの各
プライマーおよび0.2mMの各4種のヌクレオチドとともに鋳型としてのゲノ
ムDNAおよびrTthポリメラーゼ(パーキン エルマー(Perkin E
lmer))を使用してPCRを実施した。周期条件は94℃1分間、56℃1
分間ならびに72℃1分および20秒間の30周期であった。5’PCRプライ
マーは以下の配列:
5’−TGCAAGCTTAAAAAGGAAAAAATGAACAGC−3’
(配列番号79)
とともにHindIII部位を含有し、また、3’プライマーは以下の配列:
5’−TAAGGATCCCTTCCCTTCAAAACATCCTTG−3’
(配列番号80)
とともにBamHI部位を含有した。生じる1.1kbのPCRフラグメントを
HindIIIおよびBamHIで消化し、そしてpCMV発現ベクターのHi
ndIII−BamHI部位にクローン化した。配列決定された3種の生じるク
ローンは、ProからAlaへのアミノ酸43、LysからAsnへのアミノ酸
97、およびIleからPheへのアミノ酸130の変化を伴う3種の潜在的多
形を含有した。その後、ヒトTDAG8の核酸(配列番号81)およびアミノ酸
(配列番号82)の配列を決定した。
8.H9
H9を得るため、製造元により供給される緩衝液系、0.25μMの各プライ
マーおよび0.2mMの各4種のヌクレオチドとともに鋳型としての下垂体cD
NAおよびrTthポリメラーゼ(パーキン エルマー(Perkin Elm
er))を使用してPCRを実施した。周期条件は94℃1分間、62℃1分間
および72℃2分間の30周期であった。5’PCRプライマーは以下の配列:
5’−GGAAAGCTTAACGATCCCCAGGAGCAACAT−3’
(配列番号15)
とともにHindIII部位を含有し、また、3’プライマーは以下の配列:
5’−CTGGGATCCTACGAGAGCATTTTTCACACAG−3
’(配列番号16)
とともにBamHI部位を含有した。生じる1.9kbのPCRフラグメントを
HindIIIおよびBamHIで消化し、そしてpCMV発現ベクターのHi
ndIII−BamHI部位にクローン化した。H9は、アミノ酸P320S、
S493Nおよびアミノ酸G448Aの変化を伴う3種の潜在的多形を含有した
。その後、ヒトH9の核酸(配列番号139)およびアミノ酸(配列番号140
)の配列を決定しかつ確かめた。
実施例2
非内在性の構成的に活性化されるGPCRの調製
当業者は、核酸配列の突然変異のための技術を選択する能力があると信じられ
る。上に開示された非内在性の変種数種のヒトGPCRを創製するのに利用され
たアプローチを下に提示する。下に開示される突然変異はアルゴリズムのアプロ
ーチに基づき、それにより(TM6/IC3の界面近くのGPCRのTM6領域
に配置される)保存されるプロリン残基からの(GPCRのIC3領域に配置さ
れる)16番目のアミノ酸が、最も好ましくはリシンアミノ酸残基に突然変異さ
れる。
1.トランスフォーマー部位特異的[Transformer Site−Di
rected](商標)突然変異誘発
非内在性のヒトGPCRの調製は、製造元の説明書に従い、トランスフォーマ
ー部位特異的[Transformer Site−Directed](商標
)突然変異誘発キット(クロンテック(Clontech))を使用してヒトG
PCRで達成することができる。2種の突然変異誘発プライマー、最も好ましく
はリシン突然変異を創製するリシン突然変異誘発オリゴヌクレオチド、および選
択マーカーオリゴヌクレオチドを利用する。便宜上、ヒトGPCRに組み込まれ
るべきコドン突然変異を標準的形態でもまた示す(表E):
【0060】
【表6】
【0061】
指定される配列プライマーを使用して、上の方法に従い、以下のGPCRを突
然変異した(表F)。
【0062】
【表7】
【0063】
その後、非内在性のヒトGPCRを配列決定し、また、形成されかつ確かめら
れた核酸およびアミノ酸の配列を、下に表Gに要約されるとおり、本特許明細書
への付随する「配列表」付録に列挙する:
【0064】
【表8】
【0065】
2.非内在性ヒトGPCRの創製のための代替アプローチ
a.AT1
1.F239K突然変異
F239突然変異(核酸配列について配列番号89、およびアミノ酸配列につ
いて配列番号90を参照されたい)を創製することにより、非内在性の構成的に
活性化されるヒトAT1受容体の調製を達成した。突然変異誘発は、製造元の説
明書に従い、トランスフォーマー部位特異的突然変異誘発[Transform
er Site−Directed Mutagenesis](商標)キット
(クロンテック(Clontech))を使用して実施した。2種の突然変異誘
発プライマー、リシン突然変異誘発オリゴヌクレオチド(配列番号91)および
選択マーカーオリゴヌクレオチド(配列番号92)を使用した。これらはそれぞ
れ以下の配列を有した:
5’−CCAAGAAATGATGATATTAAAAAGATAATTATG
GC−3’(配列番号91)
5’−CTCCTTCGGTCCTCCTATCGTTGTCAGAAGT−3
’(配列番号92)
2.N111A突然変異
非内在性のヒトAT1受容体の調製もまた、N111A突然変異(核酸配列に
ついて配列番号93、およびアミノ酸配列について配列番号94を参照されたい
)を創製することにより達成した。10%DMSO、0.25μMの各プライマ
ー、および0.5mMの各4種のヌクレオチドを補充された、製造元により提供
される緩衝液系を用いてpfuポリメラーゼ(ストラタジーン(Stratag
ene))を使用して、2回のPCR反応を実施した。使用された5’PCRの
センスプライマーは以下の配列:
5’−CCCAAGCTTCCCCAGGTGTATTTGAT−3’(配列番
号95)
を有し、また、アンチセンスプライマーは以下の配列:
5’−CCTGCAGGCGAAACTGACTCTGGCTGAAG−3’(
配列番号96)
を有した。生じる400bpのPCRフラグメントをHindIII部位で消化
し、そしてpCMVベクターのHindIII−SmaI部位にサブクローニン
グした(5’構築物)。使用された3’PCRのセンスプライマーは以下の配列
:
5’−CTGTACGCTAGTGTGTTTCTACTCACGTGTCTC
AGCATTGAT−3’(配列番号97)
を有し、また、アンチセンスプライマーは以下の配列:
5’−GTTGGATCCACATAATGCATTTTCTC−3’(配列番
号98)
を有した。生じる880bpのPCRフラグメントをBamHIで消化し、そし
て5’構築物のPst(T4ポリメラーゼにより平滑化された)およびBamH
I部位に挿入して完全長のN111A構築物を生成した。周期条件は、94℃1
分間、60℃1分間および72℃1分間(5’PCR)もしくは1.5分間(3
’PCR)の25周期であった。
3.AT2K255IC3突然変異
AT2K255IC3の「ドメインスワップ(domain swap)」突然変異(核酸
配列について配列番号99を、およびアミノ酸配列について配列番号100を参
照されたい)を創製することにより、非内在性の構成的に活性化されるヒトAT
1の調製を達成した。AT1のIC3に隣接する制限部位を生成させて、アンジ
オテンシンIIタイプ2受容体(AT2)からの対応するIC3でのIC3の置
き換えを助長した。これは2回のPCR反応を実施することにより達成した。セ
ンスプライマーとして配列番号63を、およびアンチセンスプライマーとして以
下の配列:
5’−TCCGAATTCCAAAATAACTTGTAAGAATGATCA
GAAA−3’(配列番号101)
を利用することにより、5’非翻訳領域からIC3の開始までをコードする5’
PCRフラグメント(フラグメントA)を生成させた。センスプライマーとして
以下の配列:
5’−AGATCTTAAGAAGATAATTATGGCAATTGTGCT
−3’(配列番号102)
およびアンチセンスプライマーとして配列番号64を使用することにより、IC
3の終了から3’非翻訳領域までをコードする3’PCRフラグメント(フラグ
メントB)を生成させた。PCR条件は、10%DMSO、0.25μMの各プ
ライマーおよび0.5mMの各4種のヌクレオチドを補充された、製造元により
供給される緩衝液系を用いて、鋳型としての内在性のAT1 cDNAクローン
およびpfuポリメラーゼ(ストラタジーン(Stratagene))を使用
して、94℃1分間、55℃1分間および72℃1.5分間の30周期であった
。フラグメントA(720bp)をHindIIIおよびEcoRIで消化しそ
してサブクローニングした。フラグメントBはBamHIで消化し、そしてクロ
ーン化されたPCRフラグメントの5’にEcoRI部位をもつpCMVベクタ
ーにサブクローニングした。
【0066】
以下の配列:
5’AATTCGAAAACACTTACTGAAGACGAATAGCTAT
GGGAAGAACAGGATAACCCGTGACCAAG−3’(センス;
配列番号103)
5’TTAACTTGGTCACGGGTTATCCTGTTCTTCCCAT
AGCTATTCGTCTTCAGTAAGTGTTTTCG−3’(アンチセ
ンス;配列番号104)
を有する2種の合成オリゴヌクレオチドをアニーリングすることにより、L25
5K突然変異を伴いAT2のIC3をコードしそして5’にEcoRI付着端お
よび3’にAfIII付着端を含有するDNAフラグメント(フラグメントC)
を生成させた。
【0067】
フラグメントCを、EcoRIおよびAfIII部位によりフラグメントBの
前に挿入した。その後、生じるクローンをEcoRI部位によりフラグメントA
と連結して、AT2K255IC3をもつAT1を生成させた。
4.A243+突然変異
A243+突然変異(核酸配列について配列番号105、およびアミノ酸配列
について配列番号106を参照されたい)を創製することにより、非内在性のヒ
トAT1受容体の調製もまた達成した。A243+突然変異は以下のPCRに基
づく戦略を使用して構築した。すなわち、10%DMSO、0.25μMの各プ
ライマーおよび0.5mMの各4種のヌクレオチドを補充された、製造元により
提供される緩衝液系とともにpfuポリメラーゼ(ストラタジーン(Strat
agene))を使用して2回のPCR反応を実施した。利用された5’PCR
のセンスプライマーは以下の配列:
5’−CCCAAGCTTCCCCAGGTGTATTTGAT−3’(配列番
号107)
を有し、また、アンチセンスプライマーは以下の配列:
5’−AAGCACAATTGCTGCATAATTATCTTAAAAATA
TCATC−3’(配列番号108)
を有した。利用された3’PCRのセンスプライマーは、Ala挿入を含有する
以下の配列:
5’−AAGATAATTATGGCAGCAATTGTGCTTTTCTTT
TTCTTT−3’(配列番号109)
そしてアンチセンスプライマー:
5’−GTTGGATCCACATAATGCATTTTCTC−3’(配列番
号110)
を有した。周期条件は、94℃1分間、54℃1分間および72℃1.5分間の
25周期であった。その後、5’および3’のPCRのアリコートを共鋳型(co-
template)として使用して、5’PCRのセンスプライマーおよび3’PCRの
アンチセンスプライマーを使用する二次的PCRを実施した。PCR条件は、伸
長時間が2.5分であったことを除き一次PCRと同一であった。生じるPCR
フラグメントをHindIIIおよびBamHIで消化し、そしてpCMVベク
ターにサブクローニングした(配列番号105を参照されたい)。
4.CCKB
V322K突然変異(核酸配列について配列番号111、およびアミノ酸配列
について配列番号112を参照されたい)を創製することにより、非内在性の構
成的に活性化されるヒトCCKB受容体の調製を達成した。突然変異誘発は、実
施例1からの野性型CCKBを使用する増幅を介してPCRにより実施した。
【0068】
配列番号75、およびV322K突然変異を含んで成るアンチセンスプライマ
ー:
5’−CAGCAGCATGCGCTTCACGCGCTTCTTAGCCCA
G−3’(配列番号113)
を使用することにより第一のPCRフラグメント(1kb)を増幅した。V32
2K突然変異を含んで成るセンスプライマー:
5’−AGAAGCGCGTGAAGCGCATGCTGCTGGTGATCG
TT−3’(配列番号114)および配列番号76を使用することにより第二の
PCRフラグメント(0.44kb)を増幅した。その後、配列番号75および
配列番号76、ならびに上で示された系および条件を使用して、V332Kを含
んで成るCCKBを増幅するための鋳型として2種の生じるPCRフラグメント
を使用した。V332K突然変異を含有する生じる1.44kbのPCRフラグ
メントをHindIIIおよびEcoRIで消化し、そしてpCMV発現ベクタ
ーのHindIII−EcoRI部位にクローン化した(配列番号111を参照
されたい)。
3.クイックチェンジ[QuikChange](商標)部位特異的[Site
−Directed](商標)突然変異誘発
クイックチェンジ[QuikChange](商標)部位特異的[Site−
Directed](商標)突然変異誘発キット(ストラタジーン(Strat
agene)、製造元の説明書に従う)を使用することにより、非内在性のヒト
GPCRの調製もまた達成することができる。内在性GPCRを鋳型として好ま
しく使用し、また、2種の突然変異誘発プライマー、ならびに最も好ましくはリ
シン突然変異誘発オリゴヌクレオチドおよび選択マーカーオリゴヌクレオチド(
キットに包含される)を利用する。便宜上、ヒトGPCRに組み込まれたコドン
の突然変異およびそれぞれのオリゴヌクレオチドを標準的形態で示す(表H):
【0069】
【表9】
【0070】
実施例3
受容体の発現
タンパク質の発現のために多様な細胞が当該技術に使用可能であるが、哺乳動
物細胞を利用することが最も好ましい。これの主要な理由は実地的なことに基づ
く。すなわち、例えばGPCRの発現のための酵母細胞の利用が可能な一方で、
受容体共役型の遺伝子的機構を包含しなくてもよい(事実、酵母の場合は包含し
ない)非哺乳動物細胞および哺乳動物の系について発展した分泌経路をプロトコ
ルに導入し、従って、非哺乳動物細胞で得られる結果は、潜在的に有用な一方で
哺乳動物細胞から得られるものと同じくらい好ましくはない。哺乳動物細胞のう
ち、COS−7、293および293T細胞がとりわけ好ましいが、利用される
特定の哺乳動物細胞は当業者の特定のニーズに基づくことが可能である。
【0071】
第1日に、150mmプレートあたり1×107個の293T細胞をプレート
培養した。第2日に2本の反応チューブを準備した(各チューブについて後に続
く比率はプレート1枚あたりである)。すなわち、チューブAは、1.2mlの
血清を含まないDMEM(アーヴィン サイエンティフィック(Irvine
Scientific)、カリフォルニア州アーヴィン)中に20μgのDNA
(例えばpCMVベクター;受容体cDNAを含むpCMVベクター、など)を
混合することにより準備し;チューブBは1.2mlの血清を含まないDMEM
中に120μlのリポフェクタミン(ギブコ(Gibco)BRL)を混合する
ことにより準備した。チューブAおよびBは反転(数回)により混合し、次いで
室温で30〜45分間インキュベートした。混合状態を「トランスフェクション
混合物」と称する。プレート培養された293T細胞を1×PBSで洗浄し、次
いで10mlの血清を含まないDMEMを添加した。細胞に2.4mlのトラン
スフェクション混合物を添加し、次いで37℃/5%CO2で4時間インキュベ
ートした。トランスフェクション混合物を吸引により除去し、次いで25mlの
DMEM/10%ウシ胎児血清を添加した。細胞を37℃/5%CO2でインキ
ュベートした。72時間のインキュベーション後に細胞を収穫し、そして分析に
利用した。
実施例4
非内在性GPCRの構成的活性の測定のためのアッセイ
非内在性のヒトGPCRの構成的活性の評価には多様なアプローチが利用可能
である。以下は具体的説明であり;当業者は、当業者のニーズに優先的に利益を
もたらす技術を決定する能力があると信じられる。
1.膜結合アッセイ:[35S]GTPγSアッセイ
Gタンパク質共役型受容体がリガンド結合もしくは構成的活性化のいずれかの
結果としてその活性状態にある場合、受容体はGタンパク質に共役しそしてGD
Pの放出およびGTPのGタンパク質へのその後の結合を刺激する。Gタンパク
質−受容体複合体のαサブユニットがGTPアーゼとして作用し、そしてGTP
をGDPにゆっくりと加水分解し、この点で受容体が通常非活性化される。構成
的に活性化された受容体はGDPをGTPと交換し続ける。構成的に活性化され
る受容体を発現する膜への[35S]GTPγSの高められた結合を立証するのに
、加水分解不可能なGTP類似物[35S]GTPγSを利用することができる。
構成的活性化を測定するための[35S]GTPγS結合の使用の利点は:(a)
それが全部のGタンパク質共役型受容体に包括的に応用可能である;(b)それ
は膜表面の近接であり、細胞内カスケードに影響を及ぼす分子を拾い上げること
をより少なくありそうにすることである。
【0072】
該アッセイは、直接的に関連する受容体を発現する膜への[35S]GTPγS
の結合を刺激するGタンパク質共役型受容体の能力を利用する。従って、該アッ
セイは、既知の、オーファンのおよび構成的に活性化されるGタンパク質共役型
受容体に対する候補化合物をスクリーニングするための直接同定法で使用するこ
とができる。該アッセイは包括的であり、また、全部のGタンパク質共役型受容
体での薬物発見に対する応用性を有する。
【0073】
[35S]GTPγSアッセイは、20mM HEPES、および1mMと約2
0mMとの間のMgCl2(この量は結果の至適化のため調節することができる
が、20mMが好ましい)pH7.4、約0.3nMと約1.2nMとの間の[
35S]GTPγS(この量は結果の至適化のため調節することができるが、1.
2が好ましい)および12.5ないし75μgの膜タンパク質(例えば受容体を
発現するCOS−7細胞;この量は至適化のため調節することができるが、75
μgが好ましい)および1μM GDP(この量は至適化のため変更することが
できる)を含む結合緩衝液中で1時間インキュベートすることが可能である。そ
の後、コムギ胚芽アグルチニンビーズ(25μl;アマーシャム(Amersh
am))を添加し、そして混合物を室温で別の30分間インキュベートする。そ
の後、チューブを1500×gで室温で5分間遠心分離し、そしてその後シンチ
レーション計数器で計数する。
【0074】
より少なく高価なしかし同等に応用可能な代替物が同定されており、これはま
た大スケールのスクリーニングのニーズにも合致する。フラッシュプレート[F
lash plate](商標)およびワラック[Wallac](商標)シン
チストリップを利用して、高スループットの[35S]GTPγS結合アッセイの
全体構成を定めることができる。さらに、この技術を使用すれば、[35S]GT
PγS結合を介して効力をモニターするのと同一時点で受容体へのトリチウム化
されたリガンドの結合を同時にモニターするために、該アッセイを既知のGPC
Rに利用することができる。トリチウム標識プローブおよび35S標識プローブの
双方を見るようにワラック(Wallac)ベータ計数器がエネルギーウィンド
ウを切り替えることができるために、これが可能である。このアッセイはまた、
受容体の活性化をもたらす他の型の膜活性化事象を検出するのにも使用してよい
。例えば、該アッセイは多様な受容体(Gタンパク質共役型受容体およびチロシ
ンキナーゼ受容体の双方)の32Pホスホリル化をモニターするのに使用してよい
。膜がウェルの底部に遠心分離されている場合、結合された[35S]GTPγS
もしくは32P−ホスホリル化された受容体が、ウェルの被覆されているシンチラ
ントを活性化することができる。この原理を立証するのにシンチ[Scinti
](商標)ストリップ(ワラック(Wallac))が使用されている。加えて
、該アッセイはまた、放射活性に標識されたリガンドを使用して受容体へのリガ
ンド結合を測定するための有用性も有する。類似の様式で、放射標識された結合
されたリガンドがウェルの底部に遠心分離されている場合、シンチストリップの
標識は放射標識されたリガンドに接近して活性化および検出をもたらす。
2.アデニリルシクラーゼ
細胞に基づくアッセイのため設計されたフラッシュプレート[Flash P
late](商標)アデニリルシクラーゼキット(ニュー イングランド ニュ
ークリア(New England Nuclear);カタログ番号SMP0
04A)を、粗原形質膜との使用のため改変することができる。フラッシュプレ
ート(Flash Plate)のウェルはシンチラントのコーティングを含有
し、このコーティングはcAMPを認識する特異的抗体もまた含有する。cAM
P抗体への放射活性のcAMPトレーサーの結合についての直接の競争により、
ウェル中で生成されたcAMPを定量した。以下は、受容体を発現する膜中のc
AMPレベルの変化の測定のための簡潔なプロトコルとしてはたらく。
【0075】
トランスフェクションされた細胞をトランスフェクションのおよそ3日後に収
穫する。20mM HEPES、pH7.4および10mM MgCl2を含有
する緩衝液に懸濁された細胞の均質化により膜を調製した。均質化は、ブリンク
マン(Brinkman)ポリトロン[Polytron](商標)を使用し氷
上でおよそ10秒間実施する。生じるホモジェネートを4℃で49,000×g
で15分間遠心分離する。その後、生じるペレットを20mM HEPES、p
H7.4および0.1mM EDTAを含有する緩衝液に再懸濁し、10秒間均
質化し、次いで49,000×gで4℃で15分間遠心分離する。生じるペレッ
トは利用されるまで−80℃で保存することができる。測定の日に膜ペレットを
室温でゆっくりと融解させ、20mM HEPES、pH7.4および10mM
MgCl2(これらの量は至適化することができるが、本明細書に列挙される
値が好ましい)を含有する緩衝液に再懸濁して0.60mg/mlの最終タンパ
ク質濃度を生じる(再懸濁された膜は使用まで氷上に置いた)。
【0076】
cAMP標準および検出緩衝液(11mlの検出緩衝液に対し2μCiのトレ
ーサー[125I]cAMP(100μl)を含んで成る)を調製し、そして製造
元の説明書に従って維持する。アッセイ緩衝液はスクリーニングのため新たに調
製し、そして20mM HEPES、pH7.4、10mM MgCl2、20
mM(シグマ(Sigma))、0.1単位/mlのクレアチンホスホキナーゼ
(シグマ(Sigma))、50μM GTP(シグマ(Sigma))および
0.2mM ATP(シグマ(Sigma))を含有し;アッセイ緩衝液は利用
されるまで氷上で保存することができる。NEN フラッシュプレート(Fla
sh Plate)への50μlのアッセイ緩衝液の添加、次いで50μlの膜
懸濁液の添加によりアッセイを開始する。結果として生じるアッセイ混合物を室
温で60分間インキュベートし、次いで100μlの検出緩衝液を添加する。そ
の後、プレートを追加の2〜4時間インキュベートし、次いでワラック(Wal
lac)マイクロベータ[MicroBeta](商標)シンチレーション計数
器で計数する。各アッセイプレート内に含有される標準cAMP曲線からウェル
あたりのcAMPの値を外挿する。
C.レポーターに基づくアッセイ
1.CREBレポーターアッセイ(Gs会合型受容体)
Gs刺激を検出する方法はcAMP依存性の様式で活性化される転写因子CR
EBの既知の特性に依存する。293もしくは293T細胞中でのGs共役型の
活性についてアッセイするのに、パスディテクト[PathDetect](商
標)CREBトランスレポーティング(CREB trans−Reporti
ng)系(ストラタジーン(Stratagene)、カタログ番号21901
0)を利用することができる。細胞は、哺乳動物トランスフェクションキット(
ストラタジーン(Stratagene)、カタログ番号200285)を製造
元の説明書に従って使用して、この上の系のプラスミド成分、および内在性もし
くは突然変異体の受容体をコードする指定された発現プラスミドでトランスフェ
クションする。簡潔には、400ngのpFR−Luc(Gal4認識配列を含
有するルシフェラーゼレポータープラスミド)、40ngのFA2−CREB(
Gal4のDNA結合ドメインを含有するGal4−CREB融合タンパク質)
、80ngのpCMV−受容体発現プラスミド(受容体を含んで成る)および2
0ngのCMV−SEAP(分泌型アルカリホスファターゼ発現プラスミド;ト
ランスフェクションされた細胞の培地中でアルカリホスファターゼ活性を測定し
て、サンプル間のトランスフェクション効率の変動について制御する)を、キッ
トの説明書に従ってリン酸カルシウム沈殿中に組み合わせる。沈殿物の半分を9
6穴プレートの3個のウェルに同等に配分し、細胞上で一夜保ち、そして翌朝に
新鮮培地で置き換える。トランスフェクションの開始48時間後に細胞を処理し
、そして例えばルシフェラーゼ活性についてアッセイする。
2.AP1レポーターアッセイ(Gq会合型受容体)
Gq刺激を検出する方法は、それらのプロモーター中にAP1要素を含有する
遺伝子の活性化を引き起こすGq依存性ホスホリパーゼCの既知の特性に依存す
る。リン酸カルシウム沈殿物の成分が410ngのpAP1−Luc、80ng
のpCMV−受容体発現プラスミドおよび20ngのCMV−SEAPであった
ことを除いて、CREBレポーターアッセイに関して上に示されたプロトコルに
従い、パスディテクト[Pathdetect](商標)AP−1シスレポーテ
ィング(AP−1 cis−Reporting)系(ストラタジーン(Str
atagene)、カタログ番号219073)を利用することができる。
3.CRE−Lucレポーターアッセイ
293および293T細胞をウェルあたり細胞2×104個の密度で96穴プ
レート上でプレート培養し、そして翌日、製造元の説明書に従ってリポフェクタ
ミン試薬(BRL)を使用してトランスフェクションした。DNA/脂質混合物
を以下のとおり各6個のウェルのトランスフェクションのため調製する。すなわ
ち、100μlのDMEM中の260ngのプラスミドDNAを、100μlの
DMEM中2μlの脂質と穏やかに混合した(260ngのプラスミドDNAは
、200ngの8xCRE−Lucレポータープラスミド(プラスミドの一部分
の表示については下および図1を参照されたい)、50ngの内在性受容体もし
くは非内在性受容体を含んで成るpCMVまたはpCMV単独、ならびに10n
gのGPRS発現プラスミド(pcDNA3(インヴィトロジェン(Invit
rogen)中のGPRS)より成った)。8XCRE−Lucレポータープラ
スミドは以下のとおり調製した。すなわち、pβgal−基本ベクター(Bas
ic Vector)(クロンテック(Clontech))のBglV−Hi
ndIII部位にラットソマトスタチンプロモーター(−71/+51)をクロ
ーン化することにより、ベクターSRIF−β−galを得た。アデノウイルス
鋳型AdpCF126CCRE8(7 Human Gene Therapy
1883(1996)を参照されたい)からのPCRにより、8コピーのcA
MP応答要素を得、そしてKpn−BglV部位でSRIF−β−galベクタ
ーにクローン化して8xCRE−β−galレポーターベクターをもたらした。
8xCRE−Lucレポータープラスミドは、8xCRE−β−galレポータ
ーベクター中のβ−ガラクトシダーゼ遺伝子を、HindIII−BamHI部
位でpGL3−基本ベクター(プロメガ(Promega))から得られたルシ
フェラーゼ遺伝子で置き換えることにより生成した。室温で30分のインキュベ
ーション後に、400μlのDMEMでDNA/脂質混合物を希釈し、そして1
00μlの希釈された混合物を各ウェルに添加した。細胞培養インキュベーター
中での4時間のインキュベーション後に、10%FCSを含む100μlのDM
EMを各ウェルに添加した。翌日、トランスフェクションされた細胞を、10%
FCSを含むウェルあたり200μlのDMEMで変更した。8時間後、PBS
での1回の洗浄後に、ウェルをウェルあたり100μlのフェノールレッドを含
まないDMEMに変更した。翌日、製造元の説明書に従って、ルックライト[L
ucLite](商標)レポーター遺伝子アッセイキット(パッカード(Pac
kard))を使用してルシフェラーゼ活性を測定し、そして1450マイクロ
ベータ[MicroBeta](商標)シンチレーションおよび発光計数器(ワ
ラック(Wallac))で読み取った。
4.SRF−Lucレポーターアッセイ
Gq刺激を検出するための一方法は、それらのプロモーター中に血清応答因子
を含有する遺伝子の活性化を引き起こすGq依存性のホスホリパーゼCの既知の
特性に依存する。例えばCOS7細胞でのGq共役型の活性についてアッセイす
るのに、パスディテクト[Pathdetect](商標)SRF−Lucレポ
ーティング系(ストラタジーン(Stratagene))を利用することがで
きる。製造元の説明書に従って哺乳動物トランスフェクション[Mammali
an Transfection](商標)キット(ストラタジーン(Stra
tagene)、カタログ番号200285)を使用して、系のプラスミド成分
および内在性もしくは非内在性のGPCRをコードする指定された発現プラスミ
ドで細胞をトランスフェクションする。簡潔には、410ngのSRF−Luc
、80ngのpCMV受容体発現プラスミドおよび20ngのCMV−SEAP
(分泌型アルカリホスファターゼ発現プラスミド;トランスフェクションされた
細胞の培地中でアルカリホスファターゼ活性を測定して、サンプル間のトランス
フェクションの効率の変動について制御する)を、製造元の説明書に従ってリン
酸カルシウム沈殿物中で組み合わせる。沈殿物の半分を96穴プレートの3個の
ウェルに同等に配分し、血清を含まない培地中で細胞上で24時間保つ。指定さ
れた場合は、最後の5時間、細胞を1μMのアンジオテンシンとともにインキュ
ベートする。その後細胞を溶解し、そして、製造元の説明書に従ってルックライ
ト[Luclite](商標)キット(パッカード(Packard)、カタロ
グ番号6016911)および「トライルックス(Trilux)1450マイ
クロベータ(MicroBeta)」液体シンチレーションおよび発光計数器(
ワラック(Wallac))を使用してルシフェラーゼ活性についてアッセイす
る。データはグラフパッド プリズム[GraphPad Prism](商標
)2.0a(グラフパッド ソフトウェア インク(GraphPad Sof
tware Inc.))を使用して解析することができる。
5.細胞内IP3蓄積アッセイ
第1日に、受容体(内在性および/もしくは非内在性)を含んで成る細胞を2
4穴プレート上でプレート培養することができる(通常、ウェルあたり細胞1×
105個、とは言えこの数字は至適化することができる)。第2日に、ウェルあ
たり50μlの血清を含まないDMEM中の0.25μgのDNAおよびウェル
あたり50μlの血清を含まないDMEM中の2μlのリポフェクタミンを最初
に混合することにより、細胞をトランスフェクションすることができる。溶液は
穏やかに混合し、そして室温で15〜30分間インキュベートする。0.5ml
のPBSで細胞を洗浄し、そして400μlの血清を含まない培地をトランスフ
ェクション培地と混合しかつ細胞に添加する。その後、細胞を37℃/5%CO
2で3〜4時間インキュベートし、そしてその後、トランスフェクション培地を
除去しかつウェルあたり1mlの通常の成長培地で置き換える。第3日に、細胞
を3H−ミオイノシトールで標識する。簡潔には、培地を除去し、そして細胞を
0.5mlのPBSで洗浄する。その後、0.5mlのイノシトールを含まない
/血清を含まない培地(ギブコ(GIBCO)BRL)を、ウェルあたり0.2
5μCiの3H−ミオイノシトールとともにウェルあたりに添加し、そして細胞
を37℃/5%CO2で16〜18時間一夜インキュベートする。第4日に、細
胞を0.5mlのPBSで洗浄し、そして、イノシトールを含まない/血清を含
まない培地、10μMパージリン、10mM塩化リチウムを含有する0.45m
lのアッセイ培地、もしくは0.4mlのアッセイ培地および10μMの最終濃
度までの50μlの10×ケタンセリン(ket)を添加する。その後細胞を3
7℃で30分間インキュベートする。その後細胞を0.5mlのPBSで洗浄し
、そしてウェルあたり200μlの新鮮/氷冷停止溶液(1M KOH;18m
Mホウ酸ナトリウム;3.8mM EDTA)を添加する。溶液を5〜10分間
もしくは細胞が溶解されるまで氷上で保ち、そしてその後、200μlの新鮮/
氷冷中和溶液(7.5%HCl)により中和する。その後、ライセートを1.5
mlのエッペンドルフチューブに移し、そしてチューブあたり1mlのクロロホ
ルム/メタノール(1:2)を添加する。溶液を15秒間ボルテックス攪拌し、
そして上層をバイオラッド(BioRad)AG1−X8(商標)陰イオン交換
樹脂(100〜200メッシュ)に適用する。最初に樹脂を1:1.25W/V
の水で洗浄し、そして0.9mlの上層をカラムに負荷する。カラムを10ml
の5mMミオイノシトールおよび10mlの5mMホウ酸ナトリウム/60mM
ギ酸ナトリウムで洗浄する。2mlの0.1Mギ酸/1Mギ酸アンモニウムを用
い、10mlのシンチレーションカクテルを含有するシンチレーションバイアル
中にイノシトールトリスリン酸を溶出する。10mlの0.1Mギ酸/3Mギ酸
アンモニウムで洗浄することによりカラムを再生し、そしてddH2Oで2回す
すぎ、そして水中で4℃で保存する。
【0077】
例示的結果を下の表Iに提示する:
【0078】
【表10】
【0079】
C.細胞に基づく検出アッセイ(例−TDAG8)
293細胞をプレートあたり細胞1.3×107個の密度で150mmプレー
ト上でプレート培養し、そしてプレートあたり12μgのそれぞれのDNAおよ
び60μlのリポフェクタミン試薬(BRL)を使用してトランスフェクション
した。トランスフェクションされた細胞は、トランスフェクション後24時間に
実施されるアッセイのため血清を含有する培地中で成長させた。トランスフェク
ション後48時間に実施される検出アッセイ(血清および血清を含まない培地を
比較するアッセイ;図3を参照されたい)のため、初期培地を血清もしくは血清
を含まない培地のいずれかに変更した。血清を含まない培地は、単にダルベッコ
の改変イーグル(DME)高グルコース培地(アーヴィン サイエンティフィッ
ク(Irvine Scientific)#9024)から構成された。上の
DME培地に加えて、血清を含む培地は以下、すなわち10%ウシ胎児血清(ハ
イクロン(Hyclone)#SH30071.03)、1%の100mMピル
ビン酸ナトリウム(アーヴィン サイエンティフィック(Irvine Sci
entific)#9334)、1%の20mM L−グルタミン(アーヴィン
サイエンティフィック(Irvine Scientific)#9317)
および1%のペニシリン−ストレプトマイシン溶液(アーヴィン サイエンティ
フィック(Irvine Scientific)#9366)を含有した。
【0080】
96穴のアデニリルシクラーゼ活性化フラッシュプレート[Flashpla
te](商標)を使用した(NEN:#SMP004A)。最初に、アッセイの
ための標準50μlを、ウェルあたり50pmolから0pmolまでのcAM
P濃度の範囲にわたるプレート(二重(in duplicate))に添加した。標準cAM
P(NEN:#SMP004A)を水で再構成し、そして1×PBS(アーヴィ
ン サイエンティフィック(Irvine Scientific)#9240
)を使用して連続的希釈物を作成した。次に、50μlの刺激緩衝液(NEN:
#SMP004A)を全部のウェルに添加した。cAMPの活性化もしくは不活
性化を測定するのに化合物を使用する場合は、水で希釈された各化合物10μl
を三重でそのそれぞれのウェルに添加した。使用された多様な最終濃度は1μM
から1mMまでの範囲にわたる。アデノシン5’−三リン酸、ATP(リサーチ
バイオケミカルズ インターナショナル(Research Biochem
icals International:#A−141)およびアデノシン5
’−二リン酸、ADP(シグマ(Sigma):#A2754)をアッセイで使
用した。次に、それぞれのcDNA(CMVもしくはTDAG8)でトランスフ
ェクションされた293細胞を、トランスフェクション後24(血清培地中での
アッセイの検出)もしくは48(血清および血清を含まない培地を比較するアッ
セイの検出)時間で収穫した。培地を吸引し、そして細胞を1×PBSで1回洗
浄した。その後、5mlの1×PBSを3mlの細胞解離緩衝液(シグマ(Si
gma):#C−1544)とともに細胞に添加した。解離された細胞を遠心分
離チューブに移し、そして室温で5分間遠心分離した。上清を除去し、そして、
1ミリリットルあたり細胞2×106個の最終濃度を得るように、細胞ペレット
を適切な量の1×PBSに再懸濁した。化合物を含有するウェルに、1×PBS
中の細胞50μl(ウェルあたり細胞1×105個)を添加した。プレートを室
温で15分間振とう機でインキュベートした。トレーサーcAMPを含有する検
出緩衝液を調製した。11mlの検出緩衝液(NEN:#SMP004A)中で
50μl(1μCiに等しい)の[125I]cAMP(NEN:#SMP004
A)を添加した。インキュベーション後に、トレーサーcAMPを含有するこの
検出緩衝液50μlを各ウェルに添加した。プレートを振とう機に設置し、そし
て室温で2時間インキュベートした。最後に、プレートのウェルからの溶液を吸
引し、そしてワラック(Wallac)マイクロベータ[MicroBeta]
(商標)シンチレーション計数器を使用してフラッシュプレートを計数した。
【0081】
図2Aにおいて、ATPおよびADPは内在性のTDAG8に結合し、それぞ
れ約59%および約55%のcAMPの増大をもたらす。図2Bは、内在性のT
DAG8へのATPおよびADPの結合を明示し、ここでは内在性TDAG8が
トランスフェクションされ、そして血清および血清を含まない培地中で成長され
た。血清培地中で成長された内在性のTDAG8へのATPの結合は、化合物を
伴わない内在性のTDAG8に比較して、約65%のcAMPの増大を明示し;
血清を含まない培地中では約68%の増大が存在した。血清中の内在性TDAG
8へのADPの結合は約61%の増大を明示する一方、血清を含まない培地中で
はADP結合が約62%増大の増大を明示する。ATPおよびADPは、それぞ
れ139.8μMおよび120.5μMのEC50値で内在性のTDAG8に結
合する(データは示されない)。
【0082】
図2Bに提示される結果は、血清および血清を含まない培地を比較した場合に
実質的に同一の結果を示すが、われわれの選択は血清に基づく培地を使用するこ
とである。とは言え血清を含まない培地もまた利用することが可能である。
実施例6
GPCR融合タンパク質の調製
構成的に活性化されるGPCR−Gタンパク質融合構築物の設計を以下のとお
り達成した。すなわち、ラットGタンパク質Gsα(長形態;伊藤(Itoh,
H.)ら、83 PNAS 3776(1986))の5’および3’双方の端
をそれにHindIII(5’−AAGCTT−3’)配列を包含するように工
作した。正しい配列(隣接するHindIII配列を包含する)の確認後に、そ
のベクターのHindIII制限部位を使用してサブクローニングすることによ
り、配列全体をpcDNA3.1(−)(インヴィトロジェン(Invitro
gen)、カタログ番号V795−20)にsnuttleした。pcDNA3
.1(−)へのサブクローニング後にGsαの配列について正しい向きを決定し
た。HindIII配列でラットGsα遺伝子を含有する改変されたpcDNA
3.1(−)をその後確認し;このベクターは今や「普遍的」Gsαタンパク質
ベクターとして利用可能となった。pcDNA3.1(−)ベクターは、Hin
dIII部位の上流に多様な公知の制限部位を含有し、従ってGsタンパク質の
上流に内在性の構成的に活性なGPCRのコーディング配列を挿入する能力を有
益に提供する。他の「普遍的」Gタンパク質ベクターを創製するのにこの同一の
アプローチを利用することができ、そして、もちろん、当業者に既知の他の商業
的に入手可能なもしくは占有のベクターを利用することができる。重要な基準は
、GPCRの配列がGタンパク質の配列の上流にかつ同じ読み枠で存在すること
である。
【0083】
TDAG8はGsを介して共役する一方、H9はGzを介して共役する。以下
の例示的GPCR融合タンパク質についてGsαへの融合を達成した。
【0084】
TDAG8(I225K)−Gsα融合タンパク質構築物は以下のとおり作成
した。すなわち、プライマーを以下のとおり設計した:
5’−gatcTCTAGAATGAACAGCACATGTATTGAAG−
3’(配列番号125;センス)
5’−ctagGGTACCCGCTCAAGGACCTCTAATTCCAT
AG−3’(配列番号126;アンチセンス)。
【0085】
小文字のヌクレオチドはGタンパク質とTDAG8との間の制限部位中のスペ
ーサーとして包含される。該センスおよびアンチセンスプライマーはそれぞれX
baIおよびKpnIの制限部位を包含した。
【0086】
その後、それぞれについて以下のプロトコルを使用し、PCRを利用して、上
に開示されたGsα普遍的ベクター内の融合についてそれぞれの受容体配列を保
証した。すなわち、2μlの各プライマー(センスおよびアンチセンス)、3μ
Lの10mM dNTP、10μLの10×タックプラス[TaqPlus](
商標)プレシジョン(Precision)緩衝液、1μLのタックプラス[T
aqPlus](商標)プレシジョン(Precision)ポリメラーゼ(ス
トラタジーン(Stratagene):#600211)ならびに80μLの
水を含有する別個のチューブに、100ngのTDAG8のcDNAを添加した
。TDAG8についての反応温度および周期時間は以下のとおりであった。すな
わち、最初の変性段階はそれを94℃で5分間、そして、94℃30秒間;55
℃30秒間;72℃2分間の周期を行った。最後の伸長時間は72℃で10分間
行った。順向きのPCR産物(PCR product for)を1%アガロースゲル上で泳動
し、そしてその後精製した(データは示されない)。精製された産物をXbaI
およびKpnI(ニュー イングランド バイオラブス(New Englan
d Biolabs))で消化し、そして所望の挿入物を精製しかつそれぞれの
制限部位でGs普遍的ベクターに連結した。形質転換後に陽性のクローンを単離
し、そして制限酵素消化により決定し;293細胞を使用する発現は下に示され
るプロトコルに従って達成した。TDAG8:Gs融合タンパク質についてのそ
れぞれの陽性のクローンを配列決定して正しさを確かめた。
【0087】
非内在性の構成的に活性化されるTDAG8(I225K)を含んで成るGP
CR融合タンパク質を上のとおり分析し、そして構成的活性化について確かめた
。
【0088】
H9(F236K)−Gsα融合タンパク質構築物は以下のとおり作成した。
すなわち、プライマーを以下のとおり設計した:
5’−TTAgatatcGGGGCCCACCCTAGCGGT−3’(配列
番号145;センス)
5’−ggtaccCCCACAGCCATTTCATCAGGATC−3’(
配列番号146;アンチセンス)。
【0089】
小文字のヌクレオチドはGタンパク質とH9との間の制限部位中のスペーサー
として包含される。センスおよびアンチセンスプライマーはそれぞれEcoRV
およびKpnIの制限部位を包含し、その結果、スペーサー(制限部位に帰され
る)がGタンパク質とH9との間に存在する。
【0090】
その後、それぞれについて以下のプロトコルを使用し、PCRを利用して、上
に開示されたGsα普遍的ベクター内の融合についてそれぞれの受容体配列を保
証した。すなわち、100ngの各プライマー(センスおよびアンチセンス)な
らびに45μLのPCRスーパーミックス[Supermix](商標)(ギブ
コ(Gibco)−Brl、ライフテック(LifeTech))を含有する別
個のチューブに、80ngのH9のcDNAを添加した(50μLの総反応体積
)。H9についての反応温度および周期時間は以下のとおりであった。すなわち
、最初の変性段階はそれを94℃で1、そして、94℃30秒間;55℃30秒
間、72℃2分間の周期を行った。最後の伸長時間は72℃で7分間行った。順
向きのPCR産物を1%アガロースゲル上で泳動し、そしてその後精製した(デ
ータは示されない)。精製された産物をpCRII−TOPO(商標)系にクロ
ーン化し、次いで陽性のクローンを同定した。陽性のクローンを単離し、Eco
RVおよびKpnI(ニュー イングランド バイオラブス(New Engl
and Biolabs))で消化し、そして所望の挿入物を単離し、精製しか
つそれぞれの制限部位でGs普遍的ベクターに連結した。形質転換後に陽性のク
ローンを単離し、そして制限酵素消化により決定し;293細胞を使用する発現
は下に示されたプロトコルに従って達成した。H9(F236K):Gs融合タ
ンパク質についての各陽性のクローンを配列決定して正しさを確かめた。膜は利
用されるまで凍結(−80℃)した。
【0091】
(H9はGzと共役するのであるが)Gsタンパク質により媒介されるcAM
P応答を測定する能力を確かめるため、NENアデニルシクラーゼ活性化フラッ
シュプレート[Flashplate](商標)アッセイキット(96穴の形式
)に基づき、以下のcAMP膜アッセイを利用した。「結合緩衝液」は10mM
HEPES、100mM NaClmおよび10mM MgCl(pH7.4
)より成った。「再生緩衝液」は結合緩衝液中で調製し、そして20mMホスホ
クレアチン、20Uのクレアチンホスホキナーゼ、20μM GTP、0.2m
M ATPおよび0.6mM IBMXより成った。「cAMP標準」は以下の
とおり結合緩衝液中で調製した:
【0092】
【表11】
【0093】
凍結された膜(対照としてのpCMVおよび非内在性のH(−Gs融合タンパ
ク質の双方)を(溶液中まで室温で氷上で)融解した。膜を懸濁液中までポリト
ロンで均質化した(2×15秒)。ブラッドフォード(Bradford)アッ
セイプロトコル(下を参照されたい)を使用して膜タンパク質濃度を測定した。
再生緩衝液中で膜の濃度を0.5mg/mlに希釈した(最終アッセイ濃度−ウ
ェルあたり25μg)。その後、50μlの結合緩衝液を各ウェルに添加した。
対照には、ウェルあたり50μlのcAMP標準をウェル11および12A−G
に、結合緩衝液単独を12Hに添加した(96穴の形式で)。その後、ウェルあ
たり50μlのタンパク質をウェルに添加し、そして室温で(振とう機上で)6
0分間インキュベートした。検出緩衝液(下を参照されたい)中100μlの[
125I]cAMPを各ウェルに添加した(最終−11mlの検出緩衝液中に50
μlの[125I]cAMP)。これらを室温で2時間インキュベートした。プレ
ートを8チャンネルのマニホールドで吸引し、そしてプレートの蓋で封止した。
結果(結合されたcAMPのpmol)を「プロット#15」でワラック[Wa
llac](商標)1450で読み取った。結果を図3に提示する。
【0094】
図3に提示される結果は、Gs共役型融合がシクラーゼ反応を「駆動する」こ
とが可能であり、その結果H9(F236K)の構成的活性化の測定が実現可能
であったことを示す。これらの結果に基づけば、反作用薬、アゴニストおよび部
分的アゴニストである候補化合物の直接同定がシクラーゼに基づくアッセイを使
用して可能である。
実施例6
プロトコル:[35S]GTPγSを使用する反作用薬およびアゴニストの直接同
定
われわれは、全く理解されていない理由上、例えば反作用薬のような候補化合
物の直接同定に内在性の構成的に活性のGPCRを利用したが、アッセイ間の変
動が悪化されたようになる可能性がある。その場合、好ましくは、上に開示され
たようなGPCR融合タンパク質を非内在性の構成的に活性化されるGPCRと
ともにもまた利用する。われわれは、こうしたタンパク質を使用する場合にアッ
セイ間の変動が実質的に安定化されるようであり、それにより有効なS/N比が
得られることを決定した。これは候補化合物のより確固たる同定を見込むという
有益な結果を有する。従って、直接同定のためにはGPCR融合タンパク質を使
用すること、また、利用される場合は以下のアッセイプロトコルを利用すること
が好ましい。
膜の調製
目的の、および反作用薬、アゴニストもしくは部分的アゴニストとしての候補
化合物の直接同定での使用のための非内在性の構成的に活性のオーファンGPC
R融合タンパク質を含んで成る膜は、以下のように好ましく調製する:
a.材料
「膜掻き取り緩衝液」は20mM HEPESおよび10mM EDTA、p
H7.4より構成され;「膜洗浄緩衝液」は20mM HEPESおよび0.1
mM EDTA、pH7.4から構成され;「結合緩衝液」は20mM HEP
ES、100mM NaClおよび10mM MgCl2、pH7.4から構成
される。
b.手順
全部の材料は処置の間中氷上に保つ。最初に、細胞のコンフルエントな単層か
ら培地を吸引し、次いで10mlの冷PBSですすぎ、次いで吸引する。その後
、5mlの膜掻き取り緩衝液を添加して細胞を掻き取り;これに次いで細胞抽出
物を50ml遠心管に移す(20,000rpmで4℃で17分間遠心分離され
る)。その後、上清を吸引し、そしてペレットを30mlの膜洗浄緩衝液に再懸
濁し、次いで20,000rpmで4℃で17分間遠心分離する。上清をその後
吸引し、そしてペレットを結合緩衝液に再懸濁する。その後、ブリンクマン(B
rinkman)ポリトロン[polytron](商標)ホモジェナイザーを
使用してこれを均質化する(全部の物質が懸濁液中にあるまで15〜20秒破裂
させる)。これを本明細書で「膜タンパク質」と称する。
ブラッドフォード(Bradford)タンパク質アッセイ
均質化の後に、ブラッドフォード(Bradford)タンパク質アッセイを
使用して膜のタンパク質濃度を測定する(タンパク質を約1.5mg/mlに希
釈し、等分し、そして後の使用のため凍結する(−80℃)ことができ;凍結さ
れる場合、使用のためのプロトコルは以下のとおりである。すなわち、アッセイ
の日に、凍結された膜タンパク質を室温で融解し、次いでボルテックス攪拌し、
そしてその後、約5〜10秒間約12×1,000rpmでポリトロンで均質化
し;複数の調製のためには、異なる調製物の均質化の間にホモジェナイザーを徹
底的に洗浄すべきであることが言及される)。
a.材料
結合緩衝液(上に従う);ブラッドフォード(Bradford)色素試薬;
ブラッドフォード(Bradford)タンパク質標準を製造元の説明書に従っ
て(バイオラッド(BioRad)カタログ番号500−0006)利用する。
b.手順
二重のチューブを準備する(1本は膜を包含し、そして1本は対照の「ブラン
ク」として)。それぞれは800μlの結合緩衝液を含有した。その後、10μ
lのブラッドフォード(Bradford)タンパク質標準(1mg/ml)を
各チューブに添加し、そしてその後10μlの膜タンパク質を1本のチューブに
だけ添加する(ブランクでない)。その後、200μlのブラッドフォード(B
radford)色素試薬を各チューブに添加し、次いでそれぞれをボルテック
ス攪拌する。5分後にチューブを再度ボルテックス攪拌し、そして、その中の物
質をキュベットに移す。その後、波長595でCECIL 3041分光光度計
を使用してキュベットを読み取る。
直接同定アッセイ
a.材料
GDP緩衝液は37.5mlの結合緩衝液および2mgのGDP(シグマ(S
igma)、カタログ番号G−7127)より成り、次いで0.2μM GDP
を得るため結合緩衝液で一連の希釈を行い(各ウェル中でのGDPの最終濃度は
0.1μM GDPであった);候補化合物を含んで成る各ウェルは、100μ
lのGDP緩衝液(最終濃度、0.1μM GDP)、50μlの結合緩衝液中
の膜タンパク質、および50μlの結合緩衝液中の[35S]GTPγS(0.6
nM)(10mlの結合緩衝液あたり2.5μlの[35S]GTPγS)より成
る200μlの最終体積を有する。
b.手順
候補化合物は96穴プレートの形式(これらは−80℃で凍結させることがで
きる)を使用して好ましくスクリーニングする。膜タンパク質(もしくは、対照
として、GPCR融合タンパク質を除外する発現ベクターを含む膜)を懸濁液中
まで短く均質化する。その後、上に示されたブラッドフォード(Bradfor
d)タンパク質アッセイを使用してタンパク質濃度を測定する。その後、膜タン
パク質(および対照)を結合緩衝液で0.25mg/mlに希釈する(最終アッ
セイ濃度、ウェルあたり12.5μg)。その後、100μlのGDP緩衝液を
ワラック(Wallac)シンチストリップ[Scintistrip](商標
)(ワラック(Wallac))の各ウェルに添加する。その後、5μlのピン
ツール(pin-tool)を使用して、こうしたウェルに5μlの候補化合物を移す(す
なわち、200μlの総アッセイ体積中の5μlは1:40の比であり、その結
果候補化合物の最終スクリーニング濃度は10μMである)。再度、汚染を回避
するため、各移送段階の後にはピンツールを水(1×)、エタノール(1×)お
よび水(2×)を含んで成る3個の水槽ですすぐべきであり、過剰の液体は各す
すぎの後にツールから振りそして紙およびキムワイプでぬぐって乾かすべきであ
る。その後、50μlの膜タンパク質を各ウェルに添加し(GPCR融合タンパ
ク質を含まない膜を含んで成る対照ウェルもまた利用する)、そして室温で5〜
10分間前インキュベートする。その後、50μlの結合緩衝液中の[35S]G
TPγS(0.6nM)を各ウェルに添加し、次いで室温で振とう機で60分間
インキュベートする(再度、この実施例において、プレートをホイルで覆った)
。その後、22℃で4000RPMで15分間のプレートの回転によりアッセイ
を停止する。その後、プレートを8チャンネルのマニホールドで吸引し、そして
プレートの蓋で封止する。その後、(製造元の説明書に従って)「Prot.#
37」の設定を使用してワラック(Wallacc)1450でプレートを読み
取る。
実施例7
プロトコル:確認アッセイ
上に示されたような直接同定された候補化合物の確認を提供するための独立の
アッセイのアプローチを使用して、その後確認アッセイを利用することが好まし
い。この場合、好ましい確認アッセイはシクラーゼに基づくアッセイである。
【0095】
改変されたフラッシュプレート[Flash Plate](商標)アデニリ
ルシクラーゼキット(ニュー イングランド ニュークリア(New Engl
and Nuclear);カタログ番号SMP004A)を、以下のプロトコ
ルに従って、非内在性の構成的に活性化されるオーファンGPCRに対する反作
用薬およびアゴニストとして直接同定された候補化合物の確認に、好ましく利用
する。
【0096】
トランスフェクションされた細胞をトランスフェクション後およそ3日に収穫
する。20mM HEPES、pH7.4および10mM MgCl2を含有す
る緩衝液中に懸濁された細胞の均質化により膜を調製する。均質化は、ブリンク
マン(Brinkman)ポリトロン[Polytron](商標)を使用して
氷上でおよそ10秒間実施する。生じるホモジェネートは49,000×gで4
℃で15分間遠心分離する。その後、生じるペレットを、20mM HEPES
、pH7.4および0.1mM EDTAを含有する緩衝液に再懸濁し、10秒
間均質化し、次いで49,000×gで4℃で15分間遠心分離する。生じるペ
レットは利用されるまで−80℃で保存することができる。直接同定スクリーニ
ングの日に、膜ペレットを室温でゆっくりと融解し、20mM HEPES、p
H7.4および10mM MgCl2を含有する緩衝液に再懸濁して、0.60
mg/mlの最終タンパク質濃度を生じる(再懸濁された膜は使用まで氷上に置
く)。
【0097】
cAMP標準および検出緩衝液(11mlの検出緩衝液に対して2μCiのト
レーサー[125I]cAMP(100μl)を含んで成る)は、製造元の説明書
に従って調製しかつ維持する。アッセイ緩衝液はスクリーニングのために新たに
調製し、そして20mM HEPES、pH7.4、10mM MgCl2、2
0mMホスホクレアチン(シグマ(Sigma))、0.1単位/mlのクレア
チンホスホキナーゼ(シグマ(Sigma))、50μM GTP(シグマ(S
igma))および0.2mM ATP(シグマ(Sigma))を含有し;ア
ッセイ緩衝液は利用されるまで氷上で保存することができる。
【0098】
上に従って同定された候補化合物(凍結されている場合は室温で融解させる)
を、40μMの膜タンパク質(ウェルあたり30μg)および50μlのアッセ
イ緩衝液と一緒に、好ましくは96穴プレートのウェルに添加する(ウェルあた
り3μl;12μMの最終アッセイ濃度)。その後、この混合状態を穏やかに振
とうしながら室温で30分間インキュベートする。
【0099】
インキュベーション後に、各ウェルに100μlの検出緩衝液を添加し、次い
で2〜24時間インキュベートする。その後、プレートを、(製造元の説明書に
従い)「Prot.#31」を使用してワラック(Wallac)マイクロベー
タ[MicroBeta](商標)プレートリーダーで計数する。
【0100】
本特許文書で挙げられる特許、出願および印刷された刊行物のそれぞれはこれ
によりそっくりそのまま引用により組み込まれることが意図される。
【0101】
当業者は、多数の変更および改変を、本発明の技術思想から離れることなく本
発明の好ましい態様に対し行ってよいことを認識するであろう。全部のこうした
変形物は本発明の範囲内にあることが意図される。
【0102】
多様な発現ベクターが当業者に利用可能であるが、内在性および非内在性双方
のヒトGPCRに対する利用の目的上、利用されるベクターはpCMVであるこ
とが最も好ましい。このベクターは、特許手続上の微生物の寄託の国際的承認に
関するブダペスト条約(the Budapest Treaty for t
he International Recognition of the
Deposit of Microorganisms for the Pu
rpose of Patent Procedure)の規定の下に、199
8年10月13日にアメリカン タイプ カルチャー コレクション(Amer
ican Type Culture Collection)(ATCC)(
10801 University Blvd.,米国バージニア州マナサス
20110−2209)に寄託された。該DNAはATCCにより試験され、そ
してそうであることが決定された。ATCCはpCMVに対し以下の寄託番号:
ATCC#203351を割り当てている。
(配列表)
【特許請求の範囲】
【請求項1】
明細書に記載のcDNA。
【請求項1】
明細書に記載のcDNA。
【図1】

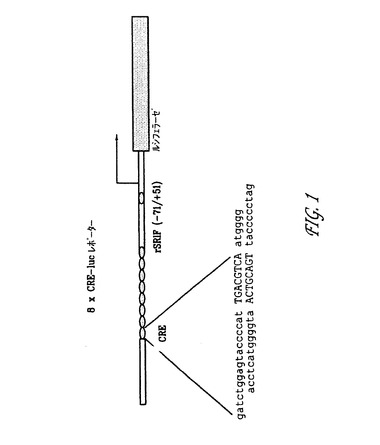
【図2】
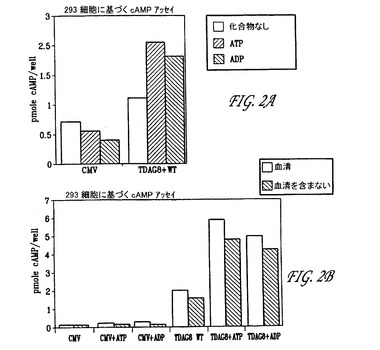
【図3】
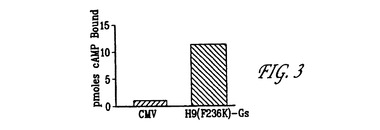
【図2】
【図3】
【公開番号】特開2010−142235(P2010−142235A)
【公開日】平成22年7月1日(2010.7.1)
【国際特許分類】
【出願番号】特願2010−87(P2010−87)
【出願日】平成22年1月4日(2010.1.4)
【分割の表示】特願2000−576021(P2000−576021)の分割
【原出願日】平成11年10月13日(1999.10.13)
【出願人】(500478097)アリーナ ファーマシューティカルズ, インコーポレイテッド (97)
【Fターム(参考)】
【公開日】平成22年7月1日(2010.7.1)
【国際特許分類】
【出願日】平成22年1月4日(2010.1.4)
【分割の表示】特願2000−576021(P2000−576021)の分割
【原出願日】平成11年10月13日(1999.10.13)
【出願人】(500478097)アリーナ ファーマシューティカルズ, インコーポレイテッド (97)
【Fターム(参考)】
[ Back to top ]