
非加水分解性ヌクレオシド二又は三リン酸誘導体及びその使用
本発明は、非加水分解性ヌクレオシドポリリン酸誘導体、例えば、2MeS−アデノシン−β,γ−CH2−5’−O−(1−ボラノ三リン酸)、2MeS−アデノシン−β,γ−CCl2−5’−O−(1−ボラノ三リン酸)、2−MeS−アデノシン−5’−ジクロロメチレン−二リン酸、2−MeS−アデノシン−5’−ジフルオロメチレン−二リン酸及び2MeS−アデノシン−5’−O−(1−ボラノ二リン酸)、並びにその医薬組成物を提供する。これらの化合物は、2型糖尿病等のP2Y受容体によって調節される疾患又は障害の予防又は治療及び疼痛管理に有用である。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、非加水分解性ヌクレオシドポリリン酸誘導体及びそれらを含む医薬組成物に関する。該化合物は、2型糖尿病等のP2Y受容体によって調節される疾患又は障害の予防又は治療及び疼痛管理に有用である。
【背景技術】
【0002】
リガンド作動性イオンチャネル(P2XR)及びGタンパク質共役受容体(P2YR)からなるP2受容体(P2R)スーパーファミリーは、主として、細胞外ヌクレオチドATP、ADP、UTP又はUDPによって活性化される(Jacobsonら、2002)。加えて、P2受容体は、いくつかのジヌクレオシドポリリン酸(ジヌクレオチド)によって活性化される(WO2003/0207825;Shaverら、2005)。
【0003】
P2YRは、正常及び病態生理学的条件下の両方で、多くの組織及び器官中での種々の機能の調節にそれらが関与することから、魅力的な医薬標的であり(Williams及びJarvis、2000;Guileら、2001、Fischer、1999)、このことがP2YRアゴニストを潜在的な薬物としている。現在、薬物として提案されているP2YRアゴニストは、酵素的及び化学的に不安定なヌクレオチド骨格からなる(Williams及びJarvis、2000;Fischer、1999;Abbracchioら、2006;Jacobsonら、2002;Laxman及びBeavo、2007)。
【0004】
ヌクレオチドベースの薬物候補の固有の不安定性を克服するためのアプローチは、(i)対応するヌクレオチドよりも代謝的に安定なジヌクレオチド、(ii)非ヌクレオチドP2Rリガンド、(iii)ヌクレオチドプロドラッグ、及び(iv)等価体ベースの非加水分解性ヌクレオチドの使用を含む。
【0005】
第1のアプローチはかなり有望であり、実際にヒトの前臨床試験においていくつかのジヌクレオチドが投与されている。例えば、Ap4A、Up4U及びUp4dCは、麻酔中の血圧を低下させるため、並びにドライアイ疾患、嚢胞性線維症及び網膜剥離の治療としてそれぞれ有効であると立証された(Kikutaら、1999;Maminishkisら、2002;Mundasadら、2001;Yerxaら、2002)。
【0006】
第2のアプローチは、クロピドグレル(Plavix(登録商標)、Sanofi−Synthelabo/BMS)という、二次的な血管イベントの予防に使用される血小板抗凝集剤の場合に成功を収めており(Chow及びZiegelstein、2007)、これは、現在利用可能な唯一のP2YR標的薬物である。P2Y12受容体アンタゴニストとして作用するクロピドグレル(Angiolilloら、2006a及び2006b)は、非ヌクレオチドである。
【0007】
第3のアプローチは、マスクされたトリエステルヌクレオチドプロドラッグの調製に関与する。これらのプロドラッグ、例えば、抗HIVヌクレオシド類似体d4Tは、膜可溶性であると立証され、細胞内で活性ヌクレオチドを放出した(McGuiganら、1993、1996a及び1996b;WO/2002/055521)。
【0008】
生物学的等価体アプローチによって、ヌクレオチドベースの薬物候補、酵素阻害剤又は受容体リガンドいずれかの安定性を改善するための試みは、ごくわずかしか報告されていない(Blackburnら、1987;Cusackら、1987;Heら、1997;Kowalskaら、2007;Linら、2001;Misiuraら、2005;Romaniuk及びEckstein、1981;Stingelinら、1980)。
【0009】
真性糖尿病は、西欧諸国において最も蔓延している慢性疾患の1つであり、人口の最大5%を侵している。これは、脂質及びタンパク質代謝における追加の異常を伴う慢性高血糖(インスリン分泌、インスリン作用又は両方の組合せにおける欠陥に起因する)を特徴とする障害の混成群である。その慢性的代謝異常に加えて、糖尿病は、種々の器官、特に、眼、神経、血管、心臓及び腎臓に関与する長期合併症に関連し、これらは、失明、切断、心臓血管疾患及び末期腎不全をもたらし得る。糖尿病性合併症の発症は、血中グルコースの慢性的上昇と関係しているように思われる。現在のところ糖尿病の治癒法はないが、有効な血糖管理は、糖尿病性合併症の発生率を低下させ、その重症度を軽減することができる。
【0010】
非インスリン依存性真性糖尿病(NIDDM)とも称される2型糖尿病は、糖尿病に罹患している患者の約95%を侵しており、インスリン抵抗性及び相対的インスリン欠乏が共存する複雑な多遺伝子性疾患であるように思われる。よって、インスリン分泌の改善が主な治療目的である。インスリン放出の欠乏は、グルコースに対する第1相インスリン応答の欠如によってだけでなく、インスリン放出の規模が正常な分泌能力の10〜20%まで全体的に低減することによっても現れる。2型糖尿病に罹患している患者は、種々の経口抗糖尿病剤、インスリン注射又は両方の組合せによって治療される。現在利用可能な経口抗糖尿薬物は、膵臓ベータ細胞からのインスリン分泌を増加させること、末梢インスリン抵抗性を低減させること、又は腸からの糖質の吸収を遅らせることのいずれかを目的としている。
【0011】
2型糖尿病に罹患している患者の約半分は経口剤で治療され、該経口剤のかなりの割合がインスリン分泌を刺激する作用物質である。インスリン分泌促進物質の選定は、スルホニル尿素及び関連化合物(「グリニド系薬」)に限定され、これらは、膜ATP感受性カリウムチャネルの制御サブユニットと結合し、その閉鎖を誘導することによりインスリン分泌を誘発する。しかしながら、スルホニル尿素は、その特異的標的である膵臓ベータ細胞への起こり得る長期的な悪影響に加えて、いくつかの望ましくない作用を有する。これらの副作用は、低グルコース濃度でのインスリン分泌の刺激による低血糖のリスク、多数の患者において正常な血糖を達成することの難しさ、年間5〜10%という十分な血糖管理の二次的な失敗率、及び心臓血管系への起こり得る負の作用を含む。
【0012】
膵臓ベータ細胞におけるP2YRの存在は、文書による十分な裏付けがあり、それらの活性化は、刺激性のグルコース濃度でのインスリン分泌の刺激をもたらす。グルコースによって引き起こされるインスリン放出をP2YRアゴニストが増強する機序は、環状AMP/タンパク質キナーゼAシグナル伝達経路に関与し得、該経路は、グルコースのK+ATPチャネル非依存性作用の有効性を増大させると報告されている。
【0013】
種々のP2R選択的リガンドは、インビボにおいて、インスリン分泌を増加させ、血糖を減少させることを示した。リガンドのリストは、2−MeS−アデノシンに迅速に分解し、よって膵十二指腸動脈に直接注射された2−メチルチオ−ATP、及び酵素加水分解に安定であり、よって静脈内又は経口のいずれかで投与されたアデノシン5’−O−(2−チオ)二リン酸を含む。
【0014】
現在の合成P2受容体アゴニストは殆どすべて、ATP又はUTP薬理作用団の修飾物である。プリン(ピリミジン)環系、リボース部分又は三リン酸鎖は、1つ又は複数の位置で修飾される(Fischer、1999)。以前、本発明者らは、2−チオエーテル−5’−O−(1−チオ三リン酸)アデノシン誘導体等、C−2位に長いチオエーテル置換を有するATP誘導体の合成について報告した(Fischerら、1999)。
【0015】
US7,319,093に対応するWO2003/034978は、ATP類似体(アデノシン−5’−α−ボラノ−三リン酸類似体)のボラノリン酸等価体に基づく一連の強力且つ選択的なP2Y1Rアゴニストを開示している(Nahumら、2002;Majorら、2004;Tulapurkarら、2004;Farretら、2006)。これらの類似体は、生理的pHにおいて高度に安定であり、pH1.4及び37℃において比較的安定であることが立証された。さらに、これらのアゴニストは、エクト−ヌクレオシド三リン酸ジホスホヒドロラーゼ(e−NTPDアーゼ)による加水分解に対して比較的耐性があり、灌流ラット膵臓において極めて強力なインスリン分泌促進物質であることが立証された。最も有効なアゴニストは、2−MeS−ATP−α−B、1であり、これは、28nMのEC50を有する基礎分泌と比較して、9倍のインスリン分泌増強を引き起こした。2−MeS−ATP−α−Bのインスリン放出作用はグルコース依存性であり、この化合物が2型糖尿病の治療用の薬物候補となり得ることを示唆しているが、アルカリホスファターゼに不安定であるという観察により、この化合物を薬物として使用するには不適格と判定された。
【発明の概要】
【課題を解決するための手段】
【0016】
一態様において、本発明は、一般式Iの化合物:
【化1】

[式中、
Xは、9位を介して結合した式Iaのアデニン残基:
【化2】
であり、ここで、
R1は、H、ハロゲン、O−ヒドロカルビル、S−ヒドロカルビル、NR4R5、ヘテロアリール、非置換ヒドロカルビル、又はハロゲン、CN、SCN、NO2、OR4、SR4、NR4R5若しくはヘテロアリールで置換されているヒドロカルビルであり、ここで、R4及びR5は、それぞれ独立に、H又はヒドロカルビルであるか、或いはR4及びR5は、それらが結合した窒素原子と一緒になって、酸素、窒素又は硫黄から選択される1〜2個のさらなるヘテロ原子を場合によって含有する5又は6員の飽和又は不飽和複素環を形成し、該追加の窒素は、置換されていないか、又はハロゲン、ヒドロキシル若しくはフェニルによって置換されているアルキルで置換されており、
R2及びR3は、それぞれ独立に、H又はヒドロカルビルであるか、
或いは、Xは、1位を介して結合した式Ibのウラシル残基:
【化3】
であり、ここで、
R6は、H、ハロゲン、O−ヒドロカルビル、S−ヒドロカルビル、NR8R9、ヘテロアリール、非置換ヒドロカルビル、又はハロゲン、CN、SCN、NO2、OR8、SR8、NR8R9若しくはヘテロアリールで置換されているヒドロカルビルであり、ここで、R8及びR9は、それぞれ独立に、H又はヒドロカルビルであるか、或いはR8及びR9は、それらが結合した窒素原子と一緒になって、酸素、窒素又は硫黄から選択される1〜2個のさらなるヘテロ原子を場合によって含有する5又は6員の飽和又は不飽和複素環を形成し、該追加の窒素は、置換されていないか、又はハロゲン、ヒドロキシル若しくはフェニルによって置換されているアルキルで置換されており、
R7はO又はSであり、
Yは、H、OH又はNH2であり、
Z1、Z2及びZ3は、それぞれ独立に、O−又はBH3−であり、
W1及びW2は、それぞれ独立に、O、CH2、C(Hal)2又はNHであり、ここで、Halは、ハロゲン、好ましくはF又はClであり、
nは0又は1であり、但し、nが0であり、W2がOである場合、Z1はBH3−であり、nが1である場合、W1及びW2の少なくとも一方はOではなく、
mは3又は4であり、
B+は、薬学的に許容されるカチオンを表す]
(nが0であり、Z1及びZ3がそれぞれO−であり、W2がCH2又はNHである化合物、並びにnが1であり、Z1〜Z3がそれぞれO−である化合物を除く)
並びにそのジアステレオ異性体に関する。
【0017】
別の態様において、本発明は、一般式Iの化合物(nが0であり、Z1及びZ3がそれぞれO−であり、W2がCH2又はNHである化合物、並びにnが1であり、Z1〜Z3がそれぞれO−である化合物を除く)又は薬学的に許容されるその塩、及び薬学的に許容される担体又は希釈剤を含む医薬組成物に関する。
【0018】
本発明は、一般式Iの化合物を含む、2型糖尿病又は疼痛等のP2Y受容体によって調節される疾患、障害又は状態の治療用の医薬組成物をさらに提供する。
【0019】
よって、さらなる態様において、本発明は、P2Y受容体によって調節される疾患、障害又は状態の治療用の医薬組成物の調製のための、一般式Iの化合物又は薬学的に許容されるその塩の使用に関する。
【0020】
一層さらなる態様において、本発明は、P2Y受容体によって調節される疾患、障害又は状態の治療用の、一般式Iの化合物又は薬学的に許容されるその塩に関する。
【0021】
またさらなる態様において、本発明は、必要とする個体における、2型糖尿病又は疼痛等のP2Y受容体によって調節される疾患、障害又は状態の治療方法であって、前記個体に、有効量の一般式Iの化合物又は薬学的に許容されるその塩を投与するステップを含む方法を提供する。
【図面の簡単な説明】
【0022】
【図1】図1A:81MHzの31P NMRによってモニターした際の、胃液様条件下(pH1.4及び37℃のKCl/HCl緩衝液中)における、本明細書において2と指定される化合物の加水分解を示す図である。化合物2の31P NMRスペクトルの変化が時間の関数として示されている。図1B:81MHzの31P NMRによってモニターした際の、胃液様条件下(pH1.4及び37℃のKCl/HCl緩衝液中)における、本明細書において2と指定される化合物の加水分解を示す図である。65時間のt1/2を示す、上記加水分解反応のt1/2の測定が示されている。
【図2】図2A:HPLCによってモニターした際の、胃液様条件下(pH1.4及び37℃のKCl/HCl緩衝液中)における、本明細書において3Bと指定される化合物の加水分解を示す図である。t=19時間における3BのHPLCクロマトグラムを示している。図2B:HPLCによってモニターした際の、胃液様条件下(pH1.4及び37℃のKCl/HCl緩衝液中)における、本明細書において3Bと指定される化合物の加水分解を示す図である。t=71時間における3BのHPLCクロマトグラムを示している。図2C:HPLCによってモニターした際の、胃液様条件下(pH1.4及び37℃のKCl/HCl緩衝液中)における、本明細書において3B及び4Bと指定される化合物の加水分解を示す図である。3B及び4Bについてそれぞれ19及び14.5時間のt1/2を示す、上記加水分解反応のt1/2の測定を示している。
【図3】ATPについて3.6時間のt1/2を示す、HPLCによってモニターした際の、37℃のヒト血清中におけるATP、ADP及びAMPの酵素加水分解を示す図である。
【図4】図4A:HPLCによってモニターした際の、37℃のヒト血清中におけるβ,γ−CH2−2MeS−ATP、2の酵素加水分解を示す図である。t=8時間におけるヒト血清中の加水分解混合物のHPLCクロマトグラムを示している。図4B:HPLCによってモニターした際の、37℃のヒト血清中におけるβ,γ−CH2−2MeS−ATP、2の酵素加水分解を示す図である。t=15時間におけるヒト血清中の加水分解混合物のHPLCクロマトグラムを示している。図4C:HPLCによってモニターした際の、37℃のヒト血清中におけるβ,γ−CH2−2MeS−ATP、2の酵素加水分解を示す図である。12.7時間のt1/2を示す、上記加水分解反応のk(t1/2)の測定を示している。
【図5】2−MeS−アデノシン−5’−O−(1−ボラノ二リン酸)、19が、ラットにおけるグルコース負荷後に血糖を低減させることを示す図である。絶食させたウィスター系ラット(n=5)を、例12に記載されている通り、グルコース負荷の10分後に2.5mg/kg又は生理食塩水の静脈注射(IV)で処置した。30分前に、陽性対照としてグリベンクラミド(0.25mg/kg)を経口で与えた。
【発明を実施するための形態】
【0023】
本発明は、一態様において、非加水分解性ヌクレオシド二又は三リン酸誘導体に関し、これらは、以上で定義された通り、本明細書における一般式IのP2Y受容体サブタイプ選択的アゴニストである。
【0024】
本明細書において使用される場合、用語「ハロゲン」は、フルオロ、クロロ、ブロモ及びヨードを含み、好ましくは、フルオロ又はクロロである。
【0025】
用語「ヒドロカルビル」は、異なる基R1〜R9の定義のいずれかにおいて、飽和若しくは不飽和、直鎖若しくは分枝鎖、環式若しくは非環式、又は芳香族であってよい炭素及び水素原子のみを含有する基を指し、C1〜C8アルキル、C2〜C8アルケニル、C2〜C8アルキニル、C3〜C10シクロアルキル、C3〜C10シクロアルケニル、C6〜C14アリール、(C1〜C8)アルキル(C6〜C14)アリール及び(C6〜C14)アリール(C1〜C8)アルキルを含む。
【0026】
用語「C1〜C8アルキル」は、典型的には、1〜8個の炭素原子を有する直鎖又は分枝鎖の炭化水素基を意味し、例えば、メチル、エチル、n−プロピル、イソプロピル、n−ブチル、sec−ブチル、イソブチル、tert−ブチル、n−ペンチル、2,2−ジメチルプロピル、n−ヘキシル、n−ヘプチル、n−オクチル等を含む。C1〜C6アルキル基が好ましく、最も好ましくはメチルである。用語「C2〜C8アルケニル」及び「C2〜C8アルキニル」は、典型的には、2〜8個の炭素原子及び1個の二重又は三重結合を有する直鎖及び分枝鎖の炭化水素基をそれそれ意味し、エテニル、3−ブテン−1−イル、2−エテニルブチル、3−オクテン−1−イル等及びプロピニル、2−ブチン−1−イル、3−ペンチン−1−イル等を含む。C2〜C6アルケニル基が好ましい。用語「C3〜C10シクロアルキル」は、シクロプロピル、シクロブチル、シクロペンチル、シクロヘキシル、アダマンチル、ビシクロ[3.2.1]オクチル、ビシクロ[2.2.1]ヘプチル等の環式又は二環式ヒドロカルビル基を意味する。用語「C6〜C14アリール」は、フェニル及びナフチル等の炭素環式芳香族基を表し、用語「アリル(C1〜C8)アルキル」は、ベンジル及びフェネチル等のアリールアルキル基を表す。
【0027】
基R1が、O−ヒドロカルビル又はS−ヒドロカルビル基であるか、又はOR4若しくはSR4基[ここで、R4はヒドロカルビルである]で置換されているヒドロカルビルである場合、前記ヒドロカルビルの各1個は、好ましくはC1〜C6アルキル、最も好ましくはメチル、又はアリール、最も好ましくはフェニル、又はアラルキル、最も好ましくはベンジル基である。
【0028】
基R2及びR3の一方又は両方がヒドロカルビルである場合、これらのヒドロカルビルのそれぞれは、好ましくはC1〜C6アルキル、最も好ましくはメチル、又はアリール、最も好ましくはフェニル、又はアラルキル、最も好ましくはベンジル基である。
【0029】
基R6が、O−ヒドロカルビル又はS−ヒドロカルビル基であるか、又はOR8若しくはSR8基[ここで、R8はヒドロカルビルである]で置換されているヒドロカルビルである場合、前記ヒドロカルビルの各1個は、好ましくはC1〜C6アルキル、最も好ましくはメチル、又はアリール、最も好ましくはフェニル、又はアラルキル、最も好ましくはベンジル基である。
【0030】
基NR4R5において、R4及びR5は、それぞれ独立に、H又は上記で定義された通りのヒドロカルビルであるか、或いはそれらが結合したN原子と一緒になって、窒素、酸素及び硫黄から選択される1又は2個のさらなるヘテロ原子を場合によって含有する、飽和又は不飽和の、好ましくは5又は6員の複素環を形成する。そのような環は、例えばピペラジン環中、環の第2の窒素原子において、例えば、1若しくは2個のC1〜C6アルキル基で、又は1個のアルキル若しくはヒドロキシアルキル基で置換されていてよい。基NR4R5の例は、アミノ、ジメチルアミノ、ジエチルアミノ、エチルメチルアミノ、フェニルメチル−アミノ、ピロリジノ、ピペリジノ、テトラヒドロピリジノ、ピペラジノ、エチルピペラジノ、ヒドロキシエチルピペラジノ、モルホリノ、チオモルホリノ、チアゾリノ等を含むがこれらに限定されない。
【0031】
基NR8R9において、R8及びR9は、それぞれ独立に、H又は上記で定義された通りのヒドロカルビルであるか、或いはそれらが結合したN原子と一緒になって、窒素、酸素及び硫黄から選択される1又は2個のさらなるヘテロ原子を場合によって含有する、飽和又は不飽和の、好ましくは5又は6員の複素環を形成する。そのような環は、例えばピペラジン環中、環の第2の窒素原子において、例えば、1若しくは2個のC1〜C6アルキル基で、又は1個のアルキル若しくはヒドロキシアルキル基で置換されていてよい。基NR8R9の例は、アミノ、ジメチルアミノ、ジエチルアミノ、エチルメチルアミノ、フェニルメチル−アミノ、ピロリジノ、ピペリジノ、テトラヒドロピリジノ、ピペラジノ、エチルピペラジノ、ヒドロキシエチルピペラジノ、モルホリノ、チオモルホリノ、チアゾリノ等を含むがこれらに限定されない。
【0032】
用語「ヘテロアリール」は、N、O及びSからなる群から選択される1〜3個のヘテロ原子を含有し、芳香族性の不飽和を有する単又は多環式環から誘導された基を指す。ヘテロアリールの非限定的な例は、ピロリル、フリル、チエニル、ピラゾリル、イミダゾリル、オキサゾリル、イソオキサゾリル、チアゾリル、イソチアゾリル、ピリジル、1,3−ベンゾジオキシニル、ピラジニル、ピリミジニル、1,3,4−トリアジニル、1,2,3−トリアジニル、1,3,5−トリアジニル、チアジニル、キノリニル、イソキノリニル、ベンゾフリル、イソベンゾフリル、インドリル、イミダゾ[1,2−a]ピリジル、ピリド[1,2−a]ピリミジニル、ベンズイミダゾリル、ベンズチアゾリル、ベンゾオキサゾリルを含む。ヘテロアリール環は、置換されていてよい。多環式芳香族複素環が置換されている場合、該置換は、ヘテロ環中又は炭素環中であり得ることを理解されたい。
【0033】
本明細書に記載されている化合物、式Iの化合物、出発化合物及び中間体、並びに既知の化合物の両方は、本明細書において、太字のアラビア数字1〜22によって識別される。全化学構造は、本明細書の附属書A、スキーム1〜5において描写されている。化合物2は名称β,γ−CH2−2MeS−アデノシン−5’−三リン酸によっても識別され、化合物3は名称アデノシン−β,γ−CH2−5’−O−(1−ボラノ三リン酸)によっても識別され、化合物4は名称2MeS−アデノシン−β,γ−CH2−5’−O−(1−ボラノ三リン酸)によっても識別され、化合物17は名称2−MeS−アデノシン−5’−ジクロロメチレン−二リン酸によっても識別され、化合物18は名称2−MeS−アデノシン−5’−ジフルオロメチレン−二リン酸によっても識別され、化合物19は名称2MeS−アデノシン−5’−O−(Pα−ボラノ)二リン酸によっても識別され、化合物20は名称アデノシン−β,γ−CCl2−5’−O−(1−ボラノ三リン酸)によっても識別され、化合物21は名称2MeS−アデノシン−β,γ−CCl2−5’−O−(1−ボラノ三リン酸)によっても識別され、化合物22は名称2MeS−アデノシン−β,γ−CF2−5’−O−(1−ボラノ三リン酸)によっても識別される。
【0034】
一実施形態において、本発明の化合物は、0〜2個のBH3−基を含有する二リン酸誘導体(ここで、nは0である)である。好ましい実施形態において、該化合物は、ボラノ基を含まないか、或いは該化合物は、唯1個のボラノ基を位置αに含み、ここで、Z1はBH3−であり、Z2はO−であるか、若しくは位置βに含み、ここで、Z3はBH3−であり、Z1はO−である;又は2個のボラノ基を位置α及びβに含む、すなわち、Z1及びZ3はBH3−である。
【0035】
別の実施形態において、本発明の化合物は、1〜3個のBH3−基を含有する三リン酸誘導体(すなわち、nが1である)である。好ましい実施形態において、該化合物は、唯1個のボラノ基を位置αに含み、ここで、Z1はBH3−であり、Z2及びZ3はO−である、位置βに含み、ここで、Z2はBH3−であり、Z1及びZ3はO−であるか、若しくは位置γに含み、ここで、Z3はBH3−であり、Z1及びZ2はO−である;2個のボラノ基を位置α及びβに含み、ここで、Z1及びZ2はBH3−であり、Z3はO−である、位置α及びγに含み、ここで、Z1及びZ3はBH3−であり、Z2はO−である、若しくは位置β及びγに含み、ここで、Z2及びZ3はBH3−であり、Z1はO−である;又は3個のボラノ基を位置α、β及びγに含み、ここで、Z1〜Z3はBH3−である。
【0036】
一実施形態において、Xはアデニン残基である、すなわち、本発明の化合物はATP又はADP誘導体である。好ましくは、該化合物は、Xがアデニン残基であり、R1がH又はS−アルキル、好ましくはS−メチルであり、R2及びR3が、それぞれ独立にHであり、YがOHであり、nが1であり、Z1がBH3−であり、Z2及びZ3がO−であり、W1がOであり、W2が、CH2、CF2又はCCl2である化合物;Xがアデニン残基であり、R1がH又はS−アルキル、好ましくはS−メチルであり、R2及びR3が、それぞれ独立にHであり、YがOHであり、nが0であり、Z1及びZ3がO−であり、W2がCF2又はCCl2である化合物;並びに、Xがアデニン残基であり、R1がH又はS−アルキル、好ましくはS−メチルであり、R2及びR3が、それぞれ独立にHであり、YがOHであり、nが0であり、Z1がBH3−であり、W2がOである化合物である。
【0037】
好ましい一実施形態において、本発明の化合物は、一般式Iの化合物[式中、Xはアデニン残基であり、R1はHであり、R2及びR3はHであり、YはOHであり、nは1であり、Z1はBH3−であり、Z2及びZ3はO−であり、W1はOであり、W2はCH2である](化合物3)である。Pαにおけるキラル中心により、この化合物は、2個のジアステレオ異性体(化合物3A及び3B)の対を有する。
【0038】
別の好ましい実施形態において、本発明の化合物は、一般式Iの化合物[式中、Xはアデニン残基であり、R1はSMeであり、R2及びR3はHであり、YはOHであり、nは1であり、Z1はBH3−であり、Z2及びZ3はO−であり、W1はOであり、W2はCH2である](化合物4)である。より好ましくは、本発明の化合物は、半分取逆相Gemini 5uカラム(C−18 110A、250×10mm、5ミクロン)及び定組成溶離[100mMの酢酸トリエチルアンモニウム(TEAA)、pH7(A):MeOH(B)、85:15]を流速5ml/分で使用して、ジアステレオ異性体の混合物から分離した際に、5.57分の保持時間(Rt)を有する異性体であることを特徴とする、化合物4のジアステレオ異性体B(化合物4B)である。
【0039】
さらなる好ましい実施形態において、本発明の化合物は、一般式Iの化合物[式中、Xはアデニン残基であり、ここで、R1はSMeであり、R2及びR3はHであり、YはOHであり、nは0であり、Z1及びZ3はO−であり、W2はCCl2である](化合物17)である。
【0040】
別の好ましい実施形態において、本発明の化合物は、一般式Iの化合物[式中、Xはアデニン残基であり、ここで、R1はSMeであり、R2及びR3はHであり、YはOHであり、nは0であり、Z1及びZ3はO−であり、W2はCF2である](化合物18)である。
【0041】
また別の実施形態において、本発明の化合物は、一般式Iの化合物[式中、Xはアデニン残基であり、ここで、R1はSMeであり、R2及びR3はHであり、YはOHであり、nは0であり、Z1はBH3−であり、Z3はO−であり、W2はOである](化合物19)である。より好ましくは、本発明の化合物は、半分取逆相Gemini 5uカラム(C−18 110A、250×10mm、5ミクロン)及び定組成溶離[100mMのTEAA、pH7(A)アセトニトリル(B)、88:12]を流速1ml/分で使用して、ジアステレオ異性体の混合物から分離した際に、8.073分の保持時間(Rt)を有する異性体であることを特徴とする、化合物19のジアステレオ異性体A(化合物19A)である。
【0042】
さらに別の好ましい実施形態において、本発明の化合物は、一般式Iの化合物[式中、Xはアデニン残基であり、R1、R2及びR3はHであり、YはOHであり、nは1であり、Z1はBH3−であり、Z2及びZ3はO−であり、W1はOであり、W2はCCl2である](化合物20)である。この化合物は、2個のジアステレオ異性体(化合物20A及び20B)を有する。
【0043】
またさらなる好ましい実施形態において、本発明の化合物は、一般式Iの化合物[式中、Xはアデニン残基であり、R1はSMeであり、R2及びR3はHであり、YはOHであり、nは1であり、Z1はBH3−であり、Z2及びZ3はO−であり、W1はOであり、W2はCCl2である](化合物21)である。この化合物は、2個のジアステレオ異性体(化合物21A及び21B)を有する。
【0044】
一層さらなる好ましい実施形態において、本発明の化合物は、一般式Iの化合物[式中、Xはアデニン残基であり、R1はSMeであり、R2及びR3はHであり、YはOHであり、nは1であり、Z1はBH3−であり、Z2及びZ3はO−であり、W1はOであり、W2はCF2である](化合物22)である。この化合物は、2個のジアステレオ異性体(化合物22A及び22B)を有する。
【0045】
別の実施形態において、Xはウラシル残基である、すなわち、本発明の化合物はUTP又はUDP誘導体である。好ましくは、該化合物は、Xがウラシル残基であり、R6がH又はS−アルキル、好ましくはS−メチルであり、R7がO又はSであり、YがOHであり、nが1であり、Z1がBH3−であり、Z2及びZ3がO−であり、W1がOであり、W2が、CH2、CF2又はCCl2である化合物;並びに、Xがウラシル残基であり、R6がH又はS−アルキル、好ましくはS−メチルであり、R7がO又はSであり、YがOHであり、nが0であり、Z1及びZ3がO−であり、W2がCF2又はCCl2である化合物である。
【0046】
本発明は、上記で定義された通りの式Iの化合物、そのジアステレオ異性体及び薬学的に許容されるその塩を包含する。
【0047】
一実施形態において、カチオンBは、Na+、K+及びLi+等であるがこれらに限定されないアルカリ金属の無機カチオンである。
【0048】
別の実施形態において、カチオンBは、アンモニウム(NH4+)であるか、又は式R4N+のアミンから誘導された有機カチオンであり、ここで、Rの各1個は、独立に、H、C1〜C22、好ましくは、メチル、エチル、プロピル、イソプロピル、ブチル等のC1〜C6アルキル、フェニル、又はピリジル、イミダゾリル、ピリミジニル等のヘテロアリールから選択されるか、或いは2個のRは、それらが結合した窒素原子と一緒になって、ピロリジン(pyrrolydine)、ピペリジン及びモルホリン等、N、S及びOから選択されるさらなるヘテロ原子を場合によって含有する3〜7員環を形成する。
【0049】
さらなる実施形態において、カチオンBは、カチオン性脂質又はカチオン性脂質の混合物である。カチオン性脂質は、多くの場合、送達剤として使用する前に中性脂質と混合される。中性脂質は、レシチン;ホスファチジル−エタノールアミン;ジオレオイルホスファチジルエタノールアミン、ジパルミトイルホスファチジルエタノールアミン、パルミトイルオレオイルホスファチジルエタノールアミン及びジステアロイルホスファチジルエタノールアミン等のジアシルホスファチジルエタノールアミン類;ホスファチジルコリン;ジオレオイルホスファチジルコリン、ジパルミトイルホスファチジルコリン、パルミトイルオレオイルホスファチジルコリン及びジステアロイルホスファチジルコリン等のジアシルホスファチジルコリン類;脂肪酸エステル;グリセロールエステル;スフィンゴ脂質;カルジオリピン;セレブロシド;セラミド;並びにそれらの混合物を含むがこれらに限定されない。中性脂質は、コレステロール及び他の3βヒドロキシ−ステロールも含む。
【0050】
本明細書において企図されている他の中性脂質は、ホスファチジルグリセロール;ジオレオイルホスファチジルグリセロール、ジパルミトイルホスファチジルグリセロール及びジステアロイルホスファチジルグリセロール等のジアシルホスファチジルグリセロール類;ホスファチジルセリン;ジオレオイル−又はジパルミトイルホスファチジルセリン等のジアシルホスファチジルセリン類;並びにジホスファチジルグリセロールを含む。
【0051】
カチオン性脂質化合物の例は、Lipofectin(登録商標)(Life Technologies、Burlington、Ontario)(カチオン性脂質N−[1−(2,3−ジオレイルオキシ)プロピル]−N,N,N−トリメチルアンモニウムクロリド及びジオレオイルホスファチジル−エタノールアミンの1:1(w/w)配合物);Lipofectamine(商標)(Life Technologies、Burlington、Ontario)(ポリカチオン性脂質2,3−ジオレイルオキシ−N−[2(スペルミン−カルボキサミド)エチル]−N,N−ジメチル−1−プロパンアミン−イウムトリフルオロアセテート及びジオレオイルホスファチジル−エタノールアミンの3:1(w/w)配合物)、Lipofectamine Plus(Life Technologies、Burlington、Ontario)(Lipofectamine and Plus試薬)、Lipofectamine2000(Life Technologies、Burlington、Ontario)(カチオン性脂質)、Effectene(Qiagen、Mississauga、Ontario)(非リポソーム脂質配合物)、Metafectene(Biontex、Munich、Germany)(ポリカチオン性脂質)、Eu−fectins(Promega Biosciences、San Luis Obispo、Calif.)(エタノールカチオン性脂質番号1〜12:C52H106N6O4・4CF3CO2H、C88H178N8O4S2・4CF3CO2H、C40H84NO3P・CF3CO2H、C50H103N7O3・4CF3CO2H、C55H116N8O2・6CF3CO2H、C49H102N6O3・4CF3CO2H、C44H89N5O3・2CF3CO2H、C100H206N12O4S2・8CF3CO2H、C162H330N22O9・13CF3CO2H、C43H88N4O2・2CF3CO2H、C43H88N4O3・2CF3CO2H、C41H78NO8P);Cytofectene(Bio−Rad、Hercules、Calif.)(カチオン性脂質及び中性脂質の混合物)、GenePORTER(登録商標)(Gene Therapy Systems、San Diego、Calif.)(中性脂質(Dope)及びカチオン性脂質の配合物)並びにFuGENE6(Roche Molecular Biochemicals、Indianapolis、Ind.)(多成分脂質ベースの非リポソーム試薬)を含むがこれらに限定されない。
【0052】
非加水分解性ヌクレオシドポリリン酸類似体は、ヌクレオチド加水分解酵素のプローブ及び阻害剤として広く使用されてきた(Labatailleら、1995;Yanachkovら、1997;Speltaら、2003)。ATP中のβ,γ架橋酸素をメチレン基で置き換えること(すなわち、β,γ−CH2−ATP)は、ヌクレオチドホスホヒドロラーゼによる加水分解に対する著しい抵抗性を付与する。例えば、β,γ−CH2−ATPはグリセロールキナーゼの阻害剤として同定され(Bystromら、1997)、3’−アジド−3’−デオキシ−チミジン−5’−β,γ−CF2−TP中の5’−β,γ−CF2−TP部分(AZT−5’−β,γ−CF2−TP)は、AZTを、血清及び細胞抽出物中で安定なヒト免疫不全逆転写酵素(HIV−RT)の強力阻害剤にした(Wangら、2004)。同様に、β,γ−CH2−ATPは、界面活性剤で可溶化したe−NTPDアーゼを阻害した(Picherら、1996)。その上、β,γ−CH2−ATPは、e−NPP及びe−NTPDアーゼによって触媒されるATP加水分解を選択的に阻害した(Josephら、2004)。
【0053】
β,γ−CH2−ATP及び類似体は、いくつかのP2受容体サブタイプについて代謝的に安定なリガンドとして評価されている(Speltaら、2003;El−Tayebら、2005;Chen及びLin、1997;Yegutkin及びBurnstock、2000;Zimmermann、2000;Josephら、2003)。例えば、β,γ−CH2−ATPは、強力なP2X1Rアゴニストである(Burnstockら、1994;Janssensら、1996)が、P2X2/3Rに対しては弱いアゴニストである(Speltaら、2003)ことが分かった。β,γ−CH2−ATPはP2Y1Rを活性化しなかった(Burnstockら、1994;Janssensら、1996)が、2−MeS−ADPによって誘発された応答を阻害するP2Y1Rに対しては弱い競合的アンタゴニストであった(Sakら、2000)。
【0054】
酵素アッセイにおいては加水分解に安定であったが、β,γ−CH2−ATPは、1321N1星状細胞腫及びC6神経膠腫細胞中、e−NPP(β,γ−CH2−ATP→AMP)及びCD73(AMP→アデノシン)による連続触媒を含む緊密な共役反応により、アデノシンに迅速に代謝された(Josephら、2004)。
【0055】
本発明者らは、NTPDアーゼによって媒介される加水分解に対するβ,γ−CH2−ATP中の安定化等価体としてのβ,γ−メチレン基の利点については承知していたが、これは不安定なα,β−リン酸ジエステル結合を保護し得ないことを認識した。さらに、本発明者らは、このメチレン等価体が、β,γ−CH2−ATPについて上述した通り、P2Y1Rにおけるヌクレオチドの活性を低減させるのではないかと疑った。そこで、ATP中におけるこの加水分解に不安定な結合を保護するために選択されたβ,γ−CH2−基に加えて、αリン酸をボラノリン酸部分で置換して、ATPのα,β−リン酸ジエステル結合をNTPDアーゼ(Nahumら、2002)及びNPP(Nahumら、2006)による加水分解に対して安定化した。β,γ−メチレン基の作用に対抗し、P2Y1Rにおける効力を増強するために、本発明者らは、ATPのC2位をSMe基で置換した(Fischerら、1993)。
【0056】
ヌクレオチド中でピロホスホネート結合を形成するための、いくつかの化学的方法が開発された。β,γ架橋酸素がメチレン基で置換されているヌクレオチド類似体は、ヌクレオシド−一リン酸(NMP)の5’−リン酸の活性化によってホスホリル供与体を形成し、続いてメチレンビスホスホネート塩(ホスホリルアクセプター)と反応させることにより、慣習的に調製される。ヌクレオシド−5’−一リン酸及びメチレンビスホスホネートの無水物は、カルボニルジイミダゾール(CDI)(Padyukovaら、1999)、トリフルオロ酢酸無水物及びN−メチルイミダゾール(Mohamady及びJakeman、2005)又はジシクロヘキシルカルボジイミド(DCC)(Myersら、1963)によるNMPの活性化、続いてメチレンビスホスホン酸又はその塩との縮合によって調製した。
【0057】
2−MeS−β,γ−CH2−ATP、2は、3ステップ合成:最初に2−MeS−AMPの調製、次いでカルボニルジイミダゾールによるAMP類似体の活性化、最後にメチレン−ジホスホン酸との反応(Cusackら、1987)で予め得た。これらの反応のための条件及び生成物収率は報告されていなかった。そこで、本発明者らは、以下の例1で詳細に記載され、スキーム1で描写される通り、この化合物の合成を改善し、簡潔なワンポット合成を提案しようと試みた。
【0058】
5’−OHにおける2−MeS−アデノシン(Macfarlane、1992)の選択的反応を確実にするために、本発明者らは、2’,3’−メトキシメチリデン−2−MeS−アデノシン、5aを出発材料として使用した。よって、最初に5aを、Proton Sponge(商標)(Aldrich)(1,8−ビス(ジメチルアミノ)ナフタレン)の存在下、0℃で3時間、トリメチルリン酸(TMP)中のPOCl3で処理し、続いてビス(トリブチルアンモニウム)メチレン−ジホスホネート及びトリブチルアミンを0℃で添加した。最後に、0.5MのTEAB中での加水分解及びメトキシメチリデン基の脱保護により、2を9%の全収率で生成した。
【0059】
2の全収率が低かったため、本発明者らは、非保護ヌクレオシド5bを出発材料として使用した。実際に、5bをTMP中のPOCl3により(Proton Sponge(商標)の存在下)0℃で2時間処理し、続いてビス(トリブチルアンモニウム)メチレン−ジホスホネート及びトリブチルアミンを0℃で25分間添加し、0.5MのTEAB中で加水分解することにより、生成物2を20%の全収率で得た。主な副生成物は2−MeS−AMPであり、2’,3’−環状リン酸−2−MeS−(β,γ−CH2−ATP)は得られず(すなわち、+20ppmにおいてシグナルは観察されなかった)、2’,3’−ヒドロキシルの保護は必要ないことを示している。
【0060】
以前、本発明者らは、類似体1の効率的な4ステップワンポット合成を開発した(Nahumら、2002)。今回、本発明者らは、例2〜3で詳細に記載され、スキーム2で描写される通り、3及び4の調製用に合成を修正した。合成法におけるホスフィチル化及びホウ素化試薬の使用は、保護されたヌクレオシド出発材料の使用を必要とする。この目的のために、本発明者らは、ヌクレオシド2’,3’−ヒドロキシルを、合成全体を通して安定であり続けるメトキシ−メチリデン基で保護し、最終ステップにおいて効率的に除去した。
【0061】
最初の合成ステップは、化合物9の5’−OHのホスフィチル化を含んでいた。この目的のために、本発明者らは、いくつかのホスフィチル化試薬を試した。このようにして、9を[(iPr)2N]2PClにより0℃で数時間処理したが、殆どの出発材料は室温で14時間経った後も消費されていなかった。クロロベンゾジオキサホスホリンを用いると、殆どの出発材料9は室温で15分後に消費された。しかしながら、1.5当量のメチレン−ビスホスホネートを室温で10分間及び10当量のBH3・SMe2を室温で30分間添加した場合には、微量の生成物3しか得られなかった。最終的に、PCl3が最良のホスフィチル化試薬であることが分かった。出発材料9は、30分未満で消費された。さらに、PCl3の高反応性により、メチレン−ビスホスホネート塩とのカップリングはかなり迅速(11分)であった。最後に、BH3・SMe2を0℃で添加し、次いで、反応混合物を室温で30分間撹拌した。これらの条件は、81MHz 31P NMRの粗反応混合物に基づいて、生成物3を39%の収率で提供した。3に加えて、AMP−α−BH3及びアデノシン−5’−H−ホスホネートが、それぞれ1:0.46:約1の比率で副生成物として得られた。これらの副生成物を、31P NMR及びMS(エレクトロンスプレーイオン化)の両方によって同定した。
【0062】
生成物4は、同じ手法で、LC分離後、28%の全収率で5aから得られた。
【0063】
生成物の同一性及び純度は、1H及び31P NMR、高分解能高速原子衝撃(FAB)MS、並びに2つの溶媒系中でのHPLCによって確立した。生成物3及び4の31P NMRスペクトルは、典型的なPαシグナルを約83ppmの多重項として示した。3及び4の1H NMRスペクトルは、ボラン水素原子を約0.4ppmの非常に広域のシグナルとして示した。
【0064】
Pαにおけるキラル中心により、類似体3及び4は、それぞれ2個のジアステレオ異性体の対として得られる。1H及び31P NMRスペクトルの両方において、3及び4の2個のジアステレオ異性体についての化学シフトの間にはわずかな差異があった。例えば、3のジアステレオ異性体について、H8に対し、8.59及び8.56ppmの2セットのシグナルが観察された。これらの異性体を、その保持時間中に逆相HPLCにより約2分の差異で十分に分離した。最初に溶離した異性体をA異性体と指定し、他方をB異性体と指定した。
【0065】
P2Y1Rアゴニスト2〜4の薬物候補としての適合性を調査するために、本発明者らはその加水分解安定性を評価した。詳細には、β,γ−CH2−ATP類似体2〜4の加水分解安定性を、31P NMR分光法又はHPLC−MSのいずれかによって、胃液の酸性度を模した条件、すなわち、pH1.4/37℃でモニターした。
【0066】
以下の例4〜5に示すように、31P NMRスペクトルに基づき、これらの条件下で、化合物2は、比較的高い安定性及びその濃度に関する擬1次速度指数関数的減衰速度式を呈し、ここで、pH1.4/37℃において測定されたその半減期は65時間であった。同様に、HPLCに基づいて、化合物3(異性体B)は、擬1次速度指数関数的減衰速度式を呈した(化合物3の加水分解は、スキーム3において描写されている)。化合物3Bについて、pH1.4/37℃において測定された半減期は19時間であった。同様に、同じ手法で測定された化合物4の半減期は14.5時間であった。2及び3の加水分解速度定数は、同じ条件下(5.9時間のt1/2)での2−MeS−ATP−α−BH3の加水分解速度定数と比較して、約3〜10倍の化学安定性の改善を表す。
【0067】
以前、本発明者らは、化合物1はアルカリホスファターゼによる加水分解を起こしやすく、大部分が2−MeS−AMP−α−BH3に分解するが、少量の2−MeS−ADP−α−BH3も検出され得ることを見いだした。具体的には、1をアルカリホスファターゼ中、37℃で12分間インキュベートした後、1は40%しか残っておらず、一方、100分後、HPLC−MSによって微量の1しか検出できなかった。
【0068】
そこで、化合物2〜4のアルカリホスファターゼに対する加水分解抵抗性と1のそれを比較するために、本発明者らは、種々の類似体を酵素とともに37℃で30分間インキュベートした。例6に示すように、酵素反応混合物のHPLC分析は、化合物2〜4がこれらの条件下において完全に無傷のままであることを示した。
【0069】
治療目的のためのヌクレオシド−5’−三リン酸の使用法は、細胞外培地におけるその迅速な脱リン酸化によって限定される。合成ヌクレオチドの細胞外濃度は、エクト−ATPアーゼによる加水分解(及びエクト−ヌクレオチド二リン酸キナーゼによる合成;細胞外ATPの制御を参照)によって制御される(Zimmermann、2000;Yegutkinら、2001及び2002;Lazarowskiら、1997及び2000)。Zimmermann(2000)に記載されている通り、4つの主なエクト−ヌクレオチダーゼのファミリー;(i)エクト−ヌクレオシド5’−三リン酸ジホスホヒドロラーゼ(e−NTPDアーゼ)、(ii)エクト−ヌクレオチドピロホスファターゼ(e−NPP)、(iii)グリコシルホスファチジルイノシトール(GPI)アンカー型エクト−5’−ヌクレオチダーゼ及び(iv)GPIアンカー型アルカリホスファターゼ(AP)が同定されている。e−NTPDアーゼ1〜3は、細胞表面酵素であり、細胞外ATPをADPに及びADPをAMPに分解して無機リン酸を放出し、一方、e−NPP1〜3は、ATPをAMP及びピロリン酸塩に直接加水分解する。細胞外AMPは、その後、エクト−アルカリホスファターゼによりアデノシンに分解することができる。血清は脱リン酸酵素を含有し、したがって、インビボにおいて細胞外環境の良好なモデル系を提供する。
【0070】
ホスホン酸で修飾されたdNTP類似体は、ヒト血清中(Arzumanovら、1996;Dyatkinaら、1996;Shirokova及びDyatkina、1996)及び筋肉切片標本中(Cusackら、1987)において、脱リン酸酵素に対する安定性の増強を見せた。よって、後者の標本では、60分のインキュベーション後に、エクト−ヌクレオチダーゼによるβ,γ−CH2−ATP及び2−MeS−β,γ−CH2−ATPの分解は検出されず、その間にATPは完全に脱リン酸化された(Cusackら、1987)。
【0071】
ヒト血清中における化合物2〜4の半減期を測定するために、これらの化合物を、ヒト血清及びRPMI−1640中、37℃で1〜最大144時間インキュベートし、それらの加水分解を同じ条件下におけるATPの加水分解と比較した。例7に示すように、ATPは3.6時間の半減期でADP及びAMPに加水分解され、一方、同じ条件下において、化合物2、3A及び3Bは、それぞれ12.7、14.1及び47.1時間の半減期で、対応するヌクレオシド−一リン酸(ボラノリン酸)に大部分が加水分解された。異なる評価方法を使用して、ATPは7.7時間の半減期で加水分解され、一方、同じ条件下において、化合物4Bは71.9時間の半減期で加水分解された。これらの値は、ATPの代謝的安定性の3.5〜20倍の置換依存性増強を表す。
【0072】
例11に記載されている実験において、ヒト星状細胞腫細胞で発現されている式Iの種々の化合物のGタンパク質共役P2YR P2Y1、P2Y2、P2Y4及びP2Y6における活性を検査した。最初に検査した化合物は化合物2〜4であり、示されている通り、化合物2及び4Bは、2−MeS−ADPについての0.004μMと比較して、それぞれ0.08及び17.2μMのEC50を有するP2Y1Rのアゴニストであり、P2Y6Rに対して100μMのわずかなアゴニスト作用を有していた。試験されたP2YRにおいて化合物3A、3B及び4Aが有していた活性は、微々たるものであった。
【0073】
化合物2は、化合物4Bと比較して、より強力且つ選択的なP2Y1Rアゴニストであることが分かったが、関連系において2−MeS−ADP(EC50 4nM)又は2−MeS−ATPよりも約1桁効力が弱かった(rP2Y1Rを発現しているHEK293細胞においてEC50 1nM。EC50はCa2+動員によって測定した)(Majorら、2004)。2の比較的低減された効力は、より高いpKa値のホスホネート対ホスフェート(8.4対6.5)と関係し得る(Blackburnら、1981)。特に、アッセイ条件下、pH7.4(及び恐らく受容体結合ポケット内)では、91%の2−MeS−ADP(ATP)がイオン化されるのに対し、化合物2(ホスホネート部分)は9%しかイオン化されない。この2の低度のイオン化の結果として、以下に記載する通り、受容体との相互作用がより弱いことになる。この仮説が正しいかを評価するために、本発明者らは、次いで、P2Y1Rにおける2−MeS−ADP−α,β−CCl2(又はCF2)(それぞれ17及び18)、2−MeS−ADPαB(異性体19A)及び2−MeS−ATPαB−β,γ−CCl2(異性体21A及び21B)のアゴニスト作用を検査した。化合物17、18及び21、並びに、特にCF2類似体における末端ホスホアネート(phosphoanate)のpKa値は約6.7であり、これらの類似体は、P2Y1受容体との相互作用が改善しており、よって該受容体における活性が著しく改善しているはずであることを示唆している。それにもかかわらず、例11においてさらに示すように、これらの化合物の各1個が化合物2と比較して効力が弱く選択性が弱いP2Y1Rアゴニストであることが分かった一方で、P2Y1Rに対して最も強力且つ選択的であることが分かったのは化合物19Aであった(化合物17、18、19A、21A及び21Bについて、それぞれ3.1、0.98、0.038、0.57及び1.2μMのEC50)。
【0074】
以前、本発明者らは、2−BuS−ATP:P2Y1R複合体のモデルを算出し、このヌクレオチド類似体のPβ,γがP2Y1R結合部位内の正電荷を有するLys240及びArg128と相互作用することを見いだした(Major及びFischer、2004)。よって、本発明者らは、化合物2のホスホネート部分のpKaが高いほど、P2Y1R結合ポケットとの重要なイオン相互作用の喪失をもたらし得、結果的にEC50値を低減させ得ると推測している。
【0075】
PCP対POPの角度及びC−P対O−Pの結合の長さにおける差異による幾何学的考察は、2対2−MeS−ADP(ATP)の分子認識においても役割を果たし得るが、これらの差異は依然としてかなり小さく(それぞれ、PCP及びPOPの角度−117.0及び128.7;並びにC−P及びO−Pの結合の長さ−1.79及び1.63Å)、2の親和性及び活性を決定する主なパラメーターはホスホネート基のpKa値であることを示唆している。
【0076】
化合物3A及び3Bが事実上不活性であったのに対し、化合物2、4B、17、18、19A、21A及び21BがP2Y1Rにおいて活性であったという事実は、2−MeS−アデニン部分対アデニンのP2Y1R結合ポケットとの相互作用の改善によるものであり得る(Majorら、2004、Major及びFischer、2004)。特に、P2Y1RにおけるATP類似体の三リン酸部分について観察された強い認識ネットワークに加えて、それよりも弱いが別の重要な相互作用のネットワークがアデニン環について観察された(Major及びFischer、2004)。これらの相互作用は、受容体との親和性を改善し、受容体サブタイプ選択性を決定するため、重要である。N1、N6及びN7との特異的H結合相互作用は、Arg310、Ser314及び恐らくTyr58によって提供される。これらの相互作用は、C2におけるSMe基の存在下で、電子的作用により増強される。すなわち、チオメチル基はアデニンN1位における電子密度を増大させ、よって、H結合アクセプターにとしてのその効力を増大させる。加えて、アデニン環のPhe131とのπスタッキング相互作用は、C2のSMe基による置換時には、この誘導体において、アデニン環がπスタッキング電荷移動錯体における電荷供与体分子として機能するため、さらに増強される。その上、この置換基は、アデニン部分と受容体との間により強固な適合を生み出す。具体的には、化合物2、4B、17、18、19A、21A及び21BにおけるC2−チオメチル基は、Leu104、Pro105、Ile130及びLeu135を含むP2Y1R疎水性ポケットと疎水性相互作用を形成する。
【0077】
本発明者らは、C2置換ATP−α−B類似体はP2Y2Rにより十分な耐性がないことを報告した(Tulapurkarら、2004)。2−Cl−及び2−MeS−ATP−α−Bは、P2Y2Rにおいて非常に弱いアゴニストであることが分かった。このことを考慮しても、P2Y2Rにおけるホスホネート2及び4Aの不活性についての本発明者らの所見は、これらの先の報告と一致する。P2Y4/6−Rにおける化合物2〜4の不活性は、これらの受容体がウリジンヌクレオチドアゴニストに対して選択的であるため、予期された。
【0078】
手短に述べると、ATP又はADP類似体の相対的な効力は、通常、加水分解に対するそれらの抵抗性と関係している(Adams、1994;Burnstock及びKennedy、1985;Evans及びKennedy、1994)ため、本発明者らは、新規非加水分解性P2Y1Rアゴニストを開発した。化合物2、4B、17、18、19A、21A及び21BのEC50値は、対応するリン酸類似体、2−MeS−ATP及び1AのEC50値よりも1〜3桁大きい範囲内である(Nahumら、2002;Majorら、2004)。さらに、前者の類似体の大きな利点は、ヒト血清中及び胃液の過酷な条件におけるその著しく高い生存率である。これらの特徴は、これらの類似体を、P2Y1Rが関与する健康障害のための魅力的且つ選択的な治療候補にする。
【0079】
別の態様において、本発明は、一般式Iの化合物(nが0であり、Z1及びZ3がそれぞれO−であり、W2がCH2又はNHである化合物、並びにnが1であり、Z1〜Z3がそれぞれO−である化合物を除く)又は薬学的に許容されるその塩、及び薬学的に許容される担体又は希釈剤を含む医薬組成物に関する。
【0080】
さらなる態様において、本発明は、一般式Iの化合物を含む、P2Y受容体によって調節される疾患、障害又は状態の治療用の医薬組成物を提供する。そのような使用のための好ましい化合物は、化合物2、4、より好ましくは4B、17、18、19、より好ましくは19A、21A及び21B、又は薬学的に許容されるその塩を含む。
【0081】
P2Y受容体によって調節される疾患又は障害は、癌、血小板凝集に関連する障害、心臓血管疾患若しくは障害、粘液の水和、分泌及び排泄の障害に関連する疾患、又は2型糖尿病であり得る。
【0082】
一般式Iの化合物によって治療され得る癌の種類は、白血病、リンパ腫、多発性骨髄腫、黒色腫、前立腺癌、脳腫瘍、結腸癌、卵巣癌、乳癌、皮膚癌、肺癌、食道癌及び膀胱癌であり得るがこれらに限定されない。
【0083】
心臓血管疾患又は障害は、虚血/再灌流損傷、心筋梗塞及び長期心不全であり得るがこれらに限定されない。
【0084】
粘液の水和、分泌及び排泄の障害に関連する疾患は、慢性閉塞性肺疾患、肺炎、気管支炎、嚢胞性線維症、原発性線毛機能不全、副鼻腔炎、中耳炎、ドライアイ疾患、緑内障、鼻涙管閉塞、浮腫性網膜障害、網膜変性、膣の乾燥、口内乾燥、胃食道逆流及び便秘を含むがこれらに限定されない。
【0085】
前述のWO03/034978において開示されている通り、ATP類似体のボラノリン酸等価体に基づく選択的P2Y1Rアゴニストは、灌流ラット膵臓における極めて強力なインスリン分泌促進物質であることが分かっており、ここで、最も有効なアゴニストは、2−MeS−ATP−α−B、1であり、これは、28nMのEC50を有する基礎分泌と比較して、9倍のインスリン分泌増強を引き起こした。よって、高度に選択的なP2Y1Rアゴニストである一般式Iの化合物、好ましくは化合物2、4B、17、18、19A、21A及び21Bを含む医薬組成物は、2型糖尿病の治療用のインスリン分泌促進物質として使用され得る。
【0086】
よって、好ましい実施形態において、P2Y受容体によって調節される疾患又は障害は、2型糖尿病である。
【0087】
疼痛もP2Y1受容体によって少なくとも部分的に調節されるため、一般式Iの化合物を含む医薬組成物は、疼痛管理のためにさらに使用され得る。
【0088】
一般式Iの化合物を含む医薬組成物は、例えば、Remington:The Science and Practice of Pharmacy、第19版、1995において記載されている通りの従来の技術によって調製できる。組成物は、従来の形態、例えば、カプセル剤、錠剤、液剤又は懸濁剤、乳剤、クリーム、噴霧剤等で出現し得る。
【0089】
投与経路は、活性化合物を適切な又は望ましい作用部位へ有効に輸送する任意の経路であってよく、経口経路が好ましい。固体担体が経口投与に使用される場合、製剤は錠剤化され、粉末又はペレット形態で硬ゼラチンカプセル剤に入れられてよく、又は舐剤の形態であってよい。液体担体が使用される場合、製剤は、シロップ剤、乳剤又は軟ゼラチンカプセル剤の形態であってよい。タルク及び/又は炭水化物担体又は結合剤等を有する錠剤、糖衣丸又はカプセル剤は、経口適用に特に適している。錠剤、糖衣丸又はカプセル剤に好ましい担体は、ラクトース、コーンスターチ及び/又はジャガイモデンプンを含む。
【0090】
さらなる態様において、本発明は、必要とする個体における、2型糖尿病又は疼痛等のP2Y受容体によって調節される疾患、障害又は状態の治療方法であって、前記個体に、有効量の一般式Iの化合物又は薬学的に許容されるその塩を投与するステップを含む方法を提供する。
【0091】
ここで、下記の非限定的な例によって本発明を説明する。
【実施例】
【0092】
実験
概略
空気及び水分感受性の反応はすべて、ゴム隔膜で密閉された、火炎乾燥しアルゴンフラッシュした二口フラスコ内で行い、試薬はシリンジを用いて導入した。反応の進行を、プレコートしたMerckシリカゲルプレート(60F−254)上でのTLCによってモニターした。UV光によって可視化を達成した。Bruker DPX−300、DMX−600又はAC−200分光計を使用する核磁気共鳴により、化合物を特徴付けた。1H NMRスペクトルは、200、300又は600MHzで計測した。Bruker AC−200及びDMX−600分光計上で85%のH3PO4を外部標準として使用する、D2O中での31P NMRにより、ヌクレオチドも特徴付けた。高分解能質量スペクトルは、化学イオン化により、オートスペック−E FISION VG質量分析計で記録した。ヌクレオチドは、Q−TOFマイクロ機器(Waters、UK)上でのESI(エレクトロンスプレーイオン化)で分析した。ヌクレオチドの一次精製は、1MのNaHCO3中、4℃で1日間膨張させたセファデックスDEAE−A25のカラムを使用するLC(Isco UA−6)システムで遂行した。樹脂は、使用前に脱イオン水で洗浄した。LC分離を、280nmのUV検出によりモニターした。緩衝液勾配0〜0.8MのNH4HCO3(500mlの水:500mlの緩衝液)を適用した。ヌクレオチドの最終精製及びジアステレオマー対の分離は、半分取逆相カラム(Gemini 5u C−18 110A、250×10.00mm、5ミクロン、Phenomenex、Torrance、USA)を使用するHPLC(Merck−日立)システムで遂行した。ヌクレオチドの純度は、以下に記載する通りの2つの溶媒系中、分析用逆相カラムシステム(Gemini 5u、C−18、110A、150×4.60mm、5ミクロン、Phenomenex、Torrance、CA、USA)で評価した。
【0093】
市販の試薬はすべて、特に断りのない限り、さらに精製することなく使用した。水分感受性の反応におけるすべての反応物質は、真空オーブン内で終夜乾燥させた。RPMI(Roswell Park Memorial Institute)1640緩衝液は、Sigma−Aldrichから入手した。2’,3’−O−メトキシメチリデンアデノシン誘導体は、Nahumら(2002)によって記載されている通りに調製した。2’,3’−O−メトキシメチリデン−2−MeS−アデノシンは、シリカゲルカラム(25+Mカラム)及び下記の勾配スキームを流速12.5ml/分で使用するMPLCシステム(Biotage、Kungsgatan、Uppsala、Sweden)上で分離した:3カラム体積(CV)の100:0の(A)CHCl3(A):(B)EtOH、5CVの100:0〜90:10のA:Bの勾配及び4CVの90:10のA:B。化学安定性の評価及びpH計測は、OrionマイクロコンビネーションpH電極及びHanna Instruments pH計測器を用いて実施した。
【0094】
細胞内カルシウム計測
シチメンチョウP2Y1、ヒトP2Y2、ヒトP2Y4又はラットP2Y6を安定発現しているヒト1321N1星状細胞腫細胞を、5%(v/v)のウシ胎仔血清、100ユニット/mlのペニシリン、100μg/mlのストレプトマイシン及び500μg/mlのジェネティシン(G−418、Life Technologies,Inc)を含有するダルベッコ変法イーグル培地中で成長させた。細胞内遊離カルシウム濃度[Ca2+]iにおける変化を、前述した通り、フラ−2を充填した細胞懸濁液の二重励起蛍光分光分析によって検出した(Garradら、1998;Grynkiewiczら、1985)。1mMのCaCl2及び1mMのMgCl2を含有する10mMのヘペス緩衝生理食塩水(pH7.4)中で細胞をアッセイした。微量遠心管中で細胞を沈殿させ、2mlの緩衝液に再懸濁させた。濃度応答データを、プリズム曲線当てはめプログラム(GraphPAD Software、San Diego、CA)により分析した。3つの実験は、各P2Y受容体サブタイプについて別の日に実行した。
【0095】
(例1)
β,γ−CH2−2MeS−アデノシン−5’−三リン酸、2の合成
β,γ−CH2−2MeS−アデノシン−5’−三リン酸、2は、スキーム1に描写され、以下に記載されている通りの2つの方法によって調製した。
【0096】
方法A.ビス(トリブチルアンモニウム)メチレンジホスホネート塩を上記で記載した通りに調製した。1,8−ビス(ジメチルアミノ)ナフタレン(117mg、0.57mmol、1.5当量)を、N2下、火炎乾燥した二口フラスコ内のトリメチルリン酸(2ml)中の2’,3’−O−メトキシメチリデン−2−MeS−アデノシン、5a(130mg、0.37mmol)に0℃で添加し、透明な溶液になるまで20分間反応物を撹拌した。POCl3(67μl、1.09mmol、3当量)を0℃で添加した。溶液を0℃で3時間撹拌した。乾燥DMF(4.3ml)中のビス(トリブチルアンモニウム)メチレンジホスホネート塩の0.5M溶液(386mg、2.19mmol、6当量)及びトリブチルアミン(360μl、1.46mmol、4当量)を0℃で添加し、反応混合物を1.6分間撹拌した。酢酸アンモニウムの0.25M溶液(10ml)を室温で添加し、反応混合物を30分間撹拌し、次いで凍結乾燥させた。得られた残留物を、活性化したセファデックスDEAE−A25カラム(0〜0.8MのNH4HCO3、全容積1l)に塗布した。関連画分を収集し、凍結乾燥させ、脱イオン水を用いる凍結乾燥の繰り返しによって過剰なNH4HCO3を除去して、生成物8aを白色固体として得た。生成物5を、pH2.3になるまで18%のHCl溶液で処理し、次いで室温で3時間撹拌した。最後に、混合物を24%のNH4OH溶液で処理し、pHを9に調整した。溶液を45分間撹拌し、次いで凍結乾燥させた。残留物をHPLCカラム上で分離して、純粋な2を得た。分離は、半分取逆相Gemini 5u C−18 110Aカラム(250×10.00mm、5ミクロン)及び85:15の(A)100mMの酢酸トリエチルアンモニウム(TEAA)、pH7〜(B)MeOHを流速5ml/分で適用することにより溶媒系Iを使用する定組成溶離を使用して達成した。関連画分(保持時間=12.09分)を凍結乾燥させた。凍結乾燥サイクルの繰り返しによって過剰な緩衝液を除去し、固体残留物を毎回脱イオン水に溶解した。最後に、純粋生成物1をセファデックス−CM C−25 Na+型カラムに通過させることにより、ヌクレオチドトリエチルアンモニウム対イオンをNa+イオンと交換した。LC分離後、生成物2が10%(23mg)の収率で得られた。半分取カラム上での保持時間:12.09分。2についてのスペクトルデータは、Mohamady及びJakeman(2005)によって記載されているデータと一致していた。
【0097】
方法B:ビス(トリブチルアンモニウム)メチレンジホスホネート塩を上記で記載した通りに調製した。1,8−ビス(ジメチルアミノ)ナフタレン(41mg、0.19mmol、2当量)を、N2下、火炎乾燥した二口フラスコ内のトリメチルリン酸(1ml)中の2−MeS−アデノシン、5b(30mg、0.09mmol)に0℃で添加し、透明な溶液になるまで20分間反応物を撹拌した。POCl3(26μl、0.28mmol、3当量)を0℃で添加した。溶液を0℃で2時間撹拌した。乾燥DMF(480μl)中のビス(トリブチルアンモニウム)メチレンジホスホネート塩の1M溶液(101mg、0.57mmol、6当量)及びトリブチルアミン(91μl、0.38mmol、4当量)を0℃で添加し、反応混合物を1.6分間撹拌した。次いで、0.5Mの重炭酸トリエチルアンモニウム(TEAB)溶液(10ml)を室温で添加し、反応混合物を30分間撹拌し、次いで凍結乾燥させた。得られた残留物を、活性化したセファデックスDEAE−A25カラム(0〜0.8MのNH4HCO3、全容積1l)に塗布した。関連画分を収集し、凍結乾燥させ、脱イオン水を用いる凍結乾燥の繰り返しによって過剰なNH4HCO3を除去して、生成物2を白色固体として得た。残留物をHPLCカラム上で分離して、純粋な2を得た。分離は、半分取逆相Gemini 5u C−18 110Aカラム(250×10.00mm、5ミクロン)及び流速5ml/分で20分間にわたる勾配(92:8〜70:30のA:B)の溶媒系I(上記を参照)を使用して達成した。関連画分(保持時間=11.94分)を凍結乾燥させた。凍結乾燥サイクルの繰り返しによって過剰な緩衝液を除去し、固体残留物を毎回脱イオン水に溶解した。最後に、純粋生成物1をセファデックス−CM C−25 Na+型カラムに通過させることにより、ヌクレオチドトリエチルアンモニウム対イオンをNa+イオンと交換した。LC分離後、生成物2が10%(11mg)の収率で得られた。2についてのスペクトルデータは、Mohamady及びJakeman(2005)によって記載されているデータと一致していた。
【0098】
(例2)
アデノシン−β,γ−CH2−5’−O−(1−ボラノ三リン酸)、3の合成、分離及び特徴付け
アデノシン−β,γ−CH2−5’−O−(1−ボラノ三リン酸)、3の合成
ビス(トリブチルアンモニウム)メチレンジホスホネート塩は、Bu3N(2当量)をEtOH中のメチレンジホスホン遊離酸に添加し、室温で2時間撹拌し、続いて減圧下で溶媒を除去して白色固体を得ることによって調製した。スキーム2で描写される通り、2’,3’−O−メトキシメチリデンアデノシン、9(100mg、0.32mmol)を、N2下、火炎乾燥した二口フラスコ内のトリメチルリン酸(2.5ml)に溶解した。1,8−ビス(ジメチルアミノ)ナフタレン(138mg、0.65mmol、2当量)を0℃で添加し、透明な溶液になるまで20分間反応物を撹拌した。PCl3(56μl、0.65mmol、2当量)を0℃で添加すると、白色固体が沈殿した。懸濁液を0℃で30分間撹拌した。次いで、乾燥DMF(1.8m)中のビス(トリブチルアンモニウム)メチレンジホスホネート塩の1M溶液(642mg、1.94mmol、6当量)及びトリブチルアミン(308μl、1.29mmol、4当量)を0℃で添加し、反応混合物を11分間撹拌した。THF中のBH3・SMe2錯体の2M溶液(2.2ml、3.9mmol、10当量)を0℃で添加すると、反応混合物は透明になった。溶液を0℃で5分間、次いで室温で30分間撹拌した。最後に、0.5MのTEAB溶液(10ml)を室温で添加し、混合物を60分間撹拌し、次いで凍結乾燥させた。得られた残留物を、活性化したセファデックスDEAE−A25カラム(0〜0.8MのNH4HCO3、全容積1l)に塗布した。関連画分を収集し、凍結乾燥させ、脱イオン水を用いる凍結乾燥サイクルの繰り返しによって過剰なNH4HCO3を除去した。生成物13aが白色固体として得られた。生成物13aを、pH2.3になるまで18%のHCl溶液で処理し、次いで室温で3時間撹拌した。最後に、混合物を24%のNH4OH溶液で処理し、pHを9に調整した。溶液を室温で45分間撹拌し、次いで凍結乾燥させた。生成物3のジアステレオマー対を、以下に記載する条件下、HPLCカラム上で分離した。最後に、精製された異性体3A及び3Bをセファデックス−CM C−25Na+型カラムに通過させて、トリエチルアンモニウム対イオンをNa+イオンと交換した。
【0099】
アデノシン−β,γ−CH2−5’−O−(1−ボラノ三リン酸)、3の分離
3のジアステレオマー対の分離は、半分取逆相Gemini 5uカラム(C−18 110A、250×10.00mm、5ミクロン)及び89:11のA:B、流速5ml/分で溶媒系I(例1を参照)を使用する定組成溶離を使用して達成し、続いて、2個のジアステレオ異性体の最終分離は、流速1ml/分で20分間にわたる90:10〜70:30のA:Bの勾配の溶媒系I(例1を参照)を適用することにより分析用Gemini 5uカラム(C−18 110A、150×4.60mm)を使用して達成した。同じ異性体[保持時間=6.33分(異性体A)、7.73分(異性体B)]を含有する画分を収集し、凍結乾燥させた。凍結乾燥サイクルの繰り返しによって過剰な緩衝液を除去し、固体残留物を毎回脱イオン水に溶解した。LC分離後、ジアステレオ異性体3A及び3Bが36%(66mg)の全収率で得られた。
【0100】
アデノシン−β,γ−CH2−5’−O−(1−ボラノ三リン酸)、3Aの特徴付け
半分取カラム上での保持時間:7.64分。
【化4】
TLC(NH4OH:H2O:イソプロパノール 2:8:11)、Rf=0.23。分析用カラム上で得られた純度データ:流速1ml/分で10分間にわたる90:10〜70:30のA:Bの勾配の溶媒系I(例1を参照)を使用して、保持時間:3.55分(純度100%)。溶媒系II、流速1ml/分で10分間にわたる90:10〜80:20の(A)0.01MのKH2PO4、pH=4.5〜(B)MeOHの勾配を使用して、保持時間:2.53分(純度95.5%)。
【0101】
アデノシン−β,γ−CH2−5’−O−(1−ボラノ三リン酸)、3Bの特徴付け
半分取カラム上での保持時間:9.67分。
【化5】
TLC(NH4OH:H2O:イソプロパノール 2:8:11)、Rf=0.23。分析用カラム上で得られた純度データ:流速1ml/分で10分間にわたる90:10〜70:30のA:Bの勾配の溶媒系I(例1を参照)を使用して、保持時間:4.09分(純度92.6%)。流速1ml/分で10分間にわたる95:10〜80:20のA:Bの勾配の溶媒系II(上記を参照)を使用して、保持時間:3.66分(純度95.5%)。
【0102】
(例3)
2MeS−アデノシン−β,γ−CH2−5’−O−(1−ボラノ三リン酸)、4の合成、分離及び特徴付け
2MeS−アデノシン−β,γ−CH2−5’−O−(1−ボラノ三リン酸)、4の合成
生成物4は、生成物3について例2に記載され、以下のスキーム2で描写されるのと同じ手法で、LC分離後、28%の全収率で5aから得られた。
【0103】
2MeS−アデノシン−β,γ−CH2−5’−O−(1−ボラノ三リン酸)、4の分離
4のジアステレオ異性体の分離は、半分取逆相Gemini 5uカラム(C−18 110A、250×10.00mm、5ミクロン)及び75:25のA:B、流速5ml/分で溶媒系I(例1を参照)を適用することにより定組成溶離を使用して達成した。2個のジアステレオ異性体の最終分離は、分析用Gemini 5uカラム(C−18 110A、150×4.6mm)及び流速1ml/分で20分間にわたる82:18〜74:26のA:Bの勾配の溶媒系I(例1を参照)を使用して遂行した。同じ異性体[保持時間=9.79分(異性体A)、11.53分(異性体B)]を含有する画分を収集し、凍結乾燥させた。凍結乾燥サイクルの繰り返しによって過剰な緩衝液を除去し、固体残留物を毎回脱イオン水に溶解した。LC分離後、ジアステレオ異性体4A及び4Bが28%(38mg)の全収率で得られた。
【0104】
2MeS−アデノシン−β,γ−CH2−5’−O−(1−ボラノ三リン酸)、4Aの特徴付け
半分取カラム上での保持時間:5.29分。
【化6】
TLC(NH4OH:H2O:イソプロパノール 2:8:11)、Rf=0.44。分析用カラム上で得られた純度データ:流速1ml/分で10分間にわたる80:20〜60:40のA:Bの勾配の溶媒系I(例1を参照)を使用して、保持時間:4.24分(純度94.3%)。流速1ml/分で10分間にわたる75:25〜65:35のA:Bの勾配の溶媒系II(例2を参照)を使用して、保持時間:2.99分(純度99.5%)。
【0105】
2MeS−アデノシン−β,γ−CH2−5’−O−(1−ボラノ三リン酸)、4Bの特徴付け
半分取カラム上での保持時間:5.57分。
【化7】
TLC(NH4OH:H2O:イソプロパノール 2:8:11)、Rf=0.44。流速1ml/分で10分間にわたる70:30〜40:60のA:Bの(A)100mMのTEAA、pH7〜(B)CH3CNの勾配を使用して、保持時間:2.12分(純度94%)。流速1ml/分で10分間にわたる50:50〜40:60のA:Bの勾配の溶媒系II(例2を参照)を使用して、保持時間:1.38分(純度100%)。
【0106】
(例4)
31P NMRによって評価した化合物2の化学安定性
pH1.4及び37℃における2の安定性を31P NMRによって評価して、生じ得る脱リン酸化生成物をモニターした。化合物2(1.5mg)を0.2MのHCl/KCl(0.35ml)及びD2O(40μl)に溶解した。0.2MのHCl(20μl)を添加することにより、最終pHを1.4に調整した。溶液を37℃の油浴中で維持した。スペクトルを12時間間隔で11日間記録した。すべての実験におけるスキャン数は500であった。リン酸エステル加水分解の割合は、β,γ−CH2−2MeS−ATPのPαシグナル(−10.5ppm)及び加水分解生成物2MeS−AMP、9のPαシグナル(0.7ppm)の積分に基づく。それぞれのNMRシグナルの積分における変化を時間とともに計測することにより、加水分解率を決定した。
【0107】
図1Aに示すように、これらの条件下で、化合物2は比較的高い安定性を呈した。特に、出発材料2に加えて、時間とともに2−MeS−AMPの量の増加が観察された。よって、0ppmのシグナル(2−MeS−AMPのPα)が次第に現れ、一方で、−11ppmのシグナル(2のPα)が時間とともに減少した。2−MeS−AMPのPαの31P NMRシグナルの時間に伴う強度変化を(β,γ−CH2−2−MeS−ATP及び2−MeS−AMPの全Pα積分の割合として)、2の濃度に関する擬1次速度指数関数的減衰速度式に当てはめた。2についてpH1.4/37℃で測定された半減期は、図1Bに示す通り、65時間であった。
【0108】
(例5)
HPLCによって評価した化合物3及び4の化学安定性
37℃の適切な緩衝溶液(0.2MのHCl/KCl、pH=1.4)中における3(異性体B)の安定性を、7〜17時間間隔で5日間のHPLC−エレクトロスプレーイオン化(ESI)MSによって評価し、時間に伴う3BピークのHPLC積分変化に基づくその加水分解率を、図2A〜2Bに示す通り、擬1次速度指数関数的減衰速度式に当てはめた。3Bに加えて、スキーム3で描写される通り、分解生成物6、7及び8が加水分解混合物中で同定された。例えば、19時間後、50%の3Bが分解されて、37%のAMP−α−B及びAMP−α−H(それぞれ6及び7。いずれも同じ保持時間で出現するが、MSはこれらの化合物の識別を可能にした)並びに13%のアデノシン(図2A)が生じた。時間に伴う加水分解混合物の組成変化を図2Cに描写する。3Bの半減期は19時間であった。
【0109】
37℃の適切な緩衝溶液(pH=1.4)中における4(異性体B)の安定性をHPLCによって評価して、生じ得る脱リン酸化生成物をモニターした。化合物4(1.6mg)を0.2MのHCl/KCl緩衝液(0.4ml)に溶解し、0.2MのHCl(15μl)を添加することにより、最終pHを1.4に調整した。溶液を37℃の油浴中で維持し、Gemini分析用カラム(5u C−18 110A、150×4.60mm)及び流速1ml/分で15分間にわたる89:11のA:B、次いで20分間にわたる82:18〜74:26のA:Bの溶媒系I(例1を参照)での勾配溶離を使用するHPLC−MSによってその組成を分析した。12時間間隔で5日間、検体を採取した。分解生成物6、7及び8のHPLCピークの積分における変化を時間とともに計測することにより、4の加水分解率を決定した。図2Cに示すように、化合物4Bの半減期は14.5時間であった。
【0110】
(例6)
アルカリホスファターゼに対する化合物2〜4の酵素安定性
酵素活性は、UV−可視分光光度計を405nmで使用し、ヌクレオチド誘導体からのp−ニトロフェノールの放出により計測した。酵素加水分解に対するヌクレオチドの相対活性及び抵抗性は、37℃で測定した。手短に述べると、32.5μlのヌクレオチド誘導体(0.1Mのトリス−HCl及び0.1MのMgCl2中77μg/ml、pH7.5)及び6μlの脱イオン水を、子ウシ腸アルカリホスファターゼ(Fermentas Inc.、Glen Burnie、MD、1ユニット/μl、37℃で6.25μl。最終pH=9.8)とともにインキュベートした。30分後、80℃における30分間のインキュベーションにより、反応を停止させた。ヌクレオチド誘導体の安定性をHPLCによって評価して、生じ得る脱リン酸化生成物をモニターした。混合物を、流速1ml/分で20分間にわたる、3A及び3Bについては90:10〜70:30のA:B、4A、4B及び2については82:18〜50:50の溶媒系I(例1を参照)での勾配溶離を使用するGemini分析用カラム(5u C−18 110A、150×4.60mm)上で分離した。それぞれのHPLCピークの積分における変化を時間とともに計測することにより加水分解率を決定し、また観察される通り、類似体2〜4はこれらの条件下において完全に無傷のままであった。
【0111】
(例7)
ヒト血清中におけるATP及び化合物2〜4の安定性
ヒト血清の調製:健康な志願者から採取した血液を血液バンク(Tel−Hashomer hospital、Israel)から入手し、4℃で12時間保存し、プラスチックチューブ中、1500g、室温で15分間遠心分離した。血清を分離し、−80℃で保存した。
【0112】
ヒト血清中における2〜3の安定性の評価、方法A
脱イオン水中40mMのヌクレオチド誘導体溶液(4.5μl)、ヒト血清(180μl)及びRPMI−1640(540μl)を含有するアッセイ混合物を、37℃で1、4、8、16、24、48、72及び96時間インキュベートした。次いで、検体を0.6Mの塩酸(430μl)で処理し、2分間遠心分離し(13,000g、4℃)、4MのKOHの添加によって中和し、2分間遠心分離し(13,000g、4℃)、凍結乾燥させた。生じ得る脱リン酸化生成物をモニターするために、ヌクレオチドの安定性をHPLCによって評価した。混合物を、勾配溶離[0.01MのKH2PO4 pH=4.5(A)/アセトニトリル(B)、2、3A及び3Bについては100:0→60:40のA:B、20分間;ATPについては100:0→95:5のA:B、10分間]及び流速1ml/分のGemini分析用カラム(5u C−18 110A、150×4.60mm)上で分離した。それぞれのHPLCピークの積分における変化を時間とともに計測することにより、加水分解率を決定した。
【0113】
ヒト血清中における4の安定性の評価、方法B
脱イオン水中40mMのヌクレオチド誘導体溶液(4.5μl)、ヒト血清(180μl)及びRPMI−1640(540μl)を含有するアッセイ混合物を、37℃で1、4、8、16、24、48、72、96、120及び144時間インキュベートした。次いで、検体を80℃に30分間加熱し、CMセファデックス(1〜2mg)で2時間処理し、6分間遠心分離し(12,000rpm)、クロロホルム(2×500μl)で抽出した。水層を凍結乾燥させた。生じ得る脱リン酸化生成物をモニターするために、ヌクレオチドの安定性をHPLCによって評価した。混合物を、勾配溶離[4A及び4Bについては、100mMのTEAA、pH7(A)/MeOH(B)、79:21のA:B、15分間;ATPについては、100mMのTEAA、pH7(A)/アセトニトリル(acetonitryl)(B)、A:B、10分間、100:0→90:10のA:B、10分間、90:10→80:20のA:B、4分間、80:20のA:B、1分間]及び流速1ml/分のGemini分析用カラム(5u C−18 110A、150×4.60mm)上で分離した。それぞれのHPLCピークの積分における変化を時間とともに計測することにより、加水分解率を決定した。
【0114】
図3に示すように、ATPは3.6時間の半減期でADP及びAMPに加水分解され(方法A)、一方、同じ条件下において、化合物2、3A及び3Bは、それぞれ12.7、14.1及び47.1時間の半減期で、対応するヌクレオシド−一リン酸(ボラノリン酸)に大部分が加水分解された(化合物2の加水分解率に関するデータを図4A〜4Cに示す)。化合物4Bは、71.9時間の半減期で加水分解され(方法B)、一方、同じ条件下において、ATPは7.7時間の半減期で加水分解された。これらの値は、ATPの代謝的安定性の3.5〜20倍の置換依存性増強を表す。
【0115】
(例8)
2−MeS−アデノシン−5’−ジハロゲノメチレン−二リン酸、17〜18の合成
2−MeS−アデノシン−5’−ジクロロメチレン−二リン酸及び2−MeS−アデノシン−5’−ジフルオロメチレン−二リン酸、それぞれ17及び18を調製するために、最初に5’−O−トシル−2’,3’−O−アセトニド−2MeS−アデノシン、16を、スキーム4で描写され、以下に記載する通りに調製した。
【0116】
2’,3’アセトニド−2−MeS−アデノシン、15(Nahumら、2002)(97mg、0.27mmol)を、N2下、火炎乾燥した二口フラスコ内の乾燥ジクロロメタン(1ml)に溶解した。乾燥ジクロロメタン(2ml)中のDMAPの溶液(134mg、1.09mmol、4当量)及び乾燥ジクロロメタン(0.5ml)中のTsClの溶液(156mg、0.82mmol、3当量)を添加し、得られた溶液を、N2下、室温で12時間撹拌した。反応物をジクロロメタン(50ml)で希釈し、NaHCO3の飽和溶液(3×30ml)で抽出した。有機層を除去し、Na2SO4で乾燥させ、濾過し、減圧下で蒸発させて白色固体を得、これを、シリカゲルカラム[25+Mカラム、下記の勾配スキームを使用:流速25ml/分で、CHCl3(A):EtOH(B)、0:0、3CV、A:B、CHCl3(A):EtOH(B)、0:0→0:10、5CV、A:B、CHCl3(A):EtOH(B)、10:0、4CV]を使用するMPLCシステム上で分離した。化合物16が52%の収率で得られた。
【化8】
【0117】
次いで、化合物16(42mg、0.08mmol)を、N2下、火炎乾燥した二口フラスコ内の乾燥DMF(0.2ml)に溶解した。乾燥DMF(0.3ml)中のトリス(テトラブチルアンモニウム)ジハロゲンメチレンジホスホネート(dihalogenemethylenediphosphonate)の溶液(0.16mmol、2当量)を添加し、溶液を室温で72時間撹拌した。TFA原液(2ml)を添加し、反応物を、N2通気下、室温で10分間撹拌した。溶媒を減圧下で除去して黄色固体を得、これを、活性化したセファデックスDEAE−A25カラム(0〜0.3MのNH4HCO3、全容積1.4l)上で分離した。化合物17又は18のいずれかを含有する関連画分を収集し、凍結乾燥させ、脱イオン水を用いる凍結乾燥サイクルの繰り返しによって過剰なNH4HCO3を除去した。
【0118】
(例9)
2−SMe−アデノシン−5’−O−(Pα−ボラノ)二リン酸、19の合成、分離及び特徴付け
スキーム5で描写される通り、2’,3’−メトキシメチリデンヌクレオシド(490.4mg、1.38mmol)を、アルゴン下、火炎乾燥し隔膜で密閉したフラスコ内のDMF(3ml)/ピリジン(0.6ml、5当量)に溶解した。次いで、ジオキサン(1ml)中のサリチルホスホクロリダイトの新たに調製した溶液(307mg、1.1当量)を、シリンジを介してフラスコに移した。室温で10分間撹拌後、DMF中のビス(トリ−n−ブチルアンモニウム)ピロリン酸の新たに調製した1M溶液(2.1ml、1.5当量)及びトリ−n−ブチルアミン(1.3ml、4当量)を、隔膜を通して同時に注入した。THF中のBH3:SMe2錯体の2M溶液(7ml、10当量)をフラスコに添加し、混合物を室温で15分間撹拌した。次いで、エチレンジアミン(0.5ml、5当量)を、シリンジによってフラスコに注入した。60分間撹拌後、脱イオン水(4ml)をフラスコに添加した。10分後、反応混合物を蒸発させた。残留物を脱イオン水によって希釈し、エチルエーテルで抽出した。次いで、水層を凍結乾燥させ、得られた残留物を、活性化したセファデックスDEAE−A25カラム(0〜0.4MのNH4HCO3、全容積900ml)に塗布した。関連画分を収集し、凍結乾燥させ、脱イオン水を用いる凍結乾燥の繰り返しによって過剰なNH4HCO3を除去して、化合物19をアンモニウム塩として得た。メトキシメチリデン保護基を酸加水分解によって除去した(pH2.3に達するまで10%のHCl溶液を添加した)。室温で3時間後、NH4OH溶液(pH11)の添加によってpHを9まで迅速に上昇させ、溶液を室温で40分間維持した。LC分離後、化合物19が46%の収率で得られた。ジアステレオ異性体の最終精製及び分離は、HPLC、TEAA:アセトニトリル 88:12を用いる定組成溶離によって遂行した。
【0119】
2MeS−アデノシン−5’−O−(Pα−ボラノ)二リン酸、19Aの特徴付け
保持時間:8.073分。
【化9】
分析用カラム上で得られた純度データ:溶媒系III(100mMのTEAA、pH7(A)/アセトニトリル(B)、88:12のA:B、流速1ml/分)を使用して、保持時間:4.113分(純度98%)。溶媒系IV(0.01MのKH2PO4、pH4.5(A)/アセトニトリル(B)、90:10のA:B流速1ml/分)を使用して、保持時間:3.158分(純度98.5%)。
【0120】
2MeS−アデノシン−5’−O−(Pα−ボラノ)二リン酸、19Bの特徴付け
保持時間:9.127分。
【化10】
分析用カラム上で得られた純度データ:溶媒系III(上記を参照)を使用して、保持時間:4.720分(純度95%)。溶媒系IV(上記を参照)を使用して、保持時間:3.764分(純度94%)。
【0121】
(例10)
アルカリホスファターゼに対する及びヒト血清中における、2MeS−アデノシン−5’−O−(Pα−ボラノ)二リン酸、19の安定性
アルカリホスファターゼに対する化合物19(異性体A)の安定性を、例6に記載されている通りに計測し、そのt1/2は、ADPについては4時間であるのに対し、約6時間であることが分かった。
【0122】
加えて、この化合物の安定性をヒト血清中において計測し、判明したところでは、この化合物は、ADPについては約2時間であるのに対し、24時間超の半減期で加水分解された。詳細には、24時間後に加水分解された化合物の割合は、約25〜40%だけであった。
【0123】
(例11)
P2Y1/6受容体の潜在的なアゴニストとしての化合物2、4B、17、18、19A、21A及び21B
化合物2、4B、17、18、19A、21(異性体A及びB)の活性を、ヒト1321N1星状細胞腫細胞で発現されているGタンパク質共役P2YR、P2Y1、P2Y2、P2Y4及びP2Y6において、上記の実験に記載されている通り、細胞内カルシウム計測に基づいて検査した。詳細には、化合物2、4B及び19の活性はP2Y1R、P2Y2R、P2Y4R及びP2Y6Rにおいて検査し、化合物17、18及び21の活性はP2Y1Rのみにおいて検査した。実験は、Columbia−Missouri University、Columbia、MO、USAのGary A.Weisman教授によって実行された。
【0124】
以下の表1に示すように、化合物19Aは、2−MeS−ADPについては0.004μMであるのと比較して、0.038μMのEC50を有する、最も強力且つ選択的なP2Y1Rのアゴニストであることが分かった。化合物2及び4Bは、それぞれ0.08及び17.2μMのEC50を有するP2Y1Rのアゴニストであり、P2Y6Rに対して100μMのわずかなアゴニスト作用を有していた。化合物17、18、21A及び21Bは、それぞれ3.1、0.98、0.57及び1.2μMのEC50を有するP2Y1Rのアゴニストであった。
【表1】
【0125】
(例12)
インスリン分泌促進物質としての本発明の化合物の効能のインビボ研究
パラダイム
この実験の目的は、カニューレを挿入したウィスター系ラットへのグルコースの単回経口強制飼養(経口)投与後の、インスリン分泌増強分子としての本発明の化合物の効能を、カニューレを挿入したラット細静脈内へのカニューレを介する試験化合物投与後の血中グルコース及びインスリンレベルを計測することにより、インビボで研究することである。
【0126】
合計約40匹の健康な10〜13週齢ウィスター系ラットを使用する。動物を処置開始前に少なくとも4日間気候順化させ、薬用でない市販の滅菌げっ歯類飼料を自由に摂食させる。飲料用水道水は自由に利用可能とする。
【0127】
処置の約48時間前、ラットを計量し、重量が均一な個体群(動物の約90%)を挿管用にする。詳細には、2.5%イソフルラン97.5%乾燥空気吸入によって動物に麻酔をかけ、P52カニューレを外科的に挿入し、頸静脈内に固定し、挿管後(及びその後は、各血液収集の直後)に0.3〜0.5mlの5%へパリン化生理食塩水で洗い流す。ラットの留置カニューレによって生じる固着又は凝固等の技術的問題の場合には、別のラットにカニューレを挿入し、予め割り当てられた研究中のラットと置き換える。
【0128】
処置日に、ラットの各1匹のカニューレを確認し、ラットを計量し、ラットの各1匹のグルコースレベルを尾静脈によって確認する。グルコースレベル及び重量が均一な個体群を3つの群に分割し、ここで、第1の群を試験化合物で処置し、第2の群を生理食塩水で処置した陰性対照群とし、第3の群を、グリベンクラミド(グリブリドとしても知られる)で処置した陽性対照群とする。後者は、II型糖尿病の治療に使用されるスルホニル尿素に分類される抗糖尿病薬物であり、これは、現在、WHO Model List of Essential Medicinesにおいて2つしかない経口糖尿病薬の1つである。グリベンクラミドは、膵臓ベータ細胞においてATP感受性カリウムチャネルを阻害することによって作用し、細胞膜脱分極及び電位依存性カルシウムチャネルの開口を引き起こし、それが、インスリン放出を刺激するベータ細胞内への細胞内カルシウムの増加の誘因となる。
【0129】
実験に関与するすべてのラットに、体重1kgにつき2gのグルコース負荷を(経口で)投与し、ここで、ラット1匹当たりのグルコース投与の全容積は、0.67g/mlの溶液から体重1kgにつき3mlである。グルコース投与の10分後、第1の群のラットに試験化合物を投与し、陰性対照群のラットに生理食塩水を投与する。いずれの場合も、投与はカニューレを介して静脈内へ実施する。試験化合物の投与量レベルは体重1kgにつき2.5mgであり、投与容積は体重1kgにつき1mlであり、投与される生理食塩水の容積は体重1kgにつき1mlである。陽性対照群のラットには、グルコース投与の30分前にグリベンクラミドを(経口で)投与する。グリベンクラミドの投与量レベルは体重1kgにつき1mgであり、投与容積は0.2mg/mlの溶液から体重1kgにつき5mlである。投薬後、採血までの間、ラットをケージに戻しておく。
【0130】
グルコース及びインスリンレベルは、グルコース投与(及び陽性対照群の場合はグリベンクラミド投与)の30分前;グルコース投与の直前及び5分後;グルコース投与の15分後、すなわち、試験化合物又は生理食塩水(それぞれ第1及び陰性対照群の場合)の投与の5分後;次いで、グルコース投与の30、45、60、120及び150分後に計測する。
【0131】
グルコースレベル計測のために、血液検体を各ラットから尾静脈を介して抜き取り、グルコメーターで直ちに試験する。インスリンレベル計測のために、血液検体を各ラットから頸動脈カニューレを介して抜き取る。各処置ラットから収集する血液の容積は150μlである。インスリンレベルのために抜き取られた血液検体を、Z血清/ゲルを加えた0.8mlチューブ中に収集する。血液を室温で少なくとも30分間放置して凝固させ、凝固後、これを約4℃で遠心分離する(3000×g、15分)。血清を取り出し、2つの0.2mlフラットキャップPCRタブに等分し(アリコート当たり少なくとも25μl)、次いで、分析するまで−20℃で冷凍保存する。臨床的観察は、各個別の動物の投薬後及び出血期間内に実施する。全血中のグルコースレベルの血液分析は、出血中に、血中グルコースモニタリングシステムをこのシステムに適した試験紙上で使用して、その場で行う。血清中のインスリンレベルの血液分析は、ラット/マウスインスリンキットを使用して行う。
【0132】
この実験の生存段階は、最終血液検体収集及び血清の取り出しに続いて、グルコース投与の150分後に完了する。
【0133】
試験化合物で処置したラットは、グルコース投与の15分後、すなわち、試験化合物の投与の5分後から開始して、採取された血液検体中に著しく低レベルのグルコースを有することが予期される。予期されるグルコースのレベルは、事実上、絶食後に計測されるグルコースレベルに極めて類似している。
【0134】
グルコース投与の約15分後、すなわち、健康な個体においてグルコース投与後に通常計測されるインスリンレベルの増加の約15分前には既に、試験化合物で処置したラットのインスリンレベルに著しい増加が見られることがさらに予期される。増加したインスリンのレベルは、30〜45分間維持され、次いで、血液中における試験化合物の安定性に応じ、ある速度で減少することが予期される。
【0135】
結果
予備研究において、2MeS−アデノシン−5’−O−(Pα−ボラノ)二リン酸、19(2.5mg/kg)を、以上に記載した通り、絶食させたウィスター系ラット(n=5)に静脈内投与し、一方、生理食塩水を陰性対照群のラットに投与し、グリベンクラミド(0.25mg/kg)を陽性対照群のラットにグルコース投与の30分前に与えた。図5に示すように、化合物19は、グリベンクラミドと同様に、生理食塩水で処置したラットにおいて計測されるグルコースレベルに対し、計測されるグルコースレベルを低減させた。
【化11】
【化12】
反応条件:
方法A:5aから出発し、a)トリメチルリン酸、POCl3、Proton Sponge(商標)、0℃、3時間;b)乾燥DMF中0.5Mのビス(トリブチルアンモニウム)メチレンジホスホネート、Bu3N、0℃、1.6分;c)0.5MのTEAB、pH=7、室温、0.5時間;並びにd)1)18%のHCl、pH2.3、室温、3時間;及び2)24%のNH4OH、pH9、室温、45分。
方法B:5bから出発し、a)トリメチルリン酸、POCl3、Proton Sponge(商標)、0℃、2時間;b)乾燥DMF中1Mのビス(トリブチルアンモニウム)メチレンジホスホネート、Bu3N、0℃、25分;及びc)0.5MのTEAB、pH7、室温、0.5時間。
【化13】
反応条件:a)トリメチルリン酸、PCl3、Proton Sponge(商標)、0℃、30分;b)乾燥DMF中1Mのビス(トリブチルアンモニウム)メチレンジホスホネート、Bu3N、0℃、11分;c)THF中2MのBH3・SMe、0℃、5分、次いで室温、30分;d)1MのTEAB、pH7、室温、0.5時間;並びにe)1)18%のHCl、pH2.3、室温、3時間;及び2)24%のNH4OH、pH9、室温、45分。
【化14】
【化15】
【化16】
【0136】
【技術分野】
【0001】
本発明は、非加水分解性ヌクレオシドポリリン酸誘導体及びそれらを含む医薬組成物に関する。該化合物は、2型糖尿病等のP2Y受容体によって調節される疾患又は障害の予防又は治療及び疼痛管理に有用である。
【背景技術】
【0002】
リガンド作動性イオンチャネル(P2XR)及びGタンパク質共役受容体(P2YR)からなるP2受容体(P2R)スーパーファミリーは、主として、細胞外ヌクレオチドATP、ADP、UTP又はUDPによって活性化される(Jacobsonら、2002)。加えて、P2受容体は、いくつかのジヌクレオシドポリリン酸(ジヌクレオチド)によって活性化される(WO2003/0207825;Shaverら、2005)。
【0003】
P2YRは、正常及び病態生理学的条件下の両方で、多くの組織及び器官中での種々の機能の調節にそれらが関与することから、魅力的な医薬標的であり(Williams及びJarvis、2000;Guileら、2001、Fischer、1999)、このことがP2YRアゴニストを潜在的な薬物としている。現在、薬物として提案されているP2YRアゴニストは、酵素的及び化学的に不安定なヌクレオチド骨格からなる(Williams及びJarvis、2000;Fischer、1999;Abbracchioら、2006;Jacobsonら、2002;Laxman及びBeavo、2007)。
【0004】
ヌクレオチドベースの薬物候補の固有の不安定性を克服するためのアプローチは、(i)対応するヌクレオチドよりも代謝的に安定なジヌクレオチド、(ii)非ヌクレオチドP2Rリガンド、(iii)ヌクレオチドプロドラッグ、及び(iv)等価体ベースの非加水分解性ヌクレオチドの使用を含む。
【0005】
第1のアプローチはかなり有望であり、実際にヒトの前臨床試験においていくつかのジヌクレオチドが投与されている。例えば、Ap4A、Up4U及びUp4dCは、麻酔中の血圧を低下させるため、並びにドライアイ疾患、嚢胞性線維症及び網膜剥離の治療としてそれぞれ有効であると立証された(Kikutaら、1999;Maminishkisら、2002;Mundasadら、2001;Yerxaら、2002)。
【0006】
第2のアプローチは、クロピドグレル(Plavix(登録商標)、Sanofi−Synthelabo/BMS)という、二次的な血管イベントの予防に使用される血小板抗凝集剤の場合に成功を収めており(Chow及びZiegelstein、2007)、これは、現在利用可能な唯一のP2YR標的薬物である。P2Y12受容体アンタゴニストとして作用するクロピドグレル(Angiolilloら、2006a及び2006b)は、非ヌクレオチドである。
【0007】
第3のアプローチは、マスクされたトリエステルヌクレオチドプロドラッグの調製に関与する。これらのプロドラッグ、例えば、抗HIVヌクレオシド類似体d4Tは、膜可溶性であると立証され、細胞内で活性ヌクレオチドを放出した(McGuiganら、1993、1996a及び1996b;WO/2002/055521)。
【0008】
生物学的等価体アプローチによって、ヌクレオチドベースの薬物候補、酵素阻害剤又は受容体リガンドいずれかの安定性を改善するための試みは、ごくわずかしか報告されていない(Blackburnら、1987;Cusackら、1987;Heら、1997;Kowalskaら、2007;Linら、2001;Misiuraら、2005;Romaniuk及びEckstein、1981;Stingelinら、1980)。
【0009】
真性糖尿病は、西欧諸国において最も蔓延している慢性疾患の1つであり、人口の最大5%を侵している。これは、脂質及びタンパク質代謝における追加の異常を伴う慢性高血糖(インスリン分泌、インスリン作用又は両方の組合せにおける欠陥に起因する)を特徴とする障害の混成群である。その慢性的代謝異常に加えて、糖尿病は、種々の器官、特に、眼、神経、血管、心臓及び腎臓に関与する長期合併症に関連し、これらは、失明、切断、心臓血管疾患及び末期腎不全をもたらし得る。糖尿病性合併症の発症は、血中グルコースの慢性的上昇と関係しているように思われる。現在のところ糖尿病の治癒法はないが、有効な血糖管理は、糖尿病性合併症の発生率を低下させ、その重症度を軽減することができる。
【0010】
非インスリン依存性真性糖尿病(NIDDM)とも称される2型糖尿病は、糖尿病に罹患している患者の約95%を侵しており、インスリン抵抗性及び相対的インスリン欠乏が共存する複雑な多遺伝子性疾患であるように思われる。よって、インスリン分泌の改善が主な治療目的である。インスリン放出の欠乏は、グルコースに対する第1相インスリン応答の欠如によってだけでなく、インスリン放出の規模が正常な分泌能力の10〜20%まで全体的に低減することによっても現れる。2型糖尿病に罹患している患者は、種々の経口抗糖尿病剤、インスリン注射又は両方の組合せによって治療される。現在利用可能な経口抗糖尿薬物は、膵臓ベータ細胞からのインスリン分泌を増加させること、末梢インスリン抵抗性を低減させること、又は腸からの糖質の吸収を遅らせることのいずれかを目的としている。
【0011】
2型糖尿病に罹患している患者の約半分は経口剤で治療され、該経口剤のかなりの割合がインスリン分泌を刺激する作用物質である。インスリン分泌促進物質の選定は、スルホニル尿素及び関連化合物(「グリニド系薬」)に限定され、これらは、膜ATP感受性カリウムチャネルの制御サブユニットと結合し、その閉鎖を誘導することによりインスリン分泌を誘発する。しかしながら、スルホニル尿素は、その特異的標的である膵臓ベータ細胞への起こり得る長期的な悪影響に加えて、いくつかの望ましくない作用を有する。これらの副作用は、低グルコース濃度でのインスリン分泌の刺激による低血糖のリスク、多数の患者において正常な血糖を達成することの難しさ、年間5〜10%という十分な血糖管理の二次的な失敗率、及び心臓血管系への起こり得る負の作用を含む。
【0012】
膵臓ベータ細胞におけるP2YRの存在は、文書による十分な裏付けがあり、それらの活性化は、刺激性のグルコース濃度でのインスリン分泌の刺激をもたらす。グルコースによって引き起こされるインスリン放出をP2YRアゴニストが増強する機序は、環状AMP/タンパク質キナーゼAシグナル伝達経路に関与し得、該経路は、グルコースのK+ATPチャネル非依存性作用の有効性を増大させると報告されている。
【0013】
種々のP2R選択的リガンドは、インビボにおいて、インスリン分泌を増加させ、血糖を減少させることを示した。リガンドのリストは、2−MeS−アデノシンに迅速に分解し、よって膵十二指腸動脈に直接注射された2−メチルチオ−ATP、及び酵素加水分解に安定であり、よって静脈内又は経口のいずれかで投与されたアデノシン5’−O−(2−チオ)二リン酸を含む。
【0014】
現在の合成P2受容体アゴニストは殆どすべて、ATP又はUTP薬理作用団の修飾物である。プリン(ピリミジン)環系、リボース部分又は三リン酸鎖は、1つ又は複数の位置で修飾される(Fischer、1999)。以前、本発明者らは、2−チオエーテル−5’−O−(1−チオ三リン酸)アデノシン誘導体等、C−2位に長いチオエーテル置換を有するATP誘導体の合成について報告した(Fischerら、1999)。
【0015】
US7,319,093に対応するWO2003/034978は、ATP類似体(アデノシン−5’−α−ボラノ−三リン酸類似体)のボラノリン酸等価体に基づく一連の強力且つ選択的なP2Y1Rアゴニストを開示している(Nahumら、2002;Majorら、2004;Tulapurkarら、2004;Farretら、2006)。これらの類似体は、生理的pHにおいて高度に安定であり、pH1.4及び37℃において比較的安定であることが立証された。さらに、これらのアゴニストは、エクト−ヌクレオシド三リン酸ジホスホヒドロラーゼ(e−NTPDアーゼ)による加水分解に対して比較的耐性があり、灌流ラット膵臓において極めて強力なインスリン分泌促進物質であることが立証された。最も有効なアゴニストは、2−MeS−ATP−α−B、1であり、これは、28nMのEC50を有する基礎分泌と比較して、9倍のインスリン分泌増強を引き起こした。2−MeS−ATP−α−Bのインスリン放出作用はグルコース依存性であり、この化合物が2型糖尿病の治療用の薬物候補となり得ることを示唆しているが、アルカリホスファターゼに不安定であるという観察により、この化合物を薬物として使用するには不適格と判定された。
【発明の概要】
【課題を解決するための手段】
【0016】
一態様において、本発明は、一般式Iの化合物:
【化1】
[式中、
Xは、9位を介して結合した式Iaのアデニン残基:
【化2】
であり、ここで、
R1は、H、ハロゲン、O−ヒドロカルビル、S−ヒドロカルビル、NR4R5、ヘテロアリール、非置換ヒドロカルビル、又はハロゲン、CN、SCN、NO2、OR4、SR4、NR4R5若しくはヘテロアリールで置換されているヒドロカルビルであり、ここで、R4及びR5は、それぞれ独立に、H又はヒドロカルビルであるか、或いはR4及びR5は、それらが結合した窒素原子と一緒になって、酸素、窒素又は硫黄から選択される1〜2個のさらなるヘテロ原子を場合によって含有する5又は6員の飽和又は不飽和複素環を形成し、該追加の窒素は、置換されていないか、又はハロゲン、ヒドロキシル若しくはフェニルによって置換されているアルキルで置換されており、
R2及びR3は、それぞれ独立に、H又はヒドロカルビルであるか、
或いは、Xは、1位を介して結合した式Ibのウラシル残基:
【化3】
であり、ここで、
R6は、H、ハロゲン、O−ヒドロカルビル、S−ヒドロカルビル、NR8R9、ヘテロアリール、非置換ヒドロカルビル、又はハロゲン、CN、SCN、NO2、OR8、SR8、NR8R9若しくはヘテロアリールで置換されているヒドロカルビルであり、ここで、R8及びR9は、それぞれ独立に、H又はヒドロカルビルであるか、或いはR8及びR9は、それらが結合した窒素原子と一緒になって、酸素、窒素又は硫黄から選択される1〜2個のさらなるヘテロ原子を場合によって含有する5又は6員の飽和又は不飽和複素環を形成し、該追加の窒素は、置換されていないか、又はハロゲン、ヒドロキシル若しくはフェニルによって置換されているアルキルで置換されており、
R7はO又はSであり、
Yは、H、OH又はNH2であり、
Z1、Z2及びZ3は、それぞれ独立に、O−又はBH3−であり、
W1及びW2は、それぞれ独立に、O、CH2、C(Hal)2又はNHであり、ここで、Halは、ハロゲン、好ましくはF又はClであり、
nは0又は1であり、但し、nが0であり、W2がOである場合、Z1はBH3−であり、nが1である場合、W1及びW2の少なくとも一方はOではなく、
mは3又は4であり、
B+は、薬学的に許容されるカチオンを表す]
(nが0であり、Z1及びZ3がそれぞれO−であり、W2がCH2又はNHである化合物、並びにnが1であり、Z1〜Z3がそれぞれO−である化合物を除く)
並びにそのジアステレオ異性体に関する。
【0017】
別の態様において、本発明は、一般式Iの化合物(nが0であり、Z1及びZ3がそれぞれO−であり、W2がCH2又はNHである化合物、並びにnが1であり、Z1〜Z3がそれぞれO−である化合物を除く)又は薬学的に許容されるその塩、及び薬学的に許容される担体又は希釈剤を含む医薬組成物に関する。
【0018】
本発明は、一般式Iの化合物を含む、2型糖尿病又は疼痛等のP2Y受容体によって調節される疾患、障害又は状態の治療用の医薬組成物をさらに提供する。
【0019】
よって、さらなる態様において、本発明は、P2Y受容体によって調節される疾患、障害又は状態の治療用の医薬組成物の調製のための、一般式Iの化合物又は薬学的に許容されるその塩の使用に関する。
【0020】
一層さらなる態様において、本発明は、P2Y受容体によって調節される疾患、障害又は状態の治療用の、一般式Iの化合物又は薬学的に許容されるその塩に関する。
【0021】
またさらなる態様において、本発明は、必要とする個体における、2型糖尿病又は疼痛等のP2Y受容体によって調節される疾患、障害又は状態の治療方法であって、前記個体に、有効量の一般式Iの化合物又は薬学的に許容されるその塩を投与するステップを含む方法を提供する。
【図面の簡単な説明】
【0022】
【図1】図1A:81MHzの31P NMRによってモニターした際の、胃液様条件下(pH1.4及び37℃のKCl/HCl緩衝液中)における、本明細書において2と指定される化合物の加水分解を示す図である。化合物2の31P NMRスペクトルの変化が時間の関数として示されている。図1B:81MHzの31P NMRによってモニターした際の、胃液様条件下(pH1.4及び37℃のKCl/HCl緩衝液中)における、本明細書において2と指定される化合物の加水分解を示す図である。65時間のt1/2を示す、上記加水分解反応のt1/2の測定が示されている。
【図2】図2A:HPLCによってモニターした際の、胃液様条件下(pH1.4及び37℃のKCl/HCl緩衝液中)における、本明細書において3Bと指定される化合物の加水分解を示す図である。t=19時間における3BのHPLCクロマトグラムを示している。図2B:HPLCによってモニターした際の、胃液様条件下(pH1.4及び37℃のKCl/HCl緩衝液中)における、本明細書において3Bと指定される化合物の加水分解を示す図である。t=71時間における3BのHPLCクロマトグラムを示している。図2C:HPLCによってモニターした際の、胃液様条件下(pH1.4及び37℃のKCl/HCl緩衝液中)における、本明細書において3B及び4Bと指定される化合物の加水分解を示す図である。3B及び4Bについてそれぞれ19及び14.5時間のt1/2を示す、上記加水分解反応のt1/2の測定を示している。
【図3】ATPについて3.6時間のt1/2を示す、HPLCによってモニターした際の、37℃のヒト血清中におけるATP、ADP及びAMPの酵素加水分解を示す図である。
【図4】図4A:HPLCによってモニターした際の、37℃のヒト血清中におけるβ,γ−CH2−2MeS−ATP、2の酵素加水分解を示す図である。t=8時間におけるヒト血清中の加水分解混合物のHPLCクロマトグラムを示している。図4B:HPLCによってモニターした際の、37℃のヒト血清中におけるβ,γ−CH2−2MeS−ATP、2の酵素加水分解を示す図である。t=15時間におけるヒト血清中の加水分解混合物のHPLCクロマトグラムを示している。図4C:HPLCによってモニターした際の、37℃のヒト血清中におけるβ,γ−CH2−2MeS−ATP、2の酵素加水分解を示す図である。12.7時間のt1/2を示す、上記加水分解反応のk(t1/2)の測定を示している。
【図5】2−MeS−アデノシン−5’−O−(1−ボラノ二リン酸)、19が、ラットにおけるグルコース負荷後に血糖を低減させることを示す図である。絶食させたウィスター系ラット(n=5)を、例12に記載されている通り、グルコース負荷の10分後に2.5mg/kg又は生理食塩水の静脈注射(IV)で処置した。30分前に、陽性対照としてグリベンクラミド(0.25mg/kg)を経口で与えた。
【発明を実施するための形態】
【0023】
本発明は、一態様において、非加水分解性ヌクレオシド二又は三リン酸誘導体に関し、これらは、以上で定義された通り、本明細書における一般式IのP2Y受容体サブタイプ選択的アゴニストである。
【0024】
本明細書において使用される場合、用語「ハロゲン」は、フルオロ、クロロ、ブロモ及びヨードを含み、好ましくは、フルオロ又はクロロである。
【0025】
用語「ヒドロカルビル」は、異なる基R1〜R9の定義のいずれかにおいて、飽和若しくは不飽和、直鎖若しくは分枝鎖、環式若しくは非環式、又は芳香族であってよい炭素及び水素原子のみを含有する基を指し、C1〜C8アルキル、C2〜C8アルケニル、C2〜C8アルキニル、C3〜C10シクロアルキル、C3〜C10シクロアルケニル、C6〜C14アリール、(C1〜C8)アルキル(C6〜C14)アリール及び(C6〜C14)アリール(C1〜C8)アルキルを含む。
【0026】
用語「C1〜C8アルキル」は、典型的には、1〜8個の炭素原子を有する直鎖又は分枝鎖の炭化水素基を意味し、例えば、メチル、エチル、n−プロピル、イソプロピル、n−ブチル、sec−ブチル、イソブチル、tert−ブチル、n−ペンチル、2,2−ジメチルプロピル、n−ヘキシル、n−ヘプチル、n−オクチル等を含む。C1〜C6アルキル基が好ましく、最も好ましくはメチルである。用語「C2〜C8アルケニル」及び「C2〜C8アルキニル」は、典型的には、2〜8個の炭素原子及び1個の二重又は三重結合を有する直鎖及び分枝鎖の炭化水素基をそれそれ意味し、エテニル、3−ブテン−1−イル、2−エテニルブチル、3−オクテン−1−イル等及びプロピニル、2−ブチン−1−イル、3−ペンチン−1−イル等を含む。C2〜C6アルケニル基が好ましい。用語「C3〜C10シクロアルキル」は、シクロプロピル、シクロブチル、シクロペンチル、シクロヘキシル、アダマンチル、ビシクロ[3.2.1]オクチル、ビシクロ[2.2.1]ヘプチル等の環式又は二環式ヒドロカルビル基を意味する。用語「C6〜C14アリール」は、フェニル及びナフチル等の炭素環式芳香族基を表し、用語「アリル(C1〜C8)アルキル」は、ベンジル及びフェネチル等のアリールアルキル基を表す。
【0027】
基R1が、O−ヒドロカルビル又はS−ヒドロカルビル基であるか、又はOR4若しくはSR4基[ここで、R4はヒドロカルビルである]で置換されているヒドロカルビルである場合、前記ヒドロカルビルの各1個は、好ましくはC1〜C6アルキル、最も好ましくはメチル、又はアリール、最も好ましくはフェニル、又はアラルキル、最も好ましくはベンジル基である。
【0028】
基R2及びR3の一方又は両方がヒドロカルビルである場合、これらのヒドロカルビルのそれぞれは、好ましくはC1〜C6アルキル、最も好ましくはメチル、又はアリール、最も好ましくはフェニル、又はアラルキル、最も好ましくはベンジル基である。
【0029】
基R6が、O−ヒドロカルビル又はS−ヒドロカルビル基であるか、又はOR8若しくはSR8基[ここで、R8はヒドロカルビルである]で置換されているヒドロカルビルである場合、前記ヒドロカルビルの各1個は、好ましくはC1〜C6アルキル、最も好ましくはメチル、又はアリール、最も好ましくはフェニル、又はアラルキル、最も好ましくはベンジル基である。
【0030】
基NR4R5において、R4及びR5は、それぞれ独立に、H又は上記で定義された通りのヒドロカルビルであるか、或いはそれらが結合したN原子と一緒になって、窒素、酸素及び硫黄から選択される1又は2個のさらなるヘテロ原子を場合によって含有する、飽和又は不飽和の、好ましくは5又は6員の複素環を形成する。そのような環は、例えばピペラジン環中、環の第2の窒素原子において、例えば、1若しくは2個のC1〜C6アルキル基で、又は1個のアルキル若しくはヒドロキシアルキル基で置換されていてよい。基NR4R5の例は、アミノ、ジメチルアミノ、ジエチルアミノ、エチルメチルアミノ、フェニルメチル−アミノ、ピロリジノ、ピペリジノ、テトラヒドロピリジノ、ピペラジノ、エチルピペラジノ、ヒドロキシエチルピペラジノ、モルホリノ、チオモルホリノ、チアゾリノ等を含むがこれらに限定されない。
【0031】
基NR8R9において、R8及びR9は、それぞれ独立に、H又は上記で定義された通りのヒドロカルビルであるか、或いはそれらが結合したN原子と一緒になって、窒素、酸素及び硫黄から選択される1又は2個のさらなるヘテロ原子を場合によって含有する、飽和又は不飽和の、好ましくは5又は6員の複素環を形成する。そのような環は、例えばピペラジン環中、環の第2の窒素原子において、例えば、1若しくは2個のC1〜C6アルキル基で、又は1個のアルキル若しくはヒドロキシアルキル基で置換されていてよい。基NR8R9の例は、アミノ、ジメチルアミノ、ジエチルアミノ、エチルメチルアミノ、フェニルメチル−アミノ、ピロリジノ、ピペリジノ、テトラヒドロピリジノ、ピペラジノ、エチルピペラジノ、ヒドロキシエチルピペラジノ、モルホリノ、チオモルホリノ、チアゾリノ等を含むがこれらに限定されない。
【0032】
用語「ヘテロアリール」は、N、O及びSからなる群から選択される1〜3個のヘテロ原子を含有し、芳香族性の不飽和を有する単又は多環式環から誘導された基を指す。ヘテロアリールの非限定的な例は、ピロリル、フリル、チエニル、ピラゾリル、イミダゾリル、オキサゾリル、イソオキサゾリル、チアゾリル、イソチアゾリル、ピリジル、1,3−ベンゾジオキシニル、ピラジニル、ピリミジニル、1,3,4−トリアジニル、1,2,3−トリアジニル、1,3,5−トリアジニル、チアジニル、キノリニル、イソキノリニル、ベンゾフリル、イソベンゾフリル、インドリル、イミダゾ[1,2−a]ピリジル、ピリド[1,2−a]ピリミジニル、ベンズイミダゾリル、ベンズチアゾリル、ベンゾオキサゾリルを含む。ヘテロアリール環は、置換されていてよい。多環式芳香族複素環が置換されている場合、該置換は、ヘテロ環中又は炭素環中であり得ることを理解されたい。
【0033】
本明細書に記載されている化合物、式Iの化合物、出発化合物及び中間体、並びに既知の化合物の両方は、本明細書において、太字のアラビア数字1〜22によって識別される。全化学構造は、本明細書の附属書A、スキーム1〜5において描写されている。化合物2は名称β,γ−CH2−2MeS−アデノシン−5’−三リン酸によっても識別され、化合物3は名称アデノシン−β,γ−CH2−5’−O−(1−ボラノ三リン酸)によっても識別され、化合物4は名称2MeS−アデノシン−β,γ−CH2−5’−O−(1−ボラノ三リン酸)によっても識別され、化合物17は名称2−MeS−アデノシン−5’−ジクロロメチレン−二リン酸によっても識別され、化合物18は名称2−MeS−アデノシン−5’−ジフルオロメチレン−二リン酸によっても識別され、化合物19は名称2MeS−アデノシン−5’−O−(Pα−ボラノ)二リン酸によっても識別され、化合物20は名称アデノシン−β,γ−CCl2−5’−O−(1−ボラノ三リン酸)によっても識別され、化合物21は名称2MeS−アデノシン−β,γ−CCl2−5’−O−(1−ボラノ三リン酸)によっても識別され、化合物22は名称2MeS−アデノシン−β,γ−CF2−5’−O−(1−ボラノ三リン酸)によっても識別される。
【0034】
一実施形態において、本発明の化合物は、0〜2個のBH3−基を含有する二リン酸誘導体(ここで、nは0である)である。好ましい実施形態において、該化合物は、ボラノ基を含まないか、或いは該化合物は、唯1個のボラノ基を位置αに含み、ここで、Z1はBH3−であり、Z2はO−であるか、若しくは位置βに含み、ここで、Z3はBH3−であり、Z1はO−である;又は2個のボラノ基を位置α及びβに含む、すなわち、Z1及びZ3はBH3−である。
【0035】
別の実施形態において、本発明の化合物は、1〜3個のBH3−基を含有する三リン酸誘導体(すなわち、nが1である)である。好ましい実施形態において、該化合物は、唯1個のボラノ基を位置αに含み、ここで、Z1はBH3−であり、Z2及びZ3はO−である、位置βに含み、ここで、Z2はBH3−であり、Z1及びZ3はO−であるか、若しくは位置γに含み、ここで、Z3はBH3−であり、Z1及びZ2はO−である;2個のボラノ基を位置α及びβに含み、ここで、Z1及びZ2はBH3−であり、Z3はO−である、位置α及びγに含み、ここで、Z1及びZ3はBH3−であり、Z2はO−である、若しくは位置β及びγに含み、ここで、Z2及びZ3はBH3−であり、Z1はO−である;又は3個のボラノ基を位置α、β及びγに含み、ここで、Z1〜Z3はBH3−である。
【0036】
一実施形態において、Xはアデニン残基である、すなわち、本発明の化合物はATP又はADP誘導体である。好ましくは、該化合物は、Xがアデニン残基であり、R1がH又はS−アルキル、好ましくはS−メチルであり、R2及びR3が、それぞれ独立にHであり、YがOHであり、nが1であり、Z1がBH3−であり、Z2及びZ3がO−であり、W1がOであり、W2が、CH2、CF2又はCCl2である化合物;Xがアデニン残基であり、R1がH又はS−アルキル、好ましくはS−メチルであり、R2及びR3が、それぞれ独立にHであり、YがOHであり、nが0であり、Z1及びZ3がO−であり、W2がCF2又はCCl2である化合物;並びに、Xがアデニン残基であり、R1がH又はS−アルキル、好ましくはS−メチルであり、R2及びR3が、それぞれ独立にHであり、YがOHであり、nが0であり、Z1がBH3−であり、W2がOである化合物である。
【0037】
好ましい一実施形態において、本発明の化合物は、一般式Iの化合物[式中、Xはアデニン残基であり、R1はHであり、R2及びR3はHであり、YはOHであり、nは1であり、Z1はBH3−であり、Z2及びZ3はO−であり、W1はOであり、W2はCH2である](化合物3)である。Pαにおけるキラル中心により、この化合物は、2個のジアステレオ異性体(化合物3A及び3B)の対を有する。
【0038】
別の好ましい実施形態において、本発明の化合物は、一般式Iの化合物[式中、Xはアデニン残基であり、R1はSMeであり、R2及びR3はHであり、YはOHであり、nは1であり、Z1はBH3−であり、Z2及びZ3はO−であり、W1はOであり、W2はCH2である](化合物4)である。より好ましくは、本発明の化合物は、半分取逆相Gemini 5uカラム(C−18 110A、250×10mm、5ミクロン)及び定組成溶離[100mMの酢酸トリエチルアンモニウム(TEAA)、pH7(A):MeOH(B)、85:15]を流速5ml/分で使用して、ジアステレオ異性体の混合物から分離した際に、5.57分の保持時間(Rt)を有する異性体であることを特徴とする、化合物4のジアステレオ異性体B(化合物4B)である。
【0039】
さらなる好ましい実施形態において、本発明の化合物は、一般式Iの化合物[式中、Xはアデニン残基であり、ここで、R1はSMeであり、R2及びR3はHであり、YはOHであり、nは0であり、Z1及びZ3はO−であり、W2はCCl2である](化合物17)である。
【0040】
別の好ましい実施形態において、本発明の化合物は、一般式Iの化合物[式中、Xはアデニン残基であり、ここで、R1はSMeであり、R2及びR3はHであり、YはOHであり、nは0であり、Z1及びZ3はO−であり、W2はCF2である](化合物18)である。
【0041】
また別の実施形態において、本発明の化合物は、一般式Iの化合物[式中、Xはアデニン残基であり、ここで、R1はSMeであり、R2及びR3はHであり、YはOHであり、nは0であり、Z1はBH3−であり、Z3はO−であり、W2はOである](化合物19)である。より好ましくは、本発明の化合物は、半分取逆相Gemini 5uカラム(C−18 110A、250×10mm、5ミクロン)及び定組成溶離[100mMのTEAA、pH7(A)アセトニトリル(B)、88:12]を流速1ml/分で使用して、ジアステレオ異性体の混合物から分離した際に、8.073分の保持時間(Rt)を有する異性体であることを特徴とする、化合物19のジアステレオ異性体A(化合物19A)である。
【0042】
さらに別の好ましい実施形態において、本発明の化合物は、一般式Iの化合物[式中、Xはアデニン残基であり、R1、R2及びR3はHであり、YはOHであり、nは1であり、Z1はBH3−であり、Z2及びZ3はO−であり、W1はOであり、W2はCCl2である](化合物20)である。この化合物は、2個のジアステレオ異性体(化合物20A及び20B)を有する。
【0043】
またさらなる好ましい実施形態において、本発明の化合物は、一般式Iの化合物[式中、Xはアデニン残基であり、R1はSMeであり、R2及びR3はHであり、YはOHであり、nは1であり、Z1はBH3−であり、Z2及びZ3はO−であり、W1はOであり、W2はCCl2である](化合物21)である。この化合物は、2個のジアステレオ異性体(化合物21A及び21B)を有する。
【0044】
一層さらなる好ましい実施形態において、本発明の化合物は、一般式Iの化合物[式中、Xはアデニン残基であり、R1はSMeであり、R2及びR3はHであり、YはOHであり、nは1であり、Z1はBH3−であり、Z2及びZ3はO−であり、W1はOであり、W2はCF2である](化合物22)である。この化合物は、2個のジアステレオ異性体(化合物22A及び22B)を有する。
【0045】
別の実施形態において、Xはウラシル残基である、すなわち、本発明の化合物はUTP又はUDP誘導体である。好ましくは、該化合物は、Xがウラシル残基であり、R6がH又はS−アルキル、好ましくはS−メチルであり、R7がO又はSであり、YがOHであり、nが1であり、Z1がBH3−であり、Z2及びZ3がO−であり、W1がOであり、W2が、CH2、CF2又はCCl2である化合物;並びに、Xがウラシル残基であり、R6がH又はS−アルキル、好ましくはS−メチルであり、R7がO又はSであり、YがOHであり、nが0であり、Z1及びZ3がO−であり、W2がCF2又はCCl2である化合物である。
【0046】
本発明は、上記で定義された通りの式Iの化合物、そのジアステレオ異性体及び薬学的に許容されるその塩を包含する。
【0047】
一実施形態において、カチオンBは、Na+、K+及びLi+等であるがこれらに限定されないアルカリ金属の無機カチオンである。
【0048】
別の実施形態において、カチオンBは、アンモニウム(NH4+)であるか、又は式R4N+のアミンから誘導された有機カチオンであり、ここで、Rの各1個は、独立に、H、C1〜C22、好ましくは、メチル、エチル、プロピル、イソプロピル、ブチル等のC1〜C6アルキル、フェニル、又はピリジル、イミダゾリル、ピリミジニル等のヘテロアリールから選択されるか、或いは2個のRは、それらが結合した窒素原子と一緒になって、ピロリジン(pyrrolydine)、ピペリジン及びモルホリン等、N、S及びOから選択されるさらなるヘテロ原子を場合によって含有する3〜7員環を形成する。
【0049】
さらなる実施形態において、カチオンBは、カチオン性脂質又はカチオン性脂質の混合物である。カチオン性脂質は、多くの場合、送達剤として使用する前に中性脂質と混合される。中性脂質は、レシチン;ホスファチジル−エタノールアミン;ジオレオイルホスファチジルエタノールアミン、ジパルミトイルホスファチジルエタノールアミン、パルミトイルオレオイルホスファチジルエタノールアミン及びジステアロイルホスファチジルエタノールアミン等のジアシルホスファチジルエタノールアミン類;ホスファチジルコリン;ジオレオイルホスファチジルコリン、ジパルミトイルホスファチジルコリン、パルミトイルオレオイルホスファチジルコリン及びジステアロイルホスファチジルコリン等のジアシルホスファチジルコリン類;脂肪酸エステル;グリセロールエステル;スフィンゴ脂質;カルジオリピン;セレブロシド;セラミド;並びにそれらの混合物を含むがこれらに限定されない。中性脂質は、コレステロール及び他の3βヒドロキシ−ステロールも含む。
【0050】
本明細書において企図されている他の中性脂質は、ホスファチジルグリセロール;ジオレオイルホスファチジルグリセロール、ジパルミトイルホスファチジルグリセロール及びジステアロイルホスファチジルグリセロール等のジアシルホスファチジルグリセロール類;ホスファチジルセリン;ジオレオイル−又はジパルミトイルホスファチジルセリン等のジアシルホスファチジルセリン類;並びにジホスファチジルグリセロールを含む。
【0051】
カチオン性脂質化合物の例は、Lipofectin(登録商標)(Life Technologies、Burlington、Ontario)(カチオン性脂質N−[1−(2,3−ジオレイルオキシ)プロピル]−N,N,N−トリメチルアンモニウムクロリド及びジオレオイルホスファチジル−エタノールアミンの1:1(w/w)配合物);Lipofectamine(商標)(Life Technologies、Burlington、Ontario)(ポリカチオン性脂質2,3−ジオレイルオキシ−N−[2(スペルミン−カルボキサミド)エチル]−N,N−ジメチル−1−プロパンアミン−イウムトリフルオロアセテート及びジオレオイルホスファチジル−エタノールアミンの3:1(w/w)配合物)、Lipofectamine Plus(Life Technologies、Burlington、Ontario)(Lipofectamine and Plus試薬)、Lipofectamine2000(Life Technologies、Burlington、Ontario)(カチオン性脂質)、Effectene(Qiagen、Mississauga、Ontario)(非リポソーム脂質配合物)、Metafectene(Biontex、Munich、Germany)(ポリカチオン性脂質)、Eu−fectins(Promega Biosciences、San Luis Obispo、Calif.)(エタノールカチオン性脂質番号1〜12:C52H106N6O4・4CF3CO2H、C88H178N8O4S2・4CF3CO2H、C40H84NO3P・CF3CO2H、C50H103N7O3・4CF3CO2H、C55H116N8O2・6CF3CO2H、C49H102N6O3・4CF3CO2H、C44H89N5O3・2CF3CO2H、C100H206N12O4S2・8CF3CO2H、C162H330N22O9・13CF3CO2H、C43H88N4O2・2CF3CO2H、C43H88N4O3・2CF3CO2H、C41H78NO8P);Cytofectene(Bio−Rad、Hercules、Calif.)(カチオン性脂質及び中性脂質の混合物)、GenePORTER(登録商標)(Gene Therapy Systems、San Diego、Calif.)(中性脂質(Dope)及びカチオン性脂質の配合物)並びにFuGENE6(Roche Molecular Biochemicals、Indianapolis、Ind.)(多成分脂質ベースの非リポソーム試薬)を含むがこれらに限定されない。
【0052】
非加水分解性ヌクレオシドポリリン酸類似体は、ヌクレオチド加水分解酵素のプローブ及び阻害剤として広く使用されてきた(Labatailleら、1995;Yanachkovら、1997;Speltaら、2003)。ATP中のβ,γ架橋酸素をメチレン基で置き換えること(すなわち、β,γ−CH2−ATP)は、ヌクレオチドホスホヒドロラーゼによる加水分解に対する著しい抵抗性を付与する。例えば、β,γ−CH2−ATPはグリセロールキナーゼの阻害剤として同定され(Bystromら、1997)、3’−アジド−3’−デオキシ−チミジン−5’−β,γ−CF2−TP中の5’−β,γ−CF2−TP部分(AZT−5’−β,γ−CF2−TP)は、AZTを、血清及び細胞抽出物中で安定なヒト免疫不全逆転写酵素(HIV−RT)の強力阻害剤にした(Wangら、2004)。同様に、β,γ−CH2−ATPは、界面活性剤で可溶化したe−NTPDアーゼを阻害した(Picherら、1996)。その上、β,γ−CH2−ATPは、e−NPP及びe−NTPDアーゼによって触媒されるATP加水分解を選択的に阻害した(Josephら、2004)。
【0053】
β,γ−CH2−ATP及び類似体は、いくつかのP2受容体サブタイプについて代謝的に安定なリガンドとして評価されている(Speltaら、2003;El−Tayebら、2005;Chen及びLin、1997;Yegutkin及びBurnstock、2000;Zimmermann、2000;Josephら、2003)。例えば、β,γ−CH2−ATPは、強力なP2X1Rアゴニストである(Burnstockら、1994;Janssensら、1996)が、P2X2/3Rに対しては弱いアゴニストである(Speltaら、2003)ことが分かった。β,γ−CH2−ATPはP2Y1Rを活性化しなかった(Burnstockら、1994;Janssensら、1996)が、2−MeS−ADPによって誘発された応答を阻害するP2Y1Rに対しては弱い競合的アンタゴニストであった(Sakら、2000)。
【0054】
酵素アッセイにおいては加水分解に安定であったが、β,γ−CH2−ATPは、1321N1星状細胞腫及びC6神経膠腫細胞中、e−NPP(β,γ−CH2−ATP→AMP)及びCD73(AMP→アデノシン)による連続触媒を含む緊密な共役反応により、アデノシンに迅速に代謝された(Josephら、2004)。
【0055】
本発明者らは、NTPDアーゼによって媒介される加水分解に対するβ,γ−CH2−ATP中の安定化等価体としてのβ,γ−メチレン基の利点については承知していたが、これは不安定なα,β−リン酸ジエステル結合を保護し得ないことを認識した。さらに、本発明者らは、このメチレン等価体が、β,γ−CH2−ATPについて上述した通り、P2Y1Rにおけるヌクレオチドの活性を低減させるのではないかと疑った。そこで、ATP中におけるこの加水分解に不安定な結合を保護するために選択されたβ,γ−CH2−基に加えて、αリン酸をボラノリン酸部分で置換して、ATPのα,β−リン酸ジエステル結合をNTPDアーゼ(Nahumら、2002)及びNPP(Nahumら、2006)による加水分解に対して安定化した。β,γ−メチレン基の作用に対抗し、P2Y1Rにおける効力を増強するために、本発明者らは、ATPのC2位をSMe基で置換した(Fischerら、1993)。
【0056】
ヌクレオチド中でピロホスホネート結合を形成するための、いくつかの化学的方法が開発された。β,γ架橋酸素がメチレン基で置換されているヌクレオチド類似体は、ヌクレオシド−一リン酸(NMP)の5’−リン酸の活性化によってホスホリル供与体を形成し、続いてメチレンビスホスホネート塩(ホスホリルアクセプター)と反応させることにより、慣習的に調製される。ヌクレオシド−5’−一リン酸及びメチレンビスホスホネートの無水物は、カルボニルジイミダゾール(CDI)(Padyukovaら、1999)、トリフルオロ酢酸無水物及びN−メチルイミダゾール(Mohamady及びJakeman、2005)又はジシクロヘキシルカルボジイミド(DCC)(Myersら、1963)によるNMPの活性化、続いてメチレンビスホスホン酸又はその塩との縮合によって調製した。
【0057】
2−MeS−β,γ−CH2−ATP、2は、3ステップ合成:最初に2−MeS−AMPの調製、次いでカルボニルジイミダゾールによるAMP類似体の活性化、最後にメチレン−ジホスホン酸との反応(Cusackら、1987)で予め得た。これらの反応のための条件及び生成物収率は報告されていなかった。そこで、本発明者らは、以下の例1で詳細に記載され、スキーム1で描写される通り、この化合物の合成を改善し、簡潔なワンポット合成を提案しようと試みた。
【0058】
5’−OHにおける2−MeS−アデノシン(Macfarlane、1992)の選択的反応を確実にするために、本発明者らは、2’,3’−メトキシメチリデン−2−MeS−アデノシン、5aを出発材料として使用した。よって、最初に5aを、Proton Sponge(商標)(Aldrich)(1,8−ビス(ジメチルアミノ)ナフタレン)の存在下、0℃で3時間、トリメチルリン酸(TMP)中のPOCl3で処理し、続いてビス(トリブチルアンモニウム)メチレン−ジホスホネート及びトリブチルアミンを0℃で添加した。最後に、0.5MのTEAB中での加水分解及びメトキシメチリデン基の脱保護により、2を9%の全収率で生成した。
【0059】
2の全収率が低かったため、本発明者らは、非保護ヌクレオシド5bを出発材料として使用した。実際に、5bをTMP中のPOCl3により(Proton Sponge(商標)の存在下)0℃で2時間処理し、続いてビス(トリブチルアンモニウム)メチレン−ジホスホネート及びトリブチルアミンを0℃で25分間添加し、0.5MのTEAB中で加水分解することにより、生成物2を20%の全収率で得た。主な副生成物は2−MeS−AMPであり、2’,3’−環状リン酸−2−MeS−(β,γ−CH2−ATP)は得られず(すなわち、+20ppmにおいてシグナルは観察されなかった)、2’,3’−ヒドロキシルの保護は必要ないことを示している。
【0060】
以前、本発明者らは、類似体1の効率的な4ステップワンポット合成を開発した(Nahumら、2002)。今回、本発明者らは、例2〜3で詳細に記載され、スキーム2で描写される通り、3及び4の調製用に合成を修正した。合成法におけるホスフィチル化及びホウ素化試薬の使用は、保護されたヌクレオシド出発材料の使用を必要とする。この目的のために、本発明者らは、ヌクレオシド2’,3’−ヒドロキシルを、合成全体を通して安定であり続けるメトキシ−メチリデン基で保護し、最終ステップにおいて効率的に除去した。
【0061】
最初の合成ステップは、化合物9の5’−OHのホスフィチル化を含んでいた。この目的のために、本発明者らは、いくつかのホスフィチル化試薬を試した。このようにして、9を[(iPr)2N]2PClにより0℃で数時間処理したが、殆どの出発材料は室温で14時間経った後も消費されていなかった。クロロベンゾジオキサホスホリンを用いると、殆どの出発材料9は室温で15分後に消費された。しかしながら、1.5当量のメチレン−ビスホスホネートを室温で10分間及び10当量のBH3・SMe2を室温で30分間添加した場合には、微量の生成物3しか得られなかった。最終的に、PCl3が最良のホスフィチル化試薬であることが分かった。出発材料9は、30分未満で消費された。さらに、PCl3の高反応性により、メチレン−ビスホスホネート塩とのカップリングはかなり迅速(11分)であった。最後に、BH3・SMe2を0℃で添加し、次いで、反応混合物を室温で30分間撹拌した。これらの条件は、81MHz 31P NMRの粗反応混合物に基づいて、生成物3を39%の収率で提供した。3に加えて、AMP−α−BH3及びアデノシン−5’−H−ホスホネートが、それぞれ1:0.46:約1の比率で副生成物として得られた。これらの副生成物を、31P NMR及びMS(エレクトロンスプレーイオン化)の両方によって同定した。
【0062】
生成物4は、同じ手法で、LC分離後、28%の全収率で5aから得られた。
【0063】
生成物の同一性及び純度は、1H及び31P NMR、高分解能高速原子衝撃(FAB)MS、並びに2つの溶媒系中でのHPLCによって確立した。生成物3及び4の31P NMRスペクトルは、典型的なPαシグナルを約83ppmの多重項として示した。3及び4の1H NMRスペクトルは、ボラン水素原子を約0.4ppmの非常に広域のシグナルとして示した。
【0064】
Pαにおけるキラル中心により、類似体3及び4は、それぞれ2個のジアステレオ異性体の対として得られる。1H及び31P NMRスペクトルの両方において、3及び4の2個のジアステレオ異性体についての化学シフトの間にはわずかな差異があった。例えば、3のジアステレオ異性体について、H8に対し、8.59及び8.56ppmの2セットのシグナルが観察された。これらの異性体を、その保持時間中に逆相HPLCにより約2分の差異で十分に分離した。最初に溶離した異性体をA異性体と指定し、他方をB異性体と指定した。
【0065】
P2Y1Rアゴニスト2〜4の薬物候補としての適合性を調査するために、本発明者らはその加水分解安定性を評価した。詳細には、β,γ−CH2−ATP類似体2〜4の加水分解安定性を、31P NMR分光法又はHPLC−MSのいずれかによって、胃液の酸性度を模した条件、すなわち、pH1.4/37℃でモニターした。
【0066】
以下の例4〜5に示すように、31P NMRスペクトルに基づき、これらの条件下で、化合物2は、比較的高い安定性及びその濃度に関する擬1次速度指数関数的減衰速度式を呈し、ここで、pH1.4/37℃において測定されたその半減期は65時間であった。同様に、HPLCに基づいて、化合物3(異性体B)は、擬1次速度指数関数的減衰速度式を呈した(化合物3の加水分解は、スキーム3において描写されている)。化合物3Bについて、pH1.4/37℃において測定された半減期は19時間であった。同様に、同じ手法で測定された化合物4の半減期は14.5時間であった。2及び3の加水分解速度定数は、同じ条件下(5.9時間のt1/2)での2−MeS−ATP−α−BH3の加水分解速度定数と比較して、約3〜10倍の化学安定性の改善を表す。
【0067】
以前、本発明者らは、化合物1はアルカリホスファターゼによる加水分解を起こしやすく、大部分が2−MeS−AMP−α−BH3に分解するが、少量の2−MeS−ADP−α−BH3も検出され得ることを見いだした。具体的には、1をアルカリホスファターゼ中、37℃で12分間インキュベートした後、1は40%しか残っておらず、一方、100分後、HPLC−MSによって微量の1しか検出できなかった。
【0068】
そこで、化合物2〜4のアルカリホスファターゼに対する加水分解抵抗性と1のそれを比較するために、本発明者らは、種々の類似体を酵素とともに37℃で30分間インキュベートした。例6に示すように、酵素反応混合物のHPLC分析は、化合物2〜4がこれらの条件下において完全に無傷のままであることを示した。
【0069】
治療目的のためのヌクレオシド−5’−三リン酸の使用法は、細胞外培地におけるその迅速な脱リン酸化によって限定される。合成ヌクレオチドの細胞外濃度は、エクト−ATPアーゼによる加水分解(及びエクト−ヌクレオチド二リン酸キナーゼによる合成;細胞外ATPの制御を参照)によって制御される(Zimmermann、2000;Yegutkinら、2001及び2002;Lazarowskiら、1997及び2000)。Zimmermann(2000)に記載されている通り、4つの主なエクト−ヌクレオチダーゼのファミリー;(i)エクト−ヌクレオシド5’−三リン酸ジホスホヒドロラーゼ(e−NTPDアーゼ)、(ii)エクト−ヌクレオチドピロホスファターゼ(e−NPP)、(iii)グリコシルホスファチジルイノシトール(GPI)アンカー型エクト−5’−ヌクレオチダーゼ及び(iv)GPIアンカー型アルカリホスファターゼ(AP)が同定されている。e−NTPDアーゼ1〜3は、細胞表面酵素であり、細胞外ATPをADPに及びADPをAMPに分解して無機リン酸を放出し、一方、e−NPP1〜3は、ATPをAMP及びピロリン酸塩に直接加水分解する。細胞外AMPは、その後、エクト−アルカリホスファターゼによりアデノシンに分解することができる。血清は脱リン酸酵素を含有し、したがって、インビボにおいて細胞外環境の良好なモデル系を提供する。
【0070】
ホスホン酸で修飾されたdNTP類似体は、ヒト血清中(Arzumanovら、1996;Dyatkinaら、1996;Shirokova及びDyatkina、1996)及び筋肉切片標本中(Cusackら、1987)において、脱リン酸酵素に対する安定性の増強を見せた。よって、後者の標本では、60分のインキュベーション後に、エクト−ヌクレオチダーゼによるβ,γ−CH2−ATP及び2−MeS−β,γ−CH2−ATPの分解は検出されず、その間にATPは完全に脱リン酸化された(Cusackら、1987)。
【0071】
ヒト血清中における化合物2〜4の半減期を測定するために、これらの化合物を、ヒト血清及びRPMI−1640中、37℃で1〜最大144時間インキュベートし、それらの加水分解を同じ条件下におけるATPの加水分解と比較した。例7に示すように、ATPは3.6時間の半減期でADP及びAMPに加水分解され、一方、同じ条件下において、化合物2、3A及び3Bは、それぞれ12.7、14.1及び47.1時間の半減期で、対応するヌクレオシド−一リン酸(ボラノリン酸)に大部分が加水分解された。異なる評価方法を使用して、ATPは7.7時間の半減期で加水分解され、一方、同じ条件下において、化合物4Bは71.9時間の半減期で加水分解された。これらの値は、ATPの代謝的安定性の3.5〜20倍の置換依存性増強を表す。
【0072】
例11に記載されている実験において、ヒト星状細胞腫細胞で発現されている式Iの種々の化合物のGタンパク質共役P2YR P2Y1、P2Y2、P2Y4及びP2Y6における活性を検査した。最初に検査した化合物は化合物2〜4であり、示されている通り、化合物2及び4Bは、2−MeS−ADPについての0.004μMと比較して、それぞれ0.08及び17.2μMのEC50を有するP2Y1Rのアゴニストであり、P2Y6Rに対して100μMのわずかなアゴニスト作用を有していた。試験されたP2YRにおいて化合物3A、3B及び4Aが有していた活性は、微々たるものであった。
【0073】
化合物2は、化合物4Bと比較して、より強力且つ選択的なP2Y1Rアゴニストであることが分かったが、関連系において2−MeS−ADP(EC50 4nM)又は2−MeS−ATPよりも約1桁効力が弱かった(rP2Y1Rを発現しているHEK293細胞においてEC50 1nM。EC50はCa2+動員によって測定した)(Majorら、2004)。2の比較的低減された効力は、より高いpKa値のホスホネート対ホスフェート(8.4対6.5)と関係し得る(Blackburnら、1981)。特に、アッセイ条件下、pH7.4(及び恐らく受容体結合ポケット内)では、91%の2−MeS−ADP(ATP)がイオン化されるのに対し、化合物2(ホスホネート部分)は9%しかイオン化されない。この2の低度のイオン化の結果として、以下に記載する通り、受容体との相互作用がより弱いことになる。この仮説が正しいかを評価するために、本発明者らは、次いで、P2Y1Rにおける2−MeS−ADP−α,β−CCl2(又はCF2)(それぞれ17及び18)、2−MeS−ADPαB(異性体19A)及び2−MeS−ATPαB−β,γ−CCl2(異性体21A及び21B)のアゴニスト作用を検査した。化合物17、18及び21、並びに、特にCF2類似体における末端ホスホアネート(phosphoanate)のpKa値は約6.7であり、これらの類似体は、P2Y1受容体との相互作用が改善しており、よって該受容体における活性が著しく改善しているはずであることを示唆している。それにもかかわらず、例11においてさらに示すように、これらの化合物の各1個が化合物2と比較して効力が弱く選択性が弱いP2Y1Rアゴニストであることが分かった一方で、P2Y1Rに対して最も強力且つ選択的であることが分かったのは化合物19Aであった(化合物17、18、19A、21A及び21Bについて、それぞれ3.1、0.98、0.038、0.57及び1.2μMのEC50)。
【0074】
以前、本発明者らは、2−BuS−ATP:P2Y1R複合体のモデルを算出し、このヌクレオチド類似体のPβ,γがP2Y1R結合部位内の正電荷を有するLys240及びArg128と相互作用することを見いだした(Major及びFischer、2004)。よって、本発明者らは、化合物2のホスホネート部分のpKaが高いほど、P2Y1R結合ポケットとの重要なイオン相互作用の喪失をもたらし得、結果的にEC50値を低減させ得ると推測している。
【0075】
PCP対POPの角度及びC−P対O−Pの結合の長さにおける差異による幾何学的考察は、2対2−MeS−ADP(ATP)の分子認識においても役割を果たし得るが、これらの差異は依然としてかなり小さく(それぞれ、PCP及びPOPの角度−117.0及び128.7;並びにC−P及びO−Pの結合の長さ−1.79及び1.63Å)、2の親和性及び活性を決定する主なパラメーターはホスホネート基のpKa値であることを示唆している。
【0076】
化合物3A及び3Bが事実上不活性であったのに対し、化合物2、4B、17、18、19A、21A及び21BがP2Y1Rにおいて活性であったという事実は、2−MeS−アデニン部分対アデニンのP2Y1R結合ポケットとの相互作用の改善によるものであり得る(Majorら、2004、Major及びFischer、2004)。特に、P2Y1RにおけるATP類似体の三リン酸部分について観察された強い認識ネットワークに加えて、それよりも弱いが別の重要な相互作用のネットワークがアデニン環について観察された(Major及びFischer、2004)。これらの相互作用は、受容体との親和性を改善し、受容体サブタイプ選択性を決定するため、重要である。N1、N6及びN7との特異的H結合相互作用は、Arg310、Ser314及び恐らくTyr58によって提供される。これらの相互作用は、C2におけるSMe基の存在下で、電子的作用により増強される。すなわち、チオメチル基はアデニンN1位における電子密度を増大させ、よって、H結合アクセプターにとしてのその効力を増大させる。加えて、アデニン環のPhe131とのπスタッキング相互作用は、C2のSMe基による置換時には、この誘導体において、アデニン環がπスタッキング電荷移動錯体における電荷供与体分子として機能するため、さらに増強される。その上、この置換基は、アデニン部分と受容体との間により強固な適合を生み出す。具体的には、化合物2、4B、17、18、19A、21A及び21BにおけるC2−チオメチル基は、Leu104、Pro105、Ile130及びLeu135を含むP2Y1R疎水性ポケットと疎水性相互作用を形成する。
【0077】
本発明者らは、C2置換ATP−α−B類似体はP2Y2Rにより十分な耐性がないことを報告した(Tulapurkarら、2004)。2−Cl−及び2−MeS−ATP−α−Bは、P2Y2Rにおいて非常に弱いアゴニストであることが分かった。このことを考慮しても、P2Y2Rにおけるホスホネート2及び4Aの不活性についての本発明者らの所見は、これらの先の報告と一致する。P2Y4/6−Rにおける化合物2〜4の不活性は、これらの受容体がウリジンヌクレオチドアゴニストに対して選択的であるため、予期された。
【0078】
手短に述べると、ATP又はADP類似体の相対的な効力は、通常、加水分解に対するそれらの抵抗性と関係している(Adams、1994;Burnstock及びKennedy、1985;Evans及びKennedy、1994)ため、本発明者らは、新規非加水分解性P2Y1Rアゴニストを開発した。化合物2、4B、17、18、19A、21A及び21BのEC50値は、対応するリン酸類似体、2−MeS−ATP及び1AのEC50値よりも1〜3桁大きい範囲内である(Nahumら、2002;Majorら、2004)。さらに、前者の類似体の大きな利点は、ヒト血清中及び胃液の過酷な条件におけるその著しく高い生存率である。これらの特徴は、これらの類似体を、P2Y1Rが関与する健康障害のための魅力的且つ選択的な治療候補にする。
【0079】
別の態様において、本発明は、一般式Iの化合物(nが0であり、Z1及びZ3がそれぞれO−であり、W2がCH2又はNHである化合物、並びにnが1であり、Z1〜Z3がそれぞれO−である化合物を除く)又は薬学的に許容されるその塩、及び薬学的に許容される担体又は希釈剤を含む医薬組成物に関する。
【0080】
さらなる態様において、本発明は、一般式Iの化合物を含む、P2Y受容体によって調節される疾患、障害又は状態の治療用の医薬組成物を提供する。そのような使用のための好ましい化合物は、化合物2、4、より好ましくは4B、17、18、19、より好ましくは19A、21A及び21B、又は薬学的に許容されるその塩を含む。
【0081】
P2Y受容体によって調節される疾患又は障害は、癌、血小板凝集に関連する障害、心臓血管疾患若しくは障害、粘液の水和、分泌及び排泄の障害に関連する疾患、又は2型糖尿病であり得る。
【0082】
一般式Iの化合物によって治療され得る癌の種類は、白血病、リンパ腫、多発性骨髄腫、黒色腫、前立腺癌、脳腫瘍、結腸癌、卵巣癌、乳癌、皮膚癌、肺癌、食道癌及び膀胱癌であり得るがこれらに限定されない。
【0083】
心臓血管疾患又は障害は、虚血/再灌流損傷、心筋梗塞及び長期心不全であり得るがこれらに限定されない。
【0084】
粘液の水和、分泌及び排泄の障害に関連する疾患は、慢性閉塞性肺疾患、肺炎、気管支炎、嚢胞性線維症、原発性線毛機能不全、副鼻腔炎、中耳炎、ドライアイ疾患、緑内障、鼻涙管閉塞、浮腫性網膜障害、網膜変性、膣の乾燥、口内乾燥、胃食道逆流及び便秘を含むがこれらに限定されない。
【0085】
前述のWO03/034978において開示されている通り、ATP類似体のボラノリン酸等価体に基づく選択的P2Y1Rアゴニストは、灌流ラット膵臓における極めて強力なインスリン分泌促進物質であることが分かっており、ここで、最も有効なアゴニストは、2−MeS−ATP−α−B、1であり、これは、28nMのEC50を有する基礎分泌と比較して、9倍のインスリン分泌増強を引き起こした。よって、高度に選択的なP2Y1Rアゴニストである一般式Iの化合物、好ましくは化合物2、4B、17、18、19A、21A及び21Bを含む医薬組成物は、2型糖尿病の治療用のインスリン分泌促進物質として使用され得る。
【0086】
よって、好ましい実施形態において、P2Y受容体によって調節される疾患又は障害は、2型糖尿病である。
【0087】
疼痛もP2Y1受容体によって少なくとも部分的に調節されるため、一般式Iの化合物を含む医薬組成物は、疼痛管理のためにさらに使用され得る。
【0088】
一般式Iの化合物を含む医薬組成物は、例えば、Remington:The Science and Practice of Pharmacy、第19版、1995において記載されている通りの従来の技術によって調製できる。組成物は、従来の形態、例えば、カプセル剤、錠剤、液剤又は懸濁剤、乳剤、クリーム、噴霧剤等で出現し得る。
【0089】
投与経路は、活性化合物を適切な又は望ましい作用部位へ有効に輸送する任意の経路であってよく、経口経路が好ましい。固体担体が経口投与に使用される場合、製剤は錠剤化され、粉末又はペレット形態で硬ゼラチンカプセル剤に入れられてよく、又は舐剤の形態であってよい。液体担体が使用される場合、製剤は、シロップ剤、乳剤又は軟ゼラチンカプセル剤の形態であってよい。タルク及び/又は炭水化物担体又は結合剤等を有する錠剤、糖衣丸又はカプセル剤は、経口適用に特に適している。錠剤、糖衣丸又はカプセル剤に好ましい担体は、ラクトース、コーンスターチ及び/又はジャガイモデンプンを含む。
【0090】
さらなる態様において、本発明は、必要とする個体における、2型糖尿病又は疼痛等のP2Y受容体によって調節される疾患、障害又は状態の治療方法であって、前記個体に、有効量の一般式Iの化合物又は薬学的に許容されるその塩を投与するステップを含む方法を提供する。
【0091】
ここで、下記の非限定的な例によって本発明を説明する。
【実施例】
【0092】
実験
概略
空気及び水分感受性の反応はすべて、ゴム隔膜で密閉された、火炎乾燥しアルゴンフラッシュした二口フラスコ内で行い、試薬はシリンジを用いて導入した。反応の進行を、プレコートしたMerckシリカゲルプレート(60F−254)上でのTLCによってモニターした。UV光によって可視化を達成した。Bruker DPX−300、DMX−600又はAC−200分光計を使用する核磁気共鳴により、化合物を特徴付けた。1H NMRスペクトルは、200、300又は600MHzで計測した。Bruker AC−200及びDMX−600分光計上で85%のH3PO4を外部標準として使用する、D2O中での31P NMRにより、ヌクレオチドも特徴付けた。高分解能質量スペクトルは、化学イオン化により、オートスペック−E FISION VG質量分析計で記録した。ヌクレオチドは、Q−TOFマイクロ機器(Waters、UK)上でのESI(エレクトロンスプレーイオン化)で分析した。ヌクレオチドの一次精製は、1MのNaHCO3中、4℃で1日間膨張させたセファデックスDEAE−A25のカラムを使用するLC(Isco UA−6)システムで遂行した。樹脂は、使用前に脱イオン水で洗浄した。LC分離を、280nmのUV検出によりモニターした。緩衝液勾配0〜0.8MのNH4HCO3(500mlの水:500mlの緩衝液)を適用した。ヌクレオチドの最終精製及びジアステレオマー対の分離は、半分取逆相カラム(Gemini 5u C−18 110A、250×10.00mm、5ミクロン、Phenomenex、Torrance、USA)を使用するHPLC(Merck−日立)システムで遂行した。ヌクレオチドの純度は、以下に記載する通りの2つの溶媒系中、分析用逆相カラムシステム(Gemini 5u、C−18、110A、150×4.60mm、5ミクロン、Phenomenex、Torrance、CA、USA)で評価した。
【0093】
市販の試薬はすべて、特に断りのない限り、さらに精製することなく使用した。水分感受性の反応におけるすべての反応物質は、真空オーブン内で終夜乾燥させた。RPMI(Roswell Park Memorial Institute)1640緩衝液は、Sigma−Aldrichから入手した。2’,3’−O−メトキシメチリデンアデノシン誘導体は、Nahumら(2002)によって記載されている通りに調製した。2’,3’−O−メトキシメチリデン−2−MeS−アデノシンは、シリカゲルカラム(25+Mカラム)及び下記の勾配スキームを流速12.5ml/分で使用するMPLCシステム(Biotage、Kungsgatan、Uppsala、Sweden)上で分離した:3カラム体積(CV)の100:0の(A)CHCl3(A):(B)EtOH、5CVの100:0〜90:10のA:Bの勾配及び4CVの90:10のA:B。化学安定性の評価及びpH計測は、OrionマイクロコンビネーションpH電極及びHanna Instruments pH計測器を用いて実施した。
【0094】
細胞内カルシウム計測
シチメンチョウP2Y1、ヒトP2Y2、ヒトP2Y4又はラットP2Y6を安定発現しているヒト1321N1星状細胞腫細胞を、5%(v/v)のウシ胎仔血清、100ユニット/mlのペニシリン、100μg/mlのストレプトマイシン及び500μg/mlのジェネティシン(G−418、Life Technologies,Inc)を含有するダルベッコ変法イーグル培地中で成長させた。細胞内遊離カルシウム濃度[Ca2+]iにおける変化を、前述した通り、フラ−2を充填した細胞懸濁液の二重励起蛍光分光分析によって検出した(Garradら、1998;Grynkiewiczら、1985)。1mMのCaCl2及び1mMのMgCl2を含有する10mMのヘペス緩衝生理食塩水(pH7.4)中で細胞をアッセイした。微量遠心管中で細胞を沈殿させ、2mlの緩衝液に再懸濁させた。濃度応答データを、プリズム曲線当てはめプログラム(GraphPAD Software、San Diego、CA)により分析した。3つの実験は、各P2Y受容体サブタイプについて別の日に実行した。
【0095】
(例1)
β,γ−CH2−2MeS−アデノシン−5’−三リン酸、2の合成
β,γ−CH2−2MeS−アデノシン−5’−三リン酸、2は、スキーム1に描写され、以下に記載されている通りの2つの方法によって調製した。
【0096】
方法A.ビス(トリブチルアンモニウム)メチレンジホスホネート塩を上記で記載した通りに調製した。1,8−ビス(ジメチルアミノ)ナフタレン(117mg、0.57mmol、1.5当量)を、N2下、火炎乾燥した二口フラスコ内のトリメチルリン酸(2ml)中の2’,3’−O−メトキシメチリデン−2−MeS−アデノシン、5a(130mg、0.37mmol)に0℃で添加し、透明な溶液になるまで20分間反応物を撹拌した。POCl3(67μl、1.09mmol、3当量)を0℃で添加した。溶液を0℃で3時間撹拌した。乾燥DMF(4.3ml)中のビス(トリブチルアンモニウム)メチレンジホスホネート塩の0.5M溶液(386mg、2.19mmol、6当量)及びトリブチルアミン(360μl、1.46mmol、4当量)を0℃で添加し、反応混合物を1.6分間撹拌した。酢酸アンモニウムの0.25M溶液(10ml)を室温で添加し、反応混合物を30分間撹拌し、次いで凍結乾燥させた。得られた残留物を、活性化したセファデックスDEAE−A25カラム(0〜0.8MのNH4HCO3、全容積1l)に塗布した。関連画分を収集し、凍結乾燥させ、脱イオン水を用いる凍結乾燥の繰り返しによって過剰なNH4HCO3を除去して、生成物8aを白色固体として得た。生成物5を、pH2.3になるまで18%のHCl溶液で処理し、次いで室温で3時間撹拌した。最後に、混合物を24%のNH4OH溶液で処理し、pHを9に調整した。溶液を45分間撹拌し、次いで凍結乾燥させた。残留物をHPLCカラム上で分離して、純粋な2を得た。分離は、半分取逆相Gemini 5u C−18 110Aカラム(250×10.00mm、5ミクロン)及び85:15の(A)100mMの酢酸トリエチルアンモニウム(TEAA)、pH7〜(B)MeOHを流速5ml/分で適用することにより溶媒系Iを使用する定組成溶離を使用して達成した。関連画分(保持時間=12.09分)を凍結乾燥させた。凍結乾燥サイクルの繰り返しによって過剰な緩衝液を除去し、固体残留物を毎回脱イオン水に溶解した。最後に、純粋生成物1をセファデックス−CM C−25 Na+型カラムに通過させることにより、ヌクレオチドトリエチルアンモニウム対イオンをNa+イオンと交換した。LC分離後、生成物2が10%(23mg)の収率で得られた。半分取カラム上での保持時間:12.09分。2についてのスペクトルデータは、Mohamady及びJakeman(2005)によって記載されているデータと一致していた。
【0097】
方法B:ビス(トリブチルアンモニウム)メチレンジホスホネート塩を上記で記載した通りに調製した。1,8−ビス(ジメチルアミノ)ナフタレン(41mg、0.19mmol、2当量)を、N2下、火炎乾燥した二口フラスコ内のトリメチルリン酸(1ml)中の2−MeS−アデノシン、5b(30mg、0.09mmol)に0℃で添加し、透明な溶液になるまで20分間反応物を撹拌した。POCl3(26μl、0.28mmol、3当量)を0℃で添加した。溶液を0℃で2時間撹拌した。乾燥DMF(480μl)中のビス(トリブチルアンモニウム)メチレンジホスホネート塩の1M溶液(101mg、0.57mmol、6当量)及びトリブチルアミン(91μl、0.38mmol、4当量)を0℃で添加し、反応混合物を1.6分間撹拌した。次いで、0.5Mの重炭酸トリエチルアンモニウム(TEAB)溶液(10ml)を室温で添加し、反応混合物を30分間撹拌し、次いで凍結乾燥させた。得られた残留物を、活性化したセファデックスDEAE−A25カラム(0〜0.8MのNH4HCO3、全容積1l)に塗布した。関連画分を収集し、凍結乾燥させ、脱イオン水を用いる凍結乾燥の繰り返しによって過剰なNH4HCO3を除去して、生成物2を白色固体として得た。残留物をHPLCカラム上で分離して、純粋な2を得た。分離は、半分取逆相Gemini 5u C−18 110Aカラム(250×10.00mm、5ミクロン)及び流速5ml/分で20分間にわたる勾配(92:8〜70:30のA:B)の溶媒系I(上記を参照)を使用して達成した。関連画分(保持時間=11.94分)を凍結乾燥させた。凍結乾燥サイクルの繰り返しによって過剰な緩衝液を除去し、固体残留物を毎回脱イオン水に溶解した。最後に、純粋生成物1をセファデックス−CM C−25 Na+型カラムに通過させることにより、ヌクレオチドトリエチルアンモニウム対イオンをNa+イオンと交換した。LC分離後、生成物2が10%(11mg)の収率で得られた。2についてのスペクトルデータは、Mohamady及びJakeman(2005)によって記載されているデータと一致していた。
【0098】
(例2)
アデノシン−β,γ−CH2−5’−O−(1−ボラノ三リン酸)、3の合成、分離及び特徴付け
アデノシン−β,γ−CH2−5’−O−(1−ボラノ三リン酸)、3の合成
ビス(トリブチルアンモニウム)メチレンジホスホネート塩は、Bu3N(2当量)をEtOH中のメチレンジホスホン遊離酸に添加し、室温で2時間撹拌し、続いて減圧下で溶媒を除去して白色固体を得ることによって調製した。スキーム2で描写される通り、2’,3’−O−メトキシメチリデンアデノシン、9(100mg、0.32mmol)を、N2下、火炎乾燥した二口フラスコ内のトリメチルリン酸(2.5ml)に溶解した。1,8−ビス(ジメチルアミノ)ナフタレン(138mg、0.65mmol、2当量)を0℃で添加し、透明な溶液になるまで20分間反応物を撹拌した。PCl3(56μl、0.65mmol、2当量)を0℃で添加すると、白色固体が沈殿した。懸濁液を0℃で30分間撹拌した。次いで、乾燥DMF(1.8m)中のビス(トリブチルアンモニウム)メチレンジホスホネート塩の1M溶液(642mg、1.94mmol、6当量)及びトリブチルアミン(308μl、1.29mmol、4当量)を0℃で添加し、反応混合物を11分間撹拌した。THF中のBH3・SMe2錯体の2M溶液(2.2ml、3.9mmol、10当量)を0℃で添加すると、反応混合物は透明になった。溶液を0℃で5分間、次いで室温で30分間撹拌した。最後に、0.5MのTEAB溶液(10ml)を室温で添加し、混合物を60分間撹拌し、次いで凍結乾燥させた。得られた残留物を、活性化したセファデックスDEAE−A25カラム(0〜0.8MのNH4HCO3、全容積1l)に塗布した。関連画分を収集し、凍結乾燥させ、脱イオン水を用いる凍結乾燥サイクルの繰り返しによって過剰なNH4HCO3を除去した。生成物13aが白色固体として得られた。生成物13aを、pH2.3になるまで18%のHCl溶液で処理し、次いで室温で3時間撹拌した。最後に、混合物を24%のNH4OH溶液で処理し、pHを9に調整した。溶液を室温で45分間撹拌し、次いで凍結乾燥させた。生成物3のジアステレオマー対を、以下に記載する条件下、HPLCカラム上で分離した。最後に、精製された異性体3A及び3Bをセファデックス−CM C−25Na+型カラムに通過させて、トリエチルアンモニウム対イオンをNa+イオンと交換した。
【0099】
アデノシン−β,γ−CH2−5’−O−(1−ボラノ三リン酸)、3の分離
3のジアステレオマー対の分離は、半分取逆相Gemini 5uカラム(C−18 110A、250×10.00mm、5ミクロン)及び89:11のA:B、流速5ml/分で溶媒系I(例1を参照)を使用する定組成溶離を使用して達成し、続いて、2個のジアステレオ異性体の最終分離は、流速1ml/分で20分間にわたる90:10〜70:30のA:Bの勾配の溶媒系I(例1を参照)を適用することにより分析用Gemini 5uカラム(C−18 110A、150×4.60mm)を使用して達成した。同じ異性体[保持時間=6.33分(異性体A)、7.73分(異性体B)]を含有する画分を収集し、凍結乾燥させた。凍結乾燥サイクルの繰り返しによって過剰な緩衝液を除去し、固体残留物を毎回脱イオン水に溶解した。LC分離後、ジアステレオ異性体3A及び3Bが36%(66mg)の全収率で得られた。
【0100】
アデノシン−β,γ−CH2−5’−O−(1−ボラノ三リン酸)、3Aの特徴付け
半分取カラム上での保持時間:7.64分。
【化4】
TLC(NH4OH:H2O:イソプロパノール 2:8:11)、Rf=0.23。分析用カラム上で得られた純度データ:流速1ml/分で10分間にわたる90:10〜70:30のA:Bの勾配の溶媒系I(例1を参照)を使用して、保持時間:3.55分(純度100%)。溶媒系II、流速1ml/分で10分間にわたる90:10〜80:20の(A)0.01MのKH2PO4、pH=4.5〜(B)MeOHの勾配を使用して、保持時間:2.53分(純度95.5%)。
【0101】
アデノシン−β,γ−CH2−5’−O−(1−ボラノ三リン酸)、3Bの特徴付け
半分取カラム上での保持時間:9.67分。
【化5】
TLC(NH4OH:H2O:イソプロパノール 2:8:11)、Rf=0.23。分析用カラム上で得られた純度データ:流速1ml/分で10分間にわたる90:10〜70:30のA:Bの勾配の溶媒系I(例1を参照)を使用して、保持時間:4.09分(純度92.6%)。流速1ml/分で10分間にわたる95:10〜80:20のA:Bの勾配の溶媒系II(上記を参照)を使用して、保持時間:3.66分(純度95.5%)。
【0102】
(例3)
2MeS−アデノシン−β,γ−CH2−5’−O−(1−ボラノ三リン酸)、4の合成、分離及び特徴付け
2MeS−アデノシン−β,γ−CH2−5’−O−(1−ボラノ三リン酸)、4の合成
生成物4は、生成物3について例2に記載され、以下のスキーム2で描写されるのと同じ手法で、LC分離後、28%の全収率で5aから得られた。
【0103】
2MeS−アデノシン−β,γ−CH2−5’−O−(1−ボラノ三リン酸)、4の分離
4のジアステレオ異性体の分離は、半分取逆相Gemini 5uカラム(C−18 110A、250×10.00mm、5ミクロン)及び75:25のA:B、流速5ml/分で溶媒系I(例1を参照)を適用することにより定組成溶離を使用して達成した。2個のジアステレオ異性体の最終分離は、分析用Gemini 5uカラム(C−18 110A、150×4.6mm)及び流速1ml/分で20分間にわたる82:18〜74:26のA:Bの勾配の溶媒系I(例1を参照)を使用して遂行した。同じ異性体[保持時間=9.79分(異性体A)、11.53分(異性体B)]を含有する画分を収集し、凍結乾燥させた。凍結乾燥サイクルの繰り返しによって過剰な緩衝液を除去し、固体残留物を毎回脱イオン水に溶解した。LC分離後、ジアステレオ異性体4A及び4Bが28%(38mg)の全収率で得られた。
【0104】
2MeS−アデノシン−β,γ−CH2−5’−O−(1−ボラノ三リン酸)、4Aの特徴付け
半分取カラム上での保持時間:5.29分。
【化6】
TLC(NH4OH:H2O:イソプロパノール 2:8:11)、Rf=0.44。分析用カラム上で得られた純度データ:流速1ml/分で10分間にわたる80:20〜60:40のA:Bの勾配の溶媒系I(例1を参照)を使用して、保持時間:4.24分(純度94.3%)。流速1ml/分で10分間にわたる75:25〜65:35のA:Bの勾配の溶媒系II(例2を参照)を使用して、保持時間:2.99分(純度99.5%)。
【0105】
2MeS−アデノシン−β,γ−CH2−5’−O−(1−ボラノ三リン酸)、4Bの特徴付け
半分取カラム上での保持時間:5.57分。
【化7】
TLC(NH4OH:H2O:イソプロパノール 2:8:11)、Rf=0.44。流速1ml/分で10分間にわたる70:30〜40:60のA:Bの(A)100mMのTEAA、pH7〜(B)CH3CNの勾配を使用して、保持時間:2.12分(純度94%)。流速1ml/分で10分間にわたる50:50〜40:60のA:Bの勾配の溶媒系II(例2を参照)を使用して、保持時間:1.38分(純度100%)。
【0106】
(例4)
31P NMRによって評価した化合物2の化学安定性
pH1.4及び37℃における2の安定性を31P NMRによって評価して、生じ得る脱リン酸化生成物をモニターした。化合物2(1.5mg)を0.2MのHCl/KCl(0.35ml)及びD2O(40μl)に溶解した。0.2MのHCl(20μl)を添加することにより、最終pHを1.4に調整した。溶液を37℃の油浴中で維持した。スペクトルを12時間間隔で11日間記録した。すべての実験におけるスキャン数は500であった。リン酸エステル加水分解の割合は、β,γ−CH2−2MeS−ATPのPαシグナル(−10.5ppm)及び加水分解生成物2MeS−AMP、9のPαシグナル(0.7ppm)の積分に基づく。それぞれのNMRシグナルの積分における変化を時間とともに計測することにより、加水分解率を決定した。
【0107】
図1Aに示すように、これらの条件下で、化合物2は比較的高い安定性を呈した。特に、出発材料2に加えて、時間とともに2−MeS−AMPの量の増加が観察された。よって、0ppmのシグナル(2−MeS−AMPのPα)が次第に現れ、一方で、−11ppmのシグナル(2のPα)が時間とともに減少した。2−MeS−AMPのPαの31P NMRシグナルの時間に伴う強度変化を(β,γ−CH2−2−MeS−ATP及び2−MeS−AMPの全Pα積分の割合として)、2の濃度に関する擬1次速度指数関数的減衰速度式に当てはめた。2についてpH1.4/37℃で測定された半減期は、図1Bに示す通り、65時間であった。
【0108】
(例5)
HPLCによって評価した化合物3及び4の化学安定性
37℃の適切な緩衝溶液(0.2MのHCl/KCl、pH=1.4)中における3(異性体B)の安定性を、7〜17時間間隔で5日間のHPLC−エレクトロスプレーイオン化(ESI)MSによって評価し、時間に伴う3BピークのHPLC積分変化に基づくその加水分解率を、図2A〜2Bに示す通り、擬1次速度指数関数的減衰速度式に当てはめた。3Bに加えて、スキーム3で描写される通り、分解生成物6、7及び8が加水分解混合物中で同定された。例えば、19時間後、50%の3Bが分解されて、37%のAMP−α−B及びAMP−α−H(それぞれ6及び7。いずれも同じ保持時間で出現するが、MSはこれらの化合物の識別を可能にした)並びに13%のアデノシン(図2A)が生じた。時間に伴う加水分解混合物の組成変化を図2Cに描写する。3Bの半減期は19時間であった。
【0109】
37℃の適切な緩衝溶液(pH=1.4)中における4(異性体B)の安定性をHPLCによって評価して、生じ得る脱リン酸化生成物をモニターした。化合物4(1.6mg)を0.2MのHCl/KCl緩衝液(0.4ml)に溶解し、0.2MのHCl(15μl)を添加することにより、最終pHを1.4に調整した。溶液を37℃の油浴中で維持し、Gemini分析用カラム(5u C−18 110A、150×4.60mm)及び流速1ml/分で15分間にわたる89:11のA:B、次いで20分間にわたる82:18〜74:26のA:Bの溶媒系I(例1を参照)での勾配溶離を使用するHPLC−MSによってその組成を分析した。12時間間隔で5日間、検体を採取した。分解生成物6、7及び8のHPLCピークの積分における変化を時間とともに計測することにより、4の加水分解率を決定した。図2Cに示すように、化合物4Bの半減期は14.5時間であった。
【0110】
(例6)
アルカリホスファターゼに対する化合物2〜4の酵素安定性
酵素活性は、UV−可視分光光度計を405nmで使用し、ヌクレオチド誘導体からのp−ニトロフェノールの放出により計測した。酵素加水分解に対するヌクレオチドの相対活性及び抵抗性は、37℃で測定した。手短に述べると、32.5μlのヌクレオチド誘導体(0.1Mのトリス−HCl及び0.1MのMgCl2中77μg/ml、pH7.5)及び6μlの脱イオン水を、子ウシ腸アルカリホスファターゼ(Fermentas Inc.、Glen Burnie、MD、1ユニット/μl、37℃で6.25μl。最終pH=9.8)とともにインキュベートした。30分後、80℃における30分間のインキュベーションにより、反応を停止させた。ヌクレオチド誘導体の安定性をHPLCによって評価して、生じ得る脱リン酸化生成物をモニターした。混合物を、流速1ml/分で20分間にわたる、3A及び3Bについては90:10〜70:30のA:B、4A、4B及び2については82:18〜50:50の溶媒系I(例1を参照)での勾配溶離を使用するGemini分析用カラム(5u C−18 110A、150×4.60mm)上で分離した。それぞれのHPLCピークの積分における変化を時間とともに計測することにより加水分解率を決定し、また観察される通り、類似体2〜4はこれらの条件下において完全に無傷のままであった。
【0111】
(例7)
ヒト血清中におけるATP及び化合物2〜4の安定性
ヒト血清の調製:健康な志願者から採取した血液を血液バンク(Tel−Hashomer hospital、Israel)から入手し、4℃で12時間保存し、プラスチックチューブ中、1500g、室温で15分間遠心分離した。血清を分離し、−80℃で保存した。
【0112】
ヒト血清中における2〜3の安定性の評価、方法A
脱イオン水中40mMのヌクレオチド誘導体溶液(4.5μl)、ヒト血清(180μl)及びRPMI−1640(540μl)を含有するアッセイ混合物を、37℃で1、4、8、16、24、48、72及び96時間インキュベートした。次いで、検体を0.6Mの塩酸(430μl)で処理し、2分間遠心分離し(13,000g、4℃)、4MのKOHの添加によって中和し、2分間遠心分離し(13,000g、4℃)、凍結乾燥させた。生じ得る脱リン酸化生成物をモニターするために、ヌクレオチドの安定性をHPLCによって評価した。混合物を、勾配溶離[0.01MのKH2PO4 pH=4.5(A)/アセトニトリル(B)、2、3A及び3Bについては100:0→60:40のA:B、20分間;ATPについては100:0→95:5のA:B、10分間]及び流速1ml/分のGemini分析用カラム(5u C−18 110A、150×4.60mm)上で分離した。それぞれのHPLCピークの積分における変化を時間とともに計測することにより、加水分解率を決定した。
【0113】
ヒト血清中における4の安定性の評価、方法B
脱イオン水中40mMのヌクレオチド誘導体溶液(4.5μl)、ヒト血清(180μl)及びRPMI−1640(540μl)を含有するアッセイ混合物を、37℃で1、4、8、16、24、48、72、96、120及び144時間インキュベートした。次いで、検体を80℃に30分間加熱し、CMセファデックス(1〜2mg)で2時間処理し、6分間遠心分離し(12,000rpm)、クロロホルム(2×500μl)で抽出した。水層を凍結乾燥させた。生じ得る脱リン酸化生成物をモニターするために、ヌクレオチドの安定性をHPLCによって評価した。混合物を、勾配溶離[4A及び4Bについては、100mMのTEAA、pH7(A)/MeOH(B)、79:21のA:B、15分間;ATPについては、100mMのTEAA、pH7(A)/アセトニトリル(acetonitryl)(B)、A:B、10分間、100:0→90:10のA:B、10分間、90:10→80:20のA:B、4分間、80:20のA:B、1分間]及び流速1ml/分のGemini分析用カラム(5u C−18 110A、150×4.60mm)上で分離した。それぞれのHPLCピークの積分における変化を時間とともに計測することにより、加水分解率を決定した。
【0114】
図3に示すように、ATPは3.6時間の半減期でADP及びAMPに加水分解され(方法A)、一方、同じ条件下において、化合物2、3A及び3Bは、それぞれ12.7、14.1及び47.1時間の半減期で、対応するヌクレオシド−一リン酸(ボラノリン酸)に大部分が加水分解された(化合物2の加水分解率に関するデータを図4A〜4Cに示す)。化合物4Bは、71.9時間の半減期で加水分解され(方法B)、一方、同じ条件下において、ATPは7.7時間の半減期で加水分解された。これらの値は、ATPの代謝的安定性の3.5〜20倍の置換依存性増強を表す。
【0115】
(例8)
2−MeS−アデノシン−5’−ジハロゲノメチレン−二リン酸、17〜18の合成
2−MeS−アデノシン−5’−ジクロロメチレン−二リン酸及び2−MeS−アデノシン−5’−ジフルオロメチレン−二リン酸、それぞれ17及び18を調製するために、最初に5’−O−トシル−2’,3’−O−アセトニド−2MeS−アデノシン、16を、スキーム4で描写され、以下に記載する通りに調製した。
【0116】
2’,3’アセトニド−2−MeS−アデノシン、15(Nahumら、2002)(97mg、0.27mmol)を、N2下、火炎乾燥した二口フラスコ内の乾燥ジクロロメタン(1ml)に溶解した。乾燥ジクロロメタン(2ml)中のDMAPの溶液(134mg、1.09mmol、4当量)及び乾燥ジクロロメタン(0.5ml)中のTsClの溶液(156mg、0.82mmol、3当量)を添加し、得られた溶液を、N2下、室温で12時間撹拌した。反応物をジクロロメタン(50ml)で希釈し、NaHCO3の飽和溶液(3×30ml)で抽出した。有機層を除去し、Na2SO4で乾燥させ、濾過し、減圧下で蒸発させて白色固体を得、これを、シリカゲルカラム[25+Mカラム、下記の勾配スキームを使用:流速25ml/分で、CHCl3(A):EtOH(B)、0:0、3CV、A:B、CHCl3(A):EtOH(B)、0:0→0:10、5CV、A:B、CHCl3(A):EtOH(B)、10:0、4CV]を使用するMPLCシステム上で分離した。化合物16が52%の収率で得られた。
【化8】
【0117】
次いで、化合物16(42mg、0.08mmol)を、N2下、火炎乾燥した二口フラスコ内の乾燥DMF(0.2ml)に溶解した。乾燥DMF(0.3ml)中のトリス(テトラブチルアンモニウム)ジハロゲンメチレンジホスホネート(dihalogenemethylenediphosphonate)の溶液(0.16mmol、2当量)を添加し、溶液を室温で72時間撹拌した。TFA原液(2ml)を添加し、反応物を、N2通気下、室温で10分間撹拌した。溶媒を減圧下で除去して黄色固体を得、これを、活性化したセファデックスDEAE−A25カラム(0〜0.3MのNH4HCO3、全容積1.4l)上で分離した。化合物17又は18のいずれかを含有する関連画分を収集し、凍結乾燥させ、脱イオン水を用いる凍結乾燥サイクルの繰り返しによって過剰なNH4HCO3を除去した。
【0118】
(例9)
2−SMe−アデノシン−5’−O−(Pα−ボラノ)二リン酸、19の合成、分離及び特徴付け
スキーム5で描写される通り、2’,3’−メトキシメチリデンヌクレオシド(490.4mg、1.38mmol)を、アルゴン下、火炎乾燥し隔膜で密閉したフラスコ内のDMF(3ml)/ピリジン(0.6ml、5当量)に溶解した。次いで、ジオキサン(1ml)中のサリチルホスホクロリダイトの新たに調製した溶液(307mg、1.1当量)を、シリンジを介してフラスコに移した。室温で10分間撹拌後、DMF中のビス(トリ−n−ブチルアンモニウム)ピロリン酸の新たに調製した1M溶液(2.1ml、1.5当量)及びトリ−n−ブチルアミン(1.3ml、4当量)を、隔膜を通して同時に注入した。THF中のBH3:SMe2錯体の2M溶液(7ml、10当量)をフラスコに添加し、混合物を室温で15分間撹拌した。次いで、エチレンジアミン(0.5ml、5当量)を、シリンジによってフラスコに注入した。60分間撹拌後、脱イオン水(4ml)をフラスコに添加した。10分後、反応混合物を蒸発させた。残留物を脱イオン水によって希釈し、エチルエーテルで抽出した。次いで、水層を凍結乾燥させ、得られた残留物を、活性化したセファデックスDEAE−A25カラム(0〜0.4MのNH4HCO3、全容積900ml)に塗布した。関連画分を収集し、凍結乾燥させ、脱イオン水を用いる凍結乾燥の繰り返しによって過剰なNH4HCO3を除去して、化合物19をアンモニウム塩として得た。メトキシメチリデン保護基を酸加水分解によって除去した(pH2.3に達するまで10%のHCl溶液を添加した)。室温で3時間後、NH4OH溶液(pH11)の添加によってpHを9まで迅速に上昇させ、溶液を室温で40分間維持した。LC分離後、化合物19が46%の収率で得られた。ジアステレオ異性体の最終精製及び分離は、HPLC、TEAA:アセトニトリル 88:12を用いる定組成溶離によって遂行した。
【0119】
2MeS−アデノシン−5’−O−(Pα−ボラノ)二リン酸、19Aの特徴付け
保持時間:8.073分。
【化9】
分析用カラム上で得られた純度データ:溶媒系III(100mMのTEAA、pH7(A)/アセトニトリル(B)、88:12のA:B、流速1ml/分)を使用して、保持時間:4.113分(純度98%)。溶媒系IV(0.01MのKH2PO4、pH4.5(A)/アセトニトリル(B)、90:10のA:B流速1ml/分)を使用して、保持時間:3.158分(純度98.5%)。
【0120】
2MeS−アデノシン−5’−O−(Pα−ボラノ)二リン酸、19Bの特徴付け
保持時間:9.127分。
【化10】
分析用カラム上で得られた純度データ:溶媒系III(上記を参照)を使用して、保持時間:4.720分(純度95%)。溶媒系IV(上記を参照)を使用して、保持時間:3.764分(純度94%)。
【0121】
(例10)
アルカリホスファターゼに対する及びヒト血清中における、2MeS−アデノシン−5’−O−(Pα−ボラノ)二リン酸、19の安定性
アルカリホスファターゼに対する化合物19(異性体A)の安定性を、例6に記載されている通りに計測し、そのt1/2は、ADPについては4時間であるのに対し、約6時間であることが分かった。
【0122】
加えて、この化合物の安定性をヒト血清中において計測し、判明したところでは、この化合物は、ADPについては約2時間であるのに対し、24時間超の半減期で加水分解された。詳細には、24時間後に加水分解された化合物の割合は、約25〜40%だけであった。
【0123】
(例11)
P2Y1/6受容体の潜在的なアゴニストとしての化合物2、4B、17、18、19A、21A及び21B
化合物2、4B、17、18、19A、21(異性体A及びB)の活性を、ヒト1321N1星状細胞腫細胞で発現されているGタンパク質共役P2YR、P2Y1、P2Y2、P2Y4及びP2Y6において、上記の実験に記載されている通り、細胞内カルシウム計測に基づいて検査した。詳細には、化合物2、4B及び19の活性はP2Y1R、P2Y2R、P2Y4R及びP2Y6Rにおいて検査し、化合物17、18及び21の活性はP2Y1Rのみにおいて検査した。実験は、Columbia−Missouri University、Columbia、MO、USAのGary A.Weisman教授によって実行された。
【0124】
以下の表1に示すように、化合物19Aは、2−MeS−ADPについては0.004μMであるのと比較して、0.038μMのEC50を有する、最も強力且つ選択的なP2Y1Rのアゴニストであることが分かった。化合物2及び4Bは、それぞれ0.08及び17.2μMのEC50を有するP2Y1Rのアゴニストであり、P2Y6Rに対して100μMのわずかなアゴニスト作用を有していた。化合物17、18、21A及び21Bは、それぞれ3.1、0.98、0.57及び1.2μMのEC50を有するP2Y1Rのアゴニストであった。
【表1】
【0125】
(例12)
インスリン分泌促進物質としての本発明の化合物の効能のインビボ研究
パラダイム
この実験の目的は、カニューレを挿入したウィスター系ラットへのグルコースの単回経口強制飼養(経口)投与後の、インスリン分泌増強分子としての本発明の化合物の効能を、カニューレを挿入したラット細静脈内へのカニューレを介する試験化合物投与後の血中グルコース及びインスリンレベルを計測することにより、インビボで研究することである。
【0126】
合計約40匹の健康な10〜13週齢ウィスター系ラットを使用する。動物を処置開始前に少なくとも4日間気候順化させ、薬用でない市販の滅菌げっ歯類飼料を自由に摂食させる。飲料用水道水は自由に利用可能とする。
【0127】
処置の約48時間前、ラットを計量し、重量が均一な個体群(動物の約90%)を挿管用にする。詳細には、2.5%イソフルラン97.5%乾燥空気吸入によって動物に麻酔をかけ、P52カニューレを外科的に挿入し、頸静脈内に固定し、挿管後(及びその後は、各血液収集の直後)に0.3〜0.5mlの5%へパリン化生理食塩水で洗い流す。ラットの留置カニューレによって生じる固着又は凝固等の技術的問題の場合には、別のラットにカニューレを挿入し、予め割り当てられた研究中のラットと置き換える。
【0128】
処置日に、ラットの各1匹のカニューレを確認し、ラットを計量し、ラットの各1匹のグルコースレベルを尾静脈によって確認する。グルコースレベル及び重量が均一な個体群を3つの群に分割し、ここで、第1の群を試験化合物で処置し、第2の群を生理食塩水で処置した陰性対照群とし、第3の群を、グリベンクラミド(グリブリドとしても知られる)で処置した陽性対照群とする。後者は、II型糖尿病の治療に使用されるスルホニル尿素に分類される抗糖尿病薬物であり、これは、現在、WHO Model List of Essential Medicinesにおいて2つしかない経口糖尿病薬の1つである。グリベンクラミドは、膵臓ベータ細胞においてATP感受性カリウムチャネルを阻害することによって作用し、細胞膜脱分極及び電位依存性カルシウムチャネルの開口を引き起こし、それが、インスリン放出を刺激するベータ細胞内への細胞内カルシウムの増加の誘因となる。
【0129】
実験に関与するすべてのラットに、体重1kgにつき2gのグルコース負荷を(経口で)投与し、ここで、ラット1匹当たりのグルコース投与の全容積は、0.67g/mlの溶液から体重1kgにつき3mlである。グルコース投与の10分後、第1の群のラットに試験化合物を投与し、陰性対照群のラットに生理食塩水を投与する。いずれの場合も、投与はカニューレを介して静脈内へ実施する。試験化合物の投与量レベルは体重1kgにつき2.5mgであり、投与容積は体重1kgにつき1mlであり、投与される生理食塩水の容積は体重1kgにつき1mlである。陽性対照群のラットには、グルコース投与の30分前にグリベンクラミドを(経口で)投与する。グリベンクラミドの投与量レベルは体重1kgにつき1mgであり、投与容積は0.2mg/mlの溶液から体重1kgにつき5mlである。投薬後、採血までの間、ラットをケージに戻しておく。
【0130】
グルコース及びインスリンレベルは、グルコース投与(及び陽性対照群の場合はグリベンクラミド投与)の30分前;グルコース投与の直前及び5分後;グルコース投与の15分後、すなわち、試験化合物又は生理食塩水(それぞれ第1及び陰性対照群の場合)の投与の5分後;次いで、グルコース投与の30、45、60、120及び150分後に計測する。
【0131】
グルコースレベル計測のために、血液検体を各ラットから尾静脈を介して抜き取り、グルコメーターで直ちに試験する。インスリンレベル計測のために、血液検体を各ラットから頸動脈カニューレを介して抜き取る。各処置ラットから収集する血液の容積は150μlである。インスリンレベルのために抜き取られた血液検体を、Z血清/ゲルを加えた0.8mlチューブ中に収集する。血液を室温で少なくとも30分間放置して凝固させ、凝固後、これを約4℃で遠心分離する(3000×g、15分)。血清を取り出し、2つの0.2mlフラットキャップPCRタブに等分し(アリコート当たり少なくとも25μl)、次いで、分析するまで−20℃で冷凍保存する。臨床的観察は、各個別の動物の投薬後及び出血期間内に実施する。全血中のグルコースレベルの血液分析は、出血中に、血中グルコースモニタリングシステムをこのシステムに適した試験紙上で使用して、その場で行う。血清中のインスリンレベルの血液分析は、ラット/マウスインスリンキットを使用して行う。
【0132】
この実験の生存段階は、最終血液検体収集及び血清の取り出しに続いて、グルコース投与の150分後に完了する。
【0133】
試験化合物で処置したラットは、グルコース投与の15分後、すなわち、試験化合物の投与の5分後から開始して、採取された血液検体中に著しく低レベルのグルコースを有することが予期される。予期されるグルコースのレベルは、事実上、絶食後に計測されるグルコースレベルに極めて類似している。
【0134】
グルコース投与の約15分後、すなわち、健康な個体においてグルコース投与後に通常計測されるインスリンレベルの増加の約15分前には既に、試験化合物で処置したラットのインスリンレベルに著しい増加が見られることがさらに予期される。増加したインスリンのレベルは、30〜45分間維持され、次いで、血液中における試験化合物の安定性に応じ、ある速度で減少することが予期される。
【0135】
結果
予備研究において、2MeS−アデノシン−5’−O−(Pα−ボラノ)二リン酸、19(2.5mg/kg)を、以上に記載した通り、絶食させたウィスター系ラット(n=5)に静脈内投与し、一方、生理食塩水を陰性対照群のラットに投与し、グリベンクラミド(0.25mg/kg)を陽性対照群のラットにグルコース投与の30分前に与えた。図5に示すように、化合物19は、グリベンクラミドと同様に、生理食塩水で処置したラットにおいて計測されるグルコースレベルに対し、計測されるグルコースレベルを低減させた。
【化11】
【化12】
反応条件:
方法A:5aから出発し、a)トリメチルリン酸、POCl3、Proton Sponge(商標)、0℃、3時間;b)乾燥DMF中0.5Mのビス(トリブチルアンモニウム)メチレンジホスホネート、Bu3N、0℃、1.6分;c)0.5MのTEAB、pH=7、室温、0.5時間;並びにd)1)18%のHCl、pH2.3、室温、3時間;及び2)24%のNH4OH、pH9、室温、45分。
方法B:5bから出発し、a)トリメチルリン酸、POCl3、Proton Sponge(商標)、0℃、2時間;b)乾燥DMF中1Mのビス(トリブチルアンモニウム)メチレンジホスホネート、Bu3N、0℃、25分;及びc)0.5MのTEAB、pH7、室温、0.5時間。
【化13】
反応条件:a)トリメチルリン酸、PCl3、Proton Sponge(商標)、0℃、30分;b)乾燥DMF中1Mのビス(トリブチルアンモニウム)メチレンジホスホネート、Bu3N、0℃、11分;c)THF中2MのBH3・SMe、0℃、5分、次いで室温、30分;d)1MのTEAB、pH7、室温、0.5時間;並びにe)1)18%のHCl、pH2.3、室温、3時間;及び2)24%のNH4OH、pH9、室温、45分。
【化14】
【化15】
【化16】
【0136】
【特許請求の範囲】
【請求項1】
一般式Iの化合物:
【化1】
[式中、
Xは、9位を介して結合した式Iaのアデニン残基:
【化2】
であり、ここで、
R1は、H、ハロゲン、O−ヒドロカルビル、S−ヒドロカルビル、NR4R5、ヘテロアリール、非置換ヒドロカルビル、又はハロゲン、CN、SCN、NO2、OR4、SR4、NR4R5若しくはヘテロアリールで置換されているヒドロカルビルであり、ここで、R4及びR5は、それぞれ独立に、H又はヒドロカルビルであるか、或いはR4及びR5は、それらが結合した窒素原子と一緒になって、酸素、窒素又は硫黄から選択される1〜2個のさらなるヘテロ原子を場合によって含有する5又は6員の飽和又は不飽和複素環を形成し、前記追加の窒素は、置換されていないか、又はハロゲン、ヒドロキシル若しくはフェニルによって置換されているアルキルで置換されており、
R2及びR3は、それぞれ独立に、H又はヒドロカルビルであるか、
或いは、Xは、1位を介して結合した式Ibのウラシル残基:
【化3】
であり、ここで、
R6は、H、ハロゲン、O−ヒドロカルビル、S−ヒドロカルビル、NR8R9、ヘテロアリール、非置換ヒドロカルビル、又はハロゲン、CN、SCN、NO2、OR8、SR8、NR8R9若しくはヘテロアリールで置換されているヒドロカルビルであり、ここで、R8及びR9は、それぞれ独立に、H又はヒドロカルビルであるか、或いはR8及びR9は、それらが結合した窒素原子と一緒になって、酸素、窒素又は硫黄から選択される1〜2個のさらなるヘテロ原子を場合によって含有する5又は6員の飽和又は不飽和複素環を形成し、前記追加の窒素は、置換されていないか、又はハロゲン、ヒドロキシル若しくはフェニルによって置換されているアルキルで置換されており、
R7はO又はSであり、
Yは、H、OH又はNH2であり、
Z1、Z2及びZ3は、それぞれ独立に、O−又はBH3−であり、
W1及びW2は、それぞれ独立に、O、CH2、C(Hal)2又はNHであり、ここで、Halは、ハロゲン、好ましくはF又はClであり、
nは0又は1であり、但し、nが0であり、W2がOである場合、Z1はBH3−であり、nが1である場合、W1及びW2の少なくとも一方はOではなく、
mは3又は4であり、
B+は、薬学的に許容されるカチオンを表す]
(nが0であり、Z1及びZ3がそれぞれO−であり、W2がCH2又はNHである化合物、並びにnが1であり、Z1〜Z3がそれぞれO−である化合物を除く)
並びにそのジアステレオ異性体。
【請求項2】
nが0であり、Z1及びZ3がOであるか、又はnが0であり、Z1及びZ3の少なくとも一方がBH3−であるか、又はnが1であり、Z1〜Z3の少なくとも1つがBH3−である、請求項1に記載の化合物。
【請求項3】
nが0であり、唯1個のボラノ基を位置αに含む、すなわち、Z1がBH3−であり、Z2がO−である;若しくは位置βに含む、すなわち、Z3がBH3−であり、Z1がO−である;又は2個のボラノ基を位置α、βに含む、すなわち、Z1及びZ3がBH3−である、請求項2に記載の化合物。
【請求項4】
nが1であり、唯1個のボラノ基を位置αに含む、すなわち、Z1がBH3−であり、Z2及びZ3がO−である;位置βに含む、すなわち、Z2がBH3−であり、Z1及びZ3がO−である;若しくは位置γに含む、すなわち、Z3がBH3−であり、Z1及びZ2がO−である;2個のボラノ基を位置α及びβに含む、すなわち、Z1及びZ2がBH3−であり、Z3がO−である;位置α及びγに含む、すなわち、Z1及びZ3がBH3−であり、Z2がO−である;若しくは位置β及びγに含む、すなわち、Z2及びZ3がBH3−であり、Z1がO−である;又は3個のボラノ基を位置α、β及びγに含む、すなわち、Z1〜Z3がBH3−である、請求項2に記載の化合物。
【請求項5】
Xがアデニン残基であり、ここで、R1が、H、ハロゲン、O−ヒドロカルビル又はS−ヒドロカルビルであり、R2及びR3が、それぞれ独立に、H又はヒドロカルビルであり、YがOHであり、nが1であり、Z1がBH3−であり、Z2及びZ3がO−であり、W1がOであり、W2が、CH2、CF2又はCCl2である、請求項1に記載の化合物。
【請求項6】
Xがアデニン残基であり、ここで、R1がH又はNR4R5であり、R4及びR5が、それぞれ独立に、H又はヒドロカルビルであるか、或いはR4及びR5が、それらが結合した窒素原子と一緒になって、酸素、窒素又は硫黄から選択される1〜2個のさらなるヘテロ原子を場合によって含有する飽和又は不飽和複素環を形成し、R2及びR3が、それぞれ独立に、H又はヒドロカルビルであり、YがOHであり、nが1であり、Z1がBH3−であり、Z2及びZ3がO−であり、W1がOであり、W2が、CH2、CF2又はCCl2である、請求項1に記載の化合物。
【請求項7】
Xがアデニン残基であり、ここで、R1が、H、ハロゲン、O−ヒドロカルビル又はS−ヒドロカルビルであり、R2及びR3が、それぞれ独立に、H又はヒドロカルビルであり、YがOHであり、nが0であり、(i)Z1及びZ3がO−であり、W2がCF2又はCCl2であるか、或いは(ii)Z1がBH3−であり、W2がOである、請求項1に記載の化合物。
【請求項8】
Xがアデニン残基であり、ここで、R1がH又はNR4R5であり、R4及びR5が、それぞれ独立に、H又はヒドロカルビルであるか、或いはR4及びR5が、それらが結合した窒素原子と一緒になって、酸素、窒素又は硫黄から選択される1〜2個のさらなるヘテロ原子を場合によって含有する飽和又は不飽和複素環を形成し、R2及びR3が、それぞれ独立に、H又はヒドロカルビルであり、YがOHであり、nが0であり、Z1及びZ3がO−であり、W2がCF2又はCCl2である、請求項1に記載の化合物。
【請求項9】
Xがアデニン残基であり、ここで、R1がHであり、R2及びR3がHであり、YがOHであり、nが1であり、Z1がBH3−であり、Z2及びZ3がO−であり、W1がOであり、W2がCH2である(化合物3)、請求項5に記載の化合物。
【請求項10】
Xがアデニン残基であり、ここで、R1がSMeであり、R2及びR3がHであり、YがOHであり、nが1であり、Z1がBH3−であり、Z2及びZ3がO−であり、W1がOであり、W2がCH2である(化合物4)、請求項5に記載の化合物。
【請求項11】
半分取逆相Gemini 5uカラム(C−18 110A、250×10mm、5ミクロン)及び定組成溶離[100mMの酢酸トリエチルアンモニウム、pH7:MeOH、85:15]を流速5ml/分で使用して、ジアステレオ異性体の混合物から分離した際に、5.57分の保持時間(Rt)を有する異性体であることを特徴とする(化合物4B)、請求項10に記載の化合物。
【請求項12】
Xがアデニン残基であり、ここで、R1がSMeであり、R2及びR3がHであり、YがOHであり、nが1であり、Z1がBH3−であり、Z2及びZ3がO−であり、W1がOであり、W2がCCl2又はCF2である(それぞれ化合物21及び22)、請求項5に記載の化合物。
【請求項13】
Xがアデニン残基であり、ここで、R1がSMeであり、R2及びR3がHであり、YがOHであり、nが0であり、Z1及びZ3がO−であり、W2がCCl2又はCF2である(それぞれ化合物17及び18)、請求項7に記載の化合物。
【請求項14】
Xがアデニン残基であり、ここで、R1がSMeであり、R2及びR3がHであり、YがOHであり、nが0であり、Z1がBH3−であり、Z3がO−であり、W2がOである(化合物19)、請求項7に記載の化合物。
【請求項15】
半分取逆相Gemini 5uカラム(C−18 110A、250×10mm、5ミクロン)及び定組成溶離[100mMの酢酸トリエチルアンモニウム、pH7:アセトニトリル、88:12]を流速1ml/分で使用して、ジアステレオ異性体の混合物から分離した際に、8.073分の保持時間(Rt)を有する異性体であることを特徴とする(化合物19A)、請求項14に記載の化合物。
【請求項16】
Xがウラシル残基であり、ここで、R6が、H、ハロゲン、O−ヒドロカルビル又はS−ヒドロカルビルであり、R7がO又はSであり、YがOHであり、nが1であり、Z1がBH3−であり、Z2及びZ3がO−であり、W1がOであり、W2が、CH2、CF2又はCCl2である、請求項1に記載の化合物。
【請求項17】
Xがウラシル残基であり、ここで、R6がH又はNR8R9であり、R8及びR9が、それぞれ独立に、H又はヒドロカルビルであるか、或いはR8及びR9が、それらが結合した窒素原子と一緒になって、酸素、窒素又は硫黄から選択される1〜2個のさらなるヘテロ原子を場合によって含有する飽和又は不飽和複素環を形成し、R7がO又はSであり、YがOHであり、nが1であり、Z1がBH3−であり、Z2及びZ3がO−であり、W1がOであり、W2が、CH2、CF2又はCCl2である、請求項1に記載の化合物。
【請求項18】
Xがウラシル残基であり、ここで、R6が、H、ハロゲン、O−ヒドロカルビル又はS−ヒドロカルビルであり、R7がO又はSであり、YがOHであり、nが0であり、Z1及びZ3がO−であり、W2がCF2又はCCl2である、請求項1に記載の化合物。
【請求項19】
Xがウラシル残基であり、ここで、R6がH又はNR8R9であり、R8及びR9が、それぞれ独立に、H又はヒドロカルビルであるか、或いはR8及びR9が、それらが結合した窒素原子と一緒になって、酸素、窒素又は硫黄から選択される1〜2個のさらなるヘテロ原子を場合によって含有する飽和又は不飽和複素環を形成し、R7がO又はSであり、YがOHであり、nが0であり、Z1及びZ3がO−であり、W2がCF2又はCCl2である、請求項1に記載の化合物。
【請求項20】
Bが、アルカリ金属のカチオン、NH4+、式R4N+の有機カチオン[式中、Rの各1個は、独立に、H又はC1〜C22、好ましくはC1〜C6アルキルである]、カチオン性脂質又はカチオン性脂質の混合物である、請求項1に記載の化合物。
【請求項21】
請求項1に記載の一般式Iの化合物又は薬学的に許容されるその塩、及び薬学的に許容される担体又は希釈剤を含む医薬組成物。
【請求項22】
請求項1に記載の一般式Iの化合物又は薬学的に許容されるその塩、及び薬学的に許容される担体又は希釈剤を含む、P2Y受容体によって調節される疾患、障害又は状態の治療用の医薬組成物。
【請求項23】
前記P2Y受容体によって調節される疾患又は障害が2型糖尿病である、請求項22に記載の医薬組成物。
【請求項24】
疼痛管理用である、請求項22に記載の医薬組成物。
【請求項25】
化合物4B、17、18、19A、21A若しくは21Bから選択される化合物又は一般式Iの化合物[式中、Xはアデニン残基であり、ここで、R1はSMeであり、R2及びR3はHであり、YはOHであり、nは1であり、Z1〜Z3はO−であり、W1はOであり、W2はCH2である](化合物2)を含む、請求項22から24までのいずれか一項に記載の医薬組成物。
【請求項26】
P2Y受容体によって調節される疾患、障害又は状態の治療用の医薬組成物の調製のための、請求項1に記載の一般式Iの化合物又は薬学的に許容されるその塩の使用。
【請求項27】
P2Y受容体によって調節される疾患、障害又は状態の治療用の、請求項1に記載の一般式Iの化合物又は薬学的に許容されるその塩。
【請求項28】
必要とする個体における、P2Y受容体によって調節される疾患、障害又は状態の治療方法であって、前記個体に、有効量の請求項1に記載の一般式Iの化合物又は薬学的に許容されるその塩を投与するステップを含む方法。
【請求項29】
前記P2Y受容体によって調節される疾患又は障害が2型糖尿病である、請求項28に記載の方法。
【請求項30】
必要とする個体における疼痛を管理するための、請求項28に記載の方法。
【請求項1】
一般式Iの化合物:
【化1】
[式中、
Xは、9位を介して結合した式Iaのアデニン残基:
【化2】
であり、ここで、
R1は、H、ハロゲン、O−ヒドロカルビル、S−ヒドロカルビル、NR4R5、ヘテロアリール、非置換ヒドロカルビル、又はハロゲン、CN、SCN、NO2、OR4、SR4、NR4R5若しくはヘテロアリールで置換されているヒドロカルビルであり、ここで、R4及びR5は、それぞれ独立に、H又はヒドロカルビルであるか、或いはR4及びR5は、それらが結合した窒素原子と一緒になって、酸素、窒素又は硫黄から選択される1〜2個のさらなるヘテロ原子を場合によって含有する5又は6員の飽和又は不飽和複素環を形成し、前記追加の窒素は、置換されていないか、又はハロゲン、ヒドロキシル若しくはフェニルによって置換されているアルキルで置換されており、
R2及びR3は、それぞれ独立に、H又はヒドロカルビルであるか、
或いは、Xは、1位を介して結合した式Ibのウラシル残基:
【化3】
であり、ここで、
R6は、H、ハロゲン、O−ヒドロカルビル、S−ヒドロカルビル、NR8R9、ヘテロアリール、非置換ヒドロカルビル、又はハロゲン、CN、SCN、NO2、OR8、SR8、NR8R9若しくはヘテロアリールで置換されているヒドロカルビルであり、ここで、R8及びR9は、それぞれ独立に、H又はヒドロカルビルであるか、或いはR8及びR9は、それらが結合した窒素原子と一緒になって、酸素、窒素又は硫黄から選択される1〜2個のさらなるヘテロ原子を場合によって含有する5又は6員の飽和又は不飽和複素環を形成し、前記追加の窒素は、置換されていないか、又はハロゲン、ヒドロキシル若しくはフェニルによって置換されているアルキルで置換されており、
R7はO又はSであり、
Yは、H、OH又はNH2であり、
Z1、Z2及びZ3は、それぞれ独立に、O−又はBH3−であり、
W1及びW2は、それぞれ独立に、O、CH2、C(Hal)2又はNHであり、ここで、Halは、ハロゲン、好ましくはF又はClであり、
nは0又は1であり、但し、nが0であり、W2がOである場合、Z1はBH3−であり、nが1である場合、W1及びW2の少なくとも一方はOではなく、
mは3又は4であり、
B+は、薬学的に許容されるカチオンを表す]
(nが0であり、Z1及びZ3がそれぞれO−であり、W2がCH2又はNHである化合物、並びにnが1であり、Z1〜Z3がそれぞれO−である化合物を除く)
並びにそのジアステレオ異性体。
【請求項2】
nが0であり、Z1及びZ3がOであるか、又はnが0であり、Z1及びZ3の少なくとも一方がBH3−であるか、又はnが1であり、Z1〜Z3の少なくとも1つがBH3−である、請求項1に記載の化合物。
【請求項3】
nが0であり、唯1個のボラノ基を位置αに含む、すなわち、Z1がBH3−であり、Z2がO−である;若しくは位置βに含む、すなわち、Z3がBH3−であり、Z1がO−である;又は2個のボラノ基を位置α、βに含む、すなわち、Z1及びZ3がBH3−である、請求項2に記載の化合物。
【請求項4】
nが1であり、唯1個のボラノ基を位置αに含む、すなわち、Z1がBH3−であり、Z2及びZ3がO−である;位置βに含む、すなわち、Z2がBH3−であり、Z1及びZ3がO−である;若しくは位置γに含む、すなわち、Z3がBH3−であり、Z1及びZ2がO−である;2個のボラノ基を位置α及びβに含む、すなわち、Z1及びZ2がBH3−であり、Z3がO−である;位置α及びγに含む、すなわち、Z1及びZ3がBH3−であり、Z2がO−である;若しくは位置β及びγに含む、すなわち、Z2及びZ3がBH3−であり、Z1がO−である;又は3個のボラノ基を位置α、β及びγに含む、すなわち、Z1〜Z3がBH3−である、請求項2に記載の化合物。
【請求項5】
Xがアデニン残基であり、ここで、R1が、H、ハロゲン、O−ヒドロカルビル又はS−ヒドロカルビルであり、R2及びR3が、それぞれ独立に、H又はヒドロカルビルであり、YがOHであり、nが1であり、Z1がBH3−であり、Z2及びZ3がO−であり、W1がOであり、W2が、CH2、CF2又はCCl2である、請求項1に記載の化合物。
【請求項6】
Xがアデニン残基であり、ここで、R1がH又はNR4R5であり、R4及びR5が、それぞれ独立に、H又はヒドロカルビルであるか、或いはR4及びR5が、それらが結合した窒素原子と一緒になって、酸素、窒素又は硫黄から選択される1〜2個のさらなるヘテロ原子を場合によって含有する飽和又は不飽和複素環を形成し、R2及びR3が、それぞれ独立に、H又はヒドロカルビルであり、YがOHであり、nが1であり、Z1がBH3−であり、Z2及びZ3がO−であり、W1がOであり、W2が、CH2、CF2又はCCl2である、請求項1に記載の化合物。
【請求項7】
Xがアデニン残基であり、ここで、R1が、H、ハロゲン、O−ヒドロカルビル又はS−ヒドロカルビルであり、R2及びR3が、それぞれ独立に、H又はヒドロカルビルであり、YがOHであり、nが0であり、(i)Z1及びZ3がO−であり、W2がCF2又はCCl2であるか、或いは(ii)Z1がBH3−であり、W2がOである、請求項1に記載の化合物。
【請求項8】
Xがアデニン残基であり、ここで、R1がH又はNR4R5であり、R4及びR5が、それぞれ独立に、H又はヒドロカルビルであるか、或いはR4及びR5が、それらが結合した窒素原子と一緒になって、酸素、窒素又は硫黄から選択される1〜2個のさらなるヘテロ原子を場合によって含有する飽和又は不飽和複素環を形成し、R2及びR3が、それぞれ独立に、H又はヒドロカルビルであり、YがOHであり、nが0であり、Z1及びZ3がO−であり、W2がCF2又はCCl2である、請求項1に記載の化合物。
【請求項9】
Xがアデニン残基であり、ここで、R1がHであり、R2及びR3がHであり、YがOHであり、nが1であり、Z1がBH3−であり、Z2及びZ3がO−であり、W1がOであり、W2がCH2である(化合物3)、請求項5に記載の化合物。
【請求項10】
Xがアデニン残基であり、ここで、R1がSMeであり、R2及びR3がHであり、YがOHであり、nが1であり、Z1がBH3−であり、Z2及びZ3がO−であり、W1がOであり、W2がCH2である(化合物4)、請求項5に記載の化合物。
【請求項11】
半分取逆相Gemini 5uカラム(C−18 110A、250×10mm、5ミクロン)及び定組成溶離[100mMの酢酸トリエチルアンモニウム、pH7:MeOH、85:15]を流速5ml/分で使用して、ジアステレオ異性体の混合物から分離した際に、5.57分の保持時間(Rt)を有する異性体であることを特徴とする(化合物4B)、請求項10に記載の化合物。
【請求項12】
Xがアデニン残基であり、ここで、R1がSMeであり、R2及びR3がHであり、YがOHであり、nが1であり、Z1がBH3−であり、Z2及びZ3がO−であり、W1がOであり、W2がCCl2又はCF2である(それぞれ化合物21及び22)、請求項5に記載の化合物。
【請求項13】
Xがアデニン残基であり、ここで、R1がSMeであり、R2及びR3がHであり、YがOHであり、nが0であり、Z1及びZ3がO−であり、W2がCCl2又はCF2である(それぞれ化合物17及び18)、請求項7に記載の化合物。
【請求項14】
Xがアデニン残基であり、ここで、R1がSMeであり、R2及びR3がHであり、YがOHであり、nが0であり、Z1がBH3−であり、Z3がO−であり、W2がOである(化合物19)、請求項7に記載の化合物。
【請求項15】
半分取逆相Gemini 5uカラム(C−18 110A、250×10mm、5ミクロン)及び定組成溶離[100mMの酢酸トリエチルアンモニウム、pH7:アセトニトリル、88:12]を流速1ml/分で使用して、ジアステレオ異性体の混合物から分離した際に、8.073分の保持時間(Rt)を有する異性体であることを特徴とする(化合物19A)、請求項14に記載の化合物。
【請求項16】
Xがウラシル残基であり、ここで、R6が、H、ハロゲン、O−ヒドロカルビル又はS−ヒドロカルビルであり、R7がO又はSであり、YがOHであり、nが1であり、Z1がBH3−であり、Z2及びZ3がO−であり、W1がOであり、W2が、CH2、CF2又はCCl2である、請求項1に記載の化合物。
【請求項17】
Xがウラシル残基であり、ここで、R6がH又はNR8R9であり、R8及びR9が、それぞれ独立に、H又はヒドロカルビルであるか、或いはR8及びR9が、それらが結合した窒素原子と一緒になって、酸素、窒素又は硫黄から選択される1〜2個のさらなるヘテロ原子を場合によって含有する飽和又は不飽和複素環を形成し、R7がO又はSであり、YがOHであり、nが1であり、Z1がBH3−であり、Z2及びZ3がO−であり、W1がOであり、W2が、CH2、CF2又はCCl2である、請求項1に記載の化合物。
【請求項18】
Xがウラシル残基であり、ここで、R6が、H、ハロゲン、O−ヒドロカルビル又はS−ヒドロカルビルであり、R7がO又はSであり、YがOHであり、nが0であり、Z1及びZ3がO−であり、W2がCF2又はCCl2である、請求項1に記載の化合物。
【請求項19】
Xがウラシル残基であり、ここで、R6がH又はNR8R9であり、R8及びR9が、それぞれ独立に、H又はヒドロカルビルであるか、或いはR8及びR9が、それらが結合した窒素原子と一緒になって、酸素、窒素又は硫黄から選択される1〜2個のさらなるヘテロ原子を場合によって含有する飽和又は不飽和複素環を形成し、R7がO又はSであり、YがOHであり、nが0であり、Z1及びZ3がO−であり、W2がCF2又はCCl2である、請求項1に記載の化合物。
【請求項20】
Bが、アルカリ金属のカチオン、NH4+、式R4N+の有機カチオン[式中、Rの各1個は、独立に、H又はC1〜C22、好ましくはC1〜C6アルキルである]、カチオン性脂質又はカチオン性脂質の混合物である、請求項1に記載の化合物。
【請求項21】
請求項1に記載の一般式Iの化合物又は薬学的に許容されるその塩、及び薬学的に許容される担体又は希釈剤を含む医薬組成物。
【請求項22】
請求項1に記載の一般式Iの化合物又は薬学的に許容されるその塩、及び薬学的に許容される担体又は希釈剤を含む、P2Y受容体によって調節される疾患、障害又は状態の治療用の医薬組成物。
【請求項23】
前記P2Y受容体によって調節される疾患又は障害が2型糖尿病である、請求項22に記載の医薬組成物。
【請求項24】
疼痛管理用である、請求項22に記載の医薬組成物。
【請求項25】
化合物4B、17、18、19A、21A若しくは21Bから選択される化合物又は一般式Iの化合物[式中、Xはアデニン残基であり、ここで、R1はSMeであり、R2及びR3はHであり、YはOHであり、nは1であり、Z1〜Z3はO−であり、W1はOであり、W2はCH2である](化合物2)を含む、請求項22から24までのいずれか一項に記載の医薬組成物。
【請求項26】
P2Y受容体によって調節される疾患、障害又は状態の治療用の医薬組成物の調製のための、請求項1に記載の一般式Iの化合物又は薬学的に許容されるその塩の使用。
【請求項27】
P2Y受容体によって調節される疾患、障害又は状態の治療用の、請求項1に記載の一般式Iの化合物又は薬学的に許容されるその塩。
【請求項28】
必要とする個体における、P2Y受容体によって調節される疾患、障害又は状態の治療方法であって、前記個体に、有効量の請求項1に記載の一般式Iの化合物又は薬学的に許容されるその塩を投与するステップを含む方法。
【請求項29】
前記P2Y受容体によって調節される疾患又は障害が2型糖尿病である、請求項28に記載の方法。
【請求項30】
必要とする個体における疼痛を管理するための、請求項28に記載の方法。
【図1】

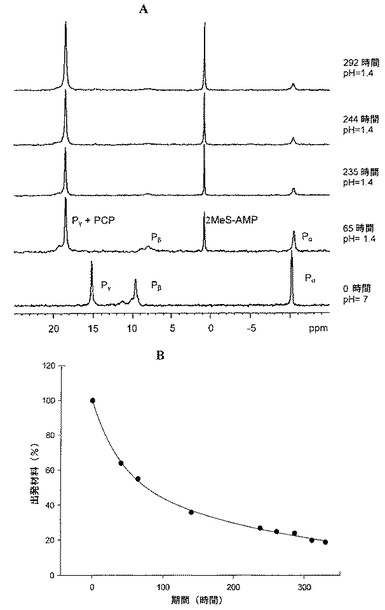
【図2−1】
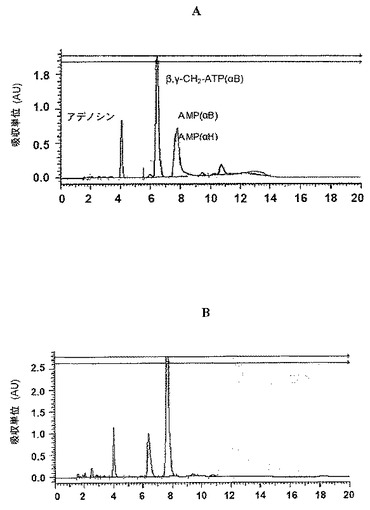
【図2−2】

【図3】
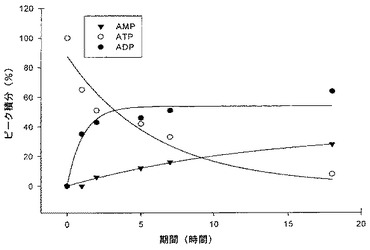
【図4−1】
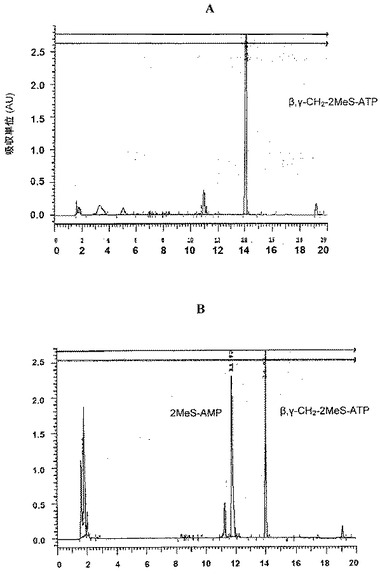
【図4−2】
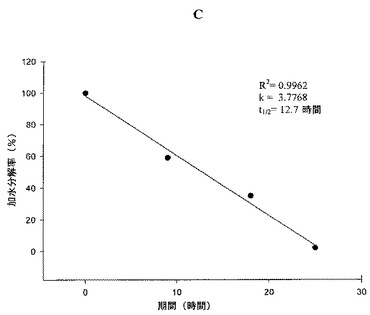
【図5】
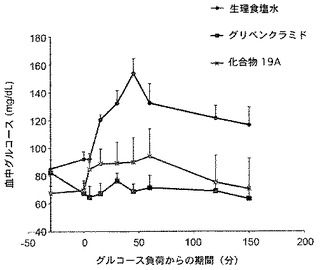
【図2−1】
【図2−2】
【図3】
【図4−1】
【図4−2】
【図5】
【公表番号】特表2011−504489(P2011−504489A)
【公表日】平成23年2月10日(2011.2.10)
【国際特許分類】
【出願番号】特願2010−534597(P2010−534597)
【出願日】平成20年11月23日(2008.11.23)
【国際出願番号】PCT/IL2008/001535
【国際公開番号】WO2009/066298
【国際公開日】平成21年5月28日(2009.5.28)
【出願人】(504164206)バーイラン ユニバーシティー (7)
【Fターム(参考)】
【公表日】平成23年2月10日(2011.2.10)
【国際特許分類】
【出願日】平成20年11月23日(2008.11.23)
【国際出願番号】PCT/IL2008/001535
【国際公開番号】WO2009/066298
【国際公開日】平成21年5月28日(2009.5.28)
【出願人】(504164206)バーイラン ユニバーシティー (7)
【Fターム(参考)】
[ Back to top ]