
非対称ナノチューブ形成性非対称双頭型脂質分子、同脂質分子により形成された非対称ナノチューブ、および同非対称ナノチューブを用いた薬剤カプセル化物
【課題】入手しやすい原料から簡便に合成でき、かつマイルドな条件で非対称ナノチューブを選択的かつ高収率に製造できる新規な非対称双頭型脂質分子及び当該脂質分子により形成される長期保存安定性の高い非対称ナノチューブの提供。また、当該非対称ナノチューブにドキソルビシンなどのカチオン性薬剤を選択的、高濃度にカプセル化したカチオン性薬剤−非対称ナノチューブ複合体を有効成分として用いた、刺激応答性徐放機能の高いカプセル化薬剤組成物の提供。
【解決手段】下記一般式(1)で表される新規な非対称ナノチューブ形成能を有する非対称双頭型脂質分子又はその塩。

(式中、nは8〜16の整数であり、mは1〜4の整数を表す。また、式中の波線で表記した2−グルコサミンの1位水酸基はα、βアノマーのいずれか、あるいはその平衡混合物を表す。)。
【解決手段】下記一般式(1)で表される新規な非対称ナノチューブ形成能を有する非対称双頭型脂質分子又はその塩。
(式中、nは8〜16の整数であり、mは1〜4の整数を表す。また、式中の波線で表記した2−グルコサミンの1位水酸基はα、βアノマーのいずれか、あるいはその平衡混合物を表す。)。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、薬剤のカプセル化に好適な中空繊維状の非対称ナノチューブ構造を形成可能な非対称双頭型脂質分子とその製造方法に関し、さらに前記脂質分子が自己集合により形成する中空繊維状有機ナノチューブ、および当該ナノチューブを用いた薬剤カプセル化への用途に関する。
【背景技術】
【0002】
ある種の脂質分子等の有機分子は、その溶媒中での自己集合によって中空繊維状構造(有機ナノチューブとよぶ)を形成することが知られている(非特許文献1、2)。これらは、外径約10nm〜1000nm、長さ約100nm〜数mmのチューブ構造を形成し、その内部に微少な中空空間を有するため、種々の材料(タンパク質、薬剤、DNA等)をカプセル化することができる。この有機ナノチューブは、水素結合やファンデルワールス力などの弱い相互作用による自己集合作用で形成されているため、構成分子の構造に依存して室温から100℃の範囲のある温度やpH等の固有の条件によって可逆的にチューブ構造を分解することが可能である。この温度やpHは生体中の環境温度やpHの範囲を含むものであるから、生体内でこれを分解するように調節可能であることを意味する。またチューブの分解がなくても、内部にカプセル化された材料がチューブ両端の開口部より徐々に放出される特性も有する(非特許文献3、4)。
これらの諸特性を用いることで、薬剤送達用および薬剤徐放用のカプセルやタンパク質などの固定マトリクスおよび保存カプセル等への応用が期待されている(非特許文献5、6)。しかし、従来の大多数の有機ナノチューブは、内外表面が同じ親水部で被覆されているため、薬剤やタンパク質などの材料を有機ナノチューブ内部へ選択的かつ効率的にカプセル化することは困難であった(非特許文献7,8、9)。具体的には、諸材料のカプセル化に際し、有機ナノチューブ内部の水を減圧下での凍結乾燥などで除去した後、材料を分散・溶解させた水溶液を添加し再水和させる(いわゆる毛細管現象)という煩雑な操作が必要とされていた(非特許文献7、8)。
【0003】
本発明者らは、従来から容易かつ効率的な薬剤のカプセル化が可能な有機ナノチューブとして、内外表面がそれぞれ異なる官能基で被覆されているという特徴を持つ様々な有機ナノチューブ(以降、非対称ナノチューブという)の研究開発を行ってきた(非特許文献10、11、12、13)。これら非対称ナノチューブは、いずれも疎水部アルキレン鎖の両端に異なる二つの親水部、具体的には、片端に1−グルコピラノシルアミン、もう一端にカルボキシル基又はアミノ基がそれぞれ連結された非対称な双頭型の脂質分子(以降、「非対称双頭型脂質」とよぶ)により形成されている(特許文献1)。これらの非対称ナノチューブは、通常室温で混合するだけでナノチューブ内表面とは逆の電荷を有する材料をカプセル化することができる。片端がカルボキシル基のカルボン酸系脂質分子を用いた場合はチューブ内表面が選択的にカルボキシル基で被覆されることにより負に荷電し、反対にアミノ基を有しているアミン系脂質分子を用いた場合は正に荷電するため、前者は正電荷を帯びた薬剤を包接し輸送する薬剤送達カプセル又は薬剤キャリアとして好ましく、後者は負電荷を帯びた薬剤輸送用の薬剤キャリアの他、細胞内への遺伝子輸送など核酸キャリアとしての用途も期待される。
本発明者らは、これまでにこれらの非対称ナノチューブを用いて、タンパク質、蛍光分子、オリゴDNAなどの選択的なカプセル化やin vitroでの放出挙動などの研究を報告している(非特許文献10、11、12、13)。このような様々な非対称ナノチューブが提供され、これら非対称ナノチューブによるカプセル化によって、生体高分子や薬剤などの不安定な材料でも、簡便かつ選択的、効率的なカプセル化が可能となってきた。
しかしながら、これらの非対称ナノチューブのカプセル用材料としての実用化には、非対称ナノチューブの水和状態での安定性向上や封入性能の向上など解決すべき問題点があり、さらに、非対称ナノチューブ製造が困難な場合や、チューブの原料となる非対称双頭型脂質分子の合成工程が煩雑な場合や精製が困難な場合があるなどの問題点があった。
すなわち、実用的な薬剤カプセル化用非対称ナノチューブとしては、以下の各点を全て満たす非対称ナノチューブの開発が必須である。
1.原料となる非対称双頭型脂質分子の合成・精製工程が簡便であること。
2.非対称ナノチューブの製造が、室温で、又は少なくとも温和な条件で、かつ選択的に行うことができること。
3.非対称ナノチューブが水和状態での長期保存安定性を有すること。
4.包接対象化合物を効率的に高濃度でカプセル化でき、かつ簡単に放出可能であること。
これまでには、部分的にこれらの問題点を解決できているものがあっても、これら全ての点を解決できた非対称ナノチューブの報告は、カルボン酸系脂質分子でもアミン系脂質分子を用いた場合でも見られなかった。
【0004】
一方で、従来は薬剤カプセルとして主にリポソーム系の薬剤カプセルが用いられていたが、カプセル化にpH勾配を用い、その後の精製が必要なため、調製が複雑で長時間必要であった。そこで、簡単に、効率的に薬剤をカプセル化でき、薬剤を安定に保持すると共に、癌細胞などターゲット細胞内では速やかに徐放できるカプセル化剤の開発が望まれていた。特に、ドキソルビシン、イダルビシン、エピルビシン、ダウノルビシン、ピラルビシンなどのアントラサイクリン系抗癌剤は、毒性、特に心臓毒性が強いため、癌細胞に直接効果的に送達し、かつ徐放が可能な優れたカプセル化剤が求められていた。
上述のように、カルボン酸系脂質分子により形成される非対称ナノチューブは、糖親水部により外表面が覆われ、内表面はカルボキシル基により構成されて負の電荷を有するため、ドキソルビシンなどのアミノ基を有する抗癌剤送達のための有効なカプセル剤として期待される。さらにこの様なナノチューブは、pHなどの外部環境によって内表面のカルボキシル基がイオン的/非イオン的な変化又はチューブの形態変化による機能的な薬剤の放出も期待できる。
しかし、本発明者らの報告しているカルボン酸系の非対称双頭型脂質分子である1−グルコピラノシルアミノ基が長鎖ジカルボン酸の一端でアミド結合を介して結合した脂質分子が形成する非対称ナノチューブ(非特許文献10)では、非対称ナノチューブの製造過程でマイクロチューブやテープ状の集合体も同時に生成するため、遠心分離などによるナノチューブの分離精製工程が必要であり、得られたナノチューブが水性溶媒中での分散性が悪く、また室温や冷蔵でも長期保存により結晶化しやすく長期的安定性が乏しい欠点があった。なお、同じ脂質分子を用い、製造過程で低分子有機化合物をインターカレートさせた非対称ナノチューブ(特許文献3)についても同様の長期的安定性が乏しい欠点がある。
また、カルボキシル基を親水部として有する脂質分子にオリゴグリシン残基を導入することで、得られる非対称ナノチューブの水分散性を高めることができ、水性溶媒中での長期保存性も高まった(非特許文献14)が、同脂質からの非対称ナノチューブの製造には、脂質分子を、ミリQ水(超純水)に分散して100℃前後に加熱するか、あるいは脂質を等量の水酸化ナトリウムを含む水溶液に60℃以上に加熱して溶解し、その後、室温で中和する工程が必要であるという煩雑で高度な技術を要するばかりか、ナノチューブ原料の脂質分子の合成に7段階以上の合成過程と過程毎のカラムクロマトグラフィー等による精製が必要なため、大量に合成することは極めて困難であり、実用的ではない。
簡便な非対称双頭型脂質分子の合成を目指す試みの1つとして、グリシン残基を含まないカルボン酸系非対称双頭型脂質分子において、他端のグルコピラノシルアミン残基として2−グルコサミンを用いたナノチューブを作製した(非特許文献15)。その脂質分子自体の合成はきわめて簡便で、2−グルコサミンと長鎖ジカルボン酸という市販の化合物からの1段階合成と再結晶で合成可能であったが、自己集合させても分散性に乏しく、ナノチューブとその断片状のものの混合物しか得られず、ナノチューブの詳細な解析結果からみて、内外表面を被覆する官能基が同じで非対称ナノチューブが形成できていない(未発表データ)。
また、1−グルコピラノシル基を親水部として有するカルボン酸系脂質分子の疎水部に重合性官能基であるジアセチレン残基を導入した場合も、ナノチューブは形成する(特許文献2)が、このナノチューブも詳細な構造解析によると非対称ナノチューブが形成されていなかった(未発表データ)。
【0005】
以上の技術的な背景から、上記の全ての課題を解決できる内表面がカルボキシル基で被覆された非対称有機ナノチューブ及びそのための新規な非対称双頭型脂質分子の提供が強く期待されていた。すなわち、入手しやすい原料から簡便に合成できる非対称双頭型脂質分子であって、マイルドな条件で非対称ナノチューブを、選択的かつ高収率に製造できて、得られる非対称ナノチューブが長期保存安定性を有すると共に、ドキソルビシンなどのカチオン性薬剤を効率的に高濃度でカプセル化でき、しかもpH条件変化などで簡単に放出可能なカプセル化物を製造できる非対称双頭型脂質分子の提供が切望されていた。
【先行技術文献】
【特許文献】
【0006】
【特許文献1】特許第4174702号公報
【特許文献2】特開2005−239632
【特許文献3】特開2008−264897
【非特許文献】
【0007】
【非特許文献1】Toshimi Shimizu, Mitsutoshi Masuda, Hiroyuki Minamikawa,Chemical Review,2004,105,1401-1443.
【非特許文献2】“有機・無機・金属ナノチューブ 非カーボンナノチューブ系の最新技術と応用展開”、清水敏美、木島 剛 編集、フロンティア出版、東京、2008.
【非特許文献3】増田光俊、亀田直弘、ネットワークポリマー、2010,31,191-200.
【非特許文献4】Naohiro Kameta、Hiroyuki Minamikawa,Mitsutoshi Masuda,Go Mizuno,Toshimi Shimizu,Soft Matter,2008,4,1681-1687.
【非特許文献5】Chia-Chun Chen,Yao-Chung Liu,Chia-Hsuan Wu,Chun-Chia Yeh,Ming-Tsan Su,Yi-Chun Wu,Advanced Materials,2005,17,404-407.
【非特許文献6】Mohammand Reza Abidian,Dong-Hwan Kim,David C Martin,Advanced Materials,2006,18,405―409.
【非特許文献7】Bo Yang,Shoko Kamiya,Yoshiki Shimizu,Naoto Koshizaki,Toshimi Shimizu,Chemistry of Materials,2004,16、2826-2831.
【非特許文献8】Hiroharu Yui,Yoshiki Shimizu,Shoko Kamiya,Ichiro Yamashita,Mitsutoshi Masuda,Kohzo Ito,Toshimi Shimizu,Chemistry Letters,2005,34,232-233.
【非特許文献9】“環状・筒状超分子新素材の応用技術”、高田十志和 編集、シーエムシー出版、東京、2006,P138-149.
【非特許文献10】Mitsutoshi Masuda,Toshimi Shimizu,Langmuir,2004,20,5969-5977.
【非特許文献11】Naohiro Kameta,Mitsutoshi Masuda,Hiroyuki Minamikawa,Nikolay V.Gotev, Jeong A.Rim,Jong H.Jung, Toshimi Shimizu,Advanced Materials,2005,17,2732-2736.
【非特許文献12】Naohiro Kameta,Go Mizuno,Mitsutosi Masuda, Hiroyuki Minamikawa, Masaki Kogiso, Toshimi Shimizu,Chemistry Letters,2007,36,896-897.
【非特許文献13】Naohiro Kameta,Kaname Yoshida,Mitsutoshi Masuda,Toshimi Shimizu,Chemistry of Materials,2009,21,5892-5898.
【非特許文献14】Soo Jing Lee,Naohiro Kameta, Hiroyuki Minamikawa,Mitsutoshi Masuda,Toshimi Shimizu,第58回高分子討論会予稿集,発表番号1Pe047、“Function of Metal Cation-Doped Nanotube by using Unsymmetrical Bolaamphiphiles”.
【非特許文献15】増田光俊、和田百代、清水敏美、第59回高分子年次大会、発表番号2Pd002、「アノマー混合物からなるグルコサミン系双頭型脂質の有機ナノチューブ形成」.
【非特許文献16】Naohiro Kameta,Mitsutoshi Masuda,Hiroyuki Minamikawa,Toshimi Shimizu,Langmuir,2007,23,4634-4641.
【非特許文献17】George Fotakis and John A.Timbrell,Toxocology Letters,2006,160,171-177.
【発明の開示】
【発明が解決しようとする課題】
【0008】
本発明は、入手しやすい原料から簡便に合成でき、かつマイルドな条件で非対称ナノチューブを選択的かつ高収率に製造できる非対称双頭型脂質分子を提供しようとするものであり、また、当該非対称双頭型脂質分子の自己集合で得られる非対称ナノチューブとして、高純度かつ比較的高濃度の長期保存でも安定性を有すると共に、ドキソルビシンなどのアミノ基を有する薬剤を選択的かつ効率的に高濃度でカプセル化でき、かつpH条件変化などで簡単に放出可能なカプセル化物を製造できる非対称ナノチューブを提供することを目的とする。そして、当該非対称ナノチューブにより、ドキソルビシンなどのカチオン性薬剤が選択的、高濃度にカプセル化された、刺激応答性徐放機能の高いカプセル化薬剤組成物を提供することを目的とするものである。
【課題を解決するための手段】
【0009】
本発明者らは上記の課題を解決すべく鋭意研究を重ねた結果、本発明者らが以前に開発した、脂質分子自体の合成はきわめて簡便であったが、非対称有機ナノチューブを形成できない、片側に2−グルコサミンのアミノ基と結合させた長鎖ジカルボン酸からなる非対称双頭型脂質分子(非特許文献15)の誘導体を製造することに思い至った。具体的には、当該非対称双頭型脂質分子の末端のカルボキシル基に対して、さらにオリゴグリシンを脱水縮合反応により連結して、下記一般式(1)であらわされる脂質分子(化合物−1)を合成したところ、精製工程も不要で穏和な条件で簡単に製造できたばかりでなく、得られた脂質分子は水分散性もよく、非対称ナノチューブのみを高選択的に形成し、さらに得られた非対称ナノチューブは、ミリQ水中、比較的高濃度である5mg/ml程度での、室温あるいは冷蔵下での6ヶ月を超える長期間の分散保存でも安定にチューブ形態を保持したまま分散状態を保持できることを見いだした。本発明で得た脂質からのナノチューブの製造が、ミリQ水からの加熱・冷却のみならず、中和によって完全に室温で達成できたことは驚くべきことである。すなわち本脂質は等モル等量の水酸化ナトリウム水溶液に室温で速やかに溶解し、この溶液を室温で塩酸等によって中和することで非対称ナノチューブを製造可能であった。さらに、脂質分子の合成も市販の原料から数えても僅か3段階の反応で完了し、全ての精製行程において反応の進行で生じる固体のろ過と、得られた固体の洗浄で十分な純度の脂質を与えることができた点もきわめて驚くべき特徴である。これらの優れた非対称ナノチューブとしての特性は、従来のどのカルボン酸系非対称ナノチューブにおいても達成できなかったばかりか、アミン系非対称ナノチューブなども含めた非対称有機ナノチューブ全てにおいてもはじめてである。
そして、これにより得た非対称ナノチューブはドキソルビシンを効率的にカプセル化する薬剤カプセルとしても機能することを確認し、本発明を完成させた。
【0010】
すなわち、本発明は以下の一般式(1)で表される非対称双頭型脂質分子(化合物−1ともいう。)又はその塩
(式中、nは8〜16の整数を表し、mは1〜4の整数を表し、波線で表記した2−グルコサミンの1位水酸基はα、βアノマーのいずれか、あるいはその平衡混合物を表す。)
とその合成方法、および室温下での同脂質のナトリウム塩からの中和によることを特徴とする自己集合によって製造される、高純度で、長期安定性を有する非対称ナノチューブを提供するものであり、上記脂質分子から得られた非対称ナノチューブとアミノ基を有する薬剤又はカチオン性薬剤のカプセル化物を提供するものである。
【0011】
具体的には、本発明は以下の通りである。
〔1〕 下記一般式(1)で表される非対称双頭型脂質分子又はその塩;
(式中、nは8〜16の整数であり、mは1〜4の整数を表す。また、式中の波線で表記した2−グルコサミンの1位水酸基はα、βアノマーのいずれか、あるいはその平衡混合物を表す。)。
〔2〕 下記一般式(2)で表されるα,ω−長鎖ジカルボン酸
HOOC−(CH2)n−COOH 一般式(2)
(式中、nは8〜16の整数を表す。)
を水性メタノール中で、2−グルコサミンと縮合させ、生じた固体をろ過、洗浄することで、下記一般式(3)で表される中間体−3
(式中nは8〜16の整数を表す。また、式中の波線で表記した2−グルコサミンの1位水酸基はα、βアノマーのいずれか、あるいはその平衡混合物を表す。)
を製造し、
当該中間体−3と、オリゴグリシンのC端をエステルなどで保護した下記一般式(4)で表されるオリゴグリシンエステル
(式中mは1〜4の整数を表す。またRはアルコール性水酸基を有する化合物の残基であり、例えば、メチル基、エチル基、三級ブチル基、ベンジル基である。)
を加えて縮合させ、下記一般式(5)で表される中間体−5
(式中nは8〜16の整数を表し、mは1〜4の整数を表す。またRはアルコール性水酸基を有する化合物の残基であり、例えば、メチル基、エチル基、三級ブチル基、ベンジル基である。また式中の波線で表記した2−グルコサミンの1位水酸基はα、βアノマーのいずれか、あるいはその平衡混合物を表す。)
を得、これをアルカリ加水分解することで脱保護し、下記一般式(1)で表される非対称双頭型脂質分子又はその塩
(式中、nは8〜16の整数であり、mは1〜4の整数を表す。また、式中の波線で表記した2−グルコサミンの1位水酸基はα、βアノマーのいずれか、あるいはその平衡混合物を表す。)
を製造する方法。
〔3〕 前記〔1〕に記載の非対称双頭型脂質分子又はその塩から形成された、内表面がカルボキシル基で被覆され、外表面が2−グルコサミド基で被覆された構造を有することを特徴とする非対称ナノチューブ。
〔4〕 前記〔1〕に記載の非対称双頭型脂質分子を水に分散し、加熱溶解後冷却するか、又は室温で水又はアルカリ性溶液に分散溶解後、酸で中和することを特徴とする、非対称ナノチューブの製造方法。
〔5〕 さらに、凍結乾燥処理又は超音波処理を施すことで、短尺チューブ状とすることを特徴とする、前記〔4〕に記載の非対称ナノチューブの製造方法。
〔6〕 前記〔3〕の非対称ナノチューブがカチオン性薬剤を包接していることを特徴とする、カチオン性薬剤−非対称ナノチューブ複合体。
〔7〕 前記〔6〕に記載のカチオン性薬剤−非対称ナノチューブ複合体を有効成分とする、カチオン性薬剤組成物。
〔8〕 カチオン性薬剤がアミノ基を有するアントラサイクリン系抗癌剤である、前記〔7〕に記載のカチオン性薬剤組成物。
【発明の効果】
【0012】
本発明によれば、内表面がカルボキシル基、外表面が糖残基で被覆された非対称ナノチューブにおいて従来困難であった、室温下のpH変化といった温和な条件で選択的かつ高収率で製造することができる。また高い分散性を有するため、水性溶媒中で高濃度でも結晶化を起こすことなく、長期保存における安定性も非常に高く、5mg/mlといった比較的高濃度の水分散液でも2ヶ月以上安定に保存可能である。また、この非対称ナノチューブを構成する非対称双頭型脂質分子は新規化合物であって、その原料となる2−グルコサミン、長鎖ジカルボン酸及びオリゴグリシン類は安価であり、わずか3段階の穏和な条件での合成反応により製造でき、精製にはカラムクロマトグラフィーを使う必要がなく、全て再沈殿工程で高純度の脂質を得ることができる。
この非対称ナノチューブ形成性脂質分子の合成および精製が簡便であり、非対称ナノチューブも温和な条件で簡単に製造できる点は、薬剤キャリアとしてのカプセルの大量合成および最終的な薬剤組成物の低価格化の点でも重要である。
そして、本発明の非対称ナノチューブの長期保存安定性や高濃度での良分散性は、抗癌剤治療においてそれぞれ薬剤の安全性の点や患者の負担軽減を図るために経静脈投与する全液量を可能な限り減らす点で必要不可欠である。また本非対称ナノチューブは外径が僅か15nmで、長さを超音波処理などの物理的な擾乱で100nm〜2μmまで調節可能であるためEPR(Enhanced Permiability and Retation)効果によって、ガン組織へ送達する量を高めることが可能となる。このように、本発明の非対称ナノチューブが備えているこれら安定性や良分散性といった特性は、薬剤キャリアとしての優れた特性の1つである。
また、本発明の非対称ナノチューブは内側がカルボキシル基に覆われ、中性付近のpHでは負の電荷を帯びているので、ドキソルビシンのようなアミノ基を有する薬剤やカチオン性薬剤を混合するだけで、薬剤のカプセル化が可能であるという優れた特性を有する。調製が複雑な従来のリポソーム系の薬剤カプセルは治療時に任意の比率で混合することは不可能であり、あらかじめ薬剤をカプセル化したもののみが市販されていたが、本発明では、薬剤カプセルとしてのナノチューブを医療現場で速やかに調整することや、ナノチューブ分散液と薬剤を治療時に任意の割合で混合することでカプセル化が可能である。上記したように原料の脂質合成やナノチューブの調製が極めて容易なため、安価な薬剤カプセルとして提供することも可能である。
そして、本発明の非対称ナノチューブはpH変化などの外部の刺激に応じ、チューブ内表面を被覆するカルボキシル基のイオン化状態の変化又はチューブ構造そのものの形態変化などによって、条件選択的かつ効率的に薬剤を放出することができる。このため、上記のEPR効果によるがん組織へのナノチューブの選択的送達に加え、がん組織特有の低pH環境での効率的な薬剤放出が可能である。さらに、がん細胞内に取り込まれたナノチューブ複合体の場合は、エンドソーム内での低いpH環境による速やかな放出が可能となる。
これによって、医療用のドラッグデリバリーシステムにおける機能性薬剤徐放性キャリアとして用いることができるほか、化粧品分野、食品分野などにおける、乳化剤、分散剤、安定剤用キャリアなどとしての利用も期待できる。
【図面の簡単な説明】
【0013】
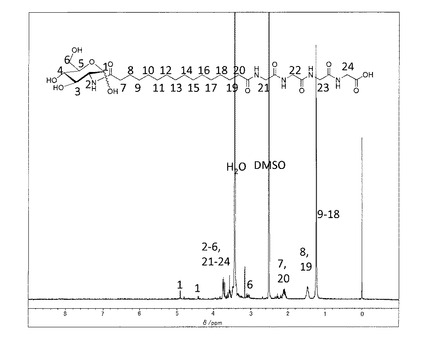
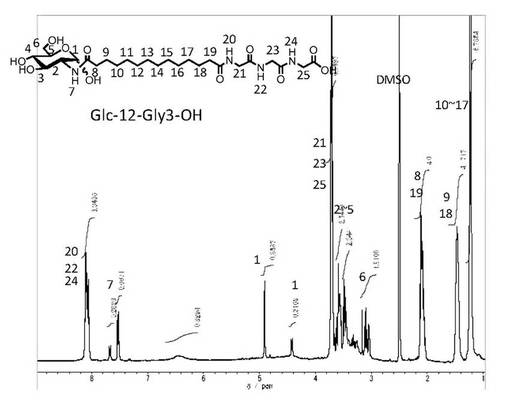
【図1】中間体−3(n=10)の1H−NMR(DMSO−d6と1滴D2O、室温)
【図2】化合物−1(n=10,m=2)の1H−NMR(DMSO−d6と1滴D2O、室温)
【図3】化合物−1(n=10,m=3)の1H−NMR(DMSO−d6と1滴D2O、室温)
【図4】化合物−1(n=10,m=3)の1H−NMR(DMSO−d6と1滴D2O、室温)
【図5】中間体−3(n=12)の1H−NMR(DMSO−d6と1滴D2O、室温)
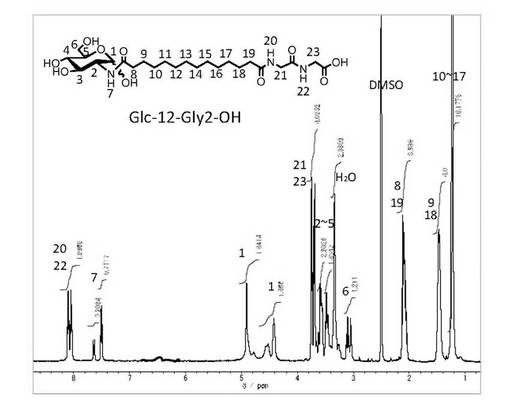
【図6】化合物−1(n=12,m=2)の1H−NMR(DMSO−d6、室温)
【図7】化合物−1(n=12,m=3)の1H−NMR(DMSO−d6、室温)
【図8】化合物−1(n=12,m=4)の1H−NMR(DMSO−d6と1滴D2O、室温)
【図9】中間体−3(n=14)の1H−NMR(DMSO−d6と1滴D2O、室温)
【図10】化合物−1(n=12,m=3)の1H−NMR(DMSO−d6、室温)
【図11】化合物−1(n=14,m=4)の1H−NMR(DMSO−d6と1滴D2O、室温)
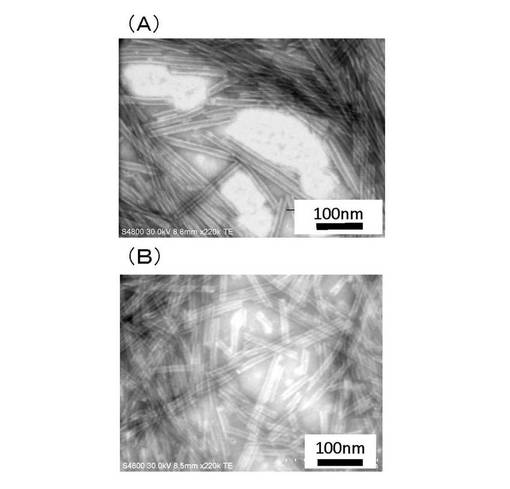
【図12】化合物−1(n=10,m=2の場合)の(A)加熱冷却および(B)pH調製法による自己集合で得られた非対称ナノチューブの電子顕微鏡観察像(透過像)。
【図13】化合物−1(n=10,m=3の場合)の(A)加熱冷却および(B)pH調製法による自己集合で得られた非対称ナノチューブの電子顕微鏡観察像(透過像)。
【図14】化合物−1(n=10,m=4の場合)の(A)加熱冷却および(B)pH調製法による自己集合で得られた非対称ナノチューブの電子顕微鏡観察像(透過像)。
【図15】化合物−1(n=12,m=2の場合)の(A)加熱冷却および(B)pH調製法による自己集合で得られた非対称ナノチューブの電子顕微鏡観察像(透過像)。左図中の挿入図は 加熱冷却によって調製されたナノチューブからなるヒドロゲル。
【図16】化合物−1(n=12,m=3の場合)の(A)加熱冷却および(B)pH調製法による自己集合で得られた非対称ナノチューブの電子顕微鏡観察像(透過像)。
【図17】化合物−1(n=12,m=4の場合)の(A)加熱冷却および(B)pH調製法による自己集合で得られた非対称ナノチューブの電子顕微鏡観察像(透過像)。
【図18】化合物−1(n=14,m=3の場合)のpH調製法による自己集合で得られた非対称ナノチューブの電子顕微鏡観察像(透過像)。
【図19】加熱冷却によって得た化合物−1(n=12,m=4)から得た非対称ナノチューブの超音波処理後の電子顕微鏡観察像(透過像)。左図中の挿入図は標記ナノチューブ分散液の様子。
【図20】加熱冷却によって得た化合物−1(n=12,m=4)から得た非対称ナノチューブの超音波処理、凍結乾燥、再水和後の電子顕微鏡観察像(透過像)。挿入図(左)は標記分散液の様子。挿入図(右図)は、電子顕微鏡観察H像の拡大像。
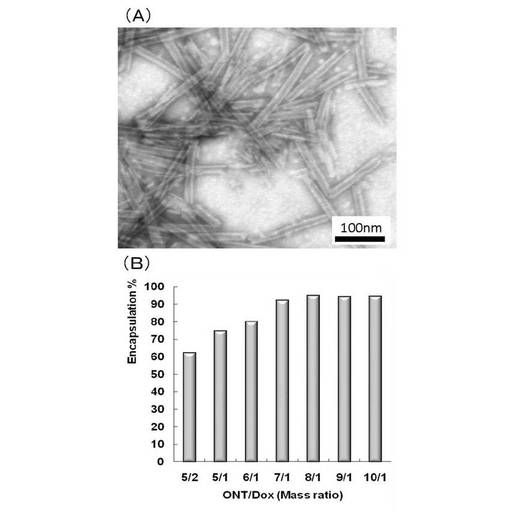
【図21】(A)ドキソルビシンをカプセル化した非対称ナノチューブ(化合物−1でn=12,m=4の場合)の電子顕微鏡観察像と(B)非対称ナノチューブによるドキソルビシンのカプセル化率。
【図22】非対称ナノチューブ(化合物−1でn=12,m=4の場合。カプセル化はONT/DOX=7/1で行ったもの)からのドキソルビシンの放出実験。図中の括弧内は用いたバッファーのpHを表す。
【図23】ドキソルビシンナノチューブのHela細胞への抗癌特性(MTSアッセイ)。
【発明を実施するための形態】
【0014】
1.本発明の非対称双頭型脂質分子について
(1−1)本発明の非対称双頭型脂質分子の特徴:一般式(1)の化合物(化合物−1)
本発明の非対称ナノチューブ形成性の脂質分子は文献未載の新規化合物であり、その形状は、長鎖カルボン酸の片端にアミド基を介し2−グルコサミン残基が連結し、もう一端にオリゴグリシンのN端が縮合した非対称双頭型脂質分子である。
本発明の非対称双頭型脂質分子は、合成が非常に簡便で、かつ外径15nm前後、内径約8−9nmの非対称なナノチューブ構造を、室温でのpH変化により選択的かつ高純度に製造できる。そのために、本発明の脂質分子のアルキレン鎖の鎖長は8〜16の範囲である必要があり、脂質合成時の分散性の良さや精製の容易さからは、10〜14の整数が好ましく、偶数であることがより好ましい。またオリゴグリシン残基の数は、1〜4の範囲であればよいが、ナノチューブ製造時の精製のしやすさ及び得られるチューブの純度の観点から2〜4が望ましい。また、本発明の非対称双頭型脂質分子は、水溶性の製薬上許容される塩の状態であってもよい。例えば、ナトリウム塩、アンモニウム塩などが好ましい。
親水部として用いられる2−グルコサミンは、D体でもL体のいずれを用いてもよいが入手の容易さからD体が好適である。本発明の実施の態様ではD体を用いたので、以下、D体を用いた場合で説明するが、本発明はこれに限られるものではない。
すなわち、本願発明の非対称双頭型脂質分子は,下記一般式(1)で表される。
(式中、nは8〜16の整数であり、mは1〜4の整数を表す。また、式中の波線で表記した2−グルコサミンの1位水酸基はα、βアノマーのいずれか、あるいはその平衡混合物を表す。)
【0015】
(1−2)本発明の非対称双頭型脂質分子の製造方法
前記一般式(1)で表される非対称双頭型脂質分子又はその塩は、例えば以下に示す方法により容易に製造することができる。
下記一般式(2)で表されるα,ω−長鎖ジカルボン酸
HOOC−(CH2)n−COOH 一般式(2)
(式中、nは8〜16の整数を表す。)
を水性メタノール中で、2−グルコサミンと縮合させ、生じた固体をろ過、洗浄することで、下記一般式(3)で表される中間体−3
(式中nは8〜16の整数を表す。また式中の水酸基の波線は、式中の波線で表記した2−グルコサミンの1位水酸基はα、βアノマーのいずれか、あるいはその平衡混合物を表す。)
を製造することができる。
次にこの一般式(3)の中間体−3とオリゴグリシンのC端をエステルなどで保護した下記一般式(4)で表されるオリゴグリシンエステル
(式中mは1〜4の整数を表す。またRはアルコール性水酸基を有する化合物の残基であり、例えば、メチル基、エチル基、三級ブチル基、ベンジル基である。)
を加えて縮合させ,下記一般式(5)で表される中間体−5
(式中nは8〜16の整数を表し、mは1〜4の整数を表す。またRはアルコール性水酸基を有する化合物の残基であり、例えば、メチル基、エチル基、ベンジル基、三級ブチル基、である。また式中の波線で表記した2−グルコサミンの1位水酸基はα、βアノマーのいずれか、あるいはその平衡混合物を表す。)
を得た。
次いで、これをアルカリ加水分解することで脱保護し、下記一般式(1)で表される非対称双頭型脂質分子(化合物−1ともいう。)又はその塩
(式中、nは8〜16の整数であり、mは1〜4の整数を表す。また、式中の波線で表記した2−グルコサミンの1位水酸基はα、βアノマーのいずれか、あるいはその平衡混合物を表す。)
を合成することができる。
【0016】
(1−3)中間体−3(一般式(3))を得る縮合工程について
本発明の中間体−3(一般式(3))は、典型的には前記非特許文献15に記載された脂質分子を含むので、非特許文献15に記載の方法に従って、又はそれと同様の方法で製造することができる。
(ア)原料の下記一般式(2)で表されるα,ω−長鎖ジカルボン酸について
HOOC−(CH2)n−COOH 一般式(2)
(式中、nは8〜16の整数を表す。)
原料の長鎖ジカルボン酸としては、中央の疎水部アルキレン鎖の炭素数nが8〜16のものを用いることができる。具体的には、ジカルボン酸セバシン酸、ウンデカン二酸、ドデカン二酸、トリデカン二酸、テトラデカン二酸、ペンタデカン二酸、ヘキサデカン二酸、ヘプタデカン二酸、オクタデカン二酸を用いることができる。これらジカルボン酸は塩又はモノエステルの状態で用いることもできるが、モノエステルの場合はグルコサミンの縮合後に加水分解が必要となる。
また、これらジカルボン酸の酸無水物や酸クロライドを用いる場合は、縮合剤なしでも縮合反応が起こるが、この場合も縮合後に水を添加して上記活性体の加水分解が必要となる。
【0017】
(イ)縮合剤について
中間体−3(一般式(3))を得る縮合反応で用いる縮合剤としては、ペプチド合成に用いられる種々の縮合剤を用いることができる。具体的には、「4−(4,6−ジメトキシ−1,3,5−トリアジン−2−イル)−4−メチルモルホリニウムクロリド」(DMT−MM)、1−エチル−3−(3−ジメチルアミノプロピル)カルボジイミド塩酸塩、ジシクロヘキシルカルボジイミド、ジイソプロピルカルボジイミドなどを用いることができる。またこのような縮合剤を用いなくても、該当するカルボン酸の酸無水物や酸クロライドを用いることもできる。しかし、水中やメタノール中の反応でも極めて高収率の縮合物を与えるDMT−MMが望ましい。また同反応は、メタノール、エタノール、イソプロパノール、ジメチルホルムアミド、ジメチルアセトアミド、N−メチルピロリドン、ジメチルスルホキシドなどを用いることができるが、選択的に片側のカルボン酸とのみ縮合反応を行い、速やかに目的とする中間体−3を沈殿させることで単離・精製するためには、ジカルボン酸、2−グルコサミン、中間体−3に適度な溶解度を有する5〜40体積%の水を含有するメタノール、エタノール、イソプロパノールが好適である。
【0018】
(ウ)2−グルコサミンについて
本発明のアミン成分は、2−グルコサミンであり、1位の水酸基はαアノマーであってもβアノマーであっても、もしくはその平衡混合物、又は塩酸塩などの塩であってもよい。
例えば、2−グルコサミンの塩酸塩を用いる場合、水に溶解後、ナトリウムメトキシドなどでアミンに変換して反応に用いる。あるいは、あらかじめα,ω−長鎖ジカルボン酸の二ナトリウム塩等を原料として用いれば、2−グルコサミン塩酸塩の中和は必要がない。
なお、2−グルコサミンは、D体でもL体のいずれを用いてもよいが入手の容易さからD体が好適であるため、本発明の実施の態様ではD体を用いている。したがって、一般式(1)などのアミン成分として表示されている2−グルコサミンはD体が示されている。
【0019】
(エ)縮合反応について
一般式(2)の長鎖ジカルボン酸及び2−グルコサミンの縮合反応は、前記縮合剤を添加し、又は添加することなく、水性溶媒中、好ましくは水含有メタノール溶液中で行わせる。
その際の反応温度としては0度ないし50度の範囲が選ばれ、反応時間は通常10分ないし30時間の範囲である。
上記縮合反応では、中間体−3のみが反応中に純度良く沈殿するため、特に精製する必要はないが、必要に応じて、再度メタノールや水含有メタノールなどを用いた加熱・冷却による再沈殿によりさらに高純度のものとすることができる。
このような反応では、一般式(2)のジカルボン酸の両端に2−グルコサミンが反応する可能性もあるが、得られた反応生成物の1H−NMRスペクトル(重ジメチルスルホキシド中、室温)の2.2〜2.3ppmのカルボキシル基に隣接するメチレンのシグナルと2.1ppmの糖アミドに隣接するメチレンのシグナルの積分比からみて、きわめて選択的に中間体−3が得られることが確認できている。
このように、本発明によれば、約25〜65%の収率で目的の中間体−3が得られる。
【0020】
(オ)中間体−3について
本発明の中間体−3は、下記一般式(3)で表される脂質分子であり、
(式中nは8〜16の整数を表す。また式中の水酸基の波線は、式中の波線で表記した2−グルコサミンの1位水酸基はα、βアノマーのいずれか、あるいはその平衡混合物を表す。)
典型的なn=10,12の脂質分子については、非特許文献15に記載されている。
中間体−3自体も、本発明の非対称双頭型脂質分子と同様に、カルボン酸系の非対称双頭型脂質分子であるが、自己集合させても、ナノチューブとその断片状のものの混合物しか得られず、非対称ナノチューブが形成できなかった点は上記[0004]で述べたとおりである。
本発明の中間体−3は、赤外線吸収スペクトルでは3700〜3300cm−1に糖水酸基に由来する特性吸収、2917〜2922cm−1、および2848〜2852cm−1にオリゴメチレン鎖に由来する特性吸収、1675〜1600cm−1にカルボキシル基に由来する吸収、1640〜1620cm−1にアミドカルボニル基に由来する吸収、1100〜1000cm−1に糖骨格に由来する吸収を示す。
さらに1H−NMR(DMSO−d6(重水素化ジメチルスルホキシド)と1滴D2O、室温)において、 δ値が7.7(βアノマーのNH), 7.5(αアノマーのNH), 4.9(αアノマーの1位のプロトン), 4.4(βアノマーの一位のプロトン), 3.2−3.7(糖のピラノース環のメチン、メチレン、2〜6位), 3.04−3.13(糖ピラノース環のメチレン、6位), 2.28(α体のカルボキシル基の隣のメチレン), 2.2(β体のカルボキシル基の隣のメチレン), 2.1(アミドに隣接したメチレン), 1.5(アミドの二つ隣のメチレン), 1.2(その他のメチレン)のシグナルが観測できることから、中間体−3であると同定できる。
【0021】
(1−4)中間体−3から中間体−5を経て本発明の非対称双頭型脂質分子(化合物−1)を得る工程について
(ア)中間体−5を得るための工程について
中間体−3を下記一般式(4)のオリゴグリシンエステルと脱水縮合することで中間体−5を得ることができる。
(式中mは1〜4の整数を表す。またRはアルコール性水酸基を有する化合物の残基であり、例えば、メチル基、エチル基、三級ブチル基、ベンジル基である。)
一般式(4)のオリゴグリシンエステルとしては、グリシンもしくはグリシルグリシンなどグリシンの重合物のメチルエステル、エチルエステル、三級ブチルエステル、ベンジルエステルなどのエステルが用いられる。アルカリ加水分解の容易さからエチルエステルあるいはメチルエステルが好適である。
【0022】
中間体−3と一般式(4)のオリゴグリシンエステルとを脱水縮合する反応においても、前記(1−3)(イ)と同様の縮合剤を用いることができる。具体的には、DMT−MM、1−エチル−3−(3−ジメチルアミノプロピル)カルボジイミド塩酸塩、ジシクロヘキシルカルボジイミド、ジイソプロピルカルボジイミドなどを用いることができるが、高収率の縮合物を与えるDMT−MMあるいは1−エチル−3−(3−ジメチルアミノプロピル)カルボジイミド塩酸塩が望ましい。また同反応はジメチルフォルムアミド(DMF)、ジメチルアセトアミド、N―メチルピロリドン、メタノール、エタノール、イソプロパノール、ジメチルスルホキシドなどを用いることができるが、原料の溶解性の点からDMFが好適である。
ここでの縮合反応も同様に、反応温度としては0度ないし50度の範囲が選ばれ、反応時間は通常10分ないし30時間の範囲である。
中間体−5を得るための縮合反応後にDMFを留去して得た固体を洗浄し不純物を溶解成分として除去するためには、メタノール、エタノール、イソプロパノール等が用いられるが、中間体−5の溶解性を抑制させるためにメタノールを用いることが望ましい。
【0023】
(イ)中間体−5について
中間体−5は、下記一般式(5)に示されるように、本願目的非対称双頭型脂質分子の末端カルボキシル基が保護基(メチル基、エチル基、三級ブチル基又はベンジル基)により保護された化合物として得られる。
(式中nは8〜16の整数を表し、mは1〜4の整数を表す。またRはアルコール性水酸基を有する化合物の残基であり、例えば、メチル基、エチル基、三級ブチル基、ベンジル基である。また式中の波線で表記した2−グルコサミンの1位水酸基はα、βアノマーのいずれか、あるいはその平衡混合物を表す。)
上記縮合反応の系においては、中間体−5のみが溶解性が低く、選択的に沈殿するため、ろ過後の沈殿固体の洗浄を除けば特に精製する必要はないが、必要に応じて、再度メタノールや水含有メタノールなどを用いた加熱・冷却による再沈殿によりさらに高純度のものとすることができる。
【0024】
(ウ)目的化合物−1を得るための加水分解工程について
中間体−5を加水分解により脱保護することで簡単に本発明の非対称双頭型脂質分子(化合物−1)を得ることができる。その際の加水分解では、溶解度と反応性の観点から、メタノール/水混合溶媒が好適である。反応温度としては20度ないし80度の範囲が選ばれ、反応時間は通常30分ないし6時間の範囲である。また中間体−5のRがベンジル基の場合は、DMF、あるいはメタノール、メタノール/水中などでのパラジウム炭素を触媒とした接触水素化還元での脱ベンジルエステルもよい。またRが三級ブチル基の場合は、DMF中で酸処理による脱エステルも可能である。
また加水分解反応も定量的に進行し、副反応もほとんど見られないことから、反応後の酸の添加による中和で純度よく目的とする化合物−1を得ることができるが、必要に応じてメタノールやエタノール、DMFなどで加熱冷却による精製を行ってもよい。
【0025】
(エ)本発明の非対称双頭型脂質分子(化合物−1)の同定
このような化合物の元素分析の実測値が誤差範囲内で計算値と一致し、また赤外線吸収スペクトルでは3700〜3300cm−1に糖水酸基に由来する特性吸収、2917−2922cm−1、および2848−2852cm−1にオリゴメチレン鎖に由来する特性吸収、1675−1600cm−1にカルボキシル基に由来する吸収、1650〜1620cm−1にグリシン残基やグルコサミンに連結したアミドカルボニル基に由来する吸収、1100〜1000cm−1に糖骨格に由来する吸収を示す。さらに1H−NMR(DMSO−d6と1滴D2OあるいはDMSO−d6のみ、室温)において、 δ値が8.2付近(グリシンのNH),7.6付近(グルコサミンのNH), 4.9付近(αアノマーの1位のプロトン), 4.4付近(βアノマーの一位のプロトン), 3.2−3.7付近(糖のピラノース環、2〜6位。グリシンのメチレン), 3.0−3.1付近(糖ピラノース環、6位のメチレン), 2.2−2.1付近(アミドカルボニル基の隣のメチレン), 1.5付近(アミドの二つ隣のメチレン), 1.2付近(その他のメチレン)のシグナルが観測できる。このことから、当該化合物は、目的の化合物−1であると同定できる。
【0026】
2.本発明の非対称ナノチューブについて
(2−1)本発明の非対称ナノチューブの製造方法
(ア)加熱溶解・冷却法
本発明の非対称双頭型脂質分子を用いて非対称ナノチューブを製造する方法の1つは、一般的な自己集合性脂質分子の自己集合のための加熱溶解・冷却法を適用することができる。
本発明の非対称双頭型脂質分子(0.5mg〜5mg)を1ミリリットルの純水(ミリQ水など)に分散し、80〜100℃に加熱溶解後、冷却(0.1℃〜10℃毎分)することで非対称ナノチューブからなるヒドロゲルが得られる。この時の電子顕微鏡観察から、長さが10μm以上あるナノチューブの絡み合った構造が確認できている。
本発明の非対称ナノチューブは構造安定性が高いので、凍結乾燥処理あるいは超音波処理などの物理的擾乱にも耐えられる。これらの物理的擾乱処理によって、収率良く軸比の短い、かつ良分散性の非対称ナノチューブに変換することが可能である。
得られた非対称ナノチューブの形態、大きさなどは、分散液を電子顕微鏡用のグリッドに滴下し、内部を電子線不透過な染色剤で染色することで、確認することができる。この様にして外径が約15nm、内径が約8〜9nm、長さが、100nm〜2μmの非対称ナノチューブを得ることができた。
【0027】
(イ)pH調整法
本発明の非対称双頭型脂質分子を室温で等モルから2等量程度の水酸化ナトリウム水溶液に分散・溶解し、つぎに室温で塩酸等の酸溶液を等モルから2等量程度添加してpH5〜6程度に調整することで同様に非対称ナノチューブを得ることができる。上記水酸化物水溶液としては、水酸化ナトリウム以外にもリチウム、カリウム、ルビジウム、セシウムなどの水酸化物水溶液も用いることができる。また上記酸溶液としては塩酸以外にもリン酸、ギ酸、酢酸、クエン酸、アスコルビン酸、硫酸等を用いることができる。
なお、原料として非対称双頭型脂質分子の塩(例えばナトリウム塩)などを用いる場合は、上記水酸化ナトリウム水溶液でもよいが、中和により多くの酸を必要とする。このため、より好ましくは室温で同塩を純水(ミリQ水など)に分散・溶解し、つぎに上記と同様室温で塩酸等の算用液を等モルから1.5等量程度添加してpH5〜6程度に調製することで同様に調製すればよい。
このpH調整法を適用する場合も、超音波処理、凍結乾燥などで軸比の短い非対称ナノチューブに変換可能であり、そのサイズはいずれもほぼ同じである。特に後者では、再水和時の添加する水の量によって良分散な状態のまま脂質濃度を10mg/ml程度まで上げることができる。またそれ以上(例えば50mg/ml)に濃度を上げることも可能であるが、ゲル状に変化する。
【0028】
(2−2)本発明の非対称ナノチューブの安定性
こうして得られた非対称ナノチューブの分散液は、そのまま室温あるいは冷蔵庫で上記のいずれの濃度で冷蔵保存しても約6ヶ月以上、沈殿を与えることなく良好な分散液として保存可能である。さらに分散液を凍結乾燥すれば1年以上の室温保存が可能で、ミリQ水等を添加して再水和すると同様の分散液を得ることができる。
【0029】
(2−3)本発明の非対称ナノチューブの構造
チューブ状構造であることの確認は、ネガティブ染色後の電子顕微鏡観察によってチューブ内空間を可視化することで可能となる(非特許文献1、10)。すなわち、チューブの内空間に電子線を透過させない染色剤を毛細管現象によって導入し、電子顕微鏡観察による透過像を得ると、染色剤が分布するチューブ内空間が暗い像を与え、チューブの壁は電子線が透過しやすい有機物で出来ているため、薄いコントラストを与える。これによって確認可能である。
また非対称ナノチューブ構造(すなわちチューブ内表面にカルボキシル基また外表面に2−グルコサミド基を配列するように配向する構造)の確認は、ナノチューブの粉末X線回折パターンから得られるナノチューブの膜厚の周期dと分子モデルから推定される分子長Lの関係から推測できる。すなわち非対称双頭型脂質が非対称ナノチューブ構造を形成しているときは、ナノチューブの膜周期dと分子長Lがほぼ同じか、あるいはdが若干小さくなることが知られている(非特許文献9、10)。そこで、以下のようにdとLを比較検討した結果、いずれの場合にも上記の関係を満たすことが明らかとなった(表1)。
以上から、本発明の有機ナノチューブは非対称ナノチューブであることがわかった。
【0030】
<表1> 非対称ナノチューブのXRDより得られる膜周期dと分子モデルから得られる分子長Lの関係
【0031】
表1には、記述していないが、本発明の化合物−1のn=14,16の場合も、n=10,12の場合と同様のチューブ形態とサイズを示すことを確認した。一般には鎖長が長くなるほど、非対称ナノチューブ構造を取りやすくなることが以前の研究より明かにされており(非特許文献17)、分子の鎖長のみが異なる一連の脂質群では、自己集合で得られるナノチューブ構造がほぼ同じ形態やサイズ(内径、外径、壁厚)を有する場合は、分子の配列様式も同様であることも知られている(非特許文献10)。したがって、n=14,16の場合も、n=10,12の場合と同様の非対称ナノチューブ構造を形成していることが示唆される。
【0032】
3.本発明の非対称ナノチューブへのカプセル化
(3−1)カプセル化できる対象物質について
本発明の非対称ナノチューブは、その内側がカルボキシル基で被覆されているため、中に好ましく包接される薬剤は、カルボキシル基とイオン結合可能なアミノ基などのカチオン性の官能基を有する化合物が好ましく、安定なカチオン性薬剤−非対称ナノチューブ複合体を形成する。また、チューブ内が負の電荷に満たされた環境となっているため、特にイオン結合可能な官能基を有している化合物でなくても、物質全体が正の電荷を帯びたカチオン性物質であって、サイズが0.1〜5nm程度の物質であれば包接可能であり、このようなカチオン性物質を包接した非対称ナノチューブもカチオン性物質−非対称ナノチューブ複合体を形成しているといえる。
カチオン性薬剤−非対称ナノチューブ複合体を形成する具体的なカチオン性薬剤としては、ドキソルビシン、イダルビシン、エピルビシン、ダウノルビシン、ピラルビシンなどのアントラサイクリン系抗癌剤やその他のアミノ基を有する薬剤が特に好ましく用いられる。
また、シスプラチン、ネダプラチン、カルボプラチン、オキサリプラチンなどのカチオン性薬剤も好ましい。
また、このような医薬品としての薬理作用を有する薬剤のみならず、化粧品用、食品用などのカチオン性の乳化剤、分散剤、安定剤、保湿剤等各種添加剤に対しても適用可能である。
【0033】
(3−2)非対称ナノチューブへのカプセル化方法
カプセル化したいカチオン性物質を純水(ミリQ水)に0.1mg/mL〜50mg/の濃度になるように溶解又は懸濁したものと、本発明の非対称ナノチューブ分散液(濃度 0.1mg/mL〜20mg/mL)とを等量混合し、10分〜3時間程度室温で放置する。そのことで、30〜95%程度のカプセル化率を達成できるため、例えば抗癌剤用組成物として用いる場合など、有効量を含有させるための組成物総量を低く抑えることができる。
またカプセル化されなかった物質は、上記混合溶液を200nmの多孔質膜でろ過することによって、効率よく取り除くことができる。これによって、カチオン性物質をカプセル化したチューブのみを固体として単離することができるので、これを任意の濃度でミリQ水に再分散させることで濃度を調整することも可能である。またカチオン性物質をカプセル化したチューブを凍結乾燥して得た固体を同様に任意の濃度でミリQ水に分散させることでも濃度を調整することができる。
薬剤のカプセル化率は、上記の手法でカプセル化した後、ナノチューブを濾別し、これをミリQ水に再分散して得られた溶液の薬剤固有のUV吸収を測定することで、定量することができる。
例えばドキソルビシンの場合、ドキソルビシン溶液200μL(それぞれミリQ水1mLあたりに2.0mg,1.0mg,0.714mg,0.625mg,0.555mg,0.50mg溶解したもの)とナノチューブ分散液(200μL、5mg/mL)を混合し、30分室温で放置した。これより200μLを取り、200nmの多孔質膜でろ過し、さらにミリQ水(400μL)で洗浄した。濾別された固体(ナノチューブ)はろ紙ごと200μLのミリQ水に再分散した。ドキソルビシンのUV吸収(480nm)によって、ろ過前の溶液とろ過後の溶液の吸収を比較して、カプセル化率を求めた。
本発明の薬剤カプセル化率は、ドキソルビシンの場合で62〜95%程度と、きわめて高濃度にカプセル化することができる。
【0034】
(3−3)薬剤のナノチューブからの放出について
本発明においてカプセル化された薬剤は、pH変化などの外部の刺激に応じ、チューブ内表面を被覆するカルボキシル基のイオン化状態の変化又はチューブ構造そのものの形態変化などによって、条件選択的かつ効率的に薬剤を放出することができる。
具体的には、ミリQ水の分散液にバッファー溶液を加えpH値を4〜6程度とすることで、内表面のカルボキシル基がプロトン化するため、放出の加速が可能である。またpHを7〜8程度にすることでも徐放することが可能である。
また、本発明の非対称ナノチューブとして、超音波処理などにより長さを50nm〜500nm程度にまでサイズダウンしたナノチューブを用いることで、EPR(Enhanced Permiability and Retation)効果により、ターゲットとなるガン細胞、組織へ送達する量を高めることが可能である。
ナノチューブからの薬剤放出量は、チューブ分散液を透析膜チューブ(分画分子量1200〜20000程度)の内水相に密閉し、膜を通過して外水相に放出された薬剤をUVなどの分光分析によって計測することで定量的に計測可能である。
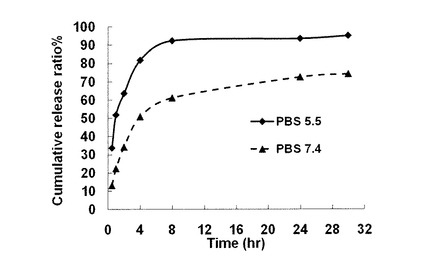
例えば、ドキソルビシンを内包化した非対称ナノチューブ(ナノチューブ/Dox=8/1)のPBSバッファー中(pH7.4)の場合は、これを透析膜(分画分子量14000)に密閉し、同様にPBSバッファー中(pH7.4)の外水相に入れて撹拌しながら外水相のドキソルビシンの放出量をUV吸収によってモニターした。
その場合、PBSバッファー中(pH7.4)中では、1時間あたり10〜12%程度、4時間目では約50%程度の徐放効果を確認できた。
一方、弱酸性状態(pH5.5)では、むしろ一時間あたり33%、4時間目で82%以上を放出し、8時間ではほぼ全量放出してしまうことから、非対称ナノチューブ内表面を覆うカルボキシル基から生じたカルボキシラートのプロトン化が刺激となって抗癌剤の放出を加速できることが確認できた。この低pH条件で抗癌剤放出が加速される点は、ナノチューブを薬剤カプセルとして利用する場合、極めて有効性であると考えられる。一般に、ガン組織およびガン細胞は、正常細胞に比べて弱酸性(低pH)であるという特性を有するからである。
【0035】
4.カプセル化された薬剤組成物
(4−1)薬剤組成物について
本発明の非対称ナノチューブは、上述のように薬剤キャリアとして優れた特性を有しているため、治療用の薬剤を有効量含む非対称ナノチューブ複合体を有効成分として、薬理学的に許容される担体と共に薬剤組成物とすることができる。なお、薬剤組成物とは、例えば抗癌剤など癌の予防、治療又は処置のための医薬組成物を含む。また、水性環境中に徐々に薬剤を放出させたい場合などにも有効であることから、この薬剤組成物としては、化粧品用、食品用として配合されるカチオン性薬剤組成物も包含される。
本発明のカプセル内の薬剤として典型的な薬剤は、上述のドキソルビシン、イダルビシン、エピルビシン、ダウノルビシン、ピラルビシンなどのアントラサイクリン系抗癌剤やその他のアミノ基を有する薬剤である。その他、シスプラチン、ネダプラチン、カルボプラチン、オキサリプラチンなどのカチオン性薬剤も同様に用いることができる。
ここで、薬学的に許容される担体とは、希釈剤又は賦形剤、例えば、水又は生理的許容される緩衝液を含む。
本発明の薬剤組成物は、錠剤などの経口投与も可能であるが、静脈経由若しくは直接患部への注射、外用剤などの非経口的に投与することもできる。
本発明の薬剤組成物を患者に投与する際の投与量は、用いる薬剤の種類、医療対象の患者の年齢、体重及び病状、投与経路などに従い適宜決めることができるが、典型的には、1回の投与当たりの薬剤の有効量としては、1μg〜1g、好ましくは100μg〜10mgの範囲である。
【0036】
(4−2)薬剤組成物の機能について
本発明の非対称ナノチューブによる薬剤カプセル化物は、中性付近での徐々に放出することができる徐放作用と、酸性条件での加速された薬剤放出効果と2つの効果が期待できる。すなわち、癌細胞の場合、若干酸性状態のがん組織付近での選択的な薬剤放出効果と共に、がん細胞内部での速やかな薬剤放出による高い抗癌活性の発現という二つの効果が期待できる。
なお、本発明で「細胞」というとき、典型的にはヒトを含む哺乳動物細胞を指す。インビトロでの培養細胞であってもよいが、薬剤治療用の場合、生体を構成する組織、臓器内の細胞や、血液など体液中の血球細胞やリンパ球細胞などの免疫細胞、及び各種の癌細胞が対象となる。
【0037】
(4−3)抗癌活性の測定
一例として、本発明のドキソルビシンを内包化した非対称ナノチューブを用いた抗癌剤組成物についての抗癌活性を、以下のように確認した。
抗癌活性については、ドキソルビシンを内包化した非対称ナノチューブを培養液中に分散させたHela細胞に添加、24時間培養後、細胞数あるいはそれにかわるもの(細胞内タンパク質量、ミトコンドリアの活性など)を測定し、対照実験として、ドキソルビシンのみの溶液と比較することで行うことが出来る。その際の測定法としては、典型的には細胞のミトコンドリアの活性を測定する方法(MTTアッセイ法、非特許文献18)を用いることができる。
本実施例では、細胞にドキソルビシンを内包化した非対称ナノチューブを添加・培養後、MTSアッセイ法による細胞のミトコンドリアの活性に従って定量した。MTTアッセイもMTSアッセイも細胞中のミトコンドリア中の酵素により生成するホルマザンという色素の濃度測定によって、活性な細胞の数を定量化するものである。MTTアッセイでは、薬剤反応後に不溶性のホルマザンが生成するため、吸光度を測定する前に溶液の処理が必要となる。これに比べてMTSアッセイでは、不溶性のホルマザンの生成が無く、後処理も不要なため、操作がより簡便で迅速に測定できるものである。
【実施例】
【0038】
次に本発明を実施例によってさらに詳細に説明するが、本発明はこれらの例によって何
ら限定されるものではない。
本発明におけるその他の用語や概念は、当該分野において慣用的に使用される用語の意味に基づくものであり、本発明を実施するために使用する様々な技術は、特にその出典を明示した技術を除いては、公知の文献等に基づいて当業者であれば容易かつ確実に実施可能である。また、各種の分析などは、使用した分析機器又は試薬、キットの取り扱い説明書、カタログなどに記載の方法を準用して行った。
なお、本明細書中に引用した技術文献、特許公報及び特許出願明細書中の記載内容は、本発明の記載内容として参照されるものとする。
【0039】
(実施例1)非対称ナノチューブ形成性非対称双頭型脂質分子(式(1)化合物―1、n=10,m=2)の合成
一般式(1)で表される非対称双頭型脂質分子のうち、n=10,m=2の場合の脂質分子(化合物−1(n=10,m=2))を、下記反応式(1)に示される中間体−5を経由した合成方法に従って合成した。
(なお、反応式中、「DMT−MM」は「4−(4,6−ジメトキシ−1,3,5−トリアジン−2−イル)−4−メチルモルホリニウムクロリド」の略であり、「MeONa」は「ナトリウムメトキシド」の略である。以下同様。)
【0040】
(1−1)中間体−3(n=10)の合成
ドデカン二酸(11.9g,46mmol)をメタノール(400 ml)に溶解した。この溶液に、水( ml)に溶したD(+)−グルコサミン塩酸塩 (5g,23mmol)とナトリウムメトキシド (1.25g,23mmol)を加えた。メタノール(20ml)に溶解したDMT−MM(6.4g,23mmol)を加え、室温で攪拌した。TLCで反応をモニターしながら、DMT−MM(9.6g,35mmol)とナトリウムメトキシド (2.5g,46mmol)を加え、30時間反応した。反応終了後、反応液を濾過し、残渣を水とメタノールで洗浄した。エタノール、水1/1で再沈澱を行い、生成物(5.79g)を得た。収率25%。
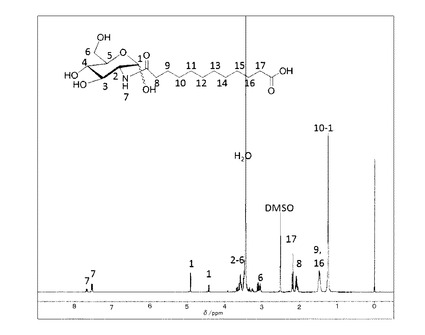
精製後の1H−NMR(DMSO−d6と1滴D2O)のチャートを図1に示す。
1H−NMR(DMSO−d6と1滴D2O)7.68(dd,J=8.4Hz,NH), 7.52(d,J=8.1Hz,NH),4.92(d,J=3.4Hz,0.7H,H−1α),4.43(d,J=8.1Hz,0.3H,H−1β),3.24−3.69(m,5H),3.04−3.13(m,1H),2.28(t,J=7.4Hz,0.3H,−CH2−COOHα),2.18(t,J=7.4Hz,1.7H,−CH2COOHβ),2.09(t,J=7.5Hz,−CH2−CONH,2H),1.47(d,J=6.8Hz,−CH2CH2CO,4H), 1.24(s,12H,−CH2)。
【0041】
(1−2)化合物−1の合成(n=10,m=2)
上記(1−1)で得られた中間体−3(n=10)(0.5g, 1.2mmol)をDMFに加熱溶解し、室温まで冷却したのちグリシルグリシン塩酸塩メチルエステル(0.22g, 1.23mmol)とDMT−MM(0.44g, 1.35mmol)、トリエチルアミン(1.25mmol)を添加した。室温で2時間撹拌した後、DMFを留去した。得られた固体にメタノールを添加し、不溶の白色固体を濾別することで中間体−5(n=10,m=2)を得た。これを加熱したメタノール/水溶媒に分散させ、水酸化ナトリウム水溶液(1.5ml、1mol/L)を60℃で添加して加水分解を行った。反応後溶媒を留去し、メタノールを添加し、固体を分散させた後、塩酸を添加してpHを4に調整し、ろ過することで目的とする化合物−1を白色固体として得た(0.35g、58%:中間体−3(n=10)より)。精製後の1H−NMR(DMSO−d6と1滴D2O)のチャートを図2に示す。
元素分析値(C22H39N3O10H2Oとして)
C H N
計算値(%) 50.47 7.89 8.03
実測値(%) 50.59 7.75 7.99
【0042】
(実施例2)非対称ナノチューブ形成性非対称双頭型脂質分子(式(1)化合物−1、n=10,m=3)の合成
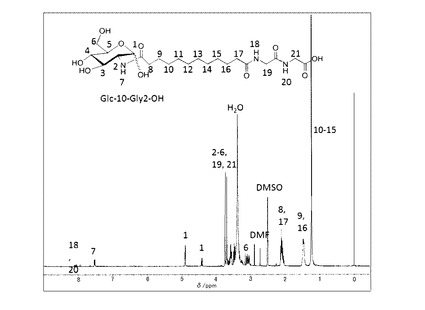
前記実施例(1−1)で得られた中間体−3(n=10)(0.5g, 1.2mmol)とグリシルグリシルグリシン塩酸塩メチルエステル(0.29g, 1.23mmol)とDMT−MM(0.44g, 1.35mmol)、トリエチルアミン(1.25mmol)を添加した。室温で2時間撹拌した後、DMFを留去した。得られた固体にメタノールを添加し、不溶の白色固体を濾別することで中間体−5(n=10,m=3)を得た。これを加熱したメタノール/水溶媒に分散させ、水酸化ナトリウム水溶液(1.5ml、1mol/L)を60℃で添加して加水分解を行った。反応後溶媒を留去し、メタノールを添加し、固体を分散させた後、塩酸を添加してpHを4に調整し、ろ過することで目的とする化合物−1を白色固体として得た(0.43g、63%:中間体−3(n=10)より)。精製後の1H−NMR(DMSO−d6と1滴D2O)のチャートを図3に示す。
元素分析値(C24H42N4O11H2Oとして)
C H N
計算値(%) 51.05 7.85 9.92
実測値(%) 50.93 7.95 9.88
【0043】
(実施例3)非対称ナノチューブ形成性非対称双頭型脂質分子(式(1)化合物−1、n=10,m=4)の合成
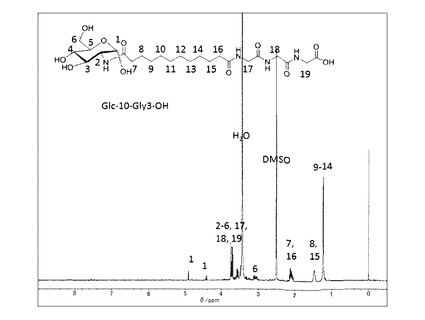
前記実施例(1−1)で得られた中間体−3(n=10)(0.5g, 1.2mmol)とグリシルグリシルグリシルグリシン塩酸塩メチルエステル(0.65g, 1.23mmol)とDMT−MM(0.44g, 1.35mmol)、トリエチルアミン(1.25mmol)を添加した。室温で2時間撹拌した後、DMFを留去した。得られた固体にメタノールを添加し、不溶の白色固体を濾別することで中間体−5(n=10,m=4)を得た。これを加熱したメタノール/水溶媒に分散させ、水酸化ナトリウム水溶液(1.5ml、1mol/L)を60℃で添加して加水分解を行った。反応後溶媒を留去し、メタノールを添加し、固体を分散させた後、塩酸を添加してpHを4に調整し、ろ過することで目的とする化合物−1を白色固体として得た(0.48g、65%:中間体−3(n=10)より)。精製後の1H−NMR(DMSO−d6と1滴D2O)のチャートを図4に示す。
元素分析値(C26H45N5O12H2Oとして)
C H N
計算値(%) 50.3948.97 7.43 10.98
実測値(%) 48.75 7.11 11.15
【0044】
(実施例4) 非対称ナノチューブ形成性非対称双頭型脂質分子(式(1)化合物−1、n=12,m=2)の合成
(4−1)中間体−3(n=12)の合成
テトラデカン二酸(4.8g, 18mmol)をメタノール(200ml)に溶解した。その後100mlの水中で水酸化ナトリウム水溶液(0.4mol/L)を添加した。この溶液に、D(+)−グルコサミン塩酸塩 (2g,9.2mmol)を加えた。これにDMT−MM(3.1g,9.5mmol)を加え、室温で30時間撹拌した。反応終了後、反応液をろ過し、残渣を水とメタノールで洗浄した。pH4に調整しなから、エタノール、水1/1で再沈殿を行い、生成物(2.2g)を得た。収率64%。精製後の1H−NMR(DMSO−d6と1滴D2O)のチャートを図5に示す。
1H−NMR(DMSO−d6と1滴D2O)7.68(dd,J=8.4Hz,NH), 7.52(d,J=8.1Hz,NH),4.92(d,J=3.4Hz,0.7H,H−1α),4.43(d,J=8.1Hz,0.3H,H−1β),3.24−3.69(m,5H),3.04−3.13(m,1H),2.28(t,J=7.4Hz,0.3H,−CH2−COOHα),2.18(t,J=7.4Hz,1.7H,−CH2COOHβ),2.09(t,J=7.5Hz,−CH2−CONH,2H),1.47(d,J=6.8Hz,−CH2CH2CO,4H), 1.24(s,16H,−CH2)。
【0045】
(4−2)化合物−1(n=12,m=2)の合成
前記(4−1)で得られた中間体−3(n=12) (0.4 g, 0.95 mmol)をDMFに加熱溶解し、室温まで冷却したのち塩酸グリシルグリシンメチルエステル(200mg,1.0mmol)とDMT−MM(340mg,1.1 mmol)、トリエチルアミン(1.0mmol)を添加した。室温で2時間撹拌した後、DMFを留去した。得られた固体にメタノールを添加し、不溶の白色固体を濾別することで中間体−5(n=12,m=2)を得た。
これを加熱したメタノール/水溶媒に分散させ、水酸化ナトリウム水溶液(1mol/L,1.5ml)を60℃で添加して加水分解を行った。反応後溶媒を留去し、メタノールを添加し、固体を分散させた後、塩酸を添加してpHを4に調整し、ろ過することで目的とする化合物−1(n=12,m=2)を白色固体として得た(355mg、70%)。精製後の1H−NMR(DMSO−d6と1滴D2O)のチャートを図6に示す。
元素分析値(C24H43N3O10H2Oとして)
C H N
計算値(%) 52.26 8.22 7.64
実測値(%) 52.29 8.18 7.48
【0046】
(実施例5) 非対称ナノチューブ形成性非対称双頭型脂質分子(式(1)化合物−1、(n=12,m=3))の合成
前記実施例(4−1)で得られた中間体−3(n=12) (0.2 g, 0.48 mmol)をDMFに加熱溶解し、室温まで冷却したのち塩酸グリシルグリシルグリシンエチルエステル(114mg,0.52mmol)とDMT−MM(170mg,0.52 mmol)、トリエチルアミン(0.52mmol)を添加した。室温で2時間撹拌した後、DMFを留去した。得られた固体にメタノールを添加し、不溶の白色固体を濾別することで中間体−5(n=12,m=3)を得た。
これを加熱したメタノール/水溶媒に分散させ、水酸化ナトリウム水溶液(1mol/L,0.75ml)を60℃で添加して加水分解を行った。反応後溶媒を留去し、メタノールを添加し、固体を分散させた後、塩酸を添加してpHを4に調整し、ろ過することで目的とする化合物−1(n=12,m=3)を白色固体として得た(210mg、 68%)。精製後の1H−NMR(DMSO−d6)のチャートを図7に示す。
元素分析値(C26H46N4O11H2Oとして)
C H N
計算値(%) 51.30 7.95 9.20
実測値(%) 51.15 7.99 9.05
【0047】
(実施例6) 非対称ナノチューブ形成性非対称双頭型脂質分子(式(1)化合物−1、(n=12,m=4))の合成
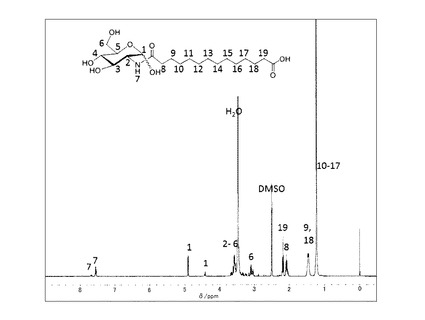
前記実施例(4−1)で得られた中間体−3(n=12)(0.4 g, 0.95 mmol)をDMFに加熱溶解し、室温まで冷却したのち塩酸グリシルグリシンエチルエステル(200mg,1.0mmol)とDMT−MM(340mg,1.0 mmol)、トリエチルアミン(1.0mmol)を添加した。室温で2時間撹拌した後、DMFを留去した。得られた固体にメタノールを添加し、不溶の白色固体を濾別することで中間体−5(n=12,m=4)を得た。
これを加熱したメタノール/水溶媒に分散させ、水酸化ナトリウム水溶液(1.5mol/L,1ml)を60℃で添加して加水分解を行った。反応後溶媒を留去し、メタノールを添加し、固体を分散させた後、塩酸を添加してpHを4に調整し、ろ過することで目的とする化合物−1(n=12,m=4)を白色固体として得た(357mg、 58%)。精製後の1H−NMR(DMSO−d6と1滴D2O)のチャートを図8に示す。
元素分析値(C28H49N5O12H2Oとして)
C H N
計算値(%) 49.18 7.81 10.24
実測値(%) 49.09 7.63 10.37
【0048】
(実施例7) 非対称ナノチューブ形成性非対称双頭型脂質分子(式(1)化合物−1(n=14,m=3)の合成
(7−1)中間体−3(n=14)の合成
ヘキサデカン二酸(1.3g,4.6mmol)をメタノール(90ml)に溶解した。この溶液に、水(2ml)に溶解したD(+)−グルコサミン塩酸塩 (0.5g,2.3mmol)とナトリウムメトキシド (0.125g,2.3mmol)を加えた。メタノール(1ml)に溶解したDMT−MM(0.641g,2.3mmol)を加え、室温で撹拌した。TLCで反応をモニターしながら、DMT−MM(0.96g,3.5mmol)とナトリウムメトキシド(0.25g,4.6mmol)を加え、29時間反応した。反応終了後、反応液をろ過し、残渣を水とメタノールで洗浄した。エタノール、水1/1で再沈殿を行い、生成物(0.34g)を得た。収率33パーセント。精製後の1H−NMR(DMSO−d6と1滴D2O)のチャートを図9に示す。
1H−NMR(DMSO−d6と1滴D2O)7.70(dd,J=8.5Hz,NH), 7.53(d,J=8.0Hz,NH),4.92(d,J=3.3Hz,0.7H,H−1α),4.43(d,J=8.2Hz,0.3H,H−1β),3.24−3.69(m,5H),3.03−3.14(m,1H),2.28(t,J=7.3Hz,0.3H,−CH2−COOHα),2.18(t,J=7.3Hz,1.7H,−CH2COOHβ),2.10(t,J=7.6Hz,−CH2−CONH,2H),1.48(d,J=6.8Hz,−CH2CH2CO,4H), 1.24(s,20H,−CH2)。
【0049】
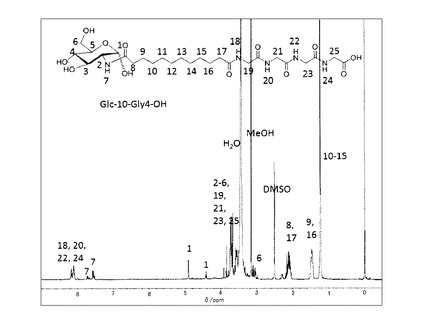
(7−2)非対称双頭型脂質分子(式(1)の化合物−1(n=14,m=3))の合成
上記(7−1)で得られた中間体−3(n=14) (400 mg, 0.9 mmol)をDMFに加熱溶解し、室温まで冷却したのち塩酸グリシルグリシルグリシンエチルエステル(250mg,1.0mmol)とDMT−MM(340mg,1.1 mmol)、トリエチルアミン(1.0mmol)を添加した。室温で2時間撹拌した後、DMFを留去した。得られた固体にメタノールを添加し、不溶の白色固体を濾別することで中間体−5(n=12,m=3)を得た。
これを加熱したメタノール/水溶媒に分散させ、水酸化ナトリウム水溶液(1mol/L,1.5ml)を80℃で添加して加水分解を行った。反応後溶媒を留去し、メタノールを添加し、固体を分散させた後、塩酸を添加してpHを4に調整し、ろ過することで目的とする化合物−1(n=14,m=3)を白色固体として得た(460mg、 78%)。精製後の1H−NMR(DMSO−d6)のチャートを図10に示す。
元素分析値(C28H50N4O11H2Oとして)
C H N
計算値(%) 52.82 8.23 8.80
実測値(%) 52.99 8.09 8.62
【0050】
(実施例8) 非対称ナノチューブ形成性非対称双頭型脂質分子(式(1)の化合物−1(n=14,m=4))の合成
前記実施例(7−1)で得られた中間体−3(n=14) (0.1g, 0.22mmol)をDMFに加熱溶解し、室温まで冷却したのち塩酸グリシルグリシルグリシルグリシンエチルエステル(61mg,0.23mmol)とDMT−MM(68mg,0.24 mmol)を添加した。室温で19時間撹拌した後、DMFを留去した。得られた固体にメタノールを添加し、不溶の白色固体を濾別することで中間体−5(n=14,m=4)を得た。
得られた中間体−5(n=14,m=4)(50mg,0.07mmol)を加熱したメタノール/水溶媒に分散させ、水酸化ナトリウム水溶液(0.1mol/L,2.7ml)を65℃で添加して加水分解を行った。塩酸を添加してpHを4に調整した後溶媒を留去した。メタノールを添加し、不溶の白色固体を濾別することで目的とする化合物−1(n=14,m=4)を白色固体として得た(25mg、41%)。精製後の1H−NMR(DMSO−d6と1滴D2O)のチャートを図11に示す。
本実施例で得られた化合物ー1(n=14、m=4)の1H−NMR(DMSO−d6と1滴D2O、室温)4.91(d,J=3.3Hz,0.7H,H−1α),4.42(d,J=8.2Hz,0.3H,H−1β),3.34−3.76(m,13H),3.04−3.14(m,1H),2.28(t,J=7.3Hz,0.3H,−CH2−COOHα),2.07−2.14(m,3.7H,−CH2COOHβ,−CH2−CONH),1.47(d,J=6.8Hz,−CH2CH2CO,4H), 1.23(s,20H,−CH2)。
元素分析値(C30H53N5O12H2Oとして)
C H N
計算値(%) 51.94 7.99 10.09
実測値(%) 51.88 8.12 10.01
【0051】
(実施例9) 非対称双頭型脂質分子(化合物−1)の自己集合による非対称ナノチューブの形成
まず、前記実施例1において製造された、本発明の非対称双頭型脂質分子のうち、化合物−1(n=10,m=2)を用いて、以下の(1)分散液の加熱による溶解、その後の冷却による方法、及び(2)室温でのナトリウム塩からのpHを調整による方法の2つの方法により非対称ナノチューブを形成させた。
(9−1)加熱・冷却法による方法
化合物−1(n=10,m=2)(5mg)を水(ミリQ水、1ml)に分散、加熱溶解(100℃程度)した。溶液を室温に冷却することで透明でナノチューブの含まれたゲル状物を得た。リンタングステン酸でネガティブ染色後、走査型電子顕微鏡観察(透過モード)から外径約15nm、内径約8nm、長さ5μm以上のナノチューブ構造を確認した(図12A)。
【0052】
(9−2)pH調整法による方法
化合物−1(n=10,m=2)(5mg)を水(ミリQ水、1ml)に分散した。室温下、溶液に水酸化ナトリウム水溶液(1モル/L、0.01mL)を加えて溶解し、さらに塩酸水溶液(1モル/L、0.01mL)を加えてpHを5に調製することでナノチューブの含まれた分散液を得た。リンタングステン酸でネガティブ染色後、走査型電子顕微鏡観察(透過モード)から外径約15nm、内径約8nm、長さ1μm以上のナノチューブ構造を確認した(図12B)。
【0053】
(9−3)その他の非対称双頭型脂質分子における非対称性ナノチューブ形成性の確認
さらに、前記実施例2〜8で製造された非対称双頭型脂質分子についても、上記(9−1)及び(9−2)と同様の方法で自己集合させることで、同様なサイズのナノチューブ構造が形成することを確認した。
前記実施例1で製造された化合物−1(n=10,m=2の場合)の加熱冷却およびpH調製法による自己集合で得られた非対称ナノチューブの電子顕微鏡観察像(透過像)をそれぞれ図12AおよびBに示す。
前記実施例2で製造された化合物−1(n=10,m=3の場合)の加熱冷却およびpH調製法による自己集合で得られた非対称ナノチューブの電子顕微鏡観察像(透過像)をそれぞれ図13AおよびBに示す。
前記実施例3で製造された化合物−1(n=10,m=4の場合)の加熱冷却およびpH調製法による自己集合で得られた非対称ナノチューブの電子顕微鏡観察像(透過像)をそれぞれ図14AおよびBに示す。
前記実施例4で製造された化合物−1(n12,m=2の場合)の加熱冷却およびpH調製法による自己集合で得られた非対称ナノチューブの電子顕微鏡観察像(透過像)をそれぞれ図15AおよびBに示す。また、挿入図は、チューブが絡まり合って良く分散した透明なゲル状物を与えており、チューブの分散性が高いことを示す。
前記実施例5で製造された化合物−1(n=12,m=3の場合)の加熱冷却およびpH調製法による自己集合で得られた非対称ナノチューブの電子顕微鏡観察像(透過像)をそれぞれ図16AおよびBに示す。
前記実施例6で製造された化合物−1(n=12,m=4の場合)のpH調製法による自己集合で得られた非対称ナノチューブの電子顕微鏡観察像(透過像)をそれぞれ図17AおよびBに示す。
前記実施例7で製造された化合物−1(n=14,m=3の場合)のpH調製法による自己集合で得られた非対称ナノチューブの電子顕微鏡観察像(透過像)を図18に示す。
【0054】
(実施例10) 非対称ナノチューブの短尺化と凍結乾燥
(10−1) 非対称ナノチューブの短尺化
前記実施例(9−3)において、化合物−1(n=12、m=4)に対して加熱冷却法を適用して形成された非対称ナノチューブを、超音波処理することで短尺化したナノチューブを得た。すなわち上記ナノチューブ溶液(5mg/mL)を、プローブ型超音波処理機で4℃、1分間処理することで得られた溶液は透明な分散液であった。この溶液を走査型電子顕微鏡で観察したところ、長さが100〜500nmの短尺状ナノチューブが得られることがわかった。さらに時間を2分にすると50〜200nmの長さに短尺化した。また同処理によって得た分散液を60日以上、4度で保存してもナノチューブ構造が安定であることを確認した。図19に加熱冷却によって得た化合物−1(n=12,m=4)から得た非対称ナノチューブの超音波処理後の電子顕微鏡観察像を示す。挿入図は、加熱冷却によって調製されたナノチューブ分散液を示す。
【0055】
(10−2) 非対称ナノチューブの凍結乾燥
ここでは、本発明の非対称ナノチューブが凍結乾燥により長期保存が可能であることを示すために、上記(10−1)で得られた非対称ナノチューブの超音波処理物を凍結乾燥後再水和して非対称ナノチューブ分散液を得た。
具体的には、上記(10−1)で得られた非対称ナノチューブの超音波処理物を凍結乾燥し、30日間室温で保持した後、ミリQ水を添加することで再水和し、高濃度(5mg/mL)の分散液を得た(図21)。この分散液は60日以上、4度で安定であることを確認した。図20に加熱冷却によって得た化合物−1(n=12,m=4)から得た非対称ナノチューブの超音波処理、凍結乾燥、再水和後の電子顕微鏡観察像を示す。
【0056】
(実施例11) 薬剤内包化実験
前記実施例10で製造した非対称性ナノチューブを用いてドキソルビシンの内包化を以下の実験によりおこなった。
ドキソルビシン溶液200μL(それぞれ2,1,0.714,0.625,0.555,0.5mg/mLのミリQ水に溶解したもの)と上記非対称ナノチューブの分散液(200μL、5mg/mL)を混合し、30分室温で放置する。200μLを取り、200nmの多孔質膜でろ過し、さらにミリQ水で洗浄する(400μL)。固体はろ紙ごと200μLのミリQ水に再分散した。ドキソルビシンのUV吸収(480nm)によって、ろ過前の溶液とろ過後の溶液の吸収を比較して、カプセル化率を求めた。図21にドキソルビシンをカプセル化後の非対称ナノチューブの電子顕微鏡による観察像とそのカプセル化率を示す。
その結果、図21(B)のように、ONT/DOX=10/1〜7/1では、90%以上がカプセル化されていることから、混合するだけで高いカプセル化率を示すことが示された。また図21(A)の画像からも明らかなように、ドキソルビシンをカプセル化後もナノチューブの形態は変化していない。そして、溶液中での凝集は認められず、高い分散性を維持していることも確認した。
【0057】
(実施例12) 薬剤放出実験
次に、前記(実施例11)でドキソルビシンを内包化した非対称性ナノチューブからのドキソルビシンの放出実験を、PBSバッファー(pH7.4とpH5.5)中での透析法によって行った。その結果、図22に示したように、中性付近(pH7.4)では、10時間後でやっと全内包化量のうち約62%程度が放出され、32時間たっても放出量は全体の70%にとどまっているのに対して、弱酸性状態(pH5.5)では、4時間目で82%以上を放出し、8時間ではほぼ全量放出してしまうことがわかり、非対称ナノチューブ内表面を覆うカルボキシル基から生じたカルボキシラートのプロトン化が刺激となって抗癌剤の放出を加速できることを示している。この低pH条件で抗癌剤放出が加速される点は、一般に、ガン組織および細胞は、正常細胞に比べて酸性(低pH)であるという特性を有するから、ガン組織での選択的な薬剤放出が期待でき、ナノチューブを薬剤カプセルとして利用することの有効性は極めて高いと考えられる。
【0058】
(実施例13) 抗癌剤組成物としての機能
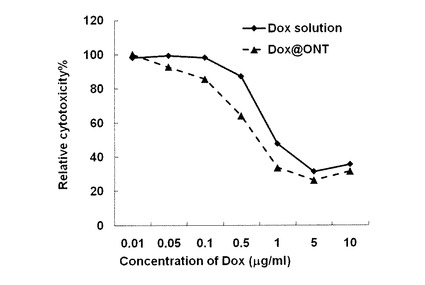
Hela細胞を用いてドキソルビシン内包化非対称ナノチューブ(化合物−1でn=12,m=4の場合。カプセル化はONT/DOX=7/1で行ったもの。)に対して、MTSアッセイによりHela細胞への細胞毒性(抗癌活性)を調べた。その結果、図23に示したように、フリーのドキソルビシンよりも高い活性を示すことがわかった。これは、ドキソルビシンをカプセル化した非対称ナノチューブが、癌細胞内のエンドソームに取り込まれ、エンドソーム特有の低いpH環境にさらされることでチューブ内表面のカルボキシレートがプロトン化され、ドキソルビシンを速やかに放出したためと考えられる。
このように、本発明の非対称ナノチューブによるドキソルビシンなどカチオン性の抗癌剤カプセル化物は、実施例12に記述したような、若干酸性状態のがん組織付近での選択的な薬剤放出効果と共に、本実施例におけるがん細胞内での速やかな薬剤放出による高い抗癌活性の発現という二つの効果が期待できる。すなわち、本発明の非対称ナノチューブによるカチオン性抗癌剤カプセル化物は、優れた抗癌剤組成物としての機能を有していることが示された。
【技術分野】
【0001】
本発明は、薬剤のカプセル化に好適な中空繊維状の非対称ナノチューブ構造を形成可能な非対称双頭型脂質分子とその製造方法に関し、さらに前記脂質分子が自己集合により形成する中空繊維状有機ナノチューブ、および当該ナノチューブを用いた薬剤カプセル化への用途に関する。
【背景技術】
【0002】
ある種の脂質分子等の有機分子は、その溶媒中での自己集合によって中空繊維状構造(有機ナノチューブとよぶ)を形成することが知られている(非特許文献1、2)。これらは、外径約10nm〜1000nm、長さ約100nm〜数mmのチューブ構造を形成し、その内部に微少な中空空間を有するため、種々の材料(タンパク質、薬剤、DNA等)をカプセル化することができる。この有機ナノチューブは、水素結合やファンデルワールス力などの弱い相互作用による自己集合作用で形成されているため、構成分子の構造に依存して室温から100℃の範囲のある温度やpH等の固有の条件によって可逆的にチューブ構造を分解することが可能である。この温度やpHは生体中の環境温度やpHの範囲を含むものであるから、生体内でこれを分解するように調節可能であることを意味する。またチューブの分解がなくても、内部にカプセル化された材料がチューブ両端の開口部より徐々に放出される特性も有する(非特許文献3、4)。
これらの諸特性を用いることで、薬剤送達用および薬剤徐放用のカプセルやタンパク質などの固定マトリクスおよび保存カプセル等への応用が期待されている(非特許文献5、6)。しかし、従来の大多数の有機ナノチューブは、内外表面が同じ親水部で被覆されているため、薬剤やタンパク質などの材料を有機ナノチューブ内部へ選択的かつ効率的にカプセル化することは困難であった(非特許文献7,8、9)。具体的には、諸材料のカプセル化に際し、有機ナノチューブ内部の水を減圧下での凍結乾燥などで除去した後、材料を分散・溶解させた水溶液を添加し再水和させる(いわゆる毛細管現象)という煩雑な操作が必要とされていた(非特許文献7、8)。
【0003】
本発明者らは、従来から容易かつ効率的な薬剤のカプセル化が可能な有機ナノチューブとして、内外表面がそれぞれ異なる官能基で被覆されているという特徴を持つ様々な有機ナノチューブ(以降、非対称ナノチューブという)の研究開発を行ってきた(非特許文献10、11、12、13)。これら非対称ナノチューブは、いずれも疎水部アルキレン鎖の両端に異なる二つの親水部、具体的には、片端に1−グルコピラノシルアミン、もう一端にカルボキシル基又はアミノ基がそれぞれ連結された非対称な双頭型の脂質分子(以降、「非対称双頭型脂質」とよぶ)により形成されている(特許文献1)。これらの非対称ナノチューブは、通常室温で混合するだけでナノチューブ内表面とは逆の電荷を有する材料をカプセル化することができる。片端がカルボキシル基のカルボン酸系脂質分子を用いた場合はチューブ内表面が選択的にカルボキシル基で被覆されることにより負に荷電し、反対にアミノ基を有しているアミン系脂質分子を用いた場合は正に荷電するため、前者は正電荷を帯びた薬剤を包接し輸送する薬剤送達カプセル又は薬剤キャリアとして好ましく、後者は負電荷を帯びた薬剤輸送用の薬剤キャリアの他、細胞内への遺伝子輸送など核酸キャリアとしての用途も期待される。
本発明者らは、これまでにこれらの非対称ナノチューブを用いて、タンパク質、蛍光分子、オリゴDNAなどの選択的なカプセル化やin vitroでの放出挙動などの研究を報告している(非特許文献10、11、12、13)。このような様々な非対称ナノチューブが提供され、これら非対称ナノチューブによるカプセル化によって、生体高分子や薬剤などの不安定な材料でも、簡便かつ選択的、効率的なカプセル化が可能となってきた。
しかしながら、これらの非対称ナノチューブのカプセル用材料としての実用化には、非対称ナノチューブの水和状態での安定性向上や封入性能の向上など解決すべき問題点があり、さらに、非対称ナノチューブ製造が困難な場合や、チューブの原料となる非対称双頭型脂質分子の合成工程が煩雑な場合や精製が困難な場合があるなどの問題点があった。
すなわち、実用的な薬剤カプセル化用非対称ナノチューブとしては、以下の各点を全て満たす非対称ナノチューブの開発が必須である。
1.原料となる非対称双頭型脂質分子の合成・精製工程が簡便であること。
2.非対称ナノチューブの製造が、室温で、又は少なくとも温和な条件で、かつ選択的に行うことができること。
3.非対称ナノチューブが水和状態での長期保存安定性を有すること。
4.包接対象化合物を効率的に高濃度でカプセル化でき、かつ簡単に放出可能であること。
これまでには、部分的にこれらの問題点を解決できているものがあっても、これら全ての点を解決できた非対称ナノチューブの報告は、カルボン酸系脂質分子でもアミン系脂質分子を用いた場合でも見られなかった。
【0004】
一方で、従来は薬剤カプセルとして主にリポソーム系の薬剤カプセルが用いられていたが、カプセル化にpH勾配を用い、その後の精製が必要なため、調製が複雑で長時間必要であった。そこで、簡単に、効率的に薬剤をカプセル化でき、薬剤を安定に保持すると共に、癌細胞などターゲット細胞内では速やかに徐放できるカプセル化剤の開発が望まれていた。特に、ドキソルビシン、イダルビシン、エピルビシン、ダウノルビシン、ピラルビシンなどのアントラサイクリン系抗癌剤は、毒性、特に心臓毒性が強いため、癌細胞に直接効果的に送達し、かつ徐放が可能な優れたカプセル化剤が求められていた。
上述のように、カルボン酸系脂質分子により形成される非対称ナノチューブは、糖親水部により外表面が覆われ、内表面はカルボキシル基により構成されて負の電荷を有するため、ドキソルビシンなどのアミノ基を有する抗癌剤送達のための有効なカプセル剤として期待される。さらにこの様なナノチューブは、pHなどの外部環境によって内表面のカルボキシル基がイオン的/非イオン的な変化又はチューブの形態変化による機能的な薬剤の放出も期待できる。
しかし、本発明者らの報告しているカルボン酸系の非対称双頭型脂質分子である1−グルコピラノシルアミノ基が長鎖ジカルボン酸の一端でアミド結合を介して結合した脂質分子が形成する非対称ナノチューブ(非特許文献10)では、非対称ナノチューブの製造過程でマイクロチューブやテープ状の集合体も同時に生成するため、遠心分離などによるナノチューブの分離精製工程が必要であり、得られたナノチューブが水性溶媒中での分散性が悪く、また室温や冷蔵でも長期保存により結晶化しやすく長期的安定性が乏しい欠点があった。なお、同じ脂質分子を用い、製造過程で低分子有機化合物をインターカレートさせた非対称ナノチューブ(特許文献3)についても同様の長期的安定性が乏しい欠点がある。
また、カルボキシル基を親水部として有する脂質分子にオリゴグリシン残基を導入することで、得られる非対称ナノチューブの水分散性を高めることができ、水性溶媒中での長期保存性も高まった(非特許文献14)が、同脂質からの非対称ナノチューブの製造には、脂質分子を、ミリQ水(超純水)に分散して100℃前後に加熱するか、あるいは脂質を等量の水酸化ナトリウムを含む水溶液に60℃以上に加熱して溶解し、その後、室温で中和する工程が必要であるという煩雑で高度な技術を要するばかりか、ナノチューブ原料の脂質分子の合成に7段階以上の合成過程と過程毎のカラムクロマトグラフィー等による精製が必要なため、大量に合成することは極めて困難であり、実用的ではない。
簡便な非対称双頭型脂質分子の合成を目指す試みの1つとして、グリシン残基を含まないカルボン酸系非対称双頭型脂質分子において、他端のグルコピラノシルアミン残基として2−グルコサミンを用いたナノチューブを作製した(非特許文献15)。その脂質分子自体の合成はきわめて簡便で、2−グルコサミンと長鎖ジカルボン酸という市販の化合物からの1段階合成と再結晶で合成可能であったが、自己集合させても分散性に乏しく、ナノチューブとその断片状のものの混合物しか得られず、ナノチューブの詳細な解析結果からみて、内外表面を被覆する官能基が同じで非対称ナノチューブが形成できていない(未発表データ)。
また、1−グルコピラノシル基を親水部として有するカルボン酸系脂質分子の疎水部に重合性官能基であるジアセチレン残基を導入した場合も、ナノチューブは形成する(特許文献2)が、このナノチューブも詳細な構造解析によると非対称ナノチューブが形成されていなかった(未発表データ)。
【0005】
以上の技術的な背景から、上記の全ての課題を解決できる内表面がカルボキシル基で被覆された非対称有機ナノチューブ及びそのための新規な非対称双頭型脂質分子の提供が強く期待されていた。すなわち、入手しやすい原料から簡便に合成できる非対称双頭型脂質分子であって、マイルドな条件で非対称ナノチューブを、選択的かつ高収率に製造できて、得られる非対称ナノチューブが長期保存安定性を有すると共に、ドキソルビシンなどのカチオン性薬剤を効率的に高濃度でカプセル化でき、しかもpH条件変化などで簡単に放出可能なカプセル化物を製造できる非対称双頭型脂質分子の提供が切望されていた。
【先行技術文献】
【特許文献】
【0006】
【特許文献1】特許第4174702号公報
【特許文献2】特開2005−239632
【特許文献3】特開2008−264897
【非特許文献】
【0007】
【非特許文献1】Toshimi Shimizu, Mitsutoshi Masuda, Hiroyuki Minamikawa,Chemical Review,2004,105,1401-1443.
【非特許文献2】“有機・無機・金属ナノチューブ 非カーボンナノチューブ系の最新技術と応用展開”、清水敏美、木島 剛 編集、フロンティア出版、東京、2008.
【非特許文献3】増田光俊、亀田直弘、ネットワークポリマー、2010,31,191-200.
【非特許文献4】Naohiro Kameta、Hiroyuki Minamikawa,Mitsutoshi Masuda,Go Mizuno,Toshimi Shimizu,Soft Matter,2008,4,1681-1687.
【非特許文献5】Chia-Chun Chen,Yao-Chung Liu,Chia-Hsuan Wu,Chun-Chia Yeh,Ming-Tsan Su,Yi-Chun Wu,Advanced Materials,2005,17,404-407.
【非特許文献6】Mohammand Reza Abidian,Dong-Hwan Kim,David C Martin,Advanced Materials,2006,18,405―409.
【非特許文献7】Bo Yang,Shoko Kamiya,Yoshiki Shimizu,Naoto Koshizaki,Toshimi Shimizu,Chemistry of Materials,2004,16、2826-2831.
【非特許文献8】Hiroharu Yui,Yoshiki Shimizu,Shoko Kamiya,Ichiro Yamashita,Mitsutoshi Masuda,Kohzo Ito,Toshimi Shimizu,Chemistry Letters,2005,34,232-233.
【非特許文献9】“環状・筒状超分子新素材の応用技術”、高田十志和 編集、シーエムシー出版、東京、2006,P138-149.
【非特許文献10】Mitsutoshi Masuda,Toshimi Shimizu,Langmuir,2004,20,5969-5977.
【非特許文献11】Naohiro Kameta,Mitsutoshi Masuda,Hiroyuki Minamikawa,Nikolay V.Gotev, Jeong A.Rim,Jong H.Jung, Toshimi Shimizu,Advanced Materials,2005,17,2732-2736.
【非特許文献12】Naohiro Kameta,Go Mizuno,Mitsutosi Masuda, Hiroyuki Minamikawa, Masaki Kogiso, Toshimi Shimizu,Chemistry Letters,2007,36,896-897.
【非特許文献13】Naohiro Kameta,Kaname Yoshida,Mitsutoshi Masuda,Toshimi Shimizu,Chemistry of Materials,2009,21,5892-5898.
【非特許文献14】Soo Jing Lee,Naohiro Kameta, Hiroyuki Minamikawa,Mitsutoshi Masuda,Toshimi Shimizu,第58回高分子討論会予稿集,発表番号1Pe047、“Function of Metal Cation-Doped Nanotube by using Unsymmetrical Bolaamphiphiles”.
【非特許文献15】増田光俊、和田百代、清水敏美、第59回高分子年次大会、発表番号2Pd002、「アノマー混合物からなるグルコサミン系双頭型脂質の有機ナノチューブ形成」.
【非特許文献16】Naohiro Kameta,Mitsutoshi Masuda,Hiroyuki Minamikawa,Toshimi Shimizu,Langmuir,2007,23,4634-4641.
【非特許文献17】George Fotakis and John A.Timbrell,Toxocology Letters,2006,160,171-177.
【発明の開示】
【発明が解決しようとする課題】
【0008】
本発明は、入手しやすい原料から簡便に合成でき、かつマイルドな条件で非対称ナノチューブを選択的かつ高収率に製造できる非対称双頭型脂質分子を提供しようとするものであり、また、当該非対称双頭型脂質分子の自己集合で得られる非対称ナノチューブとして、高純度かつ比較的高濃度の長期保存でも安定性を有すると共に、ドキソルビシンなどのアミノ基を有する薬剤を選択的かつ効率的に高濃度でカプセル化でき、かつpH条件変化などで簡単に放出可能なカプセル化物を製造できる非対称ナノチューブを提供することを目的とする。そして、当該非対称ナノチューブにより、ドキソルビシンなどのカチオン性薬剤が選択的、高濃度にカプセル化された、刺激応答性徐放機能の高いカプセル化薬剤組成物を提供することを目的とするものである。
【課題を解決するための手段】
【0009】
本発明者らは上記の課題を解決すべく鋭意研究を重ねた結果、本発明者らが以前に開発した、脂質分子自体の合成はきわめて簡便であったが、非対称有機ナノチューブを形成できない、片側に2−グルコサミンのアミノ基と結合させた長鎖ジカルボン酸からなる非対称双頭型脂質分子(非特許文献15)の誘導体を製造することに思い至った。具体的には、当該非対称双頭型脂質分子の末端のカルボキシル基に対して、さらにオリゴグリシンを脱水縮合反応により連結して、下記一般式(1)であらわされる脂質分子(化合物−1)を合成したところ、精製工程も不要で穏和な条件で簡単に製造できたばかりでなく、得られた脂質分子は水分散性もよく、非対称ナノチューブのみを高選択的に形成し、さらに得られた非対称ナノチューブは、ミリQ水中、比較的高濃度である5mg/ml程度での、室温あるいは冷蔵下での6ヶ月を超える長期間の分散保存でも安定にチューブ形態を保持したまま分散状態を保持できることを見いだした。本発明で得た脂質からのナノチューブの製造が、ミリQ水からの加熱・冷却のみならず、中和によって完全に室温で達成できたことは驚くべきことである。すなわち本脂質は等モル等量の水酸化ナトリウム水溶液に室温で速やかに溶解し、この溶液を室温で塩酸等によって中和することで非対称ナノチューブを製造可能であった。さらに、脂質分子の合成も市販の原料から数えても僅か3段階の反応で完了し、全ての精製行程において反応の進行で生じる固体のろ過と、得られた固体の洗浄で十分な純度の脂質を与えることができた点もきわめて驚くべき特徴である。これらの優れた非対称ナノチューブとしての特性は、従来のどのカルボン酸系非対称ナノチューブにおいても達成できなかったばかりか、アミン系非対称ナノチューブなども含めた非対称有機ナノチューブ全てにおいてもはじめてである。
そして、これにより得た非対称ナノチューブはドキソルビシンを効率的にカプセル化する薬剤カプセルとしても機能することを確認し、本発明を完成させた。
【0010】
すなわち、本発明は以下の一般式(1)で表される非対称双頭型脂質分子(化合物−1ともいう。)又はその塩
(式中、nは8〜16の整数を表し、mは1〜4の整数を表し、波線で表記した2−グルコサミンの1位水酸基はα、βアノマーのいずれか、あるいはその平衡混合物を表す。)
とその合成方法、および室温下での同脂質のナトリウム塩からの中和によることを特徴とする自己集合によって製造される、高純度で、長期安定性を有する非対称ナノチューブを提供するものであり、上記脂質分子から得られた非対称ナノチューブとアミノ基を有する薬剤又はカチオン性薬剤のカプセル化物を提供するものである。
【0011】
具体的には、本発明は以下の通りである。
〔1〕 下記一般式(1)で表される非対称双頭型脂質分子又はその塩;
(式中、nは8〜16の整数であり、mは1〜4の整数を表す。また、式中の波線で表記した2−グルコサミンの1位水酸基はα、βアノマーのいずれか、あるいはその平衡混合物を表す。)。
〔2〕 下記一般式(2)で表されるα,ω−長鎖ジカルボン酸
HOOC−(CH2)n−COOH 一般式(2)
(式中、nは8〜16の整数を表す。)
を水性メタノール中で、2−グルコサミンと縮合させ、生じた固体をろ過、洗浄することで、下記一般式(3)で表される中間体−3
(式中nは8〜16の整数を表す。また、式中の波線で表記した2−グルコサミンの1位水酸基はα、βアノマーのいずれか、あるいはその平衡混合物を表す。)
を製造し、
当該中間体−3と、オリゴグリシンのC端をエステルなどで保護した下記一般式(4)で表されるオリゴグリシンエステル
(式中mは1〜4の整数を表す。またRはアルコール性水酸基を有する化合物の残基であり、例えば、メチル基、エチル基、三級ブチル基、ベンジル基である。)
を加えて縮合させ、下記一般式(5)で表される中間体−5
(式中nは8〜16の整数を表し、mは1〜4の整数を表す。またRはアルコール性水酸基を有する化合物の残基であり、例えば、メチル基、エチル基、三級ブチル基、ベンジル基である。また式中の波線で表記した2−グルコサミンの1位水酸基はα、βアノマーのいずれか、あるいはその平衡混合物を表す。)
を得、これをアルカリ加水分解することで脱保護し、下記一般式(1)で表される非対称双頭型脂質分子又はその塩
(式中、nは8〜16の整数であり、mは1〜4の整数を表す。また、式中の波線で表記した2−グルコサミンの1位水酸基はα、βアノマーのいずれか、あるいはその平衡混合物を表す。)
を製造する方法。
〔3〕 前記〔1〕に記載の非対称双頭型脂質分子又はその塩から形成された、内表面がカルボキシル基で被覆され、外表面が2−グルコサミド基で被覆された構造を有することを特徴とする非対称ナノチューブ。
〔4〕 前記〔1〕に記載の非対称双頭型脂質分子を水に分散し、加熱溶解後冷却するか、又は室温で水又はアルカリ性溶液に分散溶解後、酸で中和することを特徴とする、非対称ナノチューブの製造方法。
〔5〕 さらに、凍結乾燥処理又は超音波処理を施すことで、短尺チューブ状とすることを特徴とする、前記〔4〕に記載の非対称ナノチューブの製造方法。
〔6〕 前記〔3〕の非対称ナノチューブがカチオン性薬剤を包接していることを特徴とする、カチオン性薬剤−非対称ナノチューブ複合体。
〔7〕 前記〔6〕に記載のカチオン性薬剤−非対称ナノチューブ複合体を有効成分とする、カチオン性薬剤組成物。
〔8〕 カチオン性薬剤がアミノ基を有するアントラサイクリン系抗癌剤である、前記〔7〕に記載のカチオン性薬剤組成物。
【発明の効果】
【0012】
本発明によれば、内表面がカルボキシル基、外表面が糖残基で被覆された非対称ナノチューブにおいて従来困難であった、室温下のpH変化といった温和な条件で選択的かつ高収率で製造することができる。また高い分散性を有するため、水性溶媒中で高濃度でも結晶化を起こすことなく、長期保存における安定性も非常に高く、5mg/mlといった比較的高濃度の水分散液でも2ヶ月以上安定に保存可能である。また、この非対称ナノチューブを構成する非対称双頭型脂質分子は新規化合物であって、その原料となる2−グルコサミン、長鎖ジカルボン酸及びオリゴグリシン類は安価であり、わずか3段階の穏和な条件での合成反応により製造でき、精製にはカラムクロマトグラフィーを使う必要がなく、全て再沈殿工程で高純度の脂質を得ることができる。
この非対称ナノチューブ形成性脂質分子の合成および精製が簡便であり、非対称ナノチューブも温和な条件で簡単に製造できる点は、薬剤キャリアとしてのカプセルの大量合成および最終的な薬剤組成物の低価格化の点でも重要である。
そして、本発明の非対称ナノチューブの長期保存安定性や高濃度での良分散性は、抗癌剤治療においてそれぞれ薬剤の安全性の点や患者の負担軽減を図るために経静脈投与する全液量を可能な限り減らす点で必要不可欠である。また本非対称ナノチューブは外径が僅か15nmで、長さを超音波処理などの物理的な擾乱で100nm〜2μmまで調節可能であるためEPR(Enhanced Permiability and Retation)効果によって、ガン組織へ送達する量を高めることが可能となる。このように、本発明の非対称ナノチューブが備えているこれら安定性や良分散性といった特性は、薬剤キャリアとしての優れた特性の1つである。
また、本発明の非対称ナノチューブは内側がカルボキシル基に覆われ、中性付近のpHでは負の電荷を帯びているので、ドキソルビシンのようなアミノ基を有する薬剤やカチオン性薬剤を混合するだけで、薬剤のカプセル化が可能であるという優れた特性を有する。調製が複雑な従来のリポソーム系の薬剤カプセルは治療時に任意の比率で混合することは不可能であり、あらかじめ薬剤をカプセル化したもののみが市販されていたが、本発明では、薬剤カプセルとしてのナノチューブを医療現場で速やかに調整することや、ナノチューブ分散液と薬剤を治療時に任意の割合で混合することでカプセル化が可能である。上記したように原料の脂質合成やナノチューブの調製が極めて容易なため、安価な薬剤カプセルとして提供することも可能である。
そして、本発明の非対称ナノチューブはpH変化などの外部の刺激に応じ、チューブ内表面を被覆するカルボキシル基のイオン化状態の変化又はチューブ構造そのものの形態変化などによって、条件選択的かつ効率的に薬剤を放出することができる。このため、上記のEPR効果によるがん組織へのナノチューブの選択的送達に加え、がん組織特有の低pH環境での効率的な薬剤放出が可能である。さらに、がん細胞内に取り込まれたナノチューブ複合体の場合は、エンドソーム内での低いpH環境による速やかな放出が可能となる。
これによって、医療用のドラッグデリバリーシステムにおける機能性薬剤徐放性キャリアとして用いることができるほか、化粧品分野、食品分野などにおける、乳化剤、分散剤、安定剤用キャリアなどとしての利用も期待できる。
【図面の簡単な説明】
【0013】
【図1】中間体−3(n=10)の1H−NMR(DMSO−d6と1滴D2O、室温)
【図2】化合物−1(n=10,m=2)の1H−NMR(DMSO−d6と1滴D2O、室温)
【図3】化合物−1(n=10,m=3)の1H−NMR(DMSO−d6と1滴D2O、室温)
【図4】化合物−1(n=10,m=3)の1H−NMR(DMSO−d6と1滴D2O、室温)
【図5】中間体−3(n=12)の1H−NMR(DMSO−d6と1滴D2O、室温)
【図6】化合物−1(n=12,m=2)の1H−NMR(DMSO−d6、室温)
【図7】化合物−1(n=12,m=3)の1H−NMR(DMSO−d6、室温)
【図8】化合物−1(n=12,m=4)の1H−NMR(DMSO−d6と1滴D2O、室温)
【図9】中間体−3(n=14)の1H−NMR(DMSO−d6と1滴D2O、室温)
【図10】化合物−1(n=12,m=3)の1H−NMR(DMSO−d6、室温)
【図11】化合物−1(n=14,m=4)の1H−NMR(DMSO−d6と1滴D2O、室温)
【図12】化合物−1(n=10,m=2の場合)の(A)加熱冷却および(B)pH調製法による自己集合で得られた非対称ナノチューブの電子顕微鏡観察像(透過像)。
【図13】化合物−1(n=10,m=3の場合)の(A)加熱冷却および(B)pH調製法による自己集合で得られた非対称ナノチューブの電子顕微鏡観察像(透過像)。
【図14】化合物−1(n=10,m=4の場合)の(A)加熱冷却および(B)pH調製法による自己集合で得られた非対称ナノチューブの電子顕微鏡観察像(透過像)。
【図15】化合物−1(n=12,m=2の場合)の(A)加熱冷却および(B)pH調製法による自己集合で得られた非対称ナノチューブの電子顕微鏡観察像(透過像)。左図中の挿入図は 加熱冷却によって調製されたナノチューブからなるヒドロゲル。
【図16】化合物−1(n=12,m=3の場合)の(A)加熱冷却および(B)pH調製法による自己集合で得られた非対称ナノチューブの電子顕微鏡観察像(透過像)。
【図17】化合物−1(n=12,m=4の場合)の(A)加熱冷却および(B)pH調製法による自己集合で得られた非対称ナノチューブの電子顕微鏡観察像(透過像)。
【図18】化合物−1(n=14,m=3の場合)のpH調製法による自己集合で得られた非対称ナノチューブの電子顕微鏡観察像(透過像)。
【図19】加熱冷却によって得た化合物−1(n=12,m=4)から得た非対称ナノチューブの超音波処理後の電子顕微鏡観察像(透過像)。左図中の挿入図は標記ナノチューブ分散液の様子。
【図20】加熱冷却によって得た化合物−1(n=12,m=4)から得た非対称ナノチューブの超音波処理、凍結乾燥、再水和後の電子顕微鏡観察像(透過像)。挿入図(左)は標記分散液の様子。挿入図(右図)は、電子顕微鏡観察H像の拡大像。
【図21】(A)ドキソルビシンをカプセル化した非対称ナノチューブ(化合物−1でn=12,m=4の場合)の電子顕微鏡観察像と(B)非対称ナノチューブによるドキソルビシンのカプセル化率。
【図22】非対称ナノチューブ(化合物−1でn=12,m=4の場合。カプセル化はONT/DOX=7/1で行ったもの)からのドキソルビシンの放出実験。図中の括弧内は用いたバッファーのpHを表す。
【図23】ドキソルビシンナノチューブのHela細胞への抗癌特性(MTSアッセイ)。
【発明を実施するための形態】
【0014】
1.本発明の非対称双頭型脂質分子について
(1−1)本発明の非対称双頭型脂質分子の特徴:一般式(1)の化合物(化合物−1)
本発明の非対称ナノチューブ形成性の脂質分子は文献未載の新規化合物であり、その形状は、長鎖カルボン酸の片端にアミド基を介し2−グルコサミン残基が連結し、もう一端にオリゴグリシンのN端が縮合した非対称双頭型脂質分子である。
本発明の非対称双頭型脂質分子は、合成が非常に簡便で、かつ外径15nm前後、内径約8−9nmの非対称なナノチューブ構造を、室温でのpH変化により選択的かつ高純度に製造できる。そのために、本発明の脂質分子のアルキレン鎖の鎖長は8〜16の範囲である必要があり、脂質合成時の分散性の良さや精製の容易さからは、10〜14の整数が好ましく、偶数であることがより好ましい。またオリゴグリシン残基の数は、1〜4の範囲であればよいが、ナノチューブ製造時の精製のしやすさ及び得られるチューブの純度の観点から2〜4が望ましい。また、本発明の非対称双頭型脂質分子は、水溶性の製薬上許容される塩の状態であってもよい。例えば、ナトリウム塩、アンモニウム塩などが好ましい。
親水部として用いられる2−グルコサミンは、D体でもL体のいずれを用いてもよいが入手の容易さからD体が好適である。本発明の実施の態様ではD体を用いたので、以下、D体を用いた場合で説明するが、本発明はこれに限られるものではない。
すなわち、本願発明の非対称双頭型脂質分子は,下記一般式(1)で表される。
(式中、nは8〜16の整数であり、mは1〜4の整数を表す。また、式中の波線で表記した2−グルコサミンの1位水酸基はα、βアノマーのいずれか、あるいはその平衡混合物を表す。)
【0015】
(1−2)本発明の非対称双頭型脂質分子の製造方法
前記一般式(1)で表される非対称双頭型脂質分子又はその塩は、例えば以下に示す方法により容易に製造することができる。
下記一般式(2)で表されるα,ω−長鎖ジカルボン酸
HOOC−(CH2)n−COOH 一般式(2)
(式中、nは8〜16の整数を表す。)
を水性メタノール中で、2−グルコサミンと縮合させ、生じた固体をろ過、洗浄することで、下記一般式(3)で表される中間体−3
(式中nは8〜16の整数を表す。また式中の水酸基の波線は、式中の波線で表記した2−グルコサミンの1位水酸基はα、βアノマーのいずれか、あるいはその平衡混合物を表す。)
を製造することができる。
次にこの一般式(3)の中間体−3とオリゴグリシンのC端をエステルなどで保護した下記一般式(4)で表されるオリゴグリシンエステル
(式中mは1〜4の整数を表す。またRはアルコール性水酸基を有する化合物の残基であり、例えば、メチル基、エチル基、三級ブチル基、ベンジル基である。)
を加えて縮合させ,下記一般式(5)で表される中間体−5
(式中nは8〜16の整数を表し、mは1〜4の整数を表す。またRはアルコール性水酸基を有する化合物の残基であり、例えば、メチル基、エチル基、ベンジル基、三級ブチル基、である。また式中の波線で表記した2−グルコサミンの1位水酸基はα、βアノマーのいずれか、あるいはその平衡混合物を表す。)
を得た。
次いで、これをアルカリ加水分解することで脱保護し、下記一般式(1)で表される非対称双頭型脂質分子(化合物−1ともいう。)又はその塩
(式中、nは8〜16の整数であり、mは1〜4の整数を表す。また、式中の波線で表記した2−グルコサミンの1位水酸基はα、βアノマーのいずれか、あるいはその平衡混合物を表す。)
を合成することができる。
【0016】
(1−3)中間体−3(一般式(3))を得る縮合工程について
本発明の中間体−3(一般式(3))は、典型的には前記非特許文献15に記載された脂質分子を含むので、非特許文献15に記載の方法に従って、又はそれと同様の方法で製造することができる。
(ア)原料の下記一般式(2)で表されるα,ω−長鎖ジカルボン酸について
HOOC−(CH2)n−COOH 一般式(2)
(式中、nは8〜16の整数を表す。)
原料の長鎖ジカルボン酸としては、中央の疎水部アルキレン鎖の炭素数nが8〜16のものを用いることができる。具体的には、ジカルボン酸セバシン酸、ウンデカン二酸、ドデカン二酸、トリデカン二酸、テトラデカン二酸、ペンタデカン二酸、ヘキサデカン二酸、ヘプタデカン二酸、オクタデカン二酸を用いることができる。これらジカルボン酸は塩又はモノエステルの状態で用いることもできるが、モノエステルの場合はグルコサミンの縮合後に加水分解が必要となる。
また、これらジカルボン酸の酸無水物や酸クロライドを用いる場合は、縮合剤なしでも縮合反応が起こるが、この場合も縮合後に水を添加して上記活性体の加水分解が必要となる。
【0017】
(イ)縮合剤について
中間体−3(一般式(3))を得る縮合反応で用いる縮合剤としては、ペプチド合成に用いられる種々の縮合剤を用いることができる。具体的には、「4−(4,6−ジメトキシ−1,3,5−トリアジン−2−イル)−4−メチルモルホリニウムクロリド」(DMT−MM)、1−エチル−3−(3−ジメチルアミノプロピル)カルボジイミド塩酸塩、ジシクロヘキシルカルボジイミド、ジイソプロピルカルボジイミドなどを用いることができる。またこのような縮合剤を用いなくても、該当するカルボン酸の酸無水物や酸クロライドを用いることもできる。しかし、水中やメタノール中の反応でも極めて高収率の縮合物を与えるDMT−MMが望ましい。また同反応は、メタノール、エタノール、イソプロパノール、ジメチルホルムアミド、ジメチルアセトアミド、N−メチルピロリドン、ジメチルスルホキシドなどを用いることができるが、選択的に片側のカルボン酸とのみ縮合反応を行い、速やかに目的とする中間体−3を沈殿させることで単離・精製するためには、ジカルボン酸、2−グルコサミン、中間体−3に適度な溶解度を有する5〜40体積%の水を含有するメタノール、エタノール、イソプロパノールが好適である。
【0018】
(ウ)2−グルコサミンについて
本発明のアミン成分は、2−グルコサミンであり、1位の水酸基はαアノマーであってもβアノマーであっても、もしくはその平衡混合物、又は塩酸塩などの塩であってもよい。
例えば、2−グルコサミンの塩酸塩を用いる場合、水に溶解後、ナトリウムメトキシドなどでアミンに変換して反応に用いる。あるいは、あらかじめα,ω−長鎖ジカルボン酸の二ナトリウム塩等を原料として用いれば、2−グルコサミン塩酸塩の中和は必要がない。
なお、2−グルコサミンは、D体でもL体のいずれを用いてもよいが入手の容易さからD体が好適であるため、本発明の実施の態様ではD体を用いている。したがって、一般式(1)などのアミン成分として表示されている2−グルコサミンはD体が示されている。
【0019】
(エ)縮合反応について
一般式(2)の長鎖ジカルボン酸及び2−グルコサミンの縮合反応は、前記縮合剤を添加し、又は添加することなく、水性溶媒中、好ましくは水含有メタノール溶液中で行わせる。
その際の反応温度としては0度ないし50度の範囲が選ばれ、反応時間は通常10分ないし30時間の範囲である。
上記縮合反応では、中間体−3のみが反応中に純度良く沈殿するため、特に精製する必要はないが、必要に応じて、再度メタノールや水含有メタノールなどを用いた加熱・冷却による再沈殿によりさらに高純度のものとすることができる。
このような反応では、一般式(2)のジカルボン酸の両端に2−グルコサミンが反応する可能性もあるが、得られた反応生成物の1H−NMRスペクトル(重ジメチルスルホキシド中、室温)の2.2〜2.3ppmのカルボキシル基に隣接するメチレンのシグナルと2.1ppmの糖アミドに隣接するメチレンのシグナルの積分比からみて、きわめて選択的に中間体−3が得られることが確認できている。
このように、本発明によれば、約25〜65%の収率で目的の中間体−3が得られる。
【0020】
(オ)中間体−3について
本発明の中間体−3は、下記一般式(3)で表される脂質分子であり、
(式中nは8〜16の整数を表す。また式中の水酸基の波線は、式中の波線で表記した2−グルコサミンの1位水酸基はα、βアノマーのいずれか、あるいはその平衡混合物を表す。)
典型的なn=10,12の脂質分子については、非特許文献15に記載されている。
中間体−3自体も、本発明の非対称双頭型脂質分子と同様に、カルボン酸系の非対称双頭型脂質分子であるが、自己集合させても、ナノチューブとその断片状のものの混合物しか得られず、非対称ナノチューブが形成できなかった点は上記[0004]で述べたとおりである。
本発明の中間体−3は、赤外線吸収スペクトルでは3700〜3300cm−1に糖水酸基に由来する特性吸収、2917〜2922cm−1、および2848〜2852cm−1にオリゴメチレン鎖に由来する特性吸収、1675〜1600cm−1にカルボキシル基に由来する吸収、1640〜1620cm−1にアミドカルボニル基に由来する吸収、1100〜1000cm−1に糖骨格に由来する吸収を示す。
さらに1H−NMR(DMSO−d6(重水素化ジメチルスルホキシド)と1滴D2O、室温)において、 δ値が7.7(βアノマーのNH), 7.5(αアノマーのNH), 4.9(αアノマーの1位のプロトン), 4.4(βアノマーの一位のプロトン), 3.2−3.7(糖のピラノース環のメチン、メチレン、2〜6位), 3.04−3.13(糖ピラノース環のメチレン、6位), 2.28(α体のカルボキシル基の隣のメチレン), 2.2(β体のカルボキシル基の隣のメチレン), 2.1(アミドに隣接したメチレン), 1.5(アミドの二つ隣のメチレン), 1.2(その他のメチレン)のシグナルが観測できることから、中間体−3であると同定できる。
【0021】
(1−4)中間体−3から中間体−5を経て本発明の非対称双頭型脂質分子(化合物−1)を得る工程について
(ア)中間体−5を得るための工程について
中間体−3を下記一般式(4)のオリゴグリシンエステルと脱水縮合することで中間体−5を得ることができる。
(式中mは1〜4の整数を表す。またRはアルコール性水酸基を有する化合物の残基であり、例えば、メチル基、エチル基、三級ブチル基、ベンジル基である。)
一般式(4)のオリゴグリシンエステルとしては、グリシンもしくはグリシルグリシンなどグリシンの重合物のメチルエステル、エチルエステル、三級ブチルエステル、ベンジルエステルなどのエステルが用いられる。アルカリ加水分解の容易さからエチルエステルあるいはメチルエステルが好適である。
【0022】
中間体−3と一般式(4)のオリゴグリシンエステルとを脱水縮合する反応においても、前記(1−3)(イ)と同様の縮合剤を用いることができる。具体的には、DMT−MM、1−エチル−3−(3−ジメチルアミノプロピル)カルボジイミド塩酸塩、ジシクロヘキシルカルボジイミド、ジイソプロピルカルボジイミドなどを用いることができるが、高収率の縮合物を与えるDMT−MMあるいは1−エチル−3−(3−ジメチルアミノプロピル)カルボジイミド塩酸塩が望ましい。また同反応はジメチルフォルムアミド(DMF)、ジメチルアセトアミド、N―メチルピロリドン、メタノール、エタノール、イソプロパノール、ジメチルスルホキシドなどを用いることができるが、原料の溶解性の点からDMFが好適である。
ここでの縮合反応も同様に、反応温度としては0度ないし50度の範囲が選ばれ、反応時間は通常10分ないし30時間の範囲である。
中間体−5を得るための縮合反応後にDMFを留去して得た固体を洗浄し不純物を溶解成分として除去するためには、メタノール、エタノール、イソプロパノール等が用いられるが、中間体−5の溶解性を抑制させるためにメタノールを用いることが望ましい。
【0023】
(イ)中間体−5について
中間体−5は、下記一般式(5)に示されるように、本願目的非対称双頭型脂質分子の末端カルボキシル基が保護基(メチル基、エチル基、三級ブチル基又はベンジル基)により保護された化合物として得られる。
(式中nは8〜16の整数を表し、mは1〜4の整数を表す。またRはアルコール性水酸基を有する化合物の残基であり、例えば、メチル基、エチル基、三級ブチル基、ベンジル基である。また式中の波線で表記した2−グルコサミンの1位水酸基はα、βアノマーのいずれか、あるいはその平衡混合物を表す。)
上記縮合反応の系においては、中間体−5のみが溶解性が低く、選択的に沈殿するため、ろ過後の沈殿固体の洗浄を除けば特に精製する必要はないが、必要に応じて、再度メタノールや水含有メタノールなどを用いた加熱・冷却による再沈殿によりさらに高純度のものとすることができる。
【0024】
(ウ)目的化合物−1を得るための加水分解工程について
中間体−5を加水分解により脱保護することで簡単に本発明の非対称双頭型脂質分子(化合物−1)を得ることができる。その際の加水分解では、溶解度と反応性の観点から、メタノール/水混合溶媒が好適である。反応温度としては20度ないし80度の範囲が選ばれ、反応時間は通常30分ないし6時間の範囲である。また中間体−5のRがベンジル基の場合は、DMF、あるいはメタノール、メタノール/水中などでのパラジウム炭素を触媒とした接触水素化還元での脱ベンジルエステルもよい。またRが三級ブチル基の場合は、DMF中で酸処理による脱エステルも可能である。
また加水分解反応も定量的に進行し、副反応もほとんど見られないことから、反応後の酸の添加による中和で純度よく目的とする化合物−1を得ることができるが、必要に応じてメタノールやエタノール、DMFなどで加熱冷却による精製を行ってもよい。
【0025】
(エ)本発明の非対称双頭型脂質分子(化合物−1)の同定
このような化合物の元素分析の実測値が誤差範囲内で計算値と一致し、また赤外線吸収スペクトルでは3700〜3300cm−1に糖水酸基に由来する特性吸収、2917−2922cm−1、および2848−2852cm−1にオリゴメチレン鎖に由来する特性吸収、1675−1600cm−1にカルボキシル基に由来する吸収、1650〜1620cm−1にグリシン残基やグルコサミンに連結したアミドカルボニル基に由来する吸収、1100〜1000cm−1に糖骨格に由来する吸収を示す。さらに1H−NMR(DMSO−d6と1滴D2OあるいはDMSO−d6のみ、室温)において、 δ値が8.2付近(グリシンのNH),7.6付近(グルコサミンのNH), 4.9付近(αアノマーの1位のプロトン), 4.4付近(βアノマーの一位のプロトン), 3.2−3.7付近(糖のピラノース環、2〜6位。グリシンのメチレン), 3.0−3.1付近(糖ピラノース環、6位のメチレン), 2.2−2.1付近(アミドカルボニル基の隣のメチレン), 1.5付近(アミドの二つ隣のメチレン), 1.2付近(その他のメチレン)のシグナルが観測できる。このことから、当該化合物は、目的の化合物−1であると同定できる。
【0026】
2.本発明の非対称ナノチューブについて
(2−1)本発明の非対称ナノチューブの製造方法
(ア)加熱溶解・冷却法
本発明の非対称双頭型脂質分子を用いて非対称ナノチューブを製造する方法の1つは、一般的な自己集合性脂質分子の自己集合のための加熱溶解・冷却法を適用することができる。
本発明の非対称双頭型脂質分子(0.5mg〜5mg)を1ミリリットルの純水(ミリQ水など)に分散し、80〜100℃に加熱溶解後、冷却(0.1℃〜10℃毎分)することで非対称ナノチューブからなるヒドロゲルが得られる。この時の電子顕微鏡観察から、長さが10μm以上あるナノチューブの絡み合った構造が確認できている。
本発明の非対称ナノチューブは構造安定性が高いので、凍結乾燥処理あるいは超音波処理などの物理的擾乱にも耐えられる。これらの物理的擾乱処理によって、収率良く軸比の短い、かつ良分散性の非対称ナノチューブに変換することが可能である。
得られた非対称ナノチューブの形態、大きさなどは、分散液を電子顕微鏡用のグリッドに滴下し、内部を電子線不透過な染色剤で染色することで、確認することができる。この様にして外径が約15nm、内径が約8〜9nm、長さが、100nm〜2μmの非対称ナノチューブを得ることができた。
【0027】
(イ)pH調整法
本発明の非対称双頭型脂質分子を室温で等モルから2等量程度の水酸化ナトリウム水溶液に分散・溶解し、つぎに室温で塩酸等の酸溶液を等モルから2等量程度添加してpH5〜6程度に調整することで同様に非対称ナノチューブを得ることができる。上記水酸化物水溶液としては、水酸化ナトリウム以外にもリチウム、カリウム、ルビジウム、セシウムなどの水酸化物水溶液も用いることができる。また上記酸溶液としては塩酸以外にもリン酸、ギ酸、酢酸、クエン酸、アスコルビン酸、硫酸等を用いることができる。
なお、原料として非対称双頭型脂質分子の塩(例えばナトリウム塩)などを用いる場合は、上記水酸化ナトリウム水溶液でもよいが、中和により多くの酸を必要とする。このため、より好ましくは室温で同塩を純水(ミリQ水など)に分散・溶解し、つぎに上記と同様室温で塩酸等の算用液を等モルから1.5等量程度添加してpH5〜6程度に調製することで同様に調製すればよい。
このpH調整法を適用する場合も、超音波処理、凍結乾燥などで軸比の短い非対称ナノチューブに変換可能であり、そのサイズはいずれもほぼ同じである。特に後者では、再水和時の添加する水の量によって良分散な状態のまま脂質濃度を10mg/ml程度まで上げることができる。またそれ以上(例えば50mg/ml)に濃度を上げることも可能であるが、ゲル状に変化する。
【0028】
(2−2)本発明の非対称ナノチューブの安定性
こうして得られた非対称ナノチューブの分散液は、そのまま室温あるいは冷蔵庫で上記のいずれの濃度で冷蔵保存しても約6ヶ月以上、沈殿を与えることなく良好な分散液として保存可能である。さらに分散液を凍結乾燥すれば1年以上の室温保存が可能で、ミリQ水等を添加して再水和すると同様の分散液を得ることができる。
【0029】
(2−3)本発明の非対称ナノチューブの構造
チューブ状構造であることの確認は、ネガティブ染色後の電子顕微鏡観察によってチューブ内空間を可視化することで可能となる(非特許文献1、10)。すなわち、チューブの内空間に電子線を透過させない染色剤を毛細管現象によって導入し、電子顕微鏡観察による透過像を得ると、染色剤が分布するチューブ内空間が暗い像を与え、チューブの壁は電子線が透過しやすい有機物で出来ているため、薄いコントラストを与える。これによって確認可能である。
また非対称ナノチューブ構造(すなわちチューブ内表面にカルボキシル基また外表面に2−グルコサミド基を配列するように配向する構造)の確認は、ナノチューブの粉末X線回折パターンから得られるナノチューブの膜厚の周期dと分子モデルから推定される分子長Lの関係から推測できる。すなわち非対称双頭型脂質が非対称ナノチューブ構造を形成しているときは、ナノチューブの膜周期dと分子長Lがほぼ同じか、あるいはdが若干小さくなることが知られている(非特許文献9、10)。そこで、以下のようにdとLを比較検討した結果、いずれの場合にも上記の関係を満たすことが明らかとなった(表1)。
以上から、本発明の有機ナノチューブは非対称ナノチューブであることがわかった。
【0030】
<表1> 非対称ナノチューブのXRDより得られる膜周期dと分子モデルから得られる分子長Lの関係
【0031】
表1には、記述していないが、本発明の化合物−1のn=14,16の場合も、n=10,12の場合と同様のチューブ形態とサイズを示すことを確認した。一般には鎖長が長くなるほど、非対称ナノチューブ構造を取りやすくなることが以前の研究より明かにされており(非特許文献17)、分子の鎖長のみが異なる一連の脂質群では、自己集合で得られるナノチューブ構造がほぼ同じ形態やサイズ(内径、外径、壁厚)を有する場合は、分子の配列様式も同様であることも知られている(非特許文献10)。したがって、n=14,16の場合も、n=10,12の場合と同様の非対称ナノチューブ構造を形成していることが示唆される。
【0032】
3.本発明の非対称ナノチューブへのカプセル化
(3−1)カプセル化できる対象物質について
本発明の非対称ナノチューブは、その内側がカルボキシル基で被覆されているため、中に好ましく包接される薬剤は、カルボキシル基とイオン結合可能なアミノ基などのカチオン性の官能基を有する化合物が好ましく、安定なカチオン性薬剤−非対称ナノチューブ複合体を形成する。また、チューブ内が負の電荷に満たされた環境となっているため、特にイオン結合可能な官能基を有している化合物でなくても、物質全体が正の電荷を帯びたカチオン性物質であって、サイズが0.1〜5nm程度の物質であれば包接可能であり、このようなカチオン性物質を包接した非対称ナノチューブもカチオン性物質−非対称ナノチューブ複合体を形成しているといえる。
カチオン性薬剤−非対称ナノチューブ複合体を形成する具体的なカチオン性薬剤としては、ドキソルビシン、イダルビシン、エピルビシン、ダウノルビシン、ピラルビシンなどのアントラサイクリン系抗癌剤やその他のアミノ基を有する薬剤が特に好ましく用いられる。
また、シスプラチン、ネダプラチン、カルボプラチン、オキサリプラチンなどのカチオン性薬剤も好ましい。
また、このような医薬品としての薬理作用を有する薬剤のみならず、化粧品用、食品用などのカチオン性の乳化剤、分散剤、安定剤、保湿剤等各種添加剤に対しても適用可能である。
【0033】
(3−2)非対称ナノチューブへのカプセル化方法
カプセル化したいカチオン性物質を純水(ミリQ水)に0.1mg/mL〜50mg/の濃度になるように溶解又は懸濁したものと、本発明の非対称ナノチューブ分散液(濃度 0.1mg/mL〜20mg/mL)とを等量混合し、10分〜3時間程度室温で放置する。そのことで、30〜95%程度のカプセル化率を達成できるため、例えば抗癌剤用組成物として用いる場合など、有効量を含有させるための組成物総量を低く抑えることができる。
またカプセル化されなかった物質は、上記混合溶液を200nmの多孔質膜でろ過することによって、効率よく取り除くことができる。これによって、カチオン性物質をカプセル化したチューブのみを固体として単離することができるので、これを任意の濃度でミリQ水に再分散させることで濃度を調整することも可能である。またカチオン性物質をカプセル化したチューブを凍結乾燥して得た固体を同様に任意の濃度でミリQ水に分散させることでも濃度を調整することができる。
薬剤のカプセル化率は、上記の手法でカプセル化した後、ナノチューブを濾別し、これをミリQ水に再分散して得られた溶液の薬剤固有のUV吸収を測定することで、定量することができる。
例えばドキソルビシンの場合、ドキソルビシン溶液200μL(それぞれミリQ水1mLあたりに2.0mg,1.0mg,0.714mg,0.625mg,0.555mg,0.50mg溶解したもの)とナノチューブ分散液(200μL、5mg/mL)を混合し、30分室温で放置した。これより200μLを取り、200nmの多孔質膜でろ過し、さらにミリQ水(400μL)で洗浄した。濾別された固体(ナノチューブ)はろ紙ごと200μLのミリQ水に再分散した。ドキソルビシンのUV吸収(480nm)によって、ろ過前の溶液とろ過後の溶液の吸収を比較して、カプセル化率を求めた。
本発明の薬剤カプセル化率は、ドキソルビシンの場合で62〜95%程度と、きわめて高濃度にカプセル化することができる。
【0034】
(3−3)薬剤のナノチューブからの放出について
本発明においてカプセル化された薬剤は、pH変化などの外部の刺激に応じ、チューブ内表面を被覆するカルボキシル基のイオン化状態の変化又はチューブ構造そのものの形態変化などによって、条件選択的かつ効率的に薬剤を放出することができる。
具体的には、ミリQ水の分散液にバッファー溶液を加えpH値を4〜6程度とすることで、内表面のカルボキシル基がプロトン化するため、放出の加速が可能である。またpHを7〜8程度にすることでも徐放することが可能である。
また、本発明の非対称ナノチューブとして、超音波処理などにより長さを50nm〜500nm程度にまでサイズダウンしたナノチューブを用いることで、EPR(Enhanced Permiability and Retation)効果により、ターゲットとなるガン細胞、組織へ送達する量を高めることが可能である。
ナノチューブからの薬剤放出量は、チューブ分散液を透析膜チューブ(分画分子量1200〜20000程度)の内水相に密閉し、膜を通過して外水相に放出された薬剤をUVなどの分光分析によって計測することで定量的に計測可能である。
例えば、ドキソルビシンを内包化した非対称ナノチューブ(ナノチューブ/Dox=8/1)のPBSバッファー中(pH7.4)の場合は、これを透析膜(分画分子量14000)に密閉し、同様にPBSバッファー中(pH7.4)の外水相に入れて撹拌しながら外水相のドキソルビシンの放出量をUV吸収によってモニターした。
その場合、PBSバッファー中(pH7.4)中では、1時間あたり10〜12%程度、4時間目では約50%程度の徐放効果を確認できた。
一方、弱酸性状態(pH5.5)では、むしろ一時間あたり33%、4時間目で82%以上を放出し、8時間ではほぼ全量放出してしまうことから、非対称ナノチューブ内表面を覆うカルボキシル基から生じたカルボキシラートのプロトン化が刺激となって抗癌剤の放出を加速できることが確認できた。この低pH条件で抗癌剤放出が加速される点は、ナノチューブを薬剤カプセルとして利用する場合、極めて有効性であると考えられる。一般に、ガン組織およびガン細胞は、正常細胞に比べて弱酸性(低pH)であるという特性を有するからである。
【0035】
4.カプセル化された薬剤組成物
(4−1)薬剤組成物について
本発明の非対称ナノチューブは、上述のように薬剤キャリアとして優れた特性を有しているため、治療用の薬剤を有効量含む非対称ナノチューブ複合体を有効成分として、薬理学的に許容される担体と共に薬剤組成物とすることができる。なお、薬剤組成物とは、例えば抗癌剤など癌の予防、治療又は処置のための医薬組成物を含む。また、水性環境中に徐々に薬剤を放出させたい場合などにも有効であることから、この薬剤組成物としては、化粧品用、食品用として配合されるカチオン性薬剤組成物も包含される。
本発明のカプセル内の薬剤として典型的な薬剤は、上述のドキソルビシン、イダルビシン、エピルビシン、ダウノルビシン、ピラルビシンなどのアントラサイクリン系抗癌剤やその他のアミノ基を有する薬剤である。その他、シスプラチン、ネダプラチン、カルボプラチン、オキサリプラチンなどのカチオン性薬剤も同様に用いることができる。
ここで、薬学的に許容される担体とは、希釈剤又は賦形剤、例えば、水又は生理的許容される緩衝液を含む。
本発明の薬剤組成物は、錠剤などの経口投与も可能であるが、静脈経由若しくは直接患部への注射、外用剤などの非経口的に投与することもできる。
本発明の薬剤組成物を患者に投与する際の投与量は、用いる薬剤の種類、医療対象の患者の年齢、体重及び病状、投与経路などに従い適宜決めることができるが、典型的には、1回の投与当たりの薬剤の有効量としては、1μg〜1g、好ましくは100μg〜10mgの範囲である。
【0036】
(4−2)薬剤組成物の機能について
本発明の非対称ナノチューブによる薬剤カプセル化物は、中性付近での徐々に放出することができる徐放作用と、酸性条件での加速された薬剤放出効果と2つの効果が期待できる。すなわち、癌細胞の場合、若干酸性状態のがん組織付近での選択的な薬剤放出効果と共に、がん細胞内部での速やかな薬剤放出による高い抗癌活性の発現という二つの効果が期待できる。
なお、本発明で「細胞」というとき、典型的にはヒトを含む哺乳動物細胞を指す。インビトロでの培養細胞であってもよいが、薬剤治療用の場合、生体を構成する組織、臓器内の細胞や、血液など体液中の血球細胞やリンパ球細胞などの免疫細胞、及び各種の癌細胞が対象となる。
【0037】
(4−3)抗癌活性の測定
一例として、本発明のドキソルビシンを内包化した非対称ナノチューブを用いた抗癌剤組成物についての抗癌活性を、以下のように確認した。
抗癌活性については、ドキソルビシンを内包化した非対称ナノチューブを培養液中に分散させたHela細胞に添加、24時間培養後、細胞数あるいはそれにかわるもの(細胞内タンパク質量、ミトコンドリアの活性など)を測定し、対照実験として、ドキソルビシンのみの溶液と比較することで行うことが出来る。その際の測定法としては、典型的には細胞のミトコンドリアの活性を測定する方法(MTTアッセイ法、非特許文献18)を用いることができる。
本実施例では、細胞にドキソルビシンを内包化した非対称ナノチューブを添加・培養後、MTSアッセイ法による細胞のミトコンドリアの活性に従って定量した。MTTアッセイもMTSアッセイも細胞中のミトコンドリア中の酵素により生成するホルマザンという色素の濃度測定によって、活性な細胞の数を定量化するものである。MTTアッセイでは、薬剤反応後に不溶性のホルマザンが生成するため、吸光度を測定する前に溶液の処理が必要となる。これに比べてMTSアッセイでは、不溶性のホルマザンの生成が無く、後処理も不要なため、操作がより簡便で迅速に測定できるものである。
【実施例】
【0038】
次に本発明を実施例によってさらに詳細に説明するが、本発明はこれらの例によって何
ら限定されるものではない。
本発明におけるその他の用語や概念は、当該分野において慣用的に使用される用語の意味に基づくものであり、本発明を実施するために使用する様々な技術は、特にその出典を明示した技術を除いては、公知の文献等に基づいて当業者であれば容易かつ確実に実施可能である。また、各種の分析などは、使用した分析機器又は試薬、キットの取り扱い説明書、カタログなどに記載の方法を準用して行った。
なお、本明細書中に引用した技術文献、特許公報及び特許出願明細書中の記載内容は、本発明の記載内容として参照されるものとする。
【0039】
(実施例1)非対称ナノチューブ形成性非対称双頭型脂質分子(式(1)化合物―1、n=10,m=2)の合成
一般式(1)で表される非対称双頭型脂質分子のうち、n=10,m=2の場合の脂質分子(化合物−1(n=10,m=2))を、下記反応式(1)に示される中間体−5を経由した合成方法に従って合成した。
(なお、反応式中、「DMT−MM」は「4−(4,6−ジメトキシ−1,3,5−トリアジン−2−イル)−4−メチルモルホリニウムクロリド」の略であり、「MeONa」は「ナトリウムメトキシド」の略である。以下同様。)
【0040】
(1−1)中間体−3(n=10)の合成
ドデカン二酸(11.9g,46mmol)をメタノール(400 ml)に溶解した。この溶液に、水( ml)に溶したD(+)−グルコサミン塩酸塩 (5g,23mmol)とナトリウムメトキシド (1.25g,23mmol)を加えた。メタノール(20ml)に溶解したDMT−MM(6.4g,23mmol)を加え、室温で攪拌した。TLCで反応をモニターしながら、DMT−MM(9.6g,35mmol)とナトリウムメトキシド (2.5g,46mmol)を加え、30時間反応した。反応終了後、反応液を濾過し、残渣を水とメタノールで洗浄した。エタノール、水1/1で再沈澱を行い、生成物(5.79g)を得た。収率25%。
精製後の1H−NMR(DMSO−d6と1滴D2O)のチャートを図1に示す。
1H−NMR(DMSO−d6と1滴D2O)7.68(dd,J=8.4Hz,NH), 7.52(d,J=8.1Hz,NH),4.92(d,J=3.4Hz,0.7H,H−1α),4.43(d,J=8.1Hz,0.3H,H−1β),3.24−3.69(m,5H),3.04−3.13(m,1H),2.28(t,J=7.4Hz,0.3H,−CH2−COOHα),2.18(t,J=7.4Hz,1.7H,−CH2COOHβ),2.09(t,J=7.5Hz,−CH2−CONH,2H),1.47(d,J=6.8Hz,−CH2CH2CO,4H), 1.24(s,12H,−CH2)。
【0041】
(1−2)化合物−1の合成(n=10,m=2)
上記(1−1)で得られた中間体−3(n=10)(0.5g, 1.2mmol)をDMFに加熱溶解し、室温まで冷却したのちグリシルグリシン塩酸塩メチルエステル(0.22g, 1.23mmol)とDMT−MM(0.44g, 1.35mmol)、トリエチルアミン(1.25mmol)を添加した。室温で2時間撹拌した後、DMFを留去した。得られた固体にメタノールを添加し、不溶の白色固体を濾別することで中間体−5(n=10,m=2)を得た。これを加熱したメタノール/水溶媒に分散させ、水酸化ナトリウム水溶液(1.5ml、1mol/L)を60℃で添加して加水分解を行った。反応後溶媒を留去し、メタノールを添加し、固体を分散させた後、塩酸を添加してpHを4に調整し、ろ過することで目的とする化合物−1を白色固体として得た(0.35g、58%:中間体−3(n=10)より)。精製後の1H−NMR(DMSO−d6と1滴D2O)のチャートを図2に示す。
元素分析値(C22H39N3O10H2Oとして)
C H N
計算値(%) 50.47 7.89 8.03
実測値(%) 50.59 7.75 7.99
【0042】
(実施例2)非対称ナノチューブ形成性非対称双頭型脂質分子(式(1)化合物−1、n=10,m=3)の合成
前記実施例(1−1)で得られた中間体−3(n=10)(0.5g, 1.2mmol)とグリシルグリシルグリシン塩酸塩メチルエステル(0.29g, 1.23mmol)とDMT−MM(0.44g, 1.35mmol)、トリエチルアミン(1.25mmol)を添加した。室温で2時間撹拌した後、DMFを留去した。得られた固体にメタノールを添加し、不溶の白色固体を濾別することで中間体−5(n=10,m=3)を得た。これを加熱したメタノール/水溶媒に分散させ、水酸化ナトリウム水溶液(1.5ml、1mol/L)を60℃で添加して加水分解を行った。反応後溶媒を留去し、メタノールを添加し、固体を分散させた後、塩酸を添加してpHを4に調整し、ろ過することで目的とする化合物−1を白色固体として得た(0.43g、63%:中間体−3(n=10)より)。精製後の1H−NMR(DMSO−d6と1滴D2O)のチャートを図3に示す。
元素分析値(C24H42N4O11H2Oとして)
C H N
計算値(%) 51.05 7.85 9.92
実測値(%) 50.93 7.95 9.88
【0043】
(実施例3)非対称ナノチューブ形成性非対称双頭型脂質分子(式(1)化合物−1、n=10,m=4)の合成
前記実施例(1−1)で得られた中間体−3(n=10)(0.5g, 1.2mmol)とグリシルグリシルグリシルグリシン塩酸塩メチルエステル(0.65g, 1.23mmol)とDMT−MM(0.44g, 1.35mmol)、トリエチルアミン(1.25mmol)を添加した。室温で2時間撹拌した後、DMFを留去した。得られた固体にメタノールを添加し、不溶の白色固体を濾別することで中間体−5(n=10,m=4)を得た。これを加熱したメタノール/水溶媒に分散させ、水酸化ナトリウム水溶液(1.5ml、1mol/L)を60℃で添加して加水分解を行った。反応後溶媒を留去し、メタノールを添加し、固体を分散させた後、塩酸を添加してpHを4に調整し、ろ過することで目的とする化合物−1を白色固体として得た(0.48g、65%:中間体−3(n=10)より)。精製後の1H−NMR(DMSO−d6と1滴D2O)のチャートを図4に示す。
元素分析値(C26H45N5O12H2Oとして)
C H N
計算値(%) 50.3948.97 7.43 10.98
実測値(%) 48.75 7.11 11.15
【0044】
(実施例4) 非対称ナノチューブ形成性非対称双頭型脂質分子(式(1)化合物−1、n=12,m=2)の合成
(4−1)中間体−3(n=12)の合成
テトラデカン二酸(4.8g, 18mmol)をメタノール(200ml)に溶解した。その後100mlの水中で水酸化ナトリウム水溶液(0.4mol/L)を添加した。この溶液に、D(+)−グルコサミン塩酸塩 (2g,9.2mmol)を加えた。これにDMT−MM(3.1g,9.5mmol)を加え、室温で30時間撹拌した。反応終了後、反応液をろ過し、残渣を水とメタノールで洗浄した。pH4に調整しなから、エタノール、水1/1で再沈殿を行い、生成物(2.2g)を得た。収率64%。精製後の1H−NMR(DMSO−d6と1滴D2O)のチャートを図5に示す。
1H−NMR(DMSO−d6と1滴D2O)7.68(dd,J=8.4Hz,NH), 7.52(d,J=8.1Hz,NH),4.92(d,J=3.4Hz,0.7H,H−1α),4.43(d,J=8.1Hz,0.3H,H−1β),3.24−3.69(m,5H),3.04−3.13(m,1H),2.28(t,J=7.4Hz,0.3H,−CH2−COOHα),2.18(t,J=7.4Hz,1.7H,−CH2COOHβ),2.09(t,J=7.5Hz,−CH2−CONH,2H),1.47(d,J=6.8Hz,−CH2CH2CO,4H), 1.24(s,16H,−CH2)。
【0045】
(4−2)化合物−1(n=12,m=2)の合成
前記(4−1)で得られた中間体−3(n=12) (0.4 g, 0.95 mmol)をDMFに加熱溶解し、室温まで冷却したのち塩酸グリシルグリシンメチルエステル(200mg,1.0mmol)とDMT−MM(340mg,1.1 mmol)、トリエチルアミン(1.0mmol)を添加した。室温で2時間撹拌した後、DMFを留去した。得られた固体にメタノールを添加し、不溶の白色固体を濾別することで中間体−5(n=12,m=2)を得た。
これを加熱したメタノール/水溶媒に分散させ、水酸化ナトリウム水溶液(1mol/L,1.5ml)を60℃で添加して加水分解を行った。反応後溶媒を留去し、メタノールを添加し、固体を分散させた後、塩酸を添加してpHを4に調整し、ろ過することで目的とする化合物−1(n=12,m=2)を白色固体として得た(355mg、70%)。精製後の1H−NMR(DMSO−d6と1滴D2O)のチャートを図6に示す。
元素分析値(C24H43N3O10H2Oとして)
C H N
計算値(%) 52.26 8.22 7.64
実測値(%) 52.29 8.18 7.48
【0046】
(実施例5) 非対称ナノチューブ形成性非対称双頭型脂質分子(式(1)化合物−1、(n=12,m=3))の合成
前記実施例(4−1)で得られた中間体−3(n=12) (0.2 g, 0.48 mmol)をDMFに加熱溶解し、室温まで冷却したのち塩酸グリシルグリシルグリシンエチルエステル(114mg,0.52mmol)とDMT−MM(170mg,0.52 mmol)、トリエチルアミン(0.52mmol)を添加した。室温で2時間撹拌した後、DMFを留去した。得られた固体にメタノールを添加し、不溶の白色固体を濾別することで中間体−5(n=12,m=3)を得た。
これを加熱したメタノール/水溶媒に分散させ、水酸化ナトリウム水溶液(1mol/L,0.75ml)を60℃で添加して加水分解を行った。反応後溶媒を留去し、メタノールを添加し、固体を分散させた後、塩酸を添加してpHを4に調整し、ろ過することで目的とする化合物−1(n=12,m=3)を白色固体として得た(210mg、 68%)。精製後の1H−NMR(DMSO−d6)のチャートを図7に示す。
元素分析値(C26H46N4O11H2Oとして)
C H N
計算値(%) 51.30 7.95 9.20
実測値(%) 51.15 7.99 9.05
【0047】
(実施例6) 非対称ナノチューブ形成性非対称双頭型脂質分子(式(1)化合物−1、(n=12,m=4))の合成
前記実施例(4−1)で得られた中間体−3(n=12)(0.4 g, 0.95 mmol)をDMFに加熱溶解し、室温まで冷却したのち塩酸グリシルグリシンエチルエステル(200mg,1.0mmol)とDMT−MM(340mg,1.0 mmol)、トリエチルアミン(1.0mmol)を添加した。室温で2時間撹拌した後、DMFを留去した。得られた固体にメタノールを添加し、不溶の白色固体を濾別することで中間体−5(n=12,m=4)を得た。
これを加熱したメタノール/水溶媒に分散させ、水酸化ナトリウム水溶液(1.5mol/L,1ml)を60℃で添加して加水分解を行った。反応後溶媒を留去し、メタノールを添加し、固体を分散させた後、塩酸を添加してpHを4に調整し、ろ過することで目的とする化合物−1(n=12,m=4)を白色固体として得た(357mg、 58%)。精製後の1H−NMR(DMSO−d6と1滴D2O)のチャートを図8に示す。
元素分析値(C28H49N5O12H2Oとして)
C H N
計算値(%) 49.18 7.81 10.24
実測値(%) 49.09 7.63 10.37
【0048】
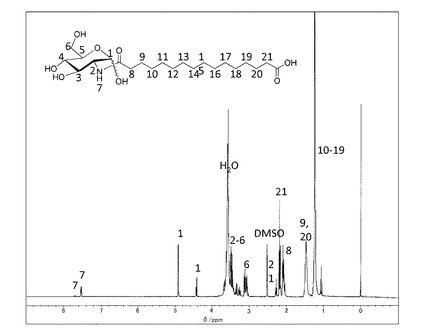
(実施例7) 非対称ナノチューブ形成性非対称双頭型脂質分子(式(1)化合物−1(n=14,m=3)の合成
(7−1)中間体−3(n=14)の合成
ヘキサデカン二酸(1.3g,4.6mmol)をメタノール(90ml)に溶解した。この溶液に、水(2ml)に溶解したD(+)−グルコサミン塩酸塩 (0.5g,2.3mmol)とナトリウムメトキシド (0.125g,2.3mmol)を加えた。メタノール(1ml)に溶解したDMT−MM(0.641g,2.3mmol)を加え、室温で撹拌した。TLCで反応をモニターしながら、DMT−MM(0.96g,3.5mmol)とナトリウムメトキシド(0.25g,4.6mmol)を加え、29時間反応した。反応終了後、反応液をろ過し、残渣を水とメタノールで洗浄した。エタノール、水1/1で再沈殿を行い、生成物(0.34g)を得た。収率33パーセント。精製後の1H−NMR(DMSO−d6と1滴D2O)のチャートを図9に示す。
1H−NMR(DMSO−d6と1滴D2O)7.70(dd,J=8.5Hz,NH), 7.53(d,J=8.0Hz,NH),4.92(d,J=3.3Hz,0.7H,H−1α),4.43(d,J=8.2Hz,0.3H,H−1β),3.24−3.69(m,5H),3.03−3.14(m,1H),2.28(t,J=7.3Hz,0.3H,−CH2−COOHα),2.18(t,J=7.3Hz,1.7H,−CH2COOHβ),2.10(t,J=7.6Hz,−CH2−CONH,2H),1.48(d,J=6.8Hz,−CH2CH2CO,4H), 1.24(s,20H,−CH2)。
【0049】
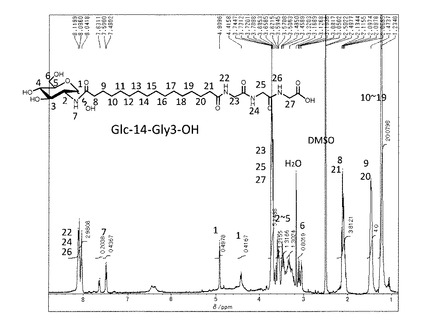
(7−2)非対称双頭型脂質分子(式(1)の化合物−1(n=14,m=3))の合成
上記(7−1)で得られた中間体−3(n=14) (400 mg, 0.9 mmol)をDMFに加熱溶解し、室温まで冷却したのち塩酸グリシルグリシルグリシンエチルエステル(250mg,1.0mmol)とDMT−MM(340mg,1.1 mmol)、トリエチルアミン(1.0mmol)を添加した。室温で2時間撹拌した後、DMFを留去した。得られた固体にメタノールを添加し、不溶の白色固体を濾別することで中間体−5(n=12,m=3)を得た。
これを加熱したメタノール/水溶媒に分散させ、水酸化ナトリウム水溶液(1mol/L,1.5ml)を80℃で添加して加水分解を行った。反応後溶媒を留去し、メタノールを添加し、固体を分散させた後、塩酸を添加してpHを4に調整し、ろ過することで目的とする化合物−1(n=14,m=3)を白色固体として得た(460mg、 78%)。精製後の1H−NMR(DMSO−d6)のチャートを図10に示す。
元素分析値(C28H50N4O11H2Oとして)
C H N
計算値(%) 52.82 8.23 8.80
実測値(%) 52.99 8.09 8.62
【0050】
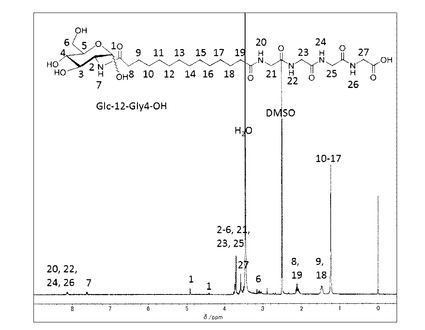
(実施例8) 非対称ナノチューブ形成性非対称双頭型脂質分子(式(1)の化合物−1(n=14,m=4))の合成
前記実施例(7−1)で得られた中間体−3(n=14) (0.1g, 0.22mmol)をDMFに加熱溶解し、室温まで冷却したのち塩酸グリシルグリシルグリシルグリシンエチルエステル(61mg,0.23mmol)とDMT−MM(68mg,0.24 mmol)を添加した。室温で19時間撹拌した後、DMFを留去した。得られた固体にメタノールを添加し、不溶の白色固体を濾別することで中間体−5(n=14,m=4)を得た。
得られた中間体−5(n=14,m=4)(50mg,0.07mmol)を加熱したメタノール/水溶媒に分散させ、水酸化ナトリウム水溶液(0.1mol/L,2.7ml)を65℃で添加して加水分解を行った。塩酸を添加してpHを4に調整した後溶媒を留去した。メタノールを添加し、不溶の白色固体を濾別することで目的とする化合物−1(n=14,m=4)を白色固体として得た(25mg、41%)。精製後の1H−NMR(DMSO−d6と1滴D2O)のチャートを図11に示す。
本実施例で得られた化合物ー1(n=14、m=4)の1H−NMR(DMSO−d6と1滴D2O、室温)4.91(d,J=3.3Hz,0.7H,H−1α),4.42(d,J=8.2Hz,0.3H,H−1β),3.34−3.76(m,13H),3.04−3.14(m,1H),2.28(t,J=7.3Hz,0.3H,−CH2−COOHα),2.07−2.14(m,3.7H,−CH2COOHβ,−CH2−CONH),1.47(d,J=6.8Hz,−CH2CH2CO,4H), 1.23(s,20H,−CH2)。
元素分析値(C30H53N5O12H2Oとして)
C H N
計算値(%) 51.94 7.99 10.09
実測値(%) 51.88 8.12 10.01
【0051】
(実施例9) 非対称双頭型脂質分子(化合物−1)の自己集合による非対称ナノチューブの形成
まず、前記実施例1において製造された、本発明の非対称双頭型脂質分子のうち、化合物−1(n=10,m=2)を用いて、以下の(1)分散液の加熱による溶解、その後の冷却による方法、及び(2)室温でのナトリウム塩からのpHを調整による方法の2つの方法により非対称ナノチューブを形成させた。
(9−1)加熱・冷却法による方法
化合物−1(n=10,m=2)(5mg)を水(ミリQ水、1ml)に分散、加熱溶解(100℃程度)した。溶液を室温に冷却することで透明でナノチューブの含まれたゲル状物を得た。リンタングステン酸でネガティブ染色後、走査型電子顕微鏡観察(透過モード)から外径約15nm、内径約8nm、長さ5μm以上のナノチューブ構造を確認した(図12A)。
【0052】
(9−2)pH調整法による方法
化合物−1(n=10,m=2)(5mg)を水(ミリQ水、1ml)に分散した。室温下、溶液に水酸化ナトリウム水溶液(1モル/L、0.01mL)を加えて溶解し、さらに塩酸水溶液(1モル/L、0.01mL)を加えてpHを5に調製することでナノチューブの含まれた分散液を得た。リンタングステン酸でネガティブ染色後、走査型電子顕微鏡観察(透過モード)から外径約15nm、内径約8nm、長さ1μm以上のナノチューブ構造を確認した(図12B)。
【0053】
(9−3)その他の非対称双頭型脂質分子における非対称性ナノチューブ形成性の確認
さらに、前記実施例2〜8で製造された非対称双頭型脂質分子についても、上記(9−1)及び(9−2)と同様の方法で自己集合させることで、同様なサイズのナノチューブ構造が形成することを確認した。
前記実施例1で製造された化合物−1(n=10,m=2の場合)の加熱冷却およびpH調製法による自己集合で得られた非対称ナノチューブの電子顕微鏡観察像(透過像)をそれぞれ図12AおよびBに示す。
前記実施例2で製造された化合物−1(n=10,m=3の場合)の加熱冷却およびpH調製法による自己集合で得られた非対称ナノチューブの電子顕微鏡観察像(透過像)をそれぞれ図13AおよびBに示す。
前記実施例3で製造された化合物−1(n=10,m=4の場合)の加熱冷却およびpH調製法による自己集合で得られた非対称ナノチューブの電子顕微鏡観察像(透過像)をそれぞれ図14AおよびBに示す。
前記実施例4で製造された化合物−1(n12,m=2の場合)の加熱冷却およびpH調製法による自己集合で得られた非対称ナノチューブの電子顕微鏡観察像(透過像)をそれぞれ図15AおよびBに示す。また、挿入図は、チューブが絡まり合って良く分散した透明なゲル状物を与えており、チューブの分散性が高いことを示す。
前記実施例5で製造された化合物−1(n=12,m=3の場合)の加熱冷却およびpH調製法による自己集合で得られた非対称ナノチューブの電子顕微鏡観察像(透過像)をそれぞれ図16AおよびBに示す。
前記実施例6で製造された化合物−1(n=12,m=4の場合)のpH調製法による自己集合で得られた非対称ナノチューブの電子顕微鏡観察像(透過像)をそれぞれ図17AおよびBに示す。
前記実施例7で製造された化合物−1(n=14,m=3の場合)のpH調製法による自己集合で得られた非対称ナノチューブの電子顕微鏡観察像(透過像)を図18に示す。
【0054】
(実施例10) 非対称ナノチューブの短尺化と凍結乾燥
(10−1) 非対称ナノチューブの短尺化
前記実施例(9−3)において、化合物−1(n=12、m=4)に対して加熱冷却法を適用して形成された非対称ナノチューブを、超音波処理することで短尺化したナノチューブを得た。すなわち上記ナノチューブ溶液(5mg/mL)を、プローブ型超音波処理機で4℃、1分間処理することで得られた溶液は透明な分散液であった。この溶液を走査型電子顕微鏡で観察したところ、長さが100〜500nmの短尺状ナノチューブが得られることがわかった。さらに時間を2分にすると50〜200nmの長さに短尺化した。また同処理によって得た分散液を60日以上、4度で保存してもナノチューブ構造が安定であることを確認した。図19に加熱冷却によって得た化合物−1(n=12,m=4)から得た非対称ナノチューブの超音波処理後の電子顕微鏡観察像を示す。挿入図は、加熱冷却によって調製されたナノチューブ分散液を示す。
【0055】
(10−2) 非対称ナノチューブの凍結乾燥
ここでは、本発明の非対称ナノチューブが凍結乾燥により長期保存が可能であることを示すために、上記(10−1)で得られた非対称ナノチューブの超音波処理物を凍結乾燥後再水和して非対称ナノチューブ分散液を得た。
具体的には、上記(10−1)で得られた非対称ナノチューブの超音波処理物を凍結乾燥し、30日間室温で保持した後、ミリQ水を添加することで再水和し、高濃度(5mg/mL)の分散液を得た(図21)。この分散液は60日以上、4度で安定であることを確認した。図20に加熱冷却によって得た化合物−1(n=12,m=4)から得た非対称ナノチューブの超音波処理、凍結乾燥、再水和後の電子顕微鏡観察像を示す。
【0056】
(実施例11) 薬剤内包化実験
前記実施例10で製造した非対称性ナノチューブを用いてドキソルビシンの内包化を以下の実験によりおこなった。
ドキソルビシン溶液200μL(それぞれ2,1,0.714,0.625,0.555,0.5mg/mLのミリQ水に溶解したもの)と上記非対称ナノチューブの分散液(200μL、5mg/mL)を混合し、30分室温で放置する。200μLを取り、200nmの多孔質膜でろ過し、さらにミリQ水で洗浄する(400μL)。固体はろ紙ごと200μLのミリQ水に再分散した。ドキソルビシンのUV吸収(480nm)によって、ろ過前の溶液とろ過後の溶液の吸収を比較して、カプセル化率を求めた。図21にドキソルビシンをカプセル化後の非対称ナノチューブの電子顕微鏡による観察像とそのカプセル化率を示す。
その結果、図21(B)のように、ONT/DOX=10/1〜7/1では、90%以上がカプセル化されていることから、混合するだけで高いカプセル化率を示すことが示された。また図21(A)の画像からも明らかなように、ドキソルビシンをカプセル化後もナノチューブの形態は変化していない。そして、溶液中での凝集は認められず、高い分散性を維持していることも確認した。
【0057】
(実施例12) 薬剤放出実験
次に、前記(実施例11)でドキソルビシンを内包化した非対称性ナノチューブからのドキソルビシンの放出実験を、PBSバッファー(pH7.4とpH5.5)中での透析法によって行った。その結果、図22に示したように、中性付近(pH7.4)では、10時間後でやっと全内包化量のうち約62%程度が放出され、32時間たっても放出量は全体の70%にとどまっているのに対して、弱酸性状態(pH5.5)では、4時間目で82%以上を放出し、8時間ではほぼ全量放出してしまうことがわかり、非対称ナノチューブ内表面を覆うカルボキシル基から生じたカルボキシラートのプロトン化が刺激となって抗癌剤の放出を加速できることを示している。この低pH条件で抗癌剤放出が加速される点は、一般に、ガン組織および細胞は、正常細胞に比べて酸性(低pH)であるという特性を有するから、ガン組織での選択的な薬剤放出が期待でき、ナノチューブを薬剤カプセルとして利用することの有効性は極めて高いと考えられる。
【0058】
(実施例13) 抗癌剤組成物としての機能
Hela細胞を用いてドキソルビシン内包化非対称ナノチューブ(化合物−1でn=12,m=4の場合。カプセル化はONT/DOX=7/1で行ったもの。)に対して、MTSアッセイによりHela細胞への細胞毒性(抗癌活性)を調べた。その結果、図23に示したように、フリーのドキソルビシンよりも高い活性を示すことがわかった。これは、ドキソルビシンをカプセル化した非対称ナノチューブが、癌細胞内のエンドソームに取り込まれ、エンドソーム特有の低いpH環境にさらされることでチューブ内表面のカルボキシレートがプロトン化され、ドキソルビシンを速やかに放出したためと考えられる。
このように、本発明の非対称ナノチューブによるドキソルビシンなどカチオン性の抗癌剤カプセル化物は、実施例12に記述したような、若干酸性状態のがん組織付近での選択的な薬剤放出効果と共に、本実施例におけるがん細胞内での速やかな薬剤放出による高い抗癌活性の発現という二つの効果が期待できる。すなわち、本発明の非対称ナノチューブによるカチオン性抗癌剤カプセル化物は、優れた抗癌剤組成物としての機能を有していることが示された。
【特許請求の範囲】
【請求項1】
下記一般式(1)で表される非対称双頭型脂質分子又はその塩;
(式中、nは8〜16の整数であり、mは1〜4の整数を表す。また、式中の波線で表記した2−グルコサミンの1位水酸基はα、βアノマーのいずれか、あるいはその平衡混合物を表す。)。
【請求項2】
下記一般式(2)で表されるα,ω−長鎖ジカルボン酸
HOOC−(CH2)n−COOH 一般式(2)
(式中、nは8〜16の整数を表す。)
を水性メタノール中で、2−グルコサミンと縮合させ、生じた固体をろ過、洗浄することで、下記一般式(3)で表される中間体−3
(式中nは8〜16の整数を表す。また、式中の波線で表記した2−グルコサミンの1位水酸基はα、βアノマーのいずれか、あるいはその平衡混合物を表す。)
を製造し、
当該中間体−3と、オリゴグリシンのC端をエステルなどで保護した下記一般式(4)で表されるオリゴグリシンエステル
(式中mは1〜4の整数を表す。またRはアルコール性水酸基を有する化合物の残基であり、例えば、メチル基、エチル基、三級ブチル基、ベンジル基である。)
を加えて縮合させ,下記一般式(5)で表される中間体−5
(式中nは8〜16の整数を表し、mは1〜4の整数を表す。またRはアルコール性水酸基を有する化合物の残基であり、例えば、メチル基、エチル基、三級ブチル基、ベンジル基である。また式中の波線で表記した2−グルコサミンの1位水酸基はα、βアノマーのいずれか、あるいはその平衡混合物を表す。)
を得、これをアルカリ加水分解することで脱保護し、下記一般式(1)で表される非対称双頭型脂質分子又はその塩
(式中、nは8〜16の整数であり、mは1〜4の整数を表す。また、式中の波線で表記した2−グルコサミンの1位水酸基はα、βアノマーのいずれか、あるいはその平衡混合物を表す。)
を製造する方法。
【請求項3】
請求項1に記載の非対称双頭型脂質分子又はその塩から形成された、内表面がカルボキシル基で被覆され、外表面が2−グルコサミド基で被覆された構造を有することを特徴とする非対称ナノチューブ。
【請求項4】
請求項1に記載の非対称双頭型脂質分子を水に分散し、加熱溶解後冷却するか、又は室温で水又はアルカリ性溶液に分散溶解後、酸で中和することを特徴とする、非対称ナノチューブの製造方法。
【請求項5】
さらに、凍結乾燥処理又は超音波処理を施すことで、短尺チューブ状とすることを特徴とする、請求項4に記載の非対称ナノチューブの製造方法。
【請求項6】
請求項3の非対称ナノチューブがカチオン性薬剤を包接していることを特徴とする、カチオン性薬剤−非対称ナノチューブ複合体。
【請求項7】
請求項6に記載のカチオン性薬剤−非対称ナノチューブ複合体を有効成分とする、カチオン性薬剤組成物。
【請求項8】
カチオン性薬剤がアミノ基を有するアントラサイクリン系抗癌剤である、請求項7に記載のカチオン性薬剤組成物。
【請求項1】
下記一般式(1)で表される非対称双頭型脂質分子又はその塩;
(式中、nは8〜16の整数であり、mは1〜4の整数を表す。また、式中の波線で表記した2−グルコサミンの1位水酸基はα、βアノマーのいずれか、あるいはその平衡混合物を表す。)。
【請求項2】
下記一般式(2)で表されるα,ω−長鎖ジカルボン酸
HOOC−(CH2)n−COOH 一般式(2)
(式中、nは8〜16の整数を表す。)
を水性メタノール中で、2−グルコサミンと縮合させ、生じた固体をろ過、洗浄することで、下記一般式(3)で表される中間体−3
(式中nは8〜16の整数を表す。また、式中の波線で表記した2−グルコサミンの1位水酸基はα、βアノマーのいずれか、あるいはその平衡混合物を表す。)
を製造し、
当該中間体−3と、オリゴグリシンのC端をエステルなどで保護した下記一般式(4)で表されるオリゴグリシンエステル
(式中mは1〜4の整数を表す。またRはアルコール性水酸基を有する化合物の残基であり、例えば、メチル基、エチル基、三級ブチル基、ベンジル基である。)
を加えて縮合させ,下記一般式(5)で表される中間体−5
(式中nは8〜16の整数を表し、mは1〜4の整数を表す。またRはアルコール性水酸基を有する化合物の残基であり、例えば、メチル基、エチル基、三級ブチル基、ベンジル基である。また式中の波線で表記した2−グルコサミンの1位水酸基はα、βアノマーのいずれか、あるいはその平衡混合物を表す。)
を得、これをアルカリ加水分解することで脱保護し、下記一般式(1)で表される非対称双頭型脂質分子又はその塩
(式中、nは8〜16の整数であり、mは1〜4の整数を表す。また、式中の波線で表記した2−グルコサミンの1位水酸基はα、βアノマーのいずれか、あるいはその平衡混合物を表す。)
を製造する方法。
【請求項3】
請求項1に記載の非対称双頭型脂質分子又はその塩から形成された、内表面がカルボキシル基で被覆され、外表面が2−グルコサミド基で被覆された構造を有することを特徴とする非対称ナノチューブ。
【請求項4】
請求項1に記載の非対称双頭型脂質分子を水に分散し、加熱溶解後冷却するか、又は室温で水又はアルカリ性溶液に分散溶解後、酸で中和することを特徴とする、非対称ナノチューブの製造方法。
【請求項5】
さらに、凍結乾燥処理又は超音波処理を施すことで、短尺チューブ状とすることを特徴とする、請求項4に記載の非対称ナノチューブの製造方法。
【請求項6】
請求項3の非対称ナノチューブがカチオン性薬剤を包接していることを特徴とする、カチオン性薬剤−非対称ナノチューブ複合体。
【請求項7】
請求項6に記載のカチオン性薬剤−非対称ナノチューブ複合体を有効成分とする、カチオン性薬剤組成物。
【請求項8】
カチオン性薬剤がアミノ基を有するアントラサイクリン系抗癌剤である、請求項7に記載のカチオン性薬剤組成物。
【図1】

【図2】
【図3】
【図4】
【図5】
【図8】
【図9】
【図10】
【図11】
【図22】
【図23】
【図6】
【図7】
【図12】

【図13】

【図14】

【図15】

【図16】

【図17】
【図18】

【図19】

【図20】

【図21】
【図2】
【図3】
【図4】
【図5】
【図8】
【図9】
【図10】
【図11】
【図22】
【図23】
【図6】
【図7】
【図12】
【図13】
【図14】
【図15】
【図16】
【図17】
【図18】
【図19】
【図20】
【図21】
【公開番号】特開2012−51828(P2012−51828A)
【公開日】平成24年3月15日(2012.3.15)
【国際特許分類】
【出願番号】特願2010−194544(P2010−194544)
【出願日】平成22年8月31日(2010.8.31)
【出願人】(301021533)独立行政法人産業技術総合研究所 (6,529)
【Fターム(参考)】
【公開日】平成24年3月15日(2012.3.15)
【国際特許分類】
【出願日】平成22年8月31日(2010.8.31)
【出願人】(301021533)独立行政法人産業技術総合研究所 (6,529)
【Fターム(参考)】
[ Back to top ]