
非晶質酸化物薄膜の製造方法及び薄膜トランジスタ
【課題】非晶質酸化物薄膜の膜質を向上する。
【解決手段】有機成分とInとを含有する第1酸化物前駆体膜4に対して有機成分の熱分解温度未満で有機成分の結合状態を選択的に変化させ、フーリエ変換型赤外分光で測定したときに得られる赤外線吸収スペクトルにおいて、赤外線の波数1380cm−1以上1520cm−1以下の範囲を赤外線の波数1380cm−1以上1450cm−1以下の範囲と赤外線の波数1450cm−1超1520cm−1以下の範囲とに分割したときに、赤外線の波数1380cm−1以上1450cm-1以下の範囲に位置するピークが、赤外線の波数1350cm−1以上1750cm−1以下の範囲における赤外線吸収スペクトルの中で最大値を示す第2酸化物前駆体膜6を得る前処理工程と、第2酸化物前駆体膜中に残存する有機成分を除去して、第2酸化物前駆体膜6を非晶質酸化物薄膜8へ変化させる後処理工程とを有する。
【解決手段】有機成分とInとを含有する第1酸化物前駆体膜4に対して有機成分の熱分解温度未満で有機成分の結合状態を選択的に変化させ、フーリエ変換型赤外分光で測定したときに得られる赤外線吸収スペクトルにおいて、赤外線の波数1380cm−1以上1520cm−1以下の範囲を赤外線の波数1380cm−1以上1450cm−1以下の範囲と赤外線の波数1450cm−1超1520cm−1以下の範囲とに分割したときに、赤外線の波数1380cm−1以上1450cm-1以下の範囲に位置するピークが、赤外線の波数1350cm−1以上1750cm−1以下の範囲における赤外線吸収スペクトルの中で最大値を示す第2酸化物前駆体膜6を得る前処理工程と、第2酸化物前駆体膜中に残存する有機成分を除去して、第2酸化物前駆体膜6を非晶質酸化物薄膜8へ変化させる後処理工程とを有する。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、非晶質酸化物薄膜の製造方法及び薄膜トランジスタに関する。
【背景技術】
【0002】
非晶質酸化物薄膜、特にIn−Ga−Zn−O系(以下、「IGZO」と呼称する)を始めとした透明非晶質酸化物半導体は、「TAOS(Transparent Amorphous Oxide Semiconductor)」と称され、様々な研究機関やメーカーが研究開発を行い、技術躍進を遂げている。特に、日本や韓国、台湾のパネルメーカーが中心となって大型のディスプレイの試作が活発に行われており、信頼性や安定性の議論、更には量産化に向けた技術検討も進み、真空成膜法による製造においてはかなり実用化に近いレベルにまで達してきている。
【0003】
一方で、大面積化のトレンドの中で製造コストを下げる狙いとして液相プロセス(以下、「液相法」と呼称する)によるTAOS−TFT(Thin Film Transistor:薄膜トランジスタ)作製の研究開発も盛んであり、例えば、近年では液相法で作製したTAOS−TFT駆動の液晶ディスプレイが報告される等、液相法によるTFT作製に対する世の中の期待は益々高まっている。しかしながら、液相法においては、真空成膜法と比較してプロセス温度がまだまだ高いこと、良質な薄膜を得ることが困難であるということが、今後乗り越えなければならない課題として挙げられる。
【0004】
そこで、特許文献1には、基板上に形成した半導体前駆体膜を加熱処理する共に電磁波を照射して、半導体前駆体膜を非晶質の金属酸化物半導体膜に変換する非晶質酸化物薄膜の製造方法が開示されている。また、この文献には、半導体前駆体膜の形成後半導体変換処理前(加熱処理前)に、酸素プラズマやUVオゾン洗浄などのドライ洗浄プロセスによって洗浄し、薄膜中及び薄膜表面に存在し不純物の原因となる有機物を分解及び洗浄して、金属成分以外の有機成分を排除することが好ましいということも開示されている。
【先行技術文献】
【特許文献】
【0005】
【特許文献1】特開2010−258058号公報
【発明の概要】
【発明が解決しようとする課題】
【0006】
しかしながら、特許文献1の製造方法では、半導体変換処理前に、半導体前駆体膜中の金属成分以外の有機成分を全て排除することになる。本発明者らは鋭意研究を重ねた結果、半導体変換処理前、すなわち加熱処理前の有機成分の結合状態がTFT特性のオン電流を向上する等非晶質酸化物薄膜の膜質の向上に繋がることを見出した。
【0007】
本発明は上記事実に鑑みてなされたものであり、膜質を向上する非晶質酸化物薄膜の製造方法及び薄膜トランジスタを提供することを目的とする。
【課題を解決するための手段】
【0008】
本発明の上記課題は下記の手段によって解決された。
<1>有機成分とInとを含有する第1酸化物前駆体膜に対して、前記有機成分の熱分解温度未満で前記有機成分の結合状態を選択的に変化させ、フーリエ変換型赤外分光で測定したときに得られる赤外線吸収スペクトルにおいて、赤外線の波数1380cm−1以上1520cm−1以下の範囲を、赤外線の波数1380cm−1以上1450cm−1以下の範囲と赤外線の波数1450cm−1超1520cm−1以下の範囲とに分割したときに、赤外線の波数1380cm−1以上1450cm-1以下の範囲に位置するピークが、赤外線の波数1350cm−1以上1750cm−1以下の範囲における赤外線吸収スペクトルの中で最大値を示す第2酸化物前駆体膜を得る前処理工程と、前記第2酸化物前駆体膜中に残存する前記有機成分を除去して、前記第2酸化物前駆体膜を、前記Inを含有する非晶質酸化物薄膜へ変化させる後処理工程と、を有する非晶質酸化物薄膜の製造方法。
<2>前記前処理工程における前記有機成分の結合状態の選択的な変化は、前記第2酸化物前駆体膜に対してフーリエ変換型赤外分光で測定したときに得られる赤外線吸収スペクトルにおいて、赤外線の波数1500cm−1以上1600cm−1以下の範囲にある前記有機成分由来のピークの減少に相当する変化を含む、<1>に記載の非晶質酸化物薄膜の製造方法。
<3>前記前処理工程における前記有機成分の結合状態の選択的な変化は、前記第2酸化物前駆体膜に対してフーリエ変換型赤外分光で測定したときに得られる赤外線吸収スペクトルにおいて、赤外線の波数2750cm−1以上3050cm−1以下の範囲にある前記有機成分由来のピークの減少に相当する変化を含む、<1>又は<2>に記載の非晶質酸化物薄膜の製造方法。
<4>前記第2酸化物前駆体膜に対してフーリエ変換型赤外分光で測定したときに得られる赤外線吸収スペクトルにおいて、赤外線の波数1500cm−1以上1600cm−1以下の範囲にあるピークの減少率が、前記前処理工程前の1500cm−1以上1600cm−1以下の範囲にあるピークを1としたとき0.37以下である、<2>に記載の非晶質酸化物薄膜の製造方法。
<5>前記第2酸化物前駆体膜に対してフーリエ変換型赤外分光で測定したときに得られる赤外線吸収スペクトルにおいて、赤外線の波数1500cm−1以上1600cm−1以下の範囲にあるピークの減少率が、前記前処理工程前の1500cm−1以上1600cm−1以下の範囲にあるピークを1としたとき0.11以下である、<4>に記載の非晶質酸化物薄膜の製造方法。
<6>前記前処理工程は、光処理工程である、<1>〜<5>の何れか1つに記載の非晶質酸化物薄膜の製造方法。
<7>前記光処理工程では、前記第1酸化物前駆体膜に対して紫外線を照射する、<6>に記載の非晶質酸化物薄膜の製造方法。
<8>前記前処理工程では、前記第2酸化物前駆体膜に対してフーリエ変換型赤外分光で測定したときに得られる赤外線吸収スペクトルにおいて、赤外線の波数1380cm−1以上1520cm−1以下の範囲に位置するピークが、光処理の時間に伴って、赤外線の波数1350cm−1以上1750cm−1以下の範囲における赤外線吸収スペクトルの中で最大値を示し始めるときに必要な光処理のエネルギー総量に対して、4倍以上のエネルギー総量を用いて光処理を行う、<6>又は<7>に記載の非晶質酸化物薄膜の製造方法。
<9>前記前処理工程では、前記第2酸化物前駆体膜に対してフーリエ変換型赤外分光で測定したときに得られる赤外線吸収スペクトルにおいて、赤外線の波数1380cm−1以上1520cm−1以下の範囲に位置するピークが、光処理の時間に伴って、赤外線の波数1350cm−1以上1750cm−1以下の範囲における赤外線吸収スペクトルの中で最大値を示し始めるときに必要な光処理のエネルギー総量に対して、6倍以下のエネルギー総量を用いて光処理を行う、<8>に記載の非晶質酸化物薄膜の製造方法。
<10>前記後処理工程は、425℃以上の温度で熱処理する熱処理工程である、<1>〜<9>の何れか1つに記載の非晶質酸化物薄膜の製造方法。
<11>前記第1酸化物前駆体膜は、前記Inの他、Ga、Zn及びSnから選ばれる少なくとも一つを含む無機成分を含有する、<1>〜<10>の何れか1つに記載の非晶質酸化物薄膜の製造方法。
<12>前記無機成分の組成が、結晶化温度以上の前記熱処理工程でInGaZnO4-δ(前記δは酸素不定比量である)が結晶相として単相で現れるような組成である、<11>に記載の非晶質酸化物薄膜の製造方法。
<13>前記前処理工程の前に、金属アルコキシド又は有機酸塩を含有する溶液を用いて液相法により前記第1酸化物前駆体膜を形成する形成工程、をさらに有する<1>〜<12>の何れか1つに記載の非晶質酸化物薄膜の製造方法。
<14><1>〜<13>の何れか1つに記載の非晶質酸化物薄膜の製造方法で作製された非晶質酸化物薄膜を活性層として用いた薄膜トランジスタ。
<15><1>〜<13>の何れか1つに記載の非晶質酸化物薄膜の製造方法で作製された非晶質酸化物薄膜を絶縁層として用いた薄膜トランジスタ。
<16><1>〜<13>の何れか1つに記載の非晶質酸化物薄膜の製造方法で作製された非晶質酸化物薄膜を導電層として用いた薄膜トランジスタ。
【発明の効果】
【0009】
本発明によれば、膜質を向上する非晶質酸化物薄膜の製造方法及び薄膜トランジスタを提供することができる。
【図面の簡単な説明】
【0010】
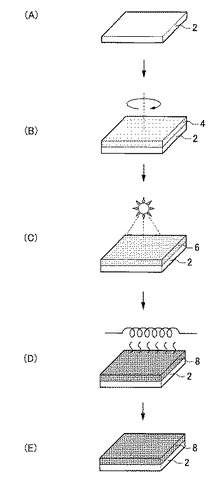
【図1】図1は、本発明の実施形態に係る非晶質酸化物薄膜の製造方法の工程図である。
【図2】図2(A)は、本発明の実施形態に係るTFTであって、トップゲート構造でトップコンタクト型のTFTの一例を示す模式図である。図2(B)は、本発明の実施形態に係るTFTであって、トップゲート構造でボトムコンタクト型のTFTの一例を示す模式図である。図2(C)は、本発明の実施形態に係るTFTであって、ボトムゲート構造でトップコンタクト型のTFTの一例を示す模式図である。図2(D)は、本発明の実施形態に係るTFTであって、ボトムゲート構造でボトムコンタクト型のTFTの一例を示す模式図である。
【図3】図3は、本発明の電気光学装置の一実施形態の液晶表示装置について、その一部分の概略断面図である。
【図4】図4は、図3に示す液晶表示装置の電気配線の概略構成図である。
【図5】図5は、本発明の電気光学装置の一実施形態のアクティブマトリックス方式の有機EL表示装置について、その一部分の概略断面図である。
【図6】図6は、図5に示す電気光学装置の電気配線の概略構成図である。
【図7】図7は、本発明のセンサの一実施形態であるX線センサについて、その一部分の概略断面図である。
【図8】図8は、図7に示すセンサの電気配線の概略構成図である。
【図9】図9は、実施例の乾燥ゲルに対するTG−DTAの測定結果を示す図である。
【図10】図10は、実施例の第1酸化物前駆体膜に対するXRDの測定結果である。
【図11】図11は、各第2酸化物前駆体膜に対してFT−IR(フーリエ変換型赤外分光)スペクトル測定した測定結果を示す図である。
【図12】図12は、図11に示す赤外線の波数1350cm−1以上1750cm−1以下の範囲における赤外線吸収スペクトルの拡大図である。
【図13】図13は、複数の第1酸化物前駆体膜を各種温度で熱処理した後の各膜に対してFT−IR(フーリエ変換型赤外分光)スペクトル測定した測定結果を示す図である。
【図14】UVオゾン処理後に熱処理を施したサンプルのXRD測定結果を示す図である。
【図15】UVオゾン処理を施していないサンプルのXRD測定結果を示す図である。
【図16】図16は、本発明の実施例に係る薄膜トランジスタの製造方法の工程図を示す図である。
【図17】図17は、熱処理温度を400℃に固定して、UV照射時間を0、1、3、5、10、20、30、60、90分と変化させたときの薄膜トランジスタのVg−Id特性を示す図である。
【図18】図18は、熱処理温度を425℃に固定して、UV照射時間を0、1、3、5、10、20、30、60、90分と変化させたときの薄膜トランジスタのVg−Id特性を示す図である。
【図19】図19は、熱処理温度を450℃に固定して、UV照射時間を0、1、3、5、10、20、30、60、90分と変化させたときの薄膜トランジスタのVg−Id特性を示す図である。
【図20】図20は、熱処理温度を500℃に固定して、UV照射時間を0、1、3、5、10、20、30、60、90分と変化させたときの薄膜トランジスタのVg−Id特性を示す図である。
【図21】図21は、熱処理温度を600℃に固定して、UV照射時間を0、1、3、5、10、20、30、60、90分と変化させたときの薄膜トランジスタのVg−Id特性を示す図である。
【図22】図22は、図17〜図21に示すVg−Id特性から求めたオンオフ比とUV照射時間との関係を示す図である。
【図23】図23は、図22の部分拡大図である。
【図24】図24は、図17〜図21に示すVg−Id特性から求めた移動度とUV照射時間との関係を示す図である。
【図25】図25は、実施例4のTFTと比較例1のTFTのVg−Id特性を示す図である。
【図26】図26は、実施例4のTFTと比較例1のTFTの熱処理温度(400℃のときは除く)と移動度との関係をプロットしたグラフ図である。
【図27】図27は、各測定サンプルのXPSデータを示す図である。
【図28】図28は、各第2酸化物前駆体膜に対してFT−IRスペクトル測定した測定結果を示す図である。
【図29】図29は、図28に示す赤外線の波数1350cm−1以上1750cm−1以下の範囲における赤外線吸収スペクトルの拡大図である。
【図30】図30は、本発明の実施例に係る薄膜トランジスタの製造方法の工程図を示す図である。
【図31】図31は、実施例により得られた薄膜トランジスタのVg−Id特性を示す図である。
【図32】図32は、UVオゾン処理を省略した薄膜トランジスタのVg−Id特性を示す図である。
【図33】図33は、熱処理温度と薄膜トランジスタの移動度との関係をプロットしたグラフ図である。
【図34】図34は、各乾燥ゲルのFT−IRスペクトルを示す図である。
【発明を実施するための形態】
【0011】
以下、添付の図面を参照しながら、本発明の実施形態に係る非晶質酸化物薄膜の製造方法及び薄膜トランジスタについて具体的に説明する。なお、図中、同一又は対応する機能を有する部材(構成要素)には同じ符号を付して適宜説明を省略する。
【0012】
1.非晶質酸化物薄膜の製造方法
本発明の実施形態に係る非晶質酸化物薄膜の製造方法は、有機成分とInとを含有する第1酸化物前駆体膜に対して、前記有機成分の熱分解温度未満で前記有機成分の結合状態を選択的に変化させ、フーリエ変換型赤外分光で測定したときに得られる赤外線吸収スペクトルにおいて、赤外線の波数1380cm−1以上1520cm−1以下の範囲に位置するピークが、赤外線の波数1350cm−1以上1750cm−1以下の範囲における赤外線吸収スペクトルの中で最大値を示す第2酸化物前駆体膜を得る前処理工程と、前記第2酸化物前駆体膜中に残存する前記有機成分を除去して、前記第2酸化物前駆体膜を、前記Inを含有する非晶質酸化物薄膜へ変化させる後処理工程と、を有する。
なお、上記「熱分解温度」は、第1酸化物前駆体膜に対してTG−DTAの測定を実施したときに、発熱反応に伴う重量減少が終了する温度であり、例えば425℃である。また、上記「有機成分を除去」とは、有機成分全部を除去するだけでなく、有機成分の一部を除去してもよい。
そして、このような製造方法を用いることで、得られる非晶質酸化物薄膜の膜質を向上することができる。
【0013】
以下、本発明の実施形態に係る非晶質酸化物薄膜の具体的な製造方法を説明する。図1は、本発明の実施形態に係る非晶質酸化物薄膜の製造方法の工程図である。
【0014】
<基板用意工程>
まず、図1(A)に示すように、非晶質酸化物薄膜を成膜するための基板2を用意する。基板2の形状、構造、大きさ等については特に制限はなく、目的に応じて適宜選択することが出来る。基板2の構造は単層構造であってもよいし、積層構造であってもよい。
基板2の材質としては特に限定はなく、例えばガラス、YSZ(イットリウム安定化ジルコニウム)等の無機基板、樹脂基板や、その複合材料等を用いることが出来る。中でも軽量である点、可撓性を有する点から樹脂基板やその複合材料が好ましい。具体的には、ポリブチレンテレフタレート、ポリエチレンテレフタレート、ポリエチレンナフタレート、ポリブチレンナフタレート、ポリスチレン、ポリカーボネート、ポリスルホン、ポリエーテルスルホン、ポリアリレート、アリルジグリコールカーボネート、ポリアミド、ポリイミド、ポリアミドイミド、ポリエーテルイミド、ポリベンズアゾール、ポリフェニレンサルファイド、ポリシクロオレフィン、ノルボルネン樹脂、ポリクロロトリフルオロエチレン等のフッ素樹脂、液晶ポリマー、アクリル樹脂、エポキシ樹脂、シリコーン樹脂、アイオノマー樹脂、シアネート樹脂、架橋フマル酸ジエステル、環状ポリオレフィン、芳香族エーテル、マレイミドーオレフィン、セルロース、エピスルフィド化合物等の合成樹脂基板、酸化珪素粒子との複合プラスチック材料、金属ナノ粒子、無機酸化物ナノ粒子、無機窒化物ナノ粒子等との複合プラスチック材料、カーボン繊維、カーボンナノチューブとの複合プラスチック材料、ガラスフェレーク、ガラスファイバー、ガラスビーズとの複合プラスチック材料、粘土鉱物や雲母派生結晶構造を有する粒子との複合プラスチック材料、薄いガラスと上記単独有機材料との間に少なくとも1回の接合界面を有する積層プラスチック材料、無機層と有機層を交互に積層することで、少なくとも1回以上の接合界面を有するバリア性能を有する複合材料、ステンレス基板或いはステンレスと異種金属を積層した金属多層基板、アルミニウム基板或いは表面に酸化処理(例えば陽極酸化処理)を施すことで表面の絶縁性を向上させた酸化皮膜付きのアルミニウム基板等を用いることが出来る。また、樹脂基板は、耐熱性、寸法安定性、耐溶剤性、電気絶縁性、加工性、低通気性、又は低吸湿性等に優れていることが好ましい。前記樹脂基板は、水分や酸素の透過を防止するためのガスバリア層や、樹脂基板の平坦性や下部電極との密着性を向上するためのアンダーコート層等を備えていてもよい。
【0015】
また、本発明における基板2の厚みに特に制限はないが、50μm以上1000μm以下が好ましく、50μm以上500μm以下であることがより好ましい。 基板2の厚みが50μm以上であると、基板2自体の平坦性がより向上する。また、基板2の厚みが500μm以下であると、基板2自体の可撓性がより向上し、フレキシブルデバイス用基板としての使用がより容易となる。
【0016】
<形成工程>
次に、図1(B)に示すように、例えばスピンコート、インクジェット、ディスペンサー、スクリーン印刷、凸版印刷又は凹版印刷等の液相法を用いて、基板2上に有機成分とInを含有する第1酸化物前駆体膜4、すなわち本実施形態に係る非晶質酸化物薄膜の前駆体膜を形成する。
この第1酸化物前駆体膜4は、Inの他、Ga、Zn及びSnから選ばれる少なくとも一つを含む無機成分を含有していることが好ましく、In、Ga及びZnを含む無機成分を含有していることがより好ましい。第1酸化物前駆体膜4中の無機成分が、In、Ga及びZnを含んでいる場合、その無機成分の組成は、後述する後処理工程で仮に結晶化温度以上の温度で熱処理を行ったときに、InGaZnO4-δ(前記δは酸素不定比量である)が結晶相として単相で現れるような組成であることが好ましい。この組成は、例えばIn:Ga:Zn=2−x:x:1(xは0.8以上1.05以下)のような組成である。上記のような組成で従来の製造方法により得られる非晶質酸化物薄膜は、薄膜トランジスタの活性層として用いた場合に、移動度等のTFT特性が低く、実用化し難い状態にあったものが、本実施形態の製造方法を経ることで、上記のような組成でも実用化に耐えうるTFT特性に向上することができる。また、上記組成の範囲外では、TFT特性は従来の製造方法でも良好ではあるが、本実施形態の製造方法を経ることで、TFT特性をより向上することができる。
液相法に用いる溶液は、様々な溶液を用いることができるが、金属アルコキシド又は有機酸塩を含有する溶液を用いることが好ましい。特に、硝酸塩や塩化物とは異なり不要な不純物成分(硝酸や塩素など)を除去するプロセスが不要という観点と有毒なガスの発生を抑制するという観点から金属アルコキシドを含有する溶液を用いることが好ましい。なお、基板2と前駆体膜4の間には、後述するような電極や絶縁膜等他の薄膜が配置されていてもよい。
有機酸塩としては、β − ジケトン錯体基、β−ケトカルボン酸エステル錯体基、β−ケトカルボン酸錯体基及びケトオキシ基(ケトオキシ錯体基)が挙げられる。β−ジケトン錯体基としては、例えば2 ,4−ペンタンジオン(アセチルアセトンあるいはアセトアセトンともいう)、1,1,1,5,5,5−ヘキサメチル−2,4−ペンタンジオン、2,2,6,6−テトラメチル−3,5−ヘプタンジオン、1,1,1−トリフルオロ−2 ,4−ペンタンジオン等を挙げることができる。β−ケトカルボン酸エステル錯体基としては、例えばアセト酢酸メチルエステル、アセト酢酸エチルエステル、アセト酢酸プロピルエステル、トリメチルアセト酢酸エチル、トリフルオロアセト酢酸メチル等を挙げることができる。β−ケトカルボン酸としては、例えば、アセト酢酸、トリメチルアセト酢酸等を挙げることができる。またケトオキシとしては、例えばアセトオキシ基(またはアセトキシ基)、プロピオニルオキシ基、ブチリロキシ基、アクリロイルオキシ基、メタクリロイルオキシ基等を挙げることができる。これらの基の炭素原子数は18以下が好ましい。また直鎖または分岐のもの、また水素原子をフッ素原子にしたものでもよい。
一方で、金属アルコキシド溶液としては、少なくとも下記一般式Iで表される金属アルコキシド化合物を含有するものである。
M(OR)n ・・・式[I]
ここで、式[I]中のMは金属元素で、nは1以上の整数である。
【0017】
特に、金属アルコキシド溶液として、下記一般式II及び一般式IIIで表される金属アルコキシド化合物を含有し、1〜100mPa・sの粘度を有するものであることが好ましい。
Zn(OR1)2 ・・・[II]
M(OR2)3 ・・・[III]
このような金属アルコキシド溶液であれば、溶液自体を加熱することにより非晶質状態でも良好な半導体特性を有する金属酸化物ナノ粒子を得ることができるので、簡便に半導体薄膜を形成することができるからである。
【0018】
本実施形態では、溶液中に少なくとも上記一般式II中でMがInである金属アルコキシド化合物が存在している。また、電気特性の観点から、MがGaである金属アルコキシド化合物とMがInである金属アルコキシド化合物とが溶液中に存在していることが好ましい。また、材料コストの観点から、MがAlである金属アルコキシド化合物とMがInである金属アルコキシド化合物とが溶液中に存在していることが好ましい。
【0019】
上記一般式I〜IIIで表される金属アルコキシド化合物は、金属アルコキシド溶液中で、それぞれ単独で存在していてもよく、その一部が連結して複合アルコキシドを形成していてもよい。上記一般式II及び一般式IIIの金属アルコキシド化合物のとき、金属アルコキシド溶液中の各化合物及び/又は複合アルコキシドにおいて、比率Zn/Mは、0.2〜10の範囲であることが好ましい。0.2未満では原料コストが高くなり、一方、10を超えるとZn成分が過剰となって酸化亜鉛の結晶が生じて複合酸化物ナノ粒子を形成し難い。Zn/Mの範囲は、電気特性の観点から0.2〜1.5の範囲であることが好ましく、0.5〜1.3の範囲であることが更に好ましい。
【0020】
上記一般式I〜III中、R、R1及びR2はそれぞれ同一でも異なっていてもよく、炭素数が1〜20、好ましくは1〜6のアルキル基を表し、置換されていても無置換であってもよい。炭素数が20を超えると、金属分子間が長くなって薄膜形成時に金属酸化物中にアルキル基が残存する場合があるため好ましくない。置換基としては、炭素数1〜4のアルキル基、アミノ基(アミノ基の水素原子は炭素数1〜4のアルキル基で置換されていてもよい)、炭素数1〜4のアルコキシ基、水酸基などを挙げることができる。
【0021】
このような一般式I〜IIIに相当する化合物の例としては、亜鉛エトキシド、亜鉛エトキシエトキシド、亜鉛ジメチルアミノエトキシド、亜鉛メトキシエトキシド、インジウムイソプロポキシド、インジウム−n−ブトキシド、インジウムメトキシエトキシド、インジウムジエチルアミノエトキシド、ガリウムエトキシド、ガリウムイソプロポキシド、アルミニウムイソプロポキシド、アルミニウム−n−ブトキシド、アルミニウム−s−ブトキシド、アルミニウム−t−ブトキシド、アルミニウムエトキシド、アルミニウムエトキシエトキシエトキシド、鉄イソプロポキシドなどを挙げることができる。
【0022】
また、金属アルコキシド溶液では、一般式II及びIIIの金属アルコキシド化合物を、ZnとMの合計濃度として好ましくは0.5〜20質量%、より好ましくは1〜10質量%で含有する。0.5質量%未満では、均一な薄膜ができない場合があり、20質量%を超えると充分に薄い薄膜を構成することができない場合がある。
【0023】
また、ナノ粒子分散液の粘度は、塗布手段により異なるが、例えばインクジェットやディスペンサーの場合、吐出性の観点から1〜100mPa・s、好ましくは1〜20mPa・sであることが望ましい。ナノ粒子分散液の上記粘度は、市販の粘度計、例えば振動式粘度計VISCOMATE(CBCマテリアルズ株式会社製)で測定することができる。
【0024】
金属アルコキシド溶液は、上記金属アルコキシド化合物を溶解するための適当な溶媒を含む。この溶媒としては、水、アルコール類、アミノアルコール類、グリコール類などを挙げることができ、分散液の安定性、乾燥性の観点から下記一般式IVで表される高沸点溶媒を少なくとも1種含むものであることが更に好ましい。
R3−OH ・・・[IV]
【0025】
ここで、R3は炭素原子数2〜12の置換又は未置換のアルキル基、アルケニル基、シクロアルキル基、又はアリール基を表す。
アルキル基としては、メチル基、エチル基、プロピル基、イソプロピル基、ブチル基、ペンチル基、ヘキシル基、オクチル基などが挙げられる。アルケニル基としては、ブテニル基、プロペニル基などが、シクロアルキル基としては、シクロヘキシル基などが、アリール基としては、フェニル基などがそれぞれ挙げられる。
アルキル基やアルケニル基への好ましい置換基の例としては、アルコキシ基(メトキシ基、エトキシ基、イソプロポキシ基、メトキシエトキシ基、フェノキシ基など)、水酸基、アミノ基などが挙げられる。シクロアルキル基やアリール基への好ましい置換基の例としては、アルキル基(メチル基、エチル基、ヒドロキシエチル基など)、アルコキシ基(メトキシ基、エトキシ基、イソプロポキシ基、メトキシエトキシ基、フェノキシ基など)、水酸基、アミノ基などが挙げられる。
特に好ましい置換基としては、ジアルキルアミノ基(ジメチルアミノ基、ジエチルアミノ基、ジ−n−プロピルアミノ基、ジイソプロピルアミノ基、ジ−n−ブチルアミノ基、ジ-t-ブチルアミノ基など)が挙げられる。
【0026】
また一般式IVで表される上記高沸点溶媒の沸点は、120℃〜250℃であり、乾燥時の負荷軽減の観点から好ましくは130℃〜200℃である。沸点が120℃未満では乾燥速度が速く十分な平滑性を得にくいため好ましくなく、250℃を超えると薄膜を形成する際に残存しやすくなるため、好ましくない。
【0027】
一般式IVに相当する高沸点溶媒としては、例えば、2−エトキシエタノール、2−(メトキシエトキシ)エタノール、2−(エトキシエトキシ)エタノール、2−イソプロポキシエタノール、1−エトキシ−2−プロパノール、2−ジエチルアミノエタノール、2-ジプロピルアミノエタノール、シクロヘキサノール、エチレングリコール、ジエチレングリコール、ベンジルアルコールなどを挙げることができる。
これらの分散媒と併用できる溶媒としては、例えばジオキサン、ジメチルホルムアミド、ジメチルスルホキシド、アセチルアセトンなどが挙げられる。
また、金属アルコキシド溶液は、帯電防止剤、可塑剤、高分子バインダー、増粘剤等の各種添加剤を目的に応じて添加し、物性調整した後に塗布用の溶液として用いてもよい。
【0028】
<前処理工程>
次に、図1(C)に示すように、上述した形成工程により得られた有機成分とInとを含有する第1酸化物前駆体膜4に対して、前記有機成分の熱分解温度未満で前記有機成分の結合状態を選択的に変化させ、フーリエ変換型赤外分光で測定したときに得られる赤外線吸収スペクトルにおいて、赤外線の波数1380cm−1以上1520cm−1以下の範囲を、赤外線の波数1380cm−1以上1450cm−1以下の範囲と赤外線の波数1450cm−1超1520cm−1以下の範囲とに分割したときに、赤外線の波数1380cm−1以上1450cm-1以下の範囲に位置するピークが、赤外線の波数1350cm−1以上1750cm−1以下の範囲における赤外線吸収スペクトルの中で最大値を示す第2酸化物前駆体膜6を得る前処理工程を行う。
なお、有機成分の熱分解温度未満としているのは、有機成分が熱分解してしまうと、前処理工程後には有機物が残存しておらず、そのまま後述する後処理工程を行っても膜質の向上が図り難いからである。また、赤外線の波数1380cm−1以上1520cm−1以下の範囲に位置するピークは、In前駆体原料に由来するものである。
このような前処理工程を行うと、後述する後処理工程後に得られる非晶質酸化物薄膜について、移動度が高く膜質の向上に繋がる。また、この非晶質酸化物薄膜をTFTの活性層として用いた場合に、オンオフ比が高く膜質の向上に繋がる。具体的には、赤外線の波数1380cm−1以上1450cm-1以下の範囲に位置するピークが、赤外線の波数1350cm−1以上1750cm−1以下の範囲における赤外線吸収スペクトルの中で最大値を示し始めるときから、移動度が約1桁以上に高くなり、且つ、オンオフ比が急激に高くなる。
【0029】
前処理工程としては、上述のような機能を果たせば手段は問わないが、例えば光処理工程又はプラズマ処理工程から選ばれる少なくとも一つが挙げられ、特に処理コストが抑えられるという観点から光処理工程が好ましい。また、この光処理工程では、第1酸化物前駆体膜4に対して紫外線を照射することが好ましい。紫外線を照射することが好ましい理由としては、紫外線のような短波長の光が有機成分の結合状態を変化させ易いからである。例えば低圧水銀ランプを用いた場合、主成分として185nm(645KJ/mol)と254nm(472KJ/mol)の紫外線が存在し、C-H(413.4KJ/mol)やC=C(607KJ/mol)やC-O(351.5KJ/mol)などの様々な有機成分の結合を分解することができる。そのため、紫外線の波長は100nm以上450nm以下であることが好ましく、150nm以上300nm以下であることがより好ましい。光源は、低圧水銀灯、重水素ランプ、キセノンエキシマランプ、メタルハライドランプ、エキシマーレーザーなどを用いることができる。
また、光処理のみでも有機成分の結合状態を変化させ得るが、最終的に得たい薄膜が酸化物であるため、光処理工程の他、オゾンや酸素ラジカルに代表される活性酸素や酸素プラズマ等と併用することでより膜質の向上を図ることができる。オゾンの場合は、酸素存在下にて紫外線を照射することで発生するが、オゾナイザー等で積極的に導入しても構わない。
なお、本実施形態においてフーリエ変換型赤外分光で用いる測定装置は、装置名がBRUKER ALPHAであり、測定配置がダイヤモンドATR配置である。
【0030】
また前処理工程では、第2酸化物前駆体膜6に対してフーリエ変換型赤外分光で測定したときに得られる赤外線吸収スペクトルにおいて、赤外線の波数1380cm−1以上1520cm−1以下の範囲に位置するピークが、光処理の時間に伴って、赤外線の波数1350cm−1以上1750cm−1以下の範囲における赤外線吸収スペクトルの中で最大値を示し始めるときに必要な光処理のエネルギー総量に対して、4倍以上のエネルギー総量を用いて光処理を行うことが好ましい。
このように4倍以上のエネルギー総量を用いて光処理を行うことで、後述する後処理工程後に得られる非晶質酸化物薄膜をTFTの活性層として用いた場合に、4倍未満のエネルギー総量を用いる場合に比べて、オンオフ比が高く、且つ、安定化(飽和)する。また、4倍未満のエネルギー総量を用いる場合に比べて、移動度が高く、且つ、安定化(飽和)する。
【0031】
さらに前処理工程では、第2酸化物前駆体膜6に対してフーリエ変換型赤外分光で測定したときに得られる赤外線吸収スペクトルにおいて、赤外線の波数1380cm−1以上1520cm−1以下の範囲に位置するピークが、光処理の時間に伴って、赤外線の波数1350cm−1以上1750cm−1以下の範囲における赤外線吸収スペクトルの中で最大値を示し始めるときに必要な光処理のエネルギー総量に対して、4倍以上6倍以下のエネルギー総量を用いて光処理を行うことがより好ましい。
6倍超のエネルギー総量を用いると移動度が安定化(飽和)するものの、第2酸化物前駆体膜6に対して長時間の光処理が行われることになる。4倍以上6倍以下のエネルギー総量を用いて光処理を行うことで、長時間の光処理を避け、溶液中の金属が酸化され電気伝導率が悪化することを抑制することができる。また、基板2が上述した樹脂基板や樹脂の複合材料で構成された基板である場合に、光処理によって基板2が変形することを抑制できる。
【0032】
前処理工程における有機成分の結合状態の選択的な変化は、第2酸化物前駆体膜6に対してフーリエ変換型赤外分光で測定したときに得られる赤外線吸収スペクトルにおいて、第1酸化物前駆体膜4の赤外線吸収スペクトルと比較した場合に、赤外線の波数1500cm−1以上1600cm−1以下の範囲にある有機成分由来のピークの減少に相当する変化を含むことが好ましい。この範囲の一部ピークは、CO系のピークであると想定される。ここで、前処理工程として光処理を単に行っても、形成工程で用いる溶液や第1酸化物前駆体膜4中の有機成分によって、減少するピークの種類は変化し得る。このように前処理工程によって選択的に減少するピークの種類を、赤外線の波数1500cm−1以上1600cm−1以下の範囲にあるピークに特定することで、確実に膜質の向上を図ることができる。
また、前処理工程における有機成分の結合状態の選択的な変化は、第2酸化物前駆体膜6に対してフーリエ変換型赤外分光で測定したときに得られる赤外線吸収スペクトルにおいて、第1酸化物前駆体膜4の赤外線吸収スペクトルと比較した場合に、赤外線の波数1500cm−1以上1600cm−1以下の範囲にあるピークの減少とは別個に又は減少と共に、照射した赤外線の波数が2750cm−1以上3050cm−1以下の範囲にある有機成分由来のピークの減少に相当する変化を含むことが好ましい。この範囲のピークは、CH系のピークであると想定される。このように前処理工程によって選択的に減少するピークの種類を、照射した赤外線の波数が2750cm−1以上3050cm−1以下の範囲にあるピークに特定することで、確実に膜質の向上を図ることができる。
【0033】
また、前処理工程後且つ後処理工程前で、第2酸化物前駆体膜6に対してフーリエ変換型赤外分光で測定したときに得られる赤外線吸収スペクトルにおいて、赤外線の波数1500cm−1以上1600cm−1以下の範囲にあるピークの減少率が、前処理工程前の1500cm−1以上1600cm−1以下の範囲にあるピークを1としたとき0.37以下であることが好ましく、0.11以下であることがより好ましい。ピークの減少率が0.37以下であると、第2酸化物前駆体膜6に対してフーリエ変換型赤外分光で測定したときに得られる赤外線吸収スペクトルにおいて、赤外線の波数1380cm−1以上1450cm-1以下の範囲に位置するピークが、赤外線の波数1350cm−1以上1750cm−1以下の範囲における赤外線吸収スペクトルの中で最大値を示し易いからである。 なお、このピークの減少率は、例えば光照射の波長、照射時間や照射量等によって調整することができる。
【0034】
<後処理工程>
次に、図1(D)に示すように、前処理工程後の第2酸化物前駆体膜中6に残存する有機成分を除去して、第2酸化物前駆体膜6を、Inを含有する非晶質金属酸化物薄膜8へ変化させる後処理工程を行う。
この後処理工程は、上述のような機能を果たせば手段は問わないが、具体的に熱処理工程が挙げられる。
熱処理工程の方法としては、電気炉やマッフル炉で加熱する方法や、赤外線ランプやホットプレートタイプの加熱方法等が挙げられる。
この熱処理工程では、425℃以上の温度で第2酸化物前駆体膜中6を熱処理することが好ましい。425℃未満で熱処理する場合に比べて、当該熱処理により得られる非晶質酸化物薄膜8の移動度を約3桁も高くすることができるからである。
また、第2酸化物前駆体膜6がIn、Ga及びZnを全て含有している場合、第2酸化物前駆体膜6を非晶質酸化物薄膜8へ変化させる過程で、非晶質の状態を保つという観点から、800℃未満、特に700℃以下の温度で熱処理することが好ましい。特に、基板2の溶解や変形を抑制するという観点から、600℃以下の温度で熱処理することが好ましい。
また、熱処理の雰囲気としては、酸化物を得やすいという観点から酸化性雰囲気で行うことが好ましい。なお、酸化性雰囲気としてはまず、大気(酸素分圧21%,(雰囲気全体に含まれる水分含有量が露点温度換算で16℃(絶対湿度13.6g/m−3)))が用いることができる。また、酸素分圧が全圧の20%以上を占める雰囲気を用いることが好ましく、例えば酸素分圧100%の雰囲気(ガスボンベからフロー。雰囲気全体に含まれる水分含有量が露点温度換算で−36℃以下(絶対湿度0.21g/m−3以下))を用いることもできる。
【0035】
<その他の工程>
最後に、図1(E)に示すように、後処理工程にて得られた非晶質酸化物薄膜8をエッチングしたり、非晶質酸化物薄膜8上に薄膜を形成したりする等その他の工程を適宜行う。
【0036】
以上の工程を経ることにより、膜質を向上した非晶質酸化物薄膜8を得ることができる。そして、このような非晶質酸化物薄膜8は薄膜トランジスタの活性層や導電層、絶縁層、中でも活性層として有用である。
【0037】
2.薄膜トランジスタ
本発明の実施形態に係る薄膜トランジスタ(以下、TFTと称す)は、ゲート電極、ゲート絶縁膜、活性層、ソース電極及びドレイン電極を有し、ゲート電極に電圧を印加して、活性層に流れる電流を制御し、ソース電極とドレイン電極間の電流をスイッチングする機能を有するアクテイブ素子である。そして、本発明の実施形態に係る薄膜トランジスタでは、活性層、絶縁層(ゲート絶縁膜や保護層等)及び導電層(ゲード電極やソース電極、ドレイン電極等)の少なくともいずれか1つとして上述の非晶質酸化物薄膜8が用いられる。
【0038】
TFTの素子構造としては、ゲート電極の位置に基づいた、いわゆる逆スタガ構造(ボトムゲート型とも呼ばれる)及びスタガ構造(トップゲート型とも呼ばれる)のいずれの態様であってもよい。また、活性層とソース電極及びドレイン電極(適宜、「ソース・ドレイン電極」という。)との接触部分に基づき、いわゆるトップコンタクト型、ボトムコンタクト型のいずれの態様であってもよい。
なお、トップゲート型とは、ゲート絶縁膜の上側にゲート電極が配置され、ゲート絶縁膜の下側に活性層が形成された形態であり、ボトムゲート型とは、ゲート絶縁膜の下側にゲート電極が配置され、ゲート絶縁膜の上側に活性層が形成された形態である。また、ボトムコンタクト型とは、ソース・ドレイン電極が活性層よりも先に形成されて活性層の下面がソース・ドレイン電極に接触する形態であり、トップコンタクト型とは、活性層がソース・ドレイン電極よりも先に形成されて活性層の上面がソース・ドレイン電極に接触する形態である。
【0039】
図2(A)は、本発明の実施形態に係るTFTであって、トップゲート構造でトップコンタクト型のTFTの一例を示す模式図である。図2(A)に示すTFT10では、基板12の一方の主面上に活性層14が積層されている。そして、この活性層14上にソース電極16及びドレイン電極18が互いに離間して設置され、更にこれらの上にゲート絶縁膜20と、ゲート電極22とが順に積層されている。
【0040】
図2(B)は、本発明の実施形態に係るTFTであって、トップゲート構造でボトムコンタクト型のTFTの一例を示す模式図である。図2(B)に示すTFT30では、基板12の一方の主面上にソース電極16及びドレイン電極18が互いに離間して設置されている。そして、活性層14と、ゲート絶縁膜20と、ゲート電極22と、が順に積層されている。
【0041】
図2(C)は、本発明の実施形態に係るTFTであって、ボトムゲート構造でトップコンタクト型のTFTの一例を示す模式図である。図2(C)に示すTFT40では、基板12の一方の主面上にゲート電極22と、ゲート絶縁膜20と、活性層14と、が順に積層されている。そして、この活性層14の表面上にソース電極16及びドレイン電極18が互いに離間して設置されている。
【0042】
図2(D)は、本発明の実施形態に係るTFTであって、ボトムゲート構造でボトムコンタクト型のTFTの一例を示す模式図である。図2(D)に示すTFT50では、基板12の一方の主面上にゲート電極22と、ゲート絶縁膜20と、が順に積層されている。そして、このゲート絶縁膜20の表面上にソース電極16及びドレイン電極18が互いに離間して設置され、更にこれらの上に、活性層14が積層されている。
【0043】
なお、本実施形態に係るTFTは、上記以外にも、様々な構成をとることが可能であり、適宜、活性層上に保護層や基板上に絶縁層等を備える構成であってもよい。
【0044】
以下、各構成要素について詳述する。なお、代表例として図2(A)に示すトップゲート構造でトップコンタクト型のTFT10であって、活性層14として本実施形態に係る非晶質酸化物薄膜8を用いる場合について具体的に説明するが、本発明は他の形態のTFTを製造する場合についても同様に適用することができる。
【0045】
<TFTの詳細構成>
−基板−
まず、TFT10を形成するための基板12を用意する。基板12は、上述した基板2と同一のものを用いることができる。
【0046】
−活性層−
次に、基板12上に、トランジスタとして主に活性層14を形成する。
活性層14は非晶質酸化物薄膜8を用いる。したがって、活性層14は、上述したような本発明の実施形態に係る非晶質酸化物薄膜8の製造方法を用いて形成する。ただし、後処理工程は、有機物を確実に除去するという観点から活性層14の形成直後であることが好ましいが、ソース電極16及びドレイン電極18の形成後であったり、ゲート電極22形成後であったり、TFT10完成後であったりしてもよい。
活性層14として用いる非晶質酸化物薄膜8の材料としては、Inを含有したIn−O系の酸化物半導体を用いる。この酸化物半導体には、Inの他Zn、Ga及びMgのうち少なくとも1種を含む酸化物がより好ましく、Inの他Ga及びZnのうちの少なくとも1種を含む酸化物がさらに好ましい。
特に、In、Ga及びZnを全て含む酸化物がより好ましい。In−Ga−Zn−O系酸化物半導体としては、結晶状態における組成がInGaO3(ZnO)m(mは6未満の自然数)で表される酸化物半導体が好ましく、特に、InGaZnO4(以下、「IGZO」とも言う。)がより好ましい。この組成の酸化物半導体の特徴としては、電気伝導度が増加するにつれ、電子移動度が増加する傾向を示す。
ただし、IGZOの組成比は、厳密にIn:Ga:Zn=1:1:1となる必要はない。また、活性層14は、上記のような酸化物半導体を主成分として含有していれば良く、その他に不純物等を含有していても良い。ここで、「主成分」とは、活性層14を構成する構成成分のうち、最も多く含有されている成分を表す。
【0047】
活性層14の形成後には、デバイスに応じて当該薄膜を適宜パターンニングする。パターンニングはフォトリソグラフィー及びエッチングにより行うことが出来る。具体的には、残存させる部分にフォトリソグラフィーによりレジストパターンを形成し、塩酸、硝酸、希硫酸、又は燐酸、硝酸及び酢酸の混合液等の酸溶液によりエッチングすることによりパターンを形成する。また、活性層14上にはソース・ドレイン電極エッチング時に活性層14を保護するための保護膜があってもよい。保護膜は活性層14と連続で成膜してもよいし、活性層14のパターンニング後に形成してもよい。さらに、活性層のパターニングは、後述するソース・ドレイン電極を形成した後に行ってもよい。パターンニングによる活性層14へのダメージを抑制することができるからである。
【0048】
活性層14の層構造は、2層以上から構成されていても良く、活性層14が低抵抗層と高抵抗層より形成され、低抵抗層がゲート絶縁膜20と接し、高抵抗層がソース電極16及びドレイン電極18の少なくとも一方と電気的に接していることが好ましい。
活性層14の厚みは、特に限定されないが、1nm以上100nm以下であり、より好ましくは、2.5nm以上50nm以下である。
【0049】
−ソース・ドレイン電極−
活性層14の上にソース・ドレイン電極16,18を形成するための導電膜を形成する。
この導電膜は高い導電性を有するものを用い、例えばAl,Mo,Cr,Ta,Ti,Au,Au等の金属、Al−Nd、Ag合金、酸化錫、酸化亜鉛、酸化インジウム、酸化インジウム錫(ITO)、酸化亜鉛インジウム(IZO)等の金属酸化物導電膜等を用いて形成することが出来る。ソース・ドレイン電極16,18としてはこれらの導電膜を単層構造又は2層以上の積層構造として用いることが出来る。
【0050】
導電膜の形成は、例えば印刷方式、コーティング方式等の湿式方式、真空蒸着法、スパッタリング法、イオンプレーティング法等の物理的方式、CVD、プラズマCVD法等の化学的方式等の中から使用する材料との適性を考慮して適宜選択した方法に従って成膜する。
成膜する導電膜の膜厚は、成膜性やエッチングやリフトオフ法によるパターンニング性、導電性等を考慮すると、10nm以上1000nm以下とすることが好ましく、50nm以上500nm以下とすることがより好ましい。
次いで、成膜した導電膜をエッチング又はリフトオフ法により所定の形状にパターンニングし、ソース電極及びドレイン電極16,18を形成する。この際、ソース・ドレイン電極16,18に接続する配線を同時にパターンニングすることが好ましい。
【0051】
−ゲート絶縁膜−
ソース・ドレイン電極16,18及び配線を形成した後、ゲート絶縁膜20を形成する。
ゲート絶縁膜20は、高い絶縁性を有するものが好ましく、例えばSiO2,SiNx,SiON,Al2O3,Y2O3,Ta2O5,HfO2等の絶縁膜、又はこれらの化合物を少なくとも二つ以上含む絶縁膜としてもよい。ゲート絶縁膜20は、印刷方式、コーティング方式等の湿式方式、真空蒸着法、スパッタリング法、イオンプレーティング法等の物理的方式、CVD、プラズマCVD法等の化学的方式等の中から使用する材料との適性を考慮して適宜選択した方法に従って成膜する。
次に、ゲート絶縁膜20は、必要に応じて、フォトリソグラフィー及びエッチングによって所定の形状にパターンニングを行う。
なお、ゲート絶縁膜20は、リーク電流の低下及び電圧耐性の向上のための厚みを有する必要がある一方、ゲート絶縁膜の厚みが大きすぎると駆動電圧の上昇を招いてしまう。 ゲート絶縁膜は材質にもよるが、ゲート絶縁膜の厚みは10nm以上10μm以下が好ましく、50nm以上1000nm以下がより好ましく、100nm以上400nm以下が特に好ましい。
【0052】
−ゲート電極−
ゲート絶縁膜20を形成した後、ゲート電極22を形成する。
ゲート電極22は、高い導電性を有するものを用い、例えばAl,Mo,Cr,Ta,Ti,Au,Au等の金属、Al−Nd、Ag合金、酸化錫、酸化亜鉛、酸化インジウム、酸化インジウム錫(ITO)、酸化亜鉛インジウム(IZO)等の金属酸化物導電膜等を用いて形成することが出来る。ゲート電極22としては、これらの導電膜を単層構造又は2層以上の積層構造として用いることが出来る。
【0053】
ゲート電極22は、例えば印刷方式、コーティング方式等の湿式方式、真空蒸着法、スパッタリング法、イオンプレーティング法等の物理的方式、CVD、プラズマCVD法等の化学的方式等の中から使用する材料との適性を考慮して適宜選択した方法に従って成膜する。成膜する導電膜の膜厚は成膜性、エッチングやリフトオフ法によるパターンニング性、導電性等を考慮すると、10nm以上1000nm以下とすることが好ましく、50nm以上500nm以下とすることがより好ましい。
成膜後、導電膜をエッチング又はリフトオフ法により所定の形状にパターンニングし、ゲート電極22を形成する。この際、ゲート電極22及びゲート配線を同時にパターンニングすることが好ましい。
【0054】
以上の手順により、図2(A)に示すような、オンオフ比が高く移動度も高い等TFT特性が良好なTFT10を作製することができる。なお、本発明の実施形態に係る非晶質酸化物薄膜8の製造方法は活性層14の形成に適用する場合を説明したが、ゲート絶縁膜20やゲート電極22、ソース電極16、ドレイン電極18の形成に用い、活性層14の形成は従来の方法を用いることもできる。また、活性層14の形成と共に、ゲート絶縁膜20、ゲート電極22、ソース電極16又はドレイン電極18の何れかの形成において、本発明の実施形態に係る非晶質酸化物薄膜8の製造方法を用いることもできる。
後述するように、本実施形態による液相法で作製したものと、真空成膜で作製したものとが、酸化物の電気物性を支配していると考えられる酸素の結合状態が類似しているため、半導体膜としての膜質の向上に限らず、ゲート電極22、ソース電極16又はドレイン電極18等の導電体膜の膜質の向上(移動度の向上)や、ゲート絶縁膜20等の絶縁体膜の膜質の向上(リーク電流の抑制)に繋がるからである。
【0055】
3.応用
以上で説明した本実施形態のTFTの用途には特に限定はないが、例えば電気光学装置における駆動素子、特に大面積デバイスに用いる場合に好適である。
更に実施形態のTFTは、樹脂基板を用いた低温プロセスで作製可能なデバイスに特に好適であり(例えばフレキシブルディスプレイ等)、X線センサなどの各種センサ、MEMS(Micro Electro Mechanical System)等、種々の電子デバイスにおける駆動素子(駆動回路)として、好適に用いられるものである。
【0056】
4.電気光学装置及びセンサ
【0057】
電気光学装置又はセンサは、本実施形態のTFTを備えて構成される。
電気光学装置の例としては、例えば液晶表示装置、有機EL(Electro Luminescence)表示装置、無機EL表示装置等の表示装置がある。
センサの例としては、CCD(Charge Coupled Device)又はCMOS(Complementary Metal Oxide Semiconductor)等のイメージセンサや、X線センサ等が好適である。
本実施形態のTFTを用いた電気光学装置およびセンサは、いずれも特性の面内均一性が高い。なお、ここで言う「特性」とは、電気光学装置(表示装置)の場合には表示特性、センサの場合には感度特性である。
以下、本実施形態によって製造されるTFTを備えた電気光学装置又はセンサの代表例として、液晶表示装置、有機EL表示装置、X線センサについて説明する。
【0058】
5.液晶表示装置
図3に、本発明の電気光学装置の一実施形態の液晶表示装置について、その一部分の概略断面図を示し、図4にその電気配線の概略構成図を示す。
【0059】
図3に示すように、本実施形態の液晶表示装置100は、図2(A)に示したトップゲート構造でトップコンタクト型のTFT10と、TFT10のパッシベーション層102で保護されたゲート電極22上に画素下部電極104およびその対向上部電極106で挟まれた液晶層108と、各画素に対応させて異なる色を発色させるためのRGBカラーフィルタ110とを備え、TFT10の基板12側およびRGBカラーフィルタ110上にそれぞれ偏光板112a、112bを備えた構成である。
【0060】
また、図4に示すように、本実施形態の液晶表示装置100は、互いに平行な複数のゲート配線112と、該ゲート配線112と交差する、互いに平行なデータ配線114とを備えている。ここでゲート配線112とデータ配線114は電気的に絶縁されている。ゲート配線112とデータ配線114との交差部付近に、TFT10が備えられている。
【0061】
TFT10のゲート電極22は、ゲート配線112に接続されており、TFT10のソース電極16はデータ配線114に接続されている。また、TFT10のドレイン電極18はゲート絶縁膜20に設けられたコンタクトホール116を介して(コンタクトホール116に導電体が埋め込まれて)画素下部電極104に接続されている。この画素下部電極104は、接地された対向上部電極106とともにキャパシタ118を構成している。
【0062】
図3に示した本実施形態の液晶装置においては、トップゲート構造のTFT10を備えるものとしたが、本発明の表示装置である液晶装置において用いられるTFTはトップゲート構造に限定されることなく、ボトムゲート構造のTFTであってもよい。
【0063】
本実施形態のTFTは、オンオフ比が非常に高いことから、安定性のある液晶表示装置を得ることができる。
【0064】
6.有機EL表示装置
図5に、本発明の電気光学装置の一実施形態のアクティブマトリックス方式の有機EL表示装置について、その一部分の概略断面図を示し、図6に電気配線の概略構成図を示す。
【0065】
有機EL表示装置の駆動方式には、単純マトリックス方式とアクティブマトリックス方式の2種類がある。単純マトリックス方式は低コストで作製できるメリットがあるが、走査線を1本ずつ選択して画素を発光させることから、走査線数と走査線あたりの発光時間は反比例する。そのため高精細化、大画面化が困難となっている。アクティブマトリックス方式は画素ごとにトランジスタやキャパシタを形成するため製造コストが高くなるが、単純マトリックス方式のように走査線数を増やせないという問題はないため高精細化、大画面化に適している。
【0066】
本実施形態のアクティブマトリックス方式の有機EL表示装置200は、図2(A)に示したトップゲート構造のTFT10が、パッシベーション層202を備えた基板12上に、駆動用TFT204およびスイッチング用TFT206として備えられ、該TFT204および206上に下部電極208および上部電極210に挟まれた有機発光層212からなる有機EL発光素子214を備え、上面もパッシベーション層216により保護された構成となっている。
【0067】
また、図6に示すように、本実施形態の有機EL表示装置200は、互いに平行な複数のゲート配線220と、該ゲート配線220と交差する、互いに平行なデータ配線222および駆動配線224とを備えている。ここで、ゲート配線220とデータ配線222、駆動配線224とは電気的に絶縁されている。スイッチング用TFT10bのゲート電極22は、ゲート配線220に接続されており、スイッチング用TFT10bのソース電極16はデータ配線222に接続されている。また、スイッチング用TFT10bのドレイン電極18は駆動用TFT10のゲート電極22に接続されるとともに、キャパシタ226を用いることで駆動用TFT10aをオン状態に保つ。駆動用TFT10aのソース電極16は駆動配線224に接続され、ドレイン電極18は有機EL発光素子214に接続される。
【0068】
図5に示した本実施形態の有機EL装置においては、トップゲート構造のTFT10aおよび10bを備えるものとしたが、本発明の表示装置である有機EL装置において用いられるTFTは、トップゲート構造に限定されることなく、ボトムゲート構造のTFTであってもよい。
【0069】
本実施形態により製造されるTFTは、オンオフ比が非常に高いことから、安定性のある有機EL表示装置が得られる。
【0070】
なお、図5に示した有機EL表示装置において、上部電極210を透明電極としてトップエミッション型としてもよいし、下部電極208およびTFTの各電極を透明電極とすることによりボトムエミッション型としてもよい。
【0071】
7.X線センサ
図7に、本発明のセンサの一実施形態であるX線センサについて、その一部分の概略断面図を示し、図8にその電気配線の概略構成図を示す。
【0072】
図7は、より具体的にはX線センサアレイの一部を拡大した概略断面図である。本実施形態のX線センサ300は基板12上に形成されたTFT10およびキャパシタ310と、キャパシタ310上に形成された電荷収集用電極302と、X線変換層304と、上部電極306とを備えて構成される。TFT10上にはパッシベーション膜308が設けられている。
【0073】
キャパシタ310は、キャパシタ用下部電極312とキャパシタ用上部電極314とで絶縁膜316を挟んだ構造となっている。キャパシタ用上部電極314は絶縁膜316に設けられたコンタクトホール318を介し、TFT10のソース電極16およびドレイン電極18のいずれか一方(図7においてはドレイン電極18)と接続されている。
【0074】
電荷収集用電極302は、キャパシタ310におけるキャパシタ用上部電極314上に設けられており、キャパシタ用上部電極314に接している。
X線変換層304はアモルファスセレンからなる層であり、TFT10およびキャパシタ310を覆うように設けられている。
上部電極306はX線変換層304上に設けられており、X線変換層304に接している。
【0075】
図8に示すように、本実施形態のX線センサ300は、互いに平行な複数のゲート配線320と、ゲート配線320と交差する、互いに平行な複数のデータ配線322とを備えている。ここでゲート配線320とデータ配線322は電気的に絶縁されている。ゲート配線320とデータ配線322との交差部付近に、TFT10が備えられている。
【0076】
TFT10のゲート電極22は、ゲート配線320に接続されており、TFT10のソース電極16はデータ配線322に接続されている。また、TFT10のドレイン電極18は電荷収集用電極302に接続されており、さらにこの電荷収集用電極302は、キャパシタ310に接続されている。
【0077】
本実施形態のX線センサ300において、X線は図7中、上部(上部電極306側)から照射され、X線変換層304で電子−正孔対を生成する。このX線変換層304に上部電極306によって高電界を印加しておくことにより、生成した電荷はキャパシタ310に蓄積され、TFT10を順次走査することによって読み出される。
【0078】
本実施形態のX線センサ300は、面内均一性の高い、信頼性に優れたTFT10を備えるため、均一性に優れた画像を得ることができる。
【0079】
なお、図7に示した本実施形態のX線センサにおいては、トップゲート構造のTFTを備えるものとしたが、本発明のセンサにおいて用いられるTFTはトップゲート構造に限定されることなく、ボトムゲート構造のTFTであってもよい。
【0080】
<変形例>
なお、本発明を特定の実施形態について詳細に説明したが、本発明はかかる実施形態に限定されるものではなく、本発明の範囲内にて他の種々の実施形態が可能であることは当業者にとって明らかであり、例えば上述の複数の実施形態は、適宜、組み合わせて実施可能である。また、以下の変形例同士を、適宜、組み合わせてもよい。
【0081】
例えば、上述した基板用意工程、形成工程、その他の工程は、全部又は一部を適宜省略することができる。
また、後処理工程の際に、例えば熱処理と同時に光照射を行ってもよい。
【実施例】
【0082】
以下に実施例を説明するが、本発明はこれら実施例により何ら限定されるものではない。
【0083】
<金属アルコキシド系の実施例>
−第1酸化物前駆体膜及び第2酸化物前駆体膜の作製及び検討−
酢酸亜鉛2水和物2.2g、ガリウムイソプロポキシド2.47g、インジウムイソプロポキシ2.74gを秤量し、ジエチルエタノールアミン中で150℃の温度にて攪拌し、淡黄色の金属アルコキシド系原料液(IGZO溶液)を得た。In,Ga,Znの組成比は、ICP測定により1.0:1.0:0.9という結果が得られた。そして、得られたIGZO溶液の乾燥ゲルに対してTG−DTAを実施した。
【0084】
図9は、実施例の乾燥ゲルに対するTG−DTAの測定結果を示す図である。
図9に示す測定結果のように、室温以上約400℃以下まで発熱反応に伴う重量減少が急激であった。また、400℃超425℃未満は重量減少が緩やかとなり、約425℃で発熱反応に伴う重量減少が終了していることが分かった。上記400℃及び425℃の温度は、乾燥ゲルの有機成分の熱分解温度に相当するものと考えられる。
【0085】
次に、石英基板上に、上記IGZO溶液を1000rpmの速度で二回スピンコートした後、室温で乾燥させることによりゲル状態の第1酸化物前駆体膜を成膜した。そして、得られた第1酸化物前駆体膜をXRDにて評価を行った。
【0086】
図10は、実施例の第1酸化物前駆体膜に対するXRDの測定結果である。
図10に示すように、室温乾燥させたのみの第1酸化物前駆体膜では、非晶質を示すパターンしか得られなかった。また、この第1酸化物前駆体膜のXRD焼成温度依存を測定すると、1000℃焼成の膜ではInGaZnO4の結晶構造を有しており、800℃焼成での膜では微弱なピークがあり一部結晶質があると推定されるものの非晶質と混在している状態であり、700℃焼成での膜では明確なピークが見られずにごく一部の結晶質があると推定されるものの非晶質と混在している状態であり、600℃以下までの膜では図10の結果のみから分析すると完全に非晶質として存在することが確認された。
【0087】
次に、室温乾燥させたのみの複数の第1酸化物前駆体膜に対して、互いに1分、3分、5分、10分、30分、60分、90分と照射時間を変えて前処理工程としてのUVオゾン処理を行い、第2酸化物前駆体膜を得た。このUVオゾン処理では低圧水銀ランプ(波長185nm及び254nm)を用いており、254nmの強度が約5mW/cm2である。
【0088】
このUVオゾン処理の前後の膜に対してXRF測定を行ったところ、In,Ga,Znの組成比及び塗布量は処理前後で変化していなかった。
また、このUVオゾン処理を行った後の第2酸化物前駆体膜に対して、装置名:BRUKER ALPHAの装置でダイヤモンドATR配置を用いて、FT−IR(フーリエ変換型赤外分光)スペクトル測定を実施した。また、比較のために、室温乾燥させたのみの複数の第1酸化物前駆体膜をUVオゾン処理の代わりに各種温度で熱処理し、その後の膜に対してFT−IR(フーリエ変換型赤外分光)スペクトル測定を実施した。
【0089】
図11は、各第2酸化物前駆体膜に対してFT−IR(フーリエ変換型赤外分光)スペクトル測定した測定結果を示す図である。図12は、図11に示す赤外線の波数1350cm−1以上1750cm−1以下の範囲における赤外線吸収スペクトルの拡大図である。
図13は、複数の第1酸化物前駆体膜を各種温度で熱処理した後の各膜に対してFT−IR(フーリエ変換型赤外分光)スペクトル測定した測定結果を示す図である。なお、図11では、90分の間UVオゾン処理した後の第2酸化物前駆体膜に対するFT−IR(フーリエ変換型赤外分光)スペクトルの測定結果は、60分の測定結果と同様であったので図示を省略している。
【0090】
図11に示すように、処理時間と共にFT−IRスペクトルの一部ピークの形状が変化していくことが確認された。UVオゾン処理と熱処理でのFT−IRスペクトルの形状変化を図11及び図13を参照しながら比較すると、熱処理では、ピークが全体的に減少しているが、UVオゾン処理では選択的にピークの形状(有機成分の結合状態)が変化していることが分かる。また、UVオゾン処理後に熱処理を施したサンプル(図14参照)ではUVオゾン処理を施していないサンプル(図15参照)と同様に600度以下ではXRD測定結果からは完全に非晶質状態であることが確認された。
さらに、図12に示すように、UVオゾン処理の照射時間が5分未満では、赤外線の波数1500cm−1以上1600cm−1以下の範囲にあるピークが、赤外線の波数1350cm−1以上1750cm−1以下の範囲(I)における赤外線吸収スペクトルの中で最大値を示している。一方で、UVオゾン処理の照射時間が5分以上では、第2酸化物前駆体膜に対してフーリエ変換型赤外分光で測定したときに得られる赤外線吸収スペクトルにおいて、赤外線の波数1380cm−1以上1520cm−1以下の範囲(II)に位置する(ダブル)ピークが、赤外線の波数1350cm−1以上1750cm−1以下の範囲(I)における赤外線吸収スペクトルの中で最大値を示すようになることが分かった。
より具体的には、UVオゾン処理の照射時間が5分以上では、第2酸化物前駆体膜に対してフーリエ変換型赤外分光で測定したときに得られる赤外線吸収スペクトルにおいて、赤外線の波数1380cm−1以上1520cm−1以下の範囲(II)を、赤外線の波数1380cm−1以上1450cm−1以下の範囲(IV)と赤外線の波数1450cm−1超1520cm−1以下の範囲(III)とに分割したときに、赤外線の波数1380cm−1以上1450cm-1以下の範囲(IV)に位置するピークが、赤外線の波数1350cm−1以上1750cm−1以下の範囲(I)における赤外線吸収スペクトルの中で最大値を示すようになる。
なお、赤外線の波数1380cm−1以上1450cm−1以下の範囲(IV)と、赤外線の波数1450cm−1超1520cm−1以下の範囲(III)とには、両範囲を跨ぐようにダブルピークが見られ、このダブルピークの一つが上記範囲(IV)に位置するピークである。
【0091】
また、図11に示すように、UVオゾン処理におけるピークの形状変化(有機成分の結合状態)の選択的な変化として、UVオゾン処理の照射時間が長くなると共に、赤外線の波数2750cm−1以上3050cm−1以下の範囲にある有機成分由来のピークの減少も含むことが分かった。同様に、UVオゾン処理におけるピークの形状変化(有機成分の結合状態)の選択的な変化として、UVオゾン処理の照射時間が長くなると共に、赤外線の波数1500cm−1以上1600cm−1以下の範囲にある有機成分由来のピークの減少も含むことが分かった。
以下、表1に照射時間及びエネルギー総量と、赤外線の波数1500cm−1以上1600cm−1以下の範囲にある有機成分由来のピークの減少率との相関関係を示す。
【0092】
【表1】

【0093】
−薄膜トランジスタの作製−
次に、UVオゾン処理の照射時間によって変化する第2酸化物前駆体膜の状態が熱処理後の薄膜へデバイス特性としてどのような影響を及ぼすか評価を行うために、ボトムゲートートップコンタクト型の薄膜トランジスタを作製した。
【0094】
図16は、本発明の実施例に係る薄膜トランジスタの製造方法の工程図を示す図である。
【0095】
本発明の実施例に係る薄膜トランジスタの基板には、P+Si/Si熱酸化膜(100nm)付基板を用い、ゲート電極としてP+Siを、ゲート絶縁膜としてSi熱酸化膜を用いた。
次に、図16(A)〜(C)に示す過程において、上記の第1酸化物前駆体膜から第2酸化物前駆体膜を得る方法と同一の方法を用いて活性層を作製した。具体的に、図16(A)に示すように、IGZO溶液(上記原料液と同じ溶液で、In:Ga:Zn=1.0:1.0:0.9)を用い、スピンコート法により最終生成物の膜厚が50nm程度になるように活性層の成膜を行った(熱処理により有機成分が減少して膜厚が変化するため)。成膜後に、図16(B)に示すように前処理工程としてUVオゾン処理を各種条件にて実施し、図16(C)に示すように後処理工程として400℃以上600℃以下の間で15分間熱処理を実施した。その後図16(D)に示すようにフォトリソグラフィーとウェットエッチングにより活性層をパターニングした。レジストにはTSMR8900−LB、エッチング液にはシュウ酸を用いた。現像には現像液としてTMAH5%溶液を用いている。ソース、ドレイン(100nm)電極にはMoを、図16(E)及び図16(F)に示すようにリフトオフにより形成した。レジストにはAZ5214Eを用いた。
【0096】
−トランジスタ特性及び移動度の評価−
次に、得られた薄膜トランジスタに対して、半導体パラメータ・アナライザー4156C(アジレントテクノロジー社製)を用い、トランジスタ特性(Vg−Id特性)及び移動度μの測定を行った。Vg−Id特性の測定は、ドレイン電圧(Vd)を5Vに固定し、ゲート電圧(Vg)を−20V〜+40Vの範囲内で変化させ、各ゲート電圧(Vg)におけるドレイン電流(Id)を測定することにより行った。
【0097】
図17〜図21は、実施例により得られた薄膜トランジスタのVg−Id特性を示す図である。具体的に、図17は、熱処理温度を400℃に固定して、UV照射時間を0、1、3、5、10、20、30、60、90分と変化させたときの薄膜トランジスタのVg−Id特性を示す図である。図18は、熱処理温度を425℃に固定して、UV照射時間を0、1、3、5、10、20、30、60、90分と変化させたときの薄膜トランジスタのVg−Id特性を示す図である。図19は、熱処理温度を450℃に固定して、UV照射時間を0、1、3、5、10、20、30、60、90分と変化させたときの薄膜トランジスタのVg−Id特性を示す図である。図20は、熱処理温度を500℃に固定して、UV照射時間を0、1、3、5、10、20、30、60、90分と変化させたときの薄膜トランジスタのVg−Id特性を示す図である。図21は、熱処理温度を600℃に固定して、UV照射時間を0、1、3、5、10、20、30、60、90分と変化させたときの薄膜トランジスタのVg−Id特性を示す図である。
なお、熱処理温度に関わらず、UV照射時間を0分としたときの薄膜トランジスタを比較例1のTFTとし、UV照射時間を1分としたときの薄膜トランジスタを比較例2のTFTとし、UV照射時間を3分としたときの薄膜トランジスタを比較例3のTFTと呼称する。また、熱処理温度に関わらず、UV照射時間を5分としたときの薄膜トランジスタを実施例1のTFTとし、UV照射時間を10分としたときの薄膜トランジスタを実施例2のTFTとし、UV照射時間を20分としたときの薄膜トランジスタを実施例3のTFTと呼称する。さらに、熱処理温度に関わらず、UV照射時間を30分としたときの薄膜トランジスタを実施例4のTFTとし、UV照射時間を60分としたときの薄膜トランジスタを実施例5のTFTとし、UV照射時間を90分としたときの薄膜トランジスタを実施例6のTFTと呼称する。
【0098】
また、図17〜図21に示すVg−Id特性から求めたオンオフ比とUV照射時間との関係を以下の表2にまとめ、図17〜図21に示すVg−Id特性から求めた移動度とUV照射時間との関係を以下の表3にまとめた。また、図17〜図21に示すVg−Id特性から求めたオンオフ比とUV照射時間との関係を図22に示し、図22の部分拡大図を図23に示した。さらに、図17〜図21に示すVg−Id特性から求めた移動度とUV照射時間との関係を図24に示した。
【0099】
【表2】
【0100】
【表3】
【0101】
図17〜図21に示すVg−Id特性から、UVオゾン処理時間が長くなると共に良好なVg−Id特性が得られていることが分かった。
特に、図22及び図23に示すように、照射時間を5分以上とした実施例1〜6のTFTは、比較例1〜3のTFTに比べてオンオフ比が急激に高くなり膜質の向上に繋がることが分かった。
【0102】
さらに、表3及び図24に示すように、照射時間を10分以上とした実施例2〜6のTFTは、実施例1のTFTに比べて移動度が高くなり膜質の向上に繋がることが分かった。すなわち、第2酸化物前駆体膜(活性層)に対して、フーリエ変換型赤外分光で測定したときに得られる赤外線吸収スペクトルにおいて、赤外線の波数1380cm−1以上1520cm−1以下の範囲(II)を、赤外線の波数1380cm−1以上1450cm−1以下の範囲(IV)と赤外線の波数1450cm−1超1520cm−1以下の範囲(III)とに分割したときに、赤外線の波数1380cm−1以上1450cm-1以下の範囲(IV)に位置するピークが、赤外線の波数1350cm−1以上1750cm−1以下の範囲(I)における赤外線吸収スペクトルの中で最大値を示し始めるときから(照射時間5分以上:エネルギー総量1.5J/cm2以上:ピーク減少率0.37以下に対応)、UV照射時間が5分未満の場合に比べて、オンオフ比が急激に高くなることが分かった。
また、表3に示すように、照射時間を5分以上とした実施例1〜6のTFTは、比較例1〜3のTFTに比べて移動度が高くなり膜質の向上に繋がることが分かった。すなわち、赤外線の波数1380cm−1以上1520cm−1以下の範囲(II)に位置するピークが、赤外線の波数1350cm−1以上1750cm−1以下の範囲(I)における赤外線吸収スペクトルの中で最大値を示し始めるときから(照射時間5分以上:エネルギー総量1.5J/cm2以上:ピーク減少率0.37以下に対応)、UV照射時間が5分未満の場合に比べて、移動度が約1桁以上高くなることが分かった。
【0103】
さらにまた、図22に示すように、照射時間を20分以上とした実施例3〜6のTFTは、実施例1や実施例2のTFTに比べてオンオフ比が高く、且つ、オンオフ比が安定化(飽和)していることが分かった。また、図24に示すように、照射時間を20分以上とした実施例3〜6のTFTは、実施例1や実施例2のTFTに比べて移動度が急激に高くなり、実施例3〜6のTFT間で移動度が安定化(飽和)していることが分かった。すなわち、前処理工程で、第2酸化物前駆体膜(活性層)に対してフーリエ変換型赤外分光で測定したときに得られる赤外線吸収スペクトルにおいて、赤外線の波数1380cm−1以上1520cm−1以下の範囲(II)に位置するピークが、UVオゾン処理の処理時間(照射時間)に伴って、赤外線の波数1350cm−1以上1750cm−1以下の範囲(I)における赤外線吸収スペクトルの中で最大値を示し始めるときに必要な光処理のエネルギー総量(実施例では1.5J/cm2)に対して、4倍(実施例では6.0J/cm2)以上のエネルギー総量を用いて光処理を行うことで、4倍未満のエネルギー総量を用いる場合に比べて、オンオフ比が高く、且つオンオフ比が安定化(飽和)し、移動度が急激に高くなる。
【0104】
また、図24に示すように、照射時間を20分以上30分以下とした実施例3〜4のTFTは、実施例1や実施例2及び実施例5や実施例6のTFTに比べて、移動度がより高くなることが分かった。すなわち、前処理工程で、第2酸化物前駆体膜(活性層)に対してフーリエ変換型赤外分光で測定したときに得られる赤外線吸収スペクトルにおいて、赤外線の波数1380cm−1以上1520cm−1以下の範囲(II)に位置するピークが、UVオゾン処理の処理時間(照射時間)に伴って、赤外線の波数1350cm−1以上1750cm−1以下の範囲(I)における赤外線吸収スペクトルの中で最大値を示し始めるときに必要な光処理のエネルギー総量(実施例では1.5J/cm2)に対して、4倍以上6倍(実施例では9.0J/cm2)以下のエネルギー総量を用いて光処理を行うことで、4倍未満又は6倍超のエネルギー総量を用いる場合に比べて、移動度がより高くなることが分かった。
また、6倍超のエネルギー総量を用いると移動度が安定化(飽和)するものの、第2酸化物前駆体膜(活性層)に対して長時間のUVオゾン処理が行われることになる。4倍以上6倍以下のエネルギー総量を用いてUVオゾン処理を行うことで、長時間のUVオゾン処理(UVの照射)を避け、溶液中の金属が酸化され活性層の電気伝導率が悪化することを抑制することができると考えられる。また、基板が上述した樹脂基板や樹脂の複合材料で構成された基板である場合に、UVオゾン処理によって基板が変形することを抑制できると考えられる。
【0105】
次に、良好な特性が得られた照射時間30分のUVオゾン処理を施した実施例4のTFTと、UVオゾン処理の有無での移動度の比較を検討した。
【0106】
図25は、実施例4のTFTと比較例1のTFTのVg−Id特性を示す図である。なお、図25では熱処理温度を425℃と600℃にしたときのデータのみ示している。
図26は、実施例4のTFTと比較例1のTFTの熱処理温度(400℃のときは除く)と移動度との関係をプロットしたグラフ図である。
【0107】
図25に示す結果から、UVオゾン処理が有るか否かで例えばオン電流が劇的に変化し、UVオゾン処理を施していない比較例1のTFTでは、特性向上が見込めず熱処理温度の低下とともにオン電流等のTFT特性が劣化していくことが確認された。
また、図26に示すように、UVオゾン処理を施した実施例4のTFTでは、比較例1のTFTに比べて一桁以上移動度が向上し、425℃〜600℃の熱処理にてその移動度がほぼ同水準の値を示した。
【0108】
−XPS評価−
次に、前処理としてのUVオゾン処理が熱処理後の物性に与える関係を調査するためにXPS測定を行った。このXPS測定には、島津製作所製AXIS−ULTRAを用いた。測定サンプルとして、上記IGZO溶液を30分UVオゾン処理した後、425℃,450℃又は500℃でそれぞれ熱処理したものを用いた。比較用の測定サンプルとして、上記IGZO溶液をそのまま425℃,450℃又は500℃でそれぞれ熱処理したものを用いた。同様に、比較用の測定サンプルとして、IGZO溶液と同じIGZO組成のスパッタ膜を用いた。
【0109】
図27は、各測定サンプルのXPSデータを示す図である。
【0110】
図27に示す結果から、30分のUVオゾン処理を施したサンプルは、酸素の結合状態がスパッタ膜に近づいていることが分かる。これはM−O(M:In,Ga,Znの何れか)の結合状態が強くなっていることを意味し、この強い結合状態が移動度向上の原因となっていると考えられる。液相プロセスにおいては、真空プロセスでは含まれないCやH等が多量に含まれているため、通常M−O結合の形成を阻害しやすい。特に真空プロセスではキャリア濃度を下げる働きをする、Ga、Al、Ta、Ti、Wなどはキャリアの発生源となる酸素の取り込みや放出を行うことが困難であるため、一度M−OHのような結合を形成してしまうと、その結合状態を再度変化させてM−O結合を形成させることが困難である。そのため、通常液相プロセスにおいては、Gaの添加量を極力少なくする傾向にあるが、本実施形態においては、UVオゾン処理等の前処理工程を行い、前駆体膜を改質することで、M−OHなどの結合を極力経由せずに、M−O結合が形成されるのを向上させることが可能となる。
【0111】
また、図27に示す結果から、30分のUVオゾン処理を施した各サンプル(非晶質酸化物薄膜)は、XPSによる酸素(O1s)の結合エネルギーにおいて最大スペクトル強度をえられる結合エネルギーの位置が、同じ金属組成にて真空プロセスにより作製したサンプル(非晶質酸化物薄膜)と同じ位置(530eV付近)に存在していることが分かった。
【0112】
<有機酸塩系の実施例>
−第1酸化物前駆体膜及び第2酸化物前駆体膜の作製及び検討−
以上の実施例では、金属アルコキシド系のIGZO溶液を用いて各種サンプルを作製したが、以下の実施例では有機酸塩系のIGZO溶液を用いて各種サンプルを作製した。
【0113】
各々0.2mol/kgで調液を行った酢酸亜鉛−メトキシエタノール溶液、ガリウムアセチルアセトナート-プロピオン酸溶液、インジウムアセチルアセトナート-プロピオン酸溶液を準備し、In:Ga:Zn=1:1:1となるように混合して、有機酸塩系原料液(IGZO溶液)を得た。
次に、石英基板上に1000rpmの速度で、1回スピンコートした後、室温で乾燥させることによりゲル状態の第1酸化物前駆体膜を成膜した。そして、得られた第1酸化物前駆体膜をXRDにて評価を行ったところ図10とほぼ同様の結果となった。
【0114】
次に、室温乾燥させたのみの複数の第1酸化物前駆体膜に対して、互いに1分、3分、5分、10分、30分、60分、90分と照射時間を変えて前処理工程としてのUVオゾン処理を行い、第2酸化物前駆体膜を得た。このUVオゾン処理では低圧水銀ランプ(波長185nm及び254nm)を用いており、254nmの強度が約5mW/cm2である。
【0115】
そして、UVオゾン処理を行った後の第2酸化物前駆体膜に対して、装置名:BRUKER ALPHAの装置でダイヤモンドATR配置を用いて、ATR配置を用いて、FT−IRスペクトル測定を実施した。比較として、UVオゾン処理を行っていない第1酸化物前駆体膜のままのFT−IRスペクトル測定も実施した。
【0116】
図28は、各第2酸化物前駆体膜に対してFT−IRスペクトル測定した測定結果を示す図である。図29は、図28に示す赤外線の波数1350cm−1以上1750cm−1以下の範囲における赤外線吸収スペクトルの拡大図である。
【0117】
図28に示すように、UV処理時間に伴い1350cm−1以上1750cm−1以下におけるピーク形状が選択的に変化していることが確認された。
また図29に示すように、UVオゾン処理の照射時間が5分未満では、赤外線の波数1500cm−1以上1600cm−1以下の範囲にあるピークが、赤外線の波数1350cm−1以上1750cm−1以下の範囲(I)における赤外線吸収スペクトルの中で最大値を示している。一方で、UVオゾン処理の照射時間が5分以上では、第2酸化物前駆体膜に対してフーリエ変換型赤外分光で測定したときに得られる赤外線吸収スペクトルにおいて、赤外線の波数1380cm−1以上1520cm−1以下の範囲(II)に位置する(ダブル)ピークが、赤外線の波数1350cm−1以上1750cm−1以下の範囲(I)における赤外線吸収スペクトルの中で最大値を示すようになることが分かった。
具体的に、UVオゾン処理の照射時間が5分以上では、第2酸化物前駆体膜に対してフーリエ変換型赤外分光で測定したときに得られる赤外線吸収スペクトルにおいて、赤外線の波数1380cm−1以上1520cm−1以下の範囲(II)を、赤外線の波数1380cm−1以上1450cm−1以下の範囲(IV)と赤外線の波数1450cm−1超1520cm−1以下の範囲(III)とに分割したときに、赤外線の波数1380cm−1以上1450cm-1以下の範囲(IV)に位置するピークが、赤外線の波数1350cm−1以上1750cm−1以下の範囲(I)における赤外線吸収スペクトルの中で最大値を示すようになることも分かった。
なお、赤外線の波数1380cm−1以上1450cm−1以下の範囲(IV)と、赤外線の波数1450cm−1超1520cm−1以下の範囲(III)とには、両範囲を跨ぐようにダブルピークが見られ、このダブルピークの一つが上記範囲(IV)に位置するピークである。
【0118】
−薄膜トランジスタの作製−
次に、UVオゾン処理の照射時間によって変化する第2酸化物前駆体膜の状態が熱処理後の薄膜へデバイス特性としてどのような影響を及ぼすか評価を行うために、ボトムゲートートップコンタクト型の薄膜トランジスタを作製した。
【0119】
図30は、本発明の実施例に係る薄膜トランジスタの製造方法の工程図を示す図である。
【0120】
本発明の実施例に係る薄膜トランジスタの基板には、P+Si/Si熱酸化膜(100nm)付基板を用い、ゲート電極としてP+Siを、ゲート絶縁膜としてSi熱酸化膜を用いた。
次に、図30(A)〜(C)に示す過程において、上記の第1酸化物前駆体膜から第2酸化物前駆体膜を得る方法と同一の方法を用いて活性層を作製した。具体的に、図30(A)に示すように、IGZO溶液(上記金属アルコキシド系のIGZO溶液と同じ溶液で、In:Ga:Zn=1.0:1.0:0.9)を用い、スピンコート法により最終生成物の膜厚が50nm程度になるように活性層の成膜を行った(熱処理により有機成分が減少して膜厚が変化するため)。成膜後に、図30(B)に示すように前処理としてUVオゾン処理を5分間実施し、図30(C)に示すように後処理として375℃〜600℃の間で15分間熱処理を実施した。その後、図30(D)に示すようにフォトリソグラフィーを用いてリフトオフパターンを形成した。レジストにはAZ5214Eを用いた。その後電極Moを100nm成膜し、レジスト剥離を行って図30(E)に示すようにSD電極パターンを形成した。そして、活性層のパターニングとしてレジストにはTSMR8900−LBを用い、Arプラズマによるドライエッチングを用いてパターニングをした。レジスト剥離には、酸素アッシングを用いて図30(F)に示すような薄膜トランジスタを作製した。
【0121】
次に、得られた薄膜トランジスタに対して、半導体パラメータ・アナライザー4156
C(アジレントテクノロジー社製)を用い、トランジスタ特性(Vg−Id特性)及び移動度μの測定を行った。Vg−Id特性の測定は、ドレイン電圧(Vd)を5Vに固定し、ゲート電圧(Vg)を−20V〜+40Vの範囲内で変化させ、各ゲート電圧(Vg)におけるドレイン電流(Id)を測定することにより行った。
【0122】
−トランジスタ特性及び移動度の評価−
次に、得られた薄膜トランジスタに対して、半導体パラメータ・アナライザー4156C(アジレントテクノロジー社製)を用い、トランジスタ特性(Vg−Id特性)及び移動度μの測定を行った。Vg−Id特性の測定は、ドレイン電圧(Vd)を5Vに固定し、ゲート電圧(Vg)を−20V〜+40Vの範囲内で変化させ、各ゲート電圧(Vg)におけるドレイン電流(Id)を測定することにより行った。なお、比較のために、上記薄膜トランジスタにおいて図30(B)に示すUVオゾン処理を省略したものに対してもトランジスタ特性(Vg−Id特性)及び移動度μの測定を行った。
【0123】
図31は、実施例により得られた薄膜トランジスタのVg−Id特性を示す図である。また、図32は、UVオゾン処理を省略した薄膜トランジスタのVg−Id特性を示す図である。さらに、図33は、熱処理温度と薄膜トランジスタの移動度との関係をプロットしたグラフ図である。
【0124】
図31と図32を比較すると、図25に示す結果と同様に、UVオゾン処理の有無で例えばオン電流が劇的に変化していることが分かった。また、図33に示す移動度の結果と図26に示す移動度の結果を比較すると、図33に示す移動度の方が約1桁高いことが分かった。これは、溶液が異なるからではなく、電極形成前に活性層をパターニングするか電極形成後に活性層をパターニングするかの違いによるものだと考えられ、電極形成後に活性層をパターニングした方が活性層にダメージを与え難く、この結果移動度が高くなったと想定される。
【0125】
−ピークの特定−
次に、図11及び図12並びに図28及び図29に示す、赤外線の波数1380cm−1以上1520cm−1以下の範囲(II)に位置するピークでUVオゾン処理の処理時間が5分以上に見られるダブルピークの由来(起因)について検討した。
【0126】
まず、各々0.2mol/kgで調液を行った亜鉛アセチルアセトナート−メタノール溶液、ガリウムアセチルアセトナート-プロピオン酸溶液、インジウムアセチルアセトナート-プロピオン酸溶液を準備した。そして、各々の溶液を石英基板上に1000rpmの速度で1回スピンコートした後室温で乾燥させて乾燥ゲルを得た。各乾燥ゲルに対してUVオゾン処理を30分実施したものと、UVオゾン処理前の乾燥ゲルとのFT−IRスペクトルを比較した。
【0127】
図34は、各乾燥ゲルのFT−IRスペクトルを示す図である。
【0128】
図34に示すように、Inを含んだ乾燥ゲルのUVオゾン処理後のFT−IRスペクトルは、図11及び図12並びに図28及び図29に現れていたダブルピークと一致した。したがって、図11及び図12並びに図28及び図29に現れていた1380cm−1以上1520cm−1のダブルピークは、In前駆体原料に由来していることが分かった。
【符号の説明】
【0129】
4 第1酸化物前駆体膜
6 第2酸化物前駆体膜
8 非晶質酸化物薄膜
12 基板
14 活性層
16 ソース電極(導電層)
18 ドレイン電極(導電層)
20 ゲート絶縁膜(絶縁層)
22 ゲート電極(導電層)
10、30、40、50、204、206 薄膜トランジスタ
【技術分野】
【0001】
本発明は、非晶質酸化物薄膜の製造方法及び薄膜トランジスタに関する。
【背景技術】
【0002】
非晶質酸化物薄膜、特にIn−Ga−Zn−O系(以下、「IGZO」と呼称する)を始めとした透明非晶質酸化物半導体は、「TAOS(Transparent Amorphous Oxide Semiconductor)」と称され、様々な研究機関やメーカーが研究開発を行い、技術躍進を遂げている。特に、日本や韓国、台湾のパネルメーカーが中心となって大型のディスプレイの試作が活発に行われており、信頼性や安定性の議論、更には量産化に向けた技術検討も進み、真空成膜法による製造においてはかなり実用化に近いレベルにまで達してきている。
【0003】
一方で、大面積化のトレンドの中で製造コストを下げる狙いとして液相プロセス(以下、「液相法」と呼称する)によるTAOS−TFT(Thin Film Transistor:薄膜トランジスタ)作製の研究開発も盛んであり、例えば、近年では液相法で作製したTAOS−TFT駆動の液晶ディスプレイが報告される等、液相法によるTFT作製に対する世の中の期待は益々高まっている。しかしながら、液相法においては、真空成膜法と比較してプロセス温度がまだまだ高いこと、良質な薄膜を得ることが困難であるということが、今後乗り越えなければならない課題として挙げられる。
【0004】
そこで、特許文献1には、基板上に形成した半導体前駆体膜を加熱処理する共に電磁波を照射して、半導体前駆体膜を非晶質の金属酸化物半導体膜に変換する非晶質酸化物薄膜の製造方法が開示されている。また、この文献には、半導体前駆体膜の形成後半導体変換処理前(加熱処理前)に、酸素プラズマやUVオゾン洗浄などのドライ洗浄プロセスによって洗浄し、薄膜中及び薄膜表面に存在し不純物の原因となる有機物を分解及び洗浄して、金属成分以外の有機成分を排除することが好ましいということも開示されている。
【先行技術文献】
【特許文献】
【0005】
【特許文献1】特開2010−258058号公報
【発明の概要】
【発明が解決しようとする課題】
【0006】
しかしながら、特許文献1の製造方法では、半導体変換処理前に、半導体前駆体膜中の金属成分以外の有機成分を全て排除することになる。本発明者らは鋭意研究を重ねた結果、半導体変換処理前、すなわち加熱処理前の有機成分の結合状態がTFT特性のオン電流を向上する等非晶質酸化物薄膜の膜質の向上に繋がることを見出した。
【0007】
本発明は上記事実に鑑みてなされたものであり、膜質を向上する非晶質酸化物薄膜の製造方法及び薄膜トランジスタを提供することを目的とする。
【課題を解決するための手段】
【0008】
本発明の上記課題は下記の手段によって解決された。
<1>有機成分とInとを含有する第1酸化物前駆体膜に対して、前記有機成分の熱分解温度未満で前記有機成分の結合状態を選択的に変化させ、フーリエ変換型赤外分光で測定したときに得られる赤外線吸収スペクトルにおいて、赤外線の波数1380cm−1以上1520cm−1以下の範囲を、赤外線の波数1380cm−1以上1450cm−1以下の範囲と赤外線の波数1450cm−1超1520cm−1以下の範囲とに分割したときに、赤外線の波数1380cm−1以上1450cm-1以下の範囲に位置するピークが、赤外線の波数1350cm−1以上1750cm−1以下の範囲における赤外線吸収スペクトルの中で最大値を示す第2酸化物前駆体膜を得る前処理工程と、前記第2酸化物前駆体膜中に残存する前記有機成分を除去して、前記第2酸化物前駆体膜を、前記Inを含有する非晶質酸化物薄膜へ変化させる後処理工程と、を有する非晶質酸化物薄膜の製造方法。
<2>前記前処理工程における前記有機成分の結合状態の選択的な変化は、前記第2酸化物前駆体膜に対してフーリエ変換型赤外分光で測定したときに得られる赤外線吸収スペクトルにおいて、赤外線の波数1500cm−1以上1600cm−1以下の範囲にある前記有機成分由来のピークの減少に相当する変化を含む、<1>に記載の非晶質酸化物薄膜の製造方法。
<3>前記前処理工程における前記有機成分の結合状態の選択的な変化は、前記第2酸化物前駆体膜に対してフーリエ変換型赤外分光で測定したときに得られる赤外線吸収スペクトルにおいて、赤外線の波数2750cm−1以上3050cm−1以下の範囲にある前記有機成分由来のピークの減少に相当する変化を含む、<1>又は<2>に記載の非晶質酸化物薄膜の製造方法。
<4>前記第2酸化物前駆体膜に対してフーリエ変換型赤外分光で測定したときに得られる赤外線吸収スペクトルにおいて、赤外線の波数1500cm−1以上1600cm−1以下の範囲にあるピークの減少率が、前記前処理工程前の1500cm−1以上1600cm−1以下の範囲にあるピークを1としたとき0.37以下である、<2>に記載の非晶質酸化物薄膜の製造方法。
<5>前記第2酸化物前駆体膜に対してフーリエ変換型赤外分光で測定したときに得られる赤外線吸収スペクトルにおいて、赤外線の波数1500cm−1以上1600cm−1以下の範囲にあるピークの減少率が、前記前処理工程前の1500cm−1以上1600cm−1以下の範囲にあるピークを1としたとき0.11以下である、<4>に記載の非晶質酸化物薄膜の製造方法。
<6>前記前処理工程は、光処理工程である、<1>〜<5>の何れか1つに記載の非晶質酸化物薄膜の製造方法。
<7>前記光処理工程では、前記第1酸化物前駆体膜に対して紫外線を照射する、<6>に記載の非晶質酸化物薄膜の製造方法。
<8>前記前処理工程では、前記第2酸化物前駆体膜に対してフーリエ変換型赤外分光で測定したときに得られる赤外線吸収スペクトルにおいて、赤外線の波数1380cm−1以上1520cm−1以下の範囲に位置するピークが、光処理の時間に伴って、赤外線の波数1350cm−1以上1750cm−1以下の範囲における赤外線吸収スペクトルの中で最大値を示し始めるときに必要な光処理のエネルギー総量に対して、4倍以上のエネルギー総量を用いて光処理を行う、<6>又は<7>に記載の非晶質酸化物薄膜の製造方法。
<9>前記前処理工程では、前記第2酸化物前駆体膜に対してフーリエ変換型赤外分光で測定したときに得られる赤外線吸収スペクトルにおいて、赤外線の波数1380cm−1以上1520cm−1以下の範囲に位置するピークが、光処理の時間に伴って、赤外線の波数1350cm−1以上1750cm−1以下の範囲における赤外線吸収スペクトルの中で最大値を示し始めるときに必要な光処理のエネルギー総量に対して、6倍以下のエネルギー総量を用いて光処理を行う、<8>に記載の非晶質酸化物薄膜の製造方法。
<10>前記後処理工程は、425℃以上の温度で熱処理する熱処理工程である、<1>〜<9>の何れか1つに記載の非晶質酸化物薄膜の製造方法。
<11>前記第1酸化物前駆体膜は、前記Inの他、Ga、Zn及びSnから選ばれる少なくとも一つを含む無機成分を含有する、<1>〜<10>の何れか1つに記載の非晶質酸化物薄膜の製造方法。
<12>前記無機成分の組成が、結晶化温度以上の前記熱処理工程でInGaZnO4-δ(前記δは酸素不定比量である)が結晶相として単相で現れるような組成である、<11>に記載の非晶質酸化物薄膜の製造方法。
<13>前記前処理工程の前に、金属アルコキシド又は有機酸塩を含有する溶液を用いて液相法により前記第1酸化物前駆体膜を形成する形成工程、をさらに有する<1>〜<12>の何れか1つに記載の非晶質酸化物薄膜の製造方法。
<14><1>〜<13>の何れか1つに記載の非晶質酸化物薄膜の製造方法で作製された非晶質酸化物薄膜を活性層として用いた薄膜トランジスタ。
<15><1>〜<13>の何れか1つに記載の非晶質酸化物薄膜の製造方法で作製された非晶質酸化物薄膜を絶縁層として用いた薄膜トランジスタ。
<16><1>〜<13>の何れか1つに記載の非晶質酸化物薄膜の製造方法で作製された非晶質酸化物薄膜を導電層として用いた薄膜トランジスタ。
【発明の効果】
【0009】
本発明によれば、膜質を向上する非晶質酸化物薄膜の製造方法及び薄膜トランジスタを提供することができる。
【図面の簡単な説明】
【0010】
【図1】図1は、本発明の実施形態に係る非晶質酸化物薄膜の製造方法の工程図である。
【図2】図2(A)は、本発明の実施形態に係るTFTであって、トップゲート構造でトップコンタクト型のTFTの一例を示す模式図である。図2(B)は、本発明の実施形態に係るTFTであって、トップゲート構造でボトムコンタクト型のTFTの一例を示す模式図である。図2(C)は、本発明の実施形態に係るTFTであって、ボトムゲート構造でトップコンタクト型のTFTの一例を示す模式図である。図2(D)は、本発明の実施形態に係るTFTであって、ボトムゲート構造でボトムコンタクト型のTFTの一例を示す模式図である。
【図3】図3は、本発明の電気光学装置の一実施形態の液晶表示装置について、その一部分の概略断面図である。
【図4】図4は、図3に示す液晶表示装置の電気配線の概略構成図である。
【図5】図5は、本発明の電気光学装置の一実施形態のアクティブマトリックス方式の有機EL表示装置について、その一部分の概略断面図である。
【図6】図6は、図5に示す電気光学装置の電気配線の概略構成図である。
【図7】図7は、本発明のセンサの一実施形態であるX線センサについて、その一部分の概略断面図である。
【図8】図8は、図7に示すセンサの電気配線の概略構成図である。
【図9】図9は、実施例の乾燥ゲルに対するTG−DTAの測定結果を示す図である。
【図10】図10は、実施例の第1酸化物前駆体膜に対するXRDの測定結果である。
【図11】図11は、各第2酸化物前駆体膜に対してFT−IR(フーリエ変換型赤外分光)スペクトル測定した測定結果を示す図である。
【図12】図12は、図11に示す赤外線の波数1350cm−1以上1750cm−1以下の範囲における赤外線吸収スペクトルの拡大図である。
【図13】図13は、複数の第1酸化物前駆体膜を各種温度で熱処理した後の各膜に対してFT−IR(フーリエ変換型赤外分光)スペクトル測定した測定結果を示す図である。
【図14】UVオゾン処理後に熱処理を施したサンプルのXRD測定結果を示す図である。
【図15】UVオゾン処理を施していないサンプルのXRD測定結果を示す図である。
【図16】図16は、本発明の実施例に係る薄膜トランジスタの製造方法の工程図を示す図である。
【図17】図17は、熱処理温度を400℃に固定して、UV照射時間を0、1、3、5、10、20、30、60、90分と変化させたときの薄膜トランジスタのVg−Id特性を示す図である。
【図18】図18は、熱処理温度を425℃に固定して、UV照射時間を0、1、3、5、10、20、30、60、90分と変化させたときの薄膜トランジスタのVg−Id特性を示す図である。
【図19】図19は、熱処理温度を450℃に固定して、UV照射時間を0、1、3、5、10、20、30、60、90分と変化させたときの薄膜トランジスタのVg−Id特性を示す図である。
【図20】図20は、熱処理温度を500℃に固定して、UV照射時間を0、1、3、5、10、20、30、60、90分と変化させたときの薄膜トランジスタのVg−Id特性を示す図である。
【図21】図21は、熱処理温度を600℃に固定して、UV照射時間を0、1、3、5、10、20、30、60、90分と変化させたときの薄膜トランジスタのVg−Id特性を示す図である。
【図22】図22は、図17〜図21に示すVg−Id特性から求めたオンオフ比とUV照射時間との関係を示す図である。
【図23】図23は、図22の部分拡大図である。
【図24】図24は、図17〜図21に示すVg−Id特性から求めた移動度とUV照射時間との関係を示す図である。
【図25】図25は、実施例4のTFTと比較例1のTFTのVg−Id特性を示す図である。
【図26】図26は、実施例4のTFTと比較例1のTFTの熱処理温度(400℃のときは除く)と移動度との関係をプロットしたグラフ図である。
【図27】図27は、各測定サンプルのXPSデータを示す図である。
【図28】図28は、各第2酸化物前駆体膜に対してFT−IRスペクトル測定した測定結果を示す図である。
【図29】図29は、図28に示す赤外線の波数1350cm−1以上1750cm−1以下の範囲における赤外線吸収スペクトルの拡大図である。
【図30】図30は、本発明の実施例に係る薄膜トランジスタの製造方法の工程図を示す図である。
【図31】図31は、実施例により得られた薄膜トランジスタのVg−Id特性を示す図である。
【図32】図32は、UVオゾン処理を省略した薄膜トランジスタのVg−Id特性を示す図である。
【図33】図33は、熱処理温度と薄膜トランジスタの移動度との関係をプロットしたグラフ図である。
【図34】図34は、各乾燥ゲルのFT−IRスペクトルを示す図である。
【発明を実施するための形態】
【0011】
以下、添付の図面を参照しながら、本発明の実施形態に係る非晶質酸化物薄膜の製造方法及び薄膜トランジスタについて具体的に説明する。なお、図中、同一又は対応する機能を有する部材(構成要素)には同じ符号を付して適宜説明を省略する。
【0012】
1.非晶質酸化物薄膜の製造方法
本発明の実施形態に係る非晶質酸化物薄膜の製造方法は、有機成分とInとを含有する第1酸化物前駆体膜に対して、前記有機成分の熱分解温度未満で前記有機成分の結合状態を選択的に変化させ、フーリエ変換型赤外分光で測定したときに得られる赤外線吸収スペクトルにおいて、赤外線の波数1380cm−1以上1520cm−1以下の範囲に位置するピークが、赤外線の波数1350cm−1以上1750cm−1以下の範囲における赤外線吸収スペクトルの中で最大値を示す第2酸化物前駆体膜を得る前処理工程と、前記第2酸化物前駆体膜中に残存する前記有機成分を除去して、前記第2酸化物前駆体膜を、前記Inを含有する非晶質酸化物薄膜へ変化させる後処理工程と、を有する。
なお、上記「熱分解温度」は、第1酸化物前駆体膜に対してTG−DTAの測定を実施したときに、発熱反応に伴う重量減少が終了する温度であり、例えば425℃である。また、上記「有機成分を除去」とは、有機成分全部を除去するだけでなく、有機成分の一部を除去してもよい。
そして、このような製造方法を用いることで、得られる非晶質酸化物薄膜の膜質を向上することができる。
【0013】
以下、本発明の実施形態に係る非晶質酸化物薄膜の具体的な製造方法を説明する。図1は、本発明の実施形態に係る非晶質酸化物薄膜の製造方法の工程図である。
【0014】
<基板用意工程>
まず、図1(A)に示すように、非晶質酸化物薄膜を成膜するための基板2を用意する。基板2の形状、構造、大きさ等については特に制限はなく、目的に応じて適宜選択することが出来る。基板2の構造は単層構造であってもよいし、積層構造であってもよい。
基板2の材質としては特に限定はなく、例えばガラス、YSZ(イットリウム安定化ジルコニウム)等の無機基板、樹脂基板や、その複合材料等を用いることが出来る。中でも軽量である点、可撓性を有する点から樹脂基板やその複合材料が好ましい。具体的には、ポリブチレンテレフタレート、ポリエチレンテレフタレート、ポリエチレンナフタレート、ポリブチレンナフタレート、ポリスチレン、ポリカーボネート、ポリスルホン、ポリエーテルスルホン、ポリアリレート、アリルジグリコールカーボネート、ポリアミド、ポリイミド、ポリアミドイミド、ポリエーテルイミド、ポリベンズアゾール、ポリフェニレンサルファイド、ポリシクロオレフィン、ノルボルネン樹脂、ポリクロロトリフルオロエチレン等のフッ素樹脂、液晶ポリマー、アクリル樹脂、エポキシ樹脂、シリコーン樹脂、アイオノマー樹脂、シアネート樹脂、架橋フマル酸ジエステル、環状ポリオレフィン、芳香族エーテル、マレイミドーオレフィン、セルロース、エピスルフィド化合物等の合成樹脂基板、酸化珪素粒子との複合プラスチック材料、金属ナノ粒子、無機酸化物ナノ粒子、無機窒化物ナノ粒子等との複合プラスチック材料、カーボン繊維、カーボンナノチューブとの複合プラスチック材料、ガラスフェレーク、ガラスファイバー、ガラスビーズとの複合プラスチック材料、粘土鉱物や雲母派生結晶構造を有する粒子との複合プラスチック材料、薄いガラスと上記単独有機材料との間に少なくとも1回の接合界面を有する積層プラスチック材料、無機層と有機層を交互に積層することで、少なくとも1回以上の接合界面を有するバリア性能を有する複合材料、ステンレス基板或いはステンレスと異種金属を積層した金属多層基板、アルミニウム基板或いは表面に酸化処理(例えば陽極酸化処理)を施すことで表面の絶縁性を向上させた酸化皮膜付きのアルミニウム基板等を用いることが出来る。また、樹脂基板は、耐熱性、寸法安定性、耐溶剤性、電気絶縁性、加工性、低通気性、又は低吸湿性等に優れていることが好ましい。前記樹脂基板は、水分や酸素の透過を防止するためのガスバリア層や、樹脂基板の平坦性や下部電極との密着性を向上するためのアンダーコート層等を備えていてもよい。
【0015】
また、本発明における基板2の厚みに特に制限はないが、50μm以上1000μm以下が好ましく、50μm以上500μm以下であることがより好ましい。 基板2の厚みが50μm以上であると、基板2自体の平坦性がより向上する。また、基板2の厚みが500μm以下であると、基板2自体の可撓性がより向上し、フレキシブルデバイス用基板としての使用がより容易となる。
【0016】
<形成工程>
次に、図1(B)に示すように、例えばスピンコート、インクジェット、ディスペンサー、スクリーン印刷、凸版印刷又は凹版印刷等の液相法を用いて、基板2上に有機成分とInを含有する第1酸化物前駆体膜4、すなわち本実施形態に係る非晶質酸化物薄膜の前駆体膜を形成する。
この第1酸化物前駆体膜4は、Inの他、Ga、Zn及びSnから選ばれる少なくとも一つを含む無機成分を含有していることが好ましく、In、Ga及びZnを含む無機成分を含有していることがより好ましい。第1酸化物前駆体膜4中の無機成分が、In、Ga及びZnを含んでいる場合、その無機成分の組成は、後述する後処理工程で仮に結晶化温度以上の温度で熱処理を行ったときに、InGaZnO4-δ(前記δは酸素不定比量である)が結晶相として単相で現れるような組成であることが好ましい。この組成は、例えばIn:Ga:Zn=2−x:x:1(xは0.8以上1.05以下)のような組成である。上記のような組成で従来の製造方法により得られる非晶質酸化物薄膜は、薄膜トランジスタの活性層として用いた場合に、移動度等のTFT特性が低く、実用化し難い状態にあったものが、本実施形態の製造方法を経ることで、上記のような組成でも実用化に耐えうるTFT特性に向上することができる。また、上記組成の範囲外では、TFT特性は従来の製造方法でも良好ではあるが、本実施形態の製造方法を経ることで、TFT特性をより向上することができる。
液相法に用いる溶液は、様々な溶液を用いることができるが、金属アルコキシド又は有機酸塩を含有する溶液を用いることが好ましい。特に、硝酸塩や塩化物とは異なり不要な不純物成分(硝酸や塩素など)を除去するプロセスが不要という観点と有毒なガスの発生を抑制するという観点から金属アルコキシドを含有する溶液を用いることが好ましい。なお、基板2と前駆体膜4の間には、後述するような電極や絶縁膜等他の薄膜が配置されていてもよい。
有機酸塩としては、β − ジケトン錯体基、β−ケトカルボン酸エステル錯体基、β−ケトカルボン酸錯体基及びケトオキシ基(ケトオキシ錯体基)が挙げられる。β−ジケトン錯体基としては、例えば2 ,4−ペンタンジオン(アセチルアセトンあるいはアセトアセトンともいう)、1,1,1,5,5,5−ヘキサメチル−2,4−ペンタンジオン、2,2,6,6−テトラメチル−3,5−ヘプタンジオン、1,1,1−トリフルオロ−2 ,4−ペンタンジオン等を挙げることができる。β−ケトカルボン酸エステル錯体基としては、例えばアセト酢酸メチルエステル、アセト酢酸エチルエステル、アセト酢酸プロピルエステル、トリメチルアセト酢酸エチル、トリフルオロアセト酢酸メチル等を挙げることができる。β−ケトカルボン酸としては、例えば、アセト酢酸、トリメチルアセト酢酸等を挙げることができる。またケトオキシとしては、例えばアセトオキシ基(またはアセトキシ基)、プロピオニルオキシ基、ブチリロキシ基、アクリロイルオキシ基、メタクリロイルオキシ基等を挙げることができる。これらの基の炭素原子数は18以下が好ましい。また直鎖または分岐のもの、また水素原子をフッ素原子にしたものでもよい。
一方で、金属アルコキシド溶液としては、少なくとも下記一般式Iで表される金属アルコキシド化合物を含有するものである。
M(OR)n ・・・式[I]
ここで、式[I]中のMは金属元素で、nは1以上の整数である。
【0017】
特に、金属アルコキシド溶液として、下記一般式II及び一般式IIIで表される金属アルコキシド化合物を含有し、1〜100mPa・sの粘度を有するものであることが好ましい。
Zn(OR1)2 ・・・[II]
M(OR2)3 ・・・[III]
このような金属アルコキシド溶液であれば、溶液自体を加熱することにより非晶質状態でも良好な半導体特性を有する金属酸化物ナノ粒子を得ることができるので、簡便に半導体薄膜を形成することができるからである。
【0018】
本実施形態では、溶液中に少なくとも上記一般式II中でMがInである金属アルコキシド化合物が存在している。また、電気特性の観点から、MがGaである金属アルコキシド化合物とMがInである金属アルコキシド化合物とが溶液中に存在していることが好ましい。また、材料コストの観点から、MがAlである金属アルコキシド化合物とMがInである金属アルコキシド化合物とが溶液中に存在していることが好ましい。
【0019】
上記一般式I〜IIIで表される金属アルコキシド化合物は、金属アルコキシド溶液中で、それぞれ単独で存在していてもよく、その一部が連結して複合アルコキシドを形成していてもよい。上記一般式II及び一般式IIIの金属アルコキシド化合物のとき、金属アルコキシド溶液中の各化合物及び/又は複合アルコキシドにおいて、比率Zn/Mは、0.2〜10の範囲であることが好ましい。0.2未満では原料コストが高くなり、一方、10を超えるとZn成分が過剰となって酸化亜鉛の結晶が生じて複合酸化物ナノ粒子を形成し難い。Zn/Mの範囲は、電気特性の観点から0.2〜1.5の範囲であることが好ましく、0.5〜1.3の範囲であることが更に好ましい。
【0020】
上記一般式I〜III中、R、R1及びR2はそれぞれ同一でも異なっていてもよく、炭素数が1〜20、好ましくは1〜6のアルキル基を表し、置換されていても無置換であってもよい。炭素数が20を超えると、金属分子間が長くなって薄膜形成時に金属酸化物中にアルキル基が残存する場合があるため好ましくない。置換基としては、炭素数1〜4のアルキル基、アミノ基(アミノ基の水素原子は炭素数1〜4のアルキル基で置換されていてもよい)、炭素数1〜4のアルコキシ基、水酸基などを挙げることができる。
【0021】
このような一般式I〜IIIに相当する化合物の例としては、亜鉛エトキシド、亜鉛エトキシエトキシド、亜鉛ジメチルアミノエトキシド、亜鉛メトキシエトキシド、インジウムイソプロポキシド、インジウム−n−ブトキシド、インジウムメトキシエトキシド、インジウムジエチルアミノエトキシド、ガリウムエトキシド、ガリウムイソプロポキシド、アルミニウムイソプロポキシド、アルミニウム−n−ブトキシド、アルミニウム−s−ブトキシド、アルミニウム−t−ブトキシド、アルミニウムエトキシド、アルミニウムエトキシエトキシエトキシド、鉄イソプロポキシドなどを挙げることができる。
【0022】
また、金属アルコキシド溶液では、一般式II及びIIIの金属アルコキシド化合物を、ZnとMの合計濃度として好ましくは0.5〜20質量%、より好ましくは1〜10質量%で含有する。0.5質量%未満では、均一な薄膜ができない場合があり、20質量%を超えると充分に薄い薄膜を構成することができない場合がある。
【0023】
また、ナノ粒子分散液の粘度は、塗布手段により異なるが、例えばインクジェットやディスペンサーの場合、吐出性の観点から1〜100mPa・s、好ましくは1〜20mPa・sであることが望ましい。ナノ粒子分散液の上記粘度は、市販の粘度計、例えば振動式粘度計VISCOMATE(CBCマテリアルズ株式会社製)で測定することができる。
【0024】
金属アルコキシド溶液は、上記金属アルコキシド化合物を溶解するための適当な溶媒を含む。この溶媒としては、水、アルコール類、アミノアルコール類、グリコール類などを挙げることができ、分散液の安定性、乾燥性の観点から下記一般式IVで表される高沸点溶媒を少なくとも1種含むものであることが更に好ましい。
R3−OH ・・・[IV]
【0025】
ここで、R3は炭素原子数2〜12の置換又は未置換のアルキル基、アルケニル基、シクロアルキル基、又はアリール基を表す。
アルキル基としては、メチル基、エチル基、プロピル基、イソプロピル基、ブチル基、ペンチル基、ヘキシル基、オクチル基などが挙げられる。アルケニル基としては、ブテニル基、プロペニル基などが、シクロアルキル基としては、シクロヘキシル基などが、アリール基としては、フェニル基などがそれぞれ挙げられる。
アルキル基やアルケニル基への好ましい置換基の例としては、アルコキシ基(メトキシ基、エトキシ基、イソプロポキシ基、メトキシエトキシ基、フェノキシ基など)、水酸基、アミノ基などが挙げられる。シクロアルキル基やアリール基への好ましい置換基の例としては、アルキル基(メチル基、エチル基、ヒドロキシエチル基など)、アルコキシ基(メトキシ基、エトキシ基、イソプロポキシ基、メトキシエトキシ基、フェノキシ基など)、水酸基、アミノ基などが挙げられる。
特に好ましい置換基としては、ジアルキルアミノ基(ジメチルアミノ基、ジエチルアミノ基、ジ−n−プロピルアミノ基、ジイソプロピルアミノ基、ジ−n−ブチルアミノ基、ジ-t-ブチルアミノ基など)が挙げられる。
【0026】
また一般式IVで表される上記高沸点溶媒の沸点は、120℃〜250℃であり、乾燥時の負荷軽減の観点から好ましくは130℃〜200℃である。沸点が120℃未満では乾燥速度が速く十分な平滑性を得にくいため好ましくなく、250℃を超えると薄膜を形成する際に残存しやすくなるため、好ましくない。
【0027】
一般式IVに相当する高沸点溶媒としては、例えば、2−エトキシエタノール、2−(メトキシエトキシ)エタノール、2−(エトキシエトキシ)エタノール、2−イソプロポキシエタノール、1−エトキシ−2−プロパノール、2−ジエチルアミノエタノール、2-ジプロピルアミノエタノール、シクロヘキサノール、エチレングリコール、ジエチレングリコール、ベンジルアルコールなどを挙げることができる。
これらの分散媒と併用できる溶媒としては、例えばジオキサン、ジメチルホルムアミド、ジメチルスルホキシド、アセチルアセトンなどが挙げられる。
また、金属アルコキシド溶液は、帯電防止剤、可塑剤、高分子バインダー、増粘剤等の各種添加剤を目的に応じて添加し、物性調整した後に塗布用の溶液として用いてもよい。
【0028】
<前処理工程>
次に、図1(C)に示すように、上述した形成工程により得られた有機成分とInとを含有する第1酸化物前駆体膜4に対して、前記有機成分の熱分解温度未満で前記有機成分の結合状態を選択的に変化させ、フーリエ変換型赤外分光で測定したときに得られる赤外線吸収スペクトルにおいて、赤外線の波数1380cm−1以上1520cm−1以下の範囲を、赤外線の波数1380cm−1以上1450cm−1以下の範囲と赤外線の波数1450cm−1超1520cm−1以下の範囲とに分割したときに、赤外線の波数1380cm−1以上1450cm-1以下の範囲に位置するピークが、赤外線の波数1350cm−1以上1750cm−1以下の範囲における赤外線吸収スペクトルの中で最大値を示す第2酸化物前駆体膜6を得る前処理工程を行う。
なお、有機成分の熱分解温度未満としているのは、有機成分が熱分解してしまうと、前処理工程後には有機物が残存しておらず、そのまま後述する後処理工程を行っても膜質の向上が図り難いからである。また、赤外線の波数1380cm−1以上1520cm−1以下の範囲に位置するピークは、In前駆体原料に由来するものである。
このような前処理工程を行うと、後述する後処理工程後に得られる非晶質酸化物薄膜について、移動度が高く膜質の向上に繋がる。また、この非晶質酸化物薄膜をTFTの活性層として用いた場合に、オンオフ比が高く膜質の向上に繋がる。具体的には、赤外線の波数1380cm−1以上1450cm-1以下の範囲に位置するピークが、赤外線の波数1350cm−1以上1750cm−1以下の範囲における赤外線吸収スペクトルの中で最大値を示し始めるときから、移動度が約1桁以上に高くなり、且つ、オンオフ比が急激に高くなる。
【0029】
前処理工程としては、上述のような機能を果たせば手段は問わないが、例えば光処理工程又はプラズマ処理工程から選ばれる少なくとも一つが挙げられ、特に処理コストが抑えられるという観点から光処理工程が好ましい。また、この光処理工程では、第1酸化物前駆体膜4に対して紫外線を照射することが好ましい。紫外線を照射することが好ましい理由としては、紫外線のような短波長の光が有機成分の結合状態を変化させ易いからである。例えば低圧水銀ランプを用いた場合、主成分として185nm(645KJ/mol)と254nm(472KJ/mol)の紫外線が存在し、C-H(413.4KJ/mol)やC=C(607KJ/mol)やC-O(351.5KJ/mol)などの様々な有機成分の結合を分解することができる。そのため、紫外線の波長は100nm以上450nm以下であることが好ましく、150nm以上300nm以下であることがより好ましい。光源は、低圧水銀灯、重水素ランプ、キセノンエキシマランプ、メタルハライドランプ、エキシマーレーザーなどを用いることができる。
また、光処理のみでも有機成分の結合状態を変化させ得るが、最終的に得たい薄膜が酸化物であるため、光処理工程の他、オゾンや酸素ラジカルに代表される活性酸素や酸素プラズマ等と併用することでより膜質の向上を図ることができる。オゾンの場合は、酸素存在下にて紫外線を照射することで発生するが、オゾナイザー等で積極的に導入しても構わない。
なお、本実施形態においてフーリエ変換型赤外分光で用いる測定装置は、装置名がBRUKER ALPHAであり、測定配置がダイヤモンドATR配置である。
【0030】
また前処理工程では、第2酸化物前駆体膜6に対してフーリエ変換型赤外分光で測定したときに得られる赤外線吸収スペクトルにおいて、赤外線の波数1380cm−1以上1520cm−1以下の範囲に位置するピークが、光処理の時間に伴って、赤外線の波数1350cm−1以上1750cm−1以下の範囲における赤外線吸収スペクトルの中で最大値を示し始めるときに必要な光処理のエネルギー総量に対して、4倍以上のエネルギー総量を用いて光処理を行うことが好ましい。
このように4倍以上のエネルギー総量を用いて光処理を行うことで、後述する後処理工程後に得られる非晶質酸化物薄膜をTFTの活性層として用いた場合に、4倍未満のエネルギー総量を用いる場合に比べて、オンオフ比が高く、且つ、安定化(飽和)する。また、4倍未満のエネルギー総量を用いる場合に比べて、移動度が高く、且つ、安定化(飽和)する。
【0031】
さらに前処理工程では、第2酸化物前駆体膜6に対してフーリエ変換型赤外分光で測定したときに得られる赤外線吸収スペクトルにおいて、赤外線の波数1380cm−1以上1520cm−1以下の範囲に位置するピークが、光処理の時間に伴って、赤外線の波数1350cm−1以上1750cm−1以下の範囲における赤外線吸収スペクトルの中で最大値を示し始めるときに必要な光処理のエネルギー総量に対して、4倍以上6倍以下のエネルギー総量を用いて光処理を行うことがより好ましい。
6倍超のエネルギー総量を用いると移動度が安定化(飽和)するものの、第2酸化物前駆体膜6に対して長時間の光処理が行われることになる。4倍以上6倍以下のエネルギー総量を用いて光処理を行うことで、長時間の光処理を避け、溶液中の金属が酸化され電気伝導率が悪化することを抑制することができる。また、基板2が上述した樹脂基板や樹脂の複合材料で構成された基板である場合に、光処理によって基板2が変形することを抑制できる。
【0032】
前処理工程における有機成分の結合状態の選択的な変化は、第2酸化物前駆体膜6に対してフーリエ変換型赤外分光で測定したときに得られる赤外線吸収スペクトルにおいて、第1酸化物前駆体膜4の赤外線吸収スペクトルと比較した場合に、赤外線の波数1500cm−1以上1600cm−1以下の範囲にある有機成分由来のピークの減少に相当する変化を含むことが好ましい。この範囲の一部ピークは、CO系のピークであると想定される。ここで、前処理工程として光処理を単に行っても、形成工程で用いる溶液や第1酸化物前駆体膜4中の有機成分によって、減少するピークの種類は変化し得る。このように前処理工程によって選択的に減少するピークの種類を、赤外線の波数1500cm−1以上1600cm−1以下の範囲にあるピークに特定することで、確実に膜質の向上を図ることができる。
また、前処理工程における有機成分の結合状態の選択的な変化は、第2酸化物前駆体膜6に対してフーリエ変換型赤外分光で測定したときに得られる赤外線吸収スペクトルにおいて、第1酸化物前駆体膜4の赤外線吸収スペクトルと比較した場合に、赤外線の波数1500cm−1以上1600cm−1以下の範囲にあるピークの減少とは別個に又は減少と共に、照射した赤外線の波数が2750cm−1以上3050cm−1以下の範囲にある有機成分由来のピークの減少に相当する変化を含むことが好ましい。この範囲のピークは、CH系のピークであると想定される。このように前処理工程によって選択的に減少するピークの種類を、照射した赤外線の波数が2750cm−1以上3050cm−1以下の範囲にあるピークに特定することで、確実に膜質の向上を図ることができる。
【0033】
また、前処理工程後且つ後処理工程前で、第2酸化物前駆体膜6に対してフーリエ変換型赤外分光で測定したときに得られる赤外線吸収スペクトルにおいて、赤外線の波数1500cm−1以上1600cm−1以下の範囲にあるピークの減少率が、前処理工程前の1500cm−1以上1600cm−1以下の範囲にあるピークを1としたとき0.37以下であることが好ましく、0.11以下であることがより好ましい。ピークの減少率が0.37以下であると、第2酸化物前駆体膜6に対してフーリエ変換型赤外分光で測定したときに得られる赤外線吸収スペクトルにおいて、赤外線の波数1380cm−1以上1450cm-1以下の範囲に位置するピークが、赤外線の波数1350cm−1以上1750cm−1以下の範囲における赤外線吸収スペクトルの中で最大値を示し易いからである。 なお、このピークの減少率は、例えば光照射の波長、照射時間や照射量等によって調整することができる。
【0034】
<後処理工程>
次に、図1(D)に示すように、前処理工程後の第2酸化物前駆体膜中6に残存する有機成分を除去して、第2酸化物前駆体膜6を、Inを含有する非晶質金属酸化物薄膜8へ変化させる後処理工程を行う。
この後処理工程は、上述のような機能を果たせば手段は問わないが、具体的に熱処理工程が挙げられる。
熱処理工程の方法としては、電気炉やマッフル炉で加熱する方法や、赤外線ランプやホットプレートタイプの加熱方法等が挙げられる。
この熱処理工程では、425℃以上の温度で第2酸化物前駆体膜中6を熱処理することが好ましい。425℃未満で熱処理する場合に比べて、当該熱処理により得られる非晶質酸化物薄膜8の移動度を約3桁も高くすることができるからである。
また、第2酸化物前駆体膜6がIn、Ga及びZnを全て含有している場合、第2酸化物前駆体膜6を非晶質酸化物薄膜8へ変化させる過程で、非晶質の状態を保つという観点から、800℃未満、特に700℃以下の温度で熱処理することが好ましい。特に、基板2の溶解や変形を抑制するという観点から、600℃以下の温度で熱処理することが好ましい。
また、熱処理の雰囲気としては、酸化物を得やすいという観点から酸化性雰囲気で行うことが好ましい。なお、酸化性雰囲気としてはまず、大気(酸素分圧21%,(雰囲気全体に含まれる水分含有量が露点温度換算で16℃(絶対湿度13.6g/m−3)))が用いることができる。また、酸素分圧が全圧の20%以上を占める雰囲気を用いることが好ましく、例えば酸素分圧100%の雰囲気(ガスボンベからフロー。雰囲気全体に含まれる水分含有量が露点温度換算で−36℃以下(絶対湿度0.21g/m−3以下))を用いることもできる。
【0035】
<その他の工程>
最後に、図1(E)に示すように、後処理工程にて得られた非晶質酸化物薄膜8をエッチングしたり、非晶質酸化物薄膜8上に薄膜を形成したりする等その他の工程を適宜行う。
【0036】
以上の工程を経ることにより、膜質を向上した非晶質酸化物薄膜8を得ることができる。そして、このような非晶質酸化物薄膜8は薄膜トランジスタの活性層や導電層、絶縁層、中でも活性層として有用である。
【0037】
2.薄膜トランジスタ
本発明の実施形態に係る薄膜トランジスタ(以下、TFTと称す)は、ゲート電極、ゲート絶縁膜、活性層、ソース電極及びドレイン電極を有し、ゲート電極に電圧を印加して、活性層に流れる電流を制御し、ソース電極とドレイン電極間の電流をスイッチングする機能を有するアクテイブ素子である。そして、本発明の実施形態に係る薄膜トランジスタでは、活性層、絶縁層(ゲート絶縁膜や保護層等)及び導電層(ゲード電極やソース電極、ドレイン電極等)の少なくともいずれか1つとして上述の非晶質酸化物薄膜8が用いられる。
【0038】
TFTの素子構造としては、ゲート電極の位置に基づいた、いわゆる逆スタガ構造(ボトムゲート型とも呼ばれる)及びスタガ構造(トップゲート型とも呼ばれる)のいずれの態様であってもよい。また、活性層とソース電極及びドレイン電極(適宜、「ソース・ドレイン電極」という。)との接触部分に基づき、いわゆるトップコンタクト型、ボトムコンタクト型のいずれの態様であってもよい。
なお、トップゲート型とは、ゲート絶縁膜の上側にゲート電極が配置され、ゲート絶縁膜の下側に活性層が形成された形態であり、ボトムゲート型とは、ゲート絶縁膜の下側にゲート電極が配置され、ゲート絶縁膜の上側に活性層が形成された形態である。また、ボトムコンタクト型とは、ソース・ドレイン電極が活性層よりも先に形成されて活性層の下面がソース・ドレイン電極に接触する形態であり、トップコンタクト型とは、活性層がソース・ドレイン電極よりも先に形成されて活性層の上面がソース・ドレイン電極に接触する形態である。
【0039】
図2(A)は、本発明の実施形態に係るTFTであって、トップゲート構造でトップコンタクト型のTFTの一例を示す模式図である。図2(A)に示すTFT10では、基板12の一方の主面上に活性層14が積層されている。そして、この活性層14上にソース電極16及びドレイン電極18が互いに離間して設置され、更にこれらの上にゲート絶縁膜20と、ゲート電極22とが順に積層されている。
【0040】
図2(B)は、本発明の実施形態に係るTFTであって、トップゲート構造でボトムコンタクト型のTFTの一例を示す模式図である。図2(B)に示すTFT30では、基板12の一方の主面上にソース電極16及びドレイン電極18が互いに離間して設置されている。そして、活性層14と、ゲート絶縁膜20と、ゲート電極22と、が順に積層されている。
【0041】
図2(C)は、本発明の実施形態に係るTFTであって、ボトムゲート構造でトップコンタクト型のTFTの一例を示す模式図である。図2(C)に示すTFT40では、基板12の一方の主面上にゲート電極22と、ゲート絶縁膜20と、活性層14と、が順に積層されている。そして、この活性層14の表面上にソース電極16及びドレイン電極18が互いに離間して設置されている。
【0042】
図2(D)は、本発明の実施形態に係るTFTであって、ボトムゲート構造でボトムコンタクト型のTFTの一例を示す模式図である。図2(D)に示すTFT50では、基板12の一方の主面上にゲート電極22と、ゲート絶縁膜20と、が順に積層されている。そして、このゲート絶縁膜20の表面上にソース電極16及びドレイン電極18が互いに離間して設置され、更にこれらの上に、活性層14が積層されている。
【0043】
なお、本実施形態に係るTFTは、上記以外にも、様々な構成をとることが可能であり、適宜、活性層上に保護層や基板上に絶縁層等を備える構成であってもよい。
【0044】
以下、各構成要素について詳述する。なお、代表例として図2(A)に示すトップゲート構造でトップコンタクト型のTFT10であって、活性層14として本実施形態に係る非晶質酸化物薄膜8を用いる場合について具体的に説明するが、本発明は他の形態のTFTを製造する場合についても同様に適用することができる。
【0045】
<TFTの詳細構成>
−基板−
まず、TFT10を形成するための基板12を用意する。基板12は、上述した基板2と同一のものを用いることができる。
【0046】
−活性層−
次に、基板12上に、トランジスタとして主に活性層14を形成する。
活性層14は非晶質酸化物薄膜8を用いる。したがって、活性層14は、上述したような本発明の実施形態に係る非晶質酸化物薄膜8の製造方法を用いて形成する。ただし、後処理工程は、有機物を確実に除去するという観点から活性層14の形成直後であることが好ましいが、ソース電極16及びドレイン電極18の形成後であったり、ゲート電極22形成後であったり、TFT10完成後であったりしてもよい。
活性層14として用いる非晶質酸化物薄膜8の材料としては、Inを含有したIn−O系の酸化物半導体を用いる。この酸化物半導体には、Inの他Zn、Ga及びMgのうち少なくとも1種を含む酸化物がより好ましく、Inの他Ga及びZnのうちの少なくとも1種を含む酸化物がさらに好ましい。
特に、In、Ga及びZnを全て含む酸化物がより好ましい。In−Ga−Zn−O系酸化物半導体としては、結晶状態における組成がInGaO3(ZnO)m(mは6未満の自然数)で表される酸化物半導体が好ましく、特に、InGaZnO4(以下、「IGZO」とも言う。)がより好ましい。この組成の酸化物半導体の特徴としては、電気伝導度が増加するにつれ、電子移動度が増加する傾向を示す。
ただし、IGZOの組成比は、厳密にIn:Ga:Zn=1:1:1となる必要はない。また、活性層14は、上記のような酸化物半導体を主成分として含有していれば良く、その他に不純物等を含有していても良い。ここで、「主成分」とは、活性層14を構成する構成成分のうち、最も多く含有されている成分を表す。
【0047】
活性層14の形成後には、デバイスに応じて当該薄膜を適宜パターンニングする。パターンニングはフォトリソグラフィー及びエッチングにより行うことが出来る。具体的には、残存させる部分にフォトリソグラフィーによりレジストパターンを形成し、塩酸、硝酸、希硫酸、又は燐酸、硝酸及び酢酸の混合液等の酸溶液によりエッチングすることによりパターンを形成する。また、活性層14上にはソース・ドレイン電極エッチング時に活性層14を保護するための保護膜があってもよい。保護膜は活性層14と連続で成膜してもよいし、活性層14のパターンニング後に形成してもよい。さらに、活性層のパターニングは、後述するソース・ドレイン電極を形成した後に行ってもよい。パターンニングによる活性層14へのダメージを抑制することができるからである。
【0048】
活性層14の層構造は、2層以上から構成されていても良く、活性層14が低抵抗層と高抵抗層より形成され、低抵抗層がゲート絶縁膜20と接し、高抵抗層がソース電極16及びドレイン電極18の少なくとも一方と電気的に接していることが好ましい。
活性層14の厚みは、特に限定されないが、1nm以上100nm以下であり、より好ましくは、2.5nm以上50nm以下である。
【0049】
−ソース・ドレイン電極−
活性層14の上にソース・ドレイン電極16,18を形成するための導電膜を形成する。
この導電膜は高い導電性を有するものを用い、例えばAl,Mo,Cr,Ta,Ti,Au,Au等の金属、Al−Nd、Ag合金、酸化錫、酸化亜鉛、酸化インジウム、酸化インジウム錫(ITO)、酸化亜鉛インジウム(IZO)等の金属酸化物導電膜等を用いて形成することが出来る。ソース・ドレイン電極16,18としてはこれらの導電膜を単層構造又は2層以上の積層構造として用いることが出来る。
【0050】
導電膜の形成は、例えば印刷方式、コーティング方式等の湿式方式、真空蒸着法、スパッタリング法、イオンプレーティング法等の物理的方式、CVD、プラズマCVD法等の化学的方式等の中から使用する材料との適性を考慮して適宜選択した方法に従って成膜する。
成膜する導電膜の膜厚は、成膜性やエッチングやリフトオフ法によるパターンニング性、導電性等を考慮すると、10nm以上1000nm以下とすることが好ましく、50nm以上500nm以下とすることがより好ましい。
次いで、成膜した導電膜をエッチング又はリフトオフ法により所定の形状にパターンニングし、ソース電極及びドレイン電極16,18を形成する。この際、ソース・ドレイン電極16,18に接続する配線を同時にパターンニングすることが好ましい。
【0051】
−ゲート絶縁膜−
ソース・ドレイン電極16,18及び配線を形成した後、ゲート絶縁膜20を形成する。
ゲート絶縁膜20は、高い絶縁性を有するものが好ましく、例えばSiO2,SiNx,SiON,Al2O3,Y2O3,Ta2O5,HfO2等の絶縁膜、又はこれらの化合物を少なくとも二つ以上含む絶縁膜としてもよい。ゲート絶縁膜20は、印刷方式、コーティング方式等の湿式方式、真空蒸着法、スパッタリング法、イオンプレーティング法等の物理的方式、CVD、プラズマCVD法等の化学的方式等の中から使用する材料との適性を考慮して適宜選択した方法に従って成膜する。
次に、ゲート絶縁膜20は、必要に応じて、フォトリソグラフィー及びエッチングによって所定の形状にパターンニングを行う。
なお、ゲート絶縁膜20は、リーク電流の低下及び電圧耐性の向上のための厚みを有する必要がある一方、ゲート絶縁膜の厚みが大きすぎると駆動電圧の上昇を招いてしまう。 ゲート絶縁膜は材質にもよるが、ゲート絶縁膜の厚みは10nm以上10μm以下が好ましく、50nm以上1000nm以下がより好ましく、100nm以上400nm以下が特に好ましい。
【0052】
−ゲート電極−
ゲート絶縁膜20を形成した後、ゲート電極22を形成する。
ゲート電極22は、高い導電性を有するものを用い、例えばAl,Mo,Cr,Ta,Ti,Au,Au等の金属、Al−Nd、Ag合金、酸化錫、酸化亜鉛、酸化インジウム、酸化インジウム錫(ITO)、酸化亜鉛インジウム(IZO)等の金属酸化物導電膜等を用いて形成することが出来る。ゲート電極22としては、これらの導電膜を単層構造又は2層以上の積層構造として用いることが出来る。
【0053】
ゲート電極22は、例えば印刷方式、コーティング方式等の湿式方式、真空蒸着法、スパッタリング法、イオンプレーティング法等の物理的方式、CVD、プラズマCVD法等の化学的方式等の中から使用する材料との適性を考慮して適宜選択した方法に従って成膜する。成膜する導電膜の膜厚は成膜性、エッチングやリフトオフ法によるパターンニング性、導電性等を考慮すると、10nm以上1000nm以下とすることが好ましく、50nm以上500nm以下とすることがより好ましい。
成膜後、導電膜をエッチング又はリフトオフ法により所定の形状にパターンニングし、ゲート電極22を形成する。この際、ゲート電極22及びゲート配線を同時にパターンニングすることが好ましい。
【0054】
以上の手順により、図2(A)に示すような、オンオフ比が高く移動度も高い等TFT特性が良好なTFT10を作製することができる。なお、本発明の実施形態に係る非晶質酸化物薄膜8の製造方法は活性層14の形成に適用する場合を説明したが、ゲート絶縁膜20やゲート電極22、ソース電極16、ドレイン電極18の形成に用い、活性層14の形成は従来の方法を用いることもできる。また、活性層14の形成と共に、ゲート絶縁膜20、ゲート電極22、ソース電極16又はドレイン電極18の何れかの形成において、本発明の実施形態に係る非晶質酸化物薄膜8の製造方法を用いることもできる。
後述するように、本実施形態による液相法で作製したものと、真空成膜で作製したものとが、酸化物の電気物性を支配していると考えられる酸素の結合状態が類似しているため、半導体膜としての膜質の向上に限らず、ゲート電極22、ソース電極16又はドレイン電極18等の導電体膜の膜質の向上(移動度の向上)や、ゲート絶縁膜20等の絶縁体膜の膜質の向上(リーク電流の抑制)に繋がるからである。
【0055】
3.応用
以上で説明した本実施形態のTFTの用途には特に限定はないが、例えば電気光学装置における駆動素子、特に大面積デバイスに用いる場合に好適である。
更に実施形態のTFTは、樹脂基板を用いた低温プロセスで作製可能なデバイスに特に好適であり(例えばフレキシブルディスプレイ等)、X線センサなどの各種センサ、MEMS(Micro Electro Mechanical System)等、種々の電子デバイスにおける駆動素子(駆動回路)として、好適に用いられるものである。
【0056】
4.電気光学装置及びセンサ
【0057】
電気光学装置又はセンサは、本実施形態のTFTを備えて構成される。
電気光学装置の例としては、例えば液晶表示装置、有機EL(Electro Luminescence)表示装置、無機EL表示装置等の表示装置がある。
センサの例としては、CCD(Charge Coupled Device)又はCMOS(Complementary Metal Oxide Semiconductor)等のイメージセンサや、X線センサ等が好適である。
本実施形態のTFTを用いた電気光学装置およびセンサは、いずれも特性の面内均一性が高い。なお、ここで言う「特性」とは、電気光学装置(表示装置)の場合には表示特性、センサの場合には感度特性である。
以下、本実施形態によって製造されるTFTを備えた電気光学装置又はセンサの代表例として、液晶表示装置、有機EL表示装置、X線センサについて説明する。
【0058】
5.液晶表示装置
図3に、本発明の電気光学装置の一実施形態の液晶表示装置について、その一部分の概略断面図を示し、図4にその電気配線の概略構成図を示す。
【0059】
図3に示すように、本実施形態の液晶表示装置100は、図2(A)に示したトップゲート構造でトップコンタクト型のTFT10と、TFT10のパッシベーション層102で保護されたゲート電極22上に画素下部電極104およびその対向上部電極106で挟まれた液晶層108と、各画素に対応させて異なる色を発色させるためのRGBカラーフィルタ110とを備え、TFT10の基板12側およびRGBカラーフィルタ110上にそれぞれ偏光板112a、112bを備えた構成である。
【0060】
また、図4に示すように、本実施形態の液晶表示装置100は、互いに平行な複数のゲート配線112と、該ゲート配線112と交差する、互いに平行なデータ配線114とを備えている。ここでゲート配線112とデータ配線114は電気的に絶縁されている。ゲート配線112とデータ配線114との交差部付近に、TFT10が備えられている。
【0061】
TFT10のゲート電極22は、ゲート配線112に接続されており、TFT10のソース電極16はデータ配線114に接続されている。また、TFT10のドレイン電極18はゲート絶縁膜20に設けられたコンタクトホール116を介して(コンタクトホール116に導電体が埋め込まれて)画素下部電極104に接続されている。この画素下部電極104は、接地された対向上部電極106とともにキャパシタ118を構成している。
【0062】
図3に示した本実施形態の液晶装置においては、トップゲート構造のTFT10を備えるものとしたが、本発明の表示装置である液晶装置において用いられるTFTはトップゲート構造に限定されることなく、ボトムゲート構造のTFTであってもよい。
【0063】
本実施形態のTFTは、オンオフ比が非常に高いことから、安定性のある液晶表示装置を得ることができる。
【0064】
6.有機EL表示装置
図5に、本発明の電気光学装置の一実施形態のアクティブマトリックス方式の有機EL表示装置について、その一部分の概略断面図を示し、図6に電気配線の概略構成図を示す。
【0065】
有機EL表示装置の駆動方式には、単純マトリックス方式とアクティブマトリックス方式の2種類がある。単純マトリックス方式は低コストで作製できるメリットがあるが、走査線を1本ずつ選択して画素を発光させることから、走査線数と走査線あたりの発光時間は反比例する。そのため高精細化、大画面化が困難となっている。アクティブマトリックス方式は画素ごとにトランジスタやキャパシタを形成するため製造コストが高くなるが、単純マトリックス方式のように走査線数を増やせないという問題はないため高精細化、大画面化に適している。
【0066】
本実施形態のアクティブマトリックス方式の有機EL表示装置200は、図2(A)に示したトップゲート構造のTFT10が、パッシベーション層202を備えた基板12上に、駆動用TFT204およびスイッチング用TFT206として備えられ、該TFT204および206上に下部電極208および上部電極210に挟まれた有機発光層212からなる有機EL発光素子214を備え、上面もパッシベーション層216により保護された構成となっている。
【0067】
また、図6に示すように、本実施形態の有機EL表示装置200は、互いに平行な複数のゲート配線220と、該ゲート配線220と交差する、互いに平行なデータ配線222および駆動配線224とを備えている。ここで、ゲート配線220とデータ配線222、駆動配線224とは電気的に絶縁されている。スイッチング用TFT10bのゲート電極22は、ゲート配線220に接続されており、スイッチング用TFT10bのソース電極16はデータ配線222に接続されている。また、スイッチング用TFT10bのドレイン電極18は駆動用TFT10のゲート電極22に接続されるとともに、キャパシタ226を用いることで駆動用TFT10aをオン状態に保つ。駆動用TFT10aのソース電極16は駆動配線224に接続され、ドレイン電極18は有機EL発光素子214に接続される。
【0068】
図5に示した本実施形態の有機EL装置においては、トップゲート構造のTFT10aおよび10bを備えるものとしたが、本発明の表示装置である有機EL装置において用いられるTFTは、トップゲート構造に限定されることなく、ボトムゲート構造のTFTであってもよい。
【0069】
本実施形態により製造されるTFTは、オンオフ比が非常に高いことから、安定性のある有機EL表示装置が得られる。
【0070】
なお、図5に示した有機EL表示装置において、上部電極210を透明電極としてトップエミッション型としてもよいし、下部電極208およびTFTの各電極を透明電極とすることによりボトムエミッション型としてもよい。
【0071】
7.X線センサ
図7に、本発明のセンサの一実施形態であるX線センサについて、その一部分の概略断面図を示し、図8にその電気配線の概略構成図を示す。
【0072】
図7は、より具体的にはX線センサアレイの一部を拡大した概略断面図である。本実施形態のX線センサ300は基板12上に形成されたTFT10およびキャパシタ310と、キャパシタ310上に形成された電荷収集用電極302と、X線変換層304と、上部電極306とを備えて構成される。TFT10上にはパッシベーション膜308が設けられている。
【0073】
キャパシタ310は、キャパシタ用下部電極312とキャパシタ用上部電極314とで絶縁膜316を挟んだ構造となっている。キャパシタ用上部電極314は絶縁膜316に設けられたコンタクトホール318を介し、TFT10のソース電極16およびドレイン電極18のいずれか一方(図7においてはドレイン電極18)と接続されている。
【0074】
電荷収集用電極302は、キャパシタ310におけるキャパシタ用上部電極314上に設けられており、キャパシタ用上部電極314に接している。
X線変換層304はアモルファスセレンからなる層であり、TFT10およびキャパシタ310を覆うように設けられている。
上部電極306はX線変換層304上に設けられており、X線変換層304に接している。
【0075】
図8に示すように、本実施形態のX線センサ300は、互いに平行な複数のゲート配線320と、ゲート配線320と交差する、互いに平行な複数のデータ配線322とを備えている。ここでゲート配線320とデータ配線322は電気的に絶縁されている。ゲート配線320とデータ配線322との交差部付近に、TFT10が備えられている。
【0076】
TFT10のゲート電極22は、ゲート配線320に接続されており、TFT10のソース電極16はデータ配線322に接続されている。また、TFT10のドレイン電極18は電荷収集用電極302に接続されており、さらにこの電荷収集用電極302は、キャパシタ310に接続されている。
【0077】
本実施形態のX線センサ300において、X線は図7中、上部(上部電極306側)から照射され、X線変換層304で電子−正孔対を生成する。このX線変換層304に上部電極306によって高電界を印加しておくことにより、生成した電荷はキャパシタ310に蓄積され、TFT10を順次走査することによって読み出される。
【0078】
本実施形態のX線センサ300は、面内均一性の高い、信頼性に優れたTFT10を備えるため、均一性に優れた画像を得ることができる。
【0079】
なお、図7に示した本実施形態のX線センサにおいては、トップゲート構造のTFTを備えるものとしたが、本発明のセンサにおいて用いられるTFTはトップゲート構造に限定されることなく、ボトムゲート構造のTFTであってもよい。
【0080】
<変形例>
なお、本発明を特定の実施形態について詳細に説明したが、本発明はかかる実施形態に限定されるものではなく、本発明の範囲内にて他の種々の実施形態が可能であることは当業者にとって明らかであり、例えば上述の複数の実施形態は、適宜、組み合わせて実施可能である。また、以下の変形例同士を、適宜、組み合わせてもよい。
【0081】
例えば、上述した基板用意工程、形成工程、その他の工程は、全部又は一部を適宜省略することができる。
また、後処理工程の際に、例えば熱処理と同時に光照射を行ってもよい。
【実施例】
【0082】
以下に実施例を説明するが、本発明はこれら実施例により何ら限定されるものではない。
【0083】
<金属アルコキシド系の実施例>
−第1酸化物前駆体膜及び第2酸化物前駆体膜の作製及び検討−
酢酸亜鉛2水和物2.2g、ガリウムイソプロポキシド2.47g、インジウムイソプロポキシ2.74gを秤量し、ジエチルエタノールアミン中で150℃の温度にて攪拌し、淡黄色の金属アルコキシド系原料液(IGZO溶液)を得た。In,Ga,Znの組成比は、ICP測定により1.0:1.0:0.9という結果が得られた。そして、得られたIGZO溶液の乾燥ゲルに対してTG−DTAを実施した。
【0084】
図9は、実施例の乾燥ゲルに対するTG−DTAの測定結果を示す図である。
図9に示す測定結果のように、室温以上約400℃以下まで発熱反応に伴う重量減少が急激であった。また、400℃超425℃未満は重量減少が緩やかとなり、約425℃で発熱反応に伴う重量減少が終了していることが分かった。上記400℃及び425℃の温度は、乾燥ゲルの有機成分の熱分解温度に相当するものと考えられる。
【0085】
次に、石英基板上に、上記IGZO溶液を1000rpmの速度で二回スピンコートした後、室温で乾燥させることによりゲル状態の第1酸化物前駆体膜を成膜した。そして、得られた第1酸化物前駆体膜をXRDにて評価を行った。
【0086】
図10は、実施例の第1酸化物前駆体膜に対するXRDの測定結果である。
図10に示すように、室温乾燥させたのみの第1酸化物前駆体膜では、非晶質を示すパターンしか得られなかった。また、この第1酸化物前駆体膜のXRD焼成温度依存を測定すると、1000℃焼成の膜ではInGaZnO4の結晶構造を有しており、800℃焼成での膜では微弱なピークがあり一部結晶質があると推定されるものの非晶質と混在している状態であり、700℃焼成での膜では明確なピークが見られずにごく一部の結晶質があると推定されるものの非晶質と混在している状態であり、600℃以下までの膜では図10の結果のみから分析すると完全に非晶質として存在することが確認された。
【0087】
次に、室温乾燥させたのみの複数の第1酸化物前駆体膜に対して、互いに1分、3分、5分、10分、30分、60分、90分と照射時間を変えて前処理工程としてのUVオゾン処理を行い、第2酸化物前駆体膜を得た。このUVオゾン処理では低圧水銀ランプ(波長185nm及び254nm)を用いており、254nmの強度が約5mW/cm2である。
【0088】
このUVオゾン処理の前後の膜に対してXRF測定を行ったところ、In,Ga,Znの組成比及び塗布量は処理前後で変化していなかった。
また、このUVオゾン処理を行った後の第2酸化物前駆体膜に対して、装置名:BRUKER ALPHAの装置でダイヤモンドATR配置を用いて、FT−IR(フーリエ変換型赤外分光)スペクトル測定を実施した。また、比較のために、室温乾燥させたのみの複数の第1酸化物前駆体膜をUVオゾン処理の代わりに各種温度で熱処理し、その後の膜に対してFT−IR(フーリエ変換型赤外分光)スペクトル測定を実施した。
【0089】
図11は、各第2酸化物前駆体膜に対してFT−IR(フーリエ変換型赤外分光)スペクトル測定した測定結果を示す図である。図12は、図11に示す赤外線の波数1350cm−1以上1750cm−1以下の範囲における赤外線吸収スペクトルの拡大図である。
図13は、複数の第1酸化物前駆体膜を各種温度で熱処理した後の各膜に対してFT−IR(フーリエ変換型赤外分光)スペクトル測定した測定結果を示す図である。なお、図11では、90分の間UVオゾン処理した後の第2酸化物前駆体膜に対するFT−IR(フーリエ変換型赤外分光)スペクトルの測定結果は、60分の測定結果と同様であったので図示を省略している。
【0090】
図11に示すように、処理時間と共にFT−IRスペクトルの一部ピークの形状が変化していくことが確認された。UVオゾン処理と熱処理でのFT−IRスペクトルの形状変化を図11及び図13を参照しながら比較すると、熱処理では、ピークが全体的に減少しているが、UVオゾン処理では選択的にピークの形状(有機成分の結合状態)が変化していることが分かる。また、UVオゾン処理後に熱処理を施したサンプル(図14参照)ではUVオゾン処理を施していないサンプル(図15参照)と同様に600度以下ではXRD測定結果からは完全に非晶質状態であることが確認された。
さらに、図12に示すように、UVオゾン処理の照射時間が5分未満では、赤外線の波数1500cm−1以上1600cm−1以下の範囲にあるピークが、赤外線の波数1350cm−1以上1750cm−1以下の範囲(I)における赤外線吸収スペクトルの中で最大値を示している。一方で、UVオゾン処理の照射時間が5分以上では、第2酸化物前駆体膜に対してフーリエ変換型赤外分光で測定したときに得られる赤外線吸収スペクトルにおいて、赤外線の波数1380cm−1以上1520cm−1以下の範囲(II)に位置する(ダブル)ピークが、赤外線の波数1350cm−1以上1750cm−1以下の範囲(I)における赤外線吸収スペクトルの中で最大値を示すようになることが分かった。
より具体的には、UVオゾン処理の照射時間が5分以上では、第2酸化物前駆体膜に対してフーリエ変換型赤外分光で測定したときに得られる赤外線吸収スペクトルにおいて、赤外線の波数1380cm−1以上1520cm−1以下の範囲(II)を、赤外線の波数1380cm−1以上1450cm−1以下の範囲(IV)と赤外線の波数1450cm−1超1520cm−1以下の範囲(III)とに分割したときに、赤外線の波数1380cm−1以上1450cm-1以下の範囲(IV)に位置するピークが、赤外線の波数1350cm−1以上1750cm−1以下の範囲(I)における赤外線吸収スペクトルの中で最大値を示すようになる。
なお、赤外線の波数1380cm−1以上1450cm−1以下の範囲(IV)と、赤外線の波数1450cm−1超1520cm−1以下の範囲(III)とには、両範囲を跨ぐようにダブルピークが見られ、このダブルピークの一つが上記範囲(IV)に位置するピークである。
【0091】
また、図11に示すように、UVオゾン処理におけるピークの形状変化(有機成分の結合状態)の選択的な変化として、UVオゾン処理の照射時間が長くなると共に、赤外線の波数2750cm−1以上3050cm−1以下の範囲にある有機成分由来のピークの減少も含むことが分かった。同様に、UVオゾン処理におけるピークの形状変化(有機成分の結合状態)の選択的な変化として、UVオゾン処理の照射時間が長くなると共に、赤外線の波数1500cm−1以上1600cm−1以下の範囲にある有機成分由来のピークの減少も含むことが分かった。
以下、表1に照射時間及びエネルギー総量と、赤外線の波数1500cm−1以上1600cm−1以下の範囲にある有機成分由来のピークの減少率との相関関係を示す。
【0092】
【表1】
【0093】
−薄膜トランジスタの作製−
次に、UVオゾン処理の照射時間によって変化する第2酸化物前駆体膜の状態が熱処理後の薄膜へデバイス特性としてどのような影響を及ぼすか評価を行うために、ボトムゲートートップコンタクト型の薄膜トランジスタを作製した。
【0094】
図16は、本発明の実施例に係る薄膜トランジスタの製造方法の工程図を示す図である。
【0095】
本発明の実施例に係る薄膜トランジスタの基板には、P+Si/Si熱酸化膜(100nm)付基板を用い、ゲート電極としてP+Siを、ゲート絶縁膜としてSi熱酸化膜を用いた。
次に、図16(A)〜(C)に示す過程において、上記の第1酸化物前駆体膜から第2酸化物前駆体膜を得る方法と同一の方法を用いて活性層を作製した。具体的に、図16(A)に示すように、IGZO溶液(上記原料液と同じ溶液で、In:Ga:Zn=1.0:1.0:0.9)を用い、スピンコート法により最終生成物の膜厚が50nm程度になるように活性層の成膜を行った(熱処理により有機成分が減少して膜厚が変化するため)。成膜後に、図16(B)に示すように前処理工程としてUVオゾン処理を各種条件にて実施し、図16(C)に示すように後処理工程として400℃以上600℃以下の間で15分間熱処理を実施した。その後図16(D)に示すようにフォトリソグラフィーとウェットエッチングにより活性層をパターニングした。レジストにはTSMR8900−LB、エッチング液にはシュウ酸を用いた。現像には現像液としてTMAH5%溶液を用いている。ソース、ドレイン(100nm)電極にはMoを、図16(E)及び図16(F)に示すようにリフトオフにより形成した。レジストにはAZ5214Eを用いた。
【0096】
−トランジスタ特性及び移動度の評価−
次に、得られた薄膜トランジスタに対して、半導体パラメータ・アナライザー4156C(アジレントテクノロジー社製)を用い、トランジスタ特性(Vg−Id特性)及び移動度μの測定を行った。Vg−Id特性の測定は、ドレイン電圧(Vd)を5Vに固定し、ゲート電圧(Vg)を−20V〜+40Vの範囲内で変化させ、各ゲート電圧(Vg)におけるドレイン電流(Id)を測定することにより行った。
【0097】
図17〜図21は、実施例により得られた薄膜トランジスタのVg−Id特性を示す図である。具体的に、図17は、熱処理温度を400℃に固定して、UV照射時間を0、1、3、5、10、20、30、60、90分と変化させたときの薄膜トランジスタのVg−Id特性を示す図である。図18は、熱処理温度を425℃に固定して、UV照射時間を0、1、3、5、10、20、30、60、90分と変化させたときの薄膜トランジスタのVg−Id特性を示す図である。図19は、熱処理温度を450℃に固定して、UV照射時間を0、1、3、5、10、20、30、60、90分と変化させたときの薄膜トランジスタのVg−Id特性を示す図である。図20は、熱処理温度を500℃に固定して、UV照射時間を0、1、3、5、10、20、30、60、90分と変化させたときの薄膜トランジスタのVg−Id特性を示す図である。図21は、熱処理温度を600℃に固定して、UV照射時間を0、1、3、5、10、20、30、60、90分と変化させたときの薄膜トランジスタのVg−Id特性を示す図である。
なお、熱処理温度に関わらず、UV照射時間を0分としたときの薄膜トランジスタを比較例1のTFTとし、UV照射時間を1分としたときの薄膜トランジスタを比較例2のTFTとし、UV照射時間を3分としたときの薄膜トランジスタを比較例3のTFTと呼称する。また、熱処理温度に関わらず、UV照射時間を5分としたときの薄膜トランジスタを実施例1のTFTとし、UV照射時間を10分としたときの薄膜トランジスタを実施例2のTFTとし、UV照射時間を20分としたときの薄膜トランジスタを実施例3のTFTと呼称する。さらに、熱処理温度に関わらず、UV照射時間を30分としたときの薄膜トランジスタを実施例4のTFTとし、UV照射時間を60分としたときの薄膜トランジスタを実施例5のTFTとし、UV照射時間を90分としたときの薄膜トランジスタを実施例6のTFTと呼称する。
【0098】
また、図17〜図21に示すVg−Id特性から求めたオンオフ比とUV照射時間との関係を以下の表2にまとめ、図17〜図21に示すVg−Id特性から求めた移動度とUV照射時間との関係を以下の表3にまとめた。また、図17〜図21に示すVg−Id特性から求めたオンオフ比とUV照射時間との関係を図22に示し、図22の部分拡大図を図23に示した。さらに、図17〜図21に示すVg−Id特性から求めた移動度とUV照射時間との関係を図24に示した。
【0099】
【表2】
【0100】
【表3】
【0101】
図17〜図21に示すVg−Id特性から、UVオゾン処理時間が長くなると共に良好なVg−Id特性が得られていることが分かった。
特に、図22及び図23に示すように、照射時間を5分以上とした実施例1〜6のTFTは、比較例1〜3のTFTに比べてオンオフ比が急激に高くなり膜質の向上に繋がることが分かった。
【0102】
さらに、表3及び図24に示すように、照射時間を10分以上とした実施例2〜6のTFTは、実施例1のTFTに比べて移動度が高くなり膜質の向上に繋がることが分かった。すなわち、第2酸化物前駆体膜(活性層)に対して、フーリエ変換型赤外分光で測定したときに得られる赤外線吸収スペクトルにおいて、赤外線の波数1380cm−1以上1520cm−1以下の範囲(II)を、赤外線の波数1380cm−1以上1450cm−1以下の範囲(IV)と赤外線の波数1450cm−1超1520cm−1以下の範囲(III)とに分割したときに、赤外線の波数1380cm−1以上1450cm-1以下の範囲(IV)に位置するピークが、赤外線の波数1350cm−1以上1750cm−1以下の範囲(I)における赤外線吸収スペクトルの中で最大値を示し始めるときから(照射時間5分以上:エネルギー総量1.5J/cm2以上:ピーク減少率0.37以下に対応)、UV照射時間が5分未満の場合に比べて、オンオフ比が急激に高くなることが分かった。
また、表3に示すように、照射時間を5分以上とした実施例1〜6のTFTは、比較例1〜3のTFTに比べて移動度が高くなり膜質の向上に繋がることが分かった。すなわち、赤外線の波数1380cm−1以上1520cm−1以下の範囲(II)に位置するピークが、赤外線の波数1350cm−1以上1750cm−1以下の範囲(I)における赤外線吸収スペクトルの中で最大値を示し始めるときから(照射時間5分以上:エネルギー総量1.5J/cm2以上:ピーク減少率0.37以下に対応)、UV照射時間が5分未満の場合に比べて、移動度が約1桁以上高くなることが分かった。
【0103】
さらにまた、図22に示すように、照射時間を20分以上とした実施例3〜6のTFTは、実施例1や実施例2のTFTに比べてオンオフ比が高く、且つ、オンオフ比が安定化(飽和)していることが分かった。また、図24に示すように、照射時間を20分以上とした実施例3〜6のTFTは、実施例1や実施例2のTFTに比べて移動度が急激に高くなり、実施例3〜6のTFT間で移動度が安定化(飽和)していることが分かった。すなわち、前処理工程で、第2酸化物前駆体膜(活性層)に対してフーリエ変換型赤外分光で測定したときに得られる赤外線吸収スペクトルにおいて、赤外線の波数1380cm−1以上1520cm−1以下の範囲(II)に位置するピークが、UVオゾン処理の処理時間(照射時間)に伴って、赤外線の波数1350cm−1以上1750cm−1以下の範囲(I)における赤外線吸収スペクトルの中で最大値を示し始めるときに必要な光処理のエネルギー総量(実施例では1.5J/cm2)に対して、4倍(実施例では6.0J/cm2)以上のエネルギー総量を用いて光処理を行うことで、4倍未満のエネルギー総量を用いる場合に比べて、オンオフ比が高く、且つオンオフ比が安定化(飽和)し、移動度が急激に高くなる。
【0104】
また、図24に示すように、照射時間を20分以上30分以下とした実施例3〜4のTFTは、実施例1や実施例2及び実施例5や実施例6のTFTに比べて、移動度がより高くなることが分かった。すなわち、前処理工程で、第2酸化物前駆体膜(活性層)に対してフーリエ変換型赤外分光で測定したときに得られる赤外線吸収スペクトルにおいて、赤外線の波数1380cm−1以上1520cm−1以下の範囲(II)に位置するピークが、UVオゾン処理の処理時間(照射時間)に伴って、赤外線の波数1350cm−1以上1750cm−1以下の範囲(I)における赤外線吸収スペクトルの中で最大値を示し始めるときに必要な光処理のエネルギー総量(実施例では1.5J/cm2)に対して、4倍以上6倍(実施例では9.0J/cm2)以下のエネルギー総量を用いて光処理を行うことで、4倍未満又は6倍超のエネルギー総量を用いる場合に比べて、移動度がより高くなることが分かった。
また、6倍超のエネルギー総量を用いると移動度が安定化(飽和)するものの、第2酸化物前駆体膜(活性層)に対して長時間のUVオゾン処理が行われることになる。4倍以上6倍以下のエネルギー総量を用いてUVオゾン処理を行うことで、長時間のUVオゾン処理(UVの照射)を避け、溶液中の金属が酸化され活性層の電気伝導率が悪化することを抑制することができると考えられる。また、基板が上述した樹脂基板や樹脂の複合材料で構成された基板である場合に、UVオゾン処理によって基板が変形することを抑制できると考えられる。
【0105】
次に、良好な特性が得られた照射時間30分のUVオゾン処理を施した実施例4のTFTと、UVオゾン処理の有無での移動度の比較を検討した。
【0106】
図25は、実施例4のTFTと比較例1のTFTのVg−Id特性を示す図である。なお、図25では熱処理温度を425℃と600℃にしたときのデータのみ示している。
図26は、実施例4のTFTと比較例1のTFTの熱処理温度(400℃のときは除く)と移動度との関係をプロットしたグラフ図である。
【0107】
図25に示す結果から、UVオゾン処理が有るか否かで例えばオン電流が劇的に変化し、UVオゾン処理を施していない比較例1のTFTでは、特性向上が見込めず熱処理温度の低下とともにオン電流等のTFT特性が劣化していくことが確認された。
また、図26に示すように、UVオゾン処理を施した実施例4のTFTでは、比較例1のTFTに比べて一桁以上移動度が向上し、425℃〜600℃の熱処理にてその移動度がほぼ同水準の値を示した。
【0108】
−XPS評価−
次に、前処理としてのUVオゾン処理が熱処理後の物性に与える関係を調査するためにXPS測定を行った。このXPS測定には、島津製作所製AXIS−ULTRAを用いた。測定サンプルとして、上記IGZO溶液を30分UVオゾン処理した後、425℃,450℃又は500℃でそれぞれ熱処理したものを用いた。比較用の測定サンプルとして、上記IGZO溶液をそのまま425℃,450℃又は500℃でそれぞれ熱処理したものを用いた。同様に、比較用の測定サンプルとして、IGZO溶液と同じIGZO組成のスパッタ膜を用いた。
【0109】
図27は、各測定サンプルのXPSデータを示す図である。
【0110】
図27に示す結果から、30分のUVオゾン処理を施したサンプルは、酸素の結合状態がスパッタ膜に近づいていることが分かる。これはM−O(M:In,Ga,Znの何れか)の結合状態が強くなっていることを意味し、この強い結合状態が移動度向上の原因となっていると考えられる。液相プロセスにおいては、真空プロセスでは含まれないCやH等が多量に含まれているため、通常M−O結合の形成を阻害しやすい。特に真空プロセスではキャリア濃度を下げる働きをする、Ga、Al、Ta、Ti、Wなどはキャリアの発生源となる酸素の取り込みや放出を行うことが困難であるため、一度M−OHのような結合を形成してしまうと、その結合状態を再度変化させてM−O結合を形成させることが困難である。そのため、通常液相プロセスにおいては、Gaの添加量を極力少なくする傾向にあるが、本実施形態においては、UVオゾン処理等の前処理工程を行い、前駆体膜を改質することで、M−OHなどの結合を極力経由せずに、M−O結合が形成されるのを向上させることが可能となる。
【0111】
また、図27に示す結果から、30分のUVオゾン処理を施した各サンプル(非晶質酸化物薄膜)は、XPSによる酸素(O1s)の結合エネルギーにおいて最大スペクトル強度をえられる結合エネルギーの位置が、同じ金属組成にて真空プロセスにより作製したサンプル(非晶質酸化物薄膜)と同じ位置(530eV付近)に存在していることが分かった。
【0112】
<有機酸塩系の実施例>
−第1酸化物前駆体膜及び第2酸化物前駆体膜の作製及び検討−
以上の実施例では、金属アルコキシド系のIGZO溶液を用いて各種サンプルを作製したが、以下の実施例では有機酸塩系のIGZO溶液を用いて各種サンプルを作製した。
【0113】
各々0.2mol/kgで調液を行った酢酸亜鉛−メトキシエタノール溶液、ガリウムアセチルアセトナート-プロピオン酸溶液、インジウムアセチルアセトナート-プロピオン酸溶液を準備し、In:Ga:Zn=1:1:1となるように混合して、有機酸塩系原料液(IGZO溶液)を得た。
次に、石英基板上に1000rpmの速度で、1回スピンコートした後、室温で乾燥させることによりゲル状態の第1酸化物前駆体膜を成膜した。そして、得られた第1酸化物前駆体膜をXRDにて評価を行ったところ図10とほぼ同様の結果となった。
【0114】
次に、室温乾燥させたのみの複数の第1酸化物前駆体膜に対して、互いに1分、3分、5分、10分、30分、60分、90分と照射時間を変えて前処理工程としてのUVオゾン処理を行い、第2酸化物前駆体膜を得た。このUVオゾン処理では低圧水銀ランプ(波長185nm及び254nm)を用いており、254nmの強度が約5mW/cm2である。
【0115】
そして、UVオゾン処理を行った後の第2酸化物前駆体膜に対して、装置名:BRUKER ALPHAの装置でダイヤモンドATR配置を用いて、ATR配置を用いて、FT−IRスペクトル測定を実施した。比較として、UVオゾン処理を行っていない第1酸化物前駆体膜のままのFT−IRスペクトル測定も実施した。
【0116】
図28は、各第2酸化物前駆体膜に対してFT−IRスペクトル測定した測定結果を示す図である。図29は、図28に示す赤外線の波数1350cm−1以上1750cm−1以下の範囲における赤外線吸収スペクトルの拡大図である。
【0117】
図28に示すように、UV処理時間に伴い1350cm−1以上1750cm−1以下におけるピーク形状が選択的に変化していることが確認された。
また図29に示すように、UVオゾン処理の照射時間が5分未満では、赤外線の波数1500cm−1以上1600cm−1以下の範囲にあるピークが、赤外線の波数1350cm−1以上1750cm−1以下の範囲(I)における赤外線吸収スペクトルの中で最大値を示している。一方で、UVオゾン処理の照射時間が5分以上では、第2酸化物前駆体膜に対してフーリエ変換型赤外分光で測定したときに得られる赤外線吸収スペクトルにおいて、赤外線の波数1380cm−1以上1520cm−1以下の範囲(II)に位置する(ダブル)ピークが、赤外線の波数1350cm−1以上1750cm−1以下の範囲(I)における赤外線吸収スペクトルの中で最大値を示すようになることが分かった。
具体的に、UVオゾン処理の照射時間が5分以上では、第2酸化物前駆体膜に対してフーリエ変換型赤外分光で測定したときに得られる赤外線吸収スペクトルにおいて、赤外線の波数1380cm−1以上1520cm−1以下の範囲(II)を、赤外線の波数1380cm−1以上1450cm−1以下の範囲(IV)と赤外線の波数1450cm−1超1520cm−1以下の範囲(III)とに分割したときに、赤外線の波数1380cm−1以上1450cm-1以下の範囲(IV)に位置するピークが、赤外線の波数1350cm−1以上1750cm−1以下の範囲(I)における赤外線吸収スペクトルの中で最大値を示すようになることも分かった。
なお、赤外線の波数1380cm−1以上1450cm−1以下の範囲(IV)と、赤外線の波数1450cm−1超1520cm−1以下の範囲(III)とには、両範囲を跨ぐようにダブルピークが見られ、このダブルピークの一つが上記範囲(IV)に位置するピークである。
【0118】
−薄膜トランジスタの作製−
次に、UVオゾン処理の照射時間によって変化する第2酸化物前駆体膜の状態が熱処理後の薄膜へデバイス特性としてどのような影響を及ぼすか評価を行うために、ボトムゲートートップコンタクト型の薄膜トランジスタを作製した。
【0119】
図30は、本発明の実施例に係る薄膜トランジスタの製造方法の工程図を示す図である。
【0120】
本発明の実施例に係る薄膜トランジスタの基板には、P+Si/Si熱酸化膜(100nm)付基板を用い、ゲート電極としてP+Siを、ゲート絶縁膜としてSi熱酸化膜を用いた。
次に、図30(A)〜(C)に示す過程において、上記の第1酸化物前駆体膜から第2酸化物前駆体膜を得る方法と同一の方法を用いて活性層を作製した。具体的に、図30(A)に示すように、IGZO溶液(上記金属アルコキシド系のIGZO溶液と同じ溶液で、In:Ga:Zn=1.0:1.0:0.9)を用い、スピンコート法により最終生成物の膜厚が50nm程度になるように活性層の成膜を行った(熱処理により有機成分が減少して膜厚が変化するため)。成膜後に、図30(B)に示すように前処理としてUVオゾン処理を5分間実施し、図30(C)に示すように後処理として375℃〜600℃の間で15分間熱処理を実施した。その後、図30(D)に示すようにフォトリソグラフィーを用いてリフトオフパターンを形成した。レジストにはAZ5214Eを用いた。その後電極Moを100nm成膜し、レジスト剥離を行って図30(E)に示すようにSD電極パターンを形成した。そして、活性層のパターニングとしてレジストにはTSMR8900−LBを用い、Arプラズマによるドライエッチングを用いてパターニングをした。レジスト剥離には、酸素アッシングを用いて図30(F)に示すような薄膜トランジスタを作製した。
【0121】
次に、得られた薄膜トランジスタに対して、半導体パラメータ・アナライザー4156
C(アジレントテクノロジー社製)を用い、トランジスタ特性(Vg−Id特性)及び移動度μの測定を行った。Vg−Id特性の測定は、ドレイン電圧(Vd)を5Vに固定し、ゲート電圧(Vg)を−20V〜+40Vの範囲内で変化させ、各ゲート電圧(Vg)におけるドレイン電流(Id)を測定することにより行った。
【0122】
−トランジスタ特性及び移動度の評価−
次に、得られた薄膜トランジスタに対して、半導体パラメータ・アナライザー4156C(アジレントテクノロジー社製)を用い、トランジスタ特性(Vg−Id特性)及び移動度μの測定を行った。Vg−Id特性の測定は、ドレイン電圧(Vd)を5Vに固定し、ゲート電圧(Vg)を−20V〜+40Vの範囲内で変化させ、各ゲート電圧(Vg)におけるドレイン電流(Id)を測定することにより行った。なお、比較のために、上記薄膜トランジスタにおいて図30(B)に示すUVオゾン処理を省略したものに対してもトランジスタ特性(Vg−Id特性)及び移動度μの測定を行った。
【0123】
図31は、実施例により得られた薄膜トランジスタのVg−Id特性を示す図である。また、図32は、UVオゾン処理を省略した薄膜トランジスタのVg−Id特性を示す図である。さらに、図33は、熱処理温度と薄膜トランジスタの移動度との関係をプロットしたグラフ図である。
【0124】
図31と図32を比較すると、図25に示す結果と同様に、UVオゾン処理の有無で例えばオン電流が劇的に変化していることが分かった。また、図33に示す移動度の結果と図26に示す移動度の結果を比較すると、図33に示す移動度の方が約1桁高いことが分かった。これは、溶液が異なるからではなく、電極形成前に活性層をパターニングするか電極形成後に活性層をパターニングするかの違いによるものだと考えられ、電極形成後に活性層をパターニングした方が活性層にダメージを与え難く、この結果移動度が高くなったと想定される。
【0125】
−ピークの特定−
次に、図11及び図12並びに図28及び図29に示す、赤外線の波数1380cm−1以上1520cm−1以下の範囲(II)に位置するピークでUVオゾン処理の処理時間が5分以上に見られるダブルピークの由来(起因)について検討した。
【0126】
まず、各々0.2mol/kgで調液を行った亜鉛アセチルアセトナート−メタノール溶液、ガリウムアセチルアセトナート-プロピオン酸溶液、インジウムアセチルアセトナート-プロピオン酸溶液を準備した。そして、各々の溶液を石英基板上に1000rpmの速度で1回スピンコートした後室温で乾燥させて乾燥ゲルを得た。各乾燥ゲルに対してUVオゾン処理を30分実施したものと、UVオゾン処理前の乾燥ゲルとのFT−IRスペクトルを比較した。
【0127】
図34は、各乾燥ゲルのFT−IRスペクトルを示す図である。
【0128】
図34に示すように、Inを含んだ乾燥ゲルのUVオゾン処理後のFT−IRスペクトルは、図11及び図12並びに図28及び図29に現れていたダブルピークと一致した。したがって、図11及び図12並びに図28及び図29に現れていた1380cm−1以上1520cm−1のダブルピークは、In前駆体原料に由来していることが分かった。
【符号の説明】
【0129】
4 第1酸化物前駆体膜
6 第2酸化物前駆体膜
8 非晶質酸化物薄膜
12 基板
14 活性層
16 ソース電極(導電層)
18 ドレイン電極(導電層)
20 ゲート絶縁膜(絶縁層)
22 ゲート電極(導電層)
10、30、40、50、204、206 薄膜トランジスタ
【特許請求の範囲】
【請求項1】
有機成分とInとを含有する第1酸化物前駆体膜に対して、前記有機成分の熱分解温度未満で前記有機成分の結合状態を選択的に変化させ、フーリエ変換型赤外分光で測定したときに得られる赤外線吸収スペクトルにおいて、赤外線の波数1380cm−1以上1520cm−1以下の範囲を、赤外線の波数1380cm−1以上1450cm−1以下の範囲と赤外線の波数1450cm−1超1520cm−1以下の範囲とに分割したときに、赤外線の波数1380cm−1以上1450cm-1以下の範囲に位置するピークが、赤外線の波数1350cm−1以上1750cm−1以下の範囲における赤外線吸収スペクトルの中で最大値を示す第2酸化物前駆体膜を得る前処理工程と、
前記第2酸化物前駆体膜中に残存する前記有機成分を除去して、前記第2酸化物前駆体膜を、前記Inを含有する非晶質酸化物薄膜へ変化させる後処理工程と、
を有する非晶質酸化物薄膜の製造方法。
【請求項2】
前記前処理工程における前記有機成分の結合状態の選択的な変化は、前記第2酸化物前駆体膜に対してフーリエ変換型赤外分光で測定したときに得られる赤外線吸収スペクトルにおいて、赤外線の波数1500cm−1以上1600cm−1以下の範囲にある前記有機成分由来のピークの減少に相当する変化を含む、
請求項1に記載の非晶質酸化物薄膜の製造方法。
【請求項3】
前記前処理工程における前記有機成分の結合状態の選択的な変化は、前記第2酸化物前駆体膜に対してフーリエ変換型赤外分光で測定したときに得られる赤外線吸収スペクトルにおいて、赤外線の波数2750cm−1以上3050cm−1以下の範囲にある前記有機成分由来のピークの減少に相当する変化を含む、
請求項1又は請求項2に記載の非晶質酸化物薄膜の製造方法。
【請求項4】
前記第2酸化物前駆体膜に対してフーリエ変換型赤外分光で測定したときに得られる赤外線吸収スペクトルにおいて、赤外線の波数1500cm−1以上1600cm−1以下の範囲にあるピークの減少率が、前記前処理工程前の1500cm−1以上1600cm−1以下の範囲にあるピークを1としたとき0.37以下である、
請求項2に記載の非晶質酸化物薄膜の製造方法。
【請求項5】
前記第2酸化物前駆体膜に対してフーリエ変換型赤外分光で測定したときに得られる赤外線吸収スペクトルにおいて、赤外線の波数1500cm−1以上1600cm−1以下の範囲にあるピークの減少率が、前記前処理工程前の1500cm−1以上1600cm−1以下の範囲にあるピークを1としたとき0.11以下である、
請求項4に記載の非晶質酸化物薄膜の製造方法。
【請求項6】
前記前処理工程は、光処理工程である、
請求項1〜請求項5の何れか1項に記載の非晶質酸化物薄膜の製造方法。
【請求項7】
前記光処理工程では、前記第1酸化物前駆体膜に対して紫外線を照射する、
請求項6に記載の非晶質酸化物薄膜の製造方法。
【請求項8】
前記前処理工程では、前記第2酸化物前駆体膜に対してフーリエ変換型赤外分光で測定したときに得られる赤外線吸収スペクトルにおいて、赤外線の波数1380cm−1以上1520cm−1以下の範囲に位置するピークが、光処理の時間に伴って、赤外線の波数1350cm−1以上1750cm−1以下の範囲における赤外線吸収スペクトルの中で最大値を示し始めるときに必要な光処理のエネルギー総量に対して、4倍以上のエネルギー総量を用いて光処理を行う、
請求項6又は請求項7に記載の非晶質酸化物薄膜の製造方法。
【請求項9】
前記前処理工程では、前記第2酸化物前駆体膜に対してフーリエ変換型赤外分光で測定したときに得られる赤外線吸収スペクトルにおいて、赤外線の波数1380cm−1以上1520cm−1以下の範囲に位置するピークが、光処理の時間に伴って、赤外線の波数1350cm−1以上1750cm−1以下の範囲における赤外線吸収スペクトルの中で最大値を示し始めるときに必要な光処理のエネルギー総量に対して、6倍以下のエネルギー総量を用いて光処理を行う、
請求項8に記載の非晶質酸化物薄膜の製造方法。
【請求項10】
前記後処理工程は、425℃以上の温度で熱処理する熱処理工程である、
請求項1〜請求項9の何れか1項に記載の非晶質酸化物薄膜の製造方法。
【請求項11】
前記第1酸化物前駆体膜は、前記Inの他、Ga、Zn及びSnから選ばれる少なくとも一つを含む無機成分を含有する、
請求項1〜請求項10の何れか1項に記載の非晶質酸化物薄膜の製造方法。
【請求項12】
前記無機成分の組成が、結晶化温度以上の前記熱処理工程でInGaZnO4-δ(前記δは酸素不定比量である)が結晶相として単相で現れるような組成である、
請求項11に記載の非晶質酸化物薄膜の製造方法。
【請求項13】
前記前処理工程の前に、金属アルコキシド又は有機酸塩を含有する溶液を用いて液相法により前記第1酸化物前駆体膜を形成する形成工程、
をさらに有する請求項1〜請求項12の何れか1項に記載の非晶質酸化物薄膜の製造方法。
【請求項14】
請求項1〜請求項13の何れか1項に記載の非晶質酸化物薄膜の製造方法で作製された非晶質酸化物薄膜を活性層として用いた薄膜トランジスタ。
【請求項15】
請求項1〜請求項13の何れか1項に記載の非晶質酸化物薄膜の製造方法で作製された非晶質酸化物薄膜を絶縁層として用いた薄膜トランジスタ。
【請求項16】
請求項1〜請求項13の何れか1項に記載の非晶質酸化物薄膜の製造方法で作製された非晶質酸化物薄膜を導電層として用いた薄膜トランジスタ。
【請求項1】
有機成分とInとを含有する第1酸化物前駆体膜に対して、前記有機成分の熱分解温度未満で前記有機成分の結合状態を選択的に変化させ、フーリエ変換型赤外分光で測定したときに得られる赤外線吸収スペクトルにおいて、赤外線の波数1380cm−1以上1520cm−1以下の範囲を、赤外線の波数1380cm−1以上1450cm−1以下の範囲と赤外線の波数1450cm−1超1520cm−1以下の範囲とに分割したときに、赤外線の波数1380cm−1以上1450cm-1以下の範囲に位置するピークが、赤外線の波数1350cm−1以上1750cm−1以下の範囲における赤外線吸収スペクトルの中で最大値を示す第2酸化物前駆体膜を得る前処理工程と、
前記第2酸化物前駆体膜中に残存する前記有機成分を除去して、前記第2酸化物前駆体膜を、前記Inを含有する非晶質酸化物薄膜へ変化させる後処理工程と、
を有する非晶質酸化物薄膜の製造方法。
【請求項2】
前記前処理工程における前記有機成分の結合状態の選択的な変化は、前記第2酸化物前駆体膜に対してフーリエ変換型赤外分光で測定したときに得られる赤外線吸収スペクトルにおいて、赤外線の波数1500cm−1以上1600cm−1以下の範囲にある前記有機成分由来のピークの減少に相当する変化を含む、
請求項1に記載の非晶質酸化物薄膜の製造方法。
【請求項3】
前記前処理工程における前記有機成分の結合状態の選択的な変化は、前記第2酸化物前駆体膜に対してフーリエ変換型赤外分光で測定したときに得られる赤外線吸収スペクトルにおいて、赤外線の波数2750cm−1以上3050cm−1以下の範囲にある前記有機成分由来のピークの減少に相当する変化を含む、
請求項1又は請求項2に記載の非晶質酸化物薄膜の製造方法。
【請求項4】
前記第2酸化物前駆体膜に対してフーリエ変換型赤外分光で測定したときに得られる赤外線吸収スペクトルにおいて、赤外線の波数1500cm−1以上1600cm−1以下の範囲にあるピークの減少率が、前記前処理工程前の1500cm−1以上1600cm−1以下の範囲にあるピークを1としたとき0.37以下である、
請求項2に記載の非晶質酸化物薄膜の製造方法。
【請求項5】
前記第2酸化物前駆体膜に対してフーリエ変換型赤外分光で測定したときに得られる赤外線吸収スペクトルにおいて、赤外線の波数1500cm−1以上1600cm−1以下の範囲にあるピークの減少率が、前記前処理工程前の1500cm−1以上1600cm−1以下の範囲にあるピークを1としたとき0.11以下である、
請求項4に記載の非晶質酸化物薄膜の製造方法。
【請求項6】
前記前処理工程は、光処理工程である、
請求項1〜請求項5の何れか1項に記載の非晶質酸化物薄膜の製造方法。
【請求項7】
前記光処理工程では、前記第1酸化物前駆体膜に対して紫外線を照射する、
請求項6に記載の非晶質酸化物薄膜の製造方法。
【請求項8】
前記前処理工程では、前記第2酸化物前駆体膜に対してフーリエ変換型赤外分光で測定したときに得られる赤外線吸収スペクトルにおいて、赤外線の波数1380cm−1以上1520cm−1以下の範囲に位置するピークが、光処理の時間に伴って、赤外線の波数1350cm−1以上1750cm−1以下の範囲における赤外線吸収スペクトルの中で最大値を示し始めるときに必要な光処理のエネルギー総量に対して、4倍以上のエネルギー総量を用いて光処理を行う、
請求項6又は請求項7に記載の非晶質酸化物薄膜の製造方法。
【請求項9】
前記前処理工程では、前記第2酸化物前駆体膜に対してフーリエ変換型赤外分光で測定したときに得られる赤外線吸収スペクトルにおいて、赤外線の波数1380cm−1以上1520cm−1以下の範囲に位置するピークが、光処理の時間に伴って、赤外線の波数1350cm−1以上1750cm−1以下の範囲における赤外線吸収スペクトルの中で最大値を示し始めるときに必要な光処理のエネルギー総量に対して、6倍以下のエネルギー総量を用いて光処理を行う、
請求項8に記載の非晶質酸化物薄膜の製造方法。
【請求項10】
前記後処理工程は、425℃以上の温度で熱処理する熱処理工程である、
請求項1〜請求項9の何れか1項に記載の非晶質酸化物薄膜の製造方法。
【請求項11】
前記第1酸化物前駆体膜は、前記Inの他、Ga、Zn及びSnから選ばれる少なくとも一つを含む無機成分を含有する、
請求項1〜請求項10の何れか1項に記載の非晶質酸化物薄膜の製造方法。
【請求項12】
前記無機成分の組成が、結晶化温度以上の前記熱処理工程でInGaZnO4-δ(前記δは酸素不定比量である)が結晶相として単相で現れるような組成である、
請求項11に記載の非晶質酸化物薄膜の製造方法。
【請求項13】
前記前処理工程の前に、金属アルコキシド又は有機酸塩を含有する溶液を用いて液相法により前記第1酸化物前駆体膜を形成する形成工程、
をさらに有する請求項1〜請求項12の何れか1項に記載の非晶質酸化物薄膜の製造方法。
【請求項14】
請求項1〜請求項13の何れか1項に記載の非晶質酸化物薄膜の製造方法で作製された非晶質酸化物薄膜を活性層として用いた薄膜トランジスタ。
【請求項15】
請求項1〜請求項13の何れか1項に記載の非晶質酸化物薄膜の製造方法で作製された非晶質酸化物薄膜を絶縁層として用いた薄膜トランジスタ。
【請求項16】
請求項1〜請求項13の何れか1項に記載の非晶質酸化物薄膜の製造方法で作製された非晶質酸化物薄膜を導電層として用いた薄膜トランジスタ。
【図1】

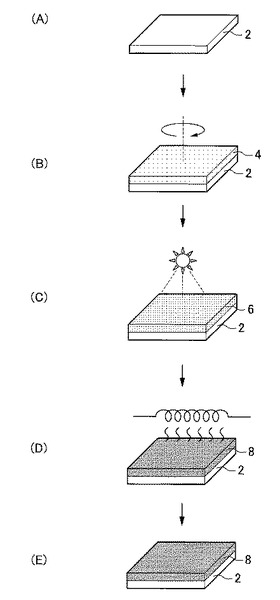
【図2】
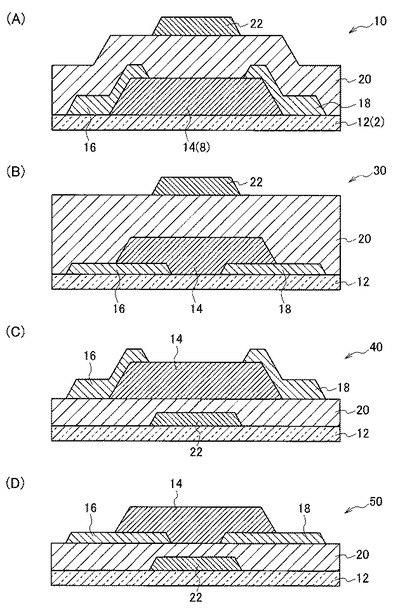
【図3】
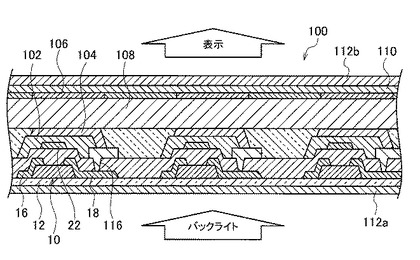
【図4】
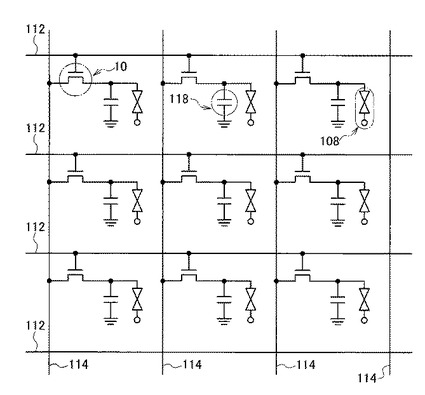
【図5】
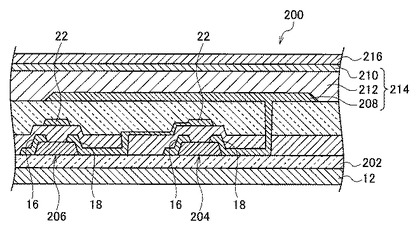
【図6】
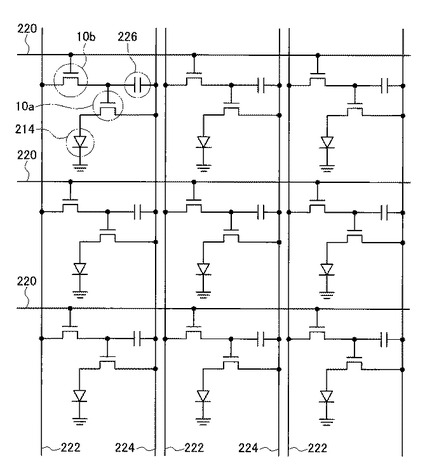
【図7】
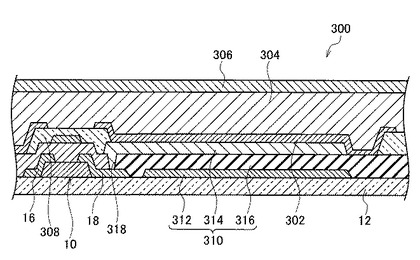
【図8】
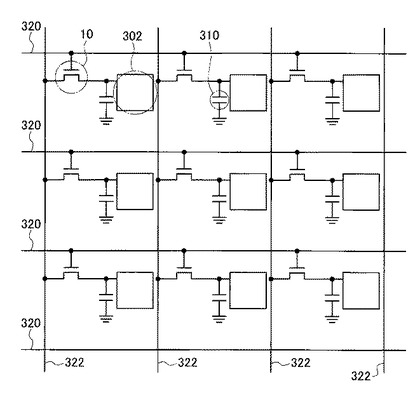
【図9】
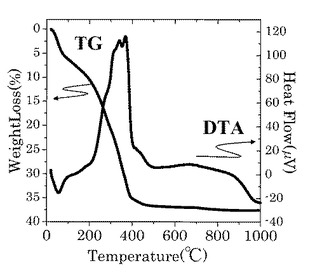
【図10】
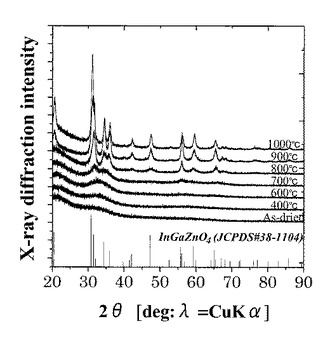
【図12】
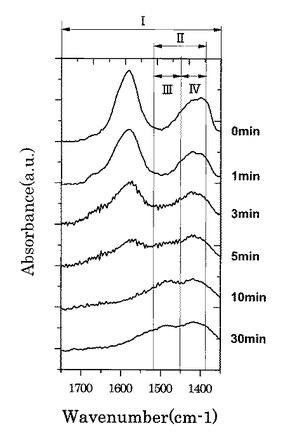
【図14】
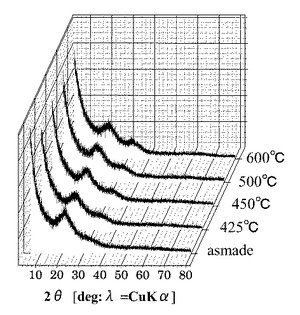
【図15】
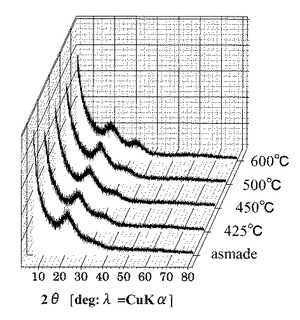
【図16】
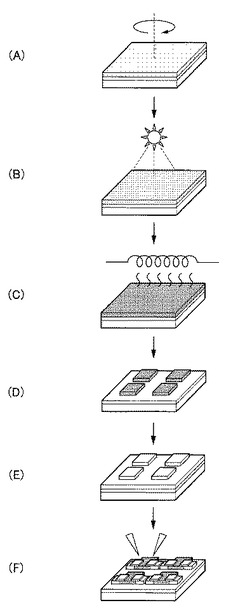
【図17】
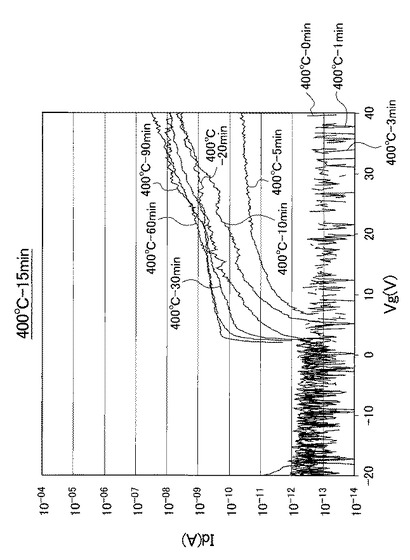
【図18】
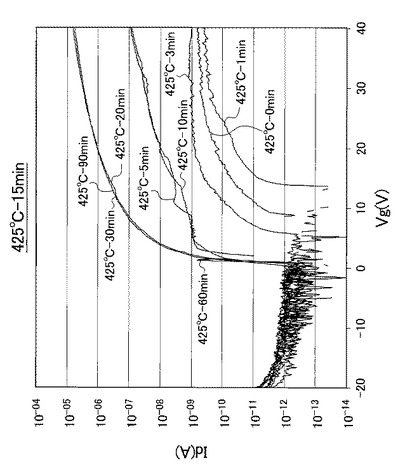
【図19】
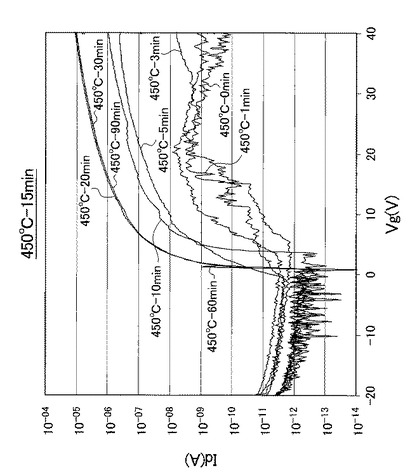
【図20】
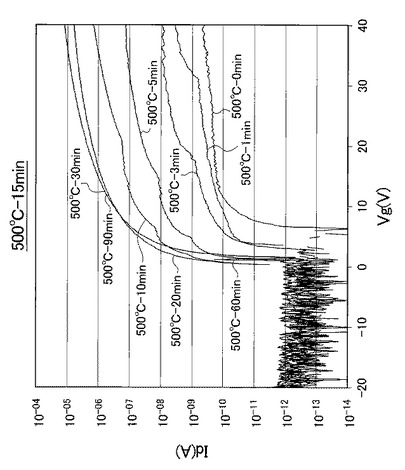
【図21】
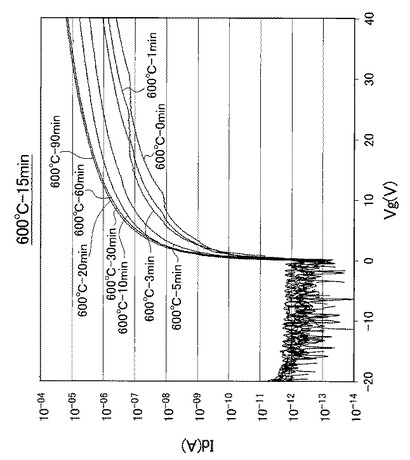
【図22】
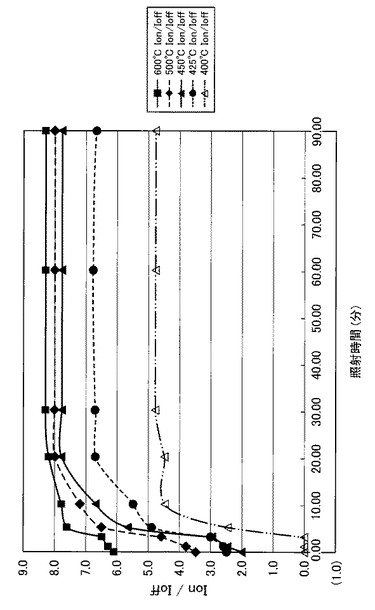
【図23】
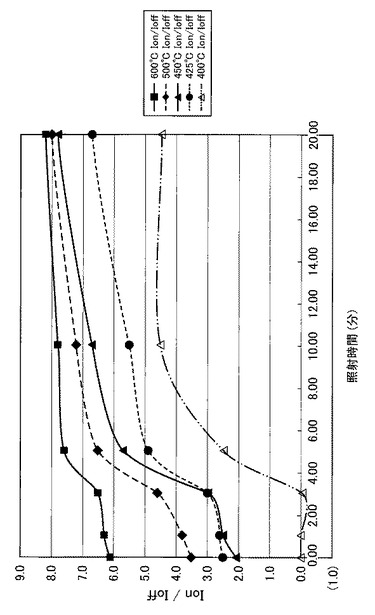
【図24】
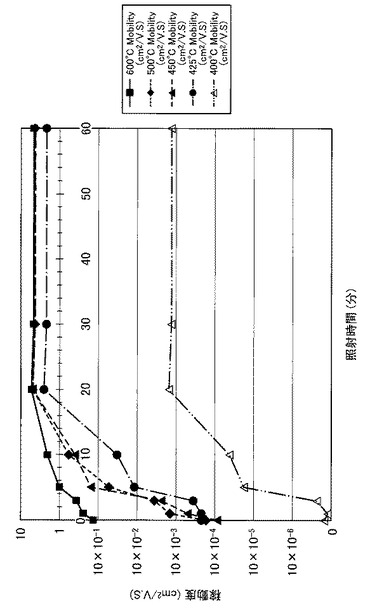
【図25】
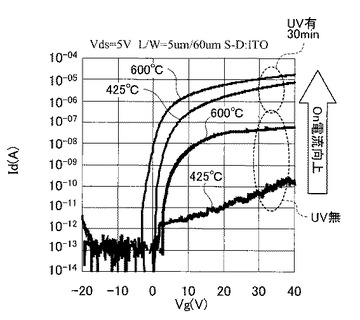
【図26】
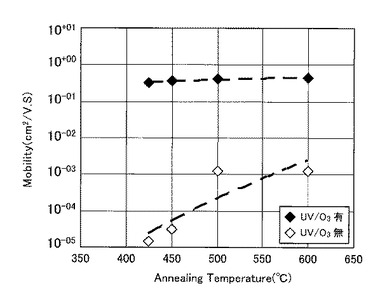
【図27】
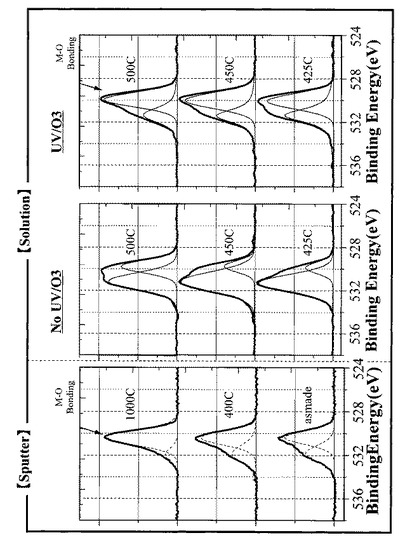
【図28】
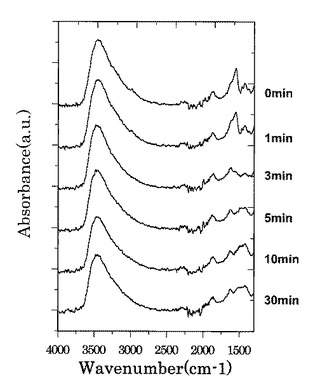
【図29】
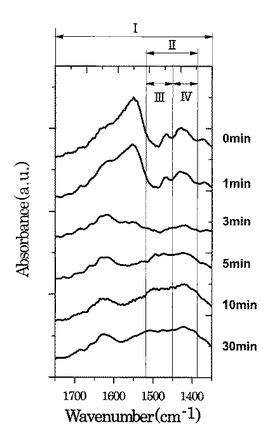
【図30】
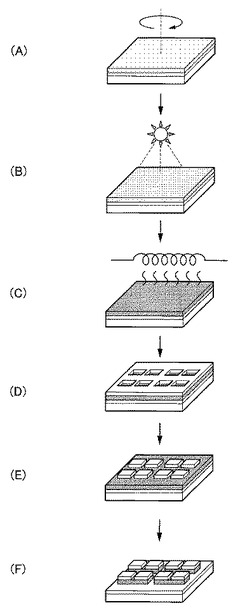
【図33】
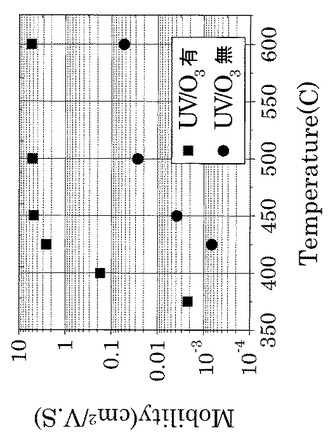
【図34】
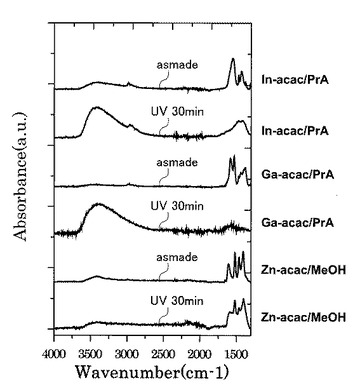
【図11】
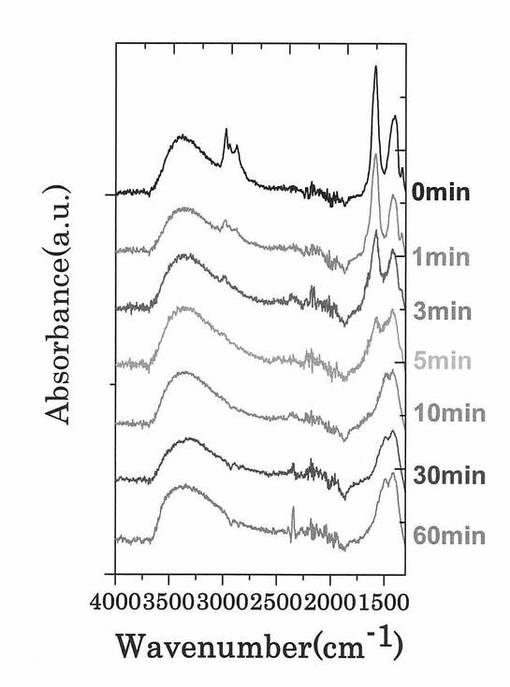
【図13】
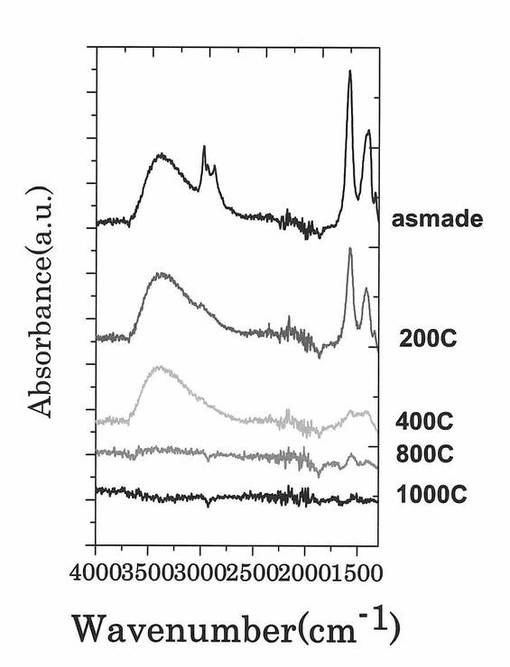
【図31】
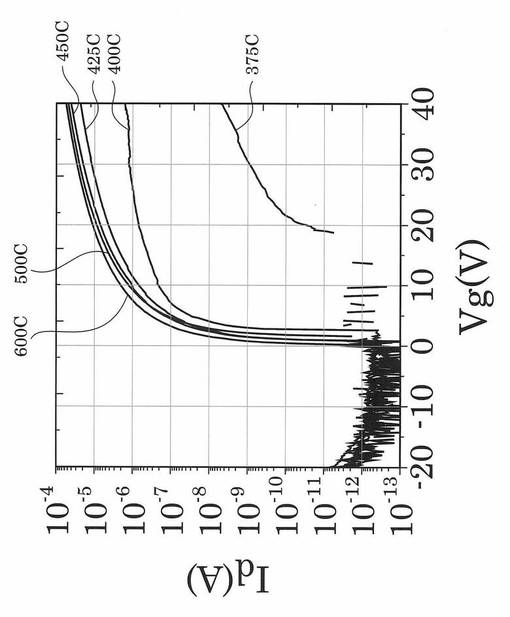
【図32】
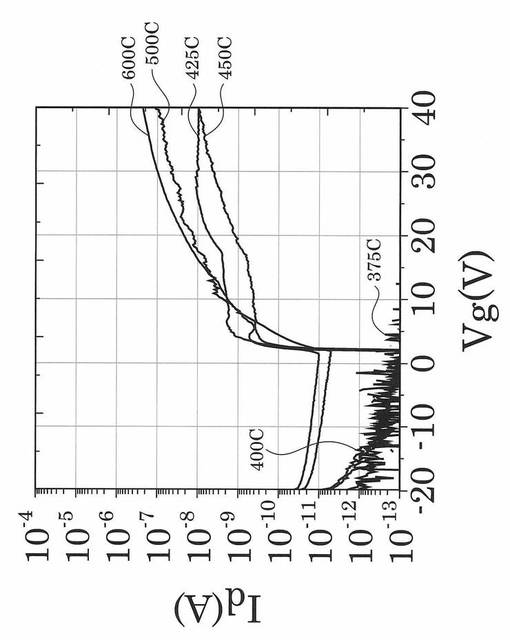
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【図12】
【図14】
【図15】
【図16】
【図17】
【図18】
【図19】
【図20】
【図21】
【図22】
【図23】
【図24】
【図25】
【図26】
【図27】
【図28】
【図29】
【図30】
【図33】
【図34】
【図11】
【図13】
【図31】
【図32】
【公開番号】特開2013−18696(P2013−18696A)
【公開日】平成25年1月31日(2013.1.31)
【国際特許分類】
【出願番号】特願2012−11270(P2012−11270)
【出願日】平成24年1月23日(2012.1.23)
【出願人】(306037311)富士フイルム株式会社 (25,513)
【出願人】(503360115)独立行政法人科学技術振興機構 (1,734)
【Fターム(参考)】
【公開日】平成25年1月31日(2013.1.31)
【国際特許分類】
【出願日】平成24年1月23日(2012.1.23)
【出願人】(306037311)富士フイルム株式会社 (25,513)
【出願人】(503360115)独立行政法人科学技術振興機構 (1,734)
【Fターム(参考)】
[ Back to top ]