
風疹ウイルスに特異的な抗体
本発明は、風疹ウイルス(Rubella virus:RuV)に結合し、中和する新規な抗体配列を提供する。この新規な配列は、RuV感染の医学的管理、特に、このウイルスを検出するために、又は医薬組成物を製造するために使用することができる。このような抗体配列によって認識されるRuV特異的抗原は、ペプチドを提示する新規なファージライブラリーを用いて同定することができる。
【発明の詳細な説明】
【技術分野】
【0001】
発明の分野
本発明は、ウイルスに結合し、ウイルスに特異的な活性を中和するファージディスプレイライブラリーから単離された新規な抗体配列に関する。
【背景技術】
【0002】
発明の背景
ファージディスプレイ技術は、細胞の表面に発現され、標的と相互作用後に生物学的機能を発揮し得るポリペプチド(抗体フラグメント、生物活性ペプチド、酵素など)をクローニングし、選択し、操作するためのバクテリア細胞(例えば、大腸菌細胞)を感染させる線状ファージ(M13など)のゲノムの小さなサイズ及び適応性を利用する。
【0003】
いくつかのクローニング及び発現戦略、ベクター、ライブラリー、ファージを繁殖させる方法、スクリーニングアッセイは、異なる用途のために開発されている。論文(Bradbury A and Marks J,2004;Mancini N et al.,2004;Conrad U and Scheller J,2005;Hust M and Dubel S,2005)及び書籍(“Phage display:A Practical Approach”,vol.266,ed.Clackson and Lowman H,Oxford Univ.Press,2004;“Phage Display:A Laboratory Manual”,ed.Burton D et al.,CSHL Press,2001)に概説されている。
【0004】
ファージディスプレイライブラリーは、組換えファージの集団によって形成され、各々は、その表面上にタンパク質配列のレパートリーの1つの要素を提示する。特定のタンパク質を発現するファージは、反復親和性及び/又は活性に基づく選択プロセス(「パンニング」)によってライブラリーから単離することができる。例えば、タンパク質は、生物学的及び機能的アッセイにおいて、抗原に対する特定の結合親和性又は活性に基づいて、単離され、特徴付けることができる、可変重鎖/軽鎖ヘテロ二量体(通常、Fabと称される)又は一本鎖フラグメント可変部(scFv)の形態で抗体フラグメントであってもよい。
【0005】
特に、スクリーニングプロセスは、病原体及び生物学的標的物に高親和性及び特異性を有する抗体フラグメントを同定するために開発されつつあり、時として、関連する生物学的活性は、このような結合特性と関係している。実際に、全体の治療アプローチ(受動免疫療法又は受動血清療法と称される)は、ヒト又は非ヒト治療標的物に指向される抗体及び抗体フラグメントの抗原結合特性に基づいている(Dunman P and Nesin M,2003;Keller M and Stiehm E,2000)。受動免疫療法は、病原性抗原(毒素、ヒトタンパク質、ウイルス、又は寄生生物など)に対する所定の結合特性を有する治療用抗体を含む医薬組成物の個体への投与からなる。
【0006】
受動免疫療法は臨床診療に導入され、広範囲な疾患(例えば、感染症、免疫を媒介した疾患及び癌)の治療のための機会を急速に拡張する。このアプローチは、標的化された分子をブロックし、及び/又は排除するために必要とされる量で及び/又は特異性をもって、免疫系がそれらを生産することができない患者において特に効果的であり得る(Chatenoud L,2005;Laffly E and Sodoyer R,2005)。
【0007】
治療用抗体を用いて標的化され得る病原性抗原のうち、ヒト細胞に影響を与えるウイルスが特に重要である。このような抗体の投与は、患者におけるウイルスの繁殖を阻害し、集団におけるウイルス感染の大発生を潜在的にブロックすることができる。あるいは、抗体は、免疫応答性個体の健康に重篤及び/又は持続的な結果を通常な有しないものを含む感染症に非常に感受性となる多かれ少なかれ延長された期間、免疫系が弱まった患者(例えば、免疫抑制された個体、高齢者、又は移植された個体)に投与されてもよい。
【0008】
風疹ウイルス(RuV、三日麻疹)は、このようなウイルスの一例である。RuVは、RNAゲノム、2つの非構造的タンパク質、宿主由来の脂質二重層に組み合わせられる3つのウイルス構造タンパク質(E1、E2、及びC)によって形成されるビリオンエンベロープを提示するトガウイルスファミリーのメンバーである(Banatvala J and Brown D,2004;Lee J and Bowden D,2000;“Rubella Viruses”Perspective in Medical Virology,Ed.Banatvala J and Peckham C,vol.15,2006,Elsevier)。
【0009】
RuVは、感染した個体の鼻咽頭分泌物、血液、排泄物、尿に存在し、呼吸噴霧を介して、ヒトからヒトへと伝染される。Ruvは、神経向性ウイルスであり、初期には、神経系細胞(例えば、乏突起膠細胞)に感染するが、RuV菌株は、種々の細胞型、特に神経及び関節組織(例えば、滑膜細胞)において感染し、複製し、持続する能力において変化する。ウイルス性アルトロトロピスム(arthrotropism)は、関節症状とのRuV感染の関係を説明することができる(Lund K and Chantler J,2000;Masuko−Hongo K et al.,2003)。RuVは、感染した細胞における細胞変性効果、アポトーシス、細胞周期停止を誘導する。実際には、RuVタンパク質は、タンパク質−タンパク質相互作用、遺伝子発現、及びタンパク質リン酸化と干渉することによって、宿主の細胞シグナル伝達及び代謝を修飾する(Hofmann J et al.,1999;Cooray S et al.,2005;Atreya C et al.,2004;Domegan L and Atkins G,2002;Adamo M et al.,2008;Figuereido A et al.,2000)。
【0010】
RuVは、一般に、軽度の疾病に関与し、曝露後の16〜20日間で発症し、主に顔及び四肢に観られる微熱及び多発を有する。また、RuV感染は、全くの無症状である場合がある。RuVは、成人では、合併症、例えば感染後の脳障害、血小板減少性紫斑病、出血兆候、又は関節炎をめったに引き起こさない(Banatvala J and Brown D,2004)。また、RuV感染は、一連の眼障害、例えば、フックス虹彩毛様体炎に関与している(De Groot−Mijnes J et al.,2006)。
【0011】
しかしながら、RuV感染は、感染した患者が妊娠女性、特に、発展途上国及び移民集団である場合には、主要な関心事になる。母体感染が必ずしも胎児感染に続かないとしても、胎盤障壁を横断し、胎児組織を感染する能力を有する。妊娠の最初の12週間でのRuV感染は、80〜90%の症例において、胎児の死亡、又は様々な異なる以上をもたらし、通常、Congenital Rubella Syndrome(CRS)の定義下で分類され、例えば、心臓欠陥及び眼の欠陥、難聴、及び精神発達遅滞などが挙げられる。妊娠のその次の週に感染が見られる場合にはその危険性は減少するが、ウイルス性奇形発生を起因とした胎児障害も存在する場合がある(Hinman A et al.,2002;Lee J and Bowden D,2000;Atreya C et al.,2004)。さらに、偽陽性からの最近のRuV感染に起因する抗体、又は感染若しくはワクチン接種後の何カ月も持続している抗体に起因する抗体を区別することを可能にする血清診断アッセイは、十分に信頼できるわけではない(Andrews J,2004;Mendelson E et al.,2006)。
【0012】
RuVワクチン接種は、工業世界において幅広く確立され、CRS及びRuV誘導の胎児死亡の減少に非常に効果的である。それにもかかわらず、最近の大発生における野生型RuVゲノムの遺伝的特徴は、さらに、初期介入について、世界保健機関によって注意深く調査されている。この調査は、参照RuVの遺伝子型及び菌株の定義、並びに世界的な大発生において同定されたRuV菌株の感染的特徴及び遺伝的特徴の分析及び分類を伴う(WHO2005;WHO 2006;Reef S et al.,2002;Zheng D et al.,2003)。
【0013】
実際に、いくつかの試験によって、発展途上国の個体の無視できない割合(少なくとも5%)は、RuVに対する低い又は検出できない抗体濃度を有することが示された。これらの対象は、ワクチン接種してから数カ月又は数年後の集団、及びワクチン接種プログラムへのアクセスが限定された最近の移民又は難民において検出された。このようにして、RuV感染、及びCRSの可能性を防止するために、思春期に近づいている特定の女子、妊娠可能年齢の女性における特異免疫の衰退に起因した、RuV感染に対する追加的処置については、非常に大多数の候補がある(Pebody R et al.,2000;Nicoara C et al.,1999;Matter L et al.,1997;Ki M et al.,2002;Ushida M et al.,2003;Banerji A et al.,2005;Rota M et al.,2007;Mendelson E et al.,2006)。疫学の研究は、RuVの日常的な子供及び成人のワクチン接種、調査、並びに選択的な再免疫化のためにプログラムを確立することの重要性を示唆している(Greenaway C et al.,2007;Kremer J et al.,2007;Semerikov V et al.,2000;Banatvala J and Brown D,2004;Chakravarti A and Jain M,2006;Am.Acad.Pediatr.Com.Infect.Dis.,2007)。
【0014】
RuV病因及び免疫生物学は、ヒト及び動物モデルにおけるRuV感染及びワクチン接種への免疫応答の機序及び効率に関連して研究されている。RuV感染に対する細胞系モデルが確立されている(Cusi M et al.,1995;Duncan R et al.,1999;Garbutt M et al.,1999;Cordoba P et al.,2000a)。一次RuV感染及びRuV特異的免疫応答の初期検出に対する高感度試験が開発されている(Takahashi S et al.,1998;Tzeng W et al.,2005;Giessauf A et al.2004;Wilson K et al.2006)。しかしながら、ワクチン接種は、有効な免疫応答を備えるになるには数週間又は数カ月を要する場合があり、胎児は、その期間中、RuV感染に対してなおも脆弱である。RuV感染は、免疫抑制された患者においては危険であり得て、ワクチンの使用は必ずしも望ましいわけではない。
【0015】
RuVに対して有用であり得る化学物質は、まだ開発の初期段階にある(Mugnaini C et al.,2007)。しかしながら、化学療法及び抗ウイルス治療は、RuVなどのウイルスに対する免疫保護を非常に喪失させ得て、再度のワクチン接種を必要とする場合があることが知られている(Yu J et al.,2007;Bekker V et al.,2006;Van Tilburg C et al.,2006;Nilsson A et al.,2002)。有意な抗RuV力価を有するヒト抗体の生産物は、感染を治療又は予防するために投与することができる(Keller M and Stiehm E,2000;Krause I et al.,2002;Kawamura N et al.,2000)が、同様のアプローチの有効性は、組換え抗体フラグメント又はモノクローナル抗体などの、ウイルスを中和するためのより特異的な免疫学的生産物を用いることによって改善されたであろう。
【0016】
RuV、特にこのウイルスの構造タンパク質に対するヒト抗体応答は、RuVに対してワクチン接種された個体又は野生型RuV菌株によって感染した個体の血清を用いて評価され、異なる対象における特異性及び応答の中和特性において実質的な差異を示した(Zhang T et al.,1992;Mauracher C et al.,1992;Mitchell L et al.,1992;Zrein M et al.,1993;Mitchell L et al.,1993,Thomas H et al.,1993;Banerji A et al.,2005)。赤血球凝集能及び異なる抗体の中和ドメインは、構造タンパク質E1及びE2として与えられた(Mendelson E et al.,2006;Cordoba P et al.,2000a;Green K and Dorsett P,1986)。また、RuV免疫病理学は、RuV由来の扁桃腺フラグメントを移植された(Perrenoud G et al.,2004)、又は腹腔を介したRuVを用いて感染させた(Wang Z et al.,2003)、重症複合型免疫不全症(SCID)マウスを用いた動物モデルにおいて試験された。
【0017】
RuVに結合し、(数例において)RuVを中和することができるいくつかのマウスモノクローナル抗体を生じた。抗体は、細胞におけるウイルス感染及び複製を試験するために、異なるRuV関連標的(ウイルス様粒子、タンパク質抽出物、組換えタンパク質、ペプチド、細胞培養条件で感染させた細胞)を用いて試験された(Green K and Dorsett P,1986;Terry G et al.,1988;Wolinsky J et al.,1991;Chaye H et al.,1992;Claus C et al.,2006;Qiu Z et al.,1994;Ou D et al.,1992;Wolinsky J et al.,1993;Robinson K et al.,1995;Cordoba P et al.,2000a;Cordoba P et al.,2000b;Cordoba P et al.,1997;Starkey W et al.,1995;Giessauf A et al.,2005;Orellana A et al.,1999;Lee J et al.,1999;Hofmann J et al.,2000;EP299673;WO93/14206;WO91/02748;WO95/09232)。
【0018】
文献では、ヒトモノクローナル抗RuV抗体は非常に少なく、関連した抗RuV中和抗体を有するものがないことが報告されている。このような抗体は、細胞融合及び/又はEBV感染によって不死化されたヒトB細胞を用いて同定された(WO07/011698、クローンR335.6.4を参照されたい;Steenbakkers P et al.,1992;Hilfenhaus J et al.,1986)。RuVに対するヒトFab抗体フラグメントが同定されている(Williamson R et al.,1993)。
【0019】
ワクチン接種は主な改善をもたらすが、疾患は持続する。RuVに効率的に結合し、中和する新規なヒト抗体及び抗体フラグメントの同定及び生産は、集団におけるこの感染症の診断、治療及び/又は予防のための改善された処置を確立するためになおも特に重要である。
【発明の概要】
【課題を解決するための手段】
【0020】
本発明は、RuVに結合し、中和し、RuV感染を検出、治療、阻害、予防、及び/又は改善するために使用可能な新規な抗体配列を提供する。
【0021】
ヒト抗体フラグメントの一団は、組換えファージ上に提示され、RuV特異的結合特異性は、ファージライブラリーにおいて検出されている。2つの抗体フラグメントの重鎖及び軽鎖可変領域をコードし、RuVを中和する抗体を有するDNA配列が同定され、DDF−RuV1及びDDF−RuV2と命名された。RuV特異的生物活性に関連する対応するタンパク質配列及び相補性決定領域(CDR)が決定された。これらのFabの結合活性は、さらに、組換えファージ上に提示さえる新規なペプチドライブラリーを用いて試験することができる。新規なタンパク質は、単離されたCDR、並びにDDF−RuV1及びDDF−RuV2に対する可変領域との同一性パーセントに基づいて定義される。
【0022】
本発明の核酸は、適切な技術、及び組換えファージ、原核宿主細胞、又は有核宿主細胞を用いて、全長の抗体、抗体フラグメント、生物活性なペプチド、又は機能性タンパク質(特に融合タンパク質)の任意の他のフォーマットの形態で、RuV特異的結合特性及び中和特性を有する組換えタンパク質を生産するために使用することができる。
【0023】
本発明のタンパク質は、RuV感染の治療検出、治療、阻害、予防、及び/又は改善のために用いることができる。特に、RuV感染の管理における治療、予防、及び/又は診断の利用性を有する組成物は、それらのRuV特異的結合特性及び中和特性を与えるこれらのタンパク質を用いて調製可能である。これらの組成物は、抗ウイルス化合物又は静脈内免疫グロブリン(IVIg)調製物に基づいているRuV治療を補足又は置換するために用いられてもよく、眼内又は局所投与に適していてもよい。
【0024】
本発明の更なる態様は、単離されたDNA及びタンパク質配列、ベクター、組換えファージ、及び宿主細胞、並びに医学的方法、組成物、及び使用を含み、下記の説明に提供される。
【図面の簡単な説明】
【0025】
【図1】DDF−RuV1及びDDF−RuV2、又は負の対照として使用された無関係なヒトFab(e137)を発現している組換えファージの調製物に対するRuV結合活性の特異性。結合活性は、プレートコーティング用の指示された抗原を用いたELISAにおいて測定された。(A)RuVの臨床単離物で感染させた線維芽細胞株(VERO細胞)からの全タンパク質抽出物をプレートコーティングに使用した。各Fabについての2つのカラムは、2つの異なる実験を意味する。(B)RuVの臨床単離物で感染させた線維芽細胞株からのタンパク質抽出物、未感染細胞からのタンパク質抽出物、又は非特異的な精製タンパク質(ウシ血清アルブミン、BSA)をプレートコーティングに使用した。
【図2】(A)DDF−RuV1の重鎖に関する可変領域のDNA(下段ケース)及びタンパク質(上段ケース)の整列(DDF−RuV1 VH;配列番号1及び2)。予測されるCDR(DDF−RuV1 HCDR1、HCDR2、及びHCDR3;配列番号3、4、及び5)に下線を引く。(B)DDF−RuV1の軽鎖に関する可変領域のDNA(下段ケース)及びタンパク質(上段ケース)の整列(DDF−RuV1 VL;配列番号6及び7)。予測されるCDR(DDF−RuV1 LCDR1、LCDR2、及びLCDR3)に下線を引く。
【図3】(A)DDF−RuV2の重鎖に関する可変領域のDNA(下段ケース)及びタンパク質(上段ケース)の整列(DDF−RuV1 VH;配列番号8及び9)。予測されるCDR(DDF−RuV2 HCDR1、HCDR2、及びHCDR3;配列番号10、11、及び12)に下線を引く。(B)DDF−RuV2の軽鎖に関する可変領域のDNA(下段ケース)及びタンパク質(上段ケース)の整列(DDF−RuV2 HC;配列番号13及び14)。予測されるCDR(DDF−RuV2 LCDR1、LCDR2、及びLCDR3)に下線を引く。
【図4】部分的に精製されたヒト組換えFabとして発現され、ファージコートタンパク質に提示された場合のヒトFab DDF−RuV1(A)及びDDF−RuV2(B)に対するRuV中和活性。投薬量応答分析は、免疫蛍光法によって測定されるRuV感染細胞の数に基づいた。パーセント値は、いずれのFabなしに前培養されたRuVを用いて得られたRuV感染細胞(負の対照)に対するデータと比較することによって計算された。
【図5】(A)pDLac−RuV1−FLAGhisベクターを用いて発現されるヒトFab DDF−RuV1の重鎖のタンパク質配列(DDF−RuV1 HCtag;配列番号15)。最初にクローニングされたこの重鎖の改変領域(配列番号2)に下線を引く。PelB配列は、アミノ酸1と22との間に含まれる。アミノ酸147〜252は、ヒトIgガンマ−1鎖C領域のアミノ酸1〜106に対応する(SwissProt Acc.No.:P01857)。アミノ酸253〜267は、FLAGhis配列に対応する。(B)pDLac−RuV1−FLAGhisベクターを用いて発現されるヒトFab DDF−RuV1の軽鎖のタンパク質配列(DDF−RuV1 LC;配列番号16)。最初にクローニングされたこの軽鎖の可変領域(配列番号7)に下線を引く。PelB配列は、アミノ酸1と22との間のアミノ酸に含まれる。アミノ酸135〜240は、Igカッパ鎖C領域のアミノ酸1〜106に対応する(SwissProt Acc.No.:P01834)。(C)pDLac−RuV2−FLAGhisベクターを用いて発現されるヒトFab DDF−RuV2の重鎖のタンパク質配列(DDF−RuV2 HCtag;配列番号17)。最初にクローニングされたこの重鎖の可変領域(配列番号9)に下線を引く。PelB配列は、アミノ酸1と22との間のアミノ酸に含まれる。アミノ酸146〜251は、ヒトIgガンマ−1鎖C領域のアミノ酸1〜106に対応する(SwissProt Acc.No.:P01857)。アミノ酸252〜266は、FLAGhis配列に対応する。(D)pDLac−RuV2−FLAGhisベクターを用いて発現されるヒトFab DDF−RuV2の軽鎖のタンパク質配列(DDF−RuV2 LC;配列番号18)。最初にクローニングされたこの軽鎖の可変領域(配列番号14)に下線を引く。PelB配列は、アミノ酸1と22との間のアミノ酸に含まれる。アミノ酸129〜234は、Igカッパ鎖C領域のアミノ酸1〜106に対応する(SwissProt Acc.No.:P01834)。配列は、重鎖又は軽鎖の定常領域内に設計されたプライマーを用いたインサートの配列決定によって確かめられた。
【図6】(A)Ph.D.−12ファージディスプレイ・ペプチドライブラリーキット(New England Biolabsウェブサイト:http://www.neb.com/nebecomm/products/productE8110.aspから入手可能)についえの指示マニュアルにおいて、生産者によって提供されるランダム12マーのペプチド−cp3融合ライブラリーの配列。M12 XhoI−フォワード(配列番号19)及びM13 SpeI−リバース(配列番号20)プライマーにおける関連制限部位の位置を矢印で示す。M13 KpnI−フォワード(配列番号21)及びM13 EagI−リバース(配列番号22)プライマーにおける関連制限部位の位置に下線を引く。(B)pDDcXSmer−orcp3ライブラリー内でクローニングされた場合のPh.D.−12ライブラリー(PhD12マー)のコーディング配列(WO07/007154の図3から修飾される;配列番号25)。最初のファージミドによって提供される配列の位置(PelBシグナルペプチド、DISリンカー、及びp3*5’末端)が示される。関連制限部位に下線を引く。14個のアミノ酸のリンカー配列は、ペプチドライブラリーとcp3*のN末端近傍(by)との間に位置される。(C)pDDcKE12mer−orcp3ライブラリー内でクローニングされた場合のPh.D.−12ライブラリー(PhD12マー)のコーディング配列(WO07/007154の図3から修飾される;配列番号26)。最初のファージミドによって提供される配列の位置(PelBシグナルペプチド、DISリンカー、及びp3*5’末端)が示される。関連制限部位に下線を引く。18個のアミノ酸のリンカー配列は、ペプチドライブラリーとcp3*のN末端近傍(by)との間に位置される。KpnI及びEagI部位は、XhoI及びSpeIを用いたpDDcの消化、並びにHAtag配列、KpnI部位、EagI部位を含む2つのアニール化されたオリゴヌクレオチドの挿入によって導入され、XhoI及びSpeI部位に適合可能な一本鎖5’末端を含む(配列番号23及び24)。また、ベクターは、DDカセットにクローニングされるZeocinマーカー遺伝子内のEagI部位を排除するために、PCRによって修飾した。
【図7】pDDcXSmer−orcp3ライブラリー、及びpDDcXSmer−orcp3/cp8ライブラリーの生成の略図(WO07/007154の図5から修飾される)。ファージミドマップは、コートタンパク質(cp3*及びcp8*)の位置、ファージミド骨格内のアンピシリンマーカー遺伝子(Ampr)及び複製起源(ColE1及びF1(+))を示す。下記の要素は、DDカセットに存在する(pDDcXSmer−orcp3/cp8ライブラリーを生成するために使用されるBglI/SfiI部位間においてクローニングされた):Ph.Dペプチドライブラリー(Lib)、PelB配列に融合されたpLacZプロモーター(Lpe)、スタッファー配列(St)及びZeocinマーカー遺伝子(Zeor)。関連制限部位の位置を矢印で示す。pDDcKE12mer−orcp3ライブラリー及びpDDcKE12mer−orcp3/cp8ライブラリーは、追加のKpnI及びEagI制限部位を提示する(図6Cを参照されたい)。
【図8】いずれかのパンニングなしのpDDcXSmer−orcp3/cp8ライブラリー(A)、3ラウンドのパンニング後のpDDcXSmer−orcp3/cp8ライブラリー(B)3ラウンドのパンニング後のpDDcKE12mer−orcp3/cp8ライブラリー(C)から無作為に選択されるクローンの消化。単離された細菌クローンは、NheI及びSpeIを用いて消化されたファージミドDNAのミニプレップを行うために使用された。反応物は、分離用の1%アガロースゲル上にロードされ、得られたフラグメント(NS)と対応する未消化のDNA(C)と比較した。3.8及び0.6Kbフラグメントを示すクローンは、cp3*に配向されたDDカセットを有する。2.6及び1.8Kbフラグメントを示すクローンは、cp8*に配向されたDDカセットを有する。矢印は、マーカーDNAによって位置される。
【図9】下記のライブラリー:pDDcXSmer−orcp3(A;配列番号27〜34)、pDDcKE12mer−orcp3(B;配列番号35〜42)において無作為に選択されたクローン由来の配列例。アンバー停止コドン(tag)は、XL1−Blue E.coli菌株内に抑制され、アミノ酸(Q)をコードする。配列は、cp3*内で設計されたプライマーを用いたインサートの配列決定によって確かめられた。
【図10】抗HAtagコロニースクリーニング。抗HAtag抗体に対するパンニングの3ラウンド後に得られた細菌コロニーは、メンブレンに転写され、アルカリ条件で溶解した。最初のコロニーにおけるHAtagの発現を検出するために、メンブレンをマウス抗HAtag抗体(パンニングのために使用された抗体)、次に、PBS/0.1%Tween−20(1:1000希釈)中のセイヨウワサビのペルオキシダーゼに結合した抗マウスIgG1とともにインキュベートした。PBS/0.1%Tween−20による3回の洗浄後、メンブレンは、Supersignal West Picoケミルミネッセンス基質(Pierce)を用いた増大したケミルミネッセンス検出に供された。
【発明を実施するための形態】
【0026】
発明の詳細な説明
WO07/007154に記載されたpDDファージミド及び関連した方法により、1つの又は他の2つの所定のファージミドコートタンパク質に融合されたタンパク質配列のクローニング、発現、及び選択が可能となる。このアプローチにより、組換えファージの表面に別個に発現されるか又は提示され、結果として、異なる効率を有するファージディスプレイライブラリーから選択され得るタンパク質配列の同定の選択を可能にする。
【0027】
本ケースでは、ファージライブラリーは、ヒト重鎖及び軽鎖の免疫グロブリンの可変領域をクローニングすることによって、pDDファージミドに構築された。このライブラリーは、RuV感染細胞のタンパク質抽出物に対してパンニングされ、選択されたクローンは、その後、RuVを中和する活性を示すクローンを決定するために細胞系圧死において試験された。2つの最大の有望なクローン(DDF−RuV1とDDF−RuV2と命名される)をコードするDNA配列を決定し、次に、細菌発現用の適切なベクターにクローニングされた。
【0028】
本発明は、RuVに結合及び中和することができ、Fab DDF−RuV1又はDDF−RuV2において同定された特定のCDR(相補性決定領域)を含む新規なタンパク質配列を提供する。特に、本発明のHCDR3(重鎖可変領域のCDR3)の各々(配列番号5及び配列番号12)は、それぞれDDF−RuV1及びDDF−RuV2の抗原結合部分を特徴とする。
【0029】
HCDR3は、抗体の抗原結合部分を特徴付け、その結果、生物活性(例えば、RuVの結合及び中和)を与えるものと考えられる。抗体のいくつか又は全てのCDRは、一般に、完全な抗原結合表面を得るために要求されるとしても、HCDR3は、抗体間の最大の相違を示すCDRである。実際に、HCDR3の配列及び長さの多様性は、大部分の抗体に対する特異性を決定するために基本的である(Xu J及びDavies M,2000;Barrios Y et al.2004;Bond C et al.,2003)。
【0030】
RuV結合部分としての本発明の特定のHCDR3を含むタンパク質は、それ(又はなし)と、このようなHCDR3が抗体タンパク質フレームワーク内で同定された同じFab由来の他のCDRとを組み合わせることによって生じさせることができる(Knappik A et al.,2000)。CDRの組み合わせは、抗体構造には関連しないタンパク質フレームワーク内であっても、元々の結合活性を破壊せずに、元々の結合性を維持する非常に短いタンパク質において互いに連結することができる(Kiss C et al.,2006;Smith J et al.,1995)。
【0031】
一態様では、本発明は、Fab DDF−RuV1のHCDR3(配列番号5)と少なくとも90%同一性を有する配列を含むタンパク質を提供する。HCDR1及びHCDR2(配列番号3及び配列番号4、図2A)と一緒に、このHCDR3は、DDF−RuV1 Fabの重鎖の可変領域(DDF−RuV1 VH;配列番号2)に含まれる。このFabを形成する軽鎖の可変領域(DDF−RuV1 VL:配列番号7)、並びにその特定のLCDR(軽鎖可変領域のCDR)が決定されている(図2B)。
【0032】
別の態様では、本発明は、Fab DDF−RuV2のHCDR3(配列番号12)と少なくとも90%同一性を有する配列を含むタンパク質を提供する。HCDR1及びHCDR2(配列番号10及び配列番号11、図3A)と一緒に、このHCDR3は、DDF−RuV2の重鎖の可変領域(DDF−RuV2 VH;配列番号9)に含まれる。このFabを形成する軽鎖の可変領域(DDF−RuV1 VL;配列番号14)、並びにその特異的LCDRが決定されている(図3B)。
【0033】
本発明のタンパク質がDDF−RuV1の配列に基づく場合、それらは、配列番号5と少なくとも90%の同一性を有する配列を含まなければならない。特に、それらは、配列番号2と少なくとも90%同一性を有する配列を含まなければならない。より特には、このようなタンパク質はまた、配列番号3及び配列番号4からなる群から選択される1以上の配列を含む。タンパク質、特に、DDF−RuV1に基づく抗体及び抗体フラグメントは、配列番号7と少なくとも90%同一性を有する配列をさらに含むことができる。
【0034】
あるいは、本発明のタンパク質がDDF−RuV2の配列に基づく場合、それらは、配列番号12と少なくとも90%の同一性を有する配列を含まなければならない。特に、それらは、配列番号9と少なくとも90%同一性を有する配列を含まなければならない。より特には、このようなタンパク質はまた、配列番号10及び配列番号11からなる群から選択される1以上の配列を含む。タンパク質、特に、DDF−RuV2に基づく抗体及び抗体フラグメントは、配列番号14と少なくとも90%同一性を有する配列をさらに含むことができる。
【0035】
本発明の更なる態様は、両方のFabの重鎖及び軽鎖の可変領域をコードするDNA配列、特に、クローニングされ、DDF−RuV1(重鎖については配列番号1;軽鎖については配列番号6)、並びにDDF−RuV2(重鎖については配列番号8;軽鎖については配列番号13)の可変領域が決定されている元々のDNA配列と少なくとも90%同一性を有するものである。
【0036】
これらのDNA配列(又は選択された部分、例えば図2及び図3に示された単離されたHCDRとLCDRをコードするもの)は、組換え抗体(例えば、完全、アフィニティー成熟、CDRグラフト、または図5においてDDF−RuV1及びDDF−RuV2のタグ化されたバージョンについて示されたフラグメント)、又はRuV結合性及び中和性を与える融合タンパク質についての既知のフォーマットの1つ内にそれらを発現するための他のベクターに移すことができる。
【0037】
同一性レベルが指示されるところはどこでも、この同一性レベルは、本発明の関連配列の全長に基づいて決定されなければならない。
【0038】
重鎖及び軽鎖を形成するDDF−RuV1又はDDF−RuV2の可変領域(又は、選択された部分、例えば、単離されたHCDR及びLCDR)は、特定のアイソタイプを有する抗体、特に完全なヒト組換え抗体内に含むことができる。この抗体は、2つの軽鎖及び2つの重鎖によって形成される四量体複合体の天然の構造における軽鎖及び重鎖可変領域として、DDF−RuV1又はDDF−RuV2のVL及びVH配列を含んでもよい。完全なヒト抗体が望まれる場合、固体は、ヒトIgG1、IgG2、IgG3、IgG4、IgM、IgA及びIgE定常領域からなる群から選択される重鎖定常領域をさらに含まなければならない。例えば、IgGアイソタイプは、ほとんど全ての承認された治療用抗体の抗体フォーマットである(Laffly E及びSodoyer R,2005)。しかしながら、ヒトIgG1から単離された抗原結合部分は、ヒトIgA配列に移され、得られた組換え抗体は元々のIgG1の活性を維持し、最近示されるように、抗体はHIV感染を阻害することができる(Mantis Nら,2007)。
【0039】
あるいは、DDF−RuV1又はDDF−RuV2を形成する重鎖及び軽鎖の可変領域(又は、選択された部分、例えば単離されたHCDR及びLCDE)は、機能性抗体フラグメントについての任意の他のタンパク質フォーマットにおいて組み合わせることができ、異なる名称で文献に記載されているように、例えば、Scfv(一本鎖フラグメント可変)、Fab(可変重鎖/軽鎖ヘテロ二量体)、ダイアボディー、ペプタボディー、VHH(重鎖抗体の可変ドメイン)単離された重鎖又は軽鎖、二重特異性抗体、非/臨床用途のための他の操作された抗体変異体などがある(Jain M et al.,2007;Laffly E and Sodoyer R,2005)。例えば、DDF−RuV1又はDDF−RuV2の組換え改変体は、pDLac−FLAGhis(PCT/IB2008/000266)と呼ばれるpDD適合可能な発現ベクターにクローニングされ、タグ化されたFab(DDF−RuV1については、配列番号15〜16、図5A〜B;DDF−RuV2については、配列番号17〜18、図5C〜D)の形態で発現可能である。
【0040】
代替の抗体及び抗体フラグメントは、軽鎖をシャッフルするためのプロセスを介して、DDF−RuV1又はDDF−RuV2の配列を用いて生じさせることができる。実際に、異なる抗体及び抗体フラグメントを生じさせ、例えば、共通のファージディスプレイ技術又はWO07/007154に記載されたものを用いて、VL配列のライブラリーと合わせられる単一の重鎖可変ドメインVH(例えば、DDF−RuV1又はDDF−RuV2のいずれか1つ)を用いたRuV特異的活性について試験することができる。このアプローチは、親和性、安定性、特異性、及び/又は組換え産物に基づいて改善された特性を用いてVH/VLを決定してもよい(Rojas G et al.,2004;Suzuki K et al.,2007)。
【0041】
さらに、抗体は、特に臨床用途(良好な薬物動態プロファイル又は抗原に対するより高い親和性)のために、改善された特徴を有する抗体を持つために、特定の一で修飾されてもよいことは知られている。これらの変化は、DDF−RuV1又はDDF−RuV2のいずれかのCDR及び又はフレームワークのいて行うことが可能である。配列は、親和性成熟の使用を作る抗体の合理的な設計のための任意の専用技術及び他の方法を適用することによって測定することができる(Kim S et al.,2005;Jain M et al.,2007)。
【0042】
また、新規な生物活性ペプチドを開発するための抗体に基づくストラテジーは、L−アミノ酸及び/又はD−アミノ酸を含むCDR誘導ペプチドを合成する実行可能性を示した。これらの代替の分子は、より適切な薬理学的プロフィールを有する元々の活性を維持することができる(Levi M et al.,2000;Wijkhuisen A et al.,2003)。このようにして、各々の新規なHCDR3、並びにそれらに非常に類似した配列、それらを含む融合タンパク質、それらから誘導される合成ペプチド(例えば、D−アミノ酸を含むか又はレトロー−インバーソ型形態である)が試験され、RuV結合タンパク質として使用することができる。
【0043】
また、本発明のタンパク質は、RuVビリオンの抗原を中和することを特徴付けるために使用されてもよい。実際に、DDF−RuV1及びDDF−RuV2は、ELISAにおけるRuV感染細胞株由来の細胞抽出物へのそれらの特異的結合に奇異真して初期にクローニングされ(図1)、その後、RuV感染を中和するそれらの能力は、RuV臨床菌株を用いたインビトロの中和アッセイによって測定された(図4)。結論として、本発明のタンパク質は、RuV感染を中和することができ、任意の関連した結合アッセイ(例えば、実施例に記載されるELISA)によって測定されるように、本発明のタンパク質と比較する他のRuV結合タンパク質(完全な句及び体、抗体フラグメント、生物活性ペプチド、又は融合タンパク質の形態で)を特定するために用いることができる。このような競合タンパク質は、場合により、DDF−RuV1又はDDF−RuV2配列において同定されたものとは、部分的に又は完全に異なっている可能性があるHCDR及びLCDRとともに、上記で定義されたHCDR3配列の何れかを単に含んでもよい。
【0044】
本発明のタンパク質は、抗体、抗体フラグメント、生物活性なペプチド、又はRuVに結合及び中和する融合タンパク質として提供される。これらの代替のタンパク質は、DDF−RuV1及びDDF−RuV2 Fabについて決定されるこのような特性を増大しない場合に維持されなければならない。
【0045】
融合タンパク質の特定のケースでは、異種配列(単数又は複数)は、このような部分の正しい発現及び生物活性を負に影響を与えないで、RuVに特異的な結合及び中和する部分(例えば、特定のHCDR3又は抗体フラグメントの可変領域に対してN末端位又はC末端位で位置しなければならない。
【0046】
用語「異種タンパク質」は、タンパク質配列がRuVに特異的に結合及び中和する部分(例えば、抗体フラグメント)に対してN末端位又はC末端位において天然に存在しないことを指示する。このタンパク質配列をコードするDNA配列は、一般に、組換えDNA技術によって融合され、少なくとも5個のアミノ酸をコードする配列を含む。この異種配列は、一般に、特定の診断及び/又は治療用途に関連した追加の特性を提供するために選択される。このような追加の特性の例には、検出又は精製のための良好な手段、追加の結合部分又は生物学的リガンド、又は融合タンパク質の翻訳後修飾(例えば、リン酸化、グリコシル化、ユビキチン化、SUMO化、又は細胞内タンパク質分解的切断)が挙げられる。
【0047】
タンパク質部分、リガンド、適切なリンカーを選択及び設計する手段、並びに融合タンパク質の構築、精製、検出及び使用についての方法及びストラテジーは、文献に提供され(Nilsson J et al.,1997;“Applications Of Chimeric Genes And Hybrid Proteins”Methods Enzymol.Vol.326−328,Academic Press,2000;WO01/77137))、臨床及び研究所において利用可能である。例えば、融合タンパク質は、インビボ及び/又はインビトロでの融合タンパク質の同定、又はその精製を促進することができる市販の抗体(タグ、例えばポリヒスチジン、FLAG、c−Myc、又はHAタグが含まれる;図5)によって認識される配列を含んでもよい。他のタンパク質配列は、直接的蛍光分析(緑色蛍光タンパク質など)によるか、又は特異的な基質又は酵素(酵素分解部位などの使用)によって容易に同定することができる。
【0048】
治療活性は、別の抗ウイルスタンパク質あるいは細胞代謝及び/又は活性を変更するタンパク質などの別の治療タンパク質との融合によって改善され得る。RuV特異的抗体、抗体フラグメント、融合タンパク質の安定性は、周知の担体タンパク質、例えばファージコートタンパク質(cp3又はcp8)、マルトース結合タンパク質(MBP)、ウシ血清アルブミン(BSA)、又はグルタチオン−S−トランスフェラーゼ(GST)をm値いて改善されてもよい。
【0049】
あるいは(又は、異種タンパク質配列への融合に加えて)、本発明のRuV特異的タンパク質の活性は、治療薬又は診断薬などの異なる化合物との結合を用いて改善されてもよい。これらの試薬の例には、化学リンカー又はポリマーを用いて結合することができる検出可能な標識(例えば、放射性同位体、蛍光化合物、コロイド金属、ケミルミネッセンス化合物、生物発酵化合物、又は酵素)である。本発明のタンパク質のRuV特異的な生物活性はまた、診断又は治療用途における代謝及び/又は安定性を変更するポリマーなdの化合物との融合によって改善されてもよい。
【0050】
本発明のタンパク質(例えば、抗体、抗体フラグメント、生物活性ペプチド、融合タンパク質、又はそれらの結合された改変体)をスクリーニングし、元々のDDF−RuV1及びDDF−RuV2と競合するそれらの能力、可能であれば、実施例又は文献に記載された任意の関連アッセイによって測定される、RuV感染を中和する類似の又はより優れた能力を示すために特徴付けることができる。
【0051】
また、文献は、RuV抗原、並びに各Fabによって認識される特定のエピトープが測定され、過去に測定されたもの比較されるものを用いるいくつかの技術の例を愛知今日する。例えば、RuVタンパク質、関連した一部切断された改変体又は合成ペプチドを用いたELISA、免疫沈降、又はウェスタンブロットは、E1又はE2タンパク質内の関連エピトープを決定するために用いた。特に、本発明の躯体、抗体フラグメント、他のタンパク質は、抗体フラグメント(Williamson R et al.,1993)、マウス又はヒトモノクローナル抗体(Green K and Dorsett P,1986;Cordoba P et al.,1997;Chaye H et al.,1992;Qiu Z et al.,1994;Ou D et al.,1992;Steenbakkers P et al.,1992;Hilfenhaus J et al.,1986;WO07/011698;WO93/14206;WO95/09232)、ヒト血清(Giessauf A et al.,2004;Starkey W et al.,1995;Zrein M et al.,1993;Mitchell L et al.,1992)についてのRuV特異的生物活性及びエピトープを特徴付けるために使用されるアッセイにおいて試験することができる。次に、本発明のタンパク質のRuV関連予防的、診断的、及び治療的使用のためのより広範囲な特徴付け及び評価は、本発明の背景に要約されるように、RuV病因及び免疫学を研究するための文献(Cusi M et al.,1995;Duncan R et al.,1999;Cordoba P et al.,2000;Lee and Bowden,2000;Garbutt M et al.,1999;Ou D et al.,1992;Tzeng W et al.,2005;Perrenoud G et al.,2004;Wang Z et al.,2003)に開示されているインビトロ又はインビボアッセイ(組織又は細胞系アッセイ、齧歯類において確立した感染モデル)の1以上を用いて行うことができる。
【0052】
本発明の更なる目的は、上記で定義される抗体、抗体フラグメント、融合タンパク質又は単離されたCDRのいずれかをコードする核酸である。実施例は、このような配列、特に、DDF−RuV1及びDDF−RuV2の完全な可変領域(配列番号1、6、8、及び13)をコードするものとして提供する。核酸は、配列番号1、配列番号6、配列番号8、及び/又は配列番号13と少なくとも90%同一性を有していなくてはならない。
【0053】
このような配列、特に、特定のCDRに関連したもの含まれる配列(図2及び3)は、場合によりプロモーターに連結されたベクター及び発現カセットに含まれ、又はpDD系ファージミドにクローニングされ得る。この特定のタイプのベクターは、cp3又はcp8ファージコートタンパク質のいずれかとの融合タンパク質としてそれらの発現を可能にし、結果として、このようなファージミドを含み、それらの表面上で本発明のFabを発現する組換えファージを生産することができる。このようにして、本発明のタンパク質を発現するファージミドベクターを含む組換えファージは、RuV感染を検出及び/又は中和するための手段として使用可能である。
【0054】
本発明の配列がクローニングされ、対応する組換えファージによって特徴付けられたpDD系ファージミドがベクターに(部分的に又は全体として)移され得るDNA配列を含み、そこでは、元々のFab、又はそれらから誘導されるタンパク質配列は、宿主細胞内で組換えタンパク質として適切に発現され得る(pDLac−FLAGhisシステムを用いた実施例に示される)。
【0055】
ファージディスプレイ技術を用いて単離される免疫グロブリン可変鎖(特にヒト免疫グロブリン可変鎖)の場合、重要な修飾は、好ましいアイソタイプ及び定常領域を有する完全な免疫グロブリンタンパク質へ選択されたFab又はscFVの変換である。このようにして、完全なヒト抗体が望まれる場合、発現ベクターは、IgG1、IgG2、IgG3、IgG4、IgM、IgA及びIgE定常領域からなる群から選択される重鎖定常領域をさらに含まなければならない。この種の修飾は、例えば、ファージディスプレイライブラリー由来の一本鎖Fv又はFabから構築される全てのアイソタイプの完全なヒトモノクローナル抗体を生じ、哺乳動物細胞又は昆虫細胞において発現を可能にする。文献(Persic L et al.,1997 Guttieri M et al.,2003)に広く記載されているように、ベクターは、抗体を発現するために具体的に設計され、所望のアイソタイプ(例えば、ヒトIgGガンマ1)の定常(Fc)領域へのこの配列の融合を可能にする。
【0056】
抗体又は融合タンパク質は、原核生物(例えば、Escherichia coli;Sorensen H and Mortensen K,2005;Venturi M et al.,2002)、植物(Ma J et al.,2005)、又は真核細胞において組換えタンパク質として発現することができ、それらは、一時的又は安定に形質転換した細胞として高レベルの発現を可能にする(Dinnis D and James D,2005)。これは、特に、抗体の特徴がより厳しい機能的及び/又はインビボアッセイを用いて実行しなければならない場合に要求される。
【0057】
文献は、記事及び書籍の章(”Phage display:A Practical Approach”,vol.266,ed.Clackson and Lowman H,Oxford Univ.Press,2004;”Phage Display:A Laboratory Manual”,ed.Burton D et al.,CSHL Press,2001;Corisdeo S and Wang B,2004;Benhar I,2001)に概説されるように、原核宿主細胞において、Fab又は抗体フラグメントについての同様のフォーマットとしてタンパク質を発現するための異なるストラテジーを提供する。
【0058】
タンパク質、特に抗体が、真核宿主細胞(特に、哺乳動物細胞株)において発現される場合、異なるベクター及び発現系は、トランスフェクトされた細胞株の安定なプールを生じるように設計されている(Aldrich T et al.,2003;Bianchi A and McGrew J,2003)。高レベルの、最適化された、安定な組換え抗体の発現は、細胞培養条件の最適化(Grunberg J et al.,2003;Yoon S et al.,2004)、より高レベルの抗体生産及び分泌を有するクローンの選択又は操作(Bohm E et al., 2004; Butler M, 2005)によって達成される(Schlatter S et al.,2005)。
【0059】
対象とする配列(例えば、重鎖及び軽鎖の関連する可変領域)をコードする核酸配列は、ベクターの発現カセット、又は区別されるベクターの発現カセットに適切にクローニングされなければならず、この場合、それらは、適切な調節配列(例えば、プロモーター、転写ターミーネーター)に操作可能に連結される。発現カセットには、プロモーター、リボソーム結合部位(必要に応じて)、開始コドン、リーダー/分泌配列が含まれ、それらは、したがって、所望のタンパク質をコードするDNAを誘導体スルファターゼ得るモノ又はビシストロン転写物の発現を誘導することができる。
【0060】
ベクターは、構造的に活性であるか又は誘導可能であるように選択される転写開始/末端調節配列の調節下で、原核又は真核宿主細胞において本発明のタンパク質の発現を可能にしなければならない。このようなタンパク質を生産するための方法には、タンパク質発現に適切である条件下で、それらのコーディング配列を含む発現ベクターを用いて形質転換された宿主細胞を培養し、宿主細胞培養物からタンパク質を回収することが含まれる。本発明の核酸を含む宿主細胞は、原核又は真核宿主細胞であってもよく、所望の組換えタンパク質の分泌を可能にする。次に、このような細胞に実質的に富んでいる細胞株は、単離され、安定な細胞株を提供することができる。
【0061】
これらの核酸、組換えファージ、宿主細胞を生じ、共通の組換えDNA技術を適用することによって本発明のタンパク質を生産するために使用することができる。要約すると、所望のDNA配列は、制限酵素を用いたファージミドの消化によって抽出されるか、又はポリメラーゼ連鎖反応(PCR)のための鋳型、重鎖及び軽鎖の完全な可変領域又はそれらの一部だけ(例えばHCDR3)を特異的に増幅するPCRプライマーとして元々のファージミドを用いて増幅することができる。次に、このようなDNAフラグメントは、組換えタンパク質のクローニング法及び生産法についての多数の書籍及び概説に記載されるように、原核又は真核宿主細胞内に更なる修飾及び/又は発現のためにより適したベクターに移すことができ、シリーズのいくつかのタイトルであるオックスフォード大学プレスによって公開されている“A Practical Approach”( “DNA Cloning 2:Expression Systems”,1995;“DNA Cloning 4:Mammalian Systems”,1996;”Protein Expression”,1999;”Protein Purification Techniques”,2001)が含まれる。
【0062】
提示され、選択されたタンパク質配列をコードするDNA配列は、適切なエピソーム又は非相同的若しくは相同的統合ベクターに挿入されると、任意の適切な手段(形質転換、トランスフェクション、コンジュゲーション、原形質融合、エレクトロポレーション、リン酸カルシウム沈殿、直接的なマイクロインジェクションなど)によって適切な宿主細胞に導入可能である。特定のプラスミド又はウイルスベクターを選択する際の重要な因子には、ベクターを含む受領細胞が認識され、ベクターを含まない受領細胞から選択される容易性;特定の細胞において望まれるベクターのコピー数;異なる種の宿主細胞間でベクターを「シャトル」することができることが望まれるかどうかが含まれる。
【0063】
真核宿主(例えば、酵母、昆虫又は哺乳動物細胞)については、異なる転写及び翻訳調節配列が使用されてもよく、これは、宿主の性質に依存する。それらは、ウイルス起源、例えば、アデノウイルス、ウシパピローマウイルス、シミアンウイルスなどから誘導されてもよく、この場合、調節シグナルは、高レベルの発現を有する特定の遺伝子と関連付けられる。実施例は、ヘルペスウイルスのTKプロモーター、SV40初期プロモーター、酵母gal4遺伝子プロモーターなどである。転写初期調節シグナルが選択されてもよく、一時的(又は構造的)抑制及び活性、結果として、遺伝子発現の調節を可能にする。更なるクローニング工程において、抗体又は融合タンパク質をコードする配列が適用され、DNA及びタンパク質レベルの両方でのみDNAレベルの特定の修飾について他のベクターに再度クローニングすることができる。これらの変化は、例えば、コドン使用頻度及び制限部位が特定のベクター及び宿主細胞を用いた組換えタンパク質をクローニングし、発現するのに最も適しているDNA配列を選択するためのソフトウェアを用いて測定することができる(Rodi D et al.,2002;Grote A et al.,2005;Carton J et al.,2007)。また、タンパク質配列は、所望の抗体フォーマット(Scfv、Fab、完全なヒト抗体など)、又は1以上の内部アミノ酸の挿入、置換、若しくは欠失に関連して付加することができる。
【0064】
また、これらの技術は、抗体の治療的特性の更なる構造及び機能的特徴付け及び最適化のために使用され(Kim Sら,2005)、あるいは、それらの安定なインビボでの送達を可能にするベクターを生じさせるために使用可能である(Fang Jら,2005)。例えば、また、組換え抗体は、可変領域に融合されるべき特定のFc領域を選択することによって(Furebring Cら,2002;Logtenberg T,2007)、組換え一本鎖抗体フラグメントを生じさせることによって(Gilliland Lら,1996)、安定化ペプチド配列を融合することによって(WO01/49713)、又は放射化合物若しくはポリマーを化学的に修飾された残基に添加することによって(Chapman Aら,1999)、構造及び/又は活性のレベルで調節可能である。
【0065】
導入されたDNAによって安定に形質転換された細胞は、発現ベクターを含む宿主細胞の選択を可能にする1以上のマーカーを導入することによっても選択することができる。また、マーカーは、栄養要求宿主への光合成、殺生物耐性、例えば抗生物質、又は重金属、例えば銅などを与えてもよく、必要に応じて、開裂可能であり、又は抑制されてもよい。選択可能なマーカー遺伝子は、発現されるべきDNA遺伝子配列に直接的に連結されるか、又は同時トランスフェクションによって同じ細胞に導入されてもよい。また、追加の転写調節因子は、最適発現に必要であってもよい。
【0066】
宿主細胞は、原核又は真核のいずれかであってもよい。原核宿主細胞の中では、好ましいものは、B.subtilis及びE.coliである。真核宿主細胞の中では、好ましいものは、酵母、昆虫細胞(バキュロウイルス系発現システムを使用)、又は哺乳動物細胞、例えばヒト、サル、昆虫(バキュロウイルス系発現システムを使用)及びチャイニーズ・ハムスター・オバレイ(CHO)細胞であり、これは、それらが、正しいフォールディング又は正しい位置でグリコシル化のある種の形態を提供するためである。また、酵母細胞は、グリコシル化を含む翻訳後ペプチド修飾を実行することができる。強力なプロモーター配列、酵母における所望のタンパク質の生産のために利用可能な高コピー数のプラスミドを利用する多数の組換えDNAストラテジーが存在する。酵母は、クローニングされた哺乳動物遺伝子産物においてリーダー配列を認識し、リーダー配列(即ち、プレペプチド)を有するペプチドを分泌する。
【0067】
組換えポリペプチドの長期、高収率な生産に関して、安定な発現が好ましい。例えば、対象とするポリペプチドを安定に発現する細胞株は、同じであるか又は別個のベクターに、複製のウイルス起源及び/又は内因性発現要素、及び選択可能なマーカー遺伝子を含んでもよい。ベクターの導入後、細胞は、それらが選択培地にスイッチされる前に強化培地において1〜2日間増殖させることができる。選択可能なマーカーの目的は、選択への耐性を付与することであり、その存在は、誘導される配列を首尾よく発現する細胞の増殖及び回収を可能にする。安定に形質転換された細胞の耐性クローンは、細胞型に適切な組織培養技術を用いて増殖可能である。次に、このような細胞において実質的に富んだ細胞株が単離され、安定な細胞を提供することができる。宿主細胞は、組換えタンパク質の発現レベルに基づいてさらに選択可能である。
【0068】
RuVを結合及び中和可能である抗体、抗体フラグメント、生物活性ペプチド、融合タンパク質、上記定義された任意の他の化合物は、細胞培養物又は合成調製物からの組換えタンパク質としてそれらを精製可能にする十分に確立した技術(即ち、抽出、沈殿、クロマトグラフィー、電気泳動など)を用いて提供することができる。これらの調製物は、組換えタンパク質の十分な量(ミクログラムからミリグラムの範囲)を提供して、RuVに関連した予防、診断、及び治療用途のためにより広範囲な特徴付け及び評価を行わなければならない。
【0069】
抗体精製の方法は、カラム内に含まれる固相化したゲルマトリックス(Nisnevitch M and Firer M,2001;Huse K et al.,2002;Horenstein A et al.,2003)、特に、基質、例えばプロテインA、プロテインG、または合成基質に対する抗体の一般的な親和に基づいて(Verdoliva A et al.,2002;Roque A et al.,2004)、並びに抗原又はエピトープに基づくアフィニティークロマトグラフィー(Murray A et al.,2002;Jensen L et al.,2004)を利用することができる。洗浄後、タンパク質は、pH又はイオン強度の変化によってゲルから溶出される。あるいは、HPLC(高速液体クロマトグラフィー)を用いることができる。溶出は、タンパク質精製に一般に使用される水−アセトニトリル系溶媒を用いて実行可能である。
【0070】
組換えタンパク質の調製物は、インビトロ又はインビボアッセイ(生化学、組織又は細胞系アッセイ、齧歯類において確立した疾患モデル、親和性測定のための生物物理学的方法、エピトープマッピングなど)において、特に、RuV病因及び免疫学を兼有するための文献に開示されたものの1以上を用いて、例えば、国際的な健康協定(WHO2005;WHO2006)に従って定義される野生型及び参照RuV菌株を用いて試験することができる。
【0071】
RuVを結合及び中和することができるものとして上記で定義された抗体、抗体フラグメント、生物活性ペプチド、融合タンパク質、及び任意の他の化合物は、RuV感染を検出、治療、阻害、予防、及び/又は改善するために使用することができる。この目的で、このような化合物は、RuV感染の医療管理のための診断、治療、又は予防用組成物を調製するために使用することができる。
【0072】
これらの組成物は、ヒトDDF−RuV1及びDDF−RuV2の配列及び活性に基づいて上記で定義された抗体、抗体フラグメント、融合タンパク質、生物活性ペプチド、任意の他の化合物を含んでもよい。組成物は、異なるRuV中和抗体又は抗体フラグメント、静脈内免疫グロブリン(IVIg)調製物、及び/又は抗ウイルス化合物をさらに含んでもよい。異なるRuV中和抗体又は抗体フラグメントは、異なる中和エピトープ、例えば文献に既に記載されているものによって特徴付けられなければならない。実際には、ぶんけんは、多くの例を示し、そこでは、ウイルス又はヒト標的に指向される2以上の抗体が医薬組成物に組み合わせられる場合、得られる組成物は、単なる相加効果ではなく、特定の相乗効果に起因して、改善された治療効果を有してもよい(Logtenberg T,2007)。
【0073】
医薬組成物は、場合により、任意の医薬として許容されるビヒクル又は担体を含んでもよい。これらの組成物は、任意の付加的な治療薬又は予防薬、例えば、ワクチン、静脈内免疫グロブリン調製物、又は抗ウイルス化合物をさらに含んでもよい(又は一緒に投与されてもよい)。また、最近の文献は、ヒトモノクローナル抗体は、この静脈内免疫グロブリン調製物に補足する(可能であれば、置換する)ために用いられ、このような医薬組成物の頻度及び/又は投薬量を減少させる機会を与えることができる(Bayry Jら,2007)。
【0074】
任意のタンパク質(例えば、抗体、抗体フラグメント、融合タンパク質、生物活性ペプチド)、上記で定義される核酸を含む組成物は、RuVに関連した診断、治療、又は予防の目的のある個体に投与することができる。これらの組成物は、RuVビリオンを標的化することによって、治療される患者内のウイルスの繁殖を阻害し、潜在的には、集団におけるウイルス感染の発生をブロックすることができる治療的化合物(特に、治療用抗体又は治療用抗体フラグメント)を提供するRuV特異的受動免疫のための手段として投与することができる。
【0075】
特定の使用に依存して、組成物は、長期間又は短期間、ヒト対象(幼児、妊娠女性、年配の個体、又はRuVに感染し、又はRuV感染した個体との接触、入院、又は免疫抑制/化学療法的治療に起因してRuVに危険であると考えられる任意の他の個体)に化合物を提供されなければならない。この目的で、組成物は、筋内、静脈内、皮下、局所、粘膜、噴霧器、吸引、点眼、非/生分解性マトリックス材料中、又はミクロビーズなどの微粒子薬物送達システムを用いることにより投与することができる。
【0076】
医薬組成物は、特に、眼内、ウイルス血症に関連した皮膚疹でのウイルスの存在があれば、眼内又は局所投与のために、十分な期間、その化合物が活性を発揮することができる化合物の治療的又は予防的な有効量を対象に提供しなければならない。抗体又は抗体フラグメントを含む組成物が、創傷、皮膚、粘膜、又は角膜に局所投与される場合に効果的であることが証明された(Brereton H et al.,2005;Castle P et al.,2002;Streit M et al.,2006;Nwanegbo E et al.,2007)。
【0077】
所望の効果は、RuV感染、再活性化、及び/又は再感染を調節することによって、RuV感染の臨床症状の少なくとも幾つかを低減させることによって、患者の状態を改善することである。例えば、組成物は、投与経路、個人の状態に依存して、約0.005〜約50mg/kg/体重の有効量で投与されなければならない。
【0078】
診断的使用を有する組成物の場合において、化合物は、生物学的試料中にウイルスを検出するための臨床及び研究所において一般に確立した技術(例えば、ELISA又は他の血清学的検定)を用いて、又は投与後の少なくとも1、2、5、10、24時間、若しくはそれを超えてインビボで対象に投与される場合に、検出されなければならない。RuVの検出は、免疫コンピテント及び免疫不全宿主の両方の危険な集団においてRuV感染をモニターするために確立された既知の手段及び手法に代えて又は結びつけて、本発明のタンパク質を用いて実行することができる。
【0079】
RuV又はRuVに関連した疾患の治療、予防、又は診断のための方法は、上記で定義されるタンパク質又は核酸の投与を含むことが可能である。この方法は、異なるRuV中和抗体又は抗体フラグメント、静脈内免疫グロブリン(IVIg)調製物、及び/又は抗ウイルス化合物の投与をさらに含んでもよい。
【0080】
臨床開発及び使用は、抗体薬物動態及び薬理学の特徴付け(Lobo Eら,2004)、臨床前及び臨床安全データ(Tabrizi M及びRiskos L,2007)、並びにヒトにおける治療的及びインビボでの診断的使用のために治療的組換え抗体の商業生産スケール調合及び分析的特徴付けのための正式な公的要求(Harris Rら,2004)に基づくべきである。
【0081】
ここで、本発明は、下記の実施例によって記載されるが、多少なりとも、本発明を制限するものとして解釈されてはならない。
【実施例】
【0082】
実施例1:ELISAにおけるRuVタンパク質抽出物に結合するヒトFabの発現及び分泌
材料&方法
ライブラリー構築
ヒトIgG1の重鎖及び軽鎖をコードするcDNAは、文献(Burioni et al.,1998;”Phage Display:A laboratory Manual”,Burton DR et al.,CSHL Press,2001)に従って、RuV血清陽性反応の個体から得たリンパ球から得られた。ファージライブラリーは、PCT特許出願WO07/007154に記載される技術に従って、pDDベクターと適合するクローニングカセットを用いて構築された。Fabは、DDカセット内にクローニングされ、ライブラリーの組換えファージの表面に発現された。pDDに基づくFabライブラリーのパンニングを介したヒトFabの選択、陽性クローンの配列決定は、文献(Burioni Rら,1998)に記載されるように行った。
【0083】
Fabの特異的なCDRは、IMGT/V−QUEST(Giudicelli Vら,2004)、European Bioinformatics Instituteによって提供されるもの、FASTA(http://www.ebi.ac.uk/fasta33/index.html)を用いて検出可能なものなどのヒト抗体のタンパク質配列を含む他のデータベースによって提供される予測及び配列整列を比較することによって特定された。
【0084】
細胞培養の細胞からのタンパク質抽出物の精製
VERO細胞(ATCC Acc.No.CCL−81)は、単層として増殖するサル線維芽細胞株であり、通常、RuVを含むヒトウイルスを試験し、繁殖させるために使用される。RuVに対する中和抗体は、VERO細胞を用いて特徴付けられている(Cordoba Pら,2000b)。
【0085】
RuV臨床単離物(H2)を用いて感染させたVERO細胞は、ファージディスプレイライブラリーをパンニングし、ELISAにおいてFabを試験するためのRuV特異的材料の調製のために使用した。VERO細胞は、10%の不活化されたウシ胎児血清(FBS)、50μg/mlのペニシリン、100μg/mlのストレプトマイシン、及び2mMのL−グルタミンを含む修飾されてイーグル培地において維持される。
【0086】
細胞(RuV感染又は未感染VERO)を回収し、250mlの溶解バッファー(50mM Tris−HCl pH8.0;150mM NaCl;0.02%アジ化ナトリウム;0.5% Triton−X)に再懸濁させ、氷上で20分間インキュベートし、次に、12000rpmで2分間、4℃で遠心分離した。得られた上清のタンパク質濃縮物は、BCAタンパク質アッセイキット(Pierce)を用いて2重に測定し、既知濃度でウシ血清アルブミン(BSA)の連続希釈に関してレポートした(0mg/ml=ブランク〜2.000mg/ml=最大値)。全ての試料の吸光度は、分光計セットを用いて540nmで測定した。
【0087】
タンパク質抽出物を用いたパンニング及びELISA
ELISAのためのタンパク質コーティングは、下記の抗原:VERo細胞の溶解物、RuV感染又は未感染(臨床単離物H2)、ウシ血清アルブミン(BSA)を用いて96ウェルプレートで行った。各試料は、カーボネートバッファー(1ウェルあたり25μlの最終体積に全タンパク質の100ナノグラム)で希釈し、プレートを一晩、4℃でインキュベートした。蒸留水で洗浄後、プレートを1%BSAを含むPBSにおいて1時間37℃でインキュベートすることによってブロックした。
【0088】
ELISAは、文献(Bugli Fら,2001)に開示されているプロトコールを用いて下記のFab:DDF−RuV1、DDF−RuV2、およびe137、C型肝炎ウイルスに特異的な無関係なFabを含む未希釈の試料40μlを用いて行った。Fabは、二重のウェルで試験した。各Fabを用いて、1時間37℃でインキュベートし、0.1%Tween−20を含むPBSで5回洗浄後、40μlのヤギ抗ヒトFab、ペルオキシダーゼ結合体(Sigma;Cat no.A0293)を添加し、1時間37℃でインキュベートした。プレート洗浄は上述のように繰り返し、酵素反応は、各ウェルに40μlの基質(TMB基質キット;Pierce)を添加することによって発色させた。ELISA反応は、15分間37℃で発色させた。酵素火星は、停止溶液(H2SO4)の添加によって停止させ、分光計セットを用いて、450ナノメートルで吸光度を測定した。
【0089】
ELISA及び中和アッセイのためのFab調製
プロトコールは、pDD又は他のファージミドを用いた文献(“Molecular Cloning:A Laboratory Manual”,Sambrook et al., Cold Spring Harbor Press,NY,1989;Burioni R et al.,1998; Bugli F et al.,2001;WO07/007154)に記載されるのと同じであった。
【0090】
要約すると、ライブラリーの個々のE.coliクローンは、抗生物質を含み、IPTGを補足されたSuper Broth(SB;3.5%バクト−トリプトン、2%酵母抽出物、0.5%NaCl)培地200mlで増殖させ、回収し、リン酸緩衝生理食塩水(PBS)で洗浄した。溶解は、調節されたパルスを用いた4℃での超音波処理によって、ペリプラスム空間に限定された。ペリプラスム抽出物中のFabは、45分間4℃でJA−10ローターで12,000rpmの超遠心分離によって部分的に精製した。生産物をろ過し、Centrionフィルターを用いて10倍に濃縮した。
【0091】
部分的に精製されたFabの濃縮物は、96ウェルプレート(Costar;Cat no.3690)の表面に結合させたImmunoPureヤギ抗ヒトIgGF(ab’)2(Pierce;Cat.no.31132)を用いたサンドイッチELISAによってペリプラスム抽出物で測定した。37℃での1時間のインキュベーション後、脱イオン水でプレートを6回洗浄し、2%BSAを含む170μl/ウェルPBSを用いてブロックした。37℃でさらに1時間のインキュベーション後、1%BSAを含むPBS中の各Fabの連続して3倍希釈物の50μl、又は既知濃度の対照のヒトFab(Cappel Labs;Cat.No.6001−0100)を各ウェルに添加し、37℃で1時間インキュベートした。次に、プレートをTPBS(0.05%Tween−20を有するPBS)で6回洗浄した。次に、抗体結合は、アルカリホスファターゼを結合したヤギ抗ヒト抗体(Pierce;Cat.No.31312)を50μl添加することによって測定し、37℃で1時間インキュベートした。プレート洗浄は、上述されるようにTPBSを用いて繰り返し、100μlのp−ニトロフェニルリン酸2ナトリウム(Sigma)を各ウェルに添加した。ELISA反応は、60分間発色させ、結果は、既知の濃度で希釈した対照ヒトFabに対してプロットした。全ての試料の吸光度は、590nmで分光計セットを用いて測定した。
【0092】
結果
組換えファージのライブラリーは、pDD技術(WO07/007154)に従って生じさせ、RuVの臨床単離物を用いて感染させた線維芽細胞株から得たタンパク質抽出物について評価した。また、5ラウンドのパンニングは、RuVよって感染していない同じ細胞株からの対照タンパク質抽出物を用いて同ライブラリーを用いて並行して行った。
【0093】
第3ラウンドによって、対照タンパク質抽出物に対して評価した試料のファージ力価は、104を下回り、一方、RuV抽出物に対して評価した試料のファージ力価は、第4ラウンドで104を超え、第5ラウンドでは105に到達した。この値は、RuV抗原に結合するそれらの表面Fabに発現している組換えファージにおけるライブラリーの進行的濃縮を示す。
【0094】
パンニングの第5ラウンド後に得られた14個のクローンは、ELISAにおいて個別に試験され、陽性であることが確かめられた。選択されたクローンにおけるHCDR3のPCR及び配列分析により、ここで命名されたDDF−RuV1及びDDF−RuV2のヒトFabを特徴付ける2つの重鎖が同定された。DDF−RuV1又はDDF−RuV2を発現している組換えファージの反応性は、RuV特異的タンパク質抽出物、並びに無関係な抗原(ウシ血清アルブミン、未感染細胞からのタンパク質抽出物)に対して試験され、ELISAフォーマットにおいてそれらの結合活性が確かめられた(図1)。また、結合の特異性は、対照のヒトFabを用いて確認された。
【0095】
これらのFabの完全な重鎖及び軽鎖可変領域のDNA配列、並びにDDF−RuV1及びDDF−RuV2に対して、対応するCDRを決定した(それぞれ、図2及び3)。これらのFabはまた、タンパク質発現のためのE.coli系システムを用いて、更なるアッセイのための十分な組換えタンパク質を得るための他のベクターに再度クローニングされた。
【0096】
実施例2:RuV感染させた細胞培養物について試験されたDDF−RuV1及びDDF−RuV2の特性
材料&方法
RuV特異的ヒトFabの中和アッセイ
免疫蛍光に基づくアッセイは、37℃で72時間のインキュベーション後には、通常、コンフルエントな単層を形成する条件下で、プレートに植菌された104〜105個のVERO細胞を用いて、Costerの24ウェルプレートで行った。RuV臨床単離物であるH2を感染に使用した。
【0097】
Fabは、実施例1に指示されるように部分的に精製し、維持培地に懸濁させた50TCID50(50%組織培養感染用量)での等体積のRuV細胞不含ストックを用いて種々の濃度(0.01、0.1、1、10及び50μg/ml)で混合した。対照は、Fabなし(ブランク対照)、又はヒトC型肝炎ウイルスに特異的な無関係なFab(e137;Bugli Fら,2001)の存在で、等体積の維持培地及びウイルスから構成された。
【0098】
37℃でのインキュベーションの1時間後、250μlのウイルス−フラグメントFab混合物又は対照混合物は、培地が除かれたウェル(2重)に植菌された。プレートを2時間37℃でインキュベートし、中和されていないウイルスの吸着を可能にする。植菌物を除去し、1.5mlの維持培地を添加した。細胞培養条件で1週間のインキュベーション後、細胞をPBSで洗浄し、冷メタノール−アセトン溶液(1:2比)で10分間、室温で固定した。固定された細胞は、30分間、37℃で、湿気雰囲気でRuVのタンパク質E1に特異的な市販のモノクローナル抗体(Chemicon;Cat.No.MAB925)とともにインキュベートし、PBSで洗浄し、最後に、30分間、37℃で、湿気雰囲気で抗マウスIgG FITC結合体(Sigma)とともにインキュベートした。スライドをPBSで洗浄し、Evans Blue色素で対比染色し、グリセロールを積層した。各Fabの中和活性は、蛍光顕微鏡(Olympus)によって観察される単一の蛍光を発する細胞をカウントし、対照試料の細胞(Fab前インキュベーションなし)と比較して、RuV陽性細胞の数の減少の割合を計算することによって測定された。
【0099】
結果
DDF−RuV1及びDDF−RuV2に関するRuV中和活性の分析は、部分的に精製したFabの調製物の使用及び免疫蛍光に基づくアッセイによって行った。
【0100】
RuV感染細胞の減少に関するデータは、DDF−RuV1及びDDF−RuV2が、RuVとともにプレインキュベートされた場合に中和活性とともに与えられることを指示する。実際に、対照(FabなしのRuVまたは無関係なFabとのRuV)と比較すると、これらのRabの添加は、投薬量に依存して感染細胞の数を減少する(図4)。
【0101】
本明細書において提示された実験的証拠は、RuV感染に関連した診断、治療、又は予防的応用のためのDDF−RuV1及びDDF−RuV2Fab(又はそれらの特異的なHCDR3に基づく代替のタンパク質配列であって、類似の性質を示す)候補化合物を作製する。
【0102】
DDF−RuV1及びDDF−RuV2は、細菌宿主細胞における組換えタンパク質として発現することができる。特に、これらのFabは、pDLac−FLAGhisベクターなどのDDカセット(PCT/IB2008/000266)に存在する制限部位と適合するベクターにそれらのコーディング配列を再度クローニングすることによって発現可能である。このベクターは、ヘテロ二量体複合体を形成する適切な定常領域、重鎖のC末端での6−His及びFagを含む可溶性Fabの発現を可能にする(図5)。このアプローチは、Fabのより直接的なクローニング、検出(例えば、ELISAにおける)及び精製(例えば、固相化した金属親和性カラムに基づく手法を用い、免疫親和性精製と比較してより簡易でありよりコストが抑えられる)を可能にする。
【0103】
実施例3:RuV特異的な中和エピトープを決定するためのpDDに基づくペプチドライブラリーの構築及び評価
RuV中和Fabは、パンニングのためのRuVで感染させたVERO細胞のタンパク質抽出物を用いて、ヒトFabのpDD系ライブラリーをパンニングすることによって初期に同定された。その後、RuV特異的な中和活性は、細胞系モデルにおいて試験されたが、実際にはDDF−RuV1及び/又はDDF−RuV2によるウイルスエピトープは特定されなかった。
【0104】
類似のエピトープは、過去における異なるアプローチを用いて想定され、合成ペプチドELISA(Giessauf Aら,2004)、タンパク質欠失突然変異体(Chaye Hら,1992)、マウスモノクローナル抗体のパネルを用いた競合分析(Green K及びDorsett P,1986)、又はこれらの技術の組み合わせ(Wolinsky Jら,1993)が含まれる。
【0105】
この範囲で、pDD技術は、WO07/007154における血球凝集素エピトープタグ(HAtag)を利用することによって例示されるように、2つの機能性ファージコートタンパク質の1つ又は他方のいずれかのファージ表面に提示されるペプチドライブラリーを構築するために適用することができる。pDD技術を用いて提示された非ランダム又はランダムペプチドのライブラリーは、抗体を特異的に結合するペプチドを提示するファージを選択するためにスクリーニングすることができる。ライブラリーにおいて選択されるペプチド配列モチーフは、抗体特異的なエピトープの特定を可能にする(Zhong Gら,1997;Yip Yら,1999a)。
【0106】
異なるファージディスプレイフォーマットを用いて提示させたペプチド配列をスクリーニングし、比較するという重要性は、文献に示されている。第1の例では、モノクローナル抗体のエピトープは、異なる長さのペプチドを各々含み、異なるファージコートタンパク質に融合された2つのファージディスプレイライブラリーを用いて、ファージによって提示されたエピトープ(又は「ファゴトープ」)内で選択された。スクリーニングの結果は、異なるペプチド配列は、2つのファージディスプレイプラットフォームを用いて選択されたということであり、これは、ファゴトープの特徴付けが、ファージコートタンパク質における比較タイプのペプチド提示を必要とし得ることを示唆する(O’Connor Kら,2005)。同様の証拠はまた、有機(例えば、ヒトの毛髪又は皮膚)又は無機(例えば、ポリプロピレン)支持体に結合するペプチドの場合において見出された。文献(Scholle Mら,2005)に記載された方法、又はそれに従って修飾された方法などの、異なる長さを有するペプチドの市販のファージによって提示されたライブラリーをスクリーニングし、比較することによって、得られる配列が顕著に異なる(WO07/03551、WO06/094093、WO06/094094、WO06/094095)。
【0107】
2つの異なるライブラリーは、pDDファージミド(pDDc)と市販のライブラリー(Ph.D.12ファージディスプレイライブラリーキット;New England BioLabs,cat,no.E8110S)との組み合わせによって構築され、それは、抗体の線状又は立体的エピトープを決定するための過去においてすでに使用され(Li Yら,2007;Petit Mら,2003)、異なる長さのペプチドライブラリーを生じるために修飾された(WO07/03551、WO06/094093、WO06/094094、WO06/094095)。このライブラリーは、64個の可能なコドンのうちほんの32個を含む。それは、第3の位置がT又はGに限定されているためであって、単一コドンを有する残基の相対的頻度を増加させ、3つの終始コドンの2つ(第3のものは適切なE.coli菌株において抑制され、グルタミンに翻訳され得る)を除去するためである。
【0108】
このライブラリーに含まれるファージミド由来の複製の二本鎖形態は、XhoI制限部位を含むフォワードプライマー、SpeI制限部位を含むリバースプライマーを用いたPCR増幅のための鋳型として使用された。第2のPCR増幅は、鋳型と同じライブラリー、KpnI制限部位を含むフォワードプライマー、EagI制限部位を含むリバースプライマーを用いて行われた(図6A)。
【0109】
PCR増幅に起因するDNAフラグメントが消化され、各々が約108の力価を有し、初期には、cp3にだけ融合する、2つの異なるpDD系ペプチドライブラリーを生じるためのpDDcベクターにクローニングされた。第1のケースでは、pDDcXSmer−orcp3ライブラリーは、XhoI及びSpeI制限部位におけるPCR増幅産物をクローニングすることによって生じた(図6B)。第2のケースでは、pDDcKE12mer−orcp3ライブラリーは、中間のpDDcKEベクターにPCR増幅をクローニングすることによって生じさせ、それには、XhoI/SpeI部位の間のKpnI及びEagI制限部位、HAコーディング領域の5’/3’末端を含む(図6C;WO07/007154)。
【0110】
クローニング部位としてSpeIの使用は、特定の効果を提供することを気づくことは重要である。それは、この制限酵素は、元々にペプチドライブラリーに存在する2つの連続コドン:スレオニンをコードするACT、セリンをコードするAGTに対応する配列(ACTAGT)内で切断するためである。他の2つの可能なフレームは、ライブラリー内で可能にしないため(即ち、A及びCは、コドンにおける第3の位置として存在しない)、DDカセット内でライブラリーをクローニングするためのSpeIの利用は、pDDベクターのSpeI部位がpDDcの場合のように同じコーディングフレームを有する場合に、低い頻度でペプチドのより短い変異体の導入を可能にする(図6B及び6C)。
【0111】
このようなアプローチは、AlwNI(CAGNNNCTGを認識)、NdeI(CATATGを認識)、HindIII(AAGCTTを認識)、又はPvuII(CAGCTGを認識)などの類似の特性(第1塩基としてA又はC、第2塩基としてA又はC、第3塩基としてG又はT、6又は9塩基対を認識する)を有する認識部位を有する他の酵素を利用するこの種のライブラリーの変異体を生じるために使用することができる。また、類似の部位は、元々のライブラリーにおける追加の非/ランダムコドンを無作為に統合するために使用されてもよく、ライブラリーの複雑性をさらに改善する。
【0112】
cp3又はcp8に融合したタンパク質を提示するために、2つのpDD系ペプチドライブラリーは、(例えば、XL1−blue E.coli菌株において)維持及び増幅することが必要である。ファージミドは精製され、BglI(DDカセットを抽出するために)で消化され、次に、BglI消化されたpDDcベクターに再度連結される。連結反応は、細菌を形質転換するために使用され、その結果、2つのライブラリーが選択され、Zeocin耐性細菌細胞として維持された。この場合、ファージミドが増幅され得て、ヘルパーファージによる感染後に組換えファージに含まれ、単一のクローンは、所望のスクリーニング後に分析可能である。
【0113】
この方法では、DDカセットのペプチドライブラリーがクローニングされ、対応するpDDcXSmer−orcp3/cp8及びpDDcKE12mer−orcp3/cp8ライブラリーにおける2つの可能な配向のいずれか1つにスクリーニングされる(図7;WO07/007154)。次にライブラリーは、E.coliで増幅され、組換えファージはパンニング実験の結果を比較することによって試験された。パンニングは、組換えファージを生じるためのヘルパーファージの使用を伴う。このファージの集団が単離され、文献に記載されるように、マイクロウェルプレートに被覆された対象の標的への結合親和性に基づいて試験することができる。対象の標的に対する3〜5つの連続的なパンニングラウンド後、特異的結合は、対照と比較されると、溶出されたファージの数の増加によって明示される。
【0114】
本ケースでは、2つのライブラリーは、市販の抗HAtag抗体(マウスモノクローナル抗体HA.11、クローン6B12;Covance)などの周知な線状エピトープを有する抗体に対して試験された。このアプローチは、ファージディスプレイプラットフォームを改善するための技術を評価するために従来使用されていた(Stratmann T及びKang A,2005;Vanhercke Tra,2005)。
【0115】
ライブラリーのタイトルにおける減少があるとしても(BglI消化、再連結、E.coli形質転換に関連する手法による)、関連した標的(抗HAtagモノクローナル抗体)又は負の対照(BSA)に対するパンニングの比較は、pDDcXSmer−orcp3/cp8(表I)及びpDDcKE12mer−orcp3/cp8(表II)ライブラリーに関して、計算されたインプット及びアウトプットについて有意な相違があることを示す。さらに、パンニングの第3ラウンドは、抗HAtagのアウトプットに対する102の特定濃縮をBSAの1つと比較することを示す。
【0116】
DDカセット及びペプチドが2つのライブラリーの各クローンに存在する配向は、制限分析によって容易に測定可能である(図7)。消化は、提示されるべき配列を有するDDカセットの末端近くを切断する酵素(例えば、XhoI又はSpeI)と、pDdベクターの骨格にあるコートタンパク質をコードする配列の末端を切断する酵素(例えば、cp3*についてはNheI)とを組み合わせることによって行われる。得られる制限パターンは、コートタンパク質をコードする配列だけを含むDNAフラグメント(DDカセットがcp3*に配向する場合)、又はDDカセット及びコートタンパク質をコードする配列を含むDNAフラグメント(DDカセットがcp8*に配向する場合)を示す。
【0117】
NheI/SpeI消化は、上記されるパンニング前及び/又は後に、2つのライブラリーから無作為に選択されるクローンのパネルにおいて行われた。消化から得られたDNAフラグメントが電気泳動によって分離されると、約0.6kbのフラグメントは、DDカセットがcp3*に配向される場合に検出され、約1.8kbのフラグメントは、DDカセットがcp8*に配向される場合に検出される。この分析は、全ての状況において、cp8*配向を有するクローンの僅かな有病率を示した(図8)。
【0118】
pDDcXSmer−orcp3/cp8及びpDDcKE12mer−orcp3/cp8ライブラリーにおいてクローニングされたペプチドの配列は、無作為に選択されたクローンについて決定され、良好なレベルの変動、pDDcXSmer−orcp3/cp8の場合には、7〜12個のアミノ酸の範囲のより短い配列を有するペプチドの存在の両方を確実にした(図9)。同じ分析は、抗HAtag抗体に対するpDDcKE12mer−orcp3/cp8をパンニング後に得られたクローンにおいてなされ、HAtag(テトラペプチドYPYD)を含むペプチドの存在が選択されたクローンにおいて確かめられた。
【0119】
ライブラリーにおけるHAtag特異的ペプチドをコードするクローンの存在及び選択の更なる確認として、パンニング選択後に生じた細菌クローンはニトロセルロース様メンブレン上に移され、アルカリ条件で溶解された。この方法では、細菌タンパク質は、ニトロセルロースペーパーに固相化され、次に、抗HAtagとともにインキュベートされる。HAtagを提示する多量のクローンを確認する(図10)。
【0120】
生物活性ペプチド(例えば、抗体、阻害剤又はプロテアーゼの認識部位を特徴付けるエピトープ、膜受容体に結合するペプチド、シグナルタンパク質、又は転写因子)が同定されるpDD系ペプチドライブラリーを生じ、パンニングするための本ストラテジーは、異なる使用又はクローニングストラテジーに適合することができる。ファージに提示されるランダムペプチドライブラリーの複雑性を改善及び評価するための条件は、文献に記載されている(Noren K et al.,2001;Rodi D et al.,2002)。
【0121】
例えば、オリジナルなペプチドライブラリー(その長さ及び複雑性を維持することが望ましい場合)は、クローニング及び形質転換プロセスのより良好な効率を可能にし得る基準の組み合わせを満足する制限酵素の異なる組み合わせを用いてクローニングされ得る。例えば、制限酵素のBstEII、MluI、AgeI、EcoRV、Sall、SnaBI、及びBg1IIは、pDDcベクター及びPh.D.12ライブラリーに切断するものがなく、(EagIと比較して)メチル化不感覚である。
【0122】
この実施例は、複数の融合タンパク質を含むpDD系ペプチドライブラリーを提供し、各融合タンパク質は、pDDベクターのDDリンカーを含む配列によって連結されるウイルス表面タンパク質(cp3*又はcp8*)及びランダムペプチド配列を含む。pDDベクター及びDDカセットの構造、機能、配列特徴は、WO07/007154において特定される。
【0123】
初期のペプチドライブラリーは、一般式NNK(Nは任意の塩基であり、KはG又はTである)を有するコドンを含むべきである。上述されるpDDベクターにおけるコドン使用及びフレームと適合され得る部位を認識する制限酵素による消化後、ライブラリーは、元々の配列の完全又はより短い変異体として実際にクローニングされる。これらのペプチド変異体は、一般には遺伝的パッケージ、特に細菌細胞又は線状ファージの表面に提示され得る。
【0124】
異なる長さを有し、pDDベクターのDDカセット内に2つの機能性ファージコートタンパク質の1つ又は他方のいずれかと融合されたペプチドを提示するためのファージミドのライブラリーを構築する方法は、下記を含む:
a)少なくとも制限酵素を用いてDDカセット内にライブラリーをクローニングするために使用される制限部位(例えば、SpeI)に対応する、少なくともコドンの組み合わせを含むDNAライブラリー(例えば、Ph.D.12ペプチドライブラリー)の消化;
b)得られたDNAライブラリーと、少なくとも同じ制限酵素を用いて消化されたファージミド(例えば、pDDc)との連結;
c)細菌細胞の形質転換のためのファージミドの第1ライブラリーの使用、及び細菌細胞の第1ライブラリーの生成;
d)細菌細胞の第1ライブラリーからのファージミドの単離;
e)DDカセットのDDリンカーに存在する制限酵素を用いて前記ファージミドの消化;
f)得られたDDカセットと、同じ制限酵素を用いて消化したpDDベクターとの連結;
g)細菌細胞の形質転換のためのファージミドの第2ライブラリーの使用、及び細菌細胞の第2ライブラリーの生成;
d)細菌細胞の第1ライブラリーからのファージミドの単離。
【0125】
このプロセスの最後に、DDカセットの調節配列、pDDベクターの1つ又は他の機能性ファージコートタンパク質(例えば、cp3*又はcp8*)の配列への操作可能に連結されたpDD系ペプチドライブラリーが生じる。このようなファージミドを含む細菌細胞のライブラリーは、パンニング、又は任意の他の種のネガティブ/ポジティブ選択のために使用される組換えファージの集団を得るために、ヘルパーファージを用いて感染させることができる。
【0126】
このクローニングアプローチは、コートされたペプチドの長さの修飾に2つのファージコートタンパク質(WO07/007154におけるpDD技術について指示されるように)の融合を組み合わせる。この方法は、所望の標的に対してスクリーニングされ得るペプチドをコードするファージミドの新規なライブラリー、及びペプチドの新規なライブラリーを提供する。実際に、この方法に従って得られる複数のファージミドは、E.coliの形質転換のために使用することができる。次に、得られる細胞は、組換えファージのライブラリーを生成するためのヘルパーファージを用いて感染させ得る。
【0127】
pDD系ペプチドライブラリーは、ファージによる提示されるランダムの元々の集団から選択される特定のペプチドをコードするDNA配列を増幅及び/又は配列決定するためのプライマーをさらに含むキットに(ファージミド又は細菌細胞として)含まれてもよい。
【0128】
pDD系ペプチドライブラリーは、特定の生物活性を有するペプチドを選択するためにスクリーニングされ得る組換えファージを生成するか、又は特定のリガンド分子に結合するペプチドを単離及び生成するために使用可能である。インビトロでのパンニングの場合、このようなライブラリーは、エピトープマッピング(O’Connor Kら,2005;Zhong Gら,1997;Yip Yら,1999a)、プロテアーゼ特異性のマッピング及びプロテアーゼ阻害剤の設計(Diamond S,2008;Sedlacek R及びChen E,2005)、あるいは細胞表面の抗原のスクリーニング(Mutuberriaら,2004)のために使用可能である。インビボのパンニングの場合、このようなライブラリー(並びに、cp3又はcp8のいずれかと融合するだけでなく、異なる長さを有するペプチドを提示するpDDcXSmer−orcp3/cp8ライブラリーのような特定のもの)は、細胞外空間内、及び細胞膜上でタンパク質−リガンド相互作用の同定を可能にする(Finger Aら,2002;Yip Yら,1999b)。
【0129】
【表1】

【0130】
【表2】
【0131】
【化1】
【0132】
【化2】
【0133】
【化3】
【0134】
【化4】
【0135】
【化5】
【0136】
【化6】
【技術分野】
【0001】
発明の分野
本発明は、ウイルスに結合し、ウイルスに特異的な活性を中和するファージディスプレイライブラリーから単離された新規な抗体配列に関する。
【背景技術】
【0002】
発明の背景
ファージディスプレイ技術は、細胞の表面に発現され、標的と相互作用後に生物学的機能を発揮し得るポリペプチド(抗体フラグメント、生物活性ペプチド、酵素など)をクローニングし、選択し、操作するためのバクテリア細胞(例えば、大腸菌細胞)を感染させる線状ファージ(M13など)のゲノムの小さなサイズ及び適応性を利用する。
【0003】
いくつかのクローニング及び発現戦略、ベクター、ライブラリー、ファージを繁殖させる方法、スクリーニングアッセイは、異なる用途のために開発されている。論文(Bradbury A and Marks J,2004;Mancini N et al.,2004;Conrad U and Scheller J,2005;Hust M and Dubel S,2005)及び書籍(“Phage display:A Practical Approach”,vol.266,ed.Clackson and Lowman H,Oxford Univ.Press,2004;“Phage Display:A Laboratory Manual”,ed.Burton D et al.,CSHL Press,2001)に概説されている。
【0004】
ファージディスプレイライブラリーは、組換えファージの集団によって形成され、各々は、その表面上にタンパク質配列のレパートリーの1つの要素を提示する。特定のタンパク質を発現するファージは、反復親和性及び/又は活性に基づく選択プロセス(「パンニング」)によってライブラリーから単離することができる。例えば、タンパク質は、生物学的及び機能的アッセイにおいて、抗原に対する特定の結合親和性又は活性に基づいて、単離され、特徴付けることができる、可変重鎖/軽鎖ヘテロ二量体(通常、Fabと称される)又は一本鎖フラグメント可変部(scFv)の形態で抗体フラグメントであってもよい。
【0005】
特に、スクリーニングプロセスは、病原体及び生物学的標的物に高親和性及び特異性を有する抗体フラグメントを同定するために開発されつつあり、時として、関連する生物学的活性は、このような結合特性と関係している。実際に、全体の治療アプローチ(受動免疫療法又は受動血清療法と称される)は、ヒト又は非ヒト治療標的物に指向される抗体及び抗体フラグメントの抗原結合特性に基づいている(Dunman P and Nesin M,2003;Keller M and Stiehm E,2000)。受動免疫療法は、病原性抗原(毒素、ヒトタンパク質、ウイルス、又は寄生生物など)に対する所定の結合特性を有する治療用抗体を含む医薬組成物の個体への投与からなる。
【0006】
受動免疫療法は臨床診療に導入され、広範囲な疾患(例えば、感染症、免疫を媒介した疾患及び癌)の治療のための機会を急速に拡張する。このアプローチは、標的化された分子をブロックし、及び/又は排除するために必要とされる量で及び/又は特異性をもって、免疫系がそれらを生産することができない患者において特に効果的であり得る(Chatenoud L,2005;Laffly E and Sodoyer R,2005)。
【0007】
治療用抗体を用いて標的化され得る病原性抗原のうち、ヒト細胞に影響を与えるウイルスが特に重要である。このような抗体の投与は、患者におけるウイルスの繁殖を阻害し、集団におけるウイルス感染の大発生を潜在的にブロックすることができる。あるいは、抗体は、免疫応答性個体の健康に重篤及び/又は持続的な結果を通常な有しないものを含む感染症に非常に感受性となる多かれ少なかれ延長された期間、免疫系が弱まった患者(例えば、免疫抑制された個体、高齢者、又は移植された個体)に投与されてもよい。
【0008】
風疹ウイルス(RuV、三日麻疹)は、このようなウイルスの一例である。RuVは、RNAゲノム、2つの非構造的タンパク質、宿主由来の脂質二重層に組み合わせられる3つのウイルス構造タンパク質(E1、E2、及びC)によって形成されるビリオンエンベロープを提示するトガウイルスファミリーのメンバーである(Banatvala J and Brown D,2004;Lee J and Bowden D,2000;“Rubella Viruses”Perspective in Medical Virology,Ed.Banatvala J and Peckham C,vol.15,2006,Elsevier)。
【0009】
RuVは、感染した個体の鼻咽頭分泌物、血液、排泄物、尿に存在し、呼吸噴霧を介して、ヒトからヒトへと伝染される。Ruvは、神経向性ウイルスであり、初期には、神経系細胞(例えば、乏突起膠細胞)に感染するが、RuV菌株は、種々の細胞型、特に神経及び関節組織(例えば、滑膜細胞)において感染し、複製し、持続する能力において変化する。ウイルス性アルトロトロピスム(arthrotropism)は、関節症状とのRuV感染の関係を説明することができる(Lund K and Chantler J,2000;Masuko−Hongo K et al.,2003)。RuVは、感染した細胞における細胞変性効果、アポトーシス、細胞周期停止を誘導する。実際には、RuVタンパク質は、タンパク質−タンパク質相互作用、遺伝子発現、及びタンパク質リン酸化と干渉することによって、宿主の細胞シグナル伝達及び代謝を修飾する(Hofmann J et al.,1999;Cooray S et al.,2005;Atreya C et al.,2004;Domegan L and Atkins G,2002;Adamo M et al.,2008;Figuereido A et al.,2000)。
【0010】
RuVは、一般に、軽度の疾病に関与し、曝露後の16〜20日間で発症し、主に顔及び四肢に観られる微熱及び多発を有する。また、RuV感染は、全くの無症状である場合がある。RuVは、成人では、合併症、例えば感染後の脳障害、血小板減少性紫斑病、出血兆候、又は関節炎をめったに引き起こさない(Banatvala J and Brown D,2004)。また、RuV感染は、一連の眼障害、例えば、フックス虹彩毛様体炎に関与している(De Groot−Mijnes J et al.,2006)。
【0011】
しかしながら、RuV感染は、感染した患者が妊娠女性、特に、発展途上国及び移民集団である場合には、主要な関心事になる。母体感染が必ずしも胎児感染に続かないとしても、胎盤障壁を横断し、胎児組織を感染する能力を有する。妊娠の最初の12週間でのRuV感染は、80〜90%の症例において、胎児の死亡、又は様々な異なる以上をもたらし、通常、Congenital Rubella Syndrome(CRS)の定義下で分類され、例えば、心臓欠陥及び眼の欠陥、難聴、及び精神発達遅滞などが挙げられる。妊娠のその次の週に感染が見られる場合にはその危険性は減少するが、ウイルス性奇形発生を起因とした胎児障害も存在する場合がある(Hinman A et al.,2002;Lee J and Bowden D,2000;Atreya C et al.,2004)。さらに、偽陽性からの最近のRuV感染に起因する抗体、又は感染若しくはワクチン接種後の何カ月も持続している抗体に起因する抗体を区別することを可能にする血清診断アッセイは、十分に信頼できるわけではない(Andrews J,2004;Mendelson E et al.,2006)。
【0012】
RuVワクチン接種は、工業世界において幅広く確立され、CRS及びRuV誘導の胎児死亡の減少に非常に効果的である。それにもかかわらず、最近の大発生における野生型RuVゲノムの遺伝的特徴は、さらに、初期介入について、世界保健機関によって注意深く調査されている。この調査は、参照RuVの遺伝子型及び菌株の定義、並びに世界的な大発生において同定されたRuV菌株の感染的特徴及び遺伝的特徴の分析及び分類を伴う(WHO2005;WHO 2006;Reef S et al.,2002;Zheng D et al.,2003)。
【0013】
実際に、いくつかの試験によって、発展途上国の個体の無視できない割合(少なくとも5%)は、RuVに対する低い又は検出できない抗体濃度を有することが示された。これらの対象は、ワクチン接種してから数カ月又は数年後の集団、及びワクチン接種プログラムへのアクセスが限定された最近の移民又は難民において検出された。このようにして、RuV感染、及びCRSの可能性を防止するために、思春期に近づいている特定の女子、妊娠可能年齢の女性における特異免疫の衰退に起因した、RuV感染に対する追加的処置については、非常に大多数の候補がある(Pebody R et al.,2000;Nicoara C et al.,1999;Matter L et al.,1997;Ki M et al.,2002;Ushida M et al.,2003;Banerji A et al.,2005;Rota M et al.,2007;Mendelson E et al.,2006)。疫学の研究は、RuVの日常的な子供及び成人のワクチン接種、調査、並びに選択的な再免疫化のためにプログラムを確立することの重要性を示唆している(Greenaway C et al.,2007;Kremer J et al.,2007;Semerikov V et al.,2000;Banatvala J and Brown D,2004;Chakravarti A and Jain M,2006;Am.Acad.Pediatr.Com.Infect.Dis.,2007)。
【0014】
RuV病因及び免疫生物学は、ヒト及び動物モデルにおけるRuV感染及びワクチン接種への免疫応答の機序及び効率に関連して研究されている。RuV感染に対する細胞系モデルが確立されている(Cusi M et al.,1995;Duncan R et al.,1999;Garbutt M et al.,1999;Cordoba P et al.,2000a)。一次RuV感染及びRuV特異的免疫応答の初期検出に対する高感度試験が開発されている(Takahashi S et al.,1998;Tzeng W et al.,2005;Giessauf A et al.2004;Wilson K et al.2006)。しかしながら、ワクチン接種は、有効な免疫応答を備えるになるには数週間又は数カ月を要する場合があり、胎児は、その期間中、RuV感染に対してなおも脆弱である。RuV感染は、免疫抑制された患者においては危険であり得て、ワクチンの使用は必ずしも望ましいわけではない。
【0015】
RuVに対して有用であり得る化学物質は、まだ開発の初期段階にある(Mugnaini C et al.,2007)。しかしながら、化学療法及び抗ウイルス治療は、RuVなどのウイルスに対する免疫保護を非常に喪失させ得て、再度のワクチン接種を必要とする場合があることが知られている(Yu J et al.,2007;Bekker V et al.,2006;Van Tilburg C et al.,2006;Nilsson A et al.,2002)。有意な抗RuV力価を有するヒト抗体の生産物は、感染を治療又は予防するために投与することができる(Keller M and Stiehm E,2000;Krause I et al.,2002;Kawamura N et al.,2000)が、同様のアプローチの有効性は、組換え抗体フラグメント又はモノクローナル抗体などの、ウイルスを中和するためのより特異的な免疫学的生産物を用いることによって改善されたであろう。
【0016】
RuV、特にこのウイルスの構造タンパク質に対するヒト抗体応答は、RuVに対してワクチン接種された個体又は野生型RuV菌株によって感染した個体の血清を用いて評価され、異なる対象における特異性及び応答の中和特性において実質的な差異を示した(Zhang T et al.,1992;Mauracher C et al.,1992;Mitchell L et al.,1992;Zrein M et al.,1993;Mitchell L et al.,1993,Thomas H et al.,1993;Banerji A et al.,2005)。赤血球凝集能及び異なる抗体の中和ドメインは、構造タンパク質E1及びE2として与えられた(Mendelson E et al.,2006;Cordoba P et al.,2000a;Green K and Dorsett P,1986)。また、RuV免疫病理学は、RuV由来の扁桃腺フラグメントを移植された(Perrenoud G et al.,2004)、又は腹腔を介したRuVを用いて感染させた(Wang Z et al.,2003)、重症複合型免疫不全症(SCID)マウスを用いた動物モデルにおいて試験された。
【0017】
RuVに結合し、(数例において)RuVを中和することができるいくつかのマウスモノクローナル抗体を生じた。抗体は、細胞におけるウイルス感染及び複製を試験するために、異なるRuV関連標的(ウイルス様粒子、タンパク質抽出物、組換えタンパク質、ペプチド、細胞培養条件で感染させた細胞)を用いて試験された(Green K and Dorsett P,1986;Terry G et al.,1988;Wolinsky J et al.,1991;Chaye H et al.,1992;Claus C et al.,2006;Qiu Z et al.,1994;Ou D et al.,1992;Wolinsky J et al.,1993;Robinson K et al.,1995;Cordoba P et al.,2000a;Cordoba P et al.,2000b;Cordoba P et al.,1997;Starkey W et al.,1995;Giessauf A et al.,2005;Orellana A et al.,1999;Lee J et al.,1999;Hofmann J et al.,2000;EP299673;WO93/14206;WO91/02748;WO95/09232)。
【0018】
文献では、ヒトモノクローナル抗RuV抗体は非常に少なく、関連した抗RuV中和抗体を有するものがないことが報告されている。このような抗体は、細胞融合及び/又はEBV感染によって不死化されたヒトB細胞を用いて同定された(WO07/011698、クローンR335.6.4を参照されたい;Steenbakkers P et al.,1992;Hilfenhaus J et al.,1986)。RuVに対するヒトFab抗体フラグメントが同定されている(Williamson R et al.,1993)。
【0019】
ワクチン接種は主な改善をもたらすが、疾患は持続する。RuVに効率的に結合し、中和する新規なヒト抗体及び抗体フラグメントの同定及び生産は、集団におけるこの感染症の診断、治療及び/又は予防のための改善された処置を確立するためになおも特に重要である。
【発明の概要】
【課題を解決するための手段】
【0020】
本発明は、RuVに結合し、中和し、RuV感染を検出、治療、阻害、予防、及び/又は改善するために使用可能な新規な抗体配列を提供する。
【0021】
ヒト抗体フラグメントの一団は、組換えファージ上に提示され、RuV特異的結合特異性は、ファージライブラリーにおいて検出されている。2つの抗体フラグメントの重鎖及び軽鎖可変領域をコードし、RuVを中和する抗体を有するDNA配列が同定され、DDF−RuV1及びDDF−RuV2と命名された。RuV特異的生物活性に関連する対応するタンパク質配列及び相補性決定領域(CDR)が決定された。これらのFabの結合活性は、さらに、組換えファージ上に提示さえる新規なペプチドライブラリーを用いて試験することができる。新規なタンパク質は、単離されたCDR、並びにDDF−RuV1及びDDF−RuV2に対する可変領域との同一性パーセントに基づいて定義される。
【0022】
本発明の核酸は、適切な技術、及び組換えファージ、原核宿主細胞、又は有核宿主細胞を用いて、全長の抗体、抗体フラグメント、生物活性なペプチド、又は機能性タンパク質(特に融合タンパク質)の任意の他のフォーマットの形態で、RuV特異的結合特性及び中和特性を有する組換えタンパク質を生産するために使用することができる。
【0023】
本発明のタンパク質は、RuV感染の治療検出、治療、阻害、予防、及び/又は改善のために用いることができる。特に、RuV感染の管理における治療、予防、及び/又は診断の利用性を有する組成物は、それらのRuV特異的結合特性及び中和特性を与えるこれらのタンパク質を用いて調製可能である。これらの組成物は、抗ウイルス化合物又は静脈内免疫グロブリン(IVIg)調製物に基づいているRuV治療を補足又は置換するために用いられてもよく、眼内又は局所投与に適していてもよい。
【0024】
本発明の更なる態様は、単離されたDNA及びタンパク質配列、ベクター、組換えファージ、及び宿主細胞、並びに医学的方法、組成物、及び使用を含み、下記の説明に提供される。
【図面の簡単な説明】
【0025】
【図1】DDF−RuV1及びDDF−RuV2、又は負の対照として使用された無関係なヒトFab(e137)を発現している組換えファージの調製物に対するRuV結合活性の特異性。結合活性は、プレートコーティング用の指示された抗原を用いたELISAにおいて測定された。(A)RuVの臨床単離物で感染させた線維芽細胞株(VERO細胞)からの全タンパク質抽出物をプレートコーティングに使用した。各Fabについての2つのカラムは、2つの異なる実験を意味する。(B)RuVの臨床単離物で感染させた線維芽細胞株からのタンパク質抽出物、未感染細胞からのタンパク質抽出物、又は非特異的な精製タンパク質(ウシ血清アルブミン、BSA)をプレートコーティングに使用した。
【図2】(A)DDF−RuV1の重鎖に関する可変領域のDNA(下段ケース)及びタンパク質(上段ケース)の整列(DDF−RuV1 VH;配列番号1及び2)。予測されるCDR(DDF−RuV1 HCDR1、HCDR2、及びHCDR3;配列番号3、4、及び5)に下線を引く。(B)DDF−RuV1の軽鎖に関する可変領域のDNA(下段ケース)及びタンパク質(上段ケース)の整列(DDF−RuV1 VL;配列番号6及び7)。予測されるCDR(DDF−RuV1 LCDR1、LCDR2、及びLCDR3)に下線を引く。
【図3】(A)DDF−RuV2の重鎖に関する可変領域のDNA(下段ケース)及びタンパク質(上段ケース)の整列(DDF−RuV1 VH;配列番号8及び9)。予測されるCDR(DDF−RuV2 HCDR1、HCDR2、及びHCDR3;配列番号10、11、及び12)に下線を引く。(B)DDF−RuV2の軽鎖に関する可変領域のDNA(下段ケース)及びタンパク質(上段ケース)の整列(DDF−RuV2 HC;配列番号13及び14)。予測されるCDR(DDF−RuV2 LCDR1、LCDR2、及びLCDR3)に下線を引く。
【図4】部分的に精製されたヒト組換えFabとして発現され、ファージコートタンパク質に提示された場合のヒトFab DDF−RuV1(A)及びDDF−RuV2(B)に対するRuV中和活性。投薬量応答分析は、免疫蛍光法によって測定されるRuV感染細胞の数に基づいた。パーセント値は、いずれのFabなしに前培養されたRuVを用いて得られたRuV感染細胞(負の対照)に対するデータと比較することによって計算された。
【図5】(A)pDLac−RuV1−FLAGhisベクターを用いて発現されるヒトFab DDF−RuV1の重鎖のタンパク質配列(DDF−RuV1 HCtag;配列番号15)。最初にクローニングされたこの重鎖の改変領域(配列番号2)に下線を引く。PelB配列は、アミノ酸1と22との間に含まれる。アミノ酸147〜252は、ヒトIgガンマ−1鎖C領域のアミノ酸1〜106に対応する(SwissProt Acc.No.:P01857)。アミノ酸253〜267は、FLAGhis配列に対応する。(B)pDLac−RuV1−FLAGhisベクターを用いて発現されるヒトFab DDF−RuV1の軽鎖のタンパク質配列(DDF−RuV1 LC;配列番号16)。最初にクローニングされたこの軽鎖の可変領域(配列番号7)に下線を引く。PelB配列は、アミノ酸1と22との間のアミノ酸に含まれる。アミノ酸135〜240は、Igカッパ鎖C領域のアミノ酸1〜106に対応する(SwissProt Acc.No.:P01834)。(C)pDLac−RuV2−FLAGhisベクターを用いて発現されるヒトFab DDF−RuV2の重鎖のタンパク質配列(DDF−RuV2 HCtag;配列番号17)。最初にクローニングされたこの重鎖の可変領域(配列番号9)に下線を引く。PelB配列は、アミノ酸1と22との間のアミノ酸に含まれる。アミノ酸146〜251は、ヒトIgガンマ−1鎖C領域のアミノ酸1〜106に対応する(SwissProt Acc.No.:P01857)。アミノ酸252〜266は、FLAGhis配列に対応する。(D)pDLac−RuV2−FLAGhisベクターを用いて発現されるヒトFab DDF−RuV2の軽鎖のタンパク質配列(DDF−RuV2 LC;配列番号18)。最初にクローニングされたこの軽鎖の可変領域(配列番号14)に下線を引く。PelB配列は、アミノ酸1と22との間のアミノ酸に含まれる。アミノ酸129〜234は、Igカッパ鎖C領域のアミノ酸1〜106に対応する(SwissProt Acc.No.:P01834)。配列は、重鎖又は軽鎖の定常領域内に設計されたプライマーを用いたインサートの配列決定によって確かめられた。
【図6】(A)Ph.D.−12ファージディスプレイ・ペプチドライブラリーキット(New England Biolabsウェブサイト:http://www.neb.com/nebecomm/products/productE8110.aspから入手可能)についえの指示マニュアルにおいて、生産者によって提供されるランダム12マーのペプチド−cp3融合ライブラリーの配列。M12 XhoI−フォワード(配列番号19)及びM13 SpeI−リバース(配列番号20)プライマーにおける関連制限部位の位置を矢印で示す。M13 KpnI−フォワード(配列番号21)及びM13 EagI−リバース(配列番号22)プライマーにおける関連制限部位の位置に下線を引く。(B)pDDcXSmer−orcp3ライブラリー内でクローニングされた場合のPh.D.−12ライブラリー(PhD12マー)のコーディング配列(WO07/007154の図3から修飾される;配列番号25)。最初のファージミドによって提供される配列の位置(PelBシグナルペプチド、DISリンカー、及びp3*5’末端)が示される。関連制限部位に下線を引く。14個のアミノ酸のリンカー配列は、ペプチドライブラリーとcp3*のN末端近傍(by)との間に位置される。(C)pDDcKE12mer−orcp3ライブラリー内でクローニングされた場合のPh.D.−12ライブラリー(PhD12マー)のコーディング配列(WO07/007154の図3から修飾される;配列番号26)。最初のファージミドによって提供される配列の位置(PelBシグナルペプチド、DISリンカー、及びp3*5’末端)が示される。関連制限部位に下線を引く。18個のアミノ酸のリンカー配列は、ペプチドライブラリーとcp3*のN末端近傍(by)との間に位置される。KpnI及びEagI部位は、XhoI及びSpeIを用いたpDDcの消化、並びにHAtag配列、KpnI部位、EagI部位を含む2つのアニール化されたオリゴヌクレオチドの挿入によって導入され、XhoI及びSpeI部位に適合可能な一本鎖5’末端を含む(配列番号23及び24)。また、ベクターは、DDカセットにクローニングされるZeocinマーカー遺伝子内のEagI部位を排除するために、PCRによって修飾した。
【図7】pDDcXSmer−orcp3ライブラリー、及びpDDcXSmer−orcp3/cp8ライブラリーの生成の略図(WO07/007154の図5から修飾される)。ファージミドマップは、コートタンパク質(cp3*及びcp8*)の位置、ファージミド骨格内のアンピシリンマーカー遺伝子(Ampr)及び複製起源(ColE1及びF1(+))を示す。下記の要素は、DDカセットに存在する(pDDcXSmer−orcp3/cp8ライブラリーを生成するために使用されるBglI/SfiI部位間においてクローニングされた):Ph.Dペプチドライブラリー(Lib)、PelB配列に融合されたpLacZプロモーター(Lpe)、スタッファー配列(St)及びZeocinマーカー遺伝子(Zeor)。関連制限部位の位置を矢印で示す。pDDcKE12mer−orcp3ライブラリー及びpDDcKE12mer−orcp3/cp8ライブラリーは、追加のKpnI及びEagI制限部位を提示する(図6Cを参照されたい)。
【図8】いずれかのパンニングなしのpDDcXSmer−orcp3/cp8ライブラリー(A)、3ラウンドのパンニング後のpDDcXSmer−orcp3/cp8ライブラリー(B)3ラウンドのパンニング後のpDDcKE12mer−orcp3/cp8ライブラリー(C)から無作為に選択されるクローンの消化。単離された細菌クローンは、NheI及びSpeIを用いて消化されたファージミドDNAのミニプレップを行うために使用された。反応物は、分離用の1%アガロースゲル上にロードされ、得られたフラグメント(NS)と対応する未消化のDNA(C)と比較した。3.8及び0.6Kbフラグメントを示すクローンは、cp3*に配向されたDDカセットを有する。2.6及び1.8Kbフラグメントを示すクローンは、cp8*に配向されたDDカセットを有する。矢印は、マーカーDNAによって位置される。
【図9】下記のライブラリー:pDDcXSmer−orcp3(A;配列番号27〜34)、pDDcKE12mer−orcp3(B;配列番号35〜42)において無作為に選択されたクローン由来の配列例。アンバー停止コドン(tag)は、XL1−Blue E.coli菌株内に抑制され、アミノ酸(Q)をコードする。配列は、cp3*内で設計されたプライマーを用いたインサートの配列決定によって確かめられた。
【図10】抗HAtagコロニースクリーニング。抗HAtag抗体に対するパンニングの3ラウンド後に得られた細菌コロニーは、メンブレンに転写され、アルカリ条件で溶解した。最初のコロニーにおけるHAtagの発現を検出するために、メンブレンをマウス抗HAtag抗体(パンニングのために使用された抗体)、次に、PBS/0.1%Tween−20(1:1000希釈)中のセイヨウワサビのペルオキシダーゼに結合した抗マウスIgG1とともにインキュベートした。PBS/0.1%Tween−20による3回の洗浄後、メンブレンは、Supersignal West Picoケミルミネッセンス基質(Pierce)を用いた増大したケミルミネッセンス検出に供された。
【発明を実施するための形態】
【0026】
発明の詳細な説明
WO07/007154に記載されたpDDファージミド及び関連した方法により、1つの又は他の2つの所定のファージミドコートタンパク質に融合されたタンパク質配列のクローニング、発現、及び選択が可能となる。このアプローチにより、組換えファージの表面に別個に発現されるか又は提示され、結果として、異なる効率を有するファージディスプレイライブラリーから選択され得るタンパク質配列の同定の選択を可能にする。
【0027】
本ケースでは、ファージライブラリーは、ヒト重鎖及び軽鎖の免疫グロブリンの可変領域をクローニングすることによって、pDDファージミドに構築された。このライブラリーは、RuV感染細胞のタンパク質抽出物に対してパンニングされ、選択されたクローンは、その後、RuVを中和する活性を示すクローンを決定するために細胞系圧死において試験された。2つの最大の有望なクローン(DDF−RuV1とDDF−RuV2と命名される)をコードするDNA配列を決定し、次に、細菌発現用の適切なベクターにクローニングされた。
【0028】
本発明は、RuVに結合及び中和することができ、Fab DDF−RuV1又はDDF−RuV2において同定された特定のCDR(相補性決定領域)を含む新規なタンパク質配列を提供する。特に、本発明のHCDR3(重鎖可変領域のCDR3)の各々(配列番号5及び配列番号12)は、それぞれDDF−RuV1及びDDF−RuV2の抗原結合部分を特徴とする。
【0029】
HCDR3は、抗体の抗原結合部分を特徴付け、その結果、生物活性(例えば、RuVの結合及び中和)を与えるものと考えられる。抗体のいくつか又は全てのCDRは、一般に、完全な抗原結合表面を得るために要求されるとしても、HCDR3は、抗体間の最大の相違を示すCDRである。実際に、HCDR3の配列及び長さの多様性は、大部分の抗体に対する特異性を決定するために基本的である(Xu J及びDavies M,2000;Barrios Y et al.2004;Bond C et al.,2003)。
【0030】
RuV結合部分としての本発明の特定のHCDR3を含むタンパク質は、それ(又はなし)と、このようなHCDR3が抗体タンパク質フレームワーク内で同定された同じFab由来の他のCDRとを組み合わせることによって生じさせることができる(Knappik A et al.,2000)。CDRの組み合わせは、抗体構造には関連しないタンパク質フレームワーク内であっても、元々の結合活性を破壊せずに、元々の結合性を維持する非常に短いタンパク質において互いに連結することができる(Kiss C et al.,2006;Smith J et al.,1995)。
【0031】
一態様では、本発明は、Fab DDF−RuV1のHCDR3(配列番号5)と少なくとも90%同一性を有する配列を含むタンパク質を提供する。HCDR1及びHCDR2(配列番号3及び配列番号4、図2A)と一緒に、このHCDR3は、DDF−RuV1 Fabの重鎖の可変領域(DDF−RuV1 VH;配列番号2)に含まれる。このFabを形成する軽鎖の可変領域(DDF−RuV1 VL:配列番号7)、並びにその特定のLCDR(軽鎖可変領域のCDR)が決定されている(図2B)。
【0032】
別の態様では、本発明は、Fab DDF−RuV2のHCDR3(配列番号12)と少なくとも90%同一性を有する配列を含むタンパク質を提供する。HCDR1及びHCDR2(配列番号10及び配列番号11、図3A)と一緒に、このHCDR3は、DDF−RuV2の重鎖の可変領域(DDF−RuV2 VH;配列番号9)に含まれる。このFabを形成する軽鎖の可変領域(DDF−RuV1 VL;配列番号14)、並びにその特異的LCDRが決定されている(図3B)。
【0033】
本発明のタンパク質がDDF−RuV1の配列に基づく場合、それらは、配列番号5と少なくとも90%の同一性を有する配列を含まなければならない。特に、それらは、配列番号2と少なくとも90%同一性を有する配列を含まなければならない。より特には、このようなタンパク質はまた、配列番号3及び配列番号4からなる群から選択される1以上の配列を含む。タンパク質、特に、DDF−RuV1に基づく抗体及び抗体フラグメントは、配列番号7と少なくとも90%同一性を有する配列をさらに含むことができる。
【0034】
あるいは、本発明のタンパク質がDDF−RuV2の配列に基づく場合、それらは、配列番号12と少なくとも90%の同一性を有する配列を含まなければならない。特に、それらは、配列番号9と少なくとも90%同一性を有する配列を含まなければならない。より特には、このようなタンパク質はまた、配列番号10及び配列番号11からなる群から選択される1以上の配列を含む。タンパク質、特に、DDF−RuV2に基づく抗体及び抗体フラグメントは、配列番号14と少なくとも90%同一性を有する配列をさらに含むことができる。
【0035】
本発明の更なる態様は、両方のFabの重鎖及び軽鎖の可変領域をコードするDNA配列、特に、クローニングされ、DDF−RuV1(重鎖については配列番号1;軽鎖については配列番号6)、並びにDDF−RuV2(重鎖については配列番号8;軽鎖については配列番号13)の可変領域が決定されている元々のDNA配列と少なくとも90%同一性を有するものである。
【0036】
これらのDNA配列(又は選択された部分、例えば図2及び図3に示された単離されたHCDRとLCDRをコードするもの)は、組換え抗体(例えば、完全、アフィニティー成熟、CDRグラフト、または図5においてDDF−RuV1及びDDF−RuV2のタグ化されたバージョンについて示されたフラグメント)、又はRuV結合性及び中和性を与える融合タンパク質についての既知のフォーマットの1つ内にそれらを発現するための他のベクターに移すことができる。
【0037】
同一性レベルが指示されるところはどこでも、この同一性レベルは、本発明の関連配列の全長に基づいて決定されなければならない。
【0038】
重鎖及び軽鎖を形成するDDF−RuV1又はDDF−RuV2の可変領域(又は、選択された部分、例えば、単離されたHCDR及びLCDR)は、特定のアイソタイプを有する抗体、特に完全なヒト組換え抗体内に含むことができる。この抗体は、2つの軽鎖及び2つの重鎖によって形成される四量体複合体の天然の構造における軽鎖及び重鎖可変領域として、DDF−RuV1又はDDF−RuV2のVL及びVH配列を含んでもよい。完全なヒト抗体が望まれる場合、固体は、ヒトIgG1、IgG2、IgG3、IgG4、IgM、IgA及びIgE定常領域からなる群から選択される重鎖定常領域をさらに含まなければならない。例えば、IgGアイソタイプは、ほとんど全ての承認された治療用抗体の抗体フォーマットである(Laffly E及びSodoyer R,2005)。しかしながら、ヒトIgG1から単離された抗原結合部分は、ヒトIgA配列に移され、得られた組換え抗体は元々のIgG1の活性を維持し、最近示されるように、抗体はHIV感染を阻害することができる(Mantis Nら,2007)。
【0039】
あるいは、DDF−RuV1又はDDF−RuV2を形成する重鎖及び軽鎖の可変領域(又は、選択された部分、例えば単離されたHCDR及びLCDE)は、機能性抗体フラグメントについての任意の他のタンパク質フォーマットにおいて組み合わせることができ、異なる名称で文献に記載されているように、例えば、Scfv(一本鎖フラグメント可変)、Fab(可変重鎖/軽鎖ヘテロ二量体)、ダイアボディー、ペプタボディー、VHH(重鎖抗体の可変ドメイン)単離された重鎖又は軽鎖、二重特異性抗体、非/臨床用途のための他の操作された抗体変異体などがある(Jain M et al.,2007;Laffly E and Sodoyer R,2005)。例えば、DDF−RuV1又はDDF−RuV2の組換え改変体は、pDLac−FLAGhis(PCT/IB2008/000266)と呼ばれるpDD適合可能な発現ベクターにクローニングされ、タグ化されたFab(DDF−RuV1については、配列番号15〜16、図5A〜B;DDF−RuV2については、配列番号17〜18、図5C〜D)の形態で発現可能である。
【0040】
代替の抗体及び抗体フラグメントは、軽鎖をシャッフルするためのプロセスを介して、DDF−RuV1又はDDF−RuV2の配列を用いて生じさせることができる。実際に、異なる抗体及び抗体フラグメントを生じさせ、例えば、共通のファージディスプレイ技術又はWO07/007154に記載されたものを用いて、VL配列のライブラリーと合わせられる単一の重鎖可変ドメインVH(例えば、DDF−RuV1又はDDF−RuV2のいずれか1つ)を用いたRuV特異的活性について試験することができる。このアプローチは、親和性、安定性、特異性、及び/又は組換え産物に基づいて改善された特性を用いてVH/VLを決定してもよい(Rojas G et al.,2004;Suzuki K et al.,2007)。
【0041】
さらに、抗体は、特に臨床用途(良好な薬物動態プロファイル又は抗原に対するより高い親和性)のために、改善された特徴を有する抗体を持つために、特定の一で修飾されてもよいことは知られている。これらの変化は、DDF−RuV1又はDDF−RuV2のいずれかのCDR及び又はフレームワークのいて行うことが可能である。配列は、親和性成熟の使用を作る抗体の合理的な設計のための任意の専用技術及び他の方法を適用することによって測定することができる(Kim S et al.,2005;Jain M et al.,2007)。
【0042】
また、新規な生物活性ペプチドを開発するための抗体に基づくストラテジーは、L−アミノ酸及び/又はD−アミノ酸を含むCDR誘導ペプチドを合成する実行可能性を示した。これらの代替の分子は、より適切な薬理学的プロフィールを有する元々の活性を維持することができる(Levi M et al.,2000;Wijkhuisen A et al.,2003)。このようにして、各々の新規なHCDR3、並びにそれらに非常に類似した配列、それらを含む融合タンパク質、それらから誘導される合成ペプチド(例えば、D−アミノ酸を含むか又はレトロー−インバーソ型形態である)が試験され、RuV結合タンパク質として使用することができる。
【0043】
また、本発明のタンパク質は、RuVビリオンの抗原を中和することを特徴付けるために使用されてもよい。実際に、DDF−RuV1及びDDF−RuV2は、ELISAにおけるRuV感染細胞株由来の細胞抽出物へのそれらの特異的結合に奇異真して初期にクローニングされ(図1)、その後、RuV感染を中和するそれらの能力は、RuV臨床菌株を用いたインビトロの中和アッセイによって測定された(図4)。結論として、本発明のタンパク質は、RuV感染を中和することができ、任意の関連した結合アッセイ(例えば、実施例に記載されるELISA)によって測定されるように、本発明のタンパク質と比較する他のRuV結合タンパク質(完全な句及び体、抗体フラグメント、生物活性ペプチド、又は融合タンパク質の形態で)を特定するために用いることができる。このような競合タンパク質は、場合により、DDF−RuV1又はDDF−RuV2配列において同定されたものとは、部分的に又は完全に異なっている可能性があるHCDR及びLCDRとともに、上記で定義されたHCDR3配列の何れかを単に含んでもよい。
【0044】
本発明のタンパク質は、抗体、抗体フラグメント、生物活性なペプチド、又はRuVに結合及び中和する融合タンパク質として提供される。これらの代替のタンパク質は、DDF−RuV1及びDDF−RuV2 Fabについて決定されるこのような特性を増大しない場合に維持されなければならない。
【0045】
融合タンパク質の特定のケースでは、異種配列(単数又は複数)は、このような部分の正しい発現及び生物活性を負に影響を与えないで、RuVに特異的な結合及び中和する部分(例えば、特定のHCDR3又は抗体フラグメントの可変領域に対してN末端位又はC末端位で位置しなければならない。
【0046】
用語「異種タンパク質」は、タンパク質配列がRuVに特異的に結合及び中和する部分(例えば、抗体フラグメント)に対してN末端位又はC末端位において天然に存在しないことを指示する。このタンパク質配列をコードするDNA配列は、一般に、組換えDNA技術によって融合され、少なくとも5個のアミノ酸をコードする配列を含む。この異種配列は、一般に、特定の診断及び/又は治療用途に関連した追加の特性を提供するために選択される。このような追加の特性の例には、検出又は精製のための良好な手段、追加の結合部分又は生物学的リガンド、又は融合タンパク質の翻訳後修飾(例えば、リン酸化、グリコシル化、ユビキチン化、SUMO化、又は細胞内タンパク質分解的切断)が挙げられる。
【0047】
タンパク質部分、リガンド、適切なリンカーを選択及び設計する手段、並びに融合タンパク質の構築、精製、検出及び使用についての方法及びストラテジーは、文献に提供され(Nilsson J et al.,1997;“Applications Of Chimeric Genes And Hybrid Proteins”Methods Enzymol.Vol.326−328,Academic Press,2000;WO01/77137))、臨床及び研究所において利用可能である。例えば、融合タンパク質は、インビボ及び/又はインビトロでの融合タンパク質の同定、又はその精製を促進することができる市販の抗体(タグ、例えばポリヒスチジン、FLAG、c−Myc、又はHAタグが含まれる;図5)によって認識される配列を含んでもよい。他のタンパク質配列は、直接的蛍光分析(緑色蛍光タンパク質など)によるか、又は特異的な基質又は酵素(酵素分解部位などの使用)によって容易に同定することができる。
【0048】
治療活性は、別の抗ウイルスタンパク質あるいは細胞代謝及び/又は活性を変更するタンパク質などの別の治療タンパク質との融合によって改善され得る。RuV特異的抗体、抗体フラグメント、融合タンパク質の安定性は、周知の担体タンパク質、例えばファージコートタンパク質(cp3又はcp8)、マルトース結合タンパク質(MBP)、ウシ血清アルブミン(BSA)、又はグルタチオン−S−トランスフェラーゼ(GST)をm値いて改善されてもよい。
【0049】
あるいは(又は、異種タンパク質配列への融合に加えて)、本発明のRuV特異的タンパク質の活性は、治療薬又は診断薬などの異なる化合物との結合を用いて改善されてもよい。これらの試薬の例には、化学リンカー又はポリマーを用いて結合することができる検出可能な標識(例えば、放射性同位体、蛍光化合物、コロイド金属、ケミルミネッセンス化合物、生物発酵化合物、又は酵素)である。本発明のタンパク質のRuV特異的な生物活性はまた、診断又は治療用途における代謝及び/又は安定性を変更するポリマーなdの化合物との融合によって改善されてもよい。
【0050】
本発明のタンパク質(例えば、抗体、抗体フラグメント、生物活性ペプチド、融合タンパク質、又はそれらの結合された改変体)をスクリーニングし、元々のDDF−RuV1及びDDF−RuV2と競合するそれらの能力、可能であれば、実施例又は文献に記載された任意の関連アッセイによって測定される、RuV感染を中和する類似の又はより優れた能力を示すために特徴付けることができる。
【0051】
また、文献は、RuV抗原、並びに各Fabによって認識される特定のエピトープが測定され、過去に測定されたもの比較されるものを用いるいくつかの技術の例を愛知今日する。例えば、RuVタンパク質、関連した一部切断された改変体又は合成ペプチドを用いたELISA、免疫沈降、又はウェスタンブロットは、E1又はE2タンパク質内の関連エピトープを決定するために用いた。特に、本発明の躯体、抗体フラグメント、他のタンパク質は、抗体フラグメント(Williamson R et al.,1993)、マウス又はヒトモノクローナル抗体(Green K and Dorsett P,1986;Cordoba P et al.,1997;Chaye H et al.,1992;Qiu Z et al.,1994;Ou D et al.,1992;Steenbakkers P et al.,1992;Hilfenhaus J et al.,1986;WO07/011698;WO93/14206;WO95/09232)、ヒト血清(Giessauf A et al.,2004;Starkey W et al.,1995;Zrein M et al.,1993;Mitchell L et al.,1992)についてのRuV特異的生物活性及びエピトープを特徴付けるために使用されるアッセイにおいて試験することができる。次に、本発明のタンパク質のRuV関連予防的、診断的、及び治療的使用のためのより広範囲な特徴付け及び評価は、本発明の背景に要約されるように、RuV病因及び免疫学を研究するための文献(Cusi M et al.,1995;Duncan R et al.,1999;Cordoba P et al.,2000;Lee and Bowden,2000;Garbutt M et al.,1999;Ou D et al.,1992;Tzeng W et al.,2005;Perrenoud G et al.,2004;Wang Z et al.,2003)に開示されているインビトロ又はインビボアッセイ(組織又は細胞系アッセイ、齧歯類において確立した感染モデル)の1以上を用いて行うことができる。
【0052】
本発明の更なる目的は、上記で定義される抗体、抗体フラグメント、融合タンパク質又は単離されたCDRのいずれかをコードする核酸である。実施例は、このような配列、特に、DDF−RuV1及びDDF−RuV2の完全な可変領域(配列番号1、6、8、及び13)をコードするものとして提供する。核酸は、配列番号1、配列番号6、配列番号8、及び/又は配列番号13と少なくとも90%同一性を有していなくてはならない。
【0053】
このような配列、特に、特定のCDRに関連したもの含まれる配列(図2及び3)は、場合によりプロモーターに連結されたベクター及び発現カセットに含まれ、又はpDD系ファージミドにクローニングされ得る。この特定のタイプのベクターは、cp3又はcp8ファージコートタンパク質のいずれかとの融合タンパク質としてそれらの発現を可能にし、結果として、このようなファージミドを含み、それらの表面上で本発明のFabを発現する組換えファージを生産することができる。このようにして、本発明のタンパク質を発現するファージミドベクターを含む組換えファージは、RuV感染を検出及び/又は中和するための手段として使用可能である。
【0054】
本発明の配列がクローニングされ、対応する組換えファージによって特徴付けられたpDD系ファージミドがベクターに(部分的に又は全体として)移され得るDNA配列を含み、そこでは、元々のFab、又はそれらから誘導されるタンパク質配列は、宿主細胞内で組換えタンパク質として適切に発現され得る(pDLac−FLAGhisシステムを用いた実施例に示される)。
【0055】
ファージディスプレイ技術を用いて単離される免疫グロブリン可変鎖(特にヒト免疫グロブリン可変鎖)の場合、重要な修飾は、好ましいアイソタイプ及び定常領域を有する完全な免疫グロブリンタンパク質へ選択されたFab又はscFVの変換である。このようにして、完全なヒト抗体が望まれる場合、発現ベクターは、IgG1、IgG2、IgG3、IgG4、IgM、IgA及びIgE定常領域からなる群から選択される重鎖定常領域をさらに含まなければならない。この種の修飾は、例えば、ファージディスプレイライブラリー由来の一本鎖Fv又はFabから構築される全てのアイソタイプの完全なヒトモノクローナル抗体を生じ、哺乳動物細胞又は昆虫細胞において発現を可能にする。文献(Persic L et al.,1997 Guttieri M et al.,2003)に広く記載されているように、ベクターは、抗体を発現するために具体的に設計され、所望のアイソタイプ(例えば、ヒトIgGガンマ1)の定常(Fc)領域へのこの配列の融合を可能にする。
【0056】
抗体又は融合タンパク質は、原核生物(例えば、Escherichia coli;Sorensen H and Mortensen K,2005;Venturi M et al.,2002)、植物(Ma J et al.,2005)、又は真核細胞において組換えタンパク質として発現することができ、それらは、一時的又は安定に形質転換した細胞として高レベルの発現を可能にする(Dinnis D and James D,2005)。これは、特に、抗体の特徴がより厳しい機能的及び/又はインビボアッセイを用いて実行しなければならない場合に要求される。
【0057】
文献は、記事及び書籍の章(”Phage display:A Practical Approach”,vol.266,ed.Clackson and Lowman H,Oxford Univ.Press,2004;”Phage Display:A Laboratory Manual”,ed.Burton D et al.,CSHL Press,2001;Corisdeo S and Wang B,2004;Benhar I,2001)に概説されるように、原核宿主細胞において、Fab又は抗体フラグメントについての同様のフォーマットとしてタンパク質を発現するための異なるストラテジーを提供する。
【0058】
タンパク質、特に抗体が、真核宿主細胞(特に、哺乳動物細胞株)において発現される場合、異なるベクター及び発現系は、トランスフェクトされた細胞株の安定なプールを生じるように設計されている(Aldrich T et al.,2003;Bianchi A and McGrew J,2003)。高レベルの、最適化された、安定な組換え抗体の発現は、細胞培養条件の最適化(Grunberg J et al.,2003;Yoon S et al.,2004)、より高レベルの抗体生産及び分泌を有するクローンの選択又は操作(Bohm E et al., 2004; Butler M, 2005)によって達成される(Schlatter S et al.,2005)。
【0059】
対象とする配列(例えば、重鎖及び軽鎖の関連する可変領域)をコードする核酸配列は、ベクターの発現カセット、又は区別されるベクターの発現カセットに適切にクローニングされなければならず、この場合、それらは、適切な調節配列(例えば、プロモーター、転写ターミーネーター)に操作可能に連結される。発現カセットには、プロモーター、リボソーム結合部位(必要に応じて)、開始コドン、リーダー/分泌配列が含まれ、それらは、したがって、所望のタンパク質をコードするDNAを誘導体スルファターゼ得るモノ又はビシストロン転写物の発現を誘導することができる。
【0060】
ベクターは、構造的に活性であるか又は誘導可能であるように選択される転写開始/末端調節配列の調節下で、原核又は真核宿主細胞において本発明のタンパク質の発現を可能にしなければならない。このようなタンパク質を生産するための方法には、タンパク質発現に適切である条件下で、それらのコーディング配列を含む発現ベクターを用いて形質転換された宿主細胞を培養し、宿主細胞培養物からタンパク質を回収することが含まれる。本発明の核酸を含む宿主細胞は、原核又は真核宿主細胞であってもよく、所望の組換えタンパク質の分泌を可能にする。次に、このような細胞に実質的に富んでいる細胞株は、単離され、安定な細胞株を提供することができる。
【0061】
これらの核酸、組換えファージ、宿主細胞を生じ、共通の組換えDNA技術を適用することによって本発明のタンパク質を生産するために使用することができる。要約すると、所望のDNA配列は、制限酵素を用いたファージミドの消化によって抽出されるか、又はポリメラーゼ連鎖反応(PCR)のための鋳型、重鎖及び軽鎖の完全な可変領域又はそれらの一部だけ(例えばHCDR3)を特異的に増幅するPCRプライマーとして元々のファージミドを用いて増幅することができる。次に、このようなDNAフラグメントは、組換えタンパク質のクローニング法及び生産法についての多数の書籍及び概説に記載されるように、原核又は真核宿主細胞内に更なる修飾及び/又は発現のためにより適したベクターに移すことができ、シリーズのいくつかのタイトルであるオックスフォード大学プレスによって公開されている“A Practical Approach”( “DNA Cloning 2:Expression Systems”,1995;“DNA Cloning 4:Mammalian Systems”,1996;”Protein Expression”,1999;”Protein Purification Techniques”,2001)が含まれる。
【0062】
提示され、選択されたタンパク質配列をコードするDNA配列は、適切なエピソーム又は非相同的若しくは相同的統合ベクターに挿入されると、任意の適切な手段(形質転換、トランスフェクション、コンジュゲーション、原形質融合、エレクトロポレーション、リン酸カルシウム沈殿、直接的なマイクロインジェクションなど)によって適切な宿主細胞に導入可能である。特定のプラスミド又はウイルスベクターを選択する際の重要な因子には、ベクターを含む受領細胞が認識され、ベクターを含まない受領細胞から選択される容易性;特定の細胞において望まれるベクターのコピー数;異なる種の宿主細胞間でベクターを「シャトル」することができることが望まれるかどうかが含まれる。
【0063】
真核宿主(例えば、酵母、昆虫又は哺乳動物細胞)については、異なる転写及び翻訳調節配列が使用されてもよく、これは、宿主の性質に依存する。それらは、ウイルス起源、例えば、アデノウイルス、ウシパピローマウイルス、シミアンウイルスなどから誘導されてもよく、この場合、調節シグナルは、高レベルの発現を有する特定の遺伝子と関連付けられる。実施例は、ヘルペスウイルスのTKプロモーター、SV40初期プロモーター、酵母gal4遺伝子プロモーターなどである。転写初期調節シグナルが選択されてもよく、一時的(又は構造的)抑制及び活性、結果として、遺伝子発現の調節を可能にする。更なるクローニング工程において、抗体又は融合タンパク質をコードする配列が適用され、DNA及びタンパク質レベルの両方でのみDNAレベルの特定の修飾について他のベクターに再度クローニングすることができる。これらの変化は、例えば、コドン使用頻度及び制限部位が特定のベクター及び宿主細胞を用いた組換えタンパク質をクローニングし、発現するのに最も適しているDNA配列を選択するためのソフトウェアを用いて測定することができる(Rodi D et al.,2002;Grote A et al.,2005;Carton J et al.,2007)。また、タンパク質配列は、所望の抗体フォーマット(Scfv、Fab、完全なヒト抗体など)、又は1以上の内部アミノ酸の挿入、置換、若しくは欠失に関連して付加することができる。
【0064】
また、これらの技術は、抗体の治療的特性の更なる構造及び機能的特徴付け及び最適化のために使用され(Kim Sら,2005)、あるいは、それらの安定なインビボでの送達を可能にするベクターを生じさせるために使用可能である(Fang Jら,2005)。例えば、また、組換え抗体は、可変領域に融合されるべき特定のFc領域を選択することによって(Furebring Cら,2002;Logtenberg T,2007)、組換え一本鎖抗体フラグメントを生じさせることによって(Gilliland Lら,1996)、安定化ペプチド配列を融合することによって(WO01/49713)、又は放射化合物若しくはポリマーを化学的に修飾された残基に添加することによって(Chapman Aら,1999)、構造及び/又は活性のレベルで調節可能である。
【0065】
導入されたDNAによって安定に形質転換された細胞は、発現ベクターを含む宿主細胞の選択を可能にする1以上のマーカーを導入することによっても選択することができる。また、マーカーは、栄養要求宿主への光合成、殺生物耐性、例えば抗生物質、又は重金属、例えば銅などを与えてもよく、必要に応じて、開裂可能であり、又は抑制されてもよい。選択可能なマーカー遺伝子は、発現されるべきDNA遺伝子配列に直接的に連結されるか、又は同時トランスフェクションによって同じ細胞に導入されてもよい。また、追加の転写調節因子は、最適発現に必要であってもよい。
【0066】
宿主細胞は、原核又は真核のいずれかであってもよい。原核宿主細胞の中では、好ましいものは、B.subtilis及びE.coliである。真核宿主細胞の中では、好ましいものは、酵母、昆虫細胞(バキュロウイルス系発現システムを使用)、又は哺乳動物細胞、例えばヒト、サル、昆虫(バキュロウイルス系発現システムを使用)及びチャイニーズ・ハムスター・オバレイ(CHO)細胞であり、これは、それらが、正しいフォールディング又は正しい位置でグリコシル化のある種の形態を提供するためである。また、酵母細胞は、グリコシル化を含む翻訳後ペプチド修飾を実行することができる。強力なプロモーター配列、酵母における所望のタンパク質の生産のために利用可能な高コピー数のプラスミドを利用する多数の組換えDNAストラテジーが存在する。酵母は、クローニングされた哺乳動物遺伝子産物においてリーダー配列を認識し、リーダー配列(即ち、プレペプチド)を有するペプチドを分泌する。
【0067】
組換えポリペプチドの長期、高収率な生産に関して、安定な発現が好ましい。例えば、対象とするポリペプチドを安定に発現する細胞株は、同じであるか又は別個のベクターに、複製のウイルス起源及び/又は内因性発現要素、及び選択可能なマーカー遺伝子を含んでもよい。ベクターの導入後、細胞は、それらが選択培地にスイッチされる前に強化培地において1〜2日間増殖させることができる。選択可能なマーカーの目的は、選択への耐性を付与することであり、その存在は、誘導される配列を首尾よく発現する細胞の増殖及び回収を可能にする。安定に形質転換された細胞の耐性クローンは、細胞型に適切な組織培養技術を用いて増殖可能である。次に、このような細胞において実質的に富んだ細胞株が単離され、安定な細胞を提供することができる。宿主細胞は、組換えタンパク質の発現レベルに基づいてさらに選択可能である。
【0068】
RuVを結合及び中和可能である抗体、抗体フラグメント、生物活性ペプチド、融合タンパク質、上記定義された任意の他の化合物は、細胞培養物又は合成調製物からの組換えタンパク質としてそれらを精製可能にする十分に確立した技術(即ち、抽出、沈殿、クロマトグラフィー、電気泳動など)を用いて提供することができる。これらの調製物は、組換えタンパク質の十分な量(ミクログラムからミリグラムの範囲)を提供して、RuVに関連した予防、診断、及び治療用途のためにより広範囲な特徴付け及び評価を行わなければならない。
【0069】
抗体精製の方法は、カラム内に含まれる固相化したゲルマトリックス(Nisnevitch M and Firer M,2001;Huse K et al.,2002;Horenstein A et al.,2003)、特に、基質、例えばプロテインA、プロテインG、または合成基質に対する抗体の一般的な親和に基づいて(Verdoliva A et al.,2002;Roque A et al.,2004)、並びに抗原又はエピトープに基づくアフィニティークロマトグラフィー(Murray A et al.,2002;Jensen L et al.,2004)を利用することができる。洗浄後、タンパク質は、pH又はイオン強度の変化によってゲルから溶出される。あるいは、HPLC(高速液体クロマトグラフィー)を用いることができる。溶出は、タンパク質精製に一般に使用される水−アセトニトリル系溶媒を用いて実行可能である。
【0070】
組換えタンパク質の調製物は、インビトロ又はインビボアッセイ(生化学、組織又は細胞系アッセイ、齧歯類において確立した疾患モデル、親和性測定のための生物物理学的方法、エピトープマッピングなど)において、特に、RuV病因及び免疫学を兼有するための文献に開示されたものの1以上を用いて、例えば、国際的な健康協定(WHO2005;WHO2006)に従って定義される野生型及び参照RuV菌株を用いて試験することができる。
【0071】
RuVを結合及び中和することができるものとして上記で定義された抗体、抗体フラグメント、生物活性ペプチド、融合タンパク質、及び任意の他の化合物は、RuV感染を検出、治療、阻害、予防、及び/又は改善するために使用することができる。この目的で、このような化合物は、RuV感染の医療管理のための診断、治療、又は予防用組成物を調製するために使用することができる。
【0072】
これらの組成物は、ヒトDDF−RuV1及びDDF−RuV2の配列及び活性に基づいて上記で定義された抗体、抗体フラグメント、融合タンパク質、生物活性ペプチド、任意の他の化合物を含んでもよい。組成物は、異なるRuV中和抗体又は抗体フラグメント、静脈内免疫グロブリン(IVIg)調製物、及び/又は抗ウイルス化合物をさらに含んでもよい。異なるRuV中和抗体又は抗体フラグメントは、異なる中和エピトープ、例えば文献に既に記載されているものによって特徴付けられなければならない。実際には、ぶんけんは、多くの例を示し、そこでは、ウイルス又はヒト標的に指向される2以上の抗体が医薬組成物に組み合わせられる場合、得られる組成物は、単なる相加効果ではなく、特定の相乗効果に起因して、改善された治療効果を有してもよい(Logtenberg T,2007)。
【0073】
医薬組成物は、場合により、任意の医薬として許容されるビヒクル又は担体を含んでもよい。これらの組成物は、任意の付加的な治療薬又は予防薬、例えば、ワクチン、静脈内免疫グロブリン調製物、又は抗ウイルス化合物をさらに含んでもよい(又は一緒に投与されてもよい)。また、最近の文献は、ヒトモノクローナル抗体は、この静脈内免疫グロブリン調製物に補足する(可能であれば、置換する)ために用いられ、このような医薬組成物の頻度及び/又は投薬量を減少させる機会を与えることができる(Bayry Jら,2007)。
【0074】
任意のタンパク質(例えば、抗体、抗体フラグメント、融合タンパク質、生物活性ペプチド)、上記で定義される核酸を含む組成物は、RuVに関連した診断、治療、又は予防の目的のある個体に投与することができる。これらの組成物は、RuVビリオンを標的化することによって、治療される患者内のウイルスの繁殖を阻害し、潜在的には、集団におけるウイルス感染の発生をブロックすることができる治療的化合物(特に、治療用抗体又は治療用抗体フラグメント)を提供するRuV特異的受動免疫のための手段として投与することができる。
【0075】
特定の使用に依存して、組成物は、長期間又は短期間、ヒト対象(幼児、妊娠女性、年配の個体、又はRuVに感染し、又はRuV感染した個体との接触、入院、又は免疫抑制/化学療法的治療に起因してRuVに危険であると考えられる任意の他の個体)に化合物を提供されなければならない。この目的で、組成物は、筋内、静脈内、皮下、局所、粘膜、噴霧器、吸引、点眼、非/生分解性マトリックス材料中、又はミクロビーズなどの微粒子薬物送達システムを用いることにより投与することができる。
【0076】
医薬組成物は、特に、眼内、ウイルス血症に関連した皮膚疹でのウイルスの存在があれば、眼内又は局所投与のために、十分な期間、その化合物が活性を発揮することができる化合物の治療的又は予防的な有効量を対象に提供しなければならない。抗体又は抗体フラグメントを含む組成物が、創傷、皮膚、粘膜、又は角膜に局所投与される場合に効果的であることが証明された(Brereton H et al.,2005;Castle P et al.,2002;Streit M et al.,2006;Nwanegbo E et al.,2007)。
【0077】
所望の効果は、RuV感染、再活性化、及び/又は再感染を調節することによって、RuV感染の臨床症状の少なくとも幾つかを低減させることによって、患者の状態を改善することである。例えば、組成物は、投与経路、個人の状態に依存して、約0.005〜約50mg/kg/体重の有効量で投与されなければならない。
【0078】
診断的使用を有する組成物の場合において、化合物は、生物学的試料中にウイルスを検出するための臨床及び研究所において一般に確立した技術(例えば、ELISA又は他の血清学的検定)を用いて、又は投与後の少なくとも1、2、5、10、24時間、若しくはそれを超えてインビボで対象に投与される場合に、検出されなければならない。RuVの検出は、免疫コンピテント及び免疫不全宿主の両方の危険な集団においてRuV感染をモニターするために確立された既知の手段及び手法に代えて又は結びつけて、本発明のタンパク質を用いて実行することができる。
【0079】
RuV又はRuVに関連した疾患の治療、予防、又は診断のための方法は、上記で定義されるタンパク質又は核酸の投与を含むことが可能である。この方法は、異なるRuV中和抗体又は抗体フラグメント、静脈内免疫グロブリン(IVIg)調製物、及び/又は抗ウイルス化合物の投与をさらに含んでもよい。
【0080】
臨床開発及び使用は、抗体薬物動態及び薬理学の特徴付け(Lobo Eら,2004)、臨床前及び臨床安全データ(Tabrizi M及びRiskos L,2007)、並びにヒトにおける治療的及びインビボでの診断的使用のために治療的組換え抗体の商業生産スケール調合及び分析的特徴付けのための正式な公的要求(Harris Rら,2004)に基づくべきである。
【0081】
ここで、本発明は、下記の実施例によって記載されるが、多少なりとも、本発明を制限するものとして解釈されてはならない。
【実施例】
【0082】
実施例1:ELISAにおけるRuVタンパク質抽出物に結合するヒトFabの発現及び分泌
材料&方法
ライブラリー構築
ヒトIgG1の重鎖及び軽鎖をコードするcDNAは、文献(Burioni et al.,1998;”Phage Display:A laboratory Manual”,Burton DR et al.,CSHL Press,2001)に従って、RuV血清陽性反応の個体から得たリンパ球から得られた。ファージライブラリーは、PCT特許出願WO07/007154に記載される技術に従って、pDDベクターと適合するクローニングカセットを用いて構築された。Fabは、DDカセット内にクローニングされ、ライブラリーの組換えファージの表面に発現された。pDDに基づくFabライブラリーのパンニングを介したヒトFabの選択、陽性クローンの配列決定は、文献(Burioni Rら,1998)に記載されるように行った。
【0083】
Fabの特異的なCDRは、IMGT/V−QUEST(Giudicelli Vら,2004)、European Bioinformatics Instituteによって提供されるもの、FASTA(http://www.ebi.ac.uk/fasta33/index.html)を用いて検出可能なものなどのヒト抗体のタンパク質配列を含む他のデータベースによって提供される予測及び配列整列を比較することによって特定された。
【0084】
細胞培養の細胞からのタンパク質抽出物の精製
VERO細胞(ATCC Acc.No.CCL−81)は、単層として増殖するサル線維芽細胞株であり、通常、RuVを含むヒトウイルスを試験し、繁殖させるために使用される。RuVに対する中和抗体は、VERO細胞を用いて特徴付けられている(Cordoba Pら,2000b)。
【0085】
RuV臨床単離物(H2)を用いて感染させたVERO細胞は、ファージディスプレイライブラリーをパンニングし、ELISAにおいてFabを試験するためのRuV特異的材料の調製のために使用した。VERO細胞は、10%の不活化されたウシ胎児血清(FBS)、50μg/mlのペニシリン、100μg/mlのストレプトマイシン、及び2mMのL−グルタミンを含む修飾されてイーグル培地において維持される。
【0086】
細胞(RuV感染又は未感染VERO)を回収し、250mlの溶解バッファー(50mM Tris−HCl pH8.0;150mM NaCl;0.02%アジ化ナトリウム;0.5% Triton−X)に再懸濁させ、氷上で20分間インキュベートし、次に、12000rpmで2分間、4℃で遠心分離した。得られた上清のタンパク質濃縮物は、BCAタンパク質アッセイキット(Pierce)を用いて2重に測定し、既知濃度でウシ血清アルブミン(BSA)の連続希釈に関してレポートした(0mg/ml=ブランク〜2.000mg/ml=最大値)。全ての試料の吸光度は、分光計セットを用いて540nmで測定した。
【0087】
タンパク質抽出物を用いたパンニング及びELISA
ELISAのためのタンパク質コーティングは、下記の抗原:VERo細胞の溶解物、RuV感染又は未感染(臨床単離物H2)、ウシ血清アルブミン(BSA)を用いて96ウェルプレートで行った。各試料は、カーボネートバッファー(1ウェルあたり25μlの最終体積に全タンパク質の100ナノグラム)で希釈し、プレートを一晩、4℃でインキュベートした。蒸留水で洗浄後、プレートを1%BSAを含むPBSにおいて1時間37℃でインキュベートすることによってブロックした。
【0088】
ELISAは、文献(Bugli Fら,2001)に開示されているプロトコールを用いて下記のFab:DDF−RuV1、DDF−RuV2、およびe137、C型肝炎ウイルスに特異的な無関係なFabを含む未希釈の試料40μlを用いて行った。Fabは、二重のウェルで試験した。各Fabを用いて、1時間37℃でインキュベートし、0.1%Tween−20を含むPBSで5回洗浄後、40μlのヤギ抗ヒトFab、ペルオキシダーゼ結合体(Sigma;Cat no.A0293)を添加し、1時間37℃でインキュベートした。プレート洗浄は上述のように繰り返し、酵素反応は、各ウェルに40μlの基質(TMB基質キット;Pierce)を添加することによって発色させた。ELISA反応は、15分間37℃で発色させた。酵素火星は、停止溶液(H2SO4)の添加によって停止させ、分光計セットを用いて、450ナノメートルで吸光度を測定した。
【0089】
ELISA及び中和アッセイのためのFab調製
プロトコールは、pDD又は他のファージミドを用いた文献(“Molecular Cloning:A Laboratory Manual”,Sambrook et al., Cold Spring Harbor Press,NY,1989;Burioni R et al.,1998; Bugli F et al.,2001;WO07/007154)に記載されるのと同じであった。
【0090】
要約すると、ライブラリーの個々のE.coliクローンは、抗生物質を含み、IPTGを補足されたSuper Broth(SB;3.5%バクト−トリプトン、2%酵母抽出物、0.5%NaCl)培地200mlで増殖させ、回収し、リン酸緩衝生理食塩水(PBS)で洗浄した。溶解は、調節されたパルスを用いた4℃での超音波処理によって、ペリプラスム空間に限定された。ペリプラスム抽出物中のFabは、45分間4℃でJA−10ローターで12,000rpmの超遠心分離によって部分的に精製した。生産物をろ過し、Centrionフィルターを用いて10倍に濃縮した。
【0091】
部分的に精製されたFabの濃縮物は、96ウェルプレート(Costar;Cat no.3690)の表面に結合させたImmunoPureヤギ抗ヒトIgGF(ab’)2(Pierce;Cat.no.31132)を用いたサンドイッチELISAによってペリプラスム抽出物で測定した。37℃での1時間のインキュベーション後、脱イオン水でプレートを6回洗浄し、2%BSAを含む170μl/ウェルPBSを用いてブロックした。37℃でさらに1時間のインキュベーション後、1%BSAを含むPBS中の各Fabの連続して3倍希釈物の50μl、又は既知濃度の対照のヒトFab(Cappel Labs;Cat.No.6001−0100)を各ウェルに添加し、37℃で1時間インキュベートした。次に、プレートをTPBS(0.05%Tween−20を有するPBS)で6回洗浄した。次に、抗体結合は、アルカリホスファターゼを結合したヤギ抗ヒト抗体(Pierce;Cat.No.31312)を50μl添加することによって測定し、37℃で1時間インキュベートした。プレート洗浄は、上述されるようにTPBSを用いて繰り返し、100μlのp−ニトロフェニルリン酸2ナトリウム(Sigma)を各ウェルに添加した。ELISA反応は、60分間発色させ、結果は、既知の濃度で希釈した対照ヒトFabに対してプロットした。全ての試料の吸光度は、590nmで分光計セットを用いて測定した。
【0092】
結果
組換えファージのライブラリーは、pDD技術(WO07/007154)に従って生じさせ、RuVの臨床単離物を用いて感染させた線維芽細胞株から得たタンパク質抽出物について評価した。また、5ラウンドのパンニングは、RuVよって感染していない同じ細胞株からの対照タンパク質抽出物を用いて同ライブラリーを用いて並行して行った。
【0093】
第3ラウンドによって、対照タンパク質抽出物に対して評価した試料のファージ力価は、104を下回り、一方、RuV抽出物に対して評価した試料のファージ力価は、第4ラウンドで104を超え、第5ラウンドでは105に到達した。この値は、RuV抗原に結合するそれらの表面Fabに発現している組換えファージにおけるライブラリーの進行的濃縮を示す。
【0094】
パンニングの第5ラウンド後に得られた14個のクローンは、ELISAにおいて個別に試験され、陽性であることが確かめられた。選択されたクローンにおけるHCDR3のPCR及び配列分析により、ここで命名されたDDF−RuV1及びDDF−RuV2のヒトFabを特徴付ける2つの重鎖が同定された。DDF−RuV1又はDDF−RuV2を発現している組換えファージの反応性は、RuV特異的タンパク質抽出物、並びに無関係な抗原(ウシ血清アルブミン、未感染細胞からのタンパク質抽出物)に対して試験され、ELISAフォーマットにおいてそれらの結合活性が確かめられた(図1)。また、結合の特異性は、対照のヒトFabを用いて確認された。
【0095】
これらのFabの完全な重鎖及び軽鎖可変領域のDNA配列、並びにDDF−RuV1及びDDF−RuV2に対して、対応するCDRを決定した(それぞれ、図2及び3)。これらのFabはまた、タンパク質発現のためのE.coli系システムを用いて、更なるアッセイのための十分な組換えタンパク質を得るための他のベクターに再度クローニングされた。
【0096】
実施例2:RuV感染させた細胞培養物について試験されたDDF−RuV1及びDDF−RuV2の特性
材料&方法
RuV特異的ヒトFabの中和アッセイ
免疫蛍光に基づくアッセイは、37℃で72時間のインキュベーション後には、通常、コンフルエントな単層を形成する条件下で、プレートに植菌された104〜105個のVERO細胞を用いて、Costerの24ウェルプレートで行った。RuV臨床単離物であるH2を感染に使用した。
【0097】
Fabは、実施例1に指示されるように部分的に精製し、維持培地に懸濁させた50TCID50(50%組織培養感染用量)での等体積のRuV細胞不含ストックを用いて種々の濃度(0.01、0.1、1、10及び50μg/ml)で混合した。対照は、Fabなし(ブランク対照)、又はヒトC型肝炎ウイルスに特異的な無関係なFab(e137;Bugli Fら,2001)の存在で、等体積の維持培地及びウイルスから構成された。
【0098】
37℃でのインキュベーションの1時間後、250μlのウイルス−フラグメントFab混合物又は対照混合物は、培地が除かれたウェル(2重)に植菌された。プレートを2時間37℃でインキュベートし、中和されていないウイルスの吸着を可能にする。植菌物を除去し、1.5mlの維持培地を添加した。細胞培養条件で1週間のインキュベーション後、細胞をPBSで洗浄し、冷メタノール−アセトン溶液(1:2比)で10分間、室温で固定した。固定された細胞は、30分間、37℃で、湿気雰囲気でRuVのタンパク質E1に特異的な市販のモノクローナル抗体(Chemicon;Cat.No.MAB925)とともにインキュベートし、PBSで洗浄し、最後に、30分間、37℃で、湿気雰囲気で抗マウスIgG FITC結合体(Sigma)とともにインキュベートした。スライドをPBSで洗浄し、Evans Blue色素で対比染色し、グリセロールを積層した。各Fabの中和活性は、蛍光顕微鏡(Olympus)によって観察される単一の蛍光を発する細胞をカウントし、対照試料の細胞(Fab前インキュベーションなし)と比較して、RuV陽性細胞の数の減少の割合を計算することによって測定された。
【0099】
結果
DDF−RuV1及びDDF−RuV2に関するRuV中和活性の分析は、部分的に精製したFabの調製物の使用及び免疫蛍光に基づくアッセイによって行った。
【0100】
RuV感染細胞の減少に関するデータは、DDF−RuV1及びDDF−RuV2が、RuVとともにプレインキュベートされた場合に中和活性とともに与えられることを指示する。実際に、対照(FabなしのRuVまたは無関係なFabとのRuV)と比較すると、これらのRabの添加は、投薬量に依存して感染細胞の数を減少する(図4)。
【0101】
本明細書において提示された実験的証拠は、RuV感染に関連した診断、治療、又は予防的応用のためのDDF−RuV1及びDDF−RuV2Fab(又はそれらの特異的なHCDR3に基づく代替のタンパク質配列であって、類似の性質を示す)候補化合物を作製する。
【0102】
DDF−RuV1及びDDF−RuV2は、細菌宿主細胞における組換えタンパク質として発現することができる。特に、これらのFabは、pDLac−FLAGhisベクターなどのDDカセット(PCT/IB2008/000266)に存在する制限部位と適合するベクターにそれらのコーディング配列を再度クローニングすることによって発現可能である。このベクターは、ヘテロ二量体複合体を形成する適切な定常領域、重鎖のC末端での6−His及びFagを含む可溶性Fabの発現を可能にする(図5)。このアプローチは、Fabのより直接的なクローニング、検出(例えば、ELISAにおける)及び精製(例えば、固相化した金属親和性カラムに基づく手法を用い、免疫親和性精製と比較してより簡易でありよりコストが抑えられる)を可能にする。
【0103】
実施例3:RuV特異的な中和エピトープを決定するためのpDDに基づくペプチドライブラリーの構築及び評価
RuV中和Fabは、パンニングのためのRuVで感染させたVERO細胞のタンパク質抽出物を用いて、ヒトFabのpDD系ライブラリーをパンニングすることによって初期に同定された。その後、RuV特異的な中和活性は、細胞系モデルにおいて試験されたが、実際にはDDF−RuV1及び/又はDDF−RuV2によるウイルスエピトープは特定されなかった。
【0104】
類似のエピトープは、過去における異なるアプローチを用いて想定され、合成ペプチドELISA(Giessauf Aら,2004)、タンパク質欠失突然変異体(Chaye Hら,1992)、マウスモノクローナル抗体のパネルを用いた競合分析(Green K及びDorsett P,1986)、又はこれらの技術の組み合わせ(Wolinsky Jら,1993)が含まれる。
【0105】
この範囲で、pDD技術は、WO07/007154における血球凝集素エピトープタグ(HAtag)を利用することによって例示されるように、2つの機能性ファージコートタンパク質の1つ又は他方のいずれかのファージ表面に提示されるペプチドライブラリーを構築するために適用することができる。pDD技術を用いて提示された非ランダム又はランダムペプチドのライブラリーは、抗体を特異的に結合するペプチドを提示するファージを選択するためにスクリーニングすることができる。ライブラリーにおいて選択されるペプチド配列モチーフは、抗体特異的なエピトープの特定を可能にする(Zhong Gら,1997;Yip Yら,1999a)。
【0106】
異なるファージディスプレイフォーマットを用いて提示させたペプチド配列をスクリーニングし、比較するという重要性は、文献に示されている。第1の例では、モノクローナル抗体のエピトープは、異なる長さのペプチドを各々含み、異なるファージコートタンパク質に融合された2つのファージディスプレイライブラリーを用いて、ファージによって提示されたエピトープ(又は「ファゴトープ」)内で選択された。スクリーニングの結果は、異なるペプチド配列は、2つのファージディスプレイプラットフォームを用いて選択されたということであり、これは、ファゴトープの特徴付けが、ファージコートタンパク質における比較タイプのペプチド提示を必要とし得ることを示唆する(O’Connor Kら,2005)。同様の証拠はまた、有機(例えば、ヒトの毛髪又は皮膚)又は無機(例えば、ポリプロピレン)支持体に結合するペプチドの場合において見出された。文献(Scholle Mら,2005)に記載された方法、又はそれに従って修飾された方法などの、異なる長さを有するペプチドの市販のファージによって提示されたライブラリーをスクリーニングし、比較することによって、得られる配列が顕著に異なる(WO07/03551、WO06/094093、WO06/094094、WO06/094095)。
【0107】
2つの異なるライブラリーは、pDDファージミド(pDDc)と市販のライブラリー(Ph.D.12ファージディスプレイライブラリーキット;New England BioLabs,cat,no.E8110S)との組み合わせによって構築され、それは、抗体の線状又は立体的エピトープを決定するための過去においてすでに使用され(Li Yら,2007;Petit Mら,2003)、異なる長さのペプチドライブラリーを生じるために修飾された(WO07/03551、WO06/094093、WO06/094094、WO06/094095)。このライブラリーは、64個の可能なコドンのうちほんの32個を含む。それは、第3の位置がT又はGに限定されているためであって、単一コドンを有する残基の相対的頻度を増加させ、3つの終始コドンの2つ(第3のものは適切なE.coli菌株において抑制され、グルタミンに翻訳され得る)を除去するためである。
【0108】
このライブラリーに含まれるファージミド由来の複製の二本鎖形態は、XhoI制限部位を含むフォワードプライマー、SpeI制限部位を含むリバースプライマーを用いたPCR増幅のための鋳型として使用された。第2のPCR増幅は、鋳型と同じライブラリー、KpnI制限部位を含むフォワードプライマー、EagI制限部位を含むリバースプライマーを用いて行われた(図6A)。
【0109】
PCR増幅に起因するDNAフラグメントが消化され、各々が約108の力価を有し、初期には、cp3にだけ融合する、2つの異なるpDD系ペプチドライブラリーを生じるためのpDDcベクターにクローニングされた。第1のケースでは、pDDcXSmer−orcp3ライブラリーは、XhoI及びSpeI制限部位におけるPCR増幅産物をクローニングすることによって生じた(図6B)。第2のケースでは、pDDcKE12mer−orcp3ライブラリーは、中間のpDDcKEベクターにPCR増幅をクローニングすることによって生じさせ、それには、XhoI/SpeI部位の間のKpnI及びEagI制限部位、HAコーディング領域の5’/3’末端を含む(図6C;WO07/007154)。
【0110】
クローニング部位としてSpeIの使用は、特定の効果を提供することを気づくことは重要である。それは、この制限酵素は、元々にペプチドライブラリーに存在する2つの連続コドン:スレオニンをコードするACT、セリンをコードするAGTに対応する配列(ACTAGT)内で切断するためである。他の2つの可能なフレームは、ライブラリー内で可能にしないため(即ち、A及びCは、コドンにおける第3の位置として存在しない)、DDカセット内でライブラリーをクローニングするためのSpeIの利用は、pDDベクターのSpeI部位がpDDcの場合のように同じコーディングフレームを有する場合に、低い頻度でペプチドのより短い変異体の導入を可能にする(図6B及び6C)。
【0111】
このようなアプローチは、AlwNI(CAGNNNCTGを認識)、NdeI(CATATGを認識)、HindIII(AAGCTTを認識)、又はPvuII(CAGCTGを認識)などの類似の特性(第1塩基としてA又はC、第2塩基としてA又はC、第3塩基としてG又はT、6又は9塩基対を認識する)を有する認識部位を有する他の酵素を利用するこの種のライブラリーの変異体を生じるために使用することができる。また、類似の部位は、元々のライブラリーにおける追加の非/ランダムコドンを無作為に統合するために使用されてもよく、ライブラリーの複雑性をさらに改善する。
【0112】
cp3又はcp8に融合したタンパク質を提示するために、2つのpDD系ペプチドライブラリーは、(例えば、XL1−blue E.coli菌株において)維持及び増幅することが必要である。ファージミドは精製され、BglI(DDカセットを抽出するために)で消化され、次に、BglI消化されたpDDcベクターに再度連結される。連結反応は、細菌を形質転換するために使用され、その結果、2つのライブラリーが選択され、Zeocin耐性細菌細胞として維持された。この場合、ファージミドが増幅され得て、ヘルパーファージによる感染後に組換えファージに含まれ、単一のクローンは、所望のスクリーニング後に分析可能である。
【0113】
この方法では、DDカセットのペプチドライブラリーがクローニングされ、対応するpDDcXSmer−orcp3/cp8及びpDDcKE12mer−orcp3/cp8ライブラリーにおける2つの可能な配向のいずれか1つにスクリーニングされる(図7;WO07/007154)。次にライブラリーは、E.coliで増幅され、組換えファージはパンニング実験の結果を比較することによって試験された。パンニングは、組換えファージを生じるためのヘルパーファージの使用を伴う。このファージの集団が単離され、文献に記載されるように、マイクロウェルプレートに被覆された対象の標的への結合親和性に基づいて試験することができる。対象の標的に対する3〜5つの連続的なパンニングラウンド後、特異的結合は、対照と比較されると、溶出されたファージの数の増加によって明示される。
【0114】
本ケースでは、2つのライブラリーは、市販の抗HAtag抗体(マウスモノクローナル抗体HA.11、クローン6B12;Covance)などの周知な線状エピトープを有する抗体に対して試験された。このアプローチは、ファージディスプレイプラットフォームを改善するための技術を評価するために従来使用されていた(Stratmann T及びKang A,2005;Vanhercke Tra,2005)。
【0115】
ライブラリーのタイトルにおける減少があるとしても(BglI消化、再連結、E.coli形質転換に関連する手法による)、関連した標的(抗HAtagモノクローナル抗体)又は負の対照(BSA)に対するパンニングの比較は、pDDcXSmer−orcp3/cp8(表I)及びpDDcKE12mer−orcp3/cp8(表II)ライブラリーに関して、計算されたインプット及びアウトプットについて有意な相違があることを示す。さらに、パンニングの第3ラウンドは、抗HAtagのアウトプットに対する102の特定濃縮をBSAの1つと比較することを示す。
【0116】
DDカセット及びペプチドが2つのライブラリーの各クローンに存在する配向は、制限分析によって容易に測定可能である(図7)。消化は、提示されるべき配列を有するDDカセットの末端近くを切断する酵素(例えば、XhoI又はSpeI)と、pDdベクターの骨格にあるコートタンパク質をコードする配列の末端を切断する酵素(例えば、cp3*についてはNheI)とを組み合わせることによって行われる。得られる制限パターンは、コートタンパク質をコードする配列だけを含むDNAフラグメント(DDカセットがcp3*に配向する場合)、又はDDカセット及びコートタンパク質をコードする配列を含むDNAフラグメント(DDカセットがcp8*に配向する場合)を示す。
【0117】
NheI/SpeI消化は、上記されるパンニング前及び/又は後に、2つのライブラリーから無作為に選択されるクローンのパネルにおいて行われた。消化から得られたDNAフラグメントが電気泳動によって分離されると、約0.6kbのフラグメントは、DDカセットがcp3*に配向される場合に検出され、約1.8kbのフラグメントは、DDカセットがcp8*に配向される場合に検出される。この分析は、全ての状況において、cp8*配向を有するクローンの僅かな有病率を示した(図8)。
【0118】
pDDcXSmer−orcp3/cp8及びpDDcKE12mer−orcp3/cp8ライブラリーにおいてクローニングされたペプチドの配列は、無作為に選択されたクローンについて決定され、良好なレベルの変動、pDDcXSmer−orcp3/cp8の場合には、7〜12個のアミノ酸の範囲のより短い配列を有するペプチドの存在の両方を確実にした(図9)。同じ分析は、抗HAtag抗体に対するpDDcKE12mer−orcp3/cp8をパンニング後に得られたクローンにおいてなされ、HAtag(テトラペプチドYPYD)を含むペプチドの存在が選択されたクローンにおいて確かめられた。
【0119】
ライブラリーにおけるHAtag特異的ペプチドをコードするクローンの存在及び選択の更なる確認として、パンニング選択後に生じた細菌クローンはニトロセルロース様メンブレン上に移され、アルカリ条件で溶解された。この方法では、細菌タンパク質は、ニトロセルロースペーパーに固相化され、次に、抗HAtagとともにインキュベートされる。HAtagを提示する多量のクローンを確認する(図10)。
【0120】
生物活性ペプチド(例えば、抗体、阻害剤又はプロテアーゼの認識部位を特徴付けるエピトープ、膜受容体に結合するペプチド、シグナルタンパク質、又は転写因子)が同定されるpDD系ペプチドライブラリーを生じ、パンニングするための本ストラテジーは、異なる使用又はクローニングストラテジーに適合することができる。ファージに提示されるランダムペプチドライブラリーの複雑性を改善及び評価するための条件は、文献に記載されている(Noren K et al.,2001;Rodi D et al.,2002)。
【0121】
例えば、オリジナルなペプチドライブラリー(その長さ及び複雑性を維持することが望ましい場合)は、クローニング及び形質転換プロセスのより良好な効率を可能にし得る基準の組み合わせを満足する制限酵素の異なる組み合わせを用いてクローニングされ得る。例えば、制限酵素のBstEII、MluI、AgeI、EcoRV、Sall、SnaBI、及びBg1IIは、pDDcベクター及びPh.D.12ライブラリーに切断するものがなく、(EagIと比較して)メチル化不感覚である。
【0122】
この実施例は、複数の融合タンパク質を含むpDD系ペプチドライブラリーを提供し、各融合タンパク質は、pDDベクターのDDリンカーを含む配列によって連結されるウイルス表面タンパク質(cp3*又はcp8*)及びランダムペプチド配列を含む。pDDベクター及びDDカセットの構造、機能、配列特徴は、WO07/007154において特定される。
【0123】
初期のペプチドライブラリーは、一般式NNK(Nは任意の塩基であり、KはG又はTである)を有するコドンを含むべきである。上述されるpDDベクターにおけるコドン使用及びフレームと適合され得る部位を認識する制限酵素による消化後、ライブラリーは、元々の配列の完全又はより短い変異体として実際にクローニングされる。これらのペプチド変異体は、一般には遺伝的パッケージ、特に細菌細胞又は線状ファージの表面に提示され得る。
【0124】
異なる長さを有し、pDDベクターのDDカセット内に2つの機能性ファージコートタンパク質の1つ又は他方のいずれかと融合されたペプチドを提示するためのファージミドのライブラリーを構築する方法は、下記を含む:
a)少なくとも制限酵素を用いてDDカセット内にライブラリーをクローニングするために使用される制限部位(例えば、SpeI)に対応する、少なくともコドンの組み合わせを含むDNAライブラリー(例えば、Ph.D.12ペプチドライブラリー)の消化;
b)得られたDNAライブラリーと、少なくとも同じ制限酵素を用いて消化されたファージミド(例えば、pDDc)との連結;
c)細菌細胞の形質転換のためのファージミドの第1ライブラリーの使用、及び細菌細胞の第1ライブラリーの生成;
d)細菌細胞の第1ライブラリーからのファージミドの単離;
e)DDカセットのDDリンカーに存在する制限酵素を用いて前記ファージミドの消化;
f)得られたDDカセットと、同じ制限酵素を用いて消化したpDDベクターとの連結;
g)細菌細胞の形質転換のためのファージミドの第2ライブラリーの使用、及び細菌細胞の第2ライブラリーの生成;
d)細菌細胞の第1ライブラリーからのファージミドの単離。
【0125】
このプロセスの最後に、DDカセットの調節配列、pDDベクターの1つ又は他の機能性ファージコートタンパク質(例えば、cp3*又はcp8*)の配列への操作可能に連結されたpDD系ペプチドライブラリーが生じる。このようなファージミドを含む細菌細胞のライブラリーは、パンニング、又は任意の他の種のネガティブ/ポジティブ選択のために使用される組換えファージの集団を得るために、ヘルパーファージを用いて感染させることができる。
【0126】
このクローニングアプローチは、コートされたペプチドの長さの修飾に2つのファージコートタンパク質(WO07/007154におけるpDD技術について指示されるように)の融合を組み合わせる。この方法は、所望の標的に対してスクリーニングされ得るペプチドをコードするファージミドの新規なライブラリー、及びペプチドの新規なライブラリーを提供する。実際に、この方法に従って得られる複数のファージミドは、E.coliの形質転換のために使用することができる。次に、得られる細胞は、組換えファージのライブラリーを生成するためのヘルパーファージを用いて感染させ得る。
【0127】
pDD系ペプチドライブラリーは、ファージによる提示されるランダムの元々の集団から選択される特定のペプチドをコードするDNA配列を増幅及び/又は配列決定するためのプライマーをさらに含むキットに(ファージミド又は細菌細胞として)含まれてもよい。
【0128】
pDD系ペプチドライブラリーは、特定の生物活性を有するペプチドを選択するためにスクリーニングされ得る組換えファージを生成するか、又は特定のリガンド分子に結合するペプチドを単離及び生成するために使用可能である。インビトロでのパンニングの場合、このようなライブラリーは、エピトープマッピング(O’Connor Kら,2005;Zhong Gら,1997;Yip Yら,1999a)、プロテアーゼ特異性のマッピング及びプロテアーゼ阻害剤の設計(Diamond S,2008;Sedlacek R及びChen E,2005)、あるいは細胞表面の抗原のスクリーニング(Mutuberriaら,2004)のために使用可能である。インビボのパンニングの場合、このようなライブラリー(並びに、cp3又はcp8のいずれかと融合するだけでなく、異なる長さを有するペプチドを提示するpDDcXSmer−orcp3/cp8ライブラリーのような特定のもの)は、細胞外空間内、及び細胞膜上でタンパク質−リガンド相互作用の同定を可能にする(Finger Aら,2002;Yip Yら,1999b)。
【0129】
【表1】
【0130】
【表2】
【0131】
【化1】
【0132】
【化2】
【0133】
【化3】
【0134】
【化4】
【0135】
【化5】
【0136】
【化6】
【特許請求の範囲】
【請求項1】
配列番号5と少なくとも90%の同一性を有する配列を含むタンパク質。
【請求項2】
前記タンパク質が、配列番号2と少なくとも90%の同一性を有する配列を含む、請求項1に記載のタンパク質。
【請求項3】
前記タンパク質が、配列番号7と少なくとも70%の同一性を有する配列をさらに含む、請求項1又は2に記載のタンパク質。
【請求項4】
前記タンパク質が、抗体、抗体フラグメント、生物活性ペプチド、又は融合タンパク質である、請求項1〜3のいずれか1項に記載のタンパク質。
【請求項5】
前記抗体フラグメントが、可変重鎖/軽鎖ヘテロ二量体又は一本鎖フラグメント可変部である、請求項4に記載のタンパク質。
【請求項6】
前記タンパク質が、風疹ウイルス(Rubella virus:RuV)に結合し、中和する、請求項1〜5のいずれか1項に記載のタンパク質。
【請求項7】
請求項1〜6のいずれか1項に記載のタンパク質をコードする核酸分子。
【請求項8】
前記核酸が、配列番号1と少なくとも90%の同一性を有する、請求項7に記載の核酸。
【請求項9】
請求項7又は8に記載の核酸を含むベクター。
【請求項10】
請求項7〜9のいずれか1項に記載の核酸を含む組換えファージ、原核宿主細胞、又は有核宿主動物。
【請求項11】
請求項1〜6のいずれか1項に記載のタンパク質を製造するための、請求項7〜9のいずれか1項に記載の核酸、又は請求項10に記載の組換えファージ若しくは宿主細胞の使用。
【請求項12】
RuV感染を検出、治療、阻害、予防及び/又は改善するための組成物を製造するための請求項1〜6のいずれか1項に記載のタンパク質の使用。
【請求項13】
請求項1〜6のいずれか1項に記載のタンパク質を含む治療用、予防用、又は診断用組成物。
【請求項14】
前記組成物が眼内又は局所投与用である、請求項13に記載の組成物。
【請求項15】
異なるRuV中和抗体、異なるRuV中和抗体フラグメント、静注用免疫グロブリン調製物、及び/又は抗ウイルス化合物をさらに含む、請求項13又は14に記載の組成物。
【請求項1】
配列番号5と少なくとも90%の同一性を有する配列を含むタンパク質。
【請求項2】
前記タンパク質が、配列番号2と少なくとも90%の同一性を有する配列を含む、請求項1に記載のタンパク質。
【請求項3】
前記タンパク質が、配列番号7と少なくとも70%の同一性を有する配列をさらに含む、請求項1又は2に記載のタンパク質。
【請求項4】
前記タンパク質が、抗体、抗体フラグメント、生物活性ペプチド、又は融合タンパク質である、請求項1〜3のいずれか1項に記載のタンパク質。
【請求項5】
前記抗体フラグメントが、可変重鎖/軽鎖ヘテロ二量体又は一本鎖フラグメント可変部である、請求項4に記載のタンパク質。
【請求項6】
前記タンパク質が、風疹ウイルス(Rubella virus:RuV)に結合し、中和する、請求項1〜5のいずれか1項に記載のタンパク質。
【請求項7】
請求項1〜6のいずれか1項に記載のタンパク質をコードする核酸分子。
【請求項8】
前記核酸が、配列番号1と少なくとも90%の同一性を有する、請求項7に記載の核酸。
【請求項9】
請求項7又は8に記載の核酸を含むベクター。
【請求項10】
請求項7〜9のいずれか1項に記載の核酸を含む組換えファージ、原核宿主細胞、又は有核宿主動物。
【請求項11】
請求項1〜6のいずれか1項に記載のタンパク質を製造するための、請求項7〜9のいずれか1項に記載の核酸、又は請求項10に記載の組換えファージ若しくは宿主細胞の使用。
【請求項12】
RuV感染を検出、治療、阻害、予防及び/又は改善するための組成物を製造するための請求項1〜6のいずれか1項に記載のタンパク質の使用。
【請求項13】
請求項1〜6のいずれか1項に記載のタンパク質を含む治療用、予防用、又は診断用組成物。
【請求項14】
前記組成物が眼内又は局所投与用である、請求項13に記載の組成物。
【請求項15】
異なるRuV中和抗体、異なるRuV中和抗体フラグメント、静注用免疫グロブリン調製物、及び/又は抗ウイルス化合物をさらに含む、請求項13又は14に記載の組成物。
【図1】

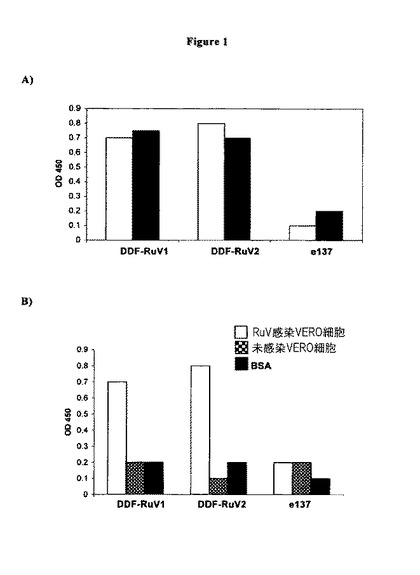
【図2】

【図3】

【図4】
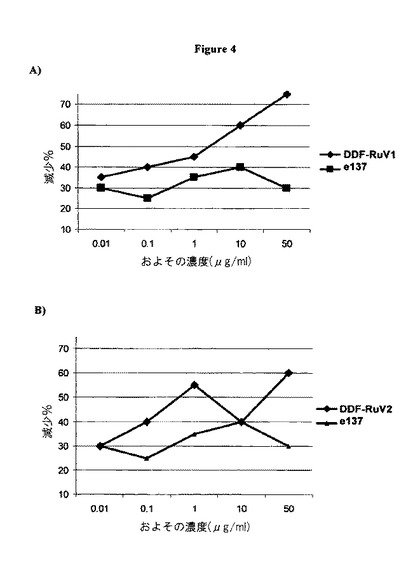
【図5】

【図6】

【図7】
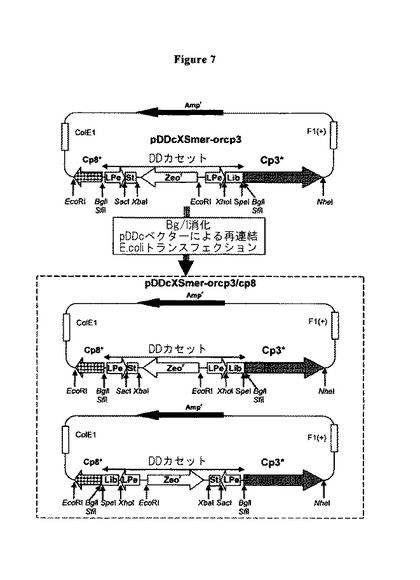
【図8A)】

【図8B)】
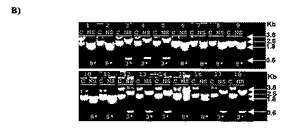
【図8C)】

【図9】
【図10】

【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8A)】
【図8B)】
【図8C)】
【図9】
【図10】
【公表番号】特表2010−519916(P2010−519916A)
【公表日】平成22年6月10日(2010.6.10)
【国際特許分類】
【出願番号】特願2009−552123(P2009−552123)
【出願日】平成20年3月6日(2008.3.6)
【国際出願番号】PCT/EP2008/001791
【国際公開番号】WO2008/107187
【国際公開日】平成20年9月12日(2008.9.12)
【出願人】(508178478)リボバックス バイオテクノロジーズ ソシエテ アノニム (6)
【Fターム(参考)】
【公表日】平成22年6月10日(2010.6.10)
【国際特許分類】
【出願日】平成20年3月6日(2008.3.6)
【国際出願番号】PCT/EP2008/001791
【国際公開番号】WO2008/107187
【国際公開日】平成20年9月12日(2008.9.12)
【出願人】(508178478)リボバックス バイオテクノロジーズ ソシエテ アノニム (6)
【Fターム(参考)】
[ Back to top ]