
食欲調整剤、治療薬、健康食品、動物用飼料、及び食欲調整方法

【課題】第1次摂食・代謝調節中枢に働きかける食欲調整剤を提供する。
【解決手段】食欲調整剤であり、視床下部弓状核のグルコース抑制性ニューロン(Glucose−Inhibited neuron)のNKA(Na++K+−ATPase)活性を調整するNKA活動調節剤を含むことを特徴とする。この調節剤は、NKAアクティベータであり、グルコース抑制性ニューロンの[Ca2+]i濃度を低下させる。これにより、従来とは作用機構の違う食欲促進剤の提供が可能となる。
【解決手段】食欲調整剤であり、視床下部弓状核のグルコース抑制性ニューロン(Glucose−Inhibited neuron)のNKA(Na++K+−ATPase)活性を調整するNKA活動調節剤を含むことを特徴とする。この調節剤は、NKAアクティベータであり、グルコース抑制性ニューロンの[Ca2+]i濃度を低下させる。これにより、従来とは作用機構の違う食欲促進剤の提供が可能となる。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、食欲調整剤、治療薬、健康食品、動物用飼料、及び食欲調整方法に係り、特に視床下部弓状核に作用する食欲調整剤、治療薬、健康食品、動物用飼料、及び食欲調整方法に関する。
【背景技術】
【0002】
近年、摂食とエネルギー代謝の調節メカニズムの研究が国内外で盛んに行われている。これは、世界的な肥満の増加と、肥満が多くの生活習慣病の起点になることが明らかにされたためであった。
図14を参照して、近年、明らかになっている摂食とエネルギー代謝の調節メカニズムについて説明する。
全身の栄養・代謝状態は、栄養素、ホルモンや自律神経の迷走神経求心路を媒介として脳に伝えられる。
これらの情報は、視床下部を中心とした中枢で情報が統合された後、自律神経遠心路や視床下部下垂体ホルモンを経由して脂肪細胞などの器官にシグナルが送られ、末梢代謝が制御されている。この一連の過程に障害が起こると、代謝恒常性の破綻や肥満をもたらし、ひいてはメタボリック・シンドロームの成因となると考えられる。
【0003】
従来、高等ほ乳類において、栄養・代謝状態を脳に伝える因子として、グルコース、インスリン、コレシストキニン(CCK)等が知られていた。
また、近年、脂肪細胞由来のレブチン、アディポネクチン、また胃由来のグレリンといった新たな摂食代謝調節因子が発見されている。
これらの因子は、栄養・代謝状態の末梢代謝シグナルであり、血液脳関門(Blood Brain Barrier、BBB)を通過して直接脳に取り込まれるか、又は、迷走神経求心路を経由して脳に情報伝達されることが分かっている。
このような末梢代謝シグナルが脳に入ると、主として視床下部弓状核(Arcuate nucleus、ARC)で受容され視床下部のネットワークにおいて統合的に処理される。
具体的には、脳において、まず視床下部弓状核等を中心とした第1次摂食・代謝調節中枢に作用し、次いで視床下部内の神経ネットワークにより統合的に情報処理される。
その情報処理の結果は、自律神経、下垂体ホルモン分泌、高次中枢に出力され、最終的に摂食行動やエネルギー代謝が調節されると考えられている。
このように、高等ほ乳類では、視床下部からの出力が自律神経活動、下垂体ホルモン分泌、高次機能などを調節することにより、摂食と代謝を調節している。
すなわち、血液脳関門を経た入力、視床下部弓状核での情報統合、自律神経を経た出力が特に重要である。
【0004】
ここで、特許文献1を参照すると、従来の肥満症治療のための薬学的組成物として、食事行動を修正する化合物、そのプロドラッグまたは前記化合物または前記プロドラッグの薬学的に許容可能な塩;および、β−アドレナリン作働剤と前記プロドラッグの薬学的に許容可能な塩を含む薬学的組成物が記載されている(以下、従来技術1とする。)。
特許文献1の薬学的組成物に含まれるβ−アドレナリン作働剤は、アデニル酸サイクラーゼを活性化するため、抗肥満症剤として有用である。また、さらに特許文献1の段落[0013][0015]等を参照すると、食事行動を修正する化合物として、上述のレプチンのような摂食代謝調節因子を食欲抑制剤として用いることが示されている。
すなわち、特許文献1の薬学的組成物は、神経伝達物質とそのシグナル伝達系に働きかけること、及びホルモンやアディポカインを利用して、動物、特にヒトの肥満症を処置するのに有用性を有する薬学的組成物および方法を提供していた。
なお、従来技術1は、食欲抑制剤として、脳内神経伝達物質でもあるセロトニン作働剤やドーパミンアゴニストも用いるように記載していた。これらの脳内神経伝達物質は、向精神薬の標的となっており、脳全体に作用するため、第1次摂食・代謝調節中枢にだけ直接作用するものではなかった。
【先行技術文献】
【特許文献】
【0005】
【特許文献1】特開平11−228447号公報
【発明の概要】
【発明が解決しようとする課題】
【0006】
しかしながら、従来技術1のように、神経伝達物質に働きかけることにより、「脳全体に作用する」ような特異性がない薬学的組成物を用いると、食欲/摂食行動を直接、選択的に変化させることができないため、副作用が大きいという問題があった。
【0007】
このため、摂食行動に関わる「食欲」そのものを変更するような、視床下部弓状核等を中心とした第1次摂食・代謝調節中枢に直接作用する食欲調整組成物が求められていた。
【0008】
本発明は、このような状況に鑑みてなされたものであり、上述の課題を解消することを課題とする。
【課題を解決するための手段】
【0009】
本発明の食欲調整剤は、視床下部弓状核におけるグルコース抑制性ニューロン(Glucose−Inhibited neuron)のNKA(Na++K+−ATPase)活性を調整するNKA活動調節剤を含むことを特徴とする。
本発明の食欲調整剤は、前記NKA活動調節剤は、NKAインヒビターであり、前記グルコース抑制性ニューロンの[Ca2+]i濃度を増加させることを特徴とする。
本発明の食欲調整剤は、前記NKAインヒビターは、ニューロペプチドYの発現を惹起することを特徴とする。
本発明の食欲調整剤は、前記NKAインヒビターは、強心配糖体であり、該強心配糖体は、ウワバイン(ouabain)又はジゴキシン(digoxin)を含むことを特徴とする。
本発明の食欲調整剤は、前記NKAインヒビターは、NKAのαサブユニットの活動を調整することを特徴とする。
本発明の食欲調整剤は、前記NKAインヒビターは、強心配糖体としての投与の濃度の1/10以下の量を投与することを特徴とする。
本発明の治療薬は、前記食欲調整剤を用いたことを特徴とする食欲不振、抗癌剤の副作用による食欲の低下、加齢、過労、低栄養、神経性食思不振症、うつ病を含む精神疾患、自律神経失調症、拒食症を含む摂食障害のいずれかに対する治療のための治療薬であることを特徴とする。
本発明の食欲調整剤は、前記NKA活動調節剤は、NKAアクティベータであり、前記グルコース抑制性ニューロンにおける低グルコース誘発[Ca2+]i増加を抑制又は阻止することを特徴とする。
本発明の食欲調整剤は、前記NKAアクティベータは、ニューロペプチドYの発現を抑制することを特徴とする。
本発明の食欲調整剤は、前記NKAアクティベータは、NKAのαサブユニットに結合することを特徴とする。
本発明の食欲調整剤は、前記NKAアクティベータは、抗体SSA412であり、前記グルコース抑制性ニューロンにおける低グルコース誘発[Ca2+]i増加を抑制又は阻止することを特徴とする。
本発明の治療薬は、前記食欲調整剤を用いたことを特徴とする、過食、過食による肥満、過食による糖尿病、メタボリックシンドロームのいずれかに対する治療のための治療薬であることを特徴とする。
本発明の健康食品は、前記食欲調整剤を用いたことを特徴とする。
本発明の動物用飼料は、前記食欲調整剤を用いたことを特徴とする。
本発明の食欲調整方法は、視床下部弓状核のグルコース抑制性ニューロン(Glucose−Inhibited neuron)のNKA(Na++K+−ATPase)活性を調整し、前記グルコース抑制性ニューロンの出力を調整することを特徴とする。
【発明の効果】
【0010】
本発明によれば、視床下部弓状核のグルコース抑制性ニューロン(Glucose−Inhibited neuron、GIニューロン)のNKA(Na++K+−ATPase)活性を調整するNKA活動調節剤を用いることで、第1次摂食・代謝調節中枢に直接作用する食欲調整剤を提供することができる。
【図面の簡単な説明】
【0011】
【図1】本発明の実施の形態に係る視床下部弓状核のGIニューロンの食欲調整のモデルを示す概念図である。
【図2】本発明の実施の形態の実施例におけるGIニューロンの同定、[Na+]i又は[Ca2+]i応答測定の概念図である。
【図3A】本発明の実施の形態の実施例において、GIニューロンとして定義したニューロンの[Ca2+]i応答のグラフである。
【図3B】本発明の実施の形態の実施例において、GIニューロンの8.3mMグルコース(8.3G)と2.8mMグルコース(2.8G)における[Ca2+]i応答の平均を示すグラフである。
【図3C】本発明の実施の形態の実施例において、非GIニューロンとして定義したニューロンの[Ca2+]i応答のグラフである。
【図3D】本発明の実施の形態の実施例において、非GIニューロンの8.3Gと2.8Gにおける[Ca2+]i応答の平均を示すグラフである。
【図4A】本発明の実施の形態の実施例において、グルコース濃度低下とウワバインによるGIニューロンの[Na+]iの応答の変化を示すグラフである。
【図4B】本発明の実施の形態の実施例において、[Na+]iとSBFI蛍光レシオの関係を表わす較正曲線の図である。
【図4C】本発明の実施の形態の実施例において、グルコース濃度低下による[Na+]iの増加を示し、同じニューロンが[Ca2+]i増加を示すことから、GIニューロンであることを示すグラフである。
【図4D】本発明の実施の形態の実施例において、グルコース濃度低下に対して[Na+]iの変化が見られず、同じニューロンが[Ca2+]iの変化を示さないことから、非GIニューロンであることを示すグラフである。
【図4E】本発明の実施の形態の実施例において、GIニューロン及び非GIニューロンにおけるグルコース濃度低下に誘発された[Na+]iの変化の平均を示すグラフである。
【図5A】本発明の実施の形態の実施例において、8.3Gでウワバインを投与した際の[Ca2+]i応答を示すグラフである。
【図5B】本発明の実施の形態の実施例において、ウワバインを投与した際の[Ca2+]i応答の平均を示すグラフである。
【図5C】本発明の実施の形態の実施例において、8.3Gで、K+を含まない(K+−free)溶液を用いた際の[Ca2+]i応答を示すグラフである。
【図5D】本発明の実施の形態の実施例において、K+−free溶液を投与した際の[Ca2+]i応答の平均を示すグラフである。
【図5E】本発明の実施の形態の実施例において、ウワバインを投与しない状態で、同一のGIニューロンをグルコース濃度低下による反復刺激をした場合の[Ca2+]i応答のグラフである。
【図5F】本発明の実施の形態の実施例において、8.3G及び2.8Gにおける、グルコース濃度低下1回目刺激(Amp1)又はグルコース濃度低下2回目刺激(Amp2)における平均[Ca2+]i応答の平均を示すグラフである。
【図5G】本発明の実施の形態の実施例において、ウワバインが継続的に存在していた場合のグルコース濃度低下反復二回目刺激により誘発された[Ca2+]i応答を示すグラフである。
【図5H】本発明の実施の形態の実施例において、グルコース濃度低下1回目刺激(Amp1)の後、100μMのウワバイン存在下でグルコース濃度低下2回目刺激(Amp2)を与えた際の[Ca2+]i応答の平均を示すグラフである。
【図5I】本発明の実施の形態の実施例において、GIニューロン又は非GIニューロンにおいて、ウワバイン濃度の変化に対して反応した細胞数の割合を示すグラフである。
【図5J】本発明の実施の形態の実施例において、GIニューロン又は非GIニューロンにおいて、ウワバイン濃度の変化に対する[Ca2+]i応答の平均を示すグラフである。
【図5K】本発明の実施の形態の実施例において、グルコース濃度低下によるGIニューロンの活性化においてウサギIgGを加えたコントロール実験の結果を示すグラフである。
【図5L】本発明の実施の形態の実施例において、グルコース濃度低下によるGIニューロンの活性化においてSSA412を加えた実験の結果を示すグラフである。
【図5M】本発明の実施の形態の実施例において、グルコース濃度低下によるGIニューロンの活性化において、ウサギIgG又はSSA412を加えた際の[Ca2+]i応答の平均示すグラフである。
【図6A】本発明の実施の形態の実施例において、グルコキナーゼの拮抗阻害剤である10mMのMHを投与した際の[Ca2+]i応答を示すグラフである。
【図6B】本発明の実施の形態の実施例において、代謝されないグルコース・アナログである10mMの2DGを投与した際の[Ca2+]i応答を示すグラフである。
【図6C】本発明の実施の形態の実施例において、GIニューロン又は非GIニューロンにおいて、MHに対する[Ca2+]i応答を示した細胞数を示すグラフである。
【図6D】本発明の実施の形態の実施例において、GIニューロン又は非GIニューロンにおいて、2DGに対する[Ca2+]i応答を示した細胞数を示すグラフである。
【図6E】本発明の実施の形態の実施例において、GIニューロン又は非GIニューロンにおいて、MHを投与した際の[Ca2+]i応答の平均を示すグラフである。
【図6F】本発明の実施の形態の実施例において、GIニューロン又は非GIニューロンにおいて、2DGを投与した際のGI及び非GIニューロンの平均[Ca2+]i応答の平均を示すグラフである。
【図7A】本発明の実施の形態の実施例において、グルコース濃度低下によるNAD(P)H自己蛍光低下を示し、同じニューロンが[Ca2+]i増加を示すことから、GIニューロンであることを示すグラフである。
【図7B】本発明の実施の形態の実施例において、GIニューロンの8.3G及び2.8GにおけるNAD(P)H自己蛍光の平均を示すグラフである。
【図8A】本発明の実施の形態の実施例において、100μMのKCNを投与した際の[Ca2+]i応答を示すグラフである。
【図8B】本発明の実施の形態の実施例において、KCNを投与した際の[Ca2+]i応答の平均を示すグラフである。
【図8C】本発明の実施の形態の実施例において、1μMのFCCPを投与した際の[Ca2+]i応答を示すグラフである。
【図8D】本発明の実施の形態の実施例において、FCCPを投与した際の[Ca2+]i応答の平均を示すグラフである。
【図9A】本発明の実施の形態の実施例において、Ca2+を含まない(Ca2+−free)溶液を投与した[Ca2+]i応答を示すグラフである。
【図9B】本発明の実施の形態の実施例において、各VDCCの非特異性のブロッカーである高濃度Ni2+(300μM)を投与した際の[Ca2+]i応答を示すグラフである。
【図9C】本発明の実施の形態の実施例において、N型Ca2+チャンネル・ブロッカーであるω−コノトキシンGVIA(300nM)を投与した際の[Ca2+]i応答を示すグラフである。
【図9D】本発明の実施の形態の実施例において、L形Ca2+チャンネル・ブロッカーであるニフェジピン10μMを投与した際の[Ca2+]i応答を示すグラフである。
【図9E】本発明の実施の形態の実施例において、図9A〜Dの[Ca2+]i応答の振幅の平均を、コントロール実験と比べて相対的に表現したグラフである。
【図10】本発明の実施の形態の実施例において、自由摂食下と24時間絶食下において、視床下部弓状核のNKA活性の変化を示すグラフである。
【図11】本発明の実施の形態の実施例において、自由摂食下と24時間絶食下において、GEニューロンが主要な働きをしている視床下部腹内側部でのNKA活性の変化を示すグラフである。
【図12A】本発明の実施の形態の実施例において、5mMグルコース(5G)−HKRB溶液、ウワバイン0.5nM、K+−free溶液(10μl)を脳室内投与した際の、摂食量の変化の平均を示したグラフである。
【図12B】本発明の実施の形態の実施例において、5G−HKRB溶液、MH、さらにMHとSSA412(SSA412)、MHとウサギIgG(Rabbit IgG)を脳室内投与して、摂食量の変化の平均を示したグラフである。
【図13A】本発明の実施の形態の実施例において、ウワバインの脳室内投与後の視床下部弓状核のNPYのmRNA発現量を示すグラフである。
【図13B】本発明の実施の形態の実施例において、ウワバインの脳室内投与後の視床下部弓状核のPOMCのmRNA発現量を示すグラフである。
【図14】従来の摂食調節メカニズムについての概念図である。
【発明を実施するための形態】
【0012】
<実施の形態>
上述したように、副作用の少ない摂食調節用の化合物が必要とされていた。このため、本発明の発明者らは、視床下部弓状核等を中心とした第1次摂食・代謝調節中枢に直接働きかけて、「食欲」そのものを調節するための食欲調節剤を探し求めることを考えた。
この際、本発明の発明者らは、視床下部弓状核(ARC)にあるグルコース抑制性(Glucose−inhibited、GI)の神経細胞(Neuron、ニューロン)に着目した。以下、このグルコース抑制製の神経細胞をGIニューロンと呼ぶ。
ここで、グルコース(glucose)は、細胞活動に対して不可欠なエネルギー基質であるため、一般的には、グルコース濃度の上昇は細胞活動の上昇をもたらす。
しかしながら、GIニューロンは、このグルコースの働きに対する逆説的な例外である。すなわち、GIニューロンは、外側視床下野の視床下部弓状核で最初に発見され、グルコース濃度の上昇により活動が抑制されるという特徴がある。つまり、GIニューロンの電気的活性及び細胞内Ca活性は、グルコース濃度低下(LG)に応じて増加する。
また、視床下部弓状核のGIニューロンは、強力な食欲促進作用をもつニューロペプチドY(Neuropeptide Y、NPY)を含んでおり、空腹・飢餓による摂食亢進に関連していることが示唆されている。
【0013】
このGIニューロンの制御機構については、従来、不明であった。しかしながら、1974年に、Oomuraが、細胞膜にある能動輸送系であるナトリウムポンプの抑制が、GIニューロンのグルコース濃度低下による活性化を媒介するという仮説を提唱していた(Oomura et al.、1974、「Glucose inhibition of the glucose−sensitive neurone in the rat lateral hypothalamus.」、Nature、247、p.284−286、参照)。この仮説において、Oomuraは、「したがって、グルコースによるこれらのニューロンの抑制は、透過性変化によらないが、それが強心性配糖体あるいは抗代謝物質で有効にブロックされるので、エネルギー依存のナトリウムポンプの活性化による薄膜過分極で引き起こされるように見える。」と述べている。
【0014】
しかしながら、GIニューロンでは、上述のOomuraの仮説のように、ナトリウムポンプであるNa+/K+−ATPアーゼ(NKA)の関与、更にcl−チャネルおよびK+チャンネルが関与の仮説が提唱されていたものの、直接的な証拠はなかった。つまり、グルコース濃度の低下に誘発されたニューロンの活性化の機構は、実際には殆ど分かっていなかった。
このため、本発明の発明者らは鋭意実験と検討を行い、グルコース濃度低下と連動したNKA活性の低下が、GIニューロンの活性化と摂食に重大な役割をしていることを見いだした。つまり、NKA活動調節剤により、食欲をコントロールすることが可能になる。
【0015】
ここで、NKAは、αとβの2種のサブユニットからなる膜貫通タンパク(EC3.6.3.9)である。
NKAは、動物細胞において、ATP(Adenosine TriPhosphate)の加水分解により、細胞内からナトリウムイオン(Na+)を汲み出し、カリウムイオン(K+)を取り込むのでナトリウム−カリウムポンプとも呼ばれている。
この酵素は、ラットやヒト等のすべての動物細胞でみられる。NKAは、1つのATP分子を加水分解して、細胞から3つのNa+を排出し、2つのK+を組み入れる能動輸送系である。
細胞は、NKAによって、細胞内イオン濃度、浸透圧及び含水量を調節している。NKAによる3Na+排出、2K+組み入れの電荷の差し引きにより形成される膜電位(Em)は、神経細胞では細胞膜の過分極に関連している。
また、NKAは、特定の種類の細胞では、グルコースやアミノ酸の能動輸送に必要なNa+濃度勾配を形成する役割がある。
細胞は、グルコース代謝で産生した数10%ものATPを、NKAを始めとするイオン能動輸送の過程に用いている。
【0016】
次に、図1を参照して、本発明の発明者らが明らかにした視床下部弓状核のGIニューロンの食欲調整のモデルについて説明する。
上述のように、本発明の発明者らは、グルコース濃度低下によりNKA活性が抑制されると、これによりGIニューロンが活性化され、摂食活動を亢進させることを明らかにした。
すなわち、グルコース濃度が低下すると:
(1)グルコース代謝の低下
(2)細胞内エネルギー産生の低下
(3)NKAの活性低下による細胞の脱分極
(4)電位依存性Ca2+チャネルの活性化
という一連の流れにより、GIニューロンが活性化され、摂食活動が亢進する。
【0017】
より詳しく説明すると、グルコース濃度低下は、GIニューロンのグルコース代謝及びATPを含むエネルギー生産を減少させ、これにより膜電位変化を伴う起電性におけるNKA活性の低下に結びつく。
このようにNKA活性が引下すると、細胞膜が脱分極し、電圧依存性Ca2+チャンネル(Voltage−Dependent Calcium Channel、VDCC)を開口する。
すると、細胞内へのCa2+流入が誘発され、結果としてCa2+に依存したGIニューロンの活性化を引き起こすことになる。
ここで、Ca2+に依存したGIニューロンの活性化は、GIニューロンがNPYを含有することから、NPYニューロンの活性化に繋がっている(Muroya S., Yada T., Shioda S., Takigawa M.、1999、「Glucose−sensitive neurons in the rat arcuate nucleus contain neuropeptide Y.」、Neuroscience Lett.264(1−3)、p.113−116、及びKohno D、et al、2003、「G hrelin directiy interacts with neuropeptide−Y−containing neurons in the rat arcuate nucleus:Ca2+ signaling via protein kinase A and N−type channel−dependentmechanisms and cross−talk with leptin and orexin.」、Diabetes、52、p.948−956、参照)。また、NPYは、摂食促進、すなわち「食欲」に関わっていることが分かっている。
すなわち、NKAは、低グルコースによるニューロンの活性化に関与し、GIニューロンの活性化によるNPY依存性食欲促進に関わっている。
【0018】
このように、本発明の発明者らは、グルコース濃度低下は細胞内のグルコース代謝及びエネルギー生産を減少させ、これにより、起電性NKAの活性が低下し、GIニューロンの脱分極及び電圧依存性Ca2+の流入が導かれることを見いだした。
また、本発明の発明者らは、NKAによるGIニューロンの調節が摂食行動に関連づけられるという新規概念を見いだした。つまり、本発明の発明者らの研究は、グルコース/エネルギー代謝の低下を、摂食行動を刺激する視床下部弓状核GIニューロンの興奮に変換するという、NKAの注目すべき潜在的な役割を明らかにした。さらに、身体中の細胞におけるユビキタスな細胞基本活動維持分子であるNKAが、摂食行動に関連する視床下部弓状核の食欲促進のニューロンを調節する特異的な生理機能を発揮することを見いだした。
このため、この機構を調整することで、摂食行動を調整するための新たな薬剤の開発が可能になる。
つまり、本実施形態においては、NKA活動を調整するNKA活動調節剤として、NKAアクティベータ/インヒビターを用いることで、肥満及び/又は糖尿病に関連した摂食亢進症を改善する、新規手法を提供する。
【0019】
よって、視床下部弓状核のGIニューロンのNKAを活性化すると、食欲を司るGIニューロンの活動を抑制し、「食欲」を抑えることができる。すなわち、第1次摂食・代謝調節中枢による「食欲」を抑制することが可能になる。
このため、本発明の実施の形態に係る食欲調整剤において、GIニューロンのNKAの活性を活性化する各種のNKAアクティベータとなる組成物を用いることができる。このNKAアクティベータを含む食欲調節剤は、食欲抑制剤として用いることができる。
NKAアクティベータとして用いる組成物としては、例えば、抗体SSA412(Xu, K. Y.、2005、「Activation of (Na++K+)−ATPase.」、Biochem Biophys Res Commun.、338、p.1669−1677、参照)を用いることができる。視床下部弓状核では、生理的な各種摂食代謝調節因子により食欲を調整する必要があるため、比較的、血液脳関門の物質透過性が高い(リーキー)ため、抗体でも通過できる可能性がある。また、ファージ等を用いた分子量の小さい抗体を作成することで、血液脳関門を通過させることも考えられる。加えて、血液脳関門を通過させるため、この抗体の活性化部位を用いた低分子のペプチドを合成することもできる。
なお、この抗体SSA412は、上述の文献に記載されているように、心筋細胞においてはNKAを活性化して[Ca2+]iを増加させるという働きがある。ところが、本発明の発明者らが明らかにしたところ、GIニューロンでは、SSA412は、[Ca2+]iを減少させる、すなわちCa2+の細胞内流入を阻害する方向に働くため、上述の文献からは想起できなかった。このため、抗体SSA412は、本発明の実施の形態に係る食欲抑制剤として自明ではない。
【0020】
なお、NKAアクティベータは、任意のものを用いることができる。たとえば、血液脳関門を通過し、NKAを活性化するペプチドホルモンを用いることができる。また、NKAを活性化する各種低分子化合物や薬剤を用いることができる。この際に、SSA412と同様にNKAのαサブユニットに結合してNKA活性を上げるようにすることができる。ここで、本実施形態のNKAアクティベータは、GIニューロンのNKAに特異的に結合して活性化する薬剤を用いることが特に好適である。例えば、他の細胞のNKAでは影響を及ぼさないように、ドラッグ・デリバリー・システムを用いた各種薬剤を用いることができる。たとえば、リポソームにNKAを活性化する薬剤を封入し、脳特異的に作用するように構成できる。加えて、siRNA、PNA等も用いることができる。
さらに弓状核ではNKAのαサブユニットのうちα1の発現が他の組織に比べて数倍高いことが報告されている(例えば、Antonelli MC et al、「Localization of Na, K−ATPase isoforms in the hypothalamus of the rat.」、1995、Cellular Molecular Biology、41、p.79−85を参照。)。このため、NKAのα1サブユニットのみに対するNKAインヒビター/アクティベータを用いることで、身体の他の部位への副作用が少ない食欲亢進剤/食欲抑制剤を提供することが可能になる。
さらに、GIニューロンの活性を押さえるためには、全身の他のNKA活性に関連しない程度の低容量の薬剤を用いるだけでも効果的である。このため、NKA活性を亢進させる各種の薬剤を、副作用の少ない低容量で用いることが可能となる。
後述する実施例における図5I、図5Jのように、実際に、ラットにおいて、脳室内投与を行った際に、通常実験で用いられる濃度の1/10程度でもGIニューロンの応答を行わせることができる。このため、経口/通常の静脈注射による投与であっても、強心剤として用いられるウワバインやジゴキシンの1/10の量で、食欲増進の効果があると考えられる。すなわち、NKAインヒビターは、強心配糖体としての投与の濃度のログオーダーで−1(1/10)以下の量を投与することが好適である。
このため、通常より低い濃度のウワバインを用いても、食欲を増進させられると考えられる。
【0021】
以上のように構成することで、以下のような機能を得ることができる。
まず、従来より、中枢セロトニン・ノルアドレナリン・カンナビノイド系に作動する食欲抑制薬が開発され臨床使用されている。
その多くが、肺高血圧症、心弁膜症等などの循環系の障害、うつ病や自殺者の増加を起したために中止されている。
現在日本で唯一臨床使用可能な食欲抑制薬である、ノルアドレナリン系作動薬であるマジンドールは、副作用があり抗肥満効果は限定的であった。
このように、脳の神経伝達に作用する物質は重篤な副作用を伴っており、これは従来技術1の薬学的組成物も同様であった。
よって、別の機序により有効に食欲を制御する肥満症治療薬が待望されている。
【0022】
これに対して、本発明の実施の形態に係る食欲抑制剤は、神経伝達物質ではなく、グルコース抑制性ニューロンの機能に着目した。すなわち、上述したように、グルコースのシグナル伝達機構の中心として、NKAの作用に介入して摂食を調節する。
これにより、本発明の実施の形態に係る食欲抑制剤は、従来技術1とは異なり、「食欲促進ニューロン」を直接抑制することができる。さらには、この食欲促進ニューロンは同時に全身エネルギー代謝を調節していることから、視床下部からの出力経路を修飾し、それにより脂肪細胞・肝臓の代謝を調節し、更なる抗肥満作用があることが期待される。同時に、脂肪細胞由来物質の視床下部へのフィードバック入力をも修飾し、統合的な食欲抑制が可能となる。
また、脳内において、GIニューロンは、視床下部弓状核の、第1次摂食・代謝調節中枢をはじめとする数か所にのみ特異的に存在するため、副作用を抑えられる。
このため、ごく少量の薬物を用いて、食欲を根本的に抑制できると考えられ、副作用の少ない食欲抑制や肥満解消を実現させることができる。よって、本発明の実施の形態に係る食欲抑制剤は、効果的な摂食抑制作用が得られ、抗肥満効果が期待でき、メタボリック・シンドロームの解消に資することができる。
これにより、本発明の実施の形態に係る食欲抑制剤は、食物摂取量を制限するために、糖尿病等にも用いることができる。
【0023】
また、視床下部GIニューロンのNKAを抑制することで、食欲を司るGIニューロンの活動を活性化させ、第1次摂食・代謝調節中枢による「食欲」を亢進させることが可能になる。
このため、本発明の実施の形態に係食欲調節剤において、GIニューロンのNKAの活性を抑制する各種のNKAインヒビターとなる組成物を用いることができる。このNKAインヒビターを含む食欲調節剤は、食欲亢進剤として用いることができる。
このNKAインヒビターとしては、例えば、強心配糖体であるウワバイン(ouabain)、又はジゴキシン(digoxin)を用いることができる。ウワバインは、キョウチクトウ科(Apocynaceae)の種子から抽出される強心配糖体の一種であり、NKAの作用を阻害する。ウワバイン又はジゴキシンは、NKAの活性を抑制することで、細胞膜の内外にNa+、K+イオンの濃度差を引き起こしやすくし、これにより膜電位が変化する脱分極(depolarization)による心臓の拍動を助けるため、従来は強心剤として用いられている。
ウワバイン又はジゴキシンは、NKAを抑制する作用が強いものの、本発明者らの予備的な実験により、心臓等に作用の出ない低容量にてGIニューロンの活性化を引き起こすことが分かっている。
これにより、例えば、抗癌剤の副作用による食欲の低下、消化管手術後の食欲低下、加齢による食欲の低下、低栄養、神経性食思不振症、摂食障害を伴う、うつ病等の精神疾患・自律神経失調症等のQOL(Quality Of Life)の低下に係る疾病の治療として、食欲を亢進させることができる。
よって、食欲の不振による体力の低下を抑えることができる。
なお、NKAインヒビターとして、他の強心配糖体、ペプチドホルモン、siRNA、PNA等を用いることができる。また、NKAインヒビターも血液脳関門を透過し、視床下部弓状核のGIニューロンに特異的に結合するようなものを用いることが望ましい。
また、ヒトに投与する場合、ヒトに用いられる強心配糖体のジゴキシンとこのアナログとして、「ハーフジゴキシンKY」、「ジゴキシンKY」(京都薬品製、トーアイエイヨー、アイテラス)、「ジゴキシン「AFP」」(アルフレッサ製)、「ジゴキシン」(中外製薬製)、メチルジゴキシン「ラニラピッド」(中外製薬製)、デスラノシド「ジギラノゲン」(アイロム製)等の製剤を用いることができる。作用機構が同じであるため、本実施の形態におけるジゴキシンとウワバインの効果は、同様となる。
【0024】
このように、本発明の実施の形態に係る視床下部弓状核のGIニューロンのNKA活性を調整する調整剤を用いることで、NKAを活性化させて食欲抑制させ、NKAを抑制させて食欲増進させるという、2つの効果を目的に応じて得ることができる。
これにより、食欲を自在に調整して、適正な体重や体組成等の身体状態を得ることができるようになる。
【0025】
以上のように、摂食亢進性グルコース抑制性ニューロンを制御する鍵となる分子して、NKAの修飾により摂食を制御できる。
これにより、NKA活性化物質の開発による新規抗肥満薬である治療剤を開発することができる。このプロトタイプとなる物質として、SSAを用いることができる。
一方、NKA抑制物質により、術後・抗がん剤使用・高齢者の食欲低下に伴うQOL低下の改善の治療剤を開発することもできる。
【0026】
また、本発明の実施の形態に係る食欲亢進剤と食欲抑制剤とを同一のパッケージに入れて提供することも可能である。これにより、患者の意志で任意の間隔で食欲を調整できる。つまり、食事のタイミングを自在に調節して、各種疾病やダイエット等に適宜用いることができる。このため、血糖値を所定の間隔に保つ必要がある糖尿病患者や、間食が止められない肥満患者等に用いることができる。また、抗癌剤の副作用等により食欲が低下している患者が、病院食を提供する間隔で食欲がでるように調整することができる。
このように本発明の実施の形態に係る食欲亢進剤と食欲抑制剤とは、同一のNKAの活性の抑制/亢進機構により動作するので、それぞれが拮抗すると考えられる。また、同一の機構により食欲亢進/食欲抑制を行うため、薬剤耐性により使用量が増えることが抑えられると考えられる。また、体内の応答を過度に刺激することも少ないと考えら得る。
このため、副作用が少なく、健康食品や動物用飼料としても健康被害がなく用いることが期待できる。
【0027】
また、本発明の実施の形態に係る食欲抑制剤又は食欲亢進剤は、任意の製剤上許容しうる担体(例えば、生理食塩水、補助薬を含む等張液、例えばD−ソルビトール、D−マンノース、D−マンニトール、塩化ナトリウム等が挙げられ、適当な溶解補助剤、例えばアルコール、具体的にはエタノール、ポリアルコール、例えばプロピレングリコール、ポリエチレングリコール、非イオン性界面活性剤、例えばポリソルベート80(TM)、HCO−50等を挙げることができるが、それらに限定されない)と共に投与することができる。また、適切な賦形剤等を含んでもよい。
【0028】
また、本発明の実施の形態に係る食欲抑制剤又は食欲亢進剤は、製剤上許容しうる担体を調製するために、適切な薬学的に許容可能なキャリアを含み得る。
このキャリアとしては、シリコーン、コラーゲン、ゼラチン等の生体親和性材料を含んでもよい。あるいはまた、種々の乳濁液であってもよい。
さらには、例えば、希釈剤、香料、防腐剤、賦形剤、崩壊剤、滑沢剤、結合剤、乳化剤、可塑剤などから選択される1または2以上の製剤用添加物を含有させてもよい。
【0029】
本発明の実施の形態に係る食欲抑制剤又は食欲亢進剤は、経口投与のための投与に適した投与形態において、当該分野で周知の製剤上許容しうる担体を用いて処方され得る。
本発明の実施の形態に係る医薬組成物の投与経路は、特に限定されないが、非経口的に投与することが好ましい。
非経口投与としては、例えば、静脈内、動脈内、皮下、真皮内、筋肉内または腹腔内の投与が挙げられる。
【0030】
本発明の実施の形態に係る食欲調整剤は、脊椎動物の脳に存在する視床下部弓状核のグルコース抑制性ニューロンを作用対象としている。
この脊椎動物は特に限定されるものではなく、例えば、ヒト、家畜動物種、野生動物を含む。
このため、本発明の実施の形態に係る食欲抑制剤又は食欲亢進剤は、広く動物の治療、家畜の発育増進等の対象とすることができる。
また、疾病の予防や健康増進のため、健康食品のような食物、動物用の飼料、又は食餌に含ませることもできる。
【0031】
また、本発明の実施の形態に係る食欲抑制剤の治療対象としては、メタボリック・シンドロームのような、摂食変調を伴う疾病、糖尿病、高血圧等を含む。また、本発明の実施の形態に係る食欲亢進剤の治療対象としては、摂食障害を含む、痩身による体力低下に関する各種疾病に用いることができる。
【0032】
また、本発明の実施の形態に係る治療用組成物を上述の治療に用いるために、投与間隔および投与量は、疾患の状況、さらに対象の状態などの種々の条件に応じて適宜選択および変更することが可能である。
【0033】
本発明の実施の形態に係る食欲抑制剤又は食欲亢進剤の1回の投与量および投与回数は、投与の目的により、さらに患者の年齢および体重、症状および疾患の重篤度などの種々の条件に応じて適宜選択および変更することが可能である。
投与回数および期間は、1日1回約2〜4週間程度投与して状態をモニターし、その状態により再度あるいは繰り返し投与を行う。
【0034】
本発明の実施の形態に係る食欲抑制剤又は食欲亢進剤は、他の組成物等と併用することも可能である。また、他の組成物と同時に本発明の組成物を投与してもよく、また間隔を空けて投与してもよいが、その投与順序は特に問わない。
【0035】
また、本発明の実施の形態において、疾患が改善または軽減される期間は特に限定されないが、一時的な改善または軽減であってもよいし、一定期間の改善または軽減であってもよい。
【実施例】
【0036】
〔本発明の実施の形態に係る視床下部弓状核のGIニューロンのNKA活性の調整剤の実施例〕
以下で、本発明の実施の形態に係る治療用組成物について、具体的な実験を基にして、実施例としてさらに具体的に説明する。しかしながら、この実施例は一例にすぎず、これに限定されるものではない。
【0037】
上述の実施の形態で説明したように、本発明の発明者らは、鋭意実験と検討を行い、GIニューロンとNKAの関係について調べた。
まず、図2の実験の概念図を参照して、本実施例における、脳のニューロン同定/機能解析方法について説明する。本実施例では、摂食機能が完成した6週齢ラットの視床下部スライスを作製し、その中から弓状核を切離した。その後、機械的分散及び酵素処理により、ニューロンを単離した。そして、蛍光画像解析装置で、細胞内のNa+([Na+]i)及びCa2+濃度([Ca2+]i)を測定した。この際、実験に応じて、細胞外液を灌流して測定した。
【0038】
より具体的には、本発明の発明者らは、視床下部弓状核からGIニューロンを単離し、グルコース濃度低下と、NKA活性を抑制する薬剤であるNKAインヒビターであるウワバインを用いた実験を行った。この結果、グルコース濃度低下及びNKA抑制の両方が、視床下部弓状核のGIニューロンの[Na+]i及び[Ca2+]iの増加を引き起こすことを見いだした。
これに対して、NKAアクティベータとして動作する抗体であるSSA412によって処理をしたところ、グルコース濃度低下に誘発された[Ca2+]iの増加は起こらなかった。
また、本発明の発明者らは、NKAの上流の機構についても調べた。すると、グルコース濃度低下は、GIニューロンのNAD(P)H自己蛍光を減少させた。また、グルコキナーゼインヒビターを用いて処理を行ったところ、グルコース濃度増加により活動しなくなったGIニューロンの[Ca2+]iを増加させた。これにより、NKAがグルコース代謝の経路により制御されていることが示された。
さらに、本発明の発明者らは、NKAのパスウェイの下流の機構についても調べた。電圧依存性Ca2+チャンネル(VDCC)ブロッカーを用いてGIニューロンを処理した。すると、グルコース濃度低下に誘発された[Ca2+]i増加が抑制された。これにより、GIニューロンにおけるNKAの下流に、脱分極を介した機構があると推測できた。
この上で、本発明の発明者らは、NKAによる摂食行動の調節について、すなわちNKAの活性の調節により食欲を変化させられるかについて調べた。具体的には、ラットを絶食状態にして、視床下部弓状核のNKAの活性を調べた。すると、絶食により、視床下部弓状核のNKAの活性が減少した。また、NKAインヒビターであるウワバインを脳室内に注射したところ、摂食活動が亢進し、ニューロペプチドYの発現も惹起した。
これに加えて、グルコース代謝を抑制する薬剤であるグルコキナーゼインヒビターをラットの脳室内に注射したところ、摂食活動を喚起したが、NKAアクティベータであるSSA412を脳室内へ注射したところ、惹起された摂食活動が打ち消された。
このように、本発明の発明者らは、NKAの抑制がGIニューロンのグルコース濃度低下に誘発された活性化を媒介するという仮説の具体的な作用機構を証明した。
よって、本実施例により、エネルギー状態の低下をGIニューロンの刺激と摂食行動に結びつける視床下部弓状核のNKAの新規機能を明らかにし、視床下部弓状核のGIニューロンのNKA活性の調整剤を実現した。これにより、従来技術1とは異なり、副作用の少ない食欲抑制剤/食欲亢進剤をそれぞれ同一の作用機構を元に実現できる。
以下で、具体的な各実験と実施の例について、図面とデータを参照して説明する。
【0039】
(視床下部弓状核からの単一のニューロンの調製)
実験に用いたラットは、オスの成熟SDラットを、12時間の明/暗サイクル及び従来の食物(CE−2; クレア社製、東京、日本)及び水で無制限に飼育した。この研究のための動物プロトコルは、自治医科大学の動物保護委員会で承認された。
視床下部弓状核の単一のニューロンは、5〜7週のラット(Sprague−Dawley rat、SDラット)から単離された。ラットはウレタン(カルバミン酸エチル;1g/kg、i.p.)により麻酔をかけられ、脳を摘出された。そして、両側視床下部弓状核を含んでいる冠状断脳切片が準備された。その後、視床下部弓状核は両側から切離された。切り分けられた組織は、下記の成分からなるHEPESバッファーを用いたクレブス・リンガー重炭酸塩緩衝液(HEPES−buffered Krebs−Ringer bicarbonate buffer、HKRB)により洗浄した:
HKRBの組成(単位、mM): NaCl(129)、NaHCO3(5.0)、KCl(3.7)、KH2PO4、1.2、CaCl2(1.8)、MgSO4(1.2)、10mMグルコースを含んでいるHEPES・pH7.4(10)
【0040】
その後、切除された視床下部弓状核は、5〜10分間温和な機械的な分散(粉砕)をした後、20ユニット/mlのパパイン(papain)(シグマ・ケミカル社製、セントルイス、ミズーリ州、米国)、0.015mg/mlのデオキシリボヌクレアーゼ、0.75mg/mlのウシ血清アルブミン、及び1mmol/Lシステインを加えたHKRBを用いて、36℃のウォーターバスで15分振とう培養し、酵素処理を行った。この細胞懸濁液は5分間100×gで遠心分離された。
上澄みの溶液を捨てた細胞の沈殿物はHKRBで再懸濁され、カバーガラス上に分散して置かれた。細胞は6時間以内、飽和湿度の皿で、室温、遮光にて維持された。その後、単一のニューロンはカバーガラスに配置された。
【0041】
(単一の視床下部弓状核ニューロンの[Ca2+]i、[Na+]i及び[NAD(P)H]iの測定)
[Ca2+]i、[Na+]i、及び[NAD(P)H]iは、レシオメトリック・マイクロ蛍光イメージング法にて測定された。
イメージインテンシファイア付電荷結合素子(ICCD)カメラにより検出された。また、レシオ(比率)・イメージは、蛍光画像解析装置であるARGUS−50システム又はAquacosmosシステム(浜松ホトニクス社製、浜松、日本)を用いて取得した。
下記で、各測定のより具体的な方法を説明する。
【0042】
[Ca2+]iの測定は、単一ニューロンの調製後、2〜6時間静置し、レシオメトリックfura−2イメージングを用いて行った。この際、2μMのCa2+感受性色素fura−2/アセトキシメチル・エステル(AM)(株式会社ケミカル同仁製、熊本、日本)を負荷して、室温で1時間培養された。その後、細胞は倒立顕微鏡のステージ上の保温装置付チェンバーに取り付けられ、32℃で1ml/分でHKRBにより灌流された。
細胞は、340及び380nmで交互に12.0秒ごとに励起された、
510nm(それぞれF340及びF380)の発光信号はICCDカメラにより検出された。
また、レシオ・イメージ(F340/F380)はARGUS−50あるいはAquacosmosシステムで取得した。
データは従来に報告された手順により、形態学的及び生理学的なニューロン判定基準を満たす細胞から得られた。
薬剤に応じた[Ca2+]i増加のピーク振幅が、自発的な変動の130%以上だった場合、それらは[Ca2+]i応答であるとした。
薬剤に応じた[Ca2+]i増加の振幅は、ピーク[Ca2+]iレシオから刺激前の基準となる[Ca2+]iレシオを引くことにより計算された。
阻害剤の影響を調査する実験では、刺激性の薬剤に誘発された[Ca2+]i増加の振幅が30%以上阻害剤で抑えられた場合、抑制と判定した。
【0043】
[Na+]iの測定は、単一ニューロンの調製後2時間静置し、32℃で1.5〜2.0時間、Na+感受性色素であるナトリウム結合イソフタル酸ベンゾフラン(SBFI)(5μM)/AM(1μM)(モレキュラー・プローブズ社製、ユージーン、オレゴン、アメリカ)及び非イオンの分散剤であるプクロニック(Pluronic)F−127(0.0125%)(モレキュラー・プローブズ社)を負荷して培養された。
その後、細胞は倒立顕微鏡のステージ上の保温装置付チェンバーに取り付けられ、32℃で1ml/分でHKRBにより灌流された。
SBFI蛍光測定法のための手順は、fura−2のための標準的な手順に従った。
HEPESバッファーを用いたNa+を含まない(Na+−free)溶液は、NaClの代わりにN−メチルDグルカミン塩化物(N−methyl−D−glucamine)を用いることにより準備された。
単離細胞の[Na+]i値は較正曲線に従って計算された。
当該構成曲線は、膜内外のNa+およびK+濃度を平衡化するイオン透過担体グラミシジンD(MPバイオメディカルズ社製、アーヴィン、CA、アメリカ)10μg/mlを含んだ各種のNa+(5、10、15、30、60、100 mM)及びK+濃度(Na++K+=135、グルコン酸塩105、CaCl2 2.5、MgCl2 1.2、HEPES 10、及びグルコース8.3(単位、mM))を含む溶液に段階的に灌流することで、観察されたレシオを設定した。
【0044】
(同一ニューロンの[NAD(P)H]iおよび[Ca2+]iの測定)
以前に報告された同様の方法により、NAD(P)Hの自己蛍光が最初に測定された。
細胞は360nmで励起された。また、470nmの蛍光発光が検出された。各時点における蛍光強度F360は、測定開始時の蛍光強度F360(0)に対する割合(F360/F360(0))により計算された。
[NAD(P)H]iを測定した後に、同一ニューロンにおいて[Ca2+]i測定のために、細胞に室温で1時間、2μM fura−2を負荷した。また、上述した手順で[Ca2+]iが測定された。
【0045】
(同一ニューロンの[Na+]iおよび[Ca2+]iの測定)
上に述べられるような[Na+]iの測定の後に、同一ニューロンにおいて、[Ca2+]iの測定を行った。Ca2+およびNa+測定のための2つの励起波長を分離するために、Ca2+感受性色素として、fluo−4/AM(モレキュラー・プローブズ社製)を使用して測定された。
[Na+]iを測定した後に、同一ニューロンにおいて[Ca2+]i測定のために、細胞に室温で1時間間2μM fluo−4が負荷された。
細胞は、490nmで12秒ごとに励起された。また、520nmの蛍光発光が検出された。
各時点における蛍光強度F490は、測定開始時の蛍光強度F490(0)に対する割合(F490/F490(0))により計算された。
【0046】
(SSA412と[Ca2+]iの測定)
最初に、[Ca2+]i測定は、灌流を行わない状態で、8.3mMグルコースを含んだ、30μlの小滴を用いて10分間、行なわれた。
第1の測定の後、20μl小滴が静かに除去された。また、グルコースのないSSA412又はコントロールのウサギIgGを含んでいる20μlのHKRBが加えられた。これにより、2.8のmMグルコースを含んでいる30μlのHKRBとなった。
この30μlのHKRBを使用して、灌流を行わない状態で、次の10分において、第2の[Ca2+]i測定が行なわれた。
最後に、GIニューロンを明確に識別するために、同一ニューロンの[Ca2+]iが上述した灌流条件下で測定された。
【0047】
(NKA活性の決定)
8週間の年齢の自由に食物を与えられた(n=7)又は24時間の絶食条件下の(n=7)ラットから、左右の視床下部弓状核およびVMHを取得し、直ちに液体窒素中で凍結した。
ウワバイン−センシティブATPアーゼアッセイの手順は、従来のKyteの方法(Kyte、1972、「J. The titration of the cardiac glycoside binding site of the (Na+ + K+)−adenosine triphosphatase.」、J Biol Chem 247、p.7634−7641)を修正して用いた。
【0048】
組織試料はそれぞれ、ティッシュティアラー(Tissue Tearor、ドレメル社製、ラシーン、ウィスコンシン州、アメリカ)を使用して、20mM Tris/HCl緩衝液、pH 7.4の0.5mlでホモジナイズされた。その上で、ガラス繊維布ホモジナイザーで均質化された。
ホモジェネートのタンパク濃度は、BCAプロテインアッセイ(ピアス社製、ロックフォード、イリノイ州、アメリカ)で決定された。
ホモジェネートされたサンプルは、分析のための、ウワバインを入れた/ウワバインを入れない2つのグループに分割された。
【0049】
酵素活性は、Na+およびK+がある状態で、ウワバインを入れた/ウワバインを入れないグループについて、MgATPのウワバイン感受性の加水分解として定義した。
タンパク質、MgATP、Na+、K+、及びウワバインの最終的な濃度は、それぞれ20μg/ml、3mM、120mM、20mM、及び2mMだった。
その反応は、37℃で30分の培養の後、MgATPを加えることにより始められ、0.75mlの反応停止溶液と混合することにより停止された。
発色剤(0.02ml)が加えられ、30分間室温で現像させた。
その後、遊離したリン酸塩の濃度が、分光測光器で700nmの波長の測定により決定された。
【0050】
(脳室内投与および食餌実験)
9〜10週のSDラットにおいて、ガイド・カニューレを脳の第三脳室に挿入した。この箇所は、正中線上でブレグマの尾側の0.7mm、及び頭蓋骨表面からの7.0mm下に位置している。
手術の後、ラットを7日間回復させ、非特異性のストレス応答を最小限にするために、毎日5分間のハンドリングを行った。
脳室内投与は、午前中(10:00−12:00)に開始し、食物摂取させた2時間後の摂取量を測定した。
【0051】
(NPY及びPOMCのmRNA量の測定)
10週の年齢のSDラットの脳室内に、5mMグルコースと一緒にウワバイン又はHKRBが投与された。視床下部弓状核は、投与の3時間後に摘出された。
mRNAは以前に報告されたのと同様な方法によりリアルタイムPCRで測定された。
トータルRNAは、Trizol(インヴィトロゲン社、ゲーサーズバーグ、メリーランンド州、アメリカ)及びRNAseフリーのDNAse(プロメガ社、マディソン、ウィスコンシン州、アメリカ)により単離された。
その後、トータルRNAをReverTra Ace(登録商標)(東洋紡績株式会社製、大阪、日本)による相補的DNA(cDNA)へ転換した後、SYBR premix Ex taq IIポリメラーゼ(タカラバイオ社製、大津、日本)が用いられ、リアルタイムPCRにより測定された。
蓄積した生成物は実時間(リアルタイム)で測定された。
また、最初に生成物を検出することができる平均のサイクルしきい値は、同じプレート上で実行されたプライマー・ペアおよびcDNAサンプル当たり5つの独立した反応の反復試験試料に対して決定された。
NPY又はPOMC(Pro−opiomelanocortin)の各cDNAサンプルは、ハウスキーピング遺伝子であるグリセリンアルデヒド−3−リン酸脱水素酵素(GAPDH)の生成物のプライマー・セットを使用して標準化された。
各cDNAに対応するプライマーは以下の通りだった:
GAPDH − フォーワードプライマー(配列番号1)、リバースプライマー(配列番号2)
NPY − フォーワードプライマー(配列番号3)、リバースプライマー(配列番号4);
POMC − フォーワードプライマー(配列番号5)、リバースプライマー(配列番号6)
【0052】
(統計)
データはすべて平均±s.e.m(standard error of the mean、SEM)として示される。
ペア又はペアでないスチューデントt検定は、2つのグループの実験で使用された。
次に、Bonfferoniのポスト・ホック・テストの後、4つのグループの実験において、階乗分散分析が使用された。
P<0.05の場合、統計的に有意であると考慮した。
【0053】
(GIニューロンの定義)
次に図3A〜図3Dを参照して、GIニューロンの定義について検討した際の実験結果について説明する。
グルコース濃度低下は、視床下部弓状核から単離された362のニューロンのうち137ニューロン(37.8%)の[Ca2+]iを増加させた。
図3Aは、本実施例において、GIニューロンとして定義したニューロンの[Ca2+]i応答の具体例を示している。本実施例では、単離された視床下部弓状核のニューロンにおいて、グルコース濃度低下とグルタミン酸ナトリウム(glutamate、100μM)とに応答して[Ca2+]iを増加させたニューロンを、GIニューロンと定義した。
図3Bは、GIニューロン(n=137)において、8.3G及び2.8Gでの[Ca2+]i応答の平均を示している。「**」は、P<0.01であることを示す。
この実験により、8.3Gから2.8Gまでグルコース濃度を低下させた際に[Ca2+]i増加応答したニューロンについて、GIニューロンとして定義した。これらのニューロンは、GIニューロンとして所定の応答基準を満たしている。
【0054】
これに対して、図3Cは、非GIニューロンとして定義されたニューロンの[Ca2+]i応答の具体例を示している。視床下部弓状核から取得したニューロンにおいて、グルコース濃度低下により、[Ca2+]iの増加を示さなかったニューロンを非GIニューロンと定義した。非GIニューロンでは、グルコース濃度低下において[Ca2+]iは変化がないものの、通常の興奮物質であるグルタミン酸ナトリウムには応答する。
図3Dは、非GIニューロン(n=123)において、8.3G及び2.8Gでの[Ca2+]i応答の平均を示している。このように、8.3Gと2.8Gでは有意差がなかった。
【0055】
また、以下の実験において、グルコース濃度低下に応答して[Ca2+]iを減少させるグルコース興奮性(GE)ニューロンは、分析から除外された。
【0056】
(GIニューロンにおけるグルコース濃度低下による[Na+]i増加応答)
ここで、本発明の発明者らは、NKA活性の指標である[Na+]iの反応を、SBFIを使用してマイクロ蛍光分析測定を行い、NKA活性を評価した。図4A〜Eを参照して、このNKA活性の実験結果について説明する。
図4Aは、グルコース濃度低下とウワバインによるGIニューロンの[Na+]i応答を示すグラフである。グルコース濃度低下およびウワバインは、視床下部弓状核から単離されたニューロンの[Na+]iを増加させた。このニューロンとしては、上述のように、摂食機能がほぼ成熟する5〜7週のラットの視床下部弓状核からニューロンを単離した。結果は、7つのニューロン(n=7)のうち代表的な反応について示した。
図4Bは、SBFI蛍光レシオに[Na+]iを関連づける較正曲線を示している。この構成曲線は、1μg/mlグラミシジンD及びNa+濃度を段階的に変化させることで作成した。
グルコース濃度低下は、[Na+]iの増加を誘発した。また、NKAの特異的インヒビターのウワバインは、同じニューロンの[Na+]i増加を誘発した。
また、グルコース濃度低下に誘発された[Na+]i増加は、7.5±1.1mM(n=7)であった。
【0057】
図4Cは、もう一つの一連の実験である、GIニューロンにおけるグルコース濃度低下による[Na+]iの変化と、同一ニューロンでの[Ca2+]iの変化を示すグラフである。グルコース濃度低下は、[Na+]i増加を誘発し、同じニューロンで[Ca2+]i増加を引き起こした。すなわち、グルコース濃度低下により、GIニューロンにおいて[Na+]iが増加した(n=13)。
【0058】
図4Dは、非GIニューロンにおけるグルコース濃度低下による[Na+]iの変化と、同一ニューロンでの[Ca2+]iの変化を示すグラフである。
グルコース濃度低下は、2.8Gに対する[Ca2+]i応答の欠如を示した非GIニューロンにおいては、[Na+]iを顕著に変化させなかった(n=8)。
【0059】
図4Eは、GIニューロン又は非GIニューロンにおけるグルコース濃度低下に誘発された[Na+]iの変化の平均を示すグラフである。「**」は、P<0.01であることを示す。
すなわち、GIニューロンにおけるグルコース濃度低下に誘発された[Na+]i増加の変化量は、非GIニューロンにおける変化量に対して、有意に大きかった。
【0060】
これらのデータにより、GIニューロンにおいて、グルコース濃度低下が、NKA活性を選択的に低減するという概念が立証された。
【0061】
(GIニューロンにおけるNKA活性低下によるGIニューロンの活性化)
次に、本発明の発明者らは、NKA活性の低下が、実際にGIニューロンの活性化に関連しているかどうかを調べた。図5A〜Mを参照して、NKAインヒビターであるウワバインと、NKAを働かせなくするためにK+を含まない溶液を投与した実験結果について説明する。
図5Aは、2.8Gで反応したGIニューロンにおいて、8.3Gでウワバイン(100μM)を投与した際の[Ca2+]i応答を示すグラフである(n=15)。このようにウワバインの投与により、GIニューロンの[Ca2+]i増加を誘発することができた。
図5Bは、ウワバインを投与しない群と投与した群における、8.3Gでの[Ca2+]i応答の平均を示すグラフである。「**」は、P<0.01であることを示す。
図5Cは、8.3Gで、K+を含まない(K+−free)溶液を投入した際の[Ca2+]i応答を示すグラフである(n=12)。このように、K+−free溶液にした場合には、細胞膜内外の電位差を変化させられないため、ウワバインと同様にNKAが機能しなくなる。この場合でも、GIニューロンの[Ca2+]i増加を誘発した。
図5Dは、K+−free溶液を投与した際の[Ca2+]i応答の平均を示すグラフである。「**」は、P<0.01であることを示す。
このように、GIニューロンにおいて、[Ca2+]i反応を安定化させる8.3Gグルコース濃度において、NKAをブロックする条件となる100μMウワバインの投与および外部K+の除去を行うことで、[Ca2+]i増加を誘発した。すなわち、NKA活性を低下させることで、GIニューロンの[Ca2+]i増加を誘発し、GIニューロンを活性化することができる。
【0062】
次に、本発明の発明者らは、ウワバインの継続的な投与にてNKAが既にブロックされていた場合、グルコース濃度低下に誘発された[Ca2+]i増加応答を防ぐことができるかどうか調べた。
図5Eは、まずはウワバインを投与しない状態で、GIニューロンに対してグルコース濃度低下による反復刺激をした場合の[Ca2+]i応答を観測したグラフである。2.8Gによるグルコース濃度低下1回目刺激(Amp1)、及びグルコース濃度低下2回目刺激(Amp2)において、それぞれグルコース濃度低下による反復刺激は[Ca2+]i増加を反復して誘発することが分かる。
図5Fは、8.3Gと、2.8Gのグルコース濃度低下1回目刺激(Amp1)又はグルコース濃度低下2回目刺激(Amp2)における[Ca2+]i応答の平均を示すグラフである。Amp2に対する[Ca2+]i応答の振幅は、Amp1と同様であった(n=9)。すなわち、Amp1、Amp2とも、それぞれ8.3Gに対してP<0.01(**)であった。
これに対して、ウワバイン非投与下におけるコントロール実験においては、グルコース濃度低下2回目刺激(Amp2)で誘発された[Ca2+]iの増加は、グルコース濃度低下1回目刺激(Amp1)で誘発された[Ca2+]iの増加と有意差が認められなかった。
【0063】
図5Gは、ウワバインが継続的に存在していた場合のグルコース濃度低下の反復刺激により誘発された[Ca2+]i応答を示すグラフである。このように、グルコース濃度低下1回目刺激(Amp1)で誘発された[Ca2+]i増加の後、100μMのウワバインが継続的に存在している状態でグルコース濃度低下2回目刺激(Amp2)を行った場合、[Ca2+]iの増加応答は抑えられた。
図5Hは、8.3Gと、2.8Gのグルコース濃度低下1回目刺激(Amp1)及び100μMのウワバインが継続的に存在している状態でグルコース濃度低下2回目刺激(Amp2)を与えた際の、[Ca2+]i応答の平均を示すグラフである(n=10)。100μMウワバインが継続的にある状態でのAmp2は、同じ実験のAmp1と比較して、有意に小さかった(*、P<0.05)。また、図5Hと図5Fとを比較すると、コントロール実験(図5FのAmp2)と比べても、図5HのAmp2は有意に小さかった(#、P<0.05)。
これらの結果により、GIニューロンにおいてNKA活性を抑制させることによって、グルコース濃度低下に誘発された場合と同様の[Ca2+]i増加が誘発されることが分かる。
すなわち、GIニューロンにおいて、低グルコース刺激による[Ca2+]i増加はNKAの活性に依存していることが示された。
【0064】
(ウワバインの濃度と、GIニューロンの活性化の関係)
次に、図5I〜Jを参照して、2.8Gで反応したGIニューロンと、反応しなかった非GIニューロンにおいて、8.3Gで、濃度を変えてウワバインを投与した際の[Ca2+]i応答について説明する。この応答の実験は、ウワバインの濃度の他は、図5A、Bの実験と同様に行った。
図5Iは、ウワバイン濃度に対して反応した細胞数の割合を示すグラフである。横軸は、ウワバインの濃度(μ mol/L、μM)を示し、縦軸はウワバインにより応答が増加したニューロンの割合(%)を示す。黒丸は、GIニューロンを示す。また、白丸は、非GIニューロンを示す。
図5Jは、[Ca2+]i増加応答の平均を示すグラフである。横軸は、ウワバインの濃度(μ mol/L、μM)を示し、縦軸は[Ca2+]i増加応答の平均を示す。黒丸はGIニューロン、白丸は非GIニューロンを示す。「#」は、P<0.01であることを示す。
このように、ウワバインの濃度が100μMの1/1000の0.1μMであっても、顕著に応答を増加させることが分かる。また、100μMの1/100の1μMにて[Ca2+]i増加応答が最大化する。さらに、100μMの1/10の10μMにて、GIニューロンと非GIニューロンとの差がなくなることが分かる。ここで、100μMのウワバインを脳室内投与すると、通常の強心剤として用いられる投与量と同等であると考えられる。すなわち、通常の投与量の1/10で、グルコース濃度低下によるGIニューロンの活性化を抑えることが可能である。
【0065】
(NKAの活性化による、GIニューロンの不活性化)
上述の結果により、NKAを活性化させる操作は、グルコース濃度低下に対する[Ca2+]i増加応答を減ずる可能性がある。
そこで、本発明の発明者らは、NAKのαサブユニットと特異的に作用し、NKAを活性化させるNKAアクティベータとなるSSA412抗体を用いて、NKA活性増加とGIニューロンの不活性化について検討した。
この実験では、灌流を行わない状態で8.3Gから2.8Gにグルコース濃度を低下させ、SSA412を2μM又はコントロールのウサギIgGを2μM投入し、単離されたニューロンの[Ca2+]iを測定した。
【0066】
図5Kは、ウサギIgGを加えたコントロール実験の結果である。
まずは、8.3Gの[Ca2+]i応答を測定した(図5K左欄)。
その後、コントロールとなる2μMのウサギIgGを加えて、2.8Gの[Ca2+]i応答を測定した(図5K中央欄)。2.8Gにおいて、[Ca2+]iの増加応答を示したニューロンが存在した。
さらにウサギIgGを洗浄し、溶液を灌流させた状態で[Ca2+]i応答を測定した(図5K右欄、n=12)。2.8Gで[Ca2+]iは増加し、これらがGIニューロンにおける反応であったことが確認できた。
【0067】
図5Lは、NKAアクティベータであるSSA412を加えた実験の結果である。
コントロール実験と同様に、まずは8.3Gの[Ca2+]i応答を測定した(図5L左欄)。
その後、2μMのSSA412を加えて、2.8Gの[Ca2+]i応答を測定した(図5L中央欄)。2.8Gにおいて、[Ca2+]iは増加応答を示さなかった。
その後、SSA412を洗浄し、溶液を灌流させた状態で[Ca2+]i応答を測定した(図5L右欄、n=8)。2.8Gで、[Ca2+]iは増加し、これらがGIニューロンにおける変化であったことが確認できた。
すなわち、GIニューロンにおいて、SSA412存在下では[Ca2+]i増加が誘発されず、GIニューロンが活性化しないことが示された。
【0068】
図5Mは、コントロールのウサギIgG、又はNKAアクティベータであるSSA412を加えた際の[Ca2+]i応答の平均を示す。「*」は、P<0.05であることを示す。また「**」は、P<0.01であることを示す。
この結果を参照すると、2.8GにおけるSSA412存在下での[Ca2+]iは、8.3Gの[Ca2+]iとの有意差を認められなかった。さらに、コントロールであるIgG存在下での[Ca2+]iに対して、SSA412存在下での[Ca2+]iは有意に低かった(P<0.05)。
すなわち、GIニューロンにおけるグルコース濃度低下に対する[Ca2+]i反応は、SSA412の存在によって抑制された。
【0069】
これらの結果は、SSA412によるNKAの活性化により、グルコース濃度低下がGIニューロンの[Ca2+]i増加を誘発するのを抑制することを示している。
NKA活性に対するインヒビターとアクティベータとで操作した上述のデータは、視床下部弓状核GIニューロンが、NKAにより、グルコース感知によるニューロン活性化と結びつけられていることを強く示唆する。
【0070】
(GIニューロン活性化の上流でのグルコースによるシグナル伝達機構)
次に、本発明の発明者らは、GIニューロン活性化の上流でのグルコースによるシグナル伝達機構を検討した。
膵臓形態のグルコキナーゼは、グルコースをリン酸化する際の律速酵素であり、ランゲルハンス島β細胞のグルコース反応及び視床下部のGEニューロンの応答に重要な役割を果たしている。また、GIニューロンにも密接に関連している可能性が推測される。
以下で図6A〜図8Dを参照して、このグルコースによるシグナル伝達機構に関する実験の結果について説明する。
【0071】
図6Aは、GIニューロンにおいて、灌流液内のグルコース濃度を8.3Gで維持させた状態で、グルコキナーゼの拮抗阻害剤である10mMマンノヘプツロース(Mannoheptulose、MH)を投与した実験例を示すグラフである(n=14)。この結果として、8.3Gにおいて、10mMのMHを投与すると[Ca2+]iを増加させた。すなわち、グルコース濃度低下と同様の効果が得られた。
図6Bは、GIニューロンにおいて灌流液内のグルコース濃度を8.3Gで維持させた状態で、代謝されないグルコース・アナログである2−デオキシ−D−グルコース(2DG)を投与した実験例を示すグラフである(n=13)。10mMの2DGを投与した結果、MHと類似した結果が得られた。すなわち、8.3Gで、10mMの2DGは、[Ca2+]i増加を誘発し、グルコース濃度低下と同様の効果が得られた。
【0072】
図6Cは、MHに対する[Ca2+]i増加応答の発生率を示しており、GIニューロン(黒塗りのバー)で77.8%(14/18)、非GIニューロン(オープンバー)では26.7%(4/15)であった。
図6Dは、2DGに対する[Ca2+]i応答の割合を示しており、GIニューロン(黒塗りのバー)で76.9%(10/13)、非GIニューロン(オープンバー)では16.7%(1/6)であった。
図6Eは、MHを投与した際のGI及び非GIニューロンの[Ca2+]i応答の平均を示している。「**」はP<0.01であることを示す。
図6Fは、2DGを投与した際のGI及び非GIニューロンの[Ca2+]i応答の平均を示している。「**」はP<0.01であることを示す。
このように、MH及び2DGに対する[Ca2+]i応答の割合と振幅は、非GIニューロンよりもGIニューロンにおいて有意に大きかった。
これらのデータにより、グルコキナーゼを媒介としたグルコース代謝が、GIニューロンを調節することが明確となった。
【0073】
次に、本発明の発明者らは、GIニューロンにおいて、グルコース低下による反応が細胞内のエネルギー生産に関連しているかについて調べた。図7を参照して、この実験について説明する。
図7Aにおいて、左図は、視床下部弓状核から単離したニューロンにおいて、NAD(P)H自己蛍光を測定した実験例である(n=6)。これらのニューロンのNAD(P)H自己蛍光は、2.8Gでは減少し、8.3Gで増加した。右図は、この同一ニューロンにおける2.8Gに対する[Ca2+]i増加応答を示しており、GIニューロンであることを確認した。
図7Bは、GIニューロンにおける8.3G及び2.8GにおけるNAD(P)H自己蛍光(の平均)を示している。「**」は、P<0.01であることを示す。平均のNAD(P)H自己蛍光レベルは、グルコース濃度が8.3Gの場合より、2.8Gの場合において有意に低かった。
これらのデータは、グルコース濃度低下がGIニューロンにおいてNAD(P)Hを減少させることを示している。
【0074】
更に、本発明の発明者らは、GIニューロンにおいてエネルギー生産を引き下げる条件により、GIニューロンが活性化するか否かを検証するための実験を行った。具体的には、GIニューロンに対して、8.3Gのグルコースにより[Ca2+]iを安定化した後、電子伝達系の阻害剤である100μMのシアン化カリウム(KCN)、又は酸化的リン酸化の脱共役剤である1μMのカルボニルシアニドp−トリフルオロメトキシフェニルヒドラゾン(FCCP)の投与をした。
図8Aは、8.3Gにおいて、100μMのKCNを投与した際の[Ca2+]i応答を示すグラフである。
図8Bは、KCNを投与した際の[Ca2+]i応答の平均を示す(n=8)。「**」はP<0.01であることを示す。
図8Cは、8.3Gにおいてで、1μMのFCCPを投与した際の[Ca2+]i応答を示すグラフである。
図8Dは、FCCPを投与した際の[Ca2+]i応答の平均を示す(n=10)。「**」はP<0.01であることを示す。
このように、KCNとFCCPは、両方とも[Ca2+]iを増加させた。これは、グルコース濃度低下時の反応と同様の反応である。
これらの結果により、グルコース濃度低下に誘発された[Ca2+]i増加は、細胞内エネルギー産生率の低下と、これにより必然的に起こるNKAの抑制にて媒介されると考えられる。
【0075】
(GIニューロン活動性の下流のVDCCによるシグナル伝達機構)
次に、本発明者らは、NKAの抑制とGIニューロンの活性化をどのように結びつけて考えることができるかについて考察した。
具体的なNKAの抑制とGIニューロンの活性化における作用機構としては、[Na+]i増加に関連したNKAの電気的な活性の抑制により細胞膜が脱分極され、これにより、電位依存Ca2+チャンネルを介してCa2+流入を刺激する、と推測される。
このため、本発明者らは、L形及びN型のVDCCとGIニューロン活性について実験により確かめた。L形及びN型のVDCCは、グルコース感知ニューロン、NPYニューロン、及び低血糖下でグルカゴンを分泌する膵臓のα−細胞の調節に密接に関連している。以下で図9A〜図9Eを参照して、グルコース濃度低下に誘発された[Ca2+]i増加に対して、各VDCCに関連する試薬を投与した実験を示している。
図9Aは、Ca2+を取り除いた(Ca2+−free)溶液にした際の[Ca2+]i応答を示すグラフである(n=8)。[Ca2+]i増加は、Ca2+−free溶液では抑制されている。
図9Bは、各VDCCの非特異性のブロッカーである高濃度Ni2+(300μM)を投与した際の[Ca2+]i応答を示すグラフである(n=13)。Ni2+投与によっても、[Ca2+]i増加は抑制されている。
図9Cは、N型Ca2+チャンネル・ブロッカーであるω−コノトキシンGVIA(ω−conotoxin)500nMを投与した際の[Ca2+]iを応答示すグラフである(n=7)。グルコース濃度低下に誘発された[Ca2+]i増加は、ω−コノトキシンGVIAによっても、抑制されている。
図9Dは、L形Ca2+チャンネル・ブロッカーであるニフェジピン(nifedipine)10μMを投与した際の[Ca2+]i応答を示すグラフである(n=9)。グルコース濃度低下に誘発された[Ca2+]i増加は、ニフェジピンによっても抑制されている。
図9Eは、図9A〜Dの試験条件下のグルコース濃度低下に対する[Ca2+]i応答の振幅の平均を、コントロール実験と比べて相対的に表現したものである。「**」は、P<0.01であることを示す。すなわち、グルコース濃度低下に誘発された[Ca2+]i増加は、Ca2+−free溶液、Ni2+(300μM)投与、ω−コノトキシンGVIA(500nM)投与、及びニフェジピン(10μM)投与により有意に減少した。
これらのデータは、グルコース濃度低下に誘発された[Ca2+]i増加は、主としてN型及びL形VDCCによるCa2+流入を介して行われることを示している。
そして、グルコース濃度低下に誘発されたNKAの抑制が、細胞膜を脱分極化してVDCCを開くことを示唆している。
【0076】
なお、グルコース濃度低下に誘発された[Ca2+]i増加において、VDCCをブロックした後に残った反応の一部は、グルコース濃度低下に誘発された[Na+]i増加が引き金となって起きる、形質膜Na+/Ca2+エクスチェンジャー(NCX)の逆モードで引き起こされることも、可能性としては考えられる。
しかしながら、本発明の発明者らは、VDCCを抑制した条件下でも[Na+]i増加を観察した。このため、上述の可能性はないと考えられる。
【0077】
(NKA活性による摂食行動の生理学的調節)
本発明の発明者らは、更に、GIニューロンの活性化を調節するNKAの重大な役割を明らかにする生体外のデータに基づき、NKA活性が摂食行動の生理学的調節に関係するか否かについて、調査した。
以下、図10、図11を参照して、実際にラット個体を用いて、NKA活性の低下が絶食又はグルコース濃度低下の感知と摂食との刺激に関連しているかについて調べた実験について説明する。
図10は、視床下部弓状核のNKA活性を計測した結果を示す(n=7)。自由に食物が与えられたラット(Control)をオープンバーで示し、断食させたラット(Fasted)を塗りつぶしたバーで示す。「**」はP<0.01であることを示す。この実験結果においては、任意に食物が与えられたラットと比較して、24時間絶食させたラットにおいて視床下部弓状核のNKA活性は48.3%と著しく減少した。
図11は、GEニューロンが主要な働きをしている視床下部腹内側部(ventromedial hypothalamus、VMH)でのNKA活性を計測した結果を示す(n=7)。視床下部弓状核とは異なり、VMHにおいては、コントロール(Control)及び絶食下(Fasted)においてもNKA特性は不変だった。
したがって、絶食により、視床下部弓状核のNKA活性は選択的に著しく低減されるため、NKA活性を低下させることで摂食を促進することが可能であると示唆される。
【0078】
次に、ラットにNKAインヒビターのウワバイン(0.5nmol/5μl)とK+−free溶液(10μl)とを脳室内投与して、摂食量を比較した。
図12Aは、この摂食量の比較を示すグラフである。ウワバイン(0.5のnmol/5μl)(n=8)及び10μlのK+−free溶液(n=5)の脳室内投与により、コントロールHKRB(n=5)と比較して、脳室内投与の2時間後に、摂食量を著しく増加させた。HKRBは、GIニューロンを興奮させないために5mMグルコース(5G)を含んでいる。「*」は、P<0.05であることを示す。
【0079】
次に、グルコキナーゼ阻害剤であるMH、NKAアクティベータであるSSA412、コントロールのウサギIgGを脳室内投与して、摂食量を比較した。
図12Bは、この摂食量の比較を示すグラフである。「*」は、P<0.05、「**」はP<0.01であることを示す。
まず、脳室内にMHを投与せず、SSA412(n=7)又はウサギIgG(n=6)のみを注射した場合、摂食量は変化なかった。
ここで、脳室内にMHを投与すると、ニューロンのグルコース利用が低減され、従ってグルコース濃度低下および飢餓/絶食の条件を反映する状況に置かれるため、摂食量が有意に増加した。
これに対して、MHに誘発された摂食行動は、10μg/5μlのSSA412の脳室内前投与により打ち消された(n=8)。
しかしながら、コントロールの10μg/5μlのウサギIgGの脳室内前投与では打ち消されなかった(n=6)。
このように、NKAアクティベータであるSSA412により、実際に摂食量を変化させ、食欲を減らすことができる可能性がある。
【0080】
さらに、本発明の発明者らは、視床下部弓状核のNPY又はPOMCのmRNA発現量についての測定を行った。
図13Aは、ウワバインの脳室内投与後3時間における、視床下部弓状核のNPYのmRNA発現量を示すグラフである(n=6)。ウワバインの脳室内投与の後、コントロール(Vehicle)と比較して、視床下部弓状核のNPYのmRNA発現量は有意に増加した。「**」は、P<0.01であることを示す。
図13Bは、ウワバインの脳室内投与後3時間における、視床下部弓状核のPOMC(Pro−opiomelanocortin)のmRNA発現量を示すグラフである(n=6)。POMCは、脳下垂体前葉に含まれ、ACTH(Adrenocorticotropic hormone)やMSH(melanocyte−stimulating hormones)等に分解されるペプチドホルモン前駆体であり、摂食抑制として機能する。ウワバインの脳室内投与を行っても、POMCのmRNA発現量は変わらなかった。
【0081】
これらの結果は、飢餓/絶食条件下でグルコース濃度低下に誘発された視床下部弓状核のNKA活性の低下が、NPYを含むGIニューロンを活性化し、これにより摂食行動を刺激することを示している。
【0082】
この摂食行動の病態生理学の意味として、GIニューロンを含む視床下部弓状核NPYニューロンが、過食・内臓肥満症の2型糖尿病Goto−Kakizaki(GK)ラットでアップレギュレートされることが考えられる。
このモデルでは、グルコース利用の低下は、グルコース濃度上昇に誘発されるインスリン分泌促進の障害及びグルコース濃度上昇に誘発されるGIニューロン、すなわちNPYニューロンの活動抑制の障害と共通の原因でありえる。グルコース利用が低下していても、NKA活性化剤によりGIニューロンを活性化すれば、過食を是正し、その結果、内臓肥満と糖尿病を改善する効果が期待できる。
摂食行動異常をもつ肥満と糖尿病は、GKラットとヒト患者で共通の機序により生じると推測できるため、SSA412を含むNKA活性化物質を本実施例の食欲抑制剤として用いることにより、肥満及び2型糖尿病の治療にも用いることが考えられる。
【0083】
以上のように、本実施例において、本発明の発明者らは、以前からの推測はあるものの証明されていない仮説である、NKA活性の低下がグルコース低下によるGIニューロンの活性化に関連していることを証明した。
【0084】
なお、上記実施の形態の構成及び動作は例であって、本発明の趣旨を逸脱しない範囲で適宜変更して実行することができることは言うまでもない。
【産業上の利用可能性】
【0085】
本発明によれば、視床下部弓状核GIニューロンのNKAの作用を調整することで、第1次摂食・代謝調節中枢に直接働きかけて、「食欲」そのものを調節することができる。よって、従来とは作用機構の違う食欲促進剤、食欲抑制剤の提供が可能となる。このため、産業条利用可能である。
【技術分野】
【0001】
本発明は、食欲調整剤、治療薬、健康食品、動物用飼料、及び食欲調整方法に係り、特に視床下部弓状核に作用する食欲調整剤、治療薬、健康食品、動物用飼料、及び食欲調整方法に関する。
【背景技術】
【0002】
近年、摂食とエネルギー代謝の調節メカニズムの研究が国内外で盛んに行われている。これは、世界的な肥満の増加と、肥満が多くの生活習慣病の起点になることが明らかにされたためであった。
図14を参照して、近年、明らかになっている摂食とエネルギー代謝の調節メカニズムについて説明する。
全身の栄養・代謝状態は、栄養素、ホルモンや自律神経の迷走神経求心路を媒介として脳に伝えられる。
これらの情報は、視床下部を中心とした中枢で情報が統合された後、自律神経遠心路や視床下部下垂体ホルモンを経由して脂肪細胞などの器官にシグナルが送られ、末梢代謝が制御されている。この一連の過程に障害が起こると、代謝恒常性の破綻や肥満をもたらし、ひいてはメタボリック・シンドロームの成因となると考えられる。
【0003】
従来、高等ほ乳類において、栄養・代謝状態を脳に伝える因子として、グルコース、インスリン、コレシストキニン(CCK)等が知られていた。
また、近年、脂肪細胞由来のレブチン、アディポネクチン、また胃由来のグレリンといった新たな摂食代謝調節因子が発見されている。
これらの因子は、栄養・代謝状態の末梢代謝シグナルであり、血液脳関門(Blood Brain Barrier、BBB)を通過して直接脳に取り込まれるか、又は、迷走神経求心路を経由して脳に情報伝達されることが分かっている。
このような末梢代謝シグナルが脳に入ると、主として視床下部弓状核(Arcuate nucleus、ARC)で受容され視床下部のネットワークにおいて統合的に処理される。
具体的には、脳において、まず視床下部弓状核等を中心とした第1次摂食・代謝調節中枢に作用し、次いで視床下部内の神経ネットワークにより統合的に情報処理される。
その情報処理の結果は、自律神経、下垂体ホルモン分泌、高次中枢に出力され、最終的に摂食行動やエネルギー代謝が調節されると考えられている。
このように、高等ほ乳類では、視床下部からの出力が自律神経活動、下垂体ホルモン分泌、高次機能などを調節することにより、摂食と代謝を調節している。
すなわち、血液脳関門を経た入力、視床下部弓状核での情報統合、自律神経を経た出力が特に重要である。
【0004】
ここで、特許文献1を参照すると、従来の肥満症治療のための薬学的組成物として、食事行動を修正する化合物、そのプロドラッグまたは前記化合物または前記プロドラッグの薬学的に許容可能な塩;および、β−アドレナリン作働剤と前記プロドラッグの薬学的に許容可能な塩を含む薬学的組成物が記載されている(以下、従来技術1とする。)。
特許文献1の薬学的組成物に含まれるβ−アドレナリン作働剤は、アデニル酸サイクラーゼを活性化するため、抗肥満症剤として有用である。また、さらに特許文献1の段落[0013][0015]等を参照すると、食事行動を修正する化合物として、上述のレプチンのような摂食代謝調節因子を食欲抑制剤として用いることが示されている。
すなわち、特許文献1の薬学的組成物は、神経伝達物質とそのシグナル伝達系に働きかけること、及びホルモンやアディポカインを利用して、動物、特にヒトの肥満症を処置するのに有用性を有する薬学的組成物および方法を提供していた。
なお、従来技術1は、食欲抑制剤として、脳内神経伝達物質でもあるセロトニン作働剤やドーパミンアゴニストも用いるように記載していた。これらの脳内神経伝達物質は、向精神薬の標的となっており、脳全体に作用するため、第1次摂食・代謝調節中枢にだけ直接作用するものではなかった。
【先行技術文献】
【特許文献】
【0005】
【特許文献1】特開平11−228447号公報
【発明の概要】
【発明が解決しようとする課題】
【0006】
しかしながら、従来技術1のように、神経伝達物質に働きかけることにより、「脳全体に作用する」ような特異性がない薬学的組成物を用いると、食欲/摂食行動を直接、選択的に変化させることができないため、副作用が大きいという問題があった。
【0007】
このため、摂食行動に関わる「食欲」そのものを変更するような、視床下部弓状核等を中心とした第1次摂食・代謝調節中枢に直接作用する食欲調整組成物が求められていた。
【0008】
本発明は、このような状況に鑑みてなされたものであり、上述の課題を解消することを課題とする。
【課題を解決するための手段】
【0009】
本発明の食欲調整剤は、視床下部弓状核におけるグルコース抑制性ニューロン(Glucose−Inhibited neuron)のNKA(Na++K+−ATPase)活性を調整するNKA活動調節剤を含むことを特徴とする。
本発明の食欲調整剤は、前記NKA活動調節剤は、NKAインヒビターであり、前記グルコース抑制性ニューロンの[Ca2+]i濃度を増加させることを特徴とする。
本発明の食欲調整剤は、前記NKAインヒビターは、ニューロペプチドYの発現を惹起することを特徴とする。
本発明の食欲調整剤は、前記NKAインヒビターは、強心配糖体であり、該強心配糖体は、ウワバイン(ouabain)又はジゴキシン(digoxin)を含むことを特徴とする。
本発明の食欲調整剤は、前記NKAインヒビターは、NKAのαサブユニットの活動を調整することを特徴とする。
本発明の食欲調整剤は、前記NKAインヒビターは、強心配糖体としての投与の濃度の1/10以下の量を投与することを特徴とする。
本発明の治療薬は、前記食欲調整剤を用いたことを特徴とする食欲不振、抗癌剤の副作用による食欲の低下、加齢、過労、低栄養、神経性食思不振症、うつ病を含む精神疾患、自律神経失調症、拒食症を含む摂食障害のいずれかに対する治療のための治療薬であることを特徴とする。
本発明の食欲調整剤は、前記NKA活動調節剤は、NKAアクティベータであり、前記グルコース抑制性ニューロンにおける低グルコース誘発[Ca2+]i増加を抑制又は阻止することを特徴とする。
本発明の食欲調整剤は、前記NKAアクティベータは、ニューロペプチドYの発現を抑制することを特徴とする。
本発明の食欲調整剤は、前記NKAアクティベータは、NKAのαサブユニットに結合することを特徴とする。
本発明の食欲調整剤は、前記NKAアクティベータは、抗体SSA412であり、前記グルコース抑制性ニューロンにおける低グルコース誘発[Ca2+]i増加を抑制又は阻止することを特徴とする。
本発明の治療薬は、前記食欲調整剤を用いたことを特徴とする、過食、過食による肥満、過食による糖尿病、メタボリックシンドロームのいずれかに対する治療のための治療薬であることを特徴とする。
本発明の健康食品は、前記食欲調整剤を用いたことを特徴とする。
本発明の動物用飼料は、前記食欲調整剤を用いたことを特徴とする。
本発明の食欲調整方法は、視床下部弓状核のグルコース抑制性ニューロン(Glucose−Inhibited neuron)のNKA(Na++K+−ATPase)活性を調整し、前記グルコース抑制性ニューロンの出力を調整することを特徴とする。
【発明の効果】
【0010】
本発明によれば、視床下部弓状核のグルコース抑制性ニューロン(Glucose−Inhibited neuron、GIニューロン)のNKA(Na++K+−ATPase)活性を調整するNKA活動調節剤を用いることで、第1次摂食・代謝調節中枢に直接作用する食欲調整剤を提供することができる。
【図面の簡単な説明】
【0011】
【図1】本発明の実施の形態に係る視床下部弓状核のGIニューロンの食欲調整のモデルを示す概念図である。
【図2】本発明の実施の形態の実施例におけるGIニューロンの同定、[Na+]i又は[Ca2+]i応答測定の概念図である。
【図3A】本発明の実施の形態の実施例において、GIニューロンとして定義したニューロンの[Ca2+]i応答のグラフである。
【図3B】本発明の実施の形態の実施例において、GIニューロンの8.3mMグルコース(8.3G)と2.8mMグルコース(2.8G)における[Ca2+]i応答の平均を示すグラフである。
【図3C】本発明の実施の形態の実施例において、非GIニューロンとして定義したニューロンの[Ca2+]i応答のグラフである。
【図3D】本発明の実施の形態の実施例において、非GIニューロンの8.3Gと2.8Gにおける[Ca2+]i応答の平均を示すグラフである。
【図4A】本発明の実施の形態の実施例において、グルコース濃度低下とウワバインによるGIニューロンの[Na+]iの応答の変化を示すグラフである。
【図4B】本発明の実施の形態の実施例において、[Na+]iとSBFI蛍光レシオの関係を表わす較正曲線の図である。
【図4C】本発明の実施の形態の実施例において、グルコース濃度低下による[Na+]iの増加を示し、同じニューロンが[Ca2+]i増加を示すことから、GIニューロンであることを示すグラフである。
【図4D】本発明の実施の形態の実施例において、グルコース濃度低下に対して[Na+]iの変化が見られず、同じニューロンが[Ca2+]iの変化を示さないことから、非GIニューロンであることを示すグラフである。
【図4E】本発明の実施の形態の実施例において、GIニューロン及び非GIニューロンにおけるグルコース濃度低下に誘発された[Na+]iの変化の平均を示すグラフである。
【図5A】本発明の実施の形態の実施例において、8.3Gでウワバインを投与した際の[Ca2+]i応答を示すグラフである。
【図5B】本発明の実施の形態の実施例において、ウワバインを投与した際の[Ca2+]i応答の平均を示すグラフである。
【図5C】本発明の実施の形態の実施例において、8.3Gで、K+を含まない(K+−free)溶液を用いた際の[Ca2+]i応答を示すグラフである。
【図5D】本発明の実施の形態の実施例において、K+−free溶液を投与した際の[Ca2+]i応答の平均を示すグラフである。
【図5E】本発明の実施の形態の実施例において、ウワバインを投与しない状態で、同一のGIニューロンをグルコース濃度低下による反復刺激をした場合の[Ca2+]i応答のグラフである。
【図5F】本発明の実施の形態の実施例において、8.3G及び2.8Gにおける、グルコース濃度低下1回目刺激(Amp1)又はグルコース濃度低下2回目刺激(Amp2)における平均[Ca2+]i応答の平均を示すグラフである。
【図5G】本発明の実施の形態の実施例において、ウワバインが継続的に存在していた場合のグルコース濃度低下反復二回目刺激により誘発された[Ca2+]i応答を示すグラフである。
【図5H】本発明の実施の形態の実施例において、グルコース濃度低下1回目刺激(Amp1)の後、100μMのウワバイン存在下でグルコース濃度低下2回目刺激(Amp2)を与えた際の[Ca2+]i応答の平均を示すグラフである。
【図5I】本発明の実施の形態の実施例において、GIニューロン又は非GIニューロンにおいて、ウワバイン濃度の変化に対して反応した細胞数の割合を示すグラフである。
【図5J】本発明の実施の形態の実施例において、GIニューロン又は非GIニューロンにおいて、ウワバイン濃度の変化に対する[Ca2+]i応答の平均を示すグラフである。
【図5K】本発明の実施の形態の実施例において、グルコース濃度低下によるGIニューロンの活性化においてウサギIgGを加えたコントロール実験の結果を示すグラフである。
【図5L】本発明の実施の形態の実施例において、グルコース濃度低下によるGIニューロンの活性化においてSSA412を加えた実験の結果を示すグラフである。
【図5M】本発明の実施の形態の実施例において、グルコース濃度低下によるGIニューロンの活性化において、ウサギIgG又はSSA412を加えた際の[Ca2+]i応答の平均示すグラフである。
【図6A】本発明の実施の形態の実施例において、グルコキナーゼの拮抗阻害剤である10mMのMHを投与した際の[Ca2+]i応答を示すグラフである。
【図6B】本発明の実施の形態の実施例において、代謝されないグルコース・アナログである10mMの2DGを投与した際の[Ca2+]i応答を示すグラフである。
【図6C】本発明の実施の形態の実施例において、GIニューロン又は非GIニューロンにおいて、MHに対する[Ca2+]i応答を示した細胞数を示すグラフである。
【図6D】本発明の実施の形態の実施例において、GIニューロン又は非GIニューロンにおいて、2DGに対する[Ca2+]i応答を示した細胞数を示すグラフである。
【図6E】本発明の実施の形態の実施例において、GIニューロン又は非GIニューロンにおいて、MHを投与した際の[Ca2+]i応答の平均を示すグラフである。
【図6F】本発明の実施の形態の実施例において、GIニューロン又は非GIニューロンにおいて、2DGを投与した際のGI及び非GIニューロンの平均[Ca2+]i応答の平均を示すグラフである。
【図7A】本発明の実施の形態の実施例において、グルコース濃度低下によるNAD(P)H自己蛍光低下を示し、同じニューロンが[Ca2+]i増加を示すことから、GIニューロンであることを示すグラフである。
【図7B】本発明の実施の形態の実施例において、GIニューロンの8.3G及び2.8GにおけるNAD(P)H自己蛍光の平均を示すグラフである。
【図8A】本発明の実施の形態の実施例において、100μMのKCNを投与した際の[Ca2+]i応答を示すグラフである。
【図8B】本発明の実施の形態の実施例において、KCNを投与した際の[Ca2+]i応答の平均を示すグラフである。
【図8C】本発明の実施の形態の実施例において、1μMのFCCPを投与した際の[Ca2+]i応答を示すグラフである。
【図8D】本発明の実施の形態の実施例において、FCCPを投与した際の[Ca2+]i応答の平均を示すグラフである。
【図9A】本発明の実施の形態の実施例において、Ca2+を含まない(Ca2+−free)溶液を投与した[Ca2+]i応答を示すグラフである。
【図9B】本発明の実施の形態の実施例において、各VDCCの非特異性のブロッカーである高濃度Ni2+(300μM)を投与した際の[Ca2+]i応答を示すグラフである。
【図9C】本発明の実施の形態の実施例において、N型Ca2+チャンネル・ブロッカーであるω−コノトキシンGVIA(300nM)を投与した際の[Ca2+]i応答を示すグラフである。
【図9D】本発明の実施の形態の実施例において、L形Ca2+チャンネル・ブロッカーであるニフェジピン10μMを投与した際の[Ca2+]i応答を示すグラフである。
【図9E】本発明の実施の形態の実施例において、図9A〜Dの[Ca2+]i応答の振幅の平均を、コントロール実験と比べて相対的に表現したグラフである。
【図10】本発明の実施の形態の実施例において、自由摂食下と24時間絶食下において、視床下部弓状核のNKA活性の変化を示すグラフである。
【図11】本発明の実施の形態の実施例において、自由摂食下と24時間絶食下において、GEニューロンが主要な働きをしている視床下部腹内側部でのNKA活性の変化を示すグラフである。
【図12A】本発明の実施の形態の実施例において、5mMグルコース(5G)−HKRB溶液、ウワバイン0.5nM、K+−free溶液(10μl)を脳室内投与した際の、摂食量の変化の平均を示したグラフである。
【図12B】本発明の実施の形態の実施例において、5G−HKRB溶液、MH、さらにMHとSSA412(SSA412)、MHとウサギIgG(Rabbit IgG)を脳室内投与して、摂食量の変化の平均を示したグラフである。
【図13A】本発明の実施の形態の実施例において、ウワバインの脳室内投与後の視床下部弓状核のNPYのmRNA発現量を示すグラフである。
【図13B】本発明の実施の形態の実施例において、ウワバインの脳室内投与後の視床下部弓状核のPOMCのmRNA発現量を示すグラフである。
【図14】従来の摂食調節メカニズムについての概念図である。
【発明を実施するための形態】
【0012】
<実施の形態>
上述したように、副作用の少ない摂食調節用の化合物が必要とされていた。このため、本発明の発明者らは、視床下部弓状核等を中心とした第1次摂食・代謝調節中枢に直接働きかけて、「食欲」そのものを調節するための食欲調節剤を探し求めることを考えた。
この際、本発明の発明者らは、視床下部弓状核(ARC)にあるグルコース抑制性(Glucose−inhibited、GI)の神経細胞(Neuron、ニューロン)に着目した。以下、このグルコース抑制製の神経細胞をGIニューロンと呼ぶ。
ここで、グルコース(glucose)は、細胞活動に対して不可欠なエネルギー基質であるため、一般的には、グルコース濃度の上昇は細胞活動の上昇をもたらす。
しかしながら、GIニューロンは、このグルコースの働きに対する逆説的な例外である。すなわち、GIニューロンは、外側視床下野の視床下部弓状核で最初に発見され、グルコース濃度の上昇により活動が抑制されるという特徴がある。つまり、GIニューロンの電気的活性及び細胞内Ca活性は、グルコース濃度低下(LG)に応じて増加する。
また、視床下部弓状核のGIニューロンは、強力な食欲促進作用をもつニューロペプチドY(Neuropeptide Y、NPY)を含んでおり、空腹・飢餓による摂食亢進に関連していることが示唆されている。
【0013】
このGIニューロンの制御機構については、従来、不明であった。しかしながら、1974年に、Oomuraが、細胞膜にある能動輸送系であるナトリウムポンプの抑制が、GIニューロンのグルコース濃度低下による活性化を媒介するという仮説を提唱していた(Oomura et al.、1974、「Glucose inhibition of the glucose−sensitive neurone in the rat lateral hypothalamus.」、Nature、247、p.284−286、参照)。この仮説において、Oomuraは、「したがって、グルコースによるこれらのニューロンの抑制は、透過性変化によらないが、それが強心性配糖体あるいは抗代謝物質で有効にブロックされるので、エネルギー依存のナトリウムポンプの活性化による薄膜過分極で引き起こされるように見える。」と述べている。
【0014】
しかしながら、GIニューロンでは、上述のOomuraの仮説のように、ナトリウムポンプであるNa+/K+−ATPアーゼ(NKA)の関与、更にcl−チャネルおよびK+チャンネルが関与の仮説が提唱されていたものの、直接的な証拠はなかった。つまり、グルコース濃度の低下に誘発されたニューロンの活性化の機構は、実際には殆ど分かっていなかった。
このため、本発明の発明者らは鋭意実験と検討を行い、グルコース濃度低下と連動したNKA活性の低下が、GIニューロンの活性化と摂食に重大な役割をしていることを見いだした。つまり、NKA活動調節剤により、食欲をコントロールすることが可能になる。
【0015】
ここで、NKAは、αとβの2種のサブユニットからなる膜貫通タンパク(EC3.6.3.9)である。
NKAは、動物細胞において、ATP(Adenosine TriPhosphate)の加水分解により、細胞内からナトリウムイオン(Na+)を汲み出し、カリウムイオン(K+)を取り込むのでナトリウム−カリウムポンプとも呼ばれている。
この酵素は、ラットやヒト等のすべての動物細胞でみられる。NKAは、1つのATP分子を加水分解して、細胞から3つのNa+を排出し、2つのK+を組み入れる能動輸送系である。
細胞は、NKAによって、細胞内イオン濃度、浸透圧及び含水量を調節している。NKAによる3Na+排出、2K+組み入れの電荷の差し引きにより形成される膜電位(Em)は、神経細胞では細胞膜の過分極に関連している。
また、NKAは、特定の種類の細胞では、グルコースやアミノ酸の能動輸送に必要なNa+濃度勾配を形成する役割がある。
細胞は、グルコース代謝で産生した数10%ものATPを、NKAを始めとするイオン能動輸送の過程に用いている。
【0016】
次に、図1を参照して、本発明の発明者らが明らかにした視床下部弓状核のGIニューロンの食欲調整のモデルについて説明する。
上述のように、本発明の発明者らは、グルコース濃度低下によりNKA活性が抑制されると、これによりGIニューロンが活性化され、摂食活動を亢進させることを明らかにした。
すなわち、グルコース濃度が低下すると:
(1)グルコース代謝の低下
(2)細胞内エネルギー産生の低下
(3)NKAの活性低下による細胞の脱分極
(4)電位依存性Ca2+チャネルの活性化
という一連の流れにより、GIニューロンが活性化され、摂食活動が亢進する。
【0017】
より詳しく説明すると、グルコース濃度低下は、GIニューロンのグルコース代謝及びATPを含むエネルギー生産を減少させ、これにより膜電位変化を伴う起電性におけるNKA活性の低下に結びつく。
このようにNKA活性が引下すると、細胞膜が脱分極し、電圧依存性Ca2+チャンネル(Voltage−Dependent Calcium Channel、VDCC)を開口する。
すると、細胞内へのCa2+流入が誘発され、結果としてCa2+に依存したGIニューロンの活性化を引き起こすことになる。
ここで、Ca2+に依存したGIニューロンの活性化は、GIニューロンがNPYを含有することから、NPYニューロンの活性化に繋がっている(Muroya S., Yada T., Shioda S., Takigawa M.、1999、「Glucose−sensitive neurons in the rat arcuate nucleus contain neuropeptide Y.」、Neuroscience Lett.264(1−3)、p.113−116、及びKohno D、et al、2003、「G hrelin directiy interacts with neuropeptide−Y−containing neurons in the rat arcuate nucleus:Ca2+ signaling via protein kinase A and N−type channel−dependentmechanisms and cross−talk with leptin and orexin.」、Diabetes、52、p.948−956、参照)。また、NPYは、摂食促進、すなわち「食欲」に関わっていることが分かっている。
すなわち、NKAは、低グルコースによるニューロンの活性化に関与し、GIニューロンの活性化によるNPY依存性食欲促進に関わっている。
【0018】
このように、本発明の発明者らは、グルコース濃度低下は細胞内のグルコース代謝及びエネルギー生産を減少させ、これにより、起電性NKAの活性が低下し、GIニューロンの脱分極及び電圧依存性Ca2+の流入が導かれることを見いだした。
また、本発明の発明者らは、NKAによるGIニューロンの調節が摂食行動に関連づけられるという新規概念を見いだした。つまり、本発明の発明者らの研究は、グルコース/エネルギー代謝の低下を、摂食行動を刺激する視床下部弓状核GIニューロンの興奮に変換するという、NKAの注目すべき潜在的な役割を明らかにした。さらに、身体中の細胞におけるユビキタスな細胞基本活動維持分子であるNKAが、摂食行動に関連する視床下部弓状核の食欲促進のニューロンを調節する特異的な生理機能を発揮することを見いだした。
このため、この機構を調整することで、摂食行動を調整するための新たな薬剤の開発が可能になる。
つまり、本実施形態においては、NKA活動を調整するNKA活動調節剤として、NKAアクティベータ/インヒビターを用いることで、肥満及び/又は糖尿病に関連した摂食亢進症を改善する、新規手法を提供する。
【0019】
よって、視床下部弓状核のGIニューロンのNKAを活性化すると、食欲を司るGIニューロンの活動を抑制し、「食欲」を抑えることができる。すなわち、第1次摂食・代謝調節中枢による「食欲」を抑制することが可能になる。
このため、本発明の実施の形態に係る食欲調整剤において、GIニューロンのNKAの活性を活性化する各種のNKAアクティベータとなる組成物を用いることができる。このNKAアクティベータを含む食欲調節剤は、食欲抑制剤として用いることができる。
NKAアクティベータとして用いる組成物としては、例えば、抗体SSA412(Xu, K. Y.、2005、「Activation of (Na++K+)−ATPase.」、Biochem Biophys Res Commun.、338、p.1669−1677、参照)を用いることができる。視床下部弓状核では、生理的な各種摂食代謝調節因子により食欲を調整する必要があるため、比較的、血液脳関門の物質透過性が高い(リーキー)ため、抗体でも通過できる可能性がある。また、ファージ等を用いた分子量の小さい抗体を作成することで、血液脳関門を通過させることも考えられる。加えて、血液脳関門を通過させるため、この抗体の活性化部位を用いた低分子のペプチドを合成することもできる。
なお、この抗体SSA412は、上述の文献に記載されているように、心筋細胞においてはNKAを活性化して[Ca2+]iを増加させるという働きがある。ところが、本発明の発明者らが明らかにしたところ、GIニューロンでは、SSA412は、[Ca2+]iを減少させる、すなわちCa2+の細胞内流入を阻害する方向に働くため、上述の文献からは想起できなかった。このため、抗体SSA412は、本発明の実施の形態に係る食欲抑制剤として自明ではない。
【0020】
なお、NKAアクティベータは、任意のものを用いることができる。たとえば、血液脳関門を通過し、NKAを活性化するペプチドホルモンを用いることができる。また、NKAを活性化する各種低分子化合物や薬剤を用いることができる。この際に、SSA412と同様にNKAのαサブユニットに結合してNKA活性を上げるようにすることができる。ここで、本実施形態のNKAアクティベータは、GIニューロンのNKAに特異的に結合して活性化する薬剤を用いることが特に好適である。例えば、他の細胞のNKAでは影響を及ぼさないように、ドラッグ・デリバリー・システムを用いた各種薬剤を用いることができる。たとえば、リポソームにNKAを活性化する薬剤を封入し、脳特異的に作用するように構成できる。加えて、siRNA、PNA等も用いることができる。
さらに弓状核ではNKAのαサブユニットのうちα1の発現が他の組織に比べて数倍高いことが報告されている(例えば、Antonelli MC et al、「Localization of Na, K−ATPase isoforms in the hypothalamus of the rat.」、1995、Cellular Molecular Biology、41、p.79−85を参照。)。このため、NKAのα1サブユニットのみに対するNKAインヒビター/アクティベータを用いることで、身体の他の部位への副作用が少ない食欲亢進剤/食欲抑制剤を提供することが可能になる。
さらに、GIニューロンの活性を押さえるためには、全身の他のNKA活性に関連しない程度の低容量の薬剤を用いるだけでも効果的である。このため、NKA活性を亢進させる各種の薬剤を、副作用の少ない低容量で用いることが可能となる。
後述する実施例における図5I、図5Jのように、実際に、ラットにおいて、脳室内投与を行った際に、通常実験で用いられる濃度の1/10程度でもGIニューロンの応答を行わせることができる。このため、経口/通常の静脈注射による投与であっても、強心剤として用いられるウワバインやジゴキシンの1/10の量で、食欲増進の効果があると考えられる。すなわち、NKAインヒビターは、強心配糖体としての投与の濃度のログオーダーで−1(1/10)以下の量を投与することが好適である。
このため、通常より低い濃度のウワバインを用いても、食欲を増進させられると考えられる。
【0021】
以上のように構成することで、以下のような機能を得ることができる。
まず、従来より、中枢セロトニン・ノルアドレナリン・カンナビノイド系に作動する食欲抑制薬が開発され臨床使用されている。
その多くが、肺高血圧症、心弁膜症等などの循環系の障害、うつ病や自殺者の増加を起したために中止されている。
現在日本で唯一臨床使用可能な食欲抑制薬である、ノルアドレナリン系作動薬であるマジンドールは、副作用があり抗肥満効果は限定的であった。
このように、脳の神経伝達に作用する物質は重篤な副作用を伴っており、これは従来技術1の薬学的組成物も同様であった。
よって、別の機序により有効に食欲を制御する肥満症治療薬が待望されている。
【0022】
これに対して、本発明の実施の形態に係る食欲抑制剤は、神経伝達物質ではなく、グルコース抑制性ニューロンの機能に着目した。すなわち、上述したように、グルコースのシグナル伝達機構の中心として、NKAの作用に介入して摂食を調節する。
これにより、本発明の実施の形態に係る食欲抑制剤は、従来技術1とは異なり、「食欲促進ニューロン」を直接抑制することができる。さらには、この食欲促進ニューロンは同時に全身エネルギー代謝を調節していることから、視床下部からの出力経路を修飾し、それにより脂肪細胞・肝臓の代謝を調節し、更なる抗肥満作用があることが期待される。同時に、脂肪細胞由来物質の視床下部へのフィードバック入力をも修飾し、統合的な食欲抑制が可能となる。
また、脳内において、GIニューロンは、視床下部弓状核の、第1次摂食・代謝調節中枢をはじめとする数か所にのみ特異的に存在するため、副作用を抑えられる。
このため、ごく少量の薬物を用いて、食欲を根本的に抑制できると考えられ、副作用の少ない食欲抑制や肥満解消を実現させることができる。よって、本発明の実施の形態に係る食欲抑制剤は、効果的な摂食抑制作用が得られ、抗肥満効果が期待でき、メタボリック・シンドロームの解消に資することができる。
これにより、本発明の実施の形態に係る食欲抑制剤は、食物摂取量を制限するために、糖尿病等にも用いることができる。
【0023】
また、視床下部GIニューロンのNKAを抑制することで、食欲を司るGIニューロンの活動を活性化させ、第1次摂食・代謝調節中枢による「食欲」を亢進させることが可能になる。
このため、本発明の実施の形態に係食欲調節剤において、GIニューロンのNKAの活性を抑制する各種のNKAインヒビターとなる組成物を用いることができる。このNKAインヒビターを含む食欲調節剤は、食欲亢進剤として用いることができる。
このNKAインヒビターとしては、例えば、強心配糖体であるウワバイン(ouabain)、又はジゴキシン(digoxin)を用いることができる。ウワバインは、キョウチクトウ科(Apocynaceae)の種子から抽出される強心配糖体の一種であり、NKAの作用を阻害する。ウワバイン又はジゴキシンは、NKAの活性を抑制することで、細胞膜の内外にNa+、K+イオンの濃度差を引き起こしやすくし、これにより膜電位が変化する脱分極(depolarization)による心臓の拍動を助けるため、従来は強心剤として用いられている。
ウワバイン又はジゴキシンは、NKAを抑制する作用が強いものの、本発明者らの予備的な実験により、心臓等に作用の出ない低容量にてGIニューロンの活性化を引き起こすことが分かっている。
これにより、例えば、抗癌剤の副作用による食欲の低下、消化管手術後の食欲低下、加齢による食欲の低下、低栄養、神経性食思不振症、摂食障害を伴う、うつ病等の精神疾患・自律神経失調症等のQOL(Quality Of Life)の低下に係る疾病の治療として、食欲を亢進させることができる。
よって、食欲の不振による体力の低下を抑えることができる。
なお、NKAインヒビターとして、他の強心配糖体、ペプチドホルモン、siRNA、PNA等を用いることができる。また、NKAインヒビターも血液脳関門を透過し、視床下部弓状核のGIニューロンに特異的に結合するようなものを用いることが望ましい。
また、ヒトに投与する場合、ヒトに用いられる強心配糖体のジゴキシンとこのアナログとして、「ハーフジゴキシンKY」、「ジゴキシンKY」(京都薬品製、トーアイエイヨー、アイテラス)、「ジゴキシン「AFP」」(アルフレッサ製)、「ジゴキシン」(中外製薬製)、メチルジゴキシン「ラニラピッド」(中外製薬製)、デスラノシド「ジギラノゲン」(アイロム製)等の製剤を用いることができる。作用機構が同じであるため、本実施の形態におけるジゴキシンとウワバインの効果は、同様となる。
【0024】
このように、本発明の実施の形態に係る視床下部弓状核のGIニューロンのNKA活性を調整する調整剤を用いることで、NKAを活性化させて食欲抑制させ、NKAを抑制させて食欲増進させるという、2つの効果を目的に応じて得ることができる。
これにより、食欲を自在に調整して、適正な体重や体組成等の身体状態を得ることができるようになる。
【0025】
以上のように、摂食亢進性グルコース抑制性ニューロンを制御する鍵となる分子して、NKAの修飾により摂食を制御できる。
これにより、NKA活性化物質の開発による新規抗肥満薬である治療剤を開発することができる。このプロトタイプとなる物質として、SSAを用いることができる。
一方、NKA抑制物質により、術後・抗がん剤使用・高齢者の食欲低下に伴うQOL低下の改善の治療剤を開発することもできる。
【0026】
また、本発明の実施の形態に係る食欲亢進剤と食欲抑制剤とを同一のパッケージに入れて提供することも可能である。これにより、患者の意志で任意の間隔で食欲を調整できる。つまり、食事のタイミングを自在に調節して、各種疾病やダイエット等に適宜用いることができる。このため、血糖値を所定の間隔に保つ必要がある糖尿病患者や、間食が止められない肥満患者等に用いることができる。また、抗癌剤の副作用等により食欲が低下している患者が、病院食を提供する間隔で食欲がでるように調整することができる。
このように本発明の実施の形態に係る食欲亢進剤と食欲抑制剤とは、同一のNKAの活性の抑制/亢進機構により動作するので、それぞれが拮抗すると考えられる。また、同一の機構により食欲亢進/食欲抑制を行うため、薬剤耐性により使用量が増えることが抑えられると考えられる。また、体内の応答を過度に刺激することも少ないと考えら得る。
このため、副作用が少なく、健康食品や動物用飼料としても健康被害がなく用いることが期待できる。
【0027】
また、本発明の実施の形態に係る食欲抑制剤又は食欲亢進剤は、任意の製剤上許容しうる担体(例えば、生理食塩水、補助薬を含む等張液、例えばD−ソルビトール、D−マンノース、D−マンニトール、塩化ナトリウム等が挙げられ、適当な溶解補助剤、例えばアルコール、具体的にはエタノール、ポリアルコール、例えばプロピレングリコール、ポリエチレングリコール、非イオン性界面活性剤、例えばポリソルベート80(TM)、HCO−50等を挙げることができるが、それらに限定されない)と共に投与することができる。また、適切な賦形剤等を含んでもよい。
【0028】
また、本発明の実施の形態に係る食欲抑制剤又は食欲亢進剤は、製剤上許容しうる担体を調製するために、適切な薬学的に許容可能なキャリアを含み得る。
このキャリアとしては、シリコーン、コラーゲン、ゼラチン等の生体親和性材料を含んでもよい。あるいはまた、種々の乳濁液であってもよい。
さらには、例えば、希釈剤、香料、防腐剤、賦形剤、崩壊剤、滑沢剤、結合剤、乳化剤、可塑剤などから選択される1または2以上の製剤用添加物を含有させてもよい。
【0029】
本発明の実施の形態に係る食欲抑制剤又は食欲亢進剤は、経口投与のための投与に適した投与形態において、当該分野で周知の製剤上許容しうる担体を用いて処方され得る。
本発明の実施の形態に係る医薬組成物の投与経路は、特に限定されないが、非経口的に投与することが好ましい。
非経口投与としては、例えば、静脈内、動脈内、皮下、真皮内、筋肉内または腹腔内の投与が挙げられる。
【0030】
本発明の実施の形態に係る食欲調整剤は、脊椎動物の脳に存在する視床下部弓状核のグルコース抑制性ニューロンを作用対象としている。
この脊椎動物は特に限定されるものではなく、例えば、ヒト、家畜動物種、野生動物を含む。
このため、本発明の実施の形態に係る食欲抑制剤又は食欲亢進剤は、広く動物の治療、家畜の発育増進等の対象とすることができる。
また、疾病の予防や健康増進のため、健康食品のような食物、動物用の飼料、又は食餌に含ませることもできる。
【0031】
また、本発明の実施の形態に係る食欲抑制剤の治療対象としては、メタボリック・シンドロームのような、摂食変調を伴う疾病、糖尿病、高血圧等を含む。また、本発明の実施の形態に係る食欲亢進剤の治療対象としては、摂食障害を含む、痩身による体力低下に関する各種疾病に用いることができる。
【0032】
また、本発明の実施の形態に係る治療用組成物を上述の治療に用いるために、投与間隔および投与量は、疾患の状況、さらに対象の状態などの種々の条件に応じて適宜選択および変更することが可能である。
【0033】
本発明の実施の形態に係る食欲抑制剤又は食欲亢進剤の1回の投与量および投与回数は、投与の目的により、さらに患者の年齢および体重、症状および疾患の重篤度などの種々の条件に応じて適宜選択および変更することが可能である。
投与回数および期間は、1日1回約2〜4週間程度投与して状態をモニターし、その状態により再度あるいは繰り返し投与を行う。
【0034】
本発明の実施の形態に係る食欲抑制剤又は食欲亢進剤は、他の組成物等と併用することも可能である。また、他の組成物と同時に本発明の組成物を投与してもよく、また間隔を空けて投与してもよいが、その投与順序は特に問わない。
【0035】
また、本発明の実施の形態において、疾患が改善または軽減される期間は特に限定されないが、一時的な改善または軽減であってもよいし、一定期間の改善または軽減であってもよい。
【実施例】
【0036】
〔本発明の実施の形態に係る視床下部弓状核のGIニューロンのNKA活性の調整剤の実施例〕
以下で、本発明の実施の形態に係る治療用組成物について、具体的な実験を基にして、実施例としてさらに具体的に説明する。しかしながら、この実施例は一例にすぎず、これに限定されるものではない。
【0037】
上述の実施の形態で説明したように、本発明の発明者らは、鋭意実験と検討を行い、GIニューロンとNKAの関係について調べた。
まず、図2の実験の概念図を参照して、本実施例における、脳のニューロン同定/機能解析方法について説明する。本実施例では、摂食機能が完成した6週齢ラットの視床下部スライスを作製し、その中から弓状核を切離した。その後、機械的分散及び酵素処理により、ニューロンを単離した。そして、蛍光画像解析装置で、細胞内のNa+([Na+]i)及びCa2+濃度([Ca2+]i)を測定した。この際、実験に応じて、細胞外液を灌流して測定した。
【0038】
より具体的には、本発明の発明者らは、視床下部弓状核からGIニューロンを単離し、グルコース濃度低下と、NKA活性を抑制する薬剤であるNKAインヒビターであるウワバインを用いた実験を行った。この結果、グルコース濃度低下及びNKA抑制の両方が、視床下部弓状核のGIニューロンの[Na+]i及び[Ca2+]iの増加を引き起こすことを見いだした。
これに対して、NKAアクティベータとして動作する抗体であるSSA412によって処理をしたところ、グルコース濃度低下に誘発された[Ca2+]iの増加は起こらなかった。
また、本発明の発明者らは、NKAの上流の機構についても調べた。すると、グルコース濃度低下は、GIニューロンのNAD(P)H自己蛍光を減少させた。また、グルコキナーゼインヒビターを用いて処理を行ったところ、グルコース濃度増加により活動しなくなったGIニューロンの[Ca2+]iを増加させた。これにより、NKAがグルコース代謝の経路により制御されていることが示された。
さらに、本発明の発明者らは、NKAのパスウェイの下流の機構についても調べた。電圧依存性Ca2+チャンネル(VDCC)ブロッカーを用いてGIニューロンを処理した。すると、グルコース濃度低下に誘発された[Ca2+]i増加が抑制された。これにより、GIニューロンにおけるNKAの下流に、脱分極を介した機構があると推測できた。
この上で、本発明の発明者らは、NKAによる摂食行動の調節について、すなわちNKAの活性の調節により食欲を変化させられるかについて調べた。具体的には、ラットを絶食状態にして、視床下部弓状核のNKAの活性を調べた。すると、絶食により、視床下部弓状核のNKAの活性が減少した。また、NKAインヒビターであるウワバインを脳室内に注射したところ、摂食活動が亢進し、ニューロペプチドYの発現も惹起した。
これに加えて、グルコース代謝を抑制する薬剤であるグルコキナーゼインヒビターをラットの脳室内に注射したところ、摂食活動を喚起したが、NKAアクティベータであるSSA412を脳室内へ注射したところ、惹起された摂食活動が打ち消された。
このように、本発明の発明者らは、NKAの抑制がGIニューロンのグルコース濃度低下に誘発された活性化を媒介するという仮説の具体的な作用機構を証明した。
よって、本実施例により、エネルギー状態の低下をGIニューロンの刺激と摂食行動に結びつける視床下部弓状核のNKAの新規機能を明らかにし、視床下部弓状核のGIニューロンのNKA活性の調整剤を実現した。これにより、従来技術1とは異なり、副作用の少ない食欲抑制剤/食欲亢進剤をそれぞれ同一の作用機構を元に実現できる。
以下で、具体的な各実験と実施の例について、図面とデータを参照して説明する。
【0039】
(視床下部弓状核からの単一のニューロンの調製)
実験に用いたラットは、オスの成熟SDラットを、12時間の明/暗サイクル及び従来の食物(CE−2; クレア社製、東京、日本)及び水で無制限に飼育した。この研究のための動物プロトコルは、自治医科大学の動物保護委員会で承認された。
視床下部弓状核の単一のニューロンは、5〜7週のラット(Sprague−Dawley rat、SDラット)から単離された。ラットはウレタン(カルバミン酸エチル;1g/kg、i.p.)により麻酔をかけられ、脳を摘出された。そして、両側視床下部弓状核を含んでいる冠状断脳切片が準備された。その後、視床下部弓状核は両側から切離された。切り分けられた組織は、下記の成分からなるHEPESバッファーを用いたクレブス・リンガー重炭酸塩緩衝液(HEPES−buffered Krebs−Ringer bicarbonate buffer、HKRB)により洗浄した:
HKRBの組成(単位、mM): NaCl(129)、NaHCO3(5.0)、KCl(3.7)、KH2PO4、1.2、CaCl2(1.8)、MgSO4(1.2)、10mMグルコースを含んでいるHEPES・pH7.4(10)
【0040】
その後、切除された視床下部弓状核は、5〜10分間温和な機械的な分散(粉砕)をした後、20ユニット/mlのパパイン(papain)(シグマ・ケミカル社製、セントルイス、ミズーリ州、米国)、0.015mg/mlのデオキシリボヌクレアーゼ、0.75mg/mlのウシ血清アルブミン、及び1mmol/Lシステインを加えたHKRBを用いて、36℃のウォーターバスで15分振とう培養し、酵素処理を行った。この細胞懸濁液は5分間100×gで遠心分離された。
上澄みの溶液を捨てた細胞の沈殿物はHKRBで再懸濁され、カバーガラス上に分散して置かれた。細胞は6時間以内、飽和湿度の皿で、室温、遮光にて維持された。その後、単一のニューロンはカバーガラスに配置された。
【0041】
(単一の視床下部弓状核ニューロンの[Ca2+]i、[Na+]i及び[NAD(P)H]iの測定)
[Ca2+]i、[Na+]i、及び[NAD(P)H]iは、レシオメトリック・マイクロ蛍光イメージング法にて測定された。
イメージインテンシファイア付電荷結合素子(ICCD)カメラにより検出された。また、レシオ(比率)・イメージは、蛍光画像解析装置であるARGUS−50システム又はAquacosmosシステム(浜松ホトニクス社製、浜松、日本)を用いて取得した。
下記で、各測定のより具体的な方法を説明する。
【0042】
[Ca2+]iの測定は、単一ニューロンの調製後、2〜6時間静置し、レシオメトリックfura−2イメージングを用いて行った。この際、2μMのCa2+感受性色素fura−2/アセトキシメチル・エステル(AM)(株式会社ケミカル同仁製、熊本、日本)を負荷して、室温で1時間培養された。その後、細胞は倒立顕微鏡のステージ上の保温装置付チェンバーに取り付けられ、32℃で1ml/分でHKRBにより灌流された。
細胞は、340及び380nmで交互に12.0秒ごとに励起された、
510nm(それぞれF340及びF380)の発光信号はICCDカメラにより検出された。
また、レシオ・イメージ(F340/F380)はARGUS−50あるいはAquacosmosシステムで取得した。
データは従来に報告された手順により、形態学的及び生理学的なニューロン判定基準を満たす細胞から得られた。
薬剤に応じた[Ca2+]i増加のピーク振幅が、自発的な変動の130%以上だった場合、それらは[Ca2+]i応答であるとした。
薬剤に応じた[Ca2+]i増加の振幅は、ピーク[Ca2+]iレシオから刺激前の基準となる[Ca2+]iレシオを引くことにより計算された。
阻害剤の影響を調査する実験では、刺激性の薬剤に誘発された[Ca2+]i増加の振幅が30%以上阻害剤で抑えられた場合、抑制と判定した。
【0043】
[Na+]iの測定は、単一ニューロンの調製後2時間静置し、32℃で1.5〜2.0時間、Na+感受性色素であるナトリウム結合イソフタル酸ベンゾフラン(SBFI)(5μM)/AM(1μM)(モレキュラー・プローブズ社製、ユージーン、オレゴン、アメリカ)及び非イオンの分散剤であるプクロニック(Pluronic)F−127(0.0125%)(モレキュラー・プローブズ社)を負荷して培養された。
その後、細胞は倒立顕微鏡のステージ上の保温装置付チェンバーに取り付けられ、32℃で1ml/分でHKRBにより灌流された。
SBFI蛍光測定法のための手順は、fura−2のための標準的な手順に従った。
HEPESバッファーを用いたNa+を含まない(Na+−free)溶液は、NaClの代わりにN−メチルDグルカミン塩化物(N−methyl−D−glucamine)を用いることにより準備された。
単離細胞の[Na+]i値は較正曲線に従って計算された。
当該構成曲線は、膜内外のNa+およびK+濃度を平衡化するイオン透過担体グラミシジンD(MPバイオメディカルズ社製、アーヴィン、CA、アメリカ)10μg/mlを含んだ各種のNa+(5、10、15、30、60、100 mM)及びK+濃度(Na++K+=135、グルコン酸塩105、CaCl2 2.5、MgCl2 1.2、HEPES 10、及びグルコース8.3(単位、mM))を含む溶液に段階的に灌流することで、観察されたレシオを設定した。
【0044】
(同一ニューロンの[NAD(P)H]iおよび[Ca2+]iの測定)
以前に報告された同様の方法により、NAD(P)Hの自己蛍光が最初に測定された。
細胞は360nmで励起された。また、470nmの蛍光発光が検出された。各時点における蛍光強度F360は、測定開始時の蛍光強度F360(0)に対する割合(F360/F360(0))により計算された。
[NAD(P)H]iを測定した後に、同一ニューロンにおいて[Ca2+]i測定のために、細胞に室温で1時間、2μM fura−2を負荷した。また、上述した手順で[Ca2+]iが測定された。
【0045】
(同一ニューロンの[Na+]iおよび[Ca2+]iの測定)
上に述べられるような[Na+]iの測定の後に、同一ニューロンにおいて、[Ca2+]iの測定を行った。Ca2+およびNa+測定のための2つの励起波長を分離するために、Ca2+感受性色素として、fluo−4/AM(モレキュラー・プローブズ社製)を使用して測定された。
[Na+]iを測定した後に、同一ニューロンにおいて[Ca2+]i測定のために、細胞に室温で1時間間2μM fluo−4が負荷された。
細胞は、490nmで12秒ごとに励起された。また、520nmの蛍光発光が検出された。
各時点における蛍光強度F490は、測定開始時の蛍光強度F490(0)に対する割合(F490/F490(0))により計算された。
【0046】
(SSA412と[Ca2+]iの測定)
最初に、[Ca2+]i測定は、灌流を行わない状態で、8.3mMグルコースを含んだ、30μlの小滴を用いて10分間、行なわれた。
第1の測定の後、20μl小滴が静かに除去された。また、グルコースのないSSA412又はコントロールのウサギIgGを含んでいる20μlのHKRBが加えられた。これにより、2.8のmMグルコースを含んでいる30μlのHKRBとなった。
この30μlのHKRBを使用して、灌流を行わない状態で、次の10分において、第2の[Ca2+]i測定が行なわれた。
最後に、GIニューロンを明確に識別するために、同一ニューロンの[Ca2+]iが上述した灌流条件下で測定された。
【0047】
(NKA活性の決定)
8週間の年齢の自由に食物を与えられた(n=7)又は24時間の絶食条件下の(n=7)ラットから、左右の視床下部弓状核およびVMHを取得し、直ちに液体窒素中で凍結した。
ウワバイン−センシティブATPアーゼアッセイの手順は、従来のKyteの方法(Kyte、1972、「J. The titration of the cardiac glycoside binding site of the (Na+ + K+)−adenosine triphosphatase.」、J Biol Chem 247、p.7634−7641)を修正して用いた。
【0048】
組織試料はそれぞれ、ティッシュティアラー(Tissue Tearor、ドレメル社製、ラシーン、ウィスコンシン州、アメリカ)を使用して、20mM Tris/HCl緩衝液、pH 7.4の0.5mlでホモジナイズされた。その上で、ガラス繊維布ホモジナイザーで均質化された。
ホモジェネートのタンパク濃度は、BCAプロテインアッセイ(ピアス社製、ロックフォード、イリノイ州、アメリカ)で決定された。
ホモジェネートされたサンプルは、分析のための、ウワバインを入れた/ウワバインを入れない2つのグループに分割された。
【0049】
酵素活性は、Na+およびK+がある状態で、ウワバインを入れた/ウワバインを入れないグループについて、MgATPのウワバイン感受性の加水分解として定義した。
タンパク質、MgATP、Na+、K+、及びウワバインの最終的な濃度は、それぞれ20μg/ml、3mM、120mM、20mM、及び2mMだった。
その反応は、37℃で30分の培養の後、MgATPを加えることにより始められ、0.75mlの反応停止溶液と混合することにより停止された。
発色剤(0.02ml)が加えられ、30分間室温で現像させた。
その後、遊離したリン酸塩の濃度が、分光測光器で700nmの波長の測定により決定された。
【0050】
(脳室内投与および食餌実験)
9〜10週のSDラットにおいて、ガイド・カニューレを脳の第三脳室に挿入した。この箇所は、正中線上でブレグマの尾側の0.7mm、及び頭蓋骨表面からの7.0mm下に位置している。
手術の後、ラットを7日間回復させ、非特異性のストレス応答を最小限にするために、毎日5分間のハンドリングを行った。
脳室内投与は、午前中(10:00−12:00)に開始し、食物摂取させた2時間後の摂取量を測定した。
【0051】
(NPY及びPOMCのmRNA量の測定)
10週の年齢のSDラットの脳室内に、5mMグルコースと一緒にウワバイン又はHKRBが投与された。視床下部弓状核は、投与の3時間後に摘出された。
mRNAは以前に報告されたのと同様な方法によりリアルタイムPCRで測定された。
トータルRNAは、Trizol(インヴィトロゲン社、ゲーサーズバーグ、メリーランンド州、アメリカ)及びRNAseフリーのDNAse(プロメガ社、マディソン、ウィスコンシン州、アメリカ)により単離された。
その後、トータルRNAをReverTra Ace(登録商標)(東洋紡績株式会社製、大阪、日本)による相補的DNA(cDNA)へ転換した後、SYBR premix Ex taq IIポリメラーゼ(タカラバイオ社製、大津、日本)が用いられ、リアルタイムPCRにより測定された。
蓄積した生成物は実時間(リアルタイム)で測定された。
また、最初に生成物を検出することができる平均のサイクルしきい値は、同じプレート上で実行されたプライマー・ペアおよびcDNAサンプル当たり5つの独立した反応の反復試験試料に対して決定された。
NPY又はPOMC(Pro−opiomelanocortin)の各cDNAサンプルは、ハウスキーピング遺伝子であるグリセリンアルデヒド−3−リン酸脱水素酵素(GAPDH)の生成物のプライマー・セットを使用して標準化された。
各cDNAに対応するプライマーは以下の通りだった:
GAPDH − フォーワードプライマー(配列番号1)、リバースプライマー(配列番号2)
NPY − フォーワードプライマー(配列番号3)、リバースプライマー(配列番号4);
POMC − フォーワードプライマー(配列番号5)、リバースプライマー(配列番号6)
【0052】
(統計)
データはすべて平均±s.e.m(standard error of the mean、SEM)として示される。
ペア又はペアでないスチューデントt検定は、2つのグループの実験で使用された。
次に、Bonfferoniのポスト・ホック・テストの後、4つのグループの実験において、階乗分散分析が使用された。
P<0.05の場合、統計的に有意であると考慮した。
【0053】
(GIニューロンの定義)
次に図3A〜図3Dを参照して、GIニューロンの定義について検討した際の実験結果について説明する。
グルコース濃度低下は、視床下部弓状核から単離された362のニューロンのうち137ニューロン(37.8%)の[Ca2+]iを増加させた。
図3Aは、本実施例において、GIニューロンとして定義したニューロンの[Ca2+]i応答の具体例を示している。本実施例では、単離された視床下部弓状核のニューロンにおいて、グルコース濃度低下とグルタミン酸ナトリウム(glutamate、100μM)とに応答して[Ca2+]iを増加させたニューロンを、GIニューロンと定義した。
図3Bは、GIニューロン(n=137)において、8.3G及び2.8Gでの[Ca2+]i応答の平均を示している。「**」は、P<0.01であることを示す。
この実験により、8.3Gから2.8Gまでグルコース濃度を低下させた際に[Ca2+]i増加応答したニューロンについて、GIニューロンとして定義した。これらのニューロンは、GIニューロンとして所定の応答基準を満たしている。
【0054】
これに対して、図3Cは、非GIニューロンとして定義されたニューロンの[Ca2+]i応答の具体例を示している。視床下部弓状核から取得したニューロンにおいて、グルコース濃度低下により、[Ca2+]iの増加を示さなかったニューロンを非GIニューロンと定義した。非GIニューロンでは、グルコース濃度低下において[Ca2+]iは変化がないものの、通常の興奮物質であるグルタミン酸ナトリウムには応答する。
図3Dは、非GIニューロン(n=123)において、8.3G及び2.8Gでの[Ca2+]i応答の平均を示している。このように、8.3Gと2.8Gでは有意差がなかった。
【0055】
また、以下の実験において、グルコース濃度低下に応答して[Ca2+]iを減少させるグルコース興奮性(GE)ニューロンは、分析から除外された。
【0056】
(GIニューロンにおけるグルコース濃度低下による[Na+]i増加応答)
ここで、本発明の発明者らは、NKA活性の指標である[Na+]iの反応を、SBFIを使用してマイクロ蛍光分析測定を行い、NKA活性を評価した。図4A〜Eを参照して、このNKA活性の実験結果について説明する。
図4Aは、グルコース濃度低下とウワバインによるGIニューロンの[Na+]i応答を示すグラフである。グルコース濃度低下およびウワバインは、視床下部弓状核から単離されたニューロンの[Na+]iを増加させた。このニューロンとしては、上述のように、摂食機能がほぼ成熟する5〜7週のラットの視床下部弓状核からニューロンを単離した。結果は、7つのニューロン(n=7)のうち代表的な反応について示した。
図4Bは、SBFI蛍光レシオに[Na+]iを関連づける較正曲線を示している。この構成曲線は、1μg/mlグラミシジンD及びNa+濃度を段階的に変化させることで作成した。
グルコース濃度低下は、[Na+]iの増加を誘発した。また、NKAの特異的インヒビターのウワバインは、同じニューロンの[Na+]i増加を誘発した。
また、グルコース濃度低下に誘発された[Na+]i増加は、7.5±1.1mM(n=7)であった。
【0057】
図4Cは、もう一つの一連の実験である、GIニューロンにおけるグルコース濃度低下による[Na+]iの変化と、同一ニューロンでの[Ca2+]iの変化を示すグラフである。グルコース濃度低下は、[Na+]i増加を誘発し、同じニューロンで[Ca2+]i増加を引き起こした。すなわち、グルコース濃度低下により、GIニューロンにおいて[Na+]iが増加した(n=13)。
【0058】
図4Dは、非GIニューロンにおけるグルコース濃度低下による[Na+]iの変化と、同一ニューロンでの[Ca2+]iの変化を示すグラフである。
グルコース濃度低下は、2.8Gに対する[Ca2+]i応答の欠如を示した非GIニューロンにおいては、[Na+]iを顕著に変化させなかった(n=8)。
【0059】
図4Eは、GIニューロン又は非GIニューロンにおけるグルコース濃度低下に誘発された[Na+]iの変化の平均を示すグラフである。「**」は、P<0.01であることを示す。
すなわち、GIニューロンにおけるグルコース濃度低下に誘発された[Na+]i増加の変化量は、非GIニューロンにおける変化量に対して、有意に大きかった。
【0060】
これらのデータにより、GIニューロンにおいて、グルコース濃度低下が、NKA活性を選択的に低減するという概念が立証された。
【0061】
(GIニューロンにおけるNKA活性低下によるGIニューロンの活性化)
次に、本発明の発明者らは、NKA活性の低下が、実際にGIニューロンの活性化に関連しているかどうかを調べた。図5A〜Mを参照して、NKAインヒビターであるウワバインと、NKAを働かせなくするためにK+を含まない溶液を投与した実験結果について説明する。
図5Aは、2.8Gで反応したGIニューロンにおいて、8.3Gでウワバイン(100μM)を投与した際の[Ca2+]i応答を示すグラフである(n=15)。このようにウワバインの投与により、GIニューロンの[Ca2+]i増加を誘発することができた。
図5Bは、ウワバインを投与しない群と投与した群における、8.3Gでの[Ca2+]i応答の平均を示すグラフである。「**」は、P<0.01であることを示す。
図5Cは、8.3Gで、K+を含まない(K+−free)溶液を投入した際の[Ca2+]i応答を示すグラフである(n=12)。このように、K+−free溶液にした場合には、細胞膜内外の電位差を変化させられないため、ウワバインと同様にNKAが機能しなくなる。この場合でも、GIニューロンの[Ca2+]i増加を誘発した。
図5Dは、K+−free溶液を投与した際の[Ca2+]i応答の平均を示すグラフである。「**」は、P<0.01であることを示す。
このように、GIニューロンにおいて、[Ca2+]i反応を安定化させる8.3Gグルコース濃度において、NKAをブロックする条件となる100μMウワバインの投与および外部K+の除去を行うことで、[Ca2+]i増加を誘発した。すなわち、NKA活性を低下させることで、GIニューロンの[Ca2+]i増加を誘発し、GIニューロンを活性化することができる。
【0062】
次に、本発明の発明者らは、ウワバインの継続的な投与にてNKAが既にブロックされていた場合、グルコース濃度低下に誘発された[Ca2+]i増加応答を防ぐことができるかどうか調べた。
図5Eは、まずはウワバインを投与しない状態で、GIニューロンに対してグルコース濃度低下による反復刺激をした場合の[Ca2+]i応答を観測したグラフである。2.8Gによるグルコース濃度低下1回目刺激(Amp1)、及びグルコース濃度低下2回目刺激(Amp2)において、それぞれグルコース濃度低下による反復刺激は[Ca2+]i増加を反復して誘発することが分かる。
図5Fは、8.3Gと、2.8Gのグルコース濃度低下1回目刺激(Amp1)又はグルコース濃度低下2回目刺激(Amp2)における[Ca2+]i応答の平均を示すグラフである。Amp2に対する[Ca2+]i応答の振幅は、Amp1と同様であった(n=9)。すなわち、Amp1、Amp2とも、それぞれ8.3Gに対してP<0.01(**)であった。
これに対して、ウワバイン非投与下におけるコントロール実験においては、グルコース濃度低下2回目刺激(Amp2)で誘発された[Ca2+]iの増加は、グルコース濃度低下1回目刺激(Amp1)で誘発された[Ca2+]iの増加と有意差が認められなかった。
【0063】
図5Gは、ウワバインが継続的に存在していた場合のグルコース濃度低下の反復刺激により誘発された[Ca2+]i応答を示すグラフである。このように、グルコース濃度低下1回目刺激(Amp1)で誘発された[Ca2+]i増加の後、100μMのウワバインが継続的に存在している状態でグルコース濃度低下2回目刺激(Amp2)を行った場合、[Ca2+]iの増加応答は抑えられた。
図5Hは、8.3Gと、2.8Gのグルコース濃度低下1回目刺激(Amp1)及び100μMのウワバインが継続的に存在している状態でグルコース濃度低下2回目刺激(Amp2)を与えた際の、[Ca2+]i応答の平均を示すグラフである(n=10)。100μMウワバインが継続的にある状態でのAmp2は、同じ実験のAmp1と比較して、有意に小さかった(*、P<0.05)。また、図5Hと図5Fとを比較すると、コントロール実験(図5FのAmp2)と比べても、図5HのAmp2は有意に小さかった(#、P<0.05)。
これらの結果により、GIニューロンにおいてNKA活性を抑制させることによって、グルコース濃度低下に誘発された場合と同様の[Ca2+]i増加が誘発されることが分かる。
すなわち、GIニューロンにおいて、低グルコース刺激による[Ca2+]i増加はNKAの活性に依存していることが示された。
【0064】
(ウワバインの濃度と、GIニューロンの活性化の関係)
次に、図5I〜Jを参照して、2.8Gで反応したGIニューロンと、反応しなかった非GIニューロンにおいて、8.3Gで、濃度を変えてウワバインを投与した際の[Ca2+]i応答について説明する。この応答の実験は、ウワバインの濃度の他は、図5A、Bの実験と同様に行った。
図5Iは、ウワバイン濃度に対して反応した細胞数の割合を示すグラフである。横軸は、ウワバインの濃度(μ mol/L、μM)を示し、縦軸はウワバインにより応答が増加したニューロンの割合(%)を示す。黒丸は、GIニューロンを示す。また、白丸は、非GIニューロンを示す。
図5Jは、[Ca2+]i増加応答の平均を示すグラフである。横軸は、ウワバインの濃度(μ mol/L、μM)を示し、縦軸は[Ca2+]i増加応答の平均を示す。黒丸はGIニューロン、白丸は非GIニューロンを示す。「#」は、P<0.01であることを示す。
このように、ウワバインの濃度が100μMの1/1000の0.1μMであっても、顕著に応答を増加させることが分かる。また、100μMの1/100の1μMにて[Ca2+]i増加応答が最大化する。さらに、100μMの1/10の10μMにて、GIニューロンと非GIニューロンとの差がなくなることが分かる。ここで、100μMのウワバインを脳室内投与すると、通常の強心剤として用いられる投与量と同等であると考えられる。すなわち、通常の投与量の1/10で、グルコース濃度低下によるGIニューロンの活性化を抑えることが可能である。
【0065】
(NKAの活性化による、GIニューロンの不活性化)
上述の結果により、NKAを活性化させる操作は、グルコース濃度低下に対する[Ca2+]i増加応答を減ずる可能性がある。
そこで、本発明の発明者らは、NAKのαサブユニットと特異的に作用し、NKAを活性化させるNKAアクティベータとなるSSA412抗体を用いて、NKA活性増加とGIニューロンの不活性化について検討した。
この実験では、灌流を行わない状態で8.3Gから2.8Gにグルコース濃度を低下させ、SSA412を2μM又はコントロールのウサギIgGを2μM投入し、単離されたニューロンの[Ca2+]iを測定した。
【0066】
図5Kは、ウサギIgGを加えたコントロール実験の結果である。
まずは、8.3Gの[Ca2+]i応答を測定した(図5K左欄)。
その後、コントロールとなる2μMのウサギIgGを加えて、2.8Gの[Ca2+]i応答を測定した(図5K中央欄)。2.8Gにおいて、[Ca2+]iの増加応答を示したニューロンが存在した。
さらにウサギIgGを洗浄し、溶液を灌流させた状態で[Ca2+]i応答を測定した(図5K右欄、n=12)。2.8Gで[Ca2+]iは増加し、これらがGIニューロンにおける反応であったことが確認できた。
【0067】
図5Lは、NKAアクティベータであるSSA412を加えた実験の結果である。
コントロール実験と同様に、まずは8.3Gの[Ca2+]i応答を測定した(図5L左欄)。
その後、2μMのSSA412を加えて、2.8Gの[Ca2+]i応答を測定した(図5L中央欄)。2.8Gにおいて、[Ca2+]iは増加応答を示さなかった。
その後、SSA412を洗浄し、溶液を灌流させた状態で[Ca2+]i応答を測定した(図5L右欄、n=8)。2.8Gで、[Ca2+]iは増加し、これらがGIニューロンにおける変化であったことが確認できた。
すなわち、GIニューロンにおいて、SSA412存在下では[Ca2+]i増加が誘発されず、GIニューロンが活性化しないことが示された。
【0068】
図5Mは、コントロールのウサギIgG、又はNKAアクティベータであるSSA412を加えた際の[Ca2+]i応答の平均を示す。「*」は、P<0.05であることを示す。また「**」は、P<0.01であることを示す。
この結果を参照すると、2.8GにおけるSSA412存在下での[Ca2+]iは、8.3Gの[Ca2+]iとの有意差を認められなかった。さらに、コントロールであるIgG存在下での[Ca2+]iに対して、SSA412存在下での[Ca2+]iは有意に低かった(P<0.05)。
すなわち、GIニューロンにおけるグルコース濃度低下に対する[Ca2+]i反応は、SSA412の存在によって抑制された。
【0069】
これらの結果は、SSA412によるNKAの活性化により、グルコース濃度低下がGIニューロンの[Ca2+]i増加を誘発するのを抑制することを示している。
NKA活性に対するインヒビターとアクティベータとで操作した上述のデータは、視床下部弓状核GIニューロンが、NKAにより、グルコース感知によるニューロン活性化と結びつけられていることを強く示唆する。
【0070】
(GIニューロン活性化の上流でのグルコースによるシグナル伝達機構)
次に、本発明の発明者らは、GIニューロン活性化の上流でのグルコースによるシグナル伝達機構を検討した。
膵臓形態のグルコキナーゼは、グルコースをリン酸化する際の律速酵素であり、ランゲルハンス島β細胞のグルコース反応及び視床下部のGEニューロンの応答に重要な役割を果たしている。また、GIニューロンにも密接に関連している可能性が推測される。
以下で図6A〜図8Dを参照して、このグルコースによるシグナル伝達機構に関する実験の結果について説明する。
【0071】
図6Aは、GIニューロンにおいて、灌流液内のグルコース濃度を8.3Gで維持させた状態で、グルコキナーゼの拮抗阻害剤である10mMマンノヘプツロース(Mannoheptulose、MH)を投与した実験例を示すグラフである(n=14)。この結果として、8.3Gにおいて、10mMのMHを投与すると[Ca2+]iを増加させた。すなわち、グルコース濃度低下と同様の効果が得られた。
図6Bは、GIニューロンにおいて灌流液内のグルコース濃度を8.3Gで維持させた状態で、代謝されないグルコース・アナログである2−デオキシ−D−グルコース(2DG)を投与した実験例を示すグラフである(n=13)。10mMの2DGを投与した結果、MHと類似した結果が得られた。すなわち、8.3Gで、10mMの2DGは、[Ca2+]i増加を誘発し、グルコース濃度低下と同様の効果が得られた。
【0072】
図6Cは、MHに対する[Ca2+]i増加応答の発生率を示しており、GIニューロン(黒塗りのバー)で77.8%(14/18)、非GIニューロン(オープンバー)では26.7%(4/15)であった。
図6Dは、2DGに対する[Ca2+]i応答の割合を示しており、GIニューロン(黒塗りのバー)で76.9%(10/13)、非GIニューロン(オープンバー)では16.7%(1/6)であった。
図6Eは、MHを投与した際のGI及び非GIニューロンの[Ca2+]i応答の平均を示している。「**」はP<0.01であることを示す。
図6Fは、2DGを投与した際のGI及び非GIニューロンの[Ca2+]i応答の平均を示している。「**」はP<0.01であることを示す。
このように、MH及び2DGに対する[Ca2+]i応答の割合と振幅は、非GIニューロンよりもGIニューロンにおいて有意に大きかった。
これらのデータにより、グルコキナーゼを媒介としたグルコース代謝が、GIニューロンを調節することが明確となった。
【0073】
次に、本発明の発明者らは、GIニューロンにおいて、グルコース低下による反応が細胞内のエネルギー生産に関連しているかについて調べた。図7を参照して、この実験について説明する。
図7Aにおいて、左図は、視床下部弓状核から単離したニューロンにおいて、NAD(P)H自己蛍光を測定した実験例である(n=6)。これらのニューロンのNAD(P)H自己蛍光は、2.8Gでは減少し、8.3Gで増加した。右図は、この同一ニューロンにおける2.8Gに対する[Ca2+]i増加応答を示しており、GIニューロンであることを確認した。
図7Bは、GIニューロンにおける8.3G及び2.8GにおけるNAD(P)H自己蛍光(の平均)を示している。「**」は、P<0.01であることを示す。平均のNAD(P)H自己蛍光レベルは、グルコース濃度が8.3Gの場合より、2.8Gの場合において有意に低かった。
これらのデータは、グルコース濃度低下がGIニューロンにおいてNAD(P)Hを減少させることを示している。
【0074】
更に、本発明の発明者らは、GIニューロンにおいてエネルギー生産を引き下げる条件により、GIニューロンが活性化するか否かを検証するための実験を行った。具体的には、GIニューロンに対して、8.3Gのグルコースにより[Ca2+]iを安定化した後、電子伝達系の阻害剤である100μMのシアン化カリウム(KCN)、又は酸化的リン酸化の脱共役剤である1μMのカルボニルシアニドp−トリフルオロメトキシフェニルヒドラゾン(FCCP)の投与をした。
図8Aは、8.3Gにおいて、100μMのKCNを投与した際の[Ca2+]i応答を示すグラフである。
図8Bは、KCNを投与した際の[Ca2+]i応答の平均を示す(n=8)。「**」はP<0.01であることを示す。
図8Cは、8.3Gにおいてで、1μMのFCCPを投与した際の[Ca2+]i応答を示すグラフである。
図8Dは、FCCPを投与した際の[Ca2+]i応答の平均を示す(n=10)。「**」はP<0.01であることを示す。
このように、KCNとFCCPは、両方とも[Ca2+]iを増加させた。これは、グルコース濃度低下時の反応と同様の反応である。
これらの結果により、グルコース濃度低下に誘発された[Ca2+]i増加は、細胞内エネルギー産生率の低下と、これにより必然的に起こるNKAの抑制にて媒介されると考えられる。
【0075】
(GIニューロン活動性の下流のVDCCによるシグナル伝達機構)
次に、本発明者らは、NKAの抑制とGIニューロンの活性化をどのように結びつけて考えることができるかについて考察した。
具体的なNKAの抑制とGIニューロンの活性化における作用機構としては、[Na+]i増加に関連したNKAの電気的な活性の抑制により細胞膜が脱分極され、これにより、電位依存Ca2+チャンネルを介してCa2+流入を刺激する、と推測される。
このため、本発明者らは、L形及びN型のVDCCとGIニューロン活性について実験により確かめた。L形及びN型のVDCCは、グルコース感知ニューロン、NPYニューロン、及び低血糖下でグルカゴンを分泌する膵臓のα−細胞の調節に密接に関連している。以下で図9A〜図9Eを参照して、グルコース濃度低下に誘発された[Ca2+]i増加に対して、各VDCCに関連する試薬を投与した実験を示している。
図9Aは、Ca2+を取り除いた(Ca2+−free)溶液にした際の[Ca2+]i応答を示すグラフである(n=8)。[Ca2+]i増加は、Ca2+−free溶液では抑制されている。
図9Bは、各VDCCの非特異性のブロッカーである高濃度Ni2+(300μM)を投与した際の[Ca2+]i応答を示すグラフである(n=13)。Ni2+投与によっても、[Ca2+]i増加は抑制されている。
図9Cは、N型Ca2+チャンネル・ブロッカーであるω−コノトキシンGVIA(ω−conotoxin)500nMを投与した際の[Ca2+]iを応答示すグラフである(n=7)。グルコース濃度低下に誘発された[Ca2+]i増加は、ω−コノトキシンGVIAによっても、抑制されている。
図9Dは、L形Ca2+チャンネル・ブロッカーであるニフェジピン(nifedipine)10μMを投与した際の[Ca2+]i応答を示すグラフである(n=9)。グルコース濃度低下に誘発された[Ca2+]i増加は、ニフェジピンによっても抑制されている。
図9Eは、図9A〜Dの試験条件下のグルコース濃度低下に対する[Ca2+]i応答の振幅の平均を、コントロール実験と比べて相対的に表現したものである。「**」は、P<0.01であることを示す。すなわち、グルコース濃度低下に誘発された[Ca2+]i増加は、Ca2+−free溶液、Ni2+(300μM)投与、ω−コノトキシンGVIA(500nM)投与、及びニフェジピン(10μM)投与により有意に減少した。
これらのデータは、グルコース濃度低下に誘発された[Ca2+]i増加は、主としてN型及びL形VDCCによるCa2+流入を介して行われることを示している。
そして、グルコース濃度低下に誘発されたNKAの抑制が、細胞膜を脱分極化してVDCCを開くことを示唆している。
【0076】
なお、グルコース濃度低下に誘発された[Ca2+]i増加において、VDCCをブロックした後に残った反応の一部は、グルコース濃度低下に誘発された[Na+]i増加が引き金となって起きる、形質膜Na+/Ca2+エクスチェンジャー(NCX)の逆モードで引き起こされることも、可能性としては考えられる。
しかしながら、本発明の発明者らは、VDCCを抑制した条件下でも[Na+]i増加を観察した。このため、上述の可能性はないと考えられる。
【0077】
(NKA活性による摂食行動の生理学的調節)
本発明の発明者らは、更に、GIニューロンの活性化を調節するNKAの重大な役割を明らかにする生体外のデータに基づき、NKA活性が摂食行動の生理学的調節に関係するか否かについて、調査した。
以下、図10、図11を参照して、実際にラット個体を用いて、NKA活性の低下が絶食又はグルコース濃度低下の感知と摂食との刺激に関連しているかについて調べた実験について説明する。
図10は、視床下部弓状核のNKA活性を計測した結果を示す(n=7)。自由に食物が与えられたラット(Control)をオープンバーで示し、断食させたラット(Fasted)を塗りつぶしたバーで示す。「**」はP<0.01であることを示す。この実験結果においては、任意に食物が与えられたラットと比較して、24時間絶食させたラットにおいて視床下部弓状核のNKA活性は48.3%と著しく減少した。
図11は、GEニューロンが主要な働きをしている視床下部腹内側部(ventromedial hypothalamus、VMH)でのNKA活性を計測した結果を示す(n=7)。視床下部弓状核とは異なり、VMHにおいては、コントロール(Control)及び絶食下(Fasted)においてもNKA特性は不変だった。
したがって、絶食により、視床下部弓状核のNKA活性は選択的に著しく低減されるため、NKA活性を低下させることで摂食を促進することが可能であると示唆される。
【0078】
次に、ラットにNKAインヒビターのウワバイン(0.5nmol/5μl)とK+−free溶液(10μl)とを脳室内投与して、摂食量を比較した。
図12Aは、この摂食量の比較を示すグラフである。ウワバイン(0.5のnmol/5μl)(n=8)及び10μlのK+−free溶液(n=5)の脳室内投与により、コントロールHKRB(n=5)と比較して、脳室内投与の2時間後に、摂食量を著しく増加させた。HKRBは、GIニューロンを興奮させないために5mMグルコース(5G)を含んでいる。「*」は、P<0.05であることを示す。
【0079】
次に、グルコキナーゼ阻害剤であるMH、NKAアクティベータであるSSA412、コントロールのウサギIgGを脳室内投与して、摂食量を比較した。
図12Bは、この摂食量の比較を示すグラフである。「*」は、P<0.05、「**」はP<0.01であることを示す。
まず、脳室内にMHを投与せず、SSA412(n=7)又はウサギIgG(n=6)のみを注射した場合、摂食量は変化なかった。
ここで、脳室内にMHを投与すると、ニューロンのグルコース利用が低減され、従ってグルコース濃度低下および飢餓/絶食の条件を反映する状況に置かれるため、摂食量が有意に増加した。
これに対して、MHに誘発された摂食行動は、10μg/5μlのSSA412の脳室内前投与により打ち消された(n=8)。
しかしながら、コントロールの10μg/5μlのウサギIgGの脳室内前投与では打ち消されなかった(n=6)。
このように、NKAアクティベータであるSSA412により、実際に摂食量を変化させ、食欲を減らすことができる可能性がある。
【0080】
さらに、本発明の発明者らは、視床下部弓状核のNPY又はPOMCのmRNA発現量についての測定を行った。
図13Aは、ウワバインの脳室内投与後3時間における、視床下部弓状核のNPYのmRNA発現量を示すグラフである(n=6)。ウワバインの脳室内投与の後、コントロール(Vehicle)と比較して、視床下部弓状核のNPYのmRNA発現量は有意に増加した。「**」は、P<0.01であることを示す。
図13Bは、ウワバインの脳室内投与後3時間における、視床下部弓状核のPOMC(Pro−opiomelanocortin)のmRNA発現量を示すグラフである(n=6)。POMCは、脳下垂体前葉に含まれ、ACTH(Adrenocorticotropic hormone)やMSH(melanocyte−stimulating hormones)等に分解されるペプチドホルモン前駆体であり、摂食抑制として機能する。ウワバインの脳室内投与を行っても、POMCのmRNA発現量は変わらなかった。
【0081】
これらの結果は、飢餓/絶食条件下でグルコース濃度低下に誘発された視床下部弓状核のNKA活性の低下が、NPYを含むGIニューロンを活性化し、これにより摂食行動を刺激することを示している。
【0082】
この摂食行動の病態生理学の意味として、GIニューロンを含む視床下部弓状核NPYニューロンが、過食・内臓肥満症の2型糖尿病Goto−Kakizaki(GK)ラットでアップレギュレートされることが考えられる。
このモデルでは、グルコース利用の低下は、グルコース濃度上昇に誘発されるインスリン分泌促進の障害及びグルコース濃度上昇に誘発されるGIニューロン、すなわちNPYニューロンの活動抑制の障害と共通の原因でありえる。グルコース利用が低下していても、NKA活性化剤によりGIニューロンを活性化すれば、過食を是正し、その結果、内臓肥満と糖尿病を改善する効果が期待できる。
摂食行動異常をもつ肥満と糖尿病は、GKラットとヒト患者で共通の機序により生じると推測できるため、SSA412を含むNKA活性化物質を本実施例の食欲抑制剤として用いることにより、肥満及び2型糖尿病の治療にも用いることが考えられる。
【0083】
以上のように、本実施例において、本発明の発明者らは、以前からの推測はあるものの証明されていない仮説である、NKA活性の低下がグルコース低下によるGIニューロンの活性化に関連していることを証明した。
【0084】
なお、上記実施の形態の構成及び動作は例であって、本発明の趣旨を逸脱しない範囲で適宜変更して実行することができることは言うまでもない。
【産業上の利用可能性】
【0085】
本発明によれば、視床下部弓状核GIニューロンのNKAの作用を調整することで、第1次摂食・代謝調節中枢に直接働きかけて、「食欲」そのものを調節することができる。よって、従来とは作用機構の違う食欲促進剤、食欲抑制剤の提供が可能となる。このため、産業条利用可能である。
【特許請求の範囲】
【請求項1】
視床下部弓状核におけるグルコース抑制性ニューロン(Glucose−Inhibited neuron)のNKA(Na++K+−ATPase)活性を調整するNKA活動調節剤を含む食欲調整剤。
【請求項2】
前記NKA活動調節剤は、NKAインヒビターであり、
前記グルコース抑制性ニューロンの[Ca2+]i濃度を増加させる
ことを特徴とする請求項1に記載の食欲調整剤。
【請求項3】
前記NKAインヒビターは、ニューロペプチドYの発現を惹起する
ことを特徴とする請求項2に記載の食欲調整剤。
【請求項4】
前記NKAインヒビターは、強心配糖体であり、該強心配糖体は、ウワバイン(ouabain)又はジゴキシン(digoxin)を含む
ことを特徴とする請求項2又は3に記載の食欲調整剤。
【請求項5】
前記NKAインヒビターは、NKAのαサブユニットの活動を調整する
ことを特徴とする請求項2乃至4のいずれか1項に記載の食欲調整剤。
【請求項6】
前記NKAインヒビターは、強心配糖体としての投与の濃度の1/10以下の量を投与する
ことを特徴とする請求項2乃至5のいずれか1項に記載の食欲調整剤。
【請求項7】
請求項2乃至6のいずれか1項に記載の食欲調整剤を用いた
ことを特徴とする食欲不振、抗癌剤の副作用による食欲の低下、加齢、過労、低栄養、神経性食思不振症、うつ病を含む精神疾患、自律神経失調症、拒食症を含む摂食障害のいずれかに対する治療のための治療薬。
【請求項8】
前記NKA活動調節剤は、NKAアクティベータであり、
前記グルコース抑制性ニューロンにおける低グルコース誘発[Ca2+]i増加を抑制又は阻止する
ことを特徴とする請求項1に記載の食欲調整剤。
【請求項9】
前記NKAアクティベータは、ニューロペプチドYの発現を抑制する
ことを特徴とする請求項8に記載の食欲調整剤。
【請求項10】
前記NKAアクティベータは、NKAのαサブユニットに結合する
ことを特徴とする請求項8又は9に記載の食欲調整剤。
【請求項11】
前記NKAアクティベータは、抗体SSA412であり、
前記グルコース抑制性ニューロンにおける低グルコース誘発[Ca2+]i増加を抑制又は阻止する
ことを特徴とする請求項8乃至10のいずれか1項に記載の食欲調整剤。
【請求項12】
請求項8乃至11のいずれか1項に記載の食欲調整剤を用いた
ことを特徴とする、過食、過食による肥満、過食による糖尿病、メタボリックシンドロームのいずれかに対する治療のための治療薬。
【請求項13】
請求項1に記載の食欲調整剤を用いた
ことを特徴とする健康食品。
【請求項14】
請求項1に記載の食欲調整剤を用いた
ことを特徴とする動物用飼料。
【請求項15】
視床下部弓状核のグルコース抑制性ニューロン(Glucose−Inhibited neuron)のNKA(Na++K+−ATPase)活性を調整し、
前記グルコース抑制性ニューロンの出力を調整する
ことを特徴とする食欲調整方法。
【請求項1】
視床下部弓状核におけるグルコース抑制性ニューロン(Glucose−Inhibited neuron)のNKA(Na++K+−ATPase)活性を調整するNKA活動調節剤を含む食欲調整剤。
【請求項2】
前記NKA活動調節剤は、NKAインヒビターであり、
前記グルコース抑制性ニューロンの[Ca2+]i濃度を増加させる
ことを特徴とする請求項1に記載の食欲調整剤。
【請求項3】
前記NKAインヒビターは、ニューロペプチドYの発現を惹起する
ことを特徴とする請求項2に記載の食欲調整剤。
【請求項4】
前記NKAインヒビターは、強心配糖体であり、該強心配糖体は、ウワバイン(ouabain)又はジゴキシン(digoxin)を含む
ことを特徴とする請求項2又は3に記載の食欲調整剤。
【請求項5】
前記NKAインヒビターは、NKAのαサブユニットの活動を調整する
ことを特徴とする請求項2乃至4のいずれか1項に記載の食欲調整剤。
【請求項6】
前記NKAインヒビターは、強心配糖体としての投与の濃度の1/10以下の量を投与する
ことを特徴とする請求項2乃至5のいずれか1項に記載の食欲調整剤。
【請求項7】
請求項2乃至6のいずれか1項に記載の食欲調整剤を用いた
ことを特徴とする食欲不振、抗癌剤の副作用による食欲の低下、加齢、過労、低栄養、神経性食思不振症、うつ病を含む精神疾患、自律神経失調症、拒食症を含む摂食障害のいずれかに対する治療のための治療薬。
【請求項8】
前記NKA活動調節剤は、NKAアクティベータであり、
前記グルコース抑制性ニューロンにおける低グルコース誘発[Ca2+]i増加を抑制又は阻止する
ことを特徴とする請求項1に記載の食欲調整剤。
【請求項9】
前記NKAアクティベータは、ニューロペプチドYの発現を抑制する
ことを特徴とする請求項8に記載の食欲調整剤。
【請求項10】
前記NKAアクティベータは、NKAのαサブユニットに結合する
ことを特徴とする請求項8又は9に記載の食欲調整剤。
【請求項11】
前記NKAアクティベータは、抗体SSA412であり、
前記グルコース抑制性ニューロンにおける低グルコース誘発[Ca2+]i増加を抑制又は阻止する
ことを特徴とする請求項8乃至10のいずれか1項に記載の食欲調整剤。
【請求項12】
請求項8乃至11のいずれか1項に記載の食欲調整剤を用いた
ことを特徴とする、過食、過食による肥満、過食による糖尿病、メタボリックシンドロームのいずれかに対する治療のための治療薬。
【請求項13】
請求項1に記載の食欲調整剤を用いた
ことを特徴とする健康食品。
【請求項14】
請求項1に記載の食欲調整剤を用いた
ことを特徴とする動物用飼料。
【請求項15】
視床下部弓状核のグルコース抑制性ニューロン(Glucose−Inhibited neuron)のNKA(Na++K+−ATPase)活性を調整し、
前記グルコース抑制性ニューロンの出力を調整する
ことを特徴とする食欲調整方法。
【図1】


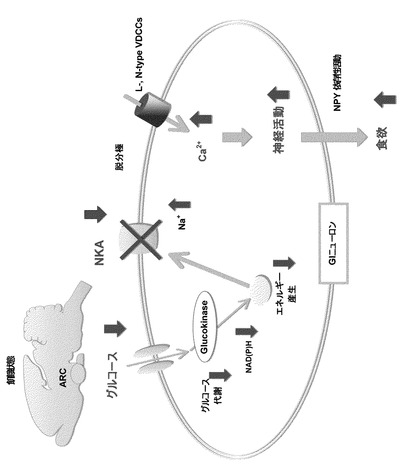
【図2】
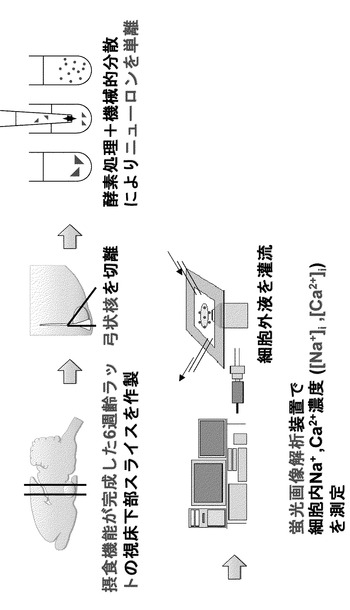
【図3A】
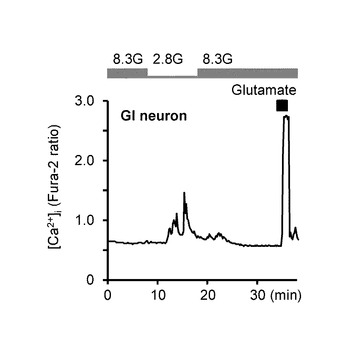
【図3B】
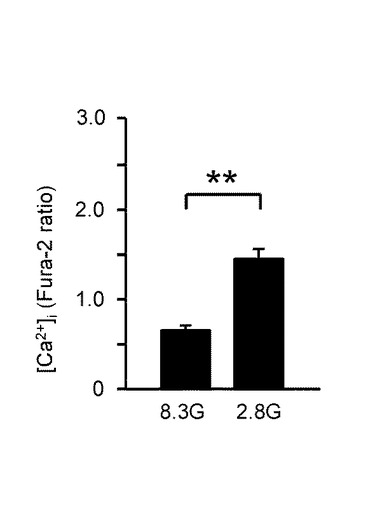
【図3C】

【図3D】
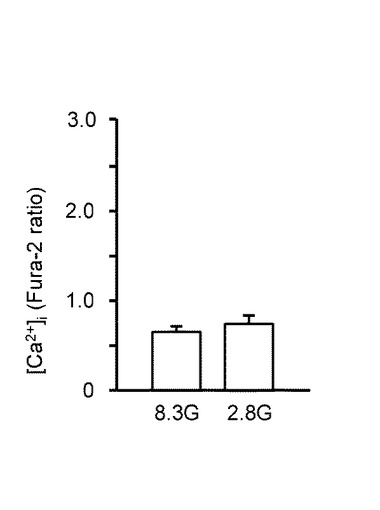
【図4A】
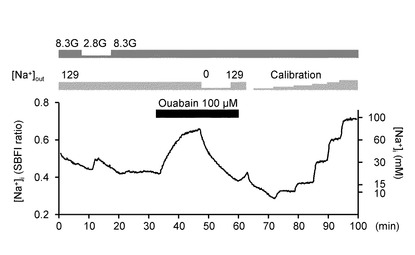
【図4B】
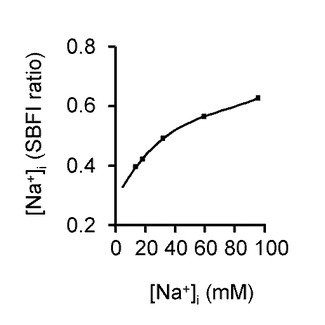
【図4C】
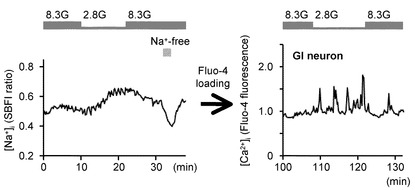
【図4D】
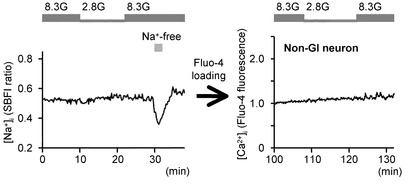
【図4E】

【図5A】
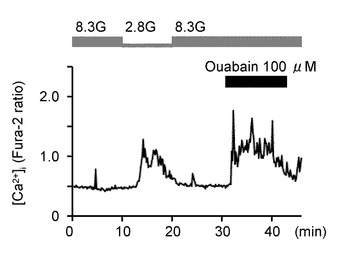
【図5B】
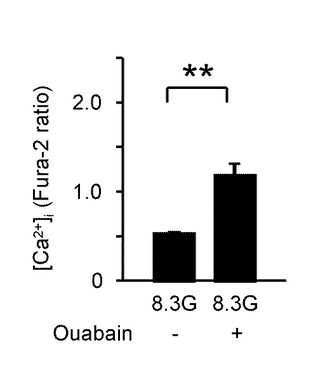
【図5C】
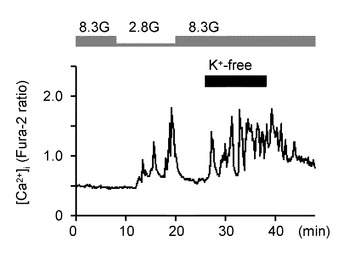
【図5D】
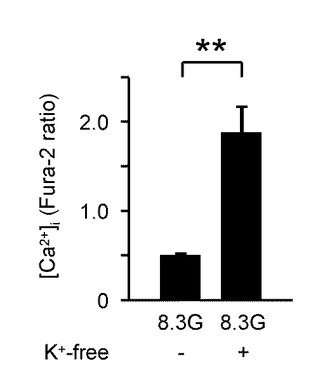
【図5E】
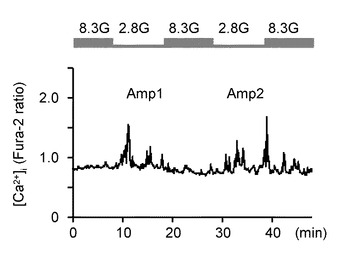
【図5F】

【図5G】

【図5H】
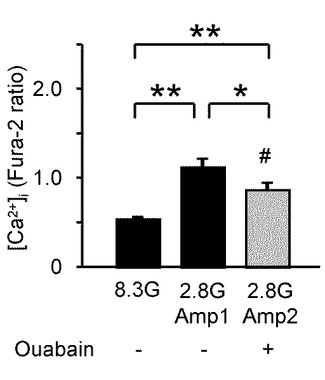
【図5I】
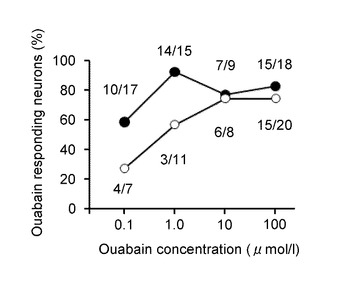
【図5J】
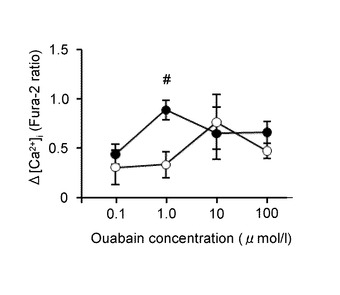
【図5K】

【図5L】

【図5M】
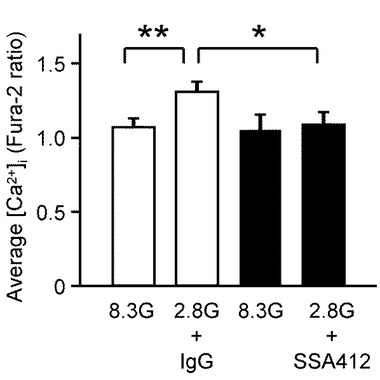
【図6A】
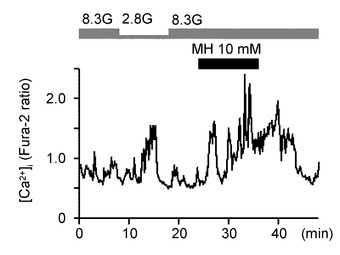
【図6B】
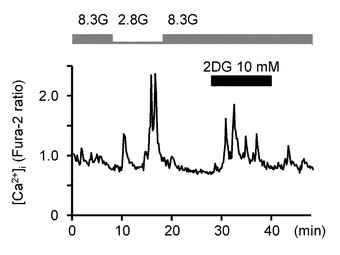
【図6C】
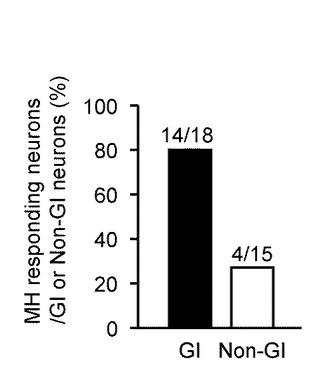
【図6D】
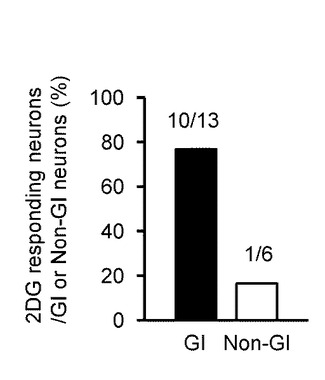
【図6E】
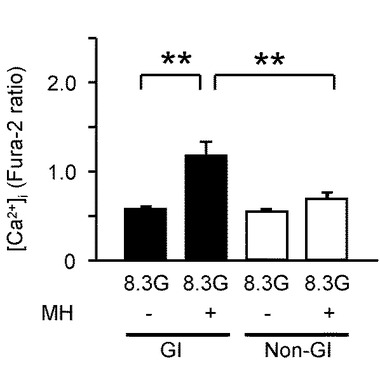
【図6F】
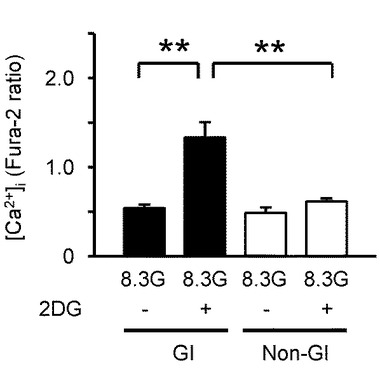
【図7A】
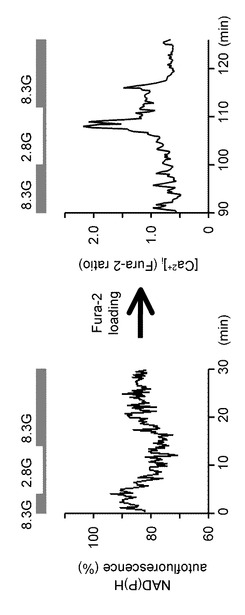
【図7B】
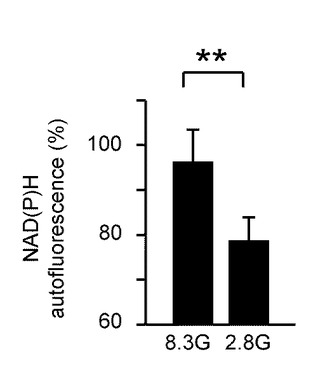
【図8A】
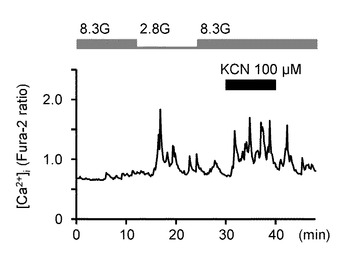
【図8B】
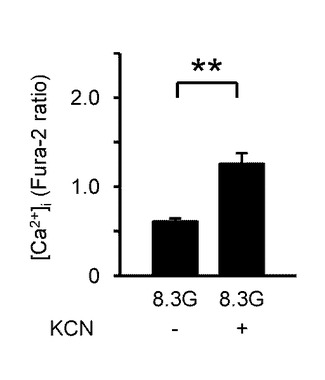
【図8C】

【図8D】
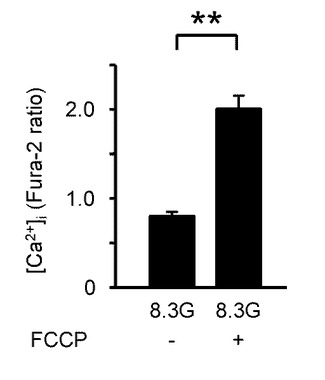
【図9A】
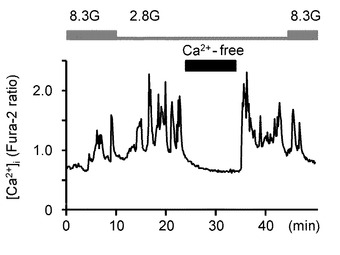
【図9B】
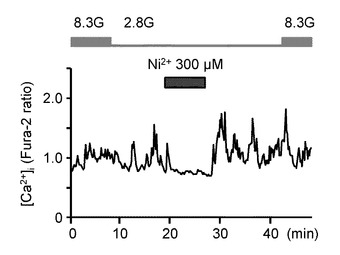
【図9C】
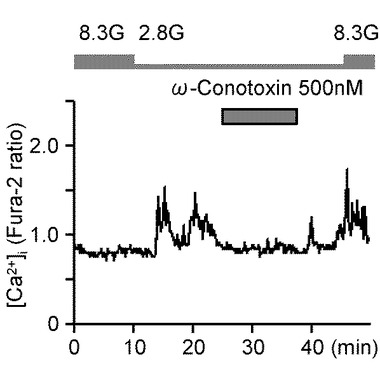
【図9D】

【図9E】
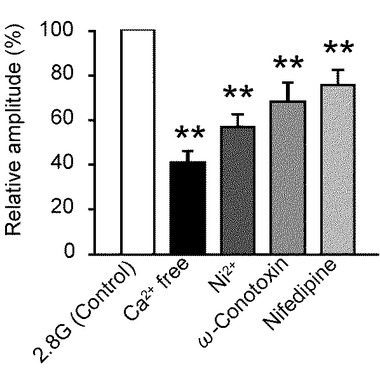
【図10】
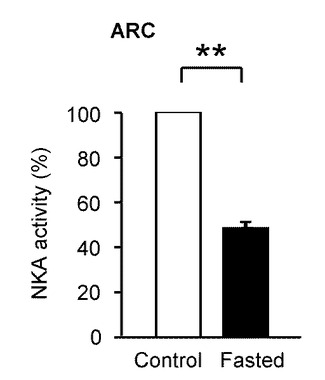
【図11】
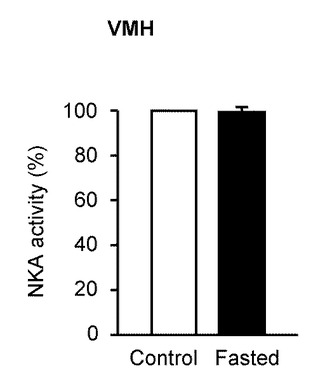
【図12A】
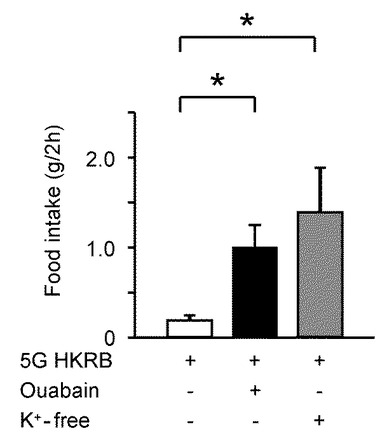
【図12B】
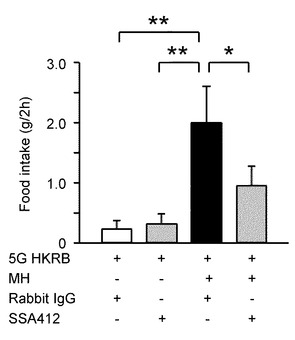
【図13A】
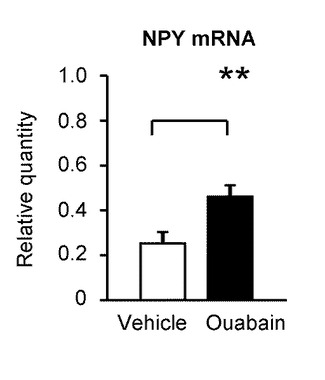
【図13B】
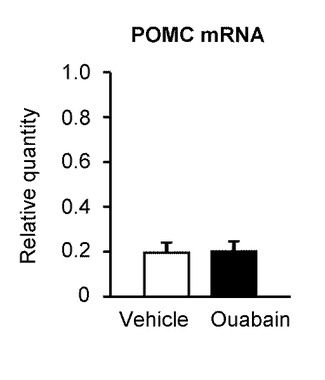
【図14】
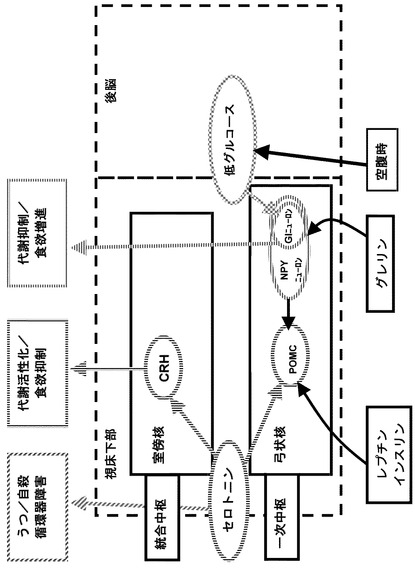
【図2】
【図3A】
【図3B】
【図3C】
【図3D】
【図4A】
【図4B】
【図4C】
【図4D】
【図4E】
【図5A】
【図5B】
【図5C】
【図5D】
【図5E】
【図5F】
【図5G】
【図5H】
【図5I】
【図5J】
【図5K】
【図5L】
【図5M】
【図6A】
【図6B】
【図6C】
【図6D】
【図6E】
【図6F】
【図7A】
【図7B】
【図8A】
【図8B】
【図8C】
【図8D】
【図9A】
【図9B】
【図9C】
【図9D】
【図9E】
【図10】
【図11】
【図12A】
【図12B】
【図13A】
【図13B】
【図14】
【公開番号】特開2013−40127(P2013−40127A)
【公開日】平成25年2月28日(2013.2.28)
【国際特許分類】
【出願番号】特願2011−177985(P2011−177985)
【出願日】平成23年8月16日(2011.8.16)
【出願人】(505246789)学校法人自治医科大学 (49)
【Fターム(参考)】
【公開日】平成25年2月28日(2013.2.28)
【国際特許分類】
【出願日】平成23年8月16日(2011.8.16)
【出願人】(505246789)学校法人自治医科大学 (49)
【Fターム(参考)】
[ Back to top ]