
飲食品中のジアセチル前駆体の定量法
【課題】飲食品中のジアセチル前駆体(アセトヒドロキシ酸)を直接定量できる、簡易で正確な方法を提供すること。
【解決手段】飲食品中のジアセチル前駆体を定量する方法であって、飲食品の試料に、マグネシウムイオンおよび還元型ニコチンアミドアデニンジヌクレオチドリン酸(NADPH)の存在下でアセトヒドロキシ酸レダクトイソメラーゼを作用させ、酸化型ニコチンアミドアデニンジヌクレオチドリン酸(NADP)の増加またはNADPHの減少を指標としてジアセチル前駆体を測定することを含む、方法。
【解決手段】飲食品中のジアセチル前駆体を定量する方法であって、飲食品の試料に、マグネシウムイオンおよび還元型ニコチンアミドアデニンジヌクレオチドリン酸(NADPH)の存在下でアセトヒドロキシ酸レダクトイソメラーゼを作用させ、酸化型ニコチンアミドアデニンジヌクレオチドリン酸(NADP)の増加またはNADPHの減少を指標としてジアセチル前駆体を測定することを含む、方法。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、飲食品中のジアセチル前駆体の定量法に関する。
【背景技術】
【0002】
多くの飲食品において、ジアセチル臭はそれらの特性を大きく左右する重要な香味(臭い)成分である。例えばヨーグルトなどの発酵乳製品では、ジアセチル臭は良い臭いの代表的成分の1つとして重要視されるが、酒類ではあってはならない臭いとして嫌われている。発酵飲食品中のジアセチル臭は、ジアセチルとその同族体、2,3−ペンタンジオン(両者を併せてビシナルジケトンとも称する)の一定濃度(弁別閾値)以上の存在により検知される。ビシナルジケトンは、発酵中の微生物により生成される、その前駆体であるアセトヒドロキシ酸(ジアセチルの前駆体であるアセト乳酸、ならびに、2,3−ペンタジオンの前駆体であるアセトヒドロキシ酪酸の総称)から生成されるが、天然物中のアセトヒドロキシ酸は不安定であり、自動的に分解してビシナルケトンを含むアセトイン(ならびに、その同族体であるアセチルエチルカルビノール)を生成する。したがって、飲食品中におけるジアセチル臭の発生は、多くの場合、製品化後に顕在化する。そこで、飲食品のジアセチル臭の調節には、アセトヒドロキシ酸濃度を測定して調節することが必要となる。飲食品、特に発酵飲食品を試料としてアセトヒドロキシ酸を定量分析する場合には、その特徴を踏まえた条件設定(場合によっては改変)の必要がある。飲食品試料の場合の、第一の特徴として、多種類の共存物がアセトヒドロキシ酸よりもはるかに多量に混在している点があげられる。したがって、アセトヒドロキシ酸の定量分析に先立って、アセトヒドロキシ酸を安定に保ち得る条件下での、共存物内の妨害物質の除去が必要である。
【0003】
発酵飲食品の場合に於いては、アセトヒドロキシ酸と同様に、関係微生物により生成される有機酸群の濃度が高く、アセトヒドロキシ酸が分解しやすい酸性であるものが多いという特徴が挙げられる。即ち、発酵飲食品製造の場合にはそのほとんどが静置発酵で行われる。そのために、当初発酵系の中に存在した酵素は急速に微生物によって消費され、系内は急速に嫌気的となり有機酸が生成するのである。
【0004】
アセトヒドロキシ酸は中性条件下では比較的安定な物質であるが、発酵食品のほとんどに於いてそうである酸性条件下では特に不安定で、ジアセチル類やアセトイン類に変化しやすい。また、その反応は温度が高いほど迅速に進行する。したがって、試料採取後直ちに分析を行うことのできない場合には、直ちに微生物等の共存物をできるだけ除いて、中和し、冷却(凍結)保存する必要がある。したがって、試料の添加により反応時の最適pHが変わらぬよう、供試試料のpH調整、緩衝液の濃度増強等の確認・処理が必要である。
【0005】
発酵飲食品中の有機酸中で特に出現頻度が高く、また濃度が高いという点で注目すべきものは乳酸である。乳酸はある濃度以上で、アセトヒドロキシ酸の定量分析を妨害するので、分析に際してはその低濃度化が必要である。
【0006】
日本酒(清酒)製造、あるいは、ある種の上面酵母発酵によるビール醸造に於いては、野生酵母による汚染を防ぐために、種菌あるいはもろみに、乳酸(あるいは乳酸菌)を添加して乳酸酸性条件下(数mM濃度)で発酵させる方法が採られている。また、酒類に対する微生物汚染は、乳酸菌の増殖による場合が多く、その場合、アセトヒドロキシ酸の高濃度生成とともに、乳酸濃度が1mMを超える場合も珍しくない。また、発酵乳製品製造の場合には、香味向上のために、チーズや発酵バター製造の初期に乳酸発酵をさせるのが常法となっている。このように、発酵飲食品製造に於いては、高濃度の乳酸の生成が常に起こると考えていなければならない。
【0007】
発酵飲食品の製造に用いられる微生物は比較的限られているが、その中でもSaccharomyces属を主体とする酵母(酒類の製造など)と乳酸菌群(発酵乳製品や漬物類など)が圧倒的に広く用いられている。
【0008】
Saccharomyces属酵母は、アセト乳酸の分解によって生成するアセトイン濃度の、場合によっては、数十倍濃度のアセトインを、別の代謝系であるアセトアルデヒドの縮合により(アルコール発酵の際に)生成する。また、乳酸菌群の中には、ジアセチルを生合成するものもあるとの説もある。また、乳酸菌は、上述のごとく、酵母によるアルコール発酵の際の、汚染微生物の代表的な菌であり、アルコール飲料の品質を大きく阻害する。ジアセチル臭は乳酸菌に汚染されたアルコール飲料のメルクマールとされる臭いである。
【0009】
発酵中に生成するアセトヒドロキシ酸の一部は、ジアセチル類、アセトイン類へ発酵中にも分解して生成する。発酵飲食品中のアセトヒドロキシ酸の定量において、これらの物質との分別定量はもっとも困難であり、手間を要し、また、正確性に欠け、現在に於いてもそれらを回避できる正確なアセトヒドロキシ酸の定量法の開発研究が世界各所で行われているのが現状である(非特許文献1、非特許文献4)。
【0010】
食品分析の分野におけるアセトヒドロキシ酸の定量分析は、一般的には、アセトイン類やジアセチル類(ビシナルジケトン)に転換後、定量分析されている。前述のようにアセトヒドロキシ酸は、食品の通常の発酵条件下であっても自動的に分解するほどに不安定な物質だからである。いまだに天然物中のアセトヒドロキシ酸は純品として抽出、精製されたことはなく、またアセトヒドロキシ酸の存在自体、合成品のアセトヒドロキシ酸の各種条件下でのジアセチル類やアセトイン類に変化する挙動と、天然物中におけるアセトヒドロキシ酸がそれらに変化する挙動との一致性から、その存在が推定されているにすぎない。
【0011】
飲食品のアセトヒドロキシ酸の定量分析の具体例を挙げると、例えばビールについて行われており、試料ビールに空気を含ませてから60℃で90分間放置し、ジアセチルに転換してから、そのジアセチルを蒸留比色法、あるいは、ガスクロマトグラフィーで定量分析するという公定法が定められている(非特許文献2)。
【0012】
しかし、この方法では、既存のジアセチルもあわせて定量分析されるという問題があり、既存のジアセチル分を差し引くための更なる分析操作が必要で、煩雑な定量法となっている。
【0013】
また、ビールにおけるアセトヒドロキシ酸のジアセチルへの転換率は100%ではなく、幾分かのアセトインも同時に生ずるとの議論もある(非特許文献3)。
【0014】
さらに、上記ビールの公定法を清酒醪中のアセトヒドロキシ酸の定量分析に適用すると、60%から80%程度に相当する値しか示されず(非特許文献4)、この方法は清酒醪などの飲食品全般にそのまま適用できるものではない。
【0015】
このように、従来のアセトヒドロキシ酸の定量分析は煩雑である上に、アセトインやジアセチルに転換される割合が、分析条件や分析対象である食品によって異なるため、現在でもジアセチル分析を対象とした公定法を持たない業界は多く、ましてや、アセトヒドロキシ酸分析のための公定法を持つ業界は、ビール業界をのぞいて皆無といってよい。
【0016】
従って、食品分析の分野では、アセトヒドロキシ酸を直接定量する方法が求められており、実際、アセトヒドロキシ酸の分解物であるジアセチル、アセトインまたはそれらの同族体からアセトヒドロキシ酸を分別定量する研究が世界中で多く行われている。
【0017】
しかし、飲食品には共存物が多く、特に発酵飲食品では、微生物によって生成される様々なタンパク質(酵素)などによる測定値への影響が考えられ、また、飲食品中の天然物であるアセトヒドロキシ酸はすぐに分解してしまうことから、直接定量法は開発されていなかった。
【0018】
食品中以外でのアセトヒドロキシ酸の測定例としては、合成品であるアセトヒドロキシ酸を基質とし、NADPHとマグネシウムイオンの存在下でケトール酸レダクトイソメラーゼを作用させて、NADPHの340nmでの吸光度の低下を測定する方法が知られている(非特許文献1、以下Arfinの方法という)。
【0019】
Arfinの方法を応用した特許出願として、例えば、植物の真菌病を処置するためのケトール酸レダクトイソメラーゼ阻害剤を特定するための方法が挙げられる(特許文献1)。特許文献1は、ケトール酸レダクトイソメラーゼの酵素活性を阻害することによって真菌の成長および/または病理発生を阻害する化合物を特定するために、宿主生物で発現させ精製したケトール酸レダクトイソメラーゼを用いてArfinの方法を適用することを開示するが、Arfinの方法に用いる基質は合成品であり、種々の物質が混在する天然物中のアセトヒドロキシ酸を定量分析するものではなく、また、特許文献1は、飲食品中の天然物であるアセトヒドロキシ酸について何ら開示していない。
【0020】
【特許文献1】特表2005−501912号明細書
【非特許文献1】S.M.Arfin and H.E.Umberger,J.Biol.Chem.,244,1118−1127,1969
【非特許文献2】改訂BCOJビール分析法、8.16.2、p.2/4、(2)、ビール酒造組合国際技術委員会編、財団法人日本醸造協会、平成10年
【非特許文献3】The American Society of Brewing Chemists,Methods of Analysis of the American Society of Brewing Chemists,8th revised edition,1992,Beer 25,p.1 of 5
【非特許文献4】Kobayashi,K.,Kusaka,K.,Takahashi,T. and Sato,K.,J.Biosci.Biotechnol.99, 502−507,2005
【発明の開示】
【発明が解決しようとする課題】
【0021】
本発明の目的は、飲食品中のアセトヒドロキシ酸を直接定量できる、簡易で正確な方法を提供することにある。
【課題を解決するための手段】
【0022】
本発明者らは、アセトヒドロキシ酸(アセト乳酸とアセトヒドロキシ酪酸の総称)をアセトイン(同族体であるアセチルエチルカルビノールも含む)やジアセチル(同族体である2,3−ペンタンジオンも含む)に変換するために要する時間と手間を省き、分析の効率化を図るため、アセトヒドロキシ酸をそのままの形で定量分析しようと考え、ピルビン酸を初発基質とし、アセトヒドロキシ酸合成を経て、ロイシン、バリン、イソロイシンを生合成する経路の中で、アセトヒドロキシ酸を基質とするアセトヒドロキシ酸レダクトイソメラーゼを用いること、具体的にはNADPHの存在下で当該酵素を作用させて、NADPの増加またはNADPHの減少を測定する方法を見出した。また、試料中のアセトヒドロキシ酸の測定を妨害する物質とアセトヒドロキシ酸を分離する方法を見出し、本発明を完成した。
【0023】
本発明の特徴は、要約すると以下の通りである。
(1)飲食品中のジアセチル前駆体を定量する方法であって、飲食品の試料に、マグネシウムイオンおよび還元型ニコチンアミドアデニンジヌクレオチドリン酸(NADPH)の存在下でアセトヒドロキシ酸レダクトイソメラーゼを作用させ、酸化型ニコチンアミドアデニンジヌクレオチドリン酸(NADP)の増加またはNADPHの減少を指標としてジアセチル前駆体を測定することを含む、前記方法。
(2)飲食品が発酵物である、(1)に記載の方法。
(3)飲食品が発酵酒または発酵乳製品である、(1)または(2)に記載の方法。
(4)飲食品が、清酒醪、ビール類製造用発酵液、果汁発酵液、サワークリーム、カテージチーズ、ヨーグルト、発酵バター、スターターディスティレート、焼酎用モロミ、漬物類、醤油、味噌、味醂または酢である、(1)〜(3)のいずれかに記載の方法。
【0024】
(5)アセトヒドロキシ酸レダクトイソメラーゼが、真核微生物由来の酵素である、(1)〜(4)のいずれかに記載の方法。
(6)アセトヒドロキシ酸レダクトイソメラーゼが、アスペルギルス属由来の酵素である、(1)〜(5)のいずれかに記載の方法。
(7)アセトヒドロキシ酸レダクトイソメラーゼが、アスペルギルス・オリゼ由来の酵素である、(1)〜(6)のいずれかに記載の方法。
(8)アスペルギルス・オリゼが、アスペルギルス・オリゼRIB40株(FERM P−18273)である、(7)に記載の方法。
【0025】
(9)アセトヒドロキシ酸レダクトイソメラーゼが、配列番号2に示すアミノ酸配列を含むタンパク質、または該アミノ酸配列において1もしくは数個のアミノ酸が欠失、置換もしくは付加されたアミノ酸配列を含み、かつアセトヒドロキシ酸レダクトイソメラーゼ活性を有するタンパク質である、(1)〜(8)のいずれかに記載の方法。
(10)NADPの増加またはNADPHの減少を、340nmの吸光度に基づいて測定する、(1)〜(9)のいずれかに記載の方法。
(11)飲食品の試料から乳酸を除去することをさらに含む、(1)〜(10)のいずれかに記載の方法。
(12)乳酸の除去を、弱塩基性陰イオン交換樹脂を用いて行う、(11)に記載の方法。
【0026】
(13)アセトヒドロキシ酸レダクトイソメラーゼを含む、飲食品中のジアセチル前駆体を定量するためのキット。
(14)定量が(1)〜(12)のいずれかに記載の方法によって行われるものである、(13)に記載のキット。
(15)アセトヒドロキシ酸レダクトイソメラーゼが、配列番号2に示すアミノ酸配列を含むタンパク質、または該アミノ酸配列において1もしくは数個のアミノ酸が欠失、置換もしくは付加されたアミノ酸配列を含み、かつアセトヒドロキシ酸レダクトイソメラーゼ活性を有するタンパク質である、(13)または(14)に記載のキット。
(16)飲食品の試料から乳酸を除去するための弱塩基性陰イオン交換樹脂カラムをさらに含む、(13)〜(15)のいずれかに記載のキット。
【0027】
本明細書において「飲食品」とは、その半製品、中間生成物、原料も含むことを意味する。
【0028】
本明細書において「発酵物」とは、固形状、ゲル状、液体状などのあらゆる形態の発酵飲食品を含み、発酵工程を経た半製品、中間生成物、原料も含み、また製品化のために加熱殺菌をすることを含む方法で生産される場合、加熱殺菌前の発酵物も含むことを意味する。
【0029】
本明細書において「1もしくは数個のアミノ酸が欠失、置換もしくは付加されたアミノ酸配列」とは、具体的には、配列番号2に示すアミノ酸配列の1〜10個、好ましくは1〜5個のアミノ酸、さらに好ましくは1〜3個のアミノ酸が変異により欠失、または他のアミノ酸に置換、あるいは付加された配列をいう。変異は、人為的に導入された変異または天然に存在する変異でもよい。
【0030】
本明細書において「アスペルギルス・オリゼRIB40株」は、独立行政法人産業技術総合研究所、独立行政法人製品評価技術基盤機構および独立行政法人酒類総合研究所により、独立行政法人産業技術総合研究所特許生物寄託センター(茨城県つくば市東1丁目1番地1)に寄託された、寄託番号FERM P−18273が付与された株をいう。
【0031】
本明細書において「受領番号NITE AP−541を付与された形質転換株」は、千葉県により、独立行政法人製品評価技術基盤機構特許微生物寄託センター(千葉県木更津市かずさ鎌足2−5−8)に寄託された株である。この株は、千葉県産業支援技術研究所(千葉県千葉市若葉区加曽利町889番地)からも分譲可能である。
【発明の効果】
【0032】
本発明によれば、飲食品中の易分解性のジアセチル前駆体を直接定量することができる。従来法のようにジアセチル前駆体をアセトインまたはジアセチルに変換する必要がなく、酸処理、熱処理等の手間と時間が大幅に削減できる。また、その変換率が問題になることもなく、試料中に既存のアセトインまたはジアセチルを測りこんでその分を後で差し引くなどの操作がいらず、簡易であり、ジアセチル前駆体を正確に定量できる。さらに、本発明によれば、試料からジアセチル前駆体の測定を妨害する物質(例えば乳酸など)を除去してジアセチル前駆体を定量することができる。
【発明を実施するための最良の形態】
【0033】
本発明の飲食品中のジアセチル前駆体を定量する方法は、飲食品の試料に、マグネシウムイオンおよびNADPHの存在下でアセトヒドロキシ酸レダクトイソメラーゼを作用させ、NADPの増加またはNADPHの減少を指標としてジアセチル前駆体を測定することを含むことを特徴とする。
【0034】
マグネシウムイオン、NADPHの存在下でアセトヒドロキシ酸レダクトイソメラーゼを作用させ、NADPの増加またはNADPHの減少を340nmの吸光度に基づいて測定してジアセチル前駆体(アセトヒドロキシ酸)を定量する方法は、以前から酵素学の分野で行われている方法であるが、易分解性であるために測定が難しいとされていた、飲食品中のジアセチル前駆体に適用し、測定することができることを証明したのは本発明者らが初めてである。
【0035】
飲食品は、ジアセチル、アセトインまたはそれらの同族体の存在を調べたい飲食品であれば、酒類、乳製品など、どんなものでもよく、例えば発酵物が挙げられるが、これに限定されない。具体的には、発酵酒、発酵乳製品、ビール類製造用発酵液、果汁発酵液、サワークリーム、カテージチーズ、ヨーグルト、発酵バター、焼酎用モロミ、漬物類、醤油、味噌、味醂、酢などが挙げられ、本発明の定量方法は種々の飲食品に適用できる。
【0036】
なお、飲食品の試料によって、試料を添加することによりpHが変化しないように測定に用いる緩衝液の緩衝能を高めたり、発酵飲食品試料中の菌体を除去したり、除タンパク質処理等を行うことは、必要に応じて行う。
【0037】
本発明の定量対象であるジアセチル前駆体(アセトヒドロキシ酸)は、生体内で分岐鎖アミノ酸(ロイシン、バリン、イソロイシン)合成経路上の中間体であり、その経路上でアセトヒドロキシ酸レダクトイソメラーゼが次の物質への変換を触媒する。
【0038】
アセトヒドロキシ酸レダクトイソメラーゼは、酵素番号ECクラス1.1.1.86に分類されるケトールアシッドレダクトイソメラーゼであり、植物ならびに細菌、酵母菌、糸状菌などの微生物においてよく特性付けされている酵素である。この酵素は、分岐鎖アミノ酸についての生合成経路の第二酵素であり、基質であるアセトヒドロキシ酸であるアセト乳酸とアセトヒドロキシ酪酸から、それぞれ、2,3−ジヒドロキシ−3−イソ吉草酸または2,3−ジヒドロキシ−3−メチル吉草酸への変換を触媒する。この反応は、マグネシウムイオン(Mg2+)の存在を必要とし、また、メチルまたはエチル基の転移、その後のNADPHによる還元という2段階で進行する。
【0039】
本発明に用いるアセトヒドロキシ酸レダクトイソメラーゼは、アセトヒドロキシ酸変換活性を有し、マグネシウムイオン存在下で、NADPHの酸化と共役するメチルまたはエチル基の転移を触媒する酵素系を提供できる生物に由来したあらゆるものを用いることができ、特に限定されない。例えば酵母、糸状菌などの真核微生物由来のものが好適に用いられる。
【0040】
本発明においては、好ましくはアスペルギルス属に属する微生物(例えばアスペルギルス・オリゼ、アスペルギルス・ニガー、アスペルギルス・ニデュランス、アスペルギルス・ジャポニカスなど)、より好ましくはアスペルギルス・オリゼ、さらに好ましくはアスペルギルス・オリゼRIB40株由来のアセトヒドロキシ酸レダクトイソメラーゼを用いることが好ましい。特に、配列番号2に示すアミノ酸配列を含むアセトヒドロキシ酸レダクトイソメラーゼ、または該アミノ酸配列において1もしくは数個のアミノ酸が欠失、置換もしくは付加されたアミノ酸配列を含み、かつアセトヒドロキシ酸レダクトイソメラーゼ活性を有するものが好ましい。配列番号2に示すアミノ酸配列をコードする核酸配列を配列番号1に示す。
【0041】
アセトヒドロキシ酸レダクトイソメラーゼは、自然環境から単離され、精製または不完全精製されたものでもよいし、上記微生物が自然生産する酵素を精製してもよく、また化学合成によっても製造できる。アセトヒドロキシ酸レダクトイソメラーゼの製造法は、当業者によく知られている。
【0042】
また、アセトヒドロキシ酸レダクトイソメラーゼは、例えば以下のように、遺伝子工学的手法により作製した形質転換株で過剰発現させ、製造することができる。
【0043】
アセトヒドロキシ酸レダクトイソメラーゼのアミノ酸配列または塩基配列から、プライマーを設計、合成する。アミノ酸配列、塩基配列はシークエンサー等で同定してもよいが、種々の文献に開示された配列情報を利用してもよい。
【0044】
例えば微生物から精製した染色体DNAを鋳型として、合成したプライマーを用いてポリメラーゼ連鎖反応(PCR)によりアセトヒドロキシ酸レダクトイソメラーゼをコードする遺伝子を増幅する。増幅した遺伝子のDNA断片を精製して適当な制限酵素で消化し、例えば電気泳動して対象の遺伝子のバンドを切り出してDNA断片を抽出する。
【0045】
一方、遺伝子組換えに用いるベクターも同様に制限酵素で切断し、上記DNA断片を挿入する。ベクターには、プロモーター配列のほか、ターミネーターや選択マーカー、リボソーム結合部位、複製開始点などを配置してもよい。
【0046】
好適なプロモーターとしては、例えばβ−ラクタマーゼプロモーター、ラクトースプロモーター、トリプトファンプロモーター、解糖系酵素(例えばα−アミラーゼ、プロテアーゼ、デヒドロゲナーゼなど)をコードする遺伝子類のプロモーター、具体的にはエノラーゼ、グリセルアルデヒド−3−ホスフェートデヒドロゲナーゼ、ヘキソキナーゼ、ピルベートデカルボキシラーゼ、ピルベートキナーゼなどに対するプロモーター、窒素代謝に関連する分解酵素などに対するプロモーター、SV40ウイルスのプロモーター、植物のプロモーター(例えばカリフラワーモザイクウイルス(CaMV)35Sプロモーター、ノパリン合成酵素遺伝子のプロモーター(Pnos)、トウモロコシ由来ユビキチンプロモーターなど)が挙げられる。
【0047】
形質転換可能な大腸菌を上記ベクターで形質転換し、培養後、プラスミドDNAを抽出する。プラスミドDNAを制限酵素で消化し精製して、例えば塩化カルシウム法、ポリエチレングリコール法、電気パルス法などで宿主細胞に形質転換し、形質転換株を得る。宿主は、細菌、酵母、真菌、昆虫細胞、植物細胞、動物細胞などを含む。
【0048】
以上のようにして作製された、アセトヒドロキシ酸レダクトイソメラーゼを高生産する形質転換株として、独立行政法人製品評価技術基盤機構特許微生物寄託センターに寄託された受領番号NITE AP−541の株が挙げられる。この株は、後述の実施例3で作製されたものである。
【0049】
形質転換株を培養し、宿主細胞または培地から、産生されたアセトヒドロキシ酸レダクトイソメラーゼを精製する。
【0050】
精製されたアセトヒドロキシ酸レダクトイソメラーゼは不安定であるため、例えば、グリセロール、硫酸アンモニウム、マグネシウムイオン、NADPHを適宜添加し、−20℃以下で保存することで、実用的に充分な保存安定性を与えることができる。
【0051】
本発明の飲食品中のジアセチル前駆体の定量法は、例えば以下の条件で行うことができる。
【0052】
本定量法では、反応系中の基質(ジアセチル前駆体)濃度が、例えば0mM〜0.1mM、好ましくは0mM〜0.01mMになるようにする。緩衝液は、例えば10mM〜200mMのTris−塩酸緩衝液、リン酸緩衝液、HEPES-水酸化ナトリウム緩衝液、TRICINE-水酸化ナトリウム緩衝液、ホウ酸緩衝液などが好ましく、pHは7.5〜10.5が好ましい。
【0053】
マグネシウムイオンとして、塩化マグネシウム、硫酸マグネシウムなどを、例えば2mM〜20mMになるよう添加する。また、NADPHは、例えば0.1mM〜0.5mMになるよう添加するが、NADPHの反応系中の濃度は、反応系に含まれる基質濃度を超える濃度にする。
【0054】
アセトヒドロキシ酸レダクトイソメラーゼは、例えば5〜20pkatal単位を添加する。ここでいう単位(katal)とは1秒間に1molの基質を変換する酵素量である。反応温度は約30〜42℃とし、反応時間は約10〜120分とし、NADPの増加またはNADPHの減少を340nmの吸光度でみる。吸光度は吸光度計で0分(反応前)に測定しておき、酵素添加後、上記適当な条件にて反応させた後、反応後の吸光度を測定し、0分時の吸光度からの減少値を算出してジアセチル前駆体を定量する。
【0055】
アセトヒドロキシ酸の定量において、酵素反応系は上述の限りではなく、適宜、反応時間、酵素添加量、基質添加量等を変更することができる。なお、ここでいう反応時間とは、反応系中のアセトヒドロキシ酸のすべてがアセトヒドロキシ酸レダクトイソメラーゼと接触するに要する時間である。
【0056】
本定量法では、飲食品の試料から、アセトヒドロキシ酸の測定を妨害する物質(例えば乳酸など)を除去してもよい。特にヨーグルトなどの発酵乳飲食品などに有効である。
【0057】
アセトヒドロキシ酸の分離は、弱塩基性陰イオン交換樹脂および水と任意の割合で混合するアルコール水溶液を用いて行うことが好ましい。アセトヒドロキシ酸は、中性から弱アルカリ性のアルコール水溶液中で弱塩基性陰イオン交換樹脂に吸着されるからである。弱塩基性陰イオン交換樹脂は、カラムに充填して用いてもよいし、試料の入っている容器に該樹脂を懸濁し、アセトヒドロキシ酸を樹脂に吸着させ、濾過や遠心分離等により樹脂を回収してもよい。また、アルコール水溶液として、例えば1価のアルコール水溶液を用いることが好ましい。1価のアルコール水溶液として、具体的にはメタノール、エタノール、1−プロパノール、2−プロパノール、tert-ブタノール水溶液が挙げられ、特に限定されないが、より好ましくはメタノール水溶液が用いられる。
【0058】
アセトヒドロキシ酸の分離を行うには、まず、飲食品の試料を好ましくはpH6〜10、より好ましくはpH7〜9に調整し、メタノール水溶液を加える。メタノールの濃度は、体積パーセント濃度で50〜100%が好ましく、75〜100%がより好ましい。試料とメタノール水溶液の混合液を遠心分離する。硬い固体試料の場合は遠心分離前にホモジナイザー等で十分にホモジナイズすることが好ましい。上澄みを弱塩基性陰イオン交換樹脂カラムにアプライし、メタノール水溶液をカラムに通液する。次に、好ましくは約0.05〜5.0M、より好ましくは約0.1〜1.0Mのアルカリ金属の水酸化物(例えば水酸化ナトリウム、水酸化リチウム、水酸化カリウムなど)、より好ましくは水酸化ナトリウムを含む溶出液を通液し、アセトヒドロキシ酸を溶出する。
【0059】
本発明によれば、飲食品中のジアセチル前駆体を定量するためのキットを作製することができる。キットは、アセトヒドロキシ酸レダクトイソメラーゼを含む。アセトヒドロキシ酸レダクトイソメラーゼは、配列番号2に示すアミノ酸配列を含むタンパク質、または該アミノ酸配列において1もしくは数個のアミノ酸が欠失、置換もしくは付加されたアミノ酸配列を含み、かつアセトヒドロキシ酸レダクトイソメラーゼ活性を有するタンパク質のものが好ましい。また、キットには、試料の前処理に用いられるもの(例えば弱塩基性陰イオン交換樹脂カラム、溶出液など)、アセトヒドロキシ酸定量に用いられるもの(例えば緩衝液、試料の遠心分離用チューブ、標準添加法に用いるアセト乳酸など)、取扱説明書などを含めてもよい。
【実施例】
【0060】
以下、実施例により本発明を具体的に説明するが、これらの実施例は本発明を限定するものではない。なお、本発明によるジアセチル前駆体の定量方法を、便宜的に「本酵素法」ということがある。
【0061】
実施例における各種遺伝子操作は、Current protocols in molecular biology(edited by F.M.Ausubel et al.,1987)に記載されている方法に従った。
【0062】
また、アセトヒドロキシ酸レダクトイソメラーゼの基質となるアセトヒドロキシ酸であるアセト乳酸とアセトヒドロキシ酪酸の合成方法は、アセト乳酸については、Krampitzの方法(L.O.Krampitz,Arch.Biochem.,17,81−85,1948)に従い、2−アセトキシ−2−メチルアセト酢酸エチル(Sigma−Aldrich社製)0.182g(0.9mmol)に対し水9ml、2N水酸化ナトリウム水溶液1mlを順次添加し、25℃で1時間攪拌して加水分解することで調製した。
【0063】
〔実施例1:アスペルギルス・オリゼ由来アセトヒドロキシ酸レダクトイソメラーゼの精製〕
1x106個胞子/mlとなるようにアスペルギルス・オリゼRIB40株胞子懸濁液をCzapek Dox培地(Nakajima,K.,et al.,Curr.Genet.,37,322−327,2000)に添加した。添加後30℃、48時間振盪培養した。菌糸体をNo.2濾紙(アドバンテック)にて濾別・回収した。回収した菌体に菌体重量の5倍量となる1mM PMSFを含む10mMトリス−塩酸緩衝液、pH8.0、2mM MgSO4、3mM 2−メルカプトエタノールを加え、フードプロセッサーにて破砕し、12000g、4℃、20分間遠心分離し、得られた上清を粗酵素液とした。粗酵素液に50%飽和となるように硫酸アンモニウムを加えた後、12000g、4℃、20分間遠心分離し、上清画分を得た。上清画分を10mMトリス−塩酸緩衝液、pH8.0、2mM MgSO4、3mM 2−メルカプトエタノール、50%飽和硫酸アンモニウムにて平衡化したOctyl−Cellulofine(生化学工業)に供し、吸着画分を50−0%飽和硫酸アンモニウムの直線濃度勾配により溶出させた。分画された溶液に対してArfinの方法に従ってアセトヒドロキシ酸レダクトイソメラーゼ活性を測定し、活性画分を追跡した。具体的な活性測定方法は後記実施例6に示す。得られた活性画分に対して、50%飽和硫酸アンモニウムとなるように硫酸アンモニウムを添加し、再度10mMトリス−塩酸緩衝液、pH8.0、2mM MgSO4、3mM 2−メルカプトエタノール、50%飽和硫酸アンモニウムにて平衡化したOctyl−Cellulofine(生化学工業)に供し、吸着画分を50−0%飽和硫酸アンモニウムの直線濃度勾配により溶出させた。得られた活性画分を10mMリン酸緩衝液、pH6.8、2mM MgSO4、3mM 2−メルカプトエタノール、5mM硫酸アンモニウムに対して透析し、12000g、4℃、20分間遠心分離し、上清画分を得た。上清画分を10mMリン酸緩衝液、pH6.8、2mM MgSO4、3mM 2−メルカプトエタノール、5mM 硫酸アンモニウムにて平衡化したDEAE−Sephadex A50(GE Healthcare)に供し、吸着画分を0−0.15M NaClの直線濃度勾配にて溶出させた。得られた活性画分を10mMリン酸緩衝液、pH6.8、2mM MgSO4、3mM 2−メルカプトエタノール、5mM 硫酸アンモニウムに対して透析し、10mMリン酸緩衝液、pH6.8、2mM MgSO4、3mM 2−メルカプトエタノール、5mM硫酸アンモニウムにて平衡化したGIGAPITE(生化学工業)に供し、吸着画分を10−300mMのリン酸直線濃度勾配にて溶出させた。得られた活性画分をSDS−PAGE(Laemmli,U.K.,Nature,227,680−685,1970)に供し電気泳動的に均一であることが確認されたため、ここで得られた活性画分をアスペルギルス・オリゼ由来アセトヒドロキシ酸レダクトイソメラーゼ精製標品とした。
【0064】
〔実施例2:アスペルギルス・オリゼ由来アセトヒドロキシ酸レダクトイソメラーゼのアミノ末端アミノ酸配列の決定〕
上記で得られた精製酵素溶液500μlに対して、250μlの100%(w/v)冷トリクロロ酢酸を加え、よく混合した後、氷中に20分間以上放置した。サンプルを15000xg、4℃、20分間遠心分離し、上清を完全に除いた後、沈殿を15μlのSDS化溶液(5%(w/v)SDS、5%(v/v)2−メルカプトエタノール、0.12M Tris−塩酸緩衝液(pH8.8)、0.05%(w/v)ブロモフェノールブルー、10%(w/v)グリセロール)に溶解させた後、沸騰湯浴中に5分間放置し、SDS化を行い、これをサンプル溶液とした。サンプル溶液をSDS−PAGEに供し、泳動後ゲル内のタンパク質をPVDF膜に転写した。PVDF膜上より目的のバンドを切り抜き、そのままアミノ末端アミノ酸配列の解析を行った。アミノ末端アミノ酸配列を解析した結果、アミノ末端より「VKNISFAGHE」であった。得られたアミノ酸配列をアスペルギルス・オリゼの全ゲノム配列情報(特開2005−176602号公報)に照らし合わしたところ、予想されたアスペルギルス・オリゼ由来のアセトヒドロキシ酸レダクトイソメラーゼアミノ酸配列(配列番号2)の、アミノ末端より数えて52番目より始まる「VKNISFAGHE」と一致した。
【0065】
〔実施例3:アスペルギルス・オリゼ由来アセトヒドロキシ酸レダクトイソメラーゼのアスペルギルス・オリゼを宿主とした過剰発現系の構築〕
次に、実施例2で求めたアスペルギルス・オリゼ由来アセトヒドロキシ酸レダクトイソメラーゼのアミノ末端アミノ酸配列情報と、アスペルギルス・オリゼの全ゲノム配列情報(特開2005−176602号公報)を基に、オリゴヌクレオチド5’−CTCCCGTCCATATGGTCAAGAAC−3’(配列番号3)と5’−CTTTTTCGCATGCGGATTTGGGAAAAAG−3’(配列番号4)を合成した。この1組のプライマーセットを用い、アスペルギルス・オリゼRIB40株の染色体DNAを鋳型としてPCRを行い、アスペルギルス・オリゼ由来アセトヒドロキシ酸レダクトイソメラーゼをコードする遺伝子を増幅した。アスペルギルス・オリゼ染色体DNA溶液(50μg/ml)は公知の方法に従って調製した。PCR用装置はPeltier Thermal Cycler PTC−200(MJ RESEARCH)を用いた。染色体DNA溶液は1μl用い、ポリメラーゼはPlatinum Taq DNA Polymerase(Invitrogen)を用いた。増幅反応は94℃、3分間鋳型DNAを変性し、94℃、45秒間、56℃、30秒間、72℃、1分10秒間保持するサイクルを30サイクル行った後、72℃、10分間で完全伸長させ、4℃で保持した。得られたPCR増幅断片をアガロースゲル電気泳動に供したところ、およそ1094塩基対からなる断片の増幅が確認された。
【0066】
得られたPCR増幅断片をNde I(NEB)、Sph I(NEB)で消化し、アガロースゲル電気泳動に供しエチジウムブロマイドで染色後、UV照射下で1068塩基対からなる断片を切り出した。ゲル中よりWizard SV Gel and PCR Clean−Up System(Promega)を用いてDNAを抽出し、これを挿入DNA断片とした。次に塩基配列中にエノラーゼのプロモーター配列(P−enoA142)を有するプラスミドpNEN142 DNA5μgをプロモーター配列直後にある制限酵素Nde I、Sph I認識配列の位置でNde I(NEB)、Sph I(NEB)で消化し、アガロースゲル電気泳動に供しエチジウムブロマイドで染色後、UV照射下で9296塩基対からなる断片を切り出した。ゲル中よりWizard SV Gel and PCR Clean−Up System(Promega)を用いてDNAを抽出し、これをベクターDNA断片とした。次にベクターDNAと挿入DNA断片をT4 DNA ligase(宝酒造)により連結させ、連結DNA溶液を得た。この連結DNA溶液を用い情報により形質転換可能な大腸菌DH5α(宝酒造)を形質転換し、その形質転換大腸菌より定法によりプラスミドDNAを抽出し、アスペルギルス・オリゼ形質転換用プラスミドDNA(pNEN−AoilvC−delation)を得た。このプラスミドDNA10μgをBamH I(NEB)で消化し、定法によりフェノール抽出、エタノール沈殿処理を行った後、10μlのTE緩衝液に溶解し形質転換用DNA溶液とした。アスペルギルス・オリゼniaD300株(RIB40株由来の硝酸還元酵素遺伝子(niaD)欠損変異株)を形質転換用DNA溶液を用いて形質転換した。形質転換法は、プロトプラスト化した後ポリエチレングリコール及び塩化カルシウムを用いる方法(Mol.Gen.Genet,218,99−104,1989)によって行い、アスペルギルス・オリゼ形質転換株(受領番号NITE AP−541)を得た。
【0067】
〔実施例4:アスペルギルス・オリゼを宿主としたアスペルギルス・オリゼ由来アセトヒドロキシ酸レダクトイソメラーゼ過剰発現系からの酵素の精製〕
実施例3で構築し得られたアスペルギルス・オリゼ形質転換株胞子懸濁液を、1x106個胞子/mlとなるようにYPM培地(2%peptone、1%Yeast extract、2%maltose)に添加した。添加後30℃、24時間振盪培養した。菌糸体をNo.2濾紙(アドバンテック)にて濾別・回収した。以後の精製過程は実施例1と同様の方法で行った。
【0068】
〔実施例5:アスペルギルス・オリゼ由来アセトヒドロキシ酸レダクトイソメラーゼの保存〕
実施例1及び実施例4で得られたアスペルギルス・オリゼ由来アセトヒドロキシ酸レダクトイソメラーゼ精製標品の保存方法を検討した。精製酵素標品を含む溶液を10mMリン酸緩衝液、pH6.8、20mM MgSO4、30mM 2−メルカプトエタノール、5mM硫酸アンモニウムに対して透析し、さらに透析後の溶液に対して等量の100%(v/v)グリセロールを添加し、−20℃にて保存した。本条件で保存することでアセトヒドロキシ酸レダクトイソメラーゼの活性は少なくとも1年間は維持されることを確認した。
【0069】
〔実施例6:アセトヒドロキシ酸定量法へのアセトヒドロキシ酸レダクトイソメラーゼの有効性の検証〕
実施例1及び実施例4で得られたアスペルギルス・オリゼ由来アセトヒドロキシ酸レダクトイソメラーゼ精製標品を用い、アセトヒドロキシ酸の分析が可能かどうかを調べた。
【0070】
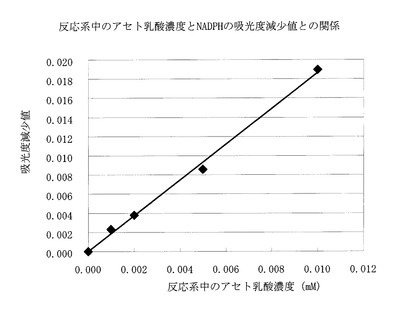
具体的にはArfinの方法に従い、830μlの50mM Tris−塩酸緩衝液(pH9.0)、10μlの10mM NADPH、10μlの1M MgSO4、50μlのアスペルギルス・オリゼ由来アセトヒドロキシ酸レダクトイソメラーゼ精製酵素溶液に、100μlの各種濃度(0.1、0.05、0.02、0.01mM)のアセト乳酸溶液を添加し、37℃で10分間保温し、その間の340nmの吸光度減少値を測定し、アセト乳酸濃度との相関性を調べた。
【0071】
結果は、図1に示すごとく、反応系中のアセト乳酸濃度と吸光度の減少値とは比例関係を示し、本酵素法でのアセト乳酸の定量は可能と判断された。
【0072】
〔実施例7:アセトヒドロキシ酸レダクトイソメラーゼの基質特異性〕
飲食品中のアセトヒドロキシ酸と共存し、多くのアセト乳酸定量分析法を妨害することの多い、ジアセチル、アセトインが本酵素法による定量にどう影響するかについて調べた。
【0073】
具体的には、実施例6における酵素反応系のアセト乳酸区分に、ジアセチルおよびアセトインを表1に示した濃度となるように添加し、37℃で10分間保温し、その間の340nmの吸光度の減少値を測定した。なお、ジアセチルは和光純薬工業製のものを、アセトインは東京化成工業製のものをエーテルで洗浄して用いた。
【0074】
結果は表1に示すごとく、ジアセチルあるいはアセトインを添加した場合にあっても、アセト乳酸の吸光度減少値に実質的な差異は見られなかった。このことから、本酵素法では試料中に存在するジアセチルやアセトインに関係なく、アセトヒドロキシ酸のみを定量することが出来ることが示された。
【0075】
【表1】

【0076】
〔実施例8:清酒醪中に含まれるアセトヒドロキシ酸の定量〕
アセトヒドロキシ酸は、発酵飲食品製造の場合、主として製造工程中使用微生物により生成された後、製造工程中、あるいは、製品化後に自動的に分解して、ジアセチルを代表とするビシナルジケトン類(ジアセチルとその同族体、2,3−ペンタンジオンの総称)となる。それらは、特徴的な強い臭気を有し、酒類などの発酵飲食品の品質に対して大きな影響を及ぼしている。しかし、両者の分別定量が困難なことについては、前述した通りである。そこで、酒類の代表である清酒の、製造工程中の半製品である清酒醪について、本酵素法によりアセトヒドロキシ酸の定量が可能であるかどうか調べることにした。
【0077】
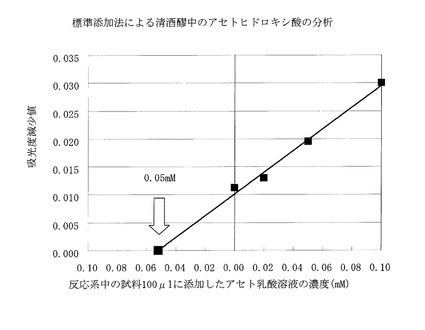
清酒醸造中の、留添(とめぞえ)後2日目の清酒醪を対象として、直ちにpH7に調整して低温下に保持して実験室に持ち帰り、遠心分離を行ってその上清を採り、実施例6と同一の酵素反応系を用いて、標準添加法により定量分析を行った。具体的には、試料中のアセト乳酸濃度がそれぞれ一定量(0.1、0.05、0.02mM)増加するようにアセト乳酸を添加した上清試料100μlを用い、実施例6と同様にして定量した。結果は図2に示すように、醪中のアセトヒドロキシ酸濃度は0.05mMと算定された。
【0078】
この結果より、清酒醪中のアセトヒドロキシ酸は、従来用いられてきた煩雑で、不正確な蒸留比色法によらずとも、本酵素法により、簡便、正確に定量できることがわかった。
【0079】
〔実施例9:発酵乳製品中に含まれるアセトヒドロキシ酸の定量〕
代表的な発酵乳製品であるヨーグルトの乳清中のアセトヒドロキシ酸の定量が可能であるかどうか調べた。
【0080】
当実験室で調製したヨーグルト乳清を水酸化ナトリウム水溶液でpH7に中和したのち、遠心式フィルターユニット(Centriprep YM−3 Millipore社製)で濾過、除タンパクし、得られた溶液について、実施例8と同様の方法で定量分析を行った。その結果、10分後の吸光度について吸光度の減少が認められなかった。従って何らかの物質が測定を妨害しており、前処理をしなければ測定できないと考えられた。
【0081】
〔実施例10:アセトヒドロキシ酸測定における乳酸の阻害について〕
乳清中に含まれる成分の中で、ヨーグルトの乳清中に多く含まれ、測定を妨害する可能性の高い乳酸の阻害について調べた。
【0082】
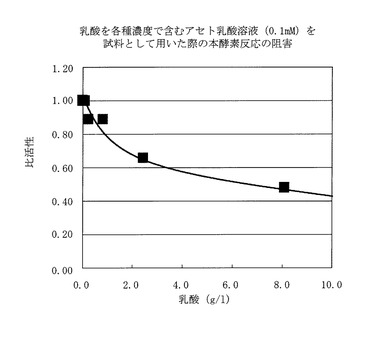
具体的には、実施例6における酵素反応系に、アセト乳酸を0.1mM、水酸化ナトリウムでpH7まで中和した乳酸を各種濃度(乳酸として0〜8.1g/l)で含む溶液を添加し、37℃10分間保温し、その間の340nmの吸光度の減少値を測定した。なお、乳酸は和光純薬工業製のものを用いた。
【0083】
結果は図3に示すごとく、乳酸を添加した場合、乳酸なしの場合の活性を1.0とすると乳酸8.1g/lの濃度では0.44となった。従って、明らかに本酵素反応は、高濃度の乳酸の存在下で阻害を受け、乳酸を10g/l以上含む一般のヨーグルトでは、測定感度を向上させるために乳酸の除去が必要になると考えられた。
【0084】
〔実施例11:乳酸溶液中の乳酸の除去〕
乳清試料中の乳酸を除去するに際し、乳酸水溶液中のアセトヒドロキシ酸を除去する手法を検討した。
【0085】
検討の結果、メタノール中では、乳酸は弱塩基性陰イオン交換樹脂に殆ど吸着されず、逆にアセトヒドロキシ酸は弱塩基性陰イオン交換樹脂に吸着されることを見いだし、この原理を用いて乳酸溶液からアセトヒドロキシ酸の分離が可能であることが確認された。
【0086】
具体的には、pH9に中和した乳酸水溶液(1.0%)25mlに、メタノール(特級:和光純薬製)を75ml添加し、一定濃度(0.2mM)になるようにアセト乳酸を添加した溶液について、弱塩基性陰イオン交換樹脂ダウエックスSD−2(ダウ・ケミカル製)を充填したポリプロピレン製カラム(ムロマックミニカラムL:室町ケミカル製)に、温度5℃のもとで、高速液体クロマトグラフ用ポンプを用い、1.0ml/分の流速で上記溶液を通液した。次いで、アンモニアを0.01%含むメタノールを50ml、次いでメタノールを50ml、それぞれ1.0ml/分の流速で通液した。最後に、35℃に加温した、メタノール+水=1+1(体積比)に1.0M濃度で水酸化ナトリウムを含む溶液30mlを1.0ml/分の速度で通液し、アセトヒドロキシ酸を溶出させた。最後に、溶出液をpH9になるまで中和し、全体を水で50mlに定容した。本法による乳酸の除去率は約70%で、アセト乳酸の回収率は約93%であった。なお、元の乳酸水溶液は25mlで、最終的に定容されるのは50mlであるので、元の乳酸水溶液中のアセト乳酸濃度は、2倍に希釈されることになる。
【0087】
〔実施例12:ヨーグルト乳清中に含まれるアセトヒドロキシ酸の定量〕
実施例11の結果をもとに、ヨーグルト乳清中のアセトヒドロキシ酸の定量を試みた。
【0088】
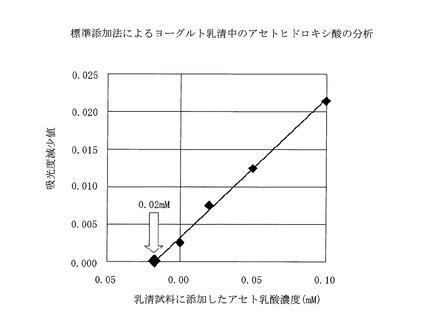
あらかじめ当実験室で調製したヨーグルト乳清(乳酸1.0%含む)25mlをpH9に中和したのち、メタノールを75ml加え、8000rpmで10分間遠心分離をした。上澄みを実施例11と同様の方法で処理し乳酸の除去を行い、得られた試料溶液を、孔径0.45μmのシリンジフィルター(DISMIC−25AS:アドバンテック東洋製)でろ過したのち、実施例8と同様の方法で定量分析を行った。結果は図4に示すように、反応に用いた溶液中のアセトヒドロキシ酸濃度は0.02mMと算定され、元の乳清中のアセトヒドロキシ酸濃度については、0.02×2(希釈倍率)より0.04mMと推定される。
【産業上の利用可能性】
【0089】
本発明によれば、飲食品中のジアセチル前駆体を直接的かつ簡易・正確に定量でき、飲食品製造においての品質管理等に応用可能性がある。
【図面の簡単な説明】
【0090】
【図1】反応系中のアセト乳酸濃度とNADPHの吸光度減少値との関係を示す。
【図2】標準添加法による清酒醪中のアセトヒドロキシ酸の分析を示す。
【図3】乳酸を各種濃度で含むアセト乳酸溶液(0.1mM)を試料として用いた際の本酵素反応の阻害を示す。
【図4】標準添加法によるヨーグルト乳清中のアセトヒドロキシ酸の分析を示す。
【技術分野】
【0001】
本発明は、飲食品中のジアセチル前駆体の定量法に関する。
【背景技術】
【0002】
多くの飲食品において、ジアセチル臭はそれらの特性を大きく左右する重要な香味(臭い)成分である。例えばヨーグルトなどの発酵乳製品では、ジアセチル臭は良い臭いの代表的成分の1つとして重要視されるが、酒類ではあってはならない臭いとして嫌われている。発酵飲食品中のジアセチル臭は、ジアセチルとその同族体、2,3−ペンタンジオン(両者を併せてビシナルジケトンとも称する)の一定濃度(弁別閾値)以上の存在により検知される。ビシナルジケトンは、発酵中の微生物により生成される、その前駆体であるアセトヒドロキシ酸(ジアセチルの前駆体であるアセト乳酸、ならびに、2,3−ペンタジオンの前駆体であるアセトヒドロキシ酪酸の総称)から生成されるが、天然物中のアセトヒドロキシ酸は不安定であり、自動的に分解してビシナルケトンを含むアセトイン(ならびに、その同族体であるアセチルエチルカルビノール)を生成する。したがって、飲食品中におけるジアセチル臭の発生は、多くの場合、製品化後に顕在化する。そこで、飲食品のジアセチル臭の調節には、アセトヒドロキシ酸濃度を測定して調節することが必要となる。飲食品、特に発酵飲食品を試料としてアセトヒドロキシ酸を定量分析する場合には、その特徴を踏まえた条件設定(場合によっては改変)の必要がある。飲食品試料の場合の、第一の特徴として、多種類の共存物がアセトヒドロキシ酸よりもはるかに多量に混在している点があげられる。したがって、アセトヒドロキシ酸の定量分析に先立って、アセトヒドロキシ酸を安定に保ち得る条件下での、共存物内の妨害物質の除去が必要である。
【0003】
発酵飲食品の場合に於いては、アセトヒドロキシ酸と同様に、関係微生物により生成される有機酸群の濃度が高く、アセトヒドロキシ酸が分解しやすい酸性であるものが多いという特徴が挙げられる。即ち、発酵飲食品製造の場合にはそのほとんどが静置発酵で行われる。そのために、当初発酵系の中に存在した酵素は急速に微生物によって消費され、系内は急速に嫌気的となり有機酸が生成するのである。
【0004】
アセトヒドロキシ酸は中性条件下では比較的安定な物質であるが、発酵食品のほとんどに於いてそうである酸性条件下では特に不安定で、ジアセチル類やアセトイン類に変化しやすい。また、その反応は温度が高いほど迅速に進行する。したがって、試料採取後直ちに分析を行うことのできない場合には、直ちに微生物等の共存物をできるだけ除いて、中和し、冷却(凍結)保存する必要がある。したがって、試料の添加により反応時の最適pHが変わらぬよう、供試試料のpH調整、緩衝液の濃度増強等の確認・処理が必要である。
【0005】
発酵飲食品中の有機酸中で特に出現頻度が高く、また濃度が高いという点で注目すべきものは乳酸である。乳酸はある濃度以上で、アセトヒドロキシ酸の定量分析を妨害するので、分析に際してはその低濃度化が必要である。
【0006】
日本酒(清酒)製造、あるいは、ある種の上面酵母発酵によるビール醸造に於いては、野生酵母による汚染を防ぐために、種菌あるいはもろみに、乳酸(あるいは乳酸菌)を添加して乳酸酸性条件下(数mM濃度)で発酵させる方法が採られている。また、酒類に対する微生物汚染は、乳酸菌の増殖による場合が多く、その場合、アセトヒドロキシ酸の高濃度生成とともに、乳酸濃度が1mMを超える場合も珍しくない。また、発酵乳製品製造の場合には、香味向上のために、チーズや発酵バター製造の初期に乳酸発酵をさせるのが常法となっている。このように、発酵飲食品製造に於いては、高濃度の乳酸の生成が常に起こると考えていなければならない。
【0007】
発酵飲食品の製造に用いられる微生物は比較的限られているが、その中でもSaccharomyces属を主体とする酵母(酒類の製造など)と乳酸菌群(発酵乳製品や漬物類など)が圧倒的に広く用いられている。
【0008】
Saccharomyces属酵母は、アセト乳酸の分解によって生成するアセトイン濃度の、場合によっては、数十倍濃度のアセトインを、別の代謝系であるアセトアルデヒドの縮合により(アルコール発酵の際に)生成する。また、乳酸菌群の中には、ジアセチルを生合成するものもあるとの説もある。また、乳酸菌は、上述のごとく、酵母によるアルコール発酵の際の、汚染微生物の代表的な菌であり、アルコール飲料の品質を大きく阻害する。ジアセチル臭は乳酸菌に汚染されたアルコール飲料のメルクマールとされる臭いである。
【0009】
発酵中に生成するアセトヒドロキシ酸の一部は、ジアセチル類、アセトイン類へ発酵中にも分解して生成する。発酵飲食品中のアセトヒドロキシ酸の定量において、これらの物質との分別定量はもっとも困難であり、手間を要し、また、正確性に欠け、現在に於いてもそれらを回避できる正確なアセトヒドロキシ酸の定量法の開発研究が世界各所で行われているのが現状である(非特許文献1、非特許文献4)。
【0010】
食品分析の分野におけるアセトヒドロキシ酸の定量分析は、一般的には、アセトイン類やジアセチル類(ビシナルジケトン)に転換後、定量分析されている。前述のようにアセトヒドロキシ酸は、食品の通常の発酵条件下であっても自動的に分解するほどに不安定な物質だからである。いまだに天然物中のアセトヒドロキシ酸は純品として抽出、精製されたことはなく、またアセトヒドロキシ酸の存在自体、合成品のアセトヒドロキシ酸の各種条件下でのジアセチル類やアセトイン類に変化する挙動と、天然物中におけるアセトヒドロキシ酸がそれらに変化する挙動との一致性から、その存在が推定されているにすぎない。
【0011】
飲食品のアセトヒドロキシ酸の定量分析の具体例を挙げると、例えばビールについて行われており、試料ビールに空気を含ませてから60℃で90分間放置し、ジアセチルに転換してから、そのジアセチルを蒸留比色法、あるいは、ガスクロマトグラフィーで定量分析するという公定法が定められている(非特許文献2)。
【0012】
しかし、この方法では、既存のジアセチルもあわせて定量分析されるという問題があり、既存のジアセチル分を差し引くための更なる分析操作が必要で、煩雑な定量法となっている。
【0013】
また、ビールにおけるアセトヒドロキシ酸のジアセチルへの転換率は100%ではなく、幾分かのアセトインも同時に生ずるとの議論もある(非特許文献3)。
【0014】
さらに、上記ビールの公定法を清酒醪中のアセトヒドロキシ酸の定量分析に適用すると、60%から80%程度に相当する値しか示されず(非特許文献4)、この方法は清酒醪などの飲食品全般にそのまま適用できるものではない。
【0015】
このように、従来のアセトヒドロキシ酸の定量分析は煩雑である上に、アセトインやジアセチルに転換される割合が、分析条件や分析対象である食品によって異なるため、現在でもジアセチル分析を対象とした公定法を持たない業界は多く、ましてや、アセトヒドロキシ酸分析のための公定法を持つ業界は、ビール業界をのぞいて皆無といってよい。
【0016】
従って、食品分析の分野では、アセトヒドロキシ酸を直接定量する方法が求められており、実際、アセトヒドロキシ酸の分解物であるジアセチル、アセトインまたはそれらの同族体からアセトヒドロキシ酸を分別定量する研究が世界中で多く行われている。
【0017】
しかし、飲食品には共存物が多く、特に発酵飲食品では、微生物によって生成される様々なタンパク質(酵素)などによる測定値への影響が考えられ、また、飲食品中の天然物であるアセトヒドロキシ酸はすぐに分解してしまうことから、直接定量法は開発されていなかった。
【0018】
食品中以外でのアセトヒドロキシ酸の測定例としては、合成品であるアセトヒドロキシ酸を基質とし、NADPHとマグネシウムイオンの存在下でケトール酸レダクトイソメラーゼを作用させて、NADPHの340nmでの吸光度の低下を測定する方法が知られている(非特許文献1、以下Arfinの方法という)。
【0019】
Arfinの方法を応用した特許出願として、例えば、植物の真菌病を処置するためのケトール酸レダクトイソメラーゼ阻害剤を特定するための方法が挙げられる(特許文献1)。特許文献1は、ケトール酸レダクトイソメラーゼの酵素活性を阻害することによって真菌の成長および/または病理発生を阻害する化合物を特定するために、宿主生物で発現させ精製したケトール酸レダクトイソメラーゼを用いてArfinの方法を適用することを開示するが、Arfinの方法に用いる基質は合成品であり、種々の物質が混在する天然物中のアセトヒドロキシ酸を定量分析するものではなく、また、特許文献1は、飲食品中の天然物であるアセトヒドロキシ酸について何ら開示していない。
【0020】
【特許文献1】特表2005−501912号明細書
【非特許文献1】S.M.Arfin and H.E.Umberger,J.Biol.Chem.,244,1118−1127,1969
【非特許文献2】改訂BCOJビール分析法、8.16.2、p.2/4、(2)、ビール酒造組合国際技術委員会編、財団法人日本醸造協会、平成10年
【非特許文献3】The American Society of Brewing Chemists,Methods of Analysis of the American Society of Brewing Chemists,8th revised edition,1992,Beer 25,p.1 of 5
【非特許文献4】Kobayashi,K.,Kusaka,K.,Takahashi,T. and Sato,K.,J.Biosci.Biotechnol.99, 502−507,2005
【発明の開示】
【発明が解決しようとする課題】
【0021】
本発明の目的は、飲食品中のアセトヒドロキシ酸を直接定量できる、簡易で正確な方法を提供することにある。
【課題を解決するための手段】
【0022】
本発明者らは、アセトヒドロキシ酸(アセト乳酸とアセトヒドロキシ酪酸の総称)をアセトイン(同族体であるアセチルエチルカルビノールも含む)やジアセチル(同族体である2,3−ペンタンジオンも含む)に変換するために要する時間と手間を省き、分析の効率化を図るため、アセトヒドロキシ酸をそのままの形で定量分析しようと考え、ピルビン酸を初発基質とし、アセトヒドロキシ酸合成を経て、ロイシン、バリン、イソロイシンを生合成する経路の中で、アセトヒドロキシ酸を基質とするアセトヒドロキシ酸レダクトイソメラーゼを用いること、具体的にはNADPHの存在下で当該酵素を作用させて、NADPの増加またはNADPHの減少を測定する方法を見出した。また、試料中のアセトヒドロキシ酸の測定を妨害する物質とアセトヒドロキシ酸を分離する方法を見出し、本発明を完成した。
【0023】
本発明の特徴は、要約すると以下の通りである。
(1)飲食品中のジアセチル前駆体を定量する方法であって、飲食品の試料に、マグネシウムイオンおよび還元型ニコチンアミドアデニンジヌクレオチドリン酸(NADPH)の存在下でアセトヒドロキシ酸レダクトイソメラーゼを作用させ、酸化型ニコチンアミドアデニンジヌクレオチドリン酸(NADP)の増加またはNADPHの減少を指標としてジアセチル前駆体を測定することを含む、前記方法。
(2)飲食品が発酵物である、(1)に記載の方法。
(3)飲食品が発酵酒または発酵乳製品である、(1)または(2)に記載の方法。
(4)飲食品が、清酒醪、ビール類製造用発酵液、果汁発酵液、サワークリーム、カテージチーズ、ヨーグルト、発酵バター、スターターディスティレート、焼酎用モロミ、漬物類、醤油、味噌、味醂または酢である、(1)〜(3)のいずれかに記載の方法。
【0024】
(5)アセトヒドロキシ酸レダクトイソメラーゼが、真核微生物由来の酵素である、(1)〜(4)のいずれかに記載の方法。
(6)アセトヒドロキシ酸レダクトイソメラーゼが、アスペルギルス属由来の酵素である、(1)〜(5)のいずれかに記載の方法。
(7)アセトヒドロキシ酸レダクトイソメラーゼが、アスペルギルス・オリゼ由来の酵素である、(1)〜(6)のいずれかに記載の方法。
(8)アスペルギルス・オリゼが、アスペルギルス・オリゼRIB40株(FERM P−18273)である、(7)に記載の方法。
【0025】
(9)アセトヒドロキシ酸レダクトイソメラーゼが、配列番号2に示すアミノ酸配列を含むタンパク質、または該アミノ酸配列において1もしくは数個のアミノ酸が欠失、置換もしくは付加されたアミノ酸配列を含み、かつアセトヒドロキシ酸レダクトイソメラーゼ活性を有するタンパク質である、(1)〜(8)のいずれかに記載の方法。
(10)NADPの増加またはNADPHの減少を、340nmの吸光度に基づいて測定する、(1)〜(9)のいずれかに記載の方法。
(11)飲食品の試料から乳酸を除去することをさらに含む、(1)〜(10)のいずれかに記載の方法。
(12)乳酸の除去を、弱塩基性陰イオン交換樹脂を用いて行う、(11)に記載の方法。
【0026】
(13)アセトヒドロキシ酸レダクトイソメラーゼを含む、飲食品中のジアセチル前駆体を定量するためのキット。
(14)定量が(1)〜(12)のいずれかに記載の方法によって行われるものである、(13)に記載のキット。
(15)アセトヒドロキシ酸レダクトイソメラーゼが、配列番号2に示すアミノ酸配列を含むタンパク質、または該アミノ酸配列において1もしくは数個のアミノ酸が欠失、置換もしくは付加されたアミノ酸配列を含み、かつアセトヒドロキシ酸レダクトイソメラーゼ活性を有するタンパク質である、(13)または(14)に記載のキット。
(16)飲食品の試料から乳酸を除去するための弱塩基性陰イオン交換樹脂カラムをさらに含む、(13)〜(15)のいずれかに記載のキット。
【0027】
本明細書において「飲食品」とは、その半製品、中間生成物、原料も含むことを意味する。
【0028】
本明細書において「発酵物」とは、固形状、ゲル状、液体状などのあらゆる形態の発酵飲食品を含み、発酵工程を経た半製品、中間生成物、原料も含み、また製品化のために加熱殺菌をすることを含む方法で生産される場合、加熱殺菌前の発酵物も含むことを意味する。
【0029】
本明細書において「1もしくは数個のアミノ酸が欠失、置換もしくは付加されたアミノ酸配列」とは、具体的には、配列番号2に示すアミノ酸配列の1〜10個、好ましくは1〜5個のアミノ酸、さらに好ましくは1〜3個のアミノ酸が変異により欠失、または他のアミノ酸に置換、あるいは付加された配列をいう。変異は、人為的に導入された変異または天然に存在する変異でもよい。
【0030】
本明細書において「アスペルギルス・オリゼRIB40株」は、独立行政法人産業技術総合研究所、独立行政法人製品評価技術基盤機構および独立行政法人酒類総合研究所により、独立行政法人産業技術総合研究所特許生物寄託センター(茨城県つくば市東1丁目1番地1)に寄託された、寄託番号FERM P−18273が付与された株をいう。
【0031】
本明細書において「受領番号NITE AP−541を付与された形質転換株」は、千葉県により、独立行政法人製品評価技術基盤機構特許微生物寄託センター(千葉県木更津市かずさ鎌足2−5−8)に寄託された株である。この株は、千葉県産業支援技術研究所(千葉県千葉市若葉区加曽利町889番地)からも分譲可能である。
【発明の効果】
【0032】
本発明によれば、飲食品中の易分解性のジアセチル前駆体を直接定量することができる。従来法のようにジアセチル前駆体をアセトインまたはジアセチルに変換する必要がなく、酸処理、熱処理等の手間と時間が大幅に削減できる。また、その変換率が問題になることもなく、試料中に既存のアセトインまたはジアセチルを測りこんでその分を後で差し引くなどの操作がいらず、簡易であり、ジアセチル前駆体を正確に定量できる。さらに、本発明によれば、試料からジアセチル前駆体の測定を妨害する物質(例えば乳酸など)を除去してジアセチル前駆体を定量することができる。
【発明を実施するための最良の形態】
【0033】
本発明の飲食品中のジアセチル前駆体を定量する方法は、飲食品の試料に、マグネシウムイオンおよびNADPHの存在下でアセトヒドロキシ酸レダクトイソメラーゼを作用させ、NADPの増加またはNADPHの減少を指標としてジアセチル前駆体を測定することを含むことを特徴とする。
【0034】
マグネシウムイオン、NADPHの存在下でアセトヒドロキシ酸レダクトイソメラーゼを作用させ、NADPの増加またはNADPHの減少を340nmの吸光度に基づいて測定してジアセチル前駆体(アセトヒドロキシ酸)を定量する方法は、以前から酵素学の分野で行われている方法であるが、易分解性であるために測定が難しいとされていた、飲食品中のジアセチル前駆体に適用し、測定することができることを証明したのは本発明者らが初めてである。
【0035】
飲食品は、ジアセチル、アセトインまたはそれらの同族体の存在を調べたい飲食品であれば、酒類、乳製品など、どんなものでもよく、例えば発酵物が挙げられるが、これに限定されない。具体的には、発酵酒、発酵乳製品、ビール類製造用発酵液、果汁発酵液、サワークリーム、カテージチーズ、ヨーグルト、発酵バター、焼酎用モロミ、漬物類、醤油、味噌、味醂、酢などが挙げられ、本発明の定量方法は種々の飲食品に適用できる。
【0036】
なお、飲食品の試料によって、試料を添加することによりpHが変化しないように測定に用いる緩衝液の緩衝能を高めたり、発酵飲食品試料中の菌体を除去したり、除タンパク質処理等を行うことは、必要に応じて行う。
【0037】
本発明の定量対象であるジアセチル前駆体(アセトヒドロキシ酸)は、生体内で分岐鎖アミノ酸(ロイシン、バリン、イソロイシン)合成経路上の中間体であり、その経路上でアセトヒドロキシ酸レダクトイソメラーゼが次の物質への変換を触媒する。
【0038】
アセトヒドロキシ酸レダクトイソメラーゼは、酵素番号ECクラス1.1.1.86に分類されるケトールアシッドレダクトイソメラーゼであり、植物ならびに細菌、酵母菌、糸状菌などの微生物においてよく特性付けされている酵素である。この酵素は、分岐鎖アミノ酸についての生合成経路の第二酵素であり、基質であるアセトヒドロキシ酸であるアセト乳酸とアセトヒドロキシ酪酸から、それぞれ、2,3−ジヒドロキシ−3−イソ吉草酸または2,3−ジヒドロキシ−3−メチル吉草酸への変換を触媒する。この反応は、マグネシウムイオン(Mg2+)の存在を必要とし、また、メチルまたはエチル基の転移、その後のNADPHによる還元という2段階で進行する。
【0039】
本発明に用いるアセトヒドロキシ酸レダクトイソメラーゼは、アセトヒドロキシ酸変換活性を有し、マグネシウムイオン存在下で、NADPHの酸化と共役するメチルまたはエチル基の転移を触媒する酵素系を提供できる生物に由来したあらゆるものを用いることができ、特に限定されない。例えば酵母、糸状菌などの真核微生物由来のものが好適に用いられる。
【0040】
本発明においては、好ましくはアスペルギルス属に属する微生物(例えばアスペルギルス・オリゼ、アスペルギルス・ニガー、アスペルギルス・ニデュランス、アスペルギルス・ジャポニカスなど)、より好ましくはアスペルギルス・オリゼ、さらに好ましくはアスペルギルス・オリゼRIB40株由来のアセトヒドロキシ酸レダクトイソメラーゼを用いることが好ましい。特に、配列番号2に示すアミノ酸配列を含むアセトヒドロキシ酸レダクトイソメラーゼ、または該アミノ酸配列において1もしくは数個のアミノ酸が欠失、置換もしくは付加されたアミノ酸配列を含み、かつアセトヒドロキシ酸レダクトイソメラーゼ活性を有するものが好ましい。配列番号2に示すアミノ酸配列をコードする核酸配列を配列番号1に示す。
【0041】
アセトヒドロキシ酸レダクトイソメラーゼは、自然環境から単離され、精製または不完全精製されたものでもよいし、上記微生物が自然生産する酵素を精製してもよく、また化学合成によっても製造できる。アセトヒドロキシ酸レダクトイソメラーゼの製造法は、当業者によく知られている。
【0042】
また、アセトヒドロキシ酸レダクトイソメラーゼは、例えば以下のように、遺伝子工学的手法により作製した形質転換株で過剰発現させ、製造することができる。
【0043】
アセトヒドロキシ酸レダクトイソメラーゼのアミノ酸配列または塩基配列から、プライマーを設計、合成する。アミノ酸配列、塩基配列はシークエンサー等で同定してもよいが、種々の文献に開示された配列情報を利用してもよい。
【0044】
例えば微生物から精製した染色体DNAを鋳型として、合成したプライマーを用いてポリメラーゼ連鎖反応(PCR)によりアセトヒドロキシ酸レダクトイソメラーゼをコードする遺伝子を増幅する。増幅した遺伝子のDNA断片を精製して適当な制限酵素で消化し、例えば電気泳動して対象の遺伝子のバンドを切り出してDNA断片を抽出する。
【0045】
一方、遺伝子組換えに用いるベクターも同様に制限酵素で切断し、上記DNA断片を挿入する。ベクターには、プロモーター配列のほか、ターミネーターや選択マーカー、リボソーム結合部位、複製開始点などを配置してもよい。
【0046】
好適なプロモーターとしては、例えばβ−ラクタマーゼプロモーター、ラクトースプロモーター、トリプトファンプロモーター、解糖系酵素(例えばα−アミラーゼ、プロテアーゼ、デヒドロゲナーゼなど)をコードする遺伝子類のプロモーター、具体的にはエノラーゼ、グリセルアルデヒド−3−ホスフェートデヒドロゲナーゼ、ヘキソキナーゼ、ピルベートデカルボキシラーゼ、ピルベートキナーゼなどに対するプロモーター、窒素代謝に関連する分解酵素などに対するプロモーター、SV40ウイルスのプロモーター、植物のプロモーター(例えばカリフラワーモザイクウイルス(CaMV)35Sプロモーター、ノパリン合成酵素遺伝子のプロモーター(Pnos)、トウモロコシ由来ユビキチンプロモーターなど)が挙げられる。
【0047】
形質転換可能な大腸菌を上記ベクターで形質転換し、培養後、プラスミドDNAを抽出する。プラスミドDNAを制限酵素で消化し精製して、例えば塩化カルシウム法、ポリエチレングリコール法、電気パルス法などで宿主細胞に形質転換し、形質転換株を得る。宿主は、細菌、酵母、真菌、昆虫細胞、植物細胞、動物細胞などを含む。
【0048】
以上のようにして作製された、アセトヒドロキシ酸レダクトイソメラーゼを高生産する形質転換株として、独立行政法人製品評価技術基盤機構特許微生物寄託センターに寄託された受領番号NITE AP−541の株が挙げられる。この株は、後述の実施例3で作製されたものである。
【0049】
形質転換株を培養し、宿主細胞または培地から、産生されたアセトヒドロキシ酸レダクトイソメラーゼを精製する。
【0050】
精製されたアセトヒドロキシ酸レダクトイソメラーゼは不安定であるため、例えば、グリセロール、硫酸アンモニウム、マグネシウムイオン、NADPHを適宜添加し、−20℃以下で保存することで、実用的に充分な保存安定性を与えることができる。
【0051】
本発明の飲食品中のジアセチル前駆体の定量法は、例えば以下の条件で行うことができる。
【0052】
本定量法では、反応系中の基質(ジアセチル前駆体)濃度が、例えば0mM〜0.1mM、好ましくは0mM〜0.01mMになるようにする。緩衝液は、例えば10mM〜200mMのTris−塩酸緩衝液、リン酸緩衝液、HEPES-水酸化ナトリウム緩衝液、TRICINE-水酸化ナトリウム緩衝液、ホウ酸緩衝液などが好ましく、pHは7.5〜10.5が好ましい。
【0053】
マグネシウムイオンとして、塩化マグネシウム、硫酸マグネシウムなどを、例えば2mM〜20mMになるよう添加する。また、NADPHは、例えば0.1mM〜0.5mMになるよう添加するが、NADPHの反応系中の濃度は、反応系に含まれる基質濃度を超える濃度にする。
【0054】
アセトヒドロキシ酸レダクトイソメラーゼは、例えば5〜20pkatal単位を添加する。ここでいう単位(katal)とは1秒間に1molの基質を変換する酵素量である。反応温度は約30〜42℃とし、反応時間は約10〜120分とし、NADPの増加またはNADPHの減少を340nmの吸光度でみる。吸光度は吸光度計で0分(反応前)に測定しておき、酵素添加後、上記適当な条件にて反応させた後、反応後の吸光度を測定し、0分時の吸光度からの減少値を算出してジアセチル前駆体を定量する。
【0055】
アセトヒドロキシ酸の定量において、酵素反応系は上述の限りではなく、適宜、反応時間、酵素添加量、基質添加量等を変更することができる。なお、ここでいう反応時間とは、反応系中のアセトヒドロキシ酸のすべてがアセトヒドロキシ酸レダクトイソメラーゼと接触するに要する時間である。
【0056】
本定量法では、飲食品の試料から、アセトヒドロキシ酸の測定を妨害する物質(例えば乳酸など)を除去してもよい。特にヨーグルトなどの発酵乳飲食品などに有効である。
【0057】
アセトヒドロキシ酸の分離は、弱塩基性陰イオン交換樹脂および水と任意の割合で混合するアルコール水溶液を用いて行うことが好ましい。アセトヒドロキシ酸は、中性から弱アルカリ性のアルコール水溶液中で弱塩基性陰イオン交換樹脂に吸着されるからである。弱塩基性陰イオン交換樹脂は、カラムに充填して用いてもよいし、試料の入っている容器に該樹脂を懸濁し、アセトヒドロキシ酸を樹脂に吸着させ、濾過や遠心分離等により樹脂を回収してもよい。また、アルコール水溶液として、例えば1価のアルコール水溶液を用いることが好ましい。1価のアルコール水溶液として、具体的にはメタノール、エタノール、1−プロパノール、2−プロパノール、tert-ブタノール水溶液が挙げられ、特に限定されないが、より好ましくはメタノール水溶液が用いられる。
【0058】
アセトヒドロキシ酸の分離を行うには、まず、飲食品の試料を好ましくはpH6〜10、より好ましくはpH7〜9に調整し、メタノール水溶液を加える。メタノールの濃度は、体積パーセント濃度で50〜100%が好ましく、75〜100%がより好ましい。試料とメタノール水溶液の混合液を遠心分離する。硬い固体試料の場合は遠心分離前にホモジナイザー等で十分にホモジナイズすることが好ましい。上澄みを弱塩基性陰イオン交換樹脂カラムにアプライし、メタノール水溶液をカラムに通液する。次に、好ましくは約0.05〜5.0M、より好ましくは約0.1〜1.0Mのアルカリ金属の水酸化物(例えば水酸化ナトリウム、水酸化リチウム、水酸化カリウムなど)、より好ましくは水酸化ナトリウムを含む溶出液を通液し、アセトヒドロキシ酸を溶出する。
【0059】
本発明によれば、飲食品中のジアセチル前駆体を定量するためのキットを作製することができる。キットは、アセトヒドロキシ酸レダクトイソメラーゼを含む。アセトヒドロキシ酸レダクトイソメラーゼは、配列番号2に示すアミノ酸配列を含むタンパク質、または該アミノ酸配列において1もしくは数個のアミノ酸が欠失、置換もしくは付加されたアミノ酸配列を含み、かつアセトヒドロキシ酸レダクトイソメラーゼ活性を有するタンパク質のものが好ましい。また、キットには、試料の前処理に用いられるもの(例えば弱塩基性陰イオン交換樹脂カラム、溶出液など)、アセトヒドロキシ酸定量に用いられるもの(例えば緩衝液、試料の遠心分離用チューブ、標準添加法に用いるアセト乳酸など)、取扱説明書などを含めてもよい。
【実施例】
【0060】
以下、実施例により本発明を具体的に説明するが、これらの実施例は本発明を限定するものではない。なお、本発明によるジアセチル前駆体の定量方法を、便宜的に「本酵素法」ということがある。
【0061】
実施例における各種遺伝子操作は、Current protocols in molecular biology(edited by F.M.Ausubel et al.,1987)に記載されている方法に従った。
【0062】
また、アセトヒドロキシ酸レダクトイソメラーゼの基質となるアセトヒドロキシ酸であるアセト乳酸とアセトヒドロキシ酪酸の合成方法は、アセト乳酸については、Krampitzの方法(L.O.Krampitz,Arch.Biochem.,17,81−85,1948)に従い、2−アセトキシ−2−メチルアセト酢酸エチル(Sigma−Aldrich社製)0.182g(0.9mmol)に対し水9ml、2N水酸化ナトリウム水溶液1mlを順次添加し、25℃で1時間攪拌して加水分解することで調製した。
【0063】
〔実施例1:アスペルギルス・オリゼ由来アセトヒドロキシ酸レダクトイソメラーゼの精製〕
1x106個胞子/mlとなるようにアスペルギルス・オリゼRIB40株胞子懸濁液をCzapek Dox培地(Nakajima,K.,et al.,Curr.Genet.,37,322−327,2000)に添加した。添加後30℃、48時間振盪培養した。菌糸体をNo.2濾紙(アドバンテック)にて濾別・回収した。回収した菌体に菌体重量の5倍量となる1mM PMSFを含む10mMトリス−塩酸緩衝液、pH8.0、2mM MgSO4、3mM 2−メルカプトエタノールを加え、フードプロセッサーにて破砕し、12000g、4℃、20分間遠心分離し、得られた上清を粗酵素液とした。粗酵素液に50%飽和となるように硫酸アンモニウムを加えた後、12000g、4℃、20分間遠心分離し、上清画分を得た。上清画分を10mMトリス−塩酸緩衝液、pH8.0、2mM MgSO4、3mM 2−メルカプトエタノール、50%飽和硫酸アンモニウムにて平衡化したOctyl−Cellulofine(生化学工業)に供し、吸着画分を50−0%飽和硫酸アンモニウムの直線濃度勾配により溶出させた。分画された溶液に対してArfinの方法に従ってアセトヒドロキシ酸レダクトイソメラーゼ活性を測定し、活性画分を追跡した。具体的な活性測定方法は後記実施例6に示す。得られた活性画分に対して、50%飽和硫酸アンモニウムとなるように硫酸アンモニウムを添加し、再度10mMトリス−塩酸緩衝液、pH8.0、2mM MgSO4、3mM 2−メルカプトエタノール、50%飽和硫酸アンモニウムにて平衡化したOctyl−Cellulofine(生化学工業)に供し、吸着画分を50−0%飽和硫酸アンモニウムの直線濃度勾配により溶出させた。得られた活性画分を10mMリン酸緩衝液、pH6.8、2mM MgSO4、3mM 2−メルカプトエタノール、5mM硫酸アンモニウムに対して透析し、12000g、4℃、20分間遠心分離し、上清画分を得た。上清画分を10mMリン酸緩衝液、pH6.8、2mM MgSO4、3mM 2−メルカプトエタノール、5mM 硫酸アンモニウムにて平衡化したDEAE−Sephadex A50(GE Healthcare)に供し、吸着画分を0−0.15M NaClの直線濃度勾配にて溶出させた。得られた活性画分を10mMリン酸緩衝液、pH6.8、2mM MgSO4、3mM 2−メルカプトエタノール、5mM 硫酸アンモニウムに対して透析し、10mMリン酸緩衝液、pH6.8、2mM MgSO4、3mM 2−メルカプトエタノール、5mM硫酸アンモニウムにて平衡化したGIGAPITE(生化学工業)に供し、吸着画分を10−300mMのリン酸直線濃度勾配にて溶出させた。得られた活性画分をSDS−PAGE(Laemmli,U.K.,Nature,227,680−685,1970)に供し電気泳動的に均一であることが確認されたため、ここで得られた活性画分をアスペルギルス・オリゼ由来アセトヒドロキシ酸レダクトイソメラーゼ精製標品とした。
【0064】
〔実施例2:アスペルギルス・オリゼ由来アセトヒドロキシ酸レダクトイソメラーゼのアミノ末端アミノ酸配列の決定〕
上記で得られた精製酵素溶液500μlに対して、250μlの100%(w/v)冷トリクロロ酢酸を加え、よく混合した後、氷中に20分間以上放置した。サンプルを15000xg、4℃、20分間遠心分離し、上清を完全に除いた後、沈殿を15μlのSDS化溶液(5%(w/v)SDS、5%(v/v)2−メルカプトエタノール、0.12M Tris−塩酸緩衝液(pH8.8)、0.05%(w/v)ブロモフェノールブルー、10%(w/v)グリセロール)に溶解させた後、沸騰湯浴中に5分間放置し、SDS化を行い、これをサンプル溶液とした。サンプル溶液をSDS−PAGEに供し、泳動後ゲル内のタンパク質をPVDF膜に転写した。PVDF膜上より目的のバンドを切り抜き、そのままアミノ末端アミノ酸配列の解析を行った。アミノ末端アミノ酸配列を解析した結果、アミノ末端より「VKNISFAGHE」であった。得られたアミノ酸配列をアスペルギルス・オリゼの全ゲノム配列情報(特開2005−176602号公報)に照らし合わしたところ、予想されたアスペルギルス・オリゼ由来のアセトヒドロキシ酸レダクトイソメラーゼアミノ酸配列(配列番号2)の、アミノ末端より数えて52番目より始まる「VKNISFAGHE」と一致した。
【0065】
〔実施例3:アスペルギルス・オリゼ由来アセトヒドロキシ酸レダクトイソメラーゼのアスペルギルス・オリゼを宿主とした過剰発現系の構築〕
次に、実施例2で求めたアスペルギルス・オリゼ由来アセトヒドロキシ酸レダクトイソメラーゼのアミノ末端アミノ酸配列情報と、アスペルギルス・オリゼの全ゲノム配列情報(特開2005−176602号公報)を基に、オリゴヌクレオチド5’−CTCCCGTCCATATGGTCAAGAAC−3’(配列番号3)と5’−CTTTTTCGCATGCGGATTTGGGAAAAAG−3’(配列番号4)を合成した。この1組のプライマーセットを用い、アスペルギルス・オリゼRIB40株の染色体DNAを鋳型としてPCRを行い、アスペルギルス・オリゼ由来アセトヒドロキシ酸レダクトイソメラーゼをコードする遺伝子を増幅した。アスペルギルス・オリゼ染色体DNA溶液(50μg/ml)は公知の方法に従って調製した。PCR用装置はPeltier Thermal Cycler PTC−200(MJ RESEARCH)を用いた。染色体DNA溶液は1μl用い、ポリメラーゼはPlatinum Taq DNA Polymerase(Invitrogen)を用いた。増幅反応は94℃、3分間鋳型DNAを変性し、94℃、45秒間、56℃、30秒間、72℃、1分10秒間保持するサイクルを30サイクル行った後、72℃、10分間で完全伸長させ、4℃で保持した。得られたPCR増幅断片をアガロースゲル電気泳動に供したところ、およそ1094塩基対からなる断片の増幅が確認された。
【0066】
得られたPCR増幅断片をNde I(NEB)、Sph I(NEB)で消化し、アガロースゲル電気泳動に供しエチジウムブロマイドで染色後、UV照射下で1068塩基対からなる断片を切り出した。ゲル中よりWizard SV Gel and PCR Clean−Up System(Promega)を用いてDNAを抽出し、これを挿入DNA断片とした。次に塩基配列中にエノラーゼのプロモーター配列(P−enoA142)を有するプラスミドpNEN142 DNA5μgをプロモーター配列直後にある制限酵素Nde I、Sph I認識配列の位置でNde I(NEB)、Sph I(NEB)で消化し、アガロースゲル電気泳動に供しエチジウムブロマイドで染色後、UV照射下で9296塩基対からなる断片を切り出した。ゲル中よりWizard SV Gel and PCR Clean−Up System(Promega)を用いてDNAを抽出し、これをベクターDNA断片とした。次にベクターDNAと挿入DNA断片をT4 DNA ligase(宝酒造)により連結させ、連結DNA溶液を得た。この連結DNA溶液を用い情報により形質転換可能な大腸菌DH5α(宝酒造)を形質転換し、その形質転換大腸菌より定法によりプラスミドDNAを抽出し、アスペルギルス・オリゼ形質転換用プラスミドDNA(pNEN−AoilvC−delation)を得た。このプラスミドDNA10μgをBamH I(NEB)で消化し、定法によりフェノール抽出、エタノール沈殿処理を行った後、10μlのTE緩衝液に溶解し形質転換用DNA溶液とした。アスペルギルス・オリゼniaD300株(RIB40株由来の硝酸還元酵素遺伝子(niaD)欠損変異株)を形質転換用DNA溶液を用いて形質転換した。形質転換法は、プロトプラスト化した後ポリエチレングリコール及び塩化カルシウムを用いる方法(Mol.Gen.Genet,218,99−104,1989)によって行い、アスペルギルス・オリゼ形質転換株(受領番号NITE AP−541)を得た。
【0067】
〔実施例4:アスペルギルス・オリゼを宿主としたアスペルギルス・オリゼ由来アセトヒドロキシ酸レダクトイソメラーゼ過剰発現系からの酵素の精製〕
実施例3で構築し得られたアスペルギルス・オリゼ形質転換株胞子懸濁液を、1x106個胞子/mlとなるようにYPM培地(2%peptone、1%Yeast extract、2%maltose)に添加した。添加後30℃、24時間振盪培養した。菌糸体をNo.2濾紙(アドバンテック)にて濾別・回収した。以後の精製過程は実施例1と同様の方法で行った。
【0068】
〔実施例5:アスペルギルス・オリゼ由来アセトヒドロキシ酸レダクトイソメラーゼの保存〕
実施例1及び実施例4で得られたアスペルギルス・オリゼ由来アセトヒドロキシ酸レダクトイソメラーゼ精製標品の保存方法を検討した。精製酵素標品を含む溶液を10mMリン酸緩衝液、pH6.8、20mM MgSO4、30mM 2−メルカプトエタノール、5mM硫酸アンモニウムに対して透析し、さらに透析後の溶液に対して等量の100%(v/v)グリセロールを添加し、−20℃にて保存した。本条件で保存することでアセトヒドロキシ酸レダクトイソメラーゼの活性は少なくとも1年間は維持されることを確認した。
【0069】
〔実施例6:アセトヒドロキシ酸定量法へのアセトヒドロキシ酸レダクトイソメラーゼの有効性の検証〕
実施例1及び実施例4で得られたアスペルギルス・オリゼ由来アセトヒドロキシ酸レダクトイソメラーゼ精製標品を用い、アセトヒドロキシ酸の分析が可能かどうかを調べた。
【0070】
具体的にはArfinの方法に従い、830μlの50mM Tris−塩酸緩衝液(pH9.0)、10μlの10mM NADPH、10μlの1M MgSO4、50μlのアスペルギルス・オリゼ由来アセトヒドロキシ酸レダクトイソメラーゼ精製酵素溶液に、100μlの各種濃度(0.1、0.05、0.02、0.01mM)のアセト乳酸溶液を添加し、37℃で10分間保温し、その間の340nmの吸光度減少値を測定し、アセト乳酸濃度との相関性を調べた。
【0071】
結果は、図1に示すごとく、反応系中のアセト乳酸濃度と吸光度の減少値とは比例関係を示し、本酵素法でのアセト乳酸の定量は可能と判断された。
【0072】
〔実施例7:アセトヒドロキシ酸レダクトイソメラーゼの基質特異性〕
飲食品中のアセトヒドロキシ酸と共存し、多くのアセト乳酸定量分析法を妨害することの多い、ジアセチル、アセトインが本酵素法による定量にどう影響するかについて調べた。
【0073】
具体的には、実施例6における酵素反応系のアセト乳酸区分に、ジアセチルおよびアセトインを表1に示した濃度となるように添加し、37℃で10分間保温し、その間の340nmの吸光度の減少値を測定した。なお、ジアセチルは和光純薬工業製のものを、アセトインは東京化成工業製のものをエーテルで洗浄して用いた。
【0074】
結果は表1に示すごとく、ジアセチルあるいはアセトインを添加した場合にあっても、アセト乳酸の吸光度減少値に実質的な差異は見られなかった。このことから、本酵素法では試料中に存在するジアセチルやアセトインに関係なく、アセトヒドロキシ酸のみを定量することが出来ることが示された。
【0075】
【表1】
【0076】
〔実施例8:清酒醪中に含まれるアセトヒドロキシ酸の定量〕
アセトヒドロキシ酸は、発酵飲食品製造の場合、主として製造工程中使用微生物により生成された後、製造工程中、あるいは、製品化後に自動的に分解して、ジアセチルを代表とするビシナルジケトン類(ジアセチルとその同族体、2,3−ペンタンジオンの総称)となる。それらは、特徴的な強い臭気を有し、酒類などの発酵飲食品の品質に対して大きな影響を及ぼしている。しかし、両者の分別定量が困難なことについては、前述した通りである。そこで、酒類の代表である清酒の、製造工程中の半製品である清酒醪について、本酵素法によりアセトヒドロキシ酸の定量が可能であるかどうか調べることにした。
【0077】
清酒醸造中の、留添(とめぞえ)後2日目の清酒醪を対象として、直ちにpH7に調整して低温下に保持して実験室に持ち帰り、遠心分離を行ってその上清を採り、実施例6と同一の酵素反応系を用いて、標準添加法により定量分析を行った。具体的には、試料中のアセト乳酸濃度がそれぞれ一定量(0.1、0.05、0.02mM)増加するようにアセト乳酸を添加した上清試料100μlを用い、実施例6と同様にして定量した。結果は図2に示すように、醪中のアセトヒドロキシ酸濃度は0.05mMと算定された。
【0078】
この結果より、清酒醪中のアセトヒドロキシ酸は、従来用いられてきた煩雑で、不正確な蒸留比色法によらずとも、本酵素法により、簡便、正確に定量できることがわかった。
【0079】
〔実施例9:発酵乳製品中に含まれるアセトヒドロキシ酸の定量〕
代表的な発酵乳製品であるヨーグルトの乳清中のアセトヒドロキシ酸の定量が可能であるかどうか調べた。
【0080】
当実験室で調製したヨーグルト乳清を水酸化ナトリウム水溶液でpH7に中和したのち、遠心式フィルターユニット(Centriprep YM−3 Millipore社製)で濾過、除タンパクし、得られた溶液について、実施例8と同様の方法で定量分析を行った。その結果、10分後の吸光度について吸光度の減少が認められなかった。従って何らかの物質が測定を妨害しており、前処理をしなければ測定できないと考えられた。
【0081】
〔実施例10:アセトヒドロキシ酸測定における乳酸の阻害について〕
乳清中に含まれる成分の中で、ヨーグルトの乳清中に多く含まれ、測定を妨害する可能性の高い乳酸の阻害について調べた。
【0082】
具体的には、実施例6における酵素反応系に、アセト乳酸を0.1mM、水酸化ナトリウムでpH7まで中和した乳酸を各種濃度(乳酸として0〜8.1g/l)で含む溶液を添加し、37℃10分間保温し、その間の340nmの吸光度の減少値を測定した。なお、乳酸は和光純薬工業製のものを用いた。
【0083】
結果は図3に示すごとく、乳酸を添加した場合、乳酸なしの場合の活性を1.0とすると乳酸8.1g/lの濃度では0.44となった。従って、明らかに本酵素反応は、高濃度の乳酸の存在下で阻害を受け、乳酸を10g/l以上含む一般のヨーグルトでは、測定感度を向上させるために乳酸の除去が必要になると考えられた。
【0084】
〔実施例11:乳酸溶液中の乳酸の除去〕
乳清試料中の乳酸を除去するに際し、乳酸水溶液中のアセトヒドロキシ酸を除去する手法を検討した。
【0085】
検討の結果、メタノール中では、乳酸は弱塩基性陰イオン交換樹脂に殆ど吸着されず、逆にアセトヒドロキシ酸は弱塩基性陰イオン交換樹脂に吸着されることを見いだし、この原理を用いて乳酸溶液からアセトヒドロキシ酸の分離が可能であることが確認された。
【0086】
具体的には、pH9に中和した乳酸水溶液(1.0%)25mlに、メタノール(特級:和光純薬製)を75ml添加し、一定濃度(0.2mM)になるようにアセト乳酸を添加した溶液について、弱塩基性陰イオン交換樹脂ダウエックスSD−2(ダウ・ケミカル製)を充填したポリプロピレン製カラム(ムロマックミニカラムL:室町ケミカル製)に、温度5℃のもとで、高速液体クロマトグラフ用ポンプを用い、1.0ml/分の流速で上記溶液を通液した。次いで、アンモニアを0.01%含むメタノールを50ml、次いでメタノールを50ml、それぞれ1.0ml/分の流速で通液した。最後に、35℃に加温した、メタノール+水=1+1(体積比)に1.0M濃度で水酸化ナトリウムを含む溶液30mlを1.0ml/分の速度で通液し、アセトヒドロキシ酸を溶出させた。最後に、溶出液をpH9になるまで中和し、全体を水で50mlに定容した。本法による乳酸の除去率は約70%で、アセト乳酸の回収率は約93%であった。なお、元の乳酸水溶液は25mlで、最終的に定容されるのは50mlであるので、元の乳酸水溶液中のアセト乳酸濃度は、2倍に希釈されることになる。
【0087】
〔実施例12:ヨーグルト乳清中に含まれるアセトヒドロキシ酸の定量〕
実施例11の結果をもとに、ヨーグルト乳清中のアセトヒドロキシ酸の定量を試みた。
【0088】
あらかじめ当実験室で調製したヨーグルト乳清(乳酸1.0%含む)25mlをpH9に中和したのち、メタノールを75ml加え、8000rpmで10分間遠心分離をした。上澄みを実施例11と同様の方法で処理し乳酸の除去を行い、得られた試料溶液を、孔径0.45μmのシリンジフィルター(DISMIC−25AS:アドバンテック東洋製)でろ過したのち、実施例8と同様の方法で定量分析を行った。結果は図4に示すように、反応に用いた溶液中のアセトヒドロキシ酸濃度は0.02mMと算定され、元の乳清中のアセトヒドロキシ酸濃度については、0.02×2(希釈倍率)より0.04mMと推定される。
【産業上の利用可能性】
【0089】
本発明によれば、飲食品中のジアセチル前駆体を直接的かつ簡易・正確に定量でき、飲食品製造においての品質管理等に応用可能性がある。
【図面の簡単な説明】
【0090】
【図1】反応系中のアセト乳酸濃度とNADPHの吸光度減少値との関係を示す。
【図2】標準添加法による清酒醪中のアセトヒドロキシ酸の分析を示す。
【図3】乳酸を各種濃度で含むアセト乳酸溶液(0.1mM)を試料として用いた際の本酵素反応の阻害を示す。
【図4】標準添加法によるヨーグルト乳清中のアセトヒドロキシ酸の分析を示す。
【特許請求の範囲】
【請求項1】
飲食品中のジアセチル前駆体を定量する方法であって、飲食品の試料に、マグネシウムイオンおよび還元型ニコチンアミドアデニンジヌクレオチドリン酸(NADPH)の存在下でアセトヒドロキシ酸レダクトイソメラーゼを作用させ、酸化型ニコチンアミドアデニンジヌクレオチドリン酸(NADP)の増加またはNADPHの減少を指標としてジアセチル前駆体を測定することを含む、前記方法。
【請求項2】
飲食品が発酵物である、請求項1に記載の方法。
【請求項3】
飲食品が発酵酒または発酵乳製品である、請求項1または2に記載の方法。
【請求項4】
飲食品が、清酒醪、ビール類製造用発酵液、果汁発酵液、サワークリーム、カテージチーズ、ヨーグルト、発酵バター、スターターディスティレート、焼酎用モロミ、漬物類、醤油、味噌、味醂または酢である、請求項1〜3のいずれか1項に記載の方法。
【請求項5】
アセトヒドロキシ酸レダクトイソメラーゼが、真核微生物由来の酵素である、請求項1〜4のいずれか1項に記載の方法。
【請求項6】
アセトヒドロキシ酸レダクトイソメラーゼが、アスペルギルス属由来の酵素である、請求項1〜5のいずれか1項に記載の方法。
【請求項7】
アセトヒドロキシ酸レダクトイソメラーゼが、アスペルギルス・オリゼ由来の酵素である、請求項1〜6のいずれか1項に記載の方法。
【請求項8】
アスペルギルス・オリゼが、アスペルギルス・オリゼRIB40株(FERM P−18273)である、請求項7に記載の方法。
【請求項9】
アセトヒドロキシ酸レダクトイソメラーゼが、配列番号2に示すアミノ酸配列を含むタンパク質、または該アミノ酸配列において1もしくは数個のアミノ酸が欠失、置換もしくは付加されたアミノ酸配列を含み、かつアセトヒドロキシ酸レダクトイソメラーゼ活性を有するタンパク質である、請求項1〜8のいずれか1項に記載の方法。
【請求項10】
NADPの増加またはNADPHの減少を、340nmの吸光度に基づいて測定する、請求項1〜9のいずれか1項に記載の方法。
【請求項11】
飲食品の試料から乳酸を除去することをさらに含む、請求項1〜10のいずれか1項に記載の方法。
【請求項12】
乳酸の除去を、弱塩基性陰イオン交換樹脂を用いて行う、請求項11に記載の方法。
【請求項13】
アセトヒドロキシ酸レダクトイソメラーゼを含む、飲食品中のジアセチル前駆体を定量するためのキット。
【請求項14】
定量が請求項1〜12のいずれかに記載の方法によって行われるものである、請求項13に記載のキット。
【請求項15】
アセトヒドロキシ酸レダクトイソメラーゼが、配列番号2に示すアミノ酸配列を含むタンパク質、または該アミノ酸配列において1もしくは数個のアミノ酸が欠失、置換もしくは付加されたアミノ酸配列を含み、かつアセトヒドロキシ酸レダクトイソメラーゼ活性を有するタンパク質である、請求項13または14に記載のキット。
【請求項16】
飲食品の試料から乳酸を除去するための弱塩基性陰イオン交換樹脂カラムをさらに含む、請求項13〜15のいずれか1項に記載のキット。
【請求項1】
飲食品中のジアセチル前駆体を定量する方法であって、飲食品の試料に、マグネシウムイオンおよび還元型ニコチンアミドアデニンジヌクレオチドリン酸(NADPH)の存在下でアセトヒドロキシ酸レダクトイソメラーゼを作用させ、酸化型ニコチンアミドアデニンジヌクレオチドリン酸(NADP)の増加またはNADPHの減少を指標としてジアセチル前駆体を測定することを含む、前記方法。
【請求項2】
飲食品が発酵物である、請求項1に記載の方法。
【請求項3】
飲食品が発酵酒または発酵乳製品である、請求項1または2に記載の方法。
【請求項4】
飲食品が、清酒醪、ビール類製造用発酵液、果汁発酵液、サワークリーム、カテージチーズ、ヨーグルト、発酵バター、スターターディスティレート、焼酎用モロミ、漬物類、醤油、味噌、味醂または酢である、請求項1〜3のいずれか1項に記載の方法。
【請求項5】
アセトヒドロキシ酸レダクトイソメラーゼが、真核微生物由来の酵素である、請求項1〜4のいずれか1項に記載の方法。
【請求項6】
アセトヒドロキシ酸レダクトイソメラーゼが、アスペルギルス属由来の酵素である、請求項1〜5のいずれか1項に記載の方法。
【請求項7】
アセトヒドロキシ酸レダクトイソメラーゼが、アスペルギルス・オリゼ由来の酵素である、請求項1〜6のいずれか1項に記載の方法。
【請求項8】
アスペルギルス・オリゼが、アスペルギルス・オリゼRIB40株(FERM P−18273)である、請求項7に記載の方法。
【請求項9】
アセトヒドロキシ酸レダクトイソメラーゼが、配列番号2に示すアミノ酸配列を含むタンパク質、または該アミノ酸配列において1もしくは数個のアミノ酸が欠失、置換もしくは付加されたアミノ酸配列を含み、かつアセトヒドロキシ酸レダクトイソメラーゼ活性を有するタンパク質である、請求項1〜8のいずれか1項に記載の方法。
【請求項10】
NADPの増加またはNADPHの減少を、340nmの吸光度に基づいて測定する、請求項1〜9のいずれか1項に記載の方法。
【請求項11】
飲食品の試料から乳酸を除去することをさらに含む、請求項1〜10のいずれか1項に記載の方法。
【請求項12】
乳酸の除去を、弱塩基性陰イオン交換樹脂を用いて行う、請求項11に記載の方法。
【請求項13】
アセトヒドロキシ酸レダクトイソメラーゼを含む、飲食品中のジアセチル前駆体を定量するためのキット。
【請求項14】
定量が請求項1〜12のいずれかに記載の方法によって行われるものである、請求項13に記載のキット。
【請求項15】
アセトヒドロキシ酸レダクトイソメラーゼが、配列番号2に示すアミノ酸配列を含むタンパク質、または該アミノ酸配列において1もしくは数個のアミノ酸が欠失、置換もしくは付加されたアミノ酸配列を含み、かつアセトヒドロキシ酸レダクトイソメラーゼ活性を有するタンパク質である、請求項13または14に記載のキット。
【請求項16】
飲食品の試料から乳酸を除去するための弱塩基性陰イオン交換樹脂カラムをさらに含む、請求項13〜15のいずれか1項に記載のキット。
【図1】

【図2】
【図3】
【図4】
【図2】
【図3】
【図4】
【公開番号】特開2008−263977(P2008−263977A)
【公開日】平成20年11月6日(2008.11.6)
【国際特許分類】
【出願番号】特願2008−84562(P2008−84562)
【出願日】平成20年3月27日(2008.3.27)
【出願人】(591014710)千葉県 (49)
【Fターム(参考)】
【公開日】平成20年11月6日(2008.11.6)
【国際特許分類】
【出願日】平成20年3月27日(2008.3.27)
【出願人】(591014710)千葉県 (49)
【Fターム(参考)】
[ Back to top ]