
骨髄腫骨疾患の分子決定因子およびその用途
【課題】多発性骨髄腫における溶解性骨疾患の治療・予防のための組成物の提供。
【解決手段】Dickkopf−1(DKK1)(登録番号NM012242)のヒトホモログに対するアンチセンスオリゴヌクレオチド、抗DKK1抗体、又はDKK1と相互作用する可溶性LRP受容体、を含む、多発性骨髄腫患者における溶解性骨疾患を治療するための、あるいは、患者における、骨への乳がん転位又は骨への前立腺がん転移によって引き起こされる、悪性腫瘍関連骨減少を調節するための医薬組成物。
【解決手段】Dickkopf−1(DKK1)(登録番号NM012242)のヒトホモログに対するアンチセンスオリゴヌクレオチド、抗DKK1抗体、又はDKK1と相互作用する可溶性LRP受容体、を含む、多発性骨髄腫患者における溶解性骨疾患を治療するための、あるいは、患者における、骨への乳がん転位又は骨への前立腺がん転移によって引き起こされる、悪性腫瘍関連骨減少を調節するための医薬組成物。
【発明の詳細な説明】
【技術分野】
【0001】
国庫補助の説明
本発明は、部分的に、米国立がん研究所助成金CA55819およびCA97513の下に連邦政府からの助成金を用いて開発された。このため、米国政府は本発明に一定の権利を有する。
【0002】
関連出願へのクロスリファレンス
本出願は、2002年12月5日出願で現在放棄されている米国特許仮出願第60/431,040号の利益を主張する。
【背景技術】
【0003】
本発明は一般的に多発性骨髄腫の研究に関する。より詳細には、本発明は全遺伝子発現プロファイリング比較による骨髄腫骨疾患の分子決定因子の同定に関する。
【0004】
関連技術
多発性骨髄腫(MM)は、米国で年間約15,000人が罹患する、最終分化した形質細胞(PC)の稀でさらに不治の悪性腫瘍であって、二番目に多い造血器悪性腫瘍に当たる。多発性骨髄腫は、白人人口においてリンパ性悪性腫瘍全体の13%に相当し、および黒人人口においてリンパ性悪性腫瘍の31%に相当する。悪性形質細胞は、骨髄へホーミングしおよび骨髄で増殖し、正常造血の消失による貧血および免疫抑制を引き起こす。
【0005】
多発性骨髄腫はまた、全身性骨粗鬆症および局所性骨破壊を伴い、消耗性骨痛および、骨折、脊髄圧迫および高カルシウム血症への感受性に繋がる。骨髄腫は、溶解性骨疾患を常に伴う唯一の血液悪性腫瘍であって、および局所性骨破壊は形質細胞に隣接する部分に限られており、悪性形質細胞が破骨細胞機能および/または骨芽細胞アネルギーを促進する因子を分泌することを示唆する。骨疾患の有病率はしばしば骨の併発を伴わないくすぶり型骨髄腫から、孤立性形質細胞腫、骨塩密度の全身性損失または限局性溶解性骨病変が患者の約80%にみられるびまん性または限局性多発性骨髄腫まで、骨髄腫の症状によって異なる。
【0006】
近年、溶解性骨疾患は、骨髄腫の結果であるだけでなく、疾患進行を促進することに複雑に関与していることが明らかになっている。骨代謝回転速度における変化は、意義不明単クローン性免疫グロブリン血症(MGUS)からの顕性骨髄腫への最大3年までの臨床的進行を予測する。最初は破骨細胞と骨芽細胞活性とは連動している一方、その連動は疾患進行に伴って失われる。破骨細胞活性は上昇したままでありおよび骨芽細胞活性は低下し、結果として溶解性骨疾患を伴う。5T2マウス骨髄腫および原発性ヒト骨髄腫のSCID−huモデルでの研究は、破骨細胞活性の阻害は骨髄腫増殖の阻害および骨髄腫腫瘍組織量の減少を伴うことを実証した。これらの研究は、ビスホスホネートを用いた骨吸収の阻害は抗骨髄腫作用を有したという報告を支持する。
【0007】
骨髄腫関連溶解性骨疾患における破骨細胞の生物学は集中的に調べられている一方、骨芽細胞活性における疾患関連変化およびその基礎となる機構についてはほとんど知られていない。骨髄腫において、間葉幹細胞が造骨系譜へ分化する能力が損なわれていると示唆されている。しかし、そのような障害を担う機構は解明されていない。
【0008】
正常な健常ドナー由来の骨髄形質細胞および新たに診断された多発性骨髄腫由来の悪性骨髄形質細胞の全遺伝子発現プロファイリング(GEP)比較が、多発性骨髄腫の悪性形質転換に関与する疾患遺伝子候補および妨害された経路を同定するための強力な方法に当たることが示されている(非特許文献1)。
【先行技術文献】
【非特許文献】
【0009】
【非特許文献1】Zhan et al.,Global gene expressi on profiling of multiple myeloma,monoclo nal gammopathy of undetermined significa nce,and normal bone marrow plasma cells. Blood 99: 1745−1757 (2002)。
【発明の概要】
【発明が解決しようとする課題】
【0010】
先行技術は、多発性骨髄腫骨疾患の原因となり得る悪性形質細胞で発現されている遺伝子を同定するための比較分析、および、多発性骨髄腫骨疾患を診断および治療するための方法に欠けている。本発明は本分野におけるこの年来の必要及び要望を満たす。
【課題を解決するための手段】
【0011】
溶解性骨疾患の分子決定因子を同定するため、溶解性病変の放射線的証拠を示さない(n=28)新たに診断された多発性骨髄腫(newly diagnosed multiple myeloma) に由来するCD138濃縮形質細胞における最大12,000個の遺伝子の発現プロファイルを、3個以上の溶解性病変を有するもの(n=47)と比較した。骨芽細胞分化におけるWNTシグナル伝達の決定的な役割と合致して、2種類の分泌型WNTシグナル伝達拮抗因子である、可溶性frizzled関連タンパク質3(SFRP−3/FRZB)およびDickkopf−1(DKK−1)のヒトホモログが、溶解性骨病変を有する47例中40例で発現されていたが、骨病変を欠く28例中の16例だけで発現されていた(P<0.05)。免疫組織化学は、遺伝子発現が高い例に由来する形質細胞で、高レベルのDKK−1およびFRZBを示した。重要なことに、DKK−1およびFRZBは、正常骨髄ドナー45例、または、骨疾患を欠く関連する形質細胞悪性腫瘍であるワルデンシュトレーム型マクログロブリン血症10例に由来する形質細胞では発現されていなかった。
【0012】
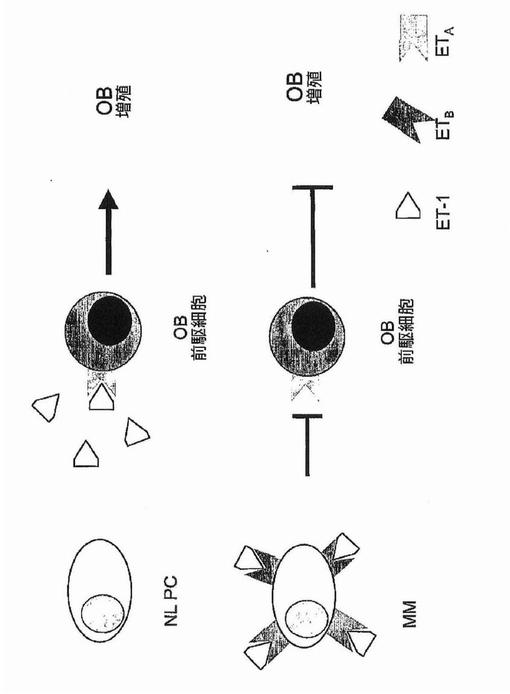
高いDKK−1を有する多発性骨髄腫患者に由来した血清は、Wntシグナル伝達および骨芽細胞分化の両方をin vitroで遮断した。重要なことに、DKK−1およびFRZB抗体との血清のプレインキュベートは、この機能を阻害した。DKK−1発現の調節および次にはアポトーシスにおけるJUNの主要な役割と合致して、髄外疾患および原発性難治性疾患に由来する形質細胞は、JUNおよびDKK−1の低発現を有した。
【0013】
多発性骨髄腫形質細胞は、in vivo処理後にDKK−1およびFRZB遺伝子発現の大幅なアップレギュレーションを示した。DKK−1およびFRZBは、多発性骨髄腫形質細胞において、その疾患を治療するために用いられる遺伝毒性薬での患者の治療後に、アップレギュレートすることができ、そのようにして多発性骨髄腫細胞アポトーシスにおけるDKK−1の役割を助長する。in vitro由来破骨細胞(OC)と共培養した原発性多発性骨髄腫細胞はアポトーシスを欠き、しかもこれはJUN、FOS、FOSB、およびDKK−1のダウンレギュレーションと密接に相関した。
【0014】
本発明で開示される結果は、DKK−1およびFRZBの産生および/または分泌を遮断することが多発性骨髄腫患者における骨減少を防ぐかまたは逆転させる可能性があることを示す。他の用途は、DKK−1およびFRZB阻害因子を一般人口における骨減少を防ぐために用いることを含む。加えて、Wntシグナル伝達は近年、造血幹細胞の自己再生能力にとって決定的に重要であることが示されている。さらに、HSC増殖に必要な骨髄ニッチは、成熟骨芽細胞によって形成される。Wntシグナル伝達に対するDKK1およびFRZBによる遮断は、直接的におよび間接的に肝星細胞(HSC)増殖を障害し、およびそのようにして、多発性骨髄腫で見られる免疫抑制および貧血の部分的な原因となり得る。このように、DKK1および/またはFRZBの遮断はまた、骨髄腫を有する患者の大部分に見られる造血の欠陥を、防ぐかまたは逆転することができる。
【0015】
本発明のその他の態様、特性、および利点は、本発明の現在好ましい実施形態の下記の説明から明らかになる。これらの実施形態は開示の目的のために与えられる。
【0016】
本発明の上述した特性、利点、および目的、およびその他明らかになる内容が達成されおよび詳細に理解可能となるように、上記に簡潔に要約した、より具体的な説明および本発明の一部の実施形態が、添付の図面に図解されている。これらの図面は本明細書の一部を成す。しかし、添付の図面は本発明の好ましい実施形態を図解し、およびしたがって実施形態の範囲を制限しないと考えられることに注意する。
【図面の簡単な説明】
【0017】
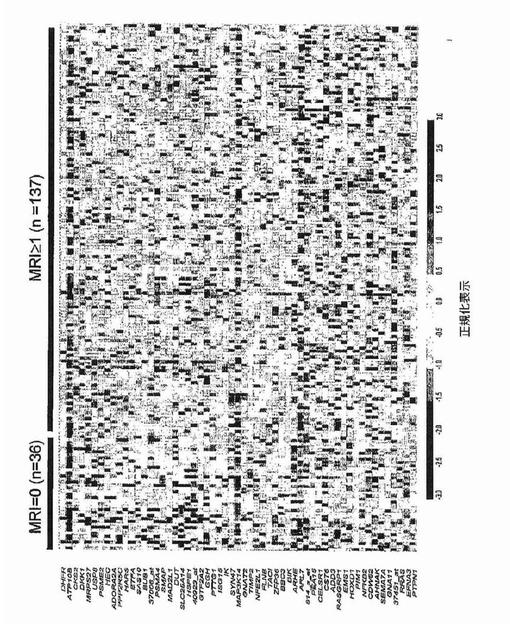
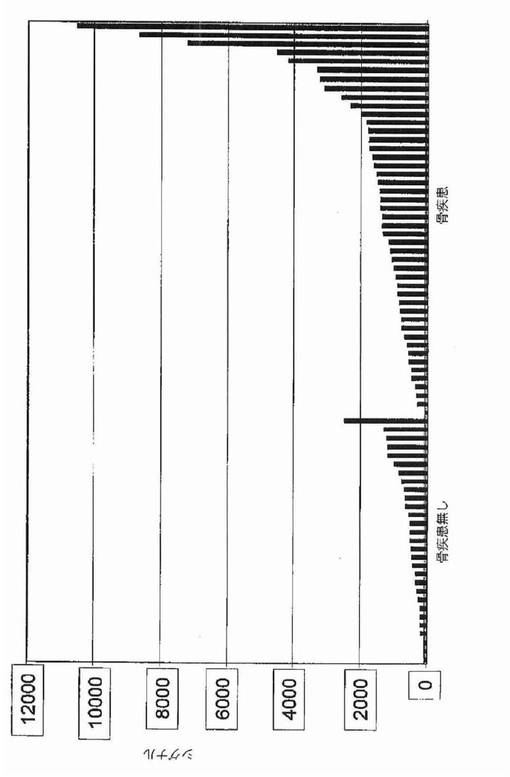
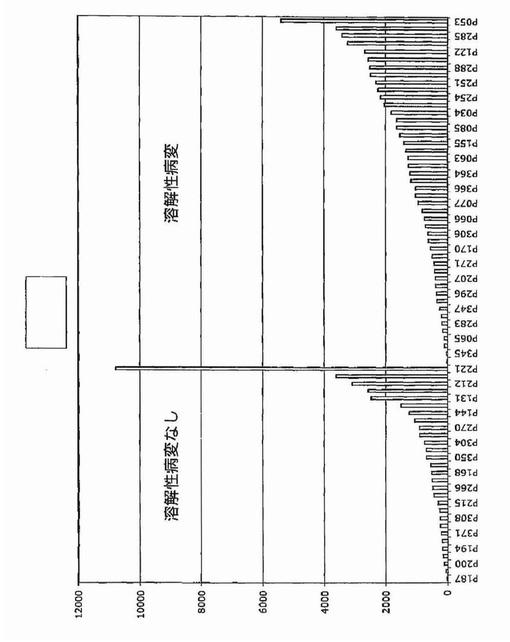
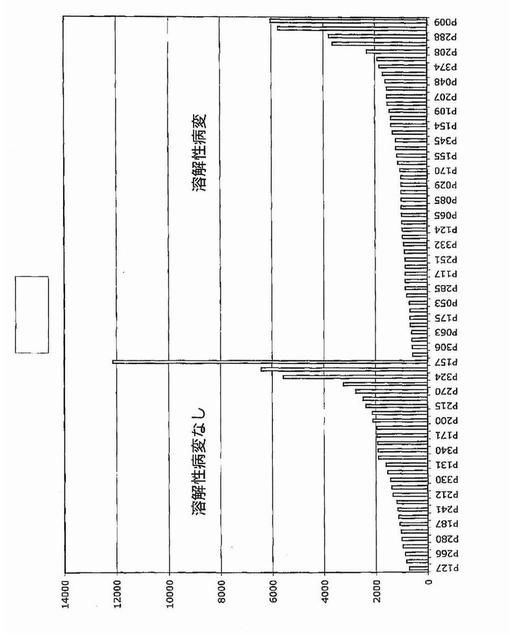
【図1A】図1Aおよび1Bは、骨髄腫における骨病変を反映する全遺伝子発現パターンを示す。図1Aは、MRI限局性病変が0個(n=36)および1個以上(n=137)の患者に由来する悪性形質細胞において有意に差がある発現がされているとロジスティック回帰分析によって同定された(P<0.0001)57遺伝子の正規化発現レベルのクラスタビューを示す。1個以上のMRI病変がある患者に由来する形質細胞における発現上昇を示している28遺伝子は、有意性の階層に基づいて上から下へ並べられている。同様にMRI病変が無い患者において有意な上昇を示す30遺伝子は、有意性階層に基づいて下から上へ並べられている。遺伝子名(遺伝子名が無い場合はアフィメトリクス・プローブセット識別子)は左に列記する。正規化発現スケールは、データ表示の下に示す通り、−30(青)から+30(赤)の範囲にわたる。順列調整後に有意なままである4個の遺伝子を下線で示す。
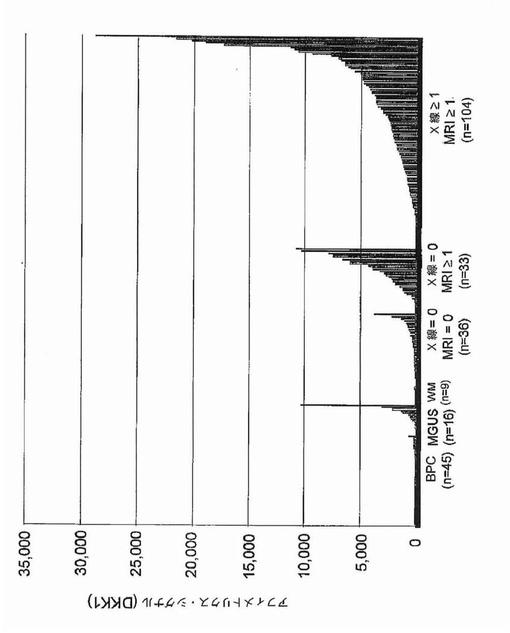
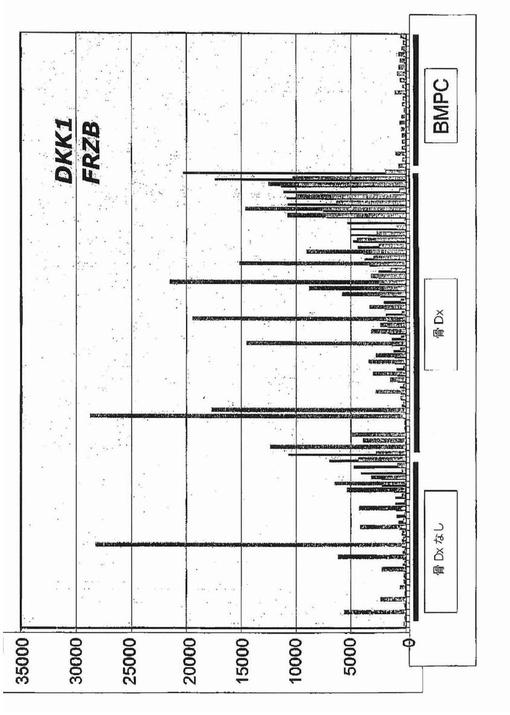
【図1B】図1Bは、X軸上に示す正常骨髄(BPC)、意義不明単クローン性免疫グロブリン血症(MGUS)、ワルデンシュトレーム型マクログロブリン血症(WM)、および多発性骨髄腫(MM)の患者に由来する形質細胞におけるDKK1遺伝子発現の棒グラフを示す。MM標本は:MRI病変なし/X線病変なし、MRI病変1個以上/X線病変なし、およびMRI病変1個以上/X線病変1個以上、の3つの骨病変群に分けられている。MAS5.01から得られる遺伝子発現の定量尺度であるアフィメトリクス・シグナルをy軸上に示す。各標本中のDKK1遺伝子発現レベルを棒で示し、棒の高さは遺伝子発現強度に比例する。標本は、DKK1遺伝子発現が最低から最高へ、x軸の左から右へ並べられている。各群の標本の数は、各群の名称の下に示す。MM部分群間の比較についての統計値は本文中に示す。
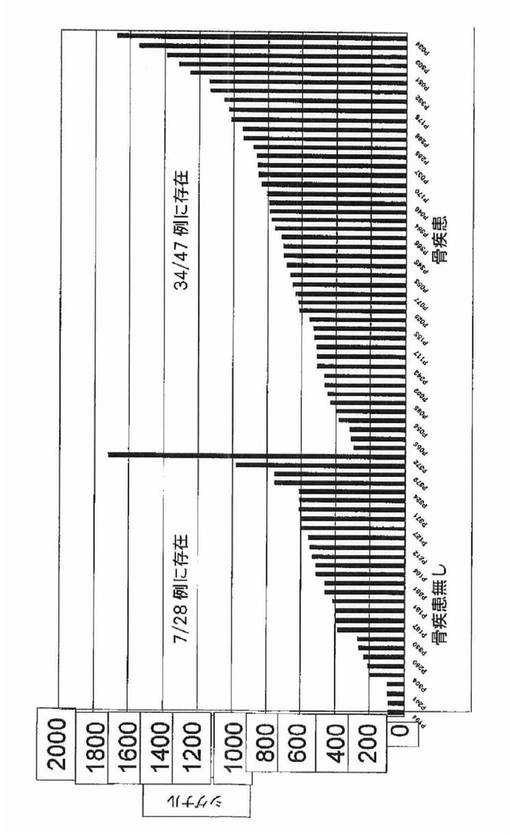
【図2】図2は、RHAMMが骨病変を有する多発性骨髄腫患者においてアップレギュレートされたことを示す。
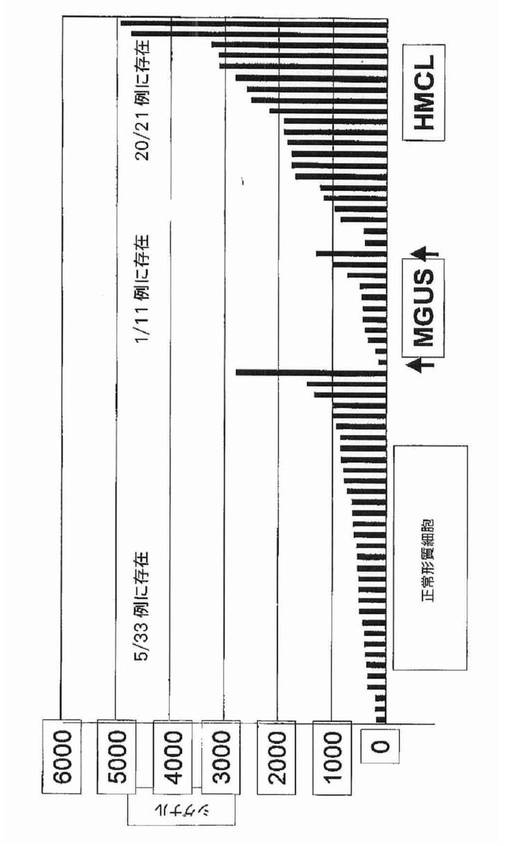
【図3】図3は、RHAMMは正常形質細胞および意義不明単クローン性免疫グロブリン血症(MGUS)にはほとんど存在しないが、事実上すべてのヒト骨髄腫細胞株に存在したことを示す。
【図4】図4は、securinが骨疾患を有する多発性骨髄腫患者においてアップレギュレートされたことを示す。
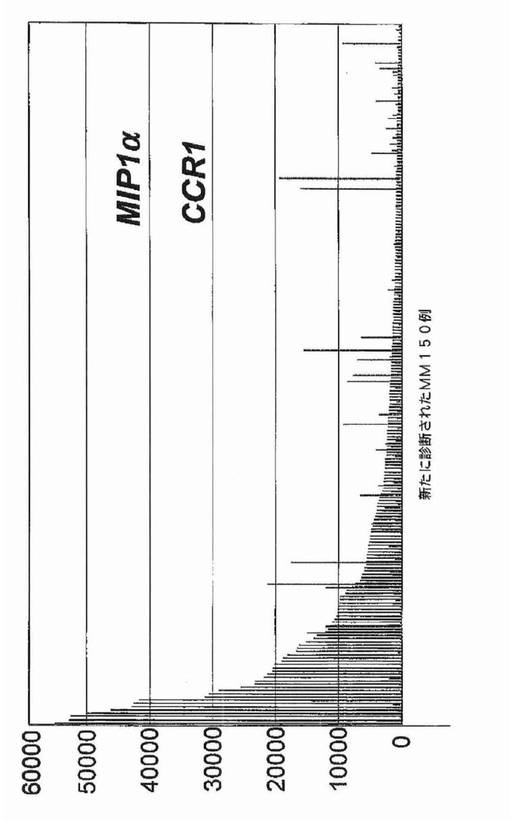
【図5】図5は、MIP−1αおよびCCR1が多発性骨髄腫において「スパイク」 遺伝子であったことを示すが、それらは溶解性病変と相関しなかった。黒棒:CCR 1;灰色棒:MIP−1α。
【図6】図6は、MIP−1αが正常形質細胞(PC)において低レベルで発現されたことを示す。
【図7】図7は、骨病変を有する多発性骨髄腫におけるWNT拮抗因子DKK−1の発現を示す。
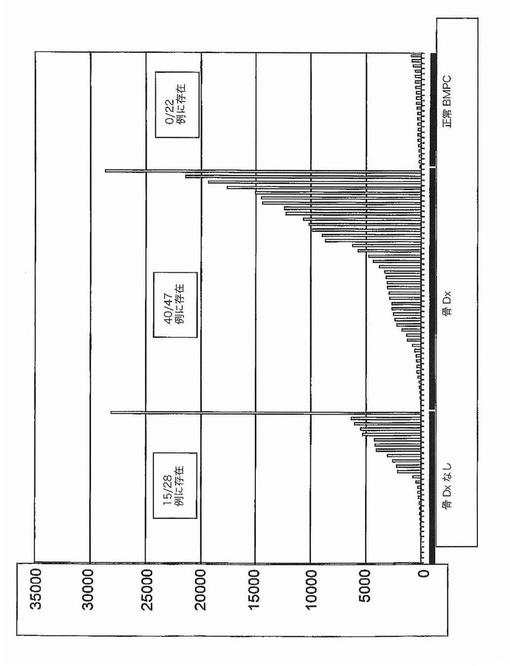
【図8】図8は、溶解性骨病変を有する多発性骨髄腫におけるWNTおとり受容体FRZBの発現を示す。
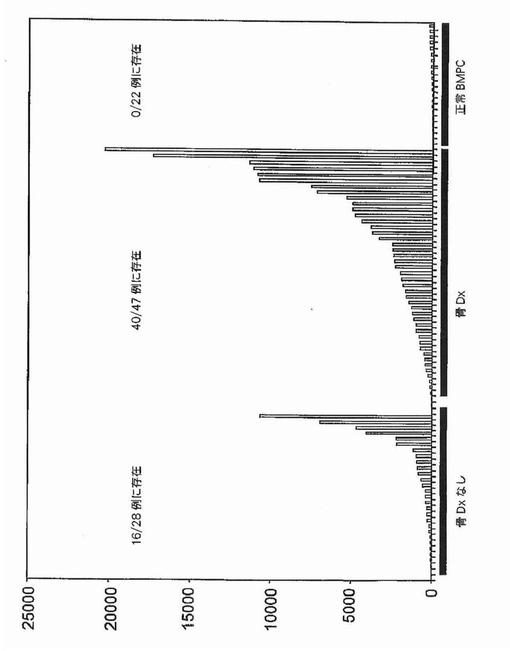
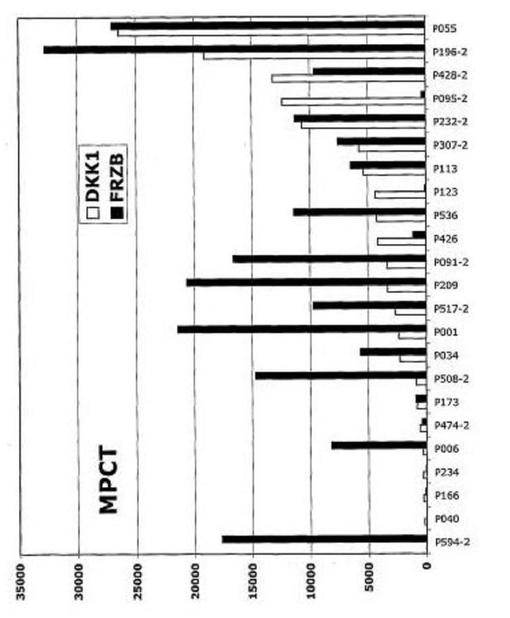
【図9】図9は、溶解性骨病変を有する多発性骨髄腫におけるDKK−1およびFRZBの発現を示す。黒棒:DKK−1;灰色棒:FRZB。
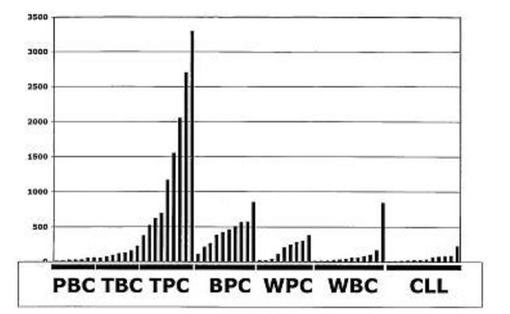
【図10】図10は、扁桃形質細胞でFRZBが発現されたことを示す。PBC、TBC=扁桃B細胞、TPC=扁桃形質細胞、BPC=骨髄形質細胞、WPC、WBC、CLL。
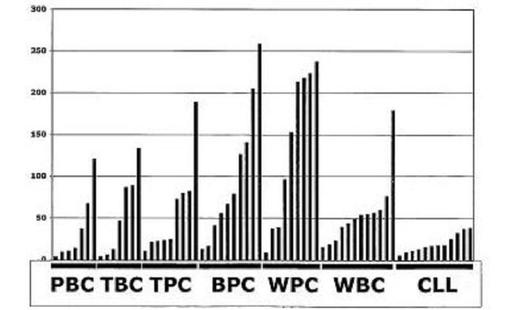
【図11】図11は、DKK−1が正常B細胞または形質細胞で発現されなかったことを示す。PBC、TBC=扁桃B細胞、TPC=扁桃形質細胞、BPC=骨髄形質細胞、WPC、WBC、CLL。
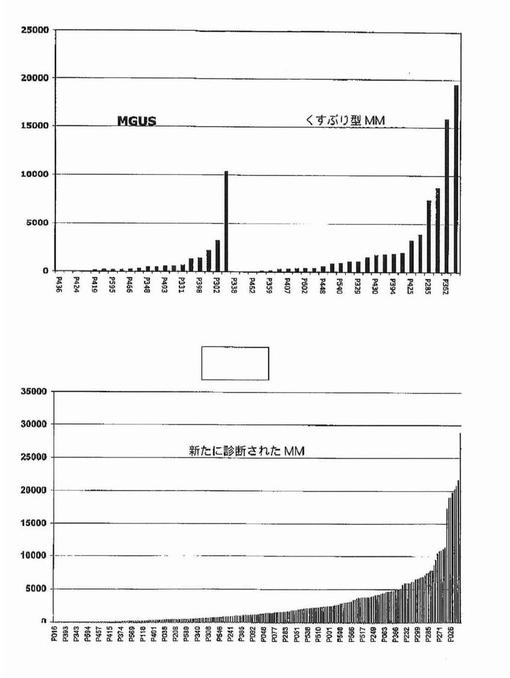
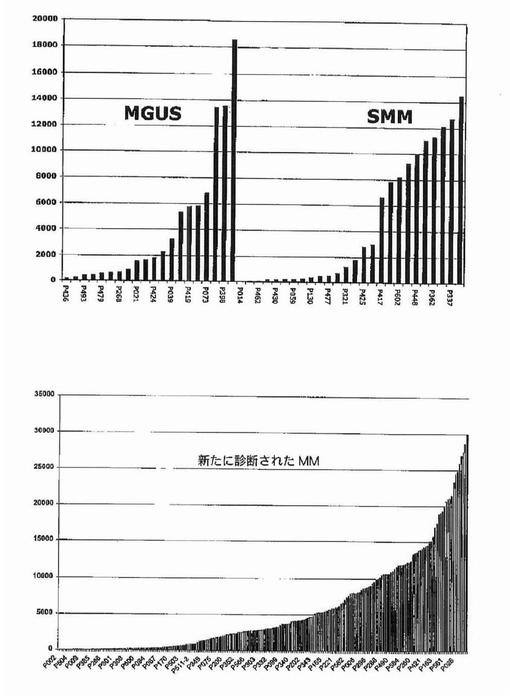
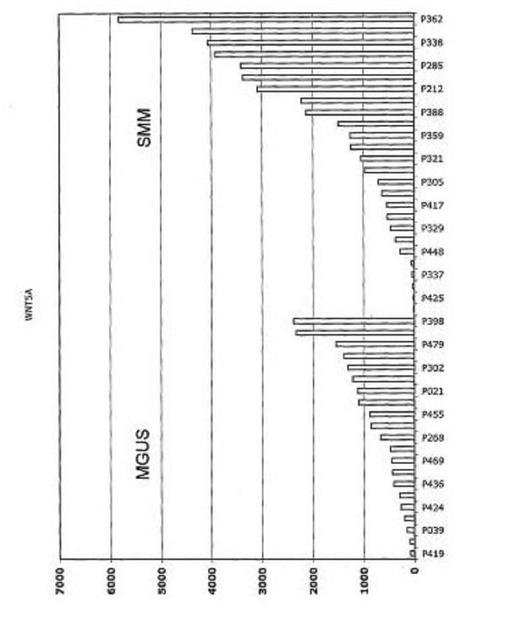
【図12】図12は、意義不明単クローン性免疫グロブリン血症(MGUS)におけるDKK−1発現が、くすぶり型多発性骨髄腫(SMM)および新たに診断された多発性骨髄腫(MM)と比較して低かったことを示す。
【図13】図13は、FRZBが意義不明単クローン性免疫グロブリン血症(MGUS)において上昇したこと、およびくすぶり型多発性骨髄腫(SMM)および新たに診断された多発性骨髄腫(MM)において発現がより高かったことを示す。
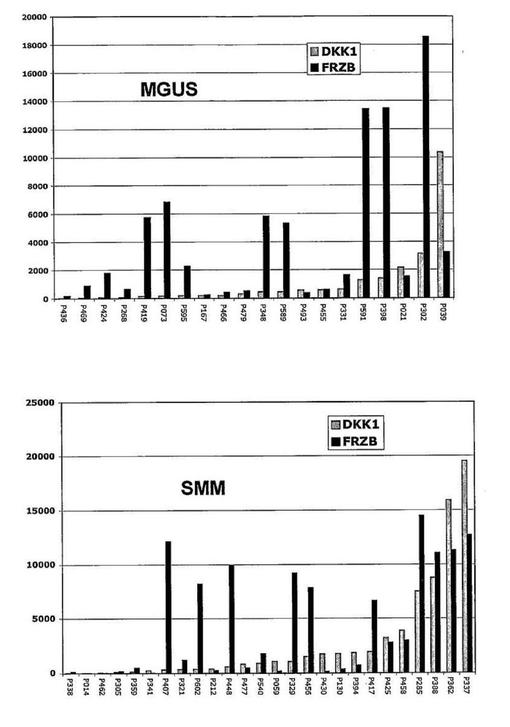
【図14】図14は、意義不明単クローン性免疫グロブリン血症(MGUS)およびくすぶり型多発性骨髄腫(SMM)におけるDKK−1およびFRZBの発現を示す。
【図15】図15は、髄外疾患におけるDKK−1の低発現を示す。
【図16】図16は、DKK−1およびFRZBの発現が、髄質PCT由来の形質細胞において、腸骨稜由来のものよりも高い傾向があることを示す。PCT、FNA。
【図17】図17は、髄質PCTの穿刺吸引検体におけるDKK−1およびFRZBの発現を示す。
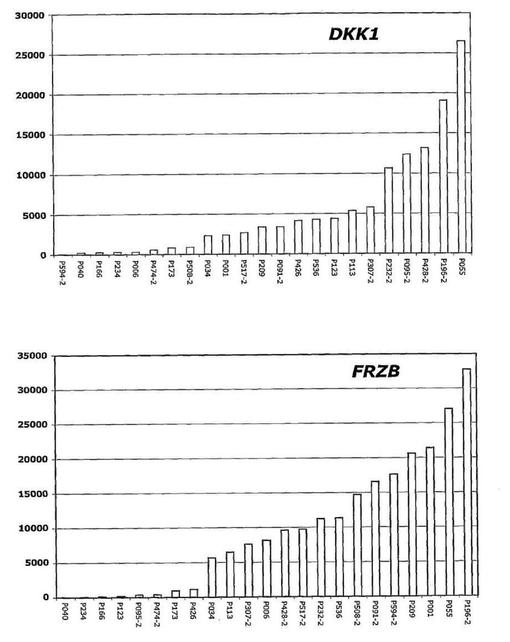
【図18】図18は、髄質形質細胞腫におけるDKK−1およびFRZBの高発現を示す。
【図19】図19は、骨減少症を伴う多発性骨髄腫におけるDKK−1のより高い発現を示す。
【図20】図20は、DKK−1がワルデンシュトレーム型マクログロブリン血症に由来する形質細胞で発現されていなかったことを示す。
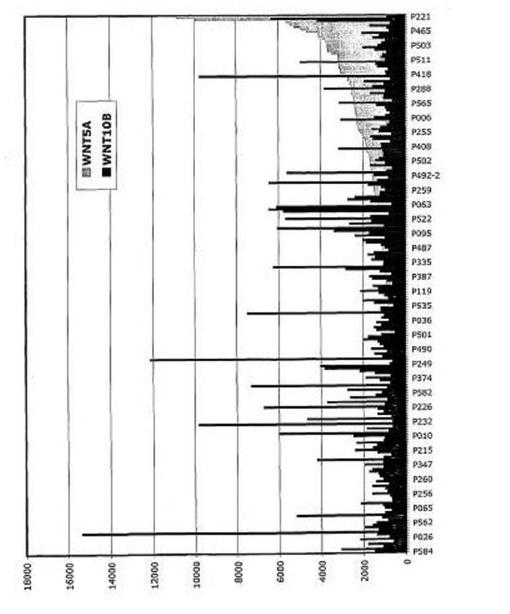
【図21】図21は、WNT5Aが新たに診断された多発性骨髄腫で上昇していたことを示す。
【図22】図22は、WNT5Aが溶解性病変を有する多発性骨髄腫でより高い傾向があることを示す。
【図23】図23は、WNT5Aが意義不明単クローン性免疫グロブリン血症(MGUS)およびくすぶり型多発性骨髄腫(SMM)でも上昇していたことを示す。
【図24】図24は、WNT10Bが溶解性病変を有する多発性骨髄腫より低い傾向があることを示す。
【図25】図25は、WNT5AおよびWNT10Bが逆相関する傾向があることを示す。黒棒:WNT10B、灰色棒:WNT5A。
【図26】図26は、DKK−1がSK−LMS細胞株に存在したことを示す。
【図27】図27は、原発性多発性骨髄腫がDKK−1タンパク質を合成したことを示す。
【図28】図28は、再発性および原発性難治性多発性骨髄腫におけるDKK−1低発現を示す。
【図29】図29は、エンドセリン受容体Bは新たに診断された多発性骨髄腫の3分の1において「スパイク」遺伝子であったことを示す。
【図30】図30は、意義不明単クローン性免疫グロブリン血症(MGUS)およびくすぶり型多発性骨髄腫におけるエンドセリン受容体Bの発現を示す。正常形質細胞はエンドセリン受容体Bを発現しない。
【図31】図31は、骨形成におけるエンドセリン受容体Bの関与を示す。
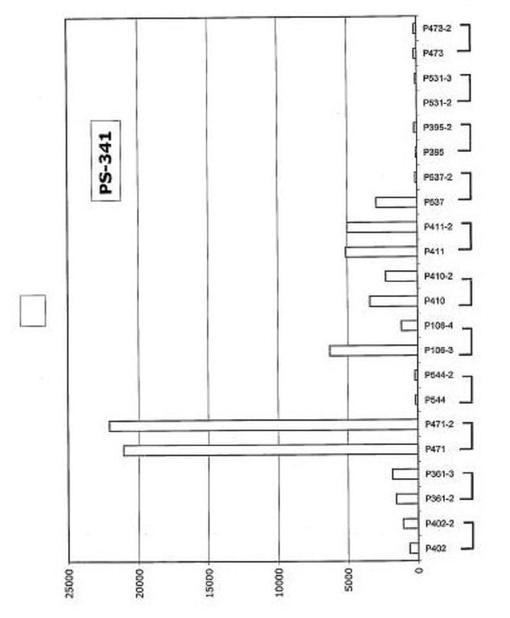
【図32】図32は、PS−341を用いた治療後のDKK−1発現を示す。
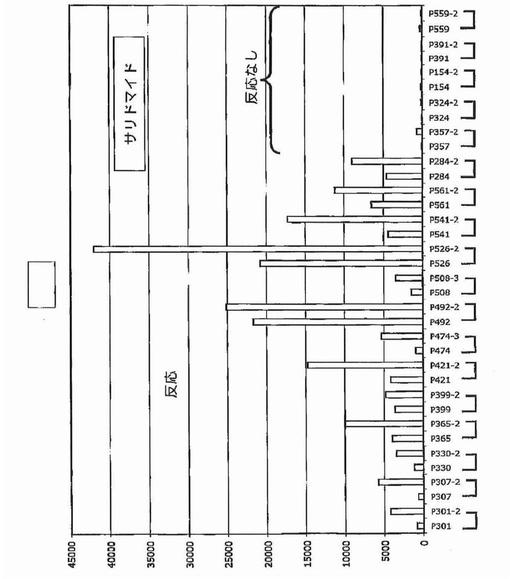
【図33】図33は、新たに診断された多発性骨髄腫におけるサロミド(サリドマイド)を用いた治療後のDKK−1発現を示す。
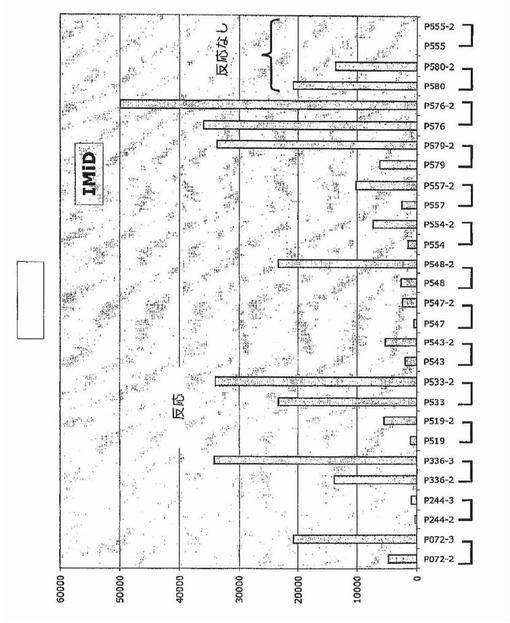
【図34】図34は、IMiDを用いた治療後のDKK−1発現を示す。
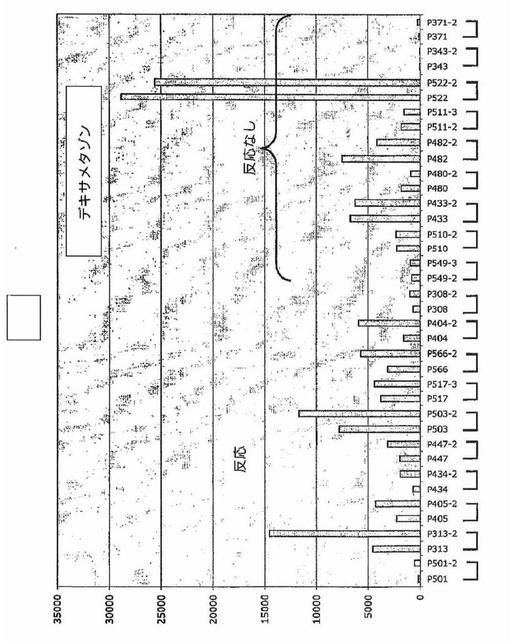
【図35】図35は、新たに診断された多発性骨髄腫における、デキサメタゾン処理後のDKK−1発現を示す。
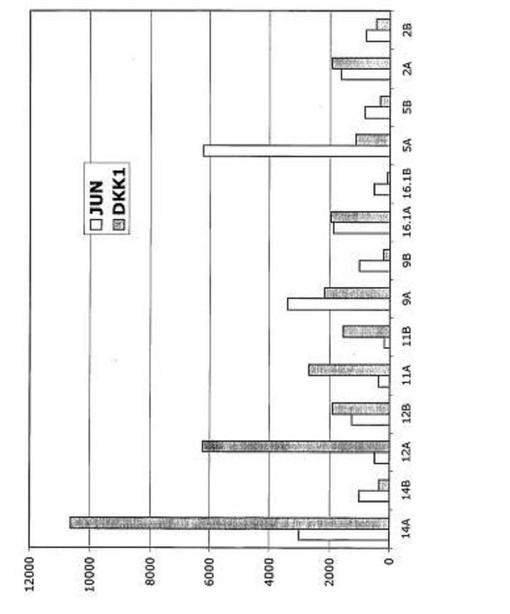
【図36】図36は、破骨細胞との共培養後の多発性骨髄腫細胞におけるJUNおよびFOSのダウンレギュレーションを示す。
【図37】図37は、破骨細胞共培養におけるJUNおよびDKK−1ダウンレギュレーションを示す。
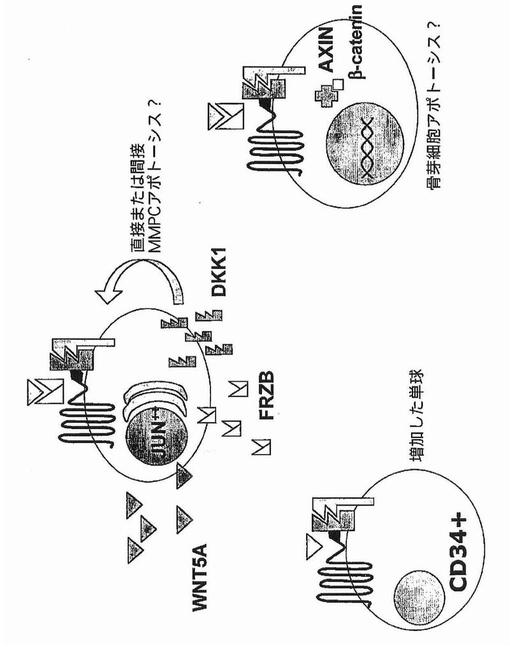
【図38】図38は、多発性骨髄腫骨疾患におけるWNTシグナル伝達を示す。
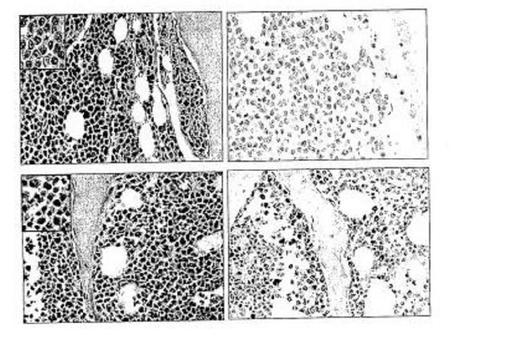
【図39】図39は、疾患進行に伴い発現が消失する、軽度骨髄腫におけるDKK1の過剰発現を示す。DKK1の発現は骨髄腫骨髄生検の免疫組織化学検査によって調べた。DKK1遺伝子発現が高い(a〜b)および低い(c〜d)骨髄腫患者に由来する骨髄生検の連続切片(倍率550x)を示す。スライドは、H&E(aおよびc)または抗DKK1および二次抗体(bおよびd)を用いて染色している。二次抗体単独の使用では細胞を染色できなかった(データ記載せず)。拡大像(倍率1,200x)は各H&E像の左上隅にある。図aは多量の細胞質を有し核小体が見えない軽度の形態を示している形質細胞との関与の間質性パターンを伴う骨髄腫を示す。図bは骨に非常に近い抗DKK1抗体を用いた間質性パターン中の形質細胞のポジティブ染色を示す。図cは大型化した核および顕著な核小体を有する重度の形態を示している形質細胞との結節状または閉塞性パターンを伴う骨髄腫を示す。図dは抗DKK1抗体を用いて形質がポジティブ染色されないことを示す。
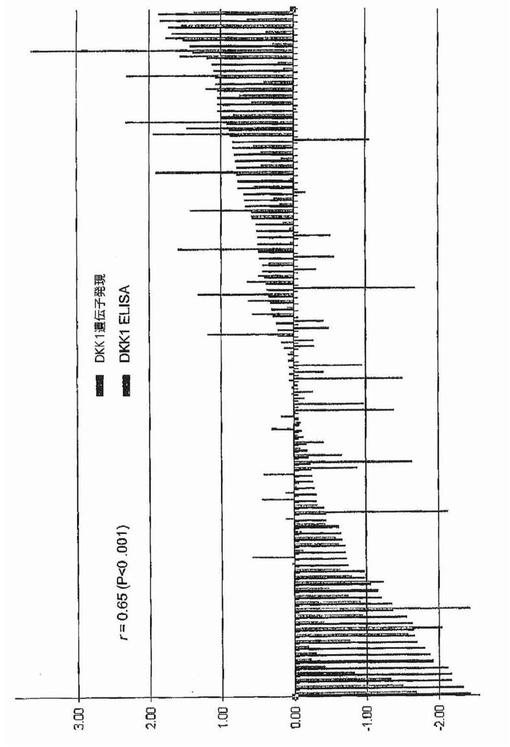
【図40A】図40Aおよび40Bは、骨髄形質中のDKK1タンパク質がDKK1遺伝子発現とおよび骨病変の存在と高度に相関することを示す。図40Aは、DKK1mRNAの発現がマイクロアレイによっておよびDKK1タンパク質がELISAによって、新たに診断された骨髄腫の計107例で検出されたことを示す。両方の分析の結果は平均値0および変動1となるように底2の対数で変換および正規化した。棒のそれぞれは各標本における遺伝子発現およびタンパク質発現の相対的関係を示す。骨髄腫形質細胞中のDKK1mRNAと骨髄形質中のタンパク質との間には有意な相関があった(r=0.65,P<0.001)。
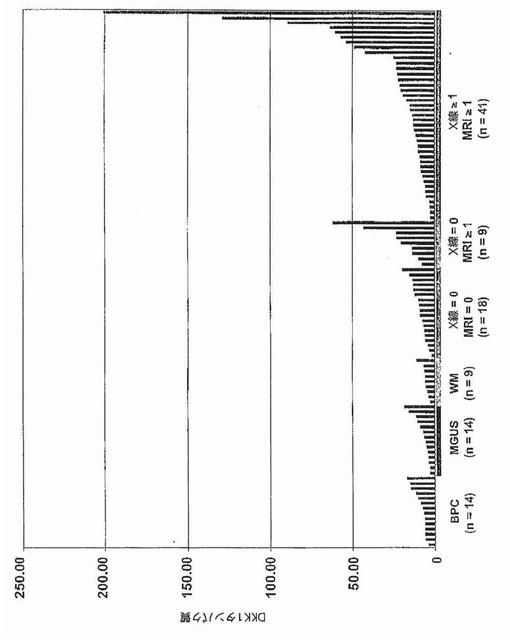
【図40B】図40Bは、骨髄形質中のDKK1タンパク質レベルの棒グラフ表示を示す。正常ドナー(BPC)、意義不明単クローン性免疫グロブリン血症(MGUS)、ワルデンシュトレーム型マクログロブリン血症(WM)、および多発性骨髄腫(MM)患者由来の形質細胞をx軸上に示す。MM標本は:MRI病変なし/X線病変なし、MRI病変1個以上/X線病変なし、およびMRI病変1個以上/X線病変1個以上、の3つの骨病変群に分けられている。DKK1タンパク質濃度(ng/ml)をy軸上に示す。より低い範囲でのDKK1タンパク質レベルの比較を可能にするため、200ng/mlを最大値とした。この結果、DKK1濃度が476ng/mlである1標本の切り捨てが生じた。各標本中のDKK1タンパク質レベルを棒で示し、棒の高さはDKK1タンパク質レベルに比例する。標本は、DKK1タンパク質レベルが最低から最高へ、x軸の左から右へ並べられている。各群の標本の数を各群の下に示す。
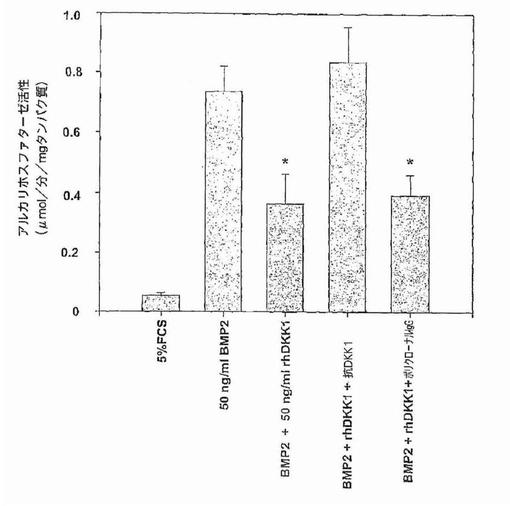
【図41A】図41Aおよび41Bは、組み換えDKK1およびMM形質が、BMP−2処理C2C12細胞におけるアルカリホスファターゼ産生を、DKK1に依存する方法で遮断することができることを示す。図41Aは、C2C12細胞において、5パーセントウシ胎仔血清単独またはBMP2、BMP2+DKK1、BMP2+DKK1+抗DKK1、またはBMP−2+DKK1+ポリクローナルIgGを加えての存在下での5日間の培養後に測定した、骨芽細胞分化のマーカーであるアルカリホスファターゼレベルを示す(y軸)。棒のそれぞれは3連の実験の平均値(±SEM)を表す。アルカリホスファターゼの活性はBMP−2の存在下で上昇したこと、および組み換えDKK1との共培養によるこのタンパク質の顕著な減少に注意する。同じく、抗DKK1抗体はDKK1の抑制活性を遮断できたが、ポリクローナルIgGはできなかったことに注意する。
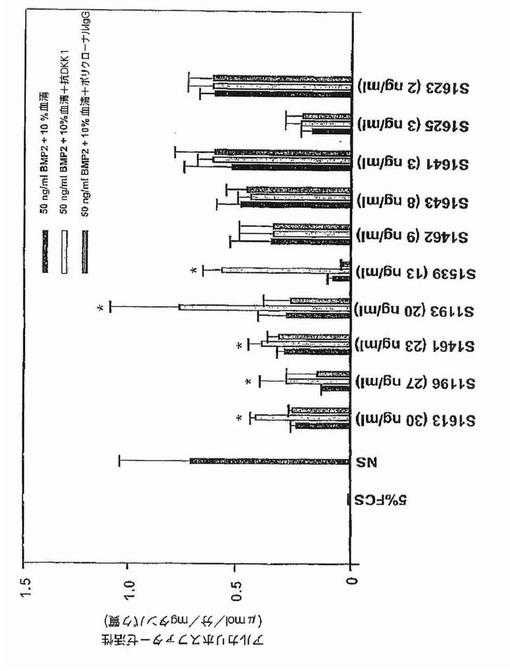
【図41B】図41Bは、C2C12細胞における、5パーセントウシ胎仔血清単独または50ng/mlBMP−2+正常骨髄形質(NS)10パーセントまたはBMP−2+新たに診断された骨髄腫の患者10名に由来する骨髄腫骨髄形質10パーセント(同定された標本を提供)、またはBMP2+骨髄腫患者形質10パーセント+抗DKK1またはヤギポリクローナルIgG中で5日間これらの細胞を培養後に測定したアルカリホスファターゼレベル(y軸)を示す。棒のそれぞれは3連の実験の平均値(±SEM)を表す。各骨髄形質標本からのDKK1濃度はELISAによって測定し、および1:10希釈後の培養中の終濃度をx軸上に示す。DKK1が12ng/mlより大である標本はアルカリホスファターゼ産生に影響を有したことに注意する。星印はBMP2+正常ヒト骨髄形質10パーセント中のアルカリホスファターゼと比較してP<0.05を示す。
【発明を実施するための形態】
【0018】
本発明は、分泌型WNTシグナル伝達拮抗因子DKK−1およびFRZBが多発性骨髄腫にみられる骨破壊を媒介することを実証する。Wntシグナル伝達が骨芽細胞増殖および分化に絶対的に必須であることの新たな証拠と共に、これらのデータは、これらの因子が骨減少に続く正常な補償的骨産生を抑制することによって骨芽細胞アネルギーを生じることおよび多発性骨髄腫骨疾患に寄与することを強く示唆する。
【0019】
破骨細胞活性を刺激することにおける多発性骨髄腫形質細胞の役割は集中的に研究されており、およびいくつかの鍵となる関連が確立されている。ここに示すデータは、多発性骨髄腫における骨芽細胞機能不全の、可能な機構的説明の証拠を初めて提供する。これらは、最近の研究がWNTシグナル伝達の阻害が骨芽細胞機能における欠陥を引き起こすことを示した点で有意義である。分泌型DKK−1およびFRZBは、多発性骨髄腫でみられる全身性骨粗鬆症、および形質細胞病巣近傍の悪化した局所性骨破壊の両方を説明し得る。
【0020】
重要なことに、DKK−1およびFRZBは、独立した経路を介しWNTシグナル伝達を阻害するように作用し、このことはそれらの同時発現が相乗作用を有することを示す。
したがって、これらの遺伝子は、骨疾患の程度および骨疾患を発症する将来のリスクを予測するのに用いることができる可能性がある。さらに、これらのタンパク質の阻害因子は、骨疾患を阻害するのに用いることができる可能性がある。また、これらの因子が一般集団における骨粗鬆症に関与している可能性もある。
【0021】
WNTシグナル伝達経路
Wnt遺伝子は、脊椎動物胚発生中に空間的におよび組織限定パターンで発現される分泌型ポリペプチドの大きなファミリーを構成する。マウスでの突然変異分析は、体軸、中枢神経系および四肢のパターン化、および器官形成中の誘導現象の調節といったさまざまな発生段階を調節することにおけるWntの重要性を示している。分泌型増殖因子のWntファミリーは、Frizzled(Fz)受容体およびその共受容体であるLDL受容体関連タンパク質5または6(LPR5またはLRP6)を介して、おそらくWntによって誘導されるFz−LPR5/LRP6複合体形成を通じて、シグナル伝達を開始する。
【0022】
Wntの分泌型拮抗因子は、Frizzled(Fz)関連タンパク質(FRPs)、Cerberus、Wnt阻害因子(WIF)およびDickkopf(DKK)を含む。Frizzled(Fz)関連タンパク質、CerberusおよびWnt阻害因子は全部が、Wntに結合および封鎖することによって作用することが示されている。Fzの分子模倣または他の機構によるWnt封鎖によって作用を発揮するWnt拮抗因子とは異なり、Dickkopf−1(DKK−1)は受容体複合体のLPR5/LRP6成分に結合することによって正規のWntシグナル伝達を特異的に阻害する。
【0023】
DKK−1は脊椎動物頭部オーガナイザーから分泌される頭部誘導因子であり、およびWntシグナル伝達に拮抗することによって全部発生を誘導する。DKK−1はLRP6に対する高親和性リガンドであり、およびWntによって誘導されるFz−LRP6複合体形成を妨げることによってWntシグナル伝達を阻害する。DKK−1はWntともFzとも結合せず、Wnt−Fz相互作用にも影響しない。頭部誘導およびWntシグナル伝達阻害におけるDKK−1機能は、LPR5/LRP6と結合しおよびFz−LPR5/LRP6会合を破壊する能力と密接に相関する。LPR5/LRP6機能およびDKK−1阻害は、Wnt/Fzベータカテニン経路に特異的であるように見える。これらの知見はしたがって、Wntシグナル調節のための新規の機構を明らかにする。
【0024】
WNTシグナル伝達および骨芽細胞分化
近年の研究は、Wntシグナル伝達経路が骨芽細胞分化および機能に決定的であることを示している。低密度リポタンパク質受容体関連タンパク質5(LRP5)の遺伝子にターゲッティング破壊を有するマウスは低骨量表現型を発生する。LRP5は骨芽細胞で発現され、および骨芽細胞における最適なWntシグナル伝達に必要である。In vivoおよびin vitro分析は、この表現型は生後に明らかになり、さらにそれはCbfa1非依存的な態様で骨芽細胞増殖および機能の低下に対して二次的なものである。
【0025】
ヒトでは、LRP5における突然変異は、常染色体劣性疾患である骨粗鬆症−偽神経膠腫症候群(OPPG)を引き起こす。骨粗鬆症−偽神経膠腫症候群のキャリヤーは、年齢および性別が一致する対照と比較した場合に骨量低下がある。
【0026】
重要なことに、LRPにおける別々のおよび異なる突然変異は、高骨量表現型を結果として生じる。骨粗鬆症−偽神経膠腫突然変異とは対照的に、高骨量の性質は機能突然変異の利益である。その変異を有する対象者において骨吸収のマーカーは正常であった一方、オステオカルシンといった骨形成のマーカーは顕著に増加していた。Wntによるシグナル伝達の既知の標的であるフィブロネクチンのレベルもまた上昇していた。In vitro試験は、Dickkopf−1(DKK−1)によるWntシグナル伝達の正常な阻害は本突然変異の存在下では不完全であったこと、およびこのことが抑制されないWnt活性によるシグナル伝達増加を結果として生じたことを示した。これらの知見は、高骨量でのLRP5機能変化の役割を実証し、および骨粗鬆症の予防または治療のための標的候補としてDKKを指し示す。
【0027】
多発性骨髄腫におけるWNT シグナル伝達および骨疾患
骨芽細胞機能におけるDKK−1の役割の間接的証拠が、高骨量表現型と連鎖しているLRP−5における機能突然変異の利益の同定によって与えられている。加えて、FRZB(SFRP−3)のホモログである分泌型firzzled関連タンパク質(SFRP−1)のターゲッティング破壊は、骨芽細胞の減少および骨細胞アポトーシスおよび骨梁形成増加に繋がる。
【0028】
骨量に影響する量的形質遺伝子座(QTL)がLRP−5領域に局在化されており、一般の人口は骨粗鬆症を発症する異なるリスクを有することを示唆する。多発性骨髄腫骨疾患は、DKK−1/FRZB発現とLRP−5対立遺伝子によって与えられる低骨量の遺伝的素因との組み合わせ作用によって影響され得ると考えられる。多発性骨髄腫の症例をLRP−5対立遺伝子変異について遺伝子型分析することができ、およびこの情報を骨疾患、およびDKK−1およびFRZB発現と相関させることができる。
【0029】
意義不明単クローン性免疫グロブリン血症(MGUS)は、多発性骨髄腫を発症する傾向がある形質細胞疾患であり、明らかな骨疾患を欠くことによって多発性骨髄腫から区別される。DKK−1および/またはFRZB発現が意義不明単クローン性免疫グロブリン血症の3分の1で発見されることの意義は不明であるが、しかしこれらの症例が多発性骨髄腫を発症するより高いリスクにある可能性があることを示唆し得る。多発性骨髄腫と同様に、この素因はまた遺伝するLRP5対立遺伝子と関連する可能性がある。代替的に、これらの意義不明単クローン性免疫グロブリン血症の例は、放射線スキャンではまだ明らかでない基礎となる発症前骨疾患を有するであろう。
【0030】
ここに示すデータは、多発性骨髄腫形質細胞によるDKK−1発現がどのように多発性骨髄腫疾患増殖調節および骨破壊に結びつき得るか、およびこれらの二つの現象がどのように1つの分子によって統合され得るかについてのモデルを示唆する。そのモデルでは、原発性多発性骨髄腫は高レベルのDKKを発現し、およびこれらのレベルはその疾患を治療するのに用いられる薬物療法で上昇させることができる。高レベルのDKK−1は多発性骨髄腫細胞のアポトーシスを誘導する可能性が高く、および、細胞増殖が高率のDKK−1誘導性アポトーシスによって緩和されるため、本疾患の初期における相対的に遅い進行を説明し得る。しかし、疾患が進行するにつれ、JUNおよびDKK−1に、破骨細胞に誘導される低下が生じ、これは最終的に髄外疾患に見られるような、JUNおよびDKK−1発現の構成的消失に発展する。
【0031】
このように、DKK−1発現を多発性骨髄腫形質細胞の視点から見るならば、高レベルのDKK−1発現は本疾患を肯定する性質と見なすことができる。しかし、DKK−1誘導性アポトーシスに対して極めて感受性である間葉性細胞系譜については、高レベルのこの分泌産物は、前駆骨芽細胞のおよびおそらく間葉幹細胞さえの大規模なプログラム細胞死もまた誘導する点で、二通りの効果を有するであろう。疾患初期における高レベルのDKK−1は間葉幹細胞の永久的損失に繋がる可能性があると考えられ、この概念は、寛解誘導後または疾患進行中に破骨細胞が多発性骨髄腫形質細胞によるDKK−1分泌を抑制する可能性が高い際に、骨修復の欠如が観察されることによって支持される。したがって、この知識の活用は、多発性骨髄腫形質細胞に対するDKK−1の作用を強調するが同時に骨芽細胞またはその前駆細胞に対するDKKの骨損傷作用を妨げる、多発性骨髄腫のための新しい治療法の開発に繋がり得る。
【0032】
本発明の一実施形態では、WNTシグナル伝達拮抗因子の発現レベルを調べることによって、多発性骨髄腫患者において骨疾患を発症する可能性を判定する方法が提供される。その拮抗因子の、正常個体における発現と比較して増加した発現は、その患者が骨疾患を発症する可能性を有することを示す。好ましくは、そのWNTシグナル伝達拮抗因子は可溶性frizzled関連タンパク質3(SFRP−3/FRZB)、またはDickkopf−1(DKK1)のヒトホモログである。一般的に、これらのタンパク質の発現レベルは、核酸またはタンパク質レベルで測定することができる。
【0033】
別の一実施形態では、WNTシグナル伝達拮抗因子の発現を阻害することによって、多発性骨髄腫患者において骨疾患を治療する方法が提供される。好ましくは、そのWNTシグナル伝達拮抗因子は可溶性frizzled関連タンパク質3(SFRP−3/FRZB)、またはDickkopf−1(DKK1)のヒトホモログである。一般的に、これらの拮抗因子の発現は、核酸またはタンパク質レベルで阻害することができる。
【0034】
さらに別の一実施形態では、WNTシグナル伝達拮抗因子の発現を阻害することによって、個体において骨減少を防ぐ方法が提供される。好ましくは、そのWNTシグナル伝達拮抗因子は可溶性frizzled関連タンパク質3(SFRP−3/FRZB)、またはDickkopf−1(DKK1)のヒトホモログである。一般的に、これらの拮抗因子の発現は、核酸またはタンパク質レベルで阻害することができる。
【0035】
さらに別の一実施形態では、DKK1遺伝子(登録番号NM012242)の発現またはDKK1遺伝子によって発現されるタンパク質の活性を阻害する工程を含む、個体において骨減少を調節する方法が提供される。DKK1遺伝子発現は、たとえば、アンチセンスオリゴヌクレオチド、または抗DKK1抗体または可溶性LRP受容体によることを含む、当業者に既知である任意の方法によって阻害される。
【0036】
さらに別の一実施形態では、DKK1タンパク質の阻害薬を個体に投与する工程を含む、個体において骨減少を調節する方法が提供される。一般的に、この方法はその個体が多発性骨髄腫、骨粗鬆症、閉経後骨粗鬆症または悪性腫瘍関連骨減少といった疾患を有する場合に有効である。一般的に、悪性腫瘍関連骨減少は、骨への乳がん転移または骨への前立腺がん転移によって引き起こされる。
【0037】
下記の実施例は本発明のさまざまな実施例を説明する目的のために与えられ、および本発明をいかなる方法でも制限しないことが意図される。当業者は、本発明は目的を実施および上記の結果および利点、およびここに内在する目的、結果および利点を得るためによく適合されていることを容易に理解し得る。請求項の範囲によって定義される通りの本発明の精神の内に包含される、その中の変化および他の用途を当業者は考えることができる。
【実施例】
【0038】
実施例1
患者
新たに診断された多発性骨髄腫の患者174名、意義不明単クローン性免疫グロブリン血症の患者16名、ワルデンシュトレーム型マクログロブリン血症の患者9名、および健常者45名を試験した。米国アーカンソー医科大学の施設内倫理委員会は本研究を承認し、およびすべての被験者はインフォームド・コンセントを書面で提供した。表1は多発性骨髄腫の患者の特性を示す。
【0039】
【表1】

【0040】
実施例2
骨画像処理
画像は、遺伝子発現データの予備知識無しで、Canon PACS(画像保存およびカタログ化システム)を用いて検査した。MRIスキャンは1.5テスラGE Signaスキャナで実施した。X線は米国放射線医学会(ACR)規格に準拠してフィルムからデジタル化した。MRIスキャンおよびX線はACRのDICOM(医用デジタル画像処理および通信)規格を用いてCanon PACSシステムに接続した。画像処理は取扱説明書に従って実施した。MRI画像は、ガドリニウムT1前および後強調処理およびSTIR(短τ反転回復)強調処理を用いて作成した。
【0041】
実施例3
形質細胞単離および遺伝子発現プロファイリング
フィコール・ハイパック勾配遠心分離後に、骨髄から得られた形質細胞を単核細胞画分から、モノクローナルマウス抗ヒトCD138抗体(Miltenyi−Biotec,Auburn,CA)を用いる免疫磁気ビーズ選択によって単離した。CD138+/CD45−およびCD38+/CD45−マーカーを用いた二色フローサイトメトリー、免疫細胞化学法による免疫グロブリン軽鎖の存在、およびライト−ギムザ染色による形態で示された通り、遺伝子発現プロファイリングに使用した細胞の90パーセント超が形質細胞であった。総RNAはRneasyミニキット(Qiagen,Valencia,CA)を用いて単離した。標識化cRNAの調製および約10,000遺伝子を含むU95Av2マイクロアレイ(Affymetrix,Santa Clara,CA)へのハイブリダイゼーションは以前に記載した通りに実施した(Zhan et al.,2002; Zhan et al.,2003)。RNA増幅は不要であった。
【0042】
実施例4
免疫組織化学
ヒト総DKK1タンパク質(R&D Systems,Minneapolis,MN)に対して免疫化したヤギに由来する抗体をTris緩衝液で1:200希釈し、およびホルマリン固定してパラフィン包埋された骨髄生検切片に2時間室温にて加えた。隣接する切片をH&Eで染色した。抗原−抗体反応はDAB(ビオチニル化抗ヤギ抗体[Vector Laboratories,Burlingame,CA] [1: 400 希釈]およびストレプトアビジン−ホースラディッシュペルオキシダーゼ[Dako]染色後)を用いて発色させ、およびヘマトキシリン−2を用いて対比染色した。
【0043】
実施例5
酵素結合免疫吸着測定法(ELISA)
Nunc−Immuno MaxiSorp表面マイクロタイタープレートを、1xリン酸緩衝生理食塩水、pH7.2に溶解した1mg/mlの抗DKK1抗体50mlを用いて4℃にて一夜被覆し、および4パーセントウシ血清アルブミンを用いてブロックした。骨髄形質を希釈緩衝液(1×リン酸緩衝生理食塩水+0.1Tween−20+1パーセントウシ血清アルブミン)で1:50希釈した。ウェル当たり計50μlを負荷し、および4℃にて一夜インキュベートし、洗浄し、および緩衝液で0.2mg/mlに希釈したビオチニル化ヤギ抗ヒトDKK1 IgG(R&D Systems)とインキュベートし、その後ストレプトアビジン−ホースラディッシュペルオキシダーゼ(Vector Laboratories)の1:10,000希釈50μlを加え、すべて取扱説明書に従った。発色はOPD基質系(Dako)を用いて取扱説明書に基づいて達成した。組み換えヒトDKK1(R&D Systems)の連続希釈を用いて標準曲線を作成した。内因性DKK1を発現しない細胞株T293、および安定にトランスフェクションされたDKK1を有するT293(Fedi,et al.,1999)を用いて本ELISA測定法のバリデーションを行った。
【0044】
実施例6
骨芽細胞分化測定法
C2C12間葉性前駆細胞(American Type Tissue Culture,Reston,VA)は、10パーセント熱不活化ウシ胎仔血清を添加したDMEM(Invitrogen,Carlsbad,CA)中で培養した。C2C12細胞におけるアルカリホスファターゼ活性は記載の通りに測定した(Gallea,et al.,2001;Spinella−Jaegle,et al.,2001)。細胞溶解物はタンパク質含量についてマイクロBCA測定法キット(Pierce,Rockford,IL)を用いて分析した。各実験は3連で実施した。
【0045】
実施例7
統計解析
多発性骨髄腫患者における骨疾患は、ロジスティック回帰を用いてモデル化した。考慮した独立変数は、新たに診断された多発性骨髄腫174例からバージョン5.01MAS(Affymetrix,Santa Clara,CA)を用いて測定した最大10,000個の遺伝子(12,625個のプローブセット)からの遺伝子発現強度値(アベレージ・ディファレンス・コール)であった。各プローブセットについて、遺伝子発現の定量尺度である「シグナル」は、ロジスティック回帰モデルおよび順列調整分析への入力前にlog2に変換した。骨髄腫において骨疾患に関連し得る遺伝子については、先行する仮説は無かった。結果として、12,625個のプローブセットのそれぞれについて骨疾患の一変量モデルが用いられた。候補遺伝子は、順列調整された有意水準でのt検定(WestfallおよびYoung,1993)を用いて選別した。ウェストフォールおよびヤングの分析を用いて、複数の単変量仮説検定に合わせて調節した。DKK1シグナルおよびDKK1タンパク質レベルの群差は、ウィルコクソンの順位和検定を用いて検定した。骨疾患の状態による患者の特徴における有意差は、フィッシャーの正確確率検定またはカイ二乗検定のどちらかを用いて検定した。ロジスティック回帰によって同定された遺伝子の発現強度は、Clusterview(Golub,et.al.,1999)を用いて視覚化した。スピアマンの相関係数を用いて、遺伝子発現およびタンパク質レベルの相関を測定した。骨芽細胞分化における対照と各実験条件との間の有意差は、ウィルコクソンの順位和検定を用いて検定した;独自のC2C12実験それぞれについて別個の比較を実施した。両側p値0.05未満を有意とし、および両側p値0.10未満を傾向差とした。
【0046】
実施例8
骨髄腫細胞の発現プロファイリング
過剰発現されおよび骨病変の存在に伴っていた遺伝子を同定するために、骨病変を有するかまたは有しない患者に由来するマイクロアレイデータの比較を実施した。MRIで定義される骨の限局性病変は、放射線で同定可能な溶解性病変の前に生じ得るので、骨病変を評価するためにT1強調およびSTIR強調画像処理を用いた。骨病変の無い患者(n=36)およびMRIで定義される限局性病変1個以上(1+)を有する患者(n=137)の骨髄に由来する精製形質細胞中の遺伝子約10,000個の遺伝子発現パターンを、ロジスティック回帰分析によってモデル化した。そのモデルによって、2群の患者で発現に差のあった(P<0.0001)57個の遺伝子が同定された(図1A)。これらの遺伝子57個をさらに、順列調整された有意性でt検定によって分析した(WestfallおよびYoung,1993)。これらの統計検定は、その57個の遺伝子のうち4個が、MRI病変1個以上を有する患者で過剰発現されていたことを示した:ジヒドロ葉酸還元酵素(DHFR)、プロテアソーム活性化因子サブユニット(PSME2)、CDC28プロテインキナーゼ2(CKS2)、およびdickkopfホモログ1(DKK1)。Wnt/β−カテニンシグナル伝達拮抗因子の遺伝子DKK1が4個のうちで唯一分泌型因子をコードすること、およびWnt/β−カテニンシグナル伝達は骨の生物学に関与することを考慮して、DKK1に関する追加の試験を実施した。骨髄腫の患者173名からの結果の分析は、MRI病変1個以上およびX線病変なしの患者についてのDKK1シグナルは、MRI病変なしおよびX線病変なしの患者と比較して有意に異なるが(中央値シグナル:2,220対285;p<0.001)、MRI病変1個以上およびX線病変1個以上の患者と比較して有意差がない(中央値シグナル:2,220対1,865;p=0.63)ことを示した(図1B、表2)。
【0047】
意義不明単クローン性免疫グロブリン血症(MGUS)は、溶解性骨病変を伴わない形質細胞疾患であり、および多発性骨髄腫に先行し得る。MGUSの16例中15例で、DKK1は骨髄形質細胞によって、骨のMRI病変またはX線病変が無い多発性骨髄腫におけるレベルと同等のレベルで発現されていた(図1B)。DKK1は、正常ドナー45名、および骨病変を欠く骨の形質細胞悪性腫瘍であるワルデンシュトレーム型マクログロブリン血症の患者9名に由来する形質細胞では検出不能であった(図1B)。
【0048】
【表2】
【0049】
実施例9
全遺伝子発現は、多発性骨髄腫において溶解性骨病変と関連するDKK−1およびFRZBを明らかにする
溶解性骨疾患の分子決定因子をさらに特定するために、骨検査で溶解性病変の放射線的証拠を示さない新たに診断された多発性骨髄腫患者(n=28)に由来するCD138濃縮形質細胞における最大12,000個の遺伝子の発現プロファイルを、3個以上の溶解性病変を有する患者(n=47)と比較した。アブソルート・コール(遺伝子発現の定性尺度)のカイ二乗検定を用いて、疾患の2つの型を区別した遺伝子30個を同定した(P<0.05)。シグナル・コール(遺伝子発現の定量尺度)のウィルコクソンの順位和(WRS)検定は、104個の遺伝子(アップレギュレート49個およびダウンレギュレート55個)が2つの疾患亜型で差があった(P<0.001)ことを明らかにした。
【0050】
カイ二乗検定は、RHAMM前がん遺伝子をその2群間の最も有意な識別子として同定した。それは骨疾患の患者47名中34名と比較して、骨疾患のない患者28名中7名にしか発現されていなかった(図2)。予想される通り、意義不明単クローン性免疫グロブリン血症11例中の1例に由来する形質細胞しかRHAMMを発現しなかった(図3)。WRSはRHAMMを溶解性病変群と非溶解性病変群の間の14番目に有意な識別子として順位づけた。NCALDは、神経細胞シグナル伝達に関与するカルシウム結合タンパク質であるが、非溶解性病変群の11/28例(40%)に存在したが溶解性病変群の2/47例(4%)にしか存在しなかった。カイ二乗分析によって同定された他の注目すべき遺伝子は、Wntシグナル伝達の拮抗因子である、溶解性病変群の40/47例(85%)および非溶解性病変群の15/28(53%)に存在したFRZBを含んだ。CBFA2/AML1BはMIP1α発現と結びつけられており、および非溶解性病変群の50%および溶解性病変群の79%に存在した。
【0051】
染色体分離に関与するPTTG1(securin)はWRSによって最も有意な識別遺伝子(P=4x10−6)と同定された。それは非溶解性病変群の11%で存在するとされたが溶解性病変群の50%で存在した(図4)。WRS検定での他の注目すべき遺伝子は、溶解性病変群でより低レベルに発現された(P=3x10−5)TSC−22ホモログDSIPIを含んだ。DSIPIはまた、破骨細胞とのex−vivo共培養後の多発性骨髄腫形質細胞12例中12例でダウンレギュレートされた。
【0052】
加えて、非溶解性病変群に対して溶解性病変群でより高頻度に見出された(p<0.05)4個のいわゆる「スパイク遺伝子」が同定された:非溶解性病変群0/28例および溶解性病変群7/47例でスパイクを示すIL6(p=0.032);非溶解性病変群0/28例および溶解性病変群7/47例でスパイクを示すオステオナイドジェン(NID2)(p=0.032);非溶解性病変群1/28例および溶解性病変群11/47例でスパイクを示すGタンパク質シグナル伝達調節因子(RGS13)(p=0.023);および非溶解性病変群1/28例および溶解性病変群1/47例でスパイクを示すピリミジン受容体P2Y(P2RY6)(p=0.023)。
【0053】
このように、これらのデータは遺伝子発現パターンが骨疾患と関連し得るということを示唆する。溶解性骨疾患の出現および意義不明単クローン性免疫グロブリン血症から顕性多発性骨髄腫への転換の予測指標として潜在的に有用であることに加えて、それらはまた潜在的介入のための標的を特定し得る。
【0054】
実施例10
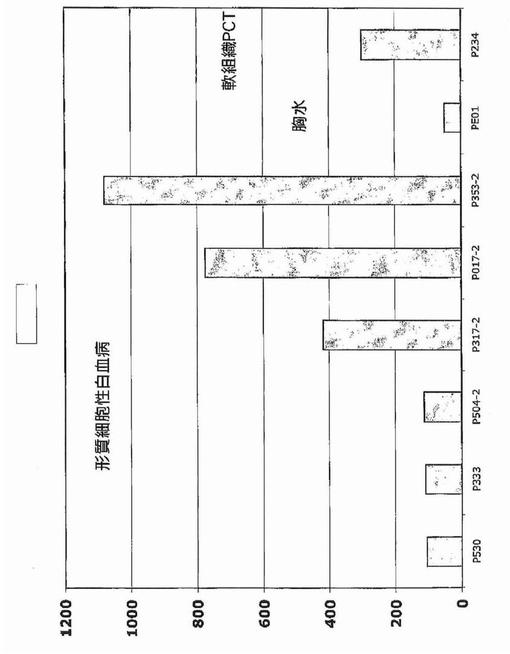
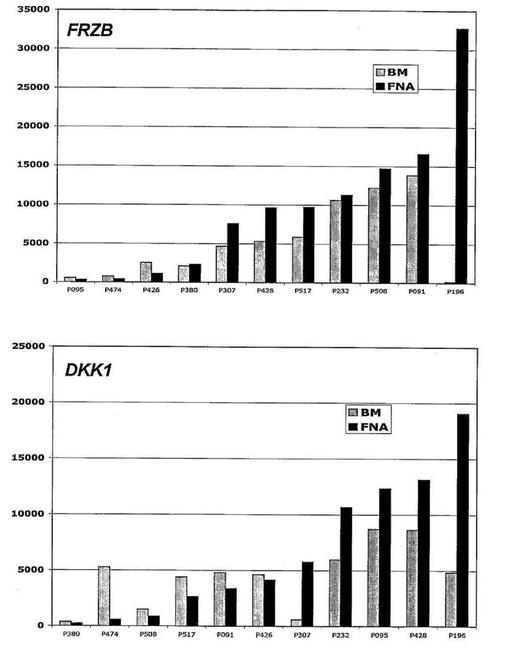
DDK1およびFRZBは無作為の骨髄由来よりも限局性病変由来の形質細胞でより高レベルに発現される傾向がある
溶解性病変に対するDKK−1およびFRZBの関係を考慮して、無作為の腸骨稜の骨髄吸引液由来の形質細胞と、脊椎の限局性病変のCT補助穿刺吸引で得られたものとで、DKK−1およびFRZB発現を比較した。これらの結果は、限局性病変由来の形質細胞における有意に高いレベルでの発現を示した。
【0055】
実施例11
DKK−1およびFRZBはワルデンシュトレーム型マクログロブリン血症由来の形質細胞で発現されていない
ワルデンシュトレーム型マクログロブリン血症は、モノクローナルIgM異常蛋白血症および骨髄、リンパ節および脾臓のリンパ形質細胞浸潤によって特徴づけられる稀な形質細胞疾患である。その臨床症状は、臨床経過がそうであるように非常に変わりやすく、しかし多発性骨髄腫とは異なり骨病変は稀である。ワルデンシュトレーム型マクログロブリン血症10例に由来するCD138濃縮骨髄形質細胞の全遺伝子発現プロファイリングは著しい異常を示したが(Zhanetal.,2002)、これらの細胞は、正常骨髄形質細胞のように、FRZBおよびDKKの発現を欠いた(図20)。
【0056】
実施例12
FRZBおよびエンドセリン受容体BはDKK−1と相関する
エンドセリン1は21アミノ酸の血管収縮因子である。エンドセリンの2種類の受容体である受容体AおよびBが同定されている。乳がんおよび前立腺がん細胞はエンドセリン1を産生することができ、および、エンドセリン1およびエンドセリン受容体Aの濃度上昇が骨転移を伴う進行前立腺がんで見出されている。エンドセリン1を産生した乳がん細胞は雌マウスで骨芽細胞転移を生じた。条件付け培地および外因性エンドセリン1はマウス頭頂骨培養において骨芽細胞増殖および新しい骨形成を刺激した(図31)。これらの結果は、エンドセリンが骨形成に関連することを示唆する。
【0057】
表3は、エンドセリン受容体B(ENDRB)の発現がDKK−1の発現と相関したことを示す。エンドセリン受容体Bは新たに診断された多発性骨髄腫の3分の1において「スパイク」遺伝子であった(図29)。エンドセリン受容体Bはまた、意義不明単クローン性免疫グロブリン血症(MGUS)およびくすぶり型多発性骨髄腫の部分集合で発現されていたが、正常形質細胞では発現されていなかった(図30)。
【0058】
【表3】
【0059】
実施例13
In Vivo薬物治療はDKK−1をアップレギュレートする
DKK−1発現はUV照射およびいくつかの他の遺伝毒性刺激によって大幅にアップレギュレートされることが示されている。多発性骨髄腫形質細胞もまたこの疾患を治療するのに用いられる薬物に反応してその遺伝子をアップレギュレートするかどうか調べるために、多発性骨髄腫形質細胞の遺伝子発現プロファイリングを、サリドマイド(図33)、ImiD(図34)、PS−341(図32)、またはデキサメタゾン(図35)を用いた48時間のin vivo処理の前後に実施した。これらのデータは、DKK−1およびFRZB発現を多くの例で大幅にアップレギュレートすることができ、およびそのようにして多発性骨髄腫形質細胞のアポトーシスを誘発することにおけるDKK−1の直接的役割を支持することを示した。最初の予備試験で試験したすべての物質に対して一次性に不応性であった新たに診断された患者がDKK−1の低レベルを示したこと、および薬物治療後にDKK−1またはFRZBの発現増大を示さなかったことに注目するのは興味深く、アポトーシスof多発性骨髄腫形質細胞のアポトーシスを促進することにおけるDKK−1発現の役割を支持する。この概念を支持して、DKK−1およびFRZBは、30例のHMCLおよび数例の髄外疾患で、低レベルから検出不能レベルにて発現されていた(図15)。
【0060】
実施例14
破骨細胞との多発性骨髄腫の共培養の結果としてJUN、FOS、およびDKK−1の大幅なダウンレギュレーションが生じる
骨髄腫細胞と破骨細胞の間の密接な関係は、消耗性溶解性骨破壊の多発性骨髄腫との関連によって臨床的に現れている。溶解性骨病変の発生は、骨髄腫形質細胞との直接的および間接的相互作用を介して破骨細胞の活性化によって引き起こされる。骨髄腫細胞の生存および増殖および疾患過程の維持における破骨細胞の決定的役割は、臨床的に調べられており、および原発性ヒト骨髄腫に関するSCID−huモデルといった実験モデルにおいてin vivoで実証されている。
【0061】
多発性骨髄腫形質細胞/破骨細胞相互作用の分子的結果を調査するために、CD138濃縮多発性骨髄腫形質細胞を、多発性骨髄腫の末梢血幹細胞(PBSC)または健常ドナー由来PBSCおよびMNCから得られた破骨細胞と共培養するex vivo系を開発した。正常ドナーまたは多発性骨髄腫患者に由来する末梢血幹細胞から得られたヒト破骨細胞と共培養したCD138濃縮多発性骨髄腫形質細胞は、アネキシンVフローサイトメトリー、BrdU標識化指数および[3H]TdR取り込みによって示される生存度および増殖活性を50日間の長期にわたって維持した。共培養の前後の形質細胞の純度レベルは、CD38/CD45フローサイトメトリーによって測定された通り、95%より大であった。
【0062】
12例の多発性骨髄腫形質細胞における最大12,000個の遺伝子の発現のマイクロアレイ分析を、4日間の共培養の前および後に実施した。12例の多発性骨髄腫形質細胞対および150例の新たに診断された多発性骨髄腫形質細胞の、7,913個のプローブセット(遺伝子)を用いた階層的クラスター分析は、共培養前標本が3つの主要なクラスター群内に分布した一方、共培養後標本は主要な枝のうち2つに互いに密にクラスター化したことを明らかにした。共培養後のその顕著な遺伝子発現変化の分析は、95個のプローブセット(遺伝子)が、共培養後に多発性骨髄腫形質細胞12例中少なくとも8例で2〜50倍変化したことを示した(アップレギュレート77個、およびダウンレギュレート18個)。健常ドナー5名に由来するCD138濃縮形質細胞はその同じ遺伝子のうち多数で同様の変化を示し、多発性骨髄腫形質細胞は破骨細胞に対して反応の変化を示さないことを示唆した。しかし、正常形質細胞は、その悪性対応物とは対照的に、破骨細胞との長期共培養で生存しなかった。
【0063】
最も著しい変化は、ケモカインGRO1、GR02、GR03、SCYA2、SCYA8、SCYA18、およびIL8のアップレギュレーションにあった。他の注目すべき遺伝子は、ケモカイン受容体CCR1、オステオポンチン(SPP1)、インテグリンITGB2およびITGB5、マトリクスメタロプロテイナーゼ9(MMP9)、カテプシンK(CTSK)およびカテプシンL(CTSL)を含んだ。驚くべきことに、多数の破骨細胞関連遺伝子が、77個のアップレギュレートされた遺伝子に含まれた。ダウンレギュレートされた遺伝子は、サイクリンB(CCNB1)、サイクリンB特異的ユビキチンリガーゼUBE2C、TSC−22ホモログDSIPI、およびJUN、JUND、FOS、およびFOSBを含んだ。
【0064】
遺伝子発現変化はまた、単独で培養された、および多発性骨髄腫形質細胞との共培養後の、破骨細胞10例で試験した。24個の遺伝子(アップレギュレート14個およびダウンレギュレート10個)が、共培養後の破骨細胞10例中少なくとも7例で2〜10倍変化した。多発性骨髄腫患者からまたは健常ドナーから得られた破骨細胞と共に培養した多発性骨髄腫形質細胞の間に遺伝子発現の有意差は無く、多発性骨髄腫破骨細胞は正常ドナーから得られたものと質的に異ならないことを示唆した。
【0065】
共培養実験から得られた培地で多発性骨髄腫形質細胞を培養した際に、遺伝子発現に有意差は観察されず、接触が重要であることを示唆した。共培養実験における多発性骨髄腫形質細胞の破骨細胞に対する低い比(1000:1)を考慮すると、すべての形質細胞が破骨細胞と同時に接触できる可能性は低い。したがって、破骨細胞と接触している多発性骨髄腫形質細胞と他の多発性骨髄腫形質細胞との間の何らかの細胞間情報伝達が起こる可能性が高い。
【0066】
破骨細胞が多発性骨髄腫骨疾患で主要な役割を果たすことおよび多発性骨髄腫に抗アポトーシス性シグナルを提供することが知られている。近年の研究は、JUNが直接的にDKK−1発現を調節すること、および、JUNおよびDKK−1がアポトーシスを調節することを示している。
【0067】
JUNおよびDKK−1を調節することによって破骨細胞が多発性骨髄腫形質細胞のアポトーシスを阻害し得るかどうかを判定するため、原発性多発性骨髄腫12例に由来する精製した形質細胞について、in vitroで得られた破骨細胞との48時間の共培養の前および後に、遺伝子発現プロファイリングを実施した。共培養中の多発性骨髄腫形質細胞は単独で培養された細胞よりも有意に高い長期生存度を有した。破骨細胞共培養の前後の多発性骨髄腫形質細胞の遺伝子発現プロファイリングは、JUN、FOS、およびFOSBは全例で2倍より大きくダウンレギュレートされた40個の遺伝子のうちの3個であることを明らかにした(n=12/12)。共培養後の多発性骨髄腫形質細胞で顕著に調節された95個の遺伝子を用いたHMCLおよび原発性多発性骨髄腫細胞の階層的クラスター分析は、HMCL、破骨細胞と共培養した原発性多発性骨髄腫、および新たに診断された多発性骨髄腫の部分集合の間に、これらの細胞型が相対的に低レベルのc−JUNおよびc−FOSを有した点で著しい類似性を明らかにした。
【0068】
重要なことに、原発性多発性骨髄腫細胞は単独で培養した際に高い程度の自発的アポトーシスを示す一方、破骨細胞の存在下で培養した多発性骨髄腫形質細胞は無限に生存することができる。これらのデータはJUNとDKK−1の間の関連を支持し、およびまた、髄外疾患および常に髄外疾患に由来するHMCLはJUNおよびDKKの両方を欠くため、多発性骨髄腫におけるJUNおよびDKK発現の消失は疾患進行と関連し得ることを示唆する。多発性骨髄腫増殖および挙動に対する破骨細胞の主要な影響の1つが、JUNおよびDKK−1をダウンレギュレートすることであってこれが直接的に形質細胞アポトーシスに影響を与えると推測するのは興味深い。HMCLおよび原発性多発性骨髄腫/破骨細胞共培養のDKK−1を用いた処理は、多発性骨髄腫形質細胞のアポトーシスを結果として生じることが予想される。DKK−1は破骨細胞に対して作用を有しない可能性が高く、それはこれらの細胞がWnt共受容体LRP−5を発現しないためである。正常骨髄由来形質細胞もまたDKK−1を発現せず、およびそれらの長命性を説明するのに役立つであろう。
【0069】
実施例15
形質細胞によるDKK1タンパク質の合成
多発性骨髄腫65例の骨髄生検由来の連続切片を、DKK1の存在に関して染色した。これらの例中の形質細胞は、遺伝子発現データと一致する形でDKK1を含んだ(図39)。正常ドナー5例由来の生検を用いた同様の実験は、どの細胞にもDKK1を検出できなかった。DKK1陽性骨髄腫には、間質性増殖パターンを伴う軽度の形態(核小体が見えない多量の細胞質)を有する強い傾向があった。この染色は骨に隣接する形質細胞で最も強かった。DKK1陰性骨髄腫は結節状または閉塞性増殖パターンを伴う重度の形態(大型化した核および顕著な核小体)を有する傾向があった。間質性増殖パターンについての生検では、DKK1は存在した(細胞のさまざまな割合で)かまたは存在しなかった。対照的に、より悪性の結節状増殖パターンを有する骨髄腫では一律にDKK1は存在しなかった。重要なことに、間質性および結節状増殖の両方を伴う例では、間質性細胞は陽性でありおよび結節状細胞は陰性であった。
【0070】
実施例16
骨髄形質中のDKK1タンパク質
酵素結合免疫吸着測定(ELISA)は、遺伝子発現データもまた利用可能である新たに診断された多発性骨髄腫患者173例のうち107例に由来する骨髄形質中のDKK1タンパク質の濃度は24.02ng/ml(S.D.49.58)であったことを示した。対照的に、DKK1は健常ドナー14例では8.9ng/ml(S.D.4.2)、MGUS14例では7.5ng/ml(S.D.4.5)、およびワルデンシュトレーム型マクログロブリン血症9例では5.5ng/ml(S.D.2.4)であった。DKK1遺伝子発現および骨髄形質中のDKK1のレベルは、骨髄腫107例で正に相関していた(r=0.65、P<0.001)(図40A)。また、骨髄形質および末梢血形質中のDKK1タンパク質レベル間に、両方の標本を同時に採取した骨髄腫41例において強い相関があった(r=0.57、P<0.001)。
【0071】
骨髄形質中のDKK1タンパク質レベルおよび骨病変の存在の両方が測定された患者68例のうち、MRI病変1個以上およびX線病変なしの患者におけるDKK1タンパク質は、MRI病変なしおよびX線病変なしの患者と比較して有意に差があったが(中央値レベル:20ng/ml対9ng/ml;p=0.002)、MRI病変1個以上およびX線病変1個以上の患者と比較して有意差は無かった(中央値レベル:20ng/ml対14ng/ml;p=0.36)(図40B、表2)。
【0072】
実施例17
in vitro骨芽細胞分化に対する骨髄血清の作用
骨形成タンパク質−2は、運命決定されていない間葉性前駆細胞株C2C12 (Katagiri,et al.,1994)の骨芽細胞への分化を、Wnt/b−カテニンシグナル伝達(Bain,et al.,2003; Roman−Roman,et al.,2002)が関与する経路を介して誘導することができる。骨芽細胞分化の特異的マーカーであるアルカリホスファターゼは、5パーセントウシ胎仔血清中で5日間培養したC2C12細胞において検出不能であった(図41A)。C2C12細胞の、50ng/mlのBMP−2を用いた5日間の処理は、C2C12細胞がアルカリホスファターゼを産生するように誘導したが、一方、アルカリホスファターゼはBMP−2および50ng/ml組み換えヒトDKK1と一緒に培養したC2C12細胞によって産生されなかった。アルカリホスファターゼ産生に対するこのin vitro 作用は、ポリクローナル 抗DKK1 抗体によって中和されたが、非特異的ポリクローナルヤギIgGでは中和されなかった。骨髄腫患者5例に由来する、DKK1濃度が12ng/mlを超える骨髄血清は、BMP−2で処理したC2C12細胞によるアルカリホスファターゼの産生を阻害し、およびこの作用は抗DKK1抗体によって逆転されたが非特異的IgGでは逆転されなかった(図41B)。対照的に、50ng/mlBMP−2および正常ドナーの骨髄由来の血清10パーセントで処理したC2C12細胞は、細胞によるアルカリホスファターゼの産生を誘導した(図41B)。
【0073】
下記の参考文献をここで引用した:
Zhan et al.,Global gene expression profiling of multiple myeloma,monoclonal gammopathy of undetermined significance,and normal bone marrow plasma cells.Blood 99: 1745−1757 (2002).
Zhan et al.,Gene expression profiling of human plasma cell differentiation and classification of multiple myeloma based on similarities to distinct stages of late−stage B−cell development.Blood 101:1128−1140 (2003).
Fedi et al.Isolation and biochemical characterization of the humanDkk−1 homologue,a novel inhibitor of mammalian Wnt signaling.J Biol Chem 274: 19465−72 (1999).
Gallea et al.Bone 28 : 491−8 (2001).
Spinella−Jaegle et al.Bone 29: 323−30 (200
1).
Westfall and Young.Resampling−based multiple testing: Examples and methods for p−value adjustment.Hoboken,NJ: Wiley−Interscience,360 (1993).
Golub et al.Science 286: 531−7 (1999).
Katagiri et al.J Cell Biol 127: 1755−66 (1994).
Bain et al.Biochem Biophys Res Commun 301: 84−91(2003).
Roman−Roman et al.American Society of Bone Mineral Research,2002.
本明細書中で言及した特許または出版物はいずれも、本発明が関係する分野の当業者のレベルを示す。さらに、これらの特許および出版物は、個々の出版物が参照により本開示に含まれると具体的および個別に示されたのと同等に、参照により本開示に含まれる。
【技術分野】
【0001】
国庫補助の説明
本発明は、部分的に、米国立がん研究所助成金CA55819およびCA97513の下に連邦政府からの助成金を用いて開発された。このため、米国政府は本発明に一定の権利を有する。
【0002】
関連出願へのクロスリファレンス
本出願は、2002年12月5日出願で現在放棄されている米国特許仮出願第60/431,040号の利益を主張する。
【背景技術】
【0003】
本発明は一般的に多発性骨髄腫の研究に関する。より詳細には、本発明は全遺伝子発現プロファイリング比較による骨髄腫骨疾患の分子決定因子の同定に関する。
【0004】
関連技術
多発性骨髄腫(MM)は、米国で年間約15,000人が罹患する、最終分化した形質細胞(PC)の稀でさらに不治の悪性腫瘍であって、二番目に多い造血器悪性腫瘍に当たる。多発性骨髄腫は、白人人口においてリンパ性悪性腫瘍全体の13%に相当し、および黒人人口においてリンパ性悪性腫瘍の31%に相当する。悪性形質細胞は、骨髄へホーミングしおよび骨髄で増殖し、正常造血の消失による貧血および免疫抑制を引き起こす。
【0005】
多発性骨髄腫はまた、全身性骨粗鬆症および局所性骨破壊を伴い、消耗性骨痛および、骨折、脊髄圧迫および高カルシウム血症への感受性に繋がる。骨髄腫は、溶解性骨疾患を常に伴う唯一の血液悪性腫瘍であって、および局所性骨破壊は形質細胞に隣接する部分に限られており、悪性形質細胞が破骨細胞機能および/または骨芽細胞アネルギーを促進する因子を分泌することを示唆する。骨疾患の有病率はしばしば骨の併発を伴わないくすぶり型骨髄腫から、孤立性形質細胞腫、骨塩密度の全身性損失または限局性溶解性骨病変が患者の約80%にみられるびまん性または限局性多発性骨髄腫まで、骨髄腫の症状によって異なる。
【0006】
近年、溶解性骨疾患は、骨髄腫の結果であるだけでなく、疾患進行を促進することに複雑に関与していることが明らかになっている。骨代謝回転速度における変化は、意義不明単クローン性免疫グロブリン血症(MGUS)からの顕性骨髄腫への最大3年までの臨床的進行を予測する。最初は破骨細胞と骨芽細胞活性とは連動している一方、その連動は疾患進行に伴って失われる。破骨細胞活性は上昇したままでありおよび骨芽細胞活性は低下し、結果として溶解性骨疾患を伴う。5T2マウス骨髄腫および原発性ヒト骨髄腫のSCID−huモデルでの研究は、破骨細胞活性の阻害は骨髄腫増殖の阻害および骨髄腫腫瘍組織量の減少を伴うことを実証した。これらの研究は、ビスホスホネートを用いた骨吸収の阻害は抗骨髄腫作用を有したという報告を支持する。
【0007】
骨髄腫関連溶解性骨疾患における破骨細胞の生物学は集中的に調べられている一方、骨芽細胞活性における疾患関連変化およびその基礎となる機構についてはほとんど知られていない。骨髄腫において、間葉幹細胞が造骨系譜へ分化する能力が損なわれていると示唆されている。しかし、そのような障害を担う機構は解明されていない。
【0008】
正常な健常ドナー由来の骨髄形質細胞および新たに診断された多発性骨髄腫由来の悪性骨髄形質細胞の全遺伝子発現プロファイリング(GEP)比較が、多発性骨髄腫の悪性形質転換に関与する疾患遺伝子候補および妨害された経路を同定するための強力な方法に当たることが示されている(非特許文献1)。
【先行技術文献】
【非特許文献】
【0009】
【非特許文献1】Zhan et al.,Global gene expressi on profiling of multiple myeloma,monoclo nal gammopathy of undetermined significa nce,and normal bone marrow plasma cells. Blood 99: 1745−1757 (2002)。
【発明の概要】
【発明が解決しようとする課題】
【0010】
先行技術は、多発性骨髄腫骨疾患の原因となり得る悪性形質細胞で発現されている遺伝子を同定するための比較分析、および、多発性骨髄腫骨疾患を診断および治療するための方法に欠けている。本発明は本分野におけるこの年来の必要及び要望を満たす。
【課題を解決するための手段】
【0011】
溶解性骨疾患の分子決定因子を同定するため、溶解性病変の放射線的証拠を示さない(n=28)新たに診断された多発性骨髄腫(newly diagnosed multiple myeloma) に由来するCD138濃縮形質細胞における最大12,000個の遺伝子の発現プロファイルを、3個以上の溶解性病変を有するもの(n=47)と比較した。骨芽細胞分化におけるWNTシグナル伝達の決定的な役割と合致して、2種類の分泌型WNTシグナル伝達拮抗因子である、可溶性frizzled関連タンパク質3(SFRP−3/FRZB)およびDickkopf−1(DKK−1)のヒトホモログが、溶解性骨病変を有する47例中40例で発現されていたが、骨病変を欠く28例中の16例だけで発現されていた(P<0.05)。免疫組織化学は、遺伝子発現が高い例に由来する形質細胞で、高レベルのDKK−1およびFRZBを示した。重要なことに、DKK−1およびFRZBは、正常骨髄ドナー45例、または、骨疾患を欠く関連する形質細胞悪性腫瘍であるワルデンシュトレーム型マクログロブリン血症10例に由来する形質細胞では発現されていなかった。
【0012】
高いDKK−1を有する多発性骨髄腫患者に由来した血清は、Wntシグナル伝達および骨芽細胞分化の両方をin vitroで遮断した。重要なことに、DKK−1およびFRZB抗体との血清のプレインキュベートは、この機能を阻害した。DKK−1発現の調節および次にはアポトーシスにおけるJUNの主要な役割と合致して、髄外疾患および原発性難治性疾患に由来する形質細胞は、JUNおよびDKK−1の低発現を有した。
【0013】
多発性骨髄腫形質細胞は、in vivo処理後にDKK−1およびFRZB遺伝子発現の大幅なアップレギュレーションを示した。DKK−1およびFRZBは、多発性骨髄腫形質細胞において、その疾患を治療するために用いられる遺伝毒性薬での患者の治療後に、アップレギュレートすることができ、そのようにして多発性骨髄腫細胞アポトーシスにおけるDKK−1の役割を助長する。in vitro由来破骨細胞(OC)と共培養した原発性多発性骨髄腫細胞はアポトーシスを欠き、しかもこれはJUN、FOS、FOSB、およびDKK−1のダウンレギュレーションと密接に相関した。
【0014】
本発明で開示される結果は、DKK−1およびFRZBの産生および/または分泌を遮断することが多発性骨髄腫患者における骨減少を防ぐかまたは逆転させる可能性があることを示す。他の用途は、DKK−1およびFRZB阻害因子を一般人口における骨減少を防ぐために用いることを含む。加えて、Wntシグナル伝達は近年、造血幹細胞の自己再生能力にとって決定的に重要であることが示されている。さらに、HSC増殖に必要な骨髄ニッチは、成熟骨芽細胞によって形成される。Wntシグナル伝達に対するDKK1およびFRZBによる遮断は、直接的におよび間接的に肝星細胞(HSC)増殖を障害し、およびそのようにして、多発性骨髄腫で見られる免疫抑制および貧血の部分的な原因となり得る。このように、DKK1および/またはFRZBの遮断はまた、骨髄腫を有する患者の大部分に見られる造血の欠陥を、防ぐかまたは逆転することができる。
【0015】
本発明のその他の態様、特性、および利点は、本発明の現在好ましい実施形態の下記の説明から明らかになる。これらの実施形態は開示の目的のために与えられる。
【0016】
本発明の上述した特性、利点、および目的、およびその他明らかになる内容が達成されおよび詳細に理解可能となるように、上記に簡潔に要約した、より具体的な説明および本発明の一部の実施形態が、添付の図面に図解されている。これらの図面は本明細書の一部を成す。しかし、添付の図面は本発明の好ましい実施形態を図解し、およびしたがって実施形態の範囲を制限しないと考えられることに注意する。
【図面の簡単な説明】
【0017】
【図1A】図1Aおよび1Bは、骨髄腫における骨病変を反映する全遺伝子発現パターンを示す。図1Aは、MRI限局性病変が0個(n=36)および1個以上(n=137)の患者に由来する悪性形質細胞において有意に差がある発現がされているとロジスティック回帰分析によって同定された(P<0.0001)57遺伝子の正規化発現レベルのクラスタビューを示す。1個以上のMRI病変がある患者に由来する形質細胞における発現上昇を示している28遺伝子は、有意性の階層に基づいて上から下へ並べられている。同様にMRI病変が無い患者において有意な上昇を示す30遺伝子は、有意性階層に基づいて下から上へ並べられている。遺伝子名(遺伝子名が無い場合はアフィメトリクス・プローブセット識別子)は左に列記する。正規化発現スケールは、データ表示の下に示す通り、−30(青)から+30(赤)の範囲にわたる。順列調整後に有意なままである4個の遺伝子を下線で示す。
【図1B】図1Bは、X軸上に示す正常骨髄(BPC)、意義不明単クローン性免疫グロブリン血症(MGUS)、ワルデンシュトレーム型マクログロブリン血症(WM)、および多発性骨髄腫(MM)の患者に由来する形質細胞におけるDKK1遺伝子発現の棒グラフを示す。MM標本は:MRI病変なし/X線病変なし、MRI病変1個以上/X線病変なし、およびMRI病変1個以上/X線病変1個以上、の3つの骨病変群に分けられている。MAS5.01から得られる遺伝子発現の定量尺度であるアフィメトリクス・シグナルをy軸上に示す。各標本中のDKK1遺伝子発現レベルを棒で示し、棒の高さは遺伝子発現強度に比例する。標本は、DKK1遺伝子発現が最低から最高へ、x軸の左から右へ並べられている。各群の標本の数は、各群の名称の下に示す。MM部分群間の比較についての統計値は本文中に示す。
【図2】図2は、RHAMMが骨病変を有する多発性骨髄腫患者においてアップレギュレートされたことを示す。
【図3】図3は、RHAMMは正常形質細胞および意義不明単クローン性免疫グロブリン血症(MGUS)にはほとんど存在しないが、事実上すべてのヒト骨髄腫細胞株に存在したことを示す。
【図4】図4は、securinが骨疾患を有する多発性骨髄腫患者においてアップレギュレートされたことを示す。
【図5】図5は、MIP−1αおよびCCR1が多発性骨髄腫において「スパイク」 遺伝子であったことを示すが、それらは溶解性病変と相関しなかった。黒棒:CCR 1;灰色棒:MIP−1α。
【図6】図6は、MIP−1αが正常形質細胞(PC)において低レベルで発現されたことを示す。
【図7】図7は、骨病変を有する多発性骨髄腫におけるWNT拮抗因子DKK−1の発現を示す。
【図8】図8は、溶解性骨病変を有する多発性骨髄腫におけるWNTおとり受容体FRZBの発現を示す。
【図9】図9は、溶解性骨病変を有する多発性骨髄腫におけるDKK−1およびFRZBの発現を示す。黒棒:DKK−1;灰色棒:FRZB。
【図10】図10は、扁桃形質細胞でFRZBが発現されたことを示す。PBC、TBC=扁桃B細胞、TPC=扁桃形質細胞、BPC=骨髄形質細胞、WPC、WBC、CLL。
【図11】図11は、DKK−1が正常B細胞または形質細胞で発現されなかったことを示す。PBC、TBC=扁桃B細胞、TPC=扁桃形質細胞、BPC=骨髄形質細胞、WPC、WBC、CLL。
【図12】図12は、意義不明単クローン性免疫グロブリン血症(MGUS)におけるDKK−1発現が、くすぶり型多発性骨髄腫(SMM)および新たに診断された多発性骨髄腫(MM)と比較して低かったことを示す。
【図13】図13は、FRZBが意義不明単クローン性免疫グロブリン血症(MGUS)において上昇したこと、およびくすぶり型多発性骨髄腫(SMM)および新たに診断された多発性骨髄腫(MM)において発現がより高かったことを示す。
【図14】図14は、意義不明単クローン性免疫グロブリン血症(MGUS)およびくすぶり型多発性骨髄腫(SMM)におけるDKK−1およびFRZBの発現を示す。
【図15】図15は、髄外疾患におけるDKK−1の低発現を示す。
【図16】図16は、DKK−1およびFRZBの発現が、髄質PCT由来の形質細胞において、腸骨稜由来のものよりも高い傾向があることを示す。PCT、FNA。
【図17】図17は、髄質PCTの穿刺吸引検体におけるDKK−1およびFRZBの発現を示す。
【図18】図18は、髄質形質細胞腫におけるDKK−1およびFRZBの高発現を示す。
【図19】図19は、骨減少症を伴う多発性骨髄腫におけるDKK−1のより高い発現を示す。
【図20】図20は、DKK−1がワルデンシュトレーム型マクログロブリン血症に由来する形質細胞で発現されていなかったことを示す。
【図21】図21は、WNT5Aが新たに診断された多発性骨髄腫で上昇していたことを示す。
【図22】図22は、WNT5Aが溶解性病変を有する多発性骨髄腫でより高い傾向があることを示す。
【図23】図23は、WNT5Aが意義不明単クローン性免疫グロブリン血症(MGUS)およびくすぶり型多発性骨髄腫(SMM)でも上昇していたことを示す。
【図24】図24は、WNT10Bが溶解性病変を有する多発性骨髄腫より低い傾向があることを示す。
【図25】図25は、WNT5AおよびWNT10Bが逆相関する傾向があることを示す。黒棒:WNT10B、灰色棒:WNT5A。
【図26】図26は、DKK−1がSK−LMS細胞株に存在したことを示す。
【図27】図27は、原発性多発性骨髄腫がDKK−1タンパク質を合成したことを示す。
【図28】図28は、再発性および原発性難治性多発性骨髄腫におけるDKK−1低発現を示す。
【図29】図29は、エンドセリン受容体Bは新たに診断された多発性骨髄腫の3分の1において「スパイク」遺伝子であったことを示す。
【図30】図30は、意義不明単クローン性免疫グロブリン血症(MGUS)およびくすぶり型多発性骨髄腫におけるエンドセリン受容体Bの発現を示す。正常形質細胞はエンドセリン受容体Bを発現しない。
【図31】図31は、骨形成におけるエンドセリン受容体Bの関与を示す。
【図32】図32は、PS−341を用いた治療後のDKK−1発現を示す。
【図33】図33は、新たに診断された多発性骨髄腫におけるサロミド(サリドマイド)を用いた治療後のDKK−1発現を示す。
【図34】図34は、IMiDを用いた治療後のDKK−1発現を示す。
【図35】図35は、新たに診断された多発性骨髄腫における、デキサメタゾン処理後のDKK−1発現を示す。
【図36】図36は、破骨細胞との共培養後の多発性骨髄腫細胞におけるJUNおよびFOSのダウンレギュレーションを示す。
【図37】図37は、破骨細胞共培養におけるJUNおよびDKK−1ダウンレギュレーションを示す。
【図38】図38は、多発性骨髄腫骨疾患におけるWNTシグナル伝達を示す。
【図39】図39は、疾患進行に伴い発現が消失する、軽度骨髄腫におけるDKK1の過剰発現を示す。DKK1の発現は骨髄腫骨髄生検の免疫組織化学検査によって調べた。DKK1遺伝子発現が高い(a〜b)および低い(c〜d)骨髄腫患者に由来する骨髄生検の連続切片(倍率550x)を示す。スライドは、H&E(aおよびc)または抗DKK1および二次抗体(bおよびd)を用いて染色している。二次抗体単独の使用では細胞を染色できなかった(データ記載せず)。拡大像(倍率1,200x)は各H&E像の左上隅にある。図aは多量の細胞質を有し核小体が見えない軽度の形態を示している形質細胞との関与の間質性パターンを伴う骨髄腫を示す。図bは骨に非常に近い抗DKK1抗体を用いた間質性パターン中の形質細胞のポジティブ染色を示す。図cは大型化した核および顕著な核小体を有する重度の形態を示している形質細胞との結節状または閉塞性パターンを伴う骨髄腫を示す。図dは抗DKK1抗体を用いて形質がポジティブ染色されないことを示す。
【図40A】図40Aおよび40Bは、骨髄形質中のDKK1タンパク質がDKK1遺伝子発現とおよび骨病変の存在と高度に相関することを示す。図40Aは、DKK1mRNAの発現がマイクロアレイによっておよびDKK1タンパク質がELISAによって、新たに診断された骨髄腫の計107例で検出されたことを示す。両方の分析の結果は平均値0および変動1となるように底2の対数で変換および正規化した。棒のそれぞれは各標本における遺伝子発現およびタンパク質発現の相対的関係を示す。骨髄腫形質細胞中のDKK1mRNAと骨髄形質中のタンパク質との間には有意な相関があった(r=0.65,P<0.001)。
【図40B】図40Bは、骨髄形質中のDKK1タンパク質レベルの棒グラフ表示を示す。正常ドナー(BPC)、意義不明単クローン性免疫グロブリン血症(MGUS)、ワルデンシュトレーム型マクログロブリン血症(WM)、および多発性骨髄腫(MM)患者由来の形質細胞をx軸上に示す。MM標本は:MRI病変なし/X線病変なし、MRI病変1個以上/X線病変なし、およびMRI病変1個以上/X線病変1個以上、の3つの骨病変群に分けられている。DKK1タンパク質濃度(ng/ml)をy軸上に示す。より低い範囲でのDKK1タンパク質レベルの比較を可能にするため、200ng/mlを最大値とした。この結果、DKK1濃度が476ng/mlである1標本の切り捨てが生じた。各標本中のDKK1タンパク質レベルを棒で示し、棒の高さはDKK1タンパク質レベルに比例する。標本は、DKK1タンパク質レベルが最低から最高へ、x軸の左から右へ並べられている。各群の標本の数を各群の下に示す。
【図41A】図41Aおよび41Bは、組み換えDKK1およびMM形質が、BMP−2処理C2C12細胞におけるアルカリホスファターゼ産生を、DKK1に依存する方法で遮断することができることを示す。図41Aは、C2C12細胞において、5パーセントウシ胎仔血清単独またはBMP2、BMP2+DKK1、BMP2+DKK1+抗DKK1、またはBMP−2+DKK1+ポリクローナルIgGを加えての存在下での5日間の培養後に測定した、骨芽細胞分化のマーカーであるアルカリホスファターゼレベルを示す(y軸)。棒のそれぞれは3連の実験の平均値(±SEM)を表す。アルカリホスファターゼの活性はBMP−2の存在下で上昇したこと、および組み換えDKK1との共培養によるこのタンパク質の顕著な減少に注意する。同じく、抗DKK1抗体はDKK1の抑制活性を遮断できたが、ポリクローナルIgGはできなかったことに注意する。
【図41B】図41Bは、C2C12細胞における、5パーセントウシ胎仔血清単独または50ng/mlBMP−2+正常骨髄形質(NS)10パーセントまたはBMP−2+新たに診断された骨髄腫の患者10名に由来する骨髄腫骨髄形質10パーセント(同定された標本を提供)、またはBMP2+骨髄腫患者形質10パーセント+抗DKK1またはヤギポリクローナルIgG中で5日間これらの細胞を培養後に測定したアルカリホスファターゼレベル(y軸)を示す。棒のそれぞれは3連の実験の平均値(±SEM)を表す。各骨髄形質標本からのDKK1濃度はELISAによって測定し、および1:10希釈後の培養中の終濃度をx軸上に示す。DKK1が12ng/mlより大である標本はアルカリホスファターゼ産生に影響を有したことに注意する。星印はBMP2+正常ヒト骨髄形質10パーセント中のアルカリホスファターゼと比較してP<0.05を示す。
【発明を実施するための形態】
【0018】
本発明は、分泌型WNTシグナル伝達拮抗因子DKK−1およびFRZBが多発性骨髄腫にみられる骨破壊を媒介することを実証する。Wntシグナル伝達が骨芽細胞増殖および分化に絶対的に必須であることの新たな証拠と共に、これらのデータは、これらの因子が骨減少に続く正常な補償的骨産生を抑制することによって骨芽細胞アネルギーを生じることおよび多発性骨髄腫骨疾患に寄与することを強く示唆する。
【0019】
破骨細胞活性を刺激することにおける多発性骨髄腫形質細胞の役割は集中的に研究されており、およびいくつかの鍵となる関連が確立されている。ここに示すデータは、多発性骨髄腫における骨芽細胞機能不全の、可能な機構的説明の証拠を初めて提供する。これらは、最近の研究がWNTシグナル伝達の阻害が骨芽細胞機能における欠陥を引き起こすことを示した点で有意義である。分泌型DKK−1およびFRZBは、多発性骨髄腫でみられる全身性骨粗鬆症、および形質細胞病巣近傍の悪化した局所性骨破壊の両方を説明し得る。
【0020】
重要なことに、DKK−1およびFRZBは、独立した経路を介しWNTシグナル伝達を阻害するように作用し、このことはそれらの同時発現が相乗作用を有することを示す。
したがって、これらの遺伝子は、骨疾患の程度および骨疾患を発症する将来のリスクを予測するのに用いることができる可能性がある。さらに、これらのタンパク質の阻害因子は、骨疾患を阻害するのに用いることができる可能性がある。また、これらの因子が一般集団における骨粗鬆症に関与している可能性もある。
【0021】
WNTシグナル伝達経路
Wnt遺伝子は、脊椎動物胚発生中に空間的におよび組織限定パターンで発現される分泌型ポリペプチドの大きなファミリーを構成する。マウスでの突然変異分析は、体軸、中枢神経系および四肢のパターン化、および器官形成中の誘導現象の調節といったさまざまな発生段階を調節することにおけるWntの重要性を示している。分泌型増殖因子のWntファミリーは、Frizzled(Fz)受容体およびその共受容体であるLDL受容体関連タンパク質5または6(LPR5またはLRP6)を介して、おそらくWntによって誘導されるFz−LPR5/LRP6複合体形成を通じて、シグナル伝達を開始する。
【0022】
Wntの分泌型拮抗因子は、Frizzled(Fz)関連タンパク質(FRPs)、Cerberus、Wnt阻害因子(WIF)およびDickkopf(DKK)を含む。Frizzled(Fz)関連タンパク質、CerberusおよびWnt阻害因子は全部が、Wntに結合および封鎖することによって作用することが示されている。Fzの分子模倣または他の機構によるWnt封鎖によって作用を発揮するWnt拮抗因子とは異なり、Dickkopf−1(DKK−1)は受容体複合体のLPR5/LRP6成分に結合することによって正規のWntシグナル伝達を特異的に阻害する。
【0023】
DKK−1は脊椎動物頭部オーガナイザーから分泌される頭部誘導因子であり、およびWntシグナル伝達に拮抗することによって全部発生を誘導する。DKK−1はLRP6に対する高親和性リガンドであり、およびWntによって誘導されるFz−LRP6複合体形成を妨げることによってWntシグナル伝達を阻害する。DKK−1はWntともFzとも結合せず、Wnt−Fz相互作用にも影響しない。頭部誘導およびWntシグナル伝達阻害におけるDKK−1機能は、LPR5/LRP6と結合しおよびFz−LPR5/LRP6会合を破壊する能力と密接に相関する。LPR5/LRP6機能およびDKK−1阻害は、Wnt/Fzベータカテニン経路に特異的であるように見える。これらの知見はしたがって、Wntシグナル調節のための新規の機構を明らかにする。
【0024】
WNTシグナル伝達および骨芽細胞分化
近年の研究は、Wntシグナル伝達経路が骨芽細胞分化および機能に決定的であることを示している。低密度リポタンパク質受容体関連タンパク質5(LRP5)の遺伝子にターゲッティング破壊を有するマウスは低骨量表現型を発生する。LRP5は骨芽細胞で発現され、および骨芽細胞における最適なWntシグナル伝達に必要である。In vivoおよびin vitro分析は、この表現型は生後に明らかになり、さらにそれはCbfa1非依存的な態様で骨芽細胞増殖および機能の低下に対して二次的なものである。
【0025】
ヒトでは、LRP5における突然変異は、常染色体劣性疾患である骨粗鬆症−偽神経膠腫症候群(OPPG)を引き起こす。骨粗鬆症−偽神経膠腫症候群のキャリヤーは、年齢および性別が一致する対照と比較した場合に骨量低下がある。
【0026】
重要なことに、LRPにおける別々のおよび異なる突然変異は、高骨量表現型を結果として生じる。骨粗鬆症−偽神経膠腫突然変異とは対照的に、高骨量の性質は機能突然変異の利益である。その変異を有する対象者において骨吸収のマーカーは正常であった一方、オステオカルシンといった骨形成のマーカーは顕著に増加していた。Wntによるシグナル伝達の既知の標的であるフィブロネクチンのレベルもまた上昇していた。In vitro試験は、Dickkopf−1(DKK−1)によるWntシグナル伝達の正常な阻害は本突然変異の存在下では不完全であったこと、およびこのことが抑制されないWnt活性によるシグナル伝達増加を結果として生じたことを示した。これらの知見は、高骨量でのLRP5機能変化の役割を実証し、および骨粗鬆症の予防または治療のための標的候補としてDKKを指し示す。
【0027】
多発性骨髄腫におけるWNT シグナル伝達および骨疾患
骨芽細胞機能におけるDKK−1の役割の間接的証拠が、高骨量表現型と連鎖しているLRP−5における機能突然変異の利益の同定によって与えられている。加えて、FRZB(SFRP−3)のホモログである分泌型firzzled関連タンパク質(SFRP−1)のターゲッティング破壊は、骨芽細胞の減少および骨細胞アポトーシスおよび骨梁形成増加に繋がる。
【0028】
骨量に影響する量的形質遺伝子座(QTL)がLRP−5領域に局在化されており、一般の人口は骨粗鬆症を発症する異なるリスクを有することを示唆する。多発性骨髄腫骨疾患は、DKK−1/FRZB発現とLRP−5対立遺伝子によって与えられる低骨量の遺伝的素因との組み合わせ作用によって影響され得ると考えられる。多発性骨髄腫の症例をLRP−5対立遺伝子変異について遺伝子型分析することができ、およびこの情報を骨疾患、およびDKK−1およびFRZB発現と相関させることができる。
【0029】
意義不明単クローン性免疫グロブリン血症(MGUS)は、多発性骨髄腫を発症する傾向がある形質細胞疾患であり、明らかな骨疾患を欠くことによって多発性骨髄腫から区別される。DKK−1および/またはFRZB発現が意義不明単クローン性免疫グロブリン血症の3分の1で発見されることの意義は不明であるが、しかしこれらの症例が多発性骨髄腫を発症するより高いリスクにある可能性があることを示唆し得る。多発性骨髄腫と同様に、この素因はまた遺伝するLRP5対立遺伝子と関連する可能性がある。代替的に、これらの意義不明単クローン性免疫グロブリン血症の例は、放射線スキャンではまだ明らかでない基礎となる発症前骨疾患を有するであろう。
【0030】
ここに示すデータは、多発性骨髄腫形質細胞によるDKK−1発現がどのように多発性骨髄腫疾患増殖調節および骨破壊に結びつき得るか、およびこれらの二つの現象がどのように1つの分子によって統合され得るかについてのモデルを示唆する。そのモデルでは、原発性多発性骨髄腫は高レベルのDKKを発現し、およびこれらのレベルはその疾患を治療するのに用いられる薬物療法で上昇させることができる。高レベルのDKK−1は多発性骨髄腫細胞のアポトーシスを誘導する可能性が高く、および、細胞増殖が高率のDKK−1誘導性アポトーシスによって緩和されるため、本疾患の初期における相対的に遅い進行を説明し得る。しかし、疾患が進行するにつれ、JUNおよびDKK−1に、破骨細胞に誘導される低下が生じ、これは最終的に髄外疾患に見られるような、JUNおよびDKK−1発現の構成的消失に発展する。
【0031】
このように、DKK−1発現を多発性骨髄腫形質細胞の視点から見るならば、高レベルのDKK−1発現は本疾患を肯定する性質と見なすことができる。しかし、DKK−1誘導性アポトーシスに対して極めて感受性である間葉性細胞系譜については、高レベルのこの分泌産物は、前駆骨芽細胞のおよびおそらく間葉幹細胞さえの大規模なプログラム細胞死もまた誘導する点で、二通りの効果を有するであろう。疾患初期における高レベルのDKK−1は間葉幹細胞の永久的損失に繋がる可能性があると考えられ、この概念は、寛解誘導後または疾患進行中に破骨細胞が多発性骨髄腫形質細胞によるDKK−1分泌を抑制する可能性が高い際に、骨修復の欠如が観察されることによって支持される。したがって、この知識の活用は、多発性骨髄腫形質細胞に対するDKK−1の作用を強調するが同時に骨芽細胞またはその前駆細胞に対するDKKの骨損傷作用を妨げる、多発性骨髄腫のための新しい治療法の開発に繋がり得る。
【0032】
本発明の一実施形態では、WNTシグナル伝達拮抗因子の発現レベルを調べることによって、多発性骨髄腫患者において骨疾患を発症する可能性を判定する方法が提供される。その拮抗因子の、正常個体における発現と比較して増加した発現は、その患者が骨疾患を発症する可能性を有することを示す。好ましくは、そのWNTシグナル伝達拮抗因子は可溶性frizzled関連タンパク質3(SFRP−3/FRZB)、またはDickkopf−1(DKK1)のヒトホモログである。一般的に、これらのタンパク質の発現レベルは、核酸またはタンパク質レベルで測定することができる。
【0033】
別の一実施形態では、WNTシグナル伝達拮抗因子の発現を阻害することによって、多発性骨髄腫患者において骨疾患を治療する方法が提供される。好ましくは、そのWNTシグナル伝達拮抗因子は可溶性frizzled関連タンパク質3(SFRP−3/FRZB)、またはDickkopf−1(DKK1)のヒトホモログである。一般的に、これらの拮抗因子の発現は、核酸またはタンパク質レベルで阻害することができる。
【0034】
さらに別の一実施形態では、WNTシグナル伝達拮抗因子の発現を阻害することによって、個体において骨減少を防ぐ方法が提供される。好ましくは、そのWNTシグナル伝達拮抗因子は可溶性frizzled関連タンパク質3(SFRP−3/FRZB)、またはDickkopf−1(DKK1)のヒトホモログである。一般的に、これらの拮抗因子の発現は、核酸またはタンパク質レベルで阻害することができる。
【0035】
さらに別の一実施形態では、DKK1遺伝子(登録番号NM012242)の発現またはDKK1遺伝子によって発現されるタンパク質の活性を阻害する工程を含む、個体において骨減少を調節する方法が提供される。DKK1遺伝子発現は、たとえば、アンチセンスオリゴヌクレオチド、または抗DKK1抗体または可溶性LRP受容体によることを含む、当業者に既知である任意の方法によって阻害される。
【0036】
さらに別の一実施形態では、DKK1タンパク質の阻害薬を個体に投与する工程を含む、個体において骨減少を調節する方法が提供される。一般的に、この方法はその個体が多発性骨髄腫、骨粗鬆症、閉経後骨粗鬆症または悪性腫瘍関連骨減少といった疾患を有する場合に有効である。一般的に、悪性腫瘍関連骨減少は、骨への乳がん転移または骨への前立腺がん転移によって引き起こされる。
【0037】
下記の実施例は本発明のさまざまな実施例を説明する目的のために与えられ、および本発明をいかなる方法でも制限しないことが意図される。当業者は、本発明は目的を実施および上記の結果および利点、およびここに内在する目的、結果および利点を得るためによく適合されていることを容易に理解し得る。請求項の範囲によって定義される通りの本発明の精神の内に包含される、その中の変化および他の用途を当業者は考えることができる。
【実施例】
【0038】
実施例1
患者
新たに診断された多発性骨髄腫の患者174名、意義不明単クローン性免疫グロブリン血症の患者16名、ワルデンシュトレーム型マクログロブリン血症の患者9名、および健常者45名を試験した。米国アーカンソー医科大学の施設内倫理委員会は本研究を承認し、およびすべての被験者はインフォームド・コンセントを書面で提供した。表1は多発性骨髄腫の患者の特性を示す。
【0039】
【表1】
【0040】
実施例2
骨画像処理
画像は、遺伝子発現データの予備知識無しで、Canon PACS(画像保存およびカタログ化システム)を用いて検査した。MRIスキャンは1.5テスラGE Signaスキャナで実施した。X線は米国放射線医学会(ACR)規格に準拠してフィルムからデジタル化した。MRIスキャンおよびX線はACRのDICOM(医用デジタル画像処理および通信)規格を用いてCanon PACSシステムに接続した。画像処理は取扱説明書に従って実施した。MRI画像は、ガドリニウムT1前および後強調処理およびSTIR(短τ反転回復)強調処理を用いて作成した。
【0041】
実施例3
形質細胞単離および遺伝子発現プロファイリング
フィコール・ハイパック勾配遠心分離後に、骨髄から得られた形質細胞を単核細胞画分から、モノクローナルマウス抗ヒトCD138抗体(Miltenyi−Biotec,Auburn,CA)を用いる免疫磁気ビーズ選択によって単離した。CD138+/CD45−およびCD38+/CD45−マーカーを用いた二色フローサイトメトリー、免疫細胞化学法による免疫グロブリン軽鎖の存在、およびライト−ギムザ染色による形態で示された通り、遺伝子発現プロファイリングに使用した細胞の90パーセント超が形質細胞であった。総RNAはRneasyミニキット(Qiagen,Valencia,CA)を用いて単離した。標識化cRNAの調製および約10,000遺伝子を含むU95Av2マイクロアレイ(Affymetrix,Santa Clara,CA)へのハイブリダイゼーションは以前に記載した通りに実施した(Zhan et al.,2002; Zhan et al.,2003)。RNA増幅は不要であった。
【0042】
実施例4
免疫組織化学
ヒト総DKK1タンパク質(R&D Systems,Minneapolis,MN)に対して免疫化したヤギに由来する抗体をTris緩衝液で1:200希釈し、およびホルマリン固定してパラフィン包埋された骨髄生検切片に2時間室温にて加えた。隣接する切片をH&Eで染色した。抗原−抗体反応はDAB(ビオチニル化抗ヤギ抗体[Vector Laboratories,Burlingame,CA] [1: 400 希釈]およびストレプトアビジン−ホースラディッシュペルオキシダーゼ[Dako]染色後)を用いて発色させ、およびヘマトキシリン−2を用いて対比染色した。
【0043】
実施例5
酵素結合免疫吸着測定法(ELISA)
Nunc−Immuno MaxiSorp表面マイクロタイタープレートを、1xリン酸緩衝生理食塩水、pH7.2に溶解した1mg/mlの抗DKK1抗体50mlを用いて4℃にて一夜被覆し、および4パーセントウシ血清アルブミンを用いてブロックした。骨髄形質を希釈緩衝液(1×リン酸緩衝生理食塩水+0.1Tween−20+1パーセントウシ血清アルブミン)で1:50希釈した。ウェル当たり計50μlを負荷し、および4℃にて一夜インキュベートし、洗浄し、および緩衝液で0.2mg/mlに希釈したビオチニル化ヤギ抗ヒトDKK1 IgG(R&D Systems)とインキュベートし、その後ストレプトアビジン−ホースラディッシュペルオキシダーゼ(Vector Laboratories)の1:10,000希釈50μlを加え、すべて取扱説明書に従った。発色はOPD基質系(Dako)を用いて取扱説明書に基づいて達成した。組み換えヒトDKK1(R&D Systems)の連続希釈を用いて標準曲線を作成した。内因性DKK1を発現しない細胞株T293、および安定にトランスフェクションされたDKK1を有するT293(Fedi,et al.,1999)を用いて本ELISA測定法のバリデーションを行った。
【0044】
実施例6
骨芽細胞分化測定法
C2C12間葉性前駆細胞(American Type Tissue Culture,Reston,VA)は、10パーセント熱不活化ウシ胎仔血清を添加したDMEM(Invitrogen,Carlsbad,CA)中で培養した。C2C12細胞におけるアルカリホスファターゼ活性は記載の通りに測定した(Gallea,et al.,2001;Spinella−Jaegle,et al.,2001)。細胞溶解物はタンパク質含量についてマイクロBCA測定法キット(Pierce,Rockford,IL)を用いて分析した。各実験は3連で実施した。
【0045】
実施例7
統計解析
多発性骨髄腫患者における骨疾患は、ロジスティック回帰を用いてモデル化した。考慮した独立変数は、新たに診断された多発性骨髄腫174例からバージョン5.01MAS(Affymetrix,Santa Clara,CA)を用いて測定した最大10,000個の遺伝子(12,625個のプローブセット)からの遺伝子発現強度値(アベレージ・ディファレンス・コール)であった。各プローブセットについて、遺伝子発現の定量尺度である「シグナル」は、ロジスティック回帰モデルおよび順列調整分析への入力前にlog2に変換した。骨髄腫において骨疾患に関連し得る遺伝子については、先行する仮説は無かった。結果として、12,625個のプローブセットのそれぞれについて骨疾患の一変量モデルが用いられた。候補遺伝子は、順列調整された有意水準でのt検定(WestfallおよびYoung,1993)を用いて選別した。ウェストフォールおよびヤングの分析を用いて、複数の単変量仮説検定に合わせて調節した。DKK1シグナルおよびDKK1タンパク質レベルの群差は、ウィルコクソンの順位和検定を用いて検定した。骨疾患の状態による患者の特徴における有意差は、フィッシャーの正確確率検定またはカイ二乗検定のどちらかを用いて検定した。ロジスティック回帰によって同定された遺伝子の発現強度は、Clusterview(Golub,et.al.,1999)を用いて視覚化した。スピアマンの相関係数を用いて、遺伝子発現およびタンパク質レベルの相関を測定した。骨芽細胞分化における対照と各実験条件との間の有意差は、ウィルコクソンの順位和検定を用いて検定した;独自のC2C12実験それぞれについて別個の比較を実施した。両側p値0.05未満を有意とし、および両側p値0.10未満を傾向差とした。
【0046】
実施例8
骨髄腫細胞の発現プロファイリング
過剰発現されおよび骨病変の存在に伴っていた遺伝子を同定するために、骨病変を有するかまたは有しない患者に由来するマイクロアレイデータの比較を実施した。MRIで定義される骨の限局性病変は、放射線で同定可能な溶解性病変の前に生じ得るので、骨病変を評価するためにT1強調およびSTIR強調画像処理を用いた。骨病変の無い患者(n=36)およびMRIで定義される限局性病変1個以上(1+)を有する患者(n=137)の骨髄に由来する精製形質細胞中の遺伝子約10,000個の遺伝子発現パターンを、ロジスティック回帰分析によってモデル化した。そのモデルによって、2群の患者で発現に差のあった(P<0.0001)57個の遺伝子が同定された(図1A)。これらの遺伝子57個をさらに、順列調整された有意性でt検定によって分析した(WestfallおよびYoung,1993)。これらの統計検定は、その57個の遺伝子のうち4個が、MRI病変1個以上を有する患者で過剰発現されていたことを示した:ジヒドロ葉酸還元酵素(DHFR)、プロテアソーム活性化因子サブユニット(PSME2)、CDC28プロテインキナーゼ2(CKS2)、およびdickkopfホモログ1(DKK1)。Wnt/β−カテニンシグナル伝達拮抗因子の遺伝子DKK1が4個のうちで唯一分泌型因子をコードすること、およびWnt/β−カテニンシグナル伝達は骨の生物学に関与することを考慮して、DKK1に関する追加の試験を実施した。骨髄腫の患者173名からの結果の分析は、MRI病変1個以上およびX線病変なしの患者についてのDKK1シグナルは、MRI病変なしおよびX線病変なしの患者と比較して有意に異なるが(中央値シグナル:2,220対285;p<0.001)、MRI病変1個以上およびX線病変1個以上の患者と比較して有意差がない(中央値シグナル:2,220対1,865;p=0.63)ことを示した(図1B、表2)。
【0047】
意義不明単クローン性免疫グロブリン血症(MGUS)は、溶解性骨病変を伴わない形質細胞疾患であり、および多発性骨髄腫に先行し得る。MGUSの16例中15例で、DKK1は骨髄形質細胞によって、骨のMRI病変またはX線病変が無い多発性骨髄腫におけるレベルと同等のレベルで発現されていた(図1B)。DKK1は、正常ドナー45名、および骨病変を欠く骨の形質細胞悪性腫瘍であるワルデンシュトレーム型マクログロブリン血症の患者9名に由来する形質細胞では検出不能であった(図1B)。
【0048】
【表2】
【0049】
実施例9
全遺伝子発現は、多発性骨髄腫において溶解性骨病変と関連するDKK−1およびFRZBを明らかにする
溶解性骨疾患の分子決定因子をさらに特定するために、骨検査で溶解性病変の放射線的証拠を示さない新たに診断された多発性骨髄腫患者(n=28)に由来するCD138濃縮形質細胞における最大12,000個の遺伝子の発現プロファイルを、3個以上の溶解性病変を有する患者(n=47)と比較した。アブソルート・コール(遺伝子発現の定性尺度)のカイ二乗検定を用いて、疾患の2つの型を区別した遺伝子30個を同定した(P<0.05)。シグナル・コール(遺伝子発現の定量尺度)のウィルコクソンの順位和(WRS)検定は、104個の遺伝子(アップレギュレート49個およびダウンレギュレート55個)が2つの疾患亜型で差があった(P<0.001)ことを明らかにした。
【0050】
カイ二乗検定は、RHAMM前がん遺伝子をその2群間の最も有意な識別子として同定した。それは骨疾患の患者47名中34名と比較して、骨疾患のない患者28名中7名にしか発現されていなかった(図2)。予想される通り、意義不明単クローン性免疫グロブリン血症11例中の1例に由来する形質細胞しかRHAMMを発現しなかった(図3)。WRSはRHAMMを溶解性病変群と非溶解性病変群の間の14番目に有意な識別子として順位づけた。NCALDは、神経細胞シグナル伝達に関与するカルシウム結合タンパク質であるが、非溶解性病変群の11/28例(40%)に存在したが溶解性病変群の2/47例(4%)にしか存在しなかった。カイ二乗分析によって同定された他の注目すべき遺伝子は、Wntシグナル伝達の拮抗因子である、溶解性病変群の40/47例(85%)および非溶解性病変群の15/28(53%)に存在したFRZBを含んだ。CBFA2/AML1BはMIP1α発現と結びつけられており、および非溶解性病変群の50%および溶解性病変群の79%に存在した。
【0051】
染色体分離に関与するPTTG1(securin)はWRSによって最も有意な識別遺伝子(P=4x10−6)と同定された。それは非溶解性病変群の11%で存在するとされたが溶解性病変群の50%で存在した(図4)。WRS検定での他の注目すべき遺伝子は、溶解性病変群でより低レベルに発現された(P=3x10−5)TSC−22ホモログDSIPIを含んだ。DSIPIはまた、破骨細胞とのex−vivo共培養後の多発性骨髄腫形質細胞12例中12例でダウンレギュレートされた。
【0052】
加えて、非溶解性病変群に対して溶解性病変群でより高頻度に見出された(p<0.05)4個のいわゆる「スパイク遺伝子」が同定された:非溶解性病変群0/28例および溶解性病変群7/47例でスパイクを示すIL6(p=0.032);非溶解性病変群0/28例および溶解性病変群7/47例でスパイクを示すオステオナイドジェン(NID2)(p=0.032);非溶解性病変群1/28例および溶解性病変群11/47例でスパイクを示すGタンパク質シグナル伝達調節因子(RGS13)(p=0.023);および非溶解性病変群1/28例および溶解性病変群1/47例でスパイクを示すピリミジン受容体P2Y(P2RY6)(p=0.023)。
【0053】
このように、これらのデータは遺伝子発現パターンが骨疾患と関連し得るということを示唆する。溶解性骨疾患の出現および意義不明単クローン性免疫グロブリン血症から顕性多発性骨髄腫への転換の予測指標として潜在的に有用であることに加えて、それらはまた潜在的介入のための標的を特定し得る。
【0054】
実施例10
DDK1およびFRZBは無作為の骨髄由来よりも限局性病変由来の形質細胞でより高レベルに発現される傾向がある
溶解性病変に対するDKK−1およびFRZBの関係を考慮して、無作為の腸骨稜の骨髄吸引液由来の形質細胞と、脊椎の限局性病変のCT補助穿刺吸引で得られたものとで、DKK−1およびFRZB発現を比較した。これらの結果は、限局性病変由来の形質細胞における有意に高いレベルでの発現を示した。
【0055】
実施例11
DKK−1およびFRZBはワルデンシュトレーム型マクログロブリン血症由来の形質細胞で発現されていない
ワルデンシュトレーム型マクログロブリン血症は、モノクローナルIgM異常蛋白血症および骨髄、リンパ節および脾臓のリンパ形質細胞浸潤によって特徴づけられる稀な形質細胞疾患である。その臨床症状は、臨床経過がそうであるように非常に変わりやすく、しかし多発性骨髄腫とは異なり骨病変は稀である。ワルデンシュトレーム型マクログロブリン血症10例に由来するCD138濃縮骨髄形質細胞の全遺伝子発現プロファイリングは著しい異常を示したが(Zhanetal.,2002)、これらの細胞は、正常骨髄形質細胞のように、FRZBおよびDKKの発現を欠いた(図20)。
【0056】
実施例12
FRZBおよびエンドセリン受容体BはDKK−1と相関する
エンドセリン1は21アミノ酸の血管収縮因子である。エンドセリンの2種類の受容体である受容体AおよびBが同定されている。乳がんおよび前立腺がん細胞はエンドセリン1を産生することができ、および、エンドセリン1およびエンドセリン受容体Aの濃度上昇が骨転移を伴う進行前立腺がんで見出されている。エンドセリン1を産生した乳がん細胞は雌マウスで骨芽細胞転移を生じた。条件付け培地および外因性エンドセリン1はマウス頭頂骨培養において骨芽細胞増殖および新しい骨形成を刺激した(図31)。これらの結果は、エンドセリンが骨形成に関連することを示唆する。
【0057】
表3は、エンドセリン受容体B(ENDRB)の発現がDKK−1の発現と相関したことを示す。エンドセリン受容体Bは新たに診断された多発性骨髄腫の3分の1において「スパイク」遺伝子であった(図29)。エンドセリン受容体Bはまた、意義不明単クローン性免疫グロブリン血症(MGUS)およびくすぶり型多発性骨髄腫の部分集合で発現されていたが、正常形質細胞では発現されていなかった(図30)。
【0058】
【表3】
【0059】
実施例13
In Vivo薬物治療はDKK−1をアップレギュレートする
DKK−1発現はUV照射およびいくつかの他の遺伝毒性刺激によって大幅にアップレギュレートされることが示されている。多発性骨髄腫形質細胞もまたこの疾患を治療するのに用いられる薬物に反応してその遺伝子をアップレギュレートするかどうか調べるために、多発性骨髄腫形質細胞の遺伝子発現プロファイリングを、サリドマイド(図33)、ImiD(図34)、PS−341(図32)、またはデキサメタゾン(図35)を用いた48時間のin vivo処理の前後に実施した。これらのデータは、DKK−1およびFRZB発現を多くの例で大幅にアップレギュレートすることができ、およびそのようにして多発性骨髄腫形質細胞のアポトーシスを誘発することにおけるDKK−1の直接的役割を支持することを示した。最初の予備試験で試験したすべての物質に対して一次性に不応性であった新たに診断された患者がDKK−1の低レベルを示したこと、および薬物治療後にDKK−1またはFRZBの発現増大を示さなかったことに注目するのは興味深く、アポトーシスof多発性骨髄腫形質細胞のアポトーシスを促進することにおけるDKK−1発現の役割を支持する。この概念を支持して、DKK−1およびFRZBは、30例のHMCLおよび数例の髄外疾患で、低レベルから検出不能レベルにて発現されていた(図15)。
【0060】
実施例14
破骨細胞との多発性骨髄腫の共培養の結果としてJUN、FOS、およびDKK−1の大幅なダウンレギュレーションが生じる
骨髄腫細胞と破骨細胞の間の密接な関係は、消耗性溶解性骨破壊の多発性骨髄腫との関連によって臨床的に現れている。溶解性骨病変の発生は、骨髄腫形質細胞との直接的および間接的相互作用を介して破骨細胞の活性化によって引き起こされる。骨髄腫細胞の生存および増殖および疾患過程の維持における破骨細胞の決定的役割は、臨床的に調べられており、および原発性ヒト骨髄腫に関するSCID−huモデルといった実験モデルにおいてin vivoで実証されている。
【0061】
多発性骨髄腫形質細胞/破骨細胞相互作用の分子的結果を調査するために、CD138濃縮多発性骨髄腫形質細胞を、多発性骨髄腫の末梢血幹細胞(PBSC)または健常ドナー由来PBSCおよびMNCから得られた破骨細胞と共培養するex vivo系を開発した。正常ドナーまたは多発性骨髄腫患者に由来する末梢血幹細胞から得られたヒト破骨細胞と共培養したCD138濃縮多発性骨髄腫形質細胞は、アネキシンVフローサイトメトリー、BrdU標識化指数および[3H]TdR取り込みによって示される生存度および増殖活性を50日間の長期にわたって維持した。共培養の前後の形質細胞の純度レベルは、CD38/CD45フローサイトメトリーによって測定された通り、95%より大であった。
【0062】
12例の多発性骨髄腫形質細胞における最大12,000個の遺伝子の発現のマイクロアレイ分析を、4日間の共培養の前および後に実施した。12例の多発性骨髄腫形質細胞対および150例の新たに診断された多発性骨髄腫形質細胞の、7,913個のプローブセット(遺伝子)を用いた階層的クラスター分析は、共培養前標本が3つの主要なクラスター群内に分布した一方、共培養後標本は主要な枝のうち2つに互いに密にクラスター化したことを明らかにした。共培養後のその顕著な遺伝子発現変化の分析は、95個のプローブセット(遺伝子)が、共培養後に多発性骨髄腫形質細胞12例中少なくとも8例で2〜50倍変化したことを示した(アップレギュレート77個、およびダウンレギュレート18個)。健常ドナー5名に由来するCD138濃縮形質細胞はその同じ遺伝子のうち多数で同様の変化を示し、多発性骨髄腫形質細胞は破骨細胞に対して反応の変化を示さないことを示唆した。しかし、正常形質細胞は、その悪性対応物とは対照的に、破骨細胞との長期共培養で生存しなかった。
【0063】
最も著しい変化は、ケモカインGRO1、GR02、GR03、SCYA2、SCYA8、SCYA18、およびIL8のアップレギュレーションにあった。他の注目すべき遺伝子は、ケモカイン受容体CCR1、オステオポンチン(SPP1)、インテグリンITGB2およびITGB5、マトリクスメタロプロテイナーゼ9(MMP9)、カテプシンK(CTSK)およびカテプシンL(CTSL)を含んだ。驚くべきことに、多数の破骨細胞関連遺伝子が、77個のアップレギュレートされた遺伝子に含まれた。ダウンレギュレートされた遺伝子は、サイクリンB(CCNB1)、サイクリンB特異的ユビキチンリガーゼUBE2C、TSC−22ホモログDSIPI、およびJUN、JUND、FOS、およびFOSBを含んだ。
【0064】
遺伝子発現変化はまた、単独で培養された、および多発性骨髄腫形質細胞との共培養後の、破骨細胞10例で試験した。24個の遺伝子(アップレギュレート14個およびダウンレギュレート10個)が、共培養後の破骨細胞10例中少なくとも7例で2〜10倍変化した。多発性骨髄腫患者からまたは健常ドナーから得られた破骨細胞と共に培養した多発性骨髄腫形質細胞の間に遺伝子発現の有意差は無く、多発性骨髄腫破骨細胞は正常ドナーから得られたものと質的に異ならないことを示唆した。
【0065】
共培養実験から得られた培地で多発性骨髄腫形質細胞を培養した際に、遺伝子発現に有意差は観察されず、接触が重要であることを示唆した。共培養実験における多発性骨髄腫形質細胞の破骨細胞に対する低い比(1000:1)を考慮すると、すべての形質細胞が破骨細胞と同時に接触できる可能性は低い。したがって、破骨細胞と接触している多発性骨髄腫形質細胞と他の多発性骨髄腫形質細胞との間の何らかの細胞間情報伝達が起こる可能性が高い。
【0066】
破骨細胞が多発性骨髄腫骨疾患で主要な役割を果たすことおよび多発性骨髄腫に抗アポトーシス性シグナルを提供することが知られている。近年の研究は、JUNが直接的にDKK−1発現を調節すること、および、JUNおよびDKK−1がアポトーシスを調節することを示している。
【0067】
JUNおよびDKK−1を調節することによって破骨細胞が多発性骨髄腫形質細胞のアポトーシスを阻害し得るかどうかを判定するため、原発性多発性骨髄腫12例に由来する精製した形質細胞について、in vitroで得られた破骨細胞との48時間の共培養の前および後に、遺伝子発現プロファイリングを実施した。共培養中の多発性骨髄腫形質細胞は単独で培養された細胞よりも有意に高い長期生存度を有した。破骨細胞共培養の前後の多発性骨髄腫形質細胞の遺伝子発現プロファイリングは、JUN、FOS、およびFOSBは全例で2倍より大きくダウンレギュレートされた40個の遺伝子のうちの3個であることを明らかにした(n=12/12)。共培養後の多発性骨髄腫形質細胞で顕著に調節された95個の遺伝子を用いたHMCLおよび原発性多発性骨髄腫細胞の階層的クラスター分析は、HMCL、破骨細胞と共培養した原発性多発性骨髄腫、および新たに診断された多発性骨髄腫の部分集合の間に、これらの細胞型が相対的に低レベルのc−JUNおよびc−FOSを有した点で著しい類似性を明らかにした。
【0068】
重要なことに、原発性多発性骨髄腫細胞は単独で培養した際に高い程度の自発的アポトーシスを示す一方、破骨細胞の存在下で培養した多発性骨髄腫形質細胞は無限に生存することができる。これらのデータはJUNとDKK−1の間の関連を支持し、およびまた、髄外疾患および常に髄外疾患に由来するHMCLはJUNおよびDKKの両方を欠くため、多発性骨髄腫におけるJUNおよびDKK発現の消失は疾患進行と関連し得ることを示唆する。多発性骨髄腫増殖および挙動に対する破骨細胞の主要な影響の1つが、JUNおよびDKK−1をダウンレギュレートすることであってこれが直接的に形質細胞アポトーシスに影響を与えると推測するのは興味深い。HMCLおよび原発性多発性骨髄腫/破骨細胞共培養のDKK−1を用いた処理は、多発性骨髄腫形質細胞のアポトーシスを結果として生じることが予想される。DKK−1は破骨細胞に対して作用を有しない可能性が高く、それはこれらの細胞がWnt共受容体LRP−5を発現しないためである。正常骨髄由来形質細胞もまたDKK−1を発現せず、およびそれらの長命性を説明するのに役立つであろう。
【0069】
実施例15
形質細胞によるDKK1タンパク質の合成
多発性骨髄腫65例の骨髄生検由来の連続切片を、DKK1の存在に関して染色した。これらの例中の形質細胞は、遺伝子発現データと一致する形でDKK1を含んだ(図39)。正常ドナー5例由来の生検を用いた同様の実験は、どの細胞にもDKK1を検出できなかった。DKK1陽性骨髄腫には、間質性増殖パターンを伴う軽度の形態(核小体が見えない多量の細胞質)を有する強い傾向があった。この染色は骨に隣接する形質細胞で最も強かった。DKK1陰性骨髄腫は結節状または閉塞性増殖パターンを伴う重度の形態(大型化した核および顕著な核小体)を有する傾向があった。間質性増殖パターンについての生検では、DKK1は存在した(細胞のさまざまな割合で)かまたは存在しなかった。対照的に、より悪性の結節状増殖パターンを有する骨髄腫では一律にDKK1は存在しなかった。重要なことに、間質性および結節状増殖の両方を伴う例では、間質性細胞は陽性でありおよび結節状細胞は陰性であった。
【0070】
実施例16
骨髄形質中のDKK1タンパク質
酵素結合免疫吸着測定(ELISA)は、遺伝子発現データもまた利用可能である新たに診断された多発性骨髄腫患者173例のうち107例に由来する骨髄形質中のDKK1タンパク質の濃度は24.02ng/ml(S.D.49.58)であったことを示した。対照的に、DKK1は健常ドナー14例では8.9ng/ml(S.D.4.2)、MGUS14例では7.5ng/ml(S.D.4.5)、およびワルデンシュトレーム型マクログロブリン血症9例では5.5ng/ml(S.D.2.4)であった。DKK1遺伝子発現および骨髄形質中のDKK1のレベルは、骨髄腫107例で正に相関していた(r=0.65、P<0.001)(図40A)。また、骨髄形質および末梢血形質中のDKK1タンパク質レベル間に、両方の標本を同時に採取した骨髄腫41例において強い相関があった(r=0.57、P<0.001)。
【0071】
骨髄形質中のDKK1タンパク質レベルおよび骨病変の存在の両方が測定された患者68例のうち、MRI病変1個以上およびX線病変なしの患者におけるDKK1タンパク質は、MRI病変なしおよびX線病変なしの患者と比較して有意に差があったが(中央値レベル:20ng/ml対9ng/ml;p=0.002)、MRI病変1個以上およびX線病変1個以上の患者と比較して有意差は無かった(中央値レベル:20ng/ml対14ng/ml;p=0.36)(図40B、表2)。
【0072】
実施例17
in vitro骨芽細胞分化に対する骨髄血清の作用
骨形成タンパク質−2は、運命決定されていない間葉性前駆細胞株C2C12 (Katagiri,et al.,1994)の骨芽細胞への分化を、Wnt/b−カテニンシグナル伝達(Bain,et al.,2003; Roman−Roman,et al.,2002)が関与する経路を介して誘導することができる。骨芽細胞分化の特異的マーカーであるアルカリホスファターゼは、5パーセントウシ胎仔血清中で5日間培養したC2C12細胞において検出不能であった(図41A)。C2C12細胞の、50ng/mlのBMP−2を用いた5日間の処理は、C2C12細胞がアルカリホスファターゼを産生するように誘導したが、一方、アルカリホスファターゼはBMP−2および50ng/ml組み換えヒトDKK1と一緒に培養したC2C12細胞によって産生されなかった。アルカリホスファターゼ産生に対するこのin vitro 作用は、ポリクローナル 抗DKK1 抗体によって中和されたが、非特異的ポリクローナルヤギIgGでは中和されなかった。骨髄腫患者5例に由来する、DKK1濃度が12ng/mlを超える骨髄血清は、BMP−2で処理したC2C12細胞によるアルカリホスファターゼの産生を阻害し、およびこの作用は抗DKK1抗体によって逆転されたが非特異的IgGでは逆転されなかった(図41B)。対照的に、50ng/mlBMP−2および正常ドナーの骨髄由来の血清10パーセントで処理したC2C12細胞は、細胞によるアルカリホスファターゼの産生を誘導した(図41B)。
【0073】
下記の参考文献をここで引用した:
Zhan et al.,Global gene expression profiling of multiple myeloma,monoclonal gammopathy of undetermined significance,and normal bone marrow plasma cells.Blood 99: 1745−1757 (2002).
Zhan et al.,Gene expression profiling of human plasma cell differentiation and classification of multiple myeloma based on similarities to distinct stages of late−stage B−cell development.Blood 101:1128−1140 (2003).
Fedi et al.Isolation and biochemical characterization of the humanDkk−1 homologue,a novel inhibitor of mammalian Wnt signaling.J Biol Chem 274: 19465−72 (1999).
Gallea et al.Bone 28 : 491−8 (2001).
Spinella−Jaegle et al.Bone 29: 323−30 (200
1).
Westfall and Young.Resampling−based multiple testing: Examples and methods for p−value adjustment.Hoboken,NJ: Wiley−Interscience,360 (1993).
Golub et al.Science 286: 531−7 (1999).
Katagiri et al.J Cell Biol 127: 1755−66 (1994).
Bain et al.Biochem Biophys Res Commun 301: 84−91(2003).
Roman−Roman et al.American Society of Bone Mineral Research,2002.
本明細書中で言及した特許または出版物はいずれも、本発明が関係する分野の当業者のレベルを示す。さらに、これらの特許および出版物は、個々の出版物が参照により本開示に含まれると具体的および個別に示されたのと同等に、参照により本開示に含まれる。
【特許請求の範囲】
【請求項1】
多発性骨髄腫患者における溶解性骨疾患を治療するための医薬組成物であって、
前記医薬組成物が、Dickkopf−1(DKK1)(登録番号NM012242)のヒトホモログに対するアンチセンスオリゴヌクレオチド、抗DKK1抗体、又はDKK1と相互作用する可溶性LRP受容体、を含むことを特徴とする医薬組成物。
【請求項2】
患者における悪性腫瘍関連骨減少を調節する医薬組成物であって、
前記医薬組成物が、DKK1に対するアンチセンスオリゴヌクレオチド、抗DKK1抗体、又はDKK1と相互作用する可溶性LRP受容体、を含むことを特徴とする医薬組成物。
【請求項3】
前記悪性腫瘍関連骨減少が、骨への乳がん転位又は骨への前立腺がん転移によって引き起こされることを特徴とする請求項2に記載の医薬組成物。
【請求項1】
多発性骨髄腫患者における溶解性骨疾患を治療するための医薬組成物であって、
前記医薬組成物が、Dickkopf−1(DKK1)(登録番号NM012242)のヒトホモログに対するアンチセンスオリゴヌクレオチド、抗DKK1抗体、又はDKK1と相互作用する可溶性LRP受容体、を含むことを特徴とする医薬組成物。
【請求項2】
患者における悪性腫瘍関連骨減少を調節する医薬組成物であって、
前記医薬組成物が、DKK1に対するアンチセンスオリゴヌクレオチド、抗DKK1抗体、又はDKK1と相互作用する可溶性LRP受容体、を含むことを特徴とする医薬組成物。
【請求項3】
前記悪性腫瘍関連骨減少が、骨への乳がん転位又は骨への前立腺がん転移によって引き起こされることを特徴とする請求項2に記載の医薬組成物。
【図1A】

【図1B】
【図2】
【図3】
【図4】
【図5】
【図6】

【図7】
【図8】
【図9】
【図10】
【図11】
【図12】
【図13】
【図14】
【図15】
【図16】
【図17】
【図18】
【図19】
【図20】

【図21】
【図22】
【図23】
【図24】
【図25】
【図26】

【図27】
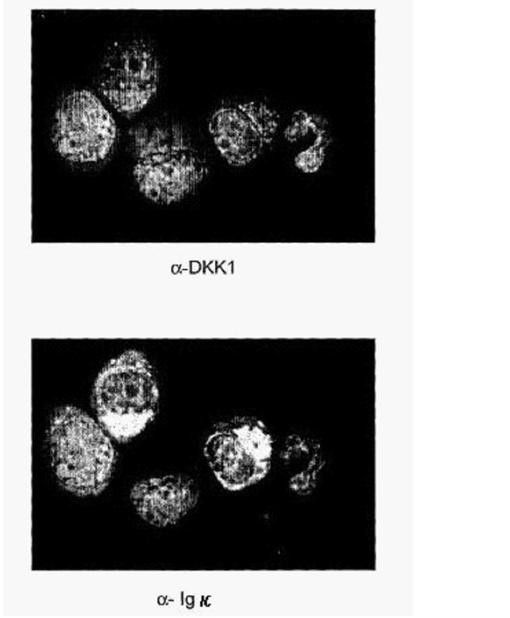
【図28】
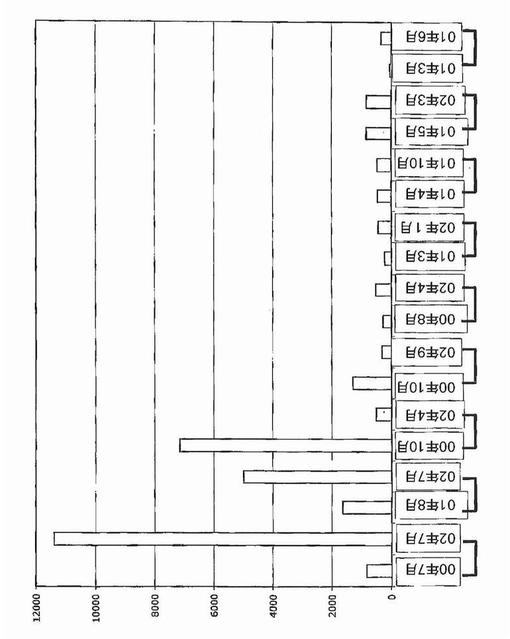
【図29】
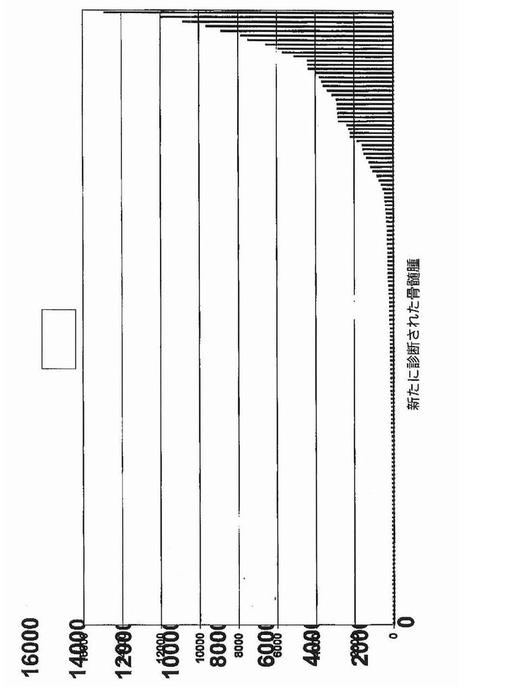
【図30】
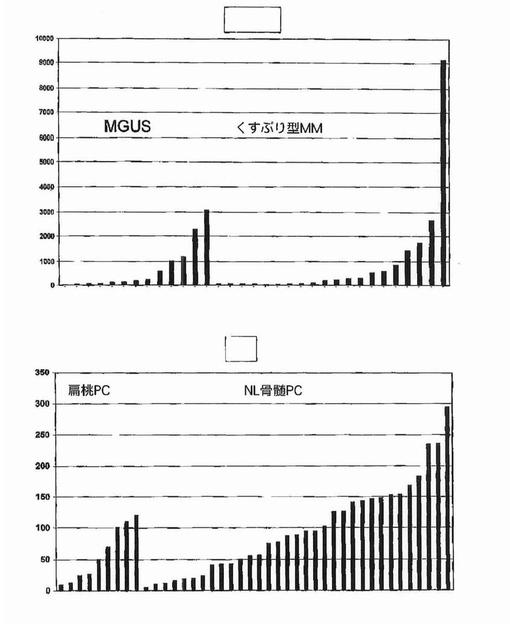
【図31】
【図32】
【図33】
【図34】
【図35】
【図36】

【図37】
【図38】
【図39】
【図40A】
【図40B】
【図41A】
【図41B】
【図1B】
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【図11】
【図12】
【図13】
【図14】
【図15】
【図16】
【図17】
【図18】
【図19】
【図20】
【図21】
【図22】
【図23】
【図24】
【図25】
【図26】
【図27】
【図28】
【図29】
【図30】
【図31】
【図32】
【図33】
【図34】
【図35】
【図36】
【図37】
【図38】
【図39】
【図40A】
【図40B】
【図41A】
【図41B】
【公開番号】特開2012−46535(P2012−46535A)
【公開日】平成24年3月8日(2012.3.8)
【国際特許分類】
【出願番号】特願2011−223234(P2011−223234)
【出願日】平成23年10月7日(2011.10.7)
【分割の表示】特願2004−559239(P2004−559239)の分割
【原出願日】平成15年12月4日(2003.12.4)
【出願人】(500517271)ザ ボード オブ トラスティーズ オブ ザ ユニヴァーシティー オブ アーカンソー システム (4)
【Fターム(参考)】
【公開日】平成24年3月8日(2012.3.8)
【国際特許分類】
【出願日】平成23年10月7日(2011.10.7)
【分割の表示】特願2004−559239(P2004−559239)の分割
【原出願日】平成15年12月4日(2003.12.4)
【出願人】(500517271)ザ ボード オブ トラスティーズ オブ ザ ユニヴァーシティー オブ アーカンソー システム (4)
【Fターム(参考)】
[ Back to top ]