
高含量イブプロフェンナトリウム顆粒、その調製、および非発泡性固体剤形を調製する際のその使用
本明細書に明記される成分から形成されるラセミイブプロフェンナトリウム2水和物の顆粒は、極めて望ましい特性を有し、実践においてしばしば直面する動作困難に遭うことなく、従来のロータリープレス錠剤化設備において効果的に使用され得る。また、湿潤造粒過程によるその調製、湿潤顆粒組成物、ロータリープレスを利用する固体剤形の調製に適合される製剤、固体剤形、およびロータリープレスにおいて固体剤形を調製する方法についても説明する。

【発明の詳細な説明】
【技術分野】
【0001】
本発明は、イブプロフェンナトリウム塩2水和物から作製されるイブプロフェン薬物の高含量顆粒、このような顆粒を生成するための処理技術、および経口投与用の非発泡性固体剤形を調製する際のこのような顆粒の使用に関する。
【背景技術】
【0002】
既刊文献によると、イブプロフェンナトリウムから作製される固体剤形は、医薬的反応速度において優れている(すなわち、この固体剤形は、イブプロフェンの他の固体剤形よりも急速に血流に入り、ピークに達する)。しかしながら、本分野において集中的に研究されているにもかかわらず、従来の手法を使用してイブプロフェンナトリウムから固体剤形を形成することは、依然として困難である。その困難の1つとして、イブプロフェンナトリウム2水和物が、流動性が低い特徴を有することから、コロイド状シリカまたはタルク等の従来の凝固阻止剤と混合する場合であっても容易に凝固する傾向があることが挙げられる。流動性向上剤とともに製剤化されるにもかかわらず流動性が低い特徴は、生成される錠剤およびカプレット等の固体剤形の異常な重量変動をもたらす可能性がある。イブプロフェンナトリウムの錠剤およびカプレットを形成する際の別の困難として、ロータリープレスのパンチ表面上にフィルムコーティング形成するイブプロフェンナトリウム2水和物の傾向が挙げられる。このようなフィルムコーティングは、過剰でない場合であっても、形成される錠剤またはカプレットの輝きつまり光沢を低減させるため、望ましくない。過剰なコーティングがパンチ表面上に形成される場合、いくつかの動作上の困難に直面する。まず第1に、形成される錠剤またはカプレットに欠陥が発生する。第2に、異常なパンチ動作がもたらされる。第3に、錠剤またはカプレットの許容不可能な重量の変動にも直面し得る。次いで、これらの動作上の困難のいずれかによって、パンチ表面を清浄するために設備を停止することが必要となる。
【0003】
ゆえに、上述の種々の困難に直面することなく、従来のロータリープレス錠剤化設備を使用して、より容易に、錠剤またはカプレット等の固体剤形に変換できるイブプロフェンナトリウム組成物を形成する方式を発見できるのであれば、相当有利になるであろう。また、顆粒で充填されるハードシェルカプセルの投与量レベルをより均一に維持することができるように、イブプロフェンナトリウム2水和物から高度に流動可能な顆粒を生成する方式を発見できるのであれば、相当有利になるであろう。
【0004】
本発明は、前述の必要性の全てに効果的に対処する。
【発明の概要】
【0005】
本発明によると、湿潤造粒過程によって、ラセミイブプロフェンナトリウム2水和物(別称、2−(4−イソブチルフェニル)プロピオン酸ナトリウム2水和物、および以下本明細書において、より単純にイブプロフェンナトリウム2水和物と呼ばれる)、炭酸ナトリウム、およびポリビニルピロリドン(別称、2−ピロリドン、1−エテニル−、ホモポリマー)から形成される顆粒が、極めて望ましい特性を有すること、ならびに適切に製剤化される際に、顆粒が、上述の種々の困難に直面することなく、従来のロータリープレス錠剤化装置において効果的に利用可能であることが発見された。さらに、造粒動作およびロータリープレス錠剤化動作では、顆粒の炭酸ナトリウムの量が3wt%以下のみであっ
ても、本発明により可能になる有利な結果を達成できることが発見された。当然ながら、顆粒が2wt%の炭酸ナトリウムのみを含有する本発明の顆粒から優れた錠剤を生成することが可能であることが発見された。
【0006】
また、このような極めて少量の炭酸ナトリウムが、顆粒と、顆粒から形成されて得られた錠剤との形成に効果的であるという事実は、高齢者が消費する錠剤に関して利点を有する。周知のように、人は歳を取るにつれて、消化管の酸性度が低下する傾向にあるため、本発明の錠剤におけるこのような低レベルの炭酸ナトリウムによって、このような人の消化過程をあまり悪化させない。
【0007】
その過程の実施形態のうちの1つでは、本発明は、
A) イブプロフェンナトリウム2水和物の高度に分散可能な自由流動顆粒を調製する過程であって、その方法は、
● (i)乾燥ベースで少なくとも80重量部のイブプロフェンナトリウム2水和物と、
(ii)乾燥ベースで1重量部から4重量部の炭酸ナトリウムと、(iii)乾燥ベ
ースで1重量部から15重量部の非架橋ポリビニルピロリドンと、(iv)湿潤混合
物を形成するための、(i)、(ii)、(iii)、および(iv)の全重量に基
づく8重量部から12重量部の水とから構成される成分を、高せん断造粒器にまとめ
ることと、
● 湿潤顆粒を形成するために、上記高せん断造粒器において上記湿潤混合物を造粒する
ことと、
● 110℃における重量損失の測定により決定可能である約11wt%から15wt%
の範囲、好ましくは、約12wt%から15wt%の範囲の含水量を有する乾燥顆粒
を形成するように、湿潤顆粒を乾燥することと、
● 16メッシュを超える粒径を有する乾燥顆粒を篩過することによって除去することと
、
を含む、過程を提供する。
【0008】
本発明の好適な過程の実施形態は、以下の通りである:
B) 上記高せん断造粒器における上記湿潤混合物の造粒は、上記湿潤混合物の乾燥後に、乾燥顆粒が示差走査熱量測定中に約100℃から約102℃の範囲でピークに達する相転移を呈するように、ある時間の間行なわれ、固体から固体への相転移に対応するDSCピークのサイズが、グラム当たり少なくとも約150ジュールである、A)における過程。
【0009】
その組成物の実施形態の中で、本発明は以下を提供する:
C) 高含量のイブプロフェンナトリウム2水和物を有する高度に分散可能な自由流動顆粒組成物であって、(i)乾燥ベースで80重量部から98重量部のイブプロフェンナトリウム2水和物と、(ii)乾燥ベースで1重量部から4重量部の無水炭酸ナトリウムと、(iii)乾燥ベースで1重量部から15重量部の非架橋ポリビニルピロリドンとを含む粉末形状の成分から形成される組成物。
D) ロータリープレスを使用する固体剤形の調製に適合されるイブプロフェンナトリウム2水和物製剤であって、
● 約40wt%から約100wt%のC)における顆粒組成物、
● 0wt%から約25wt%の微結晶性セルロース、リン酸水素カルシウム、またはそ
の両方、
● 0wt%から約8wt%のクロスポビドンまたはクロスカルメロースナトリウム、
● 0wt%から約0.5wt%のコロイド状シリカ、
● 0wt%から約10wt%のでんぷん、および
● 0wt%から約2wt%のステアリン酸、ステアリン酸マグネシウム、またはその両
方、
を含む成分から形成される製剤。
【0010】
本発明の好適な過程の実施形態は、以下の通りである:
E) 上記製剤は、示差走査熱量測定中に約100℃から約102℃の範囲でピークに達する相転移を呈し、固体から固体への相転移に対応するDSCピークのサイズが、グラム当たり少なくとも約150ジュールである、D)における製剤。
【0011】
本発明に関する他の実施形態、特徴、および利点は、以下の説明、添付の図面、および添付の請求項によって、またさらに明らかになる。
【図面の簡単な説明】
【0012】
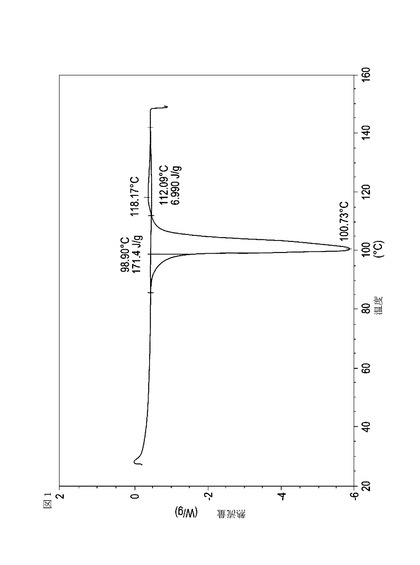
【図1】実施例11において生成された本発明の顆粒に関する示差走査熱量測定曲線である。
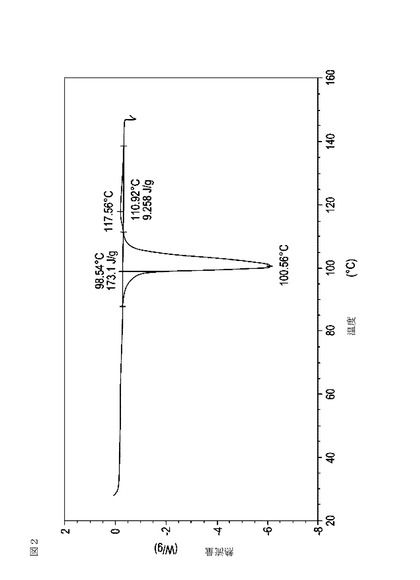
【図2】実施例12において生成された本発明の顆粒に関する示差走査熱量測定曲線である。
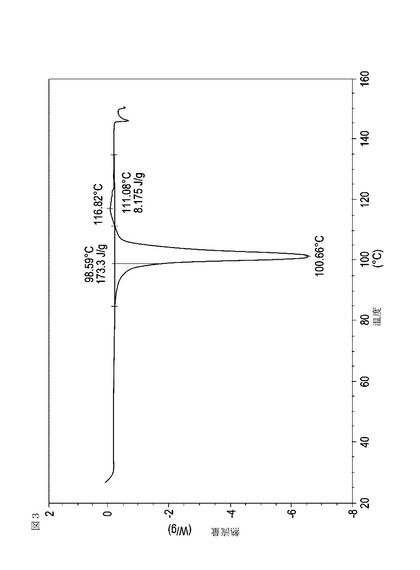
【図3】実施例13において生成された本発明の顆粒に関する示差走査熱量測定曲線である。
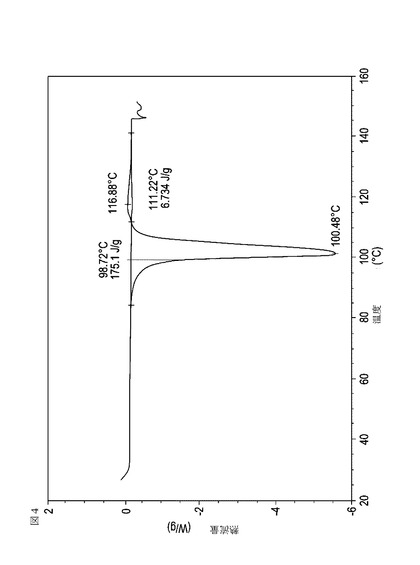
【図4】実施例14において生成された本発明の顆粒に関する示差走査熱量測定曲線である。
【図5】乾燥機入口温度が70℃に設定された状態で、顆粒の乾燥温度が35℃に到達した際に試料採取された、実施例15において生成された本発明の顆粒に関する示差走査熱量測定曲線である。
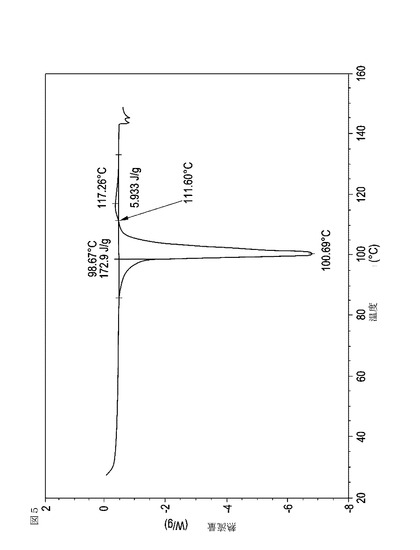
【図6】乾燥機入口温度が70℃に設定された状態で、顆粒の乾燥温度が40℃に到達した際に試料採取された、実施例15において生成された本発明の顆粒に関する示差走査熱量測定曲線である。
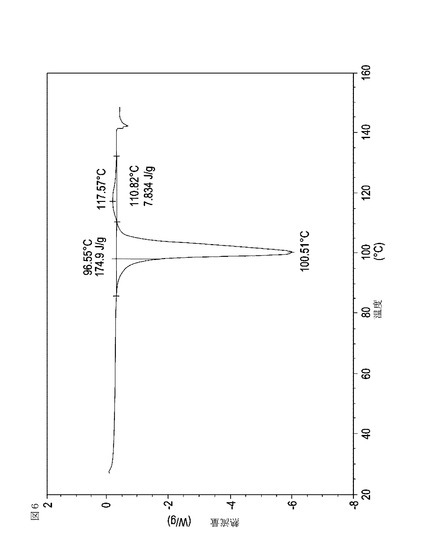
【図7】乾燥機入口温度が70℃に設定された状態で、顆粒の乾燥温度が45℃に到達した際に試料採取された、実施例15において生成された本発明の顆粒に関する示差走査熱量測定曲線である。
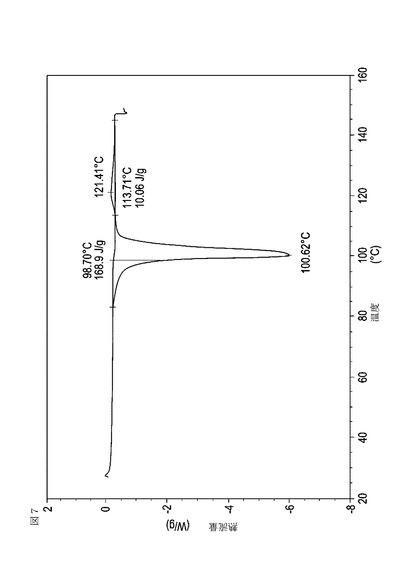
【図8】乾燥機入口温度が70℃に設定された状態で、顆粒の乾燥温度が48℃に到達した際に試料採取された、実施例15において生成された本発明の顆粒に関する示差走査熱量測定曲線である。
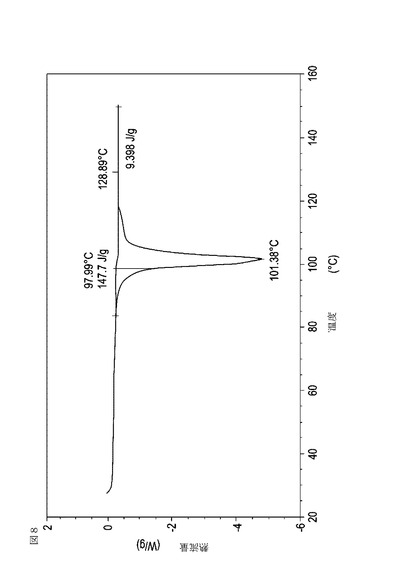
【図9】乾燥機入口温度が70℃に設定された状態で、顆粒の乾燥温度が40℃に到達した際に試料採取された、実施例16において生成された本発明の顆粒に関する示差走査熱量測定曲線である。
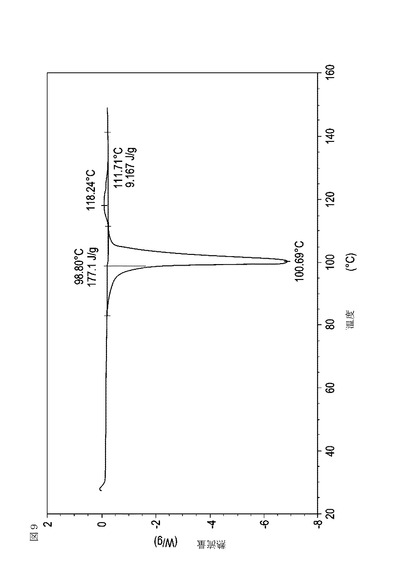
【図10】乾燥機入口温度が70℃に設定された状態で、図10の試料が取られた10分後に試料採取された、実施例16において生成された本発明の顆粒に関する示差走査熱量測定曲線である。
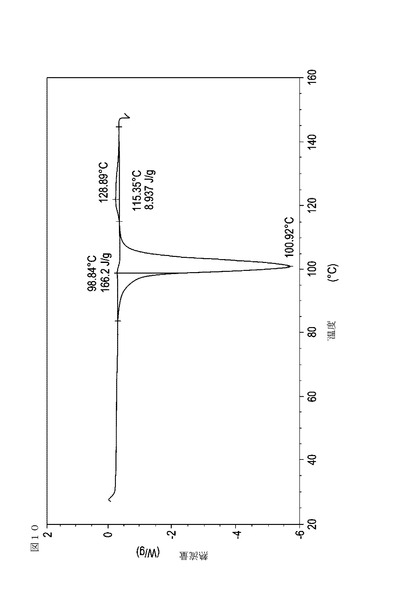
【図11】乾燥機入口温度が70℃に設定された状態で、図10の試料が取られた30分後に試料採取された、実施例16において生成された本発明の顆粒に関する示差走査熱量測定曲線である。
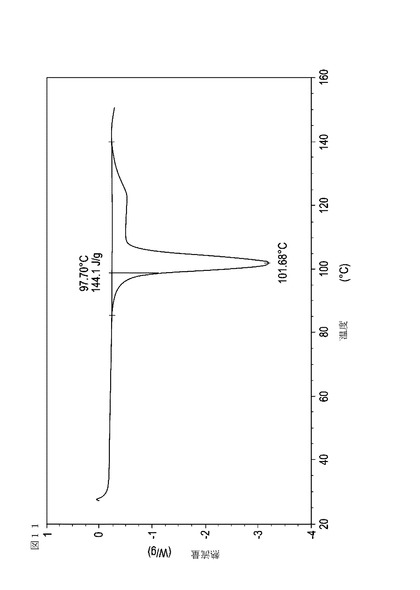
【図12】乾燥機入口温度が70℃に設定された状態で、顆粒が60℃の温度に達成した際に試料採取された、実施例17において生成された本発明の顆粒に関する示差走査熱量測定曲線である。
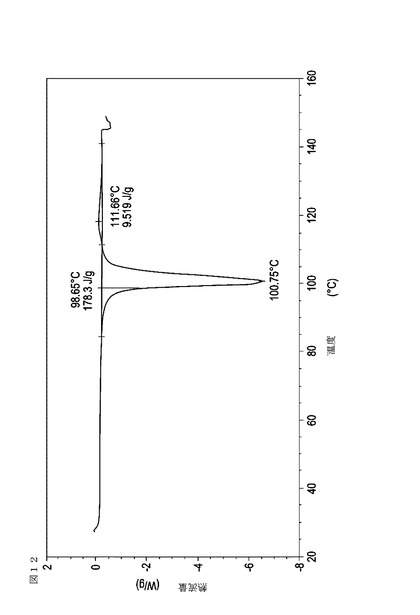
【図13】70℃から60℃に乾燥機入口空気温度を低下させることによって、顆粒が20分間60℃の温度にあった際に試料採取された、実施例17において生成された本発明の顆粒に関する示差走査熱量測定曲線である。
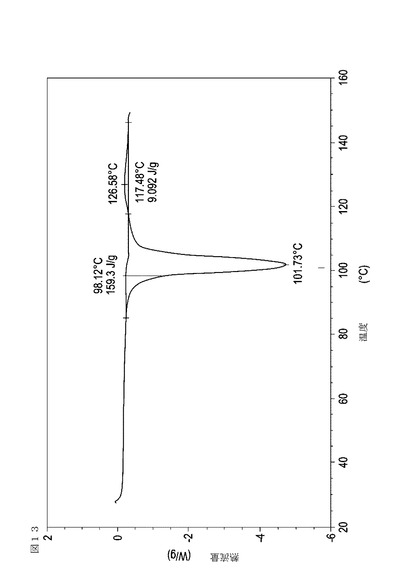
【図14】70℃から60℃に乾燥機入口空気温度を低下させることによって、顆粒が60分間60℃の温度にあった際に試料採取された、実施例17において生成された本発明の顆粒に関する示差走査熱量測定曲線である。
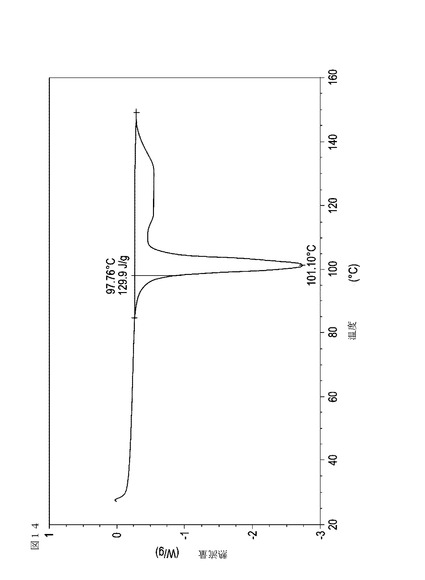
【図15】実施例18において生成された本発明の顆粒に関する示差走査熱量測定曲線である。
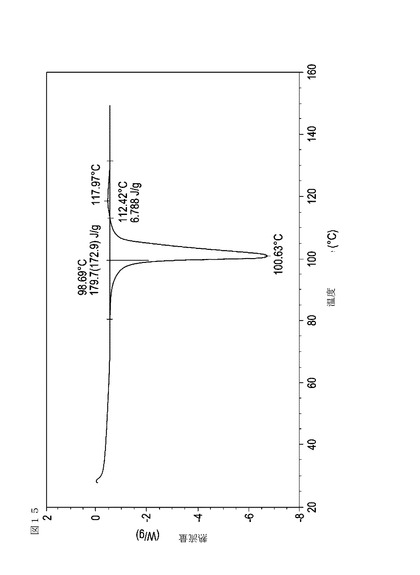
【図16】実施例19において生成された本発明の顆粒に関する示差走査熱量測定曲線である。
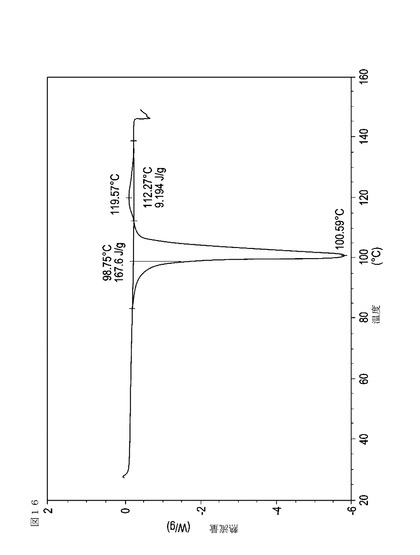
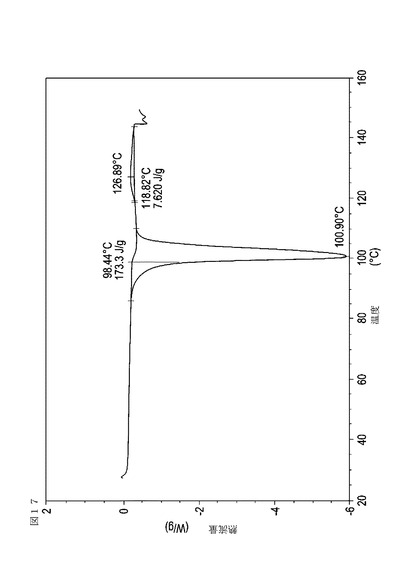
【図17】実施例20において生成された本発明の顆粒に関する示差走査熱量測定曲線である。
【発明を実施するための形態】
【0013】
主成分
【0014】
上述のように、本発明の顆粒において、ならびに錠剤、カプレット、およびハードシェルカプセル等の本発明の固体剤形において、3つの主成分が存在する。これらの成分は、イブプロフェンナトリウム2水和物、炭酸ナトリウム、およびポリビニルピロリドンである。これらの成分の各々は、当技術分野において周知であり、これらのそれぞれの材料の調製方法も周知であり、文献において報告されている。全てのこのような成分は、NF、USP、または複数公定書グレードであるべきである。言い換えると、このような成分は、医薬的に許容可能な純度、等級、および/または品質を有するべきである。
本発明の顆粒の調製
【0015】
本発明の顆粒を調製するために、上記の3つの主成分を、高せん断造粒器において水で湿らせる間に、相互に対して適切な比率で混合する。実際は、上記の3つの成分の全体的な混合を形成するまえに、水中でポリビニルピロリドンの溶液を形成することが望ましい。これにより、本発明の顆粒内にポリビニルピロリドンのより均一な分布が提供される。高せん断造粒器における混合および造粒の後に、オーブンにおけるパン乾燥等の従来の乾燥手順によって、または流動層乾燥によって、湿潤顆粒から水を除去する。典型的には、その乾燥は、最大約100℃の温度でもたらされる。好ましくは、顆粒の乾燥のためのオーブン温度は、約60℃から約70℃の範囲である。流動層乾燥機の場合、入口温度も、好ましくは、約60℃から約70℃の範囲である。特に好適な乾燥動作では、出口空気の温度は、出口空気温度が約40℃に達する際に乾燥が停止するように監視される。このように、顆粒の全含水量は、約12wt%から約15wt%の好適な範囲であり、より好ましくは、約13.5wt%であり、これは、ほぼイブプロフェンナトリウム2水和物における水和水の量である。次いで、この乾燥生成物は、過剰サイズの材料(例えば、16メッシュを超える)を除去するように篩過される。このような過剰サイズの材料は、典型的には、粉砕され、造粒動作に再利用される。
【0016】
さらに詳細には、イブプロフェンナトリウム2水和物を高せん断造粒器内に装薬し、その後、水中でポリビニルピロリドン溶液を事前形成することが好ましい。これらの成分の混合の後、必要量の炭酸ナトリウムを添加し、この混合物に、追加の高せん断造粒を供する。この造粒は、造粒中に生成される過剰な熱を除去するように、必要に応じて冷却を使用して、約15℃から約35℃の範囲の温度で行われる。炭酸ナトリウムの添加前の高せん断造粒器におけるチョッパのせん断速度は、典型的には、約3分から約6分の範囲の時間の間、約1000rpmから約2000rpmの範囲である。次いで、チョッパ速度は、約3分から約6分の範囲の追加の時間の間、約200rpmから約500rpmの低せん断速度まで低下する。炭酸ナトリウムの添加後、そのチョッパについてこの同じ2段階動作が繰り返される(すなわち、約3分から約6分の間、約1000rpmから約2000rpm、次いで、約3分から約6分の間、約200rpmから約500rpm)。
【0017】
高せん断造粒器の設計に依存して、該装置は、造粒器における混合物の運動をもたらすための別の機構を含んでもよく、その機構は、パドルとして知られている。パドルが動作する速度は、あまり重要ではないため、変動することができる。160リットルのFluid Air Pharmx High Shear Granulator,Model PX150を使用する場合、第1段階中に100rpmでパドルを動作し、第2段階において30rpmで動作する2段階方式でパドルを動作することが便利であることが分かった。これらの段階は、チョッパの2段階動作と一致する。
【0018】
顆粒を形成する際に使用する炭酸ナトリウムは、NFまたはUSPグレードであるべきであり、最初に水和化し得るが、好ましくは、無水粉末の形状である。医薬的に許容可能なグレードのポリビニルピロリドンは、本発明の実施に使用され得る種々の形状で利用可能である。しかしながら、30から120の範囲のK値を有するポリビニルピロリドンを利用することが好ましく、90のK値を有するポリビニルピロリドンを利用することがさらに好ましい。イブプロフェンナトリウム2水和物は、医薬的に許容可能なグレードのラセミ混合物であり、好ましくは、粉末形状で使用される。
【0019】
顆粒を形成する際に使用する炭酸ナトリウムの量は、典型的には、全混合物の約1重量%から4重量%の範囲である。好ましくは、その量は、全混合物の約2重量%から約3重量の範囲である。顆粒を形成する際、ポリビニルピロリドンの量は、典型的には、全混合物の約1重量%から10重量%の範囲である。好ましくは、ポリビニルピロリドンの量は、全混合物の約2重量%から4重量%の範囲である。好ましくは、全混合物の100重量%に対する残量は、イブプロフェンナトリウム2水和物から構成される。必要に応じて、3つの主成分のいずれの成分と不利に相互作用しない限り、あるいは3つの主成分から生成される顆粒または固体剤形の調製を妨げない限り、他の従来の医薬的に許容可能な賦形剤を使用することができる。しかしながら、このような使用によって、顆粒内のイブプロフェンナトリウム2水和物の濃度が低下し、生産コストおよび記録管理の増加がもたらされることから、このような賦形剤を顆粒に含めることは、望ましくはない。
【0020】
また、使用する水は、規制要件を満たすのに十分な純度を有する。顆粒を形成する際に使用する水の量は、典型的には、成分の湿潤混合物の全重量の約5wt%から約15wt%の範囲である。好ましくは、顆粒形成に使用する水の量は、成分の湿潤混合物の全重量の約8wt%から約12wt%の範囲である。使用する水が少な過ぎると、不十分な造粒が発生する。一方、使用する水が多過ぎると、混合物は、パン生地の硬さになる。いずれの場合であっても、乾燥後、生成物は、望ましい平均(中間)粒径を持たない。これに関し、乾燥顆粒の平均粒径は、典型的には、約150ミクロンから約600ミクロンの範囲、好ましくは、約200ミクロンから約300ミクロンの範囲である。
【0021】
湿潤形状における本発明の特に好適な顆粒は、特定の量の以下の成分から調製される:イブプロフェンナトリウム2水和物、85.95wt%;非架橋ポリビニルピロリドン(特にK−90)、2.25wt%;無水炭酸ナトリウム粉末、1.80wt%;精製水、10wt%。乾燥形状における本発明の特に好適な顆粒は、以下の組成を有する:イブプロフェンナトリウム2水和物として95.50wt%;2.50wt%のポリビニルピロリドン(特にK−90);および無水炭酸ナトリウムとして2.00wt%。
好適な顆粒および顆粒製剤
【0022】
上に示すように、本発明は、好適な実施形態において、示差走査熱量測定中に、約100℃から約102℃の範囲でピークに達する相転移を呈し、固体から固体への相転移に対応するDSCピークのサイズは、グラム当たり少なくとも約150ジュールであることを特徴とする顆粒および顆粒製剤を提供する。このピークの存在によって、顆粒は、上記で
E)と示す製剤のように適切に製剤化される場合に、ロータリープレスにおいて優れた処理特徴を有する固体製剤を生成し、それによって、高品質の固体剤形が生成され、現在の規制要件への順守が確実になる。顆粒およびその結果生じた製剤が、示差走査熱量測定(DSC)中に前述の熱的特徴を有することを確実にする種々の方式が存在する。例えば、顆粒試料が、このような熱的特徴を呈さない場合:
● 前述のDSCの熱的特徴を達成するのに十分な温度と時間で顆粒を加熱することがで
きる。提案する加熱条件は、約5分から約50分の間、約30℃から約70℃で加熱
することを含む;
● 湿潤造粒を行う時間を延長することができる。ベンチスケールの動作では、湿潤造粒
のこの全時間は、少なくとも約12分から16分であることが分かっている。この動
作をより大きなスケールで行う場合、湿潤造粒の全時間は、所望のDSCの熱的特徴
を達成するために12分から16分とは少なくともいくらか異なり得るため、より大
きな規模で動作する場合に、湿潤造粒の適切な時間を決定するために、必要に応じて
、いくつかのパイロット実験を行うべきである;
● 顆粒が依然として湿潤である場合に造粒過程において成分を含めることを回避するこ
とによって、前述のDSCの熱的特徴が達成されなくなる。これに関して悪影響をも
たらすと考えられる成分は、でんぷん、グリコール酸でんぷんナトリウム、およびス
テアリン酸マグネシウムである。以下に提示する実施例から分かるように、顆粒が乾
燥すると、固体剤形の生成にお使用するための製剤における成分として、微結晶性セ
ルロースを利用することができる。当然ながら、微結晶性セルロースが、このような
製剤において使用するために好適な成分である。
【0023】
以下の実施例は、例証目的のために提示される。実施例は、実施例に記載される詳細および材料に本発明を限定することを意図しない。
実施例1
顆粒の調製
【0024】
95.5wt%のイブプロフェンナトリウム水和物、2wt%の炭酸ナトリウム、および2.5wt%のポリビニルピロリドン(Plasdone K−90、International Specialty Products Inc.,Wayne,NJ)から構成される顆粒を、本発明の湿潤造粒過程によって調製した。使用する過程は、水中でPlasdone K−90を溶解することと、高せん断増強駆動部(GlobePharma,Inc.,New Brunswick,NJにより製造されるMAXI−BLEND LAB V−BLENDER)を装備するV字型ブレンダにおいて、その溶液にイブプロフェンナトリウム2水和物を添加し、高いせん断を使用してブレンダを動作することによって顆粒を形成することと、篩過された炭酸ナトリウム粉末をブレンダ内の湿潤混合物に添加することと、増強駆動部を利用してさらに5分間混合することを伴った。ブレンダの含有物をパン内に排出した後、添加された全ての水が除去されるまで、50度に維持したオーブンで顆粒を乾燥した。次いで、その生成物を、ステンレス鋼の16メッシュのU.S.A.規格の篩を通して篩過した。
実施例2
錠剤の調製
実施例2A
【0025】
実施例1で形成した顆粒を使用して、ロータリープレスを使用する錠剤調製のために、完全に製剤化されたブレンドを調製した。本動作では、乾式混合によって、顆粒を、微結晶性セルロース(MCC)およびコロイド状シリカと混合した。10ステーションロータリープレスにおいて十分処理された製剤と、得られた錠剤とは、良好な破砕性、溶解、お
よび分解を示した。最終ブレンドは、75wt%のイブプロフェンナトリウム水和物、2.37wt%のポリビニルピロリドン、1.58wt%の炭酸ナトリウム、20.95wt%の微結晶性セルロース、および0.1wt%のコロイド状シリカから構成された。
【0026】
実施例1で形成した高活性含有顆粒と、実施例2Aで形成した錠剤製剤とに関する特徴について表1に提示する。流動性指数によって、顆粒と完全に製剤化されたブレンドとの両方が、良好な流動特徴を有することが示された。加えて、錠剤は、全ての所望の性能基準を満たした。
【表1】
実施例2B
【0027】
実施例1で形成した顆粒を使用して、実施例2Aで説明したように、ロータリープレスを使用する錠剤調製のために完全に製剤化されたブレンドを調製したが、乾式混合によって顆粒をクロスポビドンと混合したことを例外とする。10ステーションロータリープレスにおいて十分処理された製剤と、得られた錠剤とは、許容可能な破砕性および良好な溶解性を示した。最終ブレンドは、93.6wt%のイブプロフェンナトリウム水和物、2.45wt%のポリビニルピロリドン、1.96wt%の炭酸ナトリウム、および2wt%のクロスポビドンから構成された。
実施例2C
【0028】
実施例1で形成した顆粒を使用して、実施例2Aで説明したように、ロータリープレスを使用する錠剤調製のために完全に製剤化されたブレンドを調製したが、乾式混合によって顆粒をNaクロスカルメロースと混合したことを例外とする。10ステーションロータリープレス内に十分流入した製剤と、得られた錠剤とは、許容可能な破砕性および良好な溶解性を示した。最終ブレンドは、93.6wt%のイブプロフェンナトリウム水和物、2.45wt%のポリビニルピロリドン、1.96wt%の炭酸ナトリウム、および2wt%のNaクロスカルメロースから構成された。
実施例3〜10
【0029】
これらの実施例では、ベンチスケール高せん断造粒器、Model PX1,2−Liter Fluid Air High Shear Granulatorにおいて、湿潤造粒技法を使用して工程実行を行った。本装置は、ボウルに配置されるインペラおよびチョッパを含む。インペラは、ボウル内で造粒されるブレンドを、ボウル内において円軌道で回転させ、チョッパは、処理中にブレンド中に形成される大粒子を破壊する。該造粒器は、圧縮空気によって動作され、本動作では、一部の圧縮空気が、ボウルに進入し、マシュマロの硬さに似た硬さを有するブレンド、すなわち、軟らかく、かつ低密度を有するブレンドの形成に寄与する。さらに混合すると、閉じ込められた空気の一部が放出される。そのブレンドは、湿潤外観を有する。得られた顆粒の外観は、同一の製剤を使用して
ツインシェルブレンダで処理された顆粒とは大幅に異なる。ツインシェルブレンダからの顆粒は、本装置で生成される顆粒よりも大幅に乾燥して見える。
【0030】
典型的な動作では、イブプロフェンナトリウム2水和物をボウルに添加した後に、蓋をボウル上に置き、チョッパ速度およびインペラ速度を、それぞれ2500RPMおよび300RPMに設定する。混合の作動直後に、ポビドンK−90水溶液を、蓋の穴を通して造粒器に注入する。典型的には、ボウルのカバーの小さい穴を通してボウルに液体を移行するのに約100秒かかる。ポビドン溶液の移行後、高せん断混合を事前設定した時間継続する。次いで、チョッパおよびインペラの両方の混合速度を、元の設定の約半分まで低下させ、この低せん断混合を、さらに数分間継続する。次いで、無水炭酸ナトリウム粉末(20メッシュスクリーンで前もって篩過されている)をボウルに添加し、さらなる高せん断−低せん断混合周期の間、混合を継続した。工程実行1で生成された湿潤顆粒を、流動層乾燥機に添加する前に、ビーカー内に排出した。プラスチック袋に回収された乾燥材料によって、16メッシュを超える多量の顆粒の存在が示される。
【0031】
本動作は、2つの段階を伴った。第1段階では、イブプロフェンナトリウム2水和物およびポリビニルピロリドン(K−90溶液)を造粒器に添加し、混合した。第2段階では、微粉化した無水炭酸ナトリウムを、第1段階からの造粒器含有物に添加し、得られた混合物を顆粒に処理した。
【0032】
実施例3〜10の第1段階における湿潤造粒動作の条件について表2に要約する。表3は、第2段階に使用した湿潤造粒動作の条件について要約する。表2および表3では、記号表示「CPR」は、「チョッパ」を示し、「IMP」は、「インペラ」を示し、「n/a」は、「該当なし」を示す。
【表2】
【表3】
【0033】
実施例4では、高せん断チョッパの砕塊効率を、造粒器内の塊の存在を確認することによって、種々の混合間隔において測定した。その元の粒子形状におけるイブプロフェンナトリウム2水和物の砕塊が効率的ではなく、ポビドン溶液の添加後に、砕塊が大幅により効率的になるという結論に達した。高せん断混合周期で約120秒後に、検出可能な塊は発見されなかった。その造粒器動作の後、造粒器ボウルの含有物を、カバーの穴に適用される真空管を使用して、流動層乾燥機が生成する吸引力により、空気圧で流動層乾燥機に移行した。回収された乾燥材料により、サイズが16メッシュを超える顆粒の含有物が極めて少量であることが示された。実施例5〜10におけるこれらの発見に起因して、砕塊ステップを省略し、流動層乾燥機への真空移行を用いた。
【0034】
実施例5は、順調に進み、生成された顆粒の見た目が良好であった。次いで、過程のロバスト性を確認するべきであることが決定された。過程のロバスト性には2つの態様が存在する。第1の態様は、同一品質を有する顆粒を一貫して生成する過程能力である。過程のロバスト性の第2の態様は、その過程を当業者によって独立して実行可能であるか否かである。したがって、当業者は、実施例6〜10の動作を実行した。
【0035】
実施例6では、表1および表2に要約した条件に加え、乾燥機パラメータについて記録した。乾燥機動作に関連するデータについて表4に要約する。全事例において、流動層乾燥機への真空移行を使用した。
【表4】
【0036】
実施例4〜10で生成した顆粒の特性について評価を行った。これらの評価を行う際、各工程実行からの全ての顆粒を個々に篩過し、16メッシュスクリーン上に保持された材料の量を記録した。次いで、光散乱法を使用して、篩過された材料の粒径分布を測定した。110℃の設定温度における水分残量を使用して、2つの別々の研究所で相互に独立して顆粒の含水量を判断した。これらの評価の結果について表5に要約し、表5において「STDEV」は、「標準偏差」を示し、「RSTD」は、相対標準偏差を示す。
【表5】
【0037】
また、顆粒の分画における活性含有物の分布についても評価した。本評価は、工程実行2および工程実行8からの約100グラムの顆粒を、ステンレス鋼の篩の束を通して別々に篩過することを伴った。(a)wt%の単位および(b)論理値のパーセントとして報告されるHPLCにより決定された分画の活性含有物におけるサイズ分画について表6に提示し、表6において、「μ」は、ミクロンを示す。表6における結果により、含有物が高度に活性であるほど、小さいサイズの粒子に発見されるという傾向があることが分かる。
【表6】
【0038】
顆粒の評価により、粒径分布の観点から、変動サイズ分画における活性含有物は、および粒子破砕性の観点から、本過程は、極めてロバストであることが示される。
【0039】
錠剤形成において上記実施例のうちのいくつかで生成した顆粒の性能を評価するために、実験を行った。その元の密封袋中の実施例9および10からの顆粒を50℃のオーブンに20時間置き、その顆粒を室温まで冷却した後に、これらの顆粒をまとめて混合して、より大きなブレンドを形成した。次いで、混合した顆粒を賦形剤と混合して、75wt%のイブプロフェンナトリウム2水和物を含有する製剤を生成した。次いで、本製剤を、ロータリープレス上で錠剤形状に変換した。ロータリープレス上でブレンドを錠剤にプレスした際にパンチコーティングが発生しなかったことが認められた。また、実施例5からの顆粒を製剤化し、ロータリープレス上で錠剤にプレスした際にもパンチコーティングが発
生しなかったことが認められた。
【0040】
錠剤の生成に使用した製剤(場合によっては、以下本明細書において製剤DTHと示す)の組成は、75%のイブプロフェンナトリウム2水和物、21.37%の微結晶性セルロース(MCC PH102;FMC Corporation)、1.96%のPVP
K−90(Plasdone K−90,International Specialty Products Inc.,Wayne,NJ)、1.57%の無水炭酸ナトリウム、および0.10%のコロイド状シリカ(Aerosil 200;Evonik
Industries,formerly Degussa Corporation)である。この錠剤製剤は、顆粒を微結晶性セルロースおよびコロイド状シリカと混合することによって調製される。DTH調製に使用する顆粒の100重量部毎に、21.21重量部の微結晶性セルロースおよび0.127重量部のコロイド状シリカが組み込まれる。
【0041】
製剤DTHを調製するためには、以下の手順が推奨される:16メッシュスクリーンを通して別々に篩過することによって、顆粒および微結晶性セルロースを砕塊する。全ての3つの成分の重さを別々に量り、コロイド状シリカと微結晶性セルロースの一部分とを混合することによって、コロイド状シリカおよび微結晶性セルロースの事前ブレンドを調製し、この混合物を20メッシュの篩に通過させる。その顆粒、その残りの微結晶性セルロース、およびその事前ブレンドを、低せん断ブレンダ内に排出し、10分間混合する。
【0042】
製剤DTHは、本発明の好適な製剤を構成するが、本発明をこの特定の製剤に限定するものとして解釈されるべきではないことを理解されたい。例えば、良好な結果は、クロスカルメロースナトリウム成分を製剤DTHから排除することによって達成され得る。本開示に基づいて本発明の顆粒を利用する他の製剤について、当業者は想到し得る。
【0043】
実施例3〜10で生成した顆粒試料の大部分の示差走査熱量測定(DSC)走査によって、100℃前後でピークに達する主な固体状態転移の直前に発熱反応の発生が示された。該発熱転移は、短時間、上昇温度(50℃)条件にそれらの顆粒を維持した後に消滅した。その転移の位置および上昇温度条件下に顆粒を維持した後の異常な消滅により、発熱ピークがそれらの顆粒における応力に関連することが示された。
【0044】
実施例11〜20は、乾燥条件に関連すると考えられる応力を引き起こすものおよび応力の排除方法を判断するために行われた実験研究を伴う。乾燥条件の研究に加え、湿潤造粒過程における混合時間を、概して、10分から16分に増加した。これらの実験は、0.5kgのバッチサイズで行なった。
実施例11〜20
【0045】
実施例11〜20で使用した手順は、成分の湿潤造粒、顆粒の乾燥、および顆粒の分析を伴った。実施例20を除く全ての実施例の造粒動作では、イブプロフェンナトリウム2水和物の同一の特定のロットからの試料を使用した。実施例20では、イブプロフェンナトリウム2水和物は、イブプロフェン生成物ナトリウム流から回収されたイブプロフェンナトリウム2水和物の小ベンチスケール結晶化からの生成物の複合混合物であった。この後者のベンチスケールイブプロフェンナトリウム2水和物は、実施例11〜19で使用したイブプロフェンナトリウム2水和物のロットの粒径よりも数倍大きい粒径を有した。使用した造粒処理ステップは、(i)イブプロフェンナトリウム2水和物(Na IBU)を高せん断造粒器の造粒器ボウル内に排出することと、(ii)Na IBUを砕塊することと、(iii)ポビドン溶液を添加して、高せん断下で造粒を推進することと、(iv)無水炭酸ナトリウム粉末をブレンド内に添加し、混合を再開することと、(v)乾燥
のために顆粒を流動層乾燥機に移行することとを伴った。実施例12においてのみ、砕塊ステップを使用した。実施例12では、PVP溶液の添加前に、チョッパを2500RPMで動作し、300RPMのインペラ速度によって、イブプロフェンナトリウム2水和物を90秒間砕塊した。砕塊の終了時に、PVP溶液を造粒器内に注入し、チョッパおよびインペラ速度を、設定された時間の間、同一速度で維持した。次いで、低下したせん断で造粒可能になるように速度を低下させた。その後、造粒器を停止し、蓋を除去した。次いで、炭酸ナトリウム(<20メッシュ)をブレンド上に振りかけた。蓋を戻して、2500RPMのチョッパ速度および300RPMのインペラ速度で約5分間混合し、1000RPMのチョッパ速度および150RPMのインペラ速度でさらに5分間混合した。表8は、高せん断造粒について、砕塊動作について、およびポリビニルピロリドン(PVP)溶液の添加に関する動作パラメータについて要約する。前述のように、表8では、「CPR」は、「チョッパ」を示し、「IMP」は、「インペラ」を示す。
【表7】
【0046】
表9は、粉末状無水炭酸ナトリウムを、造粒器内の含有物に添加した後に、湿潤造粒過程で使用した動作条件について要約する。このように、湿潤造粒手順は、完了した。表9では、前述のように、「CPR」は、「チョッパ」を示し、「IMP」は、「インペラ」を示す。用語の「n/a」は、「該当なし」を示す。
【表8】
【0047】
例えば14を例外とするが、造粒器動作の終了時に、湿潤顆粒の全てを、空気圧で即座に流動層乾燥機に移行した。実施例14では、乾燥するために空気圧で流動層乾燥機に移行する前に30分間、湿潤顆粒を造粒器に維持した。このような保持時間の使用は、湿潤
顆粒における応力が、乾燥前のある時間受けさせることによって放出され得るか否かを判断する目的のものである。
【0048】
表10は、本研究において使用した乾燥パラメータについて概説する。乾燥のための基本事例は、70℃の入口温度と、110℃における重量損失により決定された含水量の対照として、40℃の生成物温度とを使用した。実施例11、12、13、14、および20は、70℃に設定された入口温度で行ない、乾燥動作は、生成物温度は40℃に達すると停止させた。実施例15は、70℃に設定された入口温度で行なったが、試料採取は、生成物温度が35℃、40℃、45℃、および48℃に達した際に実行した。生成物温度を対照として使用する代わりに、実施例16および17は、乾燥時間を対照として使用した。実施例16では、70℃の入口温度で、生成物温度が40℃に到達した際に、分析用の第1の試料を回収し、乾燥を同一の入口温度で継続し、追加の試料を、乾燥の追加の10分および30分後にそれぞれ回収した。実施例17では、70℃の入口温度で、生成物温度が40℃に到達した際に、第1の試料を回収した。この時点において、入口温度は、60℃まで低下し、追加の試料を、それぞれ20分および60分の追加の乾燥時間の後に回収した。実施例18は、80℃の入口温度を用い、実施例19は、60℃の入口温度を使用した。表10は、これらの動作の要約を示す。
【表9】
DSC相転移および含水量
【0049】
表11は、変動する流動層乾燥条件下で回収された顆粒の水分残量によって決定された、示差走査熱量計データおよび含水量を提供する。本データによって、13.0%以上の含水量を有するほぼ全ての顆粒が、>170ジュール/グラムの転移熱を有することが示される。また、過剰乾燥によって、相転移熱が低くなり得ることも明らかである。それにもかかわらず、約100℃から約102℃の範囲でピークに達し、かつ固体から固体への相転移に対応するDSCピークのサイズがグラム当たり少なくとも約150ジュールであるDSC熱処理曲線を提供する過剰に乾燥された試料は、固体剤形の形成に利用する場合に満足できる結果を提示する。
【表10】
粒径分布
【0050】
顆粒の粒径分布により、全ての顆粒が狭い粒径分布を有することが示されるが、イブプロフェン生成物ナトリウム流(PSS)から結晶化された複合イブプロフェンナトリウム2水和物から調製される実施例20を例外とする。実施例11〜19に使用したイブプロフェンナトリウム2水和物ロットから生成された顆粒の平均中間粒径は、172ミクロンであり、標準偏差は、12ミクロンであった。実施例20で使用した複合イブプロフェンナトリウム二水和の中間粒径は、324ミクロンであり、その調製された顆粒の中間粒径は、232ミクロンであった。
【0051】
実施例11から19からの顆粒の中間粒径および粒径分布の類似性により、本過程が粒径分布に関して極めてロバストであることが示される。実施例11〜19と実施例20との中間粒径および粒径分布の違いにより、顆粒の粒径が、出発イブプロフェンナトリウム2水和物の粒径の関数であることが提案される。
変動する篩サイズ分画における篩分析およびアッセイ含有量
【0052】
実施例12および18からの20グラムの顆粒を、20、30、40、60、80、100、および200メッシュのステンレス鋼の篩から成る直径が12インチの篩の束上に置いた。20分間振動させた後、各篩上に保持された顆粒の重量を量り、選択されたサイズ分画の保持顆粒を、HPLCアッセイ測定のために提示した。理論的にwt%で表わされるアッセイ含有量について、以下の表12に提示した。本データにより、約90wt%の粒子が、40〜200のメッシュ範囲にあり、概して、粒径が大きくなればなるほど、より高いアッセイ含有量に対応することが示される。また、大部分の粒子(40〜200のメッシュ範囲)に関するアッセイ含有量における違いは、ごくわずかであることも示される。
【表11】
【0053】
本発明に関連して生成した顆粒を利用する利点を実証するために、実施例11、12、13、18、19、および20で生成された顆粒を、他の賦形剤と個々に混合して製剤、この場合、錠剤の形成に使用する製剤DTHを生成した。次いで、個々の製剤DTHブレンドを、10ステーションロータリープレス(Minipress II;GlobePharma,Inc.,New Brunswick,NJ)内に順番に供給して、約600mgのイブプロフェンに相当するナトリウム塩を含有する錠剤を調製した。主な圧縮を約10キロニュートン(KN)に維持し、事前圧縮を約1.5KNに維持し、生成速度を、1分あたり100〜120個の錠剤に維持した。錠剤試料を工程実行中に回収した。約30個の錠剤を有する錠剤試料の少なくとも1つの組をブレンド毎に回収した。全12個の錠剤試料の組を、錠剤調製過程によって処理された7つの製剤DTHブレンドから回収した。
【0054】
これらの錠剤の溶解は、10分における59%の平均理論溶解、20分における98%の溶解、13.9分における80%溶解を示す。被験製剤DTH錠剤の溶解速度は、規制の溶解速度よりも大幅に良好であった。
【0055】
回収された全12組の錠剤について、100液滴の平均破砕性は、0.14wt%であった。
【0056】
次に、図面を参照すると、図1〜17は、実施例11から20で生成され、かつ表11に関連して論じられた本発明の顆粒に関する示差走査熱量測定曲線であり、図面の簡単な説明において要約されている。その表に関連して述べたように、図8、図11、および図14の試料は、使用可能であると考えられたが、乾燥ステップ中に過剰乾燥されたために好ましくはない。
【0057】
本発明の好適な一種類の顆粒の組成と、本発明の錠剤を生成するための一種類の1つの好適な製剤の組成とについて表13に説明する。本発明のこのような好適な顆粒と本好適な錠剤製剤とに関するバッチサイズおよび組成について表14に説明する。
【表12】
【表13】
【0058】
40kgの乾燥バッチサイズスケールにおける本発明の一種類の好適な顆粒の調製のための典型的な手順は、以下の通りである:
1. 初期調製
a. 高せん断ブレンダを使用して水中でポリビニルピロリドン(PVP K−90
)を溶解する(13.33kgの精製水中に3kg)ことによって、いくつか
の工程実行に十分なPVP原液(18.37wt%)を調製する。
b. 手動で20メッシュスクリーンに通すか、または20メッシュスクリーンを装
備する粉砕機を使用するかのいずれかで、炭酸ナトリウムを篩過する。
2. 造粒
a. 38.20kgのイブプロフェンナトリウム2水和物の重量を量り、高せん断
造粒器内に装薬する。
b. 5.44kgのPVP溶液の重量を量り、高せん断下で動作中の造粒器に装薬
する。4分後、低せん断動作に切り替え、さらに4分間動作を継続する。
c. 篩過した無水炭酸ナトリウム(0.80kg)の重量を量り、高せん断下にあ
る造粒器に4分間添加し、その後、さらに4分間低せん断下にある造粒器に添
加する。
d. 顆粒を空気圧で流動層乾燥機に移行し、顆粒の静かな流動化を維持し、60℃
に設定された入口温度で乾燥を開始する。
3. 乾燥手順
a. 60℃に入口温度を設定し、ボウル温度および出口温度、試料乾燥機を定期的
に監視し、生成物の温度が約40℃に到達する際に乾燥を停止する。
b. 標的としての13.4wt%の含水量を含んで回収された試料から、含水量(
カールフィッシャーまたは110℃における水分残量)を測定する。
4. 粒径調整
a. 乾燥顆粒を16メッシュ篩に通過させて、>16メッシュの粒子を除去する。
>16メッシュ材料の重量を記録する。
b. >16メッシュ顆粒をサイズ低減器に通して粉砕し、<16メッシュ部分に添
加する。
c. 品質管理のために数百グラム保持し、残りの<16メッシュ生成物の重量を量
る。
d. 錠剤の調製に使用しない場合、ポリエチレン裏当てを含むファイバードラムに
顆粒を保管する。
5. 分析
a. 16、20、40、60、80、100、および200メッシュスクリーンを
使用して篩過することによって粒子分布を行う。
b. 含水量を測定する(カールフィッシャーまたは110℃における水分残量)。
【0059】
本発明の一種類の好適な製剤は、上述のように作製される好適な顆粒から形成され、以下の表に示す組成を有する。
【表14】
また、上記製剤は、10ステーションロータリープレス(Minipress II;GlobePharma,Inc.,New Brunswick,NJ)を使用して高品質錠剤を生成し、約400mgのイブプロフェンに相当するナトリウム塩を含有する錠剤を調製した。この製剤から作製される錠剤の溶解は、20分で80%を超える平均理論溶解を示した。これは、60分で80%の規制要件よりも大幅に優れている。これらの錠剤について、100液滴の平均破砕性は、0.23wt%であった。
【0060】
本発明の好適な錠剤製剤(製剤75)からの、本発明の一種類の好適な錠剤の調製のための典型的な手順は、以下の通りである:
1. 混合に利用可能な上述のように生成された顆粒の量を適切な変換係数と乗算するこ
とによって、3つの賦形剤(微結晶性セルロース、クロスカルメロースナトリウム、
およびコロイド状シリカ)の必要な量を決定する。例えば、製剤DTHの調製のため
に40kgの顆粒を混合する場合、微結晶性セルロース(MCC)の必要量は、8.
34kg(40kgx0.2084、後者の数字は、MCCの変換係数)であり、ク
ロスカルメロースナトリウムの必要量は、2.56kg(40kgx0.0637、
後者の数字は、クロスカルメロースナトリウムの変換係数)であり、コロイド状シリ
カの必要量は、0.051kg(40kgx0.00127、後者の数字は、コロイ
ド状シリカの変換係数)である。
2. MCCおよびクロスカルメロースナトリウムを別々に16メッシュスクリーンに通
して篩過し、これらの2つの別々の賦形剤のそれぞれの必要とされる量を取っておく
。約500グラムのMCCをプラスチック袋に取っておく(例えば、2〜5リットル
サイズの袋を使用する)。
3. コロイド状シリカの重量を量って、MCCを含有する袋に添加し、その袋の含有物
を混合し、任意の塊を手で破壊し、その混合物を、20メッシュスクリーンを通して
スクリーンにかける。
4. 顆粒、MCC、クロスカルメロースナトリウム、および事前ブレンドを、ツインシ
ェルブレンダ内に装薬し、低せん断下で10分間混合する。
5. ポリエチレン裏当てを含むファイバードラム内にこのブレンドを排出し、500グ
ラムの試料を保持する。
6. 分析
a. 16、20、40、60、80、100、および200メッシュスクリー
ンから成る束の篩を使用して篩過することによる粒子分布。
b. 含水量(カールフィッシャーまたは110℃における水分残量)。
c. 流動特徴(Flodexおよび流動性指数)。
7. 従来の条件下で動作するロータリープレスを使用して、製剤DTHから錠剤を調製
する。
【0061】
以下は、固体剤形の調製に適切な本発明の我々の例証的製剤である。
製剤AA)は、
● 粉末形状の成分から形成される顆粒組成物を含む成分であって、(i)乾燥ベースで
80重量部から98重量部のイブプロフェンナトリウム2水和物と、(ii)乾燥ベ
ースで1重量部から4重量部の無水炭酸ナトリウムと、(iii)乾燥ベースで1重
量部から15重量部の非架橋ポリビニルピロリドンとを含む成分から形成される約4
0wt%から約100wt%の製剤;
● 0wt%から約25wt%の微結晶性セルロース、リン酸水素カルシウム、またはそ
の両方;
● 0wt%から約8wt%のクロスポビドンまたはクロスカルメロースナトリウムと;● 0wt%から約0.5wt%のコロイド状シリカ;
● 0wt%から約10wt%のでんぷん;および
● 0wt%から約2wt%のステアリン酸、ステアリン酸マグネシウム、またはその両
方;
を含む、製剤である。
【0062】
製剤BB)は、顆粒組成物の量が、約70wt%から約100wt%であり、微結晶性セルロースの量が0wt%から約20wt%であり、クロスポビドンまたはクロスカルメロースナトリウムの量が0wt%から約8wt%であり、コロイド状シリカの量が約0.05wt%から約0.2wt%である製剤である。
【0063】
製剤CC)は、顆粒組成物の量が、約75wt%から約100wt%の範囲であり、微結晶性セルロースの量が、0wt%から約20wt%であり、クロスポビドンまたはクロスカルメロースナトリウムの量が、0wt%から約6wt%であり、コロイド状シリカの量が、0wt%から約0.2wt%である製剤である。
【0064】
製剤DD)は、顆粒組成物の量が約60wt%から約90wt%の範囲であり、微結晶性セルロース、リン酸水素カルシウム2水和物、またはその両方の量が、約10wt%から約30wt%であり、でんぷんの量が、0wt%から約6wt%であり、クロスポビドンまたはクロスカルメロースナトリウムの量が、0wt%から約6wt%であり、コロイド状シリカの量が、0wt%から約0.25wt%であり、ステアリン酸、ステアリン酸マグネシウム、またなその両方の量が、0wt%から約2wt%である製剤である。
【0065】
製剤EE)は、顆粒組成物の量が約85wt%から約100wt%の範囲であり、微結
晶性セルロース、リン酸水素カルシウム2水和物、またはその両方の量が、0wt%から約10wt%であり、でんぷんの量が、0wt%から約6wt%であり、クロスポビドンまたはクロスカルメロースナトリウムの量が、0wt%から約5wt%であり、コロイド状シリカの量が、0wt%から約0.25wt%であり、ステアリン酸、ステアリン酸マグネシウム、またなその両方の量が、0wt%から約2wt%である製剤である。
【0066】
また、本発明は、AA)、BB)、CC)、DD)、またはEE)のうちのいずれかの製剤から形成される固体剤形も提供する。また、本発明によって、粉末形状の成分であって、(i)乾燥ベースで80重量部から98重量部のイブプロフェンナトリウム2水和物と、(ii)乾燥ベースで1重量部から4重量部の無水炭酸ナトリウムと、(iii)乾燥ベースで1重量部から15重量部の非架橋ポリビニルピロリドンとを含む成分から形成される顆粒組成物を含有するハードシェルカプセルを含む剤形も提供される。加えて、本発明は、イブプロフェンナトリウム2水和物の固体剤形を調製する方法を提供し、本方法は、直前の文に説明する顆粒組成物をロータリープレスにおいて圧縮することを含む。
【0067】
本明細書および本明細書の請求項の任意の場所において化学名または化学式で言及される成分は、単数または複数で言及されているか否かにかかわらず、化学名または化学式で言及される別の物質(例えば別の成分、溶媒等)と接触する前に存在するものとして識別される。得られた混合物または溶液の中で、どんな化学変化、変形、および/または反応が発生することは、もしそれらが発生しなくても、このような変化、変形、および/または反応が、本開示に従って要求された条件下において特定の成分を一つにまとめた当然の結果であるので、問題ではない。したがって、これらの成分は、所望の動作の実行または所望の組成物の形成に関連して一つにまとめられる要素として識別される。
また、以下の請求項は、現在時制(「備える」、「である」等)で物質、成分、および/または要素に言及し得るが、物質、成分、または要素は、本開示に従って1つもしくは複数の他の物質、成分、および/または要素と最初に接触、混合、または混合される直前の時点に存在したものとして言及されている。
【0068】
他に明示している場合を除き、本明細書において使用する場合、および使用する際、冠詞「ある」(「a」または「an」)は、その冠詞が言及する単一の要素に請求を限定するように意図されず、また、限定するものとして解釈されるべきではない。むしろ、本明細書において使用する場合、および使用する際、本文が他に明示している場合を除き、冠詞「ある」(「a」または「an」)は1つもしくは複数のこのような要素を包むように意図される。
【0069】
本発明は、本明細書に記載の材料および/または手順を備え得るか、それらから成り得るか、またはそれらから本質的に成り得る。
【0070】
本発明は、その実践においてかなりの変動を受け易い。ゆえに、前述の説明は、上記に提示された特定の例示に本発明を限定するように意図されず、また、限定するものとして解釈されるべきではない。
【技術分野】
【0001】
本発明は、イブプロフェンナトリウム塩2水和物から作製されるイブプロフェン薬物の高含量顆粒、このような顆粒を生成するための処理技術、および経口投与用の非発泡性固体剤形を調製する際のこのような顆粒の使用に関する。
【背景技術】
【0002】
既刊文献によると、イブプロフェンナトリウムから作製される固体剤形は、医薬的反応速度において優れている(すなわち、この固体剤形は、イブプロフェンの他の固体剤形よりも急速に血流に入り、ピークに達する)。しかしながら、本分野において集中的に研究されているにもかかわらず、従来の手法を使用してイブプロフェンナトリウムから固体剤形を形成することは、依然として困難である。その困難の1つとして、イブプロフェンナトリウム2水和物が、流動性が低い特徴を有することから、コロイド状シリカまたはタルク等の従来の凝固阻止剤と混合する場合であっても容易に凝固する傾向があることが挙げられる。流動性向上剤とともに製剤化されるにもかかわらず流動性が低い特徴は、生成される錠剤およびカプレット等の固体剤形の異常な重量変動をもたらす可能性がある。イブプロフェンナトリウムの錠剤およびカプレットを形成する際の別の困難として、ロータリープレスのパンチ表面上にフィルムコーティング形成するイブプロフェンナトリウム2水和物の傾向が挙げられる。このようなフィルムコーティングは、過剰でない場合であっても、形成される錠剤またはカプレットの輝きつまり光沢を低減させるため、望ましくない。過剰なコーティングがパンチ表面上に形成される場合、いくつかの動作上の困難に直面する。まず第1に、形成される錠剤またはカプレットに欠陥が発生する。第2に、異常なパンチ動作がもたらされる。第3に、錠剤またはカプレットの許容不可能な重量の変動にも直面し得る。次いで、これらの動作上の困難のいずれかによって、パンチ表面を清浄するために設備を停止することが必要となる。
【0003】
ゆえに、上述の種々の困難に直面することなく、従来のロータリープレス錠剤化設備を使用して、より容易に、錠剤またはカプレット等の固体剤形に変換できるイブプロフェンナトリウム組成物を形成する方式を発見できるのであれば、相当有利になるであろう。また、顆粒で充填されるハードシェルカプセルの投与量レベルをより均一に維持することができるように、イブプロフェンナトリウム2水和物から高度に流動可能な顆粒を生成する方式を発見できるのであれば、相当有利になるであろう。
【0004】
本発明は、前述の必要性の全てに効果的に対処する。
【発明の概要】
【0005】
本発明によると、湿潤造粒過程によって、ラセミイブプロフェンナトリウム2水和物(別称、2−(4−イソブチルフェニル)プロピオン酸ナトリウム2水和物、および以下本明細書において、より単純にイブプロフェンナトリウム2水和物と呼ばれる)、炭酸ナトリウム、およびポリビニルピロリドン(別称、2−ピロリドン、1−エテニル−、ホモポリマー)から形成される顆粒が、極めて望ましい特性を有すること、ならびに適切に製剤化される際に、顆粒が、上述の種々の困難に直面することなく、従来のロータリープレス錠剤化装置において効果的に利用可能であることが発見された。さらに、造粒動作およびロータリープレス錠剤化動作では、顆粒の炭酸ナトリウムの量が3wt%以下のみであっ
ても、本発明により可能になる有利な結果を達成できることが発見された。当然ながら、顆粒が2wt%の炭酸ナトリウムのみを含有する本発明の顆粒から優れた錠剤を生成することが可能であることが発見された。
【0006】
また、このような極めて少量の炭酸ナトリウムが、顆粒と、顆粒から形成されて得られた錠剤との形成に効果的であるという事実は、高齢者が消費する錠剤に関して利点を有する。周知のように、人は歳を取るにつれて、消化管の酸性度が低下する傾向にあるため、本発明の錠剤におけるこのような低レベルの炭酸ナトリウムによって、このような人の消化過程をあまり悪化させない。
【0007】
その過程の実施形態のうちの1つでは、本発明は、
A) イブプロフェンナトリウム2水和物の高度に分散可能な自由流動顆粒を調製する過程であって、その方法は、
● (i)乾燥ベースで少なくとも80重量部のイブプロフェンナトリウム2水和物と、
(ii)乾燥ベースで1重量部から4重量部の炭酸ナトリウムと、(iii)乾燥ベ
ースで1重量部から15重量部の非架橋ポリビニルピロリドンと、(iv)湿潤混合
物を形成するための、(i)、(ii)、(iii)、および(iv)の全重量に基
づく8重量部から12重量部の水とから構成される成分を、高せん断造粒器にまとめ
ることと、
● 湿潤顆粒を形成するために、上記高せん断造粒器において上記湿潤混合物を造粒する
ことと、
● 110℃における重量損失の測定により決定可能である約11wt%から15wt%
の範囲、好ましくは、約12wt%から15wt%の範囲の含水量を有する乾燥顆粒
を形成するように、湿潤顆粒を乾燥することと、
● 16メッシュを超える粒径を有する乾燥顆粒を篩過することによって除去することと
、
を含む、過程を提供する。
【0008】
本発明の好適な過程の実施形態は、以下の通りである:
B) 上記高せん断造粒器における上記湿潤混合物の造粒は、上記湿潤混合物の乾燥後に、乾燥顆粒が示差走査熱量測定中に約100℃から約102℃の範囲でピークに達する相転移を呈するように、ある時間の間行なわれ、固体から固体への相転移に対応するDSCピークのサイズが、グラム当たり少なくとも約150ジュールである、A)における過程。
【0009】
その組成物の実施形態の中で、本発明は以下を提供する:
C) 高含量のイブプロフェンナトリウム2水和物を有する高度に分散可能な自由流動顆粒組成物であって、(i)乾燥ベースで80重量部から98重量部のイブプロフェンナトリウム2水和物と、(ii)乾燥ベースで1重量部から4重量部の無水炭酸ナトリウムと、(iii)乾燥ベースで1重量部から15重量部の非架橋ポリビニルピロリドンとを含む粉末形状の成分から形成される組成物。
D) ロータリープレスを使用する固体剤形の調製に適合されるイブプロフェンナトリウム2水和物製剤であって、
● 約40wt%から約100wt%のC)における顆粒組成物、
● 0wt%から約25wt%の微結晶性セルロース、リン酸水素カルシウム、またはそ
の両方、
● 0wt%から約8wt%のクロスポビドンまたはクロスカルメロースナトリウム、
● 0wt%から約0.5wt%のコロイド状シリカ、
● 0wt%から約10wt%のでんぷん、および
● 0wt%から約2wt%のステアリン酸、ステアリン酸マグネシウム、またはその両
方、
を含む成分から形成される製剤。
【0010】
本発明の好適な過程の実施形態は、以下の通りである:
E) 上記製剤は、示差走査熱量測定中に約100℃から約102℃の範囲でピークに達する相転移を呈し、固体から固体への相転移に対応するDSCピークのサイズが、グラム当たり少なくとも約150ジュールである、D)における製剤。
【0011】
本発明に関する他の実施形態、特徴、および利点は、以下の説明、添付の図面、および添付の請求項によって、またさらに明らかになる。
【図面の簡単な説明】
【0012】
【図1】実施例11において生成された本発明の顆粒に関する示差走査熱量測定曲線である。
【図2】実施例12において生成された本発明の顆粒に関する示差走査熱量測定曲線である。
【図3】実施例13において生成された本発明の顆粒に関する示差走査熱量測定曲線である。
【図4】実施例14において生成された本発明の顆粒に関する示差走査熱量測定曲線である。
【図5】乾燥機入口温度が70℃に設定された状態で、顆粒の乾燥温度が35℃に到達した際に試料採取された、実施例15において生成された本発明の顆粒に関する示差走査熱量測定曲線である。
【図6】乾燥機入口温度が70℃に設定された状態で、顆粒の乾燥温度が40℃に到達した際に試料採取された、実施例15において生成された本発明の顆粒に関する示差走査熱量測定曲線である。
【図7】乾燥機入口温度が70℃に設定された状態で、顆粒の乾燥温度が45℃に到達した際に試料採取された、実施例15において生成された本発明の顆粒に関する示差走査熱量測定曲線である。
【図8】乾燥機入口温度が70℃に設定された状態で、顆粒の乾燥温度が48℃に到達した際に試料採取された、実施例15において生成された本発明の顆粒に関する示差走査熱量測定曲線である。
【図9】乾燥機入口温度が70℃に設定された状態で、顆粒の乾燥温度が40℃に到達した際に試料採取された、実施例16において生成された本発明の顆粒に関する示差走査熱量測定曲線である。
【図10】乾燥機入口温度が70℃に設定された状態で、図10の試料が取られた10分後に試料採取された、実施例16において生成された本発明の顆粒に関する示差走査熱量測定曲線である。
【図11】乾燥機入口温度が70℃に設定された状態で、図10の試料が取られた30分後に試料採取された、実施例16において生成された本発明の顆粒に関する示差走査熱量測定曲線である。
【図12】乾燥機入口温度が70℃に設定された状態で、顆粒が60℃の温度に達成した際に試料採取された、実施例17において生成された本発明の顆粒に関する示差走査熱量測定曲線である。
【図13】70℃から60℃に乾燥機入口空気温度を低下させることによって、顆粒が20分間60℃の温度にあった際に試料採取された、実施例17において生成された本発明の顆粒に関する示差走査熱量測定曲線である。
【図14】70℃から60℃に乾燥機入口空気温度を低下させることによって、顆粒が60分間60℃の温度にあった際に試料採取された、実施例17において生成された本発明の顆粒に関する示差走査熱量測定曲線である。
【図15】実施例18において生成された本発明の顆粒に関する示差走査熱量測定曲線である。
【図16】実施例19において生成された本発明の顆粒に関する示差走査熱量測定曲線である。
【図17】実施例20において生成された本発明の顆粒に関する示差走査熱量測定曲線である。
【発明を実施するための形態】
【0013】
主成分
【0014】
上述のように、本発明の顆粒において、ならびに錠剤、カプレット、およびハードシェルカプセル等の本発明の固体剤形において、3つの主成分が存在する。これらの成分は、イブプロフェンナトリウム2水和物、炭酸ナトリウム、およびポリビニルピロリドンである。これらの成分の各々は、当技術分野において周知であり、これらのそれぞれの材料の調製方法も周知であり、文献において報告されている。全てのこのような成分は、NF、USP、または複数公定書グレードであるべきである。言い換えると、このような成分は、医薬的に許容可能な純度、等級、および/または品質を有するべきである。
本発明の顆粒の調製
【0015】
本発明の顆粒を調製するために、上記の3つの主成分を、高せん断造粒器において水で湿らせる間に、相互に対して適切な比率で混合する。実際は、上記の3つの成分の全体的な混合を形成するまえに、水中でポリビニルピロリドンの溶液を形成することが望ましい。これにより、本発明の顆粒内にポリビニルピロリドンのより均一な分布が提供される。高せん断造粒器における混合および造粒の後に、オーブンにおけるパン乾燥等の従来の乾燥手順によって、または流動層乾燥によって、湿潤顆粒から水を除去する。典型的には、その乾燥は、最大約100℃の温度でもたらされる。好ましくは、顆粒の乾燥のためのオーブン温度は、約60℃から約70℃の範囲である。流動層乾燥機の場合、入口温度も、好ましくは、約60℃から約70℃の範囲である。特に好適な乾燥動作では、出口空気の温度は、出口空気温度が約40℃に達する際に乾燥が停止するように監視される。このように、顆粒の全含水量は、約12wt%から約15wt%の好適な範囲であり、より好ましくは、約13.5wt%であり、これは、ほぼイブプロフェンナトリウム2水和物における水和水の量である。次いで、この乾燥生成物は、過剰サイズの材料(例えば、16メッシュを超える)を除去するように篩過される。このような過剰サイズの材料は、典型的には、粉砕され、造粒動作に再利用される。
【0016】
さらに詳細には、イブプロフェンナトリウム2水和物を高せん断造粒器内に装薬し、その後、水中でポリビニルピロリドン溶液を事前形成することが好ましい。これらの成分の混合の後、必要量の炭酸ナトリウムを添加し、この混合物に、追加の高せん断造粒を供する。この造粒は、造粒中に生成される過剰な熱を除去するように、必要に応じて冷却を使用して、約15℃から約35℃の範囲の温度で行われる。炭酸ナトリウムの添加前の高せん断造粒器におけるチョッパのせん断速度は、典型的には、約3分から約6分の範囲の時間の間、約1000rpmから約2000rpmの範囲である。次いで、チョッパ速度は、約3分から約6分の範囲の追加の時間の間、約200rpmから約500rpmの低せん断速度まで低下する。炭酸ナトリウムの添加後、そのチョッパについてこの同じ2段階動作が繰り返される(すなわち、約3分から約6分の間、約1000rpmから約2000rpm、次いで、約3分から約6分の間、約200rpmから約500rpm)。
【0017】
高せん断造粒器の設計に依存して、該装置は、造粒器における混合物の運動をもたらすための別の機構を含んでもよく、その機構は、パドルとして知られている。パドルが動作する速度は、あまり重要ではないため、変動することができる。160リットルのFluid Air Pharmx High Shear Granulator,Model PX150を使用する場合、第1段階中に100rpmでパドルを動作し、第2段階において30rpmで動作する2段階方式でパドルを動作することが便利であることが分かった。これらの段階は、チョッパの2段階動作と一致する。
【0018】
顆粒を形成する際に使用する炭酸ナトリウムは、NFまたはUSPグレードであるべきであり、最初に水和化し得るが、好ましくは、無水粉末の形状である。医薬的に許容可能なグレードのポリビニルピロリドンは、本発明の実施に使用され得る種々の形状で利用可能である。しかしながら、30から120の範囲のK値を有するポリビニルピロリドンを利用することが好ましく、90のK値を有するポリビニルピロリドンを利用することがさらに好ましい。イブプロフェンナトリウム2水和物は、医薬的に許容可能なグレードのラセミ混合物であり、好ましくは、粉末形状で使用される。
【0019】
顆粒を形成する際に使用する炭酸ナトリウムの量は、典型的には、全混合物の約1重量%から4重量%の範囲である。好ましくは、その量は、全混合物の約2重量%から約3重量の範囲である。顆粒を形成する際、ポリビニルピロリドンの量は、典型的には、全混合物の約1重量%から10重量%の範囲である。好ましくは、ポリビニルピロリドンの量は、全混合物の約2重量%から4重量%の範囲である。好ましくは、全混合物の100重量%に対する残量は、イブプロフェンナトリウム2水和物から構成される。必要に応じて、3つの主成分のいずれの成分と不利に相互作用しない限り、あるいは3つの主成分から生成される顆粒または固体剤形の調製を妨げない限り、他の従来の医薬的に許容可能な賦形剤を使用することができる。しかしながら、このような使用によって、顆粒内のイブプロフェンナトリウム2水和物の濃度が低下し、生産コストおよび記録管理の増加がもたらされることから、このような賦形剤を顆粒に含めることは、望ましくはない。
【0020】
また、使用する水は、規制要件を満たすのに十分な純度を有する。顆粒を形成する際に使用する水の量は、典型的には、成分の湿潤混合物の全重量の約5wt%から約15wt%の範囲である。好ましくは、顆粒形成に使用する水の量は、成分の湿潤混合物の全重量の約8wt%から約12wt%の範囲である。使用する水が少な過ぎると、不十分な造粒が発生する。一方、使用する水が多過ぎると、混合物は、パン生地の硬さになる。いずれの場合であっても、乾燥後、生成物は、望ましい平均(中間)粒径を持たない。これに関し、乾燥顆粒の平均粒径は、典型的には、約150ミクロンから約600ミクロンの範囲、好ましくは、約200ミクロンから約300ミクロンの範囲である。
【0021】
湿潤形状における本発明の特に好適な顆粒は、特定の量の以下の成分から調製される:イブプロフェンナトリウム2水和物、85.95wt%;非架橋ポリビニルピロリドン(特にK−90)、2.25wt%;無水炭酸ナトリウム粉末、1.80wt%;精製水、10wt%。乾燥形状における本発明の特に好適な顆粒は、以下の組成を有する:イブプロフェンナトリウム2水和物として95.50wt%;2.50wt%のポリビニルピロリドン(特にK−90);および無水炭酸ナトリウムとして2.00wt%。
好適な顆粒および顆粒製剤
【0022】
上に示すように、本発明は、好適な実施形態において、示差走査熱量測定中に、約100℃から約102℃の範囲でピークに達する相転移を呈し、固体から固体への相転移に対応するDSCピークのサイズは、グラム当たり少なくとも約150ジュールであることを特徴とする顆粒および顆粒製剤を提供する。このピークの存在によって、顆粒は、上記で
E)と示す製剤のように適切に製剤化される場合に、ロータリープレスにおいて優れた処理特徴を有する固体製剤を生成し、それによって、高品質の固体剤形が生成され、現在の規制要件への順守が確実になる。顆粒およびその結果生じた製剤が、示差走査熱量測定(DSC)中に前述の熱的特徴を有することを確実にする種々の方式が存在する。例えば、顆粒試料が、このような熱的特徴を呈さない場合:
● 前述のDSCの熱的特徴を達成するのに十分な温度と時間で顆粒を加熱することがで
きる。提案する加熱条件は、約5分から約50分の間、約30℃から約70℃で加熱
することを含む;
● 湿潤造粒を行う時間を延長することができる。ベンチスケールの動作では、湿潤造粒
のこの全時間は、少なくとも約12分から16分であることが分かっている。この動
作をより大きなスケールで行う場合、湿潤造粒の全時間は、所望のDSCの熱的特徴
を達成するために12分から16分とは少なくともいくらか異なり得るため、より大
きな規模で動作する場合に、湿潤造粒の適切な時間を決定するために、必要に応じて
、いくつかのパイロット実験を行うべきである;
● 顆粒が依然として湿潤である場合に造粒過程において成分を含めることを回避するこ
とによって、前述のDSCの熱的特徴が達成されなくなる。これに関して悪影響をも
たらすと考えられる成分は、でんぷん、グリコール酸でんぷんナトリウム、およびス
テアリン酸マグネシウムである。以下に提示する実施例から分かるように、顆粒が乾
燥すると、固体剤形の生成にお使用するための製剤における成分として、微結晶性セ
ルロースを利用することができる。当然ながら、微結晶性セルロースが、このような
製剤において使用するために好適な成分である。
【0023】
以下の実施例は、例証目的のために提示される。実施例は、実施例に記載される詳細および材料に本発明を限定することを意図しない。
実施例1
顆粒の調製
【0024】
95.5wt%のイブプロフェンナトリウム水和物、2wt%の炭酸ナトリウム、および2.5wt%のポリビニルピロリドン(Plasdone K−90、International Specialty Products Inc.,Wayne,NJ)から構成される顆粒を、本発明の湿潤造粒過程によって調製した。使用する過程は、水中でPlasdone K−90を溶解することと、高せん断増強駆動部(GlobePharma,Inc.,New Brunswick,NJにより製造されるMAXI−BLEND LAB V−BLENDER)を装備するV字型ブレンダにおいて、その溶液にイブプロフェンナトリウム2水和物を添加し、高いせん断を使用してブレンダを動作することによって顆粒を形成することと、篩過された炭酸ナトリウム粉末をブレンダ内の湿潤混合物に添加することと、増強駆動部を利用してさらに5分間混合することを伴った。ブレンダの含有物をパン内に排出した後、添加された全ての水が除去されるまで、50度に維持したオーブンで顆粒を乾燥した。次いで、その生成物を、ステンレス鋼の16メッシュのU.S.A.規格の篩を通して篩過した。
実施例2
錠剤の調製
実施例2A
【0025】
実施例1で形成した顆粒を使用して、ロータリープレスを使用する錠剤調製のために、完全に製剤化されたブレンドを調製した。本動作では、乾式混合によって、顆粒を、微結晶性セルロース(MCC)およびコロイド状シリカと混合した。10ステーションロータリープレスにおいて十分処理された製剤と、得られた錠剤とは、良好な破砕性、溶解、お
よび分解を示した。最終ブレンドは、75wt%のイブプロフェンナトリウム水和物、2.37wt%のポリビニルピロリドン、1.58wt%の炭酸ナトリウム、20.95wt%の微結晶性セルロース、および0.1wt%のコロイド状シリカから構成された。
【0026】
実施例1で形成した高活性含有顆粒と、実施例2Aで形成した錠剤製剤とに関する特徴について表1に提示する。流動性指数によって、顆粒と完全に製剤化されたブレンドとの両方が、良好な流動特徴を有することが示された。加えて、錠剤は、全ての所望の性能基準を満たした。
【表1】
実施例2B
【0027】
実施例1で形成した顆粒を使用して、実施例2Aで説明したように、ロータリープレスを使用する錠剤調製のために完全に製剤化されたブレンドを調製したが、乾式混合によって顆粒をクロスポビドンと混合したことを例外とする。10ステーションロータリープレスにおいて十分処理された製剤と、得られた錠剤とは、許容可能な破砕性および良好な溶解性を示した。最終ブレンドは、93.6wt%のイブプロフェンナトリウム水和物、2.45wt%のポリビニルピロリドン、1.96wt%の炭酸ナトリウム、および2wt%のクロスポビドンから構成された。
実施例2C
【0028】
実施例1で形成した顆粒を使用して、実施例2Aで説明したように、ロータリープレスを使用する錠剤調製のために完全に製剤化されたブレンドを調製したが、乾式混合によって顆粒をNaクロスカルメロースと混合したことを例外とする。10ステーションロータリープレス内に十分流入した製剤と、得られた錠剤とは、許容可能な破砕性および良好な溶解性を示した。最終ブレンドは、93.6wt%のイブプロフェンナトリウム水和物、2.45wt%のポリビニルピロリドン、1.96wt%の炭酸ナトリウム、および2wt%のNaクロスカルメロースから構成された。
実施例3〜10
【0029】
これらの実施例では、ベンチスケール高せん断造粒器、Model PX1,2−Liter Fluid Air High Shear Granulatorにおいて、湿潤造粒技法を使用して工程実行を行った。本装置は、ボウルに配置されるインペラおよびチョッパを含む。インペラは、ボウル内で造粒されるブレンドを、ボウル内において円軌道で回転させ、チョッパは、処理中にブレンド中に形成される大粒子を破壊する。該造粒器は、圧縮空気によって動作され、本動作では、一部の圧縮空気が、ボウルに進入し、マシュマロの硬さに似た硬さを有するブレンド、すなわち、軟らかく、かつ低密度を有するブレンドの形成に寄与する。さらに混合すると、閉じ込められた空気の一部が放出される。そのブレンドは、湿潤外観を有する。得られた顆粒の外観は、同一の製剤を使用して
ツインシェルブレンダで処理された顆粒とは大幅に異なる。ツインシェルブレンダからの顆粒は、本装置で生成される顆粒よりも大幅に乾燥して見える。
【0030】
典型的な動作では、イブプロフェンナトリウム2水和物をボウルに添加した後に、蓋をボウル上に置き、チョッパ速度およびインペラ速度を、それぞれ2500RPMおよび300RPMに設定する。混合の作動直後に、ポビドンK−90水溶液を、蓋の穴を通して造粒器に注入する。典型的には、ボウルのカバーの小さい穴を通してボウルに液体を移行するのに約100秒かかる。ポビドン溶液の移行後、高せん断混合を事前設定した時間継続する。次いで、チョッパおよびインペラの両方の混合速度を、元の設定の約半分まで低下させ、この低せん断混合を、さらに数分間継続する。次いで、無水炭酸ナトリウム粉末(20メッシュスクリーンで前もって篩過されている)をボウルに添加し、さらなる高せん断−低せん断混合周期の間、混合を継続した。工程実行1で生成された湿潤顆粒を、流動層乾燥機に添加する前に、ビーカー内に排出した。プラスチック袋に回収された乾燥材料によって、16メッシュを超える多量の顆粒の存在が示される。
【0031】
本動作は、2つの段階を伴った。第1段階では、イブプロフェンナトリウム2水和物およびポリビニルピロリドン(K−90溶液)を造粒器に添加し、混合した。第2段階では、微粉化した無水炭酸ナトリウムを、第1段階からの造粒器含有物に添加し、得られた混合物を顆粒に処理した。
【0032】
実施例3〜10の第1段階における湿潤造粒動作の条件について表2に要約する。表3は、第2段階に使用した湿潤造粒動作の条件について要約する。表2および表3では、記号表示「CPR」は、「チョッパ」を示し、「IMP」は、「インペラ」を示し、「n/a」は、「該当なし」を示す。
【表2】
【表3】
【0033】
実施例4では、高せん断チョッパの砕塊効率を、造粒器内の塊の存在を確認することによって、種々の混合間隔において測定した。その元の粒子形状におけるイブプロフェンナトリウム2水和物の砕塊が効率的ではなく、ポビドン溶液の添加後に、砕塊が大幅により効率的になるという結論に達した。高せん断混合周期で約120秒後に、検出可能な塊は発見されなかった。その造粒器動作の後、造粒器ボウルの含有物を、カバーの穴に適用される真空管を使用して、流動層乾燥機が生成する吸引力により、空気圧で流動層乾燥機に移行した。回収された乾燥材料により、サイズが16メッシュを超える顆粒の含有物が極めて少量であることが示された。実施例5〜10におけるこれらの発見に起因して、砕塊ステップを省略し、流動層乾燥機への真空移行を用いた。
【0034】
実施例5は、順調に進み、生成された顆粒の見た目が良好であった。次いで、過程のロバスト性を確認するべきであることが決定された。過程のロバスト性には2つの態様が存在する。第1の態様は、同一品質を有する顆粒を一貫して生成する過程能力である。過程のロバスト性の第2の態様は、その過程を当業者によって独立して実行可能であるか否かである。したがって、当業者は、実施例6〜10の動作を実行した。
【0035】
実施例6では、表1および表2に要約した条件に加え、乾燥機パラメータについて記録した。乾燥機動作に関連するデータについて表4に要約する。全事例において、流動層乾燥機への真空移行を使用した。
【表4】
【0036】
実施例4〜10で生成した顆粒の特性について評価を行った。これらの評価を行う際、各工程実行からの全ての顆粒を個々に篩過し、16メッシュスクリーン上に保持された材料の量を記録した。次いで、光散乱法を使用して、篩過された材料の粒径分布を測定した。110℃の設定温度における水分残量を使用して、2つの別々の研究所で相互に独立して顆粒の含水量を判断した。これらの評価の結果について表5に要約し、表5において「STDEV」は、「標準偏差」を示し、「RSTD」は、相対標準偏差を示す。
【表5】
【0037】
また、顆粒の分画における活性含有物の分布についても評価した。本評価は、工程実行2および工程実行8からの約100グラムの顆粒を、ステンレス鋼の篩の束を通して別々に篩過することを伴った。(a)wt%の単位および(b)論理値のパーセントとして報告されるHPLCにより決定された分画の活性含有物におけるサイズ分画について表6に提示し、表6において、「μ」は、ミクロンを示す。表6における結果により、含有物が高度に活性であるほど、小さいサイズの粒子に発見されるという傾向があることが分かる。
【表6】
【0038】
顆粒の評価により、粒径分布の観点から、変動サイズ分画における活性含有物は、および粒子破砕性の観点から、本過程は、極めてロバストであることが示される。
【0039】
錠剤形成において上記実施例のうちのいくつかで生成した顆粒の性能を評価するために、実験を行った。その元の密封袋中の実施例9および10からの顆粒を50℃のオーブンに20時間置き、その顆粒を室温まで冷却した後に、これらの顆粒をまとめて混合して、より大きなブレンドを形成した。次いで、混合した顆粒を賦形剤と混合して、75wt%のイブプロフェンナトリウム2水和物を含有する製剤を生成した。次いで、本製剤を、ロータリープレス上で錠剤形状に変換した。ロータリープレス上でブレンドを錠剤にプレスした際にパンチコーティングが発生しなかったことが認められた。また、実施例5からの顆粒を製剤化し、ロータリープレス上で錠剤にプレスした際にもパンチコーティングが発
生しなかったことが認められた。
【0040】
錠剤の生成に使用した製剤(場合によっては、以下本明細書において製剤DTHと示す)の組成は、75%のイブプロフェンナトリウム2水和物、21.37%の微結晶性セルロース(MCC PH102;FMC Corporation)、1.96%のPVP
K−90(Plasdone K−90,International Specialty Products Inc.,Wayne,NJ)、1.57%の無水炭酸ナトリウム、および0.10%のコロイド状シリカ(Aerosil 200;Evonik
Industries,formerly Degussa Corporation)である。この錠剤製剤は、顆粒を微結晶性セルロースおよびコロイド状シリカと混合することによって調製される。DTH調製に使用する顆粒の100重量部毎に、21.21重量部の微結晶性セルロースおよび0.127重量部のコロイド状シリカが組み込まれる。
【0041】
製剤DTHを調製するためには、以下の手順が推奨される:16メッシュスクリーンを通して別々に篩過することによって、顆粒および微結晶性セルロースを砕塊する。全ての3つの成分の重さを別々に量り、コロイド状シリカと微結晶性セルロースの一部分とを混合することによって、コロイド状シリカおよび微結晶性セルロースの事前ブレンドを調製し、この混合物を20メッシュの篩に通過させる。その顆粒、その残りの微結晶性セルロース、およびその事前ブレンドを、低せん断ブレンダ内に排出し、10分間混合する。
【0042】
製剤DTHは、本発明の好適な製剤を構成するが、本発明をこの特定の製剤に限定するものとして解釈されるべきではないことを理解されたい。例えば、良好な結果は、クロスカルメロースナトリウム成分を製剤DTHから排除することによって達成され得る。本開示に基づいて本発明の顆粒を利用する他の製剤について、当業者は想到し得る。
【0043】
実施例3〜10で生成した顆粒試料の大部分の示差走査熱量測定(DSC)走査によって、100℃前後でピークに達する主な固体状態転移の直前に発熱反応の発生が示された。該発熱転移は、短時間、上昇温度(50℃)条件にそれらの顆粒を維持した後に消滅した。その転移の位置および上昇温度条件下に顆粒を維持した後の異常な消滅により、発熱ピークがそれらの顆粒における応力に関連することが示された。
【0044】
実施例11〜20は、乾燥条件に関連すると考えられる応力を引き起こすものおよび応力の排除方法を判断するために行われた実験研究を伴う。乾燥条件の研究に加え、湿潤造粒過程における混合時間を、概して、10分から16分に増加した。これらの実験は、0.5kgのバッチサイズで行なった。
実施例11〜20
【0045】
実施例11〜20で使用した手順は、成分の湿潤造粒、顆粒の乾燥、および顆粒の分析を伴った。実施例20を除く全ての実施例の造粒動作では、イブプロフェンナトリウム2水和物の同一の特定のロットからの試料を使用した。実施例20では、イブプロフェンナトリウム2水和物は、イブプロフェン生成物ナトリウム流から回収されたイブプロフェンナトリウム2水和物の小ベンチスケール結晶化からの生成物の複合混合物であった。この後者のベンチスケールイブプロフェンナトリウム2水和物は、実施例11〜19で使用したイブプロフェンナトリウム2水和物のロットの粒径よりも数倍大きい粒径を有した。使用した造粒処理ステップは、(i)イブプロフェンナトリウム2水和物(Na IBU)を高せん断造粒器の造粒器ボウル内に排出することと、(ii)Na IBUを砕塊することと、(iii)ポビドン溶液を添加して、高せん断下で造粒を推進することと、(iv)無水炭酸ナトリウム粉末をブレンド内に添加し、混合を再開することと、(v)乾燥
のために顆粒を流動層乾燥機に移行することとを伴った。実施例12においてのみ、砕塊ステップを使用した。実施例12では、PVP溶液の添加前に、チョッパを2500RPMで動作し、300RPMのインペラ速度によって、イブプロフェンナトリウム2水和物を90秒間砕塊した。砕塊の終了時に、PVP溶液を造粒器内に注入し、チョッパおよびインペラ速度を、設定された時間の間、同一速度で維持した。次いで、低下したせん断で造粒可能になるように速度を低下させた。その後、造粒器を停止し、蓋を除去した。次いで、炭酸ナトリウム(<20メッシュ)をブレンド上に振りかけた。蓋を戻して、2500RPMのチョッパ速度および300RPMのインペラ速度で約5分間混合し、1000RPMのチョッパ速度および150RPMのインペラ速度でさらに5分間混合した。表8は、高せん断造粒について、砕塊動作について、およびポリビニルピロリドン(PVP)溶液の添加に関する動作パラメータについて要約する。前述のように、表8では、「CPR」は、「チョッパ」を示し、「IMP」は、「インペラ」を示す。
【表7】
【0046】
表9は、粉末状無水炭酸ナトリウムを、造粒器内の含有物に添加した後に、湿潤造粒過程で使用した動作条件について要約する。このように、湿潤造粒手順は、完了した。表9では、前述のように、「CPR」は、「チョッパ」を示し、「IMP」は、「インペラ」を示す。用語の「n/a」は、「該当なし」を示す。
【表8】
【0047】
例えば14を例外とするが、造粒器動作の終了時に、湿潤顆粒の全てを、空気圧で即座に流動層乾燥機に移行した。実施例14では、乾燥するために空気圧で流動層乾燥機に移行する前に30分間、湿潤顆粒を造粒器に維持した。このような保持時間の使用は、湿潤
顆粒における応力が、乾燥前のある時間受けさせることによって放出され得るか否かを判断する目的のものである。
【0048】
表10は、本研究において使用した乾燥パラメータについて概説する。乾燥のための基本事例は、70℃の入口温度と、110℃における重量損失により決定された含水量の対照として、40℃の生成物温度とを使用した。実施例11、12、13、14、および20は、70℃に設定された入口温度で行ない、乾燥動作は、生成物温度は40℃に達すると停止させた。実施例15は、70℃に設定された入口温度で行なったが、試料採取は、生成物温度が35℃、40℃、45℃、および48℃に達した際に実行した。生成物温度を対照として使用する代わりに、実施例16および17は、乾燥時間を対照として使用した。実施例16では、70℃の入口温度で、生成物温度が40℃に到達した際に、分析用の第1の試料を回収し、乾燥を同一の入口温度で継続し、追加の試料を、乾燥の追加の10分および30分後にそれぞれ回収した。実施例17では、70℃の入口温度で、生成物温度が40℃に到達した際に、第1の試料を回収した。この時点において、入口温度は、60℃まで低下し、追加の試料を、それぞれ20分および60分の追加の乾燥時間の後に回収した。実施例18は、80℃の入口温度を用い、実施例19は、60℃の入口温度を使用した。表10は、これらの動作の要約を示す。
【表9】
DSC相転移および含水量
【0049】
表11は、変動する流動層乾燥条件下で回収された顆粒の水分残量によって決定された、示差走査熱量計データおよび含水量を提供する。本データによって、13.0%以上の含水量を有するほぼ全ての顆粒が、>170ジュール/グラムの転移熱を有することが示される。また、過剰乾燥によって、相転移熱が低くなり得ることも明らかである。それにもかかわらず、約100℃から約102℃の範囲でピークに達し、かつ固体から固体への相転移に対応するDSCピークのサイズがグラム当たり少なくとも約150ジュールであるDSC熱処理曲線を提供する過剰に乾燥された試料は、固体剤形の形成に利用する場合に満足できる結果を提示する。
【表10】
粒径分布
【0050】
顆粒の粒径分布により、全ての顆粒が狭い粒径分布を有することが示されるが、イブプロフェン生成物ナトリウム流(PSS)から結晶化された複合イブプロフェンナトリウム2水和物から調製される実施例20を例外とする。実施例11〜19に使用したイブプロフェンナトリウム2水和物ロットから生成された顆粒の平均中間粒径は、172ミクロンであり、標準偏差は、12ミクロンであった。実施例20で使用した複合イブプロフェンナトリウム二水和の中間粒径は、324ミクロンであり、その調製された顆粒の中間粒径は、232ミクロンであった。
【0051】
実施例11から19からの顆粒の中間粒径および粒径分布の類似性により、本過程が粒径分布に関して極めてロバストであることが示される。実施例11〜19と実施例20との中間粒径および粒径分布の違いにより、顆粒の粒径が、出発イブプロフェンナトリウム2水和物の粒径の関数であることが提案される。
変動する篩サイズ分画における篩分析およびアッセイ含有量
【0052】
実施例12および18からの20グラムの顆粒を、20、30、40、60、80、100、および200メッシュのステンレス鋼の篩から成る直径が12インチの篩の束上に置いた。20分間振動させた後、各篩上に保持された顆粒の重量を量り、選択されたサイズ分画の保持顆粒を、HPLCアッセイ測定のために提示した。理論的にwt%で表わされるアッセイ含有量について、以下の表12に提示した。本データにより、約90wt%の粒子が、40〜200のメッシュ範囲にあり、概して、粒径が大きくなればなるほど、より高いアッセイ含有量に対応することが示される。また、大部分の粒子(40〜200のメッシュ範囲)に関するアッセイ含有量における違いは、ごくわずかであることも示される。
【表11】
【0053】
本発明に関連して生成した顆粒を利用する利点を実証するために、実施例11、12、13、18、19、および20で生成された顆粒を、他の賦形剤と個々に混合して製剤、この場合、錠剤の形成に使用する製剤DTHを生成した。次いで、個々の製剤DTHブレンドを、10ステーションロータリープレス(Minipress II;GlobePharma,Inc.,New Brunswick,NJ)内に順番に供給して、約600mgのイブプロフェンに相当するナトリウム塩を含有する錠剤を調製した。主な圧縮を約10キロニュートン(KN)に維持し、事前圧縮を約1.5KNに維持し、生成速度を、1分あたり100〜120個の錠剤に維持した。錠剤試料を工程実行中に回収した。約30個の錠剤を有する錠剤試料の少なくとも1つの組をブレンド毎に回収した。全12個の錠剤試料の組を、錠剤調製過程によって処理された7つの製剤DTHブレンドから回収した。
【0054】
これらの錠剤の溶解は、10分における59%の平均理論溶解、20分における98%の溶解、13.9分における80%溶解を示す。被験製剤DTH錠剤の溶解速度は、規制の溶解速度よりも大幅に良好であった。
【0055】
回収された全12組の錠剤について、100液滴の平均破砕性は、0.14wt%であった。
【0056】
次に、図面を参照すると、図1〜17は、実施例11から20で生成され、かつ表11に関連して論じられた本発明の顆粒に関する示差走査熱量測定曲線であり、図面の簡単な説明において要約されている。その表に関連して述べたように、図8、図11、および図14の試料は、使用可能であると考えられたが、乾燥ステップ中に過剰乾燥されたために好ましくはない。
【0057】
本発明の好適な一種類の顆粒の組成と、本発明の錠剤を生成するための一種類の1つの好適な製剤の組成とについて表13に説明する。本発明のこのような好適な顆粒と本好適な錠剤製剤とに関するバッチサイズおよび組成について表14に説明する。
【表12】
【表13】
【0058】
40kgの乾燥バッチサイズスケールにおける本発明の一種類の好適な顆粒の調製のための典型的な手順は、以下の通りである:
1. 初期調製
a. 高せん断ブレンダを使用して水中でポリビニルピロリドン(PVP K−90
)を溶解する(13.33kgの精製水中に3kg)ことによって、いくつか
の工程実行に十分なPVP原液(18.37wt%)を調製する。
b. 手動で20メッシュスクリーンに通すか、または20メッシュスクリーンを装
備する粉砕機を使用するかのいずれかで、炭酸ナトリウムを篩過する。
2. 造粒
a. 38.20kgのイブプロフェンナトリウム2水和物の重量を量り、高せん断
造粒器内に装薬する。
b. 5.44kgのPVP溶液の重量を量り、高せん断下で動作中の造粒器に装薬
する。4分後、低せん断動作に切り替え、さらに4分間動作を継続する。
c. 篩過した無水炭酸ナトリウム(0.80kg)の重量を量り、高せん断下にあ
る造粒器に4分間添加し、その後、さらに4分間低せん断下にある造粒器に添
加する。
d. 顆粒を空気圧で流動層乾燥機に移行し、顆粒の静かな流動化を維持し、60℃
に設定された入口温度で乾燥を開始する。
3. 乾燥手順
a. 60℃に入口温度を設定し、ボウル温度および出口温度、試料乾燥機を定期的
に監視し、生成物の温度が約40℃に到達する際に乾燥を停止する。
b. 標的としての13.4wt%の含水量を含んで回収された試料から、含水量(
カールフィッシャーまたは110℃における水分残量)を測定する。
4. 粒径調整
a. 乾燥顆粒を16メッシュ篩に通過させて、>16メッシュの粒子を除去する。
>16メッシュ材料の重量を記録する。
b. >16メッシュ顆粒をサイズ低減器に通して粉砕し、<16メッシュ部分に添
加する。
c. 品質管理のために数百グラム保持し、残りの<16メッシュ生成物の重量を量
る。
d. 錠剤の調製に使用しない場合、ポリエチレン裏当てを含むファイバードラムに
顆粒を保管する。
5. 分析
a. 16、20、40、60、80、100、および200メッシュスクリーンを
使用して篩過することによって粒子分布を行う。
b. 含水量を測定する(カールフィッシャーまたは110℃における水分残量)。
【0059】
本発明の一種類の好適な製剤は、上述のように作製される好適な顆粒から形成され、以下の表に示す組成を有する。
【表14】
また、上記製剤は、10ステーションロータリープレス(Minipress II;GlobePharma,Inc.,New Brunswick,NJ)を使用して高品質錠剤を生成し、約400mgのイブプロフェンに相当するナトリウム塩を含有する錠剤を調製した。この製剤から作製される錠剤の溶解は、20分で80%を超える平均理論溶解を示した。これは、60分で80%の規制要件よりも大幅に優れている。これらの錠剤について、100液滴の平均破砕性は、0.23wt%であった。
【0060】
本発明の好適な錠剤製剤(製剤75)からの、本発明の一種類の好適な錠剤の調製のための典型的な手順は、以下の通りである:
1. 混合に利用可能な上述のように生成された顆粒の量を適切な変換係数と乗算するこ
とによって、3つの賦形剤(微結晶性セルロース、クロスカルメロースナトリウム、
およびコロイド状シリカ)の必要な量を決定する。例えば、製剤DTHの調製のため
に40kgの顆粒を混合する場合、微結晶性セルロース(MCC)の必要量は、8.
34kg(40kgx0.2084、後者の数字は、MCCの変換係数)であり、ク
ロスカルメロースナトリウムの必要量は、2.56kg(40kgx0.0637、
後者の数字は、クロスカルメロースナトリウムの変換係数)であり、コロイド状シリ
カの必要量は、0.051kg(40kgx0.00127、後者の数字は、コロイ
ド状シリカの変換係数)である。
2. MCCおよびクロスカルメロースナトリウムを別々に16メッシュスクリーンに通
して篩過し、これらの2つの別々の賦形剤のそれぞれの必要とされる量を取っておく
。約500グラムのMCCをプラスチック袋に取っておく(例えば、2〜5リットル
サイズの袋を使用する)。
3. コロイド状シリカの重量を量って、MCCを含有する袋に添加し、その袋の含有物
を混合し、任意の塊を手で破壊し、その混合物を、20メッシュスクリーンを通して
スクリーンにかける。
4. 顆粒、MCC、クロスカルメロースナトリウム、および事前ブレンドを、ツインシ
ェルブレンダ内に装薬し、低せん断下で10分間混合する。
5. ポリエチレン裏当てを含むファイバードラム内にこのブレンドを排出し、500グ
ラムの試料を保持する。
6. 分析
a. 16、20、40、60、80、100、および200メッシュスクリー
ンから成る束の篩を使用して篩過することによる粒子分布。
b. 含水量(カールフィッシャーまたは110℃における水分残量)。
c. 流動特徴(Flodexおよび流動性指数)。
7. 従来の条件下で動作するロータリープレスを使用して、製剤DTHから錠剤を調製
する。
【0061】
以下は、固体剤形の調製に適切な本発明の我々の例証的製剤である。
製剤AA)は、
● 粉末形状の成分から形成される顆粒組成物を含む成分であって、(i)乾燥ベースで
80重量部から98重量部のイブプロフェンナトリウム2水和物と、(ii)乾燥ベ
ースで1重量部から4重量部の無水炭酸ナトリウムと、(iii)乾燥ベースで1重
量部から15重量部の非架橋ポリビニルピロリドンとを含む成分から形成される約4
0wt%から約100wt%の製剤;
● 0wt%から約25wt%の微結晶性セルロース、リン酸水素カルシウム、またはそ
の両方;
● 0wt%から約8wt%のクロスポビドンまたはクロスカルメロースナトリウムと;● 0wt%から約0.5wt%のコロイド状シリカ;
● 0wt%から約10wt%のでんぷん;および
● 0wt%から約2wt%のステアリン酸、ステアリン酸マグネシウム、またはその両
方;
を含む、製剤である。
【0062】
製剤BB)は、顆粒組成物の量が、約70wt%から約100wt%であり、微結晶性セルロースの量が0wt%から約20wt%であり、クロスポビドンまたはクロスカルメロースナトリウムの量が0wt%から約8wt%であり、コロイド状シリカの量が約0.05wt%から約0.2wt%である製剤である。
【0063】
製剤CC)は、顆粒組成物の量が、約75wt%から約100wt%の範囲であり、微結晶性セルロースの量が、0wt%から約20wt%であり、クロスポビドンまたはクロスカルメロースナトリウムの量が、0wt%から約6wt%であり、コロイド状シリカの量が、0wt%から約0.2wt%である製剤である。
【0064】
製剤DD)は、顆粒組成物の量が約60wt%から約90wt%の範囲であり、微結晶性セルロース、リン酸水素カルシウム2水和物、またはその両方の量が、約10wt%から約30wt%であり、でんぷんの量が、0wt%から約6wt%であり、クロスポビドンまたはクロスカルメロースナトリウムの量が、0wt%から約6wt%であり、コロイド状シリカの量が、0wt%から約0.25wt%であり、ステアリン酸、ステアリン酸マグネシウム、またなその両方の量が、0wt%から約2wt%である製剤である。
【0065】
製剤EE)は、顆粒組成物の量が約85wt%から約100wt%の範囲であり、微結
晶性セルロース、リン酸水素カルシウム2水和物、またはその両方の量が、0wt%から約10wt%であり、でんぷんの量が、0wt%から約6wt%であり、クロスポビドンまたはクロスカルメロースナトリウムの量が、0wt%から約5wt%であり、コロイド状シリカの量が、0wt%から約0.25wt%であり、ステアリン酸、ステアリン酸マグネシウム、またなその両方の量が、0wt%から約2wt%である製剤である。
【0066】
また、本発明は、AA)、BB)、CC)、DD)、またはEE)のうちのいずれかの製剤から形成される固体剤形も提供する。また、本発明によって、粉末形状の成分であって、(i)乾燥ベースで80重量部から98重量部のイブプロフェンナトリウム2水和物と、(ii)乾燥ベースで1重量部から4重量部の無水炭酸ナトリウムと、(iii)乾燥ベースで1重量部から15重量部の非架橋ポリビニルピロリドンとを含む成分から形成される顆粒組成物を含有するハードシェルカプセルを含む剤形も提供される。加えて、本発明は、イブプロフェンナトリウム2水和物の固体剤形を調製する方法を提供し、本方法は、直前の文に説明する顆粒組成物をロータリープレスにおいて圧縮することを含む。
【0067】
本明細書および本明細書の請求項の任意の場所において化学名または化学式で言及される成分は、単数または複数で言及されているか否かにかかわらず、化学名または化学式で言及される別の物質(例えば別の成分、溶媒等)と接触する前に存在するものとして識別される。得られた混合物または溶液の中で、どんな化学変化、変形、および/または反応が発生することは、もしそれらが発生しなくても、このような変化、変形、および/または反応が、本開示に従って要求された条件下において特定の成分を一つにまとめた当然の結果であるので、問題ではない。したがって、これらの成分は、所望の動作の実行または所望の組成物の形成に関連して一つにまとめられる要素として識別される。
また、以下の請求項は、現在時制(「備える」、「である」等)で物質、成分、および/または要素に言及し得るが、物質、成分、または要素は、本開示に従って1つもしくは複数の他の物質、成分、および/または要素と最初に接触、混合、または混合される直前の時点に存在したものとして言及されている。
【0068】
他に明示している場合を除き、本明細書において使用する場合、および使用する際、冠詞「ある」(「a」または「an」)は、その冠詞が言及する単一の要素に請求を限定するように意図されず、また、限定するものとして解釈されるべきではない。むしろ、本明細書において使用する場合、および使用する際、本文が他に明示している場合を除き、冠詞「ある」(「a」または「an」)は1つもしくは複数のこのような要素を包むように意図される。
【0069】
本発明は、本明細書に記載の材料および/または手順を備え得るか、それらから成り得るか、またはそれらから本質的に成り得る。
【0070】
本発明は、その実践においてかなりの変動を受け易い。ゆえに、前述の説明は、上記に提示された特定の例示に本発明を限定するように意図されず、また、限定するものとして解釈されるべきではない。
【特許請求の範囲】
【請求項1】
イブプロフェンナトリウム2水和物の高度に分散可能な自由流動顆粒を調製する過程であって、その方法が、
● (i)乾燥ベースで少なくとも80重量部のイブプロフェンナトリウム2水和物と、
(ii)乾燥ベースで1重量部から4重量部の炭酸ナトリウムと、(iii)乾燥ベ
ースで1重量部から15重量部の非架橋ポリビニルピロリドンと、(iv)湿潤混合
物を形成するための、(i)、(ii)、(iii)、および(iv)の全重量に基
づく8重量部から12重量部の水とから構成される成分を、高せん断造粒器内に一つ
にまとめることと、
● 湿潤顆粒を形成するために、前記高せん断造粒器において前記湿潤混合物を造粒する
ことと、
● 110℃における重量損失の測定により決定可能である約12.5wt%から15w
t%の範囲の含水量を有する乾燥顆粒を形成するように、湿潤顆粒を乾燥することと
、
● 16メッシュを超える粒径を有する乾燥顆粒を篩過することによって除去することと
、
を含む、過程。
【請求項2】
前記高せん断造粒器における前記湿潤混合物の前記造粒が、前記湿潤混合物の乾燥後に、前記乾燥顆粒が示差走査熱量測定中に約100℃から約102℃の範囲でピークに達する相転移を呈するように、ある時間の間行われ、固体から固体への相転移に対応するDSCピークのサイズが、グラム当たり少なくとも約150ジュールである、請求項1に記載の過程。
【請求項3】
前記成分(iii)および(iv)を事前に混合して溶液を形成し、前記溶液と(i)および(ii)とを前記高せん断造粒器内に一つにまとめ、任意選択により、前記溶液を、成分(i)を含有する前記高せん断造粒器内に導入し、得られた混合物を前記造粒器において混合した後に、成分(ii)を前記造粒器内に導入し、次いで、得られた混合物を造粒する、請求項2に記載の過程。
【請求項4】
前記造粒が、造粒中に生成された過剰な熱を除去するために、必要に応じて冷却を使用して、約15℃から約35℃の範囲の温度で行われる、請求項1〜3のいずれかに記載の過程。
【請求項5】
前記湿潤顆粒が、110℃における重量損失の測定により決定可能である約12wt%から約15wt%の範囲の含水量まで乾燥される、請求項1〜2のいずれかに記載の過程。
【請求項6】
前記湿潤造粒の完了時に、前記顆粒が、空気圧で流動層乾燥機に移行され、その中で迅速に乾燥される、請求項1〜3のいずれかに記載の過程。
【請求項7】
前記乾燥が、60℃から約70℃の範囲の入口空気温度で動作する流動層乾燥機において行われ、前記乾燥機の動作が、出口空気の温度が約40℃に到達する際に停止される、請求項1〜3のいずれかに記載の過程。
【請求項8】
前記造粒が、造粒中に生成された過剰な熱を除去するために、必要に応じて冷却を使用して、約15℃から約35℃の範囲の温度で行われ、前記湿潤造粒の完了時に、前記顆粒が、空気圧で流動層乾燥機に移行され、60℃から約70℃の範囲の入口空気温度を有する前記乾燥機を動作することによって、その中で迅速に乾燥され、前記乾燥機の動作が、
出口空気の温度が約40℃に到達する際に停止される、請求項1〜3のいずれかに記載の過程。
【請求項9】
高含量のイブプロフェンナトリウム2水和物を有する高度に分散可能な自由流動顆粒組成物であって、粉末形状の成分であって、(i)乾燥ベースで80重量部から98重量部のイブプロフェンナトリウム2水和物と、(ii)乾燥ベースで1重量部から4重量部の無水炭酸ナトリウムと、(iii)乾燥ベースで1重量部から15重量部の非架橋ポリビニルピロリドンとを含む成分から形成される、組成物。
【請求項10】
前記組成物が、(i)乾燥ベースで約95重量部のイブプロフェンナトリウム2水和物と、(ii)乾燥ベースで約2重量部から3重量部の無水炭酸ナトリウムと、(iii)乾燥ベースで約2重量部から3重量部の非架橋ポリビニルピロリドンとを含む成分から形成される、請求項9に記載の顆粒組成物。
【請求項11】
前記乾燥顆粒が、示差走査熱量測定中に約100℃から約102℃の範囲でピークに達する相転移を呈し、固体から固体への相転移に対応するDSCピークのサイズは、グラム当たり少なくとも約150ジュールである、請求項9または10のいずれかに記載の顆粒組成物。
【請求項12】
前記顆粒は、110℃における重量損失の測定により決定可能である約11wt%から約15wt%の範囲の含水量を有する、請求項9〜11のいずれかに記載の顆粒組成物。
【請求項13】
高含量のイブプロフェンナトリウム2水和物を有する湿潤顆粒組成物であって、粉末形状の成分であって、(i)乾燥ベースで少なくとも80wt%の量のイブプロフェンナトリウム2水和物と、(ii)乾燥ベースで1wt%から4wt%の範囲の量の無水炭酸ナトリウムと、(iii)乾燥ベースで1wt%から15wt%の量の非架橋ポリビニルピロリドンと、(iv)(i)、(ii)、(iii)、および(iv)の全重量に基づく8wt%から12wt%の範囲の量の水とを含む成分から形成される、組成物。
【請求項14】
前記組成物が、(i)乾燥ベースで約95wt%の量のイブプロフェンナトリウム2水和物と、(ii)乾燥ベースで2wt%から3wt%の量の無水炭酸ナトリウムと、(iii)乾燥ベースで3wt%から2wt%の量の非架橋ポリビニルピロリドンと、(iv)(i)、(ii)、(iii)、および(iv)の全重量に基づく約8wt%から12wt%の範囲の量の水とを含む成分から形成される、請求項13に記載の湿潤顆粒組成物。
【請求項15】
ロータリープレスを使用する固体剤形の調製に適合されるイブプロフェンナトリウム2水和物製剤であって、
● 約40wt%から約100wt%の請求項9〜11のいずれかに記載の顆粒組成物、● 0wt%から約25wt%の微結晶性セルロース、リン酸水素カルシウム、またはそ
の両方、
● 0wt%から約8wt%のクロスポビドンまたはクロスカルメロースナトリウム、
● 0wt%から約0.5wt%のコロイド状シリカ、
● 0wt%から約10wt%のでんぷん、および
● 0wt%から約2wt%のステアリン酸、ステアリン酸マグネシウム、またはその両
方、
を含む成分から形成される、製剤。
【請求項16】
前記顆粒組成物の量が、約70wt%から約100wt%の範囲にあり、微結晶性セルロースの量が、0wt%から約20wt%であり、クロスポビドンまたはクロスカルメロ
ースナトリウムの量が、0wt%から約8wt%であり、コロイド状シリカの量は、0wt%から約0.2wt%である、請求項15に記載の製剤。
【請求項17】
前記顆粒組成物の量が、約90wt%から約100wt%の範囲にあり、微結晶性セルロースの量が、0wt%から約10wt%であり、クロスポビドンまたはクロスカルメロースナトリウムの量が、0wt%から約6wt%であり、コロイド状シリカの量が、0wt%から約0.2wt%である、請求項15に記載の製剤。
【請求項18】
前記顆粒組成物の量が、約60wt%から約90wt%の範囲にあり、微結晶性セルロース、リン酸水素カルシウム2水和物、またはその両方の量が、約10wt%から約30wt%であり、クロスポビドンまたはクロスカルメロースナトリウムの量が、0wt%から約6wt%であり、でんぷんの量は、0wt%から約6wt%であり、ステアリン酸、ステアリン酸マグネシウム、またはその両方の量が、0wt%から約2wt%であり、コロイド状シリカの量が、0wt%から約0.25wt%である、請求項15に記載の製剤。
【請求項19】
前記顆粒組成物の量が、約85wt%から約100wt%の範囲にあり、微結晶性セルロース、リン酸水素カルシウム2水和物、またはその両方の量が、約0wt%から約10wt%であり、でんぷんの量が、0wt%から約6wt%であり、クロスポビドンまたはクロスカルメロースナトリウムの量が、0wt%から約5wt%であり、コロイド状シリカの量が、0wt%から約0.25wt%であり、ステアリン酸、ステアリン酸マグネシウム、またはその両方の量が、0wt%から約2wt%である、請求項15に記載の製剤。
【請求項20】
請求項15〜19のいずれかに記載の製剤から形成される固体剤形。
【請求項21】
請求項9〜11のいずれかに記載の顆粒組成物を含有するハードシェルカプセルを備える剤形。
【請求項22】
イブプロフェンナトリウム2水和物の固体剤形を調製する方法であって、請求項9〜11のいずれかに記載の顆粒組成物をロータリープレスにおいて圧縮することを含む、方法。
【請求項1】
イブプロフェンナトリウム2水和物の高度に分散可能な自由流動顆粒を調製する過程であって、その方法が、
● (i)乾燥ベースで少なくとも80重量部のイブプロフェンナトリウム2水和物と、
(ii)乾燥ベースで1重量部から4重量部の炭酸ナトリウムと、(iii)乾燥ベ
ースで1重量部から15重量部の非架橋ポリビニルピロリドンと、(iv)湿潤混合
物を形成するための、(i)、(ii)、(iii)、および(iv)の全重量に基
づく8重量部から12重量部の水とから構成される成分を、高せん断造粒器内に一つ
にまとめることと、
● 湿潤顆粒を形成するために、前記高せん断造粒器において前記湿潤混合物を造粒する
ことと、
● 110℃における重量損失の測定により決定可能である約12.5wt%から15w
t%の範囲の含水量を有する乾燥顆粒を形成するように、湿潤顆粒を乾燥することと
、
● 16メッシュを超える粒径を有する乾燥顆粒を篩過することによって除去することと
、
を含む、過程。
【請求項2】
前記高せん断造粒器における前記湿潤混合物の前記造粒が、前記湿潤混合物の乾燥後に、前記乾燥顆粒が示差走査熱量測定中に約100℃から約102℃の範囲でピークに達する相転移を呈するように、ある時間の間行われ、固体から固体への相転移に対応するDSCピークのサイズが、グラム当たり少なくとも約150ジュールである、請求項1に記載の過程。
【請求項3】
前記成分(iii)および(iv)を事前に混合して溶液を形成し、前記溶液と(i)および(ii)とを前記高せん断造粒器内に一つにまとめ、任意選択により、前記溶液を、成分(i)を含有する前記高せん断造粒器内に導入し、得られた混合物を前記造粒器において混合した後に、成分(ii)を前記造粒器内に導入し、次いで、得られた混合物を造粒する、請求項2に記載の過程。
【請求項4】
前記造粒が、造粒中に生成された過剰な熱を除去するために、必要に応じて冷却を使用して、約15℃から約35℃の範囲の温度で行われる、請求項1〜3のいずれかに記載の過程。
【請求項5】
前記湿潤顆粒が、110℃における重量損失の測定により決定可能である約12wt%から約15wt%の範囲の含水量まで乾燥される、請求項1〜2のいずれかに記載の過程。
【請求項6】
前記湿潤造粒の完了時に、前記顆粒が、空気圧で流動層乾燥機に移行され、その中で迅速に乾燥される、請求項1〜3のいずれかに記載の過程。
【請求項7】
前記乾燥が、60℃から約70℃の範囲の入口空気温度で動作する流動層乾燥機において行われ、前記乾燥機の動作が、出口空気の温度が約40℃に到達する際に停止される、請求項1〜3のいずれかに記載の過程。
【請求項8】
前記造粒が、造粒中に生成された過剰な熱を除去するために、必要に応じて冷却を使用して、約15℃から約35℃の範囲の温度で行われ、前記湿潤造粒の完了時に、前記顆粒が、空気圧で流動層乾燥機に移行され、60℃から約70℃の範囲の入口空気温度を有する前記乾燥機を動作することによって、その中で迅速に乾燥され、前記乾燥機の動作が、
出口空気の温度が約40℃に到達する際に停止される、請求項1〜3のいずれかに記載の過程。
【請求項9】
高含量のイブプロフェンナトリウム2水和物を有する高度に分散可能な自由流動顆粒組成物であって、粉末形状の成分であって、(i)乾燥ベースで80重量部から98重量部のイブプロフェンナトリウム2水和物と、(ii)乾燥ベースで1重量部から4重量部の無水炭酸ナトリウムと、(iii)乾燥ベースで1重量部から15重量部の非架橋ポリビニルピロリドンとを含む成分から形成される、組成物。
【請求項10】
前記組成物が、(i)乾燥ベースで約95重量部のイブプロフェンナトリウム2水和物と、(ii)乾燥ベースで約2重量部から3重量部の無水炭酸ナトリウムと、(iii)乾燥ベースで約2重量部から3重量部の非架橋ポリビニルピロリドンとを含む成分から形成される、請求項9に記載の顆粒組成物。
【請求項11】
前記乾燥顆粒が、示差走査熱量測定中に約100℃から約102℃の範囲でピークに達する相転移を呈し、固体から固体への相転移に対応するDSCピークのサイズは、グラム当たり少なくとも約150ジュールである、請求項9または10のいずれかに記載の顆粒組成物。
【請求項12】
前記顆粒は、110℃における重量損失の測定により決定可能である約11wt%から約15wt%の範囲の含水量を有する、請求項9〜11のいずれかに記載の顆粒組成物。
【請求項13】
高含量のイブプロフェンナトリウム2水和物を有する湿潤顆粒組成物であって、粉末形状の成分であって、(i)乾燥ベースで少なくとも80wt%の量のイブプロフェンナトリウム2水和物と、(ii)乾燥ベースで1wt%から4wt%の範囲の量の無水炭酸ナトリウムと、(iii)乾燥ベースで1wt%から15wt%の量の非架橋ポリビニルピロリドンと、(iv)(i)、(ii)、(iii)、および(iv)の全重量に基づく8wt%から12wt%の範囲の量の水とを含む成分から形成される、組成物。
【請求項14】
前記組成物が、(i)乾燥ベースで約95wt%の量のイブプロフェンナトリウム2水和物と、(ii)乾燥ベースで2wt%から3wt%の量の無水炭酸ナトリウムと、(iii)乾燥ベースで3wt%から2wt%の量の非架橋ポリビニルピロリドンと、(iv)(i)、(ii)、(iii)、および(iv)の全重量に基づく約8wt%から12wt%の範囲の量の水とを含む成分から形成される、請求項13に記載の湿潤顆粒組成物。
【請求項15】
ロータリープレスを使用する固体剤形の調製に適合されるイブプロフェンナトリウム2水和物製剤であって、
● 約40wt%から約100wt%の請求項9〜11のいずれかに記載の顆粒組成物、● 0wt%から約25wt%の微結晶性セルロース、リン酸水素カルシウム、またはそ
の両方、
● 0wt%から約8wt%のクロスポビドンまたはクロスカルメロースナトリウム、
● 0wt%から約0.5wt%のコロイド状シリカ、
● 0wt%から約10wt%のでんぷん、および
● 0wt%から約2wt%のステアリン酸、ステアリン酸マグネシウム、またはその両
方、
を含む成分から形成される、製剤。
【請求項16】
前記顆粒組成物の量が、約70wt%から約100wt%の範囲にあり、微結晶性セルロースの量が、0wt%から約20wt%であり、クロスポビドンまたはクロスカルメロ
ースナトリウムの量が、0wt%から約8wt%であり、コロイド状シリカの量は、0wt%から約0.2wt%である、請求項15に記載の製剤。
【請求項17】
前記顆粒組成物の量が、約90wt%から約100wt%の範囲にあり、微結晶性セルロースの量が、0wt%から約10wt%であり、クロスポビドンまたはクロスカルメロースナトリウムの量が、0wt%から約6wt%であり、コロイド状シリカの量が、0wt%から約0.2wt%である、請求項15に記載の製剤。
【請求項18】
前記顆粒組成物の量が、約60wt%から約90wt%の範囲にあり、微結晶性セルロース、リン酸水素カルシウム2水和物、またはその両方の量が、約10wt%から約30wt%であり、クロスポビドンまたはクロスカルメロースナトリウムの量が、0wt%から約6wt%であり、でんぷんの量は、0wt%から約6wt%であり、ステアリン酸、ステアリン酸マグネシウム、またはその両方の量が、0wt%から約2wt%であり、コロイド状シリカの量が、0wt%から約0.25wt%である、請求項15に記載の製剤。
【請求項19】
前記顆粒組成物の量が、約85wt%から約100wt%の範囲にあり、微結晶性セルロース、リン酸水素カルシウム2水和物、またはその両方の量が、約0wt%から約10wt%であり、でんぷんの量が、0wt%から約6wt%であり、クロスポビドンまたはクロスカルメロースナトリウムの量が、0wt%から約5wt%であり、コロイド状シリカの量が、0wt%から約0.25wt%であり、ステアリン酸、ステアリン酸マグネシウム、またはその両方の量が、0wt%から約2wt%である、請求項15に記載の製剤。
【請求項20】
請求項15〜19のいずれかに記載の製剤から形成される固体剤形。
【請求項21】
請求項9〜11のいずれかに記載の顆粒組成物を含有するハードシェルカプセルを備える剤形。
【請求項22】
イブプロフェンナトリウム2水和物の固体剤形を調製する方法であって、請求項9〜11のいずれかに記載の顆粒組成物をロータリープレスにおいて圧縮することを含む、方法。
【図1】

【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【図11】
【図12】
【図13】
【図14】
【図15】
【図16】
【図17】
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【図11】
【図12】
【図13】
【図14】
【図15】
【図16】
【図17】
【公表番号】特表2011−528712(P2011−528712A)
【公表日】平成23年11月24日(2011.11.24)
【国際特許分類】
【出願番号】特願2011−520090(P2011−520090)
【出願日】平成21年7月13日(2009.7.13)
【国際出願番号】PCT/US2009/050405
【国際公開番号】WO2010/011522
【国際公開日】平成22年1月28日(2010.1.28)
【出願人】(594066006)アルベマール・コーポレーシヨン (155)
【Fターム(参考)】
【公表日】平成23年11月24日(2011.11.24)
【国際特許分類】
【出願日】平成21年7月13日(2009.7.13)
【国際出願番号】PCT/US2009/050405
【国際公開番号】WO2010/011522
【国際公開日】平成22年1月28日(2010.1.28)
【出願人】(594066006)アルベマール・コーポレーシヨン (155)
【Fターム(参考)】
[ Back to top ]