
高度に可溶性の薬物のための徐放性マトリックス系
【課題】薬物の持続放出が得られる経口用徐放性固体剤形を提供する。
【解決手段】可溶性が約10g/lより大きい治療上有効な量の薬物と、pH改変剤と、ならびにヘテロ多糖ガムと周囲の液体に晒された際に該ヘテロ多糖ガムと架橋することができるホモ多糖ガムとを含有するゲル化剤を含む徐放性マトリックスとを含有し、ヒト患者への経口投与後に上記薬物の持続放出が得られる経口用徐放性固体剤形。
【解決手段】可溶性が約10g/lより大きい治療上有効な量の薬物と、pH改変剤と、ならびにヘテロ多糖ガムと周囲の液体に晒された際に該ヘテロ多糖ガムと架橋することができるホモ多糖ガムとを含有するゲル化剤を含む徐放性マトリックスとを含有し、ヒト患者への経口投与後に上記薬物の持続放出が得られる経口用徐放性固体剤形。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、経口用徐放性固体剤形に関する。
【背景技術】
【0002】
発明の背景
制御放出製品の利点は医薬分野ではよく知られ、この利点には、比較的長い期間にわたって薬物の所望血中レベルを維持し、同様の効果を達成するために必要な投与回数を減らすことによって患者のコンプライアンスを高める能力が含まれる。これらの利点は多様な方法によって達成されてきた。例えば、制御放出薬における使用に関して種々のヒドロゲルが記載されており、この中には合成物もあるが、大部分は半合成または天然起源のものである。合成物質および非合成物質の両方を含むものはわずかしかない。しかしながら、この系の中には、特殊な工程および製造装置を必要とするものもあり、さらにこれらの系には可変性の薬物放出に影響を受けやすいものもある。
【0003】
経口制御放出送達系は、放出速度および放出プロフィールが生理学および時間治療学の必要要件と一致しうるように理想的に適合させる必要がある。
【0004】
ほとんどの場合、経口送達系の放出速度は、放出の機構、例えば0次、1次、2次、擬1次機構などに従って分類されているが、多くの医薬化合物が他の複雑な機構により薬物を放出する。
【0005】
1次機構は、反応速度が反応基質濃度に依存する(従って、反応物の1次作用率(first power)に依存する)状況を指す。かかる機構においては、基質は直接に1つ以上の生成物に分解する。
【0006】
2次機構は、実験的に決定した反応速度が2つの反応物のそれぞれの濃度と比例関係にあるか、または1つの反応物の濃度の2次作用率と比例関係にある場合に起こる。
【0007】
擬1次反応は、一般的には、1次機構が支配するように挙動する2次反応として定義され、例えば、他の物質と比較して1つの反応物質の量が過剰に存在するかまたは一定濃度に維持されるように操作される場合に、この擬1次反応は起こる。このような状況においては、反応速度は操作される物質により決定される。
【0008】
0次機構は、反応速度が反応物質濃度に依存しない(従って、反応物の0次作用率(zero power)に依存する)状況を指し、律速因子は反応物質(例えば薬物)濃度以外のものである。0次機構の律速因子は、例えば、反応物質の可溶性または光化学反応の光強度でありうる。
【0009】
しかし上述のように、多くの化学反応は、0次、1次または2次反応などの単純な反応ではなく、2つ以上の反応の組み合わせを包含する。
【0010】
さらに、他の因子、例えば、温度、pH、食物影響変動性(food effect variability)、イオンおよびイオン強度依存性、粘度依存性、コロージョン/エロージョン(侵食)変動性、内容物均一性(content uniformity)問題、流動性および重量均一性問題、維持容量(carrying capacity)および物理的強度問題、加水分解、光化学分解、成分間相互作用(薬物と、製剤中の他の成分、例えば緩衝剤、保存剤などとの相互作用など)、低誘電率の溶媒の濃度(反応が反対の電荷のイオンを含む場合)などが反応速度に影響を及ぼす可能性がある。
【0011】
多くの制御放出製剤および徐放性製剤が既知であるが、ある程度〜高度に可溶性の薬物に関しては、そのような製剤中に配合する際に製剤上の困難性を有する。可溶性薬物を含有する徐放性製剤は、「用量ダンピング(dose dumping)」の影響を受けやすい。この事象は、有効成分の放出が遅延するが、放出が開始した時にその速度が極めて高い場合に起こる。この放出速度の上昇は、治療効果の低減または毒性の増大をもたらす可能性のある血液の血漿変動に関連している。これらは、徐放性製剤が解決すると考えられる問題と同じである。
【0012】
さらに、特定の徐放性製剤が、可溶性〜高度に可溶性の薬物の所望の持続放出を提供するかどうかを容易に推測できない場合もある。一般的に、摂取された場合にそのような薬物の所望の生物学的利用能を提供する徐放性製剤を得るためにかなりの実験を行う必要があることがわかっている。
【0013】
可溶性〜高度に可溶性の薬物の所望の持続放出を提供する制御放出製剤と関連した予測不可能性を補うために、二相性または多相性反応動力学を有する製剤を提供することが望ましいと考えられる場合もある。二相性放出または多相性放出は、開始速度が高く、続いて剤形が小腸の上部を通過する、吸収が最大となるところでより緩慢な速度であり、最後に剤形が小腸のより一層終末を通過する、吸収が小腸上部の場合よりも少ないところで再び速度が高くなることを特徴としうる。
【0014】
二相性放出はいくつかの理由で利点を有すると考えられ、そのような理由としては、限定するものではないが、二相性放出によって、製造者が(製剤が胃内に位置する場合に)迅速な作用開始によって胃腸管における薬物の吸収速度の変化を補償することができるという事実、ならびに(例えば製剤が大腸に位置する場合に)比較的迅速な放出速度によって比較的緩慢な吸収を補償することができるという事実、が挙げられる。
【0015】
現在のところ、二相性放出製剤は種々のいくつかの方法で提供されている。
【0016】
例えば、国際特許公開番号WO87/00044号には、二相性放出の特徴を有すると主張されている治療用製剤が記載されている。WO87/00044号には、固体剤形中の治療上有効な薬物のための担体基剤物質が記載され、この剤形は、薬物の初期の急速な放出、続いて一定期間にわたる実質的に一定の放出速度、そしてその後の、前に観察された一定速度よりも大きい放出速度を特徴とする二相性制御放出プロフィールを提供すると主張されている。担体ベースの物質は、メトキシ含量が19〜30%、ヒドロキシプロポキシ含量が4〜12%、粘度が40〜19,000cps、平均分子量が20,000〜140,000である二相性ヒドロキシプロピルメチルセルロースエーテルを含有し、これは該公報に記載のアッセイ法によって二相性放出プロフィールを示す。二相性ヒドロキシプロピルメチルセルロースは、有効成分および所望の薬物放出の期間に依存して総配合物の5〜99重量%を含有する。
【0017】
A. C. Shahら(”Gel-Matrix Systems Exhibiting Bimodal Controlled Release For Oral Drug Delivery”, Journal of Controlled Release, 9(1989), pp. 169-175)はさらに、特定の「タイプ」のヒドロキシプロピルメチルセルロースエーテルが二相性薬物放出プロフィールを示すことが見出されたことを報告している。しかしながらこの研究においては、一連のヒドロキシプロピルメチルセルロースエーテルポリマーが、ポリマー−薬物マトリックス錠剤からの二相性および非二相性放出プロフィールを示し、これによりポリマーの供給者(従って、製造方法、イオン構成、置換基の分布の変動、または分子量分布の変動)に左右されると考えられることがわかった。
【0018】
P. Giunchediら(”Ketoprofen Pulsatile Absorption From ‘Multiple Unit’ Hydrophilic Matrices” International Journal of Pharmaceutics, 77(1991), pp. 177-181)は、ケトプロフェンの経口用遅延放出製剤を記載している。該製剤は、それぞれが50mgの薬物を含有する同じ組成物の4種の親水性マトリックスから構成され、ヒドロキシプロピルメチルセルロース(Methocel. RTM.)を用いて調製され、そしてゼラチンカプセル中に入れられた複合ユニット製剤を含む。拍動血漿レベル(投与の2時間後と8時間後に2つのピークがある)が得られるものと主張されたが、in vitro試験ではかなり一定な薬物放出が示された。
【0019】
U. Conteら(”A New Ibuprofen Pulsed Release Oral Dosage Form”, Drug Development And Industrial Pharmacy, 15(14-16), pp 2583-2596 (1989))は、2つの層が投与量の薬物を含み、中間層が薬物層を分離させる制御要素として作用する3層錠剤から律動的(pulsed)放出パターンが得られたことを報告している。この制御要素は、水膨張性ポリマー(ヒドロキシプロピルメチルセルロース)の混合物である。外側被膜として不浸透性ポリマーで錠剤をコーティングしている。超崩壊剤(デンプングリコレートナトリウムおよび架橋したポリビニルピロリドン)を薬物層に含有させている。
【0020】
K. A. Kahnら(”Pharmaceutical Aspects And In-Vivo Performance Of Brufen Retard-An Ibuprofen SR Matrix Tablet”, Proced. Intern. Symp. Control. Rel. Bioact. Mater., 18(1991), Controlled Release Society, Inc.)は、二相性放出パターンを示すと主張されているイブプロフェン800mgを含有する製剤を記載している。ここで使用された放出遅延剤はキサンタンガムである。成分は、適切なキサンタンガム含量と混合され、その後打錠して錠剤にし、被膜コーティングされている。含有するキサンタンガムの量と薬物放出速度との間に逆相関があった。薬物粒子サイズおよび錠剤1つ当たりの被膜コーティングの量の増大は、薬物放出速度に有意に影響を及ぼさなかった。キサンタンガムの粒子サイズの増大がより顕著な突発性(burst)作用を引き起こし、被膜コーティングの適用によってこの突発性作用が解消された。薬物放出の迅速な開始は、ゲル層形成の変化に関連していると仮定されており、この場合、粒子が大きくなるほどゆっくりとゲル形成し、そして凝集マトリックスが形成しうる前に徐々に崩壊する(slough)。
【0021】
本発明者らの米国特許第4,994,276号、第5,128,143号および第5,135,757号(参照により本明細書に引用する)において、本発明者らは、共働作用を示す様々な大きさの多糖(例えば、キサンタンガムなどのヘテロ多糖と、該ヘテロ多糖と架橋することができる多糖ガム、例えばイナゴマメ(locust bean)ガムとの組み合わせ)を含む制御放出賦形剤を報告した。これは、薬物および潤滑剤粉末の添加の後に直圧縮を行うか、慣例的な湿式顆粒化を行うか、またはこれらの2つの組み合わせを行うかのいずれかで経口用固体剤形に加工することができる。この製剤からの薬物の放出は、0次機構または1次機構に従って進行するものである。
【0022】
本発明者らの米国特許第5,472,711号および第5,478,574号(参照により本明細書に引用する)において、本発明者らは、有効量の薬学的に許容される界面活性剤を上述の賦形剤と混合することによって、in vitroにおいて治療上有効な薬物の多相性または二相性制御放出を提供することができる製剤を報告している。
【0023】
本発明で使用する高度に可溶性の薬物の例としては、ジルチアゼム、すなわちカルシウムアンタゴニスト活性を有するベンゾチアジン誘導体である。ジルチアゼムは、高血圧症および扁桃炎の治療に広く使用されている。従って、許容可能な放出プロフィールを示す徐放性ジルチアゼムの製造には大いに関心が寄せられていた。例えば、米国特許第4,894,240号および第5,364,620号(Geogheganら)は、1日1回の投与に適したジルチアゼムペレット製剤を記載している。この製剤は、周りを不溶性多層膜が包囲する、有機酸と会合したジルチアゼムのコアを含む。該膜によって、ペレットから投与後24時間にわたって制御吸収が可能な速度でジルチアゼムが放出される。
【0024】
徐放性ジルチアゼム製剤の製造に関して他の方法が従来技術において記載されている。例えば、米国特許第5,419,917号(Chenら)は、薬学的に有効なイオン性化合物を用いてヒドロゲルからのジルチアゼムの放出速度を制御する組成物を記載している。
【0025】
本発明で使用する高度に可溶性の薬物の他の例は、オキシブチニンである。オキシブチニンは泌尿器疾患(例えば過運動性膀胱(hyperactive bladder))の治療に広く使用されている。本発明者らの米国特許第5,399,359号には、ゲル化剤を含む徐放性マトリックス中に分散させた薬学的に有効な量のオキシブチニン、有効量の薬学的に許容される水溶性カチオン性架橋剤(製剤が周囲の液体、例えば胃腸液に晒された際にゲル化剤と架橋する)、及び不活性希釈剤とを含有するオキシブチニン徐放性製剤が開示されている。
【発明の開示】
【発明が解決しようとする課題】
【0026】
発明の目的
本発明の目的は、可溶性〜高度に可溶性の治療上有効な薬物のための生体利用可能な徐放性製剤を提供することである。
【0027】
本発明のさらなる目的は、可溶性〜高度に可溶性の薬物の多相性または二相性制御放出を示しうる製剤を提供することである。
【0028】
本発明のさらなる目的は、可溶性〜高度に可溶性の薬物の生体利用可能な徐放性製剤の製造方法を提供することである。
【0029】
本発明のさらに他の目的は、可溶性〜高度に可溶性の治療上有効な薬物の経口用徐放性固体剤形の製造に使用しうる徐放性マトリックスを提供することである。
【0030】
本発明のさらなる目的は、薬物と混合すると、治療上有効な薬物血中レベルを例えば12または24時間にわたって提供する徐放性製剤を得るのに適した徐放性マトリックスを提供することである。
【0031】
本発明のさらなる目的は、市販の徐放性製剤、例えばCardizem CDと同様の血漿プロフィールを示す、ジルチアゼムの徐放性マトリックス製剤を提供することである。
【0032】
本発明のさらなる目的は、市販の徐放性製剤、例えばディトロパン(Ditropan) XLと同様の血漿プロフィールを示す、オキシブチニンの徐放性マトリックス製剤を提供することである。
【課題を解決するための手段】
【0033】
発明の概要
上述の目的および他の目的は、部分的には、ゲル剤を含む剤形にpH改変剤を組み込むことによって該剤形からの薬物の放出が促進され、生物学的利用能が高まるという驚くべき発見に関する本発明により達成される。
【0034】
特定の実施形態において、経口用徐放性固体剤形には、可溶性が約10g/lより大きい治療上有効な量の薬物;pH改変剤;およびゲル化剤を含む徐放性マトリックスが含まれる。該ゲル化剤は、ヘテロ多糖ガムと、周囲の液体に晒された際に該ヘテロ多糖ガムを架橋することができるホモ多糖ガムとを含む。好ましくは、該剤形は、少なくとも約12時間、好ましくは少なくとも約24時間にわたって薬物の持続放出を提供する。
【0035】
特定の実施形態において、上記剤形には、a)薬物の多相性放出を提供しうる薬学的に許容される界面活性剤;b)例えば単糖類、二糖類、多価アルコールまたはこれらの混合物より選択される不活性希釈剤;c)ゲル化剤の水和を緩慢にする疎水性物質;および/またはd)該徐放性製剤が周囲の液体に晒された際に形成されるゲルからの放出速度を変更するのに適した有効量の薬学的に許容されるイオン化ゲル強度増強剤、がさらに含まれる。好ましい実施形態において、本発明の製剤は錠剤を包含する。
【0036】
本発明の好ましい実施形態において、薬物とゲル化剤との比は、好ましくは約10:1〜約1:10、より好ましくは約5:1〜約1:5、最も好ましくは約1.25:1〜約2:1である。
【0037】
また本発明は、高度に水溶性の薬物の徐放性製剤の製造方法に関する。該方法には、ヘテロ多糖ガムと周囲の液体に晒された際に該ヘテロ多糖ガムを架橋することができるホモ多糖ガムとを含有するゲル化剤を含み、そして任意にイオン化ゲル強度増強剤、任意に不活性希釈剤、および任意に疎水性物質を含んでいてもよいマトリックスを調製し、その後で可溶性〜高度に可溶性の薬物、pH改変剤、そして任意に薬学的に許容される界面活性剤を添加することが含まれる。その後得られた混合物を、薬物とゲル化剤との比が約10:1〜約1:10、より好ましくは約5:1〜約1:5、最も好ましくは約1.25:1〜約2:1である生成物が得られ、錠剤が周囲の液体に晒された際にゲルマトリックスが形成され、そして各錠剤が治療上有効な量の薬物を含有するように、打錠する。得られる錠剤は、少なくとも約12時間、好ましくは約24時間にわたって治療上有効な薬物血中レベルを提供する。
【0038】
本発明はさらに、上述の経口用固体剤形を経口投与することにより患者を治療する方法に関する。
【0039】
本発明の特定の好ましい実施形態において、上記マトリックスは、例えば約10〜約99重量%のゲル化剤と、約0〜約20重量%のイオン化ゲル強度増強剤と、約1〜約89重量%の不活性医薬希釈剤と、約1〜約20%の疎水性物質とを含有する、予め顆粒化した徐放性賦形剤から調製しうる。
【0040】
他の好ましい実施形態において、マトリックスと不活性希釈剤との混合物は、該マトリックスを破壊することなくマトリックスの水和を緩慢にするのに十分な量の疎水性物質の分散液または溶液を用いて顆粒化し、その後で薬物を添加する。
【0041】
本発明の他の好ましい実施形態において、薬物の第1部を賦形剤の顆粒化の際に導入し、薬物の第2部を外部で顆粒化して(extragranularly)導入するか、または顆粒化工程の後に導入する。このような実施形態は薬物の初期の急速な放出を提供する。
【0042】
好ましい実施形態において、薬物は高度に可溶性である、すなわち、可溶性が約100g/lより大きい。
【0043】
他の好ましい実施形態において、薬物は、カルシウムチャネル遮断剤、好ましくはベンゾチアジン、最も好ましくはジルチアゼムまたはその薬学的に許容される塩を含む。
【0044】
他の好ましい実施形態において、薬物は、鎮痙薬、好ましくはオキシブチニンまたはその薬学的に許容される塩を含む。
【0045】
「徐放性(持続放出)」とは、本発明の目的のために、治療上有利な薬物血中レベル(ただし毒性レベル以下)を長い時間、例えば少なくとも約12時間または少なくとも約24時間にわたって維持するような制御速度にて、製剤から治療上有効な薬物が放出されることを意味する。
【0046】
「生体利用可能」とは、本発明の目的のために、治療上有効な薬物が徐放性製剤から吸収され、体内の薬物作用を意図する部位において、好ましくは参照基準の80%以内(AUCの比較を基準とする)で利用可能となることを意味する。
【0047】
「可溶性」とは、治療上有効な薬物が1リットル当たり約10グラム(g/l)より大きい水溶性を有することを意味する。
【0048】
「高度に可溶性」とは、治療上有効な薬物が1リットル当たり約100グラム(g/l)より大きい水溶性を有することを意味する。
【0049】
「周囲の液体」という用語は、本発明の目的のために、例えば、水溶液または胃腸液を包含する液体を意味する。
【0050】
「pH改変剤」という用語は、本発明の目的のために、薬物のイオン化を低減させることにより、ヒドロゲルマトリックスから溶液中への薬物の放出を促進するあらゆる物質を意味する。
【0051】
「Cmax」という用語は、本発明の目的のために、本発明の剤形の投与後に達成される薬物の最大血漿濃度を意味する。
【0052】
「Tmax」という用語は、本発明の目的のために、剤形の投与後から薬物のCmaxが達成されるまでの経過時間を意味する。
【0053】
「W50」という用語は、本発明の目的のために、Cmaxの50%の高さにおける血漿濃度曲線の幅で測定される時間の長さを意味する。
【0054】
本発明の目的のために、剤形は二相性薬物動力学を有しうるものであり、それゆえ開示された剤形に関して複数のCmax、TmaxおよびW50が存在しうる。
【発明を実施するための最良の形態】
【0055】
本発明に添付した図面は、本発明の実施形態を例示するものであり、特許請求の範囲により包含される本発明の範囲を限定するものではない。
【0056】
詳細な説明
本発明の徐放性マトリックスは、共働作用を示すヘテロ多糖およびホモ多糖両方からなるゲル化剤を含みうる様々な大きさの賦形剤でありうる(既に本発明者らの米国特許第4,994,276号、第5,128,143号および第5,135,757号において報告されている)。その共働作用とは、例えば、2種以上の多糖ガムの組み合わせによって、いずれかのガムを単独で用いて予測されるよりも高い粘度および急速な水和が得られ、形成するゲルがより急速に形成され、かつ堅固であることが挙げられる。
【0057】
本発明で使用する「ヘテロ多糖」という用語は、2種以上の糖単位を含有する水溶性多糖と定義され、該ヘテロ多糖は分枝鎖状またはらせん状立体構造を有し、優れた水深性(water-wicking)および大きな沈降濃縮特性を有する。
【0058】
特に好ましいヘテロ多糖は、高分子量(>106)のヘテロ多糖であるキサンタンガムである。他の好ましいヘテロ多糖には、キサンタンガムの誘導体(デアシル化キサンタンガムなど)、カルボキシメチルエーテル、およびプロピレングリコールエステルが挙げられる。
【0059】
本発明で使用するホモ多糖ガムはヘテロ多糖と架橋することができ、例えばガラクトマンナン、すなわち単にマンノースとガラクトースからなる多糖が挙げられる。ガラクトマンナンは、未置換マンノース領域の割合が高くなるほど、よりヘテロ多糖と相互作用することが見出されている。イナゴマメガムは、ガラクトースに対するマンノースの割合が高く、グアーおよびヒドロキシプロピルグアーなどの他のガラクトマンナンと比較して特に好ましい。
【0060】
本発明の制御放出製剤の制御放出特性は、様々な大きさの多糖物質の約10〜約90重量%以上の量のヘテロ多糖ガムは、許容される遅延放出生成物を提供するが、ヘテロ多糖ガムとホモ多糖物質との比が約1:1.5の場合に最適であり得る。本発明によれば、水溶液に晒された際に共働作用を示すことが知られている任意のホモ多糖ガムとの組み合わせを使用しうる。本発明のガムの組み合わせに関して存在するタイプの共働作用は、2種の同一な多糖間または2種のヘテロ多糖間で起こる可能性もある。本発明で使用しうる他の許容されるゲル化剤には、当技術分野で公知のゲル化剤が含まれる。例としては、植物ガム(アルギネート、カラゲーニン、ペクチン、グアーガムなど)、加工デンプン、ヒドロキシプロピルメチルセルロース、メチルセルロース、および他のセルロース物質(ナトリウムカルボキシメチルセルロースおよびヒドロキシプロピルセルロースなど)が挙げられる。このリストは限定を意味するものではない。
【0061】
徐放性賦形剤の不活性希釈剤は、好ましくは薬学的に許容される糖(単糖類、二糖類もしくは多価アルコールなど)、および/または上記の任意の混合物を含む。適切な不活性医薬充填剤の例としては、スクロース、デキストロース、ラクトース、微晶質セルロース、フルクトース、キシリトール、ソルビトール、デンプン、これらの混合物などが挙げられる。しかしながら、可溶性医薬充填剤、例えばラクトース、デキストロース、スクロース、またはこれらの混合物などを使用するのが好ましい。あるいはまた、不活性希釈剤または充填剤は、以下で記載する既製の直圧縮希釈剤(direct compression diluent)を含んでもよい。
【0062】
例えば、湿式顆粒化工程を行うことなく、徐放性賦形剤の成分を乾式混合することが可能である。この方法は、例えば有効成分を徐放性賦形剤の成分に直接添加する際に湿式顆粒化を行うべき場合に利用しうる。一方、この方法は、湿式顆粒化工程を全く考慮しない場合にも使用してよい。混合物を湿式顆粒化工程を行わずに製造すべき場合であって、最終混合物を打錠すべき場合には、不活性希釈剤の全部または一部が既製の直圧縮希釈剤を含有することが好ましい。かかる直圧縮希釈剤は、医薬分野で広く使用されており、多様な商業源から入手可能である。そのような既製の直圧縮賦形剤の例としては、Emcocel(登録商標)(微晶質セルロース、N.F.)、Emdex(登録商標)(デキストレート、N.F.)、およびTab-Fine(登録商標)(スクロース、フルクトースおよびデキストロースを含むいくつかの直圧縮糖)が挙げられ、これらは全てPenwest Pharmaceuticals Co., Patterson, New Yorkから市販されている。他の直圧縮希釈剤としては、Sheffield Chemical, Union, N.J. 07083からの無水ラクトース(ラクトースN.F.、無水直打錠);Degussa, D-600 Frankfurt(Main) GermanyからのElcems(登録商標)G-250(粉末セルロース、N.F.);Foremost Whey Products, Banaboo, WI 53913からのFast-Flo Lactose(登録商標)(ラクトース、N.F.、噴霧乾燥);Grain Processing Corp., Muscatine, IA 52761からのMaltrin(登録商標)(凝集マルトデキストリン);Roquet Corp., 645 5th Ave., New York, N.Y. 10022からのNeosorb 60(登録商標)(ソルビトール、N.F.、直圧縮);Ingredient Technology, Inc., Pennsauken, N.J. 08110からのNu-Tab(登録商標)(圧縮可能な糖、N.F.);GAF Corp., New York, N.Y. 10020からのPolyplasdone XL(登録商標)(Crospovidone, N.F.、架橋されたポリビニルピロリドン);Generichem Corp., Little Falls, N.J. 07424からのPrimojel(登録商標)(デンプングリコレートナトリウム、N.F.、カルボキシメチルデンプン);Penwest Pharmaceuticals Co., Patterson, N.Y. 10512からのSolka Floc(登録商標)(セルロース塊);Foremost Whey Products, Baraboo, WI 53913およびDMV Corp., Vehgel, HollandからのSpray-dried lactose(登録商標)(ラクトース、N.F.、噴霧乾燥);ならびにColorcon, Inc., West Point, PA 19486からのSta-Rx 1500(登録商標)(Starch 1500)(プレゲル化デンプン、N.F.、圧縮可能)が挙げられる。
【0063】
一般的には、上記製剤は、例えば、湿式顆粒化し、噴霧乾燥したラクトースにより直圧縮希釈剤として、または当業者に公知の方法により予め混合してある直圧縮希釈剤として調製しうる。本発明の目的のために、これらの特別に処理した不活性希釈剤を、「直圧縮」不活性希釈剤と呼ぶことにする。
【0064】
特定の実施形態において、徐放性賦形剤の成分は既製品であってもよい。しかしながら他の実施形態においては、有効な薬物を賦形剤成分に添加し、混合物を溶融顆粒化して顆粒を形成させうる。最終的に、界面活性剤を使用する場合には、可溶化または分散化ジルチアゼムまたはオキシブチニンを含む界面活性剤を成分の混合物に直接添加しうる。
【0065】
本発明のさらなる実施形態において、本発明の徐放性医薬賦形剤と共に使用する直圧縮不活性希釈剤は、米国特許出願第08/370,576号(1995年1月9日出願、発明の名称「Pharmaceutical Excipient Having Improved Compressibility」、発明者J. Staniforth, B. SherwoodおよびE. Hunter、参照によりこの全文を本明細書に引用する)に開示されているような増強(augmented)微晶質セルロースである。その開示されている増強微晶質セルロースは、Penwest Pharmaceuticals Co.より「Prosolv」という商標で市販されている。
【0066】
有効量の薬学的に許容される界面活性剤は、上述の賦形剤の成分に添加してもよいし、または薬物を添加する時点で添加してもよく、それにより薬物の生物学的利用能が高まる。適切な界面活性剤の例としては、固体剤形の約15重量%までのドキュセートナトリウムである。特に好ましい界面活性剤は、固体剤形の約15重量%までのラウリル硫酸ナトリウムである。
【0067】
一実施形態において、界面活性剤は水などの適切な溶媒中に溶解し、その後、徐放性賦形剤と薬物とを混合した混合物に添加する。これにより、界面活性剤は賦形剤の粒子を湿潤させ、その結果、溶媒が蒸発した場合には、沈殿する薬物の粒子が小さくなり凝集しない。賦形剤中に適切に微細にかつ均質に分散した薬物および界面活性剤の顆粒が得られる。
【0068】
本発明の特定の実施形態において、例えば薬物がジルチアゼムまたはオキシブチニンである場合には、界面活性剤を、例えば最終生成物の約1〜約5重量%、または約1〜15重量%の量で包含させる。しかしながら包含させる界面活性剤の上限は15%よりも高くてよい。1つの限定因子は、最終生成物が薬学的に許容される製剤を提供すべきことである。例えば錠剤の場合には、包含される界面活性剤量の上限は、薬学的に許容される錠剤、例えば摩損度が約1%未満で硬度が6〜8kgである錠剤の製造法により決定される。
【0069】
本発明で使用しうる界面活性剤には、一般的に、薬学的に許容されるアニオン性界面活性剤、カチオン性界面活性剤、両性(両親媒性/両染性(amphophilic))界面活性剤、および非イオン性界面活性剤が含まれる。好適な薬学的に許容されるアニオン性界面活性剤には、例えば、一価アルキルカルボン酸塩、アシルラクチレート(acyl lactylate)、アルキルエーテルカルボン酸塩、N-アシルサルコシネート、多価アルキル炭酸塩、N-アシルグルタミン酸塩、脂肪酸-ポリペプチド縮合物、スルホン酸エステル、アルキル硫酸塩(ラウリル硫酸ナトリウム(SLS)など)、エトキシル化アルキル硫酸塩、エステル結合スルホン酸塩(ドキュセートナトリウムまたはコハク酸ジオクチルナトリウム(DSS)など)、αオレフィンスルホン酸塩、およびリン酸化エトキシル化アルコールが挙げられる。
【0070】
適切な薬学的に許容されるカチオン性界面活性剤には、例えば、モノアルキル四級アンモニウム塩、ジアルキル四級アンモニウム化合物、アミドアミンおよびアミンイミドが含まれる。
【0071】
適切な薬学的に許容される両性(両親媒性/両染性)界面活性剤には、例えば、N-置換アルキルアミド、N-アルキルベタイン、スルホベタイン、およびN-アルキルβアミノプロピオン酸塩が含まれる。
【0072】
本発明に関連して使用するための他の好適な界面活性剤には、エステルまたはエーテルとしてのポリエチレングリコールが含まれる。例として、ポリエトキシル化ヒマシ油、ポリエトキシル化硬化ヒマシ油、ヒマシ油から誘導したポリエトキシル化脂肪酸、ヒマシ油から誘導したポリエトキシル化脂肪酸または硬化ヒマシ油から誘導したポリエトキシル化脂肪酸が挙げられる。使用しうる市販の界面活性剤は、Cremophor、Myrj、Polyoxy 40 stearate、Emerest 2675、Lipal 395およびPEG 3350という商標で知られている。
【0073】
pH改変剤は、マトリックスからの薬物の放出を促進し、最終剤形の約1〜約50重量%;約1〜約25重量%;約1〜約15重量%;または約1〜約10重量%で含有される。好ましい実施形態において、pH改変剤は、クエン酸、コハク酸、フマル酸、リンゴ酸、マレイン酸、グルタル酸または乳酸などの有機酸である。
【0074】
本発明に関連して任意に使用してもよいイオン化ゲル強度増強剤は一価または多価金属カチオンでありうる。好ましい塩は、種々のアルカリ金属および/またはアルカリ土類金属の硫酸塩、塩化物、ホウ酸塩、臭化物、クエン酸塩、酢酸塩、乳酸塩などを含む、無機塩である。適切なイオン化ゲル強度増強剤の具体的な例としては、硫酸カルシウム、塩化ナトリウム、硫酸カリウム、炭酸ナトリウム、塩化リチウム、リン酸三カリウム、ホウ酸ナトリウム、臭化カリウム、フッ化カリウム、重炭酸ナトリウム、塩化カルシウム、塩化マグネシウム、クエン酸ナトリウム、酢酸ナトリウム、乳酸カルシウム、硫酸マグネシウム、およびフッ化ナトリウムが挙げられる。多価金属カチオンを使用してもよい。しかし好ましいイオン化ゲル強度増強剤は二価である。特に好ましい塩は硫酸カルシウムおよび塩化ナトリウムである。本発明のイオン化ゲル強度増強剤は、ゲル化剤の架橋(例えばヘテロ多糖ガムとホモ多糖ガムとの架橋)に起因する所望のゲル強度の増大を得るのに有効な量で添加される。他の実施形態において、イオン化ゲル強度増強剤は、徐放性賦形剤の約1〜約20重量%の量、かつ最終剤形の0.5〜約16重量%の量で本発明の徐放性賦形剤中に含有させる。
【0075】
本発明の特定の実施形態において、本発明の徐放性マトリックスは、ヘテロ多糖ガムとホモ多糖ガムとを含む約10〜約99重量%のゲル化剤、約0〜約20重量%のイオン化ゲル強度増強剤、および約1〜約89重量%の不活性医薬希釈剤を含む徐放性賦形剤を含有する。他の実施形態において、徐放性賦形剤は、約10〜約75%のゲル化剤、約2〜約15%のイオン化ゲル強度増強剤、および約30〜約75%の不活性希釈剤を含有する。さらに他の実施形態において、徐放性賦形剤は、約30〜約75%のゲル化剤、約5〜約10%のイオン化ゲル強度増強剤、および約15〜約65%の不活性希釈剤を含有する。
【0076】
本発明の徐放性賦形剤は、(任意にイオン化ゲル強度増強剤を含有しても含有しなくてもよく)、親水性マトリックスを破壊することなくガムの水和を緩慢にする疎水性物質の組み込みによってさらに変更しうる。これは、本発明の他の実施形態で達成されるものであり、該実施形態では徐放性賦形剤を疎水性物質の溶液または分散液を用いて顆粒化した後で薬物の組み込みを行う。疎水性ポリマーは、アルキルセルロース(エチルセルロースなど)、他の疎水性セルロース物質、アクリル酸エステルまたはメタクリル酸エステルから誘導されるポリマーまたはコポリマー、アクリル酸エステルとメタクリル酸エステルとのコポリマー、ゼイン、ワックス、シェラック、硬化植物油、および当業者に公知の他のあらゆる薬学的に許容される疎水性物質から選択しうる。徐放性賦形剤中に組み込む疎水性物質の量は、周囲の液体に晒された際に形成される親水性マトリックスを破壊せずにガムの水和を緩慢にするのに有効な量である。本発明の特定の好ましい実施形態において、疎水性物質は、約1〜約20重量%の量で徐放性賦形剤中に含有される。疎水性物質のための溶媒は、水性もしくは有機溶媒、またはこれらの混合物でありうる。
【0077】
本発明の徐放性賦形剤が既製品である場合の実施形態において、例えば高剪断力ミキサー中で、薬物とその賦形剤とを混合することができる。特定の特に好ましい実施形態において、薬物は、循環系疾患および高血圧症の治療に有用である、治療上有効なベンゾチアジンである。特に好ましいジヒドロキシピリジンはジルチアゼムである。ジルチアゼムの有効な製剤は、一般的に約30〜約500mg、好ましくは約120mg〜約480mgの日用量を含有する。本発明の特定の好ましい実施形態において、剤形は、ジルチアゼムの調剤を、24時間用製剤については120mg、180mg、240mgまたは300mgの量で、そして12時間用製剤については60mg、90mgおよび120mgの量で含有する。
【0078】
特定の他の、特に好ましい実施形態において、薬物は、泌尿器疾患の治療に有用なオキシブチニンである。オキシブチニンの有用な製剤は、一般的に、約2.5mg〜約50mgの日用量、例えば12時間用製剤については約2.5mg〜約25mg、24時間用製剤については約5mg〜約50mgを含有する。本発明の特定の好ましい実施形態において、剤形は、24時間用製剤について5mg、10mgまたは15mgの量でオキシブチニンの調剤を含有する。
【0079】
有効量の一般的に許容される医薬潤滑剤(カルシウム石鹸またはマグネシウム石鹸など)は、好ましくは、成分(薬物など)の混合物を打錠して経口用固体剤形(例えば錠剤)にする前に該混合物に添加する。適切な潤滑剤の例は、固体剤形の約0.5〜約3重量%の量のステアリン酸マグネシウムである。特に好ましい潤滑剤は、ステアリルフマル酸ナトリウム(sodium stearyl fumarate), NF(Penwest Pharmaceuticals Co.からPruv(登録商標)の商標で市販されている)である。
【0080】
本発明の徐放性賦形剤は、種々の粒子サイズ分布の範囲で均一な充填特性を有し、直圧縮に続いて薬物および潤滑剤粉末の添加を行うかまたは慣用的な湿式顆粒化法により最終剤形(例えば錠剤)に加工することができる。
【0081】
本発明に従って調製される特定の賦形剤系の性質および特徴は、部分的には、ホモ多糖成分およびヘテロ多糖成分の個々の特性(ポリマー可溶性、ガラス転移温度など)、ならびに変化する溶出の液体−賦形剤相互作用における種々のホモ多糖とヘテロ多糖との共働作用、ホモ多糖およびヘテロ多糖と不活性多糖要素との共働作用に依存している。
【0082】
ゲル化剤(すなわち、キサンタンガムとイナゴマメガムとの混合物)と、不活性希釈剤と、イオン化ゲル強度増強剤および疎水性ポリマー(あってもなくてもよい)との組み合わせによって、製剤の製造者が、混合物を打錠して遅延放出錠剤を形成する前に、所望の有効薬物、pH改変剤、界面活性剤および任意に潤滑剤と賦形剤とを混合するだけでよい、すぐに使用できる徐放性賦形剤製品が提供される。ガムと純粋な(すなわち微晶質)スクロース、ラクトース、デキストロースなどとを顆粒化または凝塊形成して賦形剤を形成することが好ましいが、該賦形剤は、ガムと可溶性賦形剤(例えば圧縮可能なスクロース、ラクトースまたはデキストロース)との物理的混合物を含みうる。顆粒形態は、流動性および圧縮性を最適化しうるという事実、有効薬物を用いた打錠、カプセル中への配合、押し出し加工および球状化(speronize)によるペレット形成を行いうる事実などを含むいくつかの利点を有する。
【0083】
本発明に従って調製される医薬賦形剤は、許容される賦形剤製品を得るためのあらゆる凝塊形成法に従って調製しうる。湿式顆粒化法では、所望の量のヘテロ多糖ガム、ホモ多糖ガムおよび不活性希釈剤を一緒に混合し、その後、水、プロピレングリコール、グリセロール、アルコールなどの湿潤剤を添加して湿潤塊を調製する。次にこの湿潤塊を乾燥する。続いて乾燥した塊を慣用的な装置を用いて粉砕し、顆粒にする。それにより、賦形剤製品はすぐ使用可能なものとなる。
【0084】
既製の徐放性賦形剤は、好ましくは流動し、直圧縮可能なものである。従って、該賦形剤は、治療上有効な薬物および任意に潤滑剤と所望の割合で混合しうる(乾式顆粒化法)。あるいは、賦形剤の全部または一部について、活性成分と共に湿式顆粒化法を行い、その後で打錠してもよい。製造すべき最終生成物が錠剤の場合には、その後に、一定のバッチの錠剤を製造するのに十分な量の完全な混合物を、慣用的な製造規模の打錠機を用いて、通常の圧縮圧、すなわち約2000〜1600lbs/平方インチで打錠を行う。しかしながら、混合物は、後で胃液に晒された際に水和するのが困難な程度にまで圧縮するべきではない。
【0085】
錠剤の製造方法としての直圧縮法の限界の1つは、錠剤のサイズである。有効成分が多い場合には、医薬製剤の製造者は、正しい圧縮強度を有し基準に達するサイズの錠剤を得るために、他の賦形剤と共に有効成分に湿式顆粒化を行うことを選択してもよい。通常は、湿式顆粒化に必要とされる充填剤/結合剤または賦形剤の量は、直圧縮の場合よりも少なく、それは湿式顆粒化の工程が錠剤の所望の物理的特性にある程度寄与しているためである。
【0086】
薬物がジルチアゼムの場合には、丸型錠剤の平均錠剤サイズは好ましくは約300mg〜750mgであり、そしてカプセル形状錠剤の平均錠剤サイズは約700mg〜1000mgである。
【0087】
本発明の顆粒化賦形剤の平均粒子サイズは、好ましくは約50ミクロン〜約400ミクロン、好ましくは約185ミクロン〜約265ミクロンである。顆粒の粒子サイズは厳密に重要なものではなく、重要なパラメーターは、顆粒の平均粒子サイズが薬学的に許容される錠剤を形成する直圧縮賦形剤の形成を可能にする必要があるということである。本発明の顆粒の所望のタップ密度および充填密度は、通常約0.3〜約0.8g/mlであり、平均密度は約0.5〜約0.7g/mlである。最良の結果では、本発明の顆粒から形成される錠剤は、約5〜約20kgの硬度を有する。本発明に従って調製される顆粒の平均流量は、好ましくは約25〜約40g/秒である。試験ロータリー型打錠機を用いて圧縮した錠剤は、不活性糖成分とはほとんど別の強度プロフィールを有することが見出されている。錠剤表面の大部分の走査型電子顕微鏡写真によって、錠剤表面および破断面の両方において圧縮における過剰な塑性変形に関する質的証拠が提供されている。また該写真は、最初の溶媒侵入および溶液排出が起こりうる表面孔の証拠を示している。
【0088】
本発明の特定の実施形態において、錠剤は、製剤の薬物の放出をさらに変更することができるように十分な量の疎水性ポリマーを用いてコーティングする。錠剤コーティング中に含まれる疎水性ポリマーは、任意に徐放性賦形剤と共に顆粒化してもよい疎水性ポリマー物質と比較して同じ物質でもよいし、異なる物質でもよい。
【0089】
本発明の他の実施形態において、錠剤コーティングは、疎水性ポリマーコーティングに加えてまたはその代わりに腸溶コーティング物質を含みうる。適切な腸溶ポリマーの例としては、酢酸フタル酸セルロース、フタル酸ヒドロキシプロピルメチルセルロース、フタル酸ポリビニルアセテート、メタクリル酸コポリマー、シェラック、コハク酸ヒドロキシプロピルメチルセルロース、酢酸トリメリト酸セルロース、およびこれらの任意の混合物が挙げられる。適切な市販の腸溶物質の例は、EudraditTM L30D55の商標で入手可能である。
【0090】
さらなる実施形態において、剤形は、上述のコーティングに加えてまたはその代わりに親水性コーティングでコーティングしてもよい。かかる親水性コーティングに使用しうる適切な物質の例は、ヒドロキシプロピルメチルセルロース(例えば、Colorcon, West Point, Pennsylvaniaより市販されているOpadry(登録商標))である。
【0091】
コーティングは、当業者に公知の薬学的に許容される方法で適用しうる。例えば一実施形態において、コーティングは流動床によりまたはコーティングパン中で適用する。例えば、コーティングした錠剤は、例えばコーティングパン中で約60〜70℃にて約3〜4時間乾燥しうる。疎水性ポリマーまたは腸溶コーティングの溶媒は、有機溶媒、水性溶媒、または有機溶媒と水性溶媒との混合物でありうる。有機溶媒は、水を含有していてもしていなくてもよい、例えば、イソプロピルアルコール、エタノールなどでありうる。
【0092】
本発明のさらなる実施形態において、本発明に従って製造する錠剤にサポートプラットフォーム(support platform)を適用する。適切なサポートプラットフォームは当業者に周知である。適切なサポートプラットフォームの例は、例えば米国特許第4,839,177号(参照により本明細書に引用する)に記載されている。この特許において、錠剤をサポートプラットフォームで部分的にコーティングしており、このサポートプラットフォームは水性液体に不溶性のポリマー物質からなるものである。サポートプラットフォームは、例えば、治療上有効な薬物の運搬の間に非浸透性を維持するように設計されうる。サポートプラットフォームは、例えば錠剤表面の一部に加圧コーティングを施すことにより、錠剤表面の全部もしくは一部にサポートプラットフォームを含むポリマー物質を噴霧コーティングすることにより、またはポリマー物質の溶液中に錠剤を浸すことによって、錠剤に適用しうる。
【0093】
サポートプラットフォームは、圧縮で適用される場合には例えば約2mmの厚さ、噴霧コーティングまたは液浸により適用される場合には約10μの厚さを有しうる。一般的には、疎水性ポリマーまたは腸溶コーティングが錠剤に適用される本発明の実施形態において、錠剤を、重量が約1〜約20%、特定の実施形態では好ましくは約5〜約10%増加する程度にコーティングする。
【0094】
本発明の疎水性コーティングおよびサポートプラットフォームに有用な物質には、アクリル酸の誘導体(例えば、アクリル酸エステル、メタクリル酸エステル、およびそれらのコポリマー)、セルロースおよびそれらの誘導体(例えばエチルセルロース)、ポリビニルアルコールなどが含まれる。
【0095】
本発明の特定の実施形態において、錠剤コアには、疎水性もしくは腸溶コーティング中、または、錠剤コアの外部表面上にコーティングされたさらなるオーバーコーティング中(疎水性もしくは腸溶コーティングを含まない)に、あるいは疎水性もしくは腸溶コーティング物質を含むベースコーティングの表面上にコーティングされる第2コーティング層として、包含されるさらなる用量の薬物が含まれる。これは、例えば、治療上有効な薬物の充填量が、最初に製剤が胃液に晒された際に有効薬物の治療上有効な血中レベルを提供するのに要求される場合に、望ましいものでありうる。コーティング層中に含まれる薬物の充填量は、例えば、製剤中に含まれる薬物の合計量の約10〜約40%でありうる。
【0096】
本発明の好ましい実施形態において、最終製剤は、薬物がジルチアゼムの場合には二相性または多相性血漿レベルを提供する。
【0097】
好ましい実施形態において、薬物がジルチアゼムの場合には、本発明の製剤は、患者への経口投与後約4〜約10時間において、ジルチアゼムの初回ピーク血漿濃度時間(Tmax#1)を示す。特定の好ましい実施形態において、初回ピーク血漿濃度時間は、経口投与後約6〜約8時間で起こる。好ましい実施形態において、初回Tmaxにおけるジルチアゼムの最大血漿濃度(Cmax#1)は、本発明の経口用徐放性剤形中240mgの量のジルチアゼムの投与当たり、約50〜100ng/mlである。
【0098】
本発明のさらなる好ましい実施形態において、徐放性ジルチアゼム製剤は、第2ピーク血漿濃度(Cmax#2)を提供する。この第2ピーク血漿濃度は、患者への剤形の経口投与後約10〜約16時間(Tmax#2)において起こる。特定の好ましい実施形態において、第2ピーク血漿濃度(Cmax#2)は、ヒトへの剤形の経口投与後約12〜約14時間(Tmax#2)において起こる。好ましい実施形態において、Cmax#2におけるジルチアゼムの最大血漿濃度は、24時間にわたり投与された240mgのジルチアゼム当たり約60〜約90ng/mlである。
【0099】
特定の好ましい実施形態において、徐放性ジルチアゼム製剤は、約0.5〜約4時間、好ましくは約1〜約3時間であるCmax#1のW50を示す。Cmax#1のW50は、本発明の目的のために、Cmax#1とCmax#2との間のCminとして得られた最低値を基準にして、第1Cmax(Cmax#1)の高さの50%における血漿濃度曲線の幅として定義される。
【0100】
特定の好ましい実施形態において、徐放性ジルチアゼム製剤は、約0.5〜約8時間、好ましくは約2〜約6時間であるCmax#2のW50を示す。Cmax#2のW50は、本発明の目的のために、Cmax#1とCmax#2との間のCminとして得られた最低値を基準にして、第2Cmax(Cmax#2)の高さの50%における血漿濃度曲線の幅として定義される。
【0101】
特定の好ましい実施形態において、本発明の徐放性ジルチアゼム製剤は、Cmax#1とCmax#2との比が約0.5:1〜約1.5:1、好ましくは約0.7:1〜約1.2:1である。
【0102】
本発明の経口用徐放性製剤中のジルチアゼムの量を基準として、ジルチアゼムの種々の投与量に関して、12または24時間にわたるCmax#1、Cmax#2、Tmax#1およびTmax#2を容易に決定することができるだろう。
【0103】
本発明の特定の好ましい実施形態において、薬物がオキシブチニンの場合には、該製剤は、約5〜約15時間、好ましくは約8〜約12時間においてオキシブチニンのピーク血漿濃度時間(Tmax)を提供する。
【0104】
本発明に組み込むのに適した可溶性〜高度に可溶性の薬物の例としては、抗ヒスタミン(例えば、マレイン酸アザタジン、マレイン酸ブロムフェニルアミン、マレイン酸カルビノキサミン、マレイン酸クロルフェニルアミン、マレイン酸d‐クロルフェニルアミン、塩酸ジフェンヒドラミン、コハク酸ドキシラミン、塩酸メトジラジン、プロメタジン、酒石酸トリメプラジン、クエン酸トリペレナミン、塩酸トリペレナミン、および塩酸トリプロリジン);抗生物質(例えば、ペニシリンVカリウム、クロキサシリンナトリウム、ジクロキサシリンナトリウム、ナフシリンナトリウム、オキサシリンナトリウム、カルベニシリンインダニルナトリウム、塩酸オキシテトラサイクリン、塩酸テトラサイクリン、リン酸クリンダマイシン、塩酸クリンダマイシン、パルミチン酸クリンダマイシンhcl、リンコマイシンhcl、ノボビオシンナトリウム、ニトロフラントインナトリウム、塩酸メトロニダゾル);抗結核剤(例えば、イソニアジド);コリン作動物質(例えば、塩化アンベノニウム、塩化ベタネコル(bethanecol chloride)、臭化ネオスチグミン、臭化ピリドスチグミン);抗ムスカリン剤(例えば、臭化メチルアニソトロピン、臭化クリジニウム、塩酸ジシクロミン、グリコピロレート、メチル硫酸ヘキソシクリウム、臭化メチルホマトロピン、硫酸ヒヨスチアミン、臭化メタンテリン、臭化水素酸ヒオスシン、臭化オキシフェノニウム、臭化プロパンテリン、塩化トリジヘキセチル);交感神経興奮剤(例えば、メタンスルホン酸ビオルテロール、エフェドリン、塩酸エフェドリン、硫酸エフェドリン、硫酸オルシプレナリン、塩酸フェニルプロパノールアミン、塩酸プソイドエフェドリン、塩酸リトドリン、硫酸サルブタモール、硫酸テルブタリン);交感神経遮断剤(例えば、塩酸フェノキシベンズアミン);他の自律神経作用薬(例えば、ニコチン);鉄製剤(例えば、グルコン酸第一鉄、硫酸第一鉄);ヘモスタティック(例えば、アミノカプロン酸);強心剤(例えば、塩酸アセブトロール、リン酸ジソピラミド、酢酸フレカイニド、塩酸プロカインアミド、塩酸プロプラノロール、グルコン酸キニジン、マレイン酸チモロール、塩酸トカイニド、塩酸ベラパミル);抗高血圧剤(例えば、カプトリル、塩酸クロニジン、塩酸ヒドララジン、塩酸メカミラミン、酒石酸メトプロロール);血管拡張剤(例えば、塩酸パパベリン);非ステロイド性抗炎症剤(例えば、サリチル酸コリン、サリチル酸マグネシウム、メクロフェナム酸ナトリウム、ナプロキセンナトリウム、トルメチンナトリウム);鎮痙薬(例えば、フェノバルビタールナトリウム、フェニトインナトリウム、トロキシドン、エトスクシミド、バルプロ酸ナトリウム);精神安定剤(例えば、マレイン酸アセトフェナジン、塩酸クロルプロマジン、塩酸フルフェナジン、1,2-エタンジスルホン酸(edisylate)プロクロルペラジン、塩酸プロメタジン、塩酸チオリダジン、塩酸トリフルオロペラジン、クエン酸リチウム、塩酸モリンドン、塩酸チオチキセン);刺激剤(塩酸ベンザムフェタミン(benzamphetamine)、硫酸デキストロアンフェタミン、リン酸デキストロアンフェタミン、塩酸ジエチルプロピオン、塩酸フェンフルラミン、塩酸メタンフェタミン、塩酸メチルフェニデート、酒石酸フェンジメトラジン、塩酸フェンメトラジン、クエン酸カフェイン);バルビツル剤(例えば、アミロバルビタール(amylobarbital)ナトリウム、ブタバルビタールナトリウム、セコバルビタールナトリウム);鎮静剤(例えば、塩酸ヒドロキシジン、メトプリロン(methprylon));去痰剤(例えば、ヨウ化カリウム);制吐薬(例えば、塩酸ベンザキナミド(benzaquinamide)、塩酸メトクロプロパミド(metoclopropamide)、塩酸トリメトベンズアミド);胃腸薬(例えば、塩酸ラニチジン);重金属アンタゴニスト(例えば、ペニシラミン、塩酸ペニシラミン);抗甲状腺薬(例えば、メチマゾール);尿生殖器平滑筋弛緩薬(例えば、塩酸フラボキセイト);ビタミン剤(例えば、塩酸チアミン、アスコルビン酸);分類されていない薬剤(例えば、塩酸アマンタジン、コルヒチン、エチドロン酸二ナトリウム、ロイコボリンカルシウム、メチレンブルー、塩化カリウム、塩化プラリドキシム)が挙げられる。このリストは限定を意味するものでない。
【実施例】
【0105】
以下の実施例は本発明の種々の態様を説明するものである。これらは、いかなる意味においても特許請求の範囲を限定するものと解されるべきではない。
【0106】
実施例1〜2
製剤における薬物:ガム比の効果
実施例1および2においては、本発明の徐放性賦形剤を最初に調製し、次いで薬物(この場合はジルチアゼム)およびpH改変剤(この場合はフマル酸)を添加し、続いて最終混合物を錠剤に製剤化する。
【0107】
徐放性賦形剤を、必要量のキサンタンガム、イナゴマメガム、およびデキストロースを、高速ミキサー/造粒機において3分間ドライブレンドして調製する。チョッパー(chopper)/圧縮機(impeller)を作動させている間に、ドライブレンドした混合物に水を添加し、さらに3分間顆粒化する。次いで顆粒を、LOD(乾燥工程のロス)が約10重量%未満(例えば4〜7%のLOD)となるように液体ベッドドライヤーで乾燥させる。顆粒を20メッシュの金網を用いて粉砕し、造粒機中に分散する。実施例1および2の顆粒の成分を下記の表1に示す。
【表1】

【0108】
次に、所望の量のジルチアゼム、フマル酸および好適な量の水を、圧縮機型のミキサーで5分間にわたり混合してスラリーを形成する。続いてこのスラリーを徐放性賦形剤に1分間隔で、低速で圧縮機を作動させながら造粒機に添加する。次に混合物を、高速で作動させたチョッパーおよび圧縮機を用いて2分間にわたり顆粒化する(好適な顆粒を形成するために、さらに水と顆粒化時間を増してもよい)。続いて得られた顆粒を、液体ベッドドライヤーでLODが5%未満となるまで乾燥し、2000〜3000rpmで作動しているハンマーで粉砕する。次に粉砕した顆粒をドデシル硫酸ナトリウムとともにV-Blender中に配置し、10分間にわたりブレンドする。続いて好適な錠剤化用潤滑剤(Pruv(登録商標)、ステアリルフマル酸ナトリウム、NF、Penwest Pharmaceuticals Co.より市販されている)を添加し、混合物をさらに3分間にわたりブレンドする。続いて得られた顆粒を、カプセル型パンチを用いて錠剤へと圧縮する。この最終混合物を約768mgの錠剤とする。実施例1および2の錠剤の成分を以下の表2に示す。
【表2】
【0109】
最終産物の錠剤は、錠剤として重量768.0mgであり、硬度15Kpである。
【0110】
実施例1および2の錠剤についての溶出試験を、自動USP溶出装置(Paddle type II、100rpm)中で900MLの水中で行い、放出された薬物の量をUV分析により調べた。in vitroでの溶出の結果を、図1と以下の表3に示す。
【表3】
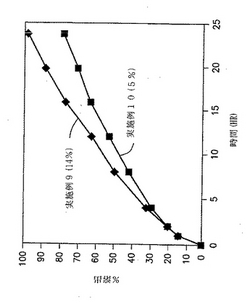
【0111】
図1および表3に示した結果から、ジルチアゼムの放出速度は、製剤中のガムの量が増加するにつれて遅くなることが明らかである。
【0112】
実施例3〜4
ガム:デキストロース比の効果
実施例3および4においては、徐放性賦形剤は実施例1および2に示した工程により調製する。実施例3および4の徐放性賦形剤の成分を、以下の表4に示す。
【表4】
【0113】
その後、ジルチアゼム錠剤を以下の通り調製する。
【0114】
所望の量のジルチアゼム、フマル酸および徐放性賦形剤を造粒機に配置し、低速で3分間にわたり混合する。続いて水を2分間隔で、低速で圧縮機を作動させながら造粒機に添加する(好適な顆粒を形成するために、さらに水と顆粒化時間を増してもよい)。続いて得られた顆粒を、液体ベッドドライヤーでLODが5%未満となるまで乾燥し、2000〜3000rpmで作動しているハンマーで金網#0050を用いて粉砕する。次に粉砕した顆粒をドデシル硫酸ナトリウムとともにV-Blender中に配置し、10分間にわたりブレンドする。続いて好適な錠剤化用潤滑剤(Pruv、ステアリルフマル酸ナトリウム、NF、Penwest Pharmaceuticals Co.より市販されている)を添加し、混合物をさらに5分間にわたりブレンドする。続いて、得られた顆粒を、カプセル型パンチを用いて錠剤へと圧縮する。この最終混合物を約750mgの錠剤とする。実施例3および4の錠剤の成分を以下の表5に示す。
【表5】
【0115】
最終産物の錠剤は、錠剤として重量750.0mgであり、硬度15Kpである。
【0116】
実施例3および4の錠剤についての溶出試験を、自動USP溶出装置(Paddle type III、15CPM)中で250MLのバッファー(pH6)中で行い、放出された薬物の量をUV分析により調べた。in vitroでの溶出の結果を、図2と以下の表6に示す。
【表6】
【0117】
図2および表6に示した結果から、デキストロースの量に対するガムの量が増加するにつれて、それに対応して薬物放出の遅れが見られた。
【0118】
実施例5〜6
界面活性剤の型の効果
実施例5および6において、徐放性賦形剤は実施例1および2に示した工程により調製する。実施例5および6の徐放性賦形剤の成分を、以下の表7に示す。
【表7】
【0119】
その後、ジルチアゼム錠剤を以下の通り調製する。
【0120】
所望の量のジルチアゼム、フマル酸および好適な量の水を、圧縮機型のミキサーで5分間にわたり混合してスラリーを形成する。続いてこのスラリーを徐放性賦形剤に1分間隔で、低速で圧縮機を作動させながら造粒機において添加する。次に混合物を、高速で作動させたチョッパーおよび圧縮機を用いて2分間にわたり顆粒化する(好適な顆粒を形成するために、さらに水と顆粒化時間を増してもよい)。続いて得られた顆粒を、液体ベッドドライヤーでLODが5%未満となるまで乾燥し、2000〜3000rpmで作動しているハンマーで粉砕する。次に粉砕した実施例5の顆粒をドデシル硫酸ナトリウムとともにV-Blender中に配置し、粉砕した実施例6の顆粒をドキュセートナトリウムとともにV-Blender中に配置し、10分間にわたりブレンドする。続いて好適な錠剤化用潤滑剤(Pruv、ステアリルフマル酸ナトリウム、NF、Penwest Pharmaceuticals Co.より市販されている)を各例に対して添加し、混合物をさらに3分間にわたりブレンドする。続いて得られた顆粒を、カプセル型パンチを用いて錠剤へと圧縮する。この最終混合物を約848mgの錠剤とする。実施例5および6の錠剤の成分を以下の表8に示す。
【表8】
【0121】
最終産物の錠剤は、錠剤として重量848.0mgであり、硬度15Kpである。
【0122】
実施例1および2の錠剤についての溶出試験を、自動USP溶出装置(Paddle type II、100rpm)中で900MLの水中で行い、放出された薬物の量をUV分析により調べた。in vitroでの溶出の結果を、図3と以下の表9に示す。
【表9】
【0123】
図3および表9に示した結果から、ジルチアゼムの放出速度は、等量比のドデシル硫酸ナトリウムおよびドキュセートナトリウムについては同じであることが明らかである。しかしながら、ドデシル硫酸ナトリウムを用いた方が製剤化工程には好適であった。
【0124】
実施例7〜8
界面活性剤のレベルの効果
実施例7および8において、徐放性賦形剤は実施例1および2に示した工程により調製する。実施例7および8の徐放性賦形剤の成分を、以下の表10に示す。
【表10】
【0125】
その後、ジルチアゼム錠剤を以下の通り調製する。
【0126】
所望の量のジルチアゼム、フマル酸および好適な量の水を、圧縮機型のミキサーで5分間にわたり混合してスラリーを形成する。続いてこのスラリーを徐放性賦形剤に1分間隔で、低速で圧縮機を作動させながら造粒機に添加する。次に混合物を、高速で作動させたチョッパーおよび圧縮機を用いて2分間にわたり顆粒化する(好適な顆粒を形成するために、さらに水と顆粒化時間を増してもよい)。続いて得られた顆粒を、液体ベッドドライヤーでLODが5%未満となるまで乾燥し、2000〜3000rpmで作動しているハンマーで粉砕する。次に粉砕した顆粒をドデシル硫酸ナトリウムとともにV-Blender中に配置し、10分間にわたりブレンドする。続いて好適な錠剤化用潤滑剤(Pruv、ステアリルフマル酸ナトリウム、NF、Penwest Pharmaceuticals Co.より市販されている)を添加し、混合物をさらに3分間にわたりブレンドする。続いて得られた顆粒を、カプセル型パンチを用いて錠剤へと圧縮する。この最終混合物を約768mgの錠剤とする。実施例7および8の錠剤の成分を以下の表11に示す。
【表11】
【0127】
実施例7の最終産物の錠剤は、錠剤として重量768.0mgであり、硬度15Kpである。
【0128】
実施例8の最終産物の錠剤は、錠剤として重量728.0mgであり、硬度15Kpである。
【0129】
実施例7および8の錠剤についての溶出試験を、自動USP溶出装置(Paddle type II、100rpm)中で900MLの水中で行い、放出された薬物の量をUV分析により調べた。結果を、図4と以下の表12に示す。
【表12】
【0130】
図4および表12に示した結果から、ジルチアゼムの溶出速度は、界面活性剤のレベルに反比例していることが明らかである。
【0131】
実施例9〜10
フマル酸レベルの効果
実施例9および10において、徐放性賦形剤は実施例1および2に示した工程により調製する。実施例9および10の徐放性賦形剤の成分を、以下の表13に示す。
【表13】
【0132】
その後、ジルチアゼム錠剤を以下の通り調製する。
【0133】
所望の量のジルチアゼム、フマル酸および好適な量の水を、圧縮機型のミキサーで5分間にわたり混合してスラリーを形成する。続いてこのスラリーを徐放性賦形剤に1分間隔で、低速で圧縮機を作動させながら造粒機に添加する。次に混合物を、高速で作動させたチョッパーおよび圧縮機を用いて2分間にわたり顆粒化する(好適な顆粒を形成するために、さらに水と顆粒化時間を増してもよい)。続いて得られた顆粒を、液体ベッドドライヤーでLODが5%未満となるまで乾燥し、2000〜3000rpmで作動しているハンマーで粉砕する。次に粉砕した顆粒をドデシル硫酸ナトリウムとともにV-Blender中に配置し、10分間にわたりブレンドする。続いて好適な錠剤化用潤滑剤(Pruv、ステアリルフマル酸ナトリウム、NF、Penwest Pharmaceuticals Co.より市販されている)を添加し、混合物をさらに3分間にわたりブレンドする。続いて得られた顆粒を、カプセル型パンチを用いて錠剤へと圧縮する。この最終混合物を約848mgの錠剤とする。実施例9および10の錠剤の成分を以下の表14に示す。
【表14】
【0134】
実施例9の最終産物の錠剤は、錠剤として重量848.0mgであり、硬度15Kpである。
【0135】
実施例10の最終産物の錠剤は、錠剤として重量768.0mgであり、硬度15Kpである。
【0136】
実施例9および10の錠剤についての溶出試験を、自動USP溶出装置(Paddle type II、100rpm)中で900MLの水中で行い、放出された薬物の量をUV分析により調べた。結果を、図5と以下の表15に示す。
【表15】
【0137】
図5および表15に示した結果から、製剤中のフマル酸の量を増加させることにより、放出速度が増加することが明らかである。
【0138】
実施例11〜12
外部顆粒化した顆粒の薬物への添加
実施例11および12においては、徐放性賦形剤は実施例1および2に示した工程により調製する。実施例11および12の徐放性賦形剤の成分を、以下の表16に示す。
【表16】
【0139】
その後、ジルチアゼム錠剤を以下の通り調製する。
【0140】
実施例11においては、所望の量のジルチアゼム、フマル酸および好適な量の水を、圧縮機型のミキサーで5分間にわたり混合してスラリーを形成する。続いてこのスラリーを徐放性賦形剤に1分間隔で、低速で圧縮機を作動させながら造粒機に添加する。次に混合物を、高速で作動させたチョッパーおよび圧縮機を用いて2分間にわたり顆粒化する(好適な顆粒を形成するために、さらに水と顆粒化時間を増してもよい)。続いて得られた顆粒を、液体ベッドドライヤーでLODが5%未満となるまで乾燥し、2000〜3000rpmで作動しているハンマーで粉砕する。次に粉砕した顆粒をドデシル硫酸ナトリウムとともにV-Blender中に配置し、10分間にわたりブレンドする。続いて好適な錠剤化用潤滑剤(Pruv、ステアリルフマル酸ナトリウム、NF、Penwest Pharmaceuticals Co.より市販されている)を添加し、混合物をさらに3分間にわたりブレンドする。続いて得られた顆粒を、カプセル型パンチを用いて錠剤へと圧縮する。この最終混合物を約848mgの錠剤とする。
【0141】
実施例12においては、ジルチアゼムの一部、フマル酸および好適な量の水を、圧縮機型のミキサーで5分間にわたり混合してスラリーを形成する。続いてこのスラリーを徐放性賦形剤に1分間隔で、低速で圧縮機を作動させながら造粒機に添加する。次に混合物を、高速で作動させたチョッパーおよび圧縮機を用いて2分間にわたり顆粒化する(好適な顆粒を形成するために、さらに水と顆粒化時間を増してもよい)。続いて得られた顆粒を、液体ベッドドライヤーでLODが5%未満となるまで乾燥し、2000〜3000rpmで作動しているハンマーで粉砕する。次に粉砕した顆粒をドデシル硫酸ナトリウムおよびジルチアゼムの残部とともにV-Blender中に配置し、10分間にわたりブレンドする。続いて好適な錠剤化用潤滑剤(Pruv、ステアリルフマル酸ナトリウム、NF、Penwest Pharmaceuticals Co.より市販されている)を添加し、混合物をさらに3分間にわたりブレンドする。続いて得られた顆粒を、カプセル型パンチを用いて錠剤へと圧縮する。この最終混合物を約848mgの錠剤とする。実施例11および12の錠剤の成分を下記の表17に示す。
【表17】
【0142】
実施例11の最終産物の錠剤は、錠剤として重量848.0mgであり、硬度15Kpである。
【0143】
実施例12の最終産物の錠剤は、錠剤として重量848.0mgであり、硬度15Kpである。
【0144】
実施例11および12の錠剤についての溶出試験を、自動USP溶出装置(Paddle type II、100rpm)中で900MLの水中で行い、放出された薬物の量をUV分析により調べた。結果を、図6と以下の表18に示す。
【表18】
【0145】
図6および表18に示した結果は、外部で顆粒化した顆粒としてのジルチアゼムの添加により約35%の初期破裂が生じることを示す。実施例11および12の剤形からのジルチアゼムの%放出速度を経時的に示す図7によっても示される通り、特定の割合の薬物を外部で顆粒化して添加すると、初期の急速な放出がもたらされることは明白である。
【0146】
実施例13〜18
Eudragit L30D55 w/NaOH(メタクリル酸コポリマー水性分散液)を有するコーティング錠剤の効果
実施例13〜18においては、徐放性賦形剤は実施例1および2に示した工程により調製する。実施例13〜18の徐放性賦形剤の成分を、以下の表19に示す。
【表19】
【0147】
その後、ジルチアゼム錠剤を以下の通り調製する。
【0148】
所望の量のジルチアゼム、フマル酸および徐放性賦形剤を、造粒機中で5分間にわたり低速で混合する。続いて好適な量の水を2分間隔で、低速で圧縮機を作動させながら添加する。次に得られたスラリーを、高速で作動させたチョッパーおよび圧縮機を用いて7.5分間にわたり顆粒化する(好適な顆粒を形成するために、さらに水と顆粒化時間を増してもよい)。続いて得られた顆粒を、液体ベッドドライヤーでLODが5%未満となるまで乾燥し、2000〜3000rpmで作動しているハンマーで、金網#0050を用いて粉砕する。次に粉砕した顆粒をドデシル硫酸ナトリウムとともにV-Blender中に配置し、10分間にわたりブレンドする。続いて好適な錠剤化用潤滑剤(Pruv、ステアリルフマル酸ナトリウム、NF、Penwest Pharmaceuticals Co.より市販されている)を添加し、混合物をさらに5分間にわたりブレンドする。続いて得られた顆粒を、カプセル型パンチを用いて錠剤へと圧縮する。この最終混合物を約750mgの錠剤とする。実施例13〜18の錠剤の成分を下記の表20に示す。
【表20】
【0149】
最終産物の錠剤は、錠剤として重量750.0mgであり、硬度15Kpである。
【0150】
続いてコア錠剤を、例えば、全錠剤の重量を基準として3%、5%、7%および9%(それぞれ実施例15〜18)の重量増加となるまでEudragit L30D55 w/NaOHの水性分散液によりコーティングした。
【0151】
該水性分散液は以下の手順により調製した。
【0152】
1.0Nの水酸化ナトリウムを、メスフラスコ中の50mlの精製水に4.0gの水酸化ナトリウムを添加し5〜15分間攪拌して調製する。次いで精製水を必要量添加して再度攪拌する。
【0153】
タルク懸濁物を、クエン酸トリエチル9.31gを精製水202.54gに攪拌しながら添加する。攪拌を続けながらタルク22.2gを容器に3分間隔で添加する。容器を懸濁物が形成されるまで攪拌する。
【0154】
次いで、Eudragitを#40のメッシュ篩を通してEudragit懸濁物を調製し、計量して294.52gとする。滴下器(dropper)を用いて、1.0N水酸化ナトリウム溶液1.78gをEudragitに攪拌しながら添加する。この混合物を30〜60分間攪拌する。
【0155】
Eudragit懸濁物を攪拌しながら、タルク懸濁物を5分間かけて添加し、30〜60分間攪拌する。
【0156】
実施例13〜18の錠剤についての溶出試験を行った。自動USP溶出装置(Paddle type III、15CPM)中で250MLのバッファー(pH6)中で行い、放出された薬物の量をUV分析により調べた。結果を図8と以下の表21に示す。
【表21】
【0157】
図8および表21に示した結果から、コーティングの量(重量による)が増加するにつれて放出速度は低くなることは明らかである。
【0158】
実施例19〜20
Eudragit RS30D/RL30D(50/50)(アンモニオメタクリル酸コポリマー水性分散液)を有するコーティング錠剤の効果
実施例19および20においては、徐放性賦形剤は実施例1および2に示した工程により調製する。実施例19および20の徐放性賦形剤の成分を、以下の表22に示す。
【表22】
【0159】
その後、ジルチアゼム錠剤を以下の通り調製する。
【0160】
所望の量のジルチアゼム、フマル酸および徐放性賦形剤を、造粒機中で3分間にわたり低速で混合する。続いて好適な量の水を2分間隔で、低速で圧縮機を作動させながら添加する。次に得られたスラリーを、高速で作動させたチョッパーおよび圧縮機を用いて6分間にわたり顆粒化する(好適な顆粒を形成するために、さらに水と顆粒化時間を増してもよい)。続いて得られた顆粒を、液体ベッドドライヤーでLODが5%未満となるまで乾燥し、2000〜3000rpmで作動しているハンマーで金網#0050を用いて粉砕する。次に粉砕した顆粒をドデシル硫酸ナトリウムとともにV-Blender中に配置し、10分間にわたりブレンドする。続いて好適な錠剤化用潤滑剤(Pruv、ステアリルフマル酸ナトリウム、NF、Penwest Pharmaceuticals Co.より市販されている)を添加し、混合物をさらに3分間にわたりブレンドする。続いて得られた顆粒を、カプセル型パンチを用いて錠剤へと圧縮する。この最終混合物を約50mgの錠剤とする。
【0161】
実施例19および20の錠剤の成分を図9および下記の表23に示す。
【表23】
【0162】
最終産物の錠剤は、錠剤として重量750.0mgであり、硬度15Kpである。
【0163】
続いてコア錠剤を、全錠剤の重量を基準として8%の重量増加となるまでEudragit RS30D/RL30D(50/50)の水性分散液によりコーティングした。
【0164】
該水性分散液は以下の手順により調製した。
【0165】
100gのEudragit RSを100gのEudragit RLと混合してEudragit RS/RL懸濁物を調製する。
【0166】
タルク懸濁物を、クエン酸トリエチル12.0gを精製水338.0gに攪拌しながらゆっくりと添加する。攪拌を続けながらタルク50.0gを容器に3分間隔で添加する。容器を懸濁物が形成されるまで攪拌する。
【0167】
Eudragit懸濁物を攪拌しながら、タルク懸濁物を5分間かけて添加する。得られた混合物を30〜60分間攪拌し、40メッシュ篩を通してふるい分ける。
【0168】
実施例19および20の錠剤についての溶出試験を行った。自動USP溶出装置(Paddle type II、100rpm)中で900MLの0.1N HCL中で行い、放出された薬物の量をUV分析により調べた。結果を図9と以下の表24に示す。
【表24】
【0169】
図9および表24に示した結果から、コーティングが放出速度を低下させることは明らかである。
【0170】
実施例21〜23
エチルセルロースを有するコーティング錠剤の効果
実施例21〜23においては、徐放性賦形剤は実施例1および2に示した工程により調製する。実施例21〜23の徐放性賦形剤の成分を、以下の表25に示す。
【表25】
【0171】
その後、ジルチアゼム錠剤を以下の通り調製する。
【0172】
所望の量のジルチアゼム、フマル酸および徐放性賦形剤を、造粒機中で3分間にわたり低速で混合する。続いて好適な量の水を2分間隔で、低速で圧縮機を作動させながら添加する。次に得られたスラリーを、高速で作動させたチョッパーおよび圧縮機を用いて3分間にわたり顆粒化する(好適な顆粒を形成するために、さらに水と顆粒化時間を増してもよい)。続いて得られた顆粒を、液体ベッドドライヤーでLODが5%未満となるまで乾燥し、2000〜3000rpmで作動しているハンマーで金網#0050を用いて粉砕する。次に粉砕した顆粒をドデシル硫酸ナトリウムとともにV-Blender中に配置し、10分間にわたりブレンドする。続いて好適な錠剤化用潤滑剤(Pruv、ステアリルフマル酸ナトリウム、NF、Penwest Pharmaceuticals Co.より市販されている)を添加し、混合物をさらに3分間にわたりブレンドする。続いて得られた顆粒を、カプセル型パンチを用いて錠剤へと圧縮する。この最終混合物を約750mgの錠剤とする。
【0173】
実施例21〜23の錠剤の成分を下記の表26に示す。
【表26】
【0174】
最終産物の錠剤は、錠剤として重量750.0mgであり、硬度15Kpである。
【0175】
続いてコア錠剤を、全錠剤の重量を基準として4%および6%(それぞれ実施例22および23)の重量増加となるまでエチルセルロース/Opadry(80/20)の水性分散液によりコーティングした。該水性分散液は以下の手順により調製した。
【0176】
最初に、60gのOpadryを340gの水と好適な容器中で混合する。混合を続けながら、944gのエチルセルロースをOpadry分散液に添加する。得られた混合物を30〜60分間にわたり攪拌する。
【0177】
実施例1および2の錠剤についての溶出試験を行った。自動USP溶出装置(Paddle type III、15CPM)中で250MLのバッファー(pH6)中で行い、放出された薬物の量をUV分析により調べた。結果を図10と以下の表27に示す。
【表27】
【0178】
図10および表27に示した結果から、コーティングの量が増えるにつれて放出速度が低下することは明らかである。
【0179】
実施例24〜25
外部の顆粒への賦形剤の添加の効果
実施例24および25においては、徐放性賦形剤は実施例1および2に示した方法により調製する。実施例24および25の徐放性賦形剤の成分を、以下の表28に示す。
【表28】
【0180】
続いて、表29の成分と下記の工程により、錠剤を製造した。
【表29】
【0181】
実施例24の最終産物の錠剤は、錠剤として重量813.0mgであり、硬度15Kpである。
【0182】
実施例25の最終産物の錠剤は、錠剤として重量750.0mgであり、硬度15Kpである。
【0183】
実施例24および25の製剤の調製工程は下記の通りである。
【0184】
所望の量の(1)、(2)、および(4)を造粒機中に分散させ、低速で3分間混合する。圧縮機を低速で作動させながら、(7)を2分間隔で添加する。この混合物をチョッパーおよび圧縮機を高速で作動させて7.5分間にわたり顆粒化する(好適な顆粒を形成するために、さらに水と顆粒化時間を増してもよい)。顆粒化した混合物を、液体ベッドドライヤーでLODが5%未満となるまで乾燥する。乾燥顆粒を2000〜3000rpmで作動しているハンマーで金網#0050により粉砕する。粉砕した顆粒および(5)または(3と5)を、V-Blender中に配置し、10分間にわたりブレンドする。(6)をV-Blenderに添加し、5分間にわたりブレンドする。この最終混合物を、カプセル型パンチを用いて錠剤へと圧縮する。
【0185】
Eudragit L30D55 w/NaOHのコーティング分散液は以下の手順により調製した。
【0186】
A. 1.0Nの水酸化ナトリウム溶液を以下のように調製した。100mlのメスフラスコ中に4.0gの水酸化ナトリウムを添加し、次いで50mlの精製水およびマグネチックスターラーバーをフラスコ中に加え、フラスコの内容物を5〜15分間攪拌した。スターラーバーを取り出し、十分な容量として混合した。
【0187】
B. タルク懸濁物を以下のように調製した。精製水202.54gを好適な容器中に計り取り、クエン酸トリエチル9.31gを、精製水を攪拌しながらゆっくりと添加した。次にタルク22.22gを2分間隔で、混合物を攪拌しながら容器に添加した(混合物を懸濁物が形成されるまで攪拌した)。
【0188】
C. Eudragit L30D55懸濁物を以下のように調製した。Eudragit L30D55を#40のメッシュ篩を通し、篩い分けしたEudragit L30D55を294.52g計り取り、好適な容器中に配置し、滴下器(dropper)を用いて、1.0N水酸化ナトリウム溶液(ステップA)3.56gを、混合物を攪拌しながら添加した。この混合物を30〜60分間攪拌した。
【0189】
D. 最終コーティング懸濁物を以下のように調製した。Eudragit L30D55懸濁物(ステップC)にタルク懸濁物(ステップB)を5分間かけて添加しながら攪拌した。この混合物を30〜60分間攪拌した。
【0190】
錠剤を、全錠剤の重量を基準として4%の重量増加となるまでコーティングした。錠剤を、透明なゼラチンカプセル中にコーティングした該錠剤を配置してカプセル化した。
【0191】
実施例24の血漿中でのプロフィール
実施例24の錠剤について、2通りの無作為オープンラベルクロスオーバーデザインを用いて、健康なボランティア(各12名の被験者)においてin vivoの研究を行った。ボランティアに断食状態で投薬し、CARDIZEN CD(登録商標)と比較した。結果を図11と下記の表30に示す。
【表30】
【0192】
比
240mgの実施例24とCARDIZEN CDとの間の曲線の下部にある領域の比は1.16:1であった。240mgの実施例25とCARDIZEN CDとの間の平均Cmaxの比は1.16:1であった。
【0193】
結果
図11および実施例24では、二相性の(Bi-Modal)in vivoプラズマレベルが示され、CARDIZEN CDもまた、2種の異なる工程で調製されたビーズ製剤の混合により二相性のin vivoプラズマレベルを示した。
【0194】
実施例25の血漿中でのプロフィール
実施例25の錠剤について、2通りの無作為オープンラベルクロスオーバーデザインを用いて、健康なボランティア(各12名の被験者)においてin vivoの研究を行った。ボランティアに断食状態で投薬し、CARDIZEN CD(登録商標)と比較した。結果を図12と下記の表31に示す。
【表31】
【0195】
比
240mgの実施例25とCARDIZEN CDとの間の曲線の下部にある領域の比は1.16:1であった。240mgの実施例25とCARDIZEN CDとの間の平均Cmaxの比は1.26:1であった。
【0196】
結果
図12および実施例25では、二相性のin vivoプラズマレベルが示され、CARDIZEN CDもまた、2種の異なる工程で調製されたビーズ製剤の混合により二相性のin vivoプラズマレベルを示した。
【0197】
実施例26および27
異なる賦形剤の効果
実施例26および27において、徐放性賦形剤を実施例1および3で記述した方法により調製する。実施例25および26の徐放性賦形剤の成分を下記表32に示す。
【表32】
【0198】
表33の製剤
その後、ジルチアゼム錠剤を次の通り調製する。
【0199】
所望の量のジルチアゼム、フマル酸、および徐放性賦形剤を造粒機に配置し、低速で3分間にわたり混合する。圧縮機を低速で作動させながら、水を2分間隔で添加する(好適な顆粒を形成するために、さらに水および顆粒化時間を増しても良い)。続いて得られた顆粒を、LODが5%未満になるまで液体ベッドドライヤーで乾燥し、金網#0050を使用して2000〜3000rpmで作動しているハンマーを用いて粉砕する。次に、粉砕した顆粒をラウリル硫酸ナトリウムとともにV-Blender中に配置し10分間にわたりブレンドする。好適な錠剤化用潤滑剤(Pruv、ステアリルフマル酸ナトリウム, NF, Penwest Pharmaceuticals Co. より市販されている)を添加し、混合物をさらに5分間にわたりブレンドする。続いて得られた顆粒を、カプセル型パンチを用いて錠剤へと圧縮する。最終混合物を約750mgの錠剤とする。実施例26および27の錠剤の成分を下記の表33に示す。
【表33】
【0200】
実施例26の最終産物の錠剤は、錠剤として重量750.0mgであり、硬度15Kpである。
【0201】
実施例27の最終産物の錠剤は、錠剤として重量750.0mgであり、硬度15Kpである。
【0202】
次いで、実施例26および27の錠剤に対する溶出試験を、自動USP溶出装置(Paddle type III, 15CPM)中で250MLのバッファー(pH 6)中で実施し、放出された薬剤の量をUV分析により調べた。in-vitro溶出試験結果を図13および下記の表34に示す。
【表34】
【0203】
結論
実施例26は実施例25よりも遅い溶出プロフィールを有していた。
【0204】
結果
異なるグレードの賦形剤を使用することにより溶出速度を改変できる。
【0205】
実施例28〜29
製剤中のガム:薬剤比の効果
実施例28および29において、本発明の徐放性賦形剤を最初に調製し、次いで薬物(この場合、オキシブチニン)およびpH改変剤(この場合、コハク酸)を添加し、続いて最終混合物を錠剤に製剤化する。
【0206】
キサンタンガム、イナゴマメガム、デキストロース、および硫酸カルシウムを高せん断力のミキサー/造粒機の中に配合し、エチルセルロースをエタノールを含む容器中に配合し、エチルセルロース/エタノール混合物をキサンタンガム、イナゴマメガム、デキストロース、硫酸カルシウム混合物に配合し、そして好適な顆粒を形成するために顆粒化し、液体ベッドドライヤーで混合物を乾燥し、好適な顆粒を形成するために乾燥した物質を粉砕することによって、徐放性賦形剤を調製する。実施例28および29の徐放性賦形剤の成分を下記の表35に示す。
【表35】
【0207】
次に、所望の量のオキシブチニンおよびステアリルフマル酸ナトリウムを25メッシュの篩で篩分けし、篩分けしたオキシブチニンおよび徐放性賦形剤をV-blenderに配合し、10分間にわたりブレンドし、篩分けしたステアリルフマル酸ナトリウムを、オキシブチニンおよび徐放性賦形剤のブレンド混合物に添加してさらに5分間にわたりブレンドし、次いでブレンドされた最終の目的生成物を5/16”の丸型ツールを使用して、錠剤へと圧縮する。この最終混合物を約179.4mgの錠剤とする。実施例28および29の錠剤の成分を下記の表36および37に示す。
【表36】
【表37】
【0208】
最終産物の錠剤は、錠剤として重量179.4mgであり、硬度5Kpである。
【0209】
続いて、実施例28および29の錠剤に対する溶出試験を行った。in-vitro溶出試験結果を下記の表38に示す。
【表38】
【0210】
実施例28の製剤は1:5の薬剤:ガム比を有し、実施例29の製剤は1:8.3の薬剤:ガム比を有する。表38に示される結果から、オキシブチニンの放出速度は、製剤中の薬剤:ガム比が増加するにつれてより遅くなることが明らかである。
【0211】
実施例30および31
ガム:デキストロース比の効果
実施例30および31において、実施例28および29に記載の方法により徐放性賦形剤を調製する。実施例30および31の徐放性賦形剤の成分を下記の表39に示す。
【表39】
【0212】
その後、オキシブチニン錠剤を次のように調製する。
【0213】
所望の量のオキシブチニンおよびステアリルフマル酸ナトリウムを25メッシュの篩で篩分けし、篩分けしたオキシブチニンおよび徐放性賦形剤をV-blenderに配合し、10分間にわたりブレンドし、篩分けしたステアリルフマル酸ナトリウムをオキシブチニンおよび徐放性賦形剤のブレンド混合物に添加してさらに5分間にわたりブレンドし、次いでブレンドした最終の目的生成物を5/16”の丸型ツールを使用して錠剤へと圧縮する。この最終混合物を約179.4mgの錠剤とする。実施例30および31の錠剤の成分を下記の表40および41に示す。
【表40】
【表41】
【0214】
最終産物の錠剤は、錠剤として重量179.4mgであり、硬度5Kpである。
【0215】
続いて、実施例30および31の錠剤に対する溶出試験を行った。in-vitro溶出試験結果を下記の表38に示す。
【表42】
【0216】
表42に示された結果から、デキストロース量に対してガムの量が増加するにつれて、これに対応するオキシブチニン放出の減少が見られることが明らかである。
【0217】
実施例32〜35
コハク酸の効果
実施例32および33において、実施例28および29に記載の方法により徐放性賦形剤を調製する。実施例32および33の徐放性賦形剤の成分を下記の表43に示す。
【表43】
【0218】
その後、オキシブチニン錠剤を次のように調製する。
【0219】
所望の量のコハク酸、オキシブチニンおよびステアリルフマル酸ナトリウムを25メッシュの篩で篩分けし、篩分けしたコハク酸および徐放性賦形剤をV-blenderに配合し、10分間にわたりブレンドし、篩分けしたオキシブチニンを、コハク酸および徐放性賦形剤のブレンド混合物に添加してさらに5分間にわたりブレンドし、篩分けしたステアリルフマル酸ナトリウムを、オキシブチニン、コハク酸および徐放性賦形剤のブレンド混合物に添加してさらに5分間にわたりブレンドし、次いでブレンドした最終の目的生成物を5/16”の丸型ツールを使用して錠剤へと圧縮する。実施例32の最終混合物を約251.0mgの錠剤とし、実施例33の最終混合物を約296.0mgの錠剤とする。実施例32および33の錠剤の成分を下記の表44および45に示す。
【表44】
【0220】
最終産物の錠剤は、錠剤として重量251.0mgであり、硬度8Kpである。
【表45】
【0221】
最終産物の錠剤は、錠剤として重量296.0mgであり、硬度8Kpである。
【0222】
続いて、実施例32および33の錠剤に対する溶出試験を行った。in-vitro溶出試験結果を下記の表46に示す。
【表46】
【0223】
表46に示される結果から、コハク酸の添加が薬物の溶出性を促進し、それゆえ、放出速度を増加させることは明らかである。
【0224】
実施例34および35において、実施例28および29に記載の方法により徐放性賦形剤を調製する。実施例34および35の徐放性賦形剤の成分を下記の表47に示す。
【表47】
【0225】
その後、オキシブチニン錠剤を次のように調製する。
【0226】
所望の量の徐放性賦形剤、コハク酸、およびオキシブチニンを造粒機中に配合する。それらを低速で圧縮機によって、チョッパーブレイド(the chopper blade)はオフにした状態で3分間にわたり乾燥混合する。水を1分間隔で添加し、次いで混合物を高速で3分間にわたり顆粒化する(好適な顆粒を形成するために、さらに水および顆粒化時間を増しても良い)。次に、混合物をLODが5%未満になるまで液体ベッドドライヤーで乾燥する。乾燥した顆粒を、2000〜3000rpmで作動しているブレードで粉砕する。粉砕した顆粒およびステアリルフマル酸ナトリウムをV-Blender中に配置し10分間にわたりブレンドする。次いでブレンドした混合物を5/16”の丸型ツールを使用して錠剤へと圧縮する。実施例34の最終混合物を約296.0mgの錠剤とし、実施例35の最終混合物を約266.0mgの錠剤とする。実施例34および35の錠剤の成分を下記の表48および49に示す。
【表48】
【0227】
最終産物の錠剤は、錠剤として重量296.0mgであり、硬度8Kpである。
【表49】
【0228】
最終産物の錠剤は、錠剤として重量266.0mgであり、硬度8Kpである。
【0229】
続いて、実施例34および35の錠剤に対する溶出試験を行った。in-vitro溶出試験結果を下記の表50に示す。
【表50】
【0230】
表50に示される結果から、製剤中のコハク酸の量が多いほど、放出速度が速くなることは明らかである。
【0231】
実施例36
エチルセルロース(SURELEASE(登録商標))/OPADRY(登録商標)(80/20)水性分散液による錠剤のコーティングの効果
エチルセルロース/Opadryコーティングで製剤する方法は次の通りである。
【0232】
先ず初めに、好適な容器に340gの水を量り、混合しながら水の中にOpadryを60g添加する。混合を続ける。Opadry分散液を混合しながら、933gのエチルセルロース分散液(Surelease)を添加し、30〜60分間攪拌する。最終的な分散液を用いて、錠剤の総重量に基づき3〜5%の重量増加まで錠剤をコーティングする。
【0233】
実施例36においては、実施例28および29に記載の方法により徐放性賦形剤を調製する。実施例36の徐放性賦形剤の成分を下記の表51に示す。
【表51】
【0234】
その後、オキシブチニン錠剤を次のように調製する。
【0235】
所望の量の徐放性賦形剤、コハク酸、およびオキシブチニンを造粒機中に配合する。それらを低速で圧縮機によって、チョッパーブレイドはオフにした状態で3分間にわたり乾燥混合する。水を1分間隔で添加し、次いで、混合物を高速で3分間にわたり顆粒化する(好適な顆粒を形成するために、さらに水および顆粒化時間を増しても良い)。次に、混合物をLODが5%未満になるまで液体ベッドドライヤーで乾燥する。乾燥した顆粒を、2000〜3000rpmで作動しているブレードで粉砕する。粉砕した顆粒およびステアリルフマル酸ナトリウムをV-Blender中に配置し10分間にわたりブレンドする。次いでブレンドした混合物を5/16”の丸型ツールを使用して錠剤へと圧縮する。実施例36の最終混合物を約296.0mgの錠剤とする。実施例36の成分を下記の表52に示す。
【表52】
【0236】
最終産物の錠剤は、錠剤として重量296.0mgであり、硬度8Kpであった。
【0237】
続いて、実施例36の錠剤に対する溶出試験を行った。in-vitro溶出試験結果を下記の表53に示す。
【表53】
【0238】
表53に示される結果から、コーテイングの量(重量による)が増加するにつれて、放出速度は減少することは明らかである。
【0239】
実施例37
フマル酸の効果
実施例37において、実施例28および29に記載の方法により徐放性賦形剤を調製する。実施例37の徐放性賦形剤の成分を下記の表54に示す。
【表54】
【0240】
最終産物の錠剤は、213.7mgの錠剤重量を有する。
【0241】
続いて、実施例37の錠剤に対する溶出試験を、ディトロパン XLと比較して行った。in-vitro溶出試験結果を下記の表55に示す。
【表55】
【0242】
pH改変剤にフマル酸を含む場合における他のオキシブチニン製剤を下記の表56に示す。
【表56】
【0243】
上記で与えられた実施例は限定的であることを意味しない。本発明の多くの他の変形例が当業者には明らかであり、それらは添付した特許請求の範囲の範囲内にあるものとして意図されている。
【図面の簡単な説明】
【0244】
【図1】実施例1および2に関する溶出(経時平均溶出%)のグラフを示す。
【図2】実施例3および4に関する溶出(経時平均溶出%)のグラフを示す。
【図3】実施例5および6に関する溶出(経時平均溶出%)のグラフを示す。
【図4】実施例7および8に関する溶出(経時平均溶出%)のグラフを示す。
【図5】実施例9および10に関する溶出(経時平均溶出%)のグラフを示す。
【図6】実施例11および12に関する溶出(経時平均溶出%)のグラフを示す。
【図7】実施例11および12に関する経時放出速度(%)のグラフを示す。
【図8】実施例13および18に関する溶出(経時平均溶出%)のグラフを示す。
【図9】実施例19および20に関する溶出(経時平均溶出%)のグラフを示す。
【図10】実施例21〜23に関する溶出(経時平均溶出%)のグラフを示す。
【図11】実施例24と参照基準(Cardizem CD 240mg)に関する経時平均ジルチアゼム血漿濃度(ng/ml)のグラフを示す。
【図12】実施例25と参照基準(Cardizem CD 240mg)に関する経時平均ジルチアゼム血漿濃度(ng/ml)のグラフを示す。
【図13】実施例26および27に関する溶出(経時平均溶出%)のグラフを示す。
【図14】実施例37と参照基準(ディトロパン XL)の溶出(経時平均溶出%)を比較したグラフを示す。
【技術分野】
【0001】
本発明は、経口用徐放性固体剤形に関する。
【背景技術】
【0002】
発明の背景
制御放出製品の利点は医薬分野ではよく知られ、この利点には、比較的長い期間にわたって薬物の所望血中レベルを維持し、同様の効果を達成するために必要な投与回数を減らすことによって患者のコンプライアンスを高める能力が含まれる。これらの利点は多様な方法によって達成されてきた。例えば、制御放出薬における使用に関して種々のヒドロゲルが記載されており、この中には合成物もあるが、大部分は半合成または天然起源のものである。合成物質および非合成物質の両方を含むものはわずかしかない。しかしながら、この系の中には、特殊な工程および製造装置を必要とするものもあり、さらにこれらの系には可変性の薬物放出に影響を受けやすいものもある。
【0003】
経口制御放出送達系は、放出速度および放出プロフィールが生理学および時間治療学の必要要件と一致しうるように理想的に適合させる必要がある。
【0004】
ほとんどの場合、経口送達系の放出速度は、放出の機構、例えば0次、1次、2次、擬1次機構などに従って分類されているが、多くの医薬化合物が他の複雑な機構により薬物を放出する。
【0005】
1次機構は、反応速度が反応基質濃度に依存する(従って、反応物の1次作用率(first power)に依存する)状況を指す。かかる機構においては、基質は直接に1つ以上の生成物に分解する。
【0006】
2次機構は、実験的に決定した反応速度が2つの反応物のそれぞれの濃度と比例関係にあるか、または1つの反応物の濃度の2次作用率と比例関係にある場合に起こる。
【0007】
擬1次反応は、一般的には、1次機構が支配するように挙動する2次反応として定義され、例えば、他の物質と比較して1つの反応物質の量が過剰に存在するかまたは一定濃度に維持されるように操作される場合に、この擬1次反応は起こる。このような状況においては、反応速度は操作される物質により決定される。
【0008】
0次機構は、反応速度が反応物質濃度に依存しない(従って、反応物の0次作用率(zero power)に依存する)状況を指し、律速因子は反応物質(例えば薬物)濃度以外のものである。0次機構の律速因子は、例えば、反応物質の可溶性または光化学反応の光強度でありうる。
【0009】
しかし上述のように、多くの化学反応は、0次、1次または2次反応などの単純な反応ではなく、2つ以上の反応の組み合わせを包含する。
【0010】
さらに、他の因子、例えば、温度、pH、食物影響変動性(food effect variability)、イオンおよびイオン強度依存性、粘度依存性、コロージョン/エロージョン(侵食)変動性、内容物均一性(content uniformity)問題、流動性および重量均一性問題、維持容量(carrying capacity)および物理的強度問題、加水分解、光化学分解、成分間相互作用(薬物と、製剤中の他の成分、例えば緩衝剤、保存剤などとの相互作用など)、低誘電率の溶媒の濃度(反応が反対の電荷のイオンを含む場合)などが反応速度に影響を及ぼす可能性がある。
【0011】
多くの制御放出製剤および徐放性製剤が既知であるが、ある程度〜高度に可溶性の薬物に関しては、そのような製剤中に配合する際に製剤上の困難性を有する。可溶性薬物を含有する徐放性製剤は、「用量ダンピング(dose dumping)」の影響を受けやすい。この事象は、有効成分の放出が遅延するが、放出が開始した時にその速度が極めて高い場合に起こる。この放出速度の上昇は、治療効果の低減または毒性の増大をもたらす可能性のある血液の血漿変動に関連している。これらは、徐放性製剤が解決すると考えられる問題と同じである。
【0012】
さらに、特定の徐放性製剤が、可溶性〜高度に可溶性の薬物の所望の持続放出を提供するかどうかを容易に推測できない場合もある。一般的に、摂取された場合にそのような薬物の所望の生物学的利用能を提供する徐放性製剤を得るためにかなりの実験を行う必要があることがわかっている。
【0013】
可溶性〜高度に可溶性の薬物の所望の持続放出を提供する制御放出製剤と関連した予測不可能性を補うために、二相性または多相性反応動力学を有する製剤を提供することが望ましいと考えられる場合もある。二相性放出または多相性放出は、開始速度が高く、続いて剤形が小腸の上部を通過する、吸収が最大となるところでより緩慢な速度であり、最後に剤形が小腸のより一層終末を通過する、吸収が小腸上部の場合よりも少ないところで再び速度が高くなることを特徴としうる。
【0014】
二相性放出はいくつかの理由で利点を有すると考えられ、そのような理由としては、限定するものではないが、二相性放出によって、製造者が(製剤が胃内に位置する場合に)迅速な作用開始によって胃腸管における薬物の吸収速度の変化を補償することができるという事実、ならびに(例えば製剤が大腸に位置する場合に)比較的迅速な放出速度によって比較的緩慢な吸収を補償することができるという事実、が挙げられる。
【0015】
現在のところ、二相性放出製剤は種々のいくつかの方法で提供されている。
【0016】
例えば、国際特許公開番号WO87/00044号には、二相性放出の特徴を有すると主張されている治療用製剤が記載されている。WO87/00044号には、固体剤形中の治療上有効な薬物のための担体基剤物質が記載され、この剤形は、薬物の初期の急速な放出、続いて一定期間にわたる実質的に一定の放出速度、そしてその後の、前に観察された一定速度よりも大きい放出速度を特徴とする二相性制御放出プロフィールを提供すると主張されている。担体ベースの物質は、メトキシ含量が19〜30%、ヒドロキシプロポキシ含量が4〜12%、粘度が40〜19,000cps、平均分子量が20,000〜140,000である二相性ヒドロキシプロピルメチルセルロースエーテルを含有し、これは該公報に記載のアッセイ法によって二相性放出プロフィールを示す。二相性ヒドロキシプロピルメチルセルロースは、有効成分および所望の薬物放出の期間に依存して総配合物の5〜99重量%を含有する。
【0017】
A. C. Shahら(”Gel-Matrix Systems Exhibiting Bimodal Controlled Release For Oral Drug Delivery”, Journal of Controlled Release, 9(1989), pp. 169-175)はさらに、特定の「タイプ」のヒドロキシプロピルメチルセルロースエーテルが二相性薬物放出プロフィールを示すことが見出されたことを報告している。しかしながらこの研究においては、一連のヒドロキシプロピルメチルセルロースエーテルポリマーが、ポリマー−薬物マトリックス錠剤からの二相性および非二相性放出プロフィールを示し、これによりポリマーの供給者(従って、製造方法、イオン構成、置換基の分布の変動、または分子量分布の変動)に左右されると考えられることがわかった。
【0018】
P. Giunchediら(”Ketoprofen Pulsatile Absorption From ‘Multiple Unit’ Hydrophilic Matrices” International Journal of Pharmaceutics, 77(1991), pp. 177-181)は、ケトプロフェンの経口用遅延放出製剤を記載している。該製剤は、それぞれが50mgの薬物を含有する同じ組成物の4種の親水性マトリックスから構成され、ヒドロキシプロピルメチルセルロース(Methocel. RTM.)を用いて調製され、そしてゼラチンカプセル中に入れられた複合ユニット製剤を含む。拍動血漿レベル(投与の2時間後と8時間後に2つのピークがある)が得られるものと主張されたが、in vitro試験ではかなり一定な薬物放出が示された。
【0019】
U. Conteら(”A New Ibuprofen Pulsed Release Oral Dosage Form”, Drug Development And Industrial Pharmacy, 15(14-16), pp 2583-2596 (1989))は、2つの層が投与量の薬物を含み、中間層が薬物層を分離させる制御要素として作用する3層錠剤から律動的(pulsed)放出パターンが得られたことを報告している。この制御要素は、水膨張性ポリマー(ヒドロキシプロピルメチルセルロース)の混合物である。外側被膜として不浸透性ポリマーで錠剤をコーティングしている。超崩壊剤(デンプングリコレートナトリウムおよび架橋したポリビニルピロリドン)を薬物層に含有させている。
【0020】
K. A. Kahnら(”Pharmaceutical Aspects And In-Vivo Performance Of Brufen Retard-An Ibuprofen SR Matrix Tablet”, Proced. Intern. Symp. Control. Rel. Bioact. Mater., 18(1991), Controlled Release Society, Inc.)は、二相性放出パターンを示すと主張されているイブプロフェン800mgを含有する製剤を記載している。ここで使用された放出遅延剤はキサンタンガムである。成分は、適切なキサンタンガム含量と混合され、その後打錠して錠剤にし、被膜コーティングされている。含有するキサンタンガムの量と薬物放出速度との間に逆相関があった。薬物粒子サイズおよび錠剤1つ当たりの被膜コーティングの量の増大は、薬物放出速度に有意に影響を及ぼさなかった。キサンタンガムの粒子サイズの増大がより顕著な突発性(burst)作用を引き起こし、被膜コーティングの適用によってこの突発性作用が解消された。薬物放出の迅速な開始は、ゲル層形成の変化に関連していると仮定されており、この場合、粒子が大きくなるほどゆっくりとゲル形成し、そして凝集マトリックスが形成しうる前に徐々に崩壊する(slough)。
【0021】
本発明者らの米国特許第4,994,276号、第5,128,143号および第5,135,757号(参照により本明細書に引用する)において、本発明者らは、共働作用を示す様々な大きさの多糖(例えば、キサンタンガムなどのヘテロ多糖と、該ヘテロ多糖と架橋することができる多糖ガム、例えばイナゴマメ(locust bean)ガムとの組み合わせ)を含む制御放出賦形剤を報告した。これは、薬物および潤滑剤粉末の添加の後に直圧縮を行うか、慣例的な湿式顆粒化を行うか、またはこれらの2つの組み合わせを行うかのいずれかで経口用固体剤形に加工することができる。この製剤からの薬物の放出は、0次機構または1次機構に従って進行するものである。
【0022】
本発明者らの米国特許第5,472,711号および第5,478,574号(参照により本明細書に引用する)において、本発明者らは、有効量の薬学的に許容される界面活性剤を上述の賦形剤と混合することによって、in vitroにおいて治療上有効な薬物の多相性または二相性制御放出を提供することができる製剤を報告している。
【0023】
本発明で使用する高度に可溶性の薬物の例としては、ジルチアゼム、すなわちカルシウムアンタゴニスト活性を有するベンゾチアジン誘導体である。ジルチアゼムは、高血圧症および扁桃炎の治療に広く使用されている。従って、許容可能な放出プロフィールを示す徐放性ジルチアゼムの製造には大いに関心が寄せられていた。例えば、米国特許第4,894,240号および第5,364,620号(Geogheganら)は、1日1回の投与に適したジルチアゼムペレット製剤を記載している。この製剤は、周りを不溶性多層膜が包囲する、有機酸と会合したジルチアゼムのコアを含む。該膜によって、ペレットから投与後24時間にわたって制御吸収が可能な速度でジルチアゼムが放出される。
【0024】
徐放性ジルチアゼム製剤の製造に関して他の方法が従来技術において記載されている。例えば、米国特許第5,419,917号(Chenら)は、薬学的に有効なイオン性化合物を用いてヒドロゲルからのジルチアゼムの放出速度を制御する組成物を記載している。
【0025】
本発明で使用する高度に可溶性の薬物の他の例は、オキシブチニンである。オキシブチニンは泌尿器疾患(例えば過運動性膀胱(hyperactive bladder))の治療に広く使用されている。本発明者らの米国特許第5,399,359号には、ゲル化剤を含む徐放性マトリックス中に分散させた薬学的に有効な量のオキシブチニン、有効量の薬学的に許容される水溶性カチオン性架橋剤(製剤が周囲の液体、例えば胃腸液に晒された際にゲル化剤と架橋する)、及び不活性希釈剤とを含有するオキシブチニン徐放性製剤が開示されている。
【発明の開示】
【発明が解決しようとする課題】
【0026】
発明の目的
本発明の目的は、可溶性〜高度に可溶性の治療上有効な薬物のための生体利用可能な徐放性製剤を提供することである。
【0027】
本発明のさらなる目的は、可溶性〜高度に可溶性の薬物の多相性または二相性制御放出を示しうる製剤を提供することである。
【0028】
本発明のさらなる目的は、可溶性〜高度に可溶性の薬物の生体利用可能な徐放性製剤の製造方法を提供することである。
【0029】
本発明のさらに他の目的は、可溶性〜高度に可溶性の治療上有効な薬物の経口用徐放性固体剤形の製造に使用しうる徐放性マトリックスを提供することである。
【0030】
本発明のさらなる目的は、薬物と混合すると、治療上有効な薬物血中レベルを例えば12または24時間にわたって提供する徐放性製剤を得るのに適した徐放性マトリックスを提供することである。
【0031】
本発明のさらなる目的は、市販の徐放性製剤、例えばCardizem CDと同様の血漿プロフィールを示す、ジルチアゼムの徐放性マトリックス製剤を提供することである。
【0032】
本発明のさらなる目的は、市販の徐放性製剤、例えばディトロパン(Ditropan) XLと同様の血漿プロフィールを示す、オキシブチニンの徐放性マトリックス製剤を提供することである。
【課題を解決するための手段】
【0033】
発明の概要
上述の目的および他の目的は、部分的には、ゲル剤を含む剤形にpH改変剤を組み込むことによって該剤形からの薬物の放出が促進され、生物学的利用能が高まるという驚くべき発見に関する本発明により達成される。
【0034】
特定の実施形態において、経口用徐放性固体剤形には、可溶性が約10g/lより大きい治療上有効な量の薬物;pH改変剤;およびゲル化剤を含む徐放性マトリックスが含まれる。該ゲル化剤は、ヘテロ多糖ガムと、周囲の液体に晒された際に該ヘテロ多糖ガムを架橋することができるホモ多糖ガムとを含む。好ましくは、該剤形は、少なくとも約12時間、好ましくは少なくとも約24時間にわたって薬物の持続放出を提供する。
【0035】
特定の実施形態において、上記剤形には、a)薬物の多相性放出を提供しうる薬学的に許容される界面活性剤;b)例えば単糖類、二糖類、多価アルコールまたはこれらの混合物より選択される不活性希釈剤;c)ゲル化剤の水和を緩慢にする疎水性物質;および/またはd)該徐放性製剤が周囲の液体に晒された際に形成されるゲルからの放出速度を変更するのに適した有効量の薬学的に許容されるイオン化ゲル強度増強剤、がさらに含まれる。好ましい実施形態において、本発明の製剤は錠剤を包含する。
【0036】
本発明の好ましい実施形態において、薬物とゲル化剤との比は、好ましくは約10:1〜約1:10、より好ましくは約5:1〜約1:5、最も好ましくは約1.25:1〜約2:1である。
【0037】
また本発明は、高度に水溶性の薬物の徐放性製剤の製造方法に関する。該方法には、ヘテロ多糖ガムと周囲の液体に晒された際に該ヘテロ多糖ガムを架橋することができるホモ多糖ガムとを含有するゲル化剤を含み、そして任意にイオン化ゲル強度増強剤、任意に不活性希釈剤、および任意に疎水性物質を含んでいてもよいマトリックスを調製し、その後で可溶性〜高度に可溶性の薬物、pH改変剤、そして任意に薬学的に許容される界面活性剤を添加することが含まれる。その後得られた混合物を、薬物とゲル化剤との比が約10:1〜約1:10、より好ましくは約5:1〜約1:5、最も好ましくは約1.25:1〜約2:1である生成物が得られ、錠剤が周囲の液体に晒された際にゲルマトリックスが形成され、そして各錠剤が治療上有効な量の薬物を含有するように、打錠する。得られる錠剤は、少なくとも約12時間、好ましくは約24時間にわたって治療上有効な薬物血中レベルを提供する。
【0038】
本発明はさらに、上述の経口用固体剤形を経口投与することにより患者を治療する方法に関する。
【0039】
本発明の特定の好ましい実施形態において、上記マトリックスは、例えば約10〜約99重量%のゲル化剤と、約0〜約20重量%のイオン化ゲル強度増強剤と、約1〜約89重量%の不活性医薬希釈剤と、約1〜約20%の疎水性物質とを含有する、予め顆粒化した徐放性賦形剤から調製しうる。
【0040】
他の好ましい実施形態において、マトリックスと不活性希釈剤との混合物は、該マトリックスを破壊することなくマトリックスの水和を緩慢にするのに十分な量の疎水性物質の分散液または溶液を用いて顆粒化し、その後で薬物を添加する。
【0041】
本発明の他の好ましい実施形態において、薬物の第1部を賦形剤の顆粒化の際に導入し、薬物の第2部を外部で顆粒化して(extragranularly)導入するか、または顆粒化工程の後に導入する。このような実施形態は薬物の初期の急速な放出を提供する。
【0042】
好ましい実施形態において、薬物は高度に可溶性である、すなわち、可溶性が約100g/lより大きい。
【0043】
他の好ましい実施形態において、薬物は、カルシウムチャネル遮断剤、好ましくはベンゾチアジン、最も好ましくはジルチアゼムまたはその薬学的に許容される塩を含む。
【0044】
他の好ましい実施形態において、薬物は、鎮痙薬、好ましくはオキシブチニンまたはその薬学的に許容される塩を含む。
【0045】
「徐放性(持続放出)」とは、本発明の目的のために、治療上有利な薬物血中レベル(ただし毒性レベル以下)を長い時間、例えば少なくとも約12時間または少なくとも約24時間にわたって維持するような制御速度にて、製剤から治療上有効な薬物が放出されることを意味する。
【0046】
「生体利用可能」とは、本発明の目的のために、治療上有効な薬物が徐放性製剤から吸収され、体内の薬物作用を意図する部位において、好ましくは参照基準の80%以内(AUCの比較を基準とする)で利用可能となることを意味する。
【0047】
「可溶性」とは、治療上有効な薬物が1リットル当たり約10グラム(g/l)より大きい水溶性を有することを意味する。
【0048】
「高度に可溶性」とは、治療上有効な薬物が1リットル当たり約100グラム(g/l)より大きい水溶性を有することを意味する。
【0049】
「周囲の液体」という用語は、本発明の目的のために、例えば、水溶液または胃腸液を包含する液体を意味する。
【0050】
「pH改変剤」という用語は、本発明の目的のために、薬物のイオン化を低減させることにより、ヒドロゲルマトリックスから溶液中への薬物の放出を促進するあらゆる物質を意味する。
【0051】
「Cmax」という用語は、本発明の目的のために、本発明の剤形の投与後に達成される薬物の最大血漿濃度を意味する。
【0052】
「Tmax」という用語は、本発明の目的のために、剤形の投与後から薬物のCmaxが達成されるまでの経過時間を意味する。
【0053】
「W50」という用語は、本発明の目的のために、Cmaxの50%の高さにおける血漿濃度曲線の幅で測定される時間の長さを意味する。
【0054】
本発明の目的のために、剤形は二相性薬物動力学を有しうるものであり、それゆえ開示された剤形に関して複数のCmax、TmaxおよびW50が存在しうる。
【発明を実施するための最良の形態】
【0055】
本発明に添付した図面は、本発明の実施形態を例示するものであり、特許請求の範囲により包含される本発明の範囲を限定するものではない。
【0056】
詳細な説明
本発明の徐放性マトリックスは、共働作用を示すヘテロ多糖およびホモ多糖両方からなるゲル化剤を含みうる様々な大きさの賦形剤でありうる(既に本発明者らの米国特許第4,994,276号、第5,128,143号および第5,135,757号において報告されている)。その共働作用とは、例えば、2種以上の多糖ガムの組み合わせによって、いずれかのガムを単独で用いて予測されるよりも高い粘度および急速な水和が得られ、形成するゲルがより急速に形成され、かつ堅固であることが挙げられる。
【0057】
本発明で使用する「ヘテロ多糖」という用語は、2種以上の糖単位を含有する水溶性多糖と定義され、該ヘテロ多糖は分枝鎖状またはらせん状立体構造を有し、優れた水深性(water-wicking)および大きな沈降濃縮特性を有する。
【0058】
特に好ましいヘテロ多糖は、高分子量(>106)のヘテロ多糖であるキサンタンガムである。他の好ましいヘテロ多糖には、キサンタンガムの誘導体(デアシル化キサンタンガムなど)、カルボキシメチルエーテル、およびプロピレングリコールエステルが挙げられる。
【0059】
本発明で使用するホモ多糖ガムはヘテロ多糖と架橋することができ、例えばガラクトマンナン、すなわち単にマンノースとガラクトースからなる多糖が挙げられる。ガラクトマンナンは、未置換マンノース領域の割合が高くなるほど、よりヘテロ多糖と相互作用することが見出されている。イナゴマメガムは、ガラクトースに対するマンノースの割合が高く、グアーおよびヒドロキシプロピルグアーなどの他のガラクトマンナンと比較して特に好ましい。
【0060】
本発明の制御放出製剤の制御放出特性は、様々な大きさの多糖物質の約10〜約90重量%以上の量のヘテロ多糖ガムは、許容される遅延放出生成物を提供するが、ヘテロ多糖ガムとホモ多糖物質との比が約1:1.5の場合に最適であり得る。本発明によれば、水溶液に晒された際に共働作用を示すことが知られている任意のホモ多糖ガムとの組み合わせを使用しうる。本発明のガムの組み合わせに関して存在するタイプの共働作用は、2種の同一な多糖間または2種のヘテロ多糖間で起こる可能性もある。本発明で使用しうる他の許容されるゲル化剤には、当技術分野で公知のゲル化剤が含まれる。例としては、植物ガム(アルギネート、カラゲーニン、ペクチン、グアーガムなど)、加工デンプン、ヒドロキシプロピルメチルセルロース、メチルセルロース、および他のセルロース物質(ナトリウムカルボキシメチルセルロースおよびヒドロキシプロピルセルロースなど)が挙げられる。このリストは限定を意味するものではない。
【0061】
徐放性賦形剤の不活性希釈剤は、好ましくは薬学的に許容される糖(単糖類、二糖類もしくは多価アルコールなど)、および/または上記の任意の混合物を含む。適切な不活性医薬充填剤の例としては、スクロース、デキストロース、ラクトース、微晶質セルロース、フルクトース、キシリトール、ソルビトール、デンプン、これらの混合物などが挙げられる。しかしながら、可溶性医薬充填剤、例えばラクトース、デキストロース、スクロース、またはこれらの混合物などを使用するのが好ましい。あるいはまた、不活性希釈剤または充填剤は、以下で記載する既製の直圧縮希釈剤(direct compression diluent)を含んでもよい。
【0062】
例えば、湿式顆粒化工程を行うことなく、徐放性賦形剤の成分を乾式混合することが可能である。この方法は、例えば有効成分を徐放性賦形剤の成分に直接添加する際に湿式顆粒化を行うべき場合に利用しうる。一方、この方法は、湿式顆粒化工程を全く考慮しない場合にも使用してよい。混合物を湿式顆粒化工程を行わずに製造すべき場合であって、最終混合物を打錠すべき場合には、不活性希釈剤の全部または一部が既製の直圧縮希釈剤を含有することが好ましい。かかる直圧縮希釈剤は、医薬分野で広く使用されており、多様な商業源から入手可能である。そのような既製の直圧縮賦形剤の例としては、Emcocel(登録商標)(微晶質セルロース、N.F.)、Emdex(登録商標)(デキストレート、N.F.)、およびTab-Fine(登録商標)(スクロース、フルクトースおよびデキストロースを含むいくつかの直圧縮糖)が挙げられ、これらは全てPenwest Pharmaceuticals Co., Patterson, New Yorkから市販されている。他の直圧縮希釈剤としては、Sheffield Chemical, Union, N.J. 07083からの無水ラクトース(ラクトースN.F.、無水直打錠);Degussa, D-600 Frankfurt(Main) GermanyからのElcems(登録商標)G-250(粉末セルロース、N.F.);Foremost Whey Products, Banaboo, WI 53913からのFast-Flo Lactose(登録商標)(ラクトース、N.F.、噴霧乾燥);Grain Processing Corp., Muscatine, IA 52761からのMaltrin(登録商標)(凝集マルトデキストリン);Roquet Corp., 645 5th Ave., New York, N.Y. 10022からのNeosorb 60(登録商標)(ソルビトール、N.F.、直圧縮);Ingredient Technology, Inc., Pennsauken, N.J. 08110からのNu-Tab(登録商標)(圧縮可能な糖、N.F.);GAF Corp., New York, N.Y. 10020からのPolyplasdone XL(登録商標)(Crospovidone, N.F.、架橋されたポリビニルピロリドン);Generichem Corp., Little Falls, N.J. 07424からのPrimojel(登録商標)(デンプングリコレートナトリウム、N.F.、カルボキシメチルデンプン);Penwest Pharmaceuticals Co., Patterson, N.Y. 10512からのSolka Floc(登録商標)(セルロース塊);Foremost Whey Products, Baraboo, WI 53913およびDMV Corp., Vehgel, HollandからのSpray-dried lactose(登録商標)(ラクトース、N.F.、噴霧乾燥);ならびにColorcon, Inc., West Point, PA 19486からのSta-Rx 1500(登録商標)(Starch 1500)(プレゲル化デンプン、N.F.、圧縮可能)が挙げられる。
【0063】
一般的には、上記製剤は、例えば、湿式顆粒化し、噴霧乾燥したラクトースにより直圧縮希釈剤として、または当業者に公知の方法により予め混合してある直圧縮希釈剤として調製しうる。本発明の目的のために、これらの特別に処理した不活性希釈剤を、「直圧縮」不活性希釈剤と呼ぶことにする。
【0064】
特定の実施形態において、徐放性賦形剤の成分は既製品であってもよい。しかしながら他の実施形態においては、有効な薬物を賦形剤成分に添加し、混合物を溶融顆粒化して顆粒を形成させうる。最終的に、界面活性剤を使用する場合には、可溶化または分散化ジルチアゼムまたはオキシブチニンを含む界面活性剤を成分の混合物に直接添加しうる。
【0065】
本発明のさらなる実施形態において、本発明の徐放性医薬賦形剤と共に使用する直圧縮不活性希釈剤は、米国特許出願第08/370,576号(1995年1月9日出願、発明の名称「Pharmaceutical Excipient Having Improved Compressibility」、発明者J. Staniforth, B. SherwoodおよびE. Hunter、参照によりこの全文を本明細書に引用する)に開示されているような増強(augmented)微晶質セルロースである。その開示されている増強微晶質セルロースは、Penwest Pharmaceuticals Co.より「Prosolv」という商標で市販されている。
【0066】
有効量の薬学的に許容される界面活性剤は、上述の賦形剤の成分に添加してもよいし、または薬物を添加する時点で添加してもよく、それにより薬物の生物学的利用能が高まる。適切な界面活性剤の例としては、固体剤形の約15重量%までのドキュセートナトリウムである。特に好ましい界面活性剤は、固体剤形の約15重量%までのラウリル硫酸ナトリウムである。
【0067】
一実施形態において、界面活性剤は水などの適切な溶媒中に溶解し、その後、徐放性賦形剤と薬物とを混合した混合物に添加する。これにより、界面活性剤は賦形剤の粒子を湿潤させ、その結果、溶媒が蒸発した場合には、沈殿する薬物の粒子が小さくなり凝集しない。賦形剤中に適切に微細にかつ均質に分散した薬物および界面活性剤の顆粒が得られる。
【0068】
本発明の特定の実施形態において、例えば薬物がジルチアゼムまたはオキシブチニンである場合には、界面活性剤を、例えば最終生成物の約1〜約5重量%、または約1〜15重量%の量で包含させる。しかしながら包含させる界面活性剤の上限は15%よりも高くてよい。1つの限定因子は、最終生成物が薬学的に許容される製剤を提供すべきことである。例えば錠剤の場合には、包含される界面活性剤量の上限は、薬学的に許容される錠剤、例えば摩損度が約1%未満で硬度が6〜8kgである錠剤の製造法により決定される。
【0069】
本発明で使用しうる界面活性剤には、一般的に、薬学的に許容されるアニオン性界面活性剤、カチオン性界面活性剤、両性(両親媒性/両染性(amphophilic))界面活性剤、および非イオン性界面活性剤が含まれる。好適な薬学的に許容されるアニオン性界面活性剤には、例えば、一価アルキルカルボン酸塩、アシルラクチレート(acyl lactylate)、アルキルエーテルカルボン酸塩、N-アシルサルコシネート、多価アルキル炭酸塩、N-アシルグルタミン酸塩、脂肪酸-ポリペプチド縮合物、スルホン酸エステル、アルキル硫酸塩(ラウリル硫酸ナトリウム(SLS)など)、エトキシル化アルキル硫酸塩、エステル結合スルホン酸塩(ドキュセートナトリウムまたはコハク酸ジオクチルナトリウム(DSS)など)、αオレフィンスルホン酸塩、およびリン酸化エトキシル化アルコールが挙げられる。
【0070】
適切な薬学的に許容されるカチオン性界面活性剤には、例えば、モノアルキル四級アンモニウム塩、ジアルキル四級アンモニウム化合物、アミドアミンおよびアミンイミドが含まれる。
【0071】
適切な薬学的に許容される両性(両親媒性/両染性)界面活性剤には、例えば、N-置換アルキルアミド、N-アルキルベタイン、スルホベタイン、およびN-アルキルβアミノプロピオン酸塩が含まれる。
【0072】
本発明に関連して使用するための他の好適な界面活性剤には、エステルまたはエーテルとしてのポリエチレングリコールが含まれる。例として、ポリエトキシル化ヒマシ油、ポリエトキシル化硬化ヒマシ油、ヒマシ油から誘導したポリエトキシル化脂肪酸、ヒマシ油から誘導したポリエトキシル化脂肪酸または硬化ヒマシ油から誘導したポリエトキシル化脂肪酸が挙げられる。使用しうる市販の界面活性剤は、Cremophor、Myrj、Polyoxy 40 stearate、Emerest 2675、Lipal 395およびPEG 3350という商標で知られている。
【0073】
pH改変剤は、マトリックスからの薬物の放出を促進し、最終剤形の約1〜約50重量%;約1〜約25重量%;約1〜約15重量%;または約1〜約10重量%で含有される。好ましい実施形態において、pH改変剤は、クエン酸、コハク酸、フマル酸、リンゴ酸、マレイン酸、グルタル酸または乳酸などの有機酸である。
【0074】
本発明に関連して任意に使用してもよいイオン化ゲル強度増強剤は一価または多価金属カチオンでありうる。好ましい塩は、種々のアルカリ金属および/またはアルカリ土類金属の硫酸塩、塩化物、ホウ酸塩、臭化物、クエン酸塩、酢酸塩、乳酸塩などを含む、無機塩である。適切なイオン化ゲル強度増強剤の具体的な例としては、硫酸カルシウム、塩化ナトリウム、硫酸カリウム、炭酸ナトリウム、塩化リチウム、リン酸三カリウム、ホウ酸ナトリウム、臭化カリウム、フッ化カリウム、重炭酸ナトリウム、塩化カルシウム、塩化マグネシウム、クエン酸ナトリウム、酢酸ナトリウム、乳酸カルシウム、硫酸マグネシウム、およびフッ化ナトリウムが挙げられる。多価金属カチオンを使用してもよい。しかし好ましいイオン化ゲル強度増強剤は二価である。特に好ましい塩は硫酸カルシウムおよび塩化ナトリウムである。本発明のイオン化ゲル強度増強剤は、ゲル化剤の架橋(例えばヘテロ多糖ガムとホモ多糖ガムとの架橋)に起因する所望のゲル強度の増大を得るのに有効な量で添加される。他の実施形態において、イオン化ゲル強度増強剤は、徐放性賦形剤の約1〜約20重量%の量、かつ最終剤形の0.5〜約16重量%の量で本発明の徐放性賦形剤中に含有させる。
【0075】
本発明の特定の実施形態において、本発明の徐放性マトリックスは、ヘテロ多糖ガムとホモ多糖ガムとを含む約10〜約99重量%のゲル化剤、約0〜約20重量%のイオン化ゲル強度増強剤、および約1〜約89重量%の不活性医薬希釈剤を含む徐放性賦形剤を含有する。他の実施形態において、徐放性賦形剤は、約10〜約75%のゲル化剤、約2〜約15%のイオン化ゲル強度増強剤、および約30〜約75%の不活性希釈剤を含有する。さらに他の実施形態において、徐放性賦形剤は、約30〜約75%のゲル化剤、約5〜約10%のイオン化ゲル強度増強剤、および約15〜約65%の不活性希釈剤を含有する。
【0076】
本発明の徐放性賦形剤は、(任意にイオン化ゲル強度増強剤を含有しても含有しなくてもよく)、親水性マトリックスを破壊することなくガムの水和を緩慢にする疎水性物質の組み込みによってさらに変更しうる。これは、本発明の他の実施形態で達成されるものであり、該実施形態では徐放性賦形剤を疎水性物質の溶液または分散液を用いて顆粒化した後で薬物の組み込みを行う。疎水性ポリマーは、アルキルセルロース(エチルセルロースなど)、他の疎水性セルロース物質、アクリル酸エステルまたはメタクリル酸エステルから誘導されるポリマーまたはコポリマー、アクリル酸エステルとメタクリル酸エステルとのコポリマー、ゼイン、ワックス、シェラック、硬化植物油、および当業者に公知の他のあらゆる薬学的に許容される疎水性物質から選択しうる。徐放性賦形剤中に組み込む疎水性物質の量は、周囲の液体に晒された際に形成される親水性マトリックスを破壊せずにガムの水和を緩慢にするのに有効な量である。本発明の特定の好ましい実施形態において、疎水性物質は、約1〜約20重量%の量で徐放性賦形剤中に含有される。疎水性物質のための溶媒は、水性もしくは有機溶媒、またはこれらの混合物でありうる。
【0077】
本発明の徐放性賦形剤が既製品である場合の実施形態において、例えば高剪断力ミキサー中で、薬物とその賦形剤とを混合することができる。特定の特に好ましい実施形態において、薬物は、循環系疾患および高血圧症の治療に有用である、治療上有効なベンゾチアジンである。特に好ましいジヒドロキシピリジンはジルチアゼムである。ジルチアゼムの有効な製剤は、一般的に約30〜約500mg、好ましくは約120mg〜約480mgの日用量を含有する。本発明の特定の好ましい実施形態において、剤形は、ジルチアゼムの調剤を、24時間用製剤については120mg、180mg、240mgまたは300mgの量で、そして12時間用製剤については60mg、90mgおよび120mgの量で含有する。
【0078】
特定の他の、特に好ましい実施形態において、薬物は、泌尿器疾患の治療に有用なオキシブチニンである。オキシブチニンの有用な製剤は、一般的に、約2.5mg〜約50mgの日用量、例えば12時間用製剤については約2.5mg〜約25mg、24時間用製剤については約5mg〜約50mgを含有する。本発明の特定の好ましい実施形態において、剤形は、24時間用製剤について5mg、10mgまたは15mgの量でオキシブチニンの調剤を含有する。
【0079】
有効量の一般的に許容される医薬潤滑剤(カルシウム石鹸またはマグネシウム石鹸など)は、好ましくは、成分(薬物など)の混合物を打錠して経口用固体剤形(例えば錠剤)にする前に該混合物に添加する。適切な潤滑剤の例は、固体剤形の約0.5〜約3重量%の量のステアリン酸マグネシウムである。特に好ましい潤滑剤は、ステアリルフマル酸ナトリウム(sodium stearyl fumarate), NF(Penwest Pharmaceuticals Co.からPruv(登録商標)の商標で市販されている)である。
【0080】
本発明の徐放性賦形剤は、種々の粒子サイズ分布の範囲で均一な充填特性を有し、直圧縮に続いて薬物および潤滑剤粉末の添加を行うかまたは慣用的な湿式顆粒化法により最終剤形(例えば錠剤)に加工することができる。
【0081】
本発明に従って調製される特定の賦形剤系の性質および特徴は、部分的には、ホモ多糖成分およびヘテロ多糖成分の個々の特性(ポリマー可溶性、ガラス転移温度など)、ならびに変化する溶出の液体−賦形剤相互作用における種々のホモ多糖とヘテロ多糖との共働作用、ホモ多糖およびヘテロ多糖と不活性多糖要素との共働作用に依存している。
【0082】
ゲル化剤(すなわち、キサンタンガムとイナゴマメガムとの混合物)と、不活性希釈剤と、イオン化ゲル強度増強剤および疎水性ポリマー(あってもなくてもよい)との組み合わせによって、製剤の製造者が、混合物を打錠して遅延放出錠剤を形成する前に、所望の有効薬物、pH改変剤、界面活性剤および任意に潤滑剤と賦形剤とを混合するだけでよい、すぐに使用できる徐放性賦形剤製品が提供される。ガムと純粋な(すなわち微晶質)スクロース、ラクトース、デキストロースなどとを顆粒化または凝塊形成して賦形剤を形成することが好ましいが、該賦形剤は、ガムと可溶性賦形剤(例えば圧縮可能なスクロース、ラクトースまたはデキストロース)との物理的混合物を含みうる。顆粒形態は、流動性および圧縮性を最適化しうるという事実、有効薬物を用いた打錠、カプセル中への配合、押し出し加工および球状化(speronize)によるペレット形成を行いうる事実などを含むいくつかの利点を有する。
【0083】
本発明に従って調製される医薬賦形剤は、許容される賦形剤製品を得るためのあらゆる凝塊形成法に従って調製しうる。湿式顆粒化法では、所望の量のヘテロ多糖ガム、ホモ多糖ガムおよび不活性希釈剤を一緒に混合し、その後、水、プロピレングリコール、グリセロール、アルコールなどの湿潤剤を添加して湿潤塊を調製する。次にこの湿潤塊を乾燥する。続いて乾燥した塊を慣用的な装置を用いて粉砕し、顆粒にする。それにより、賦形剤製品はすぐ使用可能なものとなる。
【0084】
既製の徐放性賦形剤は、好ましくは流動し、直圧縮可能なものである。従って、該賦形剤は、治療上有効な薬物および任意に潤滑剤と所望の割合で混合しうる(乾式顆粒化法)。あるいは、賦形剤の全部または一部について、活性成分と共に湿式顆粒化法を行い、その後で打錠してもよい。製造すべき最終生成物が錠剤の場合には、その後に、一定のバッチの錠剤を製造するのに十分な量の完全な混合物を、慣用的な製造規模の打錠機を用いて、通常の圧縮圧、すなわち約2000〜1600lbs/平方インチで打錠を行う。しかしながら、混合物は、後で胃液に晒された際に水和するのが困難な程度にまで圧縮するべきではない。
【0085】
錠剤の製造方法としての直圧縮法の限界の1つは、錠剤のサイズである。有効成分が多い場合には、医薬製剤の製造者は、正しい圧縮強度を有し基準に達するサイズの錠剤を得るために、他の賦形剤と共に有効成分に湿式顆粒化を行うことを選択してもよい。通常は、湿式顆粒化に必要とされる充填剤/結合剤または賦形剤の量は、直圧縮の場合よりも少なく、それは湿式顆粒化の工程が錠剤の所望の物理的特性にある程度寄与しているためである。
【0086】
薬物がジルチアゼムの場合には、丸型錠剤の平均錠剤サイズは好ましくは約300mg〜750mgであり、そしてカプセル形状錠剤の平均錠剤サイズは約700mg〜1000mgである。
【0087】
本発明の顆粒化賦形剤の平均粒子サイズは、好ましくは約50ミクロン〜約400ミクロン、好ましくは約185ミクロン〜約265ミクロンである。顆粒の粒子サイズは厳密に重要なものではなく、重要なパラメーターは、顆粒の平均粒子サイズが薬学的に許容される錠剤を形成する直圧縮賦形剤の形成を可能にする必要があるということである。本発明の顆粒の所望のタップ密度および充填密度は、通常約0.3〜約0.8g/mlであり、平均密度は約0.5〜約0.7g/mlである。最良の結果では、本発明の顆粒から形成される錠剤は、約5〜約20kgの硬度を有する。本発明に従って調製される顆粒の平均流量は、好ましくは約25〜約40g/秒である。試験ロータリー型打錠機を用いて圧縮した錠剤は、不活性糖成分とはほとんど別の強度プロフィールを有することが見出されている。錠剤表面の大部分の走査型電子顕微鏡写真によって、錠剤表面および破断面の両方において圧縮における過剰な塑性変形に関する質的証拠が提供されている。また該写真は、最初の溶媒侵入および溶液排出が起こりうる表面孔の証拠を示している。
【0088】
本発明の特定の実施形態において、錠剤は、製剤の薬物の放出をさらに変更することができるように十分な量の疎水性ポリマーを用いてコーティングする。錠剤コーティング中に含まれる疎水性ポリマーは、任意に徐放性賦形剤と共に顆粒化してもよい疎水性ポリマー物質と比較して同じ物質でもよいし、異なる物質でもよい。
【0089】
本発明の他の実施形態において、錠剤コーティングは、疎水性ポリマーコーティングに加えてまたはその代わりに腸溶コーティング物質を含みうる。適切な腸溶ポリマーの例としては、酢酸フタル酸セルロース、フタル酸ヒドロキシプロピルメチルセルロース、フタル酸ポリビニルアセテート、メタクリル酸コポリマー、シェラック、コハク酸ヒドロキシプロピルメチルセルロース、酢酸トリメリト酸セルロース、およびこれらの任意の混合物が挙げられる。適切な市販の腸溶物質の例は、EudraditTM L30D55の商標で入手可能である。
【0090】
さらなる実施形態において、剤形は、上述のコーティングに加えてまたはその代わりに親水性コーティングでコーティングしてもよい。かかる親水性コーティングに使用しうる適切な物質の例は、ヒドロキシプロピルメチルセルロース(例えば、Colorcon, West Point, Pennsylvaniaより市販されているOpadry(登録商標))である。
【0091】
コーティングは、当業者に公知の薬学的に許容される方法で適用しうる。例えば一実施形態において、コーティングは流動床によりまたはコーティングパン中で適用する。例えば、コーティングした錠剤は、例えばコーティングパン中で約60〜70℃にて約3〜4時間乾燥しうる。疎水性ポリマーまたは腸溶コーティングの溶媒は、有機溶媒、水性溶媒、または有機溶媒と水性溶媒との混合物でありうる。有機溶媒は、水を含有していてもしていなくてもよい、例えば、イソプロピルアルコール、エタノールなどでありうる。
【0092】
本発明のさらなる実施形態において、本発明に従って製造する錠剤にサポートプラットフォーム(support platform)を適用する。適切なサポートプラットフォームは当業者に周知である。適切なサポートプラットフォームの例は、例えば米国特許第4,839,177号(参照により本明細書に引用する)に記載されている。この特許において、錠剤をサポートプラットフォームで部分的にコーティングしており、このサポートプラットフォームは水性液体に不溶性のポリマー物質からなるものである。サポートプラットフォームは、例えば、治療上有効な薬物の運搬の間に非浸透性を維持するように設計されうる。サポートプラットフォームは、例えば錠剤表面の一部に加圧コーティングを施すことにより、錠剤表面の全部もしくは一部にサポートプラットフォームを含むポリマー物質を噴霧コーティングすることにより、またはポリマー物質の溶液中に錠剤を浸すことによって、錠剤に適用しうる。
【0093】
サポートプラットフォームは、圧縮で適用される場合には例えば約2mmの厚さ、噴霧コーティングまたは液浸により適用される場合には約10μの厚さを有しうる。一般的には、疎水性ポリマーまたは腸溶コーティングが錠剤に適用される本発明の実施形態において、錠剤を、重量が約1〜約20%、特定の実施形態では好ましくは約5〜約10%増加する程度にコーティングする。
【0094】
本発明の疎水性コーティングおよびサポートプラットフォームに有用な物質には、アクリル酸の誘導体(例えば、アクリル酸エステル、メタクリル酸エステル、およびそれらのコポリマー)、セルロースおよびそれらの誘導体(例えばエチルセルロース)、ポリビニルアルコールなどが含まれる。
【0095】
本発明の特定の実施形態において、錠剤コアには、疎水性もしくは腸溶コーティング中、または、錠剤コアの外部表面上にコーティングされたさらなるオーバーコーティング中(疎水性もしくは腸溶コーティングを含まない)に、あるいは疎水性もしくは腸溶コーティング物質を含むベースコーティングの表面上にコーティングされる第2コーティング層として、包含されるさらなる用量の薬物が含まれる。これは、例えば、治療上有効な薬物の充填量が、最初に製剤が胃液に晒された際に有効薬物の治療上有効な血中レベルを提供するのに要求される場合に、望ましいものでありうる。コーティング層中に含まれる薬物の充填量は、例えば、製剤中に含まれる薬物の合計量の約10〜約40%でありうる。
【0096】
本発明の好ましい実施形態において、最終製剤は、薬物がジルチアゼムの場合には二相性または多相性血漿レベルを提供する。
【0097】
好ましい実施形態において、薬物がジルチアゼムの場合には、本発明の製剤は、患者への経口投与後約4〜約10時間において、ジルチアゼムの初回ピーク血漿濃度時間(Tmax#1)を示す。特定の好ましい実施形態において、初回ピーク血漿濃度時間は、経口投与後約6〜約8時間で起こる。好ましい実施形態において、初回Tmaxにおけるジルチアゼムの最大血漿濃度(Cmax#1)は、本発明の経口用徐放性剤形中240mgの量のジルチアゼムの投与当たり、約50〜100ng/mlである。
【0098】
本発明のさらなる好ましい実施形態において、徐放性ジルチアゼム製剤は、第2ピーク血漿濃度(Cmax#2)を提供する。この第2ピーク血漿濃度は、患者への剤形の経口投与後約10〜約16時間(Tmax#2)において起こる。特定の好ましい実施形態において、第2ピーク血漿濃度(Cmax#2)は、ヒトへの剤形の経口投与後約12〜約14時間(Tmax#2)において起こる。好ましい実施形態において、Cmax#2におけるジルチアゼムの最大血漿濃度は、24時間にわたり投与された240mgのジルチアゼム当たり約60〜約90ng/mlである。
【0099】
特定の好ましい実施形態において、徐放性ジルチアゼム製剤は、約0.5〜約4時間、好ましくは約1〜約3時間であるCmax#1のW50を示す。Cmax#1のW50は、本発明の目的のために、Cmax#1とCmax#2との間のCminとして得られた最低値を基準にして、第1Cmax(Cmax#1)の高さの50%における血漿濃度曲線の幅として定義される。
【0100】
特定の好ましい実施形態において、徐放性ジルチアゼム製剤は、約0.5〜約8時間、好ましくは約2〜約6時間であるCmax#2のW50を示す。Cmax#2のW50は、本発明の目的のために、Cmax#1とCmax#2との間のCminとして得られた最低値を基準にして、第2Cmax(Cmax#2)の高さの50%における血漿濃度曲線の幅として定義される。
【0101】
特定の好ましい実施形態において、本発明の徐放性ジルチアゼム製剤は、Cmax#1とCmax#2との比が約0.5:1〜約1.5:1、好ましくは約0.7:1〜約1.2:1である。
【0102】
本発明の経口用徐放性製剤中のジルチアゼムの量を基準として、ジルチアゼムの種々の投与量に関して、12または24時間にわたるCmax#1、Cmax#2、Tmax#1およびTmax#2を容易に決定することができるだろう。
【0103】
本発明の特定の好ましい実施形態において、薬物がオキシブチニンの場合には、該製剤は、約5〜約15時間、好ましくは約8〜約12時間においてオキシブチニンのピーク血漿濃度時間(Tmax)を提供する。
【0104】
本発明に組み込むのに適した可溶性〜高度に可溶性の薬物の例としては、抗ヒスタミン(例えば、マレイン酸アザタジン、マレイン酸ブロムフェニルアミン、マレイン酸カルビノキサミン、マレイン酸クロルフェニルアミン、マレイン酸d‐クロルフェニルアミン、塩酸ジフェンヒドラミン、コハク酸ドキシラミン、塩酸メトジラジン、プロメタジン、酒石酸トリメプラジン、クエン酸トリペレナミン、塩酸トリペレナミン、および塩酸トリプロリジン);抗生物質(例えば、ペニシリンVカリウム、クロキサシリンナトリウム、ジクロキサシリンナトリウム、ナフシリンナトリウム、オキサシリンナトリウム、カルベニシリンインダニルナトリウム、塩酸オキシテトラサイクリン、塩酸テトラサイクリン、リン酸クリンダマイシン、塩酸クリンダマイシン、パルミチン酸クリンダマイシンhcl、リンコマイシンhcl、ノボビオシンナトリウム、ニトロフラントインナトリウム、塩酸メトロニダゾル);抗結核剤(例えば、イソニアジド);コリン作動物質(例えば、塩化アンベノニウム、塩化ベタネコル(bethanecol chloride)、臭化ネオスチグミン、臭化ピリドスチグミン);抗ムスカリン剤(例えば、臭化メチルアニソトロピン、臭化クリジニウム、塩酸ジシクロミン、グリコピロレート、メチル硫酸ヘキソシクリウム、臭化メチルホマトロピン、硫酸ヒヨスチアミン、臭化メタンテリン、臭化水素酸ヒオスシン、臭化オキシフェノニウム、臭化プロパンテリン、塩化トリジヘキセチル);交感神経興奮剤(例えば、メタンスルホン酸ビオルテロール、エフェドリン、塩酸エフェドリン、硫酸エフェドリン、硫酸オルシプレナリン、塩酸フェニルプロパノールアミン、塩酸プソイドエフェドリン、塩酸リトドリン、硫酸サルブタモール、硫酸テルブタリン);交感神経遮断剤(例えば、塩酸フェノキシベンズアミン);他の自律神経作用薬(例えば、ニコチン);鉄製剤(例えば、グルコン酸第一鉄、硫酸第一鉄);ヘモスタティック(例えば、アミノカプロン酸);強心剤(例えば、塩酸アセブトロール、リン酸ジソピラミド、酢酸フレカイニド、塩酸プロカインアミド、塩酸プロプラノロール、グルコン酸キニジン、マレイン酸チモロール、塩酸トカイニド、塩酸ベラパミル);抗高血圧剤(例えば、カプトリル、塩酸クロニジン、塩酸ヒドララジン、塩酸メカミラミン、酒石酸メトプロロール);血管拡張剤(例えば、塩酸パパベリン);非ステロイド性抗炎症剤(例えば、サリチル酸コリン、サリチル酸マグネシウム、メクロフェナム酸ナトリウム、ナプロキセンナトリウム、トルメチンナトリウム);鎮痙薬(例えば、フェノバルビタールナトリウム、フェニトインナトリウム、トロキシドン、エトスクシミド、バルプロ酸ナトリウム);精神安定剤(例えば、マレイン酸アセトフェナジン、塩酸クロルプロマジン、塩酸フルフェナジン、1,2-エタンジスルホン酸(edisylate)プロクロルペラジン、塩酸プロメタジン、塩酸チオリダジン、塩酸トリフルオロペラジン、クエン酸リチウム、塩酸モリンドン、塩酸チオチキセン);刺激剤(塩酸ベンザムフェタミン(benzamphetamine)、硫酸デキストロアンフェタミン、リン酸デキストロアンフェタミン、塩酸ジエチルプロピオン、塩酸フェンフルラミン、塩酸メタンフェタミン、塩酸メチルフェニデート、酒石酸フェンジメトラジン、塩酸フェンメトラジン、クエン酸カフェイン);バルビツル剤(例えば、アミロバルビタール(amylobarbital)ナトリウム、ブタバルビタールナトリウム、セコバルビタールナトリウム);鎮静剤(例えば、塩酸ヒドロキシジン、メトプリロン(methprylon));去痰剤(例えば、ヨウ化カリウム);制吐薬(例えば、塩酸ベンザキナミド(benzaquinamide)、塩酸メトクロプロパミド(metoclopropamide)、塩酸トリメトベンズアミド);胃腸薬(例えば、塩酸ラニチジン);重金属アンタゴニスト(例えば、ペニシラミン、塩酸ペニシラミン);抗甲状腺薬(例えば、メチマゾール);尿生殖器平滑筋弛緩薬(例えば、塩酸フラボキセイト);ビタミン剤(例えば、塩酸チアミン、アスコルビン酸);分類されていない薬剤(例えば、塩酸アマンタジン、コルヒチン、エチドロン酸二ナトリウム、ロイコボリンカルシウム、メチレンブルー、塩化カリウム、塩化プラリドキシム)が挙げられる。このリストは限定を意味するものでない。
【実施例】
【0105】
以下の実施例は本発明の種々の態様を説明するものである。これらは、いかなる意味においても特許請求の範囲を限定するものと解されるべきではない。
【0106】
実施例1〜2
製剤における薬物:ガム比の効果
実施例1および2においては、本発明の徐放性賦形剤を最初に調製し、次いで薬物(この場合はジルチアゼム)およびpH改変剤(この場合はフマル酸)を添加し、続いて最終混合物を錠剤に製剤化する。
【0107】
徐放性賦形剤を、必要量のキサンタンガム、イナゴマメガム、およびデキストロースを、高速ミキサー/造粒機において3分間ドライブレンドして調製する。チョッパー(chopper)/圧縮機(impeller)を作動させている間に、ドライブレンドした混合物に水を添加し、さらに3分間顆粒化する。次いで顆粒を、LOD(乾燥工程のロス)が約10重量%未満(例えば4〜7%のLOD)となるように液体ベッドドライヤーで乾燥させる。顆粒を20メッシュの金網を用いて粉砕し、造粒機中に分散する。実施例1および2の顆粒の成分を下記の表1に示す。
【表1】
【0108】
次に、所望の量のジルチアゼム、フマル酸および好適な量の水を、圧縮機型のミキサーで5分間にわたり混合してスラリーを形成する。続いてこのスラリーを徐放性賦形剤に1分間隔で、低速で圧縮機を作動させながら造粒機に添加する。次に混合物を、高速で作動させたチョッパーおよび圧縮機を用いて2分間にわたり顆粒化する(好適な顆粒を形成するために、さらに水と顆粒化時間を増してもよい)。続いて得られた顆粒を、液体ベッドドライヤーでLODが5%未満となるまで乾燥し、2000〜3000rpmで作動しているハンマーで粉砕する。次に粉砕した顆粒をドデシル硫酸ナトリウムとともにV-Blender中に配置し、10分間にわたりブレンドする。続いて好適な錠剤化用潤滑剤(Pruv(登録商標)、ステアリルフマル酸ナトリウム、NF、Penwest Pharmaceuticals Co.より市販されている)を添加し、混合物をさらに3分間にわたりブレンドする。続いて得られた顆粒を、カプセル型パンチを用いて錠剤へと圧縮する。この最終混合物を約768mgの錠剤とする。実施例1および2の錠剤の成分を以下の表2に示す。
【表2】
【0109】
最終産物の錠剤は、錠剤として重量768.0mgであり、硬度15Kpである。
【0110】
実施例1および2の錠剤についての溶出試験を、自動USP溶出装置(Paddle type II、100rpm)中で900MLの水中で行い、放出された薬物の量をUV分析により調べた。in vitroでの溶出の結果を、図1と以下の表3に示す。
【表3】
【0111】
図1および表3に示した結果から、ジルチアゼムの放出速度は、製剤中のガムの量が増加するにつれて遅くなることが明らかである。
【0112】
実施例3〜4
ガム:デキストロース比の効果
実施例3および4においては、徐放性賦形剤は実施例1および2に示した工程により調製する。実施例3および4の徐放性賦形剤の成分を、以下の表4に示す。
【表4】
【0113】
その後、ジルチアゼム錠剤を以下の通り調製する。
【0114】
所望の量のジルチアゼム、フマル酸および徐放性賦形剤を造粒機に配置し、低速で3分間にわたり混合する。続いて水を2分間隔で、低速で圧縮機を作動させながら造粒機に添加する(好適な顆粒を形成するために、さらに水と顆粒化時間を増してもよい)。続いて得られた顆粒を、液体ベッドドライヤーでLODが5%未満となるまで乾燥し、2000〜3000rpmで作動しているハンマーで金網#0050を用いて粉砕する。次に粉砕した顆粒をドデシル硫酸ナトリウムとともにV-Blender中に配置し、10分間にわたりブレンドする。続いて好適な錠剤化用潤滑剤(Pruv、ステアリルフマル酸ナトリウム、NF、Penwest Pharmaceuticals Co.より市販されている)を添加し、混合物をさらに5分間にわたりブレンドする。続いて、得られた顆粒を、カプセル型パンチを用いて錠剤へと圧縮する。この最終混合物を約750mgの錠剤とする。実施例3および4の錠剤の成分を以下の表5に示す。
【表5】
【0115】
最終産物の錠剤は、錠剤として重量750.0mgであり、硬度15Kpである。
【0116】
実施例3および4の錠剤についての溶出試験を、自動USP溶出装置(Paddle type III、15CPM)中で250MLのバッファー(pH6)中で行い、放出された薬物の量をUV分析により調べた。in vitroでの溶出の結果を、図2と以下の表6に示す。
【表6】
【0117】
図2および表6に示した結果から、デキストロースの量に対するガムの量が増加するにつれて、それに対応して薬物放出の遅れが見られた。
【0118】
実施例5〜6
界面活性剤の型の効果
実施例5および6において、徐放性賦形剤は実施例1および2に示した工程により調製する。実施例5および6の徐放性賦形剤の成分を、以下の表7に示す。
【表7】
【0119】
その後、ジルチアゼム錠剤を以下の通り調製する。
【0120】
所望の量のジルチアゼム、フマル酸および好適な量の水を、圧縮機型のミキサーで5分間にわたり混合してスラリーを形成する。続いてこのスラリーを徐放性賦形剤に1分間隔で、低速で圧縮機を作動させながら造粒機において添加する。次に混合物を、高速で作動させたチョッパーおよび圧縮機を用いて2分間にわたり顆粒化する(好適な顆粒を形成するために、さらに水と顆粒化時間を増してもよい)。続いて得られた顆粒を、液体ベッドドライヤーでLODが5%未満となるまで乾燥し、2000〜3000rpmで作動しているハンマーで粉砕する。次に粉砕した実施例5の顆粒をドデシル硫酸ナトリウムとともにV-Blender中に配置し、粉砕した実施例6の顆粒をドキュセートナトリウムとともにV-Blender中に配置し、10分間にわたりブレンドする。続いて好適な錠剤化用潤滑剤(Pruv、ステアリルフマル酸ナトリウム、NF、Penwest Pharmaceuticals Co.より市販されている)を各例に対して添加し、混合物をさらに3分間にわたりブレンドする。続いて得られた顆粒を、カプセル型パンチを用いて錠剤へと圧縮する。この最終混合物を約848mgの錠剤とする。実施例5および6の錠剤の成分を以下の表8に示す。
【表8】
【0121】
最終産物の錠剤は、錠剤として重量848.0mgであり、硬度15Kpである。
【0122】
実施例1および2の錠剤についての溶出試験を、自動USP溶出装置(Paddle type II、100rpm)中で900MLの水中で行い、放出された薬物の量をUV分析により調べた。in vitroでの溶出の結果を、図3と以下の表9に示す。
【表9】
【0123】
図3および表9に示した結果から、ジルチアゼムの放出速度は、等量比のドデシル硫酸ナトリウムおよびドキュセートナトリウムについては同じであることが明らかである。しかしながら、ドデシル硫酸ナトリウムを用いた方が製剤化工程には好適であった。
【0124】
実施例7〜8
界面活性剤のレベルの効果
実施例7および8において、徐放性賦形剤は実施例1および2に示した工程により調製する。実施例7および8の徐放性賦形剤の成分を、以下の表10に示す。
【表10】
【0125】
その後、ジルチアゼム錠剤を以下の通り調製する。
【0126】
所望の量のジルチアゼム、フマル酸および好適な量の水を、圧縮機型のミキサーで5分間にわたり混合してスラリーを形成する。続いてこのスラリーを徐放性賦形剤に1分間隔で、低速で圧縮機を作動させながら造粒機に添加する。次に混合物を、高速で作動させたチョッパーおよび圧縮機を用いて2分間にわたり顆粒化する(好適な顆粒を形成するために、さらに水と顆粒化時間を増してもよい)。続いて得られた顆粒を、液体ベッドドライヤーでLODが5%未満となるまで乾燥し、2000〜3000rpmで作動しているハンマーで粉砕する。次に粉砕した顆粒をドデシル硫酸ナトリウムとともにV-Blender中に配置し、10分間にわたりブレンドする。続いて好適な錠剤化用潤滑剤(Pruv、ステアリルフマル酸ナトリウム、NF、Penwest Pharmaceuticals Co.より市販されている)を添加し、混合物をさらに3分間にわたりブレンドする。続いて得られた顆粒を、カプセル型パンチを用いて錠剤へと圧縮する。この最終混合物を約768mgの錠剤とする。実施例7および8の錠剤の成分を以下の表11に示す。
【表11】
【0127】
実施例7の最終産物の錠剤は、錠剤として重量768.0mgであり、硬度15Kpである。
【0128】
実施例8の最終産物の錠剤は、錠剤として重量728.0mgであり、硬度15Kpである。
【0129】
実施例7および8の錠剤についての溶出試験を、自動USP溶出装置(Paddle type II、100rpm)中で900MLの水中で行い、放出された薬物の量をUV分析により調べた。結果を、図4と以下の表12に示す。
【表12】
【0130】
図4および表12に示した結果から、ジルチアゼムの溶出速度は、界面活性剤のレベルに反比例していることが明らかである。
【0131】
実施例9〜10
フマル酸レベルの効果
実施例9および10において、徐放性賦形剤は実施例1および2に示した工程により調製する。実施例9および10の徐放性賦形剤の成分を、以下の表13に示す。
【表13】
【0132】
その後、ジルチアゼム錠剤を以下の通り調製する。
【0133】
所望の量のジルチアゼム、フマル酸および好適な量の水を、圧縮機型のミキサーで5分間にわたり混合してスラリーを形成する。続いてこのスラリーを徐放性賦形剤に1分間隔で、低速で圧縮機を作動させながら造粒機に添加する。次に混合物を、高速で作動させたチョッパーおよび圧縮機を用いて2分間にわたり顆粒化する(好適な顆粒を形成するために、さらに水と顆粒化時間を増してもよい)。続いて得られた顆粒を、液体ベッドドライヤーでLODが5%未満となるまで乾燥し、2000〜3000rpmで作動しているハンマーで粉砕する。次に粉砕した顆粒をドデシル硫酸ナトリウムとともにV-Blender中に配置し、10分間にわたりブレンドする。続いて好適な錠剤化用潤滑剤(Pruv、ステアリルフマル酸ナトリウム、NF、Penwest Pharmaceuticals Co.より市販されている)を添加し、混合物をさらに3分間にわたりブレンドする。続いて得られた顆粒を、カプセル型パンチを用いて錠剤へと圧縮する。この最終混合物を約848mgの錠剤とする。実施例9および10の錠剤の成分を以下の表14に示す。
【表14】
【0134】
実施例9の最終産物の錠剤は、錠剤として重量848.0mgであり、硬度15Kpである。
【0135】
実施例10の最終産物の錠剤は、錠剤として重量768.0mgであり、硬度15Kpである。
【0136】
実施例9および10の錠剤についての溶出試験を、自動USP溶出装置(Paddle type II、100rpm)中で900MLの水中で行い、放出された薬物の量をUV分析により調べた。結果を、図5と以下の表15に示す。
【表15】
【0137】
図5および表15に示した結果から、製剤中のフマル酸の量を増加させることにより、放出速度が増加することが明らかである。
【0138】
実施例11〜12
外部顆粒化した顆粒の薬物への添加
実施例11および12においては、徐放性賦形剤は実施例1および2に示した工程により調製する。実施例11および12の徐放性賦形剤の成分を、以下の表16に示す。
【表16】
【0139】
その後、ジルチアゼム錠剤を以下の通り調製する。
【0140】
実施例11においては、所望の量のジルチアゼム、フマル酸および好適な量の水を、圧縮機型のミキサーで5分間にわたり混合してスラリーを形成する。続いてこのスラリーを徐放性賦形剤に1分間隔で、低速で圧縮機を作動させながら造粒機に添加する。次に混合物を、高速で作動させたチョッパーおよび圧縮機を用いて2分間にわたり顆粒化する(好適な顆粒を形成するために、さらに水と顆粒化時間を増してもよい)。続いて得られた顆粒を、液体ベッドドライヤーでLODが5%未満となるまで乾燥し、2000〜3000rpmで作動しているハンマーで粉砕する。次に粉砕した顆粒をドデシル硫酸ナトリウムとともにV-Blender中に配置し、10分間にわたりブレンドする。続いて好適な錠剤化用潤滑剤(Pruv、ステアリルフマル酸ナトリウム、NF、Penwest Pharmaceuticals Co.より市販されている)を添加し、混合物をさらに3分間にわたりブレンドする。続いて得られた顆粒を、カプセル型パンチを用いて錠剤へと圧縮する。この最終混合物を約848mgの錠剤とする。
【0141】
実施例12においては、ジルチアゼムの一部、フマル酸および好適な量の水を、圧縮機型のミキサーで5分間にわたり混合してスラリーを形成する。続いてこのスラリーを徐放性賦形剤に1分間隔で、低速で圧縮機を作動させながら造粒機に添加する。次に混合物を、高速で作動させたチョッパーおよび圧縮機を用いて2分間にわたり顆粒化する(好適な顆粒を形成するために、さらに水と顆粒化時間を増してもよい)。続いて得られた顆粒を、液体ベッドドライヤーでLODが5%未満となるまで乾燥し、2000〜3000rpmで作動しているハンマーで粉砕する。次に粉砕した顆粒をドデシル硫酸ナトリウムおよびジルチアゼムの残部とともにV-Blender中に配置し、10分間にわたりブレンドする。続いて好適な錠剤化用潤滑剤(Pruv、ステアリルフマル酸ナトリウム、NF、Penwest Pharmaceuticals Co.より市販されている)を添加し、混合物をさらに3分間にわたりブレンドする。続いて得られた顆粒を、カプセル型パンチを用いて錠剤へと圧縮する。この最終混合物を約848mgの錠剤とする。実施例11および12の錠剤の成分を下記の表17に示す。
【表17】
【0142】
実施例11の最終産物の錠剤は、錠剤として重量848.0mgであり、硬度15Kpである。
【0143】
実施例12の最終産物の錠剤は、錠剤として重量848.0mgであり、硬度15Kpである。
【0144】
実施例11および12の錠剤についての溶出試験を、自動USP溶出装置(Paddle type II、100rpm)中で900MLの水中で行い、放出された薬物の量をUV分析により調べた。結果を、図6と以下の表18に示す。
【表18】
【0145】
図6および表18に示した結果は、外部で顆粒化した顆粒としてのジルチアゼムの添加により約35%の初期破裂が生じることを示す。実施例11および12の剤形からのジルチアゼムの%放出速度を経時的に示す図7によっても示される通り、特定の割合の薬物を外部で顆粒化して添加すると、初期の急速な放出がもたらされることは明白である。
【0146】
実施例13〜18
Eudragit L30D55 w/NaOH(メタクリル酸コポリマー水性分散液)を有するコーティング錠剤の効果
実施例13〜18においては、徐放性賦形剤は実施例1および2に示した工程により調製する。実施例13〜18の徐放性賦形剤の成分を、以下の表19に示す。
【表19】
【0147】
その後、ジルチアゼム錠剤を以下の通り調製する。
【0148】
所望の量のジルチアゼム、フマル酸および徐放性賦形剤を、造粒機中で5分間にわたり低速で混合する。続いて好適な量の水を2分間隔で、低速で圧縮機を作動させながら添加する。次に得られたスラリーを、高速で作動させたチョッパーおよび圧縮機を用いて7.5分間にわたり顆粒化する(好適な顆粒を形成するために、さらに水と顆粒化時間を増してもよい)。続いて得られた顆粒を、液体ベッドドライヤーでLODが5%未満となるまで乾燥し、2000〜3000rpmで作動しているハンマーで、金網#0050を用いて粉砕する。次に粉砕した顆粒をドデシル硫酸ナトリウムとともにV-Blender中に配置し、10分間にわたりブレンドする。続いて好適な錠剤化用潤滑剤(Pruv、ステアリルフマル酸ナトリウム、NF、Penwest Pharmaceuticals Co.より市販されている)を添加し、混合物をさらに5分間にわたりブレンドする。続いて得られた顆粒を、カプセル型パンチを用いて錠剤へと圧縮する。この最終混合物を約750mgの錠剤とする。実施例13〜18の錠剤の成分を下記の表20に示す。
【表20】
【0149】
最終産物の錠剤は、錠剤として重量750.0mgであり、硬度15Kpである。
【0150】
続いてコア錠剤を、例えば、全錠剤の重量を基準として3%、5%、7%および9%(それぞれ実施例15〜18)の重量増加となるまでEudragit L30D55 w/NaOHの水性分散液によりコーティングした。
【0151】
該水性分散液は以下の手順により調製した。
【0152】
1.0Nの水酸化ナトリウムを、メスフラスコ中の50mlの精製水に4.0gの水酸化ナトリウムを添加し5〜15分間攪拌して調製する。次いで精製水を必要量添加して再度攪拌する。
【0153】
タルク懸濁物を、クエン酸トリエチル9.31gを精製水202.54gに攪拌しながら添加する。攪拌を続けながらタルク22.2gを容器に3分間隔で添加する。容器を懸濁物が形成されるまで攪拌する。
【0154】
次いで、Eudragitを#40のメッシュ篩を通してEudragit懸濁物を調製し、計量して294.52gとする。滴下器(dropper)を用いて、1.0N水酸化ナトリウム溶液1.78gをEudragitに攪拌しながら添加する。この混合物を30〜60分間攪拌する。
【0155】
Eudragit懸濁物を攪拌しながら、タルク懸濁物を5分間かけて添加し、30〜60分間攪拌する。
【0156】
実施例13〜18の錠剤についての溶出試験を行った。自動USP溶出装置(Paddle type III、15CPM)中で250MLのバッファー(pH6)中で行い、放出された薬物の量をUV分析により調べた。結果を図8と以下の表21に示す。
【表21】
【0157】
図8および表21に示した結果から、コーティングの量(重量による)が増加するにつれて放出速度は低くなることは明らかである。
【0158】
実施例19〜20
Eudragit RS30D/RL30D(50/50)(アンモニオメタクリル酸コポリマー水性分散液)を有するコーティング錠剤の効果
実施例19および20においては、徐放性賦形剤は実施例1および2に示した工程により調製する。実施例19および20の徐放性賦形剤の成分を、以下の表22に示す。
【表22】
【0159】
その後、ジルチアゼム錠剤を以下の通り調製する。
【0160】
所望の量のジルチアゼム、フマル酸および徐放性賦形剤を、造粒機中で3分間にわたり低速で混合する。続いて好適な量の水を2分間隔で、低速で圧縮機を作動させながら添加する。次に得られたスラリーを、高速で作動させたチョッパーおよび圧縮機を用いて6分間にわたり顆粒化する(好適な顆粒を形成するために、さらに水と顆粒化時間を増してもよい)。続いて得られた顆粒を、液体ベッドドライヤーでLODが5%未満となるまで乾燥し、2000〜3000rpmで作動しているハンマーで金網#0050を用いて粉砕する。次に粉砕した顆粒をドデシル硫酸ナトリウムとともにV-Blender中に配置し、10分間にわたりブレンドする。続いて好適な錠剤化用潤滑剤(Pruv、ステアリルフマル酸ナトリウム、NF、Penwest Pharmaceuticals Co.より市販されている)を添加し、混合物をさらに3分間にわたりブレンドする。続いて得られた顆粒を、カプセル型パンチを用いて錠剤へと圧縮する。この最終混合物を約50mgの錠剤とする。
【0161】
実施例19および20の錠剤の成分を図9および下記の表23に示す。
【表23】
【0162】
最終産物の錠剤は、錠剤として重量750.0mgであり、硬度15Kpである。
【0163】
続いてコア錠剤を、全錠剤の重量を基準として8%の重量増加となるまでEudragit RS30D/RL30D(50/50)の水性分散液によりコーティングした。
【0164】
該水性分散液は以下の手順により調製した。
【0165】
100gのEudragit RSを100gのEudragit RLと混合してEudragit RS/RL懸濁物を調製する。
【0166】
タルク懸濁物を、クエン酸トリエチル12.0gを精製水338.0gに攪拌しながらゆっくりと添加する。攪拌を続けながらタルク50.0gを容器に3分間隔で添加する。容器を懸濁物が形成されるまで攪拌する。
【0167】
Eudragit懸濁物を攪拌しながら、タルク懸濁物を5分間かけて添加する。得られた混合物を30〜60分間攪拌し、40メッシュ篩を通してふるい分ける。
【0168】
実施例19および20の錠剤についての溶出試験を行った。自動USP溶出装置(Paddle type II、100rpm)中で900MLの0.1N HCL中で行い、放出された薬物の量をUV分析により調べた。結果を図9と以下の表24に示す。
【表24】
【0169】
図9および表24に示した結果から、コーティングが放出速度を低下させることは明らかである。
【0170】
実施例21〜23
エチルセルロースを有するコーティング錠剤の効果
実施例21〜23においては、徐放性賦形剤は実施例1および2に示した工程により調製する。実施例21〜23の徐放性賦形剤の成分を、以下の表25に示す。
【表25】
【0171】
その後、ジルチアゼム錠剤を以下の通り調製する。
【0172】
所望の量のジルチアゼム、フマル酸および徐放性賦形剤を、造粒機中で3分間にわたり低速で混合する。続いて好適な量の水を2分間隔で、低速で圧縮機を作動させながら添加する。次に得られたスラリーを、高速で作動させたチョッパーおよび圧縮機を用いて3分間にわたり顆粒化する(好適な顆粒を形成するために、さらに水と顆粒化時間を増してもよい)。続いて得られた顆粒を、液体ベッドドライヤーでLODが5%未満となるまで乾燥し、2000〜3000rpmで作動しているハンマーで金網#0050を用いて粉砕する。次に粉砕した顆粒をドデシル硫酸ナトリウムとともにV-Blender中に配置し、10分間にわたりブレンドする。続いて好適な錠剤化用潤滑剤(Pruv、ステアリルフマル酸ナトリウム、NF、Penwest Pharmaceuticals Co.より市販されている)を添加し、混合物をさらに3分間にわたりブレンドする。続いて得られた顆粒を、カプセル型パンチを用いて錠剤へと圧縮する。この最終混合物を約750mgの錠剤とする。
【0173】
実施例21〜23の錠剤の成分を下記の表26に示す。
【表26】
【0174】
最終産物の錠剤は、錠剤として重量750.0mgであり、硬度15Kpである。
【0175】
続いてコア錠剤を、全錠剤の重量を基準として4%および6%(それぞれ実施例22および23)の重量増加となるまでエチルセルロース/Opadry(80/20)の水性分散液によりコーティングした。該水性分散液は以下の手順により調製した。
【0176】
最初に、60gのOpadryを340gの水と好適な容器中で混合する。混合を続けながら、944gのエチルセルロースをOpadry分散液に添加する。得られた混合物を30〜60分間にわたり攪拌する。
【0177】
実施例1および2の錠剤についての溶出試験を行った。自動USP溶出装置(Paddle type III、15CPM)中で250MLのバッファー(pH6)中で行い、放出された薬物の量をUV分析により調べた。結果を図10と以下の表27に示す。
【表27】
【0178】
図10および表27に示した結果から、コーティングの量が増えるにつれて放出速度が低下することは明らかである。
【0179】
実施例24〜25
外部の顆粒への賦形剤の添加の効果
実施例24および25においては、徐放性賦形剤は実施例1および2に示した方法により調製する。実施例24および25の徐放性賦形剤の成分を、以下の表28に示す。
【表28】
【0180】
続いて、表29の成分と下記の工程により、錠剤を製造した。
【表29】
【0181】
実施例24の最終産物の錠剤は、錠剤として重量813.0mgであり、硬度15Kpである。
【0182】
実施例25の最終産物の錠剤は、錠剤として重量750.0mgであり、硬度15Kpである。
【0183】
実施例24および25の製剤の調製工程は下記の通りである。
【0184】
所望の量の(1)、(2)、および(4)を造粒機中に分散させ、低速で3分間混合する。圧縮機を低速で作動させながら、(7)を2分間隔で添加する。この混合物をチョッパーおよび圧縮機を高速で作動させて7.5分間にわたり顆粒化する(好適な顆粒を形成するために、さらに水と顆粒化時間を増してもよい)。顆粒化した混合物を、液体ベッドドライヤーでLODが5%未満となるまで乾燥する。乾燥顆粒を2000〜3000rpmで作動しているハンマーで金網#0050により粉砕する。粉砕した顆粒および(5)または(3と5)を、V-Blender中に配置し、10分間にわたりブレンドする。(6)をV-Blenderに添加し、5分間にわたりブレンドする。この最終混合物を、カプセル型パンチを用いて錠剤へと圧縮する。
【0185】
Eudragit L30D55 w/NaOHのコーティング分散液は以下の手順により調製した。
【0186】
A. 1.0Nの水酸化ナトリウム溶液を以下のように調製した。100mlのメスフラスコ中に4.0gの水酸化ナトリウムを添加し、次いで50mlの精製水およびマグネチックスターラーバーをフラスコ中に加え、フラスコの内容物を5〜15分間攪拌した。スターラーバーを取り出し、十分な容量として混合した。
【0187】
B. タルク懸濁物を以下のように調製した。精製水202.54gを好適な容器中に計り取り、クエン酸トリエチル9.31gを、精製水を攪拌しながらゆっくりと添加した。次にタルク22.22gを2分間隔で、混合物を攪拌しながら容器に添加した(混合物を懸濁物が形成されるまで攪拌した)。
【0188】
C. Eudragit L30D55懸濁物を以下のように調製した。Eudragit L30D55を#40のメッシュ篩を通し、篩い分けしたEudragit L30D55を294.52g計り取り、好適な容器中に配置し、滴下器(dropper)を用いて、1.0N水酸化ナトリウム溶液(ステップA)3.56gを、混合物を攪拌しながら添加した。この混合物を30〜60分間攪拌した。
【0189】
D. 最終コーティング懸濁物を以下のように調製した。Eudragit L30D55懸濁物(ステップC)にタルク懸濁物(ステップB)を5分間かけて添加しながら攪拌した。この混合物を30〜60分間攪拌した。
【0190】
錠剤を、全錠剤の重量を基準として4%の重量増加となるまでコーティングした。錠剤を、透明なゼラチンカプセル中にコーティングした該錠剤を配置してカプセル化した。
【0191】
実施例24の血漿中でのプロフィール
実施例24の錠剤について、2通りの無作為オープンラベルクロスオーバーデザインを用いて、健康なボランティア(各12名の被験者)においてin vivoの研究を行った。ボランティアに断食状態で投薬し、CARDIZEN CD(登録商標)と比較した。結果を図11と下記の表30に示す。
【表30】
【0192】
比
240mgの実施例24とCARDIZEN CDとの間の曲線の下部にある領域の比は1.16:1であった。240mgの実施例25とCARDIZEN CDとの間の平均Cmaxの比は1.16:1であった。
【0193】
結果
図11および実施例24では、二相性の(Bi-Modal)in vivoプラズマレベルが示され、CARDIZEN CDもまた、2種の異なる工程で調製されたビーズ製剤の混合により二相性のin vivoプラズマレベルを示した。
【0194】
実施例25の血漿中でのプロフィール
実施例25の錠剤について、2通りの無作為オープンラベルクロスオーバーデザインを用いて、健康なボランティア(各12名の被験者)においてin vivoの研究を行った。ボランティアに断食状態で投薬し、CARDIZEN CD(登録商標)と比較した。結果を図12と下記の表31に示す。
【表31】
【0195】
比
240mgの実施例25とCARDIZEN CDとの間の曲線の下部にある領域の比は1.16:1であった。240mgの実施例25とCARDIZEN CDとの間の平均Cmaxの比は1.26:1であった。
【0196】
結果
図12および実施例25では、二相性のin vivoプラズマレベルが示され、CARDIZEN CDもまた、2種の異なる工程で調製されたビーズ製剤の混合により二相性のin vivoプラズマレベルを示した。
【0197】
実施例26および27
異なる賦形剤の効果
実施例26および27において、徐放性賦形剤を実施例1および3で記述した方法により調製する。実施例25および26の徐放性賦形剤の成分を下記表32に示す。
【表32】
【0198】
表33の製剤
その後、ジルチアゼム錠剤を次の通り調製する。
【0199】
所望の量のジルチアゼム、フマル酸、および徐放性賦形剤を造粒機に配置し、低速で3分間にわたり混合する。圧縮機を低速で作動させながら、水を2分間隔で添加する(好適な顆粒を形成するために、さらに水および顆粒化時間を増しても良い)。続いて得られた顆粒を、LODが5%未満になるまで液体ベッドドライヤーで乾燥し、金網#0050を使用して2000〜3000rpmで作動しているハンマーを用いて粉砕する。次に、粉砕した顆粒をラウリル硫酸ナトリウムとともにV-Blender中に配置し10分間にわたりブレンドする。好適な錠剤化用潤滑剤(Pruv、ステアリルフマル酸ナトリウム, NF, Penwest Pharmaceuticals Co. より市販されている)を添加し、混合物をさらに5分間にわたりブレンドする。続いて得られた顆粒を、カプセル型パンチを用いて錠剤へと圧縮する。最終混合物を約750mgの錠剤とする。実施例26および27の錠剤の成分を下記の表33に示す。
【表33】
【0200】
実施例26の最終産物の錠剤は、錠剤として重量750.0mgであり、硬度15Kpである。
【0201】
実施例27の最終産物の錠剤は、錠剤として重量750.0mgであり、硬度15Kpである。
【0202】
次いで、実施例26および27の錠剤に対する溶出試験を、自動USP溶出装置(Paddle type III, 15CPM)中で250MLのバッファー(pH 6)中で実施し、放出された薬剤の量をUV分析により調べた。in-vitro溶出試験結果を図13および下記の表34に示す。
【表34】
【0203】
結論
実施例26は実施例25よりも遅い溶出プロフィールを有していた。
【0204】
結果
異なるグレードの賦形剤を使用することにより溶出速度を改変できる。
【0205】
実施例28〜29
製剤中のガム:薬剤比の効果
実施例28および29において、本発明の徐放性賦形剤を最初に調製し、次いで薬物(この場合、オキシブチニン)およびpH改変剤(この場合、コハク酸)を添加し、続いて最終混合物を錠剤に製剤化する。
【0206】
キサンタンガム、イナゴマメガム、デキストロース、および硫酸カルシウムを高せん断力のミキサー/造粒機の中に配合し、エチルセルロースをエタノールを含む容器中に配合し、エチルセルロース/エタノール混合物をキサンタンガム、イナゴマメガム、デキストロース、硫酸カルシウム混合物に配合し、そして好適な顆粒を形成するために顆粒化し、液体ベッドドライヤーで混合物を乾燥し、好適な顆粒を形成するために乾燥した物質を粉砕することによって、徐放性賦形剤を調製する。実施例28および29の徐放性賦形剤の成分を下記の表35に示す。
【表35】
【0207】
次に、所望の量のオキシブチニンおよびステアリルフマル酸ナトリウムを25メッシュの篩で篩分けし、篩分けしたオキシブチニンおよび徐放性賦形剤をV-blenderに配合し、10分間にわたりブレンドし、篩分けしたステアリルフマル酸ナトリウムを、オキシブチニンおよび徐放性賦形剤のブレンド混合物に添加してさらに5分間にわたりブレンドし、次いでブレンドされた最終の目的生成物を5/16”の丸型ツールを使用して、錠剤へと圧縮する。この最終混合物を約179.4mgの錠剤とする。実施例28および29の錠剤の成分を下記の表36および37に示す。
【表36】
【表37】
【0208】
最終産物の錠剤は、錠剤として重量179.4mgであり、硬度5Kpである。
【0209】
続いて、実施例28および29の錠剤に対する溶出試験を行った。in-vitro溶出試験結果を下記の表38に示す。
【表38】
【0210】
実施例28の製剤は1:5の薬剤:ガム比を有し、実施例29の製剤は1:8.3の薬剤:ガム比を有する。表38に示される結果から、オキシブチニンの放出速度は、製剤中の薬剤:ガム比が増加するにつれてより遅くなることが明らかである。
【0211】
実施例30および31
ガム:デキストロース比の効果
実施例30および31において、実施例28および29に記載の方法により徐放性賦形剤を調製する。実施例30および31の徐放性賦形剤の成分を下記の表39に示す。
【表39】
【0212】
その後、オキシブチニン錠剤を次のように調製する。
【0213】
所望の量のオキシブチニンおよびステアリルフマル酸ナトリウムを25メッシュの篩で篩分けし、篩分けしたオキシブチニンおよび徐放性賦形剤をV-blenderに配合し、10分間にわたりブレンドし、篩分けしたステアリルフマル酸ナトリウムをオキシブチニンおよび徐放性賦形剤のブレンド混合物に添加してさらに5分間にわたりブレンドし、次いでブレンドした最終の目的生成物を5/16”の丸型ツールを使用して錠剤へと圧縮する。この最終混合物を約179.4mgの錠剤とする。実施例30および31の錠剤の成分を下記の表40および41に示す。
【表40】
【表41】
【0214】
最終産物の錠剤は、錠剤として重量179.4mgであり、硬度5Kpである。
【0215】
続いて、実施例30および31の錠剤に対する溶出試験を行った。in-vitro溶出試験結果を下記の表38に示す。
【表42】
【0216】
表42に示された結果から、デキストロース量に対してガムの量が増加するにつれて、これに対応するオキシブチニン放出の減少が見られることが明らかである。
【0217】
実施例32〜35
コハク酸の効果
実施例32および33において、実施例28および29に記載の方法により徐放性賦形剤を調製する。実施例32および33の徐放性賦形剤の成分を下記の表43に示す。
【表43】
【0218】
その後、オキシブチニン錠剤を次のように調製する。
【0219】
所望の量のコハク酸、オキシブチニンおよびステアリルフマル酸ナトリウムを25メッシュの篩で篩分けし、篩分けしたコハク酸および徐放性賦形剤をV-blenderに配合し、10分間にわたりブレンドし、篩分けしたオキシブチニンを、コハク酸および徐放性賦形剤のブレンド混合物に添加してさらに5分間にわたりブレンドし、篩分けしたステアリルフマル酸ナトリウムを、オキシブチニン、コハク酸および徐放性賦形剤のブレンド混合物に添加してさらに5分間にわたりブレンドし、次いでブレンドした最終の目的生成物を5/16”の丸型ツールを使用して錠剤へと圧縮する。実施例32の最終混合物を約251.0mgの錠剤とし、実施例33の最終混合物を約296.0mgの錠剤とする。実施例32および33の錠剤の成分を下記の表44および45に示す。
【表44】
【0220】
最終産物の錠剤は、錠剤として重量251.0mgであり、硬度8Kpである。
【表45】
【0221】
最終産物の錠剤は、錠剤として重量296.0mgであり、硬度8Kpである。
【0222】
続いて、実施例32および33の錠剤に対する溶出試験を行った。in-vitro溶出試験結果を下記の表46に示す。
【表46】
【0223】
表46に示される結果から、コハク酸の添加が薬物の溶出性を促進し、それゆえ、放出速度を増加させることは明らかである。
【0224】
実施例34および35において、実施例28および29に記載の方法により徐放性賦形剤を調製する。実施例34および35の徐放性賦形剤の成分を下記の表47に示す。
【表47】
【0225】
その後、オキシブチニン錠剤を次のように調製する。
【0226】
所望の量の徐放性賦形剤、コハク酸、およびオキシブチニンを造粒機中に配合する。それらを低速で圧縮機によって、チョッパーブレイド(the chopper blade)はオフにした状態で3分間にわたり乾燥混合する。水を1分間隔で添加し、次いで混合物を高速で3分間にわたり顆粒化する(好適な顆粒を形成するために、さらに水および顆粒化時間を増しても良い)。次に、混合物をLODが5%未満になるまで液体ベッドドライヤーで乾燥する。乾燥した顆粒を、2000〜3000rpmで作動しているブレードで粉砕する。粉砕した顆粒およびステアリルフマル酸ナトリウムをV-Blender中に配置し10分間にわたりブレンドする。次いでブレンドした混合物を5/16”の丸型ツールを使用して錠剤へと圧縮する。実施例34の最終混合物を約296.0mgの錠剤とし、実施例35の最終混合物を約266.0mgの錠剤とする。実施例34および35の錠剤の成分を下記の表48および49に示す。
【表48】
【0227】
最終産物の錠剤は、錠剤として重量296.0mgであり、硬度8Kpである。
【表49】
【0228】
最終産物の錠剤は、錠剤として重量266.0mgであり、硬度8Kpである。
【0229】
続いて、実施例34および35の錠剤に対する溶出試験を行った。in-vitro溶出試験結果を下記の表50に示す。
【表50】
【0230】
表50に示される結果から、製剤中のコハク酸の量が多いほど、放出速度が速くなることは明らかである。
【0231】
実施例36
エチルセルロース(SURELEASE(登録商標))/OPADRY(登録商標)(80/20)水性分散液による錠剤のコーティングの効果
エチルセルロース/Opadryコーティングで製剤する方法は次の通りである。
【0232】
先ず初めに、好適な容器に340gの水を量り、混合しながら水の中にOpadryを60g添加する。混合を続ける。Opadry分散液を混合しながら、933gのエチルセルロース分散液(Surelease)を添加し、30〜60分間攪拌する。最終的な分散液を用いて、錠剤の総重量に基づき3〜5%の重量増加まで錠剤をコーティングする。
【0233】
実施例36においては、実施例28および29に記載の方法により徐放性賦形剤を調製する。実施例36の徐放性賦形剤の成分を下記の表51に示す。
【表51】
【0234】
その後、オキシブチニン錠剤を次のように調製する。
【0235】
所望の量の徐放性賦形剤、コハク酸、およびオキシブチニンを造粒機中に配合する。それらを低速で圧縮機によって、チョッパーブレイドはオフにした状態で3分間にわたり乾燥混合する。水を1分間隔で添加し、次いで、混合物を高速で3分間にわたり顆粒化する(好適な顆粒を形成するために、さらに水および顆粒化時間を増しても良い)。次に、混合物をLODが5%未満になるまで液体ベッドドライヤーで乾燥する。乾燥した顆粒を、2000〜3000rpmで作動しているブレードで粉砕する。粉砕した顆粒およびステアリルフマル酸ナトリウムをV-Blender中に配置し10分間にわたりブレンドする。次いでブレンドした混合物を5/16”の丸型ツールを使用して錠剤へと圧縮する。実施例36の最終混合物を約296.0mgの錠剤とする。実施例36の成分を下記の表52に示す。
【表52】
【0236】
最終産物の錠剤は、錠剤として重量296.0mgであり、硬度8Kpであった。
【0237】
続いて、実施例36の錠剤に対する溶出試験を行った。in-vitro溶出試験結果を下記の表53に示す。
【表53】
【0238】
表53に示される結果から、コーテイングの量(重量による)が増加するにつれて、放出速度は減少することは明らかである。
【0239】
実施例37
フマル酸の効果
実施例37において、実施例28および29に記載の方法により徐放性賦形剤を調製する。実施例37の徐放性賦形剤の成分を下記の表54に示す。
【表54】
【0240】
最終産物の錠剤は、213.7mgの錠剤重量を有する。
【0241】
続いて、実施例37の錠剤に対する溶出試験を、ディトロパン XLと比較して行った。in-vitro溶出試験結果を下記の表55に示す。
【表55】
【0242】
pH改変剤にフマル酸を含む場合における他のオキシブチニン製剤を下記の表56に示す。
【表56】
【0243】
上記で与えられた実施例は限定的であることを意味しない。本発明の多くの他の変形例が当業者には明らかであり、それらは添付した特許請求の範囲の範囲内にあるものとして意図されている。
【図面の簡単な説明】
【0244】
【図1】実施例1および2に関する溶出(経時平均溶出%)のグラフを示す。
【図2】実施例3および4に関する溶出(経時平均溶出%)のグラフを示す。
【図3】実施例5および6に関する溶出(経時平均溶出%)のグラフを示す。
【図4】実施例7および8に関する溶出(経時平均溶出%)のグラフを示す。
【図5】実施例9および10に関する溶出(経時平均溶出%)のグラフを示す。
【図6】実施例11および12に関する溶出(経時平均溶出%)のグラフを示す。
【図7】実施例11および12に関する経時放出速度(%)のグラフを示す。
【図8】実施例13および18に関する溶出(経時平均溶出%)のグラフを示す。
【図9】実施例19および20に関する溶出(経時平均溶出%)のグラフを示す。
【図10】実施例21〜23に関する溶出(経時平均溶出%)のグラフを示す。
【図11】実施例24と参照基準(Cardizem CD 240mg)に関する経時平均ジルチアゼム血漿濃度(ng/ml)のグラフを示す。
【図12】実施例25と参照基準(Cardizem CD 240mg)に関する経時平均ジルチアゼム血漿濃度(ng/ml)のグラフを示す。
【図13】実施例26および27に関する溶出(経時平均溶出%)のグラフを示す。
【図14】実施例37と参照基準(ディトロパン XL)の溶出(経時平均溶出%)を比較したグラフを示す。
【特許請求の範囲】
【請求項1】
以下の成分:
可溶性が約10g/lより大きい、治療上有効な量の薬物;
pH改変剤;
ヘテロ多糖ガム及び周囲の液体に晒された際に該ヘテロ多糖ガムを架橋することができるホモ多糖ガムとを含有するゲル化剤を含む徐放性マトリックス;
を含有する経口用徐放性固体剤形であって、ヒト患者への経口投与後に上記薬物の持続放出を提供する、上記剤形。
【請求項2】
経口投与後少なくとも約12時間にわたって上記薬物の持続放出を提供する、請求項1記載の経口用徐放性固体剤形。
【請求項3】
経口投与後少なくとも約24時間にわたって上記薬物の持続放出を提供する、請求項1記載の経口用徐放性固体剤形。
【請求項4】
上記薬物の可溶性が約100g/lより大きい、請求項1記載の経口用徐放性固体剤形。
【請求項5】
上記薬物の可溶性が約1000g/lより大きい、請求項1記載の経口用徐放性固体剤形。
【請求項6】
不活性医薬希釈剤をさらに含有する、請求項1記載の経口用徐放性固体剤形。
【請求項7】
上記不活性希釈剤が、単糖類、二糖類、多価アルコール、およびこれらの混合物からなる群より選択される、請求項6記載の経口用徐放性固体剤形。
【請求項8】
上記不活性希釈剤と上記ゲル化剤との比が約1:3〜約3:1である、請求項6記載の経口用徐放性固体剤形。
【請求項9】
上記薬物と上記ゲル化剤との比が約1:5〜約5:1である、請求項1記載の経口用徐放性固体剤形。
【請求項10】
周囲の液体に晒された際に、上記ゲル化剤と架橋してゲル強度を増大させることができるイオン化ゲル強度増強剤をさらに含有する、請求項1記載の経口用徐放性固体剤形。
【請求項11】
上記イオン化ゲル強度増強剤が、アルカリ金属またはアルカリ土類金属の硫酸塩、塩化物、ホウ酸塩、臭化物、クエン酸塩、酢酸塩または乳酸塩を含む、請求項10記載の経口用固体剤形。
【請求項12】
上記イオン化ゲル強度増強剤が硫酸カルシウムを含む、請求項11記載の経口用固体剤形。
【請求項13】
上記ヘテロ多糖ガムがキサンタンガムを含み、上記ホモ多糖ガムがイナゴマメガムを含む、請求項1記載の経口用固体剤形。
【請求項14】
上記pH改変剤が有機酸である、請求項1記載の経口用固体剤形。
【請求項15】
上記有機酸が、クエン酸、コハク酸、フマル酸、リンゴ酸、マレイン酸、グルタル酸、乳酸、およびこれらの混合物からなる群より選択される、請求項14記載の経口用徐放性固体剤形。
【請求項16】
上記有機酸がフマル酸である、請求項15記載の経口用徐放性固体剤形。
【請求項17】
上記pH改変剤が約1%〜約10%の量で存在する、請求項1記載の経口用固体剤形。
【請求項18】
界面活性剤をさらに含有する、請求項1記載の経口用固体剤形。
【請求項19】
上記界面活性剤が、アニオン性界面活性剤、カチオン性界面活性剤、両性(両親媒性/両染性)界面活性剤、および非イオン性界面活性剤からなる群より選択される、請求項18記載の経口用固体剤形。
【請求項20】
上記界面活性剤が、ラウリル硫酸ナトリウムおよびドキュセートの薬学的に有効な塩からなる群より選択される、請求項18記載の経口用固体剤形。
【請求項21】
上記徐放性マトリックスが疎水性物質をさらに含む、請求項1記載の経口用固体剤形。
【請求項22】
上記疎水性物質が、アルキルセルロース、アクリル酸エステルとメタクリル酸エステルとのコポリマー、ワックス、シェラック、ゼイン、硬化植物油、およびこれらの混合物からなる群より選択され、周囲の液体に晒された際に上記ゲル化剤の水和を緩慢にするのに有効な量で含まれる、請求項21記載の経口用固体剤形。
【請求項23】
上記疎水性物質がエチルセルロースである、請求項21記載の経口用固体剤形。
【請求項24】
上記徐放性マトリックスが約1〜約20重量%の疎水性物質を含む、請求項5記載の経口用固体剤形。
【請求項25】
約1〜約10重量%の微晶質セルロースをさらに含有する、請求項1記載の経口用固体剤形。
【請求項26】
上記薬物がベンゾチアジンである、請求項1記載の経口用固体剤形。
【請求項27】
上記ベンゾチアジンがジルチアゼムまたはその薬学的に有効な塩である、請求項26記載の経口用固体剤形。
【請求項28】
ヒト患者への経口投与後少なくとも約12時間にわたって上記ジルチアゼムの持続放出を提供する、請求項27記載の経口用固体剤形。
【請求項29】
上記ジルチアゼムが約60mg〜約120mgの量で存在する、請求項28記載の経口用固体剤形。
【請求項30】
ヒト患者への経口投与後少なくとも約24時間にわたって上記ジルチアゼムの持続放出を提供する、請求項27記載の経口用固体剤形。
【請求項31】
上記ジルチアゼムが約120mg〜約300mgの量で存在する、請求項30記載の経口用固体剤形。
【請求項32】
上記薬物が鎮痙剤である、請求項1記載の経口用固体剤形。
【請求項33】
上記鎮痙薬がオキシブチニンまたはその薬学的に許容される塩である、請求項32記載の経口用固体剤形。
【請求項34】
上記鎮痙剤が塩化オキシブチニンである、請求項33記載の経口用固体剤形。
【請求項35】
ヒト患者への経口投与後少なくとも12時間にわたって上記オキシブチニンの持続放出を提供する、請求項33記載の経口用固体剤形。
【請求項36】
上記オキシブチニンが約2.5mg〜約25mgの量で存在する、請求項35記載の経口用固体剤形。
【請求項37】
ヒト患者への経口投与後少なくとも約24時間にわたって上記オキシブチニンの持続放出を提供する、請求項33記載の経口用固体剤形。
【請求項38】
上記オキシブチニンが約5mg〜約50mgの量で存在する、請求項37記載の経口用固体剤形。
【請求項39】
錠剤である、請求項1記載の経口用固体剤形。
【請求項40】
顆粒形態である、請求項1記載の経口用固体剤形。
【請求項41】
上記薬物の一部が顆粒の外側に存在する、請求項40記載の経口用固体剤形。
【請求項42】
有効量の上記薬物を提供するのに十分な量の上記顆粒が、薬学的に許容されるカプセル中に入れられる、請求項40記載の経口用固体剤形。
【請求項43】
上記錠剤の表面の少なくとも一部が、重量の約1〜約20重量%が増加する程度に疎水性物質でコーティングされる、請求項39記載の経口用固体剤形。
【請求項44】
上記疎水性物質が、アルキルセルロース、アクリル酸とメタクリル酸とのコポリマー、ワックス、シェラック、ゼイン、硬化植物油、およびこれらの混合物からなる群より選択される、請求項43記載の経口用固体剤形。
【請求項45】
上記薬物の二相性吸収プロフィールを提供する、請求項18記載の経口用徐放性固体剤形。
【請求項46】
以下の成分:
治療効果を得るのに有効な量の、可溶性が10g/Lより大きいカルシウムチャネル遮断剤;
pH改変剤;
薬学的に許容される界面活性剤;
ヘテロ多糖ガムと周囲の液体に晒された際に該ヘテロ多糖ガムを架橋することができるホモ多糖ガムとを含有するゲル化剤を含む徐放性賦形剤;
を含有する経口用徐放性固体剤形であって、上記カルシウムチャネル遮断剤の二相性吸収プロフィールを提供し、かつヒト患者への経口投与後少なくとも約12時間にわたって該カルシウムチャネル遮断剤の持続放出を提供する、上記剤形。
【請求項47】
上記カルシウムチャネル遮断剤がベンゾチアジンである、請求項46記載の経口用固体剤形。
【請求項48】
上記ベンゾチアジンがジルチアゼムまたはその薬学的に許容される塩である、請求項46記載の経口用固体剤形。
【請求項49】
請求項48記載の経口用固体剤形であって、上記ジルチアゼムの初回ピーク濃度(Cmax#1)が上記剤形の経口投与後約4〜約10時間で得られ、続いて第2のピーク濃度(Cmax#2)が、上記剤形の経口投与後約10〜約16時間で起こり、そしてヒト患者への経口投与後少なくとも約24時間にわたって治療効果が得られる、上記剤形。
【請求項50】
ジルチアゼムの第1ピーク血漿濃度の出現時間(Tmax#1)が、上記剤形の患者への経口投与後約6〜約8時間である、請求項49記載の剤形。
【請求項51】
ジルチアゼムの第1Tmaxにおける最大血漿濃度(Cmax#1)が、240mg用量のジルチアゼムの投与当たり約50〜約100ng/mlである、請求項49記載の剤形。
【請求項52】
第2ピーク血漿濃度(Cmax#2)が、上記剤形の患者への経口投与後約12〜約14時間(Tmax#2)において起こる、請求項46記載の剤形。
【請求項53】
ジルチアゼムのCmax#2における最大血漿濃度が、240mgのジルチアゼム当たり約60〜約90ng/mlである、請求項52記載の剤形。
【請求項54】
Cmax#1とCmax#2との間のCminとして得られた最低値を基準にして、Cmax#1の高さの50%における血漿濃度曲線の幅が、約0.5〜約4.0時間である、請求項46記載の剤形。。
【請求項55】
Cmax#1とCmax#2との間のCminとして得られた最低値を基準にして、Cmax#1の高さの50%における血漿濃度曲線の幅が、約1〜約3時間である、請求項46記載の剤形。
【請求項56】
Cmax#1とCmax#2との間のCminとして得られた最低値を基準にして、Cmax#2の高さの50%における血漿濃度曲線の幅が、約0.5〜約8時間である、請求項46記載の剤形。
【請求項57】
Cmax#1とCmax#2との間のCminとして得られた最低値を基準にして、Cmax#2の高さの50%における血漿濃度曲線の幅が、約2〜約6時間である、請求項46記載の剤形。
【請求項58】
Cmax#1とCmax#2との比が、約0.5:1〜約1.5:1である、請求項49記載の剤形。
【請求項59】
Cmax#1とCmax#2との比が、約0.7:1〜約1.2:1である、請求項58記載の剤形。
【請求項60】
高血圧、狭心症、動脈瘤、不整脈および頭痛からなる群より選択させる病状を患うヒト患者の治療用である、請求項46記載の剤形。
【請求項61】
以下の成分:
治療効果を得るために有効な量のオキシブチニンまたはその薬学的に許容される塩;
pH改変剤;
ヘテロ多糖ガム及び該ヘテロ多糖ガムを架橋することのできるホモ多糖ガムとを含有するゲル化剤を含む徐放性賦形剤;
を含有する経口用徐放性固体剤形であって、ヒト患者への投与後少なくとも約24時間にわたって治療効果が得られる、上記剤形。
【請求項62】
ピーク血漿濃度時間(Tmax)が約5〜約15時間である、請求項61記載の経口用徐放性固体剤形。
【請求項63】
ピーク血漿濃度時間(Tmax)が約8〜約12時間である、請求項62記載の経口用徐放性固体剤形。
【請求項64】
失禁症を患うヒト患者の治療用である、請求項61記載の剤形。
【請求項1】
以下の成分:
可溶性が約10g/lより大きい、治療上有効な量の薬物;
pH改変剤;
ヘテロ多糖ガム及び周囲の液体に晒された際に該ヘテロ多糖ガムを架橋することができるホモ多糖ガムとを含有するゲル化剤を含む徐放性マトリックス;
を含有する経口用徐放性固体剤形であって、ヒト患者への経口投与後に上記薬物の持続放出を提供する、上記剤形。
【請求項2】
経口投与後少なくとも約12時間にわたって上記薬物の持続放出を提供する、請求項1記載の経口用徐放性固体剤形。
【請求項3】
経口投与後少なくとも約24時間にわたって上記薬物の持続放出を提供する、請求項1記載の経口用徐放性固体剤形。
【請求項4】
上記薬物の可溶性が約100g/lより大きい、請求項1記載の経口用徐放性固体剤形。
【請求項5】
上記薬物の可溶性が約1000g/lより大きい、請求項1記載の経口用徐放性固体剤形。
【請求項6】
不活性医薬希釈剤をさらに含有する、請求項1記載の経口用徐放性固体剤形。
【請求項7】
上記不活性希釈剤が、単糖類、二糖類、多価アルコール、およびこれらの混合物からなる群より選択される、請求項6記載の経口用徐放性固体剤形。
【請求項8】
上記不活性希釈剤と上記ゲル化剤との比が約1:3〜約3:1である、請求項6記載の経口用徐放性固体剤形。
【請求項9】
上記薬物と上記ゲル化剤との比が約1:5〜約5:1である、請求項1記載の経口用徐放性固体剤形。
【請求項10】
周囲の液体に晒された際に、上記ゲル化剤と架橋してゲル強度を増大させることができるイオン化ゲル強度増強剤をさらに含有する、請求項1記載の経口用徐放性固体剤形。
【請求項11】
上記イオン化ゲル強度増強剤が、アルカリ金属またはアルカリ土類金属の硫酸塩、塩化物、ホウ酸塩、臭化物、クエン酸塩、酢酸塩または乳酸塩を含む、請求項10記載の経口用固体剤形。
【請求項12】
上記イオン化ゲル強度増強剤が硫酸カルシウムを含む、請求項11記載の経口用固体剤形。
【請求項13】
上記ヘテロ多糖ガムがキサンタンガムを含み、上記ホモ多糖ガムがイナゴマメガムを含む、請求項1記載の経口用固体剤形。
【請求項14】
上記pH改変剤が有機酸である、請求項1記載の経口用固体剤形。
【請求項15】
上記有機酸が、クエン酸、コハク酸、フマル酸、リンゴ酸、マレイン酸、グルタル酸、乳酸、およびこれらの混合物からなる群より選択される、請求項14記載の経口用徐放性固体剤形。
【請求項16】
上記有機酸がフマル酸である、請求項15記載の経口用徐放性固体剤形。
【請求項17】
上記pH改変剤が約1%〜約10%の量で存在する、請求項1記載の経口用固体剤形。
【請求項18】
界面活性剤をさらに含有する、請求項1記載の経口用固体剤形。
【請求項19】
上記界面活性剤が、アニオン性界面活性剤、カチオン性界面活性剤、両性(両親媒性/両染性)界面活性剤、および非イオン性界面活性剤からなる群より選択される、請求項18記載の経口用固体剤形。
【請求項20】
上記界面活性剤が、ラウリル硫酸ナトリウムおよびドキュセートの薬学的に有効な塩からなる群より選択される、請求項18記載の経口用固体剤形。
【請求項21】
上記徐放性マトリックスが疎水性物質をさらに含む、請求項1記載の経口用固体剤形。
【請求項22】
上記疎水性物質が、アルキルセルロース、アクリル酸エステルとメタクリル酸エステルとのコポリマー、ワックス、シェラック、ゼイン、硬化植物油、およびこれらの混合物からなる群より選択され、周囲の液体に晒された際に上記ゲル化剤の水和を緩慢にするのに有効な量で含まれる、請求項21記載の経口用固体剤形。
【請求項23】
上記疎水性物質がエチルセルロースである、請求項21記載の経口用固体剤形。
【請求項24】
上記徐放性マトリックスが約1〜約20重量%の疎水性物質を含む、請求項5記載の経口用固体剤形。
【請求項25】
約1〜約10重量%の微晶質セルロースをさらに含有する、請求項1記載の経口用固体剤形。
【請求項26】
上記薬物がベンゾチアジンである、請求項1記載の経口用固体剤形。
【請求項27】
上記ベンゾチアジンがジルチアゼムまたはその薬学的に有効な塩である、請求項26記載の経口用固体剤形。
【請求項28】
ヒト患者への経口投与後少なくとも約12時間にわたって上記ジルチアゼムの持続放出を提供する、請求項27記載の経口用固体剤形。
【請求項29】
上記ジルチアゼムが約60mg〜約120mgの量で存在する、請求項28記載の経口用固体剤形。
【請求項30】
ヒト患者への経口投与後少なくとも約24時間にわたって上記ジルチアゼムの持続放出を提供する、請求項27記載の経口用固体剤形。
【請求項31】
上記ジルチアゼムが約120mg〜約300mgの量で存在する、請求項30記載の経口用固体剤形。
【請求項32】
上記薬物が鎮痙剤である、請求項1記載の経口用固体剤形。
【請求項33】
上記鎮痙薬がオキシブチニンまたはその薬学的に許容される塩である、請求項32記載の経口用固体剤形。
【請求項34】
上記鎮痙剤が塩化オキシブチニンである、請求項33記載の経口用固体剤形。
【請求項35】
ヒト患者への経口投与後少なくとも12時間にわたって上記オキシブチニンの持続放出を提供する、請求項33記載の経口用固体剤形。
【請求項36】
上記オキシブチニンが約2.5mg〜約25mgの量で存在する、請求項35記載の経口用固体剤形。
【請求項37】
ヒト患者への経口投与後少なくとも約24時間にわたって上記オキシブチニンの持続放出を提供する、請求項33記載の経口用固体剤形。
【請求項38】
上記オキシブチニンが約5mg〜約50mgの量で存在する、請求項37記載の経口用固体剤形。
【請求項39】
錠剤である、請求項1記載の経口用固体剤形。
【請求項40】
顆粒形態である、請求項1記載の経口用固体剤形。
【請求項41】
上記薬物の一部が顆粒の外側に存在する、請求項40記載の経口用固体剤形。
【請求項42】
有効量の上記薬物を提供するのに十分な量の上記顆粒が、薬学的に許容されるカプセル中に入れられる、請求項40記載の経口用固体剤形。
【請求項43】
上記錠剤の表面の少なくとも一部が、重量の約1〜約20重量%が増加する程度に疎水性物質でコーティングされる、請求項39記載の経口用固体剤形。
【請求項44】
上記疎水性物質が、アルキルセルロース、アクリル酸とメタクリル酸とのコポリマー、ワックス、シェラック、ゼイン、硬化植物油、およびこれらの混合物からなる群より選択される、請求項43記載の経口用固体剤形。
【請求項45】
上記薬物の二相性吸収プロフィールを提供する、請求項18記載の経口用徐放性固体剤形。
【請求項46】
以下の成分:
治療効果を得るのに有効な量の、可溶性が10g/Lより大きいカルシウムチャネル遮断剤;
pH改変剤;
薬学的に許容される界面活性剤;
ヘテロ多糖ガムと周囲の液体に晒された際に該ヘテロ多糖ガムを架橋することができるホモ多糖ガムとを含有するゲル化剤を含む徐放性賦形剤;
を含有する経口用徐放性固体剤形であって、上記カルシウムチャネル遮断剤の二相性吸収プロフィールを提供し、かつヒト患者への経口投与後少なくとも約12時間にわたって該カルシウムチャネル遮断剤の持続放出を提供する、上記剤形。
【請求項47】
上記カルシウムチャネル遮断剤がベンゾチアジンである、請求項46記載の経口用固体剤形。
【請求項48】
上記ベンゾチアジンがジルチアゼムまたはその薬学的に許容される塩である、請求項46記載の経口用固体剤形。
【請求項49】
請求項48記載の経口用固体剤形であって、上記ジルチアゼムの初回ピーク濃度(Cmax#1)が上記剤形の経口投与後約4〜約10時間で得られ、続いて第2のピーク濃度(Cmax#2)が、上記剤形の経口投与後約10〜約16時間で起こり、そしてヒト患者への経口投与後少なくとも約24時間にわたって治療効果が得られる、上記剤形。
【請求項50】
ジルチアゼムの第1ピーク血漿濃度の出現時間(Tmax#1)が、上記剤形の患者への経口投与後約6〜約8時間である、請求項49記載の剤形。
【請求項51】
ジルチアゼムの第1Tmaxにおける最大血漿濃度(Cmax#1)が、240mg用量のジルチアゼムの投与当たり約50〜約100ng/mlである、請求項49記載の剤形。
【請求項52】
第2ピーク血漿濃度(Cmax#2)が、上記剤形の患者への経口投与後約12〜約14時間(Tmax#2)において起こる、請求項46記載の剤形。
【請求項53】
ジルチアゼムのCmax#2における最大血漿濃度が、240mgのジルチアゼム当たり約60〜約90ng/mlである、請求項52記載の剤形。
【請求項54】
Cmax#1とCmax#2との間のCminとして得られた最低値を基準にして、Cmax#1の高さの50%における血漿濃度曲線の幅が、約0.5〜約4.0時間である、請求項46記載の剤形。。
【請求項55】
Cmax#1とCmax#2との間のCminとして得られた最低値を基準にして、Cmax#1の高さの50%における血漿濃度曲線の幅が、約1〜約3時間である、請求項46記載の剤形。
【請求項56】
Cmax#1とCmax#2との間のCminとして得られた最低値を基準にして、Cmax#2の高さの50%における血漿濃度曲線の幅が、約0.5〜約8時間である、請求項46記載の剤形。
【請求項57】
Cmax#1とCmax#2との間のCminとして得られた最低値を基準にして、Cmax#2の高さの50%における血漿濃度曲線の幅が、約2〜約6時間である、請求項46記載の剤形。
【請求項58】
Cmax#1とCmax#2との比が、約0.5:1〜約1.5:1である、請求項49記載の剤形。
【請求項59】
Cmax#1とCmax#2との比が、約0.7:1〜約1.2:1である、請求項58記載の剤形。
【請求項60】
高血圧、狭心症、動脈瘤、不整脈および頭痛からなる群より選択させる病状を患うヒト患者の治療用である、請求項46記載の剤形。
【請求項61】
以下の成分:
治療効果を得るために有効な量のオキシブチニンまたはその薬学的に許容される塩;
pH改変剤;
ヘテロ多糖ガム及び該ヘテロ多糖ガムを架橋することのできるホモ多糖ガムとを含有するゲル化剤を含む徐放性賦形剤;
を含有する経口用徐放性固体剤形であって、ヒト患者への投与後少なくとも約24時間にわたって治療効果が得られる、上記剤形。
【請求項62】
ピーク血漿濃度時間(Tmax)が約5〜約15時間である、請求項61記載の経口用徐放性固体剤形。
【請求項63】
ピーク血漿濃度時間(Tmax)が約8〜約12時間である、請求項62記載の経口用徐放性固体剤形。
【請求項64】
失禁症を患うヒト患者の治療用である、請求項61記載の剤形。
【図1】

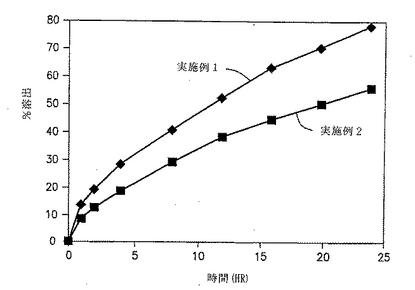
【図2】
【図3】
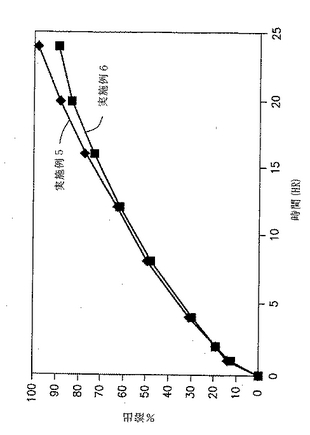
【図4】
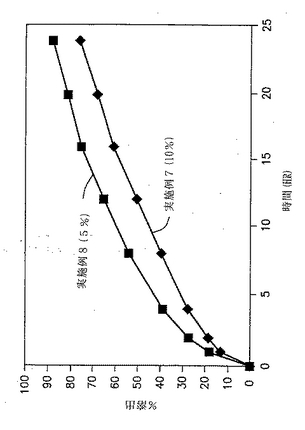
【図5】
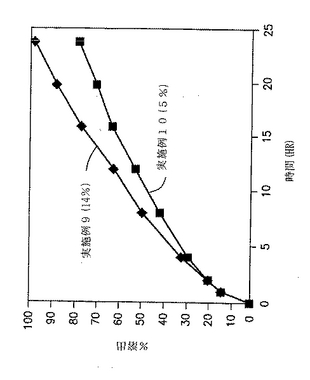
【図6】
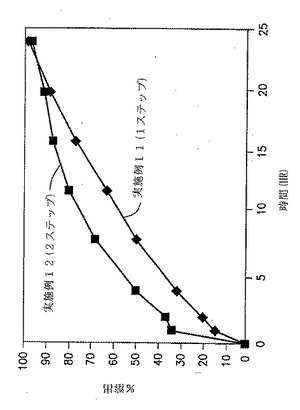
【図7】
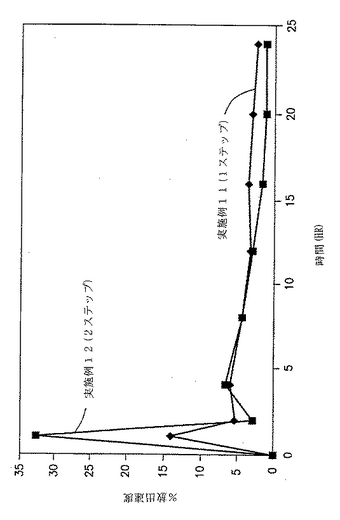
【図8】
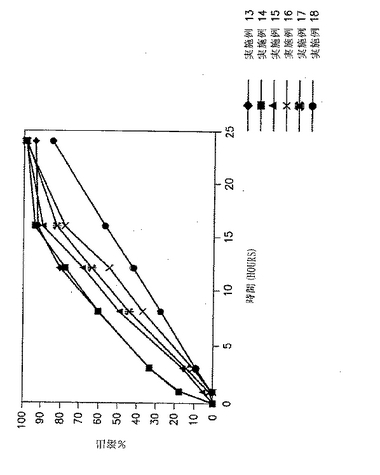
【図9】
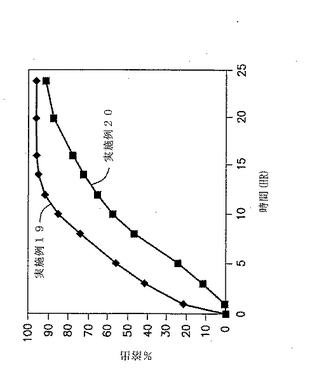
【図10】
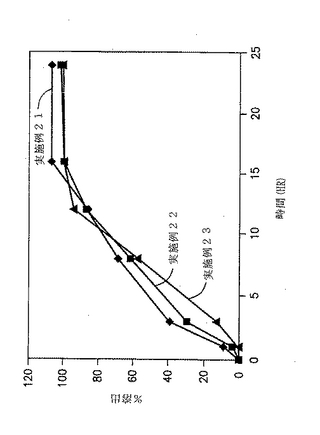
【図11】
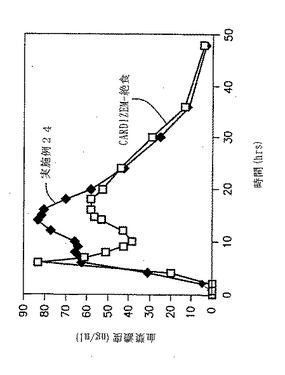
【図12】
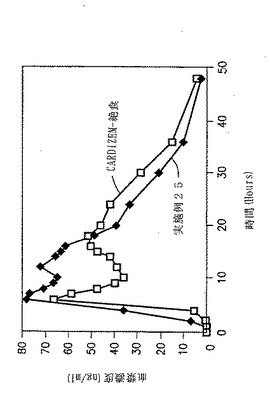
【図13】
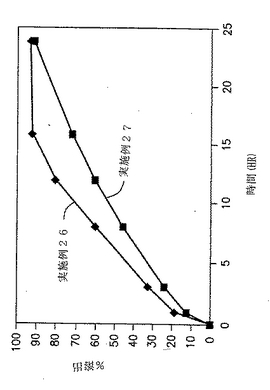
【図14】
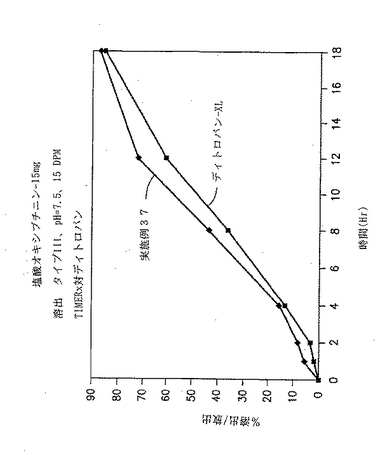
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【図11】
【図12】
【図13】
【図14】
【公開番号】特開2006−306893(P2006−306893A)
【公開日】平成18年11月9日(2006.11.9)
【国際特許分類】
【出願番号】特願2006−211366(P2006−211366)
【出願日】平成18年8月2日(2006.8.2)
【分割の表示】特願2001−526152(P2001−526152)の分割
【原出願日】平成12年9月29日(2000.9.29)
【出願人】(500175152)ペンウェスト ファーマシューティカルズ カンパニー (9)
【Fターム(参考)】
【公開日】平成18年11月9日(2006.11.9)
【国際特許分類】
【出願日】平成18年8月2日(2006.8.2)
【分割の表示】特願2001−526152(P2001−526152)の分割
【原出願日】平成12年9月29日(2000.9.29)
【出願人】(500175152)ペンウェスト ファーマシューティカルズ カンパニー (9)
【Fターム(参考)】
[ Back to top ]