
高度の歪み構造を有するインゲナン骨格の合成に係わる方法
【課題】 インゲノールの有用な合成経路を提供すること。
【解決手段】 ジオレフィン化合物の一方のオレフィン部位をスピロ環で固定して三環性ジオレフィン化合物を得る工程と、該三環性ジオレフィン化合物のジオレフィン部位をオレフィンメタセシス反応で閉環することにより四環性のノルインゲナン骨格を合成する方法、特に光学活性を示すノルインゲナン骨格を合成する方法、並びに該方法を用いたインゲノールの合成方法を提供する。
【解決手段】 ジオレフィン化合物の一方のオレフィン部位をスピロ環で固定して三環性ジオレフィン化合物を得る工程と、該三環性ジオレフィン化合物のジオレフィン部位をオレフィンメタセシス反応で閉環することにより四環性のノルインゲナン骨格を合成する方法、特に光学活性を示すノルインゲナン骨格を合成する方法、並びに該方法を用いたインゲノールの合成方法を提供する。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、インゲノールの合成に係わる技術に関する。より詳しくは、オレフィンメタセシスによる閉環反応を鍵反応として、インゲノール合成経路の中間体として有用なノルインゲナン骨格(炭素数19)を合成する技術に関する。
【背景技術】
【0002】
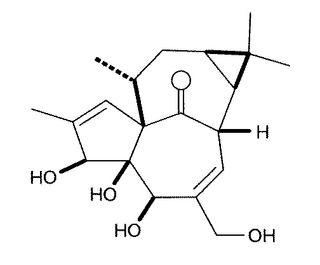
図3に示す化学構造のインゲノール(ingenol)は、トウダイクサ科の植物チュウテンカクより単離されたジテルペン(炭素数20)であり、その不飽和脂肪酸エステルは、様々な生理活性を持つことから、現在注目されている。
【0003】
例えば、特許文献1には、13−オキシインゲノール誘導体が、ヒト静脈内皮細胞による細胞接着分子(VCAM-1)の産生阻止作用を示し、癌転移抑制剤、抗炎症剤、動脈硬化、移植拒絶反応、慢性関節リウマチ、サルコイドーシス等の治療薬としての利用が期待される旨が開示されている。その他、インゲノールの誘導体は、発癌プロモーター作用、抗白血病作用、抗HIV活性などの生理活性を示すことが知られている。
【0004】
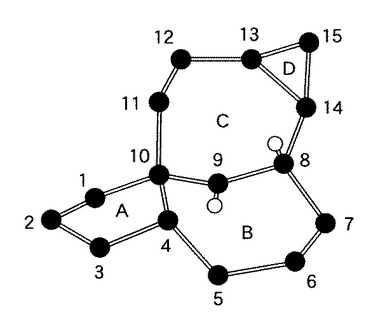
ここで、インゲノールは、transに閉環し強く歪んだ四環性のインゲナン骨格(inside,outside-ビシクロ[4.4.1]ウンデカン骨格)を持っている。具体的には、図4に示されたインゲノールの炭素環骨格は、図中のB環とC環が互いにブリッジしているので、橋頭位の10位から1位への炭素−炭素結合がBC環部の外側を向いている。これに対して、もう一方の橋頭位の8位上の水素原子は、BC環の内側に入り込んでいる。このためインゲナン骨格は、高度に歪んでいる。
【0005】
このような高度の歪み構造を有するインゲナンの炭素環骨格は、合成が困難であるので、多くの有機合成化学者の関心を集めている。これまでのところ、例えば、Winklerのグループが分子内光[2+2]付加環化反応及び四員環の開裂反応を用いて、インゲノールの最初の全合成を達成している(非特許文献1参照)。また、谷野らのグループが、インゲノールの全合成を成功させている(非特許文献2参照)。これらのグループの合成により得られたインゲノールは、いずれもラセミ体である。
【特許文献1】特開平07−258168号公報。
【非特許文献1】Winkler,J.D.;Rouse,M.B.; Greaney,M.F.; Harrison,Sean.J.;Jeon,Y.T.J.Am.Chem.Soc.2002,124,9726-9728。
【非特許文献2】Tanino,K.;Onuki,K.;Asano,K.;Miyashita,M.;Nakamura,T.;Takahashi,Y.;Kuwajima,I.J.AM.CHEM.SOC.2003,125,1498-1500。
【発明の開示】
【発明が解決しようとする課題】
【0006】
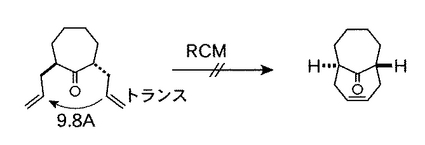
本願発明者らは、インゲナンの炭素環骨格の効率的な合成経路を模索する中で、インゲナン骨格を直接、環化反応により合成する経路に着目し、長年鋭意研究を続けたところ、オレフィンメタセシスによる閉環反応(以下「環化オレフィンメタセシス(略称RCM)」)では、環化に寄与する二つのオレフィン部位が空間的に近く配置されている必要があることを突き止めた。例えば、図5に示すように、二つのオレフィン部位がtrans体を構成する2,7-ジアリルシクロヘプタノンでは、環化オレフィンメタセシスによる閉環が困難である。
【0007】
そこで、本発明は、環化オレフィンメタセシスを有効に利用して、インゲナン骨格を直接、環化反応により合成する経路を見出すことにより、インゲノールの有用な合成経路を提供することを主な目的とする。
【課題を解決するための手段】
【0008】
本発明では、ジオレフィン化合物の一方のオレフィン部位をスピロ環で固定して三環性ジオレフィン化合物を得る工程と、該三環性ジオレフィン化合物のジオレフィン部位をオレフィンメタセシス反応で閉環する工程と、を少なくとも行うことによって四環性のノルインゲナン骨格を合成する方法、特に、薬剤開発において有用となる光学活性を有するノルインゲナン骨格を合成する方法を提供し、並びに、これらの方法を用いたインゲノール合成方法を提供する。
【0009】
ジオレフィン化合物の一方のオレフィン部位がスピロ環で固定された構造を有する三環性ジオレフィン化合物は、二つのオレフィン部位(二重結合部位)の距離が、オレフィンメタセシス反応が可能なまでに近づいているので、該オレフィンメタセシス反応による閉環を進行させることが可能となる。
【0010】
また、本発明では、上記方法によって得られるノルインゲナン骨格の第6位炭素に、アルデヒド基又はアルデヒド基に変換可能な基(例えば、メチル基、エチル基などのアルキル基、カルボキシル基、−CH2OR基など)を結合させておくように工夫する。アルキル基の場合は、例えば、セレン酸化によってアルデヒド基へ変換でき、カルボキシル基の場合は、還元反応によりアルデヒド基へ変換できる。−CH2OR基の場合は、R基を除去して−CH2OH基とした後、二酸化マンガンなどで酸化することによりアルデヒド基へ変換できる。このようにして得られるノルインゲナン骨格の第6位炭素にアルデヒド基を有する四環性アルデヒド化合物は、Winklerのインゲノール合成の中間体と一致するので、公知のインゲノール合成経路に連結できる。
【発明の効果】
【0011】
本発明によれば、環化オレフィンメタセシスを有効に利用して、インゲナン骨格を直接、環化反応により合成することができる。これにより、インゲノール、とりわけ光学活性体であるインゲノールの新規、かつ有用な合成経路を提供することができる。
【実施例】
【0012】
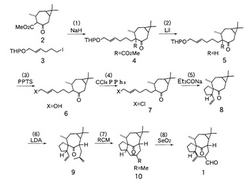
以下、本発明に係るインゲナン骨格を合成する方法の実施例について、添付図面を参照しながら、順を追って反応経路を説明する。なお、添付図面に示された実施例は、本発明に係わる方法の一実施例を示したものであり、これにより本発明の範囲が狭く解釈されることはない。図1は、本発明に係る「ノルインゲナン骨格を合成する方法」の合成経路を示す図である。
【0013】
(1)アルキル化。
【0014】
5mlの枝付きフラスコに水素化ナトリウムの60%鉱油分散物238.4mg(6.0mmol)を秤り取り、窒素雰囲気下とした後、無水N,N-ジメチルホルムアミド(N,N-dimethyl formamide、以下「DMF」)10mlを加え激しく撹拌して懸濁させた。この溶液に、図1に示したケトン(Funkケトエステル)2(Bicyclo[5.1.0]octane-4-carboxylic acid, 3,8,8-trimethyl-5-oxo-, methyl ester, [1R-(1α,3α,4β,7α)]-)を1.07g(4.8mmol)、無水DMF(2.0+1.0ml)に溶解して加えた。反応混合物を室温にて1時間撹拌した後、図1のヨウ化物3(2H-Pyran, tetrahydro-2-[[(2E)-6-iodo-2-hexenyl]oxy]-)を1.72g、無水DMF(1.0+ 1.0+1.0ml)に溶解して加え、100℃に昇温して6時間撹拌した。この反応溶液を室温まで放冷した後、水10mlを加え、酢酸エチル(3×15ml)で抽出した。抽出液を合わせて飽和塩化ナトリウム水溶液10mlで洗浄し、乾燥した後、減圧濃縮した。得られた油状物をカラムクロマトグラフィー[シリカゲルBW-820MH(100 g)、ヘキサン‐エーテル(10:1‐5:1)]で精製したところ、アルキル化物4(Bicyclo[5.1.0]octane-4-carboxylic acid, 3,8,8-trimethyl-5-oxo-4-[(4E)-6-[tetrahydro-2H-pyranyl-2-oxy]-4-hexenyl]-, methyl ester, (1R,3S,4R,7S)-)が1.46g(収率75%)が、無色油状物として得られた。なお、上記ケトン2は、天然テルペンから容易に安く得られるという利点がある。アルキル化物4の構造分析データは、次の通りである。
【0015】
[α]D21+95.4 (c 1.0, CHCl3); IR (neat) 2942, 2360, 1739, 1703, 1642, 1202, 1118, 1023 cm-1; 1H NMR (270MHz, CDCl3)δ5.66 (dt, J= 15.4, 5.6Hz, 1H), 5.57 (dt, J = 15.4, 5.4Hz, 1H) 4.62 (dd, J = 4.1, 2.7Hz, 1H), 4.17 (dd, J = 11.3, 5.6Hz, 1 H), 3.90(dd, J = 11.3, 5.6Hz, 1H), 3.86 (m, 1H), 3.71(s, 3 H), 3.50(m, 1 H), 2.66(dd, J = 14.7, 8.3 Hz, 1H), 2.27(dd, J = 14.7, 8.9 Hz, 1 H), 2.10-1.99 (m, 3H), 1.88-1.35 (m, 11H), 1.22(m, 1H), 1.12 (d, J = 6.8Hz, 3H), 1.06 (s, 3H), 1.02(s, 3H), 0.78 (ddd, J = 8.5, 8.5, 6.8 Hz,1H), 0.64(ddd, J = 8.9, 8.5, 8.3Hz, 1H); MS (FAB) m/z 407 (M + H)+; HRMS (ESI) m/z calcd. for C24H38NaO5(M + Na)+ 429.2617, found 429.2599 (Δ-1.8 mmu).
【0016】
(2)脱メトキシカルボニル化。
【0017】
30mlのナス型フラスコに、上記アルキル化物3を130.6mg(0.3mmol)秤り取り、窒素雰囲気下とした後、無水DMF3mlに溶解した。この溶液にヨウ化リチウム649.7mg(4.9mmol)を無水DMF3mlに溶解して加えた。この反応混合物を4時間還流したあと、室温まで放冷してトリエチルアミン0.1mlと水3mlを加え、得られた水混合物をヘキサン (3×10ml)で抽出した。抽出液を飽和塩化ナトリウム水溶液で洗浄し、乾燥した後、減圧濃縮した。得られた油状物をカラムクロマトグラフィー[シリカゲルBW-820MH(30g)、ヘキサン‐エーテル(10:1→8:1→5:1) ]により精製したところ、ケトン5(Bicyclo[5.1.0]octane, 3,8,8-trimethyl-5-oxo-4-[(4E)-6-[tetrahydro-2H-pyranyl-2-oxy]-4-hexenyl]-, (1R,3S,4R,7S)-)が87.3mg(収率78%)、無色油状物として得られた。このケトン5の構造分析データは、次の通りである。
【0018】
[α]D21+74.7(c0.96,CHCl3);IR(neat)2939,2359,1704,1454,1381,1200,1118,1076,1023,969cm-1; 1H NMR (270 MHz, CDCl3)δ5.69 (br dt, J =15.5, 6.2Hz, 1H), 5.56(ddd, J= 15.6, 6.2, 5.8Hz, 1H), 4.63(br t, J=4.0 Hz, 1H), 4.17 (dd, J =11.9, 5.8Hz, 1H), 3.91 (dd, J = 11.9, 6.2 Hz, 1H), 3.85 (m, 1H), 3.49 (m, 1H), 2.50 (dd, J =11.5, 7.2Hz, 1H), 2.41 (m, 1H), 2.10-2.02(m, 4H), 1.86-1.52 (m, 8H), 1.43-1.16 (m, 4H), 1.08 (s, 3 H), 1,07 (s, 3H), 0.81 (d, J = 6.5 Hz, 3H), 0.68 (m, 1H), 0.55 (m, 1H); MS (FAB) m/z 349 (M + H)+; HRMS (ESI) m/z calcd. for C22H36NaO3(M + Na)+ 371.2562, found 371.2569 (Δ+0.7 mmu).
【0019】
(3)脱保護。
【0020】
200mlのナス型フラスコに、上記ケトン5を1.03g(3.0mmol)秤り取り、p-トルエンスルホン酸ピリジニウム(PPTS)55mg(0.22mmol)をエタノール50mlに溶解して加え、55℃に昇温し10時間反応させた。この反応が十分進んでいなかったため、さらにp-トルエンスルホン酸ピリジニウム20.8mg(0.083mmol)を加え10時間反応させた。反応混合物を減圧濃縮し、得られた油状物をカラムクロマトグラフィー [シリカゲルBW-820MH(30g)、ヘキサン‐エーテル (1:1) ] により精製したところ、アリルアルコール6(Bicyclo[5.1.0]octane, 3,8,8-trimethyl-5-oxo-4-[(4E)-6-hydroxy-4-hexenyl]-, (1R,3S,4R,7S)-)が、761.2mg(収率97%)無色油状物として得られた。なお、このアリルアルコール6の構造分析データは、次の通りである。
【0021】
[α]D21+104 (c1.01, CHCl3); IR (neat) 3420, 2931, 1699, 1457, 969 cm-1; 1H NMR (270 MHz, CDCl3)δ5.73-5.57 (m, 2H), 4.08 (m, 2H), 2.51 (dd, J = 11.3, 7.0 Hz, 1H), 2.41 (dt, J = 8.6, 4.3 Hz, 1H), 2.11-1.99 (m, 4H), 1.83 (m, 1H), 1.64 (m, 1H), 1.42-1.15 (m, 5H), 1.08 (s, 3H), 1.07(s, 3H), 0.81 (d, J = 6.8Hz, 3H), 0.69 (ddd, J = 10.9, 10.6, 5.9 Hz, 1H), 0.56(ddd, J =10.6, 9.2, 7.0Hz, 1H); MS (FAB) m/z 287 (M + Na)+; HRMS (ESI) m/z calcd. for C17H29O2(M + H)+ 265.2167, found 265.2177 (Δ+1.0mmu).
【0022】
(4)クロロ化。
【0023】
200mlの三つ口フラスコに、上記アリルアルコール6を46.5mg(0.18mmol)秤り取り、窒素雰囲気下とした後、四塩化炭素(CCl4)1mlとトリフェニルホスフィン(PPh3)96.3mg(0.37mmol)を加え、7時間還流した。反応混合物を室温まで放冷した後、ヘキサン3mlを加え、綿栓ろ過してオキシトリフェニルホスフィンを除いた。残渣をヘキサンで洗浄し、ろ液と洗液を合わせて減圧濃縮して得られた油状物をカラムクロマトグラフィー [シリカゲルBW-820MH(4g)、ヘキサン‐エーテル(20:1)]により精製したところ、塩化物7(Bicyclo[5.1.0]octane, 3,8,8-trimethyl-5-oxo-4-[(4E)-6-chloro-4-hexenyl]-, (1R,3S,4R,7S)-)が、47.1mg(収率95%)無色油状物として得られた。なお、この塩化物7の構造分析データは、次の通りである。
【0024】
[α]D21+95.9 (c0.95, CHCl3); IR (neat) 2938, 2864, 1703, 1455, 1380, 1249, 966 cm-1; 1H NMR (270 MHz, CDCl3)δ5.73 (dt, J= 15.3, 6.3Hz, 1H ), 5.70 (dt, J = 15.3, 6.9 Hz, 1H), 4.02 (d, J = 6.9 Hz, 2H), 2.51 (dd, J = 11.5, 6.8Hz, 1H), 2.41 (dt, J = 8.8, 4.4Hz, 1H), 2.10-2.01 (m, 3H), 1.85 (m, 1 H), 1.65 (m, 1H), 1.42-1.10 (m, 5H), 1.08 (s, 3 H), 1,07 (s, 3 H), 0.81 (d, J = 7.0 Hz, 3H), 0.69 (ddd, J = 9.9, 9.4, 5.9 Hz, 1 H), 0.56 (ddd, J = 10.7, 9.4, 6.8Hz, 1H); MS (FAB) m/z 283 (M + H)+; HRMS (ESI) m/z calcd. for C17H28ClO (M + H)+ 283.1828, found 283.1801 (Δ-2.7mmu).
【0025】
(5)スピロ環化。
【0026】
5mlの枝付きフラスコに、上記塩化物7を24.2mg(0.086mmol)を秤り取り、窒素雰囲気下とした後、無水キシレン1.5mlに溶解した。ここに、3-エチル-3-ペンタノール0.07ml(0.50mmol)と水素化ナトリウム60%鉱油分散物9.3mg(0.23mmol)を加え、直ちに昇温し30分還流した。反応混合物を室温まで放冷した後、飽和塩化アンモニウム水溶液1mlを加え、得られた水混合物をヘキサン(5ml×3)で抽出した。この抽出液を飽和塩化ナトリウム水溶液3mlで洗浄し、乾燥した後、減圧濃縮した。得られた油状物をカラムクロマトグラフィー[シリカゲルFL-60D(2g)、ヘキサン‐エーテル(200:1→100:1) ]により精製したところ、スピロケトン8(Spiro[bicyclo[5.1.0]octane-4,1'-cyclopentan]-3-one, 2'-ethenyl-5,8,8-trimethyl-, (1R,1'R,2S,2'S,5R,7R)-)だけが、11.1mg(収率53%)が無色油状物として得られた。なお、このスピロケトン8の構造分析データは、次の通りである。
【0027】
[α]D21+135(c1.0, CHCl3); IR (neat) 2954, 1696, 1634, 1456, 1378, 1286, 1160, 993, 912 cm-1; 1H NMR (500 MHz, CDCl3)δ5.59(ddd, J=16.9, 10.0, 8.7Hz, 1H), 5.00(dd, J=16.9, 1.6Hz, 1H), 4.92(dd, J = 10.0, 1.6Hz, 1H), 2.69 (ddd, J = 8.7, 7.5, 2.0Hz, 1H), 2.30(dd, J = 11.8, 6.8Hz, 1H), 2.21(dd, J = 11.8, 10.7 Hz, 1H), 1.99(m, 1H), 1.97(dd, J = 9.9, 4.1 Hz, 1H), 1.89(m, 1 H), 1.85(dt, J=14.7, 6.2Hz, 1H), 1.78(td, J = 6.7, 2.2Hz, 1H), 1,71(m, 1H), 1.65(dd, J=14.7, 10.2Hz, 1H), 1.54(dd, J =4.9, 3.0Hz, 1H), 1.51(m, 1H), 1.07(s, 3H), 1,05(s, 3H), 0.91(d, J = 6.8Hz, 3H), 0.69(ddd, J=10.2, 9.6, 6.2Hz, 1H), 0.55(ddd, J=10.7, 9.6, 6.8Hz, 1H); 13C NMR (67.8MHz, CDCl3)δ211.1, 140.5, 114.9, 68.8, 50.1, 39.4, 34.8, 30.8, 28.8, 28.7, 26.3, 23.1, 21.2, 21.1, 20.7, 15.3, 14.8; MS (FAB) m/z 247 (M +H)+; HRMS (ESI) m/zcalcd. for C17H26NaO (M + Na)+ 269.1881, found 269.1861(Δ-2.0mmu).
【0028】
このスピロ環化に係わる反応条件では、スピロ環化を円滑に進行させるため、塩基と溶媒の選択を工夫し、塩基と原料の副反応を抑制するために、塩基の立体障害を大きくした。また、本反応の収率及び立体選択性は、塩化物7の分子内の配位に依存していると考えられることから、配位結合が強くなるように、反応溶媒の極性を低下させ、また、配位元素である塩素と強く結合するナトリウムを用いた。これにより、反応収率を向上させることができた。
【0029】
(6)メタリル化。
【0030】
20mlのナシ型フラスコに、上記スピロケトン8を21.7mg(0.09mmol)秤り取り、窒素雰囲気下とした後、無水THF0.08mlを加え溶解した。反応混合物を0℃まで冷却しヘキサメチルリン酸トリアミド0.02ml(0.46mmol)と予め調製したリチウムジイソプロピルアミド(Lithium diisopropylamide,LDA)溶液を加え、40分撹拌した。反応混合物を−10℃に冷却し、ヨウ化メタリル37.0mg(0.2mmol)を加え、さらに2時間反応した後、飽和チオ硫酸ナトリウム水溶液1mlと飽和塩化アンモニウム水溶液1mlを加え、30分撹拌した。水混合物をエーテル(3×5ml)で抽出し、抽出液を飽和塩化ナトリウム水溶液5mlで洗浄し、乾燥した後、減圧濃縮した。得られた混合物をカラムクロマトグラフィー [シリカゲルFL-60D(4g)、ヘキサン‐エーテル(100:1)]により精製したところ、メタリルスピロケトン9(Spiro[bicyclo[5.1.0]octane-4,1'-cyclopentan]-3-one, 2'-ethenyl-5,8,8-trimethyl-2-(2-methyl-2-propenyl)-,(1R,1'R,2S,2'S,5R,7R)-)が、15.7mg(収率59%)無色結晶として得られた。本手順において、LDA溶液は、30mlの枝付きフラスコにジイソプロピルアミン0.05ml、無水THF0.5mlを加え0℃に冷却した。これに1.5Mのn−ブチルリチウム0.18ml(0.27mmol)を加えて30分撹拌し、全量を反応に用いた。なお、メタリルスピロケトン9の構造分析データは、次の通りである。
【0031】
mp 87.0-91.0 °C (hexane-ether-MeOH); [α]D21 +44.5 (c0.26, CHCl3); IR (neat) 2954, 2358, 1685, 1454, 1377 cm-1; 1H NMR (270 MHz, CDCl3)δ5.61 (ddd, J = 16.9, 9.9, 9.8Hz, 1H), 5.05 (dd, J = 16.9, 1.9Hz, 1H), 4.97 (dd, J = 9.9, 1.9 Hz, 1 H), 4.75 (br s, 1H), 4.68 (br s, 1H), 2.75 (br t, J = 7.4Hz, 1H), 2.42 (ddd, J = 10.9, 9.9, 3.0 Hz, 1 H), 2.29 (dd, J = 12.1, 10.9Hz, 1H), 2.07 (dd, J = 12.1, 3.0Hz, 1H), 2.01-1.43 (m, 9H), 1.65(s, 3 H), 0.98 (s, 3 H), 0.95 (s, 3H), 0.91(d, J = 6.5Hz, 3H), 0.67(ddd, J = 9.5, 9.4, 6.1Hz, 1H), 0.10 (dd, J = 9.9, 9.5Hz, 1H);13C NMR (67.8 MHz, CDCl3)δ212.1, 143.3, 140.7, 114.9, 112.1, 68.5, 49.8, 46.6, 39.4, 34.6, 31.1, 29.7, 28.9, 28.6, 27.8, 26.7, 23.1, 22.9, 20.9, 16.0, 14.7; HRMS (FAB) m/zcalcd. for C21H32NaO (M + Na)+ 323.2351, found 323.2372 (Δ+2.1mmu).
【0032】
(7)閉環オレフィンメタセシス(RCM)。
【0033】
50mlの枝付きフラスコに、上記メタリルスピロケトン9を6.3mg(0.021mmol)秤り取り、窒素雰囲気下とした後、無水トルエン10mlに溶解した。ここに、次の「化1」で示す第二世代のGrubbs触媒4.5mg(5.3μmol)をトルエン3mlに溶解して加え、30分還流した。
【0034】
【化1】

【0035】
続いて、これを室温まで放冷し、そのままカラムクロマトグラフィー [シリカゲルBW-820MH(3g)、ベンゼン]により触媒を取り除き、さらにカラムクロマトグラフィー [シリカゲルBW-820MH(4g)、ベンゼン‐ヘキサン(1:1)]により精製したところ、テトラシクロケトン10(1H-2,8a-Methanocyclopenta[a]cyclopropa[e]cyclodecene, 1a,2,3,5a,6,7,8,9,10,10a-decahydro-1,1,4,9-tetramethyl-11-oxo-,(1aR,2S,5aS,8aR,9R,10aR)-)が、5.0mg(収率87%)無色結晶として得られた。なお、テトラシクロケトン10の構造分析データは、次の通りである。なお、図1に示されたテトラシクロケトン10の第6位炭素に結合しているR基がメチル基である場合が例示されている。このR基は、アルデヒド基に変換可能な他のアルキル基(例えば、エチル基)やカルボキシル基、−CH2OR基など)、あるいはアルデヒド基自体を結合させておくようにしてもよい。
【0036】
[α]D21+24.4 (c0.16, CHCl3); IR (neat) 2950, 1724, 1455, 1379 cm-1; 1H NMR (500 MHz, CDCl3)δ4.88 (br s, 1H), 3.19 (ddd, J = 12.5, 12.2, 3.2Hz, 1H), 3.13 (br s, 1H), 2.33 (m, 1H), 2.30 (m, 1H), 2.06 (br d, J = 16.7Hz, 1H), 1.89 (m, 1H), 1.85 (m, 1H), 1.80 (ddd, J = 14.7, 9.4, 2.6Hz, 1H), 1.65 (m, 1H), 1.64 (s, 3H), 1.58 (m, 1H), 1.47 (m, 1H), 1.44 (m, 1H), 1.26 (ddd, J = 13.3, 10.1, 7.6Hz, 1H), 1.12 (s, 3H), 1,03 (s, 3H), 0.94(d, J= 6.8Hz, 3H), 0.69(ddd, J = 9.4, 8.8, 6.2 Hz, 1 H), 0.60(dd, J = 12.2, 8.8Hz, 1H); 13C NMR (67.8 MHz, CDCl3)δ211.3, 139.4, 132.3, 71.9, 45.7, 45.5, 39.6, 36.1, 35.0, 29.5, 28.7, 27.8, 26.2, 25.3, 23.6, 23.2. 22.8, 15.4, 15.3; HRMS (FAB) m/zcalcd. for C19H28NaO (M + Na)+ 295.2038, found 295.2023 (Δ-1.5 mmu)。
【0037】
(8)セレン酸化。
【0038】
このセレン酸化は、テトラシクロケトン10の第6位炭素に結合しているアルキル基をアルデヒド基に変換するための反応である。まず、5mlの枝付きフラスコに、上記テトラシクロケトン10を1.8mg(6.6μmol)を秤り取り、無水ジオキサン0.5mlに溶解した。ここに、二酸化セレン11.8mg(0.11mmol)を加えて1時間還流した。室温まで放冷し、セライトを用いて反応混合物をろ過し、濃縮した。これをエーテル5mlに溶解し、飽和炭酸水素ナトリウム水溶液、水、飽和塩化ナトリウム水溶液で洗浄し乾燥後、減圧濃縮した。得られた混合物をカラムクロマトグラフィー [シリカゲルBW-820MH(0.5g)、ヘキサン‐エーテル(5:1)]により精製したところ、アルデヒド体1(1H-2,8a-Methanocyclopenta[a]cyclopropa[e]cyclodecene-4-carboxaldehyde,1a,2,3,5a,6,7,8,9,10,10a-decahydro-1,1,9-trimethyl-11-oxo-,(1aR,2S,5aS,8aR,9R,10aR)-)が、1.6mg(収率85%)が無色結晶として得られた。なお、第6位炭素にアルデヒド基を有するアルデヒド体1(図1参照)の構造分析データは、次の通りである。
【0039】
mp 145.5-147.5 ℃(hexane); [α]D21 +29.5 (c0.078, CHCl3); IR (neat) 2940, 1722, 1687, 1594, 1458 cm-1; 1H NMR (500 MHz, CDCl3)δ9.28 (s, 1H), 6.23 (d, J = 1.9Hz, 1H), 3.36(br s, 1H), 3.09(ddd, J = 14.0, 10.9, 3.1Hz, 1H), 2.58(dddd, J = 17.1, 13.4, 3.7, 1.7Hz, 1H), 2.38(m, 1H), 2.30(m, 1H), 2.02 (m, 1H), 1.94(ddd, J= 15.6, 5.3, 5.3 Hz, 1H), 1.85 (m, 1H), 1.79(m, 1 H), 1.70 (m, 1H), 1.45-1.25 (m, 3H), 1.10 (s, 3H), 1,05 (s, 3H), 0.98 (d, J = 6.9Hz, 3H), 0.72 (m, 2H); 13C NMR (100 MHz, C6D6)δ208.1, 193.8, 159.3, 146.5, 72.0, 46.9, 45.0, 39.8, 34.6, 29.6, 29.0, 28.4, 28.1, 25.8, 23.7, 23.3, 22.8, 15.4, 15.0; HRMS (FAB) m/z calcd. for C19H26NaO2 (M + Na)+309.1831, found 309.1853 (Δ+2.2mmu).
【0040】
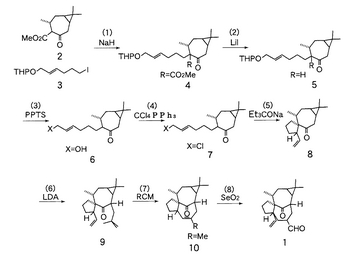
このアルデヒド体1は、炭素数19のノルインゲナン骨格を有している。このアルデヒド体1は、既述の非特許文献2(Winklerら)の9728頁に掲載されたScheme3の反応経路中の番号20の化合物と同一である。従って、以後は、Winklerらの前記Scheme3の反応経路に従えば、図3に示された化学構造を有するインゲノールを光学活性体として合成できる。なお、図2には、理解のため、Winklerらによって提案された前記Scheme3の反応経路を転載する。
【産業上の利用可能性】
【0041】
本発明によれば、生理活性を有するため注目されているが、合成が困難であったインゲノールを光学活性体として合成することができる。従って、このインゲノールに係わる薬剤開発に、特に有用である。
【図面の簡単な説明】
【0042】
【図1】本発明に係る「ノルインゲナン骨格を合成する方法」の合成経路を示す図である。
【図2】同合成経路により得られるアルデヒド体1を中間体とするWinklerらによるインゲノール合成経路を示す図(非特許文献1より転載)である。
【図3】インゲノールの化学構造を示す図である。
【図4】インゲナン骨格の炭素環骨格を示す図である。
【図5】二つのオレフィン部位がtrans体を構成する2,7−ジアリルシクロヘプタノンが、環化オレフィンメタセシスによる閉環が困難であることを示す図である。
【符号の説明】
【0043】
1 アルデヒド体(ノルインゲナン骨格、Winkler合成経路の中間体)
2 ケトン(Funkケトエステル)
3 ヨウ化物
4 アルキル化物
5 ケトン
6 アリルアルコール
7 塩化物
8 スピロケトン
9 メタリルスピロケトン
10 テトラシクロケトン(ノルインゲナン骨格)
【技術分野】
【0001】
本発明は、インゲノールの合成に係わる技術に関する。より詳しくは、オレフィンメタセシスによる閉環反応を鍵反応として、インゲノール合成経路の中間体として有用なノルインゲナン骨格(炭素数19)を合成する技術に関する。
【背景技術】
【0002】
図3に示す化学構造のインゲノール(ingenol)は、トウダイクサ科の植物チュウテンカクより単離されたジテルペン(炭素数20)であり、その不飽和脂肪酸エステルは、様々な生理活性を持つことから、現在注目されている。
【0003】
例えば、特許文献1には、13−オキシインゲノール誘導体が、ヒト静脈内皮細胞による細胞接着分子(VCAM-1)の産生阻止作用を示し、癌転移抑制剤、抗炎症剤、動脈硬化、移植拒絶反応、慢性関節リウマチ、サルコイドーシス等の治療薬としての利用が期待される旨が開示されている。その他、インゲノールの誘導体は、発癌プロモーター作用、抗白血病作用、抗HIV活性などの生理活性を示すことが知られている。
【0004】
ここで、インゲノールは、transに閉環し強く歪んだ四環性のインゲナン骨格(inside,outside-ビシクロ[4.4.1]ウンデカン骨格)を持っている。具体的には、図4に示されたインゲノールの炭素環骨格は、図中のB環とC環が互いにブリッジしているので、橋頭位の10位から1位への炭素−炭素結合がBC環部の外側を向いている。これに対して、もう一方の橋頭位の8位上の水素原子は、BC環の内側に入り込んでいる。このためインゲナン骨格は、高度に歪んでいる。
【0005】
このような高度の歪み構造を有するインゲナンの炭素環骨格は、合成が困難であるので、多くの有機合成化学者の関心を集めている。これまでのところ、例えば、Winklerのグループが分子内光[2+2]付加環化反応及び四員環の開裂反応を用いて、インゲノールの最初の全合成を達成している(非特許文献1参照)。また、谷野らのグループが、インゲノールの全合成を成功させている(非特許文献2参照)。これらのグループの合成により得られたインゲノールは、いずれもラセミ体である。
【特許文献1】特開平07−258168号公報。
【非特許文献1】Winkler,J.D.;Rouse,M.B.; Greaney,M.F.; Harrison,Sean.J.;Jeon,Y.T.J.Am.Chem.Soc.2002,124,9726-9728。
【非特許文献2】Tanino,K.;Onuki,K.;Asano,K.;Miyashita,M.;Nakamura,T.;Takahashi,Y.;Kuwajima,I.J.AM.CHEM.SOC.2003,125,1498-1500。
【発明の開示】
【発明が解決しようとする課題】
【0006】
本願発明者らは、インゲナンの炭素環骨格の効率的な合成経路を模索する中で、インゲナン骨格を直接、環化反応により合成する経路に着目し、長年鋭意研究を続けたところ、オレフィンメタセシスによる閉環反応(以下「環化オレフィンメタセシス(略称RCM)」)では、環化に寄与する二つのオレフィン部位が空間的に近く配置されている必要があることを突き止めた。例えば、図5に示すように、二つのオレフィン部位がtrans体を構成する2,7-ジアリルシクロヘプタノンでは、環化オレフィンメタセシスによる閉環が困難である。
【0007】
そこで、本発明は、環化オレフィンメタセシスを有効に利用して、インゲナン骨格を直接、環化反応により合成する経路を見出すことにより、インゲノールの有用な合成経路を提供することを主な目的とする。
【課題を解決するための手段】
【0008】
本発明では、ジオレフィン化合物の一方のオレフィン部位をスピロ環で固定して三環性ジオレフィン化合物を得る工程と、該三環性ジオレフィン化合物のジオレフィン部位をオレフィンメタセシス反応で閉環する工程と、を少なくとも行うことによって四環性のノルインゲナン骨格を合成する方法、特に、薬剤開発において有用となる光学活性を有するノルインゲナン骨格を合成する方法を提供し、並びに、これらの方法を用いたインゲノール合成方法を提供する。
【0009】
ジオレフィン化合物の一方のオレフィン部位がスピロ環で固定された構造を有する三環性ジオレフィン化合物は、二つのオレフィン部位(二重結合部位)の距離が、オレフィンメタセシス反応が可能なまでに近づいているので、該オレフィンメタセシス反応による閉環を進行させることが可能となる。
【0010】
また、本発明では、上記方法によって得られるノルインゲナン骨格の第6位炭素に、アルデヒド基又はアルデヒド基に変換可能な基(例えば、メチル基、エチル基などのアルキル基、カルボキシル基、−CH2OR基など)を結合させておくように工夫する。アルキル基の場合は、例えば、セレン酸化によってアルデヒド基へ変換でき、カルボキシル基の場合は、還元反応によりアルデヒド基へ変換できる。−CH2OR基の場合は、R基を除去して−CH2OH基とした後、二酸化マンガンなどで酸化することによりアルデヒド基へ変換できる。このようにして得られるノルインゲナン骨格の第6位炭素にアルデヒド基を有する四環性アルデヒド化合物は、Winklerのインゲノール合成の中間体と一致するので、公知のインゲノール合成経路に連結できる。
【発明の効果】
【0011】
本発明によれば、環化オレフィンメタセシスを有効に利用して、インゲナン骨格を直接、環化反応により合成することができる。これにより、インゲノール、とりわけ光学活性体であるインゲノールの新規、かつ有用な合成経路を提供することができる。
【実施例】
【0012】
以下、本発明に係るインゲナン骨格を合成する方法の実施例について、添付図面を参照しながら、順を追って反応経路を説明する。なお、添付図面に示された実施例は、本発明に係わる方法の一実施例を示したものであり、これにより本発明の範囲が狭く解釈されることはない。図1は、本発明に係る「ノルインゲナン骨格を合成する方法」の合成経路を示す図である。
【0013】
(1)アルキル化。
【0014】
5mlの枝付きフラスコに水素化ナトリウムの60%鉱油分散物238.4mg(6.0mmol)を秤り取り、窒素雰囲気下とした後、無水N,N-ジメチルホルムアミド(N,N-dimethyl formamide、以下「DMF」)10mlを加え激しく撹拌して懸濁させた。この溶液に、図1に示したケトン(Funkケトエステル)2(Bicyclo[5.1.0]octane-4-carboxylic acid, 3,8,8-trimethyl-5-oxo-, methyl ester, [1R-(1α,3α,4β,7α)]-)を1.07g(4.8mmol)、無水DMF(2.0+1.0ml)に溶解して加えた。反応混合物を室温にて1時間撹拌した後、図1のヨウ化物3(2H-Pyran, tetrahydro-2-[[(2E)-6-iodo-2-hexenyl]oxy]-)を1.72g、無水DMF(1.0+ 1.0+1.0ml)に溶解して加え、100℃に昇温して6時間撹拌した。この反応溶液を室温まで放冷した後、水10mlを加え、酢酸エチル(3×15ml)で抽出した。抽出液を合わせて飽和塩化ナトリウム水溶液10mlで洗浄し、乾燥した後、減圧濃縮した。得られた油状物をカラムクロマトグラフィー[シリカゲルBW-820MH(100 g)、ヘキサン‐エーテル(10:1‐5:1)]で精製したところ、アルキル化物4(Bicyclo[5.1.0]octane-4-carboxylic acid, 3,8,8-trimethyl-5-oxo-4-[(4E)-6-[tetrahydro-2H-pyranyl-2-oxy]-4-hexenyl]-, methyl ester, (1R,3S,4R,7S)-)が1.46g(収率75%)が、無色油状物として得られた。なお、上記ケトン2は、天然テルペンから容易に安く得られるという利点がある。アルキル化物4の構造分析データは、次の通りである。
【0015】
[α]D21+95.4 (c 1.0, CHCl3); IR (neat) 2942, 2360, 1739, 1703, 1642, 1202, 1118, 1023 cm-1; 1H NMR (270MHz, CDCl3)δ5.66 (dt, J= 15.4, 5.6Hz, 1H), 5.57 (dt, J = 15.4, 5.4Hz, 1H) 4.62 (dd, J = 4.1, 2.7Hz, 1H), 4.17 (dd, J = 11.3, 5.6Hz, 1 H), 3.90(dd, J = 11.3, 5.6Hz, 1H), 3.86 (m, 1H), 3.71(s, 3 H), 3.50(m, 1 H), 2.66(dd, J = 14.7, 8.3 Hz, 1H), 2.27(dd, J = 14.7, 8.9 Hz, 1 H), 2.10-1.99 (m, 3H), 1.88-1.35 (m, 11H), 1.22(m, 1H), 1.12 (d, J = 6.8Hz, 3H), 1.06 (s, 3H), 1.02(s, 3H), 0.78 (ddd, J = 8.5, 8.5, 6.8 Hz,1H), 0.64(ddd, J = 8.9, 8.5, 8.3Hz, 1H); MS (FAB) m/z 407 (M + H)+; HRMS (ESI) m/z calcd. for C24H38NaO5(M + Na)+ 429.2617, found 429.2599 (Δ-1.8 mmu).
【0016】
(2)脱メトキシカルボニル化。
【0017】
30mlのナス型フラスコに、上記アルキル化物3を130.6mg(0.3mmol)秤り取り、窒素雰囲気下とした後、無水DMF3mlに溶解した。この溶液にヨウ化リチウム649.7mg(4.9mmol)を無水DMF3mlに溶解して加えた。この反応混合物を4時間還流したあと、室温まで放冷してトリエチルアミン0.1mlと水3mlを加え、得られた水混合物をヘキサン (3×10ml)で抽出した。抽出液を飽和塩化ナトリウム水溶液で洗浄し、乾燥した後、減圧濃縮した。得られた油状物をカラムクロマトグラフィー[シリカゲルBW-820MH(30g)、ヘキサン‐エーテル(10:1→8:1→5:1) ]により精製したところ、ケトン5(Bicyclo[5.1.0]octane, 3,8,8-trimethyl-5-oxo-4-[(4E)-6-[tetrahydro-2H-pyranyl-2-oxy]-4-hexenyl]-, (1R,3S,4R,7S)-)が87.3mg(収率78%)、無色油状物として得られた。このケトン5の構造分析データは、次の通りである。
【0018】
[α]D21+74.7(c0.96,CHCl3);IR(neat)2939,2359,1704,1454,1381,1200,1118,1076,1023,969cm-1; 1H NMR (270 MHz, CDCl3)δ5.69 (br dt, J =15.5, 6.2Hz, 1H), 5.56(ddd, J= 15.6, 6.2, 5.8Hz, 1H), 4.63(br t, J=4.0 Hz, 1H), 4.17 (dd, J =11.9, 5.8Hz, 1H), 3.91 (dd, J = 11.9, 6.2 Hz, 1H), 3.85 (m, 1H), 3.49 (m, 1H), 2.50 (dd, J =11.5, 7.2Hz, 1H), 2.41 (m, 1H), 2.10-2.02(m, 4H), 1.86-1.52 (m, 8H), 1.43-1.16 (m, 4H), 1.08 (s, 3 H), 1,07 (s, 3H), 0.81 (d, J = 6.5 Hz, 3H), 0.68 (m, 1H), 0.55 (m, 1H); MS (FAB) m/z 349 (M + H)+; HRMS (ESI) m/z calcd. for C22H36NaO3(M + Na)+ 371.2562, found 371.2569 (Δ+0.7 mmu).
【0019】
(3)脱保護。
【0020】
200mlのナス型フラスコに、上記ケトン5を1.03g(3.0mmol)秤り取り、p-トルエンスルホン酸ピリジニウム(PPTS)55mg(0.22mmol)をエタノール50mlに溶解して加え、55℃に昇温し10時間反応させた。この反応が十分進んでいなかったため、さらにp-トルエンスルホン酸ピリジニウム20.8mg(0.083mmol)を加え10時間反応させた。反応混合物を減圧濃縮し、得られた油状物をカラムクロマトグラフィー [シリカゲルBW-820MH(30g)、ヘキサン‐エーテル (1:1) ] により精製したところ、アリルアルコール6(Bicyclo[5.1.0]octane, 3,8,8-trimethyl-5-oxo-4-[(4E)-6-hydroxy-4-hexenyl]-, (1R,3S,4R,7S)-)が、761.2mg(収率97%)無色油状物として得られた。なお、このアリルアルコール6の構造分析データは、次の通りである。
【0021】
[α]D21+104 (c1.01, CHCl3); IR (neat) 3420, 2931, 1699, 1457, 969 cm-1; 1H NMR (270 MHz, CDCl3)δ5.73-5.57 (m, 2H), 4.08 (m, 2H), 2.51 (dd, J = 11.3, 7.0 Hz, 1H), 2.41 (dt, J = 8.6, 4.3 Hz, 1H), 2.11-1.99 (m, 4H), 1.83 (m, 1H), 1.64 (m, 1H), 1.42-1.15 (m, 5H), 1.08 (s, 3H), 1.07(s, 3H), 0.81 (d, J = 6.8Hz, 3H), 0.69 (ddd, J = 10.9, 10.6, 5.9 Hz, 1H), 0.56(ddd, J =10.6, 9.2, 7.0Hz, 1H); MS (FAB) m/z 287 (M + Na)+; HRMS (ESI) m/z calcd. for C17H29O2(M + H)+ 265.2167, found 265.2177 (Δ+1.0mmu).
【0022】
(4)クロロ化。
【0023】
200mlの三つ口フラスコに、上記アリルアルコール6を46.5mg(0.18mmol)秤り取り、窒素雰囲気下とした後、四塩化炭素(CCl4)1mlとトリフェニルホスフィン(PPh3)96.3mg(0.37mmol)を加え、7時間還流した。反応混合物を室温まで放冷した後、ヘキサン3mlを加え、綿栓ろ過してオキシトリフェニルホスフィンを除いた。残渣をヘキサンで洗浄し、ろ液と洗液を合わせて減圧濃縮して得られた油状物をカラムクロマトグラフィー [シリカゲルBW-820MH(4g)、ヘキサン‐エーテル(20:1)]により精製したところ、塩化物7(Bicyclo[5.1.0]octane, 3,8,8-trimethyl-5-oxo-4-[(4E)-6-chloro-4-hexenyl]-, (1R,3S,4R,7S)-)が、47.1mg(収率95%)無色油状物として得られた。なお、この塩化物7の構造分析データは、次の通りである。
【0024】
[α]D21+95.9 (c0.95, CHCl3); IR (neat) 2938, 2864, 1703, 1455, 1380, 1249, 966 cm-1; 1H NMR (270 MHz, CDCl3)δ5.73 (dt, J= 15.3, 6.3Hz, 1H ), 5.70 (dt, J = 15.3, 6.9 Hz, 1H), 4.02 (d, J = 6.9 Hz, 2H), 2.51 (dd, J = 11.5, 6.8Hz, 1H), 2.41 (dt, J = 8.8, 4.4Hz, 1H), 2.10-2.01 (m, 3H), 1.85 (m, 1 H), 1.65 (m, 1H), 1.42-1.10 (m, 5H), 1.08 (s, 3 H), 1,07 (s, 3 H), 0.81 (d, J = 7.0 Hz, 3H), 0.69 (ddd, J = 9.9, 9.4, 5.9 Hz, 1 H), 0.56 (ddd, J = 10.7, 9.4, 6.8Hz, 1H); MS (FAB) m/z 283 (M + H)+; HRMS (ESI) m/z calcd. for C17H28ClO (M + H)+ 283.1828, found 283.1801 (Δ-2.7mmu).
【0025】
(5)スピロ環化。
【0026】
5mlの枝付きフラスコに、上記塩化物7を24.2mg(0.086mmol)を秤り取り、窒素雰囲気下とした後、無水キシレン1.5mlに溶解した。ここに、3-エチル-3-ペンタノール0.07ml(0.50mmol)と水素化ナトリウム60%鉱油分散物9.3mg(0.23mmol)を加え、直ちに昇温し30分還流した。反応混合物を室温まで放冷した後、飽和塩化アンモニウム水溶液1mlを加え、得られた水混合物をヘキサン(5ml×3)で抽出した。この抽出液を飽和塩化ナトリウム水溶液3mlで洗浄し、乾燥した後、減圧濃縮した。得られた油状物をカラムクロマトグラフィー[シリカゲルFL-60D(2g)、ヘキサン‐エーテル(200:1→100:1) ]により精製したところ、スピロケトン8(Spiro[bicyclo[5.1.0]octane-4,1'-cyclopentan]-3-one, 2'-ethenyl-5,8,8-trimethyl-, (1R,1'R,2S,2'S,5R,7R)-)だけが、11.1mg(収率53%)が無色油状物として得られた。なお、このスピロケトン8の構造分析データは、次の通りである。
【0027】
[α]D21+135(c1.0, CHCl3); IR (neat) 2954, 1696, 1634, 1456, 1378, 1286, 1160, 993, 912 cm-1; 1H NMR (500 MHz, CDCl3)δ5.59(ddd, J=16.9, 10.0, 8.7Hz, 1H), 5.00(dd, J=16.9, 1.6Hz, 1H), 4.92(dd, J = 10.0, 1.6Hz, 1H), 2.69 (ddd, J = 8.7, 7.5, 2.0Hz, 1H), 2.30(dd, J = 11.8, 6.8Hz, 1H), 2.21(dd, J = 11.8, 10.7 Hz, 1H), 1.99(m, 1H), 1.97(dd, J = 9.9, 4.1 Hz, 1H), 1.89(m, 1 H), 1.85(dt, J=14.7, 6.2Hz, 1H), 1.78(td, J = 6.7, 2.2Hz, 1H), 1,71(m, 1H), 1.65(dd, J=14.7, 10.2Hz, 1H), 1.54(dd, J =4.9, 3.0Hz, 1H), 1.51(m, 1H), 1.07(s, 3H), 1,05(s, 3H), 0.91(d, J = 6.8Hz, 3H), 0.69(ddd, J=10.2, 9.6, 6.2Hz, 1H), 0.55(ddd, J=10.7, 9.6, 6.8Hz, 1H); 13C NMR (67.8MHz, CDCl3)δ211.1, 140.5, 114.9, 68.8, 50.1, 39.4, 34.8, 30.8, 28.8, 28.7, 26.3, 23.1, 21.2, 21.1, 20.7, 15.3, 14.8; MS (FAB) m/z 247 (M +H)+; HRMS (ESI) m/zcalcd. for C17H26NaO (M + Na)+ 269.1881, found 269.1861(Δ-2.0mmu).
【0028】
このスピロ環化に係わる反応条件では、スピロ環化を円滑に進行させるため、塩基と溶媒の選択を工夫し、塩基と原料の副反応を抑制するために、塩基の立体障害を大きくした。また、本反応の収率及び立体選択性は、塩化物7の分子内の配位に依存していると考えられることから、配位結合が強くなるように、反応溶媒の極性を低下させ、また、配位元素である塩素と強く結合するナトリウムを用いた。これにより、反応収率を向上させることができた。
【0029】
(6)メタリル化。
【0030】
20mlのナシ型フラスコに、上記スピロケトン8を21.7mg(0.09mmol)秤り取り、窒素雰囲気下とした後、無水THF0.08mlを加え溶解した。反応混合物を0℃まで冷却しヘキサメチルリン酸トリアミド0.02ml(0.46mmol)と予め調製したリチウムジイソプロピルアミド(Lithium diisopropylamide,LDA)溶液を加え、40分撹拌した。反応混合物を−10℃に冷却し、ヨウ化メタリル37.0mg(0.2mmol)を加え、さらに2時間反応した後、飽和チオ硫酸ナトリウム水溶液1mlと飽和塩化アンモニウム水溶液1mlを加え、30分撹拌した。水混合物をエーテル(3×5ml)で抽出し、抽出液を飽和塩化ナトリウム水溶液5mlで洗浄し、乾燥した後、減圧濃縮した。得られた混合物をカラムクロマトグラフィー [シリカゲルFL-60D(4g)、ヘキサン‐エーテル(100:1)]により精製したところ、メタリルスピロケトン9(Spiro[bicyclo[5.1.0]octane-4,1'-cyclopentan]-3-one, 2'-ethenyl-5,8,8-trimethyl-2-(2-methyl-2-propenyl)-,(1R,1'R,2S,2'S,5R,7R)-)が、15.7mg(収率59%)無色結晶として得られた。本手順において、LDA溶液は、30mlの枝付きフラスコにジイソプロピルアミン0.05ml、無水THF0.5mlを加え0℃に冷却した。これに1.5Mのn−ブチルリチウム0.18ml(0.27mmol)を加えて30分撹拌し、全量を反応に用いた。なお、メタリルスピロケトン9の構造分析データは、次の通りである。
【0031】
mp 87.0-91.0 °C (hexane-ether-MeOH); [α]D21 +44.5 (c0.26, CHCl3); IR (neat) 2954, 2358, 1685, 1454, 1377 cm-1; 1H NMR (270 MHz, CDCl3)δ5.61 (ddd, J = 16.9, 9.9, 9.8Hz, 1H), 5.05 (dd, J = 16.9, 1.9Hz, 1H), 4.97 (dd, J = 9.9, 1.9 Hz, 1 H), 4.75 (br s, 1H), 4.68 (br s, 1H), 2.75 (br t, J = 7.4Hz, 1H), 2.42 (ddd, J = 10.9, 9.9, 3.0 Hz, 1 H), 2.29 (dd, J = 12.1, 10.9Hz, 1H), 2.07 (dd, J = 12.1, 3.0Hz, 1H), 2.01-1.43 (m, 9H), 1.65(s, 3 H), 0.98 (s, 3 H), 0.95 (s, 3H), 0.91(d, J = 6.5Hz, 3H), 0.67(ddd, J = 9.5, 9.4, 6.1Hz, 1H), 0.10 (dd, J = 9.9, 9.5Hz, 1H);13C NMR (67.8 MHz, CDCl3)δ212.1, 143.3, 140.7, 114.9, 112.1, 68.5, 49.8, 46.6, 39.4, 34.6, 31.1, 29.7, 28.9, 28.6, 27.8, 26.7, 23.1, 22.9, 20.9, 16.0, 14.7; HRMS (FAB) m/zcalcd. for C21H32NaO (M + Na)+ 323.2351, found 323.2372 (Δ+2.1mmu).
【0032】
(7)閉環オレフィンメタセシス(RCM)。
【0033】
50mlの枝付きフラスコに、上記メタリルスピロケトン9を6.3mg(0.021mmol)秤り取り、窒素雰囲気下とした後、無水トルエン10mlに溶解した。ここに、次の「化1」で示す第二世代のGrubbs触媒4.5mg(5.3μmol)をトルエン3mlに溶解して加え、30分還流した。
【0034】
【化1】
【0035】
続いて、これを室温まで放冷し、そのままカラムクロマトグラフィー [シリカゲルBW-820MH(3g)、ベンゼン]により触媒を取り除き、さらにカラムクロマトグラフィー [シリカゲルBW-820MH(4g)、ベンゼン‐ヘキサン(1:1)]により精製したところ、テトラシクロケトン10(1H-2,8a-Methanocyclopenta[a]cyclopropa[e]cyclodecene, 1a,2,3,5a,6,7,8,9,10,10a-decahydro-1,1,4,9-tetramethyl-11-oxo-,(1aR,2S,5aS,8aR,9R,10aR)-)が、5.0mg(収率87%)無色結晶として得られた。なお、テトラシクロケトン10の構造分析データは、次の通りである。なお、図1に示されたテトラシクロケトン10の第6位炭素に結合しているR基がメチル基である場合が例示されている。このR基は、アルデヒド基に変換可能な他のアルキル基(例えば、エチル基)やカルボキシル基、−CH2OR基など)、あるいはアルデヒド基自体を結合させておくようにしてもよい。
【0036】
[α]D21+24.4 (c0.16, CHCl3); IR (neat) 2950, 1724, 1455, 1379 cm-1; 1H NMR (500 MHz, CDCl3)δ4.88 (br s, 1H), 3.19 (ddd, J = 12.5, 12.2, 3.2Hz, 1H), 3.13 (br s, 1H), 2.33 (m, 1H), 2.30 (m, 1H), 2.06 (br d, J = 16.7Hz, 1H), 1.89 (m, 1H), 1.85 (m, 1H), 1.80 (ddd, J = 14.7, 9.4, 2.6Hz, 1H), 1.65 (m, 1H), 1.64 (s, 3H), 1.58 (m, 1H), 1.47 (m, 1H), 1.44 (m, 1H), 1.26 (ddd, J = 13.3, 10.1, 7.6Hz, 1H), 1.12 (s, 3H), 1,03 (s, 3H), 0.94(d, J= 6.8Hz, 3H), 0.69(ddd, J = 9.4, 8.8, 6.2 Hz, 1 H), 0.60(dd, J = 12.2, 8.8Hz, 1H); 13C NMR (67.8 MHz, CDCl3)δ211.3, 139.4, 132.3, 71.9, 45.7, 45.5, 39.6, 36.1, 35.0, 29.5, 28.7, 27.8, 26.2, 25.3, 23.6, 23.2. 22.8, 15.4, 15.3; HRMS (FAB) m/zcalcd. for C19H28NaO (M + Na)+ 295.2038, found 295.2023 (Δ-1.5 mmu)。
【0037】
(8)セレン酸化。
【0038】
このセレン酸化は、テトラシクロケトン10の第6位炭素に結合しているアルキル基をアルデヒド基に変換するための反応である。まず、5mlの枝付きフラスコに、上記テトラシクロケトン10を1.8mg(6.6μmol)を秤り取り、無水ジオキサン0.5mlに溶解した。ここに、二酸化セレン11.8mg(0.11mmol)を加えて1時間還流した。室温まで放冷し、セライトを用いて反応混合物をろ過し、濃縮した。これをエーテル5mlに溶解し、飽和炭酸水素ナトリウム水溶液、水、飽和塩化ナトリウム水溶液で洗浄し乾燥後、減圧濃縮した。得られた混合物をカラムクロマトグラフィー [シリカゲルBW-820MH(0.5g)、ヘキサン‐エーテル(5:1)]により精製したところ、アルデヒド体1(1H-2,8a-Methanocyclopenta[a]cyclopropa[e]cyclodecene-4-carboxaldehyde,1a,2,3,5a,6,7,8,9,10,10a-decahydro-1,1,9-trimethyl-11-oxo-,(1aR,2S,5aS,8aR,9R,10aR)-)が、1.6mg(収率85%)が無色結晶として得られた。なお、第6位炭素にアルデヒド基を有するアルデヒド体1(図1参照)の構造分析データは、次の通りである。
【0039】
mp 145.5-147.5 ℃(hexane); [α]D21 +29.5 (c0.078, CHCl3); IR (neat) 2940, 1722, 1687, 1594, 1458 cm-1; 1H NMR (500 MHz, CDCl3)δ9.28 (s, 1H), 6.23 (d, J = 1.9Hz, 1H), 3.36(br s, 1H), 3.09(ddd, J = 14.0, 10.9, 3.1Hz, 1H), 2.58(dddd, J = 17.1, 13.4, 3.7, 1.7Hz, 1H), 2.38(m, 1H), 2.30(m, 1H), 2.02 (m, 1H), 1.94(ddd, J= 15.6, 5.3, 5.3 Hz, 1H), 1.85 (m, 1H), 1.79(m, 1 H), 1.70 (m, 1H), 1.45-1.25 (m, 3H), 1.10 (s, 3H), 1,05 (s, 3H), 0.98 (d, J = 6.9Hz, 3H), 0.72 (m, 2H); 13C NMR (100 MHz, C6D6)δ208.1, 193.8, 159.3, 146.5, 72.0, 46.9, 45.0, 39.8, 34.6, 29.6, 29.0, 28.4, 28.1, 25.8, 23.7, 23.3, 22.8, 15.4, 15.0; HRMS (FAB) m/z calcd. for C19H26NaO2 (M + Na)+309.1831, found 309.1853 (Δ+2.2mmu).
【0040】
このアルデヒド体1は、炭素数19のノルインゲナン骨格を有している。このアルデヒド体1は、既述の非特許文献2(Winklerら)の9728頁に掲載されたScheme3の反応経路中の番号20の化合物と同一である。従って、以後は、Winklerらの前記Scheme3の反応経路に従えば、図3に示された化学構造を有するインゲノールを光学活性体として合成できる。なお、図2には、理解のため、Winklerらによって提案された前記Scheme3の反応経路を転載する。
【産業上の利用可能性】
【0041】
本発明によれば、生理活性を有するため注目されているが、合成が困難であったインゲノールを光学活性体として合成することができる。従って、このインゲノールに係わる薬剤開発に、特に有用である。
【図面の簡単な説明】
【0042】
【図1】本発明に係る「ノルインゲナン骨格を合成する方法」の合成経路を示す図である。
【図2】同合成経路により得られるアルデヒド体1を中間体とするWinklerらによるインゲノール合成経路を示す図(非特許文献1より転載)である。
【図3】インゲノールの化学構造を示す図である。
【図4】インゲナン骨格の炭素環骨格を示す図である。
【図5】二つのオレフィン部位がtrans体を構成する2,7−ジアリルシクロヘプタノンが、環化オレフィンメタセシスによる閉環が困難であることを示す図である。
【符号の説明】
【0043】
1 アルデヒド体(ノルインゲナン骨格、Winkler合成経路の中間体)
2 ケトン(Funkケトエステル)
3 ヨウ化物
4 アルキル化物
5 ケトン
6 アリルアルコール
7 塩化物
8 スピロケトン
9 メタリルスピロケトン
10 テトラシクロケトン(ノルインゲナン骨格)
【特許請求の範囲】
【請求項1】
ジオレフィン化合物の一方のオレフィン部位をスピロ環で固定して三環性ジオレフィン化合物を得る工程と、該三環性ジオレフィン化合物のジオレフィン部位をオレフィンメタセシス反応で閉環する工程と、を少なくとも行うことによって四環性のノルインゲナン骨格を合成する方法。
【請求項2】
前記ノルインゲナン骨格の第6位炭素には、アルデヒド基又はアルデヒド基に変換可能な基が結合していることを特徴とする請求項1記載の方法。
【請求項3】
前記インゲナン骨格は、光学活性を示すこと特徴とする請求項1又は2に記載の方法。
【請求項4】
請求項1から3のいずれか一項に記載された方法を少なくとも用いることを特徴とするインゲノールの製造方法。
【請求項1】
ジオレフィン化合物の一方のオレフィン部位をスピロ環で固定して三環性ジオレフィン化合物を得る工程と、該三環性ジオレフィン化合物のジオレフィン部位をオレフィンメタセシス反応で閉環する工程と、を少なくとも行うことによって四環性のノルインゲナン骨格を合成する方法。
【請求項2】
前記ノルインゲナン骨格の第6位炭素には、アルデヒド基又はアルデヒド基に変換可能な基が結合していることを特徴とする請求項1記載の方法。
【請求項3】
前記インゲナン骨格は、光学活性を示すこと特徴とする請求項1又は2に記載の方法。
【請求項4】
請求項1から3のいずれか一項に記載された方法を少なくとも用いることを特徴とするインゲノールの製造方法。
【図1】

【図2】
【図3】
【図4】
【図5】
【図2】
【図3】
【図4】
【図5】
【公開番号】特開2006−56794(P2006−56794A)
【公開日】平成18年3月2日(2006.3.2)
【国際特許分類】
【出願番号】特願2004−237933(P2004−237933)
【出願日】平成16年8月18日(2004.8.18)
【新規性喪失の例外の表示】特許法第30条第1項適用申請有り 平成16年3月11日 社団法人日本化学会発行の「日本化学会第84春季年会 講演予稿集2」に発表
【出願人】(504171134)国立大学法人 筑波大学 (510)
【Fターム(参考)】
【公開日】平成18年3月2日(2006.3.2)
【国際特許分類】
【出願日】平成16年8月18日(2004.8.18)
【新規性喪失の例外の表示】特許法第30条第1項適用申請有り 平成16年3月11日 社団法人日本化学会発行の「日本化学会第84春季年会 講演予稿集2」に発表
【出願人】(504171134)国立大学法人 筑波大学 (510)
【Fターム(参考)】
[ Back to top ]