
高生産性細胞の樹立のための発現ベクター及び高生産性細胞
【課題】 目的タンパク質遺伝子を高レベルで発現する形質転換細胞を効率的に樹立するのに効果的な発現ベクターを提供する。
【解決手段】 mRNA不安定化配列を含む薬剤選択マーカー遺伝子発現カセット、少なくとも1つの遺伝子発現安定化エレメント、及び目的タンパク質の遺伝子発現カセットを有する発現ベクター。
【解決手段】 mRNA不安定化配列を含む薬剤選択マーカー遺伝子発現カセット、少なくとも1つの遺伝子発現安定化エレメント、及び目的タンパク質の遺伝子発現カセットを有する発現ベクター。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、遺伝子組換え技術によって、目的とするタンパク質の遺伝子を挿入した発現ベクターを宿主細胞に導入し、得られた形質転換細胞の中から目的とするタンパク質の遺伝子を高レベルで発現する細胞を効率よく樹立するための発現ベクター、及び形質転換された高生産性細胞に関する。本発明の発現ベクターは、遺伝子工学的手法により、動物細胞、特に哺乳類動物細胞において医薬品などの有用タンパク質を生産するために有用である。
【背景技術】
【0002】
組換えタンパク質を生産する発現システムの開発は、研究または治療に供されるタンパク質の供給源を提供するうえで重要である。発現システムとしては、大腸菌などの原核細胞に基づくもの、ならびに酵母(Saccharomyces属,Pichia属、Kluyveromyces属など)及び哺乳類細胞などの動物細胞を含む真核細胞に基づくものの両者が用いられている。これらの中でも動物細胞、特に哺乳類細胞に基づく発現システムが治療用タンパク質の製造には好ましい。なぜなら、ヒトをはじめとする哺乳類動物において起こるタンパク質の翻訳後修飾は、時にその生物活性に深く寄与するため、タンパク質の投与対象と類似した翻訳後修飾が可能な哺乳類細胞に基づく発現システムを利用した方が、治療用タンパク質の有効性を増強しうるからである。
【0003】
組換えタンパク質を産生する細胞を樹立する方法としては、一般に、目的タンパク質の遺伝子を発現する遺伝子構築物を宿主細胞へ遺伝子導入し、得られた形質転換細胞の中からこの遺伝子構築物が宿主細胞のゲノム上に安定導入された細胞を選別する。この際、前記遺伝子構築物に、薬剤選択マーカーとなる薬剤耐性遺伝子を目的タンパク質の遺伝子と同一プロモーター下あるいは別個のプロモーター下で発現するように挿入しておき、薬剤選択によって生き残った細胞を目的タンパク質の遺伝子が安定導入された細胞として選別する。目的タンパク質の発現レベルは、目的タンパク質をコードする遺伝子が宿主細胞ゲノム内のどの領域に導入されるかによって大きく変動するが、導入領域の制御は、通常不可能である。従って、遺伝子導入しても大半の細胞は目的タンパク質遺伝子を発現しないか又は発現量が低い。このため、従来、目的タンパク質遺伝子を高レベルで発現する形質転換細胞の取得には、1000〜数千の細胞サンプルから1〜2個の細胞株を選別する作業を数ヶ月〜1年かけて繰り返し行っており、目的タンパク質遺伝子を高レベルで発現する形質転換細胞の選別に膨大な労力と時間を要していた。
【0004】
そこで、目的タンパク質遺伝子を高レベルで発現する形質転換細胞を効率的に選別するため、薬剤選択マーカーの発現や機能を減弱化することに基づく方法が開発されてきた。薬剤選択マーカーの発現や機能を減弱化すると、薬剤選択マーカー遺伝子が宿主細胞ゲノム内の低発現領域に導入された形質転換細胞は、薬剤選択マーカー(薬剤耐性遺伝子)を十分に発現することができないので、死滅してしまい、高発現領域に導入された形質転換細胞のみが薬剤選択によって生き残る。この生き残った形質転換細胞では、薬剤選択マーカー遺伝子に隣接して目的タンパク質遺伝子を有する可能性が高いため、目的タンパク質遺伝子も宿主細胞ゲノム内の高発現部位に導入されており、目的タンパク質遺伝子が高レベルで発現されている可能性が高い。このことを利用して、目的タンパク質遺伝子を高レベルで発現する形質転換細胞を効率良く選別することができると考えられる。
【0005】
薬剤選択マーカーの発現や機能を減弱化する1つの方法としては、薬剤選択マーカーの発現に用いるプロモーターとして、転写活性の弱いプロモーターであるHerpes simplex virus thymidine kinase(HSV−tk)プロモーターを利用する方法(非特許文献1)や、野生型の転写活性レベルよりも減弱化された変異型プロモーターを用いて薬剤選択マーカーの発現を抑える方法(特許文献1、2)が挙げられる。しかしながら、本発明者らがこれらの方法を追試したところ、いずれの方法も薬剤選択マーカーの発現を減弱化するという面で効果がほとんど見られなかった。このことから、これらの方法を用いて目的タンパク質遺伝子を高レベルで発現する形質転換細胞を効率的に選別するということは難しいと考えられた。
【0006】
薬剤選択マーカーの発現や機能を減弱化する別の方法としては、ネオマイシンホスホトランスフェラーゼなどの薬剤選択マーカー遺伝子のコード配列に変異を導入して薬剤選択マーカー自体の機能を減弱化する方法(特許文献3、4)が挙げられる。本発明者らがこの方法を追試したところ、この方法を用いることにより薬剤選択マーカーの機能を多少減弱化することができ、薬剤選択の結果として少量の発現量の向上した細胞株を取得することができた。しかしながら、目的タンパク質遺伝子を高レベルで発現する形質転換細胞を選別する点での効率は良くなく、この方法による顕著な効果は見られなかった。
【0007】
高生産性細胞の樹立には細胞選択技術の改良の他に、目的遺伝子の発現を安定的に維持するため、宿主細胞ゲノム上の目的遺伝子導入部位の周囲のゲノム環境からの影響を排除する工夫が必要である。組換えタンパク質の発現は、組換えタンパク質をコードする遺伝子が宿主細胞ゲノム内のどの部位に導入されるかによって大きく変動するが、導入部位の制御は通常、不可能である。従って、スクリーニング時には十分な発現をしていた細胞でも培養を継続していくうちに次第に発現が低下してしまうこともある。このような位置効果を克服するため、導入遺伝子への隣接する染色体や調節エレメントの影響を緩和する染色体エレメント(シス作用性DNAエレメント)が組換えタンパク質の製造に利用されつつある。このような機能を持った核酸配列の1つがインスレーターと呼ばれる配列で、ニワトリのβ−グロビンLCRの1.2kb長のDNaseI Hypersensitive部位(cHS4)などがよく機能解析され、組換えタンパク質の発現に利用されているが(非特許文献2)、CHO−K1細胞におけるタンパク質発現の増加はそれほど大きなものではないとされている(非特許文献3)。
【先行技術文献】
【特許文献】
【0008】
【特許文献1】US5,627,033
【特許文献2】WO2005/024015
【特許文献3】WO2001/032901
【特許文献4】WO2004/050884
【特許文献5】特開2001−37478号公報
【非特許文献】
【0009】
【非特許文献1】Niwa H、Yamamura K,Miyazaki J.(1991)Gene 108:193−200
【非特許文献2】Pikaart MJ,Recillas−Targa F,Felsenfeld G.(1998)Genes Dev 12:2852−62
【非特許文献3】Izumi M,Gilbert DM.(1999)J Cell Biochem 76:280−289
【非特許文献4】Bakheet T,Frevel M,Williams B.R.G、 Greer W,Khabar K.S.A.(2001)Nucleic Acids Research 29:246−254
【非特許文献5】Lagnado C.A,Brown C.L,Goodall G.J.(1994) Molecular and Cellular Biology 14:7984−7995
【非特許文献6】Zubiaga A.M,Belasco J.G,Greenberg M.E.(1995) Molecular and Cellular Biology 15:2219−2230
【非特許文献7】Blasco MA(2007)Nat Rev Genet 8:299−309
【非特許文献8】Gonzalo S,Jaco I,Fraga MF,Chen T,Li E,Esteller M,Blasco MA(2006)Nat Cell Biol 8:416−424
【非特許文献9】Perrod S,Gasser SM(2003)Cell Mol Life Sci 60:2303−2318
【非特許文献10】Wakimoto BT(1998)Cell 93:321−324
【非特許文献11】Bao L,Zhou M,Cui Y(2008)Nucleic Acids Research,36,D83−D87
【発明の概要】
【発明が解決しようとする課題】
【0010】
本発明の目的は、目的タンパク質遺伝子を高レベルで発現する形質転換細胞を効率的に樹立するのに効果的な発現ベクター、及び高生産性細胞を提供することにある。
【課題を解決するための手段】
【0011】
本発明者らは、上記課題を解決するため、mRNA不安定化配列を導入した薬剤選択マーカー発現カセット、及び遺伝子発現安定化エレメントを発明した。そして、mRNA不安定化配列を有する種々の薬剤選択マーカー発現カセットと遺伝子発現安定化エレメントを具備した導入遺伝子の発現ベクターを構築し、前記発現ベクターで形質転換された宿主細胞の遺伝子発現を詳しく解析したところ、目的タンパク質の高生産性細胞の割合が飛躍的に上昇することを見出し、本発明を完成させるに到った。
【0012】
すなわち、本発明によれば、以下が提供される:
(1) mRNA不安定化配列を含む薬剤選択マーカー遺伝子発現カセット、少なくとも1つの遺伝子発現安定化エレメント、及び目的タンパク質の遺伝子発現カセットを有することを特徴とする発現ベクター。
(2) mRNA不安定化配列が、サイトカイン、インターロイキン、又は癌原遺伝子の3’非翻訳領域に存在するATリッチ配列に由来することを特徴とする(1)に記載の発現ベクター。
(3) mRNA不安定化配列が、TTATTTA(A/T)(A/T)のモチーフ配列を有することを特徴とする(1)に記載の発現ベクター。
(4) モチーフ配列が、2回以上繰り返されていることを特徴とする(3)に記載の発現ベクター。
(5) モチーフ配列の繰り返し間に1塩基以上のスペーサー配列を含むことを特徴とする(4)に記載の発現ベクター。
(6) mRNA不安定化配列中に1ないし数塩基の置換、挿入または欠失を含むことを特徴とする(2)〜(5)のいずれか一項に記載の発現ベクター。
(7) 遺伝子発現安定化エレメントがチャイニーズハムスターゲノム由来であることを特徴とする(1)〜(6)のいずれかに記載の発現ベクター。
(8)遺伝子発現安定化エレメントが、以下の(a)〜(g)のいずれか一つ又はそれらの任意の組合せからなることを特徴とする(1)〜(7)のいずれかに記載の発現ベクター。
(a)配列番号26で示す配列からなるDNA;
(b)配列番号26で示す配列の部分配列からなるDNAであって、配列番号26で示す配列のうち41820番目から41839番目までの塩基で示す領域の配列を含むDNA;
(c)配列番号26で示す配列の部分配列からなるDNAであって、配列番号26で示す配列のうち41821番目から41840番目までの塩基で示す領域の配列を含むDNA;
(d)配列番号26で示す配列の部分配列からなるDNAであって、配列番号26で示す配列のうち45182番目から45200番目までの塩基で示す領域の配列を含むDNA;
(e)配列番号26で示す配列の部分配列からなるDNAであって、配列番号26で示す配列のうち91094番目から91113番目までの塩基で示す領域の配列を含むDNA;
(f)配列番号26で示す配列の部分配列からなるDNAであって、宿主細胞中で外来遺伝子発現カセットと近接するように配置された際に、外来遺伝子発現カセットに含まれる外来遺伝子からの目的組換えタンパク質の発現を増大または安定化させることができるDNA;及び
(g)前記(a)〜(f)のいずれかのDNAと相補的な塩基配列からなるDNAとストリンジェントな条件下でハイブリダイズし、かつ遺伝子発現安定化機能を有するDNA
(h)前記(a)〜(g)のいずれかのDNAと相補的な塩基配列からなるDNA
(9) 遺伝子発現安定化エレメントが、以下の(i)〜(k)のいずれか一つ又はそれらの任意の組合せからなることを特徴とする(8)に記載の発現ベクター。
(i)配列番号26で示す配列のうち41601番目から46746番目までの塩基で示す領域の配列を含むDNA;
(j)(i)のDNAと相補的な塩基配列からなるDNAとストリンジェントな条件下でハイブリダイズし、かつ遺伝子発現安定化機能を有するDNA;及び
(k)前記(i)又は(j)のDNAと相補的な塩基配列からなるDNA
(10) 遺伝子発現安定化エレメントが、以下の(l)〜(n)のいずれか一つ又はそれらの任意の組合せからなることを特徴とする(9)に記載の発現ベクター。
(l)配列番号26で示す配列のうち41601番目から42700番目までの塩基で示す領域の配列を含むDNA;
(m)(l)のDNAと相補的な塩基配列からなるDNAとストリンジェントな条件下でハイブリダイズし、かつ遺伝子発現安定化機能を有するDNA;及び
(n)(l)又は(m)のDNAと相補的な塩基配列からなるDNA
(11) 遺伝子発現安定化エレメントが、目的タンパク質の遺伝子発現カセットの上流に配置されていることを特徴とする、(1)〜(10)のいずれかに記載の発現ベクター。
(12) 遺伝子発現安定化エレメントが、目的タンパク質の遺伝子発現カセットの上流及び下流の両方に配置されていることを特徴とする、(1)〜(10)のいずれかに記載の発現ベクター。
(13) 遺伝子発現安定化エレメントが、目的タンパク質の遺伝子発現カセット及び薬剤選択マーカー遺伝子発現カセットの上流及び下流の両方に配置されていることを特徴とする(1)〜(10)のいずれかに記載の発現ベクター。
(14) 薬剤選択マーカー遺伝子が、タンパク質合成阻害系抗生物質耐性遺伝子であることを特徴とする(1)〜(13)のいずれかに記載の発現ベクター。
(15) 薬剤選択マーカー遺伝子が、ピューロマイシン−N−アセチルトランスフェラーゼ、ハイグロマイシン−B−ホスホトランスフェラーゼ、及びネオマイシンホスホトランスフェラーゼからなる群より選択されることを特徴とする(14)に記載の発現カベクター。
(16) 目的タンパク質の遺伝子発現カセットが、所望の目的タンパク質遺伝子を挿入するためのマルチクローニングサイトを備えていることを特徴とする(1)〜(15)のいずれかに記載の発現ベクター。
(17) 目的タンパク質が、抗体の重鎖及び/又は軽鎖ポリペプチドであることを特徴とする(1)〜(16)のいずれかに記載の発現ベクター。
(18) (1)〜(17)のいずれか一項に記載の発現ベクターで宿主細胞を形質転換して得られることを特徴とする形質転換細胞。
(19) 宿主細胞が動物細胞であることを特徴とする(18)に記載の形質転換細胞。
(20) 動物細胞が哺乳類細胞であることを特徴とする(19)に記載の形質転換細胞。
(21) 哺乳類細胞がチャイニーズハムスター卵巣(CHO)細胞であることを特徴とする(20)に記載の形質転換細胞。
(22) チャイニーズハムスター卵巣(CHO)細胞が無血清馴化されていることを特徴とする(21)に記載の形質転換細胞。
(23) (18)〜(22)のいずれか一項に記載の形質転換細胞を薬剤選択する工程を含むことを特徴とする、目的タンパク質遺伝子を高レベルで発現する細胞群を選別する方法。
(24) (18)〜(22)のいずれか一項に記載の形質転換細胞からなる細胞群であって、目的タンパク質遺伝子を高レベルで発現することを特徴とする細胞群。
(25) (18)〜(22)のいずれか一項に記載の形質転換細胞を用いることを特徴とする、タンパク質を生産する方法。
(26) タンパク質が、抗体であることを特徴とする(25)に記載の方法。
(27) タンパク質が、ワクチンであることを特徴とする(25)に記載の方法。
(28) (18)〜(22)のいずれか一項に記載の形質転換細胞を用いることを特徴とする、アミノ酸を生産する方法。
(29) (18)〜(22)のいずれかに記載の形質転換細胞を用いることを特徴とする、ヌクレオチドを生産する方法。
【発明の効果】
【0013】
本発明の薬剤選択マーカー遺伝子発現カセットは、mRNA不安定化配列が挿入されているため、mRNA不安定化配列を有さない従来の薬剤選択マーカー遺伝子発現カセットに比べて薬剤選択マーカーの発現が減弱化されており、宿主細胞ゲノム内の高発現領域に組み込まれないと、薬剤存在下で形質転換細胞の生存が困難となる。このため、薬剤選択マーカー遺伝子及び目的タンパク質遺伝子が宿主細胞ゲノム内の高発現領域に組み込まれた形質転換細胞のみが薬剤選択によって生き残ることができる。その結果、目的タンパク質遺伝子を高レベルで発現する高発現細胞を効率的に選別することができる。
【0014】
また本発明の遺伝子発現安定化エレメントは、細胞ゲノムにおける組換えタンパク質遺伝子に対する隣接染色体や調節エレメントの影響を低減させて組換えタンパク質遺伝子の遺伝子発現を安定化し、高発現を長期間維持することができる。そこで、本発明の弱化された薬剤選択マーカー発現カセットと遺伝子発現安定化エレメントを併用することにより、その相乗効果によって、薬剤選択によって選択された細胞において目的遺伝子を非常に高度に発現する細胞を濃縮し、かつそれらの高発現細胞における発現の安定的な維持ができる。本発明により、それら非常に高いレベルのタンパク質生産性を示す高生産細胞株を迅速、容易、且つ効率的に樹立することができる。
【図面の簡単な説明】
【0015】
【図1】pBS−CMV−SNAPm2の構築を示す図である。
【図2】pPUR・N4の構築を示す図である。以下、図中、Purとは、ピューロマイシン耐性遺伝子のことである。また、図中、iPCRとは、Inverse PCRのことである。
【図3】pBS−CMV−SNAPm−Pur及びpBS−CMV−SNAPm−Pur・N4の構築を示す図である。
【図4】pBS−CMV−SNAPm−Pur及びpBS−CMV−SNAPm−Pur・N4で形質転換された細胞集団のFACS解析の結果を示す図である。
【図5】pBS−CMV−SNAPm−Pur及びpBS−CMV−SNAPm−Pur・N4で形質転換された細胞集団のうち、SNAPmを高発現する細胞の割合をプロットしたグラフである。
【図6】pBS−CMV−SNAPm−Pur及びpBS−CMV−SNAPm−Pur・N4で形質転換された細胞集団を薬剤選択した際の明視野像である。
【図7】pBS−CMV−SNAPm−Purのコロニーを単離し、FACS解析した結果を示す図である。
【図8】pBS−CMV−SNAPm−Pur・N4のコロニーを単離し、FACS解析した結果を示す図である。
【図9】pEF1α−KOD3G8HC−RE2の構築を示す図である。以下、図中、Neoとは、ネオマイシン耐性遺伝子のことである。
【図10】pEF1α−SNAP26m−Pur・N4及びpEF1α−SNAP26m−Pur−RE2の構築を示す図である。
【図11】pEF1α−SNAP26m−Pur−RE2及びpEF1α−SNAP26m−Pur・N4で形質転換された細胞集団のFACS解析の結果を示す図である。
【図12】pEF1α−SNAP26m−Pur−RE2及びpEF1α−SNAP26m−Pur・N4で形質転換された細胞集団のうち、SNAPmを高発現する細胞の割合をプロットしたグラフである。
【図13】pEF1α−KOD3G8LC−Hyg−RE2の構築を示す図である。以下、図中、Hygとは、ハイグロマイシン耐性遺伝子のことである。
【図14】pEF1α−SNAP26m−Hyg−RE2及びpEF1α−SNAP26m−Hyg・N4の構築を示す図である。
【図15】pEF1α−SNAP26m−Hyg−RE2及びpEF1α−SNAP26m−Hyg・N4で形質転換された細胞集団のFACS解析の結果を示す図である。
【図16】pEF1α−SNAP26m−Hyg−RE2及びpEF1α−SNAP26m−Hyg・N4で形質転換された細胞集団のうち、SNAPmを高発現する細胞の割合をプロットしたグラフである。
【図17】pEF1α−SNAP26m−Neo・N4の構築を示す図である。
【図18】pEF1α−SNAP26m−Neo−RE2及びpEF1α−SNAP26m−Neo・N4で形質転換された細胞集団のFACS解析の結果を示す図である。
【図19】pEF1α−SNAP26m−Neo−RE2及びpEF1α−SNAP26m−Neo・N4で形質転換された細胞集団のうち、SNAPmを高発現する細胞の割合をプロットしたグラフである。
【図20】pEF1α−SNAP26m−Pur・N2、pEF1α−SNAP26m−Pur・N6及びpEF1α−SNAP26m−Pur・N8の構築を示す図である。
【図21】pEF1α−SNAP26m−Pur−RE2、pEF1α−SNAP26m−Pur・N2、pEF1α−SNAP26m−Pur・N4、pEF1α−SNAP26m−Pur・N6及びpEF1α−SNAP26m−Pur・N8で形質転換された細胞集団のFACS解析の結果を示す図である。
【図22】pEF1α−SNAP26m−Pur−RE2、pEF1α−SNAP26m−Pur・N2、pEF1α−SNAP26m−Pur・N4、pEF1α−SNAP26m−Pur・N6及びpEF1α−SNAP26m−Pur・N8で形質転換された細胞集団のうち、SNAPmを高発現する細胞の割合をプロットしたグラフである。
【図23】配列番号26の配列とCTCFの結合配列の予測部位を示す図である。
【図24】核酸配列断片の遺伝子発現安定化効果の確認に用いた基本コンストラクトpBS−CMV−SNAPmの構築を示す図である。
【図25】配列番号26の配列、CTCFBS、被験配列の核酸断片の位置関係を示す図である。
【図26】各核酸配列の断片を含むSNAPm発現コンストラクトを安定導入されたCHO細胞をFACS解析して得られたSNAPm発現強度と細胞数の分布を示すグラフである。
【図27】各核酸配列の断片を含むSNAPm発現コンストラクトを安定導入されたCHO細胞のFACS解析によって、10以上のSNAPmの蛍光シグナルを呈した細胞の割合(%)をプロットしたグラフである。
【図28】CHO5のデリーションコンストラクト構築と配列番号26の当該被験配列の位置関係を示す図である。
【図29】CHO5、及びCHO5デリーションコンストラクトを安定導入して得られたCHO細胞のFACS解析の結果、10以上のSNAPmの蛍光シグナルを呈した細胞の割合(%)をプロットしたグラフである。
【図30】CHO5Δ3の3’デリーションコンストラクト構築のためのInverse PCR法に用いたプライマーの位置関係とデリーションによって得られた被験挿入配列を示す図である。
【図31】CHO5Δ3、及びCHO5Δ3の3’デリーションコンストラクトを安定導入して得られたCHO細胞のFACS解析の結果、10以上のSNAPmの蛍光シグナルを呈した細胞の割合(%)をプロットしたグラフである。
【図32】pEF1α−KOD3G8HC−1.1k/1.1kの構築を示す図である。
【図33】pEF1α−SNAP26m−Pur・N4−1.1k/1.1k、pEF1α−SNAP26m−Pur−1.1k/1.1kの構築を示す図である。
【図34】pEF1α−SNAP26m−Pur−RE2、pEF1α−SNAP26m−Pur−1.1k/1.1k、pEF1α−SNAP26m−Pur・N4及びpEF1α−SNAP26m−Pur・N4−1.1k/1.1kで形質転換された細胞集団のFACS解析の結果を示す図である。
【図35】pEF1α−SNAP26m−Pur−RE2、pEF1α−SNAP26m−Pur−1.1k/1.1k、pEF1α−SNAP26m−Pur・N4及びpEF1α−SNAP26m−Pur・N4−1.1k/1.1kで形質転換された細胞集団のうち、SNAPmを高発現する細胞の割合をプロットしたグラフである。
【図36】pEF1α−SNAP26m−Hyg・N4−1.1k/1.1k、pEF1α−SNAP26m−Hyg−1.1k/1.1kの構築を示す図である。
【図37】pEF1α−SNAP26m−Hyg−RE2、pEF1α−SNAP26m−Hyg−1.1k/1.1k、pEF1α−SNAP26m−Hyg・N4及びpEF1α−SNAP26m−Hyg・N4−1.1k/1.1kで形質転換された細胞集団のFACS解析の結果を示す図である。
【図38】pEF1α−SNAP26m−Hyg−RE2、pEF1α−SNAP26m−Hyg−1.1k/1.1k、pEF1α−SNAP26m−Hyg・N4及びpEF1α−SNAP26m−Hyg・N4−1.1k/1.1kで形質転換された細胞集団のうち、SNAPmを高発現する細胞の割合をプロットしたグラフである。
【図39】pEF1α−SNAP26m−Neo・N4−1.1k/1.1k、pEF1α−SNAP26m−Neo−1.1k/1.1kの構築を示す図である。
【図40】pEF1α−SNAP26m−Neo−RE2、pEF1α−SNAP26m−Neo−1.1k/1.1k、pEF1α−SNAP26m−Neo・N4及びpEF1α−SNAP26m−Neo・N4−1.1k/1.1kで形質転換された細胞集団のFACS解析の結果を示す図である。
【図41】pEF1α−SNAP26m−Neo−RE2、pEF1α−SNAP26m−Neo−1.1k/1.1k、pEF1α−SNAP26m−Neo・N4及びpEF1α−SNAP26m−Neo・N4−1.1k/1.1kで形質転換された細胞集団のうち、SNAPmを高発現する細胞の割合をプロットしたグラフである。
【図42】pEF1α−SNAP26m−Pur・N2−1.1k/1.1k、pEF1α−SNAP26m−Pur・N6−1.1k/1.1k、pEF1α−SNAP26m−Pur・N8−1.1k/1.1kの構築を示す図である。
【図43】pEF1α−SNAP26m−Pur−1.1k/1.1k、pEF1α−SNAP26m−Pur・N2−1.1k/1.1k、pEF1α−SNAP26m−Pur・N4−1.1k/1.1k、pEF1α−SNAP26m−Pur・N6−1.1k/1.1k及びpEF1α−SNAP26m−Pur・N8−1.1k/1.1kで形質転換された細胞集団のFACS解析の結果を示す図である。
【図44】pEF1α−SNAP26m−Pur−1.1k/1.1k、pEF1α−SNAP26m−Pur・N2−1.1k/1.1k、pEF1α−SNAP26m−Pur・N4−1.1k/1.1k、pEF1α−SNAP26m−Pur・N6−1.1k/1.1k及びpEF1α−SNAP26m−Pur・N8−1.1k/1.1kで形質転換された細胞集団のうち、SNAPmを高発現する細胞の割合をプロットしたグラフである。
【図45】pEF1α−KOD3G8HC−Neo・N4−1.1k/1.1kの構築を示す図である。
【図46】pEF1α−KOD3G8LC−Hyg−1.1k/1.1kの構築を示す図である。
【図47】pEF1α−KOD3G8LC−Hyg・N4−1.1k/1.1kの構築を示す図である。
【図48】pEF1α−KOD3G8HC−Pur・N4−1.1k/1.1kの構築を示す図である。
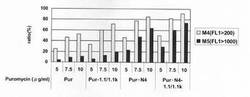
【図49】遺伝子発現安定化エレメントを含まないもの(−/Neo,−/Hyg)、遺伝子発現安定化エレメントCHO5Δ3−3を発現カセットの上流および下流の両方に挿入したもの(1.1k/Neo,1.1k/Hyg)、遺伝子発現安定化エレメントCHO5Δ3−3を発現カセットの上流および下流の両方に挿入し、薬剤耐性遺伝子(H鎖:Neo、L鎖:Hyg)にmRNA不安定化配列(N4配列)を付加したもの(1.1k/Neo・N4,1.1k/Hyg・N4)、遺伝子発現安定化エレメントCHO5Δ3−3を発現カセットの上流および下流の両方に挿入し、薬剤耐性遺伝子(H鎖:Pur、L鎖:Hyg)にmRNA不安定化配列(N4配列)を付加したもの(1.1k/Pur・N4,1.1k/Hyg・N4)について、ポリクローンの培養上清を用いてELISAを行い算出した、抗体生産量をプロットしたグラフである。
【図50】6ウェルプレートの培養上清を用いてELISAを行い算出した、各クローンの抗体生産量をプロットしたグラフである。遺伝子発現安定化エレメントを含まないもの(−/Neo,−/Hyg)と遺伝子発現安定化エレメントCHO5Δ3−3を発現カセットの上流および下流の両方に挿入したもの(1.1k/Neo,1.1k/Hyg)、および、遺伝子発現安定化エレメントを含まないもの(−/Neo,−/Hyg)と遺伝子発現安定化エレメントCHO5Δ3−3を発現カセットの上流および下流の両方に挿入し、薬剤耐性遺伝子(H鎖:Neo、L鎖:Hyg)にmRNA不安定化配列(N4配列)を付加したもの(1.1k/Neo・N4,1.1k/Hyg・N4)、および、遺伝子発現安定化エレメントを含まないもの(−/Neo,−/Hyg)と遺伝子発現安定化エレメントCHO5Δ3−3を発現カセットの上流および下流の両方に挿入し、薬剤耐性遺伝子(H鎖:Pur、L鎖:Hyg)にmRNA不安定化配列(N4配列)を付加したもの(1.1k/Pur・N4,1.1k/Hyg・N4)について、それぞれ生産量上位5クローンを同一グラフ上にプロットしている。
【図51】遺伝子安定化エレメントとmRNA不安定化配列を含まないもの(−/Neo,−/Hyg)、遺伝子安定化エレメントとmRNA不安定化配列を付加し、高い抗体生産性が認められたもの(1.1k/Pur・N4,1.1k/Hyg・N4)について、96ウェルプレートにおける2週間培養での抗体生産性の度数分布をプロットしたグラフである。
【図52】遺伝子発現安定化エレメントCHO5Δ3−3を発現カセットの上流および下流の両方に挿入したもの(1.1k/Neo,1.1k/Hyg)、遺伝子発現安定化エレメントCHO5Δ3−3を発現カセットの上流および下流の両方に挿入し、薬剤耐性遺伝子(H鎖:Pur、L鎖:Hyg)にmRNA不安定化配列(N4配列)を付加したもの(1.1k/Pur・N4,1.1k/Hyg・N4)について無血清馴化されたCHO−K1細胞へ遺伝子導入し、薬剤選択後にポリクローンの培養上清を用いてELISAを行い算出した、抗体生産量をプロットしたグラフである。
【図53】pEF1α−KOD3G8LC,HC−Pur・N4−1.1k/1.1kの構築を示す図である。
【図54】2コンストラクト抗体発現系、シングルベクター抗体発現系について、ポリクローンの培養上清を用いてELISAを行い算出した、抗体生産量をプロットしたグラフである。
【図55】6ウェルプレートの培養上清を用いてELISAを行い算出した、各クローンの抗体生産量をプロットしたグラフである。2コンストラクト抗体発現系、シングルベクター抗体発現系、それぞれ生産量上位5クローンを同一グラフ上にプロットしている。
【発明を実施するための形態】
【0016】
以下に本発明を詳細に説明する。
【0017】
タンパク質の機能解析や有用タンパク質の生産のために、遺伝子組み換え技術を用いて、細胞、特に動物細胞に、目的とするタンパク質の遺伝子を導入し、組換えタンパク質として発現させる方法が広く用いられている。具体的には、プラスミドベクターに代表されるベクターに目的タンパク質遺伝子を挿入し、細胞にベクターを取り込ませた後、組換えにより目的タンパク質遺伝子を細胞ゲノムに組み込み、細胞を形質転換させる方法がよく用いられている。
【0018】
細胞のゲノムは染色体上に存在するが、染色体はヒストンのアセチル化や高度な凝集(ヘテロクロマチン)等により遺伝子の発現が制御されており、挿入される染色体の場所によって遺伝子の発現量は大きく異なる。ベクターを用いて細胞を形質転換する方法においては、細胞ゲノムのどの場所に目的タンパク質遺伝子が導入されるかを制御することはできず、そのため形質転換細胞における目的タンパク質遺伝子の発現量、ひいては目的タンパク質の生産量は、細胞株によって大きく異なることになる。
【0019】
また、遺伝子組み換えがうまくいかず、目的タンパク質遺伝子が細胞ゲノムに組み込まれていない場合も多い。
【0020】
そこで、従前、予め発現ベクターに薬剤耐性(薬剤選択マーカー)遺伝子を挿入しておき、細胞にこのベクターを遺伝子導入した後、培地中に薬剤を投与して(薬剤選択)、形質転換が成功した細胞を選択し、その後、生じた細胞群から高い発現を示す細胞株を繰り返し選別する作業が行われてきた。しかしながら、この方法では多数の細胞株を用意する必要があり、また長期間の培養を必要とする細胞の選別作業を繰り返し行うため、多大の労力と時間を要してきた。加えて、薬剤耐性遺伝子は多くの場合、少量の発現でも薬剤に対する耐性を示すため、生存した細胞の中には、薬剤耐性遺伝子及び目的タンパク質遺伝子が細胞ゲノム内の低発現領域に組込まれており目的タンパク質遺伝子の発現量が少ない細胞株も混在しており、更に労力と時間に拍車をかけてきた。
【0021】
また、選別時には十分な発現をしていた細胞でも、培養を継続していくうちに次第に発現が低下するものが少なからず見られる。これは、遺伝子サイレンシングにより発現が抑制されるためであると考えられる。目的とするタンパク質遺伝子のゲノム導入位置の制御が通常不可能であるため、遺伝子サイレンシングの効果による発現量低下を予測することは極めて困難である。そこで、このような位置効果を緩和し、発現を高レベルで安定的に維持する技術が必要とされてきた。
【0022】
そこで、本発明はこれらの問題を解決するため、薬剤選択を利用して高発現を示す細胞を取得できるようにし、かつ遺伝子発現安定化エレメントによって、遺伝子の導入部位に左右されることなく高い発現のレベルを維持できるようにするものであり、迅速・簡便かつ効率的に目的タンパク質生産性の高い細胞株を取得できる点で極めて顕著な効果を有するものである。
【0023】
本発明ではまず、従来公知の薬剤耐性(薬剤選択マーカー)遺伝子を使用した選別において、mRNA不安定化配列を利用することによって薬剤耐性遺伝子の発現量を大きく抑え、宿主細胞ゲノム内の高発現領域に薬剤耐性遺伝子が組込まれた形質転換細胞のみが薬剤選択後に生き残るようにしている。薬剤耐性遺伝子の近隣には目的タンパク質の遺伝子が組込まれているため、生き残った細胞では、目的タンパク質遺伝子も高発現領域に組込まれている可能性が高い。従って、本発明によれば、(i)遺伝子組み換えがうまくいかず、目的タンパク質遺伝子が細胞ゲノムに組み込まれていない細胞、(ii)遺伝子組み換えに成功したが、薬剤耐性遺伝子及び目的タンパク質遺伝子が宿主細胞ゲノムの低発現領域に組込まれている細胞、及び(iii)遺伝子組み換えに成功し、しかも薬剤耐性遺伝子及び目的タンパク質遺伝子が宿主細胞ゲノムの高発現領域に組込まれている細胞の中から、(iii)の遺伝子組み換えに成功し、しかも薬剤耐性遺伝子及び目的タンパク質遺伝子が宿主細胞ゲノムの高発現領域に組込まれている細胞のみを薬剤選択によって効率的に選別することができる。従来公知の選別方法では、(i)〜(iii)の細胞から(ii)及び(iii)の細胞を薬剤選択によって選別することができるが、(ii)及び(iii)の細胞から(iii)の細胞のみを薬剤選択によって選別することはできず、薬剤選択後に生き残った形質転換細胞について目的タンパク質遺伝子の発現レベルを別途確認して、(iii)の高発現細胞を選別していた。本発明の方法は、薬剤選択のみによって(i)〜(iii)の細胞から(iii)の高発現細胞を一気に選別することができるため、効率的であり、結果として高発現細胞を迅速に選別することができる。
また、本発明では、遺伝子発現安定化エレメントを利用することにより、選別した細胞の継続培養時の遺伝子発現量の低下を防止している。本発明による遺伝子発現安定化エレメントは、目的タンパク質の発現の安定的維持のみならず、薬剤耐性遺伝子に効果を示す場合には、薬剤耐性遺伝子の発現に対する隣接染色体や調節エレメントの影響を低減させ、薬剤耐性遺伝子本来の機能を発揮する助けとなり、薬剤選択との相乗効果を示すものともなる。
【0024】
本発明の第一側面によれば、mRNA不安定化配列を含む薬剤選択マーカー遺伝子発現カセット、少なくとも1つの遺伝子発現安定化エレメント、及び目的タンパク質の遺伝子発現カセットを有することを特徴とする発現ベクターが提供される。
【0025】
本発明において、発現カセットとは、プロモーターから遺伝子コード配列、ターミネーター配列(ポリアデニレーションシグナル)までの遺伝子発現の単位をいう。加えて、イントロン、スペーサー配列、翻訳増強領域等を含むこともある。したがって、例えば「薬剤選択マーカー発現カセット」という場合、前記発現カセットの遺伝子コード配列が薬剤選択マーカー遺伝子のコード配列であるものをいう。また、「目的タンパク質の遺伝子発現カセット」という場合、前記発現カセットの遺伝子コード配列が目的とするタンパク質の遺伝子のコード配列であるものをいう。
【0026】
(薬剤選択マーカー発現カセット)
本発明で使用する薬剤選択マーカー遺伝子発現カセットは、mRNA不安定化配列を有することを除いては、従来公知の薬剤選択マーカー遺伝子発現カセットと同様の構成を有する。そこで、まず、mRNA不安定化配列について説明する。
本発明は、mRNA不安定化配列が極めて効果的に薬剤耐性遺伝子の発現を減弱化することができることを見出したことに基づくものである。本発明のmRNA不安定化配列を含む薬剤選択マーカー遺伝子発現カセットを搭載した発現ベクターを宿主細胞へ遺伝子導入した場合、mRNA不安定化配列を含まない場合と比較して、薬剤選択後の生存細胞数、あるいはそのコロニーは1/10〜1/100に減少する。これはmRNA不安定配列の存在により薬剤選択マーカーのmRNAが不安定化し、薬剤選択マーカーの発現量が下がるため、宿主細胞のゲノム中で転写活性の高い高発現領域に当該発現カセットが組み込まれた細胞でなければ薬剤選択下での生存が困難になるためと考えられる。
【0027】
「mRNA不安定化配列」とは、この配列を有するDNAから転写されたmRNAの細胞内半減期を低減させる機能を有するヌクレオチド配列のことであり、自然界では初期応答遺伝子などに存在することが見出されている。
【0028】
mRNA不安定化配列は、いくつかの遺伝子ではコード配列中、あるいは5’UTR(非翻訳領域)に存在するものもあるが、多くは3’UTRに存在する。3’UTRに存在するmRNA不安定化配列としては、AU−rich element(ARE)、histone mRNA 3’−terminal stem−loop、iron−responsive element(IRE)、insulin−like growth factor II(IGF−II)、long stem−loopなどが知られている。これらの中でもAREは、恒常的にmRNAを不安定化できることから、本発明に用いるmRNA不安定化配列として好ましい。
【0029】
AREとは、mRNAにおける「AUが豊富なエレメント」、すなわちアデニン(A)及びウラシル(U)を高い割合で含む配列もしくは領域を意味する。またAREとは、前記エレメントをコードするDNAにおける「ATが豊富なエレメント」、すなわちアデニン(A)及びチミン(T)を高い割合で含む配列もしくは領域を指すためにも用いられる。
【0030】
AREは、造血細胞増殖因子遺伝子、成長因子遺伝子、インターロイキン遺伝子、インターフェロン等のサイトカイン遺伝子、及びいくつかの癌原遺伝子(プロトオンコジーン)などに見られる(非特許文献4)。本発明の薬剤選択マーカー遺伝子発現カセットでは、これらの遺伝子中のAREに相当する核酸配列をそのまま利用してもよいが、ARE中のモチーフ配列として知られるTATTTAT(非特許文献5)やTTATTTA(T/A)(T/A)(非特許文献6)などを利用する方が、不要な制限酵素認識配列を発現ベクターに挿入してしまうことを避けることができ、利便性が高い。
【0031】
AREを持つ遺伝子の例としては、造血細胞増殖因子についてはGranulocyte−monocyte colony sitimulationg factor(GM−CSF),インターロイキンについてはInterleukin−1β、2,3、4、6、8、10、11、インターフェロンについてはInterferon−α、癌原遺伝子についてはc−fos,c−myc,c−jun,c−myb,Pim−1などが例示される。このほかにもTumor necrosis factor,Cyclin D1,Cyclooxygenase,Plasminogen activator inhibitor type2など多くの遺伝子がAREを持つことが知られており、例示された遺伝子に限定されない。
【0032】
本発明の薬剤選択マーカー遺伝子発現カセットにおいて使用することができるmRNA不安定化配列としては、下記のものが例示される。
【0033】
ATTTのモチーフ配列の1ないし2回以上の繰り返し。
【0034】
ATTTAのモチーフ配列の1ないし2回以上の繰り返し。
【0035】
TATTTATのモチーフ配列の1ないし2回以上の繰り返し。
【0036】
TTATTTA(T/A)(T/A)のモチーフ配列の1ないし2回以上の繰り返し。(T/A)はTまたはAのいずれかである。
【0037】
これらの配列は、1ないし数塩基の置換/挿入/欠失を含んでもよく、DNAの複製の誤りや突然変異などの自然変異によるもの、人為的な変異導入によるもの等が想定される。また、モチーフ配列の繰り返し間には1〜100塩基程度のスペーサー配列またはリンカー配列を含んでもよい。加えて、モチーフ配列の逆位を含むものであってもよい。
【0038】
これらのモチーフ配列の繰り返し数は、1回でもよいが、2回以上、より好ましくは4回以上とすることにより、mRNAの不安定化効果を一層高めることができ、選別効率を顕著に増大させることができる。
【0039】
繰り返しの回数の上限は特に限定されないが、細胞へのベクターの取り込みの観点から、発現ベクターが最大10kbp〜25kbp長となるようなものが望ましい。ただし、繰り返し配列数が増えるにしたがって、mRNAの不安定化の効果は飽和に向かい、一定の繰り返し数を超えるとそれ以上の顕著な効果を望めなくなる。上限値を定めるものではないが、10回以上の繰り返し配列は実用上意味がない。
【0040】
本発明の薬剤選択マーカー遺伝子発現カセットにおけるプロモーターとしては、動物細胞、特に哺乳類細胞で発現可能なプロモーターであれば特に限定されるものではないが、ヒトやマウスのサイトメガロウイルス(CMV)プロモーター、サルウイルス40(SV40)プロモーター、ヒトヘルペス単純ウイルスのチミジンキナーゼ遺伝子(HSV−tk)プロモーター、などのウイルスに由来するものや、マウスphosphoglycerate−kinase1遺伝子(PGK)プロモーターなどの非ウイルス性の細胞遺伝子に由来するプロモーター、及び由来の異なるプロモーターのハイブリッドが挙げられる。ここで、プロモーターは、プロモーターのコア領域に限るものではなく、エンハンサー領域を含む場合もある。薬剤選択マーカーの発現を減弱化するため、転写活性が低いものがより好ましい。転写活性の低いプロモーターとして変異型のプロモーターなどを利用してもよいし、コザック配列を置換してもよい。さらに、mRNA不安定化配列のモチーフ配列の繰り返しの回数を少なくし、転写活性の弱いプロモーターを併用するのも1つの想定される実施態様である。
【0041】
本発明の薬剤選択マーカー発現カセットを用いた薬剤選択に使用される薬剤としては、タンパク質合成阻害系抗生物質としてブラストサイジン(Blasticidin)、ジェネティシン(Geneticin)(G418)、ハイグロマイシン(Hygromycin;Hyg)、ピューロマイシン(Puromycin;Pur)が例示される。その他の選択用薬剤としてメトトレキセート(Methotrexate;MTX)、MSX(methionine sulphoximine)などが例示される。また、本発明の薬剤選択マーカー遺伝子発現カセットにおける薬剤選択マーカー遺伝子としては、薬剤選択に一般的に使用される薬剤に対して耐性を示す遺伝子が好適に用いられる。タンパク質合成阻害系抗生物質を薬剤として用いる場合には、タンパク質合成阻害系抗生物質耐性遺伝子を用いることが好ましい。特に限定されるものではないが、ネオマイシン耐性遺伝子(ジェネティシン耐性遺伝子としての機能も有する)として、Tn5由来ネオマイシンホスホトランスフェラーゼ(aminoglycoside 3’−phosphotransferase)(Neo)、ハイグロマイシン耐性遺伝子として、E.coli由来ハイグロマイシン−B−ホスホトランスフェラーゼ(Hph、実施例および図面ではHygと記載)、ピューロマイシン耐性遺伝子として、Streptomyces由来ピューロマイシン−N−アセチルトランスフェラーゼ遺伝子(pac、実施例および図面ではPurと記載)、ブラストサイジン耐性遺伝子として、Bacillus cereus由来ブラストサイジン耐性遺伝子(bsr)などが例示される。これらの中でも、ピューロマイシン−N−アセチルトランスフェラーゼ、ハイグロマイシン‐B−ホスホトランスフェラーゼ、ネオマイシンホスホトランスフェラーゼが好ましく、より好ましくはピューロマイシン−N−アセチルトランスフェラーゼ、ハイグロマイシン‐B−ホスホトランスフェラーゼであり、さらに好ましくはピューロマイシン−N−アセチルトランスフェラーゼである。
【0042】
本発明の薬剤選択マーカー遺伝子発現カセットにおけるポリアデニレーションシグナル(ターミネーター配列)としては、SV40ウイルス由来late polyAシグナル、early polyAシグナル、HSV−tk由来polyAシグナル、ウシ成長因子遺伝子由来polyAシグナル、ウサギβ−グロビン遺伝子由来polyAシグナルなどが例示されるが、特に限定されるものではない。
【0043】
本発明の薬剤選択マーカー遺伝子発現カセット中でのプロモーター、薬剤選択マーカー遺伝子、mRNA不安定化配列、及びポリアデニレーションシグナルの配置順序は、薬剤選択マーカー遺伝子の発現が可能である限り特に限定されないが、一般的にプロモーター、薬剤選択マーカー遺伝子、mRNA不安定化配列、及びポリアデニレーションシグナルをこの順序で上流から下流に向かって配置する。これらの四つの要素は、相互に直接連結されている必要はなく、所望により、イントロン、スペーサー配列、翻訳増強領域等を間に有していてもよい。
【0044】
(遺伝子発現安定化エレメント)
本発明における「遺伝子発現安定化エレメント」とは、目的タンパク質の生産性を高めるために、目的遺伝子発現カセットが宿主ゲノムに挿入された部位近傍のゲノムの転写の活性化の状況に影響を受けてしまう、という位置効果を解消し、遺伝子発現を安定化する機能を有する核酸領域を意味する。
【0045】
遺伝子発現を安定化する機能を有する核酸配列としては、インスレーター、スキャフォールド/マトリックス結合領域(S/MAR)、遺伝子座調節領域(LCR)、遍在作用性クロマチンオープニングエレメント(UCOE)などが利用できる。
【0046】
インスレーターとしては、ニワトリβ-グロビンLCRに由来する1.2kb DNaseI Hypersensitive site(cHS4),バフンウニ由来UR1などが例示される。S/MARとしては、ニワトリリゾチーム5’MARエレメント、ヒトβ−グロビンMARエレメントやヒトインターフェロンβSARエレメントなどが例示される。
【0047】
本発明の発現ベクターは、少なくとも一つの遺伝子発現安定化エレメントを有する。これら遺伝子発現を安定化する機能を有する核酸配列を目的タンパク質の発現カセットの上流若しくは下流、あるいは上流及び下流に配置することによって、遺伝子発現の安定的維持、及び目的タンパク質の生産性を向上させることが可能である。
【0048】
目的遺伝子を宿主細胞ゲノムへ導入するための遺伝子構築物として、プラスミドベクターが一般に用いられるが、プラスミドベクターの長さは最大10ないし25kbpであることから、遺伝子発現安定化エレメントの鎖長は出来るだけ短い方が好ましい。好ましい長さとしては5kbp以下、より好ましくは1kbp程度である。
【0049】
本発明の好ましい実施態様における遺伝子発現安定化エレメントは、特許文献5に記載のCHO細胞DR1000L−4N株(CHO細胞−4N株)から本発明者らによって同定された核酸分子である。当該株は、外来遺伝子としてヒト顆粒球マクロファージコロニー刺激因子(hGM−CSF)遺伝子を、DHFR遺伝子を持つ発現ベクターに導入し、このベクターを、DHFR遺伝子を欠損したCHO細胞DG44株に導入し、導入株をIMDM培地(核酸含有)に10%牛胎児血清を添加した培地で培養して形質転換体を得、この形質転換体を、次いでIMDM培地(核酸不含)、10%透析牛胎児血清を添加した培地で選択し、さらに50nM、100nMのメトトレキセート(MTX)を含み、核酸を含まないIMDM培地を用いて10から100倍程度に増加するまで培養し、MTX濃度を上昇させることにより得られたテロメアタイプのクローン細胞である。
【0050】
真核細胞のゲノムは、真正染色質と異質染色質の2つのクラスの染色質に分けられる。セントロメアとテロメアの配列は、構成的な異質染色質の主要な部分である。特に、テロメアは、TTAGGGの繰り返し配列とサブテロメア領域からなり、際立った異質染色質の立体構造をとる、遺伝子の乏しい領域である(非特許文献7,8)。テロメアは、ゲノムの安定性に対するその寄与や保護的な役割に加え、テロメア位置効果として知られる現象によって、近くに導入された遺伝子の発現にも影響を与える(非特許文献9,10)。
【0051】
ところが、DR1000L−4N株は、テロメアタイプのクローン細胞であるにもかかわらず、導入された外来遺伝子は導入部位付近の異質染色体からの不活性化の影響を受けることなく、長期間の培養において、安定したhGM−CSFの高い産生を維持した。これは、外来遺伝子の導入された部位の近傍に、不活性化の進行を強力に遮断するような境界エレメントが存在し、遺伝子発現を安定化していることを示唆する。
【0052】
本発明者らは、DR1000L−4N株において外来遺伝子導入部位の近傍の配列を分析した結果、配列表の配列番号26で示す約91kbpの配列からなるDNAが遺伝子発現の安定化に関与していることを見出した。
【0053】
本発明の発現ベクターに含まれる遺伝子発現安定化エレメントの長さは短い方が好ましい。そこで、より小さいサイズでも遺伝子発現安定化機能を有する領域を絞り込むため、発明者らは更なる研究を行った。
【0054】
配列番号26に示す約91kbpの配列を更に分析するにあたり、通常、ショットガン法による約3kbp程度の断片のライブラリを作成し、各断片を1つずつアッセイする方法が取られる。しかしながら、各断片が遺伝子安定化機能を有するかどうかを解析する際に、細胞の形質転換や培養に時間がかかるため、1アッセイ当たり数週間から数ヶ月の期間を要する。従って、全アッセイを行うためには膨大な時間と労力を要することになる。
【0055】
そこで、本発明者らは、配列番号26の配列中、遺伝子発現配列の境界エレメントが存在することが期待される部位を中心に解析する方法を見出した。そして、配列番号26からなるDNAのうち42kbp、45kbp、及び91kbp付近の領域が遺伝子発現安定化機能を有することを見出すに至った。
【0056】
この配列をさらに詳細に分析したところ、この配列の中でも、(i)41820番目から41839番目までの塩基で示す領域、(ii)41821番目から41840番目までの塩基で示す領域、(iii)45182番目から45200番目までの塩基で示す領域、及び(iv)91094番目から91113番目までの塩基で示す領域の4つの領域が、インスレーター結合タンパク質CTCFの結合配列モチーフに相当することを見出した。従って、これらの4つの領域の少なくともいずれかを含む、これらの領域の近傍が特に遺伝子発現の安定化に関与していると考えられた。
【0057】
本発明における実施態様では、遺伝子発現安定化エレメントとして、下記(a)〜(e)のいずれか又はその組み合わせが使用できる。
(a)配列番号26で示す配列からなるDNA;
(b)配列番号26で示す配列の部分配列からなるDNAであって、配列番号26で示す配列のうち41820番目から41839番目までの塩基で示す領域の配列を少なくとも含むDNA;
(c)配列番号26で示す配列の部分配列からなるDNAであって、配列番号26で示す配列のうち41821番目から41840番目までの塩基で示す領域の配列を少なくとも含むDNA;
(d)配列番号26で示す配列の部分配列からなるDNAであって、配列番号26で示す配列のうち45182番目から45200番目までの塩基で示す領域の配列を少なくとも含むDNA;及び
(e)配列番号26で示す配列の部分配列からなるDNAであって、配列番号26で示す配列のうち91094番目から91113番目までの塩基で示す領域の配列を少なくとも含むDNA。
【0058】
これらのエレメントは、CHOゲノムから単離同定されたものであるが、これらのエレメントと相同なエレメントは、他の哺乳類細胞ゲノムにも存在することが予想される。従って、(f)配列番号26で示す配列の部分配列からなるDNAであって、宿主細胞中で外来遺伝子発現カセットと近接するように配置された際に、外来遺伝子発現カセットに含まれる外来遺伝子からの目的組換えタンパク質の発現を増大または安定化させることができるDNAも、遺伝子発現安定化エレメントとして使用することができる。このような相同エレメントは、この技術分野の周知の技術、例えば、種間ハイブリダイゼーションまたはPCRなどによって容易に単離同定されることができる。
【0059】
また、(g)前記(a)〜(f)のいずれかのDNAと相補的な塩基配列からなるDNAとストリンジェントな条件でハイブリダイズし、かつ遺伝子発現安定化機能を有するDNAも当然、遺伝子発現安定化エレメントとして使用することができる。本発明の遺伝子発現安定化エレメントは、これらの(a)〜(g)のいずれか一つ又はそれらの任意の組合せからなる。さらに、(h)前記(a)〜(g)のいずれかのDNAと相補的な塩基配列からなるDNAも、遺伝子発現安定化機能を有し、遺伝子発現安定化エレメントとして使用することができるため、本発明に含まれる。
【0060】
ストリンジェントな条件とは、用いられるプローブ・標識方法によっても異なるが、例えば、0.1%SDSを含む0.2×SSC中40〜50℃または0.1%SDSを含む2×SSC中55〜65℃の条件下ハイブリダイズする塩基配列である。さらにストリンジェントな条件は、結合する核酸の融解温度(Tm値)に基づいて決定することができる。また、ハイブリダイズ後の洗浄条件として、「6×SSC、0.1%SDS、Tm値よりも約15〜30℃低い温度」程度の条件をあげることができる。より高いストリンジェントな条件としては「2×SSC、0.1%SDS、Tm値よりも約5〜15℃低い温度」程度、更に高いストリンジェントな条件としては「1×SSC、0.1%SDS、Tm値よりも約5〜15℃低い温度」程度の洗浄条件をあげることができる。更に高いストリンジェントな条件としては「0.1×SSC、0.1%SDS」程度の洗浄条件が例示される。
【0061】
本発明の遺伝子発現安定化エレメントの好ましい態様では、前記(b)または(c)のDNAは、配列番号26で示す配列のうち41601番目から46746番目までの塩基で示す領域、又は配列番号26で示す配列のうち41601番目から46746番目までの配列と相補的な塩基配列からなるDNAとストリンジェントな条件でハイブリダイズし、かつ遺伝子発現安定化機能を有する塩基配列領域である。また、これらの領域からなるDNAと相補的な塩基配列も含まれる。本発明の遺伝子発現安定化エレメントのさらに好ましい態様では、前記(b)または(c)のDNAは、配列番号26で示す配列のうち41601番目から42700番目までの塩基で示す領域、又は配列番号26で示す配列のうち41601番目から42700番目までの配列と相補的な塩基配列からなるDNAとストリンジェントな条件でハイブリダイズし、かつ遺伝子発現安定化機能を有する塩基配列領域であり、又はこれらの領域からなるDNAと相補的な塩基配列である。
【0062】
本発明の遺伝子発現安定化エレメントがその発現安定化効果を奏するためには、エレメントが宿主細胞ゲノム中で外来遺伝子発現カセットの導入部位に近接する位置に配置されることが必要である。本発明において、用語「近接する」とは、特に制限されるものではないが、好ましくは本発明の遺伝子発現安定化エレメントと外来遺伝子発現カセットとの間の距離が5000bp以下、さらに好ましくは500bp以下であることを意味する。
【0063】
このような近接配置は、本発明のエレメントを含む核酸配列断片と、外来遺伝子の発現カセットを含む核酸配列断片との混合物をエレクトロポレーションやトランスフェクションなどによって宿主細胞内へ導入し、外来遺伝子の発現量の高い宿主細胞クローンを選択することによって実現することもできるが、近接配置を確実に達成させるためには、本発明の遺伝子発現安定化エレメント、及びそれに近接して配置された外来遺伝子発現カセットを含む外来遺伝子発現ベクターをあらかじめ作成しておき、この発現ベクターで宿主細胞を形質転換させることが望ましい。
【0064】
(目的タンパク質の遺伝子発現カセット)
本発明における目的タンパク質遺伝子の発現カセットは、プロモーター、遺伝子コード配列、及びターミネーター配列(ポリアデニレーションシグナル)からなる。加えて、イントロンやスペーサー配列、翻訳増強領域を含むこともある。目的タンパク質遺伝子の発現プロモーターとしてはできるだけ転写活性の高いものが好ましく、ヒトやマウスのサイトメガロウイルス(CMV)プロモーター、サルウイルス40(SV40)プロモーターなどのウイルスに由来するものや、ヒトやマウス、さらにはCHO由来のElongation Factor 1(EF1α)遺伝子、ユビキチン遺伝子、β−アクチン遺伝子など、非ウイルス性の細胞遺伝子に由来するプロモーター、及び由来の異なるプロモーター/エンハンサーのハイブリッド、例えばCAGプロモーターなどが挙げられる。
【0065】
本発明では、目的タンパク質の遺伝子発現カセットは、薬剤選択マーカー発現カセットと独立した形で与えられる。真核細胞の場合、目的タンパク質遺伝子と薬剤選択マーカー遺伝子を内在性リボソームエントリー部位(Internal ribosome entry sites、IRES)でつなぐポリシストロン性の発現を行う手法もあるが、目的タンパク質遺伝子と薬剤選択マーカー遺伝子のコード配列が1つのmRNAとして合成されるため、前記mRNA不安定化配列を挿入すると、目的タンパク質遺伝子のmRNAが減少してしまい、高発現細胞を得られない。そのため、本発明では、目的タンパク質の遺伝子発現カセットは、薬剤選択マーカー発現カセットと独立して配置されることが好ましい。目的タンパク質の遺伝子発現カセットと薬剤選択マーカー発現カセットは、別個のベクターの形で利用されてもよいが、目的タンパク質の遺伝子発現カセットと薬剤選択マーカー発現カセットが同一のベクター上に配置されることがより好ましい。
【0066】
目的タンパク質の遺伝子発現カセットには、該タンパク質遺伝子のコード配列の代わりに、複数の制限酵素認識配列からなるマルチクローニングサイトを配置することができる。外来遺伝子のcDNAやコード配列を導入する際に、クローニング作業が簡便であり、好ましい。
【0067】
他の実施態様としては、目的タンパク質が抗体の重鎖及び/又は軽鎖のポリペプチドである、目的タンパク質の遺伝子発現カセットである。この場合、目的タンパク質の発現カセットは、複数配置してもよく、あるいは複数のポリペプチド遺伝子をIRESでつなぎ、ポリシストロン性の1つの発現カセットの形でもよい。
【0068】
(発現ベクター)
本発明において、発現ベクターとは、遺伝子工学に利用されるベクターである。発現ベクターは、プラスミドベクターであることができる。しかし、これに限らず、例えば、ウイルスベクター、コスミドベクター、細菌人工染色体(BAC)、酵母人工染色体(YAC)及び他の非プラスミドベクターも使用できる。
【0069】
本発明における発現ベクターの実施態様の1つは、目的タンパク質遺伝子発現カセットと薬剤選択マーカー遺伝子発現カセットの上流若しくは下流に、遺伝子発現安定化エレメントが配置された発現ベクターである。好ましくは、目的タンパク質遺伝子発現カセットと薬剤選択マーカー遺伝子発現カセットの上流に、遺伝子発現安定化エレメントが配置された発現ベクターである。この場合、目的タンパク質遺伝子発現カセットと薬剤選択マーカー遺伝子発現カセットのどちらが上流であってもよいが、好ましくは目的タンパク質遺伝子発現カセットが上流に配置されたものである。
【0070】
本発明における別の実施態様は、薬剤選択マーカー遺伝子発現カセットと目的タンパク質の遺伝子発現カセットの間に遺伝子安定化エレメントが配置されたベクターである。目的タンパク質遺伝子発現カセットの下流であって薬剤選択マーカー遺伝子発現カセットの上流、若しくは薬剤選択マーカー遺伝子発現カセットの下流であって目的タンパク質遺伝子発現カセットの上流のいずれの態様も考えられるが、好ましくは目的タンパク質遺伝子発現カセットの下流であって薬剤選択マーカー遺伝子発現カセットの上流に遺伝子発現安定化エレメントが配置されたベクターである。
【0071】
本発明における発現ベクターの好ましい実施態様は、目的タンパク質遺伝子発現カセットと薬剤選択マーカー遺伝子発現カセットの上流及び下流に、それらの発現カセットを挟むように遺伝子発現安定化エレメントが配置されたベクターである。目的タンパク質遺伝子発現カセットと薬剤選択マーカー遺伝子発現カセットの順序は問わないが、目的タンパク質遺伝子発現カセットが上流に配置されていることが好ましい。また、目的タンパク質遺伝子発現カセットと薬剤選択マーカー遺伝子発現カセットそれぞれの上流及び下流に遺伝子発現安定化エレメントが配置されていてもよい。
【0072】
本発明における好ましい別の実施態様は、目的タンパク質遺伝子発現カセットの上流及び下流に遺伝子発現安定化エレメントが配置され、さらにその下流に薬剤選択マーカー遺伝子発現カセットが配置されたベクターである。
【0073】
更には、薬剤選択マーカー遺伝子発現カセットの下流に目的タンパク質の遺伝子発現カセットが配置され、薬剤選択マーカー遺伝子発現カセットと目的タンパク質の遺伝子発現カセットの間及び目的タンパク質遺伝子発現カセットの下流に遺伝子発現安定化エレメントが配置されている発現ベクターも実施しうる。
【0074】
遺伝子発現安定化エレメントと薬剤選択マーカー遺伝子発現カセット若しくは目的タンパク質の遺伝子発現カセットとの間は、隣接していてもよいし、スペーサー配列等が挿入されていてもよい。
【0075】
前記薬剤選択マーカー遺伝子発現カセットと目的遺伝子タンパク質の発現カセットが同一のベクター上に配置される場合、両者は隣接してもよいし、間にスペーサー配列が挿入されていてもよい。目的タンパク質遺伝子の発現カセットは薬剤選択マーカー遺伝子発現カセットの上流、下流のいずれに配置されても良いが、隣接して挿入される際には、目的タンパク質遺伝子の発現カセットが上流になるように配置される方が好ましい。さらに、スペーサー配列として、隣接する発現カセットの転写干渉を抑制するためのインスレーター配列が挿入されていても良い。
【0076】
薬剤選択マーカー遺伝子発現カセットは、1つのベクターに2つ以上含まれていても良い。この場合、同一の薬剤選択マーカーが選ばれる場合もあるし、それぞれ異なる薬剤選択マーカーが選ばれる場合もある。また、各薬剤選択マーカーは隣接して配置されても良いし、離れて配置されていても良い。
【0077】
目的のタンパク質が抗体の重鎖、軽鎖のように複数のポリペプチドからなる場合、目的タンパク質の発現カセットを複数配置してもよく、あるいは複数のポリペプチド遺伝子をIRESでつなぎ、ポリシストロン性の1つの発現カセットの形にしてもよい。
【0078】
また、複数のポリペプチド発現カセットをそれぞれ異なるベクターに挿入し、それぞれのベクターを1つの宿主細胞へ導入して形質転換細胞を作製し、目的タンパク質を生産することができる。この場合、各ベクターに薬剤選択マーカー遺伝子発現カセットを配置することができ、それぞれ同一の薬剤選択マーカーを用いることもできるが、異なる薬剤選択マーカーを用いる方が、各ポリペプチドが全て高発現を示す細胞を効率よく取得する点で有利であり、好ましい。
【0079】
抗体の重鎖ポリペプチド遺伝子と軽鎖ポリペプチド遺伝子をそれぞれ異なるベクターに挿入して発現させ、抗体を生産した場合と、抗体の重鎖ポリペプチド遺伝子と軽鎖ポリペプチド遺伝子とを同一のベクターに挿入して発現させ、抗体を生産した場合の比較において、抗体の生産性はどちらも変わらない。したがって、いずれの方法を用いても高い生産性を示す細胞株を取得することができるが、抗体の重鎖ポリペプチド遺伝子と軽鎖ポリペプチド遺伝子とを同一のベクターに挿入して発現させた方が、同一ベクター上の一種類の薬剤耐性遺伝子を使うことになり導入遺伝子数の差異を軽減でき、抗体の重鎖ポリペプチドと軽鎖ポリペプチドの生産バランスがとれ、重鎖・軽鎖の2つのポリペプチドが2本ずつから成る抗体としての生産性が安定するため、好ましい。
【0080】
本発明の第二の側面によれば、本発明の第一の側面による発現ベクターで宿主細胞を形質転換して得られることを特徴とする形質転換細胞が提供される。
【0081】
本発明の形質転換細胞における宿主細胞としては、組換えタンパク質の産生に一般的に用いられるチャイニーズハムスター卵巣細胞(CHO)の他、マウスミエローマ細胞(NSO)、ベビーハムスターキドニー細胞(BHK)、ヒト繊維肉腫細胞(HT1080)、COS細胞などの哺乳類由来の細胞が例示されるが、これらに限定されるものではなく、ヒト、マウス、ラット、ハムスター、モルモット、ウサギ、イヌ、ウシ、ウマ、ヒツジ、サル、ブタなどの動物由来の細胞などを広く導入対象とすることができる。また、大腸菌などの細菌細胞や酵母、昆虫細胞などを用いたタンパク質生産システムにも応用しうる。
一般にバイオ医薬品などの工業生産には、ウイルスなどの有害成分の混入を排除するため、動物成分を含まない培地、より好ましくは化学的に組成の明らかな(Chemically Defined)な培地で生産細胞を培養することが求められる。このため、一般的に使用されるウシ胎児血清などを含有した培地で高生産細胞をスクリーニングした後、1〜2ヶ月程度かけて動物成分非含有培地やChemically Definedとされる培地へ細胞を適応させること(無血清馴化)が必要とされる。しかしながら、せっかく取得した高生産性細胞も上手く馴化できないケースもある。そこで、あらかじめ無血清馴化された細胞に遺伝子導入することは、無血清馴化工程を省略して生産細胞の樹立時間を短縮でき、非常に有効である。このような課題達成のため、本発明に用いられる宿主細胞としては、CHO細胞等における無血清馴化済み細胞がさらに例示される。
【0082】
宿主細胞の形質転換方法は、当業者であれば適宜選択しうるが、例えばリポフェクション法、リン酸カルシウム法、エレクトロポレーション法、DEAEデキストラン法、マイクロインジェクション等が挙げられる。
【0083】
本発明の第三の側面によれば、本発明の第二の側面による形質転換細胞を薬剤選択する工程を含むことを特徴とする、目的タンパク質遺伝子を高レベルで発現する細胞群を選別する方法が提供される。また、本発明の第四の側面によれば、本発明の第二の側面による形質転換細胞からなる細胞群であって、目的タンパク質遺伝子を高レベルで発現することを特徴とする細胞群が提供される。
【0084】
薬剤選択に使用する薬剤は、薬剤選択マーカー遺伝子発現カセット中の薬剤選択マーカー遺伝子の種類に応じて決定される。また、使用する薬剤の濃度としては、一般に使用される濃度の範囲で高発現細胞の濃縮が可能であるが、少し高めの濃度が好ましい。最適な濃度は、宿主細胞や用いる培地の種類によって変化する。これらの濃度の設定方法は当業者にとっては公知であり、適宜設定しうる。例えば、産業用の生産細胞として用いられるチャイニーズハムスター卵巣(CHO)細胞を10%ウシ胎児血清を含むHam‘s F12培地で培養し、ピューロマイシン耐性遺伝子発現カセットを利用した場合のピューロマイシンの濃度としては、5μg/ml以上、より好ましくは7.5μg/ml以上、さらに好ましくは10μg/ml以上である。同様の培養条件でハイグロマイシン耐性遺伝子発現カセットを利用した場合のハイグロマイシンBの濃度としては、200μg/ml以上、より好ましくは600μg/ml以上、さらに好ましくは800μg/ml以上である。同様の培養条件でネオマイシン耐性遺伝子発現カセットを利用した場合のG418の濃度としては、好ましくは400μg/ml以上、より好ましくは800μg/ml以上、さらに好ましくは1,000μg/ml以上である。
【0085】
本発明の選別方法では、薬剤選択により、mRNAの不安定化を伴わない従来の薬剤選択を行った細胞群に比べて、目的タンパク質遺伝子を高レベルで発現する細胞の割合が有意に高くなる。従って、従来は、薬剤選択後の膨大なサンプルから目的タンパク質遺伝子を高レベルで発現する細胞株を選別する作業を繰り返し行わなければならなかったが、本発明によれば、薬剤選択マーカーのmRNA不安定化を含む薬剤選択のみによって、選別の候補となるサンプル数を大幅に絞ることができる。本発明の選別方法を用いれば、薬剤選択後も生存する細胞群(ポリクローン)の状態で目的タンパク質生産量が有意に上昇していて、これらの細胞群から細胞を単離し、培養することにより、容易に高発現を示す細胞株(モノクローン)を取得することができる。本発明の実施態様の一例として、4.0×105個の細胞に本発明の薬剤選択マーカー遺伝子発現カセットが挿入されたベクターを遺伝子導入した後、薬剤選択を行って生じた細胞群から100の細胞株をランダムに選択して、個々の細胞株の発現量を調べることにより目的タンパク質の高発現株を容易に取得することができる。
【0086】
細胞の発現レベルはレポーター遺伝子の発現量により調べることができるが、一例として、SNAP26m遺伝子(Covalys社)やGFP(Green Fluorescent Protein)遺伝子等の蛍光タンパク質遺伝子を含む発現カセットを本発明の発現ベクターに導入し、FACS(fluorescence activated cell sorting)等のフローサイトメーターを用いて蛍光強度を解析することにより調べることができる。別の例として、ルシフェラーゼ遺伝子を含む発現カセットを本発明による発現ベクターに導入後、細胞溶解液にD−ルシフェリンを添加してルミノメーターで発光量を測定することにより調べることができる。また、レポーター遺伝子に限らず、ELISA(Enzyme−Linked Immunosorbent Assay)、酵素免疫測定法(EIA)等を利用することで抗体等の目的タンパク質の発現を調べることもできる。
【0087】
本発明により、細胞選別の工程及び時間を大幅に短縮することができると共に、高発現を示す細胞の割合が上昇することにより、短時間で効率的に目的タンパク質遺伝子を高レベルで発現する細胞を取得することができ、従来技術と比べて極めて顕著な効果を示す。
【0088】
例えばFACSCalibur(ベクトン・ディッキンソン社製)により、蛍光強度FL1にてSNAP26mの発現量を測定した場合、FL1が200以上若しくは1000以上を示す細胞の割合は、本発明の薬剤選択マーカー遺伝子発現カセットを有さない細胞に比べて、本発明による細胞では1.1倍以上、また多くの場合1.5倍から2倍以上大きくなる効果が認められた。また、FL1が200以上を示す細胞の割合の増加率よりもFL1が1000以上を示す細胞の割合の増加率の方が高い傾向が見られており、本発明により選別される細胞群が、本発明の薬剤選択マーカー遺伝子発現カセットを有さない細胞に比べて高い発現を示すという効果を裏付けている。
【0089】
さらに、本発明におけるmRNA不安定化配列及び遺伝子安定化エレメントを有する発現ベクターにより形質転換された細胞では、mRNA不安定化配列及び遺伝子安定化エレメントを有さない発現ベクターを使用した場合に比べ、高生産性を示す細胞の割合が7〜8倍向上する。遺伝子発現安定化エレメントのみを有する発現ベクターにより形質転換された細胞では、高生産性を示す細胞の割合が2倍程度向上し優れた効果を有しているが、本発明はmRNA不安定化配列及び遺伝子発現安定化エレメントの相乗効果によって更に顕著な効果を有する。
【0090】
本発明の第五の側面によれば、本発明の第二の側面による形質転換細胞を用いることを特徴とする、タンパク質を生産する方法が提供される。
【0091】
一つの実施態様では、生産されるタンパク質は、抗体である。この場合、抗体の重鎖ポリペプチド遺伝子と軽鎖ポリペプチド遺伝子は同一のベクター(シングルベクター)に挿入されていても良いし、それぞれ異なるベクターに挿入されて2コンストラクトとされていても良い。
【0092】
哺乳類の抗体には、IgM、IgD、IgG、IgA、IgEの5種類のクラスが存在することが明らかとなっているが、ヒトの各種疾患の診断、予防及び治療には血中半減期が長く、各種エフェクター機能を有する等の機能特性からヒトIgGクラスの抗体が主として利用されている。抗体は、形質転換細胞の培養上清よりプロテインAカラムを用いて精製することができる。また、その他に通常、蛋白質の精製で用いられる精製方法を使用することができる。例えば、ゲル濾過、イオン交換クロマトグラフィー及び限外濾過等を組み合わせて行い、精製することができる。精製したヒト化抗体のH鎖、L鎖或いは抗体分子全体の分子量は、二次元電気泳動等により測定することができる。
【0093】
本発明の方法により生産された抗体は、医薬として使用することができる。かかる医薬は、治療薬として単独で投与することも可能ではあるが、通常は薬理学的に許容される一つあるいはそれ以上の担体と一緒に混合し、製剤学の技術分野にて公知の任意の方法により製造した医薬製剤として提供されるのが好ましい。
【0094】
投与経路は、治療に際して最も効果的なものを使用するのが望ましく、経口投与、または口腔内、気道内、直腸内、皮下、筋肉内および静脈内等の非経口投与をあげることができ、抗体製剤の場合、好ましくは静脈内投与をあげることができる。投与形態としては、噴霧剤、カプセル剤、錠剤、顆粒剤、シロップ剤、乳剤、座剤、注射剤、軟膏、テープ剤等があげられる。
【0095】
経口投与に適当な製剤としては、乳剤、シロップ剤、カプセル剤、錠剤、散剤、顆粒剤等があげられる。乳剤およびシロップ剤のような液体調製物は、水、ショ糖、ソルビトール、果糖等の糖類、ポリエチレングリコール、プロピレングリコール等のグリコール類、ごま油、オリーブ油、大豆油等の油類、p−ヒドロキシ安息香酸エステル類等の防腐剤、ストロベリーフレーバー、ペパーミント等のフレーバー類等を添加剤として用いて製造できる。カプセル剤、錠剤、散剤、顆粒剤等は、乳糖、ブドウ糖、ショ糖、マンニトール等の賦形剤、デンプン、アルギン酸ナトリウム等の崩壊剤、ステアリン酸マグネシウム、タルク等の滑沢剤、ポリビニルアルコール、ヒドロキシプロピルセルロース、ゼラチン等の結合剤、脂肪酸エステル等の界面活性剤、グリセリン等の可塑剤等を添加剤として用いて製造できる。
【0096】
非経口投与に適当な製剤としては、注射剤、座剤、噴霧剤等があげられる。注射剤は、塩溶液、ブドウ糖溶液、あるいは両者の混合物からなる担体等を用いて調製される。または、ヒト化抗体を常法に従って凍結乾燥し、これに塩化ナトリウムを加えることによって粉末注射剤を調製することもできる。座剤はカカオ脂、水素化脂肪またはカルボン酸等の担体を用いて調製される。また、噴霧剤は該化合物そのもの、ないしは受容者の口腔および気道粘膜を刺激せず、かつ該化合物を微細な粒子として分散させ吸収を容易にさせる担体等を用いて調製される。
【0097】
担体として具体的には乳糖、グリセリン等が例示される。該化合物および用いる担体の性質により、エアロゾル、ドライパウダー等の製剤が可能である。また、これらの非経口剤においても経口剤で添加剤として例示した成分を添加することもできる。
【0098】
別の実施態様では、生産されるタンパク質は、ワクチンである。
【0099】
ワクチンとしては、病原体のエピトープのアミノ酸配列からなるタンパク質をワクチンとして使用することができる。また、ウイルス体の構成タンパク質すべてを生産するのみならず、エンベロープタンパク等を生産し、ワクチンとして使用することができる。
【0100】
ワクチンは任意の経路の投与のための製剤とすることができる。粘膜型投与、例えば、経口経路、経鼻経路、経気道経路、経膣経路、経直腸経路の他、非粘膜経路による投与、例えば皮下、静脈内もしくは筋肉内投与のためのワクチン製剤が挙げられる。
【0101】
本発明の第六の側面によれば、本発明の第二の側面による形質転換細胞を用いることを特徴とする、アミノ酸を生産する方法が提供される。また、本発明の第七の側面によれば、本発明の第二の側面による形質転換細胞を用いることを特徴とする、ヌクレオチドを生産する方法が提供される。
【0102】
アミノ酸の生産は、まず形質転換細胞にポリペプチドを生産させ、そのポリペプチドを加水分解することによって行うことができる。また、アミノ酸の生体外合成に必要な酵素などを形質転換細胞に生産させることによって行うこともできる。ヌクレオチドの生産も、ヌクレオチドの生体外合成に必要な酵素などを形質転換細胞に生産させることによって行うことができる。
【実施例】
【0103】
以下、実施例を例示することによって、本発明の効果をより一層明確なものとする。
【実施例1】
【0104】
ピューロマイシン耐性遺伝子におけるmRNA不安定化配列の効果
(1)pBS−CMV−SNAPm−Pur及びpBS−CMV−SNAPm−Pur・N4の構築
図1に示すスキームに従って、プラスミドpBS−CMV−SNAPm2を構築した。即ち、まずEmerald Lucベクター(pELuc−test)(東洋紡績社製)の制限酵素ClaI、EcoRIサイトにCMVプロモーター(配列番号1)が導入されたpELuc−CMVプラスミドから制限酵素EcoRI、NotIでEmerald Luc(ELuc)遺伝子を切出した。一方、SNAPm発現プラスミドpSNAPm(Covalys社製)からSNAP26m遺伝子を配列番号2,3のプライマーを用いてPCRで増幅し、pELuc−CMVプラスミドの制限酵素EcoRI、NotIサイトに導入してpSNAP26m−CMVを構築した。続いて、pSNAP26m−CMVより、CMVプロモーター/SNAP26m/SV40 polyAを配列番号4,5のプライマーを用いてPCRで増幅し、部分消化によって、プラスミドpBluescript II KS(−)(ストラタジーン社製)の制限酵素ClaI、BamHIサイトへ導入し、プラスミドpBS−CMV−SNAPm2を構築した。
【0105】
続いて、図2に示すスキームに従って、pPUR(クロンテック社製)のPuromycin耐性遺伝子のコード配列の3‘末端に、配列番号6、7のプライマーとKOD −Plus− Mutagenesis Kit(東洋紡績社製)を用いたInverse PCR法によって、ARE配列モチーフTTATTTATTの4回繰り返し配列(N4)を挿入してpPUR・N4を構築した。
【0106】
つづいて、pPUR,pPUR・N4を鋳型として、配列番号8、9のプライマーとKOD −Plus− ver.2(東洋紡績社製)を用いたPCRによって、SV40 プロモーター−Pur−SV40 polyA、SV40プロモーター−Pur・N4−SV40 polyAの薬剤発現カセット断片を増幅した。pBS−CMV−SNAPm2と前記増幅断片をそれぞれ制限酵素BamHI、SacIで部分消化して連結し、pBS−CMV−SNAPm-Pur,pBS−CMV−SNAPm-Pur・N4を構築した(図3)。
【実施例2】
【0107】
(2)pBS−CMV−SNAPm−Pur及びpBS−CMV−SNAPm−Pur・N4の細胞導入(1)
CHO−K1細胞(理研バイオリソースセンター、No.RCB0285)を1×105cells/mlに調整し、12ウェルプレートに2mlずつ前日に播種し、一晩培養して、トランスフェクション用のCHO−K1細胞を準備した。この際、培地として10%牛胎児血清を添加したHam’s F12培地(日水製薬社製)を使用した。
【0108】
トランスフェクションは、3μl GeneJuice Transfection Reagent(メルク社製)を100μl Opti−MEM I Reduced−Serum Medium(GIBCO社製)で希釈し、1μgのプラスミドとしてpBS−CMV−SNAPm−PurあるいはpBS−CMV−SNAPm−Pur・N4にこの希釈液103μlを加え、10分間放置した後、この混合液を前記CHO−K1細胞に加え、24時間インキュベートすることにより行った。翌日、培地を除去し、2.5g/l−Trypsin・1mmol/l−EDTA Solution(ナカライテスク社製)で処理して細胞を分散させ、90mmペトリディッシュに移し、6μg/ml Puromycin(InvivoGen社製)を含むHam’s F12+10%FBS培地中で3週間選択培養を行った。選択培養中、3〜4日ごとに培地を交換した。3週間の選択培養終了後、2.5g/l−Trypsin・1mmol/l−EDTA Solutionで処理して細胞を分散し、1ウェル当たり2×105細胞を12ウェルプレートに播き込み、翌日、0.5mlのHam’s F12培地に2μM SNAP−Cell−505(Covalys社製)を加え、60分間、37℃でインキュベートした。その後、Ham’s F12培地で3回リンスし、培地交換をしながら、10分間のインキュベートを3回行い、未反応の蛍光色素を除去した。この細胞を2.5g/l−Trypsin/1mmol/l−EDTA Solutionで処理して細胞を分散し、D−PBS(−)(ナカライテスク社製)に細胞を懸濁し、フローサイトメーターBD FACSCalibur(Becton,Dickinson社製)でSNAP26mの発現強度を解析した。
【0109】
図4は、薬剤選択後のpBS−CMV−SNAPm−Pur及びpBS−CMV−SNAPm−Pur・N4で形質転換された細胞集団のFACS解析の結果を示す。横軸が蛍光強度(FL1)、縦軸がカウント数である。また、図5に蛍光強度FL1がそれぞれ200以上、1000以上の細胞の割合のプロットを示す。この結果、mRNA不安定化因子であるN4が挿入されたpBS−CMV−SNAPm−Pur・N4で形質転換された細胞集団には、薬剤選択後、SNAPmの高発現を示す細胞が有意に濃縮されていることが分かる。
【実施例3】
【0110】
細胞株でのピューロマイシン耐性遺伝子におけるmRNA不安定化配列の効果
(1)pBS−CMV−SNAPm−Pur及びpBS−CMV−SNAPm−Pur・N4の細胞導入(2)
つづいて、実施例2同様に遺伝子導入を行い、9μg/ml Puromycinを含むHam’s F12+10%FBS培地中で2週間選択培養を行った。ここで形成されたコロニーを顕微鏡下でピペットの先でpBS−CMV−SNAPm−Purについては48クローン、pBS−CMV−SNAPm−Pur・N4については20クローンをかきとり、12ウェルプレートに播種した。増殖が認められた、それぞれ36クローン、10クローンについて、実施例2と同様にSNAP−Cell−505で標識処理をし、FACS解析を実施した。
【0111】
図6に、薬剤選択後の細胞の明視野像を示す。この結果、pBS−CMV−SNAPm−Purに比べ、pBS−CMV−SNAPm−Pur・N4については生存細胞数、コロニー数が顕著に減少し、選択効果が高いことが示唆される。
【0112】
図7にpBS−CMV−SNAPm−Pur形質転換細胞クローンにおける、ピーク強度が1000を超える、特にSNAPm発現の高いクローンのFACS解析の結果を示す。図8にpBS−CMV−SNAPm−Pur・N4形質転換細胞クローンにおける、ピーク強度が1000を超える、特にSNAPm発現の高いクローンのFACS解析の結果を示す。この結果、pBS−CMV−SNAPm−Purでは高発現クローンは36クローン中1クローン(2.8%)しか認められなかった。しかしながら、pBS−CMV−SNAPm−Pur・N4は取得されたクローンは少ないものの、SNAPm高発現クローンのヒット率が40%(10クローン中4クローン)と非常に選択効果が高いことが確認された。また、この結果から、実施例2で認められたように、薬剤選択後の細胞集団をFACS解析することによって高発現細胞の割合の上昇していることと、高発現クローンの取得が効率化されていることに相関が確認されたことから、以後の解析は細胞集団のFACS解析によって効果の確認を実施した。
【実施例4】
【0113】
EF1αプロモーター使用下でのピューロマイシン耐性遺伝子におけるmRNA不安定化配列の効果
(1)pEF1α−SNAP26m−Pur・N4及びpEF1α−SNAP26m−Pur−RE2の構築
次に、目的遺伝子発現カセットのプロモーターをEF1αプロモーターに置換した場合も、上述と同様の効果が認められるか検討するため、プラスミドを構築した。本実験では、図9に示すスキームに従って構築されたプラスミドpEF1α−KOD3G8HC−RE2を利用した。即ち、まず、CMVプロモーターをEF−1αプロモーターに置換したpCI−neoプラスミド(プロメガ社製)の制限酵素XbaI−NotIサイトに抗KOD抗体の重鎖を挿入したプラスミドのpEF1α−KOD3G8HCを鋳型に、KOD −Plus− Mutagenesis Kitを用いたInverse PCR法によって、配列番号10、11のプライマーを使って発現カセット上流に制限酵素SalIおよびNheIサイトを付加したpEF1α−KOD3G8HC−REを構築した。なお、抗KOD抗体の重鎖としては、Thermococcus kodakaraensis KOD1株由来のDNAポリメラーゼに特異的な抗体を産生するマウスハイブリドーマ細胞系3G8(入手先:独立行政法人産業技術総合研究所 特許生物寄託センター、寄託番号:FERM BP−6056)から得られた抗体のものを使用した。更に本プラスミドを鋳型に、KOD −Plus− Mutagenesis Kitを用いたInverse PCR法によって、配列番号12、13のプライマーを使って発現カセット下流に制限酵素BsiWIおよびXhoIサイトを付加した構築されたプラスミドがpEF1α−KOD3G8HC−RE2である。
【0114】
このpEF1α−KOD3G8HC−RE2を用いて、図10に示すスキームに従って、プラスミドpEF1α−SNAP26m−Pur・N4およびpEF1α−SNAP26m−Pur−RE2を構築した。即ち、プラスミドpEF1α−KOD3G8HC−RE2から制限酵素EcoRI、NotIで抗KOD抗体の重鎖遺伝子を切出した。一方、SNAPm発現プラスミドpSNAPmからSNAP26m遺伝子を配列番号2,3のプライマーを用いてPCRで増幅し、pEF1α−KOD3G8HC−RE2プラスミドの制限酵素EcoRI、NotIサイトに導入してpEF1α−SNAP26m−Neo−RE2を構築した。本プラスミドから制限酵素AflII、BstBIでネオマイシン耐性遺伝子を切出した。一方、pPUR・N4から下流にN4配列を付加したPuromycin耐性遺伝子を配列番号14,15のプライマーを用いてPCRで増幅し、pEF1α−SNAP26m−RE2プラスミドの制限酵素AflII、BstBIサイトに導入してpEF1α−SNAP26m−Pur・N4を構築した。続いて、本プラスミドを鋳型に、KOD −Plus− Mutagenesis Kitを用いたInverse PCR法によって、配列番号16、17のプライマーを使ってPuromycin耐性遺伝子下流のN4配列を削除したプラスミドpEF1α−SNAP26m−Pur−RE2を構築した。
【実施例5】
【0115】
(2)pEF1α−SNAP26m−Pur−RE2及びpEF1α−SNAP26m−Pur・N4の細胞導入
実施例2に記載の方法でトランスフェクション用のCHO−K1細胞を準備した。一方、実施例4で構築したSNAP26m発現コンストラクトを制限酵素AhdI(New England Biolabs社製)でリニアライズ(直鎖状化)した。トランスフェクションは、3μl GeneJuice Transfection Reagentを100μl Opti−MEM I Reduced−Serum Mediumで希釈した後、前記のリニア化したプラスミド1μgを加え、10分間放置した後、この混合液を前記CHO−K1細胞に加え、24時間インキュベートすることにより行った。翌日、培地を除去し、2.5g/l−Trypsin・1mmol/l−EDTA Solutionで処理して細胞を分散させ、90mmペトリディッシュに移し、5、7.5、あるいは10μg/ml Puromycinをそれぞれ含むHam’s F12+10%FBS培地中で1週間選択培養を行った。選択培養中、3〜4日ごとに培地を交換した。選択培養終了後、実施例2に記載の方法で、細胞集団のSNAP26mの発現強度を解析した。
【0116】
図11は、薬剤選択後のpEF1α−SNAP26m−Pur−RE2及びpEF1α−SNAP26m−Pur・N4で形質転換された細胞集団のFACS解析の結果を示す。また、図12に蛍光強度FL1が200以上、1000以上の細胞の割合のプロットを示す。この結果、薬剤選択中のPuromycin濃度に関わらず、mRNA不安定化因子であるN4が挿入されたpEF1α−SNAP26m−Pur・N4で形質転換された細胞集団には、薬剤選択後、SNAPmの高発現を示す細胞が有意に濃縮されていることが分かる。
【実施例6】
【0117】
ハイグロマイシン耐性遺伝子におけるmRNA不安定化配列の効果
(1)pEF1α−SNAP26m−Hyg‐RE2及びpEF1α−SNAP26m−Hyg・N4の構築
ハイグロマシン耐性遺伝子を用いた場合もmRNA不安定化因子の効果が認められるか検討するため、プラスミドを構築した。この構築には、図13に示すスキームに従って構築されたプラスミドpEF1α−KOD3G8LC−Hyg−RE2を利用した。即ち、まず、CMVプロモーターをEF1αプロモーターに変更したpCI−neoプラスミドの制限酵素XbaI−NotIサイトに抗KOD抗体の軽鎖を挿入したプラスミドのpEF1α−KOD3G8LCを鋳型に、KOD −Plus− Mutagenesis Kitを用いたInverse PCR法によって、配列番号10、11のプライマーを使って発現カセット上流に制限酵素SalIおよびNheIサイトを付加したpEF1α−KOD3G8LC−REを構築した。なお、抗KOD抗体の軽鎖としては、Thermococcus kodakaraensis KOD1株由来のDNAポリメラーゼに特異的な抗体を産生するマウスハイブリドーマ細胞系3G8(入手先:独立行政法人産業技術総合研究所 特許生物寄託センター、寄託番号:FERM BP−6056)から得られた抗体のものを使用した。更に本プラスミドを鋳型に、KOD −Plus− Mutagenesis Kitを用いたInverse PCR法によって、配列番号12、13のプライマーを使って発現カセット下流に制限酵素BsiWIおよびXhoIサイトを付加して構築されたプラスミドがpEF1α−KOD3G8LC−RE2である。
【0118】
このpEF1α−KOD3G8LC−RE2をAflII、BstBIで処理し、ネオマイシン耐性遺伝子を切出した。一方、pTK−Hyg(クロンテック社製)からハイグロマイシン耐性遺伝子を配列番号18、19のプライマーを使ってPCRで増幅し、pEF1α−KOD3G8LC−RE2の制限酵素AflII、BstBIサイトに導入してpEF1α−KOD3G8LC−Hyg−RE2を構築した。続いて、図14に示すスキームに従って、プラスミドpEF1α−SNAP26m−Hyg−RE2及びpEF1α−SNAP26m−Hyg・N4を構築した。即ち、プラスミドpEF1α−KOD3G8LC−Hyg−RE2から制限酵素MluI、NotIで抗KOD抗体の軽鎖遺伝子を切出した。一方、SNAPm発現プラスミドpSNAPmからSNAP26m遺伝子を配列番号3,20のプライマーを用いてPCRで増幅し、pEF1α−KOD3G8LC−Hyg−RE2プラスミドの制限酵素MluI、NotIサイトに導入してpEF1α−SNAP26m−Hyg−RE2を構築した。続いて、本プラスミドを鋳型に、KOD −Plus− Mutagenesis Kitを用いたInverse PCR法によって、配列番号21、22のプライマーを使ってHygromycin耐性遺伝子下流にN4配列を付加したpEF1α−SNAP26m−Hyg・N4を構築した。
【実施例7】
【0119】
(2)pEF1α−SNAP26m−Hyg−RE2及びpEF1α−SNAP26m−Hyg・N4の細胞導入
実施例2に記載の方法でトランスフェクション用のCHO−K1細胞を準備した。一方、実施例6で構築したSNAP26m発現コンストラクトを制限酵素AhdIでリニアライズした。トランスフェクションは、実施例5に記載の方法で行い、トランスフェクションの翌日、培地を除去し、2.5g/l−Trypsin・1mmol/l−EDTA Solutionで処理して細胞を分散させ、90mmペトリディッシュに移し、800μg/mlハイグロマイシン HygroGold(InvivoGen社製)を含むHam’s F12+10%FBS培地中で1週間選択培養を行った。選択培養中、3〜4日ごとに培地を交換した。選択培養終了後、実施例2に記載の方法で、細胞集団のSNAP26mの発現強度を解析した。
【0120】
図15は、薬剤選択後のpEF1α−SNAP26m−Hyg−RE2及びpEF1α−SNAP26m−Hyg・N4で形質転換された細胞集団のFACS解析の結果を示す。また、図16に蛍光強度FL1が200以上、1000以上の細胞の割合のプロットを示す。この結果、mRNA不安定化因子であるN4が挿入されたpEF1α−SNAP26m−Hyg・N4で形質転換された細胞集団には、薬剤選択後、SNAPmの高発現を示す細胞が有意に濃縮されていることが分かる。
【実施例8】
【0121】
ネオマイシン耐性遺伝子におけるmRNA不安定化配列の効果
(1)pEF1α−SNAP26m−Neo・N4の構築
ネオマイシン耐性遺伝子を用いた場合もmRNA不安定化因子の効果が認められるか検討するため、図17に示すスキームに従って、プラスミドpEF1α−SNAP26m−Neo・N4を構築した。即ち、実施例4に記載のpEF1α−SNAP26m−Neo−RE2を鋳型に、KOD −Plus− Mutagenesis Kitを用いたInverse PCR法によって、配列番号21、23のプライマーを使ってネオマイシン耐性遺伝子下流にN4配列を付加したpEF1α−SNAP26m−Neo・N4を構築した。
【実施例9】
【0122】
(2)pEF1α−SNAP26m−Neo−RE2及びpEF1α−SNAP26m−Neo・N4の細胞導入
実施例2に記載の方法でトランスフェクション用のCHO−K1細胞を準備した。一方、pEF1α−SNAP26m−Neo−RE2及びpEF1α−SNAP26m−Neo・N4を制限酵素AhdIでリニアライズした。トランスフェクションは、実施例5に記載の方法で行い、トランスフェクションの翌日、培地を除去し、2.5g/l−Trypsin・1mmol/l−EDTA Solutionで処理して細胞を分散させ、90mmペトリディッシュに移し、1mg/mlジェネティシン G418(ナカライテスク社製)を含むHam’s F12+10%FBS培地中で1週間選択培養を行った。選択培養中、3〜4日ごとに培地を交換した。選択培養終了後、実施例2に記載の方法で、細胞集団のSNAP26mの発現強度を解析した。
【0123】
図18は、薬剤選択後のpEF1α−SNAP26m−Neo−RE2及びpEF1α−SNAP26m−Neo・N4で形質転換された細胞集団のFACS解析の結果を示す。また、図19に蛍光強度FL1が200以上、1000以上の細胞の割合のプロットを示す。この結果、mRNA不安定化因子であるN4が挿入されたpEF1α−SNAP26m−Neo・N4で形質転換された細胞集団には、薬剤選択後、SNAPmの高発現を示す細胞が有意に濃縮されていることが分かる。
【実施例10】
【0124】
ARE配列モチーフの繰り返し配列数の検討
図20に示すスキームに従って、プラスミドpEF1α−SNAP26m−Pur・N2、pEF1α−SNAP26m−Pur・N6およびpEF1α−SNAP26m−Pur・N8を構築した。実施例4に記載のpEF1α−SNAP26m−Pur・N4を鋳型に、KOD−Plus−Mutagenesis Kitを用いたInverse PCR法によって、配列番号16、24のプライマーを使ってPuromycin耐性遺伝子の下流にN2配列(ARE配列モチーフTTATTTATTの2回繰り返し配列)を付加したプラスミドpEF1α−SNAP26m−Pur・N2を構築した。また、同様の方法で、配列番号7、24のプライマーを使ってPuromycin耐性遺伝子の下流にN6配列(ARE配列モチーフTTATTTATTの6回繰り返し配列)を付加したプラスミドpEF1α−SNAP26m−Pur・N6を構築し、配列番号7、25のプライマーを使ってPuromycin耐性遺伝子の下流にN8配列(ARE配列モチーフTTATTTATTの8回繰り返し配列)を付加したプラスミドpEF1α−SNAP26m−Pur・N8を構築した。
【0125】
実施例2に記載の方法でトランスフェクション用のCHO−K1細胞を準備した。一方、pEF1α−SNAP26m−Pur−RE2、pEF1α−SNAP26m−Pur・N2、pEF1α−SNAP26m−Pur・N4、pEF1α−SNAP26m−Pur・N6及びpEF1α−SNAP26m−Pur・N8の各SNAP26m発現コンストラクトを制限酵素AhdIでリニアライズした。トランスフェクションは、実施例5に記載の方法で行い、トランスフェクションの翌日、培地を除去し、2.5g/l−Trypsin・1mmol/l−EDTA Solutionで処理して細胞を分散させ、90mmペトリディッシュに移し、7.5μg/ml Puromycinを含むHam’s F12+10%FBS培地中で1週間選択培養を行った。選択培養中、3〜4日ごとに培地を交換した。選択培養終了後、実施例2に記載の方法で、細胞集団のSNAP26mの発現強度を解析した。
【0126】
図21は、薬剤選択後のpEF1α−SNAP26m−Pur−RE2、pEF1α−SNAP26m−Pur・N2、pEF1α−SNAP26m−Pur・N4、pEF1α−SNAP26m−Pur・N6及びpEF1α−SNAP26m−Pur・N8で形質転換された細胞集団のFACS解析の結果を示す。また、図22に蛍光強度FL1が200以上、1000以上の細胞の割合のプロットを示す。この結果、繰り返し配列数の多い、N6あるいはN8が挿入されたプラスミドのpEF1α−SNAP26m−Pur・N6あるいはpEF1α−SNAP26m−Pur・N8で形質転換したとき、pEF1α−SNAP26m−Pur・N4と比較して、SNAPm高発現細胞の割合が更に多く、高発現細胞の選択効率が高いとの結果であった。
【実施例11】
【0127】
核酸配列の決定
CHO細胞DR1000L−4N株(CHO細胞−4N株)より作成されたBACライブラリーのうち、hGM−CSF発現カセットを含むクローンCg0031N14を単離した。このクローンCg0031N14に由来するBACより作成したショットガンクローンの5’,3’ワンパスシーケンスを実施し、Phred/Phrap/Consedによる配列情報のアッセンブルを行い、hGM−CSF発現カセットが導入されたCHO細胞の近傍のゲノム配列約91kbpの配列を決定した。この配列を、配列表の配列番号26に示す。
【実施例12】
【0128】
核酸配列の解析
前記決定された核酸配列中にインスレーター配列が存在するかどうかを、インスレーター結合タンパク質CTCFの結合配列モチーフを検索することによって調査した。解析ツールとしてIn silico CTCFBS prediction tool(http://insulatordb.utmem.edu/)(非特許文献11)を使用した。前記ツールで抽出されたモチーフ配列を表1及び図23に示す。
【0129】
【表1】

【実施例13】
【0130】
導入遺伝子発現の安定化効果の確認
実施例1の途中で構築されたpSNAP26m−CMVより、CMVプロモーター/SNAP26m/SV40 polyAを配列番号27,28のプライマーを用いてPCRで増幅し、部分消化によって、プラスミドpBluescript II SK(−)の制限酵素BamHI、SacIサイトへ導入し、プラスミドpBS−CMV−SNAPmを構築した(図24)。
【0131】
次に、実施例11で作成したショットガンライブラリーのクローンうち、41820位、45182位のCTCFの結合配列モチーフの付近のCHOゲノム配列を含むクローンを抽出し、CHOゲノムに由来する導入配列(図25参照)をPCRで増幅し、pBS−CMV−SNAP26mの制限酵素KpnI、XhoIまたはXhoI、ClaIサイトに導入した。ショットガンライブラリークローンと、当該導入配列が組み込まれたSNAPm発現コンストラクトの対応関係を表2に示す。なお、表2中のSNAPm発現コンストラクトの欄の(−)は、CHOのゲノム配列が導入されていないpBS−CMV−SNAPmである。
【0132】
【表2】
【0133】
これらのSNAPm発現コンストラクトとpPURを制限酵素AhdIでリニアライズした後、それぞれ重量比が9:1となるように混合して混合プラスミドを調製した。
【0134】
一方、1×105cells/mlに調整したCHO−K1細胞を12ウェルプレートに2mlずつ前日に播種し、一晩培養して、トランスフェクション用のCHO−K1細胞を準備した。この際、培地として10%牛胎児血清を添加したHam’s F12培地を使用した。
【0135】
トランスフェクションは、3μl GeneJuice Transfection Reagentを100μl Opti−MEM I Reduced−Serum Mediumで希釈し、前記の混合プラスミド1μgにこの希釈液103μlを加え、10分間放置した後、この混合液を前記CHO−K1細胞に加え、24時間インキュベートすることにより行った。翌日、培地を除去し、2.5g/l−Trypsin・1mmol/l−EDTA Solutionで処理して細胞を分散させ、90mmペトリディッシュに移し、6μg/ml Puromycinを含むHam’s F12+10%FBS培地中で3週間選択培養を行った。選択培養中、3〜4日ごとに培地を交換した。選択培養終了後、2.5g/l−Trypsin・1mmol/l−EDTA Solutionで処理して細胞を分散し、1ウェル当たり2×105細胞を12ウェルプレートに播き込み、翌日、0.5mlのHam’s F12培地に2μM SNAP−Cell−505を加え、60分間、37℃でインキュベートした。その後、Ham’s F12培地で3回リンスし、培地交換をしながら、10分間のインキュベートを3回行い、未反応の蛍光色素を除去した。この細胞を2.5g/l−Trypsin・1mmol/l−EDTA Solutionで処理して細胞を分散し、D−PBS(−)に細胞を懸濁し、フローサイトメーターBD FACSCaliburでSNAPmの発現強度を解析した。
【0136】
図26は、被検配列CHO1〜CHO6がそれぞれ導入されたSNAPm発現コンストラクトで形質転換された細胞集団のFACS解析の結果を示す。また、図27は、未処理のCHO細胞(図26左上の(−))の解析結果から10以上のシグナル(図26中のM2)を示す細胞をSNAPmの発現細胞とし、その細胞の割合をプロットしたグラフである。図26、27から明らかな通り、CHO細胞ゲノムの一部を導入したコンストラクト、特に配列番号26の41601〜46746位を導入したCHO4及び5において、有意な発現の上昇が認められた。CHO4とCHO5の差異はサンプル間のバラツキの範囲内と考えられるが、念のため、以後の最適領域の絞り込みはCHO5に基づいて実施した。
【実施例14】
【0137】
遺伝子発現安定化能を有する配列領域の絞り込み(1)
前記で発現の上昇が最も高かったコンストラクトCHO5をもとに、KOD−Plus−Mutagenesis Kitを用いたInverse PCR法によって、配列番号39、40のプライマーを使って被験配列41601−42902位をデリーションしたCHO5Δ5を、配列番号41,42のプライマーを使って42903−44200位をデリーションしたCHO5ΔMを、配列番号43、44のプライマーを使って44201−46746位をデリーションしたCHO5Δ3を構築した(図28)。
【0138】
これらのSNAPm発現コンストラクトとpPURを制限酵素AhdIでリニアライズした後、それぞれ重量比が9:1となるように混合して混合プラスミドを調製した。
【0139】
前記の混合プラスミド1μgを前日12ウェルプレートに播種したCHO−K1細胞にトランスフェクションし、24時間インキュベートした。翌日、培地を除去し、2.5g/l−Trypsin・1mmol/l−EDTA Solutionで処理して細胞を分散させ、90mmペトリディッシュに移し、6μg/ml Puromycinを含むHam’s F12+10%FBS培地中で3週間選択培養を行った。選択培養中、3〜4日ごとに培地を交換した。選択培養終了後、2.5g/l−Trypsin・1mmol・l−EDTA Solutionで処理して細胞を分散し、1ウェルあたり2×105細胞を12ウェルプレートに播き込み、翌日、0.5mlのHam’s F12培地に2μM SNAP−Cell−505を加え、60分間、37℃でインキュベートした。その後、Ham’s F12培地で3回リンスし、培地交換をしながら、10分間のインキュベートを3回行い、未反応の蛍光色素を除去した。この細胞を2.5g/l−Trypsin・1mmol/l−EDTA Solutionで処理して細胞を分散し、D−PBS(−)に細胞を懸濁し、フローサイトメーターBD FACSCaliburでSNAPmの発現強度を解析した。
【0140】
図29は、被検配列CHO5Δ5、ΔM、Δ3がそれぞれ導入されたSNAPm発現コンストラクトで形質転換された細胞集団のFACS解析の結果、シグナル値10以上を示した細胞の割合をプロットしたものである。図29から明らかな通り、CHO5Δ3についてはデリーション前とほぼ同程度のSNAPmの発現強度が認められたのに対し、CHOΔM、Δ3では発現上昇の効果が明らかに低減した。このことから、発現の安定化には配列番号26の配列の41601〜46746位のうちの5’側〜前半部分が主に寄与していると考えられる。
【実施例15】
【0141】
遺伝子発現安定化能を有する配列領域の絞り込み(2)
配列番号44〜49のプライマーを使用し、KOD−Plus−Mutagenesis Kitを用いたInverse PCR法によって、CHO5Δ3の被験配列の3’がさらにデリーションされたSNAPm発現コンストラクトを構築した(図30及び表3)。
【0142】
【表3】
【0143】
これらのSNAP26m発現コンストラクトを実施例14と同様に処理し、SNAP26mの発現強度を解析した。
【0144】
図31は、被検配列CHO5Δ3、CHO5Δ3−1〜5のSNAPm発現コンストラクトで形質転換された細胞集団のFACS解析の結果、シグナル値10以上を示した細胞の割合をプロットしたものである。図31から明らかな通り、配列番号26の配列の41601〜42700位を含むCHO5Δ3−1からCHO5Δ3−3までについては、サンプル間のバラツキはあるものの、デリーション前とほぼ同程度のSNAPmの発現強度が認められたのに対し、かかる領域の3’側が欠失したCHO5Δ3−4及びCHO5Δ3−5では発現の上昇効果が明らかに低減した。
【実施例16】
【0145】
ピューロマイシン耐性遺伝子におけるmRNA不安定化配列と遺伝子発現安定化エレメントとの組み合わせの効果
(1)pEF1α−SNAP26m−Pur−1.1k/1.1k及びpEF1α−SNAP26m−Pur・N4−1.1k/1.1kの構築
次に、ピューロマイシン耐性遺伝子で、N4配列と実施例15で遺伝子発現安定化能の認められたCHO5Δ3−3との組合せ効果を検討するため、プラスミドを構築した。本実験では図32に記載のpEF1α−KOD3G8HC−1.1k/1.1kを利用した。即ち、まず、実施例4の途中で構築されたpEF1α−KOD3G8HC−RE2の発現カセット上流の制限酵素NheI、BglIIサイトに配列番号50、51のプライマーセットを使ってPCRで増幅した被検配列CHO5Δ3−3(以下、コンストラクト中では1.1kと表記)を挿入して、プラスミドpEF1α−KOD3G8HC−1.1kを構築した。更にプラスミドpEF1α−KOD3G8HC−1.1kの発現カセット下流の制限酵素BsiWI、XhoIサイトに配列番号52、53のプライマーセットを使ってPCRで増幅した被検配列CHO5Δ3−3を挿入して構築されたプラスミドがpEF1α−KOD3G8HC−1.1k/1.1kである。
【0146】
一方、実施例4で構築したプラスミドpEF1α−SNAP26m−Pur・N4およびpEF1α−SNAP26m−Pur−RE2を鋳型に、KOD −Plus− Mutagenesis Kitを用いたInverse PCR法によって、配列番号54、55のプライマーを使ってPuromycin耐性遺伝子内に存在する制限酵素BsiWIサイトを削除したプラスミドpEF1α−SNAP26m−Pur2・N4およびpEF1α−SNAP26m−Pur2−RE2を構築した。
【0147】
続いて、図33に示すスキームに従って、pEF1α−SNAP26m−Pur・N4−1.1k/1.1k及びpEF1α−SNAP26m−Pur−1.1k/1.1kを構築した。即ち、pEF1α−SNAP26m−Pur2・N4、pEF1α−SNAP26m−Pur2−RE2から制限酵素EcoRI、BsiWIで、SNAPm−pA−SV40 Promoter−Pur2・N4−pA、SNAPm−pA−SV40
Promoter−Pur2−pAを切出し、pEF1α−KOD3G8HC−1.1k/1.1kの制限酵素EcoRI、BsiWIサイトに導入してpEF1α−SNAP26m−Pur・N4−1.1k/1.1k、pEF1α−SNAP26m−Pur−1.1k/1.1kを構築した。
【実施例17】
【0148】
(2)ピューロマイシン耐性遺伝子での、N4配列とCHO5Δ3−3との組合せ効果の検討
実施例2に記載の方法でトランスフェクション用のCHO−K1細胞を準備した。一方、実施例4および16で構築したSNAP26m発現コンストラクトを制限酵素AhdIでリニアライズした。トランスフェクションは、実施例5に記載の方法で行い、トランスフェクションの翌日、培地を除去し、2.5g/l−Trypsin・1mmol/l−EDTA Solutionで処理して細胞を分散させ、90mmペトリディッシュに移し、それぞれ5、7.5、10μg/ml Puromycinを含むHam’s F12+10%FBS培地中で1週間選択培養を行った。選択培養中、3〜4日ごとに培地を交換した。選択培養終了後、実施例2に記載の方法で、細胞集団のSNAP26mの発現強度を解析した。
【0149】
図34は、薬剤選択後のpEF1α−SNAP26m−Pur−RE2、pEF1α−SNAP26m−Pur−1.1k/1.1k、pEF1α−SNAP26m−Pur・N4及びpEF1α−SNAP26m−Pur・N4−1.1k/1.1kで形質転換された細胞集団のFACS解析の結果を示す。また、図35に蛍光強度FL1が200以上、1000以上の細胞の割合のプロットを示す。この結果、N4配列あるいはCHO5Δ3−3の有無に関わらず、薬剤選択中のPuromycin濃度が高いほど、SNAPm高発現細胞の割合が多いことが分かる。しかしながら、CHO5Δ3−3あるいはN4配列を用いることでSNAPm高発現細胞の割合が増大し、さらにこれらを併用することによって、その相乗効果により高発現細胞の割合は最大値となることが分かる。
【実施例18】
【0150】
ハイグロマイシン耐性遺伝子におけるmRNA不安定化配列及び遺伝子発現安定化エレメントとの組み合わせの効果
(1)pEF1α−SNAP26m−Hyg−1.1k/1.1k及びpEF1α−SNAP26m−Hyg・N4−1.1k/1.1kの構築
次に、ハイグロマイシン耐性遺伝子で、N4配列と実施例15で遺伝子発現安定化能の認められたCHO5Δ3−3との組合せ効果を検討するため、図36に示すスキームに従って、pEF1α−SNAP26m−Hyg・N4−1.1k/1.1k及びpEF1α−SNAP26m−Hyg−1.1k/1.1kを構築した。即ち、pEF1α−SNAP26m−Hyg・N4、pEF1α−SNAP26m−Hyg−RE2から制限酵素MluI、BsiWIで、SNAPm−pA−SV40 プロモーター−Hyg・N4−pA、SNAP26m−pA−SV40 プロモーター−Hyg−pAを切出し、pEF1α−KOD3G8HC−1.1k/1.1kの制限酵素MluI、BsiWIサイトに導入してpEF1α−SNAP26m−Hyg・N4−1.1k/1.1k、pEF1α−SNAP26m−Hyg−1.1k/1.1kを構築した。
【実施例19】
【0151】
(2)ハイグロマイシン耐性遺伝子での、N4配列とCHO5Δ3−3との組合せ効果の検討
実施例2に記載の方法でトランスフェクション用のCHO−K1細胞を準備した。一方、実施例6および18で構築したSNAP26m発現コンストラクトを制限酵素AhdIでリニアライズした。トランスフェクションは、実施例5に記載の方法で行い、トランスフェクションの翌日、培地を除去し、2.5g/l−Trypsin・1mmol/l−EDTA Solutionで処理して細胞を分散させ、90mmペトリディッシュに移し、800μg/ml HygroGoldを含むHam’s F12+10%FBS培地中で1週間選択培養を行った。選択培養中、3〜4日ごとに培地を交換した。選択培養終了後、実施例2に記載の方法で、細胞集団のSNAP26mの発現強度を解析した。
【0152】
図37は、薬剤選択後のpEF1α−SNAP26m−Hyg−RE2、pEF1α−SNAP26m−Hyg−1.1k/1.1k、pEF1α−SNAP26m−Hyg・N4及びpEF1α−SNAP26m−Hyg・N4−1.1k/1.1kで形質転換された細胞集団のFACS解析の結果を示す。また、図38に蛍光強度FL1が200以上、1000以上の細胞の割合のプロットを示す。この結果、mRNA不安定化因子であるN4配列と遺伝子発現安定化効果を有するCHO5Δ3−3を組み合わせることで、N4配列あるいはCHO5Δ3−3を単独で用いた場合と比較してSNAPm高発現細胞の割合が更に増大しており、高生産性細胞の選択効率が向上することが分かる。
【実施例20】
【0153】
ネオマイシン耐性遺伝子におけるmRNA不安定化配列と遺伝子発現安定化エレメントとの組み合わせの効果
(1)pEF1α−SNAP26m−Neo−1.1k/1.1k及びpEF1α−SNAP26m−Neo・N4−1.1k/1.1kの構築
次に、ネオマイシン耐性遺伝子で、N4配列と実施例15で遺伝子発現安定化能の認められたCHO5Δ3−3との組合せ効果を検討するため、図39に示すスキームに従って、pEF1α−SNAP26m−Neo・N4−1.1k/1.1k及びpEF1α−SNAP26m−Neo−1.1k/1.1kを構築した。即ち、pEF1α−SNAP26m−Neo・N4、pEF1α−SNAP26m−Neo−RE2から制限酵素EcoRI、BsiWIで、SNAP26m−pA−SV40 プロモーター−Neo・N4−pA、SNAP26m−pA−SV40 プロモーター−Neo−pAを切出し、pEF1α−KOD3G8HC−1.1k/1.1kの制限酵素coRI、BsiWIサイトに導入してpEF1α−SNAP26m−Neo・N4−1.1k/1.1k、pEF1α−SNAP26m−Neo−1.1k/1.1kを構築した。
【実施例21】
【0154】
(2)ネオマイシン耐性遺伝子での、N4配列とCHO5Δ3−3との組合せ効果の検討
実施例2に記載の方法でトランスフェクション用のCHO−K1細胞を準備した。一方、実施例8および20で構築したSNAP26m発現コンストラクトを制限酵素AhdIでリニアライズした。トランスフェクションは、実施例5に記載の方法で行い、トランスフェクションの翌日、培地を除去し、2.5g/l−Trypsin・1mmol/l−EDTA Solutionで処理して細胞を分散させ、90mmペトリディッシュに移し、1mg/mlジェネティシン G418を含むHam’s F12+10%FBS培地中で1週間選択培養を行った。選択培養中、3〜4日ごとに培地を交換した。選択培養終了後、実施例2に記載の方法で、細胞集団のSNAP26mの発現強度を解析した。
【0155】
図40は、薬剤選択後のpEF1α−SNAP26m−Neo−RE2、pEF1α−SNAP26m−Neo−1.1k/1.1k、pEF1α−SNAP26m−Neo・N4及びpEF1α−SNAP26m−Neo・N4−1.1k/1.1kで形質転換された細胞集団のFACS解析の結果を示す。また、図41に蛍光強度FL1が200以上、1000以上の細胞の割合のプロットを示す。この結果、mRNA不安定化因子であるN4配列と遺伝子発現安定化効果を有するCHO5Δ3−3を組み合わせることで、N4配列あるいはCHO5Δ3−3を単独で用いた場合と比較してSNAPm高発現細胞の割合が更に増大しており、高生産性細胞の選択効率が向上することが分かる。
【実施例22】
【0156】
CHO5Δ3−3存在下でのARE配列モチーフの繰り返し配列数の検討
図42に示すスキームに従って、プラスミドpEF1α−SNAP26m−Pur・N2−1.1k/1.1k、pEF1α−SNAP26m−Pur・N6−1.1k/1.1kおよびpEF1α−SNAP26m−Pur・N8−1.1k/1.1kを構築した。まず、実施例10で構築したプラスミドpEF1α−SNAP26m−Pur・N2、pEF1α−SNAP26m−Pur・N6およびpEF1α−SNAP26m−Pur・N8を鋳型に、KOD−Plus−Mutagenesis Kitを用いたInverse PCR法によって、配列番号54、55のプライマーを使ってPuromycin耐性遺伝子内に存在する制限酵素BsiWIサイトを削除したプラスミドpEF1α−SNAP26m−Pur2・N2、pEF1α−SNAP26m−Pur2・N6およびpEF1α−SNAP26m−Pur2・N8を構築した。続いて、pEF1α−SNAP26m−Pur2・N2、pEF1α−SNAP26m−Pur2・N6およびpEF1α−SNAP26m−Pur2・N8から制限酵素EcoRI、BsiWIで、SNAP26m−pA−SV40 プロモーター−Pur2・N2−pA、SNAP26m−pA−SV40 プロモーター−Pur2・N6−pA、SNAP26m−pA−SV40 プロモーター−Pur2・N8−pAを切出し、pEF1α−KOD3G8HC−1.1k/1.1kの制限酵素EcoRI、BsiWIサイトに導入してpEF1α−SNAP26m−Pur・N2−1.1k/1.1k、pEF1α−SNAP26m−Pur・N6−1.1k/1.1k、pEF1α−SNAP26m−Pur・N8−1.1k/1.1kを構築した。
【0157】
実施例2に記載の方法でトランスフェクション用のCHO−K1細胞を準備した。一方、pEF1α−SNAP26m−Pur−1.1k/1.1k、pEF1α−SNAP26m−Pur・N2−1.1k/1.1k、pEF1α−SNAP26m−Pur・N4−1.1k/1.1k、pEF1α−SNAP26m−Pur・N6−1.1k/1.1k及びpEF1α−SNAP26m−Pur・N8−1.1k/1.1kの各SNAP26m発現コンストラクトを制限酵素AhdIでリニアライズした。トランスフェクションは、実施例5に記載の方法で行い、トランスフェクションの翌日、培地を除去し、2.5g/l−Trypsin・1mmol/l−EDTA Solutionで処理して細胞を分散させ、90mmペトリディッシュに移し、10μg/ml Puromycinを含むHam’s F12+10%FBS培地中で1週間選択培養を行った。選択培養中、3〜4日ごとに培地を交換した。選択培養終了後、実施例2に記載の方法で、細胞集団のSNAP26mの発現強度を解析した。
【0158】
図43は、薬剤選択後のpEF1α−SNAP26m−Pur−1.1k/1.1k、pEF1α−SNAP26m−Pur・N2−1.1k/1.1k、pEF1α−SNAP26m−Pur・N4−1.1k/1.1k、pEF1α−SNAP26m−Pur・N6−1.1k/1.1k及びpEF1α−SNAP26m−Pur・N8−1.1k/1.1kで形質転換された細胞集団のFACS解析の結果を示す。なお、実施例19までの条件でFACS解析に供したところ、細胞の蛍光強度FL1が強すぎて、高発現細胞の割合を正確に算出することができなかった。そのため、フローサイトメーターの蛍光感度を下げて再検討した結果を図43に示している。また、図44に蛍光強度FL1が1000以上の細胞の割合のプロットを示す。この結果、CHO5Δ3−3で発現カセットを挟んだ場合でも、繰り返し配列数の多い、N6あるいはN8が挿入されたプラスミドのpEF1α−SNAP26m−Pur・N6−1.1k/1.1kあるいはpEF1α−SNAP26m−Pur・N8−1.1k/1.1kで形質転換したとき、pEF1α−SNAP26m−Pur・N4−1.1k/1.1kの場合と比較して、SNAPm高発現細胞の割合が更に多く、高生産性細胞の選択効率が飛躍的に向上することが確認された。
【実施例23】
【0159】
抗体生産系におけるmRNA不安定化配列と遺伝子発現安定化エレメントとの組み合わせの効果
(1)pEF1α−KOD3G8HC−Neo・N4−1.1k/1.1kの構築
抗体生産系における、mRNA不安定化配列(N4配列)と実施例15で遺伝子発現安定化能の認められた遺伝子発現安定化エレメントCHO5Δ3−3との組合せ効果の検討に用いるため、図45に示すスキームに従って、pEF1α−KOD3G8HC−Neo・N4−1.1k/1.1kを構築した。即ち、実施例20に記載のpEF1α−SNAP26m−Neo・N4−1.1k/1.1kを制限酵素EcoRI、NotIで処理し、SNAP26m遺伝子を切出した。一方、実施例4に記載のpEF1α−KOD3G8HCから配列番号56,57のプライマーを用いてPCRで増幅し、制限酵素EcoRI、NotIで処理して、調製した抗KOD抗体の重鎖遺伝子を、pEF1α−SNAP26m−Neo・N4−1.1k/1.1kのEcoRI、NotIサイトに導入して、pEF1α−KOD3G8HC−Neo・N4−1.1k/1.1kを構築した。
【実施例24】
【0160】
(2)pEF1α−KOD3G8LC−Hyg−1.1k/1.1kの構築
抗体生産系における、N4配列と実施例15で遺伝子発現安定化能の認められたCHO5Δ3−3との組合せ効果の検討に用いるため、図46に示すスキームに従って、pEF1α−KOD3G8LC−Hyg−1.1k/1.1kを構築した。即ち、まず、実施例6の途中で構築されたpEF1α−KOD3G8LC−Hyg−RE2の発現カセット上流の制限酵素NheI、BglIIサイトに配列番号50、51のプライマーセットを使ってPCRで増幅した遺伝子発現安定化エレメントCHO5Δ3−3(以下、コンストラクト中では1.1kと表記)を挿入して、プラスミドpEF1α−KOD3G8LC−Hyg−1.1kを構築した。更にプラスミドpEF1α−KOD3G8LC−1.1kの発現カセット下流の制限酵素BsiWI、XhoIサイトに配列番号52、53のプライマーセットを用いてPCRで増幅した遺伝子発現安定化エレメントCHO5Δ3−3を挿入して、pEF1α−KOD3G8LC−Hyg−1.1k/1.1kを構築した。
【実施例25】
【0161】
(3)pEF1α−KOD3G8LC−Hyg・N4−1.1k/1.1kの構築
抗体生産系における、N4配列と実施例15で遺伝子発現安定化能の認められたCHO5Δ3−3との組合せ効果の検討に用いるため、図47に示すスキームに従って、pEF1α−KOD3G8LC−Hyg・N4−1.1k/1.1kを構築した。即ち、実施例18に記載のpEF1α−SNAP26m−Hyg・N4−1.1k/1.1kを制限酵素MluI、NotIで処理し、SNAP26m遺伝子を切出した。一方、実施例6に記載のpEF1α−KOD3G8LCから配列番号56,57のプライマーを用いてPCRで増幅し、制限酵素MluI、NotIで処理して、調製した抗KOD抗体の軽鎖遺伝子を、pEF1α−SNAP26m−Hyg・N4−1.1k/1.1kの制限酵素MluI、NotIサイトに導入して、pEF1α−KOD3G8LC−Hyg・N4−1.1k/1.1kを構築した。
【実施例26】
【0162】
(4)pEF1α−KOD3G8HC−Pur・N4−1.1k/1.1kの構築
抗体生産系における、N4配列と実施例15で遺伝子発現安定化能の認められたCHO5Δ3−3との組合せ効果の検討に用いるため、図48に示すスキームに従って、pEF1α−KOD3G8HC−Pur・N4−1.1k/1.1kを構築した。即ち、実施例16に記載のpEF1α−SNAP26m−Pur・N4−1.1k/1.1kを制限酵素EcoRI、NotIで処理し、SNAP26m遺伝子を切出した。一方、実施例4に記載のpEF1α−KOD3G8HCから配列番号56,57のプライマーを用いてPCRで増幅し、制限酵素EcoRI、NotIで処理して、調製した抗KOD抗体の重鎖遺伝子を、pEF1α−SNAP26m−Pur・N4−1.1k/1.1kの制限酵素EcoRI、NotIサイトに導入して、pEF1α−KOD3G8HC−Pur・N4−1.1k/1.1kを構築した。
【実施例27】
【0163】
(5)抗KOD抗体発現系での、N4配列とCHO5Δ3−3との組合せ効果のポリクローンにおける検討
抗KOD抗体の重鎖(HC)発現コンストラクトである、pEF1α−KOD3G8HC−RE2(以下、−/Neoと記載)、pEF1α−KOD3G8HC−1.1k/1.1k(以下、1.1k/Neoと記載)、pEF1α−KOD3G8HC−Neo・N4−1.1k/1.1k(以下、1.1k/Neo・N4と記載)、pEF1α−KOD3G8HC−Pur・N4−1.1k/1.1k(以下、1.1k/Pur・N4と記載)、および、抗KOD抗体の軽鎖(LC)発現コンストラクトである、pEF1α−KOD3G8LC−Hyg−RE2(以下、−/Hygと記載)、pEF1α−KOD3G8LC−Hyg−1.1k/1.1k(以下、1.1k/Hygと記載)、pEF1α−KOD3G8LC−Hyg・N4−1.1k/1.1k(以下、1.1k/Hyg・N4と記載)をそれぞれ制限酵素AhdIでリニアライズ(直鎖状化)した。そして、重鎖および軽鎖発現コンストラクトを、
(1)遺伝子発現安定化エレメントを含まないもの(−/Neoと−/Hyg)、
(2)遺伝子発現安定化エレメントCHO5Δ3−3を抗体発現カセットの上流および下流の両方に挿入したもの(1.1k/Neoと1.1k/Hyg)、
(3)遺伝子発現安定化エレメントCHO5Δ3−3を抗体発現カセットの上流および下流の両方に挿入し、薬剤耐性遺伝子(重鎖:Neo、軽鎖:Hyg)にmRNA不安定化配列(N4配列)を付加したもの(1.1k/Neo・N4と1.1k/Hyg・N4)、
(4)遺伝子発現安定化エレメントCHO5Δ3−3を抗体発現カセットの上流および下流の両方に挿入し、薬剤耐性遺伝子(重鎖:Pur、軽鎖:Hyg)にmRNA不安定化配列(N4配列)を付加したもの(1.1k/Pur・N4と1.1k/Hyg・N4)
のそれぞれについて、抗体の重鎖発現コンストラクトと軽鎖発現コンストラクトの重量比が1:1となるように混合して混合プラスミドを調製した。
【0164】
前記の混合プラスミド2μgを、前日に6ウェルプレートに播種しておいたCHO−K1細胞にトランスフェクションし、24時間インキュベートした。翌日、培地を除去し、2.5g/l−Trypsin・1mmol/l−EDTA Solutionで処理して細胞を分散させ、90mmペトリディッシュに移し、ジェネティシン G418およびHygroGold、あるいは、PuromycinおよびHygroGoldを含むHam’s F12+10%FBS培地中で2週間選択培養を行った。選択培養中、3〜4日ごとに培地を交換した。遺伝子発現安定化エレメントを含まないもの(−/Neo,−/Hyg)、および、遺伝子発現安定化エレメントCHO5Δ3−3を発現カセットの上流および下流の両方に挿入したもの(1.1k/Neo,1.1k/Hyg)では、600μg/mlジェネティシン G418・400μg/ml HygroGoldにて、選択培養をした。遺伝子発現安定化エレメントCHO5Δ3−3を発現カセットの上流および下流の両方に挿入し、薬剤耐性遺伝子(H鎖:Neo、L鎖:Hyg)にmRNA不安定化配列(N4配列)を付加したもの(1.1k/Neo・N4,1.1k/Hyg・N4)では、400μg/mlジェネティシン G418・200μg/ml HygroGoldにて選択培養をした。遺伝子発現安定化エレメントCHO5Δ3−3を発現カセットの上流および下流の両方に挿入し、薬剤耐性遺伝子(重鎖:Pur、軽鎖:Hyg)にmRNA不安定化配列(N4配列)を付加したもの(1.1k/Pur・N4,1.1k/Hyg・N4)では、7.5μg/ml Puromycin・400μg/ml HygroGoldにて選択培養をした。選択培養終了後、2.5g/l−Trypsin・1mmol/l−EDTA Solutionで処理して細胞を分散し、1ウェル当たり1.84×105細胞を6ウェルプレートに播き込んだ。3日間培養後、培養上清を回収し、ポリクローンでのKOD3G8抗体の生産量をELISA(パイロコッカス・コダカラエンシスKOD1由来のDNAポリメラーゼを固相化したもの)で測定した。
【0165】
具体的には、30μg/ml濃度のKOD1由来のDNAポリメラーゼ抗原を固相化したELISAプレートに、培養上清を添加し、35℃で2時間インキュベートした。次に、PBS−T(0.1%Tween20含有リン酸緩衝生理食塩水)で3回洗浄し、10000倍希釈した抗マウス抗体−HRP(DAKO社製)を50μl添加して35℃で1時間インキュベートした後、さらにPBS−Tで3回洗浄し、TMB+(3,3’,5,5’−tetramethylbenzidine;DAKO社製)で5分間反応させた。50μlの1N硫酸を加えて反応停止後、プレートリーダー(製品名;SPECTRA
CLASSIC、TECAN Auctria社製)(主波長450nm、副波長620nm)で吸光度を測定した。
【0166】
標準品から作成した検量線をもとに抗体濃度を算出した。なお、標準品として、Thermococcus kodakaraensis KOD1株由来のDNAポリメラーゼに特異的な抗体を産生するマウスハイブリドーマ細胞系3G8(入手先:独立行政法人産業技術総合研究所 特許生物寄託センター、寄託番号:FERM BP−6056)から得られた抗体を用いた。結果を図49に示す。図49から明らかな通り、遺伝子発現安定化エレメントを含まないもの(−/Neo,−/Hyg)と比較して、遺伝子発現安定化エレメントCHO5Δ3−3を発現カセット上流および下流の両方に挿入したもの(1.1k/Neo,1.1k/Hyg)では抗体生産量が2倍程度上昇したが、遺伝子発現安定化エレメントCHO5Δ3−3を発現カセットの上流および下流の両方に挿入し、薬剤耐性遺伝子(H鎖:Neo、L鎖:Hyg)にmRNA不安定化配列(N4配列)を付加したもの(1.1k/Neo・N4,1.1k/Hyg・N4)では抗体生産量が更に上昇し、約3倍に増加した。また、遺伝子発現安定化エレメントCHO5Δ3−3を発現カセットの上流および下流の両方に挿入し、薬剤耐性遺伝子にmRNA不安定化配列(N4配列)を付加したもので、重鎖発現コンストラクトの薬剤耐性遺伝子をNeoからPurに変更したとき、抗体生産量が顕著に増加し、遺伝子発現安定化エレメント及びmRNA不安定化配列を有さない発現コンストラクトに比べて、約5倍の抗体生産量を示した。
【実施例28】
【0167】
(6)抗KOD抗体発現系での、N4配列とCHO5Δ3−3との組合せ効果のモノクローンにおける検討
続いて、遺伝子発現安定化エレメントを含まないもの(−/Neo,−/Hyg)、遺伝子発現安定化エレメントCHO5Δ3−3を発現カセットの上流および下流の両方に挿入したもの(1.1k/Neo,1.1k/Hyg)、遺伝子発現安定化エレメントCHO5Δ3−3を発現カセットの上流および下流の両方に挿入し、薬剤耐性遺伝子(H鎖:Neo、L鎖:Hyg)にmRNA不安定化配列(N4配列)を付加したもの(1.1k/Neo・N4,1.1k/Hyg・N4)、遺伝子発現安定化エレメントCHO5Δ3−3を発現カセットの上流および下流の両方に挿入し、薬剤耐性遺伝子(H鎖:Pur、L鎖:Hyg)にmRNA不安定化配列(N4配列)を付加したもの(1.1k/Pur・N4,1.1k/Hyg・N4)のそれぞれについて、限界希釈を行い、モノクローンでの生産性の比較を行った。
【0168】
具体的には、実施例27のKOD3G8抗体を安定導入したポリクローンの細胞を2.5g/l−Trypsin・1mmol/l−EDTA Solutionで処理して細胞を分散し、0.75細胞/ウェルの濃度で96ウェルプレートに播き込んだ。2週間培養後、培養上清を回収し、KOD3G8抗体の生産量を実施例27と同様の方法でELISAにて測定した。標準品から作成した検量線をもとに各クローンの抗体生産量を算出し、遺伝子発現安定化エレメントを含まないもの(−/Neo,−/Hyg)、遺伝子発現安定化エレメントCHO5Δ3−3を発現カセットの上流および下流の両方に挿入したもの(1.1k/Neo,1.1k/Hyg)、遺伝子発現安定化エレメントCHO5Δ3−3を発現カセットの上流および下流の両方に挿入し、薬剤耐性遺伝子(H鎖:Neo、L鎖:Hyg)にmRNA不安定化配列(N4配列)を付加したもの(1.1k/Neo・N4,1.1k/Hyg・N4)、遺伝子発現安定化エレメントCHO5Δ3−3を発現カセットの上流および下流の両方に挿入し、薬剤耐性遺伝子(H鎖:Pur、L鎖:Hyg)にmRNA不安定化配列(N4配列)を付加したもの(1.1k/Pur・N4,1.1k/Hyg・N4)のそれぞれについて、発現量上位クローンを拡大培養した後、1ウェル当たり1.84×105細胞を6ウェルプレートに播き込んだ。3日間培養後、培養上清を回収し、モノクローンでのKOD3G8抗体の生産量を実施例27と同様の方法でELISAにて測定した。
【0169】
標準品から作成した検量線をもとに算出した各クローンの抗体生産量をプロットしたグラフを図50に示す。その結果、ポリクローンでの生産性を比較した場合と同様、遺伝子発現安定化エレメントを含まないもの(−/Neo,−/Hyg)と比較して、遺伝子発現安定化エレメントCHO5Δ3−3を抗体発現カセット上流および下流の両方に挿入したもの(1.1k/Neo,1.1k/Hyg)で抗体生産量が上昇したが、被験配列CHO5Δ3−3を発現カセットの上流および下流の両方に挿入し、薬剤耐性遺伝子(H鎖:Neo、L鎖:Hyg)にmRNA不安定化配列(N4配列)を付加したもの(1.1k/Neo・N4,1.1k/Hyg・N4)で抗体生産量が更に上昇していた。また、遺伝子発現安定化エレメントCHO5Δ3−3を抗体発現カセットの上流および下流の両方に挿入し、薬剤耐性遺伝子にmRNA不安定化配列(N4配列)を付加したものでは、重鎖発現コンストラクトの薬剤耐性遺伝子をNeo・N4からPur・N4に変更したとき、各細胞の抗体生産量が著しく増加し、高い抗体生産性を示すクローンの割合が顕著に大きくなった。この結果から抗体生産系における高生産性細胞の選択効率が飛躍的に向上することが確認された。
【0170】
さらに、効果を確認するため、遺伝子発現安定化エレメントもmRNA不安定化配列も含まないもの(−/Neo,−/Hyg)と、高い抗体生産量を示した遺伝子発現安定化エレメントCHO5Δ3−3を含み、さらに薬剤耐性遺伝子(H鎖:Pur、L鎖:Hyg)にmRNA不安定化配列(N4配列)を付加したもの(1.1k/Pur・N4,1.1k/Hyg・N4)について、前記96ウェルプレートにおける2週間培養後のELISA測定によって算出されたKOD3G8抗体の生産量を再度解析し、度数分布としてプロットした結果を図51に示す。この結果、KOD3G8生産量に対するクローンの分布は、1.1k/Pur・N4,1.1k/Hyg・N4において有意に高生産側へシフトしており、極めて高い抗体生産性を有する細胞の割合が顕著に増大している。遺伝子発現安定化エレメントとmRNA不安定化配列を含む薬剤選択マーカー遺伝子発現カセットの利用が、その相乗効果により、高生産細胞株取得に極めて大きく寄与していることが確認された。
【実施例29】
【0171】
無血清馴化CHO細胞を用いた、抗KOD抗体発現系での、N4配列とCHO5Δ3−3との組合せ効果のポリクローンにおける検討
EX−CELL CD CHO Serum−Free Medium for CHO Cells, Chemically Defind培地(SAFC Biosciences社)を用いて、添付のプロトコールに従って、CHO−K1細胞を無血清馴化した。無血清馴化したCHO−K1細胞2.5X105cells/mlになるようにOpti−MEM I Reduced−Serum Medium(GIBCO社製)に懸濁し、超低接着表面24ウェルプレート(Corning Life Science社)の4ウェルに1mlずつ播種した。
【0172】
抗KOD抗体の重鎖(HC)発現コンストラクトである、pEF1α−KOD3G8HC−1.1k/1.1k(以下、1.1k/Neoと記載)、pEF1α−KOD3G8HC−Pur・N4−1.1k/1.1k(以下、1.1k/Pur・N4と記載)、および、抗KOD抗体の軽鎖(LC)発現コンストラクトである、pEF1α−KOD3G8LC−Hyg−1.1k/1.1k(以下、1.1k/Hygと記載)、pEF1α−KOD3G8LC−Hyg・N4−1.1k/1.1k(以下、1.1k/Hyg・N4と記載)をそれぞれ制限酵素AhdIでリニアライズ(直鎖状化)した。そして、重鎖および軽鎖発現コンストラクトを、
(1)遺伝子発現安定化エレメントCHO5Δ3−3を抗体発現カセットの上流および下流の両方に挿入したもの(1.1k/Neoと1.1k/Hyg)、
(2)遺伝子発現安定化エレメントCHO5Δ3−3を抗体発現カセットの上流および下流の両方に挿入し、薬剤耐性遺伝子(重鎖:Pur、軽鎖:Hyg)にmRNA不安定化配列(N4配列)を付加したもの(1.1k/Pur・N4と1.1k/Hyg・N4)
のそれぞれについて、抗体の重鎖発現コンストラクトと軽鎖発現コンストラクトの重量比が1:1となるように混合して混合プラスミドを調製した。
【0173】
前記の混合プラスミド各4μgをOpti−MEM I Reduced−Serum Medium68μlで希釈した。一方で、Lipofectamine2000 15μlをOpti−MEM I Reduced−Serum Medium68μlで希釈し、前記プラスミド混合液と混合し、15分間、室温で放置した。その後、半量ずつ2ウェルの細胞に加えた。
【0174】
翌日、細胞を回収して800gX3分間遠心して上清を除去し、4mlEX−CELL CD CHO Serum−Free Medium for CHO Cells, Chemically Defind培地に懸濁し、遺伝子発現安定化エレメントCHO5Δ3−3を発現カセットの上流および下流の両方に挿入したもの(1.1k/Neo,1.1k/Hyg)には400μg/ml G418、200μg/ml HygroGold、遺伝子発現安定化エレメントCHO5Δ3−3を発現カセットの上流および下流の両方に挿入し、薬剤耐性遺伝子(H鎖:Pur、L鎖:Hyg)にmRNA不安定化配列(N4配列)を付加したもの(1.1k/Pur・N4,1.1k/Hyg・N4)には7.5μg/ml Puromycin、200μg/ml HygroGoldの抗生物質を加え、選択培養した。
【0175】
選択培養終了後、2×105cells/mlで2mlずつ6ウェルプレートに播き込んだ。3日間培養後、培養上清を回収し、ポリクローンでのKOD3G8抗体の生産量を実施例27と同様の方法でELISAにて測定した。標準品から作成した検量線をもとに算出した抗体生産量をプロットしたグラフを図52に示す。この結果、無血清馴化CHO−K1細胞を用いても1.1k/Pur・N4,1.1k/Hyg・N4における生産性は、実施例27の無血清馴化していないCHO−K1細胞の高い生産性を再現することが確認された。一般にバイオ医薬品などの工業生産には、高生産細胞をスクリーニングした後、1〜2ヶ月程度かけて無血清馴化するが、あらかじめ無血清馴化された細胞に遺伝子導入することによって無血清馴化工程を省略できることが期待される。本実施例の結果より、無血清馴化CHO細胞においても本発明の遺伝子発現安定化エレメントとmRNA不安定化配列を含む薬剤選択マーカー遺伝子発現カセットの利用によって生産性の高い細胞を取得できることが確認され、生産性の高い細胞をより短時間に樹立できることが確認された。
【実施例30】
【0176】
抗KOD抗体発現系での、2コンストラクト抗体発現系とシングルベクター抗体発現系の比較
(1)pEF1α−KOD3G8LC,HC−Pur・N4−1.1k/1.1kの構築
次に、H鎖発現カセットとL鎖発現カセットを一つのベクター上に配置した場合でも高生産細胞を取得可能か検討するため、図53に示すスキームに従ってpEF1α−KOD3G8LC,HC−Pur・N4−1.1k/1.1kを構築した。
【0177】
なお、H鎖(重鎖)発現カセットとL鎖(軽鎖)発現カセットとをそれぞれ別々のベクターに配置した場合を「2コンストラクト抗体発現系」もしくは単に「2コンストラクト」といい、H鎖発現カセットとL鎖発現カセットを一つのベクター上に配置した場合を「シングルベクター抗体発現系」もしくは単に「シングルベクター」という。
【0178】
まず、pEF1α−KOD3G8HC−Pur・N4−1.1k/1.1kにL鎖発現カセット挿入用の制限酵素サイトを挿入したpEF1α−KOD3G8HC−Pur・N4−1.1k/1.1k−Sを構築した。即ち、実施例26に記載のpEF1α−KOD3G8HC−Pur・N4−1.1k/1.1kを制限酵素BglII、MluIで処理し、EF−1αプロモーターを切り出した。一方、実施例16の途中で構築したpEF1α−KOD3G8HC−1.1kから配列番号58、59のプライマーを用いてPCRで増幅し、BglII、MluIで処理して調製したEF−1αプロモーターを、pEF1α−KOD3G8HC−Pur・N4−1.1k/1.1kのBglII、MluIサイトに導入して、EF−1αプロモーター上流にBglII、SphI、SpeIサイトを挿入したpEF1α−KOD3G8HC−Pur・N4−1.1k/1.1k−Sを構築した。
【0179】
続いて、pEF1α−KOD3G8HC−Pur・N4−1.1k/1.1k−SをBglII、SpeIで処理した。一方、実施例6の途中で構築したpEF1α−KOD3G8LC−RE2から配列番号60、61のプライマーを用いてPCRで増幅し、BglII、SpeIで処理して調製した抗KOD抗体のL鎖発現カセットを、pEF1α−KOD3G8HC−Pur・N4−1.1k/1.1k−SのBglII、SpeIサイトに導入して、シングルベクターpEF1α−KOD3G8LC,HC−Pur・N4−1.1k/1.1kを構築した。
【実施例31】
【0180】
(2)抗KOD抗体発現系での、ポリクローンにおける2コンストラクト抗体発現系とシングルベクター抗体発現系の比較
シングルベクター抗体発現系でも、2コンストラクト抗体発現系と同様に抗体生産量が高いクローンを取得可能か検討した。2コンストラクト抗体発現系として、実施例27および28で最も良好な結果が得られた、遺伝子発現安定化エレメントCHO5Δ3−3を抗体発現カセットの上流および下流の両方に挿入し、薬剤耐性遺伝子(重鎖:Pur、軽鎖:Hyg)にmRNA不安定化配列(N4配列)を付加したもの(1.1k/Pur・N4と1.1k/Hyg・N4)について抗体の重鎖発現コンストラクトと軽鎖発現コンストラクトの重量比が1:1となるように混合したプラスミド(制限酵素AhdIでリニアライズ済)をトランスフェクションに用いた。また、シングルベクター抗体発現系として、実施例30で構築したpEF1α−KOD3G8LC,HC−Pur・N4−1.1k/1.1kを制限酵素AhdIでリニアライズし、トランスフェクションに用いた。各々のプラスミド2μgを、実施例2に記載の方法で準備しておいたCHO−K1細胞にトランスフェクションし、24時間インキュベートした。翌日、培地を除去し、2.5g/l−Trypsin・1mmol/l−EDTA Solutionで処理して細胞を分散させ、90mmペトリディッシュに移し、2コンストラクト発現系では7.5μg/ml Puromycin・400μg/ml HygroGoldを含むHam’s F12+10%FBS培地中で、シングルベクター系では10μg/ml Puromycinを含むHam’s F12+10%FBS培地中で、2週間選択培養を行った。選択培養中、3〜4日ごとに培地を交換した。選択培養終了後、2.5g/l−Trypsin・1mmol/l−EDTA Solutionで処理して細胞を分散し、1ウェル当たり1.84×105細胞を6ウェルプレートに播き込んだ。3日間培養後、培養上清を回収し、ポリクローンでのKOD3G8抗体の生産量を実施例27と同様の方法でELISAにて測定した。
【0181】
標準品から作成した検量線をもとに抗体濃度を算出した。結果を図54に示す。その結果、2コンストラクト抗体発現系とシングルベクター抗体発現系間で、抗体生産量に差異はほとんど見られないとの結果であった。
【実施例32】
【0182】
(3)抗KOD抗体発現系での、モノクローンにおける2コンストラクト抗体発現系とシングルベクター抗体発現系の比較
続いて、2コンストラクト抗体発現系、シングルベクター抗体発現系のそれぞれについて、限界希釈を行い、モノクローンでの生産性の比較を行った。
【0183】
具体的には、実施例31のKOD3G8抗体を安定導入したポリクローンの細胞を2.5g/l−Trypsin・1mmol/l−EDTA Solutionで処理して細胞を分散し、0.75細胞/ウェルの濃度で96ウェルプレートに播き込んだ。2週間培養後、培養上清を回収し、実施例27と同様の方法でELISAにてKOD3G8抗体の生産量を測定した。標準品から作成した検量線をもとに各クローンの抗体生産量を算出し、2コンストラクト抗体発現系、シングルベクター抗体発現系のそれぞれについて、発現量上位クローンを選択した。選択したクローンを拡大培養した後、1ウェル当たり1.84×105細胞を6ウェルプレートに播き込んだ。3日間培養後、培養上清を回収し、モノクローンでのKOD3G8抗体の生産量を実施例27と同様の方法でELISAにて測定した。
【0184】
標準品から作成した検量線をもとに算出した発現量上位5クローンの抗体生産量をプロットしたグラフを図55に示す。その結果、ポリクローンでの結果と同様、シングルベクター抗体発現系と2コンストラクト抗体発現系間で、抗体発現量はほとんど変わらないとの結果であった。このことから、シングルベクター抗体発現系でも、2コンストラクト抗体発現系と同様に抗体生産量が高いクローンを取得可能であることがわかった。
【産業上の利用可能性】
【0185】
本発明の遺伝子発現の安定化機能を有する核酸領域と弱化された薬剤遺伝子発現カセットを具備した発現ベクターは、目的タンパク質遺伝子を高度に発現する細胞を効率よく取得することができる。従って、本発明の発現ベクターを使用した細胞発現システムは、種々の動物細胞、特に哺乳類動物細胞のタンパク質の機能解析は勿論、バイオ医薬品として創薬・医療などの産業界に寄与することが大である。
【技術分野】
【0001】
本発明は、遺伝子組換え技術によって、目的とするタンパク質の遺伝子を挿入した発現ベクターを宿主細胞に導入し、得られた形質転換細胞の中から目的とするタンパク質の遺伝子を高レベルで発現する細胞を効率よく樹立するための発現ベクター、及び形質転換された高生産性細胞に関する。本発明の発現ベクターは、遺伝子工学的手法により、動物細胞、特に哺乳類動物細胞において医薬品などの有用タンパク質を生産するために有用である。
【背景技術】
【0002】
組換えタンパク質を生産する発現システムの開発は、研究または治療に供されるタンパク質の供給源を提供するうえで重要である。発現システムとしては、大腸菌などの原核細胞に基づくもの、ならびに酵母(Saccharomyces属,Pichia属、Kluyveromyces属など)及び哺乳類細胞などの動物細胞を含む真核細胞に基づくものの両者が用いられている。これらの中でも動物細胞、特に哺乳類細胞に基づく発現システムが治療用タンパク質の製造には好ましい。なぜなら、ヒトをはじめとする哺乳類動物において起こるタンパク質の翻訳後修飾は、時にその生物活性に深く寄与するため、タンパク質の投与対象と類似した翻訳後修飾が可能な哺乳類細胞に基づく発現システムを利用した方が、治療用タンパク質の有効性を増強しうるからである。
【0003】
組換えタンパク質を産生する細胞を樹立する方法としては、一般に、目的タンパク質の遺伝子を発現する遺伝子構築物を宿主細胞へ遺伝子導入し、得られた形質転換細胞の中からこの遺伝子構築物が宿主細胞のゲノム上に安定導入された細胞を選別する。この際、前記遺伝子構築物に、薬剤選択マーカーとなる薬剤耐性遺伝子を目的タンパク質の遺伝子と同一プロモーター下あるいは別個のプロモーター下で発現するように挿入しておき、薬剤選択によって生き残った細胞を目的タンパク質の遺伝子が安定導入された細胞として選別する。目的タンパク質の発現レベルは、目的タンパク質をコードする遺伝子が宿主細胞ゲノム内のどの領域に導入されるかによって大きく変動するが、導入領域の制御は、通常不可能である。従って、遺伝子導入しても大半の細胞は目的タンパク質遺伝子を発現しないか又は発現量が低い。このため、従来、目的タンパク質遺伝子を高レベルで発現する形質転換細胞の取得には、1000〜数千の細胞サンプルから1〜2個の細胞株を選別する作業を数ヶ月〜1年かけて繰り返し行っており、目的タンパク質遺伝子を高レベルで発現する形質転換細胞の選別に膨大な労力と時間を要していた。
【0004】
そこで、目的タンパク質遺伝子を高レベルで発現する形質転換細胞を効率的に選別するため、薬剤選択マーカーの発現や機能を減弱化することに基づく方法が開発されてきた。薬剤選択マーカーの発現や機能を減弱化すると、薬剤選択マーカー遺伝子が宿主細胞ゲノム内の低発現領域に導入された形質転換細胞は、薬剤選択マーカー(薬剤耐性遺伝子)を十分に発現することができないので、死滅してしまい、高発現領域に導入された形質転換細胞のみが薬剤選択によって生き残る。この生き残った形質転換細胞では、薬剤選択マーカー遺伝子に隣接して目的タンパク質遺伝子を有する可能性が高いため、目的タンパク質遺伝子も宿主細胞ゲノム内の高発現部位に導入されており、目的タンパク質遺伝子が高レベルで発現されている可能性が高い。このことを利用して、目的タンパク質遺伝子を高レベルで発現する形質転換細胞を効率良く選別することができると考えられる。
【0005】
薬剤選択マーカーの発現や機能を減弱化する1つの方法としては、薬剤選択マーカーの発現に用いるプロモーターとして、転写活性の弱いプロモーターであるHerpes simplex virus thymidine kinase(HSV−tk)プロモーターを利用する方法(非特許文献1)や、野生型の転写活性レベルよりも減弱化された変異型プロモーターを用いて薬剤選択マーカーの発現を抑える方法(特許文献1、2)が挙げられる。しかしながら、本発明者らがこれらの方法を追試したところ、いずれの方法も薬剤選択マーカーの発現を減弱化するという面で効果がほとんど見られなかった。このことから、これらの方法を用いて目的タンパク質遺伝子を高レベルで発現する形質転換細胞を効率的に選別するということは難しいと考えられた。
【0006】
薬剤選択マーカーの発現や機能を減弱化する別の方法としては、ネオマイシンホスホトランスフェラーゼなどの薬剤選択マーカー遺伝子のコード配列に変異を導入して薬剤選択マーカー自体の機能を減弱化する方法(特許文献3、4)が挙げられる。本発明者らがこの方法を追試したところ、この方法を用いることにより薬剤選択マーカーの機能を多少減弱化することができ、薬剤選択の結果として少量の発現量の向上した細胞株を取得することができた。しかしながら、目的タンパク質遺伝子を高レベルで発現する形質転換細胞を選別する点での効率は良くなく、この方法による顕著な効果は見られなかった。
【0007】
高生産性細胞の樹立には細胞選択技術の改良の他に、目的遺伝子の発現を安定的に維持するため、宿主細胞ゲノム上の目的遺伝子導入部位の周囲のゲノム環境からの影響を排除する工夫が必要である。組換えタンパク質の発現は、組換えタンパク質をコードする遺伝子が宿主細胞ゲノム内のどの部位に導入されるかによって大きく変動するが、導入部位の制御は通常、不可能である。従って、スクリーニング時には十分な発現をしていた細胞でも培養を継続していくうちに次第に発現が低下してしまうこともある。このような位置効果を克服するため、導入遺伝子への隣接する染色体や調節エレメントの影響を緩和する染色体エレメント(シス作用性DNAエレメント)が組換えタンパク質の製造に利用されつつある。このような機能を持った核酸配列の1つがインスレーターと呼ばれる配列で、ニワトリのβ−グロビンLCRの1.2kb長のDNaseI Hypersensitive部位(cHS4)などがよく機能解析され、組換えタンパク質の発現に利用されているが(非特許文献2)、CHO−K1細胞におけるタンパク質発現の増加はそれほど大きなものではないとされている(非特許文献3)。
【先行技術文献】
【特許文献】
【0008】
【特許文献1】US5,627,033
【特許文献2】WO2005/024015
【特許文献3】WO2001/032901
【特許文献4】WO2004/050884
【特許文献5】特開2001−37478号公報
【非特許文献】
【0009】
【非特許文献1】Niwa H、Yamamura K,Miyazaki J.(1991)Gene 108:193−200
【非特許文献2】Pikaart MJ,Recillas−Targa F,Felsenfeld G.(1998)Genes Dev 12:2852−62
【非特許文献3】Izumi M,Gilbert DM.(1999)J Cell Biochem 76:280−289
【非特許文献4】Bakheet T,Frevel M,Williams B.R.G、 Greer W,Khabar K.S.A.(2001)Nucleic Acids Research 29:246−254
【非特許文献5】Lagnado C.A,Brown C.L,Goodall G.J.(1994) Molecular and Cellular Biology 14:7984−7995
【非特許文献6】Zubiaga A.M,Belasco J.G,Greenberg M.E.(1995) Molecular and Cellular Biology 15:2219−2230
【非特許文献7】Blasco MA(2007)Nat Rev Genet 8:299−309
【非特許文献8】Gonzalo S,Jaco I,Fraga MF,Chen T,Li E,Esteller M,Blasco MA(2006)Nat Cell Biol 8:416−424
【非特許文献9】Perrod S,Gasser SM(2003)Cell Mol Life Sci 60:2303−2318
【非特許文献10】Wakimoto BT(1998)Cell 93:321−324
【非特許文献11】Bao L,Zhou M,Cui Y(2008)Nucleic Acids Research,36,D83−D87
【発明の概要】
【発明が解決しようとする課題】
【0010】
本発明の目的は、目的タンパク質遺伝子を高レベルで発現する形質転換細胞を効率的に樹立するのに効果的な発現ベクター、及び高生産性細胞を提供することにある。
【課題を解決するための手段】
【0011】
本発明者らは、上記課題を解決するため、mRNA不安定化配列を導入した薬剤選択マーカー発現カセット、及び遺伝子発現安定化エレメントを発明した。そして、mRNA不安定化配列を有する種々の薬剤選択マーカー発現カセットと遺伝子発現安定化エレメントを具備した導入遺伝子の発現ベクターを構築し、前記発現ベクターで形質転換された宿主細胞の遺伝子発現を詳しく解析したところ、目的タンパク質の高生産性細胞の割合が飛躍的に上昇することを見出し、本発明を完成させるに到った。
【0012】
すなわち、本発明によれば、以下が提供される:
(1) mRNA不安定化配列を含む薬剤選択マーカー遺伝子発現カセット、少なくとも1つの遺伝子発現安定化エレメント、及び目的タンパク質の遺伝子発現カセットを有することを特徴とする発現ベクター。
(2) mRNA不安定化配列が、サイトカイン、インターロイキン、又は癌原遺伝子の3’非翻訳領域に存在するATリッチ配列に由来することを特徴とする(1)に記載の発現ベクター。
(3) mRNA不安定化配列が、TTATTTA(A/T)(A/T)のモチーフ配列を有することを特徴とする(1)に記載の発現ベクター。
(4) モチーフ配列が、2回以上繰り返されていることを特徴とする(3)に記載の発現ベクター。
(5) モチーフ配列の繰り返し間に1塩基以上のスペーサー配列を含むことを特徴とする(4)に記載の発現ベクター。
(6) mRNA不安定化配列中に1ないし数塩基の置換、挿入または欠失を含むことを特徴とする(2)〜(5)のいずれか一項に記載の発現ベクター。
(7) 遺伝子発現安定化エレメントがチャイニーズハムスターゲノム由来であることを特徴とする(1)〜(6)のいずれかに記載の発現ベクター。
(8)遺伝子発現安定化エレメントが、以下の(a)〜(g)のいずれか一つ又はそれらの任意の組合せからなることを特徴とする(1)〜(7)のいずれかに記載の発現ベクター。
(a)配列番号26で示す配列からなるDNA;
(b)配列番号26で示す配列の部分配列からなるDNAであって、配列番号26で示す配列のうち41820番目から41839番目までの塩基で示す領域の配列を含むDNA;
(c)配列番号26で示す配列の部分配列からなるDNAであって、配列番号26で示す配列のうち41821番目から41840番目までの塩基で示す領域の配列を含むDNA;
(d)配列番号26で示す配列の部分配列からなるDNAであって、配列番号26で示す配列のうち45182番目から45200番目までの塩基で示す領域の配列を含むDNA;
(e)配列番号26で示す配列の部分配列からなるDNAであって、配列番号26で示す配列のうち91094番目から91113番目までの塩基で示す領域の配列を含むDNA;
(f)配列番号26で示す配列の部分配列からなるDNAであって、宿主細胞中で外来遺伝子発現カセットと近接するように配置された際に、外来遺伝子発現カセットに含まれる外来遺伝子からの目的組換えタンパク質の発現を増大または安定化させることができるDNA;及び
(g)前記(a)〜(f)のいずれかのDNAと相補的な塩基配列からなるDNAとストリンジェントな条件下でハイブリダイズし、かつ遺伝子発現安定化機能を有するDNA
(h)前記(a)〜(g)のいずれかのDNAと相補的な塩基配列からなるDNA
(9) 遺伝子発現安定化エレメントが、以下の(i)〜(k)のいずれか一つ又はそれらの任意の組合せからなることを特徴とする(8)に記載の発現ベクター。
(i)配列番号26で示す配列のうち41601番目から46746番目までの塩基で示す領域の配列を含むDNA;
(j)(i)のDNAと相補的な塩基配列からなるDNAとストリンジェントな条件下でハイブリダイズし、かつ遺伝子発現安定化機能を有するDNA;及び
(k)前記(i)又は(j)のDNAと相補的な塩基配列からなるDNA
(10) 遺伝子発現安定化エレメントが、以下の(l)〜(n)のいずれか一つ又はそれらの任意の組合せからなることを特徴とする(9)に記載の発現ベクター。
(l)配列番号26で示す配列のうち41601番目から42700番目までの塩基で示す領域の配列を含むDNA;
(m)(l)のDNAと相補的な塩基配列からなるDNAとストリンジェントな条件下でハイブリダイズし、かつ遺伝子発現安定化機能を有するDNA;及び
(n)(l)又は(m)のDNAと相補的な塩基配列からなるDNA
(11) 遺伝子発現安定化エレメントが、目的タンパク質の遺伝子発現カセットの上流に配置されていることを特徴とする、(1)〜(10)のいずれかに記載の発現ベクター。
(12) 遺伝子発現安定化エレメントが、目的タンパク質の遺伝子発現カセットの上流及び下流の両方に配置されていることを特徴とする、(1)〜(10)のいずれかに記載の発現ベクター。
(13) 遺伝子発現安定化エレメントが、目的タンパク質の遺伝子発現カセット及び薬剤選択マーカー遺伝子発現カセットの上流及び下流の両方に配置されていることを特徴とする(1)〜(10)のいずれかに記載の発現ベクター。
(14) 薬剤選択マーカー遺伝子が、タンパク質合成阻害系抗生物質耐性遺伝子であることを特徴とする(1)〜(13)のいずれかに記載の発現ベクター。
(15) 薬剤選択マーカー遺伝子が、ピューロマイシン−N−アセチルトランスフェラーゼ、ハイグロマイシン−B−ホスホトランスフェラーゼ、及びネオマイシンホスホトランスフェラーゼからなる群より選択されることを特徴とする(14)に記載の発現カベクター。
(16) 目的タンパク質の遺伝子発現カセットが、所望の目的タンパク質遺伝子を挿入するためのマルチクローニングサイトを備えていることを特徴とする(1)〜(15)のいずれかに記載の発現ベクター。
(17) 目的タンパク質が、抗体の重鎖及び/又は軽鎖ポリペプチドであることを特徴とする(1)〜(16)のいずれかに記載の発現ベクター。
(18) (1)〜(17)のいずれか一項に記載の発現ベクターで宿主細胞を形質転換して得られることを特徴とする形質転換細胞。
(19) 宿主細胞が動物細胞であることを特徴とする(18)に記載の形質転換細胞。
(20) 動物細胞が哺乳類細胞であることを特徴とする(19)に記載の形質転換細胞。
(21) 哺乳類細胞がチャイニーズハムスター卵巣(CHO)細胞であることを特徴とする(20)に記載の形質転換細胞。
(22) チャイニーズハムスター卵巣(CHO)細胞が無血清馴化されていることを特徴とする(21)に記載の形質転換細胞。
(23) (18)〜(22)のいずれか一項に記載の形質転換細胞を薬剤選択する工程を含むことを特徴とする、目的タンパク質遺伝子を高レベルで発現する細胞群を選別する方法。
(24) (18)〜(22)のいずれか一項に記載の形質転換細胞からなる細胞群であって、目的タンパク質遺伝子を高レベルで発現することを特徴とする細胞群。
(25) (18)〜(22)のいずれか一項に記載の形質転換細胞を用いることを特徴とする、タンパク質を生産する方法。
(26) タンパク質が、抗体であることを特徴とする(25)に記載の方法。
(27) タンパク質が、ワクチンであることを特徴とする(25)に記載の方法。
(28) (18)〜(22)のいずれか一項に記載の形質転換細胞を用いることを特徴とする、アミノ酸を生産する方法。
(29) (18)〜(22)のいずれかに記載の形質転換細胞を用いることを特徴とする、ヌクレオチドを生産する方法。
【発明の効果】
【0013】
本発明の薬剤選択マーカー遺伝子発現カセットは、mRNA不安定化配列が挿入されているため、mRNA不安定化配列を有さない従来の薬剤選択マーカー遺伝子発現カセットに比べて薬剤選択マーカーの発現が減弱化されており、宿主細胞ゲノム内の高発現領域に組み込まれないと、薬剤存在下で形質転換細胞の生存が困難となる。このため、薬剤選択マーカー遺伝子及び目的タンパク質遺伝子が宿主細胞ゲノム内の高発現領域に組み込まれた形質転換細胞のみが薬剤選択によって生き残ることができる。その結果、目的タンパク質遺伝子を高レベルで発現する高発現細胞を効率的に選別することができる。
【0014】
また本発明の遺伝子発現安定化エレメントは、細胞ゲノムにおける組換えタンパク質遺伝子に対する隣接染色体や調節エレメントの影響を低減させて組換えタンパク質遺伝子の遺伝子発現を安定化し、高発現を長期間維持することができる。そこで、本発明の弱化された薬剤選択マーカー発現カセットと遺伝子発現安定化エレメントを併用することにより、その相乗効果によって、薬剤選択によって選択された細胞において目的遺伝子を非常に高度に発現する細胞を濃縮し、かつそれらの高発現細胞における発現の安定的な維持ができる。本発明により、それら非常に高いレベルのタンパク質生産性を示す高生産細胞株を迅速、容易、且つ効率的に樹立することができる。
【図面の簡単な説明】
【0015】
【図1】pBS−CMV−SNAPm2の構築を示す図である。
【図2】pPUR・N4の構築を示す図である。以下、図中、Purとは、ピューロマイシン耐性遺伝子のことである。また、図中、iPCRとは、Inverse PCRのことである。
【図3】pBS−CMV−SNAPm−Pur及びpBS−CMV−SNAPm−Pur・N4の構築を示す図である。
【図4】pBS−CMV−SNAPm−Pur及びpBS−CMV−SNAPm−Pur・N4で形質転換された細胞集団のFACS解析の結果を示す図である。
【図5】pBS−CMV−SNAPm−Pur及びpBS−CMV−SNAPm−Pur・N4で形質転換された細胞集団のうち、SNAPmを高発現する細胞の割合をプロットしたグラフである。
【図6】pBS−CMV−SNAPm−Pur及びpBS−CMV−SNAPm−Pur・N4で形質転換された細胞集団を薬剤選択した際の明視野像である。
【図7】pBS−CMV−SNAPm−Purのコロニーを単離し、FACS解析した結果を示す図である。
【図8】pBS−CMV−SNAPm−Pur・N4のコロニーを単離し、FACS解析した結果を示す図である。
【図9】pEF1α−KOD3G8HC−RE2の構築を示す図である。以下、図中、Neoとは、ネオマイシン耐性遺伝子のことである。
【図10】pEF1α−SNAP26m−Pur・N4及びpEF1α−SNAP26m−Pur−RE2の構築を示す図である。
【図11】pEF1α−SNAP26m−Pur−RE2及びpEF1α−SNAP26m−Pur・N4で形質転換された細胞集団のFACS解析の結果を示す図である。
【図12】pEF1α−SNAP26m−Pur−RE2及びpEF1α−SNAP26m−Pur・N4で形質転換された細胞集団のうち、SNAPmを高発現する細胞の割合をプロットしたグラフである。
【図13】pEF1α−KOD3G8LC−Hyg−RE2の構築を示す図である。以下、図中、Hygとは、ハイグロマイシン耐性遺伝子のことである。
【図14】pEF1α−SNAP26m−Hyg−RE2及びpEF1α−SNAP26m−Hyg・N4の構築を示す図である。
【図15】pEF1α−SNAP26m−Hyg−RE2及びpEF1α−SNAP26m−Hyg・N4で形質転換された細胞集団のFACS解析の結果を示す図である。
【図16】pEF1α−SNAP26m−Hyg−RE2及びpEF1α−SNAP26m−Hyg・N4で形質転換された細胞集団のうち、SNAPmを高発現する細胞の割合をプロットしたグラフである。
【図17】pEF1α−SNAP26m−Neo・N4の構築を示す図である。
【図18】pEF1α−SNAP26m−Neo−RE2及びpEF1α−SNAP26m−Neo・N4で形質転換された細胞集団のFACS解析の結果を示す図である。
【図19】pEF1α−SNAP26m−Neo−RE2及びpEF1α−SNAP26m−Neo・N4で形質転換された細胞集団のうち、SNAPmを高発現する細胞の割合をプロットしたグラフである。
【図20】pEF1α−SNAP26m−Pur・N2、pEF1α−SNAP26m−Pur・N6及びpEF1α−SNAP26m−Pur・N8の構築を示す図である。
【図21】pEF1α−SNAP26m−Pur−RE2、pEF1α−SNAP26m−Pur・N2、pEF1α−SNAP26m−Pur・N4、pEF1α−SNAP26m−Pur・N6及びpEF1α−SNAP26m−Pur・N8で形質転換された細胞集団のFACS解析の結果を示す図である。
【図22】pEF1α−SNAP26m−Pur−RE2、pEF1α−SNAP26m−Pur・N2、pEF1α−SNAP26m−Pur・N4、pEF1α−SNAP26m−Pur・N6及びpEF1α−SNAP26m−Pur・N8で形質転換された細胞集団のうち、SNAPmを高発現する細胞の割合をプロットしたグラフである。
【図23】配列番号26の配列とCTCFの結合配列の予測部位を示す図である。
【図24】核酸配列断片の遺伝子発現安定化効果の確認に用いた基本コンストラクトpBS−CMV−SNAPmの構築を示す図である。
【図25】配列番号26の配列、CTCFBS、被験配列の核酸断片の位置関係を示す図である。
【図26】各核酸配列の断片を含むSNAPm発現コンストラクトを安定導入されたCHO細胞をFACS解析して得られたSNAPm発現強度と細胞数の分布を示すグラフである。
【図27】各核酸配列の断片を含むSNAPm発現コンストラクトを安定導入されたCHO細胞のFACS解析によって、10以上のSNAPmの蛍光シグナルを呈した細胞の割合(%)をプロットしたグラフである。
【図28】CHO5のデリーションコンストラクト構築と配列番号26の当該被験配列の位置関係を示す図である。
【図29】CHO5、及びCHO5デリーションコンストラクトを安定導入して得られたCHO細胞のFACS解析の結果、10以上のSNAPmの蛍光シグナルを呈した細胞の割合(%)をプロットしたグラフである。
【図30】CHO5Δ3の3’デリーションコンストラクト構築のためのInverse PCR法に用いたプライマーの位置関係とデリーションによって得られた被験挿入配列を示す図である。
【図31】CHO5Δ3、及びCHO5Δ3の3’デリーションコンストラクトを安定導入して得られたCHO細胞のFACS解析の結果、10以上のSNAPmの蛍光シグナルを呈した細胞の割合(%)をプロットしたグラフである。
【図32】pEF1α−KOD3G8HC−1.1k/1.1kの構築を示す図である。
【図33】pEF1α−SNAP26m−Pur・N4−1.1k/1.1k、pEF1α−SNAP26m−Pur−1.1k/1.1kの構築を示す図である。
【図34】pEF1α−SNAP26m−Pur−RE2、pEF1α−SNAP26m−Pur−1.1k/1.1k、pEF1α−SNAP26m−Pur・N4及びpEF1α−SNAP26m−Pur・N4−1.1k/1.1kで形質転換された細胞集団のFACS解析の結果を示す図である。
【図35】pEF1α−SNAP26m−Pur−RE2、pEF1α−SNAP26m−Pur−1.1k/1.1k、pEF1α−SNAP26m−Pur・N4及びpEF1α−SNAP26m−Pur・N4−1.1k/1.1kで形質転換された細胞集団のうち、SNAPmを高発現する細胞の割合をプロットしたグラフである。
【図36】pEF1α−SNAP26m−Hyg・N4−1.1k/1.1k、pEF1α−SNAP26m−Hyg−1.1k/1.1kの構築を示す図である。
【図37】pEF1α−SNAP26m−Hyg−RE2、pEF1α−SNAP26m−Hyg−1.1k/1.1k、pEF1α−SNAP26m−Hyg・N4及びpEF1α−SNAP26m−Hyg・N4−1.1k/1.1kで形質転換された細胞集団のFACS解析の結果を示す図である。
【図38】pEF1α−SNAP26m−Hyg−RE2、pEF1α−SNAP26m−Hyg−1.1k/1.1k、pEF1α−SNAP26m−Hyg・N4及びpEF1α−SNAP26m−Hyg・N4−1.1k/1.1kで形質転換された細胞集団のうち、SNAPmを高発現する細胞の割合をプロットしたグラフである。
【図39】pEF1α−SNAP26m−Neo・N4−1.1k/1.1k、pEF1α−SNAP26m−Neo−1.1k/1.1kの構築を示す図である。
【図40】pEF1α−SNAP26m−Neo−RE2、pEF1α−SNAP26m−Neo−1.1k/1.1k、pEF1α−SNAP26m−Neo・N4及びpEF1α−SNAP26m−Neo・N4−1.1k/1.1kで形質転換された細胞集団のFACS解析の結果を示す図である。
【図41】pEF1α−SNAP26m−Neo−RE2、pEF1α−SNAP26m−Neo−1.1k/1.1k、pEF1α−SNAP26m−Neo・N4及びpEF1α−SNAP26m−Neo・N4−1.1k/1.1kで形質転換された細胞集団のうち、SNAPmを高発現する細胞の割合をプロットしたグラフである。
【図42】pEF1α−SNAP26m−Pur・N2−1.1k/1.1k、pEF1α−SNAP26m−Pur・N6−1.1k/1.1k、pEF1α−SNAP26m−Pur・N8−1.1k/1.1kの構築を示す図である。
【図43】pEF1α−SNAP26m−Pur−1.1k/1.1k、pEF1α−SNAP26m−Pur・N2−1.1k/1.1k、pEF1α−SNAP26m−Pur・N4−1.1k/1.1k、pEF1α−SNAP26m−Pur・N6−1.1k/1.1k及びpEF1α−SNAP26m−Pur・N8−1.1k/1.1kで形質転換された細胞集団のFACS解析の結果を示す図である。
【図44】pEF1α−SNAP26m−Pur−1.1k/1.1k、pEF1α−SNAP26m−Pur・N2−1.1k/1.1k、pEF1α−SNAP26m−Pur・N4−1.1k/1.1k、pEF1α−SNAP26m−Pur・N6−1.1k/1.1k及びpEF1α−SNAP26m−Pur・N8−1.1k/1.1kで形質転換された細胞集団のうち、SNAPmを高発現する細胞の割合をプロットしたグラフである。
【図45】pEF1α−KOD3G8HC−Neo・N4−1.1k/1.1kの構築を示す図である。
【図46】pEF1α−KOD3G8LC−Hyg−1.1k/1.1kの構築を示す図である。
【図47】pEF1α−KOD3G8LC−Hyg・N4−1.1k/1.1kの構築を示す図である。
【図48】pEF1α−KOD3G8HC−Pur・N4−1.1k/1.1kの構築を示す図である。
【図49】遺伝子発現安定化エレメントを含まないもの(−/Neo,−/Hyg)、遺伝子発現安定化エレメントCHO5Δ3−3を発現カセットの上流および下流の両方に挿入したもの(1.1k/Neo,1.1k/Hyg)、遺伝子発現安定化エレメントCHO5Δ3−3を発現カセットの上流および下流の両方に挿入し、薬剤耐性遺伝子(H鎖:Neo、L鎖:Hyg)にmRNA不安定化配列(N4配列)を付加したもの(1.1k/Neo・N4,1.1k/Hyg・N4)、遺伝子発現安定化エレメントCHO5Δ3−3を発現カセットの上流および下流の両方に挿入し、薬剤耐性遺伝子(H鎖:Pur、L鎖:Hyg)にmRNA不安定化配列(N4配列)を付加したもの(1.1k/Pur・N4,1.1k/Hyg・N4)について、ポリクローンの培養上清を用いてELISAを行い算出した、抗体生産量をプロットしたグラフである。
【図50】6ウェルプレートの培養上清を用いてELISAを行い算出した、各クローンの抗体生産量をプロットしたグラフである。遺伝子発現安定化エレメントを含まないもの(−/Neo,−/Hyg)と遺伝子発現安定化エレメントCHO5Δ3−3を発現カセットの上流および下流の両方に挿入したもの(1.1k/Neo,1.1k/Hyg)、および、遺伝子発現安定化エレメントを含まないもの(−/Neo,−/Hyg)と遺伝子発現安定化エレメントCHO5Δ3−3を発現カセットの上流および下流の両方に挿入し、薬剤耐性遺伝子(H鎖:Neo、L鎖:Hyg)にmRNA不安定化配列(N4配列)を付加したもの(1.1k/Neo・N4,1.1k/Hyg・N4)、および、遺伝子発現安定化エレメントを含まないもの(−/Neo,−/Hyg)と遺伝子発現安定化エレメントCHO5Δ3−3を発現カセットの上流および下流の両方に挿入し、薬剤耐性遺伝子(H鎖:Pur、L鎖:Hyg)にmRNA不安定化配列(N4配列)を付加したもの(1.1k/Pur・N4,1.1k/Hyg・N4)について、それぞれ生産量上位5クローンを同一グラフ上にプロットしている。
【図51】遺伝子安定化エレメントとmRNA不安定化配列を含まないもの(−/Neo,−/Hyg)、遺伝子安定化エレメントとmRNA不安定化配列を付加し、高い抗体生産性が認められたもの(1.1k/Pur・N4,1.1k/Hyg・N4)について、96ウェルプレートにおける2週間培養での抗体生産性の度数分布をプロットしたグラフである。
【図52】遺伝子発現安定化エレメントCHO5Δ3−3を発現カセットの上流および下流の両方に挿入したもの(1.1k/Neo,1.1k/Hyg)、遺伝子発現安定化エレメントCHO5Δ3−3を発現カセットの上流および下流の両方に挿入し、薬剤耐性遺伝子(H鎖:Pur、L鎖:Hyg)にmRNA不安定化配列(N4配列)を付加したもの(1.1k/Pur・N4,1.1k/Hyg・N4)について無血清馴化されたCHO−K1細胞へ遺伝子導入し、薬剤選択後にポリクローンの培養上清を用いてELISAを行い算出した、抗体生産量をプロットしたグラフである。
【図53】pEF1α−KOD3G8LC,HC−Pur・N4−1.1k/1.1kの構築を示す図である。
【図54】2コンストラクト抗体発現系、シングルベクター抗体発現系について、ポリクローンの培養上清を用いてELISAを行い算出した、抗体生産量をプロットしたグラフである。
【図55】6ウェルプレートの培養上清を用いてELISAを行い算出した、各クローンの抗体生産量をプロットしたグラフである。2コンストラクト抗体発現系、シングルベクター抗体発現系、それぞれ生産量上位5クローンを同一グラフ上にプロットしている。
【発明を実施するための形態】
【0016】
以下に本発明を詳細に説明する。
【0017】
タンパク質の機能解析や有用タンパク質の生産のために、遺伝子組み換え技術を用いて、細胞、特に動物細胞に、目的とするタンパク質の遺伝子を導入し、組換えタンパク質として発現させる方法が広く用いられている。具体的には、プラスミドベクターに代表されるベクターに目的タンパク質遺伝子を挿入し、細胞にベクターを取り込ませた後、組換えにより目的タンパク質遺伝子を細胞ゲノムに組み込み、細胞を形質転換させる方法がよく用いられている。
【0018】
細胞のゲノムは染色体上に存在するが、染色体はヒストンのアセチル化や高度な凝集(ヘテロクロマチン)等により遺伝子の発現が制御されており、挿入される染色体の場所によって遺伝子の発現量は大きく異なる。ベクターを用いて細胞を形質転換する方法においては、細胞ゲノムのどの場所に目的タンパク質遺伝子が導入されるかを制御することはできず、そのため形質転換細胞における目的タンパク質遺伝子の発現量、ひいては目的タンパク質の生産量は、細胞株によって大きく異なることになる。
【0019】
また、遺伝子組み換えがうまくいかず、目的タンパク質遺伝子が細胞ゲノムに組み込まれていない場合も多い。
【0020】
そこで、従前、予め発現ベクターに薬剤耐性(薬剤選択マーカー)遺伝子を挿入しておき、細胞にこのベクターを遺伝子導入した後、培地中に薬剤を投与して(薬剤選択)、形質転換が成功した細胞を選択し、その後、生じた細胞群から高い発現を示す細胞株を繰り返し選別する作業が行われてきた。しかしながら、この方法では多数の細胞株を用意する必要があり、また長期間の培養を必要とする細胞の選別作業を繰り返し行うため、多大の労力と時間を要してきた。加えて、薬剤耐性遺伝子は多くの場合、少量の発現でも薬剤に対する耐性を示すため、生存した細胞の中には、薬剤耐性遺伝子及び目的タンパク質遺伝子が細胞ゲノム内の低発現領域に組込まれており目的タンパク質遺伝子の発現量が少ない細胞株も混在しており、更に労力と時間に拍車をかけてきた。
【0021】
また、選別時には十分な発現をしていた細胞でも、培養を継続していくうちに次第に発現が低下するものが少なからず見られる。これは、遺伝子サイレンシングにより発現が抑制されるためであると考えられる。目的とするタンパク質遺伝子のゲノム導入位置の制御が通常不可能であるため、遺伝子サイレンシングの効果による発現量低下を予測することは極めて困難である。そこで、このような位置効果を緩和し、発現を高レベルで安定的に維持する技術が必要とされてきた。
【0022】
そこで、本発明はこれらの問題を解決するため、薬剤選択を利用して高発現を示す細胞を取得できるようにし、かつ遺伝子発現安定化エレメントによって、遺伝子の導入部位に左右されることなく高い発現のレベルを維持できるようにするものであり、迅速・簡便かつ効率的に目的タンパク質生産性の高い細胞株を取得できる点で極めて顕著な効果を有するものである。
【0023】
本発明ではまず、従来公知の薬剤耐性(薬剤選択マーカー)遺伝子を使用した選別において、mRNA不安定化配列を利用することによって薬剤耐性遺伝子の発現量を大きく抑え、宿主細胞ゲノム内の高発現領域に薬剤耐性遺伝子が組込まれた形質転換細胞のみが薬剤選択後に生き残るようにしている。薬剤耐性遺伝子の近隣には目的タンパク質の遺伝子が組込まれているため、生き残った細胞では、目的タンパク質遺伝子も高発現領域に組込まれている可能性が高い。従って、本発明によれば、(i)遺伝子組み換えがうまくいかず、目的タンパク質遺伝子が細胞ゲノムに組み込まれていない細胞、(ii)遺伝子組み換えに成功したが、薬剤耐性遺伝子及び目的タンパク質遺伝子が宿主細胞ゲノムの低発現領域に組込まれている細胞、及び(iii)遺伝子組み換えに成功し、しかも薬剤耐性遺伝子及び目的タンパク質遺伝子が宿主細胞ゲノムの高発現領域に組込まれている細胞の中から、(iii)の遺伝子組み換えに成功し、しかも薬剤耐性遺伝子及び目的タンパク質遺伝子が宿主細胞ゲノムの高発現領域に組込まれている細胞のみを薬剤選択によって効率的に選別することができる。従来公知の選別方法では、(i)〜(iii)の細胞から(ii)及び(iii)の細胞を薬剤選択によって選別することができるが、(ii)及び(iii)の細胞から(iii)の細胞のみを薬剤選択によって選別することはできず、薬剤選択後に生き残った形質転換細胞について目的タンパク質遺伝子の発現レベルを別途確認して、(iii)の高発現細胞を選別していた。本発明の方法は、薬剤選択のみによって(i)〜(iii)の細胞から(iii)の高発現細胞を一気に選別することができるため、効率的であり、結果として高発現細胞を迅速に選別することができる。
また、本発明では、遺伝子発現安定化エレメントを利用することにより、選別した細胞の継続培養時の遺伝子発現量の低下を防止している。本発明による遺伝子発現安定化エレメントは、目的タンパク質の発現の安定的維持のみならず、薬剤耐性遺伝子に効果を示す場合には、薬剤耐性遺伝子の発現に対する隣接染色体や調節エレメントの影響を低減させ、薬剤耐性遺伝子本来の機能を発揮する助けとなり、薬剤選択との相乗効果を示すものともなる。
【0024】
本発明の第一側面によれば、mRNA不安定化配列を含む薬剤選択マーカー遺伝子発現カセット、少なくとも1つの遺伝子発現安定化エレメント、及び目的タンパク質の遺伝子発現カセットを有することを特徴とする発現ベクターが提供される。
【0025】
本発明において、発現カセットとは、プロモーターから遺伝子コード配列、ターミネーター配列(ポリアデニレーションシグナル)までの遺伝子発現の単位をいう。加えて、イントロン、スペーサー配列、翻訳増強領域等を含むこともある。したがって、例えば「薬剤選択マーカー発現カセット」という場合、前記発現カセットの遺伝子コード配列が薬剤選択マーカー遺伝子のコード配列であるものをいう。また、「目的タンパク質の遺伝子発現カセット」という場合、前記発現カセットの遺伝子コード配列が目的とするタンパク質の遺伝子のコード配列であるものをいう。
【0026】
(薬剤選択マーカー発現カセット)
本発明で使用する薬剤選択マーカー遺伝子発現カセットは、mRNA不安定化配列を有することを除いては、従来公知の薬剤選択マーカー遺伝子発現カセットと同様の構成を有する。そこで、まず、mRNA不安定化配列について説明する。
本発明は、mRNA不安定化配列が極めて効果的に薬剤耐性遺伝子の発現を減弱化することができることを見出したことに基づくものである。本発明のmRNA不安定化配列を含む薬剤選択マーカー遺伝子発現カセットを搭載した発現ベクターを宿主細胞へ遺伝子導入した場合、mRNA不安定化配列を含まない場合と比較して、薬剤選択後の生存細胞数、あるいはそのコロニーは1/10〜1/100に減少する。これはmRNA不安定配列の存在により薬剤選択マーカーのmRNAが不安定化し、薬剤選択マーカーの発現量が下がるため、宿主細胞のゲノム中で転写活性の高い高発現領域に当該発現カセットが組み込まれた細胞でなければ薬剤選択下での生存が困難になるためと考えられる。
【0027】
「mRNA不安定化配列」とは、この配列を有するDNAから転写されたmRNAの細胞内半減期を低減させる機能を有するヌクレオチド配列のことであり、自然界では初期応答遺伝子などに存在することが見出されている。
【0028】
mRNA不安定化配列は、いくつかの遺伝子ではコード配列中、あるいは5’UTR(非翻訳領域)に存在するものもあるが、多くは3’UTRに存在する。3’UTRに存在するmRNA不安定化配列としては、AU−rich element(ARE)、histone mRNA 3’−terminal stem−loop、iron−responsive element(IRE)、insulin−like growth factor II(IGF−II)、long stem−loopなどが知られている。これらの中でもAREは、恒常的にmRNAを不安定化できることから、本発明に用いるmRNA不安定化配列として好ましい。
【0029】
AREとは、mRNAにおける「AUが豊富なエレメント」、すなわちアデニン(A)及びウラシル(U)を高い割合で含む配列もしくは領域を意味する。またAREとは、前記エレメントをコードするDNAにおける「ATが豊富なエレメント」、すなわちアデニン(A)及びチミン(T)を高い割合で含む配列もしくは領域を指すためにも用いられる。
【0030】
AREは、造血細胞増殖因子遺伝子、成長因子遺伝子、インターロイキン遺伝子、インターフェロン等のサイトカイン遺伝子、及びいくつかの癌原遺伝子(プロトオンコジーン)などに見られる(非特許文献4)。本発明の薬剤選択マーカー遺伝子発現カセットでは、これらの遺伝子中のAREに相当する核酸配列をそのまま利用してもよいが、ARE中のモチーフ配列として知られるTATTTAT(非特許文献5)やTTATTTA(T/A)(T/A)(非特許文献6)などを利用する方が、不要な制限酵素認識配列を発現ベクターに挿入してしまうことを避けることができ、利便性が高い。
【0031】
AREを持つ遺伝子の例としては、造血細胞増殖因子についてはGranulocyte−monocyte colony sitimulationg factor(GM−CSF),インターロイキンについてはInterleukin−1β、2,3、4、6、8、10、11、インターフェロンについてはInterferon−α、癌原遺伝子についてはc−fos,c−myc,c−jun,c−myb,Pim−1などが例示される。このほかにもTumor necrosis factor,Cyclin D1,Cyclooxygenase,Plasminogen activator inhibitor type2など多くの遺伝子がAREを持つことが知られており、例示された遺伝子に限定されない。
【0032】
本発明の薬剤選択マーカー遺伝子発現カセットにおいて使用することができるmRNA不安定化配列としては、下記のものが例示される。
【0033】
ATTTのモチーフ配列の1ないし2回以上の繰り返し。
【0034】
ATTTAのモチーフ配列の1ないし2回以上の繰り返し。
【0035】
TATTTATのモチーフ配列の1ないし2回以上の繰り返し。
【0036】
TTATTTA(T/A)(T/A)のモチーフ配列の1ないし2回以上の繰り返し。(T/A)はTまたはAのいずれかである。
【0037】
これらの配列は、1ないし数塩基の置換/挿入/欠失を含んでもよく、DNAの複製の誤りや突然変異などの自然変異によるもの、人為的な変異導入によるもの等が想定される。また、モチーフ配列の繰り返し間には1〜100塩基程度のスペーサー配列またはリンカー配列を含んでもよい。加えて、モチーフ配列の逆位を含むものであってもよい。
【0038】
これらのモチーフ配列の繰り返し数は、1回でもよいが、2回以上、より好ましくは4回以上とすることにより、mRNAの不安定化効果を一層高めることができ、選別効率を顕著に増大させることができる。
【0039】
繰り返しの回数の上限は特に限定されないが、細胞へのベクターの取り込みの観点から、発現ベクターが最大10kbp〜25kbp長となるようなものが望ましい。ただし、繰り返し配列数が増えるにしたがって、mRNAの不安定化の効果は飽和に向かい、一定の繰り返し数を超えるとそれ以上の顕著な効果を望めなくなる。上限値を定めるものではないが、10回以上の繰り返し配列は実用上意味がない。
【0040】
本発明の薬剤選択マーカー遺伝子発現カセットにおけるプロモーターとしては、動物細胞、特に哺乳類細胞で発現可能なプロモーターであれば特に限定されるものではないが、ヒトやマウスのサイトメガロウイルス(CMV)プロモーター、サルウイルス40(SV40)プロモーター、ヒトヘルペス単純ウイルスのチミジンキナーゼ遺伝子(HSV−tk)プロモーター、などのウイルスに由来するものや、マウスphosphoglycerate−kinase1遺伝子(PGK)プロモーターなどの非ウイルス性の細胞遺伝子に由来するプロモーター、及び由来の異なるプロモーターのハイブリッドが挙げられる。ここで、プロモーターは、プロモーターのコア領域に限るものではなく、エンハンサー領域を含む場合もある。薬剤選択マーカーの発現を減弱化するため、転写活性が低いものがより好ましい。転写活性の低いプロモーターとして変異型のプロモーターなどを利用してもよいし、コザック配列を置換してもよい。さらに、mRNA不安定化配列のモチーフ配列の繰り返しの回数を少なくし、転写活性の弱いプロモーターを併用するのも1つの想定される実施態様である。
【0041】
本発明の薬剤選択マーカー発現カセットを用いた薬剤選択に使用される薬剤としては、タンパク質合成阻害系抗生物質としてブラストサイジン(Blasticidin)、ジェネティシン(Geneticin)(G418)、ハイグロマイシン(Hygromycin;Hyg)、ピューロマイシン(Puromycin;Pur)が例示される。その他の選択用薬剤としてメトトレキセート(Methotrexate;MTX)、MSX(methionine sulphoximine)などが例示される。また、本発明の薬剤選択マーカー遺伝子発現カセットにおける薬剤選択マーカー遺伝子としては、薬剤選択に一般的に使用される薬剤に対して耐性を示す遺伝子が好適に用いられる。タンパク質合成阻害系抗生物質を薬剤として用いる場合には、タンパク質合成阻害系抗生物質耐性遺伝子を用いることが好ましい。特に限定されるものではないが、ネオマイシン耐性遺伝子(ジェネティシン耐性遺伝子としての機能も有する)として、Tn5由来ネオマイシンホスホトランスフェラーゼ(aminoglycoside 3’−phosphotransferase)(Neo)、ハイグロマイシン耐性遺伝子として、E.coli由来ハイグロマイシン−B−ホスホトランスフェラーゼ(Hph、実施例および図面ではHygと記載)、ピューロマイシン耐性遺伝子として、Streptomyces由来ピューロマイシン−N−アセチルトランスフェラーゼ遺伝子(pac、実施例および図面ではPurと記載)、ブラストサイジン耐性遺伝子として、Bacillus cereus由来ブラストサイジン耐性遺伝子(bsr)などが例示される。これらの中でも、ピューロマイシン−N−アセチルトランスフェラーゼ、ハイグロマイシン‐B−ホスホトランスフェラーゼ、ネオマイシンホスホトランスフェラーゼが好ましく、より好ましくはピューロマイシン−N−アセチルトランスフェラーゼ、ハイグロマイシン‐B−ホスホトランスフェラーゼであり、さらに好ましくはピューロマイシン−N−アセチルトランスフェラーゼである。
【0042】
本発明の薬剤選択マーカー遺伝子発現カセットにおけるポリアデニレーションシグナル(ターミネーター配列)としては、SV40ウイルス由来late polyAシグナル、early polyAシグナル、HSV−tk由来polyAシグナル、ウシ成長因子遺伝子由来polyAシグナル、ウサギβ−グロビン遺伝子由来polyAシグナルなどが例示されるが、特に限定されるものではない。
【0043】
本発明の薬剤選択マーカー遺伝子発現カセット中でのプロモーター、薬剤選択マーカー遺伝子、mRNA不安定化配列、及びポリアデニレーションシグナルの配置順序は、薬剤選択マーカー遺伝子の発現が可能である限り特に限定されないが、一般的にプロモーター、薬剤選択マーカー遺伝子、mRNA不安定化配列、及びポリアデニレーションシグナルをこの順序で上流から下流に向かって配置する。これらの四つの要素は、相互に直接連結されている必要はなく、所望により、イントロン、スペーサー配列、翻訳増強領域等を間に有していてもよい。
【0044】
(遺伝子発現安定化エレメント)
本発明における「遺伝子発現安定化エレメント」とは、目的タンパク質の生産性を高めるために、目的遺伝子発現カセットが宿主ゲノムに挿入された部位近傍のゲノムの転写の活性化の状況に影響を受けてしまう、という位置効果を解消し、遺伝子発現を安定化する機能を有する核酸領域を意味する。
【0045】
遺伝子発現を安定化する機能を有する核酸配列としては、インスレーター、スキャフォールド/マトリックス結合領域(S/MAR)、遺伝子座調節領域(LCR)、遍在作用性クロマチンオープニングエレメント(UCOE)などが利用できる。
【0046】
インスレーターとしては、ニワトリβ-グロビンLCRに由来する1.2kb DNaseI Hypersensitive site(cHS4),バフンウニ由来UR1などが例示される。S/MARとしては、ニワトリリゾチーム5’MARエレメント、ヒトβ−グロビンMARエレメントやヒトインターフェロンβSARエレメントなどが例示される。
【0047】
本発明の発現ベクターは、少なくとも一つの遺伝子発現安定化エレメントを有する。これら遺伝子発現を安定化する機能を有する核酸配列を目的タンパク質の発現カセットの上流若しくは下流、あるいは上流及び下流に配置することによって、遺伝子発現の安定的維持、及び目的タンパク質の生産性を向上させることが可能である。
【0048】
目的遺伝子を宿主細胞ゲノムへ導入するための遺伝子構築物として、プラスミドベクターが一般に用いられるが、プラスミドベクターの長さは最大10ないし25kbpであることから、遺伝子発現安定化エレメントの鎖長は出来るだけ短い方が好ましい。好ましい長さとしては5kbp以下、より好ましくは1kbp程度である。
【0049】
本発明の好ましい実施態様における遺伝子発現安定化エレメントは、特許文献5に記載のCHO細胞DR1000L−4N株(CHO細胞−4N株)から本発明者らによって同定された核酸分子である。当該株は、外来遺伝子としてヒト顆粒球マクロファージコロニー刺激因子(hGM−CSF)遺伝子を、DHFR遺伝子を持つ発現ベクターに導入し、このベクターを、DHFR遺伝子を欠損したCHO細胞DG44株に導入し、導入株をIMDM培地(核酸含有)に10%牛胎児血清を添加した培地で培養して形質転換体を得、この形質転換体を、次いでIMDM培地(核酸不含)、10%透析牛胎児血清を添加した培地で選択し、さらに50nM、100nMのメトトレキセート(MTX)を含み、核酸を含まないIMDM培地を用いて10から100倍程度に増加するまで培養し、MTX濃度を上昇させることにより得られたテロメアタイプのクローン細胞である。
【0050】
真核細胞のゲノムは、真正染色質と異質染色質の2つのクラスの染色質に分けられる。セントロメアとテロメアの配列は、構成的な異質染色質の主要な部分である。特に、テロメアは、TTAGGGの繰り返し配列とサブテロメア領域からなり、際立った異質染色質の立体構造をとる、遺伝子の乏しい領域である(非特許文献7,8)。テロメアは、ゲノムの安定性に対するその寄与や保護的な役割に加え、テロメア位置効果として知られる現象によって、近くに導入された遺伝子の発現にも影響を与える(非特許文献9,10)。
【0051】
ところが、DR1000L−4N株は、テロメアタイプのクローン細胞であるにもかかわらず、導入された外来遺伝子は導入部位付近の異質染色体からの不活性化の影響を受けることなく、長期間の培養において、安定したhGM−CSFの高い産生を維持した。これは、外来遺伝子の導入された部位の近傍に、不活性化の進行を強力に遮断するような境界エレメントが存在し、遺伝子発現を安定化していることを示唆する。
【0052】
本発明者らは、DR1000L−4N株において外来遺伝子導入部位の近傍の配列を分析した結果、配列表の配列番号26で示す約91kbpの配列からなるDNAが遺伝子発現の安定化に関与していることを見出した。
【0053】
本発明の発現ベクターに含まれる遺伝子発現安定化エレメントの長さは短い方が好ましい。そこで、より小さいサイズでも遺伝子発現安定化機能を有する領域を絞り込むため、発明者らは更なる研究を行った。
【0054】
配列番号26に示す約91kbpの配列を更に分析するにあたり、通常、ショットガン法による約3kbp程度の断片のライブラリを作成し、各断片を1つずつアッセイする方法が取られる。しかしながら、各断片が遺伝子安定化機能を有するかどうかを解析する際に、細胞の形質転換や培養に時間がかかるため、1アッセイ当たり数週間から数ヶ月の期間を要する。従って、全アッセイを行うためには膨大な時間と労力を要することになる。
【0055】
そこで、本発明者らは、配列番号26の配列中、遺伝子発現配列の境界エレメントが存在することが期待される部位を中心に解析する方法を見出した。そして、配列番号26からなるDNAのうち42kbp、45kbp、及び91kbp付近の領域が遺伝子発現安定化機能を有することを見出すに至った。
【0056】
この配列をさらに詳細に分析したところ、この配列の中でも、(i)41820番目から41839番目までの塩基で示す領域、(ii)41821番目から41840番目までの塩基で示す領域、(iii)45182番目から45200番目までの塩基で示す領域、及び(iv)91094番目から91113番目までの塩基で示す領域の4つの領域が、インスレーター結合タンパク質CTCFの結合配列モチーフに相当することを見出した。従って、これらの4つの領域の少なくともいずれかを含む、これらの領域の近傍が特に遺伝子発現の安定化に関与していると考えられた。
【0057】
本発明における実施態様では、遺伝子発現安定化エレメントとして、下記(a)〜(e)のいずれか又はその組み合わせが使用できる。
(a)配列番号26で示す配列からなるDNA;
(b)配列番号26で示す配列の部分配列からなるDNAであって、配列番号26で示す配列のうち41820番目から41839番目までの塩基で示す領域の配列を少なくとも含むDNA;
(c)配列番号26で示す配列の部分配列からなるDNAであって、配列番号26で示す配列のうち41821番目から41840番目までの塩基で示す領域の配列を少なくとも含むDNA;
(d)配列番号26で示す配列の部分配列からなるDNAであって、配列番号26で示す配列のうち45182番目から45200番目までの塩基で示す領域の配列を少なくとも含むDNA;及び
(e)配列番号26で示す配列の部分配列からなるDNAであって、配列番号26で示す配列のうち91094番目から91113番目までの塩基で示す領域の配列を少なくとも含むDNA。
【0058】
これらのエレメントは、CHOゲノムから単離同定されたものであるが、これらのエレメントと相同なエレメントは、他の哺乳類細胞ゲノムにも存在することが予想される。従って、(f)配列番号26で示す配列の部分配列からなるDNAであって、宿主細胞中で外来遺伝子発現カセットと近接するように配置された際に、外来遺伝子発現カセットに含まれる外来遺伝子からの目的組換えタンパク質の発現を増大または安定化させることができるDNAも、遺伝子発現安定化エレメントとして使用することができる。このような相同エレメントは、この技術分野の周知の技術、例えば、種間ハイブリダイゼーションまたはPCRなどによって容易に単離同定されることができる。
【0059】
また、(g)前記(a)〜(f)のいずれかのDNAと相補的な塩基配列からなるDNAとストリンジェントな条件でハイブリダイズし、かつ遺伝子発現安定化機能を有するDNAも当然、遺伝子発現安定化エレメントとして使用することができる。本発明の遺伝子発現安定化エレメントは、これらの(a)〜(g)のいずれか一つ又はそれらの任意の組合せからなる。さらに、(h)前記(a)〜(g)のいずれかのDNAと相補的な塩基配列からなるDNAも、遺伝子発現安定化機能を有し、遺伝子発現安定化エレメントとして使用することができるため、本発明に含まれる。
【0060】
ストリンジェントな条件とは、用いられるプローブ・標識方法によっても異なるが、例えば、0.1%SDSを含む0.2×SSC中40〜50℃または0.1%SDSを含む2×SSC中55〜65℃の条件下ハイブリダイズする塩基配列である。さらにストリンジェントな条件は、結合する核酸の融解温度(Tm値)に基づいて決定することができる。また、ハイブリダイズ後の洗浄条件として、「6×SSC、0.1%SDS、Tm値よりも約15〜30℃低い温度」程度の条件をあげることができる。より高いストリンジェントな条件としては「2×SSC、0.1%SDS、Tm値よりも約5〜15℃低い温度」程度、更に高いストリンジェントな条件としては「1×SSC、0.1%SDS、Tm値よりも約5〜15℃低い温度」程度の洗浄条件をあげることができる。更に高いストリンジェントな条件としては「0.1×SSC、0.1%SDS」程度の洗浄条件が例示される。
【0061】
本発明の遺伝子発現安定化エレメントの好ましい態様では、前記(b)または(c)のDNAは、配列番号26で示す配列のうち41601番目から46746番目までの塩基で示す領域、又は配列番号26で示す配列のうち41601番目から46746番目までの配列と相補的な塩基配列からなるDNAとストリンジェントな条件でハイブリダイズし、かつ遺伝子発現安定化機能を有する塩基配列領域である。また、これらの領域からなるDNAと相補的な塩基配列も含まれる。本発明の遺伝子発現安定化エレメントのさらに好ましい態様では、前記(b)または(c)のDNAは、配列番号26で示す配列のうち41601番目から42700番目までの塩基で示す領域、又は配列番号26で示す配列のうち41601番目から42700番目までの配列と相補的な塩基配列からなるDNAとストリンジェントな条件でハイブリダイズし、かつ遺伝子発現安定化機能を有する塩基配列領域であり、又はこれらの領域からなるDNAと相補的な塩基配列である。
【0062】
本発明の遺伝子発現安定化エレメントがその発現安定化効果を奏するためには、エレメントが宿主細胞ゲノム中で外来遺伝子発現カセットの導入部位に近接する位置に配置されることが必要である。本発明において、用語「近接する」とは、特に制限されるものではないが、好ましくは本発明の遺伝子発現安定化エレメントと外来遺伝子発現カセットとの間の距離が5000bp以下、さらに好ましくは500bp以下であることを意味する。
【0063】
このような近接配置は、本発明のエレメントを含む核酸配列断片と、外来遺伝子の発現カセットを含む核酸配列断片との混合物をエレクトロポレーションやトランスフェクションなどによって宿主細胞内へ導入し、外来遺伝子の発現量の高い宿主細胞クローンを選択することによって実現することもできるが、近接配置を確実に達成させるためには、本発明の遺伝子発現安定化エレメント、及びそれに近接して配置された外来遺伝子発現カセットを含む外来遺伝子発現ベクターをあらかじめ作成しておき、この発現ベクターで宿主細胞を形質転換させることが望ましい。
【0064】
(目的タンパク質の遺伝子発現カセット)
本発明における目的タンパク質遺伝子の発現カセットは、プロモーター、遺伝子コード配列、及びターミネーター配列(ポリアデニレーションシグナル)からなる。加えて、イントロンやスペーサー配列、翻訳増強領域を含むこともある。目的タンパク質遺伝子の発現プロモーターとしてはできるだけ転写活性の高いものが好ましく、ヒトやマウスのサイトメガロウイルス(CMV)プロモーター、サルウイルス40(SV40)プロモーターなどのウイルスに由来するものや、ヒトやマウス、さらにはCHO由来のElongation Factor 1(EF1α)遺伝子、ユビキチン遺伝子、β−アクチン遺伝子など、非ウイルス性の細胞遺伝子に由来するプロモーター、及び由来の異なるプロモーター/エンハンサーのハイブリッド、例えばCAGプロモーターなどが挙げられる。
【0065】
本発明では、目的タンパク質の遺伝子発現カセットは、薬剤選択マーカー発現カセットと独立した形で与えられる。真核細胞の場合、目的タンパク質遺伝子と薬剤選択マーカー遺伝子を内在性リボソームエントリー部位(Internal ribosome entry sites、IRES)でつなぐポリシストロン性の発現を行う手法もあるが、目的タンパク質遺伝子と薬剤選択マーカー遺伝子のコード配列が1つのmRNAとして合成されるため、前記mRNA不安定化配列を挿入すると、目的タンパク質遺伝子のmRNAが減少してしまい、高発現細胞を得られない。そのため、本発明では、目的タンパク質の遺伝子発現カセットは、薬剤選択マーカー発現カセットと独立して配置されることが好ましい。目的タンパク質の遺伝子発現カセットと薬剤選択マーカー発現カセットは、別個のベクターの形で利用されてもよいが、目的タンパク質の遺伝子発現カセットと薬剤選択マーカー発現カセットが同一のベクター上に配置されることがより好ましい。
【0066】
目的タンパク質の遺伝子発現カセットには、該タンパク質遺伝子のコード配列の代わりに、複数の制限酵素認識配列からなるマルチクローニングサイトを配置することができる。外来遺伝子のcDNAやコード配列を導入する際に、クローニング作業が簡便であり、好ましい。
【0067】
他の実施態様としては、目的タンパク質が抗体の重鎖及び/又は軽鎖のポリペプチドである、目的タンパク質の遺伝子発現カセットである。この場合、目的タンパク質の発現カセットは、複数配置してもよく、あるいは複数のポリペプチド遺伝子をIRESでつなぎ、ポリシストロン性の1つの発現カセットの形でもよい。
【0068】
(発現ベクター)
本発明において、発現ベクターとは、遺伝子工学に利用されるベクターである。発現ベクターは、プラスミドベクターであることができる。しかし、これに限らず、例えば、ウイルスベクター、コスミドベクター、細菌人工染色体(BAC)、酵母人工染色体(YAC)及び他の非プラスミドベクターも使用できる。
【0069】
本発明における発現ベクターの実施態様の1つは、目的タンパク質遺伝子発現カセットと薬剤選択マーカー遺伝子発現カセットの上流若しくは下流に、遺伝子発現安定化エレメントが配置された発現ベクターである。好ましくは、目的タンパク質遺伝子発現カセットと薬剤選択マーカー遺伝子発現カセットの上流に、遺伝子発現安定化エレメントが配置された発現ベクターである。この場合、目的タンパク質遺伝子発現カセットと薬剤選択マーカー遺伝子発現カセットのどちらが上流であってもよいが、好ましくは目的タンパク質遺伝子発現カセットが上流に配置されたものである。
【0070】
本発明における別の実施態様は、薬剤選択マーカー遺伝子発現カセットと目的タンパク質の遺伝子発現カセットの間に遺伝子安定化エレメントが配置されたベクターである。目的タンパク質遺伝子発現カセットの下流であって薬剤選択マーカー遺伝子発現カセットの上流、若しくは薬剤選択マーカー遺伝子発現カセットの下流であって目的タンパク質遺伝子発現カセットの上流のいずれの態様も考えられるが、好ましくは目的タンパク質遺伝子発現カセットの下流であって薬剤選択マーカー遺伝子発現カセットの上流に遺伝子発現安定化エレメントが配置されたベクターである。
【0071】
本発明における発現ベクターの好ましい実施態様は、目的タンパク質遺伝子発現カセットと薬剤選択マーカー遺伝子発現カセットの上流及び下流に、それらの発現カセットを挟むように遺伝子発現安定化エレメントが配置されたベクターである。目的タンパク質遺伝子発現カセットと薬剤選択マーカー遺伝子発現カセットの順序は問わないが、目的タンパク質遺伝子発現カセットが上流に配置されていることが好ましい。また、目的タンパク質遺伝子発現カセットと薬剤選択マーカー遺伝子発現カセットそれぞれの上流及び下流に遺伝子発現安定化エレメントが配置されていてもよい。
【0072】
本発明における好ましい別の実施態様は、目的タンパク質遺伝子発現カセットの上流及び下流に遺伝子発現安定化エレメントが配置され、さらにその下流に薬剤選択マーカー遺伝子発現カセットが配置されたベクターである。
【0073】
更には、薬剤選択マーカー遺伝子発現カセットの下流に目的タンパク質の遺伝子発現カセットが配置され、薬剤選択マーカー遺伝子発現カセットと目的タンパク質の遺伝子発現カセットの間及び目的タンパク質遺伝子発現カセットの下流に遺伝子発現安定化エレメントが配置されている発現ベクターも実施しうる。
【0074】
遺伝子発現安定化エレメントと薬剤選択マーカー遺伝子発現カセット若しくは目的タンパク質の遺伝子発現カセットとの間は、隣接していてもよいし、スペーサー配列等が挿入されていてもよい。
【0075】
前記薬剤選択マーカー遺伝子発現カセットと目的遺伝子タンパク質の発現カセットが同一のベクター上に配置される場合、両者は隣接してもよいし、間にスペーサー配列が挿入されていてもよい。目的タンパク質遺伝子の発現カセットは薬剤選択マーカー遺伝子発現カセットの上流、下流のいずれに配置されても良いが、隣接して挿入される際には、目的タンパク質遺伝子の発現カセットが上流になるように配置される方が好ましい。さらに、スペーサー配列として、隣接する発現カセットの転写干渉を抑制するためのインスレーター配列が挿入されていても良い。
【0076】
薬剤選択マーカー遺伝子発現カセットは、1つのベクターに2つ以上含まれていても良い。この場合、同一の薬剤選択マーカーが選ばれる場合もあるし、それぞれ異なる薬剤選択マーカーが選ばれる場合もある。また、各薬剤選択マーカーは隣接して配置されても良いし、離れて配置されていても良い。
【0077】
目的のタンパク質が抗体の重鎖、軽鎖のように複数のポリペプチドからなる場合、目的タンパク質の発現カセットを複数配置してもよく、あるいは複数のポリペプチド遺伝子をIRESでつなぎ、ポリシストロン性の1つの発現カセットの形にしてもよい。
【0078】
また、複数のポリペプチド発現カセットをそれぞれ異なるベクターに挿入し、それぞれのベクターを1つの宿主細胞へ導入して形質転換細胞を作製し、目的タンパク質を生産することができる。この場合、各ベクターに薬剤選択マーカー遺伝子発現カセットを配置することができ、それぞれ同一の薬剤選択マーカーを用いることもできるが、異なる薬剤選択マーカーを用いる方が、各ポリペプチドが全て高発現を示す細胞を効率よく取得する点で有利であり、好ましい。
【0079】
抗体の重鎖ポリペプチド遺伝子と軽鎖ポリペプチド遺伝子をそれぞれ異なるベクターに挿入して発現させ、抗体を生産した場合と、抗体の重鎖ポリペプチド遺伝子と軽鎖ポリペプチド遺伝子とを同一のベクターに挿入して発現させ、抗体を生産した場合の比較において、抗体の生産性はどちらも変わらない。したがって、いずれの方法を用いても高い生産性を示す細胞株を取得することができるが、抗体の重鎖ポリペプチド遺伝子と軽鎖ポリペプチド遺伝子とを同一のベクターに挿入して発現させた方が、同一ベクター上の一種類の薬剤耐性遺伝子を使うことになり導入遺伝子数の差異を軽減でき、抗体の重鎖ポリペプチドと軽鎖ポリペプチドの生産バランスがとれ、重鎖・軽鎖の2つのポリペプチドが2本ずつから成る抗体としての生産性が安定するため、好ましい。
【0080】
本発明の第二の側面によれば、本発明の第一の側面による発現ベクターで宿主細胞を形質転換して得られることを特徴とする形質転換細胞が提供される。
【0081】
本発明の形質転換細胞における宿主細胞としては、組換えタンパク質の産生に一般的に用いられるチャイニーズハムスター卵巣細胞(CHO)の他、マウスミエローマ細胞(NSO)、ベビーハムスターキドニー細胞(BHK)、ヒト繊維肉腫細胞(HT1080)、COS細胞などの哺乳類由来の細胞が例示されるが、これらに限定されるものではなく、ヒト、マウス、ラット、ハムスター、モルモット、ウサギ、イヌ、ウシ、ウマ、ヒツジ、サル、ブタなどの動物由来の細胞などを広く導入対象とすることができる。また、大腸菌などの細菌細胞や酵母、昆虫細胞などを用いたタンパク質生産システムにも応用しうる。
一般にバイオ医薬品などの工業生産には、ウイルスなどの有害成分の混入を排除するため、動物成分を含まない培地、より好ましくは化学的に組成の明らかな(Chemically Defined)な培地で生産細胞を培養することが求められる。このため、一般的に使用されるウシ胎児血清などを含有した培地で高生産細胞をスクリーニングした後、1〜2ヶ月程度かけて動物成分非含有培地やChemically Definedとされる培地へ細胞を適応させること(無血清馴化)が必要とされる。しかしながら、せっかく取得した高生産性細胞も上手く馴化できないケースもある。そこで、あらかじめ無血清馴化された細胞に遺伝子導入することは、無血清馴化工程を省略して生産細胞の樹立時間を短縮でき、非常に有効である。このような課題達成のため、本発明に用いられる宿主細胞としては、CHO細胞等における無血清馴化済み細胞がさらに例示される。
【0082】
宿主細胞の形質転換方法は、当業者であれば適宜選択しうるが、例えばリポフェクション法、リン酸カルシウム法、エレクトロポレーション法、DEAEデキストラン法、マイクロインジェクション等が挙げられる。
【0083】
本発明の第三の側面によれば、本発明の第二の側面による形質転換細胞を薬剤選択する工程を含むことを特徴とする、目的タンパク質遺伝子を高レベルで発現する細胞群を選別する方法が提供される。また、本発明の第四の側面によれば、本発明の第二の側面による形質転換細胞からなる細胞群であって、目的タンパク質遺伝子を高レベルで発現することを特徴とする細胞群が提供される。
【0084】
薬剤選択に使用する薬剤は、薬剤選択マーカー遺伝子発現カセット中の薬剤選択マーカー遺伝子の種類に応じて決定される。また、使用する薬剤の濃度としては、一般に使用される濃度の範囲で高発現細胞の濃縮が可能であるが、少し高めの濃度が好ましい。最適な濃度は、宿主細胞や用いる培地の種類によって変化する。これらの濃度の設定方法は当業者にとっては公知であり、適宜設定しうる。例えば、産業用の生産細胞として用いられるチャイニーズハムスター卵巣(CHO)細胞を10%ウシ胎児血清を含むHam‘s F12培地で培養し、ピューロマイシン耐性遺伝子発現カセットを利用した場合のピューロマイシンの濃度としては、5μg/ml以上、より好ましくは7.5μg/ml以上、さらに好ましくは10μg/ml以上である。同様の培養条件でハイグロマイシン耐性遺伝子発現カセットを利用した場合のハイグロマイシンBの濃度としては、200μg/ml以上、より好ましくは600μg/ml以上、さらに好ましくは800μg/ml以上である。同様の培養条件でネオマイシン耐性遺伝子発現カセットを利用した場合のG418の濃度としては、好ましくは400μg/ml以上、より好ましくは800μg/ml以上、さらに好ましくは1,000μg/ml以上である。
【0085】
本発明の選別方法では、薬剤選択により、mRNAの不安定化を伴わない従来の薬剤選択を行った細胞群に比べて、目的タンパク質遺伝子を高レベルで発現する細胞の割合が有意に高くなる。従って、従来は、薬剤選択後の膨大なサンプルから目的タンパク質遺伝子を高レベルで発現する細胞株を選別する作業を繰り返し行わなければならなかったが、本発明によれば、薬剤選択マーカーのmRNA不安定化を含む薬剤選択のみによって、選別の候補となるサンプル数を大幅に絞ることができる。本発明の選別方法を用いれば、薬剤選択後も生存する細胞群(ポリクローン)の状態で目的タンパク質生産量が有意に上昇していて、これらの細胞群から細胞を単離し、培養することにより、容易に高発現を示す細胞株(モノクローン)を取得することができる。本発明の実施態様の一例として、4.0×105個の細胞に本発明の薬剤選択マーカー遺伝子発現カセットが挿入されたベクターを遺伝子導入した後、薬剤選択を行って生じた細胞群から100の細胞株をランダムに選択して、個々の細胞株の発現量を調べることにより目的タンパク質の高発現株を容易に取得することができる。
【0086】
細胞の発現レベルはレポーター遺伝子の発現量により調べることができるが、一例として、SNAP26m遺伝子(Covalys社)やGFP(Green Fluorescent Protein)遺伝子等の蛍光タンパク質遺伝子を含む発現カセットを本発明の発現ベクターに導入し、FACS(fluorescence activated cell sorting)等のフローサイトメーターを用いて蛍光強度を解析することにより調べることができる。別の例として、ルシフェラーゼ遺伝子を含む発現カセットを本発明による発現ベクターに導入後、細胞溶解液にD−ルシフェリンを添加してルミノメーターで発光量を測定することにより調べることができる。また、レポーター遺伝子に限らず、ELISA(Enzyme−Linked Immunosorbent Assay)、酵素免疫測定法(EIA)等を利用することで抗体等の目的タンパク質の発現を調べることもできる。
【0087】
本発明により、細胞選別の工程及び時間を大幅に短縮することができると共に、高発現を示す細胞の割合が上昇することにより、短時間で効率的に目的タンパク質遺伝子を高レベルで発現する細胞を取得することができ、従来技術と比べて極めて顕著な効果を示す。
【0088】
例えばFACSCalibur(ベクトン・ディッキンソン社製)により、蛍光強度FL1にてSNAP26mの発現量を測定した場合、FL1が200以上若しくは1000以上を示す細胞の割合は、本発明の薬剤選択マーカー遺伝子発現カセットを有さない細胞に比べて、本発明による細胞では1.1倍以上、また多くの場合1.5倍から2倍以上大きくなる効果が認められた。また、FL1が200以上を示す細胞の割合の増加率よりもFL1が1000以上を示す細胞の割合の増加率の方が高い傾向が見られており、本発明により選別される細胞群が、本発明の薬剤選択マーカー遺伝子発現カセットを有さない細胞に比べて高い発現を示すという効果を裏付けている。
【0089】
さらに、本発明におけるmRNA不安定化配列及び遺伝子安定化エレメントを有する発現ベクターにより形質転換された細胞では、mRNA不安定化配列及び遺伝子安定化エレメントを有さない発現ベクターを使用した場合に比べ、高生産性を示す細胞の割合が7〜8倍向上する。遺伝子発現安定化エレメントのみを有する発現ベクターにより形質転換された細胞では、高生産性を示す細胞の割合が2倍程度向上し優れた効果を有しているが、本発明はmRNA不安定化配列及び遺伝子発現安定化エレメントの相乗効果によって更に顕著な効果を有する。
【0090】
本発明の第五の側面によれば、本発明の第二の側面による形質転換細胞を用いることを特徴とする、タンパク質を生産する方法が提供される。
【0091】
一つの実施態様では、生産されるタンパク質は、抗体である。この場合、抗体の重鎖ポリペプチド遺伝子と軽鎖ポリペプチド遺伝子は同一のベクター(シングルベクター)に挿入されていても良いし、それぞれ異なるベクターに挿入されて2コンストラクトとされていても良い。
【0092】
哺乳類の抗体には、IgM、IgD、IgG、IgA、IgEの5種類のクラスが存在することが明らかとなっているが、ヒトの各種疾患の診断、予防及び治療には血中半減期が長く、各種エフェクター機能を有する等の機能特性からヒトIgGクラスの抗体が主として利用されている。抗体は、形質転換細胞の培養上清よりプロテインAカラムを用いて精製することができる。また、その他に通常、蛋白質の精製で用いられる精製方法を使用することができる。例えば、ゲル濾過、イオン交換クロマトグラフィー及び限外濾過等を組み合わせて行い、精製することができる。精製したヒト化抗体のH鎖、L鎖或いは抗体分子全体の分子量は、二次元電気泳動等により測定することができる。
【0093】
本発明の方法により生産された抗体は、医薬として使用することができる。かかる医薬は、治療薬として単独で投与することも可能ではあるが、通常は薬理学的に許容される一つあるいはそれ以上の担体と一緒に混合し、製剤学の技術分野にて公知の任意の方法により製造した医薬製剤として提供されるのが好ましい。
【0094】
投与経路は、治療に際して最も効果的なものを使用するのが望ましく、経口投与、または口腔内、気道内、直腸内、皮下、筋肉内および静脈内等の非経口投与をあげることができ、抗体製剤の場合、好ましくは静脈内投与をあげることができる。投与形態としては、噴霧剤、カプセル剤、錠剤、顆粒剤、シロップ剤、乳剤、座剤、注射剤、軟膏、テープ剤等があげられる。
【0095】
経口投与に適当な製剤としては、乳剤、シロップ剤、カプセル剤、錠剤、散剤、顆粒剤等があげられる。乳剤およびシロップ剤のような液体調製物は、水、ショ糖、ソルビトール、果糖等の糖類、ポリエチレングリコール、プロピレングリコール等のグリコール類、ごま油、オリーブ油、大豆油等の油類、p−ヒドロキシ安息香酸エステル類等の防腐剤、ストロベリーフレーバー、ペパーミント等のフレーバー類等を添加剤として用いて製造できる。カプセル剤、錠剤、散剤、顆粒剤等は、乳糖、ブドウ糖、ショ糖、マンニトール等の賦形剤、デンプン、アルギン酸ナトリウム等の崩壊剤、ステアリン酸マグネシウム、タルク等の滑沢剤、ポリビニルアルコール、ヒドロキシプロピルセルロース、ゼラチン等の結合剤、脂肪酸エステル等の界面活性剤、グリセリン等の可塑剤等を添加剤として用いて製造できる。
【0096】
非経口投与に適当な製剤としては、注射剤、座剤、噴霧剤等があげられる。注射剤は、塩溶液、ブドウ糖溶液、あるいは両者の混合物からなる担体等を用いて調製される。または、ヒト化抗体を常法に従って凍結乾燥し、これに塩化ナトリウムを加えることによって粉末注射剤を調製することもできる。座剤はカカオ脂、水素化脂肪またはカルボン酸等の担体を用いて調製される。また、噴霧剤は該化合物そのもの、ないしは受容者の口腔および気道粘膜を刺激せず、かつ該化合物を微細な粒子として分散させ吸収を容易にさせる担体等を用いて調製される。
【0097】
担体として具体的には乳糖、グリセリン等が例示される。該化合物および用いる担体の性質により、エアロゾル、ドライパウダー等の製剤が可能である。また、これらの非経口剤においても経口剤で添加剤として例示した成分を添加することもできる。
【0098】
別の実施態様では、生産されるタンパク質は、ワクチンである。
【0099】
ワクチンとしては、病原体のエピトープのアミノ酸配列からなるタンパク質をワクチンとして使用することができる。また、ウイルス体の構成タンパク質すべてを生産するのみならず、エンベロープタンパク等を生産し、ワクチンとして使用することができる。
【0100】
ワクチンは任意の経路の投与のための製剤とすることができる。粘膜型投与、例えば、経口経路、経鼻経路、経気道経路、経膣経路、経直腸経路の他、非粘膜経路による投与、例えば皮下、静脈内もしくは筋肉内投与のためのワクチン製剤が挙げられる。
【0101】
本発明の第六の側面によれば、本発明の第二の側面による形質転換細胞を用いることを特徴とする、アミノ酸を生産する方法が提供される。また、本発明の第七の側面によれば、本発明の第二の側面による形質転換細胞を用いることを特徴とする、ヌクレオチドを生産する方法が提供される。
【0102】
アミノ酸の生産は、まず形質転換細胞にポリペプチドを生産させ、そのポリペプチドを加水分解することによって行うことができる。また、アミノ酸の生体外合成に必要な酵素などを形質転換細胞に生産させることによって行うこともできる。ヌクレオチドの生産も、ヌクレオチドの生体外合成に必要な酵素などを形質転換細胞に生産させることによって行うことができる。
【実施例】
【0103】
以下、実施例を例示することによって、本発明の効果をより一層明確なものとする。
【実施例1】
【0104】
ピューロマイシン耐性遺伝子におけるmRNA不安定化配列の効果
(1)pBS−CMV−SNAPm−Pur及びpBS−CMV−SNAPm−Pur・N4の構築
図1に示すスキームに従って、プラスミドpBS−CMV−SNAPm2を構築した。即ち、まずEmerald Lucベクター(pELuc−test)(東洋紡績社製)の制限酵素ClaI、EcoRIサイトにCMVプロモーター(配列番号1)が導入されたpELuc−CMVプラスミドから制限酵素EcoRI、NotIでEmerald Luc(ELuc)遺伝子を切出した。一方、SNAPm発現プラスミドpSNAPm(Covalys社製)からSNAP26m遺伝子を配列番号2,3のプライマーを用いてPCRで増幅し、pELuc−CMVプラスミドの制限酵素EcoRI、NotIサイトに導入してpSNAP26m−CMVを構築した。続いて、pSNAP26m−CMVより、CMVプロモーター/SNAP26m/SV40 polyAを配列番号4,5のプライマーを用いてPCRで増幅し、部分消化によって、プラスミドpBluescript II KS(−)(ストラタジーン社製)の制限酵素ClaI、BamHIサイトへ導入し、プラスミドpBS−CMV−SNAPm2を構築した。
【0105】
続いて、図2に示すスキームに従って、pPUR(クロンテック社製)のPuromycin耐性遺伝子のコード配列の3‘末端に、配列番号6、7のプライマーとKOD −Plus− Mutagenesis Kit(東洋紡績社製)を用いたInverse PCR法によって、ARE配列モチーフTTATTTATTの4回繰り返し配列(N4)を挿入してpPUR・N4を構築した。
【0106】
つづいて、pPUR,pPUR・N4を鋳型として、配列番号8、9のプライマーとKOD −Plus− ver.2(東洋紡績社製)を用いたPCRによって、SV40 プロモーター−Pur−SV40 polyA、SV40プロモーター−Pur・N4−SV40 polyAの薬剤発現カセット断片を増幅した。pBS−CMV−SNAPm2と前記増幅断片をそれぞれ制限酵素BamHI、SacIで部分消化して連結し、pBS−CMV−SNAPm-Pur,pBS−CMV−SNAPm-Pur・N4を構築した(図3)。
【実施例2】
【0107】
(2)pBS−CMV−SNAPm−Pur及びpBS−CMV−SNAPm−Pur・N4の細胞導入(1)
CHO−K1細胞(理研バイオリソースセンター、No.RCB0285)を1×105cells/mlに調整し、12ウェルプレートに2mlずつ前日に播種し、一晩培養して、トランスフェクション用のCHO−K1細胞を準備した。この際、培地として10%牛胎児血清を添加したHam’s F12培地(日水製薬社製)を使用した。
【0108】
トランスフェクションは、3μl GeneJuice Transfection Reagent(メルク社製)を100μl Opti−MEM I Reduced−Serum Medium(GIBCO社製)で希釈し、1μgのプラスミドとしてpBS−CMV−SNAPm−PurあるいはpBS−CMV−SNAPm−Pur・N4にこの希釈液103μlを加え、10分間放置した後、この混合液を前記CHO−K1細胞に加え、24時間インキュベートすることにより行った。翌日、培地を除去し、2.5g/l−Trypsin・1mmol/l−EDTA Solution(ナカライテスク社製)で処理して細胞を分散させ、90mmペトリディッシュに移し、6μg/ml Puromycin(InvivoGen社製)を含むHam’s F12+10%FBS培地中で3週間選択培養を行った。選択培養中、3〜4日ごとに培地を交換した。3週間の選択培養終了後、2.5g/l−Trypsin・1mmol/l−EDTA Solutionで処理して細胞を分散し、1ウェル当たり2×105細胞を12ウェルプレートに播き込み、翌日、0.5mlのHam’s F12培地に2μM SNAP−Cell−505(Covalys社製)を加え、60分間、37℃でインキュベートした。その後、Ham’s F12培地で3回リンスし、培地交換をしながら、10分間のインキュベートを3回行い、未反応の蛍光色素を除去した。この細胞を2.5g/l−Trypsin/1mmol/l−EDTA Solutionで処理して細胞を分散し、D−PBS(−)(ナカライテスク社製)に細胞を懸濁し、フローサイトメーターBD FACSCalibur(Becton,Dickinson社製)でSNAP26mの発現強度を解析した。
【0109】
図4は、薬剤選択後のpBS−CMV−SNAPm−Pur及びpBS−CMV−SNAPm−Pur・N4で形質転換された細胞集団のFACS解析の結果を示す。横軸が蛍光強度(FL1)、縦軸がカウント数である。また、図5に蛍光強度FL1がそれぞれ200以上、1000以上の細胞の割合のプロットを示す。この結果、mRNA不安定化因子であるN4が挿入されたpBS−CMV−SNAPm−Pur・N4で形質転換された細胞集団には、薬剤選択後、SNAPmの高発現を示す細胞が有意に濃縮されていることが分かる。
【実施例3】
【0110】
細胞株でのピューロマイシン耐性遺伝子におけるmRNA不安定化配列の効果
(1)pBS−CMV−SNAPm−Pur及びpBS−CMV−SNAPm−Pur・N4の細胞導入(2)
つづいて、実施例2同様に遺伝子導入を行い、9μg/ml Puromycinを含むHam’s F12+10%FBS培地中で2週間選択培養を行った。ここで形成されたコロニーを顕微鏡下でピペットの先でpBS−CMV−SNAPm−Purについては48クローン、pBS−CMV−SNAPm−Pur・N4については20クローンをかきとり、12ウェルプレートに播種した。増殖が認められた、それぞれ36クローン、10クローンについて、実施例2と同様にSNAP−Cell−505で標識処理をし、FACS解析を実施した。
【0111】
図6に、薬剤選択後の細胞の明視野像を示す。この結果、pBS−CMV−SNAPm−Purに比べ、pBS−CMV−SNAPm−Pur・N4については生存細胞数、コロニー数が顕著に減少し、選択効果が高いことが示唆される。
【0112】
図7にpBS−CMV−SNAPm−Pur形質転換細胞クローンにおける、ピーク強度が1000を超える、特にSNAPm発現の高いクローンのFACS解析の結果を示す。図8にpBS−CMV−SNAPm−Pur・N4形質転換細胞クローンにおける、ピーク強度が1000を超える、特にSNAPm発現の高いクローンのFACS解析の結果を示す。この結果、pBS−CMV−SNAPm−Purでは高発現クローンは36クローン中1クローン(2.8%)しか認められなかった。しかしながら、pBS−CMV−SNAPm−Pur・N4は取得されたクローンは少ないものの、SNAPm高発現クローンのヒット率が40%(10クローン中4クローン)と非常に選択効果が高いことが確認された。また、この結果から、実施例2で認められたように、薬剤選択後の細胞集団をFACS解析することによって高発現細胞の割合の上昇していることと、高発現クローンの取得が効率化されていることに相関が確認されたことから、以後の解析は細胞集団のFACS解析によって効果の確認を実施した。
【実施例4】
【0113】
EF1αプロモーター使用下でのピューロマイシン耐性遺伝子におけるmRNA不安定化配列の効果
(1)pEF1α−SNAP26m−Pur・N4及びpEF1α−SNAP26m−Pur−RE2の構築
次に、目的遺伝子発現カセットのプロモーターをEF1αプロモーターに置換した場合も、上述と同様の効果が認められるか検討するため、プラスミドを構築した。本実験では、図9に示すスキームに従って構築されたプラスミドpEF1α−KOD3G8HC−RE2を利用した。即ち、まず、CMVプロモーターをEF−1αプロモーターに置換したpCI−neoプラスミド(プロメガ社製)の制限酵素XbaI−NotIサイトに抗KOD抗体の重鎖を挿入したプラスミドのpEF1α−KOD3G8HCを鋳型に、KOD −Plus− Mutagenesis Kitを用いたInverse PCR法によって、配列番号10、11のプライマーを使って発現カセット上流に制限酵素SalIおよびNheIサイトを付加したpEF1α−KOD3G8HC−REを構築した。なお、抗KOD抗体の重鎖としては、Thermococcus kodakaraensis KOD1株由来のDNAポリメラーゼに特異的な抗体を産生するマウスハイブリドーマ細胞系3G8(入手先:独立行政法人産業技術総合研究所 特許生物寄託センター、寄託番号:FERM BP−6056)から得られた抗体のものを使用した。更に本プラスミドを鋳型に、KOD −Plus− Mutagenesis Kitを用いたInverse PCR法によって、配列番号12、13のプライマーを使って発現カセット下流に制限酵素BsiWIおよびXhoIサイトを付加した構築されたプラスミドがpEF1α−KOD3G8HC−RE2である。
【0114】
このpEF1α−KOD3G8HC−RE2を用いて、図10に示すスキームに従って、プラスミドpEF1α−SNAP26m−Pur・N4およびpEF1α−SNAP26m−Pur−RE2を構築した。即ち、プラスミドpEF1α−KOD3G8HC−RE2から制限酵素EcoRI、NotIで抗KOD抗体の重鎖遺伝子を切出した。一方、SNAPm発現プラスミドpSNAPmからSNAP26m遺伝子を配列番号2,3のプライマーを用いてPCRで増幅し、pEF1α−KOD3G8HC−RE2プラスミドの制限酵素EcoRI、NotIサイトに導入してpEF1α−SNAP26m−Neo−RE2を構築した。本プラスミドから制限酵素AflII、BstBIでネオマイシン耐性遺伝子を切出した。一方、pPUR・N4から下流にN4配列を付加したPuromycin耐性遺伝子を配列番号14,15のプライマーを用いてPCRで増幅し、pEF1α−SNAP26m−RE2プラスミドの制限酵素AflII、BstBIサイトに導入してpEF1α−SNAP26m−Pur・N4を構築した。続いて、本プラスミドを鋳型に、KOD −Plus− Mutagenesis Kitを用いたInverse PCR法によって、配列番号16、17のプライマーを使ってPuromycin耐性遺伝子下流のN4配列を削除したプラスミドpEF1α−SNAP26m−Pur−RE2を構築した。
【実施例5】
【0115】
(2)pEF1α−SNAP26m−Pur−RE2及びpEF1α−SNAP26m−Pur・N4の細胞導入
実施例2に記載の方法でトランスフェクション用のCHO−K1細胞を準備した。一方、実施例4で構築したSNAP26m発現コンストラクトを制限酵素AhdI(New England Biolabs社製)でリニアライズ(直鎖状化)した。トランスフェクションは、3μl GeneJuice Transfection Reagentを100μl Opti−MEM I Reduced−Serum Mediumで希釈した後、前記のリニア化したプラスミド1μgを加え、10分間放置した後、この混合液を前記CHO−K1細胞に加え、24時間インキュベートすることにより行った。翌日、培地を除去し、2.5g/l−Trypsin・1mmol/l−EDTA Solutionで処理して細胞を分散させ、90mmペトリディッシュに移し、5、7.5、あるいは10μg/ml Puromycinをそれぞれ含むHam’s F12+10%FBS培地中で1週間選択培養を行った。選択培養中、3〜4日ごとに培地を交換した。選択培養終了後、実施例2に記載の方法で、細胞集団のSNAP26mの発現強度を解析した。
【0116】
図11は、薬剤選択後のpEF1α−SNAP26m−Pur−RE2及びpEF1α−SNAP26m−Pur・N4で形質転換された細胞集団のFACS解析の結果を示す。また、図12に蛍光強度FL1が200以上、1000以上の細胞の割合のプロットを示す。この結果、薬剤選択中のPuromycin濃度に関わらず、mRNA不安定化因子であるN4が挿入されたpEF1α−SNAP26m−Pur・N4で形質転換された細胞集団には、薬剤選択後、SNAPmの高発現を示す細胞が有意に濃縮されていることが分かる。
【実施例6】
【0117】
ハイグロマイシン耐性遺伝子におけるmRNA不安定化配列の効果
(1)pEF1α−SNAP26m−Hyg‐RE2及びpEF1α−SNAP26m−Hyg・N4の構築
ハイグロマシン耐性遺伝子を用いた場合もmRNA不安定化因子の効果が認められるか検討するため、プラスミドを構築した。この構築には、図13に示すスキームに従って構築されたプラスミドpEF1α−KOD3G8LC−Hyg−RE2を利用した。即ち、まず、CMVプロモーターをEF1αプロモーターに変更したpCI−neoプラスミドの制限酵素XbaI−NotIサイトに抗KOD抗体の軽鎖を挿入したプラスミドのpEF1α−KOD3G8LCを鋳型に、KOD −Plus− Mutagenesis Kitを用いたInverse PCR法によって、配列番号10、11のプライマーを使って発現カセット上流に制限酵素SalIおよびNheIサイトを付加したpEF1α−KOD3G8LC−REを構築した。なお、抗KOD抗体の軽鎖としては、Thermococcus kodakaraensis KOD1株由来のDNAポリメラーゼに特異的な抗体を産生するマウスハイブリドーマ細胞系3G8(入手先:独立行政法人産業技術総合研究所 特許生物寄託センター、寄託番号:FERM BP−6056)から得られた抗体のものを使用した。更に本プラスミドを鋳型に、KOD −Plus− Mutagenesis Kitを用いたInverse PCR法によって、配列番号12、13のプライマーを使って発現カセット下流に制限酵素BsiWIおよびXhoIサイトを付加して構築されたプラスミドがpEF1α−KOD3G8LC−RE2である。
【0118】
このpEF1α−KOD3G8LC−RE2をAflII、BstBIで処理し、ネオマイシン耐性遺伝子を切出した。一方、pTK−Hyg(クロンテック社製)からハイグロマイシン耐性遺伝子を配列番号18、19のプライマーを使ってPCRで増幅し、pEF1α−KOD3G8LC−RE2の制限酵素AflII、BstBIサイトに導入してpEF1α−KOD3G8LC−Hyg−RE2を構築した。続いて、図14に示すスキームに従って、プラスミドpEF1α−SNAP26m−Hyg−RE2及びpEF1α−SNAP26m−Hyg・N4を構築した。即ち、プラスミドpEF1α−KOD3G8LC−Hyg−RE2から制限酵素MluI、NotIで抗KOD抗体の軽鎖遺伝子を切出した。一方、SNAPm発現プラスミドpSNAPmからSNAP26m遺伝子を配列番号3,20のプライマーを用いてPCRで増幅し、pEF1α−KOD3G8LC−Hyg−RE2プラスミドの制限酵素MluI、NotIサイトに導入してpEF1α−SNAP26m−Hyg−RE2を構築した。続いて、本プラスミドを鋳型に、KOD −Plus− Mutagenesis Kitを用いたInverse PCR法によって、配列番号21、22のプライマーを使ってHygromycin耐性遺伝子下流にN4配列を付加したpEF1α−SNAP26m−Hyg・N4を構築した。
【実施例7】
【0119】
(2)pEF1α−SNAP26m−Hyg−RE2及びpEF1α−SNAP26m−Hyg・N4の細胞導入
実施例2に記載の方法でトランスフェクション用のCHO−K1細胞を準備した。一方、実施例6で構築したSNAP26m発現コンストラクトを制限酵素AhdIでリニアライズした。トランスフェクションは、実施例5に記載の方法で行い、トランスフェクションの翌日、培地を除去し、2.5g/l−Trypsin・1mmol/l−EDTA Solutionで処理して細胞を分散させ、90mmペトリディッシュに移し、800μg/mlハイグロマイシン HygroGold(InvivoGen社製)を含むHam’s F12+10%FBS培地中で1週間選択培養を行った。選択培養中、3〜4日ごとに培地を交換した。選択培養終了後、実施例2に記載の方法で、細胞集団のSNAP26mの発現強度を解析した。
【0120】
図15は、薬剤選択後のpEF1α−SNAP26m−Hyg−RE2及びpEF1α−SNAP26m−Hyg・N4で形質転換された細胞集団のFACS解析の結果を示す。また、図16に蛍光強度FL1が200以上、1000以上の細胞の割合のプロットを示す。この結果、mRNA不安定化因子であるN4が挿入されたpEF1α−SNAP26m−Hyg・N4で形質転換された細胞集団には、薬剤選択後、SNAPmの高発現を示す細胞が有意に濃縮されていることが分かる。
【実施例8】
【0121】
ネオマイシン耐性遺伝子におけるmRNA不安定化配列の効果
(1)pEF1α−SNAP26m−Neo・N4の構築
ネオマイシン耐性遺伝子を用いた場合もmRNA不安定化因子の効果が認められるか検討するため、図17に示すスキームに従って、プラスミドpEF1α−SNAP26m−Neo・N4を構築した。即ち、実施例4に記載のpEF1α−SNAP26m−Neo−RE2を鋳型に、KOD −Plus− Mutagenesis Kitを用いたInverse PCR法によって、配列番号21、23のプライマーを使ってネオマイシン耐性遺伝子下流にN4配列を付加したpEF1α−SNAP26m−Neo・N4を構築した。
【実施例9】
【0122】
(2)pEF1α−SNAP26m−Neo−RE2及びpEF1α−SNAP26m−Neo・N4の細胞導入
実施例2に記載の方法でトランスフェクション用のCHO−K1細胞を準備した。一方、pEF1α−SNAP26m−Neo−RE2及びpEF1α−SNAP26m−Neo・N4を制限酵素AhdIでリニアライズした。トランスフェクションは、実施例5に記載の方法で行い、トランスフェクションの翌日、培地を除去し、2.5g/l−Trypsin・1mmol/l−EDTA Solutionで処理して細胞を分散させ、90mmペトリディッシュに移し、1mg/mlジェネティシン G418(ナカライテスク社製)を含むHam’s F12+10%FBS培地中で1週間選択培養を行った。選択培養中、3〜4日ごとに培地を交換した。選択培養終了後、実施例2に記載の方法で、細胞集団のSNAP26mの発現強度を解析した。
【0123】
図18は、薬剤選択後のpEF1α−SNAP26m−Neo−RE2及びpEF1α−SNAP26m−Neo・N4で形質転換された細胞集団のFACS解析の結果を示す。また、図19に蛍光強度FL1が200以上、1000以上の細胞の割合のプロットを示す。この結果、mRNA不安定化因子であるN4が挿入されたpEF1α−SNAP26m−Neo・N4で形質転換された細胞集団には、薬剤選択後、SNAPmの高発現を示す細胞が有意に濃縮されていることが分かる。
【実施例10】
【0124】
ARE配列モチーフの繰り返し配列数の検討
図20に示すスキームに従って、プラスミドpEF1α−SNAP26m−Pur・N2、pEF1α−SNAP26m−Pur・N6およびpEF1α−SNAP26m−Pur・N8を構築した。実施例4に記載のpEF1α−SNAP26m−Pur・N4を鋳型に、KOD−Plus−Mutagenesis Kitを用いたInverse PCR法によって、配列番号16、24のプライマーを使ってPuromycin耐性遺伝子の下流にN2配列(ARE配列モチーフTTATTTATTの2回繰り返し配列)を付加したプラスミドpEF1α−SNAP26m−Pur・N2を構築した。また、同様の方法で、配列番号7、24のプライマーを使ってPuromycin耐性遺伝子の下流にN6配列(ARE配列モチーフTTATTTATTの6回繰り返し配列)を付加したプラスミドpEF1α−SNAP26m−Pur・N6を構築し、配列番号7、25のプライマーを使ってPuromycin耐性遺伝子の下流にN8配列(ARE配列モチーフTTATTTATTの8回繰り返し配列)を付加したプラスミドpEF1α−SNAP26m−Pur・N8を構築した。
【0125】
実施例2に記載の方法でトランスフェクション用のCHO−K1細胞を準備した。一方、pEF1α−SNAP26m−Pur−RE2、pEF1α−SNAP26m−Pur・N2、pEF1α−SNAP26m−Pur・N4、pEF1α−SNAP26m−Pur・N6及びpEF1α−SNAP26m−Pur・N8の各SNAP26m発現コンストラクトを制限酵素AhdIでリニアライズした。トランスフェクションは、実施例5に記載の方法で行い、トランスフェクションの翌日、培地を除去し、2.5g/l−Trypsin・1mmol/l−EDTA Solutionで処理して細胞を分散させ、90mmペトリディッシュに移し、7.5μg/ml Puromycinを含むHam’s F12+10%FBS培地中で1週間選択培養を行った。選択培養中、3〜4日ごとに培地を交換した。選択培養終了後、実施例2に記載の方法で、細胞集団のSNAP26mの発現強度を解析した。
【0126】
図21は、薬剤選択後のpEF1α−SNAP26m−Pur−RE2、pEF1α−SNAP26m−Pur・N2、pEF1α−SNAP26m−Pur・N4、pEF1α−SNAP26m−Pur・N6及びpEF1α−SNAP26m−Pur・N8で形質転換された細胞集団のFACS解析の結果を示す。また、図22に蛍光強度FL1が200以上、1000以上の細胞の割合のプロットを示す。この結果、繰り返し配列数の多い、N6あるいはN8が挿入されたプラスミドのpEF1α−SNAP26m−Pur・N6あるいはpEF1α−SNAP26m−Pur・N8で形質転換したとき、pEF1α−SNAP26m−Pur・N4と比較して、SNAPm高発現細胞の割合が更に多く、高発現細胞の選択効率が高いとの結果であった。
【実施例11】
【0127】
核酸配列の決定
CHO細胞DR1000L−4N株(CHO細胞−4N株)より作成されたBACライブラリーのうち、hGM−CSF発現カセットを含むクローンCg0031N14を単離した。このクローンCg0031N14に由来するBACより作成したショットガンクローンの5’,3’ワンパスシーケンスを実施し、Phred/Phrap/Consedによる配列情報のアッセンブルを行い、hGM−CSF発現カセットが導入されたCHO細胞の近傍のゲノム配列約91kbpの配列を決定した。この配列を、配列表の配列番号26に示す。
【実施例12】
【0128】
核酸配列の解析
前記決定された核酸配列中にインスレーター配列が存在するかどうかを、インスレーター結合タンパク質CTCFの結合配列モチーフを検索することによって調査した。解析ツールとしてIn silico CTCFBS prediction tool(http://insulatordb.utmem.edu/)(非特許文献11)を使用した。前記ツールで抽出されたモチーフ配列を表1及び図23に示す。
【0129】
【表1】
【実施例13】
【0130】
導入遺伝子発現の安定化効果の確認
実施例1の途中で構築されたpSNAP26m−CMVより、CMVプロモーター/SNAP26m/SV40 polyAを配列番号27,28のプライマーを用いてPCRで増幅し、部分消化によって、プラスミドpBluescript II SK(−)の制限酵素BamHI、SacIサイトへ導入し、プラスミドpBS−CMV−SNAPmを構築した(図24)。
【0131】
次に、実施例11で作成したショットガンライブラリーのクローンうち、41820位、45182位のCTCFの結合配列モチーフの付近のCHOゲノム配列を含むクローンを抽出し、CHOゲノムに由来する導入配列(図25参照)をPCRで増幅し、pBS−CMV−SNAP26mの制限酵素KpnI、XhoIまたはXhoI、ClaIサイトに導入した。ショットガンライブラリークローンと、当該導入配列が組み込まれたSNAPm発現コンストラクトの対応関係を表2に示す。なお、表2中のSNAPm発現コンストラクトの欄の(−)は、CHOのゲノム配列が導入されていないpBS−CMV−SNAPmである。
【0132】
【表2】
【0133】
これらのSNAPm発現コンストラクトとpPURを制限酵素AhdIでリニアライズした後、それぞれ重量比が9:1となるように混合して混合プラスミドを調製した。
【0134】
一方、1×105cells/mlに調整したCHO−K1細胞を12ウェルプレートに2mlずつ前日に播種し、一晩培養して、トランスフェクション用のCHO−K1細胞を準備した。この際、培地として10%牛胎児血清を添加したHam’s F12培地を使用した。
【0135】
トランスフェクションは、3μl GeneJuice Transfection Reagentを100μl Opti−MEM I Reduced−Serum Mediumで希釈し、前記の混合プラスミド1μgにこの希釈液103μlを加え、10分間放置した後、この混合液を前記CHO−K1細胞に加え、24時間インキュベートすることにより行った。翌日、培地を除去し、2.5g/l−Trypsin・1mmol/l−EDTA Solutionで処理して細胞を分散させ、90mmペトリディッシュに移し、6μg/ml Puromycinを含むHam’s F12+10%FBS培地中で3週間選択培養を行った。選択培養中、3〜4日ごとに培地を交換した。選択培養終了後、2.5g/l−Trypsin・1mmol/l−EDTA Solutionで処理して細胞を分散し、1ウェル当たり2×105細胞を12ウェルプレートに播き込み、翌日、0.5mlのHam’s F12培地に2μM SNAP−Cell−505を加え、60分間、37℃でインキュベートした。その後、Ham’s F12培地で3回リンスし、培地交換をしながら、10分間のインキュベートを3回行い、未反応の蛍光色素を除去した。この細胞を2.5g/l−Trypsin・1mmol/l−EDTA Solutionで処理して細胞を分散し、D−PBS(−)に細胞を懸濁し、フローサイトメーターBD FACSCaliburでSNAPmの発現強度を解析した。
【0136】
図26は、被検配列CHO1〜CHO6がそれぞれ導入されたSNAPm発現コンストラクトで形質転換された細胞集団のFACS解析の結果を示す。また、図27は、未処理のCHO細胞(図26左上の(−))の解析結果から10以上のシグナル(図26中のM2)を示す細胞をSNAPmの発現細胞とし、その細胞の割合をプロットしたグラフである。図26、27から明らかな通り、CHO細胞ゲノムの一部を導入したコンストラクト、特に配列番号26の41601〜46746位を導入したCHO4及び5において、有意な発現の上昇が認められた。CHO4とCHO5の差異はサンプル間のバラツキの範囲内と考えられるが、念のため、以後の最適領域の絞り込みはCHO5に基づいて実施した。
【実施例14】
【0137】
遺伝子発現安定化能を有する配列領域の絞り込み(1)
前記で発現の上昇が最も高かったコンストラクトCHO5をもとに、KOD−Plus−Mutagenesis Kitを用いたInverse PCR法によって、配列番号39、40のプライマーを使って被験配列41601−42902位をデリーションしたCHO5Δ5を、配列番号41,42のプライマーを使って42903−44200位をデリーションしたCHO5ΔMを、配列番号43、44のプライマーを使って44201−46746位をデリーションしたCHO5Δ3を構築した(図28)。
【0138】
これらのSNAPm発現コンストラクトとpPURを制限酵素AhdIでリニアライズした後、それぞれ重量比が9:1となるように混合して混合プラスミドを調製した。
【0139】
前記の混合プラスミド1μgを前日12ウェルプレートに播種したCHO−K1細胞にトランスフェクションし、24時間インキュベートした。翌日、培地を除去し、2.5g/l−Trypsin・1mmol/l−EDTA Solutionで処理して細胞を分散させ、90mmペトリディッシュに移し、6μg/ml Puromycinを含むHam’s F12+10%FBS培地中で3週間選択培養を行った。選択培養中、3〜4日ごとに培地を交換した。選択培養終了後、2.5g/l−Trypsin・1mmol・l−EDTA Solutionで処理して細胞を分散し、1ウェルあたり2×105細胞を12ウェルプレートに播き込み、翌日、0.5mlのHam’s F12培地に2μM SNAP−Cell−505を加え、60分間、37℃でインキュベートした。その後、Ham’s F12培地で3回リンスし、培地交換をしながら、10分間のインキュベートを3回行い、未反応の蛍光色素を除去した。この細胞を2.5g/l−Trypsin・1mmol/l−EDTA Solutionで処理して細胞を分散し、D−PBS(−)に細胞を懸濁し、フローサイトメーターBD FACSCaliburでSNAPmの発現強度を解析した。
【0140】
図29は、被検配列CHO5Δ5、ΔM、Δ3がそれぞれ導入されたSNAPm発現コンストラクトで形質転換された細胞集団のFACS解析の結果、シグナル値10以上を示した細胞の割合をプロットしたものである。図29から明らかな通り、CHO5Δ3についてはデリーション前とほぼ同程度のSNAPmの発現強度が認められたのに対し、CHOΔM、Δ3では発現上昇の効果が明らかに低減した。このことから、発現の安定化には配列番号26の配列の41601〜46746位のうちの5’側〜前半部分が主に寄与していると考えられる。
【実施例15】
【0141】
遺伝子発現安定化能を有する配列領域の絞り込み(2)
配列番号44〜49のプライマーを使用し、KOD−Plus−Mutagenesis Kitを用いたInverse PCR法によって、CHO5Δ3の被験配列の3’がさらにデリーションされたSNAPm発現コンストラクトを構築した(図30及び表3)。
【0142】
【表3】
【0143】
これらのSNAP26m発現コンストラクトを実施例14と同様に処理し、SNAP26mの発現強度を解析した。
【0144】
図31は、被検配列CHO5Δ3、CHO5Δ3−1〜5のSNAPm発現コンストラクトで形質転換された細胞集団のFACS解析の結果、シグナル値10以上を示した細胞の割合をプロットしたものである。図31から明らかな通り、配列番号26の配列の41601〜42700位を含むCHO5Δ3−1からCHO5Δ3−3までについては、サンプル間のバラツキはあるものの、デリーション前とほぼ同程度のSNAPmの発現強度が認められたのに対し、かかる領域の3’側が欠失したCHO5Δ3−4及びCHO5Δ3−5では発現の上昇効果が明らかに低減した。
【実施例16】
【0145】
ピューロマイシン耐性遺伝子におけるmRNA不安定化配列と遺伝子発現安定化エレメントとの組み合わせの効果
(1)pEF1α−SNAP26m−Pur−1.1k/1.1k及びpEF1α−SNAP26m−Pur・N4−1.1k/1.1kの構築
次に、ピューロマイシン耐性遺伝子で、N4配列と実施例15で遺伝子発現安定化能の認められたCHO5Δ3−3との組合せ効果を検討するため、プラスミドを構築した。本実験では図32に記載のpEF1α−KOD3G8HC−1.1k/1.1kを利用した。即ち、まず、実施例4の途中で構築されたpEF1α−KOD3G8HC−RE2の発現カセット上流の制限酵素NheI、BglIIサイトに配列番号50、51のプライマーセットを使ってPCRで増幅した被検配列CHO5Δ3−3(以下、コンストラクト中では1.1kと表記)を挿入して、プラスミドpEF1α−KOD3G8HC−1.1kを構築した。更にプラスミドpEF1α−KOD3G8HC−1.1kの発現カセット下流の制限酵素BsiWI、XhoIサイトに配列番号52、53のプライマーセットを使ってPCRで増幅した被検配列CHO5Δ3−3を挿入して構築されたプラスミドがpEF1α−KOD3G8HC−1.1k/1.1kである。
【0146】
一方、実施例4で構築したプラスミドpEF1α−SNAP26m−Pur・N4およびpEF1α−SNAP26m−Pur−RE2を鋳型に、KOD −Plus− Mutagenesis Kitを用いたInverse PCR法によって、配列番号54、55のプライマーを使ってPuromycin耐性遺伝子内に存在する制限酵素BsiWIサイトを削除したプラスミドpEF1α−SNAP26m−Pur2・N4およびpEF1α−SNAP26m−Pur2−RE2を構築した。
【0147】
続いて、図33に示すスキームに従って、pEF1α−SNAP26m−Pur・N4−1.1k/1.1k及びpEF1α−SNAP26m−Pur−1.1k/1.1kを構築した。即ち、pEF1α−SNAP26m−Pur2・N4、pEF1α−SNAP26m−Pur2−RE2から制限酵素EcoRI、BsiWIで、SNAPm−pA−SV40 Promoter−Pur2・N4−pA、SNAPm−pA−SV40
Promoter−Pur2−pAを切出し、pEF1α−KOD3G8HC−1.1k/1.1kの制限酵素EcoRI、BsiWIサイトに導入してpEF1α−SNAP26m−Pur・N4−1.1k/1.1k、pEF1α−SNAP26m−Pur−1.1k/1.1kを構築した。
【実施例17】
【0148】
(2)ピューロマイシン耐性遺伝子での、N4配列とCHO5Δ3−3との組合せ効果の検討
実施例2に記載の方法でトランスフェクション用のCHO−K1細胞を準備した。一方、実施例4および16で構築したSNAP26m発現コンストラクトを制限酵素AhdIでリニアライズした。トランスフェクションは、実施例5に記載の方法で行い、トランスフェクションの翌日、培地を除去し、2.5g/l−Trypsin・1mmol/l−EDTA Solutionで処理して細胞を分散させ、90mmペトリディッシュに移し、それぞれ5、7.5、10μg/ml Puromycinを含むHam’s F12+10%FBS培地中で1週間選択培養を行った。選択培養中、3〜4日ごとに培地を交換した。選択培養終了後、実施例2に記載の方法で、細胞集団のSNAP26mの発現強度を解析した。
【0149】
図34は、薬剤選択後のpEF1α−SNAP26m−Pur−RE2、pEF1α−SNAP26m−Pur−1.1k/1.1k、pEF1α−SNAP26m−Pur・N4及びpEF1α−SNAP26m−Pur・N4−1.1k/1.1kで形質転換された細胞集団のFACS解析の結果を示す。また、図35に蛍光強度FL1が200以上、1000以上の細胞の割合のプロットを示す。この結果、N4配列あるいはCHO5Δ3−3の有無に関わらず、薬剤選択中のPuromycin濃度が高いほど、SNAPm高発現細胞の割合が多いことが分かる。しかしながら、CHO5Δ3−3あるいはN4配列を用いることでSNAPm高発現細胞の割合が増大し、さらにこれらを併用することによって、その相乗効果により高発現細胞の割合は最大値となることが分かる。
【実施例18】
【0150】
ハイグロマイシン耐性遺伝子におけるmRNA不安定化配列及び遺伝子発現安定化エレメントとの組み合わせの効果
(1)pEF1α−SNAP26m−Hyg−1.1k/1.1k及びpEF1α−SNAP26m−Hyg・N4−1.1k/1.1kの構築
次に、ハイグロマイシン耐性遺伝子で、N4配列と実施例15で遺伝子発現安定化能の認められたCHO5Δ3−3との組合せ効果を検討するため、図36に示すスキームに従って、pEF1α−SNAP26m−Hyg・N4−1.1k/1.1k及びpEF1α−SNAP26m−Hyg−1.1k/1.1kを構築した。即ち、pEF1α−SNAP26m−Hyg・N4、pEF1α−SNAP26m−Hyg−RE2から制限酵素MluI、BsiWIで、SNAPm−pA−SV40 プロモーター−Hyg・N4−pA、SNAP26m−pA−SV40 プロモーター−Hyg−pAを切出し、pEF1α−KOD3G8HC−1.1k/1.1kの制限酵素MluI、BsiWIサイトに導入してpEF1α−SNAP26m−Hyg・N4−1.1k/1.1k、pEF1α−SNAP26m−Hyg−1.1k/1.1kを構築した。
【実施例19】
【0151】
(2)ハイグロマイシン耐性遺伝子での、N4配列とCHO5Δ3−3との組合せ効果の検討
実施例2に記載の方法でトランスフェクション用のCHO−K1細胞を準備した。一方、実施例6および18で構築したSNAP26m発現コンストラクトを制限酵素AhdIでリニアライズした。トランスフェクションは、実施例5に記載の方法で行い、トランスフェクションの翌日、培地を除去し、2.5g/l−Trypsin・1mmol/l−EDTA Solutionで処理して細胞を分散させ、90mmペトリディッシュに移し、800μg/ml HygroGoldを含むHam’s F12+10%FBS培地中で1週間選択培養を行った。選択培養中、3〜4日ごとに培地を交換した。選択培養終了後、実施例2に記載の方法で、細胞集団のSNAP26mの発現強度を解析した。
【0152】
図37は、薬剤選択後のpEF1α−SNAP26m−Hyg−RE2、pEF1α−SNAP26m−Hyg−1.1k/1.1k、pEF1α−SNAP26m−Hyg・N4及びpEF1α−SNAP26m−Hyg・N4−1.1k/1.1kで形質転換された細胞集団のFACS解析の結果を示す。また、図38に蛍光強度FL1が200以上、1000以上の細胞の割合のプロットを示す。この結果、mRNA不安定化因子であるN4配列と遺伝子発現安定化効果を有するCHO5Δ3−3を組み合わせることで、N4配列あるいはCHO5Δ3−3を単独で用いた場合と比較してSNAPm高発現細胞の割合が更に増大しており、高生産性細胞の選択効率が向上することが分かる。
【実施例20】
【0153】
ネオマイシン耐性遺伝子におけるmRNA不安定化配列と遺伝子発現安定化エレメントとの組み合わせの効果
(1)pEF1α−SNAP26m−Neo−1.1k/1.1k及びpEF1α−SNAP26m−Neo・N4−1.1k/1.1kの構築
次に、ネオマイシン耐性遺伝子で、N4配列と実施例15で遺伝子発現安定化能の認められたCHO5Δ3−3との組合せ効果を検討するため、図39に示すスキームに従って、pEF1α−SNAP26m−Neo・N4−1.1k/1.1k及びpEF1α−SNAP26m−Neo−1.1k/1.1kを構築した。即ち、pEF1α−SNAP26m−Neo・N4、pEF1α−SNAP26m−Neo−RE2から制限酵素EcoRI、BsiWIで、SNAP26m−pA−SV40 プロモーター−Neo・N4−pA、SNAP26m−pA−SV40 プロモーター−Neo−pAを切出し、pEF1α−KOD3G8HC−1.1k/1.1kの制限酵素coRI、BsiWIサイトに導入してpEF1α−SNAP26m−Neo・N4−1.1k/1.1k、pEF1α−SNAP26m−Neo−1.1k/1.1kを構築した。
【実施例21】
【0154】
(2)ネオマイシン耐性遺伝子での、N4配列とCHO5Δ3−3との組合せ効果の検討
実施例2に記載の方法でトランスフェクション用のCHO−K1細胞を準備した。一方、実施例8および20で構築したSNAP26m発現コンストラクトを制限酵素AhdIでリニアライズした。トランスフェクションは、実施例5に記載の方法で行い、トランスフェクションの翌日、培地を除去し、2.5g/l−Trypsin・1mmol/l−EDTA Solutionで処理して細胞を分散させ、90mmペトリディッシュに移し、1mg/mlジェネティシン G418を含むHam’s F12+10%FBS培地中で1週間選択培養を行った。選択培養中、3〜4日ごとに培地を交換した。選択培養終了後、実施例2に記載の方法で、細胞集団のSNAP26mの発現強度を解析した。
【0155】
図40は、薬剤選択後のpEF1α−SNAP26m−Neo−RE2、pEF1α−SNAP26m−Neo−1.1k/1.1k、pEF1α−SNAP26m−Neo・N4及びpEF1α−SNAP26m−Neo・N4−1.1k/1.1kで形質転換された細胞集団のFACS解析の結果を示す。また、図41に蛍光強度FL1が200以上、1000以上の細胞の割合のプロットを示す。この結果、mRNA不安定化因子であるN4配列と遺伝子発現安定化効果を有するCHO5Δ3−3を組み合わせることで、N4配列あるいはCHO5Δ3−3を単独で用いた場合と比較してSNAPm高発現細胞の割合が更に増大しており、高生産性細胞の選択効率が向上することが分かる。
【実施例22】
【0156】
CHO5Δ3−3存在下でのARE配列モチーフの繰り返し配列数の検討
図42に示すスキームに従って、プラスミドpEF1α−SNAP26m−Pur・N2−1.1k/1.1k、pEF1α−SNAP26m−Pur・N6−1.1k/1.1kおよびpEF1α−SNAP26m−Pur・N8−1.1k/1.1kを構築した。まず、実施例10で構築したプラスミドpEF1α−SNAP26m−Pur・N2、pEF1α−SNAP26m−Pur・N6およびpEF1α−SNAP26m−Pur・N8を鋳型に、KOD−Plus−Mutagenesis Kitを用いたInverse PCR法によって、配列番号54、55のプライマーを使ってPuromycin耐性遺伝子内に存在する制限酵素BsiWIサイトを削除したプラスミドpEF1α−SNAP26m−Pur2・N2、pEF1α−SNAP26m−Pur2・N6およびpEF1α−SNAP26m−Pur2・N8を構築した。続いて、pEF1α−SNAP26m−Pur2・N2、pEF1α−SNAP26m−Pur2・N6およびpEF1α−SNAP26m−Pur2・N8から制限酵素EcoRI、BsiWIで、SNAP26m−pA−SV40 プロモーター−Pur2・N2−pA、SNAP26m−pA−SV40 プロモーター−Pur2・N6−pA、SNAP26m−pA−SV40 プロモーター−Pur2・N8−pAを切出し、pEF1α−KOD3G8HC−1.1k/1.1kの制限酵素EcoRI、BsiWIサイトに導入してpEF1α−SNAP26m−Pur・N2−1.1k/1.1k、pEF1α−SNAP26m−Pur・N6−1.1k/1.1k、pEF1α−SNAP26m−Pur・N8−1.1k/1.1kを構築した。
【0157】
実施例2に記載の方法でトランスフェクション用のCHO−K1細胞を準備した。一方、pEF1α−SNAP26m−Pur−1.1k/1.1k、pEF1α−SNAP26m−Pur・N2−1.1k/1.1k、pEF1α−SNAP26m−Pur・N4−1.1k/1.1k、pEF1α−SNAP26m−Pur・N6−1.1k/1.1k及びpEF1α−SNAP26m−Pur・N8−1.1k/1.1kの各SNAP26m発現コンストラクトを制限酵素AhdIでリニアライズした。トランスフェクションは、実施例5に記載の方法で行い、トランスフェクションの翌日、培地を除去し、2.5g/l−Trypsin・1mmol/l−EDTA Solutionで処理して細胞を分散させ、90mmペトリディッシュに移し、10μg/ml Puromycinを含むHam’s F12+10%FBS培地中で1週間選択培養を行った。選択培養中、3〜4日ごとに培地を交換した。選択培養終了後、実施例2に記載の方法で、細胞集団のSNAP26mの発現強度を解析した。
【0158】
図43は、薬剤選択後のpEF1α−SNAP26m−Pur−1.1k/1.1k、pEF1α−SNAP26m−Pur・N2−1.1k/1.1k、pEF1α−SNAP26m−Pur・N4−1.1k/1.1k、pEF1α−SNAP26m−Pur・N6−1.1k/1.1k及びpEF1α−SNAP26m−Pur・N8−1.1k/1.1kで形質転換された細胞集団のFACS解析の結果を示す。なお、実施例19までの条件でFACS解析に供したところ、細胞の蛍光強度FL1が強すぎて、高発現細胞の割合を正確に算出することができなかった。そのため、フローサイトメーターの蛍光感度を下げて再検討した結果を図43に示している。また、図44に蛍光強度FL1が1000以上の細胞の割合のプロットを示す。この結果、CHO5Δ3−3で発現カセットを挟んだ場合でも、繰り返し配列数の多い、N6あるいはN8が挿入されたプラスミドのpEF1α−SNAP26m−Pur・N6−1.1k/1.1kあるいはpEF1α−SNAP26m−Pur・N8−1.1k/1.1kで形質転換したとき、pEF1α−SNAP26m−Pur・N4−1.1k/1.1kの場合と比較して、SNAPm高発現細胞の割合が更に多く、高生産性細胞の選択効率が飛躍的に向上することが確認された。
【実施例23】
【0159】
抗体生産系におけるmRNA不安定化配列と遺伝子発現安定化エレメントとの組み合わせの効果
(1)pEF1α−KOD3G8HC−Neo・N4−1.1k/1.1kの構築
抗体生産系における、mRNA不安定化配列(N4配列)と実施例15で遺伝子発現安定化能の認められた遺伝子発現安定化エレメントCHO5Δ3−3との組合せ効果の検討に用いるため、図45に示すスキームに従って、pEF1α−KOD3G8HC−Neo・N4−1.1k/1.1kを構築した。即ち、実施例20に記載のpEF1α−SNAP26m−Neo・N4−1.1k/1.1kを制限酵素EcoRI、NotIで処理し、SNAP26m遺伝子を切出した。一方、実施例4に記載のpEF1α−KOD3G8HCから配列番号56,57のプライマーを用いてPCRで増幅し、制限酵素EcoRI、NotIで処理して、調製した抗KOD抗体の重鎖遺伝子を、pEF1α−SNAP26m−Neo・N4−1.1k/1.1kのEcoRI、NotIサイトに導入して、pEF1α−KOD3G8HC−Neo・N4−1.1k/1.1kを構築した。
【実施例24】
【0160】
(2)pEF1α−KOD3G8LC−Hyg−1.1k/1.1kの構築
抗体生産系における、N4配列と実施例15で遺伝子発現安定化能の認められたCHO5Δ3−3との組合せ効果の検討に用いるため、図46に示すスキームに従って、pEF1α−KOD3G8LC−Hyg−1.1k/1.1kを構築した。即ち、まず、実施例6の途中で構築されたpEF1α−KOD3G8LC−Hyg−RE2の発現カセット上流の制限酵素NheI、BglIIサイトに配列番号50、51のプライマーセットを使ってPCRで増幅した遺伝子発現安定化エレメントCHO5Δ3−3(以下、コンストラクト中では1.1kと表記)を挿入して、プラスミドpEF1α−KOD3G8LC−Hyg−1.1kを構築した。更にプラスミドpEF1α−KOD3G8LC−1.1kの発現カセット下流の制限酵素BsiWI、XhoIサイトに配列番号52、53のプライマーセットを用いてPCRで増幅した遺伝子発現安定化エレメントCHO5Δ3−3を挿入して、pEF1α−KOD3G8LC−Hyg−1.1k/1.1kを構築した。
【実施例25】
【0161】
(3)pEF1α−KOD3G8LC−Hyg・N4−1.1k/1.1kの構築
抗体生産系における、N4配列と実施例15で遺伝子発現安定化能の認められたCHO5Δ3−3との組合せ効果の検討に用いるため、図47に示すスキームに従って、pEF1α−KOD3G8LC−Hyg・N4−1.1k/1.1kを構築した。即ち、実施例18に記載のpEF1α−SNAP26m−Hyg・N4−1.1k/1.1kを制限酵素MluI、NotIで処理し、SNAP26m遺伝子を切出した。一方、実施例6に記載のpEF1α−KOD3G8LCから配列番号56,57のプライマーを用いてPCRで増幅し、制限酵素MluI、NotIで処理して、調製した抗KOD抗体の軽鎖遺伝子を、pEF1α−SNAP26m−Hyg・N4−1.1k/1.1kの制限酵素MluI、NotIサイトに導入して、pEF1α−KOD3G8LC−Hyg・N4−1.1k/1.1kを構築した。
【実施例26】
【0162】
(4)pEF1α−KOD3G8HC−Pur・N4−1.1k/1.1kの構築
抗体生産系における、N4配列と実施例15で遺伝子発現安定化能の認められたCHO5Δ3−3との組合せ効果の検討に用いるため、図48に示すスキームに従って、pEF1α−KOD3G8HC−Pur・N4−1.1k/1.1kを構築した。即ち、実施例16に記載のpEF1α−SNAP26m−Pur・N4−1.1k/1.1kを制限酵素EcoRI、NotIで処理し、SNAP26m遺伝子を切出した。一方、実施例4に記載のpEF1α−KOD3G8HCから配列番号56,57のプライマーを用いてPCRで増幅し、制限酵素EcoRI、NotIで処理して、調製した抗KOD抗体の重鎖遺伝子を、pEF1α−SNAP26m−Pur・N4−1.1k/1.1kの制限酵素EcoRI、NotIサイトに導入して、pEF1α−KOD3G8HC−Pur・N4−1.1k/1.1kを構築した。
【実施例27】
【0163】
(5)抗KOD抗体発現系での、N4配列とCHO5Δ3−3との組合せ効果のポリクローンにおける検討
抗KOD抗体の重鎖(HC)発現コンストラクトである、pEF1α−KOD3G8HC−RE2(以下、−/Neoと記載)、pEF1α−KOD3G8HC−1.1k/1.1k(以下、1.1k/Neoと記載)、pEF1α−KOD3G8HC−Neo・N4−1.1k/1.1k(以下、1.1k/Neo・N4と記載)、pEF1α−KOD3G8HC−Pur・N4−1.1k/1.1k(以下、1.1k/Pur・N4と記載)、および、抗KOD抗体の軽鎖(LC)発現コンストラクトである、pEF1α−KOD3G8LC−Hyg−RE2(以下、−/Hygと記載)、pEF1α−KOD3G8LC−Hyg−1.1k/1.1k(以下、1.1k/Hygと記載)、pEF1α−KOD3G8LC−Hyg・N4−1.1k/1.1k(以下、1.1k/Hyg・N4と記載)をそれぞれ制限酵素AhdIでリニアライズ(直鎖状化)した。そして、重鎖および軽鎖発現コンストラクトを、
(1)遺伝子発現安定化エレメントを含まないもの(−/Neoと−/Hyg)、
(2)遺伝子発現安定化エレメントCHO5Δ3−3を抗体発現カセットの上流および下流の両方に挿入したもの(1.1k/Neoと1.1k/Hyg)、
(3)遺伝子発現安定化エレメントCHO5Δ3−3を抗体発現カセットの上流および下流の両方に挿入し、薬剤耐性遺伝子(重鎖:Neo、軽鎖:Hyg)にmRNA不安定化配列(N4配列)を付加したもの(1.1k/Neo・N4と1.1k/Hyg・N4)、
(4)遺伝子発現安定化エレメントCHO5Δ3−3を抗体発現カセットの上流および下流の両方に挿入し、薬剤耐性遺伝子(重鎖:Pur、軽鎖:Hyg)にmRNA不安定化配列(N4配列)を付加したもの(1.1k/Pur・N4と1.1k/Hyg・N4)
のそれぞれについて、抗体の重鎖発現コンストラクトと軽鎖発現コンストラクトの重量比が1:1となるように混合して混合プラスミドを調製した。
【0164】
前記の混合プラスミド2μgを、前日に6ウェルプレートに播種しておいたCHO−K1細胞にトランスフェクションし、24時間インキュベートした。翌日、培地を除去し、2.5g/l−Trypsin・1mmol/l−EDTA Solutionで処理して細胞を分散させ、90mmペトリディッシュに移し、ジェネティシン G418およびHygroGold、あるいは、PuromycinおよびHygroGoldを含むHam’s F12+10%FBS培地中で2週間選択培養を行った。選択培養中、3〜4日ごとに培地を交換した。遺伝子発現安定化エレメントを含まないもの(−/Neo,−/Hyg)、および、遺伝子発現安定化エレメントCHO5Δ3−3を発現カセットの上流および下流の両方に挿入したもの(1.1k/Neo,1.1k/Hyg)では、600μg/mlジェネティシン G418・400μg/ml HygroGoldにて、選択培養をした。遺伝子発現安定化エレメントCHO5Δ3−3を発現カセットの上流および下流の両方に挿入し、薬剤耐性遺伝子(H鎖:Neo、L鎖:Hyg)にmRNA不安定化配列(N4配列)を付加したもの(1.1k/Neo・N4,1.1k/Hyg・N4)では、400μg/mlジェネティシン G418・200μg/ml HygroGoldにて選択培養をした。遺伝子発現安定化エレメントCHO5Δ3−3を発現カセットの上流および下流の両方に挿入し、薬剤耐性遺伝子(重鎖:Pur、軽鎖:Hyg)にmRNA不安定化配列(N4配列)を付加したもの(1.1k/Pur・N4,1.1k/Hyg・N4)では、7.5μg/ml Puromycin・400μg/ml HygroGoldにて選択培養をした。選択培養終了後、2.5g/l−Trypsin・1mmol/l−EDTA Solutionで処理して細胞を分散し、1ウェル当たり1.84×105細胞を6ウェルプレートに播き込んだ。3日間培養後、培養上清を回収し、ポリクローンでのKOD3G8抗体の生産量をELISA(パイロコッカス・コダカラエンシスKOD1由来のDNAポリメラーゼを固相化したもの)で測定した。
【0165】
具体的には、30μg/ml濃度のKOD1由来のDNAポリメラーゼ抗原を固相化したELISAプレートに、培養上清を添加し、35℃で2時間インキュベートした。次に、PBS−T(0.1%Tween20含有リン酸緩衝生理食塩水)で3回洗浄し、10000倍希釈した抗マウス抗体−HRP(DAKO社製)を50μl添加して35℃で1時間インキュベートした後、さらにPBS−Tで3回洗浄し、TMB+(3,3’,5,5’−tetramethylbenzidine;DAKO社製)で5分間反応させた。50μlの1N硫酸を加えて反応停止後、プレートリーダー(製品名;SPECTRA
CLASSIC、TECAN Auctria社製)(主波長450nm、副波長620nm)で吸光度を測定した。
【0166】
標準品から作成した検量線をもとに抗体濃度を算出した。なお、標準品として、Thermococcus kodakaraensis KOD1株由来のDNAポリメラーゼに特異的な抗体を産生するマウスハイブリドーマ細胞系3G8(入手先:独立行政法人産業技術総合研究所 特許生物寄託センター、寄託番号:FERM BP−6056)から得られた抗体を用いた。結果を図49に示す。図49から明らかな通り、遺伝子発現安定化エレメントを含まないもの(−/Neo,−/Hyg)と比較して、遺伝子発現安定化エレメントCHO5Δ3−3を発現カセット上流および下流の両方に挿入したもの(1.1k/Neo,1.1k/Hyg)では抗体生産量が2倍程度上昇したが、遺伝子発現安定化エレメントCHO5Δ3−3を発現カセットの上流および下流の両方に挿入し、薬剤耐性遺伝子(H鎖:Neo、L鎖:Hyg)にmRNA不安定化配列(N4配列)を付加したもの(1.1k/Neo・N4,1.1k/Hyg・N4)では抗体生産量が更に上昇し、約3倍に増加した。また、遺伝子発現安定化エレメントCHO5Δ3−3を発現カセットの上流および下流の両方に挿入し、薬剤耐性遺伝子にmRNA不安定化配列(N4配列)を付加したもので、重鎖発現コンストラクトの薬剤耐性遺伝子をNeoからPurに変更したとき、抗体生産量が顕著に増加し、遺伝子発現安定化エレメント及びmRNA不安定化配列を有さない発現コンストラクトに比べて、約5倍の抗体生産量を示した。
【実施例28】
【0167】
(6)抗KOD抗体発現系での、N4配列とCHO5Δ3−3との組合せ効果のモノクローンにおける検討
続いて、遺伝子発現安定化エレメントを含まないもの(−/Neo,−/Hyg)、遺伝子発現安定化エレメントCHO5Δ3−3を発現カセットの上流および下流の両方に挿入したもの(1.1k/Neo,1.1k/Hyg)、遺伝子発現安定化エレメントCHO5Δ3−3を発現カセットの上流および下流の両方に挿入し、薬剤耐性遺伝子(H鎖:Neo、L鎖:Hyg)にmRNA不安定化配列(N4配列)を付加したもの(1.1k/Neo・N4,1.1k/Hyg・N4)、遺伝子発現安定化エレメントCHO5Δ3−3を発現カセットの上流および下流の両方に挿入し、薬剤耐性遺伝子(H鎖:Pur、L鎖:Hyg)にmRNA不安定化配列(N4配列)を付加したもの(1.1k/Pur・N4,1.1k/Hyg・N4)のそれぞれについて、限界希釈を行い、モノクローンでの生産性の比較を行った。
【0168】
具体的には、実施例27のKOD3G8抗体を安定導入したポリクローンの細胞を2.5g/l−Trypsin・1mmol/l−EDTA Solutionで処理して細胞を分散し、0.75細胞/ウェルの濃度で96ウェルプレートに播き込んだ。2週間培養後、培養上清を回収し、KOD3G8抗体の生産量を実施例27と同様の方法でELISAにて測定した。標準品から作成した検量線をもとに各クローンの抗体生産量を算出し、遺伝子発現安定化エレメントを含まないもの(−/Neo,−/Hyg)、遺伝子発現安定化エレメントCHO5Δ3−3を発現カセットの上流および下流の両方に挿入したもの(1.1k/Neo,1.1k/Hyg)、遺伝子発現安定化エレメントCHO5Δ3−3を発現カセットの上流および下流の両方に挿入し、薬剤耐性遺伝子(H鎖:Neo、L鎖:Hyg)にmRNA不安定化配列(N4配列)を付加したもの(1.1k/Neo・N4,1.1k/Hyg・N4)、遺伝子発現安定化エレメントCHO5Δ3−3を発現カセットの上流および下流の両方に挿入し、薬剤耐性遺伝子(H鎖:Pur、L鎖:Hyg)にmRNA不安定化配列(N4配列)を付加したもの(1.1k/Pur・N4,1.1k/Hyg・N4)のそれぞれについて、発現量上位クローンを拡大培養した後、1ウェル当たり1.84×105細胞を6ウェルプレートに播き込んだ。3日間培養後、培養上清を回収し、モノクローンでのKOD3G8抗体の生産量を実施例27と同様の方法でELISAにて測定した。
【0169】
標準品から作成した検量線をもとに算出した各クローンの抗体生産量をプロットしたグラフを図50に示す。その結果、ポリクローンでの生産性を比較した場合と同様、遺伝子発現安定化エレメントを含まないもの(−/Neo,−/Hyg)と比較して、遺伝子発現安定化エレメントCHO5Δ3−3を抗体発現カセット上流および下流の両方に挿入したもの(1.1k/Neo,1.1k/Hyg)で抗体生産量が上昇したが、被験配列CHO5Δ3−3を発現カセットの上流および下流の両方に挿入し、薬剤耐性遺伝子(H鎖:Neo、L鎖:Hyg)にmRNA不安定化配列(N4配列)を付加したもの(1.1k/Neo・N4,1.1k/Hyg・N4)で抗体生産量が更に上昇していた。また、遺伝子発現安定化エレメントCHO5Δ3−3を抗体発現カセットの上流および下流の両方に挿入し、薬剤耐性遺伝子にmRNA不安定化配列(N4配列)を付加したものでは、重鎖発現コンストラクトの薬剤耐性遺伝子をNeo・N4からPur・N4に変更したとき、各細胞の抗体生産量が著しく増加し、高い抗体生産性を示すクローンの割合が顕著に大きくなった。この結果から抗体生産系における高生産性細胞の選択効率が飛躍的に向上することが確認された。
【0170】
さらに、効果を確認するため、遺伝子発現安定化エレメントもmRNA不安定化配列も含まないもの(−/Neo,−/Hyg)と、高い抗体生産量を示した遺伝子発現安定化エレメントCHO5Δ3−3を含み、さらに薬剤耐性遺伝子(H鎖:Pur、L鎖:Hyg)にmRNA不安定化配列(N4配列)を付加したもの(1.1k/Pur・N4,1.1k/Hyg・N4)について、前記96ウェルプレートにおける2週間培養後のELISA測定によって算出されたKOD3G8抗体の生産量を再度解析し、度数分布としてプロットした結果を図51に示す。この結果、KOD3G8生産量に対するクローンの分布は、1.1k/Pur・N4,1.1k/Hyg・N4において有意に高生産側へシフトしており、極めて高い抗体生産性を有する細胞の割合が顕著に増大している。遺伝子発現安定化エレメントとmRNA不安定化配列を含む薬剤選択マーカー遺伝子発現カセットの利用が、その相乗効果により、高生産細胞株取得に極めて大きく寄与していることが確認された。
【実施例29】
【0171】
無血清馴化CHO細胞を用いた、抗KOD抗体発現系での、N4配列とCHO5Δ3−3との組合せ効果のポリクローンにおける検討
EX−CELL CD CHO Serum−Free Medium for CHO Cells, Chemically Defind培地(SAFC Biosciences社)を用いて、添付のプロトコールに従って、CHO−K1細胞を無血清馴化した。無血清馴化したCHO−K1細胞2.5X105cells/mlになるようにOpti−MEM I Reduced−Serum Medium(GIBCO社製)に懸濁し、超低接着表面24ウェルプレート(Corning Life Science社)の4ウェルに1mlずつ播種した。
【0172】
抗KOD抗体の重鎖(HC)発現コンストラクトである、pEF1α−KOD3G8HC−1.1k/1.1k(以下、1.1k/Neoと記載)、pEF1α−KOD3G8HC−Pur・N4−1.1k/1.1k(以下、1.1k/Pur・N4と記載)、および、抗KOD抗体の軽鎖(LC)発現コンストラクトである、pEF1α−KOD3G8LC−Hyg−1.1k/1.1k(以下、1.1k/Hygと記載)、pEF1α−KOD3G8LC−Hyg・N4−1.1k/1.1k(以下、1.1k/Hyg・N4と記載)をそれぞれ制限酵素AhdIでリニアライズ(直鎖状化)した。そして、重鎖および軽鎖発現コンストラクトを、
(1)遺伝子発現安定化エレメントCHO5Δ3−3を抗体発現カセットの上流および下流の両方に挿入したもの(1.1k/Neoと1.1k/Hyg)、
(2)遺伝子発現安定化エレメントCHO5Δ3−3を抗体発現カセットの上流および下流の両方に挿入し、薬剤耐性遺伝子(重鎖:Pur、軽鎖:Hyg)にmRNA不安定化配列(N4配列)を付加したもの(1.1k/Pur・N4と1.1k/Hyg・N4)
のそれぞれについて、抗体の重鎖発現コンストラクトと軽鎖発現コンストラクトの重量比が1:1となるように混合して混合プラスミドを調製した。
【0173】
前記の混合プラスミド各4μgをOpti−MEM I Reduced−Serum Medium68μlで希釈した。一方で、Lipofectamine2000 15μlをOpti−MEM I Reduced−Serum Medium68μlで希釈し、前記プラスミド混合液と混合し、15分間、室温で放置した。その後、半量ずつ2ウェルの細胞に加えた。
【0174】
翌日、細胞を回収して800gX3分間遠心して上清を除去し、4mlEX−CELL CD CHO Serum−Free Medium for CHO Cells, Chemically Defind培地に懸濁し、遺伝子発現安定化エレメントCHO5Δ3−3を発現カセットの上流および下流の両方に挿入したもの(1.1k/Neo,1.1k/Hyg)には400μg/ml G418、200μg/ml HygroGold、遺伝子発現安定化エレメントCHO5Δ3−3を発現カセットの上流および下流の両方に挿入し、薬剤耐性遺伝子(H鎖:Pur、L鎖:Hyg)にmRNA不安定化配列(N4配列)を付加したもの(1.1k/Pur・N4,1.1k/Hyg・N4)には7.5μg/ml Puromycin、200μg/ml HygroGoldの抗生物質を加え、選択培養した。
【0175】
選択培養終了後、2×105cells/mlで2mlずつ6ウェルプレートに播き込んだ。3日間培養後、培養上清を回収し、ポリクローンでのKOD3G8抗体の生産量を実施例27と同様の方法でELISAにて測定した。標準品から作成した検量線をもとに算出した抗体生産量をプロットしたグラフを図52に示す。この結果、無血清馴化CHO−K1細胞を用いても1.1k/Pur・N4,1.1k/Hyg・N4における生産性は、実施例27の無血清馴化していないCHO−K1細胞の高い生産性を再現することが確認された。一般にバイオ医薬品などの工業生産には、高生産細胞をスクリーニングした後、1〜2ヶ月程度かけて無血清馴化するが、あらかじめ無血清馴化された細胞に遺伝子導入することによって無血清馴化工程を省略できることが期待される。本実施例の結果より、無血清馴化CHO細胞においても本発明の遺伝子発現安定化エレメントとmRNA不安定化配列を含む薬剤選択マーカー遺伝子発現カセットの利用によって生産性の高い細胞を取得できることが確認され、生産性の高い細胞をより短時間に樹立できることが確認された。
【実施例30】
【0176】
抗KOD抗体発現系での、2コンストラクト抗体発現系とシングルベクター抗体発現系の比較
(1)pEF1α−KOD3G8LC,HC−Pur・N4−1.1k/1.1kの構築
次に、H鎖発現カセットとL鎖発現カセットを一つのベクター上に配置した場合でも高生産細胞を取得可能か検討するため、図53に示すスキームに従ってpEF1α−KOD3G8LC,HC−Pur・N4−1.1k/1.1kを構築した。
【0177】
なお、H鎖(重鎖)発現カセットとL鎖(軽鎖)発現カセットとをそれぞれ別々のベクターに配置した場合を「2コンストラクト抗体発現系」もしくは単に「2コンストラクト」といい、H鎖発現カセットとL鎖発現カセットを一つのベクター上に配置した場合を「シングルベクター抗体発現系」もしくは単に「シングルベクター」という。
【0178】
まず、pEF1α−KOD3G8HC−Pur・N4−1.1k/1.1kにL鎖発現カセット挿入用の制限酵素サイトを挿入したpEF1α−KOD3G8HC−Pur・N4−1.1k/1.1k−Sを構築した。即ち、実施例26に記載のpEF1α−KOD3G8HC−Pur・N4−1.1k/1.1kを制限酵素BglII、MluIで処理し、EF−1αプロモーターを切り出した。一方、実施例16の途中で構築したpEF1α−KOD3G8HC−1.1kから配列番号58、59のプライマーを用いてPCRで増幅し、BglII、MluIで処理して調製したEF−1αプロモーターを、pEF1α−KOD3G8HC−Pur・N4−1.1k/1.1kのBglII、MluIサイトに導入して、EF−1αプロモーター上流にBglII、SphI、SpeIサイトを挿入したpEF1α−KOD3G8HC−Pur・N4−1.1k/1.1k−Sを構築した。
【0179】
続いて、pEF1α−KOD3G8HC−Pur・N4−1.1k/1.1k−SをBglII、SpeIで処理した。一方、実施例6の途中で構築したpEF1α−KOD3G8LC−RE2から配列番号60、61のプライマーを用いてPCRで増幅し、BglII、SpeIで処理して調製した抗KOD抗体のL鎖発現カセットを、pEF1α−KOD3G8HC−Pur・N4−1.1k/1.1k−SのBglII、SpeIサイトに導入して、シングルベクターpEF1α−KOD3G8LC,HC−Pur・N4−1.1k/1.1kを構築した。
【実施例31】
【0180】
(2)抗KOD抗体発現系での、ポリクローンにおける2コンストラクト抗体発現系とシングルベクター抗体発現系の比較
シングルベクター抗体発現系でも、2コンストラクト抗体発現系と同様に抗体生産量が高いクローンを取得可能か検討した。2コンストラクト抗体発現系として、実施例27および28で最も良好な結果が得られた、遺伝子発現安定化エレメントCHO5Δ3−3を抗体発現カセットの上流および下流の両方に挿入し、薬剤耐性遺伝子(重鎖:Pur、軽鎖:Hyg)にmRNA不安定化配列(N4配列)を付加したもの(1.1k/Pur・N4と1.1k/Hyg・N4)について抗体の重鎖発現コンストラクトと軽鎖発現コンストラクトの重量比が1:1となるように混合したプラスミド(制限酵素AhdIでリニアライズ済)をトランスフェクションに用いた。また、シングルベクター抗体発現系として、実施例30で構築したpEF1α−KOD3G8LC,HC−Pur・N4−1.1k/1.1kを制限酵素AhdIでリニアライズし、トランスフェクションに用いた。各々のプラスミド2μgを、実施例2に記載の方法で準備しておいたCHO−K1細胞にトランスフェクションし、24時間インキュベートした。翌日、培地を除去し、2.5g/l−Trypsin・1mmol/l−EDTA Solutionで処理して細胞を分散させ、90mmペトリディッシュに移し、2コンストラクト発現系では7.5μg/ml Puromycin・400μg/ml HygroGoldを含むHam’s F12+10%FBS培地中で、シングルベクター系では10μg/ml Puromycinを含むHam’s F12+10%FBS培地中で、2週間選択培養を行った。選択培養中、3〜4日ごとに培地を交換した。選択培養終了後、2.5g/l−Trypsin・1mmol/l−EDTA Solutionで処理して細胞を分散し、1ウェル当たり1.84×105細胞を6ウェルプレートに播き込んだ。3日間培養後、培養上清を回収し、ポリクローンでのKOD3G8抗体の生産量を実施例27と同様の方法でELISAにて測定した。
【0181】
標準品から作成した検量線をもとに抗体濃度を算出した。結果を図54に示す。その結果、2コンストラクト抗体発現系とシングルベクター抗体発現系間で、抗体生産量に差異はほとんど見られないとの結果であった。
【実施例32】
【0182】
(3)抗KOD抗体発現系での、モノクローンにおける2コンストラクト抗体発現系とシングルベクター抗体発現系の比較
続いて、2コンストラクト抗体発現系、シングルベクター抗体発現系のそれぞれについて、限界希釈を行い、モノクローンでの生産性の比較を行った。
【0183】
具体的には、実施例31のKOD3G8抗体を安定導入したポリクローンの細胞を2.5g/l−Trypsin・1mmol/l−EDTA Solutionで処理して細胞を分散し、0.75細胞/ウェルの濃度で96ウェルプレートに播き込んだ。2週間培養後、培養上清を回収し、実施例27と同様の方法でELISAにてKOD3G8抗体の生産量を測定した。標準品から作成した検量線をもとに各クローンの抗体生産量を算出し、2コンストラクト抗体発現系、シングルベクター抗体発現系のそれぞれについて、発現量上位クローンを選択した。選択したクローンを拡大培養した後、1ウェル当たり1.84×105細胞を6ウェルプレートに播き込んだ。3日間培養後、培養上清を回収し、モノクローンでのKOD3G8抗体の生産量を実施例27と同様の方法でELISAにて測定した。
【0184】
標準品から作成した検量線をもとに算出した発現量上位5クローンの抗体生産量をプロットしたグラフを図55に示す。その結果、ポリクローンでの結果と同様、シングルベクター抗体発現系と2コンストラクト抗体発現系間で、抗体発現量はほとんど変わらないとの結果であった。このことから、シングルベクター抗体発現系でも、2コンストラクト抗体発現系と同様に抗体生産量が高いクローンを取得可能であることがわかった。
【産業上の利用可能性】
【0185】
本発明の遺伝子発現の安定化機能を有する核酸領域と弱化された薬剤遺伝子発現カセットを具備した発現ベクターは、目的タンパク質遺伝子を高度に発現する細胞を効率よく取得することができる。従って、本発明の発現ベクターを使用した細胞発現システムは、種々の動物細胞、特に哺乳類動物細胞のタンパク質の機能解析は勿論、バイオ医薬品として創薬・医療などの産業界に寄与することが大である。
【特許請求の範囲】
【請求項1】
mRNA不安定化配列を含む薬剤選択マーカー遺伝子発現カセット、少なくとも1つの遺伝子発現安定化エレメント、及び目的タンパク質の遺伝子発現カセットを有することを特徴とする発現ベクター。
【請求項2】
mRNA不安定化配列が、サイトカイン、インターロイキン、又は癌原遺伝子の3’非翻訳領域に存在するATリッチ配列に由来することを特徴とする請求項1に記載の発現ベクター。
【請求項3】
mRNA不安定化配列が、TTATTTA(A/T)(A/T)のモチーフ配列を有することを特徴とする請求項1に記載の発現ベクター。
【請求項4】
モチーフ配列が、2回以上繰り返されていることを特徴とする請求項3に記載の発現ベクター。
【請求項5】
モチーフ配列の繰り返し間に1塩基以上のスペーサー配列を含むことを特徴とする請求項4に記載の発現ベクター。
【請求項6】
mRNA不安定化配列中に1ないし数塩基の置換、挿入または欠失を含むことを特徴とする請求項2〜5のいずれか一項に記載の発現ベクター。
【請求項7】
遺伝子発現安定化エレメントがチャイニーズハムスターゲノム由来であることを特徴とする請求項1〜6のいずれかに記載の発現ベクター。
【請求項8】
遺伝子発現安定化エレメントが、以下の(a)〜(g)のいずれか一つ又はそれらの任意の組合せからなることを特徴とする請求項1〜7のいずれかに記載の発現ベクター。
(a)配列番号26で示す配列からなるDNA;
(b)配列番号26で示す配列の部分配列からなるDNAであって、配列番号26で示す配列のうち41820番目から41839番目までの塩基で示す領域の配列を含むDNA;
(c)配列番号26で示す配列の部分配列からなるDNAであって、配列番号26で示す配列のうち41821番目から41840番目までの塩基で示す領域の配列を含むDNA;
(d)配列番号26で示す配列の部分配列からなるDNAであって、配列番号26で示す配列のうち45182番目から45200番目までの塩基で示す領域の配列を含むDNA;
(e)配列番号26で示す配列の部分配列からなるDNAであって、配列番号26で示す配列のうち91094番目から91113番目までの塩基で示す領域の配列を含むDNA;
(f)配列番号26で示す配列の部分配列からなるDNAであって、宿主細胞中で外来遺伝子発現カセットと近接するように配置された際に、外来遺伝子発現カセットに含まれる外来遺伝子からの目的組換えタンパク質の発現を増大または安定化させることができるDNA;及び
(g)前記(a)〜(f)のいずれかのDNAと相補的な塩基配列からなるDNAとストリンジェントな条件下でハイブリダイズし、かつ遺伝子発現安定化機能を有するDNA
(h)前記(a)〜(g)のいずれかのDNAと相補的な塩基配列からなるDNA
【請求項9】
遺伝子発現安定化エレメントが、以下の(i)〜(k)のいずれか一つ又はそれらの任意の組合せからなることを特徴とする請求項8に記載の発現ベクター。
(i)配列番号26で示す配列のうち41601番目から46746番目までの塩基で示す領域の配列を含むDNA;
(j)(i)のDNAと相補的な塩基配列からなるDNAとストリンジェントな条件下でハイブリダイズし、かつ遺伝子発現安定化機能を有するDNA;及び
(k)前記(i)又は(j)のDNAと相補的な塩基配列からなるDNA
【請求項10】
遺伝子発現安定化エレメントが、以下の(l)〜(n)のいずれか一つ又はそれらの任意の組合せからなることを特徴とする請求項9に記載の発現ベクター。
(l)配列番号26で示す配列のうち41601番目から42700番目までの塩基で示す領域の配列を含むDNA;
(m)(l)のDNAと相補的な塩基配列からなるDNAとストリンジェントな条件下でハイブリダイズし、かつ遺伝子発現安定化機能を有するDNA;及び
(n)(l)又は(m)のDNAと相補的な塩基配列からなるDNA
【請求項11】
遺伝子発現安定化エレメントが、目的タンパク質の遺伝子発現カセットの上流に配置されていることを特徴とする、請求項1〜10のいずれかに記載の発現ベクター。
【請求項12】
遺伝子発現安定化エレメントが、目的タンパク質の遺伝子発現カセットの上流及び下流の両方に配置されていることを特徴とする、請求項1〜10のいずれかに記載の発現ベクター。
【請求項13】
遺伝子発現安定化エレメントが、目的タンパク質の遺伝子発現カセット及び薬剤選択マーカー遺伝子発現カセットの上流及び下流の両方に配置されていることを特徴とする請求項1〜10のいずれかに記載の発現ベクター。
【請求項14】
薬剤選択マーカー遺伝子が、タンパク質合成阻害系抗生物質耐性遺伝子であることを特徴とする請求項1〜13のいずれかに記載の発現ベクター。
【請求項15】
薬剤選択マーカー遺伝子が、ピューロマイシン−N−アセチルトランスフェラーゼ、ハイグロマイシン−B−ホスホトランスフェラーゼ、及びネオマイシンホスホトランスフェラーゼからなる群より選択されることを特徴とする請求項14に記載の発現カベクター。
【請求項16】
目的タンパク質の遺伝子発現カセットが、所望の目的タンパク質遺伝子を挿入するためのマルチクローニングサイトを備えていることを特徴とする請求項1〜15のいずれかに記載の発現ベクター。
【請求項17】
目的タンパク質が、抗体の重鎖及び/又は軽鎖ポリペプチドであることを特徴とする請求項1〜16のいずれかに記載の発現ベクター。
【請求項18】
請求項1〜17のいずれか一項に記載の発現ベクターで宿主細胞を形質転換して得られることを特徴とする形質転換細胞。
【請求項19】
宿主細胞が動物細胞であることを特徴とする請求項18に記載の形質転換細胞。
【請求項20】
動物細胞が哺乳類細胞であることを特徴とする請求項19に記載の形質転換細胞。
【請求項21】
哺乳類細胞がチャイニーズハムスター卵巣(CHO)細胞であることを特徴とする請求項20に記載の形質転換細胞。
【請求項22】
チャイニーズハムスター卵巣(CHO)細胞が無血清馴化されていることを特徴とする請求項21に記載の形質転換細胞。
【請求項23】
請求項18〜22のいずれか一項に記載の形質転換細胞を薬剤選択する工程を含むことを特徴とする、目的タンパク質遺伝子を高レベルで発現する細胞群を選別する方法。
【請求項24】
請求項18〜22のいずれか一項に記載の形質転換細胞からなる細胞群であって、目的タンパク質遺伝子を高レベルで発現することを特徴とする細胞群。
【請求項25】
請求項18〜22のいずれか一項に記載の形質転換細胞を用いることを特徴とする、タンパク質を生産する方法。
【請求項26】
タンパク質が、抗体であることを特徴とする請求項25に記載の方法。
【請求項27】
タンパク質が、ワクチンであることを特徴とする請求項25に記載の方法。
【請求項28】
請求項18〜22のいずれか一項に記載の形質転換細胞を用いることを特徴とする、アミノ酸を生産する方法。
【請求項29】
請求項18〜22のいずれかに記載の形質転換細胞を用いることを特徴とする、ヌクレオチドを生産する方法。
【請求項1】
mRNA不安定化配列を含む薬剤選択マーカー遺伝子発現カセット、少なくとも1つの遺伝子発現安定化エレメント、及び目的タンパク質の遺伝子発現カセットを有することを特徴とする発現ベクター。
【請求項2】
mRNA不安定化配列が、サイトカイン、インターロイキン、又は癌原遺伝子の3’非翻訳領域に存在するATリッチ配列に由来することを特徴とする請求項1に記載の発現ベクター。
【請求項3】
mRNA不安定化配列が、TTATTTA(A/T)(A/T)のモチーフ配列を有することを特徴とする請求項1に記載の発現ベクター。
【請求項4】
モチーフ配列が、2回以上繰り返されていることを特徴とする請求項3に記載の発現ベクター。
【請求項5】
モチーフ配列の繰り返し間に1塩基以上のスペーサー配列を含むことを特徴とする請求項4に記載の発現ベクター。
【請求項6】
mRNA不安定化配列中に1ないし数塩基の置換、挿入または欠失を含むことを特徴とする請求項2〜5のいずれか一項に記載の発現ベクター。
【請求項7】
遺伝子発現安定化エレメントがチャイニーズハムスターゲノム由来であることを特徴とする請求項1〜6のいずれかに記載の発現ベクター。
【請求項8】
遺伝子発現安定化エレメントが、以下の(a)〜(g)のいずれか一つ又はそれらの任意の組合せからなることを特徴とする請求項1〜7のいずれかに記載の発現ベクター。
(a)配列番号26で示す配列からなるDNA;
(b)配列番号26で示す配列の部分配列からなるDNAであって、配列番号26で示す配列のうち41820番目から41839番目までの塩基で示す領域の配列を含むDNA;
(c)配列番号26で示す配列の部分配列からなるDNAであって、配列番号26で示す配列のうち41821番目から41840番目までの塩基で示す領域の配列を含むDNA;
(d)配列番号26で示す配列の部分配列からなるDNAであって、配列番号26で示す配列のうち45182番目から45200番目までの塩基で示す領域の配列を含むDNA;
(e)配列番号26で示す配列の部分配列からなるDNAであって、配列番号26で示す配列のうち91094番目から91113番目までの塩基で示す領域の配列を含むDNA;
(f)配列番号26で示す配列の部分配列からなるDNAであって、宿主細胞中で外来遺伝子発現カセットと近接するように配置された際に、外来遺伝子発現カセットに含まれる外来遺伝子からの目的組換えタンパク質の発現を増大または安定化させることができるDNA;及び
(g)前記(a)〜(f)のいずれかのDNAと相補的な塩基配列からなるDNAとストリンジェントな条件下でハイブリダイズし、かつ遺伝子発現安定化機能を有するDNA
(h)前記(a)〜(g)のいずれかのDNAと相補的な塩基配列からなるDNA
【請求項9】
遺伝子発現安定化エレメントが、以下の(i)〜(k)のいずれか一つ又はそれらの任意の組合せからなることを特徴とする請求項8に記載の発現ベクター。
(i)配列番号26で示す配列のうち41601番目から46746番目までの塩基で示す領域の配列を含むDNA;
(j)(i)のDNAと相補的な塩基配列からなるDNAとストリンジェントな条件下でハイブリダイズし、かつ遺伝子発現安定化機能を有するDNA;及び
(k)前記(i)又は(j)のDNAと相補的な塩基配列からなるDNA
【請求項10】
遺伝子発現安定化エレメントが、以下の(l)〜(n)のいずれか一つ又はそれらの任意の組合せからなることを特徴とする請求項9に記載の発現ベクター。
(l)配列番号26で示す配列のうち41601番目から42700番目までの塩基で示す領域の配列を含むDNA;
(m)(l)のDNAと相補的な塩基配列からなるDNAとストリンジェントな条件下でハイブリダイズし、かつ遺伝子発現安定化機能を有するDNA;及び
(n)(l)又は(m)のDNAと相補的な塩基配列からなるDNA
【請求項11】
遺伝子発現安定化エレメントが、目的タンパク質の遺伝子発現カセットの上流に配置されていることを特徴とする、請求項1〜10のいずれかに記載の発現ベクター。
【請求項12】
遺伝子発現安定化エレメントが、目的タンパク質の遺伝子発現カセットの上流及び下流の両方に配置されていることを特徴とする、請求項1〜10のいずれかに記載の発現ベクター。
【請求項13】
遺伝子発現安定化エレメントが、目的タンパク質の遺伝子発現カセット及び薬剤選択マーカー遺伝子発現カセットの上流及び下流の両方に配置されていることを特徴とする請求項1〜10のいずれかに記載の発現ベクター。
【請求項14】
薬剤選択マーカー遺伝子が、タンパク質合成阻害系抗生物質耐性遺伝子であることを特徴とする請求項1〜13のいずれかに記載の発現ベクター。
【請求項15】
薬剤選択マーカー遺伝子が、ピューロマイシン−N−アセチルトランスフェラーゼ、ハイグロマイシン−B−ホスホトランスフェラーゼ、及びネオマイシンホスホトランスフェラーゼからなる群より選択されることを特徴とする請求項14に記載の発現カベクター。
【請求項16】
目的タンパク質の遺伝子発現カセットが、所望の目的タンパク質遺伝子を挿入するためのマルチクローニングサイトを備えていることを特徴とする請求項1〜15のいずれかに記載の発現ベクター。
【請求項17】
目的タンパク質が、抗体の重鎖及び/又は軽鎖ポリペプチドであることを特徴とする請求項1〜16のいずれかに記載の発現ベクター。
【請求項18】
請求項1〜17のいずれか一項に記載の発現ベクターで宿主細胞を形質転換して得られることを特徴とする形質転換細胞。
【請求項19】
宿主細胞が動物細胞であることを特徴とする請求項18に記載の形質転換細胞。
【請求項20】
動物細胞が哺乳類細胞であることを特徴とする請求項19に記載の形質転換細胞。
【請求項21】
哺乳類細胞がチャイニーズハムスター卵巣(CHO)細胞であることを特徴とする請求項20に記載の形質転換細胞。
【請求項22】
チャイニーズハムスター卵巣(CHO)細胞が無血清馴化されていることを特徴とする請求項21に記載の形質転換細胞。
【請求項23】
請求項18〜22のいずれか一項に記載の形質転換細胞を薬剤選択する工程を含むことを特徴とする、目的タンパク質遺伝子を高レベルで発現する細胞群を選別する方法。
【請求項24】
請求項18〜22のいずれか一項に記載の形質転換細胞からなる細胞群であって、目的タンパク質遺伝子を高レベルで発現することを特徴とする細胞群。
【請求項25】
請求項18〜22のいずれか一項に記載の形質転換細胞を用いることを特徴とする、タンパク質を生産する方法。
【請求項26】
タンパク質が、抗体であることを特徴とする請求項25に記載の方法。
【請求項27】
タンパク質が、ワクチンであることを特徴とする請求項25に記載の方法。
【請求項28】
請求項18〜22のいずれか一項に記載の形質転換細胞を用いることを特徴とする、アミノ酸を生産する方法。
【請求項29】
請求項18〜22のいずれかに記載の形質転換細胞を用いることを特徴とする、ヌクレオチドを生産する方法。
【図1】

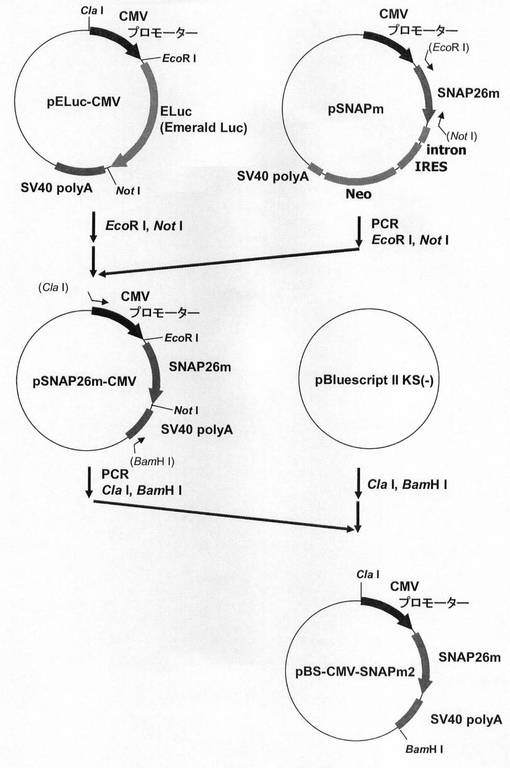
【図2】
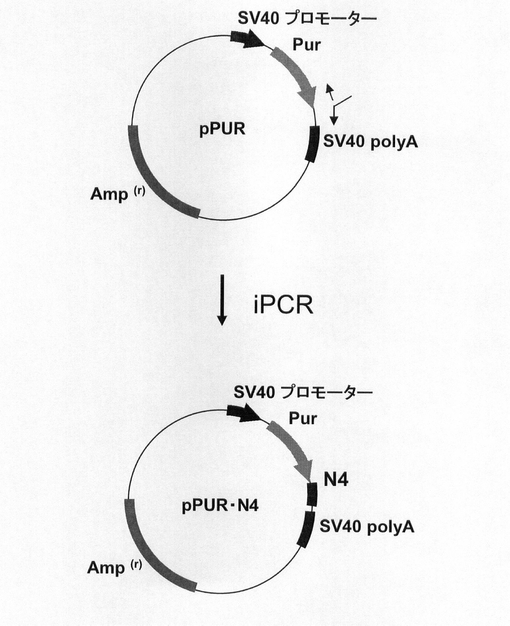
【図3】
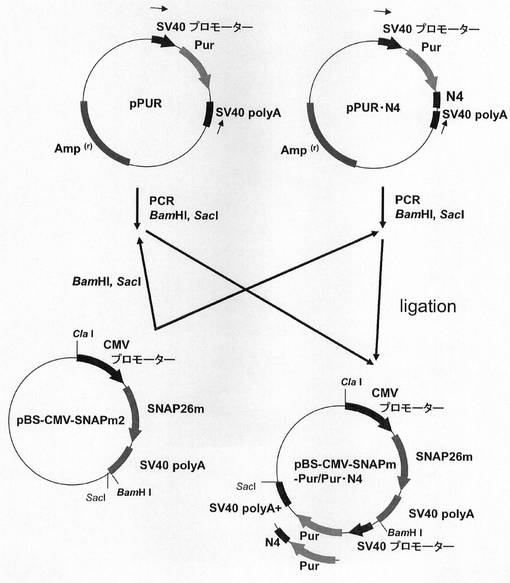
【図4】
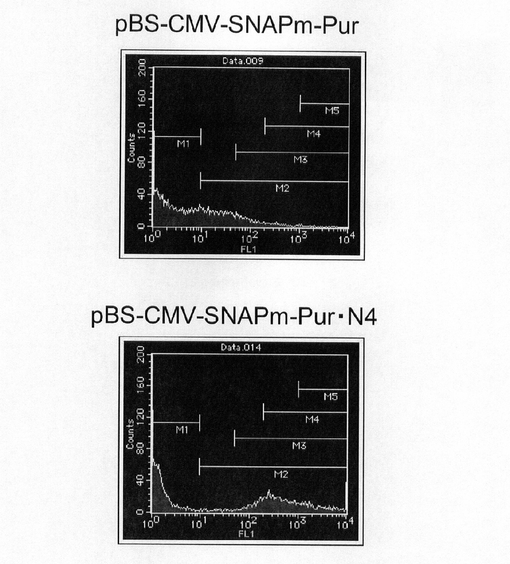
【図5】
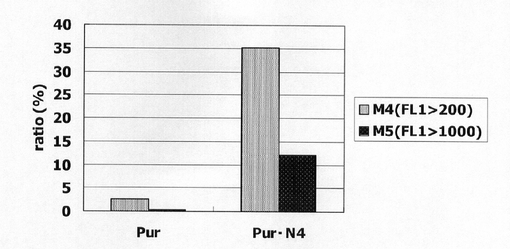
【図6】
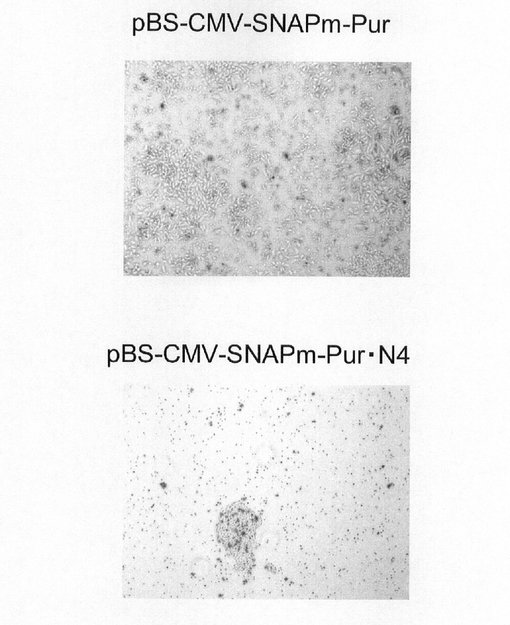
【図7】
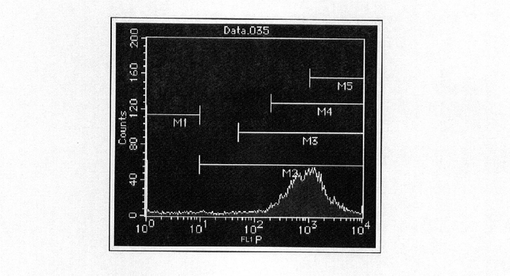
【図8】
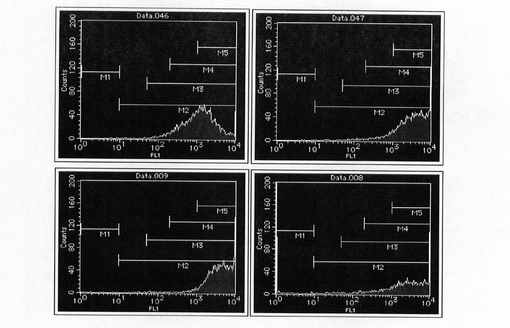
【図9】
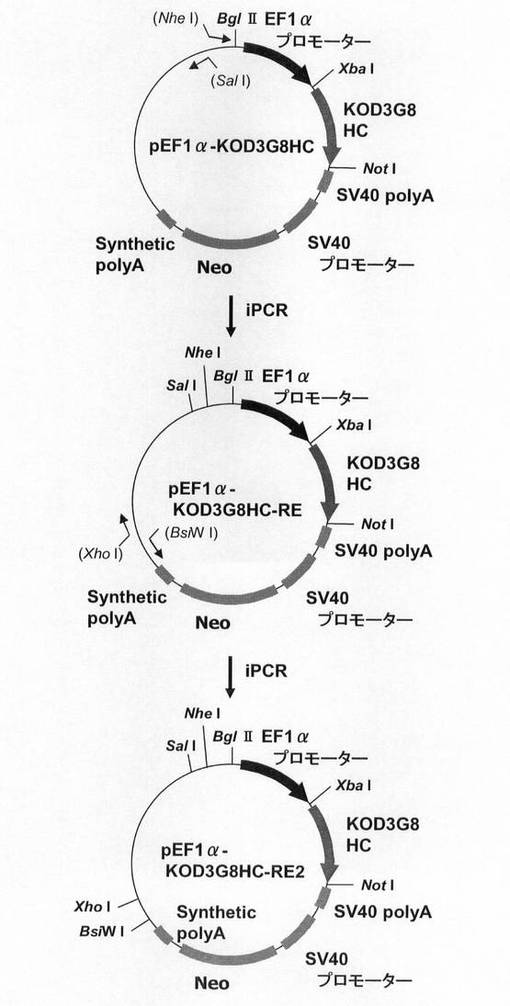
【図10】
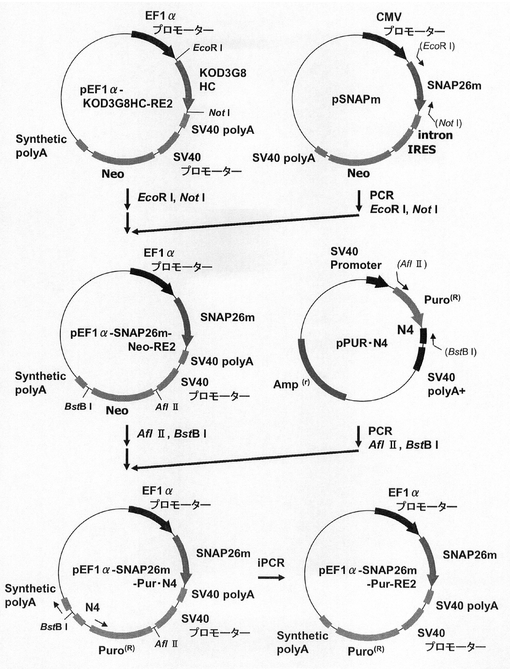
【図11】
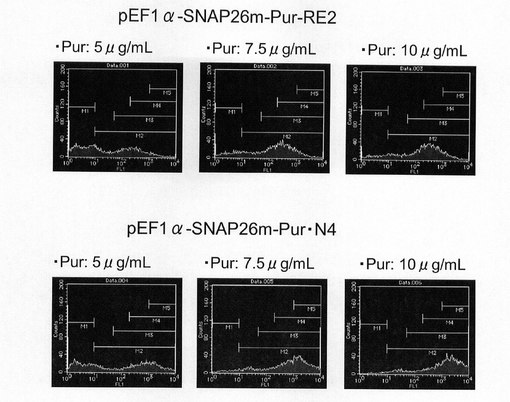
【図12】
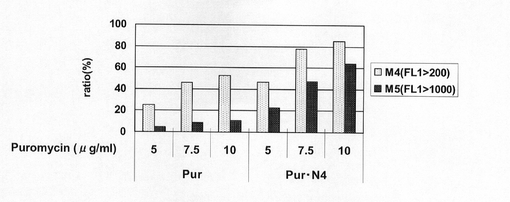
【図13】
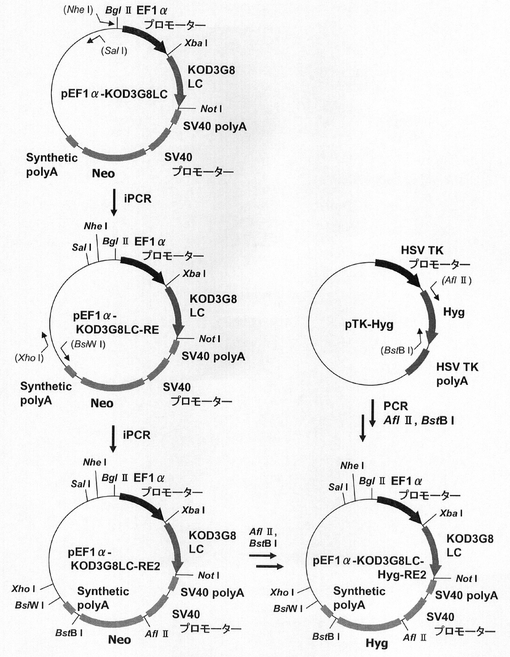
【図14】
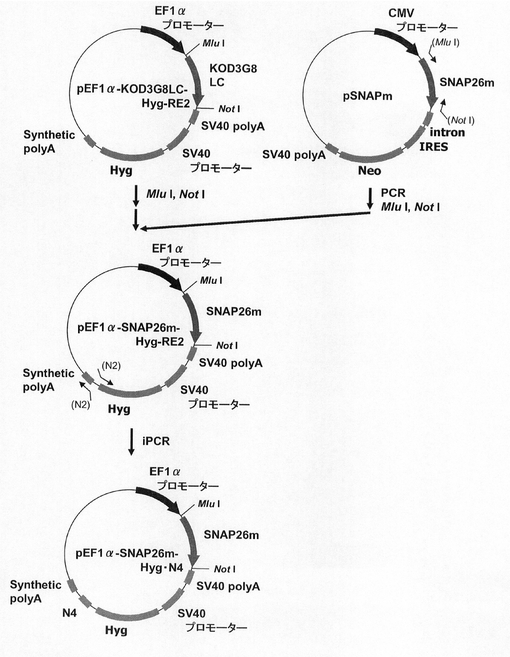
【図15】
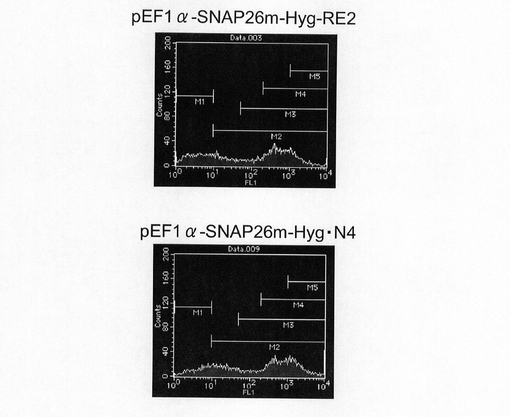
【図16】
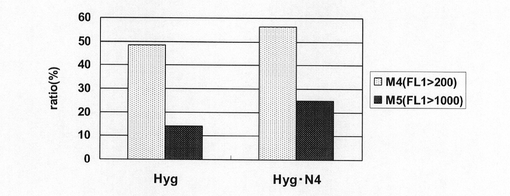
【図17】
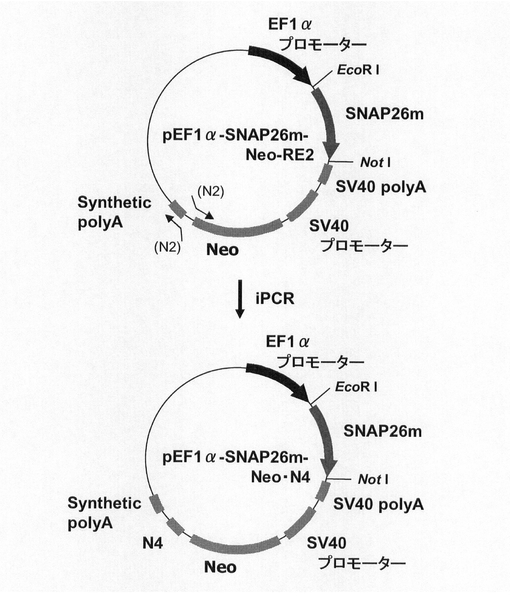
【図18】
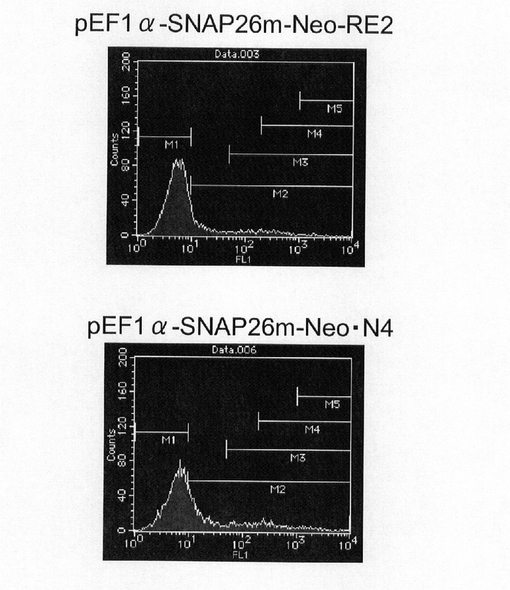
【図19】
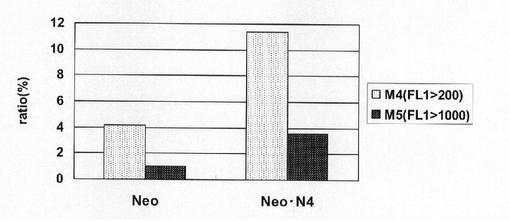
【図20】
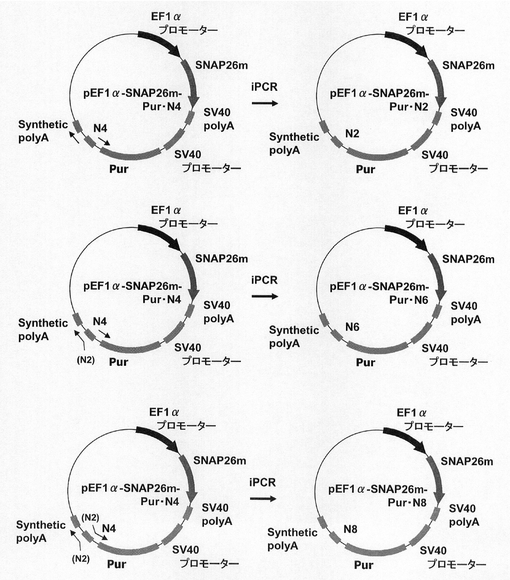
【図21】

【図22】
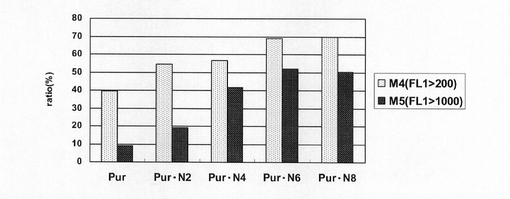
【図23】
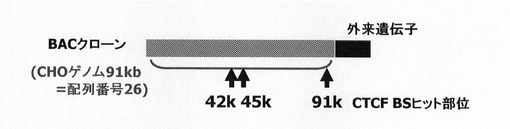
【図24】
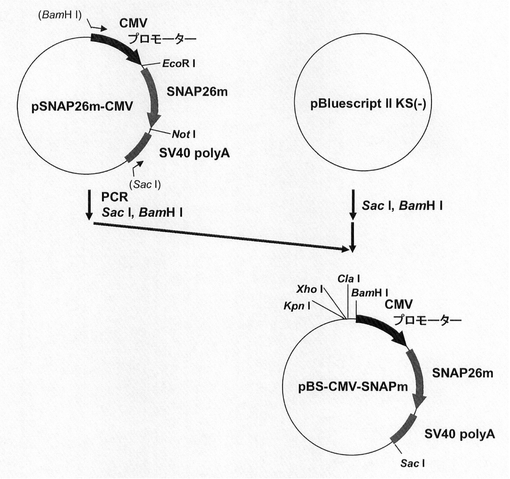
【図25】
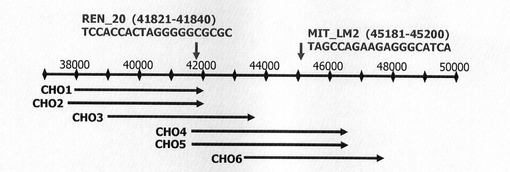
【図26】
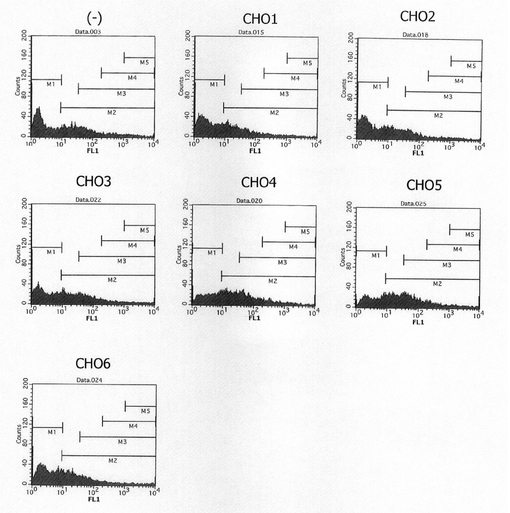
【図27】
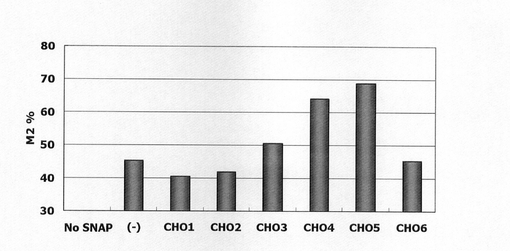
【図28】
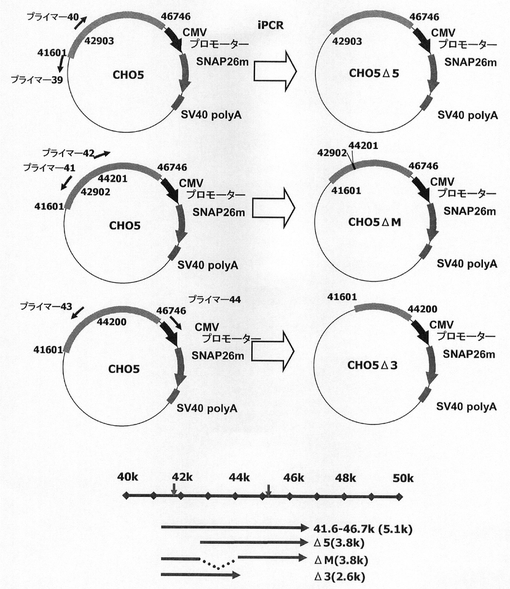
【図29】
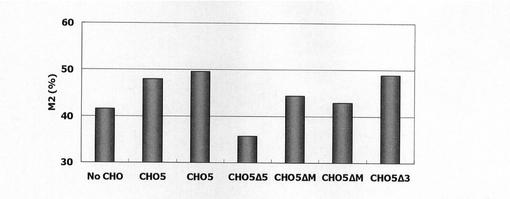
【図30】
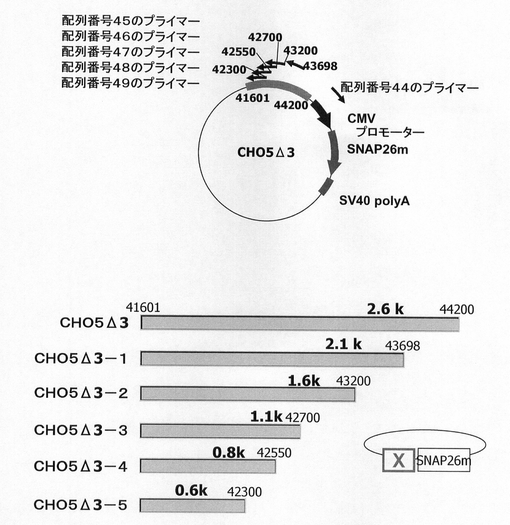
【図31】
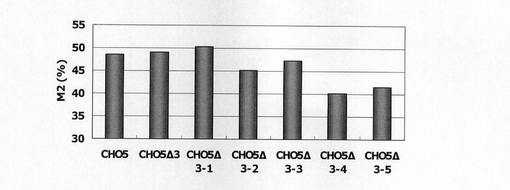
【図32】
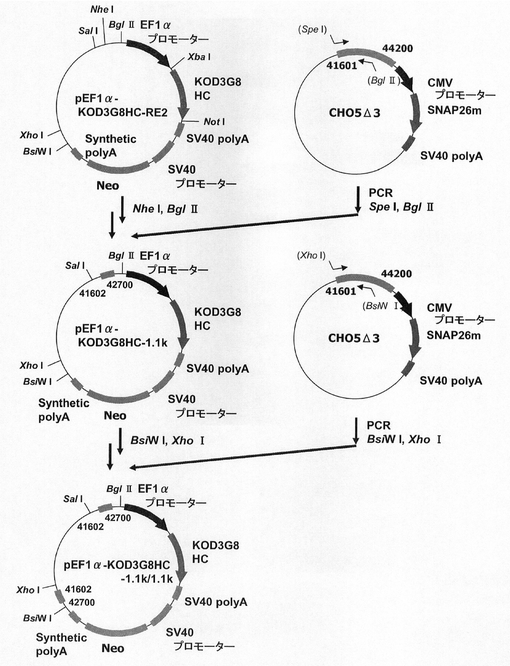
【図33】
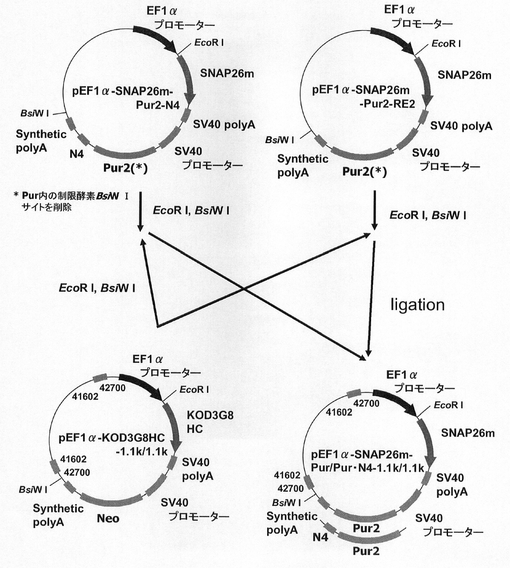
【図34】

【図35】
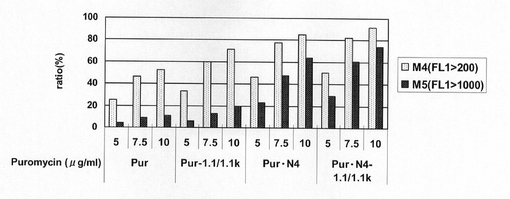
【図36】
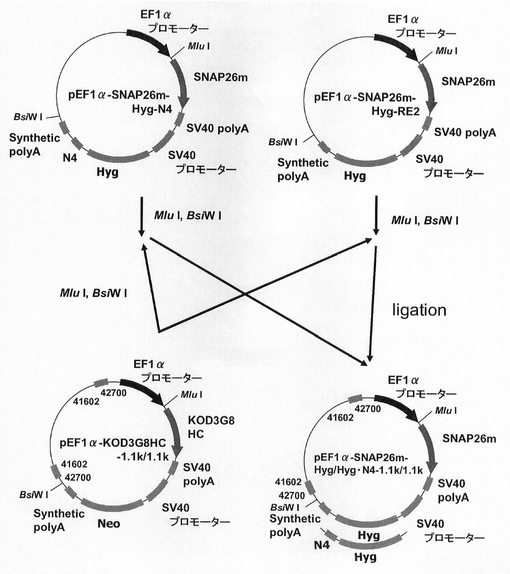
【図37】

【図38】
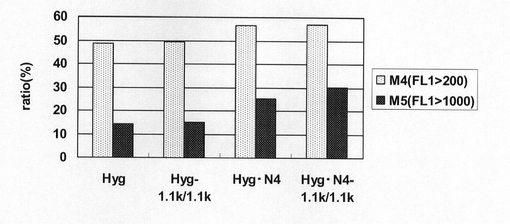
【図39】
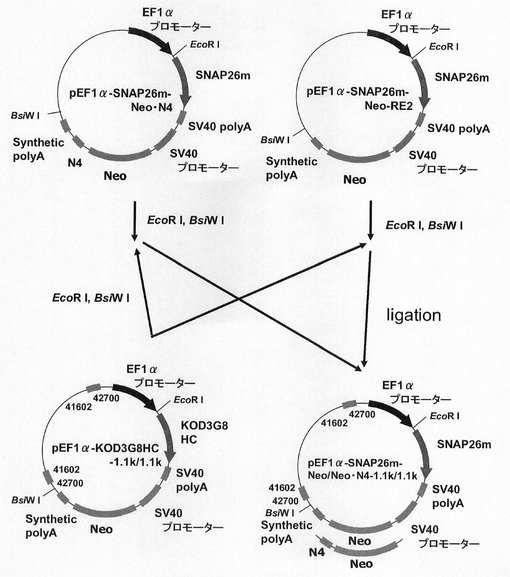
【図40】
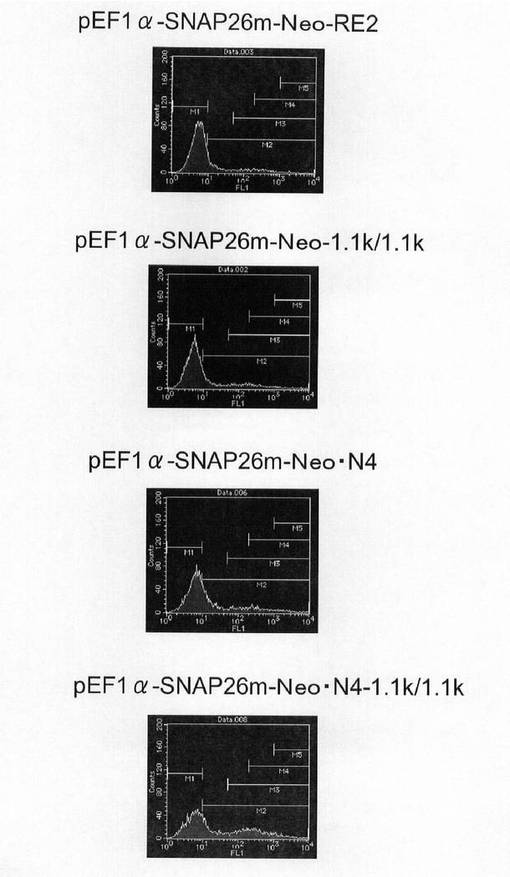
【図41】
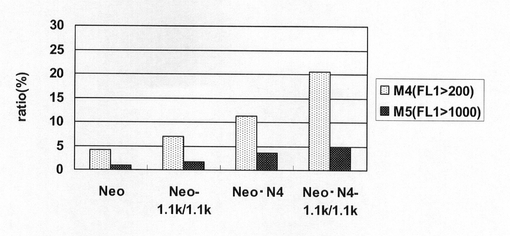
【図42】
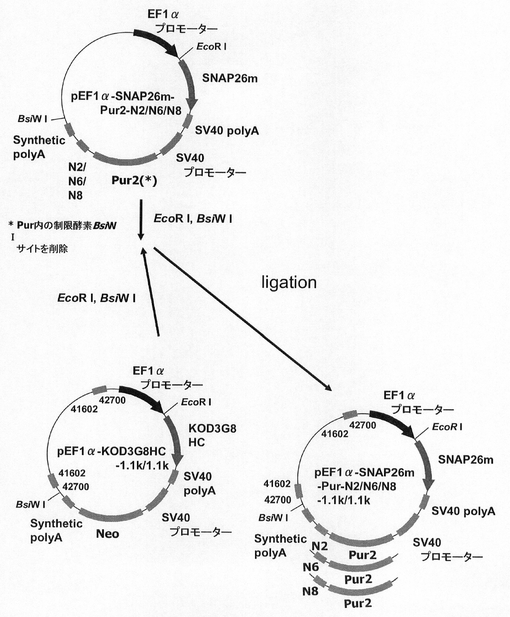
【図43】
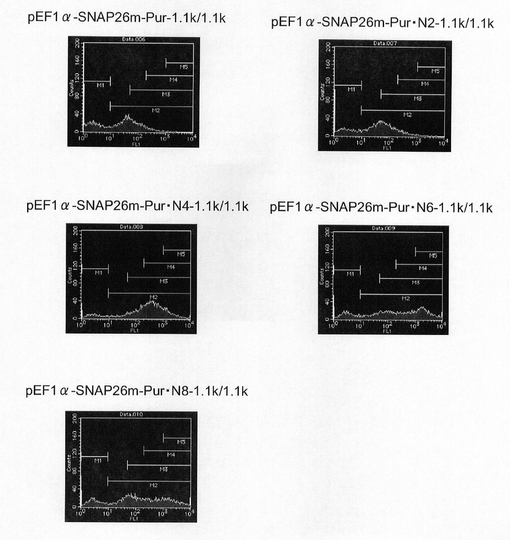
【図44】
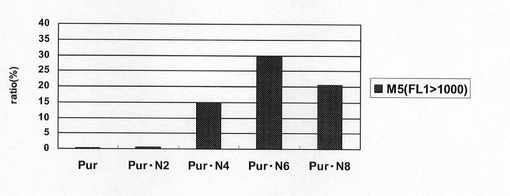
【図45】
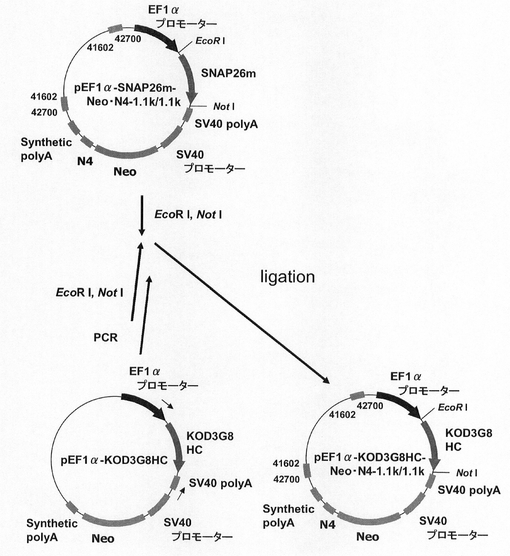
【図46】
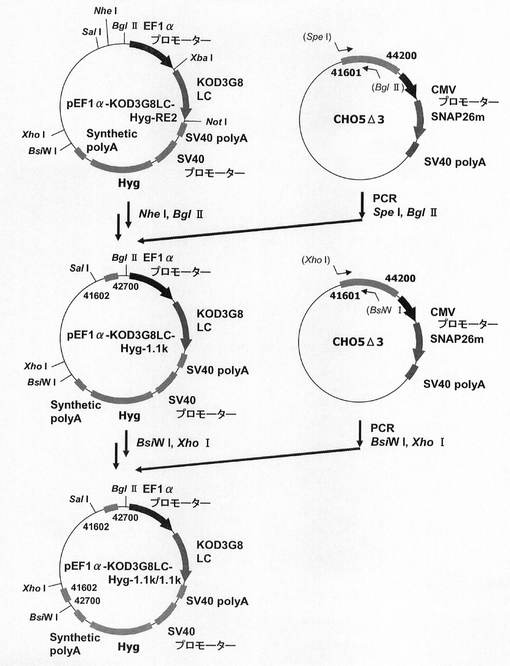
【図47】
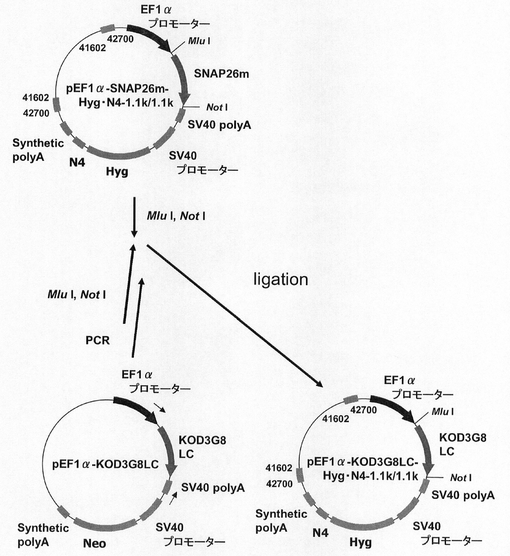
【図48】
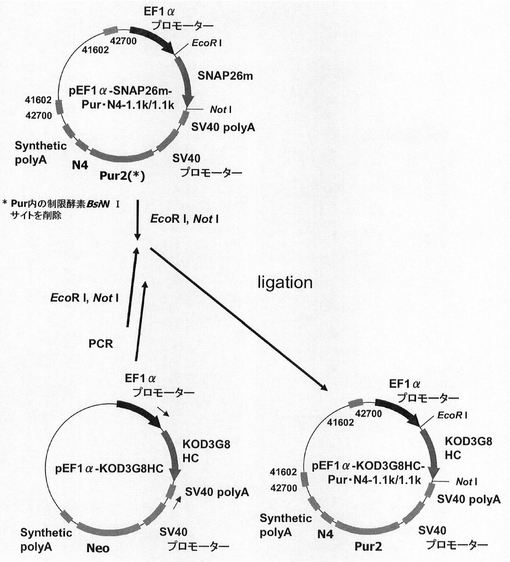
【図49】
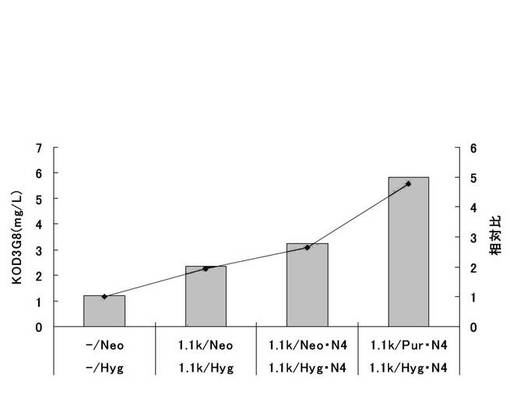
【図50】
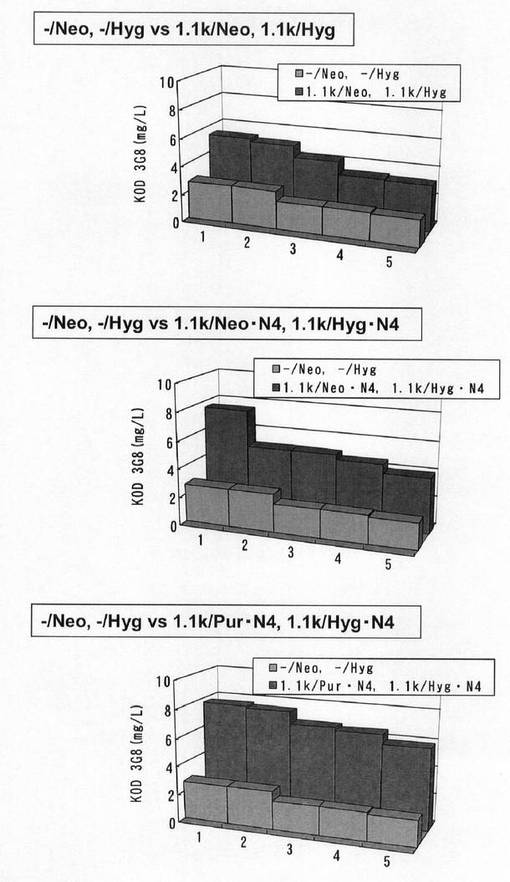
【図51】
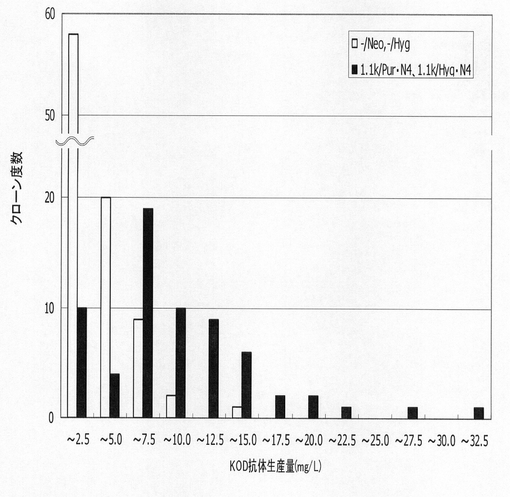
【図52】
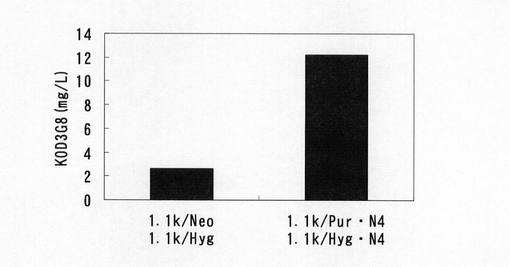
【図53】
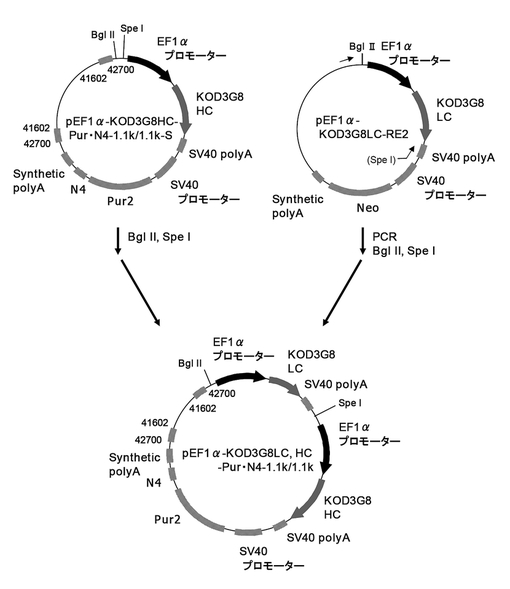
【図54】
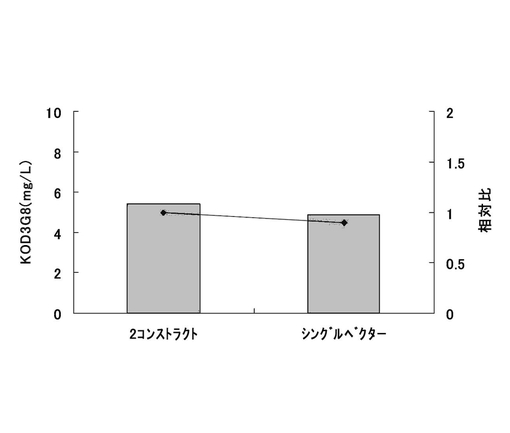
【図55】
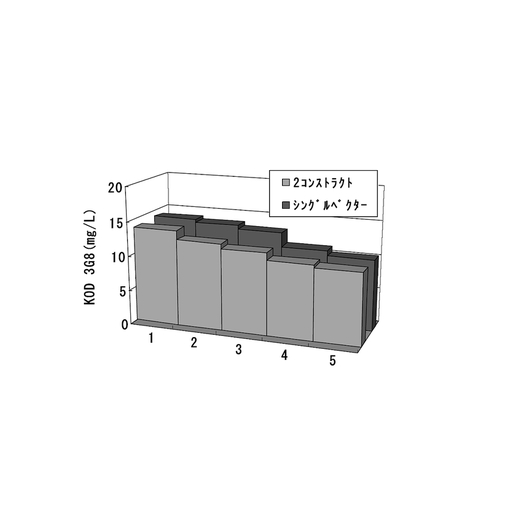
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【図11】
【図12】
【図13】
【図14】
【図15】
【図16】
【図17】
【図18】
【図19】
【図20】
【図21】
【図22】
【図23】
【図24】
【図25】
【図26】
【図27】
【図28】
【図29】
【図30】
【図31】
【図32】
【図33】
【図34】
【図35】
【図36】
【図37】
【図38】
【図39】
【図40】
【図41】
【図42】
【図43】
【図44】
【図45】
【図46】
【図47】
【図48】
【図49】
【図50】
【図51】
【図52】
【図53】
【図54】
【図55】
【公開番号】特開2011−152117(P2011−152117A)
【公開日】平成23年8月11日(2011.8.11)
【国際特許分類】
【出願番号】特願2010−27317(P2010−27317)
【出願日】平成22年2月10日(2010.2.10)
【特許番号】特許第4525863号(P4525863)
【特許公報発行日】平成22年8月18日(2010.8.18)
【出願人】(000003160)東洋紡績株式会社 (3,622)
【Fターム(参考)】
【公開日】平成23年8月11日(2011.8.11)
【国際特許分類】
【出願日】平成22年2月10日(2010.2.10)
【特許番号】特許第4525863号(P4525863)
【特許公報発行日】平成22年8月18日(2010.8.18)
【出願人】(000003160)東洋紡績株式会社 (3,622)
【Fターム(参考)】
[ Back to top ]