
高発現組換え細胞系の選択方法
本発明は、(i)poly Aが作動不能に連結された選択マーカー遺伝子を含む遺伝子発現カセットと、(ii)poly Aが作動可能に連結された目的組換え蛋白質をコードする遺伝子発現カセット、を含む発現ベクターを用いた高生産性クローンの選択方法に関する。本発明によれば、既存の細胞系選択方法より少なくとも10倍少ない数の細胞集団から高生産性細胞クローンの選択が可能である。特に、MTXの量を増加させながらの数回の増幅工程を含む一般的な段階的遺伝子増幅ストラテジーと比較して、低濃度のMTXを使用して、高生産性クローンを選択することができる。したがって、細胞系の発達期間を短縮でき、かつ高生産性クローンの選択にかかる手間及び費用を減らすことができ、それによって、MTX以外の一般的な選択マーカー遺伝子を使用するときでも、さらに効率的な蛋白質の生産が可能である。

【発明の詳細な説明】
【技術分野】
【0001】
本発明は、目的蛋白質の高発現のための細胞系(本明細書では「高生産性クローン」と称する)の選択方法に関し、さらに具体的には、(i)ポリアデニル化シグナル(poly A)が作動不能に連結された選択マーカー遺伝子を含む遺伝子発現カセットと、(ii)poly Aが作動可能に連結された目的組換え蛋白質をコードする遺伝子発現カセット、を含む発現ベクターを用いた高生産性クローンの選択方法及びその発現ベクターに関する。
【背景技術】
【0002】
生物学分野または医学分野において生産しようとする目的蛋白質は、目的蛋白質の遺伝子を発現ベクターに挿入し、前記蛋白質を生産する細胞系に形質移入し、得られた細胞系を大量に培養し、これを適切な方法で前記培養された細胞系から分離及び精製する工程を経て収得される。
【0003】
このために、産業的に使われる方法の一例としては、CHO(チャイニーズハムスター卵巣細胞(Chinese hamster ovary))dhfr(ジヒドロ葉酸リダクターゼ(dihydrofolatereductase))(−)、CHO K1、BHK(ベビーハムスター腎臓細胞(Baby Hamster Kidney))、NS0、SP2/0及びヒト細胞系等を用いた方法が含まれる(非特許文献1、非特許文献2及び非特許文献3)。
【0004】
目的とする外来(heterologous)遺伝子を発現する安定した哺乳動物細胞系作成のために、外来遺伝子は、一般的に、選択マーカー遺伝子(例えば、ネオマイシンホスホトランスフェラーゼ)と共に、所望の細胞系に形質移入によって挿入される。外来遺伝子及び選択マーカー遺伝子は、単一ベクターまたは同時に形質移入される別個のベクターによって発現され得る。形質移入の2、3日後、選択マーカーとしてネオマイシンホスホトランスフェラーゼ遺伝子を使用する場合、細胞をG418のような選択薬剤を含有する培地に移し、それらの選択条件下で数週間培養する。次に、出現する耐性細胞を単離でき、目的遺伝子産物の発現を調べる。宿主細胞ゲノムへのランダムであって非定方向的な組込みの結果として、完全に異なる割合で外来遺伝子を発現する細胞集団を収得する。これらはまた、選択マーカーを発現するが、目的とする遺伝子を発現しない非発現細胞を含むことが可能である。従って、目的とする外来遺伝子の発現が非常に高い細胞クローンを同定するためには、多数のクローンを調べて試験する必要があり、これは、時間消耗的であり、また多くの手間と費用とがかかる。
【0005】
遺伝子増幅は、組換え蛋白質の製造に使われる動物細胞培養において普遍化された技術である。遺伝子増幅は、多数の哺乳動物細胞系が有する相対的に低い生産性を顕著に改善させる。当該分野で広範囲に使われる1つの増幅技術は、ジヒドロ葉酸リダクターゼ(dhfr)系の遺伝子増幅システムであり、このdhfr欠損CHO細胞が頻繁に使われる。
【0006】
dhfr欠損CHO細胞系が、産業的に好まれる理由としては、次のような点を挙げることができる。(1)蛋白質の翻訳後修正(posttranslational modification)、すなわち、グリコシル化(glycosylation)過程やリン酸化(phosphorylation)が、他の細胞類よりもさらにヒト細胞類と類似している。(2)付着培養だけではなく、浮遊培養が可能である。(3)これらの細胞類は、無血清培地で、他の細胞類に比べて相対的に高濃度での培養が可能である。(4)微生物類に比べて、顕著に低い目的蛋白質の生産性を、dhfr/MTX(メトトレキサート(methotrexate))遺伝子増幅システムを利用して増加させることができる。(5)これらの細胞類は安定性が検証され、FDAのような監督機関から許可を容易に受けることができる。前記のような理由で、目的蛋白質を生産する組換え細胞系を作成するために、dhfr欠損CHO細胞系が産業的に広く使われている。
【0007】
しかし、目的蛋白質をコードする遺伝子をdfhr欠損CHO細胞系に形質移入した後、細胞をMTXの濃度を段階的に上昇させて処理することによって、目的蛋白質の生産性を顕著に高めることができる従来法のdhfr/MTX遺伝子増幅システムを利用する場合にさえも、MTXの濃度を段階的に高めて細胞を処理する工程で長時間かかり、かつこの工程でスクリーニングされる細胞集団の数が500〜4,000群を超えるために、高い生産性を有する細胞集団の選択工程が労働集約的であり、時間消費的及び費用消耗的になってしまう。従って、このような選択段階を簡素化するための努力が、多角的に試みられている。
【0008】
例えば、当該技術分野の研究者は、以下の方法等を利用して選択工程を簡素化させるべく努力した。(1)開始コドン近傍を改変させることにより選択マーカー遺伝子の発現を低下させる方法(特許文献1)。(2)遺伝子の変異により選択マーカー遺伝子の機能を損なわせる方法(非特許文献4及び非特許文献5)。(3)開始コドン自体を改変させ、改変開始コドンに発現しようとする遺伝子を連結させて、それにより前記遺伝子の発現レベルを上昇させる方法(非特許文献6)。(4)選択マーカー遺伝子をイントロンと連結させて、前記遺伝子を発現する方法(非特許文献7)。
【先行技術文献】
【特許文献】
【0009】
【特許文献1】米国特許第5733799号明細書
【非特許文献】
【0010】
【非特許文献1】Ogata,et al.,Appl.Microbiol.Biotechnol.,1993,38(4),520-525
【非特許文献2】Kratje,et al.,Biotechnol.Prog.,1994,10(4),410-20
【非特許文献3】Peakman,et al.,Hum.Antibodies Hybridomas,1994,5(1-2),65-74
【非特許文献4】Niwa et al.,Gene 108,193-200,1991
【非特許文献5】Sauter and Enenkel,Biotechnol.Bioeng.89,530-538,2005
【非特許文献6】van Blokland et al.,J of Biotechnology 128,237-245,2007
【非特許文献7】Lucas et al.,Nucleic Acids Res.24,1774-1779,1996
【発明の概要】
【発明が解決しようとする課題】
【0011】
本発明の目的は、ポリアデニル化シグナル(poly A)が作動不能に連結された選択マーカー遺伝子を含む発現ベクターを使用する高生産性クローンの選択方法を提供することである。
【0012】
本発明の他の目的は、前記選択方法に使用される発現ベクターを提供することである。
【0013】
本発明のさらに他の目的は、前記発現ベクターが形質移入された真核宿主細胞系を提供することである。
【課題を解決するための手段】
【0014】
前記のような目的を達成するために、本発明は、(i)ポリアデニル化シグナル(poly A)が作動不能に連結された選択マーカー遺伝子を含む遺伝子発現カセットと、(ii)poly Aが作動可能に連結された目的組換え蛋白質をコードする遺伝子発現カセット、を含む発現ベクターを使用する高生産性クローンの選択方法を提供するものである。
【0015】
また、本発明は、(i)poly Aが作動不能に連結された選択マーカー遺伝子を含む遺伝子発現カセットと、(ii)poly Aが作動可能に連結された目的組換え蛋白質をコードする遺伝子発現カセット、を含む発現ベクターを提供するものである。
【0016】
また、本発明は、前記発現ベクターで形質移入された真核宿主細胞系を提供するものである。
【発明の効果】
【0017】
本発明によれば、既存の細胞系選択方法より少なくとも10倍少ない数の細胞集団から高生産性細胞クローンの選択が可能である。特に、MTXの量を増加させながらの数回の増幅工程を含む一般的な段階的遺伝子増幅ストラテジーと比較して、低濃度のMTXを使用して、高生産性クローンを選択することができる。結果的に、細胞系の発達期間が短縮され、かつ高生産性クローンの選択にかかる手間及び費用を減らすことによって、さらに効率的な組換え蛋白質の生産が可能である。
【図面の簡単な説明】
【0018】
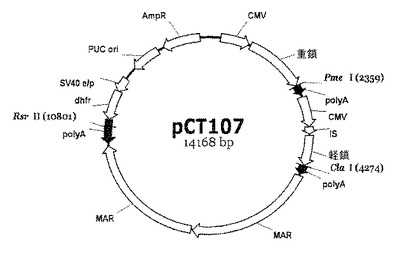
【図1】pCT107発現ベクターの開裂地図である。地図上に表示された制限酵素Pme I、Cla I及びRsr IIは、pCT107発現ベクターによってコードされる抗体の重鎖遺伝子、軽鎖遺伝子、及びDHFR遺伝子に連結されたpoly Aが作動しないようにpCT107ベクターを切断させる部位を示している。
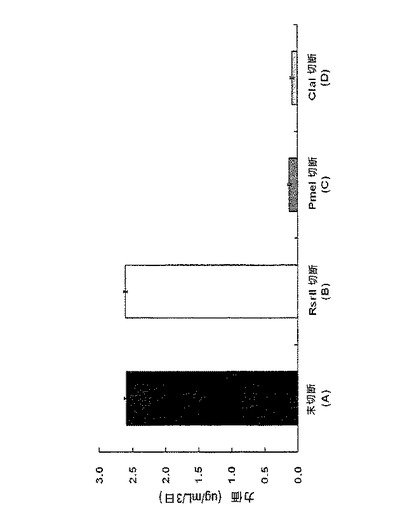
【図2】対照群(A)、実験群(B)、実験群(C)及び実験群(D)の発現ベクターでそれぞれ形質移入されたCHO細胞におけるIgG抗体の発現力価(titer)を測定した結果である。図2中、対照群(A)は、制限酵素で処理していない環形pCT107ベクターであり、 実験群(B)は、Rsr IIで処理してdhfr遺伝子に連結されたpoly Aが作動不能に操作された線形pCT107ベクターであり、 実験群(C)は、Pme Iで処理して重鎖遺伝子に連結されたpoly Aが作動不能に操作された線形pCT107ベクターであり、実験群(D)は、Cla Iで処理して軽鎖遺伝子に連結されたpoly Aが作動不能に操作された線形pCT107ベクターである。試料中に重鎖や軽鎖を含まない場合(実験群(C)及び実験群(D))では、ELISAの特性によりIgG抗体の力価が測定されていない。実験は全て5回行われ、誤差範囲は標準誤差で示した。
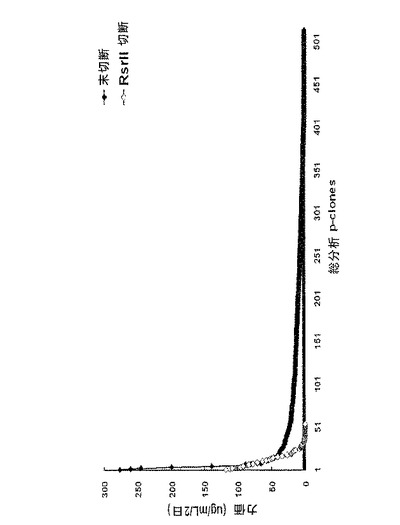
【図3】96−ウェルプレートスケールでのdhfr遺伝子に連結されたpoly Aの有無に依存して細胞クローンを選択する工程において見出された力価の比較を示すグラフである。図3中、未切断は、正常な環形pCT107ベクターであり、RsrII 切断は、Rsr IIで処理してdhfr遺伝子に連結されたpoly Aが作動不能に操作された線形pCT107ベクターである。
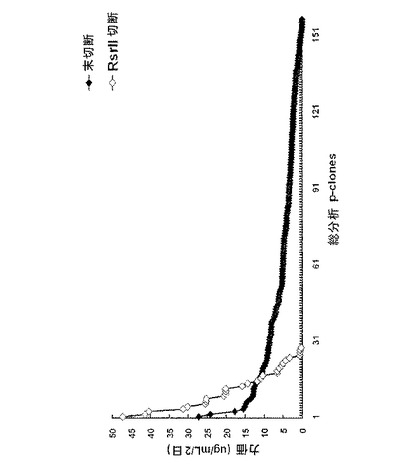
【図4】24−ウェルプレートスケールでのdhfr遺伝子に連結されたpoly Aの有無に依存して細胞クローンを選択する工程において見出された力価の比較を示すグラフである。図4中、未切断は、正常な環形pCT107ベクターであり、RsrII 切断は、Rsr IIで処理してdhfr遺伝子に連結されたpoly Aが作動不能に操作された線形pCT107ベクターである。
【図5】6−ウェルプレートスケールでのdhfr遺伝子に連結されたpoly Aの有無に依存して細胞クローンを選択する工程において見出された力価の比較を示すグラフである。図5中、未切断は、正常な環形pCT107ベクターであり、RsrII 切断は、Rsr IIで処理してdhfr遺伝子に連結されたpoly Aが作動不能に操作された線形pCT107ベクターである。
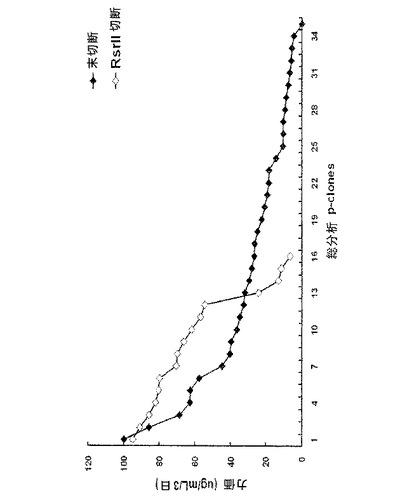
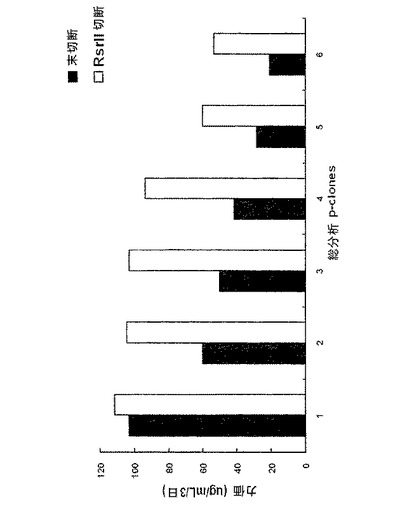
【図6】シェークフラスコを用いた回分式培養(batch culture)状態における対照群と実験群から6個のp−cloneの生産性を比較した結果を示すグラフであり、ここでp−cloneは対照群と実験群についてpoly Aの有無によって高い力価を示すものを選択したものである。図6中、未切断は正常な環形pCT107ベクターであり、RsrII 切断は、Rsr IIで処理してdhfr遺伝子に連結されたpoly Aが作動不能に操作された線形pCT107ベクターである。
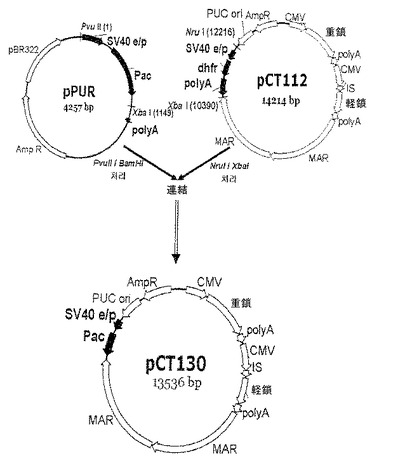
【図7】pCT130ベクターの発現ベクタークローニング工程を示す模式図である。
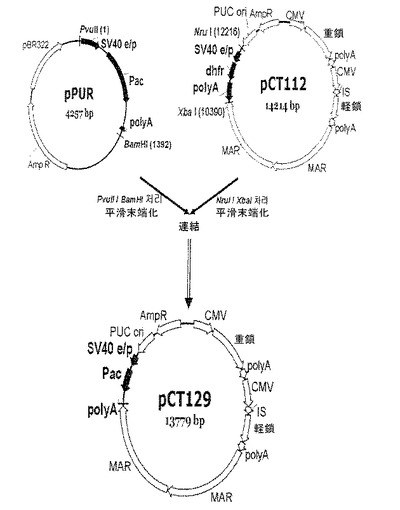
【図8】pCT129ベクターの発現ベクタークローニング工程を示す模式図である。
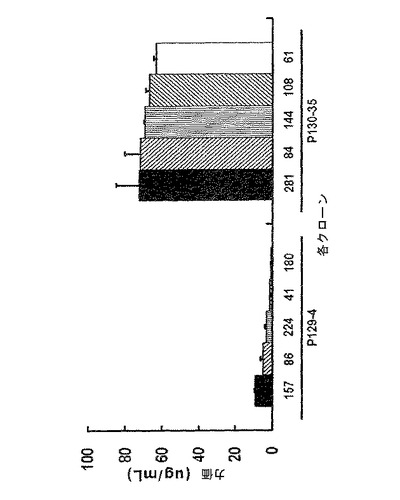
【図9】96−ウェルプレートスケールでの対照群と実験群との生産性を比較した結果を示すグラフであり、図9中、pCT129(poly Aを有する)はpoly A活性を有する対照群、pCT130(poly Aを有しない)はpoly A欠損実験群を示す。
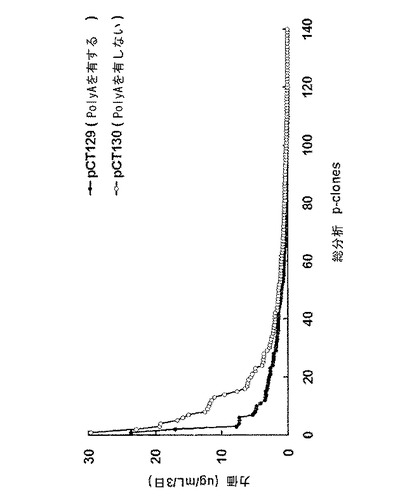
【図10】24−ウェルプレートスケールでの対照群と実験群との生産性を比較した結果を示すグラフであり、図10中、pCT129(poly Aを有する)はpoly A活性を有する対照群、pCT130(poly Aを有しない)はpoly A欠損実験群を示す。
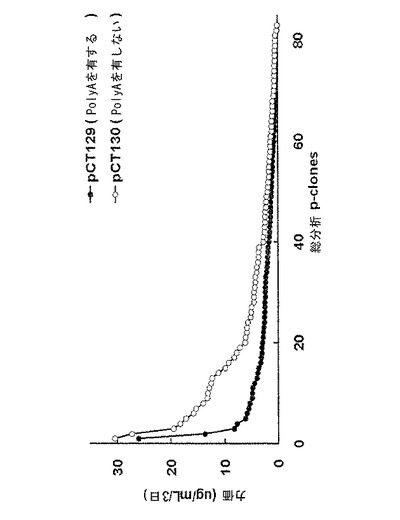
【図11】6−ウェルプレートスケールでの対照群と実験群との生産性を比較した結果を示すグラフであり、図11中、pCT129(poly Aを有する)はpoly A活性を有する対照群、pCT130(poly Aを有しない)はpoly A欠損実験群を示す。
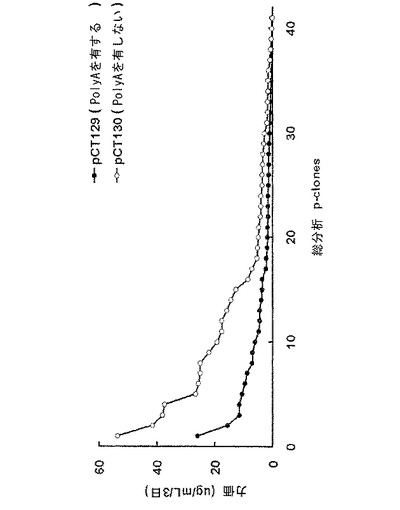
【図12】6−ウェルプレートスケールでの対照群と実験群との生産性を比較した結果を示すグラフである。
【図13】シェークフラスコを用いた回分式培養において、対照群と実験群とで生産性を比較した結果を示すグラフである。
【発明を実施するための形態】
【0019】
略語
dhfr:ジヒドロ葉酸リダクターゼ(dihydrofolate reductase)
CHO:チャイニーズハムスター卵巣細胞(Chinese Hamster Ovary)
poly A:ポリアデニル化シグナル(polyadenylation signal)
p−clone:単一細胞由来クローンではない予備クローン(preliminary clone)
MTX:メトトレキサート(methotrexate)
ELISA:酵素結合免疫吸着測定法(enzyme-linked immunosorbent assay)
【0020】
以下、本明細書中で使われる用語を定義する。
本明細書中の用語「選択マーカー遺伝子(selection marker gene)」とは、目的蛋白質の遺伝子と連結されて発現ベクターに挿入されたマーカー遺伝子であり、それによって、目的遺伝子が正常に発現される細胞を確認することができる。また、選択マーカー遺伝子によってコードされる蛋白質の抑制剤を培地に添加すれば、選択マーカー遺伝子のコピー数を増加させたり、あるいは増加した選択マーカー遺伝子に連結されている目的蛋白質の遺伝子のコピー数をともに増加させることができる。選択マーカーとしては、dhfr遺伝子、グルタミンシンテターゼ(glutamine synthetase)遺伝子、ネオマイシンホスホトランスフェラーゼ(neomycinephosphotransferase)遺伝子、ハイグロマイシンBホスホトランスフェラーゼ(hygromycin B phosphotransferase)遺伝子、ピューロマイシン−N−アセチルトランスフェラーゼ(pac、puromycin−N−acetyltransferase)遺伝子またはストレプトアロテイクス・ヒンダスタヌス(Sh、Streptoalloteichus hindustanus)ble遺伝子などがある。
【0021】
本明細書中の用語「poly A」とは、真核細胞mRNAの3’末端の特定部位を切断させ、切断された3’末端に、約100〜200個のアデニンヌクレオチド(poly A tail)配列を翻訳後に導入するシグナル配列のことである。当該ポリアデニル化シグナル(polyadenylationsignal)配列は、切断部位の約10〜30ヌクレオチド上流に位置したコンセンサス配列AATAAA及び下流に位置した配列をいう。tk poly A、SV40 late及びearly poly A、又は、BGH poly Aのようなさまざまなpoly Aシグナルが公知である。
【0022】
本明細書中の用語「作動可能に連結された(operably linked)」とは、1の核酸フラグメントが第2の核酸フラグメントと連結し、その機能又は発現が、第2の核酸フラグメントによって影響を受けることをいう。また、「作動可能に連結された」とは、例えば、遺伝子配列の転写が作動可能に連結されたプロモーター配列によって指示され、又は、遺伝子配列の翻訳が作動可能に連結された翻訳調節配列によって指示され、または遺伝子配列の翻訳後のプロセッシングが作動可能に連結されたプロセッシング配列によって指示されるような、遺伝子配列、及びプロモーター、又は他の調節配列及びプロセッシング配列との連結を意味するのに使われる。用語「作動不能に連結された」は、用語「作動可能に連結された」と反対の意味であり、一般に、当業者に公知の切断、欠失、点突然変異及びアミノ酸置換のような方法による人為的な操作によって作動不能になる連結を含む意図である。
【0023】
以下、本発明について詳細に説明する。
本発明者らは、高生産性クローンを選択する新規な方法について研究し、その結果、poly AがmRNAの転写と安定性に大きく影響を及ぼし、そして、poly Aの長さは時間の経過と共に幾分短くなった場合、mRNAが分解され始め、そしてこの理由のため、poly Aが正常に作動しなければ、mRNAの半減期は正常mRNAに比べてはるかに短い。この知見に基づき、poly Aに注目して新規な選択方法を開発するための研究を行った。
【0024】
例えば、発現ベクター内の選択マーカー遺伝子に連結されたpoly Aを作動不能に操作した後、これを宿主細胞内に形質移入し、選択マーカー遺伝子によってコードされる蛋白質の抑制剤が存在する選択条件下で培養する場合には、poly Aが正常に作動していない選択マーカー遺伝子は、安定的にmRNAを生産することができないので、前記選択条件下では、大部分の細胞が死滅するものであり、そのうち、多くのコピー数のベクターが形質移入された細胞だけ生存できる。このとき、形質移入に使用された発現ベクターには、選択マーカー遺伝子と、目的組換え蛋白質をコードする遺伝子とが共に含まれているので、選択マーカー遺伝子のコピー数と比例して、目的蛋白質をコードする遺伝子もまた、多くのコピー数で存在する。
【0025】
本発明における、高生産性クローンを選択するための新規な方法として選択マーカー遺伝子に連結したpoly Aの作動可能性を適用できることを証明するために、pCT107ベクター(図1参照)上の選択マーカー遺伝子であるdhfr遺伝子の3’末端に連結されたpoly Aを、適当な制限酵素で切断し、poly Aが作動しないように操作した。続いて、得られたベクターをCHO DG44細胞系(実験群)に形質移入し、そして、前記細胞系からの高生産性クローンの選択工程を、制限酵素を処理していないpCT107ベクターを形質移入したCHO DG44細胞系(対照群)と比較した。その結果、実験群中で、組換え蛋白質生産細胞株候補群の数字が、対照群に比較したとき、実験群において、プレート当たりの細胞が増殖したウェル数が、対照群と比較して7.6倍ほど少ないことを示し、そして、増殖細胞比率は、(1/26264):(1/814815)であり、実験群における増殖細胞比率が対照群と比較して31倍が低いということを示す(表2参照)。
【0026】
次に、対照群と実験群との間での増殖細胞の生産性を比較するために、各群で最上位の生産性を示す6個の細胞系を選択し、それらの生産性を調べた。対照群の場合、6個のp−cloneのうち1個のp−cloneだけが、約100μg/ml/3日の生産性を示し、一方、実験群の場合には、6個のp−cloneのうち4個のp−cloneが、約100μg/ml/3日の生産性を示すことを確認した(図6参照)。
【0027】
また、上述した本発明の選択方法が、CHO DG44細胞でのdhfr選択マーカーを用いたシステム以外の他のシステムにも適用されるか否かを確認するために、CHO K1細胞でのpac選択マーカーでのシステムを使用して実験した。この目的のため、pCT112ベクター(図7及び図8参照)上のdhfr転写ユニット(プロモーター−dhfr−poly A)を除去し、そして、前記ベクターにSV40プロモーター及びpac遺伝子を挿入し、それにより実験ベクター(pCT130ベクター、図7参照)を構築した。さらに、SV40プロモーター、pac遺伝子及びpoly A(pac転写ユニット)を前記ベクターに挿入し対照ベクター(pCT129ベクター、図8参照)を構築した。次に、構築したベクター夫々をCHO K1細胞に形質移入し、そして、対照細胞系と実験細胞系とを選択し、続いて選択した細胞系の生産性を調べた。図9ないし図13から理解できるように、対照群(pCT129ベクター)に由来するクローンに比べて、実験群(pCT130ベクター)に由来するクローンの生産性が高かった。特に、前記ウェルプレートに基づく選択工程で取得されたものと比較してより実質的な生産性を示すシェークフラスコ回分式培養(batch culture)では、実験群からの単一細胞由来クローンの生産性(67〜72μg/ml)が、対照群からの単一細胞由来クローンの生産性(2〜10μg/ml)よりはるかに高いということが判明した。
【0028】
これらの結果に基づき、本発明者らは、本発明の一実施の形態による選択方法が、様々な選択マーカーの使用を介した高生産性クローンの選択において、コスト及び時間を節減するのに有用であるということを見出し、この知見に基づき本発明を完成した。
【0029】
一の側面おいて、本発明は、(i)poly Aが作動不能に連結された選択マーカー遺伝子を含む遺伝子発現カセットと、(ii)poly Aが作動可能に連結された目的組換え蛋白質をコードする遺伝子発現カセット、を含む発現ベクターを使用する高生産性クローンの選択方法を提供する。
【0030】
本発明の一実施の形態において、前記発現ベクターは、(i)のpoly Aが作動不能に連結された選択マーカー遺伝子を含む遺伝子発現カセットの代わりに、poly Aを除去した選択マーカー遺伝子を含む遺伝子発現カセットを含むことができる。
【0031】
他の実施の形態において、本発明は、(i)poly Aが作動可能に連結された選択マーカー遺伝子を含む遺伝子発現カセットと、(ii)poly Aが作動可能に連結された目的組換え蛋白質をコードする遺伝子発現カセットを含み、それにより遺伝子発現カセット(i)のpoly Aが作動しないように前記発現ベクターを線形化させる工程を含む、高生産性クローンの選択方法を提供する。
【0032】
本発明の一実施の形態において、前記選択マーカー遺伝子の例として、非限定的に、dhfr遺伝子、グルタミンシンテターゼ遺伝子、ネオマイシンホスホトランスフェラーゼ遺伝子、ハイグロマイシンBホスホトランスフェラーゼ遺伝子、ピューロマイシン−N−アセチルトランスフェラーゼ(pac)遺伝子、及びストレプトアロテイクス・ヒンダスタヌス(Sh)ble遺伝子を含む。これらのマーカー遺伝子に加えて、当業者に公知の選択マーカー遺伝子であるならば、いずれも本発明で利用可能である。
【0033】
本発明の一実施の形態において、前記目的組換え蛋白質は、非限定的に、モノクローナル抗体であることが好ましい。
【0034】
本発明の一実施の形態において、前記宿主細胞の例として、非限定的に、真核宿主細胞、好ましくは、CHO細胞、ハイブリドーマ細胞、又はF2N細胞を含むヒト宿主細胞、を含む。これらの細胞に加えて、組換え蛋白質産生細胞として当業者に公知の何れの細胞系を、本発明で使用することができる。
【0035】
更なる側面において、本発明は、(i)poly Aが作動不能に連結された選択マーカー遺伝子を含む遺伝子発現カセットと、(ii)poly Aが作動可能に連結された目的組換え蛋白質をコードする遺伝子発現カセット、を含む発現ベクターを提供する。
【0036】
本発明の一実施の形態において、前記発現ベクターは、poly Aが作動不能に連結された選択マーカー遺伝子を含む前記遺伝子発現カセット(i)の代わりに、poly Aを除去した選択マーカー遺伝子を含む遺伝子発現カセットを含むことができる。
【0037】
本発明の一実施の形態において、前記発現ベクターは、poly Aが作動可能に連結された目的組換え蛋白質をコードする遺伝子発現カセット(ii)の代わりに、前記目的組換え蛋白質をコードする遺伝子の導入のためのマルチクローニングサイトを含むことができる。
【0038】
本発明の一実施の形態において、前記選択マーカー遺伝子は、非限定的に、増幅可能な選択マーカー遺伝子、好ましくは、dhfr遺伝子又はグルタミンシンテターゼ遺伝子である。
【0039】
本発明の一実施の形態において、前記選択マーカー遺伝子は、非限定的に、ネオマイシンホスホトランスフェラーゼ遺伝子、ハイグロマイシンBホスホトランスフェラーゼ遺伝子、pac遺伝子、又はSh ble遺伝子である。
【0040】
本発明の一実施の形態において、前記目的組換え蛋白質は、非限定的に、モノクローナル抗体であることが好ましい。
【0041】
更なる側面において、本発明は、前記発現ベクターで形質移入された真核宿主細胞系を提供する。
【0042】
本発明の一実施の形態において、前記宿主細胞は、動物細胞であることが好ましい。本発明の実施例では、CHO細胞系を用いたが、本発明において、これに限定されるものではなく宿主細胞を使用することができる。ハイブリドーマ細胞またはF2N細胞を含む、当業者に公知の組換え蛋白質生産細胞系を本発明で使用することができる。形質移入は、当業者に公知の方法を利用することが好ましい。
【0043】
以下、実施例を介して、本発明について詳細に説明する。ただし、下記実施例は、本発明を例示するものであり、本発明の内容が下記実施例によって限定されるものではない。
【実施例】
【0044】
実施例1.作動不能なpoly Aの誘導
poly Aの有無による遺伝子の発現程度を調べるための最初の段階として、IgG抗体発現ベクターであるpCT107ベクター(図1)内の3つの転写ユニットであるdhfr遺伝子、重鎖遺伝子及び軽鎖遺伝子の3’末端に作動可能に連結されたpoly Aを作動不能な状態にするために、前記pCT107ベクターを、それぞれRsr II、Pme I及びCla I制限酵素を用いて切断した。前記制限酵素Rsr II、Pme I及びCla Iは、それぞれdhfr遺伝子、重鎖遺伝子及び軽鎖遺伝子の3’末端と対応するpoly A間に存在する一定配列を特異的に認識して切断することによって、IgG抗体発現ベクターを線形化(linearization)させる酵素である。
【0045】
制限酵素処理方法は、下記の方式で実施される。3個のチューブを準備した後、各チューブに、(i)pCT107ベクターDNA 30μg及びRsr II(R0501S、NEB)10U、(ii)pCT107ベクターDNA 30μg及びPme I(R0560S、NEB)10U、及び(iii)pCT107ベクターDNA 30μg並びにCla I(R1097S、NEB)10Uを添加し混合し、37℃で4時間インキュベートした。次に、総試料体積の0.1倍容積のNaOACと、3倍容積の100%エタノール(EtOH)とを各チューブの総試料に添加し撹拌した後、−70℃で30分間放置し、DNAペレットを沈殿させた。次に、各チューブの内容物を、13,000rpmで10分間遠心分離させ、チューブ底にDNAペレットを沈殿させて、各チューブに残っているエタノールを除去した。続いて、同一体積の70%エタノールでペレットを洗浄した後、13,000rpmで10分間遠心分離し、チューブに残っているエタノールを除去した。そして、DNAペレットを10分間空気中で乾燥させた。乾燥させたペレットに蒸留水を添加し、常温で10分間放置した。次に、ピペットを用いてDNAペレットが完全に蒸留水に溶解されるように再懸濁させた。ナノドロップ(Nanodrop1000、Thermo scientific)を使用してDNA濃度を測定した後、アガロースゲル電気泳動を行い、ベクターDNAが、それぞれRsr II、Pme IまたはCla Iによって線形化されたか否かを確認した。発現ベクターの線形化は、pCT107ベクター中の3つの転写ユニット(重鎖遺伝子、軽鎖遺伝子及びdhfr遺伝子)に連結されたpoly Aがいずれも作動不能になったことを意味する。
【0046】
実施例2.CHO細胞系への形質移入
実施例1によって線形化されたベクターを、CHO DG44細胞系に同量で形質移入した。次の表1に示すように対照群と実験群への形質移入を行った。
【0047】
【表1】
【0048】
形質移入は、下記の方法で行った。CHO DG44細胞を、10%FBS(12105、Sigma)を補充したMEMα培地(1140076、Invitrogen)を用いて、6−ウェルプレート(well plate)に0.5×106細胞/ウェルの密度で接種し、24時間後に、FBSを含まないMEMα培地に培地を置換した。30分経過後、各ウェルに、対照群(A)、実験群(B)、実験群(C)及び実験群(D)の各ベクターDNA2.5μgと、Opti−SFM培地(12309−050、Invitrogen)500μlとを添加して十分に混合した後、LTX(15338−100、Invitrogen)6.25μlgを添加し、ボルテックスミキサーを利用して十分に撹拌した。得られた混合物を常温で30分間放置した後、各ウェルに500μlずつ添加した。4時間後に、血清が含まれたMEMα培地に再び培地を置換した。
【0049】
実施例3.ELISAを用いたIgG抗体遺伝子の発現確認
IgG抗体遺伝子の発現有無を確認するために、形質移入3日後に、ELISAを行った。まず、ヤギの抗ヒト免疫グロブリンG(Fcγ)(109−006−098、Jackson ImmunoReserarch)を、96−ウェル・マイクロタイタープレート(449824、Nunc)上に吸着させた。前記プレートを1%ウシ血清アルブミン(bovine serum albumin、BSA)を含むリン酸緩衝生理食塩水(phospate-buffered saline、PBS)で処理してブロッキングした後、連続希釈した試料を各ウェルに添加した。プレートを室温で2時間放置した後、ペルオキシダーゼ標識ヤギ抗ヒトカッパー抗体(peroxidase-labeled goat anti-human κ antibody)(I1514、Sigma)で検出のため処理した。室温で1時間放置した後、前記試料をテトラメチルベンジジン(tetramethyl benzidine、TMB)と反応させ、1N HClを加えて反応を停止させた。スタンダードとしては、骨髄種プラズマから精製したヒトIgG1 カッパー(human IgG1 kappa purified myeloma plasma)(A7164、Sigma)を250ng/mlの濃度から用いた。プレートリーダー(Spectramax plus 384、Molecular Device)を使用し、450/650nmで吸光度を測定した。
【0050】
図2から確認することができるように、対照群(A)と実験群(B)とでは、IgGが正常に発現したが、実験群(C)及び実験群(D)の場合には、IgGが正常に発現しなかった。このことは、前記対応遺伝子の3’末端に連結されたpoly Aが作動しないように操作した遺伝子発現カセットの場合、当該遺伝子がコードする蛋白質が正常に発現していないことが分かった(実験群(C)及び実験群(D))。
【0051】
また、少なくとも選択薬剤を使用せずに培養する一過性形質移入では、選択マーカー遺伝子がコードする蛋白質(DHFR蛋白質)の発現有無が、発現ベクター中の前記対応遺伝子によってコードされる蛋白質(特に、抗体)の発現レベルに影響を及ぼさないということを確認することができた。
【0052】
実施例4.poly Aが作動しないdhfr選択マーカー遺伝子を用いた細胞クローンの選択
選択マーカー遺伝子であるdhfr遺伝子の3’末端に連結されたpoly Aが作動しないように操作するために、前記実施例1と同じ処理条件下で、pCT107ベクターをRsr II制限酵素(R0501、NEB)で処理し、それにより実験群(E)を得た。得られたベクターをCHO DG44細胞系に形質移入した。対照群としては、制限酵素で処理していないpCT107ベクター(対照群(A))を用いた。前記実施例1と同じ方法で、形質移入を行った。対照群(A)のベクターと実験群(E)のベクターとを細胞系に導入し、3日後に測定したIgG抗体の発現レベルは、それぞれ3.9μg/ml及び3.8μg/mlであった。形質移入に続く培養開始後の3日目、対照群(A)細胞及び実験群(E)細胞をそれぞれの細胞を96−ウェルプレートに移し、続いて、2%ウシ胎児血清(fetal bovine serum、FBS)及び100nM MTX(813630、Bedford Labs)を含んだSFM4CHO(商標)培地(SH30549.02、HyClone)中で培養した。対照群(A)の場合、0.5×106細胞/96−ウェルプレートを接種して培養し、そして、実験群(E)の場合、2×106細胞/96−ウェルプレートを接種して培養した。
【0053】
培養後、増殖細胞が存在するウェルの数を調べた。その結果、対照群(A)の場合、総2,592個のウェル中514個のウェル(19.8%)で細胞増殖が見られ、実験群(E)の場合、総2,112個のウェル中54個のウェル(2.5%)で細胞増殖が見られた。言い換えれば、前記対照群(A)の場合、増殖細胞が96−ウェルプレート当たり19ウェルに存在し、実験群の場合、増殖細胞が96−ウェルプレート当たり2.5ウェルに存在した。前記結果を基にして、細胞増殖を示すそれぞれのウェルで増殖する細胞が、1つの細胞から由来したと仮定したとき、前記各群間での増殖細胞比率は、31:1(対照群(A):実験群(E))であって、さらに大きい違いがあるということを確認することができた(下記表2、図3〜図5)。
【0054】
【表2】
【0055】
次に、増殖細胞を有するウェル中で、高い力価を示すウェルを選択した。その結果、対照群(A)の場合、514個のウェル中から157個のウェルが選択され、実験群(E)の場合、54個のウェル中から28個のウェルが選択された。この後、前記選択されたウェル内の細胞を24−ウェルプレートに移し、培養スケールを増加させ、3日後に、前記24−ウェルプレートで高い力価を示す6の細胞集団を、対照群(A)及び実験群(E)夫々から選択し、6−ウェルプレートに接種して培養した。対照群(A)と実験群(E)夫々から選択された6の細胞集団をp−cloneと命名し、そして、FBSとMTXを含まない培地を用いて、エルレンマイヤーフラスコ中に接種して撹拌条件下で培養した。対照群(A)と実験群(E)との生産性を比較した。
【0056】
その結果、対照群(A)において、6個のp−cloneのうち1個のp−cloneだけが、約100μg/ml/3日の生産性を示したが、一方、実験群(E)の場合、6個のp−cloneのうち4個のp−cloneが、約100μg/ml/3日の生産性を示した(図6)。
【0057】
実施例5.選択マーカー遺伝子の利用のための細胞増殖条件
さまざまな細胞系で、遺伝子増幅に関与しない一般的な選択マーカー遺伝子の利用性を調べるために、各選択マーカー遺伝子に作用する薬剤を、表3に記述した条件下で各細胞系に適用し、細胞増殖の抑制を測定した。
【0058】
本実験において用いられた細胞系は、CHO K1とF2N78(ヒト細胞系)とであり、各細胞系は、CD Opti CHO培地(12681、Invitrogen)とEX−CELL 293 無血清培地(14571c、Sigma)中で培養したものであり、本実施例でも同じ培地で培養した。各細胞系5mlを、5×104細胞/mlと15×104細胞/mlの細胞密度で6−ウェルプレートの各ウェルに添加した。これは、1×106細胞/96−ウェルプレート(20ml)及び3×106細胞/96−ウェルプレート(20ml)の細胞密度に該当する。前記1×106細胞/96−ウェルプレート及び3×106細胞/96−ウェルプレートの細胞密度は、実施例6のpoly A欠損選択マーカーを用いた選択方法で用いられる細胞密度であった。2種の薬剤濃度を各細胞系に用い、3日ごとに各ウェルの半分の培地(2.5ml)を除去し、同一量の新しい培地で置換した。本実施例で用いた薬剤の種類及び詳細は、下記表4に明示した。このとき、新しい培地は、常に指定された濃度の薬剤を含んでいた。
【0059】
2週後、トリパンブルーでの細胞染色方法を介して、あらゆる薬剤処理条件(表2)において、形質移入をしていないCHO K1細胞とF2N細胞の増殖が観察されなかったことが見出された。すなわち、形質移入後、薬剤処理条件下での培養における、増殖の存在は、形質移入された選択マーカー遺伝子によって処理された薬剤が不活性化されることを示し、それにより、細胞が成長するものであることを意味する。
【0060】
【表3】
【0061】
【表4】
【0062】
実施例6.poly Aを含まないpac選択マーカー遺伝子を用いた蛋白質−生産性細胞系の確立
先に確認したとおり、poly Aが作動不能に連結されたdhfr選択マーカー遺伝子を用いた選択方法が、CHO DG44細胞系での高生産性クローン選択において、費用及び時間を低減する観点から、非常に効率的である。このような選択方法が、dhfr遺伝子以外の選択マーカーとCHO DG44細胞系以外の細胞系でも適用されるか否かを調べるために、本実験において、poly Aが完全に欠損したpac選択マーカー遺伝子を用いた選択方法をCHO K1細胞株に適用して、その効能を検討した。
【0063】
poly Aの機能を完全に除去するために、本実施例では、免疫グロブリン発現ベクター(pCT112ベクター)からdhfr転写ユニット(プロモーター−dhfr遺伝子−poly A)を完全に除去し、プロモーター−pac遺伝子またはプロモーター−pac遺伝子−poly Aを挿入することによって、pCT130ベクターとpCT129ベクターとを構築した(図7及び図8)。ベクター構築方法は、下記の通りである。pac遺伝子含有pPURベクター(631601、Clontech)からPvu II(10−642−690−0014、Roche)とXba I(R0145S、NEB)制限酵素を用いて、SV40プロモーター及びpac遺伝子のコーディング配列を単離し、Nru I(R0192L、NEB)制限酵素とXba I制限酵素を用いてdhfr転写単位を除去したpCT112ベクターにクローニングし、それにより、pCT130ベクター(図7)を構築した。また、これに対する比較群のために、pPRUベクターからPvu IIとBamH I(R0136S、NEB)制限酵素とを用いてSV40プロモーター、pac遺伝子及びpoly Aを単離し、dhfr転写ユニットを除去したpCT112ベクターにクローニングし、それにより、pCT129ベクター(図8)を構築した。
【0064】
CHO K1細胞株を、pCT129ベクター及びpCT130ベクターで形質移入し、そして2日後に、96−ウェルプレートに、下記表5のような様々な濃度のピューロマイシン(puromycin)が添加された培地を用いて細胞を接種した。本実施例で用いた培地は、CD OptiCHO培地であり、FBSは添加されなかった。pCT129ベクターを形質移入した対照群では、0.3×106細胞/プレートの接種密度と、6μg/mlピューロマイシンの選択条件が適切であり、pCT130ベクターを形質移入された実験群では、3×106細胞/プレートの接種密度と、6μg/mlピューロマイシンを用いた選択条件が適切であった。特に、本実施例では、何回もの形質移入実験を介して、poly Aを有する選択マーカーを含む対照群と、poly Aを有しない選択マーカーを含む実験群から、類似した数のp−cloneを選択し、そして、p−cloneにおける免疫グロブリンの発現レベルをそれぞれ比較した。
【0065】
【表5】
【0066】
前記表5の選択条件下で増殖する細胞が存在するウェルの抗体生産を、ELISAで測定し、96−ウェルプレートから抗体生産性の高いp−cloneを選択し(図9)、24−ウェルプレートに移した。続いて、さらに抗体の生産性を測定するために、各クローン試料を2倍段階希釈(2-fold serial dilution)し、ELISAにより分析した(図10)。前記p−cloneのうち、抗体生産性の高い41個のp−cloneを6−ウェルプレートに移した後、さらにそれらの抗体生産性を測定した(図11)。図9〜図11から分かるように、抗体生産性の高いp−cloneを選択するための2つの工程(96−ウェルプレートと24−ウェルプレートとでの選択工程)で、poly Aを含むpCT129ベクター(対照群)由来のp−cloneより、poly Aを含まないpCT130ベクター(実験群)由来のp−cloneの方から、抗体生産性の高いp−cloneがさらに多く選択されたということが理解される。
【0067】
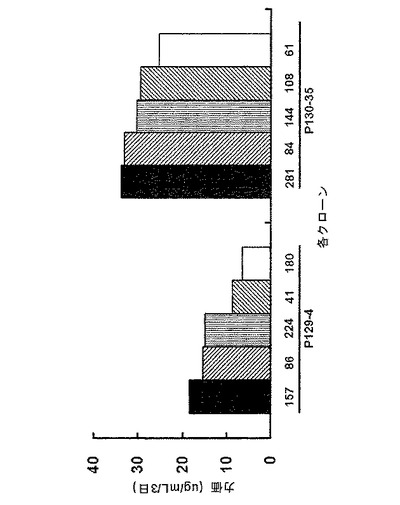
また各群から、高い生産性を有するp−clone 129−4及び130−35を選択し、制限希釈クローニング(limiting dilution cloning、LDC)を行った。それらを96−ウェルプレートで培養するときは、選択したp−cloneを2μg/mlピューロマイシンを処理し、一方、24−ウェルプレート及び6−ウェルプレートで前記クローンを培養するときは、ピューロマイシンを添加しなかった。96−ウェルプレート、24−ウェルプレート及び6−ウェルプレートスケールでの選択工程を経て、生産性の高い単一細胞クローン5個ずつ各群から選択し、6−ウェルプレートでの生産性(図12)、及びシェークフラスコ(shake flask)状態での生産性(図13)を調べた。シェークフラスコでの生産性は、同じ条件下での回分式培養(batch culture)を行うことにより重複で行った。
【0068】
図12から分かるように、pCT130ベクターに由来する前記クローンの生産性(25〜32μg/ml/3日)は、pCT129ベクターに由来する前記クローンの生産性(7〜18μg/ml/3日)よりも、約2倍高かった。これは、高い生産性のクローンが、選択マーカーにおいてpoly Aを含まないから構築され得ることを示す。また、生産性をさらに実質的な側面から検討することができるシェークフラスコを用いた回分式培養条件では、図13から分かるように、選択マーカーにおいてpoly Aを含まないpCT130ベクター(実験群)由来のクローンは、62〜72μg/mlの生産性を示し、一方、選択マーカーにおいてpoly Aを含むpCT129ベクター(対照群)由来のクローンは、2〜10μg/mlの生産性を示す。これらの細胞増殖条件において、細胞増殖率は2つの群で類似しているが、pCT130ベクター(実験群)に由来する前記クローンが、pCT129ベクター(対照群)由来の前記クローンよりも6倍以上の生産性を示す。このような顕著な差は、pCT129ベクター(対照群)由来のクローンにおいて、選択条件不在下では、IgG遺伝子の発現が容易に抑制されるか、あるいはIgG遺伝子が容易に欠失するという現象に起因すると考えることができる。
【0069】
以上の結果から、poly Aを有する選択マーカーを含む発現ベクターで形質移入された細胞系よりも、むしろ本発明の一実施の形態によるpoly Aを有しない選択マーカーを含む発現ベクターで形質移入された細胞系が、抗体発現レベルの高い細胞系を獲得できる方法であることが分かる。よって、本発明の一実施の形態による選択方法は、MTXの量を増加させながら数回増幅工程を実施することを含む従来の段階的遺伝子増幅戦略と比較して、高生産性細胞クローンを選択するための時間を短縮させることができる。これにより、高生産性を有する細胞クローン開発のための手間及び費用を低減させることができる。
【技術分野】
【0001】
本発明は、目的蛋白質の高発現のための細胞系(本明細書では「高生産性クローン」と称する)の選択方法に関し、さらに具体的には、(i)ポリアデニル化シグナル(poly A)が作動不能に連結された選択マーカー遺伝子を含む遺伝子発現カセットと、(ii)poly Aが作動可能に連結された目的組換え蛋白質をコードする遺伝子発現カセット、を含む発現ベクターを用いた高生産性クローンの選択方法及びその発現ベクターに関する。
【背景技術】
【0002】
生物学分野または医学分野において生産しようとする目的蛋白質は、目的蛋白質の遺伝子を発現ベクターに挿入し、前記蛋白質を生産する細胞系に形質移入し、得られた細胞系を大量に培養し、これを適切な方法で前記培養された細胞系から分離及び精製する工程を経て収得される。
【0003】
このために、産業的に使われる方法の一例としては、CHO(チャイニーズハムスター卵巣細胞(Chinese hamster ovary))dhfr(ジヒドロ葉酸リダクターゼ(dihydrofolatereductase))(−)、CHO K1、BHK(ベビーハムスター腎臓細胞(Baby Hamster Kidney))、NS0、SP2/0及びヒト細胞系等を用いた方法が含まれる(非特許文献1、非特許文献2及び非特許文献3)。
【0004】
目的とする外来(heterologous)遺伝子を発現する安定した哺乳動物細胞系作成のために、外来遺伝子は、一般的に、選択マーカー遺伝子(例えば、ネオマイシンホスホトランスフェラーゼ)と共に、所望の細胞系に形質移入によって挿入される。外来遺伝子及び選択マーカー遺伝子は、単一ベクターまたは同時に形質移入される別個のベクターによって発現され得る。形質移入の2、3日後、選択マーカーとしてネオマイシンホスホトランスフェラーゼ遺伝子を使用する場合、細胞をG418のような選択薬剤を含有する培地に移し、それらの選択条件下で数週間培養する。次に、出現する耐性細胞を単離でき、目的遺伝子産物の発現を調べる。宿主細胞ゲノムへのランダムであって非定方向的な組込みの結果として、完全に異なる割合で外来遺伝子を発現する細胞集団を収得する。これらはまた、選択マーカーを発現するが、目的とする遺伝子を発現しない非発現細胞を含むことが可能である。従って、目的とする外来遺伝子の発現が非常に高い細胞クローンを同定するためには、多数のクローンを調べて試験する必要があり、これは、時間消耗的であり、また多くの手間と費用とがかかる。
【0005】
遺伝子増幅は、組換え蛋白質の製造に使われる動物細胞培養において普遍化された技術である。遺伝子増幅は、多数の哺乳動物細胞系が有する相対的に低い生産性を顕著に改善させる。当該分野で広範囲に使われる1つの増幅技術は、ジヒドロ葉酸リダクターゼ(dhfr)系の遺伝子増幅システムであり、このdhfr欠損CHO細胞が頻繁に使われる。
【0006】
dhfr欠損CHO細胞系が、産業的に好まれる理由としては、次のような点を挙げることができる。(1)蛋白質の翻訳後修正(posttranslational modification)、すなわち、グリコシル化(glycosylation)過程やリン酸化(phosphorylation)が、他の細胞類よりもさらにヒト細胞類と類似している。(2)付着培養だけではなく、浮遊培養が可能である。(3)これらの細胞類は、無血清培地で、他の細胞類に比べて相対的に高濃度での培養が可能である。(4)微生物類に比べて、顕著に低い目的蛋白質の生産性を、dhfr/MTX(メトトレキサート(methotrexate))遺伝子増幅システムを利用して増加させることができる。(5)これらの細胞類は安定性が検証され、FDAのような監督機関から許可を容易に受けることができる。前記のような理由で、目的蛋白質を生産する組換え細胞系を作成するために、dhfr欠損CHO細胞系が産業的に広く使われている。
【0007】
しかし、目的蛋白質をコードする遺伝子をdfhr欠損CHO細胞系に形質移入した後、細胞をMTXの濃度を段階的に上昇させて処理することによって、目的蛋白質の生産性を顕著に高めることができる従来法のdhfr/MTX遺伝子増幅システムを利用する場合にさえも、MTXの濃度を段階的に高めて細胞を処理する工程で長時間かかり、かつこの工程でスクリーニングされる細胞集団の数が500〜4,000群を超えるために、高い生産性を有する細胞集団の選択工程が労働集約的であり、時間消費的及び費用消耗的になってしまう。従って、このような選択段階を簡素化するための努力が、多角的に試みられている。
【0008】
例えば、当該技術分野の研究者は、以下の方法等を利用して選択工程を簡素化させるべく努力した。(1)開始コドン近傍を改変させることにより選択マーカー遺伝子の発現を低下させる方法(特許文献1)。(2)遺伝子の変異により選択マーカー遺伝子の機能を損なわせる方法(非特許文献4及び非特許文献5)。(3)開始コドン自体を改変させ、改変開始コドンに発現しようとする遺伝子を連結させて、それにより前記遺伝子の発現レベルを上昇させる方法(非特許文献6)。(4)選択マーカー遺伝子をイントロンと連結させて、前記遺伝子を発現する方法(非特許文献7)。
【先行技術文献】
【特許文献】
【0009】
【特許文献1】米国特許第5733799号明細書
【非特許文献】
【0010】
【非特許文献1】Ogata,et al.,Appl.Microbiol.Biotechnol.,1993,38(4),520-525
【非特許文献2】Kratje,et al.,Biotechnol.Prog.,1994,10(4),410-20
【非特許文献3】Peakman,et al.,Hum.Antibodies Hybridomas,1994,5(1-2),65-74
【非特許文献4】Niwa et al.,Gene 108,193-200,1991
【非特許文献5】Sauter and Enenkel,Biotechnol.Bioeng.89,530-538,2005
【非特許文献6】van Blokland et al.,J of Biotechnology 128,237-245,2007
【非特許文献7】Lucas et al.,Nucleic Acids Res.24,1774-1779,1996
【発明の概要】
【発明が解決しようとする課題】
【0011】
本発明の目的は、ポリアデニル化シグナル(poly A)が作動不能に連結された選択マーカー遺伝子を含む発現ベクターを使用する高生産性クローンの選択方法を提供することである。
【0012】
本発明の他の目的は、前記選択方法に使用される発現ベクターを提供することである。
【0013】
本発明のさらに他の目的は、前記発現ベクターが形質移入された真核宿主細胞系を提供することである。
【課題を解決するための手段】
【0014】
前記のような目的を達成するために、本発明は、(i)ポリアデニル化シグナル(poly A)が作動不能に連結された選択マーカー遺伝子を含む遺伝子発現カセットと、(ii)poly Aが作動可能に連結された目的組換え蛋白質をコードする遺伝子発現カセット、を含む発現ベクターを使用する高生産性クローンの選択方法を提供するものである。
【0015】
また、本発明は、(i)poly Aが作動不能に連結された選択マーカー遺伝子を含む遺伝子発現カセットと、(ii)poly Aが作動可能に連結された目的組換え蛋白質をコードする遺伝子発現カセット、を含む発現ベクターを提供するものである。
【0016】
また、本発明は、前記発現ベクターで形質移入された真核宿主細胞系を提供するものである。
【発明の効果】
【0017】
本発明によれば、既存の細胞系選択方法より少なくとも10倍少ない数の細胞集団から高生産性細胞クローンの選択が可能である。特に、MTXの量を増加させながらの数回の増幅工程を含む一般的な段階的遺伝子増幅ストラテジーと比較して、低濃度のMTXを使用して、高生産性クローンを選択することができる。結果的に、細胞系の発達期間が短縮され、かつ高生産性クローンの選択にかかる手間及び費用を減らすことによって、さらに効率的な組換え蛋白質の生産が可能である。
【図面の簡単な説明】
【0018】
【図1】pCT107発現ベクターの開裂地図である。地図上に表示された制限酵素Pme I、Cla I及びRsr IIは、pCT107発現ベクターによってコードされる抗体の重鎖遺伝子、軽鎖遺伝子、及びDHFR遺伝子に連結されたpoly Aが作動しないようにpCT107ベクターを切断させる部位を示している。
【図2】対照群(A)、実験群(B)、実験群(C)及び実験群(D)の発現ベクターでそれぞれ形質移入されたCHO細胞におけるIgG抗体の発現力価(titer)を測定した結果である。図2中、対照群(A)は、制限酵素で処理していない環形pCT107ベクターであり、 実験群(B)は、Rsr IIで処理してdhfr遺伝子に連結されたpoly Aが作動不能に操作された線形pCT107ベクターであり、 実験群(C)は、Pme Iで処理して重鎖遺伝子に連結されたpoly Aが作動不能に操作された線形pCT107ベクターであり、実験群(D)は、Cla Iで処理して軽鎖遺伝子に連結されたpoly Aが作動不能に操作された線形pCT107ベクターである。試料中に重鎖や軽鎖を含まない場合(実験群(C)及び実験群(D))では、ELISAの特性によりIgG抗体の力価が測定されていない。実験は全て5回行われ、誤差範囲は標準誤差で示した。
【図3】96−ウェルプレートスケールでのdhfr遺伝子に連結されたpoly Aの有無に依存して細胞クローンを選択する工程において見出された力価の比較を示すグラフである。図3中、未切断は、正常な環形pCT107ベクターであり、RsrII 切断は、Rsr IIで処理してdhfr遺伝子に連結されたpoly Aが作動不能に操作された線形pCT107ベクターである。
【図4】24−ウェルプレートスケールでのdhfr遺伝子に連結されたpoly Aの有無に依存して細胞クローンを選択する工程において見出された力価の比較を示すグラフである。図4中、未切断は、正常な環形pCT107ベクターであり、RsrII 切断は、Rsr IIで処理してdhfr遺伝子に連結されたpoly Aが作動不能に操作された線形pCT107ベクターである。
【図5】6−ウェルプレートスケールでのdhfr遺伝子に連結されたpoly Aの有無に依存して細胞クローンを選択する工程において見出された力価の比較を示すグラフである。図5中、未切断は、正常な環形pCT107ベクターであり、RsrII 切断は、Rsr IIで処理してdhfr遺伝子に連結されたpoly Aが作動不能に操作された線形pCT107ベクターである。
【図6】シェークフラスコを用いた回分式培養(batch culture)状態における対照群と実験群から6個のp−cloneの生産性を比較した結果を示すグラフであり、ここでp−cloneは対照群と実験群についてpoly Aの有無によって高い力価を示すものを選択したものである。図6中、未切断は正常な環形pCT107ベクターであり、RsrII 切断は、Rsr IIで処理してdhfr遺伝子に連結されたpoly Aが作動不能に操作された線形pCT107ベクターである。
【図7】pCT130ベクターの発現ベクタークローニング工程を示す模式図である。
【図8】pCT129ベクターの発現ベクタークローニング工程を示す模式図である。
【図9】96−ウェルプレートスケールでの対照群と実験群との生産性を比較した結果を示すグラフであり、図9中、pCT129(poly Aを有する)はpoly A活性を有する対照群、pCT130(poly Aを有しない)はpoly A欠損実験群を示す。
【図10】24−ウェルプレートスケールでの対照群と実験群との生産性を比較した結果を示すグラフであり、図10中、pCT129(poly Aを有する)はpoly A活性を有する対照群、pCT130(poly Aを有しない)はpoly A欠損実験群を示す。
【図11】6−ウェルプレートスケールでの対照群と実験群との生産性を比較した結果を示すグラフであり、図11中、pCT129(poly Aを有する)はpoly A活性を有する対照群、pCT130(poly Aを有しない)はpoly A欠損実験群を示す。
【図12】6−ウェルプレートスケールでの対照群と実験群との生産性を比較した結果を示すグラフである。
【図13】シェークフラスコを用いた回分式培養において、対照群と実験群とで生産性を比較した結果を示すグラフである。
【発明を実施するための形態】
【0019】
略語
dhfr:ジヒドロ葉酸リダクターゼ(dihydrofolate reductase)
CHO:チャイニーズハムスター卵巣細胞(Chinese Hamster Ovary)
poly A:ポリアデニル化シグナル(polyadenylation signal)
p−clone:単一細胞由来クローンではない予備クローン(preliminary clone)
MTX:メトトレキサート(methotrexate)
ELISA:酵素結合免疫吸着測定法(enzyme-linked immunosorbent assay)
【0020】
以下、本明細書中で使われる用語を定義する。
本明細書中の用語「選択マーカー遺伝子(selection marker gene)」とは、目的蛋白質の遺伝子と連結されて発現ベクターに挿入されたマーカー遺伝子であり、それによって、目的遺伝子が正常に発現される細胞を確認することができる。また、選択マーカー遺伝子によってコードされる蛋白質の抑制剤を培地に添加すれば、選択マーカー遺伝子のコピー数を増加させたり、あるいは増加した選択マーカー遺伝子に連結されている目的蛋白質の遺伝子のコピー数をともに増加させることができる。選択マーカーとしては、dhfr遺伝子、グルタミンシンテターゼ(glutamine synthetase)遺伝子、ネオマイシンホスホトランスフェラーゼ(neomycinephosphotransferase)遺伝子、ハイグロマイシンBホスホトランスフェラーゼ(hygromycin B phosphotransferase)遺伝子、ピューロマイシン−N−アセチルトランスフェラーゼ(pac、puromycin−N−acetyltransferase)遺伝子またはストレプトアロテイクス・ヒンダスタヌス(Sh、Streptoalloteichus hindustanus)ble遺伝子などがある。
【0021】
本明細書中の用語「poly A」とは、真核細胞mRNAの3’末端の特定部位を切断させ、切断された3’末端に、約100〜200個のアデニンヌクレオチド(poly A tail)配列を翻訳後に導入するシグナル配列のことである。当該ポリアデニル化シグナル(polyadenylationsignal)配列は、切断部位の約10〜30ヌクレオチド上流に位置したコンセンサス配列AATAAA及び下流に位置した配列をいう。tk poly A、SV40 late及びearly poly A、又は、BGH poly Aのようなさまざまなpoly Aシグナルが公知である。
【0022】
本明細書中の用語「作動可能に連結された(operably linked)」とは、1の核酸フラグメントが第2の核酸フラグメントと連結し、その機能又は発現が、第2の核酸フラグメントによって影響を受けることをいう。また、「作動可能に連結された」とは、例えば、遺伝子配列の転写が作動可能に連結されたプロモーター配列によって指示され、又は、遺伝子配列の翻訳が作動可能に連結された翻訳調節配列によって指示され、または遺伝子配列の翻訳後のプロセッシングが作動可能に連結されたプロセッシング配列によって指示されるような、遺伝子配列、及びプロモーター、又は他の調節配列及びプロセッシング配列との連結を意味するのに使われる。用語「作動不能に連結された」は、用語「作動可能に連結された」と反対の意味であり、一般に、当業者に公知の切断、欠失、点突然変異及びアミノ酸置換のような方法による人為的な操作によって作動不能になる連結を含む意図である。
【0023】
以下、本発明について詳細に説明する。
本発明者らは、高生産性クローンを選択する新規な方法について研究し、その結果、poly AがmRNAの転写と安定性に大きく影響を及ぼし、そして、poly Aの長さは時間の経過と共に幾分短くなった場合、mRNAが分解され始め、そしてこの理由のため、poly Aが正常に作動しなければ、mRNAの半減期は正常mRNAに比べてはるかに短い。この知見に基づき、poly Aに注目して新規な選択方法を開発するための研究を行った。
【0024】
例えば、発現ベクター内の選択マーカー遺伝子に連結されたpoly Aを作動不能に操作した後、これを宿主細胞内に形質移入し、選択マーカー遺伝子によってコードされる蛋白質の抑制剤が存在する選択条件下で培養する場合には、poly Aが正常に作動していない選択マーカー遺伝子は、安定的にmRNAを生産することができないので、前記選択条件下では、大部分の細胞が死滅するものであり、そのうち、多くのコピー数のベクターが形質移入された細胞だけ生存できる。このとき、形質移入に使用された発現ベクターには、選択マーカー遺伝子と、目的組換え蛋白質をコードする遺伝子とが共に含まれているので、選択マーカー遺伝子のコピー数と比例して、目的蛋白質をコードする遺伝子もまた、多くのコピー数で存在する。
【0025】
本発明における、高生産性クローンを選択するための新規な方法として選択マーカー遺伝子に連結したpoly Aの作動可能性を適用できることを証明するために、pCT107ベクター(図1参照)上の選択マーカー遺伝子であるdhfr遺伝子の3’末端に連結されたpoly Aを、適当な制限酵素で切断し、poly Aが作動しないように操作した。続いて、得られたベクターをCHO DG44細胞系(実験群)に形質移入し、そして、前記細胞系からの高生産性クローンの選択工程を、制限酵素を処理していないpCT107ベクターを形質移入したCHO DG44細胞系(対照群)と比較した。その結果、実験群中で、組換え蛋白質生産細胞株候補群の数字が、対照群に比較したとき、実験群において、プレート当たりの細胞が増殖したウェル数が、対照群と比較して7.6倍ほど少ないことを示し、そして、増殖細胞比率は、(1/26264):(1/814815)であり、実験群における増殖細胞比率が対照群と比較して31倍が低いということを示す(表2参照)。
【0026】
次に、対照群と実験群との間での増殖細胞の生産性を比較するために、各群で最上位の生産性を示す6個の細胞系を選択し、それらの生産性を調べた。対照群の場合、6個のp−cloneのうち1個のp−cloneだけが、約100μg/ml/3日の生産性を示し、一方、実験群の場合には、6個のp−cloneのうち4個のp−cloneが、約100μg/ml/3日の生産性を示すことを確認した(図6参照)。
【0027】
また、上述した本発明の選択方法が、CHO DG44細胞でのdhfr選択マーカーを用いたシステム以外の他のシステムにも適用されるか否かを確認するために、CHO K1細胞でのpac選択マーカーでのシステムを使用して実験した。この目的のため、pCT112ベクター(図7及び図8参照)上のdhfr転写ユニット(プロモーター−dhfr−poly A)を除去し、そして、前記ベクターにSV40プロモーター及びpac遺伝子を挿入し、それにより実験ベクター(pCT130ベクター、図7参照)を構築した。さらに、SV40プロモーター、pac遺伝子及びpoly A(pac転写ユニット)を前記ベクターに挿入し対照ベクター(pCT129ベクター、図8参照)を構築した。次に、構築したベクター夫々をCHO K1細胞に形質移入し、そして、対照細胞系と実験細胞系とを選択し、続いて選択した細胞系の生産性を調べた。図9ないし図13から理解できるように、対照群(pCT129ベクター)に由来するクローンに比べて、実験群(pCT130ベクター)に由来するクローンの生産性が高かった。特に、前記ウェルプレートに基づく選択工程で取得されたものと比較してより実質的な生産性を示すシェークフラスコ回分式培養(batch culture)では、実験群からの単一細胞由来クローンの生産性(67〜72μg/ml)が、対照群からの単一細胞由来クローンの生産性(2〜10μg/ml)よりはるかに高いということが判明した。
【0028】
これらの結果に基づき、本発明者らは、本発明の一実施の形態による選択方法が、様々な選択マーカーの使用を介した高生産性クローンの選択において、コスト及び時間を節減するのに有用であるということを見出し、この知見に基づき本発明を完成した。
【0029】
一の側面おいて、本発明は、(i)poly Aが作動不能に連結された選択マーカー遺伝子を含む遺伝子発現カセットと、(ii)poly Aが作動可能に連結された目的組換え蛋白質をコードする遺伝子発現カセット、を含む発現ベクターを使用する高生産性クローンの選択方法を提供する。
【0030】
本発明の一実施の形態において、前記発現ベクターは、(i)のpoly Aが作動不能に連結された選択マーカー遺伝子を含む遺伝子発現カセットの代わりに、poly Aを除去した選択マーカー遺伝子を含む遺伝子発現カセットを含むことができる。
【0031】
他の実施の形態において、本発明は、(i)poly Aが作動可能に連結された選択マーカー遺伝子を含む遺伝子発現カセットと、(ii)poly Aが作動可能に連結された目的組換え蛋白質をコードする遺伝子発現カセットを含み、それにより遺伝子発現カセット(i)のpoly Aが作動しないように前記発現ベクターを線形化させる工程を含む、高生産性クローンの選択方法を提供する。
【0032】
本発明の一実施の形態において、前記選択マーカー遺伝子の例として、非限定的に、dhfr遺伝子、グルタミンシンテターゼ遺伝子、ネオマイシンホスホトランスフェラーゼ遺伝子、ハイグロマイシンBホスホトランスフェラーゼ遺伝子、ピューロマイシン−N−アセチルトランスフェラーゼ(pac)遺伝子、及びストレプトアロテイクス・ヒンダスタヌス(Sh)ble遺伝子を含む。これらのマーカー遺伝子に加えて、当業者に公知の選択マーカー遺伝子であるならば、いずれも本発明で利用可能である。
【0033】
本発明の一実施の形態において、前記目的組換え蛋白質は、非限定的に、モノクローナル抗体であることが好ましい。
【0034】
本発明の一実施の形態において、前記宿主細胞の例として、非限定的に、真核宿主細胞、好ましくは、CHO細胞、ハイブリドーマ細胞、又はF2N細胞を含むヒト宿主細胞、を含む。これらの細胞に加えて、組換え蛋白質産生細胞として当業者に公知の何れの細胞系を、本発明で使用することができる。
【0035】
更なる側面において、本発明は、(i)poly Aが作動不能に連結された選択マーカー遺伝子を含む遺伝子発現カセットと、(ii)poly Aが作動可能に連結された目的組換え蛋白質をコードする遺伝子発現カセット、を含む発現ベクターを提供する。
【0036】
本発明の一実施の形態において、前記発現ベクターは、poly Aが作動不能に連結された選択マーカー遺伝子を含む前記遺伝子発現カセット(i)の代わりに、poly Aを除去した選択マーカー遺伝子を含む遺伝子発現カセットを含むことができる。
【0037】
本発明の一実施の形態において、前記発現ベクターは、poly Aが作動可能に連結された目的組換え蛋白質をコードする遺伝子発現カセット(ii)の代わりに、前記目的組換え蛋白質をコードする遺伝子の導入のためのマルチクローニングサイトを含むことができる。
【0038】
本発明の一実施の形態において、前記選択マーカー遺伝子は、非限定的に、増幅可能な選択マーカー遺伝子、好ましくは、dhfr遺伝子又はグルタミンシンテターゼ遺伝子である。
【0039】
本発明の一実施の形態において、前記選択マーカー遺伝子は、非限定的に、ネオマイシンホスホトランスフェラーゼ遺伝子、ハイグロマイシンBホスホトランスフェラーゼ遺伝子、pac遺伝子、又はSh ble遺伝子である。
【0040】
本発明の一実施の形態において、前記目的組換え蛋白質は、非限定的に、モノクローナル抗体であることが好ましい。
【0041】
更なる側面において、本発明は、前記発現ベクターで形質移入された真核宿主細胞系を提供する。
【0042】
本発明の一実施の形態において、前記宿主細胞は、動物細胞であることが好ましい。本発明の実施例では、CHO細胞系を用いたが、本発明において、これに限定されるものではなく宿主細胞を使用することができる。ハイブリドーマ細胞またはF2N細胞を含む、当業者に公知の組換え蛋白質生産細胞系を本発明で使用することができる。形質移入は、当業者に公知の方法を利用することが好ましい。
【0043】
以下、実施例を介して、本発明について詳細に説明する。ただし、下記実施例は、本発明を例示するものであり、本発明の内容が下記実施例によって限定されるものではない。
【実施例】
【0044】
実施例1.作動不能なpoly Aの誘導
poly Aの有無による遺伝子の発現程度を調べるための最初の段階として、IgG抗体発現ベクターであるpCT107ベクター(図1)内の3つの転写ユニットであるdhfr遺伝子、重鎖遺伝子及び軽鎖遺伝子の3’末端に作動可能に連結されたpoly Aを作動不能な状態にするために、前記pCT107ベクターを、それぞれRsr II、Pme I及びCla I制限酵素を用いて切断した。前記制限酵素Rsr II、Pme I及びCla Iは、それぞれdhfr遺伝子、重鎖遺伝子及び軽鎖遺伝子の3’末端と対応するpoly A間に存在する一定配列を特異的に認識して切断することによって、IgG抗体発現ベクターを線形化(linearization)させる酵素である。
【0045】
制限酵素処理方法は、下記の方式で実施される。3個のチューブを準備した後、各チューブに、(i)pCT107ベクターDNA 30μg及びRsr II(R0501S、NEB)10U、(ii)pCT107ベクターDNA 30μg及びPme I(R0560S、NEB)10U、及び(iii)pCT107ベクターDNA 30μg並びにCla I(R1097S、NEB)10Uを添加し混合し、37℃で4時間インキュベートした。次に、総試料体積の0.1倍容積のNaOACと、3倍容積の100%エタノール(EtOH)とを各チューブの総試料に添加し撹拌した後、−70℃で30分間放置し、DNAペレットを沈殿させた。次に、各チューブの内容物を、13,000rpmで10分間遠心分離させ、チューブ底にDNAペレットを沈殿させて、各チューブに残っているエタノールを除去した。続いて、同一体積の70%エタノールでペレットを洗浄した後、13,000rpmで10分間遠心分離し、チューブに残っているエタノールを除去した。そして、DNAペレットを10分間空気中で乾燥させた。乾燥させたペレットに蒸留水を添加し、常温で10分間放置した。次に、ピペットを用いてDNAペレットが完全に蒸留水に溶解されるように再懸濁させた。ナノドロップ(Nanodrop1000、Thermo scientific)を使用してDNA濃度を測定した後、アガロースゲル電気泳動を行い、ベクターDNAが、それぞれRsr II、Pme IまたはCla Iによって線形化されたか否かを確認した。発現ベクターの線形化は、pCT107ベクター中の3つの転写ユニット(重鎖遺伝子、軽鎖遺伝子及びdhfr遺伝子)に連結されたpoly Aがいずれも作動不能になったことを意味する。
【0046】
実施例2.CHO細胞系への形質移入
実施例1によって線形化されたベクターを、CHO DG44細胞系に同量で形質移入した。次の表1に示すように対照群と実験群への形質移入を行った。
【0047】
【表1】
【0048】
形質移入は、下記の方法で行った。CHO DG44細胞を、10%FBS(12105、Sigma)を補充したMEMα培地(1140076、Invitrogen)を用いて、6−ウェルプレート(well plate)に0.5×106細胞/ウェルの密度で接種し、24時間後に、FBSを含まないMEMα培地に培地を置換した。30分経過後、各ウェルに、対照群(A)、実験群(B)、実験群(C)及び実験群(D)の各ベクターDNA2.5μgと、Opti−SFM培地(12309−050、Invitrogen)500μlとを添加して十分に混合した後、LTX(15338−100、Invitrogen)6.25μlgを添加し、ボルテックスミキサーを利用して十分に撹拌した。得られた混合物を常温で30分間放置した後、各ウェルに500μlずつ添加した。4時間後に、血清が含まれたMEMα培地に再び培地を置換した。
【0049】
実施例3.ELISAを用いたIgG抗体遺伝子の発現確認
IgG抗体遺伝子の発現有無を確認するために、形質移入3日後に、ELISAを行った。まず、ヤギの抗ヒト免疫グロブリンG(Fcγ)(109−006−098、Jackson ImmunoReserarch)を、96−ウェル・マイクロタイタープレート(449824、Nunc)上に吸着させた。前記プレートを1%ウシ血清アルブミン(bovine serum albumin、BSA)を含むリン酸緩衝生理食塩水(phospate-buffered saline、PBS)で処理してブロッキングした後、連続希釈した試料を各ウェルに添加した。プレートを室温で2時間放置した後、ペルオキシダーゼ標識ヤギ抗ヒトカッパー抗体(peroxidase-labeled goat anti-human κ antibody)(I1514、Sigma)で検出のため処理した。室温で1時間放置した後、前記試料をテトラメチルベンジジン(tetramethyl benzidine、TMB)と反応させ、1N HClを加えて反応を停止させた。スタンダードとしては、骨髄種プラズマから精製したヒトIgG1 カッパー(human IgG1 kappa purified myeloma plasma)(A7164、Sigma)を250ng/mlの濃度から用いた。プレートリーダー(Spectramax plus 384、Molecular Device)を使用し、450/650nmで吸光度を測定した。
【0050】
図2から確認することができるように、対照群(A)と実験群(B)とでは、IgGが正常に発現したが、実験群(C)及び実験群(D)の場合には、IgGが正常に発現しなかった。このことは、前記対応遺伝子の3’末端に連結されたpoly Aが作動しないように操作した遺伝子発現カセットの場合、当該遺伝子がコードする蛋白質が正常に発現していないことが分かった(実験群(C)及び実験群(D))。
【0051】
また、少なくとも選択薬剤を使用せずに培養する一過性形質移入では、選択マーカー遺伝子がコードする蛋白質(DHFR蛋白質)の発現有無が、発現ベクター中の前記対応遺伝子によってコードされる蛋白質(特に、抗体)の発現レベルに影響を及ぼさないということを確認することができた。
【0052】
実施例4.poly Aが作動しないdhfr選択マーカー遺伝子を用いた細胞クローンの選択
選択マーカー遺伝子であるdhfr遺伝子の3’末端に連結されたpoly Aが作動しないように操作するために、前記実施例1と同じ処理条件下で、pCT107ベクターをRsr II制限酵素(R0501、NEB)で処理し、それにより実験群(E)を得た。得られたベクターをCHO DG44細胞系に形質移入した。対照群としては、制限酵素で処理していないpCT107ベクター(対照群(A))を用いた。前記実施例1と同じ方法で、形質移入を行った。対照群(A)のベクターと実験群(E)のベクターとを細胞系に導入し、3日後に測定したIgG抗体の発現レベルは、それぞれ3.9μg/ml及び3.8μg/mlであった。形質移入に続く培養開始後の3日目、対照群(A)細胞及び実験群(E)細胞をそれぞれの細胞を96−ウェルプレートに移し、続いて、2%ウシ胎児血清(fetal bovine serum、FBS)及び100nM MTX(813630、Bedford Labs)を含んだSFM4CHO(商標)培地(SH30549.02、HyClone)中で培養した。対照群(A)の場合、0.5×106細胞/96−ウェルプレートを接種して培養し、そして、実験群(E)の場合、2×106細胞/96−ウェルプレートを接種して培養した。
【0053】
培養後、増殖細胞が存在するウェルの数を調べた。その結果、対照群(A)の場合、総2,592個のウェル中514個のウェル(19.8%)で細胞増殖が見られ、実験群(E)の場合、総2,112個のウェル中54個のウェル(2.5%)で細胞増殖が見られた。言い換えれば、前記対照群(A)の場合、増殖細胞が96−ウェルプレート当たり19ウェルに存在し、実験群の場合、増殖細胞が96−ウェルプレート当たり2.5ウェルに存在した。前記結果を基にして、細胞増殖を示すそれぞれのウェルで増殖する細胞が、1つの細胞から由来したと仮定したとき、前記各群間での増殖細胞比率は、31:1(対照群(A):実験群(E))であって、さらに大きい違いがあるということを確認することができた(下記表2、図3〜図5)。
【0054】
【表2】
【0055】
次に、増殖細胞を有するウェル中で、高い力価を示すウェルを選択した。その結果、対照群(A)の場合、514個のウェル中から157個のウェルが選択され、実験群(E)の場合、54個のウェル中から28個のウェルが選択された。この後、前記選択されたウェル内の細胞を24−ウェルプレートに移し、培養スケールを増加させ、3日後に、前記24−ウェルプレートで高い力価を示す6の細胞集団を、対照群(A)及び実験群(E)夫々から選択し、6−ウェルプレートに接種して培養した。対照群(A)と実験群(E)夫々から選択された6の細胞集団をp−cloneと命名し、そして、FBSとMTXを含まない培地を用いて、エルレンマイヤーフラスコ中に接種して撹拌条件下で培養した。対照群(A)と実験群(E)との生産性を比較した。
【0056】
その結果、対照群(A)において、6個のp−cloneのうち1個のp−cloneだけが、約100μg/ml/3日の生産性を示したが、一方、実験群(E)の場合、6個のp−cloneのうち4個のp−cloneが、約100μg/ml/3日の生産性を示した(図6)。
【0057】
実施例5.選択マーカー遺伝子の利用のための細胞増殖条件
さまざまな細胞系で、遺伝子増幅に関与しない一般的な選択マーカー遺伝子の利用性を調べるために、各選択マーカー遺伝子に作用する薬剤を、表3に記述した条件下で各細胞系に適用し、細胞増殖の抑制を測定した。
【0058】
本実験において用いられた細胞系は、CHO K1とF2N78(ヒト細胞系)とであり、各細胞系は、CD Opti CHO培地(12681、Invitrogen)とEX−CELL 293 無血清培地(14571c、Sigma)中で培養したものであり、本実施例でも同じ培地で培養した。各細胞系5mlを、5×104細胞/mlと15×104細胞/mlの細胞密度で6−ウェルプレートの各ウェルに添加した。これは、1×106細胞/96−ウェルプレート(20ml)及び3×106細胞/96−ウェルプレート(20ml)の細胞密度に該当する。前記1×106細胞/96−ウェルプレート及び3×106細胞/96−ウェルプレートの細胞密度は、実施例6のpoly A欠損選択マーカーを用いた選択方法で用いられる細胞密度であった。2種の薬剤濃度を各細胞系に用い、3日ごとに各ウェルの半分の培地(2.5ml)を除去し、同一量の新しい培地で置換した。本実施例で用いた薬剤の種類及び詳細は、下記表4に明示した。このとき、新しい培地は、常に指定された濃度の薬剤を含んでいた。
【0059】
2週後、トリパンブルーでの細胞染色方法を介して、あらゆる薬剤処理条件(表2)において、形質移入をしていないCHO K1細胞とF2N細胞の増殖が観察されなかったことが見出された。すなわち、形質移入後、薬剤処理条件下での培養における、増殖の存在は、形質移入された選択マーカー遺伝子によって処理された薬剤が不活性化されることを示し、それにより、細胞が成長するものであることを意味する。
【0060】
【表3】
【0061】
【表4】
【0062】
実施例6.poly Aを含まないpac選択マーカー遺伝子を用いた蛋白質−生産性細胞系の確立
先に確認したとおり、poly Aが作動不能に連結されたdhfr選択マーカー遺伝子を用いた選択方法が、CHO DG44細胞系での高生産性クローン選択において、費用及び時間を低減する観点から、非常に効率的である。このような選択方法が、dhfr遺伝子以外の選択マーカーとCHO DG44細胞系以外の細胞系でも適用されるか否かを調べるために、本実験において、poly Aが完全に欠損したpac選択マーカー遺伝子を用いた選択方法をCHO K1細胞株に適用して、その効能を検討した。
【0063】
poly Aの機能を完全に除去するために、本実施例では、免疫グロブリン発現ベクター(pCT112ベクター)からdhfr転写ユニット(プロモーター−dhfr遺伝子−poly A)を完全に除去し、プロモーター−pac遺伝子またはプロモーター−pac遺伝子−poly Aを挿入することによって、pCT130ベクターとpCT129ベクターとを構築した(図7及び図8)。ベクター構築方法は、下記の通りである。pac遺伝子含有pPURベクター(631601、Clontech)からPvu II(10−642−690−0014、Roche)とXba I(R0145S、NEB)制限酵素を用いて、SV40プロモーター及びpac遺伝子のコーディング配列を単離し、Nru I(R0192L、NEB)制限酵素とXba I制限酵素を用いてdhfr転写単位を除去したpCT112ベクターにクローニングし、それにより、pCT130ベクター(図7)を構築した。また、これに対する比較群のために、pPRUベクターからPvu IIとBamH I(R0136S、NEB)制限酵素とを用いてSV40プロモーター、pac遺伝子及びpoly Aを単離し、dhfr転写ユニットを除去したpCT112ベクターにクローニングし、それにより、pCT129ベクター(図8)を構築した。
【0064】
CHO K1細胞株を、pCT129ベクター及びpCT130ベクターで形質移入し、そして2日後に、96−ウェルプレートに、下記表5のような様々な濃度のピューロマイシン(puromycin)が添加された培地を用いて細胞を接種した。本実施例で用いた培地は、CD OptiCHO培地であり、FBSは添加されなかった。pCT129ベクターを形質移入した対照群では、0.3×106細胞/プレートの接種密度と、6μg/mlピューロマイシンの選択条件が適切であり、pCT130ベクターを形質移入された実験群では、3×106細胞/プレートの接種密度と、6μg/mlピューロマイシンを用いた選択条件が適切であった。特に、本実施例では、何回もの形質移入実験を介して、poly Aを有する選択マーカーを含む対照群と、poly Aを有しない選択マーカーを含む実験群から、類似した数のp−cloneを選択し、そして、p−cloneにおける免疫グロブリンの発現レベルをそれぞれ比較した。
【0065】
【表5】
【0066】
前記表5の選択条件下で増殖する細胞が存在するウェルの抗体生産を、ELISAで測定し、96−ウェルプレートから抗体生産性の高いp−cloneを選択し(図9)、24−ウェルプレートに移した。続いて、さらに抗体の生産性を測定するために、各クローン試料を2倍段階希釈(2-fold serial dilution)し、ELISAにより分析した(図10)。前記p−cloneのうち、抗体生産性の高い41個のp−cloneを6−ウェルプレートに移した後、さらにそれらの抗体生産性を測定した(図11)。図9〜図11から分かるように、抗体生産性の高いp−cloneを選択するための2つの工程(96−ウェルプレートと24−ウェルプレートとでの選択工程)で、poly Aを含むpCT129ベクター(対照群)由来のp−cloneより、poly Aを含まないpCT130ベクター(実験群)由来のp−cloneの方から、抗体生産性の高いp−cloneがさらに多く選択されたということが理解される。
【0067】
また各群から、高い生産性を有するp−clone 129−4及び130−35を選択し、制限希釈クローニング(limiting dilution cloning、LDC)を行った。それらを96−ウェルプレートで培養するときは、選択したp−cloneを2μg/mlピューロマイシンを処理し、一方、24−ウェルプレート及び6−ウェルプレートで前記クローンを培養するときは、ピューロマイシンを添加しなかった。96−ウェルプレート、24−ウェルプレート及び6−ウェルプレートスケールでの選択工程を経て、生産性の高い単一細胞クローン5個ずつ各群から選択し、6−ウェルプレートでの生産性(図12)、及びシェークフラスコ(shake flask)状態での生産性(図13)を調べた。シェークフラスコでの生産性は、同じ条件下での回分式培養(batch culture)を行うことにより重複で行った。
【0068】
図12から分かるように、pCT130ベクターに由来する前記クローンの生産性(25〜32μg/ml/3日)は、pCT129ベクターに由来する前記クローンの生産性(7〜18μg/ml/3日)よりも、約2倍高かった。これは、高い生産性のクローンが、選択マーカーにおいてpoly Aを含まないから構築され得ることを示す。また、生産性をさらに実質的な側面から検討することができるシェークフラスコを用いた回分式培養条件では、図13から分かるように、選択マーカーにおいてpoly Aを含まないpCT130ベクター(実験群)由来のクローンは、62〜72μg/mlの生産性を示し、一方、選択マーカーにおいてpoly Aを含むpCT129ベクター(対照群)由来のクローンは、2〜10μg/mlの生産性を示す。これらの細胞増殖条件において、細胞増殖率は2つの群で類似しているが、pCT130ベクター(実験群)に由来する前記クローンが、pCT129ベクター(対照群)由来の前記クローンよりも6倍以上の生産性を示す。このような顕著な差は、pCT129ベクター(対照群)由来のクローンにおいて、選択条件不在下では、IgG遺伝子の発現が容易に抑制されるか、あるいはIgG遺伝子が容易に欠失するという現象に起因すると考えることができる。
【0069】
以上の結果から、poly Aを有する選択マーカーを含む発現ベクターで形質移入された細胞系よりも、むしろ本発明の一実施の形態によるpoly Aを有しない選択マーカーを含む発現ベクターで形質移入された細胞系が、抗体発現レベルの高い細胞系を獲得できる方法であることが分かる。よって、本発明の一実施の形態による選択方法は、MTXの量を増加させながら数回増幅工程を実施することを含む従来の段階的遺伝子増幅戦略と比較して、高生産性細胞クローンを選択するための時間を短縮させることができる。これにより、高生産性を有する細胞クローン開発のための手間及び費用を低減させることができる。
【特許請求の範囲】
【請求項1】
(i)ポリアデニル化シグナル(poly A)が作動不能に連結された選択マーカー遺伝子を含む遺伝子発現カセット、及び、
(ii)poly Aが作動可能に連結された目的組換え蛋白質をコードする遺伝子発現カセット、を含む発現ベクターを用いた高生産性クローンの選択方法。
【請求項2】
前記発現ベクターが、前記(i)のpoly Aが作動不能に連結された選択マーカー遺伝子を含む遺伝子発現カセットの代わりに、poly Aを除去した選択マーカー遺伝子を含む遺伝子発現カセットを含む請求項1に記載の方法。
【請求項3】
(i)poly Aが作動可能に連結された選択マーカー遺伝子を含む遺伝子発現カセット、及び、
(ii)poly Aが作動可能に連結された目的組換え蛋白質をコードする遺伝子発現カセット、を含む発現ベクターを制限酵素で切断し、前記(i)の遺伝子発現カセットのpoly Aが作動しないように前記発現ベクターを線形化させる工程、を含む高生産性クローンの選択方法。
【請求項4】
前記選択マーカー遺伝子が、ジヒドロ葉酸レダクターゼ(dhfr)遺伝子、グルタミンシンテターゼ遺伝子、ネオマイシンホスホトランスフェラーゼ遺伝子、ハイグロマイシンBホスホトランスフェラーゼ遺伝子、ピューロマイシン−N−アセチルトランスフェラーゼ(pac)遺伝子、又はストレプトアロテイクス・ヒンダスタヌス(Sh)ble遺伝子である請求項1〜3のいずれか一項に記載の方法。
【請求項5】
前記目的組換え蛋白質が、モノクローナル抗体である請求項1〜3のいずれか一項に記載の方法。
【請求項6】
宿主細胞が、真核宿主細胞である請求項1〜3のいずれか一項に記載の方法。
【請求項7】
前記真核宿主細胞が、CHO細胞、ハイブリドーマ細胞、又はF2N細胞を含むヒト宿主細胞である請求項6に記載の方法。
【請求項8】
(i)poly Aが作動不能に連結された選択マーカー遺伝子を含む遺伝子発現カセット、及び、
(ii)poly Aが作動可能に連結された目的組換え蛋白質をコードする遺伝子発現カセット、を含む発現ベクター。
【請求項9】
前記(i)のpoly Aが作動不能に連結された選択マーカー遺伝子を含む遺伝子発現カセットの代わりに、poly Aを除去した選択マーカー遺伝子を含む遺伝子発現カセットを含む請求項8に記載の発現ベクター。
【請求項10】
前記(ii)のpoly Aが作動可能に連結された目的組換え蛋白質をコードする遺伝子発現カセットの代わりに、目的蛋白質をコードする遺伝子の導入のためのマルチクローニングサイトを含む請求項8に記載の発現ベクター。
【請求項11】
前記選択マーカー遺伝子が、増幅可能な選択マーカー遺伝子である請求項8〜10の何れか一項に記載の発現ベクター。
【請求項12】
前記増幅可能な選択マーカー遺伝子が、dhfr遺伝子またはグルタミンシンテターゼ遺伝子である請求項11に記載の発現ベクター。
【請求項13】
前記選択マーカー遺伝子が、グルタミンシンテターゼ遺伝子、ネオマイシンホスホトランスフェラーゼ遺伝子、ハイグロマイシンBホスホトランスフェラーゼ遺伝子、pac遺伝子、又はSh ble遺伝子である請求項8〜10の何れか一項に記載の発現ベクター。
【請求項14】
前記目的組換え蛋白質が、モノクローナル抗体である請求項8〜10の何れか一項に記載の発現ベクター。
【請求項15】
請求項8〜10の何れか一項に記載の発現ベクターで形質移入された真核宿主細胞。
【請求項16】
前記真核宿主細胞が、CHO細胞、ハイブリドーマ細胞、又はF2N細胞を含むヒト宿主細胞である請求項15に記載の真核宿主細胞。
【請求項1】
(i)ポリアデニル化シグナル(poly A)が作動不能に連結された選択マーカー遺伝子を含む遺伝子発現カセット、及び、
(ii)poly Aが作動可能に連結された目的組換え蛋白質をコードする遺伝子発現カセット、を含む発現ベクターを用いた高生産性クローンの選択方法。
【請求項2】
前記発現ベクターが、前記(i)のpoly Aが作動不能に連結された選択マーカー遺伝子を含む遺伝子発現カセットの代わりに、poly Aを除去した選択マーカー遺伝子を含む遺伝子発現カセットを含む請求項1に記載の方法。
【請求項3】
(i)poly Aが作動可能に連結された選択マーカー遺伝子を含む遺伝子発現カセット、及び、
(ii)poly Aが作動可能に連結された目的組換え蛋白質をコードする遺伝子発現カセット、を含む発現ベクターを制限酵素で切断し、前記(i)の遺伝子発現カセットのpoly Aが作動しないように前記発現ベクターを線形化させる工程、を含む高生産性クローンの選択方法。
【請求項4】
前記選択マーカー遺伝子が、ジヒドロ葉酸レダクターゼ(dhfr)遺伝子、グルタミンシンテターゼ遺伝子、ネオマイシンホスホトランスフェラーゼ遺伝子、ハイグロマイシンBホスホトランスフェラーゼ遺伝子、ピューロマイシン−N−アセチルトランスフェラーゼ(pac)遺伝子、又はストレプトアロテイクス・ヒンダスタヌス(Sh)ble遺伝子である請求項1〜3のいずれか一項に記載の方法。
【請求項5】
前記目的組換え蛋白質が、モノクローナル抗体である請求項1〜3のいずれか一項に記載の方法。
【請求項6】
宿主細胞が、真核宿主細胞である請求項1〜3のいずれか一項に記載の方法。
【請求項7】
前記真核宿主細胞が、CHO細胞、ハイブリドーマ細胞、又はF2N細胞を含むヒト宿主細胞である請求項6に記載の方法。
【請求項8】
(i)poly Aが作動不能に連結された選択マーカー遺伝子を含む遺伝子発現カセット、及び、
(ii)poly Aが作動可能に連結された目的組換え蛋白質をコードする遺伝子発現カセット、を含む発現ベクター。
【請求項9】
前記(i)のpoly Aが作動不能に連結された選択マーカー遺伝子を含む遺伝子発現カセットの代わりに、poly Aを除去した選択マーカー遺伝子を含む遺伝子発現カセットを含む請求項8に記載の発現ベクター。
【請求項10】
前記(ii)のpoly Aが作動可能に連結された目的組換え蛋白質をコードする遺伝子発現カセットの代わりに、目的蛋白質をコードする遺伝子の導入のためのマルチクローニングサイトを含む請求項8に記載の発現ベクター。
【請求項11】
前記選択マーカー遺伝子が、増幅可能な選択マーカー遺伝子である請求項8〜10の何れか一項に記載の発現ベクター。
【請求項12】
前記増幅可能な選択マーカー遺伝子が、dhfr遺伝子またはグルタミンシンテターゼ遺伝子である請求項11に記載の発現ベクター。
【請求項13】
前記選択マーカー遺伝子が、グルタミンシンテターゼ遺伝子、ネオマイシンホスホトランスフェラーゼ遺伝子、ハイグロマイシンBホスホトランスフェラーゼ遺伝子、pac遺伝子、又はSh ble遺伝子である請求項8〜10の何れか一項に記載の発現ベクター。
【請求項14】
前記目的組換え蛋白質が、モノクローナル抗体である請求項8〜10の何れか一項に記載の発現ベクター。
【請求項15】
請求項8〜10の何れか一項に記載の発現ベクターで形質移入された真核宿主細胞。
【請求項16】
前記真核宿主細胞が、CHO細胞、ハイブリドーマ細胞、又はF2N細胞を含むヒト宿主細胞である請求項15に記載の真核宿主細胞。
【図1】

【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【図11】
【図12】
【図13】
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【図11】
【図12】
【図13】
【公表番号】特表2012−521779(P2012−521779A)
【公表日】平成24年9月20日(2012.9.20)
【国際特許分類】
【出願番号】特願2012−503316(P2012−503316)
【出願日】平成22年3月30日(2010.3.30)
【国際出願番号】PCT/KR2010/001912
【国際公開番号】WO2010/114270
【国際公開日】平成22年10月7日(2010.10.7)
【出願人】(511237689)セルトリオン・インコーポレイテッド (1)
【Fターム(参考)】
【公表日】平成24年9月20日(2012.9.20)
【国際特許分類】
【出願日】平成22年3月30日(2010.3.30)
【国際出願番号】PCT/KR2010/001912
【国際公開番号】WO2010/114270
【国際公開日】平成22年10月7日(2010.10.7)
【出願人】(511237689)セルトリオン・インコーポレイテッド (1)
【Fターム(参考)】
[ Back to top ]