
(メタ)アクリル酸(塩)系重合体およびその製造方法
【課題】物性が高くて着色の少ない (メタ)アクリル酸(塩)系重合体とこれを高い重合率で反応釜の腐食を生じさせることなく製造し得る方法を提供する。
【解決手段】不飽和モノカルボン酸系単量体(a)と不飽和ポリアルキレングリコール系単量体(b)とを重合開始剤を用いて重合させて(メタ)アクリル酸(塩)系重合体を得る際に、前記重合開始剤として過酸化水素と還元剤とからなるレドックス系重合開始剤を特定量用い、かつ、前記重合を、重合中のpHが3.2〜7.0の範囲内となるようにして行う。前記不飽和モノカルボン酸系単量体とポリアルキレングリコール特定量の仕込み比で酸触媒の存在下においてエステル化することにより出発原料たる残存不飽和モノカルボン酸系単量体の一部を残存させるとともにエステル化後に塩基性物質で前記酸触媒と残存不飽和モノカルボン酸系単量体(a)を中和させる。
【解決手段】不飽和モノカルボン酸系単量体(a)と不飽和ポリアルキレングリコール系単量体(b)とを重合開始剤を用いて重合させて(メタ)アクリル酸(塩)系重合体を得る際に、前記重合開始剤として過酸化水素と還元剤とからなるレドックス系重合開始剤を特定量用い、かつ、前記重合を、重合中のpHが3.2〜7.0の範囲内となるようにして行う。前記不飽和モノカルボン酸系単量体とポリアルキレングリコール特定量の仕込み比で酸触媒の存在下においてエステル化することにより出発原料たる残存不飽和モノカルボン酸系単量体の一部を残存させるとともにエステル化後に塩基性物質で前記酸触媒と残存不飽和モノカルボン酸系単量体(a)を中和させる。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、セメント分散剤・顔料分散剤等に好適に使用される(メタ)アクリル酸(塩)系共重合体とその製造方法に関する。
【背景技術】
【0002】
不飽和モノカルボン酸系単量体(a)と不飽和ポリアルキレングリコール系単量体(b)とを必須成分として含む単量体成分を共重合して得られるポリカルボン酸は、従来から、セメント分散剤および顔料分散剤をはじめ、種々の分散剤に好適に用いられている。
ポリカルボン酸は、例えば、特許文献1では、ポリアルキレングリコールモノ(メタ)アクリル酸エステル単量体、(メタ)アクリル酸系単量体およびこれらと共重合可能な単量体を、ベンゾイルパーオキシド、ラウロパーオキシド等のパーオキシド、クメンハイドロパーオキシド等のハイドロパーオキシド、アゾビスイソブチロニトリルなどの脂肪族アゾ化合物を重合開始剤として用いて製造している。
【0003】
しかしながら、このような、不飽和モノカルボン酸系単量体(a)と不飽和ポリアルキレングリコール系単量体(b)との反応系においては、高温で反応させた場合、モノマーの重合率は高いものの、分子量の低いオリゴマーの生成量が増大し、分散剤としての性能を発揮しうるだけの高い分子量を有する有効なポリマー部分が減少しやすいという問題が生じる。一方、低温で反応させた場合には、モノマーの連鎖移動が抑制できるので、オリゴマー部分が少なく有効なポリマー部分の多い(メタ)アクリル酸(塩)系重合体が得られるが、モノマーの重合率が低くなる傾向がある。
この問題を解決するには、前記(メタ)アクリル酸(塩)系重合体を得る際に、低温で反応させ、かつ、モノマーの重合率を上げるために反応時間を長くすることが考えられるが、反応に長時間を要することから、生産性および作業性が悪く、工業的に実施する上でコストが問題となる。
【0004】
そこで、前記反応系において、低温でも反応性が高い重合開始剤を用いることが望まれる。
このような要望に応えるための重合開始剤として、過硫酸アンモニウムや過硫酸ナトリウム等の過硫酸塩がある。
前記反応系において過硫酸塩を開始剤として用いる公知技術の一つに、特許文献2が開示する方法がある。この方法は、重合反応液のpHを1.5〜3.5の範囲内に調整することで、物性(セメント分散性を示すペーストフロー値)の良い(メタ)アクリル酸(塩)系重合体を得るようにしている。この先行技術は、加えて、単量体の一方である不飽和ポリアルキレングリコール系単量体(b)は、ポリアルキレングリコールモノアルキルエーテルと(メタ)アクリル酸とを原料とし酸触媒を使用してエステル化することによって製造することとし、エステル化終了後に、上記酸触媒をアルカリ剤で失活させることで、重合を円滑化するようにしている。
【0005】
しかし、本発明者の検討したところによれば、特許文献2開示の上記先行方法は、開始剤としての過硫酸アンモニウム等の過硫酸塩を何らの工夫もなく使用するため、以下に述べる問題を生じている。すなわち、これらの開始剤は、重合時に硫酸が発生するため、ステンレスの反応釜では腐食するおそれがあり、グラスライニングされた反応釜を使用する必要がある。グラスライニングされた反応釜は、ステンレスの反応釜に比べ熱伝導率が低く、加熱冷却時の効率が悪い上に、衝撃に弱く、ライニング層が割れた場合の補修も困難であるため、設備の維持に多額の費用がかかるという問題を有する。ポリカルボン酸は、セメント分散剤として使用する場合、取り扱いやすくするため、通常、重合後に中和しておくことが望ましいのであるが、過硫酸塩を開始剤として用いて得られた(メタ)アクリル酸(塩)系重合体は、中和の際に塩が生成しやすく、この塩が結晶として析出して各種トラブルの原因となる恐れがある。また、過硫酸塩を開始剤として用いて得られた(メタ)アクリル酸(塩)系重合体の水溶液は茶褐色に呈色していることもあるため、着色の少ない分散剤に対する要求が高い顔料分散剤用途には不向きであった。
【0006】
本発明者の検討したところによれば、特許文献2開示の上記先行方法は、また、未だモノマーの重合率が十分でなく、未反応モノマーが残存しやすい方法であり、そのため、上記先行方法で製造した(メタ)アクリル酸(塩)系重合体は分散剤を構成するための配合割合が多くなると言う問題を有していた。
【特許文献1】特開昭59−18338号公報
【特許文献2】WO01/14438号明細書
【発明の開示】
【発明が解決しようとする課題】
【0007】
そこで、本発明の課題は、物性が高くて着色の少ない(メタ)アクリル酸(塩)系重合体を提供することと、このような高い品質の(メタ)アクリル酸(塩)系重合体を、低温でありながら高い重合率で得させることができ、しかも、反応釜の腐食を起こすことのない製造方法を提供することにある。
【課題を解決するための手段】
【0008】
本発明者は、上記課題を解決するために、種々工夫し検討し実験を重ねた。その結果、その解決方法の一つが、重合開始剤として過酸化水素と還元剤からなるレドックス系を特定量だけ用いることであり、もう一つが、単量体の一方である不飽和ポリアルキレングリコール系単量体(b)として、ポリアルキレングリコールと不飽和モノカルボン酸のエステル化で得たものを使用するが、このとき、ポリアルキレングリコールとして特定の平均付加モル数nを有するものを用いるとともに、エステル化原料の仕込み比を特定範囲に選定することとし、かつ、重合の際のpHを特許文献2開示の前記先行方法のpH範囲よりも高くすることである、ことを見出し、本発明を完成した。
【0009】
したがって、本発明にかかる(メタ)アクリル酸(塩)系重合体の製造方法は、下記一般式(1)で示される不飽和モノカルボン酸系単量体(a)と下記一般式(2)で示される不飽和ポリアルキレングリコール系単量体(b)とを重合開始剤を用いて重合させる(メタ)アクリル酸(塩)系重合体の製造方法であって、前記重合開始剤として過酸化水素と還元剤とからなるレドックス系重合開始剤を用いるとともに、前記過酸化水素の使用量が前記単量体の合計量に対して0.01〜30モル%であることを特徴とする。
【0010】
【化1】

【0011】
(ただし、式中、R1は水素原子またはメチル基を表わし、Mは水素原子、一価金属、二価金属、アンモニウム基、または有機アミン基を表す。)
【0012】
【化2】
【0013】
(ただし、式中、R1は水素またはメチル基を表し、R2は−COO(R3O)mR4を表す。前記R2を表す一般式において、R3Oは炭素数2〜18のオキシアルキレン基の1種または2種以上を表し、2種以上の場合はブロック状に付加していてもランダム状に付加していてもよい。前記R2を表す一般式において、R4は水素または炭素数1〜22のアルキル基を表し、mはオキシアルキレン基の平均付加モル数であって1〜300の整数を表す。)
【発明を実施するための最良の形態】
【0014】
(重合)
本発明にかかる(メタ)アクリル酸(塩)系重合体の製造方法においては、前記一般式(1)で示される不飽和モノカルボン酸系単量体(a)と前記一般式(2)で示される不飽和ポリアルキレングリコール系単量体(b)とを重合開始剤を用いて重合させる。
本発明で使用する上記一般式(1)で示される不飽和モノカルボン酸系単量体(a)としては、アクリル酸、メタクリル酸ならびにこれらの酸の一価金属塩、二価金属塩、アンモニウム塩および有機アミン塩を挙げることができ、これらの1種または2種以上を用いることができる。本発明では、不飽和モノカルボン酸系単量体(a)として、これらの1種を単独で使用できるほか、2種以上を混合して使用してもよい。
【0015】
上記一般式(2)で示される不飽和ポリアルキレングリコール系単量体(b)としては、ヒドロキシエチル(メタ)アクリレート、ヒドロキシプロピル(メタ)アクリレート、ポリエチレングリコールモノ(メタ)アクリレート、ポリプロピレングリコールモノ(メタ)アクリレート、ポリブチレングリコールモノ(メタ)アクリレート、ポリエチレングリコールポリプロピレングリコールモノ(メタ)アクリレート、ポリエチレングリコールポリブチレングリコールモノ(メタ)アクリレート、ポリプロピレングリコールポリブチレングリコールモノ(メタ)アクリレート、ポリエチレングリコールポリプロピレングリコールポリブチレングリコールモノ(メタ)アクリレート、メトキシポリエチレングリコールモノ(メタ)アクリレート、メトキシポリプロピレングリコールモノ(メタ)アクリレート、メトキシポリブチレングリコールモノ(メタ)アクリレート、メトキシポリエチレングリコールポリプロピレングリコールモノ(メタ)アクリレート、メトキシポリエチレングリコールポリブチレングリコールモノ(メタ)アクリレート、メトキシポリプロピレングリコールポリブチレングリコールモノ(メタ)アクリレート、メトキシポリエチレングリコールポリプロピレングリコールポリブチレングリコールモノ(メタ)アクリレート、エトキシポリエチレングリコールモノ(メタ)アクリレート、エトキシポリプロピレングリコールモノ(メタ)アクリレート、エトキシポリブチレングリコールモノ(メタ)アクリレート、エトキシポリエチレングリコールポリプロピレングリコールモノ(メタ)アクリレート、エトキシポリエチレングリコールポリブチレングリコールモノ(メタ)アクリレート、エトキシポリプロピレングリコールポリブチレングリコールモノ(メタ)アクリレート、およびエトキシポリエチレングリコールポリプロピレングリコールポリブチレングリコールモノ(メタ)アクリレート等を挙げることができる。本発明では、不飽和ポリアルキレングリコール系単量体(b)として、これらの1種を単独で使用できるほか、2種以上を混合して使用してもよい。不飽和ポリアルキレングリコール系単量体(b)のオキシアルキレン基の平均付加モル数mは1〜300の整数であり、好ましくは1〜200の整数、より好ましくは1〜110の整数、さらに好ましくは1〜100の整数、さらに好ましくは2〜100の整数、さらに好ましくは2〜50の整数、さらに好ましくは2〜20の整数である。
【0016】
重合に用いる重合性単量体としては、上記不飽和モノカルボン酸系単量体(a)と不飽和ポリアルキレングリコール系単量体(b)に加え、さらにこれらと共重合可能な他の単量体(III)を含んでいてもよい。このような他の単量体(III)としては、マレイン酸、フマル酸、シトラコン酸、メサコン酸、イタコン酸等のジカルボン酸類;これらのジカルボン酸類とHO(R5O)rR6(ただし、R5Oは炭素原子数2〜4のオキシアルキレン基の1種または2種以上の混合物を表わし、2種以上の場合はブロック状に付加していてもランダム状に付加していてもよく、rはオキシアルキレン基の平均付加モル数であり1から300の整数を表わし、R6は水素または炭素原子数1〜22、好ましくは1〜15のアルキル基を表わす。)で表わされるアルコールとのモノエステルあるいはジエステル類;(メタ)アクリルアミド、(メタ)アクリルアルキルアミド等の不飽和アミド類;酢酸ビニル、プロピオン酸ビニル等のビニルエステル類;ビニルスルホン酸、(メタ)アリルスルホン酸、スルホエチル(メタ)アクリレート、2−メチルプロパンスルホン酸(メタ)アクリルアミド、スチレンスルホン酸等の不飽和スルホン酸類およびそれらの一価金属塩、二価金属塩、アルモニウム塩、有機アミン塩類;スチレン、α−メチルスチレン等の芳香族ビニル類;炭素原子数1〜18、好ましくは1〜15の脂肪族アルコールあるいはベンジルアルコール等のフェニル基含有アルコールと(メタ)アクリル酸とのエステル類;ポリアルキレングリコールモノ(メタ)アクリレート;ポリアルキレングリコールモノ(メタ)アリルエーテル等が挙げられ、これらの1種または2種以上を用いることができる。
【0017】
共重合させる単量体(a)、(b)および(III)の重量比((a)/(b)/(III))は、特に限定はされないが、好ましくは(1〜99)/(99〜1)/(0〜50)、より好ましくは(50〜99)/(50〜1)/(0〜49)、さらに好ましくは(60〜95)/(40〜5)/(0〜30)、特に好ましくは(70〜95)/(30〜5)/(0〜10)の範囲である。
本発明の製造方法においては、使用することのできる不飽和ポリアルキレングリコール系単量体(b)は、上記記載の単量体の中の1種単独で用いても良いし2種以上を混合して使用しても良いのであるが、2種以上を混合して使用する場合には、使用用途に応じた特性を発現させることができるように、発現特性の異なる種類を適当に組み合わせて用いることが望ましく、以下の2種の組み合わせが有利である。
【0018】
すなわち、不飽和ポリアルキレングリコール系単量体(b)としては、式(2)におけるオキシアルキレン基の平均付加モル数mが、好ましくは1〜297の整数、より好ましくは1〜97の整数、さらに好ましくは1〜10の整数である第1のエステル化物(a1)と、式(2)におけるオキシアルキレン基の平均付加モル数mが、好ましくは4〜300の整数、より好ましくは4〜100の整数、さらに好ましくは11〜100の整数である第2のエステル化物(a2)との混合物(ただし、第2のエステル化物(a2)の平均付加モル数の方が第1のエステル化物(a1)の平均付加モル数よりも3以上大きいものとする)が有利である。
【0019】
このような第1のエステル化物(a1)と第2のエステル化物(a2)との混合物を使用するには、これらの第1および第2のエステル化物(a1)および(a2)を別々にエステル化反応により製造してもよいし、それぞれ相当するアルコールの混合物と(メタ)アクリル酸とのエステル化反応により製造してもよく、特に後者の方法は工業的に安価の製造方法を提供できる。この場合、第1のエステル化物(a1)と第2のエステル化物(a2)との質量比は5:95〜95:5であることが好ましく、より好ましくは10:90〜90:10である。
第1のエステル化物(a1)としては、例えば、メトキシ(ポリ)エチレングリコールモノ(メタ)アクリレート、メトキシ(ポリ)プロピレングリコールモノ(メタ)アクリレート、メトキシ(ポリ)ブチレングリコールモノ(メタ)アクリレート、メトキシ(ポリ)エチレングリコール(ポリ)プロピレングリコールモノ(メタ)アクリレート、メトキシ(ポリ)エチレングリコール(ポリ)ブチレングリコールモノ(メタ)アクリレート、メトキシ(ポリ)プロピレングリコール(ポリ)ブチレングリコールモノ(メタ)アクリレート、メトキシ(ポリ)エチレングリコール(ポリ)プロピレングリコール(ポリ)ブチレングリコールモノ(メタ)アクリレート、エトキシ(ポリ)エチレングリコールモノ(メタ)アクリレート、エトキシ(ポリ)プロピレングリコールモノ(メタ)アクリレート、エトキシ(ポリ)ブチレングリコールモノ(メタ)アクリレート、エトキシ(ポリ)エチレングリコール(ポリ)プロピレングリコールモノ(メタ)アクリレート、エトキシ(ポリ)エチレングリコール(ポリ)ブチレングリコールモノ(メタ)アクリレート、エトキシ(ポリ)プロピレングリコール(ポリ)ブチレングリコールモノ(メタ)アクリレート、エトキシ(ポリ)エチレングリコール(ポリ)プロピレングリコール(ポリ)ブチレングリコールモノ(メタ)アクリレート等が例示される。第1のエステル化物(a1)は、その側鎖の短鎖アルコールに疎水性を有することが重要である。なお、共重合のし易さの面からは、側鎖はエチレングリコール単位が多く含まれているのが好ましい。したがって、(a1)としては、平均付加モル数が好ましくは1〜297の整数、より好ましくは1〜97の整数、さらに好ましくは1〜10の整数の(アルコキシ)(ポリ)エチレングリコールモノ(メタ)アクリレートが好ましい。
【0020】
第2のエステル化物(a2)としては、例えば、メトキシポリエチレングリコールモノ(メタ)アクリレート、メトキシポリエチレングリコール(ポリ)プロピレングリコールモノ(メタ)アクリレート、メトキシポリエチレングリコール(ポリ)ブチレングリコールモノ(メタ)アクリレート、メトキシポリエチレングリコール(ポリ)プロピレングリコール(ポリ)ブチレングリコールモノ(メタ)アクリレート、エトキシポリエチレングリコールモノ(メタ)アクリレート、エトキシポリエチレングリコール(ポリ)プロピレングリコールモノ(メタ)アクリレート、エトキシポリエチレングリコール(ポリ)ブチレングリコールモノ(メタ)アクリレート、エトキシポリエチレングリコール(ポリ)プロピレングリコール(ポリ)ブチレングリコールモノ(メタ)アクリレートなどが例示される。高い減水性を得るためには、第2のエステル化物(a2)の平均付加モル数が4〜300の整数のアルコール鎖による立体反発と親水性でセメント粒子を分散させることが重要であり、好ましい。そのためには、ポリアルキレングリコール鎖にはオキシエチレン基が多く導入されることが好ましく、ポリエチレングリコール鎖が最も好ましい。よって、第2のエステル化物(a2)のアルキレングリコール鎖の平均付加モル数nは、好ましくは4〜300の整数、より好ましくは4〜100の整数、さらに好ましくは11〜100の整数である。
【0021】
本発明にかかる(メタ)アクリル酸(塩)系重合体の製造方法において、重合方法は特に限定されるものではなく、溶液重合や塊状重合などの公知の方法を採用することができる。特に、反応の制御や、重合物の取り扱いやすさの点を考慮すると、溶液重合が好ましい。
溶媒中での重合は、回分式でも連続式でも行なうことができ、その際使用される溶媒としては、水;メチルアルコール、エチルアルコール、イソプロピルアルコール等の低級アルコール;ベンゼン、トルエン、キシレン、シクロヘキサン、n−ヘキサン等の芳香族あるいは脂肪族炭化水素;酢酸エチル等のエステル化合物;アセトン、メチルエチルケトン等のケトン化合物;テトラヒドロフラン、ジオキサン等の環状エーテル化合物等が挙げられる。原料のエステル化物の単量体成分および得られる共重合体の溶解性ならびに該共重合体の使用時の便からは、水および炭素原子数1〜4の低級アルコールよりなる群から選ばれた少なくとも1種を用いることが好ましい。その場合、炭素原子数1〜4の低級アルコールの中でもメチルアルコール、エチルアルコール、イソプロピルアルコール等が特に有効である。この際、水の配合比は、好ましくは20質量%以下、さらに好ましくは10質量%以下、よりさらに好ましくは5質量%以下、最も好ましくは1質量%以下である。この割合が20質量%を超えると、該重合体が分離及び/または沈殿する恐れがある。
【0022】
本発明にかかる(メタ)アクリル酸(塩)系重合体の製造方法において用いることができる重合開始剤としては特に限定されず、例えば従来公知のもの、具体的には、ベンゾイルパーオキシド、ラウロパーオキシド等のパーオキシド、クメンハイドロパーオキシド等のハイドロパーオキシド、アゾビスイソブチロニトリルなどの脂肪族アゾ化合物、過硫酸アンモニウムや過硫酸ナトリウム等の過硫酸塩などを用いることができるが、本発明においては特に、前記重合開始剤として過酸化水素と還元剤とからなるレドックス系重合開始剤を用いることが好ましい。
すなわち、本発明にかかる(メタ)アクリル酸(塩)系重合体の製造方法の一つの好ましい形態は、一般式(1)で示される不飽和モノカルボン酸系単量体(a)と一般式(2)で示される不飽和ポリアルキレングリコール系単量体(b)とを重合開始剤を用いて重合させる(メタ)アクリル酸(塩)系重合体の製造方法であって、前記重合開始剤として過酸化水素と還元剤とからなるレドックス系重合開始剤を用いるとともに、前記過酸化水素の使用量が前記単量体の合計量に対して0.01〜30モル%であることを特徴とする。
【0023】
レドックス系重合開始剤を用いることにより、重合温度が低くても迅速に重合反応が促進でき、かつ、過酸化水素を使用し、その使用量を前記単量体の合計量に対して0.01〜30モル%にすることで、オリゴマー量を少なくし、かつ着色を防止することができる。
従来は、(メタ)アクリル酸(塩)系重合体を製造する際に使用する重合開始剤は過酸化物の単独使用が一般的であり、レドックス系重合開始剤は使用されていなかった。レドックス系重合開始剤として過硫酸アンモニウム/亜硫酸水素ナトリウムを使用すると、重合後の製品中に硫酸ナトリム等の塩類が析出するため、純度が低下する。一方、過酸化物を使用すると反応効率を高めるためにレドックス系重合開始剤を使用する場合に比較して、重合温度を高くする必要がある。このため高温反応による不純物が発生しやすく場合によっては製品が着色した。しかしながら、本発明にかかる(メタ)アクリル酸(塩)系重合体の製造方法の一つの好ましい形態では、過酸化水素と還元剤とからなるレドックス系重合開始剤を使用し、特に過酸化水素の使用量を重合性単量体の合計量に対して0.01〜30モル%、さらに好ましくは0.1〜20モル%、最も好ましくは0.5〜10モル%とすることで、低い温度でも短時間で重合ができ、しかも着色が少なく、同時にオリゴマー量も少ない(メタ)アクリル酸(塩)系重合体を製造できることを見出したのである。過酸化水素の使用量が重合性単量体の合計量の0.01モル%未満であると、未反応の単量体量が多くなり、一方、30モル%を超えると、オリゴマー部分が多い(メタ)アクリル酸(塩)系重合体が得られることとなるため、好ましくない。
【0024】
還元剤としては、モール塩に代表されるような鉄(II)、スズ(II)、チタン(III)、クロム(II)、V(II)、Cu(II)等の低原子価状態にある金属の塩類;モノエタノールアミン、ジエタノールアミン、トリエタノールアミン、ヒドロキシルアミン、塩酸ヒドロキシルアミン、ヒドラジン等のアミン化合物もしくはその塩;亜硫酸ナトリウム、亜硫酸水素ナトリウム、メタ二亜硫酸塩等のアルカリ金属亜硫酸塩;次亜リン酸、次亜リン酸ナトリウム、ヒドロ亜硫酸ナトリウム、亜二チオン酸ナトリウム等の低級酸化物もしくはその塩;ホルムアルデヒドナトリウムスルホキシラート、ヒドロキシメタンスルフィン酸ナトリウム二水和物等の−SH基、−SO2H基、−NHNH2基、−COCH(OH)−基等の基を有する有機系化合物もしくはその塩;D−フルクトース、D−グルコース等の転化糖;チオウレア、二酸化チオウレア等のチオウレア化合物;L−アスコルビン酸、L−アスコルビン酸ナトリウム、L−アスコルビン酸エステル、エリソルビン酸、エリソルビン酸ナトリウム、エリソルビン酸エステルなどが挙げられる。本発明においては、特に有機系還元剤を使用することが好ましく、有機系還元剤の中でも特に好ましくはL−アスコルビン酸である。なお、上記還元剤の使用に際してアミン化合物等の促進剤を併用することもできる。
【0025】
本発明にかかる(メタ)アクリル酸(塩)系重合体の製造方法の一つの好ましい形態では、過酸化水素の使用量は、重合性単量体の0.01〜30モル%であるが、前記還元剤の使用量は、過酸化水素に対して、0.1〜500モル%とすることが好ましく、さらに好ましくは1〜200モル%、最も好ましくは10〜100モル%とするのがよい。0.1モル%未満であると、活性ラジカルが充分に発生せず、未反応単量体が多くなり、一方、500モル%を越えると、残存する還元剤が多くなり、また、得られた(メタ)アクリル酸(塩)系重合体を中和して高濃度で保存しておく際、還元剤由来の結晶が析出することがあるため、好ましくない。
【0026】
重合温度は、用いる溶媒や重合開始剤により適宜定められるが、0〜95℃であることが好ましく、より好ましくは30〜90℃、特に好ましくは50〜85℃である。95℃を越えると熱による不純物が発生し、製品を着色させる恐れがあり、その一方0℃を下回ると重合反応に長持間を必要とし、好ましくない。本発明では、上記温度範囲でもオリゴマー部分が少なく有効なポリマー部分が多い(メタ)アクリル酸(塩)系重合体が得られる点に特徴がある。
本発明の(メタ)アクリル酸(塩)系重合体の製造方法における重合圧力は、特に限定されず、加圧、常圧(大気圧)、減圧いずれかでのよく、場合により適宜設定すればよいが、常圧が好ましい。
【0027】
本発明では、得られる重合体の分子量調節のために、チオール系連鎖移動剤を併用することもできる。この際に用いられるチオール系連鎖移動剤は、一般式HS−R7−Eg(ただし、式中、R7は炭素原子数1〜2のアルキル基を表し、Eは−OH、−COOM2、−COOR8または−SO3M2基を表し、M2は水素、一価金属、二価金属、アンモニウム基または有機アミン基を表し、R8は炭素原子数1〜10のアルキル基を表わし、gは1〜2の整数を表す。)で表わされ、例えば、メルカプトエタノール、チオグリセロール、チオグリコール酸、2−メルカプトプロピオン酸、3−メルカプトプロピオン酸、チオリンゴ酸、チオグリコール酸オクチル、3−メルカプトプロピオン酸オクチル等が挙げられ、これらの1種または2種以上を用いることができる。
(エステル化工程)
次に、本発明において用いる前記一般式(2)で示される不飽和ポリアルキレングリコール系単量体(b)の製造方法の好適な実施の形態について説明する。
【0028】
不飽和ポリアルキレングリコール系単量体(b)の製造方法としては、一般式(3)で示されるアルコキシポリアルキレングリコールと不飽和モノカルボン酸系単量体(a)とをエステル化反応させる方法が好ましい。
上記エステル化反応(エステル化工程)は、反応系(反応槽)に、原料としての一般式(3)で示されるアルコキシポリアルキレングリコールおよび不飽和モノカルボン酸系単量体(a)、酸触媒、および、必要に応じて、脱水溶剤や重合禁止剤を仕込み、これら混合物を一定温度で所定のエステル化率になるまで行う。
上記エステル化反応に原料として使用されるアルコキシポリアルキレングリコールは、下記一般式(3)で示される化合物である。
【0029】
【化3】
【0030】
上記一般式(3)において、R4は、炭素原子数1〜30の炭化水素基を表す。好ましくは炭素原子数1〜25の炭化水素基、より好ましくは炭素原子数1〜20の炭化水素基、さらに好ましくは炭素原子数1〜15の炭化水素基である。R4が炭素原子数30を超える炭化水素基である場合には、式(3)のアルコキシポリアルキレングリコールと不飽和モノカルボン酸系単量体(a)とのエステル化物を、例えば、(メタ)アクリル酸と共重合して得られる共重合体の水溶性が低下し、用途性能、例えば、セメント分散性能などが低下する。好適なR4の範囲はその使用用途により異なるものであり、例えば、セメント分散剤の原料として用いる場合には、R4は、炭素原子数1〜18の直鎖若しくは枝分かれ鎖の飽和あるいは不飽和アルキル基およびアリール基が好ましい。R4としては、具体的には、例えば、メチル基、エチル基、n−プロピル基、イソプロピル基、n−ブチル基、tert−ブチル基、ペンチル基、ヘキシル基、オクチル基、ノニル基、2−エチルヘキシル基、デシル基、ドデシル基、ウンデシル基、トリデシル基、テトラデシル基、ペンタデシル基、ヘキサデシル基、ヘプタデシル基、オクタデシル基、ノナデシル基、エイコシル基、ヘンエイコシル基、ドコシル基などのアルキル基;フェニル基などのアリール基;ベンジル基、ノニルフェニル基などのアルキルフェニル基;シクロヘキシル基などのシクロアルキル基;アルケニル基;アルキニル基などが挙げられる。これらのうち、セメント分散剤の原料として用いる場合には、上述したように、メチル基、エチル基、プロピル基、ブチル基、フェニル基が好ましいものである。
【0031】
上記一般式(3)において、R3Oは、炭素原子数2〜18、好ましくは炭素原子数2〜8、より好ましくは炭素原子数2〜4のオキシアルキレン基である。R3Oが炭素原子数18を超えるオキシアルキレン基である場合には、式(3)のアルコキシポリアルキレングリコールと不飽和モノカルボン酸系単量体(a)とのエステル化物を、例えば、(メタ)アクリル酸と共重合して得られる共重合体の水溶性が低下し、用途性能、例えば、セメント分散性能等が低下する。R3Oとしては、例えば、オキシエチレン基、オキシプロピレン基、オキシブチレン基、オキシスチレン基などが挙げられ、これらのうち、オキシエチレン基、オキシプロピレン基及びオキシブチレン基であることが好ましい。また、繰り返し単位であるR3Oは同一であってもよく異なっていてもよい。前記繰り返し単位R3Oが異なる場合、すなわち、2種以上の異なる繰り返し単位を有する場合には、各R3Oはブロック状に付加していてもよくランダム状に付加していてもよい。
【0032】
上記一般式(3)において、nは1〜300の整数であり、好ましくは1〜200の整数、より好ましくは1〜110の整数、さらに好ましくは1〜100の整数、さらに好ましくは2〜50の整数、さらに好ましくは2〜20の整数であり、R3O(オキシアルキレン基)の繰り返し単位の平均付加モル数を表わす。nが300を超える場合には、式(3)のアルコキシポリアルキレングリコールと不飽和モノカルボン酸系単量体(a)とのエステル化物の重合性が低下し、nが110を超える場合はその重合性の低下は大きい。この平均付加モル数nも、エステル化反応により得られるエステル化物の使用目的に応じて、その最適範囲は異なるものであり、例えば、セメント分散剤の原料として使用する場合には、平均付加モル数nは、好ましくは2〜100の整数、より好ましくは5〜90の整数、最も好ましくは8〜80の整数である。また、増粘剤などとして用いる場合には、平均付加モル数nは、好ましくは10〜100の整数、より好ましくは50〜90の整数である。
【0033】
エステルの製造方法において、上記一般式(3)で示されるアルコキシポリアルキレングリコールは、1種のものを単独で使用してもあるいは2種以上の混合物の形態で使用してもよい。式(3)で示されるアルコキシポリアルキレングリコール原料が2種以上の混合物での使用形態は、特に制限されるものではなく、R4、R3Oまたはnの少なくともいずれか1つが異なる2種以上の混合物での使用形態であればよいが、好ましくは(i)R4がメチル基とブチル基の2種で構成されている場合;(ii)R3Oがオキシエチレン基とオキシプロピレン基の2種で構成されている場合;(iii)nが1〜10のものと11〜100のものの2種で構成されている場合;および(i)〜(iii)を適宜組み合わせたもの等が挙げられる。
【0034】
上記エステル反応に使用することのできる不飽和モノカルボン酸系単量体(a)としては、アクリル酸およびメタクリル酸を、それぞれ単独で使用しても、あるいは混合して使用してもよく、その混合比率に関しても任意の範囲を採用することができる。
上記エステル化反応で使用される上記原料の混合比率は、化学量論的には1:1(モル比)であるが、実際には、アルコキシポリアルキレングリコールと不飽和モノカルボン酸系単量体(a)とのエステル化反応が効率良く進行する範囲であれば特に制限されるものではない。通常、一方の原料を過剰に使用してエステル化反応を速めたり、目的のエステル化物の精製面からは、蒸留留去し易いより低沸点の原料を過剰に使用することが好ましい。また、本発明では、エステル化反応時に反応生成水と脱水溶剤を共沸する際に、低沸点の不飽和モノカルボン酸系単量体(a)の一部も留去され、反応系外に持ち出されるため、アルコキシポリアルキレングリコールの使用量(仕込み量)に対して不飽和モノカルボン酸系単量体(a)の使用量(仕込み量)を化学量論的に算出される量よりも過剰に加えることが好ましい。具体的には、不飽和モノカルボン酸系単量体(a)の使用量は、通常、アルコキシポリアルキレングリコール1モルに対して、好ましくは1.0〜30モル、より好ましくは1.2〜10モル、さらに好ましくは1.5〜10モル、最も好ましくは2〜10モルである。不飽和モノカルボン酸系単量体(a)の使用量がアルコキシポリアルキレングリコール1モルに対して1.0モル未満であると、エステル化反応が円滑に進行せず、目的とするエステル化物の収率が不十分であり、逆に30モルを超えると、添加に見合う収率の向上が認められず、不経済であり、やはり好ましくない。
【0035】
上記エステル化反応においては、反応を速やかに進行させるために、酸触媒の存在下で反応を行うことが望ましい。この際に使用することができる酸触媒としては、例えば、硫酸、メタンスルホン酸、パラトルエンスルホン酸、パラトルエンスルホン酸水和物、キシレンスルホン酸、キシレンスルホン酸水和物、ナフタレンスルホン酸、ナフタレンスルホン酸水和物、トリフルオロメタンスルホン酸、「Nafion」レジン、「Amberlyst 15」レジン、リンタングステン酸、リンタングステン酸水和物、塩酸などが挙げられる。この際、酸触媒は単独で使用されてもあるいは2種以上の混合物の形態で使用されてもよい。
【0036】
これらのうち、以下に詳述する脱水溶剤と水との共沸温度、エステル化反応温度などを考慮すると、酸触媒は、常圧における沸点が高いものであることが好ましい。具体的には、本発明に好ましく使用される酸触媒の常圧における沸点は、150℃以上、より好ましくは200℃以上である。ゆえに、硫酸(常圧における沸点:317℃)、パラトルエンスルホン酸(沸点:185〜187℃/13.3Pa(0.1mmHg))、パラトルエンスルホン酸水和物及びメタンスルホン酸(沸点:167℃/1333.2Pa(10mmHg))などが好ましく使用される。さらに、本発明者は、エステル化物の品質および性能の低下の原因となる不純物のジエステルの生成原因の1つが、アルコキシポリアルキレングリコールの切断によるものであり、さらに当該切断が酸触媒によっても起こり得ることを知得した。かかる知見に基づき、当該切断のしにくい酸触媒がより望ましいことを見出した。上記点を考慮すると、本発明において特に好ましく使用される酸触媒としては、パラトルエンスルホン酸、メタンスルホン酸が挙げられる。酸触媒の使用量としては、触媒作用を有効に発現することができる範囲であれば特に制限されるものではないが、好ましくは0.4ミリ当量/g以下であり、より好ましくは0.36〜0.01ミリ当量/g、特に好ましくは0.32〜0.05ミリ当量/gの範囲内である。酸触媒の使用量が0.4ミリ当量/gを超えると、エステル化反応時に反応系内で形成されるジエステルの量が増加し、例えば、エステル化反応により得られるエステル化物(アルコキシポリアルキレングリコールモノ(メタ)アクリル酸)を用いて合成されるセメント分散剤のセメント分散能が低下する。ここで、酸触媒の使用量(ミリ当量/g)は、反応に使用した酸触媒のH+の当量数(ミリ当量)を、原料であるアルコキシポリアルキレングリコール及び不飽和モノカルボン酸系単量体(a)の合計仕込み量(g)で割った値で表される。より具体的には下記式によって算出される値である。
【0037】
酸触媒の使用量(ミリ当量/g)=
酸触媒H+の当量数(ミリ当量)/(アルコキシポリアルキレングリコールの仕込み重量(g)+不飽和モノカルボン酸系単量体の仕込み重量(g))
上記エステル化反応において、酸触媒の反応系への添加のし方は、一括、連続、または順次行ってもよいが、作業性の面からは、反応槽に、原料と共に一括で仕込むのが好ましい。
【0038】
上記エステル化反応において、酸触媒の存在下でエステル化反応を行う際に、酸触媒を水和物および/または水溶液の形態で用いてもよい。
上記態様において使用することのできる酸触媒としては、例えば、硫酸、メタンスルホン酸、パラトルエンスルホン酸、キシレンスルホン酸、ナフタレンスルホン酸、トリフルオロメタンスルホン酸、「Nafion」レジン、「Amberlyst 15」レジン、リンタングステン酸、塩酸などを水和物および/または水溶液の形態で用いるものが挙げられ、これらのうち、硫酸、パラトルエンスルホン酸、メタンスルホン酸などを水和物および/または水溶液の形態で用いるものが好ましく使用される。これらは、1種単独で使用してもよいし、2種以上を混合して使用しても良い。さらに、本発明者は、上述したように、エステル化物の品質および性能の低下の原因となる不純物のジエステルの生成原因の1つが、アルコキシポリアルキレングリコール原料の切断によるものであり、さらに当該切断が酸触媒によっても起こり得ることを知得し、かかる知見に基づき、当該切断のしにくい酸触媒がより望ましいこと見出したものである。当該酸触媒としては、具体的には、パラトルエンスルホン酸を水和物および/または水溶液の形態で用いるものである。
【0039】
上記態様による酸触媒の使用量は、所望の触媒作用を有効に発現することができる範囲であれば特に制限されるものではないが、アルコール原料の切断作用の抑制、各種用途、例えば、セメント分散剤、炭酸カルシウム、カーボンブラック、インクなどの顔料分散剤、スケール防止剤、石膏・水スラリー用分散剤、CWM用分散剤、増粘剤等に使用される重合体成分の原料となるエステル化物としての有用性、このような使用用途に要求される基本性能である分散性能などに悪影響を及ぼす原因となる分散性能の乏しい高分子量架橋ポリマーを発生させる原因となるゲル発生の防止・抑制を考慮すると、該酸触媒の使用量が、原料のアルコキシポリアルキレングリコールと不飽和モノカルボン酸系単量体(a)の合計質量に対する該酸触媒中の酸の質量の比をX(質量%)とし、該酸触媒中の水和物および/または水溶液として存在する水分の質量の比をY(質量%)とした場合に、
0<Y<1.81X−1.62
の関係を満足することが好ましい。
【0040】
なお、誤解がないように具体例を挙げて説明すれば、例えば、パラトルエンスルホン酸一水和物を例にとれば、原料の合計質量に対するパラトルエンスルホン酸の質量の比がX(質量%)であり、原料の合計質量に対する一水和物として存在する水分の質量の比がY(質量%)であるのであって、決して、酸触媒以外の酸成分(例えば、原料の(メタ)アクリル酸など)や水分(例えば、エステル化反応により生ずる生成水など)は、ここでいうXおよびYの対象物となりえない。
この際、酸触媒の使用量が上記式の関係を満足しない場合には、以下のような問題が生じる。すなわち、Y=0の場合には、酸触媒中に水和物および/または水溶液として存在する水分が存在しないこととなり、エステル化反応時に反応系内で形成されるゲルの量が増加し、エステル化反応により得られるエステル化物を用いて合成されるセメント分散剤等の用途性能、例えば、セメント分散能等が低下する。また、Y≧1.81X−1.62となる場合には、エステル化反応時に反応系内で形成されるゲルの量が増加し、エステル化反応により得られるエステル化物を用いて合成されるセメント分散剤等の用途性能、例えば、セメント分散能等が低下する。
【0041】
上記態様において、酸触媒の反応系への添加のし方は、一括、連続、または順次行ってもよいが、作業性の面からは、反応槽に、原料と共に一括で仕込むのが好ましい。
上記エステル化反応は、原料としてのアルコキシポリアルキレングリコール、不飽和モノカルボン酸系単量体(a)、またはこれらの混合物の重合を防止するため、重合禁止剤の存在下で反応を行うことが望ましい。使用できる重合禁止剤としては、公知の重合禁止剤などが挙げられ、特に制限されるものではなく、例えば、フェノチアジン、トリ−p−ニトロフェニルメチル、ジ−p−フルオロフェニルアミン、ジフェニルピクリルヒドラジル、N−(3−N−オキシアニリノ−1,3−ジメチルブチリデン)アニリンオキシド、ベンゾキノン、ハイドロキノン、メトキノン、ブチルカテコール、ニトロソベンゼン、ピクリン酸、ジチオベンゾイルジスルフィド、クペロン、塩化銅(II)などが挙げられる。これらのうち、脱水溶剤や生成水の溶解性の理由から、フェノチアジン、ハイドロキノン及びメトキノンが好ましく使用される。これらの重合禁止剤は、単独で使用してもよいほか、2種以上を混合して使用することもできる。
【0042】
また、上記のように酸触媒を水和物および/または水溶液の形で用いる場合には、フェノチアジンが、反応系内に存在する水溶液中のゲル形成物質に対しても有効に機能することができるほか、後述するように、エステル化反応終了後に、脱水溶剤を水との共沸により留去する際にも、弱いながらも重合活性のあるハイドロキノンやメトキノン等の水溶性重合禁止剤を用いなくても極めて有効に重合禁止能を発揮することができ、高分子量体の形成を効果的におさえることができる点から極めて有用である。
重合禁止剤を使用する場合、重合禁止剤の使用量は、原料としてのアルコキシポリアルキレングリコールおよび不飽和モノカルボン酸系単量体(a)の合計仕込量に対して、好ましくは0.001〜1質量%、より好ましくは0.001〜0.1質量%の範囲内である。重合禁止剤の使用量が0.001質量%未満であると、重合禁止能の発現が十分でなく、原料としてのアルコキシポリアルキレングリコール、不飽和モノカルボン酸系単量体(a)、生成物としてのエステル化物またはこれらの混合物の重合を有効に防止しにくくなるため好ましくなく、重合禁止剤の使用量が1質量%を超えると、生成物であるエステル化物中に残留する重合禁止剤量が増えるため、品質及び性能面から好ましくなく、また、過剰に添加することに見合うさらなる効果も得られず、経済的な観点からも好ましくない。
【0043】
上記エステル化反応においては、脱水溶剤中で、エステル化反応を行うことが好ましい。本明細書中、脱水溶剤とは、反応生成水と共沸する溶剤として規定されるものである。すなわち、脱水溶剤を用いることにより、エステル化反応により生成する反応生成水を効率よく共沸させることができるものである。
脱水溶剤としては、例えば、ベンゼン、トルエン、キシレン、シクロヘキサン、ジオキサン、ペンタン、ヘキサン、ヘプタン、クロロベンゼン、イソプロピルエーテルなどが挙げられ、これらを単独で、あるいは2種以上のものを混合溶剤として使用することができる。これらのうち水との共沸温度が150℃以下、より好ましくは60〜90℃の範囲であるものが好ましく、具体的には、シクロヘキサン、トルエン、ジオキサン、ベンゼン、イソプロピルエーテル、ヘキサン、ヘプタンなどが挙げられる。水との共沸温度が150℃を超える場合には、取り扱いの面(反応時の反応槽内の温度管理および共沸物の凝縮液化処理などの制御等を含む)から好ましくない。
【0044】
上記脱水溶剤は、反応系外に反応生成水と共沸させ、反応生成水を凝縮液化して分離除去しながら還流させることが望ましく、この際、脱水溶剤の使用量は、原料としてのアルコキシポリアルキレングリコールおよび不飽和モノカルボン酸系単量体(a)の合計仕込量に対して、1〜100質量%、好ましくは2〜50質量%の範囲内である。脱水溶剤の使用量が1質量%未満であると、エステル化反応中に生成する反応生成水を共沸により反応系外に十分除去できず、エステル化の平衡反応が進行しにくくなるため、好ましくなく、脱水溶剤の使用量が100質量%を超えると、過剰に添加することに見合う効果が得られず、また、反応温度を一定に維持するために多くの熱量が必要となり、経済的な観点から好ましくない。
【0045】
上記エステル化反応において脱水溶剤を使用する際には、エステル化反応中の反応温度を好ましくは30〜150℃、より好ましくは60〜140℃とし、かつ、エステル化反応中の溶剤循環速度を好ましくは0.5サイクル以上/時間、より好ましくは1〜100サイクル以上/時間とすることが望ましい。これにより、反応温度を不純物形成温度領域(130℃超の領域)まで高くして反応させる必要もなく、反応槽内で不純物が形成するのを抑えることができる。また、溶剤循環速度を速めることで、反応槽内に反応生成水を長期間滞留させることなく効率よく反応槽から共沸により留出でき、平衡反応がエステル化の方向に進むため、反応時間も短くできるものである。
【0046】
本明細書において、エステル化反応中の溶剤循環速度とは、次のように定義されるものをいう。すなわち、反応槽に仕込んだ脱水溶剤の全量(体積量)に対して、エステル化反応中に、反応槽内の脱水溶剤を反応槽から循環経路を通して再び反応槽に戻し循環させることにより、反応槽に仕込んだ脱水溶剤の全量に相当する量(体積量)が循環されたときを1サイクルと規定し、エステル化反応中の溶剤循環速度は、単位時間(1時間)あたりの当該サイクル数で表されるものとし、その単位は「サイクル/時間」とする。したがって、例えば、5時間で、反応槽に仕込んだ脱水溶剤の全量に対して、これに相当する量の15倍の量が循環されたときには、溶剤循環速度は3サイクル/時間となる。同様に、2時間で、反応系に仕込んだ脱水溶剤の全量に対して、これに相当する量の半分(0.5倍)の量が循環されたときには、溶剤循環速度は0.25サイクル/時間となる。
【0047】
なお、反応系内の脱水溶剤を反応系から留出し凝縮液化して反応系に戻し循環させる際に循環されるもの(被循環対象物)には、脱水溶剤のほか、その実施態様によっては、少量ではあるが、留出される低沸点原料(主に、不飽和モノカルボン酸系単量体原料(a))、およびこの留出原料がゲルを形成して有害な不純物となるのを防止するために添加されるゲル化防止剤(重合禁止剤または該重合禁止剤を含む溶剤等)などの各種添加剤が含まれることもあり得る。そのため、ゲル化防止剤等の添加剤を使用する場合には、これにより溶剤循環速度がエステル化反応が進むにつれて変動することを考慮して設定条件を適当に調整するのが望ましい。
【0048】
上記反応温度および溶剤循環速度は、反応槽の加熱方法(手段)およびその装置を用いて反応槽に加えられる温度(熱量)及び反応槽に仕込む原料に対する脱水溶剤の使用量などによって所望の範囲に調整することができる。なお、反応温度は、反応槽内での最大(MAX)温度である。すなわち、加熱手段として用いられる装置(例えば、外部ジャケット、内部ヒータなど)の態様により、反応槽内の温度(反応温度)は、その位置によりばらつくほか、エステル化反応が進むにつれても上がり、時間の経過によっても変動するが、反応温度が高くなることで、不純物の形成を招くため、位置的及び時間的な条件に関わらず、如何なる位置及び時間であれ、上記に規定する上限温度を超えないことが必要であることから、ここでは、最大温度をもって規定することにしたものである。
【0049】
本発明において、エステル化反応は、回分または連続いずれによっても行いうるが、回分式で行うことが好ましい。
また、エステル化反応における反応条件は、エステル化反応が円滑に進行する条件であればよく、反応温度は、好ましくは30〜150℃、より好ましくは60〜140℃、さらに好ましくは90〜135℃、特に好ましくは100〜130℃である。なお、上記反応温度は、本発明の一般的なエステル化反応の条件であり、脱水溶剤を反応系外に反応生成水と共沸させ、反応生成水を凝縮液化して分離除去しながら還流させる場合は、その1例であり、これらの範囲内に含まれるが、完全に一致するものではない。反応温度が30℃未満では、エステル化反応が進行しづらく、反応生成水の脱水(留出)にも時間がかかり、また、脱水溶剤の還流が遅くて脱水に時間がかかり、ゆえに、エステル化反応に要する時間が長くなり好ましくない。逆に、反応温度が150℃を超えると、アルコキシポリアルキレングリコール原料の切断によって過大量のジエステルが生成して、セメント分散性能のほか、各種用途における分散性能や増粘特性が低下する。また、原料の重合が生じたり、留出物への原料の混入量が増すなど、生成物であるエステル化物の性能及び品質の劣化が生じるなど、やはり好ましくない。また、反応時間は、後述するようにエステル化率が好ましくは少なくとも70%、より好ましくは少なくとも80%に達するまでであるが、通常、好ましくは1〜50時間、より好ましくは3〜40時間である。さらに、上記エステル化反応は、常圧下または減圧下いずれで行ってもよいが、設備面から、常圧下で行うことが望ましい。
【0050】
上記エステル化反応におけるエステル化率は、好ましくは70%以上、より好ましくは70〜99%、最も好ましくは80〜98%である。エステル化率が70%未満であると、製造されるエステル化物の収率が不十分であり、これを原料として得られるセメント分散剤等の用途性能、例えば、セメント分散能等が低下する。なお、本明細書において使用される「エステル化率」は、下記に示すエステル化測定条件で、エステル化の出発物質であるアルコールの減少量を測定することにより、下記式によって算出される値として定義されるものである。
<エステル化率測定条件>
解析装置; Waters製 Millennium
クロマトグラフィーマネージャー
検出器; Waters製 410 RI検出器
使用カラム;GLサイエンス製 イナートシルODS−2 3本
カラム温度;40℃
溶離液; 水 8946g
アセトニトリル 6000g
酢酸 54g
を混合して30質量%水酸化ナトリウム水溶液で
pH4.0に調整
流速; 0.6ml/min
なお、上記の式によりエステル化率を決定しているため、エステル化率が100%を越えることはない。従って、エステル化率が規定以上に達した時点でエステル化反応が終了したものとする。
【0051】
本発明においては、前記一般式(2)で示される不飽和ポリアルキレングリコール系単量体(b)が、前記一般式(3)で示されるアルコキシポリアルキレングリコールのp重量部と不飽和モノカルボン酸系単量体(a)のq重量部とを、[(p/n1/2)/q]×100≦200(ただし、nはオキシアルキレン基の平均付加モル数。)の関係を満たす条件下でエステル化反応させることにより得られるエステル化反応物の形で前記重合に供されるものであることが好ましい。
これにより、不飽和モノカルボン酸系単量体(a)をアルコキシポリアルキレングリコールに比べて過剰に存在させてエステル化反応を行うと、得られたアルコキシポリアルキレングリコールモノ(メタ)アクリル酸系単量体は不飽和モノカルボン酸系単量体(a)を含む混合物の形態で存在するので、この混合物を単離せずにそのまま、あるいは必要により不飽和モノカルボン酸系単量体(a)やこれらの単量体と共重合可能な単量体を加えて、好ましくは混合物を単離せずにそのまま共重合反応に供することにより、ポリカルボン酸系共重合体が製造できるので好ましい。すなわち、アルコキシポリアルキレングリコール及び不飽和モノカルボン酸系単量体(a)の使用量を上記したような範囲内に調節することにより、アルコキシポリアルキレングリコールモノ(メタ)アクリル酸を単離するという工程を省略することができるため、量産に適しており、産業上の観点から好ましい。
【0052】
上記の式:[(p/n1/2)/q]×100の値は、「K値」とも称し、K値は、カルボン酸の質量当たりのポリアルキレングリコール鎖の平均数を表わす尺度である。本発明において、K値は、好ましくは42〜190(42≦K値≦190)、より好ましくは45〜160(45≦K値≦160)である、この際、K値が40未満であると、得られるセメント分散剤のセメント分散性能が十分でない。逆に、K値が200を超えると、得られるセメント分散剤のセメント分散性能がやはり低下する上、エステル化反応時間が著しく増大し、生産性が大幅に低下するので好ましくない。
上記式:[(p/n1/2)/q]×100≦200において、nはオキシアルキレン基の平均付加モル数であり、1〜300の整数であるが、好ましくは1〜200の整数、より好ましくは1〜110の整数、さらに好ましくは1〜100の整数、さらに好ましくは2〜100の整数、さらに好ましくは2〜50の整数、さらに好ましくは2〜20の整数である。nが300を超える場合には、式(3)のアルコキシポリアルキレングリコールと不飽和モノカルボン酸系単量体(a)とのエステル化物の重合性が低下し、特にnが110を超える場合はその重合性の低下は大きいのでいっそう好ましくない。
【0053】
すなわち、本発明においては、前記一般式(2)で示される不飽和ポリアルキレングリコール系単量体(b)が、前記一般式(3)で示されるアルコキシポリアルキレングリコールのp重量部と不飽和モノカルボン酸系単量体(a)のq重量部とを、[(p/n1/2)/q]×100≦200(ただし、nはオキシアルキレン基の平均付加モル数であって、nは1〜110の整数、さらに好ましくは1〜100の整数、さらに好ましくは2〜100の整数、さらに好ましくは2〜50の整数、さらに好ましくは2〜20の整数である。)の関係を満たす条件下で、酸触媒の存在下においてエステル化反応させることにより出発原料たる前記不飽和モノカルボン酸系単量体(a)の一部を残存させることが好ましい。
(アルコキシポリアルキレングリコール(アルキレンオキサイド付加物)の製造)
上記エステル化工程の原料であるアルコキシポリアルキレングリコールの製造について説明する。なお、以下の説明においてアルコキシポリアルキレングリコールをアルキレンオキサイド付加物と称する。
【0054】
本発明において用いることができるアルキレンオキサイド付加物の好ましい製造方法は、水酸基含有飽和化合物にアルキレンオキサイドを付加反応させてアルキレンオキサイド付加物を得る方法であって、水酸基含有飽和化合物にアルキレンオキサイドを付加させてアルキレンオキサイド低モル付加物を得る初期工程と、前記初期工程で得られたアルキレンオキサイド低モル付加物にアルキレンオキサイドをさらに付加させる付加モル数調整工程とを含む。
なお、上記付加モル数とは、平均付加モル数のことを意味し、水酸基1モルに対する平均付加モル数のことである。
【0055】
前記初期工程においては、アルキレンオキサイドの使用量を水酸基含有飽和化合物1モルに対して平均20モル以下とすることが重要である。このアルキレンオキサイドの使用量は、好ましくは平均18モル以下、より好ましくは平均15モル以下、さらに好ましくは平均12モル以下である。
これにより、該初期工程における反応前後の容量変化を通常の製造設備で対応できる範囲(例えば、好ましくは28倍以下、より好ましくは22倍以下、さらに好ましくは16倍以下、さらにより好ましくは10倍以下)に抑えることができる。
該初期工程で用いるアルキレンオキサイドの使用量が水酸基含有飽和化合物1モルに対して平均20モルを超えると、反応前後の容量変化が大きくなり、特殊な反応器形状や特殊な攪拌装置を用いなければならなくなる。また、水酸基含有飽和化合物の仕込み量を減らして通常の設備を使用することも考えられるが、この場合、装置から混入する水分量が多くなり、ポリアルキレンオキサイド等の副生成物が増加することになるので好ましくない。
【0056】
前記初期工程におけるアルキレンオキサイドの使用量の下限値は、本発明の効果を損なわない範囲であれば特に限定されないが、水酸基含有飽和化合物1モルに対して、好ましくは平均1モル以上、より好ましくは平均2モル以上、さらに好ましくは平均3モル以上、さらにより好ましくは平均5モル以上である。
前記初期工程において用いる水酸基含有飽和化合物は、水酸基を有する飽和の化合物であれば特に限定されないが、本発明の効果を十分に発現する上で、炭化水素系の水酸基含有飽和化合物が好ましい。この場合の炭化水素系とは、化合物重量の70重量%以上、好ましくは80重量%以上、より好ましくは90重量%以上が、炭素、水素、酸素の3つの原子により構成された化合物のことをいう。さらに、本発明の効果をより十分に発現する上で好ましくは炭素数1〜30の飽和アルコールである。飽和アルコールの炭素数は、好ましくは1〜20、より好ましくは1〜15、さらに好ましくは1〜9、さらにより好ましくは1〜6である。飽和アルコールの炭素数が30を超える場合には、例えば、アルキレンオキサイド付加反応、エステル化反応またはエステル交換反応、重合反応を経て、共重合体とした場合に、かかる共重合体の水溶性が低下し、用途性能、例えば、セメント分散性能などが低下することがあるために好ましくない。
【0057】
前記初期工程において用いる水酸基含有飽和化合物として炭素数1〜30の飽和アルコールを用いる場合、具体的には、例えば、メチル基、エチル基、n−プロピル基、イソプロピル基、n−ブチル基、オクチル基、2−エチルヘキシル基、ドデシル基、ヘキサデシル基、オクタデシル基、などのアルキル基;フェニル基などのアリール基;ベンジル基、ノニルフェニル基などのアルキルフェニル基;シクロヘキシル基などのシクロアルキル基などを有する炭素数1〜30の飽和アルコールが挙げられる。これらの中でも、本発明の効果を十分に発現するためには、メタノール、エタノール、プロパノール、ブタノール、フェノールが好ましく、特にメタノールが好ましい。
【0058】
前記初期工程においては、含水率が6000ppm以下である水酸基含有飽和化合物を用いることが好ましい。より好ましくは4000ppm以下、さらに好ましくは2000ppm、さらにより好ましくは1000ppm以下、特に好ましくは500ppm以下である。このように含水率を6000ppm以下とすることにより、初期工程におけるポリアルキレンオキサイド等の副生成物の生成をさらに抑制することができると同時に、続く付加モル数調整工程においても、ポリアルキレンオキサイド等の副生成物の生成をさらに抑制することができる。含水率が6000ppmを超えると、初期工程におけるポリアルキレンオキサイド等の副生成物が増加し、得られたアルキレンオキサイド低モル付加物には多量のポリアルキレンオキサイドが含まれることとなる。そして、続く付加モル数調整工程において、不純物として含まれるポリアルキレンオキサイドがさらにアルキレンオキサイドと付加して分子量の大きなポリアルキレンオキサイドが副生することになるので、例えば、得られたアルキレンオキサイド付加物をセメント添加剤用ポリマーの原料として用いた場合に、セメント分散剤としての性能を低下させることとなり、好ましくない。
【0059】
本発明において用いることのできる前記アルキレンオキサイドとしては、特に制限されるものではないが、炭素数2〜4のアルキレンオキサイドが好ましく、具体的には、例えば、エチレンオキサイド、プロピレンオキサイド、ブチレンオキサイド等が挙げられ、これらの1種または2種以上を用いることができる。
前記付加モル数調整工程においては、前記初期工程で得られたアルキレンオキサイド低モル付加物にアルキレンオキサイドを付加させるので、水酸基含有飽和化合物に同モルのアルキレンオキサイドを直接付加反応させる場合よりも反応前後の容量変化が小さく、特殊な反応器形状や特殊な攪拌装置をもたない通常の設備で、所望の付加モル数にまでアルキレンオキサイドを付加させることができる。
【0060】
本発明においては、前記付加モル数調整工程では前記初期工程で得られたアルキレンオキサイド低モル付加物全量中の一部の量を使用することが好ましい。このように、付加モル数調整工程において初期工程で得られたアルキレンオキサイド低モル付加物の全量を用いずに一部の量を使用することによって、仕込み容量に対する生成物の容量の増大を考慮することなく、通常の設備を用いて、アルキレンオキサイドの付加モル数が高いアルキレンオキサイド付加物を製造することができる。さらに、この方法によれば、生成物の容量を考慮して原料の仕込み量を少なくする必要がないので、かかる原料の仕込み量に対する、装置から混入する水分の相対量の増加を防ぐことができ、ポリアルキレンオキサイド等の副生成物の増大が抑えられる。
【0061】
上述のように、前記付加モル数調整工程では前記初期工程で得られたアルキレンオキサイド低モル付加物全量中の一部の量を使用する場合、使用しなかった残りは、さらに付加モル数調整工程に使用してもよい。この場合、残りの全部を使用しなくてもよい。
前記初期工程で得られたアルキレンオキサイド低モル付加物の全量のうち、一部の量を使用して前記付加モル数調整工程を行い、さらに残りの少なくとも一部を用いて、1回または2回以上に分けて付加モル数調整工程を行うことが、本発明の効果を十分に発現する上で好ましい。より好ましくは、前記残りの少なくとも一部を用いて行う付加モル数調整工程が1回または2回、特に好ましくは1回である。
【0062】
付加モル数調整工程において用いるアルキレンオキサイド低モル付加物の使用量は、該工程で使用する反応器の全容量の60%以下とすることが好ましい。より好ましくは55%以下、さらに好ましくは50%以下、さらにより好ましくは45%以下、特に好ましくは40%以下である。かかる付加モル数調整工程では、反応前後の容量変化は小さいので、通常の設備を用いて、アルキレンオキサイドの付加モル数を向上させることができるのであるが、アルキレンオキサイド低モル付加物の使用量が反応器の全容量の60%を超えると、通常の設備においてはアルキレンオキサイドの使用量が制限され、所望の付加モル数にまでアルキレンオキサイドを付加させることが困難になる場合がある。
【0063】
付加モル数が非常に高いアルキレンオキサイド付加物を得ようとする場合、前記付加モル数調整工程を2回以上に分けて行うことが好ましい。具体例を挙げると、例えば、付加モル数が50以上のアルキレンオキサイド付加物を所望する場合には、一例として、第1回目の付加モル数調整工程でアルキレンオキサイドの10モル付加物を製造し、次いで、該アルキレンオキサイドの10モル付加物に40モルのアルキレンオキサイドをさらに付加させればよい。
前記付加モル数調整工程は、前記初期工程後、得られたアルキレンオキサイド低モル付加物の一部を抜き出して貯蔵タンク等に移し、前記初期工程と同一の反応器を用いて行ってもよいし、初期工程で得られたアルキレンオキサイド低モル付加物の一部を別の反応器に移し、前記初期工程と異なる反応器で行ってもよい。
【0064】
前記付加モル数調整工程においては、アルキレンオキサイドの使用量をアルキレンオキサイド低モル付加物1モルに対して平均20モル以下とすることが好ましい。このアルキレンオキサイドの使用量は、より好ましくは平均18モル以下、さらに好ましくは平均15モル以下、さらにより好ましくは平均12モル以下である。
これにより、該付加モル数調整工程における反応前後の容量変化を通常の製造設備で対応できる範囲(例えば、好ましくは28倍以下、より好ましくは22倍以下、さらに好ましくは16倍以下、さらにより好ましくは10倍以下)に抑えることができる。
該付加モル数調整工程で用いるアルキレンオキサイドの使用量がアルキレンオキサイド低モル付加物1モルに対して平均20モルを超えると、反応前後の容量変化が大きくなり、特殊な反応器形状や特殊な攪拌装置を用いなければならなくなる。また、アルキレンオキサイド低モル付加物の仕込み量を減らして通常の設備を使用することも考えられるが、この場合、装置から混入する水分量が多くなり、ポリアルキレンオキサイド等の副生成物が増加することになるので好ましくない。
【0065】
前記付加モル数調工程におけるアルキレンオキサイドの使用量の下限値は、本発明の効果を損なわない範囲であれば特に限定されないが、アルキレンオキサイド低モル付加物1モルに対して、好ましくは平均1モル以上、より好ましくは平均2モル以上、さらに好ましくは平均3モル以上、さらにより好ましくは平均5モル以上である。
アルキレンオキサイドを付加させる際には、触媒として、例えば、アルカリ金属、アルカリ土類金属、およびそれらの水酸化物が好ましく、より好ましくは、ナトリウム、ナトリウムアマルガム、ナトリウムハライド、水酸化ナトリウム、カリウム、カリウムアマルガム、カリウムハライド、水酸化カリウム等の1種または2種以上を用いることができる。これら触媒は、付加モル数調整工程では初期工程で得られた生成物中に残存する触媒が作用するので、初期工程でのみ添加するようにしてもよいが、初期工程と付加モル数調整工程の両方において添加することが好ましい。なお、触媒の使用量については特に制限はないが、アルキレンオキサイド付加物に対して10〜5000ppmとするのが好ましい。
【0066】
前記初期工程における反応温度については、特に制限はないが、好ましくは60〜180℃の範囲内、より好ましくは60〜160℃の範囲内、さらに好ましくは70〜150℃の範囲内とするのがよい。反応温度が180℃を超えると、ポリアルキレンオキサイド等の副生成物が増える傾向があり、例えば、得られたアルキレンオキサイド付加物を用いてセメント分散剤用ポリマーを得た場合、減水性能等の性能が低下する傾向がある。一方、反応温度が60℃未満であると、付加速度が遅くなり、生産性が低下するので好ましくない。
前記付加モル数調整工程における反応温度については、特に制限はないが、好ましくは80〜180℃の範囲内、より好ましくは90〜170℃の範囲内、さらに好ましくは100〜160℃の範囲内とするのがよい。反応温度が180℃を超えると、ポリアルキレンオキサイド等の副生成物が増える傾向があり、例えば、得られたアルキレンオキサイド付加物を用いてセメント分散剤用ポリマーを得た場合、減水性能等の性能が低下する傾向がある。一方、反応温度が80℃未満であると、付加速度が遅くなり、生産性が低下するので好ましくない。
【0067】
前記初期工程および前記付加モル数調整工程における付加反応は、回分式で行ってもよく、連続式で行ってもよいが、通常、水酸基含有飽和化合物もしくはアルキレンオキサイド低モル付加物、および必要に応じて前記触媒中に、アルキレンオキサイドを連続的に投入して行われる。また、前記付加反応は、窒素、アルゴン、ヘリウムなどの不活性ガス雰囲気下で行うのが好ましく、窒素雰囲気下で行うのが特に好ましい。また、前記付加反応は、加圧下で行うのが好ましい。
アルキレンオキサイド付加物の製造方法によってアルキレンオキサイド付加物を製造する場合の反応器の大きさは、特に限定されないが、反応器の容量が大きくなるほど本発明の効果がより十分に発現できるので、好ましくは100L以上、より好ましくは500L以上、さらに好ましくは1m3以上、さらにより好ましくは5m3以上の製造スケールが有効である。
(部分中和)
本発明においては、さらに、エステル化反応後に塩基性物質で酸触媒と残存不飽和モノカルボン酸系単量体(a)の0〜60モル%を中和させることが好ましい。
【0068】
エステル化反応後に脱水溶剤を留去する工程で水を加えて共沸する場合や、あるいはエステル化物を用いてさらに重合を行うためにエステル反応後に調整水を加えて生成されたエステル化物水溶液を作製する場合には、酸触媒によるエステル化物の加水分解が生じ、エステル化物の品質及び性能の低下を招くほか、加水分解により生じたもの(以下、単に加水分解生成物ともいう)がエステル化物中に残留し、当該エステル化物を用いてセメント分散剤等の各種分散剤や増粘剤等に使用される重合体を合成する場合には該加水分解生成物は重合には関与しない不純物となり、重合率(ひいては生産性)が低下し、また重合体の品質や性能の劣化にもつながる。かかる課題を解決するべく鋭意検討した結果、エステル化反応後に塩基性物質で酸触媒と残存不飽和モノカルボン酸系単量体(a)の0〜60モル%を中和させればよいことを見出した。これにより、エステル化反応後の処理過程で、加水分解生成物を生じることもなく、高純度で高品質のエステル化物を得ることができる。
【0069】
ここで、部分中和工程の好適な実施の形態につき、以下に説明する。
本発明に好適な部分中和工程では、エステル化反応終了後、好ましくは90℃以下、より好ましくは50〜0℃の範囲で酸触媒と残存不飽和モノカルボン酸系単量体(a)の0〜60モル%を塩基性物質で中和するものである。
上記部分中和工程での中和温度(反応系の液温)が、90℃を超える場合には、添加される塩基性物質が加水分解の触媒として作用し、加水分解生成物を多量に生成するようになるため好ましくない。さらに、50℃以下では、塩基性物質が加水分解の触媒として作用することはなく、加水分解生成物の発生を完全に抑えることができる。一方、0℃未満の場合には、エステル化反応液が粘稠になり、中和時の撹拌がしづらくなるほか、エステル化反応後に所定の温度まで降温するのに長時間を要するほか、室温よりも低い温度まで降温するには、新たに冷却手段(装置)を設ける必要があり、コストアップになるためあまり望ましくない。
【0070】
また、上記部分中和工程で使用することのできる塩基性物質(中和剤)としては、特に制限されるものではなく、水酸化物M(OH)nの形式をとり、水に溶解し、塩基性を示す物質であればよく、この場合のMは、アルカリ金属、アルカリ土類金属、アンモニウム基などをさす。さらに、アルカリ金属の炭酸塩や燐酸塩、アンモニア、アミン等もここでいう塩基性物質に含まれる。よって、塩基性物質としては、具体的には、例えば、水酸化ナトリウム、水酸化カリウム等のアルカリ金属の水酸化物、水酸化マグネシウム、水酸化カルシウム等のアルカリ土類金属の水酸化物、アンモニア、アミン等が挙げられるが、セメントに配合した場合に異臭が発生しないとの理由から、好ましくはアルカリ金属やアルカリ土類金属の水酸化物、炭酸塩、燐酸塩等である。また、本発明では、これら塩基性物質を1種若しくは2種以上を適当な比率で混合して使用してもよい。
【0071】
上記塩基性物質を用いて中和する酸は、酸触媒と残存不飽和モノカルボン酸系単量体(a)の0〜60モル%である。ここで、中和される不飽和モノカルボン酸系単量体(a)は残存不飽和モノカルボン酸系単量体(a)の0〜60モル%であり、好ましくは0〜55モル%、より好ましくは1〜55モル%、さらに好ましくは1〜50モル%、さらに一層好ましくは5〜50モル%、特に好ましくは5〜40モル%である。また、中和される不飽和モノカルボン酸系単量体(a)は、エステル化反応に使用した不飽和モノカルボン酸系単量体(a)の10質量%以下が好ましく、より好ましくは0.01〜5質量%の範囲である。従って、塩基性物質(中和剤)の添加量は、酸触媒1当量に対して1.0〜100当量が好ましく、より好ましくは1.0〜10当量、さらに好ましくは1.01〜2当量である。
【0072】
中和される不飽和モノカルボン酸系単量体(a)が、エステル化反応に使用した不飽和モノカルボン酸系単量体(a)の10質量%を超える場合には、おそらく、塩の形態の不飽和モノカルボン酸系単量体(a)の重合速度が塩の形態を形成していない不飽和モノカルボン酸系単量体(a)に比べて遅いために、得られたエステル化物を用いて重合する際の重合率が低下するため好ましくない。また、塩基性物質(中和剤)の添加量は、酸触媒1当量に対して1.0当量未満の場合には、酸触媒を完全に中和できず、加水分解生成物を多量に生じるようになるため好ましくない。逆に塩基性物質(中和剤)の添加量は、酸触媒1当量に対して100当量を超える場合にも、大量の不飽和モノカルボン酸系単量体(a)が中和され、やはり、塩の形態の不飽和モノカルボン酸系単量体(a)の重合速度が塩の形態を形成していない不飽和モノカルボン酸系単量体(a)に比べて遅いために、得られたエステル化物を用いて重合する際の重合率が低下するため好ましくない。
【0073】
中和すべき酸が酸触媒である理由は、上述したように、酸触媒がエステル化反応後に添加される水と強く反応し、加水分解生成物を生じさせるため、酸触媒を不活性にする必要があるためである。なお、酸成分としては、酸触媒以外に残存不飽和モノカルボン酸系単量体(a)が存在し得るが、酸触媒の方が酸強度が大きいので、酸触媒から優先的に中和される。
なお、添加される塩基性物質の形態としては、特に制限されるものではないが、アルカリ水溶液の形態とすることが、エステル化物の加水分解を防止する観点から好ましいといえる。
【0074】
特に、脱水溶剤中でエステル化反応を行う場合、塩基性物質と共に多量の水を反応系に添加するのが、エステル化物の加水分解を防止するためには好適である。すなわち、多量の水が無い反応系では、塩基性物質が脱水溶剤に難溶であるために濃い状態で系内に浮遊し、この高濃度の塩基性物質の浮遊は中和に消費されるまでの長持間にわたって消失せず、エステル化物の加水分解を引き起こす。該水の添加量は、塩基性物質の使用形態にもよるが、例えば、40〜60%のアルカリ水溶液を中和剤として添加する場合には、該アルカリ水溶液とは別に、該アルカリ水溶液の1質量部に対して通常5〜1000質量部が好ましく、より好ましくは10〜100質量部である。この場合に、水の添加量が、5質量部未満の場合には、上記理由で塩基性物質が反応系内で不均一になり、高濃度の塩基性物質がエステル化物の加水分解を引き起こし、1000質量部を超える場合には、生産性を確保するために中和槽が別途必要になるなどコスト高につながり好ましくない。
(pH調整)
本発明においては、好ましくは上記部分中和を行うことによって、重合反応の反応液のpHを3.2〜7.0の範囲内となるようにして重合反応を行うことが好ましい。重合反応の反応液のpHを3.2〜7.0の範囲内とすることにより、重合反応の重合率を高めることができるので好ましい。また、重合反応の反応液のpHを3.2〜7.0の範囲内とすることにより、重合装置(釜)の腐食を抑制することができるので好ましい。
【0075】
上記pHの範囲は、より好ましくはpH3.3〜7.0、さらに好ましくは3.5〜6.5、特に好ましくは4.0〜5.2である。
上記pH範囲を外れると、重合反応の重合率が低下し、また、重合装置(釜)の腐食がはげしいためコスト高となり、不経済である。
上記pH範囲で製造された重合体は、着色度が極めて低く、重量平均分子量Mwとピークトップ平均分子量Mpとの差であるMw−Mpも0〜9000の範囲に入るので、該重合体をセメント分散剤として使用する場合に、着色がなく、また分散性が極めて良好で、かつ減水効果が極めて高い。
【0076】
以上から、本発明にかかる(メタ)アクリル酸(塩)系重合体の製造方法における特に好ましい態様は、前記一般式(1)で示される不飽和モノカルボン酸系単量体(a)と前記一般式(2)で示される不飽和ポリアルキレングリコール系単量体(b)とを重合開始剤を用いて重合させる(メタ)アクリル酸(塩)系重合体の製造方法であって、前記一般式(2)で示される不飽和ポリアルキレングリコール系単量体(b)が、前記一般式(3)で示されるアルコキシポリアルキレングリコールのp重量部と不飽和モノカルボン酸系単量体(a)のq重量部とを、[(p/n1/2)/q]×100≦200(ただし、nはオキシアルキレン基の平均付加モル数であって、nは1〜110の整数、さらに好ましくは1〜100の整数、さらに好ましくは2〜100の整数、さらに好ましくは2〜50の整数、さらに好ましくは2〜20の整数である。)の関係を満たす条件下で、酸触媒の存在下においてエステル化反応させることにより出発原料たる前記不飽和モノカルボン酸系単量体(a)の一部を残存させるとともにエステル化反応後に塩基性物質で前記酸触媒と前記残存不飽和モノカルボン酸系単量体(a)の0〜60モル%を中和させるようにして得られるエステル化反応物の形で前記重合に供されるものであり、かつ、前記重合を、重合反応液のpHが3.2〜7.0の範囲内となるようにして行う方法である。
(溶剤留去工程:ゲル化防止)
本発明の製造方法においては、溶剤留去工程においてゲル化防止を行うことが好ましい。
【0077】
本発明において好ましく適用できるゲル化防止について、2つの実施態様を中心に説明する。
本発明者は、エステル化反応時に生成する反応生成水を留出させ、この反応生成水を含む留出物に対してゲル化防止剤を作用させることが望ましいことを見出した。以下、説明の便宜上、この態様を実施態様(A)と称することがある。
実施態様(A)により、反応系内の反応生成水を反応系外に留出してから凝縮液化し分離除去する間に、反応生成水とともに反応系外に留出されてくる低沸点の原料である(メタ)アクリル酸等により生ずるゲル状物(ポリ(メタ)アクリル酸等)の発生そのものを効果的に防止する、すなわち、製品の品質劣化や装置類の閉塞等の原因になるゲル状物を形成するのを防止することができる。
【0078】
実施態様(A)において、エステル化反応時に生成する反応生成水などの留出物に対して作用させるために用いられる上記ゲル化防止剤としては、反応生成水と共に留出されてくる低沸点の原料の留出段階、特に凝縮段階での重合反応を抑えることができ、反応槽からコンデンサへの立ち上がり管のフランジ部などで発生するゲルの形成、即ち、コンデンサのチューブや反応槽とコンデンサとの間の連結管のつまりを抑制できるものであれば特に制限されるものではない。上記ゲル化防止剤は、反応系内で同様の目的を持って使用される重合禁止剤と何ら変わるものではなく、上記に説明したと同様に使用できる。また、従来既知の各種ゲル化防止剤の中から適宜選択して使用することができる。上記ゲル化防止剤としては、具体的には、例えば、フェノチアジン、トリ−p−ニトロフェニルメチル、ジ−p−フルオロフェニルアミン、ジフェニルピクリルヒドラジル、N−(3−N−オキシアニリノ−1,3−ジメチルブチリデン)アニリンオキシド、ベンゾキノン、ハイドロキノン、メトキノン、ブチルカテコール、ニトロソベンゼン、ピクリン酸、ジチオベンゾイルジスルフィド、クペロン、塩化銅(II)などが挙げられる。これらの中でも、脱水溶剤や生成水の溶解性の理由から、フェノチアジン、ハイドロキノン、メトキノンが好ましく使用される。これらのゲル化防止剤は、単独で使用してもよいほか、2種以上を混合して使用することもできる。
【0079】
上記ゲル化防止剤の添加量としては、エステル化反応条件、特に反応系に加える熱量や反応系内に仕込む脱水溶剤量等に応じて、低沸点原料の留出量に見合う量、すなわち、共沸物の留出開始時からエステル化反応終了まで逐次留出されてくる低沸点原料に対して常にゲルの形成を効果的に防止することができる量を適宜添加すればよく、原料であるアルコキシポリアルキレングリコールおよび不飽和モノカルボン酸系単量体(a)の仕込み量に対して、好ましくは0.1〜1000質量ppm、より好ましくは1〜500質量ppmの範囲で添加することで上記目的を達成することができる。原料の仕込み量に対して0.1質量ppm未満の場合には、ゲル状物が生成する場合があり、共沸物の留出開始時からエステル化反応終了まで逐次留出されてくる低沸点原料に対して、常に重合禁止能を有効に発現させる上で不十分な量と言える。一方、原料仕込み量に対して1000質量ppmを超える場合には、ゲル形成防止(重合禁止)能を有効に発現させるには十分過ぎる量であり、過剰な添加に見合う更なる効果の発現が見込めず不経済となる。なお、添加量の全量を一時に加えたのでは、共沸物の留出開始時からエステル化反応終了まで逐次留出されてくる低沸点原料に対してゲルの形成を有効に阻止することができにくいため、共沸物の留出に呼応する形で、共沸物の留出開始時からエステル化反応終了まで逐次的に(連続的に)一定量ずつを添加し、最終的な添加量の総計が上記範囲となるように調整することが望ましい。
【0080】
上記ゲル化防止剤の作用のさせかた(作用形態や作用させる領域など)としては、反応系外に留出された低沸点原料(流体物)に対して有効に作用(接触)させることができるものであれば、特に制限されるものではなく、例えば、凝縮液化させる前のガス状の留出物に対して作用させてもよいし、凝集液化により液化した液状の留出物に対して作用させてもよい。また、上記の双方の作用方法を活用してもよい。
以下に、上記ゲル化防止剤の好適な作用方法を、作用形態ごとに例を挙げて説明する。本発明では、これらを適当に組み合わせることができるほか、従来既知の他の作用方法を適宜利用することができる。なお、下記に例示する作用方法は、当業者が本発明を容易に理解することができるように代表的なものを例示的に示したものであり、本発明がこれらに限定されるものではないことはいうまでもない。
【0081】
(i)ゲル化防止剤を液化(溶解)した状態で作用させる方法:適当な溶剤、好ましくは反応系に仕込む脱水溶剤と同種の溶剤にゲル化防止剤を溶かして液状にしたものを、反応生成水を含む留出物(好ましくは溶剤−水共沸物)を凝縮させる領域、具体的には、反応生成水を含む留出物の凝縮液化が行われるコンデンサ内部の凝縮部に、好ましくはコンデンサの上部(とりわけ塔頂部近傍)からその内部に該留出物と並流接触するように滴下ないし噴霧するものである。また、コンデンサのタイプ等によっては、ゲル化防止剤を含む溶液をコンデンサ内部に仕込んでおいて、これにガス状の留出物を吹き込むあるいは液化した留出物を流し込むようにして接触(相溶ないし分散)させるようにしてもよい。さらに上記態様では、ゲル化防止剤の作用部位をコンデンサ内部の凝縮部としたが、上記部位に加えて、反応槽とベーパーの立ち上がりラインとの間の接合部(フランジ部)やベーパーラインとコンデンサ塔頂部との間のフランジ部等のフランジ部、反応槽等に設置された温度計やのぞき窓に設けられた突起部など、ゲルが形成されやすい部位であってもよい。これらのうち、コンデンサ内部の凝縮部(とりわけ塔頂部近傍)、反応槽とベーパーの立ち上がりラインとの間のフランジ部やベーパーラインとコンデンサ塔頂部との間のフランジ部が、好ましいゲル化防止剤の作用部位である。また、上記作用部位は、一箇所でなくてもよく、必要に応じて、複数箇所を同時に設けてもよい。
【0082】
(ii)ゲル化防止剤を固化した状態で作用させる方法:粉末状のゲル化防止剤を、反応生成水を含む留出物を凝縮させる領域、具体的には、反応生成水を含む留出物の凝縮液化が行われるコンデンサ内部の凝縮部に、好ましくはコンデンサの上部(とりわけ塔頂部近傍)からコンデンサ内部に該留出物と並流接触するように投下ないし散布して降らせるものである。また、コンデンサのタイプなどによっては、一定粒度のゲル化防止剤を予めコンデンサ内部に積載ないし充填などして仕込んでおいて接触させるようにしてもよい。さらに上記態様では、ゲル化防止剤の作用部位をコンデンサ内部の凝縮部としたが、上記部位に加えて、反応槽とベーパーの立ち上がりラインとの間の接合部(フランジ部)やベーパーラインとコンデンサ塔頂部との間のフランジ部等のフランジ部、反応槽等に設置された温度計やのぞき窓に設けられた突起部など、ゲルが形成されやすい部位であってもよい。これらのうち、コンデンサ内部の凝縮部(とりわけ塔頂部近傍)、反応槽とベーパーの立ち上がりラインとの間のフランジ部やベーパーラインとコンデンサ塔頂部との間のフランジ部が好ましいゲル化防止剤の作用部位である。また、上記作用部位は、一箇所でなくてもよく、必要に応じて、複数箇所を同時に設けてもよい。
【0083】
(iii)ゲル化防止剤を気化した状態で作用させる方法:ゲル化防止剤を気化(昇華したものを含む)させて、ガス状の反応生成水を含む留出物(低沸点原料を含む)を凝縮液化させる前に、反応系(反応器)とコンデンサとを連通する配管経路内に、例えば、コンデンサ内部の凝縮部(とりわけ塔頂部近傍)、反応槽とベーパーの立ち上がりラインとの間の接合部(フランジ部)やベーパーラインとコンデンサ塔頂部との間のフランジ部等のフランジ部、反応槽等に設置された温度計やのぞき窓に設けられた突起部などのゲルが形成されやすい部位に、好ましくはコンデンサ内部の凝縮部(とりわけ塔頂部近傍)、反応槽とベーパーの立ち上がりラインとの間のフランジ部やベーパーラインとコンデンサ塔頂部との間のフランジ部に、供給して混合させるものである。
【0084】
本発明において、反応槽とベーパーの立ち上がりラインとの間のフランジ部におけるゲルの形成を抑制することを目的とする場合には、ゲル化防止剤を含ませずに脱水溶剤のみを上記フランジ部に供給することにより上記目的を達成してもよい。なお、上記において、脱水溶剤の具体例は、先に説明したものと同様である。上記の態様をエステル化反応中において適用する場合には、同じ種類の脱水溶剤を使用してもよいし、異なる種類の脱水溶剤を使用してもよい。または、以下に詳述するが、凝縮液(またはその一部)を循環させて使用してもよい。また、脱水溶剤の不存在下でエステル化反応を行う際には、別途、脱水溶剤供給機構を好ましくはフランジ部付近に設けて、脱水溶剤をフランジ部に供給すればよい。
【0085】
なお、上記(i)〜(iii)の中でも以下に説明する理由から、上記(i)を採用するのが好ましい。すなわち、経済的な観点および取り扱いの面からはより低い温度で脱水溶剤を留出し除去するのが望ましく、そのための手法としては、例えば、適量の水を用いて(特に、上記部分中和工程で薄い濃度のアルカリ水溶液で処理した場合には、大量の水が系内に既に存在しており、この水を用いてもよい)留出させる方法等が有効な手段として挙げられる。適量の水を用いて脱水溶剤と留出(共沸)させる場合には、水相側にも低沸点原料が移行し、水と共に留出されるほか、脱水溶剤の留去が漸次進につれて徐々に共沸されてくる留出物中の脱水溶剤の割合が低下し、最終的にはほとんど水(低沸点原料を含む)が留出されるようになることから、ゲル化防止剤を溶剤に溶かしても十分な効果が得られなくなることから、上記(i)の方法により、ゲル化防止剤を水と混合して作用させることが望ましく、特に、水溶性のゲル化防止剤を使用し、該水溶性ゲル化防止剤を水に溶解して作用させることがより望ましいものである。さらには、未反応の低沸点の原料を含有する留出物に有効に作用する(すなわち、低沸点の原料を含有する留出物が凝縮(液化)した際に、この液化物と速やかに接触し、ゲル化する低沸点の原料が含有されている液化物(水及び有機溶剤)に対して相溶ないし分散する)ことができるように、上記(i)の方法により、留出物の成分組成に応じてゲル化防止剤を水および/または溶剤に溶解したものを作用させることが望ましい。例えば、経時的な留出物の組成変化をセンサ等によりモニタしながら、作用させるゲル化防止剤組成(例えば、数種のゲル化防止剤を用い、溶剤、好ましくは脱水溶剤に溶解するゲル化防止剤組成と水に溶解するゲル化防止剤組成の混合比率)を変化させても良く、脱水溶剤に溶解するゲル化防止剤は脱水溶剤に溶解させたものと、水に溶解するゲル化防止剤は水に溶解させたものを別々の経路より、コンデンサ等の装置内に設けられたそれぞれの噴霧ノズルより滴下ないし噴霧するなどして作用させることが望ましいものである。また、上記(i)を採用する理由としては、単位質量あたりのゲル化防止剤に対して使用される液体の量が多くなるほど、かかるゲル化防止剤を混合した液体を液化凝縮手段の1つ(=熱交換媒体)として作用し得るとする利点も挙げられる。
【0086】
ここで、水に溶解した状態でゲル化防止剤を作用させる場合に用いることのできる水溶性のゲル化防止剤としては、例えば、ハイドロキノン、メトキノン等が好ましく使用される。
上記(i)の方法、すなわち、液化(溶解)した状態でゲル化防止剤を作用させる方法の場合に、上記ゲル化防止剤を溶解することのできる溶剤としては、例えば、ベンゼン、トルエン、キシレン、シクロヘキサン、アセトン、メチルエチルケトン、n−ヘキサン、ヘプタン等が挙げられるが、好ましくは上述したように、反応系に仕込まれる脱水溶剤と同種のものを用いるのがよい。
【0087】
すなわち、異なる溶剤を用いた場合には、これら混合溶剤を別途回収し再利用するには、多段階で分離精製処理を行う必要があり、再利用に要するコストが高くなり、使い捨てにした方が低コストである。しかし、こうした使い捨てによる混合溶剤の廃棄処理(焼却処理あるいは環境基準値以下に希釈化して廃水処理するなど)にも、一定のコストを要し、かつ少なからず大気汚染ないし水質汚染等を招くことから、今日の良く言われる地球に優しい環境づくりにいわば逆行することになる。一方、脱水溶剤と同種のものを用いる場合には、簡単な処理により低コストでの再利用が可能となり、コストおよび環境面で優れていると言える。
【0088】
上記のように、ゲル化防止剤を溶剤(好ましくは脱水溶剤)に溶解して作用させる場合、ゲル化防止剤は、ゲル状物の発生を抑制することができるように、コンデンサ内を通過する低沸点原料(ガスないし液化物)に対して、常にゲル化防止剤が存在して有効に機能するように供給されることが好ましい。
ゲル化防止剤と溶剤との混合比率は、特に制限されるものではないが、ゲル化防止剤を、溶剤100質量部に対して、通常、好ましくは0.001〜10質量部、より好ましくは0.01〜5質量部の範囲で混合する。ゲル化防止剤と溶剤との混合比率が、溶剤100質量部に対してゲル化防止剤が0.001質量部未満の場合には、使用するゲル化防止剤の添加量が上記に規定するように仕込みの原料に対して一定量であるため、結果的に使用する溶剤の量(添加される全量)が大きくなり、最初に仕込んだ脱水溶剤に対して逐次還流されることで溶剤量が増大していくため、反応系に加える熱量等の調整を行って反応生成水の留出量(留出速度)が大きく変動しないようにする必要があるなど、反応系の制御管理が複雑化する必要が生じる。また、脱水溶剤と異なる溶剤を用い、これを分離回収する場合には、その回収コストが増大し、製造コストがかさむことになる。一方、ゲル化防止剤と溶剤との混合比率が、溶剤100質量部に対してゲル化防止剤が10質量部を超える場合には、逆に使用する溶剤の量(添加される全量)が少なくなるため、単位時間当たりの添加量が制限され、低沸点原料との接触頻度が相対的に低下し、未接触のまま液状化しゲル状物を形成するのを効果的に抑制するのが困難になる。そのため、単位時間当たりに必要な添加量を確保するには、仕込みの原料に対して上記に規定する以上の大量のゲル化防止剤が必要になり、製造コストが上昇する。
【0089】
次に、ゲル化防止に関する別の実施態様(B)について説明する。
エステル化反応を脱水溶剤中で行う場合は、エステル化反応時に生成する反応生成水を脱水溶剤と共に留出させ、該反応生成水を含む留出物を凝縮液化し、該凝縮液化した凝縮液から反応生成水を分離除去し、該反応生成水を分離除去した後の脱水溶剤を含有する凝縮残液を反応槽に戻しながらエステル化反応を行う際に、該凝縮残液の一部とゲル化防止剤とを含有してなるゲル化防止剤溶液を留出物に作用させることが好ましい。この態様を実施態様(B)と称する。
実施態様(B)により反応槽内に増える凝縮残液の量を極力抑え、かつ留出物に対して(特に、留出物に対して該留出物が凝縮液化するコンデンサの壁面、とりわけ塔頂部の壁面を十分に濡らすことができるだけの)十分な量のゲル化防止剤溶液を常に供給する(コンデンサの塔頂部から降らせる)ことができる。そのため、反応槽内の反応生成水を反応槽から留出してから凝縮液化し分離除去する間に、反応生成水と共に留出されてくる低沸点の原料によるゲル状物の発生を、常に効果的に防止することができ、高品質のエスル化物を効率よく低コストで製造することができる。
【0090】
上記の実施態様(B)においては、留出物は、通常、エステル化反応により生成した反応生成水を含むほか、該反応生成水を反応槽から留出する際に一緒に留出される原料、特に不飽和モノカルボン酸系単量体(a)、さらに必要に応じて反応生成水と共沸させる目的で反応槽に加えられる脱水溶剤を含むものである。
エステル化反応終了後の脱水溶剤留去工程中に、該脱水溶剤を含む留出物に対してゲル化防止剤を作用させる場合、上記の実施態様(B)、および/または、前述の、エステル化反応時に生成する反応生成水を脱水溶剤と共に留出させた留出物にゲル化防止剤を作用させる実施態様(A)を含むものであってもよい。
【0091】
上記実施態様(B)に用いられるゲル化防止剤溶液は、留出物に作用させる溶液、より詳しくは留出物中の低沸点の原料に対してゲル化防止を目的で作用させる溶液であって、凝縮液の一部とゲル化防止剤とを含むものであるが、この際、ゲル化防止剤はそのままの形態で用いてもよいし、溶液の形態で用いてもよい。好ましくは、凝縮残液の一部と溶液形態のゲル化防止剤とを含むものである。
本明細書において、「凝縮液」ということばは、コンデンサの出口から出てきたものを意味する。また、上記実施態様(B)によると、ゲル化防止剤溶液をエステル化反応時に生成する反応生成水などの留出物に対して作用させてもよいため、このような場合には、ゲル化防止剤溶液が凝縮液に含まれる。さらにその後に水分離器で凝縮残液と分離水に分離されるため、凝縮残液及び分離水双方とも、凝縮液の定義に含まれ、これらは相互独立的に単独で使用することもできる。
【0092】
本明細書において、「凝縮液の一部」とは、凝縮液をただ単に部分的に分けたもの以外に、該凝縮液を分離して得られる凝縮残液および凝縮残液の一部も含まれる。
本明細書において、「凝縮残液」とは、水分離器で分けた溶剤側の成分をいい、「分離水」とは、水分離手段である水分離器で分けた水側の成分をいう。溶剤側の成分としては、ゲル化防止剤溶液のほか、必要に応じて使用される脱水溶剤等が含まれている。水側の成分としては、反応生成水や原料等がある。
上記コンデンサおよび水分離器は、本発明のエステル化物の製造方法において次のように使用されるものである。すなわち、本発明のエステル化物の製造方法では、エステル化反応時に生成する反応生成水を反応槽から留去する必要がある。しかし、留出物中には上記したように反応生成水以外の成分も含まれるため、直接大気中に放出することは環境汚染等の問題からできない。このため、かかる反応生成水を反応槽から留出した後に、適当に処理したり再利用したりできるようにする必要がある。そこで、コンデンサ(凝縮器)は、反応槽から留出されてなるものをそこに送り、凝縮液化するのに使われる。さらに、水分離器は、コンデンサの出口から出てきたものをそこに送り、その性質の違いを利用して2層に分離し、一方の層の水側の成分からなる分離水と、もう一方の層の溶剤側の成分からなる凝縮残液とに分けるのに使われる。
【0093】
実施態様(B)において、ゲル化防止剤溶液には、上記凝縮液の一部のほか、以下に説明するゲル化防止剤(溶液の形態を含む;以下、同様)、さらに他の添加剤、例えば、反応槽内への補充目的で適宜追加する酸触媒などが含有されていてもよい。
上述したように、ゲル化防止剤は、適当な溶剤、好ましくは脱水溶剤と同種の溶剤に溶解(ないし混合、例えば、過飽和状態で一部のゲル化防止剤が溶解せずに含まれている場合、2種以上のゲル化防止剤を用いた場合に、その一部のゲル化防止剤が溶剤に溶解せずに含まれている場合、さらにはゲル化防止剤が混合されている場合なども含む)されていることが好ましい。
【0094】
実施態様(B)において用いることができるゲル化防止剤としては、反応生成水等と共に留出されてくる低沸点の原料が、凝縮される段階で起こる重合反応を抑えることができるものであれば特に制限されるものではなく、従来既知の各種ゲル化防止剤の中から適宜選択して利用することができ、その具体例や好ましい例については、前述したものと同様である。
実施態様(B)において、上記ゲル化防止剤の使用量は、留出物の留出開始時からエステル化反応終了まで逐次留出されてくる低沸点原料に対して常にゲルの形成を効果的に防止することができる量(留出物の留出開始時からエステル化反応終了までの積算量)であることが好ましい。
【0095】
実施態様(B)において、エステル化反応に脱水溶剤を使用し、該脱水溶剤を留出し還流させる場合には、該ゲル化防止剤は、留出物に対して重合防止目的を達成した後、反応生成水を分離除去後の凝縮残液側に溶解した状態で反応槽に戻され、反応槽内に漸次蓄積される。その結果、反応により得られたエステル化物を原料として重合を行いセメント分散剤などの各種製品を製造する際に重合し難くなる。このため、ゲル化防止剤の使用量は極力抑えることが望ましい。以上の観点から、該ゲル化防止剤の使用量は、原料であるアルコキシポリアルキレングリコールおよび不飽和モノカルボン酸系単量体(a)の全使用量に対して、好ましくは0.1〜1000質量ppm、より好ましくは1〜500質量ppmの範囲である。ゲル化防止剤の使用量が、原料の全使用量に対して0.1質量ppm未満の場合には、反応生成水等を含む留出物の留出開始時からエステル化反応終了まで逐次留出されてくる低沸点原料に対して、常に重合禁止能を有効に発現させる上で不十分な量であるため、ゲル状物が生成する場合がある。一方、原料の全使用量に対して1000質量ppmを超える場合には、重合禁止能を有効に発現させるには十分過ぎる量であり、過剰な添加に見合う更なる効果の発現が見込めず不経済となるほか、得られたエステル化物を原料として重合を行いセメント分散剤などの各種製品を製造する際に重合が難しくなる。なお、ゲル化防止剤の使用量の全量を一時に加えたのでは、反応生成水を含む留出物の留出開始時からエステル化反応終了まで逐次留出されてくる低沸点原料に対してゲル状物の形成を有効に阻止することができにくいため、ゲル化防止剤の使用量が上記に規定する範囲内で連続的に加えるのが望ましい。この際、逐次留出される低沸点原料に対して、ゲル化防止剤溶液中のゲル化防止剤濃度が常に下記に規定する範囲となるように調整し連続的に加えるのがより望ましい。
【0096】
実施態様(B)において、ゲル化防止剤を溶液の形態で使用する際に用いることのできる溶剤としては、特に制限されるものではなく、例えば、ベンゼン、トルエン、キシレン、シクロヘキサン、アセトン、メチルエチルケトン、n−ヘキサン、ヘプタン等が挙げられる。また、エステル化反応に脱水溶剤を使用し、該脱水溶剤を留出し還流させる場合には、ゲル化防止剤溶液に用いた溶剤成分も凝縮残液側に含有されて反応槽に戻されるため、エステル化反応槽内で脱水溶剤として有効に作用し得るものであることが望ましい。特に、反応槽内に仕込んである脱水溶剤と異なる溶剤を用いた場合には、反応槽内の該溶剤量(濃度)の漸増により、該溶剤を含む脱水溶剤と反応生成水との共沸点(およびこれに伴う留出速度)が経時的に変動するため反応槽内部に加える熱量等の制御管理、さらには原材料の点数の増加に伴い、設備が増加し、安全・品質管理や在庫管理などが複雑化ないし煩雑化する等の点から、反応槽内に仕込んである脱水溶剤と同種の溶剤がより好ましい。
【0097】
ここでの溶剤の主な使用目的は、ゲル化防止剤の溶液化にあり、凝縮液の一部との混合が容易になされるようにし、凝縮液の一部との混合に際し撹拌装置等(例えば、撹拌槽など)を設けなくともよいようにすることにある。そして、エステル化反応に脱水溶剤を使用し、該脱水溶剤を留出し還流させる場合に、反応槽内に戻される凝縮残液量の増加を極力抑えるには、ゲル化防止剤溶液に使用する凝縮液(好ましくは凝縮残液)の一部の混合比率が高い方がよいことから、ここでの溶剤使用量は極力抑える事が望ましい。かかる観点から、上記溶液中のゲル化防止剤濃度としては、該溶液全体に対して好ましくは10質量ppm〜飽和濃度、より好ましくは100質量ppm〜飽和濃度、さらに好ましくは200質量ppm〜飽和濃度、特に好ましくは200質量ppm〜飽和濃度の95%に相当する濃度(ただし、飽和濃度は、ゲル化防止剤および溶剤の種類、温度、圧力等により変動し一義的に決まるものではないため、具体的な数値は規定していない)である。飽和溶液を用いることにより、溶剤の使用量を極力少なくすることができる。さらに、コンデンサから降らせるゲル化防止剤濃度を一定にするためには温度により変化する飽和濃度より、該飽和濃度よりも少し低濃度の方が良いため、飽和濃度の95%に相当する濃度以下で用いるのが好ましい。上記ゲル化防止剤濃度が、該溶液全体に対して10質量ppm未満の場合には、ゲル化防止剤溶液に使用する凝縮液の一部の混合比率が低下し、エステル化反応に脱水溶剤を使用し、該脱水溶剤を留出し還流させる場合には、反応槽に戻される凝縮残液の量が増える。あるいは漸増する凝縮残液をエステル化反応終了時まで貯えておける大きな保存部や時間とともに凝縮残液の一部を系外に出すための装置・手段等が必要となる。さらに溶剤の使用量も増えコストアップになる。
【0098】
ゲル化防止剤溶液に用いられるゲル化防止剤および凝縮液の一部の流量(流速)に関しては、ゲル化防止剤溶液中のゲル化防止剤の濃度および反応装置(反応槽や配管、コンデンサ等)の大きさや留出物の量等により異なるために一義的に規定することはできないが、ゲル化防止剤の量を減らして、これに代えて十分な量の凝縮液を用いることで、十分な量のゲル化防止剤溶液を留出物に作用させることができ、さらにエステル化反応に脱水溶剤を使用し、該脱水溶剤を留出し還流させる場合には、反応槽内の溶剤量の増加を極力抑えることができるように使用態様に応じて適宜決定(規定)すれば良い。
コンデンサの直径(内径)1mに対するゲル化防止剤1分間あたりの流量は、好ましくは0.01〜40リットル/分m、より好ましくは0.1〜15リットル/分m、さらに好ましくは0.1〜5リットル/分mであり、また、コンデンサの直径(内径)1mに対する凝縮液の一部の1分間あたりの流量は、好ましくは1〜1000リットル/分m、より好ましくは5〜500リットル/分m、さらに好ましくは10〜200リットル/分mである。ゲル化防止剤の流量が0.01リットル/分m未満の場合には、溶液中のゲル化防止剤濃度が低下し、常に十分な重合禁止能力を発現させることが困難となる。一方、ゲル化防止剤の流量が40リットル/分mを超える場合には、新たに加えられる溶剤量が増加するため、ゲル化防止剤の量を減らして、これに代えて十分な量の凝縮液を用いるとする本発明の主旨の達成が困難となる。また、凝縮液の一部の流量が1リットル/分m未満の場合には、留出物に対して常に十分な量の凝縮液を供給することができず、ゲル状物の発生を招くおそれがあるため好ましくない。一方、凝縮液の一部の流量が1000リットル/分mを超える場合には、これ以上の高流量で供給する事に見合う更なる効果が得られず、こうした多量の凝縮液を高流量で供給するための装置(大型のポンプや大口径ないし耐圧配管など)を設ける必要があり、不経済である。
【0099】
このように使用態様に応じて、ゲル化防止剤の流量を決定(規定)し、凝縮液の一部、好ましくは凝縮残液の一部の流量を決定(規定)した上で、流量の組み合わせは規定した流量の範囲内の組み合わせであれば全て可能であるが、本発明の主旨を十分に発揮するには、ゲル化防止剤に用いられるゲル化防止剤溶液と凝縮液の一部との混合比率は、以下の組み合わせがよい。
ゲル化防止剤1質量部に対して凝縮液の一部を好ましくは0.5〜10000質量部、より好ましくは1〜1000質量部、さらに好ましくは10〜1000質量部、特に好ましくは10〜100質量部の範囲で混合する。ゲル化防止剤1質量部に対して凝縮液の一部が0.5質量部未満の場合には、本発明の上記主旨を十分に満足させることができず好ましくない。一方、ゲル化防止剤1質量部に対して凝縮液の一部が10000質量部を超える場合には、両者を安定して混合することが困難となるためである。なお、これらの混合比率は、一定としてもあるいは可変させてもよく、本発明の上記主旨を満足するように適宜混合比率を決定すればよい。
【0100】
ゲル化防止剤溶液を作用させる方法としては、留出物、特に留出された低沸点原料に対して有効に作用させることができるものであれば、特に制限されるものではなく、従来既知の方法(手段)を適宜用いて行うことができる。好ましくはガス状の留出物を凝縮液化させる領域、具体的には、ガス状の留出物を凝縮液化する領域である熱交換器、冷却器あるいは凝縮器等(本明細書中では、これらを総称して単にコンデンサともいう)、特にガス状の留出物が凝縮液化し始めるコンデンサの塔頂部のガス入口部分において、有効にゲル化防止剤溶液が作用できるようにすることが望ましい。そのためには、ゲル化防止剤溶液が存在する領域はコンデンサ内には限られず、コンデンサの塔頂近傍、すなわち、コンデンサの塔頂ないしコンデンサ直前の留出経路内などにゲル化防止剤溶液を作用させればよく、そうすることでコンデンサの内壁を常に濡れた状態に保てることが望ましいと言える。具体例としては、(i)コンデンサの塔頂部の中央部に上向きに設置したノズル部よりコンデンサの塔頂部のガス入口部分の内壁(ここで、最初の凝縮液化が生じ、同時に低沸点原料のゲル化も生ずるためである)にゲル化防止剤溶液を噴霧したり、吹き出したり、吹き付けたり、吐出させたり、吹き上げたり、降らせたりすることでコンデンサの内壁を常に濡れた状態に保たせる方法、あるいは(ii)コンデンサ直前の留出経路(後述する第2図の配管503で形成された経路:オーバーヘッドライン)内にノズル部を設置し(第3図参照)、ここでゲル化防止剤溶液を噴霧し(または吹き出し)オーバーヘッドラインの壁を伝わせてコンデンサ内に到達させることでコンデンサの内壁を常に濡れた状態に保たせる方法などが挙げられるが、これらに限定されるものではない。さらに、エステル化反応に脱水溶剤を使用し、該脱水溶剤を留出し還流させる場合には、留出物が凝縮液化する際に、この液化物と速やかに接触し、ゲル化する低沸点の原料が含有されている脱水溶剤に対して相溶ないし分散することができるように、ゲル化防止剤は、ゲル化防止剤を脱水溶剤と同種の溶剤に溶解した形態で使用されるのが望ましい。
【0101】
また、凝縮液の一部をゲル化防止剤溶液に用いるべく還流させる方法としても、特に制限されるものではなく、従来既知の方法(手段)を適宜用いて行うことができる。具体例を以下に示す。
(i)エステル化反応に脱水溶剤を使用し、該脱水溶剤を留出し還流させる場合には、凝縮残液を反応槽に戻す際に凝縮残液の一部を抜き取って、上記ノズル部に直接的に供給し、該ノズル部でゲル化防止剤溶液とするか、あるいは上記ノズル部に供給する途中で、ゲル化防止剤と混合させてゲル化防止剤溶液とすることなどができる。具体例としては、後述する第2図に示すように、凝縮残液を反応槽(好ましくは、反応槽とベーパーの立ち上がりラインとの間のフランジ部)に戻す経路上に必要に応じて凝縮残液を一時的に貯めておく保存部(タンクなど)を設け、該保存部から該凝縮残液の一部を抜き取り、ゲル化防止剤の供給経路に抜き取った凝縮残液の一部を合流させるだけで簡単に混合されたゲル化防止剤溶液とすることができる。そのため、わざわざ両者を混合撹拌するための装置は不要である。ここで、保存部を設けるメリットとしては、ゲル化防止剤溶液用の凝縮残液の抜き取り量を一定量ないし徐々に増やす際にもその調整が便利であり、かつ反応槽に戻す凝縮残液の量を反応開始から終了までの間、常に一定量ないしは極力増加量を抑えながら還流させる事が容易に調整できる点にある。なお、保存タンクのような保存部を新たに設けなくとも、例えば、水分離器では、コンデンサで凝縮液化された凝縮液が一方の室に貯められ、水相と溶剤相の2層に分離され、下層部の水相はこの室の下部より配管を通じて逐次抜かれ、上層部の溶剤相は仕切板をオーバーフローして隣のもう一方の室に貯められるが、この溶剤相のみが貯められる室を大きくすれば、水分離器自体が保存部を兼ね備えることもできる(第4図参照)。
【0102】
保存部は必ずしも必要ではない。従来のゲル化防止剤単独使用の時と比較した場合、例えば、凝縮残液を含むゲル化防止剤溶液を使用しているため、ゲル化防止剤の量は従来と同程度あるいは少ない量で大きなゲル化防止効果を発揮できるようになった。そのため、反応槽内での溶剤量の増加も従来と同程度あるいは少ない量に抑えられている。特にエステル化時間が短い場合、反応温度の低下は少なく反応終了時間への影響も少なくてすむため、保存部を設けない方が経済的にも有利となるからである。また、反応槽内に戻される溶剤量が増加し多くなってきた場合、一部反応槽へ溶剤を戻さずに系外に抜き取ってもよい。この場合にも、系外に抜き取られる溶剤量は、大きくなくその処理コストも少ないため、わざわざ保存部を設けるよりも、経済的に有利となり、製品の性能を左右することもないからである。このように、性能面への影響、さらに費用対効果を勘案して、保存部を設けるか否か適宜判断する事が肝要であると言える。
【0103】
(ii)脱水溶剤を用いずにエステル化反応を行う場合には、本来的に留出物は反応生成水(僅かに低沸点原料を含む)だけであり、反応槽に留出物の一部を還流させることはない。従って、留出物にゲル化防止剤溶液を作用させた後の凝縮液から反応生成水(低沸点原料を含む)を分離除去した凝縮残液の全量若しくはその一部を抜き取って、上記ノズル部に直接的に供給して、該ノズル部でゲル化防止剤溶液とするか、あるいは上記ノズル部に供給する途中でゲル化防止剤と混合させてゲル化防止剤溶液とすることなどができる。なお、凝縮残液の一部を利用する場合、あとの凝縮残液は系外に抜き取れるなどすればよい。
【0104】
なお、上記に説明した作用させる方法および還流させる方法は、代表的なものを例示したに過ぎず、本発明がこれらに限定されるものではない。
本発明においては、以上のようなゲル化防止を行うことが好ましいが、さらに、溶剤留去工程として以下の形態をなすことが好ましい。
溶剤留去工程において、系内のエステル化物および脱水溶剤を含有する溶液から脱水溶剤を留出してから、凝縮液化して系外に除去するまでの装置機構に関しては、この間にゲル化防止剤を作用させるための手段(装置機構)が設けられていれば何ら制限されるものではなく、従来既知の装置機構を適当に組み合わせることができる。例えば、上述したエステル化工程において、エステル化反応中に、反応系内の脱水溶剤を反応系から留出し凝縮液化して反応系に戻し循環させるのに使用した装置機構(単に溶剤循環装置という)の一部を利用してもよく、装置設備の簡素化・小型化も図れることから望ましい実施態様の1つと言える。具体的には、ガス状の留出物を凝縮液化するための装置であるコンデンサ等に関しては先の溶剤循環装置をそのまま利用でき、凝縮液化された留出物の分離除去装置である液−液分離装置である水分離器等に関しては先の溶剤循環装置を適宜使用形態を変更して利用できる。すなわち、留出物の成分組成に応じて、当該水分離器に輸送されてくる液状の留出物を、水を系外に除去する輸送経路及び輸送装置であるポンプ等を利用して、水相部分あるいは液状の留出物の全てを系外に除去することができるほか、新たに当該水分離器等に真空ポンプ(エゼクタ)を取り付けて吸引することで、相対的に揮発性の高い成分等を選択的に、あるいは液状の留出物の全てを系外に除去するようにしてもよい。あるいは凝縮液化した留出物をコンデンサ等から別途輸送経路を設けてそのまま系外(例えば、廃棄物処理装置やリサイクル処理装置など)に取り出し、適当に処理(廃棄ないし再利用)することもできる。また、これらの装置にも、適当な制御機構が適宜設けられているのが望ましい。なお、上記に例示した装置機構に変えて、系内の脱水溶剤を留出し凝縮液化して系外に除去させるとする本来的な目的を逸脱しない限り、従来既知の他の手段及びその装置との組み合わせ、あるいは他の手段及びその装置による代替えなどによる方法を適宜採用することができることもいうまでもない。
【0105】
溶剤留去工程において、エステル化反応終了後(必要に応じて、上記部分中和工程を行い)、系内のエステル化物および脱水溶剤を含有する溶液から脱水溶剤を留去する方法に関しては、特に制限されるものではなく、上述したように水を用いて脱水溶剤と共沸させて留出し除去してもよいし、他の適当な添加剤を加えて脱水溶剤を効果的に除去するようにしてもよいほか、何等の添加剤(水を含む)を用いることなく、留出させて除去する事もできるが、エステル化反応において、酸触媒を用いることが極めて有用(すなわち、その後に部分中和しなければならないことを勘案してもその有用性は極めて高いといえる)であることから、水を用いて脱水溶剤と共沸させて留出し除去する方法が好ましい実施態様の1つと言える。なお、当該溶剤留去工程までに、酸触媒の部分中和処理が行われている際には、系内のエステル化物および脱水溶剤を含有する溶液中には、活性な酸触媒及びアルカリはなく(中和により塩になっている)、水を加えて昇温しても加水分解反応が起こらないため、脱水溶剤を留去する上で、水と共沸させる事ができる。なお、水と共沸させるほうが、より低い温度で効率よく脱水溶剤を除去することができるものである。
【0106】
溶剤留去工程において、系内の溶液中から脱水溶剤を留出させるための条件としては、系内の脱水溶剤を好適に留出(蒸発)させることができるものであれば、特に制限されるものではなく、溶剤留去中の系内温度(系内の液温(常圧下))としては、例えば、(i)水を用いる場合には、通常80〜120℃、好ましくは90〜110℃であり、(ii)水を用いない場合には、通常80〜160℃、好ましくは90〜150℃である。上記(i)ないし(ii)のいずれも場合にも、上記に規定する温度よりも低い温度の場合には、脱水溶剤を蒸発するのに十分な温度(熱量)でなく、脱水溶剤の留去に長時間を要するなど好ましくなく、一方、上記に規定する温度よりも高い温度の場合には、重合の危険性があるほか、多くの熱量が大量の低沸点原料の蒸発に消費されるため好ましくない。また、系内(装置内)圧力は、常圧下または減圧下いずれで行ってもよいが、設備面から、常圧下で行うことが望ましい。また、脱水溶剤を含む溶液から溶剤の留出を行うための装置系としては、エステル化反応で使用した装置系(反応槽)をそのまま使用するのがよい。すなわち、エステルカ反応後、別途他の装置に内容物を移し変える場合には、設備及び管理費が増加するほか、輸送時にエステル化物等が外的要因(熱、光、輸送温度、輸送圧力、活性な雰囲気ガスの介在)などにより劣化したり、輸送経路内に固着したり、逆に輸送時に装置などから不純物が溶出ないし混入するのを防止する必要があり、余分なコストが発生するなど好ましくない。
【0107】
なお、エステル化工程において、不飽和モノカルボン酸系単量体(a)の重合を防止すべく重合禁止剤の存在下に、エステル化反応を行っている場合には、当該重合禁止剤がエステル化反応後(さらには部分中和処理後)においても有効に機能するため、本溶剤留去工程において、系内の溶液中に、新たに重合禁止剤を補充する必要はないが、濃度の薄いアルカリ水溶液を用いて部分中和処理を行っている場合には、系内の溶液中に比較的多くの水が存在している。そのため、例えば、エステル化反応を行う際に使用した重合禁止剤が水に難溶ないし不溶であるような場合に限り、不飽和モノカルボン酸系単量体(a)が水に溶けて系内の溶液内で重合することがあるため、これを防止する観点から、系内の溶液に水溶性重合禁止剤を加えてから上記に規定する温度まで昇温することが望ましいものである。
【0108】
また、上記水溶性重合禁止剤としては、特に制限されるものではなく、例えば、ハイドロキノン、メトキノン、カテコール及びこれらの誘導体(例えば、p−t−ブチルカテコール等)、ハイドロキノンモノメチルエーテル等が挙げられる。なかでもハイドロキノン、メトキノンが好ましい。また、これらの水溶性重合禁止剤は、1種若しくは2種以上を混合して使用してもよい。
上記水溶性重合禁止剤の添加量としては、原料としてのアルコキシポリアルキレングリコール及び不飽和モノカルボン酸系単量体(a)の合計使用量に対して0.001〜1質量%、好ましくは0.001〜0.1質量%である。水溶性重合禁止剤の添加量が、0.001質量%未満の場合には、重合禁止能の発現が不十分な場合があり、水溶性重合禁止剤の添加量が、1質量%を超える場合には、過剰に添加することに見合う重合禁止能が得られず、不経済であり、好ましくない。
(エステル化物製造方法の具体的態様)
本発明において適用できるエステル化物の製造方法を、第1図を参照しながら説明する。
【0109】
第1図は、本発明において適用できるエステル化物の製造方法に用いられる代表的な装置構成の概略図である。
なお、以下において、原料としてのアルコキシポリアルキレングリコールを単にアルコールと称し、原料としての不飽和モノカルボン酸系単量体を単に(メタ)アクリル酸と称することがある。
第1図より、本実施形態の装置構成では、まず、所定温度まで昇温してエステル化反応し、エステル化反応後に所定温度まで降温して中和し、中和後に所定温度まで昇温し、脱水溶剤の留去を行うための熱交換手段(例えば、内部ヒータ等の直接加熱方式、外部ジャケット等の間接の熱交換方式)として加圧スチーム等を熱媒体に使用し得る外部ジャケット102を有する反応槽101が設けられている。この際、反応槽の内部の材料は、特に制限されるものではなく公知の材料が使用できるが、例えば、SUS製、好ましくは耐蝕性の面からSUS304、SUS316及びSUS316L、より好ましくはSUS316及びSUS316Lが挙げられる。または、反応槽の内部にグラスライニング加工等が施され原料及び生成物に対して不活性なものとしてもよい。該反応槽101には、アルコール原料用のステンレススチール(例えば、SUS316)製の原料貯蔵タンク103および(メタ)アクリル酸原料用の原料貯蔵タンク105、酸触媒用の触媒貯蔵タンク107、エステル化反応時の反応系(反応槽101)内の重合を防止するための重合禁止剤を貯蔵した重合禁止剤貯蔵タンク109、エステル化反応終了後の脱水溶剤の留去時の系内(反応槽101)の溶液内での重合を防止するための水溶性重合禁止剤を貯蔵した水溶性重合禁止剤貯蔵タンク110およびエステル化反応後に前記触媒を中和処理するための中和剤(中和剤水溶液)を貯蔵したカーボンスチール(例えば、高炭素鋼)製の中和剤貯蔵タンク111がそれぞれ配管113、115、117、119、120および121により連結されている。また、(メタ)アクリル酸は、重合しやすく、例えば、メタクリル酸では、長期の保存や熱等によっても重合するため微量の重合防止剤(0.1%ハイドロキノンなど)が加えられるほか、結晶化しても重合しやすくなるので、原料貯蔵タンク105内で保存する場合、ベンゼンを加え結晶化を防ぐようにしてもよいほか、第1図に示すように常時30〜40℃に保温するべく、ポンプ116を用いた外部ジャケット150(保温手段)を有する循環経路151が形成されており、(メタ)アクリル酸原料を常に30〜40℃に保持し重合しないように循環させている。(メタ)アクリル酸用の原料貯蔵タンク105、配管115およびポンプ116および循環経路151内部には、腐食性を有する(メタ)アクリル酸による腐食防止目的で、合成樹脂等の耐食性材料によるライニング加工が施されているものが使用される。同様に、触媒貯蔵タンク107およびその配管117内部にも、酸触媒による腐食防止のため、合成樹脂などの耐酸性材料によるライニング加工が施されているものが使用される。また、上記反応槽101の下部には、エステル化反応により反応槽101内部に合成されたエステル化物(あるいは、セメント分散剤等では、該エステル化物を単量体成分として該反応槽101でさらに重合を行い得られた重合体)を回収するための配管153が連結されている。さらに、上記反応槽101内には、反応温度を計測するための温度センサ(図示せず)が適当な部位(数カ所)に取り付けられている。該温度センサは、反応温度を規定の温度に保つのに必要な装置機構(例えば、反応槽101に取り付けられたジャケット102の温度)などを制御するための制御部本体(図示せず)に電気的に接続されている。
【0110】
さらに、本実施形態の装置構成では、反応系内(反応槽101内)でエステル化反応時に生成される反応生成水を含む留出物を留出し、ゲル状物の発生を防止しながら凝縮液化した後に、該反応生成水を分離除去し、残りの留出物を所定の溶剤循環速度で戻すための機構(の装置構成)として、該反応生成水を脱水溶剤とともに共沸させた留出物にゲル化防止剤を作用させて凝縮液化し、該凝縮液化した留出物から反応生成水(水相)を分離除去し、残りの凝縮物(主に脱水溶剤を含む溶剤相)を上記溶剤循環速度で還流させて反応槽101に戻す循環系が形成されている。詳しくは、反応槽101上部と向流(または並流)接触形式の縦型の多管式円管形コンデンサ125の頭頂部とが配管123により連結されている。またコンデンサ125の下底部とSUS製の水分離器127の上部とが配管129により連結されている。該水分離器127の内部には仕切板131が設けられており、該仕切板131で区切られた2つの室133、134が形成されている。このうち、コンデンサ125で凝縮液化された留出物が貯められる側の室133の下部と反応生成水の処理タンク135とが配管137により連結されている。また、該処理タンク135には廃水用の配管139が連結されている。また、水分離器127のもう一方の室134の下部と反応槽101とが配管141で連結されている。また、この配管141には、反応槽101内の反応生成水と共沸する脱水溶剤を貯蔵する脱水溶剤貯蔵タンク143と連結された配管145が合流(連結)されている。かかる合流点の手前(水分離器127側)の配管141の経路上には循環ポンプ142が設置されている。また、上記合流点の後方(反応槽101側)の配管141の経路上には流量計144が設けられている。そして、該流量計144には、計測される流量を積算し、溶剤循環速度を算出するための流量計測システム本体(図示せず)と電気的に接続されている。さらに、コンデンサ125の頭頂部には噴霧ノズル126が設けられており、この噴霧ノズル126は、留出物のゲル化防止用のゲル化防止剤を貯蔵するゲル化防止剤貯蔵タンク147と配管149により連結されている。
【0111】
本発明において、コンデンサとしては、SUS304、SUS316及びSUS316L等のSUS製や炭素鋼(CS)等、公知のものが使用できるが、好ましくは、ゲルの発生をより軽減するために、内面を鏡面仕上げやグラスライニング加工されたコンデンサを使用できるが、加工やメンテナンスにかかるコストを考慮すると、SUS304、SUS316及びSUS316L、好ましくはSUS316及びSUS316L等のSUS製のコンデンサが好ましく使用でき、このようなコンデンサを用いた場合でも、ゲルの形成を有効に防止できる。また、本発明において好ましく使用されるコンデンサの伝熱面積は、反応槽の容積などによって異なるが、例えば、反応槽30m3では、50〜500m2、好ましくは100〜200m2である。本発明において、コンデンサに使用される冷却媒体としては、水やオイルなどが挙げられる。
【0112】
また、上述したこれらの循環機構(の装置構成)、すなわち、反応系内(反応槽101内)でエステル化反応時に生成される反応生成水を含む留出物を留出し、ゲル状物を発生を防止しなから凝縮液化した後に、該反応生成水を分離除去し、残りの留出物を還流させるための機構(の装置構成)の一部は、エステル化反応後に、系内(反応槽101内)の生成物であるエステル化物を含有する溶液から脱水溶剤を含む留出物を留出し、ゲル状物の発生を防止しながら凝縮液化した後に、該脱水溶剤を含む留出物を系外に除去するための機構(の装置構成)としても利用されており、該脱水溶剤を水とともに共沸させた留出物に水溶性重合禁止剤を作用させて凝縮液化し、該凝縮液化した留出物を水相と溶剤相に分離し、それぞれを適当な方法により別々に除去する経路(取り出し経路)が形成されている。詳しくは、上述した循環機構(の装置構成)に、新たに、コンデンサ125の頭頂部に設けられた噴霧ノズル126には、脱水溶剤を含む留出物のゲル化防止用に利用される水溶性重合禁止剤を溶かした水溶液(以下、単に水溶性ゲル化防止剤ともいう)を貯蔵する水溶性ゲル化防止剤貯蔵タンク159が配管161により連結されている。さらに、水分離器127には配管157を介して、脱水溶媒を減圧吸引により除去するために真空ポンプ(エゼクタ)155が取り付けられている。
【0113】
本発明において好ましく適用できるエステル化物の製造方法では、以上の装置構成を有するエステル化物の製造装置を用いて次のように行われる。
まず、反応槽101内部に、各原料貯蔵タンク103、105、触媒貯蔵タンク107、重合禁止剤貯蔵タンク109、脱水溶剤貯蔵タンク143より配管113、115、117、119および配管145を介した配管141を通じて原料のアルコールおよび(メタ)アクリル酸、酸触媒、重合禁止剤および脱水溶剤をそれぞれ上記に規定する所定の量を送り込み(仕込み)、上記に規定するエステル化条件(反応温度、ジャケット温度、圧力)でエステル化反応を行う。エステル化反応により逐次生成する反応生成水は、反応槽101内に仕込まれた脱水溶剤と共沸され配管123を通じて留出されてくる。留出されてきたガス流体である溶剤−水共沸物は、コンデンサ125に通され凝縮液化される。この凝縮液化時に該共沸物に含まれる低沸点原料がゲル化するのを防止する目的で、ゲル化防止剤貯蔵タンク147より配管149を通じて該コンデンサ125の頭頂部に設けられた噴霧ノズル126から上記に規定する量のゲル化防止剤を連続的に滴下して、共沸物(ガス流体物および凝縮液化物の双方をいう)と並流接触させる。凝縮液化された共沸物(滴下されたゲル化防止剤を含む)は、該コンデンサ125の下部より配管129を通じて水分離器127の室133に貯められ、水相と溶剤相の2層に分離される。このうち、下層部の反応生成水は、室133の下部より配管137を通じて逐次抜かれ、反応生成水の処理タンク135に貯められる。そして該処理タンク135内で、必要に応じて、環境基準(廃水基準)値を満足するように化学的ないし生物学的に処理された後、配管139を通じて、本装置系外に廃水される。一方、上層部の溶剤相(滴下されたゲル化防止剤および低沸点原料を含む)は、仕切板131をオーバーフローして隣の室134に貯められる。そして、該溶剤相は該室134の下部よりポンプ142により配管141を通じて上記に規定する溶媒循環速度で還流され反応槽101に戻される。
【0114】
なお、本発明において、ゲル化防止剤を供給するゲル化防止剤貯蔵タンク設置の部位としては、ゲルが形成されやすい部位が好ましいものの、特に制限されず、例えば、第1図における態様、即ち、ゲル化防止剤を噴霧する噴霧ノズル126をコンデンサ125の塔頂部に設ける態様に加えて、反応槽101とコンデンサ125との間の配管123上の少なくとも1箇所にゲル化防止剤を噴霧する噴霧ノズルを設ける態様などが挙げられる。後者の態様において、配管123上にゲル化防止剤を噴霧する噴霧ノズルを設ける部位としては、例えば、コンデンサ内部の凝縮部(とりわけ塔頂部近傍)、反応槽とベーパーの立ち上がりラインとの間の接合部(フランジ部)やベーパーラインとコンデンサ塔頂部との間のフランジ部等のフランジ部、反応槽等に設置された温度計やのぞき窓に設けられた突起部など、ゲルが形成されやすい部位が挙げられ、これらのうち、コンデンサ内部の凝縮部(とりわけ塔頂部近傍)、反応槽とベーパーの立ち上がりラインとの間のフランジ部やベーパーラインとコンデンサ塔頂部との間のフランジ部が好ましい。
【0115】
エステル化反応終了(エステル化率が規定以上に達した時点で終了とする)後、降温し反応槽101の内温(液温)が上記に規定する温度(90℃)以下に下がるまで、反応槽101の外部ジャケット102に冷媒を通じて降温し、その後は所定温度以下を維持するように適宜調整しながら、中和剤貯蔵タンク111より配管121を通じて反応槽101内に、多量の水により上記に規定する濃度まで薄められたアルカリ水溶液(中和剤)を添加して酸触媒(及び(メタ)アクリル酸の一部)を部分中和する。
部分中和後、水溶性重合禁止剤を水溶性重合禁止剤タンク110より配管120を通じて反応槽101内の溶液に添加混合する。常圧下に、反応槽101の外部ジャケット102に熱媒(加圧スチーム)を通じて上記に規定する温度まで昇温することにより、反応槽101内の脱水溶剤及び部分中和処理の際に加えられている多量の水のほか未反応の低沸点原料(例えば、(メタ)アクリル酸)も共沸され配管123を通じて留出されてくる。留出されてきたガス流体である溶剤−水共沸物は、コンデンサ125に通され凝縮液化される。この場合にも未反応の低沸点原料(例えば、(メタ)アクリル酸)によりゲル状物が発生するが、ここでは、脱水溶剤が除かれていくので次第に水及び低沸点原料だけが蒸発してくるようになるため、水溶性重合禁止剤を作用させることが望ましい。この凝縮液化時に該留出物に含まれる未反応の低沸点原料がゲル化するのを防止する目的で、水溶性ゲル化防止剤貯蔵タンク159より配管161を通じて該コンデンサ125の頭頂部に設けられた噴霧ノズル126から上記に規定する量の水溶性ゲル化防止剤を連続的に滴下して、留出物(ガス流体物および凝縮液化物の双方をいう)と並流接触させる。凝縮液化された留出物(滴下された水溶性重合禁止剤を含む)は、該コンデンサ125の下部より配管129を通じて水分離器127の室133に貯められ、水相(滴下された水溶性重合禁止剤および低沸点原料を含む)と溶剤相の2層に分離される。このうち、下層部の水は、循環させずに除去する場合には、室133の下部より配管137を通じて逐次抜かれ、水の処理タンク135に貯められる。そして該処理タンク135内で、必要に応じて、環境基準(廃水基準)値を満足するように化学的ないし生物学的に処理された後、配管139を通じて、本装置系外に廃水される(また、下層部の水を循環させる場合には、室133の下部より反応槽101に連結される配管(図示せず)を設け、この配管を通じて還流すればよい。)。一方、上層部の溶剤相は、還流することなく装置系外に除去する必要上、水分離器127に取り付けられた真空ポンプ(エゼクタ)155を用いて装置系外に取り出される。なお、これらは廃棄処理されるか、あるいは系外の装置を用いて化学処理し再利用してもよい。
【0116】
最後に、系内の溶液から脱水溶剤を留去した後、反応槽101内に、配管(図示せず)により連結されている水貯蔵タンク(図示せず)または上水管(図示せず)より調整水を添加して所望のエステル化物の水溶液を得る。得られたエステル化物の水溶液は、配管153より回収(貯蔵)される。なお、得られたエステル化物をセメント分散剤等の合成に用いる場合には、該エステル化物を単量体成分の1つとして該反応槽101でさらに重合を行い、セメント分散剤の主要組成成分となり得る重合体を合成するようにしてもよい。この場合には、過剰に加えられ残っている未反応の(メタ)アクリル酸をもう一方の単量体成分として分離・除去せずにそのまま使用することが好ましい。
【0117】
以上が、本発明において好ましく適用できるエステル化物の製造方法の一実施態様を第1図を用いて説明したものであるが、本発明において好ましく適用できるエステル化物の製造方法は、当該実施態様に限定されるものではなく、エステル化反応終了後、脱水溶剤を留去する際に、該脱水溶剤を含む留出物に対して重合禁止剤を作用させることができるものであれば、その製法(手段)、装置構成などに関しては何ら制限されるものではなく、従来既知の製法、装置構成などを適宜組み合わせて利用することができる。
(重合体と用途)
本発明にかかる製造方法によって得られる重合体は、そのままでもセメント分散剤や顔料分散剤等の各種用途の主成分として用いられるが、必要に応じて、さらにアルカリ性物質で中和して得られる重合体塩をセメント分散剤等の各種用途の主成分として用いても良い。このようなアルカリ性物質としては、一価金属および二価金属の水酸化物、塩化物および炭素塩等の無機物;アンモニア;有機アミン等が好ましいものとして挙げられる。
【0118】
本発明において、(メタ)アクリル酸(塩)系重合体水溶液の中和後のpHは、特に限定されないが、3〜12が好ましい。12を越えると強アルカリ性となり、その一方3を下回ると強酸性の腐食条件となるため、高温では製造設備・貯蔵設備が腐食する恐れがある。より好ましくは5〜10、最も好ましくは6〜8である。
該単量体または(メタ)アクリル酸系重合体の中和剤としては、水酸化ナトリウム、水酸化カリウム等のアルカリ金属の水酸化物;水酸化カルシウム、水酸化マグネシウム等のアルカリ土類水酸化物;アンモニア;モノエタノールアミン、トリエタノールアミン等の有機アミンが挙げられる。これらは単独で用いてもよいし、2種以上の混合物として用いてもよい。好ましくは水酸化ナトリウム、水酸化カリウム等のアルカリ金属の水酸化物であり、より好ましくは水酸化ナトリウムである。
【0119】
中和は単量体を反応器に供給する前に予め行われてもよいし、単量体と反応器内で重合を行ないながら行ってもよい。また、重合開始から、重合終了時までの中和度は上記範囲内であれば、一定である必要は無く、具体的には、重合前半の中和度と重合後半の中和度は異なってもよい。
本発明の製造方法で得られる(メタ)アクリル酸(塩)重合体の分子量は、重量平均分子量で5,000〜10,000,000、好ましくは10,000〜5,000,000、より好ましくは20,000〜3,000,000、さらに好ましくは30,000〜1,000,000、最も好ましくは50,000〜500,000である。この重合体の重量平均分子量が5,000未満、または10,000,000を超えると分散剤として十分な性能が発揮されないといった問題が生じることがある。
【0120】
また、本発明にかかる製造方法の一つによれば、レドックス系重合開始剤を使用したため、オリゴマー部分の存在量が少ない(メタ)アクリル酸(塩)系重合体を製造することができることが判明した。レドックス系重合開始剤では、モノマーの重合率が高くかつ重合度を高く維持することができるため従来に比してオリゴマー部分が少ないのである。これは、分子量が均一な重合体が得られたことを意味するものであり、重合体の重量平均分子量の相違によって得られる重合体の特性が変化する中で、特に求める分子量のものが均一の組成で得られる点で、極めて有利である。不飽和モノカルボン酸系単量体(a)由来の構成単位(I)と不飽和ポリアルキレングリコール系単量体(b)由来の構成単位(II)との重合体において、3量体以下のものを「オリゴマー」と称すると、本発明の製造方法によれば、オリゴマー含有量が20質量%以下の(メタ)アクリル酸(塩)系重合体を製造することができる。なお、本明細書における重量平均分子量の分子量測定の項で記載した方法によって測定した数値である。
【0121】
上記方法によって製造した(メタ)アクリル酸(塩)系重合体は、重合段階で過酸化水素量を特定範囲に制限したため、還元剤としてL−アスコルビン酸を使用した場合には、L−アスコルビン酸を0.001質量%以上、好ましくは0.001〜10質量%、特に好ましくは0.001〜1質量%を含有し、該L−アスコルビン酸の還元作用により、製品を長期に保存しても、酸化による劣化や着色を抑制することができる。
すなわち、本発明にかかる(メタ)アクリル酸(塩)系重合体は、前記一般式(1)で示される不飽和モノカルボン酸系単量体(a)由来の構成単位(I)と前記一般式(2)で示される不飽和ポリアルキレングリコール系単量体(b)由来の構成単位(II)とを含む重合体であって、前記重合体に対して0.001質量%以上のL−アスコルビン酸を含んでなることを特徴とする。
【0122】
該(メタ)アクリル酸(塩)系重合体の製造方法は特に制限はないが、本発明にかかる製造方法によって好ましく製造することができる。しかも、上記単量体組成の場合には、水溶性(メタ)アクリル酸(塩)系重合体を得ることができ、種々の分散剤、特にセメント分散剤や顔料分散剤などとしての使用に優れる。
本発明における(メタ)アクリル酸(塩)系重合体は、そのままセメント分散剤として使用することができるが、従来公知のナフタレン系セメント分散剤、アミノスルホン酸系セメント分散剤、(メタ)アクリル酸(塩)系重合体系セメント分散剤およびリグニン系セメント分散剤よりなる群から選ばれた少なくとも1種のセメント分散剤を配合してもよい。これらの配合組成については、目的とする付加的機能の有無により大きく異なるものであり、上記重合体成分を100質量%(全量)ないし主成分とするものから、上記重合体成分を高付加価値成分として、従来のセメント分散剤に適量加える態様まで様々であり、一義的に規定することはできない。しかしながら、(メタ)アクリル酸(塩)系重合体の配合量は、全成分に対して、通常、5〜100質量%、好ましくは50〜100質量%である。
【0123】
また、該(メタ)アクリル酸(塩)系重合体には、従来公知のセメント分散剤の他に、空気連行剤、セメント湿潤剤、膨張剤、防水剤、遅延剤、急結剤、水溶性高分子物質、増粘剤、凝集剤、乾燥収縮低減剤、強度増進剤、硬化促進剤、消泡剤等を配合することができる。
本発明における重合体を主成分とするセメント分散剤は、少なくともセメントおよび水よりなるセメント組成物に配合することによりセメントの分散を促進する。なお、該セメント分散剤は、ポルトランドセメント、ビーライト高含有セメント、アルミナセメント、各種混合セメント等の水硬セメント、あるいは、石膏などのセメント以外の水硬材料などに用いることができる。
【実施例】
【0124】
以下、本発明の実施例により具体的に説明する。なお、実施例および比較例、製造例において、「部」は質量部を示し、各測定値は、以下の測定方法によった。
[測定方法]
(1)着色度
日本電色工業株式会社製分光式色差計(SE−2000)を用い、透過法で測定を行い、次式に基づき、YI値を測定した。
ここでX、Y、Zは、試料のXYZ表色系における三刺激値XYZの値を示す。
(2)電気泳動測定条件
測定機種:Waters製 Quanta4000 キャピラリークロマトグラフィー
使用カラム:Waters製 AccuSep 75μm×60cm
使用泳動バッファー:20mmol/gホウ酸ナトリウム
電圧:20.00KV
で測定した。
(3)重合体に含まれる還元剤量
重合体に含まれるL−アスコルビン酸量は、例えば、下記測定条件の液体クロマトグラフィー(以下、「LC」という。)により求めることができる。
【0125】
機種:Waters LCM1
検出器:Waters 410
溶離液種類:アセトニトリル/0.1N酢酸水溶液=40/60(質量%)
溶離液流量:1.0ml/min
カラム種類:GLサイエンス社製 Inertsil ODS−2 3本 4.6×250mm
(4)重合体に含まれるオリゴマー量
重合体に含まれるオリゴマー量は、例えば、前記の分子量測定条件と同条件のGPCで得られたGPCチャートから、次のようにして求めることができる。すなわち、本発明の重合体の分子量に相当するところのピークをピークAとし、該ピークAの次に高分子側にあるピークをピークBとし、該ピークAよりも低分子側の全てのピークをピークCとして、各ピーク面積を求め、次式により算出する。
(5)重合系のpH測定方法
(a)重合反応中に重合反応系から反応混合物50gを50ccガラス製容器にサンプリングする。
【0126】
(b)サンプリングした混合物を25℃に調整する。
(c)pHメーター(堀場製作所製:カニスターLAB pHメーター F−23,電極:6366−10D)を用いて25℃の混合物のpHを測定する。
なお、本発明におけるpHの測定は、WO01/14438号が開示するpH測定方法と相違し、実際の重合反応中の重合反応液からサンプリングして測定する方法であり、反応系中のpHをオンラインで実測できる点で優れている。
(6)分子量測定
(メタ)アクリル酸系重合体の重量平均分子量(Mw)は、下記の測定条件のGPC(ゲルパーミュエーションクロマトグラフィー)により測定した。
【0127】
解析ソフト:Watres製 Millennium 32 Ver.3.21
検出器:Watres製 410示差屈折計
カラム:東ソー製 TSKguardcolumn SWXL
TSKgel G4000 SWXL
TSKgel G3000 SWXL
TSKgel G2000 SWXL
流速:0.8ml/分
カラム温度:40℃
溶離液:アセトニトリル6001g,水10999g,酢酸ナトリウム三水和物115.6gの混合溶液を酢酸でpH6.0に調整したものを用いた。
【0128】
標準試料:分子量が272,500,219,300,107,000,50,000,24,000,12,600,7,100,4,250,1,470のポリエチレングリコールをそれぞれ上記溶離液を用いて0.1重量%溶液を調整し、100μL注入する。
較正曲線:上記標準試料の測定結果を3次式で近似して較正曲線を作成する。
測定未知試料:上記溶離液を用いて測定未知試料の0.5重量%溶液を調整し、100μL注入する。
[製造例1]
温度計、攪拌機、生成水分離器および還流冷却管(コンデンサ)を備えた外部ジャケット付ガラス製反応槽(内容量:30リットル)にメトキシポリ(n=25)エチレングリコール16500部、メタクリル酸4740部(K値=70)、パラトルエンスルホン酸一水和物235部、フェノチアジン5部およびシクロヘキサン1060部を仕込み、エステル化反応中、生成水分離器および還流冷却管からなる循環系から反応容器に戻される経路上に流量計を設けて、還流される溶剤の流量(体積量)を計測し、溶剤循環速度が5サイクル/時間となるように、反応容器に取り付けられたジャケット温度を135℃に設定し、必要に応じて適宜微調節しながら、反応温度120℃でエステル化反応を行った。約20時間でエステル化率が99%に達したのを確認後、49%水酸化ナトリウム水溶液135部と水4890部を加えてパラトルエンスルホン酸を中和し、ハイドロキノン8部を加えて昇温し、シクロヘキサンを水との共沸で留去した。シクロヘキサン留去後、調整水を添加して80%のエステル化物水溶液を得た。得られたエステル化物水溶液について、電気泳動測定条件に従って、キャピラリー電気泳動により不純物の定量を行った。目的のエステル化物、触媒(PTS)、原料(MAA)によるピークが認められるだけで、不純物によるピークは認められなかった。すなわち、得られたエステル化物水溶液には不純物は認められなかった。
【0129】
表1に、原料単量体の着色度を示す。
[製造例2]
温度計、攪拌機、生成水分離器および還流冷却管(コンデンサ)を備えたSUS反応槽(内容量:30リットル)にメトキシポリ(n=25)エチレングリコール16500部、メタクリル酸4740部(K値=70)、パラトルエンスルホン酸一水和物235部、フェノチアジン5部およびシクロヘキサン1060部を仕込み、エステル化反応中、生成水分離器および還流冷却管からなる循環系から反応容器に戻される経路上に流量計を設けて、還流される溶剤の流量(体積量)を計測し、溶剤循環速度が5サイクル/時間となるように、反応容器に取り付けられたジャケット温度を135℃に設定し、必要に応じて適宜微調節しながら、反応温度120℃でエステル化反応を行った。約20時間でエステル化率が99%に達したのを確認後、49%水酸化ナトリウム水溶液267部と水6527部を加えてパラトルエンスルホン酸を中和し、ハイドロキノン8部を加えて昇温し、シクロヘキサンを水との共沸で留去した。シクロヘキサン留去後、調整用の水及びMAAを添加して75%のエステル化物水溶液を得た。
[製造例3]
温度計、攪拌機、生成水分離器および還流冷却管(コンデンサ)を備えたSUS製反応槽(内容量:30リットル)にメトキシポリ(n=25)エチレングリコール16500部、メタクリル酸4740部(K値=70)、パラトルエンスルホン酸一水和物235部、フェノチアジン5部およびシクロヘキサン1060部を仕込み、エステル化反応中、生成水分離器および還流冷却管からなる循環系から反応容器に戻される経路上に流量計を設けて、還流される溶剤の流量(体積量)を計測し、溶剤循環速度が5サイクル/時間となるように、反応容器に取り付けられたジャケット温度を135℃に設定し、必要に応じて適宜微調節しながら、反応温度120℃でエステル化反応を行った。約20時間でエステル化率が99%に達したのを確認後、49%水酸化ナトリウム水溶液432部と水6375部を加えてパラトルエンスルホン酸を中和し、ハイドロキノン8部を加えて昇温し、シクロヘキサンを水との共沸で留去した。シクロヘキサン留去後、調整用の水及びMAAを添加して75%のエステル化物水溶液を得た。
[製造例4]
温度計、攪拌機、生成水分離器および還流冷却管(コンデンサ)を備えたSUS製反応槽(内容量:30リットル)にメトキシポリ(n=25)エチレングリコール16500部、メタクリル酸4740部(K値=70)、パラトルエンスルホン酸一水和物235部、フェノチアジン5部およびシクロヘキサン1060部を仕込み、エステル化反応中、生成水分離器および還流冷却管からなる循環系から反応容器に戻される経路上に流量計を設けて、還流される溶剤の流量(体積量)を計測し、溶剤循環速度が5サイクル/時間となるように、反応容器に取り付けられたジャケット温度を135℃に設定し、必要に応じて適宜微調節しながら、反応温度120℃でエステル化反応を行った。約20時間でエステル化率が99%に達したのを確認後、49%水酸化ナトリウム水溶液597部と水6223部を加えてパラトルエンスルホン酸を中和し、ハイドロキノン8部を加えて昇温し、シクロヘキサンを水との共沸で留去した。シクロヘキサン留去後、調整用の水及びMAAを添加して75%のエステル化物水溶液を得た。
[製造例5]
温度計、攪拌機、生成水分離器および還流冷却管(コンデンサ)を備えた外部ジャケット付ガラス製反応槽(内容量:30リットル)にメトキシポリ(n=9)エチレングリコール16500部、メタクリル酸9450部(K値=70)、パラトルエンスルホン酸一水和物519部、フェノチアジン5部およびシクロヘキサン1298部を仕込み、エステル化反応中、生成水分離器および還流冷却管からなる循環系から反応容器に戻される経路上に流量計を設けて、還流される溶剤の流量(体積量)を計測し、溶剤循環速度が5サイクル/時間となるように、反応容器に取り付けられたジャケット温度を135℃に設定し、必要に応じて適宜微調節しながら、反応温度120℃でエステル化反応を行った。約20時間でエステル化率が99%に達したのを確認後、49%水酸化ナトリウム水溶液223部と水19941部を加えてパラトルエンスルホン酸を中和し、ハイドロキノン8部を加えて昇温し、シクロヘキサンを水との共沸で留去した。シクロヘキサン留去後、調整用の水及びMAAを添加して55%のエステル化物水溶液を得た。
[実施例1]
温度計、攪拌機、滴下ロート、窒素導入管および還流冷却器を備えたガラス製反応器に水712.5部を仕込み、攪拌下で上記反応器を窒素置換し、窒素雰囲気下で50℃まで昇温した。
【0130】
次に、上記反応器内に、製造例1で得られた80%のエステル化物水溶液1687.5部に3−メルカプトプロピオン酸11.3を溶解させた溶液を4時間かけて滴下すると同時に、過酸化水素9.6部を水300部に溶解させた水溶液とL−アスコルビン酸12.5部を水300部に溶解させた水溶液とを5時間かけて滴下した。滴下終了後、反応混合液を50℃に1時間維持した。さらに、この反応混合液のpHを30%水酸化ナトリウム水溶液でpH=7になるように調節することにより、重量平均分子量26,900の(メタ)アクリル酸(塩)系重合体(1A)を得た。実施例1において使用した重合開始剤量は、過酸化水素の使用量は8モル%、L−アスコルビン酸の使用量は2モル%である。
【0131】
表1にレドックス系重合開始剤の使用割合、反応温度、着色度などの結果を示す。
また、重合反応中、時々反応生成物をサンプリングしてpHを測定したところ、表2のとおりであった。また、最終生成物の残存メタクリル酸(MAA)量および残存メトキシポリエチレングリコールメタクリレート(PGM25E)量を測定したところ、表2の結果が得られた。
[実施例2]
過酸化水素を4.8部、L−アスコルビン酸を6.2部を用いたこと以外は実施例1と同様の操作を行い、重量平均分子量25,900の(メタ)アクリル酸(塩)系重合体(2A)を得た。実施例2において使用した重合開始剤量は、過酸化水素の使用量は4モル%、L−アスコルビン酸の使用量は1モル%である。
【0132】
表1にレドックス系重合開始剤の使用割合、反応温度、着色度などの結果を示す。
[実施例3]
過酸化水素を2.4部、L−アスコルビン酸を3.1部を用いたこと以外は実施例1と同様の操作を行い、重量平均分子量24,000の(メタ)アクリル酸(塩)系重合体(3A)を得た。実施例3において使用した重合開始剤量は、過酸化水素の使用量は2モル%、L−アスコルビン酸の使用量は0.5モル%である。
表1にレドックス系重合開始剤の使用割合、反応温度、着色度などの結果を示す。
また、重合反応中、時々反応生成物をサンプリングしてpHを測定したところ、表2のとおりであった。また、最終生成物の残存メタクリル酸(MAA)量および残存メトキシポリエチレングリコールメタクリレート(PGM25E)量を測定したところ、表2の結果が得られた。
[実施例4]
重合温度・維持温度を80℃に変更した以外は実施例2と同様の操作を行い、重量平均分子量22,000の(メタ)アクリル酸(塩)系重合体(4A)を得た。実施例4において使用した重合開始剤量は、過酸化水素の使用量は4モル%、L−アスコルビン酸の使用量は1モル%である。
【0133】
表1にレドックス系重合開始剤の使用割合、反応温度、着色度などの結果を示す。
[実施例5]
重合温度・維持温度を80℃に変更した以外は実施例3と同様の操作を行い、重量平均分子量22,100の(メタ)アクリル酸(塩)系重合体(5A)を得た。実施例5において使用した重合開始剤量は、過酸化水素の使用量は2モル%、L−アスコルビン酸の使用量は0.5モル%である。
表1にレドックス系重合開始剤の使用割合、反応温度、着色度などの結果を示す。
[比較例1]
温度計、攪拌機、生成水分離器および還流冷却管(コンデンサ)を備えた外部ジャケット付ガラス製反応槽(内容量:30リットル)に、水8200部を仕込み、攪拌下で上記反応器を窒素置換し、窒素雰囲気下で80℃まで昇温した。次に、上記反応器内に、製造例1で得られた80%のエステル化物水溶液13100部に3−メルカプトプロピオン酸94部を溶解させた溶液を4時間かけて滴下すると同時に、過硫酸アンモニウム125部を水1000部に溶解させた水溶液を5時間かけて滴下した。滴下終了後、反応混合液を80℃に1時間維持した。さらに、この反応混合液のpHを水酸化ナトリウムでpH=7になるように調節することにより、ゲルパーミエーションクロマトグラフィーによるポリエチレングリコール換算で重量平均分子量23,800の(メタ)アクリル酸(塩)系重合体(1B)を得た。
【0134】
表1にレドックス系重合開始剤の使用割合、反応温度、着色度などの結果を示す。
また、重合反応中、時々反応生成物をサンプリングしてpHを測定したところ、表2のとおりであった。また、最終生成物の残存メタクリル酸(MAA)量および残存メトキシポリエチレングリコールメタクリレート(PGM25E)量を測定したところ、表2の結果が得られた。
[比較例2]
比較例1の方法に従って製造した(メタ)アクリル酸(塩)系重合体(1B)の水溶液をイオン交換水より希釈し、固形分が40質量%になるように調製した水溶液40gを容量50mlのガラス製のサンプル管に入れ、過酸化水素水溶液(30%)を0.37g(重合体(1B)の重合前の全モノマーの8モル%)添加した。その後、室温にて4時間攪拌を行なった後、色差計にて着色度を測定した。YI値は33.11であった。過酸化水素と還元剤の組み合わせであるレドックス系重合開始剤を使用せず、重合後に過酸化水素のみを後添加した場合には、着色を低減することができなかった。
[比較例3]
50℃で6時間攪拌した以外は、比較例2と同様の操作を行った後、色差計にて着色度を測定した。YI値は24.03であった。過酸化水素と還元剤の組み合わせであるレドックス系重合開始剤を使用せず、重合後に過酸化水素のみを後添加した場合には、着色を低減することができなかった。
[比較例4]
L−アスコルビン酸を一切用いなかったこと以外は、実施例2と同様の操作を行ったところ、重量平均分子量30,500の(メタ)アクリル酸(塩)系重合体(4B)を得ることができた。高速液体クロマトグラフィー測定により、未反応のメタクリル酸を定量したところ、添加したメタクリル酸の98.8重量%が消費され、残りの1.2重量%が未反応のまま残存していることが判った。同様に実施例2で得られた重合体中のメタクリル酸を定量したところ、残存しているメタクリル酸は検出されなかった。
[比較例5]
過酸化水素を一切用いなかったこと以外は、実施例2と同様の操作を行ったが、重合は進行せず、目的の重合体を得ることができなかった。
[比較例6]
過酸化水素を38.4部(全モノマーのモル数に対して50モル%)を用いたこと以外は、実施例2と同様の操作を行ったが、重合中発泡が激しく、うまく重合することができなかった。
[比較例7]
L−アスコルビン酸を148.8部(全モノマーのモル数に対して24モル%、過酸化水素に対して600モル%)を用いたこと以外は、実施例2と同様の操作を行ったが、製品保存中に、L−アスコルビン酸由来の結晶が析出してしまうという好ましくない現象が確認された。
【0135】
【表1】
【0136】
[結果1]
(1)実施例1〜3の結果から、過酸化水素の配合量が多いほど、YI値で示す着色度が小さく、着色の程度が過酸化水素の配合量に依存することが示唆された。なお、比較例1の着色度は39.1と実施例1〜5と比較して極めて高値であるが、製造例1で得た80%のエステル化物水溶液のYI値は10.6であり、同じ原料を使用した実施例1〜5の着色度も3.10〜8.36と極めて低値であるため、上記着色度の低下は、原料化合物に由来するものではなく、製造方法による相違であると言える。
(2)実施例2と実施例4、および実施例3と実施例5とを比較すると、重合時の温度は50℃でも80℃でもYI値で示す着色度の変化は少なく、着色の程度は反応温度にあまり依存しない傾向があった。
(3)実施例1〜5においては中和後、塩の析出は確認されなかったが、比較例1においては塩の析出が確認された。
【0137】
[実施例6]
温度計、攪拌機、滴下装置および還流冷却管(コンデンサ)を備えたガラス製反応槽(内容量:3リットル)に、水446部を仕込み、攪拌下で上記反応器を窒素置換し、窒素雰囲気下で80℃まで昇温した。次に、上記反応器内に、製造例2で得られた75%のエステル化物水溶液900部に3−メルカプトプロピオン酸6.0部を溶解させた溶液を4時間かけて滴下すると同時に、過硫酸アンモニウム7.76部を水142.24部に溶解させた水溶液を5時間かけて滴下した。滴下終了後、反応混合液を80℃に1時間維持した。さらに、この反応混合液のpHを30%水酸化ナトリウム水溶液でpH=7になるように調製し、ゲルパーミエーションクロマトグラフィーによるポリエチレングリコール換算で重量平均分子量22,900の(メタ)アクリル酸(塩)系重合体(6A)を得た。
【0138】
重合反応中、時々反応生成物をサンプリングしてpHを測定したところ、表2のとおりであった。また、最終生成物の重量平均分子量Mw、ピークトップ平均分子量Mp、Mw−Mp、残存メタクリル酸(MAA)量および残存メトキシポリエチレングリコールメタクリレート(PGM25E)量を測定したところ、表2の結果が得られた。
[実施例7]
温度計、攪拌機、滴下装置および還流冷却管(コンデンサ)を備えたガラス製反応槽(内容量:3リットル)に、水444部を仕込み、攪拌下で上記反応器を窒素置換し、窒素雰囲気下で80℃まで昇温した。次に、上記反応器内に、製造例3で得られた75%のエステル化物水溶液900部に3−メルカプトプロピオン酸5.99部を溶解させた溶液を4時間かけて滴下すると同時に、過硫酸アンモニウム7.76部を水142.24部に溶解させた水溶液を5時間かけて滴下した。滴下終了後、反応混合液を80℃に1時間維持した。さらに、この反応混合液のpHを30%水酸化ナトリウム水溶液でpH=7になるように調製し、ゲルパーミエーションクロマトグラフィーによるポリエチレングリコール換算で重量平均分子量22,300の(メタ)アクリル酸(塩)系重合体(7A)を得た。
【0139】
重合反応中、時々反応生成物をサンプリングしてpHを測定したところ、表2のとおりであった。また、最終生成物の重量平均分子量Mw、ピークトップ平均分子量Mp、Mw−Mp、残存メタクリル酸(MAA)量および残存メトキシポリエチレングリコールメタクリレート(PGM25E)量を測定したところ、表2の結果が得られた。
[実施例8]
温度計、攪拌機、滴下装置および還流冷却管(コンデンサ)を備えたガラス製反応槽(内容量:3リットル)に、水443部を仕込み、攪拌下で上記反応器を窒素置換し、窒素雰囲気下で80℃まで昇温した。次に、上記反応器内に、製造例3で得られた75%のエステル化物水溶液900部に3−メルカプトプロピオン酸5.63部を溶解させた溶液を4時間かけて滴下すると同時に、過硫酸アンモニウム7.76部を水142.24部に溶解させた水溶液を5時間かけて滴下した。滴下終了後、反応混合液を80℃に1時間維持した。さらに、この反応混合液のpHを30%水酸化ナトリウム水溶液でpH=7になるように調製し、ゲルパーミエーションクロマトグラフィーによるポリエチレングリコール換算で重量平均分子量23500の(メタ)アクリル酸(塩)系重合体(8A)を得た。
【0140】
重合反応中、時々反応生成物をサンプリングしてpHを測定したところ、表2のとおりであった。また、最終生成物の重量平均分子量Mw、ピークトップ平均分子量Mp、Mw−Mp、残存メタクリル酸(MAA)量および残存メトキシポリエチレングリコールメタクリレート(PGM25E)量を測定したところ、表2の結果が得られた。
[実施例9]
温度計、攪拌機、滴下装置および還流冷却管(コンデンサ)を備えたガラス製反応槽(内容量:3リットル)に、水300部を仕込み、攪拌下で上記反応器を窒素置換し、窒素雰囲気下で80℃まで昇温した。次に、上記反応器内に、製造例2で得られた75%のエステル化物水溶液900部に3−メルカプトプロピオン酸7.5部を溶解させた溶液を4時間かけて滴下すると同時に、過酸化水素4.82部を水145.18部に溶解させた水溶液及びL−アスコルビン酸6.24部を水143.76部に溶解させた水溶液をそれぞれ5時間かけて滴下した。滴下終了後、反応混合液を80℃に1時間維持した。さらに、この反応混合液のpHを30%水酸化ナトリウム水溶液でpH=7になるように調製し、ゲルパーミエーションクロマトグラフィーによるポリエチレングリコール換算で重量平均分子量21,100の(メタ)アクリル酸(塩)系重合体(9A)を得た。
【0141】
重合反応中、時々反応生成物をサンプリングしてpHを測定したところ、表2のとおりであった。また、最終生成物の重量平均分子量Mw、ピークトップ平均分子量Mp、Mw−Mp、残存メタクリル酸(MAA)量および残存メトキシポリエチレングリコールメタクリレート(PGM25E)量を測定したところ、表2の結果が得られた。
[実施例10]
温度計、攪拌機、滴下装置および還流冷却管(コンデンサ)を備えたガラス製反応槽(内容量:3リットル)に、水300部を仕込み、攪拌下で上記反応器を窒素置換し、窒素雰囲気下で80℃まで昇温した。次に、上記反応器内に、製造例3で得られた75%のエステル化物水溶液900部に3−メルカプトプロピオン酸7.21部を溶解させた溶液を4時間かけて滴下すると同時に、過酸化水素4.82部を水145.18部に溶解させた水溶液及びL−アスコルビン酸6.24部を水143.76部に溶解させた水溶液をそれぞれ5時間かけて滴下した。滴下終了後、反応混合液を80℃に1時間維持した。さらに、この反応混合液のpHを30%水酸化ナトリウム水溶液でpH=7になるように調製し、ゲルパーミエーションクロマトグラフィーによるポリエチレングリコール換算で重量平均分子量22,200の(メタ)アクリル酸(塩)系重合体(10A)を得た。
【0142】
重合反応中、時々反応生成物をサンプリングしてpHを測定したところ、表2のとおりであった。また、最終生成物の重量平均分子量Mw、ピークトップ平均分子量Mp、Mw−Mp、残存メタクリル酸(MAA)量および残存メトキシポリエチレングリコールメタクリレート(PGM25E)量を測定したところ、表2の結果が得られた。
[実施例11]
温度計、攪拌機、滴下装置および還流冷却管(コンデンサ)を備えたガラス製反応槽(内容量:3リットル)に、水300部を仕込み、攪拌下で上記反応器を窒素置換し、窒素雰囲気下で80℃まで昇温した。次に、上記反応器内に、製造例4で得られた75%のエステル化物水溶液900部に3−メルカプトプロピオン酸7.28部を溶解させた溶液を4時間かけて滴下すると同時に、過酸化水素4.82部を水145.18部に溶解させた水溶液及びL−アスコルビン酸6.24部を水143.76部に溶解させた水溶液をそれぞれ5時間かけて滴下した。滴下終了後、反応混合液を80℃に1時間維持した。さらに、この反応混合液のpHを30%水酸化ナトリウム水溶液でpH=7になるように調製し、ゲルパーミエーションクロマトグラフィーによるポリエチレングリコール換算で重量平均分子量22,200の(メタ)アクリル酸(塩)系重合体(11A)を得た。
【0143】
重合反応中、時々反応生成物をサンプリングしてpHを測定したところ、表2のとおりであった。また、最終生成物の重量平均分子量Mw、ピークトップ平均分子量Mp、Mw−Mp、残存メタクリル酸(MAA)量および残存メトキシポリエチレングリコールメタクリレート(PGM25E)量を測定したところ、表2の結果が得られた。
[比較例8]
WO01/14438号が開示する方法に沿って(メタ)アクリル酸(塩)系重合体を製造した。すなわち、温度計、攪拌機、滴下装置および還流冷却管(コンデンサ)を備えたガラス製反応槽(内容量:3リットル)に、水382部を仕込み、攪拌下で上記反応器を窒素置換し、窒素雰囲気下で80℃まで昇温した。次に、上記反応器内に、製造例5で得られた55%のエステル化物水溶液900部に3−メルカプトプロピオン酸2.8部を溶解させた溶液を90分かけて滴下すると同時に、過硫酸アンモニウム2.52部を水18.48部に溶解させた水溶液を120分かけて滴下した。滴下終了後、反応混合液を80℃に1時間維持した。さらに、この反応混合液のpHを30%水酸化ナトリウム水溶液でpH=7になるように調製し、ゲルパーミエーションクロマトグラフィーによるポリエチレングリコール換算で重量平均分子量43500の(メタ)アクリル酸(塩)系重合体(8B)を得た。
【0144】
重合反応中、時々反応生成物をサンプリングしてpHを測定したところ、表2のとおりであった。また、最終生成物の重量平均分子量Mw、ピークトップ平均分子量Mp、Mw−Mp、残存メタクリル酸(MAA)量および残存メトキシポリエチレングリコールメタクリレート(PGM25E)量を測定したところ、表2の結果が得られた。
【0145】
【表2】
【0146】
[結果2]
比較例1では重合中のpHは2.36〜3.11、比較例8(WO01/14438号が開示する方法に相当)では重合中のpHは2.12〜2.72となっており、低いpH範囲である。これに対し、実施例1、3や、重合時のメタクリル酸の中和率を5〜15%にした実施例6〜11では重合中のpHは3.20〜5.23となっており、比較例1や8よりも高いpH範囲となっている。
比較例1や8では、重合系のpHが低いため、SUS製の重合釜の腐食が促進されてしまう。しかしながら、実施例1、3、6〜11では、重合系のpHが高いため、腐食を効果的に抑えることができる。
【0147】
また、比較例1や8では、残存MAA、残存PGM25Eの濃度が高く、重合率が低い。これに対し、実施例1、3、6〜11ではいずれも残存MAA、残存PGM25Eの濃度が低く、重合率が高くなっていることが分かる。
【産業上の利用可能性】
【0148】
本発明によれば、従来の反応装置を用いて充分に高分子量化したポリマー部分の比率が高く、また、着色度合いの少ない重合体を得ることができる。また、中和の際に結晶が析出したり、得られる重合体中に未反応の開始剤が多く残存したりすることなく、高い重合率でしかも短時間で重合するため、生産効率に優れる。
【図面の簡単な説明】
【0149】
【図1】本発明で使用されるエステル化物の製造方法に用いられる代表的な装置構成の概略図である。
【図2】本発明で使用されるエステル化物の製造の第二の実施態様に係る代表的なゲル化防止剤供給機構の装置構成を含めた本発明の製造装置の一実施形態を表わす概略説明図である。
【図3】コンデンサ直前のオーバーヘッドライン内にノズルを設置した様子を表わす概略説明図である。
【図4】保存部を兼ね備える水分離器の概略説明図である。
【技術分野】
【0001】
本発明は、セメント分散剤・顔料分散剤等に好適に使用される(メタ)アクリル酸(塩)系共重合体とその製造方法に関する。
【背景技術】
【0002】
不飽和モノカルボン酸系単量体(a)と不飽和ポリアルキレングリコール系単量体(b)とを必須成分として含む単量体成分を共重合して得られるポリカルボン酸は、従来から、セメント分散剤および顔料分散剤をはじめ、種々の分散剤に好適に用いられている。
ポリカルボン酸は、例えば、特許文献1では、ポリアルキレングリコールモノ(メタ)アクリル酸エステル単量体、(メタ)アクリル酸系単量体およびこれらと共重合可能な単量体を、ベンゾイルパーオキシド、ラウロパーオキシド等のパーオキシド、クメンハイドロパーオキシド等のハイドロパーオキシド、アゾビスイソブチロニトリルなどの脂肪族アゾ化合物を重合開始剤として用いて製造している。
【0003】
しかしながら、このような、不飽和モノカルボン酸系単量体(a)と不飽和ポリアルキレングリコール系単量体(b)との反応系においては、高温で反応させた場合、モノマーの重合率は高いものの、分子量の低いオリゴマーの生成量が増大し、分散剤としての性能を発揮しうるだけの高い分子量を有する有効なポリマー部分が減少しやすいという問題が生じる。一方、低温で反応させた場合には、モノマーの連鎖移動が抑制できるので、オリゴマー部分が少なく有効なポリマー部分の多い(メタ)アクリル酸(塩)系重合体が得られるが、モノマーの重合率が低くなる傾向がある。
この問題を解決するには、前記(メタ)アクリル酸(塩)系重合体を得る際に、低温で反応させ、かつ、モノマーの重合率を上げるために反応時間を長くすることが考えられるが、反応に長時間を要することから、生産性および作業性が悪く、工業的に実施する上でコストが問題となる。
【0004】
そこで、前記反応系において、低温でも反応性が高い重合開始剤を用いることが望まれる。
このような要望に応えるための重合開始剤として、過硫酸アンモニウムや過硫酸ナトリウム等の過硫酸塩がある。
前記反応系において過硫酸塩を開始剤として用いる公知技術の一つに、特許文献2が開示する方法がある。この方法は、重合反応液のpHを1.5〜3.5の範囲内に調整することで、物性(セメント分散性を示すペーストフロー値)の良い(メタ)アクリル酸(塩)系重合体を得るようにしている。この先行技術は、加えて、単量体の一方である不飽和ポリアルキレングリコール系単量体(b)は、ポリアルキレングリコールモノアルキルエーテルと(メタ)アクリル酸とを原料とし酸触媒を使用してエステル化することによって製造することとし、エステル化終了後に、上記酸触媒をアルカリ剤で失活させることで、重合を円滑化するようにしている。
【0005】
しかし、本発明者の検討したところによれば、特許文献2開示の上記先行方法は、開始剤としての過硫酸アンモニウム等の過硫酸塩を何らの工夫もなく使用するため、以下に述べる問題を生じている。すなわち、これらの開始剤は、重合時に硫酸が発生するため、ステンレスの反応釜では腐食するおそれがあり、グラスライニングされた反応釜を使用する必要がある。グラスライニングされた反応釜は、ステンレスの反応釜に比べ熱伝導率が低く、加熱冷却時の効率が悪い上に、衝撃に弱く、ライニング層が割れた場合の補修も困難であるため、設備の維持に多額の費用がかかるという問題を有する。ポリカルボン酸は、セメント分散剤として使用する場合、取り扱いやすくするため、通常、重合後に中和しておくことが望ましいのであるが、過硫酸塩を開始剤として用いて得られた(メタ)アクリル酸(塩)系重合体は、中和の際に塩が生成しやすく、この塩が結晶として析出して各種トラブルの原因となる恐れがある。また、過硫酸塩を開始剤として用いて得られた(メタ)アクリル酸(塩)系重合体の水溶液は茶褐色に呈色していることもあるため、着色の少ない分散剤に対する要求が高い顔料分散剤用途には不向きであった。
【0006】
本発明者の検討したところによれば、特許文献2開示の上記先行方法は、また、未だモノマーの重合率が十分でなく、未反応モノマーが残存しやすい方法であり、そのため、上記先行方法で製造した(メタ)アクリル酸(塩)系重合体は分散剤を構成するための配合割合が多くなると言う問題を有していた。
【特許文献1】特開昭59−18338号公報
【特許文献2】WO01/14438号明細書
【発明の開示】
【発明が解決しようとする課題】
【0007】
そこで、本発明の課題は、物性が高くて着色の少ない(メタ)アクリル酸(塩)系重合体を提供することと、このような高い品質の(メタ)アクリル酸(塩)系重合体を、低温でありながら高い重合率で得させることができ、しかも、反応釜の腐食を起こすことのない製造方法を提供することにある。
【課題を解決するための手段】
【0008】
本発明者は、上記課題を解決するために、種々工夫し検討し実験を重ねた。その結果、その解決方法の一つが、重合開始剤として過酸化水素と還元剤からなるレドックス系を特定量だけ用いることであり、もう一つが、単量体の一方である不飽和ポリアルキレングリコール系単量体(b)として、ポリアルキレングリコールと不飽和モノカルボン酸のエステル化で得たものを使用するが、このとき、ポリアルキレングリコールとして特定の平均付加モル数nを有するものを用いるとともに、エステル化原料の仕込み比を特定範囲に選定することとし、かつ、重合の際のpHを特許文献2開示の前記先行方法のpH範囲よりも高くすることである、ことを見出し、本発明を完成した。
【0009】
したがって、本発明にかかる(メタ)アクリル酸(塩)系重合体の製造方法は、下記一般式(1)で示される不飽和モノカルボン酸系単量体(a)と下記一般式(2)で示される不飽和ポリアルキレングリコール系単量体(b)とを重合開始剤を用いて重合させる(メタ)アクリル酸(塩)系重合体の製造方法であって、前記重合開始剤として過酸化水素と還元剤とからなるレドックス系重合開始剤を用いるとともに、前記過酸化水素の使用量が前記単量体の合計量に対して0.01〜30モル%であることを特徴とする。
【0010】
【化1】
【0011】
(ただし、式中、R1は水素原子またはメチル基を表わし、Mは水素原子、一価金属、二価金属、アンモニウム基、または有機アミン基を表す。)
【0012】
【化2】
【0013】
(ただし、式中、R1は水素またはメチル基を表し、R2は−COO(R3O)mR4を表す。前記R2を表す一般式において、R3Oは炭素数2〜18のオキシアルキレン基の1種または2種以上を表し、2種以上の場合はブロック状に付加していてもランダム状に付加していてもよい。前記R2を表す一般式において、R4は水素または炭素数1〜22のアルキル基を表し、mはオキシアルキレン基の平均付加モル数であって1〜300の整数を表す。)
【発明を実施するための最良の形態】
【0014】
(重合)
本発明にかかる(メタ)アクリル酸(塩)系重合体の製造方法においては、前記一般式(1)で示される不飽和モノカルボン酸系単量体(a)と前記一般式(2)で示される不飽和ポリアルキレングリコール系単量体(b)とを重合開始剤を用いて重合させる。
本発明で使用する上記一般式(1)で示される不飽和モノカルボン酸系単量体(a)としては、アクリル酸、メタクリル酸ならびにこれらの酸の一価金属塩、二価金属塩、アンモニウム塩および有機アミン塩を挙げることができ、これらの1種または2種以上を用いることができる。本発明では、不飽和モノカルボン酸系単量体(a)として、これらの1種を単独で使用できるほか、2種以上を混合して使用してもよい。
【0015】
上記一般式(2)で示される不飽和ポリアルキレングリコール系単量体(b)としては、ヒドロキシエチル(メタ)アクリレート、ヒドロキシプロピル(メタ)アクリレート、ポリエチレングリコールモノ(メタ)アクリレート、ポリプロピレングリコールモノ(メタ)アクリレート、ポリブチレングリコールモノ(メタ)アクリレート、ポリエチレングリコールポリプロピレングリコールモノ(メタ)アクリレート、ポリエチレングリコールポリブチレングリコールモノ(メタ)アクリレート、ポリプロピレングリコールポリブチレングリコールモノ(メタ)アクリレート、ポリエチレングリコールポリプロピレングリコールポリブチレングリコールモノ(メタ)アクリレート、メトキシポリエチレングリコールモノ(メタ)アクリレート、メトキシポリプロピレングリコールモノ(メタ)アクリレート、メトキシポリブチレングリコールモノ(メタ)アクリレート、メトキシポリエチレングリコールポリプロピレングリコールモノ(メタ)アクリレート、メトキシポリエチレングリコールポリブチレングリコールモノ(メタ)アクリレート、メトキシポリプロピレングリコールポリブチレングリコールモノ(メタ)アクリレート、メトキシポリエチレングリコールポリプロピレングリコールポリブチレングリコールモノ(メタ)アクリレート、エトキシポリエチレングリコールモノ(メタ)アクリレート、エトキシポリプロピレングリコールモノ(メタ)アクリレート、エトキシポリブチレングリコールモノ(メタ)アクリレート、エトキシポリエチレングリコールポリプロピレングリコールモノ(メタ)アクリレート、エトキシポリエチレングリコールポリブチレングリコールモノ(メタ)アクリレート、エトキシポリプロピレングリコールポリブチレングリコールモノ(メタ)アクリレート、およびエトキシポリエチレングリコールポリプロピレングリコールポリブチレングリコールモノ(メタ)アクリレート等を挙げることができる。本発明では、不飽和ポリアルキレングリコール系単量体(b)として、これらの1種を単独で使用できるほか、2種以上を混合して使用してもよい。不飽和ポリアルキレングリコール系単量体(b)のオキシアルキレン基の平均付加モル数mは1〜300の整数であり、好ましくは1〜200の整数、より好ましくは1〜110の整数、さらに好ましくは1〜100の整数、さらに好ましくは2〜100の整数、さらに好ましくは2〜50の整数、さらに好ましくは2〜20の整数である。
【0016】
重合に用いる重合性単量体としては、上記不飽和モノカルボン酸系単量体(a)と不飽和ポリアルキレングリコール系単量体(b)に加え、さらにこれらと共重合可能な他の単量体(III)を含んでいてもよい。このような他の単量体(III)としては、マレイン酸、フマル酸、シトラコン酸、メサコン酸、イタコン酸等のジカルボン酸類;これらのジカルボン酸類とHO(R5O)rR6(ただし、R5Oは炭素原子数2〜4のオキシアルキレン基の1種または2種以上の混合物を表わし、2種以上の場合はブロック状に付加していてもランダム状に付加していてもよく、rはオキシアルキレン基の平均付加モル数であり1から300の整数を表わし、R6は水素または炭素原子数1〜22、好ましくは1〜15のアルキル基を表わす。)で表わされるアルコールとのモノエステルあるいはジエステル類;(メタ)アクリルアミド、(メタ)アクリルアルキルアミド等の不飽和アミド類;酢酸ビニル、プロピオン酸ビニル等のビニルエステル類;ビニルスルホン酸、(メタ)アリルスルホン酸、スルホエチル(メタ)アクリレート、2−メチルプロパンスルホン酸(メタ)アクリルアミド、スチレンスルホン酸等の不飽和スルホン酸類およびそれらの一価金属塩、二価金属塩、アルモニウム塩、有機アミン塩類;スチレン、α−メチルスチレン等の芳香族ビニル類;炭素原子数1〜18、好ましくは1〜15の脂肪族アルコールあるいはベンジルアルコール等のフェニル基含有アルコールと(メタ)アクリル酸とのエステル類;ポリアルキレングリコールモノ(メタ)アクリレート;ポリアルキレングリコールモノ(メタ)アリルエーテル等が挙げられ、これらの1種または2種以上を用いることができる。
【0017】
共重合させる単量体(a)、(b)および(III)の重量比((a)/(b)/(III))は、特に限定はされないが、好ましくは(1〜99)/(99〜1)/(0〜50)、より好ましくは(50〜99)/(50〜1)/(0〜49)、さらに好ましくは(60〜95)/(40〜5)/(0〜30)、特に好ましくは(70〜95)/(30〜5)/(0〜10)の範囲である。
本発明の製造方法においては、使用することのできる不飽和ポリアルキレングリコール系単量体(b)は、上記記載の単量体の中の1種単独で用いても良いし2種以上を混合して使用しても良いのであるが、2種以上を混合して使用する場合には、使用用途に応じた特性を発現させることができるように、発現特性の異なる種類を適当に組み合わせて用いることが望ましく、以下の2種の組み合わせが有利である。
【0018】
すなわち、不飽和ポリアルキレングリコール系単量体(b)としては、式(2)におけるオキシアルキレン基の平均付加モル数mが、好ましくは1〜297の整数、より好ましくは1〜97の整数、さらに好ましくは1〜10の整数である第1のエステル化物(a1)と、式(2)におけるオキシアルキレン基の平均付加モル数mが、好ましくは4〜300の整数、より好ましくは4〜100の整数、さらに好ましくは11〜100の整数である第2のエステル化物(a2)との混合物(ただし、第2のエステル化物(a2)の平均付加モル数の方が第1のエステル化物(a1)の平均付加モル数よりも3以上大きいものとする)が有利である。
【0019】
このような第1のエステル化物(a1)と第2のエステル化物(a2)との混合物を使用するには、これらの第1および第2のエステル化物(a1)および(a2)を別々にエステル化反応により製造してもよいし、それぞれ相当するアルコールの混合物と(メタ)アクリル酸とのエステル化反応により製造してもよく、特に後者の方法は工業的に安価の製造方法を提供できる。この場合、第1のエステル化物(a1)と第2のエステル化物(a2)との質量比は5:95〜95:5であることが好ましく、より好ましくは10:90〜90:10である。
第1のエステル化物(a1)としては、例えば、メトキシ(ポリ)エチレングリコールモノ(メタ)アクリレート、メトキシ(ポリ)プロピレングリコールモノ(メタ)アクリレート、メトキシ(ポリ)ブチレングリコールモノ(メタ)アクリレート、メトキシ(ポリ)エチレングリコール(ポリ)プロピレングリコールモノ(メタ)アクリレート、メトキシ(ポリ)エチレングリコール(ポリ)ブチレングリコールモノ(メタ)アクリレート、メトキシ(ポリ)プロピレングリコール(ポリ)ブチレングリコールモノ(メタ)アクリレート、メトキシ(ポリ)エチレングリコール(ポリ)プロピレングリコール(ポリ)ブチレングリコールモノ(メタ)アクリレート、エトキシ(ポリ)エチレングリコールモノ(メタ)アクリレート、エトキシ(ポリ)プロピレングリコールモノ(メタ)アクリレート、エトキシ(ポリ)ブチレングリコールモノ(メタ)アクリレート、エトキシ(ポリ)エチレングリコール(ポリ)プロピレングリコールモノ(メタ)アクリレート、エトキシ(ポリ)エチレングリコール(ポリ)ブチレングリコールモノ(メタ)アクリレート、エトキシ(ポリ)プロピレングリコール(ポリ)ブチレングリコールモノ(メタ)アクリレート、エトキシ(ポリ)エチレングリコール(ポリ)プロピレングリコール(ポリ)ブチレングリコールモノ(メタ)アクリレート等が例示される。第1のエステル化物(a1)は、その側鎖の短鎖アルコールに疎水性を有することが重要である。なお、共重合のし易さの面からは、側鎖はエチレングリコール単位が多く含まれているのが好ましい。したがって、(a1)としては、平均付加モル数が好ましくは1〜297の整数、より好ましくは1〜97の整数、さらに好ましくは1〜10の整数の(アルコキシ)(ポリ)エチレングリコールモノ(メタ)アクリレートが好ましい。
【0020】
第2のエステル化物(a2)としては、例えば、メトキシポリエチレングリコールモノ(メタ)アクリレート、メトキシポリエチレングリコール(ポリ)プロピレングリコールモノ(メタ)アクリレート、メトキシポリエチレングリコール(ポリ)ブチレングリコールモノ(メタ)アクリレート、メトキシポリエチレングリコール(ポリ)プロピレングリコール(ポリ)ブチレングリコールモノ(メタ)アクリレート、エトキシポリエチレングリコールモノ(メタ)アクリレート、エトキシポリエチレングリコール(ポリ)プロピレングリコールモノ(メタ)アクリレート、エトキシポリエチレングリコール(ポリ)ブチレングリコールモノ(メタ)アクリレート、エトキシポリエチレングリコール(ポリ)プロピレングリコール(ポリ)ブチレングリコールモノ(メタ)アクリレートなどが例示される。高い減水性を得るためには、第2のエステル化物(a2)の平均付加モル数が4〜300の整数のアルコール鎖による立体反発と親水性でセメント粒子を分散させることが重要であり、好ましい。そのためには、ポリアルキレングリコール鎖にはオキシエチレン基が多く導入されることが好ましく、ポリエチレングリコール鎖が最も好ましい。よって、第2のエステル化物(a2)のアルキレングリコール鎖の平均付加モル数nは、好ましくは4〜300の整数、より好ましくは4〜100の整数、さらに好ましくは11〜100の整数である。
【0021】
本発明にかかる(メタ)アクリル酸(塩)系重合体の製造方法において、重合方法は特に限定されるものではなく、溶液重合や塊状重合などの公知の方法を採用することができる。特に、反応の制御や、重合物の取り扱いやすさの点を考慮すると、溶液重合が好ましい。
溶媒中での重合は、回分式でも連続式でも行なうことができ、その際使用される溶媒としては、水;メチルアルコール、エチルアルコール、イソプロピルアルコール等の低級アルコール;ベンゼン、トルエン、キシレン、シクロヘキサン、n−ヘキサン等の芳香族あるいは脂肪族炭化水素;酢酸エチル等のエステル化合物;アセトン、メチルエチルケトン等のケトン化合物;テトラヒドロフラン、ジオキサン等の環状エーテル化合物等が挙げられる。原料のエステル化物の単量体成分および得られる共重合体の溶解性ならびに該共重合体の使用時の便からは、水および炭素原子数1〜4の低級アルコールよりなる群から選ばれた少なくとも1種を用いることが好ましい。その場合、炭素原子数1〜4の低級アルコールの中でもメチルアルコール、エチルアルコール、イソプロピルアルコール等が特に有効である。この際、水の配合比は、好ましくは20質量%以下、さらに好ましくは10質量%以下、よりさらに好ましくは5質量%以下、最も好ましくは1質量%以下である。この割合が20質量%を超えると、該重合体が分離及び/または沈殿する恐れがある。
【0022】
本発明にかかる(メタ)アクリル酸(塩)系重合体の製造方法において用いることができる重合開始剤としては特に限定されず、例えば従来公知のもの、具体的には、ベンゾイルパーオキシド、ラウロパーオキシド等のパーオキシド、クメンハイドロパーオキシド等のハイドロパーオキシド、アゾビスイソブチロニトリルなどの脂肪族アゾ化合物、過硫酸アンモニウムや過硫酸ナトリウム等の過硫酸塩などを用いることができるが、本発明においては特に、前記重合開始剤として過酸化水素と還元剤とからなるレドックス系重合開始剤を用いることが好ましい。
すなわち、本発明にかかる(メタ)アクリル酸(塩)系重合体の製造方法の一つの好ましい形態は、一般式(1)で示される不飽和モノカルボン酸系単量体(a)と一般式(2)で示される不飽和ポリアルキレングリコール系単量体(b)とを重合開始剤を用いて重合させる(メタ)アクリル酸(塩)系重合体の製造方法であって、前記重合開始剤として過酸化水素と還元剤とからなるレドックス系重合開始剤を用いるとともに、前記過酸化水素の使用量が前記単量体の合計量に対して0.01〜30モル%であることを特徴とする。
【0023】
レドックス系重合開始剤を用いることにより、重合温度が低くても迅速に重合反応が促進でき、かつ、過酸化水素を使用し、その使用量を前記単量体の合計量に対して0.01〜30モル%にすることで、オリゴマー量を少なくし、かつ着色を防止することができる。
従来は、(メタ)アクリル酸(塩)系重合体を製造する際に使用する重合開始剤は過酸化物の単独使用が一般的であり、レドックス系重合開始剤は使用されていなかった。レドックス系重合開始剤として過硫酸アンモニウム/亜硫酸水素ナトリウムを使用すると、重合後の製品中に硫酸ナトリム等の塩類が析出するため、純度が低下する。一方、過酸化物を使用すると反応効率を高めるためにレドックス系重合開始剤を使用する場合に比較して、重合温度を高くする必要がある。このため高温反応による不純物が発生しやすく場合によっては製品が着色した。しかしながら、本発明にかかる(メタ)アクリル酸(塩)系重合体の製造方法の一つの好ましい形態では、過酸化水素と還元剤とからなるレドックス系重合開始剤を使用し、特に過酸化水素の使用量を重合性単量体の合計量に対して0.01〜30モル%、さらに好ましくは0.1〜20モル%、最も好ましくは0.5〜10モル%とすることで、低い温度でも短時間で重合ができ、しかも着色が少なく、同時にオリゴマー量も少ない(メタ)アクリル酸(塩)系重合体を製造できることを見出したのである。過酸化水素の使用量が重合性単量体の合計量の0.01モル%未満であると、未反応の単量体量が多くなり、一方、30モル%を超えると、オリゴマー部分が多い(メタ)アクリル酸(塩)系重合体が得られることとなるため、好ましくない。
【0024】
還元剤としては、モール塩に代表されるような鉄(II)、スズ(II)、チタン(III)、クロム(II)、V(II)、Cu(II)等の低原子価状態にある金属の塩類;モノエタノールアミン、ジエタノールアミン、トリエタノールアミン、ヒドロキシルアミン、塩酸ヒドロキシルアミン、ヒドラジン等のアミン化合物もしくはその塩;亜硫酸ナトリウム、亜硫酸水素ナトリウム、メタ二亜硫酸塩等のアルカリ金属亜硫酸塩;次亜リン酸、次亜リン酸ナトリウム、ヒドロ亜硫酸ナトリウム、亜二チオン酸ナトリウム等の低級酸化物もしくはその塩;ホルムアルデヒドナトリウムスルホキシラート、ヒドロキシメタンスルフィン酸ナトリウム二水和物等の−SH基、−SO2H基、−NHNH2基、−COCH(OH)−基等の基を有する有機系化合物もしくはその塩;D−フルクトース、D−グルコース等の転化糖;チオウレア、二酸化チオウレア等のチオウレア化合物;L−アスコルビン酸、L−アスコルビン酸ナトリウム、L−アスコルビン酸エステル、エリソルビン酸、エリソルビン酸ナトリウム、エリソルビン酸エステルなどが挙げられる。本発明においては、特に有機系還元剤を使用することが好ましく、有機系還元剤の中でも特に好ましくはL−アスコルビン酸である。なお、上記還元剤の使用に際してアミン化合物等の促進剤を併用することもできる。
【0025】
本発明にかかる(メタ)アクリル酸(塩)系重合体の製造方法の一つの好ましい形態では、過酸化水素の使用量は、重合性単量体の0.01〜30モル%であるが、前記還元剤の使用量は、過酸化水素に対して、0.1〜500モル%とすることが好ましく、さらに好ましくは1〜200モル%、最も好ましくは10〜100モル%とするのがよい。0.1モル%未満であると、活性ラジカルが充分に発生せず、未反応単量体が多くなり、一方、500モル%を越えると、残存する還元剤が多くなり、また、得られた(メタ)アクリル酸(塩)系重合体を中和して高濃度で保存しておく際、還元剤由来の結晶が析出することがあるため、好ましくない。
【0026】
重合温度は、用いる溶媒や重合開始剤により適宜定められるが、0〜95℃であることが好ましく、より好ましくは30〜90℃、特に好ましくは50〜85℃である。95℃を越えると熱による不純物が発生し、製品を着色させる恐れがあり、その一方0℃を下回ると重合反応に長持間を必要とし、好ましくない。本発明では、上記温度範囲でもオリゴマー部分が少なく有効なポリマー部分が多い(メタ)アクリル酸(塩)系重合体が得られる点に特徴がある。
本発明の(メタ)アクリル酸(塩)系重合体の製造方法における重合圧力は、特に限定されず、加圧、常圧(大気圧)、減圧いずれかでのよく、場合により適宜設定すればよいが、常圧が好ましい。
【0027】
本発明では、得られる重合体の分子量調節のために、チオール系連鎖移動剤を併用することもできる。この際に用いられるチオール系連鎖移動剤は、一般式HS−R7−Eg(ただし、式中、R7は炭素原子数1〜2のアルキル基を表し、Eは−OH、−COOM2、−COOR8または−SO3M2基を表し、M2は水素、一価金属、二価金属、アンモニウム基または有機アミン基を表し、R8は炭素原子数1〜10のアルキル基を表わし、gは1〜2の整数を表す。)で表わされ、例えば、メルカプトエタノール、チオグリセロール、チオグリコール酸、2−メルカプトプロピオン酸、3−メルカプトプロピオン酸、チオリンゴ酸、チオグリコール酸オクチル、3−メルカプトプロピオン酸オクチル等が挙げられ、これらの1種または2種以上を用いることができる。
(エステル化工程)
次に、本発明において用いる前記一般式(2)で示される不飽和ポリアルキレングリコール系単量体(b)の製造方法の好適な実施の形態について説明する。
【0028】
不飽和ポリアルキレングリコール系単量体(b)の製造方法としては、一般式(3)で示されるアルコキシポリアルキレングリコールと不飽和モノカルボン酸系単量体(a)とをエステル化反応させる方法が好ましい。
上記エステル化反応(エステル化工程)は、反応系(反応槽)に、原料としての一般式(3)で示されるアルコキシポリアルキレングリコールおよび不飽和モノカルボン酸系単量体(a)、酸触媒、および、必要に応じて、脱水溶剤や重合禁止剤を仕込み、これら混合物を一定温度で所定のエステル化率になるまで行う。
上記エステル化反応に原料として使用されるアルコキシポリアルキレングリコールは、下記一般式(3)で示される化合物である。
【0029】
【化3】
【0030】
上記一般式(3)において、R4は、炭素原子数1〜30の炭化水素基を表す。好ましくは炭素原子数1〜25の炭化水素基、より好ましくは炭素原子数1〜20の炭化水素基、さらに好ましくは炭素原子数1〜15の炭化水素基である。R4が炭素原子数30を超える炭化水素基である場合には、式(3)のアルコキシポリアルキレングリコールと不飽和モノカルボン酸系単量体(a)とのエステル化物を、例えば、(メタ)アクリル酸と共重合して得られる共重合体の水溶性が低下し、用途性能、例えば、セメント分散性能などが低下する。好適なR4の範囲はその使用用途により異なるものであり、例えば、セメント分散剤の原料として用いる場合には、R4は、炭素原子数1〜18の直鎖若しくは枝分かれ鎖の飽和あるいは不飽和アルキル基およびアリール基が好ましい。R4としては、具体的には、例えば、メチル基、エチル基、n−プロピル基、イソプロピル基、n−ブチル基、tert−ブチル基、ペンチル基、ヘキシル基、オクチル基、ノニル基、2−エチルヘキシル基、デシル基、ドデシル基、ウンデシル基、トリデシル基、テトラデシル基、ペンタデシル基、ヘキサデシル基、ヘプタデシル基、オクタデシル基、ノナデシル基、エイコシル基、ヘンエイコシル基、ドコシル基などのアルキル基;フェニル基などのアリール基;ベンジル基、ノニルフェニル基などのアルキルフェニル基;シクロヘキシル基などのシクロアルキル基;アルケニル基;アルキニル基などが挙げられる。これらのうち、セメント分散剤の原料として用いる場合には、上述したように、メチル基、エチル基、プロピル基、ブチル基、フェニル基が好ましいものである。
【0031】
上記一般式(3)において、R3Oは、炭素原子数2〜18、好ましくは炭素原子数2〜8、より好ましくは炭素原子数2〜4のオキシアルキレン基である。R3Oが炭素原子数18を超えるオキシアルキレン基である場合には、式(3)のアルコキシポリアルキレングリコールと不飽和モノカルボン酸系単量体(a)とのエステル化物を、例えば、(メタ)アクリル酸と共重合して得られる共重合体の水溶性が低下し、用途性能、例えば、セメント分散性能等が低下する。R3Oとしては、例えば、オキシエチレン基、オキシプロピレン基、オキシブチレン基、オキシスチレン基などが挙げられ、これらのうち、オキシエチレン基、オキシプロピレン基及びオキシブチレン基であることが好ましい。また、繰り返し単位であるR3Oは同一であってもよく異なっていてもよい。前記繰り返し単位R3Oが異なる場合、すなわち、2種以上の異なる繰り返し単位を有する場合には、各R3Oはブロック状に付加していてもよくランダム状に付加していてもよい。
【0032】
上記一般式(3)において、nは1〜300の整数であり、好ましくは1〜200の整数、より好ましくは1〜110の整数、さらに好ましくは1〜100の整数、さらに好ましくは2〜50の整数、さらに好ましくは2〜20の整数であり、R3O(オキシアルキレン基)の繰り返し単位の平均付加モル数を表わす。nが300を超える場合には、式(3)のアルコキシポリアルキレングリコールと不飽和モノカルボン酸系単量体(a)とのエステル化物の重合性が低下し、nが110を超える場合はその重合性の低下は大きい。この平均付加モル数nも、エステル化反応により得られるエステル化物の使用目的に応じて、その最適範囲は異なるものであり、例えば、セメント分散剤の原料として使用する場合には、平均付加モル数nは、好ましくは2〜100の整数、より好ましくは5〜90の整数、最も好ましくは8〜80の整数である。また、増粘剤などとして用いる場合には、平均付加モル数nは、好ましくは10〜100の整数、より好ましくは50〜90の整数である。
【0033】
エステルの製造方法において、上記一般式(3)で示されるアルコキシポリアルキレングリコールは、1種のものを単独で使用してもあるいは2種以上の混合物の形態で使用してもよい。式(3)で示されるアルコキシポリアルキレングリコール原料が2種以上の混合物での使用形態は、特に制限されるものではなく、R4、R3Oまたはnの少なくともいずれか1つが異なる2種以上の混合物での使用形態であればよいが、好ましくは(i)R4がメチル基とブチル基の2種で構成されている場合;(ii)R3Oがオキシエチレン基とオキシプロピレン基の2種で構成されている場合;(iii)nが1〜10のものと11〜100のものの2種で構成されている場合;および(i)〜(iii)を適宜組み合わせたもの等が挙げられる。
【0034】
上記エステル反応に使用することのできる不飽和モノカルボン酸系単量体(a)としては、アクリル酸およびメタクリル酸を、それぞれ単独で使用しても、あるいは混合して使用してもよく、その混合比率に関しても任意の範囲を採用することができる。
上記エステル化反応で使用される上記原料の混合比率は、化学量論的には1:1(モル比)であるが、実際には、アルコキシポリアルキレングリコールと不飽和モノカルボン酸系単量体(a)とのエステル化反応が効率良く進行する範囲であれば特に制限されるものではない。通常、一方の原料を過剰に使用してエステル化反応を速めたり、目的のエステル化物の精製面からは、蒸留留去し易いより低沸点の原料を過剰に使用することが好ましい。また、本発明では、エステル化反応時に反応生成水と脱水溶剤を共沸する際に、低沸点の不飽和モノカルボン酸系単量体(a)の一部も留去され、反応系外に持ち出されるため、アルコキシポリアルキレングリコールの使用量(仕込み量)に対して不飽和モノカルボン酸系単量体(a)の使用量(仕込み量)を化学量論的に算出される量よりも過剰に加えることが好ましい。具体的には、不飽和モノカルボン酸系単量体(a)の使用量は、通常、アルコキシポリアルキレングリコール1モルに対して、好ましくは1.0〜30モル、より好ましくは1.2〜10モル、さらに好ましくは1.5〜10モル、最も好ましくは2〜10モルである。不飽和モノカルボン酸系単量体(a)の使用量がアルコキシポリアルキレングリコール1モルに対して1.0モル未満であると、エステル化反応が円滑に進行せず、目的とするエステル化物の収率が不十分であり、逆に30モルを超えると、添加に見合う収率の向上が認められず、不経済であり、やはり好ましくない。
【0035】
上記エステル化反応においては、反応を速やかに進行させるために、酸触媒の存在下で反応を行うことが望ましい。この際に使用することができる酸触媒としては、例えば、硫酸、メタンスルホン酸、パラトルエンスルホン酸、パラトルエンスルホン酸水和物、キシレンスルホン酸、キシレンスルホン酸水和物、ナフタレンスルホン酸、ナフタレンスルホン酸水和物、トリフルオロメタンスルホン酸、「Nafion」レジン、「Amberlyst 15」レジン、リンタングステン酸、リンタングステン酸水和物、塩酸などが挙げられる。この際、酸触媒は単独で使用されてもあるいは2種以上の混合物の形態で使用されてもよい。
【0036】
これらのうち、以下に詳述する脱水溶剤と水との共沸温度、エステル化反応温度などを考慮すると、酸触媒は、常圧における沸点が高いものであることが好ましい。具体的には、本発明に好ましく使用される酸触媒の常圧における沸点は、150℃以上、より好ましくは200℃以上である。ゆえに、硫酸(常圧における沸点:317℃)、パラトルエンスルホン酸(沸点:185〜187℃/13.3Pa(0.1mmHg))、パラトルエンスルホン酸水和物及びメタンスルホン酸(沸点:167℃/1333.2Pa(10mmHg))などが好ましく使用される。さらに、本発明者は、エステル化物の品質および性能の低下の原因となる不純物のジエステルの生成原因の1つが、アルコキシポリアルキレングリコールの切断によるものであり、さらに当該切断が酸触媒によっても起こり得ることを知得した。かかる知見に基づき、当該切断のしにくい酸触媒がより望ましいことを見出した。上記点を考慮すると、本発明において特に好ましく使用される酸触媒としては、パラトルエンスルホン酸、メタンスルホン酸が挙げられる。酸触媒の使用量としては、触媒作用を有効に発現することができる範囲であれば特に制限されるものではないが、好ましくは0.4ミリ当量/g以下であり、より好ましくは0.36〜0.01ミリ当量/g、特に好ましくは0.32〜0.05ミリ当量/gの範囲内である。酸触媒の使用量が0.4ミリ当量/gを超えると、エステル化反応時に反応系内で形成されるジエステルの量が増加し、例えば、エステル化反応により得られるエステル化物(アルコキシポリアルキレングリコールモノ(メタ)アクリル酸)を用いて合成されるセメント分散剤のセメント分散能が低下する。ここで、酸触媒の使用量(ミリ当量/g)は、反応に使用した酸触媒のH+の当量数(ミリ当量)を、原料であるアルコキシポリアルキレングリコール及び不飽和モノカルボン酸系単量体(a)の合計仕込み量(g)で割った値で表される。より具体的には下記式によって算出される値である。
【0037】
酸触媒の使用量(ミリ当量/g)=
酸触媒H+の当量数(ミリ当量)/(アルコキシポリアルキレングリコールの仕込み重量(g)+不飽和モノカルボン酸系単量体の仕込み重量(g))
上記エステル化反応において、酸触媒の反応系への添加のし方は、一括、連続、または順次行ってもよいが、作業性の面からは、反応槽に、原料と共に一括で仕込むのが好ましい。
【0038】
上記エステル化反応において、酸触媒の存在下でエステル化反応を行う際に、酸触媒を水和物および/または水溶液の形態で用いてもよい。
上記態様において使用することのできる酸触媒としては、例えば、硫酸、メタンスルホン酸、パラトルエンスルホン酸、キシレンスルホン酸、ナフタレンスルホン酸、トリフルオロメタンスルホン酸、「Nafion」レジン、「Amberlyst 15」レジン、リンタングステン酸、塩酸などを水和物および/または水溶液の形態で用いるものが挙げられ、これらのうち、硫酸、パラトルエンスルホン酸、メタンスルホン酸などを水和物および/または水溶液の形態で用いるものが好ましく使用される。これらは、1種単独で使用してもよいし、2種以上を混合して使用しても良い。さらに、本発明者は、上述したように、エステル化物の品質および性能の低下の原因となる不純物のジエステルの生成原因の1つが、アルコキシポリアルキレングリコール原料の切断によるものであり、さらに当該切断が酸触媒によっても起こり得ることを知得し、かかる知見に基づき、当該切断のしにくい酸触媒がより望ましいこと見出したものである。当該酸触媒としては、具体的には、パラトルエンスルホン酸を水和物および/または水溶液の形態で用いるものである。
【0039】
上記態様による酸触媒の使用量は、所望の触媒作用を有効に発現することができる範囲であれば特に制限されるものではないが、アルコール原料の切断作用の抑制、各種用途、例えば、セメント分散剤、炭酸カルシウム、カーボンブラック、インクなどの顔料分散剤、スケール防止剤、石膏・水スラリー用分散剤、CWM用分散剤、増粘剤等に使用される重合体成分の原料となるエステル化物としての有用性、このような使用用途に要求される基本性能である分散性能などに悪影響を及ぼす原因となる分散性能の乏しい高分子量架橋ポリマーを発生させる原因となるゲル発生の防止・抑制を考慮すると、該酸触媒の使用量が、原料のアルコキシポリアルキレングリコールと不飽和モノカルボン酸系単量体(a)の合計質量に対する該酸触媒中の酸の質量の比をX(質量%)とし、該酸触媒中の水和物および/または水溶液として存在する水分の質量の比をY(質量%)とした場合に、
0<Y<1.81X−1.62
の関係を満足することが好ましい。
【0040】
なお、誤解がないように具体例を挙げて説明すれば、例えば、パラトルエンスルホン酸一水和物を例にとれば、原料の合計質量に対するパラトルエンスルホン酸の質量の比がX(質量%)であり、原料の合計質量に対する一水和物として存在する水分の質量の比がY(質量%)であるのであって、決して、酸触媒以外の酸成分(例えば、原料の(メタ)アクリル酸など)や水分(例えば、エステル化反応により生ずる生成水など)は、ここでいうXおよびYの対象物となりえない。
この際、酸触媒の使用量が上記式の関係を満足しない場合には、以下のような問題が生じる。すなわち、Y=0の場合には、酸触媒中に水和物および/または水溶液として存在する水分が存在しないこととなり、エステル化反応時に反応系内で形成されるゲルの量が増加し、エステル化反応により得られるエステル化物を用いて合成されるセメント分散剤等の用途性能、例えば、セメント分散能等が低下する。また、Y≧1.81X−1.62となる場合には、エステル化反応時に反応系内で形成されるゲルの量が増加し、エステル化反応により得られるエステル化物を用いて合成されるセメント分散剤等の用途性能、例えば、セメント分散能等が低下する。
【0041】
上記態様において、酸触媒の反応系への添加のし方は、一括、連続、または順次行ってもよいが、作業性の面からは、反応槽に、原料と共に一括で仕込むのが好ましい。
上記エステル化反応は、原料としてのアルコキシポリアルキレングリコール、不飽和モノカルボン酸系単量体(a)、またはこれらの混合物の重合を防止するため、重合禁止剤の存在下で反応を行うことが望ましい。使用できる重合禁止剤としては、公知の重合禁止剤などが挙げられ、特に制限されるものではなく、例えば、フェノチアジン、トリ−p−ニトロフェニルメチル、ジ−p−フルオロフェニルアミン、ジフェニルピクリルヒドラジル、N−(3−N−オキシアニリノ−1,3−ジメチルブチリデン)アニリンオキシド、ベンゾキノン、ハイドロキノン、メトキノン、ブチルカテコール、ニトロソベンゼン、ピクリン酸、ジチオベンゾイルジスルフィド、クペロン、塩化銅(II)などが挙げられる。これらのうち、脱水溶剤や生成水の溶解性の理由から、フェノチアジン、ハイドロキノン及びメトキノンが好ましく使用される。これらの重合禁止剤は、単独で使用してもよいほか、2種以上を混合して使用することもできる。
【0042】
また、上記のように酸触媒を水和物および/または水溶液の形で用いる場合には、フェノチアジンが、反応系内に存在する水溶液中のゲル形成物質に対しても有効に機能することができるほか、後述するように、エステル化反応終了後に、脱水溶剤を水との共沸により留去する際にも、弱いながらも重合活性のあるハイドロキノンやメトキノン等の水溶性重合禁止剤を用いなくても極めて有効に重合禁止能を発揮することができ、高分子量体の形成を効果的におさえることができる点から極めて有用である。
重合禁止剤を使用する場合、重合禁止剤の使用量は、原料としてのアルコキシポリアルキレングリコールおよび不飽和モノカルボン酸系単量体(a)の合計仕込量に対して、好ましくは0.001〜1質量%、より好ましくは0.001〜0.1質量%の範囲内である。重合禁止剤の使用量が0.001質量%未満であると、重合禁止能の発現が十分でなく、原料としてのアルコキシポリアルキレングリコール、不飽和モノカルボン酸系単量体(a)、生成物としてのエステル化物またはこれらの混合物の重合を有効に防止しにくくなるため好ましくなく、重合禁止剤の使用量が1質量%を超えると、生成物であるエステル化物中に残留する重合禁止剤量が増えるため、品質及び性能面から好ましくなく、また、過剰に添加することに見合うさらなる効果も得られず、経済的な観点からも好ましくない。
【0043】
上記エステル化反応においては、脱水溶剤中で、エステル化反応を行うことが好ましい。本明細書中、脱水溶剤とは、反応生成水と共沸する溶剤として規定されるものである。すなわち、脱水溶剤を用いることにより、エステル化反応により生成する反応生成水を効率よく共沸させることができるものである。
脱水溶剤としては、例えば、ベンゼン、トルエン、キシレン、シクロヘキサン、ジオキサン、ペンタン、ヘキサン、ヘプタン、クロロベンゼン、イソプロピルエーテルなどが挙げられ、これらを単独で、あるいは2種以上のものを混合溶剤として使用することができる。これらのうち水との共沸温度が150℃以下、より好ましくは60〜90℃の範囲であるものが好ましく、具体的には、シクロヘキサン、トルエン、ジオキサン、ベンゼン、イソプロピルエーテル、ヘキサン、ヘプタンなどが挙げられる。水との共沸温度が150℃を超える場合には、取り扱いの面(反応時の反応槽内の温度管理および共沸物の凝縮液化処理などの制御等を含む)から好ましくない。
【0044】
上記脱水溶剤は、反応系外に反応生成水と共沸させ、反応生成水を凝縮液化して分離除去しながら還流させることが望ましく、この際、脱水溶剤の使用量は、原料としてのアルコキシポリアルキレングリコールおよび不飽和モノカルボン酸系単量体(a)の合計仕込量に対して、1〜100質量%、好ましくは2〜50質量%の範囲内である。脱水溶剤の使用量が1質量%未満であると、エステル化反応中に生成する反応生成水を共沸により反応系外に十分除去できず、エステル化の平衡反応が進行しにくくなるため、好ましくなく、脱水溶剤の使用量が100質量%を超えると、過剰に添加することに見合う効果が得られず、また、反応温度を一定に維持するために多くの熱量が必要となり、経済的な観点から好ましくない。
【0045】
上記エステル化反応において脱水溶剤を使用する際には、エステル化反応中の反応温度を好ましくは30〜150℃、より好ましくは60〜140℃とし、かつ、エステル化反応中の溶剤循環速度を好ましくは0.5サイクル以上/時間、より好ましくは1〜100サイクル以上/時間とすることが望ましい。これにより、反応温度を不純物形成温度領域(130℃超の領域)まで高くして反応させる必要もなく、反応槽内で不純物が形成するのを抑えることができる。また、溶剤循環速度を速めることで、反応槽内に反応生成水を長期間滞留させることなく効率よく反応槽から共沸により留出でき、平衡反応がエステル化の方向に進むため、反応時間も短くできるものである。
【0046】
本明細書において、エステル化反応中の溶剤循環速度とは、次のように定義されるものをいう。すなわち、反応槽に仕込んだ脱水溶剤の全量(体積量)に対して、エステル化反応中に、反応槽内の脱水溶剤を反応槽から循環経路を通して再び反応槽に戻し循環させることにより、反応槽に仕込んだ脱水溶剤の全量に相当する量(体積量)が循環されたときを1サイクルと規定し、エステル化反応中の溶剤循環速度は、単位時間(1時間)あたりの当該サイクル数で表されるものとし、その単位は「サイクル/時間」とする。したがって、例えば、5時間で、反応槽に仕込んだ脱水溶剤の全量に対して、これに相当する量の15倍の量が循環されたときには、溶剤循環速度は3サイクル/時間となる。同様に、2時間で、反応系に仕込んだ脱水溶剤の全量に対して、これに相当する量の半分(0.5倍)の量が循環されたときには、溶剤循環速度は0.25サイクル/時間となる。
【0047】
なお、反応系内の脱水溶剤を反応系から留出し凝縮液化して反応系に戻し循環させる際に循環されるもの(被循環対象物)には、脱水溶剤のほか、その実施態様によっては、少量ではあるが、留出される低沸点原料(主に、不飽和モノカルボン酸系単量体原料(a))、およびこの留出原料がゲルを形成して有害な不純物となるのを防止するために添加されるゲル化防止剤(重合禁止剤または該重合禁止剤を含む溶剤等)などの各種添加剤が含まれることもあり得る。そのため、ゲル化防止剤等の添加剤を使用する場合には、これにより溶剤循環速度がエステル化反応が進むにつれて変動することを考慮して設定条件を適当に調整するのが望ましい。
【0048】
上記反応温度および溶剤循環速度は、反応槽の加熱方法(手段)およびその装置を用いて反応槽に加えられる温度(熱量)及び反応槽に仕込む原料に対する脱水溶剤の使用量などによって所望の範囲に調整することができる。なお、反応温度は、反応槽内での最大(MAX)温度である。すなわち、加熱手段として用いられる装置(例えば、外部ジャケット、内部ヒータなど)の態様により、反応槽内の温度(反応温度)は、その位置によりばらつくほか、エステル化反応が進むにつれても上がり、時間の経過によっても変動するが、反応温度が高くなることで、不純物の形成を招くため、位置的及び時間的な条件に関わらず、如何なる位置及び時間であれ、上記に規定する上限温度を超えないことが必要であることから、ここでは、最大温度をもって規定することにしたものである。
【0049】
本発明において、エステル化反応は、回分または連続いずれによっても行いうるが、回分式で行うことが好ましい。
また、エステル化反応における反応条件は、エステル化反応が円滑に進行する条件であればよく、反応温度は、好ましくは30〜150℃、より好ましくは60〜140℃、さらに好ましくは90〜135℃、特に好ましくは100〜130℃である。なお、上記反応温度は、本発明の一般的なエステル化反応の条件であり、脱水溶剤を反応系外に反応生成水と共沸させ、反応生成水を凝縮液化して分離除去しながら還流させる場合は、その1例であり、これらの範囲内に含まれるが、完全に一致するものではない。反応温度が30℃未満では、エステル化反応が進行しづらく、反応生成水の脱水(留出)にも時間がかかり、また、脱水溶剤の還流が遅くて脱水に時間がかかり、ゆえに、エステル化反応に要する時間が長くなり好ましくない。逆に、反応温度が150℃を超えると、アルコキシポリアルキレングリコール原料の切断によって過大量のジエステルが生成して、セメント分散性能のほか、各種用途における分散性能や増粘特性が低下する。また、原料の重合が生じたり、留出物への原料の混入量が増すなど、生成物であるエステル化物の性能及び品質の劣化が生じるなど、やはり好ましくない。また、反応時間は、後述するようにエステル化率が好ましくは少なくとも70%、より好ましくは少なくとも80%に達するまでであるが、通常、好ましくは1〜50時間、より好ましくは3〜40時間である。さらに、上記エステル化反応は、常圧下または減圧下いずれで行ってもよいが、設備面から、常圧下で行うことが望ましい。
【0050】
上記エステル化反応におけるエステル化率は、好ましくは70%以上、より好ましくは70〜99%、最も好ましくは80〜98%である。エステル化率が70%未満であると、製造されるエステル化物の収率が不十分であり、これを原料として得られるセメント分散剤等の用途性能、例えば、セメント分散能等が低下する。なお、本明細書において使用される「エステル化率」は、下記に示すエステル化測定条件で、エステル化の出発物質であるアルコールの減少量を測定することにより、下記式によって算出される値として定義されるものである。
<エステル化率測定条件>
解析装置; Waters製 Millennium
クロマトグラフィーマネージャー
検出器; Waters製 410 RI検出器
使用カラム;GLサイエンス製 イナートシルODS−2 3本
カラム温度;40℃
溶離液; 水 8946g
アセトニトリル 6000g
酢酸 54g
を混合して30質量%水酸化ナトリウム水溶液で
pH4.0に調整
流速; 0.6ml/min
なお、上記の式によりエステル化率を決定しているため、エステル化率が100%を越えることはない。従って、エステル化率が規定以上に達した時点でエステル化反応が終了したものとする。
【0051】
本発明においては、前記一般式(2)で示される不飽和ポリアルキレングリコール系単量体(b)が、前記一般式(3)で示されるアルコキシポリアルキレングリコールのp重量部と不飽和モノカルボン酸系単量体(a)のq重量部とを、[(p/n1/2)/q]×100≦200(ただし、nはオキシアルキレン基の平均付加モル数。)の関係を満たす条件下でエステル化反応させることにより得られるエステル化反応物の形で前記重合に供されるものであることが好ましい。
これにより、不飽和モノカルボン酸系単量体(a)をアルコキシポリアルキレングリコールに比べて過剰に存在させてエステル化反応を行うと、得られたアルコキシポリアルキレングリコールモノ(メタ)アクリル酸系単量体は不飽和モノカルボン酸系単量体(a)を含む混合物の形態で存在するので、この混合物を単離せずにそのまま、あるいは必要により不飽和モノカルボン酸系単量体(a)やこれらの単量体と共重合可能な単量体を加えて、好ましくは混合物を単離せずにそのまま共重合反応に供することにより、ポリカルボン酸系共重合体が製造できるので好ましい。すなわち、アルコキシポリアルキレングリコール及び不飽和モノカルボン酸系単量体(a)の使用量を上記したような範囲内に調節することにより、アルコキシポリアルキレングリコールモノ(メタ)アクリル酸を単離するという工程を省略することができるため、量産に適しており、産業上の観点から好ましい。
【0052】
上記の式:[(p/n1/2)/q]×100の値は、「K値」とも称し、K値は、カルボン酸の質量当たりのポリアルキレングリコール鎖の平均数を表わす尺度である。本発明において、K値は、好ましくは42〜190(42≦K値≦190)、より好ましくは45〜160(45≦K値≦160)である、この際、K値が40未満であると、得られるセメント分散剤のセメント分散性能が十分でない。逆に、K値が200を超えると、得られるセメント分散剤のセメント分散性能がやはり低下する上、エステル化反応時間が著しく増大し、生産性が大幅に低下するので好ましくない。
上記式:[(p/n1/2)/q]×100≦200において、nはオキシアルキレン基の平均付加モル数であり、1〜300の整数であるが、好ましくは1〜200の整数、より好ましくは1〜110の整数、さらに好ましくは1〜100の整数、さらに好ましくは2〜100の整数、さらに好ましくは2〜50の整数、さらに好ましくは2〜20の整数である。nが300を超える場合には、式(3)のアルコキシポリアルキレングリコールと不飽和モノカルボン酸系単量体(a)とのエステル化物の重合性が低下し、特にnが110を超える場合はその重合性の低下は大きいのでいっそう好ましくない。
【0053】
すなわち、本発明においては、前記一般式(2)で示される不飽和ポリアルキレングリコール系単量体(b)が、前記一般式(3)で示されるアルコキシポリアルキレングリコールのp重量部と不飽和モノカルボン酸系単量体(a)のq重量部とを、[(p/n1/2)/q]×100≦200(ただし、nはオキシアルキレン基の平均付加モル数であって、nは1〜110の整数、さらに好ましくは1〜100の整数、さらに好ましくは2〜100の整数、さらに好ましくは2〜50の整数、さらに好ましくは2〜20の整数である。)の関係を満たす条件下で、酸触媒の存在下においてエステル化反応させることにより出発原料たる前記不飽和モノカルボン酸系単量体(a)の一部を残存させることが好ましい。
(アルコキシポリアルキレングリコール(アルキレンオキサイド付加物)の製造)
上記エステル化工程の原料であるアルコキシポリアルキレングリコールの製造について説明する。なお、以下の説明においてアルコキシポリアルキレングリコールをアルキレンオキサイド付加物と称する。
【0054】
本発明において用いることができるアルキレンオキサイド付加物の好ましい製造方法は、水酸基含有飽和化合物にアルキレンオキサイドを付加反応させてアルキレンオキサイド付加物を得る方法であって、水酸基含有飽和化合物にアルキレンオキサイドを付加させてアルキレンオキサイド低モル付加物を得る初期工程と、前記初期工程で得られたアルキレンオキサイド低モル付加物にアルキレンオキサイドをさらに付加させる付加モル数調整工程とを含む。
なお、上記付加モル数とは、平均付加モル数のことを意味し、水酸基1モルに対する平均付加モル数のことである。
【0055】
前記初期工程においては、アルキレンオキサイドの使用量を水酸基含有飽和化合物1モルに対して平均20モル以下とすることが重要である。このアルキレンオキサイドの使用量は、好ましくは平均18モル以下、より好ましくは平均15モル以下、さらに好ましくは平均12モル以下である。
これにより、該初期工程における反応前後の容量変化を通常の製造設備で対応できる範囲(例えば、好ましくは28倍以下、より好ましくは22倍以下、さらに好ましくは16倍以下、さらにより好ましくは10倍以下)に抑えることができる。
該初期工程で用いるアルキレンオキサイドの使用量が水酸基含有飽和化合物1モルに対して平均20モルを超えると、反応前後の容量変化が大きくなり、特殊な反応器形状や特殊な攪拌装置を用いなければならなくなる。また、水酸基含有飽和化合物の仕込み量を減らして通常の設備を使用することも考えられるが、この場合、装置から混入する水分量が多くなり、ポリアルキレンオキサイド等の副生成物が増加することになるので好ましくない。
【0056】
前記初期工程におけるアルキレンオキサイドの使用量の下限値は、本発明の効果を損なわない範囲であれば特に限定されないが、水酸基含有飽和化合物1モルに対して、好ましくは平均1モル以上、より好ましくは平均2モル以上、さらに好ましくは平均3モル以上、さらにより好ましくは平均5モル以上である。
前記初期工程において用いる水酸基含有飽和化合物は、水酸基を有する飽和の化合物であれば特に限定されないが、本発明の効果を十分に発現する上で、炭化水素系の水酸基含有飽和化合物が好ましい。この場合の炭化水素系とは、化合物重量の70重量%以上、好ましくは80重量%以上、より好ましくは90重量%以上が、炭素、水素、酸素の3つの原子により構成された化合物のことをいう。さらに、本発明の効果をより十分に発現する上で好ましくは炭素数1〜30の飽和アルコールである。飽和アルコールの炭素数は、好ましくは1〜20、より好ましくは1〜15、さらに好ましくは1〜9、さらにより好ましくは1〜6である。飽和アルコールの炭素数が30を超える場合には、例えば、アルキレンオキサイド付加反応、エステル化反応またはエステル交換反応、重合反応を経て、共重合体とした場合に、かかる共重合体の水溶性が低下し、用途性能、例えば、セメント分散性能などが低下することがあるために好ましくない。
【0057】
前記初期工程において用いる水酸基含有飽和化合物として炭素数1〜30の飽和アルコールを用いる場合、具体的には、例えば、メチル基、エチル基、n−プロピル基、イソプロピル基、n−ブチル基、オクチル基、2−エチルヘキシル基、ドデシル基、ヘキサデシル基、オクタデシル基、などのアルキル基;フェニル基などのアリール基;ベンジル基、ノニルフェニル基などのアルキルフェニル基;シクロヘキシル基などのシクロアルキル基などを有する炭素数1〜30の飽和アルコールが挙げられる。これらの中でも、本発明の効果を十分に発現するためには、メタノール、エタノール、プロパノール、ブタノール、フェノールが好ましく、特にメタノールが好ましい。
【0058】
前記初期工程においては、含水率が6000ppm以下である水酸基含有飽和化合物を用いることが好ましい。より好ましくは4000ppm以下、さらに好ましくは2000ppm、さらにより好ましくは1000ppm以下、特に好ましくは500ppm以下である。このように含水率を6000ppm以下とすることにより、初期工程におけるポリアルキレンオキサイド等の副生成物の生成をさらに抑制することができると同時に、続く付加モル数調整工程においても、ポリアルキレンオキサイド等の副生成物の生成をさらに抑制することができる。含水率が6000ppmを超えると、初期工程におけるポリアルキレンオキサイド等の副生成物が増加し、得られたアルキレンオキサイド低モル付加物には多量のポリアルキレンオキサイドが含まれることとなる。そして、続く付加モル数調整工程において、不純物として含まれるポリアルキレンオキサイドがさらにアルキレンオキサイドと付加して分子量の大きなポリアルキレンオキサイドが副生することになるので、例えば、得られたアルキレンオキサイド付加物をセメント添加剤用ポリマーの原料として用いた場合に、セメント分散剤としての性能を低下させることとなり、好ましくない。
【0059】
本発明において用いることのできる前記アルキレンオキサイドとしては、特に制限されるものではないが、炭素数2〜4のアルキレンオキサイドが好ましく、具体的には、例えば、エチレンオキサイド、プロピレンオキサイド、ブチレンオキサイド等が挙げられ、これらの1種または2種以上を用いることができる。
前記付加モル数調整工程においては、前記初期工程で得られたアルキレンオキサイド低モル付加物にアルキレンオキサイドを付加させるので、水酸基含有飽和化合物に同モルのアルキレンオキサイドを直接付加反応させる場合よりも反応前後の容量変化が小さく、特殊な反応器形状や特殊な攪拌装置をもたない通常の設備で、所望の付加モル数にまでアルキレンオキサイドを付加させることができる。
【0060】
本発明においては、前記付加モル数調整工程では前記初期工程で得られたアルキレンオキサイド低モル付加物全量中の一部の量を使用することが好ましい。このように、付加モル数調整工程において初期工程で得られたアルキレンオキサイド低モル付加物の全量を用いずに一部の量を使用することによって、仕込み容量に対する生成物の容量の増大を考慮することなく、通常の設備を用いて、アルキレンオキサイドの付加モル数が高いアルキレンオキサイド付加物を製造することができる。さらに、この方法によれば、生成物の容量を考慮して原料の仕込み量を少なくする必要がないので、かかる原料の仕込み量に対する、装置から混入する水分の相対量の増加を防ぐことができ、ポリアルキレンオキサイド等の副生成物の増大が抑えられる。
【0061】
上述のように、前記付加モル数調整工程では前記初期工程で得られたアルキレンオキサイド低モル付加物全量中の一部の量を使用する場合、使用しなかった残りは、さらに付加モル数調整工程に使用してもよい。この場合、残りの全部を使用しなくてもよい。
前記初期工程で得られたアルキレンオキサイド低モル付加物の全量のうち、一部の量を使用して前記付加モル数調整工程を行い、さらに残りの少なくとも一部を用いて、1回または2回以上に分けて付加モル数調整工程を行うことが、本発明の効果を十分に発現する上で好ましい。より好ましくは、前記残りの少なくとも一部を用いて行う付加モル数調整工程が1回または2回、特に好ましくは1回である。
【0062】
付加モル数調整工程において用いるアルキレンオキサイド低モル付加物の使用量は、該工程で使用する反応器の全容量の60%以下とすることが好ましい。より好ましくは55%以下、さらに好ましくは50%以下、さらにより好ましくは45%以下、特に好ましくは40%以下である。かかる付加モル数調整工程では、反応前後の容量変化は小さいので、通常の設備を用いて、アルキレンオキサイドの付加モル数を向上させることができるのであるが、アルキレンオキサイド低モル付加物の使用量が反応器の全容量の60%を超えると、通常の設備においてはアルキレンオキサイドの使用量が制限され、所望の付加モル数にまでアルキレンオキサイドを付加させることが困難になる場合がある。
【0063】
付加モル数が非常に高いアルキレンオキサイド付加物を得ようとする場合、前記付加モル数調整工程を2回以上に分けて行うことが好ましい。具体例を挙げると、例えば、付加モル数が50以上のアルキレンオキサイド付加物を所望する場合には、一例として、第1回目の付加モル数調整工程でアルキレンオキサイドの10モル付加物を製造し、次いで、該アルキレンオキサイドの10モル付加物に40モルのアルキレンオキサイドをさらに付加させればよい。
前記付加モル数調整工程は、前記初期工程後、得られたアルキレンオキサイド低モル付加物の一部を抜き出して貯蔵タンク等に移し、前記初期工程と同一の反応器を用いて行ってもよいし、初期工程で得られたアルキレンオキサイド低モル付加物の一部を別の反応器に移し、前記初期工程と異なる反応器で行ってもよい。
【0064】
前記付加モル数調整工程においては、アルキレンオキサイドの使用量をアルキレンオキサイド低モル付加物1モルに対して平均20モル以下とすることが好ましい。このアルキレンオキサイドの使用量は、より好ましくは平均18モル以下、さらに好ましくは平均15モル以下、さらにより好ましくは平均12モル以下である。
これにより、該付加モル数調整工程における反応前後の容量変化を通常の製造設備で対応できる範囲(例えば、好ましくは28倍以下、より好ましくは22倍以下、さらに好ましくは16倍以下、さらにより好ましくは10倍以下)に抑えることができる。
該付加モル数調整工程で用いるアルキレンオキサイドの使用量がアルキレンオキサイド低モル付加物1モルに対して平均20モルを超えると、反応前後の容量変化が大きくなり、特殊な反応器形状や特殊な攪拌装置を用いなければならなくなる。また、アルキレンオキサイド低モル付加物の仕込み量を減らして通常の設備を使用することも考えられるが、この場合、装置から混入する水分量が多くなり、ポリアルキレンオキサイド等の副生成物が増加することになるので好ましくない。
【0065】
前記付加モル数調工程におけるアルキレンオキサイドの使用量の下限値は、本発明の効果を損なわない範囲であれば特に限定されないが、アルキレンオキサイド低モル付加物1モルに対して、好ましくは平均1モル以上、より好ましくは平均2モル以上、さらに好ましくは平均3モル以上、さらにより好ましくは平均5モル以上である。
アルキレンオキサイドを付加させる際には、触媒として、例えば、アルカリ金属、アルカリ土類金属、およびそれらの水酸化物が好ましく、より好ましくは、ナトリウム、ナトリウムアマルガム、ナトリウムハライド、水酸化ナトリウム、カリウム、カリウムアマルガム、カリウムハライド、水酸化カリウム等の1種または2種以上を用いることができる。これら触媒は、付加モル数調整工程では初期工程で得られた生成物中に残存する触媒が作用するので、初期工程でのみ添加するようにしてもよいが、初期工程と付加モル数調整工程の両方において添加することが好ましい。なお、触媒の使用量については特に制限はないが、アルキレンオキサイド付加物に対して10〜5000ppmとするのが好ましい。
【0066】
前記初期工程における反応温度については、特に制限はないが、好ましくは60〜180℃の範囲内、より好ましくは60〜160℃の範囲内、さらに好ましくは70〜150℃の範囲内とするのがよい。反応温度が180℃を超えると、ポリアルキレンオキサイド等の副生成物が増える傾向があり、例えば、得られたアルキレンオキサイド付加物を用いてセメント分散剤用ポリマーを得た場合、減水性能等の性能が低下する傾向がある。一方、反応温度が60℃未満であると、付加速度が遅くなり、生産性が低下するので好ましくない。
前記付加モル数調整工程における反応温度については、特に制限はないが、好ましくは80〜180℃の範囲内、より好ましくは90〜170℃の範囲内、さらに好ましくは100〜160℃の範囲内とするのがよい。反応温度が180℃を超えると、ポリアルキレンオキサイド等の副生成物が増える傾向があり、例えば、得られたアルキレンオキサイド付加物を用いてセメント分散剤用ポリマーを得た場合、減水性能等の性能が低下する傾向がある。一方、反応温度が80℃未満であると、付加速度が遅くなり、生産性が低下するので好ましくない。
【0067】
前記初期工程および前記付加モル数調整工程における付加反応は、回分式で行ってもよく、連続式で行ってもよいが、通常、水酸基含有飽和化合物もしくはアルキレンオキサイド低モル付加物、および必要に応じて前記触媒中に、アルキレンオキサイドを連続的に投入して行われる。また、前記付加反応は、窒素、アルゴン、ヘリウムなどの不活性ガス雰囲気下で行うのが好ましく、窒素雰囲気下で行うのが特に好ましい。また、前記付加反応は、加圧下で行うのが好ましい。
アルキレンオキサイド付加物の製造方法によってアルキレンオキサイド付加物を製造する場合の反応器の大きさは、特に限定されないが、反応器の容量が大きくなるほど本発明の効果がより十分に発現できるので、好ましくは100L以上、より好ましくは500L以上、さらに好ましくは1m3以上、さらにより好ましくは5m3以上の製造スケールが有効である。
(部分中和)
本発明においては、さらに、エステル化反応後に塩基性物質で酸触媒と残存不飽和モノカルボン酸系単量体(a)の0〜60モル%を中和させることが好ましい。
【0068】
エステル化反応後に脱水溶剤を留去する工程で水を加えて共沸する場合や、あるいはエステル化物を用いてさらに重合を行うためにエステル反応後に調整水を加えて生成されたエステル化物水溶液を作製する場合には、酸触媒によるエステル化物の加水分解が生じ、エステル化物の品質及び性能の低下を招くほか、加水分解により生じたもの(以下、単に加水分解生成物ともいう)がエステル化物中に残留し、当該エステル化物を用いてセメント分散剤等の各種分散剤や増粘剤等に使用される重合体を合成する場合には該加水分解生成物は重合には関与しない不純物となり、重合率(ひいては生産性)が低下し、また重合体の品質や性能の劣化にもつながる。かかる課題を解決するべく鋭意検討した結果、エステル化反応後に塩基性物質で酸触媒と残存不飽和モノカルボン酸系単量体(a)の0〜60モル%を中和させればよいことを見出した。これにより、エステル化反応後の処理過程で、加水分解生成物を生じることもなく、高純度で高品質のエステル化物を得ることができる。
【0069】
ここで、部分中和工程の好適な実施の形態につき、以下に説明する。
本発明に好適な部分中和工程では、エステル化反応終了後、好ましくは90℃以下、より好ましくは50〜0℃の範囲で酸触媒と残存不飽和モノカルボン酸系単量体(a)の0〜60モル%を塩基性物質で中和するものである。
上記部分中和工程での中和温度(反応系の液温)が、90℃を超える場合には、添加される塩基性物質が加水分解の触媒として作用し、加水分解生成物を多量に生成するようになるため好ましくない。さらに、50℃以下では、塩基性物質が加水分解の触媒として作用することはなく、加水分解生成物の発生を完全に抑えることができる。一方、0℃未満の場合には、エステル化反応液が粘稠になり、中和時の撹拌がしづらくなるほか、エステル化反応後に所定の温度まで降温するのに長時間を要するほか、室温よりも低い温度まで降温するには、新たに冷却手段(装置)を設ける必要があり、コストアップになるためあまり望ましくない。
【0070】
また、上記部分中和工程で使用することのできる塩基性物質(中和剤)としては、特に制限されるものではなく、水酸化物M(OH)nの形式をとり、水に溶解し、塩基性を示す物質であればよく、この場合のMは、アルカリ金属、アルカリ土類金属、アンモニウム基などをさす。さらに、アルカリ金属の炭酸塩や燐酸塩、アンモニア、アミン等もここでいう塩基性物質に含まれる。よって、塩基性物質としては、具体的には、例えば、水酸化ナトリウム、水酸化カリウム等のアルカリ金属の水酸化物、水酸化マグネシウム、水酸化カルシウム等のアルカリ土類金属の水酸化物、アンモニア、アミン等が挙げられるが、セメントに配合した場合に異臭が発生しないとの理由から、好ましくはアルカリ金属やアルカリ土類金属の水酸化物、炭酸塩、燐酸塩等である。また、本発明では、これら塩基性物質を1種若しくは2種以上を適当な比率で混合して使用してもよい。
【0071】
上記塩基性物質を用いて中和する酸は、酸触媒と残存不飽和モノカルボン酸系単量体(a)の0〜60モル%である。ここで、中和される不飽和モノカルボン酸系単量体(a)は残存不飽和モノカルボン酸系単量体(a)の0〜60モル%であり、好ましくは0〜55モル%、より好ましくは1〜55モル%、さらに好ましくは1〜50モル%、さらに一層好ましくは5〜50モル%、特に好ましくは5〜40モル%である。また、中和される不飽和モノカルボン酸系単量体(a)は、エステル化反応に使用した不飽和モノカルボン酸系単量体(a)の10質量%以下が好ましく、より好ましくは0.01〜5質量%の範囲である。従って、塩基性物質(中和剤)の添加量は、酸触媒1当量に対して1.0〜100当量が好ましく、より好ましくは1.0〜10当量、さらに好ましくは1.01〜2当量である。
【0072】
中和される不飽和モノカルボン酸系単量体(a)が、エステル化反応に使用した不飽和モノカルボン酸系単量体(a)の10質量%を超える場合には、おそらく、塩の形態の不飽和モノカルボン酸系単量体(a)の重合速度が塩の形態を形成していない不飽和モノカルボン酸系単量体(a)に比べて遅いために、得られたエステル化物を用いて重合する際の重合率が低下するため好ましくない。また、塩基性物質(中和剤)の添加量は、酸触媒1当量に対して1.0当量未満の場合には、酸触媒を完全に中和できず、加水分解生成物を多量に生じるようになるため好ましくない。逆に塩基性物質(中和剤)の添加量は、酸触媒1当量に対して100当量を超える場合にも、大量の不飽和モノカルボン酸系単量体(a)が中和され、やはり、塩の形態の不飽和モノカルボン酸系単量体(a)の重合速度が塩の形態を形成していない不飽和モノカルボン酸系単量体(a)に比べて遅いために、得られたエステル化物を用いて重合する際の重合率が低下するため好ましくない。
【0073】
中和すべき酸が酸触媒である理由は、上述したように、酸触媒がエステル化反応後に添加される水と強く反応し、加水分解生成物を生じさせるため、酸触媒を不活性にする必要があるためである。なお、酸成分としては、酸触媒以外に残存不飽和モノカルボン酸系単量体(a)が存在し得るが、酸触媒の方が酸強度が大きいので、酸触媒から優先的に中和される。
なお、添加される塩基性物質の形態としては、特に制限されるものではないが、アルカリ水溶液の形態とすることが、エステル化物の加水分解を防止する観点から好ましいといえる。
【0074】
特に、脱水溶剤中でエステル化反応を行う場合、塩基性物質と共に多量の水を反応系に添加するのが、エステル化物の加水分解を防止するためには好適である。すなわち、多量の水が無い反応系では、塩基性物質が脱水溶剤に難溶であるために濃い状態で系内に浮遊し、この高濃度の塩基性物質の浮遊は中和に消費されるまでの長持間にわたって消失せず、エステル化物の加水分解を引き起こす。該水の添加量は、塩基性物質の使用形態にもよるが、例えば、40〜60%のアルカリ水溶液を中和剤として添加する場合には、該アルカリ水溶液とは別に、該アルカリ水溶液の1質量部に対して通常5〜1000質量部が好ましく、より好ましくは10〜100質量部である。この場合に、水の添加量が、5質量部未満の場合には、上記理由で塩基性物質が反応系内で不均一になり、高濃度の塩基性物質がエステル化物の加水分解を引き起こし、1000質量部を超える場合には、生産性を確保するために中和槽が別途必要になるなどコスト高につながり好ましくない。
(pH調整)
本発明においては、好ましくは上記部分中和を行うことによって、重合反応の反応液のpHを3.2〜7.0の範囲内となるようにして重合反応を行うことが好ましい。重合反応の反応液のpHを3.2〜7.0の範囲内とすることにより、重合反応の重合率を高めることができるので好ましい。また、重合反応の反応液のpHを3.2〜7.0の範囲内とすることにより、重合装置(釜)の腐食を抑制することができるので好ましい。
【0075】
上記pHの範囲は、より好ましくはpH3.3〜7.0、さらに好ましくは3.5〜6.5、特に好ましくは4.0〜5.2である。
上記pH範囲を外れると、重合反応の重合率が低下し、また、重合装置(釜)の腐食がはげしいためコスト高となり、不経済である。
上記pH範囲で製造された重合体は、着色度が極めて低く、重量平均分子量Mwとピークトップ平均分子量Mpとの差であるMw−Mpも0〜9000の範囲に入るので、該重合体をセメント分散剤として使用する場合に、着色がなく、また分散性が極めて良好で、かつ減水効果が極めて高い。
【0076】
以上から、本発明にかかる(メタ)アクリル酸(塩)系重合体の製造方法における特に好ましい態様は、前記一般式(1)で示される不飽和モノカルボン酸系単量体(a)と前記一般式(2)で示される不飽和ポリアルキレングリコール系単量体(b)とを重合開始剤を用いて重合させる(メタ)アクリル酸(塩)系重合体の製造方法であって、前記一般式(2)で示される不飽和ポリアルキレングリコール系単量体(b)が、前記一般式(3)で示されるアルコキシポリアルキレングリコールのp重量部と不飽和モノカルボン酸系単量体(a)のq重量部とを、[(p/n1/2)/q]×100≦200(ただし、nはオキシアルキレン基の平均付加モル数であって、nは1〜110の整数、さらに好ましくは1〜100の整数、さらに好ましくは2〜100の整数、さらに好ましくは2〜50の整数、さらに好ましくは2〜20の整数である。)の関係を満たす条件下で、酸触媒の存在下においてエステル化反応させることにより出発原料たる前記不飽和モノカルボン酸系単量体(a)の一部を残存させるとともにエステル化反応後に塩基性物質で前記酸触媒と前記残存不飽和モノカルボン酸系単量体(a)の0〜60モル%を中和させるようにして得られるエステル化反応物の形で前記重合に供されるものであり、かつ、前記重合を、重合反応液のpHが3.2〜7.0の範囲内となるようにして行う方法である。
(溶剤留去工程:ゲル化防止)
本発明の製造方法においては、溶剤留去工程においてゲル化防止を行うことが好ましい。
【0077】
本発明において好ましく適用できるゲル化防止について、2つの実施態様を中心に説明する。
本発明者は、エステル化反応時に生成する反応生成水を留出させ、この反応生成水を含む留出物に対してゲル化防止剤を作用させることが望ましいことを見出した。以下、説明の便宜上、この態様を実施態様(A)と称することがある。
実施態様(A)により、反応系内の反応生成水を反応系外に留出してから凝縮液化し分離除去する間に、反応生成水とともに反応系外に留出されてくる低沸点の原料である(メタ)アクリル酸等により生ずるゲル状物(ポリ(メタ)アクリル酸等)の発生そのものを効果的に防止する、すなわち、製品の品質劣化や装置類の閉塞等の原因になるゲル状物を形成するのを防止することができる。
【0078】
実施態様(A)において、エステル化反応時に生成する反応生成水などの留出物に対して作用させるために用いられる上記ゲル化防止剤としては、反応生成水と共に留出されてくる低沸点の原料の留出段階、特に凝縮段階での重合反応を抑えることができ、反応槽からコンデンサへの立ち上がり管のフランジ部などで発生するゲルの形成、即ち、コンデンサのチューブや反応槽とコンデンサとの間の連結管のつまりを抑制できるものであれば特に制限されるものではない。上記ゲル化防止剤は、反応系内で同様の目的を持って使用される重合禁止剤と何ら変わるものではなく、上記に説明したと同様に使用できる。また、従来既知の各種ゲル化防止剤の中から適宜選択して使用することができる。上記ゲル化防止剤としては、具体的には、例えば、フェノチアジン、トリ−p−ニトロフェニルメチル、ジ−p−フルオロフェニルアミン、ジフェニルピクリルヒドラジル、N−(3−N−オキシアニリノ−1,3−ジメチルブチリデン)アニリンオキシド、ベンゾキノン、ハイドロキノン、メトキノン、ブチルカテコール、ニトロソベンゼン、ピクリン酸、ジチオベンゾイルジスルフィド、クペロン、塩化銅(II)などが挙げられる。これらの中でも、脱水溶剤や生成水の溶解性の理由から、フェノチアジン、ハイドロキノン、メトキノンが好ましく使用される。これらのゲル化防止剤は、単独で使用してもよいほか、2種以上を混合して使用することもできる。
【0079】
上記ゲル化防止剤の添加量としては、エステル化反応条件、特に反応系に加える熱量や反応系内に仕込む脱水溶剤量等に応じて、低沸点原料の留出量に見合う量、すなわち、共沸物の留出開始時からエステル化反応終了まで逐次留出されてくる低沸点原料に対して常にゲルの形成を効果的に防止することができる量を適宜添加すればよく、原料であるアルコキシポリアルキレングリコールおよび不飽和モノカルボン酸系単量体(a)の仕込み量に対して、好ましくは0.1〜1000質量ppm、より好ましくは1〜500質量ppmの範囲で添加することで上記目的を達成することができる。原料の仕込み量に対して0.1質量ppm未満の場合には、ゲル状物が生成する場合があり、共沸物の留出開始時からエステル化反応終了まで逐次留出されてくる低沸点原料に対して、常に重合禁止能を有効に発現させる上で不十分な量と言える。一方、原料仕込み量に対して1000質量ppmを超える場合には、ゲル形成防止(重合禁止)能を有効に発現させるには十分過ぎる量であり、過剰な添加に見合う更なる効果の発現が見込めず不経済となる。なお、添加量の全量を一時に加えたのでは、共沸物の留出開始時からエステル化反応終了まで逐次留出されてくる低沸点原料に対してゲルの形成を有効に阻止することができにくいため、共沸物の留出に呼応する形で、共沸物の留出開始時からエステル化反応終了まで逐次的に(連続的に)一定量ずつを添加し、最終的な添加量の総計が上記範囲となるように調整することが望ましい。
【0080】
上記ゲル化防止剤の作用のさせかた(作用形態や作用させる領域など)としては、反応系外に留出された低沸点原料(流体物)に対して有効に作用(接触)させることができるものであれば、特に制限されるものではなく、例えば、凝縮液化させる前のガス状の留出物に対して作用させてもよいし、凝集液化により液化した液状の留出物に対して作用させてもよい。また、上記の双方の作用方法を活用してもよい。
以下に、上記ゲル化防止剤の好適な作用方法を、作用形態ごとに例を挙げて説明する。本発明では、これらを適当に組み合わせることができるほか、従来既知の他の作用方法を適宜利用することができる。なお、下記に例示する作用方法は、当業者が本発明を容易に理解することができるように代表的なものを例示的に示したものであり、本発明がこれらに限定されるものではないことはいうまでもない。
【0081】
(i)ゲル化防止剤を液化(溶解)した状態で作用させる方法:適当な溶剤、好ましくは反応系に仕込む脱水溶剤と同種の溶剤にゲル化防止剤を溶かして液状にしたものを、反応生成水を含む留出物(好ましくは溶剤−水共沸物)を凝縮させる領域、具体的には、反応生成水を含む留出物の凝縮液化が行われるコンデンサ内部の凝縮部に、好ましくはコンデンサの上部(とりわけ塔頂部近傍)からその内部に該留出物と並流接触するように滴下ないし噴霧するものである。また、コンデンサのタイプ等によっては、ゲル化防止剤を含む溶液をコンデンサ内部に仕込んでおいて、これにガス状の留出物を吹き込むあるいは液化した留出物を流し込むようにして接触(相溶ないし分散)させるようにしてもよい。さらに上記態様では、ゲル化防止剤の作用部位をコンデンサ内部の凝縮部としたが、上記部位に加えて、反応槽とベーパーの立ち上がりラインとの間の接合部(フランジ部)やベーパーラインとコンデンサ塔頂部との間のフランジ部等のフランジ部、反応槽等に設置された温度計やのぞき窓に設けられた突起部など、ゲルが形成されやすい部位であってもよい。これらのうち、コンデンサ内部の凝縮部(とりわけ塔頂部近傍)、反応槽とベーパーの立ち上がりラインとの間のフランジ部やベーパーラインとコンデンサ塔頂部との間のフランジ部が、好ましいゲル化防止剤の作用部位である。また、上記作用部位は、一箇所でなくてもよく、必要に応じて、複数箇所を同時に設けてもよい。
【0082】
(ii)ゲル化防止剤を固化した状態で作用させる方法:粉末状のゲル化防止剤を、反応生成水を含む留出物を凝縮させる領域、具体的には、反応生成水を含む留出物の凝縮液化が行われるコンデンサ内部の凝縮部に、好ましくはコンデンサの上部(とりわけ塔頂部近傍)からコンデンサ内部に該留出物と並流接触するように投下ないし散布して降らせるものである。また、コンデンサのタイプなどによっては、一定粒度のゲル化防止剤を予めコンデンサ内部に積載ないし充填などして仕込んでおいて接触させるようにしてもよい。さらに上記態様では、ゲル化防止剤の作用部位をコンデンサ内部の凝縮部としたが、上記部位に加えて、反応槽とベーパーの立ち上がりラインとの間の接合部(フランジ部)やベーパーラインとコンデンサ塔頂部との間のフランジ部等のフランジ部、反応槽等に設置された温度計やのぞき窓に設けられた突起部など、ゲルが形成されやすい部位であってもよい。これらのうち、コンデンサ内部の凝縮部(とりわけ塔頂部近傍)、反応槽とベーパーの立ち上がりラインとの間のフランジ部やベーパーラインとコンデンサ塔頂部との間のフランジ部が好ましいゲル化防止剤の作用部位である。また、上記作用部位は、一箇所でなくてもよく、必要に応じて、複数箇所を同時に設けてもよい。
【0083】
(iii)ゲル化防止剤を気化した状態で作用させる方法:ゲル化防止剤を気化(昇華したものを含む)させて、ガス状の反応生成水を含む留出物(低沸点原料を含む)を凝縮液化させる前に、反応系(反応器)とコンデンサとを連通する配管経路内に、例えば、コンデンサ内部の凝縮部(とりわけ塔頂部近傍)、反応槽とベーパーの立ち上がりラインとの間の接合部(フランジ部)やベーパーラインとコンデンサ塔頂部との間のフランジ部等のフランジ部、反応槽等に設置された温度計やのぞき窓に設けられた突起部などのゲルが形成されやすい部位に、好ましくはコンデンサ内部の凝縮部(とりわけ塔頂部近傍)、反応槽とベーパーの立ち上がりラインとの間のフランジ部やベーパーラインとコンデンサ塔頂部との間のフランジ部に、供給して混合させるものである。
【0084】
本発明において、反応槽とベーパーの立ち上がりラインとの間のフランジ部におけるゲルの形成を抑制することを目的とする場合には、ゲル化防止剤を含ませずに脱水溶剤のみを上記フランジ部に供給することにより上記目的を達成してもよい。なお、上記において、脱水溶剤の具体例は、先に説明したものと同様である。上記の態様をエステル化反応中において適用する場合には、同じ種類の脱水溶剤を使用してもよいし、異なる種類の脱水溶剤を使用してもよい。または、以下に詳述するが、凝縮液(またはその一部)を循環させて使用してもよい。また、脱水溶剤の不存在下でエステル化反応を行う際には、別途、脱水溶剤供給機構を好ましくはフランジ部付近に設けて、脱水溶剤をフランジ部に供給すればよい。
【0085】
なお、上記(i)〜(iii)の中でも以下に説明する理由から、上記(i)を採用するのが好ましい。すなわち、経済的な観点および取り扱いの面からはより低い温度で脱水溶剤を留出し除去するのが望ましく、そのための手法としては、例えば、適量の水を用いて(特に、上記部分中和工程で薄い濃度のアルカリ水溶液で処理した場合には、大量の水が系内に既に存在しており、この水を用いてもよい)留出させる方法等が有効な手段として挙げられる。適量の水を用いて脱水溶剤と留出(共沸)させる場合には、水相側にも低沸点原料が移行し、水と共に留出されるほか、脱水溶剤の留去が漸次進につれて徐々に共沸されてくる留出物中の脱水溶剤の割合が低下し、最終的にはほとんど水(低沸点原料を含む)が留出されるようになることから、ゲル化防止剤を溶剤に溶かしても十分な効果が得られなくなることから、上記(i)の方法により、ゲル化防止剤を水と混合して作用させることが望ましく、特に、水溶性のゲル化防止剤を使用し、該水溶性ゲル化防止剤を水に溶解して作用させることがより望ましいものである。さらには、未反応の低沸点の原料を含有する留出物に有効に作用する(すなわち、低沸点の原料を含有する留出物が凝縮(液化)した際に、この液化物と速やかに接触し、ゲル化する低沸点の原料が含有されている液化物(水及び有機溶剤)に対して相溶ないし分散する)ことができるように、上記(i)の方法により、留出物の成分組成に応じてゲル化防止剤を水および/または溶剤に溶解したものを作用させることが望ましい。例えば、経時的な留出物の組成変化をセンサ等によりモニタしながら、作用させるゲル化防止剤組成(例えば、数種のゲル化防止剤を用い、溶剤、好ましくは脱水溶剤に溶解するゲル化防止剤組成と水に溶解するゲル化防止剤組成の混合比率)を変化させても良く、脱水溶剤に溶解するゲル化防止剤は脱水溶剤に溶解させたものと、水に溶解するゲル化防止剤は水に溶解させたものを別々の経路より、コンデンサ等の装置内に設けられたそれぞれの噴霧ノズルより滴下ないし噴霧するなどして作用させることが望ましいものである。また、上記(i)を採用する理由としては、単位質量あたりのゲル化防止剤に対して使用される液体の量が多くなるほど、かかるゲル化防止剤を混合した液体を液化凝縮手段の1つ(=熱交換媒体)として作用し得るとする利点も挙げられる。
【0086】
ここで、水に溶解した状態でゲル化防止剤を作用させる場合に用いることのできる水溶性のゲル化防止剤としては、例えば、ハイドロキノン、メトキノン等が好ましく使用される。
上記(i)の方法、すなわち、液化(溶解)した状態でゲル化防止剤を作用させる方法の場合に、上記ゲル化防止剤を溶解することのできる溶剤としては、例えば、ベンゼン、トルエン、キシレン、シクロヘキサン、アセトン、メチルエチルケトン、n−ヘキサン、ヘプタン等が挙げられるが、好ましくは上述したように、反応系に仕込まれる脱水溶剤と同種のものを用いるのがよい。
【0087】
すなわち、異なる溶剤を用いた場合には、これら混合溶剤を別途回収し再利用するには、多段階で分離精製処理を行う必要があり、再利用に要するコストが高くなり、使い捨てにした方が低コストである。しかし、こうした使い捨てによる混合溶剤の廃棄処理(焼却処理あるいは環境基準値以下に希釈化して廃水処理するなど)にも、一定のコストを要し、かつ少なからず大気汚染ないし水質汚染等を招くことから、今日の良く言われる地球に優しい環境づくりにいわば逆行することになる。一方、脱水溶剤と同種のものを用いる場合には、簡単な処理により低コストでの再利用が可能となり、コストおよび環境面で優れていると言える。
【0088】
上記のように、ゲル化防止剤を溶剤(好ましくは脱水溶剤)に溶解して作用させる場合、ゲル化防止剤は、ゲル状物の発生を抑制することができるように、コンデンサ内を通過する低沸点原料(ガスないし液化物)に対して、常にゲル化防止剤が存在して有効に機能するように供給されることが好ましい。
ゲル化防止剤と溶剤との混合比率は、特に制限されるものではないが、ゲル化防止剤を、溶剤100質量部に対して、通常、好ましくは0.001〜10質量部、より好ましくは0.01〜5質量部の範囲で混合する。ゲル化防止剤と溶剤との混合比率が、溶剤100質量部に対してゲル化防止剤が0.001質量部未満の場合には、使用するゲル化防止剤の添加量が上記に規定するように仕込みの原料に対して一定量であるため、結果的に使用する溶剤の量(添加される全量)が大きくなり、最初に仕込んだ脱水溶剤に対して逐次還流されることで溶剤量が増大していくため、反応系に加える熱量等の調整を行って反応生成水の留出量(留出速度)が大きく変動しないようにする必要があるなど、反応系の制御管理が複雑化する必要が生じる。また、脱水溶剤と異なる溶剤を用い、これを分離回収する場合には、その回収コストが増大し、製造コストがかさむことになる。一方、ゲル化防止剤と溶剤との混合比率が、溶剤100質量部に対してゲル化防止剤が10質量部を超える場合には、逆に使用する溶剤の量(添加される全量)が少なくなるため、単位時間当たりの添加量が制限され、低沸点原料との接触頻度が相対的に低下し、未接触のまま液状化しゲル状物を形成するのを効果的に抑制するのが困難になる。そのため、単位時間当たりに必要な添加量を確保するには、仕込みの原料に対して上記に規定する以上の大量のゲル化防止剤が必要になり、製造コストが上昇する。
【0089】
次に、ゲル化防止に関する別の実施態様(B)について説明する。
エステル化反応を脱水溶剤中で行う場合は、エステル化反応時に生成する反応生成水を脱水溶剤と共に留出させ、該反応生成水を含む留出物を凝縮液化し、該凝縮液化した凝縮液から反応生成水を分離除去し、該反応生成水を分離除去した後の脱水溶剤を含有する凝縮残液を反応槽に戻しながらエステル化反応を行う際に、該凝縮残液の一部とゲル化防止剤とを含有してなるゲル化防止剤溶液を留出物に作用させることが好ましい。この態様を実施態様(B)と称する。
実施態様(B)により反応槽内に増える凝縮残液の量を極力抑え、かつ留出物に対して(特に、留出物に対して該留出物が凝縮液化するコンデンサの壁面、とりわけ塔頂部の壁面を十分に濡らすことができるだけの)十分な量のゲル化防止剤溶液を常に供給する(コンデンサの塔頂部から降らせる)ことができる。そのため、反応槽内の反応生成水を反応槽から留出してから凝縮液化し分離除去する間に、反応生成水と共に留出されてくる低沸点の原料によるゲル状物の発生を、常に効果的に防止することができ、高品質のエスル化物を効率よく低コストで製造することができる。
【0090】
上記の実施態様(B)においては、留出物は、通常、エステル化反応により生成した反応生成水を含むほか、該反応生成水を反応槽から留出する際に一緒に留出される原料、特に不飽和モノカルボン酸系単量体(a)、さらに必要に応じて反応生成水と共沸させる目的で反応槽に加えられる脱水溶剤を含むものである。
エステル化反応終了後の脱水溶剤留去工程中に、該脱水溶剤を含む留出物に対してゲル化防止剤を作用させる場合、上記の実施態様(B)、および/または、前述の、エステル化反応時に生成する反応生成水を脱水溶剤と共に留出させた留出物にゲル化防止剤を作用させる実施態様(A)を含むものであってもよい。
【0091】
上記実施態様(B)に用いられるゲル化防止剤溶液は、留出物に作用させる溶液、より詳しくは留出物中の低沸点の原料に対してゲル化防止を目的で作用させる溶液であって、凝縮液の一部とゲル化防止剤とを含むものであるが、この際、ゲル化防止剤はそのままの形態で用いてもよいし、溶液の形態で用いてもよい。好ましくは、凝縮残液の一部と溶液形態のゲル化防止剤とを含むものである。
本明細書において、「凝縮液」ということばは、コンデンサの出口から出てきたものを意味する。また、上記実施態様(B)によると、ゲル化防止剤溶液をエステル化反応時に生成する反応生成水などの留出物に対して作用させてもよいため、このような場合には、ゲル化防止剤溶液が凝縮液に含まれる。さらにその後に水分離器で凝縮残液と分離水に分離されるため、凝縮残液及び分離水双方とも、凝縮液の定義に含まれ、これらは相互独立的に単独で使用することもできる。
【0092】
本明細書において、「凝縮液の一部」とは、凝縮液をただ単に部分的に分けたもの以外に、該凝縮液を分離して得られる凝縮残液および凝縮残液の一部も含まれる。
本明細書において、「凝縮残液」とは、水分離器で分けた溶剤側の成分をいい、「分離水」とは、水分離手段である水分離器で分けた水側の成分をいう。溶剤側の成分としては、ゲル化防止剤溶液のほか、必要に応じて使用される脱水溶剤等が含まれている。水側の成分としては、反応生成水や原料等がある。
上記コンデンサおよび水分離器は、本発明のエステル化物の製造方法において次のように使用されるものである。すなわち、本発明のエステル化物の製造方法では、エステル化反応時に生成する反応生成水を反応槽から留去する必要がある。しかし、留出物中には上記したように反応生成水以外の成分も含まれるため、直接大気中に放出することは環境汚染等の問題からできない。このため、かかる反応生成水を反応槽から留出した後に、適当に処理したり再利用したりできるようにする必要がある。そこで、コンデンサ(凝縮器)は、反応槽から留出されてなるものをそこに送り、凝縮液化するのに使われる。さらに、水分離器は、コンデンサの出口から出てきたものをそこに送り、その性質の違いを利用して2層に分離し、一方の層の水側の成分からなる分離水と、もう一方の層の溶剤側の成分からなる凝縮残液とに分けるのに使われる。
【0093】
実施態様(B)において、ゲル化防止剤溶液には、上記凝縮液の一部のほか、以下に説明するゲル化防止剤(溶液の形態を含む;以下、同様)、さらに他の添加剤、例えば、反応槽内への補充目的で適宜追加する酸触媒などが含有されていてもよい。
上述したように、ゲル化防止剤は、適当な溶剤、好ましくは脱水溶剤と同種の溶剤に溶解(ないし混合、例えば、過飽和状態で一部のゲル化防止剤が溶解せずに含まれている場合、2種以上のゲル化防止剤を用いた場合に、その一部のゲル化防止剤が溶剤に溶解せずに含まれている場合、さらにはゲル化防止剤が混合されている場合なども含む)されていることが好ましい。
【0094】
実施態様(B)において用いることができるゲル化防止剤としては、反応生成水等と共に留出されてくる低沸点の原料が、凝縮される段階で起こる重合反応を抑えることができるものであれば特に制限されるものではなく、従来既知の各種ゲル化防止剤の中から適宜選択して利用することができ、その具体例や好ましい例については、前述したものと同様である。
実施態様(B)において、上記ゲル化防止剤の使用量は、留出物の留出開始時からエステル化反応終了まで逐次留出されてくる低沸点原料に対して常にゲルの形成を効果的に防止することができる量(留出物の留出開始時からエステル化反応終了までの積算量)であることが好ましい。
【0095】
実施態様(B)において、エステル化反応に脱水溶剤を使用し、該脱水溶剤を留出し還流させる場合には、該ゲル化防止剤は、留出物に対して重合防止目的を達成した後、反応生成水を分離除去後の凝縮残液側に溶解した状態で反応槽に戻され、反応槽内に漸次蓄積される。その結果、反応により得られたエステル化物を原料として重合を行いセメント分散剤などの各種製品を製造する際に重合し難くなる。このため、ゲル化防止剤の使用量は極力抑えることが望ましい。以上の観点から、該ゲル化防止剤の使用量は、原料であるアルコキシポリアルキレングリコールおよび不飽和モノカルボン酸系単量体(a)の全使用量に対して、好ましくは0.1〜1000質量ppm、より好ましくは1〜500質量ppmの範囲である。ゲル化防止剤の使用量が、原料の全使用量に対して0.1質量ppm未満の場合には、反応生成水等を含む留出物の留出開始時からエステル化反応終了まで逐次留出されてくる低沸点原料に対して、常に重合禁止能を有効に発現させる上で不十分な量であるため、ゲル状物が生成する場合がある。一方、原料の全使用量に対して1000質量ppmを超える場合には、重合禁止能を有効に発現させるには十分過ぎる量であり、過剰な添加に見合う更なる効果の発現が見込めず不経済となるほか、得られたエステル化物を原料として重合を行いセメント分散剤などの各種製品を製造する際に重合が難しくなる。なお、ゲル化防止剤の使用量の全量を一時に加えたのでは、反応生成水を含む留出物の留出開始時からエステル化反応終了まで逐次留出されてくる低沸点原料に対してゲル状物の形成を有効に阻止することができにくいため、ゲル化防止剤の使用量が上記に規定する範囲内で連続的に加えるのが望ましい。この際、逐次留出される低沸点原料に対して、ゲル化防止剤溶液中のゲル化防止剤濃度が常に下記に規定する範囲となるように調整し連続的に加えるのがより望ましい。
【0096】
実施態様(B)において、ゲル化防止剤を溶液の形態で使用する際に用いることのできる溶剤としては、特に制限されるものではなく、例えば、ベンゼン、トルエン、キシレン、シクロヘキサン、アセトン、メチルエチルケトン、n−ヘキサン、ヘプタン等が挙げられる。また、エステル化反応に脱水溶剤を使用し、該脱水溶剤を留出し還流させる場合には、ゲル化防止剤溶液に用いた溶剤成分も凝縮残液側に含有されて反応槽に戻されるため、エステル化反応槽内で脱水溶剤として有効に作用し得るものであることが望ましい。特に、反応槽内に仕込んである脱水溶剤と異なる溶剤を用いた場合には、反応槽内の該溶剤量(濃度)の漸増により、該溶剤を含む脱水溶剤と反応生成水との共沸点(およびこれに伴う留出速度)が経時的に変動するため反応槽内部に加える熱量等の制御管理、さらには原材料の点数の増加に伴い、設備が増加し、安全・品質管理や在庫管理などが複雑化ないし煩雑化する等の点から、反応槽内に仕込んである脱水溶剤と同種の溶剤がより好ましい。
【0097】
ここでの溶剤の主な使用目的は、ゲル化防止剤の溶液化にあり、凝縮液の一部との混合が容易になされるようにし、凝縮液の一部との混合に際し撹拌装置等(例えば、撹拌槽など)を設けなくともよいようにすることにある。そして、エステル化反応に脱水溶剤を使用し、該脱水溶剤を留出し還流させる場合に、反応槽内に戻される凝縮残液量の増加を極力抑えるには、ゲル化防止剤溶液に使用する凝縮液(好ましくは凝縮残液)の一部の混合比率が高い方がよいことから、ここでの溶剤使用量は極力抑える事が望ましい。かかる観点から、上記溶液中のゲル化防止剤濃度としては、該溶液全体に対して好ましくは10質量ppm〜飽和濃度、より好ましくは100質量ppm〜飽和濃度、さらに好ましくは200質量ppm〜飽和濃度、特に好ましくは200質量ppm〜飽和濃度の95%に相当する濃度(ただし、飽和濃度は、ゲル化防止剤および溶剤の種類、温度、圧力等により変動し一義的に決まるものではないため、具体的な数値は規定していない)である。飽和溶液を用いることにより、溶剤の使用量を極力少なくすることができる。さらに、コンデンサから降らせるゲル化防止剤濃度を一定にするためには温度により変化する飽和濃度より、該飽和濃度よりも少し低濃度の方が良いため、飽和濃度の95%に相当する濃度以下で用いるのが好ましい。上記ゲル化防止剤濃度が、該溶液全体に対して10質量ppm未満の場合には、ゲル化防止剤溶液に使用する凝縮液の一部の混合比率が低下し、エステル化反応に脱水溶剤を使用し、該脱水溶剤を留出し還流させる場合には、反応槽に戻される凝縮残液の量が増える。あるいは漸増する凝縮残液をエステル化反応終了時まで貯えておける大きな保存部や時間とともに凝縮残液の一部を系外に出すための装置・手段等が必要となる。さらに溶剤の使用量も増えコストアップになる。
【0098】
ゲル化防止剤溶液に用いられるゲル化防止剤および凝縮液の一部の流量(流速)に関しては、ゲル化防止剤溶液中のゲル化防止剤の濃度および反応装置(反応槽や配管、コンデンサ等)の大きさや留出物の量等により異なるために一義的に規定することはできないが、ゲル化防止剤の量を減らして、これに代えて十分な量の凝縮液を用いることで、十分な量のゲル化防止剤溶液を留出物に作用させることができ、さらにエステル化反応に脱水溶剤を使用し、該脱水溶剤を留出し還流させる場合には、反応槽内の溶剤量の増加を極力抑えることができるように使用態様に応じて適宜決定(規定)すれば良い。
コンデンサの直径(内径)1mに対するゲル化防止剤1分間あたりの流量は、好ましくは0.01〜40リットル/分m、より好ましくは0.1〜15リットル/分m、さらに好ましくは0.1〜5リットル/分mであり、また、コンデンサの直径(内径)1mに対する凝縮液の一部の1分間あたりの流量は、好ましくは1〜1000リットル/分m、より好ましくは5〜500リットル/分m、さらに好ましくは10〜200リットル/分mである。ゲル化防止剤の流量が0.01リットル/分m未満の場合には、溶液中のゲル化防止剤濃度が低下し、常に十分な重合禁止能力を発現させることが困難となる。一方、ゲル化防止剤の流量が40リットル/分mを超える場合には、新たに加えられる溶剤量が増加するため、ゲル化防止剤の量を減らして、これに代えて十分な量の凝縮液を用いるとする本発明の主旨の達成が困難となる。また、凝縮液の一部の流量が1リットル/分m未満の場合には、留出物に対して常に十分な量の凝縮液を供給することができず、ゲル状物の発生を招くおそれがあるため好ましくない。一方、凝縮液の一部の流量が1000リットル/分mを超える場合には、これ以上の高流量で供給する事に見合う更なる効果が得られず、こうした多量の凝縮液を高流量で供給するための装置(大型のポンプや大口径ないし耐圧配管など)を設ける必要があり、不経済である。
【0099】
このように使用態様に応じて、ゲル化防止剤の流量を決定(規定)し、凝縮液の一部、好ましくは凝縮残液の一部の流量を決定(規定)した上で、流量の組み合わせは規定した流量の範囲内の組み合わせであれば全て可能であるが、本発明の主旨を十分に発揮するには、ゲル化防止剤に用いられるゲル化防止剤溶液と凝縮液の一部との混合比率は、以下の組み合わせがよい。
ゲル化防止剤1質量部に対して凝縮液の一部を好ましくは0.5〜10000質量部、より好ましくは1〜1000質量部、さらに好ましくは10〜1000質量部、特に好ましくは10〜100質量部の範囲で混合する。ゲル化防止剤1質量部に対して凝縮液の一部が0.5質量部未満の場合には、本発明の上記主旨を十分に満足させることができず好ましくない。一方、ゲル化防止剤1質量部に対して凝縮液の一部が10000質量部を超える場合には、両者を安定して混合することが困難となるためである。なお、これらの混合比率は、一定としてもあるいは可変させてもよく、本発明の上記主旨を満足するように適宜混合比率を決定すればよい。
【0100】
ゲル化防止剤溶液を作用させる方法としては、留出物、特に留出された低沸点原料に対して有効に作用させることができるものであれば、特に制限されるものではなく、従来既知の方法(手段)を適宜用いて行うことができる。好ましくはガス状の留出物を凝縮液化させる領域、具体的には、ガス状の留出物を凝縮液化する領域である熱交換器、冷却器あるいは凝縮器等(本明細書中では、これらを総称して単にコンデンサともいう)、特にガス状の留出物が凝縮液化し始めるコンデンサの塔頂部のガス入口部分において、有効にゲル化防止剤溶液が作用できるようにすることが望ましい。そのためには、ゲル化防止剤溶液が存在する領域はコンデンサ内には限られず、コンデンサの塔頂近傍、すなわち、コンデンサの塔頂ないしコンデンサ直前の留出経路内などにゲル化防止剤溶液を作用させればよく、そうすることでコンデンサの内壁を常に濡れた状態に保てることが望ましいと言える。具体例としては、(i)コンデンサの塔頂部の中央部に上向きに設置したノズル部よりコンデンサの塔頂部のガス入口部分の内壁(ここで、最初の凝縮液化が生じ、同時に低沸点原料のゲル化も生ずるためである)にゲル化防止剤溶液を噴霧したり、吹き出したり、吹き付けたり、吐出させたり、吹き上げたり、降らせたりすることでコンデンサの内壁を常に濡れた状態に保たせる方法、あるいは(ii)コンデンサ直前の留出経路(後述する第2図の配管503で形成された経路:オーバーヘッドライン)内にノズル部を設置し(第3図参照)、ここでゲル化防止剤溶液を噴霧し(または吹き出し)オーバーヘッドラインの壁を伝わせてコンデンサ内に到達させることでコンデンサの内壁を常に濡れた状態に保たせる方法などが挙げられるが、これらに限定されるものではない。さらに、エステル化反応に脱水溶剤を使用し、該脱水溶剤を留出し還流させる場合には、留出物が凝縮液化する際に、この液化物と速やかに接触し、ゲル化する低沸点の原料が含有されている脱水溶剤に対して相溶ないし分散することができるように、ゲル化防止剤は、ゲル化防止剤を脱水溶剤と同種の溶剤に溶解した形態で使用されるのが望ましい。
【0101】
また、凝縮液の一部をゲル化防止剤溶液に用いるべく還流させる方法としても、特に制限されるものではなく、従来既知の方法(手段)を適宜用いて行うことができる。具体例を以下に示す。
(i)エステル化反応に脱水溶剤を使用し、該脱水溶剤を留出し還流させる場合には、凝縮残液を反応槽に戻す際に凝縮残液の一部を抜き取って、上記ノズル部に直接的に供給し、該ノズル部でゲル化防止剤溶液とするか、あるいは上記ノズル部に供給する途中で、ゲル化防止剤と混合させてゲル化防止剤溶液とすることなどができる。具体例としては、後述する第2図に示すように、凝縮残液を反応槽(好ましくは、反応槽とベーパーの立ち上がりラインとの間のフランジ部)に戻す経路上に必要に応じて凝縮残液を一時的に貯めておく保存部(タンクなど)を設け、該保存部から該凝縮残液の一部を抜き取り、ゲル化防止剤の供給経路に抜き取った凝縮残液の一部を合流させるだけで簡単に混合されたゲル化防止剤溶液とすることができる。そのため、わざわざ両者を混合撹拌するための装置は不要である。ここで、保存部を設けるメリットとしては、ゲル化防止剤溶液用の凝縮残液の抜き取り量を一定量ないし徐々に増やす際にもその調整が便利であり、かつ反応槽に戻す凝縮残液の量を反応開始から終了までの間、常に一定量ないしは極力増加量を抑えながら還流させる事が容易に調整できる点にある。なお、保存タンクのような保存部を新たに設けなくとも、例えば、水分離器では、コンデンサで凝縮液化された凝縮液が一方の室に貯められ、水相と溶剤相の2層に分離され、下層部の水相はこの室の下部より配管を通じて逐次抜かれ、上層部の溶剤相は仕切板をオーバーフローして隣のもう一方の室に貯められるが、この溶剤相のみが貯められる室を大きくすれば、水分離器自体が保存部を兼ね備えることもできる(第4図参照)。
【0102】
保存部は必ずしも必要ではない。従来のゲル化防止剤単独使用の時と比較した場合、例えば、凝縮残液を含むゲル化防止剤溶液を使用しているため、ゲル化防止剤の量は従来と同程度あるいは少ない量で大きなゲル化防止効果を発揮できるようになった。そのため、反応槽内での溶剤量の増加も従来と同程度あるいは少ない量に抑えられている。特にエステル化時間が短い場合、反応温度の低下は少なく反応終了時間への影響も少なくてすむため、保存部を設けない方が経済的にも有利となるからである。また、反応槽内に戻される溶剤量が増加し多くなってきた場合、一部反応槽へ溶剤を戻さずに系外に抜き取ってもよい。この場合にも、系外に抜き取られる溶剤量は、大きくなくその処理コストも少ないため、わざわざ保存部を設けるよりも、経済的に有利となり、製品の性能を左右することもないからである。このように、性能面への影響、さらに費用対効果を勘案して、保存部を設けるか否か適宜判断する事が肝要であると言える。
【0103】
(ii)脱水溶剤を用いずにエステル化反応を行う場合には、本来的に留出物は反応生成水(僅かに低沸点原料を含む)だけであり、反応槽に留出物の一部を還流させることはない。従って、留出物にゲル化防止剤溶液を作用させた後の凝縮液から反応生成水(低沸点原料を含む)を分離除去した凝縮残液の全量若しくはその一部を抜き取って、上記ノズル部に直接的に供給して、該ノズル部でゲル化防止剤溶液とするか、あるいは上記ノズル部に供給する途中でゲル化防止剤と混合させてゲル化防止剤溶液とすることなどができる。なお、凝縮残液の一部を利用する場合、あとの凝縮残液は系外に抜き取れるなどすればよい。
【0104】
なお、上記に説明した作用させる方法および還流させる方法は、代表的なものを例示したに過ぎず、本発明がこれらに限定されるものではない。
本発明においては、以上のようなゲル化防止を行うことが好ましいが、さらに、溶剤留去工程として以下の形態をなすことが好ましい。
溶剤留去工程において、系内のエステル化物および脱水溶剤を含有する溶液から脱水溶剤を留出してから、凝縮液化して系外に除去するまでの装置機構に関しては、この間にゲル化防止剤を作用させるための手段(装置機構)が設けられていれば何ら制限されるものではなく、従来既知の装置機構を適当に組み合わせることができる。例えば、上述したエステル化工程において、エステル化反応中に、反応系内の脱水溶剤を反応系から留出し凝縮液化して反応系に戻し循環させるのに使用した装置機構(単に溶剤循環装置という)の一部を利用してもよく、装置設備の簡素化・小型化も図れることから望ましい実施態様の1つと言える。具体的には、ガス状の留出物を凝縮液化するための装置であるコンデンサ等に関しては先の溶剤循環装置をそのまま利用でき、凝縮液化された留出物の分離除去装置である液−液分離装置である水分離器等に関しては先の溶剤循環装置を適宜使用形態を変更して利用できる。すなわち、留出物の成分組成に応じて、当該水分離器に輸送されてくる液状の留出物を、水を系外に除去する輸送経路及び輸送装置であるポンプ等を利用して、水相部分あるいは液状の留出物の全てを系外に除去することができるほか、新たに当該水分離器等に真空ポンプ(エゼクタ)を取り付けて吸引することで、相対的に揮発性の高い成分等を選択的に、あるいは液状の留出物の全てを系外に除去するようにしてもよい。あるいは凝縮液化した留出物をコンデンサ等から別途輸送経路を設けてそのまま系外(例えば、廃棄物処理装置やリサイクル処理装置など)に取り出し、適当に処理(廃棄ないし再利用)することもできる。また、これらの装置にも、適当な制御機構が適宜設けられているのが望ましい。なお、上記に例示した装置機構に変えて、系内の脱水溶剤を留出し凝縮液化して系外に除去させるとする本来的な目的を逸脱しない限り、従来既知の他の手段及びその装置との組み合わせ、あるいは他の手段及びその装置による代替えなどによる方法を適宜採用することができることもいうまでもない。
【0105】
溶剤留去工程において、エステル化反応終了後(必要に応じて、上記部分中和工程を行い)、系内のエステル化物および脱水溶剤を含有する溶液から脱水溶剤を留去する方法に関しては、特に制限されるものではなく、上述したように水を用いて脱水溶剤と共沸させて留出し除去してもよいし、他の適当な添加剤を加えて脱水溶剤を効果的に除去するようにしてもよいほか、何等の添加剤(水を含む)を用いることなく、留出させて除去する事もできるが、エステル化反応において、酸触媒を用いることが極めて有用(すなわち、その後に部分中和しなければならないことを勘案してもその有用性は極めて高いといえる)であることから、水を用いて脱水溶剤と共沸させて留出し除去する方法が好ましい実施態様の1つと言える。なお、当該溶剤留去工程までに、酸触媒の部分中和処理が行われている際には、系内のエステル化物および脱水溶剤を含有する溶液中には、活性な酸触媒及びアルカリはなく(中和により塩になっている)、水を加えて昇温しても加水分解反応が起こらないため、脱水溶剤を留去する上で、水と共沸させる事ができる。なお、水と共沸させるほうが、より低い温度で効率よく脱水溶剤を除去することができるものである。
【0106】
溶剤留去工程において、系内の溶液中から脱水溶剤を留出させるための条件としては、系内の脱水溶剤を好適に留出(蒸発)させることができるものであれば、特に制限されるものではなく、溶剤留去中の系内温度(系内の液温(常圧下))としては、例えば、(i)水を用いる場合には、通常80〜120℃、好ましくは90〜110℃であり、(ii)水を用いない場合には、通常80〜160℃、好ましくは90〜150℃である。上記(i)ないし(ii)のいずれも場合にも、上記に規定する温度よりも低い温度の場合には、脱水溶剤を蒸発するのに十分な温度(熱量)でなく、脱水溶剤の留去に長時間を要するなど好ましくなく、一方、上記に規定する温度よりも高い温度の場合には、重合の危険性があるほか、多くの熱量が大量の低沸点原料の蒸発に消費されるため好ましくない。また、系内(装置内)圧力は、常圧下または減圧下いずれで行ってもよいが、設備面から、常圧下で行うことが望ましい。また、脱水溶剤を含む溶液から溶剤の留出を行うための装置系としては、エステル化反応で使用した装置系(反応槽)をそのまま使用するのがよい。すなわち、エステルカ反応後、別途他の装置に内容物を移し変える場合には、設備及び管理費が増加するほか、輸送時にエステル化物等が外的要因(熱、光、輸送温度、輸送圧力、活性な雰囲気ガスの介在)などにより劣化したり、輸送経路内に固着したり、逆に輸送時に装置などから不純物が溶出ないし混入するのを防止する必要があり、余分なコストが発生するなど好ましくない。
【0107】
なお、エステル化工程において、不飽和モノカルボン酸系単量体(a)の重合を防止すべく重合禁止剤の存在下に、エステル化反応を行っている場合には、当該重合禁止剤がエステル化反応後(さらには部分中和処理後)においても有効に機能するため、本溶剤留去工程において、系内の溶液中に、新たに重合禁止剤を補充する必要はないが、濃度の薄いアルカリ水溶液を用いて部分中和処理を行っている場合には、系内の溶液中に比較的多くの水が存在している。そのため、例えば、エステル化反応を行う際に使用した重合禁止剤が水に難溶ないし不溶であるような場合に限り、不飽和モノカルボン酸系単量体(a)が水に溶けて系内の溶液内で重合することがあるため、これを防止する観点から、系内の溶液に水溶性重合禁止剤を加えてから上記に規定する温度まで昇温することが望ましいものである。
【0108】
また、上記水溶性重合禁止剤としては、特に制限されるものではなく、例えば、ハイドロキノン、メトキノン、カテコール及びこれらの誘導体(例えば、p−t−ブチルカテコール等)、ハイドロキノンモノメチルエーテル等が挙げられる。なかでもハイドロキノン、メトキノンが好ましい。また、これらの水溶性重合禁止剤は、1種若しくは2種以上を混合して使用してもよい。
上記水溶性重合禁止剤の添加量としては、原料としてのアルコキシポリアルキレングリコール及び不飽和モノカルボン酸系単量体(a)の合計使用量に対して0.001〜1質量%、好ましくは0.001〜0.1質量%である。水溶性重合禁止剤の添加量が、0.001質量%未満の場合には、重合禁止能の発現が不十分な場合があり、水溶性重合禁止剤の添加量が、1質量%を超える場合には、過剰に添加することに見合う重合禁止能が得られず、不経済であり、好ましくない。
(エステル化物製造方法の具体的態様)
本発明において適用できるエステル化物の製造方法を、第1図を参照しながら説明する。
【0109】
第1図は、本発明において適用できるエステル化物の製造方法に用いられる代表的な装置構成の概略図である。
なお、以下において、原料としてのアルコキシポリアルキレングリコールを単にアルコールと称し、原料としての不飽和モノカルボン酸系単量体を単に(メタ)アクリル酸と称することがある。
第1図より、本実施形態の装置構成では、まず、所定温度まで昇温してエステル化反応し、エステル化反応後に所定温度まで降温して中和し、中和後に所定温度まで昇温し、脱水溶剤の留去を行うための熱交換手段(例えば、内部ヒータ等の直接加熱方式、外部ジャケット等の間接の熱交換方式)として加圧スチーム等を熱媒体に使用し得る外部ジャケット102を有する反応槽101が設けられている。この際、反応槽の内部の材料は、特に制限されるものではなく公知の材料が使用できるが、例えば、SUS製、好ましくは耐蝕性の面からSUS304、SUS316及びSUS316L、より好ましくはSUS316及びSUS316Lが挙げられる。または、反応槽の内部にグラスライニング加工等が施され原料及び生成物に対して不活性なものとしてもよい。該反応槽101には、アルコール原料用のステンレススチール(例えば、SUS316)製の原料貯蔵タンク103および(メタ)アクリル酸原料用の原料貯蔵タンク105、酸触媒用の触媒貯蔵タンク107、エステル化反応時の反応系(反応槽101)内の重合を防止するための重合禁止剤を貯蔵した重合禁止剤貯蔵タンク109、エステル化反応終了後の脱水溶剤の留去時の系内(反応槽101)の溶液内での重合を防止するための水溶性重合禁止剤を貯蔵した水溶性重合禁止剤貯蔵タンク110およびエステル化反応後に前記触媒を中和処理するための中和剤(中和剤水溶液)を貯蔵したカーボンスチール(例えば、高炭素鋼)製の中和剤貯蔵タンク111がそれぞれ配管113、115、117、119、120および121により連結されている。また、(メタ)アクリル酸は、重合しやすく、例えば、メタクリル酸では、長期の保存や熱等によっても重合するため微量の重合防止剤(0.1%ハイドロキノンなど)が加えられるほか、結晶化しても重合しやすくなるので、原料貯蔵タンク105内で保存する場合、ベンゼンを加え結晶化を防ぐようにしてもよいほか、第1図に示すように常時30〜40℃に保温するべく、ポンプ116を用いた外部ジャケット150(保温手段)を有する循環経路151が形成されており、(メタ)アクリル酸原料を常に30〜40℃に保持し重合しないように循環させている。(メタ)アクリル酸用の原料貯蔵タンク105、配管115およびポンプ116および循環経路151内部には、腐食性を有する(メタ)アクリル酸による腐食防止目的で、合成樹脂等の耐食性材料によるライニング加工が施されているものが使用される。同様に、触媒貯蔵タンク107およびその配管117内部にも、酸触媒による腐食防止のため、合成樹脂などの耐酸性材料によるライニング加工が施されているものが使用される。また、上記反応槽101の下部には、エステル化反応により反応槽101内部に合成されたエステル化物(あるいは、セメント分散剤等では、該エステル化物を単量体成分として該反応槽101でさらに重合を行い得られた重合体)を回収するための配管153が連結されている。さらに、上記反応槽101内には、反応温度を計測するための温度センサ(図示せず)が適当な部位(数カ所)に取り付けられている。該温度センサは、反応温度を規定の温度に保つのに必要な装置機構(例えば、反応槽101に取り付けられたジャケット102の温度)などを制御するための制御部本体(図示せず)に電気的に接続されている。
【0110】
さらに、本実施形態の装置構成では、反応系内(反応槽101内)でエステル化反応時に生成される反応生成水を含む留出物を留出し、ゲル状物の発生を防止しながら凝縮液化した後に、該反応生成水を分離除去し、残りの留出物を所定の溶剤循環速度で戻すための機構(の装置構成)として、該反応生成水を脱水溶剤とともに共沸させた留出物にゲル化防止剤を作用させて凝縮液化し、該凝縮液化した留出物から反応生成水(水相)を分離除去し、残りの凝縮物(主に脱水溶剤を含む溶剤相)を上記溶剤循環速度で還流させて反応槽101に戻す循環系が形成されている。詳しくは、反応槽101上部と向流(または並流)接触形式の縦型の多管式円管形コンデンサ125の頭頂部とが配管123により連結されている。またコンデンサ125の下底部とSUS製の水分離器127の上部とが配管129により連結されている。該水分離器127の内部には仕切板131が設けられており、該仕切板131で区切られた2つの室133、134が形成されている。このうち、コンデンサ125で凝縮液化された留出物が貯められる側の室133の下部と反応生成水の処理タンク135とが配管137により連結されている。また、該処理タンク135には廃水用の配管139が連結されている。また、水分離器127のもう一方の室134の下部と反応槽101とが配管141で連結されている。また、この配管141には、反応槽101内の反応生成水と共沸する脱水溶剤を貯蔵する脱水溶剤貯蔵タンク143と連結された配管145が合流(連結)されている。かかる合流点の手前(水分離器127側)の配管141の経路上には循環ポンプ142が設置されている。また、上記合流点の後方(反応槽101側)の配管141の経路上には流量計144が設けられている。そして、該流量計144には、計測される流量を積算し、溶剤循環速度を算出するための流量計測システム本体(図示せず)と電気的に接続されている。さらに、コンデンサ125の頭頂部には噴霧ノズル126が設けられており、この噴霧ノズル126は、留出物のゲル化防止用のゲル化防止剤を貯蔵するゲル化防止剤貯蔵タンク147と配管149により連結されている。
【0111】
本発明において、コンデンサとしては、SUS304、SUS316及びSUS316L等のSUS製や炭素鋼(CS)等、公知のものが使用できるが、好ましくは、ゲルの発生をより軽減するために、内面を鏡面仕上げやグラスライニング加工されたコンデンサを使用できるが、加工やメンテナンスにかかるコストを考慮すると、SUS304、SUS316及びSUS316L、好ましくはSUS316及びSUS316L等のSUS製のコンデンサが好ましく使用でき、このようなコンデンサを用いた場合でも、ゲルの形成を有効に防止できる。また、本発明において好ましく使用されるコンデンサの伝熱面積は、反応槽の容積などによって異なるが、例えば、反応槽30m3では、50〜500m2、好ましくは100〜200m2である。本発明において、コンデンサに使用される冷却媒体としては、水やオイルなどが挙げられる。
【0112】
また、上述したこれらの循環機構(の装置構成)、すなわち、反応系内(反応槽101内)でエステル化反応時に生成される反応生成水を含む留出物を留出し、ゲル状物を発生を防止しなから凝縮液化した後に、該反応生成水を分離除去し、残りの留出物を還流させるための機構(の装置構成)の一部は、エステル化反応後に、系内(反応槽101内)の生成物であるエステル化物を含有する溶液から脱水溶剤を含む留出物を留出し、ゲル状物の発生を防止しながら凝縮液化した後に、該脱水溶剤を含む留出物を系外に除去するための機構(の装置構成)としても利用されており、該脱水溶剤を水とともに共沸させた留出物に水溶性重合禁止剤を作用させて凝縮液化し、該凝縮液化した留出物を水相と溶剤相に分離し、それぞれを適当な方法により別々に除去する経路(取り出し経路)が形成されている。詳しくは、上述した循環機構(の装置構成)に、新たに、コンデンサ125の頭頂部に設けられた噴霧ノズル126には、脱水溶剤を含む留出物のゲル化防止用に利用される水溶性重合禁止剤を溶かした水溶液(以下、単に水溶性ゲル化防止剤ともいう)を貯蔵する水溶性ゲル化防止剤貯蔵タンク159が配管161により連結されている。さらに、水分離器127には配管157を介して、脱水溶媒を減圧吸引により除去するために真空ポンプ(エゼクタ)155が取り付けられている。
【0113】
本発明において好ましく適用できるエステル化物の製造方法では、以上の装置構成を有するエステル化物の製造装置を用いて次のように行われる。
まず、反応槽101内部に、各原料貯蔵タンク103、105、触媒貯蔵タンク107、重合禁止剤貯蔵タンク109、脱水溶剤貯蔵タンク143より配管113、115、117、119および配管145を介した配管141を通じて原料のアルコールおよび(メタ)アクリル酸、酸触媒、重合禁止剤および脱水溶剤をそれぞれ上記に規定する所定の量を送り込み(仕込み)、上記に規定するエステル化条件(反応温度、ジャケット温度、圧力)でエステル化反応を行う。エステル化反応により逐次生成する反応生成水は、反応槽101内に仕込まれた脱水溶剤と共沸され配管123を通じて留出されてくる。留出されてきたガス流体である溶剤−水共沸物は、コンデンサ125に通され凝縮液化される。この凝縮液化時に該共沸物に含まれる低沸点原料がゲル化するのを防止する目的で、ゲル化防止剤貯蔵タンク147より配管149を通じて該コンデンサ125の頭頂部に設けられた噴霧ノズル126から上記に規定する量のゲル化防止剤を連続的に滴下して、共沸物(ガス流体物および凝縮液化物の双方をいう)と並流接触させる。凝縮液化された共沸物(滴下されたゲル化防止剤を含む)は、該コンデンサ125の下部より配管129を通じて水分離器127の室133に貯められ、水相と溶剤相の2層に分離される。このうち、下層部の反応生成水は、室133の下部より配管137を通じて逐次抜かれ、反応生成水の処理タンク135に貯められる。そして該処理タンク135内で、必要に応じて、環境基準(廃水基準)値を満足するように化学的ないし生物学的に処理された後、配管139を通じて、本装置系外に廃水される。一方、上層部の溶剤相(滴下されたゲル化防止剤および低沸点原料を含む)は、仕切板131をオーバーフローして隣の室134に貯められる。そして、該溶剤相は該室134の下部よりポンプ142により配管141を通じて上記に規定する溶媒循環速度で還流され反応槽101に戻される。
【0114】
なお、本発明において、ゲル化防止剤を供給するゲル化防止剤貯蔵タンク設置の部位としては、ゲルが形成されやすい部位が好ましいものの、特に制限されず、例えば、第1図における態様、即ち、ゲル化防止剤を噴霧する噴霧ノズル126をコンデンサ125の塔頂部に設ける態様に加えて、反応槽101とコンデンサ125との間の配管123上の少なくとも1箇所にゲル化防止剤を噴霧する噴霧ノズルを設ける態様などが挙げられる。後者の態様において、配管123上にゲル化防止剤を噴霧する噴霧ノズルを設ける部位としては、例えば、コンデンサ内部の凝縮部(とりわけ塔頂部近傍)、反応槽とベーパーの立ち上がりラインとの間の接合部(フランジ部)やベーパーラインとコンデンサ塔頂部との間のフランジ部等のフランジ部、反応槽等に設置された温度計やのぞき窓に設けられた突起部など、ゲルが形成されやすい部位が挙げられ、これらのうち、コンデンサ内部の凝縮部(とりわけ塔頂部近傍)、反応槽とベーパーの立ち上がりラインとの間のフランジ部やベーパーラインとコンデンサ塔頂部との間のフランジ部が好ましい。
【0115】
エステル化反応終了(エステル化率が規定以上に達した時点で終了とする)後、降温し反応槽101の内温(液温)が上記に規定する温度(90℃)以下に下がるまで、反応槽101の外部ジャケット102に冷媒を通じて降温し、その後は所定温度以下を維持するように適宜調整しながら、中和剤貯蔵タンク111より配管121を通じて反応槽101内に、多量の水により上記に規定する濃度まで薄められたアルカリ水溶液(中和剤)を添加して酸触媒(及び(メタ)アクリル酸の一部)を部分中和する。
部分中和後、水溶性重合禁止剤を水溶性重合禁止剤タンク110より配管120を通じて反応槽101内の溶液に添加混合する。常圧下に、反応槽101の外部ジャケット102に熱媒(加圧スチーム)を通じて上記に規定する温度まで昇温することにより、反応槽101内の脱水溶剤及び部分中和処理の際に加えられている多量の水のほか未反応の低沸点原料(例えば、(メタ)アクリル酸)も共沸され配管123を通じて留出されてくる。留出されてきたガス流体である溶剤−水共沸物は、コンデンサ125に通され凝縮液化される。この場合にも未反応の低沸点原料(例えば、(メタ)アクリル酸)によりゲル状物が発生するが、ここでは、脱水溶剤が除かれていくので次第に水及び低沸点原料だけが蒸発してくるようになるため、水溶性重合禁止剤を作用させることが望ましい。この凝縮液化時に該留出物に含まれる未反応の低沸点原料がゲル化するのを防止する目的で、水溶性ゲル化防止剤貯蔵タンク159より配管161を通じて該コンデンサ125の頭頂部に設けられた噴霧ノズル126から上記に規定する量の水溶性ゲル化防止剤を連続的に滴下して、留出物(ガス流体物および凝縮液化物の双方をいう)と並流接触させる。凝縮液化された留出物(滴下された水溶性重合禁止剤を含む)は、該コンデンサ125の下部より配管129を通じて水分離器127の室133に貯められ、水相(滴下された水溶性重合禁止剤および低沸点原料を含む)と溶剤相の2層に分離される。このうち、下層部の水は、循環させずに除去する場合には、室133の下部より配管137を通じて逐次抜かれ、水の処理タンク135に貯められる。そして該処理タンク135内で、必要に応じて、環境基準(廃水基準)値を満足するように化学的ないし生物学的に処理された後、配管139を通じて、本装置系外に廃水される(また、下層部の水を循環させる場合には、室133の下部より反応槽101に連結される配管(図示せず)を設け、この配管を通じて還流すればよい。)。一方、上層部の溶剤相は、還流することなく装置系外に除去する必要上、水分離器127に取り付けられた真空ポンプ(エゼクタ)155を用いて装置系外に取り出される。なお、これらは廃棄処理されるか、あるいは系外の装置を用いて化学処理し再利用してもよい。
【0116】
最後に、系内の溶液から脱水溶剤を留去した後、反応槽101内に、配管(図示せず)により連結されている水貯蔵タンク(図示せず)または上水管(図示せず)より調整水を添加して所望のエステル化物の水溶液を得る。得られたエステル化物の水溶液は、配管153より回収(貯蔵)される。なお、得られたエステル化物をセメント分散剤等の合成に用いる場合には、該エステル化物を単量体成分の1つとして該反応槽101でさらに重合を行い、セメント分散剤の主要組成成分となり得る重合体を合成するようにしてもよい。この場合には、過剰に加えられ残っている未反応の(メタ)アクリル酸をもう一方の単量体成分として分離・除去せずにそのまま使用することが好ましい。
【0117】
以上が、本発明において好ましく適用できるエステル化物の製造方法の一実施態様を第1図を用いて説明したものであるが、本発明において好ましく適用できるエステル化物の製造方法は、当該実施態様に限定されるものではなく、エステル化反応終了後、脱水溶剤を留去する際に、該脱水溶剤を含む留出物に対して重合禁止剤を作用させることができるものであれば、その製法(手段)、装置構成などに関しては何ら制限されるものではなく、従来既知の製法、装置構成などを適宜組み合わせて利用することができる。
(重合体と用途)
本発明にかかる製造方法によって得られる重合体は、そのままでもセメント分散剤や顔料分散剤等の各種用途の主成分として用いられるが、必要に応じて、さらにアルカリ性物質で中和して得られる重合体塩をセメント分散剤等の各種用途の主成分として用いても良い。このようなアルカリ性物質としては、一価金属および二価金属の水酸化物、塩化物および炭素塩等の無機物;アンモニア;有機アミン等が好ましいものとして挙げられる。
【0118】
本発明において、(メタ)アクリル酸(塩)系重合体水溶液の中和後のpHは、特に限定されないが、3〜12が好ましい。12を越えると強アルカリ性となり、その一方3を下回ると強酸性の腐食条件となるため、高温では製造設備・貯蔵設備が腐食する恐れがある。より好ましくは5〜10、最も好ましくは6〜8である。
該単量体または(メタ)アクリル酸系重合体の中和剤としては、水酸化ナトリウム、水酸化カリウム等のアルカリ金属の水酸化物;水酸化カルシウム、水酸化マグネシウム等のアルカリ土類水酸化物;アンモニア;モノエタノールアミン、トリエタノールアミン等の有機アミンが挙げられる。これらは単独で用いてもよいし、2種以上の混合物として用いてもよい。好ましくは水酸化ナトリウム、水酸化カリウム等のアルカリ金属の水酸化物であり、より好ましくは水酸化ナトリウムである。
【0119】
中和は単量体を反応器に供給する前に予め行われてもよいし、単量体と反応器内で重合を行ないながら行ってもよい。また、重合開始から、重合終了時までの中和度は上記範囲内であれば、一定である必要は無く、具体的には、重合前半の中和度と重合後半の中和度は異なってもよい。
本発明の製造方法で得られる(メタ)アクリル酸(塩)重合体の分子量は、重量平均分子量で5,000〜10,000,000、好ましくは10,000〜5,000,000、より好ましくは20,000〜3,000,000、さらに好ましくは30,000〜1,000,000、最も好ましくは50,000〜500,000である。この重合体の重量平均分子量が5,000未満、または10,000,000を超えると分散剤として十分な性能が発揮されないといった問題が生じることがある。
【0120】
また、本発明にかかる製造方法の一つによれば、レドックス系重合開始剤を使用したため、オリゴマー部分の存在量が少ない(メタ)アクリル酸(塩)系重合体を製造することができることが判明した。レドックス系重合開始剤では、モノマーの重合率が高くかつ重合度を高く維持することができるため従来に比してオリゴマー部分が少ないのである。これは、分子量が均一な重合体が得られたことを意味するものであり、重合体の重量平均分子量の相違によって得られる重合体の特性が変化する中で、特に求める分子量のものが均一の組成で得られる点で、極めて有利である。不飽和モノカルボン酸系単量体(a)由来の構成単位(I)と不飽和ポリアルキレングリコール系単量体(b)由来の構成単位(II)との重合体において、3量体以下のものを「オリゴマー」と称すると、本発明の製造方法によれば、オリゴマー含有量が20質量%以下の(メタ)アクリル酸(塩)系重合体を製造することができる。なお、本明細書における重量平均分子量の分子量測定の項で記載した方法によって測定した数値である。
【0121】
上記方法によって製造した(メタ)アクリル酸(塩)系重合体は、重合段階で過酸化水素量を特定範囲に制限したため、還元剤としてL−アスコルビン酸を使用した場合には、L−アスコルビン酸を0.001質量%以上、好ましくは0.001〜10質量%、特に好ましくは0.001〜1質量%を含有し、該L−アスコルビン酸の還元作用により、製品を長期に保存しても、酸化による劣化や着色を抑制することができる。
すなわち、本発明にかかる(メタ)アクリル酸(塩)系重合体は、前記一般式(1)で示される不飽和モノカルボン酸系単量体(a)由来の構成単位(I)と前記一般式(2)で示される不飽和ポリアルキレングリコール系単量体(b)由来の構成単位(II)とを含む重合体であって、前記重合体に対して0.001質量%以上のL−アスコルビン酸を含んでなることを特徴とする。
【0122】
該(メタ)アクリル酸(塩)系重合体の製造方法は特に制限はないが、本発明にかかる製造方法によって好ましく製造することができる。しかも、上記単量体組成の場合には、水溶性(メタ)アクリル酸(塩)系重合体を得ることができ、種々の分散剤、特にセメント分散剤や顔料分散剤などとしての使用に優れる。
本発明における(メタ)アクリル酸(塩)系重合体は、そのままセメント分散剤として使用することができるが、従来公知のナフタレン系セメント分散剤、アミノスルホン酸系セメント分散剤、(メタ)アクリル酸(塩)系重合体系セメント分散剤およびリグニン系セメント分散剤よりなる群から選ばれた少なくとも1種のセメント分散剤を配合してもよい。これらの配合組成については、目的とする付加的機能の有無により大きく異なるものであり、上記重合体成分を100質量%(全量)ないし主成分とするものから、上記重合体成分を高付加価値成分として、従来のセメント分散剤に適量加える態様まで様々であり、一義的に規定することはできない。しかしながら、(メタ)アクリル酸(塩)系重合体の配合量は、全成分に対して、通常、5〜100質量%、好ましくは50〜100質量%である。
【0123】
また、該(メタ)アクリル酸(塩)系重合体には、従来公知のセメント分散剤の他に、空気連行剤、セメント湿潤剤、膨張剤、防水剤、遅延剤、急結剤、水溶性高分子物質、増粘剤、凝集剤、乾燥収縮低減剤、強度増進剤、硬化促進剤、消泡剤等を配合することができる。
本発明における重合体を主成分とするセメント分散剤は、少なくともセメントおよび水よりなるセメント組成物に配合することによりセメントの分散を促進する。なお、該セメント分散剤は、ポルトランドセメント、ビーライト高含有セメント、アルミナセメント、各種混合セメント等の水硬セメント、あるいは、石膏などのセメント以外の水硬材料などに用いることができる。
【実施例】
【0124】
以下、本発明の実施例により具体的に説明する。なお、実施例および比較例、製造例において、「部」は質量部を示し、各測定値は、以下の測定方法によった。
[測定方法]
(1)着色度
日本電色工業株式会社製分光式色差計(SE−2000)を用い、透過法で測定を行い、次式に基づき、YI値を測定した。
ここでX、Y、Zは、試料のXYZ表色系における三刺激値XYZの値を示す。
(2)電気泳動測定条件
測定機種:Waters製 Quanta4000 キャピラリークロマトグラフィー
使用カラム:Waters製 AccuSep 75μm×60cm
使用泳動バッファー:20mmol/gホウ酸ナトリウム
電圧:20.00KV
で測定した。
(3)重合体に含まれる還元剤量
重合体に含まれるL−アスコルビン酸量は、例えば、下記測定条件の液体クロマトグラフィー(以下、「LC」という。)により求めることができる。
【0125】
機種:Waters LCM1
検出器:Waters 410
溶離液種類:アセトニトリル/0.1N酢酸水溶液=40/60(質量%)
溶離液流量:1.0ml/min
カラム種類:GLサイエンス社製 Inertsil ODS−2 3本 4.6×250mm
(4)重合体に含まれるオリゴマー量
重合体に含まれるオリゴマー量は、例えば、前記の分子量測定条件と同条件のGPCで得られたGPCチャートから、次のようにして求めることができる。すなわち、本発明の重合体の分子量に相当するところのピークをピークAとし、該ピークAの次に高分子側にあるピークをピークBとし、該ピークAよりも低分子側の全てのピークをピークCとして、各ピーク面積を求め、次式により算出する。
(5)重合系のpH測定方法
(a)重合反応中に重合反応系から反応混合物50gを50ccガラス製容器にサンプリングする。
【0126】
(b)サンプリングした混合物を25℃に調整する。
(c)pHメーター(堀場製作所製:カニスターLAB pHメーター F−23,電極:6366−10D)を用いて25℃の混合物のpHを測定する。
なお、本発明におけるpHの測定は、WO01/14438号が開示するpH測定方法と相違し、実際の重合反応中の重合反応液からサンプリングして測定する方法であり、反応系中のpHをオンラインで実測できる点で優れている。
(6)分子量測定
(メタ)アクリル酸系重合体の重量平均分子量(Mw)は、下記の測定条件のGPC(ゲルパーミュエーションクロマトグラフィー)により測定した。
【0127】
解析ソフト:Watres製 Millennium 32 Ver.3.21
検出器:Watres製 410示差屈折計
カラム:東ソー製 TSKguardcolumn SWXL
TSKgel G4000 SWXL
TSKgel G3000 SWXL
TSKgel G2000 SWXL
流速:0.8ml/分
カラム温度:40℃
溶離液:アセトニトリル6001g,水10999g,酢酸ナトリウム三水和物115.6gの混合溶液を酢酸でpH6.0に調整したものを用いた。
【0128】
標準試料:分子量が272,500,219,300,107,000,50,000,24,000,12,600,7,100,4,250,1,470のポリエチレングリコールをそれぞれ上記溶離液を用いて0.1重量%溶液を調整し、100μL注入する。
較正曲線:上記標準試料の測定結果を3次式で近似して較正曲線を作成する。
測定未知試料:上記溶離液を用いて測定未知試料の0.5重量%溶液を調整し、100μL注入する。
[製造例1]
温度計、攪拌機、生成水分離器および還流冷却管(コンデンサ)を備えた外部ジャケット付ガラス製反応槽(内容量:30リットル)にメトキシポリ(n=25)エチレングリコール16500部、メタクリル酸4740部(K値=70)、パラトルエンスルホン酸一水和物235部、フェノチアジン5部およびシクロヘキサン1060部を仕込み、エステル化反応中、生成水分離器および還流冷却管からなる循環系から反応容器に戻される経路上に流量計を設けて、還流される溶剤の流量(体積量)を計測し、溶剤循環速度が5サイクル/時間となるように、反応容器に取り付けられたジャケット温度を135℃に設定し、必要に応じて適宜微調節しながら、反応温度120℃でエステル化反応を行った。約20時間でエステル化率が99%に達したのを確認後、49%水酸化ナトリウム水溶液135部と水4890部を加えてパラトルエンスルホン酸を中和し、ハイドロキノン8部を加えて昇温し、シクロヘキサンを水との共沸で留去した。シクロヘキサン留去後、調整水を添加して80%のエステル化物水溶液を得た。得られたエステル化物水溶液について、電気泳動測定条件に従って、キャピラリー電気泳動により不純物の定量を行った。目的のエステル化物、触媒(PTS)、原料(MAA)によるピークが認められるだけで、不純物によるピークは認められなかった。すなわち、得られたエステル化物水溶液には不純物は認められなかった。
【0129】
表1に、原料単量体の着色度を示す。
[製造例2]
温度計、攪拌機、生成水分離器および還流冷却管(コンデンサ)を備えたSUS反応槽(内容量:30リットル)にメトキシポリ(n=25)エチレングリコール16500部、メタクリル酸4740部(K値=70)、パラトルエンスルホン酸一水和物235部、フェノチアジン5部およびシクロヘキサン1060部を仕込み、エステル化反応中、生成水分離器および還流冷却管からなる循環系から反応容器に戻される経路上に流量計を設けて、還流される溶剤の流量(体積量)を計測し、溶剤循環速度が5サイクル/時間となるように、反応容器に取り付けられたジャケット温度を135℃に設定し、必要に応じて適宜微調節しながら、反応温度120℃でエステル化反応を行った。約20時間でエステル化率が99%に達したのを確認後、49%水酸化ナトリウム水溶液267部と水6527部を加えてパラトルエンスルホン酸を中和し、ハイドロキノン8部を加えて昇温し、シクロヘキサンを水との共沸で留去した。シクロヘキサン留去後、調整用の水及びMAAを添加して75%のエステル化物水溶液を得た。
[製造例3]
温度計、攪拌機、生成水分離器および還流冷却管(コンデンサ)を備えたSUS製反応槽(内容量:30リットル)にメトキシポリ(n=25)エチレングリコール16500部、メタクリル酸4740部(K値=70)、パラトルエンスルホン酸一水和物235部、フェノチアジン5部およびシクロヘキサン1060部を仕込み、エステル化反応中、生成水分離器および還流冷却管からなる循環系から反応容器に戻される経路上に流量計を設けて、還流される溶剤の流量(体積量)を計測し、溶剤循環速度が5サイクル/時間となるように、反応容器に取り付けられたジャケット温度を135℃に設定し、必要に応じて適宜微調節しながら、反応温度120℃でエステル化反応を行った。約20時間でエステル化率が99%に達したのを確認後、49%水酸化ナトリウム水溶液432部と水6375部を加えてパラトルエンスルホン酸を中和し、ハイドロキノン8部を加えて昇温し、シクロヘキサンを水との共沸で留去した。シクロヘキサン留去後、調整用の水及びMAAを添加して75%のエステル化物水溶液を得た。
[製造例4]
温度計、攪拌機、生成水分離器および還流冷却管(コンデンサ)を備えたSUS製反応槽(内容量:30リットル)にメトキシポリ(n=25)エチレングリコール16500部、メタクリル酸4740部(K値=70)、パラトルエンスルホン酸一水和物235部、フェノチアジン5部およびシクロヘキサン1060部を仕込み、エステル化反応中、生成水分離器および還流冷却管からなる循環系から反応容器に戻される経路上に流量計を設けて、還流される溶剤の流量(体積量)を計測し、溶剤循環速度が5サイクル/時間となるように、反応容器に取り付けられたジャケット温度を135℃に設定し、必要に応じて適宜微調節しながら、反応温度120℃でエステル化反応を行った。約20時間でエステル化率が99%に達したのを確認後、49%水酸化ナトリウム水溶液597部と水6223部を加えてパラトルエンスルホン酸を中和し、ハイドロキノン8部を加えて昇温し、シクロヘキサンを水との共沸で留去した。シクロヘキサン留去後、調整用の水及びMAAを添加して75%のエステル化物水溶液を得た。
[製造例5]
温度計、攪拌機、生成水分離器および還流冷却管(コンデンサ)を備えた外部ジャケット付ガラス製反応槽(内容量:30リットル)にメトキシポリ(n=9)エチレングリコール16500部、メタクリル酸9450部(K値=70)、パラトルエンスルホン酸一水和物519部、フェノチアジン5部およびシクロヘキサン1298部を仕込み、エステル化反応中、生成水分離器および還流冷却管からなる循環系から反応容器に戻される経路上に流量計を設けて、還流される溶剤の流量(体積量)を計測し、溶剤循環速度が5サイクル/時間となるように、反応容器に取り付けられたジャケット温度を135℃に設定し、必要に応じて適宜微調節しながら、反応温度120℃でエステル化反応を行った。約20時間でエステル化率が99%に達したのを確認後、49%水酸化ナトリウム水溶液223部と水19941部を加えてパラトルエンスルホン酸を中和し、ハイドロキノン8部を加えて昇温し、シクロヘキサンを水との共沸で留去した。シクロヘキサン留去後、調整用の水及びMAAを添加して55%のエステル化物水溶液を得た。
[実施例1]
温度計、攪拌機、滴下ロート、窒素導入管および還流冷却器を備えたガラス製反応器に水712.5部を仕込み、攪拌下で上記反応器を窒素置換し、窒素雰囲気下で50℃まで昇温した。
【0130】
次に、上記反応器内に、製造例1で得られた80%のエステル化物水溶液1687.5部に3−メルカプトプロピオン酸11.3を溶解させた溶液を4時間かけて滴下すると同時に、過酸化水素9.6部を水300部に溶解させた水溶液とL−アスコルビン酸12.5部を水300部に溶解させた水溶液とを5時間かけて滴下した。滴下終了後、反応混合液を50℃に1時間維持した。さらに、この反応混合液のpHを30%水酸化ナトリウム水溶液でpH=7になるように調節することにより、重量平均分子量26,900の(メタ)アクリル酸(塩)系重合体(1A)を得た。実施例1において使用した重合開始剤量は、過酸化水素の使用量は8モル%、L−アスコルビン酸の使用量は2モル%である。
【0131】
表1にレドックス系重合開始剤の使用割合、反応温度、着色度などの結果を示す。
また、重合反応中、時々反応生成物をサンプリングしてpHを測定したところ、表2のとおりであった。また、最終生成物の残存メタクリル酸(MAA)量および残存メトキシポリエチレングリコールメタクリレート(PGM25E)量を測定したところ、表2の結果が得られた。
[実施例2]
過酸化水素を4.8部、L−アスコルビン酸を6.2部を用いたこと以外は実施例1と同様の操作を行い、重量平均分子量25,900の(メタ)アクリル酸(塩)系重合体(2A)を得た。実施例2において使用した重合開始剤量は、過酸化水素の使用量は4モル%、L−アスコルビン酸の使用量は1モル%である。
【0132】
表1にレドックス系重合開始剤の使用割合、反応温度、着色度などの結果を示す。
[実施例3]
過酸化水素を2.4部、L−アスコルビン酸を3.1部を用いたこと以外は実施例1と同様の操作を行い、重量平均分子量24,000の(メタ)アクリル酸(塩)系重合体(3A)を得た。実施例3において使用した重合開始剤量は、過酸化水素の使用量は2モル%、L−アスコルビン酸の使用量は0.5モル%である。
表1にレドックス系重合開始剤の使用割合、反応温度、着色度などの結果を示す。
また、重合反応中、時々反応生成物をサンプリングしてpHを測定したところ、表2のとおりであった。また、最終生成物の残存メタクリル酸(MAA)量および残存メトキシポリエチレングリコールメタクリレート(PGM25E)量を測定したところ、表2の結果が得られた。
[実施例4]
重合温度・維持温度を80℃に変更した以外は実施例2と同様の操作を行い、重量平均分子量22,000の(メタ)アクリル酸(塩)系重合体(4A)を得た。実施例4において使用した重合開始剤量は、過酸化水素の使用量は4モル%、L−アスコルビン酸の使用量は1モル%である。
【0133】
表1にレドックス系重合開始剤の使用割合、反応温度、着色度などの結果を示す。
[実施例5]
重合温度・維持温度を80℃に変更した以外は実施例3と同様の操作を行い、重量平均分子量22,100の(メタ)アクリル酸(塩)系重合体(5A)を得た。実施例5において使用した重合開始剤量は、過酸化水素の使用量は2モル%、L−アスコルビン酸の使用量は0.5モル%である。
表1にレドックス系重合開始剤の使用割合、反応温度、着色度などの結果を示す。
[比較例1]
温度計、攪拌機、生成水分離器および還流冷却管(コンデンサ)を備えた外部ジャケット付ガラス製反応槽(内容量:30リットル)に、水8200部を仕込み、攪拌下で上記反応器を窒素置換し、窒素雰囲気下で80℃まで昇温した。次に、上記反応器内に、製造例1で得られた80%のエステル化物水溶液13100部に3−メルカプトプロピオン酸94部を溶解させた溶液を4時間かけて滴下すると同時に、過硫酸アンモニウム125部を水1000部に溶解させた水溶液を5時間かけて滴下した。滴下終了後、反応混合液を80℃に1時間維持した。さらに、この反応混合液のpHを水酸化ナトリウムでpH=7になるように調節することにより、ゲルパーミエーションクロマトグラフィーによるポリエチレングリコール換算で重量平均分子量23,800の(メタ)アクリル酸(塩)系重合体(1B)を得た。
【0134】
表1にレドックス系重合開始剤の使用割合、反応温度、着色度などの結果を示す。
また、重合反応中、時々反応生成物をサンプリングしてpHを測定したところ、表2のとおりであった。また、最終生成物の残存メタクリル酸(MAA)量および残存メトキシポリエチレングリコールメタクリレート(PGM25E)量を測定したところ、表2の結果が得られた。
[比較例2]
比較例1の方法に従って製造した(メタ)アクリル酸(塩)系重合体(1B)の水溶液をイオン交換水より希釈し、固形分が40質量%になるように調製した水溶液40gを容量50mlのガラス製のサンプル管に入れ、過酸化水素水溶液(30%)を0.37g(重合体(1B)の重合前の全モノマーの8モル%)添加した。その後、室温にて4時間攪拌を行なった後、色差計にて着色度を測定した。YI値は33.11であった。過酸化水素と還元剤の組み合わせであるレドックス系重合開始剤を使用せず、重合後に過酸化水素のみを後添加した場合には、着色を低減することができなかった。
[比較例3]
50℃で6時間攪拌した以外は、比較例2と同様の操作を行った後、色差計にて着色度を測定した。YI値は24.03であった。過酸化水素と還元剤の組み合わせであるレドックス系重合開始剤を使用せず、重合後に過酸化水素のみを後添加した場合には、着色を低減することができなかった。
[比較例4]
L−アスコルビン酸を一切用いなかったこと以外は、実施例2と同様の操作を行ったところ、重量平均分子量30,500の(メタ)アクリル酸(塩)系重合体(4B)を得ることができた。高速液体クロマトグラフィー測定により、未反応のメタクリル酸を定量したところ、添加したメタクリル酸の98.8重量%が消費され、残りの1.2重量%が未反応のまま残存していることが判った。同様に実施例2で得られた重合体中のメタクリル酸を定量したところ、残存しているメタクリル酸は検出されなかった。
[比較例5]
過酸化水素を一切用いなかったこと以外は、実施例2と同様の操作を行ったが、重合は進行せず、目的の重合体を得ることができなかった。
[比較例6]
過酸化水素を38.4部(全モノマーのモル数に対して50モル%)を用いたこと以外は、実施例2と同様の操作を行ったが、重合中発泡が激しく、うまく重合することができなかった。
[比較例7]
L−アスコルビン酸を148.8部(全モノマーのモル数に対して24モル%、過酸化水素に対して600モル%)を用いたこと以外は、実施例2と同様の操作を行ったが、製品保存中に、L−アスコルビン酸由来の結晶が析出してしまうという好ましくない現象が確認された。
【0135】
【表1】
【0136】
[結果1]
(1)実施例1〜3の結果から、過酸化水素の配合量が多いほど、YI値で示す着色度が小さく、着色の程度が過酸化水素の配合量に依存することが示唆された。なお、比較例1の着色度は39.1と実施例1〜5と比較して極めて高値であるが、製造例1で得た80%のエステル化物水溶液のYI値は10.6であり、同じ原料を使用した実施例1〜5の着色度も3.10〜8.36と極めて低値であるため、上記着色度の低下は、原料化合物に由来するものではなく、製造方法による相違であると言える。
(2)実施例2と実施例4、および実施例3と実施例5とを比較すると、重合時の温度は50℃でも80℃でもYI値で示す着色度の変化は少なく、着色の程度は反応温度にあまり依存しない傾向があった。
(3)実施例1〜5においては中和後、塩の析出は確認されなかったが、比較例1においては塩の析出が確認された。
【0137】
[実施例6]
温度計、攪拌機、滴下装置および還流冷却管(コンデンサ)を備えたガラス製反応槽(内容量:3リットル)に、水446部を仕込み、攪拌下で上記反応器を窒素置換し、窒素雰囲気下で80℃まで昇温した。次に、上記反応器内に、製造例2で得られた75%のエステル化物水溶液900部に3−メルカプトプロピオン酸6.0部を溶解させた溶液を4時間かけて滴下すると同時に、過硫酸アンモニウム7.76部を水142.24部に溶解させた水溶液を5時間かけて滴下した。滴下終了後、反応混合液を80℃に1時間維持した。さらに、この反応混合液のpHを30%水酸化ナトリウム水溶液でpH=7になるように調製し、ゲルパーミエーションクロマトグラフィーによるポリエチレングリコール換算で重量平均分子量22,900の(メタ)アクリル酸(塩)系重合体(6A)を得た。
【0138】
重合反応中、時々反応生成物をサンプリングしてpHを測定したところ、表2のとおりであった。また、最終生成物の重量平均分子量Mw、ピークトップ平均分子量Mp、Mw−Mp、残存メタクリル酸(MAA)量および残存メトキシポリエチレングリコールメタクリレート(PGM25E)量を測定したところ、表2の結果が得られた。
[実施例7]
温度計、攪拌機、滴下装置および還流冷却管(コンデンサ)を備えたガラス製反応槽(内容量:3リットル)に、水444部を仕込み、攪拌下で上記反応器を窒素置換し、窒素雰囲気下で80℃まで昇温した。次に、上記反応器内に、製造例3で得られた75%のエステル化物水溶液900部に3−メルカプトプロピオン酸5.99部を溶解させた溶液を4時間かけて滴下すると同時に、過硫酸アンモニウム7.76部を水142.24部に溶解させた水溶液を5時間かけて滴下した。滴下終了後、反応混合液を80℃に1時間維持した。さらに、この反応混合液のpHを30%水酸化ナトリウム水溶液でpH=7になるように調製し、ゲルパーミエーションクロマトグラフィーによるポリエチレングリコール換算で重量平均分子量22,300の(メタ)アクリル酸(塩)系重合体(7A)を得た。
【0139】
重合反応中、時々反応生成物をサンプリングしてpHを測定したところ、表2のとおりであった。また、最終生成物の重量平均分子量Mw、ピークトップ平均分子量Mp、Mw−Mp、残存メタクリル酸(MAA)量および残存メトキシポリエチレングリコールメタクリレート(PGM25E)量を測定したところ、表2の結果が得られた。
[実施例8]
温度計、攪拌機、滴下装置および還流冷却管(コンデンサ)を備えたガラス製反応槽(内容量:3リットル)に、水443部を仕込み、攪拌下で上記反応器を窒素置換し、窒素雰囲気下で80℃まで昇温した。次に、上記反応器内に、製造例3で得られた75%のエステル化物水溶液900部に3−メルカプトプロピオン酸5.63部を溶解させた溶液を4時間かけて滴下すると同時に、過硫酸アンモニウム7.76部を水142.24部に溶解させた水溶液を5時間かけて滴下した。滴下終了後、反応混合液を80℃に1時間維持した。さらに、この反応混合液のpHを30%水酸化ナトリウム水溶液でpH=7になるように調製し、ゲルパーミエーションクロマトグラフィーによるポリエチレングリコール換算で重量平均分子量23500の(メタ)アクリル酸(塩)系重合体(8A)を得た。
【0140】
重合反応中、時々反応生成物をサンプリングしてpHを測定したところ、表2のとおりであった。また、最終生成物の重量平均分子量Mw、ピークトップ平均分子量Mp、Mw−Mp、残存メタクリル酸(MAA)量および残存メトキシポリエチレングリコールメタクリレート(PGM25E)量を測定したところ、表2の結果が得られた。
[実施例9]
温度計、攪拌機、滴下装置および還流冷却管(コンデンサ)を備えたガラス製反応槽(内容量:3リットル)に、水300部を仕込み、攪拌下で上記反応器を窒素置換し、窒素雰囲気下で80℃まで昇温した。次に、上記反応器内に、製造例2で得られた75%のエステル化物水溶液900部に3−メルカプトプロピオン酸7.5部を溶解させた溶液を4時間かけて滴下すると同時に、過酸化水素4.82部を水145.18部に溶解させた水溶液及びL−アスコルビン酸6.24部を水143.76部に溶解させた水溶液をそれぞれ5時間かけて滴下した。滴下終了後、反応混合液を80℃に1時間維持した。さらに、この反応混合液のpHを30%水酸化ナトリウム水溶液でpH=7になるように調製し、ゲルパーミエーションクロマトグラフィーによるポリエチレングリコール換算で重量平均分子量21,100の(メタ)アクリル酸(塩)系重合体(9A)を得た。
【0141】
重合反応中、時々反応生成物をサンプリングしてpHを測定したところ、表2のとおりであった。また、最終生成物の重量平均分子量Mw、ピークトップ平均分子量Mp、Mw−Mp、残存メタクリル酸(MAA)量および残存メトキシポリエチレングリコールメタクリレート(PGM25E)量を測定したところ、表2の結果が得られた。
[実施例10]
温度計、攪拌機、滴下装置および還流冷却管(コンデンサ)を備えたガラス製反応槽(内容量:3リットル)に、水300部を仕込み、攪拌下で上記反応器を窒素置換し、窒素雰囲気下で80℃まで昇温した。次に、上記反応器内に、製造例3で得られた75%のエステル化物水溶液900部に3−メルカプトプロピオン酸7.21部を溶解させた溶液を4時間かけて滴下すると同時に、過酸化水素4.82部を水145.18部に溶解させた水溶液及びL−アスコルビン酸6.24部を水143.76部に溶解させた水溶液をそれぞれ5時間かけて滴下した。滴下終了後、反応混合液を80℃に1時間維持した。さらに、この反応混合液のpHを30%水酸化ナトリウム水溶液でpH=7になるように調製し、ゲルパーミエーションクロマトグラフィーによるポリエチレングリコール換算で重量平均分子量22,200の(メタ)アクリル酸(塩)系重合体(10A)を得た。
【0142】
重合反応中、時々反応生成物をサンプリングしてpHを測定したところ、表2のとおりであった。また、最終生成物の重量平均分子量Mw、ピークトップ平均分子量Mp、Mw−Mp、残存メタクリル酸(MAA)量および残存メトキシポリエチレングリコールメタクリレート(PGM25E)量を測定したところ、表2の結果が得られた。
[実施例11]
温度計、攪拌機、滴下装置および還流冷却管(コンデンサ)を備えたガラス製反応槽(内容量:3リットル)に、水300部を仕込み、攪拌下で上記反応器を窒素置換し、窒素雰囲気下で80℃まで昇温した。次に、上記反応器内に、製造例4で得られた75%のエステル化物水溶液900部に3−メルカプトプロピオン酸7.28部を溶解させた溶液を4時間かけて滴下すると同時に、過酸化水素4.82部を水145.18部に溶解させた水溶液及びL−アスコルビン酸6.24部を水143.76部に溶解させた水溶液をそれぞれ5時間かけて滴下した。滴下終了後、反応混合液を80℃に1時間維持した。さらに、この反応混合液のpHを30%水酸化ナトリウム水溶液でpH=7になるように調製し、ゲルパーミエーションクロマトグラフィーによるポリエチレングリコール換算で重量平均分子量22,200の(メタ)アクリル酸(塩)系重合体(11A)を得た。
【0143】
重合反応中、時々反応生成物をサンプリングしてpHを測定したところ、表2のとおりであった。また、最終生成物の重量平均分子量Mw、ピークトップ平均分子量Mp、Mw−Mp、残存メタクリル酸(MAA)量および残存メトキシポリエチレングリコールメタクリレート(PGM25E)量を測定したところ、表2の結果が得られた。
[比較例8]
WO01/14438号が開示する方法に沿って(メタ)アクリル酸(塩)系重合体を製造した。すなわち、温度計、攪拌機、滴下装置および還流冷却管(コンデンサ)を備えたガラス製反応槽(内容量:3リットル)に、水382部を仕込み、攪拌下で上記反応器を窒素置換し、窒素雰囲気下で80℃まで昇温した。次に、上記反応器内に、製造例5で得られた55%のエステル化物水溶液900部に3−メルカプトプロピオン酸2.8部を溶解させた溶液を90分かけて滴下すると同時に、過硫酸アンモニウム2.52部を水18.48部に溶解させた水溶液を120分かけて滴下した。滴下終了後、反応混合液を80℃に1時間維持した。さらに、この反応混合液のpHを30%水酸化ナトリウム水溶液でpH=7になるように調製し、ゲルパーミエーションクロマトグラフィーによるポリエチレングリコール換算で重量平均分子量43500の(メタ)アクリル酸(塩)系重合体(8B)を得た。
【0144】
重合反応中、時々反応生成物をサンプリングしてpHを測定したところ、表2のとおりであった。また、最終生成物の重量平均分子量Mw、ピークトップ平均分子量Mp、Mw−Mp、残存メタクリル酸(MAA)量および残存メトキシポリエチレングリコールメタクリレート(PGM25E)量を測定したところ、表2の結果が得られた。
【0145】
【表2】
【0146】
[結果2]
比較例1では重合中のpHは2.36〜3.11、比較例8(WO01/14438号が開示する方法に相当)では重合中のpHは2.12〜2.72となっており、低いpH範囲である。これに対し、実施例1、3や、重合時のメタクリル酸の中和率を5〜15%にした実施例6〜11では重合中のpHは3.20〜5.23となっており、比較例1や8よりも高いpH範囲となっている。
比較例1や8では、重合系のpHが低いため、SUS製の重合釜の腐食が促進されてしまう。しかしながら、実施例1、3、6〜11では、重合系のpHが高いため、腐食を効果的に抑えることができる。
【0147】
また、比較例1や8では、残存MAA、残存PGM25Eの濃度が高く、重合率が低い。これに対し、実施例1、3、6〜11ではいずれも残存MAA、残存PGM25Eの濃度が低く、重合率が高くなっていることが分かる。
【産業上の利用可能性】
【0148】
本発明によれば、従来の反応装置を用いて充分に高分子量化したポリマー部分の比率が高く、また、着色度合いの少ない重合体を得ることができる。また、中和の際に結晶が析出したり、得られる重合体中に未反応の開始剤が多く残存したりすることなく、高い重合率でしかも短時間で重合するため、生産効率に優れる。
【図面の簡単な説明】
【0149】
【図1】本発明で使用されるエステル化物の製造方法に用いられる代表的な装置構成の概略図である。
【図2】本発明で使用されるエステル化物の製造の第二の実施態様に係る代表的なゲル化防止剤供給機構の装置構成を含めた本発明の製造装置の一実施形態を表わす概略説明図である。
【図3】コンデンサ直前のオーバーヘッドライン内にノズルを設置した様子を表わす概略説明図である。
【図4】保存部を兼ね備える水分離器の概略説明図である。
【特許請求の範囲】
【請求項1】
下記一般式(1)で示される不飽和モノカルボン酸系単量体(a)と下記一般式(2)で示される不飽和ポリアルキレングリコール系単量体(b)とを重合開始剤を用いて重合させる(メタ)アクリル酸(塩)系重合体の製造方法であって、
前記重合開始剤として過酸化水素とL−アスコルビン酸とからなるレドックス系重合開始剤を用いるとともに、前記過酸化水素の使用量が前記単量体の合計量に対して0.01〜30モル%であり、かつ、前記重合を、重合反応液のpHが3.2〜7.0の範囲内となるようにして行うこと、
を特徴とする、(メタ)アクリル酸(塩)系重合体の製造方法。
【化1】
(ただし、式中、R1は水素原子またはメチル基を表わし、Mは水素原子、一価金属、二価金属、アンモニウム基、または有機アミン基を表す。)
【化2】
(ただし、式中、R1は水素またはメチル基を表し、R2は−COO(R3O)mR4を表す。前記R2を表す一般式において、R3Oは炭素数2〜18のオキシアルキレン基の1種または2種以上を表し、2種以上の場合はブロック状に付加していてもランダム状に付加していてもよい。前記R2を表す一般式において、R4は水素または炭素数1〜22のアルキル基を表し、mはオキシアルキレン基の平均付加モル数であって1〜300の整数を表す。)
【請求項2】
2.前記L−アスコルビン酸の使用量が過酸化水素に対して0.1〜500モル%である、請求項1に記載の方法。
【請求項3】
前記重合を0〜95℃の温度で行う、請求項1または2に記載の方法。
【請求項4】
前記不飽和ポリアルキレングリコール系単量体(b)が、下記一般式(3)で示されるアルコキシポリアルキレングリコールのp重量部と不飽和モノカルボン酸系単量体(a)のq重量部とを、[(p/n1/2)/q]×100≦200(ただし、nはオキシアルキレン基の平均付加モル数。)の関係を満たす条件下でエステル化反応させることにより得られるエステル化反応物の形で前記重合に供されるものである、請求項1から3までのいずれかに記載の方法。
【化3】
(ただし、式中、R3は炭素原子数2〜18のアルキレン基を表し、R4は炭素原子数1〜30の炭化水素基を表す。一般式(3)において、nはオキシアルキレン基R3Oの平均付加モル数を表わし、1〜300の整数である。一般式(3)において、繰り返し単位であるR3Oは同一であってもよく異なっていてもよい。前記繰り返し単位R3Oが異なる場合、各R3Oはブロック状に付加していてもよくランダム状に付加していてもよい。)
【請求項5】
前記エステル化反応が、脱水溶剤の存在下で行われ、かつ、エステル化反応終了後の脱水溶剤留去工程中、前記脱水溶剤を含む留出物に対してゲル化防止剤を作用させるものである、請求項4に記載の方法。
【請求項6】
前記不飽和モノカルボン酸系単量体(a)、前記不飽和ポリアルキレングリコール系単量体(b)、およびこれらと共重合可能な他の単量体(III)を、その重量比(a)/(b)/(III)=(1〜99)/(99〜1)/(0〜50)の範囲で重合させる、請求項1から5までのいずれかに記載の方法。
【請求項7】
前記不飽和ポリアルキレングリコール系単量体(b)が、式(2)におけるオキシアルキレン基の平均付加モル数mが1〜297の整数である第1のエステル化物(a1)と、式(2)におけるオキシアルキレン基の平均付加モル数mが4〜300の整数である第2のエステル化物(a2)との混合物(ただし、第2のエステル化物(a2)の平均付加モル数の方が第1のエステル化物(a1)の平均付加モル数よりも3以上大きいものとする)である、請求項1から6までのいずれかに記載の方法。
【請求項8】
第1のエステル化物(a1)と第2のエステル化物(a2)との質量比が5:95〜95:5である、請求項7に記載の方法。
【請求項9】
nは1〜110の整数である、請求項4から8までのいずれかに記載の方法。
【請求項10】
前記不飽和ポリアルキレングリコール系単量体(b)は、酸触媒の存在下においてエステル化反応させることにより出発原料たる前記不飽和モノカルボン酸系単量体(a)の一部を残存させるとともに、エステル化反応後に塩基性物質で前記酸触媒と前記残存不飽和モノカルボン酸系単量体(a)の0〜60モル%を中和して得られるエステル化反応物の形で前記重合に供されるものである、4から9までのいずれかに記載の方法。
【請求項1】
下記一般式(1)で示される不飽和モノカルボン酸系単量体(a)と下記一般式(2)で示される不飽和ポリアルキレングリコール系単量体(b)とを重合開始剤を用いて重合させる(メタ)アクリル酸(塩)系重合体の製造方法であって、
前記重合開始剤として過酸化水素とL−アスコルビン酸とからなるレドックス系重合開始剤を用いるとともに、前記過酸化水素の使用量が前記単量体の合計量に対して0.01〜30モル%であり、かつ、前記重合を、重合反応液のpHが3.2〜7.0の範囲内となるようにして行うこと、
を特徴とする、(メタ)アクリル酸(塩)系重合体の製造方法。
【化1】
(ただし、式中、R1は水素原子またはメチル基を表わし、Mは水素原子、一価金属、二価金属、アンモニウム基、または有機アミン基を表す。)
【化2】
(ただし、式中、R1は水素またはメチル基を表し、R2は−COO(R3O)mR4を表す。前記R2を表す一般式において、R3Oは炭素数2〜18のオキシアルキレン基の1種または2種以上を表し、2種以上の場合はブロック状に付加していてもランダム状に付加していてもよい。前記R2を表す一般式において、R4は水素または炭素数1〜22のアルキル基を表し、mはオキシアルキレン基の平均付加モル数であって1〜300の整数を表す。)
【請求項2】
2.前記L−アスコルビン酸の使用量が過酸化水素に対して0.1〜500モル%である、請求項1に記載の方法。
【請求項3】
前記重合を0〜95℃の温度で行う、請求項1または2に記載の方法。
【請求項4】
前記不飽和ポリアルキレングリコール系単量体(b)が、下記一般式(3)で示されるアルコキシポリアルキレングリコールのp重量部と不飽和モノカルボン酸系単量体(a)のq重量部とを、[(p/n1/2)/q]×100≦200(ただし、nはオキシアルキレン基の平均付加モル数。)の関係を満たす条件下でエステル化反応させることにより得られるエステル化反応物の形で前記重合に供されるものである、請求項1から3までのいずれかに記載の方法。
【化3】
(ただし、式中、R3は炭素原子数2〜18のアルキレン基を表し、R4は炭素原子数1〜30の炭化水素基を表す。一般式(3)において、nはオキシアルキレン基R3Oの平均付加モル数を表わし、1〜300の整数である。一般式(3)において、繰り返し単位であるR3Oは同一であってもよく異なっていてもよい。前記繰り返し単位R3Oが異なる場合、各R3Oはブロック状に付加していてもよくランダム状に付加していてもよい。)
【請求項5】
前記エステル化反応が、脱水溶剤の存在下で行われ、かつ、エステル化反応終了後の脱水溶剤留去工程中、前記脱水溶剤を含む留出物に対してゲル化防止剤を作用させるものである、請求項4に記載の方法。
【請求項6】
前記不飽和モノカルボン酸系単量体(a)、前記不飽和ポリアルキレングリコール系単量体(b)、およびこれらと共重合可能な他の単量体(III)を、その重量比(a)/(b)/(III)=(1〜99)/(99〜1)/(0〜50)の範囲で重合させる、請求項1から5までのいずれかに記載の方法。
【請求項7】
前記不飽和ポリアルキレングリコール系単量体(b)が、式(2)におけるオキシアルキレン基の平均付加モル数mが1〜297の整数である第1のエステル化物(a1)と、式(2)におけるオキシアルキレン基の平均付加モル数mが4〜300の整数である第2のエステル化物(a2)との混合物(ただし、第2のエステル化物(a2)の平均付加モル数の方が第1のエステル化物(a1)の平均付加モル数よりも3以上大きいものとする)である、請求項1から6までのいずれかに記載の方法。
【請求項8】
第1のエステル化物(a1)と第2のエステル化物(a2)との質量比が5:95〜95:5である、請求項7に記載の方法。
【請求項9】
nは1〜110の整数である、請求項4から8までのいずれかに記載の方法。
【請求項10】
前記不飽和ポリアルキレングリコール系単量体(b)は、酸触媒の存在下においてエステル化反応させることにより出発原料たる前記不飽和モノカルボン酸系単量体(a)の一部を残存させるとともに、エステル化反応後に塩基性物質で前記酸触媒と前記残存不飽和モノカルボン酸系単量体(a)の0〜60モル%を中和して得られるエステル化反応物の形で前記重合に供されるものである、4から9までのいずれかに記載の方法。
【図1】

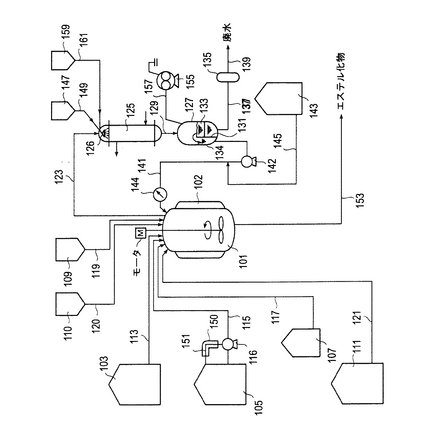
【図2】
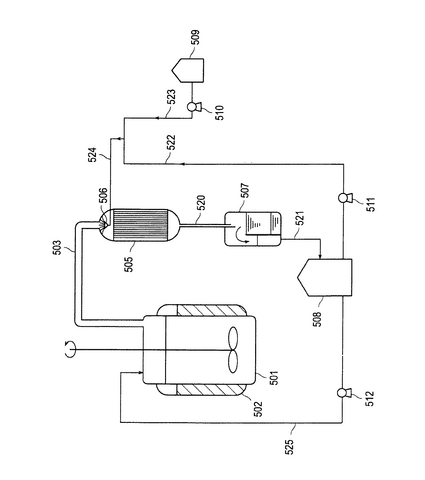
【図3】
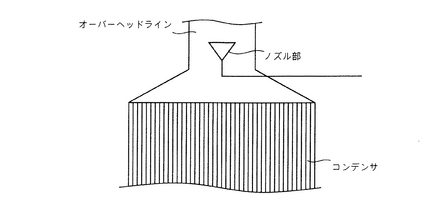
【図4】
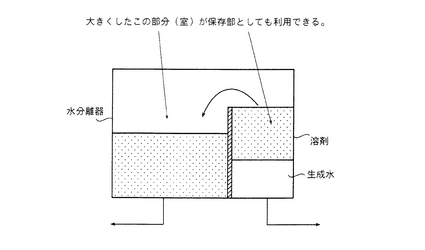
【図2】
【図3】
【図4】
【公開番号】特開2006−291213(P2006−291213A)
【公開日】平成18年10月26日(2006.10.26)
【国際特許分類】
【出願番号】特願2006−134379(P2006−134379)
【出願日】平成18年5月12日(2006.5.12)
【分割の表示】特願2003−542239(P2003−542239)の分割
【原出願日】平成14年10月24日(2002.10.24)
【出願人】(000004628)株式会社日本触媒 (2,292)
【Fターム(参考)】
【公開日】平成18年10月26日(2006.10.26)
【国際特許分類】
【出願日】平成18年5月12日(2006.5.12)
【分割の表示】特願2003−542239(P2003−542239)の分割
【原出願日】平成14年10月24日(2002.10.24)
【出願人】(000004628)株式会社日本触媒 (2,292)
【Fターム(参考)】
[ Back to top ]