
((ホスホノオキシ)メチル)ピリジニウム環を有するピリジン誘導体およびそれらを含有する抗真菌剤

【課題】優れた抗真菌作用を有し、物性、特に、水への溶解性および水溶液中での安全性、ならびに体内動態および安全性の面でも優れた抗真菌剤の提供。
【解決手段】下式(I)で表される化合物またはその塩を使用する。

(式中、R1が、水素原子、ハロゲン原子、アミノ基、C1−6アルキル基などを意味し;R2が、水素原子、C1-6アルキル基、アミノ基、またはジC1-6アルキルアミノ基を意味し;R3が、水素原子、ハロゲン原子、またはC1−6アルキル基を意味し;R4が、水素原子、ハロゲン原子、またはC1−6アルキル基を意味する。)
【解決手段】下式(I)で表される化合物またはその塩を使用する。
(式中、R1が、水素原子、ハロゲン原子、アミノ基、C1−6アルキル基などを意味し;R2が、水素原子、C1-6アルキル基、アミノ基、またはジC1-6アルキルアミノ基を意味し;R3が、水素原子、ハロゲン原子、またはC1−6アルキル基を意味し;R4が、水素原子、ハロゲン原子、またはC1−6アルキル基を意味する。)
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、新規な((ホスホノオキシ)メチル)ピリジニウム環を有するピリジン誘導体およびそれらを含有する抗真菌剤に関する。
【背景技術】
【0002】
近年、高度な化学療法等による免疫機能の低下した患者や高齢者が増加しているため、日和見感染の対策は益々重要性を増してきている。異なる弱毒菌による日和見感染が次々と起こっている事実が示すように、患者の抵抗力が低下するような基礎疾患がある限り感染症の問題は後を絶たない。従って、近い将来確実に訪れる高齢化社会においては、耐性菌の問題を含めた新たな感染症対策が重要な課題の一つとなることが見込まれている。
【0003】
抗真菌剤の分野では、従来、例えば、深在性の真菌症の治療にはポリエン系のアムホテリシンBやアゾール系のフルコナゾール、イトラコナゾール、ボリコナゾール等が開発されてきた。すでに上市されている既存薬には類似したメカニズムの薬剤が多く、現在ではアゾール耐性菌等の出現が問題となっている。
【0004】
近年、新規メカニズムの1,3−β−グルカン合成酵素阻害剤として天然物由来の環状ヘキサペプチド型のカスポファンジンやミカファンジン等が開発されてきているが、これらの薬剤には注射剤しかないことから、抗真菌剤としてはまだ充分ではない。
【0005】
このように既存の抗真菌剤では充分とはいえない状況にあり、新規なメカニズムに基づく安全性の高い薬剤の開発が切望されている。かかる新規なメカニズムに基づく抗真菌剤に関する関連技術として、特許文献1および2がある。特許文献1および2には、GPI(glycosylphosphatidyl-inositol)アンカー蛋白質の細胞壁への輸送過程を阻害することで細胞壁表層蛋白質の発現を阻害し、細胞壁assemblyを阻害するとともに真菌が細胞へ付着するのを阻害して、病原体が病原性を発揮できないようにすることにより、感染症の発症、進展、持続に対して効果を示すピリジン誘導体が記載されている。
【0006】
このような状況下において、特許文献3には、従来の抗真菌剤にはない優れた抗真菌作用を有し、物性、安全性および代謝的安定性の面でも優れた抗真菌剤として、ヘテロ環置換ピリジン誘導体が提案されている。
【0007】
一方、特許文献4または5には、それぞれ、水溶性プロドラッグとして、下記式で表わされる化合物類、N−ホスホリルオキシメチルプロドラッグが開示されている。
【0008】
【化1】
(式中、R1、R2およびR3は親第三級または第二級アミンを含む置換基であり、R4およびR5は、各々有機または無機残基であり、Xはカチオン有機または無機塩である。)
また、特許文献6には、水への溶解性、および安全性の面でも優れた抗真菌剤のプロドラッグとして、ヘテロ環およびホスホノアミノ基が置換したピリジン誘導体が提案されている。
【先行技術文献】
【特許文献】
【0009】
【特許文献1】国際公開第02/04626号パンフレット
【特許文献2】国際公開第05/033079号パンフレット
【特許文献3】国際公開第07/052615号パンフレット
【特許文献4】米国特許第6,235,728 B1
【特許文献5】特表2001−527083号公報
【特許文献6】国際公開第08/136324号パンフレット
【発明の概要】
【発明が解決しようとする課題】
【0010】
しかしながら、より優れた真菌症の治療方法を提供するためには、水への溶解性および水溶液中での安定性、ならびに安全性の観点からより優れた抗真菌剤のさらなる創製が望まれている。
かかる事情に鑑み、本発明の目的は、優れた抗真菌作用を有し、かつ、水への溶解性および水溶液中での安定性、ならびに体内動態および安全性の面でも優れた抗真菌剤を提供することにある。
【課題を解決するための手段】
【0011】
本発明者らは、上記事情に鑑み鋭意研究を重ねた結果、下式(I)
【0012】
【化2】
【0013】
で表される((ホスホノオキシ)メチル)ピリジニウム環を有するピリジン誘導体が、活性本体である親化合物のプロドラッグとして、優れた抗真菌作用を有するとともに、水への溶解性および水溶液中での安定性、ならびに体内動態および安全性の面でも優れることをも見出して、本発明を完成した。
【0014】
すなわち、本発明は、
[1]下式(I)で表される化合物またはその塩;
【化3】
式中、
R1が、水素原子、ハロゲン原子、アミノ基、C1−6アルキル基、C1−6アルコキシ基、またはC1−6アルコキシC1−6アルキル基を意味し;
R2が、水素原子、C1-6アルキル基、アミノ基、またはジC1-6アルキルアミノ基を意味し;
R3が、水素原子、ハロゲン原子、またはC1−6アルキル基を意味し;
R4が、水素原子、ハロゲン原子、またはC1−6アルキル基を意味する。
[2] R2がアミノ基である、前記[1]に記載の化合物またはその塩。
[3] R1が、水素原子である、前記[1]または[2]に記載の化合物またはその塩。
[4] R1が、アミノ基である、前記[1]ないし[2]に記載の化合物またはその塩。
[5] R3が、水素原子であり、R4が、水素原子、ハロゲン原子、またはC1−6アルキル基である、前記[1]ないし[4]のいずれか1項に記載の化合物またはその塩。
[6] 下式
【化4】
で表される2−((4−((5−(2−アミノ−3−ピリジニル)−3−イソキサゾリル)メチル)フェノキシ)メチル)−1−((ホスホノオキシ)メチル)ピリジニウムの化合物またはその塩。
[7] 前記[1]ないし[6]のいずれか1項に記載の化合物またはその塩を含有する医薬組成物。
[8] 前記[1]ないし[6]のいずれか1項に記載の化合物またはその塩を含有する医薬。
[9] 前記[1]ないし[6]のいずれか1項に記載の化合物またはその塩を有効成分として含有する抗真菌剤。
[10] 前記[1]ないし[6]のいずれか1項に記載の化合物またはその塩の薬理学的有効量を投与して、真菌感染症を予防および/または治療する方法。
[11] 抗真菌剤の製造のための、前記[1]ないし[6]のいずれか1項に記載の化合物またはその塩の使用。
を提供する。
【発明の効果】
【0015】
式(I)で表される化合物(以下、単に「本発明に係る化合物」という称する場合がある。)は、活性本体である親化合物のプロドラッグとして、1)真菌のGPI生合成阻害に基づいて細胞壁表層蛋白質の発現を阻害し、細胞壁assemblyを阻害するとともに真菌が細胞へ付着するのを阻害して、病原体が病原性を発揮できないようにすることにより、感染症の発症、進展、持続に対して効果を示し、さらに、2)物性、特に、水への溶解性および水溶液中での安定性、ならびに体内動態および安全性の面でも優れており、真菌感染症の予防または治療剤として極めて有用である。
【図面の簡単な説明】
【0016】
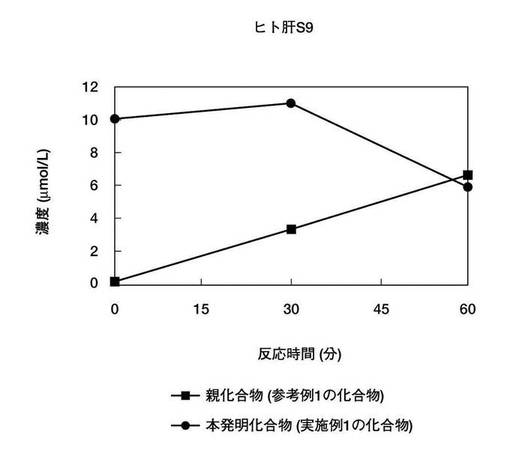
【図1】図1は、本発明の実施例1で得られた2−((4−((5−(2−アミノ−3−ピリジニル)−3−イソキサゾリル)メチル)フェノキシ)メチル)−1−((ホスホノオキシ)メチル)ピリジニウム 1トリフルオロ酢酸塩および親化合物(参考例1で示す3−(3−(4−(ピリジン−2−イルメトキシ)−ベンジル)−イソキサゾール−5−イル)−ピリジン−2−イルアミン)の、ヒト肝S9反応溶液中での各化合物濃度の経時変化を示す。
【図2】図2は、本発明の実施例1で得られた2−((4−((5−(2−アミノ−3−ピリジニル)−3−イソキサゾリル)メチル)フェノキシ)メチル)−1−((ホスホノオキシ)メチル)ピリジニウム 1トリフルオロ酢酸塩および親化合物(参考例1で示す3−(3−(4−(ピリジン−2−イルメトキシ)−ベンジル)−イソキサゾール−5−イル)−ピリジン−2−イルアミン)の、サル肝S9反応溶液中での各化合物濃度の経時変化を示す。
【図3】図3は、本発明の実施例1で得られた2−((4−((5−(2−アミノ−3−ピリジニル)−3−イソキサゾリル)メチル)フェノキシ)メチル)−1−((ホスホノオキシ)メチル)ピリジニウム 1トリフルオロ酢酸塩および親化合物(参考例1で示す3−(3−(4−(ピリジン−2−イルメトキシ)−ベンジル)−イソキサゾール−5−イル)−ピリジン−2−イルアミン)の、反応緩衝溶液中での各化合物濃度の経時変化を示す。
【発明を実施するための形態】
【0017】
以下に、本明細書において記載する記号、用語等の定義、本発明の実施の形態等を示して、本発明を詳細に説明する。なお、本発明は以下の実施の形態に限定されるものではなく、その要旨の範囲内で種々変形して実施することができる。
【0018】
本明細書中においては、化合物の構造式が便宜上一定の異性体を表すことがあるが、本発明には化合物の構造上生じ得るすべての幾何異性体、不斉炭素に基づく光学異性体、立体異性体、回転異性体、互変異性体等の異性体および異性体混合物を含み、便宜上の式の記載に限定されるものではなく、いずれか一方の異性体でも混合物でもよい。したがって、本発明の化合物には、分子内に不斉炭素原子を有し光学活性体およびラセミ体が存在することがありうるが、本発明においては限定されず、いずれもが含まれる。また、結晶多形が存在することもあるが同様に限定されず、いずれかの単一の結晶形であっても二以上の結晶形からなる混合物であってもよい。そして、本発明に係る化合物には無水物と水和物等の溶媒和物とが包含される。
【0019】
本明細書において使用する「C1−6アルキル基」とは、炭素数1〜6個の脂肪族炭化水素から任意の水素原子を1個除いて誘導される一価の基である、炭素数1〜6個の直鎖状または分枝鎖状のアルキル基を意味し、具体的には例えば、メチル基、エチル基、n−プロピル基、イソプロピル基、n−ブチル基、イソブチル基、sec−ブチル基、tert−ブチル基、n−ペンチル基、イソペンチル基、sec−ペンチル基、ネオペンチル基、1−メチルブチル基、2−メチルブチル基、1,1−ジメチルプロピル基、1,2−ジメチルプロピル基、n−ヘキシル基、イソヘキシル基、1−メチルペンチル基、2−メチルペンチル基、3−メチルペンチル基、1,1−ジメチルブチル基、1,2−ジメチルブチル基、2,2−ジメチルブチル基、1,3−ジメチルブチル基、2,3−ジメチルブチル基、3,3−ジメチルブチル基、1−エチルブチル基、2−エチルブチル基、1,1,2−トリメチルプロピル基、1,2,2−トリメチルプロピル基、1−エチル−1−メチルプロピル基、1−エチル−2−メチルプロピル基等が挙げられ、好ましくはメチル基、エチル基、n−プロピル基、イソプロピル基、n−ブチル基、イソブチル基、sec−ブチル基、tert−ブチル基等である。
【0020】
本明細書において使用する「C1−6アルコキシ基」とは、前記定義「C1−6アルキル基」の末端に酸素原子が結合した基であることを意味し、具体的には例えば、メトキシ基、エトキシ基、n−プロポキシ基、イソプロポキシ基、n−ブトキシ基、イソブトキシ基、sec−ブトキシ基、tert−ブトキシ基、n−ペンチルオキシ基、イソペンチルオキシ基、sec−ペンチルオキシ基、ネオペンチルオキシ基、1−メチルブトキシ基、2−メチルブトキシ基、1,1−ジメチルプロポキシ基、1,2−ジメチルプロポキシ基、n−ヘキシルオキシ基、イソヘキシルオキシ基、1−メチルペンチルオキシ基、2−メチルペンチルオキシ基、3−メチルペンチルオキシ基、1,1−ジメチルブトキシ基、1,2−ジメチルブトキシ基、2,2−ジメチルブトキシ基、1,3−ジメチルブトキシ基、2,3−ジメチルブトキシ基、3,3−ジメチルブトキシ基、1−エチルブトキシ基、2−エチルブトキシ基、1,1,2−トリメチルプロポキシ基、1,2,2−トリメチルプロポキシ基、1−エチル−1−メチルプロポキシ基、1−エチル−2−メチルプロポキシ基等が挙げられ、好ましくはメトキシ基、エトキシ基、n−プロポキシ基、イソプロポキシ基、n−ブトキシ基、イソブトキシ基、sec−ブトキシ基、tert−ブトキシ基等である。
【0021】
本明細書中において使用する「C1−6アルコキシC1−6アルキル基」とは、前記定義「C1−6アルキル基」中の任意の水素原子を、前記定義「C1−6アルコキシ基」で置換した基を意味し、具体的には例えば、メトキシメチル基、エトキシメチル基、n−プロポキシメチル、メトキシエチル基、エトキシエチル基等が挙げられる。
【0022】
本明細書において使用する「ハロゲン原子」とは、フッ素原子、塩素原子、臭素原子またはヨウ素原子を意味する。
【0023】
本明細書中において使用する「ジC1−6アルキルアミノ基」とは、アミノ基中の2個の水素原子を、それぞれ同一のまたは異なる、前記定義「C1−6アルキル基」で置換した基を意味し、具体的には例えば、N,N−ジメチルアミノ基、N,N−ジエチルアミノ基、N,N−ジ−n−プロピルアミノ基、N,N−ジ−イソプロピルアミノ基、N,N−ジ−n−ブチルアミノ基、N,N−ジ−イソブチルアミノ基、N,N−ジ−sec−ブチルアミノ基、N,N−ジ−tert−ブチルアミノ基、N−エチル−N−メチルアミノ基、N−n−プロピル−N−メチルアミノ基、N−イソプロピル−N−メチルアミノ基、N−n−ブチル−N−メチルアミノ基、N−イソブチル−N−メチルアミノ基、N−sec−ブチル−N−メチルアミノ基、N−tert−ブチル−N−メチルアミノ基等が挙げられ、好ましくはN,N−ジメチルアミノ基、N,N−ジエチルアミノ基、N−エチル−N−メチルアミノ基等である。
【0024】
R1は、水素原子、ハロゲン原子、アミノ基、C1−6アルキル基、C1−6アルコキシ基、またはC1−6アルコキシC1−6アルキル基を意味し、特に、水素原子またはアミノ基が好ましい。
【0025】
R2は、水素原子、C1−6アルキル基、アミノ基、またはジC1−6アルキルアミノ基を意味し、水素原子またはアミノ基が好ましい。
【0026】
R3は、水素原子、ハロゲン原子、またはC1−6アルキル基を意味し、水素原子が好ましい。
【0027】
R4は、水素原子、ハロゲン原子、またはC1−6アルキル基を意味し、水素原子が好ましい。
【0028】
本明細書において使用する「塩」とは、一価のカウンターイオンまたは二価のカウンターイオンを形成できる化合物または原子との塩を意味する。具体的には、以下のものに限定されないが、無機酸(たとえば、塩酸、臭化水素酸、リン酸、硫酸、硝酸等)との塩、有機酸(たとえば、メタンスルホン酸、エタンスルホン酸、ベンゼンスルホン酸、p−トルエンスルホン酸、フマル酸、マレイン酸、コハク酸、クエン酸、リンゴ酸、またはトリフルオロ酢酸等)との塩、または、無機塩基(たとえば、ナトリウム塩、カリウム塩、カルシウム塩、リチウム塩等)との塩、有機塩基(たとえば、メチルアミン塩、エチルアミン塩、t−ブチルアミン塩、シクロヘキシルアミン塩、N−メチル−D−グルカミン塩、リジン塩、ピペリジンまたはモルホリン等)との塩を意味する。モノ−およびビス−塩は用語「塩」に含まれる。そして、本発明に係る化合物の塩にはその塩の無水物と水和物等のその塩の溶媒和物とが包含される。
【0029】
本明細書において使用する「抗真菌剤」は、真菌感染症の予防剤および/または治療剤を意味する。
【0030】
本発明に係る化合物は、慣用されている方法により錠剤、散剤、細粒剤、顆粒剤、被覆錠剤、カプセル剤、シロップ剤、トローチ剤、吸入剤、坐剤、注射剤、軟膏剤、眼軟膏剤、テープ剤、点眼剤、点鼻剤、点耳剤、パップ剤、ローション剤等として製剤化することができる。
【0031】
製剤化には通常用いられる賦形剤、結合剤、滑沢剤、着色剤、矯味矯臭剤や、および必要により安定化剤、乳化剤、吸収促進剤、界面活性剤、pH調整剤、防腐剤、抗酸化剤等を使用することができ、一般に医薬品製剤の原料として用いられる成分を配合して常法により製剤化される。例えば経口製剤を製造するには、本発明に係る化合物と賦形剤、さらに必要に応じて結合剤、崩壊剤、滑沢剤、着色剤、矯味矯臭剤等を加えた後、常法により散剤、細粒剤、顆粒剤、錠剤、被覆錠剤、カプセル剤等とする。
【0032】
これらの成分としては、例えば、大豆油、牛脂、合成グリセライド等の動植物油;例えば、流動パラフィン、スクワラン、固形パラフィン等の炭化水素;例えば、ミリスチン酸オクチルドデシル、ミリスチン酸イソプロピル等のエステル油;例えば、セトステアリルアルコール、ベヘニルアルコール等の高級アルコール;シリコン樹脂;シリコン油;例えば、ポリオキシエチレン脂肪酸エステル、ソルビタン脂肪酸エステル、グリセリン脂肪酸エステル、ポリオキシエチレンソルビタン脂肪酸エステル、ポリオキシエチレン硬化ひまし油、ポリオキシエチレンポリオキシプロピレンブロックコポリマー等の界面活性剤;例えば、ヒドロキシエチルセルロース、ポリアクリル酸、カルボキシビニルポリマー、ポリエチレングリコール、ポリビニルピロリドン、メチルセルロース等の水溶性高分子;例えば、エタノール、イソプロパノール等の低級アルコール;例えば、グリセリン、プロピレングリコール、ジプロピレングリコール、ソルビトール等の多価アルコール;例えば、グルコース、ショ糖等の糖;例えば、無水ケイ酸、ケイ酸アルミニウムマグネシウム、ケイ酸アルミニウム等の無機粉体、精製水等が挙げられる。賦形剤としては、例えば、乳糖、コーンスターチ、白糖、ブドウ糖、マンニトール、ソルビット、結晶セルロース、二酸化ケイ素等が、結合剤としては、例えば、ポリビニルアルコール、ポリビニルエーテル、メチルセルロース、エチルセルロース、アラビアゴム、トラガント、ゼラチン、シェラック、ヒドロキシプロピルメチルセルロース、ヒドロキシプロピルセルロース、ポリビニルピロリドン、ポリプロピレングリコール・ポリオキシエチレン・ブロックポリマー、メグルミン等が、崩壊剤としては、例えば、澱粉、寒天、ゼラチン末、結晶セルロース、炭酸カルシウム、炭酸水素ナトリウム、クエン酸カルシウム、デキストリン、ペクチン、カルボキシメチルセルロース・カルシウム等が、滑沢剤としては、例えば、ステアリン酸マグネシウム、タルク、ポリエチレングリコール、シリカ、硬化植物油等が、着色剤としては医薬品に添加することが許可されているものが、矯味矯臭剤としては、例えば、ココア末、ハッカ脳、芳香散、ハッカ油、竜脳、桂皮末等が用いられる。これらの錠剤・顆粒剤には糖衣、その他、必要により適宜コーティングすることはもちろん差支えない。また、シロップ剤や注射用製剤等の液剤を製造する際には、本発明に係る化合物にpH調整剤、溶解剤、等張化剤等と、必要に応じて溶解補助剤、安定化剤等を加えて、常法により製剤化する。外用剤を製造する際の方法は限定されず、常法により製造することができる。すなわち、製剤化にあたり使用する基剤原料としては、医薬品、医薬部外品、化粧品等に通常使用される各種原料を用いることが可能である。使用する基剤原料として具体的には、例えば、動植物油、鉱物油、エステル油、ワックス類、高級アルコール類、脂肪酸類、シリコン油、界面活性剤、リン脂質類、アルコール類、多価アルコール類、水溶性高分子類、粘土鉱物類、精製水等の原料が挙げられ、さらに必要に応じ、例えば、pH調整剤、抗酸化剤、キレート剤、防腐防黴剤、着色料、香料等を添加することができるが、本発明に係る外用剤の基剤原料はこれらに限定されない。また、必要に応じて分化誘導作用を有する成分、血流促進剤、殺菌剤、消炎剤、細胞賦活剤、ビタミン類、アミノ酸、保湿剤、角質溶解剤等の成分を配合することもできる。なお、上記基剤原料の添加量は、通常外用剤の製造にあたり設定される濃度になる量である。
【0033】
本発明に係る化合物を投与する場合、その形態は特に限定されず、通常用いられる方法により経口投与でも非経口投与でもよい。例えば、錠剤、散剤、顆粒剤、カプセル剤、シロップ剤、トローチ剤、吸入剤、坐剤、注射剤、軟膏剤、眼軟膏剤、テープ剤、点眼剤、点鼻剤、点耳剤、パップ剤、ローション剤等の剤として製剤化し、投与することができる。
【0034】
本発明に係る医薬の投与量は、症状の程度、年齢、性別、体重、投与形態・塩の種類、疾患の具体的な種類等に応じて適宜選ぶことができる。
【0035】
投与量は患者の、疾患の種類、症状の程度、患者の年齢、性差、薬剤に対する感受性差等により著しく異なるが、経口剤の場合は、通常成人として1日あたり、1−10000mg、好ましくは10−2000mgを1日1−数回に分けて投与する。注射剤の場合は、通常成人として1日あたり、通常0.1mg−10000mgであり、好ましくは1mg−2000mgである。
【0036】
[一般的製造方法]
式(I)で表される化合物(以下、化合物(I)という。)の製造方法について説明する。なお、後述する製造方法では、化合物(I−1)、(I−2)および(I−3)は化合物(I)に包含される化合物の代表例として説明する。
リン酸エステルの一般的製造方法
[製造方法1] 化合物(I−1)の製造方法
【0037】
【化5】
〔式中、R1、R3、R4は前記定義と同意義を意味する(ただし、R1が、アミノ基の場合を除く)。
【0038】
化合物(I−1−1)は、後述する参考例等に記載の方法を用いて製造することができる。また、化合物(I−1−1)は国際公開WO 2007/052615 A1公報に記載された方法等により製造することもできる。
【0039】
[工程1−1]
本工程は、化合物(I−1−1)とジ−tert−ブチル ジカーバメートを塩基触媒存在下に反応させて化合物(I−1−2)を得る工程である。
化合物(I−1−1)とジ−tert−ブチル ジカーバメートを反応させる時に用いる溶媒としては、出発原料をある程度溶解するものであり、かつ、反応を阻害しないものであれば、特に制限はないが、例えば、塩化メチレン、クロロホルムなどのハロゲン化炭化水素系溶媒;テトラヒドロフラン、ジエチルエーテルなどのエーテル系溶媒;酢酸エチルなどのエステル系溶媒;アセトニトリル、テトラメチレンスルホランまたはこれらの混合溶媒などを用いることができる。ジ−tert−ブチル ジカーバメートは化合物(I−1−1)に対して2当量から20当量用いる。塩基触媒としては、例えば4−ジメチルアミノピリジンを0.001当量から0.3当量用いる。この際トリエチルアミンなどの有機塩基を1当量から2当量添加してもよい。反応温度は0℃から60℃であり、好ましくは4℃から室温である。反応時間は1時間から72時間である。
【0040】
[工程1−2]
本工程は、化合物(I−1−2)とホスホリック アシッド ジ−tert−ブチル エステル クロロメチル エステルをヨウ化ナトリウム存在下に反応させた後、酸処理により化合物(I−1)を得る工程である。
化合物(I−1−2)とホスホリック アシッド ジ−tert−ブチル エステル クロロメチル エステルをヨウ化ナトリウム存在下に反応させる時に用いる溶媒としては、出発原料をある程度溶解するものであり、かつ、反応を阻害しないものであれば、特に制限はないが、例えば、塩化メチレン、クロロホルムなどのハロゲン化炭化水素系溶媒;テトラヒドロフラン、ジエチルエーテルなどのエーテル系溶媒;酢酸エチルなどのエステル系溶媒;アセトニトリル、テトラメチレンスルホランまたはこれらの混合溶媒などを用いることができ、好ましくはテトラヒドロフランあるいはアセトニトリルを用いる。ホスホリック アシッド ジ−tert−ブチル エステル クロロメチル エステルは化合物(I−1−2)に対して1当量から10当量用いることができ、好ましくは1当量から2当量用いる。ヨウ化ナトリウムは化合物(I−1−2)に対して1当量から10当量用いることができ、好ましくは1当量から2当量用いる。反応温度は0℃から60℃であり、好ましくは4℃から室温である。反応時間は1時間から72時間である。
酸処理に用いる酸としては、例えばトリフルオロ酢酸などの有機酸、塩酸などの鉱酸を用いることができ、好ましくはトリフルオロ酢酸を用いる。酸処理に際しては、前段階の反応溶媒に酸をそのまま加えてもよいし、一旦溶媒を減圧濃縮した後、適切な溶媒たとえばジクロロメタンなどに変えて酸を加えてもよい。反応温度は−10℃から室温であり、反応時間は5分間から2時間である。
【0041】
[製造方法2] 化合物(I−2)の製造方法
【0042】
【化6】
〔式中、R2、R3、R4は前記定義と同意義を意味する(ただし、R2が、アミノ基の場合を除く)。
【0043】
化合物(I−2−1)は国際公開WO 2007/052615 A1公報に記載された方法等により製造することができる。
【0044】
[工程2−1]
本工程は、化合物(I−2−1)とジ−tert−ブチル ジカーバメートを塩基触媒存在下に反応させて化合物(I−2−2)を得る工程である。
化合物(I−2−1)とジ−tert−ブチル ジカーバメートを反応させる時に用いる溶媒としては、出発原料をある程度溶解するものであり、かつ、反応を阻害しないものであれば、特に制限はないが、例えば、塩化メチレン、クロロホルムなどのハロゲン化炭化水素系溶媒、テトラヒドロフラン、ジエチルエーテルなどのエーテル系溶媒、酢酸エチルなどのエステル系溶媒、アセトニトリル、テトラメチレンスルホランまたはこれらの混合溶媒などを用いることができる。ジ−tert−ブチル ジカーバメートは化合物(I−2−1)に対して2当量から20当量用いる。塩基触媒としては、例えば4−ジメチルアミノピリジンを0.001当量から0.3当量用いる。この際トリエチルアミンなどの有機塩基を1当量から2当量添加してもよい。反応温度は0℃から60℃であり、好ましくは4℃から室温である。反応時間は1時間から72時間である。
【0045】
[工程2−2]
本工程は、化合物(I−2−2)とホスホリック アシッド ジ−tert−ブチル エステル クロロメチル エステルをヨウ化ナトリウム存在下に反応させた後、酸処理により化合物(I−2)を得る工程である。
化合物(I−2−2)とホスホリック アシッド ジ−tert−ブチル エステル クロロメチル エステルをヨウ化ナトリウム存在下に反応させる時に用いる溶媒としては、出発原料をある程度溶解するものであり、かつ、反応を阻害しないものであれば、特に制限はないが、例えば、塩化メチレン、クロロホルムなどのハロゲン化炭化水素系溶媒;テトラヒドロフラン、ジエチルエーテルなどのエーテル系溶媒;酢酸エチルなどのエステル系溶媒;アセトニトリル、テトラメチレンスルホランまたはこれらの混合溶媒などを用いることができ、好ましくはテトラヒドロフランあるいはアセトニトリルを用いる。ホスホリック アシッド ジ−tert−ブチル エステル クロロメチル エステルは化合物(I−2−2)に対して1当量から10当量用いることができ、好ましくは1当量から2当量用いる。ヨウ化ナトリウムは化合物(I−2−2)に対して1当量から10当量用いることができ、好ましくは1当量から2当量用いる。反応温度は0℃から60℃であり、好ましくは4℃から室温である。反応時間は1時間から72時間である。
酸処理に用いる酸としては、例えばトリフルオロ酢酸などの有機酸、塩酸などの鉱酸を用いることができ、好ましくはトリフルオロ酢酸を用いる。酸処理に際しては、前段階の反応溶媒に酸をそのまま加えてもよいし、一旦溶媒を減圧濃縮した後、適切な溶媒たとえばジクロロメタンなどに変えて酸を加えてもよい。反応温度は−10℃から室温であり、反応時間は5分間から2時間である。
【0046】
[製造方法3] 化合物(I−3)の製造方法
【0047】
【化7】
〔式中、R3、R4は前記定義と同意義を意味する。
【0048】
化合物(I−3−1)は国際公開WO 2007/052615 A1公報に記載された方法等により製造することができる。
【0049】
[工程3−1]
本工程は、化合物(I−3−1)とジ−tert−ブチル ジカーバメートを塩基触媒存在下に反応させて化合物(I−3−2)を得る工程である。本工程において、化合物(I−3−2)は、一段階の反応、または一方のアミノ基のジ−tert−ブチル カーバメート体を中間体として経由した他段階の反応でも得ることができる。
化合物(I−3−1)とジ−tert−ブチル ジカーバメートを反応させる時に用いる溶媒としては、出発原料をある程度溶解するものであり、かつ、反応を阻害しないものであれば、特に制限はないが、例えば、塩化メチレン、クロロホルムなどのハロゲン化炭化水素系溶媒;テトラヒドロフラン、ジエチルエーテルなどのエーテル系溶媒;酢酸エチルなどのエステル系溶媒;アセトニトリル、テトラメチレンスルホランまたはこれらの混合溶媒などを用いることができる。ジ−tert−ブチル ジカーバメートは化合物(I−3−1)に対して2当量から20当量用いる。塩基触媒としては4−ジメチルアミノピリジンを0.001当量から0.3当量用いる。この際トリエチルアミンなどの有機塩基を1当量から4当量添加してもよい。反応温度は0℃から60℃であり、好ましくは4℃から室温である。反応時間は1時間から72時間である。
【0050】
[工程3−2]
本工程は、化合物(I−3−2)とホスホリック アシッド ジ−tert−ブチル エステル クロロメチル エステルをヨウ化ナトリウム存在下に反応させた後、酸処理により化合物(I−3)を得る工程である。
化合物(I−3−2)とホスホリック アシッド ジ−tert−ブチル エステル クロロメチル エステルをヨウ化ナトリウム存在下に反応させる時に用いる溶媒としては、出発原料をある程度溶解するものであり、かつ、反応を阻害しないものであれば、特に制限はないが、例えば、塩化メチレン、クロロホルムなどのハロゲン化炭化水素系溶媒;テトラヒドロフラン、ジエチルエーテルなどのエーテル系溶媒;酢酸エチルなどのエステル系溶媒;アセトニトリル、テトラメチレンスルホランまたはこれらの混合溶媒などを用いることができ、好ましくはテトラヒドロフランあるいはアセトニトリルを用いる。ホスホリック アシッド ジ−tert−ブチル エステル クロロメチル エステルは化合物(I−3−2)に対して1当量から10当量用いることができ、好ましくは1当量から2当量用いる。ヨウ化ナトリウムは化合物(I−3−2)に対して1当量から10当量用いることができ、好ましくは1当量から2当量用いる。反応温度は0℃から60℃であり、好ましくは4℃から室温である。反応時間は1時間から72時間である。
酸処理に用いる酸としては、例えばトリフルオロ酢酸などの有機酸、塩酸などの鉱酸を用いることができ、好ましくはトリフルオロ酢酸を用いる。酸処理に際しては、前段階の反応溶媒に酸をそのまま加えてもよいし、一旦溶媒を減圧濃縮した後、適切な溶媒たとえばジクロロメタンなどに変えて酸を加えてもよい。反応温度は−10℃から室温であり、反応時間は5分間から2時間である。
【実施例】
【0051】
本発明に係る化合物は、例えば以下の実施例、参考例および製造例等に記載した方法により製造することができる。ただし、これらは例示的なものであって、本発明に係る化合物は如何なる場合も以下の具体例に限定されるものではない。
なお、実施例、参考例および製造例等の記載中で用いる略号の意味は以下のとおりである。
TFA:トリフルオロ酢酸
【0052】
[参考例1]3−(3−(4−(ピリジン−2−イルメトキシ)−ベンジル)−イソキサゾール−5−イル)−ピリジン−2−イルアミン
【0053】
【化8】
製造例1−1−5に記載の4−(5−(2−アミノ−ピリジン−3−イル)イソキサゾール−3−イルメチル)−フェノール(4.2mg、0.016mmol)とメタノール(0.4mL)の混合物に、1N 水酸化ナトリウム水溶液(16μL、0.016mmol)を加えた後、減圧下濃縮した。残渣とN,N−ジメチルホルムアミド(0.5mL)の混合物に2−ピコリル クロリド(3.1mg、0.019mmol)を加え、室温で2時間攪拌した。反応混合物をそのまま逆相系高速液体クロマトグラフィー(アセトニトリル−水系移動相(0.1%トリフルオロ酢酸含有)を用いた)にて精製し、標記化合物(3.6mg, 39%)をジトリフルオロ酢酸塩として得た。
MS m/e(ESI) 359.16(MH+)
【0054】
また、別法として以下の方法で参考例1の化合物を得た。
製造例1−1−5に記載の4−(5−(2−アミノ−ピリジン−3−イル)イソキサゾール−3−イルメチル)−フェノール(2.97g、11.1mmol)、テトラヒドロフラン(100mL)とアセトン(100ml)の混合物に、5N 水酸化ナトリウム水溶液(2.22mL、11.1mmol)を加えた。反応混合物に超音波を30秒間照射した後、減圧下濃縮した。残渣とN,N−ジメチルホルムアミド(50mL)の混合物に2−ピコリル クロリド(3.64g、22.2mmol)を加え、60℃で2.5時間攪拌した。反応混合物を室温に戻し、水でクエンチした後、酢酸エチルにて抽出した。その有機層を水と飽和食塩水で洗浄し、無水硫酸マグネシウムにて乾燥した。その溶媒を減圧留去し、その残渣をNH−シリカゲルクロマトグラフィー(ヘプタン:酢酸エチル=1:1)にて精製し、標記化合物(2.73g, 67%)を得た。
1H−NMR Spectrum (CDCl3)δ(ppm): 4.00(2H, s), 5.20(2H,s), 5.37(2H, brs), 6.24(1H, s), 6.71(1H, dd, J=4.8,7.6Hz), 6.95−6.97(2H, m), 7.20−7.22(2H, m), 7.52(d,1H, d, J=1.9Hz), 7.69−7.74(3H, m), 8.13-8.15(1H, m), 8.60(1H, d, J=4.4Hz).
【0055】
出発物質4−(5−(2−アミノ−ピリジン−3−イル)イソキサゾール−3−イルメチル)−フェノールは以下の方法で合成した。
【0056】
[製造例1−1−1]1−ベンジルオキシ−4−((E)−2−ニトロ−ビニル)−ベンゼン
【0057】
【化9】
4−ベンジルオキシベンズアルデヒド(1.0g、4.7mmol)、ナトリウムメトキシド(28%メタノール溶液、150μL、0.74mmol)、およびメタノール(10mL)の混合物に、0℃でニトロメタン(330μL、6.1mmol)とナトリウムメトキシド(28%メタノール溶液、1.0mL、4.9mmol)を加え、室温で10分間攪拌した。反応混合物を0℃に冷却し、同温で5N 塩酸水溶液(20mL)を加えた。反応混合物を室温で15分間攪拌した。析出した固体をろ取し、標記化合物(1.2g、100%)を得た。
1H−NMR Spectrum (DMSO−d6)δ(ppm):5.20(2H,s), 7.10−7.14(2H,m), 7.32−7.48(5H,m), 7.82−7.85(2H,m), 8.12(2H,dd,J=13.5,18.2Hz).
【0058】
[製造例1−1−2]1−ベンジルオキシ−4−(2−ニトロ−エチル)−ベンゼン
【0059】
【化10】
製造例1−1−1に記載の1−ベンジルオキシ−4−((E)−2−ニトロ−ビニル)−ベンゼン(1.0g、3.9mmol)、酢酸(1mL)、およびジメチルスルホキシド(17mL)の混合物に、適宜冷却しながら室温で水素化ホウ素ナトリウム(250mg、6.3mmol)を加えた。室温で40分間攪拌し、反応混合物に水を加えた。反応混合物を酢酸エチルと水に分配した。有機層を水および飽和食塩水で洗浄し、それを無水硫酸マグネシウムで乾燥し、その溶媒を減圧下留去した。残渣をNHシリカゲルカラムクロマトグラフィー(酢酸エチル:ヘプタン=1:3)で精製し、標記化合物(710mg、70%)を得た。
1H−NMR Spectrum (CDCl3)δ(ppm):3.26(2H,t,J=7.2Hz), 4.56(2H,t,J=7.2Hz), 5.04(2H,s), 6.92(2H,d,J=8.4Hz), 7.11(2H,d,J=8.8Hz),7.30−7.42(5H,m).
【0060】
[製造例1−1−3]4−ベンジルオキシ−フェニル−アセトヒドロキシモイル クロリド
【0061】
【化11】
製造例1−1−2に記載の1−ベンジルオキシ−4−(2−ニトロ−エチル)−ベンゼン(340mg、1.3mmol)とメタノール(5mL)の混合物に、室温でリチウムメトキシド(100mg、2.6mmol)を加え、室温で15分間攪拌した。反応混合物を減圧下濃縮した。残渣に塩化メチレン(4mL)とテトラヒドロフラン(2mL)を加えた。反応混合物に−78℃でチタニウム(IV)クロリドを加え、0℃で50分間攪拌した。反応混合物を−78℃に冷却後、水(5mL)を加え、徐々に室温まで昇温させた。反応混合物を酢酸エチルと水に分配した。有機層を飽和食塩水で洗浄し、その溶媒を減圧下留去した。残渣を中性シリカゲルカラムクロマトグラフィー(酢酸エチル:ヘプタン=1:3)で精製し、標記化合物(310mg、84%)を得た。
1H−NMR Spectrum (CDCl3)δ(ppm):3.83(2H,s), 5.07(2H,s), 6.94−6.98(2H,m), 7.17−7.21(2H,m), 7.32−7.44(5H,m).
【0062】
[製造例1−1−4]3−(3−(4−ベンジルオキシ−ベンジル)−イソキサゾール−5−イル)−ピリジン−2−イルアミン
【0063】
【化12】
製造例1−1−3に記載の4−ベンジルオキシ−フェニル−アセトヒドロキシモイル クロリド(1.2g、4.4mmol)とテトラヒドロフラン(34mL)の混合物に、0℃で製造例1−2−5に記載の3−エチニル−ピリジン−2−イルアミン(260mg、2.2mmol)とトリエチルアミン(3.0mL、22mmol)を加え、室温で1時間攪拌した。反応混合物に室温で水を加え、酢酸エチル−テトラヒドロフラン(2:1)で抽出した。有機層を飽和食塩水で洗浄し、その溶媒を減圧下留去した。残渣をNHシリカゲルカラムクロマトグラフィー(酢酸エチル:ヘプタン=1:3)で精製し、標記化合物(240mg、15%)を得た。
1H−NMR Spectrum (CDCl3)δ(ppm):4.00(2H,s), 5.05(2H,s), 5.41(2H,s), 6.24(1H,s), 6.71(1H,dd,J=4.9,7.6Hz), 6.93−6.97(2H,m), 7.18−7.22(2H,m), 7.31−7.44(5H,m), 7.70(1H,dd,J=1.7,7.6Hz), 8.13(1H,dd,J=1.8,4.9Hz).
【0064】
[製造例1−1−5]4−(5−(2−アミノ−ピリジン−3−イル)イソキサゾール−3−イルメチル)−フェノール
【0065】
【化13】
製造例1−1−4に記載の3−(3−(4−ベンジルオキシ−ベンジル)−イソキサゾール−5−イル)−ピリジン−2−イルアミン(32mg、0.090mmol)とトリフルオロ酢酸(1mL)の混合物に、室温でチオアニソール(45mg、0.36mmol)を加え、同温で2時間攪拌した。飽和炭酸水素ナトリウム水溶液と酢酸エチルの混合物に反応混合物を加えた。有機層を分離し、飽和食塩水で洗浄し、その溶媒を減圧下留去した。残渣をシリカゲルカラムクロマトグラフィー(酢酸エチル:ヘプタン=4:1)で精製し、標記化合物(24mg、100%)を得た。
1H−NMR Spectrum (DMSO−d6)δ(ppm):3.90(2H,s), 6.25(2H,brs), 6.68−6.72(3H,m), 6.76(1H,s), 7.11(2H,d,J=8.6Hz), 7.87(1H,dd,J=1.5,7.7Hz), 8.10(1H,brs), 9.29(1H,s).
【0066】
出発物質3−エチニル−ピリジン−2−イルアミンは以下の方法で合成した。
【0067】
[製造例1−2−1]2,2−ジメチル−N−ピリジン−2−イル−プロピオナミド
【0068】
【化14】
2−アミノピリジン(50.0g、531mmol)の塩化メチレン(500mL)溶液に、0℃でトリエチルアミン(81.4mL、584mmol)、ピバロイル クロライド(71.9mL、584mmol)を加え、室温で4時間30分攪拌した。反応溶液を水と塩化メチレンに分配した。有機層を水と飽和食塩水で洗浄し、無水硫酸マグネシウムで乾燥し、その溶媒を減圧下留去した。得られた残渣のメタノール(300mL)溶液に、0℃で炭酸カリウム(73.4g、531mmol)を加え、室温で90分間攪拌した。反応溶液を室温で水と酢酸エチルに分配した。有機層を飽和食塩水で洗浄し、無水硫酸マグネシウムで乾燥し、その溶媒を減圧下留去した。残渣にヘプタン(300mL)を加え、析出した固体をろ取し、標記化合物(80.2g、85%)を得た。更に、ろ液を減圧下濃縮し、残渣をシリカゲルカラムクロマトグラフィー(ヘプタン:酢酸エチル=2:1)にて精製し、標記化合物(12.2g、13%)を得た。
1H−NMR Spectrum (DMSO−d6)δ(ppm):1.22(9H,s), 7.06−7.09(1H,m), 7.72−7.77(1H,m), 8.01−8.03(1H,m), 8.29−8.31(1H,m), 9.71(1H,s).
【0069】
[製造例1−2−2]N−(3−ヨード−ピリジン−2−イル)−2、2−ジメチル−プロピオナミド
【0070】
【化15】
製造例1−2−1に記載の2、2−ジメチル−N−ピリジン−2−イル−プロピオナミド(3.0g、17mmol)、N,N,N’,N’−テトラメチルエチレンジアミン(6.3mL、42mmol)、およびテトラヒドロフラン(60mL)の混合物に−78℃でn−ブチルリチウム(1.6M n−ヘキサン溶液、30mL、47mmol)を滴下し、0℃で終夜攪拌した。反応混合物に−78℃でヨウ素(6.8g、27mmol)を加え、0℃で1.5時間攪拌した。反応混合物に水と飽和チオ硫酸ナトリウム水溶液を加え、酢酸エチルで抽出した。有機層を飽和食塩水で洗浄し、その溶媒を減圧下留去した。残渣をシリカゲルカラムクロマトグラフィー(酢酸エチル:ヘプタン=2:1)にて精製し、標記化合物(2.9g、57%)を得た。
1H−NMR Spectrum (CDCl3)δ(ppm):1.38(9H,s), 6.85(1H,dd,J=4.8,7.9Hz), 7.94(1H,brs), 8.11(1H,dd,J=1.7,7.9Hz), 8.46(1H,dd,J=1.7,4.6Hz).
【0071】
[製造例1−2−3]3−ヨード−ピリジン−2−イルアミン
【0072】
【化16】
製造例1−2−2に記載のN−(3−ヨード−ピリジン−2−イル)−2、2−ジメチル−プロピオナミド(66.2g、218mmol)、5N 水酸化ナトリウム水溶液(200mL)、メタノール(200mL)の混合物を加熱還流下、1時間20分間撹拌した。反応溶液を室温に戻し、水と酢酸エチルに分配した。水層を酢酸エチルで3回抽出した。有機層を合わせ、飽和食塩水で洗浄し、それを無水硫酸ナトリウムで乾燥した。硫酸ナトリウムをろ過で除き、その溶媒を減圧下濃縮して標記化合物(41.2g、85.9%)を得た。
1H−NMR Spectrum (DMSO−d6)δ(ppm):6.00(2H,brs), 6.32(1H,dd,J=4.8Hz,7.2Hz), 7.87(1H,d,J=7.2Hz), 7.92(1H,d,J=4.8Hz).
【0073】
[製造例1−2−4]3−トリメチルシラニルエチニル−ピリジン−2−イルアミン
【0074】
【化17】
製造例1−2−3に記載の3−ヨード−ピリジン−2−イルアミン(40.2g、183mmol)、トリメチルシリルアセチレン(51.7mL、366mmol)、ヨウ化銅(I)(3.49g、18.3mmoL)、N,N−ジイソプロピルエチルアミン(63.7mL、366mmol)、N−メチルピロリジノン(200mL)の混合物にテトラキス(トリフェニルホスフィン)パラジウム(0)(10.6g、9.15mmol)を加え、窒素気流下、室温で3時間10分撹拌した。反応溶液に水を加え酢酸エチルで4回抽出した。その溶媒を減圧下濃縮した。残渣をNHシリカゲルクロマトグラフィー(ヘプタン:酢酸エチル=4:1)で精製した。得られた溶液を減圧下濃縮し、残渣をシリカゲルクロマトグラフィー(ヘプタン:酢酸エチル=2:1ついで1:1)で精製し標記化合物(28.1g、80.7%)を得た。
1H−NMR Spectrum (DMSO−d6)δ(ppm): 0.25(9H,s), 6.09(2H,brs), 6.51−6.57(1H,m), 7.50−7.55(1H,m), 7.95−7.99(1H,m).
【0075】
[製造例1−2−5]3−エチニル−ピリジン−2−イルアミン
【0076】
【化18】
製造例1−2−4に記載の3−トリメチルシラニルエチニル−ピリジン−2−イルアミン(28.1g、148mmoL)のテトラヒドロフラン(300mL)溶液にテトラブチルアンモニウム フルオリド(1M テトラヒドロフラン溶液、20mL、20mmol)、を加え室温で15分撹拌した。反応溶液に水を加え酢酸エチルで4回抽出した。有機層を無水硫酸ナトリウムで乾燥し、その溶媒を減圧下留去した。残渣をシリカゲルクロマトグラフィー(ヘプタン:酢酸エチル=1:1ついで1:2)で精製し標記化合物(16.4g、93.7%)を得た。
1H−NMR Spectrum(DMSO−d6)δ(ppm):4.43(1H,s), 6.14(2H,brs), 6.53(1H,dd,J=4.8Hz,7.2Hz), 7.53(1H,d,J=7.2Hz),7.96(1H,d,J=4.8Hz).
【0077】
[製造例1−3−1]3−トリメチルシラニルエチニル−ピリジン−2−イルアミン(製造例1−2−4の別法)
【0078】
【化19】
2−アミノ−3−ブロモピリジン(5.72g、33.1mmol)のN−メチルピロリジノン(120mL)溶液に、室温でトリメチルシリルアセチレン(9.36mL、66.2mmol)、テトラキス(トリフェニルホスフィン)パラジウム(0)(1.91g、1.66mmol)、ヨウ化銅(I)(630mg、3.31mmol)、N,N−ジイソプロピルエチルアミン(11.5mL、66.2mmol)を加え、窒素雰囲気下、70℃で6時間攪拌した。反応溶液に水を加え、酢酸エチルで抽出した。有機層を水と飽和食塩水で洗浄し、無水硫酸マグネシウムで乾燥し、その溶媒を減圧下留去した。残渣をシリカゲルカラムクロマトグラフィー(ヘプタン:酢酸エチル=2:1)で精製し、標記化合物(5.94g、94%)を得た。
1H−NMR Spectrum (DMSO−d6)δ(ppm):0.23(9H,s), 6.07(2H,brs), 6.51(1H,dd,J=4.9,7.5Hz), 7.49(1H,dd,J=1.8,7.5Hz), 7.94(1H,dd,J=1.8,4.9Hz).
【0079】
[実施例1]2−((4−((5−(2−アミノ−3−ピリジニル)−3−イソキサゾリル)メチル)フェノキシ)メチル)−1−((ホスホノオキシ)メチル)ピリジニウム 1トリフルオロ酢酸塩
【0080】
【化20】
製造例1−4−1に記載のジ−tert−ブチル−(3−(3−(4−(ピリジン−2−イルメトキシ)ベンジル)イソキサゾール−5−イル)ピリジン−2−イル)イミドジカルボネート(334mg、0.60mmol)、ホスホリック アシッド ジ−tert−ブチル エステル クロロメチル エステル(309mg、1.2mmol)、ヨウ化ナトリウム(134mg、0.90mmol)テトラヒドロフラン(0.6ml)を室温で3.5時間撹拌し、ついで、この反応溶液にトリフルオロ酢酸(2ml)を加えさらに40分室温で撹拌した。反応溶液を減圧濃縮し、残渣に重曹水と酢酸エチルを加え分液した。水層に酢酸エチルを加え再度分液した。得られた重曹水をゲルろ過(CHP20P(三菱化成)、水ついでメタノールで溶出)し、ついで溶出液を、液量が10mlくらいになるまで濃縮した。得られた溶液をODSカラム(H2O:MeOH:TFA=500:50:0.5ついで500:200:0.7)で精製した。溶媒を濃縮し、残渣を少量のアセトンに溶かした後、酢酸エチルを加え濃縮して標記化合物(53.93mg)を粉末状の固体として得た。
1H−NMR Spectrum (DMSO-d6)δ(ppm):4.02(2H,s), 5.73(2H, s), 6.33(2H, d, J=13.5Hz), 6.84(1H, dd, J=5.2, 7.6Hz), 6.89(1H, s), 7.13(2H, d, J=8.8Hz), 7.33(2H, d, J=8.8Hz), 8.07(1H, dd, J=1.6, 7.6Hz), 8.12(1H, dd, J=1.6, 5.2Hz), 8.15-8.22(1H, m), 8.28(1H, d, J=7.6Hz), 8.73(1H, ddd, J=1.6, 7.6, 7.6Hz), 9.22(1H, d=4.8Hz).
【0081】
出発物質ジ−tert−ブチル−(3−(3−(4−(ピリジン−2−イルメトキシ)ベンジル)イソキサゾール−5−イル)ピリジン−2−イル)イミドジカルボネートは以下の方法で合成した。
【0082】
[製造例1−4−1]ジ−tert−ブチル−(3−(3−(4−(ピリジン−2−イルメトキシ)ベンジル)イソキサゾール−5−イル)ピリジン−2−イル)イミドジカルボネート
【0083】
【化21】
参考例1に記載の3−(3−(4−(ピリジン−2−イルメトキシ)ベンジル)イソキサゾール−5−イル)ピリジン−2−イルアミン(300mg、0.84mmol)、ジ−tert−ブチルジカルボネート(913mg、4.2mmol)、4−ジメチルアミノピリジン(10mg、0.084mmol)、トリエチルアミン(102mg、1.0mmol)、テトラヒドロフラン(7ml)を室温で13.5時間撹拌した。反応溶液をNH−シリカゲルクロマトグラフィー(ヘプタン:酢酸エチル=1:1ついで1:2)で精製し標記化合物(334mg)を得た。
1H−NMR Spectrum (CDCl3)δ(ppm):1.24(18H, s), 3.99(2H, s), 5.18(2H, s), 6.32(1H, s), 6.90-6.95(2H, m), 7.16-7.24(3H, m), 7.41(1H, dd, 4.8, 8.0Hz), 7.51(1H, d, 8.0Hz), 7.71(1H, ddd, J=2.0, 8.0, 8.0Hz), 8.27(1H, dd, J=2.0, 8.0Hz), 8.56-8.61(2H, m).
【0084】
式(I)で表される本発明に係る化合物は、優れた抗真菌活性を有する活性体である親化合物に速やかに変換され、さらに、物性、特に水への溶解性および水溶液中での安定性、ならびに安全性の面でも優れており、真菌感染症の予防剤または治療剤として極めて有用である。
【0085】
1.水への溶解性の比較試験例
親化合物である参考例1に記載の3−(3−(4−(ピリジン−2−イルメトキシ)ベンジル)イソキサゾール−5−イル)ピリジン−2−イルアミンと実施例1の化合物を、25℃において、Britton-Robinson緩衝液(イオン強度0.3)への溶解度を比較した。表1はその結果を示す。
【0086】
【表1】
【0087】
表1に示す結果から明らかなように、実施例1の化合物は、各種pH領域において、その親化合物よりも水への溶解性が顕著に増大していることが判明した。
【0088】
2.肝S9画分での親化合物(活性体)への変換
(1).各種肝S9反応溶液の調製
ヒトおよびサル肝S9画分(タンパク濃度 0.22 mg/mL)、0.5 mmol/L塩化マグネシウム、100 mmol/L Tris-HClを含む懸濁液(pH7.4)を氷上で調製した(各種反応溶液A)。各種反応溶液A 0.27 mLに本発明に係る化合物(実施例1の化合物) 100 μmol/mL水溶液を30μL添加し(最終化合物濃度 10 μmol/L)各種肝S9反応溶液(最終タンパク濃度 0.2 mg/mL)とし、(2)の実施まで氷上で保存した。対照として、0.5 mmol/L塩化マグネシウム、100 mmol/L Tris-HCl を含む緩衝液(pH 7.4)0.27 mLに本発明に係る化合物(実施例1の化合物)100 μmol/mL水溶液を30μL添加したものを調製した(反応緩衝溶液)。
(2).各種肝S9反応溶液および反応緩衝溶液中での親化合物(参考例1の化合物)への変換
(1)の各種肝S9反応溶液および反応緩衝溶液を37℃でインキュベーションし、時間0,30および60分の時点で50 μLずつ採取しメタノール溶液100 μLを添加し反応を停止させた。
(3).本発明に係る化合物(実施例1の化合物)および親化合物(参考例1の化合物)の反応液中濃度はLC-MSにより定量した。
【0089】
2.に記載の測定法で、本発明に係る化合物(実施例1の化合物)および親化合物(参考例1の化合物)の反応液中濃度を測定した。図1ないし図3は、実施例1の化合物および参考例1の化合物の、各種肝S9反応溶液および反応緩衝溶液中での濃度の経時変化を示す。図1ないし図3に示す結果から、本発明に係る化合物(実施例1の化合物)はヒトおよびサル肝S9画分で親化合物(参考例1の化合物)に変換した。また、肝S9画分を含まない反応緩衝溶液中では、本発明に係る化合物(実施例1の化合物)から親化合物(参考例1の化合物)への変換は認められなかったことが確認された。
【0090】
3.抗カンジダ活性及び抗アスペルギルス活性
(1).菌液の調製
C. albicans CAF2−1株は、サブローデキストロース液体培地(SDB)に30℃、48時間静置培養した菌液をRPMI1640培地で希釈し、1.2x103cells/mLの菌液に調製した。A. fumigatus Tsukuba株は、−80℃凍結保存株をRPMI1640培地で希釈し、4.5x103cells/mLの菌液に調製した。
【0091】
(2).薬剤希釈プレートの作製
U底96wellプレートを用い、8検体/プレート(A〜H)の検体希釈溶液を作製した。各プレートの2〜12列目にジメチルスルホキシド溶液を10μL分注した。秤量した検体をジメチルスルホキシドに溶解し、2.5mg/mLの溶液を作製後、この溶液を準備したプレートの1列目に20μL添加し、プレート上で12段階2倍階段希釈(溶液10μL+ジメチルスルホキシド溶液10μL)した。この検体希釈溶液を1μLづつMIC測定用の平底96wellプレートに分注し、検体希釈プレートを作製した。
【0092】
(3).菌液の接種および培養
(1)で調製した菌液を、(2)で作製した被検化合物希釈液1μL/wellが入った平底96wellプレートに99μL/well接種し、35℃で42〜48時間、好気的に静置培養した。
【0093】
(4).MIC測定
一見して、コントロールと比較して菌の増殖を明らかに抑制した最小濃度を最小発育阻止濃度(MIC)とした。
【0094】
3に記載の測定法で、親化合物(参考例1の化合物)について、抗カンジダ活性及び抗アスペルギルス活性を測定した。その結果は、表2に示す。表2に示す結果から、親化合物(参考例1の化合物)は、抗カンジダ活性および抗アスペルギルス活性を有していることが確認された。
【0095】
【表2】
【産業上の利用可能性】
【0096】
本発明によれば、式(I)で表される本発明に係る化合物は、活性本体である親化合物のプロドラッグとして、1)真菌のGPI生合成阻害に基づいて細胞壁表層蛋白質の発現を阻害し、細胞壁assemblyを阻害するとともに真菌が細胞へ付着するのを阻害して、病原体が病原性を発揮できないようにすることより、感染症の発症、進展、持続に対して効果を示し、2)物性、特に、水への溶解性および水溶液中での安定性、ならびに体内動態および安全性の面でも優れており、真菌感染症の予防または治療剤として極めて有用である。
【技術分野】
【0001】
本発明は、新規な((ホスホノオキシ)メチル)ピリジニウム環を有するピリジン誘導体およびそれらを含有する抗真菌剤に関する。
【背景技術】
【0002】
近年、高度な化学療法等による免疫機能の低下した患者や高齢者が増加しているため、日和見感染の対策は益々重要性を増してきている。異なる弱毒菌による日和見感染が次々と起こっている事実が示すように、患者の抵抗力が低下するような基礎疾患がある限り感染症の問題は後を絶たない。従って、近い将来確実に訪れる高齢化社会においては、耐性菌の問題を含めた新たな感染症対策が重要な課題の一つとなることが見込まれている。
【0003】
抗真菌剤の分野では、従来、例えば、深在性の真菌症の治療にはポリエン系のアムホテリシンBやアゾール系のフルコナゾール、イトラコナゾール、ボリコナゾール等が開発されてきた。すでに上市されている既存薬には類似したメカニズムの薬剤が多く、現在ではアゾール耐性菌等の出現が問題となっている。
【0004】
近年、新規メカニズムの1,3−β−グルカン合成酵素阻害剤として天然物由来の環状ヘキサペプチド型のカスポファンジンやミカファンジン等が開発されてきているが、これらの薬剤には注射剤しかないことから、抗真菌剤としてはまだ充分ではない。
【0005】
このように既存の抗真菌剤では充分とはいえない状況にあり、新規なメカニズムに基づく安全性の高い薬剤の開発が切望されている。かかる新規なメカニズムに基づく抗真菌剤に関する関連技術として、特許文献1および2がある。特許文献1および2には、GPI(glycosylphosphatidyl-inositol)アンカー蛋白質の細胞壁への輸送過程を阻害することで細胞壁表層蛋白質の発現を阻害し、細胞壁assemblyを阻害するとともに真菌が細胞へ付着するのを阻害して、病原体が病原性を発揮できないようにすることにより、感染症の発症、進展、持続に対して効果を示すピリジン誘導体が記載されている。
【0006】
このような状況下において、特許文献3には、従来の抗真菌剤にはない優れた抗真菌作用を有し、物性、安全性および代謝的安定性の面でも優れた抗真菌剤として、ヘテロ環置換ピリジン誘導体が提案されている。
【0007】
一方、特許文献4または5には、それぞれ、水溶性プロドラッグとして、下記式で表わされる化合物類、N−ホスホリルオキシメチルプロドラッグが開示されている。
【0008】
【化1】
(式中、R1、R2およびR3は親第三級または第二級アミンを含む置換基であり、R4およびR5は、各々有機または無機残基であり、Xはカチオン有機または無機塩である。)
また、特許文献6には、水への溶解性、および安全性の面でも優れた抗真菌剤のプロドラッグとして、ヘテロ環およびホスホノアミノ基が置換したピリジン誘導体が提案されている。
【先行技術文献】
【特許文献】
【0009】
【特許文献1】国際公開第02/04626号パンフレット
【特許文献2】国際公開第05/033079号パンフレット
【特許文献3】国際公開第07/052615号パンフレット
【特許文献4】米国特許第6,235,728 B1
【特許文献5】特表2001−527083号公報
【特許文献6】国際公開第08/136324号パンフレット
【発明の概要】
【発明が解決しようとする課題】
【0010】
しかしながら、より優れた真菌症の治療方法を提供するためには、水への溶解性および水溶液中での安定性、ならびに安全性の観点からより優れた抗真菌剤のさらなる創製が望まれている。
かかる事情に鑑み、本発明の目的は、優れた抗真菌作用を有し、かつ、水への溶解性および水溶液中での安定性、ならびに体内動態および安全性の面でも優れた抗真菌剤を提供することにある。
【課題を解決するための手段】
【0011】
本発明者らは、上記事情に鑑み鋭意研究を重ねた結果、下式(I)
【0012】
【化2】
【0013】
で表される((ホスホノオキシ)メチル)ピリジニウム環を有するピリジン誘導体が、活性本体である親化合物のプロドラッグとして、優れた抗真菌作用を有するとともに、水への溶解性および水溶液中での安定性、ならびに体内動態および安全性の面でも優れることをも見出して、本発明を完成した。
【0014】
すなわち、本発明は、
[1]下式(I)で表される化合物またはその塩;
【化3】
式中、
R1が、水素原子、ハロゲン原子、アミノ基、C1−6アルキル基、C1−6アルコキシ基、またはC1−6アルコキシC1−6アルキル基を意味し;
R2が、水素原子、C1-6アルキル基、アミノ基、またはジC1-6アルキルアミノ基を意味し;
R3が、水素原子、ハロゲン原子、またはC1−6アルキル基を意味し;
R4が、水素原子、ハロゲン原子、またはC1−6アルキル基を意味する。
[2] R2がアミノ基である、前記[1]に記載の化合物またはその塩。
[3] R1が、水素原子である、前記[1]または[2]に記載の化合物またはその塩。
[4] R1が、アミノ基である、前記[1]ないし[2]に記載の化合物またはその塩。
[5] R3が、水素原子であり、R4が、水素原子、ハロゲン原子、またはC1−6アルキル基である、前記[1]ないし[4]のいずれか1項に記載の化合物またはその塩。
[6] 下式
【化4】
で表される2−((4−((5−(2−アミノ−3−ピリジニル)−3−イソキサゾリル)メチル)フェノキシ)メチル)−1−((ホスホノオキシ)メチル)ピリジニウムの化合物またはその塩。
[7] 前記[1]ないし[6]のいずれか1項に記載の化合物またはその塩を含有する医薬組成物。
[8] 前記[1]ないし[6]のいずれか1項に記載の化合物またはその塩を含有する医薬。
[9] 前記[1]ないし[6]のいずれか1項に記載の化合物またはその塩を有効成分として含有する抗真菌剤。
[10] 前記[1]ないし[6]のいずれか1項に記載の化合物またはその塩の薬理学的有効量を投与して、真菌感染症を予防および/または治療する方法。
[11] 抗真菌剤の製造のための、前記[1]ないし[6]のいずれか1項に記載の化合物またはその塩の使用。
を提供する。
【発明の効果】
【0015】
式(I)で表される化合物(以下、単に「本発明に係る化合物」という称する場合がある。)は、活性本体である親化合物のプロドラッグとして、1)真菌のGPI生合成阻害に基づいて細胞壁表層蛋白質の発現を阻害し、細胞壁assemblyを阻害するとともに真菌が細胞へ付着するのを阻害して、病原体が病原性を発揮できないようにすることにより、感染症の発症、進展、持続に対して効果を示し、さらに、2)物性、特に、水への溶解性および水溶液中での安定性、ならびに体内動態および安全性の面でも優れており、真菌感染症の予防または治療剤として極めて有用である。
【図面の簡単な説明】
【0016】
【図1】図1は、本発明の実施例1で得られた2−((4−((5−(2−アミノ−3−ピリジニル)−3−イソキサゾリル)メチル)フェノキシ)メチル)−1−((ホスホノオキシ)メチル)ピリジニウム 1トリフルオロ酢酸塩および親化合物(参考例1で示す3−(3−(4−(ピリジン−2−イルメトキシ)−ベンジル)−イソキサゾール−5−イル)−ピリジン−2−イルアミン)の、ヒト肝S9反応溶液中での各化合物濃度の経時変化を示す。
【図2】図2は、本発明の実施例1で得られた2−((4−((5−(2−アミノ−3−ピリジニル)−3−イソキサゾリル)メチル)フェノキシ)メチル)−1−((ホスホノオキシ)メチル)ピリジニウム 1トリフルオロ酢酸塩および親化合物(参考例1で示す3−(3−(4−(ピリジン−2−イルメトキシ)−ベンジル)−イソキサゾール−5−イル)−ピリジン−2−イルアミン)の、サル肝S9反応溶液中での各化合物濃度の経時変化を示す。
【図3】図3は、本発明の実施例1で得られた2−((4−((5−(2−アミノ−3−ピリジニル)−3−イソキサゾリル)メチル)フェノキシ)メチル)−1−((ホスホノオキシ)メチル)ピリジニウム 1トリフルオロ酢酸塩および親化合物(参考例1で示す3−(3−(4−(ピリジン−2−イルメトキシ)−ベンジル)−イソキサゾール−5−イル)−ピリジン−2−イルアミン)の、反応緩衝溶液中での各化合物濃度の経時変化を示す。
【発明を実施するための形態】
【0017】
以下に、本明細書において記載する記号、用語等の定義、本発明の実施の形態等を示して、本発明を詳細に説明する。なお、本発明は以下の実施の形態に限定されるものではなく、その要旨の範囲内で種々変形して実施することができる。
【0018】
本明細書中においては、化合物の構造式が便宜上一定の異性体を表すことがあるが、本発明には化合物の構造上生じ得るすべての幾何異性体、不斉炭素に基づく光学異性体、立体異性体、回転異性体、互変異性体等の異性体および異性体混合物を含み、便宜上の式の記載に限定されるものではなく、いずれか一方の異性体でも混合物でもよい。したがって、本発明の化合物には、分子内に不斉炭素原子を有し光学活性体およびラセミ体が存在することがありうるが、本発明においては限定されず、いずれもが含まれる。また、結晶多形が存在することもあるが同様に限定されず、いずれかの単一の結晶形であっても二以上の結晶形からなる混合物であってもよい。そして、本発明に係る化合物には無水物と水和物等の溶媒和物とが包含される。
【0019】
本明細書において使用する「C1−6アルキル基」とは、炭素数1〜6個の脂肪族炭化水素から任意の水素原子を1個除いて誘導される一価の基である、炭素数1〜6個の直鎖状または分枝鎖状のアルキル基を意味し、具体的には例えば、メチル基、エチル基、n−プロピル基、イソプロピル基、n−ブチル基、イソブチル基、sec−ブチル基、tert−ブチル基、n−ペンチル基、イソペンチル基、sec−ペンチル基、ネオペンチル基、1−メチルブチル基、2−メチルブチル基、1,1−ジメチルプロピル基、1,2−ジメチルプロピル基、n−ヘキシル基、イソヘキシル基、1−メチルペンチル基、2−メチルペンチル基、3−メチルペンチル基、1,1−ジメチルブチル基、1,2−ジメチルブチル基、2,2−ジメチルブチル基、1,3−ジメチルブチル基、2,3−ジメチルブチル基、3,3−ジメチルブチル基、1−エチルブチル基、2−エチルブチル基、1,1,2−トリメチルプロピル基、1,2,2−トリメチルプロピル基、1−エチル−1−メチルプロピル基、1−エチル−2−メチルプロピル基等が挙げられ、好ましくはメチル基、エチル基、n−プロピル基、イソプロピル基、n−ブチル基、イソブチル基、sec−ブチル基、tert−ブチル基等である。
【0020】
本明細書において使用する「C1−6アルコキシ基」とは、前記定義「C1−6アルキル基」の末端に酸素原子が結合した基であることを意味し、具体的には例えば、メトキシ基、エトキシ基、n−プロポキシ基、イソプロポキシ基、n−ブトキシ基、イソブトキシ基、sec−ブトキシ基、tert−ブトキシ基、n−ペンチルオキシ基、イソペンチルオキシ基、sec−ペンチルオキシ基、ネオペンチルオキシ基、1−メチルブトキシ基、2−メチルブトキシ基、1,1−ジメチルプロポキシ基、1,2−ジメチルプロポキシ基、n−ヘキシルオキシ基、イソヘキシルオキシ基、1−メチルペンチルオキシ基、2−メチルペンチルオキシ基、3−メチルペンチルオキシ基、1,1−ジメチルブトキシ基、1,2−ジメチルブトキシ基、2,2−ジメチルブトキシ基、1,3−ジメチルブトキシ基、2,3−ジメチルブトキシ基、3,3−ジメチルブトキシ基、1−エチルブトキシ基、2−エチルブトキシ基、1,1,2−トリメチルプロポキシ基、1,2,2−トリメチルプロポキシ基、1−エチル−1−メチルプロポキシ基、1−エチル−2−メチルプロポキシ基等が挙げられ、好ましくはメトキシ基、エトキシ基、n−プロポキシ基、イソプロポキシ基、n−ブトキシ基、イソブトキシ基、sec−ブトキシ基、tert−ブトキシ基等である。
【0021】
本明細書中において使用する「C1−6アルコキシC1−6アルキル基」とは、前記定義「C1−6アルキル基」中の任意の水素原子を、前記定義「C1−6アルコキシ基」で置換した基を意味し、具体的には例えば、メトキシメチル基、エトキシメチル基、n−プロポキシメチル、メトキシエチル基、エトキシエチル基等が挙げられる。
【0022】
本明細書において使用する「ハロゲン原子」とは、フッ素原子、塩素原子、臭素原子またはヨウ素原子を意味する。
【0023】
本明細書中において使用する「ジC1−6アルキルアミノ基」とは、アミノ基中の2個の水素原子を、それぞれ同一のまたは異なる、前記定義「C1−6アルキル基」で置換した基を意味し、具体的には例えば、N,N−ジメチルアミノ基、N,N−ジエチルアミノ基、N,N−ジ−n−プロピルアミノ基、N,N−ジ−イソプロピルアミノ基、N,N−ジ−n−ブチルアミノ基、N,N−ジ−イソブチルアミノ基、N,N−ジ−sec−ブチルアミノ基、N,N−ジ−tert−ブチルアミノ基、N−エチル−N−メチルアミノ基、N−n−プロピル−N−メチルアミノ基、N−イソプロピル−N−メチルアミノ基、N−n−ブチル−N−メチルアミノ基、N−イソブチル−N−メチルアミノ基、N−sec−ブチル−N−メチルアミノ基、N−tert−ブチル−N−メチルアミノ基等が挙げられ、好ましくはN,N−ジメチルアミノ基、N,N−ジエチルアミノ基、N−エチル−N−メチルアミノ基等である。
【0024】
R1は、水素原子、ハロゲン原子、アミノ基、C1−6アルキル基、C1−6アルコキシ基、またはC1−6アルコキシC1−6アルキル基を意味し、特に、水素原子またはアミノ基が好ましい。
【0025】
R2は、水素原子、C1−6アルキル基、アミノ基、またはジC1−6アルキルアミノ基を意味し、水素原子またはアミノ基が好ましい。
【0026】
R3は、水素原子、ハロゲン原子、またはC1−6アルキル基を意味し、水素原子が好ましい。
【0027】
R4は、水素原子、ハロゲン原子、またはC1−6アルキル基を意味し、水素原子が好ましい。
【0028】
本明細書において使用する「塩」とは、一価のカウンターイオンまたは二価のカウンターイオンを形成できる化合物または原子との塩を意味する。具体的には、以下のものに限定されないが、無機酸(たとえば、塩酸、臭化水素酸、リン酸、硫酸、硝酸等)との塩、有機酸(たとえば、メタンスルホン酸、エタンスルホン酸、ベンゼンスルホン酸、p−トルエンスルホン酸、フマル酸、マレイン酸、コハク酸、クエン酸、リンゴ酸、またはトリフルオロ酢酸等)との塩、または、無機塩基(たとえば、ナトリウム塩、カリウム塩、カルシウム塩、リチウム塩等)との塩、有機塩基(たとえば、メチルアミン塩、エチルアミン塩、t−ブチルアミン塩、シクロヘキシルアミン塩、N−メチル−D−グルカミン塩、リジン塩、ピペリジンまたはモルホリン等)との塩を意味する。モノ−およびビス−塩は用語「塩」に含まれる。そして、本発明に係る化合物の塩にはその塩の無水物と水和物等のその塩の溶媒和物とが包含される。
【0029】
本明細書において使用する「抗真菌剤」は、真菌感染症の予防剤および/または治療剤を意味する。
【0030】
本発明に係る化合物は、慣用されている方法により錠剤、散剤、細粒剤、顆粒剤、被覆錠剤、カプセル剤、シロップ剤、トローチ剤、吸入剤、坐剤、注射剤、軟膏剤、眼軟膏剤、テープ剤、点眼剤、点鼻剤、点耳剤、パップ剤、ローション剤等として製剤化することができる。
【0031】
製剤化には通常用いられる賦形剤、結合剤、滑沢剤、着色剤、矯味矯臭剤や、および必要により安定化剤、乳化剤、吸収促進剤、界面活性剤、pH調整剤、防腐剤、抗酸化剤等を使用することができ、一般に医薬品製剤の原料として用いられる成分を配合して常法により製剤化される。例えば経口製剤を製造するには、本発明に係る化合物と賦形剤、さらに必要に応じて結合剤、崩壊剤、滑沢剤、着色剤、矯味矯臭剤等を加えた後、常法により散剤、細粒剤、顆粒剤、錠剤、被覆錠剤、カプセル剤等とする。
【0032】
これらの成分としては、例えば、大豆油、牛脂、合成グリセライド等の動植物油;例えば、流動パラフィン、スクワラン、固形パラフィン等の炭化水素;例えば、ミリスチン酸オクチルドデシル、ミリスチン酸イソプロピル等のエステル油;例えば、セトステアリルアルコール、ベヘニルアルコール等の高級アルコール;シリコン樹脂;シリコン油;例えば、ポリオキシエチレン脂肪酸エステル、ソルビタン脂肪酸エステル、グリセリン脂肪酸エステル、ポリオキシエチレンソルビタン脂肪酸エステル、ポリオキシエチレン硬化ひまし油、ポリオキシエチレンポリオキシプロピレンブロックコポリマー等の界面活性剤;例えば、ヒドロキシエチルセルロース、ポリアクリル酸、カルボキシビニルポリマー、ポリエチレングリコール、ポリビニルピロリドン、メチルセルロース等の水溶性高分子;例えば、エタノール、イソプロパノール等の低級アルコール;例えば、グリセリン、プロピレングリコール、ジプロピレングリコール、ソルビトール等の多価アルコール;例えば、グルコース、ショ糖等の糖;例えば、無水ケイ酸、ケイ酸アルミニウムマグネシウム、ケイ酸アルミニウム等の無機粉体、精製水等が挙げられる。賦形剤としては、例えば、乳糖、コーンスターチ、白糖、ブドウ糖、マンニトール、ソルビット、結晶セルロース、二酸化ケイ素等が、結合剤としては、例えば、ポリビニルアルコール、ポリビニルエーテル、メチルセルロース、エチルセルロース、アラビアゴム、トラガント、ゼラチン、シェラック、ヒドロキシプロピルメチルセルロース、ヒドロキシプロピルセルロース、ポリビニルピロリドン、ポリプロピレングリコール・ポリオキシエチレン・ブロックポリマー、メグルミン等が、崩壊剤としては、例えば、澱粉、寒天、ゼラチン末、結晶セルロース、炭酸カルシウム、炭酸水素ナトリウム、クエン酸カルシウム、デキストリン、ペクチン、カルボキシメチルセルロース・カルシウム等が、滑沢剤としては、例えば、ステアリン酸マグネシウム、タルク、ポリエチレングリコール、シリカ、硬化植物油等が、着色剤としては医薬品に添加することが許可されているものが、矯味矯臭剤としては、例えば、ココア末、ハッカ脳、芳香散、ハッカ油、竜脳、桂皮末等が用いられる。これらの錠剤・顆粒剤には糖衣、その他、必要により適宜コーティングすることはもちろん差支えない。また、シロップ剤や注射用製剤等の液剤を製造する際には、本発明に係る化合物にpH調整剤、溶解剤、等張化剤等と、必要に応じて溶解補助剤、安定化剤等を加えて、常法により製剤化する。外用剤を製造する際の方法は限定されず、常法により製造することができる。すなわち、製剤化にあたり使用する基剤原料としては、医薬品、医薬部外品、化粧品等に通常使用される各種原料を用いることが可能である。使用する基剤原料として具体的には、例えば、動植物油、鉱物油、エステル油、ワックス類、高級アルコール類、脂肪酸類、シリコン油、界面活性剤、リン脂質類、アルコール類、多価アルコール類、水溶性高分子類、粘土鉱物類、精製水等の原料が挙げられ、さらに必要に応じ、例えば、pH調整剤、抗酸化剤、キレート剤、防腐防黴剤、着色料、香料等を添加することができるが、本発明に係る外用剤の基剤原料はこれらに限定されない。また、必要に応じて分化誘導作用を有する成分、血流促進剤、殺菌剤、消炎剤、細胞賦活剤、ビタミン類、アミノ酸、保湿剤、角質溶解剤等の成分を配合することもできる。なお、上記基剤原料の添加量は、通常外用剤の製造にあたり設定される濃度になる量である。
【0033】
本発明に係る化合物を投与する場合、その形態は特に限定されず、通常用いられる方法により経口投与でも非経口投与でもよい。例えば、錠剤、散剤、顆粒剤、カプセル剤、シロップ剤、トローチ剤、吸入剤、坐剤、注射剤、軟膏剤、眼軟膏剤、テープ剤、点眼剤、点鼻剤、点耳剤、パップ剤、ローション剤等の剤として製剤化し、投与することができる。
【0034】
本発明に係る医薬の投与量は、症状の程度、年齢、性別、体重、投与形態・塩の種類、疾患の具体的な種類等に応じて適宜選ぶことができる。
【0035】
投与量は患者の、疾患の種類、症状の程度、患者の年齢、性差、薬剤に対する感受性差等により著しく異なるが、経口剤の場合は、通常成人として1日あたり、1−10000mg、好ましくは10−2000mgを1日1−数回に分けて投与する。注射剤の場合は、通常成人として1日あたり、通常0.1mg−10000mgであり、好ましくは1mg−2000mgである。
【0036】
[一般的製造方法]
式(I)で表される化合物(以下、化合物(I)という。)の製造方法について説明する。なお、後述する製造方法では、化合物(I−1)、(I−2)および(I−3)は化合物(I)に包含される化合物の代表例として説明する。
リン酸エステルの一般的製造方法
[製造方法1] 化合物(I−1)の製造方法
【0037】
【化5】
〔式中、R1、R3、R4は前記定義と同意義を意味する(ただし、R1が、アミノ基の場合を除く)。
【0038】
化合物(I−1−1)は、後述する参考例等に記載の方法を用いて製造することができる。また、化合物(I−1−1)は国際公開WO 2007/052615 A1公報に記載された方法等により製造することもできる。
【0039】
[工程1−1]
本工程は、化合物(I−1−1)とジ−tert−ブチル ジカーバメートを塩基触媒存在下に反応させて化合物(I−1−2)を得る工程である。
化合物(I−1−1)とジ−tert−ブチル ジカーバメートを反応させる時に用いる溶媒としては、出発原料をある程度溶解するものであり、かつ、反応を阻害しないものであれば、特に制限はないが、例えば、塩化メチレン、クロロホルムなどのハロゲン化炭化水素系溶媒;テトラヒドロフラン、ジエチルエーテルなどのエーテル系溶媒;酢酸エチルなどのエステル系溶媒;アセトニトリル、テトラメチレンスルホランまたはこれらの混合溶媒などを用いることができる。ジ−tert−ブチル ジカーバメートは化合物(I−1−1)に対して2当量から20当量用いる。塩基触媒としては、例えば4−ジメチルアミノピリジンを0.001当量から0.3当量用いる。この際トリエチルアミンなどの有機塩基を1当量から2当量添加してもよい。反応温度は0℃から60℃であり、好ましくは4℃から室温である。反応時間は1時間から72時間である。
【0040】
[工程1−2]
本工程は、化合物(I−1−2)とホスホリック アシッド ジ−tert−ブチル エステル クロロメチル エステルをヨウ化ナトリウム存在下に反応させた後、酸処理により化合物(I−1)を得る工程である。
化合物(I−1−2)とホスホリック アシッド ジ−tert−ブチル エステル クロロメチル エステルをヨウ化ナトリウム存在下に反応させる時に用いる溶媒としては、出発原料をある程度溶解するものであり、かつ、反応を阻害しないものであれば、特に制限はないが、例えば、塩化メチレン、クロロホルムなどのハロゲン化炭化水素系溶媒;テトラヒドロフラン、ジエチルエーテルなどのエーテル系溶媒;酢酸エチルなどのエステル系溶媒;アセトニトリル、テトラメチレンスルホランまたはこれらの混合溶媒などを用いることができ、好ましくはテトラヒドロフランあるいはアセトニトリルを用いる。ホスホリック アシッド ジ−tert−ブチル エステル クロロメチル エステルは化合物(I−1−2)に対して1当量から10当量用いることができ、好ましくは1当量から2当量用いる。ヨウ化ナトリウムは化合物(I−1−2)に対して1当量から10当量用いることができ、好ましくは1当量から2当量用いる。反応温度は0℃から60℃であり、好ましくは4℃から室温である。反応時間は1時間から72時間である。
酸処理に用いる酸としては、例えばトリフルオロ酢酸などの有機酸、塩酸などの鉱酸を用いることができ、好ましくはトリフルオロ酢酸を用いる。酸処理に際しては、前段階の反応溶媒に酸をそのまま加えてもよいし、一旦溶媒を減圧濃縮した後、適切な溶媒たとえばジクロロメタンなどに変えて酸を加えてもよい。反応温度は−10℃から室温であり、反応時間は5分間から2時間である。
【0041】
[製造方法2] 化合物(I−2)の製造方法
【0042】
【化6】
〔式中、R2、R3、R4は前記定義と同意義を意味する(ただし、R2が、アミノ基の場合を除く)。
【0043】
化合物(I−2−1)は国際公開WO 2007/052615 A1公報に記載された方法等により製造することができる。
【0044】
[工程2−1]
本工程は、化合物(I−2−1)とジ−tert−ブチル ジカーバメートを塩基触媒存在下に反応させて化合物(I−2−2)を得る工程である。
化合物(I−2−1)とジ−tert−ブチル ジカーバメートを反応させる時に用いる溶媒としては、出発原料をある程度溶解するものであり、かつ、反応を阻害しないものであれば、特に制限はないが、例えば、塩化メチレン、クロロホルムなどのハロゲン化炭化水素系溶媒、テトラヒドロフラン、ジエチルエーテルなどのエーテル系溶媒、酢酸エチルなどのエステル系溶媒、アセトニトリル、テトラメチレンスルホランまたはこれらの混合溶媒などを用いることができる。ジ−tert−ブチル ジカーバメートは化合物(I−2−1)に対して2当量から20当量用いる。塩基触媒としては、例えば4−ジメチルアミノピリジンを0.001当量から0.3当量用いる。この際トリエチルアミンなどの有機塩基を1当量から2当量添加してもよい。反応温度は0℃から60℃であり、好ましくは4℃から室温である。反応時間は1時間から72時間である。
【0045】
[工程2−2]
本工程は、化合物(I−2−2)とホスホリック アシッド ジ−tert−ブチル エステル クロロメチル エステルをヨウ化ナトリウム存在下に反応させた後、酸処理により化合物(I−2)を得る工程である。
化合物(I−2−2)とホスホリック アシッド ジ−tert−ブチル エステル クロロメチル エステルをヨウ化ナトリウム存在下に反応させる時に用いる溶媒としては、出発原料をある程度溶解するものであり、かつ、反応を阻害しないものであれば、特に制限はないが、例えば、塩化メチレン、クロロホルムなどのハロゲン化炭化水素系溶媒;テトラヒドロフラン、ジエチルエーテルなどのエーテル系溶媒;酢酸エチルなどのエステル系溶媒;アセトニトリル、テトラメチレンスルホランまたはこれらの混合溶媒などを用いることができ、好ましくはテトラヒドロフランあるいはアセトニトリルを用いる。ホスホリック アシッド ジ−tert−ブチル エステル クロロメチル エステルは化合物(I−2−2)に対して1当量から10当量用いることができ、好ましくは1当量から2当量用いる。ヨウ化ナトリウムは化合物(I−2−2)に対して1当量から10当量用いることができ、好ましくは1当量から2当量用いる。反応温度は0℃から60℃であり、好ましくは4℃から室温である。反応時間は1時間から72時間である。
酸処理に用いる酸としては、例えばトリフルオロ酢酸などの有機酸、塩酸などの鉱酸を用いることができ、好ましくはトリフルオロ酢酸を用いる。酸処理に際しては、前段階の反応溶媒に酸をそのまま加えてもよいし、一旦溶媒を減圧濃縮した後、適切な溶媒たとえばジクロロメタンなどに変えて酸を加えてもよい。反応温度は−10℃から室温であり、反応時間は5分間から2時間である。
【0046】
[製造方法3] 化合物(I−3)の製造方法
【0047】
【化7】
〔式中、R3、R4は前記定義と同意義を意味する。
【0048】
化合物(I−3−1)は国際公開WO 2007/052615 A1公報に記載された方法等により製造することができる。
【0049】
[工程3−1]
本工程は、化合物(I−3−1)とジ−tert−ブチル ジカーバメートを塩基触媒存在下に反応させて化合物(I−3−2)を得る工程である。本工程において、化合物(I−3−2)は、一段階の反応、または一方のアミノ基のジ−tert−ブチル カーバメート体を中間体として経由した他段階の反応でも得ることができる。
化合物(I−3−1)とジ−tert−ブチル ジカーバメートを反応させる時に用いる溶媒としては、出発原料をある程度溶解するものであり、かつ、反応を阻害しないものであれば、特に制限はないが、例えば、塩化メチレン、クロロホルムなどのハロゲン化炭化水素系溶媒;テトラヒドロフラン、ジエチルエーテルなどのエーテル系溶媒;酢酸エチルなどのエステル系溶媒;アセトニトリル、テトラメチレンスルホランまたはこれらの混合溶媒などを用いることができる。ジ−tert−ブチル ジカーバメートは化合物(I−3−1)に対して2当量から20当量用いる。塩基触媒としては4−ジメチルアミノピリジンを0.001当量から0.3当量用いる。この際トリエチルアミンなどの有機塩基を1当量から4当量添加してもよい。反応温度は0℃から60℃であり、好ましくは4℃から室温である。反応時間は1時間から72時間である。
【0050】
[工程3−2]
本工程は、化合物(I−3−2)とホスホリック アシッド ジ−tert−ブチル エステル クロロメチル エステルをヨウ化ナトリウム存在下に反応させた後、酸処理により化合物(I−3)を得る工程である。
化合物(I−3−2)とホスホリック アシッド ジ−tert−ブチル エステル クロロメチル エステルをヨウ化ナトリウム存在下に反応させる時に用いる溶媒としては、出発原料をある程度溶解するものであり、かつ、反応を阻害しないものであれば、特に制限はないが、例えば、塩化メチレン、クロロホルムなどのハロゲン化炭化水素系溶媒;テトラヒドロフラン、ジエチルエーテルなどのエーテル系溶媒;酢酸エチルなどのエステル系溶媒;アセトニトリル、テトラメチレンスルホランまたはこれらの混合溶媒などを用いることができ、好ましくはテトラヒドロフランあるいはアセトニトリルを用いる。ホスホリック アシッド ジ−tert−ブチル エステル クロロメチル エステルは化合物(I−3−2)に対して1当量から10当量用いることができ、好ましくは1当量から2当量用いる。ヨウ化ナトリウムは化合物(I−3−2)に対して1当量から10当量用いることができ、好ましくは1当量から2当量用いる。反応温度は0℃から60℃であり、好ましくは4℃から室温である。反応時間は1時間から72時間である。
酸処理に用いる酸としては、例えばトリフルオロ酢酸などの有機酸、塩酸などの鉱酸を用いることができ、好ましくはトリフルオロ酢酸を用いる。酸処理に際しては、前段階の反応溶媒に酸をそのまま加えてもよいし、一旦溶媒を減圧濃縮した後、適切な溶媒たとえばジクロロメタンなどに変えて酸を加えてもよい。反応温度は−10℃から室温であり、反応時間は5分間から2時間である。
【実施例】
【0051】
本発明に係る化合物は、例えば以下の実施例、参考例および製造例等に記載した方法により製造することができる。ただし、これらは例示的なものであって、本発明に係る化合物は如何なる場合も以下の具体例に限定されるものではない。
なお、実施例、参考例および製造例等の記載中で用いる略号の意味は以下のとおりである。
TFA:トリフルオロ酢酸
【0052】
[参考例1]3−(3−(4−(ピリジン−2−イルメトキシ)−ベンジル)−イソキサゾール−5−イル)−ピリジン−2−イルアミン
【0053】
【化8】
製造例1−1−5に記載の4−(5−(2−アミノ−ピリジン−3−イル)イソキサゾール−3−イルメチル)−フェノール(4.2mg、0.016mmol)とメタノール(0.4mL)の混合物に、1N 水酸化ナトリウム水溶液(16μL、0.016mmol)を加えた後、減圧下濃縮した。残渣とN,N−ジメチルホルムアミド(0.5mL)の混合物に2−ピコリル クロリド(3.1mg、0.019mmol)を加え、室温で2時間攪拌した。反応混合物をそのまま逆相系高速液体クロマトグラフィー(アセトニトリル−水系移動相(0.1%トリフルオロ酢酸含有)を用いた)にて精製し、標記化合物(3.6mg, 39%)をジトリフルオロ酢酸塩として得た。
MS m/e(ESI) 359.16(MH+)
【0054】
また、別法として以下の方法で参考例1の化合物を得た。
製造例1−1−5に記載の4−(5−(2−アミノ−ピリジン−3−イル)イソキサゾール−3−イルメチル)−フェノール(2.97g、11.1mmol)、テトラヒドロフラン(100mL)とアセトン(100ml)の混合物に、5N 水酸化ナトリウム水溶液(2.22mL、11.1mmol)を加えた。反応混合物に超音波を30秒間照射した後、減圧下濃縮した。残渣とN,N−ジメチルホルムアミド(50mL)の混合物に2−ピコリル クロリド(3.64g、22.2mmol)を加え、60℃で2.5時間攪拌した。反応混合物を室温に戻し、水でクエンチした後、酢酸エチルにて抽出した。その有機層を水と飽和食塩水で洗浄し、無水硫酸マグネシウムにて乾燥した。その溶媒を減圧留去し、その残渣をNH−シリカゲルクロマトグラフィー(ヘプタン:酢酸エチル=1:1)にて精製し、標記化合物(2.73g, 67%)を得た。
1H−NMR Spectrum (CDCl3)δ(ppm): 4.00(2H, s), 5.20(2H,s), 5.37(2H, brs), 6.24(1H, s), 6.71(1H, dd, J=4.8,7.6Hz), 6.95−6.97(2H, m), 7.20−7.22(2H, m), 7.52(d,1H, d, J=1.9Hz), 7.69−7.74(3H, m), 8.13-8.15(1H, m), 8.60(1H, d, J=4.4Hz).
【0055】
出発物質4−(5−(2−アミノ−ピリジン−3−イル)イソキサゾール−3−イルメチル)−フェノールは以下の方法で合成した。
【0056】
[製造例1−1−1]1−ベンジルオキシ−4−((E)−2−ニトロ−ビニル)−ベンゼン
【0057】
【化9】
4−ベンジルオキシベンズアルデヒド(1.0g、4.7mmol)、ナトリウムメトキシド(28%メタノール溶液、150μL、0.74mmol)、およびメタノール(10mL)の混合物に、0℃でニトロメタン(330μL、6.1mmol)とナトリウムメトキシド(28%メタノール溶液、1.0mL、4.9mmol)を加え、室温で10分間攪拌した。反応混合物を0℃に冷却し、同温で5N 塩酸水溶液(20mL)を加えた。反応混合物を室温で15分間攪拌した。析出した固体をろ取し、標記化合物(1.2g、100%)を得た。
1H−NMR Spectrum (DMSO−d6)δ(ppm):5.20(2H,s), 7.10−7.14(2H,m), 7.32−7.48(5H,m), 7.82−7.85(2H,m), 8.12(2H,dd,J=13.5,18.2Hz).
【0058】
[製造例1−1−2]1−ベンジルオキシ−4−(2−ニトロ−エチル)−ベンゼン
【0059】
【化10】
製造例1−1−1に記載の1−ベンジルオキシ−4−((E)−2−ニトロ−ビニル)−ベンゼン(1.0g、3.9mmol)、酢酸(1mL)、およびジメチルスルホキシド(17mL)の混合物に、適宜冷却しながら室温で水素化ホウ素ナトリウム(250mg、6.3mmol)を加えた。室温で40分間攪拌し、反応混合物に水を加えた。反応混合物を酢酸エチルと水に分配した。有機層を水および飽和食塩水で洗浄し、それを無水硫酸マグネシウムで乾燥し、その溶媒を減圧下留去した。残渣をNHシリカゲルカラムクロマトグラフィー(酢酸エチル:ヘプタン=1:3)で精製し、標記化合物(710mg、70%)を得た。
1H−NMR Spectrum (CDCl3)δ(ppm):3.26(2H,t,J=7.2Hz), 4.56(2H,t,J=7.2Hz), 5.04(2H,s), 6.92(2H,d,J=8.4Hz), 7.11(2H,d,J=8.8Hz),7.30−7.42(5H,m).
【0060】
[製造例1−1−3]4−ベンジルオキシ−フェニル−アセトヒドロキシモイル クロリド
【0061】
【化11】
製造例1−1−2に記載の1−ベンジルオキシ−4−(2−ニトロ−エチル)−ベンゼン(340mg、1.3mmol)とメタノール(5mL)の混合物に、室温でリチウムメトキシド(100mg、2.6mmol)を加え、室温で15分間攪拌した。反応混合物を減圧下濃縮した。残渣に塩化メチレン(4mL)とテトラヒドロフラン(2mL)を加えた。反応混合物に−78℃でチタニウム(IV)クロリドを加え、0℃で50分間攪拌した。反応混合物を−78℃に冷却後、水(5mL)を加え、徐々に室温まで昇温させた。反応混合物を酢酸エチルと水に分配した。有機層を飽和食塩水で洗浄し、その溶媒を減圧下留去した。残渣を中性シリカゲルカラムクロマトグラフィー(酢酸エチル:ヘプタン=1:3)で精製し、標記化合物(310mg、84%)を得た。
1H−NMR Spectrum (CDCl3)δ(ppm):3.83(2H,s), 5.07(2H,s), 6.94−6.98(2H,m), 7.17−7.21(2H,m), 7.32−7.44(5H,m).
【0062】
[製造例1−1−4]3−(3−(4−ベンジルオキシ−ベンジル)−イソキサゾール−5−イル)−ピリジン−2−イルアミン
【0063】
【化12】
製造例1−1−3に記載の4−ベンジルオキシ−フェニル−アセトヒドロキシモイル クロリド(1.2g、4.4mmol)とテトラヒドロフラン(34mL)の混合物に、0℃で製造例1−2−5に記載の3−エチニル−ピリジン−2−イルアミン(260mg、2.2mmol)とトリエチルアミン(3.0mL、22mmol)を加え、室温で1時間攪拌した。反応混合物に室温で水を加え、酢酸エチル−テトラヒドロフラン(2:1)で抽出した。有機層を飽和食塩水で洗浄し、その溶媒を減圧下留去した。残渣をNHシリカゲルカラムクロマトグラフィー(酢酸エチル:ヘプタン=1:3)で精製し、標記化合物(240mg、15%)を得た。
1H−NMR Spectrum (CDCl3)δ(ppm):4.00(2H,s), 5.05(2H,s), 5.41(2H,s), 6.24(1H,s), 6.71(1H,dd,J=4.9,7.6Hz), 6.93−6.97(2H,m), 7.18−7.22(2H,m), 7.31−7.44(5H,m), 7.70(1H,dd,J=1.7,7.6Hz), 8.13(1H,dd,J=1.8,4.9Hz).
【0064】
[製造例1−1−5]4−(5−(2−アミノ−ピリジン−3−イル)イソキサゾール−3−イルメチル)−フェノール
【0065】
【化13】
製造例1−1−4に記載の3−(3−(4−ベンジルオキシ−ベンジル)−イソキサゾール−5−イル)−ピリジン−2−イルアミン(32mg、0.090mmol)とトリフルオロ酢酸(1mL)の混合物に、室温でチオアニソール(45mg、0.36mmol)を加え、同温で2時間攪拌した。飽和炭酸水素ナトリウム水溶液と酢酸エチルの混合物に反応混合物を加えた。有機層を分離し、飽和食塩水で洗浄し、その溶媒を減圧下留去した。残渣をシリカゲルカラムクロマトグラフィー(酢酸エチル:ヘプタン=4:1)で精製し、標記化合物(24mg、100%)を得た。
1H−NMR Spectrum (DMSO−d6)δ(ppm):3.90(2H,s), 6.25(2H,brs), 6.68−6.72(3H,m), 6.76(1H,s), 7.11(2H,d,J=8.6Hz), 7.87(1H,dd,J=1.5,7.7Hz), 8.10(1H,brs), 9.29(1H,s).
【0066】
出発物質3−エチニル−ピリジン−2−イルアミンは以下の方法で合成した。
【0067】
[製造例1−2−1]2,2−ジメチル−N−ピリジン−2−イル−プロピオナミド
【0068】
【化14】
2−アミノピリジン(50.0g、531mmol)の塩化メチレン(500mL)溶液に、0℃でトリエチルアミン(81.4mL、584mmol)、ピバロイル クロライド(71.9mL、584mmol)を加え、室温で4時間30分攪拌した。反応溶液を水と塩化メチレンに分配した。有機層を水と飽和食塩水で洗浄し、無水硫酸マグネシウムで乾燥し、その溶媒を減圧下留去した。得られた残渣のメタノール(300mL)溶液に、0℃で炭酸カリウム(73.4g、531mmol)を加え、室温で90分間攪拌した。反応溶液を室温で水と酢酸エチルに分配した。有機層を飽和食塩水で洗浄し、無水硫酸マグネシウムで乾燥し、その溶媒を減圧下留去した。残渣にヘプタン(300mL)を加え、析出した固体をろ取し、標記化合物(80.2g、85%)を得た。更に、ろ液を減圧下濃縮し、残渣をシリカゲルカラムクロマトグラフィー(ヘプタン:酢酸エチル=2:1)にて精製し、標記化合物(12.2g、13%)を得た。
1H−NMR Spectrum (DMSO−d6)δ(ppm):1.22(9H,s), 7.06−7.09(1H,m), 7.72−7.77(1H,m), 8.01−8.03(1H,m), 8.29−8.31(1H,m), 9.71(1H,s).
【0069】
[製造例1−2−2]N−(3−ヨード−ピリジン−2−イル)−2、2−ジメチル−プロピオナミド
【0070】
【化15】
製造例1−2−1に記載の2、2−ジメチル−N−ピリジン−2−イル−プロピオナミド(3.0g、17mmol)、N,N,N’,N’−テトラメチルエチレンジアミン(6.3mL、42mmol)、およびテトラヒドロフラン(60mL)の混合物に−78℃でn−ブチルリチウム(1.6M n−ヘキサン溶液、30mL、47mmol)を滴下し、0℃で終夜攪拌した。反応混合物に−78℃でヨウ素(6.8g、27mmol)を加え、0℃で1.5時間攪拌した。反応混合物に水と飽和チオ硫酸ナトリウム水溶液を加え、酢酸エチルで抽出した。有機層を飽和食塩水で洗浄し、その溶媒を減圧下留去した。残渣をシリカゲルカラムクロマトグラフィー(酢酸エチル:ヘプタン=2:1)にて精製し、標記化合物(2.9g、57%)を得た。
1H−NMR Spectrum (CDCl3)δ(ppm):1.38(9H,s), 6.85(1H,dd,J=4.8,7.9Hz), 7.94(1H,brs), 8.11(1H,dd,J=1.7,7.9Hz), 8.46(1H,dd,J=1.7,4.6Hz).
【0071】
[製造例1−2−3]3−ヨード−ピリジン−2−イルアミン
【0072】
【化16】
製造例1−2−2に記載のN−(3−ヨード−ピリジン−2−イル)−2、2−ジメチル−プロピオナミド(66.2g、218mmol)、5N 水酸化ナトリウム水溶液(200mL)、メタノール(200mL)の混合物を加熱還流下、1時間20分間撹拌した。反応溶液を室温に戻し、水と酢酸エチルに分配した。水層を酢酸エチルで3回抽出した。有機層を合わせ、飽和食塩水で洗浄し、それを無水硫酸ナトリウムで乾燥した。硫酸ナトリウムをろ過で除き、その溶媒を減圧下濃縮して標記化合物(41.2g、85.9%)を得た。
1H−NMR Spectrum (DMSO−d6)δ(ppm):6.00(2H,brs), 6.32(1H,dd,J=4.8Hz,7.2Hz), 7.87(1H,d,J=7.2Hz), 7.92(1H,d,J=4.8Hz).
【0073】
[製造例1−2−4]3−トリメチルシラニルエチニル−ピリジン−2−イルアミン
【0074】
【化17】
製造例1−2−3に記載の3−ヨード−ピリジン−2−イルアミン(40.2g、183mmol)、トリメチルシリルアセチレン(51.7mL、366mmol)、ヨウ化銅(I)(3.49g、18.3mmoL)、N,N−ジイソプロピルエチルアミン(63.7mL、366mmol)、N−メチルピロリジノン(200mL)の混合物にテトラキス(トリフェニルホスフィン)パラジウム(0)(10.6g、9.15mmol)を加え、窒素気流下、室温で3時間10分撹拌した。反応溶液に水を加え酢酸エチルで4回抽出した。その溶媒を減圧下濃縮した。残渣をNHシリカゲルクロマトグラフィー(ヘプタン:酢酸エチル=4:1)で精製した。得られた溶液を減圧下濃縮し、残渣をシリカゲルクロマトグラフィー(ヘプタン:酢酸エチル=2:1ついで1:1)で精製し標記化合物(28.1g、80.7%)を得た。
1H−NMR Spectrum (DMSO−d6)δ(ppm): 0.25(9H,s), 6.09(2H,brs), 6.51−6.57(1H,m), 7.50−7.55(1H,m), 7.95−7.99(1H,m).
【0075】
[製造例1−2−5]3−エチニル−ピリジン−2−イルアミン
【0076】
【化18】
製造例1−2−4に記載の3−トリメチルシラニルエチニル−ピリジン−2−イルアミン(28.1g、148mmoL)のテトラヒドロフラン(300mL)溶液にテトラブチルアンモニウム フルオリド(1M テトラヒドロフラン溶液、20mL、20mmol)、を加え室温で15分撹拌した。反応溶液に水を加え酢酸エチルで4回抽出した。有機層を無水硫酸ナトリウムで乾燥し、その溶媒を減圧下留去した。残渣をシリカゲルクロマトグラフィー(ヘプタン:酢酸エチル=1:1ついで1:2)で精製し標記化合物(16.4g、93.7%)を得た。
1H−NMR Spectrum(DMSO−d6)δ(ppm):4.43(1H,s), 6.14(2H,brs), 6.53(1H,dd,J=4.8Hz,7.2Hz), 7.53(1H,d,J=7.2Hz),7.96(1H,d,J=4.8Hz).
【0077】
[製造例1−3−1]3−トリメチルシラニルエチニル−ピリジン−2−イルアミン(製造例1−2−4の別法)
【0078】
【化19】
2−アミノ−3−ブロモピリジン(5.72g、33.1mmol)のN−メチルピロリジノン(120mL)溶液に、室温でトリメチルシリルアセチレン(9.36mL、66.2mmol)、テトラキス(トリフェニルホスフィン)パラジウム(0)(1.91g、1.66mmol)、ヨウ化銅(I)(630mg、3.31mmol)、N,N−ジイソプロピルエチルアミン(11.5mL、66.2mmol)を加え、窒素雰囲気下、70℃で6時間攪拌した。反応溶液に水を加え、酢酸エチルで抽出した。有機層を水と飽和食塩水で洗浄し、無水硫酸マグネシウムで乾燥し、その溶媒を減圧下留去した。残渣をシリカゲルカラムクロマトグラフィー(ヘプタン:酢酸エチル=2:1)で精製し、標記化合物(5.94g、94%)を得た。
1H−NMR Spectrum (DMSO−d6)δ(ppm):0.23(9H,s), 6.07(2H,brs), 6.51(1H,dd,J=4.9,7.5Hz), 7.49(1H,dd,J=1.8,7.5Hz), 7.94(1H,dd,J=1.8,4.9Hz).
【0079】
[実施例1]2−((4−((5−(2−アミノ−3−ピリジニル)−3−イソキサゾリル)メチル)フェノキシ)メチル)−1−((ホスホノオキシ)メチル)ピリジニウム 1トリフルオロ酢酸塩
【0080】
【化20】
製造例1−4−1に記載のジ−tert−ブチル−(3−(3−(4−(ピリジン−2−イルメトキシ)ベンジル)イソキサゾール−5−イル)ピリジン−2−イル)イミドジカルボネート(334mg、0.60mmol)、ホスホリック アシッド ジ−tert−ブチル エステル クロロメチル エステル(309mg、1.2mmol)、ヨウ化ナトリウム(134mg、0.90mmol)テトラヒドロフラン(0.6ml)を室温で3.5時間撹拌し、ついで、この反応溶液にトリフルオロ酢酸(2ml)を加えさらに40分室温で撹拌した。反応溶液を減圧濃縮し、残渣に重曹水と酢酸エチルを加え分液した。水層に酢酸エチルを加え再度分液した。得られた重曹水をゲルろ過(CHP20P(三菱化成)、水ついでメタノールで溶出)し、ついで溶出液を、液量が10mlくらいになるまで濃縮した。得られた溶液をODSカラム(H2O:MeOH:TFA=500:50:0.5ついで500:200:0.7)で精製した。溶媒を濃縮し、残渣を少量のアセトンに溶かした後、酢酸エチルを加え濃縮して標記化合物(53.93mg)を粉末状の固体として得た。
1H−NMR Spectrum (DMSO-d6)δ(ppm):4.02(2H,s), 5.73(2H, s), 6.33(2H, d, J=13.5Hz), 6.84(1H, dd, J=5.2, 7.6Hz), 6.89(1H, s), 7.13(2H, d, J=8.8Hz), 7.33(2H, d, J=8.8Hz), 8.07(1H, dd, J=1.6, 7.6Hz), 8.12(1H, dd, J=1.6, 5.2Hz), 8.15-8.22(1H, m), 8.28(1H, d, J=7.6Hz), 8.73(1H, ddd, J=1.6, 7.6, 7.6Hz), 9.22(1H, d=4.8Hz).
【0081】
出発物質ジ−tert−ブチル−(3−(3−(4−(ピリジン−2−イルメトキシ)ベンジル)イソキサゾール−5−イル)ピリジン−2−イル)イミドジカルボネートは以下の方法で合成した。
【0082】
[製造例1−4−1]ジ−tert−ブチル−(3−(3−(4−(ピリジン−2−イルメトキシ)ベンジル)イソキサゾール−5−イル)ピリジン−2−イル)イミドジカルボネート
【0083】
【化21】
参考例1に記載の3−(3−(4−(ピリジン−2−イルメトキシ)ベンジル)イソキサゾール−5−イル)ピリジン−2−イルアミン(300mg、0.84mmol)、ジ−tert−ブチルジカルボネート(913mg、4.2mmol)、4−ジメチルアミノピリジン(10mg、0.084mmol)、トリエチルアミン(102mg、1.0mmol)、テトラヒドロフラン(7ml)を室温で13.5時間撹拌した。反応溶液をNH−シリカゲルクロマトグラフィー(ヘプタン:酢酸エチル=1:1ついで1:2)で精製し標記化合物(334mg)を得た。
1H−NMR Spectrum (CDCl3)δ(ppm):1.24(18H, s), 3.99(2H, s), 5.18(2H, s), 6.32(1H, s), 6.90-6.95(2H, m), 7.16-7.24(3H, m), 7.41(1H, dd, 4.8, 8.0Hz), 7.51(1H, d, 8.0Hz), 7.71(1H, ddd, J=2.0, 8.0, 8.0Hz), 8.27(1H, dd, J=2.0, 8.0Hz), 8.56-8.61(2H, m).
【0084】
式(I)で表される本発明に係る化合物は、優れた抗真菌活性を有する活性体である親化合物に速やかに変換され、さらに、物性、特に水への溶解性および水溶液中での安定性、ならびに安全性の面でも優れており、真菌感染症の予防剤または治療剤として極めて有用である。
【0085】
1.水への溶解性の比較試験例
親化合物である参考例1に記載の3−(3−(4−(ピリジン−2−イルメトキシ)ベンジル)イソキサゾール−5−イル)ピリジン−2−イルアミンと実施例1の化合物を、25℃において、Britton-Robinson緩衝液(イオン強度0.3)への溶解度を比較した。表1はその結果を示す。
【0086】
【表1】
【0087】
表1に示す結果から明らかなように、実施例1の化合物は、各種pH領域において、その親化合物よりも水への溶解性が顕著に増大していることが判明した。
【0088】
2.肝S9画分での親化合物(活性体)への変換
(1).各種肝S9反応溶液の調製
ヒトおよびサル肝S9画分(タンパク濃度 0.22 mg/mL)、0.5 mmol/L塩化マグネシウム、100 mmol/L Tris-HClを含む懸濁液(pH7.4)を氷上で調製した(各種反応溶液A)。各種反応溶液A 0.27 mLに本発明に係る化合物(実施例1の化合物) 100 μmol/mL水溶液を30μL添加し(最終化合物濃度 10 μmol/L)各種肝S9反応溶液(最終タンパク濃度 0.2 mg/mL)とし、(2)の実施まで氷上で保存した。対照として、0.5 mmol/L塩化マグネシウム、100 mmol/L Tris-HCl を含む緩衝液(pH 7.4)0.27 mLに本発明に係る化合物(実施例1の化合物)100 μmol/mL水溶液を30μL添加したものを調製した(反応緩衝溶液)。
(2).各種肝S9反応溶液および反応緩衝溶液中での親化合物(参考例1の化合物)への変換
(1)の各種肝S9反応溶液および反応緩衝溶液を37℃でインキュベーションし、時間0,30および60分の時点で50 μLずつ採取しメタノール溶液100 μLを添加し反応を停止させた。
(3).本発明に係る化合物(実施例1の化合物)および親化合物(参考例1の化合物)の反応液中濃度はLC-MSにより定量した。
【0089】
2.に記載の測定法で、本発明に係る化合物(実施例1の化合物)および親化合物(参考例1の化合物)の反応液中濃度を測定した。図1ないし図3は、実施例1の化合物および参考例1の化合物の、各種肝S9反応溶液および反応緩衝溶液中での濃度の経時変化を示す。図1ないし図3に示す結果から、本発明に係る化合物(実施例1の化合物)はヒトおよびサル肝S9画分で親化合物(参考例1の化合物)に変換した。また、肝S9画分を含まない反応緩衝溶液中では、本発明に係る化合物(実施例1の化合物)から親化合物(参考例1の化合物)への変換は認められなかったことが確認された。
【0090】
3.抗カンジダ活性及び抗アスペルギルス活性
(1).菌液の調製
C. albicans CAF2−1株は、サブローデキストロース液体培地(SDB)に30℃、48時間静置培養した菌液をRPMI1640培地で希釈し、1.2x103cells/mLの菌液に調製した。A. fumigatus Tsukuba株は、−80℃凍結保存株をRPMI1640培地で希釈し、4.5x103cells/mLの菌液に調製した。
【0091】
(2).薬剤希釈プレートの作製
U底96wellプレートを用い、8検体/プレート(A〜H)の検体希釈溶液を作製した。各プレートの2〜12列目にジメチルスルホキシド溶液を10μL分注した。秤量した検体をジメチルスルホキシドに溶解し、2.5mg/mLの溶液を作製後、この溶液を準備したプレートの1列目に20μL添加し、プレート上で12段階2倍階段希釈(溶液10μL+ジメチルスルホキシド溶液10μL)した。この検体希釈溶液を1μLづつMIC測定用の平底96wellプレートに分注し、検体希釈プレートを作製した。
【0092】
(3).菌液の接種および培養
(1)で調製した菌液を、(2)で作製した被検化合物希釈液1μL/wellが入った平底96wellプレートに99μL/well接種し、35℃で42〜48時間、好気的に静置培養した。
【0093】
(4).MIC測定
一見して、コントロールと比較して菌の増殖を明らかに抑制した最小濃度を最小発育阻止濃度(MIC)とした。
【0094】
3に記載の測定法で、親化合物(参考例1の化合物)について、抗カンジダ活性及び抗アスペルギルス活性を測定した。その結果は、表2に示す。表2に示す結果から、親化合物(参考例1の化合物)は、抗カンジダ活性および抗アスペルギルス活性を有していることが確認された。
【0095】
【表2】
【産業上の利用可能性】
【0096】
本発明によれば、式(I)で表される本発明に係る化合物は、活性本体である親化合物のプロドラッグとして、1)真菌のGPI生合成阻害に基づいて細胞壁表層蛋白質の発現を阻害し、細胞壁assemblyを阻害するとともに真菌が細胞へ付着するのを阻害して、病原体が病原性を発揮できないようにすることより、感染症の発症、進展、持続に対して効果を示し、2)物性、特に、水への溶解性および水溶液中での安定性、ならびに体内動態および安全性の面でも優れており、真菌感染症の予防または治療剤として極めて有用である。
【特許請求の範囲】
【請求項1】
下式(I)で表される化合物またはその塩;
【化1】
式中、
R1が、水素原子、ハロゲン原子、アミノ基、C1−6アルキル基、C1−6アルコキシ基、またはC1−6アルコキシC1−6アルキル基を意味し;
R2が、水素原子、C1-6アルキル基、アミノ基、またはジC1-6アルキルアミノ基を意味し;
R3が、水素原子、ハロゲン原子、またはC1−6アルキル基を意味し;
R4が、水素原子、ハロゲン原子、またはC1−6アルキル基を意味する。
【請求項2】
R2がアミノ基である、請求項1に記載の化合物またはその塩。
【請求項3】
R1が、水素原子である、請求項1または2に記載の化合物またはその塩。
【請求項4】
R1が、アミノ基である、請求項1ないし2に記載の化合物またはその塩。
【請求項5】
R3が、水素原子であり、R4が、水素原子、ハロゲン原子、またはC1−6アルキル基である、請求項1ないし4のいずれか1項に記載の化合物またはその塩。
【請求項6】
下式
【化2】
で表される2−((4−((5−(2−アミノ−3−ピリジニル)−3−イソキサゾリル)メチル)フェノキシ)メチル)−1−((ホスホノオキシ)メチル)ピリジニウムの化合物またはその塩。
【請求項7】
請求項1ないし6のいずれか1項に記載の化合物またはその塩を含有する医薬組成物。
【請求項8】
請求項1ないし6のいずれか1項に記載の化合物またはその塩を含有する医薬。
【請求項9】
請求項1ないし6のいずれか1項に記載の化合物またはその塩を有効成分として含有する抗真菌剤。
【請求項10】
請求項1ないし6のいずれか1項に記載の化合物またはその塩の薬理学的有効量を投与して、真菌感染症を予防および/または治療する方法。
【請求項11】
抗真菌剤の製造のための、請求項1ないし6のいずれか1項に記載の化合物またはその塩の使用。
【請求項1】
下式(I)で表される化合物またはその塩;
【化1】
式中、
R1が、水素原子、ハロゲン原子、アミノ基、C1−6アルキル基、C1−6アルコキシ基、またはC1−6アルコキシC1−6アルキル基を意味し;
R2が、水素原子、C1-6アルキル基、アミノ基、またはジC1-6アルキルアミノ基を意味し;
R3が、水素原子、ハロゲン原子、またはC1−6アルキル基を意味し;
R4が、水素原子、ハロゲン原子、またはC1−6アルキル基を意味する。
【請求項2】
R2がアミノ基である、請求項1に記載の化合物またはその塩。
【請求項3】
R1が、水素原子である、請求項1または2に記載の化合物またはその塩。
【請求項4】
R1が、アミノ基である、請求項1ないし2に記載の化合物またはその塩。
【請求項5】
R3が、水素原子であり、R4が、水素原子、ハロゲン原子、またはC1−6アルキル基である、請求項1ないし4のいずれか1項に記載の化合物またはその塩。
【請求項6】
下式
【化2】
で表される2−((4−((5−(2−アミノ−3−ピリジニル)−3−イソキサゾリル)メチル)フェノキシ)メチル)−1−((ホスホノオキシ)メチル)ピリジニウムの化合物またはその塩。
【請求項7】
請求項1ないし6のいずれか1項に記載の化合物またはその塩を含有する医薬組成物。
【請求項8】
請求項1ないし6のいずれか1項に記載の化合物またはその塩を含有する医薬。
【請求項9】
請求項1ないし6のいずれか1項に記載の化合物またはその塩を有効成分として含有する抗真菌剤。
【請求項10】
請求項1ないし6のいずれか1項に記載の化合物またはその塩の薬理学的有効量を投与して、真菌感染症を予防および/または治療する方法。
【請求項11】
抗真菌剤の製造のための、請求項1ないし6のいずれか1項に記載の化合物またはその塩の使用。
【図1】

【図2】

【図3】

【図2】
【図3】
【公開番号】特開2012−176900(P2012−176900A)
【公開日】平成24年9月13日(2012.9.13)
【国際特許分類】
【出願番号】特願2009−149502(P2009−149502)
【出願日】平成21年6月24日(2009.6.24)
【出願人】(506137147)エーザイ・アール・アンド・ディー・マネジメント株式会社 (215)
【Fターム(参考)】
【公開日】平成24年9月13日(2012.9.13)
【国際特許分類】
【出願日】平成21年6月24日(2009.6.24)
【出願人】(506137147)エーザイ・アール・アンド・ディー・マネジメント株式会社 (215)
【Fターム(参考)】
[ Back to top ]