
((2S,4R)−4,6−ジヒドロキシテトラヒドロ−2H−ピラン−2−イル)メチルカルボキシラート及び2−デオキシリボース−5−リン酸アルドラーゼを使用の製造方法
本発明は、((2S,4R)−4,6−ジヒドロキシテトラヒドロ−2H−ピラン−2−イル)メチルカルボキシラート及びその作製方法に関する。さらに、本発明は、上記化合物が中間体として使用される、スタチン(特に、ロスバスタチン)及びその誘導体を作製するための方法に関する。

【発明の詳細な説明】
【技術分野】
【0001】
本発明は、((2S,4R)−4,6−ジヒドロキシテトラヒドロ−2H−ピラン−2−イル)メチルカルボキシラート及びその製造方法に関する。さらに、本発明は、上記化合物が中間体として使用される、スタチン(特に、ロスバスタチン)及びその誘導体の作製方法に関する。
【背景技術】
【0002】
((2S,4R)−4,6−ジヒドロキシテトラヒドロ−2H−ピラン−2−イル)メチルカルボキシラートは、スタチンの合成において中間体となり得る。スタチン(その代表例は、ロスバスタチン、セリバスタチン、アトルバスタチン、フルバスタチン、ピタバスタチン、ベルバスタチン、ダルバスタチン若しくはこれらの類縁体又はプラバスタチン、シンバスタチン、ロバスタチン若しくはこれらの類縁体から選択され得る。)は、それぞれ、芳香族又は脂環式の殻に接続されたヘプテン酸又はヘプタン酸部分(遊離の酸、塩又はラクトン)によって規定される特徴的構造を共有する。スタチンの生物活性は、その立体化学、特に、前記ヘプテン酸又はヘプタン酸部分のキラル原子における立体配置と密接に関連する。
【0003】
WO2006/134482には、アトルバスタチンを形成するための方法中に、2−デオキシリボース−5−リン酸アルドラーゼ(DERA)によって触媒されるアルドール付加工程が含まれる。
【0004】
日本国特許2005229858は、DERAの存在下で、ベンジルオキシアセトアルデヒドをアセトアルデヒドと反応させる、((4R,6S)−4,6−ジヒドロキシテトラヒドロ−2−ピロンを作製するための方法を開示する。酵素触媒の反応時間は、12時間であった。
【0005】
WO05/118794は、DERA酵素の改善を取り扱っている。極めて多様な置換基を有する2,4−ジデオキシヘキソース又は2,4,6−トリデオキシヘキソースを調製するために、単離された変異体酵素が使用され得る。
【0006】
DERA変異体は、立体特異的なアルドール反応を触媒することが記載された(Tetrahedron Letters2004,45,2439−2441)。DERA変異体は触媒活性の相対的改善を示し、従って、野生型DERAと比較して収率を改善した。酵素触媒の反応時間は6日であった。この酵素触媒から得られた1つの産物は、アトルバスタチンの合成に関して提案された。
【0007】
立体特異的アルドール反応を触媒するためのDERAは、「Proc. Nat. Acad. Sci. USA 2004, 101(16) 5788−5793」中にさらに記載されており、酵素プロセスの改善された容積生産性を示した。使用される基質の酵素活性に対する阻害的効果も記載されている。酵素触媒の反応時間は3時間であった。この酵素的触媒から得られた産物は、アトルバスタチン又はロスバスタチンの合成に関して提案された。
【0008】
2−デオキシリボース−5−リン酸アルドラーゼ(DERA)によって触媒される3つのアルデヒド基質との立体特異的アルドール反応は、全ての置換されたアセトアルデヒドをDERAに対する基質として等しく受容するわけではなく(Am. Chem. Soc;117,29,(1995)pp7585)、ある種の基質は、DERA活性に対して阻害的効果を示す。酵素的触媒の反応時間は6日であった。
【0009】
WO2007/039287A1では、ヨードラクトン合成を介したラクトン化されたスタチン側鎖中間体VIの合成が記載されており、これには、6つの有機合成工程が必要とされる。この複数工程の合成において、4番目の工程は、ヨードラクトン中間体産物の立体化学を規定するラクトン形成工程である。このラクトン形成工程は、相対的に低い立体選択性を与えるに過ぎない。I化合物、Ag化合物、グリニャール試薬及び鏡像異性体的に純粋な出発化合物のような化学量論的量で使用される幾つかの試薬は、極めて高価である。(以下に示されている)有機合成の6つの工程は、19%の総収率を得られた。
【0010】
【化1】
【先行技術文献】
【特許文献】
【0011】
【特許文献1】国際公開第2006/134482号
【特許文献2】特許第2005229858号明細書
【特許文献3】国際公開第05/118794号
【特許文献4】国際公開第2007/039287号
【非特許文献】
【0012】
【非特許文献1】Tetrahedron Letters、45、2004年、pp.2439−2441
【非特許文献2】Proc. Nat. Acad. Sci. USA 101(16)、2004年、pp.5788−5793
【非特許文献3】Am. Chem. Soc、117,29、1995年、pp.7585
【発明の概要】
【発明が解決しようとする課題】
【0013】
本発明の目的は、スタチンを効果的に作製するための構築ブロックとして中間体化合物及び方法を提供することである。
【課題を解決するための手段】
【0014】
前記目的は、少数の合成段階を必要とし、相対的に短い反応時間を示し、鏡像異性体及びジアステレオマー異性体過剰に関して高い立体化学的純度を有する産物の高総収率をもたらす方法により、((2S,4R)−4,6−ジヒドロキシテトラヒドロ−2H−ピラン−2−イル)メチルカルボキシラートを提供することによって解決される。本発明のさらなる課題は、安価な出発材料と単純な装置を用いて上記カルボキシラートを作製することである。
【0015】
本発明の一態様は、アセトアルデヒド及び式IIIR1CO2CH2CHO(R1は、下記に定義されているとおりである。)を、アルドール縮合を触媒する酵素と接触させる工程を含む、式IVの化合物
【0016】
【化2】
(R1は、それぞれ及び独立に、置換された又は置換されていない、アルキル、アルコキシ、アリール、ヘテロアリール、アリールアルキル又はヘテロアリールアルキルである。)
を調製する方法である。連続的アルドール反応を触媒する酵素を好ましく使用することによって、IVに到達するための反応工程の数は低減させることができる。
【0017】
好ましくは、基質は、式IIIの化合物(R1は、それぞれ及び独立に、置換された又は置換されていない、C1−C6アルキル又はアルコキシである。)から選択される。このような適切な酵素基質の選択によって、大幅に短縮された反応時間が可能となり、反応の顕著に改善された立体選択性が提供される。酵素反応は水性溶媒中で実施されるので、基質の適切な選択によって、基質を制御することがさらに可能となる。さらに、IVのエステル部分は、好ましくは水によって切断されないように選択される。従って、より好ましくは、R1=CH3である。特に、適切な酵素基質は、99.8%若しくはそれ以上の鏡像異性体過剰及び/又は98%若しくはそれ以上のジアステレオマー異性体過剰を有する式IVの化合物を提供するために好ましく選択される。その後、産物の精製及び単離がより容易であり、従って、収率がより高くなるので、高い鏡像異性体及びジアステレオマー異性体過剰は著しく有利である。
【0018】
酵素は、2−デオキシリボース−5−リン酸アルドラーゼ(DERA、EC4.1.2.4.)であることが好ましい。より広い基質特異性を有する酵素を見出すために、DERA酵素の異なる種類をスクリーニングすることが有用であり得る。さらに、DERA酵素は、特異的基質に対して特別に加工され得る。これらの理由のため、異なる変異体DERA酵素を検査し得る。より具体的には、前記アルドラーゼは、DERA01、DERA02、DERA03、DERA04、DERA05、DERA06、DERA07、DERA08、DERA09、DERA10、DERA11、DERA12、DERA13、DERA14、DERA15、DERA16、DERA17、DERA18、DERA19、DERA20、DERA21、DERA22及びDERA23又は前記アルドラーゼの何れかのアミノ酸配列に対して少なくとも約70%のアミノ酸配列同一性を有するアルドラーゼからなる群から選択される。より具体的には、前記アルドラーゼは、DERA01、DERA02、DERA05、DERA12及びDERA13からなる群から選択され、特に、前記アルドラーゼは配列番号2のアミノ酸配列に対して少なくとも約70%のアミノ酸配列同一性を有し、又は前記アルドラーゼは配列番号5のアミノ酸配列に対して少なくとも約80%のアミノ酸配列同一性を有し、又は前記アルドラーゼは配列番号11のアミノ酸配列に対して少なくとも約80%のアミノ酸配列同一性を有し、又は前記アルドラーゼは、配列番号25のアミノ酸配列に対して少なくとも約80%のアミノ酸配列同一性を有し、又は前記アルドラーゼは、配列番号27のアミノ酸配列に対して少なくとも約80%のアミノ酸配列同一性を有する。
【0019】
1つの好ましい実施形態において、アセトアルデヒド及び式IIIのアルデヒドR1CO2CH2CHOを接触させる前記工程は、それぞれ、アルドラーゼの生物学的に活性な形態を過剰発現する微生物又は微生物の一部とアセトアルデヒド及び前記式IIIのアルデヒドを接触させることによって達成される。前記接触工程は、アルドール縮合が触媒されるように行われる。この実施形態によれば、完全細胞触媒としてアルドラーゼを過剰発現する生物が使用される。酵素調製及び産物の精製における数個の工程が省略されるので、完全細胞触媒としてアルドラーゼを過剰発現する生物を使用する可能性は、「Proc. Nat. Acad. Sci. USA 2004, 101 (16) 5788−5793」に記載されている方法と比べて、より低い製造コストをさらに可能にする。また、細胞環境の安定化効果は、他の酵素調製物と比べて、酵素活性に対する影響をより低く抑えながら、より高い基質濃度を使用することを可能にする。これによって、より低い酵素負荷量で、より高い容積生産性が可能となり、これは、製造コストを著しく低下させる。驚くべきことに、アルドラーゼを過剰発現する生物を完全細胞触媒として使用することによって、式IVの化合物の高い鏡像異性体及びジアステレオマー異性体過剰が保持される。
【0020】
本発明の別の態様は、それぞれ、アルドラーゼの生物学的に活性な形態を過剰発現する微生物又は微生物の一部とアセトアルデヒド及び式IIIのアルデヒドR1CO2CH2CHO(R1は、下に定義されているとおりである。)を接触させる工程を含む、式IVの化合物
【0021】
【化3】
(R1は、それぞれ及び独立に、置換された又は置換されていない、アルキル、アルコキシ、アリール、ヘテロアリール、アリールアルキル又はヘテロアリールアルキルである。)を調製する方法である。前記接触工程は、アルドール縮合が触媒されるように行われる。本発明のこの実施形態によれば、完全細胞触媒としてアルドラーゼを過剰発現する生物が使用される。好ましくは、完全細胞触媒の形態の酵素は、2−デオキシリボース−5−リン酸アルドラーゼ(DERA、EC4.1.2.4)である。
【0022】
本発明の別の態様は、アルドール縮合を触媒する酵素とアセトアルデヒド及び式IIIのアルデヒドR1CO2CH2CHO(R1は、下に定義されているとおりである。)を接触させる工程を含み、前記酵素が完全細胞触媒の形態であり、前記完全細胞触媒がアルドラーゼの生物学的に活性な形態を過剰発現する微生物である、式IVの化合物
【0023】
【化4】
(R1は、それぞれ及び独立に、置換された又は置換されていない、アルキル、アルコキシ、アリール、ヘテロアリール、アリールアルキル又はヘテロアリールアルキルである。)を調製する方法である。
【0024】
本発明の別の態様は、それぞれ、アルドラーゼの生物学的に活性な形態を過剰発現する微生物又は微生物の一部とアセトアルデヒド及び式XIVのアルデヒドR4CH2CHO(R4は、上に定義されている。)を接触させる工程を含む、式XVの化合物
【0025】
【化5】
(R4=OCOR1(R1は、上に定義されているとおりである。)、塩化物、水素、それぞれ及び独立に置換された又は置換されていない、アリルオキシ及びベンジルオキシである。)
を調製する方法である。前記接触工程は、アルドール縮合が触媒されるように実施される。本発明のこの態様によれば、完全細胞触媒としてアルドラーゼを過剰発現する生物が使用される。
【0026】
好ましくは、完全細胞触媒の形態の酵素は2−デオキシリボース−5−リン酸アルドラーゼ(DERA、EC4.1.2.4)である。
【0027】
本発明の別の態様は、アルドール縮合を触媒する酵素とアセトアルデヒド及び式XIVのアルデヒドR4CH2CHO(R4は、下に定義されているとおりである。)を接触させる工程を含み、前記酵素が完全細胞触媒の形態であり、前記完全細胞触媒がアルドラーゼの生物学的に活性な形態を過剰発現する微生物である、式XVの化合物
【0028】
【化6】
(R4は、OCOR1(R1は、上に定義されているとおりである。)、塩化物、水素、それぞれ及び独立に置換された又は置換されていない、アリルオキシ及びベンジルオキシである。)
を調製する方法である。
【0029】
酵素調製及び産物の精製における幾つかの工程が省略されるので、完全細胞触媒の形態で酵素が使用されると、著しくより低いプロセスコストが達成される。また、細胞環境の安定化効果によって、他の酵素調製物と比べて、酵素活性に対する影響をより低く抑えながら、より高い基質を使用することが可能となる。さらに、完全細胞触媒の形態で酵素を使用すると、高い鏡像異性体及びジアステレオマー異性体過剰が得られる。
【0030】
本発明の方法の態様は、アルドラーゼによって触媒されるアルドール縮合に対するpHが4.5から10、好ましくは5から10の範囲に維持され、特に、5から8、好ましくは5から7のpH範囲の緩衝液を用いてpHが維持される反応条件で効果的に実施することができる。適切なpH値は、より短い反応時間をもたらす。別の態様において、適切なpHは、基質及び/又は産物の分解を低下させる。緩衝液は、pH値を一定のレベルに調整することを可能にし、これは、pH値に関して一定した反応条件に寄与する。この目的のために、緩衝液は、好ましくは、ホスファート緩衝液である。あるいは、pH制御されたポンプの補助を得て、酸又はアルカリの自動化された添加によって、正確なpHの調節を達成することができる。
【0031】
本発明の別の態様は、アセトアルデヒド及び式IIIのアルデヒドR1CO2CH2CHOを接触させる工程を含む、式IVの化合物(R1は、上に定義されているとおりである。)を形成させるために、アルドラーゼによって触媒されるアルドール縮合条件下で、式IIIの基質R1CO2CH2CHO(R1は、上に定義されているとおりである。)をアセトアルデヒドと反応させるための、アルドラーゼの使用である。特に、前記アルドラーゼは、2−デオキシリボース−5−リン酸アルドラーゼである。さらに具体的には、アルドラーゼは、上に記載されているDERA01からDERA23からなる群から選択される。特に、前記アルドラーゼは、生きた完全細胞内に含まれ、又は不活化された完全細胞内に含まれ、又は均質化された完全細胞内に含まれ、又は無細胞抽出物内に含まれ、又は精製された酵素であり、又は固定化されており、細胞外に発現されたタンパク質の形態である。
【0032】
本発明の別の態様は、酸化により、式IVの化合物を式Vの化合物へ転化する工程を含む、式Vの化合物
【0033】
【化7】
(R1は、それぞれ及び独立に、置換された又は置換されていない、アルキル、アルコキシ、アリール、ヘテロアリール、アリールアルキル又はヘテロアリールアルキルである。)
を調製する方法である。酸化のための反応試薬は、安価であり、及び高い収率を与えるべきである。従って、好ましくは、酸化は、Br2及びBaCO3を用いて行われる。
【0034】
特に、99.8%若しくはそれ以上の鏡像異性体過剰及び/又は98%若しくはそれ以上のジアステレオマー異性体過剰を有する式Vの化合物が提供される。
【0035】
本発明の別の態様は、
a)式IIの化合物R2CO2CH2CH=CHCH2O2CR2(R2は、下に定義されている。)
【0036】
【化8】
を溶媒と及びオゾンと接触させる工程、及び
b)工程a)から得られたオゾニドを加水分解する工程、
を含む、式III’のアルデヒドR2CO2CH2CHO(R2は、それぞれ及び独立に、置換された又は置換されていない、アルキル、アルコキシ、アリール、ヘテロアリール又はヘテロアリールアルキルである(n−プロピル、シクロヘキシル、フェニル、モルホリン、ピロリジン及びイミダゾールを除く。)を作製する方法。
【0037】
オゾン分解は、高い収率での前記アルデヒドの安価な作製を与える。特に、2つの所望される産物が加水分解後に得られるので、(Z)−及び/又は(E)位に、Hの他に2つの同じ置換基を有する(Z)−及び/又は(E)−アルケンは、高い分子的経済性を与えるが、(Z)−及び/又は(E)位に、Hの他に2つの異なる置換基を有する(Z)−及び/又は(E)−アルケンの転化は、1つの所望の産物及び1つの廃棄産物を与える。特に、工程a)の溶媒はジクロロメタンである。好ましくは、R2は、n−プロピル、シクロヘキシルを除くC1−C6アルキル又はアルコキシである。より好ましくは、R2は、CH3である。−50から−90℃範囲の、より好ましくは、約−80℃の温度で、工程a)を実施することが好ましい。さらに、工程a)の得られたオゾニドを硫化メチルと接触させることによって、工程b)を実施することが好ましい。特に、工程b)は、−80℃と室温の間に含まれる温度で実施される。
【0038】
式IIの化合物は、(Z)−及び/又は(E)−ブト−2−エン−1,4−ジオールを式Iの無水物R2CO2COR2(R2は、上で定義されている。)を、実施例1の工程1に又は従来技術の合成に記載されているように(J.Org.Chem.1956, 21, 328−331)反応させることによって得られる。
【0039】
本発明の別の態様は、式IV又はVの化合物
【0040】
【化9】
(R1は、それぞれ及び独立に、置換された又は置換されていない、アルキル、アルコキシ、アリール、ヘテロアリール、アリールアルキル又はヘテロアリールアルキルである。)
である。R1が、それぞれ及び独立に、置換された又は置換されていない、C1−C6アルキル又はアルコキシであり、特に、R1がCH3である式IV又はVの化合物が好ましい。
【0041】
本発明のさらに別の態様は、
a)式VIの化合物
【0042】
【化10】
を得るために、保護基R3(R3は、独立に置換された又は置換されていない、シリル、ベンジル、アルキル及びアセチルから好ましく選択される保護基であり、より好ましくは、R3は、場合によって置換された、C1−C8トリアルキルシリル、C1−C8ジアルキルシリル、C1−C8アルキルジアリールシリル(アルキルは、同一又は別異であり得る。)から選択され、より好ましくは、保護基は、tert−ブチルジメチルシリルである。)によって、4位の水酸基において、式Vの化合物
【0043】
【化11】
(R1=それぞれ及び独立に、置換された又は置換されていない、アルキル、アルコキシ、アリール、ヘテロアリール、アリールアルキル又はヘテロアリールアルキル。)
を保護する工程、
b)スタチン又は医薬として許容されるその誘導体を作製するのに十分な条件下で、式VIの前記化合物を反応させる工程、
を含む、スタチン又はその誘導体を作製する方法である。
【0044】
特に、99.8%若しくはそれ以上の鏡像異性体過剰及び/又は98%若しくはそれ以上のジアステレオマー異性体過剰を有する式VIの化合物が提供される。
【0045】
スタチン又はその誘導体を作製するための方法において、工程b)の条件はアルデヒドへのVIの転化によって、及びスタチン又はその誘導体を与えるための適切なホスホニウム塩又は他のリン誘導体とのWittigカップリングによって設定されることが好ましい。さらにより好ましくは、Wittigカップリング工程は、以下の工程を含む。
b1)式VIの化合物から式VIIIを有するアルデヒドを得る工程、
b2)式Xの化合物
【0046】
【化12】
を得るために、式IXを有するホスホニウム塩
【0047】
【化13】
(Rx、Ry及びRzは同一又は別異であり、並びに場合によって置換された、C1−C8アルキル又はC3−C6シクロアルキル又はC1−C8アルケニル又はC5−C6シクロアルケニル又はアリールから選択され、並びにXは陰イオン、好ましくは、ハロゲン又はカルボキシラート陰イオン、より好ましくは、塩化物、臭化物又はトリフルオロアセタートである。)
を得る工程、
b3)続いて、化合物Xをロスバスタチン又はその塩へ転化する工程。
【0048】
式VIの化合物から式VIIIを有するアルデヒドを得る前記工程b1)は、式VIIの化合物
【0049】
【化14】
(R3は、上に定義されている。)
を通じて実施される。
【0050】
特に、99.8%若しくはそれ以上の鏡像異性体過剰及び/又は98%若しくはそれ以上のジアステレオマー異性体過剰を有する式VIIの化合物が提供される。
【0051】
本発明のさらに別の態様は、
a)アセトアルデヒド及び式IIIR1CO2CH2CHO(R1は、上に定義されている。)のアルデヒドを、アルドール縮合を職倍する酵素と接触させる工程を含む、式IVの化合物
【0052】
【化15】
(R1は、それぞれ及び独立に、置換された又は置換されていない、アルキル、アルコキシ、アリール、ヘテロアリール、アリールアルキル又はヘテロアリールアルキルである。)
を調製する工程、
b)酸化によって、式IVの前記化合物を式Vの化合物
【0053】
【化16】
(R1は、上に定義されているとおりである。)
へ転化する工程、
c)式VIの化合物
【0054】
【化17】
を得るために、保護基R3(R3は、独立に置換された又は置換されていない、シリル、ベンジル、アルキル及びアセチルから好ましく選択される保護基であり、より好ましくは、R3は、場合によって置換された、C1−C8トリアルキルシリル、C1−C8ジアルキルアリールシリル、C1−C8アルキルジアリールシリル(アルキルは、同一又は別異であり得る。)から選択され、より好ましくは、保護基は、tert−ブチルジメチルシリルである。)によって、4位の水酸基において、式Vの前記化合物を保護する工程、
d)スタチン又は医薬として許容されるその誘導体を作製するのに十分な条件下で、式VIの前記化合物を反応させる工程、
を含み、
工程d)の条件が、アルデヒドへのVIの転化によって、及びスタチン又はその誘導体を得るために、適切なホスホニウム塩又は他のリン化合物とのWittigカップリングによって設定され、好ましくは、Wittigカップリング工程が、
b1)式VIの化合物から式VIIIを有するアルデヒドを得る工程、
b2)式Xの化合物
【0055】
【化18】
を得るために、式IXを有するホスホニウム塩
【0056】
【化19】
(Rx、Ry及びRzは同一又は別異であり、並びに場合によって置換された、C1−C8アルキル又はC3−C6シクロアルキル又はC1−C8アルケニル又はC5−C6シクロアルケニル又はアリールから選択され、
並びにXは陰イオン、好ましくは、ハロゲン又はカルボキシラート陰イオン、より好ましくは、塩化物、臭化物又はトリフルオロアセタートである。)
を得る工程、
b3)その後、化合物Xをロスバスタチン又はその塩へ転化する工程、
を含む、
スタチン又はその誘導体を作製する方法である。
【0057】
以下では、添付の図面を参照しながら、好ましい実施形態及び実施例によって本発明の方法がより詳しく記載されているが、これらの実施形態、実施例及び図面は、例示の目的で提示されているに過ぎず、いかなる意味においても本発明を限定するものではないことに留意すべきである。
【図面の簡単な説明】
【0058】
【図1】実施例6に係る酵素的反応の反応プロファイルを示す。
【発明を実施するための形態】
【0059】
本発明は、化学的には以下の一般式の((2S,4R)−4,6−ジヒドロキシテトラヒドロ−2H−ピラン−2−イル)メチルカルボキシラートである式IVの化合物を提供する。
【0060】
【化20】
(R1は、それぞれ及び独立に、置換された又は置換されていない、アルキル、アルコキシ、アリール、ヘテロアリール、アリールアルキル又はヘテロアリールアルキルである。)
【0061】
特に、R1がそれぞれ及び独立に置換された又は置換されていない、C1−C6アルキル又はアルコキシである場合、とりわけ、R1がCH3である場合の式IVの化合物の特徴は、所望の立体化学を有しており、以降の中間体をその後分離することが回避される点に存する。従って、中間体化合物IVの提供は、可能な連続的選択的酸化工程又は適切な機能的修飾(例えば、6位の水酸基の最初の酸化工程、場合によって行われる、4位の水酸基の第二の酸化工程、及びこれに加えて又はこれに代えて、R1アシル残基の切断後のメトキシ基での第三の酸化工程を含む。)を可能とする。
【0062】
本発明は、以下のスキームに示されているように、アルドラーゼによって触媒されるアルドール縮合反応において、対応するラクトールIVを形成するために、置換されたアセトアルデヒドR1CO2CH2CHO(式III)の化合物及びアセトアルデヒドを使用する酵素的プロセスを提供する。
【0063】
【化21】
(R1は、それぞれ及び独立に、置換された又は置換されていない、アルキル、アルコキシ、アリール、ヘテロアリール、アリールアルキル又はヘテロアリールアルキルから選択される。)本発明の構造IVは、2及び4位において、厳格に確定された立体異性を有するのに対して、他のキラル中心は両方の可能性で存在し得、エピマーの混合物を形成する。
【0064】
本明細書において使用される「アルドラーゼによって触媒されるアルドール縮合条件」という用語は、本明細書に記載されているように、アルドラーゼによって触媒されることができる本分野において公知のあらゆるアルドール縮合条件を表す。特に、アルドラーゼによって触媒されるアルドール縮合条件は、所望の産物の形成及び蓄積を可能とするような条件である。これらの条件には、一態様において、アルドラーゼが連続的縮合の実行を可能とするのに十分な量で提供される活性酵素である条件、別の態様において、アルドラーゼの活性の最小的阻害を示す量で、基質及びアセトアルデヒドが反応中に存在する条件、別の態様において、温度、pH、溶媒組成、撹拌及び反応の長さが所望の産物の蓄積を可能とする条件、別の態様において、前記条件が産物の安定性に対して有害な効果を持たない条件が含まれる。具体的には、これらの条件は、実施例に開示されている値によって規定される。
【0065】
式IIIの上記化合物に対するアルドラーゼ活性は、指定された酵素が単離及び/又は精製され、又は固定化され又は細胞内に存在し、又は不活化された完全細胞内に含まれ、又は均質化された細胞物質中に含まれ、又は式IIIの化合物及びアセトアルデヒドの上記反応を触媒して、IVに到達する無細胞抽出物中にあることを意味する。
【0066】
本明細書において使用される「スタチン(特に、ロスバスタチン)又は医薬として許容されるその塩を産生するのに十分な条件」という用語は、所望のスタチン化合物を得るために本分野において記載されている手段(本明細書に記載されている手段を含む。)を表す。
【0067】
本明細書において使用される「アルドラーゼの生物学的に活性な形態を過剰発現する生物」という用語は、強力なプロモーターの調節下でアルドラーゼ発現を有し、及びアルドラーゼが(野生型発現対照と比べて)高いレベルで発現されており、及び細胞内又は細胞外に蓄積されているあらゆる生物を表す。このような生物を作製する方法は、当業者に周知である。
【0068】
本発明において使用するためのアルドラーゼは、式IIIの上記化合物に対してアルドラーゼ活性を有するあらゆる化合物であり得る。本発明の一実施形態において、アルドラーゼは、2−デオキシリボース−5−リン酸アルドラーゼ(DERA)である。適切なDERA−アルドラーゼの例には、配列表に記載されているそれらのヌクレオチド配列又はそれぞれのコドン最適化されたヌクレオチド配列又はアミノ酸配列によって特定される、DERA01、DERA02、DERA03、DERA04、DERA05、DERA06、DERA07、DERA08、DERA09、DERA10、DERA11、DERA12、DERA13、DERA14、DERA15、DERA16、DERA17、DERA18、DERA19、DERA20、DERA21,DERA22及びDERA23が含まれるが、これらに限定されない。
【0069】
一般に、本分野において公知のDERAアルドラーゼの何れもが、上に列記されているDERAアルドラーゼに対する配列同一性とは無関係に、反応のために使用され得る。本発明は、30.1%に過ぎない同一性を有する2つの異なるアルドラーゼを用いて、前記反応を首尾よく実施する例を提供する。しかしながら、反応の収率は、各アルドラーゼの基質特異性及び各アルドラーゼに対する基質の阻害的効果に依存し得る。
【0070】
DERA01は、配列番号1のヌクレオチド配列又は配列番号2のアミノ酸配列を有するアルドラーゼである。DERA01(イー・コリ)は、カタログ番号91252で、Sigma Aldrich, St. Louis, MO, USAから市販されている。
【0071】
DERA02は、配列番号3若しくは4のヌクレオチド配列又は配列番号5のアミノ酸配列を有するアルドラーゼである。DERA02は、「William A. Greenberg, et al., PNAS, (2004), Vol.101 , No.16, pp. 5788」中に記載されている。
【0072】
DERA03は、配列番号6のヌクレオチド配列又は配列番号7のアミノ酸配列を有するアルドラーゼである。
【0073】
DERA04は、配列番号8のヌクレオチド配列又は配列番号9のアミノ酸配列を有するアルドラーゼである。
【0074】
DERA05は、配列番号10のヌクレオチド配列又は配列番号11のアミノ酸配列を有するアルドラーゼである。
【0075】
DERA06は、配列番号12のヌクレオチド配列又は配列番号13のアミノ酸配列を有するアルドラーゼである。
【0076】
DERA07は、配列番号14のヌクレオチド配列又は配列番号15のアミノ酸配列を有するアルドラーゼである。
【0077】
DERA08は、配列番号16のヌクレオチド配列又は配列番号17のアミノ酸配列を有するアルドラーゼである。
【0078】
DERA09は、配列番号18のヌクレオチド配列又は配列番号19のアミノ酸配列を有するアルドラーゼである。
【0079】
DERA10は、配列番号20のヌクレオチド配列又は配列番号21のアミノ酸配列を有するアルドラーゼである。
【0080】
DERA11は、配列番号22のヌクレオチド配列又は配列番号23のアミノ酸配列を有するアルドラーゼである。
【0081】
DERA12は、配列番号24のヌクレオチド配列又は配列番号25のアミノ酸配列を有するアルドラーゼである。
【0082】
DERA13は、配列番号26のヌクレオチド配列又は配列番号27のアミノ酸配列を有するアルドラーゼである。
【0083】
DERA14は、配列番号28のヌクレオチド配列又は配列番号29のアミノ酸配列を有するアルドラーゼである。
【0084】
DERA15は、配列番号30のヌクレオチド配列又は配列番号31のアミノ酸配列を有するアルドラーゼである。
【0085】
DERA16は、配列番号32のヌクレオチド配列又は配列番号33のアミノ酸配列を有するアルドラーゼである。
【0086】
DERA17は、配列番号34のヌクレオチド配列又は配列番号35のアミノ酸配列を有するアルドラーゼである。
【0087】
DERA18は、配列番号36のヌクレオチド配列又は配列番号37のアミノ酸配列を有するアルドラーゼである。
【0088】
DERA19は、配列番号38のヌクレオチド配列又は配列番号39のアミノ酸配列を有するアルドラーゼである。
【0089】
DERA20は、配列番号40のヌクレオチド配列又は配列番号41のアミノ酸配列を有するアルドラーゼである。
【0090】
DERA21は、配列番号42のヌクレオチド配列又は配列番号43のアミノ酸配列を有するアルドラーゼである。
【0091】
DERA22は、配列番号44のヌクレオチド配列又は配列番号45のアミノ酸配列を有するアルドラーゼである。
【0092】
DERA23は、配列番号46のヌクレオチド配列又は配列番号47のアミノ酸配列を有するアルドラーゼである。
【0093】
アルドラーゼは、本明細書中に記載されているアルドラーゼに対してその少なくとも約50%、好ましくは、その少なくとも約70%のアミノ酸配列同一性を有するアルドラーゼを含む。アミノ酸配列の同一性は、配列比較アルゴリズムを用いた分析によって、又は視覚的な調査によって決定される。一態様において、配列比較アルゴリズムは、設定を初期設定にしたVectorNTI9.0(InforMax)のAlignXアルゴリズムを用いて作製される。
【0094】
特に、本発明は、アセトアルデヒド及び式IIIR1CO2CH2CHO(R1は、下記に定義されている。)を、アルドール縮合を触媒する酵素と接触させる工程を含む、式IVの化合物
【0095】
【化22】
(R1は、それぞれ及び独立に、置換された又は置換されていない、アルキル、アルコキシ、アリール、ヘテロアリール、アリールアルキル又はヘテロアリールアルキルである。)
を調製する方法を提供する。
【0096】
好ましい実施形態において、アルドラーゼは、DERA01若しくはDERA02若しくはDERA05若しくはDERA12若しくはDERA13又はこれらと少なくとも約90%のアミノ酸配列同一性を有するあらゆるアルドラーゼから選択され、又は別の実施形態において、アルドラーゼは、DERA06若しくはDERA17又はこれらと少なくとも約80%のアミノ酸配列同一性を有するあらゆるアルドラーゼから好ましい実施形態において選択される。
【0097】
化合物IVは、スタチン(特に、ロスバスタチン)の合成におけるその後の使用において特に価値がある。
【0098】
本明細書中に記載されているDERAアルドラーゼは、「Sambrook and Russell, Molecular Cloning:A Laboratory Manual, 3rd Ed., Cold Spring Harbor, NY 2001」に記載されているような組換えイー・コリ中でのタンパク質発現のための標準的プロトコールなど(但し、これに限定されない。)本分野において公知のあらゆる手段によって調製することができる。クローニング条件によっては、公知のDERAアルドラーゼの修飾された様式が必要であり得、又はもたらされ得、公知のDERAアルドラーゼの修飾された様式は本発明によって包含される。
【0099】
本明細書中に記載されているDERAアルドラーゼは、あらゆる生物学的に活性な形態で使用することができる。一実施形態において、アルドラーゼは活性を有し、生きた完全細胞触媒の形態で使用され得る。一実施形態において、アルドラーゼは活性を有し、不活化された完全細胞触媒の形態で使用され得る。
【0100】
一実施形態における完全細胞触媒は、アルドラーゼの生物学的に活性な形態を過剰発現するあらゆる微生物又は微生物の一部である。前記微生物は、生きた又は休止した又は不活化された完全細胞の形態であり得る。
【0101】
これらの形態には、細胞懸濁物、細胞菌糸、細胞ペースト及び細胞が意図的に、物理的に、化学的に又は生物学的に破壊されていない微生物培養物のあらゆる他の形態が含まれ得、これらの形態には、このような微生物又はその一部の、担体に支持された、固定化された又は接着された担体がさらに含まれ得る。前記微生物は、好ましくは、細菌及び酵母から選択される。好ましくは、細菌は、エシェリヒア(Escherichia)、コリネバクテリウム(Corynebacterium)、シュードモナス(Pseudomonas)、ストレプトミセス(Streptomyces)、ロドコッカス(Rhodococcus)、バチルス(Bacillus)及びラクトバチルス(Lactobacillus)からなる群から選択され、より好ましくは、エシェリヒア・コリが使用される。酵母は、好ましくは、サッカロミセス(Saccharomyces)、ピチア(Pichia)、シゾサッカロミセス(Shizosaccharomyces)及びカンジダ(Candida)からなる属の群から選択される。
【0102】
一実施形態において、アルドラーゼは活性を有し、均質化された完全細胞触媒の形態で使用され得る。一実施形態において、アルドラーゼは活性を有し、無細胞抽出物の形態で使用され得る。一実施形態において、アルドラーゼは活性を有し、本分野において公知のあらゆる方法を用いて、精製された酵素の形態で使用され得る。別の実施形態において、アルドラーゼは活性を有し、細胞外で発現されたタンパク質の形態で使用され得る。
【0103】
スタチン産生に関して有用な中間体を作製するために使用されるアルドラーゼの最適な活性を与えるために、基質及び反応条件を選択した。
【0104】
式IIIの化合物は、最適な反応条件での、式IVの対応する化合物の産物安定性に従って選択される。特に、最高の安定性を有する産物を与えるアクセプター基質が反応のために好ましい。
【0105】
式IIIの化合物は、式VIの対応する化合物(遮蔽されたアルデヒド基を有するこれらの産物は、スタチンの調製におけるさらなる工程を可能にするWO2007/039287A1における中心的な中間体であり、特に、アルデヒド基を有する産物を与える基質が好ましい)に従っても選択される。
【0106】
式IIIの化合物は、特に、アセチルオキシアセトアルデヒド(CH3CO2CH2CHO)であり得る。
【0107】
一般に、アルドラーゼは適切な容器又は反応装置中に与えられ、式IIIの化合物及びアセトアルデヒドは、バッチ様式で又は継続的に添加される。
【0108】
具体的には、アルドラーゼは、場合によって塩(特に、50から500mMの濃度範囲のNaCl)の存在下で、(特に、0.1g/Lから30g/Lまでの濃度範囲で)水溶液中に調製される。水溶液は、水と混和性の有機溶媒(特に、2から15%V/Vの濃度のジメチルスルホキシド)を含有し得、pH4.5から9、好ましくは、pH5から9、より好ましくは、pH6から9になるように緩衝化され得る。
【0109】
適切な緩衝液は、酸、塩基、塩又はこれらの混合物及び一級、二級又は三級アミノ基を有するものを除く本分野で公知のあらゆる他の緩衝系から調製することができる。特に、10から500mMの濃度のホスファート緩衝液を使用することができる。水溶液は、前記アルドラーゼを水に添加し、無機酸、塩基、塩又はこれらの混合物の自動化された添加を用いて、反応の間にpHを維持することによっても調製することができる。
【0110】
あるいは、場合によって、塩(特に、50から500mMの濃度のNaCl)の存在下で、(特に、20g/Lから300g/L湿潤細胞重量の濃度範囲の、より具体的には、20g/Lから200g/L湿潤細胞重量の濃度範囲の)DERA過剰発現細胞、特にDERA過剰発現イー・コリ細胞の水性懸濁物中で、アルドラーゼが調製される。水性懸濁液は、水と混和性の有機溶媒(特に、2から15%V/Vの濃度範囲のジメチルスルホキシド)を含有し得、pH4.5から9、好ましくは、pH5から9、より好ましくは、pH6から9になるように緩衝化され得る。適切な緩衝液は、酸、塩基、塩又はこれらの混合物及び一級、二級又は三級アミノ基を有するものを除く本分野で公知のあらゆる他の緩衝系から調製することができる。特に、10から500mMの濃度のホスファート緩衝液を使用することができる。水性懸濁液は、前記DERA過剰発現細胞を水に添加し、無機酸、塩基、塩又はこれらの混合物の自動化された添加を用いて、反応の間にpHを維持することによっても調製することができる。
【0111】
方法の態様において、式IIIの化合物は、反応混合物へ連続的に添加され得、あるいは、式IIIの化合物は、1つのバッチ又はそれ以上のバッチ中の反応混合物に添加される。一態様において、混合物に添加される基質の総量は、添加される式IIIの化合物の総量が反応混合物の約20mmol/Lから反応混合物の約2mol/L、特に、反応混合物の約100mmol/Lから反応混合物の約1.5mol/L、より具体的には、反応混合物の約200mmol/Lから反応混合物の約700mol/Lである。アセトアルデヒドは、幾つかの手段によって添加され得る。一態様において、アセトアルデヒドは、1つのバッチ若しくはそれ以上のバッチ中であるいは連続的に、反応混合物に添加される。アセトアルデヒドは、式IIIの化合物と予め混合され、反応混合物に添加され得る。反応混合物に添加されるアセトアルデヒドの総量は、約0.1から約4モル濃度当量からアクセプター基質(化合物III)の総量まで、特に、約1から約3モル濃度当量、より好ましくは、約2から約2.5モル濃度当量である。特に、これは、所望でない産物、特に式XII及びXIIIの化合物の最小限の濃度を可能にするのに対して、式XIIの化合物は、アセトアルデヒドの一分子をIIIの一分子と反応させることによって得られ、式XIIIの化合物はアセトアルデヒドの三分子を反応させることによって得られる。
【0112】
【化23】
【0113】
好ましい実施形態において、基質は、反応のあらゆる所定の時点で、特定の流速で、プログラム可能なポンプを用いて反応混合物に継続的に添加される。流速は、基質が反応混合物中に蓄積しない最大の流速として決定される。特に、これは、所望でない産物の最小濃度を可能にする。より具体的には、この産物は、式XII及びXIIIの化合物であり得る。別の実施形態において、基質の阻害的効果は、正しい添加戦略を用いてさらに最小化することができる。
【0114】
あるいは、アルドラーゼは、式IIIの化合物又はアセトアルデヒドの少なくとも1つを含有する反応混合物に添加され得る。反応混合物は、溶媒と及びアルドラーゼ又は式IIIの化合物又はアセトアルデヒドの少なくとも1つを含むものと理解される。
【0115】
一態様において、アルドラーゼによって触媒される反応のために使用されるpHは、約5から10である。一実施形態において、アルドラーゼによって触媒される反応のために使用されるpHは、約5から約8である。具体的には、pHは、5から7の範囲の適切な緩衝液によって維持される。
【0116】
アルドラーゼ縮合中間体(特に、第一の縮合反応産物)は緩衝液と化学的な反応を起こし得るので、一般的に使用される幾つかの緩衝液は、アルドラーゼ縮合中間体(特に、第一の縮合反応産物)の利用可能性を制約することによって、先述のアルドラーゼによって触媒される反応の収率を低下させ得る。本発明者らは、ビス−トリスプロパンが前記中間体と反応することを発見した。同様に反応し得る他の緩衝液は、ビス−トリス、トリシン、トリス、ビシン又は一級、二級若しくは三級アミノ基を有するあらゆる他の緩衝液である。従って、pH値を調整するための適切な緩衝液は、この調整が必要であれば、酸、塩基、塩又はこれらの混合物、特に、リン酸及び水酸化ナトリウムを用いて作製される。
【0117】
一態様において、アルドラーゼによって触媒される反応のために使用される温度は、約20から約70℃である。一実施形態において、アルドラーゼによって触媒される反応のために使用される温度は、約25から約60℃である。一実施形態において、アルドラーゼによって触媒される反応のために使用される温度は、約30から約50℃である。
【0118】
反応は2から3時間以内に完了まで進行するので、産業的に適切である。
【0119】
反応の完了後、アセトニトリルの少なくとも約1容量を反応混合物の1容量に添加することによって、酵素は反応混合物から除去される。あるいは、酵素は、本分野において公知のあらゆる沈殿法によって除去される。一実施形態において、沈殿は、少なくとも5%m/Vの硫酸アンモニウムの添加によって行われる。あるいは、IVは、本分野において公知の塩析法によって抽出される。特に、塩析は、アセトニトリルの少なくとも約1容量から反応混合物の1容量及びNaClの5%(m/V)を添加することによって行われる。次いで、少なくとも40℃まで混合物を冷却し、液相を分離させる。次いで、アセトニトリル相を蒸発させて、IVの未精製産物を与える。あるいは、沈降技術、特に遠心を用いて、反応混合物から完全細胞触媒が除去される。別の態様において、完全細胞触媒は、ろ過技術、特に、微小ろ過によって除去され得る。
【0120】
本発明は、前記反応によって作製された純粋なラクトールを取得するための精製法も提供する。一態様において、アセトニトリルは、反応混合物から蒸発され、次いで、残存する水溶液が凍結乾燥される。別の態様において、沈降された沈殿溶液又は完全細胞触媒懸濁液の何れかの上清が凍結乾燥される。次いで、粉末化された残りがアセトニトリル/ジイソプロピルエーテル1:1中に懸濁される。不溶性の塩を除去するために、懸濁液をろ過し、移動相としてアセトニトリル/ジイソプロピルエーテル1:1を用いるシリカゲルカラムへろ液を搭載する。別の態様において、塩析抽出から得られるアセトニトリル相を蒸発させ、残りの油をアセトニトリル/ジイソプロピルエーテル1:1の最小容積中に溶解し、移動相としてアセトニトリル/ジイソプロピルエーテル1:1を用いるシリカゲルカラムに搭載する。
【0121】
特定の実施形態において、本発明は、使用されるアルドラーゼが(必要であれば、酸、塩基、塩又はこれらの混合物を用いて、特に、リン酸及び水酸化ナトリウムを用いて調整された)5から10、特に5から8のpH範囲の適切な溶媒(特に、水溶性有機溶媒と混合された水であり得る水性溶媒)中の、DERA01、DERA02であり、反応が約35から40℃の温度で進行し、転化が1から6時間で終了する、((2S,4R)−4,6−ジヒドロキシテトラヒドロ−2H−ピラン−2−イル)メチルアセタートを形成するための、アルドラーゼによって触媒されるアルドール縮合下でのCH3CO2CH2CHOのアセトアルデヒドとの反応を提供する。
【0122】
一般に、使用されるアルドラーゼは、「Sambrook, et al.(1989) Molecular cloning:A laboratory Manual 2nd Edition, New York:Cold Spring Harbor Laboratory Press, Cold Spring Harbor.」に記載されているタンパク質発現の方法によって調製される。アルドラーゼをコードする遺伝子は発現ベクター中にクローニングされ、酵素は適切な発現宿主中に発現される。
【0123】
反応収率は、反応混合物に添加された式IIIの化合物の総量に対して相対的に計算され、単離された産物のモルと反応混合物に添加された式IIIの化合物のモルとの間の比として測定される。
【0124】
本発明は、それぞれ、アルドラーゼの生物学的に活性な形態を過剰発現する微生物又は微生物の一部とアセトアルデヒド及び式XIVのアルデヒドR4CH2CHO(R4は、下記に定義されているとおりである。)を接触させる工程を含む、
式XVの化合物
【0125】
【化24】
(R4は、OCOR1(R1は、上に定義されているとおりである。)、塩化物、水素、それぞれ及び独立に置換された又は置換されていない、アリルオキシ及びベンジルオキシである。)
を調製する方法も提供する。好ましくは、R4は、アセタート、塩化物、水素であり、より好ましくは、R4はアセタートである。
【0126】
前記接触工程は、アルドール縮合が触媒されるように実施される。従って、完全細胞触媒としてアルドラーゼを過剰発現する生物が使用される。好ましくは、完全細胞触媒の形態の酵素は、上に定義されている2−デオキシリボース−5−リン酸アルドラーゼ(DERA、EC4.1.2.4)である。
【0127】
好ましくは、前記完全細胞触媒は、アルドラーゼの生物学的に活性な形態を過剰発現する細菌及び酵母から選択される。細菌は、好ましくは、エシェリヒア(Escherichia)、コリネバクテリウム(Corynebacterium)、シュードモナス(Pseudomonas)、ストレプトミセス(Streptomyces)、ロドコッカス(Rhodococcus)、バチルス(Bacillus)及びラクトバチルス(Lactobacillus)からなる属の群から選択され、より好ましくは、エシェリヒア・コリが使用される。酵母は、好ましくは、サッカロミセス(Saccharomyces)、ピチア(Pichia)、シゾサッカロミセス(Shizosaccharomyces)及びカンジダ(Candida)からなる属の群から好ましく選択される。
【0128】
本発明の特定の態様において、ロスバスタチンは、以下のスキームに開示されているように、本発明者らの式VIの化合物から開始して、WO2007/039287A1に従って調製することができる。
【0129】
【化25】
【0130】
ロスバスタチン又は他のスタチンを作製するために、式VIの化合物は、(式VIIの化合物を介した)2つの工程で、(2S,4R)−4−(保護された)−6−オキソ−テトラヒドロ−2H−ピラン−2−カルバルデヒドVIII又はその水和物VIII’へ転換される。アルデヒドVIII又はその水和物VIII’は、適切な試薬とのWittigカップリングの条件下で反応させることができ、続いて、必要であれば、水素が添加される。
【0131】
適切な試薬は、以下の式の複素環式又は脂環式誘導体(スタチンの骨格)である。
【0132】
【化26】
(Aは、結合又はOであり得、
並びにRx、Ry及びRzは同一又は別異であり、並びに場合によって置換された、C1−C8アルキル又はC3−C6シクロアルキル又はC1−C8アルケニル又はC5−C6シクロアルケニル又はアリールから選択され、
並びにXが陰イオン、好ましくは、ハロゲン又はカルボキシラート陰イオン、より好ましくは、塩化物、臭化物又はトリフルオロアセタートであり;
並びにHetは、スタチンの複素環式又は脂環式骨格を形成するように選択される。)
他のHMG−CoA還元酵素阻害剤(好ましくは、セリバスタチン、フルバスタチン、ピタバスタチン、ベルバスタチン、ダルバスタチンの中から選択される。)を同様に調製することができる。
【0133】
スタチンの複素環式又は脂環式骨格(Het)は、特に、以下から選択される。
【0134】
【化27】
【0135】
以下の実施例は、本発明の方法を例示するものであり、本発明の範囲を限定することを意図するものではない。
【実施例1】
【0136】
アセトキシアセトアルデヒド(III)
1.工程
【0137】
【化28】
トリエチルアミン(67mL、4当量)中に、1,4−ジヒドロキシブト−2−エン(10mL、0.12mol、1当量)を溶解した。溶液を0℃まで冷却し、無水酢酸(I)(34mL、3当量)を滴加した。得られた反応混合物を室温まで加温し、一晩撹拌した。
【0138】
1MH3PO4溶液(60mL)で2回、NaHCO31M溶液(60mL)で2回、溶液を洗浄した。次いで、MgSO4で溶液を脱水させ、濃縮した。高真空ポンプを70℃で使用して、AcOH及びAc2Oの微量を除去した。
【0139】
淡黄色ないし暗色の液体として、純粋なIIを得た(20.6g、100%)。1HNMR(300MHz,CDCl3)δ5.65(m,2H),4.58(m,4H),1.97(s,6H),13CNMR(75MHz,CDCl3)δ170.2,127.6,59.5,20.2。
【0140】
2.工程
【0141】
【化29】
1,4−ジアセトキシブト−2−エン(II)(1.7g、10mmol、1当量)をジクロロメタン(0.17M)中に溶解した。酸素(又は乾燥空気)散布を作動させ(約10L/h)、溶液を−80℃まで冷却した。−80℃になったら、オゾン発生装置を作動させ、溶液が青色に変化するまで、オゾンを通気した。次いで、オゾン発生装置を停止させ、青色が消失するまで、酸素(又は乾燥空気)を通気した。アルゴンを10分間通気した。散布を取り除き、−80℃で、溶液をアルゴン下に保った。硫化メチル(1.8mL、2.5当量)を滴加し、反応を室温まで加温し、20時間撹拌した。反応を濃縮して、1,4−アセトキシアセトアルデヒド(III)及びDMSOの2:1混合物を得た。収率は定量的であると思われ、1,4−ジアセトキシブト−2−エンの微量は存在しなかった。さらなる精製なしに、産物を使用した。(III)1HNMR(300MHz,CDCl3)δ9.51(s,1H),4.59(s,2H),2.10(s,3H),2.58(s,DMSO)。
【実施例2】
【0142】
アルドラーゼの調製
イー・コリK−12株由来の単離されたゲノムDNAを用いるPCR反応において、以下のオリゴヌクレオチドプライマーを用いて、エシェリヒア・コリ遺伝子deoCを増幅した。
CGGGATCCACTGATCTGAAAGCAAGCAGCC及び
GCAAGCTTGCTGCTGGCGCTCTTACC
(それぞれ、配列番号48及び49を有する。)
制限エンドヌクレアーゼBamHI及びHindIIIを用いて産物を切断し、得られた断片をアガロースゲル電気泳動上で分離し、精製した。同じ前記制限エンドヌクレアーゼを用いて、発現ベクターpQE30(Qiagene inc.,Valencia,CA,USA)を切断し、精製した。T4リガーゼ反応において、断片を集合させた。
【0143】
上記連結反応を用いて、形質転換受容性のエシェリヒア・コリDH5α細胞を形質転換した。アンピシリン耐性コロニーを培養し、プラスミドDNAを単離した。得られた構築物をpQE30DeraCと表記し、遺伝子配列を確認するために配列決定した。ベクターpQE30DeraCで形質転換受容性のエシェリヒア・コリTOP10F’株(Invitrogen corp.,Carlsbad,CA,USA)を形質転換することによって、アルドラーゼ発現生物を調製した。この方法に対して使用した方法は、「Sambrook et al. (1989) Molecular cloning:A Laboratory Manual 2nd Edition, New York:Cold Spring Harbor Laboratory Press, Cold Spring Harbor」に記載されており、当業者に周知である。
【0144】
TOP10F’PQE30DeraCの3mL一晩培養物とともに、アンピシリン(100μg/mL)が補充されたTerrific Broth培地(150mL、12g/Lバクト・トリプトン、24g/Lバクト酵母抽出物、4mL/Lグリセロール、2.31g/LKH2PO4、12.54g/LK2HPO4)に接種した。OD600が約0.8に到達するまで、細胞を増殖させた(37℃、250rpm)。IPTG(1mM最終濃度)を用いてタンパク質発現を誘導し、さらに4時間、同じ増殖条件中に細胞を放置した。遠心(10分、6000g、4℃)によって、細胞沈降物を採集した。上清を除去し、緩衝液(50mMNaH2PO4、pH7.0、300mMNaCl)の同じ容量によって交換した。沈降物を再懸濁し、遠心(10分、6000g、4℃)によって再度集めた。上清を除去し、使用前に、細胞を−20℃で保存した。DERA01を有する完全細胞触媒をこのようにして得た。
【0145】
あるいは、溶解緩衝液1L当り沈降物200gを用いて、溶解緩衝液(50mMNaH2PO4、pH7.0、300mMNaCl、2mMDTT)中に沈降物を再懸濁した。Bransonデジタル音波処理装置を用いて、細胞を音波処理し(3×15秒)、沈降(10分、20,000g、4℃)によって、細胞破片を除去した。DERA01の透明な水溶液をこのようにして得た。
【実施例3】
【0146】
((2S,4R)−4,6−ジヒドロキシテトラヒドロ−2H−ピラン−2−イル)メチルアセタート(IV)
【0147】
【化30】
DERA01の溶液600mL、反応緩衝液(50mMNaH2PO4、pH7.0、300mMNaCl、水中)200mL、アセトアルデヒド(反応緩衝液中1.05M溶液)の溶液100mL及びIII’の100mL(反応緩衝液中の500mM溶液)を撹拌された反応容器中で混合し、反応混合物1Lを得た。NaOHの1M水溶液を用いて、混合物のpH値を7.0に補正した。37℃に設定された温度制御された槽中で、混合物を3時間温置した。SynergyFusion、250×4.6mm、4μmカラムを使用して、ESIイオン化を備えた三連四重極HPLC−UV−MS/MSシステム上でのLC−MS分析を用いて、反応の間、IVの産生をモニターした。クロマトグラフィー条件は、以下のとおりであった。Tカラム=50℃、流速:1.5mL/分、Vinj=50μL。移動相を以下のように使用した。
A:水中の0.1%(m/v)NH4CH3COO、pH6.5
B:MilliQ水
C:アセトニトリル
線形勾配:
【0148】
【表1】
【0149】
保持時間=7.7±0.5分及び208の質量(M+NH4+に相当)を有するピーク面積の増大が反応の間に観察されたが、反応緩衝液で置換された反応混合物の一成分をそれぞれ有する対照の何れにもピーク面積の増大は観察されなかった。
【0150】
アセトニトリル4L及びNaCl50gの添加を用いて、反応を停止させた。懸濁液を0℃まで冷却し、遠心(10分、6000g、4℃)によって、液相を分離した。上方の相を取り出し、減圧下で蒸発させて、痰黄色の油(未精製産物)8.4gを得た。シリカゲルカラム(移動相:アセトニトリル/ジイソプロピルエーテル=1/1)上で未精製産物を精製した。その後、減圧下で溶液を蒸発させて、産物IV2.4gを得た。(IV):1HNMR(300MHz,CDCl3)δ5.39(d,1H),4.53(m,1H),4.29(m,1H),4.14(m,2H),2.11(s,3H),2.04−1.60(m,4H),13CNMR(75MHz,DMSO−d6)δ170.8,91.7,68.1,66.8,63.3,33.9,20.7
【実施例4】
【0151】
((2S,4R)−4,6−ジヒドロキシテトラヒドロ−2H−ピラン−2−イル)メチルアセタート(IV)
【0152】
【化31】
【0153】
反応緩衝液(50mMNaH2PO4、pH7.0、300mMNaCl、水中)750mL中に、DERA01を有する完全細胞触媒(300g)を懸濁し、37℃に設定した制御された温度で、撹拌された容器中において温置した。較正された蠕動式ポンプを用いて、2時間の反応時間の間に、基質溶液(630mMアセトアルデヒド(III)、反応緩衝液中の300mM溶液)750mLを添加した。NaOHの1M水溶液を用いて、pHを7.0に調節した。反応をさらに30分間継続し、次いで、遠心(10分、6000g、4℃)によって、完全細胞触媒を沈降させた。次いで、上清を凍結乾燥して淡黄色結晶(未精製産物)19.4gを得た。シリカゲルカラム(移動相:アセトニトリル/ジイソプロピルエーテル=1/1)上で未精製産物を精製した。その後、溶液を減圧下で蒸発させて、産物IV7.4gを得た。(IV):1HNMR(300MHz,CDCl3)δ5.39(d,1H),4.53(m,1H),4.29(m,1H),4.14(m,2H),2.11(s,3H),2.04−1.60(m,4H),13CNMR(75MHz,DMSO−d6)δ170.8,91.7,68.1,66.8,63.3,33.9,20.7。
【0154】
反応の間、LC−MS分析を用いて、IVの産生をモニターした。反応の間、208の質量を有するピーク面積(M+NH4+に相当)の増大が観察され、LC−MSを用いた2.5時間後の反応混合物の定量分析によって、IVの13.1g/Lの存在が示された。
【実施例5】
【0155】
((2S,4R)−4,6−ジヒドロキシテトラヒドロ−2H−ピラン−2−イル)メチルアセタート(IV)
【0156】
【化32】
【0157】
反応緩衝液(50mMNaH2PO4、pH7.0、300mMNaCl、水中)6mL中に、DERA02を有する完全細胞触媒(3g)を懸濁した。アセトアルデヒド(反応緩衝液中の2.1M溶液)の溶液2mL及びIII(反応緩衝液中の1M溶液)の2mLを反応管中に混合して、反応混合物10mLを得た。NaOHの1M水溶液を用いて、混合物のpH値を7.0に補正した。37℃に設定された温度制御された槽中で、混合物を3時間温置した。反応の間、LC−MS分析を用いて、IVの産生をモニターした。208の質量(M+NH4+に相当)を有するピーク面積の増大が反応の間に観察されたが、反応緩衝液で置換された反応混合物の一成分をそれぞれ有する対照の何れにもピーク面積の増大は観察されず、LC−MSを用いた1時間後における反応混合物の定量分析は、IVの8.7g/Lの存在を示した。
【実施例6】
【0158】
((2S,4R)−4,6−ジヒドロキシテトラヒドロ−2H−ピラン−2−イル)メチルアセタート(IV)
【0159】
【化33】
【0160】
反応緩衝液(50mMNaH2PO4、pH7.0、150mMNaCl)1070mL中に、IIIの56.6g(92%)を希釈した。NaHCO3塩を用いて、この溶液のpH値を6.2になるように調整した。DERA01を有する完全細胞触媒(700g/L)510mLを前溶液に添加し、NaHCO3塩を用いて、pH値を6.2になるように再度補正した。37℃及び一定の撹拌の800rpmに設定された、温度制御された2Lバイオリアクター内で、混合物を3時間温置した。下表に記載されている3時間の時間枠で、プログラム可能なポンプを用いて、反応緩衝液中に希釈されたアセトアルデヒド(45.4g)120mLを反応混合物に継続的に添加した。
【0161】
【表2】
【0162】
反応の間、pHは補正されず、5.5の最終値までゆっくり推移した。5.17分(化合物III)、14.04分(化合物XII)、14.44分化合物(化合物XIII)及び20.35分(化合物IV)での保持時間をモニタリングすることによって、GC分析(クロマトグラフィーカラム:DB−1 100%ジメチルポリシロキサン;温度プログラム:初期温度50℃、初期時間:5分、温度速度10℃/分、最終温度:215℃、最終時間:10分;注入装置:スプリット/スプリットレス注入装置、担体気体:ヘリウム、初期流速:10mL/分;検出装置:炎イオン化検出装置、検出装置温度:230℃;クロマトグラフィー溶液:IVの1から5mg/mLアセトニトリル)を用いて、IVの産生をモニターした。反応プロファイルは、図1に示されている。
【0163】
GCを用いた3時間後における反応混合物の定量分析は、67.7%モル濃度収率で、IVの35.3g/Lを示した。実施例6に記載されている物質に由来するさらなる合成工程中の化合物の鏡像異性体純度の分析は、99.8%又はそれ以上の鏡像異性体過剰を示し、これは、この物質の極めて高い鏡像異性体純度を示唆する。実施例6に記載されている物質に由来するさらなる合成工程中の化合物のジアステレオマー異性体純度の分析は、98%又はそれ以上のジアステレオマー異性体過剰を示し、これも、IVの極めて高いジアステレオマー異性体純度を示唆する。
【実施例7】
【0164】
((2S,4R)−4−ヒドロキシ−6−オキソテトラヒドロ−2H−ピラン−2−イル)メチルアセタート(V)
【0165】
【化34】
【0166】
水中の((2S,4R)−4,6−ジヒドロキシテトラヒドロ−2H−ピラン−2−イル)メチルアセタート(IV)(1当量)の溶液を0℃まで冷却した。炭酸バリウム(1.4当量)を添加した後、Br2(1.2当量)を滴加し、反応を室温で一晩撹拌した。溶液をNaClで飽和させ、EtOAcを用いて4回抽出した。合わせた有機相をMgSO4で脱水させ、濃縮した。フラッシュクロマトグラフィーを用いた精製(ヘキサン/アセトン=75/25から55/45)によって、((2S,4R)−4−ヒドロキシ−6−オキソテトラヒドロ−2H−ピラン−2−イル)メチルアセタート(V)を得た。
【0167】
(V):1HNMR(300MHz,アセトン−d6)δ4.88(m,1H),4.45(d,J=3.0Hz,1H),4.38(hex,J=3.0Hz,1H),4.23(dd,J=3.5Hz,J=12.0Hz,1H),4.16(dd,J=5.5Hz,J=12.1Hz,1H),2.68(dd,J=4.3Hz,J=17.5HZ,1H),2.51(dddd,J=0.8Hz,J=2.0Hz,J=3.3Hz,J=17.5Hz,1H),2.03(s,3H),1.91(m,2H),13CNMR(75MHz,アセトン−d6)δ170.8,169.7,74.2,66.5,62.7,39.1,32.3,20.6。
【実施例8】
【0168】
((2S,4R)−4−(tert−ブチルジメチルシリルオキシ)−6−オキソテトラヒドロ−2H−ピラン−2−イル)メチルアセタート(VI)
【0169】
【化35】
【0170】
((2S,4R)−4−ヒドロキシ−6−オキソテトラヒドロ−2H−ピランー2−イル)メチルアセタート(V)(1.88g、10mmol、1当量)の溶液を無水DMF(2mL、1M)中に溶解した。イミダゾール(0.88g、1.3当量)及びTBDMSCI(1.66g、1.1当量)を連続して添加し、反応が完了するまで、反応を撹拌した。水(20mL)とエーテル(20mL)の間に、反応混合物を分配した。エーテル(20mL)で1回、水相を抽出した。水(10mL)の少量、HCl1N(20mL)及び塩水(20mL)で、合わせた有機相を2回洗浄した。溶液をMgSO4で脱水させ、濃縮して、定量的収率で、((2S,4R)−4−(tert−ブチルジメチルシリルオキシ)−6−オキソテトラヒドロ−2H−ピラン−2−イル)メチルアセタート(VI)を得た。1HNMR(300MHz,CDCl3)δ4.93(m,1H),4.37(quint,J=3Hz,1H),4.30(dd,J=3Hz,J=12Hz,1H),4.21(dd,J=5Hz,J=12Hz,1H),2.62(d,J=4Hz,2H),2.11(s,3H),1.84−1.80(m,2H),0.89(s,9H),0.09(2s,6H)。13CNMR(75MHz,CDCl3)δ170.4,169.1,73.3,65.5,63.0,38.9,32.2,20.5,17.7,−5.1,−5.2。
【実施例9】
【0171】
酵素反応を介した((2S,4R)−4−(tert−ブチルジメチルシリルオキシ)−6−オキソテトラヒドロ−2H−ピラン−2−イル)メチルアセタート(VI)の(4R,6S)−4−(tert−ブチルジメチルシリルオキシ)−6−(ヒドロキシメチル)テトラヒドロ−2H−ピラン−2−オン(VII)への転化
【0172】
【化36】
【0173】
((2S,4R)−4−(tert−ブチルジメチルシリルオキシ)−6−オキソテトラヒドロ−2H−ピラン−2−イル)メチルアセタート(VI)(50g、80%純度;132.4mmol)をリン酸緩衝液(P.B.S)、pH=5.20(1.5L)に添加し、37℃まで溶液を加温した。次いで、パンクレアチン粉末(0.5当量質量;20g)を滴加した(6回、8g+3×4g)。平行して、pHをモニターし、pHを4.85と4.95の間に維持するために、1時間ごとに、NaHCO3溶液(1M)を添加することによってpHを制御した。酵素の最初の添加後9時間、反応を撹拌した。
【0174】
celite(R)を未精製混合物に添加した。celite(R)を通して、溶液をろ別した。淡黄色の液体を回収した。EtOAcの1.5Lを用いて、フィルター上の固体を洗浄した。ろ液を5分間撹拌した。2つの層を分離した。EtOAc(1.5L)を用いて、水相を1回再抽出した。40℃で減圧下において、合わせた有機相を部分的に蒸発させ、メチルシクロヘキサンから産物を再結晶させて、(4R,6S)−4−(tert−ブチルジメチルシリルオキシ)−6−(ヒドロキシメチル)テトラヒドロ−2H−ピラン−2−オン(VII)の82%を白色結晶として得た。反応の転化はほぼ定量的であり(>98%)、鏡像異性体は0.1%未満であった。
【0175】
スプリット/スプリットレス注入装置及びFID検出装置を有するBetadex120カラム上で、DCM溶液(2から3mg/mL)を用いて鏡像異性体を測定するためのGC分析を行った。
【0176】
化合物VIIのNMR分析は、検出限界での高スキャン蓄積を用いた場合でさえ、ジアステレオマー異性体が存在しないことを示し、VII中のジアステレオマー異性体のレベルが1%を下回ることを示唆している。
【実施例10】
【0177】
化学的反応を介した((2S,4R)−4−(tert−ブチルジメチルシリルオキシ)−6−オキソテトラヒドロ−2H−ピラン−2−イル)メチルアセタート(VI)の(4R,6S)−4−(tert−ブチルジメチルシリルオキシ)−6−(ヒドロキシメチル)テトラヒドロ−2H−ピラン−2−オン(VII)への転化
【0178】
【化37】
【0179】
((2S,4R)−4−(tert−ブチルジメチルシリルオキシ)−6−オキソテトラヒドロ−2H−ピラン−2−イル)メチルアセタート(VI)(0.625g、80%純度、1.65mmol)をTHF(8mL)及びMeOH(8mL)中に溶解した。スズ触媒(tBu2SnClOH)2(94mg、0.1当量)を添加し、反応を一晩撹拌した。反応を濃縮し、フラッシュクロマトグラフィーによって精製して、純粋な(4R,6S)−4−(tert−ブチルジメチルシリルオキシ)−6−(ヒドロキシメチル)テトラヒドロ−2H−ピラン−2−オン(VII)(126mg、30%)を得た。VIIは鏡像異性体の0.1%未満を含有することが明らかとなった。
【0180】
スプリット/スプリットレス注入装置及びFID検出装置を有するBetadex120カラム上で、DCM溶液(2から3mg/mL)を用いて鏡像異性体を測定するためのGC分析を行った。
【0181】
化合物VIIのNMR分析は、検出限界での高スキャン蓄積を用いた場合でさえ、ジアステレオマー異性体が存在しないことを示し、VII中のジアステレオマー異性体のレベルが1%を下回ることを示唆している。
【0182】
酵素的反応を介した及び化学的反応を介した化合物VIIへの化合物VIの転化は、望ましくない鏡像異性体が0.1%未満である優れた鏡像異性体純度を有する産物を与え、2つの方法によって調製された化合物VIIの鏡像異性体純度には、差は観察されなかった。また、調製された化合物VIIのジアステレオマー異性体純度は高く、望ましくないジアステレオマー異性体は1%未満であった。従って、鏡像異性体及びジアステレオマー異性体の過剰は、より初期の合成工程に(例えば、アルドラーゼによって触媒される化合物IIIの化合物IVへの転化に)起因する。
【技術分野】
【0001】
本発明は、((2S,4R)−4,6−ジヒドロキシテトラヒドロ−2H−ピラン−2−イル)メチルカルボキシラート及びその製造方法に関する。さらに、本発明は、上記化合物が中間体として使用される、スタチン(特に、ロスバスタチン)及びその誘導体の作製方法に関する。
【背景技術】
【0002】
((2S,4R)−4,6−ジヒドロキシテトラヒドロ−2H−ピラン−2−イル)メチルカルボキシラートは、スタチンの合成において中間体となり得る。スタチン(その代表例は、ロスバスタチン、セリバスタチン、アトルバスタチン、フルバスタチン、ピタバスタチン、ベルバスタチン、ダルバスタチン若しくはこれらの類縁体又はプラバスタチン、シンバスタチン、ロバスタチン若しくはこれらの類縁体から選択され得る。)は、それぞれ、芳香族又は脂環式の殻に接続されたヘプテン酸又はヘプタン酸部分(遊離の酸、塩又はラクトン)によって規定される特徴的構造を共有する。スタチンの生物活性は、その立体化学、特に、前記ヘプテン酸又はヘプタン酸部分のキラル原子における立体配置と密接に関連する。
【0003】
WO2006/134482には、アトルバスタチンを形成するための方法中に、2−デオキシリボース−5−リン酸アルドラーゼ(DERA)によって触媒されるアルドール付加工程が含まれる。
【0004】
日本国特許2005229858は、DERAの存在下で、ベンジルオキシアセトアルデヒドをアセトアルデヒドと反応させる、((4R,6S)−4,6−ジヒドロキシテトラヒドロ−2−ピロンを作製するための方法を開示する。酵素触媒の反応時間は、12時間であった。
【0005】
WO05/118794は、DERA酵素の改善を取り扱っている。極めて多様な置換基を有する2,4−ジデオキシヘキソース又は2,4,6−トリデオキシヘキソースを調製するために、単離された変異体酵素が使用され得る。
【0006】
DERA変異体は、立体特異的なアルドール反応を触媒することが記載された(Tetrahedron Letters2004,45,2439−2441)。DERA変異体は触媒活性の相対的改善を示し、従って、野生型DERAと比較して収率を改善した。酵素触媒の反応時間は6日であった。この酵素触媒から得られた1つの産物は、アトルバスタチンの合成に関して提案された。
【0007】
立体特異的アルドール反応を触媒するためのDERAは、「Proc. Nat. Acad. Sci. USA 2004, 101(16) 5788−5793」中にさらに記載されており、酵素プロセスの改善された容積生産性を示した。使用される基質の酵素活性に対する阻害的効果も記載されている。酵素触媒の反応時間は3時間であった。この酵素的触媒から得られた産物は、アトルバスタチン又はロスバスタチンの合成に関して提案された。
【0008】
2−デオキシリボース−5−リン酸アルドラーゼ(DERA)によって触媒される3つのアルデヒド基質との立体特異的アルドール反応は、全ての置換されたアセトアルデヒドをDERAに対する基質として等しく受容するわけではなく(Am. Chem. Soc;117,29,(1995)pp7585)、ある種の基質は、DERA活性に対して阻害的効果を示す。酵素的触媒の反応時間は6日であった。
【0009】
WO2007/039287A1では、ヨードラクトン合成を介したラクトン化されたスタチン側鎖中間体VIの合成が記載されており、これには、6つの有機合成工程が必要とされる。この複数工程の合成において、4番目の工程は、ヨードラクトン中間体産物の立体化学を規定するラクトン形成工程である。このラクトン形成工程は、相対的に低い立体選択性を与えるに過ぎない。I化合物、Ag化合物、グリニャール試薬及び鏡像異性体的に純粋な出発化合物のような化学量論的量で使用される幾つかの試薬は、極めて高価である。(以下に示されている)有機合成の6つの工程は、19%の総収率を得られた。
【0010】
【化1】
【先行技術文献】
【特許文献】
【0011】
【特許文献1】国際公開第2006/134482号
【特許文献2】特許第2005229858号明細書
【特許文献3】国際公開第05/118794号
【特許文献4】国際公開第2007/039287号
【非特許文献】
【0012】
【非特許文献1】Tetrahedron Letters、45、2004年、pp.2439−2441
【非特許文献2】Proc. Nat. Acad. Sci. USA 101(16)、2004年、pp.5788−5793
【非特許文献3】Am. Chem. Soc、117,29、1995年、pp.7585
【発明の概要】
【発明が解決しようとする課題】
【0013】
本発明の目的は、スタチンを効果的に作製するための構築ブロックとして中間体化合物及び方法を提供することである。
【課題を解決するための手段】
【0014】
前記目的は、少数の合成段階を必要とし、相対的に短い反応時間を示し、鏡像異性体及びジアステレオマー異性体過剰に関して高い立体化学的純度を有する産物の高総収率をもたらす方法により、((2S,4R)−4,6−ジヒドロキシテトラヒドロ−2H−ピラン−2−イル)メチルカルボキシラートを提供することによって解決される。本発明のさらなる課題は、安価な出発材料と単純な装置を用いて上記カルボキシラートを作製することである。
【0015】
本発明の一態様は、アセトアルデヒド及び式IIIR1CO2CH2CHO(R1は、下記に定義されているとおりである。)を、アルドール縮合を触媒する酵素と接触させる工程を含む、式IVの化合物
【0016】
【化2】
(R1は、それぞれ及び独立に、置換された又は置換されていない、アルキル、アルコキシ、アリール、ヘテロアリール、アリールアルキル又はヘテロアリールアルキルである。)
を調製する方法である。連続的アルドール反応を触媒する酵素を好ましく使用することによって、IVに到達するための反応工程の数は低減させることができる。
【0017】
好ましくは、基質は、式IIIの化合物(R1は、それぞれ及び独立に、置換された又は置換されていない、C1−C6アルキル又はアルコキシである。)から選択される。このような適切な酵素基質の選択によって、大幅に短縮された反応時間が可能となり、反応の顕著に改善された立体選択性が提供される。酵素反応は水性溶媒中で実施されるので、基質の適切な選択によって、基質を制御することがさらに可能となる。さらに、IVのエステル部分は、好ましくは水によって切断されないように選択される。従って、より好ましくは、R1=CH3である。特に、適切な酵素基質は、99.8%若しくはそれ以上の鏡像異性体過剰及び/又は98%若しくはそれ以上のジアステレオマー異性体過剰を有する式IVの化合物を提供するために好ましく選択される。その後、産物の精製及び単離がより容易であり、従って、収率がより高くなるので、高い鏡像異性体及びジアステレオマー異性体過剰は著しく有利である。
【0018】
酵素は、2−デオキシリボース−5−リン酸アルドラーゼ(DERA、EC4.1.2.4.)であることが好ましい。より広い基質特異性を有する酵素を見出すために、DERA酵素の異なる種類をスクリーニングすることが有用であり得る。さらに、DERA酵素は、特異的基質に対して特別に加工され得る。これらの理由のため、異なる変異体DERA酵素を検査し得る。より具体的には、前記アルドラーゼは、DERA01、DERA02、DERA03、DERA04、DERA05、DERA06、DERA07、DERA08、DERA09、DERA10、DERA11、DERA12、DERA13、DERA14、DERA15、DERA16、DERA17、DERA18、DERA19、DERA20、DERA21、DERA22及びDERA23又は前記アルドラーゼの何れかのアミノ酸配列に対して少なくとも約70%のアミノ酸配列同一性を有するアルドラーゼからなる群から選択される。より具体的には、前記アルドラーゼは、DERA01、DERA02、DERA05、DERA12及びDERA13からなる群から選択され、特に、前記アルドラーゼは配列番号2のアミノ酸配列に対して少なくとも約70%のアミノ酸配列同一性を有し、又は前記アルドラーゼは配列番号5のアミノ酸配列に対して少なくとも約80%のアミノ酸配列同一性を有し、又は前記アルドラーゼは配列番号11のアミノ酸配列に対して少なくとも約80%のアミノ酸配列同一性を有し、又は前記アルドラーゼは、配列番号25のアミノ酸配列に対して少なくとも約80%のアミノ酸配列同一性を有し、又は前記アルドラーゼは、配列番号27のアミノ酸配列に対して少なくとも約80%のアミノ酸配列同一性を有する。
【0019】
1つの好ましい実施形態において、アセトアルデヒド及び式IIIのアルデヒドR1CO2CH2CHOを接触させる前記工程は、それぞれ、アルドラーゼの生物学的に活性な形態を過剰発現する微生物又は微生物の一部とアセトアルデヒド及び前記式IIIのアルデヒドを接触させることによって達成される。前記接触工程は、アルドール縮合が触媒されるように行われる。この実施形態によれば、完全細胞触媒としてアルドラーゼを過剰発現する生物が使用される。酵素調製及び産物の精製における数個の工程が省略されるので、完全細胞触媒としてアルドラーゼを過剰発現する生物を使用する可能性は、「Proc. Nat. Acad. Sci. USA 2004, 101 (16) 5788−5793」に記載されている方法と比べて、より低い製造コストをさらに可能にする。また、細胞環境の安定化効果は、他の酵素調製物と比べて、酵素活性に対する影響をより低く抑えながら、より高い基質濃度を使用することを可能にする。これによって、より低い酵素負荷量で、より高い容積生産性が可能となり、これは、製造コストを著しく低下させる。驚くべきことに、アルドラーゼを過剰発現する生物を完全細胞触媒として使用することによって、式IVの化合物の高い鏡像異性体及びジアステレオマー異性体過剰が保持される。
【0020】
本発明の別の態様は、それぞれ、アルドラーゼの生物学的に活性な形態を過剰発現する微生物又は微生物の一部とアセトアルデヒド及び式IIIのアルデヒドR1CO2CH2CHO(R1は、下に定義されているとおりである。)を接触させる工程を含む、式IVの化合物
【0021】
【化3】
(R1は、それぞれ及び独立に、置換された又は置換されていない、アルキル、アルコキシ、アリール、ヘテロアリール、アリールアルキル又はヘテロアリールアルキルである。)を調製する方法である。前記接触工程は、アルドール縮合が触媒されるように行われる。本発明のこの実施形態によれば、完全細胞触媒としてアルドラーゼを過剰発現する生物が使用される。好ましくは、完全細胞触媒の形態の酵素は、2−デオキシリボース−5−リン酸アルドラーゼ(DERA、EC4.1.2.4)である。
【0022】
本発明の別の態様は、アルドール縮合を触媒する酵素とアセトアルデヒド及び式IIIのアルデヒドR1CO2CH2CHO(R1は、下に定義されているとおりである。)を接触させる工程を含み、前記酵素が完全細胞触媒の形態であり、前記完全細胞触媒がアルドラーゼの生物学的に活性な形態を過剰発現する微生物である、式IVの化合物
【0023】
【化4】
(R1は、それぞれ及び独立に、置換された又は置換されていない、アルキル、アルコキシ、アリール、ヘテロアリール、アリールアルキル又はヘテロアリールアルキルである。)を調製する方法である。
【0024】
本発明の別の態様は、それぞれ、アルドラーゼの生物学的に活性な形態を過剰発現する微生物又は微生物の一部とアセトアルデヒド及び式XIVのアルデヒドR4CH2CHO(R4は、上に定義されている。)を接触させる工程を含む、式XVの化合物
【0025】
【化5】
(R4=OCOR1(R1は、上に定義されているとおりである。)、塩化物、水素、それぞれ及び独立に置換された又は置換されていない、アリルオキシ及びベンジルオキシである。)
を調製する方法である。前記接触工程は、アルドール縮合が触媒されるように実施される。本発明のこの態様によれば、完全細胞触媒としてアルドラーゼを過剰発現する生物が使用される。
【0026】
好ましくは、完全細胞触媒の形態の酵素は2−デオキシリボース−5−リン酸アルドラーゼ(DERA、EC4.1.2.4)である。
【0027】
本発明の別の態様は、アルドール縮合を触媒する酵素とアセトアルデヒド及び式XIVのアルデヒドR4CH2CHO(R4は、下に定義されているとおりである。)を接触させる工程を含み、前記酵素が完全細胞触媒の形態であり、前記完全細胞触媒がアルドラーゼの生物学的に活性な形態を過剰発現する微生物である、式XVの化合物
【0028】
【化6】
(R4は、OCOR1(R1は、上に定義されているとおりである。)、塩化物、水素、それぞれ及び独立に置換された又は置換されていない、アリルオキシ及びベンジルオキシである。)
を調製する方法である。
【0029】
酵素調製及び産物の精製における幾つかの工程が省略されるので、完全細胞触媒の形態で酵素が使用されると、著しくより低いプロセスコストが達成される。また、細胞環境の安定化効果によって、他の酵素調製物と比べて、酵素活性に対する影響をより低く抑えながら、より高い基質を使用することが可能となる。さらに、完全細胞触媒の形態で酵素を使用すると、高い鏡像異性体及びジアステレオマー異性体過剰が得られる。
【0030】
本発明の方法の態様は、アルドラーゼによって触媒されるアルドール縮合に対するpHが4.5から10、好ましくは5から10の範囲に維持され、特に、5から8、好ましくは5から7のpH範囲の緩衝液を用いてpHが維持される反応条件で効果的に実施することができる。適切なpH値は、より短い反応時間をもたらす。別の態様において、適切なpHは、基質及び/又は産物の分解を低下させる。緩衝液は、pH値を一定のレベルに調整することを可能にし、これは、pH値に関して一定した反応条件に寄与する。この目的のために、緩衝液は、好ましくは、ホスファート緩衝液である。あるいは、pH制御されたポンプの補助を得て、酸又はアルカリの自動化された添加によって、正確なpHの調節を達成することができる。
【0031】
本発明の別の態様は、アセトアルデヒド及び式IIIのアルデヒドR1CO2CH2CHOを接触させる工程を含む、式IVの化合物(R1は、上に定義されているとおりである。)を形成させるために、アルドラーゼによって触媒されるアルドール縮合条件下で、式IIIの基質R1CO2CH2CHO(R1は、上に定義されているとおりである。)をアセトアルデヒドと反応させるための、アルドラーゼの使用である。特に、前記アルドラーゼは、2−デオキシリボース−5−リン酸アルドラーゼである。さらに具体的には、アルドラーゼは、上に記載されているDERA01からDERA23からなる群から選択される。特に、前記アルドラーゼは、生きた完全細胞内に含まれ、又は不活化された完全細胞内に含まれ、又は均質化された完全細胞内に含まれ、又は無細胞抽出物内に含まれ、又は精製された酵素であり、又は固定化されており、細胞外に発現されたタンパク質の形態である。
【0032】
本発明の別の態様は、酸化により、式IVの化合物を式Vの化合物へ転化する工程を含む、式Vの化合物
【0033】
【化7】
(R1は、それぞれ及び独立に、置換された又は置換されていない、アルキル、アルコキシ、アリール、ヘテロアリール、アリールアルキル又はヘテロアリールアルキルである。)
を調製する方法である。酸化のための反応試薬は、安価であり、及び高い収率を与えるべきである。従って、好ましくは、酸化は、Br2及びBaCO3を用いて行われる。
【0034】
特に、99.8%若しくはそれ以上の鏡像異性体過剰及び/又は98%若しくはそれ以上のジアステレオマー異性体過剰を有する式Vの化合物が提供される。
【0035】
本発明の別の態様は、
a)式IIの化合物R2CO2CH2CH=CHCH2O2CR2(R2は、下に定義されている。)
【0036】
【化8】
を溶媒と及びオゾンと接触させる工程、及び
b)工程a)から得られたオゾニドを加水分解する工程、
を含む、式III’のアルデヒドR2CO2CH2CHO(R2は、それぞれ及び独立に、置換された又は置換されていない、アルキル、アルコキシ、アリール、ヘテロアリール又はヘテロアリールアルキルである(n−プロピル、シクロヘキシル、フェニル、モルホリン、ピロリジン及びイミダゾールを除く。)を作製する方法。
【0037】
オゾン分解は、高い収率での前記アルデヒドの安価な作製を与える。特に、2つの所望される産物が加水分解後に得られるので、(Z)−及び/又は(E)位に、Hの他に2つの同じ置換基を有する(Z)−及び/又は(E)−アルケンは、高い分子的経済性を与えるが、(Z)−及び/又は(E)位に、Hの他に2つの異なる置換基を有する(Z)−及び/又は(E)−アルケンの転化は、1つの所望の産物及び1つの廃棄産物を与える。特に、工程a)の溶媒はジクロロメタンである。好ましくは、R2は、n−プロピル、シクロヘキシルを除くC1−C6アルキル又はアルコキシである。より好ましくは、R2は、CH3である。−50から−90℃範囲の、より好ましくは、約−80℃の温度で、工程a)を実施することが好ましい。さらに、工程a)の得られたオゾニドを硫化メチルと接触させることによって、工程b)を実施することが好ましい。特に、工程b)は、−80℃と室温の間に含まれる温度で実施される。
【0038】
式IIの化合物は、(Z)−及び/又は(E)−ブト−2−エン−1,4−ジオールを式Iの無水物R2CO2COR2(R2は、上で定義されている。)を、実施例1の工程1に又は従来技術の合成に記載されているように(J.Org.Chem.1956, 21, 328−331)反応させることによって得られる。
【0039】
本発明の別の態様は、式IV又はVの化合物
【0040】
【化9】
(R1は、それぞれ及び独立に、置換された又は置換されていない、アルキル、アルコキシ、アリール、ヘテロアリール、アリールアルキル又はヘテロアリールアルキルである。)
である。R1が、それぞれ及び独立に、置換された又は置換されていない、C1−C6アルキル又はアルコキシであり、特に、R1がCH3である式IV又はVの化合物が好ましい。
【0041】
本発明のさらに別の態様は、
a)式VIの化合物
【0042】
【化10】
を得るために、保護基R3(R3は、独立に置換された又は置換されていない、シリル、ベンジル、アルキル及びアセチルから好ましく選択される保護基であり、より好ましくは、R3は、場合によって置換された、C1−C8トリアルキルシリル、C1−C8ジアルキルシリル、C1−C8アルキルジアリールシリル(アルキルは、同一又は別異であり得る。)から選択され、より好ましくは、保護基は、tert−ブチルジメチルシリルである。)によって、4位の水酸基において、式Vの化合物
【0043】
【化11】
(R1=それぞれ及び独立に、置換された又は置換されていない、アルキル、アルコキシ、アリール、ヘテロアリール、アリールアルキル又はヘテロアリールアルキル。)
を保護する工程、
b)スタチン又は医薬として許容されるその誘導体を作製するのに十分な条件下で、式VIの前記化合物を反応させる工程、
を含む、スタチン又はその誘導体を作製する方法である。
【0044】
特に、99.8%若しくはそれ以上の鏡像異性体過剰及び/又は98%若しくはそれ以上のジアステレオマー異性体過剰を有する式VIの化合物が提供される。
【0045】
スタチン又はその誘導体を作製するための方法において、工程b)の条件はアルデヒドへのVIの転化によって、及びスタチン又はその誘導体を与えるための適切なホスホニウム塩又は他のリン誘導体とのWittigカップリングによって設定されることが好ましい。さらにより好ましくは、Wittigカップリング工程は、以下の工程を含む。
b1)式VIの化合物から式VIIIを有するアルデヒドを得る工程、
b2)式Xの化合物
【0046】
【化12】
を得るために、式IXを有するホスホニウム塩
【0047】
【化13】
(Rx、Ry及びRzは同一又は別異であり、並びに場合によって置換された、C1−C8アルキル又はC3−C6シクロアルキル又はC1−C8アルケニル又はC5−C6シクロアルケニル又はアリールから選択され、並びにXは陰イオン、好ましくは、ハロゲン又はカルボキシラート陰イオン、より好ましくは、塩化物、臭化物又はトリフルオロアセタートである。)
を得る工程、
b3)続いて、化合物Xをロスバスタチン又はその塩へ転化する工程。
【0048】
式VIの化合物から式VIIIを有するアルデヒドを得る前記工程b1)は、式VIIの化合物
【0049】
【化14】
(R3は、上に定義されている。)
を通じて実施される。
【0050】
特に、99.8%若しくはそれ以上の鏡像異性体過剰及び/又は98%若しくはそれ以上のジアステレオマー異性体過剰を有する式VIIの化合物が提供される。
【0051】
本発明のさらに別の態様は、
a)アセトアルデヒド及び式IIIR1CO2CH2CHO(R1は、上に定義されている。)のアルデヒドを、アルドール縮合を職倍する酵素と接触させる工程を含む、式IVの化合物
【0052】
【化15】
(R1は、それぞれ及び独立に、置換された又は置換されていない、アルキル、アルコキシ、アリール、ヘテロアリール、アリールアルキル又はヘテロアリールアルキルである。)
を調製する工程、
b)酸化によって、式IVの前記化合物を式Vの化合物
【0053】
【化16】
(R1は、上に定義されているとおりである。)
へ転化する工程、
c)式VIの化合物
【0054】
【化17】
を得るために、保護基R3(R3は、独立に置換された又は置換されていない、シリル、ベンジル、アルキル及びアセチルから好ましく選択される保護基であり、より好ましくは、R3は、場合によって置換された、C1−C8トリアルキルシリル、C1−C8ジアルキルアリールシリル、C1−C8アルキルジアリールシリル(アルキルは、同一又は別異であり得る。)から選択され、より好ましくは、保護基は、tert−ブチルジメチルシリルである。)によって、4位の水酸基において、式Vの前記化合物を保護する工程、
d)スタチン又は医薬として許容されるその誘導体を作製するのに十分な条件下で、式VIの前記化合物を反応させる工程、
を含み、
工程d)の条件が、アルデヒドへのVIの転化によって、及びスタチン又はその誘導体を得るために、適切なホスホニウム塩又は他のリン化合物とのWittigカップリングによって設定され、好ましくは、Wittigカップリング工程が、
b1)式VIの化合物から式VIIIを有するアルデヒドを得る工程、
b2)式Xの化合物
【0055】
【化18】
を得るために、式IXを有するホスホニウム塩
【0056】
【化19】
(Rx、Ry及びRzは同一又は別異であり、並びに場合によって置換された、C1−C8アルキル又はC3−C6シクロアルキル又はC1−C8アルケニル又はC5−C6シクロアルケニル又はアリールから選択され、
並びにXは陰イオン、好ましくは、ハロゲン又はカルボキシラート陰イオン、より好ましくは、塩化物、臭化物又はトリフルオロアセタートである。)
を得る工程、
b3)その後、化合物Xをロスバスタチン又はその塩へ転化する工程、
を含む、
スタチン又はその誘導体を作製する方法である。
【0057】
以下では、添付の図面を参照しながら、好ましい実施形態及び実施例によって本発明の方法がより詳しく記載されているが、これらの実施形態、実施例及び図面は、例示の目的で提示されているに過ぎず、いかなる意味においても本発明を限定するものではないことに留意すべきである。
【図面の簡単な説明】
【0058】
【図1】実施例6に係る酵素的反応の反応プロファイルを示す。
【発明を実施するための形態】
【0059】
本発明は、化学的には以下の一般式の((2S,4R)−4,6−ジヒドロキシテトラヒドロ−2H−ピラン−2−イル)メチルカルボキシラートである式IVの化合物を提供する。
【0060】
【化20】
(R1は、それぞれ及び独立に、置換された又は置換されていない、アルキル、アルコキシ、アリール、ヘテロアリール、アリールアルキル又はヘテロアリールアルキルである。)
【0061】
特に、R1がそれぞれ及び独立に置換された又は置換されていない、C1−C6アルキル又はアルコキシである場合、とりわけ、R1がCH3である場合の式IVの化合物の特徴は、所望の立体化学を有しており、以降の中間体をその後分離することが回避される点に存する。従って、中間体化合物IVの提供は、可能な連続的選択的酸化工程又は適切な機能的修飾(例えば、6位の水酸基の最初の酸化工程、場合によって行われる、4位の水酸基の第二の酸化工程、及びこれに加えて又はこれに代えて、R1アシル残基の切断後のメトキシ基での第三の酸化工程を含む。)を可能とする。
【0062】
本発明は、以下のスキームに示されているように、アルドラーゼによって触媒されるアルドール縮合反応において、対応するラクトールIVを形成するために、置換されたアセトアルデヒドR1CO2CH2CHO(式III)の化合物及びアセトアルデヒドを使用する酵素的プロセスを提供する。
【0063】
【化21】
(R1は、それぞれ及び独立に、置換された又は置換されていない、アルキル、アルコキシ、アリール、ヘテロアリール、アリールアルキル又はヘテロアリールアルキルから選択される。)本発明の構造IVは、2及び4位において、厳格に確定された立体異性を有するのに対して、他のキラル中心は両方の可能性で存在し得、エピマーの混合物を形成する。
【0064】
本明細書において使用される「アルドラーゼによって触媒されるアルドール縮合条件」という用語は、本明細書に記載されているように、アルドラーゼによって触媒されることができる本分野において公知のあらゆるアルドール縮合条件を表す。特に、アルドラーゼによって触媒されるアルドール縮合条件は、所望の産物の形成及び蓄積を可能とするような条件である。これらの条件には、一態様において、アルドラーゼが連続的縮合の実行を可能とするのに十分な量で提供される活性酵素である条件、別の態様において、アルドラーゼの活性の最小的阻害を示す量で、基質及びアセトアルデヒドが反応中に存在する条件、別の態様において、温度、pH、溶媒組成、撹拌及び反応の長さが所望の産物の蓄積を可能とする条件、別の態様において、前記条件が産物の安定性に対して有害な効果を持たない条件が含まれる。具体的には、これらの条件は、実施例に開示されている値によって規定される。
【0065】
式IIIの上記化合物に対するアルドラーゼ活性は、指定された酵素が単離及び/又は精製され、又は固定化され又は細胞内に存在し、又は不活化された完全細胞内に含まれ、又は均質化された細胞物質中に含まれ、又は式IIIの化合物及びアセトアルデヒドの上記反応を触媒して、IVに到達する無細胞抽出物中にあることを意味する。
【0066】
本明細書において使用される「スタチン(特に、ロスバスタチン)又は医薬として許容されるその塩を産生するのに十分な条件」という用語は、所望のスタチン化合物を得るために本分野において記載されている手段(本明細書に記載されている手段を含む。)を表す。
【0067】
本明細書において使用される「アルドラーゼの生物学的に活性な形態を過剰発現する生物」という用語は、強力なプロモーターの調節下でアルドラーゼ発現を有し、及びアルドラーゼが(野生型発現対照と比べて)高いレベルで発現されており、及び細胞内又は細胞外に蓄積されているあらゆる生物を表す。このような生物を作製する方法は、当業者に周知である。
【0068】
本発明において使用するためのアルドラーゼは、式IIIの上記化合物に対してアルドラーゼ活性を有するあらゆる化合物であり得る。本発明の一実施形態において、アルドラーゼは、2−デオキシリボース−5−リン酸アルドラーゼ(DERA)である。適切なDERA−アルドラーゼの例には、配列表に記載されているそれらのヌクレオチド配列又はそれぞれのコドン最適化されたヌクレオチド配列又はアミノ酸配列によって特定される、DERA01、DERA02、DERA03、DERA04、DERA05、DERA06、DERA07、DERA08、DERA09、DERA10、DERA11、DERA12、DERA13、DERA14、DERA15、DERA16、DERA17、DERA18、DERA19、DERA20、DERA21,DERA22及びDERA23が含まれるが、これらに限定されない。
【0069】
一般に、本分野において公知のDERAアルドラーゼの何れもが、上に列記されているDERAアルドラーゼに対する配列同一性とは無関係に、反応のために使用され得る。本発明は、30.1%に過ぎない同一性を有する2つの異なるアルドラーゼを用いて、前記反応を首尾よく実施する例を提供する。しかしながら、反応の収率は、各アルドラーゼの基質特異性及び各アルドラーゼに対する基質の阻害的効果に依存し得る。
【0070】
DERA01は、配列番号1のヌクレオチド配列又は配列番号2のアミノ酸配列を有するアルドラーゼである。DERA01(イー・コリ)は、カタログ番号91252で、Sigma Aldrich, St. Louis, MO, USAから市販されている。
【0071】
DERA02は、配列番号3若しくは4のヌクレオチド配列又は配列番号5のアミノ酸配列を有するアルドラーゼである。DERA02は、「William A. Greenberg, et al., PNAS, (2004), Vol.101 , No.16, pp. 5788」中に記載されている。
【0072】
DERA03は、配列番号6のヌクレオチド配列又は配列番号7のアミノ酸配列を有するアルドラーゼである。
【0073】
DERA04は、配列番号8のヌクレオチド配列又は配列番号9のアミノ酸配列を有するアルドラーゼである。
【0074】
DERA05は、配列番号10のヌクレオチド配列又は配列番号11のアミノ酸配列を有するアルドラーゼである。
【0075】
DERA06は、配列番号12のヌクレオチド配列又は配列番号13のアミノ酸配列を有するアルドラーゼである。
【0076】
DERA07は、配列番号14のヌクレオチド配列又は配列番号15のアミノ酸配列を有するアルドラーゼである。
【0077】
DERA08は、配列番号16のヌクレオチド配列又は配列番号17のアミノ酸配列を有するアルドラーゼである。
【0078】
DERA09は、配列番号18のヌクレオチド配列又は配列番号19のアミノ酸配列を有するアルドラーゼである。
【0079】
DERA10は、配列番号20のヌクレオチド配列又は配列番号21のアミノ酸配列を有するアルドラーゼである。
【0080】
DERA11は、配列番号22のヌクレオチド配列又は配列番号23のアミノ酸配列を有するアルドラーゼである。
【0081】
DERA12は、配列番号24のヌクレオチド配列又は配列番号25のアミノ酸配列を有するアルドラーゼである。
【0082】
DERA13は、配列番号26のヌクレオチド配列又は配列番号27のアミノ酸配列を有するアルドラーゼである。
【0083】
DERA14は、配列番号28のヌクレオチド配列又は配列番号29のアミノ酸配列を有するアルドラーゼである。
【0084】
DERA15は、配列番号30のヌクレオチド配列又は配列番号31のアミノ酸配列を有するアルドラーゼである。
【0085】
DERA16は、配列番号32のヌクレオチド配列又は配列番号33のアミノ酸配列を有するアルドラーゼである。
【0086】
DERA17は、配列番号34のヌクレオチド配列又は配列番号35のアミノ酸配列を有するアルドラーゼである。
【0087】
DERA18は、配列番号36のヌクレオチド配列又は配列番号37のアミノ酸配列を有するアルドラーゼである。
【0088】
DERA19は、配列番号38のヌクレオチド配列又は配列番号39のアミノ酸配列を有するアルドラーゼである。
【0089】
DERA20は、配列番号40のヌクレオチド配列又は配列番号41のアミノ酸配列を有するアルドラーゼである。
【0090】
DERA21は、配列番号42のヌクレオチド配列又は配列番号43のアミノ酸配列を有するアルドラーゼである。
【0091】
DERA22は、配列番号44のヌクレオチド配列又は配列番号45のアミノ酸配列を有するアルドラーゼである。
【0092】
DERA23は、配列番号46のヌクレオチド配列又は配列番号47のアミノ酸配列を有するアルドラーゼである。
【0093】
アルドラーゼは、本明細書中に記載されているアルドラーゼに対してその少なくとも約50%、好ましくは、その少なくとも約70%のアミノ酸配列同一性を有するアルドラーゼを含む。アミノ酸配列の同一性は、配列比較アルゴリズムを用いた分析によって、又は視覚的な調査によって決定される。一態様において、配列比較アルゴリズムは、設定を初期設定にしたVectorNTI9.0(InforMax)のAlignXアルゴリズムを用いて作製される。
【0094】
特に、本発明は、アセトアルデヒド及び式IIIR1CO2CH2CHO(R1は、下記に定義されている。)を、アルドール縮合を触媒する酵素と接触させる工程を含む、式IVの化合物
【0095】
【化22】
(R1は、それぞれ及び独立に、置換された又は置換されていない、アルキル、アルコキシ、アリール、ヘテロアリール、アリールアルキル又はヘテロアリールアルキルである。)
を調製する方法を提供する。
【0096】
好ましい実施形態において、アルドラーゼは、DERA01若しくはDERA02若しくはDERA05若しくはDERA12若しくはDERA13又はこれらと少なくとも約90%のアミノ酸配列同一性を有するあらゆるアルドラーゼから選択され、又は別の実施形態において、アルドラーゼは、DERA06若しくはDERA17又はこれらと少なくとも約80%のアミノ酸配列同一性を有するあらゆるアルドラーゼから好ましい実施形態において選択される。
【0097】
化合物IVは、スタチン(特に、ロスバスタチン)の合成におけるその後の使用において特に価値がある。
【0098】
本明細書中に記載されているDERAアルドラーゼは、「Sambrook and Russell, Molecular Cloning:A Laboratory Manual, 3rd Ed., Cold Spring Harbor, NY 2001」に記載されているような組換えイー・コリ中でのタンパク質発現のための標準的プロトコールなど(但し、これに限定されない。)本分野において公知のあらゆる手段によって調製することができる。クローニング条件によっては、公知のDERAアルドラーゼの修飾された様式が必要であり得、又はもたらされ得、公知のDERAアルドラーゼの修飾された様式は本発明によって包含される。
【0099】
本明細書中に記載されているDERAアルドラーゼは、あらゆる生物学的に活性な形態で使用することができる。一実施形態において、アルドラーゼは活性を有し、生きた完全細胞触媒の形態で使用され得る。一実施形態において、アルドラーゼは活性を有し、不活化された完全細胞触媒の形態で使用され得る。
【0100】
一実施形態における完全細胞触媒は、アルドラーゼの生物学的に活性な形態を過剰発現するあらゆる微生物又は微生物の一部である。前記微生物は、生きた又は休止した又は不活化された完全細胞の形態であり得る。
【0101】
これらの形態には、細胞懸濁物、細胞菌糸、細胞ペースト及び細胞が意図的に、物理的に、化学的に又は生物学的に破壊されていない微生物培養物のあらゆる他の形態が含まれ得、これらの形態には、このような微生物又はその一部の、担体に支持された、固定化された又は接着された担体がさらに含まれ得る。前記微生物は、好ましくは、細菌及び酵母から選択される。好ましくは、細菌は、エシェリヒア(Escherichia)、コリネバクテリウム(Corynebacterium)、シュードモナス(Pseudomonas)、ストレプトミセス(Streptomyces)、ロドコッカス(Rhodococcus)、バチルス(Bacillus)及びラクトバチルス(Lactobacillus)からなる群から選択され、より好ましくは、エシェリヒア・コリが使用される。酵母は、好ましくは、サッカロミセス(Saccharomyces)、ピチア(Pichia)、シゾサッカロミセス(Shizosaccharomyces)及びカンジダ(Candida)からなる属の群から選択される。
【0102】
一実施形態において、アルドラーゼは活性を有し、均質化された完全細胞触媒の形態で使用され得る。一実施形態において、アルドラーゼは活性を有し、無細胞抽出物の形態で使用され得る。一実施形態において、アルドラーゼは活性を有し、本分野において公知のあらゆる方法を用いて、精製された酵素の形態で使用され得る。別の実施形態において、アルドラーゼは活性を有し、細胞外で発現されたタンパク質の形態で使用され得る。
【0103】
スタチン産生に関して有用な中間体を作製するために使用されるアルドラーゼの最適な活性を与えるために、基質及び反応条件を選択した。
【0104】
式IIIの化合物は、最適な反応条件での、式IVの対応する化合物の産物安定性に従って選択される。特に、最高の安定性を有する産物を与えるアクセプター基質が反応のために好ましい。
【0105】
式IIIの化合物は、式VIの対応する化合物(遮蔽されたアルデヒド基を有するこれらの産物は、スタチンの調製におけるさらなる工程を可能にするWO2007/039287A1における中心的な中間体であり、特に、アルデヒド基を有する産物を与える基質が好ましい)に従っても選択される。
【0106】
式IIIの化合物は、特に、アセチルオキシアセトアルデヒド(CH3CO2CH2CHO)であり得る。
【0107】
一般に、アルドラーゼは適切な容器又は反応装置中に与えられ、式IIIの化合物及びアセトアルデヒドは、バッチ様式で又は継続的に添加される。
【0108】
具体的には、アルドラーゼは、場合によって塩(特に、50から500mMの濃度範囲のNaCl)の存在下で、(特に、0.1g/Lから30g/Lまでの濃度範囲で)水溶液中に調製される。水溶液は、水と混和性の有機溶媒(特に、2から15%V/Vの濃度のジメチルスルホキシド)を含有し得、pH4.5から9、好ましくは、pH5から9、より好ましくは、pH6から9になるように緩衝化され得る。
【0109】
適切な緩衝液は、酸、塩基、塩又はこれらの混合物及び一級、二級又は三級アミノ基を有するものを除く本分野で公知のあらゆる他の緩衝系から調製することができる。特に、10から500mMの濃度のホスファート緩衝液を使用することができる。水溶液は、前記アルドラーゼを水に添加し、無機酸、塩基、塩又はこれらの混合物の自動化された添加を用いて、反応の間にpHを維持することによっても調製することができる。
【0110】
あるいは、場合によって、塩(特に、50から500mMの濃度のNaCl)の存在下で、(特に、20g/Lから300g/L湿潤細胞重量の濃度範囲の、より具体的には、20g/Lから200g/L湿潤細胞重量の濃度範囲の)DERA過剰発現細胞、特にDERA過剰発現イー・コリ細胞の水性懸濁物中で、アルドラーゼが調製される。水性懸濁液は、水と混和性の有機溶媒(特に、2から15%V/Vの濃度範囲のジメチルスルホキシド)を含有し得、pH4.5から9、好ましくは、pH5から9、より好ましくは、pH6から9になるように緩衝化され得る。適切な緩衝液は、酸、塩基、塩又はこれらの混合物及び一級、二級又は三級アミノ基を有するものを除く本分野で公知のあらゆる他の緩衝系から調製することができる。特に、10から500mMの濃度のホスファート緩衝液を使用することができる。水性懸濁液は、前記DERA過剰発現細胞を水に添加し、無機酸、塩基、塩又はこれらの混合物の自動化された添加を用いて、反応の間にpHを維持することによっても調製することができる。
【0111】
方法の態様において、式IIIの化合物は、反応混合物へ連続的に添加され得、あるいは、式IIIの化合物は、1つのバッチ又はそれ以上のバッチ中の反応混合物に添加される。一態様において、混合物に添加される基質の総量は、添加される式IIIの化合物の総量が反応混合物の約20mmol/Lから反応混合物の約2mol/L、特に、反応混合物の約100mmol/Lから反応混合物の約1.5mol/L、より具体的には、反応混合物の約200mmol/Lから反応混合物の約700mol/Lである。アセトアルデヒドは、幾つかの手段によって添加され得る。一態様において、アセトアルデヒドは、1つのバッチ若しくはそれ以上のバッチ中であるいは連続的に、反応混合物に添加される。アセトアルデヒドは、式IIIの化合物と予め混合され、反応混合物に添加され得る。反応混合物に添加されるアセトアルデヒドの総量は、約0.1から約4モル濃度当量からアクセプター基質(化合物III)の総量まで、特に、約1から約3モル濃度当量、より好ましくは、約2から約2.5モル濃度当量である。特に、これは、所望でない産物、特に式XII及びXIIIの化合物の最小限の濃度を可能にするのに対して、式XIIの化合物は、アセトアルデヒドの一分子をIIIの一分子と反応させることによって得られ、式XIIIの化合物はアセトアルデヒドの三分子を反応させることによって得られる。
【0112】
【化23】
【0113】
好ましい実施形態において、基質は、反応のあらゆる所定の時点で、特定の流速で、プログラム可能なポンプを用いて反応混合物に継続的に添加される。流速は、基質が反応混合物中に蓄積しない最大の流速として決定される。特に、これは、所望でない産物の最小濃度を可能にする。より具体的には、この産物は、式XII及びXIIIの化合物であり得る。別の実施形態において、基質の阻害的効果は、正しい添加戦略を用いてさらに最小化することができる。
【0114】
あるいは、アルドラーゼは、式IIIの化合物又はアセトアルデヒドの少なくとも1つを含有する反応混合物に添加され得る。反応混合物は、溶媒と及びアルドラーゼ又は式IIIの化合物又はアセトアルデヒドの少なくとも1つを含むものと理解される。
【0115】
一態様において、アルドラーゼによって触媒される反応のために使用されるpHは、約5から10である。一実施形態において、アルドラーゼによって触媒される反応のために使用されるpHは、約5から約8である。具体的には、pHは、5から7の範囲の適切な緩衝液によって維持される。
【0116】
アルドラーゼ縮合中間体(特に、第一の縮合反応産物)は緩衝液と化学的な反応を起こし得るので、一般的に使用される幾つかの緩衝液は、アルドラーゼ縮合中間体(特に、第一の縮合反応産物)の利用可能性を制約することによって、先述のアルドラーゼによって触媒される反応の収率を低下させ得る。本発明者らは、ビス−トリスプロパンが前記中間体と反応することを発見した。同様に反応し得る他の緩衝液は、ビス−トリス、トリシン、トリス、ビシン又は一級、二級若しくは三級アミノ基を有するあらゆる他の緩衝液である。従って、pH値を調整するための適切な緩衝液は、この調整が必要であれば、酸、塩基、塩又はこれらの混合物、特に、リン酸及び水酸化ナトリウムを用いて作製される。
【0117】
一態様において、アルドラーゼによって触媒される反応のために使用される温度は、約20から約70℃である。一実施形態において、アルドラーゼによって触媒される反応のために使用される温度は、約25から約60℃である。一実施形態において、アルドラーゼによって触媒される反応のために使用される温度は、約30から約50℃である。
【0118】
反応は2から3時間以内に完了まで進行するので、産業的に適切である。
【0119】
反応の完了後、アセトニトリルの少なくとも約1容量を反応混合物の1容量に添加することによって、酵素は反応混合物から除去される。あるいは、酵素は、本分野において公知のあらゆる沈殿法によって除去される。一実施形態において、沈殿は、少なくとも5%m/Vの硫酸アンモニウムの添加によって行われる。あるいは、IVは、本分野において公知の塩析法によって抽出される。特に、塩析は、アセトニトリルの少なくとも約1容量から反応混合物の1容量及びNaClの5%(m/V)を添加することによって行われる。次いで、少なくとも40℃まで混合物を冷却し、液相を分離させる。次いで、アセトニトリル相を蒸発させて、IVの未精製産物を与える。あるいは、沈降技術、特に遠心を用いて、反応混合物から完全細胞触媒が除去される。別の態様において、完全細胞触媒は、ろ過技術、特に、微小ろ過によって除去され得る。
【0120】
本発明は、前記反応によって作製された純粋なラクトールを取得するための精製法も提供する。一態様において、アセトニトリルは、反応混合物から蒸発され、次いで、残存する水溶液が凍結乾燥される。別の態様において、沈降された沈殿溶液又は完全細胞触媒懸濁液の何れかの上清が凍結乾燥される。次いで、粉末化された残りがアセトニトリル/ジイソプロピルエーテル1:1中に懸濁される。不溶性の塩を除去するために、懸濁液をろ過し、移動相としてアセトニトリル/ジイソプロピルエーテル1:1を用いるシリカゲルカラムへろ液を搭載する。別の態様において、塩析抽出から得られるアセトニトリル相を蒸発させ、残りの油をアセトニトリル/ジイソプロピルエーテル1:1の最小容積中に溶解し、移動相としてアセトニトリル/ジイソプロピルエーテル1:1を用いるシリカゲルカラムに搭載する。
【0121】
特定の実施形態において、本発明は、使用されるアルドラーゼが(必要であれば、酸、塩基、塩又はこれらの混合物を用いて、特に、リン酸及び水酸化ナトリウムを用いて調整された)5から10、特に5から8のpH範囲の適切な溶媒(特に、水溶性有機溶媒と混合された水であり得る水性溶媒)中の、DERA01、DERA02であり、反応が約35から40℃の温度で進行し、転化が1から6時間で終了する、((2S,4R)−4,6−ジヒドロキシテトラヒドロ−2H−ピラン−2−イル)メチルアセタートを形成するための、アルドラーゼによって触媒されるアルドール縮合下でのCH3CO2CH2CHOのアセトアルデヒドとの反応を提供する。
【0122】
一般に、使用されるアルドラーゼは、「Sambrook, et al.(1989) Molecular cloning:A laboratory Manual 2nd Edition, New York:Cold Spring Harbor Laboratory Press, Cold Spring Harbor.」に記載されているタンパク質発現の方法によって調製される。アルドラーゼをコードする遺伝子は発現ベクター中にクローニングされ、酵素は適切な発現宿主中に発現される。
【0123】
反応収率は、反応混合物に添加された式IIIの化合物の総量に対して相対的に計算され、単離された産物のモルと反応混合物に添加された式IIIの化合物のモルとの間の比として測定される。
【0124】
本発明は、それぞれ、アルドラーゼの生物学的に活性な形態を過剰発現する微生物又は微生物の一部とアセトアルデヒド及び式XIVのアルデヒドR4CH2CHO(R4は、下記に定義されているとおりである。)を接触させる工程を含む、
式XVの化合物
【0125】
【化24】
(R4は、OCOR1(R1は、上に定義されているとおりである。)、塩化物、水素、それぞれ及び独立に置換された又は置換されていない、アリルオキシ及びベンジルオキシである。)
を調製する方法も提供する。好ましくは、R4は、アセタート、塩化物、水素であり、より好ましくは、R4はアセタートである。
【0126】
前記接触工程は、アルドール縮合が触媒されるように実施される。従って、完全細胞触媒としてアルドラーゼを過剰発現する生物が使用される。好ましくは、完全細胞触媒の形態の酵素は、上に定義されている2−デオキシリボース−5−リン酸アルドラーゼ(DERA、EC4.1.2.4)である。
【0127】
好ましくは、前記完全細胞触媒は、アルドラーゼの生物学的に活性な形態を過剰発現する細菌及び酵母から選択される。細菌は、好ましくは、エシェリヒア(Escherichia)、コリネバクテリウム(Corynebacterium)、シュードモナス(Pseudomonas)、ストレプトミセス(Streptomyces)、ロドコッカス(Rhodococcus)、バチルス(Bacillus)及びラクトバチルス(Lactobacillus)からなる属の群から選択され、より好ましくは、エシェリヒア・コリが使用される。酵母は、好ましくは、サッカロミセス(Saccharomyces)、ピチア(Pichia)、シゾサッカロミセス(Shizosaccharomyces)及びカンジダ(Candida)からなる属の群から好ましく選択される。
【0128】
本発明の特定の態様において、ロスバスタチンは、以下のスキームに開示されているように、本発明者らの式VIの化合物から開始して、WO2007/039287A1に従って調製することができる。
【0129】
【化25】
【0130】
ロスバスタチン又は他のスタチンを作製するために、式VIの化合物は、(式VIIの化合物を介した)2つの工程で、(2S,4R)−4−(保護された)−6−オキソ−テトラヒドロ−2H−ピラン−2−カルバルデヒドVIII又はその水和物VIII’へ転換される。アルデヒドVIII又はその水和物VIII’は、適切な試薬とのWittigカップリングの条件下で反応させることができ、続いて、必要であれば、水素が添加される。
【0131】
適切な試薬は、以下の式の複素環式又は脂環式誘導体(スタチンの骨格)である。
【0132】
【化26】
(Aは、結合又はOであり得、
並びにRx、Ry及びRzは同一又は別異であり、並びに場合によって置換された、C1−C8アルキル又はC3−C6シクロアルキル又はC1−C8アルケニル又はC5−C6シクロアルケニル又はアリールから選択され、
並びにXが陰イオン、好ましくは、ハロゲン又はカルボキシラート陰イオン、より好ましくは、塩化物、臭化物又はトリフルオロアセタートであり;
並びにHetは、スタチンの複素環式又は脂環式骨格を形成するように選択される。)
他のHMG−CoA還元酵素阻害剤(好ましくは、セリバスタチン、フルバスタチン、ピタバスタチン、ベルバスタチン、ダルバスタチンの中から選択される。)を同様に調製することができる。
【0133】
スタチンの複素環式又は脂環式骨格(Het)は、特に、以下から選択される。
【0134】
【化27】
【0135】
以下の実施例は、本発明の方法を例示するものであり、本発明の範囲を限定することを意図するものではない。
【実施例1】
【0136】
アセトキシアセトアルデヒド(III)
1.工程
【0137】
【化28】
トリエチルアミン(67mL、4当量)中に、1,4−ジヒドロキシブト−2−エン(10mL、0.12mol、1当量)を溶解した。溶液を0℃まで冷却し、無水酢酸(I)(34mL、3当量)を滴加した。得られた反応混合物を室温まで加温し、一晩撹拌した。
【0138】
1MH3PO4溶液(60mL)で2回、NaHCO31M溶液(60mL)で2回、溶液を洗浄した。次いで、MgSO4で溶液を脱水させ、濃縮した。高真空ポンプを70℃で使用して、AcOH及びAc2Oの微量を除去した。
【0139】
淡黄色ないし暗色の液体として、純粋なIIを得た(20.6g、100%)。1HNMR(300MHz,CDCl3)δ5.65(m,2H),4.58(m,4H),1.97(s,6H),13CNMR(75MHz,CDCl3)δ170.2,127.6,59.5,20.2。
【0140】
2.工程
【0141】
【化29】
1,4−ジアセトキシブト−2−エン(II)(1.7g、10mmol、1当量)をジクロロメタン(0.17M)中に溶解した。酸素(又は乾燥空気)散布を作動させ(約10L/h)、溶液を−80℃まで冷却した。−80℃になったら、オゾン発生装置を作動させ、溶液が青色に変化するまで、オゾンを通気した。次いで、オゾン発生装置を停止させ、青色が消失するまで、酸素(又は乾燥空気)を通気した。アルゴンを10分間通気した。散布を取り除き、−80℃で、溶液をアルゴン下に保った。硫化メチル(1.8mL、2.5当量)を滴加し、反応を室温まで加温し、20時間撹拌した。反応を濃縮して、1,4−アセトキシアセトアルデヒド(III)及びDMSOの2:1混合物を得た。収率は定量的であると思われ、1,4−ジアセトキシブト−2−エンの微量は存在しなかった。さらなる精製なしに、産物を使用した。(III)1HNMR(300MHz,CDCl3)δ9.51(s,1H),4.59(s,2H),2.10(s,3H),2.58(s,DMSO)。
【実施例2】
【0142】
アルドラーゼの調製
イー・コリK−12株由来の単離されたゲノムDNAを用いるPCR反応において、以下のオリゴヌクレオチドプライマーを用いて、エシェリヒア・コリ遺伝子deoCを増幅した。
CGGGATCCACTGATCTGAAAGCAAGCAGCC及び
GCAAGCTTGCTGCTGGCGCTCTTACC
(それぞれ、配列番号48及び49を有する。)
制限エンドヌクレアーゼBamHI及びHindIIIを用いて産物を切断し、得られた断片をアガロースゲル電気泳動上で分離し、精製した。同じ前記制限エンドヌクレアーゼを用いて、発現ベクターpQE30(Qiagene inc.,Valencia,CA,USA)を切断し、精製した。T4リガーゼ反応において、断片を集合させた。
【0143】
上記連結反応を用いて、形質転換受容性のエシェリヒア・コリDH5α細胞を形質転換した。アンピシリン耐性コロニーを培養し、プラスミドDNAを単離した。得られた構築物をpQE30DeraCと表記し、遺伝子配列を確認するために配列決定した。ベクターpQE30DeraCで形質転換受容性のエシェリヒア・コリTOP10F’株(Invitrogen corp.,Carlsbad,CA,USA)を形質転換することによって、アルドラーゼ発現生物を調製した。この方法に対して使用した方法は、「Sambrook et al. (1989) Molecular cloning:A Laboratory Manual 2nd Edition, New York:Cold Spring Harbor Laboratory Press, Cold Spring Harbor」に記載されており、当業者に周知である。
【0144】
TOP10F’PQE30DeraCの3mL一晩培養物とともに、アンピシリン(100μg/mL)が補充されたTerrific Broth培地(150mL、12g/Lバクト・トリプトン、24g/Lバクト酵母抽出物、4mL/Lグリセロール、2.31g/LKH2PO4、12.54g/LK2HPO4)に接種した。OD600が約0.8に到達するまで、細胞を増殖させた(37℃、250rpm)。IPTG(1mM最終濃度)を用いてタンパク質発現を誘導し、さらに4時間、同じ増殖条件中に細胞を放置した。遠心(10分、6000g、4℃)によって、細胞沈降物を採集した。上清を除去し、緩衝液(50mMNaH2PO4、pH7.0、300mMNaCl)の同じ容量によって交換した。沈降物を再懸濁し、遠心(10分、6000g、4℃)によって再度集めた。上清を除去し、使用前に、細胞を−20℃で保存した。DERA01を有する完全細胞触媒をこのようにして得た。
【0145】
あるいは、溶解緩衝液1L当り沈降物200gを用いて、溶解緩衝液(50mMNaH2PO4、pH7.0、300mMNaCl、2mMDTT)中に沈降物を再懸濁した。Bransonデジタル音波処理装置を用いて、細胞を音波処理し(3×15秒)、沈降(10分、20,000g、4℃)によって、細胞破片を除去した。DERA01の透明な水溶液をこのようにして得た。
【実施例3】
【0146】
((2S,4R)−4,6−ジヒドロキシテトラヒドロ−2H−ピラン−2−イル)メチルアセタート(IV)
【0147】
【化30】
DERA01の溶液600mL、反応緩衝液(50mMNaH2PO4、pH7.0、300mMNaCl、水中)200mL、アセトアルデヒド(反応緩衝液中1.05M溶液)の溶液100mL及びIII’の100mL(反応緩衝液中の500mM溶液)を撹拌された反応容器中で混合し、反応混合物1Lを得た。NaOHの1M水溶液を用いて、混合物のpH値を7.0に補正した。37℃に設定された温度制御された槽中で、混合物を3時間温置した。SynergyFusion、250×4.6mm、4μmカラムを使用して、ESIイオン化を備えた三連四重極HPLC−UV−MS/MSシステム上でのLC−MS分析を用いて、反応の間、IVの産生をモニターした。クロマトグラフィー条件は、以下のとおりであった。Tカラム=50℃、流速:1.5mL/分、Vinj=50μL。移動相を以下のように使用した。
A:水中の0.1%(m/v)NH4CH3COO、pH6.5
B:MilliQ水
C:アセトニトリル
線形勾配:
【0148】
【表1】
【0149】
保持時間=7.7±0.5分及び208の質量(M+NH4+に相当)を有するピーク面積の増大が反応の間に観察されたが、反応緩衝液で置換された反応混合物の一成分をそれぞれ有する対照の何れにもピーク面積の増大は観察されなかった。
【0150】
アセトニトリル4L及びNaCl50gの添加を用いて、反応を停止させた。懸濁液を0℃まで冷却し、遠心(10分、6000g、4℃)によって、液相を分離した。上方の相を取り出し、減圧下で蒸発させて、痰黄色の油(未精製産物)8.4gを得た。シリカゲルカラム(移動相:アセトニトリル/ジイソプロピルエーテル=1/1)上で未精製産物を精製した。その後、減圧下で溶液を蒸発させて、産物IV2.4gを得た。(IV):1HNMR(300MHz,CDCl3)δ5.39(d,1H),4.53(m,1H),4.29(m,1H),4.14(m,2H),2.11(s,3H),2.04−1.60(m,4H),13CNMR(75MHz,DMSO−d6)δ170.8,91.7,68.1,66.8,63.3,33.9,20.7
【実施例4】
【0151】
((2S,4R)−4,6−ジヒドロキシテトラヒドロ−2H−ピラン−2−イル)メチルアセタート(IV)
【0152】
【化31】
【0153】
反応緩衝液(50mMNaH2PO4、pH7.0、300mMNaCl、水中)750mL中に、DERA01を有する完全細胞触媒(300g)を懸濁し、37℃に設定した制御された温度で、撹拌された容器中において温置した。較正された蠕動式ポンプを用いて、2時間の反応時間の間に、基質溶液(630mMアセトアルデヒド(III)、反応緩衝液中の300mM溶液)750mLを添加した。NaOHの1M水溶液を用いて、pHを7.0に調節した。反応をさらに30分間継続し、次いで、遠心(10分、6000g、4℃)によって、完全細胞触媒を沈降させた。次いで、上清を凍結乾燥して淡黄色結晶(未精製産物)19.4gを得た。シリカゲルカラム(移動相:アセトニトリル/ジイソプロピルエーテル=1/1)上で未精製産物を精製した。その後、溶液を減圧下で蒸発させて、産物IV7.4gを得た。(IV):1HNMR(300MHz,CDCl3)δ5.39(d,1H),4.53(m,1H),4.29(m,1H),4.14(m,2H),2.11(s,3H),2.04−1.60(m,4H),13CNMR(75MHz,DMSO−d6)δ170.8,91.7,68.1,66.8,63.3,33.9,20.7。
【0154】
反応の間、LC−MS分析を用いて、IVの産生をモニターした。反応の間、208の質量を有するピーク面積(M+NH4+に相当)の増大が観察され、LC−MSを用いた2.5時間後の反応混合物の定量分析によって、IVの13.1g/Lの存在が示された。
【実施例5】
【0155】
((2S,4R)−4,6−ジヒドロキシテトラヒドロ−2H−ピラン−2−イル)メチルアセタート(IV)
【0156】
【化32】
【0157】
反応緩衝液(50mMNaH2PO4、pH7.0、300mMNaCl、水中)6mL中に、DERA02を有する完全細胞触媒(3g)を懸濁した。アセトアルデヒド(反応緩衝液中の2.1M溶液)の溶液2mL及びIII(反応緩衝液中の1M溶液)の2mLを反応管中に混合して、反応混合物10mLを得た。NaOHの1M水溶液を用いて、混合物のpH値を7.0に補正した。37℃に設定された温度制御された槽中で、混合物を3時間温置した。反応の間、LC−MS分析を用いて、IVの産生をモニターした。208の質量(M+NH4+に相当)を有するピーク面積の増大が反応の間に観察されたが、反応緩衝液で置換された反応混合物の一成分をそれぞれ有する対照の何れにもピーク面積の増大は観察されず、LC−MSを用いた1時間後における反応混合物の定量分析は、IVの8.7g/Lの存在を示した。
【実施例6】
【0158】
((2S,4R)−4,6−ジヒドロキシテトラヒドロ−2H−ピラン−2−イル)メチルアセタート(IV)
【0159】
【化33】
【0160】
反応緩衝液(50mMNaH2PO4、pH7.0、150mMNaCl)1070mL中に、IIIの56.6g(92%)を希釈した。NaHCO3塩を用いて、この溶液のpH値を6.2になるように調整した。DERA01を有する完全細胞触媒(700g/L)510mLを前溶液に添加し、NaHCO3塩を用いて、pH値を6.2になるように再度補正した。37℃及び一定の撹拌の800rpmに設定された、温度制御された2Lバイオリアクター内で、混合物を3時間温置した。下表に記載されている3時間の時間枠で、プログラム可能なポンプを用いて、反応緩衝液中に希釈されたアセトアルデヒド(45.4g)120mLを反応混合物に継続的に添加した。
【0161】
【表2】
【0162】
反応の間、pHは補正されず、5.5の最終値までゆっくり推移した。5.17分(化合物III)、14.04分(化合物XII)、14.44分化合物(化合物XIII)及び20.35分(化合物IV)での保持時間をモニタリングすることによって、GC分析(クロマトグラフィーカラム:DB−1 100%ジメチルポリシロキサン;温度プログラム:初期温度50℃、初期時間:5分、温度速度10℃/分、最終温度:215℃、最終時間:10分;注入装置:スプリット/スプリットレス注入装置、担体気体:ヘリウム、初期流速:10mL/分;検出装置:炎イオン化検出装置、検出装置温度:230℃;クロマトグラフィー溶液:IVの1から5mg/mLアセトニトリル)を用いて、IVの産生をモニターした。反応プロファイルは、図1に示されている。
【0163】
GCを用いた3時間後における反応混合物の定量分析は、67.7%モル濃度収率で、IVの35.3g/Lを示した。実施例6に記載されている物質に由来するさらなる合成工程中の化合物の鏡像異性体純度の分析は、99.8%又はそれ以上の鏡像異性体過剰を示し、これは、この物質の極めて高い鏡像異性体純度を示唆する。実施例6に記載されている物質に由来するさらなる合成工程中の化合物のジアステレオマー異性体純度の分析は、98%又はそれ以上のジアステレオマー異性体過剰を示し、これも、IVの極めて高いジアステレオマー異性体純度を示唆する。
【実施例7】
【0164】
((2S,4R)−4−ヒドロキシ−6−オキソテトラヒドロ−2H−ピラン−2−イル)メチルアセタート(V)
【0165】
【化34】
【0166】
水中の((2S,4R)−4,6−ジヒドロキシテトラヒドロ−2H−ピラン−2−イル)メチルアセタート(IV)(1当量)の溶液を0℃まで冷却した。炭酸バリウム(1.4当量)を添加した後、Br2(1.2当量)を滴加し、反応を室温で一晩撹拌した。溶液をNaClで飽和させ、EtOAcを用いて4回抽出した。合わせた有機相をMgSO4で脱水させ、濃縮した。フラッシュクロマトグラフィーを用いた精製(ヘキサン/アセトン=75/25から55/45)によって、((2S,4R)−4−ヒドロキシ−6−オキソテトラヒドロ−2H−ピラン−2−イル)メチルアセタート(V)を得た。
【0167】
(V):1HNMR(300MHz,アセトン−d6)δ4.88(m,1H),4.45(d,J=3.0Hz,1H),4.38(hex,J=3.0Hz,1H),4.23(dd,J=3.5Hz,J=12.0Hz,1H),4.16(dd,J=5.5Hz,J=12.1Hz,1H),2.68(dd,J=4.3Hz,J=17.5HZ,1H),2.51(dddd,J=0.8Hz,J=2.0Hz,J=3.3Hz,J=17.5Hz,1H),2.03(s,3H),1.91(m,2H),13CNMR(75MHz,アセトン−d6)δ170.8,169.7,74.2,66.5,62.7,39.1,32.3,20.6。
【実施例8】
【0168】
((2S,4R)−4−(tert−ブチルジメチルシリルオキシ)−6−オキソテトラヒドロ−2H−ピラン−2−イル)メチルアセタート(VI)
【0169】
【化35】
【0170】
((2S,4R)−4−ヒドロキシ−6−オキソテトラヒドロ−2H−ピランー2−イル)メチルアセタート(V)(1.88g、10mmol、1当量)の溶液を無水DMF(2mL、1M)中に溶解した。イミダゾール(0.88g、1.3当量)及びTBDMSCI(1.66g、1.1当量)を連続して添加し、反応が完了するまで、反応を撹拌した。水(20mL)とエーテル(20mL)の間に、反応混合物を分配した。エーテル(20mL)で1回、水相を抽出した。水(10mL)の少量、HCl1N(20mL)及び塩水(20mL)で、合わせた有機相を2回洗浄した。溶液をMgSO4で脱水させ、濃縮して、定量的収率で、((2S,4R)−4−(tert−ブチルジメチルシリルオキシ)−6−オキソテトラヒドロ−2H−ピラン−2−イル)メチルアセタート(VI)を得た。1HNMR(300MHz,CDCl3)δ4.93(m,1H),4.37(quint,J=3Hz,1H),4.30(dd,J=3Hz,J=12Hz,1H),4.21(dd,J=5Hz,J=12Hz,1H),2.62(d,J=4Hz,2H),2.11(s,3H),1.84−1.80(m,2H),0.89(s,9H),0.09(2s,6H)。13CNMR(75MHz,CDCl3)δ170.4,169.1,73.3,65.5,63.0,38.9,32.2,20.5,17.7,−5.1,−5.2。
【実施例9】
【0171】
酵素反応を介した((2S,4R)−4−(tert−ブチルジメチルシリルオキシ)−6−オキソテトラヒドロ−2H−ピラン−2−イル)メチルアセタート(VI)の(4R,6S)−4−(tert−ブチルジメチルシリルオキシ)−6−(ヒドロキシメチル)テトラヒドロ−2H−ピラン−2−オン(VII)への転化
【0172】
【化36】
【0173】
((2S,4R)−4−(tert−ブチルジメチルシリルオキシ)−6−オキソテトラヒドロ−2H−ピラン−2−イル)メチルアセタート(VI)(50g、80%純度;132.4mmol)をリン酸緩衝液(P.B.S)、pH=5.20(1.5L)に添加し、37℃まで溶液を加温した。次いで、パンクレアチン粉末(0.5当量質量;20g)を滴加した(6回、8g+3×4g)。平行して、pHをモニターし、pHを4.85と4.95の間に維持するために、1時間ごとに、NaHCO3溶液(1M)を添加することによってpHを制御した。酵素の最初の添加後9時間、反応を撹拌した。
【0174】
celite(R)を未精製混合物に添加した。celite(R)を通して、溶液をろ別した。淡黄色の液体を回収した。EtOAcの1.5Lを用いて、フィルター上の固体を洗浄した。ろ液を5分間撹拌した。2つの層を分離した。EtOAc(1.5L)を用いて、水相を1回再抽出した。40℃で減圧下において、合わせた有機相を部分的に蒸発させ、メチルシクロヘキサンから産物を再結晶させて、(4R,6S)−4−(tert−ブチルジメチルシリルオキシ)−6−(ヒドロキシメチル)テトラヒドロ−2H−ピラン−2−オン(VII)の82%を白色結晶として得た。反応の転化はほぼ定量的であり(>98%)、鏡像異性体は0.1%未満であった。
【0175】
スプリット/スプリットレス注入装置及びFID検出装置を有するBetadex120カラム上で、DCM溶液(2から3mg/mL)を用いて鏡像異性体を測定するためのGC分析を行った。
【0176】
化合物VIIのNMR分析は、検出限界での高スキャン蓄積を用いた場合でさえ、ジアステレオマー異性体が存在しないことを示し、VII中のジアステレオマー異性体のレベルが1%を下回ることを示唆している。
【実施例10】
【0177】
化学的反応を介した((2S,4R)−4−(tert−ブチルジメチルシリルオキシ)−6−オキソテトラヒドロ−2H−ピラン−2−イル)メチルアセタート(VI)の(4R,6S)−4−(tert−ブチルジメチルシリルオキシ)−6−(ヒドロキシメチル)テトラヒドロ−2H−ピラン−2−オン(VII)への転化
【0178】
【化37】
【0179】
((2S,4R)−4−(tert−ブチルジメチルシリルオキシ)−6−オキソテトラヒドロ−2H−ピラン−2−イル)メチルアセタート(VI)(0.625g、80%純度、1.65mmol)をTHF(8mL)及びMeOH(8mL)中に溶解した。スズ触媒(tBu2SnClOH)2(94mg、0.1当量)を添加し、反応を一晩撹拌した。反応を濃縮し、フラッシュクロマトグラフィーによって精製して、純粋な(4R,6S)−4−(tert−ブチルジメチルシリルオキシ)−6−(ヒドロキシメチル)テトラヒドロ−2H−ピラン−2−オン(VII)(126mg、30%)を得た。VIIは鏡像異性体の0.1%未満を含有することが明らかとなった。
【0180】
スプリット/スプリットレス注入装置及びFID検出装置を有するBetadex120カラム上で、DCM溶液(2から3mg/mL)を用いて鏡像異性体を測定するためのGC分析を行った。
【0181】
化合物VIIのNMR分析は、検出限界での高スキャン蓄積を用いた場合でさえ、ジアステレオマー異性体が存在しないことを示し、VII中のジアステレオマー異性体のレベルが1%を下回ることを示唆している。
【0182】
酵素的反応を介した及び化学的反応を介した化合物VIIへの化合物VIの転化は、望ましくない鏡像異性体が0.1%未満である優れた鏡像異性体純度を有する産物を与え、2つの方法によって調製された化合物VIIの鏡像異性体純度には、差は観察されなかった。また、調製された化合物VIIのジアステレオマー異性体純度は高く、望ましくないジアステレオマー異性体は1%未満であった。従って、鏡像異性体及びジアステレオマー異性体の過剰は、より初期の合成工程に(例えば、アルドラーゼによって触媒される化合物IIIの化合物IVへの転化に)起因する。
【特許請求の範囲】
【請求項1】
アセトアルデヒド及び式IIIR1CO2CH2CHO(R1は、下記に定義されているとおりである。)のアルデヒドを、アルドール縮合を触媒する酵素と接触させる工程を含む、式IVの化合物を調製する方法
【化1】
(R1は、それぞれ及び独立に、置換された又は置換されていない、アルキル、アルコキシ、アリール、ヘテロアリール、アリールアルキル又はヘテロアリールアルキルである。)。
【請求項2】
R1がそれぞれ及び独立に置換された又は置換されていない、C1−C6アルキル又はアルコキシである、好ましくはR1がCH3である、請求項1に記載の方法。
【請求項3】
式IVの化合物が98%若しくはそれ以上の鏡像異性体過剰及び/又は98%若しくはそれ以上のジアステレオマー異性体過剰を有する、請求項1又は2に記載の方法。
【請求項4】
酵素が2−デオキシリボース−5−リン酸アルドラーゼ(DERA、EC4.1.2.4)である、請求項1から3の何れか一項に記載の方法。
【請求項5】
前記酵素が以下のアルドラーゼの1つである、請求項1から4の何れかに記載の方法。
配列番号1のヌクレオチド配列若しくは配列番号2のアミノ酸配列を含むDERA01;
配列番号3若しくは配列番号4のヌクレオチド配列若しくは配列番号5のアミノ酸配列を含むDERA02;
配列番号6のヌクレオチド配列若しくは配列番号7のアミノ酸配列を含むDERA03;
配列番号8のヌクレオチド配列若しくは配列番号9のアミノ酸配列を含むDERA04;
配列番号10のヌクレオチド配列若しくは配列番号11のアミノ酸配列を含むDERA05;
配列番号12のヌクレオチド配列若しくは配列番号13のアミノ酸配列を含むDERA06;
配列番号14のヌクレオチド配列若しくは配列番号15のアミノ酸配列を含むDERA07;
配列番号16のヌクレオチド配列若しくは配列番号17のアミノ酸配列を含むDERA08;
配列番号18のヌクレオチド配列若しくは配列番号19のアミノ酸配列を含むDERA09;
配列番号20のヌクレオチド配列若しくは配列番号21のアミノ酸配列を含むDERA10;
配列番号22のヌクレオチド配列若しくは配列番号23のアミノ酸配列を含むDERA11;
配列番号24のヌクレオチド配列若しくは配列番号25のアミノ酸配列を含むDERA12;
配列番号26のヌクレオチド配列若しくは配列番号27のアミノ酸配列を含むDERA13;
配列番号28のヌクレオチド配列若しくは配列番号29のアミノ酸配列を含むDERA14;
配列番号30のヌクレオチド配列若しくは配列番号31のアミノ酸配列を含むDERA15;
配列番号32のヌクレオチド配列若しくは配列番号33のアミノ酸配列を含むDERA16;
配列番号34のヌクレオチド配列若しくは配列番号35のアミノ酸配列を含むDERA17;
DERA18は配列番号36のヌクレオチド配列若しくは配列番号37のアミノ酸配列を含むアルドラーゼであり;
配列番号38のヌクレオチド配列若しくは配列番号39のアミノ酸配列を含むDERA19;
配列番号40のヌクレオチド配列若しくは配列番号41のアミノ酸配列を含むDERA20;
配列番号42のヌクレオチド配列若しくは配列番号43のアミノ酸配列を含むDERA21;
配列番号44のヌクレオチド配列若しくは配列番号45のアミノ酸配列を含むDERA22;
配列番号46のヌクレオチド配列若しくは配列番号47のアミノ酸配列を含むDERA23;
又は前記アルドラーゼの何れかのアミノ酸配列と少なくとも約70%のアミノ酸配列同一性を有するアルドラーゼ。
【請求項6】
前記アルドラーゼが以下の群の1つから選択される、請求項4に記載の方法;
配列番号2のアミノ酸配列に対する少なくとも約70%のアミノ酸配列同一性;及び
配列番号5のアミノ酸配列に対する少なくとも80%のアミノ酸配列同一性;及び
配列番号11のアミノ酸配列に対する少なくとも80%のアミノ酸配列同一性;及び
配列番号25のアミノ酸配列に対する少なくとも80%のアミノ酸配列同一性;及び
配列番号27のアミノ酸配列に対する少なくとも80%のアミノ酸配列同一性。
【請求項7】
アルドラーゼによって触媒されるアルドール縮合に対するpHが、4.5から10、好ましくは5から10の範囲に維持され、より好ましくは、pHがpH範囲5から8の緩衝液を用いて維持される、請求項1から6の何れかに記載の方法。
【請求項8】
ホスファート緩衝液がアルドール縮合を触媒するために使用される、請求項1から7の何れか一項に記載の方法。
【請求項9】
アセトアルデヒド及び式IIIのアルデヒドを接触させる工程を含む、
式IVの化合物
【化2】
(R1は下記に定義のとおりである。)
を形成させるために、アルドラーゼによって触媒されるアルドール縮合条件下で、式IIIの基質R1CO2CH2CHO(R1は、それぞれ及び独立に、置換された又は置換されていない、アルキル、アルコキシ、アリール、ヘテロアリール、アリールアルキル又はヘテロアリールアルキルである。)をアセトアルデヒドと反応させるための、アルドラーゼの使用。
【請求項10】
前記アルドラーゼが2−デオキシリボース−5−リン酸アルドラーゼ、好ましくは、
配列番号1のヌクレオチド配列又は配列番号2のアミノ酸配列を含むDERA01;
配列番号3若しくは配列番号4のヌクレオチド配列又は配列番号5のアミノ酸配列を含むDERA02;
配列番号6のヌクレオチド配列又は配列番号7のアミノ酸配列を含むDERA03;
配列番号8のヌクレオチド配列又は配列番号9のアミノ酸配列を含むDERA04;
配列番号10のヌクレオチド配列又は配列番号11のアミノ酸配列を含むDERA05;
配列番号12のヌクレオチド配列又は配列番号13のアミノ酸配列を含むDERA06;
配列番号14のヌクレオチド配列又は配列番号15のアミノ酸配列を含むDERA07;
配列番号16のヌクレオチド配列又は配列番号17のアミノ酸配列を含むDERA08;
配列番号18のヌクレオチド配列又は配列番号19のアミノ酸配列を含むDERA09;
配列番号20のヌクレオチド配列又は配列番号21のアミノ酸配列を含むDERA10;
配列番号22のヌクレオチド配列又は配列番号23のアミノ酸配列を含むDERA11;
配列番号24のヌクレオチド配列又は配列番号25のアミノ酸配列を含むDERA12;
配列番号26のヌクレオチド配列又は配列番号27のアミノ酸配列を含むDERA13;
配列番号28のヌクレオチド配列又は配列番号29のアミノ酸配列を含むDERA14;
配列番号30のヌクレオチド配列又は配列番号31のアミノ酸配列を含むDERA15;
配列番号32のヌクレオチド配列又は配列番号33のアミノ酸配列を含むDERA16;
配列番号34のヌクレオチド配列又は配列番号35のアミノ酸配列を含むDERA17;
DERA18は配列番号36のヌクレオチド配列又は配列番号37のアミノ酸配列を含むアルドラーゼであり;
配列番号38のヌクレオチド配列又は配列番号39のアミノ酸配列を含むDERA19;
配列番号40のヌクレオチド配列又は配列番号41のアミノ酸配列を含むDERA20;
配列番号42のヌクレオチド配列又は配列番号43のアミノ酸配列を含むDERA21;
配列番号44のヌクレオチド配列又は配列番号45のアミノ酸配列を含むDERA22;
配列番号46のヌクレオチド配列又は配列番号47のアミノ酸配列を含むDERA23;
である、請求項9に記載の使用。
【請求項11】
前記アルドラーゼが生きた完全細胞内に含まれ、又は不活化された完全細胞内に含まれ、又は均質化された完全細胞内に含まれ、又は無細胞抽出物内に含まれ、又は精製された酵素であり、又は固定化されており、又は細胞外に発現されたタンパク質の形態である、請求項9又は10に記載の使用。
【請求項12】
酸化によって、式IVの化合物
【化3】
を式Vの化合物へ転化させる工程を含む、
式Vの化合物
【化4】
(R1は、請求項1に定義されている。)
を調製する方法。
【請求項13】
酸化がBr2及びBaCO3を用いて行われる、請求項12に記載の方法。
【請求項14】
a)式IIの化合物R2CO2CH2CH=CHCH2O2CR2(R2は、下記に定義されている。)を溶媒と及びオゾンと接触させる工程、並びに
b)工程a)から得られたオゾニドを加水分解する工程、
を含む、式III’のアルデヒドR2CO2CH2CHO(R2は、n−プロピル、シクロヘキシル、フェニル、モルホリン、ピロリジン及びイミダゾールを除く請求項1に定義されているR1である。)を作製する方法。
【請求項15】
工程a)の溶媒がジクロロメタンである、請求項14に記載の方法。
【請求項16】
R2が、C1−C6アルキル又はアルコキシ(n−プロピル、シクロヘキシルを除く。)であり、好ましくはR2がCH3である、請求項14又は15に記載の方法。
【請求項17】
工程a)が−50から−90℃の範囲の温度で実施され、及び/又は工程b)が室温で実施される、請求項14から16に記載の方法。
【請求項18】
工程a)の得られたオゾニドを硫化メチルと接触させることによって、工程b)が実施される、請求項14から17に記載の方法。
【請求項19】
式IV又はVに記載の化合物
【化5】
(R1は、それぞれ及び独立に、置換された又は置換されていない、アルキル、アルコキシ、アリール、ヘテロアリール、アリールアルキル又はヘテロアリールアルキルである。)。
【請求項20】
R1がC1−C6アルキル又はアルコキシ、好ましくは、R1がCH3である、請求項19に記載の化合物。
【請求項21】
a)式VIの化合物
【化6】
を得るために、保護基R3によって、4位の水酸基において、式Vの化合物
【化7】
(R1は、それぞれ及び独立に、置換された又は置換されていない、アルキル、アルコキシ、アリール、ヘテロアリール、アリールアルキル又はヘテロアリールアルキルである。)
を保護する工程、
b)スタチン又は医薬として許容されるその誘導体を作製するのに十分な条件下で、式VIの前記化合物を反応させる工程、
を含む、スタチン又はその誘導体を作製する方法。
【請求項22】
工程b)の条件が、アルデヒドへのVIの転化によって、及びスタチン又はその誘導体を得るための適切なホスホニウム塩とのこのアルデヒドのWittigカップリングによって設定され、
b1)式VIの化合物から式VIIIを有するアルデヒドを得る工程、
b2)式Xの化合物
【化8】
を得るために、式IXを有するホスホニウム塩を得る工程、
【化9】
(Rx、Ry及びRzは同一又は別異であり、並びに場合によって置換された、C1−C8アルキル又はC3−C6シクロアルキル又はC1−C8アルケニル又はC5−C6シクロアルケニル又はアリールから選択され、
並びにXは陰イオン、好ましくは、ハロゲン又はカルボキシラート陰イオン、より好ましくは、塩化物、臭化物又はトリフルオロアセタートである。)
b3)その後、化合物Xをロスバスタチン又はその塩へ転化する工程、
を好ましく含む、請求項21に記載の方法。
【請求項23】
式VIの化合物から式VIIIを有するアルデヒドを得る前記工程b1)が式VIIの化合物
【化10】
(R3は、独立に置換された又は置換されていない、シリル、ベンジル、アルキル及びアセチルから選択される保護基であり、好ましくは、R3は、場合によって置換された、C1−C8トリアルキルシリル、C1−C8ジアルキルアリールシリル、C1−C8アルキルジアリールシリルから選択され、アルキルは同一又は別異であり得、より好ましくは、保護基はtert−ブチルジメチルシリルである。)
を通じて行われる、請求項22に記載の方法。
【請求項24】
それぞれ、アルドラーゼの生物学的に活性な形態を過剰発現する微生物又は微生物の一部とアセトアルデヒド及び式IIIのアルデヒドを接触させることによって、アセトアルデヒド及び式IIIのアルデヒドを接触させる前記工程が達成される、請求項1から8の何れか一項に記載の方法。
【請求項25】
それぞれ、アルドラーゼの生物学的に活性な形態を過剰発現する微生物又は微生物の一部とアセトアルデヒド及び式XIVのアルデヒドR4CH2CHO(R4は、下記に定義されているとおりである。)を接触させる工程を含む、
式XVの化合物
【化11】
(R4は、それぞれ及び独立に置換された又は置換されていない、OCOR1(R1は、請求項1に定義されているとおりである。)、塩化物、水素、アリルオキシ及びベンジルオキシである。)
を調製する方法。
【請求項26】
アルドール縮合が触媒されるように、接触工程が行われる、請求項24から25の何れか一項に記載の方法。
【請求項27】
前記酵素が完全細胞触媒の形態であり、前記完全細胞触媒がアルドラーゼの生物学的に活性な形態を過剰発現する微生物である、請求項1から8の何れか一項に記載の方法。
【請求項28】
アセトアルデヒドと式XIVのアルデヒドR4CH2CHO(R4が下記に定義されている。)を、アルドール縮合を触媒する酵素と接触させることを含み、前記酵素が完全細胞触媒の形態であり、前記完全細胞触媒がアルドラーゼの生物学的に活性な形態を過剰発現する微生物である、
式XVの化合物
【化12】
(R4は、OCOR1(R1は、請求項1に定義されているとおりである。)、塩化物、水素、それぞれ及び独立に、置換された又は置換されていない、アリルオキシ及びベンジルオキシである。)
を調製する方法。
【請求項29】
前記微生物が、好ましくはエシェリヒア(Escherichia)、コリネバクテリウム(Corynebacterium)、シュードモナス(Pseudomonas)、ストレプトミセス(Streptomyces)、ロドコッカス(Rhodococcus)、バチルス(Bacillus)及びラクボタシラス(Lactobacillus)からなる属の群から選択される細菌であり、より好ましくは、エシェリヒア・コリ(Escherichia coli)が使用される、請求項24から28の何れか一項に記載の方法。
【請求項30】
前記微生物が、好ましくはサッカロミセス(Saccharomyces)、ピチア(Pichia)、シゾサッカロミセス(Shizosaccharomyces)及びカンジダ(Candida)からなる属の群から選択される酵母である、請求項24から28の何れか一項に記載の方法。
【請求項31】
式V、VI又はVIIの前記化合物が99.8%若しくはそれ以上の鏡像異性体過剰及び/又は98%若しくはそれ以上のジアステレオマー異性体過剰を有する、請求項1から8、12から13、21から24又は27の何れか一項に記載の方法。
【請求項32】
それぞれ、99.8%若しくはそれ以上の鏡像異性体過剰及び/又は98%若しくはそれ以上のジアステレオマー異性体過剰を有する、式V、VI又はVIIの化合物。
【請求項1】
アセトアルデヒド及び式IIIR1CO2CH2CHO(R1は、下記に定義されているとおりである。)のアルデヒドを、アルドール縮合を触媒する酵素と接触させる工程を含む、式IVの化合物を調製する方法
【化1】
(R1は、それぞれ及び独立に、置換された又は置換されていない、アルキル、アルコキシ、アリール、ヘテロアリール、アリールアルキル又はヘテロアリールアルキルである。)。
【請求項2】
R1がそれぞれ及び独立に置換された又は置換されていない、C1−C6アルキル又はアルコキシである、好ましくはR1がCH3である、請求項1に記載の方法。
【請求項3】
式IVの化合物が98%若しくはそれ以上の鏡像異性体過剰及び/又は98%若しくはそれ以上のジアステレオマー異性体過剰を有する、請求項1又は2に記載の方法。
【請求項4】
酵素が2−デオキシリボース−5−リン酸アルドラーゼ(DERA、EC4.1.2.4)である、請求項1から3の何れか一項に記載の方法。
【請求項5】
前記酵素が以下のアルドラーゼの1つである、請求項1から4の何れかに記載の方法。
配列番号1のヌクレオチド配列若しくは配列番号2のアミノ酸配列を含むDERA01;
配列番号3若しくは配列番号4のヌクレオチド配列若しくは配列番号5のアミノ酸配列を含むDERA02;
配列番号6のヌクレオチド配列若しくは配列番号7のアミノ酸配列を含むDERA03;
配列番号8のヌクレオチド配列若しくは配列番号9のアミノ酸配列を含むDERA04;
配列番号10のヌクレオチド配列若しくは配列番号11のアミノ酸配列を含むDERA05;
配列番号12のヌクレオチド配列若しくは配列番号13のアミノ酸配列を含むDERA06;
配列番号14のヌクレオチド配列若しくは配列番号15のアミノ酸配列を含むDERA07;
配列番号16のヌクレオチド配列若しくは配列番号17のアミノ酸配列を含むDERA08;
配列番号18のヌクレオチド配列若しくは配列番号19のアミノ酸配列を含むDERA09;
配列番号20のヌクレオチド配列若しくは配列番号21のアミノ酸配列を含むDERA10;
配列番号22のヌクレオチド配列若しくは配列番号23のアミノ酸配列を含むDERA11;
配列番号24のヌクレオチド配列若しくは配列番号25のアミノ酸配列を含むDERA12;
配列番号26のヌクレオチド配列若しくは配列番号27のアミノ酸配列を含むDERA13;
配列番号28のヌクレオチド配列若しくは配列番号29のアミノ酸配列を含むDERA14;
配列番号30のヌクレオチド配列若しくは配列番号31のアミノ酸配列を含むDERA15;
配列番号32のヌクレオチド配列若しくは配列番号33のアミノ酸配列を含むDERA16;
配列番号34のヌクレオチド配列若しくは配列番号35のアミノ酸配列を含むDERA17;
DERA18は配列番号36のヌクレオチド配列若しくは配列番号37のアミノ酸配列を含むアルドラーゼであり;
配列番号38のヌクレオチド配列若しくは配列番号39のアミノ酸配列を含むDERA19;
配列番号40のヌクレオチド配列若しくは配列番号41のアミノ酸配列を含むDERA20;
配列番号42のヌクレオチド配列若しくは配列番号43のアミノ酸配列を含むDERA21;
配列番号44のヌクレオチド配列若しくは配列番号45のアミノ酸配列を含むDERA22;
配列番号46のヌクレオチド配列若しくは配列番号47のアミノ酸配列を含むDERA23;
又は前記アルドラーゼの何れかのアミノ酸配列と少なくとも約70%のアミノ酸配列同一性を有するアルドラーゼ。
【請求項6】
前記アルドラーゼが以下の群の1つから選択される、請求項4に記載の方法;
配列番号2のアミノ酸配列に対する少なくとも約70%のアミノ酸配列同一性;及び
配列番号5のアミノ酸配列に対する少なくとも80%のアミノ酸配列同一性;及び
配列番号11のアミノ酸配列に対する少なくとも80%のアミノ酸配列同一性;及び
配列番号25のアミノ酸配列に対する少なくとも80%のアミノ酸配列同一性;及び
配列番号27のアミノ酸配列に対する少なくとも80%のアミノ酸配列同一性。
【請求項7】
アルドラーゼによって触媒されるアルドール縮合に対するpHが、4.5から10、好ましくは5から10の範囲に維持され、より好ましくは、pHがpH範囲5から8の緩衝液を用いて維持される、請求項1から6の何れかに記載の方法。
【請求項8】
ホスファート緩衝液がアルドール縮合を触媒するために使用される、請求項1から7の何れか一項に記載の方法。
【請求項9】
アセトアルデヒド及び式IIIのアルデヒドを接触させる工程を含む、
式IVの化合物
【化2】
(R1は下記に定義のとおりである。)
を形成させるために、アルドラーゼによって触媒されるアルドール縮合条件下で、式IIIの基質R1CO2CH2CHO(R1は、それぞれ及び独立に、置換された又は置換されていない、アルキル、アルコキシ、アリール、ヘテロアリール、アリールアルキル又はヘテロアリールアルキルである。)をアセトアルデヒドと反応させるための、アルドラーゼの使用。
【請求項10】
前記アルドラーゼが2−デオキシリボース−5−リン酸アルドラーゼ、好ましくは、
配列番号1のヌクレオチド配列又は配列番号2のアミノ酸配列を含むDERA01;
配列番号3若しくは配列番号4のヌクレオチド配列又は配列番号5のアミノ酸配列を含むDERA02;
配列番号6のヌクレオチド配列又は配列番号7のアミノ酸配列を含むDERA03;
配列番号8のヌクレオチド配列又は配列番号9のアミノ酸配列を含むDERA04;
配列番号10のヌクレオチド配列又は配列番号11のアミノ酸配列を含むDERA05;
配列番号12のヌクレオチド配列又は配列番号13のアミノ酸配列を含むDERA06;
配列番号14のヌクレオチド配列又は配列番号15のアミノ酸配列を含むDERA07;
配列番号16のヌクレオチド配列又は配列番号17のアミノ酸配列を含むDERA08;
配列番号18のヌクレオチド配列又は配列番号19のアミノ酸配列を含むDERA09;
配列番号20のヌクレオチド配列又は配列番号21のアミノ酸配列を含むDERA10;
配列番号22のヌクレオチド配列又は配列番号23のアミノ酸配列を含むDERA11;
配列番号24のヌクレオチド配列又は配列番号25のアミノ酸配列を含むDERA12;
配列番号26のヌクレオチド配列又は配列番号27のアミノ酸配列を含むDERA13;
配列番号28のヌクレオチド配列又は配列番号29のアミノ酸配列を含むDERA14;
配列番号30のヌクレオチド配列又は配列番号31のアミノ酸配列を含むDERA15;
配列番号32のヌクレオチド配列又は配列番号33のアミノ酸配列を含むDERA16;
配列番号34のヌクレオチド配列又は配列番号35のアミノ酸配列を含むDERA17;
DERA18は配列番号36のヌクレオチド配列又は配列番号37のアミノ酸配列を含むアルドラーゼであり;
配列番号38のヌクレオチド配列又は配列番号39のアミノ酸配列を含むDERA19;
配列番号40のヌクレオチド配列又は配列番号41のアミノ酸配列を含むDERA20;
配列番号42のヌクレオチド配列又は配列番号43のアミノ酸配列を含むDERA21;
配列番号44のヌクレオチド配列又は配列番号45のアミノ酸配列を含むDERA22;
配列番号46のヌクレオチド配列又は配列番号47のアミノ酸配列を含むDERA23;
である、請求項9に記載の使用。
【請求項11】
前記アルドラーゼが生きた完全細胞内に含まれ、又は不活化された完全細胞内に含まれ、又は均質化された完全細胞内に含まれ、又は無細胞抽出物内に含まれ、又は精製された酵素であり、又は固定化されており、又は細胞外に発現されたタンパク質の形態である、請求項9又は10に記載の使用。
【請求項12】
酸化によって、式IVの化合物
【化3】
を式Vの化合物へ転化させる工程を含む、
式Vの化合物
【化4】
(R1は、請求項1に定義されている。)
を調製する方法。
【請求項13】
酸化がBr2及びBaCO3を用いて行われる、請求項12に記載の方法。
【請求項14】
a)式IIの化合物R2CO2CH2CH=CHCH2O2CR2(R2は、下記に定義されている。)を溶媒と及びオゾンと接触させる工程、並びに
b)工程a)から得られたオゾニドを加水分解する工程、
を含む、式III’のアルデヒドR2CO2CH2CHO(R2は、n−プロピル、シクロヘキシル、フェニル、モルホリン、ピロリジン及びイミダゾールを除く請求項1に定義されているR1である。)を作製する方法。
【請求項15】
工程a)の溶媒がジクロロメタンである、請求項14に記載の方法。
【請求項16】
R2が、C1−C6アルキル又はアルコキシ(n−プロピル、シクロヘキシルを除く。)であり、好ましくはR2がCH3である、請求項14又は15に記載の方法。
【請求項17】
工程a)が−50から−90℃の範囲の温度で実施され、及び/又は工程b)が室温で実施される、請求項14から16に記載の方法。
【請求項18】
工程a)の得られたオゾニドを硫化メチルと接触させることによって、工程b)が実施される、請求項14から17に記載の方法。
【請求項19】
式IV又はVに記載の化合物
【化5】
(R1は、それぞれ及び独立に、置換された又は置換されていない、アルキル、アルコキシ、アリール、ヘテロアリール、アリールアルキル又はヘテロアリールアルキルである。)。
【請求項20】
R1がC1−C6アルキル又はアルコキシ、好ましくは、R1がCH3である、請求項19に記載の化合物。
【請求項21】
a)式VIの化合物
【化6】
を得るために、保護基R3によって、4位の水酸基において、式Vの化合物
【化7】
(R1は、それぞれ及び独立に、置換された又は置換されていない、アルキル、アルコキシ、アリール、ヘテロアリール、アリールアルキル又はヘテロアリールアルキルである。)
を保護する工程、
b)スタチン又は医薬として許容されるその誘導体を作製するのに十分な条件下で、式VIの前記化合物を反応させる工程、
を含む、スタチン又はその誘導体を作製する方法。
【請求項22】
工程b)の条件が、アルデヒドへのVIの転化によって、及びスタチン又はその誘導体を得るための適切なホスホニウム塩とのこのアルデヒドのWittigカップリングによって設定され、
b1)式VIの化合物から式VIIIを有するアルデヒドを得る工程、
b2)式Xの化合物
【化8】
を得るために、式IXを有するホスホニウム塩を得る工程、
【化9】
(Rx、Ry及びRzは同一又は別異であり、並びに場合によって置換された、C1−C8アルキル又はC3−C6シクロアルキル又はC1−C8アルケニル又はC5−C6シクロアルケニル又はアリールから選択され、
並びにXは陰イオン、好ましくは、ハロゲン又はカルボキシラート陰イオン、より好ましくは、塩化物、臭化物又はトリフルオロアセタートである。)
b3)その後、化合物Xをロスバスタチン又はその塩へ転化する工程、
を好ましく含む、請求項21に記載の方法。
【請求項23】
式VIの化合物から式VIIIを有するアルデヒドを得る前記工程b1)が式VIIの化合物
【化10】
(R3は、独立に置換された又は置換されていない、シリル、ベンジル、アルキル及びアセチルから選択される保護基であり、好ましくは、R3は、場合によって置換された、C1−C8トリアルキルシリル、C1−C8ジアルキルアリールシリル、C1−C8アルキルジアリールシリルから選択され、アルキルは同一又は別異であり得、より好ましくは、保護基はtert−ブチルジメチルシリルである。)
を通じて行われる、請求項22に記載の方法。
【請求項24】
それぞれ、アルドラーゼの生物学的に活性な形態を過剰発現する微生物又は微生物の一部とアセトアルデヒド及び式IIIのアルデヒドを接触させることによって、アセトアルデヒド及び式IIIのアルデヒドを接触させる前記工程が達成される、請求項1から8の何れか一項に記載の方法。
【請求項25】
それぞれ、アルドラーゼの生物学的に活性な形態を過剰発現する微生物又は微生物の一部とアセトアルデヒド及び式XIVのアルデヒドR4CH2CHO(R4は、下記に定義されているとおりである。)を接触させる工程を含む、
式XVの化合物
【化11】
(R4は、それぞれ及び独立に置換された又は置換されていない、OCOR1(R1は、請求項1に定義されているとおりである。)、塩化物、水素、アリルオキシ及びベンジルオキシである。)
を調製する方法。
【請求項26】
アルドール縮合が触媒されるように、接触工程が行われる、請求項24から25の何れか一項に記載の方法。
【請求項27】
前記酵素が完全細胞触媒の形態であり、前記完全細胞触媒がアルドラーゼの生物学的に活性な形態を過剰発現する微生物である、請求項1から8の何れか一項に記載の方法。
【請求項28】
アセトアルデヒドと式XIVのアルデヒドR4CH2CHO(R4が下記に定義されている。)を、アルドール縮合を触媒する酵素と接触させることを含み、前記酵素が完全細胞触媒の形態であり、前記完全細胞触媒がアルドラーゼの生物学的に活性な形態を過剰発現する微生物である、
式XVの化合物
【化12】
(R4は、OCOR1(R1は、請求項1に定義されているとおりである。)、塩化物、水素、それぞれ及び独立に、置換された又は置換されていない、アリルオキシ及びベンジルオキシである。)
を調製する方法。
【請求項29】
前記微生物が、好ましくはエシェリヒア(Escherichia)、コリネバクテリウム(Corynebacterium)、シュードモナス(Pseudomonas)、ストレプトミセス(Streptomyces)、ロドコッカス(Rhodococcus)、バチルス(Bacillus)及びラクボタシラス(Lactobacillus)からなる属の群から選択される細菌であり、より好ましくは、エシェリヒア・コリ(Escherichia coli)が使用される、請求項24から28の何れか一項に記載の方法。
【請求項30】
前記微生物が、好ましくはサッカロミセス(Saccharomyces)、ピチア(Pichia)、シゾサッカロミセス(Shizosaccharomyces)及びカンジダ(Candida)からなる属の群から選択される酵母である、請求項24から28の何れか一項に記載の方法。
【請求項31】
式V、VI又はVIIの前記化合物が99.8%若しくはそれ以上の鏡像異性体過剰及び/又は98%若しくはそれ以上のジアステレオマー異性体過剰を有する、請求項1から8、12から13、21から24又は27の何れか一項に記載の方法。
【請求項32】
それぞれ、99.8%若しくはそれ以上の鏡像異性体過剰及び/又は98%若しくはそれ以上のジアステレオマー異性体過剰を有する、式V、VI又はVIIの化合物。
【図1】

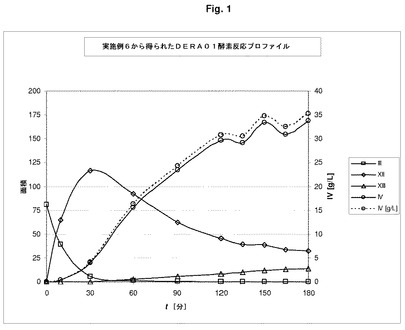
【公表番号】特表2011−510038(P2011−510038A)
【公表日】平成23年3月31日(2011.3.31)
【国際特許分類】
【出願番号】特願2010−543473(P2010−543473)
【出願日】平成21年1月20日(2009.1.20)
【国際出願番号】PCT/EP2009/050583
【国際公開番号】WO2009/092702
【国際公開日】平成21年7月30日(2009.7.30)
【出願人】(504359293)レツク・フアーマシユーテイカルズ・デー・デー (60)
【Fターム(参考)】
【公表日】平成23年3月31日(2011.3.31)
【国際特許分類】
【出願日】平成21年1月20日(2009.1.20)
【国際出願番号】PCT/EP2009/050583
【国際公開番号】WO2009/092702
【国際公開日】平成21年7月30日(2009.7.30)
【出願人】(504359293)レツク・フアーマシユーテイカルズ・デー・デー (60)
【Fターム(参考)】
[ Back to top ]