
(5S)−5−[4−(5−クロロ−ピリジン−2−イルオキシ)−ピペリジン−1−スルホニルメチル]−5−メチル−イミダゾリジン−2,4−ジオン(I)の新規結晶形Gおよびその中間体
(5S)−5−[4−(5−クロロ−ピリジン−2−イルオキシ)−ピペリジン−1−スルホニルメチル]−5−メチル−イミダゾリジン−2,4−ジオンの新規結晶多形を、そのような多形の製造方法、そのような多形を含む医薬組成物、治療におけるそのような多形の使用と共に記載する。
【発明の詳細な説明】
【技術分野】
【0001】
発明の分野
本発明は、(5S)−5−[4−(5−クロロ−ピリジン−2−イルオキシ)−ピペリジン−1−スルホニルメチル]−5−メチル−イミダゾリジン−2,4−ジオンの新規結晶多形、このような多形の製造方法、そのような多形を含む医薬組成物、および治療におけるそのような多形の使用を開示する。
【背景技術】
【0002】
発明の背景
引用によりその全体を本明細書に包含させるWO02/074767は、治療に有用なメタロプロテイナーゼ阻害剤のクラスを教示する。
【0003】
WO02/074767は、さらに、(5S)−5−[4−(5−クロロ−ピリジン−2−イルオキシ)−ピペリジン−1−スルホニルメチル]−5−メチル−イミダゾリジン−2,4−ジオンとしてその中で同定されている具体的メタロプロテイナーゼ阻害剤化合物を開示する(65頁、15〜27行;および120頁、23〜29行)。この化合物を、ここでは化合物(I)と呼ぶ。
【化1】

WO02/074767は、さらに、化合物(I)の製造方法も開示する。
【0004】
それ故に、一つの態様において、化合物(I)は、以下のスキーム(WO02/074767;87、113および120頁)に示す経路に準じるが、工程(d)において適当なアミンに置き換えて製造する:
【化2】
スキーム1の試薬および条件:a)KCN、(NH4)2CO3、EtOH/H2O、+90℃、3時間;b)キラル分割、CHIRALPAK AD、溶離剤としてメタノール;c)Cl2(g)、AcOH/H2O、<+15℃、25分;d)ジイソプロピルエチルアミン、THF。−20℃、30分。
【0005】
次いで、得られた化合物(I)を沈殿とエタノール/水の洗浄、または分取HPLCのいずれかにより精製する。
【0006】
第二の態様において、化合物(I)のラセミ体、(5RS)−5−[4−(5−クロロ−ピリジン−2−イルオキシ)−ピペリジン−1−スルホニルメチル]−5−メチル−イミダゾリジン−2,4−ジオンを、1−[4−(5−クロロ−ピリジン−2−イルオキシ)−ピペリジン−1−スルホニル]−プロパン−2−オンと過剰のカリウムシアニドおよび炭酸アンモニウムを、エタノール中で反応させ、生成物を沈殿により単離することにより製造した。次いで、化合物(I)、(5S)−エナンチオマーをキラルHPLCにより得た(WO02/074767;55および65頁)。
化合物(I)の結晶形はWO02/074767に開示されていない。
【0007】
化合物(I)は強力なメタロプロテイナーゼ阻害剤、特にMMP12の強力な阻害剤であり、それ自体治療に有用である。しかしながら、WO02/074767に記載の方法に従って製造したとき、化合物(I)は、熱力学的安定性に関して予測されない固体状態特性を示す。米国および他の国際的な保健登録認定機関(health registration authorities)の要求に従って、ヒトに投与するための化合物(I)を含む医薬製剤を製造するために、一定の物理特性を有する、安定な結晶形態のような安定な形で化合物(I)を製造する必要がある。
【0008】
多形は、特定の化合物が、同じ化学式を維持しながら異なる結晶多形に結晶化する能力として特徴付けることができる。ある物質の多形は、同じ方法で互いに結合している同じ原子を含んで化学的に同一であるが、それらの結晶多形が異なり、これは溶解速度、融点、かさ密度、安定性、流動特性などのような1種以上の物理的特性に影響し得る。本発明において特定の化合物について使用するとき、用語“多形”、“結晶多形”、“結晶形”、“結晶の多形”および“(結晶)形態”は同義語であると理解すべきである。
【0009】
本発明は、固体状態における化合物(I)の熱力学的特性を改善するための方法を提供し、それ故に、一貫したそして遊離な物理特性を有する安定な結晶多形の化合物(I)を提供する。
【0010】
図面の簡単な説明
図1は、化合物(I)形態GのX線粉末回折ダイアグラムである;
図2は、化合物(I)形態Gの示差走査熱量測定(DSC)トレースおよび熱重量分析(TGA)トレースである;
図3は、化合物(I)形態AのX線粉末回折ダイアグラムである;
図4は、化合物(I)形態BのX線粉末回折ダイアグラムである;
図5は、化合物(I)形態CのX線粉末回折ダイアグラムである;
図6は、化合物(I)形態DのX線粉末回折ダイアグラムである;
図7は、化合物(I)形態EのX線粉末回折ダイアグラムである;
図8は、化合物(I)形態FのX線粉末回折ダイアグラムである。
【発明の開示】
【0011】
発明の開示
本発明により、驚くべきことに、(5S)−5−[4−(5−クロロ−ピリジン−2−イルオキシ)−ピペリジン−1−スルホニルメチル]−5−メチル−イミダゾリジン−2,4−ジオン、化合物(I)が少なくとも7個の異なる結晶多形(多形)で存在できることが判明した。
【化3】
一つの局面において、本発明は、式(I)の化合物の7種の多形形態を提供する。
【0012】
一つの態様において、本発明は、形態Gと命名し、CuKα放射を使用して測定して10.1、16.2、16.8および19.0°2θにおける特異的ピークを含むX線粉末回折(XPRD)パターンを有することを特徴とする、(5S)−5−[4−(5−クロロ−ピリジン−2−イルオキシ)−ピペリジン−1−スルホニルメチル]−5−メチル−イミダゾリジン−2,4−ジオンの結晶多形を提供する。
【0013】
他の態様において、本発明は、形態Gと命名し、CuKα放射を使用して測定して9.7、10.1、11.5、12.8、14.1、16.2、16.8および19.0°2θにおける特異的ピークを含むX線粉末回折(XPRD)パターンを有することを特徴とする、(5S)−5−[4−(5−クロロ−ピリジン−2−イルオキシ)−ピペリジン−1−スルホニルメチル]−5−メチル−イミダゾリジン−2,4−ジオンの結晶多形を提供する。
【0014】
他の態様において、本発明は、形態Gと命名し、CuKα放射を使用して測定して図1に示すものと実質的に同じX線粉末回折(XPRD)パターンを有することを特徴とする、(5S)−5−[4−(5−クロロ−ピリジン−2−イルオキシ)−ピペリジン−1−スルホニルメチル]−5−メチル−イミダゾリジン−2,4−ジオンの結晶多形を提供する。
【0015】
他の態様において、本発明は、形態Gと命名し、図2に示すものと実質的に同じ示差走査熱量測定(DSC)トレースを有することを特徴とする、(5S)−5−[4−(5−クロロ−ピリジン−2−イルオキシ)−ピペリジン−1−スルホニルメチル]−5−メチル−イミダゾリジン−2,4−ジオンの結晶多形を提供する。
【0016】
他の態様において、本発明は、形態Aと命名し、6.8、9.8、13.7、16.4、18.4、18.7、20.4および22.6°2θにおける特異的ピークを含むX線粉末回折パターンを有することを特徴とする、(5S)−5−[4−(5−クロロ−ピリジン−2−イルオキシ)−ピペリジン−1−スルホニルメチル]−5−メチル−イミダゾリジン−2,4−ジオンの結晶多形を提供する。他の態様において、本発明は、図3に示すものと実質的に同じX線粉末回折パターンを有することを特徴とする、(5S)−5−[4−(5−クロロ−ピリジン−2−イルオキシ)−ピペリジン−1−スルホニルメチル]−5−メチル−イミダゾリジン−2,4−ジオンの結晶多形を提供する。
【0017】
他の態様において、本発明は、形態Bと命名し、6.6、7.1、8.3、9.0、13.6、14.3、16.8および17.7°2θにおける特異的ピークを含むX線粉末回折パターンを有することを特徴とする、(5S)−5−[4−(5−クロロ−ピリジン−2−イルオキシ)−ピペリジン−1−スルホニルメチル]−5−メチル−イミダゾリジン−2,4−ジオンの結晶多形を提供する。他の態様において、本発明は、形態Bと命名し、図4に示すものと実質的に同じX線粉末回折パターンを有することを特徴とする、(5S)−5−[4−(5−クロロ−ピリジン−2−イルオキシ)−ピペリジン−1−スルホニルメチル]−5−メチル−イミダゾリジン−2,4−ジオンの結晶多形を提供する。
【0018】
他の態様において、本発明は、形態Cと命名し、6.3、12.8、14.3、16.6、17.8、19.4、22.2および23.7°2θにおける特異的ピークを含むX線粉末回折パターンを有することを特徴とする、(5S)−5−[4−(5−クロロ−ピリジン−2−イルオキシ)−ピペリジン−1−スルホニルメチル]−5−メチル−イミダゾリジン−2,4−ジオンの結晶多形を提供する。他の態様において、本発明は、形態Cと命名し、図5に示すものと実質的に同じX線粉末回折パターンを有することを特徴とする、(5S)−5−[4−(5−クロロ−ピリジン−2−イルオキシ)−ピペリジン−1−スルホニルメチル]−5−メチル−イミダゾリジン−2,4−ジオンの結晶多形を提供する。
【0019】
他の態様において、本発明は、形態Dと命名し、6.6、10.9、11.2、15.6、15.9、17.7、18.2および18.4°2θにおける特異的ピークを含むX線粉末回折パターンを有することを特徴とする、(5S)−5−[4−(5−クロロ−ピリジン−2−イルオキシ)−ピペリジン−1−スルホニルメチル]−5−メチル−イミダゾリジン−2,4−ジオンの結晶多形を提供する。他の態様において、本発明は、形態Dと命名し、図6に示すものと実質的に同じX線粉末回折パターンを有することを特徴とする、(5S)−5−[4−(5−クロロ−ピリジン−2−イルオキシ)−ピペリジン−1−スルホニルメチル]−5−メチル−イミダゾリジン−2,4−ジオンの結晶多形を提供する。
【0020】
他の態様において、本発明は、形態Eと命名し、12.1、13.9、14.5、14.8、15.3、16.2、18.7および19.8°2θにおける特異的ピークを含むX線粉末回折パターンを有することを特徴とする、(5S)−5−[4−(5−クロロ−ピリジン−2−イルオキシ)−ピペリジン−1−スルホニルメチル]−5−メチル−イミダゾリジン−2,4−ジオンの結晶多形を提供する。他の態様において、本発明は、形態Eと命名し、図7に示すものと実質的に同じX線粉末回折パターンを有することを特徴とする、(5S)−5−[4−(5−クロロ−ピリジン−2−イルオキシ)−ピペリジン−1−スルホニルメチル]−5−メチル−イミダゾリジン−2,4−ジオンの結晶多形を提供する。
【0021】
他の態様において、本発明は、形態Fと命名し、7.4、9.5、13.9、14.9、17.3、18.1、20.0および20.4°2θにおける特異的ピークを含むX線粉末回折パターンを有することを特徴とする、(5S)−5−[4−(5−クロロ−ピリジン−2−イルオキシ)−ピペリジン−1−スルホニルメチル]−5−メチル−イミダゾリジン−2,4−ジオンの結晶多形を提供する。他の態様において、本発明は、形態Fと命名し、図8に示すものと実質的に同じX線粉末回折パターンを有することを特徴とする、(5S)−5−[4−(5−クロロ−ピリジン−2−イルオキシ)−ピペリジン−1−スルホニルメチル]−5−メチル−イミダゾリジン−2,4−ジオンの結晶多形を提供する。
【0022】
X線粉末回折(XPRD)パターンのピークの相対的強度は、試験下のサンプルの方位および使用した装置のタイプおよび設定によって変わることがあり、従って、本明細書に含まれるXPRDトレースの強度は、説明的であり、絶対的比較に使用することを意図しないことは理解されよう。
【0023】
本発明の結晶多形または形態は、好ましくは実質的に純粋であり、式(I)の化合物の各結晶多形または形態が10重量%未満、好ましくは5重量%未満、好ましくは3重量%未満、好ましくは1重量%未満の、本化合物の他の結晶多形または形態を含む不純物を含むことを意味する。
【0024】
故に、一つの態様において、本発明は、形態Gと命名され、CuKα放射を使用して測定して10.1、16.2、16.8および19.0°2θにおける特異的ピークを含むX線粉末回折(XPRD)パターンを有することを特徴とする、(5S)−5−[4−(5−クロロ−ピリジン−2−イルオキシ)−ピペリジン−1−スルホニルメチル]−5−メチル−イミダゾリジン−2,4−ジオンの実質的に純粋な結晶多形を提供する。
【0025】
他の態様において、本発明は、形態Gと命名され、CuKα放射を使用して測定して9.7、10.1、11.5、12.8、14.1、16.2、16.8および19.0°2θにおける特異的ピークを含むX線粉末回折(XPRD)パターンを有することを特徴とする、(5S)−5−[4−(5−クロロ−ピリジン−2−イルオキシ)−ピペリジン−1−スルホニルメチル]−5−メチル−イミダゾリジン−2,4−ジオンの実質的に純粋な結晶多形を提供する。
【0026】
他の態様において、本発明は、形態Gと命名し、CuKα放射を使用して測定して図1に示すものと実質的に同じX線粉末回折(XPRD)パターンを有することを特徴とする、(5S)−5−[4−(5−クロロ−ピリジン−2−イルオキシ)−ピペリジン−1−スルホニルメチル]−5−メチル−イミダゾリジン−2,4−ジオンの実質的に純粋な結晶多形を提供する。
【0027】
化合物(I)形態Gは、針状晶癖を示す結晶を含む、白色結晶性粉末として得られる。本物質はX線粉末回折測定で測定して、本質的に100%結晶である。本結晶構造を、単結晶X線回折により決定した。本結晶において、分子は、斜方晶系空間群(P212121)にパックされている。非対称単位セル(a=10.510Å、b=11.169Å、c=15.560Å)に4分子存在する。内部空間の欠乏をもたらす最密充填は、1.46g/mLの相対的に高い密度で認められる。
【0028】
単結晶X線回折データを使用して計算した化合物(I)の模擬X線粉末回折パターンは、図1に示す実験により決定したパターンと良く一致する。回折ピークの一は非常に合い、相対ピーク強度の差異は、選択方位効果に帰する。
【0029】
WO02/074767に開示の方法で製造したとき、(5S)−5−[4−(5−クロロ−ピリジン−2−イルオキシ)−ピペリジン−1−スルホニルメチル]−5−メチル−イミダゾリジン−2,4−ジオンは、非晶相または形態Aまたは形態Cまたはそれらの混合物で得られる。
【0030】
化合物(I)形態Aの融点は、加熱によりそれが約175℃で形態Bに変形するために観察されていない。
【0031】
化合物(I)形態Bは、形態Aを約175℃に加熱したときの固体状態転移により製造する。形態Bは約207℃で融解し、その後形態Cに再結晶し、続いて再び約210℃で融解する。
化合物(I)形態Cは約210℃で融解する。
【0032】
化合物(I)形態Dは、化合物(I)を融解物からの結晶化により製造したときに産生される。例えば、形態Dを、形態B(室温で形態Aから開始)を形態Bの融解温度で融解させ;次いで室温に急冷して、非晶物質を産生し;次いで、再び5°/分で加熱することにより製造する。加熱の間に、この非晶物質はガラス遷移温度を通り、その後形態Dとして再結晶する。形態Dは約209℃で融解する。
【0033】
化合物(I)形態Eを、形態CをpH3の水中、例えば、環境温度で数日間スラリー化したとき産生する。形態Aと同様、形態Eは、約175℃で、恐らく形態Bに熱転移する。
【0034】
化合物(I)形態Fは、形態Aまたは形態Cを、エタノール中、例えば、環境温度で数日間スラリー化したとき産生する。形態Aおよび形態Eと同様、形態Fは、約175℃で、恐らく形態Bに熱転移する。
【0035】
化合物(I)形態Gは、(5S)−5−[4−(5−クロロ−ピリジン−2−イルオキシ)−ピペリジン−1−スルホニルメチル]−5−メチル−イミダゾリジン−2,4−ジオンを水性エタノールまたは工業用変性アルコール水溶液(aqueous industrial methylated spirits)から再結晶したときに再現性よく産生される。形態Gは約201℃で融解し、使用する条件、例えば、加熱速度に依存して、その後一部または完全に形態Cに再結晶し、それは次いで約210℃で再融解する。
【0036】
化合物(I)形態AからGのいずれかを加熱したとき、融解前に、約175℃で起こり得る上記に略記した可能性のある固体状態転移以外、溶媒損失も何らかの他の熱事象も見られない。故に、形態AからGの各々は熱的に安定である。
【0037】
化合物(I)形態AからGの相対的な熱力学的安定性は、形態AからGの混合物を5日間、5から40℃の範囲の温度で、水中共インキュベートした懸濁液実験により評価した。全例で、得られた沈殿のX線粉末回折試験(XRPD)は、形態Gへの完全な変換を証明した。同じ結果が、種々の有機溶媒(エタノール、メタノール、1−プロパノール、2−プロパノール、アセトンまたは酢酸エチル)中の形態Aの懸濁液のインキュベーション後に観察された。これらの結果に基づき、化合物(I)形態Gが、試験した温度範囲で、7種の結晶多形の中で最も熱力学的に安定であると結論付けることができる。
【0038】
ここに開示の方法を使用して、化合物(I)形態Gは小規模、中規模または大規模合成法に従って再現性よく製造できる。
【0039】
事実上溶媒分子のための内部空間が示されない化合物(I)形態Gの単結晶X線構造決定から予測される通り、重量測定的蒸気収着(GVS)を使用した吸湿測定は、本物質が、高相対湿度でさえほとんど湿気取り込みがないことを示した(80%RHで<0.05%湿度取り込み)。本物質は、それ故に、欧州薬局方に定義された基準に従って、非吸湿性と有利に分離される。
【0040】
化合物(I)形態Gは、優れたそして非常に有利な固体状態特性を有する。それは結晶性、非吸湿性であり、そして200℃以下で熱的に安定であり、融解前に溶媒損失も何らかの他の熱的事象も示さない(DSCおよびTGAトレース、図2参照)。形態Gはまた、化合物(I)の既知の7種の結晶多形の中で熱力学的に最も安定である。
【0041】
化合物(I)形態Gの固体状態安定性を3条件:25℃/乾燥;25℃/60%RH;および40℃/75%RHで試験した。サンプルを、2週間、4週間、8週間および12週間後に試験し、化学的および物理的安定性を評価した。本物質は、何らかの可能性のある分解産物と比較して(5S)−5−[4−(5−クロロ−ピリジン−2−イルオキシ)−ピペリジン−1−スルホニルメチル]−5−メチル−イミダゾリジン−2,4−ジオンの含量(勾配RPLC)に;化合物(I)の形態(XRPD)に;化合物(I)の形態学(SEM)に;溶媒含量(TGA)に;または融解行動(DSC)に変化が見られなかったため、全貯蔵条件下で化学的および物理的に安定であると結論付けられた。それ故に、化合物(I)形態Gは、薬学的に適切な貯蔵条件下で、固体状態で優れたそして有利な化学および物理的安定性を有すると見なされる。
【0042】
一つの局面において、本発明は、(5S)−5−[4−(5−クロロ−ピリジン−2−イルオキシ)−ピペリジン−1−スルホニルメチル]−5−メチル−イミダゾリジン−2,4−ジオン形態Gを提供する。
【0043】
さらなる局面において、本発明は、(5S)−5−[4−(5−クロロ−ピリジン−2−イルオキシ)−ピペリジン−1−スルホニルメチル]−5−メチル−イミダゾリジン−2,4−ジオン形態Gの製造方法を提供する。故に、一つの局面において、本発明は、水性エタノールからの結晶化または再結晶を含む、(5S)−5−[4−(5−クロロ−ピリジン−2−イルオキシ)−ピペリジン−1−スルホニルメチル]−5−メチル−イミダゾリジン−2,4−ジオン形態Gの製造方法を提供する。他の局面において、本発明は、工業用変性アルコール水溶液からの結晶化または再結晶を含む、(5S)−5−[4−(5−クロロ−ピリジン−2−イルオキシ)−ピペリジン−1−スルホニルメチル]−5−メチル−イミダゾリジン−2,4−ジオン形態Gの製造方法を提供する。
【0044】
他の局面において、本発明は、以下の工程を含む、(5S)−5−[4−(5−クロロ−ピリジン−2−イルオキシ)−ピペリジン−1−スルホニルメチル]−5−メチル−イミダゾリジン−2,4−ジオン形態Gの製造方法を提供する:
i) (5S)−5−[4−(5−クロロ−ピリジン−2−イルオキシ)−ピペリジン−1−スルホニルメチル]−5−メチル−イミダゾリジン−2,4−ジオンを、工業用変性アルコール(IMS):水の2:1混合物に添加し;
ii) 本混合物を加熱還流して溶液を得て;
iii)熱溶液を濾過し;
iv) 本濾液を加熱還流し、次いでそれを約0.5℃/分の速度で約20℃まで冷却し;
v) (5S)−5−[4−(5−クロロ−ピリジン−2−イルオキシ)−ピペリジン−1−スルホニルメチル]−5−メチル−イミダゾリジン−2,4−ジオン形態Gを回収し、乾燥させる。
【0045】
さらなる局面において、本発明は、治療に使用するための(5S)−5−[4−(5−クロロ−ピリジン−2−イルオキシ)−ピペリジン−1−スルホニルメチル]−5−メチル−イミダゾリジン−2,4−ジオン形態Gを提供する。
【0046】
さらなる局面において、本発明は、MMP活性の阻害が有益である疾患または状態の処置または予防用医薬の製造において使用するための、(5S)−5−[4−(5−クロロ−ピリジン−2−イルオキシ)−ピペリジン−1−スルホニルメチル]−5−メチル−イミダゾリジン−2,4−ジオンの結晶多形を提供する。
【0047】
さらなる局面において、本発明は、MMP活性の阻害が有益である疾患または状態の処置用医薬の製造に使用するための、(5S)−5−[4−(5−クロロ−ピリジン−2−イルオキシ)−ピペリジン−1−スルホニルメチル]−5−メチル−イミダゾリジン−2,4−ジオン形態Gを提供する。
【0048】
さらなる局面において、本発明は、処置を必要とする患者に治療的有効量の(5S)−5−[4−(5−クロロ−ピリジン−2−イルオキシ)−ピペリジン−1−スルホニルメチル]−5−メチル−イミダゾリジン−2,4−ジオンの結晶多形を投与することを含む、
MMP活性が仲介する疾患または状態の処置または予防方法を提供する。
【0049】
さらなる局面において、本発明は、処置を必要とする患者に治療的有効量の(5S)−5−[4−(5−クロロ−ピリジン−2−イルオキシ)−ピペリジン−1−スルホニルメチル]−5−メチル−イミダゾリジン−2,4−ジオン形態Gを投与することを含む、MMP活性が仲介する疾患または状態の処置または予防方法を提供する。
【0050】
特に、化合物(I)は、MMP12および/またはMMP13および/またはMMP9および/またはMMP8および/またはMMP3が仲介する疾患または状態の処置;とりわけMMP12および/またはMMP9が仲介する疾患または状態の処置;特にMMP12が仲介する疾患または状態の処置に有用である。
【0051】
さらなる局面において、本発明は、(5S)−5−[4−(5−クロロ−ピリジン−2−イルオキシ)−ピペリジン−1−スルホニルメチル]−5−メチル−イミダゾリジン−2,4−ジオンの結晶多形を含む医薬組成物を提供する。
【0052】
さらなる局面において、本発明は、(5S)−5−[4−(5−クロロ−ピリジン−2−イルオキシ)−ピペリジン−1−スルホニルメチル]−5−メチル−イミダゾリジン−2,4−ジオン形態Gを含む医薬組成物を提供する。
【0053】
さらなる局面において、本発明は、処置を必要とする患者に治療的有効量の(5S)−5−[4−(5−クロロ−ピリジン−2−イルオキシ)−ピペリジン−1−スルホニルメチル]−5−メチル−イミダゾリジン−2,4−ジオンの結晶多形を含む医薬組成物を投与することを含む、メタロプロテイナーゼ活性が仲介する疾患または状態の処置方法を提供する。
【0054】
さらなる局面において、本発明は、処置を必要とする患者に治療的有効量の(5S)−5−[4−(5−クロロ−ピリジン−2−イルオキシ)−ピペリジン−1−スルホニルメチル]−5−メチル−イミダゾリジン−2,4−ジオン形態Gを含む医薬組成物を投与することを含む、メタロプロテイナーゼ活性が仲介する疾患または状態の処置方法を提供する。
【0055】
さらなる局面において、本発明は、MMP活性の阻害が有益である疾患または状態の処置のための、(5S)−5−[4−(5−クロロ−ピリジン−2−イルオキシ)−ピペリジン−1−スルホニルメチル]−5−メチル−イミダゾリジン−2,4−ジオンの結晶多形を含む医薬製剤の使用を提供する。
【0056】
さらなる局面において、本発明は、MMP活性の阻害が有益である疾患または状態の処置のための、(5S)−5−[4−(5−クロロ−ピリジン−2−イルオキシ)−ピペリジン−1−スルホニルメチル]−5−メチル−イミダゾリジン−2,4−ジオン形態Gを含む医薬製剤の使用を提供する。
【0057】
他の局面において、本発明は、炎症性疾患または状態の処置または予防のための医薬の製造における式(I)の化合物形態Gの使用;および炎症性疾患または状態を処置、またはリスクを軽減する方法であって、該疾患または状態に罹患している、またはリスクのあるヒトに治療的有効量の式(I)の化合物形態Gを投与することを含む、方法を提供する。
【0058】
化合物(I)は、間欠性および永続性両方のおよび全ての重症度、および気道過敏反応性の他の原因を含む、気管支、アレルギー性、内因性、外因性、運動誘発性、薬剤誘発性(アスピリンおよびNSAID誘発性を含む)および粉塵誘発性喘息を含む、喘息;慢性閉塞性肺疾患(COPD);感染性および好酸球性気管支炎を含む気管支炎;気腫;気管支拡張症;嚢胞性線維症;サルコイドーシス;農夫肺および関連疾患;過敏性肺炎;原因不明線維化肺胞炎、特発性間質性肺炎、抗新生物治療および結核およびアスペルギルス症および他の真菌感染を含む慢性感染に合併する線維症を含む、肺線維症;肺移植の合併症;肺脈管構造の血管炎性および血栓性障害、および肺高血圧;気道の炎症性および分泌状態と関連する慢性咳、および医原性咳;薬物性鼻炎、および血管運動性鼻炎を含む急性および慢性鼻炎;神経性鼻炎(枯草熱)を含む通年性および季節性アレルギー性鼻炎;鼻のポリープ症;一般的な風邪、および呼吸器多核体ウイルス、インフルエンザ、コロナウイルス(SARSを含む)およびアデノウイルスによる感染を含む、急性ウイルス感染を含む、気道の閉塞性疾患のような呼吸管の疾患の処置に使用できる。
【0059】
化合物(I)はまた、原発性および、例えば、先天的股関節異形成症に二次性両方の骨関節症/骨関節症と関連するまたは含む関節炎(arthritides);頚部および腰部脊椎炎、および背下部および頚部痛;リウマチ性関節炎およびスチル病;強直性脊椎炎、乾癬性関節炎、反応性関節炎および未分化脊椎関節症(spondarthropathy)を含む血清反応陰性脊椎関節症;敗血症性関節炎およびポット病およびポンセ病を含む結核のような、他の感染関連関節症(arthopathies)および骨障害;尿酸塩痛風、ピロリン酸カルシウム沈着疾患、およびカルシウムアパタイト関連腱、滑液包および滑膜炎症を含む、急性および慢性結晶誘発滑膜炎;ベーチェット病;原発性および二次性シェーグレン症候群;全身性硬化症および限局型強皮症;全身性エリテマトーデス、混合型結合組織疾患、および未分化結合組織疾患;皮膚筋炎および多発性筋炎を含む炎症性ミオパシー;リウマチ性多発筋痛症;どんな関節分布であれ特発性炎症性関節炎(arthritides)および関連症候群、およびリウマチ熱およびその全身合併症を含む若年性関節炎;巨細胞性動脈炎、高安動脈炎、チャーグ・ストラウス症候群、結節性多発性動脈炎、顕微鏡的多発動脈炎、およびウイルス感染、過敏症反応、クリオグロブリン、およびパラプロテインと関連する脈管炎を含む脈管炎;背下部疼痛;家族性地中海熱、マックル・ウェルズ症候群、および家族性アイルランド熱(Familial Hibernian Fever)、キクチ病;薬剤誘発性関節痛(arthalgias)、腱炎(tendonititides)、およびミオパシーのような骨および関節の疾患の処置にも使用できる。
【0060】
化合物(I)はまた、疼痛および傷害[例えば、運動傷害]または疾患による筋骨格障害の結合組織リモデリング:関節炎(arthitides)(例えばリウマチ性関節炎、骨関節症、痛風または結晶性関節症)、他の関節疾患(例えば椎間板変性または側頭下顎関節変性)、骨リモデリング疾患(例えば骨粗鬆症、ページェット病または骨壊死)、多発性軟骨炎、強皮症、混合型結合組織障害、脊椎関節症または歯周疾患(例えば歯周炎)の処置にも使用できる。
【0061】
化合物(I)はまた、乾癬、アトピー性皮膚炎、接触性皮膚炎または他の湿疹性皮膚炎、および遅延型過敏症反応;植物および光皮膚炎;脂漏性皮膚炎、疱疹状皮膚炎、扁平苔癬、硬化性萎縮性苔癬、壊疽性膿皮症、皮膚サルコイド、円板状エリテマトーデス、天疱瘡、類天疱瘡、表皮水疱症、蕁麻疹、血管浮腫、脈管炎、毒性紅斑、皮膚好酸球増加症、円形脱毛症、男性型禿頭、スウィート症候群、ウェーバー・クリスチャン症候群、多形性紅斑;感染性および非感染性両方の蜂巣炎;脂肪織炎;皮膚リンパ腫、非黒色腫皮膚癌および他の形成異常病巣;固定薬疹を含む薬剤誘発性障害のような皮膚の疾患の処置にも使用できる。
【0062】
化合物(I)はまた、眼瞼炎;通年性および春季アレルギー性結膜炎を含む結膜炎;虹彩炎;前部および後部ブドウ膜炎;脈絡膜炎;自己免疫性;網膜に影響する変性または炎症性障害;交感神経性眼炎を含む眼炎;サルコイドーシス;ウイルス、真菌、および細菌を含む感染のような眼の疾患の処置にも使用できる。
【0063】
化合物(I)はまた舌炎、歯肉炎、歯周炎;逆流性を含む食道炎;好酸球性胃腸炎、肥満細胞症、クローン病、潰瘍性大腸炎を含む大腸炎、直腸炎、肛門掻痒症;セリアック病、過敏性腸症候群、非炎症性下痢、および腸から離れて作用し得る食物関連アレルギー(例えば、偏頭痛、鼻炎または湿疹)のような胃腸管の疾患の処置にも使用できる。
【0064】
化合物(I)はまた、冠血管および末梢循環に影響するアテローム性動脈硬化症;心膜炎;心筋炎、心筋サルコイドを含む炎症性および自己免疫性心筋症;虚血再灌流傷害;感染性(例えば梅毒性)を含む心内膜炎、弁膜炎、および大動脈炎;脈管炎;深部静脈血栓症および静脈瘤の合併症を含む静脈炎および血栓症を含む、近位および末梢静脈の障害のような心血管系の処置にも使用できる。
【0065】
化合物(I)はまた、転移疾患および腫瘍再発、および新生物随伴症候群の予防および処置を含む;前立腺、乳房、肺、卵巣、膵臓、腸および結腸、胃、皮膚および脳腫瘍および骨髄(白血病を含む)およびホジキンおよび非ホジキンリンパ腫のようなリンパ増殖性系に影響する悪性物を含む一般的な癌の処置のような腫瘍学においても使用できる。
【0066】
特に、化合物(I)は、成人呼吸窮迫症候群(ARDS)、嚢胞性線維症、肺気腫、慢性閉塞性肺疾患(COPD)、肺高血圧、喘息、鼻炎、虚血−再灌流傷害、リウマチ性関節炎、骨関節症、癌、アテローム性動脈硬化症および胃粘膜傷害の処置に使用できる。
【0067】
より具体的に、化合物(I)は慢性閉塞性肺疾患(COPD)、喘息および鼻炎の処置に使用できる。
さらに具体的に、化合物(I)は慢性閉塞性肺疾患(COPD)の処置に使用できる。
【0068】
予防は、以前に当該疾患または状態の事象に罹患したか、他の点でリスクが増加していると見なされるヒトの処置に特に適切である。特定の疾患または状態を発症するリスクのあるヒトは、一般に、該疾患または状態の家族歴がある、または遺伝子試験またはスクリーニングにより該疾患または状態の発症に特に感受性であることが同定されているヒトを含む。
【0069】
上記の治療適応症について、投与すべき化合物の容量は、処置すべき疾患、疾患の重症度、投与形態、患者の年齢、体重および性別による。このような因子は担当医により決定され得る。しかしながら、一般に、本化合物を、ヒトに0.1mg/kgから100mg/kg(活性成分として測定)の1日量を投与したときに、満足いく結果が得られる。
【0070】
式(I)の結晶化合物は、それ自体で、または本発明の化合物を薬学的に許容される希釈剤、アジュバントまたは担体と組み合わせて含む適当な医薬製剤の形で使用できる。特に好ましいのは、有害反応、例えば、アレルギー性反応の原因となり得る物質を含まない組成物である。適当な医薬製剤の選択および製造のための慣用法は、例えば、“Pharmaceuticals - The Science of Dosage Form Designs”, M. E. Aulton, Churchill Livingstone, 1988に記載されている。
【0071】
本発明によって、好ましくは95重量%未満、より好ましくは50重量%未満の式(I)の化合物形態Gを、薬学的に許容される希釈剤または担体と組み合わせて含む、医薬製剤が提供される。
【0072】
我々は、これらの成分を混合することを含む、医薬製剤の製造方法も提供する。
【0073】
本化合物は、例えば、肺および/または気道に、溶液、懸濁液、HFAエアロゾルまたは乾燥粉末製剤の形で、例えば、Turbuhaler(登録商標)として既知の吸入デバイス中の製剤として、局所的に;または例えば、錠剤、ピル、カプセル、シロップ、粉末または顆粒の形で、経口投与により、全身的に;または例えば、非経腸溶液または懸濁液の形で、非経腸(腹腔内、静脈内、皮下または筋肉内注射を含む)投与により;または例えば、坐薬の形で、直腸投与により投与できる。
【0074】
本発明の化合物の乾燥粉末製剤および加圧HFAエアロゾルは、経口または経鼻吸入により投与できる。吸入のために、本化合物は、望ましくは微粉化する。微粉化された化合物は、好ましくは10μm未満の質量中央径を有し、噴射剤混合物に、分散剤、例えばC8−C20脂肪酸またはその塩(例えば、オレイン酸)、胆汁塩、リン脂質、アルキルサッカライド、過フッ素化またはポリエトキシル化界面活性剤、または他の薬学的に許容される分散剤の助けを借りて、懸濁し得る。
【0075】
本発明の化合物はまた乾燥粉末吸入器の手段により投与してもよい。本吸入器は1回量または多回量吸入器であってよく、呼気作動型乾燥粉末吸入器であってよい。
【0076】
一つの可能性は、微粉化した化合物と担体物質、例えば、モノ、ジまたはポリサッカライド、糖アルコール、または他のポリオールの混合である。適当な担体は、糖類、例えば、ラクトース、グルコース、ラフィノース、メレジトース、ラクチトール、マルチトール、トレハロース、スクロース、マンニトール;およびデンプンである。あるいは、微粉化した化合物を、他の物質でコーティングし得る。粉末混合物をまた、各々所望量の活性化合物を含む硬ゼラチンカプセルに分配してもよい。
【0077】
他の可能性は、微粉化した粉末の、吸入工程中に破壊する球体への加工である。この球状粉末を、多回容量吸入器、例えば、Turbuhaler(登録商標)として既知の吸入器の貯蔵部に充填し得て、ここで、投与ユニットが所望量を定量し、次いでそれが患者により吸入される。このシステムで、活性化合物は、担体物質と共にまたは無しで、患者に送達される。
【0078】
経口投与のために、活性化合物をアジュバントまたは担体、例えば、ラクトース、サッカロース、ソルビトール、マンニトール;デンプン、例えば、ジャガイモデンプン、コーンデンプンまたはアミロペクチン;セルロース誘導体;結合剤、例えば、ゼラチンまたはポリビニルピロリドン;および/または平滑剤、例えば、ステアリン酸マグネシウム、ステアリン酸カルシウム、ポリエチレングリコール、蝋、パラフィンなどと混合し、次いで、錠剤に圧縮してよい。コーティング錠が必要ならば、上記のように製造したコアを、例えば、アラビアゴム、ゼラチン、タルク、二酸化チタンなどを含み得る濃縮糖溶液でコーティングし得る。あるいは、錠剤を、易揮発性の有機溶媒に溶解した適当なポリマーでコーティングして良い。
【0079】
軟ゼラチンカプセルの製造のために、化合物を、例えば、植物油またはポリエチレングリコールと混合し得る。硬ゼラチンカプセルは、錠剤について上記の賦形剤のいずれかを使用し、化合物の顆粒を含み得る。また、薬剤の液体または半固体製剤を硬ゼラチンカプセルに充填し得る。
【0080】
経口投与用液体製剤は、化合物を含むシロップまたは懸濁液、例えば、溶液の形であり得て、バランスは糖およびエタノール、水、グリセロールとプロピレングリコールの混合物である。所望により、このような液体製剤は、着色剤、香味剤、濃化剤としてサッカリンおよび/またはカルボキシメチルセルロース、または当業者に既知の他の賦形剤を含み得る。
【0081】
本発明のさらなる局面において、我々は、化合物(I)の新規合成方法を提供する。特に、化合物(I)の結晶多形の新規合成方法を開示する。特に、化合物(I)形態Gの新規合成方法を開示する。
【0082】
化合物(I)の好ましい合成方法をスキーム2に示す。
【化4】
【0083】
スキーム2において、化合物(II)、(III)および(IV)の硫黄部分をS−ベンジル誘導体として保護する。当業者は、t−ブチルのような他の適当な保護基を、代わりに使用できることを容易に認識しよう。故に、簡便のために、以降の反応はS−ベンジル保護された化合物を使用して示すが、t−ブチルのような適当な他の保護基も使用できることは理解すべきである。
【0084】
5−クロロ−2−(ピペリジン−4−イルオキシ)−ピリジン(VI)は、蝋状固体(m.p.約43℃)であり、そのようなものとして、特に大規模でのこの物質の結晶化および単離は理想的ではない。酢酸塩(VII)のような塩の製造は、化合物をより簡便に取り扱われる固体として単離することを可能にする。酢酸塩以外の塩も使用できる。このような塩は、リン酸塩、一塩酸塩、二塩酸塩、酢酸トリメチル、酒石酸塩、クエン酸塩、フマル酸塩、マレイン酸塩、安息香酸塩、一臭化水素酸塩、二臭化水素酸塩、炭酸塩および0.5炭酸塩を含む。炭酸塩は、それらが熱に不安定であり、そのため遊離塩基が単純に加温によりイン・サイチュで遊離できるため、特に有用である。
【0085】
故に、一つの局面において、我々は、5−クロロ−2−(ピペリジン−4−イルオキシ)−ピリジン酢酸塩(VII)の仲介を含む、5−クロロ−2−(ピペリジン−4−イルオキシ)−ピリジン(VI)の単離および取り扱いの改良法を開示する。
【0086】
他の局面において、我々は、化合物(I)の製造における中間体として有用な、5−クロロ−2−(ピペリジン−4−イルオキシ)−ピリジン(VI)の新規塩を開示する。
【0087】
好ましい方法において、5−クロロ−2−(ピペリジン−4−イルオキシ)−ピリジン酢酸塩(VII)の合成は、トルエンのような溶媒中で有利に行われる。反応溶媒としてのトルエンの使用は、2,5−ジクロロピリジンと4−ヒドロキシピペリジンの反応、続く水性洗浄および塩形成を、中間体遊離塩基を単離する必要なく、同じ反応容器で実施することを可能にする。水がこの反応における重要なパラメータであるため(そして4−ヒドロキシピペリジンは吸湿性である)、反応開始前に水を共沸により除去するためのトルエンの使用は、顕著な改善を示し、数キログラム(multi-kilogram)規模でさえ、単離される一定の収率を可能にする。
【0088】
ベンジルチオアセトン(II)からの(RS)−5−メチル−5−{[(フェニルメチル)チオ]メチル}イミダゾリジン−2,4−ジオン(III)の製造は、WO02/074767に記載されている。そこに記載されている条件と比較して、我々は、有機溶媒がエタノールから2−プロパノールに代わり、使用されるカリウムシアニド量が2当量から約1.00乃至1.02当量に減少した、改善法をここに開示する。この方法で、カリウムシアニドは、反応で本質的に完全に消費され、大量の未反応カリウムシアニドを含む溶液の廃棄が避けられる。我々は、使用する炭酸アンモニウム量の約5当量から約1.1乃至1.25当量への減少が特に有益であることをさらに記載する。この方法で、最大操作圧が約9バールから約1.5乃至2.5バールに低下し、特に大規模作業のためには、安全面で著しく有益である。これらの改良したパラメータを使用して、(RS)−5−メチル−5−{[(フェニルメチル)チオ]メチル}−イミダゾリジン−2,4−ジオン(III)の合成は、数キログラム規模で日常的に実施されている。
【0089】
故に、他の局面において、我々は、(RS)−5−メチル−5−{[(フェニルメチル)チオ]メチル}イミダゾリジン−2,4−ジオン(III)をベンジルチオアセトン(II)から製造するための改善された条件を開示する。これらの改善された条件は、大規模製造に特に有利である。
【0090】
WO02/074767に記載の通り、(RS)−5−メチル−5−{[(フェニルメチル)チオ]メチル}イミダゾリジン−2,4−ジオン(III)の構成エナンチオマーへの分割は、固定相としてChiralpak ADカラムおよび溶離剤としてメタノールを使用するキラルHPLCで簡便に達成される。特に大規模作業に簡便な別法として、我々は、キラル分割を、本質的に同じ条件で行うが、模擬移動床(SMB)クロマトグラフィーを使用する方法をここに開示する。この方法で、(S)−5−メチル−5−{[(フェニルメチル)チオ]メチル}イミダゾリジン−2,4−ジオン(IV)は、数キログラム規模で得ることができる。保護されていないチオール、(RS)−5−メチル−5−チオメチル−イミダゾリジン−2,4−ジオンは、驚くべきことに安定であり、固定相としてChiralpak ADカラムおよび移動相としてイソヘキサン/エタノール/ジエチルアミンを使用したキラルHPLCにより簡便に分割される。
【0091】
キラルクロマトグラフィーの代わりとして、他のキラルイミダゾリジン−2,4−ジオン(IV)に至る経路を開示する。
【0092】
ある種のヒダントイン誘導体の分割における(S)−α−メチルベンジルアミンの使用は開示されている(WO92/08702)。我々は、ラセミ体(RS)−5−メチル−5−{[(フェニルメチル)チオ]メチル}イミダゾリジン−2,4−ジオン(III)が、キラルアミンおよび水酸化ナトリウムのような塩基の存在下、適当な溶媒からの結晶化により分割できることを発見した。キラル素子アミンの例は、1S)−(−)−α−メチルベンジルアミン、(1R)−(+)−α−メチルベンジルアミン、L−チロシンアミド、(1S)−(−)−α−(1−ナフチル)エチルアミン、(1R)−(+)−α−(1−ナフチル)エチルアミン、L−(−)−シンコニジン、D−(+)−シンコニン、(−)−キニン、(+)−β−キニジン、(1R,2S)−(−)−エフェドリン、(2R)−(−)−2−アミノ−1−ブタノール、(2R)−1−アミノ−2−プロパノール(D−アラニノール)、(1R,2S)−(−)−2−アミノ−1,2−ジフェニルエタノール、N−メチル−D−(−)−グルカミン、(2S)−(+)−2−フェニルグリシノール、ノルエフェドリン、(−)−ブルシン、(−)−ストリキニーネ、(+)−ヨヒンビン、(1S,2S)−(+)−threo−2−アミノ−1−(p−ニトロフェニル)−1,3−プロパンジオール、(L)−(+)−threo−2−アミノ−1−フェニル−1,3−プロパンジオール、cis−ミルタニルアミン、(1R,2R)−(−)−1,2−ジアミノシクロヘキサンおよび(2R)−(−)−2−アミノ−2−フェニルエタノールを含む。
【0093】
好ましい方法において、キラルアミンは(1S)−(−)−α−メチルベンジルアミンである。
故に、一つの局面において、我々は、(1S)−α−メチルベンジルアミンを使用したラセミ体(RS)−5−メチル−5−{[(フェニルメチル)チオ]メチル}イミダゾリジン−2,4−ジオン(III)の分割方法を開示する。
【0094】
好ましい方法において、キラルアミンは(1S)−(−)−α−メチルベンジルアミン(1.0〜2.0当量)であり、塩基は水酸化ナトリウム(0.4〜0.6当量)であり、そして溶媒は水(4〜8容量)である。次いで、結晶化により、高エナチオマー純度、一般に、>95%の(5S)−5−ベンジルチオメチル−5−メチル−イミダゾリジン−2,4−ジオン(S)−α−メチルベンジルアミンを得る。この物質の(5S)−5−メチル−5−{[(フェニルメチル)チオ]メチル}−イミダゾリジン−2,4−ジオン(IV)へのさらなる変換は、標準条件下、例えば、2N 塩酸を使用して、または簡単に酢酸イソプロピル、メチルイソブチルケトン(MIBK)、トルエン、t−ブチルメチルエーテル(TBME)、およびこのような溶媒の組合せを含む適当な溶媒からの結晶化により行い得る。(5S)−5−ベンジルチオメチル−5−メチル−イミダゾリジン−2,4−ジオン(S)−α−メチルベンジルアミンの(IV)への変換は、簡単に、シクロヘキサン、ジブチルエーテルまたは水のような適当な、熱溶媒への固体のスラリー化により行い得る。故に、溶液中でまたはスラリーとして共結晶を温める行動が、遊離(5S)−5−メチル−5−{[(フェニルメチル)チオ]メチル}−イミダゾリジン−2,4−ジオン(IV)の放出をもたらし、次いで、それが冷却により結晶化する。(IV)を遊離させるためにいずれかの方法を使用したとき、さらなるキラル増大が観察される。
【0095】
他の局面において、ラセミ体2−アミノ−3−ベンジルチオ−2−メチルプロピオンアミド(VIII)
【化5】
を、適当なキラル酸存在下の適当な溶媒からの結晶化により分割できる。キラル素子酸の例は、(L)−酒石酸、(R)−(−)−マンデル酸、ジベンゾイル−(L)−酒石酸[DBTA]、ジ−p−トルオイル−(L)−酒石酸[DTTA]、(L)−リンゴ酸[(2S)−(−)−2−ヒドロキシコハク酸]、(1S)−(+)−10−カンファースルホン酸[(D)−CSA]、(1R,3S)−(+)−樟脳酸[cis−樟脳酸]、(L)−グルタミン酸[(2S)−(+)−2−アミノペンタン二酸]、(L)−アスパラギン酸[(S)−(+)−アミノコハク酸]、(L)−ピログルタミン酸[(S)−(−)−2−ピロリドン−5−カルボン酸]、(L)−オルニチンヒドロクロライド[(2S)−(+)−2,5−ジアミノペンタン酸]、(L)−ヒスチジン、(L)−リシン[(2S)−(+)−2,6−ジアミノヘキサン酸]、(L)−アルギニン、N−アセチル−(L)−フェニルアラニン、N−アセチル−(L)−ロイシン、N−カルボベンジルオキシ−(L)−アラニン[(2S)−2−ベンジルオキシカルボニルアミノプロピオン酸]、(−)−メトキシ酢酸、N−アセチル−(L)−チロシンおよび(2R)−(+)−2−(4−ヒドロキシフェノキシ)プロピオン酸を含む。
【0096】
一つの好ましい方法において、キラル酸は(R)−(−)−マンデル酸である。
故に、一つの局面において、我々は、(R)−(−)−マンデル酸を使用するラセミ体2−アミノ−3−ベンジルチオ−2−メチルプロピオンアミド(VIII)の製造法を開示する。
【0097】
一つの好ましい方法において、キラル酸は(R)−(−)−マンデル酸であり、そして、溶媒はメタノールと酢酸イソプロピルの混合物である。結晶化は水の存在下で実施すべきである。この方法で、高エナンチオマー純度の(2S)−2−アミノ−3−ベンジルチオ−2−メチルプロピオンアミド(R)−マンデラート0.5水和物が得られる。この塩のエナンチオマー純度は、酢酸イソプロピルのような溶媒からの再結晶によりさらに増大され得る。
【0098】
他の好ましい方法において、キラル酸はL−酒石酸である。
故に、一つの局面において、我々は、L−酒石酸を使用したラセミ体2−アミノ−3−ベンジルチオ−2−メチルプロピオンアミド(VIII)の分割方法を開示する。
【0099】
他の好ましい方法において、キラル酸はL−酒石酸であり、そして溶媒はエタノールである。得られた(2S)−2−アミノ−3−ベンジルチオ−2−メチルプロピオンアミド(L)−タートレートの、メタノールとメチルイソブチルケトンの混合物のような適当な溶媒からの再結晶により、高エナンチオマー純度の物質が得られる。
【0100】
(2S)−2−アミノ−3−ベンジルチオ−2−メチルプロピオンアミドの(5S)−5−メチル−5−{[(フェニルメチル)チオ]メチル}イミダゾリジン−2,4−ジオン(IV)へのさらなる変換は、当業者に容易に明らかとなる方法を使用して達成できる。例えば、Tetrahedron Asymm., 2001, 12, 101;Tetrahedron, 1991, 47(12), 2133;およびChem. Ber., 1928, 1431参照。
【0101】
他の局面において、キラル5−メチル−5−{[(フェニルメチル)チオ]メチル}イミダゾリジン−2,4−ジオン(IV)を、適当なラセミ体前駆分子の生体触媒(酵素)分割を介して製造できる。ある可能な経路をスキーム3に概説する。
【化6】
【0102】
スキーム3に示す通り、(S)−2−アミノ−3−ベンジルスルファニル−2−メチルプロピオンアミド(IX)または(S)−2−アミノ−3−ベンジルスルファニル−2−メチルプロピオン酸(X)のいずれかは、望むキラルヒダントイン(IV)の適当な前駆体として働き得る。
【0103】
ラセミ体アミノアミド(VIII)の生体触媒分割は、この幾分立体的に障害された基質を受け入れることができるアミダーゼの使用を必要とする。Cα−四置換α−アミノアミドの分割のためのアミダーゼ・マイコバクテリウム・ネオオーラム(Mycobacterium neoaurum) ATCC 25795またはオクロバクテリウム・アンスロピ(Ochrobactrum anthropi) NCIMB 40321の使用は、Tetrahedron, 2001, 57, 6567-6577に開示されている。マイコバクテリウム・ネオオーラム(Mycobacterium neoaurum)は、この特定のアミノアミド(VIII)の分割に適当なアミダーゼであることが証明されていたが、オクロバクテリウム・アンスロピ(Ochrobactrum anthropi)は、驚くべきことにラセミ体加水分解をもたらした。アミノアミド(VIII)の分割に好結果で使用できる他のアミダーゼは、ロドコッカス・エリスロポリス(Rhodococcus erthoplis)およびシュードモナス・フルオレッセンス(Psseudomonas fluorescens) AL45を含む。シュードモナス・フルオレッセンス(Psseudomonas fluorescens) AL45を使用したアミノアミド(VIII)の分割は、WO2005/123932に開示されている。
【0104】
スキーム4に示す通り、これらの生体触媒分割の立体化学結果は、適当なアミダーゼの選択により簡便に制御される。ラセミ体アミノアミド(VIII)の生体触媒分割のための典型的な具体法を本明細書の実施例部分に記載し、このような方法は、本発明の具体的局面を表す。
【化7】
【0105】
別の生体触媒法において、ラセミ体ヒダントイン(III)の加水分解により、または対応するラセミ体アミノ酸から製造したラセミ体α−ウレイド酸(XI)を、ヒダントイナーゼ触媒閉環(スキーム5)に付す。適当なヒダントイナーゼは、ロシュ・ヒダントイナーゼ1およびヒダントイナーゼ2を含む。
【0106】
一つの局面において、我々は、(RS)−3−ベンジルスルファニル−2−メチル−2−ウレイド−プロピオン酸(XI)の閉環を行うためにヒダントイナーゼ酵素を使用することを含む、(5S)−5−[4−(5−クロロ−ピリジン−2−イルオキシ)−ピペリジン−1−スルホニルメチル]−5−メチル−イミダゾリジン−2,4−ジオンの合成における中間体として有用な(S)−5−メチル−5−{[(フェニルメチル)チオ]メチル}イミダゾリジン−2,4−ジオン(IV)の製造方法を開示する。さらなる局面において、ヒダントイナーゼ酵素はロシュ・ヒダントイナーゼ1またはヒダントイナーゼ2である。
【0107】
α−ウレイド酸の分割のための別の生体触媒は、EP0175312(Kanegafuchi)およびWO03/106689(Kaneka)に記載されている。
【化8】
【0108】
別の生体触媒法(スキーム6)において、アミノ酸(X)のラセミ体を、対応するトリフルオロアセチル保護されたアミノ酸(XII)に変換し、それを次いでアミノ酸アシラーゼ触媒加水分解に付す。適当なアミノ酸アシラーゼは、アスペルギルス属、L−Hog腎臓アシラーゼおよびペニシリウム属からのL−アシラーゼである。他の適当なアシラーゼは、当業者には容易に明らかとなろう。
【化9】
【0109】
驚くべきことに、化合物(XII)に対応するN−アセチルまたはN−クロロアセチルアミドのようなより伝統的な基質は、L−アミノ酸アシラーゼとの反応を何等示さなかった。当業者は、トリフルオロアセチルアミド(XII)が他の活性化アミドに置き換えられ、分割の選択性がD−アミノ酸アシラーゼの選択により逆転され、それにより、反応混合物からの(S)−アミノ酸の直接結晶化が促進されることを容易に認識するであろう。
【0110】
一つの局面において、我々は、(RS)−2−アミノ−3−ベンジルスルファニル−2−メチル−プロピオン酸の活性化アミドを適当なアシラーゼ酵素で処理することを含む、(R)−または(S)−5−メチル−5−{[(フェニルメチル)チオ]メチル}イミダゾリジン−2,4−ジオンの合成における中間体として有用な(R)−または(S)−2−アミノ−3−ベンジルスルファニル−2−メチル−プロピオン酸の製造方法を提供する。一つの特定の局面において、活性化アミドはトリフルオロアセチルアミドである。
【0111】
他の局面において、キラル5−メチル−5−{[(フェニルメチル)チオ]メチル}イミダゾリジン−2,4−ジオン(IV)を、適当なキラル(メソ)前駆分子の生体触媒(酵素)非対称化(desymmetrisation)を介して製造する。上記の酵素変換は、全て分割であり、それ故望む立体異性体の理論的最大収率は50%である。対照的に、単純なプロキラル(メソ)化合物の非対称化は、理論上100%の望む立体異性体を製造できる。ある可能な経路をスキーム7に略記する。
【化10】
【0112】
故に、ニトリル(XIII)、アミド(XIV)またはエステル(XV)のような適当なメソ−前駆体は、適当な酵素を使用して非対称化でき、それにより、上に示すキラルヒダントイン前駆体を得る。エステル(XV)について適当なR基は、C1−4アルキルを示す。
【0113】
必要なメソ−前駆体は、文献に記載のものに準じた方法を使用して製造できる。例えば、J. Org. Chem., 1995, 60(17), 5487;J. Chem. Soc., Perkin Trans. 1, 1991, 4, 2589;Synth. Comm., 2001, 1323;およびInorg. Chem., 2003, 42(9), 2950。具体的なメソ−前駆体の合成は、実施例部分に開示する。
【0114】
メソ−ニトリル(XIII)の非対称化のための可能性のある酵素は、例えば、Tetrahedron Asym., 2004, 15, 2817;Tetrahedron Asym., 2001, 12, 3367;Tetrahedron Asym., 1993, 4, 1081;およびJ. Org. Chem., 2003, 68, 2479に記載されている。
【0115】
メソ−アミド(XIV)の非対称化は、ロドコッカス・エリスロポリス(Rhodococcus erthoplis)アミダーゼを使用して達成した。得られたキラル酸アミド(XVI)を、次いで一容器連続反応(single pot sequence)でさらに変換して、優れたeeでキラルヒダントイン(IV)を得た。
【0116】
ブタ肝臓エステラーゼを使用したメソ−S−t−ブチルメチルエステルの非対称化は、以前に記載されている(J. Org. Chem., 2003, 68(13), 5403)。
【化11】
【0117】
ここで、メソ−S−ベンジルエチルエステル(XV、R=Et)もまたこの酵素の基質であることが示された。非対称化は、該文献例に従って進行し、(最初に形成された酸エステル(XVII)のクルチウス転位および続くエステル加水分解後)、(R)−アミノ酸(X)を60−80%eeで得る。
【0118】
類似の非対称化変換が、2種の異なる酵素クラスの代表、すなわち、バチルス・リケニホルミス(Bacillus licheniformis)プロテアーゼおよびアミノ酸アシラーゼを使用して達成され得ることもさらに確立された。バチルス・リケニホルミス(Bacillus licheniformis)プロテアーゼを使用した非対称化の場合、変換は、(S)−エステル酸を提供するために逆立体選択性で進行する。この(S)−エステル/酸をさらに対応するアミノ酸に変換し、その絶対配置およびキラル純度を、標準品サンプルとの比較により決定した。アミノ酸から(S)−5−メチル−5−{[(フェニルメチル)チオ]メチル}イミダゾリジン−2,4−ジオン(IV)への、文献において既知の方法に従った(例えば、Chem. Rev., 1950, 46, 403参照)。
【0119】
故に、さらなる局面において、我々は、メソ−ニトリル(XIII)、またはメソ−アミド(XIV)またはメソ−エステル(XV)の酵素的非対称化を含む、化合物(I)の合成における中間体として有用な(S)−5−メチル−5−{[(フェニルメチル)チオ]メチル}イミダゾリジン−2,4−ジオン(IV)の合成方法を開示する。特定の局面において、我々は、メソ−アミド(XIV)を、適当なアミダーゼ酵素を使用して非対称化する方法を開示する。他の特定の局面において、アミダーゼはロドコッカス・エリスロポリス(Rhodococcus erthoplis)アミダーゼである。
【0120】
上記生体触媒分割において、酵素は、適当であれば、それ自体でまたは固定化(支持)形態で使用してよい。
他の局面において、キラル5−メチル−5−{[(フェニルメチル)チオ]メチル}イミダゾリジン−2,4−ジオン(IV)を、非対称合成を介して製造し得る。
【0121】
非対称ストレッカー反応は、α,α−ジアルキルアミノ酸の合成のために重要な方法である。(R)−フェニルグリシノールは、このような方法において使用する典型的キラル助剤である(Tetrahedron, 2001, 57, 6383-6397)。故に、ベンジルチオアセトン(II)と(R)−(−)−フェニルグリシノールの縮合は、ジアステレオマー混合物としてオキサゾリジン(XVIII)を提供する。次いで、オキサゾリジン(XVIII)とトリメチルシリルシアニドの反応は、85:15比のジアステレオマー混合物としてアミノニトリル(XIX)をもたらす。この混合物の、イソヘキサンのような適当な溶媒からの再結晶は、この化合物(XIX)のジアステレオマー比を99:1を超えて高める(スキーム8):
【化12】
【0122】
次いで、塩化水素ガス存在下、アミノニトリル(XIX)の1当量の水での処理により、ラクトン(XX)を得る。シアン酸カリウムとの反応、続く臭化水素の酢酸溶液での側鎖の除去により、次いでキラルヒダントイン(XXII)を得る。
【0123】
あるいは、アミノニトリル(XIX)のクロロスルホニルイソシアネートでの処理は、ヒダントイン(XXI;R=H)および(XXI;R=CONH2)を含む混合物をもたらし、この混合物は、臭化水素の酢酸溶液で処理したとき、キラル(R)−ヒダントイン(XXII)をもたらした。
【0124】
キラル助剤として(S)−フェニルグリシノールを使用して、エナンチオマー(S)−ヒダントインを得る。
【0125】
一つの局面において、我々は、キラル助剤標識オキサゾリジン(XVIII)の開環を含む、(R)−または(S)−5−メチル−5−{[(フェニルメチル)チオ]メチル}イミダゾリジン−2,4−ジオンの合成方法を提供する。
【0126】
我々は、さらに、非対称相間移動触媒を使用したキラルヒダントインの合成を開示する。触媒的非対称工程は、本方法においてしばしば最も効果である準化学量論量のキラルコントロール要素の使用を可能にするため、この点で、とりわけ魅力的である。キラル相間移動触媒は、典型的に、穏やかな条件、単純な反応工程、安全で安価な試薬および溶媒、有機触媒(金属非含有触媒)の使用、および小規模または大規模いずれでも反応を実施する可能性を含むため、さらなる利点を提供する。しかしながら、4級立体中心(4種の異なる非水素基を有する炭素)を含む化合物の、触媒的エナンチオ選択的方法での構築は、未だ挑戦的である。このような4級立体中心の構築を可能にする適当な経路を、スキーム9に略記する。
【化13】
【0127】
最初の工程で、t−ブチル(DL)−アラニネートまたはイソプロピル(DL)−アラニネートを、ベンズアルデヒド、クロロベンズアルデヒドまたは2−ナフトアルデヒドのような適当なカルボニル誘導体と縮合させて、イミンエステル(XXIII)を得る。好ましくはt−ブチルエステルを使用する。次いでこのイミンを、適当な塩基および適当なキラル相間移動触媒の存在下、ブロモメチルスルファニルメチルベンゼンでアルキル化して、イミン(XXIV)を得る。適当な塩基は、例えば、水酸化カリウム、水素化ナトリウム、水酸化セシウムおよび水酸化ルビジウムを含む。適当な相間移動触媒は、例えば、(−)−O−アリル−N−(9−アントラセニルメチル)シンコニジニウム(cinchonidinium)ブロマイドおよび(+)−O−アリル−N−(9−アントラセニルメチル)シンコニジニウムブロマイドを含む。正しいシュード−エナンチオマーの(正反対の)相間移動触媒が、得られるイミン(XXIV)の絶対立体配置の制御を可能にする。他の適当なキラル相間移動触媒は、当業者には容易に明白となろう。
【0128】
次いでイミン(XXIV)の加水分解によりα−アミノ酸(X)を得る。正しい試薬および正しい反応条件の選択により、キラルα−アミノ酸(X)、またはそのエナンチオマーが、高エナンチオマー純度で得られる。特定の局面において、添付の実施例に記載の具体的方法を特に請求している。
【0129】
一つの局面において、我々は、適当なキラル相間移動触媒の存在下でのイミンエステル(XXIII)のアルキル化を含む、(R)−または(S)−5−メチル−5−{[(フェニルメチル)チオ]メチル}イミダゾリジン−2,4−ジオンの合成における中間体として有用な(R)−または(S)−2−アミノ−3−ベンジルスルファニル−2−メチル−プロピオン酸の製造方法を提供する。一つの特定の局面において、相間移動触媒は(−)−O−アリル−N−(9−アントラセニルメチル)シンコニジニウムブロマイドまたは(+)−O−アリル−N−(9−アントラセニルメチル)−シンコニジニウムブロマイドである。当業者は、上記方法において、硫黄原子を、ベンジル以外の基で代わりに保護できることを容易に認識するであろう。
【0130】
次いで、キラルα−アミノ酸(X)を、さらに、文献法を使用して、キラルヒダントイン(IV)に変換し得る。例えば、Rev., 1950, 46, 403を参照。
【0131】
(S)−5−メチル−5−{[(フェニルメチル)チオ]メチル}イミダゾリジン−2,4−ジオン(IV)からの酢酸水溶液中直接塩素化による((S)−4−メチル−2,5−ジオキソ−イミダゾリジン−4−イル)−メタンスルホニルクロライド(V)の製造は、WO02/074767に開示されている。スルホニルクロライド(V)の新規別法が、同時係属出願US60/782892に開示されている。
【0132】
スルホニルクロライド(V)とカップリングするために、ピペリジニルエーテル酢酸塩(VII)を、最初に、対応する遊離塩基(VI)に再変換しなければならない。この変換は、酢酸エチルまたは酢酸イソプロピルのようなエステル溶媒の存在下、炭酸ナトリウムのような塩基を使用して達成できる。好ましい方法において、この変換は、酢酸塩をトルエンに懸濁させることにより、そして塩基として水性水酸化ナトリウムを使用して、二相系で達成される。トルエンの使用は、共沸蒸留による遊離塩基(VI)のより効率的な乾燥を可能にする。これは、化合物(VI)とスルホニルクロライド(V)のカップリングが、特に水の存在に感受性のため、重要な利点である。いずれの場合も、酢酸イソプロピルまたはトルエンのいずれかの中の遊離塩基(VI)の得られた溶液を、次いで直接スルホニルクロライド(V)t、ジイソプロピルエチルアミンのような適当な塩基の存在下、テトラヒドロフランを共溶媒として使用して、反応させる。この方法で、化合物(I)は、数キログラム規模でさえ、簡便にそして効率的に製造される。
【0133】
一つの局面において、我々は、5−クロロ−2−(ピペリジン−4−イルオキシ)−ピリジン(VI)と((S)−4−メチル−2,5−ジオキソ−イミダゾリジン−4−イル)−メタンスルホニルクロライド(V)(ここで、化合物(VI)は対応する塩からの遊離塩基の遊離により製造する)の反応を含む、化合物(I)の製造法を開示する。さらに特定の局面において、化合物(VI)を、5−クロロ−2−(ピペリジン−4−イルオキシ)−ピリジン酢酸塩(VII)からの遊離塩基の遊離により製造する。
【0134】
化合物(I)の結晶多形はWO02/074767に開示されていない。我々は、いずれかの合成法で製造した化合物(I)を、溶媒として水性エタノールまたは工業用変性アルコール水溶液を使用して結晶化して、導入物質の多形形態(polymorphic modifications)とは無関係に、再現性よく化合物(I)多形Gを得ることができることを発見した。
【0135】
異なる多形形態およびそれらの結晶性は、以下の装置および方法を使用して調べた:
X線粉末回折(XPRD)
XRPD測定は、以下のいずれかを使用した:
i) Scintag Inc. XDS 2000装置で以下のパラメータ:
CuKα(1.5418Å)
45kVおよび30mA
2°≦2θ≦35°
1°/分、増加0.03°
回転石英ディスク
環境条件
約10mgの試験サンプルをサンプルホルダーに置き、石英表面に平テフロン棒を使用して塗布した;または
【0136】
ii) Panalytical X’Pert PRO MPD装置で以下のパラメータ:
CuKα(1.5418Å)
45kVおよび40mA
2°≦2θ≦40°
4°/分、増加0.016°
回転シリコンウエハー
環境条件
約2mgの試験サンプルをサンプルホルダーに置き、シリコン表面に平テフロン棒を使用して塗布した。
【0137】
熱量測定(DSC)
試験サンプルの上昇させた温度に対する熱量測定的応答を、種々の方法を使用してQ1000変調温度示差走査熱量測定(MTDSC)(TA Instruments)を使用して試験し、主要な特色は:
5℃/分の勾配速度の標準的に変調されたモード(“加熱のみ”)(変調なしで1および20℃/分もまた使用した)。温度範囲は環境温度直下から200℃以上であった。
約2mgの試験サンプルを、蓋付き(圧接なし)アルミニウムカップに入れた。
【0138】
重量分析(TGA)
試験サンプルの上昇させた温度に対する重量測定応答を、以下のパラメータを使用したQ500熱重量分析器(TGA)(TA Instruments)を使用して試験した:
加熱速度(標準):5℃/分
約2〜5mgの試験サンプルをカップに入れ、200℃の直ぐ上まで加熱した。
【0139】
湿気相互作用
湿気変化に対する試験サンプルの重量測定応答を、以下の特性のSGA 100(VTI Corporation)またはDVS 2(Surface Measurement System)重量測定的蒸気収着(GVS)装置いずれかを使用して調べた:
乾燥乃至90%RHおよび、例えば、10%RHの工程まで戻る。
平衡条件:<0.01重量%/10分(<0.001重量%/分)
約5mgの試験サンプルをカップに入れ、評価した。
【0140】
形態学
試験化合物の形態学を、500倍までの倍率で、Jeol JSM-5200走査型電子顕微鏡(SEM)を使用して試験した。
数個の粒子を粘性カーボンテープが付いたサンプルホルダーに振りまき、薄金層でコーティングし、試験した。
【0141】
一般的化学法
1H NMRおよび13C NMRスペクトルを、300 MHz Varian Unity Inovaまたは400 MHz Varian Unity Inova装置で記録した。クロロホルム−d(δH 7.27ppm)、ジメチルスルホキシド−d6(δH 2.50ppm)、アセトニトリル−d3(δH 1.95ppm)またはメタノール−d4(δH 3.31ppm)の中央ピークを内部標準として使用した。カラムクロマトグラフィーをシリカゲル(0.040−0.063mm、Merck)を使用して行った。出発物質は、特記しない限り市販されていた。全溶媒および試験試薬は研究室グレードであり、受け取ったまま使用した。特記しない限り、操作は環境温度で、典型的に20〜25℃で行った。
LC分析を、Agilent 1100 HPLC装置を使用して行った。種々のLC法を、生成物分析に使用した。
LCMS分析は、Waters 2790 HPLCと996フォトダイオードアレイ検出器およびMicroMass ZMD、単一四重極マススペクトロメーターとZ−スプレー界面を使用して行った。
【0142】
【表1】
【0143】
実施例1
5−クロロ−2−(ピペリジン−4−イルオキシ)−ピリジン酢酸塩
4−ヒドロキシピペリジン(12.1g、0.12mol、1.18mol eq.)をトルエン(120mL)に懸濁して、オレンジ色懸濁液を得た。得られた混合物を15分加熱還流した(ジャケット温度115℃)。オレンジ色溶液が85−90℃で形成され、幾分かの油汚染が撹拌シャフトおよび温度プローブに見られた。トルエン(26mL)を蒸留により除去した。反応混合物を20℃に15分にわたり冷却した。白色固体が約30〜35℃で沈殿した。別の容器で、tert−BuOK(13.4g、0.12mol、1.18mol eq.)をトルエン(150mL)に懸濁した。この懸濁液を4−ヒドロキシピペリジン混合物に20℃で添加した。次いでトルエンライン洗浄(11mL)を実施した。得られた濃厚懸濁液を50℃で30分にわたり激しく撹拌しながら加熱した。2,5−ジクロロピリジン(15g、0.10mol、1mol eq.)のトルエン(45mL)溶液を、スラリーに50℃で約1時間にわたり添加し、続いてトルエンライン洗浄(11mL)した。反応混合物を還流(約105−107℃)に70分にわたり加温し、次いで2時間加熱還流した。反応混合物を環境温度に30分にわたり冷却し、一晩撹拌した。反応混合物を水(2×75mL)で洗浄し、次いで90℃で1時間加熱した。氷酢酸(6.1g、0.10mol、1mol eq.)のトルエン(60mL)溶液を、混合物に90℃一度に添加し、続いてトルエンライン洗浄(15mL)した。添加完了後、溶液をRTに70分にわたり冷却した。必要な5−クロロ−2−(ピペリジン−4−イルオキシ)−ピリジン酢酸塩が冷却中に沈殿した。1時間、RTで撹拌後、懸濁液を濾過し、ケーキをトルエン(2×75mL)で洗浄した。真空オーブンで50℃で一晩乾燥させて、生成物を85〜95%収率で得た。
1H NMR(400 MHz, D2O)δ 8.1(1H, d), 7.8(1H, dd), 6.9(1H, d), 5.0(1H, m), 3.4(4H, m), 2.2(4H, m), 1.9(3H, s)。
【0144】
5−クロロ−2−(ピペリジン−4−イルオキシ)−ピリジンの別の塩
適当な酸(鉱酸については溶媒中;または有機酸については固体として;または、炭酸塩については、CO2ガスを溶液にバブリング)を、5−クロロ−2−(ピペリジン−4−イルオキシ)−ピリジンのトルエン溶液にRTで添加した。さらなるトルエンを添加し、得られた溶液を結晶化が起こるまで撹拌した。形成した固体を濾過により回収し、イソヘキサンで洗浄した。
【0145】
この方法に使用した酸の例は以下を含む:
・水性リン酸
1H NMR(D2O)δ 2.10(2H, m), 2.20(2H, m), 3.28(2H, m), 3.45(2H, m), 5.15(1H, m), 6.91(1H, d, J 8.8 Hz), 7.78(1H, d, 8.8 Hz), 8.10(1H, s)。
【0146】
・塩酸(プロパノール中) − 一および二塩酸塩を製造するために使用
一HCl、1H NMR(d6-DMSO)δ 1.91(2H, m), 2.14(2H, m), 3.07(2H, m), 3.21(2H, m), 5.19(1H, m), 6.90(1H, d, J 9.6 Hz), 7.83(1H, d, J 9.6 Hz), 8.21(1H, s), 9.17(2H, bs). M.p. 156℃。
二HCl、1H NMR(d6-DMSO)δ 1.93(2H, m), 2.16(2H, m), 3.08(2H, m), 3.20(2H, m), 5.20(1H, m), 5.27(1H, bs), 6.91(1H, d, J 9.2 Hz), 7.83(1H, d, J 9.2 Hz), 8.21(1H, s), 9.35(2H, bs). M.p. 131℃。
【0147】
・トリメチル酢酸
1H NMR(d6-DMSO)δ 1.10(9H, s), 1.48(2H, m), 1.93(2H, m), 2.58(2H, m), 2.95(2H, m), 4.99(1H, m), 6.83(1H, d, J 8.8 Hz), 7.77(1H, d, J 8.8 Hz), 8.18(1H, s)。
M.p. 91-96℃。
【0148】
・酒石酸
1H NMR(D2O)δ 2.10(2H, m), 2.21(2H, m), 3.29(2H, m), 3.46(2H, m), 4.53(2H, s), 5.17(1H, m), 6.93(1H, d, J 9.2 Hz), 7.80(1H, d, J 9.2 Hz), 8.12(1H, s)。
【0149】
・クエン酸
0.5クエン酸塩、1H NMR(D2O)δ 2.10(2H, m), 2.21(2H, m), 2.64(1H, d, J 15.2 Hz), 2.73(1H, d, J 15.2 Hz), 3.28(2H, m), 3.46(2H, m), 5.16(1H, bs), 6.915(1H, d, J 8.8 Hz), 7.79(1H, d, J 8.8 Hz), 8.10(1H, s). M.p. 88℃。
【0150】
・フマル酸
1H NMR(CD3OD)δ 2.05(2H, m), 2.20(2H, m), 3.23(2H, m), 3.39(2H, m), 5.30(1H, m), 6.72(2H, s), 6.83(1H, d, J 8.8 Hz), 7.70(1H, d, J 8.8 Hz), 8.11(1H, s). M.p. 128℃。
【0151】
・マレイン酸
1H NMR(d6-DMSO)δ 1.85(2H, m), 2.13(2H, m), 3.13(2H, m), 3.27(2H, m), 5.20(1H, m), 6.12(2H, s), 6.90(1H, d, J 9.2 Hz), 7.83(1H, d, J 9.2 Hz), 8.21(1H, s), 8.48(2H, bs). M.p. 96-104℃。
【0152】
・安息香酸
1H NMR(d6-DMSO)δ 1.66(2H, m), 2.02(2H, m), 2.80(2H, m), 3.09(2H, m), 5.08(1H, m), 6.86(1H, d, J 8.8 Hz), 7.40(2H, t, J 9.6), 7.48(1H, d, J 9.6 Hz), 7.79(1H, d, 8.8 Hz), 7.91(2H, d, J 9.6 Hz), 8.19(1H, s). M.p. 140℃。
【0153】
・臭化水素酸(水性) − 一および二臭化水素酸塩を製造するために使用
一HBr、1H NMR(d6-DMSO)δ 1.90(2H, m), 2.15(2H, m), 3.12(2H, m), 3.25(2H, m), 5.19(1H, m), 6.91(1H, d, J 8.8 Hz), 7.85(1H, d, J 8.8 Hz), 8.21(1H, s), 8.80(2H, bs). M.p. 198℃。
二HBr、1H NMR(d6-DMSO)δ 1.91(2H, m), 2.16(2H, m), 3.15(2H, m), 3.26(2H, m), 5.21(1H, m), 6.92(1H, d, J 9.2 Hz), 7.84(1H, d, J 9.2 Hz), 8.21(1H, s), 8.76(2H, bs). M.p. 183℃。
【0154】
・炭酸(CO2ガス)
0.5炭酸塩、1H NMR(d6-DMSO)δ 1.53(2H, m), 1.94(2H, m), 2.59-3.02(4H), 5.04(1H), 6.84(1H, d, J 8.7 Hz), 7.77(1H, d, J 8.7 Hz), 8.17(1H, s)。
【0155】
実施例2
(RS)−5−メチル−5−{[(フェニルメチル)チオ]メチル}イミダゾリジン−2,4−ジオン
適当なサイズの標準加圧反応器にベンジルチオアセトン(95%純度)(85.26g、450mmol、1mol eq.)、水(413mL)および2−プロパノール(146mL)を入れた。混合物を約15分撹拌して、均質にした。次いで炭酸アンモニウム(49.56g、509mmol、1.13mol eq.)およびカリウムシアニド(30.54g、460mmol、1.02mol eq.)を入れた。反応混合物を90℃に温め、これは約2.5バール(barg)の圧力を誘発した。反応を冷却し、LCで出発物質の消失について分析した。反応完了後、必要な生成物を結晶化させた。必要であれば、結晶化を種晶添加により誘発した。結晶化後、水(971.9mL)および濃塩酸(96.7g)を反応混合物に入れた。これによりpHが約11.9から7.4に変化した。結晶塊を濾取し、続いて酢酸イソプロピルで洗浄した。乾燥後、表題化合物を白色結晶性固体として86%収率で得た。
1H NMR(d6-DMSO)δ 10.74(1H, s), 8.00(1H, s), 7.35-7.20(5H, m), 3.76(2H, s), 2.72, 2.262(各2x1H, Abq), 1.29(3H, s)。
【0156】
実施例3
(S)−5−メチル−5−{[(フェニルメチル)チオ]メチル}イミダゾリジン−2,4−ジオン
(RS)−5−メチル−5−{[(フェニルメチル)チオ]メチル}イミダゾリジン−2,4−ジオンを、分取キラル模擬移動床クロマトグラフィー(SMB)を使用してエナンチオマー要素に分割した。WO02/074767(89頁)に記載されているものと同じキラル固定相および移動相を使用した。エナンチオマーを本質的に定量的収率で回収した。
【0157】
得られた(S)−5−ベンジルスルファニルメチル−5−メチル−イミダゾリジン−2,4−ジオン(5g)のメタノール性溶液の容量を、減圧下、35℃で減らした(約20mLまで)。水(40mL)を、内部温度を35℃に維持しながら溶液に滴下した。約半量の水を添加したら、生成物が沈殿を始めた。混合物をRTにゆっくり冷却し、次いで氷浴で2℃に冷却した。生成物(4.56g、SMB分割後、理論値の91%)を、白色結晶性固体として、2℃での濾過により回収した。5.86kg規模で、この再結晶化工程からの生成物を98%収率(5.73kg)で単離した。
【0158】
(S)−5−メチル−5−{[(フェニルメチル)チオ]メチル}イミダゾリジン−2,4−ジオンはまた、メタノール/トルエンまたはメタノール/ジブチルエーテルを含む他のメタノール混合物から結晶化できる。それは、トルエン、ジ−イソプロピルエーテル、ジブチルエーテルおよび水を含む範囲の溶媒から結晶化できる。
1H NMR(300 MHz, d6-DMSO)δ 10.74(1H, s), 8.00(1H, s), 7.35-7.20(5H, m), 3.76(2H, s), 2.72, 2.262(各2x1H, ABq), 1.29(3H, s)。
【0159】
生成物のキラル純度を、Hewlett Packard 1100 series HPLC(ダイオードアレイ検出器;Astec Chirobiotic V 50mm×4.6mmカラム装着)を使用するHCにより、移動相として70:30 イソヘキサン:エタノール;オーブン温度55℃;流速1.0mL/分;210nmで検出;注入量1μL;およびランタイム5分で確定した。(S)−および(R)−異性体の保持時間は、それぞれ2.6および3.8分であった。
【0160】
実施例4
((S)−4−メチル−2,5−ジオキソ−イミダゾリジン−4−イル)−メタンスルホニルクロライド
方法1
(S)−5−ベンジルスルファニルメチル−5−メチル−イミダゾリジン−2,4−ジオン(106.9g、427.1mmol、1.000mol eq.)を、氷酢酸(8vol eq.)および水(1vol eq.)の混合物に溶解し、約4℃に冷却した。次いで塩素ガス(96.9g、3.2mol eq.)を十分に撹拌している溶液に約1時間にわたり、反応混合物温度が、添加中、ほとんど12〜15℃に維持されるように、一定速度で添加した(ジャケット温度は、ずっと4℃を保った)。反応完了後(混合物は特徴的緑色に変わり、温度が急激に低下した)、混合物に窒素導入し、約30℃に加熱して、白色スラリーを得る。次いで溶媒の大部分を真空蒸留により除去した。トルエン(534.5mL)を添加し、真空下蒸留により同量の溶媒を除去した。トルエンの添加/蒸留をさらに一回繰り返した。次いでイソヘキサン(534.5mL)を残渣に添加し、混合物を20℃に冷却した。徹底的に撹拌(stir-out)後、生成物を濾過により回収した。回収した固体をイソヘキサン(213.8mL)で洗浄し、真空で40℃で一定重量になるまで乾燥させ、必要なスルホニルクロライドを白色結晶性固体として得た(95.5g、98.7%)。
1H NMR(300MHz, d8-THF)δ, 9.91(1H, bs), 7.57(1H, s), 4.53, 4.44(2×1H, 各ABq), 1.52(3H, s)。
【0161】
方法2
(S)−5−ベンジルスルファニルメチル−5−メチル−イミダゾリジン−2,4−ジオン(50g、199.74mmol)を氷酢酸(8vol eq.)および水(4mol eq.)の混合物に溶解し、約4℃に冷却した。次いで塩素ガスを、よく撹拌している溶液に、反応混合物温度が、添加中、ほとんど約12℃に維持されるように、一定速度で約1時間にわたり添加した(Huberコントローラーを、反応温度が12℃に維持されるように設定)。反応完了後(混合物が特徴的緑色に変わり、温度が急激に低下した)、混合物に窒素導入し、約20℃に加熱して、白色スラリーを得た。次いで溶媒の1/2乃至2/3を真空蒸留により除去した(100mbar圧で)。次いでイソオクタン(250mL、5vol eq.)を残渣に添加し、混合物20℃に冷却した。徹底的に撹拌(stir-out)後、生成物を濾過により回収した。回収した固体をイソオクタン(2×100mL)で洗浄し、真空で40−50℃で一定重量になるまで乾燥させ、必要なスルホニルクロライドを白色結晶性固体として得た(41.37g、91%)。
1H NMR(300MHz, d8-THF)δ, 9.91(1H, bs), 7.57(1H, s), 4.53, 4.44(2×1H, 各ABq), 1.52(3H, s)。
【0162】
実施例5
(S)−5−[4−(5−クロロ−ピリジン−2−イルオキシ)−ピペリジン−1−スルホニルメチル]−5−メチル−イミダゾリジン−2,4−ジオン方法1
a)5−クロロ−2−(ピペリジン−4−イルオキシ)−ピリジン
5−クロロ−2−(ピペリジン−4−イルオキシ)−ピリジンアセテート(40g、0.146mol)をイソPrOAc(664mL)中に30℃でスラリー化した。このスラリーに、Na2CO3(1.5mol/L;196mL、2mol eq.)を添加した。次いでスラリーを、30℃で15分急速に撹拌した。二相性混合物を静置し、底水性相を分離し、廃棄した。上記の塩基性洗浄工程をさらに2回繰り返した。次いで有機相を1回水(200mL)で洗浄した。得られたイソPrOAc溶液の容量を、減圧下の蒸留により約300mLに減らした。次いで溶液をイソPrOAc(400mL)と共に蒸留し、再び約300mLまで蒸留により減らした。この工程をさらに1回繰り返した。サンプルを、5−クロロ−2−(ピペリジン−4−イルオキシ)−ピリジン含量および水含量分析のために取った。溶液の重量または総容量を、iPrOAc溶液中の5−クロロ−2−(ピペリジン−4−イルオキシ)−ピリジンの濃度を計算するために測定した。
【0163】
b)(S)−5−[4−(5−クロロ−ピリジン−2−イルオキシ)−ピペリジン−1−スルホニルメチル]−5−メチル−イミダゾリジン−2,4−ジオン
ジイソプロピルエチルアミン(24.3mL、0.139mol、1mol eq.)を、パート(a)で製造したイソPrOAc溶液[約300mL;31.2g、0.146mol、1.05mol当量の5−クロロ−2−(ピペリジン−4−イルオキシ)−ピリジンに相当]に、RTで一度に添加した。次いで溶液を−15℃に冷却した。
((S)−4−メチル−2,5−ジオキソ−イミダゾリジン−4−イル)−メタンスルホニルクロライド(31.65g、0.139mol、1mol eq.)を、乾燥THF(285mL)にRTで撹拌しながら添加した。次いで得られた溶液を、5−クロロ−2−(ピペリジン−4−イルオキシ)−ピリジンのイソPrOAc溶液に、−15℃で約1.5時間にわたり滴下した。沈殿が、((S)−4−メチル−2,5−ジオキソ−イミダゾリジン−4−イル)−メタンスルホニルクロライドの添加により見られた。添加終了時、乾燥THF(32mL)を反応混合物に添加してラインを洗浄し、混合物を1時間、−15℃で洗浄した。次いでそれを20℃に1時間にわたり温め、さらに20℃で1時間撹拌した。
【0164】
反応を、急速に撹拌しながら10wt%NaHSO4(157mL)でクエンチした。約15分後、二相性混合物を静置し、底水性相を分離し、廃棄した。この酸洗浄工程をさらに1回繰り返した。次いで有機相を、急速な撹拌を使用して水(157mL)で洗浄し、完全に相分割させた後、分配した。次いで反応溶液を40℃に温め、再び水(157mL)で洗浄した。THF(95mL)を有機層に添加し、次いでそれを40℃に温め、40℃で濾過して、全ての粒子状物を除去した。次いで溶媒容量を、55℃のジャケット温度での減圧蒸留により約157mLに減らした。次いでイソPrOAc(317mL)を添加し、容量を再び約157mLに減らした。さらに2回イソPrOAc(317mL)の添加−除去を繰り返した。蒸留中に固体が沈殿し始め、懸濁液が得られた。各回容量を約157mLまで減らし、最終蒸留の後、次いで少量の溶媒のサンプルを、残存THF分析のために反応混合物から取った。1H NMRは、THFピークを示さなかった。次いで反応内容物を0℃に冷却し、生成物を濾過により回収した。反応容器をイソPrOAc(63mL)で洗浄し、この濯ぎ液を使用してフィルター上の生成物を洗浄した。生成物を、一晩、真空オーブンで40℃で乾燥させた。必要な(S)−5−[4−(5−クロロ−ピリジン−2−イルオキシ)−ピペリジン−1−スルホニルメチル]−5−メチル−イミダゾリジン−2,4−ジオンが、白色固体として、71%収率(41.8g)で得られた。
1H NMR(300MHz, d6-DMSO)δ 10.74(1H, s), 8.20(1H, d), 8.01(1H, s), 7.81(1H, dd), 6.87(1H, d), 5.09(1H, m), 3.52-3.35(4H, m), 3.13(2H, m), 2.02(2H, m), 1.72(2H, m), 1.33(3H, s)。
【0165】
実施例6
(S)−5−[4−(5−クロロ−ピリジン−2−イルオキシ)−ピペリジン−1−スルホニルメチル]−5−メチル−イミダゾリジン−2,4−ジオン方法2
a)5−クロロ−2−(ピペリジン−4−イルオキシ)−ピリジン
5−クロロ−2−(ピペリジン−4−イルオキシ)−ピリジンアセテート(70g、257mmol)を、トルエン(560mL)中、RTでスラリー化した。1M NaOH(420mL)を添加し、次いでスラリーをRTで15分急速に撹拌した。二相性混合物を静置し、底水性相を分離し、廃棄した。次いで有機相を水(2×420mL)で洗浄した。サンプルを有機相から除去し、5−クロロ−2−(ピペリジン−4−イルオキシ)−ピリジンについて分析した。
次いで得られたトルエン溶液を、減圧下での蒸留により減らし、約168mLまで減らした(5−クロロ−2−(ピペリジン−4−イルオキシ)−ピリジンアセテート負荷について2.4vol eq.)。次いで溶液をトルエン(420mL)と蒸発させて、再び約168mL(2.4vol eq.)まで蒸留により減らした。サンプルを、水含量分析のために取った。
【0166】
b)(S)−5−[4−(5−クロロ−ピリジン−2−イルオキシ)−ピペリジン−1−スルホニルメチル]−5−メチル−イミダゾリジン−2,4−ジオン
ジイソプロピルエチルアミン(38.4mL、220mmol)を、工程(a)で得た5−クロロ−2−(ピペリジン−4−イルオキシ)−ピリジンのトルエン溶液(236mmol含有)に一度に添加し、続いて乾燥THF(151mL)をライン洗浄として添加した。((S)−4−メチル−2,5−ジオキソ−イミダゾリジン−4−イル)−メタンスルホニルクロライド(48.7g、215mmol)を、RTで撹拌しながら乾燥THF(352mL)に溶解した。次いでスルホニルクロライドの得られた溶液を、5−クロロ−2−(ピペリジン−4−イルオキシ)−ピリジンおよびジイソプロピルエチルアミンのトルエン/THF溶液に、RTで1〜2時間にわたり滴下した。スルホニルクロライドの添加により沈殿が見られた。添加終了時、乾燥THF(50mL)を、ライン洗浄として反応混合物に添加した。添加完了後、反応を約30分、RTで洗浄した。
【0167】
反応を10wt%NaHSO4(251mL)で、約15分急速に撹拌しながらクエンチした。二相性混合物を静置し、底水性相を分離し、廃棄した。この酸洗浄工程をさらに1回繰り返した。次いで溶媒容量を、減圧蒸留により約220mLに減らした。次いでトルエン(300mL)を添加し、容量を約245mLに減らし、固体が、蒸留中に沈殿し始め、懸濁液が得られた。最後の蒸留後、次いで溶媒の少量のサンプルを残留THF分析のために反応混合物から取った。
次いで反応混合物の内容物を0℃に冷却し、この温度で約30分撹拌し、生成物を濾過により回収した。反応容器をトルエン(100mL)で洗浄し、この濯ぎ液をフィルター上の生成物の洗浄に使用した。生成物を、真空オーブンで、40℃で一定重量になるまで乾燥させた。(S)−5−[4−(5−クロロ−ピリジン−2−イルオキシ)−ピペリジン−1−スルホニルメチル]−5−メチル−イミダゾリジン−2,4−ジオンを、2工程で典型的に85〜88%収率で、白色固体として得た。
【0168】
実施例7
(S)−5−[4−(5−クロロ−ピリジン−2−イルオキシ)−ピペリジン−1−スルホニルメチル]−5−メチル−イミダゾリジン−2,4−ジオン形態G
工業用変性アルコール(IMS):水(10vol eq.)の2:1混合物を、(S)−5−[4−(5−クロロ−ピリジン−2−イルオキシ)−ピペリジン−1−スルホニルメチル]−5−メチル−イミダゾリジン−2,4−ジオンに添加した。混合物を加熱還流(約80〜82℃)して、溶液を得た。この溶液を還流に15分維持し、次いで濾過した。濾液を加熱還流し、この温度で15分維持した。次いで溶液を0.5℃/分の速度で20℃に冷却した。20℃で2〜3時間撹拌後、(S)−5−[4−(5−クロロ−ピリジン−2−イルオキシ)−ピペリジン−1−スルホニルメチル]−5−メチル−イミダゾリジン−2,4−ジオン形態Gを濾過により回収し、IMS:水 2:1(2.5vol eq.)で洗浄し、乾燥させた。生成物を80〜87%収率で単離した。
この方法は、導入物質の多形形態と無関係に、再現性よく多形Gをもたらすことが示された。多形A、C、Fおよび多形混合物は、この方法を使用して再結晶させて、全て形態Gとなった。
【0169】
実施例8
(1S)−(−)−α−メチルベンジルアミンを使用した(RS)−5−ベンジルスルファニルメチル−5−メチル−イミダゾリジン−2,4−ジオンの分割
NaOH(0.45eq.)の水(4vol eq.)溶液を、(1S)−α−メチルベンジルアミン(1.7eq.)および(RS)−5−ベンジルスルファニルメチル−5−メチル−イミダゾリジン−2,4−ジオン(50g)に添加した。得られた懸濁液を83℃に加熱した。混合物をゆっくり冷却し、RTより高い温度(36℃)で種晶添加した。種晶添加は必須ではないが、その反応混合物の撹拌および濾過を容易にし得る。さらに水(3vol eq.)を冷却サイクル中に添加した。反応を20℃で一晩撹拌し、その後生成物を濾過により回収し、シクロヘキサンで洗浄して、(5S)−5−ベンジルスルファニルメチル−5−メチル−イミダゾリジン−2,4−ジオン(1S)−α−メチルベンジルアミン塩を45%収率で得た。
LC(実施例3と同じ条件)は、生成物が94.5%エナンチオマー純度を有することを示した。
1H NMR(400 MHz, d6-DMSO)δ 1.23(3H, d, J=6.7 Hz), 1.28(3H, s), 2.61(1H, d, J 14.1 Hz)2.72(1H, d, J 14.1 Hz), 3.76(2H, s), 3.96(1H, q, J=6.7 Hz), 7.37-7.15(10H, m), 7.97(1H, s)。
【0170】
2−アミノ−3−ベンジルチオ−2−メチルプロピオンアミド(VIII)
カリウムシアニド(1.07Kg)および水酸化アンモニウム溶液(28%溶液の4.46Kg)を、20Lフラスコに入れた。この混合物に、予め形成した塩化アンモニウム溶液(3.47Kg水中1Kg)を、約0.5時間にわたり撹拌しながら添加した。
1−(ベンジルチオ)−2−プロパンオン(2.71Kg)を次いで添加した。混合物を約40℃で65時間撹拌した。脱イオン水(1L)を添加し、相を分離した。分離した有機相(3.13Kg)を、次工程に直接使用した。
37%塩酸(13.3Kg)および48%臭化水素酸(1.26Kg)の混合物に、上記からの物質の半量を約1時間にわたり、約5℃で添加した。次いで混合物を2時間、5℃で撹拌し、次いで約30℃に温め、一晩撹拌した。次いで混合物を冷却し、生成物を濾過により回収した。フィルターケーキを氷冷濃塩酸およびアセトンで洗浄した。得られた物質をアセトン(10L)に再懸濁し、短時間加熱還流した。次いで生成物を濾過により回収し、減圧下一晩乾燥させた。この工程を、出発物質の残りの半分で繰り返した。
上記の通り製造した物質を合わせたバッチ(1.66Kgおよび1.51Kg)に、酢酸エチル(9Kg)および予め形成させた炭酸カリウム溶液(9Kgの50wt%水溶液)を添加した。混合物を1時間撹拌し、次いで相を分離した。水性相を酢酸エチル(3Kg)で抽出した。合わせた有機相を炭酸カリウム溶液(3.1Kgの50wt%水溶液)で抽出した。得られた有機相を炭酸ナトリウムで乾燥させ、濾過し、濃縮した。得られた固体をスラリー化し、TBME中、短時間加熱還流した。冷却後、生成物を濾過により回収し、一晩真空下で乾燥させた。
2−アミノ−3−ベンジルチオ−2−メチルプロピオンアミド(VIII)(全体で2.4Kg、73.2%)を単離した。
【0171】
実施例9
(R)−(−)−マンデル酸を使用した(RS)−2−アミノ−3−ベンジルスルファニル−2−メチル−プロピオンアミドの分割
(2S)−2−アミノ−3−ベンジルスルファニル−2−メチル−プロピオンアミド(R)−マンデラート
メタノール(20mL、4vol eq.)および酢酸イソプロピル(80mL、16vol eq.)を混合して、メタノールの酢酸イソプロピルの20%v/v溶液を得た。
(RS)−2−アミノ−3−ベンジルスルファニル−2−メチル−プロピオンアミド(5g、22.2mmol)を、予め混合したMeOH/iPrOAc溶液(13vol eq.)および水(0.4mL、1mol eq.)の溶液に溶解した。得られた溶液を50℃に加熱した。(R)−(−)−マンデル酸(3.39g、22.2mmol、1.0mol eq.)を、予め混合したMeOH/iPrOAc溶液(30mL、6vol eq.)の溶液に溶解し、次いでアミド溶液に1時間にわたり管理された方法で添加した。添加中、反応温度を50℃に維持した。生成物の沈殿が添加中に開始し得る。残った予め混合したMeOH/iPrOAc溶液(約1vol eq.)をライン洗浄として使用した。
次いで懸濁液をゆっくり0℃に冷却し、この温度で徹底的に撹拌(stir-out)後、生成物を濾過により回収した。反応容器を20℃に戻し、iPrOAc(25mL、5vol eq.)で約5〜10分洗浄した。続いてこの溶液をフィルター上の生成物の洗浄に使用した。生成物を、真空オーブンで40℃で一定重量になるまで乾燥させた。(2S)−2−アミノ−3−ベンジルスルファニル−2−メチル−プロピオンアミド(R)−マンデラート0.5水和物を、典型的に47〜48%収率および97〜>99%エナンチオマー純度で白色固体として単離した。
90%を超えるエナンチオマー純度の(2S)−2−アミノ−3−ベンジルスルファニル−2−メチル−プロピオンアミド(R)−マンデラート0.5水和物を、iPrOAc(35vol eq.)からの再結晶により、エナンチオマーを増加することができ、>99.35%エナンチオマー純度の物質が、>87%回収率で得られる。
1H NMR(400 MHz, d6-DMSO)δ 1.28(s, 3H, CH3), 2.79(AB q, J=140.5, 13.8 Hz, 2H SCH2Cq), 3.76(AB q, J=16.4, 13.1 Hz, 2H, ArCH2S), 4.79(s, 1H ArCHOH), 7.41-7.18(m, 11H, ArHおよびCONHAHB), 7.56(s, 1H, CONHAHB)。
生成物のキラル純度を、Hewlett Packard 1100 series HPLC(ダイオードアレイ検出器;Chiracel ChiralPak AD 25cm×0.46cm ID×10μmカラム装着)を使用するLCにより、溶媒−0.1%v/v ジエチルアミンのエタノール溶液;アイソクラチック法;オーブン温度20℃;流速1.0mL/分;サンプル希釈剤−精製水;210nmで検出;注入量5.0μL;およびランタイム15分で確立した。(S)−および(R)−アミノアミドの保持時間は、それぞれ約5.4および11.8分であった。
【0172】
実施例10
L−酒石酸を使用した(RS)−2−アミノ−3−ベンジルスルファニル−2−メチル−プロピオンアミドの分割
(2S)−2−アミノ−3−ベンジルスルファニル−2−メチル−プロピオンアミド(2R,3R)−タートレート
方法1
(RS)−2−アミノ−3−ベンジルスルファニル−2−メチル−プロピオンアミドおよびL−酒石酸(0.25−1.0eq.)のエタノール(11.75−90vol eq.)溶液を混合した。沈殿した得られた固体を濾過により回収した。これにより、2−アミノ−3−ベンジルスルファニル−2−メチル−プロピオンアミドタートレートを、典型的に45〜90%収率および50〜92%エナンチオマー純度(ep)で得た。エタノール以外の溶媒(方法2の下を参照)も使用できた。
L−酒石酸(1eq.)およびEtOH(90vol eq.)を使用して、生成物を1:1塩として、44%収率および92.2%epで得た。
【0173】
方法2
(RS)−2−アミノ−3−ベンジルスルファニル−2−メチル−プロピオンアミドおよびL−酒石酸(0.25−1.0eq.)の混合物を、最小量の溶媒(適当な溶媒は、メタノール、エタノール、イソプロパノール、n−ブタノール、酢酸イソプロピル、酢酸エチル、トルエン、アセトニトリルおよびIMSを含み、これに限定されない)にスラリー化した。次いでこれらのスラリーを環境温度を超える温度で洗浄し、さらなる溶媒を、種々温度で完全な用芸を形成させるために添加した。完全な溶液が、35vol eq.の溶媒で還流で形成されないならば、十分な水を添加して溶液を作成した。次いでこれらの溶液をRTに冷却し、得られた固体を濾過により回収して、2−アミノ−3−ベンジルスルファニル−2−メチル−プロピオンアミドタートレートを、78〜100%収率(L−酒石酸導入量に対して、および1:1塩または2:1塩のいずれかの形成について補正)で、50〜60%の範囲のエナンチオマー純度で得た。
これらの2−アミノ−3−ベンジルスルファニル−2−メチル−プロピオンアミドL−酒石酸塩を、種々の溶媒(適当な溶媒の例は、(i)溶媒としてMeOHまたは水のいずれかおよび貧溶媒としてイソプロパノール、n−ブタノール、酢酸エチル、酢酸イソプロピル、トルエン、アセトニトリル、アセトン、THF、TBME、DCM、MIBK、ジエチルエーテル、2,2,4−トリメチルペンタンまたはIMSのいずれかの混合溶媒結晶化;または(ii)エタノール、メタノール、IMSまたは水からの直接再結晶を含み、これに限定されない)から再結晶できた。典型的に、再結晶は、1〜99%の範囲の収率で、50〜96%の範囲のエナンチオマー純度の生成物をもたらした。
MeOH/MIBK(各々20/29.5vol eq.)からの再結晶により、(2S)−2−アミノ−3−ベンジルスルファニル−2−メチル−プロピオンアミド(2R,3R)−タートレートを75%収率および96.63%のエナンチオマー純度で得た(出発物質は約50%ep)であった。
キラル純度は、実施例9のようにLCで確立した。
1H NMR(300 MHz, d6-DMSO)δ 1.35(3H, s, CH3), 2.69(1H, d, J 13.7 Hz, S-CH2), 3.00(1H, d, J 13.7 Hz, S-CH2), 3.79(2H, s, S-CH2), 3.98(2H, s, CHOH), 7.22-7.36(5H, m, Ar-H), 7.44(1H, s, NH), 7.63(1H, s, NH)。
【0174】
実施例11
生体触媒分割
1. ロドコッカス・エリスロポリス(Rhodococcus erthoplis)アミダーゼを使用
(RS)−2−アミノ−3−ベンジルスルファニル−2−メチル−プロピオンアミド塩酸塩(5g、0.019mol)を、0.1N pH8.00ホウ酸緩衝液(100mL)に溶解して、pH約4.0の溶液を得た。2N KOH溶液でpHを7.00に調節し、ロドコッカス・エリスロポリス(Rhodococcus erthoplis)アミダーゼを、架橋酵素凝集体(CLEA)(緩衝液中1mLの懸濁液、25単位/mL 特異的活性)として添加した。反応混合物を35℃で1.5時間撹拌した。HPLCは、アミドの対応するアミノ酸への25〜30%変換を示した。
濃塩酸でpHを1.00に調節し、次いで酵素CLEAを除去するために濾過した。2N KOH溶液でpHを11に調節し、DCM(1×100mL;1×40mL)で抽出した。合わせたDCM抽出物を乾燥させ、蒸発させて、(R)−2−アミノ−3−ベンジルスルファニル−2−メチル−プロピオンアミドを白色固体(2.67g)として62%収率で得た。エナンチオマー純度は、キラルHPLC(34%ee)で67%(R)−entであることが判明した。
水性層を濃塩酸で処理してpH6.80とし、pH6.80を2N KOH溶液の時折の添加により1時間維持した。得られた白色結晶を濾過により回収し、pH7.00緩衝液で洗浄して、40℃で一晩乾燥させて、(S)−2−アミノ−3−ベンジルスルファニル−2−メチル−プロピオン酸(1.2g、27%収率)を得た。エナンチオマー純度は99%(S)−ent(98%ee)であることが判明した。
【0175】
2. シュードモナス・フルオレッセンス(Psseudomonas fluorescens)AL45アミダーゼ(WO2005/123932)を使用
a) 微生物の増殖
Ps fluorescens AL45を、10L発酵槽中、酵母抽出物(2g/L)およびラクトアミド(2.5g/L)を補った5Lのミネラル塩培地 pH7.2で28℃で増殖させた。発酵槽に5L/分で空気供給し、400rpmで24時間撹拌した。細胞を遠心により回収し、回収した細胞を100mM リン酸緩衝液 pH7.2に再懸濁し、再遠心することにより洗浄した。回収した細胞を4℃で一晩貯蔵して、その後生体内変化に使用した。
【0176】
b)生体内変化条件
細胞ペレットをリン酸緩衝液(100mM、pH7.2、1L)に再懸濁し、それに(RS)−2−アミノ−3−ベンジルスルファニル−2−メチル−プロピオンアミド(5g)を添加した。混合物を撹拌し、28℃でインキュベートし、サンプルを分析のために定期的に取った。8.5時間後、定量的HPLCは、反応が50%加水分解に到達していることを示した。キラル分析(LC)は、(R)−2−アミノ−3−ベンジルスルファニル−2−メチル−プロピオンアミド96%ee;および(S)−2−アミノ−3−ベンジルスルファニル−2−メチル−プロピオン酸97.5%eeを示した。
【0177】
c)反応後処理
細胞を、生体内変化ブロスから遠心により除去し、上清(約1000mL)を20%炭酸ナトリウム溶液(最終容量約1200mL)を使用してpH10.5に調節した。アルカリ性上清をTBME(750mL)で抽出し、全てのエマルジョンは有機相に保持した。TBME相をアセトン(約250mL)と混合し、これは沈殿の形成をもたらした。同時に水性相をTBME(750mL)で再抽出し、有機相をアセトン(約250mL)と混合した。これは、少量の水性層の分離をもたらし、これを残りの水性相と合わせた。2個のTBME/アセトン相を合わせ、乾燥させ(Na2SO4)、濾過し、溶媒を除去して、(R)−2−アミノ−3−ベンジルスルファニル−2−メチル−プロピオンアミドをオフホワイト色結晶性固体(1.7g)として、68%収率で得た。エナンチオマー純度は>99%(R)−entであった。
水性相を2M HClを使用してpH6.8に調節し、凍結乾燥により約300mLまで濃縮した。融解により白色スラリーを産生した。結晶性固体を濾過により回収し、一晩、37℃で乾燥させて、(S)−2−アミノ−3−ベンジルスルファニル−2−メチル−プロピオン酸を微細白色結晶(1.8g)として72%収率で、HPLCで98%純度で得た。エナンチオマー純度は>99%(S)−entであった。
【0178】
3. マイコバクテリウム・ネオアルム(Mycobacterium neoarum)L−アミダーゼを使用
ロドコッカス・エリスロポリス(Rhodococcus erthoplis)について記載のものと同様の方法を使用して、(RS)−2−アミノ−3−ベンジルスルファニル−2−メチル−プロピオンアミドを、マイコバクテリウム・ネオアルム(Mycobacterium neoarum)からのL−特異的アミダーゼを使用して分離できた。この方法は、(R)−2−アミノ−3−ベンジルスルファニル−2−メチル−プロピオン酸および(S)−2−アミノ−3−ベンジルスルファニル−2−メチル−プロピオンアミドが〜99%eeで製造される点で、上記(1)および(2)と相補的である。
【0179】
4. (RS)−3−ベンジルスルファニル−2−メチル−2−ウレイド−プロピオン酸の分割を介した(S)−5−ベンジルスルファニルメチル−5−メチル−イミダゾリジン−2,4−ジオン
(RS)−3−ベンジルスルファニル−2−メチル−2−ウレイド−プロピオン酸
【化14】
(RS)−2−アミノ−3−ベンジルスルファニル−2−メチル−プロピオン酸(350g)を水(2.6L)およびTHF(2.6L)に懸濁し、シアン酸カリウム(504g)を添加し、反応を25℃で一晩撹拌した。さらにシアン酸カリウム(75g)を次いで添加し、反応をさらに6時間撹拌した。次いで撹拌を止め、反応を0℃で一晩冷却した。得られたスラリーを濾過し、濾液を減圧下濃縮した。濃縮物を、急速に撹拌しながら6%水性HCl溶液でpH2に酸性化した。1時間後、得られた固体を濾過により回収し、次いで水(4L)およびメタノール(4L)に連続的に再スラリー化した。真空下乾燥後、生成物をシリカクロマトグラフィーを使用して精製した。不純物をDCM/THFを使用して精製し、一方生成物ををTHF/AcOH、続いてMeOHを使用して抽出した。溶媒の除去、続いてTBMEでのトリチュレーションにより、3−ベンジルスルファニル−2−メチル−2−ウレイド−プロピオン酸(151g)をオフホワイト色固体として得た。
【0180】
(RS)−3−ベンジルスルファニル−2−メチル−2−ウレイド−プロピオン酸(9g、約90%純度、30mmolに相当)を、亜硫酸ナトリウム溶液(5mmol、100mL)に添加した。KOH(2.5g、44mmol)を添加した。溶液を、酸が溶解するまで撹拌し、次いで濾過した。pHを、氷酢酸で12.5から7.00に調節した。MnCl2.4H2O溶液(10mmolar、10mL)を添加した。溶液をN2下40℃に加熱し、D−特異的ヒダントイナーゼを添加した(緩衝液中1mL懸濁液、E. coli.組み換えロシュ・ヒダントイナーゼ2、300単位/mg、全タンパク質70mg)。反応混合物を40℃でN2下撹拌した。反応中、pHを5%v/v酢酸溶液滴定により7.0に維持した。
【表2】
【0181】
反応中、濃厚白色沈殿が形成した。混合物を環境温度に冷却し、3時間撹拌し、次いで濾過し、0.1N pH7リン酸カリウム緩衝液で洗浄した。固体を一晩40℃で乾燥させた。生成物を白色粉末(3.3g)として、42%収率、HPLCで100%化学純度で単離した。キラルHPLC分析は、生成物が100%(S)−entであることを示した。
キラル純度は、25cm×4.6mm Chiralpack ADカラム、30℃、溶離剤MeOH+0.1%v/v HCO2H、流速1mL min−1、検出220nMのUVを使用して確立した。Rt(S)−エナンチオマー4.42分;Rt(R)−エナンチオマー9.21分。
【0182】
5. アスペルギルス属からのL−アシラーゼを使用した(RS)−3−ベンジルスルファニル−2−メチル−2−(2,2,2−トリフルオロ−アセチルアミノ)−プロピオン酸の分割
(RS)−3−ベンジルスルファニル−2−メチル−2−(2,2,2−トリフルオロ−アセチルアミノ)−プロピオン酸
【化15】
(RS)−2−アミノ−3−ベンジルスルファニル−2−メチル−プロピオン酸(5g、0.022mol)をトリフルオロ酢酸無水物(15mL)およびDCM(15mL)にスラリー化した。スラリーを−20℃に冷却し、トリエチルアミン(2.5g、0.025mole)を滴下した。反応をRTに温めた。HPLCはアミドへの完全な変換を示した。混合物を水(30mL)およびDCM(30mL)で希釈した。水性層を廃棄し、有機層を水(3×50mL)で洗浄した。有機層を分離し、蒸発させて、油状物を得た。この物質をアセトニトリルに溶解し、溶媒を真空で蒸発させた。7.2g薄黄色油状物を得た。(〜100%収率)。
MS M+、321(100%)。
1H NMR(CDCl3)δ 7.4(1H, brs), 7.30(5H, m), 3.85(2H, m), 3.4(1H, d), 3.05(1H, d), 1.75(3H, s)。
【0183】
【化16】
CoCl2.6H20(0.25g、0.001mol)をホウ酸緩衝液(100mL、pH8.0、0.1N)に溶解した。溶液を脱気し、(RS)−トリフルオロアセトアミド(7.00g)を油状物として添加した。混合物をN2下45℃でpHが8.00になるまで飽和Na2CO3溶液を添加しながら撹拌した(約35mL添加)。L−アミノ酸アシラーゼ(0.7g、アスペルギルス属からのL−アシラーゼ、30,000単位g−1)をpH8.00緩衝液(5mL)に溶解し、反応に添加した。反応を30時間、45℃でN2下、pHを〜7.50に維持しながら撹拌した。HOAcでpHを7.00に調節し、沈殿した固体を濾過により回収し、pH7.00緩衝液で洗浄し、真空乾燥させた。(R)−アミノ酸(1.7g、34%)をオフホワイト色固体として単離した。キラルHPLCは、本物質が98%(R)−エナンチオマー(96%ee)であることを示した。幾分かの結晶化していないアミノ酸が濾液に残った。
濾液を濃HClでpH1に調節し、DCM(2×75mL)で抽出した。HPLCは、DCM抽出物が(R)−アミノ酸を含まないことを示した。
DCMを乾燥させ(Na2SO4)、蒸発させて、(S)−3−ベンジルスルファニル−2−メチル−2−(2,2,2−トリフルオロ−アセチルアミノ)−プロピオン酸(2.5g、35%)を蝋状オフホワイト色固体として得た。
この物質をMeOH/水/KOHを使用して(S)−アミノ酸に98%収率で加水分解した。生成物(S)−アミノ酸は、HPLCで96%eeであることが示された。
【0184】
実施例12
非対称化を使用した生体触媒分割
2−ベンジルスルファニルメチル−2−メチル−マロン酸ジエチルエステル
【化17】
冷却したジエチルメチルマロネート(79.9g)およびベンジルチオブロモメタン(110.4g)の2−メチル−THF(480mL)溶液に、カリウムブトキシド(53.6g)を少しずつ約2時間に渡り添加し、その間温度を0℃以下に維持した。次いで混合物をRTに温め、一晩撹拌した。反応を水(320mL)で希釈し、相を分離した。有機相を乾燥させ(Na2SO4)、濾過し、溶媒を真空下で除去した。得られた油状物をカラムクロマトグラフィー(溶離剤DCM/ヘキサン)を使用して精製して表題化合物を黄色油状物(109g、78%)として得た。
1H NMR(CDCl3)δ 1.23(6H, t), 1.47(3H, s), 2.98(2H, s), 3.72(2H, s), 4.17(4H, q), 7.2-7.3(5H, m)。
【0185】
2−ベンジルスルファニルメチル−2−メチル−マロンアミド
【化18】
メチルマロノニトリル(6.6g)およびテトラブチルアンモニウムブロマイド(1.06g)のDCM(50mL)中の混合物を約0℃に冷却した。この混合物にカリウムt−ブトキシド(9.2g)を添加し、続いてベンジルチオブロモメタン(17.89g)をゆっくり添加した。反応をRTで一晩温め、その時点でそれを塩水(100mL)で希釈した。相を分離し、水性相をさらに3回DCM(3×25mL)で抽出した。合わせた有機相を乾燥させ(MgSO4)、濾過し、真空下濃縮した。得られた褐色油状物をカラムクロマトグラフィー(溶離剤酢酸エチル/ヘキサン)を使用して精製した。2−ベンジルスルファニルメチル−2−メチル−マロノニトリルを無色固体(9.76g、52%)として単離した。
1H NMR(CDCl3)δ 1.82(3H, s), 2.89(2H, s), 4.00(2H, s), 7.26-7.37(5H, m)。
【0186】
2−ベンジルスルファニル−2−メチルマロノニトリル(2.3g)をt−ブタノール(30mL)に溶解した。溶液を60℃に温め、その時点で粉末水酸化カリウム(10g)を少しずつ添加した。60℃で一晩加熱後、反応を塩水で希釈し、DCM(3×25mL)で抽出した。合わせた有機相を乾燥させ(MgSO4)、濾過し、真空下濃縮して、表題化合物をオフホワイト色固体として得た。
1H NMR(d6-DMSO)δ 1.38(3H, s), 2.98(2H, s), 3.78(2H, s), 6.94(4H, s), 7.15-7.43(5H, m)。
【0187】
ニトリルヒドラターゼを使用した別製造法
(RS)−2−ベンジルスルファニル−2−メチルマロノニトリル(4g、0.19mol)をDSMO(15mL)に溶解した。ニトリルヒドラターゼ(ZyanotaseTM)含有凍結乾燥細胞(0.75g)を0.1N pH7 リン酸緩衝液(120mL)中にスラリー化し、ジニトリルのDMSO溶液を添加した。反応を38℃で2日振盪し、次いで濾過した。結晶化生成物をアセトニトリル(70mL)に溶解し、濾過して細胞を除去した。溶媒を蒸発により除去し、結晶化生成物を水で洗浄し、乾燥させて2−ベンジルスルファニルメチル−2−メチル−マロンアミド(3g、65%)を得た。反応からの水性濾液のDCM(100mL)での抽出、続いて蒸発により、さらに生成物(750mg、16%)を得た。
【0188】
非対称化を介する(S)−2−アミノ−3−ベンジルスルファニル−2−メチル−プロピオン酸
【化19】
2−ベンジルスルファニルメチル−2−メチル−マロン酸ジエチルエステル(5g)を、0.1M pH7 リン酸緩衝液にスラリー化し、バチルス・リケニホルミス(Bacillus licheniformis)プロテアーゼ(1g)を添加し、反応混合物を35℃で撹拌した。pHを、pHスタットを介して2Mアンモニアを添加することにより維持した。4日後、反応を酢酸エチル(2×50mL)で抽出して未反応ジエステルを除去し、希HClでpH4に酸性化し、1,2−ジクロロエタン(2×50mL)で抽出した。生成物含有ジクロロエタン相を乾燥させ(Na2SO4)、濾過した。ジクロロエタン相に、トリエチルアミン(1.6g)、続いてジフェニルホスホリルアジド(PhO)2P(O)N3(4.4g)を添加した。反応を一晩加熱還流し、5M HCl(50mL)と混合し、さらに6時間加熱還流した。生成物アミノ酸が抽出されている酸性水性相のキラルHPLC分析は、(S)−アミノ酸が存在し、eeが100%であることを示した。すなわち、(R)−アミノ酸が検出され得ない。
キラル純度を、25cm×4.6mm Chirobiotic Tカラム、30℃、溶離剤20%v/v水のEtOH溶液、流速1mL min−1、検出220nMのUVを使用して確立した。Rt(R)−エナンチオマー6.86分;Rt(S)−エナンチオマー8.21分。
【0189】
非対称化を介した(S)−5−ベンジルスルファニルメチル−5−メチル−イミダゾリジン−2,4−ジオン
【化20】
2−ベンジルスルファニルメチル−2−メチル−マロンアミド(2g)をDMSO(10mL)に50℃で溶解した。得られた溶液に0.1M pH7 リン酸緩衝液(100mL)に添加した。これに、ロドコッカス・エリスロポリス(Rhodococcus erthoplis)アミダーゼ(架橋酵素凝集体(CLEA)の25単位/mL懸濁液2mL)を添加した。35℃で4日撹拌後、CLEAを濾過により除去し、濾液を濃HClでpH4に酸性化し、生成物酸/アミドを酢酸イソプロピル(3×50mL)で抽出した。キラルHPLCは、モノ酸が93%のeeを有することを示した。溶媒を真空下で除去し、1,2−ジクロロエタン(50mL)に置き換えた。この溶液に、トリエチルアミン(0.79g)、続いてジフェニルホスホリルアジド(2.2g)を添加した。反応を18時間加熱還流し、冷却し、希HCl(0.1M、2×25mL)で洗浄し、真空下濃縮した。粗生成物は、(S)−エナンチオマーおよびキラルHPLCで93%eeであることが示された。生成物ヒダントインを、カラムクロマトグラフィー(溶離剤酢酸エチル)を使用して精製しし、エタノールから再結晶して、表題化合物(1g、49%)を無色固体として得た。eeは97%に増加した。
キラル純度を、25cm×4.6mm Chiralpack ADカラム、30℃、溶離剤MeOH+0.1%v/v HCO2H、流速1mL min−1、検出220nMのUVを使用して確立した。Rt(S)−エナンチオマー4.42分;Rt(R)−エナンチオマー9.21分。
【0190】
実施例13
a)(R)−2−ベンジルスルファニルメチル−2−メチル−4−フェニル−オキサゾリジン
【化21】
4Åモレキュラー・シーブ(3.6g)およびベンジルチオアセトン(10.0mmol、1.65mL、1.80g)を、(R)−(−)−フェニルグリシノール(1.00eq、10.0mmol、1.37g)のトルエン(170mmol、18.0mL、15.7g)溶液に環境温度で添加した。混合物を50℃に加熱し、この温度で24時間撹拌した。50mm 3番 焼結漏斗を通した濾過後、必要なオキサゾリジンの溶液が得られた。この溶液を続く実験にさらに精製することなく使用した。分析目的で、この溶液のサンプルを少量蒸発乾固して、明色油状物を得た。この物質を1H NMR分光学(CDCl3、400MHz)で分析し、オキサゾリジンが(2R,4R)−および(2S,4R)−異性体の54:46混合物として存在することが示された。
【0191】
1H NMR(CDCl3、400MHz):
(2R,4R)−異性体:δ 1.43 (3H, s); 2.83 (2H, AB, J 14.5Hz, Δ 119.5Hz); 3.65 (1H, t, J 7.5Hz); 3.86 (2H, AB, J 13.1Hz, Δ 40.7Hz); 4.24 (1H, t, J 7.5Hz); 4.51 (1H, brt, J 7.5Hz); 7.2-7.5 (10H, m))。
(2S,4R)−異性体:δ 1.50 (3H, s); 2.70 (2H, AB, J 13.7Hz, Δ 43.1Hz); 3.65 (1H, t, J 7.5Hz); 3.86 (2H, AB, J 12.8Hz, Δ 11.3Hz); 4.24 (1H, t, J 7.5Hz); 4.46 (1H, brt, J 7.5Hz); 7.2-7.5 (10H, m)。
【0192】
b)(R)−3−ベンジルスルファニル−2−メチル−2−((R)−1−フェニル−2−トリメチルシラニルオキシ−エチルアミノ)−プロピオニトリル
【化22】
(R)−2−ベンジルスルファニルメチル−2−メチル−4−フェニル−オキサゾリジン(1.00eq、100mmol、29.9g)のトルエン(4.25mol、449mL、391g)溶液に、−20℃でマグネシウムブロマイドジエチルエーテラート(99wt/wt%、5.00mmol、1.30g)、続いてトリメチルシリルシアニド(105mmol、14.1mL、10.4g)を添加した。混合物を−20℃で18時間撹拌し、次いで0℃に温め、水(60mL、2vol eq.)で洗浄した。溶媒をロータリーエバポレーター(浴温度44℃)を使用して60mL(2vol eq.)容量まで除き、次いでイソヘキサン(240mL、8vol eq.)で希釈した。混合物を加熱還流して、透明黄色溶液を得て、次いで一晩撹拌しながら冷却した。固体生成物を濾過により回収し、イソヘキサン(30mL、1vol eq.)で洗浄し、乾燥させて、表題化合物(21.5g、54.0mmol、54%)を得た。
1H NMR(CDCl3, 400MHz):δ 0.16 (9H, s); 0.98 (3H, s); 2.69 (2H, AB, J 14.2Hz, Δ 43.1Hz); 3.33 (1H, brs); 3.42 (1H, dd, J 10.6, 9.8Hz); 3.66 (1H, dd, J 10.6, 4.2Hz); 3.99 (2H, AB, J 13.5Hz, Δ32.4Hz); 4.03 (1H, dd, J 9.8, 4.2Hz); 7.0-7.5 (10H, m)。
【0193】
c)(3R,5R)−3−ベンジルスルファニルメチル−3−メチル−5−フェニル−モルホリン−2−オン
【化23】
塩化水素ガスを、15分間、−20℃に維持した(R)−3−ベンジルスルファニル−2−メチル−2−((R)−1−フェニル−2−トリメチルシラニルオキシ−エチルアミノ)−プロピオニトリル(1.00eq.、10.0mmol、3.99g)のDCM(622mmol、39.9mL、52.8g)溶液にバブリングした。水(10.0mmol、180μL、180mg)を添加し、塩化水素をさらに20分間バブリングした。混合物を環境温度に温め、次いで環境温度で12時間撹拌し、その後減圧下蒸発乾固した。混合物をDCM(50mL)に分散し、飽和水性炭酸水素ナトリウム(50mL)と共に30分撹拌した。有機層を分離し、乾燥させ(Na2SO4)、減圧下で蒸発乾固して、表題化合物を明褐色結晶性固体として得た。
【0194】
1H NMR(d6-DMSO, 400MHz);δ 1.66 (s, 3H); 2.80 (AB, J 13.1Hz, Δ 71.1 Hz, 2H); 3.02 (brs, 1H); 3.91 (s, 2H); 4.15 (t, J 10.4 Hz, 1H); 4.30 (1H, dd, J 10.3, 2.8 Hz); 4.40 (dd, J 10.8, 2.8 Hz, 1H); 7.2-7.4 (m, 5H)。
13C NMR(d6-DMSO, 100MHz);δ 25.4;37.1;42.7;51.8;62.6;74.8;126.7;127.3;128.0;128.4;128.4;128.8;138.5;138.9;172.0)。
IR(非希釈(neat)固体)3312, 3061, 3030, 2978, 2921, 2322(w), 1730(s), 1494, 1453, 1405, 1369, 1325, 1289, 1276, 1237, 1202(m), 1167(s), 1071, 1053(m), 1029, 758, 733, 697(s)cm-1。
MS(CI), m/z(%);350(M+Na, 30), 328(M+H, 100)。
【0195】
d)(R)−5−ベンジルスルファニルメチル−1−((R)−2−ヒドロキシ−1−フェニル−エチル)−5−メチル−イミダゾリジン−2,4−ジオンおよびカルバミン酸(R)−2−((R)−5−ベンジルスルファニルメチル−5−メチル−2,4−ジオキソ−イミダゾリジン−1−イル)−2−フェニル−エチルエステル
【化24】
クロロスルホニルイソシアネート(2.26mmol、197μL、320mg)を、(R)−3−ベンジルスルファニル−2−メチル−2−((R)−1−フェニル−2−トリメチルシラニルオキシ−エチルアミノ)−プロピオニトリル(2.26mmol、900mg)のDCM(9.00mL)溶液に−55℃で添加した。混合物を−55から−40℃で2時間撹拌し、次いで環境温度に温め、溶媒を減圧下蒸発により除去してオレンジ色泡状物を得た。この物質を、1M 塩酸(9.0mL)中、2時間加熱還流し、次いで環境温度に冷却し、DCM(10mL)で抽出した。DCM抽出物を乾燥させ(Na2SO4)、減圧下で蒸発乾固して、オレンジ色−褐色泡状物を得た。biotage 40Mカラムクロマトグラフィー(溶離剤勾配90:10 イソヘキサン:酢酸エチルから酢酸エチル)により、無色泡状物を得て、それは高真空乾燥で硬化して、ガラス状固体となった。トルエン(1.8mL)からの結晶化により、60:25:13:2比のヒダントイン(XV;R=H、n=1)、ジスルフィドアナログ(XV;R=H、n=2)と、対応するウレタン(XV;R=CONH2、n=1)および(XV;R=CONH2、n=2)の混合物が白色固体(150mg、約0.41mmol、18%)として得られた。
【0196】
ヒダントイン(XV;R=H、n=1)
1H NMR(CDCl3, 300MHz);δ 1.52 (3H, s); 2.50 (2H, AB, J 14.4Hz, Δ 51.0Hz); 3.02 (1H, t, J 6.0Hz); 3.41 (2H, AB, J 13.3Hz, Δ 44.4Hz); 3.89 (1H, ddd, J 11.5, 6.0, 4.5Hz); 4.26 (1H, dd, J 9.0, 4.5Hz); 4.55 (1H, ddd, J 11.5, 9.0, 6.0Hz); 7.1-7.5(10H, m); 8.50 (1H, brs)。
13C NMR(CDCl3, 75MHz);δ 21.7;36.5;37.3;60.6;63.4;68.8;127〜140(12ピーク);156.9;175.9。
IR 3189(br);3061(m);1765(m);720(s);1495(w);1432;1374(m);1265;1242;1199;1130;1070;1045;851;764(w);699(m)cm-1。
MS(CI), m/z(%);371(M+H, 70);251(M-PhCH2CH2OH, 100)。
【0197】
e)(R)−5−ベンジルスルファニルメチル−1−((R)−2−ヒドロキシ−1−フェニル−エチル)−5−メチル−イミダゾリジン−2,4−ジオン
【化25】
(3R,5R)−3−ベンジルスルファニルメチル−3−メチル−5−フェニル−モルホリン−2−オン(1.00eq.、2.69mmol、880mg)を酢酸(9mL)に溶解し、ステンレススチール‘ボンベ’型容器に入れた。シアン酸カリウム(26.9mmol、2.18g)を入れ、容器を直ぐに密閉し、内容物を環境温度で4日間撹拌した。混合物を水(18mL)に注ぎ、2回DCM(18mL)で抽出した。有機抽出物を乾燥させ(Na2SO4)、減圧下で蒸発乾固して、褐色油状物を得た。この物質のLC−MSによる分析は、約40:60比での(R)−5−ベンジルスルファニルメチル−1−((R)−2−ヒドロキシ−1−フェニル−エチル)−5−メチル−イミダゾリジン−2,4−ジオンおよび(3R,5R)−3−ベンジルスルファニルメチル−3−メチル−5−フェニル−モルホリン−2−オンの混合物を示した。
【0198】
f)(R)−5−ベンジルスルファニルメチル−5−メチル−イミダゾリジン−2,4−ジオン
【化26】
(R)−5−ベンジルスルファニルメチル−1−((R)−2−ヒドロキシ−1−フェニル−エチル)−5−メチル−イミダゾリジン−2,4−ジオン(1.00eq.、1.00mmol、370mg)を酢酸(140mmol、8.00mL、8.38g)に溶解した。48%臭化水素酸(133mmol、15.0mL、22.5g)を添加し、混合物を4時間加熱還流した。混合物を0℃に冷却し、氷(100g)を添加し、混合物を0.880 SG 水性アンモニア添加によりpH7に中和し、DCM(100mL)で抽出した。有機抽出物を乾燥させ(Na2SO4)、減圧下で蒸発乾固して、表題化合物を褐色ガム(100mg、0.624mmol、62%)として得た。
【0199】
実施例14
a)tert−ブチルN−ベンジリデンアラニネート
【化27】
t−ブチル(DL)−アラニネートヒドロクロライド(5g、27mmol)をDCM(100mL)にスラリー化し、硫酸マグネシウム(6.50g、51mmol)、ベンズアルデヒド(2.86g、27mmol)およびトリエチルアミン(2.73g、27mmol)を添加した。この混合物を16時間、RTで窒素雰囲気下撹拌した。硫酸マグネシウム(0.81g、0.67mmol)を混合物に添加し、反応をさらに24時間撹拌した。混合物を濾過し、濾液を水(2×50mL)で洗浄した。有機層を乾燥させ(MgSO4)、蒸発させて、表題化合物を無色油状物(5.54g、83%)として得た。
1H-NMR(300MHz, CDCl3)δ 1.47(9H, s), 1.50(3H, s), 4.01-4.08(1H, q), 7.39-7.43(3H, m), 7.76-7.79(2H, m), 8.31(1H, s)。
【0200】
b)tert−ブチル−ベンジル−N−ベンジリデン−2−メチルシステイネート
【化28】
水素化ナトリウム(0.17g、4.3mmol)を、tert−ブチルN−ベンジリデンアラニネート(0.5g、2mmol)のTHF(5mL)溶液にRTで添加した。混合物を30分撹拌し、次いでブロモメチルスルファニルメチル−ベンゼン(0.52g、2.4mmol)を添加した。混合物をRTで48時間撹拌し、次いで水(10mL)でクエンチした。水性層を酢酸エチル(2×50mL)で抽出した。酢酸エチル層を水(10mL)で洗浄し、乾燥させ(MgSO4)、蒸発乾固して、表題化合物を黄色油状物(0.60g、86%)として得た。
【0201】
キラルHPLC分析は、エナンチオマーの1:1混合物の存在を示した:
【表3】
1H-NMR(300MHz, CDCl3)δ 1.47(9H, s), 1.48(3H, s), 2.89-3.05(2H, m), 3.78(2H,s), 7.2-7.50(5H, m), 7.39-7.43(3H, m), 7.76-7.79(2H, m), 8.31(1H, s)。
【0202】
c)tert−ブチル−ベンジル−N−ベンジリデン−2−メチル−D(R)−システイネート
【化29】
粉末水酸化カリウム(5.90g、105mmol)およびキラル相間移動触媒(−)−O−アリル−N−(9−アントラセニルメチル)シンコニジニウムブロマイド(1.27g、2.1mmol)を、tert−ブチルN−ベンジリデンアラニネート(5g、21mmol)のトルエン(50mL)溶液に−20℃で添加した。混合物をこの温度で1時間撹拌した。反応混合物を次いで−30℃に冷却し、ブロモメチルスルファニルメチル−ベンゼン(11.6g、53mmol)のトルエン(50mL)溶液を1時間にわたり滴下した。混合物をこの温度で1時間撹拌し、次いでRTにした。反応をセライトを通して濾過し、トルエン(100mL)で洗浄した。溶媒を蒸発させて、粗残渣(19g)を得て、それをトルエン(100mL)に取り込み、水(2×25mL)で洗浄した。有機層を蒸発させて、表題化合物を得た。この段階でのエナンチオマー純度は測定しなかった。この物質を直接次工程に使用した。
1H-NMR(300MHz, CDCl3)δ 1.47(9H, s), 1.48(3H, s), 2.89-3.05(2H, m), 3.78(2H, s), 7.2-7.50(5H, m), 7.39-7.43(3H, m), 7.76-7.79(2H, m), 8.31(1H, s)。
【0203】
d)S−ベンジル−2−メチル−D(R)−システインヒドロクロライド
【化30】
THF(50mL)および6M 塩酸(50mL)を、工程(c)からの粗生成物に添加した。混合物をRTで1時間撹拌した。有機溶媒を蒸発させ、残った水性層を酢酸エチル(3×50mL)で洗浄した。合わせた有機層を6M 塩酸(50mL)で洗浄した。合わせた水性層を60℃で24時間加熱した。溶媒を減圧下蒸発させ、得られた物質をDCM(100mL)でスラリー化し、固体を濾過により回収した。この固体をDCM(100mL)で洗浄して、表題化合物を固体(3.6g、2工程で64%収率)として得た。キラルHPLC分析は、生成物が96.90%エナンチオマー純度であることを示した。
【0204】
LC法:
【表4】
1H-NMR(400MHz, dmso-d6)δ 1.44(3H, s), 2.87(1H, d), 2.98(1H, d), 3.8-3.85(2H, m), 7.3-7.39(5H, m), 8.50(3H, s)。
MS m/z, 226 [MH]+。
【0205】
実施例15
a)tert−ブチル−ベンジル−N−ベンジリデン−2−メチル−L(S)−システイネート
【化31】
粉末水酸化カリウム(1.17g、21mmol)およびキラル相間移動触媒(+)−O−アリル−N−(9−アントラセニルメチル)シンコニジニウムブロマイド(0.26g、0.42mmol)を、tert−ブチルN−ベンジリデンアラニネート(1g、4.2mmol)のトルエン(15mL)溶液に−30℃で添加した。ブロモメチルスルファニルメチル−ベンゼン(4.65g、21mmol)のトルエン(5mL)溶液を、この混合物に−30℃で添加した。反応を−15℃で16時間撹拌した。混合物をセライトを通して濾過し、トルエン(40mL)で洗浄した。濾液を水(5mL)で洗浄し、有機層を蒸発乾固して、暗褐色油状物を得た。エナンチオマー純度は、この段階で約91.8%(S)−entであることが判明した。粗物質を直接次段階に使用した。
【0206】
b)S−ベンジル−2−メチル−L(S)−システインヒドロクロライド
【化32】
THF(20mL)および6M 塩酸(20mL)を、工程(a)からの粗生成物に添加した。混合物をRTで1時間撹拌した。有機溶媒を蒸発させ、残った水性層をエーテル(3×20mL)で洗浄した。合わせた有機層を6M 塩酸(20mL)で逆抽出した。合わせた水性相を16時間加熱還流した。溶媒を減圧下で蒸発させて、表題化合物をクリーム色固体(1.10g、100%)として得た。エナンチオマー純度は90.12%(S)−entであることが判明した(LC;実施例14dと同じ方法)。
1H-NMR(400MHz, dmso-d6)δ 1.48(3H, s), 2.87(1H, d), 2.98(1H, d), 3.82(2H, m), 7.3-7.39(5H, m), 8.48(3H, s)。
【0207】
実施例16
a)イソプロピルアラニネートヒドロクロライド
【化33】
塩化チオニル(26.70g、224mmol)を、DL−アラニン(10g、112mmol)のイソプロパノール(400mL)溶液に−20℃で滴下した。混合物をRTにし、次いで4時間加熱還流した。溶媒を蒸発させて、表題化合物(18.8g、100%)を得た。
1H-NMR(300MHz)δ 1.20-1.22(6H+3H, 各d), 3.46-3.50(1H, q), 4.01-4.06(1H, m)。
【0208】
b)イソプロピルN−ベンジリデンアラニネート
【化34】
硫酸マグネシウム(13.50g、112mmol)およびベンズアルデヒド(4.76g、44.8mmol)を、イソプロピルアラニネートヒドロクロライド(9.40g、56mmol)のDCM(200mL)中のスラリーに添加した。トリエチルアミン(5.6g、56mmol)をこの混合物に添加し、次いでそれを16時間窒素雰囲気下で撹拌した。さらに硫酸マグネシウム(3.30g、28mmol)およびトリエチルアミン(0.56g、5.6mmol)を添加した。混合物を24時間撹拌した。混合物を濾過し、濾液を水(2×50mL)で洗浄した。有機層を乾燥させ(MgSO4)、溶媒を蒸発させて、生成物を無色油状物(9g、76%)として得た。
1H-NMR(400MHz, CDCl3,)δ 1.23-1.26(6H, m), 1.51(3H, d), 4.08-4.13(1H, q), 5.03-5.09(1H, m), 7.36-7.45(3H, m), 7.76-7.79(2H, m), 8.31(1H, s)。
【0209】
c)S−ベンジル−2−メチル−D(R)−システインヒドロクロライド
【化35】
粉末水酸化セシウム(3.82g、22.8mmol)およびキラル相間移動触媒(−)−O−アリル−N−(9−アントラセニルメチル)シンコニジニウムブロマイド(0.27g、0.45mmol)を、イソプロピルN−ベンジリデンアラニネート(1g、4.5mmol)のトルエン(10mL)溶液に−10℃で添加した。混合物をこの温度で1時間撹拌した。反応混合物を−30℃に冷却し、ブロモメチルスルファニルメチル−ベンゼン(2.47g、11.4mmol)のトルエン(10mL)溶液を滴下した。反応をこの温度で1時間撹拌し、次いでRTにした。反応混合物をセライトを通して濾過し、トルエン(10mL)で洗浄した。溶媒を蒸発させて、粗生成物をガムとして得た。この物質をTHF(20mL)に取り込み、6M 塩酸(20mL)を添加した。混合物をRTで16時間撹拌し、次いで4時間加熱還流した。混合物をRTに冷却し、酢酸エチル(20mL)で洗浄した。水性層を減圧下蒸発乾固した。残渣をトルエン(2×20mL)に取り込み、次いで減圧下蒸発乾固して、表題化合物をクリーム色固体(1.20g、100%)として得た。キラルHPLC分析は、生成物が91.72%エナンチオマー純度であることを示した。
1H-NMR(400MHz, dmso-d6)δ 1.44(3H, s), 2.87(1H, d), 2.98(1H, d), 3.8-3.85(2H, m), 7.3-7.39(5H, m), 8.50(3H, s)。
MS m/z 226 [MH]+。
【0210】
実施例17
a)tert−ブチルN−(ナフタレン−2−イル−メチリデン)アラニネート
【化36】
硫酸マグネシウム(2.65g、22mmol)、2−ナフトアルデヒド(1.63g、20mmol)およびトリエチルアミン(1.1g、10mmol)を、t−ブチルアラニネートヒドロクロライド(DL)(2g、11mmol)のDCM(40mL)中のスラリーに添加した。混合物を24時間窒素雰囲気下で撹拌した。硫酸マグネシウム(1.32g、11mmol)を添加し、反応をさらに16時間撹拌した。分析後、さらに硫酸マグネシウム(2.65g、22mmol)を添加し、混合物を3日間、RTで撹拌した。混合物を濾過し、濾液を水(2×100mL)で洗浄した。有機層を硫酸マグネシウムで乾燥させ、濾過し、溶媒を蒸発させて、表題化合物を白色固体(2.43g、78%)として得た。
1H-NMR(300MHz, CDCl3)δ 1.46(9H, s), 1.56(3H, s), 4.07-4.14(1H, q), 7.48-7.55(2H, m), 7.84-7.91(3H, m), 8.02-8.09(2H, m), 8.45(1H, s)。
【0211】
b)S−ベンジル−2−メチル−D(R)−システインヒドロクロライド
【化37】
粉末水酸化ルビジウム一水和物(1.79g、17.5mmol)およびキラル相間移動触媒(−)−O−アリル−N−(9−アントラセニルメチル)シンコニジニウムブロマイド(0.21g、0.35mmol)を、tert−ブチルN−(ナフタレン−2−イル−メチリデン)アラニネート(1g、3.5mmol)のトルエン(10mL)溶液に−30℃で添加した。混合物をこの温度で30分撹拌した。ブロモメチルスルファニルメチル−ベンゼン(1.91g、8.8mmol)のトルエン(10mL)溶液を、この混合物に滴下した。反応をこの温度で1時間撹拌し、次いでRTで3日撹拌した。混合物をセライトを通して濾過し、トルエン(50mL)で洗浄した。溶媒を蒸発させて、保護されたメチルシステインをガムとして得た。この物質を単離または特徴付けせずに、直接使用した。粗生成物をTHF(20mL)および6M 塩酸(20mL)の混合物に取りこんだ。混合物をRTで16時間撹拌した。有機溶媒を蒸発させ、残渣を酢酸エチル(20mL)で洗浄した。酢酸エチル層を6M 塩酸(20mL)で逆抽出した。次いで合わせた水性相を70℃で10時間加熱した。水性層を酢酸エチル(50mL)で洗浄し、その後減圧下蒸発乾固した。残渣をトルエン(50mL)を使用して共沸乾燥させ、表題化合物をクリーム色固体(0.90g、83%)として得た。キラルHPLC分析は、生成物が94.3%エナンチオマー純度であることを示した。
1H-NMR(300MHz, dmso-d6)δ 1.50(3H, s), 2.90(1H, d), 3.20(1H, d), 3.82(2H, s), 7.3-7.39(5H, m), 8.52(3H, s)。
MS(ES), m/z 226 [MH]+。
【図面の簡単な説明】
【0212】
【図1】化合物(I)形態GのX線粉末回折ダイアグラムである;
【図2】化合物(I)形態Gの示差走査熱量測定(DSC)トレースおよび熱重量分析(TGA)トレースである;
【図3】化合物(I)形態AのX線粉末回折ダイアグラムである;
【図4】化合物(I)形態BのX線粉末回折ダイアグラムである;
【図5】化合物(I)形態CのX線粉末回折ダイアグラムである;
【図6】化合物(I)形態DのX線粉末回折ダイアグラムである;
【図7】化合物(I)形態EのX線粉末回折ダイアグラムである;
【図8】化合物(I)形態FのX線粉末回折ダイアグラムである
【技術分野】
【0001】
発明の分野
本発明は、(5S)−5−[4−(5−クロロ−ピリジン−2−イルオキシ)−ピペリジン−1−スルホニルメチル]−5−メチル−イミダゾリジン−2,4−ジオンの新規結晶多形、このような多形の製造方法、そのような多形を含む医薬組成物、および治療におけるそのような多形の使用を開示する。
【背景技術】
【0002】
発明の背景
引用によりその全体を本明細書に包含させるWO02/074767は、治療に有用なメタロプロテイナーゼ阻害剤のクラスを教示する。
【0003】
WO02/074767は、さらに、(5S)−5−[4−(5−クロロ−ピリジン−2−イルオキシ)−ピペリジン−1−スルホニルメチル]−5−メチル−イミダゾリジン−2,4−ジオンとしてその中で同定されている具体的メタロプロテイナーゼ阻害剤化合物を開示する(65頁、15〜27行;および120頁、23〜29行)。この化合物を、ここでは化合物(I)と呼ぶ。
【化1】
WO02/074767は、さらに、化合物(I)の製造方法も開示する。
【0004】
それ故に、一つの態様において、化合物(I)は、以下のスキーム(WO02/074767;87、113および120頁)に示す経路に準じるが、工程(d)において適当なアミンに置き換えて製造する:
【化2】
スキーム1の試薬および条件:a)KCN、(NH4)2CO3、EtOH/H2O、+90℃、3時間;b)キラル分割、CHIRALPAK AD、溶離剤としてメタノール;c)Cl2(g)、AcOH/H2O、<+15℃、25分;d)ジイソプロピルエチルアミン、THF。−20℃、30分。
【0005】
次いで、得られた化合物(I)を沈殿とエタノール/水の洗浄、または分取HPLCのいずれかにより精製する。
【0006】
第二の態様において、化合物(I)のラセミ体、(5RS)−5−[4−(5−クロロ−ピリジン−2−イルオキシ)−ピペリジン−1−スルホニルメチル]−5−メチル−イミダゾリジン−2,4−ジオンを、1−[4−(5−クロロ−ピリジン−2−イルオキシ)−ピペリジン−1−スルホニル]−プロパン−2−オンと過剰のカリウムシアニドおよび炭酸アンモニウムを、エタノール中で反応させ、生成物を沈殿により単離することにより製造した。次いで、化合物(I)、(5S)−エナンチオマーをキラルHPLCにより得た(WO02/074767;55および65頁)。
化合物(I)の結晶形はWO02/074767に開示されていない。
【0007】
化合物(I)は強力なメタロプロテイナーゼ阻害剤、特にMMP12の強力な阻害剤であり、それ自体治療に有用である。しかしながら、WO02/074767に記載の方法に従って製造したとき、化合物(I)は、熱力学的安定性に関して予測されない固体状態特性を示す。米国および他の国際的な保健登録認定機関(health registration authorities)の要求に従って、ヒトに投与するための化合物(I)を含む医薬製剤を製造するために、一定の物理特性を有する、安定な結晶形態のような安定な形で化合物(I)を製造する必要がある。
【0008】
多形は、特定の化合物が、同じ化学式を維持しながら異なる結晶多形に結晶化する能力として特徴付けることができる。ある物質の多形は、同じ方法で互いに結合している同じ原子を含んで化学的に同一であるが、それらの結晶多形が異なり、これは溶解速度、融点、かさ密度、安定性、流動特性などのような1種以上の物理的特性に影響し得る。本発明において特定の化合物について使用するとき、用語“多形”、“結晶多形”、“結晶形”、“結晶の多形”および“(結晶)形態”は同義語であると理解すべきである。
【0009】
本発明は、固体状態における化合物(I)の熱力学的特性を改善するための方法を提供し、それ故に、一貫したそして遊離な物理特性を有する安定な結晶多形の化合物(I)を提供する。
【0010】
図面の簡単な説明
図1は、化合物(I)形態GのX線粉末回折ダイアグラムである;
図2は、化合物(I)形態Gの示差走査熱量測定(DSC)トレースおよび熱重量分析(TGA)トレースである;
図3は、化合物(I)形態AのX線粉末回折ダイアグラムである;
図4は、化合物(I)形態BのX線粉末回折ダイアグラムである;
図5は、化合物(I)形態CのX線粉末回折ダイアグラムである;
図6は、化合物(I)形態DのX線粉末回折ダイアグラムである;
図7は、化合物(I)形態EのX線粉末回折ダイアグラムである;
図8は、化合物(I)形態FのX線粉末回折ダイアグラムである。
【発明の開示】
【0011】
発明の開示
本発明により、驚くべきことに、(5S)−5−[4−(5−クロロ−ピリジン−2−イルオキシ)−ピペリジン−1−スルホニルメチル]−5−メチル−イミダゾリジン−2,4−ジオン、化合物(I)が少なくとも7個の異なる結晶多形(多形)で存在できることが判明した。
【化3】
一つの局面において、本発明は、式(I)の化合物の7種の多形形態を提供する。
【0012】
一つの態様において、本発明は、形態Gと命名し、CuKα放射を使用して測定して10.1、16.2、16.8および19.0°2θにおける特異的ピークを含むX線粉末回折(XPRD)パターンを有することを特徴とする、(5S)−5−[4−(5−クロロ−ピリジン−2−イルオキシ)−ピペリジン−1−スルホニルメチル]−5−メチル−イミダゾリジン−2,4−ジオンの結晶多形を提供する。
【0013】
他の態様において、本発明は、形態Gと命名し、CuKα放射を使用して測定して9.7、10.1、11.5、12.8、14.1、16.2、16.8および19.0°2θにおける特異的ピークを含むX線粉末回折(XPRD)パターンを有することを特徴とする、(5S)−5−[4−(5−クロロ−ピリジン−2−イルオキシ)−ピペリジン−1−スルホニルメチル]−5−メチル−イミダゾリジン−2,4−ジオンの結晶多形を提供する。
【0014】
他の態様において、本発明は、形態Gと命名し、CuKα放射を使用して測定して図1に示すものと実質的に同じX線粉末回折(XPRD)パターンを有することを特徴とする、(5S)−5−[4−(5−クロロ−ピリジン−2−イルオキシ)−ピペリジン−1−スルホニルメチル]−5−メチル−イミダゾリジン−2,4−ジオンの結晶多形を提供する。
【0015】
他の態様において、本発明は、形態Gと命名し、図2に示すものと実質的に同じ示差走査熱量測定(DSC)トレースを有することを特徴とする、(5S)−5−[4−(5−クロロ−ピリジン−2−イルオキシ)−ピペリジン−1−スルホニルメチル]−5−メチル−イミダゾリジン−2,4−ジオンの結晶多形を提供する。
【0016】
他の態様において、本発明は、形態Aと命名し、6.8、9.8、13.7、16.4、18.4、18.7、20.4および22.6°2θにおける特異的ピークを含むX線粉末回折パターンを有することを特徴とする、(5S)−5−[4−(5−クロロ−ピリジン−2−イルオキシ)−ピペリジン−1−スルホニルメチル]−5−メチル−イミダゾリジン−2,4−ジオンの結晶多形を提供する。他の態様において、本発明は、図3に示すものと実質的に同じX線粉末回折パターンを有することを特徴とする、(5S)−5−[4−(5−クロロ−ピリジン−2−イルオキシ)−ピペリジン−1−スルホニルメチル]−5−メチル−イミダゾリジン−2,4−ジオンの結晶多形を提供する。
【0017】
他の態様において、本発明は、形態Bと命名し、6.6、7.1、8.3、9.0、13.6、14.3、16.8および17.7°2θにおける特異的ピークを含むX線粉末回折パターンを有することを特徴とする、(5S)−5−[4−(5−クロロ−ピリジン−2−イルオキシ)−ピペリジン−1−スルホニルメチル]−5−メチル−イミダゾリジン−2,4−ジオンの結晶多形を提供する。他の態様において、本発明は、形態Bと命名し、図4に示すものと実質的に同じX線粉末回折パターンを有することを特徴とする、(5S)−5−[4−(5−クロロ−ピリジン−2−イルオキシ)−ピペリジン−1−スルホニルメチル]−5−メチル−イミダゾリジン−2,4−ジオンの結晶多形を提供する。
【0018】
他の態様において、本発明は、形態Cと命名し、6.3、12.8、14.3、16.6、17.8、19.4、22.2および23.7°2θにおける特異的ピークを含むX線粉末回折パターンを有することを特徴とする、(5S)−5−[4−(5−クロロ−ピリジン−2−イルオキシ)−ピペリジン−1−スルホニルメチル]−5−メチル−イミダゾリジン−2,4−ジオンの結晶多形を提供する。他の態様において、本発明は、形態Cと命名し、図5に示すものと実質的に同じX線粉末回折パターンを有することを特徴とする、(5S)−5−[4−(5−クロロ−ピリジン−2−イルオキシ)−ピペリジン−1−スルホニルメチル]−5−メチル−イミダゾリジン−2,4−ジオンの結晶多形を提供する。
【0019】
他の態様において、本発明は、形態Dと命名し、6.6、10.9、11.2、15.6、15.9、17.7、18.2および18.4°2θにおける特異的ピークを含むX線粉末回折パターンを有することを特徴とする、(5S)−5−[4−(5−クロロ−ピリジン−2−イルオキシ)−ピペリジン−1−スルホニルメチル]−5−メチル−イミダゾリジン−2,4−ジオンの結晶多形を提供する。他の態様において、本発明は、形態Dと命名し、図6に示すものと実質的に同じX線粉末回折パターンを有することを特徴とする、(5S)−5−[4−(5−クロロ−ピリジン−2−イルオキシ)−ピペリジン−1−スルホニルメチル]−5−メチル−イミダゾリジン−2,4−ジオンの結晶多形を提供する。
【0020】
他の態様において、本発明は、形態Eと命名し、12.1、13.9、14.5、14.8、15.3、16.2、18.7および19.8°2θにおける特異的ピークを含むX線粉末回折パターンを有することを特徴とする、(5S)−5−[4−(5−クロロ−ピリジン−2−イルオキシ)−ピペリジン−1−スルホニルメチル]−5−メチル−イミダゾリジン−2,4−ジオンの結晶多形を提供する。他の態様において、本発明は、形態Eと命名し、図7に示すものと実質的に同じX線粉末回折パターンを有することを特徴とする、(5S)−5−[4−(5−クロロ−ピリジン−2−イルオキシ)−ピペリジン−1−スルホニルメチル]−5−メチル−イミダゾリジン−2,4−ジオンの結晶多形を提供する。
【0021】
他の態様において、本発明は、形態Fと命名し、7.4、9.5、13.9、14.9、17.3、18.1、20.0および20.4°2θにおける特異的ピークを含むX線粉末回折パターンを有することを特徴とする、(5S)−5−[4−(5−クロロ−ピリジン−2−イルオキシ)−ピペリジン−1−スルホニルメチル]−5−メチル−イミダゾリジン−2,4−ジオンの結晶多形を提供する。他の態様において、本発明は、形態Fと命名し、図8に示すものと実質的に同じX線粉末回折パターンを有することを特徴とする、(5S)−5−[4−(5−クロロ−ピリジン−2−イルオキシ)−ピペリジン−1−スルホニルメチル]−5−メチル−イミダゾリジン−2,4−ジオンの結晶多形を提供する。
【0022】
X線粉末回折(XPRD)パターンのピークの相対的強度は、試験下のサンプルの方位および使用した装置のタイプおよび設定によって変わることがあり、従って、本明細書に含まれるXPRDトレースの強度は、説明的であり、絶対的比較に使用することを意図しないことは理解されよう。
【0023】
本発明の結晶多形または形態は、好ましくは実質的に純粋であり、式(I)の化合物の各結晶多形または形態が10重量%未満、好ましくは5重量%未満、好ましくは3重量%未満、好ましくは1重量%未満の、本化合物の他の結晶多形または形態を含む不純物を含むことを意味する。
【0024】
故に、一つの態様において、本発明は、形態Gと命名され、CuKα放射を使用して測定して10.1、16.2、16.8および19.0°2θにおける特異的ピークを含むX線粉末回折(XPRD)パターンを有することを特徴とする、(5S)−5−[4−(5−クロロ−ピリジン−2−イルオキシ)−ピペリジン−1−スルホニルメチル]−5−メチル−イミダゾリジン−2,4−ジオンの実質的に純粋な結晶多形を提供する。
【0025】
他の態様において、本発明は、形態Gと命名され、CuKα放射を使用して測定して9.7、10.1、11.5、12.8、14.1、16.2、16.8および19.0°2θにおける特異的ピークを含むX線粉末回折(XPRD)パターンを有することを特徴とする、(5S)−5−[4−(5−クロロ−ピリジン−2−イルオキシ)−ピペリジン−1−スルホニルメチル]−5−メチル−イミダゾリジン−2,4−ジオンの実質的に純粋な結晶多形を提供する。
【0026】
他の態様において、本発明は、形態Gと命名し、CuKα放射を使用して測定して図1に示すものと実質的に同じX線粉末回折(XPRD)パターンを有することを特徴とする、(5S)−5−[4−(5−クロロ−ピリジン−2−イルオキシ)−ピペリジン−1−スルホニルメチル]−5−メチル−イミダゾリジン−2,4−ジオンの実質的に純粋な結晶多形を提供する。
【0027】
化合物(I)形態Gは、針状晶癖を示す結晶を含む、白色結晶性粉末として得られる。本物質はX線粉末回折測定で測定して、本質的に100%結晶である。本結晶構造を、単結晶X線回折により決定した。本結晶において、分子は、斜方晶系空間群(P212121)にパックされている。非対称単位セル(a=10.510Å、b=11.169Å、c=15.560Å)に4分子存在する。内部空間の欠乏をもたらす最密充填は、1.46g/mLの相対的に高い密度で認められる。
【0028】
単結晶X線回折データを使用して計算した化合物(I)の模擬X線粉末回折パターンは、図1に示す実験により決定したパターンと良く一致する。回折ピークの一は非常に合い、相対ピーク強度の差異は、選択方位効果に帰する。
【0029】
WO02/074767に開示の方法で製造したとき、(5S)−5−[4−(5−クロロ−ピリジン−2−イルオキシ)−ピペリジン−1−スルホニルメチル]−5−メチル−イミダゾリジン−2,4−ジオンは、非晶相または形態Aまたは形態Cまたはそれらの混合物で得られる。
【0030】
化合物(I)形態Aの融点は、加熱によりそれが約175℃で形態Bに変形するために観察されていない。
【0031】
化合物(I)形態Bは、形態Aを約175℃に加熱したときの固体状態転移により製造する。形態Bは約207℃で融解し、その後形態Cに再結晶し、続いて再び約210℃で融解する。
化合物(I)形態Cは約210℃で融解する。
【0032】
化合物(I)形態Dは、化合物(I)を融解物からの結晶化により製造したときに産生される。例えば、形態Dを、形態B(室温で形態Aから開始)を形態Bの融解温度で融解させ;次いで室温に急冷して、非晶物質を産生し;次いで、再び5°/分で加熱することにより製造する。加熱の間に、この非晶物質はガラス遷移温度を通り、その後形態Dとして再結晶する。形態Dは約209℃で融解する。
【0033】
化合物(I)形態Eを、形態CをpH3の水中、例えば、環境温度で数日間スラリー化したとき産生する。形態Aと同様、形態Eは、約175℃で、恐らく形態Bに熱転移する。
【0034】
化合物(I)形態Fは、形態Aまたは形態Cを、エタノール中、例えば、環境温度で数日間スラリー化したとき産生する。形態Aおよび形態Eと同様、形態Fは、約175℃で、恐らく形態Bに熱転移する。
【0035】
化合物(I)形態Gは、(5S)−5−[4−(5−クロロ−ピリジン−2−イルオキシ)−ピペリジン−1−スルホニルメチル]−5−メチル−イミダゾリジン−2,4−ジオンを水性エタノールまたは工業用変性アルコール水溶液(aqueous industrial methylated spirits)から再結晶したときに再現性よく産生される。形態Gは約201℃で融解し、使用する条件、例えば、加熱速度に依存して、その後一部または完全に形態Cに再結晶し、それは次いで約210℃で再融解する。
【0036】
化合物(I)形態AからGのいずれかを加熱したとき、融解前に、約175℃で起こり得る上記に略記した可能性のある固体状態転移以外、溶媒損失も何らかの他の熱事象も見られない。故に、形態AからGの各々は熱的に安定である。
【0037】
化合物(I)形態AからGの相対的な熱力学的安定性は、形態AからGの混合物を5日間、5から40℃の範囲の温度で、水中共インキュベートした懸濁液実験により評価した。全例で、得られた沈殿のX線粉末回折試験(XRPD)は、形態Gへの完全な変換を証明した。同じ結果が、種々の有機溶媒(エタノール、メタノール、1−プロパノール、2−プロパノール、アセトンまたは酢酸エチル)中の形態Aの懸濁液のインキュベーション後に観察された。これらの結果に基づき、化合物(I)形態Gが、試験した温度範囲で、7種の結晶多形の中で最も熱力学的に安定であると結論付けることができる。
【0038】
ここに開示の方法を使用して、化合物(I)形態Gは小規模、中規模または大規模合成法に従って再現性よく製造できる。
【0039】
事実上溶媒分子のための内部空間が示されない化合物(I)形態Gの単結晶X線構造決定から予測される通り、重量測定的蒸気収着(GVS)を使用した吸湿測定は、本物質が、高相対湿度でさえほとんど湿気取り込みがないことを示した(80%RHで<0.05%湿度取り込み)。本物質は、それ故に、欧州薬局方に定義された基準に従って、非吸湿性と有利に分離される。
【0040】
化合物(I)形態Gは、優れたそして非常に有利な固体状態特性を有する。それは結晶性、非吸湿性であり、そして200℃以下で熱的に安定であり、融解前に溶媒損失も何らかの他の熱的事象も示さない(DSCおよびTGAトレース、図2参照)。形態Gはまた、化合物(I)の既知の7種の結晶多形の中で熱力学的に最も安定である。
【0041】
化合物(I)形態Gの固体状態安定性を3条件:25℃/乾燥;25℃/60%RH;および40℃/75%RHで試験した。サンプルを、2週間、4週間、8週間および12週間後に試験し、化学的および物理的安定性を評価した。本物質は、何らかの可能性のある分解産物と比較して(5S)−5−[4−(5−クロロ−ピリジン−2−イルオキシ)−ピペリジン−1−スルホニルメチル]−5−メチル−イミダゾリジン−2,4−ジオンの含量(勾配RPLC)に;化合物(I)の形態(XRPD)に;化合物(I)の形態学(SEM)に;溶媒含量(TGA)に;または融解行動(DSC)に変化が見られなかったため、全貯蔵条件下で化学的および物理的に安定であると結論付けられた。それ故に、化合物(I)形態Gは、薬学的に適切な貯蔵条件下で、固体状態で優れたそして有利な化学および物理的安定性を有すると見なされる。
【0042】
一つの局面において、本発明は、(5S)−5−[4−(5−クロロ−ピリジン−2−イルオキシ)−ピペリジン−1−スルホニルメチル]−5−メチル−イミダゾリジン−2,4−ジオン形態Gを提供する。
【0043】
さらなる局面において、本発明は、(5S)−5−[4−(5−クロロ−ピリジン−2−イルオキシ)−ピペリジン−1−スルホニルメチル]−5−メチル−イミダゾリジン−2,4−ジオン形態Gの製造方法を提供する。故に、一つの局面において、本発明は、水性エタノールからの結晶化または再結晶を含む、(5S)−5−[4−(5−クロロ−ピリジン−2−イルオキシ)−ピペリジン−1−スルホニルメチル]−5−メチル−イミダゾリジン−2,4−ジオン形態Gの製造方法を提供する。他の局面において、本発明は、工業用変性アルコール水溶液からの結晶化または再結晶を含む、(5S)−5−[4−(5−クロロ−ピリジン−2−イルオキシ)−ピペリジン−1−スルホニルメチル]−5−メチル−イミダゾリジン−2,4−ジオン形態Gの製造方法を提供する。
【0044】
他の局面において、本発明は、以下の工程を含む、(5S)−5−[4−(5−クロロ−ピリジン−2−イルオキシ)−ピペリジン−1−スルホニルメチル]−5−メチル−イミダゾリジン−2,4−ジオン形態Gの製造方法を提供する:
i) (5S)−5−[4−(5−クロロ−ピリジン−2−イルオキシ)−ピペリジン−1−スルホニルメチル]−5−メチル−イミダゾリジン−2,4−ジオンを、工業用変性アルコール(IMS):水の2:1混合物に添加し;
ii) 本混合物を加熱還流して溶液を得て;
iii)熱溶液を濾過し;
iv) 本濾液を加熱還流し、次いでそれを約0.5℃/分の速度で約20℃まで冷却し;
v) (5S)−5−[4−(5−クロロ−ピリジン−2−イルオキシ)−ピペリジン−1−スルホニルメチル]−5−メチル−イミダゾリジン−2,4−ジオン形態Gを回収し、乾燥させる。
【0045】
さらなる局面において、本発明は、治療に使用するための(5S)−5−[4−(5−クロロ−ピリジン−2−イルオキシ)−ピペリジン−1−スルホニルメチル]−5−メチル−イミダゾリジン−2,4−ジオン形態Gを提供する。
【0046】
さらなる局面において、本発明は、MMP活性の阻害が有益である疾患または状態の処置または予防用医薬の製造において使用するための、(5S)−5−[4−(5−クロロ−ピリジン−2−イルオキシ)−ピペリジン−1−スルホニルメチル]−5−メチル−イミダゾリジン−2,4−ジオンの結晶多形を提供する。
【0047】
さらなる局面において、本発明は、MMP活性の阻害が有益である疾患または状態の処置用医薬の製造に使用するための、(5S)−5−[4−(5−クロロ−ピリジン−2−イルオキシ)−ピペリジン−1−スルホニルメチル]−5−メチル−イミダゾリジン−2,4−ジオン形態Gを提供する。
【0048】
さらなる局面において、本発明は、処置を必要とする患者に治療的有効量の(5S)−5−[4−(5−クロロ−ピリジン−2−イルオキシ)−ピペリジン−1−スルホニルメチル]−5−メチル−イミダゾリジン−2,4−ジオンの結晶多形を投与することを含む、
MMP活性が仲介する疾患または状態の処置または予防方法を提供する。
【0049】
さらなる局面において、本発明は、処置を必要とする患者に治療的有効量の(5S)−5−[4−(5−クロロ−ピリジン−2−イルオキシ)−ピペリジン−1−スルホニルメチル]−5−メチル−イミダゾリジン−2,4−ジオン形態Gを投与することを含む、MMP活性が仲介する疾患または状態の処置または予防方法を提供する。
【0050】
特に、化合物(I)は、MMP12および/またはMMP13および/またはMMP9および/またはMMP8および/またはMMP3が仲介する疾患または状態の処置;とりわけMMP12および/またはMMP9が仲介する疾患または状態の処置;特にMMP12が仲介する疾患または状態の処置に有用である。
【0051】
さらなる局面において、本発明は、(5S)−5−[4−(5−クロロ−ピリジン−2−イルオキシ)−ピペリジン−1−スルホニルメチル]−5−メチル−イミダゾリジン−2,4−ジオンの結晶多形を含む医薬組成物を提供する。
【0052】
さらなる局面において、本発明は、(5S)−5−[4−(5−クロロ−ピリジン−2−イルオキシ)−ピペリジン−1−スルホニルメチル]−5−メチル−イミダゾリジン−2,4−ジオン形態Gを含む医薬組成物を提供する。
【0053】
さらなる局面において、本発明は、処置を必要とする患者に治療的有効量の(5S)−5−[4−(5−クロロ−ピリジン−2−イルオキシ)−ピペリジン−1−スルホニルメチル]−5−メチル−イミダゾリジン−2,4−ジオンの結晶多形を含む医薬組成物を投与することを含む、メタロプロテイナーゼ活性が仲介する疾患または状態の処置方法を提供する。
【0054】
さらなる局面において、本発明は、処置を必要とする患者に治療的有効量の(5S)−5−[4−(5−クロロ−ピリジン−2−イルオキシ)−ピペリジン−1−スルホニルメチル]−5−メチル−イミダゾリジン−2,4−ジオン形態Gを含む医薬組成物を投与することを含む、メタロプロテイナーゼ活性が仲介する疾患または状態の処置方法を提供する。
【0055】
さらなる局面において、本発明は、MMP活性の阻害が有益である疾患または状態の処置のための、(5S)−5−[4−(5−クロロ−ピリジン−2−イルオキシ)−ピペリジン−1−スルホニルメチル]−5−メチル−イミダゾリジン−2,4−ジオンの結晶多形を含む医薬製剤の使用を提供する。
【0056】
さらなる局面において、本発明は、MMP活性の阻害が有益である疾患または状態の処置のための、(5S)−5−[4−(5−クロロ−ピリジン−2−イルオキシ)−ピペリジン−1−スルホニルメチル]−5−メチル−イミダゾリジン−2,4−ジオン形態Gを含む医薬製剤の使用を提供する。
【0057】
他の局面において、本発明は、炎症性疾患または状態の処置または予防のための医薬の製造における式(I)の化合物形態Gの使用;および炎症性疾患または状態を処置、またはリスクを軽減する方法であって、該疾患または状態に罹患している、またはリスクのあるヒトに治療的有効量の式(I)の化合物形態Gを投与することを含む、方法を提供する。
【0058】
化合物(I)は、間欠性および永続性両方のおよび全ての重症度、および気道過敏反応性の他の原因を含む、気管支、アレルギー性、内因性、外因性、運動誘発性、薬剤誘発性(アスピリンおよびNSAID誘発性を含む)および粉塵誘発性喘息を含む、喘息;慢性閉塞性肺疾患(COPD);感染性および好酸球性気管支炎を含む気管支炎;気腫;気管支拡張症;嚢胞性線維症;サルコイドーシス;農夫肺および関連疾患;過敏性肺炎;原因不明線維化肺胞炎、特発性間質性肺炎、抗新生物治療および結核およびアスペルギルス症および他の真菌感染を含む慢性感染に合併する線維症を含む、肺線維症;肺移植の合併症;肺脈管構造の血管炎性および血栓性障害、および肺高血圧;気道の炎症性および分泌状態と関連する慢性咳、および医原性咳;薬物性鼻炎、および血管運動性鼻炎を含む急性および慢性鼻炎;神経性鼻炎(枯草熱)を含む通年性および季節性アレルギー性鼻炎;鼻のポリープ症;一般的な風邪、および呼吸器多核体ウイルス、インフルエンザ、コロナウイルス(SARSを含む)およびアデノウイルスによる感染を含む、急性ウイルス感染を含む、気道の閉塞性疾患のような呼吸管の疾患の処置に使用できる。
【0059】
化合物(I)はまた、原発性および、例えば、先天的股関節異形成症に二次性両方の骨関節症/骨関節症と関連するまたは含む関節炎(arthritides);頚部および腰部脊椎炎、および背下部および頚部痛;リウマチ性関節炎およびスチル病;強直性脊椎炎、乾癬性関節炎、反応性関節炎および未分化脊椎関節症(spondarthropathy)を含む血清反応陰性脊椎関節症;敗血症性関節炎およびポット病およびポンセ病を含む結核のような、他の感染関連関節症(arthopathies)および骨障害;尿酸塩痛風、ピロリン酸カルシウム沈着疾患、およびカルシウムアパタイト関連腱、滑液包および滑膜炎症を含む、急性および慢性結晶誘発滑膜炎;ベーチェット病;原発性および二次性シェーグレン症候群;全身性硬化症および限局型強皮症;全身性エリテマトーデス、混合型結合組織疾患、および未分化結合組織疾患;皮膚筋炎および多発性筋炎を含む炎症性ミオパシー;リウマチ性多発筋痛症;どんな関節分布であれ特発性炎症性関節炎(arthritides)および関連症候群、およびリウマチ熱およびその全身合併症を含む若年性関節炎;巨細胞性動脈炎、高安動脈炎、チャーグ・ストラウス症候群、結節性多発性動脈炎、顕微鏡的多発動脈炎、およびウイルス感染、過敏症反応、クリオグロブリン、およびパラプロテインと関連する脈管炎を含む脈管炎;背下部疼痛;家族性地中海熱、マックル・ウェルズ症候群、および家族性アイルランド熱(Familial Hibernian Fever)、キクチ病;薬剤誘発性関節痛(arthalgias)、腱炎(tendonititides)、およびミオパシーのような骨および関節の疾患の処置にも使用できる。
【0060】
化合物(I)はまた、疼痛および傷害[例えば、運動傷害]または疾患による筋骨格障害の結合組織リモデリング:関節炎(arthitides)(例えばリウマチ性関節炎、骨関節症、痛風または結晶性関節症)、他の関節疾患(例えば椎間板変性または側頭下顎関節変性)、骨リモデリング疾患(例えば骨粗鬆症、ページェット病または骨壊死)、多発性軟骨炎、強皮症、混合型結合組織障害、脊椎関節症または歯周疾患(例えば歯周炎)の処置にも使用できる。
【0061】
化合物(I)はまた、乾癬、アトピー性皮膚炎、接触性皮膚炎または他の湿疹性皮膚炎、および遅延型過敏症反応;植物および光皮膚炎;脂漏性皮膚炎、疱疹状皮膚炎、扁平苔癬、硬化性萎縮性苔癬、壊疽性膿皮症、皮膚サルコイド、円板状エリテマトーデス、天疱瘡、類天疱瘡、表皮水疱症、蕁麻疹、血管浮腫、脈管炎、毒性紅斑、皮膚好酸球増加症、円形脱毛症、男性型禿頭、スウィート症候群、ウェーバー・クリスチャン症候群、多形性紅斑;感染性および非感染性両方の蜂巣炎;脂肪織炎;皮膚リンパ腫、非黒色腫皮膚癌および他の形成異常病巣;固定薬疹を含む薬剤誘発性障害のような皮膚の疾患の処置にも使用できる。
【0062】
化合物(I)はまた、眼瞼炎;通年性および春季アレルギー性結膜炎を含む結膜炎;虹彩炎;前部および後部ブドウ膜炎;脈絡膜炎;自己免疫性;網膜に影響する変性または炎症性障害;交感神経性眼炎を含む眼炎;サルコイドーシス;ウイルス、真菌、および細菌を含む感染のような眼の疾患の処置にも使用できる。
【0063】
化合物(I)はまた舌炎、歯肉炎、歯周炎;逆流性を含む食道炎;好酸球性胃腸炎、肥満細胞症、クローン病、潰瘍性大腸炎を含む大腸炎、直腸炎、肛門掻痒症;セリアック病、過敏性腸症候群、非炎症性下痢、および腸から離れて作用し得る食物関連アレルギー(例えば、偏頭痛、鼻炎または湿疹)のような胃腸管の疾患の処置にも使用できる。
【0064】
化合物(I)はまた、冠血管および末梢循環に影響するアテローム性動脈硬化症;心膜炎;心筋炎、心筋サルコイドを含む炎症性および自己免疫性心筋症;虚血再灌流傷害;感染性(例えば梅毒性)を含む心内膜炎、弁膜炎、および大動脈炎;脈管炎;深部静脈血栓症および静脈瘤の合併症を含む静脈炎および血栓症を含む、近位および末梢静脈の障害のような心血管系の処置にも使用できる。
【0065】
化合物(I)はまた、転移疾患および腫瘍再発、および新生物随伴症候群の予防および処置を含む;前立腺、乳房、肺、卵巣、膵臓、腸および結腸、胃、皮膚および脳腫瘍および骨髄(白血病を含む)およびホジキンおよび非ホジキンリンパ腫のようなリンパ増殖性系に影響する悪性物を含む一般的な癌の処置のような腫瘍学においても使用できる。
【0066】
特に、化合物(I)は、成人呼吸窮迫症候群(ARDS)、嚢胞性線維症、肺気腫、慢性閉塞性肺疾患(COPD)、肺高血圧、喘息、鼻炎、虚血−再灌流傷害、リウマチ性関節炎、骨関節症、癌、アテローム性動脈硬化症および胃粘膜傷害の処置に使用できる。
【0067】
より具体的に、化合物(I)は慢性閉塞性肺疾患(COPD)、喘息および鼻炎の処置に使用できる。
さらに具体的に、化合物(I)は慢性閉塞性肺疾患(COPD)の処置に使用できる。
【0068】
予防は、以前に当該疾患または状態の事象に罹患したか、他の点でリスクが増加していると見なされるヒトの処置に特に適切である。特定の疾患または状態を発症するリスクのあるヒトは、一般に、該疾患または状態の家族歴がある、または遺伝子試験またはスクリーニングにより該疾患または状態の発症に特に感受性であることが同定されているヒトを含む。
【0069】
上記の治療適応症について、投与すべき化合物の容量は、処置すべき疾患、疾患の重症度、投与形態、患者の年齢、体重および性別による。このような因子は担当医により決定され得る。しかしながら、一般に、本化合物を、ヒトに0.1mg/kgから100mg/kg(活性成分として測定)の1日量を投与したときに、満足いく結果が得られる。
【0070】
式(I)の結晶化合物は、それ自体で、または本発明の化合物を薬学的に許容される希釈剤、アジュバントまたは担体と組み合わせて含む適当な医薬製剤の形で使用できる。特に好ましいのは、有害反応、例えば、アレルギー性反応の原因となり得る物質を含まない組成物である。適当な医薬製剤の選択および製造のための慣用法は、例えば、“Pharmaceuticals - The Science of Dosage Form Designs”, M. E. Aulton, Churchill Livingstone, 1988に記載されている。
【0071】
本発明によって、好ましくは95重量%未満、より好ましくは50重量%未満の式(I)の化合物形態Gを、薬学的に許容される希釈剤または担体と組み合わせて含む、医薬製剤が提供される。
【0072】
我々は、これらの成分を混合することを含む、医薬製剤の製造方法も提供する。
【0073】
本化合物は、例えば、肺および/または気道に、溶液、懸濁液、HFAエアロゾルまたは乾燥粉末製剤の形で、例えば、Turbuhaler(登録商標)として既知の吸入デバイス中の製剤として、局所的に;または例えば、錠剤、ピル、カプセル、シロップ、粉末または顆粒の形で、経口投与により、全身的に;または例えば、非経腸溶液または懸濁液の形で、非経腸(腹腔内、静脈内、皮下または筋肉内注射を含む)投与により;または例えば、坐薬の形で、直腸投与により投与できる。
【0074】
本発明の化合物の乾燥粉末製剤および加圧HFAエアロゾルは、経口または経鼻吸入により投与できる。吸入のために、本化合物は、望ましくは微粉化する。微粉化された化合物は、好ましくは10μm未満の質量中央径を有し、噴射剤混合物に、分散剤、例えばC8−C20脂肪酸またはその塩(例えば、オレイン酸)、胆汁塩、リン脂質、アルキルサッカライド、過フッ素化またはポリエトキシル化界面活性剤、または他の薬学的に許容される分散剤の助けを借りて、懸濁し得る。
【0075】
本発明の化合物はまた乾燥粉末吸入器の手段により投与してもよい。本吸入器は1回量または多回量吸入器であってよく、呼気作動型乾燥粉末吸入器であってよい。
【0076】
一つの可能性は、微粉化した化合物と担体物質、例えば、モノ、ジまたはポリサッカライド、糖アルコール、または他のポリオールの混合である。適当な担体は、糖類、例えば、ラクトース、グルコース、ラフィノース、メレジトース、ラクチトール、マルチトール、トレハロース、スクロース、マンニトール;およびデンプンである。あるいは、微粉化した化合物を、他の物質でコーティングし得る。粉末混合物をまた、各々所望量の活性化合物を含む硬ゼラチンカプセルに分配してもよい。
【0077】
他の可能性は、微粉化した粉末の、吸入工程中に破壊する球体への加工である。この球状粉末を、多回容量吸入器、例えば、Turbuhaler(登録商標)として既知の吸入器の貯蔵部に充填し得て、ここで、投与ユニットが所望量を定量し、次いでそれが患者により吸入される。このシステムで、活性化合物は、担体物質と共にまたは無しで、患者に送達される。
【0078】
経口投与のために、活性化合物をアジュバントまたは担体、例えば、ラクトース、サッカロース、ソルビトール、マンニトール;デンプン、例えば、ジャガイモデンプン、コーンデンプンまたはアミロペクチン;セルロース誘導体;結合剤、例えば、ゼラチンまたはポリビニルピロリドン;および/または平滑剤、例えば、ステアリン酸マグネシウム、ステアリン酸カルシウム、ポリエチレングリコール、蝋、パラフィンなどと混合し、次いで、錠剤に圧縮してよい。コーティング錠が必要ならば、上記のように製造したコアを、例えば、アラビアゴム、ゼラチン、タルク、二酸化チタンなどを含み得る濃縮糖溶液でコーティングし得る。あるいは、錠剤を、易揮発性の有機溶媒に溶解した適当なポリマーでコーティングして良い。
【0079】
軟ゼラチンカプセルの製造のために、化合物を、例えば、植物油またはポリエチレングリコールと混合し得る。硬ゼラチンカプセルは、錠剤について上記の賦形剤のいずれかを使用し、化合物の顆粒を含み得る。また、薬剤の液体または半固体製剤を硬ゼラチンカプセルに充填し得る。
【0080】
経口投与用液体製剤は、化合物を含むシロップまたは懸濁液、例えば、溶液の形であり得て、バランスは糖およびエタノール、水、グリセロールとプロピレングリコールの混合物である。所望により、このような液体製剤は、着色剤、香味剤、濃化剤としてサッカリンおよび/またはカルボキシメチルセルロース、または当業者に既知の他の賦形剤を含み得る。
【0081】
本発明のさらなる局面において、我々は、化合物(I)の新規合成方法を提供する。特に、化合物(I)の結晶多形の新規合成方法を開示する。特に、化合物(I)形態Gの新規合成方法を開示する。
【0082】
化合物(I)の好ましい合成方法をスキーム2に示す。
【化4】
【0083】
スキーム2において、化合物(II)、(III)および(IV)の硫黄部分をS−ベンジル誘導体として保護する。当業者は、t−ブチルのような他の適当な保護基を、代わりに使用できることを容易に認識しよう。故に、簡便のために、以降の反応はS−ベンジル保護された化合物を使用して示すが、t−ブチルのような適当な他の保護基も使用できることは理解すべきである。
【0084】
5−クロロ−2−(ピペリジン−4−イルオキシ)−ピリジン(VI)は、蝋状固体(m.p.約43℃)であり、そのようなものとして、特に大規模でのこの物質の結晶化および単離は理想的ではない。酢酸塩(VII)のような塩の製造は、化合物をより簡便に取り扱われる固体として単離することを可能にする。酢酸塩以外の塩も使用できる。このような塩は、リン酸塩、一塩酸塩、二塩酸塩、酢酸トリメチル、酒石酸塩、クエン酸塩、フマル酸塩、マレイン酸塩、安息香酸塩、一臭化水素酸塩、二臭化水素酸塩、炭酸塩および0.5炭酸塩を含む。炭酸塩は、それらが熱に不安定であり、そのため遊離塩基が単純に加温によりイン・サイチュで遊離できるため、特に有用である。
【0085】
故に、一つの局面において、我々は、5−クロロ−2−(ピペリジン−4−イルオキシ)−ピリジン酢酸塩(VII)の仲介を含む、5−クロロ−2−(ピペリジン−4−イルオキシ)−ピリジン(VI)の単離および取り扱いの改良法を開示する。
【0086】
他の局面において、我々は、化合物(I)の製造における中間体として有用な、5−クロロ−2−(ピペリジン−4−イルオキシ)−ピリジン(VI)の新規塩を開示する。
【0087】
好ましい方法において、5−クロロ−2−(ピペリジン−4−イルオキシ)−ピリジン酢酸塩(VII)の合成は、トルエンのような溶媒中で有利に行われる。反応溶媒としてのトルエンの使用は、2,5−ジクロロピリジンと4−ヒドロキシピペリジンの反応、続く水性洗浄および塩形成を、中間体遊離塩基を単離する必要なく、同じ反応容器で実施することを可能にする。水がこの反応における重要なパラメータであるため(そして4−ヒドロキシピペリジンは吸湿性である)、反応開始前に水を共沸により除去するためのトルエンの使用は、顕著な改善を示し、数キログラム(multi-kilogram)規模でさえ、単離される一定の収率を可能にする。
【0088】
ベンジルチオアセトン(II)からの(RS)−5−メチル−5−{[(フェニルメチル)チオ]メチル}イミダゾリジン−2,4−ジオン(III)の製造は、WO02/074767に記載されている。そこに記載されている条件と比較して、我々は、有機溶媒がエタノールから2−プロパノールに代わり、使用されるカリウムシアニド量が2当量から約1.00乃至1.02当量に減少した、改善法をここに開示する。この方法で、カリウムシアニドは、反応で本質的に完全に消費され、大量の未反応カリウムシアニドを含む溶液の廃棄が避けられる。我々は、使用する炭酸アンモニウム量の約5当量から約1.1乃至1.25当量への減少が特に有益であることをさらに記載する。この方法で、最大操作圧が約9バールから約1.5乃至2.5バールに低下し、特に大規模作業のためには、安全面で著しく有益である。これらの改良したパラメータを使用して、(RS)−5−メチル−5−{[(フェニルメチル)チオ]メチル}−イミダゾリジン−2,4−ジオン(III)の合成は、数キログラム規模で日常的に実施されている。
【0089】
故に、他の局面において、我々は、(RS)−5−メチル−5−{[(フェニルメチル)チオ]メチル}イミダゾリジン−2,4−ジオン(III)をベンジルチオアセトン(II)から製造するための改善された条件を開示する。これらの改善された条件は、大規模製造に特に有利である。
【0090】
WO02/074767に記載の通り、(RS)−5−メチル−5−{[(フェニルメチル)チオ]メチル}イミダゾリジン−2,4−ジオン(III)の構成エナンチオマーへの分割は、固定相としてChiralpak ADカラムおよび溶離剤としてメタノールを使用するキラルHPLCで簡便に達成される。特に大規模作業に簡便な別法として、我々は、キラル分割を、本質的に同じ条件で行うが、模擬移動床(SMB)クロマトグラフィーを使用する方法をここに開示する。この方法で、(S)−5−メチル−5−{[(フェニルメチル)チオ]メチル}イミダゾリジン−2,4−ジオン(IV)は、数キログラム規模で得ることができる。保護されていないチオール、(RS)−5−メチル−5−チオメチル−イミダゾリジン−2,4−ジオンは、驚くべきことに安定であり、固定相としてChiralpak ADカラムおよび移動相としてイソヘキサン/エタノール/ジエチルアミンを使用したキラルHPLCにより簡便に分割される。
【0091】
キラルクロマトグラフィーの代わりとして、他のキラルイミダゾリジン−2,4−ジオン(IV)に至る経路を開示する。
【0092】
ある種のヒダントイン誘導体の分割における(S)−α−メチルベンジルアミンの使用は開示されている(WO92/08702)。我々は、ラセミ体(RS)−5−メチル−5−{[(フェニルメチル)チオ]メチル}イミダゾリジン−2,4−ジオン(III)が、キラルアミンおよび水酸化ナトリウムのような塩基の存在下、適当な溶媒からの結晶化により分割できることを発見した。キラル素子アミンの例は、1S)−(−)−α−メチルベンジルアミン、(1R)−(+)−α−メチルベンジルアミン、L−チロシンアミド、(1S)−(−)−α−(1−ナフチル)エチルアミン、(1R)−(+)−α−(1−ナフチル)エチルアミン、L−(−)−シンコニジン、D−(+)−シンコニン、(−)−キニン、(+)−β−キニジン、(1R,2S)−(−)−エフェドリン、(2R)−(−)−2−アミノ−1−ブタノール、(2R)−1−アミノ−2−プロパノール(D−アラニノール)、(1R,2S)−(−)−2−アミノ−1,2−ジフェニルエタノール、N−メチル−D−(−)−グルカミン、(2S)−(+)−2−フェニルグリシノール、ノルエフェドリン、(−)−ブルシン、(−)−ストリキニーネ、(+)−ヨヒンビン、(1S,2S)−(+)−threo−2−アミノ−1−(p−ニトロフェニル)−1,3−プロパンジオール、(L)−(+)−threo−2−アミノ−1−フェニル−1,3−プロパンジオール、cis−ミルタニルアミン、(1R,2R)−(−)−1,2−ジアミノシクロヘキサンおよび(2R)−(−)−2−アミノ−2−フェニルエタノールを含む。
【0093】
好ましい方法において、キラルアミンは(1S)−(−)−α−メチルベンジルアミンである。
故に、一つの局面において、我々は、(1S)−α−メチルベンジルアミンを使用したラセミ体(RS)−5−メチル−5−{[(フェニルメチル)チオ]メチル}イミダゾリジン−2,4−ジオン(III)の分割方法を開示する。
【0094】
好ましい方法において、キラルアミンは(1S)−(−)−α−メチルベンジルアミン(1.0〜2.0当量)であり、塩基は水酸化ナトリウム(0.4〜0.6当量)であり、そして溶媒は水(4〜8容量)である。次いで、結晶化により、高エナチオマー純度、一般に、>95%の(5S)−5−ベンジルチオメチル−5−メチル−イミダゾリジン−2,4−ジオン(S)−α−メチルベンジルアミンを得る。この物質の(5S)−5−メチル−5−{[(フェニルメチル)チオ]メチル}−イミダゾリジン−2,4−ジオン(IV)へのさらなる変換は、標準条件下、例えば、2N 塩酸を使用して、または簡単に酢酸イソプロピル、メチルイソブチルケトン(MIBK)、トルエン、t−ブチルメチルエーテル(TBME)、およびこのような溶媒の組合せを含む適当な溶媒からの結晶化により行い得る。(5S)−5−ベンジルチオメチル−5−メチル−イミダゾリジン−2,4−ジオン(S)−α−メチルベンジルアミンの(IV)への変換は、簡単に、シクロヘキサン、ジブチルエーテルまたは水のような適当な、熱溶媒への固体のスラリー化により行い得る。故に、溶液中でまたはスラリーとして共結晶を温める行動が、遊離(5S)−5−メチル−5−{[(フェニルメチル)チオ]メチル}−イミダゾリジン−2,4−ジオン(IV)の放出をもたらし、次いで、それが冷却により結晶化する。(IV)を遊離させるためにいずれかの方法を使用したとき、さらなるキラル増大が観察される。
【0095】
他の局面において、ラセミ体2−アミノ−3−ベンジルチオ−2−メチルプロピオンアミド(VIII)
【化5】
を、適当なキラル酸存在下の適当な溶媒からの結晶化により分割できる。キラル素子酸の例は、(L)−酒石酸、(R)−(−)−マンデル酸、ジベンゾイル−(L)−酒石酸[DBTA]、ジ−p−トルオイル−(L)−酒石酸[DTTA]、(L)−リンゴ酸[(2S)−(−)−2−ヒドロキシコハク酸]、(1S)−(+)−10−カンファースルホン酸[(D)−CSA]、(1R,3S)−(+)−樟脳酸[cis−樟脳酸]、(L)−グルタミン酸[(2S)−(+)−2−アミノペンタン二酸]、(L)−アスパラギン酸[(S)−(+)−アミノコハク酸]、(L)−ピログルタミン酸[(S)−(−)−2−ピロリドン−5−カルボン酸]、(L)−オルニチンヒドロクロライド[(2S)−(+)−2,5−ジアミノペンタン酸]、(L)−ヒスチジン、(L)−リシン[(2S)−(+)−2,6−ジアミノヘキサン酸]、(L)−アルギニン、N−アセチル−(L)−フェニルアラニン、N−アセチル−(L)−ロイシン、N−カルボベンジルオキシ−(L)−アラニン[(2S)−2−ベンジルオキシカルボニルアミノプロピオン酸]、(−)−メトキシ酢酸、N−アセチル−(L)−チロシンおよび(2R)−(+)−2−(4−ヒドロキシフェノキシ)プロピオン酸を含む。
【0096】
一つの好ましい方法において、キラル酸は(R)−(−)−マンデル酸である。
故に、一つの局面において、我々は、(R)−(−)−マンデル酸を使用するラセミ体2−アミノ−3−ベンジルチオ−2−メチルプロピオンアミド(VIII)の製造法を開示する。
【0097】
一つの好ましい方法において、キラル酸は(R)−(−)−マンデル酸であり、そして、溶媒はメタノールと酢酸イソプロピルの混合物である。結晶化は水の存在下で実施すべきである。この方法で、高エナンチオマー純度の(2S)−2−アミノ−3−ベンジルチオ−2−メチルプロピオンアミド(R)−マンデラート0.5水和物が得られる。この塩のエナンチオマー純度は、酢酸イソプロピルのような溶媒からの再結晶によりさらに増大され得る。
【0098】
他の好ましい方法において、キラル酸はL−酒石酸である。
故に、一つの局面において、我々は、L−酒石酸を使用したラセミ体2−アミノ−3−ベンジルチオ−2−メチルプロピオンアミド(VIII)の分割方法を開示する。
【0099】
他の好ましい方法において、キラル酸はL−酒石酸であり、そして溶媒はエタノールである。得られた(2S)−2−アミノ−3−ベンジルチオ−2−メチルプロピオンアミド(L)−タートレートの、メタノールとメチルイソブチルケトンの混合物のような適当な溶媒からの再結晶により、高エナンチオマー純度の物質が得られる。
【0100】
(2S)−2−アミノ−3−ベンジルチオ−2−メチルプロピオンアミドの(5S)−5−メチル−5−{[(フェニルメチル)チオ]メチル}イミダゾリジン−2,4−ジオン(IV)へのさらなる変換は、当業者に容易に明らかとなる方法を使用して達成できる。例えば、Tetrahedron Asymm., 2001, 12, 101;Tetrahedron, 1991, 47(12), 2133;およびChem. Ber., 1928, 1431参照。
【0101】
他の局面において、キラル5−メチル−5−{[(フェニルメチル)チオ]メチル}イミダゾリジン−2,4−ジオン(IV)を、適当なラセミ体前駆分子の生体触媒(酵素)分割を介して製造できる。ある可能な経路をスキーム3に概説する。
【化6】
【0102】
スキーム3に示す通り、(S)−2−アミノ−3−ベンジルスルファニル−2−メチルプロピオンアミド(IX)または(S)−2−アミノ−3−ベンジルスルファニル−2−メチルプロピオン酸(X)のいずれかは、望むキラルヒダントイン(IV)の適当な前駆体として働き得る。
【0103】
ラセミ体アミノアミド(VIII)の生体触媒分割は、この幾分立体的に障害された基質を受け入れることができるアミダーゼの使用を必要とする。Cα−四置換α−アミノアミドの分割のためのアミダーゼ・マイコバクテリウム・ネオオーラム(Mycobacterium neoaurum) ATCC 25795またはオクロバクテリウム・アンスロピ(Ochrobactrum anthropi) NCIMB 40321の使用は、Tetrahedron, 2001, 57, 6567-6577に開示されている。マイコバクテリウム・ネオオーラム(Mycobacterium neoaurum)は、この特定のアミノアミド(VIII)の分割に適当なアミダーゼであることが証明されていたが、オクロバクテリウム・アンスロピ(Ochrobactrum anthropi)は、驚くべきことにラセミ体加水分解をもたらした。アミノアミド(VIII)の分割に好結果で使用できる他のアミダーゼは、ロドコッカス・エリスロポリス(Rhodococcus erthoplis)およびシュードモナス・フルオレッセンス(Psseudomonas fluorescens) AL45を含む。シュードモナス・フルオレッセンス(Psseudomonas fluorescens) AL45を使用したアミノアミド(VIII)の分割は、WO2005/123932に開示されている。
【0104】
スキーム4に示す通り、これらの生体触媒分割の立体化学結果は、適当なアミダーゼの選択により簡便に制御される。ラセミ体アミノアミド(VIII)の生体触媒分割のための典型的な具体法を本明細書の実施例部分に記載し、このような方法は、本発明の具体的局面を表す。
【化7】
【0105】
別の生体触媒法において、ラセミ体ヒダントイン(III)の加水分解により、または対応するラセミ体アミノ酸から製造したラセミ体α−ウレイド酸(XI)を、ヒダントイナーゼ触媒閉環(スキーム5)に付す。適当なヒダントイナーゼは、ロシュ・ヒダントイナーゼ1およびヒダントイナーゼ2を含む。
【0106】
一つの局面において、我々は、(RS)−3−ベンジルスルファニル−2−メチル−2−ウレイド−プロピオン酸(XI)の閉環を行うためにヒダントイナーゼ酵素を使用することを含む、(5S)−5−[4−(5−クロロ−ピリジン−2−イルオキシ)−ピペリジン−1−スルホニルメチル]−5−メチル−イミダゾリジン−2,4−ジオンの合成における中間体として有用な(S)−5−メチル−5−{[(フェニルメチル)チオ]メチル}イミダゾリジン−2,4−ジオン(IV)の製造方法を開示する。さらなる局面において、ヒダントイナーゼ酵素はロシュ・ヒダントイナーゼ1またはヒダントイナーゼ2である。
【0107】
α−ウレイド酸の分割のための別の生体触媒は、EP0175312(Kanegafuchi)およびWO03/106689(Kaneka)に記載されている。
【化8】
【0108】
別の生体触媒法(スキーム6)において、アミノ酸(X)のラセミ体を、対応するトリフルオロアセチル保護されたアミノ酸(XII)に変換し、それを次いでアミノ酸アシラーゼ触媒加水分解に付す。適当なアミノ酸アシラーゼは、アスペルギルス属、L−Hog腎臓アシラーゼおよびペニシリウム属からのL−アシラーゼである。他の適当なアシラーゼは、当業者には容易に明らかとなろう。
【化9】
【0109】
驚くべきことに、化合物(XII)に対応するN−アセチルまたはN−クロロアセチルアミドのようなより伝統的な基質は、L−アミノ酸アシラーゼとの反応を何等示さなかった。当業者は、トリフルオロアセチルアミド(XII)が他の活性化アミドに置き換えられ、分割の選択性がD−アミノ酸アシラーゼの選択により逆転され、それにより、反応混合物からの(S)−アミノ酸の直接結晶化が促進されることを容易に認識するであろう。
【0110】
一つの局面において、我々は、(RS)−2−アミノ−3−ベンジルスルファニル−2−メチル−プロピオン酸の活性化アミドを適当なアシラーゼ酵素で処理することを含む、(R)−または(S)−5−メチル−5−{[(フェニルメチル)チオ]メチル}イミダゾリジン−2,4−ジオンの合成における中間体として有用な(R)−または(S)−2−アミノ−3−ベンジルスルファニル−2−メチル−プロピオン酸の製造方法を提供する。一つの特定の局面において、活性化アミドはトリフルオロアセチルアミドである。
【0111】
他の局面において、キラル5−メチル−5−{[(フェニルメチル)チオ]メチル}イミダゾリジン−2,4−ジオン(IV)を、適当なキラル(メソ)前駆分子の生体触媒(酵素)非対称化(desymmetrisation)を介して製造する。上記の酵素変換は、全て分割であり、それ故望む立体異性体の理論的最大収率は50%である。対照的に、単純なプロキラル(メソ)化合物の非対称化は、理論上100%の望む立体異性体を製造できる。ある可能な経路をスキーム7に略記する。
【化10】
【0112】
故に、ニトリル(XIII)、アミド(XIV)またはエステル(XV)のような適当なメソ−前駆体は、適当な酵素を使用して非対称化でき、それにより、上に示すキラルヒダントイン前駆体を得る。エステル(XV)について適当なR基は、C1−4アルキルを示す。
【0113】
必要なメソ−前駆体は、文献に記載のものに準じた方法を使用して製造できる。例えば、J. Org. Chem., 1995, 60(17), 5487;J. Chem. Soc., Perkin Trans. 1, 1991, 4, 2589;Synth. Comm., 2001, 1323;およびInorg. Chem., 2003, 42(9), 2950。具体的なメソ−前駆体の合成は、実施例部分に開示する。
【0114】
メソ−ニトリル(XIII)の非対称化のための可能性のある酵素は、例えば、Tetrahedron Asym., 2004, 15, 2817;Tetrahedron Asym., 2001, 12, 3367;Tetrahedron Asym., 1993, 4, 1081;およびJ. Org. Chem., 2003, 68, 2479に記載されている。
【0115】
メソ−アミド(XIV)の非対称化は、ロドコッカス・エリスロポリス(Rhodococcus erthoplis)アミダーゼを使用して達成した。得られたキラル酸アミド(XVI)を、次いで一容器連続反応(single pot sequence)でさらに変換して、優れたeeでキラルヒダントイン(IV)を得た。
【0116】
ブタ肝臓エステラーゼを使用したメソ−S−t−ブチルメチルエステルの非対称化は、以前に記載されている(J. Org. Chem., 2003, 68(13), 5403)。
【化11】
【0117】
ここで、メソ−S−ベンジルエチルエステル(XV、R=Et)もまたこの酵素の基質であることが示された。非対称化は、該文献例に従って進行し、(最初に形成された酸エステル(XVII)のクルチウス転位および続くエステル加水分解後)、(R)−アミノ酸(X)を60−80%eeで得る。
【0118】
類似の非対称化変換が、2種の異なる酵素クラスの代表、すなわち、バチルス・リケニホルミス(Bacillus licheniformis)プロテアーゼおよびアミノ酸アシラーゼを使用して達成され得ることもさらに確立された。バチルス・リケニホルミス(Bacillus licheniformis)プロテアーゼを使用した非対称化の場合、変換は、(S)−エステル酸を提供するために逆立体選択性で進行する。この(S)−エステル/酸をさらに対応するアミノ酸に変換し、その絶対配置およびキラル純度を、標準品サンプルとの比較により決定した。アミノ酸から(S)−5−メチル−5−{[(フェニルメチル)チオ]メチル}イミダゾリジン−2,4−ジオン(IV)への、文献において既知の方法に従った(例えば、Chem. Rev., 1950, 46, 403参照)。
【0119】
故に、さらなる局面において、我々は、メソ−ニトリル(XIII)、またはメソ−アミド(XIV)またはメソ−エステル(XV)の酵素的非対称化を含む、化合物(I)の合成における中間体として有用な(S)−5−メチル−5−{[(フェニルメチル)チオ]メチル}イミダゾリジン−2,4−ジオン(IV)の合成方法を開示する。特定の局面において、我々は、メソ−アミド(XIV)を、適当なアミダーゼ酵素を使用して非対称化する方法を開示する。他の特定の局面において、アミダーゼはロドコッカス・エリスロポリス(Rhodococcus erthoplis)アミダーゼである。
【0120】
上記生体触媒分割において、酵素は、適当であれば、それ自体でまたは固定化(支持)形態で使用してよい。
他の局面において、キラル5−メチル−5−{[(フェニルメチル)チオ]メチル}イミダゾリジン−2,4−ジオン(IV)を、非対称合成を介して製造し得る。
【0121】
非対称ストレッカー反応は、α,α−ジアルキルアミノ酸の合成のために重要な方法である。(R)−フェニルグリシノールは、このような方法において使用する典型的キラル助剤である(Tetrahedron, 2001, 57, 6383-6397)。故に、ベンジルチオアセトン(II)と(R)−(−)−フェニルグリシノールの縮合は、ジアステレオマー混合物としてオキサゾリジン(XVIII)を提供する。次いで、オキサゾリジン(XVIII)とトリメチルシリルシアニドの反応は、85:15比のジアステレオマー混合物としてアミノニトリル(XIX)をもたらす。この混合物の、イソヘキサンのような適当な溶媒からの再結晶は、この化合物(XIX)のジアステレオマー比を99:1を超えて高める(スキーム8):
【化12】
【0122】
次いで、塩化水素ガス存在下、アミノニトリル(XIX)の1当量の水での処理により、ラクトン(XX)を得る。シアン酸カリウムとの反応、続く臭化水素の酢酸溶液での側鎖の除去により、次いでキラルヒダントイン(XXII)を得る。
【0123】
あるいは、アミノニトリル(XIX)のクロロスルホニルイソシアネートでの処理は、ヒダントイン(XXI;R=H)および(XXI;R=CONH2)を含む混合物をもたらし、この混合物は、臭化水素の酢酸溶液で処理したとき、キラル(R)−ヒダントイン(XXII)をもたらした。
【0124】
キラル助剤として(S)−フェニルグリシノールを使用して、エナンチオマー(S)−ヒダントインを得る。
【0125】
一つの局面において、我々は、キラル助剤標識オキサゾリジン(XVIII)の開環を含む、(R)−または(S)−5−メチル−5−{[(フェニルメチル)チオ]メチル}イミダゾリジン−2,4−ジオンの合成方法を提供する。
【0126】
我々は、さらに、非対称相間移動触媒を使用したキラルヒダントインの合成を開示する。触媒的非対称工程は、本方法においてしばしば最も効果である準化学量論量のキラルコントロール要素の使用を可能にするため、この点で、とりわけ魅力的である。キラル相間移動触媒は、典型的に、穏やかな条件、単純な反応工程、安全で安価な試薬および溶媒、有機触媒(金属非含有触媒)の使用、および小規模または大規模いずれでも反応を実施する可能性を含むため、さらなる利点を提供する。しかしながら、4級立体中心(4種の異なる非水素基を有する炭素)を含む化合物の、触媒的エナンチオ選択的方法での構築は、未だ挑戦的である。このような4級立体中心の構築を可能にする適当な経路を、スキーム9に略記する。
【化13】
【0127】
最初の工程で、t−ブチル(DL)−アラニネートまたはイソプロピル(DL)−アラニネートを、ベンズアルデヒド、クロロベンズアルデヒドまたは2−ナフトアルデヒドのような適当なカルボニル誘導体と縮合させて、イミンエステル(XXIII)を得る。好ましくはt−ブチルエステルを使用する。次いでこのイミンを、適当な塩基および適当なキラル相間移動触媒の存在下、ブロモメチルスルファニルメチルベンゼンでアルキル化して、イミン(XXIV)を得る。適当な塩基は、例えば、水酸化カリウム、水素化ナトリウム、水酸化セシウムおよび水酸化ルビジウムを含む。適当な相間移動触媒は、例えば、(−)−O−アリル−N−(9−アントラセニルメチル)シンコニジニウム(cinchonidinium)ブロマイドおよび(+)−O−アリル−N−(9−アントラセニルメチル)シンコニジニウムブロマイドを含む。正しいシュード−エナンチオマーの(正反対の)相間移動触媒が、得られるイミン(XXIV)の絶対立体配置の制御を可能にする。他の適当なキラル相間移動触媒は、当業者には容易に明白となろう。
【0128】
次いでイミン(XXIV)の加水分解によりα−アミノ酸(X)を得る。正しい試薬および正しい反応条件の選択により、キラルα−アミノ酸(X)、またはそのエナンチオマーが、高エナンチオマー純度で得られる。特定の局面において、添付の実施例に記載の具体的方法を特に請求している。
【0129】
一つの局面において、我々は、適当なキラル相間移動触媒の存在下でのイミンエステル(XXIII)のアルキル化を含む、(R)−または(S)−5−メチル−5−{[(フェニルメチル)チオ]メチル}イミダゾリジン−2,4−ジオンの合成における中間体として有用な(R)−または(S)−2−アミノ−3−ベンジルスルファニル−2−メチル−プロピオン酸の製造方法を提供する。一つの特定の局面において、相間移動触媒は(−)−O−アリル−N−(9−アントラセニルメチル)シンコニジニウムブロマイドまたは(+)−O−アリル−N−(9−アントラセニルメチル)−シンコニジニウムブロマイドである。当業者は、上記方法において、硫黄原子を、ベンジル以外の基で代わりに保護できることを容易に認識するであろう。
【0130】
次いで、キラルα−アミノ酸(X)を、さらに、文献法を使用して、キラルヒダントイン(IV)に変換し得る。例えば、Rev., 1950, 46, 403を参照。
【0131】
(S)−5−メチル−5−{[(フェニルメチル)チオ]メチル}イミダゾリジン−2,4−ジオン(IV)からの酢酸水溶液中直接塩素化による((S)−4−メチル−2,5−ジオキソ−イミダゾリジン−4−イル)−メタンスルホニルクロライド(V)の製造は、WO02/074767に開示されている。スルホニルクロライド(V)の新規別法が、同時係属出願US60/782892に開示されている。
【0132】
スルホニルクロライド(V)とカップリングするために、ピペリジニルエーテル酢酸塩(VII)を、最初に、対応する遊離塩基(VI)に再変換しなければならない。この変換は、酢酸エチルまたは酢酸イソプロピルのようなエステル溶媒の存在下、炭酸ナトリウムのような塩基を使用して達成できる。好ましい方法において、この変換は、酢酸塩をトルエンに懸濁させることにより、そして塩基として水性水酸化ナトリウムを使用して、二相系で達成される。トルエンの使用は、共沸蒸留による遊離塩基(VI)のより効率的な乾燥を可能にする。これは、化合物(VI)とスルホニルクロライド(V)のカップリングが、特に水の存在に感受性のため、重要な利点である。いずれの場合も、酢酸イソプロピルまたはトルエンのいずれかの中の遊離塩基(VI)の得られた溶液を、次いで直接スルホニルクロライド(V)t、ジイソプロピルエチルアミンのような適当な塩基の存在下、テトラヒドロフランを共溶媒として使用して、反応させる。この方法で、化合物(I)は、数キログラム規模でさえ、簡便にそして効率的に製造される。
【0133】
一つの局面において、我々は、5−クロロ−2−(ピペリジン−4−イルオキシ)−ピリジン(VI)と((S)−4−メチル−2,5−ジオキソ−イミダゾリジン−4−イル)−メタンスルホニルクロライド(V)(ここで、化合物(VI)は対応する塩からの遊離塩基の遊離により製造する)の反応を含む、化合物(I)の製造法を開示する。さらに特定の局面において、化合物(VI)を、5−クロロ−2−(ピペリジン−4−イルオキシ)−ピリジン酢酸塩(VII)からの遊離塩基の遊離により製造する。
【0134】
化合物(I)の結晶多形はWO02/074767に開示されていない。我々は、いずれかの合成法で製造した化合物(I)を、溶媒として水性エタノールまたは工業用変性アルコール水溶液を使用して結晶化して、導入物質の多形形態(polymorphic modifications)とは無関係に、再現性よく化合物(I)多形Gを得ることができることを発見した。
【0135】
異なる多形形態およびそれらの結晶性は、以下の装置および方法を使用して調べた:
X線粉末回折(XPRD)
XRPD測定は、以下のいずれかを使用した:
i) Scintag Inc. XDS 2000装置で以下のパラメータ:
CuKα(1.5418Å)
45kVおよび30mA
2°≦2θ≦35°
1°/分、増加0.03°
回転石英ディスク
環境条件
約10mgの試験サンプルをサンプルホルダーに置き、石英表面に平テフロン棒を使用して塗布した;または
【0136】
ii) Panalytical X’Pert PRO MPD装置で以下のパラメータ:
CuKα(1.5418Å)
45kVおよび40mA
2°≦2θ≦40°
4°/分、増加0.016°
回転シリコンウエハー
環境条件
約2mgの試験サンプルをサンプルホルダーに置き、シリコン表面に平テフロン棒を使用して塗布した。
【0137】
熱量測定(DSC)
試験サンプルの上昇させた温度に対する熱量測定的応答を、種々の方法を使用してQ1000変調温度示差走査熱量測定(MTDSC)(TA Instruments)を使用して試験し、主要な特色は:
5℃/分の勾配速度の標準的に変調されたモード(“加熱のみ”)(変調なしで1および20℃/分もまた使用した)。温度範囲は環境温度直下から200℃以上であった。
約2mgの試験サンプルを、蓋付き(圧接なし)アルミニウムカップに入れた。
【0138】
重量分析(TGA)
試験サンプルの上昇させた温度に対する重量測定応答を、以下のパラメータを使用したQ500熱重量分析器(TGA)(TA Instruments)を使用して試験した:
加熱速度(標準):5℃/分
約2〜5mgの試験サンプルをカップに入れ、200℃の直ぐ上まで加熱した。
【0139】
湿気相互作用
湿気変化に対する試験サンプルの重量測定応答を、以下の特性のSGA 100(VTI Corporation)またはDVS 2(Surface Measurement System)重量測定的蒸気収着(GVS)装置いずれかを使用して調べた:
乾燥乃至90%RHおよび、例えば、10%RHの工程まで戻る。
平衡条件:<0.01重量%/10分(<0.001重量%/分)
約5mgの試験サンプルをカップに入れ、評価した。
【0140】
形態学
試験化合物の形態学を、500倍までの倍率で、Jeol JSM-5200走査型電子顕微鏡(SEM)を使用して試験した。
数個の粒子を粘性カーボンテープが付いたサンプルホルダーに振りまき、薄金層でコーティングし、試験した。
【0141】
一般的化学法
1H NMRおよび13C NMRスペクトルを、300 MHz Varian Unity Inovaまたは400 MHz Varian Unity Inova装置で記録した。クロロホルム−d(δH 7.27ppm)、ジメチルスルホキシド−d6(δH 2.50ppm)、アセトニトリル−d3(δH 1.95ppm)またはメタノール−d4(δH 3.31ppm)の中央ピークを内部標準として使用した。カラムクロマトグラフィーをシリカゲル(0.040−0.063mm、Merck)を使用して行った。出発物質は、特記しない限り市販されていた。全溶媒および試験試薬は研究室グレードであり、受け取ったまま使用した。特記しない限り、操作は環境温度で、典型的に20〜25℃で行った。
LC分析を、Agilent 1100 HPLC装置を使用して行った。種々のLC法を、生成物分析に使用した。
LCMS分析は、Waters 2790 HPLCと996フォトダイオードアレイ検出器およびMicroMass ZMD、単一四重極マススペクトロメーターとZ−スプレー界面を使用して行った。
【0142】
【表1】
【0143】
実施例1
5−クロロ−2−(ピペリジン−4−イルオキシ)−ピリジン酢酸塩
4−ヒドロキシピペリジン(12.1g、0.12mol、1.18mol eq.)をトルエン(120mL)に懸濁して、オレンジ色懸濁液を得た。得られた混合物を15分加熱還流した(ジャケット温度115℃)。オレンジ色溶液が85−90℃で形成され、幾分かの油汚染が撹拌シャフトおよび温度プローブに見られた。トルエン(26mL)を蒸留により除去した。反応混合物を20℃に15分にわたり冷却した。白色固体が約30〜35℃で沈殿した。別の容器で、tert−BuOK(13.4g、0.12mol、1.18mol eq.)をトルエン(150mL)に懸濁した。この懸濁液を4−ヒドロキシピペリジン混合物に20℃で添加した。次いでトルエンライン洗浄(11mL)を実施した。得られた濃厚懸濁液を50℃で30分にわたり激しく撹拌しながら加熱した。2,5−ジクロロピリジン(15g、0.10mol、1mol eq.)のトルエン(45mL)溶液を、スラリーに50℃で約1時間にわたり添加し、続いてトルエンライン洗浄(11mL)した。反応混合物を還流(約105−107℃)に70分にわたり加温し、次いで2時間加熱還流した。反応混合物を環境温度に30分にわたり冷却し、一晩撹拌した。反応混合物を水(2×75mL)で洗浄し、次いで90℃で1時間加熱した。氷酢酸(6.1g、0.10mol、1mol eq.)のトルエン(60mL)溶液を、混合物に90℃一度に添加し、続いてトルエンライン洗浄(15mL)した。添加完了後、溶液をRTに70分にわたり冷却した。必要な5−クロロ−2−(ピペリジン−4−イルオキシ)−ピリジン酢酸塩が冷却中に沈殿した。1時間、RTで撹拌後、懸濁液を濾過し、ケーキをトルエン(2×75mL)で洗浄した。真空オーブンで50℃で一晩乾燥させて、生成物を85〜95%収率で得た。
1H NMR(400 MHz, D2O)δ 8.1(1H, d), 7.8(1H, dd), 6.9(1H, d), 5.0(1H, m), 3.4(4H, m), 2.2(4H, m), 1.9(3H, s)。
【0144】
5−クロロ−2−(ピペリジン−4−イルオキシ)−ピリジンの別の塩
適当な酸(鉱酸については溶媒中;または有機酸については固体として;または、炭酸塩については、CO2ガスを溶液にバブリング)を、5−クロロ−2−(ピペリジン−4−イルオキシ)−ピリジンのトルエン溶液にRTで添加した。さらなるトルエンを添加し、得られた溶液を結晶化が起こるまで撹拌した。形成した固体を濾過により回収し、イソヘキサンで洗浄した。
【0145】
この方法に使用した酸の例は以下を含む:
・水性リン酸
1H NMR(D2O)δ 2.10(2H, m), 2.20(2H, m), 3.28(2H, m), 3.45(2H, m), 5.15(1H, m), 6.91(1H, d, J 8.8 Hz), 7.78(1H, d, 8.8 Hz), 8.10(1H, s)。
【0146】
・塩酸(プロパノール中) − 一および二塩酸塩を製造するために使用
一HCl、1H NMR(d6-DMSO)δ 1.91(2H, m), 2.14(2H, m), 3.07(2H, m), 3.21(2H, m), 5.19(1H, m), 6.90(1H, d, J 9.6 Hz), 7.83(1H, d, J 9.6 Hz), 8.21(1H, s), 9.17(2H, bs). M.p. 156℃。
二HCl、1H NMR(d6-DMSO)δ 1.93(2H, m), 2.16(2H, m), 3.08(2H, m), 3.20(2H, m), 5.20(1H, m), 5.27(1H, bs), 6.91(1H, d, J 9.2 Hz), 7.83(1H, d, J 9.2 Hz), 8.21(1H, s), 9.35(2H, bs). M.p. 131℃。
【0147】
・トリメチル酢酸
1H NMR(d6-DMSO)δ 1.10(9H, s), 1.48(2H, m), 1.93(2H, m), 2.58(2H, m), 2.95(2H, m), 4.99(1H, m), 6.83(1H, d, J 8.8 Hz), 7.77(1H, d, J 8.8 Hz), 8.18(1H, s)。
M.p. 91-96℃。
【0148】
・酒石酸
1H NMR(D2O)δ 2.10(2H, m), 2.21(2H, m), 3.29(2H, m), 3.46(2H, m), 4.53(2H, s), 5.17(1H, m), 6.93(1H, d, J 9.2 Hz), 7.80(1H, d, J 9.2 Hz), 8.12(1H, s)。
【0149】
・クエン酸
0.5クエン酸塩、1H NMR(D2O)δ 2.10(2H, m), 2.21(2H, m), 2.64(1H, d, J 15.2 Hz), 2.73(1H, d, J 15.2 Hz), 3.28(2H, m), 3.46(2H, m), 5.16(1H, bs), 6.915(1H, d, J 8.8 Hz), 7.79(1H, d, J 8.8 Hz), 8.10(1H, s). M.p. 88℃。
【0150】
・フマル酸
1H NMR(CD3OD)δ 2.05(2H, m), 2.20(2H, m), 3.23(2H, m), 3.39(2H, m), 5.30(1H, m), 6.72(2H, s), 6.83(1H, d, J 8.8 Hz), 7.70(1H, d, J 8.8 Hz), 8.11(1H, s). M.p. 128℃。
【0151】
・マレイン酸
1H NMR(d6-DMSO)δ 1.85(2H, m), 2.13(2H, m), 3.13(2H, m), 3.27(2H, m), 5.20(1H, m), 6.12(2H, s), 6.90(1H, d, J 9.2 Hz), 7.83(1H, d, J 9.2 Hz), 8.21(1H, s), 8.48(2H, bs). M.p. 96-104℃。
【0152】
・安息香酸
1H NMR(d6-DMSO)δ 1.66(2H, m), 2.02(2H, m), 2.80(2H, m), 3.09(2H, m), 5.08(1H, m), 6.86(1H, d, J 8.8 Hz), 7.40(2H, t, J 9.6), 7.48(1H, d, J 9.6 Hz), 7.79(1H, d, 8.8 Hz), 7.91(2H, d, J 9.6 Hz), 8.19(1H, s). M.p. 140℃。
【0153】
・臭化水素酸(水性) − 一および二臭化水素酸塩を製造するために使用
一HBr、1H NMR(d6-DMSO)δ 1.90(2H, m), 2.15(2H, m), 3.12(2H, m), 3.25(2H, m), 5.19(1H, m), 6.91(1H, d, J 8.8 Hz), 7.85(1H, d, J 8.8 Hz), 8.21(1H, s), 8.80(2H, bs). M.p. 198℃。
二HBr、1H NMR(d6-DMSO)δ 1.91(2H, m), 2.16(2H, m), 3.15(2H, m), 3.26(2H, m), 5.21(1H, m), 6.92(1H, d, J 9.2 Hz), 7.84(1H, d, J 9.2 Hz), 8.21(1H, s), 8.76(2H, bs). M.p. 183℃。
【0154】
・炭酸(CO2ガス)
0.5炭酸塩、1H NMR(d6-DMSO)δ 1.53(2H, m), 1.94(2H, m), 2.59-3.02(4H), 5.04(1H), 6.84(1H, d, J 8.7 Hz), 7.77(1H, d, J 8.7 Hz), 8.17(1H, s)。
【0155】
実施例2
(RS)−5−メチル−5−{[(フェニルメチル)チオ]メチル}イミダゾリジン−2,4−ジオン
適当なサイズの標準加圧反応器にベンジルチオアセトン(95%純度)(85.26g、450mmol、1mol eq.)、水(413mL)および2−プロパノール(146mL)を入れた。混合物を約15分撹拌して、均質にした。次いで炭酸アンモニウム(49.56g、509mmol、1.13mol eq.)およびカリウムシアニド(30.54g、460mmol、1.02mol eq.)を入れた。反応混合物を90℃に温め、これは約2.5バール(barg)の圧力を誘発した。反応を冷却し、LCで出発物質の消失について分析した。反応完了後、必要な生成物を結晶化させた。必要であれば、結晶化を種晶添加により誘発した。結晶化後、水(971.9mL)および濃塩酸(96.7g)を反応混合物に入れた。これによりpHが約11.9から7.4に変化した。結晶塊を濾取し、続いて酢酸イソプロピルで洗浄した。乾燥後、表題化合物を白色結晶性固体として86%収率で得た。
1H NMR(d6-DMSO)δ 10.74(1H, s), 8.00(1H, s), 7.35-7.20(5H, m), 3.76(2H, s), 2.72, 2.262(各2x1H, Abq), 1.29(3H, s)。
【0156】
実施例3
(S)−5−メチル−5−{[(フェニルメチル)チオ]メチル}イミダゾリジン−2,4−ジオン
(RS)−5−メチル−5−{[(フェニルメチル)チオ]メチル}イミダゾリジン−2,4−ジオンを、分取キラル模擬移動床クロマトグラフィー(SMB)を使用してエナンチオマー要素に分割した。WO02/074767(89頁)に記載されているものと同じキラル固定相および移動相を使用した。エナンチオマーを本質的に定量的収率で回収した。
【0157】
得られた(S)−5−ベンジルスルファニルメチル−5−メチル−イミダゾリジン−2,4−ジオン(5g)のメタノール性溶液の容量を、減圧下、35℃で減らした(約20mLまで)。水(40mL)を、内部温度を35℃に維持しながら溶液に滴下した。約半量の水を添加したら、生成物が沈殿を始めた。混合物をRTにゆっくり冷却し、次いで氷浴で2℃に冷却した。生成物(4.56g、SMB分割後、理論値の91%)を、白色結晶性固体として、2℃での濾過により回収した。5.86kg規模で、この再結晶化工程からの生成物を98%収率(5.73kg)で単離した。
【0158】
(S)−5−メチル−5−{[(フェニルメチル)チオ]メチル}イミダゾリジン−2,4−ジオンはまた、メタノール/トルエンまたはメタノール/ジブチルエーテルを含む他のメタノール混合物から結晶化できる。それは、トルエン、ジ−イソプロピルエーテル、ジブチルエーテルおよび水を含む範囲の溶媒から結晶化できる。
1H NMR(300 MHz, d6-DMSO)δ 10.74(1H, s), 8.00(1H, s), 7.35-7.20(5H, m), 3.76(2H, s), 2.72, 2.262(各2x1H, ABq), 1.29(3H, s)。
【0159】
生成物のキラル純度を、Hewlett Packard 1100 series HPLC(ダイオードアレイ検出器;Astec Chirobiotic V 50mm×4.6mmカラム装着)を使用するHCにより、移動相として70:30 イソヘキサン:エタノール;オーブン温度55℃;流速1.0mL/分;210nmで検出;注入量1μL;およびランタイム5分で確定した。(S)−および(R)−異性体の保持時間は、それぞれ2.6および3.8分であった。
【0160】
実施例4
((S)−4−メチル−2,5−ジオキソ−イミダゾリジン−4−イル)−メタンスルホニルクロライド
方法1
(S)−5−ベンジルスルファニルメチル−5−メチル−イミダゾリジン−2,4−ジオン(106.9g、427.1mmol、1.000mol eq.)を、氷酢酸(8vol eq.)および水(1vol eq.)の混合物に溶解し、約4℃に冷却した。次いで塩素ガス(96.9g、3.2mol eq.)を十分に撹拌している溶液に約1時間にわたり、反応混合物温度が、添加中、ほとんど12〜15℃に維持されるように、一定速度で添加した(ジャケット温度は、ずっと4℃を保った)。反応完了後(混合物は特徴的緑色に変わり、温度が急激に低下した)、混合物に窒素導入し、約30℃に加熱して、白色スラリーを得る。次いで溶媒の大部分を真空蒸留により除去した。トルエン(534.5mL)を添加し、真空下蒸留により同量の溶媒を除去した。トルエンの添加/蒸留をさらに一回繰り返した。次いでイソヘキサン(534.5mL)を残渣に添加し、混合物を20℃に冷却した。徹底的に撹拌(stir-out)後、生成物を濾過により回収した。回収した固体をイソヘキサン(213.8mL)で洗浄し、真空で40℃で一定重量になるまで乾燥させ、必要なスルホニルクロライドを白色結晶性固体として得た(95.5g、98.7%)。
1H NMR(300MHz, d8-THF)δ, 9.91(1H, bs), 7.57(1H, s), 4.53, 4.44(2×1H, 各ABq), 1.52(3H, s)。
【0161】
方法2
(S)−5−ベンジルスルファニルメチル−5−メチル−イミダゾリジン−2,4−ジオン(50g、199.74mmol)を氷酢酸(8vol eq.)および水(4mol eq.)の混合物に溶解し、約4℃に冷却した。次いで塩素ガスを、よく撹拌している溶液に、反応混合物温度が、添加中、ほとんど約12℃に維持されるように、一定速度で約1時間にわたり添加した(Huberコントローラーを、反応温度が12℃に維持されるように設定)。反応完了後(混合物が特徴的緑色に変わり、温度が急激に低下した)、混合物に窒素導入し、約20℃に加熱して、白色スラリーを得た。次いで溶媒の1/2乃至2/3を真空蒸留により除去した(100mbar圧で)。次いでイソオクタン(250mL、5vol eq.)を残渣に添加し、混合物20℃に冷却した。徹底的に撹拌(stir-out)後、生成物を濾過により回収した。回収した固体をイソオクタン(2×100mL)で洗浄し、真空で40−50℃で一定重量になるまで乾燥させ、必要なスルホニルクロライドを白色結晶性固体として得た(41.37g、91%)。
1H NMR(300MHz, d8-THF)δ, 9.91(1H, bs), 7.57(1H, s), 4.53, 4.44(2×1H, 各ABq), 1.52(3H, s)。
【0162】
実施例5
(S)−5−[4−(5−クロロ−ピリジン−2−イルオキシ)−ピペリジン−1−スルホニルメチル]−5−メチル−イミダゾリジン−2,4−ジオン方法1
a)5−クロロ−2−(ピペリジン−4−イルオキシ)−ピリジン
5−クロロ−2−(ピペリジン−4−イルオキシ)−ピリジンアセテート(40g、0.146mol)をイソPrOAc(664mL)中に30℃でスラリー化した。このスラリーに、Na2CO3(1.5mol/L;196mL、2mol eq.)を添加した。次いでスラリーを、30℃で15分急速に撹拌した。二相性混合物を静置し、底水性相を分離し、廃棄した。上記の塩基性洗浄工程をさらに2回繰り返した。次いで有機相を1回水(200mL)で洗浄した。得られたイソPrOAc溶液の容量を、減圧下の蒸留により約300mLに減らした。次いで溶液をイソPrOAc(400mL)と共に蒸留し、再び約300mLまで蒸留により減らした。この工程をさらに1回繰り返した。サンプルを、5−クロロ−2−(ピペリジン−4−イルオキシ)−ピリジン含量および水含量分析のために取った。溶液の重量または総容量を、iPrOAc溶液中の5−クロロ−2−(ピペリジン−4−イルオキシ)−ピリジンの濃度を計算するために測定した。
【0163】
b)(S)−5−[4−(5−クロロ−ピリジン−2−イルオキシ)−ピペリジン−1−スルホニルメチル]−5−メチル−イミダゾリジン−2,4−ジオン
ジイソプロピルエチルアミン(24.3mL、0.139mol、1mol eq.)を、パート(a)で製造したイソPrOAc溶液[約300mL;31.2g、0.146mol、1.05mol当量の5−クロロ−2−(ピペリジン−4−イルオキシ)−ピリジンに相当]に、RTで一度に添加した。次いで溶液を−15℃に冷却した。
((S)−4−メチル−2,5−ジオキソ−イミダゾリジン−4−イル)−メタンスルホニルクロライド(31.65g、0.139mol、1mol eq.)を、乾燥THF(285mL)にRTで撹拌しながら添加した。次いで得られた溶液を、5−クロロ−2−(ピペリジン−4−イルオキシ)−ピリジンのイソPrOAc溶液に、−15℃で約1.5時間にわたり滴下した。沈殿が、((S)−4−メチル−2,5−ジオキソ−イミダゾリジン−4−イル)−メタンスルホニルクロライドの添加により見られた。添加終了時、乾燥THF(32mL)を反応混合物に添加してラインを洗浄し、混合物を1時間、−15℃で洗浄した。次いでそれを20℃に1時間にわたり温め、さらに20℃で1時間撹拌した。
【0164】
反応を、急速に撹拌しながら10wt%NaHSO4(157mL)でクエンチした。約15分後、二相性混合物を静置し、底水性相を分離し、廃棄した。この酸洗浄工程をさらに1回繰り返した。次いで有機相を、急速な撹拌を使用して水(157mL)で洗浄し、完全に相分割させた後、分配した。次いで反応溶液を40℃に温め、再び水(157mL)で洗浄した。THF(95mL)を有機層に添加し、次いでそれを40℃に温め、40℃で濾過して、全ての粒子状物を除去した。次いで溶媒容量を、55℃のジャケット温度での減圧蒸留により約157mLに減らした。次いでイソPrOAc(317mL)を添加し、容量を再び約157mLに減らした。さらに2回イソPrOAc(317mL)の添加−除去を繰り返した。蒸留中に固体が沈殿し始め、懸濁液が得られた。各回容量を約157mLまで減らし、最終蒸留の後、次いで少量の溶媒のサンプルを、残存THF分析のために反応混合物から取った。1H NMRは、THFピークを示さなかった。次いで反応内容物を0℃に冷却し、生成物を濾過により回収した。反応容器をイソPrOAc(63mL)で洗浄し、この濯ぎ液を使用してフィルター上の生成物を洗浄した。生成物を、一晩、真空オーブンで40℃で乾燥させた。必要な(S)−5−[4−(5−クロロ−ピリジン−2−イルオキシ)−ピペリジン−1−スルホニルメチル]−5−メチル−イミダゾリジン−2,4−ジオンが、白色固体として、71%収率(41.8g)で得られた。
1H NMR(300MHz, d6-DMSO)δ 10.74(1H, s), 8.20(1H, d), 8.01(1H, s), 7.81(1H, dd), 6.87(1H, d), 5.09(1H, m), 3.52-3.35(4H, m), 3.13(2H, m), 2.02(2H, m), 1.72(2H, m), 1.33(3H, s)。
【0165】
実施例6
(S)−5−[4−(5−クロロ−ピリジン−2−イルオキシ)−ピペリジン−1−スルホニルメチル]−5−メチル−イミダゾリジン−2,4−ジオン方法2
a)5−クロロ−2−(ピペリジン−4−イルオキシ)−ピリジン
5−クロロ−2−(ピペリジン−4−イルオキシ)−ピリジンアセテート(70g、257mmol)を、トルエン(560mL)中、RTでスラリー化した。1M NaOH(420mL)を添加し、次いでスラリーをRTで15分急速に撹拌した。二相性混合物を静置し、底水性相を分離し、廃棄した。次いで有機相を水(2×420mL)で洗浄した。サンプルを有機相から除去し、5−クロロ−2−(ピペリジン−4−イルオキシ)−ピリジンについて分析した。
次いで得られたトルエン溶液を、減圧下での蒸留により減らし、約168mLまで減らした(5−クロロ−2−(ピペリジン−4−イルオキシ)−ピリジンアセテート負荷について2.4vol eq.)。次いで溶液をトルエン(420mL)と蒸発させて、再び約168mL(2.4vol eq.)まで蒸留により減らした。サンプルを、水含量分析のために取った。
【0166】
b)(S)−5−[4−(5−クロロ−ピリジン−2−イルオキシ)−ピペリジン−1−スルホニルメチル]−5−メチル−イミダゾリジン−2,4−ジオン
ジイソプロピルエチルアミン(38.4mL、220mmol)を、工程(a)で得た5−クロロ−2−(ピペリジン−4−イルオキシ)−ピリジンのトルエン溶液(236mmol含有)に一度に添加し、続いて乾燥THF(151mL)をライン洗浄として添加した。((S)−4−メチル−2,5−ジオキソ−イミダゾリジン−4−イル)−メタンスルホニルクロライド(48.7g、215mmol)を、RTで撹拌しながら乾燥THF(352mL)に溶解した。次いでスルホニルクロライドの得られた溶液を、5−クロロ−2−(ピペリジン−4−イルオキシ)−ピリジンおよびジイソプロピルエチルアミンのトルエン/THF溶液に、RTで1〜2時間にわたり滴下した。スルホニルクロライドの添加により沈殿が見られた。添加終了時、乾燥THF(50mL)を、ライン洗浄として反応混合物に添加した。添加完了後、反応を約30分、RTで洗浄した。
【0167】
反応を10wt%NaHSO4(251mL)で、約15分急速に撹拌しながらクエンチした。二相性混合物を静置し、底水性相を分離し、廃棄した。この酸洗浄工程をさらに1回繰り返した。次いで溶媒容量を、減圧蒸留により約220mLに減らした。次いでトルエン(300mL)を添加し、容量を約245mLに減らし、固体が、蒸留中に沈殿し始め、懸濁液が得られた。最後の蒸留後、次いで溶媒の少量のサンプルを残留THF分析のために反応混合物から取った。
次いで反応混合物の内容物を0℃に冷却し、この温度で約30分撹拌し、生成物を濾過により回収した。反応容器をトルエン(100mL)で洗浄し、この濯ぎ液をフィルター上の生成物の洗浄に使用した。生成物を、真空オーブンで、40℃で一定重量になるまで乾燥させた。(S)−5−[4−(5−クロロ−ピリジン−2−イルオキシ)−ピペリジン−1−スルホニルメチル]−5−メチル−イミダゾリジン−2,4−ジオンを、2工程で典型的に85〜88%収率で、白色固体として得た。
【0168】
実施例7
(S)−5−[4−(5−クロロ−ピリジン−2−イルオキシ)−ピペリジン−1−スルホニルメチル]−5−メチル−イミダゾリジン−2,4−ジオン形態G
工業用変性アルコール(IMS):水(10vol eq.)の2:1混合物を、(S)−5−[4−(5−クロロ−ピリジン−2−イルオキシ)−ピペリジン−1−スルホニルメチル]−5−メチル−イミダゾリジン−2,4−ジオンに添加した。混合物を加熱還流(約80〜82℃)して、溶液を得た。この溶液を還流に15分維持し、次いで濾過した。濾液を加熱還流し、この温度で15分維持した。次いで溶液を0.5℃/分の速度で20℃に冷却した。20℃で2〜3時間撹拌後、(S)−5−[4−(5−クロロ−ピリジン−2−イルオキシ)−ピペリジン−1−スルホニルメチル]−5−メチル−イミダゾリジン−2,4−ジオン形態Gを濾過により回収し、IMS:水 2:1(2.5vol eq.)で洗浄し、乾燥させた。生成物を80〜87%収率で単離した。
この方法は、導入物質の多形形態と無関係に、再現性よく多形Gをもたらすことが示された。多形A、C、Fおよび多形混合物は、この方法を使用して再結晶させて、全て形態Gとなった。
【0169】
実施例8
(1S)−(−)−α−メチルベンジルアミンを使用した(RS)−5−ベンジルスルファニルメチル−5−メチル−イミダゾリジン−2,4−ジオンの分割
NaOH(0.45eq.)の水(4vol eq.)溶液を、(1S)−α−メチルベンジルアミン(1.7eq.)および(RS)−5−ベンジルスルファニルメチル−5−メチル−イミダゾリジン−2,4−ジオン(50g)に添加した。得られた懸濁液を83℃に加熱した。混合物をゆっくり冷却し、RTより高い温度(36℃)で種晶添加した。種晶添加は必須ではないが、その反応混合物の撹拌および濾過を容易にし得る。さらに水(3vol eq.)を冷却サイクル中に添加した。反応を20℃で一晩撹拌し、その後生成物を濾過により回収し、シクロヘキサンで洗浄して、(5S)−5−ベンジルスルファニルメチル−5−メチル−イミダゾリジン−2,4−ジオン(1S)−α−メチルベンジルアミン塩を45%収率で得た。
LC(実施例3と同じ条件)は、生成物が94.5%エナンチオマー純度を有することを示した。
1H NMR(400 MHz, d6-DMSO)δ 1.23(3H, d, J=6.7 Hz), 1.28(3H, s), 2.61(1H, d, J 14.1 Hz)2.72(1H, d, J 14.1 Hz), 3.76(2H, s), 3.96(1H, q, J=6.7 Hz), 7.37-7.15(10H, m), 7.97(1H, s)。
【0170】
2−アミノ−3−ベンジルチオ−2−メチルプロピオンアミド(VIII)
カリウムシアニド(1.07Kg)および水酸化アンモニウム溶液(28%溶液の4.46Kg)を、20Lフラスコに入れた。この混合物に、予め形成した塩化アンモニウム溶液(3.47Kg水中1Kg)を、約0.5時間にわたり撹拌しながら添加した。
1−(ベンジルチオ)−2−プロパンオン(2.71Kg)を次いで添加した。混合物を約40℃で65時間撹拌した。脱イオン水(1L)を添加し、相を分離した。分離した有機相(3.13Kg)を、次工程に直接使用した。
37%塩酸(13.3Kg)および48%臭化水素酸(1.26Kg)の混合物に、上記からの物質の半量を約1時間にわたり、約5℃で添加した。次いで混合物を2時間、5℃で撹拌し、次いで約30℃に温め、一晩撹拌した。次いで混合物を冷却し、生成物を濾過により回収した。フィルターケーキを氷冷濃塩酸およびアセトンで洗浄した。得られた物質をアセトン(10L)に再懸濁し、短時間加熱還流した。次いで生成物を濾過により回収し、減圧下一晩乾燥させた。この工程を、出発物質の残りの半分で繰り返した。
上記の通り製造した物質を合わせたバッチ(1.66Kgおよび1.51Kg)に、酢酸エチル(9Kg)および予め形成させた炭酸カリウム溶液(9Kgの50wt%水溶液)を添加した。混合物を1時間撹拌し、次いで相を分離した。水性相を酢酸エチル(3Kg)で抽出した。合わせた有機相を炭酸カリウム溶液(3.1Kgの50wt%水溶液)で抽出した。得られた有機相を炭酸ナトリウムで乾燥させ、濾過し、濃縮した。得られた固体をスラリー化し、TBME中、短時間加熱還流した。冷却後、生成物を濾過により回収し、一晩真空下で乾燥させた。
2−アミノ−3−ベンジルチオ−2−メチルプロピオンアミド(VIII)(全体で2.4Kg、73.2%)を単離した。
【0171】
実施例9
(R)−(−)−マンデル酸を使用した(RS)−2−アミノ−3−ベンジルスルファニル−2−メチル−プロピオンアミドの分割
(2S)−2−アミノ−3−ベンジルスルファニル−2−メチル−プロピオンアミド(R)−マンデラート
メタノール(20mL、4vol eq.)および酢酸イソプロピル(80mL、16vol eq.)を混合して、メタノールの酢酸イソプロピルの20%v/v溶液を得た。
(RS)−2−アミノ−3−ベンジルスルファニル−2−メチル−プロピオンアミド(5g、22.2mmol)を、予め混合したMeOH/iPrOAc溶液(13vol eq.)および水(0.4mL、1mol eq.)の溶液に溶解した。得られた溶液を50℃に加熱した。(R)−(−)−マンデル酸(3.39g、22.2mmol、1.0mol eq.)を、予め混合したMeOH/iPrOAc溶液(30mL、6vol eq.)の溶液に溶解し、次いでアミド溶液に1時間にわたり管理された方法で添加した。添加中、反応温度を50℃に維持した。生成物の沈殿が添加中に開始し得る。残った予め混合したMeOH/iPrOAc溶液(約1vol eq.)をライン洗浄として使用した。
次いで懸濁液をゆっくり0℃に冷却し、この温度で徹底的に撹拌(stir-out)後、生成物を濾過により回収した。反応容器を20℃に戻し、iPrOAc(25mL、5vol eq.)で約5〜10分洗浄した。続いてこの溶液をフィルター上の生成物の洗浄に使用した。生成物を、真空オーブンで40℃で一定重量になるまで乾燥させた。(2S)−2−アミノ−3−ベンジルスルファニル−2−メチル−プロピオンアミド(R)−マンデラート0.5水和物を、典型的に47〜48%収率および97〜>99%エナンチオマー純度で白色固体として単離した。
90%を超えるエナンチオマー純度の(2S)−2−アミノ−3−ベンジルスルファニル−2−メチル−プロピオンアミド(R)−マンデラート0.5水和物を、iPrOAc(35vol eq.)からの再結晶により、エナンチオマーを増加することができ、>99.35%エナンチオマー純度の物質が、>87%回収率で得られる。
1H NMR(400 MHz, d6-DMSO)δ 1.28(s, 3H, CH3), 2.79(AB q, J=140.5, 13.8 Hz, 2H SCH2Cq), 3.76(AB q, J=16.4, 13.1 Hz, 2H, ArCH2S), 4.79(s, 1H ArCHOH), 7.41-7.18(m, 11H, ArHおよびCONHAHB), 7.56(s, 1H, CONHAHB)。
生成物のキラル純度を、Hewlett Packard 1100 series HPLC(ダイオードアレイ検出器;Chiracel ChiralPak AD 25cm×0.46cm ID×10μmカラム装着)を使用するLCにより、溶媒−0.1%v/v ジエチルアミンのエタノール溶液;アイソクラチック法;オーブン温度20℃;流速1.0mL/分;サンプル希釈剤−精製水;210nmで検出;注入量5.0μL;およびランタイム15分で確立した。(S)−および(R)−アミノアミドの保持時間は、それぞれ約5.4および11.8分であった。
【0172】
実施例10
L−酒石酸を使用した(RS)−2−アミノ−3−ベンジルスルファニル−2−メチル−プロピオンアミドの分割
(2S)−2−アミノ−3−ベンジルスルファニル−2−メチル−プロピオンアミド(2R,3R)−タートレート
方法1
(RS)−2−アミノ−3−ベンジルスルファニル−2−メチル−プロピオンアミドおよびL−酒石酸(0.25−1.0eq.)のエタノール(11.75−90vol eq.)溶液を混合した。沈殿した得られた固体を濾過により回収した。これにより、2−アミノ−3−ベンジルスルファニル−2−メチル−プロピオンアミドタートレートを、典型的に45〜90%収率および50〜92%エナンチオマー純度(ep)で得た。エタノール以外の溶媒(方法2の下を参照)も使用できた。
L−酒石酸(1eq.)およびEtOH(90vol eq.)を使用して、生成物を1:1塩として、44%収率および92.2%epで得た。
【0173】
方法2
(RS)−2−アミノ−3−ベンジルスルファニル−2−メチル−プロピオンアミドおよびL−酒石酸(0.25−1.0eq.)の混合物を、最小量の溶媒(適当な溶媒は、メタノール、エタノール、イソプロパノール、n−ブタノール、酢酸イソプロピル、酢酸エチル、トルエン、アセトニトリルおよびIMSを含み、これに限定されない)にスラリー化した。次いでこれらのスラリーを環境温度を超える温度で洗浄し、さらなる溶媒を、種々温度で完全な用芸を形成させるために添加した。完全な溶液が、35vol eq.の溶媒で還流で形成されないならば、十分な水を添加して溶液を作成した。次いでこれらの溶液をRTに冷却し、得られた固体を濾過により回収して、2−アミノ−3−ベンジルスルファニル−2−メチル−プロピオンアミドタートレートを、78〜100%収率(L−酒石酸導入量に対して、および1:1塩または2:1塩のいずれかの形成について補正)で、50〜60%の範囲のエナンチオマー純度で得た。
これらの2−アミノ−3−ベンジルスルファニル−2−メチル−プロピオンアミドL−酒石酸塩を、種々の溶媒(適当な溶媒の例は、(i)溶媒としてMeOHまたは水のいずれかおよび貧溶媒としてイソプロパノール、n−ブタノール、酢酸エチル、酢酸イソプロピル、トルエン、アセトニトリル、アセトン、THF、TBME、DCM、MIBK、ジエチルエーテル、2,2,4−トリメチルペンタンまたはIMSのいずれかの混合溶媒結晶化;または(ii)エタノール、メタノール、IMSまたは水からの直接再結晶を含み、これに限定されない)から再結晶できた。典型的に、再結晶は、1〜99%の範囲の収率で、50〜96%の範囲のエナンチオマー純度の生成物をもたらした。
MeOH/MIBK(各々20/29.5vol eq.)からの再結晶により、(2S)−2−アミノ−3−ベンジルスルファニル−2−メチル−プロピオンアミド(2R,3R)−タートレートを75%収率および96.63%のエナンチオマー純度で得た(出発物質は約50%ep)であった。
キラル純度は、実施例9のようにLCで確立した。
1H NMR(300 MHz, d6-DMSO)δ 1.35(3H, s, CH3), 2.69(1H, d, J 13.7 Hz, S-CH2), 3.00(1H, d, J 13.7 Hz, S-CH2), 3.79(2H, s, S-CH2), 3.98(2H, s, CHOH), 7.22-7.36(5H, m, Ar-H), 7.44(1H, s, NH), 7.63(1H, s, NH)。
【0174】
実施例11
生体触媒分割
1. ロドコッカス・エリスロポリス(Rhodococcus erthoplis)アミダーゼを使用
(RS)−2−アミノ−3−ベンジルスルファニル−2−メチル−プロピオンアミド塩酸塩(5g、0.019mol)を、0.1N pH8.00ホウ酸緩衝液(100mL)に溶解して、pH約4.0の溶液を得た。2N KOH溶液でpHを7.00に調節し、ロドコッカス・エリスロポリス(Rhodococcus erthoplis)アミダーゼを、架橋酵素凝集体(CLEA)(緩衝液中1mLの懸濁液、25単位/mL 特異的活性)として添加した。反応混合物を35℃で1.5時間撹拌した。HPLCは、アミドの対応するアミノ酸への25〜30%変換を示した。
濃塩酸でpHを1.00に調節し、次いで酵素CLEAを除去するために濾過した。2N KOH溶液でpHを11に調節し、DCM(1×100mL;1×40mL)で抽出した。合わせたDCM抽出物を乾燥させ、蒸発させて、(R)−2−アミノ−3−ベンジルスルファニル−2−メチル−プロピオンアミドを白色固体(2.67g)として62%収率で得た。エナンチオマー純度は、キラルHPLC(34%ee)で67%(R)−entであることが判明した。
水性層を濃塩酸で処理してpH6.80とし、pH6.80を2N KOH溶液の時折の添加により1時間維持した。得られた白色結晶を濾過により回収し、pH7.00緩衝液で洗浄して、40℃で一晩乾燥させて、(S)−2−アミノ−3−ベンジルスルファニル−2−メチル−プロピオン酸(1.2g、27%収率)を得た。エナンチオマー純度は99%(S)−ent(98%ee)であることが判明した。
【0175】
2. シュードモナス・フルオレッセンス(Psseudomonas fluorescens)AL45アミダーゼ(WO2005/123932)を使用
a) 微生物の増殖
Ps fluorescens AL45を、10L発酵槽中、酵母抽出物(2g/L)およびラクトアミド(2.5g/L)を補った5Lのミネラル塩培地 pH7.2で28℃で増殖させた。発酵槽に5L/分で空気供給し、400rpmで24時間撹拌した。細胞を遠心により回収し、回収した細胞を100mM リン酸緩衝液 pH7.2に再懸濁し、再遠心することにより洗浄した。回収した細胞を4℃で一晩貯蔵して、その後生体内変化に使用した。
【0176】
b)生体内変化条件
細胞ペレットをリン酸緩衝液(100mM、pH7.2、1L)に再懸濁し、それに(RS)−2−アミノ−3−ベンジルスルファニル−2−メチル−プロピオンアミド(5g)を添加した。混合物を撹拌し、28℃でインキュベートし、サンプルを分析のために定期的に取った。8.5時間後、定量的HPLCは、反応が50%加水分解に到達していることを示した。キラル分析(LC)は、(R)−2−アミノ−3−ベンジルスルファニル−2−メチル−プロピオンアミド96%ee;および(S)−2−アミノ−3−ベンジルスルファニル−2−メチル−プロピオン酸97.5%eeを示した。
【0177】
c)反応後処理
細胞を、生体内変化ブロスから遠心により除去し、上清(約1000mL)を20%炭酸ナトリウム溶液(最終容量約1200mL)を使用してpH10.5に調節した。アルカリ性上清をTBME(750mL)で抽出し、全てのエマルジョンは有機相に保持した。TBME相をアセトン(約250mL)と混合し、これは沈殿の形成をもたらした。同時に水性相をTBME(750mL)で再抽出し、有機相をアセトン(約250mL)と混合した。これは、少量の水性層の分離をもたらし、これを残りの水性相と合わせた。2個のTBME/アセトン相を合わせ、乾燥させ(Na2SO4)、濾過し、溶媒を除去して、(R)−2−アミノ−3−ベンジルスルファニル−2−メチル−プロピオンアミドをオフホワイト色結晶性固体(1.7g)として、68%収率で得た。エナンチオマー純度は>99%(R)−entであった。
水性相を2M HClを使用してpH6.8に調節し、凍結乾燥により約300mLまで濃縮した。融解により白色スラリーを産生した。結晶性固体を濾過により回収し、一晩、37℃で乾燥させて、(S)−2−アミノ−3−ベンジルスルファニル−2−メチル−プロピオン酸を微細白色結晶(1.8g)として72%収率で、HPLCで98%純度で得た。エナンチオマー純度は>99%(S)−entであった。
【0178】
3. マイコバクテリウム・ネオアルム(Mycobacterium neoarum)L−アミダーゼを使用
ロドコッカス・エリスロポリス(Rhodococcus erthoplis)について記載のものと同様の方法を使用して、(RS)−2−アミノ−3−ベンジルスルファニル−2−メチル−プロピオンアミドを、マイコバクテリウム・ネオアルム(Mycobacterium neoarum)からのL−特異的アミダーゼを使用して分離できた。この方法は、(R)−2−アミノ−3−ベンジルスルファニル−2−メチル−プロピオン酸および(S)−2−アミノ−3−ベンジルスルファニル−2−メチル−プロピオンアミドが〜99%eeで製造される点で、上記(1)および(2)と相補的である。
【0179】
4. (RS)−3−ベンジルスルファニル−2−メチル−2−ウレイド−プロピオン酸の分割を介した(S)−5−ベンジルスルファニルメチル−5−メチル−イミダゾリジン−2,4−ジオン
(RS)−3−ベンジルスルファニル−2−メチル−2−ウレイド−プロピオン酸
【化14】
(RS)−2−アミノ−3−ベンジルスルファニル−2−メチル−プロピオン酸(350g)を水(2.6L)およびTHF(2.6L)に懸濁し、シアン酸カリウム(504g)を添加し、反応を25℃で一晩撹拌した。さらにシアン酸カリウム(75g)を次いで添加し、反応をさらに6時間撹拌した。次いで撹拌を止め、反応を0℃で一晩冷却した。得られたスラリーを濾過し、濾液を減圧下濃縮した。濃縮物を、急速に撹拌しながら6%水性HCl溶液でpH2に酸性化した。1時間後、得られた固体を濾過により回収し、次いで水(4L)およびメタノール(4L)に連続的に再スラリー化した。真空下乾燥後、生成物をシリカクロマトグラフィーを使用して精製した。不純物をDCM/THFを使用して精製し、一方生成物ををTHF/AcOH、続いてMeOHを使用して抽出した。溶媒の除去、続いてTBMEでのトリチュレーションにより、3−ベンジルスルファニル−2−メチル−2−ウレイド−プロピオン酸(151g)をオフホワイト色固体として得た。
【0180】
(RS)−3−ベンジルスルファニル−2−メチル−2−ウレイド−プロピオン酸(9g、約90%純度、30mmolに相当)を、亜硫酸ナトリウム溶液(5mmol、100mL)に添加した。KOH(2.5g、44mmol)を添加した。溶液を、酸が溶解するまで撹拌し、次いで濾過した。pHを、氷酢酸で12.5から7.00に調節した。MnCl2.4H2O溶液(10mmolar、10mL)を添加した。溶液をN2下40℃に加熱し、D−特異的ヒダントイナーゼを添加した(緩衝液中1mL懸濁液、E. coli.組み換えロシュ・ヒダントイナーゼ2、300単位/mg、全タンパク質70mg)。反応混合物を40℃でN2下撹拌した。反応中、pHを5%v/v酢酸溶液滴定により7.0に維持した。
【表2】
【0181】
反応中、濃厚白色沈殿が形成した。混合物を環境温度に冷却し、3時間撹拌し、次いで濾過し、0.1N pH7リン酸カリウム緩衝液で洗浄した。固体を一晩40℃で乾燥させた。生成物を白色粉末(3.3g)として、42%収率、HPLCで100%化学純度で単離した。キラルHPLC分析は、生成物が100%(S)−entであることを示した。
キラル純度は、25cm×4.6mm Chiralpack ADカラム、30℃、溶離剤MeOH+0.1%v/v HCO2H、流速1mL min−1、検出220nMのUVを使用して確立した。Rt(S)−エナンチオマー4.42分;Rt(R)−エナンチオマー9.21分。
【0182】
5. アスペルギルス属からのL−アシラーゼを使用した(RS)−3−ベンジルスルファニル−2−メチル−2−(2,2,2−トリフルオロ−アセチルアミノ)−プロピオン酸の分割
(RS)−3−ベンジルスルファニル−2−メチル−2−(2,2,2−トリフルオロ−アセチルアミノ)−プロピオン酸
【化15】
(RS)−2−アミノ−3−ベンジルスルファニル−2−メチル−プロピオン酸(5g、0.022mol)をトリフルオロ酢酸無水物(15mL)およびDCM(15mL)にスラリー化した。スラリーを−20℃に冷却し、トリエチルアミン(2.5g、0.025mole)を滴下した。反応をRTに温めた。HPLCはアミドへの完全な変換を示した。混合物を水(30mL)およびDCM(30mL)で希釈した。水性層を廃棄し、有機層を水(3×50mL)で洗浄した。有機層を分離し、蒸発させて、油状物を得た。この物質をアセトニトリルに溶解し、溶媒を真空で蒸発させた。7.2g薄黄色油状物を得た。(〜100%収率)。
MS M+、321(100%)。
1H NMR(CDCl3)δ 7.4(1H, brs), 7.30(5H, m), 3.85(2H, m), 3.4(1H, d), 3.05(1H, d), 1.75(3H, s)。
【0183】
【化16】
CoCl2.6H20(0.25g、0.001mol)をホウ酸緩衝液(100mL、pH8.0、0.1N)に溶解した。溶液を脱気し、(RS)−トリフルオロアセトアミド(7.00g)を油状物として添加した。混合物をN2下45℃でpHが8.00になるまで飽和Na2CO3溶液を添加しながら撹拌した(約35mL添加)。L−アミノ酸アシラーゼ(0.7g、アスペルギルス属からのL−アシラーゼ、30,000単位g−1)をpH8.00緩衝液(5mL)に溶解し、反応に添加した。反応を30時間、45℃でN2下、pHを〜7.50に維持しながら撹拌した。HOAcでpHを7.00に調節し、沈殿した固体を濾過により回収し、pH7.00緩衝液で洗浄し、真空乾燥させた。(R)−アミノ酸(1.7g、34%)をオフホワイト色固体として単離した。キラルHPLCは、本物質が98%(R)−エナンチオマー(96%ee)であることを示した。幾分かの結晶化していないアミノ酸が濾液に残った。
濾液を濃HClでpH1に調節し、DCM(2×75mL)で抽出した。HPLCは、DCM抽出物が(R)−アミノ酸を含まないことを示した。
DCMを乾燥させ(Na2SO4)、蒸発させて、(S)−3−ベンジルスルファニル−2−メチル−2−(2,2,2−トリフルオロ−アセチルアミノ)−プロピオン酸(2.5g、35%)を蝋状オフホワイト色固体として得た。
この物質をMeOH/水/KOHを使用して(S)−アミノ酸に98%収率で加水分解した。生成物(S)−アミノ酸は、HPLCで96%eeであることが示された。
【0184】
実施例12
非対称化を使用した生体触媒分割
2−ベンジルスルファニルメチル−2−メチル−マロン酸ジエチルエステル
【化17】
冷却したジエチルメチルマロネート(79.9g)およびベンジルチオブロモメタン(110.4g)の2−メチル−THF(480mL)溶液に、カリウムブトキシド(53.6g)を少しずつ約2時間に渡り添加し、その間温度を0℃以下に維持した。次いで混合物をRTに温め、一晩撹拌した。反応を水(320mL)で希釈し、相を分離した。有機相を乾燥させ(Na2SO4)、濾過し、溶媒を真空下で除去した。得られた油状物をカラムクロマトグラフィー(溶離剤DCM/ヘキサン)を使用して精製して表題化合物を黄色油状物(109g、78%)として得た。
1H NMR(CDCl3)δ 1.23(6H, t), 1.47(3H, s), 2.98(2H, s), 3.72(2H, s), 4.17(4H, q), 7.2-7.3(5H, m)。
【0185】
2−ベンジルスルファニルメチル−2−メチル−マロンアミド
【化18】
メチルマロノニトリル(6.6g)およびテトラブチルアンモニウムブロマイド(1.06g)のDCM(50mL)中の混合物を約0℃に冷却した。この混合物にカリウムt−ブトキシド(9.2g)を添加し、続いてベンジルチオブロモメタン(17.89g)をゆっくり添加した。反応をRTで一晩温め、その時点でそれを塩水(100mL)で希釈した。相を分離し、水性相をさらに3回DCM(3×25mL)で抽出した。合わせた有機相を乾燥させ(MgSO4)、濾過し、真空下濃縮した。得られた褐色油状物をカラムクロマトグラフィー(溶離剤酢酸エチル/ヘキサン)を使用して精製した。2−ベンジルスルファニルメチル−2−メチル−マロノニトリルを無色固体(9.76g、52%)として単離した。
1H NMR(CDCl3)δ 1.82(3H, s), 2.89(2H, s), 4.00(2H, s), 7.26-7.37(5H, m)。
【0186】
2−ベンジルスルファニル−2−メチルマロノニトリル(2.3g)をt−ブタノール(30mL)に溶解した。溶液を60℃に温め、その時点で粉末水酸化カリウム(10g)を少しずつ添加した。60℃で一晩加熱後、反応を塩水で希釈し、DCM(3×25mL)で抽出した。合わせた有機相を乾燥させ(MgSO4)、濾過し、真空下濃縮して、表題化合物をオフホワイト色固体として得た。
1H NMR(d6-DMSO)δ 1.38(3H, s), 2.98(2H, s), 3.78(2H, s), 6.94(4H, s), 7.15-7.43(5H, m)。
【0187】
ニトリルヒドラターゼを使用した別製造法
(RS)−2−ベンジルスルファニル−2−メチルマロノニトリル(4g、0.19mol)をDSMO(15mL)に溶解した。ニトリルヒドラターゼ(ZyanotaseTM)含有凍結乾燥細胞(0.75g)を0.1N pH7 リン酸緩衝液(120mL)中にスラリー化し、ジニトリルのDMSO溶液を添加した。反応を38℃で2日振盪し、次いで濾過した。結晶化生成物をアセトニトリル(70mL)に溶解し、濾過して細胞を除去した。溶媒を蒸発により除去し、結晶化生成物を水で洗浄し、乾燥させて2−ベンジルスルファニルメチル−2−メチル−マロンアミド(3g、65%)を得た。反応からの水性濾液のDCM(100mL)での抽出、続いて蒸発により、さらに生成物(750mg、16%)を得た。
【0188】
非対称化を介する(S)−2−アミノ−3−ベンジルスルファニル−2−メチル−プロピオン酸
【化19】
2−ベンジルスルファニルメチル−2−メチル−マロン酸ジエチルエステル(5g)を、0.1M pH7 リン酸緩衝液にスラリー化し、バチルス・リケニホルミス(Bacillus licheniformis)プロテアーゼ(1g)を添加し、反応混合物を35℃で撹拌した。pHを、pHスタットを介して2Mアンモニアを添加することにより維持した。4日後、反応を酢酸エチル(2×50mL)で抽出して未反応ジエステルを除去し、希HClでpH4に酸性化し、1,2−ジクロロエタン(2×50mL)で抽出した。生成物含有ジクロロエタン相を乾燥させ(Na2SO4)、濾過した。ジクロロエタン相に、トリエチルアミン(1.6g)、続いてジフェニルホスホリルアジド(PhO)2P(O)N3(4.4g)を添加した。反応を一晩加熱還流し、5M HCl(50mL)と混合し、さらに6時間加熱還流した。生成物アミノ酸が抽出されている酸性水性相のキラルHPLC分析は、(S)−アミノ酸が存在し、eeが100%であることを示した。すなわち、(R)−アミノ酸が検出され得ない。
キラル純度を、25cm×4.6mm Chirobiotic Tカラム、30℃、溶離剤20%v/v水のEtOH溶液、流速1mL min−1、検出220nMのUVを使用して確立した。Rt(R)−エナンチオマー6.86分;Rt(S)−エナンチオマー8.21分。
【0189】
非対称化を介した(S)−5−ベンジルスルファニルメチル−5−メチル−イミダゾリジン−2,4−ジオン
【化20】
2−ベンジルスルファニルメチル−2−メチル−マロンアミド(2g)をDMSO(10mL)に50℃で溶解した。得られた溶液に0.1M pH7 リン酸緩衝液(100mL)に添加した。これに、ロドコッカス・エリスロポリス(Rhodococcus erthoplis)アミダーゼ(架橋酵素凝集体(CLEA)の25単位/mL懸濁液2mL)を添加した。35℃で4日撹拌後、CLEAを濾過により除去し、濾液を濃HClでpH4に酸性化し、生成物酸/アミドを酢酸イソプロピル(3×50mL)で抽出した。キラルHPLCは、モノ酸が93%のeeを有することを示した。溶媒を真空下で除去し、1,2−ジクロロエタン(50mL)に置き換えた。この溶液に、トリエチルアミン(0.79g)、続いてジフェニルホスホリルアジド(2.2g)を添加した。反応を18時間加熱還流し、冷却し、希HCl(0.1M、2×25mL)で洗浄し、真空下濃縮した。粗生成物は、(S)−エナンチオマーおよびキラルHPLCで93%eeであることが示された。生成物ヒダントインを、カラムクロマトグラフィー(溶離剤酢酸エチル)を使用して精製しし、エタノールから再結晶して、表題化合物(1g、49%)を無色固体として得た。eeは97%に増加した。
キラル純度を、25cm×4.6mm Chiralpack ADカラム、30℃、溶離剤MeOH+0.1%v/v HCO2H、流速1mL min−1、検出220nMのUVを使用して確立した。Rt(S)−エナンチオマー4.42分;Rt(R)−エナンチオマー9.21分。
【0190】
実施例13
a)(R)−2−ベンジルスルファニルメチル−2−メチル−4−フェニル−オキサゾリジン
【化21】
4Åモレキュラー・シーブ(3.6g)およびベンジルチオアセトン(10.0mmol、1.65mL、1.80g)を、(R)−(−)−フェニルグリシノール(1.00eq、10.0mmol、1.37g)のトルエン(170mmol、18.0mL、15.7g)溶液に環境温度で添加した。混合物を50℃に加熱し、この温度で24時間撹拌した。50mm 3番 焼結漏斗を通した濾過後、必要なオキサゾリジンの溶液が得られた。この溶液を続く実験にさらに精製することなく使用した。分析目的で、この溶液のサンプルを少量蒸発乾固して、明色油状物を得た。この物質を1H NMR分光学(CDCl3、400MHz)で分析し、オキサゾリジンが(2R,4R)−および(2S,4R)−異性体の54:46混合物として存在することが示された。
【0191】
1H NMR(CDCl3、400MHz):
(2R,4R)−異性体:δ 1.43 (3H, s); 2.83 (2H, AB, J 14.5Hz, Δ 119.5Hz); 3.65 (1H, t, J 7.5Hz); 3.86 (2H, AB, J 13.1Hz, Δ 40.7Hz); 4.24 (1H, t, J 7.5Hz); 4.51 (1H, brt, J 7.5Hz); 7.2-7.5 (10H, m))。
(2S,4R)−異性体:δ 1.50 (3H, s); 2.70 (2H, AB, J 13.7Hz, Δ 43.1Hz); 3.65 (1H, t, J 7.5Hz); 3.86 (2H, AB, J 12.8Hz, Δ 11.3Hz); 4.24 (1H, t, J 7.5Hz); 4.46 (1H, brt, J 7.5Hz); 7.2-7.5 (10H, m)。
【0192】
b)(R)−3−ベンジルスルファニル−2−メチル−2−((R)−1−フェニル−2−トリメチルシラニルオキシ−エチルアミノ)−プロピオニトリル
【化22】
(R)−2−ベンジルスルファニルメチル−2−メチル−4−フェニル−オキサゾリジン(1.00eq、100mmol、29.9g)のトルエン(4.25mol、449mL、391g)溶液に、−20℃でマグネシウムブロマイドジエチルエーテラート(99wt/wt%、5.00mmol、1.30g)、続いてトリメチルシリルシアニド(105mmol、14.1mL、10.4g)を添加した。混合物を−20℃で18時間撹拌し、次いで0℃に温め、水(60mL、2vol eq.)で洗浄した。溶媒をロータリーエバポレーター(浴温度44℃)を使用して60mL(2vol eq.)容量まで除き、次いでイソヘキサン(240mL、8vol eq.)で希釈した。混合物を加熱還流して、透明黄色溶液を得て、次いで一晩撹拌しながら冷却した。固体生成物を濾過により回収し、イソヘキサン(30mL、1vol eq.)で洗浄し、乾燥させて、表題化合物(21.5g、54.0mmol、54%)を得た。
1H NMR(CDCl3, 400MHz):δ 0.16 (9H, s); 0.98 (3H, s); 2.69 (2H, AB, J 14.2Hz, Δ 43.1Hz); 3.33 (1H, brs); 3.42 (1H, dd, J 10.6, 9.8Hz); 3.66 (1H, dd, J 10.6, 4.2Hz); 3.99 (2H, AB, J 13.5Hz, Δ32.4Hz); 4.03 (1H, dd, J 9.8, 4.2Hz); 7.0-7.5 (10H, m)。
【0193】
c)(3R,5R)−3−ベンジルスルファニルメチル−3−メチル−5−フェニル−モルホリン−2−オン
【化23】
塩化水素ガスを、15分間、−20℃に維持した(R)−3−ベンジルスルファニル−2−メチル−2−((R)−1−フェニル−2−トリメチルシラニルオキシ−エチルアミノ)−プロピオニトリル(1.00eq.、10.0mmol、3.99g)のDCM(622mmol、39.9mL、52.8g)溶液にバブリングした。水(10.0mmol、180μL、180mg)を添加し、塩化水素をさらに20分間バブリングした。混合物を環境温度に温め、次いで環境温度で12時間撹拌し、その後減圧下蒸発乾固した。混合物をDCM(50mL)に分散し、飽和水性炭酸水素ナトリウム(50mL)と共に30分撹拌した。有機層を分離し、乾燥させ(Na2SO4)、減圧下で蒸発乾固して、表題化合物を明褐色結晶性固体として得た。
【0194】
1H NMR(d6-DMSO, 400MHz);δ 1.66 (s, 3H); 2.80 (AB, J 13.1Hz, Δ 71.1 Hz, 2H); 3.02 (brs, 1H); 3.91 (s, 2H); 4.15 (t, J 10.4 Hz, 1H); 4.30 (1H, dd, J 10.3, 2.8 Hz); 4.40 (dd, J 10.8, 2.8 Hz, 1H); 7.2-7.4 (m, 5H)。
13C NMR(d6-DMSO, 100MHz);δ 25.4;37.1;42.7;51.8;62.6;74.8;126.7;127.3;128.0;128.4;128.4;128.8;138.5;138.9;172.0)。
IR(非希釈(neat)固体)3312, 3061, 3030, 2978, 2921, 2322(w), 1730(s), 1494, 1453, 1405, 1369, 1325, 1289, 1276, 1237, 1202(m), 1167(s), 1071, 1053(m), 1029, 758, 733, 697(s)cm-1。
MS(CI), m/z(%);350(M+Na, 30), 328(M+H, 100)。
【0195】
d)(R)−5−ベンジルスルファニルメチル−1−((R)−2−ヒドロキシ−1−フェニル−エチル)−5−メチル−イミダゾリジン−2,4−ジオンおよびカルバミン酸(R)−2−((R)−5−ベンジルスルファニルメチル−5−メチル−2,4−ジオキソ−イミダゾリジン−1−イル)−2−フェニル−エチルエステル
【化24】
クロロスルホニルイソシアネート(2.26mmol、197μL、320mg)を、(R)−3−ベンジルスルファニル−2−メチル−2−((R)−1−フェニル−2−トリメチルシラニルオキシ−エチルアミノ)−プロピオニトリル(2.26mmol、900mg)のDCM(9.00mL)溶液に−55℃で添加した。混合物を−55から−40℃で2時間撹拌し、次いで環境温度に温め、溶媒を減圧下蒸発により除去してオレンジ色泡状物を得た。この物質を、1M 塩酸(9.0mL)中、2時間加熱還流し、次いで環境温度に冷却し、DCM(10mL)で抽出した。DCM抽出物を乾燥させ(Na2SO4)、減圧下で蒸発乾固して、オレンジ色−褐色泡状物を得た。biotage 40Mカラムクロマトグラフィー(溶離剤勾配90:10 イソヘキサン:酢酸エチルから酢酸エチル)により、無色泡状物を得て、それは高真空乾燥で硬化して、ガラス状固体となった。トルエン(1.8mL)からの結晶化により、60:25:13:2比のヒダントイン(XV;R=H、n=1)、ジスルフィドアナログ(XV;R=H、n=2)と、対応するウレタン(XV;R=CONH2、n=1)および(XV;R=CONH2、n=2)の混合物が白色固体(150mg、約0.41mmol、18%)として得られた。
【0196】
ヒダントイン(XV;R=H、n=1)
1H NMR(CDCl3, 300MHz);δ 1.52 (3H, s); 2.50 (2H, AB, J 14.4Hz, Δ 51.0Hz); 3.02 (1H, t, J 6.0Hz); 3.41 (2H, AB, J 13.3Hz, Δ 44.4Hz); 3.89 (1H, ddd, J 11.5, 6.0, 4.5Hz); 4.26 (1H, dd, J 9.0, 4.5Hz); 4.55 (1H, ddd, J 11.5, 9.0, 6.0Hz); 7.1-7.5(10H, m); 8.50 (1H, brs)。
13C NMR(CDCl3, 75MHz);δ 21.7;36.5;37.3;60.6;63.4;68.8;127〜140(12ピーク);156.9;175.9。
IR 3189(br);3061(m);1765(m);720(s);1495(w);1432;1374(m);1265;1242;1199;1130;1070;1045;851;764(w);699(m)cm-1。
MS(CI), m/z(%);371(M+H, 70);251(M-PhCH2CH2OH, 100)。
【0197】
e)(R)−5−ベンジルスルファニルメチル−1−((R)−2−ヒドロキシ−1−フェニル−エチル)−5−メチル−イミダゾリジン−2,4−ジオン
【化25】
(3R,5R)−3−ベンジルスルファニルメチル−3−メチル−5−フェニル−モルホリン−2−オン(1.00eq.、2.69mmol、880mg)を酢酸(9mL)に溶解し、ステンレススチール‘ボンベ’型容器に入れた。シアン酸カリウム(26.9mmol、2.18g)を入れ、容器を直ぐに密閉し、内容物を環境温度で4日間撹拌した。混合物を水(18mL)に注ぎ、2回DCM(18mL)で抽出した。有機抽出物を乾燥させ(Na2SO4)、減圧下で蒸発乾固して、褐色油状物を得た。この物質のLC−MSによる分析は、約40:60比での(R)−5−ベンジルスルファニルメチル−1−((R)−2−ヒドロキシ−1−フェニル−エチル)−5−メチル−イミダゾリジン−2,4−ジオンおよび(3R,5R)−3−ベンジルスルファニルメチル−3−メチル−5−フェニル−モルホリン−2−オンの混合物を示した。
【0198】
f)(R)−5−ベンジルスルファニルメチル−5−メチル−イミダゾリジン−2,4−ジオン
【化26】
(R)−5−ベンジルスルファニルメチル−1−((R)−2−ヒドロキシ−1−フェニル−エチル)−5−メチル−イミダゾリジン−2,4−ジオン(1.00eq.、1.00mmol、370mg)を酢酸(140mmol、8.00mL、8.38g)に溶解した。48%臭化水素酸(133mmol、15.0mL、22.5g)を添加し、混合物を4時間加熱還流した。混合物を0℃に冷却し、氷(100g)を添加し、混合物を0.880 SG 水性アンモニア添加によりpH7に中和し、DCM(100mL)で抽出した。有機抽出物を乾燥させ(Na2SO4)、減圧下で蒸発乾固して、表題化合物を褐色ガム(100mg、0.624mmol、62%)として得た。
【0199】
実施例14
a)tert−ブチルN−ベンジリデンアラニネート
【化27】
t−ブチル(DL)−アラニネートヒドロクロライド(5g、27mmol)をDCM(100mL)にスラリー化し、硫酸マグネシウム(6.50g、51mmol)、ベンズアルデヒド(2.86g、27mmol)およびトリエチルアミン(2.73g、27mmol)を添加した。この混合物を16時間、RTで窒素雰囲気下撹拌した。硫酸マグネシウム(0.81g、0.67mmol)を混合物に添加し、反応をさらに24時間撹拌した。混合物を濾過し、濾液を水(2×50mL)で洗浄した。有機層を乾燥させ(MgSO4)、蒸発させて、表題化合物を無色油状物(5.54g、83%)として得た。
1H-NMR(300MHz, CDCl3)δ 1.47(9H, s), 1.50(3H, s), 4.01-4.08(1H, q), 7.39-7.43(3H, m), 7.76-7.79(2H, m), 8.31(1H, s)。
【0200】
b)tert−ブチル−ベンジル−N−ベンジリデン−2−メチルシステイネート
【化28】
水素化ナトリウム(0.17g、4.3mmol)を、tert−ブチルN−ベンジリデンアラニネート(0.5g、2mmol)のTHF(5mL)溶液にRTで添加した。混合物を30分撹拌し、次いでブロモメチルスルファニルメチル−ベンゼン(0.52g、2.4mmol)を添加した。混合物をRTで48時間撹拌し、次いで水(10mL)でクエンチした。水性層を酢酸エチル(2×50mL)で抽出した。酢酸エチル層を水(10mL)で洗浄し、乾燥させ(MgSO4)、蒸発乾固して、表題化合物を黄色油状物(0.60g、86%)として得た。
【0201】
キラルHPLC分析は、エナンチオマーの1:1混合物の存在を示した:
【表3】
1H-NMR(300MHz, CDCl3)δ 1.47(9H, s), 1.48(3H, s), 2.89-3.05(2H, m), 3.78(2H,s), 7.2-7.50(5H, m), 7.39-7.43(3H, m), 7.76-7.79(2H, m), 8.31(1H, s)。
【0202】
c)tert−ブチル−ベンジル−N−ベンジリデン−2−メチル−D(R)−システイネート
【化29】
粉末水酸化カリウム(5.90g、105mmol)およびキラル相間移動触媒(−)−O−アリル−N−(9−アントラセニルメチル)シンコニジニウムブロマイド(1.27g、2.1mmol)を、tert−ブチルN−ベンジリデンアラニネート(5g、21mmol)のトルエン(50mL)溶液に−20℃で添加した。混合物をこの温度で1時間撹拌した。反応混合物を次いで−30℃に冷却し、ブロモメチルスルファニルメチル−ベンゼン(11.6g、53mmol)のトルエン(50mL)溶液を1時間にわたり滴下した。混合物をこの温度で1時間撹拌し、次いでRTにした。反応をセライトを通して濾過し、トルエン(100mL)で洗浄した。溶媒を蒸発させて、粗残渣(19g)を得て、それをトルエン(100mL)に取り込み、水(2×25mL)で洗浄した。有機層を蒸発させて、表題化合物を得た。この段階でのエナンチオマー純度は測定しなかった。この物質を直接次工程に使用した。
1H-NMR(300MHz, CDCl3)δ 1.47(9H, s), 1.48(3H, s), 2.89-3.05(2H, m), 3.78(2H, s), 7.2-7.50(5H, m), 7.39-7.43(3H, m), 7.76-7.79(2H, m), 8.31(1H, s)。
【0203】
d)S−ベンジル−2−メチル−D(R)−システインヒドロクロライド
【化30】
THF(50mL)および6M 塩酸(50mL)を、工程(c)からの粗生成物に添加した。混合物をRTで1時間撹拌した。有機溶媒を蒸発させ、残った水性層を酢酸エチル(3×50mL)で洗浄した。合わせた有機層を6M 塩酸(50mL)で洗浄した。合わせた水性層を60℃で24時間加熱した。溶媒を減圧下蒸発させ、得られた物質をDCM(100mL)でスラリー化し、固体を濾過により回収した。この固体をDCM(100mL)で洗浄して、表題化合物を固体(3.6g、2工程で64%収率)として得た。キラルHPLC分析は、生成物が96.90%エナンチオマー純度であることを示した。
【0204】
LC法:
【表4】
1H-NMR(400MHz, dmso-d6)δ 1.44(3H, s), 2.87(1H, d), 2.98(1H, d), 3.8-3.85(2H, m), 7.3-7.39(5H, m), 8.50(3H, s)。
MS m/z, 226 [MH]+。
【0205】
実施例15
a)tert−ブチル−ベンジル−N−ベンジリデン−2−メチル−L(S)−システイネート
【化31】
粉末水酸化カリウム(1.17g、21mmol)およびキラル相間移動触媒(+)−O−アリル−N−(9−アントラセニルメチル)シンコニジニウムブロマイド(0.26g、0.42mmol)を、tert−ブチルN−ベンジリデンアラニネート(1g、4.2mmol)のトルエン(15mL)溶液に−30℃で添加した。ブロモメチルスルファニルメチル−ベンゼン(4.65g、21mmol)のトルエン(5mL)溶液を、この混合物に−30℃で添加した。反応を−15℃で16時間撹拌した。混合物をセライトを通して濾過し、トルエン(40mL)で洗浄した。濾液を水(5mL)で洗浄し、有機層を蒸発乾固して、暗褐色油状物を得た。エナンチオマー純度は、この段階で約91.8%(S)−entであることが判明した。粗物質を直接次段階に使用した。
【0206】
b)S−ベンジル−2−メチル−L(S)−システインヒドロクロライド
【化32】
THF(20mL)および6M 塩酸(20mL)を、工程(a)からの粗生成物に添加した。混合物をRTで1時間撹拌した。有機溶媒を蒸発させ、残った水性層をエーテル(3×20mL)で洗浄した。合わせた有機層を6M 塩酸(20mL)で逆抽出した。合わせた水性相を16時間加熱還流した。溶媒を減圧下で蒸発させて、表題化合物をクリーム色固体(1.10g、100%)として得た。エナンチオマー純度は90.12%(S)−entであることが判明した(LC;実施例14dと同じ方法)。
1H-NMR(400MHz, dmso-d6)δ 1.48(3H, s), 2.87(1H, d), 2.98(1H, d), 3.82(2H, m), 7.3-7.39(5H, m), 8.48(3H, s)。
【0207】
実施例16
a)イソプロピルアラニネートヒドロクロライド
【化33】
塩化チオニル(26.70g、224mmol)を、DL−アラニン(10g、112mmol)のイソプロパノール(400mL)溶液に−20℃で滴下した。混合物をRTにし、次いで4時間加熱還流した。溶媒を蒸発させて、表題化合物(18.8g、100%)を得た。
1H-NMR(300MHz)δ 1.20-1.22(6H+3H, 各d), 3.46-3.50(1H, q), 4.01-4.06(1H, m)。
【0208】
b)イソプロピルN−ベンジリデンアラニネート
【化34】
硫酸マグネシウム(13.50g、112mmol)およびベンズアルデヒド(4.76g、44.8mmol)を、イソプロピルアラニネートヒドロクロライド(9.40g、56mmol)のDCM(200mL)中のスラリーに添加した。トリエチルアミン(5.6g、56mmol)をこの混合物に添加し、次いでそれを16時間窒素雰囲気下で撹拌した。さらに硫酸マグネシウム(3.30g、28mmol)およびトリエチルアミン(0.56g、5.6mmol)を添加した。混合物を24時間撹拌した。混合物を濾過し、濾液を水(2×50mL)で洗浄した。有機層を乾燥させ(MgSO4)、溶媒を蒸発させて、生成物を無色油状物(9g、76%)として得た。
1H-NMR(400MHz, CDCl3,)δ 1.23-1.26(6H, m), 1.51(3H, d), 4.08-4.13(1H, q), 5.03-5.09(1H, m), 7.36-7.45(3H, m), 7.76-7.79(2H, m), 8.31(1H, s)。
【0209】
c)S−ベンジル−2−メチル−D(R)−システインヒドロクロライド
【化35】
粉末水酸化セシウム(3.82g、22.8mmol)およびキラル相間移動触媒(−)−O−アリル−N−(9−アントラセニルメチル)シンコニジニウムブロマイド(0.27g、0.45mmol)を、イソプロピルN−ベンジリデンアラニネート(1g、4.5mmol)のトルエン(10mL)溶液に−10℃で添加した。混合物をこの温度で1時間撹拌した。反応混合物を−30℃に冷却し、ブロモメチルスルファニルメチル−ベンゼン(2.47g、11.4mmol)のトルエン(10mL)溶液を滴下した。反応をこの温度で1時間撹拌し、次いでRTにした。反応混合物をセライトを通して濾過し、トルエン(10mL)で洗浄した。溶媒を蒸発させて、粗生成物をガムとして得た。この物質をTHF(20mL)に取り込み、6M 塩酸(20mL)を添加した。混合物をRTで16時間撹拌し、次いで4時間加熱還流した。混合物をRTに冷却し、酢酸エチル(20mL)で洗浄した。水性層を減圧下蒸発乾固した。残渣をトルエン(2×20mL)に取り込み、次いで減圧下蒸発乾固して、表題化合物をクリーム色固体(1.20g、100%)として得た。キラルHPLC分析は、生成物が91.72%エナンチオマー純度であることを示した。
1H-NMR(400MHz, dmso-d6)δ 1.44(3H, s), 2.87(1H, d), 2.98(1H, d), 3.8-3.85(2H, m), 7.3-7.39(5H, m), 8.50(3H, s)。
MS m/z 226 [MH]+。
【0210】
実施例17
a)tert−ブチルN−(ナフタレン−2−イル−メチリデン)アラニネート
【化36】
硫酸マグネシウム(2.65g、22mmol)、2−ナフトアルデヒド(1.63g、20mmol)およびトリエチルアミン(1.1g、10mmol)を、t−ブチルアラニネートヒドロクロライド(DL)(2g、11mmol)のDCM(40mL)中のスラリーに添加した。混合物を24時間窒素雰囲気下で撹拌した。硫酸マグネシウム(1.32g、11mmol)を添加し、反応をさらに16時間撹拌した。分析後、さらに硫酸マグネシウム(2.65g、22mmol)を添加し、混合物を3日間、RTで撹拌した。混合物を濾過し、濾液を水(2×100mL)で洗浄した。有機層を硫酸マグネシウムで乾燥させ、濾過し、溶媒を蒸発させて、表題化合物を白色固体(2.43g、78%)として得た。
1H-NMR(300MHz, CDCl3)δ 1.46(9H, s), 1.56(3H, s), 4.07-4.14(1H, q), 7.48-7.55(2H, m), 7.84-7.91(3H, m), 8.02-8.09(2H, m), 8.45(1H, s)。
【0211】
b)S−ベンジル−2−メチル−D(R)−システインヒドロクロライド
【化37】
粉末水酸化ルビジウム一水和物(1.79g、17.5mmol)およびキラル相間移動触媒(−)−O−アリル−N−(9−アントラセニルメチル)シンコニジニウムブロマイド(0.21g、0.35mmol)を、tert−ブチルN−(ナフタレン−2−イル−メチリデン)アラニネート(1g、3.5mmol)のトルエン(10mL)溶液に−30℃で添加した。混合物をこの温度で30分撹拌した。ブロモメチルスルファニルメチル−ベンゼン(1.91g、8.8mmol)のトルエン(10mL)溶液を、この混合物に滴下した。反応をこの温度で1時間撹拌し、次いでRTで3日撹拌した。混合物をセライトを通して濾過し、トルエン(50mL)で洗浄した。溶媒を蒸発させて、保護されたメチルシステインをガムとして得た。この物質を単離または特徴付けせずに、直接使用した。粗生成物をTHF(20mL)および6M 塩酸(20mL)の混合物に取りこんだ。混合物をRTで16時間撹拌した。有機溶媒を蒸発させ、残渣を酢酸エチル(20mL)で洗浄した。酢酸エチル層を6M 塩酸(20mL)で逆抽出した。次いで合わせた水性相を70℃で10時間加熱した。水性層を酢酸エチル(50mL)で洗浄し、その後減圧下蒸発乾固した。残渣をトルエン(50mL)を使用して共沸乾燥させ、表題化合物をクリーム色固体(0.90g、83%)として得た。キラルHPLC分析は、生成物が94.3%エナンチオマー純度であることを示した。
1H-NMR(300MHz, dmso-d6)δ 1.50(3H, s), 2.90(1H, d), 3.20(1H, d), 3.82(2H, s), 7.3-7.39(5H, m), 8.52(3H, s)。
MS(ES), m/z 226 [MH]+。
【図面の簡単な説明】
【0212】
【図1】化合物(I)形態GのX線粉末回折ダイアグラムである;
【図2】化合物(I)形態Gの示差走査熱量測定(DSC)トレースおよび熱重量分析(TGA)トレースである;
【図3】化合物(I)形態AのX線粉末回折ダイアグラムである;
【図4】化合物(I)形態BのX線粉末回折ダイアグラムである;
【図5】化合物(I)形態CのX線粉末回折ダイアグラムである;
【図6】化合物(I)形態DのX線粉末回折ダイアグラムである;
【図7】化合物(I)形態EのX線粉末回折ダイアグラムである;
【図8】化合物(I)形態FのX線粉末回折ダイアグラムである
【特許請求の範囲】
【請求項1】
CuKα放射を使用して測定して10.1、16.2、16.8および19.0°2θにおける特異的ピークを含むX線粉末回折パターンを有することを特徴とする、(5S)−5−[4−(5−クロロ−ピリジン−2−イルオキシ)−ピペリジン−1−スルホニルメチル]−5−メチル−イミダゾリジン−2,4−ジオン形態G。
【請求項2】
CuKα放射を使用して測定して9.7、10.1、11.5、12.8、14.1、16.2、16.8および19.0°2θにおける特異的ピークを含むX線粉末回折パターンを有することを特徴とする、(5S)−5−[4−(5−クロロ−ピリジン−2−イルオキシ)−ピペリジン−1−スルホニルメチル]−5−メチル−イミダゾリジン−2,4−ジオン形態G。
【請求項3】
CuKα放射を使用して測定して図1に示すものと実質的に同じX線粉末回折パターンを有することを特徴とする、(5S)−5−[4−(5−クロロ−ピリジン−2−イルオキシ)−ピペリジン−1−スルホニルメチル]−5−メチル−イミダゾリジン−2,4−ジオン形態G。
【請求項4】
実質的に純粋な(5S)−5−[4−(5−クロロ−ピリジン−2−イルオキシ)−ピペリジン−1−スルホニルメチル]−5−メチル−イミダゾリジン−2,4−ジオン形態Gである、請求項1から3のいずれかに記載の化合物。
【請求項5】
少なくとも99%純粋である、請求項4に記載の化合物。
【請求項6】
少なくとも95%純粋である、請求項4に記載の化合物。
【請求項7】
治療において使用するための、請求項1から6のいずれかに記載の化合物(I)形態G。
【請求項8】
MMP活性の阻害が有益である疾患または状態の処置または予防において使用するための医薬の製造において使用するための、請求項1から6のいずれかに記載の化合物(I)形態G。
【請求項9】
疾患または状態が炎症性疾患または状態である、請求項8に記載の使用。
【請求項10】
疾患がCOPDである、請求項9に記載の使用。
【請求項11】
請求項1から6のいずれかに記載の(5S)−5−[4−(5−クロロ−ピリジン−2−イルオキシ)−ピペリジン−1−スルホニルメチル]−5−メチル−イミダゾリジン−2,4−ジオン形態Gを薬学的に許容される希釈剤または担体と組み合わせて含む、医薬組成物。
【請求項12】
処置を必要とする患者に、治療的有効量の請求項11に記載の医薬組成物を投与することを含む、メタロプロテイナーゼ活性が仲介する疾患または状態の処置方法。
【請求項13】
(5S)−5−[4−(5−クロロ−ピリジン−2−イルオキシ)−ピペリジン−1−スルホニルメチル]−5−メチル−イミダゾリジン−2,4−ジオンをアルコール水溶液からまたは工業用変性アルコール水溶液(aqueous industrial methylated spirits)から結晶化させることを含む、請求項1から6のいずれかに記載の化合物(I)形態Gの製造方法。
【請求項14】
(5S)−5−[4−(5−クロロ−ピリジン−2−イルオキシ)−ピペリジン−1−スルホニルメチル]−5−メチル−イミダゾリジン−2,4−ジオンを、5−クロロ−2−(ピペリジン−4−イルオキシ)−ピリジン(VI)と((S)−4−メチル−2,5−ジオキソ−イミダゾリジン−4−イル)−メタンスルホニルクロライド(V)の反応により製造し、ここで、化合物(VI)を、対応する塩からの遊離塩基の遊離により製造する、請求項13に記載の方法。
【請求項15】
(RS)−3−ベンジルスルファニル−2−メチル−2−ウレイド−プロピオン酸(XI)の閉環を行うためにヒダントイナーゼ酵素を使用することを含む、(5S)−5−[4−(5−クロロ−ピリジン−2−イルオキシ)−ピペリジン−1−スルホニルメチル]−5−メチル−イミダゾリジン−2,4−ジオンの合成における中間体として有用な(S)−5−メチル−5−{[(フェニルメチル)チオ]メチル}−イミダゾリジン−2,4−ジオン(IV)の製造方法。
【請求項16】
ヒダントイナーゼ酵素がロシュ・ヒダントイナーゼ1またはヒダントイナーゼ2である、請求項15に記載の方法。
【請求項17】
ラセミ体2−アミノ−3−ベンジルチオ−2−メチルプロピオンアミドおよび(R)−(−)−マンデル酸を水の存在下で結晶させることを含む、(5S)−5−[4−(5−クロロ−ピリジン−2−イルオキシ)−ピペリジン−1−スルホニルメチル]−5−メチル−イミダゾリジン−2,4−ジオンの合成における中間体として有用な(2S)−2−アミノ−3−ベンジルチオ−2−メチルプロピオンアミド(R)−(−)−マンデラート0.5水和物の製造方法。
【請求項18】
適当なアミダーゼ酵素を使用したメソ−アミド(XIV)の酵素的非対称化(desymmetrisation)を含む、(5S)−5−[4−(5−クロロ−ピリジン−2−イルオキシ)−ピペリジン−1−スルホニルメチル]−5−メチル−イミダゾリジン−2,4−ジオンの合成における中間体として有用な(S)−5−メチル−5−{[(フェニルメチル)チオ]メチル}イミダゾリジン−2,4−ジオンの製造方法。
【請求項19】
アミダーゼがロドコッカス・エリスロポリス(Rhodococcus erthoplis)アミダーゼである、請求項18に記載の方法。
【請求項1】
CuKα放射を使用して測定して10.1、16.2、16.8および19.0°2θにおける特異的ピークを含むX線粉末回折パターンを有することを特徴とする、(5S)−5−[4−(5−クロロ−ピリジン−2−イルオキシ)−ピペリジン−1−スルホニルメチル]−5−メチル−イミダゾリジン−2,4−ジオン形態G。
【請求項2】
CuKα放射を使用して測定して9.7、10.1、11.5、12.8、14.1、16.2、16.8および19.0°2θにおける特異的ピークを含むX線粉末回折パターンを有することを特徴とする、(5S)−5−[4−(5−クロロ−ピリジン−2−イルオキシ)−ピペリジン−1−スルホニルメチル]−5−メチル−イミダゾリジン−2,4−ジオン形態G。
【請求項3】
CuKα放射を使用して測定して図1に示すものと実質的に同じX線粉末回折パターンを有することを特徴とする、(5S)−5−[4−(5−クロロ−ピリジン−2−イルオキシ)−ピペリジン−1−スルホニルメチル]−5−メチル−イミダゾリジン−2,4−ジオン形態G。
【請求項4】
実質的に純粋な(5S)−5−[4−(5−クロロ−ピリジン−2−イルオキシ)−ピペリジン−1−スルホニルメチル]−5−メチル−イミダゾリジン−2,4−ジオン形態Gである、請求項1から3のいずれかに記載の化合物。
【請求項5】
少なくとも99%純粋である、請求項4に記載の化合物。
【請求項6】
少なくとも95%純粋である、請求項4に記載の化合物。
【請求項7】
治療において使用するための、請求項1から6のいずれかに記載の化合物(I)形態G。
【請求項8】
MMP活性の阻害が有益である疾患または状態の処置または予防において使用するための医薬の製造において使用するための、請求項1から6のいずれかに記載の化合物(I)形態G。
【請求項9】
疾患または状態が炎症性疾患または状態である、請求項8に記載の使用。
【請求項10】
疾患がCOPDである、請求項9に記載の使用。
【請求項11】
請求項1から6のいずれかに記載の(5S)−5−[4−(5−クロロ−ピリジン−2−イルオキシ)−ピペリジン−1−スルホニルメチル]−5−メチル−イミダゾリジン−2,4−ジオン形態Gを薬学的に許容される希釈剤または担体と組み合わせて含む、医薬組成物。
【請求項12】
処置を必要とする患者に、治療的有効量の請求項11に記載の医薬組成物を投与することを含む、メタロプロテイナーゼ活性が仲介する疾患または状態の処置方法。
【請求項13】
(5S)−5−[4−(5−クロロ−ピリジン−2−イルオキシ)−ピペリジン−1−スルホニルメチル]−5−メチル−イミダゾリジン−2,4−ジオンをアルコール水溶液からまたは工業用変性アルコール水溶液(aqueous industrial methylated spirits)から結晶化させることを含む、請求項1から6のいずれかに記載の化合物(I)形態Gの製造方法。
【請求項14】
(5S)−5−[4−(5−クロロ−ピリジン−2−イルオキシ)−ピペリジン−1−スルホニルメチル]−5−メチル−イミダゾリジン−2,4−ジオンを、5−クロロ−2−(ピペリジン−4−イルオキシ)−ピリジン(VI)と((S)−4−メチル−2,5−ジオキソ−イミダゾリジン−4−イル)−メタンスルホニルクロライド(V)の反応により製造し、ここで、化合物(VI)を、対応する塩からの遊離塩基の遊離により製造する、請求項13に記載の方法。
【請求項15】
(RS)−3−ベンジルスルファニル−2−メチル−2−ウレイド−プロピオン酸(XI)の閉環を行うためにヒダントイナーゼ酵素を使用することを含む、(5S)−5−[4−(5−クロロ−ピリジン−2−イルオキシ)−ピペリジン−1−スルホニルメチル]−5−メチル−イミダゾリジン−2,4−ジオンの合成における中間体として有用な(S)−5−メチル−5−{[(フェニルメチル)チオ]メチル}−イミダゾリジン−2,4−ジオン(IV)の製造方法。
【請求項16】
ヒダントイナーゼ酵素がロシュ・ヒダントイナーゼ1またはヒダントイナーゼ2である、請求項15に記載の方法。
【請求項17】
ラセミ体2−アミノ−3−ベンジルチオ−2−メチルプロピオンアミドおよび(R)−(−)−マンデル酸を水の存在下で結晶させることを含む、(5S)−5−[4−(5−クロロ−ピリジン−2−イルオキシ)−ピペリジン−1−スルホニルメチル]−5−メチル−イミダゾリジン−2,4−ジオンの合成における中間体として有用な(2S)−2−アミノ−3−ベンジルチオ−2−メチルプロピオンアミド(R)−(−)−マンデラート0.5水和物の製造方法。
【請求項18】
適当なアミダーゼ酵素を使用したメソ−アミド(XIV)の酵素的非対称化(desymmetrisation)を含む、(5S)−5−[4−(5−クロロ−ピリジン−2−イルオキシ)−ピペリジン−1−スルホニルメチル]−5−メチル−イミダゾリジン−2,4−ジオンの合成における中間体として有用な(S)−5−メチル−5−{[(フェニルメチル)チオ]メチル}イミダゾリジン−2,4−ジオンの製造方法。
【請求項19】
アミダーゼがロドコッカス・エリスロポリス(Rhodococcus erthoplis)アミダーゼである、請求項18に記載の方法。
【図1】

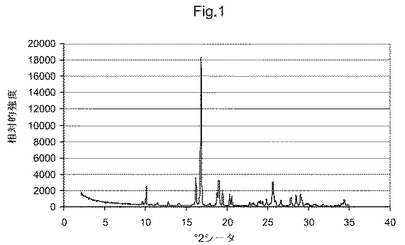
【図2】
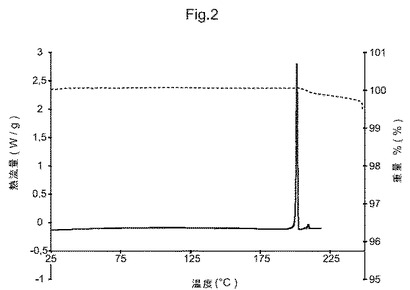
【図3】
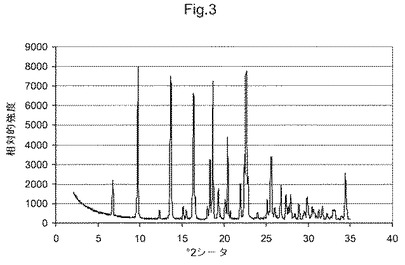
【図4】
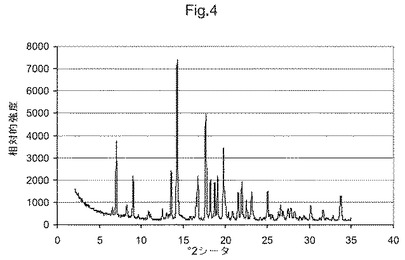
【図5】
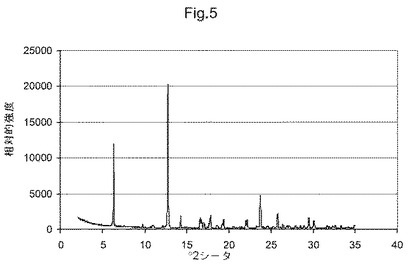
【図6】
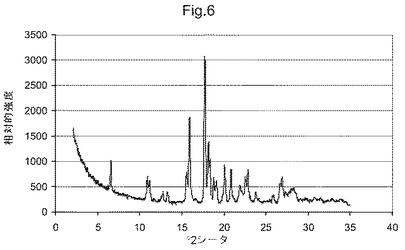
【図7】
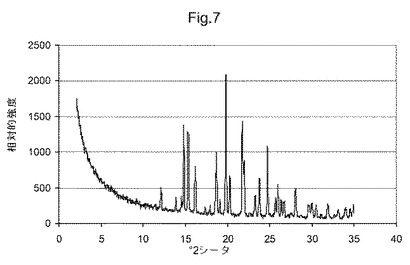
【図8】
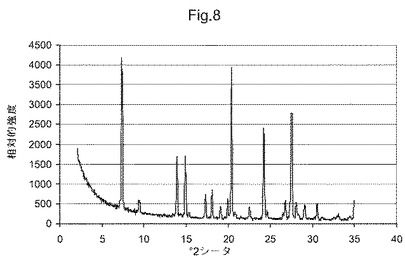
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【公表番号】特表2009−531313(P2009−531313A)
【公表日】平成21年9月3日(2009.9.3)
【国際特許分類】
【出願番号】特願2009−500326(P2009−500326)
【出願日】平成19年3月15日(2007.3.15)
【国際出願番号】PCT/SE2007/000256
【国際公開番号】WO2007/106022
【国際公開日】平成19年9月20日(2007.9.20)
【公序良俗違反の表示】
(特許庁注:以下のものは登録商標)
1.テフロン
【出願人】(391008951)アストラゼネカ・アクチエボラーグ (625)
【氏名又は名称原語表記】ASTRAZENECA AKTIEBOLAG
【Fターム(参考)】
【公表日】平成21年9月3日(2009.9.3)
【国際特許分類】
【出願日】平成19年3月15日(2007.3.15)
【国際出願番号】PCT/SE2007/000256
【国際公開番号】WO2007/106022
【国際公開日】平成19年9月20日(2007.9.20)
【公序良俗違反の表示】
(特許庁注:以下のものは登録商標)
1.テフロン
【出願人】(391008951)アストラゼネカ・アクチエボラーグ (625)
【氏名又は名称原語表記】ASTRAZENECA AKTIEBOLAG
【Fターム(参考)】
[ Back to top ]