
1−(4−(ピリジン−2−イル)ベンジル)イミダゾリジン−2,4−ジオン誘導体
本発明は、一般式(I)

[式中、R1は、H、(C1−6)アルキル(オキソ、(C1−3)アルキルオキシ、(C1−3)−アルキル−オキソカルボニル、ハロゲンまたはCNで場合によって置換されている。)、(C3−6)シクロアルキルまたは(C3−6)シクロアルキル(C1−3)アルキルであり、各シクロアルキル環は、OおよびSから選択されるヘテロ原子を場合によって含み、R2およびR3は独立して、Hもしくは(C1−3)アルキルであり、またはR2およびR3は、これらが結合している炭素原子と一緒になって(C3−5)シクロアルキル基を形成し、R4は、Hまたは1個から3個のF置換基であり、R5は、Hまたは1個から4個のF置換基であり、R6およびR7は独立して、HまたはFであり、Xは、R8、OR8、NR8R9、式(Ia)または(Ib)を表し、R8は、O、S、SOおよびSO2から選択されるヘテロ原子を場合によって含む(C5−7)シクロアルキルであり、R9は、Hまたは(C1−4)アルキルであり、R10は、H、(C1−3)アルキル、ハロゲン、オキソ、CNおよびCF3から独立して選択される1−3個の置換基を表し、Yは、CF2、O、S、SOまたはSO2である。]を有する1−(4−(ピリジン−2−イル)ベンジル)イミダゾリジン−2,4−ジオン誘導体または医薬として許容できるこの塩、これを含む医薬組成物、ならびに例えば手術前後の疼痛、慢性疼痛、神経因性疼痛、癌疼痛などの疼痛ならびに多発性硬化症に伴う疼痛および痙直の治療における前記1−(4−(ピリジン−2−イル)ベンジル)イミダゾリジン−2,4−ジオン誘導体の使用に関する。
[式中、R1は、H、(C1−6)アルキル(オキソ、(C1−3)アルキルオキシ、(C1−3)−アルキル−オキソカルボニル、ハロゲンまたはCNで場合によって置換されている。)、(C3−6)シクロアルキルまたは(C3−6)シクロアルキル(C1−3)アルキルであり、各シクロアルキル環は、OおよびSから選択されるヘテロ原子を場合によって含み、R2およびR3は独立して、Hもしくは(C1−3)アルキルであり、またはR2およびR3は、これらが結合している炭素原子と一緒になって(C3−5)シクロアルキル基を形成し、R4は、Hまたは1個から3個のF置換基であり、R5は、Hまたは1個から4個のF置換基であり、R6およびR7は独立して、HまたはFであり、Xは、R8、OR8、NR8R9、式(Ia)または(Ib)を表し、R8は、O、S、SOおよびSO2から選択されるヘテロ原子を場合によって含む(C5−7)シクロアルキルであり、R9は、Hまたは(C1−4)アルキルであり、R10は、H、(C1−3)アルキル、ハロゲン、オキソ、CNおよびCF3から独立して選択される1−3個の置換基を表し、Yは、CF2、O、S、SOまたはSO2である。]を有する1−(4−(ピリジン−2−イル)ベンジル)イミダゾリジン−2,4−ジオン誘導体または医薬として許容できるこの塩、これを含む医薬組成物、ならびに例えば手術前後の疼痛、慢性疼痛、神経因性疼痛、癌疼痛などの疼痛ならびに多発性硬化症に伴う疼痛および痙直の治療における前記1−(4−(ピリジン−2−イル)ベンジル)イミダゾリジン−2,4−ジオン誘導体の使用に関する。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、1−(4−(ピリジン−2−イル)ベンジル)イミダゾリジン−2,4−ジオン誘導体、これらを含む医薬組成物、および治療、特に疼痛の治療におけるこれらの1−(4−(ピリジン−2−イル)ベンジル)イミダゾリジン−2,4−ジオン誘導体の使用に関する。
【背景技術】
【0002】
疼痛は、実際または潜在的な組織損傷に伴う不快な感覚および感情の経験である。疼痛は元来、侵害受容性または神経障害性起源であり得る。関節炎の結果として経験される疼痛は、一般に、組織の炎症および侵害受容器の刺激により引き起こされる侵害受容性の性質のものである。侵害受容性疼痛の有病率を上げる主要な兆候は、腰背痛、骨関節炎、術後疼痛および癌関連疼痛である。侵害受容性疼痛に対する満たされていない主な要求は、効力の改善および副作用低減である。慢性疼痛市場は現在、非ステロイド性抗炎症薬(NSAID)およびシクロオキシゲナーゼCOX−2阻害剤によって独占されている。NSAIDは、軽度から中程度の疼痛を軽減するための適切な鎮痛をもたらし、通常、炎症性疼痛においてより大きな有効性を有する。個々のNSAIDは、これらの効力が変動し、これらの変動は、異なるCOX−1/COX−2選択性によって一部決定される。その結果として、患者は、彼らの疼痛が適切に治療される前にいくつかの異なる薬物で治療される必要がある場合がある。薬物治療に伴う副作用は、治療選択において、特に多くの疼痛症候群が長期慢性状態である場合、重要な因子である。
【0003】
NSAIDの最も一般的な副作用は、便秘症および消化障害であり、大部分の抗炎症薬は酸性の性質であり、胃における酸の産生を促進する。他の重篤な副作用は、胃潰瘍、粘膜損傷および消化性びらんなどの胃腸管系合併症である。NSAIDは、毎年米国において潰瘍の合併症による107,000人もの入院および16,500人もの死亡の原因と考えられている(Singh、Recent considerations in nonsteroidal anti−inflammatory drug gastropathy.Am J.Med.、1998、105:31S−38S)。COX−2阻害剤は、改善された胃腸管系副作用プロフィールを有する一方、これらの使用は、心筋梗塞および脳卒中のリスク増加、ならびに高血圧症のリスク増加を伴ってきた。
【0004】
神経系の損傷、疾患または機能不全に起因する慢性疼痛として定義される神経因性疼痛は、前記集団の約1%に存在し、最も大きい患者集団として、有痛性の糖尿病性末梢神経障害を持つもの、および帯状疱疹(ヘルペス後神経痛)の発作後に持続する神経痛を有するものが挙げられる。これは、組織損傷が存在せずに発生し得る自発的疼痛を含めて、複雑な組合せの症状を特徴とする。神経因性疼痛を患う患者は、有痛性として通常知覚される刺激(痛覚過敏)、ならびに疼痛を通常誘発しない刺激(アロディニア)の両方に対する感度も増加している。これらの症状は従来の鎮痛療法に対してしばしば難治性であり、大部分の患者は、彼らの症状の完全な軽減を達成しない。現在、抗うつ薬、抗痙攣薬およびオピオイドは、ゴールドスタンダードであるガバペンチンとともに依然として一次治療である。これらの薬物の全てが、用量制限性の顕著な副作用を有する。さらに、効力が神経因性疼痛の市場における考慮すべき問題であり、現在の治療は、総疼痛スコアにおいてベースラインから最大50%の低減を示す。その結果として、より高い効力/応答者の割合を有し、現在使用されている薬物と比較して副作用が低減されている薬剤の医学的必要が、依然として満たされていない。
【0005】
新たな臨床的証拠、ならびに大麻を自己投薬している患者からの事例報告は、カンナビノイド受容体アゴニストが、疼痛を治療する際に役割を担う場合があることを示唆している(Fox A、Bevan S.、Therapeutic potential of cannabinoid receptor agonists as analgesic agents.Expert Opin Investig Drugs、2005、14、695−703)。GW Sativex、神経因性疼痛の治療のための個別投薬を可能にする口腔粘膜スプレー製剤におけるΔ9−THCおよびカンナビジオールの1:1比は、GW Pharmaceuticalsによって始められた。Sativexの臨床研究は、難治性疼痛(慢性神経因性疼痛、腕神経叢神経損傷による疼痛、アロディニア末梢性神経因性疼痛および進行癌疼痛)、関節リウマチ、および多発性硬化症に伴う症状(疼痛、痙直、膀胱制御不良および睡眠分断)を持つ患者において効力を実証した(Barnes MP.2006.Sativex:clinical efficacy and tolerability in the treatment of symptoms of multiple sclerosis and neuropathic pain.Expert Opin.Pharmacother.7(5):607−615)。
【0006】
カンナビノイド受容体の2つのタイプが同定された。カンナビノイドCB1受容体は主に中枢神経系(CNS;脳脊髄)に位置するが、末梢ニューロンによっても発現され、他の末端組織において、より低い程度で発現される。カンナビノイドCB2受容体は、大抵免疫細胞中の末梢に主に限局されている(Howlett、A.C.ら、International Union of Pharmacology.XXVII.Classification of Cannabinoid Receptors.Pharmacol.Rev.54、161−202、2002)。テトラヒドロカンナビノール(THC)など従来のCB1受容体アゴニストおよびCB1/CB2受容体アゴニストは、動物における疼痛モデルで高い効果がある一方、人間におけるこれらの治療有用性は、向精神効果などの望ましくないCNS副作用、および濫用の可能性によって制限される(Chapman、V.およびFinn、D.P.「Analgesic effects of cannabinoids:sites and mechanism of action.」Rev.Analg.7、25−39、2003)。
【0007】
近年の文献証拠は、CB2受容体の選択的活性化が、望ましくないCNS副作用を起こすことなく疼痛および炎症を治療するための新規な戦略となり得ることを示唆している。(Guindon、J.およびHohmann、A.、「Cannabinoid CB2 receptors:a therapeutic target for the treatment of inflammatory and neuropathic pain」、Br.J.Pharmacol.、2008、153、319−334)。CB2受容体の活性化は、動物モデルにおいて急性、炎症性および神経因性の疼痛応答を阻害することが判明した(Whiteside G.T.、Lee G.P.、Valenzano K.J.「The role of the cannabinoid CB2 receptor in pain transmission and therapeutic potential of small molecule CB2 receptor agonists」、Current Med.Chem.、2007、14、917−936)。CB2ノックアウトマウスの研究も、疼痛におけるCB2受容体の役割を裏付けている(Malan TP、Jr、Ibrahim MM、Lai J、Vanderah TW、Makriyannis AおよびPorreca F.CB2 cannabinoid receptor agonists:pain relief without psychoactive effects? Curr.Opin.Pharmacol.2003;3:62−67)。
【0008】
CB2媒介抗侵害受容に寄与する細胞性機序はまだ明らかではないが、CB2受容体の活性化が、免疫細胞活性の調節を介して間接的に炎症性疼痛に影響し、炎症の局所部位で媒介物質の放出減少をもたらすことが提唱された。末梢効果に加えて、近年の学術誌は、CB2受容体アゴニストが、末梢ニューロンおよび活性化ミクログリア上に発現したCB2受容体と相互作用することによって疼痛伝達を調節することもできることを示唆している(Beltramoら、2006.CB2 receptor−mediated antihyperalgesia:possible direct involvement of neural mechanisms.Eur J Neurosci.23(6):1530−80;Romero−Sandoval&Eisenach、2007.Spinal cannabinoid receptor type 2 activation reduces hypersensitivity and spinal cord glial activation after paw incision.Anesthesiology106(4):787−94)。
【0009】
つまり、CB2受容体アゴニストは、骨関節炎、関節リウマチならびに急性の術後疼痛および神経因性疼痛などの急性および慢性疼痛状態の治療に適当であり得る。前臨床モデルにおけるCB2アゴニストによるカタレプシーの不在は、望ましくないCNS副作用のない急性および慢性疼痛の治療が有望であることを示す。
【先行技術文献】
【非特許文献】
【0010】
【非特許文献1】Singh、Recent considerations in nonsteroidal anti−inflammatory drug gastropathy.Am J.Med.、1998、105:31S−38S
【非特許文献2】Fox A、Bevan S.、Therapeutic potential of cannabinoid receptor agonists as analgesic agents.Expert Opin Investig Drugs、2005、14、695−703
【非特許文献3】Barnes MP.2006.Sativex:clinical efficacy and tolerability in the treatment of symptoms of multiple sclerosis and neuropathic pain.Expert Opin.Pharmacother.7(5):607−615
【非特許文献4】Howlett、A.C.ら、International Union of Pharmacology.XXVII.Classification of Cannabinoid Receptors.Pharmacol.Rev.54、161−202、2002
【非特許文献5】Chapman、V.およびFinn、D.P.「Analgesic effects of cannabinoids:sites and mechanism of action.」Rev.Analg.7、25−39、2003
【非特許文献6】Guindon、J.およびHohmann、A.、「Cannabinoid CB2 receptors:a therapeutic target for the treatment of inflammatory and neuropathic pain」、Br.J.Pharmacol.、2008、153、319−334
【非特許文献7】Whiteside G.T.、Lee G.P.、Valenzano K.J.「The role of the cannabinoid CB2 receptor in pain transmission and therapeutic potential of small molecule CB2 receptor agonists」、Current Med.Chem.、2007、14、917−936
【非特許文献8】Malan TP、Jr、Ibrahim MM、Lai J、Vanderah TW、Makriyannis AおよびPorreca F.CB2 cannabinoid receptor agonists:pain relief without psychoactive effects? Curr.Opin.Pharmacol.2003;3:62−67
【非特許文献9】Beltramoら、2006.CB2 receptor−mediated antihyperalgesia:possible direct involvement of neural mechanisms.Eur J Neurosci.23(6):1530−80
【非特許文献10】Romero−Sandoval&Eisenach、2007.Spinal cannabinoid receptor type 2 activation reduces hypersensitivity and spinal cord glial activation after paw incision.Anesthesiology106(4):787−94
【発明の概要】
【発明が解決しようとする課題】
【0011】
したがって、疼痛の治療における治療剤としての選択的CB2カンナビノイド受容体アゴニストが求められている。
【課題を解決するための手段】
【0012】
この目的のため、本発明は、例えば手術前後の疼痛、慢性疼痛、神経因性疼痛、癌疼痛などの疼痛ならびに多発性硬化症に伴う疼痛および痙直の治療に使用することができるカンナビノイドCB2受容体のアゴニストとして、一般式Iを有する1−(4−(ピリジン−2−イル)ベンジル)イミダゾリジン−2,4−ジオン誘導体の新規な構造部類
【0013】
【化1】
[式中、
R1は、H、(C1−6)アルキル(オキソ、(C1−3)アルキルオキシ、(C1−3)アルキルオキシ−カルボニル、ハロゲンまたはCNで場合によって置換されている。)、(C3−6)シクロアルキルまたは(C3−6)シクロアルキル(C1−3)アルキルであり、各シクロアルキル環は、OおよびSから選択されるヘテロ原子を場合によって含み、
R2およびR3は独立して、Hもしくは(C1−3)アルキルであり、または
R2およびR3は、これらが結合している炭素原子と一緒になって(C3−5)−シクロアルキル基を形成し、
R4は、Hまたは1個から3個のF置換基であり、
R5は、Hまたは1個から4個のF置換基であり、
R6およびR7は独立して、HまたはFであり、
Xは、R8、OR8、NR8R9、
【0014】
【化2】
を表し、
R8は、O、S、SOおよびSO2から選択されるヘテロ原子を場合によって含む(C5−7)シクロアルキルであり、
R9は、Hまたは(C1−4)アルキルであり、
R10は、H、(C1−3)アルキル、ハロゲン、オキソ、CNおよびCF3から独立して選択される1−3個の置換基を表し、
Yは、CF2、O、S、SOまたはSO2である。]
または医薬として許容できるこの塩を提供する。
【0015】
(C1−6)アルキルという用語は、式Iの定義で使用される場合、ヘキシル、ペンチル、ブチル、イソブチル、第三ブチル、プロピル、イソプロピル、エチルおよびメチルのように、1−6個の炭素原子を有する分枝または非分枝のアルキル基を意味する。
【0016】
(C1−4)アルキルという用語は同様に、n−ブチル、tert−ブチル、プロピル、イソプロピル、エチルおよびメチルのように、1−4個の炭素原子を有する分枝または非分枝のアルキル基を意味する。
【0017】
(C1−3)アルキルという用語は同様に、プロピル、イソプロピル、エチルおよびメチルにように、1−3個の炭素原子を有する分枝または非分枝のアルキル基を意味する。
【0018】
(C1−3)アルキルオキシおよび(C1−3)アルキルオキソカルボニルという用語における(C1−3)アルキルの意味は、上記で定義した通りである。
【0019】
(C3−6)シクロアルキルという用語は、シクロヘキシル、シクロペンチル、シクロブチルおよびシクロプロピルのように、3−6個の炭素原子を有するシクロアルキル基を意味する。
【0020】
(C3−5)シクロアルキルという用語は、シクロペンチル、シクロブチルおよびシクロプロピルにように、3−5個の炭素原子を有するシクロアルキル基を意味する。
【0021】
(C5−7)シクロアルキルという用語は同様に、5−7個の炭素原子を有するシクロアルキル基を意味する。
【0022】
好ましい(C5−7)シクロアルキルはシクロヘキシルである。
【0023】
ハロゲンという用語は、F、Cl、BrまたはIを意味する。
【0024】
R2、R3およびR5がHである式Iに従った1−(4−(ピリジン−2−イル)ベンジル)イミダゾリジン−2,4−ジオン誘導体が優先される。R1が(C1−4)アルキルである式Iに従った化合物も好ましい。さらに好ましいのは、XがNR8R9、
【0025】
【化3】
を表す式Iに従った化合物である。
【0026】
より好ましいのは、XがNR8R9であり、R8がOおよびSから選択されるヘテロ原子を場合によって含むシクロヘキシルである式Iの化合物である。
【0027】
さらに好ましいのは、R4がCR6R7X基に対してオルト位のF置換基である化合物である。
【0028】
本発明の特に好ましい1−(4−(ピリジン−2−イル)ベンジル)イミダゾリジン−2,4−ジオン誘導体は、
−3−イソブチル−1−(4−(6−(ピペリジン−1−イルメチル)ピリジン−2−イル)ベンジル)イミダゾリジン−2,4−ジオン;
−3−イソブチル−1−(4−(6−(モルホリノメチル)ピリジン−2−イル)ベンジル)イミダゾリジン−2,4−ジオン;
−1−(4−(5−フルオロ−6−((テトラヒドロ−2H−ピラン−4−イルアミノ)メチル)ピリジン−2−イル)ベンジル)−3−イソブチルイミダゾリジン−2,4−ジオン;
−1−(4−(6−((1,1−ジオキソ−1λ6−チオモルホリン−4−イル)メチル)ピリジン−2−イル)ベンジル)−3−イソブチルイミダゾリジン−2,4−ジオン;
−1−(4−(6−((1,1−ジオキソ−1λ6−チオモルホリン−4−イル)メチル)−5−フルオロピリジン−2−イル)ベンジル)−3−イソブチルイミダゾリジン−2,4−ジオン;
−3−イソブチル−1−(4−(6−((テトラヒドロ−2H−ピラン−4−イルアミノ)メチル)ピリジン−2−イル)ベンジル)イミダゾリジン−2,4−ジオン;
−3−エチル−1−(4−(6−(ピペリジン−1−イルメチル)ピリジン−2−イル)ベンジル)イミダゾリジン−2,4−ジオン;
−3−エチル−1−(4−(5−フルオロ−6−(ピペリジン−1−イルメチル)ピリジン−2−イル)ベンジル)イミダゾリジン−2,4−ジオン;
−1−(4−(6−(1,1−ジオキソ−1λ6−チオモルホリン−4−イルメチル)−5−フルオロピリジン−2−イル)ベンジル)−3−エチルイミダゾリジン−2,4−ジオン;
−1−(4−(6−(1,1−ジオキソ−1λ6−チオモルホリン−4−イルメチル)ピリジン−2−イル)ベンジル)−3−エチルイミダゾリジン−2,4−ジオン;
−1−(4−(5−フルオロ−6−((テトラヒドロ−2H−ピラン−4−イルアミノ)メチル)ピリジン−2−イル)ベンジル)−3−プロピルイミダゾリジン−2,4−ジオン;
−1−(4−(6−(1,1−ジオキソ−1λ6−チオモルホリン−4−イルメチル)−5−フルオロピリジン−2−イル)ベンジル)−3−プロピルイミダゾリジン−2,4−ジオン;
−3−(2,2−ジフルオロエチル)−1−(4−(6−(ピペリジン−1−イルメチル)ピリジン−2−イル)ベンジル)イミダゾリジン−2,4−ジオン;
−3−(2,2−ジフルオロエチル)−1−(4−(5−フルオロ−6−(ピペリジン−1−イルメチル)ピリジン−2−イル)ベンジル)イミダゾリジン−2,4−ジオン;
−3−(シクロプロピルメチル)−1−(4−(5−フルオロ−6−(ピペリジン−1−イルメチル)ピリジン−2−イル)ベンジル)イミダゾリジン−2,4−ジオン;
−3−(シクロプロピルメチル)−1−(4−(6−(ピペリジン−1−イルメチル)ピリジン−2−イル)ベンジル)イミダゾリジン−2,4−ジオン;
−3−(2−オキソプロピル)−1−(4−(6−(ピペリジン−1−イルメチル)ピリジン−2−イル)ベンジル)イミダゾリジン−2,4−ジオン;
−1−(4−(5−フルオロ−6−(ピペリジン−1−イルメチル)ピリジン−2−イル)ベンジル)−3−(2−オキソプロピル)イミダゾリジン−2,4−ジオン;
−1−(4−(6−(1,1−ジオキソ−1λ6−チオモルホリン−4−イルメチル)−5−フルオロピリジン−2−イル)ベンジル)−3−(2−オキソプロピル)イミダゾリジン−2,4−ジオン;
−3−ルソプロピル−1−(4−(6−(ピペリジン−1−イルメチル)ピリジン−2−イル)ベンジル)イミダゾリジン−2,4−ジオン;
−1−(4−(6−(1,1−ジオキソ−1λ6−チオモルホリン−4−イルメチル)−5−フルオロピリジン−2−イル)ベンジル)−3−イソプロピルイミダゾリジン−2,4−ジオン;
−1−(4−(6−1,1−ジオキソ−1λ6−チオモルホリン−4−イルメチル)ピリジン−2−イル)ベンジル)−3−イソプロピルイミダゾリジン−2,4−ジオン;
−3−シクロプロピル−1−(4−(5−フルオロ−6−(ピペリジン−1−イルメチル)ピリジン−2−イル)ベンジル)イミダゾリジン−2,4−ジオン;
−3−シクロプロピル−1−(4−(6−(1,1−ジオキソ−1λ6−チオモルホリン−4−イルメチル)−5−フルオロピリジン−2−イル)ベンジル)イミダゾリジン−2,4−ジオン;
−3−シクロブチル−1−(4−(5−フルオロ−6−((テトラヒドロ−2H−ピラン−4−イルアミノ)メチル)ピリジン−2−イル)ベンジル)イミダゾリジン−2,4−ジオン;
−3−シクロブチル−1−(4−(6−((テトラヒドロ−2H−ピラン−4−イルアミノ)メチル)ピリジン−2−イル)ベンジル)イミダゾリジン−2,4−ジオン;
−3−シクロブチル−1−(4−(6−(1,1−ジオキソ−1λ6−チオモルホリン−4−イルメチル)−5−フルオロピリジン−2−イル)ベンジル)イミダゾリジン−2,4−ジオン;
−3−シクロブチル−1−(4−(6−(1,1−ジオキソ−1λ6−チオモルホリン−4−イルメチル)ピリジン−2−イル)ベンジル)イミダゾリジン−2,4−ジオン;
−1−(4−(6−(ピペリジン−1−イルメチル)ピリジン−2−イル)ベンジル)−3−(2,2,2−トリフルオロエチル)イミダゾリジン−2,4−ジオン;
−1−(4−(6−(1,1−ジオキソ−1λ6−チオモルホリン−4−イルメチル)−5−フルオロピリジン−2−イル)ベンジル−3−(2,2,2−トリフルオロエチル)イミダゾリジン−2,4−ジオン;
−1−(4−(6−((テトラヒドロ−2H−ピラン−4−イルアミノ)メチル)ピリジン−2−イル)ベンジル)−3−(2,2,2−トリフルオロ−エチル)イミダゾリジン−2,4−ジオン;
−1−(4−(5−フルオロ−6−((テトラヒドロ−2H−ピラン−4−イルアミノ)メチル)ピリジン−2−イル)ベンジル)−3−(2,2,2−トリフルオロエチル)イミダゾリジン−2,4−ジオン
または医薬として許容できるこれらの塩である。
【0029】
式Iを有する本発明の1−(4−(ピリジン−2−イル)ベンジル)イミダゾリジン−2,4−ジオン誘導体は、有機化学の当技術分野において知られている方法によって調製することができる。本発明の化合物は、例えば、式II(式中、R4、R6、R7およびXは、すでに定義した通りの意味を有する。)の2−ブロモピリジン誘導体と、式III(式中、R1、R2、R3およびR5は、すでに定義した通りの意味を有する。)のベンジル化イミダゾリジンから調製される式IVのボロン酸誘導体との間の、炭酸カリウムおよびPd(Ph3P)4などのパラジウム(0)錯体を使用する鈴木カップリング反応から得ることができる(スキームIを参照のこと。)。
【0030】
【化4】
【0031】
XがR8を表し、R8がSおよびOから選択されるヘテロ原子を含む(C5−7)シクロアルキルである式IIの化合物は、式1(式中、R4は、すでに定義した通りの意味を有する。)の二臭化ピリジン誘導体と式2(式中、Zは、CH2、OまたはSを表す。)のニトリル誘導体との縮合から式3の中間体ケト誘導体を生成し、これを式4のヒドラジド誘導体を介して還元することにより、R6およびR7がHである式IIの化合物に対応する式IIbの化合物を得ることができる、または三フッ化ジエチルアミノ硫黄を用いて還元することにより、R6およびR7がFである式IIの化合物に対応する式IIaの化合物を生成することができることで調製することができる(スキームIIを参照のこと)。
【0032】
【化5】
【0033】
条件
A:ブチルリチウム/H2SO4。B:4−メチルベンゼンスルホニルヒドラジド。C:水素化ジイソブチルアルミニウム。D:三フッ化ジエチルアミノ硫黄。
【0034】
XがOR8表す式IIの化合物は、スキームIIIに示されている通り、式5の2−ブロモ−6−メチルピリジン誘導体の臭素化で出発し、N−ブロモコハク酸イミド/アゾ−ジ−イソブチロニトリルを用いて式6の対応する6−ブロモメチル誘導体を生成し、これを式7(式中、Zは、CH2,OまたはSである。)のアルコール誘導体と反応させることにより式IIcの化合物を生成することで調製することができる。
【0035】
【化6】
XがNHR8、
【0036】
【化7】
を表す式IIの化合物は、例えば酢酸/水素化トリアセトキシホウ素ナトリウムの使用とともに、式X−Hの適切なアミン誘導体を用いる式8のカルバルデヒド誘導体の還元的アミノ化反応を使用して調製することができる。
【0037】
【化8】
【0038】
式IIIの化合物は、還元的アミノ化条件下でのアミノ酸H2N−C(R2,R3)−COOHと式9の4−ブロモベンズアルデヒド誘導体とのカップリングによって式12のN−ベンジル誘導体が得られ、これを引き続き、ジシクロヘキシル−カルボジイミド(DCCI)、TBTUまたはPyBOPなどのアミド結合形成試薬を用いてアミンH2N−R1(式中、R1は、前に定義されている意味を有する。)とカップリングさせることによりアミド誘導体13にし、これから、カルボニルジイミダゾールを使用する閉環反応によって式IIIのイミダゾリジン−2,4−ジオン誘導体を調製できることで調製することができる。
【0039】
スキームIVに示されている代替経路において、式9の4−ブロモベンズアルデヒド誘導体を還元的アミノ化条件下で式H2N−C(R2,R3)−CONH2のアミノ酸アミドにカップリングさせることにより、式10のN−ベンジル誘導体が得られ、これは、カルボニルジイミダゾールを使用する環化、および式Hal−R1のハロゲン化物を用いる後続のアルキル化によって式IIIの化合物に変換することができる。
【0040】
【化9】
【0041】
式Iの1−(4−(ピリジン−2−イル)ベンジル)イミダゾリジン−2,4−ジオン誘導体およびこれらの塩は、少なくとも1つのキラリティー中心を含有し、したがって、エナンチオマーおよびジアステレオマーを含めた立体異性体として存在することができる。本発明は、この範囲内に、前述の立体異性体、ならびに式Iの化合物の個々のRおよびSエナンチオマーのそれぞれならびにこれらの塩、実質的に遊離塩基、即ち5%未満、好ましくは2%未満、特に1%未満の他のエナンチオマーを伴う遊離塩基、ならびに実質的に等量の2種のエナンチオマーを含有するラセミ混合物を含めた任意の比率のこうしたエナンチオマーの混合物を含める。
【0042】
純粋な立体異性体が得られる不斉合成またはキラル分離のための方法は、当技術分野においてよく知られており、例えばキラル誘導を用いる合成、もしくは市販されているキラル基質から出発する合成、または例えばキラル媒体上のクロマトグラフィーを使用するもしくはキラル対イオンを用いる結晶化による立体異性体の分離である。
【0043】
医薬として許容できる塩は、式Iの1−(4−(ピリジン−2−イル)ベンジル)イミダゾリジン−2,4−ジオン誘導体の遊離塩基を、塩酸、臭化水素酸、リン酸および硫酸などの鉱酸、または例えばアスコルビン酸、クエン酸、酒石酸、乳酸、マレイン酸、マロン酸、フマル酸、グリコール酸、コハク酸、プロピオン酸、酢酸およびメタンスルホン酸などの有機酸で処理することによって得ることができる。
【0044】
本発明の化合物は、非溶媒和で、ならびに水およびエタノールなどの医薬として許容できる溶媒との溶媒和形態で存在することができる。一般に、溶媒和形態は、本発明の目的においては非溶媒和形態と同等と考えられる。
【0045】
本発明は、医薬として許容できる助剤および場合により他の治療剤との混合で、一般式Iに従った1−(4−(ピリジン−2−イル)ベンジル)イミダゾリジン−2,4−ジオン誘導体または医薬として許容できるこの塩を含む医薬組成物をさらに提供する。「許容できる」という用語は、前記組成物の他の成分と相容性であり、この受容者に有害でないことを意味する。組成物は、例えば、全て投与用単位剤形における経口、舌下、皮下、静脈内、硬膜外、くも膜下腔内、筋肉内、経皮、肺、局所、眼球または直腸投与などに適当なものを含める。投与の好ましい経路は経口経路である。
【0046】
経口投与のため、前記活性成分は、錠剤、カプセル、粉末、顆粒、溶液および懸濁液などの個別単位として存在することができる。非経口投与のため、本発明の前記医薬組成物は、単位用量または多用量容器、例えば密封したバイアルおよびアンプル中の所定の量での例えば注入液体中に存在することができ、使用前に滅菌液体担体、例えば水の添加のみを必要とする凍結乾燥させた(凍結乾燥)状態で貯蔵することもできる。
【0047】
例えば標準的文献のGennaro、A.R.ら、Remington:The Science and Practice of Pharmacy(第20版、Lippincott Williams&Wilkins、2000、特に5部:Pharmaceutical Manufacturingを参照のこと。)に記載されている通りに、医薬として許容できるこうした助剤と混合され、前記活性薬剤は、丸薬、錠剤などの固体投与単位に圧縮することができ、またはカプセル、坐剤もしくはパッチにすることができる。医薬として許容できる液体の手段によって、前記活性薬剤は、流体組成物、例えば溶液、懸濁液、エマルジョンの形態で注入調製物として、またはスプレー、例えば経鼻スプレーとして適用することができる。
【0048】
固体投与単位を製造するため、充填剤、着色剤およびポリマーバインダーなどの従来の添加剤の使用が企図される。一般に、前記活性化合物の機能を妨げることのない任意の医薬として許容できる添加剤を使用することができる。本発明の活性薬剤を固体組成物として投与することができる適当な担体として、適当な量で使用されるラクトース、デンプンおよびセルロース誘導体など、またはこれらの混合物が挙げられる。非経口投与のため、プロピレングリコールまたはブチレングリコールなど医薬として許容できる分散剤および/または湿潤剤を含有する水性懸濁液、等張生理食塩水および滅菌注射可能溶液を使用することができる。
【0049】
本発明は、前記組成物に適当な包装材料との組合せにおいて前に記載されている通りの医薬組成物をさらに含め、前記包装材料は、前に記載されている通りの使用のための組成物使用用指示を含める。
【0050】
本発明の1−(4−(ピリジン−2−イル)ベンジル)イミダゾリジン−2,4−ジオン誘導体は、CHO細胞を使用するヒトCB2およびCB1レポーターアッセイにおいて決定された通り、CB1受容体と比較するとCB2受容体の選択的アゴニストであることが判明した。受容体結合ならびにカンナビノイド受容体調節因子のインビトロ生物活性を決定する方法は、当技術分野においてよく知られている。一般に、発現された受容体は前記化合物を用いてインキュベートすることにより試験され、結合または機能的応答の刺激もしくは阻害が測定される。
【0051】
機能的応答を測定するため、CB2またはCB1受容体遺伝子、好ましくはヒト受容体をコードする単離DNAは、適当な宿主細胞中に発現される。こうした細胞はチャイニーズハムスター卵巣細胞であってよいが、他の細胞も適当である。好ましくは、前記細胞は哺乳動物起源である。
【0052】
組換えCB2またはCB1発現細胞株を構築するための方法は、当技術分野においてよく知られている(Sambrookら、Molecular Cloning:a Laboratory Manual、Cold Spring Harbor Laboratory Press、Cold Spring Harbor、最新版)。受容体の発現は、所望のタンパク質をコードするDNAの発現によって達成される。追加配列の連結および適当な発現系の構成のための技術は全て、これまで当技術分野においてよく知られている。標準的な固相技術を使用して、所望のタンパク質をコードするDNAの一部分または全てを合成的に構築することにより、好ましくは、連結を緩めるための制限部位を含めることができる。含まれたコード配列の転写および翻訳のための適当な制御領域を、前記DNAコード配列に提供することができる。よく知られている通り、細菌などの原核宿主、ならびに酵母、植物細胞、昆虫細胞、哺乳動物細胞、および鳥細胞などの真核宿主を含めた多種多様な宿主と相容性のある発現系が現在利用可能である。
【0053】
前記受容体を発現する細胞を次いで、試験化合物を用いてインキュベートすることにより、結合または機能的応答の刺激もしくは阻害を観察する。
【0054】
別法として、発現されたCB2またはCB1受容体を含有する単離細胞膜を使用することにより、化合物の結合を測定することができる。
【0055】
結合の測定のため、放射活性または蛍光標識化合物を使用することができる。最も広範に使用される放射標識カンナビノイドプローブは、CB1およびCB2結合部位に対しておよそ同等の親和性を有する(3H)CP55940である。
【0056】
機能的なCB2またはCB1アゴニスト活性は、例えばcAMPまたはMAPキナーゼ経路における受容体媒介変化の測定など、第二メッセンジャー応答を決定することによって測定することができる。したがって、こうした方法は、宿主細胞の細胞表面上におけるCB2またはCB1受容体の発現、および試験化合物に前記細胞を曝露することを伴う。第二メッセンジャー応答を次いで測定する。第二メッセンジャー濃度は、受容体の結合に対する試験化合物の影響に依存して低減または増加する。
【0057】
曝露細胞における例えばcAMP濃度の直接測定に加えて、受容体コードDNAのトランスフェクションに加えてレポーター遺伝子をコードする第二DNAもトランスフェクトされている細胞を使用することができ、この発現は受容体活性化と相関する。一般に、レポーター遺伝子発現は、第二メッセンジャー濃度の変化に反応する任意の応答配列によって制御され得る。適当なレポーター遺伝子は、例えばLacZ、アルカリホスファターゼ、蛍ルシフェラーゼおよび緑色蛍光タンパク質である。こうしたトランス活性化アッセイの原理は当技術分野においてよく知られており、例えばStratowa、C.、Himmler、A.およびCzernilofsky、A.P.、Curr.Opin.Biotechnol.6、574(1995)に記載されている。CB2受容体に対する選択的活性アゴニスト化合物を選択するため、化合物のEC50値は、<10−5M、好ましくは<10−7Mであり、EC50(CB1)/EC50(CB2)として定義した通りのCB1受容体アゴニストに対する選択率は、>10、好ましくは>50である。
【発明の効果】
【0058】
前記化合物は、例えば手術前後の疼痛などの急性疼痛、慢性疼痛、神経因性疼痛、癌疼痛、内臓痛、頭痛などの疼痛、および多発性硬化症に伴う痙直の治療における鎮痛剤として使用することができる。
【0059】
本発明のカンナビノイドアゴニストは、(腸管の)炎症、アトピー性皮膚炎、肝疾患、呼吸器障害、アレルギー、腫瘍、てんかん、片頭痛、骨粗鬆症、循環器障害、外傷性脳損傷および脳卒中などの急性神経変性疾患、ならびにアルツハイマー病、多発性硬化症およびALSなどの緩徐性神経変性疾患を含めた他の障害の治療においても潜在的に有用であると思われる(Parcher P、Batkai S、Kunos、G、The endocannabinoid system as an emerging target of pharmacotherapy、Pharmacol Rev.2006、58(3):389−462)。
【0060】
前記化合物は、COX−2選択的阻害剤を含めて、他の薬物、例えばオピオイドおよび非ステロイド性抗炎症薬(NSAID)などの鎮痛薬と併せて使用することもできる。
【0061】
本発明の化合物は、症状を軽減するのに十分な量および十分な時間でヒトに投与することができる。例示として、ヒトへの投与量は、体重1kg当たり0.001−50mgの範囲内、好ましくは体重1kg当たり0.01−20mgの投与量であってよい。
【図面の簡単な説明】
【0062】
【図1】ラットにおける神経障害誘発機械的アロディニアにおける化合物12の急性経口投与の影響を示す。
【発明を実施するための形態】
【0063】
本発明を以下の実施例によって例示する。
【実施例】
【0064】
略語:Boc:tert−ブトキシカルボニル;CDCl3:クロロホルム−d;DBU:1,8−ジアザビシクロ(5.4.0)ウンデス−7−エン;CDI:N,N’−カルボニルジイミダゾール;DCCI:1,3−ジシクロヘキシルカルボジイミド;DCM:ジクロロメタン;DIPEA:N,N−ジイソプロピルエチルアミン;DMAP:4−ジメチルアミノピリジン;DMF:N,N−ジメチルホルムアミド;Et3NまたはTEA:トリエチルアミン;Gly:グリシニル;HPLC:高速液体クロマトグラフィー;HOAc:酢酸;HOBt:1−ヒドロキシベンゾ−トリアゾール;MeOH:メタノール;Me3SiClまたはTMSCl:クロロトリメチルシラン;MS:質量スペクトル;(PPh3)4Pd:テトラキス(トリフェニルホスフィン)パラジウム(0);PyBOP:(ベンゾトリアゾール−1−イルオキシ)トリピロリジノホスホニウムヘキサフルオロホスフェート;PyBrOP:ブロモ(トリスピロリジノ)ホスホニウムテトラフルオロホスフェート;TBTU:((ベンゾトリアゾール−1−イルオキシ)−ジメチルアミノ−メチレン)−ジメチル−アンモニウムテトラフルオロボレート;TFA:トリフルオロ酢酸;THF:テトラヒドロフラン;TLC:薄層クロマトグラフィー。
【0065】
化合物の名称は、Cambridgesoft’s Chemdraw Ultra、バージョン9.0.7.で作成した。
【0066】
実施例1
1−(4−ブロモベンジル)−3−イソブチルイミダゾリジン−2,4−ジオン
【0067】
【化10】
i)4−ブロモベンズアルデヒド(100g、0.54mol)およびグリシンアミド塩酸塩(54g、0.48mol)のメタノール/水(1500ml、5.5/1)中溶液に、水酸化ナトリウム(21.6g、0.54mol)を添加した。17時間室温で撹拌した後、反応混合物を0℃に冷却した。水素化ホウ素ナトリウム(38g、1.0mol)を添加し、清澄な液が得られるまで混合物を撹拌した。pH=3まで濃塩酸を添加することによって、反応物をクエンチした。17時間撹拌した後、混合物を飽和炭酸水素ナトリウム水溶液で中和し、生成物をジクロロメタン中に抽出した。合わせた有機相を水、ブラインで洗浄し、硫酸ナトリウム上で乾燥させ、減圧下で濃縮することにより、2−(4−ブロモベンジルアミノ)アセトアミド(92g)が得られた。
【0068】
ii)前のステップで得られた生成物(60.6g、0.25mol)のアセトニトリル(1500ml)中溶液に、CDI(81g、0.5mol)DMAP(61g、0.5mol)を添加した。16時間60℃で撹拌した後、溶液を室温に冷却し、2M塩酸水溶液中に注いだ。生成物を酢酸エチル中に抽出し、合わせた有機相を水、ブラインで洗浄し、硫酸ナトリウム上で乾燥させ、減圧下で濃縮した。残留している固体をアセトンとともに撹拌した。濾過により、1−(4−ブロモベンジル)イミダゾリジン−2,4−ジオン(45g)を白色固体として得た。生成物をさらに精製することなく以下のステップで使用した。
【0069】
iii)前のステップで得られた生成物(10g、37.2mmol)のDMF(90ml)中溶液に、炭酸カリウム(15.4g、111mmol)および1−ブロモ−2−メチルプロパン(8.08ml、74.3mmol)を室温で添加した。50℃にて窒素雰囲気下で17時間撹拌した後、反応混合物を室温に冷却し、濾過した。清澄な液を減圧下で濃縮した。カラムクロマトグラフィーにより、1−(4−ブロモベンジル)−3−イソブチルイミダゾリジン−2,4−ジオン(10.9g)を白色固体として得た。
【0070】
実施例2
実施例1に記載されているものと類似の手順に従って、以下の化合物を調製した。
2A:1−(4−ブロモベンジル)−3−メチルイミダゾリジン−2,4−ジオン
1H NMR(400MHz,CDCl3):δ7.50(d,J=8.61Hz,2H)、7.14(d,J=8.61Hz,2H)、4.52(s,2H)、3.73(s,2H)、3.06(s,3H)。
2B:1−(4−ブロモベンジル)−3−エチルイミダゾリジン−2,4−ジオン
1H NMR(400MHz,CDCl3):δ7.50(d,J=8.22Hz,2H)、7.13(d,J=8.22Hz,2H)、4.52(s,2H)、3.71(s,2H)、3.59(q,J=7.43Hz,2H)、1.24(t,J=7.43Hz,3H)。
2C:1−(4−ブロモベンジル)−3−プロピルイミダゾリジン−2,4−ジオン
1H NMR(400MHz,CDCl3):δ7.49(d,J=8.22Hz,2H)、7.13(d,J=8.22Hz,2H)、4.51(s,2H)、3.72(s,2H)、3.50(dd J=7.43および6.26Hz,2H)、1.72−1.60(m,2H)、0.93(t,J=7.43Hz,3H)。
2D:1−(4−ブロモベンジル)−3−(2,2−ジフルオロエチル)イミダゾリジン−2,4−ジオン
1H NMR(400MHz,CDCl3):δ7.51(d,J=8.22Hz,2H)、7.14(d,J=8.22Hz,2H)、6.21−5.87(tt,J=55.95,4.70および4.30Hz,1H)、4.53(s,2H)、3.90(td,J=13.70および4.30Hz,2H)、3.80(s,2H)。
2E:1−(4−ブロモベンジル)−3−(シクロプロピルメチル)イミダゾリジン−2,4−ジオン
1H NMR(400MHz,CDCl3):δ7.50(d,J=8.22Hz,2H)、7.15(d,J=8.22Hz,2H)、4.53(s,2H)、3.74(s,2H)、3.56(d,J=7.44Hz,2H)、1.23−1.12(m,1H)、0.54−0.45(m,2H)、0.38−0.32(m,2H)。
2F:1−(4−ブロモベンジル)−3−(シクロブチルメチル)イミダゾリジン−2,4−ジオン
1H NMR(400MHz,CDCl3):δ7.49(d,J=8.22Hz,2H)、7.13(d,J=8.22Hz,2H)、4.50(s,2H)、3.71(s,2H)、3.57(d,J=7.43Hz,2H)、2.75−2.62(m,1H)、2.08−1.97(m,2H)、1.92−1.70(m,4H)。
2G:1−(4−ブロモ−ベンジル)−3−(2−メトキシエチル)イミダゾリジン−2,4−ジオン
1H NMR(400MHz,CDCl3):δ7.49(d,J=8.22Hz,2H)、7.14(d,J=8.22Hz,2H)、4.52(s,2H)、3.77−3.72(m,4H)、3.60(t,J=5.87Hz,2H)、3.36(s,3H)。
2H:メチル2−(3−(4−ブロモベンジル)−2,5−ジオキソイミダゾリジン−1−イル)アセテート
1H NMR(400MHz,CDCl3):δ7.50(d,J=8.22Hz,2H)、7.15(d,J=8.22Hz,2H)、4.55(s,2H)、4.30(s,2H)、3.83(s,2H)、3.78(s,3H)。
2L:1−(4−ブロモ−ベンジル)−3−(2−オキソプロピル)イミダゾリジン−2,4−ジオン
1H NMR(400MHz,CDCl3):δ7.51(d,J=8.61Hz,2H)、7.15(d,J=8.61Hz,2H)、4.54(s,2H)、4.35(s,2H)、3.83(s,2H)、2.25(s,3H)。
【0071】
実施例3
1−(4−ブロモベンジル)−3−イソプロピルイミダゾリジン−2,4−ジオン
【0072】
【化11】
i)グリシン(8.11g、108mmol)の水(40ml)中溶液に、水酸化ナトリウム水溶液(108mmol、15ml)および4−ブロモベンズアルデヒド(20g、108mmol)のメタノール(240ml)中溶液を添加した。室温で30分撹拌した後、水素化ホウ素ナトリウム(4.09g、108mmol)を、この懸濁液に少量ずつ添加した。室温で18時間撹拌した後、反応混合物を減圧下で濃縮し、生じた水性相をジエチルエーテルで洗浄した。2M塩酸水溶液の添加によって、水性相を中和した。生じた沈殿物を濾過によって回収し、水およびジエチルエーテルで洗浄した。白色固体を乾燥させることにより、2−(4−ブロモベンジルアミノ)酢酸(14.2g)を得た。生成物をさらに精製することなく以下のステップで使用した。
【0073】
ii)前のステップで得られた生成物(1.7g、6.96mmol)のジクロロメタン(20ml)中懸濁液に、トリエチルアミン(1.94ml、13.9mmol)、イソプロピルアミン(0.65ml、7.66mmol)およびo−(7−アザベンゾトリアゾール−1−イル)−1,1,3,3−テトラメチルウロニウムヘキサフルオロホスフェート(3.18g、8.36mmol)を添加した。室温で17時間撹拌した後、反応混合物を減圧下で濃縮した。カラムクロマトグラフィーにより、2−(4−ブロモベンジルアミノ)−N−イソプロピルアセトアミド(1.95g)を得た。
【0074】
iii)前のステップで得られた生成物(1.95g、6.96mmol)のアセトニトリル中溶液に、(ジイミダゾール−1−イル)ケトン(2.26g、13.9mmol)および4−ジメチルアミノピリジン(1.70g、13.9mmol)を添加した。60℃で17時間撹拌した後、反応混合物を室温に冷却し、飽和炭酸水素ナトリウム水溶液の添加によってクエンチした。生成物を酢酸エチル中に抽出し、合わせた有機相を水、ブラインで洗浄し、硫酸ナトリウム上で乾燥させた。カラムクロマトグラフィーにより、標題化合物1−(4−ブロモベンジル)−3−イソプロピルイミダゾリジン−2,4−ジオン(1.8g)を淡黄色油として得た。
1H NMR(400MHz,CDCl3):δ7.50(d,J=8.61Hz,2H)、7.13(d,J=8.61Hz,2H)、4.49(s,2H)、4.38−4.30(m,1H)、1.43(d,J=6.65Hz,6H)。
【0075】
実施例4
実施例3に記載されているものと類似の手順に従って、以下の化合物を調製した。
4A:1−(4−ブロモベンジル)−3−シクロプロピルイミダゾリジン−2,4−ジオン
1H NMR(400MHz,CDCl3):δ7.50(d,J=8.61Hz,2H)、7.14(d,J=8.61Hz,2H)、4.48(s,2H)、3.66(s,2H)、2.65−2.56(m,1H)、0.97(d,J=5.87Hz,4H)。
4B:1−(4−ブロモベンジル)−3−シクロブチルイミダゾリジン−2,4−ジオン
1H NMR(400MHz,CDCl3):δ7.49(d,J=8.22Hz,2H)、7.14(d,J=8.22Hz,2H)、4.60−4.51(m,1H)、4.49(s,2H)、3.65(s,2H)、2.95−2.82(m,2H)、2.23−2.13(m,2H)、1.91−1.81(m,1H)、1.79−1.65(m,1H)。
4C:1−(4−ブロモベンジル)−3−(2,2,2−トリフルオロエチル)イミダゾリジン−2,4−ジオン
1H NMR(400MHz,CDCl3):δ7.52(d,J=8.61Hz,2H)、7.15(d,J=8.61Hz,2H)、4.55(s,2H)、4.16(q,J=8.61Hz,2H)、3.83(s,2H)。
4D:1−(4−ブロモベンジル)−3−(1−シクロプロピルエチル)イミダゾリジン−2,4−ジオン
1H NMR(400MHz,CDCl3):δ7.54(d,J=8.61Hz,2H)、7.20(d,J=8.61Hz,2H)、4.72(s,2H)、3.87(s,2H)、3.46−3.36(m,1H)、1.22(d,J=6.65Hz,3H)、0.85−0.79(m,1H)、0.59−0.43(m,2H)、0.38−0.21(m,2H)。
4E:(S)−1−(4−ブロモベンジル)−3−(1−メトキシプロパン−2−イル)イミダゾリジン−2,4−ジオン
1H NMR(400MHz,CDCl3):δ7.49(d,J=8.61Hz,2H)、7.13(d,J=8.61Hz,2H)、4.50(d,J=3.91Hz,2H)、4.46−4.39(m,1H)、3.69(s,2H)、3.34(s,3H)、1.18(d,J=7.04Hz,3H)。
4F:1−(4−ブロモベンジル)−3−(テトラヒドロフラン−3−イル)イミダゾリジン−2,4−ジオン
1H NMR(400MHz,CDCl3):δ7.50(d,J=8.61Hz,2H)、7.14(d,J=8.61Hz,2H)、4.78−4.69(m,1H)、4.50(s,2H)、4.19−4.11(m,1H)、4.03−3.84(m,3H)、3.70(s,2H)、2.41−2.32(m,1H)、2.26−2.14(m,1H)。
4G:1−(4−ブロモベンジル)−3−(オキサゾール−5−イルメチル)イミダゾリジン−2,4−ジオン
1H NMR(400MHz,CDCl3):δ7.83(s,1H)、7.50(d,J=8.61Hz,2H)、7.15−7.11(m,3H)、4.77(s,2H)、4.52(s,2H)、3.77(s,2H)。
【0076】
実施例5
2−ブロモ−6−(ピペリジン−1−イルメチル)ピリジン
【0077】
【化12】
i)6−ブロモピリジン−2−カルバルデヒド(25g、135mmol)のジクロロメタン(500ml)中溶液に、ピペリジン(12.6g、149mmol)を10℃でゆっくり添加した。15分間10℃で撹拌した後、酢酸(8.9g、149mmol)を添加し、続いて水素化トリアセトキシホウ素ナトリウムを少量ずつ添加する一方、温度を5−10℃に保持した。2時間室温で撹拌した後、反応混合物を飽和炭酸水素ナトリウム水溶液中に注いだ。生成物をジクロロメタン中に抽出し、合わせた有機相をブラインで洗浄し、硫酸ナトリウム上で乾燥させ、減圧下で濃縮した。カラムクロマトグラフィーにより、2−ブロモ−6−(ピペリジン−1−イルメチル)ピリジン(30g)を無色油として得た。
1H NMR(400MHz,CDCl3):δ7.54−7.43(m,2H)、7.33(d,J=7.43Hz,1H)、3.60(s,2H)、2.48−2.38(m,4H)、1.63−1.54(m,4H)、1.48−1.39(m,2H)。
【0078】
実施例6
実施例5に記載されているものと類似の手順に従って、以下の化合物を調製した。
6A:4−((6−ブロモピリジン−2−イル)メチル)チオモルホリン1,1−ジオキシド
1H NMR(400MHz,CDCl3):δ7.61−7.54(dd,J=7.83および7.43Hz,1H)、7.42(d,J=7.83Hz,1H)、7.39(d,J=7.43Hz,1H)、3.81(s,2H)、3.14−3.04(m,8H)。
6B:4−((6−ブロモピリジン−2−イル)メチル)モルホリン
1H NMR(400MHz,CDCl3):δ7.53(dd,J=7.83および7.43Hz,1H)、7.45(d,J=7.83Hz,1H)、7.37(d,J=7.43Hz,1H)、3.76−3.71(m,4H)、3.65(s,2H)、2.55−2.49(m,4H)。
6C:2−ブロモ−6−((3−メチルピペリジン−1−イル)メチル)ピリジン
1H NMR(400MHz,CDCl3):δ7.47−7.37(m,2H)、7.26(d,J=7.43Hz,1H)、3.54(s,2H)、2.76−2.64(m,2H)、1.92(td,J=10.96および3.52Hz,1H)、1.68−1.44(m,5H)、0.87−0.72(m,4H)。
6D:2−ブロモ−6−((3,3−ジフルオロピペリジン−1−イル)メチル)ピリジン
1H NMR(400MHz,CDCl3):δ7.58−7.49(m,2H)、7.47(d,J=7.43Hz,1H)、3.74(s,2H)、2.71(dd,J=11.35および10.96Hz,2H)、2.54(dd,J=5.48および5.09Hz,2H)、1.97−1.84(m,2H)、1.83−1.76(m,2H)。
6E:2−ブロモ−6−((3−フルオロピペリジン−1−イル)メチル)ピリジン
1H NMR(400MHz,CDCl3):δ7.53(dd,J=7.83および7.43Hz,1H)、7.47(d,J=7.43Hz,1H)、7.36(d,J=7.83Hz,1H)、4.76−4.56(m,1H)、3.68(s,2H)、2.82−2.71(m,1H)、2.60−2.47(m,2H)、2.43−2.36(m,1H)、1.93−1.78(m,2H)、1.73−1.50(m,2H)。
6F:2−ブロモ−6−((3−トリフルオロメチルピペリジン−1−イル)メチル)ピリジン
(m/z)=324(M+H)+
6G:1−((6−ブロモピリジン−2−イル)メチル)−ピペリジン−3−オン
1H NMR(400MHz,CDCl3):δ7.54(dd,J=7.83および7.43Hz,1H)、7.41(d,J=7.43Hz,1H)、7.38(d,J=7.83Hz,1H)、3.73(s,2H)、3.08(s,2H)、2.73(dd,J=5.48Hz,2H)、2.39(dd,J=7.04Hz,2H)、2.03−1.94(m,2H)。
6H:N−((6−ブロモピリジン−2−イル)メチル)テトラヒドロ−2H−ピラン−4−アミン
1H NMR(400MHz,CDCl3):δ7.52(dd,J=7.83および7.43Hz,1H)、7.37(d,J=7.83Hz,1H)、7.31(d,J=7.43Hz,1H)、4.02−3.96(m,2H)、3.93(s,2H)、3.39(td,J=11.74および2.35Hz,2H)、2.78−2.69(m,1H)、1.90−1.82(m,2H)、1.55−1.42(m,2H)。
6I:((6−ブロモピリジン−2−イル)メチル)シクロヘキシルアミン
1H NMR(400MHz,CDCl3):δ7.50(dd,J=7.83および7.43Hz,1H)、7.34(d,J=7.83Hz,1H)、7.31(d,J=7.43Hz,1H)、3.90(s,2H)、2.50−2.41(m,1H)、1.96−1.88(m,2H)、1.78−1.69(m,3H)、1.65−1.57(m,1H)、1.31−1.06(m,5H)。
6J:N−((6−ブロモピリジン−2−イル)メチル)テトラヒドロ−2H−ピラン−3−アミン
1H NMR(400MHz,CDCl3):δ7.51(dd,J=7.83および7.43Hz,1H)、7.36(d,J=7.83Hz,1H)、7.31(d,J=7.43Hz,1H)、3.96−3.89(m,3H)、3.83(m,1H)、3.47−3.39(m,1H)、3.27−3.20(dd,J=8.61および8.22Hz,1H)、2.71−2.63(m,1H)、2.04−1.96(m,1H)、1.76−1.43(m,3H)、1.48−1.37(m,1H)。
6K:4−((6−ブロモピリジン−2−イル)メチル)チオモルホリン
1H NMR(400MHz,CDCl3):δ7.53(dd,J=7.83および7.43Hz,1H)、7.42(d,J=7.83Hz,1H)、7.37(d,J=7.43Hz,1H)、3.67(s,2H)、2.84−2.66(m,8H)。
【0079】
実施例7
実施例5に記載されているものと類似の手順に従って、6−ブロモ−3−フルオロピリジン−2−カルバルデヒドを出発原料として使用し、以下の化合物を調製した。
7A:4−((6−ブロモ−3−フルオロピリジン−2−イル)メチル)チオモルホリン1,1−ジオキシド
1H NMR(400MHz,CDCl3):δ7.45(dd,J=8.22および3.52Hz,1H)、7.32(d,J=8.61および8.22Hz,1H)、3.89(d,J=2.74Hz,2H)、3.17(m,8H)。
7B:4−((6−ブロモ−3−フルオロピリジン−2−イル)メチル)モルホリン
1H NMR(400MHz,CDCl3):δ7.41(dd,J=8.22および3.52Hz,1H)、7.28(d,J=8.61および8.22Hz,1H)、3.73−3.69(m,6H)、2.61−2.54(m,4H)。
7C:6−ブロモ−3−フルオロ−2−((3−メチルピペリジン−1−イル)メチル)ピリジン
(m/z)=288(M+H)+
7D:6−ブロモ−3−フルオロ−2−(ピペリジン−1−イルメチル)ピリジン
1H NMR(400MHz,CDCl3):δ7.38(dd,J=8.22および3.52Hz,1H)、7.26(d,J=8.61および8.22Hz,1H)、3.69(d,J=2.74Hz,2H)、2.54−2.46(m,4H)、1.61−1.53(m,4H)、1.45−1.36(m,2H)。
7E:6−ブロモ−2−((3,3−ジメチルピペリジン−1−イル)メチル)−3−フルオロピリジン
1H NMR(400MHz,CDCl3):δ7.37(dd,J=8.22および3.52Hz,1H)、7.25(d,J=8.61および8.22Hz,1H)、3.66(d,J=2.35Hz,2H)、2.41(bs,2H)、2.10(bs,2H)、1.60−1.53(m,2H)、1.22−1.13(m,2H)、0.90(s,6H)。
7F:6−ブロモ−2−((3,3−ジフルオロピペリジン−1−イル)メチル)−3−フルオロピリジン
1H NMR(400MHz,CDCl3):δ7.41(dd,J=8.22および3.52Hz,1H)、7.29(d,J=8.61および8.22Hz,1H)、3.86(d,J=2.35Hz,2H)、2.79(dd,J=11.35および10.95Hz,2H)、2.59(dd,J=5.48および5.09Hz,2H)、1.91−1.72(m,4H)。
7G:6−ブロモ−3−フルオロ−2−((3−フルオロピペリジン−1−イル)メチル)ピリジン
1H NMR(400MHz,CDCl3):δ7.40(dd,J=8.61および3.52Hz,1H)、7.28(d,J=8.61および8.22Hz,1H)、4.72−4.53(m,1H)、3.78(bs,2H)、2.97−2.86(m,1H)、2.60−2.57(m,1H)、2.55−2.47(m,1H)、2.42−2.35(m,1H)、1.92−1.75(m,2H)、1.62−1.47(m,2H)。
7H:6−ブロモ−3−フルオロ−2−((3−トリフルオロメチルピペリジン−1−イル)メチル)ピリジン
(m/z)=342(M+H)+
7I:N−((6−ブロモ−3−フルオロピリジン−2−イル)メチル)テトラヒドロ−2H−ピラン−4−アミン
1H NMR(400MHz,CDCl3):δ7.37(dd,J=8.61および3.52Hz,1H)、7.26(d,J=8.61および8.22Hz,1H)、4.04−3.95(m,4H)、3.40(td,J=11.73および1.96Hz,2H)、2.76−2.66(m,1H)、1.90−1.81(m,2H)、1.56−1.42(m,2H)。
7J:N−((6−ブロモ−3−フルオロピリジン−2−イル)メチル)シクロヘキサンアミン
1H NMR(400MHz,CDCl3):δ7.35(dd,J=8.61および3.52Hz,1H)、7.24(d,J=8.61および8.22Hz,1H)、3.96(d,J=2.35Hz,2H)、2.50−2.40(m,1H)、1.98−1.85(m,3H)、1.79−1.70(m,2H)、1.65−1.58(m,1H)、1.32−1.09(m,5H)。
7K:1−((6−ブロモ−3−フルオロピリジン−2−イル)メチル)ピペリジン−3−カルボニトリル
1H NMR(400MHz,CDCl3):δ7.41(dd,J=8.61および3.52Hz,1H)、7.29(d,J=8.61および8.22Hz,1H)、3.77(d,J=2.35Hz,2H)、2.96−2.87(m,1H)、2.82−2.72(m,1H)、2.71−2.63(m,1H)、2.62−2.53(m,1H)、2.46−2.36(m,1H)、1.93−1.71(m,2H)、1.64−1.52(m,2H)。
7L:4−((6−ブロモ−3−フルオロピリジン−2−イル)メチル)チオモルホリン
1H NMR(400MHz,CDCl3):δ7.40(dd,J=8.61および3.52Hz,1H)、7.32(d,J=8.61および8.22Hz,1H)、3.75(d,J=2.74Hz,2H)、2.87−2.81(m,4H)、2.71−2.65(m,4H)。
【0080】
実施例8
実施例5に記載されているものと類似の手順に従って、2−ブロモ−ピリジン−4−カルバルデヒドを出発原料として使用し、以下の化合物を調製した。
2−ブロモ−4−(ピペリジン−1−イルメチル)ピリジン。
1H NMR(400MHz,CDCl3):δ8.27(d,J=5.09,1H)、7.48(s,1H)、7.23(d,J=5.09Hz,1H)、3.93(s,2H)、2.41−2.31(m,4H)、1.63−1.55(m,4H)、1.50−1.39(m,2H)。
【0081】
実施例9
6−ブロモ−3−フルオロ−2−((テトラヒドロ−2H−ピラン−4−イルオキシ)メチル)ピリジン
【0082】
【化13】
i)6−ブロモ−3−フルオロ−2−メチルピリジン(0.5gr、2.63mmol)のジクロロメタン(5ml)中溶液に、室温で、N−ブロモコハク酸イミド(937mg、5.26mmol)およびアゾ−ジ−イソブチロニトリル(86mg、0.526mmol)を添加した。55℃で17時間撹拌した後、反応混合物を水の添加によってクエンチし、生成物をジクロロメタン中に抽出した。合わせた有機相をブラインで洗浄し、硫酸ナトリウム上で乾燥させ、減圧下で濃縮した。カラムクロマトグラフィーにより、6−ブロモ−2−(ブロモメチル)−3−フルオロピリジン(388mg)を清澄な油として得た。
【0083】
ii)前のステップで得られた生成物(388mg、1.44mmol)およびテトラヒドロ−2H−ピラン−4−オール(0.206ml、2.16mmol)のテトラヒドロフラン(10ml)中溶液に、水素化ナトリウム(69.3mg、2.31mmol、油中に80%分散)を添加した。室温で1時間撹拌した後、反応混合物を水の添加によってクエンチし、生成物をジクロロメタン中に抽出した。合わせた有機相をブラインで洗浄し、硫酸ナトリウム上で乾燥させ、減圧下で濃縮した。カラムクロマトグラフィーにより、標題化合物6−ブロモ−3−フルオロ−2−((テトラヒドロ−2H−ピラン−4−イルオキシ)メチル)ピリジン(177mg)を清澄な油として得た。
1H NMR(400MHz,CDCl3):δ7.44(dd,J=8.61および3.52Hz,1H)、7.31(d,J=8.61および8.22Hz,1H)、4.67(d,J=2.35Hz,2H)、3.99−3.92(m,2H)、3.72−3.64(m,1H)、3.49−3.41(m,2H)、2.00−1.91(m,2H)、1.71−1.60(m,2H)。
【0084】
実施例10
2−ブロモ−6−(シクロヘキシルメチル)ピリジン
i)2,6−ジブロモピリジン(1.37g、5.8mmol)のTHF/ヘキサン/ジエチルエーテル(1/1/3、15ml)中溶液を、窒素雰囲気下で、2.5Mのn−ブチルリチウムのヘキサン(2.43ml、6.08mmol)中溶液に−78℃で滴下により添加した。
【0085】
10分撹拌した後、シクロヘキサンカルボニトリル(633mg、5.8mmol)のTHF/ヘキサン/ジエチルエーテル(1/1/3、4ml)中溶液を添加し、反応混合物を2.5時間−78℃で撹拌した。反応混合物を室温に温め、さらに1.5時間撹拌した。反応混合物を、2M硫酸水溶液(7ml)の添加によってクエンチした。2時間激しく撹拌した後、水を添加し、生成物をジエチルエーテル中に抽出した。合わせた有機相を飽和炭酸水素ナトリウム水溶液、ブラインで洗浄し、硫酸ナトリウム上で乾燥させ、減圧下で濃縮した。カラムクロマトグラフィーにより、(6−ブロモ−ピリジン−2−イル)(シクロヘキシル)メタノン(800mg)を清澄な油として得た。
【0086】
ii)前のステップで得られた生成物(600mg、2.24mmol)および4−メチルベンゼンスルホニルヒドラジド(458mg、2.46mmol)のエタノール(2ml)中懸濁液を、1000Cに15分間マイクロ波中で加熱した。室温に冷却した後、反応混合物を減圧下で濃縮した。カラムクロマトグラフィーにより、N’−((6−ブロモピリジン−2−イル)(シクロヘキシル)メチレン)−4−メチルベンゼンスルホノヒドラジド(608mg)を白色固体として得た。
【0087】
iii)前のステップで得られた生成物(600mg、1.38mmol)のジクロロメタン4ml中溶液に、20%水素化ジイソブチルアルミニウムのトルエン(0.97g、13.8mmol)中溶液をゆっくり添加した。室温で17時間撹拌した後、pH=10まで2M水酸化ナトリウム水溶液をゆっくり添加するすることによって、反応混合物をクエンチした。生成物を酢酸エチル中に抽出し、合わせた有機相を水、ブラインで洗浄し、硫酸ナトリウム上で乾燥させ、減圧下で濃縮した。カラムクロマトグラフィーにより、標題化合物2−ブロモ−6−(シクロヘキシルメチル)ピリジンを白色固体として得た。
1H NMR(400MHz,CDCl3):δ7.42−7.21(m,3H)、3.62(d,J=7.04Hz,2H)、1.81−0.85(m,11H)。
【0088】
実施例11
2−ブロモ−6−(ジフルオロ−(テトラヒドロ−2H−ピラン−4−イル)メチル)ピリジン
【0089】
【化14】
i)(6−ブロモ−ピリジン−2−イル)−(テトラヒドロ−2H−ピラン−4−イル)メタノンを、実施例10、ステップi)に記載されているものと類似の手順に従って、テトラヒドロ−2H−ピラン−4−カルボニトリルを出発原料として使用して調製した。
【0090】
ii)前のステップで得られた生成物(200mg、0.74mmol)のジクロロメタン(2ml)中溶液に、少量ずつ6日の期間をかけて、ジエチルアミノサルファ−トリ−フルオリド(1.49g、9.25mmol)を窒素雰囲気下で添加した。完了後、反応混合物をメタノールの添加によって慎重にクエンチし、生成物を酢酸エチル中に抽出した。合わせた有機相を水、ブラインで洗浄し、硫酸ナトリウム上で乾燥させ、減圧下で濃縮した。カラムクロマトグラフィーにより、標題化合物2−ブロモ−6−(ジフルオロ−(テトラヒドロ−2H−ピラン−4−イル)メチル)ピリジン(144mg)を清澄な油として得た。
1H NMR(400MHz,CDCl3):δ7670(t,J=7.83Hz,1H)、7.60−7.54(m,3H)、4.03(dd,J=11.35および4.30Hz,2H)、3.42(td,J=11.74および2.35Hz,2H)、2.80−2.63(m,1H)、1.72(m,4H)。
【0091】
実施例12
3−イソブチル−1−(4−(6−(ピペリジン−1−イルメチル)ピリジン−2−イル)ベンジル)イミダゾリジン−2,4−ジオン
【0092】
【化15】
i)1−(4−ブロモベンジル)−3−イソブチルイミダゾリジン−2,4−ジオン(実施例1、ステップi))(2.0g、6.2mmol)、ビス(ピナコレート)ジボロン(1.6g、6.2mmol)および酢酸カリウム(1.8g、18.5mmol)のDMF(50ml)中溶液に、窒素雰囲気下で、1,1’−ビス(ジフェニルホスフィノ)フェロセンジクロロパラジウム(II)(134mg、0.18mmol)を添加した。75℃で17時間撹拌した後、反応混合物を室温に冷却した。水を添加し、生成物を酢酸エチル中に抽出した。合わせた有機相を飽和炭酸水素ナトリウム水溶液、水、ブラインで洗浄し、硫酸ナトリウム上で乾燥させ、減圧下で濃縮することにより、3−イソブチル−1−(4−(4,4,5,5−テトラメチル−(1,3,2)ジオキサボロラン−2−イル)ベンジル)イミダゾリジン−2,4−ジオン(6.8g)が黒色油として得られた。生成物をさらに精製することなく以下のステップで使用した。
【0093】
ii)前のステップで得られた生成物(1.31g、3.52mmol)および2−ブロモ−6−(ピペリジン−1−イルメチル)ピリジン(実施例5)(748mg、2.93mmol)のトルエン/エタノール(4/1、25ml)中溶液に、2M炭酸カリウム水溶液を添加した。窒素雰囲気下で15分撹拌した後、テトラキス(トリフェニルホスフィン)パラジウム(0)(85mg、0.073mmol)を添加し、この混合物を17時間75℃にて窒素雰囲気下で撹拌した。完了後、混合物を室温に冷却し、デカライトを通して濾過した。水を濾液に添加し、生成物を酢酸エチル中に抽出した。合わせた有機相を水、ブラインで洗浄し、硫酸ナトリウム上で乾燥させ、減圧下で濃縮した。カラムクロマトグラフィーにより、3−イソブチル−1−(4−(6−(ピペリジン−1−イルメチル)ピリジン−2−イル)ベンジル)イミダゾリジン−2,4−ジオン(120mg)を白色固体として得た。
1H NMR(400MHz,CDCl3):δ8.00(d,J=8.22Hz,2H)、7.72(dd,J=8.22および7.83Hz,1H)、7.56(d,J=8.22Hz,1H)、7.43(d,J=7.83Hz,1H)、7.34(d,J=8.22Hz,2H)、4.62(s,2H)、3.74(s,2H)、3.72(s,2H)、3.36(d,J=7.43Hz,2H)、2.53−2.47(m,4H)、1.65−1.58(m,4H)、1.50−1.42(m,2H)、2.15−2.05(m,1H)、0.93(d,J=6.65Hz,6H)。
【0094】
実施例12に記載されているものと類似の手順に従って、以下の化合物を調製した。
【0095】
【表1】
【0096】
実施例89
1−(4−(6−(ピペリジン−1−イルメチル)ピリジン−2−イル)ベンジル)イミダゾリジン−2,4−ジオン
【0097】
【化16】
i)2−ブロモ−6−ピペリジン−1−イルメチル−ピリジン(実施例5)(8.0g、31.4mmol)および4−ホルミルフェニルボロン酸(6.1g、40.8mmol)のトルエン/エタノール(4/1、320ml)中溶液に、2M炭酸カリウム水溶液を添加した。窒素雰囲気下で15分撹拌した後、テトラキス(トリフェニルホスフィン)パラジウム(0)(1.2g、1.02mmol)を添加した。17時間80℃にて窒素雰囲気下で撹拌した後、混合物を室温に冷却し、デカライトを通して濾過した。水を添加し、生成物を酢酸エチル中に抽出した。合わせた有機相を水、ブラインで洗浄し、硫酸ナトリウム上で乾燥させ、減圧下で濃縮した。カラムクロマトグラフィーにより、4−(6−(ピペリジン−1−イルメチル)ピリジン−2−イル)ベンズアルデヒド(7.69g)を白色固体として得た。
【0098】
ii)グリシンメチルエステル.HCl(4.79g、38.1mmol)およびトリエチルアミン4.60ml、33.0mmol)のメタノール(70ml)中溶液を、前のステップで得られた生成物(7.12g、25.4mmol)のメタノール(70ml)中溶液に、窒素雰囲気下で滴下により添加した。1時間室温で撹拌した後、水素化トリアセトキシホウ素ナトリウム(12.9g、61.0mmol)を少量ずつ30分の間添加した。17時間撹拌した後、より多くのグリシンメチルエステル.HCl(1.6g、12.7mmol)を添加し、続いて水素化トリアセトキシホウ素ナトリウム(5.38g、25.4mmol)を添加した。さらに17時間撹拌した後、反応混合物を飽和炭酸水素ナトリウム水溶液の添加によってクエンチした。生成物をジクロロメタン中に抽出し、合わせた有機相を水、ブラインで洗浄し、相分離フィルターに通して濾過し、減圧下で濃縮した。カラムクロマトグラフィーにより、メチル2−(4−(6−(ピペリジン−1−イルメチル)ピリジン−2−イル)ベンジルアミノ)アセテート(3.48g)を白色固体としてを得た。
【0099】
iii)前のステップで得られた生成物(8.03g、22.7mmol)のジオキサン/水(1/1)(100ml)中懸濁液に、カリウムシアネート(2.76g、34.1mmol)を室温で添加した。20分撹拌した後、酢酸(4.16ml、72.7mmol)を添加し、混合物をさらに17時間室温で撹拌した。反応混合物を水の添加によってクエンチし、飽和炭酸水素ナトリウム水溶液の添加によってpH=9まで塩基性化した。生成物をジクロロメタン中に抽出し、合わせた有機相をブラインで洗浄し、相分離フィルターに通して濾過し、減圧下で濃縮することにより、メチル2−(1−(4−(6−(ピペリジン−1−イルメチル)ピリジン−2−イル)ベンジル)ウレイド)アセテート(8.2g)が油として得られた。生成物をさらに精製することなく以下のステップで使用した。
【0100】
iv)前のステップで得られた生成物(8.2g、20.68mmol)のメタノール(50ml)中溶液に、ナトリウムメトキシド(2.24g、41.4mmol)を室温で添加し、反応混合物を3時間室温にて窒素雰囲気下で撹拌した。反応混合物を水中に注ぎ、飽和塩化アンモニウム水溶液の添加によって中和した。生成物をジクロロメタン中に抽出し、合わせた有機相をブラインで洗浄し、硫酸ナトリウム上で乾燥させ、減圧下で濃縮した。カラムクロマトグラフィーにより、標題化合物1−(4−(6−(ピペリジン−1−イルメチル)ピリジン−2−イル)ベンジル)イミダゾリジン−2,4−ジオン(5.8g)を白色固体として得た。
1H NMR(400MHz,CDCl3):δ7.99(d,J=8.61Hz,2H)、7.72(dd,J=7.83および7.43Hz,1H)、7.57(d,J=7.83Hz,1H)、7.42(d,J=7.43Hz,1H)、7.34(d,J=8.61Hz,2H)、4.57(s,2H)、3.77(s,2H)、3.73(s,2H)、2.56−2.48(m,4H)、1.67−1.57(m,4H)、1.50−1.42(m,2H)。
【0101】
実施例90
1−(4−(6−(ピペリジン−1−イルメチル)ピリジン−2−イル)ベンジル)−3−(3,3,3−トリフルオロ−プロピル)イミダゾリジン−2,4−ジオン
【0102】
【化17】
i)1−(4−(6−ピペリジン−1−イルメチル−ピリジン−2−イル)ベンジル)イミダゾリジン−2,4−ジオン(実施例134)(120mg、0.33mmol)、炭酸カリウム(137mg、0.99mmol)および3−ブロモ−1,1,1−トリフルオロプロパン(117mg、0.66mmol)のDMF(2.5ml)中溶液を、17時間の間50℃で撹拌した。室温に冷却した後、反応混合物を水の添加によってクエンチした。生成物を酢酸エチル中に抽出し、合わせた有機相をブラインで洗浄し、硫酸ナトリウム上で乾燥させ、減圧下で濃縮した。カラムクロマトグラフィーにより、標題化合物(50mg)を白色固体として得た。
1H NMR(400MHz,CDCl3):δ7.99(d,J=8.61Hz,2H)、7.72(dd,J=7.83および7.43Hz,1H)、7.56(d,J=7.83Hz,1H)、7.44(d,J=7.43Hz,1H)、7.33(d,J=8.61Hz,2H)、4.62(s,2H)、3.83(t,J=7.04Hz,2H)、3.76(s,2H)、3.73(s,2H)、2.60−2.45(m,6H)、1.66−1.59(m,4H)、1.51−1.41(m,2H)。
【0103】
実施例91
実施例90に記載されているものと類似の手順に従って、以下の化合物を調製した。
91A:4−(2,5−ジオキソ−3−(4−(6−(ピペリジン−1−イルメチル)ピリジン−2−イル)ベンジル)イミダゾリジン−1−イル)ブタンニトリル
1H NMR(400MHz,CDCl3):δ8.00(d,J=8.61Hz,2H)、7.72(dd,J=7.83および7.43Hz,1H)、7.56(d,J=7.83Hz,1H)、7.43(d,J=7.43Hz,1H)、7.35(d,J=8.61Hz,2H)、4.62(s,2H)、3.78(s,2H)、3.72(s,2H)、3.69(t,J=6.65Hz,2H)、2.54−2.47(m,4H)、2.44(t,J=7.04Hz,2H)、2.05(m,2H)、1.65−1.56(m,4H)、1.50−1.40(m,2H)。
91B:(R)−メチル3−(2,5−ジオキソ−3−(4−(6−(ピペリジン−1−イルメチル)ピリジン−2−イル)ベンジル)イミダゾリジン−1−イル)−2−メチルプロパノエート
1H NMR(400MHz,CDCl3):δ7.99(d,J=8.22Hz,2H)、7.72(dd,J=7.83および7.43Hz,1H)、7.56(d,J=7.83Hz,1H)、7.43(d,J=7.43Hz,1H)、7.34(d,J=8.22Hz,2H)、4.61(s,2H)、3.83(dd,J=13.69および7.83Hz,1H)、3.75(s,2H)、3.72(s,2H)、3.69(s,3H)、3.63(dd,J=14.09および6.65Hz,1H)、3.00−2.90(m,1H)、2.54−2.46(m,4H)、1.65−1.54(m,4H)、1.51−1.41(m,2H)、1.20(d,J=7.04Hz,3H)。
91C:3−(オキセタン−2−イルメチル)−1−(4−(6−(ピペリジン−1−イルメチル)ピリジン−2−イル)ベンジル)イミダゾリジン−2,4−ジオン
1H NMR(400MHz,CDCl3):δ7.99(d,J=8.61Hz,2H)、7.72(dd,J=7.83および7.43Hz,1H)、7.56(d,J=7.83Hz,1H)、7.43(d,J=7.43Hz,1H)、7.34(d,J=8.61Hz,2H)、5.08−5.00(m,1H)、4.70−4.56(m,4H)、3.99−3.93(dd,J=14.08および7.43Hz,1H)、3.78−3.70(m,5H)、2.78−2.70(m,1H)、2.55−2.45(m,4H)、1.66−1.58(m,5H)、1.50−1.41(m,2H)。
91D:3−(2−オキソテトラヒドロフラン−3−イル)−1−(4−(6−(ピペリジン−1−イルメチル)ピリジン−2−イル)ベンジル)イミダゾリジン−2,4−ジオン
1H NMR(400MHz,CDCl3):δ8.00(d,J=8.61Hz,2H)、7.73(dd,J=7.83および7.43Hz,1H)、7.57(d,J=7.83Hz,1H)、7.44(d,J=7.43Hz,1H)、7.36(d,J=8.61Hz,2H)、4.97−4.90(m,1H)、4.65−4.56(m,5H)、4.39−4.31(m,1H)、3.82(s,2H)、3.73(s,2H)、2.84−2.72(m,1H)、2.59−2.47(m,5H)、1.66−1.57(m,4H)、1.50−1.42(m,2H)。
91E:1−(4−(6−(ピペリジン−1−イルメチル)ピリジン−2−イル)ベンジル)−3−(テトラヒドロフラン−2−イル)イミダゾリジン−2,4−ジオン
1H NMR(400MHz,CDCl3):δ7.99(d,J=8.22Hz,2H)、7.72(dd,J=7.83および7.43Hz,1H)、7.56(d,J=7.83Hz,1H)、7.43(d,J=7.43Hz,1H)、7.34(d,J=8.22Hz,2H)、4.65(d,J=15.26Hz,1H)、4.58(d,J=15.26Hz,1H)、4.30−4.22(m,1H)、3.96−3.88(m,1H)、3.81−3.64(m,6H)、3.50(dd,J=13.69および4.70Hz,1H)、2.55−2.45(m,4H)、2.08−1.84(m,3H)、1.72−1.57(m,5H)、1.50−1.42(m,2H)。
【0104】
実施例92
2−アミノ−N−シクロプロピルアセトアミドトリフルオロアセテート
i)TBTU(5.1g、16.5mmol)、DIPEA(2.9ml、16.5mmol)およびシクロプロピルアミン(2.2ml、33mmol)を、BOC−Gly−OH(2.63g、15mmol)の乾燥ジクロロメタン(10ml)中溶液に添加した。17時間撹拌した後、反応混合物を減圧下で濃縮し、水を残渣に添加した。生成物を酢酸エチル中に抽出した。合わせた有機相を2M塩酸水溶液、飽和炭酸水素ナトリウム水溶液、ブラインで洗浄し、硫酸ナトリウム上で乾燥させ、減圧下で濃縮することにより、2−tert−ブチル2−(シクロプロピルアミノ)−2−オキソエチルカルバメート(847mg)が得られた。生成物をさらに精製することなく以下のステップで使用した。
【0105】
ii)TFA(65ml、875mmol)を、前のステップで得られた生成物(34.9g、163mmol)のジクロロメタン(300ml)中溶液に添加した。17時間撹拌した後、反応混合物を減圧下で濃縮した。DCM/ジイソプロピルエーテルエーテルからの結晶化により、標題化合物2−アミノ−N−シクロプロピルアセトアミドトリフルオロアセテートが得られた。
(22.2g)。1H NMR(400MHz,MeOD):δ3.62(s,2H)、2.76−2.74(m,1H)、0.75(m,2H)、0.52(m,2H)
【0106】
実施例93
3−シクロプロピル−1−(3−フルオロ−4−(5−フルオロ−6−(ピペリジン−1−イルメチル)ピリジン−2−イル)ベンジル)イミダゾリジン−2,4−ジオン
i)3−フルオロ−4−(5−フルオロ−6−(ピペリジン−1−イルメチル)ピリジン−2−イル)ベンズアルデヒドを、実施例89、ステップi)に記載されているものと類似の手順に従って、2−フルオロ−4−ホルミルフェニルボロン酸を出発原料として使用して調製した。
【0107】
ii)前のステップで得られた生成物(1.3g、4.1mmol)のメタノール(60ml)中溶液に、0℃で、KOH(4.6mg、0.08mmol)および2−アミノ−N−シクロプロピル−アセトアミドトリフルオロアセテート(1.9g、8.22mmol)を添加した。30分後0℃で、トリアセトキシ水素化ホウ素ナトリウム(2.6g、12.3mmol)を添加した。室温で17時間撹拌した後、飽和炭酸水素ナトリウム溶液の添加によって反応混合物をクエンチし、生成物をジクロロメタン中に抽出した。合わせた有機相を水、ブラインで洗浄し、硫酸ナトリウム上で乾燥させ、減圧下で濃縮した。カラムクロマトグラフィーにより、N−シクロプロピル−2−(3−フルオロ−4−(5−フルオロ−6−(ピペリジン−1−イルメチル)ピリジン−2−イル)ベンジルアミノ)アセトアミド(0.6g)を得た。
【0108】
iii)実施例3、ステップiii)に記載されているものと類似の手順に従って、前のステップで得られた生成物(0.28g)を、標題化合物3−シクロプロピル−1−(3−フルオロ−4−(5−フルオロ−6−(ピペリジン−1−イルメチル)ピリジン−2−イル)ベンジル)イミダゾリジン−2,4−ジオン(43mg)に変換した。
1H NMR(400MHz,CDCl3):δ8.00(dd,J=8.22および7.83Hz,1H)、7.74−7.68(m,1H)、7.43(dd,J=9.00および8.61Hz,1H)、7.15(dd,J=8.22および1.96Hz,1H)、7.06(dd,J=11.74および1.57Hz,1H)、4.57(s,2H)、3.81(d,J=2.35Hz,2H)、3.71(s,2H)、2.68−2.53(m,5H)、1.65−1.50(m,4H)、1.46−1.38(m,2H)、1.00(s,2H)、0.98(s,2H)。
【0109】
実施例94
1−(3−フルオロ−4−(6−(ピペリジン−1−イルメチル)ピリジン−2−イル)ベンジル)−3−イソブチルイミダゾリジン−2,4−ジオン
i)3−フルオロ−4−(6−(ピペリジン−1−イルメチル)ピリジン−2−イル)ベンズアルデヒドを、実施例89、ステップi)に記載されているものと類似の手順に従って、2−フルオロ−4−ホルミルフェニルボロン酸を出発原料として調製した。
【0110】
ii)実施例93、ステップii)に記載されているものと類似の手順に従って、前のステップで得られた生成物(1g)を、グリシンアミド塩酸を出発原料として使用して、2−(3−フルオロ−4−(6−(ピペリジン−1−イルメチル)ピリジン−2−イル)ベンジルアミノ)−アセトアミド(0.9g)に変換した。
【0111】
iii)実施例3、ステップiii)に記載されているものと類似の手順に従って、前のステップで得られた生成物(0.9g)を、1−(3−フルオロ−4−(6−(ピペリジン−1−イルメチル)ピリジン−2−イル)ベンジル)イミダゾリジン−2,4−ジオン(0.49g)に変換した。
【0112】
iv)実施例90、ステップiに記載されているものと類似の手順に従って、前のステップで得られた生成物(0.49g)を、1−(3−フルオロ−4−(6−(ピペリジン−1−イルメチル)ピリジン−2−イル)ベンジル)−3−イソブチルイミダゾリジン−2,4−ジオン(52mg)に変換した。
1H NMR(400MHz,CDCl3):δ8.00(dd,J=8.22および7.83Hz,1H)、7.73(dd,J=7.83および7.43Hz,1H)、7.65−7.60(m,1H)、7.46(d,J=7.83Hz,1H)、7.14(dd,J=7.83および1.57Hz,1H)、7.05(dd,J=11.74および1.57Hz,1H)、4.60(s,2H)、3.77(s,2H)、3.71(s,2H)、3.37(d,J=7.43Hz,2H)、2.53−2.45(m,4H)、2.14−2.06(m,1H)、1.65−1.57(m,4H)、1.50−1.42(m,2H)、0.94(d,J=6.65Hz,6H)。
【0113】
実施例95
ヒトCB2トランスフェクトCHO細胞におけるアゴニスト誘導cAMP変化
アデニル酸シクラーゼアッセイを、ヒト組換えCB2受容体を安定に過剰発現するCHO細胞を使用して実施した。1%(v/v)ペニシリン/ストレプトマイシン(ギブコ15140−122)、10%ウシ胎児血清(FBS)および400μg/mlのジェネティシン(インビトロジェン10131−027)を含有するDMEM/HAMF12中で細胞を培養した。化合物および基準物(CP55,940)をDMSO中に溶解し、2μMのRolipram(シグマR6520)および1μMのフォルスコリン(シグマ、F3917)を含有する血清フリーの培養液中で希釈物を作製した。各希釈物10μlをアッセイプレート(384ウェル白色培養プレート、パーキンエルマー)に移動した。1%(v/v)ペニシリン/ストレプトマイシンを含有するDMEM/HAMF12中に5×105細胞/mlを含有する細胞懸濁液を、hCB2_C2−CHO細胞から調製し、これらのうち10μl(5,000細胞/ウェル)をアッセイプレートに移動し、細胞を45分間37℃でインキュベートした。均一時間分解蛍光(HTRF;CisBio)を、10μlのcAMP−XL665および10μlの抗cAMP(Eu)クリプテートを順次添加することによって読み出しとして使用し、室温で1時間インキュベーションした後、615nmおよび665nmでの蛍光をEnvision(パーキンエルマー)で測定した。結果は個々の化合物について665nm/615nm比から算出し、対照化合物について得られた値と比較した。実施例12−88、90、91、93および94からの化合物は、CB2に関してEC50≦1×10−7Mを有する。
【0114】
実施例95A
ヒトCB1トランスフェクトCHO細胞におけるアゴニスト誘導cAMP変化
アデニル酸シクラーゼアッセイを、ヒト組換えCB1受容体を安定に過剰発現するCHO細胞を使用して実施した。1%(v/v)ペニシリン/ストレプトマイシン(ギブコ15140−122)、10%ウシ胎児血清(FBS)、400μg/mlのジェネティシン(インビトロジェン10131−027)およびZeocine250μg/ml(インビトロジェン、45−0430)を含有するDMEM/HAMF12中で細胞を培養した。化合物および基準物(CP55,940)をDMSO中に溶解し、2μMのRolipram(シグマR6520)および1μMのフォルスコリン(シグマ、F3917)を含有する血清フリーの培養液中で希釈物を作製した。各希釈物10μlをアッセイプレート(384ウェル白色培養プレート、パーキンエルマー)に移動した。1%(v/v)ペニシリン/ストレプトマイシンを含有するDMEM/HAMF12中に5×105細胞/mlを含有する細胞懸濁液を、hCB1_A2−CHO細胞から調製し、これらのうち10μl(5,000細胞/ウェル)をアッセイプレートに移動し、細胞を45分間37℃でインキュベートした。均一時間分解蛍光(HTRF;CisBio)を、10μlのcAMP−XL665および10μlの抗cAMP(Eu)クリプテートを順次添加することによって読み出しとして使用し、室温で1時間インキュベーションした後、615nmおよび665nmでの蛍光をEnvision(パーキンエルマー)で測定した。結果は個々の化合物について665nm/615nm比から算出し、対照化合物について得られた値と比較した。実施例12−88、90、91、93および94からの化合物は、CB1に関してEC50≧1×10−7Mを有する。
【0115】
実施例96
神経因性疼痛のラット(Chung)モデル
このモデルにおいて、機械的アロディニアは、左L5脊髄神経の緊密な連結によって誘発される。このアッセイを用いることによって、神経因性疼痛の治療において臨床的に使用される抗痙攣薬(ガバペンチン)、抗うつ薬(デュロキセチン)およびオピオイド鎮痛薬(モルヒネ)の抗アロディニア効果を実証するのに成功した。
【0116】
雄性Wistarラット(外科手術時の体重228−301g)を前記試験に用いた。パースペックスボックス内の高くした(約40cm)メッシュ床の上にラットを置き、機械的刺激(較正フォンフライフィラメント)に対するラットの引き込み閾値を、上記した通り、増加する力(2.6−167mN)のフィラメントを使用して測定した。フォンフライフィラメントを足の足底表面に適用し、上げ下げ法を使用して閾値応答を決定した。足を鋭く引っ込めると陽性応答が認められた。15gのカットオフを試験の上限として選択した。ベースライン測定に続いて、各動物に麻酔し、L5脊髄神経を緊密に連結した。動物を少なくとも3日の期間外科手術から回復させた。薬物投与当日、足引き込み閾値を再び測定した(0分)。この読み取り直後、ラットにビヒクルまたは試験化合物を経口投与し、化合物投与後様々な時点で読み取りを測定した。
【0117】
データは平均±s.e.m.として示された。非パラメーター統計試験のKruskal−Wallis一元配置分散分析を使用して統計分析を行った。治療群のそれぞれを次いで、非パラメーターDunn試験を使用してビヒクル群に対して比較した。
【0118】
一例として、選択的CB2受容体アゴニスト3−イソブチル−1−(4−(6−(ピペリジン−1−イルメチル)ピリジン−2−イル)ベンジル)イミダゾリジン−2,4−ジオン(実施例12)の経口投与により、用量依存的様式(表1;図1)において薬物投与後120および180分でそれぞれ機械的アロディニアが減弱した。最小有効量(MED)は43.8μmol/kgであった。これらのデータは、選択的CB2受容体アゴニストが神経因性疼痛のラットモデルにおいて強力な経口抗アロディニア作用を保有していることを実証している。
【0119】
【表2】
【0120】
実施例142
ラットにおける機械的痛覚過敏
炎症性疼痛のこのラットモデルにおいて、炎症は完全フロイントアジュバント(CFA)の皮下注入によって後足に誘発される。伴われる機械的痛覚過敏を、足の機械的圧縮に対する足引き込み閾値(PWT)の低減を測定することによって定量化する。このアッセイを用いて、炎症性疼痛の治療において臨床的に使用される非ステロイド系抗炎症薬(インドメタシン)およびコキシブ(セレコキシブ)の抗痛覚過敏効果を実証するのに成功した。
【0121】
実験は、雄性Wistarラット体重(141−175g)を使用して行われた。簡潔には、後足の機械的圧縮に対するラットの足引き込み閾値(PWT)を、Randall−Sellito装置(Ugo Basile)を使用して測定した(ベースライン読み取り)。足に対する組織損傷を最小にするため、20gのカットオフを用いた。動物に次いでイソフルラン(1−3%)で軽く麻酔し、完全フロイントアジュバント(1つの足当たり0.1ml)を左後足の足底表面に皮下注入(s.c.)した。動物を次いでこれらのホームケージに戻し、放置して炎症を発生させた。CFA注入の24時間後、PWTを再び測定し(0分)、この読み取り直後、ラットにビヒクルまたは試験化合物(化合物12の4.4−43.8μmol/kg p.o.)のいずれかを経口投与した。次いで薬物投与の3時間後に読み取りを行った。
【0122】
データを平均±s.e.m.としてプロットし、非パラメーター統計試験のKruskal−Wallis一元配置分散分析を使用して群間で比較した。この試験で統計的有意性(P<0.05)が観察された場合に、非パラメーターDunn試験を使用してビヒクル群および治療群のそれぞれを比較した。機械的痛覚過敏の減弱パーセントを以下の通りに算出した。
【0123】
【数1】
【0124】
化合物12(4.4−43.8μmol/kg)の経口投与は、用量依存的様式においてCFAによって誘発された機械的痛覚過敏を逆転させた(表2)。Org266919−1のMEDは13.2μmol/kgであった。
【0125】
これらのデータは、選択的CB2受容体アゴニストが炎症性疼痛のラットモデルにおける強力な経口抗発痛作用を保有していることを実証している。
【0126】
【表3】
【技術分野】
【0001】
本発明は、1−(4−(ピリジン−2−イル)ベンジル)イミダゾリジン−2,4−ジオン誘導体、これらを含む医薬組成物、および治療、特に疼痛の治療におけるこれらの1−(4−(ピリジン−2−イル)ベンジル)イミダゾリジン−2,4−ジオン誘導体の使用に関する。
【背景技術】
【0002】
疼痛は、実際または潜在的な組織損傷に伴う不快な感覚および感情の経験である。疼痛は元来、侵害受容性または神経障害性起源であり得る。関節炎の結果として経験される疼痛は、一般に、組織の炎症および侵害受容器の刺激により引き起こされる侵害受容性の性質のものである。侵害受容性疼痛の有病率を上げる主要な兆候は、腰背痛、骨関節炎、術後疼痛および癌関連疼痛である。侵害受容性疼痛に対する満たされていない主な要求は、効力の改善および副作用低減である。慢性疼痛市場は現在、非ステロイド性抗炎症薬(NSAID)およびシクロオキシゲナーゼCOX−2阻害剤によって独占されている。NSAIDは、軽度から中程度の疼痛を軽減するための適切な鎮痛をもたらし、通常、炎症性疼痛においてより大きな有効性を有する。個々のNSAIDは、これらの効力が変動し、これらの変動は、異なるCOX−1/COX−2選択性によって一部決定される。その結果として、患者は、彼らの疼痛が適切に治療される前にいくつかの異なる薬物で治療される必要がある場合がある。薬物治療に伴う副作用は、治療選択において、特に多くの疼痛症候群が長期慢性状態である場合、重要な因子である。
【0003】
NSAIDの最も一般的な副作用は、便秘症および消化障害であり、大部分の抗炎症薬は酸性の性質であり、胃における酸の産生を促進する。他の重篤な副作用は、胃潰瘍、粘膜損傷および消化性びらんなどの胃腸管系合併症である。NSAIDは、毎年米国において潰瘍の合併症による107,000人もの入院および16,500人もの死亡の原因と考えられている(Singh、Recent considerations in nonsteroidal anti−inflammatory drug gastropathy.Am J.Med.、1998、105:31S−38S)。COX−2阻害剤は、改善された胃腸管系副作用プロフィールを有する一方、これらの使用は、心筋梗塞および脳卒中のリスク増加、ならびに高血圧症のリスク増加を伴ってきた。
【0004】
神経系の損傷、疾患または機能不全に起因する慢性疼痛として定義される神経因性疼痛は、前記集団の約1%に存在し、最も大きい患者集団として、有痛性の糖尿病性末梢神経障害を持つもの、および帯状疱疹(ヘルペス後神経痛)の発作後に持続する神経痛を有するものが挙げられる。これは、組織損傷が存在せずに発生し得る自発的疼痛を含めて、複雑な組合せの症状を特徴とする。神経因性疼痛を患う患者は、有痛性として通常知覚される刺激(痛覚過敏)、ならびに疼痛を通常誘発しない刺激(アロディニア)の両方に対する感度も増加している。これらの症状は従来の鎮痛療法に対してしばしば難治性であり、大部分の患者は、彼らの症状の完全な軽減を達成しない。現在、抗うつ薬、抗痙攣薬およびオピオイドは、ゴールドスタンダードであるガバペンチンとともに依然として一次治療である。これらの薬物の全てが、用量制限性の顕著な副作用を有する。さらに、効力が神経因性疼痛の市場における考慮すべき問題であり、現在の治療は、総疼痛スコアにおいてベースラインから最大50%の低減を示す。その結果として、より高い効力/応答者の割合を有し、現在使用されている薬物と比較して副作用が低減されている薬剤の医学的必要が、依然として満たされていない。
【0005】
新たな臨床的証拠、ならびに大麻を自己投薬している患者からの事例報告は、カンナビノイド受容体アゴニストが、疼痛を治療する際に役割を担う場合があることを示唆している(Fox A、Bevan S.、Therapeutic potential of cannabinoid receptor agonists as analgesic agents.Expert Opin Investig Drugs、2005、14、695−703)。GW Sativex、神経因性疼痛の治療のための個別投薬を可能にする口腔粘膜スプレー製剤におけるΔ9−THCおよびカンナビジオールの1:1比は、GW Pharmaceuticalsによって始められた。Sativexの臨床研究は、難治性疼痛(慢性神経因性疼痛、腕神経叢神経損傷による疼痛、アロディニア末梢性神経因性疼痛および進行癌疼痛)、関節リウマチ、および多発性硬化症に伴う症状(疼痛、痙直、膀胱制御不良および睡眠分断)を持つ患者において効力を実証した(Barnes MP.2006.Sativex:clinical efficacy and tolerability in the treatment of symptoms of multiple sclerosis and neuropathic pain.Expert Opin.Pharmacother.7(5):607−615)。
【0006】
カンナビノイド受容体の2つのタイプが同定された。カンナビノイドCB1受容体は主に中枢神経系(CNS;脳脊髄)に位置するが、末梢ニューロンによっても発現され、他の末端組織において、より低い程度で発現される。カンナビノイドCB2受容体は、大抵免疫細胞中の末梢に主に限局されている(Howlett、A.C.ら、International Union of Pharmacology.XXVII.Classification of Cannabinoid Receptors.Pharmacol.Rev.54、161−202、2002)。テトラヒドロカンナビノール(THC)など従来のCB1受容体アゴニストおよびCB1/CB2受容体アゴニストは、動物における疼痛モデルで高い効果がある一方、人間におけるこれらの治療有用性は、向精神効果などの望ましくないCNS副作用、および濫用の可能性によって制限される(Chapman、V.およびFinn、D.P.「Analgesic effects of cannabinoids:sites and mechanism of action.」Rev.Analg.7、25−39、2003)。
【0007】
近年の文献証拠は、CB2受容体の選択的活性化が、望ましくないCNS副作用を起こすことなく疼痛および炎症を治療するための新規な戦略となり得ることを示唆している。(Guindon、J.およびHohmann、A.、「Cannabinoid CB2 receptors:a therapeutic target for the treatment of inflammatory and neuropathic pain」、Br.J.Pharmacol.、2008、153、319−334)。CB2受容体の活性化は、動物モデルにおいて急性、炎症性および神経因性の疼痛応答を阻害することが判明した(Whiteside G.T.、Lee G.P.、Valenzano K.J.「The role of the cannabinoid CB2 receptor in pain transmission and therapeutic potential of small molecule CB2 receptor agonists」、Current Med.Chem.、2007、14、917−936)。CB2ノックアウトマウスの研究も、疼痛におけるCB2受容体の役割を裏付けている(Malan TP、Jr、Ibrahim MM、Lai J、Vanderah TW、Makriyannis AおよびPorreca F.CB2 cannabinoid receptor agonists:pain relief without psychoactive effects? Curr.Opin.Pharmacol.2003;3:62−67)。
【0008】
CB2媒介抗侵害受容に寄与する細胞性機序はまだ明らかではないが、CB2受容体の活性化が、免疫細胞活性の調節を介して間接的に炎症性疼痛に影響し、炎症の局所部位で媒介物質の放出減少をもたらすことが提唱された。末梢効果に加えて、近年の学術誌は、CB2受容体アゴニストが、末梢ニューロンおよび活性化ミクログリア上に発現したCB2受容体と相互作用することによって疼痛伝達を調節することもできることを示唆している(Beltramoら、2006.CB2 receptor−mediated antihyperalgesia:possible direct involvement of neural mechanisms.Eur J Neurosci.23(6):1530−80;Romero−Sandoval&Eisenach、2007.Spinal cannabinoid receptor type 2 activation reduces hypersensitivity and spinal cord glial activation after paw incision.Anesthesiology106(4):787−94)。
【0009】
つまり、CB2受容体アゴニストは、骨関節炎、関節リウマチならびに急性の術後疼痛および神経因性疼痛などの急性および慢性疼痛状態の治療に適当であり得る。前臨床モデルにおけるCB2アゴニストによるカタレプシーの不在は、望ましくないCNS副作用のない急性および慢性疼痛の治療が有望であることを示す。
【先行技術文献】
【非特許文献】
【0010】
【非特許文献1】Singh、Recent considerations in nonsteroidal anti−inflammatory drug gastropathy.Am J.Med.、1998、105:31S−38S
【非特許文献2】Fox A、Bevan S.、Therapeutic potential of cannabinoid receptor agonists as analgesic agents.Expert Opin Investig Drugs、2005、14、695−703
【非特許文献3】Barnes MP.2006.Sativex:clinical efficacy and tolerability in the treatment of symptoms of multiple sclerosis and neuropathic pain.Expert Opin.Pharmacother.7(5):607−615
【非特許文献4】Howlett、A.C.ら、International Union of Pharmacology.XXVII.Classification of Cannabinoid Receptors.Pharmacol.Rev.54、161−202、2002
【非特許文献5】Chapman、V.およびFinn、D.P.「Analgesic effects of cannabinoids:sites and mechanism of action.」Rev.Analg.7、25−39、2003
【非特許文献6】Guindon、J.およびHohmann、A.、「Cannabinoid CB2 receptors:a therapeutic target for the treatment of inflammatory and neuropathic pain」、Br.J.Pharmacol.、2008、153、319−334
【非特許文献7】Whiteside G.T.、Lee G.P.、Valenzano K.J.「The role of the cannabinoid CB2 receptor in pain transmission and therapeutic potential of small molecule CB2 receptor agonists」、Current Med.Chem.、2007、14、917−936
【非特許文献8】Malan TP、Jr、Ibrahim MM、Lai J、Vanderah TW、Makriyannis AおよびPorreca F.CB2 cannabinoid receptor agonists:pain relief without psychoactive effects? Curr.Opin.Pharmacol.2003;3:62−67
【非特許文献9】Beltramoら、2006.CB2 receptor−mediated antihyperalgesia:possible direct involvement of neural mechanisms.Eur J Neurosci.23(6):1530−80
【非特許文献10】Romero−Sandoval&Eisenach、2007.Spinal cannabinoid receptor type 2 activation reduces hypersensitivity and spinal cord glial activation after paw incision.Anesthesiology106(4):787−94
【発明の概要】
【発明が解決しようとする課題】
【0011】
したがって、疼痛の治療における治療剤としての選択的CB2カンナビノイド受容体アゴニストが求められている。
【課題を解決するための手段】
【0012】
この目的のため、本発明は、例えば手術前後の疼痛、慢性疼痛、神経因性疼痛、癌疼痛などの疼痛ならびに多発性硬化症に伴う疼痛および痙直の治療に使用することができるカンナビノイドCB2受容体のアゴニストとして、一般式Iを有する1−(4−(ピリジン−2−イル)ベンジル)イミダゾリジン−2,4−ジオン誘導体の新規な構造部類
【0013】
【化1】
[式中、
R1は、H、(C1−6)アルキル(オキソ、(C1−3)アルキルオキシ、(C1−3)アルキルオキシ−カルボニル、ハロゲンまたはCNで場合によって置換されている。)、(C3−6)シクロアルキルまたは(C3−6)シクロアルキル(C1−3)アルキルであり、各シクロアルキル環は、OおよびSから選択されるヘテロ原子を場合によって含み、
R2およびR3は独立して、Hもしくは(C1−3)アルキルであり、または
R2およびR3は、これらが結合している炭素原子と一緒になって(C3−5)−シクロアルキル基を形成し、
R4は、Hまたは1個から3個のF置換基であり、
R5は、Hまたは1個から4個のF置換基であり、
R6およびR7は独立して、HまたはFであり、
Xは、R8、OR8、NR8R9、
【0014】
【化2】
を表し、
R8は、O、S、SOおよびSO2から選択されるヘテロ原子を場合によって含む(C5−7)シクロアルキルであり、
R9は、Hまたは(C1−4)アルキルであり、
R10は、H、(C1−3)アルキル、ハロゲン、オキソ、CNおよびCF3から独立して選択される1−3個の置換基を表し、
Yは、CF2、O、S、SOまたはSO2である。]
または医薬として許容できるこの塩を提供する。
【0015】
(C1−6)アルキルという用語は、式Iの定義で使用される場合、ヘキシル、ペンチル、ブチル、イソブチル、第三ブチル、プロピル、イソプロピル、エチルおよびメチルのように、1−6個の炭素原子を有する分枝または非分枝のアルキル基を意味する。
【0016】
(C1−4)アルキルという用語は同様に、n−ブチル、tert−ブチル、プロピル、イソプロピル、エチルおよびメチルのように、1−4個の炭素原子を有する分枝または非分枝のアルキル基を意味する。
【0017】
(C1−3)アルキルという用語は同様に、プロピル、イソプロピル、エチルおよびメチルにように、1−3個の炭素原子を有する分枝または非分枝のアルキル基を意味する。
【0018】
(C1−3)アルキルオキシおよび(C1−3)アルキルオキソカルボニルという用語における(C1−3)アルキルの意味は、上記で定義した通りである。
【0019】
(C3−6)シクロアルキルという用語は、シクロヘキシル、シクロペンチル、シクロブチルおよびシクロプロピルのように、3−6個の炭素原子を有するシクロアルキル基を意味する。
【0020】
(C3−5)シクロアルキルという用語は、シクロペンチル、シクロブチルおよびシクロプロピルにように、3−5個の炭素原子を有するシクロアルキル基を意味する。
【0021】
(C5−7)シクロアルキルという用語は同様に、5−7個の炭素原子を有するシクロアルキル基を意味する。
【0022】
好ましい(C5−7)シクロアルキルはシクロヘキシルである。
【0023】
ハロゲンという用語は、F、Cl、BrまたはIを意味する。
【0024】
R2、R3およびR5がHである式Iに従った1−(4−(ピリジン−2−イル)ベンジル)イミダゾリジン−2,4−ジオン誘導体が優先される。R1が(C1−4)アルキルである式Iに従った化合物も好ましい。さらに好ましいのは、XがNR8R9、
【0025】
【化3】
を表す式Iに従った化合物である。
【0026】
より好ましいのは、XがNR8R9であり、R8がOおよびSから選択されるヘテロ原子を場合によって含むシクロヘキシルである式Iの化合物である。
【0027】
さらに好ましいのは、R4がCR6R7X基に対してオルト位のF置換基である化合物である。
【0028】
本発明の特に好ましい1−(4−(ピリジン−2−イル)ベンジル)イミダゾリジン−2,4−ジオン誘導体は、
−3−イソブチル−1−(4−(6−(ピペリジン−1−イルメチル)ピリジン−2−イル)ベンジル)イミダゾリジン−2,4−ジオン;
−3−イソブチル−1−(4−(6−(モルホリノメチル)ピリジン−2−イル)ベンジル)イミダゾリジン−2,4−ジオン;
−1−(4−(5−フルオロ−6−((テトラヒドロ−2H−ピラン−4−イルアミノ)メチル)ピリジン−2−イル)ベンジル)−3−イソブチルイミダゾリジン−2,4−ジオン;
−1−(4−(6−((1,1−ジオキソ−1λ6−チオモルホリン−4−イル)メチル)ピリジン−2−イル)ベンジル)−3−イソブチルイミダゾリジン−2,4−ジオン;
−1−(4−(6−((1,1−ジオキソ−1λ6−チオモルホリン−4−イル)メチル)−5−フルオロピリジン−2−イル)ベンジル)−3−イソブチルイミダゾリジン−2,4−ジオン;
−3−イソブチル−1−(4−(6−((テトラヒドロ−2H−ピラン−4−イルアミノ)メチル)ピリジン−2−イル)ベンジル)イミダゾリジン−2,4−ジオン;
−3−エチル−1−(4−(6−(ピペリジン−1−イルメチル)ピリジン−2−イル)ベンジル)イミダゾリジン−2,4−ジオン;
−3−エチル−1−(4−(5−フルオロ−6−(ピペリジン−1−イルメチル)ピリジン−2−イル)ベンジル)イミダゾリジン−2,4−ジオン;
−1−(4−(6−(1,1−ジオキソ−1λ6−チオモルホリン−4−イルメチル)−5−フルオロピリジン−2−イル)ベンジル)−3−エチルイミダゾリジン−2,4−ジオン;
−1−(4−(6−(1,1−ジオキソ−1λ6−チオモルホリン−4−イルメチル)ピリジン−2−イル)ベンジル)−3−エチルイミダゾリジン−2,4−ジオン;
−1−(4−(5−フルオロ−6−((テトラヒドロ−2H−ピラン−4−イルアミノ)メチル)ピリジン−2−イル)ベンジル)−3−プロピルイミダゾリジン−2,4−ジオン;
−1−(4−(6−(1,1−ジオキソ−1λ6−チオモルホリン−4−イルメチル)−5−フルオロピリジン−2−イル)ベンジル)−3−プロピルイミダゾリジン−2,4−ジオン;
−3−(2,2−ジフルオロエチル)−1−(4−(6−(ピペリジン−1−イルメチル)ピリジン−2−イル)ベンジル)イミダゾリジン−2,4−ジオン;
−3−(2,2−ジフルオロエチル)−1−(4−(5−フルオロ−6−(ピペリジン−1−イルメチル)ピリジン−2−イル)ベンジル)イミダゾリジン−2,4−ジオン;
−3−(シクロプロピルメチル)−1−(4−(5−フルオロ−6−(ピペリジン−1−イルメチル)ピリジン−2−イル)ベンジル)イミダゾリジン−2,4−ジオン;
−3−(シクロプロピルメチル)−1−(4−(6−(ピペリジン−1−イルメチル)ピリジン−2−イル)ベンジル)イミダゾリジン−2,4−ジオン;
−3−(2−オキソプロピル)−1−(4−(6−(ピペリジン−1−イルメチル)ピリジン−2−イル)ベンジル)イミダゾリジン−2,4−ジオン;
−1−(4−(5−フルオロ−6−(ピペリジン−1−イルメチル)ピリジン−2−イル)ベンジル)−3−(2−オキソプロピル)イミダゾリジン−2,4−ジオン;
−1−(4−(6−(1,1−ジオキソ−1λ6−チオモルホリン−4−イルメチル)−5−フルオロピリジン−2−イル)ベンジル)−3−(2−オキソプロピル)イミダゾリジン−2,4−ジオン;
−3−ルソプロピル−1−(4−(6−(ピペリジン−1−イルメチル)ピリジン−2−イル)ベンジル)イミダゾリジン−2,4−ジオン;
−1−(4−(6−(1,1−ジオキソ−1λ6−チオモルホリン−4−イルメチル)−5−フルオロピリジン−2−イル)ベンジル)−3−イソプロピルイミダゾリジン−2,4−ジオン;
−1−(4−(6−1,1−ジオキソ−1λ6−チオモルホリン−4−イルメチル)ピリジン−2−イル)ベンジル)−3−イソプロピルイミダゾリジン−2,4−ジオン;
−3−シクロプロピル−1−(4−(5−フルオロ−6−(ピペリジン−1−イルメチル)ピリジン−2−イル)ベンジル)イミダゾリジン−2,4−ジオン;
−3−シクロプロピル−1−(4−(6−(1,1−ジオキソ−1λ6−チオモルホリン−4−イルメチル)−5−フルオロピリジン−2−イル)ベンジル)イミダゾリジン−2,4−ジオン;
−3−シクロブチル−1−(4−(5−フルオロ−6−((テトラヒドロ−2H−ピラン−4−イルアミノ)メチル)ピリジン−2−イル)ベンジル)イミダゾリジン−2,4−ジオン;
−3−シクロブチル−1−(4−(6−((テトラヒドロ−2H−ピラン−4−イルアミノ)メチル)ピリジン−2−イル)ベンジル)イミダゾリジン−2,4−ジオン;
−3−シクロブチル−1−(4−(6−(1,1−ジオキソ−1λ6−チオモルホリン−4−イルメチル)−5−フルオロピリジン−2−イル)ベンジル)イミダゾリジン−2,4−ジオン;
−3−シクロブチル−1−(4−(6−(1,1−ジオキソ−1λ6−チオモルホリン−4−イルメチル)ピリジン−2−イル)ベンジル)イミダゾリジン−2,4−ジオン;
−1−(4−(6−(ピペリジン−1−イルメチル)ピリジン−2−イル)ベンジル)−3−(2,2,2−トリフルオロエチル)イミダゾリジン−2,4−ジオン;
−1−(4−(6−(1,1−ジオキソ−1λ6−チオモルホリン−4−イルメチル)−5−フルオロピリジン−2−イル)ベンジル−3−(2,2,2−トリフルオロエチル)イミダゾリジン−2,4−ジオン;
−1−(4−(6−((テトラヒドロ−2H−ピラン−4−イルアミノ)メチル)ピリジン−2−イル)ベンジル)−3−(2,2,2−トリフルオロ−エチル)イミダゾリジン−2,4−ジオン;
−1−(4−(5−フルオロ−6−((テトラヒドロ−2H−ピラン−4−イルアミノ)メチル)ピリジン−2−イル)ベンジル)−3−(2,2,2−トリフルオロエチル)イミダゾリジン−2,4−ジオン
または医薬として許容できるこれらの塩である。
【0029】
式Iを有する本発明の1−(4−(ピリジン−2−イル)ベンジル)イミダゾリジン−2,4−ジオン誘導体は、有機化学の当技術分野において知られている方法によって調製することができる。本発明の化合物は、例えば、式II(式中、R4、R6、R7およびXは、すでに定義した通りの意味を有する。)の2−ブロモピリジン誘導体と、式III(式中、R1、R2、R3およびR5は、すでに定義した通りの意味を有する。)のベンジル化イミダゾリジンから調製される式IVのボロン酸誘導体との間の、炭酸カリウムおよびPd(Ph3P)4などのパラジウム(0)錯体を使用する鈴木カップリング反応から得ることができる(スキームIを参照のこと。)。
【0030】
【化4】
【0031】
XがR8を表し、R8がSおよびOから選択されるヘテロ原子を含む(C5−7)シクロアルキルである式IIの化合物は、式1(式中、R4は、すでに定義した通りの意味を有する。)の二臭化ピリジン誘導体と式2(式中、Zは、CH2、OまたはSを表す。)のニトリル誘導体との縮合から式3の中間体ケト誘導体を生成し、これを式4のヒドラジド誘導体を介して還元することにより、R6およびR7がHである式IIの化合物に対応する式IIbの化合物を得ることができる、または三フッ化ジエチルアミノ硫黄を用いて還元することにより、R6およびR7がFである式IIの化合物に対応する式IIaの化合物を生成することができることで調製することができる(スキームIIを参照のこと)。
【0032】
【化5】
【0033】
条件
A:ブチルリチウム/H2SO4。B:4−メチルベンゼンスルホニルヒドラジド。C:水素化ジイソブチルアルミニウム。D:三フッ化ジエチルアミノ硫黄。
【0034】
XがOR8表す式IIの化合物は、スキームIIIに示されている通り、式5の2−ブロモ−6−メチルピリジン誘導体の臭素化で出発し、N−ブロモコハク酸イミド/アゾ−ジ−イソブチロニトリルを用いて式6の対応する6−ブロモメチル誘導体を生成し、これを式7(式中、Zは、CH2,OまたはSである。)のアルコール誘導体と反応させることにより式IIcの化合物を生成することで調製することができる。
【0035】
【化6】
XがNHR8、
【0036】
【化7】
を表す式IIの化合物は、例えば酢酸/水素化トリアセトキシホウ素ナトリウムの使用とともに、式X−Hの適切なアミン誘導体を用いる式8のカルバルデヒド誘導体の還元的アミノ化反応を使用して調製することができる。
【0037】
【化8】
【0038】
式IIIの化合物は、還元的アミノ化条件下でのアミノ酸H2N−C(R2,R3)−COOHと式9の4−ブロモベンズアルデヒド誘導体とのカップリングによって式12のN−ベンジル誘導体が得られ、これを引き続き、ジシクロヘキシル−カルボジイミド(DCCI)、TBTUまたはPyBOPなどのアミド結合形成試薬を用いてアミンH2N−R1(式中、R1は、前に定義されている意味を有する。)とカップリングさせることによりアミド誘導体13にし、これから、カルボニルジイミダゾールを使用する閉環反応によって式IIIのイミダゾリジン−2,4−ジオン誘導体を調製できることで調製することができる。
【0039】
スキームIVに示されている代替経路において、式9の4−ブロモベンズアルデヒド誘導体を還元的アミノ化条件下で式H2N−C(R2,R3)−CONH2のアミノ酸アミドにカップリングさせることにより、式10のN−ベンジル誘導体が得られ、これは、カルボニルジイミダゾールを使用する環化、および式Hal−R1のハロゲン化物を用いる後続のアルキル化によって式IIIの化合物に変換することができる。
【0040】
【化9】
【0041】
式Iの1−(4−(ピリジン−2−イル)ベンジル)イミダゾリジン−2,4−ジオン誘導体およびこれらの塩は、少なくとも1つのキラリティー中心を含有し、したがって、エナンチオマーおよびジアステレオマーを含めた立体異性体として存在することができる。本発明は、この範囲内に、前述の立体異性体、ならびに式Iの化合物の個々のRおよびSエナンチオマーのそれぞれならびにこれらの塩、実質的に遊離塩基、即ち5%未満、好ましくは2%未満、特に1%未満の他のエナンチオマーを伴う遊離塩基、ならびに実質的に等量の2種のエナンチオマーを含有するラセミ混合物を含めた任意の比率のこうしたエナンチオマーの混合物を含める。
【0042】
純粋な立体異性体が得られる不斉合成またはキラル分離のための方法は、当技術分野においてよく知られており、例えばキラル誘導を用いる合成、もしくは市販されているキラル基質から出発する合成、または例えばキラル媒体上のクロマトグラフィーを使用するもしくはキラル対イオンを用いる結晶化による立体異性体の分離である。
【0043】
医薬として許容できる塩は、式Iの1−(4−(ピリジン−2−イル)ベンジル)イミダゾリジン−2,4−ジオン誘導体の遊離塩基を、塩酸、臭化水素酸、リン酸および硫酸などの鉱酸、または例えばアスコルビン酸、クエン酸、酒石酸、乳酸、マレイン酸、マロン酸、フマル酸、グリコール酸、コハク酸、プロピオン酸、酢酸およびメタンスルホン酸などの有機酸で処理することによって得ることができる。
【0044】
本発明の化合物は、非溶媒和で、ならびに水およびエタノールなどの医薬として許容できる溶媒との溶媒和形態で存在することができる。一般に、溶媒和形態は、本発明の目的においては非溶媒和形態と同等と考えられる。
【0045】
本発明は、医薬として許容できる助剤および場合により他の治療剤との混合で、一般式Iに従った1−(4−(ピリジン−2−イル)ベンジル)イミダゾリジン−2,4−ジオン誘導体または医薬として許容できるこの塩を含む医薬組成物をさらに提供する。「許容できる」という用語は、前記組成物の他の成分と相容性であり、この受容者に有害でないことを意味する。組成物は、例えば、全て投与用単位剤形における経口、舌下、皮下、静脈内、硬膜外、くも膜下腔内、筋肉内、経皮、肺、局所、眼球または直腸投与などに適当なものを含める。投与の好ましい経路は経口経路である。
【0046】
経口投与のため、前記活性成分は、錠剤、カプセル、粉末、顆粒、溶液および懸濁液などの個別単位として存在することができる。非経口投与のため、本発明の前記医薬組成物は、単位用量または多用量容器、例えば密封したバイアルおよびアンプル中の所定の量での例えば注入液体中に存在することができ、使用前に滅菌液体担体、例えば水の添加のみを必要とする凍結乾燥させた(凍結乾燥)状態で貯蔵することもできる。
【0047】
例えば標準的文献のGennaro、A.R.ら、Remington:The Science and Practice of Pharmacy(第20版、Lippincott Williams&Wilkins、2000、特に5部:Pharmaceutical Manufacturingを参照のこと。)に記載されている通りに、医薬として許容できるこうした助剤と混合され、前記活性薬剤は、丸薬、錠剤などの固体投与単位に圧縮することができ、またはカプセル、坐剤もしくはパッチにすることができる。医薬として許容できる液体の手段によって、前記活性薬剤は、流体組成物、例えば溶液、懸濁液、エマルジョンの形態で注入調製物として、またはスプレー、例えば経鼻スプレーとして適用することができる。
【0048】
固体投与単位を製造するため、充填剤、着色剤およびポリマーバインダーなどの従来の添加剤の使用が企図される。一般に、前記活性化合物の機能を妨げることのない任意の医薬として許容できる添加剤を使用することができる。本発明の活性薬剤を固体組成物として投与することができる適当な担体として、適当な量で使用されるラクトース、デンプンおよびセルロース誘導体など、またはこれらの混合物が挙げられる。非経口投与のため、プロピレングリコールまたはブチレングリコールなど医薬として許容できる分散剤および/または湿潤剤を含有する水性懸濁液、等張生理食塩水および滅菌注射可能溶液を使用することができる。
【0049】
本発明は、前記組成物に適当な包装材料との組合せにおいて前に記載されている通りの医薬組成物をさらに含め、前記包装材料は、前に記載されている通りの使用のための組成物使用用指示を含める。
【0050】
本発明の1−(4−(ピリジン−2−イル)ベンジル)イミダゾリジン−2,4−ジオン誘導体は、CHO細胞を使用するヒトCB2およびCB1レポーターアッセイにおいて決定された通り、CB1受容体と比較するとCB2受容体の選択的アゴニストであることが判明した。受容体結合ならびにカンナビノイド受容体調節因子のインビトロ生物活性を決定する方法は、当技術分野においてよく知られている。一般に、発現された受容体は前記化合物を用いてインキュベートすることにより試験され、結合または機能的応答の刺激もしくは阻害が測定される。
【0051】
機能的応答を測定するため、CB2またはCB1受容体遺伝子、好ましくはヒト受容体をコードする単離DNAは、適当な宿主細胞中に発現される。こうした細胞はチャイニーズハムスター卵巣細胞であってよいが、他の細胞も適当である。好ましくは、前記細胞は哺乳動物起源である。
【0052】
組換えCB2またはCB1発現細胞株を構築するための方法は、当技術分野においてよく知られている(Sambrookら、Molecular Cloning:a Laboratory Manual、Cold Spring Harbor Laboratory Press、Cold Spring Harbor、最新版)。受容体の発現は、所望のタンパク質をコードするDNAの発現によって達成される。追加配列の連結および適当な発現系の構成のための技術は全て、これまで当技術分野においてよく知られている。標準的な固相技術を使用して、所望のタンパク質をコードするDNAの一部分または全てを合成的に構築することにより、好ましくは、連結を緩めるための制限部位を含めることができる。含まれたコード配列の転写および翻訳のための適当な制御領域を、前記DNAコード配列に提供することができる。よく知られている通り、細菌などの原核宿主、ならびに酵母、植物細胞、昆虫細胞、哺乳動物細胞、および鳥細胞などの真核宿主を含めた多種多様な宿主と相容性のある発現系が現在利用可能である。
【0053】
前記受容体を発現する細胞を次いで、試験化合物を用いてインキュベートすることにより、結合または機能的応答の刺激もしくは阻害を観察する。
【0054】
別法として、発現されたCB2またはCB1受容体を含有する単離細胞膜を使用することにより、化合物の結合を測定することができる。
【0055】
結合の測定のため、放射活性または蛍光標識化合物を使用することができる。最も広範に使用される放射標識カンナビノイドプローブは、CB1およびCB2結合部位に対しておよそ同等の親和性を有する(3H)CP55940である。
【0056】
機能的なCB2またはCB1アゴニスト活性は、例えばcAMPまたはMAPキナーゼ経路における受容体媒介変化の測定など、第二メッセンジャー応答を決定することによって測定することができる。したがって、こうした方法は、宿主細胞の細胞表面上におけるCB2またはCB1受容体の発現、および試験化合物に前記細胞を曝露することを伴う。第二メッセンジャー応答を次いで測定する。第二メッセンジャー濃度は、受容体の結合に対する試験化合物の影響に依存して低減または増加する。
【0057】
曝露細胞における例えばcAMP濃度の直接測定に加えて、受容体コードDNAのトランスフェクションに加えてレポーター遺伝子をコードする第二DNAもトランスフェクトされている細胞を使用することができ、この発現は受容体活性化と相関する。一般に、レポーター遺伝子発現は、第二メッセンジャー濃度の変化に反応する任意の応答配列によって制御され得る。適当なレポーター遺伝子は、例えばLacZ、アルカリホスファターゼ、蛍ルシフェラーゼおよび緑色蛍光タンパク質である。こうしたトランス活性化アッセイの原理は当技術分野においてよく知られており、例えばStratowa、C.、Himmler、A.およびCzernilofsky、A.P.、Curr.Opin.Biotechnol.6、574(1995)に記載されている。CB2受容体に対する選択的活性アゴニスト化合物を選択するため、化合物のEC50値は、<10−5M、好ましくは<10−7Mであり、EC50(CB1)/EC50(CB2)として定義した通りのCB1受容体アゴニストに対する選択率は、>10、好ましくは>50である。
【発明の効果】
【0058】
前記化合物は、例えば手術前後の疼痛などの急性疼痛、慢性疼痛、神経因性疼痛、癌疼痛、内臓痛、頭痛などの疼痛、および多発性硬化症に伴う痙直の治療における鎮痛剤として使用することができる。
【0059】
本発明のカンナビノイドアゴニストは、(腸管の)炎症、アトピー性皮膚炎、肝疾患、呼吸器障害、アレルギー、腫瘍、てんかん、片頭痛、骨粗鬆症、循環器障害、外傷性脳損傷および脳卒中などの急性神経変性疾患、ならびにアルツハイマー病、多発性硬化症およびALSなどの緩徐性神経変性疾患を含めた他の障害の治療においても潜在的に有用であると思われる(Parcher P、Batkai S、Kunos、G、The endocannabinoid system as an emerging target of pharmacotherapy、Pharmacol Rev.2006、58(3):389−462)。
【0060】
前記化合物は、COX−2選択的阻害剤を含めて、他の薬物、例えばオピオイドおよび非ステロイド性抗炎症薬(NSAID)などの鎮痛薬と併せて使用することもできる。
【0061】
本発明の化合物は、症状を軽減するのに十分な量および十分な時間でヒトに投与することができる。例示として、ヒトへの投与量は、体重1kg当たり0.001−50mgの範囲内、好ましくは体重1kg当たり0.01−20mgの投与量であってよい。
【図面の簡単な説明】
【0062】
【図1】ラットにおける神経障害誘発機械的アロディニアにおける化合物12の急性経口投与の影響を示す。
【発明を実施するための形態】
【0063】
本発明を以下の実施例によって例示する。
【実施例】
【0064】
略語:Boc:tert−ブトキシカルボニル;CDCl3:クロロホルム−d;DBU:1,8−ジアザビシクロ(5.4.0)ウンデス−7−エン;CDI:N,N’−カルボニルジイミダゾール;DCCI:1,3−ジシクロヘキシルカルボジイミド;DCM:ジクロロメタン;DIPEA:N,N−ジイソプロピルエチルアミン;DMAP:4−ジメチルアミノピリジン;DMF:N,N−ジメチルホルムアミド;Et3NまたはTEA:トリエチルアミン;Gly:グリシニル;HPLC:高速液体クロマトグラフィー;HOAc:酢酸;HOBt:1−ヒドロキシベンゾ−トリアゾール;MeOH:メタノール;Me3SiClまたはTMSCl:クロロトリメチルシラン;MS:質量スペクトル;(PPh3)4Pd:テトラキス(トリフェニルホスフィン)パラジウム(0);PyBOP:(ベンゾトリアゾール−1−イルオキシ)トリピロリジノホスホニウムヘキサフルオロホスフェート;PyBrOP:ブロモ(トリスピロリジノ)ホスホニウムテトラフルオロホスフェート;TBTU:((ベンゾトリアゾール−1−イルオキシ)−ジメチルアミノ−メチレン)−ジメチル−アンモニウムテトラフルオロボレート;TFA:トリフルオロ酢酸;THF:テトラヒドロフラン;TLC:薄層クロマトグラフィー。
【0065】
化合物の名称は、Cambridgesoft’s Chemdraw Ultra、バージョン9.0.7.で作成した。
【0066】
実施例1
1−(4−ブロモベンジル)−3−イソブチルイミダゾリジン−2,4−ジオン
【0067】
【化10】
i)4−ブロモベンズアルデヒド(100g、0.54mol)およびグリシンアミド塩酸塩(54g、0.48mol)のメタノール/水(1500ml、5.5/1)中溶液に、水酸化ナトリウム(21.6g、0.54mol)を添加した。17時間室温で撹拌した後、反応混合物を0℃に冷却した。水素化ホウ素ナトリウム(38g、1.0mol)を添加し、清澄な液が得られるまで混合物を撹拌した。pH=3まで濃塩酸を添加することによって、反応物をクエンチした。17時間撹拌した後、混合物を飽和炭酸水素ナトリウム水溶液で中和し、生成物をジクロロメタン中に抽出した。合わせた有機相を水、ブラインで洗浄し、硫酸ナトリウム上で乾燥させ、減圧下で濃縮することにより、2−(4−ブロモベンジルアミノ)アセトアミド(92g)が得られた。
【0068】
ii)前のステップで得られた生成物(60.6g、0.25mol)のアセトニトリル(1500ml)中溶液に、CDI(81g、0.5mol)DMAP(61g、0.5mol)を添加した。16時間60℃で撹拌した後、溶液を室温に冷却し、2M塩酸水溶液中に注いだ。生成物を酢酸エチル中に抽出し、合わせた有機相を水、ブラインで洗浄し、硫酸ナトリウム上で乾燥させ、減圧下で濃縮した。残留している固体をアセトンとともに撹拌した。濾過により、1−(4−ブロモベンジル)イミダゾリジン−2,4−ジオン(45g)を白色固体として得た。生成物をさらに精製することなく以下のステップで使用した。
【0069】
iii)前のステップで得られた生成物(10g、37.2mmol)のDMF(90ml)中溶液に、炭酸カリウム(15.4g、111mmol)および1−ブロモ−2−メチルプロパン(8.08ml、74.3mmol)を室温で添加した。50℃にて窒素雰囲気下で17時間撹拌した後、反応混合物を室温に冷却し、濾過した。清澄な液を減圧下で濃縮した。カラムクロマトグラフィーにより、1−(4−ブロモベンジル)−3−イソブチルイミダゾリジン−2,4−ジオン(10.9g)を白色固体として得た。
【0070】
実施例2
実施例1に記載されているものと類似の手順に従って、以下の化合物を調製した。
2A:1−(4−ブロモベンジル)−3−メチルイミダゾリジン−2,4−ジオン
1H NMR(400MHz,CDCl3):δ7.50(d,J=8.61Hz,2H)、7.14(d,J=8.61Hz,2H)、4.52(s,2H)、3.73(s,2H)、3.06(s,3H)。
2B:1−(4−ブロモベンジル)−3−エチルイミダゾリジン−2,4−ジオン
1H NMR(400MHz,CDCl3):δ7.50(d,J=8.22Hz,2H)、7.13(d,J=8.22Hz,2H)、4.52(s,2H)、3.71(s,2H)、3.59(q,J=7.43Hz,2H)、1.24(t,J=7.43Hz,3H)。
2C:1−(4−ブロモベンジル)−3−プロピルイミダゾリジン−2,4−ジオン
1H NMR(400MHz,CDCl3):δ7.49(d,J=8.22Hz,2H)、7.13(d,J=8.22Hz,2H)、4.51(s,2H)、3.72(s,2H)、3.50(dd J=7.43および6.26Hz,2H)、1.72−1.60(m,2H)、0.93(t,J=7.43Hz,3H)。
2D:1−(4−ブロモベンジル)−3−(2,2−ジフルオロエチル)イミダゾリジン−2,4−ジオン
1H NMR(400MHz,CDCl3):δ7.51(d,J=8.22Hz,2H)、7.14(d,J=8.22Hz,2H)、6.21−5.87(tt,J=55.95,4.70および4.30Hz,1H)、4.53(s,2H)、3.90(td,J=13.70および4.30Hz,2H)、3.80(s,2H)。
2E:1−(4−ブロモベンジル)−3−(シクロプロピルメチル)イミダゾリジン−2,4−ジオン
1H NMR(400MHz,CDCl3):δ7.50(d,J=8.22Hz,2H)、7.15(d,J=8.22Hz,2H)、4.53(s,2H)、3.74(s,2H)、3.56(d,J=7.44Hz,2H)、1.23−1.12(m,1H)、0.54−0.45(m,2H)、0.38−0.32(m,2H)。
2F:1−(4−ブロモベンジル)−3−(シクロブチルメチル)イミダゾリジン−2,4−ジオン
1H NMR(400MHz,CDCl3):δ7.49(d,J=8.22Hz,2H)、7.13(d,J=8.22Hz,2H)、4.50(s,2H)、3.71(s,2H)、3.57(d,J=7.43Hz,2H)、2.75−2.62(m,1H)、2.08−1.97(m,2H)、1.92−1.70(m,4H)。
2G:1−(4−ブロモ−ベンジル)−3−(2−メトキシエチル)イミダゾリジン−2,4−ジオン
1H NMR(400MHz,CDCl3):δ7.49(d,J=8.22Hz,2H)、7.14(d,J=8.22Hz,2H)、4.52(s,2H)、3.77−3.72(m,4H)、3.60(t,J=5.87Hz,2H)、3.36(s,3H)。
2H:メチル2−(3−(4−ブロモベンジル)−2,5−ジオキソイミダゾリジン−1−イル)アセテート
1H NMR(400MHz,CDCl3):δ7.50(d,J=8.22Hz,2H)、7.15(d,J=8.22Hz,2H)、4.55(s,2H)、4.30(s,2H)、3.83(s,2H)、3.78(s,3H)。
2L:1−(4−ブロモ−ベンジル)−3−(2−オキソプロピル)イミダゾリジン−2,4−ジオン
1H NMR(400MHz,CDCl3):δ7.51(d,J=8.61Hz,2H)、7.15(d,J=8.61Hz,2H)、4.54(s,2H)、4.35(s,2H)、3.83(s,2H)、2.25(s,3H)。
【0071】
実施例3
1−(4−ブロモベンジル)−3−イソプロピルイミダゾリジン−2,4−ジオン
【0072】
【化11】
i)グリシン(8.11g、108mmol)の水(40ml)中溶液に、水酸化ナトリウム水溶液(108mmol、15ml)および4−ブロモベンズアルデヒド(20g、108mmol)のメタノール(240ml)中溶液を添加した。室温で30分撹拌した後、水素化ホウ素ナトリウム(4.09g、108mmol)を、この懸濁液に少量ずつ添加した。室温で18時間撹拌した後、反応混合物を減圧下で濃縮し、生じた水性相をジエチルエーテルで洗浄した。2M塩酸水溶液の添加によって、水性相を中和した。生じた沈殿物を濾過によって回収し、水およびジエチルエーテルで洗浄した。白色固体を乾燥させることにより、2−(4−ブロモベンジルアミノ)酢酸(14.2g)を得た。生成物をさらに精製することなく以下のステップで使用した。
【0073】
ii)前のステップで得られた生成物(1.7g、6.96mmol)のジクロロメタン(20ml)中懸濁液に、トリエチルアミン(1.94ml、13.9mmol)、イソプロピルアミン(0.65ml、7.66mmol)およびo−(7−アザベンゾトリアゾール−1−イル)−1,1,3,3−テトラメチルウロニウムヘキサフルオロホスフェート(3.18g、8.36mmol)を添加した。室温で17時間撹拌した後、反応混合物を減圧下で濃縮した。カラムクロマトグラフィーにより、2−(4−ブロモベンジルアミノ)−N−イソプロピルアセトアミド(1.95g)を得た。
【0074】
iii)前のステップで得られた生成物(1.95g、6.96mmol)のアセトニトリル中溶液に、(ジイミダゾール−1−イル)ケトン(2.26g、13.9mmol)および4−ジメチルアミノピリジン(1.70g、13.9mmol)を添加した。60℃で17時間撹拌した後、反応混合物を室温に冷却し、飽和炭酸水素ナトリウム水溶液の添加によってクエンチした。生成物を酢酸エチル中に抽出し、合わせた有機相を水、ブラインで洗浄し、硫酸ナトリウム上で乾燥させた。カラムクロマトグラフィーにより、標題化合物1−(4−ブロモベンジル)−3−イソプロピルイミダゾリジン−2,4−ジオン(1.8g)を淡黄色油として得た。
1H NMR(400MHz,CDCl3):δ7.50(d,J=8.61Hz,2H)、7.13(d,J=8.61Hz,2H)、4.49(s,2H)、4.38−4.30(m,1H)、1.43(d,J=6.65Hz,6H)。
【0075】
実施例4
実施例3に記載されているものと類似の手順に従って、以下の化合物を調製した。
4A:1−(4−ブロモベンジル)−3−シクロプロピルイミダゾリジン−2,4−ジオン
1H NMR(400MHz,CDCl3):δ7.50(d,J=8.61Hz,2H)、7.14(d,J=8.61Hz,2H)、4.48(s,2H)、3.66(s,2H)、2.65−2.56(m,1H)、0.97(d,J=5.87Hz,4H)。
4B:1−(4−ブロモベンジル)−3−シクロブチルイミダゾリジン−2,4−ジオン
1H NMR(400MHz,CDCl3):δ7.49(d,J=8.22Hz,2H)、7.14(d,J=8.22Hz,2H)、4.60−4.51(m,1H)、4.49(s,2H)、3.65(s,2H)、2.95−2.82(m,2H)、2.23−2.13(m,2H)、1.91−1.81(m,1H)、1.79−1.65(m,1H)。
4C:1−(4−ブロモベンジル)−3−(2,2,2−トリフルオロエチル)イミダゾリジン−2,4−ジオン
1H NMR(400MHz,CDCl3):δ7.52(d,J=8.61Hz,2H)、7.15(d,J=8.61Hz,2H)、4.55(s,2H)、4.16(q,J=8.61Hz,2H)、3.83(s,2H)。
4D:1−(4−ブロモベンジル)−3−(1−シクロプロピルエチル)イミダゾリジン−2,4−ジオン
1H NMR(400MHz,CDCl3):δ7.54(d,J=8.61Hz,2H)、7.20(d,J=8.61Hz,2H)、4.72(s,2H)、3.87(s,2H)、3.46−3.36(m,1H)、1.22(d,J=6.65Hz,3H)、0.85−0.79(m,1H)、0.59−0.43(m,2H)、0.38−0.21(m,2H)。
4E:(S)−1−(4−ブロモベンジル)−3−(1−メトキシプロパン−2−イル)イミダゾリジン−2,4−ジオン
1H NMR(400MHz,CDCl3):δ7.49(d,J=8.61Hz,2H)、7.13(d,J=8.61Hz,2H)、4.50(d,J=3.91Hz,2H)、4.46−4.39(m,1H)、3.69(s,2H)、3.34(s,3H)、1.18(d,J=7.04Hz,3H)。
4F:1−(4−ブロモベンジル)−3−(テトラヒドロフラン−3−イル)イミダゾリジン−2,4−ジオン
1H NMR(400MHz,CDCl3):δ7.50(d,J=8.61Hz,2H)、7.14(d,J=8.61Hz,2H)、4.78−4.69(m,1H)、4.50(s,2H)、4.19−4.11(m,1H)、4.03−3.84(m,3H)、3.70(s,2H)、2.41−2.32(m,1H)、2.26−2.14(m,1H)。
4G:1−(4−ブロモベンジル)−3−(オキサゾール−5−イルメチル)イミダゾリジン−2,4−ジオン
1H NMR(400MHz,CDCl3):δ7.83(s,1H)、7.50(d,J=8.61Hz,2H)、7.15−7.11(m,3H)、4.77(s,2H)、4.52(s,2H)、3.77(s,2H)。
【0076】
実施例5
2−ブロモ−6−(ピペリジン−1−イルメチル)ピリジン
【0077】
【化12】
i)6−ブロモピリジン−2−カルバルデヒド(25g、135mmol)のジクロロメタン(500ml)中溶液に、ピペリジン(12.6g、149mmol)を10℃でゆっくり添加した。15分間10℃で撹拌した後、酢酸(8.9g、149mmol)を添加し、続いて水素化トリアセトキシホウ素ナトリウムを少量ずつ添加する一方、温度を5−10℃に保持した。2時間室温で撹拌した後、反応混合物を飽和炭酸水素ナトリウム水溶液中に注いだ。生成物をジクロロメタン中に抽出し、合わせた有機相をブラインで洗浄し、硫酸ナトリウム上で乾燥させ、減圧下で濃縮した。カラムクロマトグラフィーにより、2−ブロモ−6−(ピペリジン−1−イルメチル)ピリジン(30g)を無色油として得た。
1H NMR(400MHz,CDCl3):δ7.54−7.43(m,2H)、7.33(d,J=7.43Hz,1H)、3.60(s,2H)、2.48−2.38(m,4H)、1.63−1.54(m,4H)、1.48−1.39(m,2H)。
【0078】
実施例6
実施例5に記載されているものと類似の手順に従って、以下の化合物を調製した。
6A:4−((6−ブロモピリジン−2−イル)メチル)チオモルホリン1,1−ジオキシド
1H NMR(400MHz,CDCl3):δ7.61−7.54(dd,J=7.83および7.43Hz,1H)、7.42(d,J=7.83Hz,1H)、7.39(d,J=7.43Hz,1H)、3.81(s,2H)、3.14−3.04(m,8H)。
6B:4−((6−ブロモピリジン−2−イル)メチル)モルホリン
1H NMR(400MHz,CDCl3):δ7.53(dd,J=7.83および7.43Hz,1H)、7.45(d,J=7.83Hz,1H)、7.37(d,J=7.43Hz,1H)、3.76−3.71(m,4H)、3.65(s,2H)、2.55−2.49(m,4H)。
6C:2−ブロモ−6−((3−メチルピペリジン−1−イル)メチル)ピリジン
1H NMR(400MHz,CDCl3):δ7.47−7.37(m,2H)、7.26(d,J=7.43Hz,1H)、3.54(s,2H)、2.76−2.64(m,2H)、1.92(td,J=10.96および3.52Hz,1H)、1.68−1.44(m,5H)、0.87−0.72(m,4H)。
6D:2−ブロモ−6−((3,3−ジフルオロピペリジン−1−イル)メチル)ピリジン
1H NMR(400MHz,CDCl3):δ7.58−7.49(m,2H)、7.47(d,J=7.43Hz,1H)、3.74(s,2H)、2.71(dd,J=11.35および10.96Hz,2H)、2.54(dd,J=5.48および5.09Hz,2H)、1.97−1.84(m,2H)、1.83−1.76(m,2H)。
6E:2−ブロモ−6−((3−フルオロピペリジン−1−イル)メチル)ピリジン
1H NMR(400MHz,CDCl3):δ7.53(dd,J=7.83および7.43Hz,1H)、7.47(d,J=7.43Hz,1H)、7.36(d,J=7.83Hz,1H)、4.76−4.56(m,1H)、3.68(s,2H)、2.82−2.71(m,1H)、2.60−2.47(m,2H)、2.43−2.36(m,1H)、1.93−1.78(m,2H)、1.73−1.50(m,2H)。
6F:2−ブロモ−6−((3−トリフルオロメチルピペリジン−1−イル)メチル)ピリジン
(m/z)=324(M+H)+
6G:1−((6−ブロモピリジン−2−イル)メチル)−ピペリジン−3−オン
1H NMR(400MHz,CDCl3):δ7.54(dd,J=7.83および7.43Hz,1H)、7.41(d,J=7.43Hz,1H)、7.38(d,J=7.83Hz,1H)、3.73(s,2H)、3.08(s,2H)、2.73(dd,J=5.48Hz,2H)、2.39(dd,J=7.04Hz,2H)、2.03−1.94(m,2H)。
6H:N−((6−ブロモピリジン−2−イル)メチル)テトラヒドロ−2H−ピラン−4−アミン
1H NMR(400MHz,CDCl3):δ7.52(dd,J=7.83および7.43Hz,1H)、7.37(d,J=7.83Hz,1H)、7.31(d,J=7.43Hz,1H)、4.02−3.96(m,2H)、3.93(s,2H)、3.39(td,J=11.74および2.35Hz,2H)、2.78−2.69(m,1H)、1.90−1.82(m,2H)、1.55−1.42(m,2H)。
6I:((6−ブロモピリジン−2−イル)メチル)シクロヘキシルアミン
1H NMR(400MHz,CDCl3):δ7.50(dd,J=7.83および7.43Hz,1H)、7.34(d,J=7.83Hz,1H)、7.31(d,J=7.43Hz,1H)、3.90(s,2H)、2.50−2.41(m,1H)、1.96−1.88(m,2H)、1.78−1.69(m,3H)、1.65−1.57(m,1H)、1.31−1.06(m,5H)。
6J:N−((6−ブロモピリジン−2−イル)メチル)テトラヒドロ−2H−ピラン−3−アミン
1H NMR(400MHz,CDCl3):δ7.51(dd,J=7.83および7.43Hz,1H)、7.36(d,J=7.83Hz,1H)、7.31(d,J=7.43Hz,1H)、3.96−3.89(m,3H)、3.83(m,1H)、3.47−3.39(m,1H)、3.27−3.20(dd,J=8.61および8.22Hz,1H)、2.71−2.63(m,1H)、2.04−1.96(m,1H)、1.76−1.43(m,3H)、1.48−1.37(m,1H)。
6K:4−((6−ブロモピリジン−2−イル)メチル)チオモルホリン
1H NMR(400MHz,CDCl3):δ7.53(dd,J=7.83および7.43Hz,1H)、7.42(d,J=7.83Hz,1H)、7.37(d,J=7.43Hz,1H)、3.67(s,2H)、2.84−2.66(m,8H)。
【0079】
実施例7
実施例5に記載されているものと類似の手順に従って、6−ブロモ−3−フルオロピリジン−2−カルバルデヒドを出発原料として使用し、以下の化合物を調製した。
7A:4−((6−ブロモ−3−フルオロピリジン−2−イル)メチル)チオモルホリン1,1−ジオキシド
1H NMR(400MHz,CDCl3):δ7.45(dd,J=8.22および3.52Hz,1H)、7.32(d,J=8.61および8.22Hz,1H)、3.89(d,J=2.74Hz,2H)、3.17(m,8H)。
7B:4−((6−ブロモ−3−フルオロピリジン−2−イル)メチル)モルホリン
1H NMR(400MHz,CDCl3):δ7.41(dd,J=8.22および3.52Hz,1H)、7.28(d,J=8.61および8.22Hz,1H)、3.73−3.69(m,6H)、2.61−2.54(m,4H)。
7C:6−ブロモ−3−フルオロ−2−((3−メチルピペリジン−1−イル)メチル)ピリジン
(m/z)=288(M+H)+
7D:6−ブロモ−3−フルオロ−2−(ピペリジン−1−イルメチル)ピリジン
1H NMR(400MHz,CDCl3):δ7.38(dd,J=8.22および3.52Hz,1H)、7.26(d,J=8.61および8.22Hz,1H)、3.69(d,J=2.74Hz,2H)、2.54−2.46(m,4H)、1.61−1.53(m,4H)、1.45−1.36(m,2H)。
7E:6−ブロモ−2−((3,3−ジメチルピペリジン−1−イル)メチル)−3−フルオロピリジン
1H NMR(400MHz,CDCl3):δ7.37(dd,J=8.22および3.52Hz,1H)、7.25(d,J=8.61および8.22Hz,1H)、3.66(d,J=2.35Hz,2H)、2.41(bs,2H)、2.10(bs,2H)、1.60−1.53(m,2H)、1.22−1.13(m,2H)、0.90(s,6H)。
7F:6−ブロモ−2−((3,3−ジフルオロピペリジン−1−イル)メチル)−3−フルオロピリジン
1H NMR(400MHz,CDCl3):δ7.41(dd,J=8.22および3.52Hz,1H)、7.29(d,J=8.61および8.22Hz,1H)、3.86(d,J=2.35Hz,2H)、2.79(dd,J=11.35および10.95Hz,2H)、2.59(dd,J=5.48および5.09Hz,2H)、1.91−1.72(m,4H)。
7G:6−ブロモ−3−フルオロ−2−((3−フルオロピペリジン−1−イル)メチル)ピリジン
1H NMR(400MHz,CDCl3):δ7.40(dd,J=8.61および3.52Hz,1H)、7.28(d,J=8.61および8.22Hz,1H)、4.72−4.53(m,1H)、3.78(bs,2H)、2.97−2.86(m,1H)、2.60−2.57(m,1H)、2.55−2.47(m,1H)、2.42−2.35(m,1H)、1.92−1.75(m,2H)、1.62−1.47(m,2H)。
7H:6−ブロモ−3−フルオロ−2−((3−トリフルオロメチルピペリジン−1−イル)メチル)ピリジン
(m/z)=342(M+H)+
7I:N−((6−ブロモ−3−フルオロピリジン−2−イル)メチル)テトラヒドロ−2H−ピラン−4−アミン
1H NMR(400MHz,CDCl3):δ7.37(dd,J=8.61および3.52Hz,1H)、7.26(d,J=8.61および8.22Hz,1H)、4.04−3.95(m,4H)、3.40(td,J=11.73および1.96Hz,2H)、2.76−2.66(m,1H)、1.90−1.81(m,2H)、1.56−1.42(m,2H)。
7J:N−((6−ブロモ−3−フルオロピリジン−2−イル)メチル)シクロヘキサンアミン
1H NMR(400MHz,CDCl3):δ7.35(dd,J=8.61および3.52Hz,1H)、7.24(d,J=8.61および8.22Hz,1H)、3.96(d,J=2.35Hz,2H)、2.50−2.40(m,1H)、1.98−1.85(m,3H)、1.79−1.70(m,2H)、1.65−1.58(m,1H)、1.32−1.09(m,5H)。
7K:1−((6−ブロモ−3−フルオロピリジン−2−イル)メチル)ピペリジン−3−カルボニトリル
1H NMR(400MHz,CDCl3):δ7.41(dd,J=8.61および3.52Hz,1H)、7.29(d,J=8.61および8.22Hz,1H)、3.77(d,J=2.35Hz,2H)、2.96−2.87(m,1H)、2.82−2.72(m,1H)、2.71−2.63(m,1H)、2.62−2.53(m,1H)、2.46−2.36(m,1H)、1.93−1.71(m,2H)、1.64−1.52(m,2H)。
7L:4−((6−ブロモ−3−フルオロピリジン−2−イル)メチル)チオモルホリン
1H NMR(400MHz,CDCl3):δ7.40(dd,J=8.61および3.52Hz,1H)、7.32(d,J=8.61および8.22Hz,1H)、3.75(d,J=2.74Hz,2H)、2.87−2.81(m,4H)、2.71−2.65(m,4H)。
【0080】
実施例8
実施例5に記載されているものと類似の手順に従って、2−ブロモ−ピリジン−4−カルバルデヒドを出発原料として使用し、以下の化合物を調製した。
2−ブロモ−4−(ピペリジン−1−イルメチル)ピリジン。
1H NMR(400MHz,CDCl3):δ8.27(d,J=5.09,1H)、7.48(s,1H)、7.23(d,J=5.09Hz,1H)、3.93(s,2H)、2.41−2.31(m,4H)、1.63−1.55(m,4H)、1.50−1.39(m,2H)。
【0081】
実施例9
6−ブロモ−3−フルオロ−2−((テトラヒドロ−2H−ピラン−4−イルオキシ)メチル)ピリジン
【0082】
【化13】
i)6−ブロモ−3−フルオロ−2−メチルピリジン(0.5gr、2.63mmol)のジクロロメタン(5ml)中溶液に、室温で、N−ブロモコハク酸イミド(937mg、5.26mmol)およびアゾ−ジ−イソブチロニトリル(86mg、0.526mmol)を添加した。55℃で17時間撹拌した後、反応混合物を水の添加によってクエンチし、生成物をジクロロメタン中に抽出した。合わせた有機相をブラインで洗浄し、硫酸ナトリウム上で乾燥させ、減圧下で濃縮した。カラムクロマトグラフィーにより、6−ブロモ−2−(ブロモメチル)−3−フルオロピリジン(388mg)を清澄な油として得た。
【0083】
ii)前のステップで得られた生成物(388mg、1.44mmol)およびテトラヒドロ−2H−ピラン−4−オール(0.206ml、2.16mmol)のテトラヒドロフラン(10ml)中溶液に、水素化ナトリウム(69.3mg、2.31mmol、油中に80%分散)を添加した。室温で1時間撹拌した後、反応混合物を水の添加によってクエンチし、生成物をジクロロメタン中に抽出した。合わせた有機相をブラインで洗浄し、硫酸ナトリウム上で乾燥させ、減圧下で濃縮した。カラムクロマトグラフィーにより、標題化合物6−ブロモ−3−フルオロ−2−((テトラヒドロ−2H−ピラン−4−イルオキシ)メチル)ピリジン(177mg)を清澄な油として得た。
1H NMR(400MHz,CDCl3):δ7.44(dd,J=8.61および3.52Hz,1H)、7.31(d,J=8.61および8.22Hz,1H)、4.67(d,J=2.35Hz,2H)、3.99−3.92(m,2H)、3.72−3.64(m,1H)、3.49−3.41(m,2H)、2.00−1.91(m,2H)、1.71−1.60(m,2H)。
【0084】
実施例10
2−ブロモ−6−(シクロヘキシルメチル)ピリジン
i)2,6−ジブロモピリジン(1.37g、5.8mmol)のTHF/ヘキサン/ジエチルエーテル(1/1/3、15ml)中溶液を、窒素雰囲気下で、2.5Mのn−ブチルリチウムのヘキサン(2.43ml、6.08mmol)中溶液に−78℃で滴下により添加した。
【0085】
10分撹拌した後、シクロヘキサンカルボニトリル(633mg、5.8mmol)のTHF/ヘキサン/ジエチルエーテル(1/1/3、4ml)中溶液を添加し、反応混合物を2.5時間−78℃で撹拌した。反応混合物を室温に温め、さらに1.5時間撹拌した。反応混合物を、2M硫酸水溶液(7ml)の添加によってクエンチした。2時間激しく撹拌した後、水を添加し、生成物をジエチルエーテル中に抽出した。合わせた有機相を飽和炭酸水素ナトリウム水溶液、ブラインで洗浄し、硫酸ナトリウム上で乾燥させ、減圧下で濃縮した。カラムクロマトグラフィーにより、(6−ブロモ−ピリジン−2−イル)(シクロヘキシル)メタノン(800mg)を清澄な油として得た。
【0086】
ii)前のステップで得られた生成物(600mg、2.24mmol)および4−メチルベンゼンスルホニルヒドラジド(458mg、2.46mmol)のエタノール(2ml)中懸濁液を、1000Cに15分間マイクロ波中で加熱した。室温に冷却した後、反応混合物を減圧下で濃縮した。カラムクロマトグラフィーにより、N’−((6−ブロモピリジン−2−イル)(シクロヘキシル)メチレン)−4−メチルベンゼンスルホノヒドラジド(608mg)を白色固体として得た。
【0087】
iii)前のステップで得られた生成物(600mg、1.38mmol)のジクロロメタン4ml中溶液に、20%水素化ジイソブチルアルミニウムのトルエン(0.97g、13.8mmol)中溶液をゆっくり添加した。室温で17時間撹拌した後、pH=10まで2M水酸化ナトリウム水溶液をゆっくり添加するすることによって、反応混合物をクエンチした。生成物を酢酸エチル中に抽出し、合わせた有機相を水、ブラインで洗浄し、硫酸ナトリウム上で乾燥させ、減圧下で濃縮した。カラムクロマトグラフィーにより、標題化合物2−ブロモ−6−(シクロヘキシルメチル)ピリジンを白色固体として得た。
1H NMR(400MHz,CDCl3):δ7.42−7.21(m,3H)、3.62(d,J=7.04Hz,2H)、1.81−0.85(m,11H)。
【0088】
実施例11
2−ブロモ−6−(ジフルオロ−(テトラヒドロ−2H−ピラン−4−イル)メチル)ピリジン
【0089】
【化14】
i)(6−ブロモ−ピリジン−2−イル)−(テトラヒドロ−2H−ピラン−4−イル)メタノンを、実施例10、ステップi)に記載されているものと類似の手順に従って、テトラヒドロ−2H−ピラン−4−カルボニトリルを出発原料として使用して調製した。
【0090】
ii)前のステップで得られた生成物(200mg、0.74mmol)のジクロロメタン(2ml)中溶液に、少量ずつ6日の期間をかけて、ジエチルアミノサルファ−トリ−フルオリド(1.49g、9.25mmol)を窒素雰囲気下で添加した。完了後、反応混合物をメタノールの添加によって慎重にクエンチし、生成物を酢酸エチル中に抽出した。合わせた有機相を水、ブラインで洗浄し、硫酸ナトリウム上で乾燥させ、減圧下で濃縮した。カラムクロマトグラフィーにより、標題化合物2−ブロモ−6−(ジフルオロ−(テトラヒドロ−2H−ピラン−4−イル)メチル)ピリジン(144mg)を清澄な油として得た。
1H NMR(400MHz,CDCl3):δ7670(t,J=7.83Hz,1H)、7.60−7.54(m,3H)、4.03(dd,J=11.35および4.30Hz,2H)、3.42(td,J=11.74および2.35Hz,2H)、2.80−2.63(m,1H)、1.72(m,4H)。
【0091】
実施例12
3−イソブチル−1−(4−(6−(ピペリジン−1−イルメチル)ピリジン−2−イル)ベンジル)イミダゾリジン−2,4−ジオン
【0092】
【化15】
i)1−(4−ブロモベンジル)−3−イソブチルイミダゾリジン−2,4−ジオン(実施例1、ステップi))(2.0g、6.2mmol)、ビス(ピナコレート)ジボロン(1.6g、6.2mmol)および酢酸カリウム(1.8g、18.5mmol)のDMF(50ml)中溶液に、窒素雰囲気下で、1,1’−ビス(ジフェニルホスフィノ)フェロセンジクロロパラジウム(II)(134mg、0.18mmol)を添加した。75℃で17時間撹拌した後、反応混合物を室温に冷却した。水を添加し、生成物を酢酸エチル中に抽出した。合わせた有機相を飽和炭酸水素ナトリウム水溶液、水、ブラインで洗浄し、硫酸ナトリウム上で乾燥させ、減圧下で濃縮することにより、3−イソブチル−1−(4−(4,4,5,5−テトラメチル−(1,3,2)ジオキサボロラン−2−イル)ベンジル)イミダゾリジン−2,4−ジオン(6.8g)が黒色油として得られた。生成物をさらに精製することなく以下のステップで使用した。
【0093】
ii)前のステップで得られた生成物(1.31g、3.52mmol)および2−ブロモ−6−(ピペリジン−1−イルメチル)ピリジン(実施例5)(748mg、2.93mmol)のトルエン/エタノール(4/1、25ml)中溶液に、2M炭酸カリウム水溶液を添加した。窒素雰囲気下で15分撹拌した後、テトラキス(トリフェニルホスフィン)パラジウム(0)(85mg、0.073mmol)を添加し、この混合物を17時間75℃にて窒素雰囲気下で撹拌した。完了後、混合物を室温に冷却し、デカライトを通して濾過した。水を濾液に添加し、生成物を酢酸エチル中に抽出した。合わせた有機相を水、ブラインで洗浄し、硫酸ナトリウム上で乾燥させ、減圧下で濃縮した。カラムクロマトグラフィーにより、3−イソブチル−1−(4−(6−(ピペリジン−1−イルメチル)ピリジン−2−イル)ベンジル)イミダゾリジン−2,4−ジオン(120mg)を白色固体として得た。
1H NMR(400MHz,CDCl3):δ8.00(d,J=8.22Hz,2H)、7.72(dd,J=8.22および7.83Hz,1H)、7.56(d,J=8.22Hz,1H)、7.43(d,J=7.83Hz,1H)、7.34(d,J=8.22Hz,2H)、4.62(s,2H)、3.74(s,2H)、3.72(s,2H)、3.36(d,J=7.43Hz,2H)、2.53−2.47(m,4H)、1.65−1.58(m,4H)、1.50−1.42(m,2H)、2.15−2.05(m,1H)、0.93(d,J=6.65Hz,6H)。
【0094】
実施例12に記載されているものと類似の手順に従って、以下の化合物を調製した。
【0095】
【表1】
【0096】
実施例89
1−(4−(6−(ピペリジン−1−イルメチル)ピリジン−2−イル)ベンジル)イミダゾリジン−2,4−ジオン
【0097】
【化16】
i)2−ブロモ−6−ピペリジン−1−イルメチル−ピリジン(実施例5)(8.0g、31.4mmol)および4−ホルミルフェニルボロン酸(6.1g、40.8mmol)のトルエン/エタノール(4/1、320ml)中溶液に、2M炭酸カリウム水溶液を添加した。窒素雰囲気下で15分撹拌した後、テトラキス(トリフェニルホスフィン)パラジウム(0)(1.2g、1.02mmol)を添加した。17時間80℃にて窒素雰囲気下で撹拌した後、混合物を室温に冷却し、デカライトを通して濾過した。水を添加し、生成物を酢酸エチル中に抽出した。合わせた有機相を水、ブラインで洗浄し、硫酸ナトリウム上で乾燥させ、減圧下で濃縮した。カラムクロマトグラフィーにより、4−(6−(ピペリジン−1−イルメチル)ピリジン−2−イル)ベンズアルデヒド(7.69g)を白色固体として得た。
【0098】
ii)グリシンメチルエステル.HCl(4.79g、38.1mmol)およびトリエチルアミン4.60ml、33.0mmol)のメタノール(70ml)中溶液を、前のステップで得られた生成物(7.12g、25.4mmol)のメタノール(70ml)中溶液に、窒素雰囲気下で滴下により添加した。1時間室温で撹拌した後、水素化トリアセトキシホウ素ナトリウム(12.9g、61.0mmol)を少量ずつ30分の間添加した。17時間撹拌した後、より多くのグリシンメチルエステル.HCl(1.6g、12.7mmol)を添加し、続いて水素化トリアセトキシホウ素ナトリウム(5.38g、25.4mmol)を添加した。さらに17時間撹拌した後、反応混合物を飽和炭酸水素ナトリウム水溶液の添加によってクエンチした。生成物をジクロロメタン中に抽出し、合わせた有機相を水、ブラインで洗浄し、相分離フィルターに通して濾過し、減圧下で濃縮した。カラムクロマトグラフィーにより、メチル2−(4−(6−(ピペリジン−1−イルメチル)ピリジン−2−イル)ベンジルアミノ)アセテート(3.48g)を白色固体としてを得た。
【0099】
iii)前のステップで得られた生成物(8.03g、22.7mmol)のジオキサン/水(1/1)(100ml)中懸濁液に、カリウムシアネート(2.76g、34.1mmol)を室温で添加した。20分撹拌した後、酢酸(4.16ml、72.7mmol)を添加し、混合物をさらに17時間室温で撹拌した。反応混合物を水の添加によってクエンチし、飽和炭酸水素ナトリウム水溶液の添加によってpH=9まで塩基性化した。生成物をジクロロメタン中に抽出し、合わせた有機相をブラインで洗浄し、相分離フィルターに通して濾過し、減圧下で濃縮することにより、メチル2−(1−(4−(6−(ピペリジン−1−イルメチル)ピリジン−2−イル)ベンジル)ウレイド)アセテート(8.2g)が油として得られた。生成物をさらに精製することなく以下のステップで使用した。
【0100】
iv)前のステップで得られた生成物(8.2g、20.68mmol)のメタノール(50ml)中溶液に、ナトリウムメトキシド(2.24g、41.4mmol)を室温で添加し、反応混合物を3時間室温にて窒素雰囲気下で撹拌した。反応混合物を水中に注ぎ、飽和塩化アンモニウム水溶液の添加によって中和した。生成物をジクロロメタン中に抽出し、合わせた有機相をブラインで洗浄し、硫酸ナトリウム上で乾燥させ、減圧下で濃縮した。カラムクロマトグラフィーにより、標題化合物1−(4−(6−(ピペリジン−1−イルメチル)ピリジン−2−イル)ベンジル)イミダゾリジン−2,4−ジオン(5.8g)を白色固体として得た。
1H NMR(400MHz,CDCl3):δ7.99(d,J=8.61Hz,2H)、7.72(dd,J=7.83および7.43Hz,1H)、7.57(d,J=7.83Hz,1H)、7.42(d,J=7.43Hz,1H)、7.34(d,J=8.61Hz,2H)、4.57(s,2H)、3.77(s,2H)、3.73(s,2H)、2.56−2.48(m,4H)、1.67−1.57(m,4H)、1.50−1.42(m,2H)。
【0101】
実施例90
1−(4−(6−(ピペリジン−1−イルメチル)ピリジン−2−イル)ベンジル)−3−(3,3,3−トリフルオロ−プロピル)イミダゾリジン−2,4−ジオン
【0102】
【化17】
i)1−(4−(6−ピペリジン−1−イルメチル−ピリジン−2−イル)ベンジル)イミダゾリジン−2,4−ジオン(実施例134)(120mg、0.33mmol)、炭酸カリウム(137mg、0.99mmol)および3−ブロモ−1,1,1−トリフルオロプロパン(117mg、0.66mmol)のDMF(2.5ml)中溶液を、17時間の間50℃で撹拌した。室温に冷却した後、反応混合物を水の添加によってクエンチした。生成物を酢酸エチル中に抽出し、合わせた有機相をブラインで洗浄し、硫酸ナトリウム上で乾燥させ、減圧下で濃縮した。カラムクロマトグラフィーにより、標題化合物(50mg)を白色固体として得た。
1H NMR(400MHz,CDCl3):δ7.99(d,J=8.61Hz,2H)、7.72(dd,J=7.83および7.43Hz,1H)、7.56(d,J=7.83Hz,1H)、7.44(d,J=7.43Hz,1H)、7.33(d,J=8.61Hz,2H)、4.62(s,2H)、3.83(t,J=7.04Hz,2H)、3.76(s,2H)、3.73(s,2H)、2.60−2.45(m,6H)、1.66−1.59(m,4H)、1.51−1.41(m,2H)。
【0103】
実施例91
実施例90に記載されているものと類似の手順に従って、以下の化合物を調製した。
91A:4−(2,5−ジオキソ−3−(4−(6−(ピペリジン−1−イルメチル)ピリジン−2−イル)ベンジル)イミダゾリジン−1−イル)ブタンニトリル
1H NMR(400MHz,CDCl3):δ8.00(d,J=8.61Hz,2H)、7.72(dd,J=7.83および7.43Hz,1H)、7.56(d,J=7.83Hz,1H)、7.43(d,J=7.43Hz,1H)、7.35(d,J=8.61Hz,2H)、4.62(s,2H)、3.78(s,2H)、3.72(s,2H)、3.69(t,J=6.65Hz,2H)、2.54−2.47(m,4H)、2.44(t,J=7.04Hz,2H)、2.05(m,2H)、1.65−1.56(m,4H)、1.50−1.40(m,2H)。
91B:(R)−メチル3−(2,5−ジオキソ−3−(4−(6−(ピペリジン−1−イルメチル)ピリジン−2−イル)ベンジル)イミダゾリジン−1−イル)−2−メチルプロパノエート
1H NMR(400MHz,CDCl3):δ7.99(d,J=8.22Hz,2H)、7.72(dd,J=7.83および7.43Hz,1H)、7.56(d,J=7.83Hz,1H)、7.43(d,J=7.43Hz,1H)、7.34(d,J=8.22Hz,2H)、4.61(s,2H)、3.83(dd,J=13.69および7.83Hz,1H)、3.75(s,2H)、3.72(s,2H)、3.69(s,3H)、3.63(dd,J=14.09および6.65Hz,1H)、3.00−2.90(m,1H)、2.54−2.46(m,4H)、1.65−1.54(m,4H)、1.51−1.41(m,2H)、1.20(d,J=7.04Hz,3H)。
91C:3−(オキセタン−2−イルメチル)−1−(4−(6−(ピペリジン−1−イルメチル)ピリジン−2−イル)ベンジル)イミダゾリジン−2,4−ジオン
1H NMR(400MHz,CDCl3):δ7.99(d,J=8.61Hz,2H)、7.72(dd,J=7.83および7.43Hz,1H)、7.56(d,J=7.83Hz,1H)、7.43(d,J=7.43Hz,1H)、7.34(d,J=8.61Hz,2H)、5.08−5.00(m,1H)、4.70−4.56(m,4H)、3.99−3.93(dd,J=14.08および7.43Hz,1H)、3.78−3.70(m,5H)、2.78−2.70(m,1H)、2.55−2.45(m,4H)、1.66−1.58(m,5H)、1.50−1.41(m,2H)。
91D:3−(2−オキソテトラヒドロフラン−3−イル)−1−(4−(6−(ピペリジン−1−イルメチル)ピリジン−2−イル)ベンジル)イミダゾリジン−2,4−ジオン
1H NMR(400MHz,CDCl3):δ8.00(d,J=8.61Hz,2H)、7.73(dd,J=7.83および7.43Hz,1H)、7.57(d,J=7.83Hz,1H)、7.44(d,J=7.43Hz,1H)、7.36(d,J=8.61Hz,2H)、4.97−4.90(m,1H)、4.65−4.56(m,5H)、4.39−4.31(m,1H)、3.82(s,2H)、3.73(s,2H)、2.84−2.72(m,1H)、2.59−2.47(m,5H)、1.66−1.57(m,4H)、1.50−1.42(m,2H)。
91E:1−(4−(6−(ピペリジン−1−イルメチル)ピリジン−2−イル)ベンジル)−3−(テトラヒドロフラン−2−イル)イミダゾリジン−2,4−ジオン
1H NMR(400MHz,CDCl3):δ7.99(d,J=8.22Hz,2H)、7.72(dd,J=7.83および7.43Hz,1H)、7.56(d,J=7.83Hz,1H)、7.43(d,J=7.43Hz,1H)、7.34(d,J=8.22Hz,2H)、4.65(d,J=15.26Hz,1H)、4.58(d,J=15.26Hz,1H)、4.30−4.22(m,1H)、3.96−3.88(m,1H)、3.81−3.64(m,6H)、3.50(dd,J=13.69および4.70Hz,1H)、2.55−2.45(m,4H)、2.08−1.84(m,3H)、1.72−1.57(m,5H)、1.50−1.42(m,2H)。
【0104】
実施例92
2−アミノ−N−シクロプロピルアセトアミドトリフルオロアセテート
i)TBTU(5.1g、16.5mmol)、DIPEA(2.9ml、16.5mmol)およびシクロプロピルアミン(2.2ml、33mmol)を、BOC−Gly−OH(2.63g、15mmol)の乾燥ジクロロメタン(10ml)中溶液に添加した。17時間撹拌した後、反応混合物を減圧下で濃縮し、水を残渣に添加した。生成物を酢酸エチル中に抽出した。合わせた有機相を2M塩酸水溶液、飽和炭酸水素ナトリウム水溶液、ブラインで洗浄し、硫酸ナトリウム上で乾燥させ、減圧下で濃縮することにより、2−tert−ブチル2−(シクロプロピルアミノ)−2−オキソエチルカルバメート(847mg)が得られた。生成物をさらに精製することなく以下のステップで使用した。
【0105】
ii)TFA(65ml、875mmol)を、前のステップで得られた生成物(34.9g、163mmol)のジクロロメタン(300ml)中溶液に添加した。17時間撹拌した後、反応混合物を減圧下で濃縮した。DCM/ジイソプロピルエーテルエーテルからの結晶化により、標題化合物2−アミノ−N−シクロプロピルアセトアミドトリフルオロアセテートが得られた。
(22.2g)。1H NMR(400MHz,MeOD):δ3.62(s,2H)、2.76−2.74(m,1H)、0.75(m,2H)、0.52(m,2H)
【0106】
実施例93
3−シクロプロピル−1−(3−フルオロ−4−(5−フルオロ−6−(ピペリジン−1−イルメチル)ピリジン−2−イル)ベンジル)イミダゾリジン−2,4−ジオン
i)3−フルオロ−4−(5−フルオロ−6−(ピペリジン−1−イルメチル)ピリジン−2−イル)ベンズアルデヒドを、実施例89、ステップi)に記載されているものと類似の手順に従って、2−フルオロ−4−ホルミルフェニルボロン酸を出発原料として使用して調製した。
【0107】
ii)前のステップで得られた生成物(1.3g、4.1mmol)のメタノール(60ml)中溶液に、0℃で、KOH(4.6mg、0.08mmol)および2−アミノ−N−シクロプロピル−アセトアミドトリフルオロアセテート(1.9g、8.22mmol)を添加した。30分後0℃で、トリアセトキシ水素化ホウ素ナトリウム(2.6g、12.3mmol)を添加した。室温で17時間撹拌した後、飽和炭酸水素ナトリウム溶液の添加によって反応混合物をクエンチし、生成物をジクロロメタン中に抽出した。合わせた有機相を水、ブラインで洗浄し、硫酸ナトリウム上で乾燥させ、減圧下で濃縮した。カラムクロマトグラフィーにより、N−シクロプロピル−2−(3−フルオロ−4−(5−フルオロ−6−(ピペリジン−1−イルメチル)ピリジン−2−イル)ベンジルアミノ)アセトアミド(0.6g)を得た。
【0108】
iii)実施例3、ステップiii)に記載されているものと類似の手順に従って、前のステップで得られた生成物(0.28g)を、標題化合物3−シクロプロピル−1−(3−フルオロ−4−(5−フルオロ−6−(ピペリジン−1−イルメチル)ピリジン−2−イル)ベンジル)イミダゾリジン−2,4−ジオン(43mg)に変換した。
1H NMR(400MHz,CDCl3):δ8.00(dd,J=8.22および7.83Hz,1H)、7.74−7.68(m,1H)、7.43(dd,J=9.00および8.61Hz,1H)、7.15(dd,J=8.22および1.96Hz,1H)、7.06(dd,J=11.74および1.57Hz,1H)、4.57(s,2H)、3.81(d,J=2.35Hz,2H)、3.71(s,2H)、2.68−2.53(m,5H)、1.65−1.50(m,4H)、1.46−1.38(m,2H)、1.00(s,2H)、0.98(s,2H)。
【0109】
実施例94
1−(3−フルオロ−4−(6−(ピペリジン−1−イルメチル)ピリジン−2−イル)ベンジル)−3−イソブチルイミダゾリジン−2,4−ジオン
i)3−フルオロ−4−(6−(ピペリジン−1−イルメチル)ピリジン−2−イル)ベンズアルデヒドを、実施例89、ステップi)に記載されているものと類似の手順に従って、2−フルオロ−4−ホルミルフェニルボロン酸を出発原料として調製した。
【0110】
ii)実施例93、ステップii)に記載されているものと類似の手順に従って、前のステップで得られた生成物(1g)を、グリシンアミド塩酸を出発原料として使用して、2−(3−フルオロ−4−(6−(ピペリジン−1−イルメチル)ピリジン−2−イル)ベンジルアミノ)−アセトアミド(0.9g)に変換した。
【0111】
iii)実施例3、ステップiii)に記載されているものと類似の手順に従って、前のステップで得られた生成物(0.9g)を、1−(3−フルオロ−4−(6−(ピペリジン−1−イルメチル)ピリジン−2−イル)ベンジル)イミダゾリジン−2,4−ジオン(0.49g)に変換した。
【0112】
iv)実施例90、ステップiに記載されているものと類似の手順に従って、前のステップで得られた生成物(0.49g)を、1−(3−フルオロ−4−(6−(ピペリジン−1−イルメチル)ピリジン−2−イル)ベンジル)−3−イソブチルイミダゾリジン−2,4−ジオン(52mg)に変換した。
1H NMR(400MHz,CDCl3):δ8.00(dd,J=8.22および7.83Hz,1H)、7.73(dd,J=7.83および7.43Hz,1H)、7.65−7.60(m,1H)、7.46(d,J=7.83Hz,1H)、7.14(dd,J=7.83および1.57Hz,1H)、7.05(dd,J=11.74および1.57Hz,1H)、4.60(s,2H)、3.77(s,2H)、3.71(s,2H)、3.37(d,J=7.43Hz,2H)、2.53−2.45(m,4H)、2.14−2.06(m,1H)、1.65−1.57(m,4H)、1.50−1.42(m,2H)、0.94(d,J=6.65Hz,6H)。
【0113】
実施例95
ヒトCB2トランスフェクトCHO細胞におけるアゴニスト誘導cAMP変化
アデニル酸シクラーゼアッセイを、ヒト組換えCB2受容体を安定に過剰発現するCHO細胞を使用して実施した。1%(v/v)ペニシリン/ストレプトマイシン(ギブコ15140−122)、10%ウシ胎児血清(FBS)および400μg/mlのジェネティシン(インビトロジェン10131−027)を含有するDMEM/HAMF12中で細胞を培養した。化合物および基準物(CP55,940)をDMSO中に溶解し、2μMのRolipram(シグマR6520)および1μMのフォルスコリン(シグマ、F3917)を含有する血清フリーの培養液中で希釈物を作製した。各希釈物10μlをアッセイプレート(384ウェル白色培養プレート、パーキンエルマー)に移動した。1%(v/v)ペニシリン/ストレプトマイシンを含有するDMEM/HAMF12中に5×105細胞/mlを含有する細胞懸濁液を、hCB2_C2−CHO細胞から調製し、これらのうち10μl(5,000細胞/ウェル)をアッセイプレートに移動し、細胞を45分間37℃でインキュベートした。均一時間分解蛍光(HTRF;CisBio)を、10μlのcAMP−XL665および10μlの抗cAMP(Eu)クリプテートを順次添加することによって読み出しとして使用し、室温で1時間インキュベーションした後、615nmおよび665nmでの蛍光をEnvision(パーキンエルマー)で測定した。結果は個々の化合物について665nm/615nm比から算出し、対照化合物について得られた値と比較した。実施例12−88、90、91、93および94からの化合物は、CB2に関してEC50≦1×10−7Mを有する。
【0114】
実施例95A
ヒトCB1トランスフェクトCHO細胞におけるアゴニスト誘導cAMP変化
アデニル酸シクラーゼアッセイを、ヒト組換えCB1受容体を安定に過剰発現するCHO細胞を使用して実施した。1%(v/v)ペニシリン/ストレプトマイシン(ギブコ15140−122)、10%ウシ胎児血清(FBS)、400μg/mlのジェネティシン(インビトロジェン10131−027)およびZeocine250μg/ml(インビトロジェン、45−0430)を含有するDMEM/HAMF12中で細胞を培養した。化合物および基準物(CP55,940)をDMSO中に溶解し、2μMのRolipram(シグマR6520)および1μMのフォルスコリン(シグマ、F3917)を含有する血清フリーの培養液中で希釈物を作製した。各希釈物10μlをアッセイプレート(384ウェル白色培養プレート、パーキンエルマー)に移動した。1%(v/v)ペニシリン/ストレプトマイシンを含有するDMEM/HAMF12中に5×105細胞/mlを含有する細胞懸濁液を、hCB1_A2−CHO細胞から調製し、これらのうち10μl(5,000細胞/ウェル)をアッセイプレートに移動し、細胞を45分間37℃でインキュベートした。均一時間分解蛍光(HTRF;CisBio)を、10μlのcAMP−XL665および10μlの抗cAMP(Eu)クリプテートを順次添加することによって読み出しとして使用し、室温で1時間インキュベーションした後、615nmおよび665nmでの蛍光をEnvision(パーキンエルマー)で測定した。結果は個々の化合物について665nm/615nm比から算出し、対照化合物について得られた値と比較した。実施例12−88、90、91、93および94からの化合物は、CB1に関してEC50≧1×10−7Mを有する。
【0115】
実施例96
神経因性疼痛のラット(Chung)モデル
このモデルにおいて、機械的アロディニアは、左L5脊髄神経の緊密な連結によって誘発される。このアッセイを用いることによって、神経因性疼痛の治療において臨床的に使用される抗痙攣薬(ガバペンチン)、抗うつ薬(デュロキセチン)およびオピオイド鎮痛薬(モルヒネ)の抗アロディニア効果を実証するのに成功した。
【0116】
雄性Wistarラット(外科手術時の体重228−301g)を前記試験に用いた。パースペックスボックス内の高くした(約40cm)メッシュ床の上にラットを置き、機械的刺激(較正フォンフライフィラメント)に対するラットの引き込み閾値を、上記した通り、増加する力(2.6−167mN)のフィラメントを使用して測定した。フォンフライフィラメントを足の足底表面に適用し、上げ下げ法を使用して閾値応答を決定した。足を鋭く引っ込めると陽性応答が認められた。15gのカットオフを試験の上限として選択した。ベースライン測定に続いて、各動物に麻酔し、L5脊髄神経を緊密に連結した。動物を少なくとも3日の期間外科手術から回復させた。薬物投与当日、足引き込み閾値を再び測定した(0分)。この読み取り直後、ラットにビヒクルまたは試験化合物を経口投与し、化合物投与後様々な時点で読み取りを測定した。
【0117】
データは平均±s.e.m.として示された。非パラメーター統計試験のKruskal−Wallis一元配置分散分析を使用して統計分析を行った。治療群のそれぞれを次いで、非パラメーターDunn試験を使用してビヒクル群に対して比較した。
【0118】
一例として、選択的CB2受容体アゴニスト3−イソブチル−1−(4−(6−(ピペリジン−1−イルメチル)ピリジン−2−イル)ベンジル)イミダゾリジン−2,4−ジオン(実施例12)の経口投与により、用量依存的様式(表1;図1)において薬物投与後120および180分でそれぞれ機械的アロディニアが減弱した。最小有効量(MED)は43.8μmol/kgであった。これらのデータは、選択的CB2受容体アゴニストが神経因性疼痛のラットモデルにおいて強力な経口抗アロディニア作用を保有していることを実証している。
【0119】
【表2】
【0120】
実施例142
ラットにおける機械的痛覚過敏
炎症性疼痛のこのラットモデルにおいて、炎症は完全フロイントアジュバント(CFA)の皮下注入によって後足に誘発される。伴われる機械的痛覚過敏を、足の機械的圧縮に対する足引き込み閾値(PWT)の低減を測定することによって定量化する。このアッセイを用いて、炎症性疼痛の治療において臨床的に使用される非ステロイド系抗炎症薬(インドメタシン)およびコキシブ(セレコキシブ)の抗痛覚過敏効果を実証するのに成功した。
【0121】
実験は、雄性Wistarラット体重(141−175g)を使用して行われた。簡潔には、後足の機械的圧縮に対するラットの足引き込み閾値(PWT)を、Randall−Sellito装置(Ugo Basile)を使用して測定した(ベースライン読み取り)。足に対する組織損傷を最小にするため、20gのカットオフを用いた。動物に次いでイソフルラン(1−3%)で軽く麻酔し、完全フロイントアジュバント(1つの足当たり0.1ml)を左後足の足底表面に皮下注入(s.c.)した。動物を次いでこれらのホームケージに戻し、放置して炎症を発生させた。CFA注入の24時間後、PWTを再び測定し(0分)、この読み取り直後、ラットにビヒクルまたは試験化合物(化合物12の4.4−43.8μmol/kg p.o.)のいずれかを経口投与した。次いで薬物投与の3時間後に読み取りを行った。
【0122】
データを平均±s.e.m.としてプロットし、非パラメーター統計試験のKruskal−Wallis一元配置分散分析を使用して群間で比較した。この試験で統計的有意性(P<0.05)が観察された場合に、非パラメーターDunn試験を使用してビヒクル群および治療群のそれぞれを比較した。機械的痛覚過敏の減弱パーセントを以下の通りに算出した。
【0123】
【数1】
【0124】
化合物12(4.4−43.8μmol/kg)の経口投与は、用量依存的様式においてCFAによって誘発された機械的痛覚過敏を逆転させた(表2)。Org266919−1のMEDは13.2μmol/kgであった。
【0125】
これらのデータは、選択的CB2受容体アゴニストが炎症性疼痛のラットモデルにおける強力な経口抗発痛作用を保有していることを実証している。
【0126】
【表3】
【特許請求の範囲】
【請求項1】
一般式I
【化1】
[式中、
R1は、H、(C1−6)アルキル(オキソ、(C1−3)アルキルオキシ、(C1−3)アルキルオキシ−カルボニル、ハロゲンまたはCNで場合によって置換されている。)、(C3−6)シクロアルキルまたは(C3−6)シクロアルキル(C1−3)アルキルであり、各シクロアルキル環は、OおよびSから選択されるヘテロ原子を場合によって含み、
R2およびR3は独立して、Hもしくは(C1−3)アルキルであり、または
R2およびR3は、これらが結合している炭素原子と一緒になって(C3−5)−シクロアルキル基を形成し、
R4は、Hまたは1個から3個のF置換基であり、
R5は、Hまたは1個から4個のF置換基であり、
R6およびR7は独立して、HまたはFであり、
Xは、R8、OR8、NR8R9、
【化2】
を表し、
R8は、O、S、SOおよびSO2から選択されるヘテロ原子を場合によって含む(C5−7)シクロアルキルであり、
R9は、Hまたは(C1−4)アルキルであり、
R10は、H、(C1−3)アルキル、ハロゲン、オキソ、CNおよびCF3から独立して選択される1−3個の置換基を表し、
Yは、CF2、O、S、SOまたはSO2である。]
を有する1−(4−(ピリジン−2−イル)ベンジル)イミダゾリジン−2,4−ジオン誘導体または医薬として許容できるこの塩。
【請求項2】
R2、R3およびR5がHである、請求項1に記載の1−(4−(ピリジン−2−イル)ベンジル)イミダゾリジン−2,4−ジオン誘導体。
【請求項3】
R1が(C1−4)アルキルである、請求項1または2に記載の1−(4−(ピリジン−2−イル)ベンジル)イミダゾリジン−2,4−ジオン誘導体。
【請求項4】
XがNR8R9
【化3】
を表す、請求項1、2または3に記載の1−(4−(ピリジン−2−イル)ベンジル)イミダゾリジン−2,4−ジオン誘導体。
【請求項5】
XがNR8R9であり、R8がOおよびSから選択されるヘテロ原子を場合によって含むシクロヘキシルである、請求項4に記載の1−(4−(ピリジン−2−イル)ベンジル)イミダゾリジン−2,4−ジオン誘導体。
【請求項6】
R4がCR6R7X基に対してオルト位のF置換基である、請求項5に記載の1−(4−(ピリジン−2−イル)ベンジル)イミダゾリジン−2,4−ジオン誘導体。
【請求項7】
−3−イソブチル−1−(4−(6−(ピペリジン−1−イルメチル)ピリジン−2−イル)ベンジル)イミダゾリジン−2,4−ジオン;
−3−イソブチル−1−(4−(6−(モルホリノメチル)ピリジン−2−イル)ベンジル)イミダゾリジン−2,4−ジオン;
−1−(4−(5−フルオロ−6−((テトラヒドロ−2H−ピラン−4−イルアミノ)メチル)ピリジン−2−イル)ベンジル)−3−イソブチルイミダゾリジン−2,4−ジオン;
−1−(4−(6−((1,1−ジオキソ−1λ6−チオモルホリン−4−イル)メチル)ピリジン−2−イル)ベンジル)−3−イソブチルイミダゾリジン−2,4−ジオン;
−1−(4−(6−((1,1−ジオキソ−1λ6−チオモルホリン−4−イル)メチル)−5−フルオロピリジン−2−イル)ベンジル)−3−イソブチルイミダゾリジン−2,4−ジオン;
−3−イソブチル−1−(4−(6−((テトラヒドロ−2H−ピラン−4−イルアミノ)メチル)ピリジン−2−イル)ベンジル)イミダゾリジン−2,4−ジオン;
−3−エチル−1−(4−(6−(ピペリジン−1−イルメチル)ピリジン−2−イル)ベンジル)イミダゾリジン−2,4−ジオン;
−3−エチル−1−(4−(5−フルオロ−6−(ピペリジン−1−イルメチル)ピリジン−2−イル)ベンジル)イミダゾリジン−2,4−ジオン;
−1−(4−(6−(1,1−ジオキソ−1λ6−チオモルホリン−4−イルメチル)−5−フルオロピリジン−2−イル)ベンジル)−3−エチルイミダゾリジン−2,4−ジオン;
−1−(4−(6−(1,1−ジオキソ−1λ6−チオモルホリン−4−イルメチル)ピリジン−2−イル)ベンジル)−3−エチルイミダゾリジン−2,4−ジオン;
−1−(4−(5−フルオロ−6−((テトラヒドロ−2H−ピラン−4−イルアミノ)メチル)ピリジン−2−イル)ベンジル)−3−プロピルイミダゾリジン−2,4−ジオン;
−1−(4−(6−(1,1−ジオキソ−1λ6−チオモルホリン−4−イルメチル)−5−フルオロピリジン−2−イル)ベンジル)−3−プロピルイミダゾリジン−2,4−ジオン;
−3−(2,2−ジフルオロエチル)−1−(4−(6−(ピペリジン−1−イルメチル)ピリジン−2−イル)ベンジル)イミダゾリジン−2,4−ジオン;
−3−(2,2−ジフルオロエチル)−1−(4−(5−フルオロ−6−(ピペリジン−1−イルメチル)ピリジン−2−イル)ベンジル)イミダゾリジン−2,4−ジオン;
−3−(シクロプロピルメチル)−1−(4−(5−フルオロ−6−(ピペリジン−1−イルメチル)ピリジン−2−イル)ベンジル)イミダゾリジン−2,4−ジオン;
−3−(シクロプロピルメチル)−1−(4−(6−(ピペリジン−1−イルメチル)ピリジン−2−イル)ベンジル)イミダゾリジン−2,4−ジオン;
−3−(2−オキソプロピル)−1−(4−(6−(ピペリジン−1−イルメチル)ピリジン−2−イル)ベンジル)イミダゾリジン−2,4−ジオン;
−1−(4−(5−フルオロ−6−(ピペリジン−1−イルメチル)ピリジン−2−イル)ベンジル)−3−(2−オキソプロピル)イミダゾリジン−2,4−ジオン;
−1−(4−(6−(1,1−ジオキソ−1λ6−チオモルホリン−4−イルメチル)−5−フルオロピリジン−2−イル)ベンジル)−3−(2−オキソプロピル)イミダゾリジン−2,4−ジオン;
−3−ルソプロピル−1−(4−(6−(ピペリジン−1−イルメチル)ピリジン−2−イル)ベンジル)イミダゾリジン−2,4−ジオン;
−1−(4−(6−(1,1−ジオキソ−1λ6−チオモルホリン−4−イルメチル)−5−フルオロピリジン−2−イル)ベンジル)−3−イソプロピルイミダゾリジン−2,4−ジオン;
−1−(4−(6−1,1−ジオキソ−1λ6−チオモルホリン−4−イルメチル)ピリジン−2−イル)ベンジル)−3−イソプロピルイミダゾリジン−2,4−ジオン;
−3−シクロプロピル−1−(4−(5−フルオロ−6−(ピペリジン−1−イルメチル)ピリジン−2−イル)ベンジル)イミダゾリジン−2,4−ジオン;
−3−シクロプロピル−1−(4−(6−(1,1−ジオキソ−1λ6−チオモルホリン−4−イルメチル)−5−フルオロピリジン−2−イル)ベンジル)イミダゾリジン−2,4−ジオン;
−3−シクロブチル−1−(4−(5−フルオロ−6−((テトラヒドロ−2H−ピラン−4−イルアミノ)メチル)ピリジン−2−イル)ベンジル)イミダゾリジン−2,4−ジオン;
−3−シクロブチル−1−(4−(6−((テトラヒドロ−2H−ピラン−4−イルアミノ)メチル)ピリジン−2−イル)ベンジル)イミダゾリジン−2,4−ジオン;
−3−シクロブチル−1−(4−(6−(1,1−ジオキソ−1λ6−チオモルホリン−4−イルメチル)−5−フルオロピリジン−2−イル)ベンジル)イミダゾリジン−2,4−ジオン;
−3−シクロブチル−1−(4−(6−(1,1−ジオキソ−1λ6−チオモルホリン−4−イルメチル)ピリジン−2−イル)ベンジル)イミダゾリジン−2,4−ジオン;
−1−(4−(6−(ピペリジン−1−イルメチル)ピリジン−2−イル)ベンジル)−3−(2,2,2−トリフルオロエチル)イミダゾリジン−2,4−ジオン;
−1−(4−(6−(1,1−ジオキソ−1λ6−チオモルホリン−4−イルメチル)−5−フルオロピリジン−2−イル)ベンジル−3−(2,2,2−トリフルオロエチル)イミダゾリジン−2,4−ジオン;
−1−(4−(6−((テトラヒドロ−2H−ピラン−4−イルアミノ)メチル)ピリジン−2−イル)ベンジル)−3−(2,2,2−トリフルオロエチル)イミダゾリジン−2,4−ジオン;
−1−(4−(5−フルオロ−6−((テトラヒドロ−2H−ピラン−4−イルアミノ)メチル)ピリジン−2−イル)ベンジル)−3−(2,2,2−トリフルオロエチル)イミダゾリジン−2,4−ジオン
から選択される、請求項1に記載の1−(4−(ピリジン−2−イル)ベンジル)イミダゾリジン−2,4−ジオン誘導体または医薬として許容できるこれらの塩。
【請求項8】
治療における使用のための、請求項1から7のいずれか一項に記載の1−(4−(ピリジン−2−イル)ベンジル)イミダゾリジン−2,4−ジオン誘導体。
【請求項9】
疼痛の治療のための、請求項1から7のいずれか一項に記載の1−(4−(ピリジン−2−イル)ベンジル)イミダゾリジン−2,4−ジオン誘導体。
【請求項10】
医薬として許容できる助剤との混合で、請求項1から7のいずれか一項に記載の1−(4−(ピリジン−2−イル)ベンジル)イミダゾリジン−2,4−ジオン誘導体を含む医薬組成物。
【請求項11】
疼痛の治療用薬物の調製における、請求項1に記載の式Iの1−(4−(ピリジン−2−イル)ベンジル)イミダゾリジン−2,4−ジオン誘導体の使用。
【請求項12】
請求項1から7のいずれか一項に記載の1−(4−(ピリジン−2−イル)ベンジル)イミダゾリジン−2,4−ジオン誘導体の治療有効量を、これを必要とする患者に投与することによって、手術前後の疼痛、慢性疼痛、神経因性疼痛、癌疼痛などの疼痛ならびに多発性硬化症に伴う疼痛および痙直を治療する方法。
【請求項1】
一般式I
【化1】
[式中、
R1は、H、(C1−6)アルキル(オキソ、(C1−3)アルキルオキシ、(C1−3)アルキルオキシ−カルボニル、ハロゲンまたはCNで場合によって置換されている。)、(C3−6)シクロアルキルまたは(C3−6)シクロアルキル(C1−3)アルキルであり、各シクロアルキル環は、OおよびSから選択されるヘテロ原子を場合によって含み、
R2およびR3は独立して、Hもしくは(C1−3)アルキルであり、または
R2およびR3は、これらが結合している炭素原子と一緒になって(C3−5)−シクロアルキル基を形成し、
R4は、Hまたは1個から3個のF置換基であり、
R5は、Hまたは1個から4個のF置換基であり、
R6およびR7は独立して、HまたはFであり、
Xは、R8、OR8、NR8R9、
【化2】
を表し、
R8は、O、S、SOおよびSO2から選択されるヘテロ原子を場合によって含む(C5−7)シクロアルキルであり、
R9は、Hまたは(C1−4)アルキルであり、
R10は、H、(C1−3)アルキル、ハロゲン、オキソ、CNおよびCF3から独立して選択される1−3個の置換基を表し、
Yは、CF2、O、S、SOまたはSO2である。]
を有する1−(4−(ピリジン−2−イル)ベンジル)イミダゾリジン−2,4−ジオン誘導体または医薬として許容できるこの塩。
【請求項2】
R2、R3およびR5がHである、請求項1に記載の1−(4−(ピリジン−2−イル)ベンジル)イミダゾリジン−2,4−ジオン誘導体。
【請求項3】
R1が(C1−4)アルキルである、請求項1または2に記載の1−(4−(ピリジン−2−イル)ベンジル)イミダゾリジン−2,4−ジオン誘導体。
【請求項4】
XがNR8R9
【化3】
を表す、請求項1、2または3に記載の1−(4−(ピリジン−2−イル)ベンジル)イミダゾリジン−2,4−ジオン誘導体。
【請求項5】
XがNR8R9であり、R8がOおよびSから選択されるヘテロ原子を場合によって含むシクロヘキシルである、請求項4に記載の1−(4−(ピリジン−2−イル)ベンジル)イミダゾリジン−2,4−ジオン誘導体。
【請求項6】
R4がCR6R7X基に対してオルト位のF置換基である、請求項5に記載の1−(4−(ピリジン−2−イル)ベンジル)イミダゾリジン−2,4−ジオン誘導体。
【請求項7】
−3−イソブチル−1−(4−(6−(ピペリジン−1−イルメチル)ピリジン−2−イル)ベンジル)イミダゾリジン−2,4−ジオン;
−3−イソブチル−1−(4−(6−(モルホリノメチル)ピリジン−2−イル)ベンジル)イミダゾリジン−2,4−ジオン;
−1−(4−(5−フルオロ−6−((テトラヒドロ−2H−ピラン−4−イルアミノ)メチル)ピリジン−2−イル)ベンジル)−3−イソブチルイミダゾリジン−2,4−ジオン;
−1−(4−(6−((1,1−ジオキソ−1λ6−チオモルホリン−4−イル)メチル)ピリジン−2−イル)ベンジル)−3−イソブチルイミダゾリジン−2,4−ジオン;
−1−(4−(6−((1,1−ジオキソ−1λ6−チオモルホリン−4−イル)メチル)−5−フルオロピリジン−2−イル)ベンジル)−3−イソブチルイミダゾリジン−2,4−ジオン;
−3−イソブチル−1−(4−(6−((テトラヒドロ−2H−ピラン−4−イルアミノ)メチル)ピリジン−2−イル)ベンジル)イミダゾリジン−2,4−ジオン;
−3−エチル−1−(4−(6−(ピペリジン−1−イルメチル)ピリジン−2−イル)ベンジル)イミダゾリジン−2,4−ジオン;
−3−エチル−1−(4−(5−フルオロ−6−(ピペリジン−1−イルメチル)ピリジン−2−イル)ベンジル)イミダゾリジン−2,4−ジオン;
−1−(4−(6−(1,1−ジオキソ−1λ6−チオモルホリン−4−イルメチル)−5−フルオロピリジン−2−イル)ベンジル)−3−エチルイミダゾリジン−2,4−ジオン;
−1−(4−(6−(1,1−ジオキソ−1λ6−チオモルホリン−4−イルメチル)ピリジン−2−イル)ベンジル)−3−エチルイミダゾリジン−2,4−ジオン;
−1−(4−(5−フルオロ−6−((テトラヒドロ−2H−ピラン−4−イルアミノ)メチル)ピリジン−2−イル)ベンジル)−3−プロピルイミダゾリジン−2,4−ジオン;
−1−(4−(6−(1,1−ジオキソ−1λ6−チオモルホリン−4−イルメチル)−5−フルオロピリジン−2−イル)ベンジル)−3−プロピルイミダゾリジン−2,4−ジオン;
−3−(2,2−ジフルオロエチル)−1−(4−(6−(ピペリジン−1−イルメチル)ピリジン−2−イル)ベンジル)イミダゾリジン−2,4−ジオン;
−3−(2,2−ジフルオロエチル)−1−(4−(5−フルオロ−6−(ピペリジン−1−イルメチル)ピリジン−2−イル)ベンジル)イミダゾリジン−2,4−ジオン;
−3−(シクロプロピルメチル)−1−(4−(5−フルオロ−6−(ピペリジン−1−イルメチル)ピリジン−2−イル)ベンジル)イミダゾリジン−2,4−ジオン;
−3−(シクロプロピルメチル)−1−(4−(6−(ピペリジン−1−イルメチル)ピリジン−2−イル)ベンジル)イミダゾリジン−2,4−ジオン;
−3−(2−オキソプロピル)−1−(4−(6−(ピペリジン−1−イルメチル)ピリジン−2−イル)ベンジル)イミダゾリジン−2,4−ジオン;
−1−(4−(5−フルオロ−6−(ピペリジン−1−イルメチル)ピリジン−2−イル)ベンジル)−3−(2−オキソプロピル)イミダゾリジン−2,4−ジオン;
−1−(4−(6−(1,1−ジオキソ−1λ6−チオモルホリン−4−イルメチル)−5−フルオロピリジン−2−イル)ベンジル)−3−(2−オキソプロピル)イミダゾリジン−2,4−ジオン;
−3−ルソプロピル−1−(4−(6−(ピペリジン−1−イルメチル)ピリジン−2−イル)ベンジル)イミダゾリジン−2,4−ジオン;
−1−(4−(6−(1,1−ジオキソ−1λ6−チオモルホリン−4−イルメチル)−5−フルオロピリジン−2−イル)ベンジル)−3−イソプロピルイミダゾリジン−2,4−ジオン;
−1−(4−(6−1,1−ジオキソ−1λ6−チオモルホリン−4−イルメチル)ピリジン−2−イル)ベンジル)−3−イソプロピルイミダゾリジン−2,4−ジオン;
−3−シクロプロピル−1−(4−(5−フルオロ−6−(ピペリジン−1−イルメチル)ピリジン−2−イル)ベンジル)イミダゾリジン−2,4−ジオン;
−3−シクロプロピル−1−(4−(6−(1,1−ジオキソ−1λ6−チオモルホリン−4−イルメチル)−5−フルオロピリジン−2−イル)ベンジル)イミダゾリジン−2,4−ジオン;
−3−シクロブチル−1−(4−(5−フルオロ−6−((テトラヒドロ−2H−ピラン−4−イルアミノ)メチル)ピリジン−2−イル)ベンジル)イミダゾリジン−2,4−ジオン;
−3−シクロブチル−1−(4−(6−((テトラヒドロ−2H−ピラン−4−イルアミノ)メチル)ピリジン−2−イル)ベンジル)イミダゾリジン−2,4−ジオン;
−3−シクロブチル−1−(4−(6−(1,1−ジオキソ−1λ6−チオモルホリン−4−イルメチル)−5−フルオロピリジン−2−イル)ベンジル)イミダゾリジン−2,4−ジオン;
−3−シクロブチル−1−(4−(6−(1,1−ジオキソ−1λ6−チオモルホリン−4−イルメチル)ピリジン−2−イル)ベンジル)イミダゾリジン−2,4−ジオン;
−1−(4−(6−(ピペリジン−1−イルメチル)ピリジン−2−イル)ベンジル)−3−(2,2,2−トリフルオロエチル)イミダゾリジン−2,4−ジオン;
−1−(4−(6−(1,1−ジオキソ−1λ6−チオモルホリン−4−イルメチル)−5−フルオロピリジン−2−イル)ベンジル−3−(2,2,2−トリフルオロエチル)イミダゾリジン−2,4−ジオン;
−1−(4−(6−((テトラヒドロ−2H−ピラン−4−イルアミノ)メチル)ピリジン−2−イル)ベンジル)−3−(2,2,2−トリフルオロエチル)イミダゾリジン−2,4−ジオン;
−1−(4−(5−フルオロ−6−((テトラヒドロ−2H−ピラン−4−イルアミノ)メチル)ピリジン−2−イル)ベンジル)−3−(2,2,2−トリフルオロエチル)イミダゾリジン−2,4−ジオン
から選択される、請求項1に記載の1−(4−(ピリジン−2−イル)ベンジル)イミダゾリジン−2,4−ジオン誘導体または医薬として許容できるこれらの塩。
【請求項8】
治療における使用のための、請求項1から7のいずれか一項に記載の1−(4−(ピリジン−2−イル)ベンジル)イミダゾリジン−2,4−ジオン誘導体。
【請求項9】
疼痛の治療のための、請求項1から7のいずれか一項に記載の1−(4−(ピリジン−2−イル)ベンジル)イミダゾリジン−2,4−ジオン誘導体。
【請求項10】
医薬として許容できる助剤との混合で、請求項1から7のいずれか一項に記載の1−(4−(ピリジン−2−イル)ベンジル)イミダゾリジン−2,4−ジオン誘導体を含む医薬組成物。
【請求項11】
疼痛の治療用薬物の調製における、請求項1に記載の式Iの1−(4−(ピリジン−2−イル)ベンジル)イミダゾリジン−2,4−ジオン誘導体の使用。
【請求項12】
請求項1から7のいずれか一項に記載の1−(4−(ピリジン−2−イル)ベンジル)イミダゾリジン−2,4−ジオン誘導体の治療有効量を、これを必要とする患者に投与することによって、手術前後の疼痛、慢性疼痛、神経因性疼痛、癌疼痛などの疼痛ならびに多発性硬化症に伴う疼痛および痙直を治療する方法。
【図1】

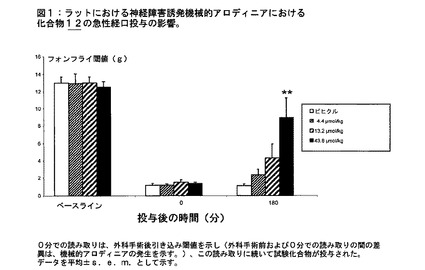
【公表番号】特表2012−510496(P2012−510496A)
【公表日】平成24年5月10日(2012.5.10)
【国際特許分類】
【出願番号】特願2011−538979(P2011−538979)
【出願日】平成21年11月30日(2009.11.30)
【国際出願番号】PCT/EP2009/066030
【国際公開番号】WO2010/063666
【国際公開日】平成22年6月10日(2010.6.10)
【出願人】(398057282)ナームローゼ・フエンノートチヤツプ・オルガノン (93)
【Fターム(参考)】
【公表日】平成24年5月10日(2012.5.10)
【国際特許分類】
【出願日】平成21年11月30日(2009.11.30)
【国際出願番号】PCT/EP2009/066030
【国際公開番号】WO2010/063666
【国際公開日】平成22年6月10日(2010.6.10)
【出願人】(398057282)ナームローゼ・フエンノートチヤツプ・オルガノン (93)
【Fターム(参考)】
[ Back to top ]