
1,2−ジヒドロピリジン化合物の結晶の製造方法
【課題】3−(2−シアノフェニル)−5−(2−ピリジル)−1−フェニル−1,2−ジヒドロピリジン−2−オンの水和物の結晶の製造方法の提供。
【解決手段】3−(2−シアノフェニル)−5−(2−ピリジル)−1−フェニル−1,2−ジヒドロピリジン−2−オンを、アルコール系溶媒、アルキルケトン系溶媒および水からなる群から選ばれる1または2の溶媒を結晶化溶媒として用いて結晶化させることを特徴とする3−(2−シアノフェニル)−5−(2−ピリジル)−1−フェニル−1,2−ジヒドロピリジン−2−オンの水和物の結晶の製造方法。
【解決手段】3−(2−シアノフェニル)−5−(2−ピリジル)−1−フェニル−1,2−ジヒドロピリジン−2−オンを、アルコール系溶媒、アルキルケトン系溶媒および水からなる群から選ばれる1または2の溶媒を結晶化溶媒として用いて結晶化させることを特徴とする3−(2−シアノフェニル)−5−(2−ピリジル)−1−フェニル−1,2−ジヒドロピリジン−2−オンの水和物の結晶の製造方法。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、AMPA(α−アミノ−3−ヒドロキシ−5−メチル−4−イソオキサゾールプロピオン酸)受容体拮抗作用および/またはカイニン酸受容体阻害作用を有する神経変性疾患等の治療剤または予防剤として有用な1,2−ジヒドロピリジン化合物(3−(2−シアノフェニル)−5−(2−ピリジル)−1−フェニル−1,2−ジヒドロピリジン−2−オン)の結晶の製造方法に関する。
【背景技術】
【0002】
1,2−ジヒドロピリジン化合物は、AMPA受容体拮抗作用および/またはカイニン酸受容体阻害作用を有する神経変性疾患等の治療剤または予防剤として有用であり、中でも3−(2−シアノフェニル)−5−(2−ピリジル)−1−フェニル−1,2−ジヒドロピリジン−2−オン(以下、化合物(1)と示す。)は顕著なAMPA受容体拮抗作用を示す(特許文献1参照)。
化合物(1)については特許文献1の実施例7に製造方法の開示があるが、「残渣をシリカゲルカラムクロマト(酢酸エチル/ヘキサン 1:2)で精製して」と記載されているのみで、得られた化合物の態様については開示されていない。
【0003】
【特許文献1】国際公開第01/96308号パンフレット
【発明の開示】
【発明が解決しようとする課題】
【0004】
結晶多形が存在する化合物を医薬品として用いる場合、医薬品として要求される均一な品質および一定の作用強度を確保するために、均一の結晶形を有する化合物を安定して供給することが必要である。また、保存中および製剤化工程(混合、造粒等)において、同一品質を維持できる結晶形が望まれる。
【0005】
医薬品原薬は、工業的に大量に取り扱うことから、爆発性、危険性の指標の1つである爆発下限濃度や最小着火エネルギーが高い結晶形が望まれる。
【0006】
一般に帯電する粉体は、他の物体への付着性も高く保護具や皮膚への付着が懸念される。
医薬品原薬が帯電性を有している場合、化合物製造時の粉砕の段階において粉砕刃に化合物が付着したり、製剤化の工程において生産機器に付着凝集したりすると、生産効率や作業性が悪化することがあり、また、帯電性を有する大量の粉体を工業的に扱う場合には粉塵爆発の起こる可能性があることから、医薬品原薬としては、帯電性がより弱い化合物(結晶)が望まれる。
また、医薬品原薬のような高い薬理活性を持つ化合物については、作業者の暴露の抑制や施設汚染防止の観点からも帯電しにくい粉体が望ましい。
【0007】
そのため、医薬の有効成分が結晶性物質として得られる場合は単一の結晶形からなり、安定した良好な物性を持ち、金属等の不純物を含まないことが望ましい。また、そのような結晶を安定して工業的規模で製造できる方法の開発も望まれている。
そこで、本発明は、化合物(1)の単一の結晶形からなる結晶およびその製造方法を提供することを目的とする。
【課題を解決するための手段】
【0008】
本発明者らは、精力的に研究を重ねた結果、化合物(1)の晶析に際して特定の結晶化溶媒を用いることにより、単一の結晶形からなる化合物(1)が得られることを見出して本発明を完成した。
すなわち、本発明は、
〔1〕 粉末X線回折において、回折角度(2θ±0.2°)8.7°に回折ピークを有する、3−(2−シアノフェニル)−5−(2−ピリジル)−1−フェニル−1,2−ジヒドロピリジン−2−オンの水和物の結晶(水和物結晶)。
〔2〕 X線回折において、回折角度(2θ±0.2°)12.5°に回折ピークを有する、3−(2−シアノフェニル)−5−(2−ピリジル)−1−フェニル−1,2−ジヒドロピリジン−2−オンの水和物の結晶(水和物結晶)。
〔3〕 粉末X線回折において、回折角度(2θ±0.2°)8.7°および12.5°に回折ピークを有する、3−(2−シアノフェニル)−5−(2−ピリジル)−1−フェニル−1,2−ジヒドロピリジン−2−オンの水和物の結晶(水和物結晶)。
〔4〕 赤外吸収スペクトル(KBr法)において、波数1588±1cm−1に吸収ピークを有する、3−(2−シアノフェニル)−5−(2−ピリジル)−1−フェニル−1,2−ジヒドロピリジン−2−オンの水和物の結晶(水和物結晶)。
〔5〕 赤外吸収スペクトル(KBr法)において、波数1588±1cm−1および751±1cm−1に吸収ピークを有する、3−(2−シアノフェニル)−5−(2−ピリジル)−1−フェニル−1,2−ジヒドロピリジン−2−オンの水和物の結晶(水和物結晶)。
〔5−2〕 赤外吸収スペクトル(KBr法)において、波数1588±1cm−1に吸収ピークを有する、前記〔1〕〜〔3〕のいずれか1記載の結晶(水和物結晶)。
〔5−3〕 赤外吸収スペクトル(KBr法)において、波数1588±1cm−1および751±1cm−1に吸収ピークを有する、前記〔1〕〜〔3〕のいずれか1記載の結晶(水和物結晶)。
〔5−4〕 パラジウムの含量が20ppm以下(好ましくは15ppm以下)である、前記〔1〕〜〔5〕、〔5−2〕および〔5−3〕のいずれか1記載の結晶(水和物結晶)。
〔6〕 13C固体NMRスペクトルにおいて、化学シフト約146.7ppmおよび約123.3ppmにピークを有する、3−(2−シアノフェニル)−5−(2−ピリジル)−1−フェニル−1,2−ジヒドロピリジン−2−オンの水和物の結晶(水和物結晶)。
〔7〕 3−(2−シアノフェニル)−5−(2−ピリジル)−1−フェニル−1,2−ジヒドロピリジン−2−オンを、アルコール系溶媒、アルキルケトン系溶媒および水からなる群から選ばれる1または2の溶媒を結晶化溶媒として用いて結晶化することを特徴とする前記〔1〕〜〔5〕、〔5−1〕、〔5−2〕、〔5−3〕、〔5−4〕および〔6〕のいずれか1記載の3−(2−シアノフェニル)−5−(2−ピリジル)−1−フェニル−1,2−ジヒドロピリジン−2−オンの水和物の結晶の製造方法。
〔8〕 結晶化溶媒が、アセトンと水の混合溶媒である前記〔7〕記載の製造方法。
〔9〕 結晶化溶媒が、アセトンと水(体積比37:3〜24:16)の混合溶媒(好適にはアセトンと水(9:1〜7:3)の混合溶媒であり、より好適にはアセトンと水(9:1)の混合溶媒に溶解後、水を加えてアセトンと水(8:2)の溶液とした混合溶媒)である前記〔7〕記載の製造方法。
〔10〕 結晶化を60〜−30℃で実施することを特徴とする前記〔7〕〜〔9〕のいずれか1記載の製造方法。
〔11〕 3−(2−シアノフェニル)−5−(2−ピリジル)−1−フェニル−1,2−ジヒドロピリジン−2−オンを結晶化溶媒に溶解した溶液を、50℃以上(好ましくは結晶化溶媒の還流温度から50℃、より好ましくは65〜55℃)で加熱後、40〜5℃/時間(好ましくは25〜15℃/時間)の冷却速度で、10〜−20℃(好ましくは10〜5℃)まで冷却することを特徴とする前記〔7〕〜〔9〕のいずれか1記載の製造方法。
〔12〕 3−(2−シアノフェニル)−5−(2−ピリジル)−1−フェニル−1,2−ジヒドロピリジン−2−オンの重量に対する容量比で10〜50倍量(v/w)の結晶化溶媒を用いることを特徴とする前記〔7〕〜〔11〕のいずれか1記載の製造方法。
(結晶化溶媒の量として好ましくは、30〜50倍量(v/w)であり、より好ましくは、結晶化溶媒としてアセトンと水(9:1)を用いる場合には約40倍量(v/w)であり、結晶化溶媒としてアセトンと水(8:2)を用いる場合には約45倍量(v/w)である。)
〔13〕 60℃以下(好ましくは55〜0℃、より好ましくは55〜35℃、さらに好ましくは約40℃)で種結晶(少量の3−(2−シアノフェニル)−5−(2−ピリジル)−1−フェニル−1,2−ジヒドロピリジン−2−オンの水和物の結晶)を加えることを特徴とする前記〔7〕〜〔12〕のいずれか1記載の製造方法。
〔14〕 結晶化の後、減圧乾燥することを特徴とする前記〔7〕〜〔13〕のいずれか1記載の製造方法。
〔15〕 結晶化および減圧乾燥の後、大気中放置することを特徴とする前記〔7〕〜〔14〕のいずれか1記載の製造方法。
〔15−1〕 結晶化の後、大気中放置することを特徴とする前記〔7〕〜〔13〕のいずれか1記載の製造方法。
〔15−2〕 減圧乾燥の後、大気中に放置することを特徴とする前記〔14〕記載の製造方法。
〔16〕 粉末X線回折において、回折角度(2θ±0.2°)10.3°に回折ピークを有する、3−(2−シアノフェニル)−5−(2−ピリジル)−1−フェニル−1,2−ジヒドロピリジン−2−オンの無水物の結晶(I型無水物結晶)。
〔17〕 粉末X線回折において、さらに、回折角度(2θ±0.2°)19.1°に回折ピークを有する、前記〔16〕記載の結晶(I型無水物結晶)。
〔18〕 13C固体NMRスペクトルにおいて、化学シフト約149.0ppmおよび約125.6ppmにピークを有する、3−(2−シアノフェニル)−5−(2−ピリジル)−1−フェニル−1,2−ジヒドロピリジン−2−オンの無水物の結晶(I型無水物結晶)。
〔19〕 粉末X線回折において、回折角度(2θ±0.2°)16.7°に回折ピークを有する、3−(2−シアノフェニル)−5−(2−ピリジル)−1−フェニル−1,2−ジヒドロピリジン−2−オンの無水物の結晶(V型無水物結晶)。
〔20〕 粉末X線回折において、さらに、回折角度(2θ±0.2°)12.9°および24.9°に回折ピークを有する、前記〔19〕記載の結晶(V型無水物結晶)。
〔21〕 赤外吸収スペクトル(KBr法)において、波数1658±1cm−1に吸収ピークを有する、3−(2−シアノフェニル)−5−(2−ピリジル)−1−フェニル−1,2−ジヒドロピリジン−2−オンの無水物の結晶(V型無水物結晶)。
〔22〕 赤外吸収スペクトル(KBr法)において、さらに、波数501±1cm−1に吸収ピークを有する、前記〔21〕記載の結晶(V型無水物結晶)。
〔23〕 13C固体NMRスペクトルにおいて、化学シフト約145.9ppmおよび約137.7ppmにピークを有する、3−(2−シアノフェニル)−5−(2−ピリジル)−1−フェニル−1,2−ジヒドロピリジン−2−オンの無水物の結晶(V型無水物結晶)。
〔24〕 粉末X線回折において、回折角度(2θ±0.2°)23.7°および25.0°に回折ピークを有する、3−(2−シアノフェニル)−5−(2−ピリジル)−1−フェニル−1,2−ジヒドロピリジン−2−オンの無水物の結晶(III型無水物結晶)。
〔25〕 粉末X線回折において、さらに、回折角度(2θ±0.2°)5.7°および9.5°に回折ピークを有する、前記〔24〕記載の結晶(III型無水物結晶)。
〔26〕 前記〔1〕記載の結晶を含有してなる医薬。
〔27〕 前記〔1〕記載の結晶を含有してなる医薬組成物。
〔28〕 前記〔1〕記載の結晶を含有してなる、急性神経変性疾患の治療剤または予防剤。
〔29〕 前記〔1〕記載の結晶を含有してなる、脳血管障害急性期、頭部外傷、脊髄損傷、低酸素による神経障害または低血糖による神経障害の治療剤または予防剤。
〔30〕 前記〔1〕記載の結晶を含有してなる、慢性神経変性疾患の治療剤または予防剤。
〔31〕 前記〔1〕記載の結晶を含有してなる、アルツハイマー病、パーキンソン病、ハンチントン舞踏病、筋萎縮性側索硬化症または脊髄小脳変性症の治療剤または予防剤。
〔32〕 前記〔1〕記載の結晶を含有してなる、てんかん、肝性脳症、末梢神経障害、パーキンソン症候群、痙性麻痺、痛み、神経痛、精神分裂病(統合失調症)、不安、薬物依存症、嘔気、嘔吐、排尿障害、緑内障による視力障害、抗生物質による聴覚障害または食中毒の治療剤または予防剤。
〔33〕 前記〔1〕記載の結晶を含有してなる、感染性脳脊髄炎、脳血管性痴呆、髄膜炎による痴呆または神経症状の治療剤または予防剤。
〔34〕 前記〔1〕記載の結晶を含有してなる、脱髄性疾患の治療剤または予防剤。
〔35〕 感染性脳脊髄炎がHIV性脳脊髄炎である前記〔33〕記載の治療剤または予防剤。
〔36〕 脱髄性疾患が脳炎、急性散在性脳脊髄炎、多発性硬化症、急性多発性根神経炎、ギラン−バレー症候群、慢性炎症性脱髄性多発神経障害、Marchifava−Bignami病、中心性橋延髄崩壊症、視神経脊髄炎、デビック病、バロ病、HIV性ミエロパシー、HTLV性ミエロパシー、進行性多巣性白質脳症または二次性脱髄性疾患である前記〔34〕記載の治療剤または予防剤。
〔37〕 二次性脱髄性疾患がCNSエリテマトーデス、結節性多発動脈炎、シェーグレン症候群、サルコイドーシスまたは乖離性脳血管炎である前記〔36〕記載の治療剤または予防剤、等を提供するものである。
【発明の効果】
【0009】
本発明により、化合物(1)を単一の結晶態様として、容易に工業的規模で製造することが可能となった。本発明の結晶は、帯電性を有さないなど良好な物性を有し、神経変性疾患等の治療剤または予防剤の有効成分として使用するのに適している。
【発明を実施するための最良の形態】
【0010】
以下本発明の内容について詳細に説明する。
本明細書において「3−(2−シアノフェニル)−5−(2−ピリジル)−1−フェニル−1,2−ジヒドロピリジン−2−オンの水和物の結晶」とは、結晶水を含む3−(2−シアノフェニル)−5−(2−ピリジル)−1−フェニル−1,2−ジヒドロピリジン−2−オンの結晶であれば、当該結晶中に含まれる結晶水の量は特に限定されず、結晶水の一部を欠損していてもよく、さらに付着水と共存している態様も含まれる。
この「3−(2−シアノフェニル)−5−(2−ピリジル)−1−フェニル−1,2−ジヒドロピリジン−2−オンの水和物の結晶」として好適には、1分子の3−(2−シアノフェニル)−5−(2−ピリジル)−1−フェニル−1,2−ジヒドロピリジン−2−オンに対して1/2〜1分子の結晶水を有し、さらに0〜1/4分子の付着水を含んでいてもよく、結晶水のうち0〜1/2分子の水和物を欠損していてもよい結晶態様を意味し、例えば、
(1)3−(2−シアノフェニル)−5−(2−ピリジル)−1−フェニル−1,2−ジヒドロピリジン−2−オンの3/4水和物の結晶、
(2)3−(2−シアノフェニル)−5−(2−ピリジル)−1−フェニル−1,2−ジヒドロピリジン−2−オンの1水和物(1/4の結晶水を欠損)の結晶、
(3)3−(2−シアノフェニル)−5−(2−ピリジル)−1−フェニル−1,2−ジヒドロピリジン−2−オンの1/2水和物の結晶および1/4の付着水が共存している態様、
(4)3−(2−シアノフェニル)−5−(2−ピリジル)−1−フェニル−1,2−ジヒドロピリジン−2−オンの1/2水和物の結晶、および
(5)3−(2−シアノフェニル)−5−(2−ピリジル)−1−フェニル−1,2−ジヒドロピリジン−2−オンの1水和物の結晶などを意味する。
【0011】
本発明の結晶は、以下の特徴を有する化合物(1)の水和物の結晶である。(粉末X線回折パターン、赤外吸収スペクトル(KBr法)の各測定条件は特に限定されないが、下記記載の粉末X線回折パターンの測定条件、赤外吸収スペクトル(KBr法)の測定条件にて測定することが好ましい。)
(1)粉末X線回折において回折角度(2θ±0.2°)8.7°に回折ピークを有することを特徴とする結晶、
(2)粉末X線回折において回折角度(2θ±0.2°)12.5°に回折ピークを有することを特徴とする結晶、
(3)粉末X線回折において回折角度(2θ±0.2°)8.7°および12.5°に回折ピークを有することを特徴とする結晶、
(4)粉末X線回折において下記図4または下記表5記載の回折角度(2θ±0.2°)に回折ピークを有することを特徴とする結晶、
(5)赤外吸収スペクトル(KBr法)において、波数1588±1cm−1に吸収ピークを有することを特徴とする結晶、
(6)赤外吸収スペクトル(KBr法)において、波数1588±1cm−1および751±1cm−1に吸収ピークを有することを特徴とする結晶、
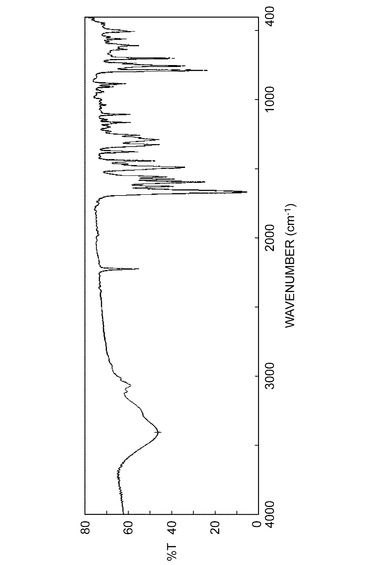
(7)赤外吸収スペクトル(KBr法)において、下記図1または下記表2記載の波数に吸収ピークを有することを特徴とする結晶である。
【0012】
これらの粉末X線回折における特徴的ピークは特許文献1に開示の製造方法によって得られた結晶(下記参考例A1、下記表4、下記図3)では観察されない。
一般に、粉末X線回折における回折角度(2θ)は±0.2°の範囲内で誤差が生じ得るから、上記の回折角度の値は±0.2°程度の範囲内の数値も含むものとして理解される必要がある。したがって、粉末X線回折におけるピークの回折角度が完全に一致する結晶だけでなく、ピークの回折角度が±0.2°程度の誤差で一致する結晶も本発明に含まれる。
【0013】
[水和物結晶]
本明細書において「回折角度(2θ±0.2°)8.7°に回折ピークを有する」とは、「回折角度(2θ)8.5°〜8.9°に回折ピークを有する」ということを意味し、「回折角度(2θ±0.2°)12.5°に回折ピークを有する」とは、「回折角度(2θ)12.3°〜12.7°に回折ピークを有する」ということを意味する。
【0014】
本明細書において「波数1588±1cm−1に吸収ピークを有する」とは、「波数1587〜1589cm−1に吸収ピークを有する」ということを意味する。
【0015】
本明細書において「波数1588±1cm−1および751±1cm−1に吸収ピークを有する」とは、「波数1587〜1589cm−1および750〜752cm−1に吸収ピークを有する」ということを意味する。
【0016】
本明細書において「化学シフト約146.7ppmにピークを有する」とは、「通常の測定条件、もしくは本明細書に記載の条件と実質的に同一の条件にて13C固体NMRスペクトル測定を行い、化学シフト146.7ppmと実質的に同等なピークを有する」ことを意味する。また、本明細書において「化学シフト約123.3ppmにピークを有する」とは、「通常の測定条件、もしくは本明細書に記載の条件と実質的に同一の条件にて13C固体NMRスペクトル測定を行い、化学シフト123.3ppmと実質的に同等なピークを有する」ことを意味する。
【0017】
[I型無水物結晶]
本明細書において「回折角度(2θ±0.2°)10.3°に回折ピークを有する」とは、「回折角度(2θ)10.1°〜10.5°に回折ピークを有する」ということを意味し、「回折角度(2θ±0.2°)19.1°に回折ピークを有する」とは、「回折角度(2θ)18.9°〜19.3°に回折ピークを有する」ということを意味する。
【0018】
本明細書において「化学シフト約149.0ppmにピークを有する」とは、「通常の測定条件、もしくは本明細書と実質的に同一の条件にて13C固体NMRスペクトル測定を行い、化学シフト149.0ppmと実質的に同等なピークを有する」ことを意味する。また、本明細書において「化学シフト約125.6ppmにピークを有する」とは、「通常の測定条件、もしくは本明細書と実質的に同一の条件にて13C固体NMRスペクトル測定を行い、化学シフト125.6ppmと実質的に同等なピークを有する」ことを意味する。
【0019】
[V型無水物結晶]
本明細書において「回折角度(2θ±0.2°)16.7°に回折ピークを有する」とは、「回折角度(2θ)16.5°〜16.9°に回折ピークを有する」ということを意味し、「回折角度(2θ±0.2°)12.9°に回折ピークを有する」とは、「回折角度(2θ)12.7°〜13.1°に回折ピークを有する」ということを意味し、「回折角度(2θ±0.2°)24.9°に回折ピークを有する」とは、「回折角度(2θ)24.7°〜25.1°に回折ピークを有する」ということを意味する。
【0020】
本明細書において「波数1658±1cm−1に吸収ピークを有する」とは、「波数1657〜1659cm−1に吸収ピークを有する」ということを意味する。
【0021】
本明細書において「波数501±1cm−1に吸収ピークを有する」とは、「波数500〜502cm−1に吸収ピークを有する」ということを意味する。
【0022】
本明細書において「化学シフト約145.9ppmにピークを有する」とは、「通常の測定条件、もしくは本明細書と実質的に同一の条件にて13C固体NMRスペクトル測定を行い、化学シフト145.9ppmと実質的に同等なピークを有する」ことを意味する。また、本明細書において「化学シフト約137.7ppmにピークを有する」とは、「通常の測定条件、もしくは本明細書と実質的に同一の条件にて13C固体NMRスペクトル測定を行い、化学シフト137.7ppmと実質的に同等なピークを有する」ことを意味する。
【0023】
[III型無水物結晶]
本明細書において「回折角度(2θ±0.2°)23.7°に回折ピークを有する」とは、「回折角度(2θ)23.5°〜23.9°に回折ピークを有する」ということを意味し、本明細書において「回折角度(2θ±0.2°)25.0°に回折ピークを有する」とは、「回折角度(2θ)24.8°〜25.2°に回折ピークを有する」ということを意味し、本明細書において「回折角度(2θ±0.2°)5.7°に回折ピークを有する」とは、「回折角度(2θ)5.5°〜5.9°に回折ピークを有する」ということを意味し、「回折角度(2θ±0.2°)9.5°に回折ピークを有する」とは、「回折角度(2θ)9.3°〜9.7°に回折ピークを有する」ということを意味する。
【0024】
本明細書において「アルキルケトン系溶媒」とは、アセトンまたはメチルエチルケトン等のジアルキルケトンである有機溶媒を意味し、好適にはアセトンである。
【0025】
本明細書において「アルコール系溶媒」とは、メタノール、エタノール、1−プロパノール、2−プロパノール等のC1−6アルコールである有機溶媒を意味し、好適にはメタノールまたは1−プロパノールである。
【0026】
本明細書において「減圧」とは、760mmHg以下なら特に限定されないが、好適には、760mmHg〜0.1mmHgであり、より好適には、50mmHg〜0.1mmHgであり、さらに好適には30〜5mmHgである。
【0027】
[水和物結晶の一般製造方法]
本発明の水和物結晶は、上記特許文献1(国際公開第01/96308号パンフレット)の実施例7または下記製造例3にしたがって化合物(1)を製造し、この化合物(1)を特定の溶媒中で加熱溶解し、攪拌下冷却して晶析することにより、工業的規模で安定して製造することができる。
【0028】
晶析に使用する化合物(1)は、どのような形態であってもよく、水和物でも無水物でもよく、非晶質でも結晶質(複数の結晶多形からなるものを含む)でもよく、これらの混合物であってもよい。
【0029】
晶析に使用する溶媒は、アルコール系溶媒、アルキルケトン系溶媒および水からなる群より選ばれる1の溶媒または2の溶媒の混合溶媒を挙げることができ、好ましくはアセトンおよび水の混合溶媒である。
アセトンおよび水の混合溶媒を使用する場合の混合比(容量比)は、好ましくは37:3〜24:16であり、より好ましくは9:1〜7:3であり、さらに好ましくは約8:2であり、最も好適にはアセトンと水(9:1)の混合溶媒に溶解後、水を加えてアセトンと水(8:2)の溶液とした混合溶媒である。
【0030】
また、溶媒の使用量は、化合物(1)が加熱により溶解する量を下限とし、結晶の収量が著しく低下しない量を上限として適宜選択することができるが、好ましくは化合物(1)の重量に対する容量比で10〜50倍量(v/w)である。結晶化溶媒の量として好ましくは、30〜50倍量(v/w)であり、より好ましくは、結晶化溶媒としてアセトンと水(9:1)を用いる場合には約40倍量(v/w)であり、結晶化溶媒としてアセトンと水(8:2)を用いる場合には約45倍量(v/w)である。
【0031】
化合物(1)を加熱して溶解する温度は、溶媒に応じて化合物(1)が溶解する温度を適宜選択すればよいが、好ましくは結晶化溶媒の還流温度から50℃であり、より好ましくは65〜55℃である。
晶析時の冷却速度を変えると、態様の異なる結晶(多形)を与えうるので、結晶の品質や粒度等への影響を考慮して適宜冷却速度を調整して実施することが望ましく、好ましくは40〜5℃/時間の速度での冷却、より好ましくは25〜15℃/時間の速度での冷却することが好ましい。
また、最終的な晶析温度は、結晶の収量と品質等から適宜選択することができるが、好ましくは10〜−25℃である。
【0032】
結晶晶析において、種結晶(少量の3−(2−シアノフェニル)−5−(2−ピリジル)−1−フェニル−1,2−ジヒドロピリジン−2−オンの水和物の結晶)を加えても、加えなくても良い。種結晶を加える温度は特に規定されないが、好ましくは60℃以下であり、より好ましくは55〜0℃、さらに好ましくは55〜35℃であり、最も好ましくは約40℃である。
【0033】
晶析した結晶を通常の濾過操作で分離し、必要に応じて溶媒で洗浄し、さらに乾燥して目的の結晶を得ることができる。結晶の洗浄に使用する溶媒は、晶析溶媒と共通であるが、好ましくはアセトン−水(9:1〜1:1)の混合溶媒であり、さらに好ましくはアセトン−水(約1:1)の混合溶媒である。
【0034】
[結晶の乾燥方法]
濾過操作で分離した結晶は、適宜、大気下に放置することにより、または加熱によって乾燥することができる。
乾燥時間は、残留溶媒が所定の量を下回るまでの時間を製造量、乾燥装置、乾燥温度等に応じて適宜選択すればよい。また、乾燥は通風下でも減圧下でも行うことができる。減圧度は、製造量、乾燥装置、乾燥温度等に応じて適宜選択すればよい。得られた結晶は、乾燥後、必要に応じて大気中に放置することもできる。
【0035】
上記の方法によって得られた結晶は単一の結晶形からなり、この結晶形は安定であって、容易に他の結晶形や非晶質に転移することがなく、また吸湿性もない等の良好な物性を有しており、製剤化にも適している。
化合物(1)の神経変性疾患等治療剤としての使用に関しては特許文献1に詳細に開示されており、同様に本発明の結晶は神経変性疾患等の治療剤の有効成分として使用することができる。特許文献1の開示のすべてを参照として本願明細書の開示に含める。
【0036】
本発明の化合物を医薬として使用する場合、通常、本発明の化合物と適当な添加剤とを混和し、製剤化したものを使用する。ただし、前記は、本発明の化合物を原体のまま医薬として使用することを否定するものではない。
上記添加剤としては、一般に医薬に使用される、賦形剤、結合剤、滑沢剤、崩壊剤、着色剤、矯味矯臭剤、乳化剤、界面活性剤、溶解補助剤、懸濁化剤、等張化剤、緩衝剤、防腐剤、抗酸化剤、安定化剤、吸収促進剤等を挙げることができ、所望により、これらを適宜組み合わせて使用することもできる。
【0037】
上記賦形剤としては、例えば乳糖、白糖、ブドウ糖、コーンスターチ、マンニトール、ソルビトール、デンプン、α化デンプン、デキストリン、結晶セルロース、軽質無水ケイ酸、ケイ酸アルミニウム、ケイ酸カルシウム、メタケイ酸アルミン酸マグネシウム、リン酸水素カルシウム等を挙げることができる。
【0038】
上記結合剤としては、例えばポリビニルアルコール、メチルセルロース、エチルセルロース、アラビアゴム、トラガント、ゼラチン、シェラック、ヒドロキシプロピルメチルセルロース、ヒドロキシプロピルセルロース、カルボキシメチルセルロースナトリウム、ポリビニルピロリドン、マクロゴール等を挙げることがでる。
【0039】
上記滑沢剤としては、例えばステアリン酸マグネシウム、ステアリン酸カルシウム、フマル酸ステアリルナトリウム、タルク、ポリエチレングリコール、コロイドシリカ等を挙げることができる。
【0040】
上記崩壊剤としては、例えば結晶セルロース、寒天、ゼラチン、炭酸カルシウム、炭酸水素ナトリウム、クエン酸カルシウム、デキストリン、ペクチン、低置換度ヒドロキシプロピルセルロース、カルボキシメチルセルロース、カルボキシメチルセルロースカルシウム、クロスカルメロースナトリウム、カルボキシメチルスターチ、カルボキシメチルスターチナトリウム等を挙げることができる。
【0041】
上記着色剤としては、三二酸化鉄、黄色三二酸化鉄、カルミン、カラメル、β−カロチン、酸化チタン、タルク、リン酸リボフラビンナトリウム、黄色アルミニウムレーキ等、医薬品に添加することが許可されているものを挙げることができる。
【0042】
上記矯味矯臭剤としては、ココア末、ハッカ脳、芳香散、ハッカ油、竜脳、桂皮末等を挙げることができる。
【0043】
上記乳化剤または界面活性剤としては、ステアリルトリエタノールアミン、ラウリル硫酸ナトリウム、ラウリルアミノプロピオン酸、レシチン、モノステアリン酸グリセリン、ショ糖脂肪酸エステル、グリセリン脂肪酸エステル等を挙げることができる。
【0044】
上記溶解補助剤としては、ポリエチレングリコール、プロピレングリコール、安息香酸ベンジル、エタノール、コレステロール、トリエタノールアミン、炭酸ナトリウム、クエン酸ナトリウム、ポリソルベート80、ニコチン酸アミド等を挙げることができる。
【0045】
上記懸濁化剤としては、前記界面活性剤のほか、ポリビニルアルコール、ポリビニルピロリドン、メチルセルロース、ヒドロキシメチルセルロース、ヒドロキシエチルセルロース、ヒドロキシプロピルセルロース等の親水性高分子を挙げることができる。
【0046】
上記等張化剤としては、ブドウ糖、塩化ナトリウム、マンニトール、ソルビトール等を挙げることができる。
【0047】
上記緩衝剤としては、リン酸塩、酢酸塩、炭酸塩、クエン酸塩等の緩衝液を挙げることができる。
【0048】
上記防腐剤としては、メチルパラベン、プロピルパラベン、クロロブタノール、ベンジルアルコール、フェネチルアルコール、デヒドロ酢酸、ソルビン酸等を挙げることができる。
【0049】
上記抗酸化剤としては、亜硫酸塩、アスコルビン酸、α−トコフェロール等を挙げることができる。
【0050】
上記安定化剤としては、一般に医薬に使用されるものを挙げることができる。
【0051】
上記吸収促進剤としては、一般に医薬に使用されるものを挙げることができる。
【0052】
また、上記製剤としては、錠剤、散剤、顆粒剤、カプセル剤、シロップ剤、トローチ剤、吸入剤のような経口剤;坐剤、軟膏剤、眼軟膏剤、テープ剤、点眼剤、点鼻剤、点耳剤、パップ剤、ローション剤のような外用剤または注射剤を挙げることができる。
【0053】
上記経口剤は、上記添加剤を適宜組み合わせて製剤化する。なお、必要に応じてこれらの表面をコーティングしてもよい。
【0054】
上記外用剤は、上記添加剤のうち、特に賦形剤、結合剤、矯味矯臭剤、乳化剤、界面活性剤、溶解補助剤、懸濁化剤、等張化剤、防腐剤、抗酸化剤、安定化剤または吸収促進剤を適宜組み合わせて製剤化する。
【0055】
上記注射剤は、上記添加剤のうち、特に乳化剤、界面活性剤、溶解補助剤、懸濁化剤、等張化剤、緩衝剤、防腐剤、抗酸化剤、安定化剤または吸収促進剤を適宜組み合わせて製剤化する。
【0056】
本発明の化合物を医薬として使用する場合、その使用量は症状や年齢等により異なるが、通常、経口剤の場合には、0.05〜10mg(好ましくは0.1〜5mg)、外用剤の場合には、0.01〜10mg(好ましくは0.05〜5mg)、注射剤の場合には、0.01〜5mgを1日に1回投与または2〜6回に分けて使用する。なお、上記経口剤および注射剤については、実際に投与する値を、また、外用剤については、実際に生体に吸収される値を示している。
【0057】
本発明の3−(2−シアノフェニル)−5−(2−ピリジル)−1−フェニル−1,2−ジヒドロピリジン−2−オンの水和物の結晶(化合物(1))を含有する、人への治療または予防等に用いるための製剤は、製剤学的に一般的に用いられている方法によって得ることができるが、具体的製剤処方の例を以下に示す。
【0058】
本発明化合物(3−(2−シアノフェニル)−5−(2−ピリジル)−1−フェニル−1,2−ジヒドロピリジン−2−オン)、乳糖、低置換度ヒドロキシプロピルセルロースを混合後、ポリビニルピロリドンを適量の精製水に溶解したものを用いて湿式造粒した。この造粒物を乾燥後、整粒し、得られた顆粒に低置換度ヒドロキシプロピルセルロースとステアリン酸マグネシウムを入れて混合後、打錠した。得られた錠剤にコーティング基剤(ヒドロキシプロピルメチルセルロース、タルク、マクロゴール6000、酸化チタンおよび黄色三二酸化鉄の混合物)の水溶液によりフィルムコートを施した。1錠あたりの各使用原料の量を下表に示す。
【0059】
【表1】

【実施例】
【0060】
以下の実施例により本発明を詳細に且つ具体的に説明するが、本発明はこれらの実施例に限定されるものではない。
【0061】
製造例1
5−(2−ピリジル)−1−フェニル−1,2−ジヒドロピリジン−2−オンの合成
【化1】
5−(2−ピリジル)−1,2−ジヒドロピリジン−2−オン(WO2004/009553)(7.33kg)、トリフェニルボロキシン(9.0kg)、酢酸銅(無水)(0.80kg)、水(0.50kg)、ピリジン(7.1kg)、N,N−ジメチルホルムアミド(66.7kg)の混合物を、反応容器内を窒素置換後、内温28℃で1時間撹拌した。
反応容器中へ9%酸素濃度に窒素で調整した空気を30L/minの速度で吹き込みながら、反応混合物を39℃から40℃(内温)で16時間撹拌し、反応混合物1Aを得た。
水(191kg)、25%アンモニア水(85.8kg)を別の反応容器に入れ、冷水で8.7℃まで冷却後、上記の反応混合物1Aを3分間かけて加えた。反応混合物を冷水にて冷却下4時間撹拌した。反応混合物中の析出物を遠心分離機で濾取し、濾滓を水65kgで洗浄した。
析出物、水(97kg)、25%アンモニア水(43.5kg)を反応容器に投入し、25℃の温水で保温して1時間撹拌した。反応混合物中の析出物を遠心分離機で濾取し、濾滓を水32.6kgで洗浄後、減圧乾燥(60℃、18時間)を行い5−(2−ピリジル)−1−フェニル−1,2−ジヒドロピリジン−2−オン 9.6kgを得た。
1H NMR (400MHz, DMSO-d6)δ 8.61-8.50 (m, 1H), 8.36 (d, 1H), 8.29 (dd,1H,), 7.90 (d, 1H), 7.80 (ddd, 1H), 7.56-7.45 (m, 5H), 7.27 (dd, 1H), 6.62 (d,1H).
【0062】
製造例2
3−ブロモ−5−(2−ピリジル)−1−フェニル−1,2−ジヒドロピリジン−2−オンの合成
【化2】
10L反応容器中に5−(2−ピリジル)−1−フェニル−1,2−ジヒドロピリジン−2−オン(200g)、N−ブロモスクシンイミド(157.7g)、酢酸エチル(4L)を加え、反応混合物を、窒素気流下30℃(外温)にて9時間20分撹拌した。この反応混合物中へ3%ハイドロサルファイト水溶液(2L)、トルエン(2L)を加えた後、55℃(外温)にて30分撹拌した。反応後、反応混合物中の水層(下層)を分離し、次いで有機層の水洗(水2L)を4回行い、撹拌減圧下溶媒を留去した。
その後さらに1,2−ジメトキシエタン(4L)を加え、減圧濃縮を行い、3−ブロモ−5−(2−ピリジル)−1−フェニル−1,2−ジヒドロピリジン−2−オン粗体を得た。
【0063】
製造例3
3−(2−シアノフェニル)−5−(2−ピリジル)−1−フェニル−1,2−ジヒドロピリジン−2−オンの合成
【化3】
上記製造例2において濃縮残渣として得られた3−ブロモ−5−(2−ピリジル)−1−フェニル−1,2−ジヒドロピリジン−2−オン粗体の全量が入った反応容器へ、2−(1,3,2−ジオキサボリナン−2−イル)ベンゾニトリル(214.9g)、酢酸パラジウム(3.44g)、トリフェニルホスフィン(16.07g)、ヨウ化第一銅(7.29g)、1,2−ジメトキシエタン(3.1L)、炭酸カリウム(158.8g)を加え、窒素雰囲気下、70℃(外温)、30分加熱撹拌し、次いで4時間、加熱還流下にて撹拌した。
その後、反応混合物中へ、70℃(外温)酢酸エチル(2.5L)を加え10分撹拌した。反応混合物を濾過し、さらに濾滓を酢酸エチル(2.5L)で洗浄した。この濾液すべてを反応容器へ移し、さらに12.5%アンモニア水(5L)を加え、60℃(外温)にて53分撹拌した。反応混合物中の下層(水層)を分離した。残った有機層中へ5%食塩水(2.5L)、25%アンモニア水(2.5L)を加え撹拌後、下層(水層)を分離し、さらに残った有機層中へ5%食塩水(5L)を加え攪拌後、下層(水層)を分離した。残った有機層を減圧濃縮し、その後アセトン4Lを加え減圧濃縮を行った。
この残渣にアセトン(7.2L)、水(0.8L)を加え、60℃(外温)で1時間10分撹拌し溶解した。次いで38℃(外温)にて18分間撹拌冷却した。反応混合物中へ、内温40℃で種結晶(3−(2−シアノフェニル)−5−(2−ピリジル)−1−フェニル−1,2−ジヒドロピリジン−2−オンの水和物の結晶)1gを加え35℃(外温)にて30分間撹拌した。その後、反応混合物を30分ごとに外温を5℃ずつ下げて、外温10℃では17時間撹拌した。
撹拌下、反応混合物中へ水(2.29L)を3時間10分かけて滴下し、滴下後、さらに1時間20分撹拌した。反応混合物を濾過し、濾滓を50%アセトン−水2Lで洗浄し、3−(2−シアノフェニル)−5−(2−ピリジル)−1−フェニル−1,2−ジヒドロピリジン−2−オン(526.28g)を湿体として得た。(乾燥重量として168.3g)
【0064】
湿体中の3−(2−シアノフェニル)−5−(2−ピリジル)−1−フェニル−1,2−ジヒドロピリジン−2−オンの乾燥重量の換算
得られた湿体4.378gを秤取し、50℃で4時間減圧乾燥を行い1.4005gの乾燥粉体を得た。
乾燥重量換算値=(1.4005/4.378)×526.28=168.3g
【0065】
湿体中の3−(2−シアノフェニル)−5−(2−ピリジル)−1−フェニル−1,2−ジヒドロピリジン−2−オンの湿体中のアセトンおよび水の重量の計算
得られた湿体の以下の条件のガスクロマトグラフィー分析により、製造例3で得られた湿体中、アセトン168mL、水186mLが含まれていることを確認した。
【0066】
ガスクロマトグラフィー分析条件:
カラム:DB−WAX(30m×0.53mm,1μm);検出器 TCD;オーブン温度 60℃(8分)、60−180℃(70℃/分)、180℃(5分);検出器温度 210℃;インレット温度 150℃;カラム流速 5.0mL/分
スプリット比 1:4;注入量 2μL
【0067】
実施例1X
3−(2−シアノフェニル)−5−(2−ピリジル)−1−フェニル−1,2−ジヒドロピリジン−2−オン(水和物結晶)の結晶化
上記製造例3において湿体として得た3−(2−シアノフェニル)−5−(2−ピリジル)−1−フェニル−1,2−ジヒドロピリジン−2−オン(526.28g)を10Lフラスコに投入し、アセトン5890mLと水490mLで調製したアセトン水のうち5.5Lを加えて加熱し、溶解後濾過した。残った上記アセトン水全量で10Lフラスコと濾滓を洗浄しながら、濾液すべてを10Lフラスコに移した。
当該混合物を外温40℃にて撹拌し、内温が40℃になってから、外温を35℃にし、次いで混合物中に3−(2−シアノフェニル)−5−(2−ピリジル)−1−フェニル−1,2−ジヒドロピリジン−2−オン水和物842mgを加えた。混合物を30分間撹拌後に、外温を30℃に変更し、さらに30分後に外温を25℃に変更し、以後30分ごとに5℃ずつ外温を下げ、外温15℃まで下げた。混合物を外温15℃にて30分間撹拌後に、さらに外温を8℃に下げて1時間撹拌した。
混合物中へ、11℃(内温)にて水842mLを1時間10分かけて滴下して加えた。滴下終了1時間後に外温を0℃に変更し、混合物を40分攪拌後、さらに外温を−20℃に下げて15時間撹拌した。
混合物中の析出物を濾取し、その析出物を50%アセトン水(1700mL)で洗浄後、50分間通風乾燥を行った。次いでこの析出物を振動乾燥機にて減圧下40℃にて11時間乾燥し、さらに60℃で3時間乾燥した。
乾燥機温度を室温まで冷却後、乾燥機内を950hpa、4時間外気を吸引し、3−(2−シアノフェニル)−5−(2−ピリジル)−1−フェニル−1,2−ジヒドロピリジン−2−オン(水和物結晶)172.4gを得た。
1H NMR (400MHz, DMSO-d6)δ 8.61-8.57 (m, 1H), 8.53-8.52 (d-like, 1H),8.47 (d, 1H), 8.01 (d, 1H), 7.92 (d, 1H), 7.86-7.81 (t-like, 1H),7.79-7.76(t-like, 1H), 7.72(d, 1H), 7.61-7.48 (m, 6H), 7.31-7.28 (m, 1H).
残留パラジウム 15ppm
【0068】
参考例A1
3−(2−シアノフェニル)−5−(2−ピリジル)−1−フェニル−1,2−ジヒドロピリジン−2−オンの無水物の結晶の製造(II型無水物結晶)
WO01/96308号、実施例7記載の反応処理後の操作方法と同様に、以下のように実施した。3−(2−シアノフェニル)−5−(2−ピリジル)−1−フェニル−1,2−ジヒドロピリジン−2−オン(別名:2−(2−オキソ−1−フェニル−5−(ピリジン−2−イル)−1,2−ジヒドロピリジン−3−イル)ベンゾニトリル)の合成方法はWO01/96308号中の実施例7および上記製造例3に記載されている。
3−(2−シアノフェニル)−5−(2−ピリジル)−1−フェニル−1,2−ジヒドロピリジン−2−オン(8g)を入れ、酢酸エチル(400mL)を加えた。これを60℃の温浴にて加熱し、さらに酢酸エチル(160mL)を加え、70℃の温浴にて加熱し固体を溶解させた。この溶液にn−ヘキサン(80mL)を加えた後、減圧下に溶媒留去して7.7gの淡黄色粉末を得た。
1H NMR (400MHz, DMSO-d6)δ 8.59-8.57 (m, 1H), 8.53 (d, 1H), 8.47 (d,1H,), 8.01 (d, 1H), 7.92 (d, 1H), 7.83 (ddd, 1H), 7.80-7.76(m,1H),7.73-7.71(d-like, 1H), 7.61-7.48 (m, 6H), 7.30 (dd, 1H).
【0069】
実施例B1
3−(2−シアノフェニル)−5−(2−ピリジル)−1−フェニル−1,2−ジヒドロピリジン−2−オンの水和物の結晶の製造(水和物結晶)
3−(2−シアノフェニル)−5−(2−ピリジル)−1−フェニル−1,2−ジヒドロピリジン−2−オン(7g)を500mLナス型容器に取り、アセトン252mLと水28mLで調製した90%アセトン水280mLを加え、水浴バスで加熱撹拌し、還流下(水浴65℃)溶解した。溶解確認後、水浴を50℃に冷却し、水35mL添加した後、内温50℃で種結晶(少量の3−(2−シアノフェニル)−5−(2−ピリジル)−1−フェニル−1,2−ジヒドロピリジン−2−オンの水和物の結晶)140mg添加して、恒温槽を用いて約35℃/時間の冷却速度で−20℃まで冷却した。−20℃で1時間撹拌した後、析出した固体を濾取し、減圧乾燥(外温30℃ 1時間、60℃、2時間)させ、得られた乾燥粉末6.3gをシャーレに移し、17時間大気中(湿度放置前55.4%、一夜後61.6%)で放置させ、3−(2−シアノフェニル)−5−(2−ピリジル)−1−フェニル−1,2−ジヒドロピリジン−2−オンの水和物の結晶 6.2gを得た。
1H NMR (400MHz, DMSO-d6)δ 8.61-8.57 (m, 1H), 8.53 (d, 1H), 8.47 (d,1H,), 8.01 (d, 1H), 7.92 (d, 1H), 7.83 (ddd, 1H), 7.78(ddd, 1H),7.73-7.71(d-like, 1H), 7.61-7.48 (m, 6H), 7.30 (dd, 1H).
【0070】
実施例C1
3−(2−シアノフェニル)−5−(2−ピリジル)−1−フェニル−1,2−ジヒドロピリジン−2−オンの無水物の結晶の製造(V型無水物結晶)
3−(2−シアノフェニル)−5−(2−ピリジル)−1−フェニル−1,2−ジヒドロピリジン−2−オン(水和物結晶)9gを500mL容器に取り、アセトン360mLを加えて、加熱還流下(水浴70℃)撹拌した。
溶解後、当該混合物を吸引濾過し、濾液を常圧、75℃で濃縮し固化させた。固化物を乳鉢で微細化後、アセトン216mLと水の54mLから調製したアセトン水溶液を加えた。
当該混合物を加熱還流下(水浴75℃)撹拌し、溶解後、さらに加熱還流下2時間40分撹拌した。次いで、当該混合物水浴の温度(外温)を10℃/時間の冷却速度で、室温まで冷却していき、室温下で16時間撹拌した。
反応混合物中の析出物を吸引濾取し、次いで減圧乾燥(外温20℃で40分、外温60℃で3時間)させ、3−(2−シアノフェニル)−5−(2−ピリジル)−1−フェニル−1,2−ジヒドロピリジン−2−オンの無水物の結晶 7.2gを得た。
1H NMR (400MHz, DMSO-d6)δ 8.61-8.57 (m, 1H), 8.53 (d, 1H), 8.47 (d,1H,), 8.01 (d, 1H), 7.92 (dd, 1H), 7.83 (ddd, 1H), 7.78(ddd, 1H), 7.72(dd, 1H),7.61-7.48 (m, 6H), 7.31-7.28 (m, 1H).
【0071】
実施例D1
3−(2−シアノフェニル)−5−(2−ピリジル)−1−フェニル−1,2−ジヒドロピリジン−2−オンの無水物の結晶の製造(I型無水物結晶)
3−(2−シアノフェニル)−5−(2−ピリジル)−1−フェニル−1,2−ジヒドロピリジン−2−オン(水和物結晶)8gを1L容器に取り、酢酸エチル480mLを加えて、加熱還流下(オイルバス)撹拌して溶解させた。加熱を止め、オイルバスにつけたまま(徐冷下)で撹拌を続けた。内温が50.9℃になった時点で、混合物中へ種結晶(3−(2−シアノフェニル)−5−(2−ピリジル)−1−フェニル−1,2−ジヒドロピリジン−2−オンの無水物の結晶)0.2gを加え、次いで、内温が31.3℃になるまで撹拌を続けた。混合物を氷浴中でさらに2時間撹拌し、析出した結晶を濾取して、次いで通風乾燥(50℃/18時間)を行い、3−(2−シアノフェニル)−5−(2−ピリジル)−1−フェニル−1,2−ジヒドロピリジン−2−オンの無水物の結晶5.8gを得た。
1H NMR (400MHz, DMSO-d6)δ 8.58 (d, 1H), 8.53 (d, 1H), 8.47 (d, 1H,),8.01 (d, 1H), 7.93 (d, 1H), 7.83 (ddd, 1H), 7.78(d, 1H), 7.72(d, 1H), 7.61-7.48(m, 6H), 7.32- 7.27 (m, 1H).
【0072】
水または水:エタノール(1:1)混液存在下での混合操作における物理的安定性
(操作方法)
各結晶約150mgをメノウ乳鉢に採り、水(もしくは水:エタノール(1:1)混液)を滴下しながら室温で数分間混合操作を行う。次いで、約60℃において2〜3時間乾燥する。
(結果)
粉末X線回折より、参考例A1で得られる結晶は、上記水または水:エタノール(1:1)混液存在下での混合操作において、結晶の態様が変化し、実施例D1と同じ結晶態様のものが増加してくることが見出された。
粉末X線回折より、実施例B1、実施例C1、実施例D1で得られる各結晶は、結晶の態様の変化は見られなく、上記水または水:エタノール(1:1)混液存在下での混合操作において物理的に安定であることが見出された。
【0073】
温度および湿度の変化が水和物結晶に及ぼす影響
(装置)
理学X線DTAシステム:RINT-2000(株式会社リガク製)
(操作方法)
実施例B1で得られた結晶(水和物結晶)をメノウ乳鉢で粉砕後13mm径ガラス板にサンプリングし、各温湿度条件下での試料について以下の条件で測定を行った。
使用X線:CuKα線
管電圧:40kV
管電流:200mA
発散スリット:1/2deg
受光スリット:0.3mm
散乱スリット:1/2deg
走査速度:2゜/分
走査ステップ:0.01゜
測定範囲(2θ):5〜40゜
【0074】
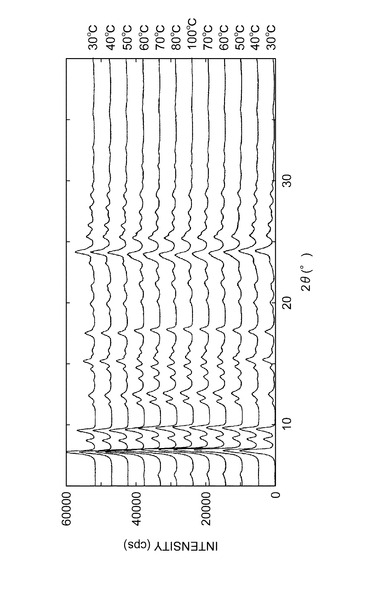
測定温度を以下のように変化させ、各温度における粉末X線回折パターンを測定した:30→40→50→60→70→80→100→70→60→50→40→30℃。
(結果)
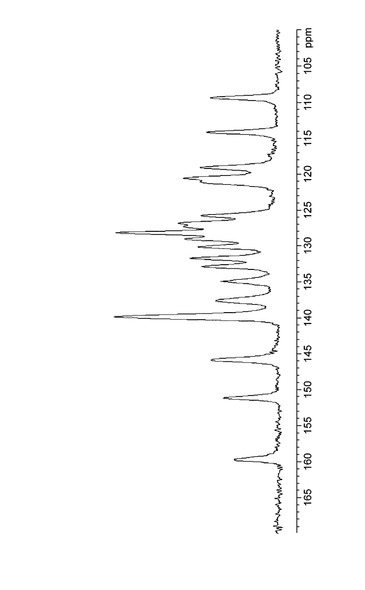
水和物結晶の上記各温度における粉末X線回析パターンを図11に表す。粉末X線回折パターンの変化より、実施例B1記載の結晶(水和物結晶)は約60℃以上で実施例E1記載と同一の結晶(III型無水物結晶)に転移するが、温度を下げると再び水和物結晶に戻ることが確認された。
【0075】
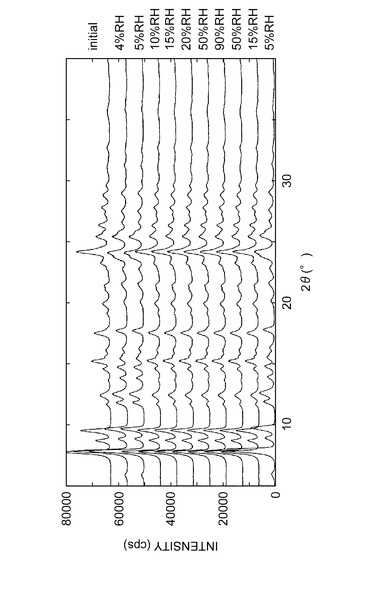
測定湿度を以下のように変化させ、各湿度における粉末X線回折パターンを測定した:4→5→10→15→20→50→90→50→15→5%RH(相対湿度)。
(結果)
水和物結晶の上記各湿度における粉末X線回析パターンを図12に表す。粉末X線回折パターンの変化より、湿度10%RH付近を境にして水和物結晶パターンとIII型無水物結晶のパターンが可逆的に観測された。実施例B1記載の結晶(水和物結晶)は約10%RH以下の湿度下でIII型無水物結晶に変化するが、約10%RH以上の湿度下にすることにより、水和物結晶になることが確認された。
【0076】
水和物結晶の温度、湿度変化による影響についてのこれらの実験、および実施例1Xの記載から、実施例1Xにおける通風乾燥前の析出物の態様が、実施例E1記載と同一の結晶(III型無水物結晶)、もしくはこのIII型無水物結晶と水和物結晶の混合物であり、これが水和物結晶を製造するための有用な中間体であることを見出した。
【0077】
最小着火エネルギーと爆発下限濃度
(操作方法)
吹上式粉塵爆発試験装置の試料皿に濃度に見合った量の水和物結晶を均等に乗せた。1.3リットル圧力タンク内に50kPaのコンプレッサエアーを溜め、電磁弁によってガラス円筒内に開放し粉塵雲を形成した。電磁弁の開放から0.1秒遅れて放電電極にエネルギーを与えた。着火の判定は、放電電極の上部100mmに記す着火目印線を火炎が超えた場合とした。
(下限濃度の測定条件)
測定室の温度:24℃、湿度:49%、圧縮空気吹き出し圧力:50kPa、放電開始時間:0.1秒、着火試験の繰り返し数:5回、着火放電エネルギー:10J
(着火エネルギーの測定条件)
測定室の温度:24℃、湿度:49%、圧縮空気吹き出し圧力:50kPa、放電開始時間:0.1秒、着火試験の繰り返し数:10回
(測定装置)
吹き上げ式粉塵爆発試験装置(株式会社環境衛生研究所 DES-10)
(結果)
爆発下限濃度:160〜170g/m3
最小着火エネルギー:50〜100mJ
粉塵濃度:1250g/m3
【0078】
帯電性
(操作方法)
各化合物約1gを秤量ビン(直径35mm)に秤り、撹拌子(フッ素樹脂(4フッ化エチレン樹脂)製、20mm)を加え、ふたをして粉を30分撹拌する。撹拌停止と同時にふたを開け、粉体の静電電位を静電電位測定器で測定する。
(装置)
スタチロン−DZ3 シシド静電気株式会社製
(結果)
参考例A1の結晶:70〜100V
実施例B1の結晶:0V
【0079】
赤外スペクトル測定
実施例B1で得られた結晶の赤外スペクトル測定は、日本薬局方の一般試験法に記載された赤外スペクトル測定法の臭化カリウム錠剤法に従い、以下の測定条件で行った。
(装置) 日本分光株式会社製FT/IR−620
測定範囲:4000〜400cm−1
分解能:4cm−1
積算回数:36
スキャンスピード:2mm/分
【0080】
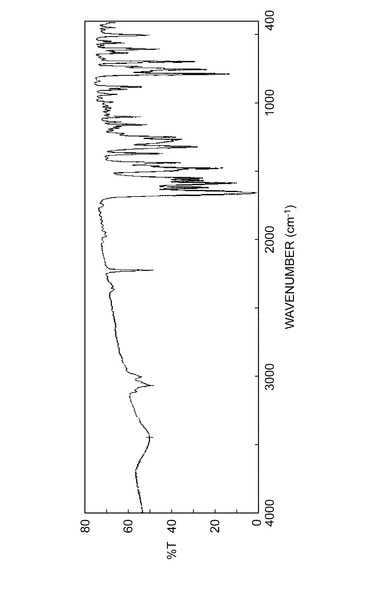
実施例B1で得られた結晶の赤外線吸収スペクトル測定(KBr法)を図1に示し、実施例C1で得られた結晶の赤外線吸収スペクトル測定(KBr法)を図2に示し、実施例B1で得られた結晶の吸収ピークの波数(cm−1)及び透過率%を表2に示し、実施例C1で得られた結晶の吸収ピークの波数(cm−1)及び透過率%を表3に示した。
【0081】
【表2】
【0082】
【表3】
【0083】
粉末X線回折パターンの測定
各実施例で得られた結晶の粉末X線回折測定は、日本薬局方の一般試験法に記載された粉末X線回折測定法に従い、以下の測定条件で行った。
(装置)
理学X線DTAシステム:RINT−2000(株式会社リガク製)
(操作方法)
試料についてメノウ乳鉢で粉砕後13mm径ガラス板にサンプリングし、以下の条件で測定を行った。
使用X線:CuKα線
管電圧:40kV
管電流:200mA
発散スリット:1/2deg
受光スリット:0.3mm
散乱スリット:1/2deg
走査速度:1゜/分
走査ステップ:0.01゜
測定範囲(2θ):5〜40゜
【0084】
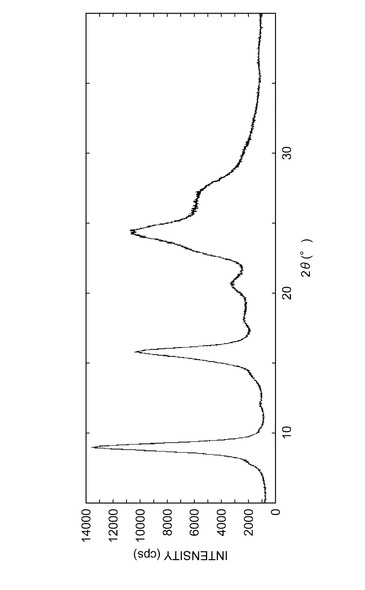
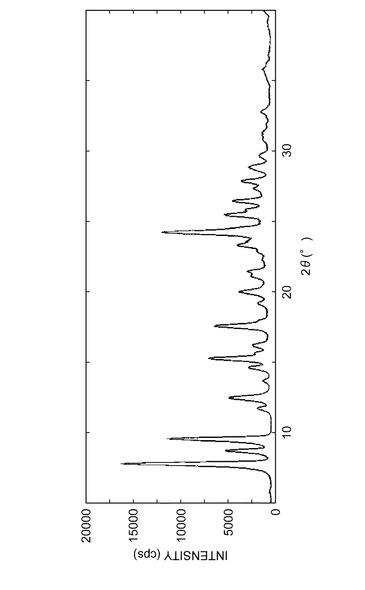
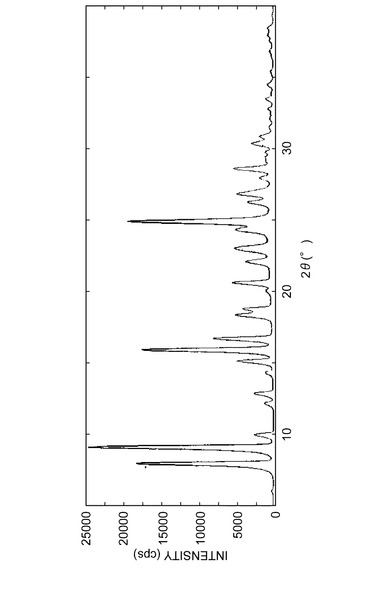
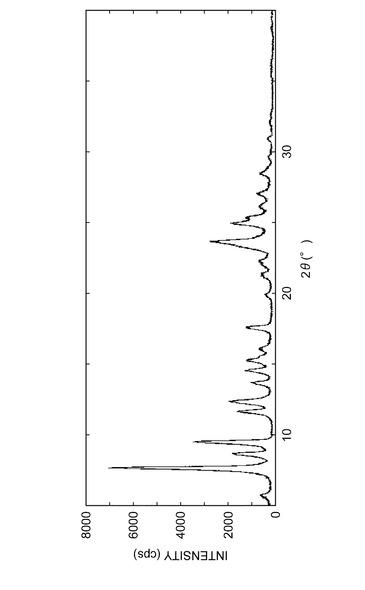
参考例A1で得られた結晶の粉末X線回折パターンを図3に示し、実施例B1で得られた結晶の粉末X線回折パターンを図4に示し、実施例C1で得られた結晶の粉末X線回折パターンを図5に示し、実施例D1で得られた結晶の粉末X線回折パターンを図6に示した。
【0085】
参考例A1で得られた結晶の回折角(2θ)のピークおよび強度を表4に示し、実施例B1で得られた結晶の回折角(2θ)のピークおよび強度を表5に示し、実施例C1で得られた結晶の回折角(2θ)のピークおよび強度を表6に示し、実施例D1で得られた結晶の回折角(2θ)のピークおよび強度を表7に示した。
【0086】
実施例B1で得られた結晶の粉末X線回折パターンである図4、表5から、実施例B1で得られた結晶の粉末X線回折において、回折角(2θ)が約12.5°の特徴的なピークを有することが分かる。
これより、参考例A1で得られた結晶の粉末X線回折パターンである図3、表4において、回折角(2θ)が約12.5のピークを有しておらず、実施例B1で得られる結晶と同じ態様の結晶は含んでいないと考えられる。
【0087】
実施例E1(III型無水物結晶)
上記実施例B1で得られた、3−(2−シアノフェニル)−5−(2−ピリジル)−1−フェニル−1,2−ジヒドロピリジン−2−オンの水和物結晶を上記粉末X線回折測定条件と同様の条件下で、粉末X線回折の測定を行った。(但し、走査速度:2゜/分、110℃付近に加温条件下で測定した)。
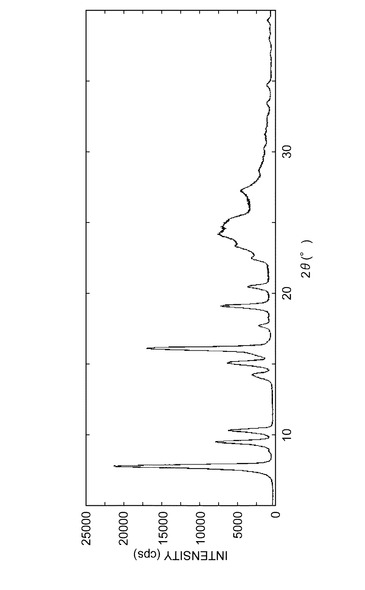
その粉末X線回折パターンを図7に示し、結晶の回折角(2θ)のピークおよび強度を表8に示した。
【0088】
【表4】
【0089】
【表5】
【0090】
【表6】
【0091】
【表7】
【0092】
【表8】
【0093】
13C固体NMRスペクトルの測定
実施例B1、C1およびD1で得られた結晶の13C固体NMRスペクトル測定を以下の条件で行った。
測定温度: 室温(〜22℃)
基準物質: グリシンのカルボニル炭素(外部基準:176.03ppm)
測定核: 13C(100.6248425MHz)
パルス繰り返し時間:
50秒(実施例C1、D1)
5秒(実施例B1)
パルスモード: CP/TOSS測定
【0094】
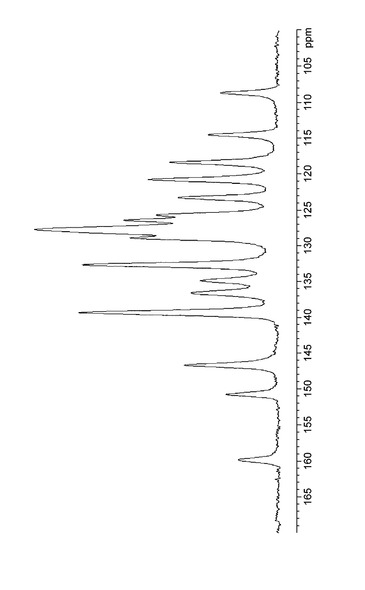
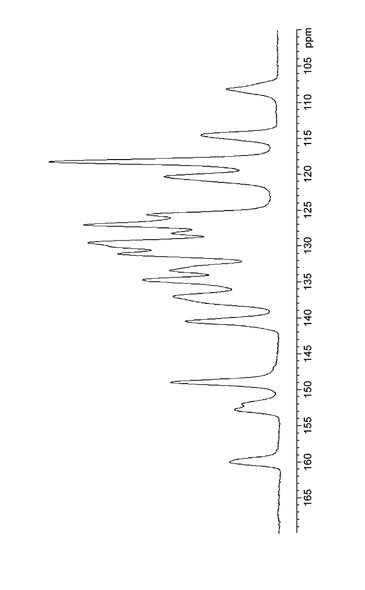
実施例B1で得られた結晶の13C固体NMRスペクトルを図8に示し、化学シフトを表9にまとめた。実施例D1で得られた結晶の13C固体NMRスペクトルを図9に示し、化学シフトを表10にした。実施例C1で得られた結晶の13C固体NMRスペクトルを図10に示し、化学シフトを表11にした。
【0095】
【表9】
【0096】
【表10】
【0097】
【表11】
【産業上の利用可能性】
【0098】
本発明の結晶は良好な物性を有し、神経変性疾患等の治療剤または予防剤の有効成分として使用するのに適している。
【図面の簡単な説明】
【0099】
【図1】実施例B1で得られた結晶の赤外線吸収スペクトル(KBr法)を表す図である。
【図2】実施例C1で得られた結晶の赤外線吸収スペクトル(KBr法)を表す図である。
【図3】参考例A1で得られた結晶の粉末X線回析パターンを表す図である。
【図4】実施例B1で得られた結晶の粉末X線回析パターンを表す図である。
【図5】実施例C1で得られた結晶の粉末X線回析パターンを表す図である。
【図6】実施例D1で得られた結晶の粉末X線回析パターンを表す図である。
【図7】実施例E1における粉末X線回析パターンを表す図である。
【図8】実施例B1で得られた結晶の13C固体核磁気共鳴(NMR)スペクトルを表す図である。
【図9】実施例D1で得られた結晶の13C固体NMRスペクトルを表す図である。
【図10】実施例C1で得られた結晶の13C固体NMRスペクトルを表す図である。
【図11】様々な温度における水和物結晶の粉末X線回析パターンを表す図である。
【図12】様々な相対湿度における水和物結晶の粉末X線回析パターンを表す図である。
【技術分野】
【0001】
本発明は、AMPA(α−アミノ−3−ヒドロキシ−5−メチル−4−イソオキサゾールプロピオン酸)受容体拮抗作用および/またはカイニン酸受容体阻害作用を有する神経変性疾患等の治療剤または予防剤として有用な1,2−ジヒドロピリジン化合物(3−(2−シアノフェニル)−5−(2−ピリジル)−1−フェニル−1,2−ジヒドロピリジン−2−オン)の結晶の製造方法に関する。
【背景技術】
【0002】
1,2−ジヒドロピリジン化合物は、AMPA受容体拮抗作用および/またはカイニン酸受容体阻害作用を有する神経変性疾患等の治療剤または予防剤として有用であり、中でも3−(2−シアノフェニル)−5−(2−ピリジル)−1−フェニル−1,2−ジヒドロピリジン−2−オン(以下、化合物(1)と示す。)は顕著なAMPA受容体拮抗作用を示す(特許文献1参照)。
化合物(1)については特許文献1の実施例7に製造方法の開示があるが、「残渣をシリカゲルカラムクロマト(酢酸エチル/ヘキサン 1:2)で精製して」と記載されているのみで、得られた化合物の態様については開示されていない。
【0003】
【特許文献1】国際公開第01/96308号パンフレット
【発明の開示】
【発明が解決しようとする課題】
【0004】
結晶多形が存在する化合物を医薬品として用いる場合、医薬品として要求される均一な品質および一定の作用強度を確保するために、均一の結晶形を有する化合物を安定して供給することが必要である。また、保存中および製剤化工程(混合、造粒等)において、同一品質を維持できる結晶形が望まれる。
【0005】
医薬品原薬は、工業的に大量に取り扱うことから、爆発性、危険性の指標の1つである爆発下限濃度や最小着火エネルギーが高い結晶形が望まれる。
【0006】
一般に帯電する粉体は、他の物体への付着性も高く保護具や皮膚への付着が懸念される。
医薬品原薬が帯電性を有している場合、化合物製造時の粉砕の段階において粉砕刃に化合物が付着したり、製剤化の工程において生産機器に付着凝集したりすると、生産効率や作業性が悪化することがあり、また、帯電性を有する大量の粉体を工業的に扱う場合には粉塵爆発の起こる可能性があることから、医薬品原薬としては、帯電性がより弱い化合物(結晶)が望まれる。
また、医薬品原薬のような高い薬理活性を持つ化合物については、作業者の暴露の抑制や施設汚染防止の観点からも帯電しにくい粉体が望ましい。
【0007】
そのため、医薬の有効成分が結晶性物質として得られる場合は単一の結晶形からなり、安定した良好な物性を持ち、金属等の不純物を含まないことが望ましい。また、そのような結晶を安定して工業的規模で製造できる方法の開発も望まれている。
そこで、本発明は、化合物(1)の単一の結晶形からなる結晶およびその製造方法を提供することを目的とする。
【課題を解決するための手段】
【0008】
本発明者らは、精力的に研究を重ねた結果、化合物(1)の晶析に際して特定の結晶化溶媒を用いることにより、単一の結晶形からなる化合物(1)が得られることを見出して本発明を完成した。
すなわち、本発明は、
〔1〕 粉末X線回折において、回折角度(2θ±0.2°)8.7°に回折ピークを有する、3−(2−シアノフェニル)−5−(2−ピリジル)−1−フェニル−1,2−ジヒドロピリジン−2−オンの水和物の結晶(水和物結晶)。
〔2〕 X線回折において、回折角度(2θ±0.2°)12.5°に回折ピークを有する、3−(2−シアノフェニル)−5−(2−ピリジル)−1−フェニル−1,2−ジヒドロピリジン−2−オンの水和物の結晶(水和物結晶)。
〔3〕 粉末X線回折において、回折角度(2θ±0.2°)8.7°および12.5°に回折ピークを有する、3−(2−シアノフェニル)−5−(2−ピリジル)−1−フェニル−1,2−ジヒドロピリジン−2−オンの水和物の結晶(水和物結晶)。
〔4〕 赤外吸収スペクトル(KBr法)において、波数1588±1cm−1に吸収ピークを有する、3−(2−シアノフェニル)−5−(2−ピリジル)−1−フェニル−1,2−ジヒドロピリジン−2−オンの水和物の結晶(水和物結晶)。
〔5〕 赤外吸収スペクトル(KBr法)において、波数1588±1cm−1および751±1cm−1に吸収ピークを有する、3−(2−シアノフェニル)−5−(2−ピリジル)−1−フェニル−1,2−ジヒドロピリジン−2−オンの水和物の結晶(水和物結晶)。
〔5−2〕 赤外吸収スペクトル(KBr法)において、波数1588±1cm−1に吸収ピークを有する、前記〔1〕〜〔3〕のいずれか1記載の結晶(水和物結晶)。
〔5−3〕 赤外吸収スペクトル(KBr法)において、波数1588±1cm−1および751±1cm−1に吸収ピークを有する、前記〔1〕〜〔3〕のいずれか1記載の結晶(水和物結晶)。
〔5−4〕 パラジウムの含量が20ppm以下(好ましくは15ppm以下)である、前記〔1〕〜〔5〕、〔5−2〕および〔5−3〕のいずれか1記載の結晶(水和物結晶)。
〔6〕 13C固体NMRスペクトルにおいて、化学シフト約146.7ppmおよび約123.3ppmにピークを有する、3−(2−シアノフェニル)−5−(2−ピリジル)−1−フェニル−1,2−ジヒドロピリジン−2−オンの水和物の結晶(水和物結晶)。
〔7〕 3−(2−シアノフェニル)−5−(2−ピリジル)−1−フェニル−1,2−ジヒドロピリジン−2−オンを、アルコール系溶媒、アルキルケトン系溶媒および水からなる群から選ばれる1または2の溶媒を結晶化溶媒として用いて結晶化することを特徴とする前記〔1〕〜〔5〕、〔5−1〕、〔5−2〕、〔5−3〕、〔5−4〕および〔6〕のいずれか1記載の3−(2−シアノフェニル)−5−(2−ピリジル)−1−フェニル−1,2−ジヒドロピリジン−2−オンの水和物の結晶の製造方法。
〔8〕 結晶化溶媒が、アセトンと水の混合溶媒である前記〔7〕記載の製造方法。
〔9〕 結晶化溶媒が、アセトンと水(体積比37:3〜24:16)の混合溶媒(好適にはアセトンと水(9:1〜7:3)の混合溶媒であり、より好適にはアセトンと水(9:1)の混合溶媒に溶解後、水を加えてアセトンと水(8:2)の溶液とした混合溶媒)である前記〔7〕記載の製造方法。
〔10〕 結晶化を60〜−30℃で実施することを特徴とする前記〔7〕〜〔9〕のいずれか1記載の製造方法。
〔11〕 3−(2−シアノフェニル)−5−(2−ピリジル)−1−フェニル−1,2−ジヒドロピリジン−2−オンを結晶化溶媒に溶解した溶液を、50℃以上(好ましくは結晶化溶媒の還流温度から50℃、より好ましくは65〜55℃)で加熱後、40〜5℃/時間(好ましくは25〜15℃/時間)の冷却速度で、10〜−20℃(好ましくは10〜5℃)まで冷却することを特徴とする前記〔7〕〜〔9〕のいずれか1記載の製造方法。
〔12〕 3−(2−シアノフェニル)−5−(2−ピリジル)−1−フェニル−1,2−ジヒドロピリジン−2−オンの重量に対する容量比で10〜50倍量(v/w)の結晶化溶媒を用いることを特徴とする前記〔7〕〜〔11〕のいずれか1記載の製造方法。
(結晶化溶媒の量として好ましくは、30〜50倍量(v/w)であり、より好ましくは、結晶化溶媒としてアセトンと水(9:1)を用いる場合には約40倍量(v/w)であり、結晶化溶媒としてアセトンと水(8:2)を用いる場合には約45倍量(v/w)である。)
〔13〕 60℃以下(好ましくは55〜0℃、より好ましくは55〜35℃、さらに好ましくは約40℃)で種結晶(少量の3−(2−シアノフェニル)−5−(2−ピリジル)−1−フェニル−1,2−ジヒドロピリジン−2−オンの水和物の結晶)を加えることを特徴とする前記〔7〕〜〔12〕のいずれか1記載の製造方法。
〔14〕 結晶化の後、減圧乾燥することを特徴とする前記〔7〕〜〔13〕のいずれか1記載の製造方法。
〔15〕 結晶化および減圧乾燥の後、大気中放置することを特徴とする前記〔7〕〜〔14〕のいずれか1記載の製造方法。
〔15−1〕 結晶化の後、大気中放置することを特徴とする前記〔7〕〜〔13〕のいずれか1記載の製造方法。
〔15−2〕 減圧乾燥の後、大気中に放置することを特徴とする前記〔14〕記載の製造方法。
〔16〕 粉末X線回折において、回折角度(2θ±0.2°)10.3°に回折ピークを有する、3−(2−シアノフェニル)−5−(2−ピリジル)−1−フェニル−1,2−ジヒドロピリジン−2−オンの無水物の結晶(I型無水物結晶)。
〔17〕 粉末X線回折において、さらに、回折角度(2θ±0.2°)19.1°に回折ピークを有する、前記〔16〕記載の結晶(I型無水物結晶)。
〔18〕 13C固体NMRスペクトルにおいて、化学シフト約149.0ppmおよび約125.6ppmにピークを有する、3−(2−シアノフェニル)−5−(2−ピリジル)−1−フェニル−1,2−ジヒドロピリジン−2−オンの無水物の結晶(I型無水物結晶)。
〔19〕 粉末X線回折において、回折角度(2θ±0.2°)16.7°に回折ピークを有する、3−(2−シアノフェニル)−5−(2−ピリジル)−1−フェニル−1,2−ジヒドロピリジン−2−オンの無水物の結晶(V型無水物結晶)。
〔20〕 粉末X線回折において、さらに、回折角度(2θ±0.2°)12.9°および24.9°に回折ピークを有する、前記〔19〕記載の結晶(V型無水物結晶)。
〔21〕 赤外吸収スペクトル(KBr法)において、波数1658±1cm−1に吸収ピークを有する、3−(2−シアノフェニル)−5−(2−ピリジル)−1−フェニル−1,2−ジヒドロピリジン−2−オンの無水物の結晶(V型無水物結晶)。
〔22〕 赤外吸収スペクトル(KBr法)において、さらに、波数501±1cm−1に吸収ピークを有する、前記〔21〕記載の結晶(V型無水物結晶)。
〔23〕 13C固体NMRスペクトルにおいて、化学シフト約145.9ppmおよび約137.7ppmにピークを有する、3−(2−シアノフェニル)−5−(2−ピリジル)−1−フェニル−1,2−ジヒドロピリジン−2−オンの無水物の結晶(V型無水物結晶)。
〔24〕 粉末X線回折において、回折角度(2θ±0.2°)23.7°および25.0°に回折ピークを有する、3−(2−シアノフェニル)−5−(2−ピリジル)−1−フェニル−1,2−ジヒドロピリジン−2−オンの無水物の結晶(III型無水物結晶)。
〔25〕 粉末X線回折において、さらに、回折角度(2θ±0.2°)5.7°および9.5°に回折ピークを有する、前記〔24〕記載の結晶(III型無水物結晶)。
〔26〕 前記〔1〕記載の結晶を含有してなる医薬。
〔27〕 前記〔1〕記載の結晶を含有してなる医薬組成物。
〔28〕 前記〔1〕記載の結晶を含有してなる、急性神経変性疾患の治療剤または予防剤。
〔29〕 前記〔1〕記載の結晶を含有してなる、脳血管障害急性期、頭部外傷、脊髄損傷、低酸素による神経障害または低血糖による神経障害の治療剤または予防剤。
〔30〕 前記〔1〕記載の結晶を含有してなる、慢性神経変性疾患の治療剤または予防剤。
〔31〕 前記〔1〕記載の結晶を含有してなる、アルツハイマー病、パーキンソン病、ハンチントン舞踏病、筋萎縮性側索硬化症または脊髄小脳変性症の治療剤または予防剤。
〔32〕 前記〔1〕記載の結晶を含有してなる、てんかん、肝性脳症、末梢神経障害、パーキンソン症候群、痙性麻痺、痛み、神経痛、精神分裂病(統合失調症)、不安、薬物依存症、嘔気、嘔吐、排尿障害、緑内障による視力障害、抗生物質による聴覚障害または食中毒の治療剤または予防剤。
〔33〕 前記〔1〕記載の結晶を含有してなる、感染性脳脊髄炎、脳血管性痴呆、髄膜炎による痴呆または神経症状の治療剤または予防剤。
〔34〕 前記〔1〕記載の結晶を含有してなる、脱髄性疾患の治療剤または予防剤。
〔35〕 感染性脳脊髄炎がHIV性脳脊髄炎である前記〔33〕記載の治療剤または予防剤。
〔36〕 脱髄性疾患が脳炎、急性散在性脳脊髄炎、多発性硬化症、急性多発性根神経炎、ギラン−バレー症候群、慢性炎症性脱髄性多発神経障害、Marchifava−Bignami病、中心性橋延髄崩壊症、視神経脊髄炎、デビック病、バロ病、HIV性ミエロパシー、HTLV性ミエロパシー、進行性多巣性白質脳症または二次性脱髄性疾患である前記〔34〕記載の治療剤または予防剤。
〔37〕 二次性脱髄性疾患がCNSエリテマトーデス、結節性多発動脈炎、シェーグレン症候群、サルコイドーシスまたは乖離性脳血管炎である前記〔36〕記載の治療剤または予防剤、等を提供するものである。
【発明の効果】
【0009】
本発明により、化合物(1)を単一の結晶態様として、容易に工業的規模で製造することが可能となった。本発明の結晶は、帯電性を有さないなど良好な物性を有し、神経変性疾患等の治療剤または予防剤の有効成分として使用するのに適している。
【発明を実施するための最良の形態】
【0010】
以下本発明の内容について詳細に説明する。
本明細書において「3−(2−シアノフェニル)−5−(2−ピリジル)−1−フェニル−1,2−ジヒドロピリジン−2−オンの水和物の結晶」とは、結晶水を含む3−(2−シアノフェニル)−5−(2−ピリジル)−1−フェニル−1,2−ジヒドロピリジン−2−オンの結晶であれば、当該結晶中に含まれる結晶水の量は特に限定されず、結晶水の一部を欠損していてもよく、さらに付着水と共存している態様も含まれる。
この「3−(2−シアノフェニル)−5−(2−ピリジル)−1−フェニル−1,2−ジヒドロピリジン−2−オンの水和物の結晶」として好適には、1分子の3−(2−シアノフェニル)−5−(2−ピリジル)−1−フェニル−1,2−ジヒドロピリジン−2−オンに対して1/2〜1分子の結晶水を有し、さらに0〜1/4分子の付着水を含んでいてもよく、結晶水のうち0〜1/2分子の水和物を欠損していてもよい結晶態様を意味し、例えば、
(1)3−(2−シアノフェニル)−5−(2−ピリジル)−1−フェニル−1,2−ジヒドロピリジン−2−オンの3/4水和物の結晶、
(2)3−(2−シアノフェニル)−5−(2−ピリジル)−1−フェニル−1,2−ジヒドロピリジン−2−オンの1水和物(1/4の結晶水を欠損)の結晶、
(3)3−(2−シアノフェニル)−5−(2−ピリジル)−1−フェニル−1,2−ジヒドロピリジン−2−オンの1/2水和物の結晶および1/4の付着水が共存している態様、
(4)3−(2−シアノフェニル)−5−(2−ピリジル)−1−フェニル−1,2−ジヒドロピリジン−2−オンの1/2水和物の結晶、および
(5)3−(2−シアノフェニル)−5−(2−ピリジル)−1−フェニル−1,2−ジヒドロピリジン−2−オンの1水和物の結晶などを意味する。
【0011】
本発明の結晶は、以下の特徴を有する化合物(1)の水和物の結晶である。(粉末X線回折パターン、赤外吸収スペクトル(KBr法)の各測定条件は特に限定されないが、下記記載の粉末X線回折パターンの測定条件、赤外吸収スペクトル(KBr法)の測定条件にて測定することが好ましい。)
(1)粉末X線回折において回折角度(2θ±0.2°)8.7°に回折ピークを有することを特徴とする結晶、
(2)粉末X線回折において回折角度(2θ±0.2°)12.5°に回折ピークを有することを特徴とする結晶、
(3)粉末X線回折において回折角度(2θ±0.2°)8.7°および12.5°に回折ピークを有することを特徴とする結晶、
(4)粉末X線回折において下記図4または下記表5記載の回折角度(2θ±0.2°)に回折ピークを有することを特徴とする結晶、
(5)赤外吸収スペクトル(KBr法)において、波数1588±1cm−1に吸収ピークを有することを特徴とする結晶、
(6)赤外吸収スペクトル(KBr法)において、波数1588±1cm−1および751±1cm−1に吸収ピークを有することを特徴とする結晶、
(7)赤外吸収スペクトル(KBr法)において、下記図1または下記表2記載の波数に吸収ピークを有することを特徴とする結晶である。
【0012】
これらの粉末X線回折における特徴的ピークは特許文献1に開示の製造方法によって得られた結晶(下記参考例A1、下記表4、下記図3)では観察されない。
一般に、粉末X線回折における回折角度(2θ)は±0.2°の範囲内で誤差が生じ得るから、上記の回折角度の値は±0.2°程度の範囲内の数値も含むものとして理解される必要がある。したがって、粉末X線回折におけるピークの回折角度が完全に一致する結晶だけでなく、ピークの回折角度が±0.2°程度の誤差で一致する結晶も本発明に含まれる。
【0013】
[水和物結晶]
本明細書において「回折角度(2θ±0.2°)8.7°に回折ピークを有する」とは、「回折角度(2θ)8.5°〜8.9°に回折ピークを有する」ということを意味し、「回折角度(2θ±0.2°)12.5°に回折ピークを有する」とは、「回折角度(2θ)12.3°〜12.7°に回折ピークを有する」ということを意味する。
【0014】
本明細書において「波数1588±1cm−1に吸収ピークを有する」とは、「波数1587〜1589cm−1に吸収ピークを有する」ということを意味する。
【0015】
本明細書において「波数1588±1cm−1および751±1cm−1に吸収ピークを有する」とは、「波数1587〜1589cm−1および750〜752cm−1に吸収ピークを有する」ということを意味する。
【0016】
本明細書において「化学シフト約146.7ppmにピークを有する」とは、「通常の測定条件、もしくは本明細書に記載の条件と実質的に同一の条件にて13C固体NMRスペクトル測定を行い、化学シフト146.7ppmと実質的に同等なピークを有する」ことを意味する。また、本明細書において「化学シフト約123.3ppmにピークを有する」とは、「通常の測定条件、もしくは本明細書に記載の条件と実質的に同一の条件にて13C固体NMRスペクトル測定を行い、化学シフト123.3ppmと実質的に同等なピークを有する」ことを意味する。
【0017】
[I型無水物結晶]
本明細書において「回折角度(2θ±0.2°)10.3°に回折ピークを有する」とは、「回折角度(2θ)10.1°〜10.5°に回折ピークを有する」ということを意味し、「回折角度(2θ±0.2°)19.1°に回折ピークを有する」とは、「回折角度(2θ)18.9°〜19.3°に回折ピークを有する」ということを意味する。
【0018】
本明細書において「化学シフト約149.0ppmにピークを有する」とは、「通常の測定条件、もしくは本明細書と実質的に同一の条件にて13C固体NMRスペクトル測定を行い、化学シフト149.0ppmと実質的に同等なピークを有する」ことを意味する。また、本明細書において「化学シフト約125.6ppmにピークを有する」とは、「通常の測定条件、もしくは本明細書と実質的に同一の条件にて13C固体NMRスペクトル測定を行い、化学シフト125.6ppmと実質的に同等なピークを有する」ことを意味する。
【0019】
[V型無水物結晶]
本明細書において「回折角度(2θ±0.2°)16.7°に回折ピークを有する」とは、「回折角度(2θ)16.5°〜16.9°に回折ピークを有する」ということを意味し、「回折角度(2θ±0.2°)12.9°に回折ピークを有する」とは、「回折角度(2θ)12.7°〜13.1°に回折ピークを有する」ということを意味し、「回折角度(2θ±0.2°)24.9°に回折ピークを有する」とは、「回折角度(2θ)24.7°〜25.1°に回折ピークを有する」ということを意味する。
【0020】
本明細書において「波数1658±1cm−1に吸収ピークを有する」とは、「波数1657〜1659cm−1に吸収ピークを有する」ということを意味する。
【0021】
本明細書において「波数501±1cm−1に吸収ピークを有する」とは、「波数500〜502cm−1に吸収ピークを有する」ということを意味する。
【0022】
本明細書において「化学シフト約145.9ppmにピークを有する」とは、「通常の測定条件、もしくは本明細書と実質的に同一の条件にて13C固体NMRスペクトル測定を行い、化学シフト145.9ppmと実質的に同等なピークを有する」ことを意味する。また、本明細書において「化学シフト約137.7ppmにピークを有する」とは、「通常の測定条件、もしくは本明細書と実質的に同一の条件にて13C固体NMRスペクトル測定を行い、化学シフト137.7ppmと実質的に同等なピークを有する」ことを意味する。
【0023】
[III型無水物結晶]
本明細書において「回折角度(2θ±0.2°)23.7°に回折ピークを有する」とは、「回折角度(2θ)23.5°〜23.9°に回折ピークを有する」ということを意味し、本明細書において「回折角度(2θ±0.2°)25.0°に回折ピークを有する」とは、「回折角度(2θ)24.8°〜25.2°に回折ピークを有する」ということを意味し、本明細書において「回折角度(2θ±0.2°)5.7°に回折ピークを有する」とは、「回折角度(2θ)5.5°〜5.9°に回折ピークを有する」ということを意味し、「回折角度(2θ±0.2°)9.5°に回折ピークを有する」とは、「回折角度(2θ)9.3°〜9.7°に回折ピークを有する」ということを意味する。
【0024】
本明細書において「アルキルケトン系溶媒」とは、アセトンまたはメチルエチルケトン等のジアルキルケトンである有機溶媒を意味し、好適にはアセトンである。
【0025】
本明細書において「アルコール系溶媒」とは、メタノール、エタノール、1−プロパノール、2−プロパノール等のC1−6アルコールである有機溶媒を意味し、好適にはメタノールまたは1−プロパノールである。
【0026】
本明細書において「減圧」とは、760mmHg以下なら特に限定されないが、好適には、760mmHg〜0.1mmHgであり、より好適には、50mmHg〜0.1mmHgであり、さらに好適には30〜5mmHgである。
【0027】
[水和物結晶の一般製造方法]
本発明の水和物結晶は、上記特許文献1(国際公開第01/96308号パンフレット)の実施例7または下記製造例3にしたがって化合物(1)を製造し、この化合物(1)を特定の溶媒中で加熱溶解し、攪拌下冷却して晶析することにより、工業的規模で安定して製造することができる。
【0028】
晶析に使用する化合物(1)は、どのような形態であってもよく、水和物でも無水物でもよく、非晶質でも結晶質(複数の結晶多形からなるものを含む)でもよく、これらの混合物であってもよい。
【0029】
晶析に使用する溶媒は、アルコール系溶媒、アルキルケトン系溶媒および水からなる群より選ばれる1の溶媒または2の溶媒の混合溶媒を挙げることができ、好ましくはアセトンおよび水の混合溶媒である。
アセトンおよび水の混合溶媒を使用する場合の混合比(容量比)は、好ましくは37:3〜24:16であり、より好ましくは9:1〜7:3であり、さらに好ましくは約8:2であり、最も好適にはアセトンと水(9:1)の混合溶媒に溶解後、水を加えてアセトンと水(8:2)の溶液とした混合溶媒である。
【0030】
また、溶媒の使用量は、化合物(1)が加熱により溶解する量を下限とし、結晶の収量が著しく低下しない量を上限として適宜選択することができるが、好ましくは化合物(1)の重量に対する容量比で10〜50倍量(v/w)である。結晶化溶媒の量として好ましくは、30〜50倍量(v/w)であり、より好ましくは、結晶化溶媒としてアセトンと水(9:1)を用いる場合には約40倍量(v/w)であり、結晶化溶媒としてアセトンと水(8:2)を用いる場合には約45倍量(v/w)である。
【0031】
化合物(1)を加熱して溶解する温度は、溶媒に応じて化合物(1)が溶解する温度を適宜選択すればよいが、好ましくは結晶化溶媒の還流温度から50℃であり、より好ましくは65〜55℃である。
晶析時の冷却速度を変えると、態様の異なる結晶(多形)を与えうるので、結晶の品質や粒度等への影響を考慮して適宜冷却速度を調整して実施することが望ましく、好ましくは40〜5℃/時間の速度での冷却、より好ましくは25〜15℃/時間の速度での冷却することが好ましい。
また、最終的な晶析温度は、結晶の収量と品質等から適宜選択することができるが、好ましくは10〜−25℃である。
【0032】
結晶晶析において、種結晶(少量の3−(2−シアノフェニル)−5−(2−ピリジル)−1−フェニル−1,2−ジヒドロピリジン−2−オンの水和物の結晶)を加えても、加えなくても良い。種結晶を加える温度は特に規定されないが、好ましくは60℃以下であり、より好ましくは55〜0℃、さらに好ましくは55〜35℃であり、最も好ましくは約40℃である。
【0033】
晶析した結晶を通常の濾過操作で分離し、必要に応じて溶媒で洗浄し、さらに乾燥して目的の結晶を得ることができる。結晶の洗浄に使用する溶媒は、晶析溶媒と共通であるが、好ましくはアセトン−水(9:1〜1:1)の混合溶媒であり、さらに好ましくはアセトン−水(約1:1)の混合溶媒である。
【0034】
[結晶の乾燥方法]
濾過操作で分離した結晶は、適宜、大気下に放置することにより、または加熱によって乾燥することができる。
乾燥時間は、残留溶媒が所定の量を下回るまでの時間を製造量、乾燥装置、乾燥温度等に応じて適宜選択すればよい。また、乾燥は通風下でも減圧下でも行うことができる。減圧度は、製造量、乾燥装置、乾燥温度等に応じて適宜選択すればよい。得られた結晶は、乾燥後、必要に応じて大気中に放置することもできる。
【0035】
上記の方法によって得られた結晶は単一の結晶形からなり、この結晶形は安定であって、容易に他の結晶形や非晶質に転移することがなく、また吸湿性もない等の良好な物性を有しており、製剤化にも適している。
化合物(1)の神経変性疾患等治療剤としての使用に関しては特許文献1に詳細に開示されており、同様に本発明の結晶は神経変性疾患等の治療剤の有効成分として使用することができる。特許文献1の開示のすべてを参照として本願明細書の開示に含める。
【0036】
本発明の化合物を医薬として使用する場合、通常、本発明の化合物と適当な添加剤とを混和し、製剤化したものを使用する。ただし、前記は、本発明の化合物を原体のまま医薬として使用することを否定するものではない。
上記添加剤としては、一般に医薬に使用される、賦形剤、結合剤、滑沢剤、崩壊剤、着色剤、矯味矯臭剤、乳化剤、界面活性剤、溶解補助剤、懸濁化剤、等張化剤、緩衝剤、防腐剤、抗酸化剤、安定化剤、吸収促進剤等を挙げることができ、所望により、これらを適宜組み合わせて使用することもできる。
【0037】
上記賦形剤としては、例えば乳糖、白糖、ブドウ糖、コーンスターチ、マンニトール、ソルビトール、デンプン、α化デンプン、デキストリン、結晶セルロース、軽質無水ケイ酸、ケイ酸アルミニウム、ケイ酸カルシウム、メタケイ酸アルミン酸マグネシウム、リン酸水素カルシウム等を挙げることができる。
【0038】
上記結合剤としては、例えばポリビニルアルコール、メチルセルロース、エチルセルロース、アラビアゴム、トラガント、ゼラチン、シェラック、ヒドロキシプロピルメチルセルロース、ヒドロキシプロピルセルロース、カルボキシメチルセルロースナトリウム、ポリビニルピロリドン、マクロゴール等を挙げることがでる。
【0039】
上記滑沢剤としては、例えばステアリン酸マグネシウム、ステアリン酸カルシウム、フマル酸ステアリルナトリウム、タルク、ポリエチレングリコール、コロイドシリカ等を挙げることができる。
【0040】
上記崩壊剤としては、例えば結晶セルロース、寒天、ゼラチン、炭酸カルシウム、炭酸水素ナトリウム、クエン酸カルシウム、デキストリン、ペクチン、低置換度ヒドロキシプロピルセルロース、カルボキシメチルセルロース、カルボキシメチルセルロースカルシウム、クロスカルメロースナトリウム、カルボキシメチルスターチ、カルボキシメチルスターチナトリウム等を挙げることができる。
【0041】
上記着色剤としては、三二酸化鉄、黄色三二酸化鉄、カルミン、カラメル、β−カロチン、酸化チタン、タルク、リン酸リボフラビンナトリウム、黄色アルミニウムレーキ等、医薬品に添加することが許可されているものを挙げることができる。
【0042】
上記矯味矯臭剤としては、ココア末、ハッカ脳、芳香散、ハッカ油、竜脳、桂皮末等を挙げることができる。
【0043】
上記乳化剤または界面活性剤としては、ステアリルトリエタノールアミン、ラウリル硫酸ナトリウム、ラウリルアミノプロピオン酸、レシチン、モノステアリン酸グリセリン、ショ糖脂肪酸エステル、グリセリン脂肪酸エステル等を挙げることができる。
【0044】
上記溶解補助剤としては、ポリエチレングリコール、プロピレングリコール、安息香酸ベンジル、エタノール、コレステロール、トリエタノールアミン、炭酸ナトリウム、クエン酸ナトリウム、ポリソルベート80、ニコチン酸アミド等を挙げることができる。
【0045】
上記懸濁化剤としては、前記界面活性剤のほか、ポリビニルアルコール、ポリビニルピロリドン、メチルセルロース、ヒドロキシメチルセルロース、ヒドロキシエチルセルロース、ヒドロキシプロピルセルロース等の親水性高分子を挙げることができる。
【0046】
上記等張化剤としては、ブドウ糖、塩化ナトリウム、マンニトール、ソルビトール等を挙げることができる。
【0047】
上記緩衝剤としては、リン酸塩、酢酸塩、炭酸塩、クエン酸塩等の緩衝液を挙げることができる。
【0048】
上記防腐剤としては、メチルパラベン、プロピルパラベン、クロロブタノール、ベンジルアルコール、フェネチルアルコール、デヒドロ酢酸、ソルビン酸等を挙げることができる。
【0049】
上記抗酸化剤としては、亜硫酸塩、アスコルビン酸、α−トコフェロール等を挙げることができる。
【0050】
上記安定化剤としては、一般に医薬に使用されるものを挙げることができる。
【0051】
上記吸収促進剤としては、一般に医薬に使用されるものを挙げることができる。
【0052】
また、上記製剤としては、錠剤、散剤、顆粒剤、カプセル剤、シロップ剤、トローチ剤、吸入剤のような経口剤;坐剤、軟膏剤、眼軟膏剤、テープ剤、点眼剤、点鼻剤、点耳剤、パップ剤、ローション剤のような外用剤または注射剤を挙げることができる。
【0053】
上記経口剤は、上記添加剤を適宜組み合わせて製剤化する。なお、必要に応じてこれらの表面をコーティングしてもよい。
【0054】
上記外用剤は、上記添加剤のうち、特に賦形剤、結合剤、矯味矯臭剤、乳化剤、界面活性剤、溶解補助剤、懸濁化剤、等張化剤、防腐剤、抗酸化剤、安定化剤または吸収促進剤を適宜組み合わせて製剤化する。
【0055】
上記注射剤は、上記添加剤のうち、特に乳化剤、界面活性剤、溶解補助剤、懸濁化剤、等張化剤、緩衝剤、防腐剤、抗酸化剤、安定化剤または吸収促進剤を適宜組み合わせて製剤化する。
【0056】
本発明の化合物を医薬として使用する場合、その使用量は症状や年齢等により異なるが、通常、経口剤の場合には、0.05〜10mg(好ましくは0.1〜5mg)、外用剤の場合には、0.01〜10mg(好ましくは0.05〜5mg)、注射剤の場合には、0.01〜5mgを1日に1回投与または2〜6回に分けて使用する。なお、上記経口剤および注射剤については、実際に投与する値を、また、外用剤については、実際に生体に吸収される値を示している。
【0057】
本発明の3−(2−シアノフェニル)−5−(2−ピリジル)−1−フェニル−1,2−ジヒドロピリジン−2−オンの水和物の結晶(化合物(1))を含有する、人への治療または予防等に用いるための製剤は、製剤学的に一般的に用いられている方法によって得ることができるが、具体的製剤処方の例を以下に示す。
【0058】
本発明化合物(3−(2−シアノフェニル)−5−(2−ピリジル)−1−フェニル−1,2−ジヒドロピリジン−2−オン)、乳糖、低置換度ヒドロキシプロピルセルロースを混合後、ポリビニルピロリドンを適量の精製水に溶解したものを用いて湿式造粒した。この造粒物を乾燥後、整粒し、得られた顆粒に低置換度ヒドロキシプロピルセルロースとステアリン酸マグネシウムを入れて混合後、打錠した。得られた錠剤にコーティング基剤(ヒドロキシプロピルメチルセルロース、タルク、マクロゴール6000、酸化チタンおよび黄色三二酸化鉄の混合物)の水溶液によりフィルムコートを施した。1錠あたりの各使用原料の量を下表に示す。
【0059】
【表1】
【実施例】
【0060】
以下の実施例により本発明を詳細に且つ具体的に説明するが、本発明はこれらの実施例に限定されるものではない。
【0061】
製造例1
5−(2−ピリジル)−1−フェニル−1,2−ジヒドロピリジン−2−オンの合成
【化1】
5−(2−ピリジル)−1,2−ジヒドロピリジン−2−オン(WO2004/009553)(7.33kg)、トリフェニルボロキシン(9.0kg)、酢酸銅(無水)(0.80kg)、水(0.50kg)、ピリジン(7.1kg)、N,N−ジメチルホルムアミド(66.7kg)の混合物を、反応容器内を窒素置換後、内温28℃で1時間撹拌した。
反応容器中へ9%酸素濃度に窒素で調整した空気を30L/minの速度で吹き込みながら、反応混合物を39℃から40℃(内温)で16時間撹拌し、反応混合物1Aを得た。
水(191kg)、25%アンモニア水(85.8kg)を別の反応容器に入れ、冷水で8.7℃まで冷却後、上記の反応混合物1Aを3分間かけて加えた。反応混合物を冷水にて冷却下4時間撹拌した。反応混合物中の析出物を遠心分離機で濾取し、濾滓を水65kgで洗浄した。
析出物、水(97kg)、25%アンモニア水(43.5kg)を反応容器に投入し、25℃の温水で保温して1時間撹拌した。反応混合物中の析出物を遠心分離機で濾取し、濾滓を水32.6kgで洗浄後、減圧乾燥(60℃、18時間)を行い5−(2−ピリジル)−1−フェニル−1,2−ジヒドロピリジン−2−オン 9.6kgを得た。
1H NMR (400MHz, DMSO-d6)δ 8.61-8.50 (m, 1H), 8.36 (d, 1H), 8.29 (dd,1H,), 7.90 (d, 1H), 7.80 (ddd, 1H), 7.56-7.45 (m, 5H), 7.27 (dd, 1H), 6.62 (d,1H).
【0062】
製造例2
3−ブロモ−5−(2−ピリジル)−1−フェニル−1,2−ジヒドロピリジン−2−オンの合成
【化2】
10L反応容器中に5−(2−ピリジル)−1−フェニル−1,2−ジヒドロピリジン−2−オン(200g)、N−ブロモスクシンイミド(157.7g)、酢酸エチル(4L)を加え、反応混合物を、窒素気流下30℃(外温)にて9時間20分撹拌した。この反応混合物中へ3%ハイドロサルファイト水溶液(2L)、トルエン(2L)を加えた後、55℃(外温)にて30分撹拌した。反応後、反応混合物中の水層(下層)を分離し、次いで有機層の水洗(水2L)を4回行い、撹拌減圧下溶媒を留去した。
その後さらに1,2−ジメトキシエタン(4L)を加え、減圧濃縮を行い、3−ブロモ−5−(2−ピリジル)−1−フェニル−1,2−ジヒドロピリジン−2−オン粗体を得た。
【0063】
製造例3
3−(2−シアノフェニル)−5−(2−ピリジル)−1−フェニル−1,2−ジヒドロピリジン−2−オンの合成
【化3】
上記製造例2において濃縮残渣として得られた3−ブロモ−5−(2−ピリジル)−1−フェニル−1,2−ジヒドロピリジン−2−オン粗体の全量が入った反応容器へ、2−(1,3,2−ジオキサボリナン−2−イル)ベンゾニトリル(214.9g)、酢酸パラジウム(3.44g)、トリフェニルホスフィン(16.07g)、ヨウ化第一銅(7.29g)、1,2−ジメトキシエタン(3.1L)、炭酸カリウム(158.8g)を加え、窒素雰囲気下、70℃(外温)、30分加熱撹拌し、次いで4時間、加熱還流下にて撹拌した。
その後、反応混合物中へ、70℃(外温)酢酸エチル(2.5L)を加え10分撹拌した。反応混合物を濾過し、さらに濾滓を酢酸エチル(2.5L)で洗浄した。この濾液すべてを反応容器へ移し、さらに12.5%アンモニア水(5L)を加え、60℃(外温)にて53分撹拌した。反応混合物中の下層(水層)を分離した。残った有機層中へ5%食塩水(2.5L)、25%アンモニア水(2.5L)を加え撹拌後、下層(水層)を分離し、さらに残った有機層中へ5%食塩水(5L)を加え攪拌後、下層(水層)を分離した。残った有機層を減圧濃縮し、その後アセトン4Lを加え減圧濃縮を行った。
この残渣にアセトン(7.2L)、水(0.8L)を加え、60℃(外温)で1時間10分撹拌し溶解した。次いで38℃(外温)にて18分間撹拌冷却した。反応混合物中へ、内温40℃で種結晶(3−(2−シアノフェニル)−5−(2−ピリジル)−1−フェニル−1,2−ジヒドロピリジン−2−オンの水和物の結晶)1gを加え35℃(外温)にて30分間撹拌した。その後、反応混合物を30分ごとに外温を5℃ずつ下げて、外温10℃では17時間撹拌した。
撹拌下、反応混合物中へ水(2.29L)を3時間10分かけて滴下し、滴下後、さらに1時間20分撹拌した。反応混合物を濾過し、濾滓を50%アセトン−水2Lで洗浄し、3−(2−シアノフェニル)−5−(2−ピリジル)−1−フェニル−1,2−ジヒドロピリジン−2−オン(526.28g)を湿体として得た。(乾燥重量として168.3g)
【0064】
湿体中の3−(2−シアノフェニル)−5−(2−ピリジル)−1−フェニル−1,2−ジヒドロピリジン−2−オンの乾燥重量の換算
得られた湿体4.378gを秤取し、50℃で4時間減圧乾燥を行い1.4005gの乾燥粉体を得た。
乾燥重量換算値=(1.4005/4.378)×526.28=168.3g
【0065】
湿体中の3−(2−シアノフェニル)−5−(2−ピリジル)−1−フェニル−1,2−ジヒドロピリジン−2−オンの湿体中のアセトンおよび水の重量の計算
得られた湿体の以下の条件のガスクロマトグラフィー分析により、製造例3で得られた湿体中、アセトン168mL、水186mLが含まれていることを確認した。
【0066】
ガスクロマトグラフィー分析条件:
カラム:DB−WAX(30m×0.53mm,1μm);検出器 TCD;オーブン温度 60℃(8分)、60−180℃(70℃/分)、180℃(5分);検出器温度 210℃;インレット温度 150℃;カラム流速 5.0mL/分
スプリット比 1:4;注入量 2μL
【0067】
実施例1X
3−(2−シアノフェニル)−5−(2−ピリジル)−1−フェニル−1,2−ジヒドロピリジン−2−オン(水和物結晶)の結晶化
上記製造例3において湿体として得た3−(2−シアノフェニル)−5−(2−ピリジル)−1−フェニル−1,2−ジヒドロピリジン−2−オン(526.28g)を10Lフラスコに投入し、アセトン5890mLと水490mLで調製したアセトン水のうち5.5Lを加えて加熱し、溶解後濾過した。残った上記アセトン水全量で10Lフラスコと濾滓を洗浄しながら、濾液すべてを10Lフラスコに移した。
当該混合物を外温40℃にて撹拌し、内温が40℃になってから、外温を35℃にし、次いで混合物中に3−(2−シアノフェニル)−5−(2−ピリジル)−1−フェニル−1,2−ジヒドロピリジン−2−オン水和物842mgを加えた。混合物を30分間撹拌後に、外温を30℃に変更し、さらに30分後に外温を25℃に変更し、以後30分ごとに5℃ずつ外温を下げ、外温15℃まで下げた。混合物を外温15℃にて30分間撹拌後に、さらに外温を8℃に下げて1時間撹拌した。
混合物中へ、11℃(内温)にて水842mLを1時間10分かけて滴下して加えた。滴下終了1時間後に外温を0℃に変更し、混合物を40分攪拌後、さらに外温を−20℃に下げて15時間撹拌した。
混合物中の析出物を濾取し、その析出物を50%アセトン水(1700mL)で洗浄後、50分間通風乾燥を行った。次いでこの析出物を振動乾燥機にて減圧下40℃にて11時間乾燥し、さらに60℃で3時間乾燥した。
乾燥機温度を室温まで冷却後、乾燥機内を950hpa、4時間外気を吸引し、3−(2−シアノフェニル)−5−(2−ピリジル)−1−フェニル−1,2−ジヒドロピリジン−2−オン(水和物結晶)172.4gを得た。
1H NMR (400MHz, DMSO-d6)δ 8.61-8.57 (m, 1H), 8.53-8.52 (d-like, 1H),8.47 (d, 1H), 8.01 (d, 1H), 7.92 (d, 1H), 7.86-7.81 (t-like, 1H),7.79-7.76(t-like, 1H), 7.72(d, 1H), 7.61-7.48 (m, 6H), 7.31-7.28 (m, 1H).
残留パラジウム 15ppm
【0068】
参考例A1
3−(2−シアノフェニル)−5−(2−ピリジル)−1−フェニル−1,2−ジヒドロピリジン−2−オンの無水物の結晶の製造(II型無水物結晶)
WO01/96308号、実施例7記載の反応処理後の操作方法と同様に、以下のように実施した。3−(2−シアノフェニル)−5−(2−ピリジル)−1−フェニル−1,2−ジヒドロピリジン−2−オン(別名:2−(2−オキソ−1−フェニル−5−(ピリジン−2−イル)−1,2−ジヒドロピリジン−3−イル)ベンゾニトリル)の合成方法はWO01/96308号中の実施例7および上記製造例3に記載されている。
3−(2−シアノフェニル)−5−(2−ピリジル)−1−フェニル−1,2−ジヒドロピリジン−2−オン(8g)を入れ、酢酸エチル(400mL)を加えた。これを60℃の温浴にて加熱し、さらに酢酸エチル(160mL)を加え、70℃の温浴にて加熱し固体を溶解させた。この溶液にn−ヘキサン(80mL)を加えた後、減圧下に溶媒留去して7.7gの淡黄色粉末を得た。
1H NMR (400MHz, DMSO-d6)δ 8.59-8.57 (m, 1H), 8.53 (d, 1H), 8.47 (d,1H,), 8.01 (d, 1H), 7.92 (d, 1H), 7.83 (ddd, 1H), 7.80-7.76(m,1H),7.73-7.71(d-like, 1H), 7.61-7.48 (m, 6H), 7.30 (dd, 1H).
【0069】
実施例B1
3−(2−シアノフェニル)−5−(2−ピリジル)−1−フェニル−1,2−ジヒドロピリジン−2−オンの水和物の結晶の製造(水和物結晶)
3−(2−シアノフェニル)−5−(2−ピリジル)−1−フェニル−1,2−ジヒドロピリジン−2−オン(7g)を500mLナス型容器に取り、アセトン252mLと水28mLで調製した90%アセトン水280mLを加え、水浴バスで加熱撹拌し、還流下(水浴65℃)溶解した。溶解確認後、水浴を50℃に冷却し、水35mL添加した後、内温50℃で種結晶(少量の3−(2−シアノフェニル)−5−(2−ピリジル)−1−フェニル−1,2−ジヒドロピリジン−2−オンの水和物の結晶)140mg添加して、恒温槽を用いて約35℃/時間の冷却速度で−20℃まで冷却した。−20℃で1時間撹拌した後、析出した固体を濾取し、減圧乾燥(外温30℃ 1時間、60℃、2時間)させ、得られた乾燥粉末6.3gをシャーレに移し、17時間大気中(湿度放置前55.4%、一夜後61.6%)で放置させ、3−(2−シアノフェニル)−5−(2−ピリジル)−1−フェニル−1,2−ジヒドロピリジン−2−オンの水和物の結晶 6.2gを得た。
1H NMR (400MHz, DMSO-d6)δ 8.61-8.57 (m, 1H), 8.53 (d, 1H), 8.47 (d,1H,), 8.01 (d, 1H), 7.92 (d, 1H), 7.83 (ddd, 1H), 7.78(ddd, 1H),7.73-7.71(d-like, 1H), 7.61-7.48 (m, 6H), 7.30 (dd, 1H).
【0070】
実施例C1
3−(2−シアノフェニル)−5−(2−ピリジル)−1−フェニル−1,2−ジヒドロピリジン−2−オンの無水物の結晶の製造(V型無水物結晶)
3−(2−シアノフェニル)−5−(2−ピリジル)−1−フェニル−1,2−ジヒドロピリジン−2−オン(水和物結晶)9gを500mL容器に取り、アセトン360mLを加えて、加熱還流下(水浴70℃)撹拌した。
溶解後、当該混合物を吸引濾過し、濾液を常圧、75℃で濃縮し固化させた。固化物を乳鉢で微細化後、アセトン216mLと水の54mLから調製したアセトン水溶液を加えた。
当該混合物を加熱還流下(水浴75℃)撹拌し、溶解後、さらに加熱還流下2時間40分撹拌した。次いで、当該混合物水浴の温度(外温)を10℃/時間の冷却速度で、室温まで冷却していき、室温下で16時間撹拌した。
反応混合物中の析出物を吸引濾取し、次いで減圧乾燥(外温20℃で40分、外温60℃で3時間)させ、3−(2−シアノフェニル)−5−(2−ピリジル)−1−フェニル−1,2−ジヒドロピリジン−2−オンの無水物の結晶 7.2gを得た。
1H NMR (400MHz, DMSO-d6)δ 8.61-8.57 (m, 1H), 8.53 (d, 1H), 8.47 (d,1H,), 8.01 (d, 1H), 7.92 (dd, 1H), 7.83 (ddd, 1H), 7.78(ddd, 1H), 7.72(dd, 1H),7.61-7.48 (m, 6H), 7.31-7.28 (m, 1H).
【0071】
実施例D1
3−(2−シアノフェニル)−5−(2−ピリジル)−1−フェニル−1,2−ジヒドロピリジン−2−オンの無水物の結晶の製造(I型無水物結晶)
3−(2−シアノフェニル)−5−(2−ピリジル)−1−フェニル−1,2−ジヒドロピリジン−2−オン(水和物結晶)8gを1L容器に取り、酢酸エチル480mLを加えて、加熱還流下(オイルバス)撹拌して溶解させた。加熱を止め、オイルバスにつけたまま(徐冷下)で撹拌を続けた。内温が50.9℃になった時点で、混合物中へ種結晶(3−(2−シアノフェニル)−5−(2−ピリジル)−1−フェニル−1,2−ジヒドロピリジン−2−オンの無水物の結晶)0.2gを加え、次いで、内温が31.3℃になるまで撹拌を続けた。混合物を氷浴中でさらに2時間撹拌し、析出した結晶を濾取して、次いで通風乾燥(50℃/18時間)を行い、3−(2−シアノフェニル)−5−(2−ピリジル)−1−フェニル−1,2−ジヒドロピリジン−2−オンの無水物の結晶5.8gを得た。
1H NMR (400MHz, DMSO-d6)δ 8.58 (d, 1H), 8.53 (d, 1H), 8.47 (d, 1H,),8.01 (d, 1H), 7.93 (d, 1H), 7.83 (ddd, 1H), 7.78(d, 1H), 7.72(d, 1H), 7.61-7.48(m, 6H), 7.32- 7.27 (m, 1H).
【0072】
水または水:エタノール(1:1)混液存在下での混合操作における物理的安定性
(操作方法)
各結晶約150mgをメノウ乳鉢に採り、水(もしくは水:エタノール(1:1)混液)を滴下しながら室温で数分間混合操作を行う。次いで、約60℃において2〜3時間乾燥する。
(結果)
粉末X線回折より、参考例A1で得られる結晶は、上記水または水:エタノール(1:1)混液存在下での混合操作において、結晶の態様が変化し、実施例D1と同じ結晶態様のものが増加してくることが見出された。
粉末X線回折より、実施例B1、実施例C1、実施例D1で得られる各結晶は、結晶の態様の変化は見られなく、上記水または水:エタノール(1:1)混液存在下での混合操作において物理的に安定であることが見出された。
【0073】
温度および湿度の変化が水和物結晶に及ぼす影響
(装置)
理学X線DTAシステム:RINT-2000(株式会社リガク製)
(操作方法)
実施例B1で得られた結晶(水和物結晶)をメノウ乳鉢で粉砕後13mm径ガラス板にサンプリングし、各温湿度条件下での試料について以下の条件で測定を行った。
使用X線:CuKα線
管電圧:40kV
管電流:200mA
発散スリット:1/2deg
受光スリット:0.3mm
散乱スリット:1/2deg
走査速度:2゜/分
走査ステップ:0.01゜
測定範囲(2θ):5〜40゜
【0074】
測定温度を以下のように変化させ、各温度における粉末X線回折パターンを測定した:30→40→50→60→70→80→100→70→60→50→40→30℃。
(結果)
水和物結晶の上記各温度における粉末X線回析パターンを図11に表す。粉末X線回折パターンの変化より、実施例B1記載の結晶(水和物結晶)は約60℃以上で実施例E1記載と同一の結晶(III型無水物結晶)に転移するが、温度を下げると再び水和物結晶に戻ることが確認された。
【0075】
測定湿度を以下のように変化させ、各湿度における粉末X線回折パターンを測定した:4→5→10→15→20→50→90→50→15→5%RH(相対湿度)。
(結果)
水和物結晶の上記各湿度における粉末X線回析パターンを図12に表す。粉末X線回折パターンの変化より、湿度10%RH付近を境にして水和物結晶パターンとIII型無水物結晶のパターンが可逆的に観測された。実施例B1記載の結晶(水和物結晶)は約10%RH以下の湿度下でIII型無水物結晶に変化するが、約10%RH以上の湿度下にすることにより、水和物結晶になることが確認された。
【0076】
水和物結晶の温度、湿度変化による影響についてのこれらの実験、および実施例1Xの記載から、実施例1Xにおける通風乾燥前の析出物の態様が、実施例E1記載と同一の結晶(III型無水物結晶)、もしくはこのIII型無水物結晶と水和物結晶の混合物であり、これが水和物結晶を製造するための有用な中間体であることを見出した。
【0077】
最小着火エネルギーと爆発下限濃度
(操作方法)
吹上式粉塵爆発試験装置の試料皿に濃度に見合った量の水和物結晶を均等に乗せた。1.3リットル圧力タンク内に50kPaのコンプレッサエアーを溜め、電磁弁によってガラス円筒内に開放し粉塵雲を形成した。電磁弁の開放から0.1秒遅れて放電電極にエネルギーを与えた。着火の判定は、放電電極の上部100mmに記す着火目印線を火炎が超えた場合とした。
(下限濃度の測定条件)
測定室の温度:24℃、湿度:49%、圧縮空気吹き出し圧力:50kPa、放電開始時間:0.1秒、着火試験の繰り返し数:5回、着火放電エネルギー:10J
(着火エネルギーの測定条件)
測定室の温度:24℃、湿度:49%、圧縮空気吹き出し圧力:50kPa、放電開始時間:0.1秒、着火試験の繰り返し数:10回
(測定装置)
吹き上げ式粉塵爆発試験装置(株式会社環境衛生研究所 DES-10)
(結果)
爆発下限濃度:160〜170g/m3
最小着火エネルギー:50〜100mJ
粉塵濃度:1250g/m3
【0078】
帯電性
(操作方法)
各化合物約1gを秤量ビン(直径35mm)に秤り、撹拌子(フッ素樹脂(4フッ化エチレン樹脂)製、20mm)を加え、ふたをして粉を30分撹拌する。撹拌停止と同時にふたを開け、粉体の静電電位を静電電位測定器で測定する。
(装置)
スタチロン−DZ3 シシド静電気株式会社製
(結果)
参考例A1の結晶:70〜100V
実施例B1の結晶:0V
【0079】
赤外スペクトル測定
実施例B1で得られた結晶の赤外スペクトル測定は、日本薬局方の一般試験法に記載された赤外スペクトル測定法の臭化カリウム錠剤法に従い、以下の測定条件で行った。
(装置) 日本分光株式会社製FT/IR−620
測定範囲:4000〜400cm−1
分解能:4cm−1
積算回数:36
スキャンスピード:2mm/分
【0080】
実施例B1で得られた結晶の赤外線吸収スペクトル測定(KBr法)を図1に示し、実施例C1で得られた結晶の赤外線吸収スペクトル測定(KBr法)を図2に示し、実施例B1で得られた結晶の吸収ピークの波数(cm−1)及び透過率%を表2に示し、実施例C1で得られた結晶の吸収ピークの波数(cm−1)及び透過率%を表3に示した。
【0081】
【表2】
【0082】
【表3】
【0083】
粉末X線回折パターンの測定
各実施例で得られた結晶の粉末X線回折測定は、日本薬局方の一般試験法に記載された粉末X線回折測定法に従い、以下の測定条件で行った。
(装置)
理学X線DTAシステム:RINT−2000(株式会社リガク製)
(操作方法)
試料についてメノウ乳鉢で粉砕後13mm径ガラス板にサンプリングし、以下の条件で測定を行った。
使用X線:CuKα線
管電圧:40kV
管電流:200mA
発散スリット:1/2deg
受光スリット:0.3mm
散乱スリット:1/2deg
走査速度:1゜/分
走査ステップ:0.01゜
測定範囲(2θ):5〜40゜
【0084】
参考例A1で得られた結晶の粉末X線回折パターンを図3に示し、実施例B1で得られた結晶の粉末X線回折パターンを図4に示し、実施例C1で得られた結晶の粉末X線回折パターンを図5に示し、実施例D1で得られた結晶の粉末X線回折パターンを図6に示した。
【0085】
参考例A1で得られた結晶の回折角(2θ)のピークおよび強度を表4に示し、実施例B1で得られた結晶の回折角(2θ)のピークおよび強度を表5に示し、実施例C1で得られた結晶の回折角(2θ)のピークおよび強度を表6に示し、実施例D1で得られた結晶の回折角(2θ)のピークおよび強度を表7に示した。
【0086】
実施例B1で得られた結晶の粉末X線回折パターンである図4、表5から、実施例B1で得られた結晶の粉末X線回折において、回折角(2θ)が約12.5°の特徴的なピークを有することが分かる。
これより、参考例A1で得られた結晶の粉末X線回折パターンである図3、表4において、回折角(2θ)が約12.5のピークを有しておらず、実施例B1で得られる結晶と同じ態様の結晶は含んでいないと考えられる。
【0087】
実施例E1(III型無水物結晶)
上記実施例B1で得られた、3−(2−シアノフェニル)−5−(2−ピリジル)−1−フェニル−1,2−ジヒドロピリジン−2−オンの水和物結晶を上記粉末X線回折測定条件と同様の条件下で、粉末X線回折の測定を行った。(但し、走査速度:2゜/分、110℃付近に加温条件下で測定した)。
その粉末X線回折パターンを図7に示し、結晶の回折角(2θ)のピークおよび強度を表8に示した。
【0088】
【表4】
【0089】
【表5】
【0090】
【表6】
【0091】
【表7】
【0092】
【表8】
【0093】
13C固体NMRスペクトルの測定
実施例B1、C1およびD1で得られた結晶の13C固体NMRスペクトル測定を以下の条件で行った。
測定温度: 室温(〜22℃)
基準物質: グリシンのカルボニル炭素(外部基準:176.03ppm)
測定核: 13C(100.6248425MHz)
パルス繰り返し時間:
50秒(実施例C1、D1)
5秒(実施例B1)
パルスモード: CP/TOSS測定
【0094】
実施例B1で得られた結晶の13C固体NMRスペクトルを図8に示し、化学シフトを表9にまとめた。実施例D1で得られた結晶の13C固体NMRスペクトルを図9に示し、化学シフトを表10にした。実施例C1で得られた結晶の13C固体NMRスペクトルを図10に示し、化学シフトを表11にした。
【0095】
【表9】
【0096】
【表10】
【0097】
【表11】
【産業上の利用可能性】
【0098】
本発明の結晶は良好な物性を有し、神経変性疾患等の治療剤または予防剤の有効成分として使用するのに適している。
【図面の簡単な説明】
【0099】
【図1】実施例B1で得られた結晶の赤外線吸収スペクトル(KBr法)を表す図である。
【図2】実施例C1で得られた結晶の赤外線吸収スペクトル(KBr法)を表す図である。
【図3】参考例A1で得られた結晶の粉末X線回析パターンを表す図である。
【図4】実施例B1で得られた結晶の粉末X線回析パターンを表す図である。
【図5】実施例C1で得られた結晶の粉末X線回析パターンを表す図である。
【図6】実施例D1で得られた結晶の粉末X線回析パターンを表す図である。
【図7】実施例E1における粉末X線回析パターンを表す図である。
【図8】実施例B1で得られた結晶の13C固体核磁気共鳴(NMR)スペクトルを表す図である。
【図9】実施例D1で得られた結晶の13C固体NMRスペクトルを表す図である。
【図10】実施例C1で得られた結晶の13C固体NMRスペクトルを表す図である。
【図11】様々な温度における水和物結晶の粉末X線回析パターンを表す図である。
【図12】様々な相対湿度における水和物結晶の粉末X線回析パターンを表す図である。
【特許請求の範囲】
【請求項1】
3−(2−シアノフェニル)−5−(2−ピリジル)−1−フェニル−1,2−ジヒドロピリジン−2−オンを、アルコール系溶媒、アルキルケトン系溶媒および水からなる群から選ばれる1または2の溶媒を結晶化溶媒として用いて結晶化させることを特徴とする回折角度(2θ±0.2°)8.7°に回折ピークを有する3−(2−シアノフェニル)−5−(2−ピリジル)−1−フェニル−1,2−ジヒドロピリジン−2−オンの水和物の結晶の製造方法。
【請求項2】
結晶化溶媒が、アセトンと水の混合溶媒である請求項1記載の製造方法。
【請求項3】
結晶化溶媒が、アセトンと水(体積比37:3〜24:16)の混合溶媒である請求項1記載の製造方法。
【請求項4】
結晶化を60〜−30℃で実施することを特徴とする請求項1〜3いずれか1項記載の製造方法。
【請求項5】
3−(2−シアノフェニル)−5−(2−ピリジル)−1−フェニル−1,2−ジヒドロピリジン−2−オンを結晶化溶媒に溶解した溶液を、50℃以上で加熱後、40〜5℃/時間の冷却速度で、10〜−20℃まで冷却することを特徴とする、請求項1〜3いずれか1項記載の製造方法。
【請求項6】
3−(2−シアノフェニル)−5−(2−ピリジル)−1−フェニル−1,2−ジヒドロピリジン−2−オンの重量に対する容量比で10〜50倍量(v/w)の結晶化溶媒を用いることを特徴とする、請求項1〜5いずれか1項記載の製造方法。
【請求項7】
60℃以下で種結晶を加えることを特徴とする、請求項1〜6いずれか1項記載の製造方法。
【請求項8】
結晶化の後、減圧乾燥することを特徴とする、請求項1〜7いずれか1項記載の製造方法。
【請求項9】
結晶化および減圧乾燥の後、大気中放置することを特徴とする、請求項1〜8いずれか1項記載の製造方法。
【請求項1】
3−(2−シアノフェニル)−5−(2−ピリジル)−1−フェニル−1,2−ジヒドロピリジン−2−オンを、アルコール系溶媒、アルキルケトン系溶媒および水からなる群から選ばれる1または2の溶媒を結晶化溶媒として用いて結晶化させることを特徴とする回折角度(2θ±0.2°)8.7°に回折ピークを有する3−(2−シアノフェニル)−5−(2−ピリジル)−1−フェニル−1,2−ジヒドロピリジン−2−オンの水和物の結晶の製造方法。
【請求項2】
結晶化溶媒が、アセトンと水の混合溶媒である請求項1記載の製造方法。
【請求項3】
結晶化溶媒が、アセトンと水(体積比37:3〜24:16)の混合溶媒である請求項1記載の製造方法。
【請求項4】
結晶化を60〜−30℃で実施することを特徴とする請求項1〜3いずれか1項記載の製造方法。
【請求項5】
3−(2−シアノフェニル)−5−(2−ピリジル)−1−フェニル−1,2−ジヒドロピリジン−2−オンを結晶化溶媒に溶解した溶液を、50℃以上で加熱後、40〜5℃/時間の冷却速度で、10〜−20℃まで冷却することを特徴とする、請求項1〜3いずれか1項記載の製造方法。
【請求項6】
3−(2−シアノフェニル)−5−(2−ピリジル)−1−フェニル−1,2−ジヒドロピリジン−2−オンの重量に対する容量比で10〜50倍量(v/w)の結晶化溶媒を用いることを特徴とする、請求項1〜5いずれか1項記載の製造方法。
【請求項7】
60℃以下で種結晶を加えることを特徴とする、請求項1〜6いずれか1項記載の製造方法。
【請求項8】
結晶化の後、減圧乾燥することを特徴とする、請求項1〜7いずれか1項記載の製造方法。
【請求項9】
結晶化および減圧乾燥の後、大気中放置することを特徴とする、請求項1〜8いずれか1項記載の製造方法。
【図1】

【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【図11】
【図12】
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【図11】
【図12】
【公開番号】特開2007−99781(P2007−99781A)
【公開日】平成19年4月19日(2007.4.19)
【国際特許分類】
【出願番号】特願2007−9518(P2007−9518)
【出願日】平成19年1月18日(2007.1.18)
【分割の表示】特願2006−528904(P2006−528904)の分割
【原出願日】平成17年7月5日(2005.7.5)
【出願人】(506137147)エーザイ・アール・アンド・ディー・マネジメント株式会社 (215)
【Fターム(参考)】
【公開日】平成19年4月19日(2007.4.19)
【国際特許分類】
【出願日】平成19年1月18日(2007.1.18)
【分割の表示】特願2006−528904(P2006−528904)の分割
【原出願日】平成17年7月5日(2005.7.5)
【出願人】(506137147)エーザイ・アール・アンド・ディー・マネジメント株式会社 (215)
【Fターム(参考)】
[ Back to top ]