
1,3−ジ置換−6−メチル−5−ニトロウラシル誘導体の製造方法
【課題】抗炎症治療薬等の原薬の中間体として有用な1,3-ジ置換-6-メチル-5-ニトロウラシル誘導体の製造方法の提供。
【解決手段】式(1)(式中、R1およびR2は、同一又は異なって、C1-6アルキル基、又はC3-7シクロアルキル基を表す。)で示される化合物と濃硫酸の混合物に、式(1)の化合物に対して1.0モル倍から2.0モル倍の濃硝酸と反応させる工程を含む、式(2)で示される化合物を製造する方法。

【解決手段】式(1)(式中、R1およびR2は、同一又は異なって、C1-6アルキル基、又はC3-7シクロアルキル基を表す。)で示される化合物と濃硫酸の混合物に、式(1)の化合物に対して1.0モル倍から2.0モル倍の濃硝酸と反応させる工程を含む、式(2)で示される化合物を製造する方法。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、医薬品原料等として有用な1,3-ジ置換-6-メチル-5-ニトロウラシル誘導体の製造方法に関する。
【背景技術】
【0002】
1,3-ジ置換-6-メチル-5-ニトロウラシル誘導体は、抗炎症治療薬や育毛・養毛剤などとして有用なピロロ[3,2-d]ピリミジン-2,6-ジオン誘導体の製造において、原料として使用できる(例えば、特許文献1、特許文献2参照)。
【0003】
1,3-ジ置換-6-メチル-5-ニトロウラシル誘導体の製造方法としては、例えば、混酸(濃硫酸と濃硝酸の混合物)に1,3-ジ置換-6-メチルウラシル誘導体を加える方法が知られている(例えば、特許文献1、特許文献3、非特許文献1参照)。しかし該製造法は収率の面で満足いくものではない(収率30%〜56%)。また、この収率を改善する方法についての記載や示唆もない。
【0004】
その他の製造方法としては、1,3,6-トリメチルウラシルを発煙硝酸と硫酸を用いてニトロ化する方法が知られている(例えば、非特許文献2参照)。しかしながら、この製造法は、酸化力がきわめて強く、かつ腐食性が強い発煙硝酸を使用しているため、安全面および設備面を考慮すると、工業生産に適していない。
【0005】
【化1】
【0006】
上記ニトロ化の原料となる1,3-ジ置換-6-メチルウラシル誘導体の製造方法としては、例えば、水酸化ナトリウム水溶液と6-メチルウラシルの混合物に硫酸ジメチルを加えて反応させる方法が知られている(例えば、非特許文献3参照)。しかしながら、当該文献記載の方法で反応を行うと、反応が途中で停止してしまい、反応完結まで適宜水酸化ナトリウムおよび硫酸ジメチルを追加していく必要があるため、この製造方法には再現性の面で問題がある。
【0007】
【化2】
【先行技術文献】
【特許文献】
【0008】
【特許文献1】国際公開第2005/042534号
【特許文献2】特開2007−204387号
【特許文献3】特開昭47−14182号
【非特許文献】
【0009】
【非特許文献1】J. Med. Chem., 15, 471(1972)
【非特許文献2】Chem. Pharm. Bull., 6, 482(1958)
【非特許文献3】Chem. Ber., 90, 728(1957)
【発明の概要】
【発明が解決しようとする課題】
【0010】
本発明の課題は、工業生産に適した1,3-ジ置換-6-メチル-5-ニトロウラシル誘導体の製造法を提供することにある。
【課題を解決するための手段】
【0011】
本発明者らは、これら先行技術文献に記載の方法において用いる試薬の仕込み順と当量について鋭意検討した結果、1.0モル倍から2.0モル倍の濃硝酸を式(1)で表される化合物と濃硫酸の混合物に添加することによって、式(2)で表される1,3-ジ置換-6-メチル-5-ニトロウラシル誘導体の収率が格段に向上すること、さらに、6-メチルウラシルと硫酸ジメチルの混合物に、塩基の水溶液を滴下して反応させることにより、高収率で再現性よく1,3,6-トリメチルウラシルを製造できることを見出し、本発明を完成するに至った。
【0012】
すなわち本発明によれば、1,3-ジ置換-6-メチル-5-ニトロウラシル誘導体の新規な製造法が提供される。
〔1〕式(1):
【0013】
【化3】
(式中、R1およびR2は、同一又は異なって、C1-6アルキル基、又はC3-7シクロアルキル基を表す。)で表される化合物と濃硫酸の混合物に、式(1)の化合物に対して1.0モル倍から2.0モル倍の濃硝酸を滴下して反応させる工程を含む、式(2):
【0014】
【化4】
[式中、R1およびR2は、前記記載と同義である。]で表される化合物の製造方法。
〔2〕R1およびR2が、同一又は異なって、C1-6アルキル基である、〔1〕に記載の製造方法。
〔3〕R1およびR2がメチル基である、〔1〕に記載の製造方法。
〔4〕6-メチルウラシルと硫酸ジメチルの混合物に、塩基の水溶液を滴下して反応させることにより1,3,6-トリメチルウラシルを得る第1工程と、1,3,6-トリメチルウラシルと濃硫酸の混合物に、式(1)の化合物に対して1.0モル倍から2.0モル倍の濃硝酸を滴下して反応させることにより、1,3,6-トリメチル-5-ニトロウラシルを得る第2工程を有する、〔3〕に記載の製造方法。
〔5〕塩基がアルカリ金属水酸化物である〔4〕に記載の製造方法。
【発明の効果】
【0015】
本発明の製造方法を用いることにより、医薬等の原薬の中間体として有用な1,3-ジ置換-6-メチル-5-ニトロウラシル誘導体を、高収率で安全かつ安価に製造することができる。特に、本発明のニトロ化工程は、通常では大過剰使用される濃硝酸の使用量も必要最小限の量となっており、さらに工業生産において安全面、設備面から問題となる発煙硝酸を使用することがない。そして、比較例2に示す製法では収率が49%であるのに対し、実施例2に示す収率が93.8%であることからも明らかなとおり、本発明における収率は従来技術と比べて大幅に改善されている。また、本発明の6-メチルウラシルの製造方法を組み合わせることにより、1,3,6-トリメチルウラシルの全収率も41.6%から89.2%へと大幅に向上している。したがって、本発明の製造方法を用いれば、カラムクロマトグラフィーによる精製を行うことなく、1,3-ジ置換-6-メチル-5-ニトロウラシル誘導体を安全かつ効率的に合成することができる。
【図面の簡単な説明】
【0016】
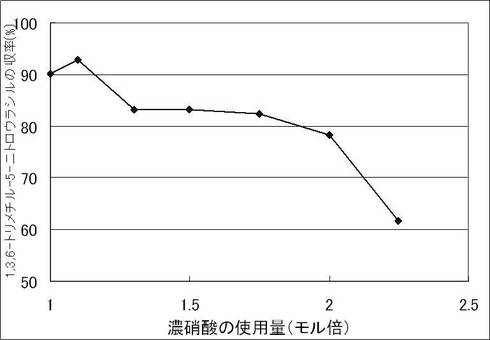
【図1】実施例3における濃硝酸の使用量と1,3,6-トリメチル-5-ニトロウラシルの収率の関係を示す。
【発明を実施するための形態】
【0017】
以下に、本発明をさらに詳細に説明する。
【0018】
「C1-6アルキル基」は、炭素数1〜6個を有する直鎖または分枝状の飽和炭化水素基を意味する。好ましくは、「C1-4アルキル基」である。「C1-6アルキル基」の具体例としては、例えば、メチル、エチル、プロピル、イソプロピル、ブチル、イソブチル、sec−ブチル、tert−ブチル、ペンチル、イソペンチル、ネオペンチル、1−エチルプロピル、ヘキシル、イソヘキシル、1,1−ジメチルブチル、2,2−ジメチルブチル、3,3−ジメチルブチル、2−エチルブチル等が挙げられる。
【0019】
「C3-7シクロアルキル基」は、炭素数3〜7個を有し、環状の飽和または不飽和炭化水素基を意味する。好ましくは、「C3-6シクロアルキル基」である。「C3-7シクロアルキル基」の具体例としては、例えば、シクロプロピル、シクロブチル、シクロペンチル、シクロヘキシル、シクロヘプチル、シクロペンテニル、シクロヘキセニル等が挙げられる。
【0020】
次に本発明の製造方法について詳細に説明する。
1,3,6-トリメチルウラシルは、6-メチルウラシル(別名:2,4-ジヒドロキシ-6-メチルピリミジン)を適当な塩基存在下、硫酸ジメチルと反応させることにより得られる。塩基としては、例えば、水酸化ナトリウムや水酸化カリウムなどのアルカリ金属水酸化物が用いられる。硫酸ジメチルの使用量は、6-メチルウラシル1モルに対して、2モルから10モルの範囲であり、好ましくは2モルから5モルの範囲である。塩基の使用量は、6-メチルウラシル1モルに対して、2モルから20モルの範囲であり、好ましくは2モルから5モルの範囲である。反応溶媒としては水が挙げられる。反応温度は通常0℃から100℃、好ましくは0℃から20℃である。また、該工程においては、6-メチルウラシルと硫酸ジメチルと水の混合物に対し、塩基の水溶液を滴下して反応させることにより、塩基による硫酸ジメチルの過剰な分解を制御することができ、工業的に好ましいプロセスとなる。
【0021】
式(2)で示される化合物は、式(1)で示される化合物と濃硫酸の混合物に、濃硝酸を添加することにより得られる。濃硫酸の使用量は、式(1)で示される化合物1モルに対して、2モルから20モルの範囲であり、好ましくは10モルから15モルの範囲である。濃硝酸の使用量は、式(1)で示される化合物1モルに対して、1.0モルから2.0モルの範囲[式(1)の化合物に対して1.0モル倍から2.0モル倍の範囲]であり、好ましくは1.0モルから1.75 モルの範囲[式(1)の化合物に対して1.0モル倍から1.75モル倍の範囲]であり、さらに好ましくは1.0モルから1.1モル の範囲[式(1)の化合物に対して1.0モル倍から1.1モル倍の範囲]である。濃硝酸の濃度は60%から70%が好ましい。反応溶媒としては無溶媒もしくは水が挙げられる。反応温度は通常0℃から30℃、好ましくは0℃から15℃である。また、該工程においては、式(2)の化合物と濃硫酸の混合物に対し、濃硝酸を滴下して反応させることにより、工業的に好ましいプロセスとなる。
【実施例】
【0022】
以下に本発明を、実施例によりさらに詳細に説明するが、本発明はこれらの具体例に限定されるものではない。尚、以下の実施例において示された化合物名は、必ずしもIUPAC命名法に従うものではない。
【0023】
HPLC測定条件に記載されている測定条件は以下の内容を表す。
カラム:Agilent Zorbax SB-Phenyl ,5μm 4.6mmφ×250mm
移動層:
A液:0.1%トリフルオロ酢酸水
B液:アセトニトリル
グラジェント条件:
【表1】
流量:1.0mL/min
検出器:UV(254nm)
カラム温度:25℃
尚、本条件における1,3,6-トリメチル-5-ニトロウラシルの保持時間は13.0分であった。
【0024】
比較例1
1,3,6-トリメチルウラシルの製造
非特許文献3の方法に準じて表記化合物を合成した。
6-メチルウラシル 25 g(0.20mol)を水(100 g)に懸濁させ、30℃で20%水酸化ナトリウム水溶液99.12g(0.50mol)を加えた。ここに、硫酸ジメチル 55.01g(0.44mol)を2時間で滴下した。2時間保温後、さらに20%水酸化ナトリウム水溶液 19.8g(0.10mol)と硫酸ジメチル 10.0g(0.08mol)を加えた。これを1時間おきに3回繰り返した。さらに14時間攪拌後、20%硫酸水溶液35.1gを加えてpHを2.7に調整し、クロロホルム 125gで4回抽出した。有機層を合わせて、溶媒を減圧濃縮した。残渣に2-プロパノール 60gを加え、60℃に昇温して溶解し、0℃まで冷却して濾取することにより、表題化合物 25.91 g(収率84.47%)を白色固体として得た。
【0025】
実施例1
1,3,6-トリメチルウラシルの製造
前記比較例1より水酸化ナトリウム水溶液と硫酸ジメチルの仕込み順を変更して反応を行なった。
6-メチルウラシル 9.75 kg (77.31 mol)を水(29.3 kg)に懸濁させ、5℃に冷却した。ここに、硫酸ジメチル 29.3 kg (232.3 mol)加えた後、20%水酸化ナトリウム水溶液 38.6 kg(193.25 mol)を4.5時間かけて滴下した。滴下終了後同温で3.5時間攪拌し、20%水酸化ナトリウム水溶液 15.4 kg(77.25 mol)を加えた。この混合物を、14.4℃まで昇温し、13時間攪拌した。20%硫酸水溶液 8.72 kgを加えてpHを1.92に調整し、クロロホルム(49 kg)を加え、攪拌後、油層と水層を分離した。この操作をさらに3回行った。すべての有機層を併せて濃縮し、残渣に2−プロパノール 49 kgを加えて、残渣が25 kgになるまでさらに濃縮した。残渣を65℃まで昇温して溶解し、さらに0℃まで冷却した。0℃で30分保温し、析出している結晶を濾取し、減圧乾燥することにより、表題化合物 11.3 kg(収率95.1%)を白色固体として得た。
1H NMR (400 MHz, CDCl3) δ 5.62 (d, 1H, J=0.8Hz), 3.41 (s, 3H), 3.34 (s, 3H), 2.24(d, 3H, J=0.8Hz).
【0026】
比較例2
1,3,6-トリメチル-5-ニトロウラシルの製造
1,3,6-トリメチルウラシル 130.22 g (0.84 mol)を96%硫酸(996.67g)に溶解させ、20℃で60% 硝酸 239.31 g (2.28 mol;1,3,6-トリメチルウラシルに対して2.7モル倍の量)を1.5時間で滴下した。滴下終了後25℃で2時間攪拌し、さらに0℃まで冷却した。この混合物を5℃に冷却した水(2.6kg)に0〜10℃の範囲で滴下し、滴下終了後、5℃で2時間攪拌した。析出している結晶を濾取し、氷冷水(650g)で1回、2-プロパノール(325g)で2回洗浄した。結晶を減圧乾燥することにより、表題化合物 93.88g(LC面百値87.45%、収率49.3%)を淡黄色固体として得た。比較例1と比較例2の全収率は41.6%であった。
【0027】
実施例2
1,3,6-トリメチル-5-ニトロウラシルの製造
前記比較例2より濃硝酸の仕込み順と当量を変更して反応を行なった。
1,3,6-トリメチルウラシル 11.34 kg (73.56 mol)を96%硫酸(86.8 kg)に溶解させ、10℃で60%硝酸 8.48 kg (80.75 mol;1,3,6-トリメチルウラシルに対して1.1モル倍の量)を1.5時間かけて滴下した。滴下終了後10℃で1時間攪拌した。この混合物を氷水(227kg)に1時間45分かけて滴下し、滴下終了後、0℃で15時間攪拌した。析出している結晶を濾取し、氷冷水(56 kg)で1回、2-プロパノール(29.6 kg)で2回洗浄した。結晶を減圧乾燥することにより、表題化合物 13.7kg(収率93.8%)を淡黄色固体として得た。実施例1と実施例2の全収率は89.2%であった。
1H NMR (400 MHz, CDCl3) δ 3.52(s, 3H), 3.40 (s, 3H), 2.42 (s, 3H).
【0028】
実施例3
1,3,6-トリメチル-5-ニトロウラシルの製造(濃硝酸の最適当量検討)
1,3,6-トリメチルウラシル 5.00 g (32.4 mmol)を96%硫酸(38.3g)に溶解させ、25〜30℃で当量を変化させて60% 硝酸を滴下した。滴下終了後7℃で4時間攪拌し、この混合物を5℃に冷却した水(100g)に0〜10℃の範囲で滴下し、滴下終了後、5℃で2時間攪拌した。析出している結晶を濾取し、氷冷水(25g)で1回、2-プロパノール(10g)で2回洗浄した。結晶を減圧乾燥することにより、表題化合物 を取得した。濃硝酸の当量と1,3,6-トリメチル-5-ニトロウラシルの収率との関係を[表2]と [図1]に示す。この結果より、濃硝酸を原料に対して2.0モル倍を超えて使用すると、生成物が顕著に分解することがわかった。
【表2】
【産業上の利用可能性】
【0029】
本発明の製造方法を用いることにより、抗炎症治療薬等の原薬の中間体として有用な1,3-ジ置換-6-メチル-5-ニトロウラシル誘導体を、高収率で安全かつ安価に製造することができる。
【技術分野】
【0001】
本発明は、医薬品原料等として有用な1,3-ジ置換-6-メチル-5-ニトロウラシル誘導体の製造方法に関する。
【背景技術】
【0002】
1,3-ジ置換-6-メチル-5-ニトロウラシル誘導体は、抗炎症治療薬や育毛・養毛剤などとして有用なピロロ[3,2-d]ピリミジン-2,6-ジオン誘導体の製造において、原料として使用できる(例えば、特許文献1、特許文献2参照)。
【0003】
1,3-ジ置換-6-メチル-5-ニトロウラシル誘導体の製造方法としては、例えば、混酸(濃硫酸と濃硝酸の混合物)に1,3-ジ置換-6-メチルウラシル誘導体を加える方法が知られている(例えば、特許文献1、特許文献3、非特許文献1参照)。しかし該製造法は収率の面で満足いくものではない(収率30%〜56%)。また、この収率を改善する方法についての記載や示唆もない。
【0004】
その他の製造方法としては、1,3,6-トリメチルウラシルを発煙硝酸と硫酸を用いてニトロ化する方法が知られている(例えば、非特許文献2参照)。しかしながら、この製造法は、酸化力がきわめて強く、かつ腐食性が強い発煙硝酸を使用しているため、安全面および設備面を考慮すると、工業生産に適していない。
【0005】
【化1】
【0006】
上記ニトロ化の原料となる1,3-ジ置換-6-メチルウラシル誘導体の製造方法としては、例えば、水酸化ナトリウム水溶液と6-メチルウラシルの混合物に硫酸ジメチルを加えて反応させる方法が知られている(例えば、非特許文献3参照)。しかしながら、当該文献記載の方法で反応を行うと、反応が途中で停止してしまい、反応完結まで適宜水酸化ナトリウムおよび硫酸ジメチルを追加していく必要があるため、この製造方法には再現性の面で問題がある。
【0007】
【化2】
【先行技術文献】
【特許文献】
【0008】
【特許文献1】国際公開第2005/042534号
【特許文献2】特開2007−204387号
【特許文献3】特開昭47−14182号
【非特許文献】
【0009】
【非特許文献1】J. Med. Chem., 15, 471(1972)
【非特許文献2】Chem. Pharm. Bull., 6, 482(1958)
【非特許文献3】Chem. Ber., 90, 728(1957)
【発明の概要】
【発明が解決しようとする課題】
【0010】
本発明の課題は、工業生産に適した1,3-ジ置換-6-メチル-5-ニトロウラシル誘導体の製造法を提供することにある。
【課題を解決するための手段】
【0011】
本発明者らは、これら先行技術文献に記載の方法において用いる試薬の仕込み順と当量について鋭意検討した結果、1.0モル倍から2.0モル倍の濃硝酸を式(1)で表される化合物と濃硫酸の混合物に添加することによって、式(2)で表される1,3-ジ置換-6-メチル-5-ニトロウラシル誘導体の収率が格段に向上すること、さらに、6-メチルウラシルと硫酸ジメチルの混合物に、塩基の水溶液を滴下して反応させることにより、高収率で再現性よく1,3,6-トリメチルウラシルを製造できることを見出し、本発明を完成するに至った。
【0012】
すなわち本発明によれば、1,3-ジ置換-6-メチル-5-ニトロウラシル誘導体の新規な製造法が提供される。
〔1〕式(1):
【0013】
【化3】
(式中、R1およびR2は、同一又は異なって、C1-6アルキル基、又はC3-7シクロアルキル基を表す。)で表される化合物と濃硫酸の混合物に、式(1)の化合物に対して1.0モル倍から2.0モル倍の濃硝酸を滴下して反応させる工程を含む、式(2):
【0014】
【化4】
[式中、R1およびR2は、前記記載と同義である。]で表される化合物の製造方法。
〔2〕R1およびR2が、同一又は異なって、C1-6アルキル基である、〔1〕に記載の製造方法。
〔3〕R1およびR2がメチル基である、〔1〕に記載の製造方法。
〔4〕6-メチルウラシルと硫酸ジメチルの混合物に、塩基の水溶液を滴下して反応させることにより1,3,6-トリメチルウラシルを得る第1工程と、1,3,6-トリメチルウラシルと濃硫酸の混合物に、式(1)の化合物に対して1.0モル倍から2.0モル倍の濃硝酸を滴下して反応させることにより、1,3,6-トリメチル-5-ニトロウラシルを得る第2工程を有する、〔3〕に記載の製造方法。
〔5〕塩基がアルカリ金属水酸化物である〔4〕に記載の製造方法。
【発明の効果】
【0015】
本発明の製造方法を用いることにより、医薬等の原薬の中間体として有用な1,3-ジ置換-6-メチル-5-ニトロウラシル誘導体を、高収率で安全かつ安価に製造することができる。特に、本発明のニトロ化工程は、通常では大過剰使用される濃硝酸の使用量も必要最小限の量となっており、さらに工業生産において安全面、設備面から問題となる発煙硝酸を使用することがない。そして、比較例2に示す製法では収率が49%であるのに対し、実施例2に示す収率が93.8%であることからも明らかなとおり、本発明における収率は従来技術と比べて大幅に改善されている。また、本発明の6-メチルウラシルの製造方法を組み合わせることにより、1,3,6-トリメチルウラシルの全収率も41.6%から89.2%へと大幅に向上している。したがって、本発明の製造方法を用いれば、カラムクロマトグラフィーによる精製を行うことなく、1,3-ジ置換-6-メチル-5-ニトロウラシル誘導体を安全かつ効率的に合成することができる。
【図面の簡単な説明】
【0016】
【図1】実施例3における濃硝酸の使用量と1,3,6-トリメチル-5-ニトロウラシルの収率の関係を示す。
【発明を実施するための形態】
【0017】
以下に、本発明をさらに詳細に説明する。
【0018】
「C1-6アルキル基」は、炭素数1〜6個を有する直鎖または分枝状の飽和炭化水素基を意味する。好ましくは、「C1-4アルキル基」である。「C1-6アルキル基」の具体例としては、例えば、メチル、エチル、プロピル、イソプロピル、ブチル、イソブチル、sec−ブチル、tert−ブチル、ペンチル、イソペンチル、ネオペンチル、1−エチルプロピル、ヘキシル、イソヘキシル、1,1−ジメチルブチル、2,2−ジメチルブチル、3,3−ジメチルブチル、2−エチルブチル等が挙げられる。
【0019】
「C3-7シクロアルキル基」は、炭素数3〜7個を有し、環状の飽和または不飽和炭化水素基を意味する。好ましくは、「C3-6シクロアルキル基」である。「C3-7シクロアルキル基」の具体例としては、例えば、シクロプロピル、シクロブチル、シクロペンチル、シクロヘキシル、シクロヘプチル、シクロペンテニル、シクロヘキセニル等が挙げられる。
【0020】
次に本発明の製造方法について詳細に説明する。
1,3,6-トリメチルウラシルは、6-メチルウラシル(別名:2,4-ジヒドロキシ-6-メチルピリミジン)を適当な塩基存在下、硫酸ジメチルと反応させることにより得られる。塩基としては、例えば、水酸化ナトリウムや水酸化カリウムなどのアルカリ金属水酸化物が用いられる。硫酸ジメチルの使用量は、6-メチルウラシル1モルに対して、2モルから10モルの範囲であり、好ましくは2モルから5モルの範囲である。塩基の使用量は、6-メチルウラシル1モルに対して、2モルから20モルの範囲であり、好ましくは2モルから5モルの範囲である。反応溶媒としては水が挙げられる。反応温度は通常0℃から100℃、好ましくは0℃から20℃である。また、該工程においては、6-メチルウラシルと硫酸ジメチルと水の混合物に対し、塩基の水溶液を滴下して反応させることにより、塩基による硫酸ジメチルの過剰な分解を制御することができ、工業的に好ましいプロセスとなる。
【0021】
式(2)で示される化合物は、式(1)で示される化合物と濃硫酸の混合物に、濃硝酸を添加することにより得られる。濃硫酸の使用量は、式(1)で示される化合物1モルに対して、2モルから20モルの範囲であり、好ましくは10モルから15モルの範囲である。濃硝酸の使用量は、式(1)で示される化合物1モルに対して、1.0モルから2.0モルの範囲[式(1)の化合物に対して1.0モル倍から2.0モル倍の範囲]であり、好ましくは1.0モルから1.75 モルの範囲[式(1)の化合物に対して1.0モル倍から1.75モル倍の範囲]であり、さらに好ましくは1.0モルから1.1モル の範囲[式(1)の化合物に対して1.0モル倍から1.1モル倍の範囲]である。濃硝酸の濃度は60%から70%が好ましい。反応溶媒としては無溶媒もしくは水が挙げられる。反応温度は通常0℃から30℃、好ましくは0℃から15℃である。また、該工程においては、式(2)の化合物と濃硫酸の混合物に対し、濃硝酸を滴下して反応させることにより、工業的に好ましいプロセスとなる。
【実施例】
【0022】
以下に本発明を、実施例によりさらに詳細に説明するが、本発明はこれらの具体例に限定されるものではない。尚、以下の実施例において示された化合物名は、必ずしもIUPAC命名法に従うものではない。
【0023】
HPLC測定条件に記載されている測定条件は以下の内容を表す。
カラム:Agilent Zorbax SB-Phenyl ,5μm 4.6mmφ×250mm
移動層:
A液:0.1%トリフルオロ酢酸水
B液:アセトニトリル
グラジェント条件:
【表1】
流量:1.0mL/min
検出器:UV(254nm)
カラム温度:25℃
尚、本条件における1,3,6-トリメチル-5-ニトロウラシルの保持時間は13.0分であった。
【0024】
比較例1
1,3,6-トリメチルウラシルの製造
非特許文献3の方法に準じて表記化合物を合成した。
6-メチルウラシル 25 g(0.20mol)を水(100 g)に懸濁させ、30℃で20%水酸化ナトリウム水溶液99.12g(0.50mol)を加えた。ここに、硫酸ジメチル 55.01g(0.44mol)を2時間で滴下した。2時間保温後、さらに20%水酸化ナトリウム水溶液 19.8g(0.10mol)と硫酸ジメチル 10.0g(0.08mol)を加えた。これを1時間おきに3回繰り返した。さらに14時間攪拌後、20%硫酸水溶液35.1gを加えてpHを2.7に調整し、クロロホルム 125gで4回抽出した。有機層を合わせて、溶媒を減圧濃縮した。残渣に2-プロパノール 60gを加え、60℃に昇温して溶解し、0℃まで冷却して濾取することにより、表題化合物 25.91 g(収率84.47%)を白色固体として得た。
【0025】
実施例1
1,3,6-トリメチルウラシルの製造
前記比較例1より水酸化ナトリウム水溶液と硫酸ジメチルの仕込み順を変更して反応を行なった。
6-メチルウラシル 9.75 kg (77.31 mol)を水(29.3 kg)に懸濁させ、5℃に冷却した。ここに、硫酸ジメチル 29.3 kg (232.3 mol)加えた後、20%水酸化ナトリウム水溶液 38.6 kg(193.25 mol)を4.5時間かけて滴下した。滴下終了後同温で3.5時間攪拌し、20%水酸化ナトリウム水溶液 15.4 kg(77.25 mol)を加えた。この混合物を、14.4℃まで昇温し、13時間攪拌した。20%硫酸水溶液 8.72 kgを加えてpHを1.92に調整し、クロロホルム(49 kg)を加え、攪拌後、油層と水層を分離した。この操作をさらに3回行った。すべての有機層を併せて濃縮し、残渣に2−プロパノール 49 kgを加えて、残渣が25 kgになるまでさらに濃縮した。残渣を65℃まで昇温して溶解し、さらに0℃まで冷却した。0℃で30分保温し、析出している結晶を濾取し、減圧乾燥することにより、表題化合物 11.3 kg(収率95.1%)を白色固体として得た。
1H NMR (400 MHz, CDCl3) δ 5.62 (d, 1H, J=0.8Hz), 3.41 (s, 3H), 3.34 (s, 3H), 2.24(d, 3H, J=0.8Hz).
【0026】
比較例2
1,3,6-トリメチル-5-ニトロウラシルの製造
1,3,6-トリメチルウラシル 130.22 g (0.84 mol)を96%硫酸(996.67g)に溶解させ、20℃で60% 硝酸 239.31 g (2.28 mol;1,3,6-トリメチルウラシルに対して2.7モル倍の量)を1.5時間で滴下した。滴下終了後25℃で2時間攪拌し、さらに0℃まで冷却した。この混合物を5℃に冷却した水(2.6kg)に0〜10℃の範囲で滴下し、滴下終了後、5℃で2時間攪拌した。析出している結晶を濾取し、氷冷水(650g)で1回、2-プロパノール(325g)で2回洗浄した。結晶を減圧乾燥することにより、表題化合物 93.88g(LC面百値87.45%、収率49.3%)を淡黄色固体として得た。比較例1と比較例2の全収率は41.6%であった。
【0027】
実施例2
1,3,6-トリメチル-5-ニトロウラシルの製造
前記比較例2より濃硝酸の仕込み順と当量を変更して反応を行なった。
1,3,6-トリメチルウラシル 11.34 kg (73.56 mol)を96%硫酸(86.8 kg)に溶解させ、10℃で60%硝酸 8.48 kg (80.75 mol;1,3,6-トリメチルウラシルに対して1.1モル倍の量)を1.5時間かけて滴下した。滴下終了後10℃で1時間攪拌した。この混合物を氷水(227kg)に1時間45分かけて滴下し、滴下終了後、0℃で15時間攪拌した。析出している結晶を濾取し、氷冷水(56 kg)で1回、2-プロパノール(29.6 kg)で2回洗浄した。結晶を減圧乾燥することにより、表題化合物 13.7kg(収率93.8%)を淡黄色固体として得た。実施例1と実施例2の全収率は89.2%であった。
1H NMR (400 MHz, CDCl3) δ 3.52(s, 3H), 3.40 (s, 3H), 2.42 (s, 3H).
【0028】
実施例3
1,3,6-トリメチル-5-ニトロウラシルの製造(濃硝酸の最適当量検討)
1,3,6-トリメチルウラシル 5.00 g (32.4 mmol)を96%硫酸(38.3g)に溶解させ、25〜30℃で当量を変化させて60% 硝酸を滴下した。滴下終了後7℃で4時間攪拌し、この混合物を5℃に冷却した水(100g)に0〜10℃の範囲で滴下し、滴下終了後、5℃で2時間攪拌した。析出している結晶を濾取し、氷冷水(25g)で1回、2-プロパノール(10g)で2回洗浄した。結晶を減圧乾燥することにより、表題化合物 を取得した。濃硝酸の当量と1,3,6-トリメチル-5-ニトロウラシルの収率との関係を[表2]と [図1]に示す。この結果より、濃硝酸を原料に対して2.0モル倍を超えて使用すると、生成物が顕著に分解することがわかった。
【表2】
【産業上の利用可能性】
【0029】
本発明の製造方法を用いることにより、抗炎症治療薬等の原薬の中間体として有用な1,3-ジ置換-6-メチル-5-ニトロウラシル誘導体を、高収率で安全かつ安価に製造することができる。
【特許請求の範囲】
【請求項1】
式(1):
【化1】
(式中、R1およびR2は、同一又は異なって、C1-6アルキル基、又はC3-7シクロアルキル基を表す。)で表される化合物と濃硫酸の混合物に、式(1)の化合物に対して1.0モル倍から2.0モル倍の濃硝酸を滴下して反応させる工程を含む、式(2):
【化2】
[式中、R1およびR2は、前記記載と同義である。]で表される化合物の製造方法。
【請求項2】
R1およびR2が、同一又は異なって、C1-6アルキル基である、請求項1に記載の製造方法。
【請求項3】
R1およびR2がメチル基である、請求項1に記載の製造方法。
【請求項4】
6-メチルウラシルと硫酸ジメチルの混合物に、塩基の水溶液を滴下して反応させることにより1,3,6-トリメチルウラシルを得る第1工程と、1,3,6-トリメチルウラシルと濃硫酸の混合物に、式(1)の化合物に対して1.0モル倍から2.0モル倍の濃硝酸を滴下して反応させることにより、1,3,6-トリメチル-5-ニトロウラシルを得る第2工程を有する、請求項3に記載の製造方法。
【請求項5】
塩基がアルカリ金属水酸化物である、請求項4に記載の製造方法。
【請求項1】
式(1):
【化1】
(式中、R1およびR2は、同一又は異なって、C1-6アルキル基、又はC3-7シクロアルキル基を表す。)で表される化合物と濃硫酸の混合物に、式(1)の化合物に対して1.0モル倍から2.0モル倍の濃硝酸を滴下して反応させる工程を含む、式(2):
【化2】
[式中、R1およびR2は、前記記載と同義である。]で表される化合物の製造方法。
【請求項2】
R1およびR2が、同一又は異なって、C1-6アルキル基である、請求項1に記載の製造方法。
【請求項3】
R1およびR2がメチル基である、請求項1に記載の製造方法。
【請求項4】
6-メチルウラシルと硫酸ジメチルの混合物に、塩基の水溶液を滴下して反応させることにより1,3,6-トリメチルウラシルを得る第1工程と、1,3,6-トリメチルウラシルと濃硫酸の混合物に、式(1)の化合物に対して1.0モル倍から2.0モル倍の濃硝酸を滴下して反応させることにより、1,3,6-トリメチル-5-ニトロウラシルを得る第2工程を有する、請求項3に記載の製造方法。
【請求項5】
塩基がアルカリ金属水酸化物である、請求項4に記載の製造方法。
【図1】

【公開番号】特開2013−6813(P2013−6813A)
【公開日】平成25年1月10日(2013.1.10)
【国際特許分類】
【出願番号】特願2011−142129(P2011−142129)
【出願日】平成23年6月27日(2011.6.27)
【出願人】(000002912)大日本住友製薬株式会社 (332)
【公開日】平成25年1月10日(2013.1.10)
【国際特許分類】
【出願日】平成23年6月27日(2011.6.27)
【出願人】(000002912)大日本住友製薬株式会社 (332)
[ Back to top ]