
1,4−ジヒドロピリジン誘導体を含有する製剤
【発明の詳細な説明】
【0001】
【産業上の利用分野】本発明は、水難溶性薬物1,4−ジヒドロピリジン誘導体を含有する新規な製剤に関する。
【0002】
【従来の技術】医薬として用いられる薬物の中には、優れた薬理作用を有しているにも拘らず、水に対して難溶性であるため消化管からの吸収が悪く、医薬品として利用されていないか又は多量に投与しなければならないものが少なくない。血管拡張作用をもつ1,4−ジヒドロピリジン誘導体もそのような薬物の1つであり、この化合物は、水に対して極めて難溶性であるため経口投与によるときは体内への吸収率があまりにも低く、従って生物学的利用率が低いという実用上の問題を有していた。
【0003】例えば、ニフェジピン(米国特許第3,644,627 号明細書)は狭心症発作治療剤として賞用されているが、吸収が遅いという欠点を有するため速吸収型(即効性)のニフェジピン経口投与製剤が種々提案されてきた。例えば、ニフェジピンとポリビニルピロリドンの混合物を有機溶媒に溶解後、溶媒を留去して得られる固形物(特開昭54−2316号公報)、ニフェジピンを液状ポリエチレングリコールに溶解して得られる溶液を充填した軟カプセル(特開昭48−28621 号公報)などが挙げられる。
【0004】一方、例えば降圧剤として使用する場合には、即効性よりも持続性が要求され、この場合には、遅吸収型の製剤が望まれる。このような遅吸収型のニフェジピン製剤をめざしたものとしては、ニフェジピン含有顆粒と、該顆粒の表面に水難溶性物質と腸溶性高分子とからなる被膜を形成させて得られる顆粒との混和顆粒剤(特開昭58−46019 号公報)が知られている。
【0005】
【発明が解決しようとする課題】従来の遅吸収型ニフェジピン製剤においては、その血中レベルが長時間に亘って一定の有効値に維持されることが要求されてきた。
【0006】しかし、薬物が本来もつ吸収性、即ち非徐放性の通常製剤に特徴的な吸収性を保持したまま、その吸収率を向上させる製剤が開発されたならば、薬物の血中レベルの維持に関連して惹起される副作用等の欠点を回避し得る点でより理想的な製剤となり得る。
【0007】本発明の目的は、このような望ましい吸収性を有し且つ吸収率を改善した水難溶性1,4−ジヒドロピリジン誘導体含有製剤を提供することである。
【0008】
【課題を解決するための手段】本発明は、一般式[I]:
【0009】
【化2】

【0010】[式中、Arは置換アリール基若しくはベンゾフラザニル基であり、R1 は置換若しくは無置換アルキル基であり、R2 は置換若しくは無置換アルキル又はアルケニル基であるか又は置換ピペリジル基であり、R3 及びR4 は置換若しくは無置換アルキル基であり、但し、5位の置換基R1 OCO−はジアルキルホスホノエート又はアルキレンジオキシホスホリル基で置き換えられてもよい]で表わされる水難溶性の薬物1,4−ジヒドロピリジン誘導体又はその酸付加塩とヒドロキシプロピルセルロースから成る固体分散物と、該1,4−ジヒドロピリジン誘導体又はその酸付加塩とヒドロキシプロピルメチルセルロースフタレートから成る固体分散物とを含み、且つ固体分散錠である製剤を提供する。
【0011】本発明製剤においては、有効成分としての前記1,4−ジヒドロピリジン誘導体の他に、結合剤(担体)としての水溶性高分子ヒドロキシプロピルセルロース及び腸溶性高分子ヒドロキシプロピルメチルセルロースフタレートが必須成分である。
【0012】上記一般式[I]中、置換若しくは無置換アルキル基としては、ジアルキルアミノ、ピペラジニル、アルコキシ、−ONO2 基などの置換基で置換された又は無置換の直鎖若しくは分枝アルキル基、好ましくは低級アルキル基、より好ましくはC1 〜C4 アルキル基が挙げられ、置換若しくは無置換アルケニル基としては、フェニル基などの置換基で置換された又は無置換のアルケニル基、好ましくは低級アルケニル基、より好ましくはC1 〜C4 アルケニル基が挙げられ、また、置換アリール基としては、ハロゲン、ニトロ、フラザニル基などの置換基を有するフェニル基などが挙げられる。
【0013】1,4−ジヒドロピリジン誘導体の具体例としては、狭心症治療剤、血管拡張剤として知られるニフェジピン及びその誘導体、例えばニソルジピン、ニカルジピン、ニモジピン、ニトレンジピン、ニルバジピン、マニジピン、、FRC8653(特開昭60−233058号)などが挙げられる。
【0014】本発明製剤は、一般式[I]の1,4−ジヒドロピリジン誘導体又はその酸付加塩1重量部に対し、ヒドロキシプロピルセルロース4重量部以上、好ましくは4〜5重量部を含む固体分散物(a)、及び該1,4−ジヒドロピリジン誘導体又はその酸付加塩1重量部に対し、ヒドロキシプロピルセルロースフタレート2重量部以上、好ましくは2〜5重量部を含む固体分散物(b)を混合し圧縮して固体分散錠に製剤化することにより得られる。固体分散物(a)と(b)の含有比は、該薬物が本来もつ体内吸収性を変えることなくその吸収率を向上させるように選択され、好ましくは(a)/(b)=2.5/7.5〜3.5/6.5(重量/重量)、より好ましくは(a)/(b)=3/7である。
【0015】本明細書中、「固体分散物」なる用語は不活性担体の中に薬物が単分子状に分散した固体を意味し、薬物は担体中に非晶質の形態で存在する。非晶質の判定は熱分析によって行い得る。
【0016】また、本発明製剤においては、好適には固体分散物(a)及び(b)の中に1種以上の賦形剤、崩壊剤又は滑沢剤、あるいはこれらの組合せ物を含ませることもできる。
【0017】賦形剤としては、例えば乳糖、マンニトール、ブドウ糖、コーンスターチ、結晶セルロース、天然又は合成ケイ酸アルミニウム等が挙げられ、メタケイ酸アルミン酸マグネシウムが好適に使用される。メタケイ酸アルミン酸マグネシウムを含有させる場合には通常固体分散物(a)又は(b)中に配合され、その配合量は、固体分散物(a)中では該薬物1重量部に対し11重量部以上、好ましくは11〜12重量部であり、また固体分散物(b)中では該薬物1重量部に対し5.5重量部以上、好ましくは5.5〜12重量部である。
【0018】崩壊剤としては、例えばアラビアゴム、アルギン酸ナトリウム、カルボキシメチルセルロースナトリウム、繊維系グルコン酸カルシウム等が挙げられ、また滑沢剤としては、例えばタルク、ステアリン酸マグネシウム等が挙げられる。
【0019】さらに必要に応じ、本発明錠剤をコーティング剤でフィルムコーティングしてもよい。コーティング剤としては、例えばヒドロキシプロピルメチルセルロース、エチルセルロース、などが挙げられ、またコーティング剤中には例えば不透明化剤(例えば、酸化チタン)や可塑剤(例えばポリエチレングリコール)を含有させることも可能である。
【0020】本発明製剤の利点は、FRC8653によって例示されるように、1,4−ジヒドロピリジン誘導体の経口投与後の最高血中濃度到達時間(tmax )が1.3時間(図8)であり、血中濃度−時間曲線下面積(AUC)が64.5ng・hr/ml(実施例5)であり、崩壊時間が4〜6分(実施例5)であり、及び100%溶出時間が150分(図7)であることから判るように、抗狭心症薬として市販されているニフェジピンの固体分散細粒(鐘紡(株))とは異なり胃内で必要以上に速く溶出しないで且つ100%溶出し、薬物が本来的にもつ吸収性(図1)を変えることなく(図8)、ヒドロキシプロピルセルロース・固体分散錠(実施例2)と同程度の高い体内吸収率を兼ね備えていることにある。本明細書中、「吸収性」なる用語は、薬物が本来もつ吸収性、即ち非徐放性の通常製剤に特徴的な吸収性を意味し、薬物血中濃度の経時変化によって表わされる。ここで、通常製剤とは非徐放性製剤を指す。
【0021】本発明製剤はまた、胃内での薬物の溶出に際してpH依存性が少ないという特性をも有している(図9)。
【0022】上述のような本発明製剤の性質は、水難溶性薬物、ヒドロキシプロピルセルロース及びヒドロキシプロピルメチルセルロースフタレートの必須三成分を一時に混合して得られる固体分散物(比較例2)又は固体分散物(a)若しくは(b)単独(実施例5)の性質と異なるものであり、本発明製剤に特徴的な性質である。特に、薬物本来の吸収性を変化させないことは、少なくとも薬物の血中濃度の維持に関連して惹起される副作用を回避し得ることを意味している。
【0023】本発明の1,4−ジヒドロピリジン誘導体含有製剤は以下の方法によって製造し得る。すなわち、この方法は、(i) 一般式[I]で表わされる1,4−ジヒドロピリジン誘導体又はその酸付加塩とヒドロキシプロピルセルロースとを含む混合物から固体分散物(a)を製造する工程、(ii) 該1,4−ジヒドロピリジン誘導体又はその酸付加塩とヒドロキシプロピルメチルセルロースフタレートとを含む混合物から固体分散物(b)を製造する工程、(iii) 前記固体分散物(a)と(b)とを混合する工程、及び、(iv) 得られた混合物を錠剤に成形する工程、から成る。
【0024】固体分散物(a)又は(b)は、前記1,4−ジヒドロピリジン誘導体又はその酸付加塩、ヒドロキシプロピルセルロース又はヒドロキシプロピルメチルセルロースフタレートを有機溶媒に溶解し賦形剤、崩壊剤等の助剤に混合した後、該溶媒を蒸発等により除去することにより得ることができる。
【0025】また固体分散物中に、賦形剤を含有させる場合には、メタケイ酸アルミン酸マグネシウムが望ましい。この場合、固体分散物(a)及び(b)中に、メタケイ酸アルミン酸マグネシウムがそれぞれ前記1,4−ジヒドロピリジン誘導体又はその酸付加塩対メタケイ酸アルミン酸マグネシウム1:11以上、好ましくは1:11〜12、及び1:5.5以上、好ましくは1:5.5〜12の重量比で含有し得る。
【0026】有機溶媒は、例えばメタノール、エタノール等の低級アルコール類、ジクロルメタン、クロロホルム等のハロゲン化炭化水素類、及びこれらの混合物が挙げられる。
【0027】また、錠剤への成形は、公知の方法で実施され得る。
【0028】尚、本発明製剤である固体分散錠は、固体分散物(a)/(b)=3/7の混合顆粒(比較例3)とも性質が異なるものであり、本発明製剤は錠剤でなければならない。
【0029】
【実施例】以下の比較例及び実施例では、一般式[I]の1,4−ジヒドロピリジン誘導体の具体例としてFRC8653(特開昭60−233058号)を取り挙げて本発明をさらに具体的に説明するが、その他の誘導体の場合にも同様に適用可能である。なお、FRC8653は、式[I]中、Arがm−ニトロフェニル基、R1 がメチル基、R2 がシンナミル基、R3 及びR4 がメチル基を表わす降圧剤である。
【0030】比較例1湿式法顆粒圧縮錠1錠 (135mg)あたり、FRC8653原体(体積平均径50μm以下)5.0mgに、賦形剤としての乳糖適量、コーンスターチ12.5mg及び結晶セルロース18.7mgと、崩壊剤としての低置換度ヒドロキシプロピルセルロース12.5mg、並びに結合剤としてのヒドロキシプロピルセルロース(日本曹達(株))(以下HPCと称する)3.7mgの水溶液を混合・撹拌し、湿式顆粒を作製し、さらに滑沢剤としてのタルク1.3mg及びステアリン酸マグネシウム1.2mgを添加し打錠した後、得られた錠剤を、コーティング剤としてヒドロキシプロピルメチルセルロース2910(6cs)(信越化学工業(株))5.0mg、エチルセルロース(ハーキュレス(株))1.25mg、可塑剤としてマクロゴール6000(日本油脂(株))0.625mg及びマクロゴール600(日本油脂(株))0.625mg、並びに不透明化剤として酸化チタン2.5mgから成る混合物をメタノール・ジクロルメタン混合溶媒に溶解した溶液でフィルムコーティングし、湿式法顆粒圧縮錠を作製した。
【0031】得られた湿式法顆粒圧縮錠の崩壊性及び溶出性を測定した結果、日局崩壊試験法による崩壊時間は3.0〜5.5分であり、日局溶出試験法・第2法による溶出率は、試験液としてFRC8653原体の37℃における水に対する飽和溶解度が5倍以上となるように0.5%(w/v)ポリソルベート80・日局崩壊試験第1液を用い、また試験液量を500mlとし、パドルの回転数を100回転としたとき、10分で約20%、30分で約35%、60分で約50%,90分で約60%及び150分で約70%であった。
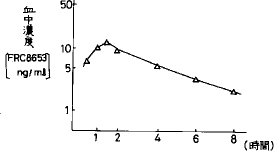
【0032】また、湿式法顆粒圧縮錠をイヌ一匹当たりFRC8653 10mg(2錠)を経口投与し、高速液体クロマトグラフィー(クロマト条件:ODSカラム、アセトニトリル・水系)を用いてFRC8653の血中濃度を測定した結果、経口投与1.2時間後に最大血中濃度5.3ng/mlとなった(図1)。さらにまた、血中濃度−時間曲線から求めた薬物動力学的パラメーターは、最高血中濃度到達時間(tmax )1.2時間、半減期(t1/2 )1.2時間及び血中濃度曲線下面積(AUC)は14.1ng・hr/mlを示した。FRC8653をエタノールとニッコール(HCO−60)(日光ケミカルズ(株))の混合溶媒(1/1)に溶解させ、その溶液を生理食塩水で50倍希釈して静注した時のAUCをもとに生物学的利用率を算出すると僅かに5.3%であった。
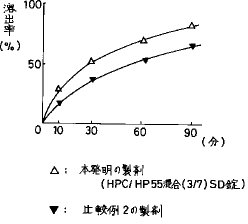
【0033】比較例21錠(135mg)あたり、FRC8653(体積平均径50μm以下)5.0mgと結合剤(担体)としてのHPC6.0mg及びヒドロキシプロピルメチルセルロースフタレート(以下HP55と称する)(信越化学工業(株)) 14.0 mgを同時にメタノール−ジクロルメタン混合溶媒110.0mgに溶解した溶液を賦形剤としての乳糖適量、結晶セルロース15.7mg、マクロゴール400(日本油脂(株))3.5mg及びメタケイ酸アルミン酸マグネシウム55.0mg、崩壊剤としてのクロスカルメロースナトリウム(旭化成工業(株))12.0mgに加え混合・撹拌し、溶媒を留去してHPC/HP55(3/7)固体分散(SD)顆粒124.0mgを作製し、更に滑沢剤としてタルク0.5mg及びステアリン酸マグネシウム0.5mgを添加し打錠した後、前記比較例と同様の条件でフィルムコーティングを行い、HPC/HP55(3/7)SD錠を作製した。その結果、図2に示す通り、単にFRC8653、HPC及びHP55の必須3成分を混合して作製した製剤は、本発明製剤(実施例5で作製したHPC/HP55混合(3/7)SD錠)と比較して有意に溶出率が悪い。よって、その吸収性は良くないものと予想される。
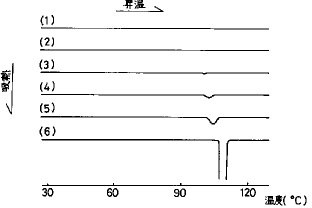
【0034】実施例1ヒドロキシプロピルセルロース(HPC)を担体とした固体分散錠(HPC・SD錠)作製におけるFRC8653に対するHPC量の決定FRC8653 1重量部に対しHPC1、2、3、4、5重量部の割合で両物質をメタノール−ジクロルメタン混合溶媒に混合・溶解し、脱溶媒し、乾燥させたのち熱分析(DSC)を行った。結果を図3に示す。FRC8653を非晶質化させるHPC量はFRC8653 1重量部あたりHPC4重量部以上であった。
【0035】実施例2HPC・SD顆粒の作製1錠(135mg)あたり、FRC8653(体積平均径50μm以下)5.0mgと結合剤(担体)としてのHPC 20.0 mgをメタノール−ジクロルメタン混合溶媒75mgに溶解した溶液を賦形剤としての乳糖適量、結晶セルロース15.7mg、マクロゴール400(日本油脂(株))3.5mg及びメタケイ酸アルミン酸マグネシウム55.0mg、崩壊剤としてのクロスカルメロースナトリウム(旭化成工業(株))12.0mgに加え、混合・撹拌し、溶媒を留去してHPC・SD顆粒 124.0 mg を作製し、更に滑沢剤としてタルク0.5mg及びステアリン酸マグネシウム0.5mgを添加し打錠した後、前記比較例と同様の条件でフィルムコーティングを行い、HPC・SD錠を作製した。
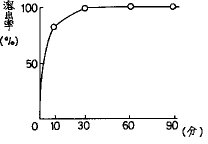
【0036】HPC・SD錠について、比較例1と同様にして崩壊時間、溶出率、血中濃度及び薬物動力学的パラメーターを求めた。その結果、崩壊時間は4.0〜5.5分となり良好な崩壊性を示し、さらに100%溶出するに要する時間は30分と短く、極めて速い溶出速度を与えた(図4)。またイヌに経口投与したときのFRC8653の血中濃度の経時変化は図5のとおりとなり、図から求めた薬物動力学的パラメーターはtmax 30分以内、t1/21.9時間及びAUC64.4ng・hr/mlであった。このように迅速な溶出は、図1との比較から明らかなように、薬物本来の吸収性から逸脱するものである。
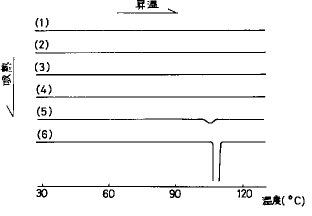
【0037】実施例3ヒドロキシプロピルセルロース(HPC)とヒドロキシプロピルメチルセルロースフタレート(HP55)を結合剤とした混合固体分散錠(HPC/HP55混合SD錠)作製におけるFRC8653に対するHP55量の決定FRC8653 1重量部に対しHP55(信越化学工業(株))1、2、3、4、5重量部の割合で両物質をメタノール−ジクロルメタンの混合溶媒に混合・溶解し、脱溶媒し、乾燥したのち熱分析を行った。結果を図6R>6に示す。FRC8653を非晶質化させるHP55量はFRC8653 1重量部あたりHP552重量部以上であった。
【0038】実施例4HP55・SD顆粒の作製FRC8653(体積平均径50μm以下)5.0mgと結合剤としてのHP55 20.0mgをメタノール−ジクロルメタン混合溶媒125mgに溶解した溶液を賦形剤としての乳糖適量、結晶セルロース15.7mg、マクロゴール4003.5mg及びメタケイ酸アルミン酸マグネシウム55.0mg、崩壊剤としてのクロスカルメロースナトリウム12.0mgに加え、混合・撹拌し、溶媒を留去してHP55・SD顆粒124.0mgを作製した。
【0039】実施例5HPC/HP55混合SD錠の作製実施例2及び4の方法に従って作製したHPC・SD顆粒及びHP55・SD顆粒を7/3、3/7、0/10(重量/重量)となるように混合し、更にHPC・SD錠(実施例2)と同様に滑沢剤を添加して打錠し、フィルムコーティングを行い、HPC/HP55混合SD錠を作製した。
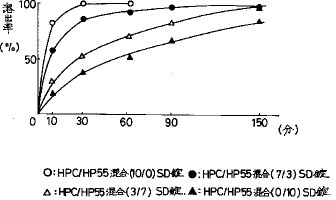
【0040】得られたHPC/HP55混合(7/3)SD錠、HPC/HP55混合(3/7)SD錠及びHPC/HP55混合(0/10)SD錠について、崩壊性及び溶出性を測定した。これらのSD錠の崩壊時間はすべて4.0〜6.0分であり良好な崩壊性を示した。溶出率については図7に示すように、経口投与後の胃内滞留時間を2.5時間と仮定し溶出率の試験時間を2.5時間までとしたとき、HPC/HP55混合(3/7)SD錠では2.5時間でFRC8653が100%溶出し、HPC/HP55混合(0/10)SD錠では2.5時間で86%溶出し、またHPC/HP55混合(7/3)SD錠では1.5時間で100%溶出した。
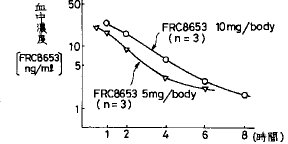
【0041】さらにまた、上記の溶出率の結果から血圧降下剤FRC8653含有製剤として適切であると予想されるHPC/HP55混合(3/7)SD錠をイヌ1匹あたり10mgを経口投与し、比較例1と同様の条件の高速液体クロマトグラフィーを用いて血中濃度を測定した(図8)。図の血中濃度−時間曲線から薬物動力学的パラメーターを決定した。その結果、tmax 1.3時間、t1/2 1.8時間及びAUC64.5ng・hr/mlとなり、tmax は湿式法顆粒圧縮錠(比較例1)のtmax と、またAUCはHPC・SD錠(実施例2)のAUCとそれぞれほぼ等しい値であった。
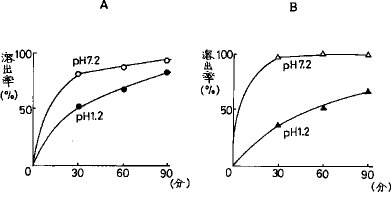
【0042】実施例6HPC/HP55混合 (3/7)SD錠の酸性及び中性領域での溶出プロファイルHPC/HP55混合(3/7)SD錠とHPC/HP55混合(0/10)SD錠(各FRC8653 5mg含有)についてpH1.2(0.5%(w/v)ポリソルベート80・日局崩壊試験第1液)及びpH7.2(0.5%(w/v)ポリソルベート80・リン酸塩緩衝液)の試験液500mlでのFRC8653の溶出挙動を測定した(図9)。
【0043】図から、HPC/HP55混合(3/7)SD錠の場合の90分後のFRC8653溶出率はpH1.2で85%、pH7.2で95%でありpH依存性が少ないのに対し、HPC/HP55混合(0/10)SD錠の場合の90分後のFRC8653溶出率はpH1.2で65%、pH7.2で100%であった。
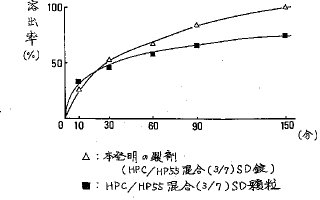
【0044】比較例3実施例2及び4の方法に従って作製したHPC・SD顆粒及びHP55・SD顆粒を3/7(重量/重量)に混合した混合顆粒124.0mgと実施例5に示したHPC/HP55混合(3/7)SD錠について比較例1と同様にして溶出率を求めた。その結果、図10に示す通り混合顆粒は150分で約75%であり、HPC/HP55混合(3/7)SD錠の100%に比べて溶出率が悪かった。よって、その吸収値は良くないものと予想される。
【0045】
【発明の効果】本発明の1,4−ジヒドロピリジン誘導体含有製剤は、胃内での薬物の溶出に際しpH依存性が少なく、薬物本来の吸収性を維持しつつHPC・SD錠と同程度の高い体内吸収率をもつという利点を有する。従って、本発明製剤によって、水に対して極めて難溶性の1,4−ジヒドロピリジン誘導体の生物学的利用率が改善され、また副作用の問題も回避されよう。
【図面の簡単な説明】
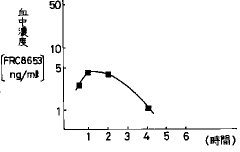
【図1】イヌに湿式法顆粒圧縮錠(比較例1)を経口投与したときのFRC8653血中濃度の経時変化を示す(FRC8653 10mg/body、n=6)。
【図2】FRC8653、HPC及びHP55を一時に混合して作製したHPC/HP55(3/7)SD錠(比較例2)の溶出曲線を示す。
【図3】FRC8653とHPCの固体分散系及びFRC8653のDSC曲線を示す。(1):FRC8653/HPC(1/5),(2):FRC8653/HPC(1/4),(3):FRC8653/HPC(1/3),(4):FRC8653/HPC(1/2),(5):FRC8653/HPC(1/1),(6):FRC8653。
【図4】HPC・SD錠のFRC8653溶出曲線を示す。
【図5】イヌにHPC・SD錠を経口投与したときのFRC8653血中濃度の経時変化を示す。
【図6】FRC8653とHP55の固体分散系及びFRC8653のDSC曲線を示す。(1):FRC8653/HP55(1/5),(2):FRC8653/HP55(1/4),(3):FRC8653/HP55(1/3),(4):FRC8653/HP55(1/2),(5):FRC8653/HP55(1/1),(6):FRC8653。
【図7】HPC・SD錠及びHPC/HP55混合SD錠のFRC8653溶出曲線を示す。
【図8】イヌにHPC/HP55混合(3/7)SD錠を経口投与したときのFRC8653血中濃度の経時変化を示す(FRC8653 10mg/body、n=6)。
【図9】HPC/HP55混合(3/7)SD錠およびHPC/HP55混合(0/10)SD錠の酸性、中性pHにおけるFRC8653溶出曲線を示す。A:HPC/HP55混合(3/7)SD錠、B:HPC/HP55混合(0/10)SD錠。
【図10】HPC/HP55混合(3/7)SD錠と該SD顆粒との溶出率の比較を示す。
【0001】
【産業上の利用分野】本発明は、水難溶性薬物1,4−ジヒドロピリジン誘導体を含有する新規な製剤に関する。
【0002】
【従来の技術】医薬として用いられる薬物の中には、優れた薬理作用を有しているにも拘らず、水に対して難溶性であるため消化管からの吸収が悪く、医薬品として利用されていないか又は多量に投与しなければならないものが少なくない。血管拡張作用をもつ1,4−ジヒドロピリジン誘導体もそのような薬物の1つであり、この化合物は、水に対して極めて難溶性であるため経口投与によるときは体内への吸収率があまりにも低く、従って生物学的利用率が低いという実用上の問題を有していた。
【0003】例えば、ニフェジピン(米国特許第3,644,627 号明細書)は狭心症発作治療剤として賞用されているが、吸収が遅いという欠点を有するため速吸収型(即効性)のニフェジピン経口投与製剤が種々提案されてきた。例えば、ニフェジピンとポリビニルピロリドンの混合物を有機溶媒に溶解後、溶媒を留去して得られる固形物(特開昭54−2316号公報)、ニフェジピンを液状ポリエチレングリコールに溶解して得られる溶液を充填した軟カプセル(特開昭48−28621 号公報)などが挙げられる。
【0004】一方、例えば降圧剤として使用する場合には、即効性よりも持続性が要求され、この場合には、遅吸収型の製剤が望まれる。このような遅吸収型のニフェジピン製剤をめざしたものとしては、ニフェジピン含有顆粒と、該顆粒の表面に水難溶性物質と腸溶性高分子とからなる被膜を形成させて得られる顆粒との混和顆粒剤(特開昭58−46019 号公報)が知られている。
【0005】
【発明が解決しようとする課題】従来の遅吸収型ニフェジピン製剤においては、その血中レベルが長時間に亘って一定の有効値に維持されることが要求されてきた。
【0006】しかし、薬物が本来もつ吸収性、即ち非徐放性の通常製剤に特徴的な吸収性を保持したまま、その吸収率を向上させる製剤が開発されたならば、薬物の血中レベルの維持に関連して惹起される副作用等の欠点を回避し得る点でより理想的な製剤となり得る。
【0007】本発明の目的は、このような望ましい吸収性を有し且つ吸収率を改善した水難溶性1,4−ジヒドロピリジン誘導体含有製剤を提供することである。
【0008】
【課題を解決するための手段】本発明は、一般式[I]:
【0009】
【化2】
【0010】[式中、Arは置換アリール基若しくはベンゾフラザニル基であり、R1 は置換若しくは無置換アルキル基であり、R2 は置換若しくは無置換アルキル又はアルケニル基であるか又は置換ピペリジル基であり、R3 及びR4 は置換若しくは無置換アルキル基であり、但し、5位の置換基R1 OCO−はジアルキルホスホノエート又はアルキレンジオキシホスホリル基で置き換えられてもよい]で表わされる水難溶性の薬物1,4−ジヒドロピリジン誘導体又はその酸付加塩とヒドロキシプロピルセルロースから成る固体分散物と、該1,4−ジヒドロピリジン誘導体又はその酸付加塩とヒドロキシプロピルメチルセルロースフタレートから成る固体分散物とを含み、且つ固体分散錠である製剤を提供する。
【0011】本発明製剤においては、有効成分としての前記1,4−ジヒドロピリジン誘導体の他に、結合剤(担体)としての水溶性高分子ヒドロキシプロピルセルロース及び腸溶性高分子ヒドロキシプロピルメチルセルロースフタレートが必須成分である。
【0012】上記一般式[I]中、置換若しくは無置換アルキル基としては、ジアルキルアミノ、ピペラジニル、アルコキシ、−ONO2 基などの置換基で置換された又は無置換の直鎖若しくは分枝アルキル基、好ましくは低級アルキル基、より好ましくはC1 〜C4 アルキル基が挙げられ、置換若しくは無置換アルケニル基としては、フェニル基などの置換基で置換された又は無置換のアルケニル基、好ましくは低級アルケニル基、より好ましくはC1 〜C4 アルケニル基が挙げられ、また、置換アリール基としては、ハロゲン、ニトロ、フラザニル基などの置換基を有するフェニル基などが挙げられる。
【0013】1,4−ジヒドロピリジン誘導体の具体例としては、狭心症治療剤、血管拡張剤として知られるニフェジピン及びその誘導体、例えばニソルジピン、ニカルジピン、ニモジピン、ニトレンジピン、ニルバジピン、マニジピン、、FRC8653(特開昭60−233058号)などが挙げられる。
【0014】本発明製剤は、一般式[I]の1,4−ジヒドロピリジン誘導体又はその酸付加塩1重量部に対し、ヒドロキシプロピルセルロース4重量部以上、好ましくは4〜5重量部を含む固体分散物(a)、及び該1,4−ジヒドロピリジン誘導体又はその酸付加塩1重量部に対し、ヒドロキシプロピルセルロースフタレート2重量部以上、好ましくは2〜5重量部を含む固体分散物(b)を混合し圧縮して固体分散錠に製剤化することにより得られる。固体分散物(a)と(b)の含有比は、該薬物が本来もつ体内吸収性を変えることなくその吸収率を向上させるように選択され、好ましくは(a)/(b)=2.5/7.5〜3.5/6.5(重量/重量)、より好ましくは(a)/(b)=3/7である。
【0015】本明細書中、「固体分散物」なる用語は不活性担体の中に薬物が単分子状に分散した固体を意味し、薬物は担体中に非晶質の形態で存在する。非晶質の判定は熱分析によって行い得る。
【0016】また、本発明製剤においては、好適には固体分散物(a)及び(b)の中に1種以上の賦形剤、崩壊剤又は滑沢剤、あるいはこれらの組合せ物を含ませることもできる。
【0017】賦形剤としては、例えば乳糖、マンニトール、ブドウ糖、コーンスターチ、結晶セルロース、天然又は合成ケイ酸アルミニウム等が挙げられ、メタケイ酸アルミン酸マグネシウムが好適に使用される。メタケイ酸アルミン酸マグネシウムを含有させる場合には通常固体分散物(a)又は(b)中に配合され、その配合量は、固体分散物(a)中では該薬物1重量部に対し11重量部以上、好ましくは11〜12重量部であり、また固体分散物(b)中では該薬物1重量部に対し5.5重量部以上、好ましくは5.5〜12重量部である。
【0018】崩壊剤としては、例えばアラビアゴム、アルギン酸ナトリウム、カルボキシメチルセルロースナトリウム、繊維系グルコン酸カルシウム等が挙げられ、また滑沢剤としては、例えばタルク、ステアリン酸マグネシウム等が挙げられる。
【0019】さらに必要に応じ、本発明錠剤をコーティング剤でフィルムコーティングしてもよい。コーティング剤としては、例えばヒドロキシプロピルメチルセルロース、エチルセルロース、などが挙げられ、またコーティング剤中には例えば不透明化剤(例えば、酸化チタン)や可塑剤(例えばポリエチレングリコール)を含有させることも可能である。
【0020】本発明製剤の利点は、FRC8653によって例示されるように、1,4−ジヒドロピリジン誘導体の経口投与後の最高血中濃度到達時間(tmax )が1.3時間(図8)であり、血中濃度−時間曲線下面積(AUC)が64.5ng・hr/ml(実施例5)であり、崩壊時間が4〜6分(実施例5)であり、及び100%溶出時間が150分(図7)であることから判るように、抗狭心症薬として市販されているニフェジピンの固体分散細粒(鐘紡(株))とは異なり胃内で必要以上に速く溶出しないで且つ100%溶出し、薬物が本来的にもつ吸収性(図1)を変えることなく(図8)、ヒドロキシプロピルセルロース・固体分散錠(実施例2)と同程度の高い体内吸収率を兼ね備えていることにある。本明細書中、「吸収性」なる用語は、薬物が本来もつ吸収性、即ち非徐放性の通常製剤に特徴的な吸収性を意味し、薬物血中濃度の経時変化によって表わされる。ここで、通常製剤とは非徐放性製剤を指す。
【0021】本発明製剤はまた、胃内での薬物の溶出に際してpH依存性が少ないという特性をも有している(図9)。
【0022】上述のような本発明製剤の性質は、水難溶性薬物、ヒドロキシプロピルセルロース及びヒドロキシプロピルメチルセルロースフタレートの必須三成分を一時に混合して得られる固体分散物(比較例2)又は固体分散物(a)若しくは(b)単独(実施例5)の性質と異なるものであり、本発明製剤に特徴的な性質である。特に、薬物本来の吸収性を変化させないことは、少なくとも薬物の血中濃度の維持に関連して惹起される副作用を回避し得ることを意味している。
【0023】本発明の1,4−ジヒドロピリジン誘導体含有製剤は以下の方法によって製造し得る。すなわち、この方法は、(i) 一般式[I]で表わされる1,4−ジヒドロピリジン誘導体又はその酸付加塩とヒドロキシプロピルセルロースとを含む混合物から固体分散物(a)を製造する工程、(ii) 該1,4−ジヒドロピリジン誘導体又はその酸付加塩とヒドロキシプロピルメチルセルロースフタレートとを含む混合物から固体分散物(b)を製造する工程、(iii) 前記固体分散物(a)と(b)とを混合する工程、及び、(iv) 得られた混合物を錠剤に成形する工程、から成る。
【0024】固体分散物(a)又は(b)は、前記1,4−ジヒドロピリジン誘導体又はその酸付加塩、ヒドロキシプロピルセルロース又はヒドロキシプロピルメチルセルロースフタレートを有機溶媒に溶解し賦形剤、崩壊剤等の助剤に混合した後、該溶媒を蒸発等により除去することにより得ることができる。
【0025】また固体分散物中に、賦形剤を含有させる場合には、メタケイ酸アルミン酸マグネシウムが望ましい。この場合、固体分散物(a)及び(b)中に、メタケイ酸アルミン酸マグネシウムがそれぞれ前記1,4−ジヒドロピリジン誘導体又はその酸付加塩対メタケイ酸アルミン酸マグネシウム1:11以上、好ましくは1:11〜12、及び1:5.5以上、好ましくは1:5.5〜12の重量比で含有し得る。
【0026】有機溶媒は、例えばメタノール、エタノール等の低級アルコール類、ジクロルメタン、クロロホルム等のハロゲン化炭化水素類、及びこれらの混合物が挙げられる。
【0027】また、錠剤への成形は、公知の方法で実施され得る。
【0028】尚、本発明製剤である固体分散錠は、固体分散物(a)/(b)=3/7の混合顆粒(比較例3)とも性質が異なるものであり、本発明製剤は錠剤でなければならない。
【0029】
【実施例】以下の比較例及び実施例では、一般式[I]の1,4−ジヒドロピリジン誘導体の具体例としてFRC8653(特開昭60−233058号)を取り挙げて本発明をさらに具体的に説明するが、その他の誘導体の場合にも同様に適用可能である。なお、FRC8653は、式[I]中、Arがm−ニトロフェニル基、R1 がメチル基、R2 がシンナミル基、R3 及びR4 がメチル基を表わす降圧剤である。
【0030】比較例1湿式法顆粒圧縮錠1錠 (135mg)あたり、FRC8653原体(体積平均径50μm以下)5.0mgに、賦形剤としての乳糖適量、コーンスターチ12.5mg及び結晶セルロース18.7mgと、崩壊剤としての低置換度ヒドロキシプロピルセルロース12.5mg、並びに結合剤としてのヒドロキシプロピルセルロース(日本曹達(株))(以下HPCと称する)3.7mgの水溶液を混合・撹拌し、湿式顆粒を作製し、さらに滑沢剤としてのタルク1.3mg及びステアリン酸マグネシウム1.2mgを添加し打錠した後、得られた錠剤を、コーティング剤としてヒドロキシプロピルメチルセルロース2910(6cs)(信越化学工業(株))5.0mg、エチルセルロース(ハーキュレス(株))1.25mg、可塑剤としてマクロゴール6000(日本油脂(株))0.625mg及びマクロゴール600(日本油脂(株))0.625mg、並びに不透明化剤として酸化チタン2.5mgから成る混合物をメタノール・ジクロルメタン混合溶媒に溶解した溶液でフィルムコーティングし、湿式法顆粒圧縮錠を作製した。
【0031】得られた湿式法顆粒圧縮錠の崩壊性及び溶出性を測定した結果、日局崩壊試験法による崩壊時間は3.0〜5.5分であり、日局溶出試験法・第2法による溶出率は、試験液としてFRC8653原体の37℃における水に対する飽和溶解度が5倍以上となるように0.5%(w/v)ポリソルベート80・日局崩壊試験第1液を用い、また試験液量を500mlとし、パドルの回転数を100回転としたとき、10分で約20%、30分で約35%、60分で約50%,90分で約60%及び150分で約70%であった。
【0032】また、湿式法顆粒圧縮錠をイヌ一匹当たりFRC8653 10mg(2錠)を経口投与し、高速液体クロマトグラフィー(クロマト条件:ODSカラム、アセトニトリル・水系)を用いてFRC8653の血中濃度を測定した結果、経口投与1.2時間後に最大血中濃度5.3ng/mlとなった(図1)。さらにまた、血中濃度−時間曲線から求めた薬物動力学的パラメーターは、最高血中濃度到達時間(tmax )1.2時間、半減期(t1/2 )1.2時間及び血中濃度曲線下面積(AUC)は14.1ng・hr/mlを示した。FRC8653をエタノールとニッコール(HCO−60)(日光ケミカルズ(株))の混合溶媒(1/1)に溶解させ、その溶液を生理食塩水で50倍希釈して静注した時のAUCをもとに生物学的利用率を算出すると僅かに5.3%であった。
【0033】比較例21錠(135mg)あたり、FRC8653(体積平均径50μm以下)5.0mgと結合剤(担体)としてのHPC6.0mg及びヒドロキシプロピルメチルセルロースフタレート(以下HP55と称する)(信越化学工業(株)) 14.0 mgを同時にメタノール−ジクロルメタン混合溶媒110.0mgに溶解した溶液を賦形剤としての乳糖適量、結晶セルロース15.7mg、マクロゴール400(日本油脂(株))3.5mg及びメタケイ酸アルミン酸マグネシウム55.0mg、崩壊剤としてのクロスカルメロースナトリウム(旭化成工業(株))12.0mgに加え混合・撹拌し、溶媒を留去してHPC/HP55(3/7)固体分散(SD)顆粒124.0mgを作製し、更に滑沢剤としてタルク0.5mg及びステアリン酸マグネシウム0.5mgを添加し打錠した後、前記比較例と同様の条件でフィルムコーティングを行い、HPC/HP55(3/7)SD錠を作製した。その結果、図2に示す通り、単にFRC8653、HPC及びHP55の必須3成分を混合して作製した製剤は、本発明製剤(実施例5で作製したHPC/HP55混合(3/7)SD錠)と比較して有意に溶出率が悪い。よって、その吸収性は良くないものと予想される。
【0034】実施例1ヒドロキシプロピルセルロース(HPC)を担体とした固体分散錠(HPC・SD錠)作製におけるFRC8653に対するHPC量の決定FRC8653 1重量部に対しHPC1、2、3、4、5重量部の割合で両物質をメタノール−ジクロルメタン混合溶媒に混合・溶解し、脱溶媒し、乾燥させたのち熱分析(DSC)を行った。結果を図3に示す。FRC8653を非晶質化させるHPC量はFRC8653 1重量部あたりHPC4重量部以上であった。
【0035】実施例2HPC・SD顆粒の作製1錠(135mg)あたり、FRC8653(体積平均径50μm以下)5.0mgと結合剤(担体)としてのHPC 20.0 mgをメタノール−ジクロルメタン混合溶媒75mgに溶解した溶液を賦形剤としての乳糖適量、結晶セルロース15.7mg、マクロゴール400(日本油脂(株))3.5mg及びメタケイ酸アルミン酸マグネシウム55.0mg、崩壊剤としてのクロスカルメロースナトリウム(旭化成工業(株))12.0mgに加え、混合・撹拌し、溶媒を留去してHPC・SD顆粒 124.0 mg を作製し、更に滑沢剤としてタルク0.5mg及びステアリン酸マグネシウム0.5mgを添加し打錠した後、前記比較例と同様の条件でフィルムコーティングを行い、HPC・SD錠を作製した。
【0036】HPC・SD錠について、比較例1と同様にして崩壊時間、溶出率、血中濃度及び薬物動力学的パラメーターを求めた。その結果、崩壊時間は4.0〜5.5分となり良好な崩壊性を示し、さらに100%溶出するに要する時間は30分と短く、極めて速い溶出速度を与えた(図4)。またイヌに経口投与したときのFRC8653の血中濃度の経時変化は図5のとおりとなり、図から求めた薬物動力学的パラメーターはtmax 30分以内、t1/21.9時間及びAUC64.4ng・hr/mlであった。このように迅速な溶出は、図1との比較から明らかなように、薬物本来の吸収性から逸脱するものである。
【0037】実施例3ヒドロキシプロピルセルロース(HPC)とヒドロキシプロピルメチルセルロースフタレート(HP55)を結合剤とした混合固体分散錠(HPC/HP55混合SD錠)作製におけるFRC8653に対するHP55量の決定FRC8653 1重量部に対しHP55(信越化学工業(株))1、2、3、4、5重量部の割合で両物質をメタノール−ジクロルメタンの混合溶媒に混合・溶解し、脱溶媒し、乾燥したのち熱分析を行った。結果を図6R>6に示す。FRC8653を非晶質化させるHP55量はFRC8653 1重量部あたりHP552重量部以上であった。
【0038】実施例4HP55・SD顆粒の作製FRC8653(体積平均径50μm以下)5.0mgと結合剤としてのHP55 20.0mgをメタノール−ジクロルメタン混合溶媒125mgに溶解した溶液を賦形剤としての乳糖適量、結晶セルロース15.7mg、マクロゴール4003.5mg及びメタケイ酸アルミン酸マグネシウム55.0mg、崩壊剤としてのクロスカルメロースナトリウム12.0mgに加え、混合・撹拌し、溶媒を留去してHP55・SD顆粒124.0mgを作製した。
【0039】実施例5HPC/HP55混合SD錠の作製実施例2及び4の方法に従って作製したHPC・SD顆粒及びHP55・SD顆粒を7/3、3/7、0/10(重量/重量)となるように混合し、更にHPC・SD錠(実施例2)と同様に滑沢剤を添加して打錠し、フィルムコーティングを行い、HPC/HP55混合SD錠を作製した。
【0040】得られたHPC/HP55混合(7/3)SD錠、HPC/HP55混合(3/7)SD錠及びHPC/HP55混合(0/10)SD錠について、崩壊性及び溶出性を測定した。これらのSD錠の崩壊時間はすべて4.0〜6.0分であり良好な崩壊性を示した。溶出率については図7に示すように、経口投与後の胃内滞留時間を2.5時間と仮定し溶出率の試験時間を2.5時間までとしたとき、HPC/HP55混合(3/7)SD錠では2.5時間でFRC8653が100%溶出し、HPC/HP55混合(0/10)SD錠では2.5時間で86%溶出し、またHPC/HP55混合(7/3)SD錠では1.5時間で100%溶出した。
【0041】さらにまた、上記の溶出率の結果から血圧降下剤FRC8653含有製剤として適切であると予想されるHPC/HP55混合(3/7)SD錠をイヌ1匹あたり10mgを経口投与し、比較例1と同様の条件の高速液体クロマトグラフィーを用いて血中濃度を測定した(図8)。図の血中濃度−時間曲線から薬物動力学的パラメーターを決定した。その結果、tmax 1.3時間、t1/2 1.8時間及びAUC64.5ng・hr/mlとなり、tmax は湿式法顆粒圧縮錠(比較例1)のtmax と、またAUCはHPC・SD錠(実施例2)のAUCとそれぞれほぼ等しい値であった。
【0042】実施例6HPC/HP55混合 (3/7)SD錠の酸性及び中性領域での溶出プロファイルHPC/HP55混合(3/7)SD錠とHPC/HP55混合(0/10)SD錠(各FRC8653 5mg含有)についてpH1.2(0.5%(w/v)ポリソルベート80・日局崩壊試験第1液)及びpH7.2(0.5%(w/v)ポリソルベート80・リン酸塩緩衝液)の試験液500mlでのFRC8653の溶出挙動を測定した(図9)。
【0043】図から、HPC/HP55混合(3/7)SD錠の場合の90分後のFRC8653溶出率はpH1.2で85%、pH7.2で95%でありpH依存性が少ないのに対し、HPC/HP55混合(0/10)SD錠の場合の90分後のFRC8653溶出率はpH1.2で65%、pH7.2で100%であった。
【0044】比較例3実施例2及び4の方法に従って作製したHPC・SD顆粒及びHP55・SD顆粒を3/7(重量/重量)に混合した混合顆粒124.0mgと実施例5に示したHPC/HP55混合(3/7)SD錠について比較例1と同様にして溶出率を求めた。その結果、図10に示す通り混合顆粒は150分で約75%であり、HPC/HP55混合(3/7)SD錠の100%に比べて溶出率が悪かった。よって、その吸収値は良くないものと予想される。
【0045】
【発明の効果】本発明の1,4−ジヒドロピリジン誘導体含有製剤は、胃内での薬物の溶出に際しpH依存性が少なく、薬物本来の吸収性を維持しつつHPC・SD錠と同程度の高い体内吸収率をもつという利点を有する。従って、本発明製剤によって、水に対して極めて難溶性の1,4−ジヒドロピリジン誘導体の生物学的利用率が改善され、また副作用の問題も回避されよう。
【図面の簡単な説明】
【図1】イヌに湿式法顆粒圧縮錠(比較例1)を経口投与したときのFRC8653血中濃度の経時変化を示す(FRC8653 10mg/body、n=6)。
【図2】FRC8653、HPC及びHP55を一時に混合して作製したHPC/HP55(3/7)SD錠(比較例2)の溶出曲線を示す。
【図3】FRC8653とHPCの固体分散系及びFRC8653のDSC曲線を示す。(1):FRC8653/HPC(1/5),(2):FRC8653/HPC(1/4),(3):FRC8653/HPC(1/3),(4):FRC8653/HPC(1/2),(5):FRC8653/HPC(1/1),(6):FRC8653。
【図4】HPC・SD錠のFRC8653溶出曲線を示す。
【図5】イヌにHPC・SD錠を経口投与したときのFRC8653血中濃度の経時変化を示す。
【図6】FRC8653とHP55の固体分散系及びFRC8653のDSC曲線を示す。(1):FRC8653/HP55(1/5),(2):FRC8653/HP55(1/4),(3):FRC8653/HP55(1/3),(4):FRC8653/HP55(1/2),(5):FRC8653/HP55(1/1),(6):FRC8653。
【図7】HPC・SD錠及びHPC/HP55混合SD錠のFRC8653溶出曲線を示す。
【図8】イヌにHPC/HP55混合(3/7)SD錠を経口投与したときのFRC8653血中濃度の経時変化を示す(FRC8653 10mg/body、n=6)。
【図9】HPC/HP55混合(3/7)SD錠およびHPC/HP55混合(0/10)SD錠の酸性、中性pHにおけるFRC8653溶出曲線を示す。A:HPC/HP55混合(3/7)SD錠、B:HPC/HP55混合(0/10)SD錠。
【図10】HPC/HP55混合(3/7)SD錠と該SD顆粒との溶出率の比較を示す。
【特許請求の範囲】
【請求項1】 一般式[I]:
【化1】
[式中、Arは置換アリール基若しくはベンゾフラザニル基であり、R1は置換若しくは無置換アルキル基であり、R2 は置換若しくは無置換アルキル又はアルケニル基であるか又は置換ピペリジル基であり、R3 及びR4 は置換若しくは無置換アルキル基であり、但し、5位の置換基R1 OCO−はジアルキルホスホノエート又はアルキレンジオキシホスホリル基で置き換えられてもよい]で表わされる水難溶性の薬物1,4−ジヒドロピリジン誘導体又はその酸付加塩とヒドロキシプロピルセルロースから成る固体分散物(a)と、該1,4−ジヒドロピリジン誘導体又はその酸付加塩とヒドロキシプロピルメチルセルロースフタレートから成る固体分散物(b)とを含み、且つ固体分散錠であることを特徴とする製剤。
【請求項2】 該1,4−ジヒドロピリジン誘導体又はその酸付加塩対ヒドロキシプロピルセルロースの重量比が1:4〜5であり、且つ該1,4−ジヒドロピリジン誘導体又はその酸付加塩対ヒドロキシプロピルメチルセルロースフタレートの重量比が1:2〜5である請求項1記載の製剤。
【請求項3】 該固体分散物(a)対該固体分散物(b)の混合比が2.5:7.5〜3.5:6.5(重量比)である請求項1又は2記載の製剤。
【請求項4】 該固体分散物(a)及び(b)がメタケイ酸アルミン酸マグネシウムを含有する請求項1記載の製剤。
【請求項5】 該固体分散物(a)及び(b)が、メタケイ酸アルミン酸マグネシウムを、それぞれ該1,4−ジヒドロピリジン誘導体又はその酸付加塩対メタケイ酸アルミン酸マグネシウム1:11〜12及び1:5.5〜12の重量比で含有する請求項4記載の製剤。
【請求項1】 一般式[I]:
【化1】
[式中、Arは置換アリール基若しくはベンゾフラザニル基であり、R1は置換若しくは無置換アルキル基であり、R2 は置換若しくは無置換アルキル又はアルケニル基であるか又は置換ピペリジル基であり、R3 及びR4 は置換若しくは無置換アルキル基であり、但し、5位の置換基R1 OCO−はジアルキルホスホノエート又はアルキレンジオキシホスホリル基で置き換えられてもよい]で表わされる水難溶性の薬物1,4−ジヒドロピリジン誘導体又はその酸付加塩とヒドロキシプロピルセルロースから成る固体分散物(a)と、該1,4−ジヒドロピリジン誘導体又はその酸付加塩とヒドロキシプロピルメチルセルロースフタレートから成る固体分散物(b)とを含み、且つ固体分散錠であることを特徴とする製剤。
【請求項2】 該1,4−ジヒドロピリジン誘導体又はその酸付加塩対ヒドロキシプロピルセルロースの重量比が1:4〜5であり、且つ該1,4−ジヒドロピリジン誘導体又はその酸付加塩対ヒドロキシプロピルメチルセルロースフタレートの重量比が1:2〜5である請求項1記載の製剤。
【請求項3】 該固体分散物(a)対該固体分散物(b)の混合比が2.5:7.5〜3.5:6.5(重量比)である請求項1又は2記載の製剤。
【請求項4】 該固体分散物(a)及び(b)がメタケイ酸アルミン酸マグネシウムを含有する請求項1記載の製剤。
【請求項5】 該固体分散物(a)及び(b)が、メタケイ酸アルミン酸マグネシウムを、それぞれ該1,4−ジヒドロピリジン誘導体又はその酸付加塩対メタケイ酸アルミン酸マグネシウム1:11〜12及び1:5.5〜12の重量比で含有する請求項4記載の製剤。
【図1】

【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【特許番号】特許第3110794号(P3110794)
【登録日】平成12年9月14日(2000.9.14)
【発行日】平成12年11月20日(2000.11.20)
【国際特許分類】
【出願番号】特願平3−134344
【出願日】平成3年6月5日(1991.6.5)
【公開番号】特開平4−360833
【公開日】平成4年12月14日(1992.12.14)
【審査請求日】平成9年10月2日(1997.10.2)
【出願人】(500307269)ユーシービージャパン株式会社 (4)
【上記1名の代理人】
【識別番号】100062007
【弁理士】
【氏名又は名称】川口 義雄
【出願人】(000000066)味の素株式会社 (887)
【上記1名の代理人】
【識別番号】100062007
【弁理士】
【氏名又は名称】川口 義雄 (外2名)
【登録日】平成12年9月14日(2000.9.14)
【発行日】平成12年11月20日(2000.11.20)
【国際特許分類】
【出願日】平成3年6月5日(1991.6.5)
【公開番号】特開平4−360833
【公開日】平成4年12月14日(1992.12.14)
【審査請求日】平成9年10月2日(1997.10.2)
【出願人】(500307269)ユーシービージャパン株式会社 (4)
【上記1名の代理人】
【識別番号】100062007
【弁理士】
【氏名又は名称】川口 義雄
【出願人】(000000066)味の素株式会社 (887)
【上記1名の代理人】
【識別番号】100062007
【弁理士】
【氏名又は名称】川口 義雄 (外2名)
[ Back to top ]