
11C標識イソキノリン誘導体、その製造方法、その前駆体、それを利用したPET用プローブ及び組織の画像化方法
【課題】Rhoキナーゼ阻害剤であるファスジルやその類縁体に存在するイソキノリン環に[11C]で標識化されたメチル基を導入した11C標識イソキノリン誘導体及びその製造方法並びにその11C標識イソキノリン誘導体を製造するのに好適に用いることができる前駆体を提供する。
【解決手段】11C標識イソキノリン誘導体として、(S)−ヘキサヒドロ−2−メチル−1−[(4−[11C]メチル−5−イソキノリニル)スルホニル]−1H−1,4−ジアゼピン等が例示される。該化合物はトリハロアセチル基などの保護基を有する前駆体を使用し、前駆体のイソキノリン環上に[11C]メチル基を導入してから塩基性条件下で保護基を脱離させることによって短時間に得ることができる。このため、PET法などにより組織を画像化する際にイメージング薬として好適に用いることができる。
【解決手段】11C標識イソキノリン誘導体として、(S)−ヘキサヒドロ−2−メチル−1−[(4−[11C]メチル−5−イソキノリニル)スルホニル]−1H−1,4−ジアゼピン等が例示される。該化合物はトリハロアセチル基などの保護基を有する前駆体を使用し、前駆体のイソキノリン環上に[11C]メチル基を導入してから塩基性条件下で保護基を脱離させることによって短時間に得ることができる。このため、PET法などにより組織を画像化する際にイメージング薬として好適に用いることができる。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、イソキノリンスルホンアミド化合物類のイソキノリン環上に[11C]メチル基を導入した11C標識イソキノリン誘導体、その製造方法、その前駆体、分子プローブ及び組織の画像化方法に関する。
【背景技術】
【0002】
陽電子放射断層画像撮影法(PET法)は、小動物からヒトまで生体内の分子を非侵襲的に画像化できる唯一の技術である。このため、近年、医療や創薬分野でのPET法活用への期待が急速に膨らんでいる。しかし、技術活用の鍵となるPETプローブの合成は、分子に組み込む放射核の寿命が短いために、困難を伴う。
【0003】
従来は、導入が容易であるという理由から、O(酸素)、N(窒素)、S(硫黄)などのヘテロ原子上への[11C]メチル化のみが行われてきた。しかし、生体内においてはこれらのヘテロ原子を手がかりとして酵素による代謝を受けやすく、代謝によって標識部位の化学結合が切断されてしまうため、得られたPET画像の信憑性に疑問が残るという問題が指摘されてきた。
【0004】
こうした問題を解決するため、本発明者らは、11CH3Iを用いた炭素母核上への[11C]メチル化について開発を行ってきた。すべての有機化合物は、その構造中に炭素原子を有していることから、低分子有機化合物のPET分子プローブ化を行う上で、短寿命放射性炭素11Cの利用は理想的な放射性核種であると考えられるからである。
【0005】
これまで、本発明者らは、有機スズ化合物を中間原料として、これにStille型カップリング反応を適用することにより、芳香環上への高速C-[11C]メチル化のみならず、オレフィンやアルキンやヘテロ芳香環上への高速C-[11C]メチル化も可能としてきた(特許文献1、2及び非特許文献1〜3)。そしてさらには、従来の有機スズ化合物への高速C-[11C]メチル化と相補的に、新たに有機ホウ素化合物を用いた高速C-[11C]メチル化反応も開発している(特許文献3、非特許文献4)。
【0006】
一方、医薬品や生理活性物質において、イソキノリン環を有する数多くの化合物が知られている。この中でもイソキノリン環の5位が環状アミノスルホニルで置換された化合物は、循環器官用剤(血管拡張剤、脳循環改善剤、狭心症治療薬、脳心血管系の血栓症の予防および治療薬、高血圧症の予防治療薬、緑内障の予防および治療薬)として有用であり(特許文献4〜7)、プロテインキナーゼ阻害作用を有することが見出されている(非特許文献5)。
【0007】
本発明者の日高らは、臨床応用された世界最初で唯一のRhoキナーゼ阻害剤であるファスジル[ヘキサヒドロ−1−(5−イソキノリンスルホニル)−1H−1,4−ジアゼピン]を発明し、くも膜下出血後の脳血管攣縮の治療薬として人類に提供したが、ファスジルもイソキノリン環の5位が環状アミノスルホニルで置換された化合物である(特許文献6)。さらに、イソキノリン環の5位が環状アミノスルホニルで置換されていて、且つ、4位にメチル基などの置換基を有する化合物も開示されている(特許文献7〜9)。
【0008】
その構造的特徴を有する化合物の中でも、本発明者である日高により見出されたH-1152P[(S)−(+)−ヘキサヒドロ−2−メチル−1−[(4−メチル−5−イソキノリニル)スルホニル]−1H−1,4−ジアゼピン]は、選択的で作用強度の強いRhoキナーゼ阻害剤として知られている(特許文献6、非特許文献6)。
【0009】
Rhoキナーゼは、低分子量GTP結合タンパク質であるRhoの標的タンパク質として同定されたセリン/スレオニン・プロテインキナーゼであり、細胞移動、細胞増殖、細胞収縮、細胞粘着、細胞の運動性など、さまざまな細胞の機能にかかわる重要な酵素である。
【0010】
ファスジルやH-1152PなどのRhoキナーゼ阻害剤は、疾患におけるRhoキナーゼの役割を検討する研究に広く用いられ、その結果、Rhoキナーゼ阻害剤は、種々の疾患、例えば、脳血管攣縮、冠動脈攣縮、高血圧、肺性高血圧、喘息、緑内障、腎障害、狭心症、心筋梗塞、動脈硬化症、再狭窄、脳卒中、心不全、心筋肥大、虚血再還流障害、血管内皮障害、炎症性腸疾患、前立腺肥大、レイノー病、神経因性疼痛、Alzheimer病、Huntington舞踏病、脊髄損傷、てんかん、多発性硬化症、骨粗しょう症、勃起不全、肝障害、がん、創傷治癒、インスリン抵抗性、糖尿病などの治療薬として有用であることが報告されている。がんにおいては、進行が早く浸潤性の強いがんや転移性のがんでRhoキナーゼの発現亢進が報告されており、Rhoキナーゼが転移・浸潤に関与することが知られている。これらのがんには、肉腫、Glioblastoma、神経膠腫、頭頸部がん、肝細胞がん、腎明細胞がん、肺がん、卵巣がん、乳がん、食道がん、大腸がん、膀胱がん、前立腺がん、悪性黒色腫が含まれる。
【0011】
イソキノリン環の5位が環状アミノスルホニルで置換されている化合物は、Rhoキナーゼ阻害作用のみならず、PKA、PKG、AURKA、CAMK2など各種プロテインキナーゼを阻害する化合物が発見されており(非特許文献7)、プロテインキナーゼ研究のツールとして利用できるのみならず、医薬としても有用である。
【0012】
したがって、プロテインキナーゼ阻害など特定の生理活性を有するイソキノリン誘導体のイソキノリン環上に高速C-[11C]メチル化反応によって[11C]メチル基を導入できれば、PET画像による解析が可能となり、例えば体内における脳内動態解析や薬剤(治療薬)としての潜在性を解析などが可能となる。さらには、それらプロテインキナーゼ阻害剤を用いた生物学的研究や動物・ヒトのPETによる臨床研究の基盤技術とすることができる。
【先行技術文献】
【特許文献】
【0013】
【特許文献1】WO/2007/046258
【特許文献2】WO/2010/074272
【特許文献3】WO2008/023780
【特許文献4】特開昭57-156463号公報
【特許文献5】特開昭58-121279号公報
【特許文献6】特開昭61-227581号公報
【特許文献7】US2008/116481
【特許文献8】WO1997/028130
【特許文献9】WO2006/115247
【非特許文献】
【0014】
【非特許文献1】Suzuki, M., et al., Chem. Eur. J., 3, 2039-2042 (1997).
【非特許文献2】Hosoya, T., et al., Org. Biomol. Chem., 4, 410-415 (2006).
【非特許文献3】Hosoya, T., et al., Org. Biomol. Chem., 2, 24-27 (2004).
【非特許文献4】Doi, H., et al., Chem. Eur. J., 15,4165-4171 (2009).
【非特許文献5】Hidaka, H et al., Biochemistry, 23, 5036-5041 (1984).
【非特許文献6】Sasaki, Y., et al., Pharmacol. Ther. 93, 225-232 (2002).
【非特許文献7】Tamura, M., et al., Biochim Biophys Acta, 1754,245-252 (2005).
【0015】
しかしながら、11Cの半減期はわずか20分であるため、11Cの導入は高速化が求められ、かつ合成の最終段階で行うことがこれまでの技術常識であった。また、測定時間(1時間程度)を確保するため、反応、精製、生体への投与までを半減期の2倍の40分以内で行うという制約もある。このため、11Cの導入に許される時間はわずか5分程度となる。さらには、サイクロトロンで製造できる11C核種は超微量(数十から数百nmolレベル;12Cの混入を考慮した値)であり、超希薄な11C核種の化合物と化学反応させるために、大過剰の被標識基質の存在下という特殊な条件下で行われる。このため、いかに短時間で効率良く生物活性有機化合物や創薬候補化合物をPET分子プローブ化できるかということが最重要課題となっている。
【0016】
本発明者の鈴木らが開発した前述の高速メチル化法(特許文献1、2及び非特許文献1〜3)は、これらの課題を解決するための強力なツールとなる方法ではあるが、完全なる汎用方法とはなり得ない。なぜならば、メチル化される基質に存在する官能基が反応を阻害したりする場合があるからである。
【0017】
このことは、前述したイソキノリン環の5位が環状アミノスルホニルで置換されている化合物の[11C]メチル化において顕著である。すなわち、上記の高速メチル化反応によってイソキノリン骨格を[11C]メチル化しようとした場合、前駆体に存在する化学的に活性な2級アミノ基がメチル化反応を阻害する。また、2級アミノ基が[11C]メチル化されてしまう。そこで通常であれば、前駆体の2級のアミノ基を保護基で保護しておき、[11C]メチル化後に脱保護することが考えられる。ところが、前述したように、11Cの導入から生体へ投与するまでの一連の操作は高速化が求められることから、合成の最終段階で[11C]メチル化反応を行うことが当業者の常識であり、[11C]メチル化後に脱保護反応を行うことは、時間的及び技術的に困難であった。このため、有用な化合物であるにもかかわらず、イソキノリン環の5位が環状アミノスルホニルで置換されている化合物については、そのイソキノリン環の炭素上への[11C]メチル基の導入は従来なされていなかった。
【発明の概要】
【発明が解決しようとする課題】
【0018】
本発明は、上記従来の実情に鑑みてなされたものであり、イソキノリン骨格に[11C]メチル基を導入した11C標識イソキノリン誘導体及びその製造方法並びにその11C標識イソキノリン誘導体を製造するのに好適に用いることができる前駆体を提供することを目的とする。
【課題を解決するための手段】
【0019】
本発明者らは上記の課題を解決するために鋭意研究を重ね、下記の一般式(1)で表される11C標識イソキノリン誘導体が、後に述べる製造法に従って下記一般式(2)で表される前駆体を使用して、短時間かつ高収率で製造できることを見出した。さらに一般式(1)の化合物を用いてPET用分子プローブを調製して生体に投与し、PETにより組織の画像化を行った。その結果、一般式(1)の化合物はPETのための放射性医薬品及び放射性診断薬として有用であることを見出し、本発明を完成した。
すなわち、本発明は下記の各発明を提供する。
【0020】
本発明は下記一般式(1)で表される11C標識イソキノリン誘導体又はその薬学上許容される塩、水和物、溶媒和物若しくはプロドラッグを提供する。
【0021】
【化1】

【0022】
(式中、
R1及びR2は、どちらか一方が[11C]メチル基を示し、他方は水素原子、シアノ基、アルキル基、ハロゲノアルキル基、アルケニル基、アルコキシ基、アルキルチオ基、ニトロ基、フルオロ基又はアリール基を示し;
R3及びR4はそれぞれ独立して水素原子、アルキル基、アルケニル基、アミノ基、アルキルアミノ基、ジアルキルアミノ基、アミノアルキル基、ハロゲノアルキル基、アルカノイル基、アミノアルカノイル基又はアルキルアミノアルカノイル基を示すか、あるいはR3及びR4は一緒になってアルキレン基又はアルケニレン基を形成し、一般式(1)中の環状ジアミノ基における任意の2つの炭素間で架橋しているか、のいずれかであり;
R5は水素原子、アミノアルカノイル基又はアルキルアミノアルカノイル基を示し;
mは1〜4の自然数を示し、nは2又は3の数を示す。)
【0023】
本発明はまた、一般式(1)の化合物を製造するのに好適な前駆体を提供し、それは下記の一般式(2)で表される。
【0024】
【化2】
【0025】
(式中、
R6及びR7は、どちらか一方がトリアルキルスズ基、ボロニル基又はジアルコキシボリル基を示し、他方は水素原子、シアノ基、アルキル基、ハロゲノアルキル基、アルケニル基、アルコキシ基、アルキルチオ基、ニトロ基、フルオロ基又はアリール基を示し;
R3及びR4は前記と同じであり;
R8はtert−ブトキシカルボニル基、トリハロアセチル基、tert−ブトキシカルボニルアミノアルカノイル基、トリハロアセチルアミノアルカノイル基、N−アルキル−N−tert−ブトキシカルボニルアミノアルカノイル基又はN−アルキル−N−トリハロアセチルアミノアルカノイル基を示し;
mは1〜4の自然数を示し、nは2又は3の数を示す。)
【0026】
また本発明は、一般式(1)の前駆体(2)におけるイソキノリン骨格上に[11C]メチル基を導入してから塩基性条件下でトリハロアセチル基などの保護基を脱離させることを特徴とする一般式(1)の11C標識イソキノリン誘導体の短時間で高収率かつ容易な製造方法を提供する。
【0027】
また本発明は、一般式(1)の11C標識イソキノリン誘導体を含有するPET用プローブを提供する。
【0028】
さらに本発明は、一般式(1)の11C標識イソキノリン誘導体を生体に投与して当該生体内における11Cの分布を調べることを特徴とする組織の画像化方法を提供する。
【図面の簡単な説明】
【0029】
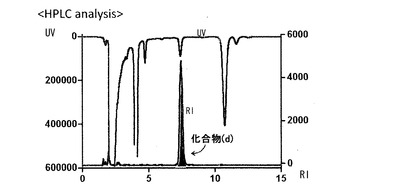
【図1】実施例3における反応ろ液の分取HPLCのチャートである。
【発明を実施するための形態】
【0030】
一般式(1)中、R1及びR2は、どちらか一方が[11C]メチル基を示し、他方は水素原子、シアノ基、アルキル基、ハロゲノアルキル基、アルケニル基、アルコキシ基、アルキルチオ基、ニトロ基、フルオロ基又はアリール基を示す。
【0031】
ここでアルキル基としては、炭素数1〜8の直鎖状、分枝鎖状、又は環状のアルキル基(C1-8アルキル基)(ここで「C1-8」とは炭素数が1〜8のいずれかであることを示す。以下同様)が挙げられ、炭素数1〜6のアルキル基が好ましく、さらに炭素数1〜3のアルキル基が好ましい。
具体例としては、メチル基、エチル基、n−プロピル基、イソプロピル基、n−ブチル基、イソブチル基、sec−ブチル基、tert−ブチル基、n−ペンチル基、イソペンチル基、n−ヘキシル基、イソヘキシル基、シクロプロピル基を挙げることができる。なかでも炭素数1〜3のものが好ましく、特にメチル基、エチル基が好ましい。
【0032】
ハロゲノアルキル基としては、ハロゲノC1-8アルキル基が好ましく、ハロゲノC1-6アルキル基がより好ましい。具体例としてはクロロメチル基、フルオロメチル基、クロロエチル基、フルオロエチル基、トリフルオロメチル基等が挙げられる。
【0033】
アルケニル基としては、炭素数2〜8の直鎖状又は分枝鎖状のアルケニル基(C2-8アルケニル基)が挙げられ、炭素数2〜6のアルケニル基が好ましい。具体例としてはビニル基、アリル基、イソプロペニル基、2−メタリル基、2−ブテニル基、3−ブテニル基を挙げることができる。なかでも炭素数2〜4のものが好ましい。
【0034】
アルコキシ基としては炭素数1〜8の直鎖状又は分枝鎖状のアルコキシ基(C1-8アルコキシ基)が挙げられ、炭素数1〜6のアルコキシ基が好ましい。具体例としては、メトキシ基、エトキシ基、n−プロポキシ基、イソプロポキシ基、n−ブトキシ基、イソブトキシ基、sec−ブトキシ基、tert−ブトキシ基を挙げることができる。
【0035】
アルキルチオ基としては、炭素数1〜8の直鎖状又は分枝鎖状のアルキルチオ基(C1-8アルキルチオ基)が挙げられ、炭素数1〜6のアルキルチオ基が好ましい。具体例としてはメチルチオ基、エチルチオ基、イソプロピルチオ基、n−プロピルチオ基等が挙げられる。
【0036】
アリール基としては、C6-14アリール基が挙げられ、フェニル基、ナフチル基が好ましく、フェニル基がより好ましい。
【0037】
R1及びR2としては、どちらか一方が[11C]メチル基を示し、他方は水素原子、C1-8アルキル基、ニトロ基、フルオロ基、シアノ基、ハロゲノC1-8アルキル基、フェニル基又はC2-8アルケニル基であるのが好ましい。また、[11C]メチル基の他方は水素原子、シアノ基、C1-6アルキル基又はハロゲノC1-6アルキル基であるのがより好ましい。さらに、[11C]メチル基の他方は水素原子又はC1-3アルキル基であるのが好ましい。
【0038】
R1はイソキノリン骨格の1位、3位及び4位のいずれに置換していてもよい。また、R2はイソキノリン骨格の6位、7位及び8位のいずれに置換していてもよい。ただし、[11C]メチル基の置換位置としてはイソキノリン環の4位が特に好ましい。
【0039】
R3及びR4は、それぞれ独立して水素原子、アルキル基、アルケニル基、アミノ基、アルキルアミノ基、ジアルキルアミノ基、アミノアルキル基、ハロゲノアルキル基、アルカノイル基、アミノアルカノイル基又はアルキルアミノアルカノイル基を示すか、あるいはR3とR4は一緒になってアルキレン基又はアルケニレン基を形成し、一般式(1)中の環状ジアミノ基における任意の2つの炭素間で架橋していてもよい。
【0040】
アルキル基、アルケニル基及びハロゲノアルキル基としては、前記R1及びR2の例として示したものが挙げられる。
アルキルアミノ基としては、C1-8アルキルアミノ基が好ましく、具体例としてはメチルアミノ基、エチルアミノ基、n−プロピルアミノ基、イソプロピルアミノ基、n−ブチルアミノ基、イソブチルアミノ基、sec−ブチルアミノ基、n−ペンチルアミノ基、n−ヘキシルアミノ基等が挙げられる。ジアルキルアミノ基としては、ジ−C1-8アルキルアミノ基が好ましく、具体例としてはジメチルアミノ基、ジエチルアミノ基、ジプロピルアミノ基、ジブチルアミノ基等が挙げられる。アミノアルキル基としては、アミノC1-8アルキル基が好ましく、具体例としてはアミノメチル基、アミノエチル基、アミノプロピル基、アミノブチル基等が挙げられる。
【0041】
アルカノイル基としては、炭素数2〜8の直鎖状又は分枝鎖状のアルカノイル基(C2-8アルカノイル基)が挙げられ、炭素数2〜6のアルカノイル基が好ましい。具体例としてはアセチル基、プロピオニル基、ブチリル基等が挙げられる。アミノアルカノイル基としては、アミノ−C2-8アルカノイル基が挙げられ、アミノ−C2-6アルカノイル基が好ましい。具体例としてはアミノアセチル基、アミノプロピオニル基、アミノブチリル基等が挙げられる。
【0042】
アルキルアミノアルカノイル基としては、C1-8アルキルアミノC2-8アルカノイル基が挙げられ、C1-4アルキルアミノC2-4アルカノイル基が好ましく、具体例としてはメチルアミノアセチル基、メチルアミノプロピオニル基等が挙げられる。アルコキシカルボニル基としては、C1-8アルコキシカルボニル基が挙げられ、例えばメトキシカルボニル基、エトキシカルボニル基が挙げられる。
【0043】
R3及びR4が一緒になって形成されるアルキレン基としては、C1-3アルキレン基、例えばメチレン基、エチレン基、トリメチレン基(−CH2CH2CH2−)が挙げられ、特にメチレン基、エチレン基が好ましい。R3及びR4が一緒になって形成されるアルケニレン基としては、C2-4アルケニレン基、例えば−CH=CH−、−CH2CH=CH−等が挙げられる。これらのアルキレン基又はアルケニレン基は、式(1)中の窒素含有飽和複素環上の任意の位置へ2つの炭素間で架橋してもよい。そのような架橋としては、架橋C1-3アルキレン基、すなわち−CH2−、−CH2CH2−、−CH2CH2CH2−による架橋が好ましい。
【0044】
R3及びR4としては、それぞれ独立して水素原子、C1-8アルキル基、C2-8アルケニル基、アミノ基、アミノC1-8アルキル基、ハロゲノC1-8アルキル基、C1-8アルキルアミノ基、ジ−C1-8アルキルアミノ基、C2-8アルカノイル基又はアミノC2-8アルカノイル基を示すか、あるいはR3とR4とが一緒になって架橋C1-3アルキレン基を形成するのが好ましい。
また、R3及びR4としては、それぞれ独立して水素原子、C1-8アルキル基、アミノ基、C1-8アルキルアミノ基、アミノC1-8アルキル基又はハロゲノC1-8アルキル基であるか、あるいはR3とR4とが一緒になって架橋C1-3のアルキレン基を形成するのがより好ましい。
さらにR3及びR4は水素原子、C1-6アルキル基又はハロゲノC1-6アルキル基であるのが好ましい。
【0045】
R5としては、水素原子、アミノアルカノイル基又はアルキルアミノアルカノイル基を示す。アミノアルカノイル基又はアルキルアミノアルカノイル基は前記と同じ。
【0046】
mは1〜4の自然数を示し、nは2又は3を示すが、mは1〜3の自然数が好ましく、また、nは2が特に好ましい。
【0047】
本発明の11C標識イソキノリン誘導体には、上記一般式(1)で示される化合物の他、その塩、水和物、溶媒和物及びプロドラッグが含まれる。ここで、プロドラッグとは、生体内で加水分解されて一般式(1)で示される化合物を生成する化合物をいう。こうしたプロドラッグには、当業者に知られたプロドラッグ化のすべての手法で製造される化合物が含まれる。例えば、アミノ基を生体内で容易に加水分解されうるアミド基等に誘導した化合物が挙げられる。具体的には一般式(1)に存在するアミノ基をアセトアミド等に誘導した化合物等が挙げられる。これらのプロドラッグは、上記一般式(1)の11C標識イソキノリン誘導体から短時間で容易に製造できるため、PET用プローブとしての適用に対しても、それほどの障害とはならない。
【0048】
また、薬学上許容される塩としては、一般式(1)に存在するアミノ基に対する酸付加塩が挙げられる。酸付加塩としては、例えば塩酸塩、臭化水素酸塩、リン酸塩、硫酸塩、硝酸塩等の無機酸塩、ギ酸塩、酢酸塩、プロピオン酸塩、マレイン酸塩、フマル酸塩、コハク酸塩、乳酸塩、リンゴ酸塩、酒石酸塩、クエン酸塩、アスコルビン酸塩、マロン酸塩、シュウ酸塩、グリコール酸塩、フタル酸塩、ベンゼンスルホン酸塩等の有機酸塩が挙げられ、また、上記の塩を組み合わせて用いることもできる。
【0049】
次に一般式(2)で表される化合物としては、式(2)中、R3-4,m及びnは、前記一般式(1)で表される11C標識イソキノリン誘導体において示したR3-4,m及びnと同じである。
【0050】
R8としては、tert−ブトキシカルボニル基、トリハロアセチル基、tert−ブトキシカルボニルアミノアルカノイル基、トリハロアセチルアミノアルカノイル基、N−アルキル−N−tert−ブトキシカルボニルアミノアルカノイル基又はN−アルキル−N−トリハロアセチルアミノアルカノイル基を示す。
【0051】
トリハロアセチル基としては、トリフルオロアセチル基、トリクロロアセチル基又はトリブロモアセチル基を用いることができるが、脱離容易なトリフルオロアセチル基が好ましい。
【0052】
トリハロアセチルアミノアルカノイル基とN−アルキル−N−トリハロアセチルアミノアルカノイル基としては、前記のアミノアルカノイル基又はアルキルアミノアルカノイル基のアミノ基をトリハロアセチル基で保護したものであり、トリハロアセチルアミノアルカノイル基としては、トリハロアセチルアミノ−C2-8アルカノイル基が挙げられ、トリハロアセチルアミノ−C2-6アルカノイル基が好ましい。具体例としてはトリフルオロアセチルアミノアセチル基、トリフルオロアセチルアミノプロピオニル基、トリフルオロアセチルアミノブチリル基等が挙げられる。N−アルキル−N−トリハロアセチルアミノアルカノイル基としては、N−C1-8アルキル−N−トリハロアセチルアミノC2-8アルカノイル基が挙げられ、N−C1-4アルキル−N−トリハロアセチルアミノC2-4アルカノイル基が好ましく、具体例としてはN−メチル−N−トリフルオロアセチルアミノアセチル基、N−メチル−N−トリフルオロアセチルアミノプロピオニル基等が挙げられる。
同様に、tert−ブトキシカルボニルアミノアルカノイル基とN−アルキル−N−tert−ブトキシカルボニルアミノアルカノイル基としては、前記のアミノアルカノイル基又はアルキルアミノアルカノイル基のアミノ基をtert−ブトキシカルボニル基で保護したものを示す。
【0053】
R6及びR7は、どちらか一方がトリアルキルスズ基、ボロニル基又はジアルコキシボリル基を示し、他方は水素原子、シアノ基、アルキル基、ハロゲノアルキル基、アルケニル基、アルコキシ基、アルキルチオ基、ニトロ基、フルオロ基又はアリール基を示す。
【0054】
R6はイソキノリン骨格の1位、3位及び4位のいずれに置換していてもよい。
また、R7はイソキノリン骨格の6位、7位及び8位のいずれに置換していてもよい。ただし、トリアルキルスズ基、ボロニル基又はジアルコキシボリル基の置換位置としてはイソキノリン環の4位が特に好ましい。
【0055】
R6及びR7としては、どちらか一方がトリアルキルスズ基、ボロニル基又はジアルコキシボリル基を示し、他方は水素原子、C1-8アルキル基、ニトロ基、フルオロ基、シアノ基、ハロゲノC1-8アルキル基、フェニル基又はC2-8アルケニル基であるのが好ましい。また、トリアルキルスズ基、ボロニル基又はジアルコキシボリル基の他方は水素原子、シアノ基、C1-6アルキル基又はハロゲノC1-6アルキル基であるのがより好ましい。さらに、トリアルキルスズ基、ボロニル基又はジアルコキシボリル基の他方は水素原子又はC1-3アルキル基であるのが好ましい。
【0056】
トリアルキルスズ基としては、トリC1-6アルキルスズ基が挙げられ、トリC2-4アルキルスズ基が好ましく、具体例としてはトリエチルスズ基、トリプロピルスズ基又はトリブチルスズ基が挙げられる。
【0057】
ジアルコキシボリル基としては、ジC1-6アルコキシボリル基が挙げられ、ジC1-3アルコキシボリル基が好ましく、具体例としてはジメトキシボリル基、ジエトキシボリル基又はジプロピルキシボリル基が挙げられる。また、二つのアルコキシ基は一緒になってアルキレン又はフェニレンを形成してもよく、具体例としては4,4,5,5−テトラメチル−1,3,2−ジオキサボロラン−2−イル基、4,4,6,6−テトラメチル−1,3,2−ジオキサボリナン−2−イル基又はベンゾ[d][1,3,2]ジオキサボロル−2−イル基が挙げられる。
【0058】
<製造法>
前記一般式(1)で表される11C標識イソキノリン誘導体は、後述する方法で製造された化合物(3)から、前記一般式(2)で表される化合物を介して、例えば、スキーム1に示す方法に従って製造することができる。
【0059】
【化3】
【0060】
(式中、R9及びR10は、どちらか一方がハロゲン原子を示し、他方は水素原子、シアノ基、アルキル基、ハロゲノアルキル基、アルケニル基、アルコキシ基、アルキルチオ基、ニトロ基、フルオロ基又はアリール基を示す。
R1-8、m及びnは前記と同じ。)
【0061】
[第1工程]
化合物(4)は、化合物(3)に適当な溶媒中、塩基存在下に、ジ−tert−ブチルジカーボネート等のtert−ブトキシカルボニル化剤や、ハロゲン化トリハロアセチル又は無水トリハロ酢酸等のトリハロアセチル化剤を反応させることにより製造することができる。
【0062】
反応溶媒としては、反応に支障のないものであればよく、例えば、テトラヒドロフラン、ジオキサン、ジエチルエーテル等のエーテル類、ベンゼン、トルエン等の炭化水素類、塩化メチレン、クロロホルム等のハロゲン化炭化水素類、N,N−ジメチルホルムアミド、N,N−ジメチルアセトアミド等の非プロトン性溶媒、ピリジン、アセトニトリル、アセトン、水又はこれらの混合物を用いることができる。
【0063】
本反応においては、適当な塩基の存在下に行うのが好ましい。かかる塩基としては、アルカリ金属炭酸水素塩(例、炭酸水素ナトリウム)、アルカリ金属炭酸塩(例、炭酸カリウム)、アルカリ金属水酸化物(例、水酸化ナトリウム、水酸化カリウム)のようなアルカリ、トリエチルアミン、トリエチレンジアミン等の有機第3級アミンを用いることができる。溶媒としてピリジンのような塩基性溶媒を使用すれば、かかる塩基は不要であり、好ましい。
【0064】
通常、本反応は室温で進行する場合が多いが、必要に応じて冷却又は加熱して、−78〜150℃、好ましくは、0〜120℃で行うことができる。反応時間は、使用する原料、溶媒、反応温度等によって異なるが、通常、5分〜70時間である。
【0065】
[第2工程]
化合物(4)のハロゲン原子を、それ自体公知の方法で、トリアルキルスズ基、ボロニル基又はジアルコキシボリル基に変換して、化合物(2)を製造することができる。
すなわち、一般式(4)で表されるハロゲン体を、テトラヒドロフラン、ジオキサン、ジエチルエーテル等のエーテル類の溶媒中で、マグネシウムを用いるグリニャール試薬化やアルキルリチウムなどの有機金属試薬を用いるリチオ化などにより活性化させ、続いてR6及びR7に対応する塩化トリアルキルスズ又は有機ホウ素化合物で処理することにより化合物(2)で表されるトリアルキルスズ体又はボロニル体を製造することができる。また、一般式(4)で表されるハロゲン体を、パラジウム触媒存在下にビス(トリアルキルスズ)体あるいはジボラン体を用いたスズ化あるいはホウ素化反応により一般式(2)で表されるトリアルキルスズ体、ボロニル体、ジアルコキシボリル体を製造することができる。本反応は、公知の方法に準じて行うことができる。
【0066】
R9及びR10で表されるハロゲンとしては、塩素、臭素、またはヨウ素が好ましい。
【0067】
[第3工程]
化合物(2)で表されるトリアルキルスズ体、ボロニル体又はジアルコキシボリル体を、非プロトン性極性溶媒中で、配位子、パラジウム錯体及び塩基の存在下に、[11C]ヨウ化メチルを反応させることにより、化合物(5)で表される[11C]メチル体を製造することができる。
[11C]メチル化の方法としては、前述した特許文献1、2又は3の方法を用いることができる。
【0068】
反応溶媒としては、ベンゼン、トルエン等の炭化水素類、塩化メチレン、クロロホルム等のハロゲン化炭化水素類、N,N−ジメチルホルムアミド、N,N−ジメチルアセトアミド、N−C1-6アルキル−2−ピロリジノン等の非プロトン性極性溶媒、ピリジン、アセトニトリル、水、又はこれらの混合物を用いることができる。なかでも、N,N−ジメチルホルムアミドやN−C1-6アルキル−2−ピロリジノンなどの非プロトン性極性溶媒が好ましい。特に好ましくは、N−C1-3アルキル−2−ピロリジノンであり、具体的にはN−メチル−2−ピロリジノン(NMP)、N−エチル−2−ピロリジノン又はN−プロピル−2−ピロリジノンが挙げられる。
【0069】
配位子としては、トリアルキルホスフィンやトリアリールホスフィンなどのホスフィン配位子又はトリアリールアルシンなどのアルシン配位子を用いることができる。通常、この反応は、配位子の嵩高さが活性の高い反応場を形成するため、特にトリ−o−トリルホスフィンや(ジ−tert−ブチル)メチルホスフィンなどのホスフィン配位子が好ましい。
【0070】
パラジウム錯体としては、通常知られているテトラキストリフェニルホスフィンパラジウムなどの0価のパラジウム錯体や塩化パラジウムや酢酸パラジウムなどの2価のパラジウム錯体を用いることができる。なかでも0価のパラジウム錯体が好ましいが、とくにトリス(ジベンジリデンアセトン)ジパラジウムが好ましい。
【0071】
塩基としては、アルカリ金属炭酸水素塩(例、炭酸水素ナトリウム)、アルカリ金属炭酸塩(例、炭酸カリウム、炭酸セシウム)、アルカリ金属リン酸塩(例、リン酸カリウム)、アルカリ金属水酸化物(例、水酸化ナトリウム、水酸化カリウム)のような無機塩基、トリエチルアミン、エチルジイソプロピルアミン等の有機第3級アミンを用いることができる。
【0072】
[第4工程]
11C標識イソキノリン誘導体(1)は、化合物(5)のtert−ブトキシカルボニル基又はトリハロアセチル基を酸処理やアルカリ処理により除去して製造することができる。反応はそれ自体公知の方法に従えばよい。例えば、tert−ブトキシカルボニル基の場合、適当な反応溶媒中で、塩酸やトリフルオロ酢酸等の酸を反応させることにより除去することができる。また、トリハロアセチル基の場合、アルカリ金属炭酸水素塩、アルカリ金属炭酸塩、アルカリ金属水酸化物等のような無機塩基を用いた加水分解反応あるいは加溶媒分解反応により除去することができる。
【0073】
スキーム1で示した化合物(3)は、特許文献7〜9に記載の方法、又はそれらの類似の方法によって合成することができる。また、以下のスキーム2〜6に示す方法によって合成することもできる。
【0074】
【化4】
【0075】
(式中、L1は水酸基又は脱離基を示し、R11は、Nの保護基を示す。また、R3、R4、R9、R10、m及びnは前記と同じ。)
【0076】
脱離基としてのL1は、後記するスルホン酸の反応性誘導体の残基を挙げることができる。Nに用いられる保護基としては、例えば、ホルミル、アセチル、ベンゾイル等のアシル基、ベンジルオキシカルボニル等のアラルキルオキシカルボニル基、tert−ブチルオキシカルボニル等のアルコキシカルボニル基、ベンジル等のアラルキル基を挙げることができる。
【0077】
一般式(7)で表されるアミンを適当な溶媒中、一般式(6)で表されるスルホン酸、又はその反応性誘導体と反応させ、必要により保護基を除去して化合物(3a)を製造することができる。
【0078】
スルホン酸の反応性誘導体としては、スルホン酸ハライド(例、スルホン酸フルオライド、スルホン酸クロライド、スルホン酸ブロマイド)、スルホン酸無水物、N−スルホニルイミダゾリド等が用いられる。特にスルホン酸ハライドが好ましい。
【0079】
【化5】
【0080】
(式中、L2は脱離基を示し、R3、R4、R9、R10、m及びnは前記と同じ。)
【0081】
脱離基としてのL2は、塩素若しくは臭素等のハロゲン又はアセチルオキシ等のアシルオキシ基、若しくはメシルオキシ、トシルオキシ等のスルホニルオキシ基を挙げることができる。
化合物(8)をアミン、グアニジン又はアンモニアと反応させることにより化合物(3a)を製造することができる。本反応は、公知の方法(Acta Chemica Scand., 45,621(1991))に準じて行うことができる。
【0082】
【化6】
【0083】
(式中、R12は水素原子又はアミノ基の保護基を示し、L2、R3、R4、R9、R10、m及びnは前記と同じ。)
【0084】
化合物(9)と化合物(10)とを反応させ、必要により、酸又はアルカリ処理して保護基を除去することにより化合物(3a)を製造することができる。本反応は、公知の方法(Acta Chemica Scand., 45, 621(1991))に準じて行うことができる。
【0085】
【化7】
【0086】
(式中、R12は水素原子又はアミノ基の保護基を示し、L2、R3、R4、R9、R10、m及びnは前記と同じ。)
化合物(3a)は、化合物(11)をトリフェニルホスフィンなどの有機リン試薬とアゾジカルボン酸ジエチル又はアゾジカルボン酸ジイソプロピルなどのアゾ試薬を用いて分子内脱水反応(いわゆる光延反応)を行い、続いて保護基R12を除去することによって製造することができる。
【0087】
【化8】
【0088】
(式中、L3は水酸基又は脱離基を示し、R13は水素原子又はC1-7アルキル基を示し、lは1〜7の自然数を示す。また、R3、R4、R9、R10、R12、m及びnは前記と同じ。)
【0089】
一般式(3a)で表されるアミンを、適当な縮合剤の存在下に一般式(12)で表されるカルボン酸に反応させ、必要により保護基を除去して化合物(3b)を製造することができる。又は一般式(3a)を適当な塩基存在下にカルボン酸の反応性誘導体と反応させ、化合物(3b)を製造する。
【0090】
縮合剤としては、ジシクロヘキシルカルボジイミドや1−エチル−3−(3−ジメチルアミノプロピル)カルボジイミド塩酸塩などのカルボジイミド縮合剤や1H−ベンゾトリアゾール−1−イルオキシトリピロリジノホスホニウム及び、ヘキサフルオロホスフェートなどのホスホニウム縮合剤などが用いられる。
【0091】
脱離基としてのL3は、カルボン酸の反応性誘導体の残基を挙げることができる。カルボン酸の反応性誘導体としては、カルボン酸ハライド(例、カルボン酸クロライド、カルボン酸ブロマイド)、カルボン酸無水物、カルボン酸アジド、活性エステルが用いられる。
【0092】
11C標識イソキノリン誘導体(1)には、不斉炭素を有し、光学異性体が存在するものもある。これらの各異性体及びこれらの混合物のいずれも本発明に包含される。光学異性体は、光学活性な原料化合物(S配置又はR配置)を用いることにより得ることができる。またラセミ体はそのままでも薬理活性を有するが、所望によりそれぞれの異性体に分割することができる。例えば、異性体混合物を公知の光学分割法、例えば、光学活性なカルボン酸(例、(+)−又は(−)−酒石酸、(+)−又は(−)−リンゴ酸)又は光学活性なスルホン酸(例、(+)−ショウノウスルホン酸)との塩を生成させ、分別結晶する方法、光学活性カラムを用いる方法によって分離することができる。
11C標識イソキノリン誘導体(1)は、公知の方法により、塩を形成させることができる。例えば塩酸塩は、本発明化合物を塩化水素のアルコール溶液又はエチルエーテル溶液に溶解することにより得ることができる。
【0093】
本発明化合物である11C標識イソキノリン誘導体(1)は、後述するように、PET用分子プローブとして好適に用いることができる。その際の投与経路としては、経口でも、非経口でも投与することができる。投与剤型として錠剤、カプセル剤、顆粒剤、散剤、注射剤、点眼剤等が挙げられ、それらは汎用される技術を組み合わせて使用することができる。ただし、11Cの半減期を考慮すると、製剤化するのに時間を要しない注射剤や点眼剤が好ましい。
注射剤、又は点眼剤とする場合は、液状媒体として例えば滅菌精製水、生理食塩水、等張液等の水性媒体、又は例えばゴマ油、大豆油、オリーブ油等の非水性媒体が用いられる。水性媒体と油性媒体の両者を含有するエマルジョンでもよい。
注射剤、点眼剤等の非経口剤は、例えば、グリセリン、プロピレングリコール、塩化ナトリウム、塩化カリウム、ソルビトール、マンニトール等の等張化剤、リン酸、リン酸塩、クエン酸、氷酢酸、ε−アミノカプロン酸、トロメタモール等の緩衝剤、塩酸、クエン酸、リン酸、氷酢酸、水酸化ナトリウム、水酸化カリウム、炭酸ナトリウム、炭酸水素ナトリウム等のpH調節剤、ポリソルベート80、ポリオキシエチレン硬化ヒマシ油60、マクロゴール等の溶解補助剤、精製卵黄レシチン、精製大豆レシチン、ポリオキシエチレン(160)ポリオキシプロピレン(30)グリコール等の乳化剤、ヒドロキシプロピルメチルセルロース、ヒドロキシプロピルセルロース等のセルロース系高分子、ポリビニルアルコール、ポリビニルピロリドン等の増粘剤、エデト酸、エデト酸ナトリウム等の安定化剤、汎用のソルビン酸、ソルビン酸カリウム、塩化ベンザルコニウム、塩化ベンゼトニウム、パラオキシ安息香酸メチル、パラオキシ安息香酸プロピル、クロロブタノール等の保存又は防腐剤、クロロブタノール、ベンジルアルコール、リドカイン等の無痛化剤を必要に応じて本発明化合物に組合わせて、調製することができる。
【0094】
尚、注射剤又は点眼剤の場合、pHは4.0〜8.0に設定することが望ましく、また、浸透圧比は1.0付近に設定することが望ましい。
【0095】
このPET用プローブを放射性医薬品や診断薬組成物等として利用することにより、PET画像によるプロテインキナーゼ阻害剤の解析が可能となり、脳内動態解析や薬剤(治療薬)としての潜在性を解析することが可能となる。さらには、生物学的研究や動物・ヒトのPETによる臨床研究の基盤技術とすることができる。
【0096】
すなわち、本発明の11C標識イソキノリン誘導体を生体に投与して該生体内における11Cの分布を調べることにより、組織の画像化を行うことができる。11Cの分布は陽電子を検出するPET法の手法を用いることができる。こうした組織の画像化の対象として例えば脳を選べば、脳内のプロテインキナーゼと結合した11C標識イソキノリン誘導体を捉えることが可能となり、ひいては脳内のプロテインキナーゼの働き等についての多くの知見を得ることができる。
【0097】
例えばH-1152PはRhoキナーゼの特異的阻害剤であることから、その11C体である本発明化合物を実験用ラット等に対し静脈注射等によって投与した後、例えば脳のPET画像を経時的に撮影することにより、脳内のRhoキナーゼの分布や、脳内のRhoキナーゼと結合した11C標識イソキノリン誘導体を捉えることが可能となり、脳内のRhoキナーゼの働き等についての多くの知見を得ることができる。
【0098】
さらに、本発明の11C標識イソキノリン誘導体を含有するPET用製剤をヒトに投与した後撮影されたPET画像は、プロテインキナーゼの分布の異常を伴う疾患やプロテインキナーゼに関連する疾患の診断に用いることができる。そのプロテインキナーゼには、Rhoキナーゼ、PKA、PKG、AURKA、CAMK2などが含まれるが、これらに限定されない。
【0099】
例えばH-1152PはRhoキナーゼの特異的阻害剤であることから、その11C体である本発明化合物をヒトに投与した後撮影されたPET画像は、Rhoキナーゼの分布の異常を伴う疾患やRhoキナーゼに関連する疾患の診断に用いることができる。
Rhoキナーゼに関連する疾患には、脳血管攣縮、冠動脈攣縮、高血圧、肺性高血圧、喘息、緑内障、腎障害、狭心症、心筋梗塞、動脈硬化症、再狭窄、脳卒中、心不全、心筋肥大、虚血再還流障害、血管内皮障害、炎症性腸疾患、前立腺肥大、レイノー病、神経因性疼痛、Alzheimer病、Huntington舞踏病、脊髄損傷、てんかん、多発性硬化症、骨粗しょう症、勃起不全、肝障害、がん、創傷治癒、インスリン抵抗性、及び糖尿病が含まれる。上記のがんには、肉腫、Glioblastoma、神経膠腫、頭頸部がん、肝細胞がん、腎明細胞がん、肺がん、卵巣がん、乳がん、食道がん、大腸がん、膀胱がん、前立腺がん、及び悪性黒色腫が含まれる。
【0100】
また、PETによる11C標識イソキノリン誘導体の体内での検出は極めて高感度であるため、これをマイクロドーズ臨床試験に適用することもできる。本臨床試験は、超微量(マイクロドーズ)の薬剤候補物質を人体に投与して、高感度微量分析を用いて薬物動態などを解析する方法であり、超微量での薬理動態を観察することから、人体に対する影響が少ない。このため、薬理試験の安全性が極めて高く、マイクロドーズ臨床試験ではフェーズI に入る前の段階で、医薬品の候補物質を安全に早く人に適用することにより、人体における薬物動態を知り、薬物動態面からの開発候補物質のスクリーニングを行うことができ、非臨床試験期間を大幅に短縮することができる。
【実施例】
【0101】
以下に実施例により本発明を具体的に説明する。尚、それらの例示は本発明をよりよく理解するためのものであり、本発明の範囲を限定するものではない。
【0102】
参考例1
4−トリフルオロアセチル−{(S)−ヘキサヒドロ−2−メチル−1−(4−ブロモ−5−イソキノリンスルホニル)}−1H−1,4−ジアゼピン(b)の合成
【0103】
【化9】
【0104】
特許文献8の方法に従い合成した(S)−ヘキサヒドロ−2−メチル−1−(4−ブロモ−5−イソキノリンスルホニル)−1H−1,4−ジアゼピン(a)をトリフルオロアセチル基により保護した。
すなわち、(a)(246.6 mg, 641.4 μmol) の乾燥ジクロロメタン溶液 (2 mL) にトリエチルアミン (134 μL, 97.3 mg, 962 μmol) を添加し、0℃に冷却したのち、トリフルオロ酢酸無水物 (134 μL, 202 mg, 962 μmol) を添加した。この混合溶液を0℃で2時間攪拌したのち、反応混合物を蒸留水(約10 mL)に加え、ジクロロメタン (3×15 mL) で抽出した。有機層を蒸留水 (15 mL)、飽和食塩水 (15 mL) で順次洗浄後、無水硫酸ナトリウムで乾燥した。乾燥剤をろ別し、ろ液を減圧下で濃縮した。得られた残査はシリカゲルカラムクロマトグラフィー (シリカゲル 5 g, 展開溶媒;ヘキサン:アセトン (2:1)) で精製し、淡黄色の油状化合物(b)(266 mg, 554 μmol, 86.4 %)を得た。
1H NMR (500 MHz, CDCl3, 25℃): δ=9.21 (s, 1H), 8.99 (s, 1H), 8.50 (dd, 4J(H,H)=1.0 Hz, 3J(H,H)=7.5 Hz, 1H), 8.21 (dd, 4J(H,H)=1.0 Hz, 3J(H,H)=8.0 Hz, 1H), 7.67-7.71 (m, 1H), 3.90-4.36 (m, 4H), 3.33-3.66 (m, 3H), 2.18-2.32 (m, 1H), 1.87-2.04 (m, 1H), 1.06 ppm (d, 3J(H,H)=7.0 Hz, 3H); HRMS (EI) m/z: [M+H]+ calcd for C9H7BrN 207.9762 found 207.9745; Anal. Calcd for C17H17BrF3N3O3S: C, 72.51; H, 3.57; N, 8.75. Found: C, 72.74; H, 3.79; N, 8.70.
【0105】
実施例1
<有機スズ前駆体の合成>
4−トリフルオロアセチル−[(S)−ヘキサヒドロ−2−メチル−1−{4−(トリ−n−ブチルスタニル)−5−イソキノリンスルホニル}]−1H−1,4−ジアゼピン(c)の合成
【0106】
【化10】
【0107】
化合物(b)から有機スズ前駆体(c)を合成した。
すなわち、アルゴン気下、乾燥20mLシュレンク型反応管に(b)(99.4 mg, 0.205 mmol)と乾燥THF (2.5 mL) を加えた。この混合溶液を-100℃に冷却したのち、tert−ブチルリチウム (279μL, 1.47 M ヘキサン溶液, 0.410 mmol) を20分間かけて滴下し、同じ温度で2時間撹拌した。この反応溶液に、ヘキサメチルホスホリックトリアミド(HMPA) (214 μL, 220 mg, 1.23 mmol)のTHF溶液(1.0 mL)と(n-C4H9)3SnCl (317 μL, 381 mg, 1.17 mmol)のTHF溶液(1.0 mL)を順に加え、室温まで徐々に昇温し、10時間撹拌した。得られた反応溶液を冷やした飽和塩化アンモニウム水溶液 (約2 mL) に加え、酢酸エチル (3×10 mL)で抽出した。有機層を飽和食塩水(20 mL)で洗浄後、無水硫酸ナトリウムで乾燥した。乾燥剤をろ別し、ろ液を減圧下で濃縮した。残査はシリカゲルカラムクロマトグラフィー (シリカゲル 10 g, 展開溶媒;ヘキサン:アセトン (2:1))で精製し、オレンジ色の油状化合物(c)(96.1 mg, 0.138 mmol, 67.5%) を得た。
1H NMR (400 MHz, CDCl3, 25℃): δ=9.15 (s, 1H), 8.74 (s, 1H), 8.25 (dd, 4J(H,H)=1.4, 3J(H,H)=7.5 Hz, 1H), 8.12 (dd, 4J(H,H)=1.2, 3J(H,H)=8.0 Hz, 1H), 7.61 (m, 1H), 4.16-4.21 (m, 1H), 2.91-3.87 (m, 6H), 1.52-1.71 (m, 2H), 1.44-1.50 (m, 6H), 1.24-1.34 (m, 6H), 1.10-1.15 (m, 6H), 0.84 (d, 3J(H,H)=5.3 Hz, 3H), 0.83 ppm (t, 3J(H,H)=7.4 Hz, 9H); HRMS (EI+, 100% acetone): m/z: calcd for C25H35F3N3O3SSn ([M-C4H9]+) 634.1376; found, 634.1359.
【0108】
実施例2
<有機ボロン酸エステル前駆体の合成>
(S)−2,2,2−トリフルオロ−1−[3−メチル−4−{4−(4,4,5,5−テトラメチル−1,3,2−ジオキサボラン−2−イル)イソキノリン−5−イルスルホニル}−1,4−ジアゼパン−1−イル]エタンの合成
特許文献3に記載の方法に従って化合物(b)から(S)−2,2,2−トリフルオロ−1−[3−メチル−4−{4−(4,4,5,5−テトラメチル−1,3,2−ジオキサボラン−2−イル)イソキノリン−5−イルスルホニル}−1,4−ジアゼパン−1−イル]エタンを合成することができる。
【0109】
実施例3
<11C標識イソキノリン誘導体(d)の合成1>
(S)−ヘキサヒドロ−2−メチル−1−[(4−[11C]メチル−5−イソキノリニル)スルホニル]−1H−1,4−ジアゼピン(d)の合成
【0110】
実施例1の化合物(c)を用いて、下記合成方法に基づき、イソキノリン骨格の4位を[11C]メチル化してから塩基性条件下でトリフルオロアセチル基を脱離させることによりイソキノリン骨格の4位のメチル基を[11C]メチル化した11C標識イソキノリン誘導体(d)を得た。詳細を以下に示す。
【0111】
【化11】
【0112】
11C核の製造は住友重機械工業社製サイクロトロンCYPRIS HM-12Sを使用し、14N(p,α)11Cの核反応により製造した。[11C]ヨウ化メチルの合成は専用の標識用合成装置を用いて、11CO2ガスを出発物質として、11CO2→11CH3OH→11CH3Iの順に変換して合成した。
【0113】
一方、トリス(ジベンジリデンアセトン)ジパラジウム(Pd2(dba)3)(1.8 mg, 1.9 μmol)および、トリ-o-トリルホスフィン(P(o-tolyl)3)(9.4 mg, 30.9μmol)のNMP(N-メチルピロリジノン)溶液(0.27 mL)を標識用合成装置の反応容器(A)に入れ、室温に設置した。反応容器(A)中の溶液は、[11C]ヨウ化メチル(11CH3I)を吹き込む10-20分前に設置した。また、有機スズ前駆体(c)(4.0 mg, 5.8μmol)、CuBr(0.5 mg, 3.9 μmol)、およびCsF(1.5 mg, 9.7 μmol)のNMP溶液(80 μL)を反応容器(B)に準備し、室温に設置した。続いて反応容器(A)に[11C]ヨウ化メチルを60-80 mL/minのガス流量で吹き込み、その後1分間静置した。得られた溶液を反応容器(B)に移送し、さらに反応容器(A)の内部を少量のNMP溶液(60 μL)を用いて洗浄して、この洗浄液も反応容器(B)に移送した。続いて、反応容器(B)中の混合溶液を80℃で5分間加熱した。続いて、2M NaOH (400μL)を加えて100℃で1分間加熱した。得られた反応溶液をメタノール(500μL)とリン酸ナトリウム緩衝液(10 mM、pH7.4, 500μL)で希釈した後、綿栓あるいはフィルターを用いてろ過した。ろ液を分取HPLCに供し(分取HPLCのチャートを図1に示した)、分取した11C-標識化合物(d)を含むフラクションをエバポレーターを用いて減圧濃縮した。濃縮液は、生理食塩水を主とする臨床用投与溶液を用いて希釈し、無菌バイヤルに入れた。本溶液の一部(20μL)を分析HPLCに供して、目的化合物の同定、純度検定、および比放射能の算出を行った。なお、11C-標識化合物(d)の同定は非標識体の化合物(d)を用い、下記の収率や分析値などは3回の実験から得られた結果である。
【0114】
11C-標識化合物(d)の総放射能:3.8±1.2 GBq、[11C]ヨウ化メチルに基づく崩壊補正放射化学収率:63±14%、合成時間:38分、放射化学的純度:99%以上、化学的純度:99%、比放射能: 97±10 GBq/μmol
分取条件:分取用カラムはWaters 社製XBridge, 5μm, C18 10mm×250mm、流速は5mL/minで、移動層はCH3OH:10mM リン酸ナトリウム緩衝液(pH=7.4) =50:50を使用した。UV検出波長254 nmおよびγ線検出器で測定した結果、11C-標識化合物(d)の保持時間は約9.2分であった。
分析条件:カラムはPhenomenex 社製Gemini NX, 5μm, 4.6mm×150mm を使用し、カラム温度30℃で分析を行った。流速は1mL/minで、移動層はCH3OH:10mM リン酸ナトリウム緩衝液(pH=7.4) =50:50を使用した。UV検出波長215 nmおよびγ線検出器で測定した結果、11C-標識化合物(d)の放射化学的純度および化学的純度はどちらも99%以上であった。
【0115】
<実験装置、実験方法及び使用した試薬>
標識用合成装置は、理化学研究所分子イメージング科学研究センターに設置してある標準型標識用自動合成装置を用いた。化学薬品は市販のものをそのまま用いた。脱水N-メチル-2-ピロリジノン(NMP)(関東化学社製)、トリス(ジベンジリデンアセトン)ジパラジウム(0)(Aldrich社製)、トリ-o-トリルホスフィン(Aldrich社製)、臭化銅(和光社製)、フッ化セシウム(和光社製)を使用した。
【0116】
実施例4
<11C標識イソキノリン誘導体(d)の合成2>
(S)−ヘキサヒドロ−2−メチル−1−[(4−[11C]メチル−5−イソキノリニル)スルホニル]−1H−1,4−ジアゼピンの合成
実施例2に記載した化合物から化合物(d)を得ることができる。
すなわち、非プロトン性極性溶媒中において、有機ボロン酸化合物のボロン酸エステルを、パラジウム錯体、ホスフィン配位子及び塩基の存在下で[11C]ヨウ化メチルとクロスカップリングさせることによって11C標識イソキノリン誘導体(d)を製造することができる。
【0117】
以下の実施例5〜10の化合物は、参考例1及び実施例1〜4に記載の方法を用いることで合成することができる。
実施例5
2,2,2−トリフルオロ−1−[4−{4−(トリブチルスタニル)イソキノリン−5−イルスルホニル}−1,4−ジアゼパン−1−イル]エタン
実施例6
2,2,2−トリフルオロ−1−[4−{4−(トリブチルスタニル)イソキノリン−5−イルスルホニル}ピペラジン−1−イル]エタン
実施例7
(S)−2,2,2−トリフルオロ−N−(2−[3−メチル−4−{4−(トリブチルスタニル)イソキノリン−5−イルスルホニル}−1,4−ジアゼパン−1−イル]−2−オキソエチル)アセタミド
実施例8
5−(1,4−ジアゼパン−1−イルスルホニル)−4−[11C]メチルイソキノリン
実施例9
4−[11C]メチル−5−(ピペラジン−1−イルスルホニル)イソキノリン
実施例10
(S)−2−アミノ−1−{3−メチル−4−(4−[11C]メチルイソキノリン−5−イルスルホニル)−1,4−ジアゼパン−1−イル}エタノン
【0118】
実施例11
<PET用製剤例>
実施例4で分取し濃縮して得た化合物(d)の油状物を例えばポリソルベート80含有生理食塩水に溶解し、さらに生理食塩水にて希釈することで静脈注射PET用製剤を調製することができる。
【0119】
実施例12
<PET撮影>
本発明の11C標識イソキノリン誘導体について、これをPET用プローブとしてPET撮影を行うためには、例えば次のような操作を行えばよい。
すなわち、HPLCによって本発明の11C標識イソキノリン誘導体(d)を分取し、実施例11に記載の製剤を調製し、これをPET用プローブとして実験用ラット等に対し静脈注射等によって投与する。そして、例えば小動物用PET装置(microPET、SIMENS社製)を用いて連続撮像を行う。さらに、非RI標識体による結合阻害効果を検討するために、非標識のイソキノリン誘導体を生理食塩水で希釈した溶液を実験用ラット等に対し静脈注射等によって投与する。また、PET撮像終了後、組織の切片を作製し、Ex vivoオートラジオグラム実験をおこなって、その分布を調べる。こうして、さまざまなPET画像及びその経時的な変化を調べることができる。
【技術分野】
【0001】
本発明は、イソキノリンスルホンアミド化合物類のイソキノリン環上に[11C]メチル基を導入した11C標識イソキノリン誘導体、その製造方法、その前駆体、分子プローブ及び組織の画像化方法に関する。
【背景技術】
【0002】
陽電子放射断層画像撮影法(PET法)は、小動物からヒトまで生体内の分子を非侵襲的に画像化できる唯一の技術である。このため、近年、医療や創薬分野でのPET法活用への期待が急速に膨らんでいる。しかし、技術活用の鍵となるPETプローブの合成は、分子に組み込む放射核の寿命が短いために、困難を伴う。
【0003】
従来は、導入が容易であるという理由から、O(酸素)、N(窒素)、S(硫黄)などのヘテロ原子上への[11C]メチル化のみが行われてきた。しかし、生体内においてはこれらのヘテロ原子を手がかりとして酵素による代謝を受けやすく、代謝によって標識部位の化学結合が切断されてしまうため、得られたPET画像の信憑性に疑問が残るという問題が指摘されてきた。
【0004】
こうした問題を解決するため、本発明者らは、11CH3Iを用いた炭素母核上への[11C]メチル化について開発を行ってきた。すべての有機化合物は、その構造中に炭素原子を有していることから、低分子有機化合物のPET分子プローブ化を行う上で、短寿命放射性炭素11Cの利用は理想的な放射性核種であると考えられるからである。
【0005】
これまで、本発明者らは、有機スズ化合物を中間原料として、これにStille型カップリング反応を適用することにより、芳香環上への高速C-[11C]メチル化のみならず、オレフィンやアルキンやヘテロ芳香環上への高速C-[11C]メチル化も可能としてきた(特許文献1、2及び非特許文献1〜3)。そしてさらには、従来の有機スズ化合物への高速C-[11C]メチル化と相補的に、新たに有機ホウ素化合物を用いた高速C-[11C]メチル化反応も開発している(特許文献3、非特許文献4)。
【0006】
一方、医薬品や生理活性物質において、イソキノリン環を有する数多くの化合物が知られている。この中でもイソキノリン環の5位が環状アミノスルホニルで置換された化合物は、循環器官用剤(血管拡張剤、脳循環改善剤、狭心症治療薬、脳心血管系の血栓症の予防および治療薬、高血圧症の予防治療薬、緑内障の予防および治療薬)として有用であり(特許文献4〜7)、プロテインキナーゼ阻害作用を有することが見出されている(非特許文献5)。
【0007】
本発明者の日高らは、臨床応用された世界最初で唯一のRhoキナーゼ阻害剤であるファスジル[ヘキサヒドロ−1−(5−イソキノリンスルホニル)−1H−1,4−ジアゼピン]を発明し、くも膜下出血後の脳血管攣縮の治療薬として人類に提供したが、ファスジルもイソキノリン環の5位が環状アミノスルホニルで置換された化合物である(特許文献6)。さらに、イソキノリン環の5位が環状アミノスルホニルで置換されていて、且つ、4位にメチル基などの置換基を有する化合物も開示されている(特許文献7〜9)。
【0008】
その構造的特徴を有する化合物の中でも、本発明者である日高により見出されたH-1152P[(S)−(+)−ヘキサヒドロ−2−メチル−1−[(4−メチル−5−イソキノリニル)スルホニル]−1H−1,4−ジアゼピン]は、選択的で作用強度の強いRhoキナーゼ阻害剤として知られている(特許文献6、非特許文献6)。
【0009】
Rhoキナーゼは、低分子量GTP結合タンパク質であるRhoの標的タンパク質として同定されたセリン/スレオニン・プロテインキナーゼであり、細胞移動、細胞増殖、細胞収縮、細胞粘着、細胞の運動性など、さまざまな細胞の機能にかかわる重要な酵素である。
【0010】
ファスジルやH-1152PなどのRhoキナーゼ阻害剤は、疾患におけるRhoキナーゼの役割を検討する研究に広く用いられ、その結果、Rhoキナーゼ阻害剤は、種々の疾患、例えば、脳血管攣縮、冠動脈攣縮、高血圧、肺性高血圧、喘息、緑内障、腎障害、狭心症、心筋梗塞、動脈硬化症、再狭窄、脳卒中、心不全、心筋肥大、虚血再還流障害、血管内皮障害、炎症性腸疾患、前立腺肥大、レイノー病、神経因性疼痛、Alzheimer病、Huntington舞踏病、脊髄損傷、てんかん、多発性硬化症、骨粗しょう症、勃起不全、肝障害、がん、創傷治癒、インスリン抵抗性、糖尿病などの治療薬として有用であることが報告されている。がんにおいては、進行が早く浸潤性の強いがんや転移性のがんでRhoキナーゼの発現亢進が報告されており、Rhoキナーゼが転移・浸潤に関与することが知られている。これらのがんには、肉腫、Glioblastoma、神経膠腫、頭頸部がん、肝細胞がん、腎明細胞がん、肺がん、卵巣がん、乳がん、食道がん、大腸がん、膀胱がん、前立腺がん、悪性黒色腫が含まれる。
【0011】
イソキノリン環の5位が環状アミノスルホニルで置換されている化合物は、Rhoキナーゼ阻害作用のみならず、PKA、PKG、AURKA、CAMK2など各種プロテインキナーゼを阻害する化合物が発見されており(非特許文献7)、プロテインキナーゼ研究のツールとして利用できるのみならず、医薬としても有用である。
【0012】
したがって、プロテインキナーゼ阻害など特定の生理活性を有するイソキノリン誘導体のイソキノリン環上に高速C-[11C]メチル化反応によって[11C]メチル基を導入できれば、PET画像による解析が可能となり、例えば体内における脳内動態解析や薬剤(治療薬)としての潜在性を解析などが可能となる。さらには、それらプロテインキナーゼ阻害剤を用いた生物学的研究や動物・ヒトのPETによる臨床研究の基盤技術とすることができる。
【先行技術文献】
【特許文献】
【0013】
【特許文献1】WO/2007/046258
【特許文献2】WO/2010/074272
【特許文献3】WO2008/023780
【特許文献4】特開昭57-156463号公報
【特許文献5】特開昭58-121279号公報
【特許文献6】特開昭61-227581号公報
【特許文献7】US2008/116481
【特許文献8】WO1997/028130
【特許文献9】WO2006/115247
【非特許文献】
【0014】
【非特許文献1】Suzuki, M., et al., Chem. Eur. J., 3, 2039-2042 (1997).
【非特許文献2】Hosoya, T., et al., Org. Biomol. Chem., 4, 410-415 (2006).
【非特許文献3】Hosoya, T., et al., Org. Biomol. Chem., 2, 24-27 (2004).
【非特許文献4】Doi, H., et al., Chem. Eur. J., 15,4165-4171 (2009).
【非特許文献5】Hidaka, H et al., Biochemistry, 23, 5036-5041 (1984).
【非特許文献6】Sasaki, Y., et al., Pharmacol. Ther. 93, 225-232 (2002).
【非特許文献7】Tamura, M., et al., Biochim Biophys Acta, 1754,245-252 (2005).
【0015】
しかしながら、11Cの半減期はわずか20分であるため、11Cの導入は高速化が求められ、かつ合成の最終段階で行うことがこれまでの技術常識であった。また、測定時間(1時間程度)を確保するため、反応、精製、生体への投与までを半減期の2倍の40分以内で行うという制約もある。このため、11Cの導入に許される時間はわずか5分程度となる。さらには、サイクロトロンで製造できる11C核種は超微量(数十から数百nmolレベル;12Cの混入を考慮した値)であり、超希薄な11C核種の化合物と化学反応させるために、大過剰の被標識基質の存在下という特殊な条件下で行われる。このため、いかに短時間で効率良く生物活性有機化合物や創薬候補化合物をPET分子プローブ化できるかということが最重要課題となっている。
【0016】
本発明者の鈴木らが開発した前述の高速メチル化法(特許文献1、2及び非特許文献1〜3)は、これらの課題を解決するための強力なツールとなる方法ではあるが、完全なる汎用方法とはなり得ない。なぜならば、メチル化される基質に存在する官能基が反応を阻害したりする場合があるからである。
【0017】
このことは、前述したイソキノリン環の5位が環状アミノスルホニルで置換されている化合物の[11C]メチル化において顕著である。すなわち、上記の高速メチル化反応によってイソキノリン骨格を[11C]メチル化しようとした場合、前駆体に存在する化学的に活性な2級アミノ基がメチル化反応を阻害する。また、2級アミノ基が[11C]メチル化されてしまう。そこで通常であれば、前駆体の2級のアミノ基を保護基で保護しておき、[11C]メチル化後に脱保護することが考えられる。ところが、前述したように、11Cの導入から生体へ投与するまでの一連の操作は高速化が求められることから、合成の最終段階で[11C]メチル化反応を行うことが当業者の常識であり、[11C]メチル化後に脱保護反応を行うことは、時間的及び技術的に困難であった。このため、有用な化合物であるにもかかわらず、イソキノリン環の5位が環状アミノスルホニルで置換されている化合物については、そのイソキノリン環の炭素上への[11C]メチル基の導入は従来なされていなかった。
【発明の概要】
【発明が解決しようとする課題】
【0018】
本発明は、上記従来の実情に鑑みてなされたものであり、イソキノリン骨格に[11C]メチル基を導入した11C標識イソキノリン誘導体及びその製造方法並びにその11C標識イソキノリン誘導体を製造するのに好適に用いることができる前駆体を提供することを目的とする。
【課題を解決するための手段】
【0019】
本発明者らは上記の課題を解決するために鋭意研究を重ね、下記の一般式(1)で表される11C標識イソキノリン誘導体が、後に述べる製造法に従って下記一般式(2)で表される前駆体を使用して、短時間かつ高収率で製造できることを見出した。さらに一般式(1)の化合物を用いてPET用分子プローブを調製して生体に投与し、PETにより組織の画像化を行った。その結果、一般式(1)の化合物はPETのための放射性医薬品及び放射性診断薬として有用であることを見出し、本発明を完成した。
すなわち、本発明は下記の各発明を提供する。
【0020】
本発明は下記一般式(1)で表される11C標識イソキノリン誘導体又はその薬学上許容される塩、水和物、溶媒和物若しくはプロドラッグを提供する。
【0021】
【化1】
【0022】
(式中、
R1及びR2は、どちらか一方が[11C]メチル基を示し、他方は水素原子、シアノ基、アルキル基、ハロゲノアルキル基、アルケニル基、アルコキシ基、アルキルチオ基、ニトロ基、フルオロ基又はアリール基を示し;
R3及びR4はそれぞれ独立して水素原子、アルキル基、アルケニル基、アミノ基、アルキルアミノ基、ジアルキルアミノ基、アミノアルキル基、ハロゲノアルキル基、アルカノイル基、アミノアルカノイル基又はアルキルアミノアルカノイル基を示すか、あるいはR3及びR4は一緒になってアルキレン基又はアルケニレン基を形成し、一般式(1)中の環状ジアミノ基における任意の2つの炭素間で架橋しているか、のいずれかであり;
R5は水素原子、アミノアルカノイル基又はアルキルアミノアルカノイル基を示し;
mは1〜4の自然数を示し、nは2又は3の数を示す。)
【0023】
本発明はまた、一般式(1)の化合物を製造するのに好適な前駆体を提供し、それは下記の一般式(2)で表される。
【0024】
【化2】
【0025】
(式中、
R6及びR7は、どちらか一方がトリアルキルスズ基、ボロニル基又はジアルコキシボリル基を示し、他方は水素原子、シアノ基、アルキル基、ハロゲノアルキル基、アルケニル基、アルコキシ基、アルキルチオ基、ニトロ基、フルオロ基又はアリール基を示し;
R3及びR4は前記と同じであり;
R8はtert−ブトキシカルボニル基、トリハロアセチル基、tert−ブトキシカルボニルアミノアルカノイル基、トリハロアセチルアミノアルカノイル基、N−アルキル−N−tert−ブトキシカルボニルアミノアルカノイル基又はN−アルキル−N−トリハロアセチルアミノアルカノイル基を示し;
mは1〜4の自然数を示し、nは2又は3の数を示す。)
【0026】
また本発明は、一般式(1)の前駆体(2)におけるイソキノリン骨格上に[11C]メチル基を導入してから塩基性条件下でトリハロアセチル基などの保護基を脱離させることを特徴とする一般式(1)の11C標識イソキノリン誘導体の短時間で高収率かつ容易な製造方法を提供する。
【0027】
また本発明は、一般式(1)の11C標識イソキノリン誘導体を含有するPET用プローブを提供する。
【0028】
さらに本発明は、一般式(1)の11C標識イソキノリン誘導体を生体に投与して当該生体内における11Cの分布を調べることを特徴とする組織の画像化方法を提供する。
【図面の簡単な説明】
【0029】
【図1】実施例3における反応ろ液の分取HPLCのチャートである。
【発明を実施するための形態】
【0030】
一般式(1)中、R1及びR2は、どちらか一方が[11C]メチル基を示し、他方は水素原子、シアノ基、アルキル基、ハロゲノアルキル基、アルケニル基、アルコキシ基、アルキルチオ基、ニトロ基、フルオロ基又はアリール基を示す。
【0031】
ここでアルキル基としては、炭素数1〜8の直鎖状、分枝鎖状、又は環状のアルキル基(C1-8アルキル基)(ここで「C1-8」とは炭素数が1〜8のいずれかであることを示す。以下同様)が挙げられ、炭素数1〜6のアルキル基が好ましく、さらに炭素数1〜3のアルキル基が好ましい。
具体例としては、メチル基、エチル基、n−プロピル基、イソプロピル基、n−ブチル基、イソブチル基、sec−ブチル基、tert−ブチル基、n−ペンチル基、イソペンチル基、n−ヘキシル基、イソヘキシル基、シクロプロピル基を挙げることができる。なかでも炭素数1〜3のものが好ましく、特にメチル基、エチル基が好ましい。
【0032】
ハロゲノアルキル基としては、ハロゲノC1-8アルキル基が好ましく、ハロゲノC1-6アルキル基がより好ましい。具体例としてはクロロメチル基、フルオロメチル基、クロロエチル基、フルオロエチル基、トリフルオロメチル基等が挙げられる。
【0033】
アルケニル基としては、炭素数2〜8の直鎖状又は分枝鎖状のアルケニル基(C2-8アルケニル基)が挙げられ、炭素数2〜6のアルケニル基が好ましい。具体例としてはビニル基、アリル基、イソプロペニル基、2−メタリル基、2−ブテニル基、3−ブテニル基を挙げることができる。なかでも炭素数2〜4のものが好ましい。
【0034】
アルコキシ基としては炭素数1〜8の直鎖状又は分枝鎖状のアルコキシ基(C1-8アルコキシ基)が挙げられ、炭素数1〜6のアルコキシ基が好ましい。具体例としては、メトキシ基、エトキシ基、n−プロポキシ基、イソプロポキシ基、n−ブトキシ基、イソブトキシ基、sec−ブトキシ基、tert−ブトキシ基を挙げることができる。
【0035】
アルキルチオ基としては、炭素数1〜8の直鎖状又は分枝鎖状のアルキルチオ基(C1-8アルキルチオ基)が挙げられ、炭素数1〜6のアルキルチオ基が好ましい。具体例としてはメチルチオ基、エチルチオ基、イソプロピルチオ基、n−プロピルチオ基等が挙げられる。
【0036】
アリール基としては、C6-14アリール基が挙げられ、フェニル基、ナフチル基が好ましく、フェニル基がより好ましい。
【0037】
R1及びR2としては、どちらか一方が[11C]メチル基を示し、他方は水素原子、C1-8アルキル基、ニトロ基、フルオロ基、シアノ基、ハロゲノC1-8アルキル基、フェニル基又はC2-8アルケニル基であるのが好ましい。また、[11C]メチル基の他方は水素原子、シアノ基、C1-6アルキル基又はハロゲノC1-6アルキル基であるのがより好ましい。さらに、[11C]メチル基の他方は水素原子又はC1-3アルキル基であるのが好ましい。
【0038】
R1はイソキノリン骨格の1位、3位及び4位のいずれに置換していてもよい。また、R2はイソキノリン骨格の6位、7位及び8位のいずれに置換していてもよい。ただし、[11C]メチル基の置換位置としてはイソキノリン環の4位が特に好ましい。
【0039】
R3及びR4は、それぞれ独立して水素原子、アルキル基、アルケニル基、アミノ基、アルキルアミノ基、ジアルキルアミノ基、アミノアルキル基、ハロゲノアルキル基、アルカノイル基、アミノアルカノイル基又はアルキルアミノアルカノイル基を示すか、あるいはR3とR4は一緒になってアルキレン基又はアルケニレン基を形成し、一般式(1)中の環状ジアミノ基における任意の2つの炭素間で架橋していてもよい。
【0040】
アルキル基、アルケニル基及びハロゲノアルキル基としては、前記R1及びR2の例として示したものが挙げられる。
アルキルアミノ基としては、C1-8アルキルアミノ基が好ましく、具体例としてはメチルアミノ基、エチルアミノ基、n−プロピルアミノ基、イソプロピルアミノ基、n−ブチルアミノ基、イソブチルアミノ基、sec−ブチルアミノ基、n−ペンチルアミノ基、n−ヘキシルアミノ基等が挙げられる。ジアルキルアミノ基としては、ジ−C1-8アルキルアミノ基が好ましく、具体例としてはジメチルアミノ基、ジエチルアミノ基、ジプロピルアミノ基、ジブチルアミノ基等が挙げられる。アミノアルキル基としては、アミノC1-8アルキル基が好ましく、具体例としてはアミノメチル基、アミノエチル基、アミノプロピル基、アミノブチル基等が挙げられる。
【0041】
アルカノイル基としては、炭素数2〜8の直鎖状又は分枝鎖状のアルカノイル基(C2-8アルカノイル基)が挙げられ、炭素数2〜6のアルカノイル基が好ましい。具体例としてはアセチル基、プロピオニル基、ブチリル基等が挙げられる。アミノアルカノイル基としては、アミノ−C2-8アルカノイル基が挙げられ、アミノ−C2-6アルカノイル基が好ましい。具体例としてはアミノアセチル基、アミノプロピオニル基、アミノブチリル基等が挙げられる。
【0042】
アルキルアミノアルカノイル基としては、C1-8アルキルアミノC2-8アルカノイル基が挙げられ、C1-4アルキルアミノC2-4アルカノイル基が好ましく、具体例としてはメチルアミノアセチル基、メチルアミノプロピオニル基等が挙げられる。アルコキシカルボニル基としては、C1-8アルコキシカルボニル基が挙げられ、例えばメトキシカルボニル基、エトキシカルボニル基が挙げられる。
【0043】
R3及びR4が一緒になって形成されるアルキレン基としては、C1-3アルキレン基、例えばメチレン基、エチレン基、トリメチレン基(−CH2CH2CH2−)が挙げられ、特にメチレン基、エチレン基が好ましい。R3及びR4が一緒になって形成されるアルケニレン基としては、C2-4アルケニレン基、例えば−CH=CH−、−CH2CH=CH−等が挙げられる。これらのアルキレン基又はアルケニレン基は、式(1)中の窒素含有飽和複素環上の任意の位置へ2つの炭素間で架橋してもよい。そのような架橋としては、架橋C1-3アルキレン基、すなわち−CH2−、−CH2CH2−、−CH2CH2CH2−による架橋が好ましい。
【0044】
R3及びR4としては、それぞれ独立して水素原子、C1-8アルキル基、C2-8アルケニル基、アミノ基、アミノC1-8アルキル基、ハロゲノC1-8アルキル基、C1-8アルキルアミノ基、ジ−C1-8アルキルアミノ基、C2-8アルカノイル基又はアミノC2-8アルカノイル基を示すか、あるいはR3とR4とが一緒になって架橋C1-3アルキレン基を形成するのが好ましい。
また、R3及びR4としては、それぞれ独立して水素原子、C1-8アルキル基、アミノ基、C1-8アルキルアミノ基、アミノC1-8アルキル基又はハロゲノC1-8アルキル基であるか、あるいはR3とR4とが一緒になって架橋C1-3のアルキレン基を形成するのがより好ましい。
さらにR3及びR4は水素原子、C1-6アルキル基又はハロゲノC1-6アルキル基であるのが好ましい。
【0045】
R5としては、水素原子、アミノアルカノイル基又はアルキルアミノアルカノイル基を示す。アミノアルカノイル基又はアルキルアミノアルカノイル基は前記と同じ。
【0046】
mは1〜4の自然数を示し、nは2又は3を示すが、mは1〜3の自然数が好ましく、また、nは2が特に好ましい。
【0047】
本発明の11C標識イソキノリン誘導体には、上記一般式(1)で示される化合物の他、その塩、水和物、溶媒和物及びプロドラッグが含まれる。ここで、プロドラッグとは、生体内で加水分解されて一般式(1)で示される化合物を生成する化合物をいう。こうしたプロドラッグには、当業者に知られたプロドラッグ化のすべての手法で製造される化合物が含まれる。例えば、アミノ基を生体内で容易に加水分解されうるアミド基等に誘導した化合物が挙げられる。具体的には一般式(1)に存在するアミノ基をアセトアミド等に誘導した化合物等が挙げられる。これらのプロドラッグは、上記一般式(1)の11C標識イソキノリン誘導体から短時間で容易に製造できるため、PET用プローブとしての適用に対しても、それほどの障害とはならない。
【0048】
また、薬学上許容される塩としては、一般式(1)に存在するアミノ基に対する酸付加塩が挙げられる。酸付加塩としては、例えば塩酸塩、臭化水素酸塩、リン酸塩、硫酸塩、硝酸塩等の無機酸塩、ギ酸塩、酢酸塩、プロピオン酸塩、マレイン酸塩、フマル酸塩、コハク酸塩、乳酸塩、リンゴ酸塩、酒石酸塩、クエン酸塩、アスコルビン酸塩、マロン酸塩、シュウ酸塩、グリコール酸塩、フタル酸塩、ベンゼンスルホン酸塩等の有機酸塩が挙げられ、また、上記の塩を組み合わせて用いることもできる。
【0049】
次に一般式(2)で表される化合物としては、式(2)中、R3-4,m及びnは、前記一般式(1)で表される11C標識イソキノリン誘導体において示したR3-4,m及びnと同じである。
【0050】
R8としては、tert−ブトキシカルボニル基、トリハロアセチル基、tert−ブトキシカルボニルアミノアルカノイル基、トリハロアセチルアミノアルカノイル基、N−アルキル−N−tert−ブトキシカルボニルアミノアルカノイル基又はN−アルキル−N−トリハロアセチルアミノアルカノイル基を示す。
【0051】
トリハロアセチル基としては、トリフルオロアセチル基、トリクロロアセチル基又はトリブロモアセチル基を用いることができるが、脱離容易なトリフルオロアセチル基が好ましい。
【0052】
トリハロアセチルアミノアルカノイル基とN−アルキル−N−トリハロアセチルアミノアルカノイル基としては、前記のアミノアルカノイル基又はアルキルアミノアルカノイル基のアミノ基をトリハロアセチル基で保護したものであり、トリハロアセチルアミノアルカノイル基としては、トリハロアセチルアミノ−C2-8アルカノイル基が挙げられ、トリハロアセチルアミノ−C2-6アルカノイル基が好ましい。具体例としてはトリフルオロアセチルアミノアセチル基、トリフルオロアセチルアミノプロピオニル基、トリフルオロアセチルアミノブチリル基等が挙げられる。N−アルキル−N−トリハロアセチルアミノアルカノイル基としては、N−C1-8アルキル−N−トリハロアセチルアミノC2-8アルカノイル基が挙げられ、N−C1-4アルキル−N−トリハロアセチルアミノC2-4アルカノイル基が好ましく、具体例としてはN−メチル−N−トリフルオロアセチルアミノアセチル基、N−メチル−N−トリフルオロアセチルアミノプロピオニル基等が挙げられる。
同様に、tert−ブトキシカルボニルアミノアルカノイル基とN−アルキル−N−tert−ブトキシカルボニルアミノアルカノイル基としては、前記のアミノアルカノイル基又はアルキルアミノアルカノイル基のアミノ基をtert−ブトキシカルボニル基で保護したものを示す。
【0053】
R6及びR7は、どちらか一方がトリアルキルスズ基、ボロニル基又はジアルコキシボリル基を示し、他方は水素原子、シアノ基、アルキル基、ハロゲノアルキル基、アルケニル基、アルコキシ基、アルキルチオ基、ニトロ基、フルオロ基又はアリール基を示す。
【0054】
R6はイソキノリン骨格の1位、3位及び4位のいずれに置換していてもよい。
また、R7はイソキノリン骨格の6位、7位及び8位のいずれに置換していてもよい。ただし、トリアルキルスズ基、ボロニル基又はジアルコキシボリル基の置換位置としてはイソキノリン環の4位が特に好ましい。
【0055】
R6及びR7としては、どちらか一方がトリアルキルスズ基、ボロニル基又はジアルコキシボリル基を示し、他方は水素原子、C1-8アルキル基、ニトロ基、フルオロ基、シアノ基、ハロゲノC1-8アルキル基、フェニル基又はC2-8アルケニル基であるのが好ましい。また、トリアルキルスズ基、ボロニル基又はジアルコキシボリル基の他方は水素原子、シアノ基、C1-6アルキル基又はハロゲノC1-6アルキル基であるのがより好ましい。さらに、トリアルキルスズ基、ボロニル基又はジアルコキシボリル基の他方は水素原子又はC1-3アルキル基であるのが好ましい。
【0056】
トリアルキルスズ基としては、トリC1-6アルキルスズ基が挙げられ、トリC2-4アルキルスズ基が好ましく、具体例としてはトリエチルスズ基、トリプロピルスズ基又はトリブチルスズ基が挙げられる。
【0057】
ジアルコキシボリル基としては、ジC1-6アルコキシボリル基が挙げられ、ジC1-3アルコキシボリル基が好ましく、具体例としてはジメトキシボリル基、ジエトキシボリル基又はジプロピルキシボリル基が挙げられる。また、二つのアルコキシ基は一緒になってアルキレン又はフェニレンを形成してもよく、具体例としては4,4,5,5−テトラメチル−1,3,2−ジオキサボロラン−2−イル基、4,4,6,6−テトラメチル−1,3,2−ジオキサボリナン−2−イル基又はベンゾ[d][1,3,2]ジオキサボロル−2−イル基が挙げられる。
【0058】
<製造法>
前記一般式(1)で表される11C標識イソキノリン誘導体は、後述する方法で製造された化合物(3)から、前記一般式(2)で表される化合物を介して、例えば、スキーム1に示す方法に従って製造することができる。
【0059】
【化3】
【0060】
(式中、R9及びR10は、どちらか一方がハロゲン原子を示し、他方は水素原子、シアノ基、アルキル基、ハロゲノアルキル基、アルケニル基、アルコキシ基、アルキルチオ基、ニトロ基、フルオロ基又はアリール基を示す。
R1-8、m及びnは前記と同じ。)
【0061】
[第1工程]
化合物(4)は、化合物(3)に適当な溶媒中、塩基存在下に、ジ−tert−ブチルジカーボネート等のtert−ブトキシカルボニル化剤や、ハロゲン化トリハロアセチル又は無水トリハロ酢酸等のトリハロアセチル化剤を反応させることにより製造することができる。
【0062】
反応溶媒としては、反応に支障のないものであればよく、例えば、テトラヒドロフラン、ジオキサン、ジエチルエーテル等のエーテル類、ベンゼン、トルエン等の炭化水素類、塩化メチレン、クロロホルム等のハロゲン化炭化水素類、N,N−ジメチルホルムアミド、N,N−ジメチルアセトアミド等の非プロトン性溶媒、ピリジン、アセトニトリル、アセトン、水又はこれらの混合物を用いることができる。
【0063】
本反応においては、適当な塩基の存在下に行うのが好ましい。かかる塩基としては、アルカリ金属炭酸水素塩(例、炭酸水素ナトリウム)、アルカリ金属炭酸塩(例、炭酸カリウム)、アルカリ金属水酸化物(例、水酸化ナトリウム、水酸化カリウム)のようなアルカリ、トリエチルアミン、トリエチレンジアミン等の有機第3級アミンを用いることができる。溶媒としてピリジンのような塩基性溶媒を使用すれば、かかる塩基は不要であり、好ましい。
【0064】
通常、本反応は室温で進行する場合が多いが、必要に応じて冷却又は加熱して、−78〜150℃、好ましくは、0〜120℃で行うことができる。反応時間は、使用する原料、溶媒、反応温度等によって異なるが、通常、5分〜70時間である。
【0065】
[第2工程]
化合物(4)のハロゲン原子を、それ自体公知の方法で、トリアルキルスズ基、ボロニル基又はジアルコキシボリル基に変換して、化合物(2)を製造することができる。
すなわち、一般式(4)で表されるハロゲン体を、テトラヒドロフラン、ジオキサン、ジエチルエーテル等のエーテル類の溶媒中で、マグネシウムを用いるグリニャール試薬化やアルキルリチウムなどの有機金属試薬を用いるリチオ化などにより活性化させ、続いてR6及びR7に対応する塩化トリアルキルスズ又は有機ホウ素化合物で処理することにより化合物(2)で表されるトリアルキルスズ体又はボロニル体を製造することができる。また、一般式(4)で表されるハロゲン体を、パラジウム触媒存在下にビス(トリアルキルスズ)体あるいはジボラン体を用いたスズ化あるいはホウ素化反応により一般式(2)で表されるトリアルキルスズ体、ボロニル体、ジアルコキシボリル体を製造することができる。本反応は、公知の方法に準じて行うことができる。
【0066】
R9及びR10で表されるハロゲンとしては、塩素、臭素、またはヨウ素が好ましい。
【0067】
[第3工程]
化合物(2)で表されるトリアルキルスズ体、ボロニル体又はジアルコキシボリル体を、非プロトン性極性溶媒中で、配位子、パラジウム錯体及び塩基の存在下に、[11C]ヨウ化メチルを反応させることにより、化合物(5)で表される[11C]メチル体を製造することができる。
[11C]メチル化の方法としては、前述した特許文献1、2又は3の方法を用いることができる。
【0068】
反応溶媒としては、ベンゼン、トルエン等の炭化水素類、塩化メチレン、クロロホルム等のハロゲン化炭化水素類、N,N−ジメチルホルムアミド、N,N−ジメチルアセトアミド、N−C1-6アルキル−2−ピロリジノン等の非プロトン性極性溶媒、ピリジン、アセトニトリル、水、又はこれらの混合物を用いることができる。なかでも、N,N−ジメチルホルムアミドやN−C1-6アルキル−2−ピロリジノンなどの非プロトン性極性溶媒が好ましい。特に好ましくは、N−C1-3アルキル−2−ピロリジノンであり、具体的にはN−メチル−2−ピロリジノン(NMP)、N−エチル−2−ピロリジノン又はN−プロピル−2−ピロリジノンが挙げられる。
【0069】
配位子としては、トリアルキルホスフィンやトリアリールホスフィンなどのホスフィン配位子又はトリアリールアルシンなどのアルシン配位子を用いることができる。通常、この反応は、配位子の嵩高さが活性の高い反応場を形成するため、特にトリ−o−トリルホスフィンや(ジ−tert−ブチル)メチルホスフィンなどのホスフィン配位子が好ましい。
【0070】
パラジウム錯体としては、通常知られているテトラキストリフェニルホスフィンパラジウムなどの0価のパラジウム錯体や塩化パラジウムや酢酸パラジウムなどの2価のパラジウム錯体を用いることができる。なかでも0価のパラジウム錯体が好ましいが、とくにトリス(ジベンジリデンアセトン)ジパラジウムが好ましい。
【0071】
塩基としては、アルカリ金属炭酸水素塩(例、炭酸水素ナトリウム)、アルカリ金属炭酸塩(例、炭酸カリウム、炭酸セシウム)、アルカリ金属リン酸塩(例、リン酸カリウム)、アルカリ金属水酸化物(例、水酸化ナトリウム、水酸化カリウム)のような無機塩基、トリエチルアミン、エチルジイソプロピルアミン等の有機第3級アミンを用いることができる。
【0072】
[第4工程]
11C標識イソキノリン誘導体(1)は、化合物(5)のtert−ブトキシカルボニル基又はトリハロアセチル基を酸処理やアルカリ処理により除去して製造することができる。反応はそれ自体公知の方法に従えばよい。例えば、tert−ブトキシカルボニル基の場合、適当な反応溶媒中で、塩酸やトリフルオロ酢酸等の酸を反応させることにより除去することができる。また、トリハロアセチル基の場合、アルカリ金属炭酸水素塩、アルカリ金属炭酸塩、アルカリ金属水酸化物等のような無機塩基を用いた加水分解反応あるいは加溶媒分解反応により除去することができる。
【0073】
スキーム1で示した化合物(3)は、特許文献7〜9に記載の方法、又はそれらの類似の方法によって合成することができる。また、以下のスキーム2〜6に示す方法によって合成することもできる。
【0074】
【化4】
【0075】
(式中、L1は水酸基又は脱離基を示し、R11は、Nの保護基を示す。また、R3、R4、R9、R10、m及びnは前記と同じ。)
【0076】
脱離基としてのL1は、後記するスルホン酸の反応性誘導体の残基を挙げることができる。Nに用いられる保護基としては、例えば、ホルミル、アセチル、ベンゾイル等のアシル基、ベンジルオキシカルボニル等のアラルキルオキシカルボニル基、tert−ブチルオキシカルボニル等のアルコキシカルボニル基、ベンジル等のアラルキル基を挙げることができる。
【0077】
一般式(7)で表されるアミンを適当な溶媒中、一般式(6)で表されるスルホン酸、又はその反応性誘導体と反応させ、必要により保護基を除去して化合物(3a)を製造することができる。
【0078】
スルホン酸の反応性誘導体としては、スルホン酸ハライド(例、スルホン酸フルオライド、スルホン酸クロライド、スルホン酸ブロマイド)、スルホン酸無水物、N−スルホニルイミダゾリド等が用いられる。特にスルホン酸ハライドが好ましい。
【0079】
【化5】
【0080】
(式中、L2は脱離基を示し、R3、R4、R9、R10、m及びnは前記と同じ。)
【0081】
脱離基としてのL2は、塩素若しくは臭素等のハロゲン又はアセチルオキシ等のアシルオキシ基、若しくはメシルオキシ、トシルオキシ等のスルホニルオキシ基を挙げることができる。
化合物(8)をアミン、グアニジン又はアンモニアと反応させることにより化合物(3a)を製造することができる。本反応は、公知の方法(Acta Chemica Scand., 45,621(1991))に準じて行うことができる。
【0082】
【化6】
【0083】
(式中、R12は水素原子又はアミノ基の保護基を示し、L2、R3、R4、R9、R10、m及びnは前記と同じ。)
【0084】
化合物(9)と化合物(10)とを反応させ、必要により、酸又はアルカリ処理して保護基を除去することにより化合物(3a)を製造することができる。本反応は、公知の方法(Acta Chemica Scand., 45, 621(1991))に準じて行うことができる。
【0085】
【化7】
【0086】
(式中、R12は水素原子又はアミノ基の保護基を示し、L2、R3、R4、R9、R10、m及びnは前記と同じ。)
化合物(3a)は、化合物(11)をトリフェニルホスフィンなどの有機リン試薬とアゾジカルボン酸ジエチル又はアゾジカルボン酸ジイソプロピルなどのアゾ試薬を用いて分子内脱水反応(いわゆる光延反応)を行い、続いて保護基R12を除去することによって製造することができる。
【0087】
【化8】
【0088】
(式中、L3は水酸基又は脱離基を示し、R13は水素原子又はC1-7アルキル基を示し、lは1〜7の自然数を示す。また、R3、R4、R9、R10、R12、m及びnは前記と同じ。)
【0089】
一般式(3a)で表されるアミンを、適当な縮合剤の存在下に一般式(12)で表されるカルボン酸に反応させ、必要により保護基を除去して化合物(3b)を製造することができる。又は一般式(3a)を適当な塩基存在下にカルボン酸の反応性誘導体と反応させ、化合物(3b)を製造する。
【0090】
縮合剤としては、ジシクロヘキシルカルボジイミドや1−エチル−3−(3−ジメチルアミノプロピル)カルボジイミド塩酸塩などのカルボジイミド縮合剤や1H−ベンゾトリアゾール−1−イルオキシトリピロリジノホスホニウム及び、ヘキサフルオロホスフェートなどのホスホニウム縮合剤などが用いられる。
【0091】
脱離基としてのL3は、カルボン酸の反応性誘導体の残基を挙げることができる。カルボン酸の反応性誘導体としては、カルボン酸ハライド(例、カルボン酸クロライド、カルボン酸ブロマイド)、カルボン酸無水物、カルボン酸アジド、活性エステルが用いられる。
【0092】
11C標識イソキノリン誘導体(1)には、不斉炭素を有し、光学異性体が存在するものもある。これらの各異性体及びこれらの混合物のいずれも本発明に包含される。光学異性体は、光学活性な原料化合物(S配置又はR配置)を用いることにより得ることができる。またラセミ体はそのままでも薬理活性を有するが、所望によりそれぞれの異性体に分割することができる。例えば、異性体混合物を公知の光学分割法、例えば、光学活性なカルボン酸(例、(+)−又は(−)−酒石酸、(+)−又は(−)−リンゴ酸)又は光学活性なスルホン酸(例、(+)−ショウノウスルホン酸)との塩を生成させ、分別結晶する方法、光学活性カラムを用いる方法によって分離することができる。
11C標識イソキノリン誘導体(1)は、公知の方法により、塩を形成させることができる。例えば塩酸塩は、本発明化合物を塩化水素のアルコール溶液又はエチルエーテル溶液に溶解することにより得ることができる。
【0093】
本発明化合物である11C標識イソキノリン誘導体(1)は、後述するように、PET用分子プローブとして好適に用いることができる。その際の投与経路としては、経口でも、非経口でも投与することができる。投与剤型として錠剤、カプセル剤、顆粒剤、散剤、注射剤、点眼剤等が挙げられ、それらは汎用される技術を組み合わせて使用することができる。ただし、11Cの半減期を考慮すると、製剤化するのに時間を要しない注射剤や点眼剤が好ましい。
注射剤、又は点眼剤とする場合は、液状媒体として例えば滅菌精製水、生理食塩水、等張液等の水性媒体、又は例えばゴマ油、大豆油、オリーブ油等の非水性媒体が用いられる。水性媒体と油性媒体の両者を含有するエマルジョンでもよい。
注射剤、点眼剤等の非経口剤は、例えば、グリセリン、プロピレングリコール、塩化ナトリウム、塩化カリウム、ソルビトール、マンニトール等の等張化剤、リン酸、リン酸塩、クエン酸、氷酢酸、ε−アミノカプロン酸、トロメタモール等の緩衝剤、塩酸、クエン酸、リン酸、氷酢酸、水酸化ナトリウム、水酸化カリウム、炭酸ナトリウム、炭酸水素ナトリウム等のpH調節剤、ポリソルベート80、ポリオキシエチレン硬化ヒマシ油60、マクロゴール等の溶解補助剤、精製卵黄レシチン、精製大豆レシチン、ポリオキシエチレン(160)ポリオキシプロピレン(30)グリコール等の乳化剤、ヒドロキシプロピルメチルセルロース、ヒドロキシプロピルセルロース等のセルロース系高分子、ポリビニルアルコール、ポリビニルピロリドン等の増粘剤、エデト酸、エデト酸ナトリウム等の安定化剤、汎用のソルビン酸、ソルビン酸カリウム、塩化ベンザルコニウム、塩化ベンゼトニウム、パラオキシ安息香酸メチル、パラオキシ安息香酸プロピル、クロロブタノール等の保存又は防腐剤、クロロブタノール、ベンジルアルコール、リドカイン等の無痛化剤を必要に応じて本発明化合物に組合わせて、調製することができる。
【0094】
尚、注射剤又は点眼剤の場合、pHは4.0〜8.0に設定することが望ましく、また、浸透圧比は1.0付近に設定することが望ましい。
【0095】
このPET用プローブを放射性医薬品や診断薬組成物等として利用することにより、PET画像によるプロテインキナーゼ阻害剤の解析が可能となり、脳内動態解析や薬剤(治療薬)としての潜在性を解析することが可能となる。さらには、生物学的研究や動物・ヒトのPETによる臨床研究の基盤技術とすることができる。
【0096】
すなわち、本発明の11C標識イソキノリン誘導体を生体に投与して該生体内における11Cの分布を調べることにより、組織の画像化を行うことができる。11Cの分布は陽電子を検出するPET法の手法を用いることができる。こうした組織の画像化の対象として例えば脳を選べば、脳内のプロテインキナーゼと結合した11C標識イソキノリン誘導体を捉えることが可能となり、ひいては脳内のプロテインキナーゼの働き等についての多くの知見を得ることができる。
【0097】
例えばH-1152PはRhoキナーゼの特異的阻害剤であることから、その11C体である本発明化合物を実験用ラット等に対し静脈注射等によって投与した後、例えば脳のPET画像を経時的に撮影することにより、脳内のRhoキナーゼの分布や、脳内のRhoキナーゼと結合した11C標識イソキノリン誘導体を捉えることが可能となり、脳内のRhoキナーゼの働き等についての多くの知見を得ることができる。
【0098】
さらに、本発明の11C標識イソキノリン誘導体を含有するPET用製剤をヒトに投与した後撮影されたPET画像は、プロテインキナーゼの分布の異常を伴う疾患やプロテインキナーゼに関連する疾患の診断に用いることができる。そのプロテインキナーゼには、Rhoキナーゼ、PKA、PKG、AURKA、CAMK2などが含まれるが、これらに限定されない。
【0099】
例えばH-1152PはRhoキナーゼの特異的阻害剤であることから、その11C体である本発明化合物をヒトに投与した後撮影されたPET画像は、Rhoキナーゼの分布の異常を伴う疾患やRhoキナーゼに関連する疾患の診断に用いることができる。
Rhoキナーゼに関連する疾患には、脳血管攣縮、冠動脈攣縮、高血圧、肺性高血圧、喘息、緑内障、腎障害、狭心症、心筋梗塞、動脈硬化症、再狭窄、脳卒中、心不全、心筋肥大、虚血再還流障害、血管内皮障害、炎症性腸疾患、前立腺肥大、レイノー病、神経因性疼痛、Alzheimer病、Huntington舞踏病、脊髄損傷、てんかん、多発性硬化症、骨粗しょう症、勃起不全、肝障害、がん、創傷治癒、インスリン抵抗性、及び糖尿病が含まれる。上記のがんには、肉腫、Glioblastoma、神経膠腫、頭頸部がん、肝細胞がん、腎明細胞がん、肺がん、卵巣がん、乳がん、食道がん、大腸がん、膀胱がん、前立腺がん、及び悪性黒色腫が含まれる。
【0100】
また、PETによる11C標識イソキノリン誘導体の体内での検出は極めて高感度であるため、これをマイクロドーズ臨床試験に適用することもできる。本臨床試験は、超微量(マイクロドーズ)の薬剤候補物質を人体に投与して、高感度微量分析を用いて薬物動態などを解析する方法であり、超微量での薬理動態を観察することから、人体に対する影響が少ない。このため、薬理試験の安全性が極めて高く、マイクロドーズ臨床試験ではフェーズI に入る前の段階で、医薬品の候補物質を安全に早く人に適用することにより、人体における薬物動態を知り、薬物動態面からの開発候補物質のスクリーニングを行うことができ、非臨床試験期間を大幅に短縮することができる。
【実施例】
【0101】
以下に実施例により本発明を具体的に説明する。尚、それらの例示は本発明をよりよく理解するためのものであり、本発明の範囲を限定するものではない。
【0102】
参考例1
4−トリフルオロアセチル−{(S)−ヘキサヒドロ−2−メチル−1−(4−ブロモ−5−イソキノリンスルホニル)}−1H−1,4−ジアゼピン(b)の合成
【0103】
【化9】
【0104】
特許文献8の方法に従い合成した(S)−ヘキサヒドロ−2−メチル−1−(4−ブロモ−5−イソキノリンスルホニル)−1H−1,4−ジアゼピン(a)をトリフルオロアセチル基により保護した。
すなわち、(a)(246.6 mg, 641.4 μmol) の乾燥ジクロロメタン溶液 (2 mL) にトリエチルアミン (134 μL, 97.3 mg, 962 μmol) を添加し、0℃に冷却したのち、トリフルオロ酢酸無水物 (134 μL, 202 mg, 962 μmol) を添加した。この混合溶液を0℃で2時間攪拌したのち、反応混合物を蒸留水(約10 mL)に加え、ジクロロメタン (3×15 mL) で抽出した。有機層を蒸留水 (15 mL)、飽和食塩水 (15 mL) で順次洗浄後、無水硫酸ナトリウムで乾燥した。乾燥剤をろ別し、ろ液を減圧下で濃縮した。得られた残査はシリカゲルカラムクロマトグラフィー (シリカゲル 5 g, 展開溶媒;ヘキサン:アセトン (2:1)) で精製し、淡黄色の油状化合物(b)(266 mg, 554 μmol, 86.4 %)を得た。
1H NMR (500 MHz, CDCl3, 25℃): δ=9.21 (s, 1H), 8.99 (s, 1H), 8.50 (dd, 4J(H,H)=1.0 Hz, 3J(H,H)=7.5 Hz, 1H), 8.21 (dd, 4J(H,H)=1.0 Hz, 3J(H,H)=8.0 Hz, 1H), 7.67-7.71 (m, 1H), 3.90-4.36 (m, 4H), 3.33-3.66 (m, 3H), 2.18-2.32 (m, 1H), 1.87-2.04 (m, 1H), 1.06 ppm (d, 3J(H,H)=7.0 Hz, 3H); HRMS (EI) m/z: [M+H]+ calcd for C9H7BrN 207.9762 found 207.9745; Anal. Calcd for C17H17BrF3N3O3S: C, 72.51; H, 3.57; N, 8.75. Found: C, 72.74; H, 3.79; N, 8.70.
【0105】
実施例1
<有機スズ前駆体の合成>
4−トリフルオロアセチル−[(S)−ヘキサヒドロ−2−メチル−1−{4−(トリ−n−ブチルスタニル)−5−イソキノリンスルホニル}]−1H−1,4−ジアゼピン(c)の合成
【0106】
【化10】
【0107】
化合物(b)から有機スズ前駆体(c)を合成した。
すなわち、アルゴン気下、乾燥20mLシュレンク型反応管に(b)(99.4 mg, 0.205 mmol)と乾燥THF (2.5 mL) を加えた。この混合溶液を-100℃に冷却したのち、tert−ブチルリチウム (279μL, 1.47 M ヘキサン溶液, 0.410 mmol) を20分間かけて滴下し、同じ温度で2時間撹拌した。この反応溶液に、ヘキサメチルホスホリックトリアミド(HMPA) (214 μL, 220 mg, 1.23 mmol)のTHF溶液(1.0 mL)と(n-C4H9)3SnCl (317 μL, 381 mg, 1.17 mmol)のTHF溶液(1.0 mL)を順に加え、室温まで徐々に昇温し、10時間撹拌した。得られた反応溶液を冷やした飽和塩化アンモニウム水溶液 (約2 mL) に加え、酢酸エチル (3×10 mL)で抽出した。有機層を飽和食塩水(20 mL)で洗浄後、無水硫酸ナトリウムで乾燥した。乾燥剤をろ別し、ろ液を減圧下で濃縮した。残査はシリカゲルカラムクロマトグラフィー (シリカゲル 10 g, 展開溶媒;ヘキサン:アセトン (2:1))で精製し、オレンジ色の油状化合物(c)(96.1 mg, 0.138 mmol, 67.5%) を得た。
1H NMR (400 MHz, CDCl3, 25℃): δ=9.15 (s, 1H), 8.74 (s, 1H), 8.25 (dd, 4J(H,H)=1.4, 3J(H,H)=7.5 Hz, 1H), 8.12 (dd, 4J(H,H)=1.2, 3J(H,H)=8.0 Hz, 1H), 7.61 (m, 1H), 4.16-4.21 (m, 1H), 2.91-3.87 (m, 6H), 1.52-1.71 (m, 2H), 1.44-1.50 (m, 6H), 1.24-1.34 (m, 6H), 1.10-1.15 (m, 6H), 0.84 (d, 3J(H,H)=5.3 Hz, 3H), 0.83 ppm (t, 3J(H,H)=7.4 Hz, 9H); HRMS (EI+, 100% acetone): m/z: calcd for C25H35F3N3O3SSn ([M-C4H9]+) 634.1376; found, 634.1359.
【0108】
実施例2
<有機ボロン酸エステル前駆体の合成>
(S)−2,2,2−トリフルオロ−1−[3−メチル−4−{4−(4,4,5,5−テトラメチル−1,3,2−ジオキサボラン−2−イル)イソキノリン−5−イルスルホニル}−1,4−ジアゼパン−1−イル]エタンの合成
特許文献3に記載の方法に従って化合物(b)から(S)−2,2,2−トリフルオロ−1−[3−メチル−4−{4−(4,4,5,5−テトラメチル−1,3,2−ジオキサボラン−2−イル)イソキノリン−5−イルスルホニル}−1,4−ジアゼパン−1−イル]エタンを合成することができる。
【0109】
実施例3
<11C標識イソキノリン誘導体(d)の合成1>
(S)−ヘキサヒドロ−2−メチル−1−[(4−[11C]メチル−5−イソキノリニル)スルホニル]−1H−1,4−ジアゼピン(d)の合成
【0110】
実施例1の化合物(c)を用いて、下記合成方法に基づき、イソキノリン骨格の4位を[11C]メチル化してから塩基性条件下でトリフルオロアセチル基を脱離させることによりイソキノリン骨格の4位のメチル基を[11C]メチル化した11C標識イソキノリン誘導体(d)を得た。詳細を以下に示す。
【0111】
【化11】
【0112】
11C核の製造は住友重機械工業社製サイクロトロンCYPRIS HM-12Sを使用し、14N(p,α)11Cの核反応により製造した。[11C]ヨウ化メチルの合成は専用の標識用合成装置を用いて、11CO2ガスを出発物質として、11CO2→11CH3OH→11CH3Iの順に変換して合成した。
【0113】
一方、トリス(ジベンジリデンアセトン)ジパラジウム(Pd2(dba)3)(1.8 mg, 1.9 μmol)および、トリ-o-トリルホスフィン(P(o-tolyl)3)(9.4 mg, 30.9μmol)のNMP(N-メチルピロリジノン)溶液(0.27 mL)を標識用合成装置の反応容器(A)に入れ、室温に設置した。反応容器(A)中の溶液は、[11C]ヨウ化メチル(11CH3I)を吹き込む10-20分前に設置した。また、有機スズ前駆体(c)(4.0 mg, 5.8μmol)、CuBr(0.5 mg, 3.9 μmol)、およびCsF(1.5 mg, 9.7 μmol)のNMP溶液(80 μL)を反応容器(B)に準備し、室温に設置した。続いて反応容器(A)に[11C]ヨウ化メチルを60-80 mL/minのガス流量で吹き込み、その後1分間静置した。得られた溶液を反応容器(B)に移送し、さらに反応容器(A)の内部を少量のNMP溶液(60 μL)を用いて洗浄して、この洗浄液も反応容器(B)に移送した。続いて、反応容器(B)中の混合溶液を80℃で5分間加熱した。続いて、2M NaOH (400μL)を加えて100℃で1分間加熱した。得られた反応溶液をメタノール(500μL)とリン酸ナトリウム緩衝液(10 mM、pH7.4, 500μL)で希釈した後、綿栓あるいはフィルターを用いてろ過した。ろ液を分取HPLCに供し(分取HPLCのチャートを図1に示した)、分取した11C-標識化合物(d)を含むフラクションをエバポレーターを用いて減圧濃縮した。濃縮液は、生理食塩水を主とする臨床用投与溶液を用いて希釈し、無菌バイヤルに入れた。本溶液の一部(20μL)を分析HPLCに供して、目的化合物の同定、純度検定、および比放射能の算出を行った。なお、11C-標識化合物(d)の同定は非標識体の化合物(d)を用い、下記の収率や分析値などは3回の実験から得られた結果である。
【0114】
11C-標識化合物(d)の総放射能:3.8±1.2 GBq、[11C]ヨウ化メチルに基づく崩壊補正放射化学収率:63±14%、合成時間:38分、放射化学的純度:99%以上、化学的純度:99%、比放射能: 97±10 GBq/μmol
分取条件:分取用カラムはWaters 社製XBridge, 5μm, C18 10mm×250mm、流速は5mL/minで、移動層はCH3OH:10mM リン酸ナトリウム緩衝液(pH=7.4) =50:50を使用した。UV検出波長254 nmおよびγ線検出器で測定した結果、11C-標識化合物(d)の保持時間は約9.2分であった。
分析条件:カラムはPhenomenex 社製Gemini NX, 5μm, 4.6mm×150mm を使用し、カラム温度30℃で分析を行った。流速は1mL/minで、移動層はCH3OH:10mM リン酸ナトリウム緩衝液(pH=7.4) =50:50を使用した。UV検出波長215 nmおよびγ線検出器で測定した結果、11C-標識化合物(d)の放射化学的純度および化学的純度はどちらも99%以上であった。
【0115】
<実験装置、実験方法及び使用した試薬>
標識用合成装置は、理化学研究所分子イメージング科学研究センターに設置してある標準型標識用自動合成装置を用いた。化学薬品は市販のものをそのまま用いた。脱水N-メチル-2-ピロリジノン(NMP)(関東化学社製)、トリス(ジベンジリデンアセトン)ジパラジウム(0)(Aldrich社製)、トリ-o-トリルホスフィン(Aldrich社製)、臭化銅(和光社製)、フッ化セシウム(和光社製)を使用した。
【0116】
実施例4
<11C標識イソキノリン誘導体(d)の合成2>
(S)−ヘキサヒドロ−2−メチル−1−[(4−[11C]メチル−5−イソキノリニル)スルホニル]−1H−1,4−ジアゼピンの合成
実施例2に記載した化合物から化合物(d)を得ることができる。
すなわち、非プロトン性極性溶媒中において、有機ボロン酸化合物のボロン酸エステルを、パラジウム錯体、ホスフィン配位子及び塩基の存在下で[11C]ヨウ化メチルとクロスカップリングさせることによって11C標識イソキノリン誘導体(d)を製造することができる。
【0117】
以下の実施例5〜10の化合物は、参考例1及び実施例1〜4に記載の方法を用いることで合成することができる。
実施例5
2,2,2−トリフルオロ−1−[4−{4−(トリブチルスタニル)イソキノリン−5−イルスルホニル}−1,4−ジアゼパン−1−イル]エタン
実施例6
2,2,2−トリフルオロ−1−[4−{4−(トリブチルスタニル)イソキノリン−5−イルスルホニル}ピペラジン−1−イル]エタン
実施例7
(S)−2,2,2−トリフルオロ−N−(2−[3−メチル−4−{4−(トリブチルスタニル)イソキノリン−5−イルスルホニル}−1,4−ジアゼパン−1−イル]−2−オキソエチル)アセタミド
実施例8
5−(1,4−ジアゼパン−1−イルスルホニル)−4−[11C]メチルイソキノリン
実施例9
4−[11C]メチル−5−(ピペラジン−1−イルスルホニル)イソキノリン
実施例10
(S)−2−アミノ−1−{3−メチル−4−(4−[11C]メチルイソキノリン−5−イルスルホニル)−1,4−ジアゼパン−1−イル}エタノン
【0118】
実施例11
<PET用製剤例>
実施例4で分取し濃縮して得た化合物(d)の油状物を例えばポリソルベート80含有生理食塩水に溶解し、さらに生理食塩水にて希釈することで静脈注射PET用製剤を調製することができる。
【0119】
実施例12
<PET撮影>
本発明の11C標識イソキノリン誘導体について、これをPET用プローブとしてPET撮影を行うためには、例えば次のような操作を行えばよい。
すなわち、HPLCによって本発明の11C標識イソキノリン誘導体(d)を分取し、実施例11に記載の製剤を調製し、これをPET用プローブとして実験用ラット等に対し静脈注射等によって投与する。そして、例えば小動物用PET装置(microPET、SIMENS社製)を用いて連続撮像を行う。さらに、非RI標識体による結合阻害効果を検討するために、非標識のイソキノリン誘導体を生理食塩水で希釈した溶液を実験用ラット等に対し静脈注射等によって投与する。また、PET撮像終了後、組織の切片を作製し、Ex vivoオートラジオグラム実験をおこなって、その分布を調べる。こうして、さまざまなPET画像及びその経時的な変化を調べることができる。
【特許請求の範囲】
【請求項1】
一般式(1)又はその薬学上許容される塩、水和物、溶媒和物若しくはプロドラッグからなる11C標識イソキノリン誘導体。
【化1】
(式中、
R1及びR2は、どちらか一方が[11C]メチル基を示し、他方は水素原子、シアノ基、アルキル基、ハロゲノアルキル基、アルケニル基、アルコキシ基、アルキルチオ基、ニトロ基、フルオロ基又はアリール基を示し;
R3及びR4はそれぞれ独立して水素原子、アルキル基、アルケニル基、アミノ基、アルキルアミノ基、ジアルキルアミノ基、アミノアルキル基、ハロゲノアルキル基、アルカノイル基、アミノアルカノイル基又はアルキルアミノアルカノイル基を示すか、あるいはR3及びR4は一緒になってアルキレン基又はアルケニレン基を形成し、一般式(1)中の環状ジアミノ基における任意の2つの炭素間で架橋しているか、のいずれかであり;
R5は水素原子、アミノアルカノイル基又はアルキルアミノアルカノイル基を示し;
mは1〜4の自然数を示し、nは2又は3を示す。)
【請求項2】
R1がイソキノリン環の4位に結合する[11C]メチル基である請求項1記載の11C標識イソキノリン誘導体。
【請求項3】
(S)−ヘキサヒドロ−2−メチル−1−[(4−[11C]メチル−5−イソキノリニル)スルホニル]−1H−1,4−ジアゼピン、5−(1,4−ジアゼパン−1−イルスルホニル)−4−[11C]メチルイソキノリン、4−[11C]メチル−5−(ピペラジン−1−イルスルホニル)イソキノリン及び(S)−2−アミノ−1−{3−メチル−4−(4−[11C]メチルイソキノリン−5−イルスルホニル)−1,4−ジアゼパン−1−イル}エタノンからなる群から選択される化合物又はその薬学上許容される塩、水和物、溶媒和物若しくはプロドラッグ。
【請求項4】
請求項1〜3のいずれか1項記載の11C標識イソキノリン誘導体を含有するPET用分子プローブ。
【請求項5】
請求項1に記載の11C標識イソキノリン誘導体を製造するための化合物であって、下記一般式(2)からなる化合物。
【化2】
(式中、
R6及びR7は、どちらか一方がトリアルキルスズ基、ボロニル基又はジアルコキシボリル基を示し、他方は水素原子、シアノ基、アルキル基、ハロゲノアルキル基、アルケニル基、アルコキシ基、アルキルチオ基、ニトロ基、フルオロ基又はアリール基を示し;
R3及びR4はそれぞれ独立して水素原子、アルキル基、アルケニル基、アミノ基、アルキルアミノ基、ジアルキルアミノ基、アミノアルキル基、ハロゲノアルキル基、アルカノイル基、アミノアルカノイル基又はアルキルアミノアルカノイル基示すか、あるいはR3及びR4は一緒になってアルキレン基又はアルケニレン基を形成し、一般式(2)中の環状ジアミノ基における任意の2つの炭素間で架橋しているかのいずれかであり;
R8はtert−ブトキシカルボニル基、トリハロアセチル基、tert−ブトキシカルボニルアミノアルカノイル基、トリハロアセチルアミノアルカノイル基、N−アルキル−N−tert−ブトキシカルボニルアミノアルカノイル基又はN−アルキル−N−トリハロアセチルアミノアルカノイル基を示し;
mは1〜4の自然数を示し、nは2又は3を示す。)
【請求項6】
R6がイソキノリン環の4位に結合するトリアルキルスズ基、ボロニル基又はジアルコキシボリル基であり、R8がトリハロアセチル基、トリハロアセチルアミノアルカノイル基、又はN−アルキル−N−トリハロアセチルアミノアルカノイル基である請求項5記載の化合物。
【請求項7】
R6がイソキノリン環の4位に結合するトリアルキルスズ基、ボロニル基又はジアルコキシボリル基であり、R3が水素原子又はメチル基であり、R4及びR7が水素原子であり、R8がトリハロアセチル基、トリハロアセチルアミノアルカノイル基、又はN−アルキル−N−トリハロアセチルアミノアルカノイル基であり、mが1又は2、nが2である請求項5記載の化合物
【請求項8】
請求項5〜7のいずれか1項記載の化合物のイソキノリン環上に[11C]メチル基を導入し、後続してR8を塩基性条件下で脱離させることを特徴とする請求項1〜3のいずれか1項記載の化合物の製造方法。
【請求項9】
請求項1〜3のいずれか1項記載の11C標識イソキノリン誘導体の製造のための請求項5〜7のいずれか1項記載の化合物の使用。
【請求項10】
請求項1〜3のいずれか1項記載の11C標識イソキノリン誘導体を生体に投与して当該生体内における11Cの分布を調べる組織の画像化方法。
【請求項11】
11Cの分布を陽電子によって調べる請求項10に記載の組織の画像化方法。
【請求項12】
検査対象が脳である請求項10記載の組織の画像化方法。
【請求項13】
前記11Cの分布を調べることによって生体内に存在するプロテインキナーゼの分布を調べる請求項10記載の組織の画像化方法。
【請求項14】
プロテインキナーゼがRhoキナーゼである請求項13記載の組織の画像化方法。
【請求項1】
一般式(1)又はその薬学上許容される塩、水和物、溶媒和物若しくはプロドラッグからなる11C標識イソキノリン誘導体。
【化1】
(式中、
R1及びR2は、どちらか一方が[11C]メチル基を示し、他方は水素原子、シアノ基、アルキル基、ハロゲノアルキル基、アルケニル基、アルコキシ基、アルキルチオ基、ニトロ基、フルオロ基又はアリール基を示し;
R3及びR4はそれぞれ独立して水素原子、アルキル基、アルケニル基、アミノ基、アルキルアミノ基、ジアルキルアミノ基、アミノアルキル基、ハロゲノアルキル基、アルカノイル基、アミノアルカノイル基又はアルキルアミノアルカノイル基を示すか、あるいはR3及びR4は一緒になってアルキレン基又はアルケニレン基を形成し、一般式(1)中の環状ジアミノ基における任意の2つの炭素間で架橋しているか、のいずれかであり;
R5は水素原子、アミノアルカノイル基又はアルキルアミノアルカノイル基を示し;
mは1〜4の自然数を示し、nは2又は3を示す。)
【請求項2】
R1がイソキノリン環の4位に結合する[11C]メチル基である請求項1記載の11C標識イソキノリン誘導体。
【請求項3】
(S)−ヘキサヒドロ−2−メチル−1−[(4−[11C]メチル−5−イソキノリニル)スルホニル]−1H−1,4−ジアゼピン、5−(1,4−ジアゼパン−1−イルスルホニル)−4−[11C]メチルイソキノリン、4−[11C]メチル−5−(ピペラジン−1−イルスルホニル)イソキノリン及び(S)−2−アミノ−1−{3−メチル−4−(4−[11C]メチルイソキノリン−5−イルスルホニル)−1,4−ジアゼパン−1−イル}エタノンからなる群から選択される化合物又はその薬学上許容される塩、水和物、溶媒和物若しくはプロドラッグ。
【請求項4】
請求項1〜3のいずれか1項記載の11C標識イソキノリン誘導体を含有するPET用分子プローブ。
【請求項5】
請求項1に記載の11C標識イソキノリン誘導体を製造するための化合物であって、下記一般式(2)からなる化合物。
【化2】
(式中、
R6及びR7は、どちらか一方がトリアルキルスズ基、ボロニル基又はジアルコキシボリル基を示し、他方は水素原子、シアノ基、アルキル基、ハロゲノアルキル基、アルケニル基、アルコキシ基、アルキルチオ基、ニトロ基、フルオロ基又はアリール基を示し;
R3及びR4はそれぞれ独立して水素原子、アルキル基、アルケニル基、アミノ基、アルキルアミノ基、ジアルキルアミノ基、アミノアルキル基、ハロゲノアルキル基、アルカノイル基、アミノアルカノイル基又はアルキルアミノアルカノイル基示すか、あるいはR3及びR4は一緒になってアルキレン基又はアルケニレン基を形成し、一般式(2)中の環状ジアミノ基における任意の2つの炭素間で架橋しているかのいずれかであり;
R8はtert−ブトキシカルボニル基、トリハロアセチル基、tert−ブトキシカルボニルアミノアルカノイル基、トリハロアセチルアミノアルカノイル基、N−アルキル−N−tert−ブトキシカルボニルアミノアルカノイル基又はN−アルキル−N−トリハロアセチルアミノアルカノイル基を示し;
mは1〜4の自然数を示し、nは2又は3を示す。)
【請求項6】
R6がイソキノリン環の4位に結合するトリアルキルスズ基、ボロニル基又はジアルコキシボリル基であり、R8がトリハロアセチル基、トリハロアセチルアミノアルカノイル基、又はN−アルキル−N−トリハロアセチルアミノアルカノイル基である請求項5記載の化合物。
【請求項7】
R6がイソキノリン環の4位に結合するトリアルキルスズ基、ボロニル基又はジアルコキシボリル基であり、R3が水素原子又はメチル基であり、R4及びR7が水素原子であり、R8がトリハロアセチル基、トリハロアセチルアミノアルカノイル基、又はN−アルキル−N−トリハロアセチルアミノアルカノイル基であり、mが1又は2、nが2である請求項5記載の化合物
【請求項8】
請求項5〜7のいずれか1項記載の化合物のイソキノリン環上に[11C]メチル基を導入し、後続してR8を塩基性条件下で脱離させることを特徴とする請求項1〜3のいずれか1項記載の化合物の製造方法。
【請求項9】
請求項1〜3のいずれか1項記載の11C標識イソキノリン誘導体の製造のための請求項5〜7のいずれか1項記載の化合物の使用。
【請求項10】
請求項1〜3のいずれか1項記載の11C標識イソキノリン誘導体を生体に投与して当該生体内における11Cの分布を調べる組織の画像化方法。
【請求項11】
11Cの分布を陽電子によって調べる請求項10に記載の組織の画像化方法。
【請求項12】
検査対象が脳である請求項10記載の組織の画像化方法。
【請求項13】
前記11Cの分布を調べることによって生体内に存在するプロテインキナーゼの分布を調べる請求項10記載の組織の画像化方法。
【請求項14】
プロテインキナーゼがRhoキナーゼである請求項13記載の組織の画像化方法。
【図1】

【公開番号】特開2013−40112(P2013−40112A)
【公開日】平成25年2月28日(2013.2.28)
【国際特許分類】
【出願番号】特願2011−176449(P2011−176449)
【出願日】平成23年8月12日(2011.8.12)
【国等の委託研究の成果に係る記載事項】(出願人による申告)平成22年度 文部科学省、科学技術試験研究委託事業「創薬候補物質探索拠点」、産業技術力強化法第19条の適用を受ける特許出願
【出願人】(503359821)独立行政法人理化学研究所 (1,056)
【出願人】(599118539)株式会社デ・ウエスタン・セラピテクス研究所 (8)
【Fターム(参考)】
【公開日】平成25年2月28日(2013.2.28)
【国際特許分類】
【出願日】平成23年8月12日(2011.8.12)
【国等の委託研究の成果に係る記載事項】(出願人による申告)平成22年度 文部科学省、科学技術試験研究委託事業「創薬候補物質探索拠点」、産業技術力強化法第19条の適用を受ける特許出願
【出願人】(503359821)独立行政法人理化学研究所 (1,056)
【出願人】(599118539)株式会社デ・ウエスタン・セラピテクス研究所 (8)
【Fターム(参考)】
[ Back to top ]