
17−アセトキシ−11β−[4−(ジメチルアミノ)−フェニル]−21−メトキシ−19−ノルプレグナ−4,9−ジエン−3,20−ジオンの合成のための工業的方法およびプロセスのキーとなる中間体
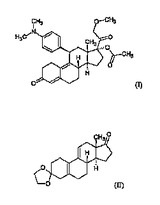
本発明は式(II)の3,3−[1,2−エタンジイル−ビス(オキシ)]−エストラ−5(10),9(11)−ジエン−17−オンからの式(I)の既知17−アセトキシ−11β−[4−(ジメチルアミノ)−フェニル]−21−メトキシ−19−ノルプレグナ−4,9−ジエン−3,20−ジオン(以後CDB−4124)の合成プロセスに関する。化合物CD−4124は抗ホルモン類に属する。本発明による方法は以下の通りである:i)式(II)の3,3−[1,2−エタンジイル−ビス(オキシ)]−エストラ−5(10),9(11)−ジエン−17−オンの5(10)位の二重結合上への過酸化水素によるエポキシドの形成;ii)得られた式(III)の5,10α−エポキシ−3,3−[1,2−エタンジイル−ビス(オキシ)]−5α−エストラ−9(11)−エン−17−オンの17位へのインサイツで形成されたシアン化水素の付加;iii)生成した式(IV)の5,10α−エポキシ−3,3,−[1,2−エタンジイル−ビス(オキシ)]−17α−ヒドロキシ−5α−エストラ−9(11)−エン−17β−カルボニトリルの17位水酸基のトリメチルクロロシランによるシリル化;iv)得られた式(V)の5,10α−エポキシ−3,3−[1,2−エタンジイル−ビス(オキシ)]−17−[トリメチルシリル−オキシ]−5α−エストラ−9(11)−エン−17β−カルボニトリルのCuCl存在下での4−(ジメチルアミノ)−フェニル臭化マグネシウムグリニヤ試薬との反応(トーシュ反応);v)得られた式(VI)の11β−[4−(ジメチルアミノ)−フェニル]−3,3−[1,2−エタンジイル−ビス(オキシ)]−5−ヒドロキシ−17α−[トリメチルシリル−(オキシ)]−5α−エストラ−9−エン−17β−カルボニトリルの5位の水酸基のトリメチルクロロシランによるシリル化;vi)得られた式(VII)の11β−[4−(ジメチルアミノ)−フェニル]−3,3−[1,2−エタンジイル−ビス(オキシ)]−5,17α−ビス−[トリメチル−シリル−(オキシ)]−5α−エストラ−9−エン−17β−カルボニトリルのジイソブチル水素化アルミニウムとの反応および反応混合物への酸の添加後;vii)得られた式(VIII)の11β−[4−(ジメチルアミノ)−フェニル]−3,3−[1,2−エタンジイル−ビス(オキシ)]−5,17α−ビス−[トリメチル−シリル−(オキシ)]−5α−エストラ−9−エン−17β−カルボアルデヒドのインサイツで形成されるメトキシ−メチルグリニヤ試薬によるメトキシ−メチル化とトリメチルシリル保護基の加水分解;viii)得られた式(IX)の17,20ξ−ジヒドロキシ−11β−[4−(ジメチルアミノ)−フェニル]−21−メトキシ−19−ノルプレグナ−4,9−ジエン−3−オンの20位の水酸基のジメチルスルホキシドと有機強酸の存在下ジシクロヘキシルカルボジイミドによる酸化(スワーン酸化)、およびある場合にはクロマトグラフィーによる精製後に;ix)得られた式(X)の11β−[4−(ジメチルアミノ)−フェニル]−17−ヒドロキシ−21−メトキシ−19−ノルプレグナ−4,9−ジエン−3,20−ジオンの17位の水酸基の過塩素酸存在下無水酢酸によるアセチル化、およびある場合には得られた式(I)の7−アセトキシ−11β−[4−(ジメチルアミノ)−フェニル]−21−メトキシ−19−ノルプレグナ−4,9−ジエン−3,20−ジオンをクロマトグラフィーにより精製する。本発明はまた式(VII)および(VIII)の新規中間体に関する。
【化1】

【化2】
【化1】
【化2】
【発明の詳細な説明】
【技術分野】
【0001】
本発明は式(I)の既知17−アセトキシ−11β−[4−(ジメチルアミノ)−フェニル]−21−メトキシ−19−ノルプレグナ−4,9−ジエン−3,20−ジオン(以下CDB−4124)の
【化1】
式(II)の3,3−[1,2−エタンジイル−ビス(オキシ)]−エストラ−5(10),9(11)−ジエン−17−オン(以後ケト−ケタール)からの合成プロセスに関する。
【化2】
【0002】
化合物CD−4124は抗ホルモン類に属する。抗ホルモンは、例えば男性および女性ホルモンあるいは副腎によってつくられるホルモンの標的臓器の結合部位への結合を阻害することによって生体のホルモンの作用を不活化し、それゆえ抗ホルモンの投与によってホルモンにより誘導されるそれらの機能は遮断される。
【0003】
プロゲステロンの合成あるいはその受容体への結合を阻害するこれらの化合物は避妊およびプロゲステロンが役割を果たす病理学的症例において用いられる可能性がある。
【0004】
理想的な抗プロゲストーゲン化合物は:
− 特異的(遮断されるべき受容体のみに結合する)、
− 受容体に対し高い親和性を持ち解離が遅い、
− 他の生物学的または薬理学的効果がない。
診療所で用いられた最初の抗プロゲスチンは1981[EP57115]に述べられていて、その名前はミフェプリストンであった。それ以来、いくつかの類縁体が合成され化合物の構造活性相関が調べられ、特に選択性、主に抗プロゲスチンと抗グルココルチコイド活性の比が調べられた。現在、既知の抗プロゲストーゲン化合物で選択性の要求を完全に満たすものはない。
【0005】
我々の発明のプロセスに従って合成できる化合物CDB−4124は、これまで行われた臨床試験により有望な化合物でありその工業的規模での経済的合成にかなっている。
【0006】
式(I)のCDB−4124の実験室的合成については、文献上、出発物質あるいは反応工程数が異なるいくつかの方法がある。種々の官能基の合成が同様な方法で行われている。これらの合成法の特徴は通例、プロセスのスケールアップの安全条件、特に溶媒(反応媒体)の可燃性を考慮していないことであり、それらの溶媒は健康に有害でありいくつかの試薬は高価である。
【0007】
最初の合成の目的は化合物/化合物から薬理試験を実施するに十分な量を合成することであった。化合物を治療に用いるため要求される純度を与えるにはさらなる開発が必要である。経済的合成の工業的実現は通常もとのプロセスの修飾あるいは改善された合成である。
【0008】
CDB−4124の最初の合成は国際公開第97/41145号パンフレットに述べられていて、その主題は11β−および21−置換19−ノルプロゲステロン誘導体およびその類縁体の合成であった。これらの化合物は有意な抗プロゲステロン活性を有する。スキーム3に化合物CDB−4124の合成を図示する。
【0009】
合成の出発物質は17α−[(ブロモ−メチル)−ジメチル−シリル−オキシ]−3,3−[1,2−エタンジイル−ビス−(オキシ)]−5(10),9(11)−ジエン−17β−カルボニトリルであり、それは商業的に入手可能な3,3−[1,2−エタンジイル−ビス−(オキシ)]−17α−ヒドロキシ−エストラ−5(10),9(11)−ジエン−17β−カルボニトリル(Davos Chemical Inc. New Jersey)から17位の水酸基の(ブロモ−メチル)−ジメチル−シリルクロリドによるシリル化によって69.5%の収率で得ることができた。精製はフラッシュカラムクロマトグラフィーで行われた。
【0010】
出発物質をテトラヒドロフラン溶液中−78℃でリチウムジイソプロピルアミドと反応し、生成物の単離は酢酸エチルでの抽出およびエーテルでの精製により行った。収率60.4%で得られた21−ブロモ化合物を酢酸カリウム(99%)と反応し、ついで21−ヒドロキシ誘導体を収率57.6%で与えるように炭酸水素カリウムで加水分解した。
【0011】
3位のケト基を保護するため20ビス−ケタール形成が用いられた(収率62.5%)。
【0012】
合成の重要工程、3および20位が保護された17α,21−ジヒドロキシ誘導体の21−モノメチル化はトリメチル−オキソニウム−(テトラフルオロ−ボレート)塩と“プロトン−スポンジ”[1,8−ビス−(ジメチルアミノ)ナフタレン]の1:1混合物の存在下で行われた。得られた生成物{3,3;20:20−ビス[1,2−エタンジイル−ビス(オキシ)]−17−ヒドロキシ−21−メトキシ−19−ノル−プレグナ−5(10),9(11)−ジエン}はジクロロメタンから収率79%で単離され次の工程に用いられた。
【0013】
得られた粗21−メトキシ誘導体の5(10)位二重結合へのエポキシド形成はヘキサフルオロアセトン3水和物の存在下過酸化水素で行われた。NMR分光法により得られた生成物は4つの型のエポキシドを含んでいた。(主生成物は66%で5α,10α−エポキシドであった)。
【0014】
得られた粗エポキシド混合物は銅(I)イオンによって触媒されるグリニヤ反応に用いられた。エーテル溶液から単離後、生成物はフラッシュカラムクロマトグラフィーで精製された。11β−[4−(ジメチルアミノ)−フェニル]誘導体のジケタール保護基の加水分解はテトラヒドロフラン中トリフルオロ酢酸−水の3:1混合物で行われた。生成物はジクロロメタン抽出、濃縮および油状残渣の水処理後、収率96.3%で得られた。
【0015】
合成の最終工程は17位の水酸基のアセチル化であり、ジクロロメタン中p−トルエンスルホン酸触媒の存在下0℃でトリフルオロ無水酢酸と酢酸の混合物で行った。反応完結後、混合物を水で希釈し、水酸化アンモニウム溶液で中和し、ジクロロメタンで抽出し塩水で洗浄した。合わせた有機層を濃縮し残渣をフラッシュカラムクロマトグラフィーで精製し化合物CDB−4124を収率75.8%で得た。
【0016】
上記の特許で述べられた合成の出発物質は17α−[(ブロモ−メチル)−ジメチル−シリル−オキシ]−3,3−[1,2−エタンジイル−ビス−(オキシ)]−5(10),9(11)−ジエン−17β−カルボニトリルであり、それはケト−ケタールからシアン化物イオンの付加とそれに続く水酸基のシリル化によって合成された。17−シリル−オキシ−ブロモ化合物は−78℃でリチウムジイソプロピルアミドによって21−ブロモ誘導体に変換された。21位へのメトキシ基の導入はいくつかの工程を通して間接的な方法で(21−ブロモ化合物、21−アセトキシ誘導体)21−ヒドロキシ化合物を経て[6当量(出発物質に対して)]のトリメチル−オキソニウム−(テトラフルオロ−ボレート)塩および“プロトン−スポンジ”を一緒に用いて(SNAP反応)行われた。この方法は長く費用がかかり、過剰の“プロトン−スポンジ”の除去が難しく、多くの場合生成物を何回も生成する必要がある。5(10)位の二重結合上のエポキシドの形成は(NMR分光法により)4つの型のエポキシドを生じ、その66%だけが望ましい5α,10α−エポキシドであった。粗生成物は34%の望ましくない生成物(β−エポキシド)を含むという事実にも関わらず、これがグリニヤ反応に用いられた。グリニヤ反応において5倍過剰の4−ブロモジメチルアニリンが用いられ、それはモノメチル誘導体および試薬の2量体化に有利に働き、それゆえ作業手続および生成物の単離が困難であり、単離した生成物の収率は比較的低かった。戦略的観点から反応系列の第7工程のエポキシ化(粗混合物を生じる)は経済的でない。11工程の合成の4工程でフラッシュクロマトグラフィーが用いられた。いくつかの場合中間体の単離および精製中にエーテルが用いられたが、それは工業的実用化においては危険である。最終生成物の収率は精製工程のために減少し、それゆえ合成の全収率はたった3.22%であった。
【0017】
国際公開第01/47945号パンフレットのスキーム1および2はさらに化合物CDB−4124の合成の2つの経路を示している。
【0018】
両合成の出発物質は3,3−[1,2−エタンジイル−ビス(オキシ)]−17α−ヒドロキシ−エストラ−5(10),9(11)−ジエン−17β−カルボニトリルであり、それは上に述べられた方法によってシリル化されたが、21−ハロゲン誘導体(クロロおよびブロモ)もまた同じ反応混合物で合成された。次の工程、臭素原子のアセトキシ基置換および加水分解、は既知の方法と同様であった。スキーム1に示された合成のさらなる工程は前の特許で述べられたものと同様である。
【0019】
スキーム2によれば21−ヒドロキシ誘導体の3−モノケタール誘導体が合成され、それに続いて21−メトキシ官能基の導入ためSNAP反応および20位へのケト基(一時的保護)の導入ためテトラヒドリドリチウムアルミニウムによる還元が行われた。得られた20−ヒドロキシ誘導体の5(10)位にエポキシド形成が行われた。エポキシドの開環およびケタール基の除去は前述の方法と同様であった。20位の水酸基の再酸化のためヨード−オキシ−安息香酸が用いられた。最終工程、望ましい17−アセトキシ生成物の合成は図式1に述べられた方法によって行われた。この合成は12工程により、全収率3.89%で達成された。
【0020】
合成の最初の2工程の組合せはよい解決ではあるけれども、例えば反応系列の後期でのエポキシド誘導体の形成(精製工程のため、収率低下の結果となる)は最新ではなく費用がかかる。さらに中間体および最終生成物の精製にフラッシュクロマトグラフィーを用いたことは不利な解決策である。いくつかの事例では生成物の単離はエーテル処理によって行われたが、それは大規模合成に用いることはできない。メトキシ基の導入は数工程で(前述のように)行われた。SNAP反応で用いられた“プロトン−スポンジ”の除去は反復精製によってのみ達成できた。テトラヒドリドリチウムアルミニウムによる還元(20位のオキソ官能基の一時的保護に用いられた)は工業的規模では特に危険である。さらにヨード−オキシ−安息香酸での酸化による20位のオキソ基の再生は高価であり、それゆえ工業的実現には適さない。
【先行技術文献】
【特許文献】
【0021】
【特許文献1】国際公開第97/41145号パンフレット
【特許文献2】国際公開第01/47945号パンフレット
【発明の概要】
【発明が解決しようとする課題】
【0022】
上述の事実によって、簡単な反応条件を用いたCDB−4124の工業規模での合成の実現に適した既知のプロセスはない。我々の目的はスケールアップが容易で、その工業的実現が安全、経済的で、活性成分の純度が局方の要求を満たすプロセスを詳述することであった。
【課題を解決するための手段】
【0023】
驚くべきことに、以下のプロセスが上述の要求を満たすことが見出された。
【0024】
i)式(II)の3,3−[1,2−エタンジイル−ビス(オキシ)]−エストラ−5(10),9(11)−ジエン−17−オンの5(10)位の二重結合への過酸化水素によるエポキシド形成;
【化3】
【0025】
ii)得られた式(III)の5,10α−エポキシ−3,3−[1,2−エタンジイル−ビス(オキシ)]−5α−エストラ−9(11)−エン−17−オンの17位へのインサイツで生じたシアン化水素の付加;
【化4】
【0026】
iii)生成した式(IV)の5,10α−エポキシ−3,3−[1,2−エタンジイル−ビス−(オキシ)]−17α−ヒドロキシ−5α−エストラ−9(11)−エン−17β−カルボニトリルの17位の水酸基のトリメチルクロロシランによるシリル化;
【化5】
【0027】
iv)得られた式(V)の5,10α−エポキシ−3,3−[1,2−エタンジイル−ビス(オキシ)]−17−[トリメチル−シリル−オキシ]−5α−エストラ−9(11)−エン−17βカルボニトリルのCuCl存在下での臭化4−(ジメチルアミノ)−フェニルマグネシウムグリニヤ試薬との反応(Teutsch反応);
【化6】
【0028】
v)生成した式(VI)の11β−[4−(ジメチル−アミノ)−フェニル]−3,3−[1,2−エタンジイル−ビス(オキシ)]−5−ヒドロキシ−17α−[トリメチルシリル−(オキシ)]−5α−エストラ−9−エン−17β−カルボニトリルの5位の水酸基のトリメチルクロロシランによるシリル化;
【化7】
【0029】
vi)得られた式(VII)の11β−[4−(ジメチルアミノ)−フェニル]−3,3−[1,2−エタンジイル−ビス(オキシ)]−5,17α−ビス−[トリメチル−シリル−(オキシ)]−5α−エストラ−9−エン−17β−カルボニトリルのジイソブチル水素化アルミニウムとの反応および反応混合物への酸添加後;
【化8】
【0030】
vii)式(VIII)の11β−[4−(ジメチルアミノ)−フェニル]−3,3−[1,2−エタンジイル−ビス(オキシ)]−5,17α−ビス−[トリメチル−シリル−(オキシ)]−5α−エストラ−9−エン−17β−カルボアルデヒドのインサイツで生成したメトキシ−メチルグリニヤ試薬によるメトキシ−メチル化と、同時にトリメチルシリル保護基の加水分解;
【化9】
【0031】
viii)得られた式(IX)の17,20ξ−ジヒドロキシ−11β−[4−(ジメチルアミノ)−フェニル]−21−メトキシ−19−ノルプレグナ−4,9−ジエン−3−オンの20位の水酸基のジメチルスルホキシドおよび有機強酸の存在下でのジシクロヘキシルカルボジイミドによる酸化(Swern酸化)、およびある場合にはクロマトグラフィーによる精製の後;
【化10】
【0032】
ix)得られた式(X)の11β−[4−(ジメチルアミノ)−フェニル]−17−ヒドロキシ−21−メトキシ−19−ノルプレグナ−4,9−ジエン−3,20−ジオンの過塩素酸存在下無水酢酸によるアセチル化、およびある場合には得られた式(I)の7−アセトキシ−11β−[4−(ジメチルアミノ)−フェニル]−21−メトキシ−19−ノルプレグナ−4,9−ジエン−3,20−ジオンをクロマトグラフィーにより精製する。
【化11】
【0033】
式(II)のケト−ケタールは好ましくは50%過酸化水素の無水ジクロロメタン溶液とピリジンおよびヘキサクロロアセトンの存在下0−1℃で20−24時間反応させた。反応完結後、混合物をジクロロメタンで稀釈し、過剰の過酸化水素を分解し、有機層を分離し、水層をジクロロメタンで2回抽出し、合わせた有機層を水で洗い、乾燥、濃縮した。油状の残渣を酢酸エチル−ジイソプロピルエーテルの1:3混合物で処理する。
【0034】
得られた式(III)のケト−ケタール−エポキシド(純度98.8%でHPLCにより5α,10α−エポキシドを95.3%含む)をさらに精製することなく工程ii)で用いる。
【0035】
工程ii)は好ましくは式(III)のケト−ケタール−エポキシドをメタノール中に懸濁し、粉末シアン化カリウムを20−25℃で加え、ついで酢酸を注意しながら加えた後反応混合物を50−55℃に暖めることによって行われる。反応混合物を1時間にわたり20−25℃に冷却し、ついでこの温度で5時間攪拌する。反応完結後、混合物を水で稀釈し、1時間攪拌して沈殿した結晶性の式(IV)のエポキシ−カルボニトリルを濾別する。生成物はさらに精製することなく工程iii)に用いることができる。
【0036】
工程iii)では式(IV)のエポキシ−カルボニトリルを好ましくはジクロロメタンに激しく攪拌して溶解し、ついで乾燥した後水含有率を検査する。乾燥した溶液にイミダゾールを加え20−25℃でトリメチルシランを1時間にわたり加える。反応完結後、溶液をジクロロメタンで稀釈し、過剰のトリメチルクロロシランを水を加えて分解する。有機層を分離し、水で洗い、乾燥し濃縮する。残渣をメタノールから結晶化し、濾過し乾燥する。そのようにして得られた式(V)のTMSO−カルボニトリル(73.3%)はさらに精製することなしに次の工程に用いることができる。
【0037】
工程iv)で生成した式(V)のTMSO−カルボニトリルを好ましくは次のように反応する。先ずマグネシウムと1,2−ジブロモ−エタンを無水テトラヒドロフランに加える。反応混合物の温度は上昇し始め活性化が有効であることを示す。次いで4−ブロモ−ジメチル−アニリンおよび少量の1,2−ジブロモ−エタンの無水テトラヒドロフランとトルエン溶液をマグネシウムを含む攪拌反応混合物に加える(グリニヤ試薬)。反応混合物の還流は活性化が有効であることを示す。ついでCuClをグリニヤ試薬溶液に加え5分間攪拌した後に混合物を8−13℃に冷却する。TMSO−カルボニトリルのジクロロメタン溶液を、温度を10−15℃に保ちながら加える。反応完結後、混合物をアルカリ金属ピロ亜硫酸塩を含む攪拌冷却した10%塩化アンモニウム溶液に加える。暖かい溶液を室温に冷却し、ジクロロメタンで稀釈して、有機層を分離し水層をジクロロメタンで抽出する。有機層を合わせて水で洗い、シリカゲルで処理し、無水硫酸ナトリウム上で乾燥し、濾過し真空で濃縮する。残渣をメタノールから結晶化する。結晶性生成物を単離し乾燥する。得られた式(VI)のA−TMSO−カルボニトリル(80.79%)はさらに精製することなく次の工程に用いることができる。
【0038】
工程v)は好ましくは得られた式(VI)のA−TMSO−カルボニトリルをジクロロメタン中に20−25℃で溶解し、ついでイミダゾールを添加した後5位の水酸基をトリメチルクロロシランでシリル化して行う。反応時間はおよそ2時間で、ついで混合物をジクロロメタンと水で稀釈し、有機層を分離し、水で洗い、無水硫酸ナトリウム上で乾燥し、濾過濃縮する。残渣をメタノールで処理し、得られた結晶性の式(VII)のA−ビス−TMSO−カルボニトリル(80.09%)を濾別し乾燥する。それはさらに精製することなく次の工程に用いることができる。
【0039】
工程vi)では得られた式(VII)のA−ビス−TMSO−カルボニトリルをメチル−tert−ブチルエーテルとテトラヒドロフランの混合物中に溶解し、(−15)−(−20)℃に冷却し1M DIBAL−H(水素化ジイソブチルアルミニウム)のシクロヘキサン溶液を、温度を保ちながら30分間かけて加え、ついで反応混合物をこの温度でさらに1時間攪拌する。反応完結後、水と酢酸の2:1混合物を(−5)−(−10)℃で加え、ついで混合物を20分間攪拌する。有機層を分離し、水、0.3M炭酸水素ナトリウム溶液および水で洗う。有機層を乾燥することなく40−45℃で濃縮し、残渣をメタノールに溶解し所定の容積に濃縮する(実施例を見よ)。結晶性の懸濁物を5−10℃に冷却し、1時間静置した後、0−(−5)℃のメタノールで洗い乾燥する。得られた式(VIII)のA−ビス−TMSO−カルボアルデヒド(83.6%)はさらに精製することなく次の工程に用いることができる。
【0040】
工程vii)で得られた式(VIII)のA−ビス−TMSO−カルボアルデヒドを鎖延長により21−メトキシ誘導体に変換する。これはマグネシウム削り屑を上述のように無水テトラヒドロフラン中で1,2−ジブロモ−エタンで活性化し、ついで塩化水銀(II)をアマルガムを生成するように加えることによって行われる。混合物をトルエンで稀釈し、ついでアマルガムの活性を実施例7、8および19で述べるように検査する。反応物質の活性を検査した後、塩化メトキシ−メチルのトルエン溶液を加える。これと並行してA−ビス−TMSO−カルボアルデヒドをトルエンに溶解し溶液をアマルガム溶液に0−5℃で30分間かけて加える。反応完結後、混合物を1Mの硫酸水素カリウム水溶液に温度を30℃より低く保ちながら加える。2時間攪拌した後、層を分離し、水相を1M炭酸水素ナトリウム溶液とジクロロメタンの混合物に加え10−15分攪拌する。有機層を分離し、水相をジクロロメタンで抽出し、有機層を合わせて乾燥し、炭で処理し、濾過して濃縮する。残渣は式(IX)の固体ジオール(84.1%)であり、さらに精製することなく次の工程に用いることができる。
【0041】
得られた式(IX)のジオールをさらに本発明の工程viii)に従って反応する。それを好ましくは無水トルエンに溶解し窒素下でジメチルスルホキシド、ピリジンおよびトリフルオロ酢酸を20−25℃で加える。ついで混合物にジシクロヘキシルカルボジイミドのトルエン溶液を加える(Swern酸化)。反応混合物を40℃で2時間攪拌し、ついで20−25℃に冷却して1Mの硫酸水素カリウム水溶液を加える。30分間攪拌した後、沈殿した結晶性の化合物を濾別し1Mの硫酸水素カリウム水溶液で洗浄する。2相の濾液を分離して、水相を1Mの水酸化ナトリウム水溶液に加え、沈殿した粗生成物を濾別、水で洗浄し乾燥する。得られた式(X)のケトン(79.5%)を精製後次の工程に用いる。
【0042】
治療への応用の純度要求を満たす純粋な式(I)のCBD−4124の合成は、2回のHPLC精製工程を含む。最初の工程は式(X)のケトンの精製である。精製した式(X)のケトンのアセチル化は粗CDB−4124に導き、後者のHPLCによる精製は純度99%の活性成分を与える。
【0043】
式(X)のケトンおよび粗CDB−4124のクロマトグラフィーは、実験室および工業的プロセスの両者において、好ましくは充填床としてシリカゲル、溶出液としてシクロヘキサン−メチル−tert−ブチルエーテル−アセトンの53:35:12の混合物を用いて行われる。シクロヘキサンの代わりにn−ヘキサンおよびn−へプタンもまた用いることができる。溶出液の溶媒成分の比率は決められた限界内で変えることができる(シクロヘキサン、n−ヘキサン、n−へプタン:40−60%;メチル−tert−ブチルエーテル:25−45%;アセトン:10−20%)。
【0044】
工程viii)の最後に式(X)のケトンを好ましくは次のように精製する:シリカゲル吸着剤(ZEOPREP C−GEL C−490L、ZEOCHEM製;粒径15−35μm;充填床長約60cm)をHPLCカラムにスラリー充填法により充填しカラムを溶出液(シクロヘキサン−メチル−tert−ブチルエーテル−アセトンの53:35:12の混合物)で平衡化する。式(X)の粗ケトンをアセトンとメチル−tert−ブチルエーテルとシクロヘキサンとの混合物に溶解して溶液に加える。そのようにして得られた溶液を濾過してカラムに注入する。UV検出を用いる。最初の画分を分離し純物質を含む画分を集めて濃縮する。他の方法によれば画分を濃縮後、残渣からジクロロメタンを蒸留して除き、生成物をジクロロメタンに溶解する。両者における不純物含有率:4%以下。このジクロロメタン溶液は次の工程に用いられる。
【0045】
式(I)のCDB−4124は式(X)の純化したケトンから本発明の工程ix)により過塩素酸の存在下無水酢酸を用いて合成される:70%過塩素酸を冷却した((−20)−(−25)℃)無水酢酸に温度を(−15)℃以下に保つような速度で加える。ついで精製した式(X)の11β−[4−(ジメチルアミノ)−フェニル]−17−ヒドロキシ−21−メトキシ−19−ノルプレグナ−4,9−ジエン−3,20−ジオンのジクロロメタン溶液を加える。反応完結後、混合物をジクロロメタンで稀釈し、(−10)℃に冷却して無水酢酸を分解するため水を加える。混合物のpHを水酸化アンモニウム溶液の添加により7−8に調整する。ついで水相を分離し、ジクロロメタンで抽出し、有機層を合わせて水洗し、乾燥し濃縮する。得られた粗最終生成物、式(I)のCDB−4124を上述の方法に従ってHPLCにより精製する。
【発明の効果】
【0046】
既知の方法に比べた本発明のプロセスの利点は以下のように要約できる。
【0047】
a)合成の出発物質[式(II)のケト−ケタール]はエストラ−4−エン−3,17−ジオンから既知の方法により容易に合成できる。
【0048】
b)我々のプロセスによれば5(10)位の二重結合上へのエポキシド形成は合成の最初の工程である。生成したエポキシドの異性体混合物から5α,10α−エポキシドのみが望ましい化合物に導き、従って他の異性体は不用な生成物である。我々のプロセスによれば物質の多量の損失をもたらすこの工程は反応系列の最初に行われ、それゆえ物質の損失は出発物質に由来し後のプロセス中間体に由来せず、そのことは大いに価値がある。我々の発明のプロセスはより経済的である。さらに既知の反応手順の後の工程で行われるエポキシ化の不都合は得られた化合物の精製がより困難なことである。
【0049】
c)我々のプロセスによれば戦略的に重要な21−メトキシ基の導入は式(VII)および(VIII)の新規中間体を経て2段階で行われる。式(VII)の新規中間体の応用は21位へのアルデヒド基の形成を可能にし得られた式(VIII)の新規化合物は簡単かつ工業的に応用可能な21位へのメトキシ基の導入を保証する。式(VII)のシリル化したシアンヒドリンは式(VIII)のA−ビス−TMSO−カルボアルデヒドを与えるようにDIBAL−Hのシクロヘキサン溶液と反応し、それは塩化または臭化メトキシ−メチルとグリニヤ型で反応し式(IX)のジオールを生成する。グリニヤ反応で生成した20−ヒドロキシ誘導体の20−ケト誘導体への酸化は(文献に従って用いられるヨウ化オキシ安息香酸に代わって)ジメチルスルホキシドと有機強酸の存在下でジシクロヘキシルカルボジイミドによるSwern酸化によって行われる。我々の発明のプロセスの利点は用いる試薬が安定でその使用が経済的なことである。
【0050】
d)文献に述べられている手順の出発物質はシリル化シアンヒドリンであり、それから21−メトキシ誘導体が4工程で合成された(必然的に遠回りの方法で)。シアンヒドリンはリチウムジイソプロピルアミドによって−78℃で21−クロロまたは21−ブロモ誘導体に変換され、ついでハロゲン置換基はアセトキシ基に交換され後者は21−ヒドロキシ誘導体を生じるように加水分解され、それは21−メトキシ誘導体を与えるようSNAP反応でトリメチル−オキソニウム−テトラフルオロボレート(“プロトン−スポンジ”を用いて)と反応された。
【0051】
e)我々の発明では水素化リチウムアルミニウムのような危険な反応物質もエーテルのような非常に燃えやすい溶媒もない。
【0052】
f)我々の発明のプロセスのさらなる利点は殆どの場合生成した中間体が精製なしに次の工程の使用に十分なほど純粋なことである。
【0053】
g)我々の発明のプロセスの最後の2工程でのみクロマトグラフィーによる精製が用いられるとはいえ、得られた活性成分CDB−4124の純度は99%であり、それは治療への応用の要求を満たす。
【0054】
h)我々の発明のプロセスのさらなる利点は個々の反応工程の収率が高いことである(48.65%;73.36%;80.79%;89.09%;83.6%;84.1%;99.56%;83.78%;)。既知の手順での全収率(3.22%および3.89%)に反して、11工程の合成の全収率は8.23%である。
【0055】
i)我々の発明のプロセスのいくつかの反応工程で用いられる反応条件は既知の手順で用いられる条件と異なり、それゆえ得られた生成物の収率および純度はより高い。例えばグリニヤ反応において(11位の置換)出発物質のステロイドと4−ブロモ−ジメチル−アニリンの比率は既知の手順で用いられる1:5に反して1:1.25である。このように反応の実現はより費用がかからず、不純物の生成が少ないので生成物の単離がより容易である。
【発明を実施するための形態】
【0056】
我々の発明によるプロセスはそれに限定されるものではないが以下の実施例に示される。
【実施例1】
【0057】
5,10α−エポキシ−3,3−[1,2−エタンジイル−ビス(オキシ)]−5α−エストラ−9(11)−エン−17−オン[式(III)の化合物]
窒素下で3,3−[1,2−エタンジイル−ビス(オキシ)−エストラ−5(10),9(11)−ジエン−17−オン(46.7g,149mmol)をピリジン(2.46ml,0.2mol−当量)とジクロロメタン(234ml)の混合物に激しく攪拌溶解し溶液を(−6)−(−8)℃に冷却した。ヘキサクロロアセトン(5.46ml,35.95mmol)を添加した後、0−(−2)℃の50%過酸化水素(60ml,1058mmol)を攪拌溶液に温度を0℃以下に保つような速度で加えた。反応混合物を1−(−1)℃で20−24時間攪拌し、ついで0−5℃のジクロロメタン(390ml)で稀釈し、過剰の過酸化水素をチオ硫酸ナトリウム5水和物(327g,1318mmol,8.87mol−当量)の氷冷水溶液(1500ml)の添加により分解した。反応混合物を1.5時間攪拌し、ついで有機相を分離した。水相をジクロロメタンで抽出し、合わせた有機層を水で洗い、無水硫酸ナトリウム上で乾燥し、濾過、濃縮した。油状の残渣を0.1%のピリジンを含む酢酸エチル−ジイソプロピルエーテルの1:3混合物(435ml)から結晶化した。そのようにして得られた生成物を乾燥し23.87g(48.66%)の標記化合物を得た。標記化合物の純度は98.5%−98.8%(HPLCにより測定)であり;α−エポキシドを95.3%含んでいた。
融点:153−155℃。
[α](25,D)=127.5°(c=1%,クロロホルム)
NMR:1H NMR(500MHz,CDCl3(TMS),δ(ppm)):0.88(3H,d,18−CH3);1.91(1H,dd,Hx−4);2.17(1H,d,Hy−4);3.86−3.98(4H,m,O−CH2−CH2−O);6.05(1H,m,H−11)
13C NMR(125MHz,CDCl3(TMS),δ(ppm)):14.8(C−18);40.3(C−4);60.1(C−10);61.6(C−5);64.1および64.3(O−CH2−CH2−O);107.0(C−3);125.7(C−11);136.7(C−9);221.1(C−17)
【実施例2】
【0058】
5,10α−エポキシ−3,3−[1,2−エタンジイル−ビス(オキシ)]−17α−ヒドロキシ−5α−エストラ−9(11)−エン−17β−カルボニトリル[式(IV)の化合物]
実施例1で得られた5,10α−エポキシ−3,3−[1,2−エタンジイル−ビス(オキシ)]−5α−エストラ−9(11)−エン−17−オン(33g,0.1mol)をメタノール(132ml)に懸濁し、ついで粉末にしたシアン化カリウム(19.5g,0.3mol)を20−25℃で加えた。酢酸(11.5ml,0.2mol)を注意深く加えた後、不均一な反応混合物15分間にわたって55℃まで加熱し、ついで1時間かけて25℃まで冷却し、この温度で5時間攪拌した。反応完結後、水132ml)を30分間で加え、得られた結晶性の生成物を濾過、水で洗浄して乾燥することなく次の工程で用いた。乾燥した試料の融点:143−144℃。
[α](25,D)=+13.5°(c=1%,クロロホルム)
NMR:1H NMR(500MHz,CDCl3(TMS),δ(ppm)):0.93(3H,s,18−CH3);3.09(1H,s,OH);3.86および3.98(4H,m,O−CH2−CH2−O);6.07(1H,m,H−11)
13C NMR(125MHz,CDCl3(TMS),δ(ppm)):16.8(C−18);60.2(C−10);61.9(C−5);64.0および64.2(O−CH2−CH2−O);77.3(C−17);106.9(C−3);120.7(C−20);125.9(C11);135.7(C−9)
【実施例3】
【0059】
5,10α−エポキシ−3,3−[1,2−エタンジイル−ビス(オキシ)]−17α−[(トリメチル−シリル)−オキシ]−5α−エストラ−9(11)−エン−17β−カルボニトリル[式(V)の化合物]
実施例2で得られた5,10α−エポキシ−3,3−[1,2−エタンジイル−ビス(オキシ)]−17α−ヒドロキシ−5α−エストラ−9(11)−エン−17βカルボニトリルをジクロロメタン(300ml)に激しく攪拌溶解し、溶液を無水硫酸ナトリウム上で乾燥し、ついで溶液から200mlのジクロロメタンを蒸留除去した。イミダゾール(10.1g,0.148mol)をそのようにして得られた溶液に加え、ついでトリメチルクロロシラン(15.5ml,0−121mol)を20−25℃で20分間にわたって滴下した。1時間攪拌後、溶液をジクロロメタン(66ml)と水(66ml)で稀釈した。有機層を分離し、水で洗い、無水硫酸ナトリウム上で乾燥し、濾過し濃縮した。残渣をメタノール(60ml)で処理し、0℃に冷却し、沈殿した結晶性の生成物を濾過し、0℃のメタノールで洗浄し40℃で真空乾燥して標記化合物31.5g(73.36%)を得た。この生成物を次の反応工程に用いた。
融点:167−170℃
[α](25,D)=+12.5°(c=1%,クロロホルム)
NMR:1H NMR(300MHz,CDCl3(TMS),δ(ppm)):0.15(9H,s,17−O−Si(CH3)3);0.83(3H,d,18−CH3);1.83(1H,dd,Hx−4);2.08(1H,d,Hy−4);3.76−3.94(4H,m,O−CH2−CH2−O);6.01(1H,m,H−11)
13C NMR(75MHz,CDCl3(TMS),δ(ppm)):0.9(17−O−Si(CH3)3);16.3(C−18);40.1(C−4);59.9(C−10);61.5(C−5);63.9および64.1(O−CH2−CH2−O);78.2(C−17);106.8(C−3);120.5(C−20);126.3(C−11);135.4(C−9)
【実施例4】
【0060】
11β−[4−(ジメチルアミノ)−フェニル]]−3,3−[1,2−エタンジイル−ビス(オキシ)]−5−ヒドロキシ−17α−[(トリメチル−シリル)−オキシ]−5α−エストラ−9−エン−17β−カルボニトリル[式(VI)の化合物]
窒素下で、マグネシウムの切り屑(3.3g,0.136mol)、無水テトラヒドロフラン(24ml)および1,2−ジブロモ−エタン(0.12ml,0.00131mol)を攪拌機、温度計、滴下ロート、ガス出入り口を備えたフラスコに20−25℃で加えた。5−10分間攪拌後、温度が上がり始め活性化が有効であることを示した。
【0061】
並行して次の溶液を25℃でつくった:無水テトラヒドロフラン(15ml)、無水トルエン(84ml)、4−ブロモ−N,N−ジメチル−アニリン(25g,0.125mol)および1,2−ジブロモ−エタン(0.16ml,0.00186mol)。この溶液から2mlをマグネシウム切り屑を含む溶液に加え、そのようにして得られた反応混合物を攪拌しながら60℃に暖めた。反応混合物の激しい還流が活性化の有効性を示したならば、残りの4−ブロモ−N,N−ジメチル−アニリン溶液を冷却後に滴下し、さらに2時間冷却して温度を14−16℃に保った。
【0062】
得られたグリニヤ試薬溶液に塩化銅(I)(0.4g,4.04mmol)を加え、ついで反応混合物を20−25℃で5分間攪拌した。8−13℃に冷却後、ジクロロメタン(180ml)中の5,10α−エポキシ−3,3−[1,2−エタンジイル−ビス(オキシ)]−17α−[(トリメチルシリル)−オキシ]−5α−エストラ−9(11)−エン−17β−カルボニトリル(42.96g,0.1mol)を攪拌冷却した溶液に温度を10−15℃に保つような速度で滴下した。ついで冷却を停止し反応混合物をさらに4時間攪拌した。
【0063】
反応完結後、混合物をピロ硫酸ナトリウム(0.4g,2.1mmol)を含む激しく攪拌した塩化アンモニウム溶液(100ml,10%水溶液)に加え、ジクロロメタン(100ml)で稀釈し、攪拌静置した。有機層を分離した後、水相をジクロロメタンで抽出し、有機層を合わせて水で洗い、無水硫酸ナトリウム上で乾燥し、濾過して濃縮した。残渣をメタノールから再結晶して44.5g(80.79%)の標記化合物を得た。
融点:243−256℃
[α](25,D)=−12.4°(c=1%,クロロホルム)
NMR:1H NMR(300MHz,CDCl3(TMS),δ(ppm)):0.24(9H,s,17−O−Si(CH3)3);0.55(3H,s,18−CH3);1.67(1H,d,Hx−4);2.02(1H,dd,Hy−4);2.91(6H,s,N−CH3);3.87−4.07(4H,m,O−CH2−CH2−O);4.29(1H,d,H−11);4.42(1H,d,OH);6.64(2H,m,H−3’およびH−5’);7.05(2H,m,H−2’およびH−6’)
13C NMR(75MHz,CDCl3(TMS),δ(ppm)):1.1(17−O−Si(CH3)3);16.9(C−18);38.8(C−11);40.7(N−CH3);47.5(C−4);64.1および64.5(O−CH2−CH2−O);70.1(C−5);78.9(C−17);108.8(C−3);112.6(C−3’およびC−5’);121.0(C−20);127.6(C−2’およびC−6’);133.9,134.0,134.1(C−9,C−10,C−1’);148.4(C−4’)
そのようにして得られた生成物をさらに精製することなく次の工程に用いた。
【実施例5】
【0064】
11β−[4−(ジメチルアミノ)−フェニル]−3,3−[1,2−エタンジイル−ビス(オキシ)]−5,17α−ビス−[(トリメチル−シリル)−オキシ]−5α−エストラ−9−エン−17β−カルボニトリル[式(VII)の化合物]
11β−[4−(ジメチルアミノ)−フェニル]−3,3−[1,2−エタンジイル−ビス(オキシ)]−5−ヒドロキシ−17α−[(トリメチル−シリル)−オキシ]−5α−エストラ−9−エン−17β−カルボニトリル(55g,0.1mol)およびイミダゾール(10.2g,0.15mol)をジクロロメタン(225ml)に20−25℃で攪拌溶解した。溶液にトリメチルクロロシラン(15.75ml,0.123mol)を20分間で滴下した。反応物質を添加中、イミダゾール塩酸が沈殿し始め反応の進行を示した。2時間攪拌後、反応混合物をジクロロメタン(100ml)および水(100ml)で稀釈し、数分間攪拌、静置し、ついで有機層を分離し、水で洗い、無水硫酸ナトリウム上で乾燥し、濾過して濃縮した。残渣をメタノールから結晶化して、濾過した生成物を真空乾燥して標記化合物55.5 g(89.09%)を得た。
融点:164−166℃。
[α](25,D)=+14,7°(c=1%,クロロホルム)
NMR:1H NMR(300MHz,DMSO−d6(TMS),δ(ppm)):0.11(9H,s,17−O−Si(CH3)3);0.22(9H,s,5−O−Si(CH3))3);0.45(3H,s,18−CH3);1.63(1H,d,Hx−4);2.07(1H,dd,Hy−4);2.84(6H,s,N−CH3);3.65−3.90(4H,m,O−CH2−CH2−O);4.21(1H,d,H−11);6.64(2H,m,H−3’およびH−5’);7.03(2H,m,H−2’およびH−6’)
13CNMR(75MHz,DMSO−d6(TMS),δ(ppm));0.9(17−O−Si(CH3)3);2.5(5−O−Si(CH3)3);16.7(C−18);37.8(C−11);40.1(N−CH3);48.6(C−4);62.7および64.0(O−CH2−CH2−O);73.0(C−5);78.5(C−17);107.6(C−3);112.3(C−3’およびC−5’);120.7(C−20);127.4(C−2’およびC−6’);132.3,133.2,134.9(C−9,C−10,C−1’);148.1(C−4’)
【実施例6】
【0065】
11β−[4−(ジメチルアミノ)−フェニル]−3,3−[1,2−エタンジイル−ビス(オキシ)]−5,17α−ビス−[(トリメチル−シリル)−オキシ]−5α−エストラ−9−エン−17β−カルボアルデヒド[式(VIII)の化合物]
窒素下で、11β−[4−(ジメチルアミノ)−フェニル]−3,3−[1,2−エタンジイル−ビス(オキシ)]−5,17α−ビス−[(トリメチル−シリル)−オキシ]−5α−エストラ−9−エン−17β−カルボニトリル(40g,62.4mmol)をメチル−tert−ブチルエーテル(220ml)とテトラヒドロフラン(17ml)の混合物中に溶解した。溶液を(−15)−(−20)℃に冷却し、ついで1M DIBAL−H(水素化ジイソブチル−アルミニウム)のシクロヘキサン溶液(160ml)を(−15)−(−20)℃で30分間かけて加えた。反応混合物を1時間攪拌し、ついで(−5)−(−10)℃の水(160ml)と酢酸(80ml)の混合物を窒素下で激しく攪拌しながら15−20分間にわたって加えた。そのようにして得られた反応混合物を20−25℃で30分間攪拌し、ついで有機層を分離し、水(200ml)、0.3M炭酸水素ナトリウム溶液(2x200ml)および水(200ml)で洗った。有機層を乾燥せずに40−45℃で真空で濃縮した。残渣をメタノール(140ml)に溶解し真空で容積30mlまで濃縮した。得られた結晶性の懸濁物を5−10℃に冷却し、1時間静置後濾過し、洗浄して60℃以下で真空で乾燥して標記化合物33.6g(83.6%)を得て、次の工程に用いた。
融点:154−158℃。
[α](25,D)=+7.7°(c=1%,クロロホルム)。
NMR;1H NMR(500MHz,CDCl3(TMS),δ(ppm)):0.12(9H,s,17−OSi(CH3)3);0.19(9H,s,5−O−Si(CH3)3);0.33(3H,s,18−CH3);2.88(6H,s,N−CH3);3.86および3.98(4H,m,O−CH2−CH2−O);4.21(1H,m,H−11);6.61(2H,m,H−3’およびH−5’);7.00(2H,m,H−2’およびH−6’);9.56(1H,s,H−20)
13C NMR(125MHz,CDCl3(TMS),δ(ppm)):1.9(17−O−Si(CH3)3);2.6(5−O−Si(CH3)3);15.7(C−18);38.8(C−11);40.7(N−CH3);63.4および64.4(O−CH2−CH2−O);73.7(C−5);91.1(C−17);108.5(C−3);112.8(C−3’およびC−5’);127.6(C−2’およびC−6’);133.6(C−10);134.3(C−1’);135.9(C−9);148.3(C−4’);203.3(C−20)
【実施例7】
【0066】
11β−[4−(ジメチルアミノ)−フェニル]−17,20ξ−ジヒドロキシ−21−メトキシ−19−ノルプレグナ−4,9−ジエン−3−オン[式(IX)の化合物]
窒素下で、マグネシウム削り屑(4.2g,173mmol)、無水テトラヒドロフラン(60ml)および1,2−ジブロモ−エタン(2.4ml,28mmol)を攪拌機、温度計、滴下ロート、還流凝縮器、ガス導入および導出口を備えた500mlの四つ首フラスコに20−25℃で加えた。数分間攪拌後、混合物は還流温度に達した。ついで反応混合物を35−40℃に冷却し塩化水銀(II)(0.23g,0.85mmol)を加え、15分間攪拌後混合物を20−25℃に冷却して無水トルエン(20ml)を加えた。塩化メトキシメチル(12.8ml,168mmol)を無水トルエン(50ml)に溶解し、そのようにして得られた溶液6mlを四つ首フラスコの混合物に加えた。数分後反応混合物の温度は35℃に上昇した。反応混合物を0−(−5)℃に冷却し、残りの塩化メトキシメチルのトルエン溶液を、温度を0−(−5)℃に保ちながら2−2.5時間かけて加えた。添加終了後、11β−[(4−ジメチルアミノ)−フェニル]−3,3−[(1,2−エタンジイル)−ビス(オキシ)]−5,17α−ビス−[トリメチル−シリル−(オキシ)]−5α−エストラ−9−エン−17β−カルボアルデヒド(20.0g,32mmol)の無水トルエン溶液(80ml)を温度を0−(−5)℃に保ちながら1時間かけて加えた。反応完結後、反応混合物を1Mの硫酸水素カリウム水溶液(200ml)に温度を30℃以下に保つような速度で加えた。混合物を20−25℃で2時間攪拌し、ついで有機層を分離して1Mの硫酸水素カリウム(1x10ml)で洗浄した。水相を合わせて1Mの炭酸水素ナトリウム溶液(225ml)とジクロロメタン(75ml)の攪拌混合物に加えた。10−15分間攪拌後、有機層を分離した。水相をジクロロメタン(5x50ml)で抽出し、有機層を合わせて無水硫酸ナトリウム(2g)上で乾燥し、濾過し、ジクロロメタン(2x20ml)で洗い、濾液を木炭(2.5g)と10分間攪拌した。木炭を濾別し、ジクロロメタン(2x20ml)で洗い濾液を濃縮して12.51g(84.1%)の標記化合物を得た。
融点:105℃(軟化)。
[α](25,D)=+157.7°(c=1%,ジクロロメタン)
NMR:1H NMR((主ジアステレオマー)、500MHz,CDCl3(TMS),δ(ppm)):0.49(3H,s,18−CH3);2.91(6H,s,N−CH3);3.37(3H,s,O−CH3);3.49(1H,m,Hx−21);3.57(1H,m,Hy−21);3.81(1H,m,H−20);4.30(1H,m,H−11);5.73(1H,s,H−4);6.67(2H,m,H−3’およびH−5’);7.04(2H,m,H−2’およびH−6’)
13C NMR((主ジアステレオマー),125MHz,CDCl3(TMS),δ(ppm)):16.5(C−18);39.5(C−11);40.7(N−CH3);59.2(O−CH3);71.9(C−20);74.9(C−21);84.4(C−17);112.8(C−3’およびC−5’);122.5(C−4);127.6(C−2’およびC−6’);128.6(C−10);132.0(C−1’);147.2(C−9);148.5(C−4’);157.1(C−5);199.7(C−3)
【実施例8】
【0067】
11β−[4−(ジメチルアミノ)−フェニル]−17,20ξ−ジヒドロキシ−21−メトキシ−19−ノルプレグナ−4,9−ジエン−3−オン[式(IX)の化合物]
窒素下で、マグネシウムの切り屑(4.2g,173mmol)、無水テトラヒドロフラン(60ml)および1,2−ジブロモ−エタン(2.4ml,28mmol)を攪拌機、温度計、滴下ロート、還流凝縮器、ガス導入および導出口を備えた500mlの四つ口フラスコに20−25℃で加えた。数分間攪拌後、混合物は還流温度に達した。ついで反応混合物を35−40℃に冷却し塩化水銀(II)(0.23g,0.85mmol)を加え、15分間攪拌後混合物を20−25℃に冷却して無水トルエン(20ml)を加えた。臭化メトキシメチル(13.7ml,168mmol)を無水トルエン(50ml)に溶かしそのようにして得られた溶液6mlを四つ首フラスコの混合物に加えた。数分後反応混合物の温度は30−35℃に上昇した。反応混合物を10−15℃に冷却し、残りの塩化メトキシメチルトルエン溶液を、温度を10−15℃に保ちながら2−2.5時間かけて加えた。添加終了後、11β−[(4−ジメチルアミノ)−フェニル]−3,3−[(1,2−エタンジイル)−ビス(オキシ)]−5,17α−ビス−[トリメチル−シリル−(オキシ)]−5α−エストラ−9−エン−17β−カルボアルデヒド(20.0g,32mmol)の無水トルエン溶液(80ml)を温度を10−15℃に保ちながら1時間かけて加えた。反応完結後、反応混合物を1Mの硫酸水素カリウム水溶液(200ml)に温度を30℃以下に保つような速度で加えた。混合物を20−25℃で2時間攪拌し、ついで有機層を分離して1Mの硫酸水素カリウム(1x10ml)で洗浄した。水相を合わせて1Mの炭酸水素ナトリウム溶液(225ml)とジクロロメタン(75ml)の攪拌混合物に加えた。10−15分間攪拌後、有機層を分離した。水相をジクロロメタン(5x50ml)で抽出し、有機層を合わせて無水硫酸ナトリウム(2g)上で乾燥し、濾過し、ジクロロメタン(2x20ml)で洗い、濾液を木炭(2.5g)と10分間攪拌した。木炭を濾別し、ジクロロメタン(2x20ml)で洗い濾液を濃縮して10.76g(72.3%)の標記化合物を得た。
生成物の少量をn−ペンタンと攪拌、融点:105℃(ガラス状構造、軟化)。
[α](25,D)=+157.7°(c=1%,ジクロロメタン)
【実施例9】
【0068】
11β−[4−(ジメチルアミノ)−フェニル]−17−ヒドロキシ−21−メトキシ−19−ノルプレグナ−4,9−ジエン−3,20−ジオン[式(X)の化合物]
窒素下で、17,20ξ−ジヒドロキシ−11β−[(4−ジメチルアミノ)−フェニル]−21−メトキシ−19−ノルプレグナ−4,9−ジエン−3−オン(11.94g,25.7mmol)と無水トルエン(68ml)を攪拌機、温度計、滴下ロート、還流凝縮器、ガス導入および導出口を備えた500mlの四つ口フラスコに加えた。そのようにして得られた溶液に無水ジメチルスルホキシド(9.9ml,139.5mmol)、ピリジン(2.9ml,36.0mmol)およびトリフルオロ酢酸(0.99ml,12.85mmol)を20−25℃で加えた。ついでジシクロヘキシルカルボジイミド(10.6g,51.4mmol)のトルエン(54ml)溶液を反応混合物に加え、そのようにして得られた混合物を40℃で攪拌した。反応時間は3時間であった。反応完結後、反応混合物を20−25℃に冷却し1Mの硫酸水素カリウム溶液(78ml)を加えた。30分間攪拌後、沈殿した結晶を濾別し1Mの硫酸水素カリウム溶液(4x19ml)で洗浄した。濾液の2相を分離し、水相を1Mの苛性ソーダ溶液(254ml)に10−20℃で加えた。30分間攪拌後、沈殿した粗生成物を濾別し、水で洗い乾燥して9,45g(79.5%)の標記化合物を得た。
融点:105−110℃。
【0069】
粗生成物を次の実施例で述べる方法に従ってHPLCにより精製した。
NMR:1H NMR(500MHz,DMSO−d6(TMS),δ(ppm)):0.20(3H,s,18−CH3);2.82(6H,s,N−CH3);3.26(3H,s,O−CH3);4.20(1H,d,Hx−21);4.35(1H,m,H−11);4.49(1H,d,Hy−21);5.37(1H,s,OH);5.67(1H,s,H−4);6.62(2H,m,H−3’およびH−5’);7.00(2H,m,H−2’およびH−6’)
13C NMR(125MHz,DMSO−d6(TMS),δ(ppm)):15.8(C−18);38.7(C−11);40.1(N−CH3);58.3(O−CH3);75.4(C−21);88.6(C−17);112.5(C−3’およびC−5’);122.0(C−4);127.2(C−2’およびC−6’);128.1(C−10);132.0(C−1’);146.6(C−9);148.2(C−4’);156.5(C−5);197.9(C−3);208.9(C−20)
【実施例10】
【0070】
粗11β−[4−(ジメチルアミノ)−フェニル]−17−ヒドロキシ−21−メトキシ−19−ノルプレグナ−4,9−ジエン−3,20−ジオン[式(X)の化合物]のHPLCによる精製(実験室規模)
シリカゲル(510g,ZEOPREP C−GEL C−490L,粒径15−35μm;充填床長約60cm)を直径5cmの軸床圧縮HPLCカラムにスラリー充填法で充填し、カラムをシクロヘキサン/メチル−tert−ブチルエーテル/アセトン溶出液45:40:15の混合物で平衡化した。10gの粗標記化合物(活性成分含量:80%)をアセトン(30ml)とメチル−tert−ブチルエーテル(80ml)およびシクロヘキサン(90ml)の混合物に溶解して攪拌溶液に加えた。そのようにして得られた溶液を濾過しカラムに注入した。生成物を85ml/分の流速で溶出し、UV検出を用いた。最初の画分はおよそ50mlであり、純粋な標記化合物を含む主画分は600mlであった。溶出した主画分の濃縮により固体の標記化合物を得た。
融点:108−110℃。
[α](25,D)=+199.2°(c=1%クロロホルム)
NMR:1H NMR(500MHz、DMSO−d6(TMS),δ(ppm)):0.20(3H,s,18−CH3);2.82(6H,s,N−CH3);3.26(3H,s,O−CH3);4.20(1H,d,Hx−21);4.35(1H,m,H−11);4.49(1H,d,Hy−21);5.37(1H,s,OH);5.67(1H,s,H−4);6.62(2H,m,H−3’およびH−5’);7.00(2H,m,H−2’およびH−6’)
13C NMR(125MHz,DMSO−d6(TMS),δ(ppm)):15.8(C−18);38.7(C−11);40.1(N−CH3);58.3(O−CH3);75.4(C−21);88.6(C−17);112.5(C−3’およびC−5’);122.0(C−4);127.2(C−2’およびC−6’);128.1(C−10);132.0(C−1’);146.6(C−9);148.2(C−4’);156.5(C−5);197.9(C−3);208.9(C−20)
【実施例11】
【0071】
17−アセトキシ−11β−[4−(ジメチルアミノ)−フェニル]−21−メトキシ−19−ノルプレグナ−4,9−ジエン−3,20−ジオン[式(I)の化合物]
70%過塩素酸(6ml)を攪拌冷却((−20)−(−25)℃)した無水酢酸(45ml)に温度を(−15)℃以下に保つような速度で加えた。ついで11β−[4−(ジメチルアミノ)−フェニル]−17−ヒドロキシ−21−メトキシ−19−ノルプレグナ−4,9−ジエン−3,20−ジオン(15.5g)のジクロロメタン(60ml)溶液を(−20)−(−25)℃で加えた。反応完結後−薄層クロマトグラフィーにより−反応混合物をジクロロメタン(50ml)で稀釈し、(−10)℃に冷却し無水酢酸を分解するためイオン交換水(52ml)を加えた。10分間攪拌後、25%水酸化アンモニウム溶液(77ml)を温度を25℃(pH=7−8)に保つような速度で加えた。ついで沈殿したカルボアミド副産物を濾別し、水相を分離し、ジクロロメタン(2x30ml)で抽出し合わせた有機層を濃縮して16.2g(95.8%)の標記化合物を得て、それを次の実施例に記載の方法によりHPLCで精製した。
NMR:1H NMR(500MHz,CDCl3(TMS),δ(pm)):0.40(3H,s,18−CH3);2.10(3H,s,O−CO−CH3);2.90(6H,s,N−CH3);3.41(3H,s,O−CH3);4.09(1H,d,Hx−21);4.38(1H,m,H−11);4.29(1H,d,Hy−21);5.77(1H,br,H−4);6.62(2H,m,H−3’およびH−5’);6.96(2H,m,H−2’およびH−6’)
13C NMR(125MHz,CDCl3(TMS),δ(ppm)):15.6(C−18);21.1(O−CO−CH3);39.3(C−11);40.6(N−CH3);59.4(O−CH3);76.0(C−21);93.9(C−17);112.8(C−3’およびC−5’);123.0(C−4);127.3(C−2’およびC−6’);129.4(C−10);131.3(C−1’);145.5(C−9);148.7(C−4’);156.4(C−5);170.7(O−CO−CH3);199.4(C−3);202.7(C−20)
【実施例12】
【0072】
粗CDB−4124のHPLCによる精製(溶出液:シクロヘキサン:メチル−tert−ブチルエーテル:アセトン=60:30:10(実験室規模)[式(I)の化合物]
シリカゲル(510g,ZEOPREP C−GEL C−490L,粒径15−35μm;充填床長約60cm)を直径5cmの軸床圧縮HPLCカラムにスラリー充填法で充填し、カラムをシクロヘキサン/メチル−tert−ブチルエーテル/アセトン溶出液60:30:10の混合物で平衡化した。前の実施例で得られた式(I)の化合物(CDB−4124)5.1g(不純物含有率:4%以下)を溶出液(100ml)に溶解し、濾過してカラムに注入した。生成物を85ml/分の流速で溶出しUV検出を用いた。最初の画分は40mlであり、純CDB−4124を含む主画分は560mlであった。溶出した主画分の濃縮により固体の標記化合物を得た。収率:4.25g(83.33%),不純物含有率:0.5%以下。
融点:118℃。
[α](25,D)=+127.2°(c=1%,クロロホルム)
NMR:1H NMR(500MHz,CDCl3(TMS),δ(ppm)):0.40(3H,s,18−CH3);2.10(3H,s,O−CO−CH3);2.90(6H,s,N−CH3);3.41(3H,s,O−CH3);4.09(1H,d,Hx−21);4.38(1H,m,H−11);4.29(1H,d,Hy−21);5.77(1H,br,H−4);6.62(2H,m,H−3’およびH−5’);6.96(2H,m.H−2’およびH−6’)
13C NMR(125MHz,CDCl3(TMS),δ(ppm)):15.6(C−18);21.1(O−CO−CH3);39.3(C−11);40.6(N−CH3);59.4(O−CH3);76.0(C−21);93.9(C−17);112.8(C−3’およびC−5’);123.0(C−4);127.3(C−2’およびC−6’);129.4(C−10);131.3(C−1’);145.5(C−9);148.7(C−4’);156.4(C−5);170.7(O−CO−CH3);199.4(C−3);202.7(C−20)
【実施例13】
【0073】
5,10α−エポキシ−3,3−[1.2−エタンジイル−ビス(オキシ)]−5α−エストラ−9(11)−エン−17−オン[式(III)の化合物]
装置:回転速度可変プロペラ攪拌機、還流凝縮器および温度計を備えた500リットルの耐酸性スチール反応器。
【0074】
窒素下で、3,3−[1,2−エタンジイル−ビス(オキシ)]−エストラ−5(10),9(11)−ジエン−17−オン(21.0kg)をピリジン(1,106L,0.2mol−当量)と無水ジクロロメタン(105.2 l)の混合物に激しく攪拌して溶解し、溶液を(−6)−(−8)℃に冷却した。ヘキサクロロアセトン(2.455L)の添加後、0−(−2)℃の50%過酸化水素(26.97L)を攪拌溶液に温度を0℃以下に保つような速度で加えた。反応混合物を1−(−1)℃で20−24時間攪拌し、ついで0−5℃のジクロロメタン(175L)で稀釈し、過剰の過酸化水素をチオ硫酸ナトリウム5水和物(8.87mol−当量)の氷冷水溶液(150L)の添加によって分解した。反応混合物を1.5時間攪拌し、ついで有機相を分離した。水相をジクロロメタンで抽出し、有機層を合わせて水で洗い、無水硫酸ナトリウム上で乾燥し、濾過して濃縮した。油状の残渣を0.1%のピリジンを含む酢酸エチル−ジイソプロピルエーテルの1:3混合物(195.6L)から結晶化した。そのようにして得られた生成物を乾燥して11.500kg(52.13%)の標記化合物を得た。標記化合物の純度は98.5−98.9%(HPLCにより測定)であり;α−エポキシドを96.2%含んでいた。
融点:151−154℃。
[α](25,D)=125.0°(c=1%,クロロホルム)
【実施例14】
【0075】
5,10α−エポキシ−3,3−[1,2−エタンジイル−ビス(オキシ)]−17α−ヒドロキシ−5α−エストラ−9(11)−エン−17β−カルボニトリル[式(IV)の化合物]
装置:回転速度可変プロペラ攪拌機、還流凝縮器および温度計を備えた250リットルのホウロウ反応器。
【0076】
実施例13で得られた5,10α−エポキシ−3,3−[1,2−エタンジイル−ビス(オキシ)]−5α−エストラ−9(11)−エン−17−オン(9.9kg)をメタノール(39.6L)に懸濁し、ついで粉末シアン化カリウム(5.85kg,0.3mol)を20−25℃で加えた。酢酸(3.48L)を注意して加えた後、不均一な反応混合物を15分間にわたり55℃に暖め、ついで1時間で25℃に冷却し、この温度でさらに5時間攪拌した。反応完結後、水(39.6L)を30分間にわたり加え、得られた結晶性の生成物を濾過し、水で洗い乾燥することなく次の工程に用いた。乾燥した試料の融点:140−143℃。
[α](25,D)=+13.0°(c=1%,クロロホルム)
【実施例15】
【0077】
5,10α−エポキシ−3,3−[1,2−エタンジイル−ビス(オキシ)]−17α−[(トリメチル−シリル)−オキシ]−5α−エストラ−9(11)−エン−17β−カルボニトリル[式(V)の化合物]
装置:回転速度可変プロペラ攪拌機、還流凝縮器および温度計を備えた250リットルのホウロウ反応器。
【0078】
実施例14で得られた5,10α−エポキシ−3,3−[1,2−エタンジイル−ビス(オキシ)]−17α−ヒドロキシ−5α−エストラ−9(11)−エン−17β−カルボニトリルをジクロロメタン(90L)に激しく攪拌して溶解し、溶液を無水硫酸ナトリウム上で乾燥し、ついで60Lのジクロロメタンを溶液から蒸留した。そのようにして得られた溶液にイミダゾール(0.303kg)を加え、ついでトリメチルクロロシラン(7.2L)を20−25℃で20分間にわたり滴下した。1時間攪拌後、溶液をジクロロメタン(19.8L)と水(19.8L)で稀釈した。有機層を分離し、水で洗い、無水硫酸ナトリウム上で乾燥し、濾過して濃縮した。残渣をメタノール(18L)で処理し、0℃に冷却し、沈殿した結晶性の生成物を濾過し、0℃のメタノールで洗浄し40℃で真空乾燥して標記化合物10.1kg(78.4%)を得た。この生成物をさらに精製することなく次の工程に用いた。
融点:167−170℃。
[α](25,D)=+12.5°(c=1%,クロロホルム)
【実施例16】
【0079】
11β−[4−(ジメチルアミノ)−フェニル]−3,3−[1,2−エタンジイル−ビス(オキシ)]−5−ヒドロキシ−17α−[(トリメチル−シリル)−オキシ]−5α−エストラ−9−エン−17β−カルボニトリル[式(VI)の化合物]
装置:可変回転速度プロペラ攪拌機、還流凝縮器および温度計を備えた250リットルホウロウ反応器。
【0080】
窒素下で、マグネシウム切り屑(0.768g)、無水テトラヒドロフラン(5.59L)および1,2−ジブロモ−エタン(27.94ml)を装置に20−25℃で加えた。5−10分攪拌後、温度を上昇し始め活性化が有効であることを示した。
【0081】
並行して次の溶液を窒素下で25℃でつくった:無水テトラヒドロフラン(3.5L)、無水トルエン(19.6L)、4−ブロモ−N,N−ジメチル−アニリン(5.8kg)および1,2−ジブロモ−エタン(34.25ml)。この溶液から400mlをマグネシウム切り屑を含む溶液に加え、そのようにして得られた反応混合物を60度に暖めた。反応混合物の激しい還流は活性化が有効であることを示し、そこで冷却後残りの4−ブロモ−N,N−ジメチル−アニリンの溶液を滴下し、さらに2時間冷却しながら温度を14−16℃に保った。
【0082】
塩化銅(I)(93.11g)を得られたグリニヤ試薬溶液に加え、ついで反応混合物を20−25℃で5分間攪拌した。8−13℃に冷却後、ジクロロメタン(42L)中の5,10α−エポキシ−3,3−[1,2−エタンジイル−ビス(オキシ)]−17α−[(トリメチルシリル)−オキシ]−5α−エストラ−9(11)−エン−17β−カルボニトリル(10.0kg)を攪拌冷却した溶液に温度を10−15℃に保つような速度で滴下した。ついで冷却を停止し反応混合物をさらに4時間攪拌した。
【0083】
反応完結後、混合物を激しく攪拌したピロ硫酸ナトリウム(93.1g)を含む塩化アンモニウム溶液(23.3L,10%水溶液)に加え、ジクロロメタン(23.3L)で稀釈し、攪拌し静置した。有機層を分離後、水相をジクロロメタンで抽出し、有機層を合わせて水で洗い、無水硫酸ナトリウム上で乾燥し、濾過して濃縮した。残渣をメタノールから再結晶して10.5kg(85.71%)の標記化合物を得た。
融点:243−256℃。
[α](25,D)=−12.4°(c=1%,クロロホルム)
そのようにして得られた生成物をさらに精製することなく次の工程に用いた。
【実施例17】
【0084】
11β−[4−(ジメチルアミノ)−フェニル]−3,3−[1,2−エタンジイル−ビス(オキシ)]−5,17α−ビス−[(トリメチル−シリル)−オキシ]−5α−エストラ−9−エン−17β−カルボニトリル[式(VII)の化合物]
装置:可変回転速度プロペラ攪拌機、還流凝縮器および温度計を備えた250リットルホウロウ反応器。
【0085】
11β−[4−(ジメチルアミノ)−フェニル]−3,3−[1,2−エタンジイル−ビス(オキシ)]−5−ヒドロキシ−17α−[(トリメチル−シリル)−オキシ]−5α−エストラ−9−エン−17β−カルボニトリル(10.45kg)とイミダゾール(1.93kg)をジクロロメタン(42.75L)に20−25℃で攪拌溶解した。トリメチルクロロシラン(3.0L)を溶液に20分間にわたって滴下した。反応剤を添加中、イミダゾール塩酸が沈殿し始め反応の進行を示した。2時間攪拌後、反応混合物をジクロロメタン(19L)と水(19L)で稀釈し、数分間攪拌し、静置、ついで有機層を分離し、水で洗い、無水硫酸ナトリウム上で乾燥し、濾過して濃縮した。残渣をメタノールから結晶化し、濾過した生成物を真空乾燥して10.25kg(87.0%)の標記化合物を得た。
融点:164−166℃。
[α](25,D)=+14.7°(c=1%,クロロホルム)
【実施例18】
【0086】
11β−[4−(ジメチルアミノ)−フェニル]−3,3−[1,2−エタンジイル−ビス(オキシ)]−5,17α−ビス−[(トリメチル−シリル)−オキシ]−5α−エストラ−9−エン−17β−カルボアルデヒド[式(VIII)の化合物]
装置:可変回転速度プロペラ攪拌機、還流凝縮器および温度計を備えた250リットルホウロウ反応器。
【0087】
窒素下で、11β−[4−(ジメチルアミノ)−フェニル)]−3,3−[1,2−エタンジイル−ビス−(オキシ)]−5,17α−ビス−[(トリメチル−シリル)−オキシ]−5α−エストラ−9−エン−17β−カルボニトリル(8kg)をメチル−tert−ブチルエーテル(44L)とテトラヒドロフラン(3.4L)の混合物に溶解した。溶液を(−15)−(−20)℃に冷却し、ついで1M DIBAL−Hのシクロヘキサン(32L)溶液を(−15)−(−20)℃で15分かけて加えた。反応混合物を1時間攪拌し、ついで(−5)−(−10)℃の水(32L)と酢酸(16L)の混合物を窒素下で激しく攪拌しながら15−20分間かけて加えた。そのようにして得られた反応混合物を20−25℃で30分間攪拌し、ついで有機層を分離し、水(40L)、0.3M炭酸水素ナトリウム(2x40L)および水(40L)で洗った。有機層を乾燥せずに40−45℃で真空乾燥した。残渣をメタノール(28L)に溶解し6Lまで真空乾燥した。得られた結晶性の懸濁液を5−10℃に冷却し、1時間静置後濾過し、洗浄し60℃以下で真空乾燥して6.95kg(86.46%)の標記化合物を得て、それをさらに精製することなく次の工程に用いた。
融点:154−158℃。
[α](25,D)=+7.7℃(c=1%,クロロホルム)
【実施例19】
【0088】
11β−[4−(ジメチルアミノ)−フェニル]−17,20ξ−ジヒドロキシ−21−メトキシ−19−ノルプレグナ−4,9−ジエン−3−オン[式(IX)の化合物]
装置:可変回転速度プロペラ攪拌機、還流凝縮器および温度計を備えた250リットルホウロウ反応器。
【0089】
窒素下で、マグネシウム切り屑(1.05kg)、無水テトラヒドロフラン(15L)および1,2−ジブロモ−エタン(600ml)を20−25℃で装置に加えた。数分間攪拌後、反応混合物は還流温度に達した。ついで反応混合物を35−40℃に冷却し、塩化水銀(II)(57.5g)を加え、15分間攪拌後反応混合物を20−25℃に冷却し無水トルエン(5L)を加えた。塩化メトキシ−メチル(3.2L)を無水トルエン(12.5L)に溶解しそのようにして得られた溶液1.5Lを反応混合物に加えた。数分後反応混合物の温度は35℃に上昇した。反応混合物を0−(−5)℃に冷却し残りの塩化メトキシメチルのトルエン溶液を温度を0−(−5)℃に保ちながら2−2.5時間にわたって加えた。添加終了後、11β−[(4−ジメチルアミノ)−フェニル]−3,3−[(1,2−エタンジイル)−ビス(オキシ)]−5,17α−ビス−[トリメチル−シリル−(オキシ)]−5α−エストラ−9−エン−17β−カルボアルデヒド(5.0kg)トルエン(20L)溶液を温度を0−(−5)℃に保ちながら1時間かけて加えた。反応完結後、反応混合物を1Mの硫酸水素カリウム水溶液(50L)に温度を30℃以下に保つような速度で加えた。混合物を20−25℃で2時間攪拌し、ついで有機層を分離し1M硫酸水素カリウム(1x2,5L)で洗浄した。水相を合わせて攪拌している1M炭酸水素ナトリウム溶液(56L)とジクロロメタン(19L)の混合物に加えた。10−15分間攪拌後、有機層を分離した。水相をジクロロメタン(5x12.5L)で抽出し、合わせた有機層を無水硫酸ナトリウム(500g)上で乾燥し、濾過し、ジクロロメタン(2x2L)で洗い濾液を木炭(625g)と10分間攪拌した。木炭を濾別し、ジクロロメタン(2x5L)で洗い、濾液を濃縮して3.3kg(88.73%)の標記化合物を得た。
融点:105℃(軟化)。
[α](25,D)=+156.2°(c=1%,ジクロロメタン)。
【実施例20】
【0090】
11β−[4−(ジメチルアミノ)−フェニル]−17−ヒドロキシ−21−メトキシ−19−ノルプレグナ−4,9−ジエン−3,20−ジオン[式(X)の化合物]
装置:可変回転速度プロペラ攪拌機、還流凝縮器および温度計を備えた250リットルホウロウ反応器。
【0091】
窒素下で、17,20ξ−ジヒドロキシ−11β−[(4−ジメチルアミノ)−フェニル]−21−メトキシ−19−ノルプレグナ−4,9−ジエン−3−オン(3.2kg)および無水トルエン(18L)を反応器に加えた。そのようにして得られた溶液に無水ジメチルスルホキシド(2.7L)、ピリジン(0.78L)およびトリフルオロ酢酸(0.265L)を20−25℃で加えた。ついでジシクロヘキシルカルボジイミド(2.84kg)のトルエン溶液(14.5L)を反応混合物に加え、そのようにして得られた混合物を40℃で攪拌した。反応時間は3時間であった。反応完結後、反応混合物を20−25℃に冷却して1M硫酸水素カリウム溶液(21L)を加えた。30分間攪拌後、沈殿した結晶を濾別し1M硫酸水素カリウム溶液(4x5L)で洗浄した。濾液の2相を分離して、水相を1M水酸化ナトリウム溶液(68L)に10−20℃で加えた。30分間攪拌後、沈殿した粗生成物を濾別し、水で洗い2,556kg(80.0%)の標記化合物を得た。
【0092】
粗生成物を次の実施例で述べる方法に従ってHPLCにより精製した。
【実施例21】
【0093】
粗11β−[4−(ジメチルアミノ)−フェニル]−17−ヒドロキシ−21−メトキシ−19−ノルプレグナ−4,9−ジエン−3,20−ジオン[式(X)の化合物]のHPLCによる精製(工業的規模)
シリカゲル(8kg,ZEOPREP C−GEL C−490L,粒径15−35μm;充填床長約60cm)を直径20cmの軸床圧縮HPLCカラムにスラリー充填法で充填し、カラムをシクロヘキサン/メチル−tert−ブチルエーテル/アセトン、53:34:12の混合流出液で平衡化した。160gの粗標記化合物(活性成分含有率:80%)をアセトン(0.48L)とメチル−tert−ブチルエーテル(1.28L)の混合物に溶解しシクロヘキサン(1.44L)を攪拌溶液に加えた。そのようにして得られた溶液を濾過してカラムに注入した。生成物は80L/時間の流速で流出しUV検出を用いた。最初の画分はおよそ1Lであり、純粋な標記化合物を含む主画分はおよそ14Lであった。固体の標記化合物流出した主画分の濃縮によって得られるが、好ましくは主画分の濃縮後残渣からジクロロメタンを蒸留除去し、生成物をジクロロメタンに溶解した。このジクロロメタン溶液を次の工程に用いた。
収率:120g(75%)の純粋な固体の標記化合物またはジクロロメタン溶液中の活性成分含量。不純物含有率:4%以下。
融点:105−110℃。
[α](25,D)=+199.2°(c=1%,クロロホルム)
【実施例22】
【0094】
粗17−アセトキシ−11β−[4−(ジメチルアミノ)−フェニル]−21−メトキシ−19−ノルプレグナ−4,9−ジエン−3,20−ジオン[式(I)の化合物]
装置:可変回転速度プロペラ攪拌機、還流凝縮器および温度計を備えた250リットルホウロウ反応器。
【0095】
70%過塩素酸(1.8L)を攪拌冷却((−20)−(−25)℃)した無水酢酸(13.5L)に温度を(−15)℃以下に保つような速度で加えた。ついで11β−[4−(ジメチルアミノ)−フェニル]−17−ヒドロキシ−21−メトキシ−19−ノルプレグナ−4,9−ジエン−3,20−ジオン(4.65kg)のジクロロメタン(18L)溶液に(−20)−(−25)℃で加えた。反応完結後−薄層クロマトグラフィーによって−反応混合物をジクロロメタン(15L)で稀釈し、(−10)℃に冷却して無水酢酸を分解するためイオン交換水(15.5L)を加えた。10分間攪拌後、25%水酸化アンモニウム溶液(23L)を温度を25℃(pH=7−8)に保つような速度で加えた。ついで沈殿したカルボアミド副産物を濾別し、水相を分離し、ジクロロメタン(2x9L)で抽出し有機層を合わせて濃縮し4.73kg(93.79%)の標記化合物(CDB−4124)を得て、それを次の実施例で述べる方法に従ってHPLCにより精製した。
【実施例23】
【0096】
粗CDB−4124のHPLC(工業的規模)による精製[式(I)の化合物]
シリカゲル(8kg, ZEOPREP C−GEL C−490L,粒径15−35μm;充填床長約60cm)を直径20cmの軸床圧縮HPLCカラムにスラリー充填法で充填し、カラムをシクロヘキサン/メチル−tert−ブチルエーテル/アセトン、53:35:12の混合流出液で平衡化した。先の実施例で得られた式(I)(CDB−4124)の粗化合物80g(不純物含有率:4%以下)を溶出液(1.6L)に溶解し、濾過してカラムに注入した。生成物を80L/時間の流速で溶出しUV検出を用いた。最初の画分は0.7L、純CDB−4124を含む主画分はおよそ10Lであった。固体の標記化合物が溶出した主画分の濃縮によって得られあるいは主画分の濃縮後生成物をメタノールに溶解しメタノール溶液として得られた。収量:固体の標記化合物またはメタノール溶液中の活性成分含有量として70g。不純物含有率:0.5%以下。
融点:118℃
[α](25,D)=+127.2°(c=1%,クロロホルム)
【技術分野】
【0001】
本発明は式(I)の既知17−アセトキシ−11β−[4−(ジメチルアミノ)−フェニル]−21−メトキシ−19−ノルプレグナ−4,9−ジエン−3,20−ジオン(以下CDB−4124)の
【化1】
式(II)の3,3−[1,2−エタンジイル−ビス(オキシ)]−エストラ−5(10),9(11)−ジエン−17−オン(以後ケト−ケタール)からの合成プロセスに関する。
【化2】
【0002】
化合物CD−4124は抗ホルモン類に属する。抗ホルモンは、例えば男性および女性ホルモンあるいは副腎によってつくられるホルモンの標的臓器の結合部位への結合を阻害することによって生体のホルモンの作用を不活化し、それゆえ抗ホルモンの投与によってホルモンにより誘導されるそれらの機能は遮断される。
【0003】
プロゲステロンの合成あるいはその受容体への結合を阻害するこれらの化合物は避妊およびプロゲステロンが役割を果たす病理学的症例において用いられる可能性がある。
【0004】
理想的な抗プロゲストーゲン化合物は:
− 特異的(遮断されるべき受容体のみに結合する)、
− 受容体に対し高い親和性を持ち解離が遅い、
− 他の生物学的または薬理学的効果がない。
診療所で用いられた最初の抗プロゲスチンは1981[EP57115]に述べられていて、その名前はミフェプリストンであった。それ以来、いくつかの類縁体が合成され化合物の構造活性相関が調べられ、特に選択性、主に抗プロゲスチンと抗グルココルチコイド活性の比が調べられた。現在、既知の抗プロゲストーゲン化合物で選択性の要求を完全に満たすものはない。
【0005】
我々の発明のプロセスに従って合成できる化合物CDB−4124は、これまで行われた臨床試験により有望な化合物でありその工業的規模での経済的合成にかなっている。
【0006】
式(I)のCDB−4124の実験室的合成については、文献上、出発物質あるいは反応工程数が異なるいくつかの方法がある。種々の官能基の合成が同様な方法で行われている。これらの合成法の特徴は通例、プロセスのスケールアップの安全条件、特に溶媒(反応媒体)の可燃性を考慮していないことであり、それらの溶媒は健康に有害でありいくつかの試薬は高価である。
【0007】
最初の合成の目的は化合物/化合物から薬理試験を実施するに十分な量を合成することであった。化合物を治療に用いるため要求される純度を与えるにはさらなる開発が必要である。経済的合成の工業的実現は通常もとのプロセスの修飾あるいは改善された合成である。
【0008】
CDB−4124の最初の合成は国際公開第97/41145号パンフレットに述べられていて、その主題は11β−および21−置換19−ノルプロゲステロン誘導体およびその類縁体の合成であった。これらの化合物は有意な抗プロゲステロン活性を有する。スキーム3に化合物CDB−4124の合成を図示する。
【0009】
合成の出発物質は17α−[(ブロモ−メチル)−ジメチル−シリル−オキシ]−3,3−[1,2−エタンジイル−ビス−(オキシ)]−5(10),9(11)−ジエン−17β−カルボニトリルであり、それは商業的に入手可能な3,3−[1,2−エタンジイル−ビス−(オキシ)]−17α−ヒドロキシ−エストラ−5(10),9(11)−ジエン−17β−カルボニトリル(Davos Chemical Inc. New Jersey)から17位の水酸基の(ブロモ−メチル)−ジメチル−シリルクロリドによるシリル化によって69.5%の収率で得ることができた。精製はフラッシュカラムクロマトグラフィーで行われた。
【0010】
出発物質をテトラヒドロフラン溶液中−78℃でリチウムジイソプロピルアミドと反応し、生成物の単離は酢酸エチルでの抽出およびエーテルでの精製により行った。収率60.4%で得られた21−ブロモ化合物を酢酸カリウム(99%)と反応し、ついで21−ヒドロキシ誘導体を収率57.6%で与えるように炭酸水素カリウムで加水分解した。
【0011】
3位のケト基を保護するため20ビス−ケタール形成が用いられた(収率62.5%)。
【0012】
合成の重要工程、3および20位が保護された17α,21−ジヒドロキシ誘導体の21−モノメチル化はトリメチル−オキソニウム−(テトラフルオロ−ボレート)塩と“プロトン−スポンジ”[1,8−ビス−(ジメチルアミノ)ナフタレン]の1:1混合物の存在下で行われた。得られた生成物{3,3;20:20−ビス[1,2−エタンジイル−ビス(オキシ)]−17−ヒドロキシ−21−メトキシ−19−ノル−プレグナ−5(10),9(11)−ジエン}はジクロロメタンから収率79%で単離され次の工程に用いられた。
【0013】
得られた粗21−メトキシ誘導体の5(10)位二重結合へのエポキシド形成はヘキサフルオロアセトン3水和物の存在下過酸化水素で行われた。NMR分光法により得られた生成物は4つの型のエポキシドを含んでいた。(主生成物は66%で5α,10α−エポキシドであった)。
【0014】
得られた粗エポキシド混合物は銅(I)イオンによって触媒されるグリニヤ反応に用いられた。エーテル溶液から単離後、生成物はフラッシュカラムクロマトグラフィーで精製された。11β−[4−(ジメチルアミノ)−フェニル]誘導体のジケタール保護基の加水分解はテトラヒドロフラン中トリフルオロ酢酸−水の3:1混合物で行われた。生成物はジクロロメタン抽出、濃縮および油状残渣の水処理後、収率96.3%で得られた。
【0015】
合成の最終工程は17位の水酸基のアセチル化であり、ジクロロメタン中p−トルエンスルホン酸触媒の存在下0℃でトリフルオロ無水酢酸と酢酸の混合物で行った。反応完結後、混合物を水で希釈し、水酸化アンモニウム溶液で中和し、ジクロロメタンで抽出し塩水で洗浄した。合わせた有機層を濃縮し残渣をフラッシュカラムクロマトグラフィーで精製し化合物CDB−4124を収率75.8%で得た。
【0016】
上記の特許で述べられた合成の出発物質は17α−[(ブロモ−メチル)−ジメチル−シリル−オキシ]−3,3−[1,2−エタンジイル−ビス−(オキシ)]−5(10),9(11)−ジエン−17β−カルボニトリルであり、それはケト−ケタールからシアン化物イオンの付加とそれに続く水酸基のシリル化によって合成された。17−シリル−オキシ−ブロモ化合物は−78℃でリチウムジイソプロピルアミドによって21−ブロモ誘導体に変換された。21位へのメトキシ基の導入はいくつかの工程を通して間接的な方法で(21−ブロモ化合物、21−アセトキシ誘導体)21−ヒドロキシ化合物を経て[6当量(出発物質に対して)]のトリメチル−オキソニウム−(テトラフルオロ−ボレート)塩および“プロトン−スポンジ”を一緒に用いて(SNAP反応)行われた。この方法は長く費用がかかり、過剰の“プロトン−スポンジ”の除去が難しく、多くの場合生成物を何回も生成する必要がある。5(10)位の二重結合上のエポキシドの形成は(NMR分光法により)4つの型のエポキシドを生じ、その66%だけが望ましい5α,10α−エポキシドであった。粗生成物は34%の望ましくない生成物(β−エポキシド)を含むという事実にも関わらず、これがグリニヤ反応に用いられた。グリニヤ反応において5倍過剰の4−ブロモジメチルアニリンが用いられ、それはモノメチル誘導体および試薬の2量体化に有利に働き、それゆえ作業手続および生成物の単離が困難であり、単離した生成物の収率は比較的低かった。戦略的観点から反応系列の第7工程のエポキシ化(粗混合物を生じる)は経済的でない。11工程の合成の4工程でフラッシュクロマトグラフィーが用いられた。いくつかの場合中間体の単離および精製中にエーテルが用いられたが、それは工業的実用化においては危険である。最終生成物の収率は精製工程のために減少し、それゆえ合成の全収率はたった3.22%であった。
【0017】
国際公開第01/47945号パンフレットのスキーム1および2はさらに化合物CDB−4124の合成の2つの経路を示している。
【0018】
両合成の出発物質は3,3−[1,2−エタンジイル−ビス(オキシ)]−17α−ヒドロキシ−エストラ−5(10),9(11)−ジエン−17β−カルボニトリルであり、それは上に述べられた方法によってシリル化されたが、21−ハロゲン誘導体(クロロおよびブロモ)もまた同じ反応混合物で合成された。次の工程、臭素原子のアセトキシ基置換および加水分解、は既知の方法と同様であった。スキーム1に示された合成のさらなる工程は前の特許で述べられたものと同様である。
【0019】
スキーム2によれば21−ヒドロキシ誘導体の3−モノケタール誘導体が合成され、それに続いて21−メトキシ官能基の導入ためSNAP反応および20位へのケト基(一時的保護)の導入ためテトラヒドリドリチウムアルミニウムによる還元が行われた。得られた20−ヒドロキシ誘導体の5(10)位にエポキシド形成が行われた。エポキシドの開環およびケタール基の除去は前述の方法と同様であった。20位の水酸基の再酸化のためヨード−オキシ−安息香酸が用いられた。最終工程、望ましい17−アセトキシ生成物の合成は図式1に述べられた方法によって行われた。この合成は12工程により、全収率3.89%で達成された。
【0020】
合成の最初の2工程の組合せはよい解決ではあるけれども、例えば反応系列の後期でのエポキシド誘導体の形成(精製工程のため、収率低下の結果となる)は最新ではなく費用がかかる。さらに中間体および最終生成物の精製にフラッシュクロマトグラフィーを用いたことは不利な解決策である。いくつかの事例では生成物の単離はエーテル処理によって行われたが、それは大規模合成に用いることはできない。メトキシ基の導入は数工程で(前述のように)行われた。SNAP反応で用いられた“プロトン−スポンジ”の除去は反復精製によってのみ達成できた。テトラヒドリドリチウムアルミニウムによる還元(20位のオキソ官能基の一時的保護に用いられた)は工業的規模では特に危険である。さらにヨード−オキシ−安息香酸での酸化による20位のオキソ基の再生は高価であり、それゆえ工業的実現には適さない。
【先行技術文献】
【特許文献】
【0021】
【特許文献1】国際公開第97/41145号パンフレット
【特許文献2】国際公開第01/47945号パンフレット
【発明の概要】
【発明が解決しようとする課題】
【0022】
上述の事実によって、簡単な反応条件を用いたCDB−4124の工業規模での合成の実現に適した既知のプロセスはない。我々の目的はスケールアップが容易で、その工業的実現が安全、経済的で、活性成分の純度が局方の要求を満たすプロセスを詳述することであった。
【課題を解決するための手段】
【0023】
驚くべきことに、以下のプロセスが上述の要求を満たすことが見出された。
【0024】
i)式(II)の3,3−[1,2−エタンジイル−ビス(オキシ)]−エストラ−5(10),9(11)−ジエン−17−オンの5(10)位の二重結合への過酸化水素によるエポキシド形成;
【化3】
【0025】
ii)得られた式(III)の5,10α−エポキシ−3,3−[1,2−エタンジイル−ビス(オキシ)]−5α−エストラ−9(11)−エン−17−オンの17位へのインサイツで生じたシアン化水素の付加;
【化4】
【0026】
iii)生成した式(IV)の5,10α−エポキシ−3,3−[1,2−エタンジイル−ビス−(オキシ)]−17α−ヒドロキシ−5α−エストラ−9(11)−エン−17β−カルボニトリルの17位の水酸基のトリメチルクロロシランによるシリル化;
【化5】
【0027】
iv)得られた式(V)の5,10α−エポキシ−3,3−[1,2−エタンジイル−ビス(オキシ)]−17−[トリメチル−シリル−オキシ]−5α−エストラ−9(11)−エン−17βカルボニトリルのCuCl存在下での臭化4−(ジメチルアミノ)−フェニルマグネシウムグリニヤ試薬との反応(Teutsch反応);
【化6】
【0028】
v)生成した式(VI)の11β−[4−(ジメチル−アミノ)−フェニル]−3,3−[1,2−エタンジイル−ビス(オキシ)]−5−ヒドロキシ−17α−[トリメチルシリル−(オキシ)]−5α−エストラ−9−エン−17β−カルボニトリルの5位の水酸基のトリメチルクロロシランによるシリル化;
【化7】
【0029】
vi)得られた式(VII)の11β−[4−(ジメチルアミノ)−フェニル]−3,3−[1,2−エタンジイル−ビス(オキシ)]−5,17α−ビス−[トリメチル−シリル−(オキシ)]−5α−エストラ−9−エン−17β−カルボニトリルのジイソブチル水素化アルミニウムとの反応および反応混合物への酸添加後;
【化8】
【0030】
vii)式(VIII)の11β−[4−(ジメチルアミノ)−フェニル]−3,3−[1,2−エタンジイル−ビス(オキシ)]−5,17α−ビス−[トリメチル−シリル−(オキシ)]−5α−エストラ−9−エン−17β−カルボアルデヒドのインサイツで生成したメトキシ−メチルグリニヤ試薬によるメトキシ−メチル化と、同時にトリメチルシリル保護基の加水分解;
【化9】
【0031】
viii)得られた式(IX)の17,20ξ−ジヒドロキシ−11β−[4−(ジメチルアミノ)−フェニル]−21−メトキシ−19−ノルプレグナ−4,9−ジエン−3−オンの20位の水酸基のジメチルスルホキシドおよび有機強酸の存在下でのジシクロヘキシルカルボジイミドによる酸化(Swern酸化)、およびある場合にはクロマトグラフィーによる精製の後;
【化10】
【0032】
ix)得られた式(X)の11β−[4−(ジメチルアミノ)−フェニル]−17−ヒドロキシ−21−メトキシ−19−ノルプレグナ−4,9−ジエン−3,20−ジオンの過塩素酸存在下無水酢酸によるアセチル化、およびある場合には得られた式(I)の7−アセトキシ−11β−[4−(ジメチルアミノ)−フェニル]−21−メトキシ−19−ノルプレグナ−4,9−ジエン−3,20−ジオンをクロマトグラフィーにより精製する。
【化11】
【0033】
式(II)のケト−ケタールは好ましくは50%過酸化水素の無水ジクロロメタン溶液とピリジンおよびヘキサクロロアセトンの存在下0−1℃で20−24時間反応させた。反応完結後、混合物をジクロロメタンで稀釈し、過剰の過酸化水素を分解し、有機層を分離し、水層をジクロロメタンで2回抽出し、合わせた有機層を水で洗い、乾燥、濃縮した。油状の残渣を酢酸エチル−ジイソプロピルエーテルの1:3混合物で処理する。
【0034】
得られた式(III)のケト−ケタール−エポキシド(純度98.8%でHPLCにより5α,10α−エポキシドを95.3%含む)をさらに精製することなく工程ii)で用いる。
【0035】
工程ii)は好ましくは式(III)のケト−ケタール−エポキシドをメタノール中に懸濁し、粉末シアン化カリウムを20−25℃で加え、ついで酢酸を注意しながら加えた後反応混合物を50−55℃に暖めることによって行われる。反応混合物を1時間にわたり20−25℃に冷却し、ついでこの温度で5時間攪拌する。反応完結後、混合物を水で稀釈し、1時間攪拌して沈殿した結晶性の式(IV)のエポキシ−カルボニトリルを濾別する。生成物はさらに精製することなく工程iii)に用いることができる。
【0036】
工程iii)では式(IV)のエポキシ−カルボニトリルを好ましくはジクロロメタンに激しく攪拌して溶解し、ついで乾燥した後水含有率を検査する。乾燥した溶液にイミダゾールを加え20−25℃でトリメチルシランを1時間にわたり加える。反応完結後、溶液をジクロロメタンで稀釈し、過剰のトリメチルクロロシランを水を加えて分解する。有機層を分離し、水で洗い、乾燥し濃縮する。残渣をメタノールから結晶化し、濾過し乾燥する。そのようにして得られた式(V)のTMSO−カルボニトリル(73.3%)はさらに精製することなしに次の工程に用いることができる。
【0037】
工程iv)で生成した式(V)のTMSO−カルボニトリルを好ましくは次のように反応する。先ずマグネシウムと1,2−ジブロモ−エタンを無水テトラヒドロフランに加える。反応混合物の温度は上昇し始め活性化が有効であることを示す。次いで4−ブロモ−ジメチル−アニリンおよび少量の1,2−ジブロモ−エタンの無水テトラヒドロフランとトルエン溶液をマグネシウムを含む攪拌反応混合物に加える(グリニヤ試薬)。反応混合物の還流は活性化が有効であることを示す。ついでCuClをグリニヤ試薬溶液に加え5分間攪拌した後に混合物を8−13℃に冷却する。TMSO−カルボニトリルのジクロロメタン溶液を、温度を10−15℃に保ちながら加える。反応完結後、混合物をアルカリ金属ピロ亜硫酸塩を含む攪拌冷却した10%塩化アンモニウム溶液に加える。暖かい溶液を室温に冷却し、ジクロロメタンで稀釈して、有機層を分離し水層をジクロロメタンで抽出する。有機層を合わせて水で洗い、シリカゲルで処理し、無水硫酸ナトリウム上で乾燥し、濾過し真空で濃縮する。残渣をメタノールから結晶化する。結晶性生成物を単離し乾燥する。得られた式(VI)のA−TMSO−カルボニトリル(80.79%)はさらに精製することなく次の工程に用いることができる。
【0038】
工程v)は好ましくは得られた式(VI)のA−TMSO−カルボニトリルをジクロロメタン中に20−25℃で溶解し、ついでイミダゾールを添加した後5位の水酸基をトリメチルクロロシランでシリル化して行う。反応時間はおよそ2時間で、ついで混合物をジクロロメタンと水で稀釈し、有機層を分離し、水で洗い、無水硫酸ナトリウム上で乾燥し、濾過濃縮する。残渣をメタノールで処理し、得られた結晶性の式(VII)のA−ビス−TMSO−カルボニトリル(80.09%)を濾別し乾燥する。それはさらに精製することなく次の工程に用いることができる。
【0039】
工程vi)では得られた式(VII)のA−ビス−TMSO−カルボニトリルをメチル−tert−ブチルエーテルとテトラヒドロフランの混合物中に溶解し、(−15)−(−20)℃に冷却し1M DIBAL−H(水素化ジイソブチルアルミニウム)のシクロヘキサン溶液を、温度を保ちながら30分間かけて加え、ついで反応混合物をこの温度でさらに1時間攪拌する。反応完結後、水と酢酸の2:1混合物を(−5)−(−10)℃で加え、ついで混合物を20分間攪拌する。有機層を分離し、水、0.3M炭酸水素ナトリウム溶液および水で洗う。有機層を乾燥することなく40−45℃で濃縮し、残渣をメタノールに溶解し所定の容積に濃縮する(実施例を見よ)。結晶性の懸濁物を5−10℃に冷却し、1時間静置した後、0−(−5)℃のメタノールで洗い乾燥する。得られた式(VIII)のA−ビス−TMSO−カルボアルデヒド(83.6%)はさらに精製することなく次の工程に用いることができる。
【0040】
工程vii)で得られた式(VIII)のA−ビス−TMSO−カルボアルデヒドを鎖延長により21−メトキシ誘導体に変換する。これはマグネシウム削り屑を上述のように無水テトラヒドロフラン中で1,2−ジブロモ−エタンで活性化し、ついで塩化水銀(II)をアマルガムを生成するように加えることによって行われる。混合物をトルエンで稀釈し、ついでアマルガムの活性を実施例7、8および19で述べるように検査する。反応物質の活性を検査した後、塩化メトキシ−メチルのトルエン溶液を加える。これと並行してA−ビス−TMSO−カルボアルデヒドをトルエンに溶解し溶液をアマルガム溶液に0−5℃で30分間かけて加える。反応完結後、混合物を1Mの硫酸水素カリウム水溶液に温度を30℃より低く保ちながら加える。2時間攪拌した後、層を分離し、水相を1M炭酸水素ナトリウム溶液とジクロロメタンの混合物に加え10−15分攪拌する。有機層を分離し、水相をジクロロメタンで抽出し、有機層を合わせて乾燥し、炭で処理し、濾過して濃縮する。残渣は式(IX)の固体ジオール(84.1%)であり、さらに精製することなく次の工程に用いることができる。
【0041】
得られた式(IX)のジオールをさらに本発明の工程viii)に従って反応する。それを好ましくは無水トルエンに溶解し窒素下でジメチルスルホキシド、ピリジンおよびトリフルオロ酢酸を20−25℃で加える。ついで混合物にジシクロヘキシルカルボジイミドのトルエン溶液を加える(Swern酸化)。反応混合物を40℃で2時間攪拌し、ついで20−25℃に冷却して1Mの硫酸水素カリウム水溶液を加える。30分間攪拌した後、沈殿した結晶性の化合物を濾別し1Mの硫酸水素カリウム水溶液で洗浄する。2相の濾液を分離して、水相を1Mの水酸化ナトリウム水溶液に加え、沈殿した粗生成物を濾別、水で洗浄し乾燥する。得られた式(X)のケトン(79.5%)を精製後次の工程に用いる。
【0042】
治療への応用の純度要求を満たす純粋な式(I)のCBD−4124の合成は、2回のHPLC精製工程を含む。最初の工程は式(X)のケトンの精製である。精製した式(X)のケトンのアセチル化は粗CDB−4124に導き、後者のHPLCによる精製は純度99%の活性成分を与える。
【0043】
式(X)のケトンおよび粗CDB−4124のクロマトグラフィーは、実験室および工業的プロセスの両者において、好ましくは充填床としてシリカゲル、溶出液としてシクロヘキサン−メチル−tert−ブチルエーテル−アセトンの53:35:12の混合物を用いて行われる。シクロヘキサンの代わりにn−ヘキサンおよびn−へプタンもまた用いることができる。溶出液の溶媒成分の比率は決められた限界内で変えることができる(シクロヘキサン、n−ヘキサン、n−へプタン:40−60%;メチル−tert−ブチルエーテル:25−45%;アセトン:10−20%)。
【0044】
工程viii)の最後に式(X)のケトンを好ましくは次のように精製する:シリカゲル吸着剤(ZEOPREP C−GEL C−490L、ZEOCHEM製;粒径15−35μm;充填床長約60cm)をHPLCカラムにスラリー充填法により充填しカラムを溶出液(シクロヘキサン−メチル−tert−ブチルエーテル−アセトンの53:35:12の混合物)で平衡化する。式(X)の粗ケトンをアセトンとメチル−tert−ブチルエーテルとシクロヘキサンとの混合物に溶解して溶液に加える。そのようにして得られた溶液を濾過してカラムに注入する。UV検出を用いる。最初の画分を分離し純物質を含む画分を集めて濃縮する。他の方法によれば画分を濃縮後、残渣からジクロロメタンを蒸留して除き、生成物をジクロロメタンに溶解する。両者における不純物含有率:4%以下。このジクロロメタン溶液は次の工程に用いられる。
【0045】
式(I)のCDB−4124は式(X)の純化したケトンから本発明の工程ix)により過塩素酸の存在下無水酢酸を用いて合成される:70%過塩素酸を冷却した((−20)−(−25)℃)無水酢酸に温度を(−15)℃以下に保つような速度で加える。ついで精製した式(X)の11β−[4−(ジメチルアミノ)−フェニル]−17−ヒドロキシ−21−メトキシ−19−ノルプレグナ−4,9−ジエン−3,20−ジオンのジクロロメタン溶液を加える。反応完結後、混合物をジクロロメタンで稀釈し、(−10)℃に冷却して無水酢酸を分解するため水を加える。混合物のpHを水酸化アンモニウム溶液の添加により7−8に調整する。ついで水相を分離し、ジクロロメタンで抽出し、有機層を合わせて水洗し、乾燥し濃縮する。得られた粗最終生成物、式(I)のCDB−4124を上述の方法に従ってHPLCにより精製する。
【発明の効果】
【0046】
既知の方法に比べた本発明のプロセスの利点は以下のように要約できる。
【0047】
a)合成の出発物質[式(II)のケト−ケタール]はエストラ−4−エン−3,17−ジオンから既知の方法により容易に合成できる。
【0048】
b)我々のプロセスによれば5(10)位の二重結合上へのエポキシド形成は合成の最初の工程である。生成したエポキシドの異性体混合物から5α,10α−エポキシドのみが望ましい化合物に導き、従って他の異性体は不用な生成物である。我々のプロセスによれば物質の多量の損失をもたらすこの工程は反応系列の最初に行われ、それゆえ物質の損失は出発物質に由来し後のプロセス中間体に由来せず、そのことは大いに価値がある。我々の発明のプロセスはより経済的である。さらに既知の反応手順の後の工程で行われるエポキシ化の不都合は得られた化合物の精製がより困難なことである。
【0049】
c)我々のプロセスによれば戦略的に重要な21−メトキシ基の導入は式(VII)および(VIII)の新規中間体を経て2段階で行われる。式(VII)の新規中間体の応用は21位へのアルデヒド基の形成を可能にし得られた式(VIII)の新規化合物は簡単かつ工業的に応用可能な21位へのメトキシ基の導入を保証する。式(VII)のシリル化したシアンヒドリンは式(VIII)のA−ビス−TMSO−カルボアルデヒドを与えるようにDIBAL−Hのシクロヘキサン溶液と反応し、それは塩化または臭化メトキシ−メチルとグリニヤ型で反応し式(IX)のジオールを生成する。グリニヤ反応で生成した20−ヒドロキシ誘導体の20−ケト誘導体への酸化は(文献に従って用いられるヨウ化オキシ安息香酸に代わって)ジメチルスルホキシドと有機強酸の存在下でジシクロヘキシルカルボジイミドによるSwern酸化によって行われる。我々の発明のプロセスの利点は用いる試薬が安定でその使用が経済的なことである。
【0050】
d)文献に述べられている手順の出発物質はシリル化シアンヒドリンであり、それから21−メトキシ誘導体が4工程で合成された(必然的に遠回りの方法で)。シアンヒドリンはリチウムジイソプロピルアミドによって−78℃で21−クロロまたは21−ブロモ誘導体に変換され、ついでハロゲン置換基はアセトキシ基に交換され後者は21−ヒドロキシ誘導体を生じるように加水分解され、それは21−メトキシ誘導体を与えるようSNAP反応でトリメチル−オキソニウム−テトラフルオロボレート(“プロトン−スポンジ”を用いて)と反応された。
【0051】
e)我々の発明では水素化リチウムアルミニウムのような危険な反応物質もエーテルのような非常に燃えやすい溶媒もない。
【0052】
f)我々の発明のプロセスのさらなる利点は殆どの場合生成した中間体が精製なしに次の工程の使用に十分なほど純粋なことである。
【0053】
g)我々の発明のプロセスの最後の2工程でのみクロマトグラフィーによる精製が用いられるとはいえ、得られた活性成分CDB−4124の純度は99%であり、それは治療への応用の要求を満たす。
【0054】
h)我々の発明のプロセスのさらなる利点は個々の反応工程の収率が高いことである(48.65%;73.36%;80.79%;89.09%;83.6%;84.1%;99.56%;83.78%;)。既知の手順での全収率(3.22%および3.89%)に反して、11工程の合成の全収率は8.23%である。
【0055】
i)我々の発明のプロセスのいくつかの反応工程で用いられる反応条件は既知の手順で用いられる条件と異なり、それゆえ得られた生成物の収率および純度はより高い。例えばグリニヤ反応において(11位の置換)出発物質のステロイドと4−ブロモ−ジメチル−アニリンの比率は既知の手順で用いられる1:5に反して1:1.25である。このように反応の実現はより費用がかからず、不純物の生成が少ないので生成物の単離がより容易である。
【発明を実施するための形態】
【0056】
我々の発明によるプロセスはそれに限定されるものではないが以下の実施例に示される。
【実施例1】
【0057】
5,10α−エポキシ−3,3−[1,2−エタンジイル−ビス(オキシ)]−5α−エストラ−9(11)−エン−17−オン[式(III)の化合物]
窒素下で3,3−[1,2−エタンジイル−ビス(オキシ)−エストラ−5(10),9(11)−ジエン−17−オン(46.7g,149mmol)をピリジン(2.46ml,0.2mol−当量)とジクロロメタン(234ml)の混合物に激しく攪拌溶解し溶液を(−6)−(−8)℃に冷却した。ヘキサクロロアセトン(5.46ml,35.95mmol)を添加した後、0−(−2)℃の50%過酸化水素(60ml,1058mmol)を攪拌溶液に温度を0℃以下に保つような速度で加えた。反応混合物を1−(−1)℃で20−24時間攪拌し、ついで0−5℃のジクロロメタン(390ml)で稀釈し、過剰の過酸化水素をチオ硫酸ナトリウム5水和物(327g,1318mmol,8.87mol−当量)の氷冷水溶液(1500ml)の添加により分解した。反応混合物を1.5時間攪拌し、ついで有機相を分離した。水相をジクロロメタンで抽出し、合わせた有機層を水で洗い、無水硫酸ナトリウム上で乾燥し、濾過、濃縮した。油状の残渣を0.1%のピリジンを含む酢酸エチル−ジイソプロピルエーテルの1:3混合物(435ml)から結晶化した。そのようにして得られた生成物を乾燥し23.87g(48.66%)の標記化合物を得た。標記化合物の純度は98.5%−98.8%(HPLCにより測定)であり;α−エポキシドを95.3%含んでいた。
融点:153−155℃。
[α](25,D)=127.5°(c=1%,クロロホルム)
NMR:1H NMR(500MHz,CDCl3(TMS),δ(ppm)):0.88(3H,d,18−CH3);1.91(1H,dd,Hx−4);2.17(1H,d,Hy−4);3.86−3.98(4H,m,O−CH2−CH2−O);6.05(1H,m,H−11)
13C NMR(125MHz,CDCl3(TMS),δ(ppm)):14.8(C−18);40.3(C−4);60.1(C−10);61.6(C−5);64.1および64.3(O−CH2−CH2−O);107.0(C−3);125.7(C−11);136.7(C−9);221.1(C−17)
【実施例2】
【0058】
5,10α−エポキシ−3,3−[1,2−エタンジイル−ビス(オキシ)]−17α−ヒドロキシ−5α−エストラ−9(11)−エン−17β−カルボニトリル[式(IV)の化合物]
実施例1で得られた5,10α−エポキシ−3,3−[1,2−エタンジイル−ビス(オキシ)]−5α−エストラ−9(11)−エン−17−オン(33g,0.1mol)をメタノール(132ml)に懸濁し、ついで粉末にしたシアン化カリウム(19.5g,0.3mol)を20−25℃で加えた。酢酸(11.5ml,0.2mol)を注意深く加えた後、不均一な反応混合物15分間にわたって55℃まで加熱し、ついで1時間かけて25℃まで冷却し、この温度で5時間攪拌した。反応完結後、水132ml)を30分間で加え、得られた結晶性の生成物を濾過、水で洗浄して乾燥することなく次の工程で用いた。乾燥した試料の融点:143−144℃。
[α](25,D)=+13.5°(c=1%,クロロホルム)
NMR:1H NMR(500MHz,CDCl3(TMS),δ(ppm)):0.93(3H,s,18−CH3);3.09(1H,s,OH);3.86および3.98(4H,m,O−CH2−CH2−O);6.07(1H,m,H−11)
13C NMR(125MHz,CDCl3(TMS),δ(ppm)):16.8(C−18);60.2(C−10);61.9(C−5);64.0および64.2(O−CH2−CH2−O);77.3(C−17);106.9(C−3);120.7(C−20);125.9(C11);135.7(C−9)
【実施例3】
【0059】
5,10α−エポキシ−3,3−[1,2−エタンジイル−ビス(オキシ)]−17α−[(トリメチル−シリル)−オキシ]−5α−エストラ−9(11)−エン−17β−カルボニトリル[式(V)の化合物]
実施例2で得られた5,10α−エポキシ−3,3−[1,2−エタンジイル−ビス(オキシ)]−17α−ヒドロキシ−5α−エストラ−9(11)−エン−17βカルボニトリルをジクロロメタン(300ml)に激しく攪拌溶解し、溶液を無水硫酸ナトリウム上で乾燥し、ついで溶液から200mlのジクロロメタンを蒸留除去した。イミダゾール(10.1g,0.148mol)をそのようにして得られた溶液に加え、ついでトリメチルクロロシラン(15.5ml,0−121mol)を20−25℃で20分間にわたって滴下した。1時間攪拌後、溶液をジクロロメタン(66ml)と水(66ml)で稀釈した。有機層を分離し、水で洗い、無水硫酸ナトリウム上で乾燥し、濾過し濃縮した。残渣をメタノール(60ml)で処理し、0℃に冷却し、沈殿した結晶性の生成物を濾過し、0℃のメタノールで洗浄し40℃で真空乾燥して標記化合物31.5g(73.36%)を得た。この生成物を次の反応工程に用いた。
融点:167−170℃
[α](25,D)=+12.5°(c=1%,クロロホルム)
NMR:1H NMR(300MHz,CDCl3(TMS),δ(ppm)):0.15(9H,s,17−O−Si(CH3)3);0.83(3H,d,18−CH3);1.83(1H,dd,Hx−4);2.08(1H,d,Hy−4);3.76−3.94(4H,m,O−CH2−CH2−O);6.01(1H,m,H−11)
13C NMR(75MHz,CDCl3(TMS),δ(ppm)):0.9(17−O−Si(CH3)3);16.3(C−18);40.1(C−4);59.9(C−10);61.5(C−5);63.9および64.1(O−CH2−CH2−O);78.2(C−17);106.8(C−3);120.5(C−20);126.3(C−11);135.4(C−9)
【実施例4】
【0060】
11β−[4−(ジメチルアミノ)−フェニル]]−3,3−[1,2−エタンジイル−ビス(オキシ)]−5−ヒドロキシ−17α−[(トリメチル−シリル)−オキシ]−5α−エストラ−9−エン−17β−カルボニトリル[式(VI)の化合物]
窒素下で、マグネシウムの切り屑(3.3g,0.136mol)、無水テトラヒドロフラン(24ml)および1,2−ジブロモ−エタン(0.12ml,0.00131mol)を攪拌機、温度計、滴下ロート、ガス出入り口を備えたフラスコに20−25℃で加えた。5−10分間攪拌後、温度が上がり始め活性化が有効であることを示した。
【0061】
並行して次の溶液を25℃でつくった:無水テトラヒドロフラン(15ml)、無水トルエン(84ml)、4−ブロモ−N,N−ジメチル−アニリン(25g,0.125mol)および1,2−ジブロモ−エタン(0.16ml,0.00186mol)。この溶液から2mlをマグネシウム切り屑を含む溶液に加え、そのようにして得られた反応混合物を攪拌しながら60℃に暖めた。反応混合物の激しい還流が活性化の有効性を示したならば、残りの4−ブロモ−N,N−ジメチル−アニリン溶液を冷却後に滴下し、さらに2時間冷却して温度を14−16℃に保った。
【0062】
得られたグリニヤ試薬溶液に塩化銅(I)(0.4g,4.04mmol)を加え、ついで反応混合物を20−25℃で5分間攪拌した。8−13℃に冷却後、ジクロロメタン(180ml)中の5,10α−エポキシ−3,3−[1,2−エタンジイル−ビス(オキシ)]−17α−[(トリメチルシリル)−オキシ]−5α−エストラ−9(11)−エン−17β−カルボニトリル(42.96g,0.1mol)を攪拌冷却した溶液に温度を10−15℃に保つような速度で滴下した。ついで冷却を停止し反応混合物をさらに4時間攪拌した。
【0063】
反応完結後、混合物をピロ硫酸ナトリウム(0.4g,2.1mmol)を含む激しく攪拌した塩化アンモニウム溶液(100ml,10%水溶液)に加え、ジクロロメタン(100ml)で稀釈し、攪拌静置した。有機層を分離した後、水相をジクロロメタンで抽出し、有機層を合わせて水で洗い、無水硫酸ナトリウム上で乾燥し、濾過して濃縮した。残渣をメタノールから再結晶して44.5g(80.79%)の標記化合物を得た。
融点:243−256℃
[α](25,D)=−12.4°(c=1%,クロロホルム)
NMR:1H NMR(300MHz,CDCl3(TMS),δ(ppm)):0.24(9H,s,17−O−Si(CH3)3);0.55(3H,s,18−CH3);1.67(1H,d,Hx−4);2.02(1H,dd,Hy−4);2.91(6H,s,N−CH3);3.87−4.07(4H,m,O−CH2−CH2−O);4.29(1H,d,H−11);4.42(1H,d,OH);6.64(2H,m,H−3’およびH−5’);7.05(2H,m,H−2’およびH−6’)
13C NMR(75MHz,CDCl3(TMS),δ(ppm)):1.1(17−O−Si(CH3)3);16.9(C−18);38.8(C−11);40.7(N−CH3);47.5(C−4);64.1および64.5(O−CH2−CH2−O);70.1(C−5);78.9(C−17);108.8(C−3);112.6(C−3’およびC−5’);121.0(C−20);127.6(C−2’およびC−6’);133.9,134.0,134.1(C−9,C−10,C−1’);148.4(C−4’)
そのようにして得られた生成物をさらに精製することなく次の工程に用いた。
【実施例5】
【0064】
11β−[4−(ジメチルアミノ)−フェニル]−3,3−[1,2−エタンジイル−ビス(オキシ)]−5,17α−ビス−[(トリメチル−シリル)−オキシ]−5α−エストラ−9−エン−17β−カルボニトリル[式(VII)の化合物]
11β−[4−(ジメチルアミノ)−フェニル]−3,3−[1,2−エタンジイル−ビス(オキシ)]−5−ヒドロキシ−17α−[(トリメチル−シリル)−オキシ]−5α−エストラ−9−エン−17β−カルボニトリル(55g,0.1mol)およびイミダゾール(10.2g,0.15mol)をジクロロメタン(225ml)に20−25℃で攪拌溶解した。溶液にトリメチルクロロシラン(15.75ml,0.123mol)を20分間で滴下した。反応物質を添加中、イミダゾール塩酸が沈殿し始め反応の進行を示した。2時間攪拌後、反応混合物をジクロロメタン(100ml)および水(100ml)で稀釈し、数分間攪拌、静置し、ついで有機層を分離し、水で洗い、無水硫酸ナトリウム上で乾燥し、濾過して濃縮した。残渣をメタノールから結晶化して、濾過した生成物を真空乾燥して標記化合物55.5 g(89.09%)を得た。
融点:164−166℃。
[α](25,D)=+14,7°(c=1%,クロロホルム)
NMR:1H NMR(300MHz,DMSO−d6(TMS),δ(ppm)):0.11(9H,s,17−O−Si(CH3)3);0.22(9H,s,5−O−Si(CH3))3);0.45(3H,s,18−CH3);1.63(1H,d,Hx−4);2.07(1H,dd,Hy−4);2.84(6H,s,N−CH3);3.65−3.90(4H,m,O−CH2−CH2−O);4.21(1H,d,H−11);6.64(2H,m,H−3’およびH−5’);7.03(2H,m,H−2’およびH−6’)
13CNMR(75MHz,DMSO−d6(TMS),δ(ppm));0.9(17−O−Si(CH3)3);2.5(5−O−Si(CH3)3);16.7(C−18);37.8(C−11);40.1(N−CH3);48.6(C−4);62.7および64.0(O−CH2−CH2−O);73.0(C−5);78.5(C−17);107.6(C−3);112.3(C−3’およびC−5’);120.7(C−20);127.4(C−2’およびC−6’);132.3,133.2,134.9(C−9,C−10,C−1’);148.1(C−4’)
【実施例6】
【0065】
11β−[4−(ジメチルアミノ)−フェニル]−3,3−[1,2−エタンジイル−ビス(オキシ)]−5,17α−ビス−[(トリメチル−シリル)−オキシ]−5α−エストラ−9−エン−17β−カルボアルデヒド[式(VIII)の化合物]
窒素下で、11β−[4−(ジメチルアミノ)−フェニル]−3,3−[1,2−エタンジイル−ビス(オキシ)]−5,17α−ビス−[(トリメチル−シリル)−オキシ]−5α−エストラ−9−エン−17β−カルボニトリル(40g,62.4mmol)をメチル−tert−ブチルエーテル(220ml)とテトラヒドロフラン(17ml)の混合物中に溶解した。溶液を(−15)−(−20)℃に冷却し、ついで1M DIBAL−H(水素化ジイソブチル−アルミニウム)のシクロヘキサン溶液(160ml)を(−15)−(−20)℃で30分間かけて加えた。反応混合物を1時間攪拌し、ついで(−5)−(−10)℃の水(160ml)と酢酸(80ml)の混合物を窒素下で激しく攪拌しながら15−20分間にわたって加えた。そのようにして得られた反応混合物を20−25℃で30分間攪拌し、ついで有機層を分離し、水(200ml)、0.3M炭酸水素ナトリウム溶液(2x200ml)および水(200ml)で洗った。有機層を乾燥せずに40−45℃で真空で濃縮した。残渣をメタノール(140ml)に溶解し真空で容積30mlまで濃縮した。得られた結晶性の懸濁物を5−10℃に冷却し、1時間静置後濾過し、洗浄して60℃以下で真空で乾燥して標記化合物33.6g(83.6%)を得て、次の工程に用いた。
融点:154−158℃。
[α](25,D)=+7.7°(c=1%,クロロホルム)。
NMR;1H NMR(500MHz,CDCl3(TMS),δ(ppm)):0.12(9H,s,17−OSi(CH3)3);0.19(9H,s,5−O−Si(CH3)3);0.33(3H,s,18−CH3);2.88(6H,s,N−CH3);3.86および3.98(4H,m,O−CH2−CH2−O);4.21(1H,m,H−11);6.61(2H,m,H−3’およびH−5’);7.00(2H,m,H−2’およびH−6’);9.56(1H,s,H−20)
13C NMR(125MHz,CDCl3(TMS),δ(ppm)):1.9(17−O−Si(CH3)3);2.6(5−O−Si(CH3)3);15.7(C−18);38.8(C−11);40.7(N−CH3);63.4および64.4(O−CH2−CH2−O);73.7(C−5);91.1(C−17);108.5(C−3);112.8(C−3’およびC−5’);127.6(C−2’およびC−6’);133.6(C−10);134.3(C−1’);135.9(C−9);148.3(C−4’);203.3(C−20)
【実施例7】
【0066】
11β−[4−(ジメチルアミノ)−フェニル]−17,20ξ−ジヒドロキシ−21−メトキシ−19−ノルプレグナ−4,9−ジエン−3−オン[式(IX)の化合物]
窒素下で、マグネシウム削り屑(4.2g,173mmol)、無水テトラヒドロフラン(60ml)および1,2−ジブロモ−エタン(2.4ml,28mmol)を攪拌機、温度計、滴下ロート、還流凝縮器、ガス導入および導出口を備えた500mlの四つ首フラスコに20−25℃で加えた。数分間攪拌後、混合物は還流温度に達した。ついで反応混合物を35−40℃に冷却し塩化水銀(II)(0.23g,0.85mmol)を加え、15分間攪拌後混合物を20−25℃に冷却して無水トルエン(20ml)を加えた。塩化メトキシメチル(12.8ml,168mmol)を無水トルエン(50ml)に溶解し、そのようにして得られた溶液6mlを四つ首フラスコの混合物に加えた。数分後反応混合物の温度は35℃に上昇した。反応混合物を0−(−5)℃に冷却し、残りの塩化メトキシメチルのトルエン溶液を、温度を0−(−5)℃に保ちながら2−2.5時間かけて加えた。添加終了後、11β−[(4−ジメチルアミノ)−フェニル]−3,3−[(1,2−エタンジイル)−ビス(オキシ)]−5,17α−ビス−[トリメチル−シリル−(オキシ)]−5α−エストラ−9−エン−17β−カルボアルデヒド(20.0g,32mmol)の無水トルエン溶液(80ml)を温度を0−(−5)℃に保ちながら1時間かけて加えた。反応完結後、反応混合物を1Mの硫酸水素カリウム水溶液(200ml)に温度を30℃以下に保つような速度で加えた。混合物を20−25℃で2時間攪拌し、ついで有機層を分離して1Mの硫酸水素カリウム(1x10ml)で洗浄した。水相を合わせて1Mの炭酸水素ナトリウム溶液(225ml)とジクロロメタン(75ml)の攪拌混合物に加えた。10−15分間攪拌後、有機層を分離した。水相をジクロロメタン(5x50ml)で抽出し、有機層を合わせて無水硫酸ナトリウム(2g)上で乾燥し、濾過し、ジクロロメタン(2x20ml)で洗い、濾液を木炭(2.5g)と10分間攪拌した。木炭を濾別し、ジクロロメタン(2x20ml)で洗い濾液を濃縮して12.51g(84.1%)の標記化合物を得た。
融点:105℃(軟化)。
[α](25,D)=+157.7°(c=1%,ジクロロメタン)
NMR:1H NMR((主ジアステレオマー)、500MHz,CDCl3(TMS),δ(ppm)):0.49(3H,s,18−CH3);2.91(6H,s,N−CH3);3.37(3H,s,O−CH3);3.49(1H,m,Hx−21);3.57(1H,m,Hy−21);3.81(1H,m,H−20);4.30(1H,m,H−11);5.73(1H,s,H−4);6.67(2H,m,H−3’およびH−5’);7.04(2H,m,H−2’およびH−6’)
13C NMR((主ジアステレオマー),125MHz,CDCl3(TMS),δ(ppm)):16.5(C−18);39.5(C−11);40.7(N−CH3);59.2(O−CH3);71.9(C−20);74.9(C−21);84.4(C−17);112.8(C−3’およびC−5’);122.5(C−4);127.6(C−2’およびC−6’);128.6(C−10);132.0(C−1’);147.2(C−9);148.5(C−4’);157.1(C−5);199.7(C−3)
【実施例8】
【0067】
11β−[4−(ジメチルアミノ)−フェニル]−17,20ξ−ジヒドロキシ−21−メトキシ−19−ノルプレグナ−4,9−ジエン−3−オン[式(IX)の化合物]
窒素下で、マグネシウムの切り屑(4.2g,173mmol)、無水テトラヒドロフラン(60ml)および1,2−ジブロモ−エタン(2.4ml,28mmol)を攪拌機、温度計、滴下ロート、還流凝縮器、ガス導入および導出口を備えた500mlの四つ口フラスコに20−25℃で加えた。数分間攪拌後、混合物は還流温度に達した。ついで反応混合物を35−40℃に冷却し塩化水銀(II)(0.23g,0.85mmol)を加え、15分間攪拌後混合物を20−25℃に冷却して無水トルエン(20ml)を加えた。臭化メトキシメチル(13.7ml,168mmol)を無水トルエン(50ml)に溶かしそのようにして得られた溶液6mlを四つ首フラスコの混合物に加えた。数分後反応混合物の温度は30−35℃に上昇した。反応混合物を10−15℃に冷却し、残りの塩化メトキシメチルトルエン溶液を、温度を10−15℃に保ちながら2−2.5時間かけて加えた。添加終了後、11β−[(4−ジメチルアミノ)−フェニル]−3,3−[(1,2−エタンジイル)−ビス(オキシ)]−5,17α−ビス−[トリメチル−シリル−(オキシ)]−5α−エストラ−9−エン−17β−カルボアルデヒド(20.0g,32mmol)の無水トルエン溶液(80ml)を温度を10−15℃に保ちながら1時間かけて加えた。反応完結後、反応混合物を1Mの硫酸水素カリウム水溶液(200ml)に温度を30℃以下に保つような速度で加えた。混合物を20−25℃で2時間攪拌し、ついで有機層を分離して1Mの硫酸水素カリウム(1x10ml)で洗浄した。水相を合わせて1Mの炭酸水素ナトリウム溶液(225ml)とジクロロメタン(75ml)の攪拌混合物に加えた。10−15分間攪拌後、有機層を分離した。水相をジクロロメタン(5x50ml)で抽出し、有機層を合わせて無水硫酸ナトリウム(2g)上で乾燥し、濾過し、ジクロロメタン(2x20ml)で洗い、濾液を木炭(2.5g)と10分間攪拌した。木炭を濾別し、ジクロロメタン(2x20ml)で洗い濾液を濃縮して10.76g(72.3%)の標記化合物を得た。
生成物の少量をn−ペンタンと攪拌、融点:105℃(ガラス状構造、軟化)。
[α](25,D)=+157.7°(c=1%,ジクロロメタン)
【実施例9】
【0068】
11β−[4−(ジメチルアミノ)−フェニル]−17−ヒドロキシ−21−メトキシ−19−ノルプレグナ−4,9−ジエン−3,20−ジオン[式(X)の化合物]
窒素下で、17,20ξ−ジヒドロキシ−11β−[(4−ジメチルアミノ)−フェニル]−21−メトキシ−19−ノルプレグナ−4,9−ジエン−3−オン(11.94g,25.7mmol)と無水トルエン(68ml)を攪拌機、温度計、滴下ロート、還流凝縮器、ガス導入および導出口を備えた500mlの四つ口フラスコに加えた。そのようにして得られた溶液に無水ジメチルスルホキシド(9.9ml,139.5mmol)、ピリジン(2.9ml,36.0mmol)およびトリフルオロ酢酸(0.99ml,12.85mmol)を20−25℃で加えた。ついでジシクロヘキシルカルボジイミド(10.6g,51.4mmol)のトルエン(54ml)溶液を反応混合物に加え、そのようにして得られた混合物を40℃で攪拌した。反応時間は3時間であった。反応完結後、反応混合物を20−25℃に冷却し1Mの硫酸水素カリウム溶液(78ml)を加えた。30分間攪拌後、沈殿した結晶を濾別し1Mの硫酸水素カリウム溶液(4x19ml)で洗浄した。濾液の2相を分離し、水相を1Mの苛性ソーダ溶液(254ml)に10−20℃で加えた。30分間攪拌後、沈殿した粗生成物を濾別し、水で洗い乾燥して9,45g(79.5%)の標記化合物を得た。
融点:105−110℃。
【0069】
粗生成物を次の実施例で述べる方法に従ってHPLCにより精製した。
NMR:1H NMR(500MHz,DMSO−d6(TMS),δ(ppm)):0.20(3H,s,18−CH3);2.82(6H,s,N−CH3);3.26(3H,s,O−CH3);4.20(1H,d,Hx−21);4.35(1H,m,H−11);4.49(1H,d,Hy−21);5.37(1H,s,OH);5.67(1H,s,H−4);6.62(2H,m,H−3’およびH−5’);7.00(2H,m,H−2’およびH−6’)
13C NMR(125MHz,DMSO−d6(TMS),δ(ppm)):15.8(C−18);38.7(C−11);40.1(N−CH3);58.3(O−CH3);75.4(C−21);88.6(C−17);112.5(C−3’およびC−5’);122.0(C−4);127.2(C−2’およびC−6’);128.1(C−10);132.0(C−1’);146.6(C−9);148.2(C−4’);156.5(C−5);197.9(C−3);208.9(C−20)
【実施例10】
【0070】
粗11β−[4−(ジメチルアミノ)−フェニル]−17−ヒドロキシ−21−メトキシ−19−ノルプレグナ−4,9−ジエン−3,20−ジオン[式(X)の化合物]のHPLCによる精製(実験室規模)
シリカゲル(510g,ZEOPREP C−GEL C−490L,粒径15−35μm;充填床長約60cm)を直径5cmの軸床圧縮HPLCカラムにスラリー充填法で充填し、カラムをシクロヘキサン/メチル−tert−ブチルエーテル/アセトン溶出液45:40:15の混合物で平衡化した。10gの粗標記化合物(活性成分含量:80%)をアセトン(30ml)とメチル−tert−ブチルエーテル(80ml)およびシクロヘキサン(90ml)の混合物に溶解して攪拌溶液に加えた。そのようにして得られた溶液を濾過しカラムに注入した。生成物を85ml/分の流速で溶出し、UV検出を用いた。最初の画分はおよそ50mlであり、純粋な標記化合物を含む主画分は600mlであった。溶出した主画分の濃縮により固体の標記化合物を得た。
融点:108−110℃。
[α](25,D)=+199.2°(c=1%クロロホルム)
NMR:1H NMR(500MHz、DMSO−d6(TMS),δ(ppm)):0.20(3H,s,18−CH3);2.82(6H,s,N−CH3);3.26(3H,s,O−CH3);4.20(1H,d,Hx−21);4.35(1H,m,H−11);4.49(1H,d,Hy−21);5.37(1H,s,OH);5.67(1H,s,H−4);6.62(2H,m,H−3’およびH−5’);7.00(2H,m,H−2’およびH−6’)
13C NMR(125MHz,DMSO−d6(TMS),δ(ppm)):15.8(C−18);38.7(C−11);40.1(N−CH3);58.3(O−CH3);75.4(C−21);88.6(C−17);112.5(C−3’およびC−5’);122.0(C−4);127.2(C−2’およびC−6’);128.1(C−10);132.0(C−1’);146.6(C−9);148.2(C−4’);156.5(C−5);197.9(C−3);208.9(C−20)
【実施例11】
【0071】
17−アセトキシ−11β−[4−(ジメチルアミノ)−フェニル]−21−メトキシ−19−ノルプレグナ−4,9−ジエン−3,20−ジオン[式(I)の化合物]
70%過塩素酸(6ml)を攪拌冷却((−20)−(−25)℃)した無水酢酸(45ml)に温度を(−15)℃以下に保つような速度で加えた。ついで11β−[4−(ジメチルアミノ)−フェニル]−17−ヒドロキシ−21−メトキシ−19−ノルプレグナ−4,9−ジエン−3,20−ジオン(15.5g)のジクロロメタン(60ml)溶液を(−20)−(−25)℃で加えた。反応完結後−薄層クロマトグラフィーにより−反応混合物をジクロロメタン(50ml)で稀釈し、(−10)℃に冷却し無水酢酸を分解するためイオン交換水(52ml)を加えた。10分間攪拌後、25%水酸化アンモニウム溶液(77ml)を温度を25℃(pH=7−8)に保つような速度で加えた。ついで沈殿したカルボアミド副産物を濾別し、水相を分離し、ジクロロメタン(2x30ml)で抽出し合わせた有機層を濃縮して16.2g(95.8%)の標記化合物を得て、それを次の実施例に記載の方法によりHPLCで精製した。
NMR:1H NMR(500MHz,CDCl3(TMS),δ(pm)):0.40(3H,s,18−CH3);2.10(3H,s,O−CO−CH3);2.90(6H,s,N−CH3);3.41(3H,s,O−CH3);4.09(1H,d,Hx−21);4.38(1H,m,H−11);4.29(1H,d,Hy−21);5.77(1H,br,H−4);6.62(2H,m,H−3’およびH−5’);6.96(2H,m,H−2’およびH−6’)
13C NMR(125MHz,CDCl3(TMS),δ(ppm)):15.6(C−18);21.1(O−CO−CH3);39.3(C−11);40.6(N−CH3);59.4(O−CH3);76.0(C−21);93.9(C−17);112.8(C−3’およびC−5’);123.0(C−4);127.3(C−2’およびC−6’);129.4(C−10);131.3(C−1’);145.5(C−9);148.7(C−4’);156.4(C−5);170.7(O−CO−CH3);199.4(C−3);202.7(C−20)
【実施例12】
【0072】
粗CDB−4124のHPLCによる精製(溶出液:シクロヘキサン:メチル−tert−ブチルエーテル:アセトン=60:30:10(実験室規模)[式(I)の化合物]
シリカゲル(510g,ZEOPREP C−GEL C−490L,粒径15−35μm;充填床長約60cm)を直径5cmの軸床圧縮HPLCカラムにスラリー充填法で充填し、カラムをシクロヘキサン/メチル−tert−ブチルエーテル/アセトン溶出液60:30:10の混合物で平衡化した。前の実施例で得られた式(I)の化合物(CDB−4124)5.1g(不純物含有率:4%以下)を溶出液(100ml)に溶解し、濾過してカラムに注入した。生成物を85ml/分の流速で溶出しUV検出を用いた。最初の画分は40mlであり、純CDB−4124を含む主画分は560mlであった。溶出した主画分の濃縮により固体の標記化合物を得た。収率:4.25g(83.33%),不純物含有率:0.5%以下。
融点:118℃。
[α](25,D)=+127.2°(c=1%,クロロホルム)
NMR:1H NMR(500MHz,CDCl3(TMS),δ(ppm)):0.40(3H,s,18−CH3);2.10(3H,s,O−CO−CH3);2.90(6H,s,N−CH3);3.41(3H,s,O−CH3);4.09(1H,d,Hx−21);4.38(1H,m,H−11);4.29(1H,d,Hy−21);5.77(1H,br,H−4);6.62(2H,m,H−3’およびH−5’);6.96(2H,m.H−2’およびH−6’)
13C NMR(125MHz,CDCl3(TMS),δ(ppm)):15.6(C−18);21.1(O−CO−CH3);39.3(C−11);40.6(N−CH3);59.4(O−CH3);76.0(C−21);93.9(C−17);112.8(C−3’およびC−5’);123.0(C−4);127.3(C−2’およびC−6’);129.4(C−10);131.3(C−1’);145.5(C−9);148.7(C−4’);156.4(C−5);170.7(O−CO−CH3);199.4(C−3);202.7(C−20)
【実施例13】
【0073】
5,10α−エポキシ−3,3−[1.2−エタンジイル−ビス(オキシ)]−5α−エストラ−9(11)−エン−17−オン[式(III)の化合物]
装置:回転速度可変プロペラ攪拌機、還流凝縮器および温度計を備えた500リットルの耐酸性スチール反応器。
【0074】
窒素下で、3,3−[1,2−エタンジイル−ビス(オキシ)]−エストラ−5(10),9(11)−ジエン−17−オン(21.0kg)をピリジン(1,106L,0.2mol−当量)と無水ジクロロメタン(105.2 l)の混合物に激しく攪拌して溶解し、溶液を(−6)−(−8)℃に冷却した。ヘキサクロロアセトン(2.455L)の添加後、0−(−2)℃の50%過酸化水素(26.97L)を攪拌溶液に温度を0℃以下に保つような速度で加えた。反応混合物を1−(−1)℃で20−24時間攪拌し、ついで0−5℃のジクロロメタン(175L)で稀釈し、過剰の過酸化水素をチオ硫酸ナトリウム5水和物(8.87mol−当量)の氷冷水溶液(150L)の添加によって分解した。反応混合物を1.5時間攪拌し、ついで有機相を分離した。水相をジクロロメタンで抽出し、有機層を合わせて水で洗い、無水硫酸ナトリウム上で乾燥し、濾過して濃縮した。油状の残渣を0.1%のピリジンを含む酢酸エチル−ジイソプロピルエーテルの1:3混合物(195.6L)から結晶化した。そのようにして得られた生成物を乾燥して11.500kg(52.13%)の標記化合物を得た。標記化合物の純度は98.5−98.9%(HPLCにより測定)であり;α−エポキシドを96.2%含んでいた。
融点:151−154℃。
[α](25,D)=125.0°(c=1%,クロロホルム)
【実施例14】
【0075】
5,10α−エポキシ−3,3−[1,2−エタンジイル−ビス(オキシ)]−17α−ヒドロキシ−5α−エストラ−9(11)−エン−17β−カルボニトリル[式(IV)の化合物]
装置:回転速度可変プロペラ攪拌機、還流凝縮器および温度計を備えた250リットルのホウロウ反応器。
【0076】
実施例13で得られた5,10α−エポキシ−3,3−[1,2−エタンジイル−ビス(オキシ)]−5α−エストラ−9(11)−エン−17−オン(9.9kg)をメタノール(39.6L)に懸濁し、ついで粉末シアン化カリウム(5.85kg,0.3mol)を20−25℃で加えた。酢酸(3.48L)を注意して加えた後、不均一な反応混合物を15分間にわたり55℃に暖め、ついで1時間で25℃に冷却し、この温度でさらに5時間攪拌した。反応完結後、水(39.6L)を30分間にわたり加え、得られた結晶性の生成物を濾過し、水で洗い乾燥することなく次の工程に用いた。乾燥した試料の融点:140−143℃。
[α](25,D)=+13.0°(c=1%,クロロホルム)
【実施例15】
【0077】
5,10α−エポキシ−3,3−[1,2−エタンジイル−ビス(オキシ)]−17α−[(トリメチル−シリル)−オキシ]−5α−エストラ−9(11)−エン−17β−カルボニトリル[式(V)の化合物]
装置:回転速度可変プロペラ攪拌機、還流凝縮器および温度計を備えた250リットルのホウロウ反応器。
【0078】
実施例14で得られた5,10α−エポキシ−3,3−[1,2−エタンジイル−ビス(オキシ)]−17α−ヒドロキシ−5α−エストラ−9(11)−エン−17β−カルボニトリルをジクロロメタン(90L)に激しく攪拌して溶解し、溶液を無水硫酸ナトリウム上で乾燥し、ついで60Lのジクロロメタンを溶液から蒸留した。そのようにして得られた溶液にイミダゾール(0.303kg)を加え、ついでトリメチルクロロシラン(7.2L)を20−25℃で20分間にわたり滴下した。1時間攪拌後、溶液をジクロロメタン(19.8L)と水(19.8L)で稀釈した。有機層を分離し、水で洗い、無水硫酸ナトリウム上で乾燥し、濾過して濃縮した。残渣をメタノール(18L)で処理し、0℃に冷却し、沈殿した結晶性の生成物を濾過し、0℃のメタノールで洗浄し40℃で真空乾燥して標記化合物10.1kg(78.4%)を得た。この生成物をさらに精製することなく次の工程に用いた。
融点:167−170℃。
[α](25,D)=+12.5°(c=1%,クロロホルム)
【実施例16】
【0079】
11β−[4−(ジメチルアミノ)−フェニル]−3,3−[1,2−エタンジイル−ビス(オキシ)]−5−ヒドロキシ−17α−[(トリメチル−シリル)−オキシ]−5α−エストラ−9−エン−17β−カルボニトリル[式(VI)の化合物]
装置:可変回転速度プロペラ攪拌機、還流凝縮器および温度計を備えた250リットルホウロウ反応器。
【0080】
窒素下で、マグネシウム切り屑(0.768g)、無水テトラヒドロフラン(5.59L)および1,2−ジブロモ−エタン(27.94ml)を装置に20−25℃で加えた。5−10分攪拌後、温度を上昇し始め活性化が有効であることを示した。
【0081】
並行して次の溶液を窒素下で25℃でつくった:無水テトラヒドロフラン(3.5L)、無水トルエン(19.6L)、4−ブロモ−N,N−ジメチル−アニリン(5.8kg)および1,2−ジブロモ−エタン(34.25ml)。この溶液から400mlをマグネシウム切り屑を含む溶液に加え、そのようにして得られた反応混合物を60度に暖めた。反応混合物の激しい還流は活性化が有効であることを示し、そこで冷却後残りの4−ブロモ−N,N−ジメチル−アニリンの溶液を滴下し、さらに2時間冷却しながら温度を14−16℃に保った。
【0082】
塩化銅(I)(93.11g)を得られたグリニヤ試薬溶液に加え、ついで反応混合物を20−25℃で5分間攪拌した。8−13℃に冷却後、ジクロロメタン(42L)中の5,10α−エポキシ−3,3−[1,2−エタンジイル−ビス(オキシ)]−17α−[(トリメチルシリル)−オキシ]−5α−エストラ−9(11)−エン−17β−カルボニトリル(10.0kg)を攪拌冷却した溶液に温度を10−15℃に保つような速度で滴下した。ついで冷却を停止し反応混合物をさらに4時間攪拌した。
【0083】
反応完結後、混合物を激しく攪拌したピロ硫酸ナトリウム(93.1g)を含む塩化アンモニウム溶液(23.3L,10%水溶液)に加え、ジクロロメタン(23.3L)で稀釈し、攪拌し静置した。有機層を分離後、水相をジクロロメタンで抽出し、有機層を合わせて水で洗い、無水硫酸ナトリウム上で乾燥し、濾過して濃縮した。残渣をメタノールから再結晶して10.5kg(85.71%)の標記化合物を得た。
融点:243−256℃。
[α](25,D)=−12.4°(c=1%,クロロホルム)
そのようにして得られた生成物をさらに精製することなく次の工程に用いた。
【実施例17】
【0084】
11β−[4−(ジメチルアミノ)−フェニル]−3,3−[1,2−エタンジイル−ビス(オキシ)]−5,17α−ビス−[(トリメチル−シリル)−オキシ]−5α−エストラ−9−エン−17β−カルボニトリル[式(VII)の化合物]
装置:可変回転速度プロペラ攪拌機、還流凝縮器および温度計を備えた250リットルホウロウ反応器。
【0085】
11β−[4−(ジメチルアミノ)−フェニル]−3,3−[1,2−エタンジイル−ビス(オキシ)]−5−ヒドロキシ−17α−[(トリメチル−シリル)−オキシ]−5α−エストラ−9−エン−17β−カルボニトリル(10.45kg)とイミダゾール(1.93kg)をジクロロメタン(42.75L)に20−25℃で攪拌溶解した。トリメチルクロロシラン(3.0L)を溶液に20分間にわたって滴下した。反応剤を添加中、イミダゾール塩酸が沈殿し始め反応の進行を示した。2時間攪拌後、反応混合物をジクロロメタン(19L)と水(19L)で稀釈し、数分間攪拌し、静置、ついで有機層を分離し、水で洗い、無水硫酸ナトリウム上で乾燥し、濾過して濃縮した。残渣をメタノールから結晶化し、濾過した生成物を真空乾燥して10.25kg(87.0%)の標記化合物を得た。
融点:164−166℃。
[α](25,D)=+14.7°(c=1%,クロロホルム)
【実施例18】
【0086】
11β−[4−(ジメチルアミノ)−フェニル]−3,3−[1,2−エタンジイル−ビス(オキシ)]−5,17α−ビス−[(トリメチル−シリル)−オキシ]−5α−エストラ−9−エン−17β−カルボアルデヒド[式(VIII)の化合物]
装置:可変回転速度プロペラ攪拌機、還流凝縮器および温度計を備えた250リットルホウロウ反応器。
【0087】
窒素下で、11β−[4−(ジメチルアミノ)−フェニル)]−3,3−[1,2−エタンジイル−ビス−(オキシ)]−5,17α−ビス−[(トリメチル−シリル)−オキシ]−5α−エストラ−9−エン−17β−カルボニトリル(8kg)をメチル−tert−ブチルエーテル(44L)とテトラヒドロフラン(3.4L)の混合物に溶解した。溶液を(−15)−(−20)℃に冷却し、ついで1M DIBAL−Hのシクロヘキサン(32L)溶液を(−15)−(−20)℃で15分かけて加えた。反応混合物を1時間攪拌し、ついで(−5)−(−10)℃の水(32L)と酢酸(16L)の混合物を窒素下で激しく攪拌しながら15−20分間かけて加えた。そのようにして得られた反応混合物を20−25℃で30分間攪拌し、ついで有機層を分離し、水(40L)、0.3M炭酸水素ナトリウム(2x40L)および水(40L)で洗った。有機層を乾燥せずに40−45℃で真空乾燥した。残渣をメタノール(28L)に溶解し6Lまで真空乾燥した。得られた結晶性の懸濁液を5−10℃に冷却し、1時間静置後濾過し、洗浄し60℃以下で真空乾燥して6.95kg(86.46%)の標記化合物を得て、それをさらに精製することなく次の工程に用いた。
融点:154−158℃。
[α](25,D)=+7.7℃(c=1%,クロロホルム)
【実施例19】
【0088】
11β−[4−(ジメチルアミノ)−フェニル]−17,20ξ−ジヒドロキシ−21−メトキシ−19−ノルプレグナ−4,9−ジエン−3−オン[式(IX)の化合物]
装置:可変回転速度プロペラ攪拌機、還流凝縮器および温度計を備えた250リットルホウロウ反応器。
【0089】
窒素下で、マグネシウム切り屑(1.05kg)、無水テトラヒドロフラン(15L)および1,2−ジブロモ−エタン(600ml)を20−25℃で装置に加えた。数分間攪拌後、反応混合物は還流温度に達した。ついで反応混合物を35−40℃に冷却し、塩化水銀(II)(57.5g)を加え、15分間攪拌後反応混合物を20−25℃に冷却し無水トルエン(5L)を加えた。塩化メトキシ−メチル(3.2L)を無水トルエン(12.5L)に溶解しそのようにして得られた溶液1.5Lを反応混合物に加えた。数分後反応混合物の温度は35℃に上昇した。反応混合物を0−(−5)℃に冷却し残りの塩化メトキシメチルのトルエン溶液を温度を0−(−5)℃に保ちながら2−2.5時間にわたって加えた。添加終了後、11β−[(4−ジメチルアミノ)−フェニル]−3,3−[(1,2−エタンジイル)−ビス(オキシ)]−5,17α−ビス−[トリメチル−シリル−(オキシ)]−5α−エストラ−9−エン−17β−カルボアルデヒド(5.0kg)トルエン(20L)溶液を温度を0−(−5)℃に保ちながら1時間かけて加えた。反応完結後、反応混合物を1Mの硫酸水素カリウム水溶液(50L)に温度を30℃以下に保つような速度で加えた。混合物を20−25℃で2時間攪拌し、ついで有機層を分離し1M硫酸水素カリウム(1x2,5L)で洗浄した。水相を合わせて攪拌している1M炭酸水素ナトリウム溶液(56L)とジクロロメタン(19L)の混合物に加えた。10−15分間攪拌後、有機層を分離した。水相をジクロロメタン(5x12.5L)で抽出し、合わせた有機層を無水硫酸ナトリウム(500g)上で乾燥し、濾過し、ジクロロメタン(2x2L)で洗い濾液を木炭(625g)と10分間攪拌した。木炭を濾別し、ジクロロメタン(2x5L)で洗い、濾液を濃縮して3.3kg(88.73%)の標記化合物を得た。
融点:105℃(軟化)。
[α](25,D)=+156.2°(c=1%,ジクロロメタン)。
【実施例20】
【0090】
11β−[4−(ジメチルアミノ)−フェニル]−17−ヒドロキシ−21−メトキシ−19−ノルプレグナ−4,9−ジエン−3,20−ジオン[式(X)の化合物]
装置:可変回転速度プロペラ攪拌機、還流凝縮器および温度計を備えた250リットルホウロウ反応器。
【0091】
窒素下で、17,20ξ−ジヒドロキシ−11β−[(4−ジメチルアミノ)−フェニル]−21−メトキシ−19−ノルプレグナ−4,9−ジエン−3−オン(3.2kg)および無水トルエン(18L)を反応器に加えた。そのようにして得られた溶液に無水ジメチルスルホキシド(2.7L)、ピリジン(0.78L)およびトリフルオロ酢酸(0.265L)を20−25℃で加えた。ついでジシクロヘキシルカルボジイミド(2.84kg)のトルエン溶液(14.5L)を反応混合物に加え、そのようにして得られた混合物を40℃で攪拌した。反応時間は3時間であった。反応完結後、反応混合物を20−25℃に冷却して1M硫酸水素カリウム溶液(21L)を加えた。30分間攪拌後、沈殿した結晶を濾別し1M硫酸水素カリウム溶液(4x5L)で洗浄した。濾液の2相を分離して、水相を1M水酸化ナトリウム溶液(68L)に10−20℃で加えた。30分間攪拌後、沈殿した粗生成物を濾別し、水で洗い2,556kg(80.0%)の標記化合物を得た。
【0092】
粗生成物を次の実施例で述べる方法に従ってHPLCにより精製した。
【実施例21】
【0093】
粗11β−[4−(ジメチルアミノ)−フェニル]−17−ヒドロキシ−21−メトキシ−19−ノルプレグナ−4,9−ジエン−3,20−ジオン[式(X)の化合物]のHPLCによる精製(工業的規模)
シリカゲル(8kg,ZEOPREP C−GEL C−490L,粒径15−35μm;充填床長約60cm)を直径20cmの軸床圧縮HPLCカラムにスラリー充填法で充填し、カラムをシクロヘキサン/メチル−tert−ブチルエーテル/アセトン、53:34:12の混合流出液で平衡化した。160gの粗標記化合物(活性成分含有率:80%)をアセトン(0.48L)とメチル−tert−ブチルエーテル(1.28L)の混合物に溶解しシクロヘキサン(1.44L)を攪拌溶液に加えた。そのようにして得られた溶液を濾過してカラムに注入した。生成物は80L/時間の流速で流出しUV検出を用いた。最初の画分はおよそ1Lであり、純粋な標記化合物を含む主画分はおよそ14Lであった。固体の標記化合物流出した主画分の濃縮によって得られるが、好ましくは主画分の濃縮後残渣からジクロロメタンを蒸留除去し、生成物をジクロロメタンに溶解した。このジクロロメタン溶液を次の工程に用いた。
収率:120g(75%)の純粋な固体の標記化合物またはジクロロメタン溶液中の活性成分含量。不純物含有率:4%以下。
融点:105−110℃。
[α](25,D)=+199.2°(c=1%,クロロホルム)
【実施例22】
【0094】
粗17−アセトキシ−11β−[4−(ジメチルアミノ)−フェニル]−21−メトキシ−19−ノルプレグナ−4,9−ジエン−3,20−ジオン[式(I)の化合物]
装置:可変回転速度プロペラ攪拌機、還流凝縮器および温度計を備えた250リットルホウロウ反応器。
【0095】
70%過塩素酸(1.8L)を攪拌冷却((−20)−(−25)℃)した無水酢酸(13.5L)に温度を(−15)℃以下に保つような速度で加えた。ついで11β−[4−(ジメチルアミノ)−フェニル]−17−ヒドロキシ−21−メトキシ−19−ノルプレグナ−4,9−ジエン−3,20−ジオン(4.65kg)のジクロロメタン(18L)溶液に(−20)−(−25)℃で加えた。反応完結後−薄層クロマトグラフィーによって−反応混合物をジクロロメタン(15L)で稀釈し、(−10)℃に冷却して無水酢酸を分解するためイオン交換水(15.5L)を加えた。10分間攪拌後、25%水酸化アンモニウム溶液(23L)を温度を25℃(pH=7−8)に保つような速度で加えた。ついで沈殿したカルボアミド副産物を濾別し、水相を分離し、ジクロロメタン(2x9L)で抽出し有機層を合わせて濃縮し4.73kg(93.79%)の標記化合物(CDB−4124)を得て、それを次の実施例で述べる方法に従ってHPLCにより精製した。
【実施例23】
【0096】
粗CDB−4124のHPLC(工業的規模)による精製[式(I)の化合物]
シリカゲル(8kg, ZEOPREP C−GEL C−490L,粒径15−35μm;充填床長約60cm)を直径20cmの軸床圧縮HPLCカラムにスラリー充填法で充填し、カラムをシクロヘキサン/メチル−tert−ブチルエーテル/アセトン、53:35:12の混合流出液で平衡化した。先の実施例で得られた式(I)(CDB−4124)の粗化合物80g(不純物含有率:4%以下)を溶出液(1.6L)に溶解し、濾過してカラムに注入した。生成物を80L/時間の流速で溶出しUV検出を用いた。最初の画分は0.7L、純CDB−4124を含む主画分はおよそ10Lであった。固体の標記化合物が溶出した主画分の濃縮によって得られあるいは主画分の濃縮後生成物をメタノールに溶解しメタノール溶液として得られた。収量:固体の標記化合物またはメタノール溶液中の活性成分含有量として70g。不純物含有率:0.5%以下。
融点:118℃
[α](25,D)=+127.2°(c=1%,クロロホルム)
【特許請求の範囲】
【請求項1】
式(I)の17−アセトキシ−11β−[4−(ジメチルアミノ)−フェニル]−21−メトキシ−19−ノルプレグナ−4,9−ジエン−3,20−ジオンを
【化1】
式(II)の3,3−[1,2−エタンジイル−ビス(オキシ)]−エストラ−5(10),9(11)−ジエン−17−オンから合成する工業的プロセスで、
【化2】
i)式(II)の3,3−[1,2−エタンジイル−ビス(オキシ)]−エストラ−5,(10),9(11)−ジエン−17−オンの5(10)位の二重結合への過酸化水素によるエポキシドの形成、
【化3】
ii)得られた式(III)の5,10α−エポキシ−3,3−[1,2−エタンジイル−ビス(オキシ)]−5α−エストラ−9(11)−エン−17−オンの17位へのインサイツで生じたシアン化水素の付加、
【化4】
iii)生じた式(IV)の5,10α−エポキシ−3,3−[1,2−エタンジイル−ビス(オキシ)]−17α−ヒドロキシ−5α−エストラ−9(11)−エン−17β−カルボニトリルの17位の水酸基のトリメチルクロロシランによるシリル化、
【化5】
iv)得られた式(V)の5,10α−エポキ−3,3−[1,2−エタンジイル−ビス(オキシ)]−17−[トリメチル−シリル−オキシ]−5α−エストラ−9(11)−エン−17β−カルボニトリルの、
【化6】
臭化4−(ジメチルアミノ)−フェニルマグネシウムグリニヤ試薬とのCuCl存在下での反応(Teutsch反応)、
v)生じた式(VI)の11β−[4−(ジメチルアミノ)−フェニル]−3,3−[1,2−エタンジイル−ビス(オキシ)−5−ヒドロキシ−17α−[トリメチルシリル−(オキシ)]−5α−エストラ−9−エン−17β−カルボニトリルの5位の水酸基のトリメチルクロロシランによるシリル化、
【化7】
vi)得られた式(VII)の11β−[4−(ジメチルアミノ)−フェニル]−3,3−[1,2−エタンジイル−ビス(オキシ)]−5,17α−ビス−[トリメチル−シリル−(オキシ)]−5α−エストラ−9−エン−17β−カルボニトリルの水素化ジイソブチルアルミニウムとの反応および反応混合物への酸付加の後、
【化8】
vii)得られた式(VIII)の11β−[4−(ジメチルアミノ)−フェニル]−3,3−[1,2−エタンジイル−ビス(オキシ)]−5,17α−ビス−[トリメチル−シリル−(オキシ)]−5α−エストラ−9−エン−17β−カルボアルデヒドのインサイツで生じたメトキシ−メチルグリニヤ試薬によるメトキシ−メチル化および同時にトリメチルシリル保護基の加水分解、
【化9】
viii)得られた式(IX)の17,20ξ−ジヒドロキシ−11β−[4−(ジメチルアミノ)−フェニル]−21−メトキシ−19−ノルプレグナ−4,9−ジエン−3−オンの20位の水酸基のジメチルスルホキシドおよび有機強酸の存在下でのジシクロヘキシルカルボジイミドによる酸化(Swern酸化)、およびある場合にはクロマトグラフィーによる精製の後、
【化10】
ix)得られた式(X)の11β−[4−(ジメチルアミノ)−フェニル]−17−ヒドロキシ−21−メトキシ−19−ノルプレグナ−4,9−ジエン−3,20−ジオンの17位の水酸基の過塩素酸存在下での無水酢酸によるアセチル化、およびある場合には得られた式(I)の7−アセトキシ−11β−[4−(ジメチルアミノ)−フェニル]−21−メトキシ−19−ノルプレグナ−4,9−ジエン−3,20−ジオンをクロマトグラフィーにより精製、
【化11】
することを特徴とする工業的プロセス。
【請求項2】
工程iv)において式(V)の5,10α−エポキシ−3,3−[1,2−エタンジイル−ビス(オキシ)]−17−[トリメチル−シリル−オキシ]−5α−エストラ−9(11)−エン−17β−カルボニトリルと比べ0.25±0.025当量過剰の臭化4−(ジメチルアミノ)−フェニルマグネシウムグリニヤ試薬を用いることを特徴とする請求項1に記載のプロセス。
【請求項3】
工程viii)の有機強酸としてトリフルオロ酢酸を用いることを特徴とする請求項1に記載のプロセス。
【請求項4】
式(VII)の11β−[4−(ジメチルアミノ)−フェニル]−3,3−[1,2−エタンジイル−ビス(オキシ)]−5,17α−ビス[トリメチル−シリル−(オキシ)]−5α−エストラ−9−エン−17β−カルボニトリル。
【請求項5】
式(VIII)の11β−[4−(ジメチルアミノ)−フェニル]−3,3−[1,2−エタンジイル−ビス(オキシ)]−5,17α−ビス−[トリメチル−シリル−(オキシ)]−5α−エストラ−9−エン−17β−カルボアルデヒド。
【請求項1】
式(I)の17−アセトキシ−11β−[4−(ジメチルアミノ)−フェニル]−21−メトキシ−19−ノルプレグナ−4,9−ジエン−3,20−ジオンを
【化1】
式(II)の3,3−[1,2−エタンジイル−ビス(オキシ)]−エストラ−5(10),9(11)−ジエン−17−オンから合成する工業的プロセスで、
【化2】
i)式(II)の3,3−[1,2−エタンジイル−ビス(オキシ)]−エストラ−5,(10),9(11)−ジエン−17−オンの5(10)位の二重結合への過酸化水素によるエポキシドの形成、
【化3】
ii)得られた式(III)の5,10α−エポキシ−3,3−[1,2−エタンジイル−ビス(オキシ)]−5α−エストラ−9(11)−エン−17−オンの17位へのインサイツで生じたシアン化水素の付加、
【化4】
iii)生じた式(IV)の5,10α−エポキシ−3,3−[1,2−エタンジイル−ビス(オキシ)]−17α−ヒドロキシ−5α−エストラ−9(11)−エン−17β−カルボニトリルの17位の水酸基のトリメチルクロロシランによるシリル化、
【化5】
iv)得られた式(V)の5,10α−エポキ−3,3−[1,2−エタンジイル−ビス(オキシ)]−17−[トリメチル−シリル−オキシ]−5α−エストラ−9(11)−エン−17β−カルボニトリルの、
【化6】
臭化4−(ジメチルアミノ)−フェニルマグネシウムグリニヤ試薬とのCuCl存在下での反応(Teutsch反応)、
v)生じた式(VI)の11β−[4−(ジメチルアミノ)−フェニル]−3,3−[1,2−エタンジイル−ビス(オキシ)−5−ヒドロキシ−17α−[トリメチルシリル−(オキシ)]−5α−エストラ−9−エン−17β−カルボニトリルの5位の水酸基のトリメチルクロロシランによるシリル化、
【化7】
vi)得られた式(VII)の11β−[4−(ジメチルアミノ)−フェニル]−3,3−[1,2−エタンジイル−ビス(オキシ)]−5,17α−ビス−[トリメチル−シリル−(オキシ)]−5α−エストラ−9−エン−17β−カルボニトリルの水素化ジイソブチルアルミニウムとの反応および反応混合物への酸付加の後、
【化8】
vii)得られた式(VIII)の11β−[4−(ジメチルアミノ)−フェニル]−3,3−[1,2−エタンジイル−ビス(オキシ)]−5,17α−ビス−[トリメチル−シリル−(オキシ)]−5α−エストラ−9−エン−17β−カルボアルデヒドのインサイツで生じたメトキシ−メチルグリニヤ試薬によるメトキシ−メチル化および同時にトリメチルシリル保護基の加水分解、
【化9】
viii)得られた式(IX)の17,20ξ−ジヒドロキシ−11β−[4−(ジメチルアミノ)−フェニル]−21−メトキシ−19−ノルプレグナ−4,9−ジエン−3−オンの20位の水酸基のジメチルスルホキシドおよび有機強酸の存在下でのジシクロヘキシルカルボジイミドによる酸化(Swern酸化)、およびある場合にはクロマトグラフィーによる精製の後、
【化10】
ix)得られた式(X)の11β−[4−(ジメチルアミノ)−フェニル]−17−ヒドロキシ−21−メトキシ−19−ノルプレグナ−4,9−ジエン−3,20−ジオンの17位の水酸基の過塩素酸存在下での無水酢酸によるアセチル化、およびある場合には得られた式(I)の7−アセトキシ−11β−[4−(ジメチルアミノ)−フェニル]−21−メトキシ−19−ノルプレグナ−4,9−ジエン−3,20−ジオンをクロマトグラフィーにより精製、
【化11】
することを特徴とする工業的プロセス。
【請求項2】
工程iv)において式(V)の5,10α−エポキシ−3,3−[1,2−エタンジイル−ビス(オキシ)]−17−[トリメチル−シリル−オキシ]−5α−エストラ−9(11)−エン−17β−カルボニトリルと比べ0.25±0.025当量過剰の臭化4−(ジメチルアミノ)−フェニルマグネシウムグリニヤ試薬を用いることを特徴とする請求項1に記載のプロセス。
【請求項3】
工程viii)の有機強酸としてトリフルオロ酢酸を用いることを特徴とする請求項1に記載のプロセス。
【請求項4】
式(VII)の11β−[4−(ジメチルアミノ)−フェニル]−3,3−[1,2−エタンジイル−ビス(オキシ)]−5,17α−ビス[トリメチル−シリル−(オキシ)]−5α−エストラ−9−エン−17β−カルボニトリル。
【請求項5】
式(VIII)の11β−[4−(ジメチルアミノ)−フェニル]−3,3−[1,2−エタンジイル−ビス(オキシ)]−5,17α−ビス−[トリメチル−シリル−(オキシ)]−5α−エストラ−9−エン−17β−カルボアルデヒド。
【公表番号】特表2010−531346(P2010−531346A)
【公表日】平成22年9月24日(2010.9.24)
【国際特許分類】
【出願番号】特願2010−514149(P2010−514149)
【出願日】平成20年6月19日(2008.6.19)
【国際出願番号】PCT/HU2008/000073
【国際公開番号】WO2009/001148
【国際公開日】平成20年12月31日(2008.12.31)
【出願人】(591180314)リヒター ゲデオン ニルバーノシャン ミーケデーレスベニュタールシャシャーグ (33)
【Fターム(参考)】
【公表日】平成22年9月24日(2010.9.24)
【国際特許分類】
【出願日】平成20年6月19日(2008.6.19)
【国際出願番号】PCT/HU2008/000073
【国際公開番号】WO2009/001148
【国際公開日】平成20年12月31日(2008.12.31)
【出願人】(591180314)リヒター ゲデオン ニルバーノシャン ミーケデーレスベニュタールシャシャーグ (33)
【Fターム(参考)】
[ Back to top ]