
18SrRNA検出用FRETプローブ
【課題】18S rRNAを高感度で定量的に検出することが可能なFRETプローブを提供する。
【解決手段】配列番号1に記載の塩基配列からなる核酸の第631〜725位の領域に、隣接してハイブリダイズ可能な第1のプローブ及び第2のプローブからなり、第1のプローブは第1の蛍光物質で標識されており、第2のプローブは第1の蛍光物質と蛍光共鳴エネルギー移動(FRET)を起こす第2の蛍光物質で標識されている、18S rRNA検出用FRETプローブ。
【解決手段】配列番号1に記載の塩基配列からなる核酸の第631〜725位の領域に、隣接してハイブリダイズ可能な第1のプローブ及び第2のプローブからなり、第1のプローブは第1の蛍光物質で標識されており、第2のプローブは第1の蛍光物質と蛍光共鳴エネルギー移動(FRET)を起こす第2の蛍光物質で標識されている、18S rRNA検出用FRETプローブ。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は18S rRNA検出用蛍光共鳴エネルギー移動(FRET)プローブに関する。
【背景技術】
【0002】
リボソームの合成過程は真核細胞において最もエネルギーが消費される過程である。真核細胞で転写される遺伝子の6割強はリボソームに関わる遺伝子であり、リボソームRNA(rRNA)の転写は細胞の分化・増殖と密接な関係にある。また細胞内のエネルギー消費調節にも大きく影響している。rRNA量の増減は、老化、寿命、細胞周期、細胞増殖、癌、栄養状態、低酸素状態等の生体の状態や疾患を大きく反映したものである。
【0003】
例えば、非特許文献1には、アルツハイマー病患者の末梢血中に含まれる18S rRNAと28S rRNAの量比が、健常人のそれと異なることが報告されている。
【0004】
真核生物のrRNAには、18S、28S及び5.8Sの3種類が存在する。18S rRNAは小サブユニットを構成し、28S及び5.8S rRNAは大サブユニットを構成する。
【0005】
rRNAの検出方法としては、in situハイブリダイゼーション(例えば、非特許文献2を参照)、アクリジンオレンジによる染色(例えば、非特許文献3を参照)、quenched autoligating FRET(QFRET)プローブによる染色(例えば、非特許文献4を参照)等が報告されている。
【0006】
非特許文献3の方法は、アクリジンオレンジが2本鎖核酸と結合した場合と1本鎖核酸と結合した場合でそれぞれ発する蛍光の波長が異なることを利用したものである。非特許文献4の方法は、2本のプローブが近接すると、プローブ間で共有結合するように2価試薬により修飾された、蛍光共鳴エネルギー移動(FRET)プローブを使用するものである。標的RNA分子上に隣接してハイブリダイズしたFRETプローブ間で共有結合が形成されることにより、FRETが起きる状況が維持される。これにより、信号の増幅が可能となり、より少ない分子の有無を定性的に判断できる。
【0007】
特許文献1には、18S、28S及び5.8S rRNAをコードするリボソームDNA(rDNA)の塩基配列及びその部分配列が記載されている。
【0008】
特許文献2には、FRET効率の高いFRETプローブを設計する方法が記載されている。
【先行技術文献】
【特許文献】
【0009】
【特許文献1】特表2009−504192号公報
【特許文献2】特開2007−37465号公報
【非特許文献】
【0010】
【非特許文献1】da Silva AM、et al、“Quantitative evaluation of the rRNA in Alzheimer’s disease”、Mech Ageing Dev.、2000、120(1−3)、57−64
【非特許文献2】Wallner G、et al、“Optimizing fluorescent in situ hybridization with rRNA−targeted oligonucleotide probes for flow cytometric identification of microorganisms”、Cytometry、1993、14(2)、136−43
【非特許文献3】Gordon RY、et al、“Acridine orange as an indicator of the cytoplasmic ribosome state”、Cytometry、1997、29(3)、215−21
【非特許文献4】Hiroshi Abe、Eric T. Kool、“Flow cytometric detection of specific RNAs in native human cells with quenched autoligating FRET probes”、PNAS、2006、103(2)、263−268
【発明の概要】
【発明が解決しようとする課題】
【0011】
しかしながら、非特許文献2の方法では、2本鎖核酸と1本鎖核酸とを染め分けることが可能であるものの、18S rRNAを選択的に検出することは不可能である。また、18S rRNAの検出を目的とした場合、細胞内に数十万分子以上存在することから、非特許文献4のQFRETプローブによる信号の増幅は必要ない。また、非特許文献4の方法では、連続的な信号の増幅により定量性が低下する場合がある。そこで、本発明は、18S rRNAを高感度で定量的に検出することが可能なFRETプローブを提供することを目的とする。
【課題を解決するための手段】
【0012】
本発明は、配列番号1に記載の塩基配列からなる核酸の第631〜725位(番目)の領域に、隣接してハイブリダイズ可能な第1のプローブ及び第2のプローブからなり、第1のプローブは第1の蛍光物質で標識されており、第2のプローブは前記第1の蛍光物質とFRETを起こす第2の蛍光物質で標識されている、18S rRNA検出用FRETプローブを提供する。
【0013】
上記本発明のFRETプローブによれば、18S rRNAを高感度で定量的に検出することができる。
【0014】
上記第1のプローブは、配列番号1に記載の塩基配列からなる核酸の第669〜687位の領域にハイブリダイズ可能な核酸又は核酸誘導体であり、上記第2のプローブは、配列番号1に記載の塩基配列からなる核酸の第645〜664位の領域にハイブリダイズ可能な核酸又は核酸誘導体であることがより好ましい。
【0015】
さらに、上記第1のプローブは、配列番号2に記載の塩基配列からなる2’−O−メチル オリゴリボヌクレオチドであり、上記第2のプローブは、配列番号3に記載の塩基配列からなる2’−O−メチル オリゴリボヌクレオチドであることが更に好ましい。
【0016】
このようなプローブは、18S rRNAを高感度で定量的に検出する性能が更に高い。
【発明の効果】
【0017】
本発明により、18S rRNAを高感度で定量的に検出することが可能なFRETプローブが提供される。
【図面の簡単な説明】
【0018】
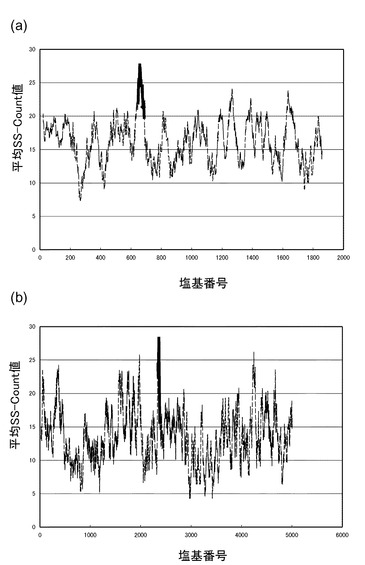
【図1】(a)は、18S rRNAの塩基配列について、横軸に塩基番号、縦軸にSS−count値の平均値をプロットしたグラフである。(b)は、28S rRNAの塩基配列について、横軸に塩基番号、縦軸にSS−count値の平均値をプロットしたグラフである。
【図2】(a)及び(b)は、実験例1の結果を示したグラフである。
【図3】(a)〜(d)は、実験例2の結果を示したグラフである。
【図4】実験例3の結果を示したグラフ及び写真である。
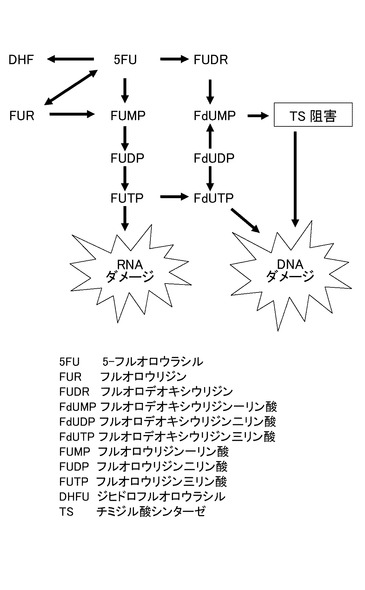
【図5】5FUとFUDRの作用機序を示す図である。
【図6】実験例4の結果を示したグラフである。
【図7】実験例5の結果を示したグラフである。
【図8】(a)及び(b)は、実験例6の結果を示したグラフである。
【図9】18S rRNAの塩基配列について、横軸に塩基番号、縦軸にSS−count値の平均値をプロットしたグラフである。
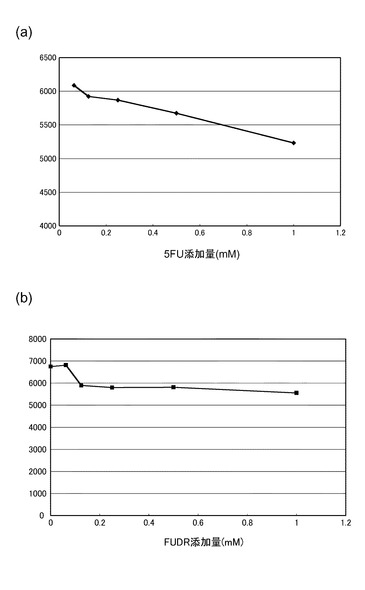
【図10】(a)〜(c)は、実験例7の結果を示したグラフである。
【発明を実施するための形態】
【0019】
(プローブの設計)
本実施形態の18S rRNA検出用FRETプローブは、配列番号1に記載の塩基配列からなる核酸の第631〜725位の領域に、隣接してハイブリダイズ可能な第1のプローブ及び第2のプローブからなり、第1のプローブは第1の蛍光物質で標識されており、第2のプローブは前記第1の蛍光物質と蛍光共鳴エネルギー移動(FRET)を起こす第2の蛍光物質で標識されている。
【0020】
上記第1のプローブ及び第2のプローブのうち、隣接して標的核酸にハイブリダイズした状態で、(標的配列の相補鎖配列で)5’側に位置する方を5’側プローブと呼び、他方を3’側プローブと呼ぶ。本明細書において、「隣接」とは、5’側プローブの3’末端塩基と3’側プローブの5’末端塩基との間が、0〜8塩基、好ましくは0〜4塩基離れて隣り合うことをいう。5’側プローブの3’末端及び3’側プローブの5’末端を、それぞれFRETを起こす組み合わせの蛍光物質で標識する。FRETを起こす蛍光物質の組み合わせのうち、エネルギーを与える方の物質をドナー蛍光物質と呼び、エネルギーを受けとる方の分子をアクセプター蛍光物質と呼ぶ。5’側プローブと3’側プローブのいずれにドナー蛍光物質を標識してもよい。以下、ドナー蛍光物質で標識した方のプローブをドナープローブと呼び、アクセプター蛍光物質で標識した方のプローブをアクセプタープローブと呼ぶ場合がある。
【0021】
本実施形態のFRETプローブを設計する場合、配列番号1に記載の塩基配列からなる核酸の第631〜725位の領域に隣接してハイブリダイズ可能なようにドナープローブとアクセプタープローブを設計する。
【0022】
ドナープローブとアクセプタープローブの融解温度(Tm値)は、ほぼ一致することが好ましい。これは、ドナープローブとアクセプタープローブをほぼ同じ確率で標的配列に結合させるためである。
【0023】
ドナープローブとアクセプタープローブのTm値は、36℃以上であることが好ましい。これは生細胞が生息可能な温度域で標的核酸とプローブがハイブリダイズしなければならないためである。また、ドナープローブとアクセプタープローブの長さは、それぞれ15〜25塩基であることが好ましく、16〜22塩基であることがより好ましい。
【0024】
FRETプローブは、蛍光物質に隣接してグアニン(G)塩基が存在しないことが好ましい。これは、蛍光物質の近傍に核酸塩基(特にグアニン)が存在すると蛍光物質が消光する場合があるためである。
【0025】
また、ドナープローブ同士、アクセプタープローブ同士、又はドナープローブ及びアクセプタープローブが相補的に結合しにくい配列であることが好ましい。これは、相補的な配列が存在すると、プローブのみで互いにハイブリダイズしてしまう場合があるためである。その結果、標的核酸に依存せずFRETを起こしたり、プローブが定量的に標的核酸にハイブリダイズできなくなる等の問題が生じる場合がある。経験的に、4塩基以上連続して相補的な配列を持たないような配列を選択するとよい。プローブ単独又はプローブ間で相補的に結合しにくい配列であるか否かは、実際にプローブを合成し、溶液中における蛍光測定により確認することができる。
【0026】
(プローブの合成)
上記のようにして設計したFRETプローブの塩基配列に基づいて、実際にFRETプローブを合成する。プローブは核酸又は核酸誘導体により構成される。より具体的には、DNAやRNA等の核酸、ペプチド核酸(PNA)、Locked Nucleic Acid(LNA)、ホスホロチオエートオリゴリボヌクレオチド等の核酸誘導体、及び、部分的にこれらの核酸や核酸誘導体が混合したものが挙げられる。また、U(ウラシル)がT(チミン)に置き換えられていてもよい。FRETプローブがRNAである場合、RNaseによる分解を抑制するために、2’−O−メチル オリゴリボヌクレオチドとすることが好ましい。
【0027】
(蛍光物質)
5’側プローブ及び3’側プローブの一方をドナー蛍光物質で修飾し、他方をアクセプター蛍光物質で修飾する。ドナー蛍光物質とアクセプター蛍光物質との間の距離は、5’側プローブ及び3’側プローブが標的核酸にハイブリダイズした状態で4塩基であることが好ましい。この距離がFRETを起こすのに適しているためである。
【0028】
一実施形態において、プローブが標的核酸にハイブリダイズした状態で、5’側プローブの3’末端と、3’側プローブの5’末端の間の距離が4塩基となるようにプローブを設計し、5’側プローブの3’末端と、3’側プローブの5’末端を蛍光物質で修飾する。別の実施形態において、蛍光物質で修飾するのはプローブの末端ではなく、より内側の塩基である。このような場合であっても、プローブが標的核酸にハイブリダイズした状態で、ドナー蛍光物質とアクセプター蛍光物質との間の距離が4塩基であれば、効率よくFRETを起こすことができる。
【0029】
ドナー蛍光物質とアクセプター蛍光物質の組み合わせとしては、例えば、表1に示すものが使用できる。
【0030】
【表1】

【0031】
(18S rRNAの検出)
一実施形態において、インビトロで18S rRNAを検出し、定量することができる。サンプルとしては、対象から抽出した全RNA等を使用できる。例えば、1μMのドナープローブおよびアクセプタープローブを溶解した緩衝液中に全RNAを混合する。プローブを標的核酸にハイブリダイズさせるためには、例えば150mM NaCl−15mM Sodium Citrate pH7.0 (SSC)溶液中、温度37℃等の条件下で20分間放置するとよい。続いて、蛍光顕微鏡、蛍光分光光度計等の装置を用いて、FRETの蛍光を検出する。より具体的には、ドナー蛍光物質の励起波長の光を照射し、アクセプター蛍光物質の蛍光を測定する。例えば、ドナー蛍光物質にAlexaFluor488(インビトロジェン社)を用い、アクセプター蛍光物質にAlexaFluor647(インビトロジェン社)を用いた場合、ドナー蛍光物質の励起波長は488nmであり、アクセプター蛍光物質の蛍光波長は630nmである。
【0032】
別の実施形態において、生きた又は固定した細胞内で18S rRNAを検出し、定量することができる。生きた細胞へのプローブの導入には、後述するストレプトリジンO(SLO)を使用する方法、マイクロインジェクション、エレクトロポレーション、リポフェクション等の方法が使用できる。固定した細胞を対象として18S rRNAを検出する場合には、細胞へのプローブの導入には、界面活性剤を用いる等の方法が使用できる。細胞内にプローブを導入する方法は条件が異なってくるので一概に同じ条件を設定できないが、ハイブリダイゼーションに限れば数分でハイブリダイズする。続いて、蛍光顕微鏡、フローサイトメーター等を用いて、FRETの蛍光を検出することができる。
【実施例】
【0033】
以下、本発明の実施例を示して、本発明を更に具体的に説明するが、本発明はこれらの実施例に限定されるものではなく、本発明の技術的思想を逸脱しない範囲での種々の変更が可能である。
【0034】
(FRETプローブの設計及び合成)
ヒト18S rRNA遺伝子(配列番号1、Genbankアクセッション番号K03432.1)及びヒト28S RNA遺伝子(配列番号4、Genbankアクセッション番号NR_003287)の塩基配列を使用し、特開2007−37465号公報に記載された方法にしたがって、18S rRNA及び28S RNA検出用FRETプローブを設計した。
【0035】
具体的には、18S rRNA及び28S RNAのそれぞれの塩基配列について、次の操作を行った。まず、長さnの塩基配列のi番目(1≦i≦n)の塩基のSS−count値S(i)を算出した。SS−count値とは、RNAの高次構造予測の指標の一つである。SS−count値は、RNAの各塩基における相補鎖を形成しない場合の数を表し、この数値が高い塩基ほど、高次構造の中で熱的に不安定で1本鎖となっている確率が高い。SS−count値は、Zuckerらによって開発されたRNAの2次構造を予測するプログラムである、mfold(Zuker M. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 2003 Jul 1;31(13):3406−15.)を使用して算出することができる。
【0036】
Lは一組のFRETプローブがハイブリダイズする標的部位全体の長さである。ここではドナープローブ20塩基、アクセプタープローブ20塩基、ドナー色素−アクセプター色素間の至適間隔4塩基の合計L=44塩基とした。
【0037】
続いて、18S及び28S rRNAの各塩基配列の塩基番号をiとし、1≦i≦n−L+1である全てのiについて、i番目の塩基からi+L−1番目の塩基までのSS−count値S(i)の平均値M(i)を算出した。図1(a)は、18S rRNAの塩基配列について、横軸に塩基番号、縦軸にM(i)をプロットしたグラフである。図1(b)は、28S rRNAの塩基配列について、図1(a)と同様にして作成したグラフである。
【0038】
図1(a)及び(b)において、塩基配列上のM(i)の値が大きい領域は、1本鎖となっている確率が高いため、FRETプローブがハイブリダイズする標的領域として適していると考えられる。図1(a)及び(b)中、FRETプローブの標的領域として選択した領域を太線で示した。
【0039】
図1(a)及び(b)に示したFRETプローブの標的領域から、ドナープローブ及びアクセプタープローブの塩基配列を選択した。FRETプローブとしては、M(i)の値が大きいだけでなく、上述した理由から、次の条件を満たす配列を選択した。
(1)ドナープローブとアクセプタープローブの融解温度(Tm値)がほぼ一致すること。
(2)蛍光物質標識に隣接してグアニン(G)塩基が存在しないこと。
(3)ドナープローブ同士、アクセプタープローブ同士、又はドナープローブ及びアクセプタープローブが相補的に結合しにくい配列であること。
【0040】
以上の方法により、18S rRNA用FRETプローブセットとして次の塩基配列を設計した。
5’−GAU CCA ACU ACG AGC UUU U−3’(18S_DnrR、配列番号2)
5’−UGC AGC AAC UUU AAU AUA CG−3’(18S_AccR、配列番号3)
【0041】
設計した塩基配列に基づき、FRETプローブを合成した。合成は株式会社日本バイオサービスに依頼した。プローブの合成には、2’−O−メチル オリゴリボヌクレオチド(2’OMe)を用いた。プローブの合成に用いるアミダイト試薬としては、いずれもGlenresearch社製の2’−OMe−A−CE−Phosphoamidite(型番#10−3100−90)、2’−OMe−C−CE−Phosphoamidite(型番#10−3110−90)、2’−OMe−G−CE−Phosphoamidite(型番#10−3121−90)、2’−OMe−U−CE−Phosphoamidite(型番#10−3130−90)を用いた。
【0042】
また、プローブ18S_DnrRの3’末端を蛍光物質AlexaFluor488(インビトロジェン社)で修飾した。また、プローブ18S_AccRの5’末端を蛍光物質AlexaFluor647(インビトロジェン社)で修飾した。より具体的には、プローブの3’末端は、プローブ合成後に蛍光物質と結合させることにより、蛍光物質で修飾した。また、プローブの5’末端は、蛍光物質修飾済みのアミダイド試薬を使用してプローブを合成することによって蛍光物質で修飾した。
【0043】
上記のプローブ18S_DnrRは、配列番号1に示す18S rRNAの塩基配列に対して相補的な配列であり、配列番号1の塩基配列の第687〜669位にハイブリダイズする。同様に、プローブ18S_AccRは、配列番号1の塩基配列の第664〜645位にハイブリダイズする。この結果、第669位に位置するドナー蛍光色素と第664位に位置するアクセプター蛍光色素が4塩基離れて隣接することになり、効率的にFRETを起こすことが可能となる。
【0044】
28S rRNA用FRETプローブセットとして次の塩基配列を設計した。
5’−ACU UCG GCC UUC AAA GUU−3’(28S_DnrR、配列番号5)
5’−UUU GAA UAU UUG CUA CUA CC−3’(28S_AccR、配列番号6)
【0045】
設計した塩基配列に基づき、FRETプローブを合成した。プローブの合成には2’OMeを用いた。また、プローブ28S_DnrRの3’末端を蛍光物質AlexaFluor488(ライフテクノロジーズジャパン株式会社)で修飾した。また、プローブ28S_AccRの5’末端を蛍光物質AlexaFluor647(ライフテクノロジーズジャパン株式会社)で修飾した。
【0046】
上記のプローブ28S_DnrRは、配列番号4に示す28S rRNAの塩基配列に対して相補的な配列であり、配列番号4の塩基配列の第2398〜2381位にハイブリダイズする。同様に、プローブ28S_AccRは、配列番号4の塩基配列の第2376〜2357位にハイブリダイズする。この結果、第2381位に位置するドナー蛍光色素と第2376位に位置するアクセプター蛍光色素が4塩基離れて隣接することになり、効率的にFRETを起こすことが可能となる。
【0047】
合成した18S及び28S rRNA用FRETプローブをそれぞれ水に溶解し、100nMの溶液を調製した。
【0048】
(実験例1)
(細胞内のrRNAのFRETによる検出)
慢性骨髄性白血病細胞株K562を実験に用いた。細胞内へのFRETプローブの導入には、ストレプトリジンO(SLO)により膜穿孔を行い、血清培地により再封入する方法を用いた(例えば、特開2007−37463号公報を参照)。
【0049】
具体的には、まず、培養したK562細胞を常法により回収して、血球計数板で細胞密度を測定し、1×106個/mlの細胞密度となるように調整した。次に、濃度調整したK562細胞1mlを遠心チューブに移し、遠心して上澄みを除去した後、等容量のリン酸緩衝液(PBS)で懸濁して洗浄し、再び遠心して上澄みを捨てた。続いて、100nMのドナープローブ及びアクセプタープローブを4μlずつ添加し、0.1mg/ml濃度のSLO(シグマアルドリッチ社)の0.01%ウシ血清アルブミン溶液を加えて全量を40μlに調整し、ピペッティングにより懸濁した。続いて37℃で5分間インキュベートした。次に、37℃に加温した血清入りの培地1mlを加えて37℃で30分間放置し、SLOを失活させ、細胞膜を再封入した。血清入りの培地としては、具体的には10%FBS(ウシ胎児血清、シグマアルドリッチ社)−RPMI 1640(シグマアルドリッチ社)を用いた。続いて、遠心して細胞を回収後、PBSに懸濁してフローサイトメトリーで測定した。フローサイトメーターとしては、FACS Aria(商品名、日本BDバイオサイエンス社)を使用した。
【0050】
図2(a)及び(b)に、フローサイトメトリーによる測定結果を示す。図2(a)はFRETプローブで18S rRNAを検出した結果を示し、(b)は、28S rRNAを検出した結果を示す。
【0051】
グラフの横軸は、アクセプター蛍光物質の励起波長(波長633nm)で励起した結果発生したアクセプター蛍光物質の蛍光(波長695nm)を検出した値(以下「AA」という場合がある。)であり、すなわち、各細胞内に存在するアクセプタープローブの量を示す。
【0052】
グラフの縦軸は、ドナー蛍光物質の励起波長(波長488nm)で励起した結果発生したアクセプター蛍光物質の蛍光(波長695nm)を検出した値(すなわちFRETによる蛍光、以下「DA」という場合がある)を、ドナー蛍光物質の励起波長(波長488nm)で励起した結果発生したドナー蛍光物質の蛍光(波長525nm)を検出した値(以下「DD」という場合がある)で割った値(DA/DD)であり、FRETを蛍光強度比で示すことにより蛍光強度変化をより明確に示したものである。
【0053】
図2の結果から明らかなように、18S rRNA及び28S rRNAのいずれを検出した場合においてもFRETが観察され、各rRNAが検出されたことが示された。また、18S rRNAの検出に使用したFRETプローブは、DA/DDの比が大きく、非常に感度が高いことが明らかとなった。
【0054】
(実験例2)
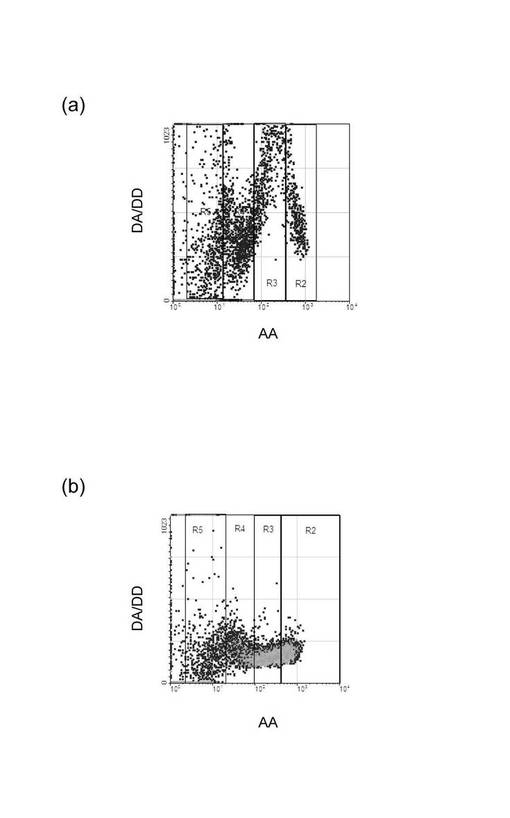
(細胞内のrRNAのFRETによる検出)
プローブの組み合わせを変更し、正しくFRETを検出できているか否かについて検討した。図3(a)〜(d)は、プローブの組み合わせを変更した点以外は実験例1と同様にして細胞内のrRNAをFRETにより検出した結果を示すグラフである。図3(a)は18S rRNA検出用ドナープローブ18S_DnrR及びアクセプタープローブ18S_AccRを組み合わせた結果であり、(b)は28S rRNA検出用ドナープローブ28S_DnrR及びアクセプタープローブ28S_AccRを組み合わせた結果であり、(c)は18S rRNA検出用ドナープローブ18S_DnrR及び28S rRNA検出用アクセプタープローブ28S_AccRを組み合わせた結果であり、(d)は28S rRNA検出用ドナープローブ28S_DnrR及び18S rRNA検出用アクセプタープローブ18S_AccRを組み合わせた結果である。図3の結果から明らかなように、正しいプローブの組み合わせの場合にFRETによる蛍光強度比の変化が観察された。このことから、各FRETプローブが標的分子に特異的に結合して蛍光を発していると考えられた。
【0055】
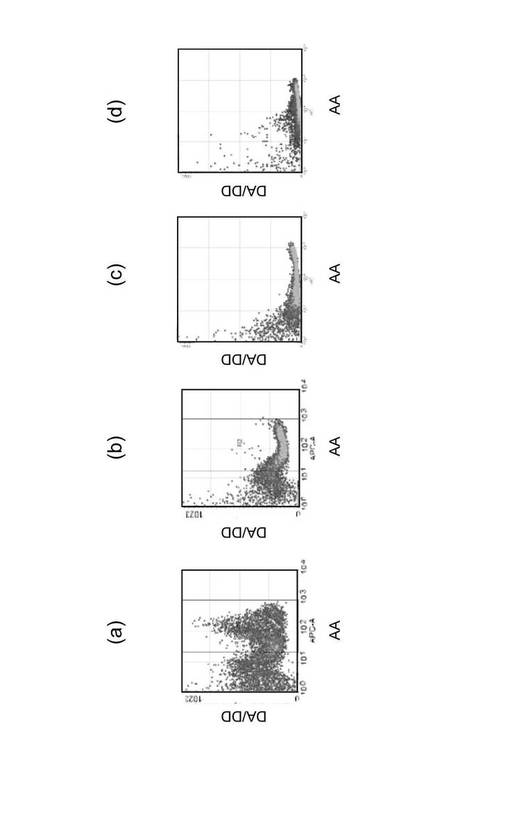
(実験例3)
(熱ショックによるrRNAの増加)
細胞を数時間、40℃程度の高温環境に晒すと、細胞の保護を目的とした一群のタンパク質が合成される。これらのタンパク質は熱ショックタンパク質と呼ばれ、温度変化に対する発現が迅速であり、かつ大量に合成されることが知られている。タンパク質合成が盛んであれば、その合成の場となるリボソームも必要となり、従ってrRNAの合成が盛んになることが推定される。そこで熱ショック(42℃、2時間)の有無により増加する細胞内のrRNAの定量的な測定を行った。
【0056】
K562細胞を106個/mlの濃度に調整し、(1)37℃で2時間インキュベーション、(2)37℃で1時間インキュベーション後に42℃で1時間インキュベーション、及び(3)42℃で2時間インキュベーションした後に、実験例1と同様にしてFRETプローブを用いて細胞内のrRNAを検出した。また、細胞の蛍光顕微鏡観察及びRT−PCRも行った。
【0057】
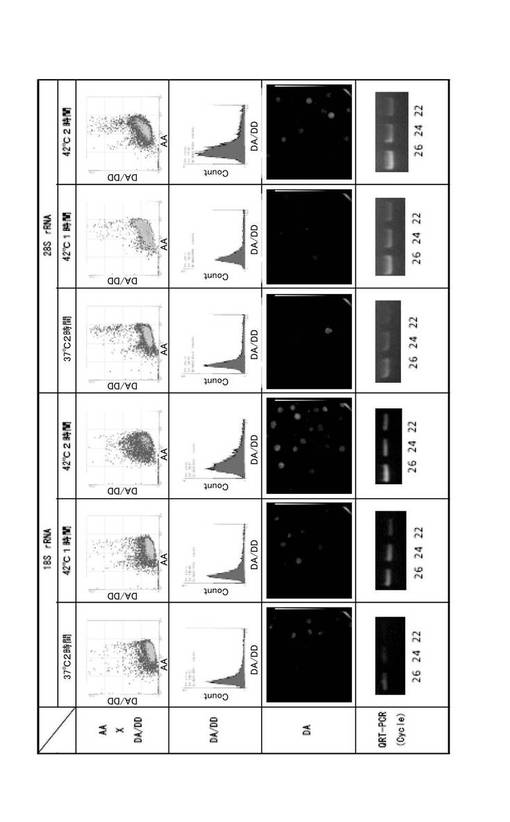
図4に測定結果をまとめた。左側3列は18S rRNAの測定結果を示し、右側3列は28S rRNAの測定結果を示す。最上行は横軸にAAを、縦軸にDA/DDを示したフローサイトメトリー測定の結果である。第2行は、横軸にDA/DDを、縦軸に細胞数をとり、最上行のフローサイトメトリーの結果を表示し直したヒストグラムである。第3行は、培養細胞のDAの蛍光を観察した蛍光顕微鏡像である。最下段は、各熱処理を施した細胞のrRNAをRT−PCRにより増幅後、アガロース電気泳動した結果を示す写真であり、各レーンの数字はPCRのサイクル数を示す。cDNAの調製には、Cells−to−cDNA(商品名、Ambion社製)を使用した。cDNA溶液は、2倍ずつ段階希釈して6〜8系列の希釈溶液を調製して鋳型とし、同時にPCR反応を行った。PCR反応は、94℃で2分熱変性した後、94℃30秒、53℃30秒及び72℃30秒を26サイクル行い、最後に72℃で5分の反応条件で行った。PCR産物を電気泳動して増幅断片のバンドを確認し、希釈系列の倍数とサイクル数からrRNA量を見積もった。PCRに使用したプライマーの塩基配列は次の通りである。
【0058】
18S rRNA 増幅用プライマー
5’−TCG AGG CCC TGT AAT TGG AA−3’(18S_Fwd、配列番号7)
5’−TTG CGC CGG TCC AAG AAT TT−3’(18S_Rev、配列番号8)
【0059】
28S rRNA 増幅用プライマー
5’−GCG CAT GAA TGG ATG AAC GA−3’(28S_Fwd、配列番号9)
5’−AAG CGA GCT TTT GCC CTT CT−3’(28S_Rev、配列番号10)
【0060】
42℃で2時間のインキュベーションにより、多くのrRNAが転写されたことが示された。これは、熱ショック関連タンパク質が盛んに合成され、タンパク質合成の場であるリボソームが必要となり、その要請に応じて多くのrRNAが転写された結果であると考えられる。フローサイトメトリー、蛍光顕微鏡観察及びRT−PCRの結果は、いずれもFRETによる蛍光強度変化とrRNA量の間に相関関係があることを示した。
【0061】
(実験例4)
(抗がん剤によるrRNA合成阻害実験)
癌細胞は細胞増殖が盛んなことから、タンパク質の合成も盛んである。タンパク質合成の場であるリボソームは精密にタンパク質の合成を制御しているため、リボソームの要求をサポートする為にrRNAの転写も盛んである。抗がん剤の一つである5FU(5−フルオロウラシル)はrDNAからrRNAを転写する酵素であるRNAポリメラーゼIの酵素活性を選択的に阻害することでrRNAの合成を阻害する薬剤として一般的に用いられている(例えば、Wilkinson DS,Tisty TD. And Hanas RJ. The Inhibition of Ribosomal RNA Synthesis and Maturation on Novikoff Hepatoma Cells by 5−Fluorouridine. Cancer Res., 1975 Nov; 35, 3014−3020.を参照。総説であれば、Longley DB, Harkin DP, Jhonston PG. 5−fluorouracil: mechanisms of action and clinical strategies. Nat Rev Cancer. 2003 May;3(5):330−8.を参照。)。
【0062】
図5は、5FUとFUDR(フルオロデオキシウリジン)の作用機序を示す図である。5FUは代謝を受けてFUTP(フルオロウリジン三リン酸)になり、RNAの中に入り込んでRNA合成を阻害する。また、5FUは代謝を受けてFdUMP(フルオロデオキシウリジン一リン酸)になり、チミジル酸シンターゼ(TS)を阻害してDNA合成を阻害する。したがって、5FUはRNA合成及びDNA合成の双方を阻害する。これに対し、FUDRはDNA合成を阻害するのみで、RNA合成には影響しない。
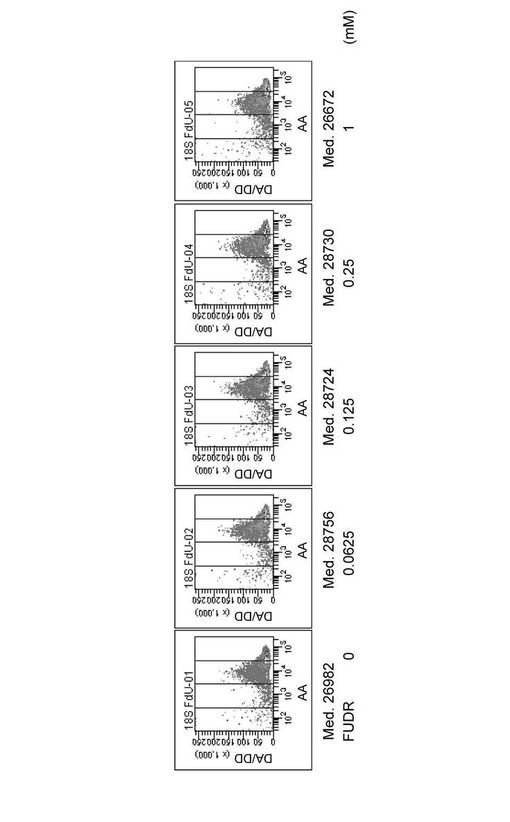
【0063】
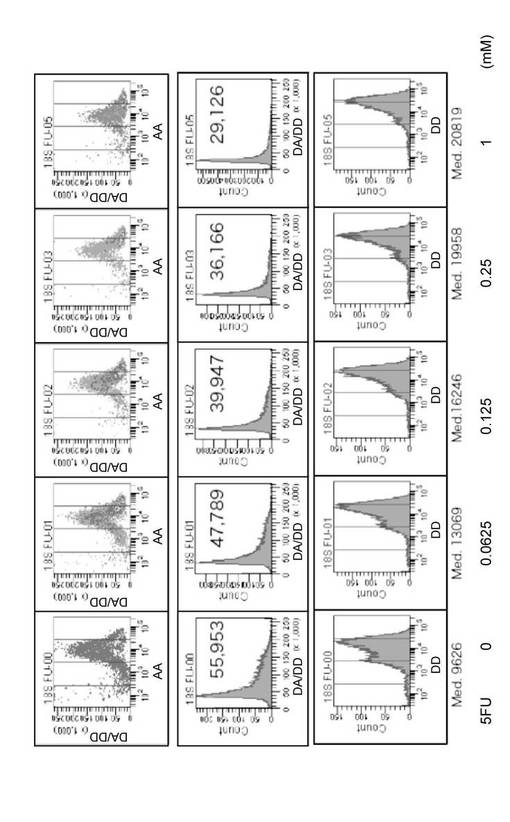
白血病細胞株HL60(1×106個)に各濃度の5FUを添加して、37℃で2時間インキュベーションした後、実験例1と同様にして、FRETプローブ18S_DnrR及び18S_AccRを用いて細胞内の18S rRNAを検出した。図6は、フローサイトメトリーの結果を示すグラフである。最左列から順に、0、0.0625、0.125、0.25及び1mMの5FUを添加した結果を示す。また、最上行は横軸にAAを、縦軸にDA/DDを示した測定の結果である。第2行は、横軸にDA/DDを、縦軸に細胞数をとり、最上行のフローサイトメトリーの結果を表示し直したヒストグラムである。また、グラフ中の数値は全細胞のDA/DDの値の平均値である。第3行は、横軸にDDを、縦軸に細胞数をとり、最上行のフローサイトメトリーの結果を表示し直したヒストグラムである。
【0064】
図6から明らかなように、5FUの添加量が増加するにしたがってDA/DDの値が低下していくことが確認された。すなわち、5FUの添加量が増加するにしたがって18S rRNAの合成が抑制された。この結果は、第2行及び第3行のヒストグラムからも明らかである。第2行のヒストグラムでは、5FUの濃度が上昇するにしたがってヒストグラムの肩(右上部分)が消失し、DA/DDの値の平均値も低下した。また、第3行のヒストグラムでは、5FUを添加しない場合には蛍光強度の異なる細胞集団が2つ存在(ピークが2つ)していたものが、5FUの添加により蛍光強度の低い細胞集団が消失して、蛍光強度の高い細胞集団に移行する過程が観察された。
【0065】
この現象は蛍光がFRETに由来するものであるかを蛍光顕微鏡で確認するために行われる「アクセプターブリーチング操作」に類似していると考えられる。つまり、rRNAが十分に存在する状況では、FRETに関与するドナープローブの蛍光強度は低いが、5FUによってRNAの合成が阻害された細胞では、rRNAの量が減少するためにFRETが解消される。その結果、細胞内に導入されたドナープローブのうち、FRETが成立しないドナープローブの割合が増加し、細胞から発せられる蛍光の強度はドナープローブのみを導入した場合の蛍光強度に近づく。蛍光顕微鏡観察では、FRETを起こしているプローブにおいて、アクセプタープローブが存在しない場合のドナープローブの蛍光強度を測定するには、アクセプター蛍光物質を退色させて、ドナー蛍光物質の蛍光を測定するが(アクセプターブリーチング操作)、フローサイトメトリーでそのようなことはできないので、実際はFRETを起こしている細胞の有無を総和として観察していると考えられる。
【0066】
このように、HL60細胞を用いた細胞内FRETによるrRNAの蛍光検出では、5FUの添加によりRNA合成が阻害された結実、RNA量が低下し、rRNAに由来するFRETの蛍光が低下した。
【0067】
(実験例5)
(抗がん剤によるrRNA合成阻害実験)
5FUの代わりにFUDRを用いた点以外は実験例4と同様の実験を行った。上述したように、FUDRはDNA合成を阻害するのみで、RNA合成には影響しない。
【0068】
HL60細胞(1×106個)に各濃度のFUDRを添加して、37℃で2時間インキュベーションした後、実験例1と同様にして、FRETプローブ18S_DnrR及び18S_AccRを用いて細胞内の18S rRNAを検出した。図7は、フローサイトメトリーの結果を示すグラフである。最左列から順に、0、0.0625、0.125、0.25及び1mMのFUDRを添加した結果を示す。グラフは、横軸にAAを、縦軸にDA/DDを示した測定の結果である。
【0069】
図7の結果から明らかなように、HL60細胞にFUDRを添加しても、18S rRNAの量は変化しなかった。これは、FUDRがDNA合成を阻害するのみで、RNA合成には影響しないことによると考えられた。
【0070】
(実験例6)
(抗がん剤によるrRNA合成阻害実験)
抗がん剤によって実際にRNAの合成が阻害されることを、別の染色法により確認した。染色には、SYTO−RNASelect(商品名、インビトロジェン社)を使用した。
【0071】
具体的には、まず、HL60細胞(2×106個)に、各濃度の5FU又はFUDRを添加し、血清を除いた培地中で37℃で2時間インキュベーションした。続いて、培地に対して1/10000量のSYTO−RNASelectを添加して30分インキュベートした。続いて、細胞を回収し、フローサイトメトリーによりSYTO−RNASelectに由来する蛍光を測定した。
【0072】
図8(a)及び(b)は、実験結果を示すグラフである。図8(a)は5FUを添加した結果であり、(b)はFUDRを添加した結果である。横軸は添加した5FU又はFUDRの濃度を示し、縦軸はSYTO−RNASelectに由来する蛍光の蛍光強度の平均値を示す。
【0073】
RNA合成を阻害する5FUの添加量が増加するにしたがって、RNAの蛍光強度が低下することが示された。ここで、SYTO−RNASelectは、rRNAを特異的に染色することはできないが、(一般的な細胞に含まれる)全RNAの9割をrRNAが占めることから、SYTO−RNASelectの蛍光は、rRNAの量を反映していると考えられる。一方、RNA合成を阻害しないFUDRを添加しても、RNAの蛍光強度には有意な変化は観察されなかった。この結果は、実験例4の結果が正しいことを支持するものであると考えられる。
【0074】
(実験例7)
(18S rRNA検出用FRETプローブの検討)
上記の検討により、18S_DnrR及び18S_AccRからなる18S rRNA検出用FRETプローブは、18S rRNAの検出感度が非常に高いことが示された。そこで、上記プローブと標的部位を変更した18S rRNA検出用FRETプローブを設計、合成し、反応性を検討した。
【0075】
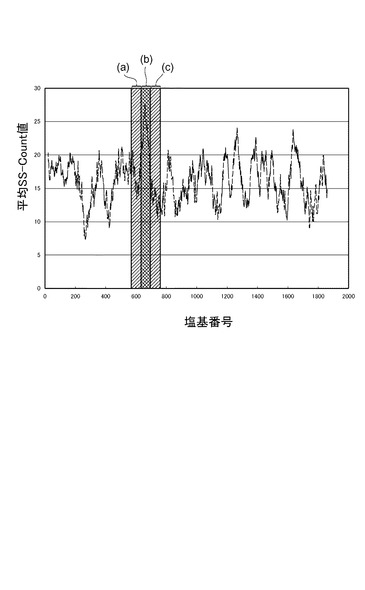
図9は、横軸に18S rRNAの塩基番号、縦軸にSS−count値S(i)の平均値をプロットしたグラフである。グラフ中(b)と表示した領域は、18S_DnrR及び18S_AccRからなる18S rRNA検出用FRETプローブの標的領域を示す。ここでは、(b)の領域に隣接する、(a)及び(c)の領域を標的領域とする18S rRNA検出用FRETプローブを設計、合成した。
【0076】
図9の(a)の領域を標的領域とする18S rRNA検出用FRETプローブの塩基配列は次の通りであった。
5’−AUU ACC GCG GCU GCU GGC A−3’(18S_refA_DnrR、配列番号11)
5’−ACU UGC CCU CCA AUG GAU CC−3’(18S_refA_AccR、配列番号12)
プローブの合成には、2’−O−メチル オリゴリボヌクレオチド(2’OMe)を用いた。プローブ18S_refA_DnrRの3’末端を蛍光物質AlexaFluor488(インビトロジェン社)で修飾した。また、プローブ18S_refA_AccRの5’末端を蛍光物質AlexaFluor647(インビトロジェン社)で修飾した。
【0077】
図9の(c)の領域を標的領域とする18S rRNA検出用FRETプローブの塩基配列は次の通りであった。
5’−AUC GAG GGG GCG CCG AGA−3’(18S_refB_DnrR、配列番号13)
5’−AGG GGC GGG GAC GGG CG−3’(18S_refB_AccR、配列番号14)
プローブの合成には、2’−O−メチル オリゴリボヌクレオチド(2’OMe)を用いた。プローブ18S_refB_DnrRの3’末端を蛍光物質AlexaFluor488(インビトロジェン社)で修飾した。また、プローブ18S_refB_AccRの5’末端を蛍光物質AlexaFluor647(インビトロジェン社)で修飾した。
【0078】
上記のプローブ18S_refA_DnrRは、配列番号1に示す18S rRNAの塩基配列に対して相補的な配列であり、配列番号1の塩基配列の第770〜752位にハイブリダイズする。同様に、プローブ18S_refA_AccRは、配列番号1の塩基配列の第747〜728位にハイブリダイズする。この結果、第752位に位置するドナー蛍光色素と第747位に位置するアクセプター蛍光色素が4塩基離れて隣接することになり、効率的にFRETを起こすことが可能となる。同様に、上記のプローブ18S_refB_DnrRは、配列番号1の塩基配列の第904〜887位にハイブリダイズする。また、プローブ18S_refB_AccRは、配列番号1の塩基配列の第882〜866位にハイブリダイズする。この結果、第887位に位置するドナー蛍光色素と第882位に位置するアクセプター蛍光色素が4塩基離れて隣接することになり、効率的にFRETを起こすことが可能となる。
【0079】
図10(a)〜(c)は、HL60細胞を用いて、実験例1と同様にして細胞内の18S rRNAをFRETにより検出した結果を示すグラフである。図10(a)は、プローブとして18S_refA_DnrR及び18S_refA_AccRを使用した結果であり、(b)は、プローブとして18S_DnrR及び18S_AccRを使用した結果であり、(c)は、プローブとして18S_refB_DnrR及び18S_refB_AccRを使用した結果である。図10の結果から明らかなように、18S_DnrR及び18S_AccRからなる18S rRNA検出用FRETプローブは、他のプローブと比較して、格段に18S rRNAの検出感度が高いことが示された。
【技術分野】
【0001】
本発明は18S rRNA検出用蛍光共鳴エネルギー移動(FRET)プローブに関する。
【背景技術】
【0002】
リボソームの合成過程は真核細胞において最もエネルギーが消費される過程である。真核細胞で転写される遺伝子の6割強はリボソームに関わる遺伝子であり、リボソームRNA(rRNA)の転写は細胞の分化・増殖と密接な関係にある。また細胞内のエネルギー消費調節にも大きく影響している。rRNA量の増減は、老化、寿命、細胞周期、細胞増殖、癌、栄養状態、低酸素状態等の生体の状態や疾患を大きく反映したものである。
【0003】
例えば、非特許文献1には、アルツハイマー病患者の末梢血中に含まれる18S rRNAと28S rRNAの量比が、健常人のそれと異なることが報告されている。
【0004】
真核生物のrRNAには、18S、28S及び5.8Sの3種類が存在する。18S rRNAは小サブユニットを構成し、28S及び5.8S rRNAは大サブユニットを構成する。
【0005】
rRNAの検出方法としては、in situハイブリダイゼーション(例えば、非特許文献2を参照)、アクリジンオレンジによる染色(例えば、非特許文献3を参照)、quenched autoligating FRET(QFRET)プローブによる染色(例えば、非特許文献4を参照)等が報告されている。
【0006】
非特許文献3の方法は、アクリジンオレンジが2本鎖核酸と結合した場合と1本鎖核酸と結合した場合でそれぞれ発する蛍光の波長が異なることを利用したものである。非特許文献4の方法は、2本のプローブが近接すると、プローブ間で共有結合するように2価試薬により修飾された、蛍光共鳴エネルギー移動(FRET)プローブを使用するものである。標的RNA分子上に隣接してハイブリダイズしたFRETプローブ間で共有結合が形成されることにより、FRETが起きる状況が維持される。これにより、信号の増幅が可能となり、より少ない分子の有無を定性的に判断できる。
【0007】
特許文献1には、18S、28S及び5.8S rRNAをコードするリボソームDNA(rDNA)の塩基配列及びその部分配列が記載されている。
【0008】
特許文献2には、FRET効率の高いFRETプローブを設計する方法が記載されている。
【先行技術文献】
【特許文献】
【0009】
【特許文献1】特表2009−504192号公報
【特許文献2】特開2007−37465号公報
【非特許文献】
【0010】
【非特許文献1】da Silva AM、et al、“Quantitative evaluation of the rRNA in Alzheimer’s disease”、Mech Ageing Dev.、2000、120(1−3)、57−64
【非特許文献2】Wallner G、et al、“Optimizing fluorescent in situ hybridization with rRNA−targeted oligonucleotide probes for flow cytometric identification of microorganisms”、Cytometry、1993、14(2)、136−43
【非特許文献3】Gordon RY、et al、“Acridine orange as an indicator of the cytoplasmic ribosome state”、Cytometry、1997、29(3)、215−21
【非特許文献4】Hiroshi Abe、Eric T. Kool、“Flow cytometric detection of specific RNAs in native human cells with quenched autoligating FRET probes”、PNAS、2006、103(2)、263−268
【発明の概要】
【発明が解決しようとする課題】
【0011】
しかしながら、非特許文献2の方法では、2本鎖核酸と1本鎖核酸とを染め分けることが可能であるものの、18S rRNAを選択的に検出することは不可能である。また、18S rRNAの検出を目的とした場合、細胞内に数十万分子以上存在することから、非特許文献4のQFRETプローブによる信号の増幅は必要ない。また、非特許文献4の方法では、連続的な信号の増幅により定量性が低下する場合がある。そこで、本発明は、18S rRNAを高感度で定量的に検出することが可能なFRETプローブを提供することを目的とする。
【課題を解決するための手段】
【0012】
本発明は、配列番号1に記載の塩基配列からなる核酸の第631〜725位(番目)の領域に、隣接してハイブリダイズ可能な第1のプローブ及び第2のプローブからなり、第1のプローブは第1の蛍光物質で標識されており、第2のプローブは前記第1の蛍光物質とFRETを起こす第2の蛍光物質で標識されている、18S rRNA検出用FRETプローブを提供する。
【0013】
上記本発明のFRETプローブによれば、18S rRNAを高感度で定量的に検出することができる。
【0014】
上記第1のプローブは、配列番号1に記載の塩基配列からなる核酸の第669〜687位の領域にハイブリダイズ可能な核酸又は核酸誘導体であり、上記第2のプローブは、配列番号1に記載の塩基配列からなる核酸の第645〜664位の領域にハイブリダイズ可能な核酸又は核酸誘導体であることがより好ましい。
【0015】
さらに、上記第1のプローブは、配列番号2に記載の塩基配列からなる2’−O−メチル オリゴリボヌクレオチドであり、上記第2のプローブは、配列番号3に記載の塩基配列からなる2’−O−メチル オリゴリボヌクレオチドであることが更に好ましい。
【0016】
このようなプローブは、18S rRNAを高感度で定量的に検出する性能が更に高い。
【発明の効果】
【0017】
本発明により、18S rRNAを高感度で定量的に検出することが可能なFRETプローブが提供される。
【図面の簡単な説明】
【0018】
【図1】(a)は、18S rRNAの塩基配列について、横軸に塩基番号、縦軸にSS−count値の平均値をプロットしたグラフである。(b)は、28S rRNAの塩基配列について、横軸に塩基番号、縦軸にSS−count値の平均値をプロットしたグラフである。
【図2】(a)及び(b)は、実験例1の結果を示したグラフである。
【図3】(a)〜(d)は、実験例2の結果を示したグラフである。
【図4】実験例3の結果を示したグラフ及び写真である。
【図5】5FUとFUDRの作用機序を示す図である。
【図6】実験例4の結果を示したグラフである。
【図7】実験例5の結果を示したグラフである。
【図8】(a)及び(b)は、実験例6の結果を示したグラフである。
【図9】18S rRNAの塩基配列について、横軸に塩基番号、縦軸にSS−count値の平均値をプロットしたグラフである。
【図10】(a)〜(c)は、実験例7の結果を示したグラフである。
【発明を実施するための形態】
【0019】
(プローブの設計)
本実施形態の18S rRNA検出用FRETプローブは、配列番号1に記載の塩基配列からなる核酸の第631〜725位の領域に、隣接してハイブリダイズ可能な第1のプローブ及び第2のプローブからなり、第1のプローブは第1の蛍光物質で標識されており、第2のプローブは前記第1の蛍光物質と蛍光共鳴エネルギー移動(FRET)を起こす第2の蛍光物質で標識されている。
【0020】
上記第1のプローブ及び第2のプローブのうち、隣接して標的核酸にハイブリダイズした状態で、(標的配列の相補鎖配列で)5’側に位置する方を5’側プローブと呼び、他方を3’側プローブと呼ぶ。本明細書において、「隣接」とは、5’側プローブの3’末端塩基と3’側プローブの5’末端塩基との間が、0〜8塩基、好ましくは0〜4塩基離れて隣り合うことをいう。5’側プローブの3’末端及び3’側プローブの5’末端を、それぞれFRETを起こす組み合わせの蛍光物質で標識する。FRETを起こす蛍光物質の組み合わせのうち、エネルギーを与える方の物質をドナー蛍光物質と呼び、エネルギーを受けとる方の分子をアクセプター蛍光物質と呼ぶ。5’側プローブと3’側プローブのいずれにドナー蛍光物質を標識してもよい。以下、ドナー蛍光物質で標識した方のプローブをドナープローブと呼び、アクセプター蛍光物質で標識した方のプローブをアクセプタープローブと呼ぶ場合がある。
【0021】
本実施形態のFRETプローブを設計する場合、配列番号1に記載の塩基配列からなる核酸の第631〜725位の領域に隣接してハイブリダイズ可能なようにドナープローブとアクセプタープローブを設計する。
【0022】
ドナープローブとアクセプタープローブの融解温度(Tm値)は、ほぼ一致することが好ましい。これは、ドナープローブとアクセプタープローブをほぼ同じ確率で標的配列に結合させるためである。
【0023】
ドナープローブとアクセプタープローブのTm値は、36℃以上であることが好ましい。これは生細胞が生息可能な温度域で標的核酸とプローブがハイブリダイズしなければならないためである。また、ドナープローブとアクセプタープローブの長さは、それぞれ15〜25塩基であることが好ましく、16〜22塩基であることがより好ましい。
【0024】
FRETプローブは、蛍光物質に隣接してグアニン(G)塩基が存在しないことが好ましい。これは、蛍光物質の近傍に核酸塩基(特にグアニン)が存在すると蛍光物質が消光する場合があるためである。
【0025】
また、ドナープローブ同士、アクセプタープローブ同士、又はドナープローブ及びアクセプタープローブが相補的に結合しにくい配列であることが好ましい。これは、相補的な配列が存在すると、プローブのみで互いにハイブリダイズしてしまう場合があるためである。その結果、標的核酸に依存せずFRETを起こしたり、プローブが定量的に標的核酸にハイブリダイズできなくなる等の問題が生じる場合がある。経験的に、4塩基以上連続して相補的な配列を持たないような配列を選択するとよい。プローブ単独又はプローブ間で相補的に結合しにくい配列であるか否かは、実際にプローブを合成し、溶液中における蛍光測定により確認することができる。
【0026】
(プローブの合成)
上記のようにして設計したFRETプローブの塩基配列に基づいて、実際にFRETプローブを合成する。プローブは核酸又は核酸誘導体により構成される。より具体的には、DNAやRNA等の核酸、ペプチド核酸(PNA)、Locked Nucleic Acid(LNA)、ホスホロチオエートオリゴリボヌクレオチド等の核酸誘導体、及び、部分的にこれらの核酸や核酸誘導体が混合したものが挙げられる。また、U(ウラシル)がT(チミン)に置き換えられていてもよい。FRETプローブがRNAである場合、RNaseによる分解を抑制するために、2’−O−メチル オリゴリボヌクレオチドとすることが好ましい。
【0027】
(蛍光物質)
5’側プローブ及び3’側プローブの一方をドナー蛍光物質で修飾し、他方をアクセプター蛍光物質で修飾する。ドナー蛍光物質とアクセプター蛍光物質との間の距離は、5’側プローブ及び3’側プローブが標的核酸にハイブリダイズした状態で4塩基であることが好ましい。この距離がFRETを起こすのに適しているためである。
【0028】
一実施形態において、プローブが標的核酸にハイブリダイズした状態で、5’側プローブの3’末端と、3’側プローブの5’末端の間の距離が4塩基となるようにプローブを設計し、5’側プローブの3’末端と、3’側プローブの5’末端を蛍光物質で修飾する。別の実施形態において、蛍光物質で修飾するのはプローブの末端ではなく、より内側の塩基である。このような場合であっても、プローブが標的核酸にハイブリダイズした状態で、ドナー蛍光物質とアクセプター蛍光物質との間の距離が4塩基であれば、効率よくFRETを起こすことができる。
【0029】
ドナー蛍光物質とアクセプター蛍光物質の組み合わせとしては、例えば、表1に示すものが使用できる。
【0030】
【表1】
【0031】
(18S rRNAの検出)
一実施形態において、インビトロで18S rRNAを検出し、定量することができる。サンプルとしては、対象から抽出した全RNA等を使用できる。例えば、1μMのドナープローブおよびアクセプタープローブを溶解した緩衝液中に全RNAを混合する。プローブを標的核酸にハイブリダイズさせるためには、例えば150mM NaCl−15mM Sodium Citrate pH7.0 (SSC)溶液中、温度37℃等の条件下で20分間放置するとよい。続いて、蛍光顕微鏡、蛍光分光光度計等の装置を用いて、FRETの蛍光を検出する。より具体的には、ドナー蛍光物質の励起波長の光を照射し、アクセプター蛍光物質の蛍光を測定する。例えば、ドナー蛍光物質にAlexaFluor488(インビトロジェン社)を用い、アクセプター蛍光物質にAlexaFluor647(インビトロジェン社)を用いた場合、ドナー蛍光物質の励起波長は488nmであり、アクセプター蛍光物質の蛍光波長は630nmである。
【0032】
別の実施形態において、生きた又は固定した細胞内で18S rRNAを検出し、定量することができる。生きた細胞へのプローブの導入には、後述するストレプトリジンO(SLO)を使用する方法、マイクロインジェクション、エレクトロポレーション、リポフェクション等の方法が使用できる。固定した細胞を対象として18S rRNAを検出する場合には、細胞へのプローブの導入には、界面活性剤を用いる等の方法が使用できる。細胞内にプローブを導入する方法は条件が異なってくるので一概に同じ条件を設定できないが、ハイブリダイゼーションに限れば数分でハイブリダイズする。続いて、蛍光顕微鏡、フローサイトメーター等を用いて、FRETの蛍光を検出することができる。
【実施例】
【0033】
以下、本発明の実施例を示して、本発明を更に具体的に説明するが、本発明はこれらの実施例に限定されるものではなく、本発明の技術的思想を逸脱しない範囲での種々の変更が可能である。
【0034】
(FRETプローブの設計及び合成)
ヒト18S rRNA遺伝子(配列番号1、Genbankアクセッション番号K03432.1)及びヒト28S RNA遺伝子(配列番号4、Genbankアクセッション番号NR_003287)の塩基配列を使用し、特開2007−37465号公報に記載された方法にしたがって、18S rRNA及び28S RNA検出用FRETプローブを設計した。
【0035】
具体的には、18S rRNA及び28S RNAのそれぞれの塩基配列について、次の操作を行った。まず、長さnの塩基配列のi番目(1≦i≦n)の塩基のSS−count値S(i)を算出した。SS−count値とは、RNAの高次構造予測の指標の一つである。SS−count値は、RNAの各塩基における相補鎖を形成しない場合の数を表し、この数値が高い塩基ほど、高次構造の中で熱的に不安定で1本鎖となっている確率が高い。SS−count値は、Zuckerらによって開発されたRNAの2次構造を予測するプログラムである、mfold(Zuker M. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 2003 Jul 1;31(13):3406−15.)を使用して算出することができる。
【0036】
Lは一組のFRETプローブがハイブリダイズする標的部位全体の長さである。ここではドナープローブ20塩基、アクセプタープローブ20塩基、ドナー色素−アクセプター色素間の至適間隔4塩基の合計L=44塩基とした。
【0037】
続いて、18S及び28S rRNAの各塩基配列の塩基番号をiとし、1≦i≦n−L+1である全てのiについて、i番目の塩基からi+L−1番目の塩基までのSS−count値S(i)の平均値M(i)を算出した。図1(a)は、18S rRNAの塩基配列について、横軸に塩基番号、縦軸にM(i)をプロットしたグラフである。図1(b)は、28S rRNAの塩基配列について、図1(a)と同様にして作成したグラフである。
【0038】
図1(a)及び(b)において、塩基配列上のM(i)の値が大きい領域は、1本鎖となっている確率が高いため、FRETプローブがハイブリダイズする標的領域として適していると考えられる。図1(a)及び(b)中、FRETプローブの標的領域として選択した領域を太線で示した。
【0039】
図1(a)及び(b)に示したFRETプローブの標的領域から、ドナープローブ及びアクセプタープローブの塩基配列を選択した。FRETプローブとしては、M(i)の値が大きいだけでなく、上述した理由から、次の条件を満たす配列を選択した。
(1)ドナープローブとアクセプタープローブの融解温度(Tm値)がほぼ一致すること。
(2)蛍光物質標識に隣接してグアニン(G)塩基が存在しないこと。
(3)ドナープローブ同士、アクセプタープローブ同士、又はドナープローブ及びアクセプタープローブが相補的に結合しにくい配列であること。
【0040】
以上の方法により、18S rRNA用FRETプローブセットとして次の塩基配列を設計した。
5’−GAU CCA ACU ACG AGC UUU U−3’(18S_DnrR、配列番号2)
5’−UGC AGC AAC UUU AAU AUA CG−3’(18S_AccR、配列番号3)
【0041】
設計した塩基配列に基づき、FRETプローブを合成した。合成は株式会社日本バイオサービスに依頼した。プローブの合成には、2’−O−メチル オリゴリボヌクレオチド(2’OMe)を用いた。プローブの合成に用いるアミダイト試薬としては、いずれもGlenresearch社製の2’−OMe−A−CE−Phosphoamidite(型番#10−3100−90)、2’−OMe−C−CE−Phosphoamidite(型番#10−3110−90)、2’−OMe−G−CE−Phosphoamidite(型番#10−3121−90)、2’−OMe−U−CE−Phosphoamidite(型番#10−3130−90)を用いた。
【0042】
また、プローブ18S_DnrRの3’末端を蛍光物質AlexaFluor488(インビトロジェン社)で修飾した。また、プローブ18S_AccRの5’末端を蛍光物質AlexaFluor647(インビトロジェン社)で修飾した。より具体的には、プローブの3’末端は、プローブ合成後に蛍光物質と結合させることにより、蛍光物質で修飾した。また、プローブの5’末端は、蛍光物質修飾済みのアミダイド試薬を使用してプローブを合成することによって蛍光物質で修飾した。
【0043】
上記のプローブ18S_DnrRは、配列番号1に示す18S rRNAの塩基配列に対して相補的な配列であり、配列番号1の塩基配列の第687〜669位にハイブリダイズする。同様に、プローブ18S_AccRは、配列番号1の塩基配列の第664〜645位にハイブリダイズする。この結果、第669位に位置するドナー蛍光色素と第664位に位置するアクセプター蛍光色素が4塩基離れて隣接することになり、効率的にFRETを起こすことが可能となる。
【0044】
28S rRNA用FRETプローブセットとして次の塩基配列を設計した。
5’−ACU UCG GCC UUC AAA GUU−3’(28S_DnrR、配列番号5)
5’−UUU GAA UAU UUG CUA CUA CC−3’(28S_AccR、配列番号6)
【0045】
設計した塩基配列に基づき、FRETプローブを合成した。プローブの合成には2’OMeを用いた。また、プローブ28S_DnrRの3’末端を蛍光物質AlexaFluor488(ライフテクノロジーズジャパン株式会社)で修飾した。また、プローブ28S_AccRの5’末端を蛍光物質AlexaFluor647(ライフテクノロジーズジャパン株式会社)で修飾した。
【0046】
上記のプローブ28S_DnrRは、配列番号4に示す28S rRNAの塩基配列に対して相補的な配列であり、配列番号4の塩基配列の第2398〜2381位にハイブリダイズする。同様に、プローブ28S_AccRは、配列番号4の塩基配列の第2376〜2357位にハイブリダイズする。この結果、第2381位に位置するドナー蛍光色素と第2376位に位置するアクセプター蛍光色素が4塩基離れて隣接することになり、効率的にFRETを起こすことが可能となる。
【0047】
合成した18S及び28S rRNA用FRETプローブをそれぞれ水に溶解し、100nMの溶液を調製した。
【0048】
(実験例1)
(細胞内のrRNAのFRETによる検出)
慢性骨髄性白血病細胞株K562を実験に用いた。細胞内へのFRETプローブの導入には、ストレプトリジンO(SLO)により膜穿孔を行い、血清培地により再封入する方法を用いた(例えば、特開2007−37463号公報を参照)。
【0049】
具体的には、まず、培養したK562細胞を常法により回収して、血球計数板で細胞密度を測定し、1×106個/mlの細胞密度となるように調整した。次に、濃度調整したK562細胞1mlを遠心チューブに移し、遠心して上澄みを除去した後、等容量のリン酸緩衝液(PBS)で懸濁して洗浄し、再び遠心して上澄みを捨てた。続いて、100nMのドナープローブ及びアクセプタープローブを4μlずつ添加し、0.1mg/ml濃度のSLO(シグマアルドリッチ社)の0.01%ウシ血清アルブミン溶液を加えて全量を40μlに調整し、ピペッティングにより懸濁した。続いて37℃で5分間インキュベートした。次に、37℃に加温した血清入りの培地1mlを加えて37℃で30分間放置し、SLOを失活させ、細胞膜を再封入した。血清入りの培地としては、具体的には10%FBS(ウシ胎児血清、シグマアルドリッチ社)−RPMI 1640(シグマアルドリッチ社)を用いた。続いて、遠心して細胞を回収後、PBSに懸濁してフローサイトメトリーで測定した。フローサイトメーターとしては、FACS Aria(商品名、日本BDバイオサイエンス社)を使用した。
【0050】
図2(a)及び(b)に、フローサイトメトリーによる測定結果を示す。図2(a)はFRETプローブで18S rRNAを検出した結果を示し、(b)は、28S rRNAを検出した結果を示す。
【0051】
グラフの横軸は、アクセプター蛍光物質の励起波長(波長633nm)で励起した結果発生したアクセプター蛍光物質の蛍光(波長695nm)を検出した値(以下「AA」という場合がある。)であり、すなわち、各細胞内に存在するアクセプタープローブの量を示す。
【0052】
グラフの縦軸は、ドナー蛍光物質の励起波長(波長488nm)で励起した結果発生したアクセプター蛍光物質の蛍光(波長695nm)を検出した値(すなわちFRETによる蛍光、以下「DA」という場合がある)を、ドナー蛍光物質の励起波長(波長488nm)で励起した結果発生したドナー蛍光物質の蛍光(波長525nm)を検出した値(以下「DD」という場合がある)で割った値(DA/DD)であり、FRETを蛍光強度比で示すことにより蛍光強度変化をより明確に示したものである。
【0053】
図2の結果から明らかなように、18S rRNA及び28S rRNAのいずれを検出した場合においてもFRETが観察され、各rRNAが検出されたことが示された。また、18S rRNAの検出に使用したFRETプローブは、DA/DDの比が大きく、非常に感度が高いことが明らかとなった。
【0054】
(実験例2)
(細胞内のrRNAのFRETによる検出)
プローブの組み合わせを変更し、正しくFRETを検出できているか否かについて検討した。図3(a)〜(d)は、プローブの組み合わせを変更した点以外は実験例1と同様にして細胞内のrRNAをFRETにより検出した結果を示すグラフである。図3(a)は18S rRNA検出用ドナープローブ18S_DnrR及びアクセプタープローブ18S_AccRを組み合わせた結果であり、(b)は28S rRNA検出用ドナープローブ28S_DnrR及びアクセプタープローブ28S_AccRを組み合わせた結果であり、(c)は18S rRNA検出用ドナープローブ18S_DnrR及び28S rRNA検出用アクセプタープローブ28S_AccRを組み合わせた結果であり、(d)は28S rRNA検出用ドナープローブ28S_DnrR及び18S rRNA検出用アクセプタープローブ18S_AccRを組み合わせた結果である。図3の結果から明らかなように、正しいプローブの組み合わせの場合にFRETによる蛍光強度比の変化が観察された。このことから、各FRETプローブが標的分子に特異的に結合して蛍光を発していると考えられた。
【0055】
(実験例3)
(熱ショックによるrRNAの増加)
細胞を数時間、40℃程度の高温環境に晒すと、細胞の保護を目的とした一群のタンパク質が合成される。これらのタンパク質は熱ショックタンパク質と呼ばれ、温度変化に対する発現が迅速であり、かつ大量に合成されることが知られている。タンパク質合成が盛んであれば、その合成の場となるリボソームも必要となり、従ってrRNAの合成が盛んになることが推定される。そこで熱ショック(42℃、2時間)の有無により増加する細胞内のrRNAの定量的な測定を行った。
【0056】
K562細胞を106個/mlの濃度に調整し、(1)37℃で2時間インキュベーション、(2)37℃で1時間インキュベーション後に42℃で1時間インキュベーション、及び(3)42℃で2時間インキュベーションした後に、実験例1と同様にしてFRETプローブを用いて細胞内のrRNAを検出した。また、細胞の蛍光顕微鏡観察及びRT−PCRも行った。
【0057】
図4に測定結果をまとめた。左側3列は18S rRNAの測定結果を示し、右側3列は28S rRNAの測定結果を示す。最上行は横軸にAAを、縦軸にDA/DDを示したフローサイトメトリー測定の結果である。第2行は、横軸にDA/DDを、縦軸に細胞数をとり、最上行のフローサイトメトリーの結果を表示し直したヒストグラムである。第3行は、培養細胞のDAの蛍光を観察した蛍光顕微鏡像である。最下段は、各熱処理を施した細胞のrRNAをRT−PCRにより増幅後、アガロース電気泳動した結果を示す写真であり、各レーンの数字はPCRのサイクル数を示す。cDNAの調製には、Cells−to−cDNA(商品名、Ambion社製)を使用した。cDNA溶液は、2倍ずつ段階希釈して6〜8系列の希釈溶液を調製して鋳型とし、同時にPCR反応を行った。PCR反応は、94℃で2分熱変性した後、94℃30秒、53℃30秒及び72℃30秒を26サイクル行い、最後に72℃で5分の反応条件で行った。PCR産物を電気泳動して増幅断片のバンドを確認し、希釈系列の倍数とサイクル数からrRNA量を見積もった。PCRに使用したプライマーの塩基配列は次の通りである。
【0058】
18S rRNA 増幅用プライマー
5’−TCG AGG CCC TGT AAT TGG AA−3’(18S_Fwd、配列番号7)
5’−TTG CGC CGG TCC AAG AAT TT−3’(18S_Rev、配列番号8)
【0059】
28S rRNA 増幅用プライマー
5’−GCG CAT GAA TGG ATG AAC GA−3’(28S_Fwd、配列番号9)
5’−AAG CGA GCT TTT GCC CTT CT−3’(28S_Rev、配列番号10)
【0060】
42℃で2時間のインキュベーションにより、多くのrRNAが転写されたことが示された。これは、熱ショック関連タンパク質が盛んに合成され、タンパク質合成の場であるリボソームが必要となり、その要請に応じて多くのrRNAが転写された結果であると考えられる。フローサイトメトリー、蛍光顕微鏡観察及びRT−PCRの結果は、いずれもFRETによる蛍光強度変化とrRNA量の間に相関関係があることを示した。
【0061】
(実験例4)
(抗がん剤によるrRNA合成阻害実験)
癌細胞は細胞増殖が盛んなことから、タンパク質の合成も盛んである。タンパク質合成の場であるリボソームは精密にタンパク質の合成を制御しているため、リボソームの要求をサポートする為にrRNAの転写も盛んである。抗がん剤の一つである5FU(5−フルオロウラシル)はrDNAからrRNAを転写する酵素であるRNAポリメラーゼIの酵素活性を選択的に阻害することでrRNAの合成を阻害する薬剤として一般的に用いられている(例えば、Wilkinson DS,Tisty TD. And Hanas RJ. The Inhibition of Ribosomal RNA Synthesis and Maturation on Novikoff Hepatoma Cells by 5−Fluorouridine. Cancer Res., 1975 Nov; 35, 3014−3020.を参照。総説であれば、Longley DB, Harkin DP, Jhonston PG. 5−fluorouracil: mechanisms of action and clinical strategies. Nat Rev Cancer. 2003 May;3(5):330−8.を参照。)。
【0062】
図5は、5FUとFUDR(フルオロデオキシウリジン)の作用機序を示す図である。5FUは代謝を受けてFUTP(フルオロウリジン三リン酸)になり、RNAの中に入り込んでRNA合成を阻害する。また、5FUは代謝を受けてFdUMP(フルオロデオキシウリジン一リン酸)になり、チミジル酸シンターゼ(TS)を阻害してDNA合成を阻害する。したがって、5FUはRNA合成及びDNA合成の双方を阻害する。これに対し、FUDRはDNA合成を阻害するのみで、RNA合成には影響しない。
【0063】
白血病細胞株HL60(1×106個)に各濃度の5FUを添加して、37℃で2時間インキュベーションした後、実験例1と同様にして、FRETプローブ18S_DnrR及び18S_AccRを用いて細胞内の18S rRNAを検出した。図6は、フローサイトメトリーの結果を示すグラフである。最左列から順に、0、0.0625、0.125、0.25及び1mMの5FUを添加した結果を示す。また、最上行は横軸にAAを、縦軸にDA/DDを示した測定の結果である。第2行は、横軸にDA/DDを、縦軸に細胞数をとり、最上行のフローサイトメトリーの結果を表示し直したヒストグラムである。また、グラフ中の数値は全細胞のDA/DDの値の平均値である。第3行は、横軸にDDを、縦軸に細胞数をとり、最上行のフローサイトメトリーの結果を表示し直したヒストグラムである。
【0064】
図6から明らかなように、5FUの添加量が増加するにしたがってDA/DDの値が低下していくことが確認された。すなわち、5FUの添加量が増加するにしたがって18S rRNAの合成が抑制された。この結果は、第2行及び第3行のヒストグラムからも明らかである。第2行のヒストグラムでは、5FUの濃度が上昇するにしたがってヒストグラムの肩(右上部分)が消失し、DA/DDの値の平均値も低下した。また、第3行のヒストグラムでは、5FUを添加しない場合には蛍光強度の異なる細胞集団が2つ存在(ピークが2つ)していたものが、5FUの添加により蛍光強度の低い細胞集団が消失して、蛍光強度の高い細胞集団に移行する過程が観察された。
【0065】
この現象は蛍光がFRETに由来するものであるかを蛍光顕微鏡で確認するために行われる「アクセプターブリーチング操作」に類似していると考えられる。つまり、rRNAが十分に存在する状況では、FRETに関与するドナープローブの蛍光強度は低いが、5FUによってRNAの合成が阻害された細胞では、rRNAの量が減少するためにFRETが解消される。その結果、細胞内に導入されたドナープローブのうち、FRETが成立しないドナープローブの割合が増加し、細胞から発せられる蛍光の強度はドナープローブのみを導入した場合の蛍光強度に近づく。蛍光顕微鏡観察では、FRETを起こしているプローブにおいて、アクセプタープローブが存在しない場合のドナープローブの蛍光強度を測定するには、アクセプター蛍光物質を退色させて、ドナー蛍光物質の蛍光を測定するが(アクセプターブリーチング操作)、フローサイトメトリーでそのようなことはできないので、実際はFRETを起こしている細胞の有無を総和として観察していると考えられる。
【0066】
このように、HL60細胞を用いた細胞内FRETによるrRNAの蛍光検出では、5FUの添加によりRNA合成が阻害された結実、RNA量が低下し、rRNAに由来するFRETの蛍光が低下した。
【0067】
(実験例5)
(抗がん剤によるrRNA合成阻害実験)
5FUの代わりにFUDRを用いた点以外は実験例4と同様の実験を行った。上述したように、FUDRはDNA合成を阻害するのみで、RNA合成には影響しない。
【0068】
HL60細胞(1×106個)に各濃度のFUDRを添加して、37℃で2時間インキュベーションした後、実験例1と同様にして、FRETプローブ18S_DnrR及び18S_AccRを用いて細胞内の18S rRNAを検出した。図7は、フローサイトメトリーの結果を示すグラフである。最左列から順に、0、0.0625、0.125、0.25及び1mMのFUDRを添加した結果を示す。グラフは、横軸にAAを、縦軸にDA/DDを示した測定の結果である。
【0069】
図7の結果から明らかなように、HL60細胞にFUDRを添加しても、18S rRNAの量は変化しなかった。これは、FUDRがDNA合成を阻害するのみで、RNA合成には影響しないことによると考えられた。
【0070】
(実験例6)
(抗がん剤によるrRNA合成阻害実験)
抗がん剤によって実際にRNAの合成が阻害されることを、別の染色法により確認した。染色には、SYTO−RNASelect(商品名、インビトロジェン社)を使用した。
【0071】
具体的には、まず、HL60細胞(2×106個)に、各濃度の5FU又はFUDRを添加し、血清を除いた培地中で37℃で2時間インキュベーションした。続いて、培地に対して1/10000量のSYTO−RNASelectを添加して30分インキュベートした。続いて、細胞を回収し、フローサイトメトリーによりSYTO−RNASelectに由来する蛍光を測定した。
【0072】
図8(a)及び(b)は、実験結果を示すグラフである。図8(a)は5FUを添加した結果であり、(b)はFUDRを添加した結果である。横軸は添加した5FU又はFUDRの濃度を示し、縦軸はSYTO−RNASelectに由来する蛍光の蛍光強度の平均値を示す。
【0073】
RNA合成を阻害する5FUの添加量が増加するにしたがって、RNAの蛍光強度が低下することが示された。ここで、SYTO−RNASelectは、rRNAを特異的に染色することはできないが、(一般的な細胞に含まれる)全RNAの9割をrRNAが占めることから、SYTO−RNASelectの蛍光は、rRNAの量を反映していると考えられる。一方、RNA合成を阻害しないFUDRを添加しても、RNAの蛍光強度には有意な変化は観察されなかった。この結果は、実験例4の結果が正しいことを支持するものであると考えられる。
【0074】
(実験例7)
(18S rRNA検出用FRETプローブの検討)
上記の検討により、18S_DnrR及び18S_AccRからなる18S rRNA検出用FRETプローブは、18S rRNAの検出感度が非常に高いことが示された。そこで、上記プローブと標的部位を変更した18S rRNA検出用FRETプローブを設計、合成し、反応性を検討した。
【0075】
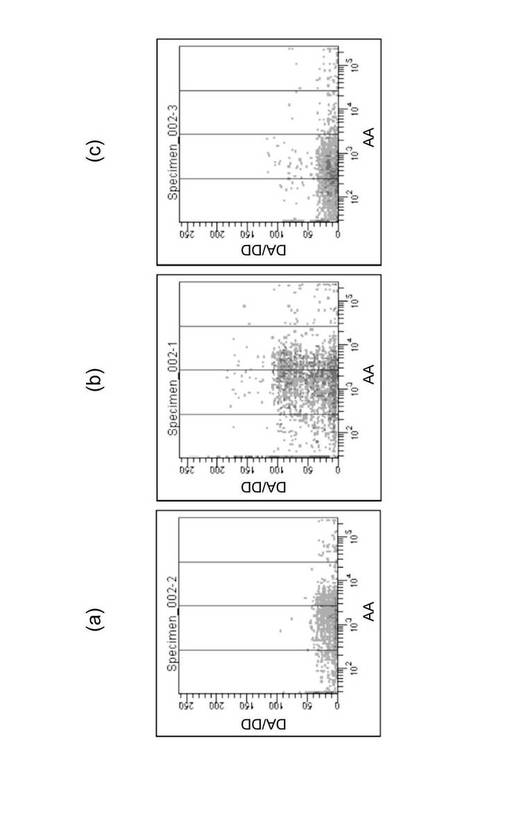
図9は、横軸に18S rRNAの塩基番号、縦軸にSS−count値S(i)の平均値をプロットしたグラフである。グラフ中(b)と表示した領域は、18S_DnrR及び18S_AccRからなる18S rRNA検出用FRETプローブの標的領域を示す。ここでは、(b)の領域に隣接する、(a)及び(c)の領域を標的領域とする18S rRNA検出用FRETプローブを設計、合成した。
【0076】
図9の(a)の領域を標的領域とする18S rRNA検出用FRETプローブの塩基配列は次の通りであった。
5’−AUU ACC GCG GCU GCU GGC A−3’(18S_refA_DnrR、配列番号11)
5’−ACU UGC CCU CCA AUG GAU CC−3’(18S_refA_AccR、配列番号12)
プローブの合成には、2’−O−メチル オリゴリボヌクレオチド(2’OMe)を用いた。プローブ18S_refA_DnrRの3’末端を蛍光物質AlexaFluor488(インビトロジェン社)で修飾した。また、プローブ18S_refA_AccRの5’末端を蛍光物質AlexaFluor647(インビトロジェン社)で修飾した。
【0077】
図9の(c)の領域を標的領域とする18S rRNA検出用FRETプローブの塩基配列は次の通りであった。
5’−AUC GAG GGG GCG CCG AGA−3’(18S_refB_DnrR、配列番号13)
5’−AGG GGC GGG GAC GGG CG−3’(18S_refB_AccR、配列番号14)
プローブの合成には、2’−O−メチル オリゴリボヌクレオチド(2’OMe)を用いた。プローブ18S_refB_DnrRの3’末端を蛍光物質AlexaFluor488(インビトロジェン社)で修飾した。また、プローブ18S_refB_AccRの5’末端を蛍光物質AlexaFluor647(インビトロジェン社)で修飾した。
【0078】
上記のプローブ18S_refA_DnrRは、配列番号1に示す18S rRNAの塩基配列に対して相補的な配列であり、配列番号1の塩基配列の第770〜752位にハイブリダイズする。同様に、プローブ18S_refA_AccRは、配列番号1の塩基配列の第747〜728位にハイブリダイズする。この結果、第752位に位置するドナー蛍光色素と第747位に位置するアクセプター蛍光色素が4塩基離れて隣接することになり、効率的にFRETを起こすことが可能となる。同様に、上記のプローブ18S_refB_DnrRは、配列番号1の塩基配列の第904〜887位にハイブリダイズする。また、プローブ18S_refB_AccRは、配列番号1の塩基配列の第882〜866位にハイブリダイズする。この結果、第887位に位置するドナー蛍光色素と第882位に位置するアクセプター蛍光色素が4塩基離れて隣接することになり、効率的にFRETを起こすことが可能となる。
【0079】
図10(a)〜(c)は、HL60細胞を用いて、実験例1と同様にして細胞内の18S rRNAをFRETにより検出した結果を示すグラフである。図10(a)は、プローブとして18S_refA_DnrR及び18S_refA_AccRを使用した結果であり、(b)は、プローブとして18S_DnrR及び18S_AccRを使用した結果であり、(c)は、プローブとして18S_refB_DnrR及び18S_refB_AccRを使用した結果である。図10の結果から明らかなように、18S_DnrR及び18S_AccRからなる18S rRNA検出用FRETプローブは、他のプローブと比較して、格段に18S rRNAの検出感度が高いことが示された。
【特許請求の範囲】
【請求項1】
配列番号1に記載の塩基配列からなる核酸の第631〜725位の領域に、隣接してハイブリダイズ可能な第1のプローブ及び第2のプローブからなり、
前記第1のプローブは第1の蛍光物質で標識されており、
前記第2のプローブは前記第1の蛍光物質と蛍光共鳴エネルギー移動(FRET)を起こす第2の蛍光物質で標識されている、
18S rRNA検出用FRETプローブ。
【請求項2】
前記第1のプローブは、配列番号1に記載の塩基配列からなる核酸の第669〜687位の領域にハイブリダイズ可能な核酸又は核酸誘導体であり、
前記第2のプローブは、配列番号1に記載の塩基配列からなる核酸の第645〜664位の領域にハイブリダイズ可能な核酸又は核酸誘導体である、請求項1に記載の18S rRNA検出用FRETプローブ。
【請求項3】
前記第1のプローブは、配列番号2に記載の塩基配列からなる2’−O−メチル オリゴリボヌクレオチドであり、
前記第2のプローブは、配列番号3に記載の塩基配列からなる2’−O−メチル オリゴリボヌクレオチドである、請求項1又は2に記載の18S rRNA検出用FRETプローブ。
【請求項1】
配列番号1に記載の塩基配列からなる核酸の第631〜725位の領域に、隣接してハイブリダイズ可能な第1のプローブ及び第2のプローブからなり、
前記第1のプローブは第1の蛍光物質で標識されており、
前記第2のプローブは前記第1の蛍光物質と蛍光共鳴エネルギー移動(FRET)を起こす第2の蛍光物質で標識されている、
18S rRNA検出用FRETプローブ。
【請求項2】
前記第1のプローブは、配列番号1に記載の塩基配列からなる核酸の第669〜687位の領域にハイブリダイズ可能な核酸又は核酸誘導体であり、
前記第2のプローブは、配列番号1に記載の塩基配列からなる核酸の第645〜664位の領域にハイブリダイズ可能な核酸又は核酸誘導体である、請求項1に記載の18S rRNA検出用FRETプローブ。
【請求項3】
前記第1のプローブは、配列番号2に記載の塩基配列からなる2’−O−メチル オリゴリボヌクレオチドであり、
前記第2のプローブは、配列番号3に記載の塩基配列からなる2’−O−メチル オリゴリボヌクレオチドである、請求項1又は2に記載の18S rRNA検出用FRETプローブ。
【図1】

【図5】
【図8】
【図9】
【図2】
【図3】
【図4】
【図6】
【図7】
【図10】
【図5】
【図8】
【図9】
【図2】
【図3】
【図4】
【図6】
【図7】
【図10】
【公開番号】特開2013−39046(P2013−39046A)
【公開日】平成25年2月28日(2013.2.28)
【国際特許分類】
【出願番号】特願2011−176289(P2011−176289)
【出願日】平成23年8月11日(2011.8.11)
【出願人】(000236436)浜松ホトニクス株式会社 (1,479)
【Fターム(参考)】
【公開日】平成25年2月28日(2013.2.28)
【国際特許分類】
【出願日】平成23年8月11日(2011.8.11)
【出願人】(000236436)浜松ホトニクス株式会社 (1,479)
【Fターム(参考)】
[ Back to top ]