
2−オキソグルタル酸デカルボキシラーゼ、それをコードするDNA、該DNAを含む組換えベクター、該組換えベクターを用いて得られた形質転換体、2−オキソグルタル酸デカルボキシラーゼの製造方法及び2−オキソグルタル酸の測定方法
【課題】2−オキソグルタル酸デカルボキシラーゼ、それをコードするDNA、該DNAを含む組換えベクター、該組換えベクターを用いて得られた形質転換体、2−オキソグルタル酸デカルボキシラーゼの製造方法及び2−オキソグルタル酸の測定方法の提供。
【解決手段】以下の(a)又は(b)に示されるアミノ酸配列からなる2−オキソグルタル酸デカルボキシラーゼ、(a)特定のアミノ酸配列、(b)特定のアミノ酸配列のうち1又は2以上のアミノ酸が欠失、置換又は付加されたアミノ酸配列であって、かつ、2−オキソグルタル酸デカルボキシラーゼ活性を有するアミノ酸配列。
【解決手段】以下の(a)又は(b)に示されるアミノ酸配列からなる2−オキソグルタル酸デカルボキシラーゼ、(a)特定のアミノ酸配列、(b)特定のアミノ酸配列のうち1又は2以上のアミノ酸が欠失、置換又は付加されたアミノ酸配列であって、かつ、2−オキソグルタル酸デカルボキシラーゼ活性を有するアミノ酸配列。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は2−オキソグルタル酸デカルボキシラーゼ、それをコードするDNA、該DNAを含む組換えベクター、該組換えベクターを用いて得られた形質転換体、2−オキソグルタル酸デカルボキシラーゼの製造方法及び2−オキソグルタル酸の測定方法に係り、特にユーグレナ属由来の2−オキソグルタル酸デカルボキシラーゼ、それをコードするDNA、該DNAを含む組換えベクター、該組換えベクターを用いて得られた形質転換体、2−オキソグルタル酸デカルボキシラーゼの製造方法及び2−オキソグルタル酸の測定方法に関する。
【背景技術】
【0002】
ユーグレナ(Euglena)は、動物学と植物学の双方に分類され、動物学上は鞭毛虫綱、植物学上はミドリムシ藻類綱に属するミドリムシ目の微生物である。ユーグレナ属の微生物は、動物学的、植物学的な特徴を兼ね備えた独特の性質を有しており、生化学、生理学等の分野で古くから研究対象として用いられてきた。
【0003】
ユーグレナは、他の微生物と比較して特殊なTCA回路を有しており、コハク酸セミアルデヒド(SSA)という物質を産生することが知られている。
コハク酸セミアルデヒドは、2−オキソグルタル酸デカルボキシラーゼ(2−OGD)により2−オキソグルタル酸(2−OG:α−ケトグルタル酸ともいう)から生成する。2−オキソグルタル酸デカルボキシラーゼの触媒反応は、下記一般式(1)のとおりである。
【0004】
【化1】

(ここで、TPPはチアミンピロリン酸を示す。)
【0005】
コハク酸セミアルデヒドの炭素鎖は、乳酸よりも炭素1つ分長いため、コハク酸セミアルデヒドの重合体はポリ乳酸と同様に生分解性プラスチックとしての性質を有していると同時に、ポリ乳酸とは炭素1つ分異なる特性を有していると考えられる。
このため、コハク酸セミアルデヒドは、現在ポリ乳酸を主体に研究、開発が進んでいる生分解性プラスチックの分野において、乳酸に代わる新たな原料として期待されている。
【0006】
さらに、2−オキソグルタル酸デカルボキシラーゼの遺伝子を他の植物等に遺伝子組換えなどで組み込むことにより、コハク酸セミアルデヒドやその重合体である生分解性プラスチックを産生する植物等を生成することも可能になる。特に、コハク酸セミアルデヒドは、濃度が高くなると自然に重合するため、宿主細胞中でコハク酸セミアルデヒドを大量に生成させて高濃度状態とすることにより、細胞中にコハク酸セミアルデヒド重合体からなる生分解性プラスチックを産生する新規な生物を作出できると期待されている。
【0007】
また、上述した2−オキソグルタル酸デカルボキシラーゼの生成物であるコハク酸セミアルデヒドの利用のみならず、2−オキソグルタル酸デカルボキシラーゼそのものを産業に利用することも期待されている。
例えば、2−オキソグルタル酸デカルボキシラーゼを用いることで、酵素反応により2−オキソグルタル酸を定量することができる。以下、詳細に説明する。
【0008】
従来から、悪性腫瘍を有する患者は、血漿中の2−オキソグルタル酸濃度が高くなることが知られている。これは、腫瘍細胞ではクエン酸回路に異常が生じて細胞中に2−オキソグルタル酸が大量に蓄積し、高濃度の2−オキソグルタル酸が血漿中に浸潤するためと考えられている。したがって、血漿中の2−オキソグルタル酸を定量することで、悪性腫瘍の有無を検出することが可能となる。
【0009】
従来、血漿中に含まれる2−オキソグルタル酸の定量は、2−オキソグルタル酸と反応する試薬を用いた高速液体クロマトグラフィー(HPLC)法により行われていた(例えば、非特許文献1参照)。
非特許文献1の方法では、2−オキソグルタル酸を含む血漿中に2−ニトロフェニルヒドラジンと2,4−ジニトロフェニルヒドラジンを混合して反応させ、生成した2−オキソグルタル酸誘導体をHPLC用のカラムに流して2−オキソグルタル酸誘導体を定量する。これにより、間接的に2−オキソグルタル酸の定量を行っている。
【0010】
しかしながら、HPLC法による定量では、反応物をカラムから溶出する時間がかかり、迅速な定量が困難であった。そこで、2−オキソグルタル酸の定量方法として、2−オキソグルタル酸を基質として特異的に反応する2−オキソグルタル酸デカルボキシラーゼを用いる方法が新たに検討されている。
【0011】
【非特許文献1】K.Michailら、「Development and validation of a liquid chromatographic method for the determination of hydroxymethylfurfural and alpha−ketoglutaric acid in human plasma」、ANALYTICA CHIMICA ACTA、(オランダ)、Elsevier Science Publishers、2007、581、p.287−297
【発明の開示】
【発明が解決しようとする課題】
【0012】
このように、2−オキソグルタル酸デカルボキシラーゼは、生成物であるコハク酸セミアルデヒドが産業上有用であるばかりでなく、基質である2−オキソグルタル酸の定量にも用いることができることから、その生化学的、構造学的特性の詳細な研究が期待されている。
しかしながら、これまで、ユーグレナ由来の2−オキソグルタル酸デカルボキシラーゼ遺伝子のクローニングに成功した例はなく、そのアミノ酸配列やDNA塩基配列についても未知であった。
【0013】
本発明の目的は、上記課題に鑑み、2−オキソグルタル酸デカルボキシラーゼ、それをコードするDNA、該DNAを含む組換えベクター、該組換えベクターを用いて得られた形質転換体、2−オキソグルタル酸デカルボキシラーゼの製造方法及び2−オキソグルタル酸の測定方法を提供することにある。
【課題を解決するための手段】
【0014】
今回発明者らは、上記目的を達成するために鋭意努力した結果、ユーグレナ由来の2−オキソグルタル酸デカルボキシラーゼの新たなアミノ酸配列及びDNA塩基配列の取得に成功し、本発明を完成させるに至った。
すなわち、本発明は以下のとおりである。
以下の(a)又は(b)に示されるアミノ酸配列からなる2−オキソグルタル酸デカルボキシラーゼ。
(a)配列番号1に記載のアミノ酸配列。
(b)配列番号1に記載のアミノ酸配列のうち1又は2以上のアミノ酸が欠失、置換又は付加されたアミノ酸配列であって、かつ、2−オキソグルタル酸デカルボキシラーゼ活性を有するアミノ酸配列。
【0015】
以下の(c)又は(d)に示されるDNA。
(c)配列番号1に記載のアミノ酸配列をコードするDNA。
(d)前記(c)に記載のDNAとストリンジェントな条件下でハイブリダイズするDNAであって、かつ、2−オキソグルタル酸デカルボキシラーゼ活性を有するタンパク質をコードするDNA。
【0016】
以下の(e)又は(f)に示されるDNA。
(e)配列番号2に記載のDNA。
(f)配列番号2に記載のDNAとストリンジェントな条件下でハイブリダイズするDNAであって、かつ、2−オキソグルタル酸デカルボキシラーゼ活性を有するタンパク質をコードするDNA。
【0017】
前記いずれかに記載のDNAを含有する組換えベクター。
【0018】
前記組換えベクターを含有する形質転換体。
【0019】
前記形質転換体を培養し、培養物から2−オキソグルタル酸デカルボキシラーゼを採取することを特徴とする2−オキソグルタル酸デカルボキシラーゼの製造方法。
【0020】
前記いずれかに記載の2−オキソグルタル酸デカルボキシラーゼと2−オキソグルタル酸とを反応させ、生成したコハク酸セミアルデヒド及び二酸化炭素の少なくとも一方の生成量を測定することで、前記2−オキソグルタル酸の定量を行うことを特徴とする2−オキソグルタル酸の測定方法。
【発明の効果】
【0021】
本発明によって、2−オキソグルタル酸デカルボキシラーゼの新規なアミノ酸配列と、これをコードするDNA塩基配列とが初めて提供された。そして、2−オキソグルタル酸デカルボキシラーゼを用いることで、その基質である2−オキソグルタル酸の定量が容易になり、また、その反応生成物であるコハク酸セミアルデヒドを用いて新たな特性を有する生分解性プラスチックを生成することが可能となる。このため、本発明は産業上極めて有用である。
【発明を実施するための最良の形態】
【0022】
以下、本発明の一実施形態について説明する。なお、以下に説明する部材、配置等は、本発明を限定するものではなく、本発明の趣旨に沿って各種改変することができることは勿論である。
【0023】
本発明の2−オキソグルタル酸デカルボキシラーゼは、チアミンピロリン酸とマグネシウムイオンの存在下で2−オキソグルタル酸を非酸化的に脱炭酸し、コハク酸セミアルデヒドを生成する脱炭酸酵素の一種である。2−オキソグルタル酸デカルボキシラーゼは、分子量約6万2000からなる4量体構造をしている。
【0024】
本発明の2−オキソグルタル酸デカルボキシラーゼは、配列番号1に記載のアミノ酸配列であるか、又は配列番号1に記載のアミノ酸配列のうち1又は2以上のアミノ酸が欠失、置換又は付加されたアミノ酸配列であって、かつ、2−オキソグルタル酸デカルボキシラーゼ活性を有するアミノ酸配列を有している。
【0025】
ここで、上記「1又は2以上のアミノ酸が欠失、置換又は付加されたアミノ酸配列」とは、例えば1〜20個、好ましくは1〜10個、より好ましくは1〜5個の任意のアミノ酸が欠失、置換又は付加されたアミノ酸配列を意味する。
なお、アミノ酸の欠失、置換、付加は、自然発生的な突然変異により、又は部位特異的突然変異により導入することができる。
【0026】
また、本発明のDNAは、配列番号1に記載のアミノ酸配列をコードするDNAであるか、又はこのDNAとストリンジェントな条件下でハイブリダイズするDNAであって、かつ、2−オキソグルタル酸デカルボキシラーゼ活性を有するタンパク質をコードするDNAである。
特に、本発明のDNAは、配列番号2に記載のDNAであるか、又は配列番号2に記載のDNAとストリンジェントな条件下でハイブリダイズするDNAであって、かつ、2−オキソグルタル酸デカルボキシラーゼ活性を有するタンパク質をコードするDNAであることが好ましい。
【0027】
「ストリンジェントな条件下でハイブリダイズする」とは、特異的なハイブリッドが形成され、非特異的なハイブリッドがほとんど形成されない条件をいう。このような条件としては、J.Sambrook et al. Molecular Cloning, A Laboratory Manual, 2nd Edition., Cold Spring Harbor Laboratory(1989)に記載されている条件が挙げられる。例えば、6×SSC(1×SSCの組成:0.15MのNaCl、0.015Mのクエン酸ナトリウム、pH7.0)、0.5%のデンハルト溶液(フィコール400+PVP+BSA=1:1:1)、0.5%のSDS、100μg/mlの熱変性サケ精子DNA、50%のホルムアミドを含む溶液中、45℃での一晩保存する条件が挙げられる。また、より高ストリンジェンシーな条件として、50℃以上、更には60℃以上、特に65℃以上の条件が挙げられる。
【0028】
なお、上述した「ストリンジェントな条件下でハイブリダイズするDNA」としては、配列番号1に記載のアミノ酸配列をコードするDNA塩基配列又は配列番号2のDNA塩基配列と一定以上の相同性を有するDNAが挙げられる。この場合の相同性としては、通常60%以上であり、好ましくは80%以上、更に好ましくは90%以上、特に好ましくは95%以上、最も好ましくは98%以上である。
【0029】
本発明の2−オキソグルタル酸デカルボキシラーゼは、例えばユーグレナ(Euglena)属の微生物から単離することができる。ここで「ユーグレナ」とは、動物学や植物学の分類でユーグレナ属に属する種、変種、変異種のすべてを含み、かつ配列番号1に記載されたアミノ酸配列を有する2−オキソグルタル酸デカルボキシラーゼを生成するすべての生物を意味する。さらに、この場合のユーグレナには、配列番号1に記載のアミノ酸配列のうち1又は2以上のアミノ酸が欠失、置換又は付加されたアミノ酸配列であって、かつ、2−オキソグルタル酸デカルボキシラーゼ活性を有するアミノ酸配列を有するポリペプチドを生成する生物も含まれる。
【0030】
ここで、ユーグレナ属(Euglena)の微生物とは、動物学では原生動物門(Protozoa)の鞭毛虫綱(Mastigophorea)、植物鞭毛虫亜綱(Phytomastigophorea)に属するミドリムシ目(Euglenida)のユーグレノイディナ亜目(Euglenoidina)に属する微生物である。一方、ユーグレナ属の微生物は、植物学ではミドリムシ植物門(Euglenophyta)のミドリムシ藻類綱(Euglenophyceae)に属するミドリムシ目(Euglenales)に属している。
【0031】
ユーグレナ属の微生物としては、具体的には、Euglena acus、Euglena caudata、Euglena chadefaudii、Euglena deses、Euglena gracilis、Euglena granulata、Euglena intermedia、Euglena mutabilis、Euglena oxyuris、Euglena proxima、Euglena spirogyra、Euglena viridis、Euglena vermiformisなどが挙げられる。このうち特に、広く研究に利用されているユーグレナ グラシリス(Euglena gracilis)が好適である。
【0032】
ユーグレナは、Cramer−Myers培地、Hutner培地、Koren−Hutner培地や、これらの一部組成を変更した改変培地などを用いて培養することができる。培養容器には、坂口フラスコ、三角フラスコ、試薬ビンなどを用いることができる。ユーグレナはCO2を資化するため、1〜5%CO2を含む空気を培地中に通過させることが好ましい。また、終濃度0.5〜1.0%(容量比)となるように培地中にエタノールを加えると良好な生育となるため好ましい。さらに、葉緑体を十分に発達させるために、培地1リットルあたり1〜5g程度のリン酸アンモニウムを加えるとよい。培養温度は、通常20〜34℃で、特に28〜30℃が好適である。ユーグレナは、培養条件にもよるが、通常、培養開始後2〜3日で対数増殖期となり、4〜5日程度で定常期に到達する。
【0033】
2−オキソグルタル酸デカルボキシラーゼは、ユーグレナから公知の精製方法を用いて精製することができる。精製方法としては、例えばクロマトグラフィー法が挙げられる。クロマトグラフィー法の具体例として、分子排斥クロマトグラフィー、吸着クロマトグラフィー、イオン交換クロマトグラフィー、アフィニティークロマトグラフィーなどが挙げられる。
【0034】
得られた精製品は、20%〜50%のグリセロールやエチレングリコール等の凍結保護剤が添加された保存液中で保存することができる。なお、長期保存安定性の観点から、2−オキソグルタル酸デカルボキシラーゼは、−20℃程度で冷凍保存されることが好ましい。
2−オキソグルタル酸デカルボキシラーゼのアミノ酸配列は、精製されたタンパク質を用いてエドマン法などの公知のアミノ酸配列決定方法で行うことができる。
【0035】
2−オキソグルタル酸デカルボキシラーゼ遺伝子のクローニングは、ユーグレナのmRNAを抽出し、cDNAを合成することで行われる。
mRNAの調製は、公知のmRNA抽出方法で行うことができる。例えば、培養後のユーグレナをフェノール・クロロホルムなどの有機溶媒で溶解し、mRNAを水層に移動させ、エタノール沈殿法によりmRNAを沈殿させる方法で行う。
【0036】
cDNAの増幅は、上述したアミノ酸配列決定方法により決定された既知配列の一部をプライマーとして、RACE法などの公知の手法で行うことができる。例えば、上記エドマン法で決定されたN末端側のアミノ酸残基を元にセンスプライマーを作成し、一方でmRNAの3'側のポリA領域にアニールするポリdT領域を含むアンカープライマーを用いて、センス・アンカープライマー間の未知領域の増幅を行う。
cDNAの増幅は、逆転写酵素を作用させることで行うことができる。逆転写酵素としては、遺伝子工学で通常用いられる酵素、例えばAMV(トリ骨髄芽球症ウイルス)、MoMuLV(モロニーマウス白血病ウイルス)などを用いることができる。
【0037】
2−オキソグルタル酸デカルボキシラーゼ遺伝子の塩基配列決定は、上記でクローニングされたcDNAを用いて公知の配列決定方法、例えばマキサム・ギルバート法やジデオキシヌクレオチド法(サンガー法)などで行うことができる。
【0038】
2−オキソグルタル酸デカルボキシラーゼをコードする二本鎖DNAを適当なベクターに組み込むことで組換えベクターを作成することができる。ベクターとしては、プラスミド、ファージなどの宿主細胞内で自立複製が可能なベクターや、宿主細胞の染色体中に組み込まれることで複製可能なベクターなどが挙げられる。これらのベクターには、2−オキソグルタル酸デカルボキシラーゼ遺伝子が組み込まれる位置の上流側や下流側にプロモータ、エンハンサー、ターミネータなどの配列を有するものを用いることができる。
このようなベクターとして、例えばpBR系、pUC系、pKC系などの大腸菌プラスミドベクター、pUB110、pC194などの枯草菌プラスミドベクター、YRp系、YEp系などの酵母プラスミドベクターなどが挙げられる。
【0039】
2−オキソグルタル酸デカルボキシラーゼをコードするDNAを組み込んだ組換えベクターは、適当な宿主細胞に導入され、これを形質転換する。宿主細胞としては、大腸菌、枯草菌、酵母、植物細胞、動物細胞、昆虫細胞などが挙げられる。形質転換は、塩化カルシウム法、エレクトロポレーション法、DEAEデキストラン法などの公知の方法で行うことができる。
組換えベクターを用いて形質転換体を作成することにより、特定の細胞内で2−オキソグルタル酸デカルボキシラーゼを大量発現させる大量発現系を構築することが可能である。
【0040】
得られた2−オキソグルタル酸デカルボキシラーゼは、2−オキソグルタル酸の定量に用いることができる。2−オキソグルタル酸は、悪性腫瘍を有する患者の血漿中から高濃度に検出されることから、腫瘍診断のマーカとして用いられている。
2−オキソグルタル酸は、反応生成物であるコハク酸セミアルデヒドの生成量を測定することにより、定量することができる。コハク酸セミアルデヒドは、例えばo−アミノベンズアルデヒドとの反応により発色させ、分光光度計などを用いて波長440nmでの吸光度を測定することで定量する。
なお、2−オキソグルタル酸は、他の反応生成物である二酸化炭素をCO2センサーなどで測定することでも定量することができる。
【実施例】
【0041】
(1)ユーグレナの培養
ユーグレナ グラシリス(Euglena gracilis)SM−ZK株(葉緑体欠損株)を、Koren−Hutner改変培地(以下、KH改変培地という)で培養した。KH改変培地は、公知のKoren−Hutner培地(Koren,L.E. and Hutner, S.H. (1967) J.Protozool., 14, Suppl., 17.)のグルタミン酸を4倍量にした培地である。
【0042】
まず、坂口フラスコにKH改変培地150mlを入れ、同じく予めKH改変培地で生育して対数増殖期にあるユーグレナ グラシリス SM−ZK株1mlを植え継ぎ、29℃、100回転/分の振とう条件下で培養し、4日後に培養を停止した。
次に、培養停止後のユーグレナ培養液を、遠心分離機(HITACHI製 himac CR200)を用いて4℃、5000rpmで15分間遠心分離し、細胞を回収した。
【0043】
(2)2−オキソグルタル酸デカルボキシラーゼの精製
(細胞の破砕)
得られた細胞約69.23gに対し、破砕バッファ140mlで懸濁した。ここで、破砕バッファの組成は、50mMのリン酸カリウム(pH7.0)、1mMのTPP、1mMのMgCl2、1mMの2‐メルカプトエタノール、残部が20%ポリエチレングリコール(MW.600)である。次に、4280S型(海上電機製)を用いてこの懸濁液を4℃で10秒×10回超音波処理し、細胞を破砕した。得られた細胞破砕液を、遠心分離機を用いて4℃、12000rpmで10分間遠心分離し、約172mlの上清を得た。
得られた上清を、恒温槽 Mini−80(TAITEC製)を用いて50℃で10分間熱処理し、さらに熱処理後の細胞破砕液を12000rpm、10分間で超遠心分離し、約146mlの上清を得た。
【0044】
(DEAE Sepharose(登録商標)精製)
上で得られた上清を、DEAE Sepharose(DEAE Sepharose Fast Flow 17−0709−01(GEヘルスケア バイオサイエンス製))を充填したカラムにアプライした。続いて、初期濃度0M〜最終濃度0.5MとなるようにKCl濃度を変化させつつ、破砕バッファにより4℃でグラジエント溶出を行った。カラムから溶出した溶液を2mlずつフラクションコレクターで採取し、各フラクションに含まれる2−オキソグルタル酸デカルボキシラーゼの量を、以下に述べる2−オキソグルタル酸デカルボキシラーゼの活性測定法を用いて調べた。その結果、活性のピークが検出されたフラクションを含む第16〜50番目のフラクションを回収し、約68mlの溶液を得た。得られた溶液を透析チューブに入れ、破砕バッファ中で4℃、16時間透析した。
【0045】
(2−オキソグルタル酸デカルボキシラーゼの活性測定法)
2−オキソグルタル酸デカルボキシラーゼの活性測定は、下記の1)〜8)に示すo−アミノベンズアルデヒド法により行った。すなわち、2−オキソグルタル酸デカルボキシラーゼと2−オキソグルタル酸の反応により生成するコハク酸セミアルデヒドをo−アミノベンズアルデヒドで発色定量することにより、溶液中の2−オキソグルタル酸デカルボキシラーゼの活性を測定した。
コハク酸セミアルデヒドの測定は、波長440nmの吸光度を測定することで行った(A440=0.48のときコハク酸セミアルデヒド濃度=0.9mM)。以下、2−オキソグルタル酸デカルボキシラーゼ活性測定においても同様の方法を用いた。
【0046】
1) 活性測定バッファ2mlに50μlの粗酵素溶液を添加(活性測定バッファの組成:50mMのリン酸カリウム(pH7.0)、1mMの2−オキソグルタル酸、1mMのMgCl2、0.2mMのTPP、0.5mMのNADP+、1mMの2−メルカプトエタノール)
2) 30℃で10分間インキュベート
3) 25% TCAを100μl添加し、反応を停止
4) 反応液500μlに0.8M KOHを200μl添加
5) 1.0M グリシン緩衝液(pH 8.8)を1μl添加
6) 25mM o−アミノベンズアルデヒドを500μl添加
7) 35℃で15分間インキュベート
8) 分光光度計を用いて波長440nmの吸光度を測定
【0047】
(Blue Sepharose精製)
Blue Sepharose(HiTrap Blue HP 17−0412−01(GEヘルスケア バイオサイエンス製))を充填したカラムに透析後の溶液をアプライした。続いて、上述したDEAE Sepharoseの精製時と同様に、0〜0.5MでKCl濃度を変化させつつ、破砕バッファにより4℃でグラジエント溶出を行った。カラムから溶出した溶液を1mlずつフラクションコレクターで採取し、各フラクションに含まれる2−オキソグルタル酸デカルボキシラーゼの量を、上述した2−オキソグルタル酸デカルボキシラーゼ活性測定法を用いて調べた。その結果、活性のピークが検出されたフラクションを含む第9〜41番目のフラクションを回収し、約34mlの溶液を得た。得られた溶液を透析チューブに入れ、破砕バッファ中で4℃、16時間透析した。
【0048】
(UNO Q精製)
次に、UNO Q(UNO Q−6カラム 720−0003(BIO−RAD製))を充填したカラムに濃縮後の溶液をアプライした。続いて、上述したDEAE Sepharoseの精製時と同様に、0〜0.5MでKCl濃度を変化させつつ、破砕バッファにより4℃でグラジエント溶出を行った。カラムから溶出した溶液を1mlずつフラクションコレクターで採取し、各フラクションに含まれる2−オキソグルタル酸デカルボキシラーゼの量を、上述した2−オキソグルタル酸デカルボキシラーゼ活性測定法を用いて調べた。その結果、活性のピークが検出されたフラクションを含む第22〜26番目のフラクションを回収し、約5mlの溶液を得た。得られた溶液を透析チューブに入れ、破砕バッファ中で4℃、16時間透析した。
【0049】
(ゲルろ過)
続いて、Superdex(登録商標) 200(HiLoad 16/60 Superdex 200 pg 17−1069−01(GEヘルスケア バイオサイエンス製))を充填したカラムに濃縮後の溶液をアプライし、バッファ(破砕バッファの中のトリスを10mMリン酸に変えた溶液)を用いて4℃で溶出させた。カラムから溶出した溶液を120mlずつフラクションコレクターで採取し、活性のピークが検出されたフラクションを含むフラクションを回収し、約6.5mlの溶液を得た。
【0050】
(ハイドロキシアパタイトゲル精製)
最後に、CHT−I Hydroxyapatite(Bio−Scale CHT 10−Iカラム 751−0025(BIO−RAD製))を充填したカラムに濃縮後の溶液をアプライした。続いて、上述したDEAE Sepharoseの精製時と同様に、0〜0.5MでKCl濃度を変化させつつ、破砕バッファにより4℃でグラジエント溶出を行った。カラムから溶出した溶液を1mlずつフラクションコレクターで採取し、活性のピークが検出されたフラクションを含むフラクションを回収し、約3.6mlの溶液を得た。
【0051】
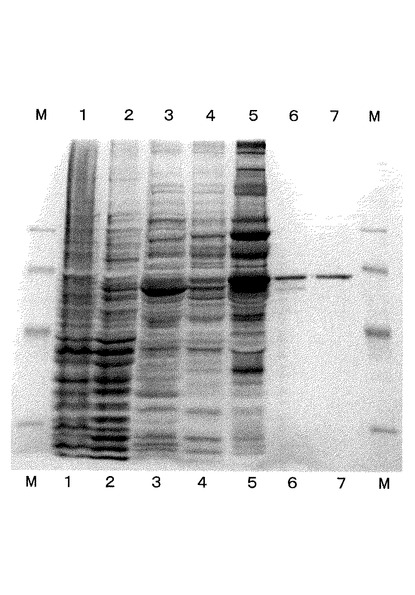
精製の結果を図1に示す。図1は各精製過程において溶液をサンプリングし、SDS−PAGEで電気泳動した結果を示す写真である。
この写真において、両端のレーンはマーカ、第1レーンは集菌直後(クルード)、第2レーンは熱処理後、第3レーンはDEAE Sepharose精製後、第4レーンはBlue Sepharose精製後、第5レーンはUNO Q精製後、第6レーンはゲルろ過(Superdex 200)後、第7レーンはハイドロキシアパタイト(Hydroxyapatite)ゲル精製後を示している。
【0052】
この図の第7レーンに示すように、精製の最終段階では目的とする2−オキソグルタル酸デカルボキシラーゼが高濃度に精製され、他のタンパク質がほとんど含まれないことがわかる。
次に、精製の各段階での総タンパク量、2−オキソグルタル酸デカルボキシラーゼの全酵素活性、比活性、収率、精製度をそれぞれ求めた。その結果を表1に示す。
【表1】
【0053】
(3)2−オキソグルタル酸デカルボキシラーゼ遺伝子のクローニング・配列決定
(N末端側アミノ酸配列の決定)
精製後の2−オキソグルタル酸デカルボキシラーゼを用い、492 Sequencer(ABI製)を使用して、エドマン法によりN末端側から十数残基分のアミノ酸配列を決定した。得られた配列(配列番号3)は以下のとおりである。
Lys−Ala−Leu−Asp−Gly−Pro−Asp−Arg−Arg−Xaa−Gln−Xaa−Thr−Xaa−Xaa
(ここで、N末端側から1番目のLysはSerであってもよい。)
【0054】
続いて、このN末端側配列を元に、ESTデータベース(Department de Biochimie, Universite de Montreal Montreal, Canada)に登録されているユーグレナ グラシリスのDNA塩基配列に対してBLASTによる相同性検索を行った。その結果、アセトラクテート シンテース(Acetolactate synthase:SEQ ID ELL00002303)のN末端側の一部の配列(1168bp)がヒットした。そのN末端側アミノ酸配列を確認したところ、得られた配列(配列番号4)は以下のとおりであった。
Ile−Ala−Leu−Asp−Gly−Pro−Asp−Arg−Asp−Arg−Met−Phe−Asn−Ala−Tyr
【0055】
(Sepasol−CTAB法によるRNA抽出)
上記「(1)ユーグレナの培養」で述べた方法でユーグレナを50mlフラスコで4日間培養し、6000rpm、10分間遠心分離して細胞約1gを得た。
得られた細胞をファルコンチューブに入れ、これにSepasol(登録商標)を加えてボルテックスで強く攪拌し、細胞を完全に破砕した。次に、破砕後の試料を50℃、10分間加温し、室温で5分間静置した。続いて、1.5mlのエッペンドルフ(登録商標)チューブに1mlずつ試料を分注し、50℃、10分間加温した後、室温で5分間静置した。
【0056】
次に、クロロホルム・イソアミルアルコール(24:1)を700μl加え、ボルテックスで攪拌した後、3分間室温に静置した。次に、4℃、15000rpmで5分間遠心分離し、上清を別のチューブに移した。このチューブに等量のイソプロパノールを加え、ボルテックスで攪拌後、室温で10分間静置した。4℃、15000rpmで5分間遠心分離し、上清を除いた。
【0057】
次に、DEPC処理水600μlに沈殿を溶解し、5MのNaClを100μl、CTAB(10%)/NaCl(0.7M)溶液を80μl加え、65℃、10分間静置した。さらに、クロロホルム・イソアミルアルコールを700μl加え、ボルテックスで攪拌後、4℃、15000rpmで5分間遠心分離して水層を別のチューブに移した。
続いて、イソプロパノール700μlを加え、ボルテックスで攪拌後、室温に10分間静置した後、4℃、15000rpmで15分間遠心分離した。上清を除き、75%エタノール1mlを加えて沈殿を洗浄し、4℃、15000rpmで5分間遠心分離した。上清を除き、沈殿を軽く乾かした後、DEPC処理水に溶解した。
【0058】
(全RNAの定量)
RNAの定量は、石英セルを用いて分光光度計により行った。波長260nmは核酸、280nmはタンパク質による吸光であるため、RNAの濃度は〔OD260nm×40=μg/ml〕で計算した。純粋な核酸があるときには、260nmの吸光度は280nmの吸光度の1.8〜2.0倍になる。
まず、全RNAを1μl分取し、1mlのDEPC処理水で希釈した。続いて、波長260nmと280nmの吸光度を測定した。OD260nmの値から全RNA濃度を算出した。その結果、全RNA濃度は2.5μg/μlであった。
【0059】
(DNaseI処理法によるゲノムDNA除去)
上記Sepasol−CTAB法により得られた試料中に含まれるゲノムDNAの除去を行った。
まず、全RNAを50μg、RNase Inhibitor(40U/μl)を20U、DNaseI(RNase−free:5U/μl)を10Uとなるように加え、さらに10×DNaseI緩衝液を1倍希釈となるように加えた。さらに、DEPC処理水を混合し、Total50μlの反応液を調製した。続いて、この反応液を37℃で20〜30分間反応させた。
【0060】
次に、50μlのDEPC処理水と100μlのフェノール/クロロホルム/イソアミルアルコール(25:24:1)溶液を加えて混合し、室温、15000rpmで5分間遠心分離した。得られた上清を新しいチューブに移し、100μlのクロロホルム/イソアミルアルコール(24:1)を加えて混合し、室温、15000rpmで5分間遠心分離した。
【0061】
上清を新しいチューブに移し、10μlの3M酢酸ナトリウムと250μlの冷エタノールを加えて−80℃で20分間静置した。次に、4℃、15000rpmで10分間遠心分離し、上清を捨て、70%エタノールで沈殿を洗浄した。4℃、15000rpmで5分間遠心分離し、上清を捨て、沈殿を乾燥させた後、10μlのDEPC処理水に溶解した。上述した全RNAの定量法を用いてRNA濃度を測定した結果、全RNA濃度は1.0μg/μlであった。
【0062】
(逆転写反応による1st strandの作製)
以下の反応系により逆転写反応を行い、1st strand cDNAを作製した。プライマーとして、mRNAの3'側ポリA配列と相補的なポリdT配列とアンカー配列の2つの配列を有する3' RACE アンカープライマー(配列番号5)を用いた。
【0063】
まず、反応混合液(1μgの全RNA、1mMのdNTP Mixture、0.25μMの3' RACE アンカープライマー(配列番号5)、100UのReverTra Ace(100U/μl)、1×RTバッファ)を20μl調製した。
続いて、サーマルサイクラー(MyCycler ThermalCycler(登録商標) 577BR 0209(BIO−RAD製))を用い、42℃で60分間、95℃で5分間、逆転写反応を行った。反応終了後のサンプルを、4℃で3分間冷却したのち、10秒間スピンダウンした。
【0064】
(1st strandを用いたPCR法による2OGD合成遺伝子の部分作製)
得られた1st strand cDNAに対して5'側センスプライマー及び3'側アンチセンスプライマーを用いてPCRを行い、以下に述べる方法で2nd strand cDNAを作製した。上述したESTデータベースでヒットした配列(配列番号4)を元にEG.2OGD 5'−1 プライマー(配列番号7)とEG.2OGD 5'−2(配列番号8)の2種類のプライマーを作成し、センスプライマーとして用いた。また、アンチセンスプライマーとして、上記3' RACE アンカープライマー(配列番号5)のアンカー配列と同一の配列である3' RACE1 プライマー(配列番号6)を用いた。
【0065】
まず、反応混合液(1μlのサンプル、1mMのdNTP Mixture、0.25μMのEG.2OGD 5'−1 プライマー、0.25μMの3' RACE1 プライマー(配列番号6)、100UのPrimeSTAR(登録商標)(TaKaRa製、100U/μl)、1×RTバッファ)を50μl調製した。続いて、上述したサーマルサイクラーを用い、98℃で10秒間、55℃で5秒間、72℃で1.5分間を30サイクル繰り返し、最後に72℃で10分間とした。
得られたPCR産物をアガロースゲル電気泳動により電気泳動し、2.1〜2.2kbpのバンドを含むゲルを切り出して回収した。
【0066】
(cDNAのシークエンスによる部分配列決定)
得られたcDNAを、Ligation−Convenience Kit(日本ジーン製)を用いて大腸菌ベクター(pZErO−2(インビトロジェン製))に組み込んだ。得られた組換えベクターを用いて大腸菌(Top10(インビトロジェン製))を形質転換し、形質転換体を得た。
得られた形質転換体を培養し、アルカリSDS法により組換えベクターを回収し、DNAシーケンサー(Applied Biosystems製 3100AVANT)を用いてDNAシーケンスを行った。
【0067】
(プライマーウォーキング法による全長配列決定)
5'−1 プライマーと5'−2 プライマーを用いて決定された配列を元に、EG.2OGD 5'−3 プライマー(配列番号9)を作製した。続いて、「RT−PCR法による2nd strandの作製」で説明した反応系及び反応温度と同様の条件でPCRを行い、DNAシーケンスにより配列を決定した。決定された配列を元に、EG.2OGD 5'−4 プライマー(配列番号10)を作製し、更にDNAシーケンスを行った。
【0068】
次に、ESTデータベースでヒットしたN末端側配列と、上述したシーケンスで得られた配列とを連結し、ユーグレナ グラシリス SM−ZK株由来の2−オキソグルタル酸デカルボキシラーゼのcDNA全長配列を得た(配列番号2)。また、この全長配列を元に、2−オキソグルタル酸デカルボキシラーゼのアミノ酸配列を得た(配列番号1)。
【0069】
(得られた全長配列の確認)
上で得られたDNAはESTデータベースで得られた部分配列と、DNAシーケンスで得られた配列を組み合わせて決定された配列であるため、これがクローニングされたcDNAの全長配列と同じであることを確かめるため、全長配列の読み直しを行った。まず、全長配列を元に、5'末端側の配列を含むプライマー(プライマーA)と、3'末端側の配列を含むプライマー(プライマーB)の2種類のプライマーを作製した。プライマーAは制限酵素NdeIの切断サイトを、プライマーBは制限酵素XhoIの切断サイトをそれぞれ有している。
上記プライマーA,Bを用いてPCRを95℃(10秒)→55℃(10秒)→72℃(2分)を計35サイクル行い、72℃、10分間に保温した後、12℃で保存した。得られたPCR産物で大腸菌(Top10)を形質転換し、プレート上に撒いて得られたコロニーのいくつかを選択して培養した。培養後にアルカリSDSによりDNAを抽出し、NdeI、XhoIを用いて切断を行い、アガロースゲル電気泳動により制限酵素で切断が確認されたサンプルを選択した。
選択したサンプルのベクターに対して、上述したDNAシーケンサーを用いてDNAシーケンスを行い、2−オキソグルタル酸デカルボキシラーゼのcDNA全長配列を得た。この全長配列と先に得られたcDNA全長配列(配列番号2)とを比較し、両者が同一であることを確認した。
【図面の簡単な説明】
【0070】
【図1】各精製過程において溶液をサンプリングし、SDS−PAGEで電気泳動した結果を示す写真である。
【技術分野】
【0001】
本発明は2−オキソグルタル酸デカルボキシラーゼ、それをコードするDNA、該DNAを含む組換えベクター、該組換えベクターを用いて得られた形質転換体、2−オキソグルタル酸デカルボキシラーゼの製造方法及び2−オキソグルタル酸の測定方法に係り、特にユーグレナ属由来の2−オキソグルタル酸デカルボキシラーゼ、それをコードするDNA、該DNAを含む組換えベクター、該組換えベクターを用いて得られた形質転換体、2−オキソグルタル酸デカルボキシラーゼの製造方法及び2−オキソグルタル酸の測定方法に関する。
【背景技術】
【0002】
ユーグレナ(Euglena)は、動物学と植物学の双方に分類され、動物学上は鞭毛虫綱、植物学上はミドリムシ藻類綱に属するミドリムシ目の微生物である。ユーグレナ属の微生物は、動物学的、植物学的な特徴を兼ね備えた独特の性質を有しており、生化学、生理学等の分野で古くから研究対象として用いられてきた。
【0003】
ユーグレナは、他の微生物と比較して特殊なTCA回路を有しており、コハク酸セミアルデヒド(SSA)という物質を産生することが知られている。
コハク酸セミアルデヒドは、2−オキソグルタル酸デカルボキシラーゼ(2−OGD)により2−オキソグルタル酸(2−OG:α−ケトグルタル酸ともいう)から生成する。2−オキソグルタル酸デカルボキシラーゼの触媒反応は、下記一般式(1)のとおりである。
【0004】
【化1】
(ここで、TPPはチアミンピロリン酸を示す。)
【0005】
コハク酸セミアルデヒドの炭素鎖は、乳酸よりも炭素1つ分長いため、コハク酸セミアルデヒドの重合体はポリ乳酸と同様に生分解性プラスチックとしての性質を有していると同時に、ポリ乳酸とは炭素1つ分異なる特性を有していると考えられる。
このため、コハク酸セミアルデヒドは、現在ポリ乳酸を主体に研究、開発が進んでいる生分解性プラスチックの分野において、乳酸に代わる新たな原料として期待されている。
【0006】
さらに、2−オキソグルタル酸デカルボキシラーゼの遺伝子を他の植物等に遺伝子組換えなどで組み込むことにより、コハク酸セミアルデヒドやその重合体である生分解性プラスチックを産生する植物等を生成することも可能になる。特に、コハク酸セミアルデヒドは、濃度が高くなると自然に重合するため、宿主細胞中でコハク酸セミアルデヒドを大量に生成させて高濃度状態とすることにより、細胞中にコハク酸セミアルデヒド重合体からなる生分解性プラスチックを産生する新規な生物を作出できると期待されている。
【0007】
また、上述した2−オキソグルタル酸デカルボキシラーゼの生成物であるコハク酸セミアルデヒドの利用のみならず、2−オキソグルタル酸デカルボキシラーゼそのものを産業に利用することも期待されている。
例えば、2−オキソグルタル酸デカルボキシラーゼを用いることで、酵素反応により2−オキソグルタル酸を定量することができる。以下、詳細に説明する。
【0008】
従来から、悪性腫瘍を有する患者は、血漿中の2−オキソグルタル酸濃度が高くなることが知られている。これは、腫瘍細胞ではクエン酸回路に異常が生じて細胞中に2−オキソグルタル酸が大量に蓄積し、高濃度の2−オキソグルタル酸が血漿中に浸潤するためと考えられている。したがって、血漿中の2−オキソグルタル酸を定量することで、悪性腫瘍の有無を検出することが可能となる。
【0009】
従来、血漿中に含まれる2−オキソグルタル酸の定量は、2−オキソグルタル酸と反応する試薬を用いた高速液体クロマトグラフィー(HPLC)法により行われていた(例えば、非特許文献1参照)。
非特許文献1の方法では、2−オキソグルタル酸を含む血漿中に2−ニトロフェニルヒドラジンと2,4−ジニトロフェニルヒドラジンを混合して反応させ、生成した2−オキソグルタル酸誘導体をHPLC用のカラムに流して2−オキソグルタル酸誘導体を定量する。これにより、間接的に2−オキソグルタル酸の定量を行っている。
【0010】
しかしながら、HPLC法による定量では、反応物をカラムから溶出する時間がかかり、迅速な定量が困難であった。そこで、2−オキソグルタル酸の定量方法として、2−オキソグルタル酸を基質として特異的に反応する2−オキソグルタル酸デカルボキシラーゼを用いる方法が新たに検討されている。
【0011】
【非特許文献1】K.Michailら、「Development and validation of a liquid chromatographic method for the determination of hydroxymethylfurfural and alpha−ketoglutaric acid in human plasma」、ANALYTICA CHIMICA ACTA、(オランダ)、Elsevier Science Publishers、2007、581、p.287−297
【発明の開示】
【発明が解決しようとする課題】
【0012】
このように、2−オキソグルタル酸デカルボキシラーゼは、生成物であるコハク酸セミアルデヒドが産業上有用であるばかりでなく、基質である2−オキソグルタル酸の定量にも用いることができることから、その生化学的、構造学的特性の詳細な研究が期待されている。
しかしながら、これまで、ユーグレナ由来の2−オキソグルタル酸デカルボキシラーゼ遺伝子のクローニングに成功した例はなく、そのアミノ酸配列やDNA塩基配列についても未知であった。
【0013】
本発明の目的は、上記課題に鑑み、2−オキソグルタル酸デカルボキシラーゼ、それをコードするDNA、該DNAを含む組換えベクター、該組換えベクターを用いて得られた形質転換体、2−オキソグルタル酸デカルボキシラーゼの製造方法及び2−オキソグルタル酸の測定方法を提供することにある。
【課題を解決するための手段】
【0014】
今回発明者らは、上記目的を達成するために鋭意努力した結果、ユーグレナ由来の2−オキソグルタル酸デカルボキシラーゼの新たなアミノ酸配列及びDNA塩基配列の取得に成功し、本発明を完成させるに至った。
すなわち、本発明は以下のとおりである。
以下の(a)又は(b)に示されるアミノ酸配列からなる2−オキソグルタル酸デカルボキシラーゼ。
(a)配列番号1に記載のアミノ酸配列。
(b)配列番号1に記載のアミノ酸配列のうち1又は2以上のアミノ酸が欠失、置換又は付加されたアミノ酸配列であって、かつ、2−オキソグルタル酸デカルボキシラーゼ活性を有するアミノ酸配列。
【0015】
以下の(c)又は(d)に示されるDNA。
(c)配列番号1に記載のアミノ酸配列をコードするDNA。
(d)前記(c)に記載のDNAとストリンジェントな条件下でハイブリダイズするDNAであって、かつ、2−オキソグルタル酸デカルボキシラーゼ活性を有するタンパク質をコードするDNA。
【0016】
以下の(e)又は(f)に示されるDNA。
(e)配列番号2に記載のDNA。
(f)配列番号2に記載のDNAとストリンジェントな条件下でハイブリダイズするDNAであって、かつ、2−オキソグルタル酸デカルボキシラーゼ活性を有するタンパク質をコードするDNA。
【0017】
前記いずれかに記載のDNAを含有する組換えベクター。
【0018】
前記組換えベクターを含有する形質転換体。
【0019】
前記形質転換体を培養し、培養物から2−オキソグルタル酸デカルボキシラーゼを採取することを特徴とする2−オキソグルタル酸デカルボキシラーゼの製造方法。
【0020】
前記いずれかに記載の2−オキソグルタル酸デカルボキシラーゼと2−オキソグルタル酸とを反応させ、生成したコハク酸セミアルデヒド及び二酸化炭素の少なくとも一方の生成量を測定することで、前記2−オキソグルタル酸の定量を行うことを特徴とする2−オキソグルタル酸の測定方法。
【発明の効果】
【0021】
本発明によって、2−オキソグルタル酸デカルボキシラーゼの新規なアミノ酸配列と、これをコードするDNA塩基配列とが初めて提供された。そして、2−オキソグルタル酸デカルボキシラーゼを用いることで、その基質である2−オキソグルタル酸の定量が容易になり、また、その反応生成物であるコハク酸セミアルデヒドを用いて新たな特性を有する生分解性プラスチックを生成することが可能となる。このため、本発明は産業上極めて有用である。
【発明を実施するための最良の形態】
【0022】
以下、本発明の一実施形態について説明する。なお、以下に説明する部材、配置等は、本発明を限定するものではなく、本発明の趣旨に沿って各種改変することができることは勿論である。
【0023】
本発明の2−オキソグルタル酸デカルボキシラーゼは、チアミンピロリン酸とマグネシウムイオンの存在下で2−オキソグルタル酸を非酸化的に脱炭酸し、コハク酸セミアルデヒドを生成する脱炭酸酵素の一種である。2−オキソグルタル酸デカルボキシラーゼは、分子量約6万2000からなる4量体構造をしている。
【0024】
本発明の2−オキソグルタル酸デカルボキシラーゼは、配列番号1に記載のアミノ酸配列であるか、又は配列番号1に記載のアミノ酸配列のうち1又は2以上のアミノ酸が欠失、置換又は付加されたアミノ酸配列であって、かつ、2−オキソグルタル酸デカルボキシラーゼ活性を有するアミノ酸配列を有している。
【0025】
ここで、上記「1又は2以上のアミノ酸が欠失、置換又は付加されたアミノ酸配列」とは、例えば1〜20個、好ましくは1〜10個、より好ましくは1〜5個の任意のアミノ酸が欠失、置換又は付加されたアミノ酸配列を意味する。
なお、アミノ酸の欠失、置換、付加は、自然発生的な突然変異により、又は部位特異的突然変異により導入することができる。
【0026】
また、本発明のDNAは、配列番号1に記載のアミノ酸配列をコードするDNAであるか、又はこのDNAとストリンジェントな条件下でハイブリダイズするDNAであって、かつ、2−オキソグルタル酸デカルボキシラーゼ活性を有するタンパク質をコードするDNAである。
特に、本発明のDNAは、配列番号2に記載のDNAであるか、又は配列番号2に記載のDNAとストリンジェントな条件下でハイブリダイズするDNAであって、かつ、2−オキソグルタル酸デカルボキシラーゼ活性を有するタンパク質をコードするDNAであることが好ましい。
【0027】
「ストリンジェントな条件下でハイブリダイズする」とは、特異的なハイブリッドが形成され、非特異的なハイブリッドがほとんど形成されない条件をいう。このような条件としては、J.Sambrook et al. Molecular Cloning, A Laboratory Manual, 2nd Edition., Cold Spring Harbor Laboratory(1989)に記載されている条件が挙げられる。例えば、6×SSC(1×SSCの組成:0.15MのNaCl、0.015Mのクエン酸ナトリウム、pH7.0)、0.5%のデンハルト溶液(フィコール400+PVP+BSA=1:1:1)、0.5%のSDS、100μg/mlの熱変性サケ精子DNA、50%のホルムアミドを含む溶液中、45℃での一晩保存する条件が挙げられる。また、より高ストリンジェンシーな条件として、50℃以上、更には60℃以上、特に65℃以上の条件が挙げられる。
【0028】
なお、上述した「ストリンジェントな条件下でハイブリダイズするDNA」としては、配列番号1に記載のアミノ酸配列をコードするDNA塩基配列又は配列番号2のDNA塩基配列と一定以上の相同性を有するDNAが挙げられる。この場合の相同性としては、通常60%以上であり、好ましくは80%以上、更に好ましくは90%以上、特に好ましくは95%以上、最も好ましくは98%以上である。
【0029】
本発明の2−オキソグルタル酸デカルボキシラーゼは、例えばユーグレナ(Euglena)属の微生物から単離することができる。ここで「ユーグレナ」とは、動物学や植物学の分類でユーグレナ属に属する種、変種、変異種のすべてを含み、かつ配列番号1に記載されたアミノ酸配列を有する2−オキソグルタル酸デカルボキシラーゼを生成するすべての生物を意味する。さらに、この場合のユーグレナには、配列番号1に記載のアミノ酸配列のうち1又は2以上のアミノ酸が欠失、置換又は付加されたアミノ酸配列であって、かつ、2−オキソグルタル酸デカルボキシラーゼ活性を有するアミノ酸配列を有するポリペプチドを生成する生物も含まれる。
【0030】
ここで、ユーグレナ属(Euglena)の微生物とは、動物学では原生動物門(Protozoa)の鞭毛虫綱(Mastigophorea)、植物鞭毛虫亜綱(Phytomastigophorea)に属するミドリムシ目(Euglenida)のユーグレノイディナ亜目(Euglenoidina)に属する微生物である。一方、ユーグレナ属の微生物は、植物学ではミドリムシ植物門(Euglenophyta)のミドリムシ藻類綱(Euglenophyceae)に属するミドリムシ目(Euglenales)に属している。
【0031】
ユーグレナ属の微生物としては、具体的には、Euglena acus、Euglena caudata、Euglena chadefaudii、Euglena deses、Euglena gracilis、Euglena granulata、Euglena intermedia、Euglena mutabilis、Euglena oxyuris、Euglena proxima、Euglena spirogyra、Euglena viridis、Euglena vermiformisなどが挙げられる。このうち特に、広く研究に利用されているユーグレナ グラシリス(Euglena gracilis)が好適である。
【0032】
ユーグレナは、Cramer−Myers培地、Hutner培地、Koren−Hutner培地や、これらの一部組成を変更した改変培地などを用いて培養することができる。培養容器には、坂口フラスコ、三角フラスコ、試薬ビンなどを用いることができる。ユーグレナはCO2を資化するため、1〜5%CO2を含む空気を培地中に通過させることが好ましい。また、終濃度0.5〜1.0%(容量比)となるように培地中にエタノールを加えると良好な生育となるため好ましい。さらに、葉緑体を十分に発達させるために、培地1リットルあたり1〜5g程度のリン酸アンモニウムを加えるとよい。培養温度は、通常20〜34℃で、特に28〜30℃が好適である。ユーグレナは、培養条件にもよるが、通常、培養開始後2〜3日で対数増殖期となり、4〜5日程度で定常期に到達する。
【0033】
2−オキソグルタル酸デカルボキシラーゼは、ユーグレナから公知の精製方法を用いて精製することができる。精製方法としては、例えばクロマトグラフィー法が挙げられる。クロマトグラフィー法の具体例として、分子排斥クロマトグラフィー、吸着クロマトグラフィー、イオン交換クロマトグラフィー、アフィニティークロマトグラフィーなどが挙げられる。
【0034】
得られた精製品は、20%〜50%のグリセロールやエチレングリコール等の凍結保護剤が添加された保存液中で保存することができる。なお、長期保存安定性の観点から、2−オキソグルタル酸デカルボキシラーゼは、−20℃程度で冷凍保存されることが好ましい。
2−オキソグルタル酸デカルボキシラーゼのアミノ酸配列は、精製されたタンパク質を用いてエドマン法などの公知のアミノ酸配列決定方法で行うことができる。
【0035】
2−オキソグルタル酸デカルボキシラーゼ遺伝子のクローニングは、ユーグレナのmRNAを抽出し、cDNAを合成することで行われる。
mRNAの調製は、公知のmRNA抽出方法で行うことができる。例えば、培養後のユーグレナをフェノール・クロロホルムなどの有機溶媒で溶解し、mRNAを水層に移動させ、エタノール沈殿法によりmRNAを沈殿させる方法で行う。
【0036】
cDNAの増幅は、上述したアミノ酸配列決定方法により決定された既知配列の一部をプライマーとして、RACE法などの公知の手法で行うことができる。例えば、上記エドマン法で決定されたN末端側のアミノ酸残基を元にセンスプライマーを作成し、一方でmRNAの3'側のポリA領域にアニールするポリdT領域を含むアンカープライマーを用いて、センス・アンカープライマー間の未知領域の増幅を行う。
cDNAの増幅は、逆転写酵素を作用させることで行うことができる。逆転写酵素としては、遺伝子工学で通常用いられる酵素、例えばAMV(トリ骨髄芽球症ウイルス)、MoMuLV(モロニーマウス白血病ウイルス)などを用いることができる。
【0037】
2−オキソグルタル酸デカルボキシラーゼ遺伝子の塩基配列決定は、上記でクローニングされたcDNAを用いて公知の配列決定方法、例えばマキサム・ギルバート法やジデオキシヌクレオチド法(サンガー法)などで行うことができる。
【0038】
2−オキソグルタル酸デカルボキシラーゼをコードする二本鎖DNAを適当なベクターに組み込むことで組換えベクターを作成することができる。ベクターとしては、プラスミド、ファージなどの宿主細胞内で自立複製が可能なベクターや、宿主細胞の染色体中に組み込まれることで複製可能なベクターなどが挙げられる。これらのベクターには、2−オキソグルタル酸デカルボキシラーゼ遺伝子が組み込まれる位置の上流側や下流側にプロモータ、エンハンサー、ターミネータなどの配列を有するものを用いることができる。
このようなベクターとして、例えばpBR系、pUC系、pKC系などの大腸菌プラスミドベクター、pUB110、pC194などの枯草菌プラスミドベクター、YRp系、YEp系などの酵母プラスミドベクターなどが挙げられる。
【0039】
2−オキソグルタル酸デカルボキシラーゼをコードするDNAを組み込んだ組換えベクターは、適当な宿主細胞に導入され、これを形質転換する。宿主細胞としては、大腸菌、枯草菌、酵母、植物細胞、動物細胞、昆虫細胞などが挙げられる。形質転換は、塩化カルシウム法、エレクトロポレーション法、DEAEデキストラン法などの公知の方法で行うことができる。
組換えベクターを用いて形質転換体を作成することにより、特定の細胞内で2−オキソグルタル酸デカルボキシラーゼを大量発現させる大量発現系を構築することが可能である。
【0040】
得られた2−オキソグルタル酸デカルボキシラーゼは、2−オキソグルタル酸の定量に用いることができる。2−オキソグルタル酸は、悪性腫瘍を有する患者の血漿中から高濃度に検出されることから、腫瘍診断のマーカとして用いられている。
2−オキソグルタル酸は、反応生成物であるコハク酸セミアルデヒドの生成量を測定することにより、定量することができる。コハク酸セミアルデヒドは、例えばo−アミノベンズアルデヒドとの反応により発色させ、分光光度計などを用いて波長440nmでの吸光度を測定することで定量する。
なお、2−オキソグルタル酸は、他の反応生成物である二酸化炭素をCO2センサーなどで測定することでも定量することができる。
【実施例】
【0041】
(1)ユーグレナの培養
ユーグレナ グラシリス(Euglena gracilis)SM−ZK株(葉緑体欠損株)を、Koren−Hutner改変培地(以下、KH改変培地という)で培養した。KH改変培地は、公知のKoren−Hutner培地(Koren,L.E. and Hutner, S.H. (1967) J.Protozool., 14, Suppl., 17.)のグルタミン酸を4倍量にした培地である。
【0042】
まず、坂口フラスコにKH改変培地150mlを入れ、同じく予めKH改変培地で生育して対数増殖期にあるユーグレナ グラシリス SM−ZK株1mlを植え継ぎ、29℃、100回転/分の振とう条件下で培養し、4日後に培養を停止した。
次に、培養停止後のユーグレナ培養液を、遠心分離機(HITACHI製 himac CR200)を用いて4℃、5000rpmで15分間遠心分離し、細胞を回収した。
【0043】
(2)2−オキソグルタル酸デカルボキシラーゼの精製
(細胞の破砕)
得られた細胞約69.23gに対し、破砕バッファ140mlで懸濁した。ここで、破砕バッファの組成は、50mMのリン酸カリウム(pH7.0)、1mMのTPP、1mMのMgCl2、1mMの2‐メルカプトエタノール、残部が20%ポリエチレングリコール(MW.600)である。次に、4280S型(海上電機製)を用いてこの懸濁液を4℃で10秒×10回超音波処理し、細胞を破砕した。得られた細胞破砕液を、遠心分離機を用いて4℃、12000rpmで10分間遠心分離し、約172mlの上清を得た。
得られた上清を、恒温槽 Mini−80(TAITEC製)を用いて50℃で10分間熱処理し、さらに熱処理後の細胞破砕液を12000rpm、10分間で超遠心分離し、約146mlの上清を得た。
【0044】
(DEAE Sepharose(登録商標)精製)
上で得られた上清を、DEAE Sepharose(DEAE Sepharose Fast Flow 17−0709−01(GEヘルスケア バイオサイエンス製))を充填したカラムにアプライした。続いて、初期濃度0M〜最終濃度0.5MとなるようにKCl濃度を変化させつつ、破砕バッファにより4℃でグラジエント溶出を行った。カラムから溶出した溶液を2mlずつフラクションコレクターで採取し、各フラクションに含まれる2−オキソグルタル酸デカルボキシラーゼの量を、以下に述べる2−オキソグルタル酸デカルボキシラーゼの活性測定法を用いて調べた。その結果、活性のピークが検出されたフラクションを含む第16〜50番目のフラクションを回収し、約68mlの溶液を得た。得られた溶液を透析チューブに入れ、破砕バッファ中で4℃、16時間透析した。
【0045】
(2−オキソグルタル酸デカルボキシラーゼの活性測定法)
2−オキソグルタル酸デカルボキシラーゼの活性測定は、下記の1)〜8)に示すo−アミノベンズアルデヒド法により行った。すなわち、2−オキソグルタル酸デカルボキシラーゼと2−オキソグルタル酸の反応により生成するコハク酸セミアルデヒドをo−アミノベンズアルデヒドで発色定量することにより、溶液中の2−オキソグルタル酸デカルボキシラーゼの活性を測定した。
コハク酸セミアルデヒドの測定は、波長440nmの吸光度を測定することで行った(A440=0.48のときコハク酸セミアルデヒド濃度=0.9mM)。以下、2−オキソグルタル酸デカルボキシラーゼ活性測定においても同様の方法を用いた。
【0046】
1) 活性測定バッファ2mlに50μlの粗酵素溶液を添加(活性測定バッファの組成:50mMのリン酸カリウム(pH7.0)、1mMの2−オキソグルタル酸、1mMのMgCl2、0.2mMのTPP、0.5mMのNADP+、1mMの2−メルカプトエタノール)
2) 30℃で10分間インキュベート
3) 25% TCAを100μl添加し、反応を停止
4) 反応液500μlに0.8M KOHを200μl添加
5) 1.0M グリシン緩衝液(pH 8.8)を1μl添加
6) 25mM o−アミノベンズアルデヒドを500μl添加
7) 35℃で15分間インキュベート
8) 分光光度計を用いて波長440nmの吸光度を測定
【0047】
(Blue Sepharose精製)
Blue Sepharose(HiTrap Blue HP 17−0412−01(GEヘルスケア バイオサイエンス製))を充填したカラムに透析後の溶液をアプライした。続いて、上述したDEAE Sepharoseの精製時と同様に、0〜0.5MでKCl濃度を変化させつつ、破砕バッファにより4℃でグラジエント溶出を行った。カラムから溶出した溶液を1mlずつフラクションコレクターで採取し、各フラクションに含まれる2−オキソグルタル酸デカルボキシラーゼの量を、上述した2−オキソグルタル酸デカルボキシラーゼ活性測定法を用いて調べた。その結果、活性のピークが検出されたフラクションを含む第9〜41番目のフラクションを回収し、約34mlの溶液を得た。得られた溶液を透析チューブに入れ、破砕バッファ中で4℃、16時間透析した。
【0048】
(UNO Q精製)
次に、UNO Q(UNO Q−6カラム 720−0003(BIO−RAD製))を充填したカラムに濃縮後の溶液をアプライした。続いて、上述したDEAE Sepharoseの精製時と同様に、0〜0.5MでKCl濃度を変化させつつ、破砕バッファにより4℃でグラジエント溶出を行った。カラムから溶出した溶液を1mlずつフラクションコレクターで採取し、各フラクションに含まれる2−オキソグルタル酸デカルボキシラーゼの量を、上述した2−オキソグルタル酸デカルボキシラーゼ活性測定法を用いて調べた。その結果、活性のピークが検出されたフラクションを含む第22〜26番目のフラクションを回収し、約5mlの溶液を得た。得られた溶液を透析チューブに入れ、破砕バッファ中で4℃、16時間透析した。
【0049】
(ゲルろ過)
続いて、Superdex(登録商標) 200(HiLoad 16/60 Superdex 200 pg 17−1069−01(GEヘルスケア バイオサイエンス製))を充填したカラムに濃縮後の溶液をアプライし、バッファ(破砕バッファの中のトリスを10mMリン酸に変えた溶液)を用いて4℃で溶出させた。カラムから溶出した溶液を120mlずつフラクションコレクターで採取し、活性のピークが検出されたフラクションを含むフラクションを回収し、約6.5mlの溶液を得た。
【0050】
(ハイドロキシアパタイトゲル精製)
最後に、CHT−I Hydroxyapatite(Bio−Scale CHT 10−Iカラム 751−0025(BIO−RAD製))を充填したカラムに濃縮後の溶液をアプライした。続いて、上述したDEAE Sepharoseの精製時と同様に、0〜0.5MでKCl濃度を変化させつつ、破砕バッファにより4℃でグラジエント溶出を行った。カラムから溶出した溶液を1mlずつフラクションコレクターで採取し、活性のピークが検出されたフラクションを含むフラクションを回収し、約3.6mlの溶液を得た。
【0051】
精製の結果を図1に示す。図1は各精製過程において溶液をサンプリングし、SDS−PAGEで電気泳動した結果を示す写真である。
この写真において、両端のレーンはマーカ、第1レーンは集菌直後(クルード)、第2レーンは熱処理後、第3レーンはDEAE Sepharose精製後、第4レーンはBlue Sepharose精製後、第5レーンはUNO Q精製後、第6レーンはゲルろ過(Superdex 200)後、第7レーンはハイドロキシアパタイト(Hydroxyapatite)ゲル精製後を示している。
【0052】
この図の第7レーンに示すように、精製の最終段階では目的とする2−オキソグルタル酸デカルボキシラーゼが高濃度に精製され、他のタンパク質がほとんど含まれないことがわかる。
次に、精製の各段階での総タンパク量、2−オキソグルタル酸デカルボキシラーゼの全酵素活性、比活性、収率、精製度をそれぞれ求めた。その結果を表1に示す。
【表1】
【0053】
(3)2−オキソグルタル酸デカルボキシラーゼ遺伝子のクローニング・配列決定
(N末端側アミノ酸配列の決定)
精製後の2−オキソグルタル酸デカルボキシラーゼを用い、492 Sequencer(ABI製)を使用して、エドマン法によりN末端側から十数残基分のアミノ酸配列を決定した。得られた配列(配列番号3)は以下のとおりである。
Lys−Ala−Leu−Asp−Gly−Pro−Asp−Arg−Arg−Xaa−Gln−Xaa−Thr−Xaa−Xaa
(ここで、N末端側から1番目のLysはSerであってもよい。)
【0054】
続いて、このN末端側配列を元に、ESTデータベース(Department de Biochimie, Universite de Montreal Montreal, Canada)に登録されているユーグレナ グラシリスのDNA塩基配列に対してBLASTによる相同性検索を行った。その結果、アセトラクテート シンテース(Acetolactate synthase:SEQ ID ELL00002303)のN末端側の一部の配列(1168bp)がヒットした。そのN末端側アミノ酸配列を確認したところ、得られた配列(配列番号4)は以下のとおりであった。
Ile−Ala−Leu−Asp−Gly−Pro−Asp−Arg−Asp−Arg−Met−Phe−Asn−Ala−Tyr
【0055】
(Sepasol−CTAB法によるRNA抽出)
上記「(1)ユーグレナの培養」で述べた方法でユーグレナを50mlフラスコで4日間培養し、6000rpm、10分間遠心分離して細胞約1gを得た。
得られた細胞をファルコンチューブに入れ、これにSepasol(登録商標)を加えてボルテックスで強く攪拌し、細胞を完全に破砕した。次に、破砕後の試料を50℃、10分間加温し、室温で5分間静置した。続いて、1.5mlのエッペンドルフ(登録商標)チューブに1mlずつ試料を分注し、50℃、10分間加温した後、室温で5分間静置した。
【0056】
次に、クロロホルム・イソアミルアルコール(24:1)を700μl加え、ボルテックスで攪拌した後、3分間室温に静置した。次に、4℃、15000rpmで5分間遠心分離し、上清を別のチューブに移した。このチューブに等量のイソプロパノールを加え、ボルテックスで攪拌後、室温で10分間静置した。4℃、15000rpmで5分間遠心分離し、上清を除いた。
【0057】
次に、DEPC処理水600μlに沈殿を溶解し、5MのNaClを100μl、CTAB(10%)/NaCl(0.7M)溶液を80μl加え、65℃、10分間静置した。さらに、クロロホルム・イソアミルアルコールを700μl加え、ボルテックスで攪拌後、4℃、15000rpmで5分間遠心分離して水層を別のチューブに移した。
続いて、イソプロパノール700μlを加え、ボルテックスで攪拌後、室温に10分間静置した後、4℃、15000rpmで15分間遠心分離した。上清を除き、75%エタノール1mlを加えて沈殿を洗浄し、4℃、15000rpmで5分間遠心分離した。上清を除き、沈殿を軽く乾かした後、DEPC処理水に溶解した。
【0058】
(全RNAの定量)
RNAの定量は、石英セルを用いて分光光度計により行った。波長260nmは核酸、280nmはタンパク質による吸光であるため、RNAの濃度は〔OD260nm×40=μg/ml〕で計算した。純粋な核酸があるときには、260nmの吸光度は280nmの吸光度の1.8〜2.0倍になる。
まず、全RNAを1μl分取し、1mlのDEPC処理水で希釈した。続いて、波長260nmと280nmの吸光度を測定した。OD260nmの値から全RNA濃度を算出した。その結果、全RNA濃度は2.5μg/μlであった。
【0059】
(DNaseI処理法によるゲノムDNA除去)
上記Sepasol−CTAB法により得られた試料中に含まれるゲノムDNAの除去を行った。
まず、全RNAを50μg、RNase Inhibitor(40U/μl)を20U、DNaseI(RNase−free:5U/μl)を10Uとなるように加え、さらに10×DNaseI緩衝液を1倍希釈となるように加えた。さらに、DEPC処理水を混合し、Total50μlの反応液を調製した。続いて、この反応液を37℃で20〜30分間反応させた。
【0060】
次に、50μlのDEPC処理水と100μlのフェノール/クロロホルム/イソアミルアルコール(25:24:1)溶液を加えて混合し、室温、15000rpmで5分間遠心分離した。得られた上清を新しいチューブに移し、100μlのクロロホルム/イソアミルアルコール(24:1)を加えて混合し、室温、15000rpmで5分間遠心分離した。
【0061】
上清を新しいチューブに移し、10μlの3M酢酸ナトリウムと250μlの冷エタノールを加えて−80℃で20分間静置した。次に、4℃、15000rpmで10分間遠心分離し、上清を捨て、70%エタノールで沈殿を洗浄した。4℃、15000rpmで5分間遠心分離し、上清を捨て、沈殿を乾燥させた後、10μlのDEPC処理水に溶解した。上述した全RNAの定量法を用いてRNA濃度を測定した結果、全RNA濃度は1.0μg/μlであった。
【0062】
(逆転写反応による1st strandの作製)
以下の反応系により逆転写反応を行い、1st strand cDNAを作製した。プライマーとして、mRNAの3'側ポリA配列と相補的なポリdT配列とアンカー配列の2つの配列を有する3' RACE アンカープライマー(配列番号5)を用いた。
【0063】
まず、反応混合液(1μgの全RNA、1mMのdNTP Mixture、0.25μMの3' RACE アンカープライマー(配列番号5)、100UのReverTra Ace(100U/μl)、1×RTバッファ)を20μl調製した。
続いて、サーマルサイクラー(MyCycler ThermalCycler(登録商標) 577BR 0209(BIO−RAD製))を用い、42℃で60分間、95℃で5分間、逆転写反応を行った。反応終了後のサンプルを、4℃で3分間冷却したのち、10秒間スピンダウンした。
【0064】
(1st strandを用いたPCR法による2OGD合成遺伝子の部分作製)
得られた1st strand cDNAに対して5'側センスプライマー及び3'側アンチセンスプライマーを用いてPCRを行い、以下に述べる方法で2nd strand cDNAを作製した。上述したESTデータベースでヒットした配列(配列番号4)を元にEG.2OGD 5'−1 プライマー(配列番号7)とEG.2OGD 5'−2(配列番号8)の2種類のプライマーを作成し、センスプライマーとして用いた。また、アンチセンスプライマーとして、上記3' RACE アンカープライマー(配列番号5)のアンカー配列と同一の配列である3' RACE1 プライマー(配列番号6)を用いた。
【0065】
まず、反応混合液(1μlのサンプル、1mMのdNTP Mixture、0.25μMのEG.2OGD 5'−1 プライマー、0.25μMの3' RACE1 プライマー(配列番号6)、100UのPrimeSTAR(登録商標)(TaKaRa製、100U/μl)、1×RTバッファ)を50μl調製した。続いて、上述したサーマルサイクラーを用い、98℃で10秒間、55℃で5秒間、72℃で1.5分間を30サイクル繰り返し、最後に72℃で10分間とした。
得られたPCR産物をアガロースゲル電気泳動により電気泳動し、2.1〜2.2kbpのバンドを含むゲルを切り出して回収した。
【0066】
(cDNAのシークエンスによる部分配列決定)
得られたcDNAを、Ligation−Convenience Kit(日本ジーン製)を用いて大腸菌ベクター(pZErO−2(インビトロジェン製))に組み込んだ。得られた組換えベクターを用いて大腸菌(Top10(インビトロジェン製))を形質転換し、形質転換体を得た。
得られた形質転換体を培養し、アルカリSDS法により組換えベクターを回収し、DNAシーケンサー(Applied Biosystems製 3100AVANT)を用いてDNAシーケンスを行った。
【0067】
(プライマーウォーキング法による全長配列決定)
5'−1 プライマーと5'−2 プライマーを用いて決定された配列を元に、EG.2OGD 5'−3 プライマー(配列番号9)を作製した。続いて、「RT−PCR法による2nd strandの作製」で説明した反応系及び反応温度と同様の条件でPCRを行い、DNAシーケンスにより配列を決定した。決定された配列を元に、EG.2OGD 5'−4 プライマー(配列番号10)を作製し、更にDNAシーケンスを行った。
【0068】
次に、ESTデータベースでヒットしたN末端側配列と、上述したシーケンスで得られた配列とを連結し、ユーグレナ グラシリス SM−ZK株由来の2−オキソグルタル酸デカルボキシラーゼのcDNA全長配列を得た(配列番号2)。また、この全長配列を元に、2−オキソグルタル酸デカルボキシラーゼのアミノ酸配列を得た(配列番号1)。
【0069】
(得られた全長配列の確認)
上で得られたDNAはESTデータベースで得られた部分配列と、DNAシーケンスで得られた配列を組み合わせて決定された配列であるため、これがクローニングされたcDNAの全長配列と同じであることを確かめるため、全長配列の読み直しを行った。まず、全長配列を元に、5'末端側の配列を含むプライマー(プライマーA)と、3'末端側の配列を含むプライマー(プライマーB)の2種類のプライマーを作製した。プライマーAは制限酵素NdeIの切断サイトを、プライマーBは制限酵素XhoIの切断サイトをそれぞれ有している。
上記プライマーA,Bを用いてPCRを95℃(10秒)→55℃(10秒)→72℃(2分)を計35サイクル行い、72℃、10分間に保温した後、12℃で保存した。得られたPCR産物で大腸菌(Top10)を形質転換し、プレート上に撒いて得られたコロニーのいくつかを選択して培養した。培養後にアルカリSDSによりDNAを抽出し、NdeI、XhoIを用いて切断を行い、アガロースゲル電気泳動により制限酵素で切断が確認されたサンプルを選択した。
選択したサンプルのベクターに対して、上述したDNAシーケンサーを用いてDNAシーケンスを行い、2−オキソグルタル酸デカルボキシラーゼのcDNA全長配列を得た。この全長配列と先に得られたcDNA全長配列(配列番号2)とを比較し、両者が同一であることを確認した。
【図面の簡単な説明】
【0070】
【図1】各精製過程において溶液をサンプリングし、SDS−PAGEで電気泳動した結果を示す写真である。
【特許請求の範囲】
【請求項1】
以下の(a)又は(b)に示されるアミノ酸配列からなる2−オキソグルタル酸デカルボキシラーゼ。
(a)配列番号1に記載のアミノ酸配列。
(b)配列番号1に記載のアミノ酸配列のうち1又は2以上のアミノ酸が欠失、置換又は付加されたアミノ酸配列であって、かつ、2−オキソグルタル酸デカルボキシラーゼ活性を有するアミノ酸配列。
【請求項2】
以下の(c)又は(d)に示されるDNA。
(c)配列番号1に記載のアミノ酸配列をコードするDNA。
(d)前記(c)に記載のDNAとストリンジェントな条件下でハイブリダイズするDNAであって、かつ、2−オキソグルタル酸デカルボキシラーゼ活性を有するタンパク質をコードするDNA。
【請求項3】
以下の(e)又は(f)に示されるDNA。
(e)配列番号2に記載のDNA。
(f)配列番号2に記載のDNAとストリンジェントな条件下でハイブリダイズするDNAであって、かつ、2−オキソグルタル酸デカルボキシラーゼ活性を有するタンパク質をコードするDNA。
【請求項4】
請求項2又は3に記載のDNAを含有する組換えベクター。
【請求項5】
請求項4に記載の組換えベクターを含有する形質転換体。
【請求項6】
請求項5に記載の形質転換体を培養し、培養物から2−オキソグルタル酸デカルボキシラーゼを採取することを特徴とする2−オキソグルタル酸デカルボキシラーゼの製造方法。
【請求項7】
請求項1に記載の2−オキソグルタル酸デカルボキシラーゼと2−オキソグルタル酸とを反応させ、生成したコハク酸セミアルデヒド及び二酸化炭素の少なくとも一方の生成量を測定することで、前記2−オキソグルタル酸の定量を行うことを特徴とする2−オキソグルタル酸の測定方法。
【請求項1】
以下の(a)又は(b)に示されるアミノ酸配列からなる2−オキソグルタル酸デカルボキシラーゼ。
(a)配列番号1に記載のアミノ酸配列。
(b)配列番号1に記載のアミノ酸配列のうち1又は2以上のアミノ酸が欠失、置換又は付加されたアミノ酸配列であって、かつ、2−オキソグルタル酸デカルボキシラーゼ活性を有するアミノ酸配列。
【請求項2】
以下の(c)又は(d)に示されるDNA。
(c)配列番号1に記載のアミノ酸配列をコードするDNA。
(d)前記(c)に記載のDNAとストリンジェントな条件下でハイブリダイズするDNAであって、かつ、2−オキソグルタル酸デカルボキシラーゼ活性を有するタンパク質をコードするDNA。
【請求項3】
以下の(e)又は(f)に示されるDNA。
(e)配列番号2に記載のDNA。
(f)配列番号2に記載のDNAとストリンジェントな条件下でハイブリダイズするDNAであって、かつ、2−オキソグルタル酸デカルボキシラーゼ活性を有するタンパク質をコードするDNA。
【請求項4】
請求項2又は3に記載のDNAを含有する組換えベクター。
【請求項5】
請求項4に記載の組換えベクターを含有する形質転換体。
【請求項6】
請求項5に記載の形質転換体を培養し、培養物から2−オキソグルタル酸デカルボキシラーゼを採取することを特徴とする2−オキソグルタル酸デカルボキシラーゼの製造方法。
【請求項7】
請求項1に記載の2−オキソグルタル酸デカルボキシラーゼと2−オキソグルタル酸とを反応させ、生成したコハク酸セミアルデヒド及び二酸化炭素の少なくとも一方の生成量を測定することで、前記2−オキソグルタル酸の定量を行うことを特徴とする2−オキソグルタル酸の測定方法。
【図1】

【公開番号】特開2009−136197(P2009−136197A)
【公開日】平成21年6月25日(2009.6.25)
【国際特許分類】
【出願番号】特願2007−314913(P2007−314913)
【出願日】平成19年12月5日(2007.12.5)
【出願人】(506141225)株式会社ユーグレナ (12)
【Fターム(参考)】
【公開日】平成21年6月25日(2009.6.25)
【国際特許分類】
【出願日】平成19年12月5日(2007.12.5)
【出願人】(506141225)株式会社ユーグレナ (12)
【Fターム(参考)】
[ Back to top ]