
2−ペンチン−1−オールの製造方法
本発明は、2−プロピン−1−オールから出発して、続く中間体(I)、(II)[式中、R1はHまたは線状C1〜6アルキルであり、R2は線状C1〜6アルキルまたは分岐C3〜6アルキルであり、R3は線状C1〜6アルキルである]を介して2−ペンチン−1−オールを製造するための方法、ならびにそうした中間体自体に関する。

【発明の詳細な説明】
【発明の詳細な説明】
【0001】
2−ペンチン−1−オールは、香料工業における中間体、例えばジャスモン酸エステル型の香りの中間体であるがゆえ、魅力的な化合物である(W.Y.Lee,S.Yang,Se Y.Jang,S.Y.Lee,Bulletin of the Korean Chemical Society 1991,12,26;T.Joshida,A.Yamaguchi,A.Kamatsu,Agricult.Biol.Chem.1966,30,370を参照)。
【0002】
本発明の目的は、工業規模で実施できる方法を見出すことであった。
【0003】
文献には、2−ペンチン−1−オールの幾つかの製造方法がすでに記載されている。過去には、ブチンおよびホルムアルデヒド(気体)から出発する方法がよく知られていた。ブチンを、例えば、臭化エチルマグネシウムで処理して対応するブチン−マグネシウム化合物にし、その後ホルムアルデヒドと反応させた。加水分解および蒸留を行った後、およそ60%の収率で2−ペンチノールを分離できた(Y.Tchai Lai,Bull.Chim Soc.France 1933,53,682;K.−E.Schulte,W.Engelhardt,Archiv Ber.Dt.Pharm.Soc.1954,287,495;JP 50049213を参照)。ブチンのリチウム塩に関して類似の反応が記載されている。例えば、米国特許第4,143,230号明細書(ここでは、1−ブチニルリチウムをパラホルムアルデヒドと反応させて2−ペンチン−1−オールにする反応が記載されている)を参照されたい。
【0004】
本発明のさらなる目的は、2−ペンチン−1−オールの製造においてホルムアルデヒド、ホルマリンまたはパラホルムアルデヒドの使用を避けることである。それは、それらが発がん物質また突然変異原であり生殖毒性があるからである。
【0005】
2−ペンチノールを合成する別の方法は、クロロブタノールから出発し、クロロブタノールをハロゲン化メチルマグネシウムで処理するものであり、収率は57〜65%である(J.Colonge,G.Descotes,Bull Soc Chim.France 1959,815;A.A.Kraevskii,B.Yu.Pyatnova,G.I,Myagkova.,I.K.Sarycheva,N.A.Preobrazhenskii,Doklady Akademii Nauk SSSR 1962,146,1349;A.A.Kraevskii,I.K.Sarycheva,N.A.Preobrazhenskii,Zhurnal Obshchei Khimii 1963,33,1831を参照)。こうした方法には、ハロゲン化物を取り扱うこと、収率が低いこと、大量の廃棄物がでることなど、幾つかの欠点がある。
【0006】
2−ペンチン−1−オールへ至る別の経路がWuによって開示されている(J.Wu,X.Kuang,Y.Tang,Huaxue Yanjiu Yu Yingyong(Chemical Research and Application)1999,11(5),521−522を参照)。最初にプロパルギルアルコールおよびリチウムアミドを添加し、その後臭化エチルを添加すると、2−ペンチン−1−オールが得られた。
【0007】
2−ペンチン−1−オールの別の合成方法では、出発物質であるプロパルギルアルコールをジヒドロピランで処理してテトラヒドロ−2−(2−プロピニルオキシ)−2H−ピラン(化合物A)にし(収率86%)、その後ナトリウムアミドおよびエチルブロミドが添加される(T.Joshida,A.Yamaguchi,A.Kamatsu,Agricult.Biol.Chem.1966,30,370を参照)。得られた2−ペンチン−1−イル−2−テトラヒドロピラニルエーテル(2−(ペンタ−2−イニルオキシ)テトラヒドロ−2H−ピラン(化合物B))をエーテルで抽出し、蒸留により精製された(収率96%)。
【0008】
145〜155℃において85%水性リン含有酸で処理することにより、この物質から2−ペンチン−1−オールが合成された(収率82%)。プロパルギルアルコールからの全収率は67.7%であった。類似の方法が米国特許第4,186,141号明細書にも記載されている。
【化1】
主な短所は、分離精製操作の後に最終生成物からの保護基の脱離が困難であることである。工業規模で作業する場合、これにはあまりにも費用がかかるであろう。
【0009】
したがって、本発明の更なる目的は、2−ペンチン−1−オールの中間体としての2−(ペンタ−2−イニルオキシ)テトラヒドロ−2H−ピランの使用を回避することである。
【0010】
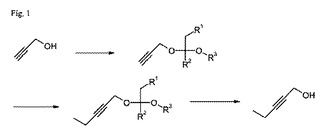
こうした必要は、2−プロピン−1−オールから出発して2−ペンチン−1−オールを製造する本発明の方法によって満たされ、その方法は以下のステップを含む。
a)酸触媒の存在下で2−プロピノールから出発して式Iのケタールを調製するステップ;
【化2】
b)アンモニアと、リチウムアミド、アルキルリチウムおよびアリールリチウムからなる群から選択されるリチウム化合物との存在下で、式Iのケタールを、塩化エチル、臭化エチルおよびヨウ化エチルからなる群から選択されるハロゲン化アルキルと反応させて式IIのケタールにするステップ;
【化3】
c)酸触媒およびプロトン性溶媒の存在下で式IIのケタールを反応させて2−ペンチン−1−オールにするステップ。
上式において、R1はHまたは線状C1〜6アルキルであり、R2は線状C1〜6アルキルまたは分岐C3〜6アルキルであり、R3は線状C1〜6アルキルである。
【0011】
この方法を図1に示す。
【図面の簡単な説明】
【0012】
【図1】図1は、2−プロピン−1−オールから出発して2−ペンチン−1−オールを製造する本発明の方法を示す。
【0013】
「線状C1〜6アルキル」という表現は、メチル、エチル、n−プロピル、n−ブチル、n−ペンチルおよびn−ヘキシルを包含する。「分岐C3〜6アルキル」という用語は、イソプロピル、イソブチル、tert−ブチル、2−ペンチル、3−ペンチル、2−メチルブチル、3−メチルブチル、3−メチル−2−ブチル、2,2−ジメチル−プロピル、2−ヘキシル、3−ヘキシル、3−メチル−2−ペンチル、3−メチル−3−ペンチル、2,2−ジメチル−ブチル、3,3−ジメチル−ブチル、2,3−ジメチル−2−ブチル、2,3−ジメチル−3−ブチルおよび3,3−ジメチル−2−ブチルを包含する。
【0014】
好ましい式IおよびIIの化合物は、R1がHまたは線状C1〜3アルキルであり、かつ/またはR2が線状C1〜3アルキルであり、かつ/またはR3が線状C1〜3アルキルである化合物であり;より好ましいのは、R1がHまたは線状C1〜3アルキルであり、R2およびR3が互いに独立に線状C1〜3アルキルである化合物である。
【0015】
さらにより好ましい式IおよびIIの化合物は、R1がHまたは線状C1〜6アルキルであり、R2が線状C1〜6アルキルまたは分岐C3〜6アルキルであり、R3が線状C1〜6アルキルである化合物である。いっそうより好ましいのは、R1がHまたはメチルであり、R2がメチルまたはエチルであり、R3がメチルである式IおよびIIの化合物である。
【0016】
もっとも好ましい式IおよびIIの化合物は、それぞれ式Ia(R1=R2=R3=CH3)、Ib(R1=H;R2=R3=CH3)、IIa(R1=R2=R3=CH3)およびIIb(R1=H;R2=R3=CH3)の化合物、すなわち、それぞれR1がHまたはメチルでありR2およびR3がメチルである式IおよびIIの化合物である。
【化4】
【0017】
式IおよびIIの化合物は、新規ではない式Ibの具体的な化合物を除いて、新規である。したがって、本発明はまた、新規化合物、すなわち式Iの化合物
【化5】
[式中、R1はHまたは線状C1〜6アルキルであり、R2は線状C1〜6アルキルまたは分岐C3〜6アルキルであり、R3は線状C1〜6アルキルであるが、但し、R2およびR3がメチルである場合にR1はHではないことを条件とする]、ならびに式IIの化合物
【化6】
[式中、R1はHまたは線状C1〜6アルキルであり、R2は線状C1〜6アルキルまたは分岐C3〜6アルキルであり、R3は線状C1〜6アルキルである]に関する。
【0018】
この方法について以下にさらに詳しく説明する。
【0019】
[ステップa)]
[触媒]
ステップa)で使用される酸触媒は、好ましくはブレンステッド酸および固体酸からなる群から選択される。
【0020】
ブレンステッド酸の好ましい例は、p−TsOH(p−トルエンスルホン酸。有機スルホン酸の中で好ましいもの)などの有機スルホン酸、H2SO4(硫酸)およびH3PO4(リン含有酸)である。H3PO4が特に好ましいが、これは、その場合に生成する副生成物が少ないからである。主な副生成物は、式Vの二量体である。
【化7】
【0021】
こうした副生成物は、さらに反応して式VIの化合物にもなりうるし、また本発明の方法のステップc)で開裂して2−ペンチン−1−オールにもなりうる。
【化8】
ブレンステッド酸は有機溶媒溶液の形で使用してもよい。H3PO4は、例えば、17重量%のアセトン溶液またはメタノール溶液として使用してよい。H2SO4は、例えば、8重量%水溶液として、またp−TsOHは一水和物(p−TsOH・H2O)として使用してよい。
【0022】
固体酸の好ましい例は、担体上のブレンステッド酸、強酸性陽イオン交換体および酸性基を有するポリマー(すなわち、官能基化されたポリマー)である。
【0023】
強酸性陽イオン交換体の例は、Amberlyst型のポリマー(例えば、Amberlyst(登録商標)15、Amberlyst(登録商標)16)、Dowex、およびポリマーに結合したp−TsOH(p−トルエンスルホン酸)である。特に好ましいのは、Amberlyst(登録商標)15および16である。Amberlyst型のポリマー、ならびにDowexはすべて、官能基化されたスチレンジビニルベンゼンコポリマーである。
【0024】
固体酸は、好ましくは細孔径が100〜500Åの範囲であり、表面積が少なくとも20m2/g(窒素BETで測定)、好ましくは表面積が20〜500m2/gの範囲(より好ましくは、25〜300m2/gの範囲、28〜300m2/gの範囲、28〜70m2/gの範囲、30〜55m2/gの範囲)、また全細孔容積が少なくとも0.10ml/g、好ましくは全細孔容積が0.10〜0.50ml/gの範囲、より好ましくは全細孔容積が0.15〜0.45ml/gの範囲、さらにより好ましくは全細孔容積が0.18〜0.42ml/gの範囲、もっとも好ましくは全細孔容積が0.20〜0.40ml/gの範囲であり;さらに酸部位(すなわちスルホン酸基)の濃度が≧4.0当量/kg(特に≧4.5当量/kg;4.5〜6.0当量/kgの範囲;4.5〜5.5当量/kgの範囲)である。
【0025】
好ましくはこうした固体酸の酸性基は、スルホン酸基(−SO3H)であるか、またはpKa≦4の任意の他の基である。
【0026】
好ましい固体酸は、担体上の有機スルホン酸、または官能性スルホン酸基を有するポリマー樹脂である。
【0027】
更なる好適な固体酸は、CO2H基で官能基化されたポリマーである。
【0028】
本発明との関連において引用されているすべての官能基化ポリマーのうちでも、巨大網状ポリマーが好ましい。
【0029】
好ましくは、本発明に用いる固定化/固体ブレンステッド酸は、pka値が≦4であり、より好ましくは≦2である。
【0030】
固定化/固体ブレンステッド酸の例は、有機スルホン酸、H2SO4およびH3PO4(すべて担体上にある)である。
【0031】
担体は有機または無機のいずれであってよい。有機担体の例はポリマーである。無機担体の例は、例えば、SiO2、Al2O3、TiO2、ZrO2およびGeO2などの酸化物担体である。
【0032】
担体上のブレンステッド酸の例は、ポリマー上のp−トルエンスルホン酸またはシリカ上のプロピルスルホン酸である。
【0033】
さらに、pKA≦4である、好ましくはpKA≦2.5である他の酸触媒も、本発明の方法のステップa)で式Iのケタールの調製を触媒するのに適している。
【0034】
[反応条件]
ステップa)は、有機溶媒の存在下でも不存在下でも実施できる。有機溶媒が存在する場合には、好ましくは、エーテル、ケトン、エステルおよびそれらの混合物からなる群から選択されるであろう。
【0035】
好ましくはステップa)は、有機溶媒の不存在下で実施する。
【0036】
ステップa)は、バッチ式でも連続式でも実施できる。
【0037】
好ましくはステップa)は、0〜35℃の範囲の温度、好ましくは3〜25℃の範囲の温度、より好ましくは5〜20℃の範囲の温度で実施する。
【0038】
プロパルギルアルコール(基質(s))と触媒(c)とのモル比は一般に、s/cが500〜500000の範囲、好ましくは1000〜150000の範囲、特に好ましくは5000〜15000の範囲である。
【0039】
[ステップb)]
[ハロゲン化アルキル]
好ましいハロゲン化アルキルは臭化エチルおよびヨウ化エチルであり、その中で臭化エチルがもっとも好ましいものである。
【0040】
[リチウム化合物]
「アルキルリチウム」という用語は、直鎖(straight)C1〜6−アルキルリチウム、ならびに分岐C3〜6−アルキルリチウムを包含する。アルキルリチウムの好ましい例として、メチルリチウムおよびn−ブチルリチウムがある。
【0041】
「アリールリチウム」の例としてフェニルリチウムがある。
【0042】
リチウムアミドは好ましいリチウム化合物である。リチウムアミドは、当業者に知られている任意の方法、例えば、Lambert Brandsmaの著書「Synthesis of Acetylenes,Allenes and Cumulenes:Methods and Techniques」(Elsevier Ltd.UK,2004)の第2.3.1.1章(そこでは、リチウムが無水液体アンモニアと硝酸鉄(III)との混合物に加えられている)にLambert Brandsmaによって記載されているようにして調製できる。
【0043】
好ましくは、リチウムアミドは欧州特許出願公開第1238944号明細書(その内容を本明細書に援用する)に記載されている方法に従って調製する。つまり、リチウム金属をアンモニアに加えて「リチウムブロンズ(lithium bronze)」を生じさせ、その後その「リチウムブロンズ」を有機溶媒の存在下で1,3−ジエンまたはアリールオレフィンと反応させる。しかし、我々の本発明の方法のステップb)では、有機溶媒が存在する必要はない。リチウムアミドの調製は、好ましくはアンモニア以外のどんな有機溶媒も存在しない状態で実施する。リチウムアミド調製用の好ましいオレフィンは、ピペリレン、イソプレン、スチレンおよびミルセンなどの1,3−オレフィンである。ピペリレンおよびスチレンが特に好ましい。驚くべきことに、そのように調製されたリチウムアミドの場合、それによって調製された化合物IIの純度が高くなることが見出された。したがって、そのように調製されたリチウムアミドを使用するのが特に有利である。
【0044】
[反応条件]
ステップb)は、有機溶媒の存在下でも不存在下でも実施できる。そのような有機溶媒は、エーテル、アルカン、オレフィン、芳香族化合物およびそれらの混合物からなる群から選択できる。
【0045】
しかし、好ましくは、ステップb)は有機溶媒の不存在下で実施する。
【0046】
一般に、ステップb)は、反応条件下でアンモニアが液体であるような温度および圧力で実施する。大気圧では、温度は−70〜−35℃の範囲である。
【0047】
好ましくは、欧州特許出願公開第1238944号明細書の方法に従って、特にピペリレン(1,3−ペンタジエン)またはスチレンを用いて調製されたリチウムアミドを使用する。
【0048】
好ましくは、リチウム化合物と式Iのケタールとのモル比は、(1.5〜3)/1の範囲、好ましくは(1.75〜2.5)/1の範囲である。
【0049】
好ましくは、式Iのケタールとハロゲン化アルキルとのモル比は1:1〜1:1.5の範囲である。
【0050】
[ステップc)]
ステップc)で使用される酸触媒は、好ましくはブレンステッド酸および固体酸からなる群から選択される。ステップa)を実施するのに適しているブレンステッド酸および固体酸と同じものが、ステップc)を実施するのにもの適している。
【0051】
一般に、pKA≦4、好ましくはpKA≦2.5である酸触媒は、保護基を開裂させる(ステップc))のに適している。
【0052】
好ましくは、ブレンステッド酸がステップc)を実施するのに使用され、より好ましくは、ブレンステッド酸はp−TsOH、H2SO4およびH3PO4からなる群から選択される。
【0053】
もっとも好ましくは、ステップc)で使用されるブレンステッド酸はH2SO4である。
【0054】
ステップc)は、好ましくは、0〜50℃の範囲の温度、より好ましくは5〜30℃の範囲の温度で実施する。
【0055】
式IIの化合物(基質(s))と酸触媒(c)とのモル比は一般に、s/c=(100〜2000)/1の範囲、好ましくは(200〜1000)/1の範囲である。
【0056】
プロトン性溶媒は、好ましくは、H2O、アルコールおよびそれらの混合物からなる群から選択される。
【0057】
アルコールの好ましい例としては、メタノールおよびエタノールがある。
【0058】
水をプロトン性溶媒として使用する場合、水と式IIのケタールとのモル比は好ましくは少なくとも1:1、より好ましくは(1.5〜3):1である。
【0059】
本発明による方法の利点は、ステップa)〜c)すべてをアンモニア以外の(有機)溶媒なしで実施できることである。さらに、中間体(式IIおよびIIIの化合物)の精製が不要である。そのため本発明の方法は大量生産に非常に適したものとなっている。
【0060】
[本発明のさらなる実施形態]
●式Iの化合物
【化9】
[式中、R1はHまたは線状C1〜6アルキルであり、R2は線状C1〜6アルキルまたは分岐C3〜6アルキルであり、R3は線状C1〜6アルキルであるが、但し、R2およびR3がメチルである場合にはR1はHではないことを条件とする]。
【0061】
好ましくはR1=R2=R3=メチル、すなわち式Iaの化合物
【化10】
●式IIの化合物
【化11】
[式中、R1はHまたは線状C1〜6アルキルであり、R2は線状C1〜6アルキルまたは分岐C3〜6アルキルであり、R3は線状C1〜6アルキルである]。
【0062】
好ましくはR1=Hまたはメチル、R2=R3=メチル、すなわち式IIaおよびIIbの化合物
【化12】
【0063】
●式I
【化13】
のケタールを製造するための方法であって、2−プロピン−1−オールを酸触媒の存在下で式III
【化14】
の化合物と反応させるステップを含み、上式において、R1はHまたは線状C1〜6アルキルであり、R2は線状C1〜6アルキルまたは分岐C3〜6アルキルであり、R3は線状C1〜6アルキルである、方法。
【0064】
この方法は、上述の方法のステップa)に対応する。好ましい条件を含め上に示した詳細はすべて、ここでも当てはまる。
【0065】
●式II
【化15】
のケタールを製造するための方法であって、下記の式Iの化合物を、アンモニアと、リチウムアミド、アルキルリチウムおよびアリールリチウムからなる群から選択されるリチウム化合物との存在下で、式IVの化合物と反応させるステップを含み、以下の式
【化16】
において、R1はHまたは線状C1〜6アルキルであり、R2は線状C1〜6アルキルまたは分岐C3〜6アルキルであり、R3は線状C1〜6アルキルであり、XはCl、BrまたはIである、方法。
【0066】
この方法は、上述の方法のステップb)に対応する。好ましい条件を含め上に示した詳細はすべて、ここでも当てはまる。
【0067】
●2−ペンチン−1−オールを製造するための方法であって、プロトン性溶媒中で酸触媒を用いて、式II
【化17】
[式中、R1はHまたは線状C1〜6アルキルであり、R2は線状C1〜6アルキルまたは分岐C3〜6アルキルであり、R3は線状C1〜6アルキルである]
の化合物からヒドロキシ保護基を開裂させて2−ペンチン−1−オールを得るステップを含む、方法。
【0068】
この方法は、上述の方法のステップc)に対応する。好ましい条件を含め上に示した詳細はすべて、ここでも当てはまる。
【0069】
これから本発明を以下の非限定例を用いてさらに示す。
【0070】
[実施例]
以下の略号を使用する。
BME 2−ブテニルメチルエーテル
GC ガスクロマトグラフィー
IPM イソプロペニルメチルエーテル
p−TsOH p−トルエンスルホン酸
THP 3,4−ジヒドロ−2H−ピラン
【0071】
[全体的説明]
プロパルギルアルコール(propargylic alcohol)(=2−プロピン−1−オール)(Aldrich N°P5.080−3,99.0% GC)、p−トルエンスルホン酸(p−TsOH,Fluka N°89760,98.5% GC)、Amberlyst 15(Fluka N°06423)、リチウム(Acros,99.0%)、スチレン(Fluka 85960,99.0% GC)、ブロモエタン(=臭化エチル;Fluka 03150,98.0%)、イソプレン(Aldrich 119551,100.0% GC)、ミルセン(Givaudan DE−396,98.0% GC)、テトラヒドロピラン(THP)(Fluka 37350,95.0% GC)は市販されており、さらなる精製を行わずに使用した。
【0072】
イソプロペニルメチルエーテル(DSM Nutritional Products Ltd,Lalden(CH),96.0% GC)、ブテニルメチルエーテル(DSM Nutritional Products Ltd,Sisseln(CH),96.2% GC)、および1,3−ペンタジエン(=ピペリレン)(SPC,China,68.5% GC)は、更なる精製を行わずに使用した。
【0073】
[NMR分光法]
NMRスペクトルはBruker Avance 300MHz分光計で記録した。1H−NMRスペクトルは300MHzで、13Cスペクトルは75MHzでそれぞれ記録した。定量スペクトル(quantitative spectra)は、DMSO−d6で記録し、1,4ジメトキシベンゼンを内標準として用いた。2つのパルス間の遅延(delay)dlは30秒に設定した。12〜25mgの試料を使用した。スペクトルはCDCl3で記録した。δはppmで示す。多重度に関しては次のような略号を使用した:S=一重項、d=二重項、t=三重項、q=四重項、dd=二重項の二重項、dq=四重項の二重項、m=多重項。
【0074】
[実施例1: 3−(l−メトキシ−l−メチル−エトキシ)−プロピンの製造]
[実施例1.1: 触媒としてのH3PO4の使用]
25μl(0.04mmol)のリン酸(アセトン中に17%(w/w))を22.6g(399mmol)のプロパルギルアルコールに22℃で加えた。溶液を滴下漏斗へ移した。58.8mlのIPM(600mmol)を別の滴下漏斗に移した。両方の溶液を45分以内に同時にガラス反応器に加えた。反応物を添加する間、内部温度を15℃から20℃の間に維持した。添加後に、反応混合物を約15分間15℃に保った。最後に反応混合物を減圧下(40℃、40mbar)で濃縮し、粗生成物をGCで分析した。純度96.0%の3−(1−メトキシ−1−メチル−エトキシ)−プロピンを47.86g得た。プロパルギルアルコールに対する収率は89.85%であった。
【0075】
[実施例1.2: 触媒としてのp−TsOH・H2Oの使用]
10mg(0.05mmol)のp−TsOHの一水和物を22℃で22.43g(396mmol)のプロパルギルアルコール中に溶かした。溶液を滴下漏斗へ移した。56.93mlのIPM(581mmol)を別の滴下漏斗に移した。両方の溶液を40分以内に同時にガラス反応器に加えた。反応物を添加する間、内部温度を15℃から20℃の間に維持した。100mlの飽和重炭酸ナトリウム溶液を5分以内に加え、その混合物を5分間激しく攪拌した。その溶液を20mlのn−ヘキサンで抽出した。最後に有機相を減圧下(40℃、40mbar)で濃縮し、粗生成物をGCで分析した。純度87.6%の未精製の3−(1−メトキシ−1−メチル−エトキシ)−プロピンを48.6g得た。プロパルギルアルコールに対する収率は83.9%であった。
【0076】
[実施例1.3: 減らした量の触媒としてのp−TsOH・H2Oの使用]
同じ量の触媒を用いて実施例1.2を繰り返したが、ただし、22.43gのプロパルギルアルコールの代わりに372g(6.6mol)のプロパルギルアルコールを、また56.93mlのIPMの代わりに959ml(9.8mol)のIPMを使用した。さらに、反応物の添加を終えた後に、反応混合物をさらに15分間15℃で攪拌した。この少量の触媒の場合、実施例1.2におけるような重炭酸ナトリウム溶液による中和は不要であった。純度89.9%の未精製の3−(1−メトキシ−1−メチル−エトキシ)−プロピンを852.8g得た。プロパルギルアルコールに対する収率は91.0%であった。
【0077】
[実施例1.4: 触媒としてのAmberlyst(登録商標)15の使用]
10℃に冷却されたジャケット付反応器中に、820mgのAmberlyst 15を加えた。372g(6.6mol)のプロパルギルアルコールと958.5mlのIPM(9.8mol)との混合物を、45分以内に反応器に加えた。反応物を加える間、内部温度を10℃に維持した。添加後に、反応混合物を約15分間10℃に保った。濾過により触媒を分離し、反応混合物を減圧下(40℃、40mbar)で濃縮した。粗生成物をGCで分析した。純度90.3%の未精製の3−(1−メトキシ−1−メチル−エトキシ)−プロピンを881.65g得た。プロパルギルアルコールに対する収率は94.6%であった。
【0078】
[実施例1.5: 触媒としてのAmberlyst(登録商標)15の使用−連続式]
プロパルギルアルコール(230.4g、4.0mol)および458.3gのIPM(6.1mol)を、10℃で4時間、100mgのAmberlyst(登録商標)15を満たした固定床反応器(長さ:10cm、直径:9mm)中に送り込んだ(プロパルギルアルコールの送り速度=1ml/min.、IPMの送り速度=2.5ml/min.)。1時間の間に、溶液343.1gを回収した。過剰のIPMを減圧下(40℃、40mbar)で蒸留し、粗生成物をGCで分析した。純度95.9%の未精製の3−(l−メトキシ−1−メチル−エトキシ)−プロピン(243.2g)を得た(プロパルギルアルコールに対する収率:91.0%)。
【0079】
[実施例1.6: 触媒としてのH2SO4の使用]
5μl(4μmol)の硫酸(8%(w/w))を22℃で22.6g(399mmol)のプロパルギルアルコールに加えた。その溶液を滴下漏斗へ移した。58.75mlのIPM(599mmol)を別の滴下漏斗に移した。両方の溶液を45分以内に同時にガラス反応器に加えた。反応物を添加する間、内部温度を15℃から20℃の間に維持した。添加後に、反応混合物を約15分間15℃に保った。最後に反応混合物を減圧下(40℃、40mbar)で濃縮し、粗生成物をGCで分析した。純度87.4%の3−(1−メトキシ−1−メチル−エトキシ)−プロピンを51.0g得た。プロパルギルアルコールに対する収率は87.2%であった。
【0080】
[実施例2: 2−メトキシ−2−プロパ−2−イニルオキシ−ブタンの製造]
[実施例2.1: 触媒としてのH3PO4の使用]
130mg(0.3μmol)のリン酸(アセトン中に19%(w/w))と22.80g(399mmol)のプロパルギルアルコールとの混合物を、滴下漏斗中に満たす。53.5g(598mmol)の2−メトキシ−ブタ−2−エン(96.2% GC)を別の滴下漏斗に移した。両方の溶液を、冷却されたフラスコ(8℃)に30分以内に同時に加えた。反応物を添加する間、内部温度を8℃から12℃の間に維持した。反応混合物を約2時間10℃に保った。反応混合物を0.5gの炭酸ナトリウムで中和し、減圧下(40℃、80mbar)で濃縮した。粗生成物をGCで分析した。純度85.5%の未精製の2−メトキシ−2−プロパ−2−イニルオキシ−ブタンを58.0g得た(プロパルギルアルコールに対する収率:96.9%)。
【0081】
[実施例3: 1−(1−メトキシ−1−メチル−エトキシ)−ペンタ−2−インの製造]
[実施例3.1: 基剤としてリチウムアミド(スチレンで調製)を使用]
883mg(126mmol)の粒状リチウムを−38℃で5分以内に107g(6.30mol)の液体アンモニアに加えた。その混合物をリチウムが浮遊しなくなるまで(およそ10分後)攪拌した。液体アンモニア中にリチウムを含む紺青色溶液を得た。14.6g(140mmol)のスチレンを20分以内に何回かに分けて加えた。リチウムアミドの形成の終了は、反応混合物の色の変化で見分けることができた。9.9g(70mmol)の3−(1−メトキシ−1−メチル−エトキシ)−プロピン(90.8% GC)を滴下漏斗で20分以内に−38℃で反応混合物に加えた。反応混合物を約1時間、−38℃に保った。14g(126mmol)のブロモエタンを滴下漏斗で20分以内に反応混合物に加えた。その後、反応混合物を1時間攪拌した。100mlのn−ヘキサンを反応混合物に加え、液体アンモニアを16時間以内に標準圧力下で蒸発させた。70mlの水を22℃で反応混合物に加え、塩がすべて溶解するまでその混合物を攪拌した。有機層を分離し、2gの無水硫酸マグネシウムで乾燥させた。溶液を減圧下(40℃、40mbar)で濃縮し、粗生成物をGCで分析した。純度84.4%の1−(1−メトキシ−1−メチル−エトキシ)−ペンタ−2−インを10.6g得た(3−(l−メトキシ−1−メチル−エトキシ)−プロピンに対する収率:81.8%)。
【0082】
[実施例3.2: 基剤としてリチウムアミド(イソプレンで調製)を使用]
259mg(37mmol)の粒状リチウムを、−40℃で5分以内に47.7g(1.9mol)の液体アンモニアに加えた。その混合物をリチウムが浮遊しなくなるまで(およそ10分後)攪拌した。液体アンモニア中にリチウムを含む紺青色溶液を得た。4.0ml(40mmol)のイソプレンを10分以内に何回かに分けて加えた。リチウムアミドの形成の終了は、反応混合物の色の変化で見分けることができた。2.9g(20mmol)の3−(1−メトキシ−1−メチル−エトキシ)−プロピン(88.9% GC)を滴下漏斗で20分以内に−40℃で反応混合物に加えた。反応混合物を約1時間、−40℃に保った。8.9g(80mmol)のブロモエタンを滴下漏斗で10分以内に反応混合物に加えた。その後、反応混合物を1時間攪拌した。20mlのn−ヘキサンを反応混合物に加え、液体アンモニアを5時間以内に標準圧力下で蒸発させた。20mlの水を22℃で反応混合物に加え、塩がすべて溶解するまでその混合物を攪拌した。有機層を分離し、2gの無水硫酸マグネシウムで乾燥させた。溶液を減圧下(40℃、40mbar)で濃縮し、粗生成物をGCで分析した。純度75.6%の3.3gの未精製の1−(1−メトキシ−1−メチル−エトキシ)−ペンタ−2−インを得た(3−(l−メトキシ−1−メチル−エトキシ)−プロピンに対する収率:80.0%)。
【0083】
[実施例3.3: 基剤としてリチウムアミド(ミルセンで調製)を使用]
0.89g(127mmol)の粒状リチウムを、−37℃で15分以内に200ml(8mol)の液体アンモニアに加えた。その混合物をリチウムが浮遊しなくなるまで(およそ15分後)攪拌した。液体アンモニア中にリチウムを含む紺青色溶液を得た。12.6g(91mmol)のミルセンを30分以内に何回かに分けて加えた。リチウムアミドの形成の終了は、反応混合物の色の変化で見分けることができた。10.0g(71mmol)の3−(1−メトキシ−1−メチル−エトキシ)−プロピン(90.8% GC)を、滴下漏斗で30分以内に−37℃で反応混合物に加えた。反応混合物を約1時間、−37℃に保った。14.0g(126mmol)のブロモエタンを滴下漏斗で30分以内に反応混合物に加えた。その後、反応混合物を1時間攪拌した。157mlのn−ヘキサンを反応混合物に加え、液体アンモニアを15時間以内に標準圧力下で蒸発させた。50mlの水を22℃で反応混合物に加え、塩がすべて溶解するまでその混合物を攪拌した。有機層を分離し、減圧下(40℃、40mbar)で濃縮した。粗生成物をGCで分析した。25.8gの未精製の1−(1−メトキシ−1−メチル−エトキシ)−ペンタ−2−インを得た(3−(1−メトキシ−1−メチル−エトキシ)−プロピンに対する収率:83.0%)。
【0084】
[実施例3.4: 基剤としてリチウムアミド(1,3−ペンタジエンで調製)を使用]
0.89g(127mmol)の粒状リチウムを、−37℃で20分以内に100ml(4mol)の液体アンモニアに加えた。その混合物をリチウムが浮遊しなくなるまで(およそ15分後)攪拌した。液体アンモニア中にリチウムを含む暗青緑色の溶液を得た。6.34g(92mmol)のピペリレン(=1,3−ペンタジエン)を30分以内に何回かに分けて加えた。リチウムアミドの形成の終了は、反応混合物の色の変化で見分けることができた。10.1g(71mmol)の3−(1−メトキシ−1−メチル−エトキシ)−プロピン(89.7% GC)を、滴下漏斗で30分以内に−37℃で反応混合物に加えた。反応混合物を約1時間、−37℃に保った。14.0g(126mmol)のブロモエタンを滴下漏斗で30分以内に反応混合物に加えた。その後、反応混合物を1時間攪拌した。100mlのn−ヘキサンを反応混合物に加え、液体アンモニアを1.5時間以内に標準圧力下で蒸発させた。50mlの水を22℃で反応混合物に加え、塩がすべて溶解するまでその混合物を激しく攪拌した。水層を50mlのn−ヘキサンで1回抽出した。有機層を5gの無水硫酸ナトリウムで乾燥させた。溶液を減圧下(40℃、60mbar)で濃縮し、粗生成物をGCで分析した。純度87.5%の11.3gの未精製の1−(1−メトキシ−1−メチル−エトキシ)−ペンタ−2−インを得た(3−(1−メトキシ−1−メチル−エトキシ)−プロピンに対する収率:85.3%)。
【0085】
[実施例3.5: 臭化エチルの代わりにヨウ化エチルを使用]
0.89g(127mmol)の粒状リチウムを、−37℃で15分以内に100ml(4mol)の液体アンモニアに加えた。その混合物をリチウムが浮遊しなくなるまで(およそ15分後)攪拌した。液体アンモニア中にリチウムを含む紺青色溶液を得た。5.94g(86.3mmol)のイソプレンを30分以内に何回かに分けて加えた。LiNH2の形成の終了は、反応混合物の脱色により見分けることができた。10.69g(70.1mmol)の3−(l−メトキシ−1−メチル−エトキシ)−プロピンを−37℃で30分以内に反応混合物に加えた。反応混合物を約1時間、−37℃に保った。ヨウ化エチル(20.05g、126mmol)を滴下漏斗で30分以内に反応混合物に加えた。その後、反応混合物を1時間攪拌した。n−ヘキサン(100ml)を反応混合物に加え、液体アンモニアを1.5時間以内に標準圧力下で蒸発させた。水(50ml)を22℃で反応混合物に加え、塩がすべて溶解するまでその混合物を激しく攪拌した。水層を50mlのn−ヘキサンで2回抽出した。有機層を5gの無水硫酸ナトリウムで乾燥させた。溶液を減圧下(40℃、60mbar)で濃縮し、粗生成物をGCで分析した。純度72.95%の未精製の1−(1−メトキシ−1−メチル−エトキシ)−ペンタ−2−イン(13.21g)を得た(3−(1−メトキシ−1−メチル−エトキシ)−プロピンに対する収率:88.0%)。
【0086】
[実施例4: 1−(1−メトキシ−1−メチル−プロポキシ)−ペンタ−2−インの製造]
[実施例4.1: 基剤としてリチウムアミド(イソプレンで調製)を使用]
890mg(127mmol)の粒状リチウムを、−40℃で5分以内に100ml(4.0mol)の液体アンモニアに加えた。その混合物をリチウムが浮遊しなくなるまで(およそ10分後)攪拌した。液体アンモニア中にリチウムを含む紺青色溶液を得た。5.94g(86.6mmol)のイソプレンを10分以内に何回かに分けて加えたが、溶液は添加の終了時に白色になる。混合物を−38℃で25分間攪拌した。9.1g(69.5mmol)の2−メトキシ−2−プロパ−2−イニルオキシ−ブタン(97.4% GC)を、滴下漏斗で25分以内に−38℃で反応混合物に加えた。反応混合物を約1時間、−38℃に保った。14g(128.5mmol)のブロモエタンを滴下漏斗で20分以内に反応混合物に加えた。その後、反応混合物を1時間攪拌した。100mlのn−ヘキサンを反応混合物に加え、液体アンモニアを1時間30分以内に標準圧力下で蒸発させた。50mlの水を40℃で反応混合物に加え、塩がすべて溶解するまでその混合物を攪拌した。混合物を分液漏斗に移し、層を分離した。水層を127mlのn−ヘキサンで抽出した。有機層を0.5gの無水硫酸ナトリウムで乾燥させ、減圧下(40℃、60mbar)で濃縮し、粗生成物(1−(1−メトキシ−1−メチル−プロポキシ)−ペンタ−2−イン)をGCで分析した。純度87.1%の10.54gの未精製の1−(1−メトキシ−1−メチル−プロポキシ)−ペンタ−2−インを得た(2−メトキシ−2−プロパ−2−イニルオキシ−ブタンに対する収率:77.6%)。
【0087】
[実施例5: 1−(1−メトキシ−1−メチル−エトキシ)−ペンタ−2−インから出発する2−ペンチン−1−オールの製造]
[実施例5.1: 触媒としてp−トルエンスルホン酸を使用]
5.0g(25.7/24.387mmol)の1−(1−メトキシ−1−メチル−エトキシ)−ペンタ−2−イン(GCによる純度:80.4%)を、20℃において100mlのn−ヘキサン(工業等級)で希釈した。20mgのp−トルエンスルホン酸一水和物および500μl(27.8mmol)の脱イオン水を、22℃で攪拌しながら溶液に加えた。その混合物を約1時間45分の間、22℃に保った。500mg(4.7mmol)の炭酸ナトリウムを加え、その後、反応混合物を5分間攪拌した。不要な塩を濾過で分離した。最後に溶液を減圧下(40℃、50mbar)で濃縮し、粗生成物をGCで分析した。2.23gの未精製の2−ペンチン−1−オール(GCによる純度:78.62%)を得た(1−(1−メトキシ−1−メチル−エトキシ)−ペンタ−2−インに対する収率:85.5%)。
【0088】
[実施例5.2: 触媒として8重量%のH2SO4水溶液を使用]
5.0g(25.7mmol)の3(80.4% GC)を20℃において100mlのn−ヘキサンで希釈した。93μl(0.1mmol)の硫酸(8%(w/w))および440μl(24.4mmol)の水を、22℃で攪拌しながら溶液に加えた。その混合物を約1時間30分の間22℃に保った。溶液を500mg(4.7mmol)の炭酸ナトリウムで中和し、不要な塩を濾過で分離した。最後に溶液を減圧下(40℃、50mbar)で濃縮し、粗生成物をGCで分析した。純度81.2%の2.26gの未精製の2−ペンチン−1−オールを得た(1−(l−メトキシ−1−メチル−エトキシ)−ペンタ−2−インに対する収率:85.3%)。
【0089】
[実施例5.3: 触媒としてAmberlyst 15を使用]
25mgのAmberlyst(登録商標)15[Amberlyst 15 WET]をアルゴン雰囲気下において22℃で、7.5g(45.6mmol)の1−(1−メトキシ−1−メチル−エトキシ)−ペンタ−2−イン(GCによる純度:95%)と3.75ml(208mmol)の脱イオン水との混合物に加えた。その混合物を約1時間30分の間攪拌しながら22℃に保った。触媒を濾過で分離した。2−ペンチン−1−オールを総量で60mlのジエチルエーテルで抽出し、有機層を5gの硫酸ナトリウム(無水)で乾燥させた。溶液を減圧下(40℃、50mbar)で濃縮し、粗生成物をGCで分析した。純度95.0%の3.85gの未精製の2−ペンチン−1−オールを得た(1−(1−メトキシ−1−メチル−エトキシ)−ペンタ−2−インに対する収率:95.3%)。
【0090】
[実施例5.4: 触媒として8重量%のH2SO4水溶液を、また更なる出発物質として二量体の1−(1−メチル−1−ペンタ−2−イニルオキシ−エトキシ)−ペンタ−2−インを使用]
213μl(0.2mmol)の8重量%の硫酸水溶液および1169μl(65mmol)の水を、22℃において攪拌しながら、1−(1−メトキシ−1−メチル−エトキシ)−ペンタ−2−イン(72.5% GC)と1−(1−メチル−1−ペンタ−2−イニルオキシ−エトキシ)−ペンタ−2−イン(3.5% GC)との混合物12.7g(合計で61.1mmolの1−(1−メトキシ−1−メチル−エトキシ)−ペンタ−2−イン)に加えた。そのエマルジョンを約1時間30分の間22℃に保った。得られた反応混合物をバルブ間蒸留(bulb−to−bulb distillation)で精製し(P=65mbar/オーブン温度 97〜100℃)、最終生成物をGCで分析した。純度が99.7%および89.4%の2−ペンチン−1−オールの2つの部分を蒸留した(蒸留後の収率は、1−(1−メトキシ−1−メチル−エトキシ)−ペンタ−2−イン + 1−(1−メチル−1−ペンタ−2−イニルオキシ−エトキシ)−ペンタ−2−インに対して94.5%であった)。
【0091】
[実施例5.5: 触媒として8重量%のH2SO4水溶液を使用]
180μl(0.16mmol)の8重量%硫酸水溶液および900μl(50mmol)の水を、22℃で攪拌しながら10.8g(59mmol)の1−(1−メトキシ−1−メチル−エトキシ)−ペンタ−2−インに加えた。そのエマルジョンを約1時間30分の間22℃に保った。得られた反応混合物をバルブ間蒸留で精製し(P=65mbar/オーブン温度 97〜100℃)、最終生成物をGCで分析した。純度が97.3%および89.5%の2−ペンチン−1−オールの2つの部分を蒸留した(蒸留後の収率は1−(1−メトキシ−1−メチル−エトキシ)−ペンタ−2−インに対して89.8%であった)。
【0092】
[実施例6: 1−(1−メトキシ−1−メチル−プロポキシ)−ペンタ−2−インから出発する2−ペンチン−1−オールの製造]
[実施例6.1: 触媒として8重量%のH2SO4水溶液を使用]
三つ口フラスコに、2.89(16.7mmol)の1−(1−メトキシ−1−メチル−プロポキシ)−ペンタ−2−イン(98.6% GC)、60μl(0.05mmol)の硫酸(8%(w/w))および285μl(15.8mmol)の水を充填した。その溶液を約1時間30分の間22℃で攪拌した。溶液を500mg(4.7mmol)の炭酸ナトリウムで中和し、次いで500mgの硫酸ナトリウム(無水)で乾燥させた。不要な塩を濾過した後、混合物を減圧下(40℃、50mbar)で濃縮し、粗生成物をGCで分析した。純度92.8%の1.35gの未精製の2−ペンチン−1−オールを得た(1−(1−メトキシ−1−メチル−プロポキシ)−ペンタ−2−インに対する収率:89.0%)。
【0093】
[実施例6.2: 触媒として8重量%のH2SO4水溶液を使用]
三つ口フラスコに、2.89(16.7mmol)の1−(1−メトキシ−1−メチル−プロポキシ)−ペンタ−2−イン、60μ1(0.05mmol)の8重量%硫酸水溶液および285μl(15.8mmol)の水を充填した。その溶液を約1時間30分の間22℃で攪拌した。溶液を500mg(4.7mmol)の炭酸ナトリウムで中和し、次いで500mgの硫酸ナトリウム(無水)で乾燥させた。不要な塩を濾過した後、混合物を減圧下(40℃、50mbar)で濃縮し、粗生成物をGCで分析した。純度92.8%の未精製の2−ペンチン−1−オール(1.35g)を得た(1−(1−メトキシ−1−メチル−プロポキシ)−ペンタ−2−インに対する収率:89.0%)。
【0094】
[比較例]
[比較例A: 保護基としてTHPを用いた2−ペンチン−1−オールの合成]
[A.1 触媒としてAmberlyst 15を用いた2−プロパ−2−イニルオキシ−テトラヒドロピランの調製]
10℃に冷却したジャケット付反応器に、150mgのAmberlyst 15を充填した。7.53g(133mmol)のプロパルギルアルコールと17.66gのTHP(199mmol)との混合物を、45分以内に加えた。反応物を加える間、内部温度を10℃に維持した。反応混合物を約15分間10℃に保った。触媒を濾過により分離し、反応混合物を減圧下(40℃、40mbar)で濃縮し、粗生成物をGCで分析した。純度87.1%の未精製の2−プロパ−2−イニルオキシ−テトラヒドロピラン(20.89g)を得た(プロパルギルアルコールに対する収率:97.6%)。
【0095】
[A.2 液体アンモニア、イソプレンで調製したリチウムアミド中での2−ペンタ−2−イニルオキシ−テトラヒドロ−ピランの調製]
0.89g(127mmol)の粒状リチウムを、−38℃で5分以内に100ml(4mol)の液体アンモニアに加えた。その混合物をリチウムが浮遊しなくなるまで(およそ10分後)攪拌した。液体アンモニア中にリチウムを含む紺青色溶液を得た。イソプレン(5.94g、86mmol)を20分以内に何回かに分けて加えた。反応混合物を約15分間、−38℃に保った。Liアミドの形成の終了は、反応混合物の脱色で見分けることができた。11.33g(70mmol)の2−プロパ−2−イニルオキシ−テトラヒドロピランを、滴下漏斗で−38℃において20分以内に反応混合物に加えた。反応混合物を約1時間、−38℃に保った。臭化エチル14.0g(126mmol)を滴下漏斗で20分以内に反応混合物に加えた。その後、反応混合物を1時間攪拌した。100mlのn−ヘキサンを反応混合物に加え、液体アンモニアを1.5時間以内に標準圧力下で蒸発させた。50mlの水を22℃で反応混合物に加え、塩がすべて溶解するまでその混合物を攪拌した。水層を50mlのn−ヘキサンで2回抽出した。有機層を5gの無水硫酸ナトリウムで乾燥させた。溶液を減圧下(40℃、60mbar)で濃縮し、粗生成物をGCで分析した。純度90.1%の未精製の2−ペンタ−2−イニルオキシ−テトラヒドロ−ピラン(12.4g)を得た(2−プロパ−2−イニルオキシ−テトラヒドロピランに対する収率:94.3%)。
【0096】
[A.3 8重量%H2SO4水溶液で触媒する2−ペンタ−2−イニルオキシ−テトラヒドロ−ピランからの2−ペンチン−1−オールの調製]
200μl(0.2mmol)の8重量%硫酸水溶液、200μl(11mmol)の水および20ml(789mmol)のメタノールを、22℃で攪拌しながら1.0g(5.4mmol)の2−ペンタ−2−イニルオキシ−テトラヒドロ−ピランに加えた。反応混合物を約2時間、還流状態(温度:65℃)に保った。反応混合物を0.5gの炭酸ナトリウムで中和した。メタノールを減圧下(40℃、60mbar)で蒸発させた。残留物を100mlの酢酸エチルで希釈し、その混合物を硫酸ナトリウム(無水)で乾燥させた。不要な塩を濾過した後、溶液を減圧下(40℃、60mbar)で蒸発させ、粗生成物をGCで分析した。純度82.2%の未精製の2−ペンチン−1−オール(0.5g)を得た(2−ペンタ−2−イニルオキシ−テトラヒドロ−ピランに対する収率:91.3%)。
【0097】
[分析データ]
【化18】
IR(ATR、cm−1):3293(w、−CCH)、2993、2944(m、−CH3、−CH2−CH3、−CH2−)、2832(w、−OCH3)、2121(w、−CCH)、1461(m、−CH2−)、1379(s)。
【0098】
1H−NMR(300MHz、CDCl3):δ=4.11(d、J=2.5Hz、2H、H3)、3.23(s、3H、H7)、2.39(t、J=2.5Hz、1H、H1)、1.38(s、6H、H5、H6)。
【0099】
13C−NMR(75MHz、CDCl3):δ=100.9(C4)、80.9(C2)、73.1(Cl、1JC、H=243Hz)、49.0(C3)、48.8(C7)、24.3(C5、C6)。
【0100】
MS(EI)m/z(相対強度(%)):113[M+−CH3、15]、97[M+−OCH3、27]、73[M+−C(CH3)2(OCH3)、55]。
【0101】
微量分析:C7H1202(MW 128.17)の計算値、C 65.60、H 9.44、O 24.97;実測値:C 64.82、H 9.08、O 25.81。
【化19】
【0102】
IR(ATR、cm−1):3294(w、−CCH)、2975、2945(m、−CH3、−CH2−CH3、−CH2−)、2832(w、−OCH3)、2121(w、−CCH)、1463(m、−CH2−)、1381(s)。
【0103】
1H−NMR(300MHz、CDCI3):δ=4.10−3.96(m、2H、H3)、3.14(s、3H、H8)、2.31(t、J=2.5Hz、1H、H1)、1.63−1.55(q、J=7.4Hz、2H、H5)、1.22(s、3H、H7)、0.84(t、J=7.5Hz、3H、H6)。
【0104】
13C−NMR(75MHz、CDCl3):δ=103.1(C4)、80.9(C2)、73.0(C1、1JC、H=258Hz)、48.6(C3)、48.5(C8)、29.4(C5)、20.7(C7)、8.5(C6)。
【0105】
MS(EI)m/z(相対強度(%)):127[M+−CH3、9]、87[M+−C(CH3)(CH2CH3)(OCH3)、91]。
【0106】
微量分析:C8H14O2(MW 142.20)の計算値、C 67.57、H 9.92、O 22.50;実測値:C 66.65、H 9.97、O 22.90。
【化20】
【0107】
IR(ATR、cm−1):2990、2940、2880(m、−CH3、−CH2−CH3、−CH2−)、2830(w、−OCH3)、2210(w、−CCH)、1459(m、−CH2−)、1379(s)。
【0108】
1H−NMR(300MHz、CDCl3):δ=4.09(t、J=2.1Hz、2H、H5)、3.23(s、3H、H9)、2.27−2.19(m、2H、H2)、1.37(s、6H、H7、H8)、1.14(t、J=7.4Hz、3H、H1)。
【0109】
13C−NMR(75MHz、CDCl3):δ=100.6(C6)、87.0(C3)、76.1(C4)、49.5(C5)、48.7(C9)、24.4(C7、C8)、13.7(C1)、12.6(C2)。
【0110】
MS(EI)m/z(相対強度(%)):141[M+−CH3、9]、125[M+−OCH3、7]、73[M+−C(CH3)2(OCH3)、65]、67[M+−CH2CCCH2CH3、19]。
【0111】
微量分析:C9H16O2(MW 156.23)の計算値、C 69.19、H 10.32、O 20.48;実測値:C 68.53、H 10.20、O 21.18。
【化21】
【0112】
IR(ATR、cm−1):2976、2941、2883、2832(s、−CH3、−CH2−CH3、−CH2−)、2832(w、−OCH3)、2210(w、−CCH)、1461(m、−CH2−)、1379(s)。
【0113】
1H NMR(300MHz、CDCl3):δ=4.01−3.99(q、J=1.1、2.1Hz、2H、H5)、3.14(s、3H、H10)、2.20−2.11(m、2H、H2)、1.63−1.55(q、J=7.5Hz、2H、H8)、1.22(s、3H、H7)、1.09−1.04(t、J=7.5Hz、3H、H1)、0.86−0.81(t、J=7.5Hz、3H、H9)。
【0114】
13C NMR(75MHz、CDCl3):δ=101.9(C3)、85.9(C6)、75.1(C4)、48.2(C10)、47.4(C5)、28.4(C8)、19.8(C7)、12.8(C1)、11.6(C2)、7.5(C9)。
【0115】
MS(EI)m/z(相対強度(%)):155[M+−CH3、1]、141[M+−CH2CH3、11]、87[M+−C(CH3)(CH2CH3)(OCH3)、55]、67[M+−CH2CCCH2CH3、34]。
【0116】
微量分析:C10H18O2(MW 170.25)の計算値、C 70.55、H 10.66、O 18.79;実測値:C 69.54、H 10.55、O 20.19。
【化22】
【0117】
IR(ATR、cm−1):3327(sbr、OH)、2977、2938、2878(s、−CH3、−CH2−CH3、−CH2−)、2230(w、−CCH)、1455(s、−CH2−)、1319(s)。
【0118】
1H−NMR(300MHz、CDCl3):δ=4.25(t、J=2.1Hz、2H、H5)、2.28−2.18(m、2H、H2)、2.02(sbr、1H、OH)、1.15(t、J=7.5Hz、3H、H1)。
【0119】
13C−NMR(75MHz、CDCl3):δ=87.8(C3)、77.7(C4)、51.3(C5)、13.8(C1)、12.4(C2)。
【0120】
MS(EI)m/z(相対強度(%)):83[M+−H、27]、65[M+−H2O、9]、55[M+−CH2CH3、36]、39[M+−CH2CH3、−OH、28]。
【0121】
微量分析:C5H8O(MW 84.12)の計算値、C 71.39、H 9.59、O 19.02;実測値:C 70.20、H 9.46、O 20.10。
【発明の詳細な説明】
【0001】
2−ペンチン−1−オールは、香料工業における中間体、例えばジャスモン酸エステル型の香りの中間体であるがゆえ、魅力的な化合物である(W.Y.Lee,S.Yang,Se Y.Jang,S.Y.Lee,Bulletin of the Korean Chemical Society 1991,12,26;T.Joshida,A.Yamaguchi,A.Kamatsu,Agricult.Biol.Chem.1966,30,370を参照)。
【0002】
本発明の目的は、工業規模で実施できる方法を見出すことであった。
【0003】
文献には、2−ペンチン−1−オールの幾つかの製造方法がすでに記載されている。過去には、ブチンおよびホルムアルデヒド(気体)から出発する方法がよく知られていた。ブチンを、例えば、臭化エチルマグネシウムで処理して対応するブチン−マグネシウム化合物にし、その後ホルムアルデヒドと反応させた。加水分解および蒸留を行った後、およそ60%の収率で2−ペンチノールを分離できた(Y.Tchai Lai,Bull.Chim Soc.France 1933,53,682;K.−E.Schulte,W.Engelhardt,Archiv Ber.Dt.Pharm.Soc.1954,287,495;JP 50049213を参照)。ブチンのリチウム塩に関して類似の反応が記載されている。例えば、米国特許第4,143,230号明細書(ここでは、1−ブチニルリチウムをパラホルムアルデヒドと反応させて2−ペンチン−1−オールにする反応が記載されている)を参照されたい。
【0004】
本発明のさらなる目的は、2−ペンチン−1−オールの製造においてホルムアルデヒド、ホルマリンまたはパラホルムアルデヒドの使用を避けることである。それは、それらが発がん物質また突然変異原であり生殖毒性があるからである。
【0005】
2−ペンチノールを合成する別の方法は、クロロブタノールから出発し、クロロブタノールをハロゲン化メチルマグネシウムで処理するものであり、収率は57〜65%である(J.Colonge,G.Descotes,Bull Soc Chim.France 1959,815;A.A.Kraevskii,B.Yu.Pyatnova,G.I,Myagkova.,I.K.Sarycheva,N.A.Preobrazhenskii,Doklady Akademii Nauk SSSR 1962,146,1349;A.A.Kraevskii,I.K.Sarycheva,N.A.Preobrazhenskii,Zhurnal Obshchei Khimii 1963,33,1831を参照)。こうした方法には、ハロゲン化物を取り扱うこと、収率が低いこと、大量の廃棄物がでることなど、幾つかの欠点がある。
【0006】
2−ペンチン−1−オールへ至る別の経路がWuによって開示されている(J.Wu,X.Kuang,Y.Tang,Huaxue Yanjiu Yu Yingyong(Chemical Research and Application)1999,11(5),521−522を参照)。最初にプロパルギルアルコールおよびリチウムアミドを添加し、その後臭化エチルを添加すると、2−ペンチン−1−オールが得られた。
【0007】
2−ペンチン−1−オールの別の合成方法では、出発物質であるプロパルギルアルコールをジヒドロピランで処理してテトラヒドロ−2−(2−プロピニルオキシ)−2H−ピラン(化合物A)にし(収率86%)、その後ナトリウムアミドおよびエチルブロミドが添加される(T.Joshida,A.Yamaguchi,A.Kamatsu,Agricult.Biol.Chem.1966,30,370を参照)。得られた2−ペンチン−1−イル−2−テトラヒドロピラニルエーテル(2−(ペンタ−2−イニルオキシ)テトラヒドロ−2H−ピラン(化合物B))をエーテルで抽出し、蒸留により精製された(収率96%)。
【0008】
145〜155℃において85%水性リン含有酸で処理することにより、この物質から2−ペンチン−1−オールが合成された(収率82%)。プロパルギルアルコールからの全収率は67.7%であった。類似の方法が米国特許第4,186,141号明細書にも記載されている。
【化1】
主な短所は、分離精製操作の後に最終生成物からの保護基の脱離が困難であることである。工業規模で作業する場合、これにはあまりにも費用がかかるであろう。
【0009】
したがって、本発明の更なる目的は、2−ペンチン−1−オールの中間体としての2−(ペンタ−2−イニルオキシ)テトラヒドロ−2H−ピランの使用を回避することである。
【0010】
こうした必要は、2−プロピン−1−オールから出発して2−ペンチン−1−オールを製造する本発明の方法によって満たされ、その方法は以下のステップを含む。
a)酸触媒の存在下で2−プロピノールから出発して式Iのケタールを調製するステップ;
【化2】
b)アンモニアと、リチウムアミド、アルキルリチウムおよびアリールリチウムからなる群から選択されるリチウム化合物との存在下で、式Iのケタールを、塩化エチル、臭化エチルおよびヨウ化エチルからなる群から選択されるハロゲン化アルキルと反応させて式IIのケタールにするステップ;
【化3】
c)酸触媒およびプロトン性溶媒の存在下で式IIのケタールを反応させて2−ペンチン−1−オールにするステップ。
上式において、R1はHまたは線状C1〜6アルキルであり、R2は線状C1〜6アルキルまたは分岐C3〜6アルキルであり、R3は線状C1〜6アルキルである。
【0011】
この方法を図1に示す。
【図面の簡単な説明】
【0012】
【図1】図1は、2−プロピン−1−オールから出発して2−ペンチン−1−オールを製造する本発明の方法を示す。
【0013】
「線状C1〜6アルキル」という表現は、メチル、エチル、n−プロピル、n−ブチル、n−ペンチルおよびn−ヘキシルを包含する。「分岐C3〜6アルキル」という用語は、イソプロピル、イソブチル、tert−ブチル、2−ペンチル、3−ペンチル、2−メチルブチル、3−メチルブチル、3−メチル−2−ブチル、2,2−ジメチル−プロピル、2−ヘキシル、3−ヘキシル、3−メチル−2−ペンチル、3−メチル−3−ペンチル、2,2−ジメチル−ブチル、3,3−ジメチル−ブチル、2,3−ジメチル−2−ブチル、2,3−ジメチル−3−ブチルおよび3,3−ジメチル−2−ブチルを包含する。
【0014】
好ましい式IおよびIIの化合物は、R1がHまたは線状C1〜3アルキルであり、かつ/またはR2が線状C1〜3アルキルであり、かつ/またはR3が線状C1〜3アルキルである化合物であり;より好ましいのは、R1がHまたは線状C1〜3アルキルであり、R2およびR3が互いに独立に線状C1〜3アルキルである化合物である。
【0015】
さらにより好ましい式IおよびIIの化合物は、R1がHまたは線状C1〜6アルキルであり、R2が線状C1〜6アルキルまたは分岐C3〜6アルキルであり、R3が線状C1〜6アルキルである化合物である。いっそうより好ましいのは、R1がHまたはメチルであり、R2がメチルまたはエチルであり、R3がメチルである式IおよびIIの化合物である。
【0016】
もっとも好ましい式IおよびIIの化合物は、それぞれ式Ia(R1=R2=R3=CH3)、Ib(R1=H;R2=R3=CH3)、IIa(R1=R2=R3=CH3)およびIIb(R1=H;R2=R3=CH3)の化合物、すなわち、それぞれR1がHまたはメチルでありR2およびR3がメチルである式IおよびIIの化合物である。
【化4】
【0017】
式IおよびIIの化合物は、新規ではない式Ibの具体的な化合物を除いて、新規である。したがって、本発明はまた、新規化合物、すなわち式Iの化合物
【化5】
[式中、R1はHまたは線状C1〜6アルキルであり、R2は線状C1〜6アルキルまたは分岐C3〜6アルキルであり、R3は線状C1〜6アルキルであるが、但し、R2およびR3がメチルである場合にR1はHではないことを条件とする]、ならびに式IIの化合物
【化6】
[式中、R1はHまたは線状C1〜6アルキルであり、R2は線状C1〜6アルキルまたは分岐C3〜6アルキルであり、R3は線状C1〜6アルキルである]に関する。
【0018】
この方法について以下にさらに詳しく説明する。
【0019】
[ステップa)]
[触媒]
ステップa)で使用される酸触媒は、好ましくはブレンステッド酸および固体酸からなる群から選択される。
【0020】
ブレンステッド酸の好ましい例は、p−TsOH(p−トルエンスルホン酸。有機スルホン酸の中で好ましいもの)などの有機スルホン酸、H2SO4(硫酸)およびH3PO4(リン含有酸)である。H3PO4が特に好ましいが、これは、その場合に生成する副生成物が少ないからである。主な副生成物は、式Vの二量体である。
【化7】
【0021】
こうした副生成物は、さらに反応して式VIの化合物にもなりうるし、また本発明の方法のステップc)で開裂して2−ペンチン−1−オールにもなりうる。
【化8】
ブレンステッド酸は有機溶媒溶液の形で使用してもよい。H3PO4は、例えば、17重量%のアセトン溶液またはメタノール溶液として使用してよい。H2SO4は、例えば、8重量%水溶液として、またp−TsOHは一水和物(p−TsOH・H2O)として使用してよい。
【0022】
固体酸の好ましい例は、担体上のブレンステッド酸、強酸性陽イオン交換体および酸性基を有するポリマー(すなわち、官能基化されたポリマー)である。
【0023】
強酸性陽イオン交換体の例は、Amberlyst型のポリマー(例えば、Amberlyst(登録商標)15、Amberlyst(登録商標)16)、Dowex、およびポリマーに結合したp−TsOH(p−トルエンスルホン酸)である。特に好ましいのは、Amberlyst(登録商標)15および16である。Amberlyst型のポリマー、ならびにDowexはすべて、官能基化されたスチレンジビニルベンゼンコポリマーである。
【0024】
固体酸は、好ましくは細孔径が100〜500Åの範囲であり、表面積が少なくとも20m2/g(窒素BETで測定)、好ましくは表面積が20〜500m2/gの範囲(より好ましくは、25〜300m2/gの範囲、28〜300m2/gの範囲、28〜70m2/gの範囲、30〜55m2/gの範囲)、また全細孔容積が少なくとも0.10ml/g、好ましくは全細孔容積が0.10〜0.50ml/gの範囲、より好ましくは全細孔容積が0.15〜0.45ml/gの範囲、さらにより好ましくは全細孔容積が0.18〜0.42ml/gの範囲、もっとも好ましくは全細孔容積が0.20〜0.40ml/gの範囲であり;さらに酸部位(すなわちスルホン酸基)の濃度が≧4.0当量/kg(特に≧4.5当量/kg;4.5〜6.0当量/kgの範囲;4.5〜5.5当量/kgの範囲)である。
【0025】
好ましくはこうした固体酸の酸性基は、スルホン酸基(−SO3H)であるか、またはpKa≦4の任意の他の基である。
【0026】
好ましい固体酸は、担体上の有機スルホン酸、または官能性スルホン酸基を有するポリマー樹脂である。
【0027】
更なる好適な固体酸は、CO2H基で官能基化されたポリマーである。
【0028】
本発明との関連において引用されているすべての官能基化ポリマーのうちでも、巨大網状ポリマーが好ましい。
【0029】
好ましくは、本発明に用いる固定化/固体ブレンステッド酸は、pka値が≦4であり、より好ましくは≦2である。
【0030】
固定化/固体ブレンステッド酸の例は、有機スルホン酸、H2SO4およびH3PO4(すべて担体上にある)である。
【0031】
担体は有機または無機のいずれであってよい。有機担体の例はポリマーである。無機担体の例は、例えば、SiO2、Al2O3、TiO2、ZrO2およびGeO2などの酸化物担体である。
【0032】
担体上のブレンステッド酸の例は、ポリマー上のp−トルエンスルホン酸またはシリカ上のプロピルスルホン酸である。
【0033】
さらに、pKA≦4である、好ましくはpKA≦2.5である他の酸触媒も、本発明の方法のステップa)で式Iのケタールの調製を触媒するのに適している。
【0034】
[反応条件]
ステップa)は、有機溶媒の存在下でも不存在下でも実施できる。有機溶媒が存在する場合には、好ましくは、エーテル、ケトン、エステルおよびそれらの混合物からなる群から選択されるであろう。
【0035】
好ましくはステップa)は、有機溶媒の不存在下で実施する。
【0036】
ステップa)は、バッチ式でも連続式でも実施できる。
【0037】
好ましくはステップa)は、0〜35℃の範囲の温度、好ましくは3〜25℃の範囲の温度、より好ましくは5〜20℃の範囲の温度で実施する。
【0038】
プロパルギルアルコール(基質(s))と触媒(c)とのモル比は一般に、s/cが500〜500000の範囲、好ましくは1000〜150000の範囲、特に好ましくは5000〜15000の範囲である。
【0039】
[ステップb)]
[ハロゲン化アルキル]
好ましいハロゲン化アルキルは臭化エチルおよびヨウ化エチルであり、その中で臭化エチルがもっとも好ましいものである。
【0040】
[リチウム化合物]
「アルキルリチウム」という用語は、直鎖(straight)C1〜6−アルキルリチウム、ならびに分岐C3〜6−アルキルリチウムを包含する。アルキルリチウムの好ましい例として、メチルリチウムおよびn−ブチルリチウムがある。
【0041】
「アリールリチウム」の例としてフェニルリチウムがある。
【0042】
リチウムアミドは好ましいリチウム化合物である。リチウムアミドは、当業者に知られている任意の方法、例えば、Lambert Brandsmaの著書「Synthesis of Acetylenes,Allenes and Cumulenes:Methods and Techniques」(Elsevier Ltd.UK,2004)の第2.3.1.1章(そこでは、リチウムが無水液体アンモニアと硝酸鉄(III)との混合物に加えられている)にLambert Brandsmaによって記載されているようにして調製できる。
【0043】
好ましくは、リチウムアミドは欧州特許出願公開第1238944号明細書(その内容を本明細書に援用する)に記載されている方法に従って調製する。つまり、リチウム金属をアンモニアに加えて「リチウムブロンズ(lithium bronze)」を生じさせ、その後その「リチウムブロンズ」を有機溶媒の存在下で1,3−ジエンまたはアリールオレフィンと反応させる。しかし、我々の本発明の方法のステップb)では、有機溶媒が存在する必要はない。リチウムアミドの調製は、好ましくはアンモニア以外のどんな有機溶媒も存在しない状態で実施する。リチウムアミド調製用の好ましいオレフィンは、ピペリレン、イソプレン、スチレンおよびミルセンなどの1,3−オレフィンである。ピペリレンおよびスチレンが特に好ましい。驚くべきことに、そのように調製されたリチウムアミドの場合、それによって調製された化合物IIの純度が高くなることが見出された。したがって、そのように調製されたリチウムアミドを使用するのが特に有利である。
【0044】
[反応条件]
ステップb)は、有機溶媒の存在下でも不存在下でも実施できる。そのような有機溶媒は、エーテル、アルカン、オレフィン、芳香族化合物およびそれらの混合物からなる群から選択できる。
【0045】
しかし、好ましくは、ステップb)は有機溶媒の不存在下で実施する。
【0046】
一般に、ステップb)は、反応条件下でアンモニアが液体であるような温度および圧力で実施する。大気圧では、温度は−70〜−35℃の範囲である。
【0047】
好ましくは、欧州特許出願公開第1238944号明細書の方法に従って、特にピペリレン(1,3−ペンタジエン)またはスチレンを用いて調製されたリチウムアミドを使用する。
【0048】
好ましくは、リチウム化合物と式Iのケタールとのモル比は、(1.5〜3)/1の範囲、好ましくは(1.75〜2.5)/1の範囲である。
【0049】
好ましくは、式Iのケタールとハロゲン化アルキルとのモル比は1:1〜1:1.5の範囲である。
【0050】
[ステップc)]
ステップc)で使用される酸触媒は、好ましくはブレンステッド酸および固体酸からなる群から選択される。ステップa)を実施するのに適しているブレンステッド酸および固体酸と同じものが、ステップc)を実施するのにもの適している。
【0051】
一般に、pKA≦4、好ましくはpKA≦2.5である酸触媒は、保護基を開裂させる(ステップc))のに適している。
【0052】
好ましくは、ブレンステッド酸がステップc)を実施するのに使用され、より好ましくは、ブレンステッド酸はp−TsOH、H2SO4およびH3PO4からなる群から選択される。
【0053】
もっとも好ましくは、ステップc)で使用されるブレンステッド酸はH2SO4である。
【0054】
ステップc)は、好ましくは、0〜50℃の範囲の温度、より好ましくは5〜30℃の範囲の温度で実施する。
【0055】
式IIの化合物(基質(s))と酸触媒(c)とのモル比は一般に、s/c=(100〜2000)/1の範囲、好ましくは(200〜1000)/1の範囲である。
【0056】
プロトン性溶媒は、好ましくは、H2O、アルコールおよびそれらの混合物からなる群から選択される。
【0057】
アルコールの好ましい例としては、メタノールおよびエタノールがある。
【0058】
水をプロトン性溶媒として使用する場合、水と式IIのケタールとのモル比は好ましくは少なくとも1:1、より好ましくは(1.5〜3):1である。
【0059】
本発明による方法の利点は、ステップa)〜c)すべてをアンモニア以外の(有機)溶媒なしで実施できることである。さらに、中間体(式IIおよびIIIの化合物)の精製が不要である。そのため本発明の方法は大量生産に非常に適したものとなっている。
【0060】
[本発明のさらなる実施形態]
●式Iの化合物
【化9】
[式中、R1はHまたは線状C1〜6アルキルであり、R2は線状C1〜6アルキルまたは分岐C3〜6アルキルであり、R3は線状C1〜6アルキルであるが、但し、R2およびR3がメチルである場合にはR1はHではないことを条件とする]。
【0061】
好ましくはR1=R2=R3=メチル、すなわち式Iaの化合物
【化10】
●式IIの化合物
【化11】
[式中、R1はHまたは線状C1〜6アルキルであり、R2は線状C1〜6アルキルまたは分岐C3〜6アルキルであり、R3は線状C1〜6アルキルである]。
【0062】
好ましくはR1=Hまたはメチル、R2=R3=メチル、すなわち式IIaおよびIIbの化合物
【化12】
【0063】
●式I
【化13】
のケタールを製造するための方法であって、2−プロピン−1−オールを酸触媒の存在下で式III
【化14】
の化合物と反応させるステップを含み、上式において、R1はHまたは線状C1〜6アルキルであり、R2は線状C1〜6アルキルまたは分岐C3〜6アルキルであり、R3は線状C1〜6アルキルである、方法。
【0064】
この方法は、上述の方法のステップa)に対応する。好ましい条件を含め上に示した詳細はすべて、ここでも当てはまる。
【0065】
●式II
【化15】
のケタールを製造するための方法であって、下記の式Iの化合物を、アンモニアと、リチウムアミド、アルキルリチウムおよびアリールリチウムからなる群から選択されるリチウム化合物との存在下で、式IVの化合物と反応させるステップを含み、以下の式
【化16】
において、R1はHまたは線状C1〜6アルキルであり、R2は線状C1〜6アルキルまたは分岐C3〜6アルキルであり、R3は線状C1〜6アルキルであり、XはCl、BrまたはIである、方法。
【0066】
この方法は、上述の方法のステップb)に対応する。好ましい条件を含め上に示した詳細はすべて、ここでも当てはまる。
【0067】
●2−ペンチン−1−オールを製造するための方法であって、プロトン性溶媒中で酸触媒を用いて、式II
【化17】
[式中、R1はHまたは線状C1〜6アルキルであり、R2は線状C1〜6アルキルまたは分岐C3〜6アルキルであり、R3は線状C1〜6アルキルである]
の化合物からヒドロキシ保護基を開裂させて2−ペンチン−1−オールを得るステップを含む、方法。
【0068】
この方法は、上述の方法のステップc)に対応する。好ましい条件を含め上に示した詳細はすべて、ここでも当てはまる。
【0069】
これから本発明を以下の非限定例を用いてさらに示す。
【0070】
[実施例]
以下の略号を使用する。
BME 2−ブテニルメチルエーテル
GC ガスクロマトグラフィー
IPM イソプロペニルメチルエーテル
p−TsOH p−トルエンスルホン酸
THP 3,4−ジヒドロ−2H−ピラン
【0071】
[全体的説明]
プロパルギルアルコール(propargylic alcohol)(=2−プロピン−1−オール)(Aldrich N°P5.080−3,99.0% GC)、p−トルエンスルホン酸(p−TsOH,Fluka N°89760,98.5% GC)、Amberlyst 15(Fluka N°06423)、リチウム(Acros,99.0%)、スチレン(Fluka 85960,99.0% GC)、ブロモエタン(=臭化エチル;Fluka 03150,98.0%)、イソプレン(Aldrich 119551,100.0% GC)、ミルセン(Givaudan DE−396,98.0% GC)、テトラヒドロピラン(THP)(Fluka 37350,95.0% GC)は市販されており、さらなる精製を行わずに使用した。
【0072】
イソプロペニルメチルエーテル(DSM Nutritional Products Ltd,Lalden(CH),96.0% GC)、ブテニルメチルエーテル(DSM Nutritional Products Ltd,Sisseln(CH),96.2% GC)、および1,3−ペンタジエン(=ピペリレン)(SPC,China,68.5% GC)は、更なる精製を行わずに使用した。
【0073】
[NMR分光法]
NMRスペクトルはBruker Avance 300MHz分光計で記録した。1H−NMRスペクトルは300MHzで、13Cスペクトルは75MHzでそれぞれ記録した。定量スペクトル(quantitative spectra)は、DMSO−d6で記録し、1,4ジメトキシベンゼンを内標準として用いた。2つのパルス間の遅延(delay)dlは30秒に設定した。12〜25mgの試料を使用した。スペクトルはCDCl3で記録した。δはppmで示す。多重度に関しては次のような略号を使用した:S=一重項、d=二重項、t=三重項、q=四重項、dd=二重項の二重項、dq=四重項の二重項、m=多重項。
【0074】
[実施例1: 3−(l−メトキシ−l−メチル−エトキシ)−プロピンの製造]
[実施例1.1: 触媒としてのH3PO4の使用]
25μl(0.04mmol)のリン酸(アセトン中に17%(w/w))を22.6g(399mmol)のプロパルギルアルコールに22℃で加えた。溶液を滴下漏斗へ移した。58.8mlのIPM(600mmol)を別の滴下漏斗に移した。両方の溶液を45分以内に同時にガラス反応器に加えた。反応物を添加する間、内部温度を15℃から20℃の間に維持した。添加後に、反応混合物を約15分間15℃に保った。最後に反応混合物を減圧下(40℃、40mbar)で濃縮し、粗生成物をGCで分析した。純度96.0%の3−(1−メトキシ−1−メチル−エトキシ)−プロピンを47.86g得た。プロパルギルアルコールに対する収率は89.85%であった。
【0075】
[実施例1.2: 触媒としてのp−TsOH・H2Oの使用]
10mg(0.05mmol)のp−TsOHの一水和物を22℃で22.43g(396mmol)のプロパルギルアルコール中に溶かした。溶液を滴下漏斗へ移した。56.93mlのIPM(581mmol)を別の滴下漏斗に移した。両方の溶液を40分以内に同時にガラス反応器に加えた。反応物を添加する間、内部温度を15℃から20℃の間に維持した。100mlの飽和重炭酸ナトリウム溶液を5分以内に加え、その混合物を5分間激しく攪拌した。その溶液を20mlのn−ヘキサンで抽出した。最後に有機相を減圧下(40℃、40mbar)で濃縮し、粗生成物をGCで分析した。純度87.6%の未精製の3−(1−メトキシ−1−メチル−エトキシ)−プロピンを48.6g得た。プロパルギルアルコールに対する収率は83.9%であった。
【0076】
[実施例1.3: 減らした量の触媒としてのp−TsOH・H2Oの使用]
同じ量の触媒を用いて実施例1.2を繰り返したが、ただし、22.43gのプロパルギルアルコールの代わりに372g(6.6mol)のプロパルギルアルコールを、また56.93mlのIPMの代わりに959ml(9.8mol)のIPMを使用した。さらに、反応物の添加を終えた後に、反応混合物をさらに15分間15℃で攪拌した。この少量の触媒の場合、実施例1.2におけるような重炭酸ナトリウム溶液による中和は不要であった。純度89.9%の未精製の3−(1−メトキシ−1−メチル−エトキシ)−プロピンを852.8g得た。プロパルギルアルコールに対する収率は91.0%であった。
【0077】
[実施例1.4: 触媒としてのAmberlyst(登録商標)15の使用]
10℃に冷却されたジャケット付反応器中に、820mgのAmberlyst 15を加えた。372g(6.6mol)のプロパルギルアルコールと958.5mlのIPM(9.8mol)との混合物を、45分以内に反応器に加えた。反応物を加える間、内部温度を10℃に維持した。添加後に、反応混合物を約15分間10℃に保った。濾過により触媒を分離し、反応混合物を減圧下(40℃、40mbar)で濃縮した。粗生成物をGCで分析した。純度90.3%の未精製の3−(1−メトキシ−1−メチル−エトキシ)−プロピンを881.65g得た。プロパルギルアルコールに対する収率は94.6%であった。
【0078】
[実施例1.5: 触媒としてのAmberlyst(登録商標)15の使用−連続式]
プロパルギルアルコール(230.4g、4.0mol)および458.3gのIPM(6.1mol)を、10℃で4時間、100mgのAmberlyst(登録商標)15を満たした固定床反応器(長さ:10cm、直径:9mm)中に送り込んだ(プロパルギルアルコールの送り速度=1ml/min.、IPMの送り速度=2.5ml/min.)。1時間の間に、溶液343.1gを回収した。過剰のIPMを減圧下(40℃、40mbar)で蒸留し、粗生成物をGCで分析した。純度95.9%の未精製の3−(l−メトキシ−1−メチル−エトキシ)−プロピン(243.2g)を得た(プロパルギルアルコールに対する収率:91.0%)。
【0079】
[実施例1.6: 触媒としてのH2SO4の使用]
5μl(4μmol)の硫酸(8%(w/w))を22℃で22.6g(399mmol)のプロパルギルアルコールに加えた。その溶液を滴下漏斗へ移した。58.75mlのIPM(599mmol)を別の滴下漏斗に移した。両方の溶液を45分以内に同時にガラス反応器に加えた。反応物を添加する間、内部温度を15℃から20℃の間に維持した。添加後に、反応混合物を約15分間15℃に保った。最後に反応混合物を減圧下(40℃、40mbar)で濃縮し、粗生成物をGCで分析した。純度87.4%の3−(1−メトキシ−1−メチル−エトキシ)−プロピンを51.0g得た。プロパルギルアルコールに対する収率は87.2%であった。
【0080】
[実施例2: 2−メトキシ−2−プロパ−2−イニルオキシ−ブタンの製造]
[実施例2.1: 触媒としてのH3PO4の使用]
130mg(0.3μmol)のリン酸(アセトン中に19%(w/w))と22.80g(399mmol)のプロパルギルアルコールとの混合物を、滴下漏斗中に満たす。53.5g(598mmol)の2−メトキシ−ブタ−2−エン(96.2% GC)を別の滴下漏斗に移した。両方の溶液を、冷却されたフラスコ(8℃)に30分以内に同時に加えた。反応物を添加する間、内部温度を8℃から12℃の間に維持した。反応混合物を約2時間10℃に保った。反応混合物を0.5gの炭酸ナトリウムで中和し、減圧下(40℃、80mbar)で濃縮した。粗生成物をGCで分析した。純度85.5%の未精製の2−メトキシ−2−プロパ−2−イニルオキシ−ブタンを58.0g得た(プロパルギルアルコールに対する収率:96.9%)。
【0081】
[実施例3: 1−(1−メトキシ−1−メチル−エトキシ)−ペンタ−2−インの製造]
[実施例3.1: 基剤としてリチウムアミド(スチレンで調製)を使用]
883mg(126mmol)の粒状リチウムを−38℃で5分以内に107g(6.30mol)の液体アンモニアに加えた。その混合物をリチウムが浮遊しなくなるまで(およそ10分後)攪拌した。液体アンモニア中にリチウムを含む紺青色溶液を得た。14.6g(140mmol)のスチレンを20分以内に何回かに分けて加えた。リチウムアミドの形成の終了は、反応混合物の色の変化で見分けることができた。9.9g(70mmol)の3−(1−メトキシ−1−メチル−エトキシ)−プロピン(90.8% GC)を滴下漏斗で20分以内に−38℃で反応混合物に加えた。反応混合物を約1時間、−38℃に保った。14g(126mmol)のブロモエタンを滴下漏斗で20分以内に反応混合物に加えた。その後、反応混合物を1時間攪拌した。100mlのn−ヘキサンを反応混合物に加え、液体アンモニアを16時間以内に標準圧力下で蒸発させた。70mlの水を22℃で反応混合物に加え、塩がすべて溶解するまでその混合物を攪拌した。有機層を分離し、2gの無水硫酸マグネシウムで乾燥させた。溶液を減圧下(40℃、40mbar)で濃縮し、粗生成物をGCで分析した。純度84.4%の1−(1−メトキシ−1−メチル−エトキシ)−ペンタ−2−インを10.6g得た(3−(l−メトキシ−1−メチル−エトキシ)−プロピンに対する収率:81.8%)。
【0082】
[実施例3.2: 基剤としてリチウムアミド(イソプレンで調製)を使用]
259mg(37mmol)の粒状リチウムを、−40℃で5分以内に47.7g(1.9mol)の液体アンモニアに加えた。その混合物をリチウムが浮遊しなくなるまで(およそ10分後)攪拌した。液体アンモニア中にリチウムを含む紺青色溶液を得た。4.0ml(40mmol)のイソプレンを10分以内に何回かに分けて加えた。リチウムアミドの形成の終了は、反応混合物の色の変化で見分けることができた。2.9g(20mmol)の3−(1−メトキシ−1−メチル−エトキシ)−プロピン(88.9% GC)を滴下漏斗で20分以内に−40℃で反応混合物に加えた。反応混合物を約1時間、−40℃に保った。8.9g(80mmol)のブロモエタンを滴下漏斗で10分以内に反応混合物に加えた。その後、反応混合物を1時間攪拌した。20mlのn−ヘキサンを反応混合物に加え、液体アンモニアを5時間以内に標準圧力下で蒸発させた。20mlの水を22℃で反応混合物に加え、塩がすべて溶解するまでその混合物を攪拌した。有機層を分離し、2gの無水硫酸マグネシウムで乾燥させた。溶液を減圧下(40℃、40mbar)で濃縮し、粗生成物をGCで分析した。純度75.6%の3.3gの未精製の1−(1−メトキシ−1−メチル−エトキシ)−ペンタ−2−インを得た(3−(l−メトキシ−1−メチル−エトキシ)−プロピンに対する収率:80.0%)。
【0083】
[実施例3.3: 基剤としてリチウムアミド(ミルセンで調製)を使用]
0.89g(127mmol)の粒状リチウムを、−37℃で15分以内に200ml(8mol)の液体アンモニアに加えた。その混合物をリチウムが浮遊しなくなるまで(およそ15分後)攪拌した。液体アンモニア中にリチウムを含む紺青色溶液を得た。12.6g(91mmol)のミルセンを30分以内に何回かに分けて加えた。リチウムアミドの形成の終了は、反応混合物の色の変化で見分けることができた。10.0g(71mmol)の3−(1−メトキシ−1−メチル−エトキシ)−プロピン(90.8% GC)を、滴下漏斗で30分以内に−37℃で反応混合物に加えた。反応混合物を約1時間、−37℃に保った。14.0g(126mmol)のブロモエタンを滴下漏斗で30分以内に反応混合物に加えた。その後、反応混合物を1時間攪拌した。157mlのn−ヘキサンを反応混合物に加え、液体アンモニアを15時間以内に標準圧力下で蒸発させた。50mlの水を22℃で反応混合物に加え、塩がすべて溶解するまでその混合物を攪拌した。有機層を分離し、減圧下(40℃、40mbar)で濃縮した。粗生成物をGCで分析した。25.8gの未精製の1−(1−メトキシ−1−メチル−エトキシ)−ペンタ−2−インを得た(3−(1−メトキシ−1−メチル−エトキシ)−プロピンに対する収率:83.0%)。
【0084】
[実施例3.4: 基剤としてリチウムアミド(1,3−ペンタジエンで調製)を使用]
0.89g(127mmol)の粒状リチウムを、−37℃で20分以内に100ml(4mol)の液体アンモニアに加えた。その混合物をリチウムが浮遊しなくなるまで(およそ15分後)攪拌した。液体アンモニア中にリチウムを含む暗青緑色の溶液を得た。6.34g(92mmol)のピペリレン(=1,3−ペンタジエン)を30分以内に何回かに分けて加えた。リチウムアミドの形成の終了は、反応混合物の色の変化で見分けることができた。10.1g(71mmol)の3−(1−メトキシ−1−メチル−エトキシ)−プロピン(89.7% GC)を、滴下漏斗で30分以内に−37℃で反応混合物に加えた。反応混合物を約1時間、−37℃に保った。14.0g(126mmol)のブロモエタンを滴下漏斗で30分以内に反応混合物に加えた。その後、反応混合物を1時間攪拌した。100mlのn−ヘキサンを反応混合物に加え、液体アンモニアを1.5時間以内に標準圧力下で蒸発させた。50mlの水を22℃で反応混合物に加え、塩がすべて溶解するまでその混合物を激しく攪拌した。水層を50mlのn−ヘキサンで1回抽出した。有機層を5gの無水硫酸ナトリウムで乾燥させた。溶液を減圧下(40℃、60mbar)で濃縮し、粗生成物をGCで分析した。純度87.5%の11.3gの未精製の1−(1−メトキシ−1−メチル−エトキシ)−ペンタ−2−インを得た(3−(1−メトキシ−1−メチル−エトキシ)−プロピンに対する収率:85.3%)。
【0085】
[実施例3.5: 臭化エチルの代わりにヨウ化エチルを使用]
0.89g(127mmol)の粒状リチウムを、−37℃で15分以内に100ml(4mol)の液体アンモニアに加えた。その混合物をリチウムが浮遊しなくなるまで(およそ15分後)攪拌した。液体アンモニア中にリチウムを含む紺青色溶液を得た。5.94g(86.3mmol)のイソプレンを30分以内に何回かに分けて加えた。LiNH2の形成の終了は、反応混合物の脱色により見分けることができた。10.69g(70.1mmol)の3−(l−メトキシ−1−メチル−エトキシ)−プロピンを−37℃で30分以内に反応混合物に加えた。反応混合物を約1時間、−37℃に保った。ヨウ化エチル(20.05g、126mmol)を滴下漏斗で30分以内に反応混合物に加えた。その後、反応混合物を1時間攪拌した。n−ヘキサン(100ml)を反応混合物に加え、液体アンモニアを1.5時間以内に標準圧力下で蒸発させた。水(50ml)を22℃で反応混合物に加え、塩がすべて溶解するまでその混合物を激しく攪拌した。水層を50mlのn−ヘキサンで2回抽出した。有機層を5gの無水硫酸ナトリウムで乾燥させた。溶液を減圧下(40℃、60mbar)で濃縮し、粗生成物をGCで分析した。純度72.95%の未精製の1−(1−メトキシ−1−メチル−エトキシ)−ペンタ−2−イン(13.21g)を得た(3−(1−メトキシ−1−メチル−エトキシ)−プロピンに対する収率:88.0%)。
【0086】
[実施例4: 1−(1−メトキシ−1−メチル−プロポキシ)−ペンタ−2−インの製造]
[実施例4.1: 基剤としてリチウムアミド(イソプレンで調製)を使用]
890mg(127mmol)の粒状リチウムを、−40℃で5分以内に100ml(4.0mol)の液体アンモニアに加えた。その混合物をリチウムが浮遊しなくなるまで(およそ10分後)攪拌した。液体アンモニア中にリチウムを含む紺青色溶液を得た。5.94g(86.6mmol)のイソプレンを10分以内に何回かに分けて加えたが、溶液は添加の終了時に白色になる。混合物を−38℃で25分間攪拌した。9.1g(69.5mmol)の2−メトキシ−2−プロパ−2−イニルオキシ−ブタン(97.4% GC)を、滴下漏斗で25分以内に−38℃で反応混合物に加えた。反応混合物を約1時間、−38℃に保った。14g(128.5mmol)のブロモエタンを滴下漏斗で20分以内に反応混合物に加えた。その後、反応混合物を1時間攪拌した。100mlのn−ヘキサンを反応混合物に加え、液体アンモニアを1時間30分以内に標準圧力下で蒸発させた。50mlの水を40℃で反応混合物に加え、塩がすべて溶解するまでその混合物を攪拌した。混合物を分液漏斗に移し、層を分離した。水層を127mlのn−ヘキサンで抽出した。有機層を0.5gの無水硫酸ナトリウムで乾燥させ、減圧下(40℃、60mbar)で濃縮し、粗生成物(1−(1−メトキシ−1−メチル−プロポキシ)−ペンタ−2−イン)をGCで分析した。純度87.1%の10.54gの未精製の1−(1−メトキシ−1−メチル−プロポキシ)−ペンタ−2−インを得た(2−メトキシ−2−プロパ−2−イニルオキシ−ブタンに対する収率:77.6%)。
【0087】
[実施例5: 1−(1−メトキシ−1−メチル−エトキシ)−ペンタ−2−インから出発する2−ペンチン−1−オールの製造]
[実施例5.1: 触媒としてp−トルエンスルホン酸を使用]
5.0g(25.7/24.387mmol)の1−(1−メトキシ−1−メチル−エトキシ)−ペンタ−2−イン(GCによる純度:80.4%)を、20℃において100mlのn−ヘキサン(工業等級)で希釈した。20mgのp−トルエンスルホン酸一水和物および500μl(27.8mmol)の脱イオン水を、22℃で攪拌しながら溶液に加えた。その混合物を約1時間45分の間、22℃に保った。500mg(4.7mmol)の炭酸ナトリウムを加え、その後、反応混合物を5分間攪拌した。不要な塩を濾過で分離した。最後に溶液を減圧下(40℃、50mbar)で濃縮し、粗生成物をGCで分析した。2.23gの未精製の2−ペンチン−1−オール(GCによる純度:78.62%)を得た(1−(1−メトキシ−1−メチル−エトキシ)−ペンタ−2−インに対する収率:85.5%)。
【0088】
[実施例5.2: 触媒として8重量%のH2SO4水溶液を使用]
5.0g(25.7mmol)の3(80.4% GC)を20℃において100mlのn−ヘキサンで希釈した。93μl(0.1mmol)の硫酸(8%(w/w))および440μl(24.4mmol)の水を、22℃で攪拌しながら溶液に加えた。その混合物を約1時間30分の間22℃に保った。溶液を500mg(4.7mmol)の炭酸ナトリウムで中和し、不要な塩を濾過で分離した。最後に溶液を減圧下(40℃、50mbar)で濃縮し、粗生成物をGCで分析した。純度81.2%の2.26gの未精製の2−ペンチン−1−オールを得た(1−(l−メトキシ−1−メチル−エトキシ)−ペンタ−2−インに対する収率:85.3%)。
【0089】
[実施例5.3: 触媒としてAmberlyst 15を使用]
25mgのAmberlyst(登録商標)15[Amberlyst 15 WET]をアルゴン雰囲気下において22℃で、7.5g(45.6mmol)の1−(1−メトキシ−1−メチル−エトキシ)−ペンタ−2−イン(GCによる純度:95%)と3.75ml(208mmol)の脱イオン水との混合物に加えた。その混合物を約1時間30分の間攪拌しながら22℃に保った。触媒を濾過で分離した。2−ペンチン−1−オールを総量で60mlのジエチルエーテルで抽出し、有機層を5gの硫酸ナトリウム(無水)で乾燥させた。溶液を減圧下(40℃、50mbar)で濃縮し、粗生成物をGCで分析した。純度95.0%の3.85gの未精製の2−ペンチン−1−オールを得た(1−(1−メトキシ−1−メチル−エトキシ)−ペンタ−2−インに対する収率:95.3%)。
【0090】
[実施例5.4: 触媒として8重量%のH2SO4水溶液を、また更なる出発物質として二量体の1−(1−メチル−1−ペンタ−2−イニルオキシ−エトキシ)−ペンタ−2−インを使用]
213μl(0.2mmol)の8重量%の硫酸水溶液および1169μl(65mmol)の水を、22℃において攪拌しながら、1−(1−メトキシ−1−メチル−エトキシ)−ペンタ−2−イン(72.5% GC)と1−(1−メチル−1−ペンタ−2−イニルオキシ−エトキシ)−ペンタ−2−イン(3.5% GC)との混合物12.7g(合計で61.1mmolの1−(1−メトキシ−1−メチル−エトキシ)−ペンタ−2−イン)に加えた。そのエマルジョンを約1時間30分の間22℃に保った。得られた反応混合物をバルブ間蒸留(bulb−to−bulb distillation)で精製し(P=65mbar/オーブン温度 97〜100℃)、最終生成物をGCで分析した。純度が99.7%および89.4%の2−ペンチン−1−オールの2つの部分を蒸留した(蒸留後の収率は、1−(1−メトキシ−1−メチル−エトキシ)−ペンタ−2−イン + 1−(1−メチル−1−ペンタ−2−イニルオキシ−エトキシ)−ペンタ−2−インに対して94.5%であった)。
【0091】
[実施例5.5: 触媒として8重量%のH2SO4水溶液を使用]
180μl(0.16mmol)の8重量%硫酸水溶液および900μl(50mmol)の水を、22℃で攪拌しながら10.8g(59mmol)の1−(1−メトキシ−1−メチル−エトキシ)−ペンタ−2−インに加えた。そのエマルジョンを約1時間30分の間22℃に保った。得られた反応混合物をバルブ間蒸留で精製し(P=65mbar/オーブン温度 97〜100℃)、最終生成物をGCで分析した。純度が97.3%および89.5%の2−ペンチン−1−オールの2つの部分を蒸留した(蒸留後の収率は1−(1−メトキシ−1−メチル−エトキシ)−ペンタ−2−インに対して89.8%であった)。
【0092】
[実施例6: 1−(1−メトキシ−1−メチル−プロポキシ)−ペンタ−2−インから出発する2−ペンチン−1−オールの製造]
[実施例6.1: 触媒として8重量%のH2SO4水溶液を使用]
三つ口フラスコに、2.89(16.7mmol)の1−(1−メトキシ−1−メチル−プロポキシ)−ペンタ−2−イン(98.6% GC)、60μl(0.05mmol)の硫酸(8%(w/w))および285μl(15.8mmol)の水を充填した。その溶液を約1時間30分の間22℃で攪拌した。溶液を500mg(4.7mmol)の炭酸ナトリウムで中和し、次いで500mgの硫酸ナトリウム(無水)で乾燥させた。不要な塩を濾過した後、混合物を減圧下(40℃、50mbar)で濃縮し、粗生成物をGCで分析した。純度92.8%の1.35gの未精製の2−ペンチン−1−オールを得た(1−(1−メトキシ−1−メチル−プロポキシ)−ペンタ−2−インに対する収率:89.0%)。
【0093】
[実施例6.2: 触媒として8重量%のH2SO4水溶液を使用]
三つ口フラスコに、2.89(16.7mmol)の1−(1−メトキシ−1−メチル−プロポキシ)−ペンタ−2−イン、60μ1(0.05mmol)の8重量%硫酸水溶液および285μl(15.8mmol)の水を充填した。その溶液を約1時間30分の間22℃で攪拌した。溶液を500mg(4.7mmol)の炭酸ナトリウムで中和し、次いで500mgの硫酸ナトリウム(無水)で乾燥させた。不要な塩を濾過した後、混合物を減圧下(40℃、50mbar)で濃縮し、粗生成物をGCで分析した。純度92.8%の未精製の2−ペンチン−1−オール(1.35g)を得た(1−(1−メトキシ−1−メチル−プロポキシ)−ペンタ−2−インに対する収率:89.0%)。
【0094】
[比較例]
[比較例A: 保護基としてTHPを用いた2−ペンチン−1−オールの合成]
[A.1 触媒としてAmberlyst 15を用いた2−プロパ−2−イニルオキシ−テトラヒドロピランの調製]
10℃に冷却したジャケット付反応器に、150mgのAmberlyst 15を充填した。7.53g(133mmol)のプロパルギルアルコールと17.66gのTHP(199mmol)との混合物を、45分以内に加えた。反応物を加える間、内部温度を10℃に維持した。反応混合物を約15分間10℃に保った。触媒を濾過により分離し、反応混合物を減圧下(40℃、40mbar)で濃縮し、粗生成物をGCで分析した。純度87.1%の未精製の2−プロパ−2−イニルオキシ−テトラヒドロピラン(20.89g)を得た(プロパルギルアルコールに対する収率:97.6%)。
【0095】
[A.2 液体アンモニア、イソプレンで調製したリチウムアミド中での2−ペンタ−2−イニルオキシ−テトラヒドロ−ピランの調製]
0.89g(127mmol)の粒状リチウムを、−38℃で5分以内に100ml(4mol)の液体アンモニアに加えた。その混合物をリチウムが浮遊しなくなるまで(およそ10分後)攪拌した。液体アンモニア中にリチウムを含む紺青色溶液を得た。イソプレン(5.94g、86mmol)を20分以内に何回かに分けて加えた。反応混合物を約15分間、−38℃に保った。Liアミドの形成の終了は、反応混合物の脱色で見分けることができた。11.33g(70mmol)の2−プロパ−2−イニルオキシ−テトラヒドロピランを、滴下漏斗で−38℃において20分以内に反応混合物に加えた。反応混合物を約1時間、−38℃に保った。臭化エチル14.0g(126mmol)を滴下漏斗で20分以内に反応混合物に加えた。その後、反応混合物を1時間攪拌した。100mlのn−ヘキサンを反応混合物に加え、液体アンモニアを1.5時間以内に標準圧力下で蒸発させた。50mlの水を22℃で反応混合物に加え、塩がすべて溶解するまでその混合物を攪拌した。水層を50mlのn−ヘキサンで2回抽出した。有機層を5gの無水硫酸ナトリウムで乾燥させた。溶液を減圧下(40℃、60mbar)で濃縮し、粗生成物をGCで分析した。純度90.1%の未精製の2−ペンタ−2−イニルオキシ−テトラヒドロ−ピラン(12.4g)を得た(2−プロパ−2−イニルオキシ−テトラヒドロピランに対する収率:94.3%)。
【0096】
[A.3 8重量%H2SO4水溶液で触媒する2−ペンタ−2−イニルオキシ−テトラヒドロ−ピランからの2−ペンチン−1−オールの調製]
200μl(0.2mmol)の8重量%硫酸水溶液、200μl(11mmol)の水および20ml(789mmol)のメタノールを、22℃で攪拌しながら1.0g(5.4mmol)の2−ペンタ−2−イニルオキシ−テトラヒドロ−ピランに加えた。反応混合物を約2時間、還流状態(温度:65℃)に保った。反応混合物を0.5gの炭酸ナトリウムで中和した。メタノールを減圧下(40℃、60mbar)で蒸発させた。残留物を100mlの酢酸エチルで希釈し、その混合物を硫酸ナトリウム(無水)で乾燥させた。不要な塩を濾過した後、溶液を減圧下(40℃、60mbar)で蒸発させ、粗生成物をGCで分析した。純度82.2%の未精製の2−ペンチン−1−オール(0.5g)を得た(2−ペンタ−2−イニルオキシ−テトラヒドロ−ピランに対する収率:91.3%)。
【0097】
[分析データ]
【化18】
IR(ATR、cm−1):3293(w、−CCH)、2993、2944(m、−CH3、−CH2−CH3、−CH2−)、2832(w、−OCH3)、2121(w、−CCH)、1461(m、−CH2−)、1379(s)。
【0098】
1H−NMR(300MHz、CDCl3):δ=4.11(d、J=2.5Hz、2H、H3)、3.23(s、3H、H7)、2.39(t、J=2.5Hz、1H、H1)、1.38(s、6H、H5、H6)。
【0099】
13C−NMR(75MHz、CDCl3):δ=100.9(C4)、80.9(C2)、73.1(Cl、1JC、H=243Hz)、49.0(C3)、48.8(C7)、24.3(C5、C6)。
【0100】
MS(EI)m/z(相対強度(%)):113[M+−CH3、15]、97[M+−OCH3、27]、73[M+−C(CH3)2(OCH3)、55]。
【0101】
微量分析:C7H1202(MW 128.17)の計算値、C 65.60、H 9.44、O 24.97;実測値:C 64.82、H 9.08、O 25.81。
【化19】
【0102】
IR(ATR、cm−1):3294(w、−CCH)、2975、2945(m、−CH3、−CH2−CH3、−CH2−)、2832(w、−OCH3)、2121(w、−CCH)、1463(m、−CH2−)、1381(s)。
【0103】
1H−NMR(300MHz、CDCI3):δ=4.10−3.96(m、2H、H3)、3.14(s、3H、H8)、2.31(t、J=2.5Hz、1H、H1)、1.63−1.55(q、J=7.4Hz、2H、H5)、1.22(s、3H、H7)、0.84(t、J=7.5Hz、3H、H6)。
【0104】
13C−NMR(75MHz、CDCl3):δ=103.1(C4)、80.9(C2)、73.0(C1、1JC、H=258Hz)、48.6(C3)、48.5(C8)、29.4(C5)、20.7(C7)、8.5(C6)。
【0105】
MS(EI)m/z(相対強度(%)):127[M+−CH3、9]、87[M+−C(CH3)(CH2CH3)(OCH3)、91]。
【0106】
微量分析:C8H14O2(MW 142.20)の計算値、C 67.57、H 9.92、O 22.50;実測値:C 66.65、H 9.97、O 22.90。
【化20】
【0107】
IR(ATR、cm−1):2990、2940、2880(m、−CH3、−CH2−CH3、−CH2−)、2830(w、−OCH3)、2210(w、−CCH)、1459(m、−CH2−)、1379(s)。
【0108】
1H−NMR(300MHz、CDCl3):δ=4.09(t、J=2.1Hz、2H、H5)、3.23(s、3H、H9)、2.27−2.19(m、2H、H2)、1.37(s、6H、H7、H8)、1.14(t、J=7.4Hz、3H、H1)。
【0109】
13C−NMR(75MHz、CDCl3):δ=100.6(C6)、87.0(C3)、76.1(C4)、49.5(C5)、48.7(C9)、24.4(C7、C8)、13.7(C1)、12.6(C2)。
【0110】
MS(EI)m/z(相対強度(%)):141[M+−CH3、9]、125[M+−OCH3、7]、73[M+−C(CH3)2(OCH3)、65]、67[M+−CH2CCCH2CH3、19]。
【0111】
微量分析:C9H16O2(MW 156.23)の計算値、C 69.19、H 10.32、O 20.48;実測値:C 68.53、H 10.20、O 21.18。
【化21】
【0112】
IR(ATR、cm−1):2976、2941、2883、2832(s、−CH3、−CH2−CH3、−CH2−)、2832(w、−OCH3)、2210(w、−CCH)、1461(m、−CH2−)、1379(s)。
【0113】
1H NMR(300MHz、CDCl3):δ=4.01−3.99(q、J=1.1、2.1Hz、2H、H5)、3.14(s、3H、H10)、2.20−2.11(m、2H、H2)、1.63−1.55(q、J=7.5Hz、2H、H8)、1.22(s、3H、H7)、1.09−1.04(t、J=7.5Hz、3H、H1)、0.86−0.81(t、J=7.5Hz、3H、H9)。
【0114】
13C NMR(75MHz、CDCl3):δ=101.9(C3)、85.9(C6)、75.1(C4)、48.2(C10)、47.4(C5)、28.4(C8)、19.8(C7)、12.8(C1)、11.6(C2)、7.5(C9)。
【0115】
MS(EI)m/z(相対強度(%)):155[M+−CH3、1]、141[M+−CH2CH3、11]、87[M+−C(CH3)(CH2CH3)(OCH3)、55]、67[M+−CH2CCCH2CH3、34]。
【0116】
微量分析:C10H18O2(MW 170.25)の計算値、C 70.55、H 10.66、O 18.79;実測値:C 69.54、H 10.55、O 20.19。
【化22】
【0117】
IR(ATR、cm−1):3327(sbr、OH)、2977、2938、2878(s、−CH3、−CH2−CH3、−CH2−)、2230(w、−CCH)、1455(s、−CH2−)、1319(s)。
【0118】
1H−NMR(300MHz、CDCl3):δ=4.25(t、J=2.1Hz、2H、H5)、2.28−2.18(m、2H、H2)、2.02(sbr、1H、OH)、1.15(t、J=7.5Hz、3H、H1)。
【0119】
13C−NMR(75MHz、CDCl3):δ=87.8(C3)、77.7(C4)、51.3(C5)、13.8(C1)、12.4(C2)。
【0120】
MS(EI)m/z(相対強度(%)):83[M+−H、27]、65[M+−H2O、9]、55[M+−CH2CH3、36]、39[M+−CH2CH3、−OH、28]。
【0121】
微量分析:C5H8O(MW 84.12)の計算値、C 71.39、H 9.59、O 19.02;実測値:C 70.20、H 9.46、O 20.10。
【特許請求の範囲】
【請求項1】
a)酸触媒の存在下で2−プロピノールから出発して式I
【化1】
のケタールを調製するステップと;
b)アンモニアと、リチウムアミド、アルキルリチウムおよびアリールリチウムからなる群から選択されるリチウム化合物との存在下で、式Iのケタールを、塩化エチル、臭化エチルおよびヨウ化エチルからなる群から選択されるハロゲン化アルキルと反応させて式II
【化2】
のケタールにするステップと;
c)酸触媒およびプロトン性溶媒の存在下で式IIのケタールを反応させて2−ペンチン−1−オールにするステップと
を含む、2−プロピン−1−オールから出発して2−ペンチン−1−オールを製造するための方法であって、上式において、R1がHまたは線状C1〜6アルキルであり、R2が線状C1〜6アルキルまたは分岐C3〜6アルキルであり、R3が線状C1〜6アルキルである方法。
【請求項2】
ステップa)で使用される前記酸触媒がブレンステッド酸および固体酸からなる群から選択される、請求項1に記載の方法。
【請求項3】
ステップc)で使用される酸触媒がブレンステッド酸および固体酸からなる群から選択される、請求項1または2に記載の方法。
【請求項4】
前記酸触媒がpKA≦4であり、好ましくは前記酸触媒がpKA≦2.5である、請求項2または3に記載の方法。
【請求項5】
前記ブレンステッド酸がp−TsOH、H2SO4およびH3PO4からなる群から選択される、請求項2または3に記載の方法。
【請求項6】
前記ブレンステッド酸がH3PO4である、請求項2に記載の方法。
【請求項7】
前記ブレンステッド酸がH2SO4である、請求項3に記載の方法。
【請求項8】
前記固体酸が、担体上のブレンステッド酸、強酸性陽イオン交換体および酸性基を有するポリマーからなる群から選択される、請求項2または3に記載の方法。
【請求項9】
ステップa)を有機溶媒の非存在下で実施する、請求項1〜8のいずれか一項に記載の方法。
【請求項10】
ステップa)を、0〜35℃の範囲の温度、好ましくは3〜25℃の範囲の温度、より好ましくは5〜20℃の範囲の温度で実施する、請求項1〜9のいずれか一項に記載の方法。
【請求項11】
ステップb)を有機溶媒の非存在下で実施する、請求項1〜10のいずれか一項に記載の方法。
【請求項12】
アンモニアが前記反応条件下で液体であるような温度および圧力でステップb)を実施する、請求項1〜11のいずれか一項に記載の方法。
【請求項13】
ステップc)を、0〜50℃の範囲の温度、好ましくは5〜30℃の範囲の温度で実施する、請求項1〜12のいずれか一項に記載の方法。
【請求項14】
R1がHまたはメチルであり、R2およびR3がメチルである、請求項1〜13のいずれか一項に記載の方法。
【請求項15】
式I
【化3】
[式中、R1はHまたは線状C1〜6アルキルであり、R2は線状C1〜6アルキルまたは分岐C3〜6アルキルであり、R3は線状C1〜6アルキルであるが、但し、R2およびR3がメチルである場合にはR1はHではないことを条件とする]
の化合物。
【請求項16】
R1=R2=R3=メチルである、請求項15に記載の式Iの化合物。
【請求項17】
式II
【化4】
[式中、R1はHまたは線状C1〜6アルキルであり、R2は線状C1〜6アルキルまたは分岐C3〜6アルキルであり、R3は線状C1〜6アルキルである]
の化合物。
【請求項18】
R1=Hまたはメチルであり、R2=R3=メチルである、請求項17に記載の式IIの化合物。
【請求項19】
式I
【化5】
のケタールを製造するための方法であって、2−プロピン−1−オールを酸触媒の存在下で式III
【化6】
の化合物と反応させるステップを含み、上式において、R1はHまたは線状C1〜6アルキルであり、R2は線状C1〜6アルキルまたは分岐C3〜6アルキルであり、R3は線状C1〜6アルキルである、方法。
【請求項20】
式II
【化7】
のケタールを製造するための方法であって、下記の式Iの化合物を、アンモニアと、リチウムアミド、アルキルリチウムおよびアリールリチウムからなる群から選択されるリチウム化合物との存在下で、式IVの化合物と反応させるステップを含み、以下の式
【化8】
において、R1はHまたは線状C1〜6アルキルであり、R2は線状C1〜6アルキルまたは分岐C3〜6アルキルであり、R3は線状C1〜6アルキルであり、XはCl、BrまたはIである、方法。
【請求項21】
XがBrである、請求項20に記載の方法。
【請求項22】
2−ペンチン−1−オールを製造するための方法であって、プロトン性溶媒中で酸触媒を用いて、式II
【化9】
[式中、R1はHまたは線状C1〜6アルキルであり、R2は線状C1〜6アルキルまたは分岐C3〜6アルキルであり、R3は線状C1〜6アルキルである]
の化合物からヒドロキシ保護基を開裂させて2−ペンチン−1−オールを得るステップを含む、方法。
【請求項23】
R1がHまたはメチルであり、R2およびR3がメチルである、請求項19〜22のいずれか一項に記載の方法。
【請求項1】
a)酸触媒の存在下で2−プロピノールから出発して式I
【化1】
のケタールを調製するステップと;
b)アンモニアと、リチウムアミド、アルキルリチウムおよびアリールリチウムからなる群から選択されるリチウム化合物との存在下で、式Iのケタールを、塩化エチル、臭化エチルおよびヨウ化エチルからなる群から選択されるハロゲン化アルキルと反応させて式II
【化2】
のケタールにするステップと;
c)酸触媒およびプロトン性溶媒の存在下で式IIのケタールを反応させて2−ペンチン−1−オールにするステップと
を含む、2−プロピン−1−オールから出発して2−ペンチン−1−オールを製造するための方法であって、上式において、R1がHまたは線状C1〜6アルキルであり、R2が線状C1〜6アルキルまたは分岐C3〜6アルキルであり、R3が線状C1〜6アルキルである方法。
【請求項2】
ステップa)で使用される前記酸触媒がブレンステッド酸および固体酸からなる群から選択される、請求項1に記載の方法。
【請求項3】
ステップc)で使用される酸触媒がブレンステッド酸および固体酸からなる群から選択される、請求項1または2に記載の方法。
【請求項4】
前記酸触媒がpKA≦4であり、好ましくは前記酸触媒がpKA≦2.5である、請求項2または3に記載の方法。
【請求項5】
前記ブレンステッド酸がp−TsOH、H2SO4およびH3PO4からなる群から選択される、請求項2または3に記載の方法。
【請求項6】
前記ブレンステッド酸がH3PO4である、請求項2に記載の方法。
【請求項7】
前記ブレンステッド酸がH2SO4である、請求項3に記載の方法。
【請求項8】
前記固体酸が、担体上のブレンステッド酸、強酸性陽イオン交換体および酸性基を有するポリマーからなる群から選択される、請求項2または3に記載の方法。
【請求項9】
ステップa)を有機溶媒の非存在下で実施する、請求項1〜8のいずれか一項に記載の方法。
【請求項10】
ステップa)を、0〜35℃の範囲の温度、好ましくは3〜25℃の範囲の温度、より好ましくは5〜20℃の範囲の温度で実施する、請求項1〜9のいずれか一項に記載の方法。
【請求項11】
ステップb)を有機溶媒の非存在下で実施する、請求項1〜10のいずれか一項に記載の方法。
【請求項12】
アンモニアが前記反応条件下で液体であるような温度および圧力でステップb)を実施する、請求項1〜11のいずれか一項に記載の方法。
【請求項13】
ステップc)を、0〜50℃の範囲の温度、好ましくは5〜30℃の範囲の温度で実施する、請求項1〜12のいずれか一項に記載の方法。
【請求項14】
R1がHまたはメチルであり、R2およびR3がメチルである、請求項1〜13のいずれか一項に記載の方法。
【請求項15】
式I
【化3】
[式中、R1はHまたは線状C1〜6アルキルであり、R2は線状C1〜6アルキルまたは分岐C3〜6アルキルであり、R3は線状C1〜6アルキルであるが、但し、R2およびR3がメチルである場合にはR1はHではないことを条件とする]
の化合物。
【請求項16】
R1=R2=R3=メチルである、請求項15に記載の式Iの化合物。
【請求項17】
式II
【化4】
[式中、R1はHまたは線状C1〜6アルキルであり、R2は線状C1〜6アルキルまたは分岐C3〜6アルキルであり、R3は線状C1〜6アルキルである]
の化合物。
【請求項18】
R1=Hまたはメチルであり、R2=R3=メチルである、請求項17に記載の式IIの化合物。
【請求項19】
式I
【化5】
のケタールを製造するための方法であって、2−プロピン−1−オールを酸触媒の存在下で式III
【化6】
の化合物と反応させるステップを含み、上式において、R1はHまたは線状C1〜6アルキルであり、R2は線状C1〜6アルキルまたは分岐C3〜6アルキルであり、R3は線状C1〜6アルキルである、方法。
【請求項20】
式II
【化7】
のケタールを製造するための方法であって、下記の式Iの化合物を、アンモニアと、リチウムアミド、アルキルリチウムおよびアリールリチウムからなる群から選択されるリチウム化合物との存在下で、式IVの化合物と反応させるステップを含み、以下の式
【化8】
において、R1はHまたは線状C1〜6アルキルであり、R2は線状C1〜6アルキルまたは分岐C3〜6アルキルであり、R3は線状C1〜6アルキルであり、XはCl、BrまたはIである、方法。
【請求項21】
XがBrである、請求項20に記載の方法。
【請求項22】
2−ペンチン−1−オールを製造するための方法であって、プロトン性溶媒中で酸触媒を用いて、式II
【化9】
[式中、R1はHまたは線状C1〜6アルキルであり、R2は線状C1〜6アルキルまたは分岐C3〜6アルキルであり、R3は線状C1〜6アルキルである]
の化合物からヒドロキシ保護基を開裂させて2−ペンチン−1−オールを得るステップを含む、方法。
【請求項23】
R1がHまたはメチルであり、R2およびR3がメチルである、請求項19〜22のいずれか一項に記載の方法。
【図1】

【公表番号】特表2013−501029(P2013−501029A)
【公表日】平成25年1月10日(2013.1.10)
【国際特許分類】
【出願番号】特願2012−523337(P2012−523337)
【出願日】平成22年8月5日(2010.8.5)
【国際出願番号】PCT/EP2010/061411
【国際公開番号】WO2011/015623
【国際公開日】平成23年2月10日(2011.2.10)
【出願人】(503220392)ディーエスエム アイピー アセッツ ビー.ブイ. (873)
【Fターム(参考)】
【公表日】平成25年1月10日(2013.1.10)
【国際特許分類】
【出願日】平成22年8月5日(2010.8.5)
【国際出願番号】PCT/EP2010/061411
【国際公開番号】WO2011/015623
【国際公開日】平成23年2月10日(2011.2.10)
【出願人】(503220392)ディーエスエム アイピー アセッツ ビー.ブイ. (873)
【Fターム(参考)】
[ Back to top ]