
2つのC2領域を有する脳特異的タンパク質
【目的】 2つのC2領域を有する脳特異的タンパク質(Doc2)およびDoc2をコードする遺伝子を提供する。さらに、該遺伝子を有する発現ベクター、該発現ベクターを導入した形質転換体、および該形質転換体を培養することによりDoc2タンパク質を製造する方法も提供する。また、Doc2タンパク質を認識する抗体、該抗体を産生し得るハイブリドーマ、および該抗体を用いるDoc2タンパク質の免疫学的測定方法を提供する。
【構成】 2つのC2領域を有する脳特異的なタンパク質(Doc2)をコードする遺伝子が得られた。この遺伝子を有する発現ベクターを導入した形質転換体を培養することによりDoc2タンパク質が得られる。Doc2はカルシウムイオン依存的にホスホリピッドと結合し得るが、膜貫通領域およびRab3A結合領域を有さないことが見いだされた。このように、Doc2は既知のタンパク質とは異なる構造を有し、このDoc2の機能を解明することにより、神経伝達物質の放出機構がより明らかになり得る。従って、Doc2は脳神経系の疾患の原因究明手段として有用である。
【構成】 2つのC2領域を有する脳特異的なタンパク質(Doc2)をコードする遺伝子が得られた。この遺伝子を有する発現ベクターを導入した形質転換体を培養することによりDoc2タンパク質が得られる。Doc2はカルシウムイオン依存的にホスホリピッドと結合し得るが、膜貫通領域およびRab3A結合領域を有さないことが見いだされた。このように、Doc2は既知のタンパク質とは異なる構造を有し、このDoc2の機能を解明することにより、神経伝達物質の放出機構がより明らかになり得る。従って、Doc2は脳神経系の疾患の原因究明手段として有用である。
【発明の詳細な説明】
【0001】
【産業上の利用分野】本発明は、2つのC2領域を有する脳特異的タンパク質;該脳特異的タンパク質の構造遺伝子;該遺伝子を含む発現ベクター;該発現ベクターを有する形質転換体;該形質転換体を用いた脳特異的タンパク質の製造方法;脳特異的タンパク質に対する抗体;該抗体を産生し得るハイブリドーマ;および該抗体を用いた試料中の脳特異的タンパク質を定量するための免疫学的測定方法に関する。
【0002】
【従来の技術】プロテインキナーゼCは最初、カルシウムイオンおよびホスファチジルセリン存在下にジアシルグリセロールにより活性化されるプロテインキナーゼとして発見された(Y. Takaiら、Biochem. Biophys. Res. Commun.、91、1218-1224 (1979)、およびY. Takaiら、J. Biol. Chem.、254、5049-5052 (1979))。タンパク質の構造解析により、プロテインキナーゼCは、C1、C2、およびC3領域と、V1、V2、V3、およびV4領域とからなり、C1領域はジアシルグリセロールと結合し、C2領域はカルシウムイオンおよびホスファチジルセリンと結合することが明らかになっている(Y. Nishizuka、Nature、334、661-665 (1988))。
【0003】C2領域を有するタンパク質としては、プロテインキナーゼC以外に、ホスホリパーゼA2、ホスホリパーゼCγ、Unc-13、Ras GAP、シナプトタグミン、およびラブフィリン3Aなどが知られている(M.L. Stahlら、Nature、332、269-272 (1988)、U.S. Dixonら、Nature、335、90-93 (1988)、J.D. Clarkら、Cell、65、1043-1051 (1991)、I.N. MaruyamaおよびS. Brenner、Proc. Natl. Acad. Sci. USA、88、5729-5733 (1991)、M.S. Perinら、Nature、345、260-263 (1990)、およびH. Shiratakiら、Mol. Cell Biol.、13、2061-2068 (1993))。このうち、プロテインキナーゼC、ホスホリパーゼA2、Unc-13、ホスホリパーゼC、およびRas GAPは、1つのC2領域を有し、一方、シナプトタグミン、およびラブフィリン3Aは2つのC2領域を有している。これらのタンパク質の中で、プロテインキナーゼC、ホスホリパーゼA2、シナプトタグミン、およびラブフィリン3Aは、無細胞系において、カルシウムイオンおよびホスファチジルセリンと結合することが証明されている。プロテインキナーゼC、ホスホリパーゼA2、ホスホリパーゼCγ、およびRas GAPは膜に存在する受容体からのシグナル伝達において重要な働きをしていることが明らかにされている。また、シナプトタグミンはC2領域の上流に膜貫通領域を有し、シナプス小胞に存在し、神経伝達物質の放出に関与している。ラブフィリン3Aは、低分子量GTP結合タンパク質の1つで神経伝達物質の放出に関与しているRab3Aの標的タンパク質として発見され、膜貫通領域を有さないが、シナプス小胞に存在している。シナプトタグミンおよびラブフィリン3Aは、神経伝達物質の放出において重要な役割を果たしていると考えられている。
【0004】最近、3種類のシナプトタグミン、I、II、およびIIIが発見され、これらがすべて2つのC2領域を有することが明らかとなった(M. Geppertら、J. Biol. Chem.、266、13548-13552 (1991)、およびM. Mizutaら、J. Biol. Chem.、269、11675-11678 (1994))。シナプトタグミンIIは脳に局在しているが、シナプトタグミンIおよびIIIは、脳以外に、副腎髄質などの他の内分泌系組織でも見いだされている。また、ラブフィリン3Aは、脳で大量に存在しているが、内分泌系組織でも少量見いだされている。
【0005】
【発明が解決しようとする課題】本発明の目的は、新たな脳特異的タンパク質、特に2つのC2領域を有し、神経伝達物質の放出に重要な役割を果たすと考えられる、脳特異的タンパク質を提供することにある。さらに本発明の目的は、上記脳特異的タンパク質の構造遺伝子;該遺伝子を含む発現ベクター;該発現ベクターを有する形質転換体;該形質転換体を用いた脳特異的タンパク質の製造方法;該脳特異的タンパク質に対する抗体;該抗体を産生し得るハイブリドーマ;および該抗体を用いた試料中の脳特異的タンパク質を定量するための免疫学的測定方法を提供することにある。
【0006】
【課題を解決するための手段】本発明者らは、C2領域に共通のアミノ酸配列をプライマーとして設定したPCR法を用いて、ラブフィリン3Aに構造が似ている遺伝子を単離することを試みた。その結果、脳特異的に発現している、C2領域を2つ有し、膜貫通領域およびRab3A結合領域を有さない新規遺伝子を発見し、この遺伝子によりコードされるタンパク質をDoc2(Double C2)と命名した。本発明者らは、大腸菌を用いてDoc2を発現し、このタンパク質がRab3Aとは結合しないが、カルシウムイオンおよびホスファチジルセリンと結合することを発見した。さらに、Doc2を特異的に認識する抗体を得て、本発明を完成するに至った。
【0007】本発明の脳特異的タンパク質は、配列表の配列番号1の9位のMetから80位のAspまでのアミノ酸配列を含む。
【0008】好ましい実施態様では、上記脳特異的タンパク質は、2つのC2領域を有する。
【0009】好ましい実施態様では、上記脳特異的タンパク質は、配列表の配列番号1の1位のMetから400位のAlaまでのアミノ酸配列を含む。
【0010】本発明のDNA配列は、上記のいずれかに記載の脳特異的タンパク質をコードする。
【0011】好ましい実施態様では、上記DNA配列は、配列表の配列番号1の149位のAから364位のCまででなる塩基配列を有する。
【0012】好ましい実施態様では、上記DNA配列は、配列表の配列番号1の125位のAから1324位のCまででなる塩基配列を有する。
【0013】本発明の発現ベクターは、上記のいずれかに記載のDNA配列を有する。
【0014】本発明の形質転換体は、上記発現ベクターを宿主に導入して得られる。
【0015】好ましい実施態様では、上記宿主は、大腸菌である。
【0016】本発明の脳特異的タンパク質の製造方法は、上記形質転換体を培養する工程および産生された脳特異的タンパク質を培養培地から回収する工程を包含する。
【0017】本発明の抗体は、上記のいずれかに記載の脳特異的タンパク質を認識する。
【0018】好ましい実施態様では、上記抗体は、配列表の配列番号1の9位のMetから80位のAspまでのアミノ酸配列に含まれる部分を認識する。
【0019】本発明のハイブリドーマは、上記のいずれかに記載の抗体を産生し得る。
【0020】好ましい実施態様では、上記ハイブリドーマは、FERM P-14577である。
【0021】本発明の脳特異的タンパク質の免疫学的測定方法は、上記のいずれかに記載の脳特異的タンパク質を含有し得る試料を、上記のいずれかに記載の抗体とともに、抗原抗体複合体を形成させる条件下でインキュベーションする工程、および該抗原抗体複合体の量を測定する工程を包含する。
【0022】次に本発明を工程の順に説明する。本発明においては、特に指示のない限り、当該分野で公知である組換えDNA法、タンパク質の分離および分析法、および免疫学的手法が採用され得る。
【0023】(1)Doc2タンパク質をコードするDNAの配列決定本発明のDoc2タンパク質をコードするDNAを含むDNA断片の配列決定方法を以下に例示する。このDNA断片の配列は、例えば、ヒト急性リンパ性白血病細胞株、ヒト脳のcDNAライブラリーを以下に示すプローブを用いてスクリーニングし、得られたDNAをDNAシークエンシングにより分析することにより、決定され得る。
【0024】(A)DNAプローブの作製Doc2タンパク質をコードする遺伝子は、例えば、ヒト脳由来のcDNAライブラリーから得ることができる。そのためには、まず、ヒト脳由来のcDNAライブラリーからDoc2タンパク質をコードする遺伝子のクローニングを行うためのプローブが、例えば、次のようにして作製される。
【0025】Doc2が2つのC2領域を有することから、C2領域に保存されたアミノ酸配列をもとに、ポリメラーゼチェーン反応(PCR)用のDNAプライマーを合成し、このプライマーを用いて、以下のようにして調製したPCR用の鋳型を増幅させて、スクリーニング用のプローブとし得る。このPCR用の鋳型としては、例えばDoc2が多く存在していることが考えられるヒト脳から得られるcDNAを用いることができる。このようなDNAは、例えば、ヒト脳から、グアニジンチオシアネート緩衝液を用いてRNAを抽出し、このRNAからcDNAを調製する方法により、得られる。RNAからのcDNAの調製は、まず、プライマーをRNAにアニーリングさせ、逆転写酵素により該プライマーからDNAを合成していくことにより、行われ得る。
【0026】このようにして得られたプローブを標識して、以下のスクリーニングに用いることができる。
【0027】(B)ライブラリーのスクリーニングDoc2をコードするDNAをスクリーニングするためのライブラリーとして、哺乳動物の脳由来またはヒト由来の様々なライブラリーを用い得る。例えば、ヒト脳のpSport1 cDNAライブラリー、ヒト急性リンパ性白血病細胞株Molt-4のpSport1cDNAライブラリー、およびマウス脳のpSport1 cDNAライブラリーが含まれる。
【0028】ライブラリーのスクリーニングは、当該技術分野で公知の方法によって、上記(A)項で得られたプローブを用いて行われ得る。このスクリーニング法には、例えば、組換えファージプラークのプラークハイブリダイゼーションおよび組換え大腸菌のコロニーハイブリダイゼーションが含まれる。
【0029】(C)cDNAの塩基配列の決定上記(B)項で得られた目的の組換えプラスミドの挿入断片の塩基配列の決定は、例えば以下のように行われる。まず、挿入断片を該断片の内部に存在する制限酵素部位を用いて切断し、それぞれのcDNA断片をそれぞれ適当なシークエンスベクター、例えば、pCR II中にサブクローニングする。次にクローニングした断片の塩基配列を、例えば自動DNAシーケンサーを用いて、Sanger法(F. Sangerら、Proc. Natl. Acad. Sci. USA, 74, 5463-5467 (1977))によって、決定し得る。これにより、塩基配列が決定される。
【0030】(2)組換え型Doc2タンパク質の発現本発明のDoc2タンパク質遺伝子は、適当なベクターに組み込まれて、Doc2タンパク質を発現させるための発現ベクターとされる。この発現ベクターを、例えば、細菌、酵母、昆虫細胞、または動物細胞に導入して、形質転換体が作製される。この形質転換体を培養することにより、Doc2タンパク質が産生され得る。
【0031】例えば、本発明のDoc2タンパク質遺伝子を含む発現ベクターを、大腸菌BL21株に導入することによって形質転換体を作製し、この形質転換体を培養することにより、例えば、N末端にオリゴヒスチジンを有するDoc2タンパク質が産生され得る。
【0032】(3)抗Doc2抗体の作製本発明のDoc2タンパク質に対する抗体は、例えば、N末端側の配列表の配列番号1の9位から80位の特異的な72個のアミノ酸配列部分またはこれを含みかつDoc2タンパク質を認識する抗体を産生させ得るタンパク質を免疫原とし、マウス、ラット、ウサギなどの動物を免疫して、その血清由来の抗体(ポリクローナル抗体)を作製し得る。または、免疫した動物の脾臓またはリンパ節から細胞を取り出し、ミエローマ細胞などの細胞と融合させてハイブリドーマを作製した後、該ハイブリドーマからモノクローナル抗体を産生させ得る。
【0033】(4)Doc2タンパク質の免疫学的測定法免疫に用いた免疫原と同一の抗原部位を有する標識した一定量のタンパク質に、濃度既知の非標識抗原、および血清由来のポリクローナル抗体またはモノクローナル抗体を加えて、抗原抗体競合反応を行わせる。非標識抗原の濃度を適当に変化させた後、抗体に結合した標識抗原と抗体に結合していない標識抗原とを適当な方法で分離して、抗体と結合した標識抗原の放射能量、酵素活性、または蛍光強度を測定する。非標識抗原の量が増すにつれ、抗体と結合する標識抗原の量は減少する。この関係をグラフにして標準曲線を得る。
【0034】次に、上記の反応系に濃度既知の非標識抗原の代わりに未知量の抗原を含む試料を加え、これを反応させた後に得られる放射能量、酵素活性、または蛍光強度を、標準曲線にあてはめれば、試料中の抗原の量を知ることができる。
【0035】(5)Doc2タンパク質の組織分布の確認本発明のDoc2タンパク質の組織分布は、例えば、mRNAの発現またはDoc2タンパク質の発現を解析することにより、確認することができる。mRNAの発現については、cDNAを用いてノーザンブロット解析により、また、Doc2タンパク質の発現については、上記(4)項で得た抗体を用いてウエスタンブロット解析により確認し得る。
【0036】(6)Doc2タンパク質のホスホリピッドとの結合の確認本発明のDoc2タンパク質のホスホリピッドへの結合は、例えば以下のようにして確認できる。種々の濃度のカルシウムイオン存在下で、Doc2タンパク質とリポソーム化したホスホリピッドとを反応させた後、リポソーム画分についてウエスタンブロット解析と同様にしてDoc2タンパク質の量を検出することにより、ホスホリピッドと結合したDoc2タンパク質を測定し得る。
【0037】
【実施例】本発明を以下の実施例によりさらに説明する。
【0038】[実施例1]Doc2 cDNAの単離およびその構造決定(1)cDNAライブラリーの作製ヒト急性リンパ性白血病細胞株Molt-4およびヒト脳のpSport1cDNAライブラリーを、GIBCO-BRL社製のcDNA合成システムおよびcDNAクローニングシステムを用いて常法に従って(T. Maniatisら、Molecular Cloning: A Laboratory Manual、第2版、Cold Spring Harbor Laboratory, New York (1989))、以下のように作製した。
【0039】まず、次のように、Molt-4細胞あるいはヒト脳よりpoly(A)+RNAを抽出した。Molt-4細胞109個またはヒト脳100μgを、1mlの4Mグアニジンチオシアネート緩衝液(4Mグアニジンチオシアネート、25mM クエン酸ナトリウム、 pH 7.0、0.5% サルコシル、および0.1M 2-メルカプトエタノール)に加えて溶解した。次に、1mlの水飽和フェノール、および0.2mlのクロロホルム:イソアミルアルコール(49:1)を加えて混和し、氷中で15分間放置した後、TOMY遠心機MRX-150(トミー精工社製)で15000回転で10分間遠心分離した。得られた沈澱物を滅菌蒸留水に溶解し、波長260nmでの吸光度を測定することにより、回収した全RNA量を計算した。Molt-4細胞より1.3mg、そしてヒト脳より1mgの全RNAがそれぞれ得られた。全RNAの100μgを、100μlのOligotex-dT30(日本合成ゴム社製、宝酒造より購入)に、反応緩衝液(10mM Tris-HCl、pH 7.5、1mM EDTA、0.5M NaCl、および0.1% SDS)中で、室温で15分間反応させて結合させた。さらに、滅菌蒸留水中で65℃で5分間反応させた後、溶出して、poly(A)+RNAとした。これを波長260nmでの吸光度を測定することにより、回収したpoly(A)+RNA量を計算した。Molt-4細胞より12μg、そしてヒト脳より9μgのpoly(A)+RNAがそれぞれ得られた。
【0040】次に、精製したpoly(A)+RNAを鋳型としてcDNAの合成を以下のように行った。まず、2μgのpoly(A)+RNAを鋳型として逆転写酵素SUPER SCRIPT II RT(ストラタジーン社製)を用いて、反応緩衝液(50mM Tris-HCl、pH 8.3、75mM KCl、3mMMgCl2、10mM DTT、500μM dNTP(dATP、dGTP、dTTP、dCTP)、50μg/ml NotIプライマー−アダプター、および20,000U/ml 逆転写酵素SUPER SCRIPT II RT)中で、37℃で1時間反応させて1本鎖DNAを合成した。なお、NotIプライマー−アダプターの配列を、配列表の配列番号2に示す。この一本鎖DNAを鋳型として、反応緩衝液(25mM Tris-HCl、pH 7.5、100mM KCl、5mM MgCl2、10mM (NH4)2SO4、0.15mM β-NAD+、250μM dNTP(dATP、dGTP、dTTP、dCTP)、1.2mM DTT、65U/ml DNAリガーゼ、250U/ml DNAポリメラーゼI、および13U/ml RNase H)中で、16℃で2時間反応させてcDNAを合成した。次に、T4 DNAポリメラーゼを、最終濃度が65 U/mlとなるように加えて、さらに16℃で5分間反応させて二本鎖DNAとした。この二本鎖DNAを、反応緩衝液(50mM Tris-HCl、pH 7.6、10mM MgCl2、1mM ATP、5% PEG 8000、1mM DTT、200μg/ml SalIアダプター、および100U/ml T4リガーゼ)中で、16℃で16時間反応させて、SalIアダプターに結合させた。なお、SalIアダプターは、配列表の配列番号3および4に示すDNAがアニーリングした二本鎖であった。こうして完成したcDNAをpSport 1ベクターのNotIとSalI部位の間に挿入して、cDNAライブラリーを作製した。
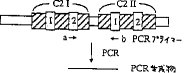
【0041】(2)Doc2 cDNAのクローニングおよび塩基配列の決定まず、C2領域の2カ所の保存されたアミノ酸配列(図1の1および2の部分、それぞれ配列表の配列番号7および8に示す)をコードする、退縮した2つのオリゴヌクレオチドを設計し(それぞれ配列表の配列番号5および6に示す)、DNA合成機(Cyclone Plus DNA Synthesizer、ミリジェン/バイオサーチ社)で合成した。合成にはミリジェン/バイオサーチ社のβ−リンク・ベータシアノエチルホスホアミダイト試薬を用いた。合成後、アンモニア水(28%、ナカライテスク社製)2mlで合成カラムから合成オリゴヌクレオチドを溶出した後、60℃、5時間処理することにより保護基を離脱させた。脱保護したオリゴヌクレオチドを、10倍量のブタノールを加えてTOMY遠心機MRX-101(トミー精工社製)で3000回転で10分間遠心分離することにより沈澱させて回収した。回収したオリゴヌクレオチドを滅菌水に溶解し、波長260nmでの吸光度を測定することによりその量を計算した。最終的に、配列表の配列番号5に示すオリゴヌクレオチドを10μg得た。また、配列表の配列番号6に示すオリゴヌクレオチドを490μg得た。合成したこれらのオリゴヌクレオチドは、それぞれC2領域の保存されている2か所のアミノ酸配列に対応しており、これらのオリゴヌクレオチド用いてPCRを行うことによりC2領域を2つ以上持つ遺伝子のみが増幅されると考えられた。
【0042】図1に示すように、設計したオリゴヌクレオチド(図1のaおよびb、それぞれ配列表の配列番号5および6に示す)をプライマーとして用いて、Molt-4cDNAライブラリーDNAを鋳型としたPCR反応を行った。PCRは宝酒造から購入したAmpliTaq Kit(Perkin-Elmer社製)を用い、宝酒造から購入したDNA Thermal Cycler(Perkin-Elmer社製)で行った。反応は、鋳型DNA(ヒト急性リンパ性白血病細胞株のpSport 1 cDNAライブラリー)0.5μg、AmpliTaq DNAポリメラーゼ 5ユニット、オリゴヌクレオチドをそれぞれ500 pmol、および10XPCR用緩衝液(100mM Tris-HCl、pH 8.3、500mM KCl、15mM MgCl2、0.1%ゼラチン)5μlを加え、さらにdNTP(dATP、dGTP、dTTP、dCTP)を最終濃度が200μMとなるように加え、滅菌蒸留水で最終的に50μlとして行った。反応は、94℃で5分間処理した後、94℃で1分間、45℃で1分間、72℃で2分間の反応サイクルを30回繰り返し、さらに72℃で5分間行った。反応生成物を、2%アガロースゲル(SeaKem Agarose、FMC社製、宝酒造より購入)上で電気泳動した後、エチジウムブロミド(500ng/ml)で5分間染色して確認した。約430bpのバンドが1μg/50μl生成した。反応生成物(430bpの生成物)の回収方法は常法に従って以下のように行った(T. Maniatisら、Molecular Cloning: A Laboratory Manual、第2版、Cold Spring Harbor Laboratory, New York (1989))。反応生成物を6%ポリアクリルアミドゲル上で電気泳動した後、エチジウムブロミド(500ng/ml)で5分間染色し、430bpのバンドをカッターを用いて切り出して、これをマクサム・ギルバート溶出緩衝液(500mM酢酸アンモニウム、10mM MgCl2、1mM EDTA、0.1% SDS)に浸して、37℃で16時間処理した。次に、当量の0.1M Tris-HCl、pH 8.0の飽和フェノール・クロロフォルム(1:1)を加えて攪拌した後、TOMY遠心機MRX-150(トミー精工社製)で12000回転で5分間遠心分離した。その水層を取り出して、2.5倍量のエタノールを加えて、-80℃で15分間処理後、再びTOMY遠心機MRX-150(トミー精工社製)で12000回転で5分間遠心分離してその沈澱物をTE緩衝液(10mM Tris-HCl、pH8.0、1mM EDTA)に溶解した。そして、波長260nmでの吸光度を測定することにより回収した430bpのバンドのDNA量を計算した。最終的に0.4μgのDNAが得られた。得られた430bpのDNA断片をInvitrogen社製のTAクローニングベクター(pCR II)に挿入し、塩基配列を決定した。塩基配列の決定は、Sanger法により、Pharmacia社のT7 Sequencing Kitを用いて行った(F. Sangerら、Proc. Natl. Acad. Sci. USA, 74, 5463-5467 (1977))。この結果、得られたPCR生成物の塩基配列が、ラブフィリン3Aと類似性があることが明らかとなった。
【0043】次に、完全長のDNAを得るために、430bpのDNA断片をマルチプライムDNA標識システム(アマシャム・ジャパン社製)により32Pで標識し、これをプローブとして、ヒト脳cDNAライブラリーをコロニーハイブリダイゼーションによりスクリーニングした。コロニーハイブリダイゼーションは常法に従って以下のように行った(T. Maniatisら、Molecular Cloning: A Laboratory Manual、第2版、ColdSpring Harbor Laboratory, New York (1989))。ヒト脳cDNAライブラリーの入った大腸菌DH5αをLB-プレート(10gトリプトン、5gイーストエキストラクト、10g NaCl、15gアガー/1L蒸留水)に播き、一晩37℃で培養した。プレートに生えた大腸菌のコロニーをナイロン膜(Hybond-N+、アマシャム・ジャパン社製)に転写した後、SDS処理(10% SDS)、アルカリ変性(0.5M NaOH、1.5M NaCl)、そして洗浄(2XSSC)の操作を行った。この膜を、32P標識430bp DNA断片をプローブとしてハイブリダイゼーションした。ハイブリダイゼーション溶液は、6XSSC(1XSSCは、0.15M NaClおよび0.015Mクエン酸ナトリウムからなる)、5Xデンハート溶液、0.5% SDS、50%ホルムアミド、および100μg/mlサケ精子DNAを用い、42℃で一晩ハイブリダイゼーションを行った。フィルターの洗浄は、2XSSC、0.5% SDSで室温10分間、1XSSC、0.5% SDSで65℃で30分間を2回、さらに0.1XSSC、0.5% SDSで65℃で30分間を2回行った後、X線フィルム(Kodak社製)に感光させ、これをフィルム現像して、プローブと反応するコロニーを同定した。最終的に2つのcDNAクローンが得られ、塩基配列の決定は、Sanger法により、Pharmacia社のT7 Sequencing Kitを用いて行った。
【0044】得られたクローンの塩基配列および内部に翻訳終止コドンを持たないオープンリーデングフレームのアミノ酸配列を配列表の配列番号1に示す。このオープンリーデングフレームの上流および下流には翻訳終止コドンが存在することより、このクローンは完全長の翻訳領域を持っていることが証明された。この遺伝子は400個のアミノ酸よりなるオープンリーデングフレームを有し、2つのC2領域を有する遺伝子であることが明らかとなった。この400個のアミノ酸をDoc2(Double C2)と命名した。この400個のアミノ酸からなるタンパク質の分子量は、計算によると44,071であった。
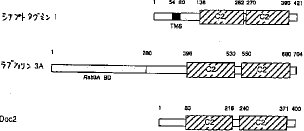
【0045】Doc2の類似性解析は、IDEASプログラム(M. I. Kanehisa、Nucleic Acids Res.、10、183-196 (1982))を用いて行い、その結果に基づく模式図を図2R>2に示す。このタンパク質のC2領域の320個のアミノ酸は、ラブフィリン3AのC2領域と61%の類似性があり、シナプトタグミンのC2領域と37%の類似性があることが明らかとなった。また、Doc2は、シナプトタグミンに存在する膜貫通領域(図2のTMS)およびラブフィリン3Aに存在するRab3A結合領域(図2のRab3A BD)を含んでいなかった。さらに、Doc2は、N末端側に配列表の配列番号1の9位から80位の72個のアミノ酸からなる、他のタンパク質と類似性のない特異的領域を有することも明らかになった。
【0046】[実施例2]組換えDoc2の発現および精製(1)組換えDoc2N末端タンパク質の発現および精製Doc2に特異的なアミノ末端側の72アミノ酸(Doc2N末端タンパク質)を持つ断片を作製するために、発現ベクターpGEX-2T-Doc2-Nを以下の方法で構築した。両端にBamHIおよびKpnI部位を持ち、翻訳開始コドンより9個目のMet以下72アミノ酸を含み、その下流に翻訳終了コドンを持つ0.21KbのDNA断片を、Doc2cDNAを鋳型としてPCR法により得た。用いた2つのオリゴヌクレオチドの配列を配列表の配列番号9および10に示す。PCRは、Ampli Taq Kit(Perkin-Elmer社製)を用い、DNA Thermal Cycler(Perkin-Elmer社製)で行った。反応は、鋳型DNA(ヒト脳よりクローニングしたDoc2のcDNAの全長1.7kbが組み込まれたpSport 1ベクター)0.5μg、Ampli Taq DNAポリメラーゼ 5ユニット、オリゴヌクレオチドをそれぞれ100pmol、および10XPCR用緩衝液(100mM Tris-HCl、pH 8.3、500mM KCl、15mM MgCl2、0.1%ゼラチン)5μlを加え、さらにdNTP(dATP、dGTP、dTTP、dCTP)を最終濃度が200μMとなるように加え、滅菌蒸留水で最終的に50μlとして行った。反応は、94℃で5分間処理した後、94℃で1分間、50℃で1分間、72℃で2分間の反応サイクルを30回繰り返し、さらに72℃で5分間行った。反応生成物を6%ポリアクリルアミドゲル上で電気泳動した後、エチジウムブロミド(500ng/ml)で5分間染色し、210bpのバンドをカッターを用いて切り出し、これをマクサム・ギルバート溶出緩衝液(500mM酢酸アンモニウム、10mM MgCl2、1mM EDTA、0.1% SDS)に浸して、37℃で16時間処理した。等量の0.1M Tris-HCl、pH 8.0飽和フェノール・クロロフォルム(1:1)を加えて攪拌した後、TOMY遠心機MRX-150(トミー精工社製)で12000回転で5分間遠心分離後、その水層を取り出し、2.5倍量のエタノールを加えて、-80℃で15分間処理後、再びTOMY遠心機MRX-150(トミー精工社製)で12000回転で5分間遠心分離してその沈澱物をTE緩衝液(10mM Tris-HCl、pH 8.0、1mM EDTA)に溶解した。そして、波長260nmでの吸光度を測定することにより、回収した210bpのバンドのDNA量を計算した。最終的に0.8μgのDNAが得られた。このDNA断片をBamHIで消化後、pGEX-2T(Pharmacia社製)のBamHI部位に挿入し、発現ベクターpGEX-2T-Doc2-Nを得た。
【0047】発現ベクターpGEX-2T-Doc2-Nを導入した大腸菌DH5α株を用いて、Doc2のアミノ末端側の72アミノ酸を、グルタチオン-S-トランスフェラーゼとの融合タンパク質として発現させた。大腸菌DH5αの培養は、LB培地(10g トリプトン、5g イーストエキストラクト、10g NaCl/1L蒸留水)で行い、Doc2N末端タンパク質の発現誘導は、0.1mM IPTG(イソプロピル-β-D-チオガラクトピラノシド)を加えて、30℃で3時間培養することにより行った。発現誘導後の大腸菌を、PBS(-)(8gNaCl、0.2g KCl、2.9g Na2HPO4、0.2g KH2PO4/1L蒸留水)に懸濁して超音波処理を行った。次いで、100,000xgで1時間、4℃の条件で遠心分離し、その上清をグルタチオンセファロースカラム(Pharmacia社製)にかけた。Doc2N末端タンパク質の結合したカラムをPBSで洗浄後、溶出緩衝液(20mM グルタチオン、50mM Tris-HCl(pH 8.0))にて、Doc2N末端タンパク質を溶出し精製した。
【0048】(2)組換えDoc2の発現および精製発現ベクターpRset-Doc2は以下の方法で構築した。両端にBamHIおよびKpnIサイトを持ち、翻訳開始コドンから翻訳終了コドンまでを含む、1.2kbのDNA断片をDoc2cDNAを鋳型としてPCR法により得た。PCRに用いた2つのオリゴヌクレオチドの配列を、配列表の配列番号11および12に示す。PCRは、AmpliTaq Kit(Perkin-Elmer社製)を用い、DNA Thermal Cycler(Perkin-Elmer社製)で行った。反応は、鋳型DNA(ヒト脳よりクローニングしたDoc2cDNAの全長1.7kbが組み込まれたpSport 1ベクター)0.5μg、Ampli Taq DNAポリメラーゼ 5ユニット、オリゴヌクレオチドをそれぞれ100pmol、および10XPCR用緩衝液(100mM Tris-HCl、pH 8.3、500mM KCl、15mM MgCl2、0.1%ゼラチン)5μlを加え、さらにdNTP(dATP、dGTP、dTTP、dCTP)を最終濃度が200μMになるように加え、滅菌蒸留水で最終的に50μlとして行った。反応は、94℃で5分間処理後、94℃で1分間、50℃で1分間、72℃で2分間の反応サイクルを30回繰り返し、さらに72℃で5分間行った。反応生成物は、4%ポリアクリルアミドゲル上で電気泳動した後、エチジウムブロミド(500ng/ml)で5分間染色し、1.2Kbのバンドをカッターを用いて切り出し、これをマクサム・ギルバート溶出緩衝液(500mM 酢酸アンモニウム、10mM MgCl2、1mM EDTA、0.1% SDS)に浸して、37℃で16時間処理した。等量の0.1M Tris-HCl、pH 8.0飽和フェノール・クロロフォルム(1:1)を加えて攪拌した後、TOMY遠心機MRXー150(トミー精工社製)で12000回転で5分間遠心分離後、その水層を取り出し、2.5倍量のエタノールを加えて、-80℃で15分処理後、再びTOMY遠心機MRX-150(トミー精工社製)で12000回転で5分間遠心分離して、その沈澱物をTE緩衝液(10mM Tris-HCl、pH 8.0、1mM EDTA)に溶解した。そして、波長260nmでの吸光度を測定することにより、回収した1.2KbのバンドのDNA量を計算した。最終的に0.2μgのDNAが得られた。このDNA断片をKpnIで消化後、pRset(Invitorogen社製)のKpnI部位に挿入し、発現ベクターpRset-Doc2を得た。
【0049】発現ベクターpRset-Doc2を導入した大腸菌BL2l株を用いて、オリゴヒスチジンをアミノ末端に持つ融合タンパク質(F.W. StudierおよびB.A. Moffatt、J. Mol. Biol., 189, 113-130 (1986))としてDoc2を発現させた。大腸菌BL2l株の培養はLB培地で行い、30℃で3時間培養した。その後大腸菌をリン酸緩衝液(20mM リン酸ナトリウム、500mM NaCl、pH 7.8)に懸濁し、超音波処理を行った。次いで、100,000xg、1時間、4℃の条件で遠心分離し、その上清を、プレーボンドレジン(Invitrogen)にかけた。Doc2タンパク質の結合したカラムをリン酸緩衝液(20mM リン酸ナトリウム、500mM NaCl、pH 6.0)で洗浄後、溶出緩衝液(500mMイミダゾール、20mM リン酸ナトリウム、500mM NaCl、pH 6.0)にて、Doc2タンパク質を溶出した。溶出したDoc2タンパク質を、20mM HEPES(pH 7.4)、1mM DTTを用いて透析した後、モノQカラム(0.5×5cm)(Pharmacia社製)にかけ、A緩衝液(20mM HEPES(pH 7.4)、1mM DTT、1%コール酸)およびB緩衝液(20mM HEPES (pH 7.4)、1mM DTT、1M NaCl、1%コール酸)を用いたNaClの塩濃度の勾配により溶出し、Doc2タンパク質を含むフラクションを集めて最終的な精製品を得た。
【0050】[実施例3]ノーザンブロット解析によるDoc2 mRNAの組織分布いくつかのヒト組織より単離したpoly(A)+ RNA 2μgを、アガロースゲル電気泳動にかけ、ナイロン膜に移したものをClonetech社より購入した。この膜を、マルチプライムDNA標識システム(アマシャム・ジャパン社製)により32Pで標識したDoc2DNA(翻訳開始点より25から989番目の塩基を標識)をプローブとして、ハイブリダイゼーションを行った。ハイブリダイゼーション溶液は、5XSSPE(1XSSPEは、0.18M NaCl、0.01M リン酸ナトリウム、pH 7.5、1mM EDTAよりなる)、10Xデンハート溶液、2% SDS、50%ホルムアミド、100μg/mlサケ精子DNAを用い、42℃で一晩ハイブリダイゼーションを行った。フィルターの洗浄は2XSSC、0.5% SDSで室温で10分間、1XSSC、0.5% SDCで65℃で30分間を2回、さらに0.1XSSC、0.5% SDSで65℃で30分間を2回行った後、X線フィルム(Kodak社製)に感光させ、それを現像して解析した。
【0051】結果を図3に示す。各レーンは、それぞれ、1:心臓、2:脳、3:胎盤、4:肺、5:肝臓、6:骨格筋、7:腎臓、8:膵臓、9:脾臓、10:胸腺、11:前立腺、12:精巣、13:卵巣、14:小腸、15:大腸、および16:末梢血リンパ球であり、RNA分子量マーカーの位置をキロベース(kb)で示した。脳において、Doc2のmRNAである2.2kbの大きさのmRNAが大量に発現しているが、他の部位にはほとんど検出されないことが明らかとなった。また、2.2kbのmRNAは、Molt-4のmRNAを用いたノーザンブロット解析でも検出された。
【0052】[実施例4]抗Doc2ポリクローナル抗体の作製実施例2で得たPBS中の精製Doc2N末端タンパク質(72個のアミノ酸)およびグルタチオン-S-トランスフェラーゼの融合タンパク質を、等量のフロイント完全アジュバント(DIFCO LABORATORIES社製、和光純薬より購入)と混ぜ、コンジュゲートとしてウサギ(日本白色種、雄)の皮下に、3週間おきに3回免疫し、最終免疫から1週間後に頸動脈より全採血して抗血清を得た。1回目の免疫では融合タンパク質1mgを用い、2回目の免疫では500μg、そして3回目の免疫では10μgを用いた。
【0053】抗体価のチェックは、実施例2で得たオリゴヒスチジンをアミノ末端に有するDoc2タンパク質を用いて以下のように行った。種々の量のDoc2タンパク質(0.1ng−100ng)をSDS緩衝液(60mM Tris-HCl、pH 6.8、2% SDS、10% グリセロール、5% 2-メルカプトエタノール、0.001%ブロモフェノールブルー)に溶解し、100℃で5分間反応させてSDS化した後、SDS-PAGEで電気泳動した。SDS-PAGEからPVDF膜(クリアブロットメンブレン-P、アトー社製)にトランスファー緩衝液(0.025M Tris-HCl、pH 8.3、0.192M グリシン、5%メタノール)を用いて電気的に移した膜を、20%牛胎児血清を含むPBS緩衝液中で、500倍希釈したウサギ抗血清とともに、室温で1時間反応した。その後、1% Tween 20(ポリオキシエチレンソルビタンモノラウレート、ナカライテスク社製)を含むPBSを用いて、室温で3回洗浄した。次に、これを、20%牛胎児血清を含むPBS緩衝液中で、1000倍希釈した西洋ワサビペルオキシダーゼ(HRP)に結合したプロテインG(ZYMED LABORATORIES社製)と、室温で1時間反応した。その後、1%Tween 20を含むPBSを用いて、室温で3回洗浄した。洗浄後、ECLウエスタンブロッティング検出システム(アマシャム・ジャパン社製)を用いて発色反応を行い、X線フィルム(Kodak社製)に感光させ、現像されたDoc2タンパク質のバンドを解析した。
【0054】上記抗血清を、常法に従い(T. Maniatisら、Molecular Cloning: A Laboratory Manual、第2版、Cold Spring Harbor Laboratory, New York (1989))、グルタチオン-S-トランスフェラーゼ(GST)結合セファロース4Bカラムにかけて、抗GTS抗体を除いた後、免疫に用いた融合タンパク質結合セファロース4BカラムにかけてDoc2に対する抗体を精製した。詳細には、以下の方法により精製した。カップリング緩衝液(0.1M NaHCO3、0.5M NaCl)に溶解したDoc2N末端側の72アミノ酸とGSTとの融合タンパク質、あるいはGST 10mgと、1mM HClで膨潤させた2gの臭化シアン活性化セファロース4Bカラム(Pharmacia社製)とを、カップリング緩衝液中で、室温で2時間反応させた。その後、カップリング緩衝液を除き、ブロッキング緩衝液(1M Tris-HCl、pH 8.0)中で室温で2時間反応させた。その後、洗浄緩衝液(0.1M 酢酸ナトリウム、pH 4.0、0.5M NaCl)で洗浄後、再び、カップリング緩衝液に懸濁し、カラムに充填し、10mM Tris-HCl(pH 7.5)で平衡化した。6mlのウサギ抗血清を9mlの10mM Tris-HCl(pH 7.5)と混ぜ、まずGST結合カラムにかけて、素通りしてきた画分を、Doc2N末端側の72アミノ酸とGSTとの融合タンパク質結合カラムにかけた。このカラムを、20mlの10mM Tris-HCl(pH 7.5)および20mlの10mM Tris-HCl、500mM NaCl(pH 7.5)で洗浄後、7mlの溶出緩衝液(100mMグリシン、pH 2.5)で溶出した。この溶出液に0.7mlの10mM Tris-HCl(pH 8.5)を加えたものを、抗Doc2ポリクローナル抗体として用いた。
【0055】[実施例5]抗Doc2モノクローナル抗体の作製実施例2で得たPBS中の精製したDoc2N末端側の72アミノ酸とGSTとの融合タンパク質(実施例4のポリクローナル抗体の作製に用いた抗原と同じもの)50μgを、最終濃度が0.25μg/μlとなるようにフロイント完全アジュバントとコンジュゲートして、3匹のマウス(Balb/c、雌)の足蹠に各50μlずつ2または3日ごとに3回接種した。最後の接種の翌日に全てのマウスから後リンパ節を摘出し、リンパ細胞をマウスミエローマ細胞Sp2と10:1の割合で混合して50%PEGを含むRPMI1640培地に加えることにより細胞融合を行った。4枚の96ウェルプレート中のHAT培地(10%FCS、0.1mM ヒポキサンチンナトリウム、0.4μM アミノプテリン、16μM チミジンを含むRPMI1640培地)に、約100細胞/ウェルになるように細胞を播いた。3日後に培養上清100μlを同量のHT培地(10%FCS、0.1mM ヒポキサンチンナトリウム、16μM チミジンを含むRPMI1640培地)と交換し、以後4日ごとに上清を同様に10%FCS、10%Origen(ボクスイブラウン)を含むRPMI1640培地と交換し維持継代した。培養開始16日目に培養上清について、GSTタンパク質、および、オリゴヒスチジンをN末端に持つDoc2タンパク質(His-Doc2タンパク質)に対するELISA試験を以下のように行った。96ウェルプレート(Maxisorb、Nunc)に抗原を0.5または1μg/ウェル入れ、37℃で30分間保存しコートした。次に抗原を捨てブロック溶液(10%FCSを含むPBS)を加え4℃で一晩ブロックした。ブロック後ブロック溶液を捨て、試験するハイブリドーマの培養上清を加えて37℃で30分間反応した。その後プレートをプレート洗浄器(Nunc)で洗浄し、ブロック液で希釈したHRP結合抗マウス免疫グロブリン抗体(Dako)を加えて37℃で30分反応した。そしてプレートをプレート洗浄器で洗浄しABTS試薬によりHRP活性を450nmの吸光度として求めた。その結果、24ウェルがGSTタンパク質に対して陰性およびHis-Doc2タンパク質に対して陽性であった。これらをHis-Doc2タンパク質に対してウエスタンブロットし、最も力価の高かった4E11を選択した。
【0056】次に、このウェルのハイブリドーマをクローン化するために、細胞を10-1細胞/ウェルとなるように限界希釈して96ウェルプレートに播き、ELISA試験によりGSTに対して陰性およびDoc2タンパク質に対して陽性を示すシングルコロニーのウェル(D3)を選択した。このハイブリドーマの培養上清が、His-Doc2タンパク質に対して特異的に反応することをウエスタンブロットにより確認した。このハイブリドーマは、工業技術院生命工学研究所に平成6年10月7日付で寄託され、受託番号FERM P-14577が付与されている。得られたハイブリドーマD3をRPMI培地に懸濁し(2×107細胞/ml)、1匹あたり500μlずつ、11日前にプリスタンを腹腔内に投与しておいたマウス(Balb/c、雌)5匹の腹腔内に投与し1週間後にマウスから腹水を回収した。回収した腹水は2,000rpm10分間遠心分離し、その上清を抗Doc2モノクローナル抗体とした。
【0057】[実施例6]ウエスタンブロット解析実施例4および5で作製したDoc2N末端側の72アミノ酸に対するポリクローナル抗体およびモノクローナル抗体を用いて、ラット組織でのDoc2タンパク質の発現をウエスタンブロットにより以下のように検討した。
【0058】ラットの種々の組織を、SDS緩衝液(60mM Tris-HCl、pH 6.8、2%SDS、10%グリセロール、5% 2-メルカプトエタノール、0.001%ブロモフェノールブルー)に溶解し、100℃で5分間反応させSDS化した後、SDS-PAGEで電気泳動した。SDS-PAGEからPVDF膜(クリアブロットメンブレン-P、アトー社製)にトランスファー緩衝液を用いて電気的に移し、この膜を、20%牛胎児血清を含むPBS緩衝液中で、1000倍希釈した抗Doc2ポリクローナル抗体あるいは2000倍希釈した抗Doc2モノクローナル抗体と、室温で1時間反応した。その後、1% Tween 20を含むPBSを用いて、室温で3回洗浄した。次に、20%牛胎児血清を含むPBS緩衝液中で、一次抗体が抗Doc2ポリクローナル抗体の場合は2000倍希釈したHRP結合プロテインG(ZYMED LABORATORIES社製)と、一次抗体が抗Doc2モノクローナル抗体の場合は5000倍希釈したHRP結合抗マウスイムノグロブリンヒツジポリクローナル抗体(アマシャム・ジャパン社製)と、それぞれ室温で1時間反応させた。その後、1% Tween 20の入ったPBSを用いて、室温で3回洗浄した。洗浄後、ECLウエスタンブロッティング検出システム(アマシャム・ジャパン社製)を用いて発色反応を行い、X線フィルム(Kodak社製)に感光させ、現像したDoc2タンパク質のバンドを解析した。
【0059】抗Doc2ポリクローナル抗体を用いた場合のウエスタンブロットの結果を図4に示す。図中の各レーンは、それぞれ、1:脳、2:心臓、3:肺、4:肝臓、5:腎臓、6:骨格筋、7:脾臓、8:胸腺、9:小腸、および10:大腸であり、タンパク質分子量マーカーの位置をキロダルトン(kD)で示した。分子量46kDのバンドが脳において検出されたが、他の臓器では検出されなかった。
【0060】抗Doc2モノクローナル抗体を用いた場合のウエスタンブロットの結果を図5に示す。図中の各レーンは、それぞれ、1:心臓、2:脳、3:小腸、4:肺、5:肝臓、6:骨格筋、7:腎臓、8:副腎髄質、9:脾臓、および10:胸腺であり、タンパク質分子量マーカーの位置をキロダルトン(kD)で示した。その結果、ポリクローナル抗体を用いた場合と同様に、分子量46kDのバンドが脳において検出されたが、他の臓器では検出されなかった。
【0061】[実施例7]Doc2とホスホリピッドとの結合解析C2領域を有するDoc2とホスホリピッド(ホスファチジルセリンおよびホスファチジルコリン)との結合解析は報告されている方法に従って行った(T. Yamaguchiら、J. Biol. Chem.、268、27164-27170 (1993))。ホスファチジルセリンおよびホスファチジルコリンはフナコシより購入した。リポソームを作製するために、ホスホリピッドを窒素ガスを用いてフイルム状に乾燥し、20mM HEPES/NaOH(pH 7.4)、150mM NaCl緩衝液に溶解した後、100,000×gで20分間、4℃にて遠心分離した。沈澱を再び同じ緩衝液に溶解した。実施例2で得た組換えHis-Doc2タンパク質20pmolを、上記のリポソームとともに、種々の濃度のカルシウムイオンを含む溶液100μl(100μgリポソーム、20mM HEPES/NaOH(pH 7.4)、150mM NaCl、10-8〜10-4Mの種々の濃度のカルシウムイオン)中で、60分間、4℃で反応させた。反応後、100,000×gで20分間、4℃にて遠心分離し、上清および沈澱を、SDS緩衝液(60mM Tris-HCl、pH 6.8、2%SDS、10%グリセロール、5% 2-メルカプトエタノール、0.001%ブロモフェノールブルー)に溶解し、100℃で5分間反応してSDS化した後、SDS-PAGEで電気泳動した。SDS-PAGEからPVDF膜に電気的に移した膜を、20%牛胎児血清を含むPBS緩衝液中で、2000倍希釈した抗Doc2モノクローナル抗体と、室温で1時間反応させた。その後、1% Tween 20を含むPBSを用いて、室温で3回洗浄した。次に、20%牛胎児血清を含むPBS緩衝液中で、2000倍希釈したHRP結合抗マウスイムノグロブリンヒツジポリクローナル抗体(アマシャム・ジャパン社製)と、室温で1時間反応させた。その後、1% Tween 20を含むPBSを用いて、室温で3回洗浄した。洗浄後、ECLウエスタンブロッティング検出システム(アマシャム・ジャパン社製)を用いて発色反応を行い、X線フィルム(Kodak社製)に感光させ、現像したDoc2タンパク質のバンドを解析した。
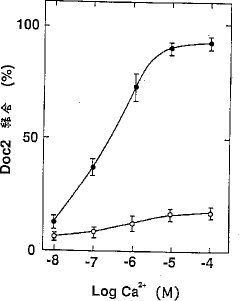
【0062】結果を図6に示す。図中の黒丸はホスファチジルセリン、および白丸はホスファチジルコリンで作製したリポソームとの結合の結果をそれぞれ示している。組換えHis-Doc2タンパク質はカルシウムイオン存在下にホスホリピッドと結合した。ホスファチジルセリンで作製したリポソームへのDoc2の結合はカルシウムイオン濃度に依存的であり、カルシウムイオンの濃度が5x10-7Mの時、半量のDoc2がリポソームへ結合した。ホスファチジルコリンでリポソームを作製した場合、Doc2はリポソームにほとんど結合しなかった。また、データには示さないが、組換えDoc2タンパク質はGTPγS-結合型のRab3Aとは結合しなかった。
【0063】
【発明の効果】本発明によれば、2つのC2領域を有する脳特異的タンパク質であるDoc2タンパク質;Doc2タンパク質の構造遺伝子;該遺伝子を含む発現ベクター;該発現ベクターを有する形質転換体;該形質転換体を用いたDoc2タンパク質の製造方法;Doc2タンパク質に対する抗体;該抗体を産生し得るハイブリドーマ;および該抗体を用いた試料中のDoc2タンパク質を定量するための免疫学的測定方法が提供される。
【0064】神経伝達物質の放出に関与するシナプトタグミンおよびラブフィリン3AのC2領域は、カルシウムイオンおよびホスホリピッド(特にホスファチジルセリン)と結合することが知られており、同様に、組換えDoc2タンパク質もカルシウムイオン依存的にホスホリピッド(特にホスファチジルセリン)と結合する。従って、Doc2は、神経伝達物質の放出に作用すると考えられる。しかし、シナプトタグミンおよびラブフィリン3Aと異なり、Doc2は、シナプトタグミンに存在する膜貫通領域およびラブフィリン3Aに存在するRab3A結合領域を有さず、一方、Doc2はそのアミノ末端側に72個のアミノ酸よりなる、他のタンパク質と類似性のないユニークな領域を有している。このようなシナプトタグミンおよびラブフィリン3Aとの構造上の類似性から考えて、Doc2のアミノ末端領域も、何らかの細胞内構成成分、例えば、ある種のタンパク質を介してシナプス小胞と結合していることが予想され、Doc2はカルシウムセンサーとして働き、プレシナプスからの神経伝達物質の放出に関与していることが予想される。このことは、プレシナプス膜に電気刺激が伝達された際、アクティブゾーンのカルシウムイオン濃度は、電位依存性のカルシウムチャンネルからのカルシウムイオンの流入により、1×10-8Mから1×10-4Mまで上昇すること(S.J. SmithおよびG.J. Augustine、Trends Neurosci.、11、458-464 (1988))から類推され得る。従って、本発明のDoc2は、その機能の解明により、神経伝達物質の放出機構を解明するのに有用である。また、本発明のDoc2遺伝子および抗Doc2抗体は、Doc2の遺伝子変異およびそのmRNAおよびタンパク質の発現状態を解析するのに有用であり、脳神経系の疾患の原因究明の新たな手段を提供し得、脳神経系の疾患の診断または治療方法の開発に有用である。
【0065】
【配列表】
【0066】
【配列番号:1】
配列の長さ:1718配列の型:核酸鎖の数:二本鎖トポロジー:直鎖状配列の種類:cDNA to mRNA起源生物名:ヒト配列の特徴特徴を表す記号:CDS存在位置:125..1324特徴を決定した方法:S配列CCACGCGTCC GGTCCTGACG GCGCTGGAGC TGAGGGGCAG TGCGGATGCC CCAGGAAGGC 60TCCTAGGAAG AGGGGACCCA CGGTGACTTC CTAAGGAAGC GCGGTTCCCA GCCAGGGGTG 120CTGC ATG AGG GGC CGC AGG GGC GAT CGC ATG ACC ATC AAC ATC CAG GAG 169 Met Arg Gly Arg Arg Gly Asp Arg Met Thr Ile Asn Ile Gln Glu 1 5 10 15CAC ATG GCC ATC AAC GTG TGC CCC GGG CCC ATC CGG CCC ATC CGC CAG 217His Met Ala Ile Asn Val Cys Pro Gly Pro Ile Arg Pro Ile Arg Gln 20 25 30ATC TCT GAC TAC TTC CCC CGG GGA CCA GGA CCT GAA GGG GGC GGC GGG 265Ile Ser Asp Tyr Phe Pro Arg Gly Pro Gly Pro Glu Gly Gly Gly Gly 35 40 45AGC GGC GGG GAG GCC CCC GCC CAT CTG GTC CCC CTG GCT CTG GCC CCC 313Ser Gly Gly Glu Ala Pro Ala His Leu Val Pro Leu Ala Leu Ala Pro 50 55 60CCT GCA GCC CTC CTT GGG GCC ACC ACG CCT GAG GAT GGT GCG GAG GTG 361Pro Ala Ala Leu Leu Gly Ala Thr Thr Pro Glu Asp Gly Ala Glu Val 65 70 75GAC AGC TAT GAC TCG GAT GAT GCC ACC GCC CTA GGC AAG CTG GAG TTT 409Asp Ser Tyr Asp Ser Asp Asp Ala Thr Ala Leu Gly Lys Leu Glu Phe 80 85 90 95GAC CTT CTC TAC GAC CGG GCC TCC TGC ACT CTG CAC GTA TGC ATC CTC 457Asp Leu Leu Tyr Asp Arg Ala Ser Cys Thr Leu His Val Cys Ile Leu 100 105 110AGG GCC AAG GGC CTC AAG CCC ATG GAT TTC AAT GGC CTC GCC GAC CCC 505Arg Ala Lys Gly Leu Lys Pro Met Asp Phe Asn Gly Leu Ala Asp Pro 115 120 125TAC GTC AAG CTG CAC TTG CTG CCT GGA GCC TGT AAG GCC AAT AAG CTA 553Tyr Val Lys Leu His Leu Leu Pro Gly Ala Cys Lys Ala Asn Lys Leu 130 135 140AAA ACG AAG ACT CAG AGG AAC ACA CTG AAT CCC GTG TGG AAT GAG GAC 601Lys Thr Lys Thr Gln Arg Asn Thr Leu Asn Pro Val Trp Asn Glu Asp 145 150 155CTG ACT TAC AGC GGG ATC ACA GAT GAC GAC ATC ACG CAC AAG GTG CTC 649Leu Thr Tyr Ser Gly Ile Thr Asp Asp Asp Ile Thr His Lys Val Leu 160 165 170 175AGG ATC GCC GTC TGT GAT GAG GAC AAG CTG AGT CAC AAT GAG TTT ATT 697Arg Ile Ala Val Cys Asp Glu Asp Lys Leu Ser His Asn Glu Phe Ile 180 185 190GGG GAG ATC CGC GTG CCC CTC CGC CGC CTC AAG CCT TCG CAG AAG AAG 745Gly Glu Ile Arg Val Pro Leu Arg Arg Leu Lys Pro Ser Gln Lys Lys 195 200 205CAT TTT AAC ATC TGC CTC GAG CGC CAA GTC CCG CTG GCG TCC CCC TCT 793His Phe Asn Ile Cys Leu Glu Arg Gln Val Pro Leu Ala Ser Pro Ser 210 215 220TCC ATG TCA GCG GCG CTG AGG GGC ATC TCC TGT TAT CTG AAG GAC TTG 841Ser Met Ser Ala Ala Leu Arg Gly Ile Ser Cys Tyr Leu Lys Asp Leu 225 230 235GAG CAG GCG GAG CAG GGG CAG GGG CTG CTG GAG GAG CGT GGC CGC ATC 889Glu Gln Ala Glu Gln Gly Gln Gly Leu Leu Glu Glu Arg Gly Arg Ile 240 245 250 255CTG CTG AGT CTC AGC TAC AGC TCG CGG CGC CGG GGA CTG CTG GTA GGC 937Leu Leu Ser Leu Ser Tyr Ser Ser Arg Arg Arg Gly Leu Leu Val Gly 260 265 270ATC TTG CGC TGC GCC CAT CTG GCT GCC ATG GAC GTC AAC GGT TAC TCG 985Ile Leu Arg Cys Ala His Leu Ala Ala Met Asp Val Asn Gly Tyr Ser 275 280 285GAC CCC TAC GTC AAG ACG TAC CTG AGG CCC GAT GTG GAC AAG AAA TCC 1033Asp Pro Tyr Val Lys Thr Tyr Leu Arg Pro Asp Val Asp Lys Lys Ser 290 295 300AAG CAT AAG ACG TGT GTG AAG AAG AAG ACT CTC AAC CCA GAA TTT AAC 1081Lys His Lys Thr Cys Val Lys Lys Lys Thr Leu Asn Pro Glu Phe Asn 305 310 315GAG GAG TTT TTC TAC GAG ATA GAG CTC TCC ACT CTG GCC ACC AAG ACC 1129Glu Glu Phe Phe Tyr Glu Ile Glu Leu Ser Thr Leu Ala Thr Lys Thr 320 325 330 335CTG GAA GTC ACC GTC TGG GAC TAT GAC ATT GGC AAA TCC AAT GAC TTC 1177Leu Glu Val Thr Val Trp Asp Tyr Asp Ile Gly Lys Ser Asn Asp Phe 340 345 350ATT GGT GGC GTG TCC CTG GGG CCA GGT GCC CGA GGC GAG GCT CGG AAG 1225Ile Gly Gly Val Ser Leu Gly Pro Gly Ala Arg Gly Glu Ala Arg Lys 355 360 365CAC TGG AGT GAC TGC CTG CAG CAG CGG GAC GCA GCC CTG GAG CGC TGG 1273His Trp Ser Asp Cys Leu Gln Gln Arg Asp Ala Ala Leu Glu Arg Trp 370 375 380CAC ACC CTG ACC AGT GAG CTG CCC CCT GCG GCC GGG GCT CTG TCC TCA 1321His Thr Leu Thr Ser Glu Leu Pro Pro Ala Ala Gly Ala Leu Ser Ser 385 390 395GCC TGAGTGGACA GCAGTGTCCC GGCACAGGCC CATCGAGCCG GGTCCAGTAC 1374Ala 400CCAACCTTCG CACGAGTGTG TTGCACGTTT ACACAGGTGG GCTGCCCCAC CCTGCACTAC 1434CTATTTTGTG AGTCTCGTGA CCCGGGTCTG TCTGCTCATG AGGGGCTGCG GAGTTCTATA 1494TTCACATATG CAAACCTCCT GCCTGACTCG CTAGTCCCTG CAAATATGCA AACCCCCCTA 1554CTACTGCACA CCCGGGCAGT GCTCAGAGCC GCCCAGGCCC CGCGCTCCTC ACTCCTGCCT 1614CTCCACGCTG CCCCGTCCCT CTCCCCCAAC AGGGAGGAGG TCGGATTAGG GAGGTTCAGA 1674GGAGGAGAAT GTCTCAAAAA AAAAAAAAAA AAAAAAAAAA AAAA 1718
【0067】
【配列番号:2】
配列の長さ:45配列の型:核酸鎖の数:一本鎖トポロジー:直鎖状配列の種類:他の核酸 合成DNA配列GATTAGTTCT AGATCGCGAC GCGGCCGCCC TTTTTTTTTT TTTTT 45
【0068】
【配列番号:3】
配列の長さ:16配列の型:核酸鎖の数:一本鎖トポロジー:直鎖状配列の種類:他の核酸 合成DNA配列TCGACCCACG CGTCCG 16
【0069】
【配列番号:4】
配列の長さ:12配列の型:核酸鎖の数:一本鎖トポロジー:直鎖状配列の種類:他の核酸 合成DNA配列CGGACGCGTG GG 12
【0070】
【配列番号:5】
配列の長さ:23配列の型:核酸鎖の数:一本鎖トポロジー:直鎖状配列の種類:他の核酸 合成DNA配列の特徴他の情報:Nはイノシンである。
配列TTNACRWANG GRTCNSWNDD NCC 23
【0071】
【配列番号:6】
配列の長さ:23配列の型:核酸鎖の数:一本鎖トポロジー:直鎖状配列の種類:他の核酸 合成DNA配列の特徴他の情報:Nはイノシンである。
配列ACNYTNAAYC CNNDNTDWAA YGA 23
【0072】
【配列番号:7】
配列の長さ:8配列の型:アミノ酸鎖の数:一本鎖トポロジー:直鎖状配列の種類:ペプチド配列の特徴他の情報:2位のXaaは任意のアミノ酸、6位のXaa破GluまたはTyrである。

【0073】
【配列番号:8】
配列の長さ:8配列の型:アミノ酸鎖の数:一本鎖トポロジー:直鎖状配列の種類:ペプチド配列の特徴他の情報:5位のXaaは任意のアミノ酸、6位のXaaはPhe、Tyr、またはTrpである。
【0074】
【配列番号:9】
配列の長さ:36配列の型:核酸鎖の数:一本鎖トポロジー:直鎖状配列の種類:他の核酸 合成DNA配列CATGGATCCG GTACCATGAC CATCAACATC CAGGAG 36
【0075】
【配列番号:10】
配列の長さ:38配列の型:核酸鎖の数:一本鎖トポロジー:直鎖状配列の種類:他の核酸 合成DNA配列CATGGATCCG GTACCTCAAT AGCTGTCCAC CTCCGCAC 38
【0076】
【配列番号:11】
配列の長さ:36配列の型:核酸鎖の数:一本鎖トポロジー:直鎖状配列の種類:他の核酸 合成DNA配列CATGGATCCG GTACCATGAG GGGCCGCAGG GGCGAT 36
【0077】
【配列番号:12】
配列の長さ:36配列の型:核酸鎖の数:一本鎖トポロジー:直鎖状配列の種類:他の核酸 合成DNA配列CATGGATCCG GTACCTCAGG CTGAGGACAG AGCCCC 36
【図面の簡単な説明】
【図1】C2領域を2つ有する遺伝子を得るためのプライマーの設計を示す図である。
【図2】本発明のDoc2タンパク質と、ラブフィリン3AおよびシナプトタグミンIとのアミノ酸配列の比較の構造の模式図である。
【図3】種々のヒト組織におけるDoc2 mRNAの発現のノーザンブロット解析の結果を示す図である。
【図4】抗Doc2ポリクローナル抗体を用いた、種々のラット組織におけるDoc2タンパク質の発現のウエスタンブロット解析の結果を示す図である。
【図5】抗Doc2モノクローナル抗体を用いた、種々のラット組織におけるDoc2タンパク質の発現のウエスタンブロット解析の結果を示す図である。
【図6】本発明のDoc2タンパク質の、種々の濃度のカルシウムイオン存在下における、ホスホリピッドからなるリポソームへの結合能を示す図である。
【0001】
【産業上の利用分野】本発明は、2つのC2領域を有する脳特異的タンパク質;該脳特異的タンパク質の構造遺伝子;該遺伝子を含む発現ベクター;該発現ベクターを有する形質転換体;該形質転換体を用いた脳特異的タンパク質の製造方法;脳特異的タンパク質に対する抗体;該抗体を産生し得るハイブリドーマ;および該抗体を用いた試料中の脳特異的タンパク質を定量するための免疫学的測定方法に関する。
【0002】
【従来の技術】プロテインキナーゼCは最初、カルシウムイオンおよびホスファチジルセリン存在下にジアシルグリセロールにより活性化されるプロテインキナーゼとして発見された(Y. Takaiら、Biochem. Biophys. Res. Commun.、91、1218-1224 (1979)、およびY. Takaiら、J. Biol. Chem.、254、5049-5052 (1979))。タンパク質の構造解析により、プロテインキナーゼCは、C1、C2、およびC3領域と、V1、V2、V3、およびV4領域とからなり、C1領域はジアシルグリセロールと結合し、C2領域はカルシウムイオンおよびホスファチジルセリンと結合することが明らかになっている(Y. Nishizuka、Nature、334、661-665 (1988))。
【0003】C2領域を有するタンパク質としては、プロテインキナーゼC以外に、ホスホリパーゼA2、ホスホリパーゼCγ、Unc-13、Ras GAP、シナプトタグミン、およびラブフィリン3Aなどが知られている(M.L. Stahlら、Nature、332、269-272 (1988)、U.S. Dixonら、Nature、335、90-93 (1988)、J.D. Clarkら、Cell、65、1043-1051 (1991)、I.N. MaruyamaおよびS. Brenner、Proc. Natl. Acad. Sci. USA、88、5729-5733 (1991)、M.S. Perinら、Nature、345、260-263 (1990)、およびH. Shiratakiら、Mol. Cell Biol.、13、2061-2068 (1993))。このうち、プロテインキナーゼC、ホスホリパーゼA2、Unc-13、ホスホリパーゼC、およびRas GAPは、1つのC2領域を有し、一方、シナプトタグミン、およびラブフィリン3Aは2つのC2領域を有している。これらのタンパク質の中で、プロテインキナーゼC、ホスホリパーゼA2、シナプトタグミン、およびラブフィリン3Aは、無細胞系において、カルシウムイオンおよびホスファチジルセリンと結合することが証明されている。プロテインキナーゼC、ホスホリパーゼA2、ホスホリパーゼCγ、およびRas GAPは膜に存在する受容体からのシグナル伝達において重要な働きをしていることが明らかにされている。また、シナプトタグミンはC2領域の上流に膜貫通領域を有し、シナプス小胞に存在し、神経伝達物質の放出に関与している。ラブフィリン3Aは、低分子量GTP結合タンパク質の1つで神経伝達物質の放出に関与しているRab3Aの標的タンパク質として発見され、膜貫通領域を有さないが、シナプス小胞に存在している。シナプトタグミンおよびラブフィリン3Aは、神経伝達物質の放出において重要な役割を果たしていると考えられている。
【0004】最近、3種類のシナプトタグミン、I、II、およびIIIが発見され、これらがすべて2つのC2領域を有することが明らかとなった(M. Geppertら、J. Biol. Chem.、266、13548-13552 (1991)、およびM. Mizutaら、J. Biol. Chem.、269、11675-11678 (1994))。シナプトタグミンIIは脳に局在しているが、シナプトタグミンIおよびIIIは、脳以外に、副腎髄質などの他の内分泌系組織でも見いだされている。また、ラブフィリン3Aは、脳で大量に存在しているが、内分泌系組織でも少量見いだされている。
【0005】
【発明が解決しようとする課題】本発明の目的は、新たな脳特異的タンパク質、特に2つのC2領域を有し、神経伝達物質の放出に重要な役割を果たすと考えられる、脳特異的タンパク質を提供することにある。さらに本発明の目的は、上記脳特異的タンパク質の構造遺伝子;該遺伝子を含む発現ベクター;該発現ベクターを有する形質転換体;該形質転換体を用いた脳特異的タンパク質の製造方法;該脳特異的タンパク質に対する抗体;該抗体を産生し得るハイブリドーマ;および該抗体を用いた試料中の脳特異的タンパク質を定量するための免疫学的測定方法を提供することにある。
【0006】
【課題を解決するための手段】本発明者らは、C2領域に共通のアミノ酸配列をプライマーとして設定したPCR法を用いて、ラブフィリン3Aに構造が似ている遺伝子を単離することを試みた。その結果、脳特異的に発現している、C2領域を2つ有し、膜貫通領域およびRab3A結合領域を有さない新規遺伝子を発見し、この遺伝子によりコードされるタンパク質をDoc2(Double C2)と命名した。本発明者らは、大腸菌を用いてDoc2を発現し、このタンパク質がRab3Aとは結合しないが、カルシウムイオンおよびホスファチジルセリンと結合することを発見した。さらに、Doc2を特異的に認識する抗体を得て、本発明を完成するに至った。
【0007】本発明の脳特異的タンパク質は、配列表の配列番号1の9位のMetから80位のAspまでのアミノ酸配列を含む。
【0008】好ましい実施態様では、上記脳特異的タンパク質は、2つのC2領域を有する。
【0009】好ましい実施態様では、上記脳特異的タンパク質は、配列表の配列番号1の1位のMetから400位のAlaまでのアミノ酸配列を含む。
【0010】本発明のDNA配列は、上記のいずれかに記載の脳特異的タンパク質をコードする。
【0011】好ましい実施態様では、上記DNA配列は、配列表の配列番号1の149位のAから364位のCまででなる塩基配列を有する。
【0012】好ましい実施態様では、上記DNA配列は、配列表の配列番号1の125位のAから1324位のCまででなる塩基配列を有する。
【0013】本発明の発現ベクターは、上記のいずれかに記載のDNA配列を有する。
【0014】本発明の形質転換体は、上記発現ベクターを宿主に導入して得られる。
【0015】好ましい実施態様では、上記宿主は、大腸菌である。
【0016】本発明の脳特異的タンパク質の製造方法は、上記形質転換体を培養する工程および産生された脳特異的タンパク質を培養培地から回収する工程を包含する。
【0017】本発明の抗体は、上記のいずれかに記載の脳特異的タンパク質を認識する。
【0018】好ましい実施態様では、上記抗体は、配列表の配列番号1の9位のMetから80位のAspまでのアミノ酸配列に含まれる部分を認識する。
【0019】本発明のハイブリドーマは、上記のいずれかに記載の抗体を産生し得る。
【0020】好ましい実施態様では、上記ハイブリドーマは、FERM P-14577である。
【0021】本発明の脳特異的タンパク質の免疫学的測定方法は、上記のいずれかに記載の脳特異的タンパク質を含有し得る試料を、上記のいずれかに記載の抗体とともに、抗原抗体複合体を形成させる条件下でインキュベーションする工程、および該抗原抗体複合体の量を測定する工程を包含する。
【0022】次に本発明を工程の順に説明する。本発明においては、特に指示のない限り、当該分野で公知である組換えDNA法、タンパク質の分離および分析法、および免疫学的手法が採用され得る。
【0023】(1)Doc2タンパク質をコードするDNAの配列決定本発明のDoc2タンパク質をコードするDNAを含むDNA断片の配列決定方法を以下に例示する。このDNA断片の配列は、例えば、ヒト急性リンパ性白血病細胞株、ヒト脳のcDNAライブラリーを以下に示すプローブを用いてスクリーニングし、得られたDNAをDNAシークエンシングにより分析することにより、決定され得る。
【0024】(A)DNAプローブの作製Doc2タンパク質をコードする遺伝子は、例えば、ヒト脳由来のcDNAライブラリーから得ることができる。そのためには、まず、ヒト脳由来のcDNAライブラリーからDoc2タンパク質をコードする遺伝子のクローニングを行うためのプローブが、例えば、次のようにして作製される。
【0025】Doc2が2つのC2領域を有することから、C2領域に保存されたアミノ酸配列をもとに、ポリメラーゼチェーン反応(PCR)用のDNAプライマーを合成し、このプライマーを用いて、以下のようにして調製したPCR用の鋳型を増幅させて、スクリーニング用のプローブとし得る。このPCR用の鋳型としては、例えばDoc2が多く存在していることが考えられるヒト脳から得られるcDNAを用いることができる。このようなDNAは、例えば、ヒト脳から、グアニジンチオシアネート緩衝液を用いてRNAを抽出し、このRNAからcDNAを調製する方法により、得られる。RNAからのcDNAの調製は、まず、プライマーをRNAにアニーリングさせ、逆転写酵素により該プライマーからDNAを合成していくことにより、行われ得る。
【0026】このようにして得られたプローブを標識して、以下のスクリーニングに用いることができる。
【0027】(B)ライブラリーのスクリーニングDoc2をコードするDNAをスクリーニングするためのライブラリーとして、哺乳動物の脳由来またはヒト由来の様々なライブラリーを用い得る。例えば、ヒト脳のpSport1 cDNAライブラリー、ヒト急性リンパ性白血病細胞株Molt-4のpSport1cDNAライブラリー、およびマウス脳のpSport1 cDNAライブラリーが含まれる。
【0028】ライブラリーのスクリーニングは、当該技術分野で公知の方法によって、上記(A)項で得られたプローブを用いて行われ得る。このスクリーニング法には、例えば、組換えファージプラークのプラークハイブリダイゼーションおよび組換え大腸菌のコロニーハイブリダイゼーションが含まれる。
【0029】(C)cDNAの塩基配列の決定上記(B)項で得られた目的の組換えプラスミドの挿入断片の塩基配列の決定は、例えば以下のように行われる。まず、挿入断片を該断片の内部に存在する制限酵素部位を用いて切断し、それぞれのcDNA断片をそれぞれ適当なシークエンスベクター、例えば、pCR II中にサブクローニングする。次にクローニングした断片の塩基配列を、例えば自動DNAシーケンサーを用いて、Sanger法(F. Sangerら、Proc. Natl. Acad. Sci. USA, 74, 5463-5467 (1977))によって、決定し得る。これにより、塩基配列が決定される。
【0030】(2)組換え型Doc2タンパク質の発現本発明のDoc2タンパク質遺伝子は、適当なベクターに組み込まれて、Doc2タンパク質を発現させるための発現ベクターとされる。この発現ベクターを、例えば、細菌、酵母、昆虫細胞、または動物細胞に導入して、形質転換体が作製される。この形質転換体を培養することにより、Doc2タンパク質が産生され得る。
【0031】例えば、本発明のDoc2タンパク質遺伝子を含む発現ベクターを、大腸菌BL21株に導入することによって形質転換体を作製し、この形質転換体を培養することにより、例えば、N末端にオリゴヒスチジンを有するDoc2タンパク質が産生され得る。
【0032】(3)抗Doc2抗体の作製本発明のDoc2タンパク質に対する抗体は、例えば、N末端側の配列表の配列番号1の9位から80位の特異的な72個のアミノ酸配列部分またはこれを含みかつDoc2タンパク質を認識する抗体を産生させ得るタンパク質を免疫原とし、マウス、ラット、ウサギなどの動物を免疫して、その血清由来の抗体(ポリクローナル抗体)を作製し得る。または、免疫した動物の脾臓またはリンパ節から細胞を取り出し、ミエローマ細胞などの細胞と融合させてハイブリドーマを作製した後、該ハイブリドーマからモノクローナル抗体を産生させ得る。
【0033】(4)Doc2タンパク質の免疫学的測定法免疫に用いた免疫原と同一の抗原部位を有する標識した一定量のタンパク質に、濃度既知の非標識抗原、および血清由来のポリクローナル抗体またはモノクローナル抗体を加えて、抗原抗体競合反応を行わせる。非標識抗原の濃度を適当に変化させた後、抗体に結合した標識抗原と抗体に結合していない標識抗原とを適当な方法で分離して、抗体と結合した標識抗原の放射能量、酵素活性、または蛍光強度を測定する。非標識抗原の量が増すにつれ、抗体と結合する標識抗原の量は減少する。この関係をグラフにして標準曲線を得る。
【0034】次に、上記の反応系に濃度既知の非標識抗原の代わりに未知量の抗原を含む試料を加え、これを反応させた後に得られる放射能量、酵素活性、または蛍光強度を、標準曲線にあてはめれば、試料中の抗原の量を知ることができる。
【0035】(5)Doc2タンパク質の組織分布の確認本発明のDoc2タンパク質の組織分布は、例えば、mRNAの発現またはDoc2タンパク質の発現を解析することにより、確認することができる。mRNAの発現については、cDNAを用いてノーザンブロット解析により、また、Doc2タンパク質の発現については、上記(4)項で得た抗体を用いてウエスタンブロット解析により確認し得る。
【0036】(6)Doc2タンパク質のホスホリピッドとの結合の確認本発明のDoc2タンパク質のホスホリピッドへの結合は、例えば以下のようにして確認できる。種々の濃度のカルシウムイオン存在下で、Doc2タンパク質とリポソーム化したホスホリピッドとを反応させた後、リポソーム画分についてウエスタンブロット解析と同様にしてDoc2タンパク質の量を検出することにより、ホスホリピッドと結合したDoc2タンパク質を測定し得る。
【0037】
【実施例】本発明を以下の実施例によりさらに説明する。
【0038】[実施例1]Doc2 cDNAの単離およびその構造決定(1)cDNAライブラリーの作製ヒト急性リンパ性白血病細胞株Molt-4およびヒト脳のpSport1cDNAライブラリーを、GIBCO-BRL社製のcDNA合成システムおよびcDNAクローニングシステムを用いて常法に従って(T. Maniatisら、Molecular Cloning: A Laboratory Manual、第2版、Cold Spring Harbor Laboratory, New York (1989))、以下のように作製した。
【0039】まず、次のように、Molt-4細胞あるいはヒト脳よりpoly(A)+RNAを抽出した。Molt-4細胞109個またはヒト脳100μgを、1mlの4Mグアニジンチオシアネート緩衝液(4Mグアニジンチオシアネート、25mM クエン酸ナトリウム、 pH 7.0、0.5% サルコシル、および0.1M 2-メルカプトエタノール)に加えて溶解した。次に、1mlの水飽和フェノール、および0.2mlのクロロホルム:イソアミルアルコール(49:1)を加えて混和し、氷中で15分間放置した後、TOMY遠心機MRX-150(トミー精工社製)で15000回転で10分間遠心分離した。得られた沈澱物を滅菌蒸留水に溶解し、波長260nmでの吸光度を測定することにより、回収した全RNA量を計算した。Molt-4細胞より1.3mg、そしてヒト脳より1mgの全RNAがそれぞれ得られた。全RNAの100μgを、100μlのOligotex-dT30(日本合成ゴム社製、宝酒造より購入)に、反応緩衝液(10mM Tris-HCl、pH 7.5、1mM EDTA、0.5M NaCl、および0.1% SDS)中で、室温で15分間反応させて結合させた。さらに、滅菌蒸留水中で65℃で5分間反応させた後、溶出して、poly(A)+RNAとした。これを波長260nmでの吸光度を測定することにより、回収したpoly(A)+RNA量を計算した。Molt-4細胞より12μg、そしてヒト脳より9μgのpoly(A)+RNAがそれぞれ得られた。
【0040】次に、精製したpoly(A)+RNAを鋳型としてcDNAの合成を以下のように行った。まず、2μgのpoly(A)+RNAを鋳型として逆転写酵素SUPER SCRIPT II RT(ストラタジーン社製)を用いて、反応緩衝液(50mM Tris-HCl、pH 8.3、75mM KCl、3mMMgCl2、10mM DTT、500μM dNTP(dATP、dGTP、dTTP、dCTP)、50μg/ml NotIプライマー−アダプター、および20,000U/ml 逆転写酵素SUPER SCRIPT II RT)中で、37℃で1時間反応させて1本鎖DNAを合成した。なお、NotIプライマー−アダプターの配列を、配列表の配列番号2に示す。この一本鎖DNAを鋳型として、反応緩衝液(25mM Tris-HCl、pH 7.5、100mM KCl、5mM MgCl2、10mM (NH4)2SO4、0.15mM β-NAD+、250μM dNTP(dATP、dGTP、dTTP、dCTP)、1.2mM DTT、65U/ml DNAリガーゼ、250U/ml DNAポリメラーゼI、および13U/ml RNase H)中で、16℃で2時間反応させてcDNAを合成した。次に、T4 DNAポリメラーゼを、最終濃度が65 U/mlとなるように加えて、さらに16℃で5分間反応させて二本鎖DNAとした。この二本鎖DNAを、反応緩衝液(50mM Tris-HCl、pH 7.6、10mM MgCl2、1mM ATP、5% PEG 8000、1mM DTT、200μg/ml SalIアダプター、および100U/ml T4リガーゼ)中で、16℃で16時間反応させて、SalIアダプターに結合させた。なお、SalIアダプターは、配列表の配列番号3および4に示すDNAがアニーリングした二本鎖であった。こうして完成したcDNAをpSport 1ベクターのNotIとSalI部位の間に挿入して、cDNAライブラリーを作製した。
【0041】(2)Doc2 cDNAのクローニングおよび塩基配列の決定まず、C2領域の2カ所の保存されたアミノ酸配列(図1の1および2の部分、それぞれ配列表の配列番号7および8に示す)をコードする、退縮した2つのオリゴヌクレオチドを設計し(それぞれ配列表の配列番号5および6に示す)、DNA合成機(Cyclone Plus DNA Synthesizer、ミリジェン/バイオサーチ社)で合成した。合成にはミリジェン/バイオサーチ社のβ−リンク・ベータシアノエチルホスホアミダイト試薬を用いた。合成後、アンモニア水(28%、ナカライテスク社製)2mlで合成カラムから合成オリゴヌクレオチドを溶出した後、60℃、5時間処理することにより保護基を離脱させた。脱保護したオリゴヌクレオチドを、10倍量のブタノールを加えてTOMY遠心機MRX-101(トミー精工社製)で3000回転で10分間遠心分離することにより沈澱させて回収した。回収したオリゴヌクレオチドを滅菌水に溶解し、波長260nmでの吸光度を測定することによりその量を計算した。最終的に、配列表の配列番号5に示すオリゴヌクレオチドを10μg得た。また、配列表の配列番号6に示すオリゴヌクレオチドを490μg得た。合成したこれらのオリゴヌクレオチドは、それぞれC2領域の保存されている2か所のアミノ酸配列に対応しており、これらのオリゴヌクレオチド用いてPCRを行うことによりC2領域を2つ以上持つ遺伝子のみが増幅されると考えられた。
【0042】図1に示すように、設計したオリゴヌクレオチド(図1のaおよびb、それぞれ配列表の配列番号5および6に示す)をプライマーとして用いて、Molt-4cDNAライブラリーDNAを鋳型としたPCR反応を行った。PCRは宝酒造から購入したAmpliTaq Kit(Perkin-Elmer社製)を用い、宝酒造から購入したDNA Thermal Cycler(Perkin-Elmer社製)で行った。反応は、鋳型DNA(ヒト急性リンパ性白血病細胞株のpSport 1 cDNAライブラリー)0.5μg、AmpliTaq DNAポリメラーゼ 5ユニット、オリゴヌクレオチドをそれぞれ500 pmol、および10XPCR用緩衝液(100mM Tris-HCl、pH 8.3、500mM KCl、15mM MgCl2、0.1%ゼラチン)5μlを加え、さらにdNTP(dATP、dGTP、dTTP、dCTP)を最終濃度が200μMとなるように加え、滅菌蒸留水で最終的に50μlとして行った。反応は、94℃で5分間処理した後、94℃で1分間、45℃で1分間、72℃で2分間の反応サイクルを30回繰り返し、さらに72℃で5分間行った。反応生成物を、2%アガロースゲル(SeaKem Agarose、FMC社製、宝酒造より購入)上で電気泳動した後、エチジウムブロミド(500ng/ml)で5分間染色して確認した。約430bpのバンドが1μg/50μl生成した。反応生成物(430bpの生成物)の回収方法は常法に従って以下のように行った(T. Maniatisら、Molecular Cloning: A Laboratory Manual、第2版、Cold Spring Harbor Laboratory, New York (1989))。反応生成物を6%ポリアクリルアミドゲル上で電気泳動した後、エチジウムブロミド(500ng/ml)で5分間染色し、430bpのバンドをカッターを用いて切り出して、これをマクサム・ギルバート溶出緩衝液(500mM酢酸アンモニウム、10mM MgCl2、1mM EDTA、0.1% SDS)に浸して、37℃で16時間処理した。次に、当量の0.1M Tris-HCl、pH 8.0の飽和フェノール・クロロフォルム(1:1)を加えて攪拌した後、TOMY遠心機MRX-150(トミー精工社製)で12000回転で5分間遠心分離した。その水層を取り出して、2.5倍量のエタノールを加えて、-80℃で15分間処理後、再びTOMY遠心機MRX-150(トミー精工社製)で12000回転で5分間遠心分離してその沈澱物をTE緩衝液(10mM Tris-HCl、pH8.0、1mM EDTA)に溶解した。そして、波長260nmでの吸光度を測定することにより回収した430bpのバンドのDNA量を計算した。最終的に0.4μgのDNAが得られた。得られた430bpのDNA断片をInvitrogen社製のTAクローニングベクター(pCR II)に挿入し、塩基配列を決定した。塩基配列の決定は、Sanger法により、Pharmacia社のT7 Sequencing Kitを用いて行った(F. Sangerら、Proc. Natl. Acad. Sci. USA, 74, 5463-5467 (1977))。この結果、得られたPCR生成物の塩基配列が、ラブフィリン3Aと類似性があることが明らかとなった。
【0043】次に、完全長のDNAを得るために、430bpのDNA断片をマルチプライムDNA標識システム(アマシャム・ジャパン社製)により32Pで標識し、これをプローブとして、ヒト脳cDNAライブラリーをコロニーハイブリダイゼーションによりスクリーニングした。コロニーハイブリダイゼーションは常法に従って以下のように行った(T. Maniatisら、Molecular Cloning: A Laboratory Manual、第2版、ColdSpring Harbor Laboratory, New York (1989))。ヒト脳cDNAライブラリーの入った大腸菌DH5αをLB-プレート(10gトリプトン、5gイーストエキストラクト、10g NaCl、15gアガー/1L蒸留水)に播き、一晩37℃で培養した。プレートに生えた大腸菌のコロニーをナイロン膜(Hybond-N+、アマシャム・ジャパン社製)に転写した後、SDS処理(10% SDS)、アルカリ変性(0.5M NaOH、1.5M NaCl)、そして洗浄(2XSSC)の操作を行った。この膜を、32P標識430bp DNA断片をプローブとしてハイブリダイゼーションした。ハイブリダイゼーション溶液は、6XSSC(1XSSCは、0.15M NaClおよび0.015Mクエン酸ナトリウムからなる)、5Xデンハート溶液、0.5% SDS、50%ホルムアミド、および100μg/mlサケ精子DNAを用い、42℃で一晩ハイブリダイゼーションを行った。フィルターの洗浄は、2XSSC、0.5% SDSで室温10分間、1XSSC、0.5% SDSで65℃で30分間を2回、さらに0.1XSSC、0.5% SDSで65℃で30分間を2回行った後、X線フィルム(Kodak社製)に感光させ、これをフィルム現像して、プローブと反応するコロニーを同定した。最終的に2つのcDNAクローンが得られ、塩基配列の決定は、Sanger法により、Pharmacia社のT7 Sequencing Kitを用いて行った。
【0044】得られたクローンの塩基配列および内部に翻訳終止コドンを持たないオープンリーデングフレームのアミノ酸配列を配列表の配列番号1に示す。このオープンリーデングフレームの上流および下流には翻訳終止コドンが存在することより、このクローンは完全長の翻訳領域を持っていることが証明された。この遺伝子は400個のアミノ酸よりなるオープンリーデングフレームを有し、2つのC2領域を有する遺伝子であることが明らかとなった。この400個のアミノ酸をDoc2(Double C2)と命名した。この400個のアミノ酸からなるタンパク質の分子量は、計算によると44,071であった。
【0045】Doc2の類似性解析は、IDEASプログラム(M. I. Kanehisa、Nucleic Acids Res.、10、183-196 (1982))を用いて行い、その結果に基づく模式図を図2R>2に示す。このタンパク質のC2領域の320個のアミノ酸は、ラブフィリン3AのC2領域と61%の類似性があり、シナプトタグミンのC2領域と37%の類似性があることが明らかとなった。また、Doc2は、シナプトタグミンに存在する膜貫通領域(図2のTMS)およびラブフィリン3Aに存在するRab3A結合領域(図2のRab3A BD)を含んでいなかった。さらに、Doc2は、N末端側に配列表の配列番号1の9位から80位の72個のアミノ酸からなる、他のタンパク質と類似性のない特異的領域を有することも明らかになった。
【0046】[実施例2]組換えDoc2の発現および精製(1)組換えDoc2N末端タンパク質の発現および精製Doc2に特異的なアミノ末端側の72アミノ酸(Doc2N末端タンパク質)を持つ断片を作製するために、発現ベクターpGEX-2T-Doc2-Nを以下の方法で構築した。両端にBamHIおよびKpnI部位を持ち、翻訳開始コドンより9個目のMet以下72アミノ酸を含み、その下流に翻訳終了コドンを持つ0.21KbのDNA断片を、Doc2cDNAを鋳型としてPCR法により得た。用いた2つのオリゴヌクレオチドの配列を配列表の配列番号9および10に示す。PCRは、Ampli Taq Kit(Perkin-Elmer社製)を用い、DNA Thermal Cycler(Perkin-Elmer社製)で行った。反応は、鋳型DNA(ヒト脳よりクローニングしたDoc2のcDNAの全長1.7kbが組み込まれたpSport 1ベクター)0.5μg、Ampli Taq DNAポリメラーゼ 5ユニット、オリゴヌクレオチドをそれぞれ100pmol、および10XPCR用緩衝液(100mM Tris-HCl、pH 8.3、500mM KCl、15mM MgCl2、0.1%ゼラチン)5μlを加え、さらにdNTP(dATP、dGTP、dTTP、dCTP)を最終濃度が200μMとなるように加え、滅菌蒸留水で最終的に50μlとして行った。反応は、94℃で5分間処理した後、94℃で1分間、50℃で1分間、72℃で2分間の反応サイクルを30回繰り返し、さらに72℃で5分間行った。反応生成物を6%ポリアクリルアミドゲル上で電気泳動した後、エチジウムブロミド(500ng/ml)で5分間染色し、210bpのバンドをカッターを用いて切り出し、これをマクサム・ギルバート溶出緩衝液(500mM酢酸アンモニウム、10mM MgCl2、1mM EDTA、0.1% SDS)に浸して、37℃で16時間処理した。等量の0.1M Tris-HCl、pH 8.0飽和フェノール・クロロフォルム(1:1)を加えて攪拌した後、TOMY遠心機MRX-150(トミー精工社製)で12000回転で5分間遠心分離後、その水層を取り出し、2.5倍量のエタノールを加えて、-80℃で15分間処理後、再びTOMY遠心機MRX-150(トミー精工社製)で12000回転で5分間遠心分離してその沈澱物をTE緩衝液(10mM Tris-HCl、pH 8.0、1mM EDTA)に溶解した。そして、波長260nmでの吸光度を測定することにより、回収した210bpのバンドのDNA量を計算した。最終的に0.8μgのDNAが得られた。このDNA断片をBamHIで消化後、pGEX-2T(Pharmacia社製)のBamHI部位に挿入し、発現ベクターpGEX-2T-Doc2-Nを得た。
【0047】発現ベクターpGEX-2T-Doc2-Nを導入した大腸菌DH5α株を用いて、Doc2のアミノ末端側の72アミノ酸を、グルタチオン-S-トランスフェラーゼとの融合タンパク質として発現させた。大腸菌DH5αの培養は、LB培地(10g トリプトン、5g イーストエキストラクト、10g NaCl/1L蒸留水)で行い、Doc2N末端タンパク質の発現誘導は、0.1mM IPTG(イソプロピル-β-D-チオガラクトピラノシド)を加えて、30℃で3時間培養することにより行った。発現誘導後の大腸菌を、PBS(-)(8gNaCl、0.2g KCl、2.9g Na2HPO4、0.2g KH2PO4/1L蒸留水)に懸濁して超音波処理を行った。次いで、100,000xgで1時間、4℃の条件で遠心分離し、その上清をグルタチオンセファロースカラム(Pharmacia社製)にかけた。Doc2N末端タンパク質の結合したカラムをPBSで洗浄後、溶出緩衝液(20mM グルタチオン、50mM Tris-HCl(pH 8.0))にて、Doc2N末端タンパク質を溶出し精製した。
【0048】(2)組換えDoc2の発現および精製発現ベクターpRset-Doc2は以下の方法で構築した。両端にBamHIおよびKpnIサイトを持ち、翻訳開始コドンから翻訳終了コドンまでを含む、1.2kbのDNA断片をDoc2cDNAを鋳型としてPCR法により得た。PCRに用いた2つのオリゴヌクレオチドの配列を、配列表の配列番号11および12に示す。PCRは、AmpliTaq Kit(Perkin-Elmer社製)を用い、DNA Thermal Cycler(Perkin-Elmer社製)で行った。反応は、鋳型DNA(ヒト脳よりクローニングしたDoc2cDNAの全長1.7kbが組み込まれたpSport 1ベクター)0.5μg、Ampli Taq DNAポリメラーゼ 5ユニット、オリゴヌクレオチドをそれぞれ100pmol、および10XPCR用緩衝液(100mM Tris-HCl、pH 8.3、500mM KCl、15mM MgCl2、0.1%ゼラチン)5μlを加え、さらにdNTP(dATP、dGTP、dTTP、dCTP)を最終濃度が200μMになるように加え、滅菌蒸留水で最終的に50μlとして行った。反応は、94℃で5分間処理後、94℃で1分間、50℃で1分間、72℃で2分間の反応サイクルを30回繰り返し、さらに72℃で5分間行った。反応生成物は、4%ポリアクリルアミドゲル上で電気泳動した後、エチジウムブロミド(500ng/ml)で5分間染色し、1.2Kbのバンドをカッターを用いて切り出し、これをマクサム・ギルバート溶出緩衝液(500mM 酢酸アンモニウム、10mM MgCl2、1mM EDTA、0.1% SDS)に浸して、37℃で16時間処理した。等量の0.1M Tris-HCl、pH 8.0飽和フェノール・クロロフォルム(1:1)を加えて攪拌した後、TOMY遠心機MRXー150(トミー精工社製)で12000回転で5分間遠心分離後、その水層を取り出し、2.5倍量のエタノールを加えて、-80℃で15分処理後、再びTOMY遠心機MRX-150(トミー精工社製)で12000回転で5分間遠心分離して、その沈澱物をTE緩衝液(10mM Tris-HCl、pH 8.0、1mM EDTA)に溶解した。そして、波長260nmでの吸光度を測定することにより、回収した1.2KbのバンドのDNA量を計算した。最終的に0.2μgのDNAが得られた。このDNA断片をKpnIで消化後、pRset(Invitorogen社製)のKpnI部位に挿入し、発現ベクターpRset-Doc2を得た。
【0049】発現ベクターpRset-Doc2を導入した大腸菌BL2l株を用いて、オリゴヒスチジンをアミノ末端に持つ融合タンパク質(F.W. StudierおよびB.A. Moffatt、J. Mol. Biol., 189, 113-130 (1986))としてDoc2を発現させた。大腸菌BL2l株の培養はLB培地で行い、30℃で3時間培養した。その後大腸菌をリン酸緩衝液(20mM リン酸ナトリウム、500mM NaCl、pH 7.8)に懸濁し、超音波処理を行った。次いで、100,000xg、1時間、4℃の条件で遠心分離し、その上清を、プレーボンドレジン(Invitrogen)にかけた。Doc2タンパク質の結合したカラムをリン酸緩衝液(20mM リン酸ナトリウム、500mM NaCl、pH 6.0)で洗浄後、溶出緩衝液(500mMイミダゾール、20mM リン酸ナトリウム、500mM NaCl、pH 6.0)にて、Doc2タンパク質を溶出した。溶出したDoc2タンパク質を、20mM HEPES(pH 7.4)、1mM DTTを用いて透析した後、モノQカラム(0.5×5cm)(Pharmacia社製)にかけ、A緩衝液(20mM HEPES(pH 7.4)、1mM DTT、1%コール酸)およびB緩衝液(20mM HEPES (pH 7.4)、1mM DTT、1M NaCl、1%コール酸)を用いたNaClの塩濃度の勾配により溶出し、Doc2タンパク質を含むフラクションを集めて最終的な精製品を得た。
【0050】[実施例3]ノーザンブロット解析によるDoc2 mRNAの組織分布いくつかのヒト組織より単離したpoly(A)+ RNA 2μgを、アガロースゲル電気泳動にかけ、ナイロン膜に移したものをClonetech社より購入した。この膜を、マルチプライムDNA標識システム(アマシャム・ジャパン社製)により32Pで標識したDoc2DNA(翻訳開始点より25から989番目の塩基を標識)をプローブとして、ハイブリダイゼーションを行った。ハイブリダイゼーション溶液は、5XSSPE(1XSSPEは、0.18M NaCl、0.01M リン酸ナトリウム、pH 7.5、1mM EDTAよりなる)、10Xデンハート溶液、2% SDS、50%ホルムアミド、100μg/mlサケ精子DNAを用い、42℃で一晩ハイブリダイゼーションを行った。フィルターの洗浄は2XSSC、0.5% SDSで室温で10分間、1XSSC、0.5% SDCで65℃で30分間を2回、さらに0.1XSSC、0.5% SDSで65℃で30分間を2回行った後、X線フィルム(Kodak社製)に感光させ、それを現像して解析した。
【0051】結果を図3に示す。各レーンは、それぞれ、1:心臓、2:脳、3:胎盤、4:肺、5:肝臓、6:骨格筋、7:腎臓、8:膵臓、9:脾臓、10:胸腺、11:前立腺、12:精巣、13:卵巣、14:小腸、15:大腸、および16:末梢血リンパ球であり、RNA分子量マーカーの位置をキロベース(kb)で示した。脳において、Doc2のmRNAである2.2kbの大きさのmRNAが大量に発現しているが、他の部位にはほとんど検出されないことが明らかとなった。また、2.2kbのmRNAは、Molt-4のmRNAを用いたノーザンブロット解析でも検出された。
【0052】[実施例4]抗Doc2ポリクローナル抗体の作製実施例2で得たPBS中の精製Doc2N末端タンパク質(72個のアミノ酸)およびグルタチオン-S-トランスフェラーゼの融合タンパク質を、等量のフロイント完全アジュバント(DIFCO LABORATORIES社製、和光純薬より購入)と混ぜ、コンジュゲートとしてウサギ(日本白色種、雄)の皮下に、3週間おきに3回免疫し、最終免疫から1週間後に頸動脈より全採血して抗血清を得た。1回目の免疫では融合タンパク質1mgを用い、2回目の免疫では500μg、そして3回目の免疫では10μgを用いた。
【0053】抗体価のチェックは、実施例2で得たオリゴヒスチジンをアミノ末端に有するDoc2タンパク質を用いて以下のように行った。種々の量のDoc2タンパク質(0.1ng−100ng)をSDS緩衝液(60mM Tris-HCl、pH 6.8、2% SDS、10% グリセロール、5% 2-メルカプトエタノール、0.001%ブロモフェノールブルー)に溶解し、100℃で5分間反応させてSDS化した後、SDS-PAGEで電気泳動した。SDS-PAGEからPVDF膜(クリアブロットメンブレン-P、アトー社製)にトランスファー緩衝液(0.025M Tris-HCl、pH 8.3、0.192M グリシン、5%メタノール)を用いて電気的に移した膜を、20%牛胎児血清を含むPBS緩衝液中で、500倍希釈したウサギ抗血清とともに、室温で1時間反応した。その後、1% Tween 20(ポリオキシエチレンソルビタンモノラウレート、ナカライテスク社製)を含むPBSを用いて、室温で3回洗浄した。次に、これを、20%牛胎児血清を含むPBS緩衝液中で、1000倍希釈した西洋ワサビペルオキシダーゼ(HRP)に結合したプロテインG(ZYMED LABORATORIES社製)と、室温で1時間反応した。その後、1%Tween 20を含むPBSを用いて、室温で3回洗浄した。洗浄後、ECLウエスタンブロッティング検出システム(アマシャム・ジャパン社製)を用いて発色反応を行い、X線フィルム(Kodak社製)に感光させ、現像されたDoc2タンパク質のバンドを解析した。
【0054】上記抗血清を、常法に従い(T. Maniatisら、Molecular Cloning: A Laboratory Manual、第2版、Cold Spring Harbor Laboratory, New York (1989))、グルタチオン-S-トランスフェラーゼ(GST)結合セファロース4Bカラムにかけて、抗GTS抗体を除いた後、免疫に用いた融合タンパク質結合セファロース4BカラムにかけてDoc2に対する抗体を精製した。詳細には、以下の方法により精製した。カップリング緩衝液(0.1M NaHCO3、0.5M NaCl)に溶解したDoc2N末端側の72アミノ酸とGSTとの融合タンパク質、あるいはGST 10mgと、1mM HClで膨潤させた2gの臭化シアン活性化セファロース4Bカラム(Pharmacia社製)とを、カップリング緩衝液中で、室温で2時間反応させた。その後、カップリング緩衝液を除き、ブロッキング緩衝液(1M Tris-HCl、pH 8.0)中で室温で2時間反応させた。その後、洗浄緩衝液(0.1M 酢酸ナトリウム、pH 4.0、0.5M NaCl)で洗浄後、再び、カップリング緩衝液に懸濁し、カラムに充填し、10mM Tris-HCl(pH 7.5)で平衡化した。6mlのウサギ抗血清を9mlの10mM Tris-HCl(pH 7.5)と混ぜ、まずGST結合カラムにかけて、素通りしてきた画分を、Doc2N末端側の72アミノ酸とGSTとの融合タンパク質結合カラムにかけた。このカラムを、20mlの10mM Tris-HCl(pH 7.5)および20mlの10mM Tris-HCl、500mM NaCl(pH 7.5)で洗浄後、7mlの溶出緩衝液(100mMグリシン、pH 2.5)で溶出した。この溶出液に0.7mlの10mM Tris-HCl(pH 8.5)を加えたものを、抗Doc2ポリクローナル抗体として用いた。
【0055】[実施例5]抗Doc2モノクローナル抗体の作製実施例2で得たPBS中の精製したDoc2N末端側の72アミノ酸とGSTとの融合タンパク質(実施例4のポリクローナル抗体の作製に用いた抗原と同じもの)50μgを、最終濃度が0.25μg/μlとなるようにフロイント完全アジュバントとコンジュゲートして、3匹のマウス(Balb/c、雌)の足蹠に各50μlずつ2または3日ごとに3回接種した。最後の接種の翌日に全てのマウスから後リンパ節を摘出し、リンパ細胞をマウスミエローマ細胞Sp2と10:1の割合で混合して50%PEGを含むRPMI1640培地に加えることにより細胞融合を行った。4枚の96ウェルプレート中のHAT培地(10%FCS、0.1mM ヒポキサンチンナトリウム、0.4μM アミノプテリン、16μM チミジンを含むRPMI1640培地)に、約100細胞/ウェルになるように細胞を播いた。3日後に培養上清100μlを同量のHT培地(10%FCS、0.1mM ヒポキサンチンナトリウム、16μM チミジンを含むRPMI1640培地)と交換し、以後4日ごとに上清を同様に10%FCS、10%Origen(ボクスイブラウン)を含むRPMI1640培地と交換し維持継代した。培養開始16日目に培養上清について、GSTタンパク質、および、オリゴヒスチジンをN末端に持つDoc2タンパク質(His-Doc2タンパク質)に対するELISA試験を以下のように行った。96ウェルプレート(Maxisorb、Nunc)に抗原を0.5または1μg/ウェル入れ、37℃で30分間保存しコートした。次に抗原を捨てブロック溶液(10%FCSを含むPBS)を加え4℃で一晩ブロックした。ブロック後ブロック溶液を捨て、試験するハイブリドーマの培養上清を加えて37℃で30分間反応した。その後プレートをプレート洗浄器(Nunc)で洗浄し、ブロック液で希釈したHRP結合抗マウス免疫グロブリン抗体(Dako)を加えて37℃で30分反応した。そしてプレートをプレート洗浄器で洗浄しABTS試薬によりHRP活性を450nmの吸光度として求めた。その結果、24ウェルがGSTタンパク質に対して陰性およびHis-Doc2タンパク質に対して陽性であった。これらをHis-Doc2タンパク質に対してウエスタンブロットし、最も力価の高かった4E11を選択した。
【0056】次に、このウェルのハイブリドーマをクローン化するために、細胞を10-1細胞/ウェルとなるように限界希釈して96ウェルプレートに播き、ELISA試験によりGSTに対して陰性およびDoc2タンパク質に対して陽性を示すシングルコロニーのウェル(D3)を選択した。このハイブリドーマの培養上清が、His-Doc2タンパク質に対して特異的に反応することをウエスタンブロットにより確認した。このハイブリドーマは、工業技術院生命工学研究所に平成6年10月7日付で寄託され、受託番号FERM P-14577が付与されている。得られたハイブリドーマD3をRPMI培地に懸濁し(2×107細胞/ml)、1匹あたり500μlずつ、11日前にプリスタンを腹腔内に投与しておいたマウス(Balb/c、雌)5匹の腹腔内に投与し1週間後にマウスから腹水を回収した。回収した腹水は2,000rpm10分間遠心分離し、その上清を抗Doc2モノクローナル抗体とした。
【0057】[実施例6]ウエスタンブロット解析実施例4および5で作製したDoc2N末端側の72アミノ酸に対するポリクローナル抗体およびモノクローナル抗体を用いて、ラット組織でのDoc2タンパク質の発現をウエスタンブロットにより以下のように検討した。
【0058】ラットの種々の組織を、SDS緩衝液(60mM Tris-HCl、pH 6.8、2%SDS、10%グリセロール、5% 2-メルカプトエタノール、0.001%ブロモフェノールブルー)に溶解し、100℃で5分間反応させSDS化した後、SDS-PAGEで電気泳動した。SDS-PAGEからPVDF膜(クリアブロットメンブレン-P、アトー社製)にトランスファー緩衝液を用いて電気的に移し、この膜を、20%牛胎児血清を含むPBS緩衝液中で、1000倍希釈した抗Doc2ポリクローナル抗体あるいは2000倍希釈した抗Doc2モノクローナル抗体と、室温で1時間反応した。その後、1% Tween 20を含むPBSを用いて、室温で3回洗浄した。次に、20%牛胎児血清を含むPBS緩衝液中で、一次抗体が抗Doc2ポリクローナル抗体の場合は2000倍希釈したHRP結合プロテインG(ZYMED LABORATORIES社製)と、一次抗体が抗Doc2モノクローナル抗体の場合は5000倍希釈したHRP結合抗マウスイムノグロブリンヒツジポリクローナル抗体(アマシャム・ジャパン社製)と、それぞれ室温で1時間反応させた。その後、1% Tween 20の入ったPBSを用いて、室温で3回洗浄した。洗浄後、ECLウエスタンブロッティング検出システム(アマシャム・ジャパン社製)を用いて発色反応を行い、X線フィルム(Kodak社製)に感光させ、現像したDoc2タンパク質のバンドを解析した。
【0059】抗Doc2ポリクローナル抗体を用いた場合のウエスタンブロットの結果を図4に示す。図中の各レーンは、それぞれ、1:脳、2:心臓、3:肺、4:肝臓、5:腎臓、6:骨格筋、7:脾臓、8:胸腺、9:小腸、および10:大腸であり、タンパク質分子量マーカーの位置をキロダルトン(kD)で示した。分子量46kDのバンドが脳において検出されたが、他の臓器では検出されなかった。
【0060】抗Doc2モノクローナル抗体を用いた場合のウエスタンブロットの結果を図5に示す。図中の各レーンは、それぞれ、1:心臓、2:脳、3:小腸、4:肺、5:肝臓、6:骨格筋、7:腎臓、8:副腎髄質、9:脾臓、および10:胸腺であり、タンパク質分子量マーカーの位置をキロダルトン(kD)で示した。その結果、ポリクローナル抗体を用いた場合と同様に、分子量46kDのバンドが脳において検出されたが、他の臓器では検出されなかった。
【0061】[実施例7]Doc2とホスホリピッドとの結合解析C2領域を有するDoc2とホスホリピッド(ホスファチジルセリンおよびホスファチジルコリン)との結合解析は報告されている方法に従って行った(T. Yamaguchiら、J. Biol. Chem.、268、27164-27170 (1993))。ホスファチジルセリンおよびホスファチジルコリンはフナコシより購入した。リポソームを作製するために、ホスホリピッドを窒素ガスを用いてフイルム状に乾燥し、20mM HEPES/NaOH(pH 7.4)、150mM NaCl緩衝液に溶解した後、100,000×gで20分間、4℃にて遠心分離した。沈澱を再び同じ緩衝液に溶解した。実施例2で得た組換えHis-Doc2タンパク質20pmolを、上記のリポソームとともに、種々の濃度のカルシウムイオンを含む溶液100μl(100μgリポソーム、20mM HEPES/NaOH(pH 7.4)、150mM NaCl、10-8〜10-4Mの種々の濃度のカルシウムイオン)中で、60分間、4℃で反応させた。反応後、100,000×gで20分間、4℃にて遠心分離し、上清および沈澱を、SDS緩衝液(60mM Tris-HCl、pH 6.8、2%SDS、10%グリセロール、5% 2-メルカプトエタノール、0.001%ブロモフェノールブルー)に溶解し、100℃で5分間反応してSDS化した後、SDS-PAGEで電気泳動した。SDS-PAGEからPVDF膜に電気的に移した膜を、20%牛胎児血清を含むPBS緩衝液中で、2000倍希釈した抗Doc2モノクローナル抗体と、室温で1時間反応させた。その後、1% Tween 20を含むPBSを用いて、室温で3回洗浄した。次に、20%牛胎児血清を含むPBS緩衝液中で、2000倍希釈したHRP結合抗マウスイムノグロブリンヒツジポリクローナル抗体(アマシャム・ジャパン社製)と、室温で1時間反応させた。その後、1% Tween 20を含むPBSを用いて、室温で3回洗浄した。洗浄後、ECLウエスタンブロッティング検出システム(アマシャム・ジャパン社製)を用いて発色反応を行い、X線フィルム(Kodak社製)に感光させ、現像したDoc2タンパク質のバンドを解析した。
【0062】結果を図6に示す。図中の黒丸はホスファチジルセリン、および白丸はホスファチジルコリンで作製したリポソームとの結合の結果をそれぞれ示している。組換えHis-Doc2タンパク質はカルシウムイオン存在下にホスホリピッドと結合した。ホスファチジルセリンで作製したリポソームへのDoc2の結合はカルシウムイオン濃度に依存的であり、カルシウムイオンの濃度が5x10-7Mの時、半量のDoc2がリポソームへ結合した。ホスファチジルコリンでリポソームを作製した場合、Doc2はリポソームにほとんど結合しなかった。また、データには示さないが、組換えDoc2タンパク質はGTPγS-結合型のRab3Aとは結合しなかった。
【0063】
【発明の効果】本発明によれば、2つのC2領域を有する脳特異的タンパク質であるDoc2タンパク質;Doc2タンパク質の構造遺伝子;該遺伝子を含む発現ベクター;該発現ベクターを有する形質転換体;該形質転換体を用いたDoc2タンパク質の製造方法;Doc2タンパク質に対する抗体;該抗体を産生し得るハイブリドーマ;および該抗体を用いた試料中のDoc2タンパク質を定量するための免疫学的測定方法が提供される。
【0064】神経伝達物質の放出に関与するシナプトタグミンおよびラブフィリン3AのC2領域は、カルシウムイオンおよびホスホリピッド(特にホスファチジルセリン)と結合することが知られており、同様に、組換えDoc2タンパク質もカルシウムイオン依存的にホスホリピッド(特にホスファチジルセリン)と結合する。従って、Doc2は、神経伝達物質の放出に作用すると考えられる。しかし、シナプトタグミンおよびラブフィリン3Aと異なり、Doc2は、シナプトタグミンに存在する膜貫通領域およびラブフィリン3Aに存在するRab3A結合領域を有さず、一方、Doc2はそのアミノ末端側に72個のアミノ酸よりなる、他のタンパク質と類似性のないユニークな領域を有している。このようなシナプトタグミンおよびラブフィリン3Aとの構造上の類似性から考えて、Doc2のアミノ末端領域も、何らかの細胞内構成成分、例えば、ある種のタンパク質を介してシナプス小胞と結合していることが予想され、Doc2はカルシウムセンサーとして働き、プレシナプスからの神経伝達物質の放出に関与していることが予想される。このことは、プレシナプス膜に電気刺激が伝達された際、アクティブゾーンのカルシウムイオン濃度は、電位依存性のカルシウムチャンネルからのカルシウムイオンの流入により、1×10-8Mから1×10-4Mまで上昇すること(S.J. SmithおよびG.J. Augustine、Trends Neurosci.、11、458-464 (1988))から類推され得る。従って、本発明のDoc2は、その機能の解明により、神経伝達物質の放出機構を解明するのに有用である。また、本発明のDoc2遺伝子および抗Doc2抗体は、Doc2の遺伝子変異およびそのmRNAおよびタンパク質の発現状態を解析するのに有用であり、脳神経系の疾患の原因究明の新たな手段を提供し得、脳神経系の疾患の診断または治療方法の開発に有用である。
【0065】
【配列表】
【0066】
【配列番号:1】
配列の長さ:1718配列の型:核酸鎖の数:二本鎖トポロジー:直鎖状配列の種類:cDNA to mRNA起源生物名:ヒト配列の特徴特徴を表す記号:CDS存在位置:125..1324特徴を決定した方法:S配列CCACGCGTCC GGTCCTGACG GCGCTGGAGC TGAGGGGCAG TGCGGATGCC CCAGGAAGGC 60TCCTAGGAAG AGGGGACCCA CGGTGACTTC CTAAGGAAGC GCGGTTCCCA GCCAGGGGTG 120CTGC ATG AGG GGC CGC AGG GGC GAT CGC ATG ACC ATC AAC ATC CAG GAG 169 Met Arg Gly Arg Arg Gly Asp Arg Met Thr Ile Asn Ile Gln Glu 1 5 10 15CAC ATG GCC ATC AAC GTG TGC CCC GGG CCC ATC CGG CCC ATC CGC CAG 217His Met Ala Ile Asn Val Cys Pro Gly Pro Ile Arg Pro Ile Arg Gln 20 25 30ATC TCT GAC TAC TTC CCC CGG GGA CCA GGA CCT GAA GGG GGC GGC GGG 265Ile Ser Asp Tyr Phe Pro Arg Gly Pro Gly Pro Glu Gly Gly Gly Gly 35 40 45AGC GGC GGG GAG GCC CCC GCC CAT CTG GTC CCC CTG GCT CTG GCC CCC 313Ser Gly Gly Glu Ala Pro Ala His Leu Val Pro Leu Ala Leu Ala Pro 50 55 60CCT GCA GCC CTC CTT GGG GCC ACC ACG CCT GAG GAT GGT GCG GAG GTG 361Pro Ala Ala Leu Leu Gly Ala Thr Thr Pro Glu Asp Gly Ala Glu Val 65 70 75GAC AGC TAT GAC TCG GAT GAT GCC ACC GCC CTA GGC AAG CTG GAG TTT 409Asp Ser Tyr Asp Ser Asp Asp Ala Thr Ala Leu Gly Lys Leu Glu Phe 80 85 90 95GAC CTT CTC TAC GAC CGG GCC TCC TGC ACT CTG CAC GTA TGC ATC CTC 457Asp Leu Leu Tyr Asp Arg Ala Ser Cys Thr Leu His Val Cys Ile Leu 100 105 110AGG GCC AAG GGC CTC AAG CCC ATG GAT TTC AAT GGC CTC GCC GAC CCC 505Arg Ala Lys Gly Leu Lys Pro Met Asp Phe Asn Gly Leu Ala Asp Pro 115 120 125TAC GTC AAG CTG CAC TTG CTG CCT GGA GCC TGT AAG GCC AAT AAG CTA 553Tyr Val Lys Leu His Leu Leu Pro Gly Ala Cys Lys Ala Asn Lys Leu 130 135 140AAA ACG AAG ACT CAG AGG AAC ACA CTG AAT CCC GTG TGG AAT GAG GAC 601Lys Thr Lys Thr Gln Arg Asn Thr Leu Asn Pro Val Trp Asn Glu Asp 145 150 155CTG ACT TAC AGC GGG ATC ACA GAT GAC GAC ATC ACG CAC AAG GTG CTC 649Leu Thr Tyr Ser Gly Ile Thr Asp Asp Asp Ile Thr His Lys Val Leu 160 165 170 175AGG ATC GCC GTC TGT GAT GAG GAC AAG CTG AGT CAC AAT GAG TTT ATT 697Arg Ile Ala Val Cys Asp Glu Asp Lys Leu Ser His Asn Glu Phe Ile 180 185 190GGG GAG ATC CGC GTG CCC CTC CGC CGC CTC AAG CCT TCG CAG AAG AAG 745Gly Glu Ile Arg Val Pro Leu Arg Arg Leu Lys Pro Ser Gln Lys Lys 195 200 205CAT TTT AAC ATC TGC CTC GAG CGC CAA GTC CCG CTG GCG TCC CCC TCT 793His Phe Asn Ile Cys Leu Glu Arg Gln Val Pro Leu Ala Ser Pro Ser 210 215 220TCC ATG TCA GCG GCG CTG AGG GGC ATC TCC TGT TAT CTG AAG GAC TTG 841Ser Met Ser Ala Ala Leu Arg Gly Ile Ser Cys Tyr Leu Lys Asp Leu 225 230 235GAG CAG GCG GAG CAG GGG CAG GGG CTG CTG GAG GAG CGT GGC CGC ATC 889Glu Gln Ala Glu Gln Gly Gln Gly Leu Leu Glu Glu Arg Gly Arg Ile 240 245 250 255CTG CTG AGT CTC AGC TAC AGC TCG CGG CGC CGG GGA CTG CTG GTA GGC 937Leu Leu Ser Leu Ser Tyr Ser Ser Arg Arg Arg Gly Leu Leu Val Gly 260 265 270ATC TTG CGC TGC GCC CAT CTG GCT GCC ATG GAC GTC AAC GGT TAC TCG 985Ile Leu Arg Cys Ala His Leu Ala Ala Met Asp Val Asn Gly Tyr Ser 275 280 285GAC CCC TAC GTC AAG ACG TAC CTG AGG CCC GAT GTG GAC AAG AAA TCC 1033Asp Pro Tyr Val Lys Thr Tyr Leu Arg Pro Asp Val Asp Lys Lys Ser 290 295 300AAG CAT AAG ACG TGT GTG AAG AAG AAG ACT CTC AAC CCA GAA TTT AAC 1081Lys His Lys Thr Cys Val Lys Lys Lys Thr Leu Asn Pro Glu Phe Asn 305 310 315GAG GAG TTT TTC TAC GAG ATA GAG CTC TCC ACT CTG GCC ACC AAG ACC 1129Glu Glu Phe Phe Tyr Glu Ile Glu Leu Ser Thr Leu Ala Thr Lys Thr 320 325 330 335CTG GAA GTC ACC GTC TGG GAC TAT GAC ATT GGC AAA TCC AAT GAC TTC 1177Leu Glu Val Thr Val Trp Asp Tyr Asp Ile Gly Lys Ser Asn Asp Phe 340 345 350ATT GGT GGC GTG TCC CTG GGG CCA GGT GCC CGA GGC GAG GCT CGG AAG 1225Ile Gly Gly Val Ser Leu Gly Pro Gly Ala Arg Gly Glu Ala Arg Lys 355 360 365CAC TGG AGT GAC TGC CTG CAG CAG CGG GAC GCA GCC CTG GAG CGC TGG 1273His Trp Ser Asp Cys Leu Gln Gln Arg Asp Ala Ala Leu Glu Arg Trp 370 375 380CAC ACC CTG ACC AGT GAG CTG CCC CCT GCG GCC GGG GCT CTG TCC TCA 1321His Thr Leu Thr Ser Glu Leu Pro Pro Ala Ala Gly Ala Leu Ser Ser 385 390 395GCC TGAGTGGACA GCAGTGTCCC GGCACAGGCC CATCGAGCCG GGTCCAGTAC 1374Ala 400CCAACCTTCG CACGAGTGTG TTGCACGTTT ACACAGGTGG GCTGCCCCAC CCTGCACTAC 1434CTATTTTGTG AGTCTCGTGA CCCGGGTCTG TCTGCTCATG AGGGGCTGCG GAGTTCTATA 1494TTCACATATG CAAACCTCCT GCCTGACTCG CTAGTCCCTG CAAATATGCA AACCCCCCTA 1554CTACTGCACA CCCGGGCAGT GCTCAGAGCC GCCCAGGCCC CGCGCTCCTC ACTCCTGCCT 1614CTCCACGCTG CCCCGTCCCT CTCCCCCAAC AGGGAGGAGG TCGGATTAGG GAGGTTCAGA 1674GGAGGAGAAT GTCTCAAAAA AAAAAAAAAA AAAAAAAAAA AAAA 1718
【0067】
【配列番号:2】
配列の長さ:45配列の型:核酸鎖の数:一本鎖トポロジー:直鎖状配列の種類:他の核酸 合成DNA配列GATTAGTTCT AGATCGCGAC GCGGCCGCCC TTTTTTTTTT TTTTT 45
【0068】
【配列番号:3】
配列の長さ:16配列の型:核酸鎖の数:一本鎖トポロジー:直鎖状配列の種類:他の核酸 合成DNA配列TCGACCCACG CGTCCG 16
【0069】
【配列番号:4】
配列の長さ:12配列の型:核酸鎖の数:一本鎖トポロジー:直鎖状配列の種類:他の核酸 合成DNA配列CGGACGCGTG GG 12
【0070】
【配列番号:5】
配列の長さ:23配列の型:核酸鎖の数:一本鎖トポロジー:直鎖状配列の種類:他の核酸 合成DNA配列の特徴他の情報:Nはイノシンである。
配列TTNACRWANG GRTCNSWNDD NCC 23
【0071】
【配列番号:6】
配列の長さ:23配列の型:核酸鎖の数:一本鎖トポロジー:直鎖状配列の種類:他の核酸 合成DNA配列の特徴他の情報:Nはイノシンである。
配列ACNYTNAAYC CNNDNTDWAA YGA 23
【0072】
【配列番号:7】
配列の長さ:8配列の型:アミノ酸鎖の数:一本鎖トポロジー:直鎖状配列の種類:ペプチド配列の特徴他の情報:2位のXaaは任意のアミノ酸、6位のXaa破GluまたはTyrである。
【0073】
【配列番号:8】
配列の長さ:8配列の型:アミノ酸鎖の数:一本鎖トポロジー:直鎖状配列の種類:ペプチド配列の特徴他の情報:5位のXaaは任意のアミノ酸、6位のXaaはPhe、Tyr、またはTrpである。
【0074】
【配列番号:9】
配列の長さ:36配列の型:核酸鎖の数:一本鎖トポロジー:直鎖状配列の種類:他の核酸 合成DNA配列CATGGATCCG GTACCATGAC CATCAACATC CAGGAG 36
【0075】
【配列番号:10】
配列の長さ:38配列の型:核酸鎖の数:一本鎖トポロジー:直鎖状配列の種類:他の核酸 合成DNA配列CATGGATCCG GTACCTCAAT AGCTGTCCAC CTCCGCAC 38
【0076】
【配列番号:11】
配列の長さ:36配列の型:核酸鎖の数:一本鎖トポロジー:直鎖状配列の種類:他の核酸 合成DNA配列CATGGATCCG GTACCATGAG GGGCCGCAGG GGCGAT 36
【0077】
【配列番号:12】
配列の長さ:36配列の型:核酸鎖の数:一本鎖トポロジー:直鎖状配列の種類:他の核酸 合成DNA配列CATGGATCCG GTACCTCAGG CTGAGGACAG AGCCCC 36
【図面の簡単な説明】
【図1】C2領域を2つ有する遺伝子を得るためのプライマーの設計を示す図である。
【図2】本発明のDoc2タンパク質と、ラブフィリン3AおよびシナプトタグミンIとのアミノ酸配列の比較の構造の模式図である。
【図3】種々のヒト組織におけるDoc2 mRNAの発現のノーザンブロット解析の結果を示す図である。
【図4】抗Doc2ポリクローナル抗体を用いた、種々のラット組織におけるDoc2タンパク質の発現のウエスタンブロット解析の結果を示す図である。
【図5】抗Doc2モノクローナル抗体を用いた、種々のラット組織におけるDoc2タンパク質の発現のウエスタンブロット解析の結果を示す図である。
【図6】本発明のDoc2タンパク質の、種々の濃度のカルシウムイオン存在下における、ホスホリピッドからなるリポソームへの結合能を示す図である。
【特許請求の範囲】
【請求項1】 配列表の配列番号1の9位のMetから80位のAspまでのアミノ酸配列を含む、脳特異的タンパク質。
【請求項2】 2つのC2領域を有する、請求項1に記載の脳特異的タンパク質。
【請求項3】 配列表の配列番号1の1位のMetから400位のAlaまでのアミノ酸配列を含む、請求項2に記載の脳特異的タンパク質。
【請求項4】 請求項1〜3のいずれかに記載の脳特異的タンパク質をコードするDNA配列。
【請求項5】 配列表の配列番号1の149位のAから364位のCまででなる塩基配列を有する、請求項4に記載のDNA配列。
【請求項6】 配列表の配列番号1の125位のAから1324位のCまででなる塩基配列を有する、請求項4に記載のDNA配列。
【請求項7】 請求項4〜6のいずれかに記載のDNA配列を有する発現ベクター。
【請求項8】 請求項7に記載の発現ベクターを宿主に導入して得られる形質転換体。
【請求項9】 前記宿主が大腸菌である、請求項8に記載の形質転換体。
【請求項10】 請求項9に記載の形質転換体を培養する工程、および産生された脳特異的タンパク質を培養培地から回収する工程を包含する、請求項1〜3のいずれかに記載の脳特異的タンパク質の製造方法。
【請求項11】 請求項1〜3のいずれかに記載の脳特異的タンパク質を認識する抗体。
【請求項12】 配列表の配列番号1の9位のMetから80位のAspまでのアミノ酸配列に含まれる部分を認識する、請求項11に記載の抗体。
【請求項13】 請求項11または12に記載の抗体を産生し得る、ハイブリドーマ。
【請求項14】 FERM P-14577である、請求項13に記載のハイブリドーマ。
【請求項15】 請求項1〜3のいずれかに記載の脳特異的タンパク質を含有し得る試料を、請求項11または12に記載の抗体とともに、抗原抗体複合体を形成させる条件下でインキュベーションする工程、および該抗原抗体複合体の量を測定する工程を包含する、脳特異的タンパク質の免疫学的測定方法。
【請求項1】 配列表の配列番号1の9位のMetから80位のAspまでのアミノ酸配列を含む、脳特異的タンパク質。
【請求項2】 2つのC2領域を有する、請求項1に記載の脳特異的タンパク質。
【請求項3】 配列表の配列番号1の1位のMetから400位のAlaまでのアミノ酸配列を含む、請求項2に記載の脳特異的タンパク質。
【請求項4】 請求項1〜3のいずれかに記載の脳特異的タンパク質をコードするDNA配列。
【請求項5】 配列表の配列番号1の149位のAから364位のCまででなる塩基配列を有する、請求項4に記載のDNA配列。
【請求項6】 配列表の配列番号1の125位のAから1324位のCまででなる塩基配列を有する、請求項4に記載のDNA配列。
【請求項7】 請求項4〜6のいずれかに記載のDNA配列を有する発現ベクター。
【請求項8】 請求項7に記載の発現ベクターを宿主に導入して得られる形質転換体。
【請求項9】 前記宿主が大腸菌である、請求項8に記載の形質転換体。
【請求項10】 請求項9に記載の形質転換体を培養する工程、および産生された脳特異的タンパク質を培養培地から回収する工程を包含する、請求項1〜3のいずれかに記載の脳特異的タンパク質の製造方法。
【請求項11】 請求項1〜3のいずれかに記載の脳特異的タンパク質を認識する抗体。
【請求項12】 配列表の配列番号1の9位のMetから80位のAspまでのアミノ酸配列に含まれる部分を認識する、請求項11に記載の抗体。
【請求項13】 請求項11または12に記載の抗体を産生し得る、ハイブリドーマ。
【請求項14】 FERM P-14577である、請求項13に記載のハイブリドーマ。
【請求項15】 請求項1〜3のいずれかに記載の脳特異的タンパク質を含有し得る試料を、請求項11または12に記載の抗体とともに、抗原抗体複合体を形成させる条件下でインキュベーションする工程、および該抗原抗体複合体の量を測定する工程を包含する、脳特異的タンパク質の免疫学的測定方法。
【図1】

【図2】
【図3】

【図4】

【図5】

【図6】
【図2】
【図3】
【図4】
【図5】
【図6】
【公開番号】特開平8−168385
【公開日】平成8年(1996)7月2日
【国際特許分類】
【出願番号】特願平6−320775
【出願日】平成6年(1994)12月22日
【出願人】(000001926)塩野義製薬株式会社 (229)
【公開日】平成8年(1996)7月2日
【国際特許分類】
【出願日】平成6年(1994)12月22日
【出願人】(000001926)塩野義製薬株式会社 (229)
[ Back to top ]