
2種のピノセンブリン、その製造方法、および、医薬組成物の製造におけるその使用
本発明は、以下の[化1]にて表される2つの結晶形ピノセンブリン(α及びβ)、その製造方法、医薬組成物の製造におけるこれらの使用に関する。これらの結晶形の間には、生物学的利用能に差がある。これらは、神経血管ユニットの保護作用によって脳虚血を治療及び予防するとともに、インビボにおける血中薬物濃度を改善するために用いられる。
【化1】

【化1】
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、2種の結晶形ピノセンブリン(pinocembrin)、および、医薬活性成分、2種の結晶形ピノセンブリンを含む医薬組成物および剤形、医薬組成物の製造および疾病の治療におけるその使用、並びに、2種のピノセンブリンの製造方法に関する。
【背景技術】
【0002】
ピノセンブリン(化学名:5,7-ジヒドロキシ‐2-フェニル-4-クロマノンは、天然に広く存在するフラボン(flavone)化合物である。その化学式は次の[化1]に示すとおりで、l-異性体、d-異性体、l-異性体又はd-異性体を多く含む混合物、又は、ラセミ化合物の形態で存在する。
【0003】
【化1】
【0004】
これまでの薬物研究によれば、ピノセンブリンは、強い静菌、抗ウイルス、および、抗真菌活性を有する。例えば、中国伝統健康管理食品である蜂蜜には、ピノセンブリンが多く含まれている。蜂蜜を頻繁に食べても歯には悪影響がないだけでなく、口腔の殺菌効果も期待できる。例えば、口内炎を和らげ、治癒を促す。中国特許CN169508A(発明の名称:神経細胞損傷に関連した疾病の治療および予防用医薬組成物の製造におけるピノセンブリンの使用;参考文献1)には、脳虚血、脳虚血の後遺症、神経細胞の損傷および機能の変質に関連した疾病を治療又は予防するための医薬組成物を製造することにおけるピノセンブリンの使用が記載されている。
【発明の概要】
【発明が解決しようとする課題】
【0005】
発明者らは、ピノセンブリンが2種の結晶形αおよびβを有することと、その製造法を見出した。また、本発明者らは、これらの2種の結晶形の間には生命体による取込み(吸収)に有意な差があることに着目した。要するに、β結晶形の取込み率(吸収率)のほうが、α結晶形よりも大きい。例えば、β結晶形の取込み率が、α結晶形の取込み率の2倍以上であり得る。薬物療法におけるこれらの生物学的活性は、血中薬物濃度差に基づいて異なってくる。
【課題を解決するための手段】
【0006】
本発明は、α結晶形ピノセンブリン、β結晶形ピノセンブリン、又は、任意の比率のα結晶形ピノセンブリンとβ結晶形ピノセンブリンの混合物を提供する。これらの結晶形又は結晶形の混合物は、結晶水又はその他の有機溶媒を含まないことが好ましい。
【0007】
また、本発明は、α結晶形ピノセンブリン、β結晶形ピノセンブリン、又は、任意の比率のα結晶形ピノセンブリンとβ結晶形ピノセンブリンの混合物の製造方法を提供する。
【0008】
また、本発明は、純粋なα結晶形ピノセンブリン、純粋なβ結晶形ピノセンブリン、又は、任意の比率のα結晶形ピノセンブリンとβ結晶形ピノセンブリンの混合物を含む医薬組成物を提供する。この医薬組成物は、1以上の医薬的に許可可能な担体を含むことができる。これらの担体に特に制限はなく、製剤に適したもので、本発明に係る結晶形ピノセンブリンに悪影響を及ぼさないものであれば使用することができる。
【0009】
また、本発明は、結晶形ピノセンブリン固体を含む剤形を提供する。剤形に特に制限はない。例えば、錠剤、カプセル、丸薬、注射剤、徐放性製剤、制御放出製剤などの剤形を有する。
【0010】
また、本発明は、治療における薬物の取込み(drug uptake)の差異を起こさせるための、α結晶形ピノセンブリン、β結晶形ピノセンブリン、又は、任意の比率のα結晶形ピノセンブリンとβ結晶形ピノセンブリンの混合物を含む結晶形ピノセンブリン固体の使用を提供する。
【0011】
また、本発明は、生命体における血中濃度を、結晶形効果に基づいて改善(向上)させるとともに、脳虚血関連疾病の治療に神経血管ユニット機能を保護するためのピノセンブリンの使用を提供する。
【発明の効果】
【0012】
本発明に係る結晶形ピノセンブリンは、生命体における血中濃度を結晶形効果(crystal form effect)に基づいて改善させるとともに、脳虚血関連疾病の治療に神経血管ユニット機能を保護するために用いられる。
【図面の簡単な説明】
【0013】
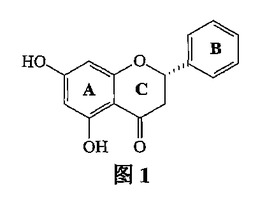
【図1】図1は、分子の相対的な配置を示す。
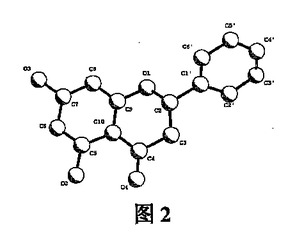
【図2】図2は、分子の立体構造の投影図である。
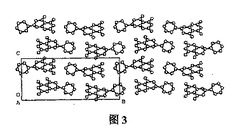
【図3】図3は、(軸に沿った)分子の単位セル積み重ねを示す。
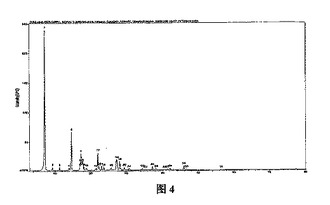
【図4】図4は、α結晶形ピノセンブリンサンプルの粉末X線回析パターンである。
【図5】図5は、α結晶形ピノセンブリンサンプルのDSCトレース(trace)である。
【図6】図6は、α結晶形ピノセンブリンサンプルの赤外線吸収スペクトルである。
【図7】図7は、β結晶形ピノセンブリンサンプルの粉末X線回析パターンである。
【図8】図8は、β結晶形ピノセンブリンサンプルのDSCトレースである。
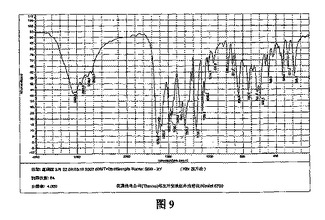
【図9】図9は、β結晶形ピノセンブリンサンプルの赤外線吸収スペクトルである。
【発明を実施するための形態】
【0014】
ピノセンブリンのα結晶形のサンプルの形態学的特徴
本発明の実施例に基づいて得られたα結晶形ピノセンブリンのサンプルをX線単一結晶回析(X-ray single crystal diffraction)によって分析すると、単斜晶対称を示し、その空間群はP21/cであり、結晶セルパラメーター(crystal cell parameter)値は、次のとおりである:a=5.189Å、b=24.149Å、c=10.472Å、α=90°、β=102.31°及び γ=90°。
【0015】
図1は、分子の相対的な配置を示す。図2は、分子の立体構造を投影したものである。図3は、軸に沿った分子の単位セル積み重ねを示す。表1は、原子座標パラメーターおよび同等の温度ファクターを示す。表2は、結合原子の結合距離(値)を示す。表3は、結合原子の結合角(値)を示す。B環の一部の炭素原子が乱れた配向状態(orientation state)有するために、4つの原子C2'、C3'、C5'、およびC6'は2つの位置を占有し、その占有率はそれぞれ0.5である。
【0016】
【化2】
【0017】
化2は、α結晶形ピノセンブリンの相対的な分子配置を示す。
【0018】
【表1】
【0019】
【表2】
【0020】
【表3】
【0021】
表2及び3においては、B環の原子C2'、C3'、C5'、および、C6'につき、一つの位置の結合強度および結合角度のみが記載されている。
【0022】
α結晶形ピノセンブリンの固体に対し粉末(多結晶)X線回析(CuKα線)を行った。回析ピークの位置(2−シータ値(°)又はd値(Å))および回析ピークの相対的な強度(ピークの高さ値(高さ%)又はピーク面積値(面積%))から次の特徴が得られた(表4、図4参照)。
【0023】
【表4】
【0024】
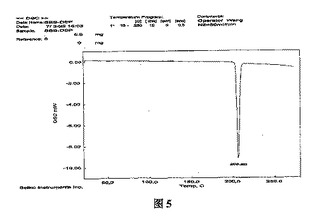
本発明の一実施例によれば、DSCによって分析されたとき、α結晶形ピノセンブリンは、約206℃の減輝転移温度(decalescence transition temperature)を有する(図5)。
【0025】
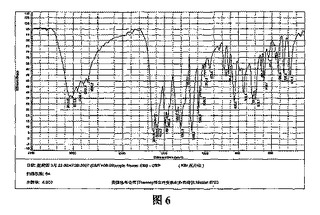
α結晶形ピノセブリン固体に対しKBrペレットを用いたIR分析を行った(図6参照)。特性ピークは次のとおりである:3090.6、3011.6、2889.1、2747.4、2636.2、1631.5、1602.5、1584.3、1487.7、1466.2、1454.5、1435.6、1354.9、1302.4、1257.0、1217.0、1168.2、1088.6、1064.9、1028.0、1014.6、1001.3、975.8、918.0、887.7、861.8、825.9、789.9、766.4、715.2、698.1、663.7、646.7、620.3、587.3、574.9、560.5、526.9及び487.9cm-1。このうち2891.1、2747.4、2636.2、1631.5および1354.9cm-1のピークは、α結晶形ピノセブリン固体の主な特性ピークである。
【0026】
本発明の実施例に基づくβ結晶形ピノセンブリンサンプルの形態学的特徴
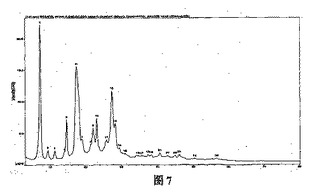
β結晶形ピノセンブリンの固体に対し粉末(多結晶)X線回析(CuKα線)を行った。回析ピークの位置(2−シータ値(°)又はd値(Å))および回析ピークの相対的な強度(ピークの高さ値(%)又はピーク面積値(%))から次の特徴が得られた(表5、図7参照)。
【0027】
【表5】
【0028】
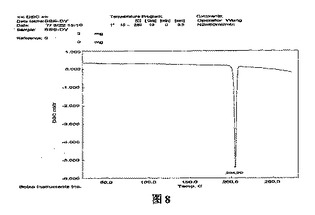
本発明の一実施例によれば、DSCによって分析されたとき、β結晶形ピノセンブリンは、約204℃の減輝転移温度を有する(図8)。
【0029】
β結晶形ピノセブリン固体に対しKBrペレットを用いたIR分析を行った(図9参照)。特性ピークは次のとおりである:3090.8、2890.0、2748.9、2638.3、1633.5、1602.9、1585.0、1487.9、1466.1、1454.3、1344.4、1302.7、1216.7、1168.2、1088.4、1065.5、1028.8、1014.3、1001.5、975.8、917.8、888.2、861.8、826.6、789.1、766.6、741.1、715.4、698.0、663.7、646.0、620.5、587.9、574.8、560.9、527.2及び488.4cm-1。このうち2890.0、2748.9、2638.3、1633.5および1344.4cm-1のピークは、β結晶形ピノセブリン固体の特性ピークである。
【0030】
本発明の実施例に基づくα結晶形ピノセンブリンサンプルの製造法
(1)サンプルをメタノール、エタノール、クロロホルム、アセトン、酢酸エチル、n‐ブタノール、イソプロパノール、アセトニトリル、THF、ジオキサン、95%エタノール、氷酢酸、ギ酸、エーテル、ジクロロメタン、トルエン、ベンゼン、n−ヘキサン、シクロヘキサン、DMF、石油エーテル、アンモニア、n−プロパノール、又は、これらの混合物からなる群から選択された溶媒に完全に溶かし、その後、
(a)その混合物を4〜50℃の温度および相対湿度10〜75%の条件下で放置して(1〜60日)再結晶化させるか、又は、
(b)水を加えて沈殿させた後、減圧濾過、凍結乾燥、又は、コールドスプレーによってα結晶形ピノセンブリンを得る。
【0031】
本発明の実施例に基づくβ結晶形ピノセンブリンサンプルの製造法
材料としてα結晶形固体サンプルを用いる。その後、
(a)粉砕(grinding)による結晶転移(crystal trasition)、又は、
(b)材料をピリジン又はDMSOの溶媒に完全に溶かし、水を加えて沈殿させ、減圧濾過、凍結乾燥、又は、コールドスプレーを行うこと
によってβ結晶形ピノセンブリンのサンプルを得る。
【0032】
ピノセンブリンサンプルの薬力学的特徴
本発明に基づく純粋なα結晶形ピノセンブリン、純粋なβ結晶形ピノセンブリン、又は、α結晶形ピノセンブリンとβ結晶形ピノセンブリンの混合物(任意の比率)は、神経血管ユニット機能(neurovascular unit function)を保護することによって、脳虚血に関連した疾病の治療、又は、脳虚血に関連した疾病の予防に役立つ。
【0033】
本発明による純粋なα結晶形および純粋なβ結晶形の間には、生物学的利用能に差が存在する。経口投与の場合、β結晶形の生物学的利用能が、α結晶形よりも2倍大きい。2種の結晶形の混合物(任意の比率)の場合、その生物学的利用能はβ結晶形の含量に基づいて異なってくる。
【0034】
投与量および剤形
純粋なα結晶形、純粋なβ結晶形、又は、それらの混合物(任意の比率)を含む医薬組成物又はその製剤の一日投与量は、ピノセンブリン結晶形固体に基づいて5〜250mgである。剤形は、錠剤、カプセル剤、丸薬、注射剤、徐放出剤、制御放出剤等を含む。
【0035】
以下の説明は、本発明をよりよく理解してもらうために設けられたものであり、本発明を限定するものではない。
【0036】
実施例に採用された装置及び実験条件:
1.単結晶X線分析
装置:MAC DIP-2030Kエリア検出器
実験条件:チューブ電圧(tube voltage):50KV、チューブ電流(tube flow):80 mA、ωスキャニング、MoKα、2θ≦50.0°、スキャン範囲:0〜180°、回転角 (pivot angle):6°、ステップ:6°、スキャン速度:1.8°/分
2.粉末X線分析
装置:リガクD/max2550粉末X線回折計
実験条件:電圧:40KV、電流:150mA、スキャン速度:8°/分
3.DSC分析
装置:セイコーインスツル株式会社製示差走査熱量計
実験条件:パージガス:N2、加熱速度:10℃/分
温度範囲:25-250℃
4.IR吸収スペクトル
装置:NicoletFT-IRスペクトルメータ:IMPACT400
実験件:KBrペレット
5.HPLC分析
装置:SHIMADZU LC-10Avp高性能液体クロマトグラフィ、SPD-M10Avpダイオード層検出器、CLASS-VPクロマトグラフィデータシステム;カラム:Alltch C18(5μ,150×4.6mm);実験条件:カラム温度;波長:290nm;移動相:メタノール/燐酸塩食塩水pH3.0(64/36);流速(流量):1.0 mL/分;注入容積:20μL;注入濃度:500.0μg/mL
【0037】
製造1:ピノセンブリンサンプルの合成
1000mlの水素化反応容器に5g(19.7mmol)の5,7-ジヒドロキシフラボン、650mlの無水エタノール、及び、1.5gの10%パラジウム(炭素上)を加えた。0.13Mpaの水素圧下で、40℃にて4時間反応させた。反応が終ったら、パラジウム炭素(palladium on carbon)をろ過した。そのろ液を濃縮し、分離し、真空下でカラムクロマトグラフィ(エタノール:酢酸エーテル:石油エーテル=2:10:100 (V:V:V)によって溶出される。)によって精製した。その溶媒を蒸発させて乾燥し、白色の無定形固体粉末3.9gを得た(純度:98.6%、HPLCによって検出)。その収率は、52%であった(参考文献2)。
【0038】
α結晶形ピノセンブリンサンプルの製造
【0039】
[実施例1]
α結晶形ピノセンブリンサンプルの製造方法
5gのピノセンブリンサンプルを20mlの95%エタノールに加え、それが完全に溶けるまで加熱した。その後、室温に冷却して、24時間放置した。白色の固体を沈殿させ、ろ過し、乾燥させた。4.5gの白色の結晶(純度:98.8%、HPLCによって検出)を得た(回収率:90%)
【0040】
それにより得られた結晶をX線単結晶回折によって分析した。それは単斜対称を示し、スペース群(space group)はP21/cであり、結晶セルパラメーター(crystal cell parameter)値はa=5.189Å、b=24.149Å、c=10.472Å、α=90°、β=102.31°、及び、γ=90°であった。
【0041】
得られた結晶に対し粉末(多結晶)X線回折(CuKα線)を行った。回折ピーク位置の特性ピーク値:2-シータ値(°)又はd値(Å)及び回折ピークの相対的な強度:ピーク高さ値(高さ%)又はピーク面積値(面積%)は表4に示し、得られたトレース(trace)は図4に示す。
【0042】
得られた結晶に対しDSC分析を行うと、その減輝転移温度は206℃であった。
【0043】
得られた結晶に対しKBrペレットを用いたIR分析を行った。その特性ピークは次のとおりである:3090.6、3011.6、2889.1、2747.4、2636.2、1631.5、1602.5、1584.3、1487.7、1466.2、1454.5、1435.6、1354.9、1302.4、1257.0、1217.0、1168.2、1088.6、1064.9、1028.0、1014.6、1001.3、975.8、918.0、887.7、861.8、825.9、789.9、766.4、715.2、698.1、663.7、646.7、620.3、587.3、574.9、560.5、526.9および487.9cm-1。
【0044】
前記スペクトルデータは、本実施例にて得られた結晶形がα結晶形であることを示した。
【0045】
[実施例2〜10]
α結晶形ピノセブリンのサンプルの製造方法2〜10
実施例1に基づいて、溶媒として酢酸エチル、クロロホルム、アセトン、アセトニトリル、THF,エーテル、ベンゼン、シクロヘキサン、又は、DMFを用いて、白色のピノセブリンの結晶を得た。実験の結果を表6に示した。その得られた結晶に対し粉末X線回折、DSC,及び、IR分析を行い、その結果によれば、結晶形がα結晶形ピノセンブリンであることが分かった。
【0046】
【表6】
【0047】
[実施例11]
α結晶形ピノセンブリンサンプルの製造方法11
室温で5gのピノセンブリンサンプルを100mlの95%エタノール及びアセトン(95%エタノール:アセトン=1:1)の混合物に完全に溶かした。その後、攪拌しながら100mlの水を加えると、白色の沈殿が現れた。その沈殿を減圧濾過し、乾燥させて4.00gの白色の結晶(純度:98.7%)を得た。その収率は、80%であった。その得られた結晶形に対し粉末X線解析、DSC,及び、IR分析を行った。その結果によれば、得られた結晶形は、α結晶形ピノセンブリンであった。
【0048】
[実施例12〜16]
αピノセンブリンサンプルの製造方法12〜16
実施例1に記載された製造方法に基づいて、溶媒として、イソプロパノール及びTHF(イソプロパノール:TFH=2:1)の混合物、アセトニトリル及びDMFの混合物(アセトニトリル:DMF=4:1)、メタノール及びアセトンの混合物(メタノール:アセトン=3:2)、エタノール及びアセトニトリルの混合物(エタノール:アセトニトリル=1:1)、及び、エタノール、アセトン、氷酢酸の混合物(エタノール:アセトン:氷酢酸=2:1:0.1)を用いて、白色のピノセンブリン結晶を得た。実験結果を表7に示す。その得られた結晶に対し粉末X線回折、DSC,及び、IR分析を行った。その結果によれば、得られた結晶形は、α結晶形ピノセンブリンであった。
【0049】
【表7】
【0050】
β結晶形ピノセンブリンサンプルの製造
【0051】
[実施例17]
β結晶形ピノセンブリンサンプルの製造方法1
10gのα結晶形ピノセンブリンをモルタルに入れて、室温で1時間同じ方向に砕けた。それにより白色の結晶が得られたが、それはα結晶形ピノセンブリンとは異なるものであった。
【0052】
得られた結晶に対し粉末(多結晶)X線回折(CuKα線)を行った。回折ピーク位置の特性ピーク値:2-シータ値(°)又はd値(Å)及び回折ピークの相対的な強度:ピーク高さ値(高さ%)又はピーク面積値(面積%)は表5に示し、得られたトレース(trace)は図7に示した。
【0053】
得られた結晶に対しDSC分析を行うと、DSCトレースにおいて示されたとおり、減輝転移温度は204℃であった。
【0054】
得られた結晶に対しKBrペレットを用いたIR分析を行った。特性ピークは次のとおりである:3090.8、2890.0、2748.9、2638.3、1633.5、1602.9、1585.0、1487.9、1466.1、1454.3、1344.4、1302.7、1216.7、1168.2、1088.4、1065.5、1028.8、1014.3、1001.5、975.8、917.8、888.2、861.8、826.6、789.1、766.6、741.1、715.4、698.0、663.7、646.0、620.5、587.9、574.8、560.9、527.2および488.4cm-1。
【0055】
前記スペクトルデータによれば、この実施例において得られた結晶形は、β結晶形ピノセンブリンであることが分かった。
【0056】
[実施例18]
β結晶形ピノセンブリンの製造方法2
室温で5gのピノセンブリンサンプルを75mlのDMSOに溶かし、攪拌しながら150mlの水を加えると、白色の沈殿が現れた。その沈殿をろ過し、乾燥して、4.2gの白色の結晶を得た(純度:98.8%、HPLCによって検出)。その収率は84.0%であった。得られた結晶に対し粉末X線回折、DSC,及び、IR分析を行った。その結果によれば、得られた結晶は、β結晶性ピノセンブリンであることが分かった。
【0057】
[実施例19]
β結晶形ピノセンブリンの製造方法3
溶媒としてピリジンを用いる点を除いて、実施例18と同じ製造方法を用いて、8.8gの白色の結晶を得た(純度:98.6%、HPLCによって検出)。その収率は88.0%であった。得られた結晶に対し粉末X線回折、DSC,及び、IR分析を行った。その結果によれば、得られた結晶形はβ結晶性ピノセンブリンであることが分かった。
【0058】
[実施例20]
α結晶形及びβ結晶形のピノセンブリン混合物(1:1)サンプルの製造方法
10gのα結晶形ピノセンブリンサンプル及びβ結晶形ピノセンブリンサンプルをそれぞれ量り、密封性容器に入れた。その容器を密封し、固体が均一に混合されるまでよく振った。α結晶形及びβ結晶形の混合物サンプル(1:1)が得られた。
【0059】
製剤(formulation)
【0060】
[実施例21]
混合医薬製剤(錠剤)の製造方法1
純粋なα結晶形サンプル、純粋なβ結晶形サンプル、又は、(α+β)結晶形の固体混合物(α:β=1:1)を賦形剤(excipient)と任意の比率で混合し、組み合わされた固体の医薬活性成分を得た。活性成分5〜60mgを含む錠剤を製造した。その組成を表8に示す。
【0061】
【表8】
【0062】
製造方法の詳細は、次のとおりである:ピノセンブリンと賦形剤を均一に混合し、適量の1%ナトリウムヒドロキシルメチルセルロース(sodium hydroxymethyl cellulose)溶液を加えて生地(dough)を製造した。その生地をスクリーンして、顆粒を得た。ぬれた顆粒(wet granule)を乾燥させて、ふるいにかけた。その後、ステアリン酸マグネシウム及びタルク粉を加えて、均一に混合した。その生成物を錠剤にした。
【0063】
[実施例22]
混合医薬製剤の製造方法2(カプセル)
純粋なα結晶形サンプル、純粋なβ結晶形サンプル、(α+β)結晶形の固体混合物(α:β=1:1又は1:3)を賦形剤と任意の比率で混合して、医薬活性成分(固体)を得た。活性成分5〜60mgを含むカプセルを製造した。そのカプセルの組成を表9に示す。
【0064】
【表9】
【0065】
製造方法の詳細は、次のとおりである:ピノセンブリンと賦形剤を均一に混合し、適量の1%ナトリウムヒドロキシルメチルセルロース溶液を加えて、ぬれた顆粒を製造した。そのぬれた顆粒を乾燥させて、ふるいにかけた。その後、ステアリン酸マグネシウムを加えて、均一に混合した。その混合物を空っぽのカプセルに充填することによって生成物を得た。或いは、顆粒化(granulation)することなく、賦形剤とピノセンブリンを均一に混合し、ふるいにかけてから、空っぽのカプセルに直接充填することで生成物を得た。
【0066】
[実施例23]
混合医薬製剤の製造方法3(注射溶液及び注射用凍結乾燥粉末)
純粋なα結晶形サンプル、純粋なβ結晶形サンプル、(α+β)結晶形の固体混合物(α:β=1:1)を賦形剤と任意の比率で混合して、医薬活性成分(固体)を得た。その後、アンプルあたり活性成分5〜60mgを含む注射液を製造した。その注射剤の組成を表10に示す。
【0067】
【表10】
【0068】
製剤1:ピノセンブリン注射液の製造
(1)40gのヒドロキシプロピル−β−シクロデキストリンを400mlの蒸留水に加えて、攪拌しながら溶かし;
(2)20mlのエタノールに1gのピノセンブリンを加え、溶かし、そして、得られた溶液を前述のヒドロキシ−β−シクロデキストリン溶液に加え;
(3)混合された溶液を40〜50℃にて20分間磁気的に攪拌した。溶液が透明になったら、0.5gの活性炭(注射用)を加えた。その後、混合物を攪拌しながら80℃に加熱し、この温度を15分間保ち、その活性炭をろ過した。そのろ液をアンプルにそれぞれ4mlずつ入れた。121℃で15分間滅菌した後、ピノセンブリン注射液を得た。
【0069】
製剤2:注射用凍結乾燥粉末の製造
(1)滅菌室において、40gのヒドロキシプロピル−β−シクロデキストリンを量り、水に溶かして150ml溶液を製造した。0.1gの活性炭を加えて、その混合物を15分間軽く沸騰させた。その後、活性炭をろ過除去した。
(2)1gのピノセンブリンを20mlの無水エタノールに溶かし、それによって得られた溶液を前述のヒドロキシプロピル−β−シクロデキストリン溶液に加えた。
(3)その混合物を40〜50℃で20分間磁気的に攪拌した。その溶液が透明になったら、ヒドロキシプロピル−β−シクロデキストリン中ピノセンブリンの包接錯体溶液(inclusion complex solution)を得た。
(4)その包接錯体溶液に水を加えて200mlに調整した。その混合物を0.22μmろ過膜を通じてろ過した。そのろ液を10mlのバイアルに入れ、凍結乾燥器内において凍結乾燥させた。そのバイアルのストッパーを密封して、注射用滅菌粉末を得た。
【0070】
製剤3:ピノセンブリン塩化ナトリウム点滴(infusion)の製造
(1)200mlの蒸留水に、20gのヒドロキシプロピル−β−シクロデキストリンを加えて、攪拌しながら溶かした。点滴用活性炭0.5gを加えた。その混合物を攪拌しながら80℃に加熱し、その温度を15分間保ち、その活性炭をろ過した。
(2)1gのピノセンブリンを量り、20mlの無水エタノールに溶かし、それによって得られた溶液を前述のヒドロキシプロピル−β−シクロデキストリン溶液に入れた。
(3)その混合物を40〜50℃で20分間磁気的に攪拌した。その溶液が透明になったら、ヒドロキシプロピル−β−シクロデキストリン中ピノセンブリンの包接錯体溶液を得た。
(4)その包接錯体溶液に水を加えて800mlに調整した。注射用塩化ナトリウム90gを加えて、溶液のpHを8〜9に調整し、水を用いてイ10000mlに希釈した。その後、10gの活性炭(注射用)を加えて、20分間攪拌した。
(5)活性炭を除去したら、その溶液を100mlのペットボトルに入れた。121℃で30分間滅菌してからその生成物を得た。
【0071】
製剤4:ピノセンブリンデキストロース点滴の製造
(1)20gのヒドロキシプロピル−β−シクロデキストリンを200mlの蒸留水に加えて、攪拌しながら溶かした。0.5gの活性炭(点滴用)を加えた。攪拌しながらその混合物を80℃に加熱し、その温度を15分間保ち、活性炭をろ過した。
(2)1gのピノセンブリンを20mlの無水エタノールに溶かし、それにより得られた溶液を前述のヒドロキシプロピル−β−シクロデキストリンの溶液に加えた。
(3)その混合物を40〜50℃で20分間磁気的に攪拌した。その溶液が透明になったら、ヒドロキシプロピル−β−シクロデキストリン中ピノセンブリンの包接錯体溶液を得た。
(4)その包接錯体溶液に水を加えて800mlに調整した。注射用グルコース500gを加えて、その溶液をpH8〜9に調整し、水を用いて10000mlに希釈した。その後、10gの活性炭(注射用)を加えて、20分間攪拌した。
(5)活性炭を除去した後、その溶液を100mlのペットボトルに入れた。121℃で30分間滅菌処理して、生成物を得た。
【0072】
[実施例24]
α及びβ結晶形ピノセンブリン固体活性成分のインビボ吸収及び血中濃度特徴
体重230g〜250gの18匹のSDラット(雄、雌が半々ずつ)をランダムに3つのグループ(一つのグループには6匹が属し、そのうち半数が雌である)に分けた。10時間断食(水は自由に与えられた)させたラットにα結晶形、β結晶形、又は、1:1(α+β)結晶形のピノセンブリン固体活性成分粉末を胃に投与した(投与量:50mg/kg)。その後、異なる時間における動脈血サンプルをとり、ピノセンブリンの含量を測定した。その結果によれば、異なる結晶形ピノセンブリンを経口投与によって同量投与したときに、血中濃度及びピーク濃度に達する時間が異なった。α結晶形ピノセンブリンの血中濃度はβ結晶形ピノセンブリンよりも明らかに低かった。
【0073】
【表11】
【0074】
[参考文献]
1.中国特許公報第CN1695608A号
2.Cheng Yonghao, etc. synthesis of 5,7-dihydricflavanone, chemical reagents, 2006, 28(7): 437
【技術分野】
【0001】
本発明は、2種の結晶形ピノセンブリン(pinocembrin)、および、医薬活性成分、2種の結晶形ピノセンブリンを含む医薬組成物および剤形、医薬組成物の製造および疾病の治療におけるその使用、並びに、2種のピノセンブリンの製造方法に関する。
【背景技術】
【0002】
ピノセンブリン(化学名:5,7-ジヒドロキシ‐2-フェニル-4-クロマノンは、天然に広く存在するフラボン(flavone)化合物である。その化学式は次の[化1]に示すとおりで、l-異性体、d-異性体、l-異性体又はd-異性体を多く含む混合物、又は、ラセミ化合物の形態で存在する。
【0003】
【化1】
【0004】
これまでの薬物研究によれば、ピノセンブリンは、強い静菌、抗ウイルス、および、抗真菌活性を有する。例えば、中国伝統健康管理食品である蜂蜜には、ピノセンブリンが多く含まれている。蜂蜜を頻繁に食べても歯には悪影響がないだけでなく、口腔の殺菌効果も期待できる。例えば、口内炎を和らげ、治癒を促す。中国特許CN169508A(発明の名称:神経細胞損傷に関連した疾病の治療および予防用医薬組成物の製造におけるピノセンブリンの使用;参考文献1)には、脳虚血、脳虚血の後遺症、神経細胞の損傷および機能の変質に関連した疾病を治療又は予防するための医薬組成物を製造することにおけるピノセンブリンの使用が記載されている。
【発明の概要】
【発明が解決しようとする課題】
【0005】
発明者らは、ピノセンブリンが2種の結晶形αおよびβを有することと、その製造法を見出した。また、本発明者らは、これらの2種の結晶形の間には生命体による取込み(吸収)に有意な差があることに着目した。要するに、β結晶形の取込み率(吸収率)のほうが、α結晶形よりも大きい。例えば、β結晶形の取込み率が、α結晶形の取込み率の2倍以上であり得る。薬物療法におけるこれらの生物学的活性は、血中薬物濃度差に基づいて異なってくる。
【課題を解決するための手段】
【0006】
本発明は、α結晶形ピノセンブリン、β結晶形ピノセンブリン、又は、任意の比率のα結晶形ピノセンブリンとβ結晶形ピノセンブリンの混合物を提供する。これらの結晶形又は結晶形の混合物は、結晶水又はその他の有機溶媒を含まないことが好ましい。
【0007】
また、本発明は、α結晶形ピノセンブリン、β結晶形ピノセンブリン、又は、任意の比率のα結晶形ピノセンブリンとβ結晶形ピノセンブリンの混合物の製造方法を提供する。
【0008】
また、本発明は、純粋なα結晶形ピノセンブリン、純粋なβ結晶形ピノセンブリン、又は、任意の比率のα結晶形ピノセンブリンとβ結晶形ピノセンブリンの混合物を含む医薬組成物を提供する。この医薬組成物は、1以上の医薬的に許可可能な担体を含むことができる。これらの担体に特に制限はなく、製剤に適したもので、本発明に係る結晶形ピノセンブリンに悪影響を及ぼさないものであれば使用することができる。
【0009】
また、本発明は、結晶形ピノセンブリン固体を含む剤形を提供する。剤形に特に制限はない。例えば、錠剤、カプセル、丸薬、注射剤、徐放性製剤、制御放出製剤などの剤形を有する。
【0010】
また、本発明は、治療における薬物の取込み(drug uptake)の差異を起こさせるための、α結晶形ピノセンブリン、β結晶形ピノセンブリン、又は、任意の比率のα結晶形ピノセンブリンとβ結晶形ピノセンブリンの混合物を含む結晶形ピノセンブリン固体の使用を提供する。
【0011】
また、本発明は、生命体における血中濃度を、結晶形効果に基づいて改善(向上)させるとともに、脳虚血関連疾病の治療に神経血管ユニット機能を保護するためのピノセンブリンの使用を提供する。
【発明の効果】
【0012】
本発明に係る結晶形ピノセンブリンは、生命体における血中濃度を結晶形効果(crystal form effect)に基づいて改善させるとともに、脳虚血関連疾病の治療に神経血管ユニット機能を保護するために用いられる。
【図面の簡単な説明】
【0013】
【図1】図1は、分子の相対的な配置を示す。
【図2】図2は、分子の立体構造の投影図である。
【図3】図3は、(軸に沿った)分子の単位セル積み重ねを示す。
【図4】図4は、α結晶形ピノセンブリンサンプルの粉末X線回析パターンである。
【図5】図5は、α結晶形ピノセンブリンサンプルのDSCトレース(trace)である。
【図6】図6は、α結晶形ピノセンブリンサンプルの赤外線吸収スペクトルである。
【図7】図7は、β結晶形ピノセンブリンサンプルの粉末X線回析パターンである。
【図8】図8は、β結晶形ピノセンブリンサンプルのDSCトレースである。
【図9】図9は、β結晶形ピノセンブリンサンプルの赤外線吸収スペクトルである。
【発明を実施するための形態】
【0014】
ピノセンブリンのα結晶形のサンプルの形態学的特徴
本発明の実施例に基づいて得られたα結晶形ピノセンブリンのサンプルをX線単一結晶回析(X-ray single crystal diffraction)によって分析すると、単斜晶対称を示し、その空間群はP21/cであり、結晶セルパラメーター(crystal cell parameter)値は、次のとおりである:a=5.189Å、b=24.149Å、c=10.472Å、α=90°、β=102.31°及び γ=90°。
【0015】
図1は、分子の相対的な配置を示す。図2は、分子の立体構造を投影したものである。図3は、軸に沿った分子の単位セル積み重ねを示す。表1は、原子座標パラメーターおよび同等の温度ファクターを示す。表2は、結合原子の結合距離(値)を示す。表3は、結合原子の結合角(値)を示す。B環の一部の炭素原子が乱れた配向状態(orientation state)有するために、4つの原子C2'、C3'、C5'、およびC6'は2つの位置を占有し、その占有率はそれぞれ0.5である。
【0016】
【化2】
【0017】
化2は、α結晶形ピノセンブリンの相対的な分子配置を示す。
【0018】
【表1】
【0019】
【表2】
【0020】
【表3】
【0021】
表2及び3においては、B環の原子C2'、C3'、C5'、および、C6'につき、一つの位置の結合強度および結合角度のみが記載されている。
【0022】
α結晶形ピノセンブリンの固体に対し粉末(多結晶)X線回析(CuKα線)を行った。回析ピークの位置(2−シータ値(°)又はd値(Å))および回析ピークの相対的な強度(ピークの高さ値(高さ%)又はピーク面積値(面積%))から次の特徴が得られた(表4、図4参照)。
【0023】
【表4】
【0024】
本発明の一実施例によれば、DSCによって分析されたとき、α結晶形ピノセンブリンは、約206℃の減輝転移温度(decalescence transition temperature)を有する(図5)。
【0025】
α結晶形ピノセブリン固体に対しKBrペレットを用いたIR分析を行った(図6参照)。特性ピークは次のとおりである:3090.6、3011.6、2889.1、2747.4、2636.2、1631.5、1602.5、1584.3、1487.7、1466.2、1454.5、1435.6、1354.9、1302.4、1257.0、1217.0、1168.2、1088.6、1064.9、1028.0、1014.6、1001.3、975.8、918.0、887.7、861.8、825.9、789.9、766.4、715.2、698.1、663.7、646.7、620.3、587.3、574.9、560.5、526.9及び487.9cm-1。このうち2891.1、2747.4、2636.2、1631.5および1354.9cm-1のピークは、α結晶形ピノセブリン固体の主な特性ピークである。
【0026】
本発明の実施例に基づくβ結晶形ピノセンブリンサンプルの形態学的特徴
β結晶形ピノセンブリンの固体に対し粉末(多結晶)X線回析(CuKα線)を行った。回析ピークの位置(2−シータ値(°)又はd値(Å))および回析ピークの相対的な強度(ピークの高さ値(%)又はピーク面積値(%))から次の特徴が得られた(表5、図7参照)。
【0027】
【表5】
【0028】
本発明の一実施例によれば、DSCによって分析されたとき、β結晶形ピノセンブリンは、約204℃の減輝転移温度を有する(図8)。
【0029】
β結晶形ピノセブリン固体に対しKBrペレットを用いたIR分析を行った(図9参照)。特性ピークは次のとおりである:3090.8、2890.0、2748.9、2638.3、1633.5、1602.9、1585.0、1487.9、1466.1、1454.3、1344.4、1302.7、1216.7、1168.2、1088.4、1065.5、1028.8、1014.3、1001.5、975.8、917.8、888.2、861.8、826.6、789.1、766.6、741.1、715.4、698.0、663.7、646.0、620.5、587.9、574.8、560.9、527.2及び488.4cm-1。このうち2890.0、2748.9、2638.3、1633.5および1344.4cm-1のピークは、β結晶形ピノセブリン固体の特性ピークである。
【0030】
本発明の実施例に基づくα結晶形ピノセンブリンサンプルの製造法
(1)サンプルをメタノール、エタノール、クロロホルム、アセトン、酢酸エチル、n‐ブタノール、イソプロパノール、アセトニトリル、THF、ジオキサン、95%エタノール、氷酢酸、ギ酸、エーテル、ジクロロメタン、トルエン、ベンゼン、n−ヘキサン、シクロヘキサン、DMF、石油エーテル、アンモニア、n−プロパノール、又は、これらの混合物からなる群から選択された溶媒に完全に溶かし、その後、
(a)その混合物を4〜50℃の温度および相対湿度10〜75%の条件下で放置して(1〜60日)再結晶化させるか、又は、
(b)水を加えて沈殿させた後、減圧濾過、凍結乾燥、又は、コールドスプレーによってα結晶形ピノセンブリンを得る。
【0031】
本発明の実施例に基づくβ結晶形ピノセンブリンサンプルの製造法
材料としてα結晶形固体サンプルを用いる。その後、
(a)粉砕(grinding)による結晶転移(crystal trasition)、又は、
(b)材料をピリジン又はDMSOの溶媒に完全に溶かし、水を加えて沈殿させ、減圧濾過、凍結乾燥、又は、コールドスプレーを行うこと
によってβ結晶形ピノセンブリンのサンプルを得る。
【0032】
ピノセンブリンサンプルの薬力学的特徴
本発明に基づく純粋なα結晶形ピノセンブリン、純粋なβ結晶形ピノセンブリン、又は、α結晶形ピノセンブリンとβ結晶形ピノセンブリンの混合物(任意の比率)は、神経血管ユニット機能(neurovascular unit function)を保護することによって、脳虚血に関連した疾病の治療、又は、脳虚血に関連した疾病の予防に役立つ。
【0033】
本発明による純粋なα結晶形および純粋なβ結晶形の間には、生物学的利用能に差が存在する。経口投与の場合、β結晶形の生物学的利用能が、α結晶形よりも2倍大きい。2種の結晶形の混合物(任意の比率)の場合、その生物学的利用能はβ結晶形の含量に基づいて異なってくる。
【0034】
投与量および剤形
純粋なα結晶形、純粋なβ結晶形、又は、それらの混合物(任意の比率)を含む医薬組成物又はその製剤の一日投与量は、ピノセンブリン結晶形固体に基づいて5〜250mgである。剤形は、錠剤、カプセル剤、丸薬、注射剤、徐放出剤、制御放出剤等を含む。
【0035】
以下の説明は、本発明をよりよく理解してもらうために設けられたものであり、本発明を限定するものではない。
【0036】
実施例に採用された装置及び実験条件:
1.単結晶X線分析
装置:MAC DIP-2030Kエリア検出器
実験条件:チューブ電圧(tube voltage):50KV、チューブ電流(tube flow):80 mA、ωスキャニング、MoKα、2θ≦50.0°、スキャン範囲:0〜180°、回転角 (pivot angle):6°、ステップ:6°、スキャン速度:1.8°/分
2.粉末X線分析
装置:リガクD/max2550粉末X線回折計
実験条件:電圧:40KV、電流:150mA、スキャン速度:8°/分
3.DSC分析
装置:セイコーインスツル株式会社製示差走査熱量計
実験条件:パージガス:N2、加熱速度:10℃/分
温度範囲:25-250℃
4.IR吸収スペクトル
装置:NicoletFT-IRスペクトルメータ:IMPACT400
実験件:KBrペレット
5.HPLC分析
装置:SHIMADZU LC-10Avp高性能液体クロマトグラフィ、SPD-M10Avpダイオード層検出器、CLASS-VPクロマトグラフィデータシステム;カラム:Alltch C18(5μ,150×4.6mm);実験条件:カラム温度;波長:290nm;移動相:メタノール/燐酸塩食塩水pH3.0(64/36);流速(流量):1.0 mL/分;注入容積:20μL;注入濃度:500.0μg/mL
【0037】
製造1:ピノセンブリンサンプルの合成
1000mlの水素化反応容器に5g(19.7mmol)の5,7-ジヒドロキシフラボン、650mlの無水エタノール、及び、1.5gの10%パラジウム(炭素上)を加えた。0.13Mpaの水素圧下で、40℃にて4時間反応させた。反応が終ったら、パラジウム炭素(palladium on carbon)をろ過した。そのろ液を濃縮し、分離し、真空下でカラムクロマトグラフィ(エタノール:酢酸エーテル:石油エーテル=2:10:100 (V:V:V)によって溶出される。)によって精製した。その溶媒を蒸発させて乾燥し、白色の無定形固体粉末3.9gを得た(純度:98.6%、HPLCによって検出)。その収率は、52%であった(参考文献2)。
【0038】
α結晶形ピノセンブリンサンプルの製造
【0039】
[実施例1]
α結晶形ピノセンブリンサンプルの製造方法
5gのピノセンブリンサンプルを20mlの95%エタノールに加え、それが完全に溶けるまで加熱した。その後、室温に冷却して、24時間放置した。白色の固体を沈殿させ、ろ過し、乾燥させた。4.5gの白色の結晶(純度:98.8%、HPLCによって検出)を得た(回収率:90%)
【0040】
それにより得られた結晶をX線単結晶回折によって分析した。それは単斜対称を示し、スペース群(space group)はP21/cであり、結晶セルパラメーター(crystal cell parameter)値はa=5.189Å、b=24.149Å、c=10.472Å、α=90°、β=102.31°、及び、γ=90°であった。
【0041】
得られた結晶に対し粉末(多結晶)X線回折(CuKα線)を行った。回折ピーク位置の特性ピーク値:2-シータ値(°)又はd値(Å)及び回折ピークの相対的な強度:ピーク高さ値(高さ%)又はピーク面積値(面積%)は表4に示し、得られたトレース(trace)は図4に示す。
【0042】
得られた結晶に対しDSC分析を行うと、その減輝転移温度は206℃であった。
【0043】
得られた結晶に対しKBrペレットを用いたIR分析を行った。その特性ピークは次のとおりである:3090.6、3011.6、2889.1、2747.4、2636.2、1631.5、1602.5、1584.3、1487.7、1466.2、1454.5、1435.6、1354.9、1302.4、1257.0、1217.0、1168.2、1088.6、1064.9、1028.0、1014.6、1001.3、975.8、918.0、887.7、861.8、825.9、789.9、766.4、715.2、698.1、663.7、646.7、620.3、587.3、574.9、560.5、526.9および487.9cm-1。
【0044】
前記スペクトルデータは、本実施例にて得られた結晶形がα結晶形であることを示した。
【0045】
[実施例2〜10]
α結晶形ピノセブリンのサンプルの製造方法2〜10
実施例1に基づいて、溶媒として酢酸エチル、クロロホルム、アセトン、アセトニトリル、THF,エーテル、ベンゼン、シクロヘキサン、又は、DMFを用いて、白色のピノセブリンの結晶を得た。実験の結果を表6に示した。その得られた結晶に対し粉末X線回折、DSC,及び、IR分析を行い、その結果によれば、結晶形がα結晶形ピノセンブリンであることが分かった。
【0046】
【表6】
【0047】
[実施例11]
α結晶形ピノセンブリンサンプルの製造方法11
室温で5gのピノセンブリンサンプルを100mlの95%エタノール及びアセトン(95%エタノール:アセトン=1:1)の混合物に完全に溶かした。その後、攪拌しながら100mlの水を加えると、白色の沈殿が現れた。その沈殿を減圧濾過し、乾燥させて4.00gの白色の結晶(純度:98.7%)を得た。その収率は、80%であった。その得られた結晶形に対し粉末X線解析、DSC,及び、IR分析を行った。その結果によれば、得られた結晶形は、α結晶形ピノセンブリンであった。
【0048】
[実施例12〜16]
αピノセンブリンサンプルの製造方法12〜16
実施例1に記載された製造方法に基づいて、溶媒として、イソプロパノール及びTHF(イソプロパノール:TFH=2:1)の混合物、アセトニトリル及びDMFの混合物(アセトニトリル:DMF=4:1)、メタノール及びアセトンの混合物(メタノール:アセトン=3:2)、エタノール及びアセトニトリルの混合物(エタノール:アセトニトリル=1:1)、及び、エタノール、アセトン、氷酢酸の混合物(エタノール:アセトン:氷酢酸=2:1:0.1)を用いて、白色のピノセンブリン結晶を得た。実験結果を表7に示す。その得られた結晶に対し粉末X線回折、DSC,及び、IR分析を行った。その結果によれば、得られた結晶形は、α結晶形ピノセンブリンであった。
【0049】
【表7】
【0050】
β結晶形ピノセンブリンサンプルの製造
【0051】
[実施例17]
β結晶形ピノセンブリンサンプルの製造方法1
10gのα結晶形ピノセンブリンをモルタルに入れて、室温で1時間同じ方向に砕けた。それにより白色の結晶が得られたが、それはα結晶形ピノセンブリンとは異なるものであった。
【0052】
得られた結晶に対し粉末(多結晶)X線回折(CuKα線)を行った。回折ピーク位置の特性ピーク値:2-シータ値(°)又はd値(Å)及び回折ピークの相対的な強度:ピーク高さ値(高さ%)又はピーク面積値(面積%)は表5に示し、得られたトレース(trace)は図7に示した。
【0053】
得られた結晶に対しDSC分析を行うと、DSCトレースにおいて示されたとおり、減輝転移温度は204℃であった。
【0054】
得られた結晶に対しKBrペレットを用いたIR分析を行った。特性ピークは次のとおりである:3090.8、2890.0、2748.9、2638.3、1633.5、1602.9、1585.0、1487.9、1466.1、1454.3、1344.4、1302.7、1216.7、1168.2、1088.4、1065.5、1028.8、1014.3、1001.5、975.8、917.8、888.2、861.8、826.6、789.1、766.6、741.1、715.4、698.0、663.7、646.0、620.5、587.9、574.8、560.9、527.2および488.4cm-1。
【0055】
前記スペクトルデータによれば、この実施例において得られた結晶形は、β結晶形ピノセンブリンであることが分かった。
【0056】
[実施例18]
β結晶形ピノセンブリンの製造方法2
室温で5gのピノセンブリンサンプルを75mlのDMSOに溶かし、攪拌しながら150mlの水を加えると、白色の沈殿が現れた。その沈殿をろ過し、乾燥して、4.2gの白色の結晶を得た(純度:98.8%、HPLCによって検出)。その収率は84.0%であった。得られた結晶に対し粉末X線回折、DSC,及び、IR分析を行った。その結果によれば、得られた結晶は、β結晶性ピノセンブリンであることが分かった。
【0057】
[実施例19]
β結晶形ピノセンブリンの製造方法3
溶媒としてピリジンを用いる点を除いて、実施例18と同じ製造方法を用いて、8.8gの白色の結晶を得た(純度:98.6%、HPLCによって検出)。その収率は88.0%であった。得られた結晶に対し粉末X線回折、DSC,及び、IR分析を行った。その結果によれば、得られた結晶形はβ結晶性ピノセンブリンであることが分かった。
【0058】
[実施例20]
α結晶形及びβ結晶形のピノセンブリン混合物(1:1)サンプルの製造方法
10gのα結晶形ピノセンブリンサンプル及びβ結晶形ピノセンブリンサンプルをそれぞれ量り、密封性容器に入れた。その容器を密封し、固体が均一に混合されるまでよく振った。α結晶形及びβ結晶形の混合物サンプル(1:1)が得られた。
【0059】
製剤(formulation)
【0060】
[実施例21]
混合医薬製剤(錠剤)の製造方法1
純粋なα結晶形サンプル、純粋なβ結晶形サンプル、又は、(α+β)結晶形の固体混合物(α:β=1:1)を賦形剤(excipient)と任意の比率で混合し、組み合わされた固体の医薬活性成分を得た。活性成分5〜60mgを含む錠剤を製造した。その組成を表8に示す。
【0061】
【表8】
【0062】
製造方法の詳細は、次のとおりである:ピノセンブリンと賦形剤を均一に混合し、適量の1%ナトリウムヒドロキシルメチルセルロース(sodium hydroxymethyl cellulose)溶液を加えて生地(dough)を製造した。その生地をスクリーンして、顆粒を得た。ぬれた顆粒(wet granule)を乾燥させて、ふるいにかけた。その後、ステアリン酸マグネシウム及びタルク粉を加えて、均一に混合した。その生成物を錠剤にした。
【0063】
[実施例22]
混合医薬製剤の製造方法2(カプセル)
純粋なα結晶形サンプル、純粋なβ結晶形サンプル、(α+β)結晶形の固体混合物(α:β=1:1又は1:3)を賦形剤と任意の比率で混合して、医薬活性成分(固体)を得た。活性成分5〜60mgを含むカプセルを製造した。そのカプセルの組成を表9に示す。
【0064】
【表9】
【0065】
製造方法の詳細は、次のとおりである:ピノセンブリンと賦形剤を均一に混合し、適量の1%ナトリウムヒドロキシルメチルセルロース溶液を加えて、ぬれた顆粒を製造した。そのぬれた顆粒を乾燥させて、ふるいにかけた。その後、ステアリン酸マグネシウムを加えて、均一に混合した。その混合物を空っぽのカプセルに充填することによって生成物を得た。或いは、顆粒化(granulation)することなく、賦形剤とピノセンブリンを均一に混合し、ふるいにかけてから、空っぽのカプセルに直接充填することで生成物を得た。
【0066】
[実施例23]
混合医薬製剤の製造方法3(注射溶液及び注射用凍結乾燥粉末)
純粋なα結晶形サンプル、純粋なβ結晶形サンプル、(α+β)結晶形の固体混合物(α:β=1:1)を賦形剤と任意の比率で混合して、医薬活性成分(固体)を得た。その後、アンプルあたり活性成分5〜60mgを含む注射液を製造した。その注射剤の組成を表10に示す。
【0067】
【表10】
【0068】
製剤1:ピノセンブリン注射液の製造
(1)40gのヒドロキシプロピル−β−シクロデキストリンを400mlの蒸留水に加えて、攪拌しながら溶かし;
(2)20mlのエタノールに1gのピノセンブリンを加え、溶かし、そして、得られた溶液を前述のヒドロキシ−β−シクロデキストリン溶液に加え;
(3)混合された溶液を40〜50℃にて20分間磁気的に攪拌した。溶液が透明になったら、0.5gの活性炭(注射用)を加えた。その後、混合物を攪拌しながら80℃に加熱し、この温度を15分間保ち、その活性炭をろ過した。そのろ液をアンプルにそれぞれ4mlずつ入れた。121℃で15分間滅菌した後、ピノセンブリン注射液を得た。
【0069】
製剤2:注射用凍結乾燥粉末の製造
(1)滅菌室において、40gのヒドロキシプロピル−β−シクロデキストリンを量り、水に溶かして150ml溶液を製造した。0.1gの活性炭を加えて、その混合物を15分間軽く沸騰させた。その後、活性炭をろ過除去した。
(2)1gのピノセンブリンを20mlの無水エタノールに溶かし、それによって得られた溶液を前述のヒドロキシプロピル−β−シクロデキストリン溶液に加えた。
(3)その混合物を40〜50℃で20分間磁気的に攪拌した。その溶液が透明になったら、ヒドロキシプロピル−β−シクロデキストリン中ピノセンブリンの包接錯体溶液(inclusion complex solution)を得た。
(4)その包接錯体溶液に水を加えて200mlに調整した。その混合物を0.22μmろ過膜を通じてろ過した。そのろ液を10mlのバイアルに入れ、凍結乾燥器内において凍結乾燥させた。そのバイアルのストッパーを密封して、注射用滅菌粉末を得た。
【0070】
製剤3:ピノセンブリン塩化ナトリウム点滴(infusion)の製造
(1)200mlの蒸留水に、20gのヒドロキシプロピル−β−シクロデキストリンを加えて、攪拌しながら溶かした。点滴用活性炭0.5gを加えた。その混合物を攪拌しながら80℃に加熱し、その温度を15分間保ち、その活性炭をろ過した。
(2)1gのピノセンブリンを量り、20mlの無水エタノールに溶かし、それによって得られた溶液を前述のヒドロキシプロピル−β−シクロデキストリン溶液に入れた。
(3)その混合物を40〜50℃で20分間磁気的に攪拌した。その溶液が透明になったら、ヒドロキシプロピル−β−シクロデキストリン中ピノセンブリンの包接錯体溶液を得た。
(4)その包接錯体溶液に水を加えて800mlに調整した。注射用塩化ナトリウム90gを加えて、溶液のpHを8〜9に調整し、水を用いてイ10000mlに希釈した。その後、10gの活性炭(注射用)を加えて、20分間攪拌した。
(5)活性炭を除去したら、その溶液を100mlのペットボトルに入れた。121℃で30分間滅菌してからその生成物を得た。
【0071】
製剤4:ピノセンブリンデキストロース点滴の製造
(1)20gのヒドロキシプロピル−β−シクロデキストリンを200mlの蒸留水に加えて、攪拌しながら溶かした。0.5gの活性炭(点滴用)を加えた。攪拌しながらその混合物を80℃に加熱し、その温度を15分間保ち、活性炭をろ過した。
(2)1gのピノセンブリンを20mlの無水エタノールに溶かし、それにより得られた溶液を前述のヒドロキシプロピル−β−シクロデキストリンの溶液に加えた。
(3)その混合物を40〜50℃で20分間磁気的に攪拌した。その溶液が透明になったら、ヒドロキシプロピル−β−シクロデキストリン中ピノセンブリンの包接錯体溶液を得た。
(4)その包接錯体溶液に水を加えて800mlに調整した。注射用グルコース500gを加えて、その溶液をpH8〜9に調整し、水を用いて10000mlに希釈した。その後、10gの活性炭(注射用)を加えて、20分間攪拌した。
(5)活性炭を除去した後、その溶液を100mlのペットボトルに入れた。121℃で30分間滅菌処理して、生成物を得た。
【0072】
[実施例24]
α及びβ結晶形ピノセンブリン固体活性成分のインビボ吸収及び血中濃度特徴
体重230g〜250gの18匹のSDラット(雄、雌が半々ずつ)をランダムに3つのグループ(一つのグループには6匹が属し、そのうち半数が雌である)に分けた。10時間断食(水は自由に与えられた)させたラットにα結晶形、β結晶形、又は、1:1(α+β)結晶形のピノセンブリン固体活性成分粉末を胃に投与した(投与量:50mg/kg)。その後、異なる時間における動脈血サンプルをとり、ピノセンブリンの含量を測定した。その結果によれば、異なる結晶形ピノセンブリンを経口投与によって同量投与したときに、血中濃度及びピーク濃度に達する時間が異なった。α結晶形ピノセンブリンの血中濃度はβ結晶形ピノセンブリンよりも明らかに低かった。
【0073】
【表11】
【0074】
[参考文献]
1.中国特許公報第CN1695608A号
2.Cheng Yonghao, etc. synthesis of 5,7-dihydricflavanone, chemical reagents, 2006, 28(7): 437
【特許請求の範囲】
【請求項1】
以下の[化1]にて表されるα結晶形ピノセンブリンであって、
【化1】
X線単一結晶回析によって分析されたとき、単斜晶対称を示し、空間群はP21/cであり、そして、結晶セルパラメーター値は、a=5.189Å、b=24.149Å、c=10.472Å、α=90°、β=102.31°及びγ=90°であるα結晶形ピノセンブリン。
【請求項2】
以下の[化2]にて表されるα結晶形ピノセンブリンであって、
【化2】
X線粉末(多結晶)回析(CuKα線)によって分析されたとき、回析ピークの位置(2−シータ値(°)又はd値(Å))、および、回析ピークの相対的な強度(ピーク高さ値(高さ%)又はピーク面積値(面積%))が、次の[表1]に示す特徴を有するα結晶形ピノセンブリン。
【表1】
【請求項3】
DSCトレースにおける減輝転移温度が、約206℃である、請求項1又は2に記載のα結晶形ピノセンブリン。
【請求項4】
赤外線吸収スペクトルが、3090.6、3011.6、2889.1、2747.4、2636.2、1631.5、1602.5、1584.3、1487.7、1466.2、1454.5、1435.6、1354.9、1302.4、1257.0、1217.0、1168.2、1088.6、1064.9、1028.0、1014.6、1001.3、975.8、918.0、887.7、861.8、825.9、789.9、766.4、715.2、698.1、663.7、646.7、620.3、587.3、574.9、560.5、526.9及び487.9cm-1、のピークを示し、前記ピークのうち2891.1、2747.4、2636.2、1631.5及び1354.9 cm-1のピークが、α結晶形ピノセンブリンの主な特性ピークである、請求項1又は2に記載のα結晶形ピノセンブリン。
【請求項5】
以下の[化3]にて表されるβ結晶形ピノセンブリンであって、
【化3】
X線粉末(多結晶)回析(CuKα線)によって分析されたとき、回析ピークの位置(2−シータ値(°)又はd値(Å))、および、回析ピークの相対的な強度(ピークの高さ値(高さ%)又はピーク面積値(面積%))が、次の[表2]に示す特徴を有するβ結晶形ピノセンブリン。
【表2】
【請求項6】
DSCトレースにおける減輝転移温度が、約204℃である、請求項5に記載のβ結晶形ピノセンブリン。
【請求項7】
赤外線吸収スペクトルが、3090.8、2890.0、2748.9、2638.3、1633.5、1602.9、1585.0、1487.9、1466.1、1454.3、1344.4、1302.7、1216.7、1168.2、1088.4、1065.5、1028.8、1014.3、1001.5、975.8、917.8、888.2、861.8、826.6、789.1、766.6、741.1、715.4、698.0、663.7、646.0、620.5、587.9、574.8、560.9、527.2、及び、488.4cm-1のピークを示し、前記ピークのうち2890.0、2748.9、2638.3、1633.5、及び、1344.4cm-1のピークがβ結晶形ピノセンブリンの主な特性ピークである、請求項5に記載のβ結晶形ピノセンブリン。
【請求項8】
α結晶形ピノセンブリンとβ結晶形ピノセンブリンとが任意の比率で混合されたピノセンブリン。
【請求項9】
前記結晶形が、結晶水、又は、結晶水以外の結晶性溶媒を含まない、請求項1〜8のいずれか一項に記載のピノセンブリン。
【請求項10】
神経血管ユニット機能を保護することによって、脳虚血関連疾病の治療又は予防のための医薬の製造に用いられる、請求項1,2,5、又は、8に記載の結晶形ピノセンブリンを含む医薬活性成分。
【請求項11】
固体結晶形ピノセンブリンに基づく一日投与量が5〜250mgである、請求項10に記載の医薬活性成分。
【請求項12】
治療的有効量の請求項1、2、5、および、8に記載の結晶性ピノセンブリンと、1以上の医薬的に許容可能な担体と、を含む医薬組成物。
【請求項13】
請求項10又は11に記載の医薬活性成分、又は、請求項12に記載の医薬組成物を含む剤形であって、前記剤形が、錠剤、カプセル剤、丸薬、注射剤、徐放性製剤、又は、制御放出製剤である剤形。
【請求項14】
神経血管ユニット機能を保護することによって、脳虚血関連疾病の治療又は予防のための医薬の製造における請求項1、2、5、或いは、8に記載の結晶形ピノセンブリン、請求項10の記載の医薬活性成分、又は、請求項12に記載の医薬組成物の使用。
【請求項15】
医薬の製造における請求項1、2、5、或いは、8に記載の結晶形ピノセンブリン、請求項10の記載の医薬活性成分、又は、請求項12に記載の医薬組成物の使用であって、生命体における前記ピノセンブリン、前記医薬活性成分、又は、前記医薬組成物の血中濃度が、ピノセンブリンの結晶形に基づいて改善される、使用。
【請求項16】
α結晶形ピノセンブリンの製造方法であって、
メタノール、エタノール、クロロホルム、アセトン、酢酸エチル、n−ブタノール、イソプロパノール、アセトニトリル、THF,ジオキサン、95%エタノール、氷酢酸、ギ酸、エーテル、ジクロロメタン、トルエン、ベンゼン、n−へキサン、シクロヘキサン、DMF,石油エーテル、アンモニア、n−プロパノール、又は、これらの混合物からなる群から選択された溶媒にサンプルを溶かし、
(a)4〜50℃の温度、及び、10〜75%の相対湿度下に前記混合物をおき、1〜60日間再結晶化させるか、又は、
(b)水を加えて沈殿化させ、減圧濾過、凍結乾燥又はコールドスプレーを通じてα結晶形ピノセンブリンを得る
ことを含む製造方法。
【請求項17】
β結晶形ピノセンブリンの製造方法であって、
材料としてα結晶形ピノセンブリンを用い、かつ、
(a)粉砕による結晶転移、又は、
(b)ピリジン又はDMSOの溶媒に前記材料を完全に溶かし、水を加えて沈殿化させ、そして、減圧濾過、凍結乾燥、又は、コールドスプレーを行うこと
によってβ結晶形ピノセンブリンを得る
ことを含む製造方法。
【請求項1】
以下の[化1]にて表されるα結晶形ピノセンブリンであって、
【化1】
X線単一結晶回析によって分析されたとき、単斜晶対称を示し、空間群はP21/cであり、そして、結晶セルパラメーター値は、a=5.189Å、b=24.149Å、c=10.472Å、α=90°、β=102.31°及びγ=90°であるα結晶形ピノセンブリン。
【請求項2】
以下の[化2]にて表されるα結晶形ピノセンブリンであって、
【化2】
X線粉末(多結晶)回析(CuKα線)によって分析されたとき、回析ピークの位置(2−シータ値(°)又はd値(Å))、および、回析ピークの相対的な強度(ピーク高さ値(高さ%)又はピーク面積値(面積%))が、次の[表1]に示す特徴を有するα結晶形ピノセンブリン。
【表1】
【請求項3】
DSCトレースにおける減輝転移温度が、約206℃である、請求項1又は2に記載のα結晶形ピノセンブリン。
【請求項4】
赤外線吸収スペクトルが、3090.6、3011.6、2889.1、2747.4、2636.2、1631.5、1602.5、1584.3、1487.7、1466.2、1454.5、1435.6、1354.9、1302.4、1257.0、1217.0、1168.2、1088.6、1064.9、1028.0、1014.6、1001.3、975.8、918.0、887.7、861.8、825.9、789.9、766.4、715.2、698.1、663.7、646.7、620.3、587.3、574.9、560.5、526.9及び487.9cm-1、のピークを示し、前記ピークのうち2891.1、2747.4、2636.2、1631.5及び1354.9 cm-1のピークが、α結晶形ピノセンブリンの主な特性ピークである、請求項1又は2に記載のα結晶形ピノセンブリン。
【請求項5】
以下の[化3]にて表されるβ結晶形ピノセンブリンであって、
【化3】
X線粉末(多結晶)回析(CuKα線)によって分析されたとき、回析ピークの位置(2−シータ値(°)又はd値(Å))、および、回析ピークの相対的な強度(ピークの高さ値(高さ%)又はピーク面積値(面積%))が、次の[表2]に示す特徴を有するβ結晶形ピノセンブリン。
【表2】
【請求項6】
DSCトレースにおける減輝転移温度が、約204℃である、請求項5に記載のβ結晶形ピノセンブリン。
【請求項7】
赤外線吸収スペクトルが、3090.8、2890.0、2748.9、2638.3、1633.5、1602.9、1585.0、1487.9、1466.1、1454.3、1344.4、1302.7、1216.7、1168.2、1088.4、1065.5、1028.8、1014.3、1001.5、975.8、917.8、888.2、861.8、826.6、789.1、766.6、741.1、715.4、698.0、663.7、646.0、620.5、587.9、574.8、560.9、527.2、及び、488.4cm-1のピークを示し、前記ピークのうち2890.0、2748.9、2638.3、1633.5、及び、1344.4cm-1のピークがβ結晶形ピノセンブリンの主な特性ピークである、請求項5に記載のβ結晶形ピノセンブリン。
【請求項8】
α結晶形ピノセンブリンとβ結晶形ピノセンブリンとが任意の比率で混合されたピノセンブリン。
【請求項9】
前記結晶形が、結晶水、又は、結晶水以外の結晶性溶媒を含まない、請求項1〜8のいずれか一項に記載のピノセンブリン。
【請求項10】
神経血管ユニット機能を保護することによって、脳虚血関連疾病の治療又は予防のための医薬の製造に用いられる、請求項1,2,5、又は、8に記載の結晶形ピノセンブリンを含む医薬活性成分。
【請求項11】
固体結晶形ピノセンブリンに基づく一日投与量が5〜250mgである、請求項10に記載の医薬活性成分。
【請求項12】
治療的有効量の請求項1、2、5、および、8に記載の結晶性ピノセンブリンと、1以上の医薬的に許容可能な担体と、を含む医薬組成物。
【請求項13】
請求項10又は11に記載の医薬活性成分、又は、請求項12に記載の医薬組成物を含む剤形であって、前記剤形が、錠剤、カプセル剤、丸薬、注射剤、徐放性製剤、又は、制御放出製剤である剤形。
【請求項14】
神経血管ユニット機能を保護することによって、脳虚血関連疾病の治療又は予防のための医薬の製造における請求項1、2、5、或いは、8に記載の結晶形ピノセンブリン、請求項10の記載の医薬活性成分、又は、請求項12に記載の医薬組成物の使用。
【請求項15】
医薬の製造における請求項1、2、5、或いは、8に記載の結晶形ピノセンブリン、請求項10の記載の医薬活性成分、又は、請求項12に記載の医薬組成物の使用であって、生命体における前記ピノセンブリン、前記医薬活性成分、又は、前記医薬組成物の血中濃度が、ピノセンブリンの結晶形に基づいて改善される、使用。
【請求項16】
α結晶形ピノセンブリンの製造方法であって、
メタノール、エタノール、クロロホルム、アセトン、酢酸エチル、n−ブタノール、イソプロパノール、アセトニトリル、THF,ジオキサン、95%エタノール、氷酢酸、ギ酸、エーテル、ジクロロメタン、トルエン、ベンゼン、n−へキサン、シクロヘキサン、DMF,石油エーテル、アンモニア、n−プロパノール、又は、これらの混合物からなる群から選択された溶媒にサンプルを溶かし、
(a)4〜50℃の温度、及び、10〜75%の相対湿度下に前記混合物をおき、1〜60日間再結晶化させるか、又は、
(b)水を加えて沈殿化させ、減圧濾過、凍結乾燥又はコールドスプレーを通じてα結晶形ピノセンブリンを得る
ことを含む製造方法。
【請求項17】
β結晶形ピノセンブリンの製造方法であって、
材料としてα結晶形ピノセンブリンを用い、かつ、
(a)粉砕による結晶転移、又は、
(b)ピリジン又はDMSOの溶媒に前記材料を完全に溶かし、水を加えて沈殿化させ、そして、減圧濾過、凍結乾燥、又は、コールドスプレーを行うこと
によってβ結晶形ピノセンブリンを得る
ことを含む製造方法。
【図1】

【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【公表番号】特表2012−508694(P2012−508694A)
【公表日】平成24年4月12日(2012.4.12)
【国際特許分類】
【出願番号】特願2011−535851(P2011−535851)
【出願日】平成20年11月13日(2008.11.13)
【国際出願番号】PCT/CN2008/073047
【国際公開番号】WO2010/054512
【国際公開日】平成22年5月20日(2010.5.20)
【出願人】(506417359)石薬集団中奇制薬技術(石家庄)有限公司 (8)
【出願人】(504466591)中国医学科学院薬物研究所 (7)
【Fターム(参考)】
【公表日】平成24年4月12日(2012.4.12)
【国際特許分類】
【出願日】平成20年11月13日(2008.11.13)
【国際出願番号】PCT/CN2008/073047
【国際公開番号】WO2010/054512
【国際公開日】平成22年5月20日(2010.5.20)
【出願人】(506417359)石薬集団中奇制薬技術(石家庄)有限公司 (8)
【出願人】(504466591)中国医学科学院薬物研究所 (7)
【Fターム(参考)】
[ Back to top ]