
2,5−ジアミノメチル−ビシクロ[2,2,1]ヘプタン及びその製造方法
【課題】異性体を実質的に含有しない高純度の2,5−ジアミノメチル−ビシクロ[2,2,1]ヘプタン、及びその製造方法を提供する。
【解決手段】異性体混合物に含まれるそれぞれの化合物のアミノ基を保護基で保護した後、異性体を分離し、次いで分離した異性体の保護基を脱保護することにより、下記式(I)又は(II)で表される、ガスクロマトグラフィーにより分析される純度が95%以上である2,5−ジアミノメチル−ビシクロ[2,2,1]ヘプタンを製造する。
【化1】

【解決手段】異性体混合物に含まれるそれぞれの化合物のアミノ基を保護基で保護した後、異性体を分離し、次いで分離した異性体の保護基を脱保護することにより、下記式(I)又は(II)で表される、ガスクロマトグラフィーにより分析される純度が95%以上である2,5−ジアミノメチル−ビシクロ[2,2,1]ヘプタンを製造する。
【化1】
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、高純度の2,5−ジアミノメチル−ビシクロ[2,2,1]ヘプタン、及びその製造方法に関する。
【背景技術】
【0002】
2,5(又は6)−ジアミノメチル−ビシクロ[2,2,1]ヘプタン(以下、「NBDA」とも記す)は、ポリイミド、ポリアミド等のモノマーとして、またエポキシ樹脂、ビスマレイミド、ジイソシアネート等の原料として、或いはそれらの硬化剤として極めて有用なジアミン化合物である。また、このNBDAを原料に用いることで、光線透過率の良好な(無色透明性の高い)ポリイミドが得られることが知られている(例えば、特許文献1及び2参照)。
【0003】
NBDAは、従来、ビシクロ[2,2,1]−5−ヘプテン−2−カルボニトリルに、パラジウム触媒及びトリフェニルホスファイト、或いは0価ニッケル錯体触媒の存在下でシアン化水素を付加させてジシアノ体とした後、接触水素化することにより製造されている(例えば、特許文献3及び4参照)。このようにして製造されるNBDAには、ビシクロ[2,2,1]ヘプタン環におけるアミノメチル基の置換位置が異なる異性体として、2,5−ジアミノメチル−ビシクロ[2,2,1]ヘプタン(以下、「2,5−NBDA」とも記す)、及び2,6−ジアミノメチル−ビシクロ[2,2,1]ヘプタン(以下、「2,6−NBDA」とも記す)が含まれている。
【0004】
これらの異性体は、蒸留等の通常の分離手段では分離することが非常に困難であることが知られている。更に、2,5−NBDAと2,6−NBDAのそれぞれにも光学異性体が存在する。このため、上記のようにして製造されるNBDAは、一般的な分離手段では分離困難な4種類の異性体を含有する異性体混合物である。
【0005】
NBDAに含まれる4種の異性体を分離する方法として、それぞれの異性体の融点差を利用し、凝固させたNBDAを徐々に昇温させながらそれぞれの異性体が多く含まれる留分に分画する方法が開示されている(例えば、特許文献5参照)。
【先行技術文献】
【特許文献】
【0006】
【特許文献1】特開平10−7906号公報
【特許文献2】国際公開第1998/029471号
【特許文献3】特許第2713612号公報
【特許文献4】特開2002−69043号公報
【特許文献5】国際公開第2002/010253号
【発明の概要】
【発明が解決しようとする課題】
【0007】
特許文献5で開示された異性体の分離方法によると、それぞれの留分に含まれる異性体の量比をある程度制御することは可能であった。しかしながら、他の異性体を実質的に含有しない高純度の化合物を分離精製することは極めて困難であった。
【0008】
本発明は、このような従来技術の有する問題点に鑑みてなされたものであり、その課題とするところは、高純度の2,5−ジアミノメチル−ビシクロ[2,2,1]ヘプタン、及びその製造方法を提供することにある。
【課題を解決するための手段】
【0009】
本発明者らは上記課題を達成すべく鋭意検討した結果、原材料となるジアミノメチル−ビシクロ[2,2,1]ヘプタンの異性体混合物に含まれるそれぞれの化合物のアミノ基を保護基で保護した後、異性体を分離し、次いで分離した異性体の保護基を脱保護することによって、上記課題を達成することが可能であることを見出し、本発明を完成するに至った。
【0010】
即ち、本発明によれば、以下に示す2,5−ジアミノメチル−ビシクロ[2,2,1]ヘプタン、及び2,5−ジアミノメチル−ビシクロ[2,2,1]ヘプタンの製造方法が提供される。
【0011】
[1]下記式(I)又は(II)で表される、ガスクロマトグラフィーにより分析される純度が95%以上である2,5−ジアミノメチル−ビシクロ[2,2,1]ヘプタン。
【0012】
【化1】
【0013】
[2]下記式(I)で表される化合物、下記式(II)で表される化合物、下記式(III)で表される化合物、及び下記式(IV)で表される化合物を含むジアミノメチル−ビシクロ[2,2,1]ヘプタンの異性体混合物に含まれるそれぞれの化合物のアミノ基を保護基で保護した後、下記式(I)又は(II)で表される化合物に由来する異性体を結晶化と固液分離を含む分離操作により分離し、次いで分離した前記異性体の前記保護基を脱保護することを含む、下記式(I)又は(II)で表される、ガスクロマトグラフィーにより分析される純度が95%以上である2,5−ジアミノメチル−ビシクロ[2,2,1]ヘプタンの製造方法。
【0014】
【化2】
【0015】
[3]前記保護基が、フタルイミド基、t−ブチルオキシカルボニル基、又はベンジルオキシカルボニル基である、前記[2]に記載の製造方法。
【発明の効果】
【0016】
本発明の2,5−ジアミノメチル−ビシクロ[2,2,1]ヘプタンは、ガスクロマトグラフィーにより分析される純度が95%以上の、異性体を実質的に含有しない高純度な化合物であり、各種の樹脂材料や化成品の原料として、特に、電子部品の構成材料として用いられるポリイミドフィルムを形成するための原料として極めて有用である。
【0017】
本発明の製造方法によれば、ガスクロマトグラフィーにより分析される純度が95%以上の、異性体を実質的に含有しない高純度な2,5−ジアミノメチル−ビシクロ[2,2,1]ヘプタンを、工業的にも適用可能な簡便な操作で製造することができる。
【図面の簡単な説明】
【0018】
【図1】実施例1で得られた2,5−diexo−ビスフタルイミドメチル−ビシクロ[2,2,1]ヘプタンの13C−NMRチャートである。
【図2】実施例1で得られた2,5−diexo−ジアミノメチル−ビシクロ[2,2,1]ヘプタンの13C−NMRチャートである。
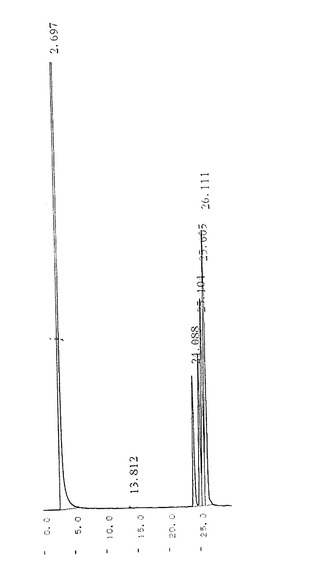
【図3】原材料であるジアミノメチル−ビシクロ[2,2,1]ヘプタンのガスクロマトグラフィー(GC)チャートである。
【図4】実施例1で得られた2,5−diexo−ジアミノメチル−ビシクロ[2,2,1]ヘプタンのガスクロマトグラフィー(GC)チャートである。
【図5】実施例2で得られた2−endo−5−exo−ビスフタルイミドメチル−ビシクロ[2,2,1]ヘプタンの13C−NMRチャートである。
【図6】実施例2で得られた2−endo−5−exo−ジアミノメチル−ビシクロ[2,2,1]ヘプタンの13C−NMRチャートである。
【図7】実施例2で得られた2−endo−5−exo−ジアミノメチル−ビシクロ[2,2,1]ヘプタンのガスクロマトグラフィー(GC)チャートである。
【発明を実施するための形態】
【0019】
以下、本発明の実施の形態について説明するが、本発明は以下の実施の形態に限定されるものではない。
【0020】
[1]2,5−ジアミノメチル−ビシクロ[2,2,1]ヘプタン:
本発明の2,5−ジアミノメチル−ビシクロ[2,2,1]ヘプタン(以下、「2,5−NBDA」とも記す)は、下記式(I)又は(II)で表されるジアミン化合物である。なお、下記式(I)で表されるジアミン化合物は、2,5−diexo−ジアミノメチル−ビシクロ[2,2,1]ヘプタン(以下、「2,5−diexo−NBDA」とも記す)である。また、下記式(II)で表されるジアミン化合物は、2−endo−5−exo−ジアミノメチル−ビシクロ[2,2,1]ヘプタン(以下、「2−endo−5−exo−NBDA」とも記す)である。
【0021】
【化3】
【0022】
前記式(I)で表される2,5−diexo−NBDA、及び前記式(II)で表される2−endo−5−exo−NBDAは、いずれも他の異性体を実質的に含有せず、純度の高いものである。より具体的には、ガスクロマトグラフィー(GC)により分析して得られるチャートの領域面積に基づき算出される2,5−diexo−NBDA及び2−endo−5−exo−NBDAの純度は、いずれも95%以上、好ましくは97.0%以上、更に好ましくは99.0%以上、特に好ましくは100%のものである。
【0023】
本発明の2,5−NBDAは、他の異性体を実質的に含有しない、これまでにない高純度のジアミン化合物である。このため、各種の樹脂材料や化成品の原料、或いは電気・電子材料、光学材料、塗料・接着剤材料、構造部材、断熱材等としての用途が期待されるが、特に、電子部品の構成材料として用いられるポリイミドフィルムを形成するための原料として有用である。
【0024】
[2]2,5−ジアミノメチル−ビシクロ[2,2,1]ヘプタンの製造方法:
本発明の2,5−NBDAの製造方法は、NBDAに含まれるそれぞれの化合物のアミノ基を保護基で保護した後、2,5−NBDAに由来する異性体を結晶化と固液分離を含む分離操作により分離し、次いで分離した異性体の保護基を脱保護することを含む製造方法である。NBDAに含まれる化合物のアミノ基を保護基で保護して得られるそれぞれの化合物どうしの結晶性の差異は、保護する前の化合物どうしの結晶性の差異に比して大きくなる。即ち、本発明の製造方法によれば、アミノ基を保護することにより、異性体どうしを工業的にも十分実用化が可能な簡便な結晶化及び固液分離を含む分離操作により分離することができる。
【0025】
NBDAは、下記式(I)で表される2,5−diexo−NBDA、下記式(II)で表される2−endo−5−exo−NBDA、下記式(III)で表される2−exo−6−exo−ジアミノメチル−ビシクロ[2,2,1]ヘプタン(以下、「2,6−diexo−NBDA」ともいう)、及び下記式(IV)で表される2−endo−6−exo−ジアミノメチル−ビシクロ[2,2,1]ヘプタン(「2−endo−6−exo−NBDA」とも記す)を含有する異性体混合物である。
【0026】
【化4】
【0027】
[2−1]NBDA:
原料となる異性体混合物であるNBDAとしては、従来公知の方法に従って製造されたものを用いることができる。具体的には、ビシクロ[2,2,1]−5−ヘプテン−2−カルボニトリルに、パラジウム触媒及びトリフェニルホスファイト、或いは0価ニッケル錯体触媒の存在下でシアン化水素を付加させてジシアノ体とした後、接触水素化することにより製造されたもの等を用いることができる。また、市販品としては三井化学社製の商品名「NBDA」がある。これらのNBDAには、通常、2,5−diexo−NBDAが約26質量%、2−endo−5−exo−NBDAが約36質量%、2,6−diexo−NBDAが約20質量%、及び2−endo−6−exo−NBDAが約18質量%の割合で含有されている。
【0028】
[2−2]アミノ基の保護:
先ず、NBDAに含まれるそれぞれの化合物のアミノ基を保護基により保護する。保護基の種類は特に限定されず、1級のアミノ基を保護することが可能な保護基であればよい。但し、試薬の取り扱い性や、保護された化合物(異性体)をそれらの結晶性の相違に基づいて分離する際の分離性の観点からは、フタルイミド基、t−ブチルオキシカルボニル基(t−BOC基)、又はベンジルオキシカルボニル基(Z−基)が好ましい。なお、アミノ基の保護基をフタルイミド基とした場合には、保護された異性体どうしの結晶性の差が大きくなるため、結晶化と固液分離により異性体をより簡便に分離しやすくなるために好ましい。
【0029】
[2−3]異性体の分離:
次に、保護基によってアミノ基が保護された異性体のうち、2,5−diexo−NBDA及び2−endo−5−exo−NBDAに由来する異性体を、結晶化と固液分離を含む分離操作によりそれぞれ分離する。保護基の種類がフタルイミド基、t−BOC基、及びZ−基である場合を例に挙げると、下記式(I−1)〜(I−3)、及び下記式(II−1)〜(II−3)で表される異性体をそれぞれ分離する。
【0030】
【化5】
【0031】
【化6】
【0032】
例えば、各種溶媒を用いた固液分離や再結晶を行うこと等により、アミノ基が保護された異性体を分離することができる。このとき、用いる溶媒の種類や温度等を適宜選択・設定することにより、2,5−diexo−NBDAに由来する異性体のみを実質的に含有する分画や、2−endo−5−exo−NBDAに由来する異性体のみを実質的に含有する分画を得ることができる。
【0033】
[2−4]脱保護:
分離した異性体の保護基を脱保護することにより、目的とする2,5−diexo−NBDA及び2−endo−5−exo−NBDAを得ることができる。保護基は、保護基の種類に応じて従来公知の方法に従って脱保護することができる。例えば、保護基がフタルイミド基である場合には、ヒドラジン等で処理することで脱保護することができる。保護基がt−BOC基である場合には、トリフルオロ酢酸等で処理することで脱保護することができる。また、保護基がZ−基である場合には、パラジウム触媒の存在下に水素添加すること等で脱保護することができる。
【0034】
なお、保護基を脱保護した後、必要に応じて精製して目的とする2,5−diexo−NBDA及び2−endo−5−exo−NBDAの純度を高めることが好ましい。精製方法は特に限定されないが、好適例として減圧蒸留、カラムによる分離、再結晶等の方法を挙げることができる。
【0035】
このようにして得られる2,5−diexo−NBDA及び2−endo−5−exo−NBDAのガスクロマトグラフィーにより分析される純度は、いずれも95%以上である。なお、純度100%の2,5−diexo−NBDA及び2−endo−5−exo−NBDAは、常温・常圧下でいずれも液状(オイル状)である。
【0036】
[2−5]2,5−NBDAの製造:
以下、保護基の種類がフタルイミド基である場合を例に挙げ、2,5−NBDAの製造方法の更なる詳細について説明する。先ず、原料となるNBDAと無水フタル酸を、p−トルエンスルホン酸等の適当な触媒の存在下、有機溶媒中で加熱して脱水縮合させる。有機溶媒の具体例としては、トルエン、DMF等を挙げることができる。なお、系外に水分を排出しながら反応させることが好ましい。
【0037】
[2−5−1]2,5−diexo−NBDAの製造:
NBDAと無水フタル酸の反応が終了した後、室温(25℃)まで冷却し、析出した結晶を濾取する。得られた結晶を、適当な有機溶媒を使用して再結晶するか、或いは有機溶媒中に分散させたスラリーを加熱するスラッジング処理を行うことによって精製することが好ましい。再結晶及びスラッジング処理に用いる有機溶媒の具体例としては、トルエン、アセトニトリル、キシレン、メチルセロソルブ、酢酸エチル等を挙げることができる。再結晶及び/又はスラッジング処理による精製を好ましくは2回以上行うことによって、前記式(I−1)で表される異性体を得ることができる。
【0038】
次に、前記式(I−1)で表される異性体を、イソプロピルアルコール、メタノール、メチルセロソルブ等の溶媒中、加熱条件下でヒドラジン一水和物と反応させると、2,5−diexo−NBDAのフタラジンジオン塩が反応系中に析出する。濾過して取り出した2,5−diexo−NBDAのフタラジンジオン塩を塩酸等の酸で処理して酸分解した後、水酸化ナトリウム等の塩基で中和すれば、2,5−diexo−NBDAの粗生成物を得ることができる。その後、有機溶媒による抽出、及び減圧蒸留すること等によって精製すれば、目的とする高純度の2,5−diexo−NBDAを得ることができる。
【0039】
[2−5−2]2−endo−5−exo−NBDAの製造:
NBDAと無水フタル酸の反応が終了した後、室温(25℃)まで冷却し、析出した結晶を濾別して濾液を得る。得られた濾液を水中に徐々に投入し、析出した結晶を濾取する。得られた結晶を乾燥した後、100℃前後に加熱した有機溶媒に溶解させる。有機溶媒としてはトルエン、キシレン、メシチレン、メチルセロソルブ、メチルエチルケトン等を用いることができるが、なかでもトルエンが好ましい。結晶を有機溶媒に溶解させて得られた溶液を室温(25℃)まで冷却し、析出した結晶を濾別する。得られた濾液を加熱条件下で濃縮した後、好ましくは0〜50℃に冷却して析出した結晶を濾取する。得られた結晶を、必要に応じてDMF等の有機溶媒を用いて再結晶することにより、前記式(II−1)で表される異性体を得ることができる。
【0040】
次に、前記式(II−1)で表される異性体を、イソプロピルアルコール、メタノール、メチルセロソルブ等の溶媒中、加熱条件下でヒドラジン一水和物と反応させ、アミノ基を保護する保護基を脱保護する。生成したフタラジンジオンの結晶を濾別した後、濾液を濃縮すれば2−endo−5−exo−NBDAの粗生成物を得ることができる。その後、減圧蒸留すること等によって精製すれば、目的とする高純度の2−endo−5−exo−NBDAを得ることができる。
【実施例】
【0041】
以下、本発明を実施例に基づいて具体的に説明するが、本発明はこれらの実施例に限定されるものではない。
【0042】
(実施例1:2,5−diexo−NBDAの製造)
(1)アミノ基の保護(フタルイミド化):
撹拌装置、滴下漏斗、温度計、コンデンサー、窒素導入管、及びディーンスターク水分離器を備え付けた300mlガラス製反応装置に、無水フタル酸325.9g(2.2mol)、p−トルエンスルホン酸0.75g、N,N−ジメチルホルムアミド1250g、及びトルエン250gを装入し、窒素雰囲気下において撹拌しながら140℃へ昇温した。トルエン還流下において140〜145℃を維持しながら、4種の異性体混合物である、ジアミノメチル−ビシクロ[2,2,1]ヘプタン(商品名:NBDA、三井化学社製)154.3g(1.0mol)、及びN,N−ジメチルホルムアミド154.3gの混合溶液を、90分かけて徐々に滴下した。途中反応により生成する水はトルエンとの共沸によりディーンスターク水分離器により系外へ排出し、トルエンは系内へ戻した。滴下終了後、同温度を6時間維持して充分に反応を進行させた後、放冷により室温まで冷却し、析出した白色結晶を濾取した。なお、NBDAのアミノ基をフタルイミド化する際の反応式を以下に示す。
【0043】
【化7】
【0044】
得られた白色結晶をHPLCで分析したところ、83.7面積%の主成分を含む混合物であることが判明した。なお、収量は97.5gであった。一方、濾液(以下、「濾液A」と記す)をHPLCで分析したところ、この濾液Aには、3種の主生成物、及び上記白色結晶と同様の保持時間で溶出するピークを含む極僅かな混合物が含まれていることが判明した。
【0045】
得られた白色結晶を400gのアセトニトリル中に分散させてスラリー状態とし、スラリー状態のまま加熱して30分間還流した。室温まで冷却した後、85.0gの白色結晶を得た。HPLCで分析した得られた白色結晶の純度(領域面積に基づく)は97.3%であった。得られた白色結晶を450gのトルエン中に分散させてスラリー状態とし、スラリー状態のまま加熱して70℃で1時間スラッジングした後、60〜65℃で熱濾過することで白色結晶を得た。HPLCで分析した得られた白色結晶の純度(領域面積に基づく)は100.0%であり、収量は73.0gであり、NBDAからの収率は17.6%であった。また、得られた白色結晶の13C−NMRチャートを図1に示す。図1に示す13C−NMRチャートから、得られた白色結晶は前記式(I−1)で表される2,5−diexo−ビスフタルイミドメチル−ビシクロ[2,2,1]ヘプタンであることが判明した。なお、13C−NMRは、日本電子社製の「ECA500」(500MHz)を使用し、重クロロホルムを溶媒として用いて測定した。
【0046】
(2)脱保護:
撹拌装置、温度計、温度計、窒素導入管、及びコンデンサーを備え付けた1000mlガラス製反応装置に、上記(1)の操作を繰り返し行って得られた2,5−diexo−ビスフタルイミドメチル−ビシクロ[2,2,1]ヘプタン124.4g(0.3mol)及びイソプロピルアルコール1200gを装入し、撹拌しながら60℃まで加熱した。同温度を維持しながら96%ヒドラジン(1水和物)90.0g(1.8mol)を1時間かけて滴下した。同温度を維持しながら更に3時間撹拌して反応を完結させた。なお、HPLCで2,5−diexo−ビスフタルイミドメチル−ビシクロ[2,2,1]ヘプタンの消失を確認することにより、反応の完結(終点)を確認した。反応終了後、室温まで冷却し、析出した2,5−diexo−NBDAのフタラジンジオン塩(以下単に「フタラジンジオン塩」と記す)を濾過して単離した。
【0047】
撹拌装置、温度計、及び滴下漏斗を備え付けた3000mlガラス製反応装置に単離したフタラジンジオン塩を装入し、水340gを加えて撹拌してスラリー状態とした。撹拌しながら14.2質量%塩化水素水溶液840g(3.26mol)を1時間かけて滴下し、塩を酸分解して2,5−diexo−NBDA塩酸塩とした。続けて30質量%水酸化ナトリウム水溶液520.8gを1時間かけて滴下し、2,5−diexo−NBDA塩酸塩を中和した後、固形分を濾過した。ロータリーエバポレーターを使用して濾液を濃縮し、塩化ナトリウム、フタラジンジオン、少量の水酸化ナトリウム、及び2,5−diexo−NBDAを含む淡黄色の塊状物を得た。なお、フタルイミド基を脱保護する際の反応式を以下に示す。
【0048】
【化8】
【0049】
得られた塊状物にトルエン600gを添加してよく撹拌し、目的物である2,5−diexo−NBDAをトルエンに溶解させた。固形分を濾別した後、ロータリーエバポレーターを使用して濾液を濃縮し、少量のトルエンを含む41.5gの淡黄色液体を得た。得られた液体について170〜175℃に加熱された油浴中、2.8〜3.5kPaの減圧度で減圧蒸留を行ったところ、無色透明の2,5−diexo−NBDA30.2gを得た。ガスクロマトグラフィーにより分析した2,5−diexo−NBDAの純度は100%であり、2,5−diexo−ビスフタルイミドメチル−ビシクロ[2,2,1]ヘプタンからの収率は65.3%であった。また、DSCにより融点を測定しようとしたところ−20℃においても結晶化しなかった。得られた2,5−diexo−NBDAの13C−NMRチャートを図2に示す。また、原材料であるNBDA及び得られた2,5−diexo−NBDAのガスクロマトグラフィー(GC)チャートを図3及び4にそれぞれ示す。なお、ガスクロマトグラフィーの条件を以下に示す。
【0050】
カラム種類:無極性カラム、商品名「ZB−1」、Phenomenex社製
カラムサイズ:内径×長さ×膜厚=0.53mm×30m×3μm
キャリアーガス:ヘリウム
カラム圧力:定圧モード20kPa
カラム温度:160℃
試料気化室及び検出部温度:280℃
【0051】
(実施例2:2−endo−5−exo−NBDAの製造)
(3)アミノ基の保護(フタルイミド化):
撹拌装置、滴下漏斗、及び温度計を備え付け、水3500gを装入したガラス製反応装置に、上記実施例1における(1)の操作で得られた濾液Aを30分かけて滴下し、結晶を析出させた。析出した結晶を濾別及び乾燥した後、100℃に加熱した800gのトルエンに溶解した。得られた溶液を放冷し、室温(25℃)まで冷却して生じた結晶を濾別した。ロータリーエバポレーターを使用し、約400mlになるまで濾液を濃縮した。濃縮した濾液を放冷したところ徐々に白色結晶が析出した。50℃まで冷却したところで濾過し、85.0gの白色結晶を得た。得られた白色結晶をHPLCで分析したところ、88.4面積%の主成分を含む混合物であることが判明した。この混合物(白色結晶)の全量を420gのN,N−ジメチルホルムアミドに溶解させて再結晶を行い、56.0gの白色結晶を得た。HPLCで分析した得られた白色結晶の純度(領域面積に基づく)は100.0%であり、NBDAからの収率は9.0%であった。また、得られた白色結晶の13C−NMRチャートを図5に示す。図5に示す13C−NMRチャートから、得られた白色結晶は前記式(II−1)で表される2−endo−5−exo−ビスフタルイミドメチル−ビシクロ[2,2,1]ヘプタンであることが判明した。
【0052】
(4)脱保護:
撹拌装置、温度計、温度計、窒素導入管、及びコンデンサーを備え付けた1000mlガラス製反応装置に、上記(3)の操作を繰り返し行って得られた2−endo−5−exo−ビスフタルイミドメチル−ビシクロ[2,2,1]ヘプタン124.4g(0.3mol)及びイソプロピルアルコール1000gを装入し、撹拌しながら60℃まで加熱した。同温度を維持しながら96%ヒドラジン(1水和物)90.1g(1.8mol)を1時間かけて滴下した。同温度を維持しながら更に3時間撹拌して反応を完結させた。なお、HPLCで2−endo−5−exo−ビスフタルイミドメチル−ビシクロ[2,2,1]ヘプタンの消失を確認することにより、反応の完結(終点)を確認した。反応終了後、室温まで冷却し、析出したフタラジンジオンを濾別した。また、目的物である2−endo−5−exo−NBDAが濾液中に存在していることをガスクロマトグラフィーによりで分析することにより確認した。なお、フタルイミド基を脱保護する際の反応式を以下に示す。
【0053】
【化9】
【0054】
ロータリーエバポレーターを使用して濾液を濃縮し、少量の溶媒及びヒドラジンを含む41.9gの淡黄色液体を得た。得られた液体について170〜175℃に加熱された油浴中、2.8〜3.5kPaの減圧度で減圧蒸留を行ったところ、無色透明の2−endo−5−exo−NBDA35.7gを得た。ガスクロマトグラフィーにより分析した2−endo−5−exo−NBDAの純度は100%であり、2−endo−5−exo−ビスフタルイミドメチル−ビシクロ[2,2,1]ヘプタンからの収率は77.2%であった。また、DSCにより融点を測定しようとしたところ−20℃においても結晶化しなかった。得られた2−endo−5−exo−NBDAの13C−NMRチャートを図6に示す。また、得られた2−endo−5−exo−NBDAのガスクロマトグラフィー(GC)チャートを図7に示す。
【産業上の利用可能性】
【0055】
本発明の2,5−ジアミノメチル−ビシクロ[2,2,1]ヘプタンは、ポリイミド、エポキシ樹脂、ウレタン樹脂、及びポリイソシアネート等の原料として有用であり、特に、電子部品の構成材料として用いられるポリイミドフィルムを形成するための原料として好適である。
【技術分野】
【0001】
本発明は、高純度の2,5−ジアミノメチル−ビシクロ[2,2,1]ヘプタン、及びその製造方法に関する。
【背景技術】
【0002】
2,5(又は6)−ジアミノメチル−ビシクロ[2,2,1]ヘプタン(以下、「NBDA」とも記す)は、ポリイミド、ポリアミド等のモノマーとして、またエポキシ樹脂、ビスマレイミド、ジイソシアネート等の原料として、或いはそれらの硬化剤として極めて有用なジアミン化合物である。また、このNBDAを原料に用いることで、光線透過率の良好な(無色透明性の高い)ポリイミドが得られることが知られている(例えば、特許文献1及び2参照)。
【0003】
NBDAは、従来、ビシクロ[2,2,1]−5−ヘプテン−2−カルボニトリルに、パラジウム触媒及びトリフェニルホスファイト、或いは0価ニッケル錯体触媒の存在下でシアン化水素を付加させてジシアノ体とした後、接触水素化することにより製造されている(例えば、特許文献3及び4参照)。このようにして製造されるNBDAには、ビシクロ[2,2,1]ヘプタン環におけるアミノメチル基の置換位置が異なる異性体として、2,5−ジアミノメチル−ビシクロ[2,2,1]ヘプタン(以下、「2,5−NBDA」とも記す)、及び2,6−ジアミノメチル−ビシクロ[2,2,1]ヘプタン(以下、「2,6−NBDA」とも記す)が含まれている。
【0004】
これらの異性体は、蒸留等の通常の分離手段では分離することが非常に困難であることが知られている。更に、2,5−NBDAと2,6−NBDAのそれぞれにも光学異性体が存在する。このため、上記のようにして製造されるNBDAは、一般的な分離手段では分離困難な4種類の異性体を含有する異性体混合物である。
【0005】
NBDAに含まれる4種の異性体を分離する方法として、それぞれの異性体の融点差を利用し、凝固させたNBDAを徐々に昇温させながらそれぞれの異性体が多く含まれる留分に分画する方法が開示されている(例えば、特許文献5参照)。
【先行技術文献】
【特許文献】
【0006】
【特許文献1】特開平10−7906号公報
【特許文献2】国際公開第1998/029471号
【特許文献3】特許第2713612号公報
【特許文献4】特開2002−69043号公報
【特許文献5】国際公開第2002/010253号
【発明の概要】
【発明が解決しようとする課題】
【0007】
特許文献5で開示された異性体の分離方法によると、それぞれの留分に含まれる異性体の量比をある程度制御することは可能であった。しかしながら、他の異性体を実質的に含有しない高純度の化合物を分離精製することは極めて困難であった。
【0008】
本発明は、このような従来技術の有する問題点に鑑みてなされたものであり、その課題とするところは、高純度の2,5−ジアミノメチル−ビシクロ[2,2,1]ヘプタン、及びその製造方法を提供することにある。
【課題を解決するための手段】
【0009】
本発明者らは上記課題を達成すべく鋭意検討した結果、原材料となるジアミノメチル−ビシクロ[2,2,1]ヘプタンの異性体混合物に含まれるそれぞれの化合物のアミノ基を保護基で保護した後、異性体を分離し、次いで分離した異性体の保護基を脱保護することによって、上記課題を達成することが可能であることを見出し、本発明を完成するに至った。
【0010】
即ち、本発明によれば、以下に示す2,5−ジアミノメチル−ビシクロ[2,2,1]ヘプタン、及び2,5−ジアミノメチル−ビシクロ[2,2,1]ヘプタンの製造方法が提供される。
【0011】
[1]下記式(I)又は(II)で表される、ガスクロマトグラフィーにより分析される純度が95%以上である2,5−ジアミノメチル−ビシクロ[2,2,1]ヘプタン。
【0012】
【化1】
【0013】
[2]下記式(I)で表される化合物、下記式(II)で表される化合物、下記式(III)で表される化合物、及び下記式(IV)で表される化合物を含むジアミノメチル−ビシクロ[2,2,1]ヘプタンの異性体混合物に含まれるそれぞれの化合物のアミノ基を保護基で保護した後、下記式(I)又は(II)で表される化合物に由来する異性体を結晶化と固液分離を含む分離操作により分離し、次いで分離した前記異性体の前記保護基を脱保護することを含む、下記式(I)又は(II)で表される、ガスクロマトグラフィーにより分析される純度が95%以上である2,5−ジアミノメチル−ビシクロ[2,2,1]ヘプタンの製造方法。
【0014】
【化2】
【0015】
[3]前記保護基が、フタルイミド基、t−ブチルオキシカルボニル基、又はベンジルオキシカルボニル基である、前記[2]に記載の製造方法。
【発明の効果】
【0016】
本発明の2,5−ジアミノメチル−ビシクロ[2,2,1]ヘプタンは、ガスクロマトグラフィーにより分析される純度が95%以上の、異性体を実質的に含有しない高純度な化合物であり、各種の樹脂材料や化成品の原料として、特に、電子部品の構成材料として用いられるポリイミドフィルムを形成するための原料として極めて有用である。
【0017】
本発明の製造方法によれば、ガスクロマトグラフィーにより分析される純度が95%以上の、異性体を実質的に含有しない高純度な2,5−ジアミノメチル−ビシクロ[2,2,1]ヘプタンを、工業的にも適用可能な簡便な操作で製造することができる。
【図面の簡単な説明】
【0018】
【図1】実施例1で得られた2,5−diexo−ビスフタルイミドメチル−ビシクロ[2,2,1]ヘプタンの13C−NMRチャートである。
【図2】実施例1で得られた2,5−diexo−ジアミノメチル−ビシクロ[2,2,1]ヘプタンの13C−NMRチャートである。
【図3】原材料であるジアミノメチル−ビシクロ[2,2,1]ヘプタンのガスクロマトグラフィー(GC)チャートである。
【図4】実施例1で得られた2,5−diexo−ジアミノメチル−ビシクロ[2,2,1]ヘプタンのガスクロマトグラフィー(GC)チャートである。
【図5】実施例2で得られた2−endo−5−exo−ビスフタルイミドメチル−ビシクロ[2,2,1]ヘプタンの13C−NMRチャートである。
【図6】実施例2で得られた2−endo−5−exo−ジアミノメチル−ビシクロ[2,2,1]ヘプタンの13C−NMRチャートである。
【図7】実施例2で得られた2−endo−5−exo−ジアミノメチル−ビシクロ[2,2,1]ヘプタンのガスクロマトグラフィー(GC)チャートである。
【発明を実施するための形態】
【0019】
以下、本発明の実施の形態について説明するが、本発明は以下の実施の形態に限定されるものではない。
【0020】
[1]2,5−ジアミノメチル−ビシクロ[2,2,1]ヘプタン:
本発明の2,5−ジアミノメチル−ビシクロ[2,2,1]ヘプタン(以下、「2,5−NBDA」とも記す)は、下記式(I)又は(II)で表されるジアミン化合物である。なお、下記式(I)で表されるジアミン化合物は、2,5−diexo−ジアミノメチル−ビシクロ[2,2,1]ヘプタン(以下、「2,5−diexo−NBDA」とも記す)である。また、下記式(II)で表されるジアミン化合物は、2−endo−5−exo−ジアミノメチル−ビシクロ[2,2,1]ヘプタン(以下、「2−endo−5−exo−NBDA」とも記す)である。
【0021】
【化3】
【0022】
前記式(I)で表される2,5−diexo−NBDA、及び前記式(II)で表される2−endo−5−exo−NBDAは、いずれも他の異性体を実質的に含有せず、純度の高いものである。より具体的には、ガスクロマトグラフィー(GC)により分析して得られるチャートの領域面積に基づき算出される2,5−diexo−NBDA及び2−endo−5−exo−NBDAの純度は、いずれも95%以上、好ましくは97.0%以上、更に好ましくは99.0%以上、特に好ましくは100%のものである。
【0023】
本発明の2,5−NBDAは、他の異性体を実質的に含有しない、これまでにない高純度のジアミン化合物である。このため、各種の樹脂材料や化成品の原料、或いは電気・電子材料、光学材料、塗料・接着剤材料、構造部材、断熱材等としての用途が期待されるが、特に、電子部品の構成材料として用いられるポリイミドフィルムを形成するための原料として有用である。
【0024】
[2]2,5−ジアミノメチル−ビシクロ[2,2,1]ヘプタンの製造方法:
本発明の2,5−NBDAの製造方法は、NBDAに含まれるそれぞれの化合物のアミノ基を保護基で保護した後、2,5−NBDAに由来する異性体を結晶化と固液分離を含む分離操作により分離し、次いで分離した異性体の保護基を脱保護することを含む製造方法である。NBDAに含まれる化合物のアミノ基を保護基で保護して得られるそれぞれの化合物どうしの結晶性の差異は、保護する前の化合物どうしの結晶性の差異に比して大きくなる。即ち、本発明の製造方法によれば、アミノ基を保護することにより、異性体どうしを工業的にも十分実用化が可能な簡便な結晶化及び固液分離を含む分離操作により分離することができる。
【0025】
NBDAは、下記式(I)で表される2,5−diexo−NBDA、下記式(II)で表される2−endo−5−exo−NBDA、下記式(III)で表される2−exo−6−exo−ジアミノメチル−ビシクロ[2,2,1]ヘプタン(以下、「2,6−diexo−NBDA」ともいう)、及び下記式(IV)で表される2−endo−6−exo−ジアミノメチル−ビシクロ[2,2,1]ヘプタン(「2−endo−6−exo−NBDA」とも記す)を含有する異性体混合物である。
【0026】
【化4】
【0027】
[2−1]NBDA:
原料となる異性体混合物であるNBDAとしては、従来公知の方法に従って製造されたものを用いることができる。具体的には、ビシクロ[2,2,1]−5−ヘプテン−2−カルボニトリルに、パラジウム触媒及びトリフェニルホスファイト、或いは0価ニッケル錯体触媒の存在下でシアン化水素を付加させてジシアノ体とした後、接触水素化することにより製造されたもの等を用いることができる。また、市販品としては三井化学社製の商品名「NBDA」がある。これらのNBDAには、通常、2,5−diexo−NBDAが約26質量%、2−endo−5−exo−NBDAが約36質量%、2,6−diexo−NBDAが約20質量%、及び2−endo−6−exo−NBDAが約18質量%の割合で含有されている。
【0028】
[2−2]アミノ基の保護:
先ず、NBDAに含まれるそれぞれの化合物のアミノ基を保護基により保護する。保護基の種類は特に限定されず、1級のアミノ基を保護することが可能な保護基であればよい。但し、試薬の取り扱い性や、保護された化合物(異性体)をそれらの結晶性の相違に基づいて分離する際の分離性の観点からは、フタルイミド基、t−ブチルオキシカルボニル基(t−BOC基)、又はベンジルオキシカルボニル基(Z−基)が好ましい。なお、アミノ基の保護基をフタルイミド基とした場合には、保護された異性体どうしの結晶性の差が大きくなるため、結晶化と固液分離により異性体をより簡便に分離しやすくなるために好ましい。
【0029】
[2−3]異性体の分離:
次に、保護基によってアミノ基が保護された異性体のうち、2,5−diexo−NBDA及び2−endo−5−exo−NBDAに由来する異性体を、結晶化と固液分離を含む分離操作によりそれぞれ分離する。保護基の種類がフタルイミド基、t−BOC基、及びZ−基である場合を例に挙げると、下記式(I−1)〜(I−3)、及び下記式(II−1)〜(II−3)で表される異性体をそれぞれ分離する。
【0030】
【化5】
【0031】
【化6】
【0032】
例えば、各種溶媒を用いた固液分離や再結晶を行うこと等により、アミノ基が保護された異性体を分離することができる。このとき、用いる溶媒の種類や温度等を適宜選択・設定することにより、2,5−diexo−NBDAに由来する異性体のみを実質的に含有する分画や、2−endo−5−exo−NBDAに由来する異性体のみを実質的に含有する分画を得ることができる。
【0033】
[2−4]脱保護:
分離した異性体の保護基を脱保護することにより、目的とする2,5−diexo−NBDA及び2−endo−5−exo−NBDAを得ることができる。保護基は、保護基の種類に応じて従来公知の方法に従って脱保護することができる。例えば、保護基がフタルイミド基である場合には、ヒドラジン等で処理することで脱保護することができる。保護基がt−BOC基である場合には、トリフルオロ酢酸等で処理することで脱保護することができる。また、保護基がZ−基である場合には、パラジウム触媒の存在下に水素添加すること等で脱保護することができる。
【0034】
なお、保護基を脱保護した後、必要に応じて精製して目的とする2,5−diexo−NBDA及び2−endo−5−exo−NBDAの純度を高めることが好ましい。精製方法は特に限定されないが、好適例として減圧蒸留、カラムによる分離、再結晶等の方法を挙げることができる。
【0035】
このようにして得られる2,5−diexo−NBDA及び2−endo−5−exo−NBDAのガスクロマトグラフィーにより分析される純度は、いずれも95%以上である。なお、純度100%の2,5−diexo−NBDA及び2−endo−5−exo−NBDAは、常温・常圧下でいずれも液状(オイル状)である。
【0036】
[2−5]2,5−NBDAの製造:
以下、保護基の種類がフタルイミド基である場合を例に挙げ、2,5−NBDAの製造方法の更なる詳細について説明する。先ず、原料となるNBDAと無水フタル酸を、p−トルエンスルホン酸等の適当な触媒の存在下、有機溶媒中で加熱して脱水縮合させる。有機溶媒の具体例としては、トルエン、DMF等を挙げることができる。なお、系外に水分を排出しながら反応させることが好ましい。
【0037】
[2−5−1]2,5−diexo−NBDAの製造:
NBDAと無水フタル酸の反応が終了した後、室温(25℃)まで冷却し、析出した結晶を濾取する。得られた結晶を、適当な有機溶媒を使用して再結晶するか、或いは有機溶媒中に分散させたスラリーを加熱するスラッジング処理を行うことによって精製することが好ましい。再結晶及びスラッジング処理に用いる有機溶媒の具体例としては、トルエン、アセトニトリル、キシレン、メチルセロソルブ、酢酸エチル等を挙げることができる。再結晶及び/又はスラッジング処理による精製を好ましくは2回以上行うことによって、前記式(I−1)で表される異性体を得ることができる。
【0038】
次に、前記式(I−1)で表される異性体を、イソプロピルアルコール、メタノール、メチルセロソルブ等の溶媒中、加熱条件下でヒドラジン一水和物と反応させると、2,5−diexo−NBDAのフタラジンジオン塩が反応系中に析出する。濾過して取り出した2,5−diexo−NBDAのフタラジンジオン塩を塩酸等の酸で処理して酸分解した後、水酸化ナトリウム等の塩基で中和すれば、2,5−diexo−NBDAの粗生成物を得ることができる。その後、有機溶媒による抽出、及び減圧蒸留すること等によって精製すれば、目的とする高純度の2,5−diexo−NBDAを得ることができる。
【0039】
[2−5−2]2−endo−5−exo−NBDAの製造:
NBDAと無水フタル酸の反応が終了した後、室温(25℃)まで冷却し、析出した結晶を濾別して濾液を得る。得られた濾液を水中に徐々に投入し、析出した結晶を濾取する。得られた結晶を乾燥した後、100℃前後に加熱した有機溶媒に溶解させる。有機溶媒としてはトルエン、キシレン、メシチレン、メチルセロソルブ、メチルエチルケトン等を用いることができるが、なかでもトルエンが好ましい。結晶を有機溶媒に溶解させて得られた溶液を室温(25℃)まで冷却し、析出した結晶を濾別する。得られた濾液を加熱条件下で濃縮した後、好ましくは0〜50℃に冷却して析出した結晶を濾取する。得られた結晶を、必要に応じてDMF等の有機溶媒を用いて再結晶することにより、前記式(II−1)で表される異性体を得ることができる。
【0040】
次に、前記式(II−1)で表される異性体を、イソプロピルアルコール、メタノール、メチルセロソルブ等の溶媒中、加熱条件下でヒドラジン一水和物と反応させ、アミノ基を保護する保護基を脱保護する。生成したフタラジンジオンの結晶を濾別した後、濾液を濃縮すれば2−endo−5−exo−NBDAの粗生成物を得ることができる。その後、減圧蒸留すること等によって精製すれば、目的とする高純度の2−endo−5−exo−NBDAを得ることができる。
【実施例】
【0041】
以下、本発明を実施例に基づいて具体的に説明するが、本発明はこれらの実施例に限定されるものではない。
【0042】
(実施例1:2,5−diexo−NBDAの製造)
(1)アミノ基の保護(フタルイミド化):
撹拌装置、滴下漏斗、温度計、コンデンサー、窒素導入管、及びディーンスターク水分離器を備え付けた300mlガラス製反応装置に、無水フタル酸325.9g(2.2mol)、p−トルエンスルホン酸0.75g、N,N−ジメチルホルムアミド1250g、及びトルエン250gを装入し、窒素雰囲気下において撹拌しながら140℃へ昇温した。トルエン還流下において140〜145℃を維持しながら、4種の異性体混合物である、ジアミノメチル−ビシクロ[2,2,1]ヘプタン(商品名:NBDA、三井化学社製)154.3g(1.0mol)、及びN,N−ジメチルホルムアミド154.3gの混合溶液を、90分かけて徐々に滴下した。途中反応により生成する水はトルエンとの共沸によりディーンスターク水分離器により系外へ排出し、トルエンは系内へ戻した。滴下終了後、同温度を6時間維持して充分に反応を進行させた後、放冷により室温まで冷却し、析出した白色結晶を濾取した。なお、NBDAのアミノ基をフタルイミド化する際の反応式を以下に示す。
【0043】
【化7】
【0044】
得られた白色結晶をHPLCで分析したところ、83.7面積%の主成分を含む混合物であることが判明した。なお、収量は97.5gであった。一方、濾液(以下、「濾液A」と記す)をHPLCで分析したところ、この濾液Aには、3種の主生成物、及び上記白色結晶と同様の保持時間で溶出するピークを含む極僅かな混合物が含まれていることが判明した。
【0045】
得られた白色結晶を400gのアセトニトリル中に分散させてスラリー状態とし、スラリー状態のまま加熱して30分間還流した。室温まで冷却した後、85.0gの白色結晶を得た。HPLCで分析した得られた白色結晶の純度(領域面積に基づく)は97.3%であった。得られた白色結晶を450gのトルエン中に分散させてスラリー状態とし、スラリー状態のまま加熱して70℃で1時間スラッジングした後、60〜65℃で熱濾過することで白色結晶を得た。HPLCで分析した得られた白色結晶の純度(領域面積に基づく)は100.0%であり、収量は73.0gであり、NBDAからの収率は17.6%であった。また、得られた白色結晶の13C−NMRチャートを図1に示す。図1に示す13C−NMRチャートから、得られた白色結晶は前記式(I−1)で表される2,5−diexo−ビスフタルイミドメチル−ビシクロ[2,2,1]ヘプタンであることが判明した。なお、13C−NMRは、日本電子社製の「ECA500」(500MHz)を使用し、重クロロホルムを溶媒として用いて測定した。
【0046】
(2)脱保護:
撹拌装置、温度計、温度計、窒素導入管、及びコンデンサーを備え付けた1000mlガラス製反応装置に、上記(1)の操作を繰り返し行って得られた2,5−diexo−ビスフタルイミドメチル−ビシクロ[2,2,1]ヘプタン124.4g(0.3mol)及びイソプロピルアルコール1200gを装入し、撹拌しながら60℃まで加熱した。同温度を維持しながら96%ヒドラジン(1水和物)90.0g(1.8mol)を1時間かけて滴下した。同温度を維持しながら更に3時間撹拌して反応を完結させた。なお、HPLCで2,5−diexo−ビスフタルイミドメチル−ビシクロ[2,2,1]ヘプタンの消失を確認することにより、反応の完結(終点)を確認した。反応終了後、室温まで冷却し、析出した2,5−diexo−NBDAのフタラジンジオン塩(以下単に「フタラジンジオン塩」と記す)を濾過して単離した。
【0047】
撹拌装置、温度計、及び滴下漏斗を備え付けた3000mlガラス製反応装置に単離したフタラジンジオン塩を装入し、水340gを加えて撹拌してスラリー状態とした。撹拌しながら14.2質量%塩化水素水溶液840g(3.26mol)を1時間かけて滴下し、塩を酸分解して2,5−diexo−NBDA塩酸塩とした。続けて30質量%水酸化ナトリウム水溶液520.8gを1時間かけて滴下し、2,5−diexo−NBDA塩酸塩を中和した後、固形分を濾過した。ロータリーエバポレーターを使用して濾液を濃縮し、塩化ナトリウム、フタラジンジオン、少量の水酸化ナトリウム、及び2,5−diexo−NBDAを含む淡黄色の塊状物を得た。なお、フタルイミド基を脱保護する際の反応式を以下に示す。
【0048】
【化8】
【0049】
得られた塊状物にトルエン600gを添加してよく撹拌し、目的物である2,5−diexo−NBDAをトルエンに溶解させた。固形分を濾別した後、ロータリーエバポレーターを使用して濾液を濃縮し、少量のトルエンを含む41.5gの淡黄色液体を得た。得られた液体について170〜175℃に加熱された油浴中、2.8〜3.5kPaの減圧度で減圧蒸留を行ったところ、無色透明の2,5−diexo−NBDA30.2gを得た。ガスクロマトグラフィーにより分析した2,5−diexo−NBDAの純度は100%であり、2,5−diexo−ビスフタルイミドメチル−ビシクロ[2,2,1]ヘプタンからの収率は65.3%であった。また、DSCにより融点を測定しようとしたところ−20℃においても結晶化しなかった。得られた2,5−diexo−NBDAの13C−NMRチャートを図2に示す。また、原材料であるNBDA及び得られた2,5−diexo−NBDAのガスクロマトグラフィー(GC)チャートを図3及び4にそれぞれ示す。なお、ガスクロマトグラフィーの条件を以下に示す。
【0050】
カラム種類:無極性カラム、商品名「ZB−1」、Phenomenex社製
カラムサイズ:内径×長さ×膜厚=0.53mm×30m×3μm
キャリアーガス:ヘリウム
カラム圧力:定圧モード20kPa
カラム温度:160℃
試料気化室及び検出部温度:280℃
【0051】
(実施例2:2−endo−5−exo−NBDAの製造)
(3)アミノ基の保護(フタルイミド化):
撹拌装置、滴下漏斗、及び温度計を備え付け、水3500gを装入したガラス製反応装置に、上記実施例1における(1)の操作で得られた濾液Aを30分かけて滴下し、結晶を析出させた。析出した結晶を濾別及び乾燥した後、100℃に加熱した800gのトルエンに溶解した。得られた溶液を放冷し、室温(25℃)まで冷却して生じた結晶を濾別した。ロータリーエバポレーターを使用し、約400mlになるまで濾液を濃縮した。濃縮した濾液を放冷したところ徐々に白色結晶が析出した。50℃まで冷却したところで濾過し、85.0gの白色結晶を得た。得られた白色結晶をHPLCで分析したところ、88.4面積%の主成分を含む混合物であることが判明した。この混合物(白色結晶)の全量を420gのN,N−ジメチルホルムアミドに溶解させて再結晶を行い、56.0gの白色結晶を得た。HPLCで分析した得られた白色結晶の純度(領域面積に基づく)は100.0%であり、NBDAからの収率は9.0%であった。また、得られた白色結晶の13C−NMRチャートを図5に示す。図5に示す13C−NMRチャートから、得られた白色結晶は前記式(II−1)で表される2−endo−5−exo−ビスフタルイミドメチル−ビシクロ[2,2,1]ヘプタンであることが判明した。
【0052】
(4)脱保護:
撹拌装置、温度計、温度計、窒素導入管、及びコンデンサーを備え付けた1000mlガラス製反応装置に、上記(3)の操作を繰り返し行って得られた2−endo−5−exo−ビスフタルイミドメチル−ビシクロ[2,2,1]ヘプタン124.4g(0.3mol)及びイソプロピルアルコール1000gを装入し、撹拌しながら60℃まで加熱した。同温度を維持しながら96%ヒドラジン(1水和物)90.1g(1.8mol)を1時間かけて滴下した。同温度を維持しながら更に3時間撹拌して反応を完結させた。なお、HPLCで2−endo−5−exo−ビスフタルイミドメチル−ビシクロ[2,2,1]ヘプタンの消失を確認することにより、反応の完結(終点)を確認した。反応終了後、室温まで冷却し、析出したフタラジンジオンを濾別した。また、目的物である2−endo−5−exo−NBDAが濾液中に存在していることをガスクロマトグラフィーによりで分析することにより確認した。なお、フタルイミド基を脱保護する際の反応式を以下に示す。
【0053】
【化9】
【0054】
ロータリーエバポレーターを使用して濾液を濃縮し、少量の溶媒及びヒドラジンを含む41.9gの淡黄色液体を得た。得られた液体について170〜175℃に加熱された油浴中、2.8〜3.5kPaの減圧度で減圧蒸留を行ったところ、無色透明の2−endo−5−exo−NBDA35.7gを得た。ガスクロマトグラフィーにより分析した2−endo−5−exo−NBDAの純度は100%であり、2−endo−5−exo−ビスフタルイミドメチル−ビシクロ[2,2,1]ヘプタンからの収率は77.2%であった。また、DSCにより融点を測定しようとしたところ−20℃においても結晶化しなかった。得られた2−endo−5−exo−NBDAの13C−NMRチャートを図6に示す。また、得られた2−endo−5−exo−NBDAのガスクロマトグラフィー(GC)チャートを図7に示す。
【産業上の利用可能性】
【0055】
本発明の2,5−ジアミノメチル−ビシクロ[2,2,1]ヘプタンは、ポリイミド、エポキシ樹脂、ウレタン樹脂、及びポリイソシアネート等の原料として有用であり、特に、電子部品の構成材料として用いられるポリイミドフィルムを形成するための原料として好適である。
【特許請求の範囲】
【請求項1】
下記式(I)又は(II)で表される、ガスクロマトグラフィーにより分析される純度が95%以上である2,5−ジアミノメチル−ビシクロ[2,2,1]ヘプタン。
【化1】
【請求項2】
下記式(I)で表される化合物、下記式(II)で表される化合物、下記式(III)で表される化合物、及び下記式(IV)で表される化合物を含むジアミノメチル−ビシクロ[2,2,1]ヘプタンの異性体混合物に含まれるそれぞれの化合物のアミノ基を保護基で保護した後、
下記式(I)又は(II)で表される化合物に由来する異性体を結晶化と固液分離を含む分離操作により分離し、次いで
分離した前記異性体の前記保護基を脱保護することを含む、下記式(I)又は(II)で表される、ガスクロマトグラフィーにより分析される純度が95%以上である2,5−ジアミノメチル−ビシクロ[2,2,1]ヘプタンの製造方法。
【化2】
【請求項3】
前記保護基が、フタルイミド基、t−ブチルオキシカルボニル基、又はベンジルオキシカルボニル基である、請求項2に記載の製造方法。
【請求項1】
下記式(I)又は(II)で表される、ガスクロマトグラフィーにより分析される純度が95%以上である2,5−ジアミノメチル−ビシクロ[2,2,1]ヘプタン。
【化1】
【請求項2】
下記式(I)で表される化合物、下記式(II)で表される化合物、下記式(III)で表される化合物、及び下記式(IV)で表される化合物を含むジアミノメチル−ビシクロ[2,2,1]ヘプタンの異性体混合物に含まれるそれぞれの化合物のアミノ基を保護基で保護した後、
下記式(I)又は(II)で表される化合物に由来する異性体を結晶化と固液分離を含む分離操作により分離し、次いで
分離した前記異性体の前記保護基を脱保護することを含む、下記式(I)又は(II)で表される、ガスクロマトグラフィーにより分析される純度が95%以上である2,5−ジアミノメチル−ビシクロ[2,2,1]ヘプタンの製造方法。
【化2】
【請求項3】
前記保護基が、フタルイミド基、t−ブチルオキシカルボニル基、又はベンジルオキシカルボニル基である、請求項2に記載の製造方法。
【図1】


【図2】

【図3】
【図4】

【図5】

【図6】

【図7】

【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【公開番号】特開2011−201787(P2011−201787A)
【公開日】平成23年10月13日(2011.10.13)
【国際特許分類】
【出願番号】特願2010−68199(P2010−68199)
【出願日】平成22年3月24日(2010.3.24)
【出願人】(000005887)三井化学株式会社 (2,318)
【Fターム(参考)】
【公開日】平成23年10月13日(2011.10.13)
【国際特許分類】
【出願日】平成22年3月24日(2010.3.24)
【出願人】(000005887)三井化学株式会社 (2,318)
【Fターム(参考)】
[ Back to top ]