
3−ヒドロキシカルボン酸又はそのエステルの脱水用触媒の再生方法、及び、(メタ)アクリル酸又はそのエステルの製造方法
【課題】3−ヒドロキシカルボン酸又はそのエステルの脱水用触媒上に付着した炭素状物質を除去する触媒の再生方法、及び、長期間にわたり安定的に(メタ)アクリル酸又はそのエステルを製造する方法を提供する。
【解決手段】3−ヒドロキシカルボン酸又はそのエステルの脱水用触媒上の炭素状物質を除去することによって触媒を再生する方法であって、該再生方法は、脱水用触媒に酸化剤を接触させて炭素状物質を除去することを特徴とする3−ヒドロキシカルボン酸又はそのエステルの脱水用触媒の再生方法。また、上記3−ヒドロキシカルボン酸又はそのエステルの脱水用触媒の再生方法を含むことを特徴とする(メタ)アクリル酸又はそのエステルの製造方法。
【解決手段】3−ヒドロキシカルボン酸又はそのエステルの脱水用触媒上の炭素状物質を除去することによって触媒を再生する方法であって、該再生方法は、脱水用触媒に酸化剤を接触させて炭素状物質を除去することを特徴とする3−ヒドロキシカルボン酸又はそのエステルの脱水用触媒の再生方法。また、上記3−ヒドロキシカルボン酸又はそのエステルの脱水用触媒の再生方法を含むことを特徴とする(メタ)アクリル酸又はそのエステルの製造方法。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、3−ヒドロキシカルボン酸又はそのエステルの脱水用触媒の再生方法、及び、(メタ)アクリル酸又はそのエステルを製造する方法に関する。
【背景技術】
【0002】
(メタ)アクリル酸は親水性樹脂や吸水性樹脂等の原料として工業的に広く利用されており、通常、(メタ)アクリル酸の製法としては、固定床多管式反応器を用い酸化物触媒の存在下、プロピレンやイソブチレンを接触気相酸化によりアクロレインやメタクロレインとし、得られたアクロレインやメタクロレインの接触気相酸化により(メタ)アクリル酸を製造する二段酸化方法が一般的である。プロピレンやイソブチレンは化石資源由来の原料であるため、地球温暖化ガスの排出削減等、環境負荷の低減の観点から再生可能資源から製造することが望まれている。
また(メタ)アクリル酸エステルは、(メタ)アクリル酸のエステル化等により製造されており、粘着剤、塗料等各種樹脂の原料として広く用いられている。
【0003】
再生可能な資源であるバイオマス等を利用して、(メタ)アクリル酸を商業的規模で経済的に製造する試みが行われている。バイオマスからの(メタ)アクリル酸の生成方法としては、天然物であり容易に入手可能な乳酸や糖類から発酵により調製される3−ヒドロキシカルボン酸等を脱水することにより(メタ)アクリル酸を調製する方法が挙げられる。
【0004】
特許文献1は、発酵等により得られたβ−ヒドロキシカルボン酸、その塩又はそのエステルを含む水溶液又は溶液を準備し、その溶液を脱水触媒の存在又は非存在の下で加熱することにより脱水を施し、不飽和カルボン酸、その塩又はそのエステルを製造する方法を開示している。特許文献2は、α−又はβ−ヒドロキシカルボン酸を含む水溶液を不活性なセラミック等や酸性の固体触媒を保持したところへ導入して加熱することによりα,β−不飽和カルボン酸を調製する方法を開示している。更にα−又はβ−ヒドロキシカルボン酸から形成されるポリマー、オリゴマー、ラクチド、ラクトン等を含む水溶液を用いることができるとの記載はあるが、具体的に実施した例の開示はない。
【先行技術文献】
【特許文献】
【0005】
【特許文献1】特表2005−521718号公報
【特許文献2】国際公開第2005/095320号
【発明の概要】
【発明が解決しようとする課題】
【0006】
上記の通り、3−ヒドロキシカルボン酸又はそのエステルを脱水する反応は各特許文献に開示されているものの、これらの文献には、反応によって触媒にもたらされる障害や、その解決方法については、開示されていない。しかし3−ヒドロキシカルボン酸又はそのエステルの脱水用触媒を用いて、脱水反応を実施すると、触媒上に炭素状物質が付着し、触媒の活性低下を招いたり、選択率の低下を引き起こす、又は炭素状物質により反応管が閉塞するという問題があり、これらの問題が(メタ)アクリル酸又はそのエステルを効率良く得るための障害となる。
上記事情に鑑み、本発明は、3−ヒドロキシカルボン酸又はそのエステルの脱水用触媒上に付着した炭素状物質を除去する触媒の再生方法、及び、長期間にわたり安定的に(メタ)アクリル酸又はそのエステルを製造する方法の提供を目的とする。
【課題を解決するための手段】
【0007】
本発明者らは、種々検討した結果、3−ヒドロキシカルボン酸又はそのエステルの脱水用触媒に付着した炭素状物質に、酸化剤を接触させることにより炭素状物質を除去し、3−ヒドロキシカルボン酸又はそのエステル脱水用触媒の活性を回復し、長期間に渡って安定的に(メタ)アクリル酸又はそのエステルを製造することができることを見出して、本発明を完成した。
【0008】
すなわち、本発明は、3−ヒドロキシカルボン酸又はそのエステルの脱水用触媒上の炭素状物質を除去することによって触媒を再生する方法であって、該再生方法は、脱水用触媒に酸化剤を接触させて炭素状物質を除去することを特徴とする3−ヒドロキシカルボン酸又はそのエステルの脱水用触媒の再生方法である。
また、本発明は、前記酸化剤は、ガス状酸化剤であることを特徴とする上記3−ヒドロキシカルボン酸又はそのエステルの脱水用触媒の再生方法である。
また、本発明は、前記再生方法は、固定床反応器内に充填された3−ヒドロキシカルボン酸又はそのエステルの脱水用触媒を再生する方法であり、該固定床反応器内にガス状酸化剤を流通させて、炭素状物質を除去することを特徴とする上記3−ヒドロキシカルボン酸又はそのエステルの脱水用触媒の再生方法である。
【0009】
更に、本発明は、上記3−ヒドロキシカルボン酸又はそのエステルの脱水用触媒の再生方法を含むことを特徴とする(メタ)アクリル酸又はそのエステルの製造方法である。
更に、本発明は、第一の脱水工程、触媒再生工程及び第二の脱水工程を含み、該第一の脱水工程は、3−ヒドロキシカルボン酸又はそのエステルと脱水用触媒との気相接触反応により3−ヒドロキシカルボン酸又はそのエステルを脱水する工程であり、該触媒再生工程は、前記再生方法により脱水用触媒を再生する工程であり、該第二の脱水工程は、3−ヒドロキシカルボン酸又はそのエステルと触媒再生工程後の脱水用触媒との気相接触反応により3−ヒドロキシカルボン酸又はそのエステルを脱水する工程であることを特徴とする上記(メタ)アクリル酸又はそのエステルの製造方法である。
【発明の効果】
【0010】
本発明の脱水用触媒の再生方法によれば、3−ヒドロキシカルボン酸又はそのエステルの脱水用触媒の活性を効率的に回復させることができる。また、当該再生方法を含む(メタ)アクリル酸又はそのエステルの製造方法により、触媒上の炭素状物質による反応管の閉塞を抑制し、3−ヒドロキシカルボン酸又はそのエステルを効率良く(メタ)アクリル酸又はそのエステルに転化することができ、(メタ)アクリル酸又はそのエステルを長期間にわたり安定して製造することができる。更に、本製法で得られた(メタ)アクリル酸を使用して吸水性樹脂の製造をすることにより、高性能の吸水性樹脂を得ることができる。
【図面の簡単な説明】
【0011】
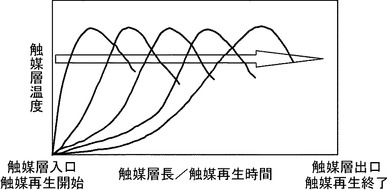
【図1】固定床反応器内において炭素状物質を触媒上から酸化分解除去するときの触媒の温度変化を説明するための図である。
【発明を実施するための形態】
【0012】
以下、本発明を詳細に説明する。
なお、以下において記載する本発明の個々の好ましい形態を2つ以上組み合わせたものもまた、本発明の好ましい形態である。
【0013】
なお、本明細書においては、特に断りのない限り、3−ヒドロキシカルボン酸又はそのエステルについては、3−ヒドロキシカルボン酸をこれらの代表として記載し、また、(メタ)アクリル酸又はそのエステルについては、(メタ)アクリル酸をこれらの代表として記載する。なお、3−ヒドロキシカルボン酸エステル及び(メタ)アクリル酸エステルは、それぞれ対応する酸を、後述の方法によりエステル化して得ることができる。
【0014】
本発明における3−ヒドロキシカルボン酸としては、3−ヒドロキシプロピオン酸(以下、3HPともいう)、3−ヒドロキシイソ酪酸等が挙げられ、好ましくは3−ヒドロキシプロピオン酸である。また、(メタ)アクリル酸としては、アクリル酸、メタクリル酸が挙げられ、好ましくはアクリル酸である。
上記3−ヒドロキシカルボン酸は、1種でも2種でも用いることができる。また、(メタ)アクリル酸は、使用した3−ヒドロキシカルボン酸の種類に応じて得られる。
【0015】
本発明は、3−ヒドロキシカルボン酸又はそのエステルの脱水用触媒(以下、単に「触媒」ともいう)上の炭素状物質に、酸化剤を接触させて、前記炭素状物質を除去することを特徴とする3−ヒドロキシカルボン酸又はそのエステルの脱水用触媒の再生方法である。
ここで本発明における「酸化剤」とは、炭素状物質を酸化剤の作用によって二酸化炭素、一酸化炭素、その他の炭素含有化合物に酸化分解するものをいう。
【0016】
本発明における触媒の再生方法は、触媒上に付着した炭素状物質に酸化剤を接触させて、この炭素状物質を触媒上から酸化分解除去する方法である。炭素状物質を酸化分解除去した後には、必要に応じて、触媒活性成分の補充処理が行われる。
【0017】
上記再生方法は、3−ヒドロキシカルボン酸又はそのエステルと、脱水用触媒との気相接触反応により3−ヒドロキシカルボン酸又はそのエステルを脱水する第一の3−ヒドロキシカルボン酸又はそのエステルの脱水工程と、この脱水工程と同様の第二の3−ヒドロキシカルボン酸又はそのエステルの脱水工程との間の、触媒再生工程に使用される方法である。
【0018】
本発明で再生対象となる触媒は、3−ヒドロキシカルボン酸又はそのエステルの脱水用触媒である。そして、この触媒上には、3−ヒドロキシカルボン酸又はそのエステルとの気相接触反応過程で生じた炭素状物質が表面に堆積している。この炭素状物質は、触媒の表面や細孔内等、触媒のあらゆる部分に付着しうる。
【0019】
上記触媒としては、ゼオライト等の結晶性メタロシリケート;結晶性メタロシリケートに、アルカリ金属、アルカリ土類金属又は遷移金属等を、イオン交換等の方法によって担持させたもの;カオリナイト、ベントナイト、モンモリロナイト等の天然又は合成粘土化合物;硫酸、ヘテロポリ酸、リン酸、リン酸塩(リン酸のアルカリ金属塩、アルカリ土類金属塩、リン酸マンガン、リン酸ジルコニウム等)、アルカリ金属又はアルカリ土類金属を、アルミナやシリカ等の担体に担持させた触媒;活性アルミナ(Al2O3)、SiO2、TiO2、ZrO2、SnO2、V2O5、SiO2−Al2O3、SiO2−TiO2、SiO2−ZrO2、TiO2−WO3、TiO2−ZrO2等の無機酸化物又は無機複合酸化物;MgSO4、Al2(SO4)3、K2SO4、AlPO4、Zr(SO4)2等の金属の硫酸塩、リン酸塩等の固体酸性物質;酸化カルシウム、酸化マグネシウム、ハイドロタルサイト等の固体塩基性物質;等が挙げられる。
好ましくは、Al2O3、SiO2、SiO2−Al2O3、TiO2、ゼオライト;ゼオライトに、アルカリ金属又はアルカリ土類金属を担持させたもの;リン酸、リン酸塩、アルカリ金属又はアルカリ土類金属をシリカ等の担体に担持させたものである。
【0020】
上記触媒は、粉体であっても成形体であっても良い。その成形体形状としては、限定されるものではなく、球状、シリンダー型、リング型、ハニカム型等が挙げられる。触媒の形態は使用する反応形式に応じて選択することができる。固定床反応器の場合は、圧力損失を小さくするために成型体が好ましく、流動床反応器の場合は、触媒を流動させるために粉体の方が好ましい。
上記触媒の物性としては、触媒活性等の点から、BET法による比表面積は、0.01〜500m2/gが好ましく、0.1〜400m2/gがより好ましい。触媒活性、触媒寿命等の点から、ハメットの酸度関数H0は、+4〜−10が好ましく、+2〜−8がより好ましい。また、触媒活性や反応器の圧力損失の点から、触媒の大きさは、長径が0.1mm〜50mmが好ましく、0.5mm〜40mmがより好ましい。
【0021】
上記酸化剤としては、過酸化水素水、有機過酸化物、硝酸、亜硝酸等が溶解した液状の酸化剤を使用しても良いし、ガス状の酸化剤を使用しても良い。好ましくは、ガス状の酸化剤である。
ガス状の酸化剤は、炭素状物質の酸化分解のために該炭素状物質に酸素元素を供給することが可能な気体分子であり、例えば、酸素(空気中の酸素も酸化剤に該当する)、オゾン、一酸化窒素、二酸化窒素、一酸化二窒素等を挙げることができる。これらの酸化剤のうち、少なくとも一種以上のガス状酸化剤が含まれていれば良く、例えば、空気と酸素との混合ガス、一酸化窒素と酸素との混合ガスを使用しても良く、窒素、二酸化炭素、アルゴン、ヘリウム、及び水蒸気等の不活性ガスから任意に選択した一種以上のガスと酸化剤との混合ガスを使用しても良い。
なお、オゾンや過酸化水素水を用いると、触媒が酸点を有する場合、無効分解によって、炭素状物質の除去効率が悪くなることがある。従って、酸点の多い触媒を用いる場合は、酸化剤として酸素を用いることがより好ましい。
具体的には、触媒の酸点が、アンモニア昇温脱離法による100〜600℃のアンモニア脱離量として0.001mmol/g以上の時は、酸素による触媒再生がより好ましい。
【0022】
触媒再生において、第一の3−ヒドロキシカルボン酸又はそのエステルの脱水工程で使用された反応器内から取り出した触媒を、酸化剤ガスに曝しても良いが、本発明では、簡便に触媒を再生するため、触媒が充填された固定床反応器内にガス状酸化剤を流通させて、前記炭素状物質を除去することが好ましい。
【0023】
触媒再生において触媒の加熱温度は、高温であるほど触媒再生時間を短縮できるが、あまり高すぎると触媒の活性や選択率が低下する恐れがある。通常好ましい範囲は300〜800℃であり、より好ましくは350〜700℃である。800℃を超えると、例えばシンタリングによる触媒表面積の低下や相転移による触媒の結晶構造変化等の触媒における物理的構造及び化学的性質が変わることになって、触媒活性や選択率が低下する恐れがある。温度は、触媒種によっても上限は異なるが、触媒調製の際に触媒を焼成する場合、焼成温度を超えない温度で加熱することが好ましい。
なお、後述の本発明の製造方法により得られる生成物である(メタ)アクリル酸は、脱水触媒上で非常にコーキングを起こしやすく、炭素状物質として触媒上に蓄積しやすいことが明らかとなった。また、原料組成物中に含まれる3−ヒドロキシカルボン酸オリゴマーは、沸点が高く蒸発しにくいため、触媒上に吸着すると更に重質化して容易に炭素状物質へと変化する。よって、これらの炭素状物質は、酸化剤を用いて酸化分解するにあたり、通常よりも高い温度が必要とされる。例えば酸化剤として酸素を用いて触媒再生を行う場合は、触媒の温度が350℃〜800℃の範囲が好ましく、より好ましくは400℃〜700℃である。
【0024】
上記加熱温度を制御するには、該触媒を加熱するための加熱器の設定温度、酸化剤濃度、及びガス流量等を調整すると良い。この場合、加熱器の設定温度及び/又は酸化剤の濃度が高いほど、触媒を加熱する温度が高くなる。そして、触媒加熱温度を連続的に測定しつつ、加熱器の設定温度及び/又は酸化剤の濃度を調整して触媒加熱温度を制御することも可能である。また、特開平5−192590号公報に開示されている触媒加熱温度の制御方法も挙げられる。
酸化剤濃度としては、温度制御や生産コスト等の点から、好ましくは1〜21体積%である。
処理時間としては、(メタ)アクリル酸の生産性等の点から、好ましくは1〜100時間、より好ましくは2〜50時間である。
【0025】
ここで、固定床反応器内の温度変化を、図1をもって説明する。図1は、炭素状物質を触媒上から酸化分解除去するときの触媒の温度変化を説明するための概略温度グラフであり、縦軸が触媒層温度、横軸が触媒再生時間及び固定床反応器内における触媒層長、矢印が固定床反応器内におけるガス流通方向を表している。図示の通り、固定床反応器内における最高の触媒加熱温度(ピーク温度)は、時間の経過と共に固定床反応器の入口から出口(ガス流通方向)に向かって移動する。すなわち、炭素状物質の酸化分解除去当初では、固定床反応器入口付近における炭素状物質の酸化分解のために酸化剤の大部分が消費され、この消費位置は、再生時間の経過と共に出口側に移動するのである。なお、図1に示すように、固定床反応器内のピーク温度位置を当該反応器のガス流通方向に向けて連続的に移動させ、触媒を当該触媒調製における焼成温度以下にて加熱することは、本実施形態の好ましい態様である。
【0026】
炭素状物質を酸化分解除去する本実施形態に係る方法では、触媒の種類によっては、炭素状物質の酸化分解過程で触媒活性成分が飛散する場合がある。このような場合には、必要とする触媒活性に応じて、触媒活性成分の補充処理が行われる。当該補充処理を行うことが好ましい触媒としては、リン化合物担持触媒が例示される。シリカやアルミナ等の担体にリン化合物が担持されたリン担持触媒は、3−ヒドロキシカルボン酸の脱水反応過程や炭素状物質の酸化分解除去過程で飛散した触媒活性成分(リン)が補充されると、活性が回復する。
【0027】
触媒活性成分の補充処理を行うためには、触媒調製の際における活性成分を担持させる方法を再実行すると良い。この方法としては、例えば、(1)含浸等の方法により、反応管から抜き出した触媒に、所定量の触媒活性成分を補充する方法、(2)特開平2−290255号公報に開示されている方法等により、反応管に充填されている状態の触媒に揮発性化合物(リン酸エステル等のリン元素を含有する化合物)を接触させる処理により、触媒活性成分を補充する方法、等が挙げられる。
上記再生方法により、触媒の再生が行われる。
【0028】
次に、本発明の(メタ)アクリル酸又はそのエステルの製造方法は、上記3−ヒドロキシカルボン酸又はそのエステルの脱水用触媒の再生方法を含むものである。
また、本発明の(メタ)アクリル酸又はそのエステルの製造方法は、第一の脱水工程、触媒再生工程及び第二の脱水工程を含み、該第一の脱水工程は、3−ヒドロキシカルボン酸又はそのエステルと脱水用触媒との気相接触反応により3−ヒドロキシカルボン酸又はそのエステルを脱水する工程であり、該触媒再生工程は、前記再生方法により脱水用触媒を再生する工程であり、該第二の脱水工程は、3−ヒドロキシカルボン酸又はそのエステルと触媒再生工程後の脱水用触媒との気相接触反応により3−ヒドロキシカルボン酸又はそのエステルを脱水する工程であることが好ましい。
【0029】
そこで、本実施形態に係る再生方法を使用する前の触媒を使用する第一の3−ヒドロキシカルボン酸又はそのエステルの脱水工程と、その再生後の触媒を使用する第二の3−ヒドロキシカルボン酸又はそのエステルの脱水工程について説明する。
第一の及び第二の3−ヒドロキシカルボン酸又はそのエステルの脱水工程では、3−ヒドロキシカルボン酸又はそのエステルの誘導体である(メタ)アクリル酸又はそのエステルを、気相接触反応により得るために、固定床反応器に反応ガスを流通させることが好ましい。
なお、本実施形態では、第一の3−ヒドロキシカルボン酸又はそのエステルの脱水工程及び第二の3−ヒドロキシカルボン酸又はそのエステルの脱水工程の、条件及び手順は同じである。
【0030】
上記第一の脱水工程は、3−ヒドロキシカルボン酸又はそのエステルと脱水用触媒との気相接触反応により、3−ヒドロキシカルボン酸又はそのエステルを脱水する工程である。
上記脱水工程では、3−ヒドロキシカルボン酸又はそのエステルを含む原料組成物を原料として用いる。そこで、3−ヒドロキシカルボン酸又はそのエステルを含む原料組成物の物性や調製方法等について、まず説明する。
【0031】
上記原料組成物に含まれる3−ヒドロキシカルボン酸又はそのエステルの濃度は、好ましくは5〜95質量%、より好ましくは10〜90質量%、更に好ましくは20〜90質量%である。
また、3−ヒドロキシカルボン酸又はそのエステルのダイマーやトリマー等のオリゴマーが含まれていても良く、そのオリゴマーは原料として使用することができる。原料組成物に含まれる上記オリゴマーは、3−ヒドロキシカルボン酸とそのエステルの合計100質量%に対して、好ましくは1〜400質量%、より好ましくは1〜300質量%含まれていてもよい。上記オリゴマー量を1質量%以上とすることにより、3−ヒドロキシカルボン酸又はそのエステルの、オリゴマー化抑制やオリゴマー量低減のための設備やエネルギーを最小限に抑えることができ、また、400質量%以下とすることにより、脱水触媒上での炭素状物質の生成及びコーキングによる閉塞を抑制し、(メタ)アクリル酸の収率を向上させることができる。
【0032】
原料組成物には、溶媒が含まれていても良い。当該溶媒としては、3−ヒドロキシカルボン酸又はそのエステルを溶解できるものであれば、特に限定されないが、例えば、水、アルコール、炭化水素、エーテル、ケトン、エステル、アミン、アミド等が挙げられる。これらは、1種でも2種以上でも用いることができる。溶媒の沸点は、気化が容易になるため3−ヒドロキシカルボン酸又はそのエステルよりも低い方が好ましい。好適には水である。
【0033】
本発明において、原料組成物中に溶媒を含有させる場合、原料組成物100質量%における溶媒の濃度は、好ましくは5〜95質量%であり、より好ましくは10〜90質量%、更に好ましくは10〜80質量%である。溶媒の濃度が5質量%以上であれば、粘度の低下により原料組成物の取り扱いが容易になり、また、3−ヒドロキシカルボン酸又はそのエステルの蒸発が促進される効果が期待できる。一方、95質量%以下とすることにより、蒸発にかかる熱量を抑制し、用役費の低減に寄与できる。
【0034】
上記原料組成物には、3−ヒドロキシカルボン酸又はそのエステル以外の成分、例えば、3−ヒドロキシカルボン酸を発酵により合成する際の副生物等が含まれていても良い。当該副生物としては、具体的には、発酵において3−ヒドロキシカルボン酸と共に副生される可能性のある、プロピオン酸、ギ酸、酢酸、酪酸、乳酸、乳酸エステル、エタノール、アミノ酸類等が例示できる。しかし、乳酸、乳酸エステルやエタノールは、その存在量が多いと、脱水反応において脱水触媒上での炭素状物質の生成を促進するため、その量は、3−ヒドロキシカルボン酸又はそのエステルに対して、それぞれ2質量%以下であることが好ましい。
【0035】
本発明で用いられる3−ヒドロキシカルボン酸は種々の源から得ることができるが、地球温暖化及び環境保護の観点から、炭素源としてリサイクル可能な生物由来資源を用いることが好ましい。また、植物由来の糖類又はセルロース等を分解して得られる糖類や、油脂から得られるグリセリンから、更に発酵により調製された3−ヒドロキシカルボン酸を用いることができる。
本発明においては、原料組成物中に含まれる3−ヒドロキシカルボン酸の少なくとも一部又は全部が、発酵により得られる3−ヒドロキシカルボン酸であることが好ましい。
【0036】
3−ヒドロキシカルボン酸は、公知の方法で入手可能であり、例えば国際公開第2008/027742号に記載されている、Streptomyces griseus ATCC21897由来beta−alanine aminotransferase遺伝子導入大腸菌を用いた、グルコースを炭素源とした発酵により得ることができる。また、国際公開第2001/016346号に記載されている、Klebsiella pneumoniae由来グリセリン脱水酵素及び大腸菌由来アルデヒド酸化酵素導入大腸菌を用いた、グリセリンを炭素源とした発酵によっても得ることができる。
【0037】
3−ヒドロキシカルボン酸の入手方法の例として上記公知文献を記載したが、本発明の方法を用いる限り、発酵に用いる細菌又は組換え細菌は特に限定されず、3−ヒドロキシカルボン酸生成能を有する生物を用いた発酵により入手した3−ヒドロキシカルボン酸であれば、本発明の製法で利用可能である。また、発酵以外にも原料とする糖類と生物とを接触させることにより生成した3−ヒドロキシカルボン酸でも、本発明の製法で(メタ)アクリル酸へ変換することができる。
糖類と生物を接触させるとは、原料として利用する糖類の存在下で微生物又はその処理物を用いて反応を行うことをも包含する。該処理物としては、アセトン、トルエン等で処理した菌体、菌死体、凍結乾燥菌体、菌体破砕物、菌体を破砕した無細胞抽出物、これらから酵素を抽出した粗酵素液、精製酵素等が挙げられる。また、常法により担体に固定化した菌体、該処理物、酵素等を用いて反応を行うことにより入手した3−ヒドロキシカルボン酸も用いることができる。
【0038】
本発明では、生物由来資源を用いて発酵により3−ヒドロキシカルボン酸を得る具体的実施形態に係る方法において、固体(特に微細な植物の部分又は細胞及び/又は細胞断片)、発酵の後に得られる3−ヒドロキシカルボン酸及び微生物等を含む水性組成物から、微生物や生物的材料等を分離するのが良い。前記分離は、固体を液状組成物から分離するための、当業者に公知の全ての方法により実施することができるが、好ましくは沈殿法、遠心分離法又は濾過法により、最も好ましくは濾過法により、分離するのがよい。
【0039】
3−ヒドロキシカルボン酸及び微生物等を含む水性組成物から微生物等を分離する処理においては、そこに含まれる微生物に処理を施すことなく行っても良いが、そこに含まれる微生物を殺菌する処理工程を含んでも良い。前記水性組成物から微生物等を殺菌する処理は、微生物を分離する前、その間又は後に行うことができる。上記殺菌処理としては、加熱処理(加熱による微生物の殺菌)、高エネルギー照射処理(例えば紫外線照射による微生物の殺菌)等が挙げられ、好ましくは加熱処理である。
上記加熱処理としては、3−ヒドロキシカルボン酸及び微生物等を含む水性組成物を、好ましくは少なくとも60秒間、より好ましくは少なくとも10分間、更に好ましくは少なくとも30分間の処理時間で、好ましくは少なくとも100℃、より好ましくは少なくとも110℃、更に好ましくは少なくとも120℃の温度で、加熱することによって実施するのが好ましい。当該加熱処理は、当業者に公知の装置(例えばオートクレーブ等)において実施するのが好ましい。
【0040】
本発明に用いる3−ヒドロキシカルボン酸は、発酵液より回収し、純度を高めたものを脱水工程で用いることが好ましい。発酵液からの3−ヒドロキシカルボン酸の回収方法としては、公知の方法が利用可能である。例えば、蒸留、蒸発、抽出、膜分離、晶析、イオン交換、電気透析等が挙げられ、これらを組み合わせても良い。更に具体的には、発酵により得られた粗製3−ヒドロキシカルボン酸を、カルシウム塩を用いて沈殿させて3−ヒドロキシカルボン酸のカルシウム塩として回収し、その後、硫酸等の酸と反応させて3−ヒドロキシカルボン酸を精製する方法や、発酵により得たアンモニウム型の3−ヒドロキシカルボン酸を電気透析又は陽イオン交換法によって3−ヒドロキシカルボン酸に化学変換させて精製する方法等が利用できる。
【0041】
また、発酵により得られたアンモニウム型の3−ヒドロキシカルボン酸に、水に不混和性のアミン溶媒を添加し加熱することで、アンモニアを除去して3−ヒドロキシカルボン酸のアミン溶液を得ることができる。そこに水を加えて加熱することで、3−ヒドロキシカルボン酸の水溶液を得ることができる。
【0042】
また、上記3−ヒドロキシカルボン酸をアルコールにてエステル化を行い、3−ヒドロキシカルボン酸エステルを合成することができる。
アルコールとしては、特に限定されないが、炭素数が1〜20のアルコールが好ましく、炭素数が1〜10のアルコールがより好ましく、炭素数1〜5のアルコールが更に好ましい。
エステル化反応は、3−ヒドロキシカルボン酸とアルコールを、エステル化触媒の存在下又は非存在下、加熱することによって行うことができる。3−ヒドロキシカルボン酸はカルボン酸のため、触媒の非存在下でもエステル化反応は進行する。しかし、生産効率の点から、エステル化触媒を用いることが好ましい。
エステル化触媒としては、公知のものが使用できるが、塩酸、硫酸、硝酸、リン酸等の鉱酸類;ゼオライトやイオン交換樹脂等の固体酸類;ヘテロポリ酸等の無機酸;p−トルエンスルホン酸等のスルホン酸類;ジブチルスズジラウレート、酸化スズ、ジブチルスズオキサイド、酢酸亜鉛、テトラアルコキシチタン等の金属化合物類等が挙げられる。
反応温度は、50℃〜300℃が好ましく、80℃〜250℃がより好ましい。エステル化反応は平衡反応のため、収率向上のために、反応蒸留や、生成物を抽出しながらの反応も効果的である。
【0043】
上述のようにして、3−ヒドロキシカルボン酸又はそのエステルを調製することができるが、発酵液中には微生物由来のタンパク質やアミノ酸といった含窒素化合物が含まれる。これらの含窒素化合物やその分解物が、3−ヒドロキシカルボン酸又はそのエステルと共に反応器に供給され、触媒と接触すると、アミノ基等の含窒素官能基が触媒上に吸着して、触媒活性を低下させることがある。また、含窒素化合物は、反応して重質化しやすく、生成した重質物が触媒上に付着して活性低下の要因となる。更に、触媒から、窒素を含んだ炭素状物質を除去する場合、分解しにくく再生温度が高くなったり、酸化剤と接触させた時に排ガス中に有害な窒素酸化物が含まれることとなり、除去装置が必要となる場合がある。
これらの悪影響を小さくするためには、原料中に含まれる窒素化合物中の窒素量が、3−ヒドロキシカルボン酸、そのエステル、及び、それらのオリゴマー類の合計100質量%に対して、0.2質量%以下であることが好ましい。より好ましくは0.1質量%以下である。窒素量の下限は、好ましくは0.1質量ppm以上である。より好ましくは0.5質量ppm以上であり、更に好ましくは1質量ppm以上である。0.1質量ppm未満とするためには、過大な精製工程が必要となり、コストアップの要因となる。
原料中の窒素化合物を低減させるには、発酵液から原料を調製する過程で、以下の方法により実施することができる。例えば、ナノ濾過膜や逆浸透膜等を用いてタンパク質やアミノ酸類と3−ヒドロキシカルボン酸を分離する方法;タンパク質と3−ヒドロキシカルボン酸を含む液を加熱し、タンパク質を変性、重質化し析出させて分離する方法;蒸留、蒸発、晶析、抽出、電気透析等を用いて、3−ヒドロキシカルボン酸と含窒素化合物を分離する方法等が挙げられる。
【0044】
また、3−ヒドロキシカルボン酸又はそのエステルは、複数の分子がエステル結合で連なった、3−ヒドロキシカルボン酸のダイマーやトリマーといったオリゴマー類を形成しやすい特性を持っている。オリゴマー類も(メタ)アクリル酸製造の原料として使用できるが、オリゴマーは沸点が高く、触媒上に吸着するとより重質化しやすいため、その濃度、分子量や加熱条件によっては、蒸発器や反応器での閉塞の原因となったり、脱水触媒上での炭素状物質の生成を促進することがある。
3−ヒドロキシカルボン酸又はそのエステルを含む原料組成物は、蒸発器内でその一部が反応し、(メタ)アクリル酸又はそのエステルや、3−ヒドロキシカルボン酸又はそのエステルのオリゴマーが生成する。また条件によっては、原料組成物中に含まれているオリゴマーや生成したオリゴマーの分解反応も起こる。脱水触媒上での炭素状物質の生成を抑制するためには、脱水触媒と接触する蒸発器出口ガスの組成を制御することが好ましい。
蒸発器出口ガスの組成としては、3−ヒドロキシカルボン酸又はそのエステルのオリゴマー類の濃度が、3−ヒドロキシカルボン酸とそのエステルの合計100質量%に対して、1〜100質量%であることが好ましい。より好ましくは1〜80質量%である。上記オリゴマー類の濃度を1質量%以上とすることにより、3−ヒドロキシカルボン酸又はそのエステルの、オリゴマー化抑制やオリゴマー濃度低減のための設備やエネルギーを最小限に抑えることができ、また、100質量%以下とすることにより、脱水触媒上での炭素状物質の生成を抑制し、触媒の活性低下を抑えることで、再生までの反応時間を長くすることが可能となり、(メタ)アクリル酸又はそのエステルの生産性を向上させることができる。
脱水触媒に供給される蒸発器出口ガス中の3−ヒドロキシカルボン酸又はそのエステルのオリゴマーは、3−ヒドロキシカルボン酸又はそのエステルの分子が2〜10個結合したものが、オリゴマー全体の70質量%以上であることが好ましく、80質量%以上であることがより好ましい。更に、3−ヒドロキシカルボン酸又はそのエステルの分子が2〜8個結合したものが、オリゴマー全体の70質量%以上であることが好ましく、80質量%以上であることがより好ましい。
【0045】
脱水触媒へ供給する原料中のオリゴマーを上記範囲に制御するためには、例えば蒸発器に供給する前の段階で、3−ヒドロキシカルボン酸を水や溶媒で希釈し低濃度で取り扱う、取扱いを低温で行う等、オリゴマーの生成を抑制する方法等が挙げられる。また、蒸発器内でオリゴマーが生成しないように、蒸発器内の滞留時間を短くする、不活性ガスを供給して蒸発しやすくする、蒸発温度を高くする等の方法が挙げられる。更に、原料組成物中にオリゴマーを含んでいても、蒸発器内でオリゴマーを分解し、低分子化した上で反応器に供給する方法が挙げられる。蒸発器内でオリゴマーを分解する方法としては、高温でオリゴマーを分解する方法、触媒と接触させてオリゴマーを分解する方法等が挙げられる。脱水触媒へ供給される蒸発器出口ガス中のオリゴマーは、蒸発器と反応器が分離した設備であれば、蒸発器出口のガスを捕集して分析する、又は反応器に触媒を充填せずに原料組成物を蒸発器に供給し、反応器出口ガスを捕集して分析することで確認することができる。蒸発器と反応器が一体となっている設備であれば、触媒を充填せずに原料組成物を供給し、反応器出口ガスを捕集して分析することで確認することができる。
【0046】
上述のようにして3−ヒドロキシカルボン酸又はそのエステルを含む原料組成物を調製することができる。
次いで、上記3−ヒドロキシカルボン酸又はそのエステルを含む原料組成物と脱水用触媒との気相接触反応により、3−ヒドロキシカルボン酸又はそのエステルを脱水する。
上記脱水用触媒としては、上述したものと同じものが挙げられる。
上記3−ヒドロキシカルボン酸又はそのエステルを含む原料組成物と脱水用触媒との気相接触反応は、3−ヒドロキシカルボン酸又はそのエステルを含む原料組成物を気化させ、気化した原料組成物を脱水用触媒と接触させることにより、行うことができる。
【0047】
3−ヒドロキシカルボン酸又はそのエステルを含む原料組成物の気化(蒸発)は、該組成物を加熱することにより実施することができる。蒸発器内の温度は、150℃〜500℃が好ましく、170℃〜480℃がより好ましく、200℃〜460℃が更に好ましく、220℃〜440℃が特に好ましく、220℃〜420℃が最も好ましい。当該温度が150℃〜500℃の範囲であれば、原料組成物が速やかに気化し、副反応による蒸発器や反応器の閉塞が生じない。また、500℃より高いと、加熱に必要なエネルギーが過大になるばかりか、該組成物がコーキングを起こし、炭素質の析出物が蒸発器や反応器内に付着して閉塞を起こす可能性がある。
【0048】
蒸発器内の圧力は、低いほど原料組成物の蒸発が起こりやすくなるため有利であるが、引き続く反応器の適正な圧力や設備等のコストも合わせて選択する必要がある。蒸発器内の圧力としては、好ましくは10kPa〜1000kPaであり、より好ましくは30kPa〜300kPaであり、更に好ましくは50kPa〜250kPaである。
【0049】
蒸発器は、液体で供給する原料組成物に効率的に熱を伝える構造が好ましい。例えば、水平管型や垂直管型の自然循環式蒸発器、強制循環式蒸発器等が挙げられる。
また、蒸発器内の原料組成物の流路に、ラシヒリング、ベルルサドル、球状成型物、金網の成型物(ディクソンパッキン、マクマホンパッキン等)、メラパック(スルザーケムテック社製)といった不規則充填物や規則充填物等の、単位充填容積当たりの表面積が大きな充填物を充填し、そこに原料組成物を供給することで、原料組成物(液体)が接する表面積を大きくして蒸発させる方法も挙げられる。こうすることにより、供給した原料組成物が、表面積の大きな充填物と接触することになり、伝熱面積が増え、蒸発効率が向上する。
上記充填物の材料としては、鉄やステンレス等の金属材料や、シリカ、セラミック等の無機材料等が使用できる。
充填物の表面積は、蒸発器に充填した際に、5cm2/cm3以上が好ましく、10cm2/cm3以上がより好ましく、20cm2/cm3以上が更に好ましく、30cm2/cm3以上が特に好ましい。また、上限は特に限定されないが、1000cm2/cm3以下が好ましく、800cm2/cm3以下がより好ましい。
【0050】
また、上昇液膜型、流下液膜型、撹拌液膜型等の薄膜式熱交換器を用いて、液体の表面積を大きくして、短時間で蒸発させる方法も挙げられる。更に、スプレーやアトマイザー等を用いて、該組成物を細かい液滴にして分散させて蒸発させる方法も例示できる。液滴の径は、1mm以下が好ましく、0.5mm以下がより好ましい。
その他、加熱した原料組成物を蒸発室に供給し、気化させるフラッシュ蒸発器を用いても良い。フラッシュ蒸発器においては、原料組成物を、常圧又は加圧下で加熱し、減圧又は常圧下の蒸発室に液を供給して、原料組成物を気化させることができる。
また、原料組成物を流動床式の蒸発器に供給して、気化させても良い。例えば、粉末状の不活性固体を不活性ガスで流動化させ、加熱された流動床式蒸発器に原料組成物を供給し、気化させても良い。
また、原料組成物の気化は、上記方法を多段階で行っても良い。例えば、フラッシュ蒸発器の後に充填物を充填した蒸発器を設置し、フラッシュ蒸発器で、原料組成物の一部を蒸発させた後、充填物を充填した蒸発器で、残りの原料組成物を蒸発させても良い。
また、原料組成物の気化には、上記方法を併用してもよい。例えば、スプレーで原料組成物を噴霧し、充填物を充填した蒸発器で原料組成物を気化させることもできる。
【0051】
蒸発させる際の好ましい形態は、加熱する際にガス(水や不活性気体等)を導入しながら蒸発させる形態が好ましい。原料組成物と共に、水や不活性気体等のガスを導入すると、原料組成物の蒸発が促進され、安定な反応を継続できるため、好ましい形態である。
ガスとしては、窒素、ヘリウム、アルゴン、二酸化炭素、水が蒸発した水蒸気等が挙げられ、これらは1種でも2種以上でも用いることができる。好適には、窒素、水蒸気である。この水蒸気には、原料組成物中に溶媒として含まれる水が気化した水蒸気も含まれる。
ガスの供給量としては、原料組成物中の3−ヒドロキシカルボン酸、そのエステル及びそれらのオリゴマーの総量に対して、水及び/又は不活性気体の総量として0.5モル倍〜100モル倍が好ましく、1モル倍〜50モル倍がより好ましい。
【0052】
熱の供給方法は、蒸気、熱媒、溶融塩、ヒーター等の熱源を用いて、熱交換により加熱する方法等が挙げられる。また、加熱した水蒸気や、窒素、アルゴン、ヘリウム、二酸化炭素等の不活性ガスを蒸発器に供給し、原料組成物に接触させて熱を与える方法等も挙げられる。また、これらを併用しても良い。
例えば、加熱した充填物の上に、原料組成物を噴霧して蒸発させる方法や、更に加熱した窒素や水蒸気を同時に供給する方法等が好適である。
【0053】
次に、脱水工程で使用する反応器としては、中に固体触媒を保持し、加熱することができればよく、例えば、固定床反応器、流動床反応器等が使用でき、固定床反応器が好ましい。
上記固定床反応器を用いる場合は、反応器内に触媒を充填して加熱しておき、そこに原料組成物の蒸気を供給すればよい。原料組成物の蒸気は、上昇流、下降流、水平流、いずれも好適に使用できる。また、熱交換の容易さから、固定床多管式連続反応器が好適に使用できる。
上記流動床反応器を用いる場合は、反応器の中に粉末状の触媒を入れ、原料組成物の蒸気や、別途供給する不活性ガス等で触媒を流動させながら、反応させることができる。触媒が流動しているため、重質分による閉塞が起こりにくい。また、触媒の一部を連続的に抜き出して、新しい触媒や再生した触媒を連続的に供給することもできる。
【0054】
脱水工程は上記蒸発の後にあれば良く、その間に別の工程があっても良い。例えば、蒸発器で気化させた原料組成物の蒸気を、所定の温度に加熱/冷却する温度調整工程を経て、反応器で脱水工程を実施しても良い。
また、蒸発器と反応器を一体化しても良い。例えば、反応管に、蒸発層として表面積の大きい充填物を充填し、当該蒸発層の下に触媒を充填することにより、蒸発層を蒸発工程、触媒層を脱水工程として連続した運転も、好ましい形態の1つである。
また、1つ乃至は複数の蒸発層と、触媒を充填した多管式の反応器を連結して運転することも、好ましい形態である。
【0055】
上記脱水工程において、触媒層の温度は、150℃〜500℃に保持することが好ましい。より好ましくは200℃〜450℃、更に好ましくは220℃〜430℃、特に好ましくは250℃〜400℃である。この温度範囲(150℃〜500℃)であると、反応速度が速く、副反応も生じにくく、(メタ)アクリル酸の収率が高くなる。
反応圧力は、特に限定されないが、原料組成物の蒸発方法、脱水反応の生産性、脱水反応後の捕集効率等を勘案して決定することができる。反応圧力としては、10kPa〜1000kPaが好ましく、より好ましくは30kPa〜300kPa、更に好ましくは50kPa〜250kPaである。
このようにして第一の脱水工程を行うことができる。また、第二の脱水工程は、上記触媒再生工程で再生した触媒を用いること以外は、第一の脱水工程と同じである。
【0056】
上述のように、本発明の(メタ)アクリル酸又はそのエステルの製造方法は、第一の脱水工程、触媒再生工程及び第二の脱水工程を含むものである。
上記再生方法により触媒の再生を行う間に、3−ヒドロキシカルボン酸又はそのエステルの脱水反応を中断させないためには、複数の固定床反応器を並列配置して、ある固定床反応器内の触媒再生の間には、他の固定床反応器を3−ヒドロキシカルボン酸又はそのエステルの脱水反応に使用すると良い。つまり、一の固定床反応器で前記触媒再生工程を行う間、他の固定床反応器で前記第一又は第二の3−ヒドロキシカルボン酸又はそのエステルの脱水工程を行うことが好ましい。
【0057】
複数の固定床反応器を使用する場合、3−ヒドロキシカルボン酸又はそのエステルの脱水反応により(メタ)アクリル酸又はそのエステルを製造する時間(反応時間)と触媒上に蓄積した炭素状物質を除去して触媒を再生する時間(再生時間)に応じた数の反応器が必要である。例えば、再生時間が反応時間よりも短ければ、3−ヒドロキシカルボン酸又はそのエステルの脱水反応に使用されている反応器1基に対して、触媒再生に使用される反応器1基が必要であり、再生時間が反応時間の2倍以下であれば、3−ヒドロキシカルボン酸又はそのエステルの脱水反応に使用されている反応器1基に対して、触媒再生に使用される反応器2基があれば足りる。そのため触媒再生に使用される反応器の数を減らせば経済的に3−ヒドロキシカルボン酸又はそのエステルの脱水反応を行えることになり、反応器数を減らすためには、再生時間をできるだけ短時間にすることが好ましいといえる。触媒再生温度や、反応器の設計温度の上限を超えない範囲で、なるべく高い温度で再生することにより、再生時間を短時間にすることができる。そのために、酸化剤の濃度や、酸化剤と共に流通させる気体成分の流量等を制御することができる。
【0058】
また、本発明において、上記工程により得られた反応生成物を冷却して、(メタ)アクリル酸又はそのエステルを含む組成物を得ることができるが、その方法としては、特に限定されるものではない。例えば、反応生成ガスを熱交換器に導入して反応生成ガスの露点以下の温度で凝縮して得る方法や、反応生成ガスを溶剤等の捕集剤に接触させて吸収する方法等により冷却して、(メタ)アクリル酸又はそのエステルを含む組成物を得ることができる。
【0059】
本発明においては、3−ヒドロキシカルボン酸又はそのエステルの脱水用触媒上の炭素状物質に、酸化剤を接触させて前記炭素状物質を除去する。しかし、前述したように3−ヒドロキシカルボン酸又はそのエステルは、ダイマー、トリマーといったオリゴマー化が進行しやすい。特に触媒層へ原料を気体で供給するための蒸発器においては、原料の3−ヒドロキシカルボン酸又はそのエステルが液体で加熱されるため、オリゴマー化が非常に進行しやすく、一部のオリゴマーが蒸発されず、蒸発器内に残存し、長時間の加熱によって炭素質へと変化する場合がある。その場合、ラインの閉塞や、炭素状物質の析出により、伝熱係数が低下する。
【0060】
このような場合、蒸発器中で生成し残存する炭素状物質にも酸化剤を接触させて、炭素状物質を除去することができる。除去方法は、触媒再生の方法と同様である。蒸発器中の炭素状物質の除去と、触媒再生は、別々に実施しても良いし、同時に実施しても良い。蒸発器は反応器と連結されているため、蒸発器中の炭素状物質の除去と触媒再生は同時に実施すると効率がよい。酸化剤を蒸発器の上流側から供給すると、まず蒸発器中の炭素状物質が除去され、引き続き反応器中の触媒が再生されることになる。また酸化剤を蒸発器と反応器のそれぞれの入口から別々に供給することもできる。
【0061】
蒸発器は、原料の速やかな蒸発や、炭素状物質除去の際の除熱を速やかに行うため、熱の移動がしやすい構造が好ましい。例えば多管式の熱交換器や、薄膜式の加熱器が挙げられる。
蒸発器中の炭素状物質を酸化剤と接触させて除去する場合、蒸発器は、例えば不活性な無機担体や充填物を充填した形式が好ましい。例えば多管式の熱交換器の管内に不活性担体を充填しておき、管内で原料を蒸発させることにより、炭素状物質が生成しても、担体表面に付着させることにより、閉塞までの時間や、伝熱係数の低下を遅らせることができる。
【0062】
上記工程を経て、3−ヒドロキシカルボン酸又はそのエステルから、(メタ)アクリル酸又は(メタ)アクリル酸エステルを得ることができる。
このようにして得られた反応生成物中には、主な反応生成物である水、(メタ)アクリル酸又は(メタ)アクリル酸エステルが含まれており、その他に副生物や原料組成物中の溶媒や不純物が含まれる場合がある。溶媒が水の場合は、(メタ)アクリル酸又はそのエステルの水溶液の状態で重合物製造の原料とすることができる。また、精製工程を加えることにより、高純度の(メタ)アクリル酸又はそのエステルにすることができる。精製工程は、蒸留、膜分離、晶析等公知の技術により実施でき、それらを組み合わせて実施しても良い。
上記のようにして得られた(メタ)アクリル酸を含む反応生成物は、捕集や精製工程の取扱いを、重合禁止剤の存在下で行うことが好ましい。重合禁止剤としては、メトキノン、酢酸マンガン、ニトロソフェノール、クペロン、N−オキシル化合物、ジブチルチオカルバミン酸銅、フェノチアジン、ハイドロキノン等が例示できる。また、必要に応じて酸素含有ガスを供給してもよい。
【0063】
このように、本発明で得られた(メタ)アクリル酸の組成物を精製することにより高純度の(メタ)アクリル酸を得ることができる。したがって、本発明の方法は、高純度の(メタ)アクリル酸の製造方法をも提供する。
【0064】
上記のガス状の反応生成物を冷却凝縮や溶剤捕集等により液化し、必要に応じて、この液化物に含まれる水や捕集溶剤を従来公知の方法(例えば、蒸留)により除去したものを、晶析方法によって高純度の(メタ)アクリル酸を得る方法を以下に示す。
ここで、粗(メタ)アクリル酸とは、冷却工程で得られた(メタ)アクリル酸を含む組成物を指し、特に(メタ)アクリル酸の水溶液が好適に用いられる。
晶析工程は、粗(メタ)アクリル酸からプロピオン酸を分離することができる従来公知の方法、例えば、特開平9−227445号公報や特表2002−519402号公報に記載された方法を用いて行うことができる。
【0065】
晶析工程は、粗(メタ)アクリル酸を晶析装置に供給して結晶化させることにより、精製(メタ)アクリル酸を得る工程である。なお、結晶化の方法としては、従来公知の結晶化方法を採用すればよく、特に限定されるものではない。結晶化は、例えば、連続式又は回分式の晶析装置を用いて、1段又は2段以上で実施することができる。得られた(メタ)アクリル酸の結晶は、必要に応じて、更に洗浄や発汗等の精製を行うことにより、更に純度の高い精製(メタ)アクリル酸を得ることができる。
【0066】
連続式の晶析装置としては、例えば、結晶化部、固液分離部及び結晶精製部が一体になった晶析装置(例えば、新日鐵化学社製のBMC(Backmixing Column Crystallizer)装置、月島機械社製の連続溶融精製システム)や、結晶化部(例えば、GMF GOUDA社製のCDC(Cooling Disk Crystallizer)装置)、固液分離部(例えば、遠心分離器、ベルトフィルター)及び結晶精製部(例えば、呉羽テクノエンジ社製のKCP(Kureha Crystal Purifier)精製装置)を組み合わせた晶析装置等を使用することができる。
回分式の晶析装置としては、例えば、Sulzer Chemtech社製の層結晶化装置(動的結晶化装置)、BEFS PROKEM社製の静的結晶化装置等を使用することができる。
【0067】
動的結晶化とは、例えば、結晶化、発汗、融解を行うための温度制御機構を備えた管状の結晶器と、発汗後の母液を回収するタンクと、結晶器に粗(メタ)アクリル酸を供給する循環ポンプとを備え、結晶器の下部に設けた貯蔵器から循環ポンプにより粗(メタ)アクリル酸を結晶器の管内上部に移送できる動的結晶化装置を使用して晶析を行う方法である。
また、静的結晶化とは、例えば、結晶化、発汗、融解を行うための温度制御機構を備えた管状の結晶器であり、下部に抜き出し弁を有する結晶器と、発汗後の母液を回収するタンクとを備えた静的結晶化装置を使用して晶析を行う方法である。
【0068】
具体的には、粗(メタ)アクリル酸を液相として結晶器に導入し、液相中の(メタ)アクリル酸を冷却面(管壁面)に凝固させる。冷却面に生成した固相の質量が、結晶器に導入した粗(メタ)アクリル酸に対して、好ましくは10〜90質量%、より好ましくは20〜80質量%になったら、直ちに、液相を結晶器から排出し、固相と液相とを分離する。液相の排出は、ポンプで汲み出す方式(動的結晶化)、結晶器から流出させる方式(静的結晶化)のいずれであってもよい。他方、固相は、結晶器から取り出した後、更に純度を向上させるために、洗浄や発汗等の精製を行ってもよい。
【0069】
動的結晶化や静的結晶化を多段で行う場合、向流の原理を採用すれば、有利に実施することができる。このとき、各段階で結晶化された(メタ)アクリル酸は、残留母液から分離され、より高い純度を有する(メタ)アクリル酸が生成する段階に供給される。他方、残留母液は、より低い純度を有する(メタ)アクリル酸が生成する段階に供給される。
なお、動的結晶化では、(メタ)アクリル酸の純度が低くなると、結晶化が困難になるが、静的結晶化では、動的結晶化に比べて、残留母液が冷却面に接触する時間が長く、また、温度の影響が伝わり易いので、(メタ)アクリル酸の純度が低下しても、結晶化が容易である。それゆえ、(メタ)アクリル酸の回収率を向上させるために、動的結晶化における最終的な残留母液を静的結晶化に付して、更に結晶化を行ってもよい。
【0070】
必要となる結晶化段数は、どの程度の純度が要求されるかに依存する。高純度の(メタ)アクリル酸を得るために必要な段数は、精製段階(動的結晶化)が、通常1〜6回、好ましくは2〜5回、より好ましくは2〜4回であり、ストリッピング段階(動的結晶化及び/又は静的結晶化)が、通常0〜5回、好ましくは0〜3回である。通常、供給される粗(メタ)アクリル酸より高い純度を有する(メタ)アクリル酸が得られる段階は、すべて精製段階であり、それ以外の段階は、すべてストリッピング段階である。ストリッピング段階は、精製段階から残留母液に含まれる(メタ)アクリル酸を回収するために実施される。なお、ストリッピング段階は、必ずしも設ける必要はなく、例えば、蒸留塔を用いて、晶析装置の残留母液から低沸点成分を分離する場合には、ストリッピング段階は省略してもよい。
【0071】
動的結晶化及び静的結晶化のいずれを採用する場合であっても、晶析工程で得られる(メタ)アクリル酸の結晶は、そのまま製品としてもよいし、必要に応じて、更に洗浄や発汗等の精製を行ってから製品としてもよい。他方、晶析工程で排出される残留母液は、系外に取り出してもよい。
【0072】
以上の方法により、(メタ)アクリル酸を製造することができる。かくして製造された(メタ)アクリル酸は、すでに公知となっているように、(メタ)アクリル酸エステル等の(メタ)アクリル酸誘導体;ポリ(メタ)アクリル酸、ポリ(メタ)アクリル酸ナトリウム等の親水性樹脂;吸水性樹脂等の合成原料として有用である。
【0073】
更に、本発明による親水性樹脂の製造方法は、上記のようなアクリル酸又はそのエステルの製造方法により得られる、アクリル酸又はそのエステルを含む単量体成分を重合することを特徴とする。すなわち、本発明の製造方法により得られたアクリル酸又はそのエステルは、吸水性樹脂や水溶性樹脂等の親水性樹脂の原料として用いることができる。
【0074】
本発明の製造方法により得られたアクリル酸を、吸水性樹脂や水溶性樹脂等の親水性樹脂を製造するための原料として用いた場合、重合反応を制御しやすく、得られた親水性樹脂の品質が安定し、吸水性能、無機材料の分散性能等の各種性能が改善される。
親水性樹脂としては、吸水性樹脂であることが好ましい。
【0075】
吸水性樹脂を製造する場合には、例えば、本発明の製造方法により得られたアクリル酸又はその塩(アクリル酸を部分中和して得た塩)を単量体成分の主成分(好ましくは70モル%以上、より好ましくは90モル%以上)とし、更に0.001〜5モル%(アクリル酸に対する値)程度の架橋剤、0.001〜2モル%(単量体成分に対する値)程度のラジカル重合開始剤を用いて、架橋重合させた後、乾燥・粉砕することにより、吸水性樹脂を得ることができる。
【0076】
ここで、吸水性樹脂とは、架橋構造を有する水膨潤性水不溶性のポリアクリル酸であって、自重の3倍以上、好ましくは10〜1,000倍の純水又は生理食塩水を吸水することにより、水溶性成分(水可溶分)が好ましくは25質量%以下、より好ましくは10質量%以下である水不溶性ヒドロゲルを生成するポリアクリル酸を意味する。
このような吸水性樹脂の具体例や物性測定法は、例えば、米国特許第6,107,358号、米国特許第6,174,978号、米国特許第6,241,928号等に記載されている。
また、生産性向上の観点から好ましい製造方法は、例えば、米国特許第6,867,269号、米国特許第6,906,159号、米国特許第7,091,253号、国際公開第2001/038402号、国際公開第2006/034806号等に記載されている。
【0077】
アクリル酸を出発原料として、中和、重合、乾燥等により、吸水性樹脂を製造する一連の工程は、例えば以下の通りである。
本発明の製造方法により得られるアクリル酸の一部は、ラインを介して、吸水性樹脂の製造プロセスに供給される。吸収性樹脂の製造プロセスにおいては、アクリル酸を中和工程、重合工程、乾燥工程に導入して、所望の処理を施すことにより、吸水性樹脂を製造する。各種物性の改善を目的として所望の処理を施してもよく、例えば、重合中又は重合後に架橋工程を介在させてもよい。
【0078】
中和工程は、任意の工程であり、例えば、所定量の塩基性物質の粉末又は水溶液と、アクリル酸やポリアクリル酸(塩)とを混合する方法が例示されるが、従来公知の方法を採用すればよく、特に限定されるものではない。なお、中和工程は、重合前又は重合後のいずれで行なってもよく、また、重合前後の両方で行なってもよい。
アクリル酸やポリアクリル酸(塩)の中和に用いられる塩基性物質としては、例えば、炭酸(水素)塩、アルカリ金属の水酸化物、アンモニア、有機アミン等、従来公知の塩基性物質を適宜用いればよい。
また、ポリアクリル酸の中和率は、特に限定されるものではなく、任意の中和率(例えば、30〜100モル%の範囲内における任意の値)となるように調整すればよい。
【0079】
重合工程における重合方法は、特に限定されるものではなく、ラジカル重合開始剤による重合、放射線重合、電子線や活性エネルギー線の照射による重合、光増感剤による紫外線重合等、従来公知の重合方法を用いればよい。また、重合開始剤、重合条件等の各種条件については、任意に選択することができる。もちろん、必要に応じて、架橋剤や他の単量体、更には水溶性連鎖移動剤や親水性高分子等、従来公知の添加剤を添加してもよい。
【0080】
重合後のアクリル酸塩系ポリマー(すなわち、吸水性樹脂)は、乾燥工程に付される。乾燥方法としては、特に限定されるものではなく、熱風乾燥機、流動層乾燥機、ナウター式乾燥機等、従来公知の乾燥手段を用いて、所望の乾燥温度、好ましくは70〜230℃で、適宜乾燥させればよい。乾燥工程を経て得られた吸水性樹脂は、そのまま用いてもよく、更に所望の形状に造粒・粉砕、表面架橋をしてから用いてもよく、還元剤、香料、バインダー等の従来公知の添加剤を添加する等、用途に応じた後処理を施してから用いてもよい。
【実施例】
【0081】
以下に実施例を挙げて本発明をより具体的に説明するが、本発明は、下記実施例によって限定されるものではなく、適宜変更して実施することも可能であり、それらはいずれも本発明の技術的範囲に包含される。
【0082】
(調製例1)
(3−ヒドロキシプロピオン酸(3HP)を含む組成物の取得方法)
Klebsiella pneumoniae ATCC25955株のゲノムDNAをテンプテレ−トとしてグリセロールデヒドラターゼ遺伝子(GD遺伝子)及びグリセロールデヒドラターゼ再活性化因子(GDR遺伝子)を含む領域を、下記の2つのプライマーを用いてPCRで増幅し、増幅断片の末端を制限酵素NdeI、BglIIで切断し、電気泳動によって切断断片を回収した。なお、GD遺伝子及びGDR遺伝子配列を増幅する以下のプライマーはGenBank Accession number:NC_009648記載のDNA配列を元に設計した。
フォワードプライマー:
5’−GCGCGCCATATGTTAATTCGCCTGACCGGCC−3’
リバースプライマー:
5’−GCGCGCAGATCTTCAGTTTCTCTCACTTAACG−3’
【0083】
pACYCDuet−1プラスミド(タカラバイオ社)をテンプレートにして下記の2つのプライマーでベクター配列を増幅し、pACYCDuet−1プラスミドのT7プロモーターの後ろにNdeIサイト及びBgl IIサイトを持ったDNA断片を増幅した。
フォワードプライマー:
5’−GAAGGAGATATACATATGGCGCGC−3’
リバースプライマー:
5’−CCGATATCCAATTGAGATCTGCGCGC−3’
【0084】
増幅断片を制限酵素BglIIとNdeIで切断し、電気泳動によって切断断片を分離して回収した。この2つのDNA断片をライゲーションし、大腸菌TOP10コンピテントセル(インビトロジェン社)に導入し、クロラムフェニコール含有プレートに広げて培養したところ、クロラムフェニコール耐性大腸菌を得ることができた。クロラムフェニコール耐性大腸菌からプラスミドDNAを抽出し、制限酵素により分子量の確認を行ったところ、目的とするGD遺伝子及びGDR遺伝子がpACYCDuet−1プラスミドに挿入されていることが確認できた。構築した組換えプラスミドをGD−GDR/pACYCDuet−1と命名し、以降の実験に用いた。
【0085】
大腸菌K−12 W3110株のゲノムDNAをテンプテレ−トとしてγ−glutamyl−γ−aminobutyraldehyde dehydrogenase遺伝子(aldH遺伝子)を下記の2つのプライマーを用いてPCR法で増幅し、増幅断片の末端を制限酵素NdeI、BglIIで切断し、電気泳動によって切断断片を回収した。なお、以下プライマーはGenBank Accession number:AB200319記載のDNA配列を元に設計した。
フォワードプライマー:
5’−GGGGGGCCATATGAATTTTCATCATCTGGCTTACTG−3’
リバースプライマー:
5’−CCCCAGATCTTCAGGCCTCCAGGCTTATCCAGATG−3’
【0086】
pUC18プラスミドをテンプレートにして下記の2つのプライマーでベクター配列を増幅し、pUC18プラスミドのlacプロモーターの後ろにNdeIサイトを持ち、lacZ遺伝子の終始コドンの位置にBamHIサイトを持ったDNA断片を増幅した。
フォワードプライマー:
5’−CCCCCCCATATGTGTTTCCTGTGTGAAATTGTTATCCGCTCACAATTCCACACAATATACGAGCC−3’
リバースプライマー:
5’−CCCCGGATCCTTAGTTAAGCCAGCCCCGACACCCGCCAACACC−3’
【0087】
増幅断片を制限酵素BamHIとNdeIで切断し、電気泳動によって切断断片を分離して回収した。この2つのDNA断片をライゲーションし、大腸菌TOP10コンピテントセルに導入し、アンピシリン含有プレートに広げて培養した。得られた形質転換体からプラスミドを抽出し、制限酵素処理により分子量の確認をしたところ、目的どおり、aldHがpUC18プラスミドに挿入されていることを確認した。構築した組換えプラスミドをaldH/pUC18と命名し、以降の実験で使用した。
【0088】
構築したGD−GDR/pACYCDuet−1及びaldH/pUC18をEscherichia coli BL21(DE3) competent cell(Merck社製)のプロトコールに従って、ヒートショック法により導入し、E.coli(GD−GDR/pACYCDuet−1、aldH/pUC18)を作出した。
E.coli(GD−GDR/pACYCDuet−1、aldH/pUC18)を、アンピシリン100ppm、クロラムフェニコール50ppm添加LB液体培地5mL(LB培地1Lあたりの組成:トリプトン10g、酵母エキス5g、NaCl10g)で37℃、16時間、振盪培養し、前培養液を得た。次に前培養液5mLを、アンピシリン100ppm、クロラムフェニコール50ppm、添加NS液体培地1Lに植菌し、37℃、攪拌速度725rpm、通気量1L/minで通気攪拌培養を行った。なお、NS液体培地の組成は、グリセリン40g/L、硫酸アンモニウム10g/L、リン酸二水素カリウム2g/L、リン酸水素二カリウム6g/L、硫酸マグネシウム7水和物1g/L、酵母エキス40g/Lである。また、培養には、バイオット社製ジャーファーメンター:BMJ−02NP2を使用し、培養中はアンモニア水を用いて培養液中のpHを7にコントロールした。培養8時間後に1M IPTG溶液を1mL、8mMアデノシルコバラミン溶液を1mL添加し、培養途中にグリセリンが枯渇しないように適時グリセリンを添加しながら100時間培養を行った。得られた培養液を遠心分離にかけ、培養液上清を回収した。
【0089】
以下記載の高速液体クロマトグラフィーを用いた分析方法で培養液上清中の生成物の確認を行ったところ、生成物である3−ヒドロキシプロピオン酸のピークを7.9分の位置に確認することができ、培養液中の3−ヒドロキシプロピオン酸の濃度は2質量%であった。
高速液体クロマトグラフィーでの分析条件:
使用カラム: YMC−pACK FA(YMC社製)
流量:1mL/min
インジェクション量:10μL
溶離液:メタノール/アセトニトリル/H2O=40/5/55(V/V/V)
内部標準:2−Hydroxy−2−methyl−n−butyric acid
検出:UV400nm
【0090】
培養上清100μLに内部標準液200μLを加えた。ヒドロキシカルボン酸ラベル化試薬(YMC社製)の試薬A液200μL、試薬B液200μLを加え、よく混合した後、60℃、20分間処理した。ヒドロキシカルボン酸ラベル化試薬(YMC社製)の試薬C液200μLを添加し、よく混合した。60℃、15分間処理後、室温まで冷えたら0.45mmフィルターに通し、LC分析サンプルとして供した。
菌体を除去した2質量%の3−ヒドロキシプロピオン酸含有培養液からWO2002/090312号記載の方法で、原料組成物として12質量%の3HP水溶液を得た。
【0091】
(実施例1)
(3HPの第一の脱水工程)
特開2009−190915号公報の方法に従い、球状のZSM−5型ゼオライト(プロトン型)を合成した。内径10mmのステンレス製反応管に、ZSM−5型ゼオライトを充填し、その上にステンレス製の1.5mmのディクソンパッキンを蒸発層として積層した。反応管を電気炉にて300℃に加熱し、上記調製例1で得られた3HP水溶液を、毎時16.7gの速度で反応管の上部に供給した。同時に、毎時3Lの速度で窒素ガスを流した。
反応管の下部から抜き出した反応ガスを、冷却捕集し反応液を得た。得られた反応液を液体クロマトグラフィーで分析した。反応後1時間目から2時間目の間の反応成績は、3HPの転化率は100%、アクリル酸の収率は95モル%であった。反応後7時間目から8時間目の間の反応成績は、3HPの転化率は95%、アクリル酸の収率は91モル%であり、経時的な触媒活性低下が観察された。
反応後、ディクソンパッキン及び触媒を抜き出してみると、褐色の着色が見られた。
【0092】
(触媒の再生工程)
反応器から抜き出したディクソンパッキン及び触媒を、磁性皿に薄く広げ、焼成炉内に設置した。その後、焼成炉内に、0.5L/分の流量で空気を流通させ、1時間かけて500℃まで昇温し1時間保持して、触媒上の炭素状物質を酸化分解して、再生を行った。冷却後、取り出したディクソンパッキンは元の銀色に、触媒は元の白色に変化していた。
【0093】
(3HPの第二の脱水工程)
触媒再生工程で得られた触媒を、再度反応器に充填し、第一の脱水工程と同様の条件で、3HPの脱水反応を実施した。反応後1時間目から2時間目の間の反応成績は、3HPの転化率は100%、アクリル酸の収率は94モル%であった。反応後7時間目から8時間目の間の反応成績は、3HPの転化率は96%、アクリル酸の収率は92モル%であった。触媒を再生することによって、第一の脱水工程とほぼ同様の反応結果を第二の脱水工程でも得ることができた。
上記第一の脱水工程後の触媒(サンプル1−1)、触媒再生後の触媒(サンプル1−2)、第二の脱水工程後の触媒(サンプル1−3)を、示差熱−熱重量測定装置にて分析を行った。分析条件は、試料30mgを10℃/分で800℃まで昇温し、流通空気量は50mL/分とした。その結果、重量減少率は、サンプル1−1で5.0%、サンプル1−2で0.5%、サンプル1−3で5.1%であった。触媒再生工程によって、炭素状物質が減少し、触媒活性が回復したことが示唆される。
【0094】
(比較例1)
実施例1の3HPの第一の脱水工程を、16時間継続して行った。反応後15時間目から16時間目の間の反応成績は、3HPの転化率は82%、アクリル酸の収率は72モル%であった。
第一の脱水工程後に、触媒再生工程を経て、第二の脱水工程を行って合計16時間反応した実施例1に比べると、3HPの転化率、アクリル酸の収率とも大きく低下した。
上記第一の脱水工程後の触媒(比較サンプル1)を、実施例1と同様に示差熱−熱重量測定装置にて分析を行った。その結果、重量減少率は、比較サンプル1で13.2%であった。比較例1では、大量の炭素状物質が触媒上に付着していることが示唆された。
【0095】
(実施例2)
触媒を市販のγ−アルミナペレット(サンゴバン社製)に変えた以外は、実施例1と同様に反応、触媒再生を行った。
(3HPの第一の脱水工程)
反応後1時間目から2時間目の間の反応成績は、3HPの転化率は91%、アクリル酸の収率は86モル%であった。反応後7時間目から8時間目の間の反応成績は、3HPの転化率は79%、アクリル酸の収率は76モル%であり、経時的な触媒活性低下が観察された。
(触媒の再生工程)
第一の脱水工程終了後、反応器の温度を450℃に上げ、窒素を5L/時間の流量で1時間流通した。その後、1.5L/時間の流量で酸化剤含有ガス(酸素2容量%、残部は窒素)を流通させたところ、触媒層の温度の上昇が観察された。次に、酸化剤の含有量を多くしたガス(酸素5容量%、残部は窒素)を流通させ、最後に更に酸化剤の含有量を多くしたガス(酸素8容量%、残部は窒素)を、炭素状物質の酸化分解による発熱が収まるまでの約24時間流通させた。
(3HPの第二の脱水工程)
触媒の再生工程終了後、引き続き、第一の脱水工程と同様の条件で、3HPの脱水反応を実施した。反応後1時間目から2時間目の間の反応成績は、3HPの転化率は91%、アクリル酸の収率は86モル%であった。反応後7時間目から8時間目の間の反応成績は、3HPの転化率は78%、アクリル酸の収率は76モル%であった。触媒を再生することによって、第一の脱水工程とほぼ同様の反応結果を第二の脱水工程でも得ることができた。
【0096】
(実施例3)
調製例1で得られた12質量%の3HP水溶液の濃縮を行った。薄膜蒸発器を用いて水の除去を行った。得られた濃縮液を、水分計、液体クロマトグラフィー及びNMRで分析したところ、水が34質量%、3HPが61質量%、3HPのオリゴマーが5質量%含まれていた。
(3HPの第一の脱水工程)
上記濃縮液を原料組成物として用い、実施例1で使用したZSM−5を触媒として脱水反応を実施した。上記濃縮液を、毎時3gの速度で反応管の上部に供給した。同時に、毎時22Lの速度で窒素ガスを流した。反応後1時間目から2時間目の間の反応成績は、3HP及びオリゴマーの転化率は97%、アクリル酸の収率(対3HP及びオリゴマー基準)は93モル%であった。反応後7時間目から8時間目の間の反応成績は、3HP及びオリゴマーの転化率は91%、アクリル酸の収率は87モル%であり、経時的な触媒活性低下が観察された。
(触媒の再生工程)
第一の脱水工程終了後、反応器の温度を450℃に上げ、窒素を5L/時間の流量で1時間流通した。その後、1.5L/時間の流量で酸化剤含有ガス(酸素2容量%、残部は窒素)を流通させたところ、触媒層の温度の上昇が観察された。次に、酸化剤の含有量を多くしたガス(酸素5容量%、残部は窒素)を流通させ、最後に更に酸化剤の含有量を多くしたガス(酸素8容量%、残部は窒素)を、炭素状物質の酸化分解による発熱が収まるまでの約30時間流通させた。
(3HPの第二の脱水工程)
第一の脱水工程と同様の条件で、3HPの脱水反応を実施した。反応後1時間目から2時間目の間の反応成績は、3HP及びオリゴマーの転化率は96%、アクリル酸の収率は92モル%であった。反応後7時間目から8時間目の間の反応成績は、3HP及びオリゴマーの転化率は90%、アクリル酸の収率は87モル%であった。触媒を再生することによって、第一の脱水工程とほぼ同様の反応結果を第二の脱水工程でも得ることができた。
【0097】
上記実施例3における第一の脱水工程後の触媒(サンプル3−1)、触媒再生後の触媒(サンプル3−2)を、実施例1と同様に示差熱−熱重量測定装置にて分析を行った。その結果、重量減少率は、サンプル3−1で6.1%、サンプル3−2で0.9%であった。
【0098】
(実施例4)
調製例1で得られた12質量%の3HP水溶液の濃縮を行った。ロータリーエバポレーターを用いて水の除去を行った。得られた濃縮液を、水分計、液体クロマトグラフィー及びNMRで分析したところ、水が34質量%、3HPが30質量%、3HPのオリゴマーが36質量%含まれていた。
(3HPの第一の脱水工程)
上記濃縮液を原料組成物として用い、実施例1で使用したZSM−5を触媒として脱水反応を実施した。上記濃縮液を、毎時3gの速度で反応管の上部に供給した。同時に、毎時22Lの速度で窒素ガスを流した。反応後1時間目から2時間目の間の反応成績は、3HP及びオリゴマーの転化率は89%、アクリル酸の収率(3HP及びオリゴマー基準)は83モル%であった。反応後7時間目から8時間目の間の反応成績は、3HP及びオリゴマーの転化率は78%、アクリル酸の収率は73モル%であり、経時的な触媒活性低下が観察された。
(触媒の再生工程)
第一の脱水工程終了後、反応器の温度を450℃に上げ、窒素を5L/時間の流量で1時間流通した。その後、1.5L/時間の流量で酸化剤含有ガス(酸素2容量%、残部は窒素)を流通させたところ、触媒層の温度の上昇が観察された。次に、酸化剤の含有量を多くしたガス(酸素5容量%、残部は窒素)を流通させ、最後に更に酸化剤の含有量を多くしたガス(酸素8容量%、残部は窒素)を、炭素状物質の酸化分解による発熱が収まるまでの約38時間流通させた。
(3HPの第二の脱水工程)
第一の脱水工程と同様の条件で、3HPの脱水反応を実施した。反応後1時間目から2時間目の間の反応成績は、3HPの転化率は82%、アクリル酸の収率は76モル%であった。反応後7時間目から8時間目の間の反応成績は、3HP及びオリゴマーの転化率は72%、アクリル酸の収率は66モル%であった。
【0099】
上記触媒再生後の触媒(サンプル4)を、実施例1と同様に示差熱−熱重量測定装置にて分析を行った。その結果、重量減少率は、サンプル4で2.7%であった。
【0100】
(実施例5)
(触媒の調製)
硝酸カリウム(2.5g)とリン酸第2アンモニウム(1.6g)を100gの水に溶解した溶液に、酸化ケイ素(30g)を加え、湯浴上で均一に加熱混合した後、加熱乾固した。次いで、空気中120℃で20時間仮焼してから10〜24メッシュに破砕し、更に空気中500℃で2時間焼成することにより、酸素を除く組成がK1Si20P0.5からなる触媒を得た。
(3HPの第一の脱水工程)
内径10mmのステンレス管に、蒸発層としてステンレス製の1.5mmディクソンパッキンを充填し、電気炉内に設置し蒸発器とした。また内径10mmのステンレス管に、上記の触媒を充填し、電気炉内に設置し反応器とした。蒸発器の出口と反応器の入口をステンレス管で連結し、周囲を電気ヒーターで加熱できるようにした。
また、蒸発器内の温度を275℃とし、反応器内の温度を300℃とし、実施例1と同じ原料組成物を毎時33.5gの速度で蒸発器の上部に供給した。同時に、毎時6Lの速度で窒素ガスを流した。蒸発器の出口ガスはそのまま反応器へ供給し、24時間継続して反応を実施した。1〜2時間目及び23〜24時間目の、反応器の出口ガスを冷却捕集し、得られた液を分析した。結果を表1に示す。
(触媒の再生工程)
第一の脱水工程終了後、反応器の温度を450℃に上げ、窒素を5L/時間の流量で1時間流通した。その後、1.5L/時間の流量で酸化剤含有ガス(酸素2容量%、残部は窒素)を流通させたところ、触媒層の温度の上昇が観察された。次に、酸化剤の含有量を多くしたガス(酸素5容量%、残部は窒素)を流通させ、最後に空気を炭素状物質の酸化分解による発熱が収まるまでの約30時間流通させた。
(3HPの第二の脱水工程)
第一の脱水工程と同様の条件で、3HPの脱水反応を実施した。その後、触媒再生−3HP脱水反応を繰り返し実施した。これらの結果を表1に示す。
脱水反応を4回繰り返しても、触媒活性に変化はなかった。
【0101】
【表1】

【0102】
(実施例6)吸水性樹脂の製造例
実施例1で得られた精製アクリル酸に重合禁止剤を60質量ppm添加した。別途、鉄を0.2質量ppm含有する苛性ソーダから得られたNaOH水溶液に対して、上記の重合禁止剤添加アクリル酸を冷却下(液温35℃)で添加することにより、75モル%中和を行った。得られた、中和率75モル%、濃度35質量%のアクリル酸ナトリウム水溶液に、内部架橋剤としてポリエチレングリコールジアクリレート0.05モル%(アクリル酸ナトリウム水溶液に対する値)を溶解させることにより、単量体成分を得た。この単量体成分350gを容積1Lの円筒容器に入れ、2L/minの割合で窒素を吹き込んで、20分間脱気した。次いで、過硫酸ナトリウム0.12g/モル(単量体成分に対する値)及びL−アスコルビン酸0.005g/モル(単量体成分に対する値)の水溶液をスターラー攪拌下で添加して、重合を開始させた。重合開始後に攪拌を停止し、静置水溶液重合を行った。単量体成分の温度が約15分(重合ピーク時間)後にピーク重合温度108℃を示した後、30分間重合を進行させた。その後、重合物を円筒容器から取り出し、含水ゲル状架橋重合体を得た。
得られた含水ゲル状架橋重合体は、45℃でミートチョッパー(孔径:8mm)により細分化した後、170℃の熱風乾燥機で、20分間加熱乾燥させた。更に、乾燥重合体(固形分:約95%)をロールミルで粉砕し、JIS標準篩で粒径600〜300μmに分級することにより、ポリアクリル酸系吸水性樹脂(中和率:75%)を得た。
本発明の製造方法により得られたアクリル酸の重合性は、プロピレンを原料とするアクリル酸の製造方法により得られたアクリル酸の重合性と同等であり、得られた吸水性樹脂は、臭気がなく、物性も同等であった。
【0103】
このように、3−ヒドロキシカルボン酸又はそのエステルの脱水用触媒上の炭素状物質に、酸化剤を接触させて炭素状物質を除去する、本発明の脱水用触媒の再生方法を用いることによって、脱水用触媒の活性を効率的に回復させることができ、また、当該再生方法を含む(メタ)アクリル酸又はそのエステルの製造方法により、触媒上の炭素状物質による反応管の閉塞を抑制し、3−ヒドロキシカルボン酸又はそのエステルを効率良く(メタ)アクリル酸又はそのエステルに転化することができ、(メタ)アクリル酸又はそのエステルを長期間にわたり安定して製造することができるという作用機序は、すべて同様であるものと考えられる。
したがって、上記実施例の結果から、本発明の技術的範囲全般において、また、本明細書において開示した種々の形態において本発明が適用でき、有利な作用効果を発揮することができるといえる。
【技術分野】
【0001】
本発明は、3−ヒドロキシカルボン酸又はそのエステルの脱水用触媒の再生方法、及び、(メタ)アクリル酸又はそのエステルを製造する方法に関する。
【背景技術】
【0002】
(メタ)アクリル酸は親水性樹脂や吸水性樹脂等の原料として工業的に広く利用されており、通常、(メタ)アクリル酸の製法としては、固定床多管式反応器を用い酸化物触媒の存在下、プロピレンやイソブチレンを接触気相酸化によりアクロレインやメタクロレインとし、得られたアクロレインやメタクロレインの接触気相酸化により(メタ)アクリル酸を製造する二段酸化方法が一般的である。プロピレンやイソブチレンは化石資源由来の原料であるため、地球温暖化ガスの排出削減等、環境負荷の低減の観点から再生可能資源から製造することが望まれている。
また(メタ)アクリル酸エステルは、(メタ)アクリル酸のエステル化等により製造されており、粘着剤、塗料等各種樹脂の原料として広く用いられている。
【0003】
再生可能な資源であるバイオマス等を利用して、(メタ)アクリル酸を商業的規模で経済的に製造する試みが行われている。バイオマスからの(メタ)アクリル酸の生成方法としては、天然物であり容易に入手可能な乳酸や糖類から発酵により調製される3−ヒドロキシカルボン酸等を脱水することにより(メタ)アクリル酸を調製する方法が挙げられる。
【0004】
特許文献1は、発酵等により得られたβ−ヒドロキシカルボン酸、その塩又はそのエステルを含む水溶液又は溶液を準備し、その溶液を脱水触媒の存在又は非存在の下で加熱することにより脱水を施し、不飽和カルボン酸、その塩又はそのエステルを製造する方法を開示している。特許文献2は、α−又はβ−ヒドロキシカルボン酸を含む水溶液を不活性なセラミック等や酸性の固体触媒を保持したところへ導入して加熱することによりα,β−不飽和カルボン酸を調製する方法を開示している。更にα−又はβ−ヒドロキシカルボン酸から形成されるポリマー、オリゴマー、ラクチド、ラクトン等を含む水溶液を用いることができるとの記載はあるが、具体的に実施した例の開示はない。
【先行技術文献】
【特許文献】
【0005】
【特許文献1】特表2005−521718号公報
【特許文献2】国際公開第2005/095320号
【発明の概要】
【発明が解決しようとする課題】
【0006】
上記の通り、3−ヒドロキシカルボン酸又はそのエステルを脱水する反応は各特許文献に開示されているものの、これらの文献には、反応によって触媒にもたらされる障害や、その解決方法については、開示されていない。しかし3−ヒドロキシカルボン酸又はそのエステルの脱水用触媒を用いて、脱水反応を実施すると、触媒上に炭素状物質が付着し、触媒の活性低下を招いたり、選択率の低下を引き起こす、又は炭素状物質により反応管が閉塞するという問題があり、これらの問題が(メタ)アクリル酸又はそのエステルを効率良く得るための障害となる。
上記事情に鑑み、本発明は、3−ヒドロキシカルボン酸又はそのエステルの脱水用触媒上に付着した炭素状物質を除去する触媒の再生方法、及び、長期間にわたり安定的に(メタ)アクリル酸又はそのエステルを製造する方法の提供を目的とする。
【課題を解決するための手段】
【0007】
本発明者らは、種々検討した結果、3−ヒドロキシカルボン酸又はそのエステルの脱水用触媒に付着した炭素状物質に、酸化剤を接触させることにより炭素状物質を除去し、3−ヒドロキシカルボン酸又はそのエステル脱水用触媒の活性を回復し、長期間に渡って安定的に(メタ)アクリル酸又はそのエステルを製造することができることを見出して、本発明を完成した。
【0008】
すなわち、本発明は、3−ヒドロキシカルボン酸又はそのエステルの脱水用触媒上の炭素状物質を除去することによって触媒を再生する方法であって、該再生方法は、脱水用触媒に酸化剤を接触させて炭素状物質を除去することを特徴とする3−ヒドロキシカルボン酸又はそのエステルの脱水用触媒の再生方法である。
また、本発明は、前記酸化剤は、ガス状酸化剤であることを特徴とする上記3−ヒドロキシカルボン酸又はそのエステルの脱水用触媒の再生方法である。
また、本発明は、前記再生方法は、固定床反応器内に充填された3−ヒドロキシカルボン酸又はそのエステルの脱水用触媒を再生する方法であり、該固定床反応器内にガス状酸化剤を流通させて、炭素状物質を除去することを特徴とする上記3−ヒドロキシカルボン酸又はそのエステルの脱水用触媒の再生方法である。
【0009】
更に、本発明は、上記3−ヒドロキシカルボン酸又はそのエステルの脱水用触媒の再生方法を含むことを特徴とする(メタ)アクリル酸又はそのエステルの製造方法である。
更に、本発明は、第一の脱水工程、触媒再生工程及び第二の脱水工程を含み、該第一の脱水工程は、3−ヒドロキシカルボン酸又はそのエステルと脱水用触媒との気相接触反応により3−ヒドロキシカルボン酸又はそのエステルを脱水する工程であり、該触媒再生工程は、前記再生方法により脱水用触媒を再生する工程であり、該第二の脱水工程は、3−ヒドロキシカルボン酸又はそのエステルと触媒再生工程後の脱水用触媒との気相接触反応により3−ヒドロキシカルボン酸又はそのエステルを脱水する工程であることを特徴とする上記(メタ)アクリル酸又はそのエステルの製造方法である。
【発明の効果】
【0010】
本発明の脱水用触媒の再生方法によれば、3−ヒドロキシカルボン酸又はそのエステルの脱水用触媒の活性を効率的に回復させることができる。また、当該再生方法を含む(メタ)アクリル酸又はそのエステルの製造方法により、触媒上の炭素状物質による反応管の閉塞を抑制し、3−ヒドロキシカルボン酸又はそのエステルを効率良く(メタ)アクリル酸又はそのエステルに転化することができ、(メタ)アクリル酸又はそのエステルを長期間にわたり安定して製造することができる。更に、本製法で得られた(メタ)アクリル酸を使用して吸水性樹脂の製造をすることにより、高性能の吸水性樹脂を得ることができる。
【図面の簡単な説明】
【0011】
【図1】固定床反応器内において炭素状物質を触媒上から酸化分解除去するときの触媒の温度変化を説明するための図である。
【発明を実施するための形態】
【0012】
以下、本発明を詳細に説明する。
なお、以下において記載する本発明の個々の好ましい形態を2つ以上組み合わせたものもまた、本発明の好ましい形態である。
【0013】
なお、本明細書においては、特に断りのない限り、3−ヒドロキシカルボン酸又はそのエステルについては、3−ヒドロキシカルボン酸をこれらの代表として記載し、また、(メタ)アクリル酸又はそのエステルについては、(メタ)アクリル酸をこれらの代表として記載する。なお、3−ヒドロキシカルボン酸エステル及び(メタ)アクリル酸エステルは、それぞれ対応する酸を、後述の方法によりエステル化して得ることができる。
【0014】
本発明における3−ヒドロキシカルボン酸としては、3−ヒドロキシプロピオン酸(以下、3HPともいう)、3−ヒドロキシイソ酪酸等が挙げられ、好ましくは3−ヒドロキシプロピオン酸である。また、(メタ)アクリル酸としては、アクリル酸、メタクリル酸が挙げられ、好ましくはアクリル酸である。
上記3−ヒドロキシカルボン酸は、1種でも2種でも用いることができる。また、(メタ)アクリル酸は、使用した3−ヒドロキシカルボン酸の種類に応じて得られる。
【0015】
本発明は、3−ヒドロキシカルボン酸又はそのエステルの脱水用触媒(以下、単に「触媒」ともいう)上の炭素状物質に、酸化剤を接触させて、前記炭素状物質を除去することを特徴とする3−ヒドロキシカルボン酸又はそのエステルの脱水用触媒の再生方法である。
ここで本発明における「酸化剤」とは、炭素状物質を酸化剤の作用によって二酸化炭素、一酸化炭素、その他の炭素含有化合物に酸化分解するものをいう。
【0016】
本発明における触媒の再生方法は、触媒上に付着した炭素状物質に酸化剤を接触させて、この炭素状物質を触媒上から酸化分解除去する方法である。炭素状物質を酸化分解除去した後には、必要に応じて、触媒活性成分の補充処理が行われる。
【0017】
上記再生方法は、3−ヒドロキシカルボン酸又はそのエステルと、脱水用触媒との気相接触反応により3−ヒドロキシカルボン酸又はそのエステルを脱水する第一の3−ヒドロキシカルボン酸又はそのエステルの脱水工程と、この脱水工程と同様の第二の3−ヒドロキシカルボン酸又はそのエステルの脱水工程との間の、触媒再生工程に使用される方法である。
【0018】
本発明で再生対象となる触媒は、3−ヒドロキシカルボン酸又はそのエステルの脱水用触媒である。そして、この触媒上には、3−ヒドロキシカルボン酸又はそのエステルとの気相接触反応過程で生じた炭素状物質が表面に堆積している。この炭素状物質は、触媒の表面や細孔内等、触媒のあらゆる部分に付着しうる。
【0019】
上記触媒としては、ゼオライト等の結晶性メタロシリケート;結晶性メタロシリケートに、アルカリ金属、アルカリ土類金属又は遷移金属等を、イオン交換等の方法によって担持させたもの;カオリナイト、ベントナイト、モンモリロナイト等の天然又は合成粘土化合物;硫酸、ヘテロポリ酸、リン酸、リン酸塩(リン酸のアルカリ金属塩、アルカリ土類金属塩、リン酸マンガン、リン酸ジルコニウム等)、アルカリ金属又はアルカリ土類金属を、アルミナやシリカ等の担体に担持させた触媒;活性アルミナ(Al2O3)、SiO2、TiO2、ZrO2、SnO2、V2O5、SiO2−Al2O3、SiO2−TiO2、SiO2−ZrO2、TiO2−WO3、TiO2−ZrO2等の無機酸化物又は無機複合酸化物;MgSO4、Al2(SO4)3、K2SO4、AlPO4、Zr(SO4)2等の金属の硫酸塩、リン酸塩等の固体酸性物質;酸化カルシウム、酸化マグネシウム、ハイドロタルサイト等の固体塩基性物質;等が挙げられる。
好ましくは、Al2O3、SiO2、SiO2−Al2O3、TiO2、ゼオライト;ゼオライトに、アルカリ金属又はアルカリ土類金属を担持させたもの;リン酸、リン酸塩、アルカリ金属又はアルカリ土類金属をシリカ等の担体に担持させたものである。
【0020】
上記触媒は、粉体であっても成形体であっても良い。その成形体形状としては、限定されるものではなく、球状、シリンダー型、リング型、ハニカム型等が挙げられる。触媒の形態は使用する反応形式に応じて選択することができる。固定床反応器の場合は、圧力損失を小さくするために成型体が好ましく、流動床反応器の場合は、触媒を流動させるために粉体の方が好ましい。
上記触媒の物性としては、触媒活性等の点から、BET法による比表面積は、0.01〜500m2/gが好ましく、0.1〜400m2/gがより好ましい。触媒活性、触媒寿命等の点から、ハメットの酸度関数H0は、+4〜−10が好ましく、+2〜−8がより好ましい。また、触媒活性や反応器の圧力損失の点から、触媒の大きさは、長径が0.1mm〜50mmが好ましく、0.5mm〜40mmがより好ましい。
【0021】
上記酸化剤としては、過酸化水素水、有機過酸化物、硝酸、亜硝酸等が溶解した液状の酸化剤を使用しても良いし、ガス状の酸化剤を使用しても良い。好ましくは、ガス状の酸化剤である。
ガス状の酸化剤は、炭素状物質の酸化分解のために該炭素状物質に酸素元素を供給することが可能な気体分子であり、例えば、酸素(空気中の酸素も酸化剤に該当する)、オゾン、一酸化窒素、二酸化窒素、一酸化二窒素等を挙げることができる。これらの酸化剤のうち、少なくとも一種以上のガス状酸化剤が含まれていれば良く、例えば、空気と酸素との混合ガス、一酸化窒素と酸素との混合ガスを使用しても良く、窒素、二酸化炭素、アルゴン、ヘリウム、及び水蒸気等の不活性ガスから任意に選択した一種以上のガスと酸化剤との混合ガスを使用しても良い。
なお、オゾンや過酸化水素水を用いると、触媒が酸点を有する場合、無効分解によって、炭素状物質の除去効率が悪くなることがある。従って、酸点の多い触媒を用いる場合は、酸化剤として酸素を用いることがより好ましい。
具体的には、触媒の酸点が、アンモニア昇温脱離法による100〜600℃のアンモニア脱離量として0.001mmol/g以上の時は、酸素による触媒再生がより好ましい。
【0022】
触媒再生において、第一の3−ヒドロキシカルボン酸又はそのエステルの脱水工程で使用された反応器内から取り出した触媒を、酸化剤ガスに曝しても良いが、本発明では、簡便に触媒を再生するため、触媒が充填された固定床反応器内にガス状酸化剤を流通させて、前記炭素状物質を除去することが好ましい。
【0023】
触媒再生において触媒の加熱温度は、高温であるほど触媒再生時間を短縮できるが、あまり高すぎると触媒の活性や選択率が低下する恐れがある。通常好ましい範囲は300〜800℃であり、より好ましくは350〜700℃である。800℃を超えると、例えばシンタリングによる触媒表面積の低下や相転移による触媒の結晶構造変化等の触媒における物理的構造及び化学的性質が変わることになって、触媒活性や選択率が低下する恐れがある。温度は、触媒種によっても上限は異なるが、触媒調製の際に触媒を焼成する場合、焼成温度を超えない温度で加熱することが好ましい。
なお、後述の本発明の製造方法により得られる生成物である(メタ)アクリル酸は、脱水触媒上で非常にコーキングを起こしやすく、炭素状物質として触媒上に蓄積しやすいことが明らかとなった。また、原料組成物中に含まれる3−ヒドロキシカルボン酸オリゴマーは、沸点が高く蒸発しにくいため、触媒上に吸着すると更に重質化して容易に炭素状物質へと変化する。よって、これらの炭素状物質は、酸化剤を用いて酸化分解するにあたり、通常よりも高い温度が必要とされる。例えば酸化剤として酸素を用いて触媒再生を行う場合は、触媒の温度が350℃〜800℃の範囲が好ましく、より好ましくは400℃〜700℃である。
【0024】
上記加熱温度を制御するには、該触媒を加熱するための加熱器の設定温度、酸化剤濃度、及びガス流量等を調整すると良い。この場合、加熱器の設定温度及び/又は酸化剤の濃度が高いほど、触媒を加熱する温度が高くなる。そして、触媒加熱温度を連続的に測定しつつ、加熱器の設定温度及び/又は酸化剤の濃度を調整して触媒加熱温度を制御することも可能である。また、特開平5−192590号公報に開示されている触媒加熱温度の制御方法も挙げられる。
酸化剤濃度としては、温度制御や生産コスト等の点から、好ましくは1〜21体積%である。
処理時間としては、(メタ)アクリル酸の生産性等の点から、好ましくは1〜100時間、より好ましくは2〜50時間である。
【0025】
ここで、固定床反応器内の温度変化を、図1をもって説明する。図1は、炭素状物質を触媒上から酸化分解除去するときの触媒の温度変化を説明するための概略温度グラフであり、縦軸が触媒層温度、横軸が触媒再生時間及び固定床反応器内における触媒層長、矢印が固定床反応器内におけるガス流通方向を表している。図示の通り、固定床反応器内における最高の触媒加熱温度(ピーク温度)は、時間の経過と共に固定床反応器の入口から出口(ガス流通方向)に向かって移動する。すなわち、炭素状物質の酸化分解除去当初では、固定床反応器入口付近における炭素状物質の酸化分解のために酸化剤の大部分が消費され、この消費位置は、再生時間の経過と共に出口側に移動するのである。なお、図1に示すように、固定床反応器内のピーク温度位置を当該反応器のガス流通方向に向けて連続的に移動させ、触媒を当該触媒調製における焼成温度以下にて加熱することは、本実施形態の好ましい態様である。
【0026】
炭素状物質を酸化分解除去する本実施形態に係る方法では、触媒の種類によっては、炭素状物質の酸化分解過程で触媒活性成分が飛散する場合がある。このような場合には、必要とする触媒活性に応じて、触媒活性成分の補充処理が行われる。当該補充処理を行うことが好ましい触媒としては、リン化合物担持触媒が例示される。シリカやアルミナ等の担体にリン化合物が担持されたリン担持触媒は、3−ヒドロキシカルボン酸の脱水反応過程や炭素状物質の酸化分解除去過程で飛散した触媒活性成分(リン)が補充されると、活性が回復する。
【0027】
触媒活性成分の補充処理を行うためには、触媒調製の際における活性成分を担持させる方法を再実行すると良い。この方法としては、例えば、(1)含浸等の方法により、反応管から抜き出した触媒に、所定量の触媒活性成分を補充する方法、(2)特開平2−290255号公報に開示されている方法等により、反応管に充填されている状態の触媒に揮発性化合物(リン酸エステル等のリン元素を含有する化合物)を接触させる処理により、触媒活性成分を補充する方法、等が挙げられる。
上記再生方法により、触媒の再生が行われる。
【0028】
次に、本発明の(メタ)アクリル酸又はそのエステルの製造方法は、上記3−ヒドロキシカルボン酸又はそのエステルの脱水用触媒の再生方法を含むものである。
また、本発明の(メタ)アクリル酸又はそのエステルの製造方法は、第一の脱水工程、触媒再生工程及び第二の脱水工程を含み、該第一の脱水工程は、3−ヒドロキシカルボン酸又はそのエステルと脱水用触媒との気相接触反応により3−ヒドロキシカルボン酸又はそのエステルを脱水する工程であり、該触媒再生工程は、前記再生方法により脱水用触媒を再生する工程であり、該第二の脱水工程は、3−ヒドロキシカルボン酸又はそのエステルと触媒再生工程後の脱水用触媒との気相接触反応により3−ヒドロキシカルボン酸又はそのエステルを脱水する工程であることが好ましい。
【0029】
そこで、本実施形態に係る再生方法を使用する前の触媒を使用する第一の3−ヒドロキシカルボン酸又はそのエステルの脱水工程と、その再生後の触媒を使用する第二の3−ヒドロキシカルボン酸又はそのエステルの脱水工程について説明する。
第一の及び第二の3−ヒドロキシカルボン酸又はそのエステルの脱水工程では、3−ヒドロキシカルボン酸又はそのエステルの誘導体である(メタ)アクリル酸又はそのエステルを、気相接触反応により得るために、固定床反応器に反応ガスを流通させることが好ましい。
なお、本実施形態では、第一の3−ヒドロキシカルボン酸又はそのエステルの脱水工程及び第二の3−ヒドロキシカルボン酸又はそのエステルの脱水工程の、条件及び手順は同じである。
【0030】
上記第一の脱水工程は、3−ヒドロキシカルボン酸又はそのエステルと脱水用触媒との気相接触反応により、3−ヒドロキシカルボン酸又はそのエステルを脱水する工程である。
上記脱水工程では、3−ヒドロキシカルボン酸又はそのエステルを含む原料組成物を原料として用いる。そこで、3−ヒドロキシカルボン酸又はそのエステルを含む原料組成物の物性や調製方法等について、まず説明する。
【0031】
上記原料組成物に含まれる3−ヒドロキシカルボン酸又はそのエステルの濃度は、好ましくは5〜95質量%、より好ましくは10〜90質量%、更に好ましくは20〜90質量%である。
また、3−ヒドロキシカルボン酸又はそのエステルのダイマーやトリマー等のオリゴマーが含まれていても良く、そのオリゴマーは原料として使用することができる。原料組成物に含まれる上記オリゴマーは、3−ヒドロキシカルボン酸とそのエステルの合計100質量%に対して、好ましくは1〜400質量%、より好ましくは1〜300質量%含まれていてもよい。上記オリゴマー量を1質量%以上とすることにより、3−ヒドロキシカルボン酸又はそのエステルの、オリゴマー化抑制やオリゴマー量低減のための設備やエネルギーを最小限に抑えることができ、また、400質量%以下とすることにより、脱水触媒上での炭素状物質の生成及びコーキングによる閉塞を抑制し、(メタ)アクリル酸の収率を向上させることができる。
【0032】
原料組成物には、溶媒が含まれていても良い。当該溶媒としては、3−ヒドロキシカルボン酸又はそのエステルを溶解できるものであれば、特に限定されないが、例えば、水、アルコール、炭化水素、エーテル、ケトン、エステル、アミン、アミド等が挙げられる。これらは、1種でも2種以上でも用いることができる。溶媒の沸点は、気化が容易になるため3−ヒドロキシカルボン酸又はそのエステルよりも低い方が好ましい。好適には水である。
【0033】
本発明において、原料組成物中に溶媒を含有させる場合、原料組成物100質量%における溶媒の濃度は、好ましくは5〜95質量%であり、より好ましくは10〜90質量%、更に好ましくは10〜80質量%である。溶媒の濃度が5質量%以上であれば、粘度の低下により原料組成物の取り扱いが容易になり、また、3−ヒドロキシカルボン酸又はそのエステルの蒸発が促進される効果が期待できる。一方、95質量%以下とすることにより、蒸発にかかる熱量を抑制し、用役費の低減に寄与できる。
【0034】
上記原料組成物には、3−ヒドロキシカルボン酸又はそのエステル以外の成分、例えば、3−ヒドロキシカルボン酸を発酵により合成する際の副生物等が含まれていても良い。当該副生物としては、具体的には、発酵において3−ヒドロキシカルボン酸と共に副生される可能性のある、プロピオン酸、ギ酸、酢酸、酪酸、乳酸、乳酸エステル、エタノール、アミノ酸類等が例示できる。しかし、乳酸、乳酸エステルやエタノールは、その存在量が多いと、脱水反応において脱水触媒上での炭素状物質の生成を促進するため、その量は、3−ヒドロキシカルボン酸又はそのエステルに対して、それぞれ2質量%以下であることが好ましい。
【0035】
本発明で用いられる3−ヒドロキシカルボン酸は種々の源から得ることができるが、地球温暖化及び環境保護の観点から、炭素源としてリサイクル可能な生物由来資源を用いることが好ましい。また、植物由来の糖類又はセルロース等を分解して得られる糖類や、油脂から得られるグリセリンから、更に発酵により調製された3−ヒドロキシカルボン酸を用いることができる。
本発明においては、原料組成物中に含まれる3−ヒドロキシカルボン酸の少なくとも一部又は全部が、発酵により得られる3−ヒドロキシカルボン酸であることが好ましい。
【0036】
3−ヒドロキシカルボン酸は、公知の方法で入手可能であり、例えば国際公開第2008/027742号に記載されている、Streptomyces griseus ATCC21897由来beta−alanine aminotransferase遺伝子導入大腸菌を用いた、グルコースを炭素源とした発酵により得ることができる。また、国際公開第2001/016346号に記載されている、Klebsiella pneumoniae由来グリセリン脱水酵素及び大腸菌由来アルデヒド酸化酵素導入大腸菌を用いた、グリセリンを炭素源とした発酵によっても得ることができる。
【0037】
3−ヒドロキシカルボン酸の入手方法の例として上記公知文献を記載したが、本発明の方法を用いる限り、発酵に用いる細菌又は組換え細菌は特に限定されず、3−ヒドロキシカルボン酸生成能を有する生物を用いた発酵により入手した3−ヒドロキシカルボン酸であれば、本発明の製法で利用可能である。また、発酵以外にも原料とする糖類と生物とを接触させることにより生成した3−ヒドロキシカルボン酸でも、本発明の製法で(メタ)アクリル酸へ変換することができる。
糖類と生物を接触させるとは、原料として利用する糖類の存在下で微生物又はその処理物を用いて反応を行うことをも包含する。該処理物としては、アセトン、トルエン等で処理した菌体、菌死体、凍結乾燥菌体、菌体破砕物、菌体を破砕した無細胞抽出物、これらから酵素を抽出した粗酵素液、精製酵素等が挙げられる。また、常法により担体に固定化した菌体、該処理物、酵素等を用いて反応を行うことにより入手した3−ヒドロキシカルボン酸も用いることができる。
【0038】
本発明では、生物由来資源を用いて発酵により3−ヒドロキシカルボン酸を得る具体的実施形態に係る方法において、固体(特に微細な植物の部分又は細胞及び/又は細胞断片)、発酵の後に得られる3−ヒドロキシカルボン酸及び微生物等を含む水性組成物から、微生物や生物的材料等を分離するのが良い。前記分離は、固体を液状組成物から分離するための、当業者に公知の全ての方法により実施することができるが、好ましくは沈殿法、遠心分離法又は濾過法により、最も好ましくは濾過法により、分離するのがよい。
【0039】
3−ヒドロキシカルボン酸及び微生物等を含む水性組成物から微生物等を分離する処理においては、そこに含まれる微生物に処理を施すことなく行っても良いが、そこに含まれる微生物を殺菌する処理工程を含んでも良い。前記水性組成物から微生物等を殺菌する処理は、微生物を分離する前、その間又は後に行うことができる。上記殺菌処理としては、加熱処理(加熱による微生物の殺菌)、高エネルギー照射処理(例えば紫外線照射による微生物の殺菌)等が挙げられ、好ましくは加熱処理である。
上記加熱処理としては、3−ヒドロキシカルボン酸及び微生物等を含む水性組成物を、好ましくは少なくとも60秒間、より好ましくは少なくとも10分間、更に好ましくは少なくとも30分間の処理時間で、好ましくは少なくとも100℃、より好ましくは少なくとも110℃、更に好ましくは少なくとも120℃の温度で、加熱することによって実施するのが好ましい。当該加熱処理は、当業者に公知の装置(例えばオートクレーブ等)において実施するのが好ましい。
【0040】
本発明に用いる3−ヒドロキシカルボン酸は、発酵液より回収し、純度を高めたものを脱水工程で用いることが好ましい。発酵液からの3−ヒドロキシカルボン酸の回収方法としては、公知の方法が利用可能である。例えば、蒸留、蒸発、抽出、膜分離、晶析、イオン交換、電気透析等が挙げられ、これらを組み合わせても良い。更に具体的には、発酵により得られた粗製3−ヒドロキシカルボン酸を、カルシウム塩を用いて沈殿させて3−ヒドロキシカルボン酸のカルシウム塩として回収し、その後、硫酸等の酸と反応させて3−ヒドロキシカルボン酸を精製する方法や、発酵により得たアンモニウム型の3−ヒドロキシカルボン酸を電気透析又は陽イオン交換法によって3−ヒドロキシカルボン酸に化学変換させて精製する方法等が利用できる。
【0041】
また、発酵により得られたアンモニウム型の3−ヒドロキシカルボン酸に、水に不混和性のアミン溶媒を添加し加熱することで、アンモニアを除去して3−ヒドロキシカルボン酸のアミン溶液を得ることができる。そこに水を加えて加熱することで、3−ヒドロキシカルボン酸の水溶液を得ることができる。
【0042】
また、上記3−ヒドロキシカルボン酸をアルコールにてエステル化を行い、3−ヒドロキシカルボン酸エステルを合成することができる。
アルコールとしては、特に限定されないが、炭素数が1〜20のアルコールが好ましく、炭素数が1〜10のアルコールがより好ましく、炭素数1〜5のアルコールが更に好ましい。
エステル化反応は、3−ヒドロキシカルボン酸とアルコールを、エステル化触媒の存在下又は非存在下、加熱することによって行うことができる。3−ヒドロキシカルボン酸はカルボン酸のため、触媒の非存在下でもエステル化反応は進行する。しかし、生産効率の点から、エステル化触媒を用いることが好ましい。
エステル化触媒としては、公知のものが使用できるが、塩酸、硫酸、硝酸、リン酸等の鉱酸類;ゼオライトやイオン交換樹脂等の固体酸類;ヘテロポリ酸等の無機酸;p−トルエンスルホン酸等のスルホン酸類;ジブチルスズジラウレート、酸化スズ、ジブチルスズオキサイド、酢酸亜鉛、テトラアルコキシチタン等の金属化合物類等が挙げられる。
反応温度は、50℃〜300℃が好ましく、80℃〜250℃がより好ましい。エステル化反応は平衡反応のため、収率向上のために、反応蒸留や、生成物を抽出しながらの反応も効果的である。
【0043】
上述のようにして、3−ヒドロキシカルボン酸又はそのエステルを調製することができるが、発酵液中には微生物由来のタンパク質やアミノ酸といった含窒素化合物が含まれる。これらの含窒素化合物やその分解物が、3−ヒドロキシカルボン酸又はそのエステルと共に反応器に供給され、触媒と接触すると、アミノ基等の含窒素官能基が触媒上に吸着して、触媒活性を低下させることがある。また、含窒素化合物は、反応して重質化しやすく、生成した重質物が触媒上に付着して活性低下の要因となる。更に、触媒から、窒素を含んだ炭素状物質を除去する場合、分解しにくく再生温度が高くなったり、酸化剤と接触させた時に排ガス中に有害な窒素酸化物が含まれることとなり、除去装置が必要となる場合がある。
これらの悪影響を小さくするためには、原料中に含まれる窒素化合物中の窒素量が、3−ヒドロキシカルボン酸、そのエステル、及び、それらのオリゴマー類の合計100質量%に対して、0.2質量%以下であることが好ましい。より好ましくは0.1質量%以下である。窒素量の下限は、好ましくは0.1質量ppm以上である。より好ましくは0.5質量ppm以上であり、更に好ましくは1質量ppm以上である。0.1質量ppm未満とするためには、過大な精製工程が必要となり、コストアップの要因となる。
原料中の窒素化合物を低減させるには、発酵液から原料を調製する過程で、以下の方法により実施することができる。例えば、ナノ濾過膜や逆浸透膜等を用いてタンパク質やアミノ酸類と3−ヒドロキシカルボン酸を分離する方法;タンパク質と3−ヒドロキシカルボン酸を含む液を加熱し、タンパク質を変性、重質化し析出させて分離する方法;蒸留、蒸発、晶析、抽出、電気透析等を用いて、3−ヒドロキシカルボン酸と含窒素化合物を分離する方法等が挙げられる。
【0044】
また、3−ヒドロキシカルボン酸又はそのエステルは、複数の分子がエステル結合で連なった、3−ヒドロキシカルボン酸のダイマーやトリマーといったオリゴマー類を形成しやすい特性を持っている。オリゴマー類も(メタ)アクリル酸製造の原料として使用できるが、オリゴマーは沸点が高く、触媒上に吸着するとより重質化しやすいため、その濃度、分子量や加熱条件によっては、蒸発器や反応器での閉塞の原因となったり、脱水触媒上での炭素状物質の生成を促進することがある。
3−ヒドロキシカルボン酸又はそのエステルを含む原料組成物は、蒸発器内でその一部が反応し、(メタ)アクリル酸又はそのエステルや、3−ヒドロキシカルボン酸又はそのエステルのオリゴマーが生成する。また条件によっては、原料組成物中に含まれているオリゴマーや生成したオリゴマーの分解反応も起こる。脱水触媒上での炭素状物質の生成を抑制するためには、脱水触媒と接触する蒸発器出口ガスの組成を制御することが好ましい。
蒸発器出口ガスの組成としては、3−ヒドロキシカルボン酸又はそのエステルのオリゴマー類の濃度が、3−ヒドロキシカルボン酸とそのエステルの合計100質量%に対して、1〜100質量%であることが好ましい。より好ましくは1〜80質量%である。上記オリゴマー類の濃度を1質量%以上とすることにより、3−ヒドロキシカルボン酸又はそのエステルの、オリゴマー化抑制やオリゴマー濃度低減のための設備やエネルギーを最小限に抑えることができ、また、100質量%以下とすることにより、脱水触媒上での炭素状物質の生成を抑制し、触媒の活性低下を抑えることで、再生までの反応時間を長くすることが可能となり、(メタ)アクリル酸又はそのエステルの生産性を向上させることができる。
脱水触媒に供給される蒸発器出口ガス中の3−ヒドロキシカルボン酸又はそのエステルのオリゴマーは、3−ヒドロキシカルボン酸又はそのエステルの分子が2〜10個結合したものが、オリゴマー全体の70質量%以上であることが好ましく、80質量%以上であることがより好ましい。更に、3−ヒドロキシカルボン酸又はそのエステルの分子が2〜8個結合したものが、オリゴマー全体の70質量%以上であることが好ましく、80質量%以上であることがより好ましい。
【0045】
脱水触媒へ供給する原料中のオリゴマーを上記範囲に制御するためには、例えば蒸発器に供給する前の段階で、3−ヒドロキシカルボン酸を水や溶媒で希釈し低濃度で取り扱う、取扱いを低温で行う等、オリゴマーの生成を抑制する方法等が挙げられる。また、蒸発器内でオリゴマーが生成しないように、蒸発器内の滞留時間を短くする、不活性ガスを供給して蒸発しやすくする、蒸発温度を高くする等の方法が挙げられる。更に、原料組成物中にオリゴマーを含んでいても、蒸発器内でオリゴマーを分解し、低分子化した上で反応器に供給する方法が挙げられる。蒸発器内でオリゴマーを分解する方法としては、高温でオリゴマーを分解する方法、触媒と接触させてオリゴマーを分解する方法等が挙げられる。脱水触媒へ供給される蒸発器出口ガス中のオリゴマーは、蒸発器と反応器が分離した設備であれば、蒸発器出口のガスを捕集して分析する、又は反応器に触媒を充填せずに原料組成物を蒸発器に供給し、反応器出口ガスを捕集して分析することで確認することができる。蒸発器と反応器が一体となっている設備であれば、触媒を充填せずに原料組成物を供給し、反応器出口ガスを捕集して分析することで確認することができる。
【0046】
上述のようにして3−ヒドロキシカルボン酸又はそのエステルを含む原料組成物を調製することができる。
次いで、上記3−ヒドロキシカルボン酸又はそのエステルを含む原料組成物と脱水用触媒との気相接触反応により、3−ヒドロキシカルボン酸又はそのエステルを脱水する。
上記脱水用触媒としては、上述したものと同じものが挙げられる。
上記3−ヒドロキシカルボン酸又はそのエステルを含む原料組成物と脱水用触媒との気相接触反応は、3−ヒドロキシカルボン酸又はそのエステルを含む原料組成物を気化させ、気化した原料組成物を脱水用触媒と接触させることにより、行うことができる。
【0047】
3−ヒドロキシカルボン酸又はそのエステルを含む原料組成物の気化(蒸発)は、該組成物を加熱することにより実施することができる。蒸発器内の温度は、150℃〜500℃が好ましく、170℃〜480℃がより好ましく、200℃〜460℃が更に好ましく、220℃〜440℃が特に好ましく、220℃〜420℃が最も好ましい。当該温度が150℃〜500℃の範囲であれば、原料組成物が速やかに気化し、副反応による蒸発器や反応器の閉塞が生じない。また、500℃より高いと、加熱に必要なエネルギーが過大になるばかりか、該組成物がコーキングを起こし、炭素質の析出物が蒸発器や反応器内に付着して閉塞を起こす可能性がある。
【0048】
蒸発器内の圧力は、低いほど原料組成物の蒸発が起こりやすくなるため有利であるが、引き続く反応器の適正な圧力や設備等のコストも合わせて選択する必要がある。蒸発器内の圧力としては、好ましくは10kPa〜1000kPaであり、より好ましくは30kPa〜300kPaであり、更に好ましくは50kPa〜250kPaである。
【0049】
蒸発器は、液体で供給する原料組成物に効率的に熱を伝える構造が好ましい。例えば、水平管型や垂直管型の自然循環式蒸発器、強制循環式蒸発器等が挙げられる。
また、蒸発器内の原料組成物の流路に、ラシヒリング、ベルルサドル、球状成型物、金網の成型物(ディクソンパッキン、マクマホンパッキン等)、メラパック(スルザーケムテック社製)といった不規則充填物や規則充填物等の、単位充填容積当たりの表面積が大きな充填物を充填し、そこに原料組成物を供給することで、原料組成物(液体)が接する表面積を大きくして蒸発させる方法も挙げられる。こうすることにより、供給した原料組成物が、表面積の大きな充填物と接触することになり、伝熱面積が増え、蒸発効率が向上する。
上記充填物の材料としては、鉄やステンレス等の金属材料や、シリカ、セラミック等の無機材料等が使用できる。
充填物の表面積は、蒸発器に充填した際に、5cm2/cm3以上が好ましく、10cm2/cm3以上がより好ましく、20cm2/cm3以上が更に好ましく、30cm2/cm3以上が特に好ましい。また、上限は特に限定されないが、1000cm2/cm3以下が好ましく、800cm2/cm3以下がより好ましい。
【0050】
また、上昇液膜型、流下液膜型、撹拌液膜型等の薄膜式熱交換器を用いて、液体の表面積を大きくして、短時間で蒸発させる方法も挙げられる。更に、スプレーやアトマイザー等を用いて、該組成物を細かい液滴にして分散させて蒸発させる方法も例示できる。液滴の径は、1mm以下が好ましく、0.5mm以下がより好ましい。
その他、加熱した原料組成物を蒸発室に供給し、気化させるフラッシュ蒸発器を用いても良い。フラッシュ蒸発器においては、原料組成物を、常圧又は加圧下で加熱し、減圧又は常圧下の蒸発室に液を供給して、原料組成物を気化させることができる。
また、原料組成物を流動床式の蒸発器に供給して、気化させても良い。例えば、粉末状の不活性固体を不活性ガスで流動化させ、加熱された流動床式蒸発器に原料組成物を供給し、気化させても良い。
また、原料組成物の気化は、上記方法を多段階で行っても良い。例えば、フラッシュ蒸発器の後に充填物を充填した蒸発器を設置し、フラッシュ蒸発器で、原料組成物の一部を蒸発させた後、充填物を充填した蒸発器で、残りの原料組成物を蒸発させても良い。
また、原料組成物の気化には、上記方法を併用してもよい。例えば、スプレーで原料組成物を噴霧し、充填物を充填した蒸発器で原料組成物を気化させることもできる。
【0051】
蒸発させる際の好ましい形態は、加熱する際にガス(水や不活性気体等)を導入しながら蒸発させる形態が好ましい。原料組成物と共に、水や不活性気体等のガスを導入すると、原料組成物の蒸発が促進され、安定な反応を継続できるため、好ましい形態である。
ガスとしては、窒素、ヘリウム、アルゴン、二酸化炭素、水が蒸発した水蒸気等が挙げられ、これらは1種でも2種以上でも用いることができる。好適には、窒素、水蒸気である。この水蒸気には、原料組成物中に溶媒として含まれる水が気化した水蒸気も含まれる。
ガスの供給量としては、原料組成物中の3−ヒドロキシカルボン酸、そのエステル及びそれらのオリゴマーの総量に対して、水及び/又は不活性気体の総量として0.5モル倍〜100モル倍が好ましく、1モル倍〜50モル倍がより好ましい。
【0052】
熱の供給方法は、蒸気、熱媒、溶融塩、ヒーター等の熱源を用いて、熱交換により加熱する方法等が挙げられる。また、加熱した水蒸気や、窒素、アルゴン、ヘリウム、二酸化炭素等の不活性ガスを蒸発器に供給し、原料組成物に接触させて熱を与える方法等も挙げられる。また、これらを併用しても良い。
例えば、加熱した充填物の上に、原料組成物を噴霧して蒸発させる方法や、更に加熱した窒素や水蒸気を同時に供給する方法等が好適である。
【0053】
次に、脱水工程で使用する反応器としては、中に固体触媒を保持し、加熱することができればよく、例えば、固定床反応器、流動床反応器等が使用でき、固定床反応器が好ましい。
上記固定床反応器を用いる場合は、反応器内に触媒を充填して加熱しておき、そこに原料組成物の蒸気を供給すればよい。原料組成物の蒸気は、上昇流、下降流、水平流、いずれも好適に使用できる。また、熱交換の容易さから、固定床多管式連続反応器が好適に使用できる。
上記流動床反応器を用いる場合は、反応器の中に粉末状の触媒を入れ、原料組成物の蒸気や、別途供給する不活性ガス等で触媒を流動させながら、反応させることができる。触媒が流動しているため、重質分による閉塞が起こりにくい。また、触媒の一部を連続的に抜き出して、新しい触媒や再生した触媒を連続的に供給することもできる。
【0054】
脱水工程は上記蒸発の後にあれば良く、その間に別の工程があっても良い。例えば、蒸発器で気化させた原料組成物の蒸気を、所定の温度に加熱/冷却する温度調整工程を経て、反応器で脱水工程を実施しても良い。
また、蒸発器と反応器を一体化しても良い。例えば、反応管に、蒸発層として表面積の大きい充填物を充填し、当該蒸発層の下に触媒を充填することにより、蒸発層を蒸発工程、触媒層を脱水工程として連続した運転も、好ましい形態の1つである。
また、1つ乃至は複数の蒸発層と、触媒を充填した多管式の反応器を連結して運転することも、好ましい形態である。
【0055】
上記脱水工程において、触媒層の温度は、150℃〜500℃に保持することが好ましい。より好ましくは200℃〜450℃、更に好ましくは220℃〜430℃、特に好ましくは250℃〜400℃である。この温度範囲(150℃〜500℃)であると、反応速度が速く、副反応も生じにくく、(メタ)アクリル酸の収率が高くなる。
反応圧力は、特に限定されないが、原料組成物の蒸発方法、脱水反応の生産性、脱水反応後の捕集効率等を勘案して決定することができる。反応圧力としては、10kPa〜1000kPaが好ましく、より好ましくは30kPa〜300kPa、更に好ましくは50kPa〜250kPaである。
このようにして第一の脱水工程を行うことができる。また、第二の脱水工程は、上記触媒再生工程で再生した触媒を用いること以外は、第一の脱水工程と同じである。
【0056】
上述のように、本発明の(メタ)アクリル酸又はそのエステルの製造方法は、第一の脱水工程、触媒再生工程及び第二の脱水工程を含むものである。
上記再生方法により触媒の再生を行う間に、3−ヒドロキシカルボン酸又はそのエステルの脱水反応を中断させないためには、複数の固定床反応器を並列配置して、ある固定床反応器内の触媒再生の間には、他の固定床反応器を3−ヒドロキシカルボン酸又はそのエステルの脱水反応に使用すると良い。つまり、一の固定床反応器で前記触媒再生工程を行う間、他の固定床反応器で前記第一又は第二の3−ヒドロキシカルボン酸又はそのエステルの脱水工程を行うことが好ましい。
【0057】
複数の固定床反応器を使用する場合、3−ヒドロキシカルボン酸又はそのエステルの脱水反応により(メタ)アクリル酸又はそのエステルを製造する時間(反応時間)と触媒上に蓄積した炭素状物質を除去して触媒を再生する時間(再生時間)に応じた数の反応器が必要である。例えば、再生時間が反応時間よりも短ければ、3−ヒドロキシカルボン酸又はそのエステルの脱水反応に使用されている反応器1基に対して、触媒再生に使用される反応器1基が必要であり、再生時間が反応時間の2倍以下であれば、3−ヒドロキシカルボン酸又はそのエステルの脱水反応に使用されている反応器1基に対して、触媒再生に使用される反応器2基があれば足りる。そのため触媒再生に使用される反応器の数を減らせば経済的に3−ヒドロキシカルボン酸又はそのエステルの脱水反応を行えることになり、反応器数を減らすためには、再生時間をできるだけ短時間にすることが好ましいといえる。触媒再生温度や、反応器の設計温度の上限を超えない範囲で、なるべく高い温度で再生することにより、再生時間を短時間にすることができる。そのために、酸化剤の濃度や、酸化剤と共に流通させる気体成分の流量等を制御することができる。
【0058】
また、本発明において、上記工程により得られた反応生成物を冷却して、(メタ)アクリル酸又はそのエステルを含む組成物を得ることができるが、その方法としては、特に限定されるものではない。例えば、反応生成ガスを熱交換器に導入して反応生成ガスの露点以下の温度で凝縮して得る方法や、反応生成ガスを溶剤等の捕集剤に接触させて吸収する方法等により冷却して、(メタ)アクリル酸又はそのエステルを含む組成物を得ることができる。
【0059】
本発明においては、3−ヒドロキシカルボン酸又はそのエステルの脱水用触媒上の炭素状物質に、酸化剤を接触させて前記炭素状物質を除去する。しかし、前述したように3−ヒドロキシカルボン酸又はそのエステルは、ダイマー、トリマーといったオリゴマー化が進行しやすい。特に触媒層へ原料を気体で供給するための蒸発器においては、原料の3−ヒドロキシカルボン酸又はそのエステルが液体で加熱されるため、オリゴマー化が非常に進行しやすく、一部のオリゴマーが蒸発されず、蒸発器内に残存し、長時間の加熱によって炭素質へと変化する場合がある。その場合、ラインの閉塞や、炭素状物質の析出により、伝熱係数が低下する。
【0060】
このような場合、蒸発器中で生成し残存する炭素状物質にも酸化剤を接触させて、炭素状物質を除去することができる。除去方法は、触媒再生の方法と同様である。蒸発器中の炭素状物質の除去と、触媒再生は、別々に実施しても良いし、同時に実施しても良い。蒸発器は反応器と連結されているため、蒸発器中の炭素状物質の除去と触媒再生は同時に実施すると効率がよい。酸化剤を蒸発器の上流側から供給すると、まず蒸発器中の炭素状物質が除去され、引き続き反応器中の触媒が再生されることになる。また酸化剤を蒸発器と反応器のそれぞれの入口から別々に供給することもできる。
【0061】
蒸発器は、原料の速やかな蒸発や、炭素状物質除去の際の除熱を速やかに行うため、熱の移動がしやすい構造が好ましい。例えば多管式の熱交換器や、薄膜式の加熱器が挙げられる。
蒸発器中の炭素状物質を酸化剤と接触させて除去する場合、蒸発器は、例えば不活性な無機担体や充填物を充填した形式が好ましい。例えば多管式の熱交換器の管内に不活性担体を充填しておき、管内で原料を蒸発させることにより、炭素状物質が生成しても、担体表面に付着させることにより、閉塞までの時間や、伝熱係数の低下を遅らせることができる。
【0062】
上記工程を経て、3−ヒドロキシカルボン酸又はそのエステルから、(メタ)アクリル酸又は(メタ)アクリル酸エステルを得ることができる。
このようにして得られた反応生成物中には、主な反応生成物である水、(メタ)アクリル酸又は(メタ)アクリル酸エステルが含まれており、その他に副生物や原料組成物中の溶媒や不純物が含まれる場合がある。溶媒が水の場合は、(メタ)アクリル酸又はそのエステルの水溶液の状態で重合物製造の原料とすることができる。また、精製工程を加えることにより、高純度の(メタ)アクリル酸又はそのエステルにすることができる。精製工程は、蒸留、膜分離、晶析等公知の技術により実施でき、それらを組み合わせて実施しても良い。
上記のようにして得られた(メタ)アクリル酸を含む反応生成物は、捕集や精製工程の取扱いを、重合禁止剤の存在下で行うことが好ましい。重合禁止剤としては、メトキノン、酢酸マンガン、ニトロソフェノール、クペロン、N−オキシル化合物、ジブチルチオカルバミン酸銅、フェノチアジン、ハイドロキノン等が例示できる。また、必要に応じて酸素含有ガスを供給してもよい。
【0063】
このように、本発明で得られた(メタ)アクリル酸の組成物を精製することにより高純度の(メタ)アクリル酸を得ることができる。したがって、本発明の方法は、高純度の(メタ)アクリル酸の製造方法をも提供する。
【0064】
上記のガス状の反応生成物を冷却凝縮や溶剤捕集等により液化し、必要に応じて、この液化物に含まれる水や捕集溶剤を従来公知の方法(例えば、蒸留)により除去したものを、晶析方法によって高純度の(メタ)アクリル酸を得る方法を以下に示す。
ここで、粗(メタ)アクリル酸とは、冷却工程で得られた(メタ)アクリル酸を含む組成物を指し、特に(メタ)アクリル酸の水溶液が好適に用いられる。
晶析工程は、粗(メタ)アクリル酸からプロピオン酸を分離することができる従来公知の方法、例えば、特開平9−227445号公報や特表2002−519402号公報に記載された方法を用いて行うことができる。
【0065】
晶析工程は、粗(メタ)アクリル酸を晶析装置に供給して結晶化させることにより、精製(メタ)アクリル酸を得る工程である。なお、結晶化の方法としては、従来公知の結晶化方法を採用すればよく、特に限定されるものではない。結晶化は、例えば、連続式又は回分式の晶析装置を用いて、1段又は2段以上で実施することができる。得られた(メタ)アクリル酸の結晶は、必要に応じて、更に洗浄や発汗等の精製を行うことにより、更に純度の高い精製(メタ)アクリル酸を得ることができる。
【0066】
連続式の晶析装置としては、例えば、結晶化部、固液分離部及び結晶精製部が一体になった晶析装置(例えば、新日鐵化学社製のBMC(Backmixing Column Crystallizer)装置、月島機械社製の連続溶融精製システム)や、結晶化部(例えば、GMF GOUDA社製のCDC(Cooling Disk Crystallizer)装置)、固液分離部(例えば、遠心分離器、ベルトフィルター)及び結晶精製部(例えば、呉羽テクノエンジ社製のKCP(Kureha Crystal Purifier)精製装置)を組み合わせた晶析装置等を使用することができる。
回分式の晶析装置としては、例えば、Sulzer Chemtech社製の層結晶化装置(動的結晶化装置)、BEFS PROKEM社製の静的結晶化装置等を使用することができる。
【0067】
動的結晶化とは、例えば、結晶化、発汗、融解を行うための温度制御機構を備えた管状の結晶器と、発汗後の母液を回収するタンクと、結晶器に粗(メタ)アクリル酸を供給する循環ポンプとを備え、結晶器の下部に設けた貯蔵器から循環ポンプにより粗(メタ)アクリル酸を結晶器の管内上部に移送できる動的結晶化装置を使用して晶析を行う方法である。
また、静的結晶化とは、例えば、結晶化、発汗、融解を行うための温度制御機構を備えた管状の結晶器であり、下部に抜き出し弁を有する結晶器と、発汗後の母液を回収するタンクとを備えた静的結晶化装置を使用して晶析を行う方法である。
【0068】
具体的には、粗(メタ)アクリル酸を液相として結晶器に導入し、液相中の(メタ)アクリル酸を冷却面(管壁面)に凝固させる。冷却面に生成した固相の質量が、結晶器に導入した粗(メタ)アクリル酸に対して、好ましくは10〜90質量%、より好ましくは20〜80質量%になったら、直ちに、液相を結晶器から排出し、固相と液相とを分離する。液相の排出は、ポンプで汲み出す方式(動的結晶化)、結晶器から流出させる方式(静的結晶化)のいずれであってもよい。他方、固相は、結晶器から取り出した後、更に純度を向上させるために、洗浄や発汗等の精製を行ってもよい。
【0069】
動的結晶化や静的結晶化を多段で行う場合、向流の原理を採用すれば、有利に実施することができる。このとき、各段階で結晶化された(メタ)アクリル酸は、残留母液から分離され、より高い純度を有する(メタ)アクリル酸が生成する段階に供給される。他方、残留母液は、より低い純度を有する(メタ)アクリル酸が生成する段階に供給される。
なお、動的結晶化では、(メタ)アクリル酸の純度が低くなると、結晶化が困難になるが、静的結晶化では、動的結晶化に比べて、残留母液が冷却面に接触する時間が長く、また、温度の影響が伝わり易いので、(メタ)アクリル酸の純度が低下しても、結晶化が容易である。それゆえ、(メタ)アクリル酸の回収率を向上させるために、動的結晶化における最終的な残留母液を静的結晶化に付して、更に結晶化を行ってもよい。
【0070】
必要となる結晶化段数は、どの程度の純度が要求されるかに依存する。高純度の(メタ)アクリル酸を得るために必要な段数は、精製段階(動的結晶化)が、通常1〜6回、好ましくは2〜5回、より好ましくは2〜4回であり、ストリッピング段階(動的結晶化及び/又は静的結晶化)が、通常0〜5回、好ましくは0〜3回である。通常、供給される粗(メタ)アクリル酸より高い純度を有する(メタ)アクリル酸が得られる段階は、すべて精製段階であり、それ以外の段階は、すべてストリッピング段階である。ストリッピング段階は、精製段階から残留母液に含まれる(メタ)アクリル酸を回収するために実施される。なお、ストリッピング段階は、必ずしも設ける必要はなく、例えば、蒸留塔を用いて、晶析装置の残留母液から低沸点成分を分離する場合には、ストリッピング段階は省略してもよい。
【0071】
動的結晶化及び静的結晶化のいずれを採用する場合であっても、晶析工程で得られる(メタ)アクリル酸の結晶は、そのまま製品としてもよいし、必要に応じて、更に洗浄や発汗等の精製を行ってから製品としてもよい。他方、晶析工程で排出される残留母液は、系外に取り出してもよい。
【0072】
以上の方法により、(メタ)アクリル酸を製造することができる。かくして製造された(メタ)アクリル酸は、すでに公知となっているように、(メタ)アクリル酸エステル等の(メタ)アクリル酸誘導体;ポリ(メタ)アクリル酸、ポリ(メタ)アクリル酸ナトリウム等の親水性樹脂;吸水性樹脂等の合成原料として有用である。
【0073】
更に、本発明による親水性樹脂の製造方法は、上記のようなアクリル酸又はそのエステルの製造方法により得られる、アクリル酸又はそのエステルを含む単量体成分を重合することを特徴とする。すなわち、本発明の製造方法により得られたアクリル酸又はそのエステルは、吸水性樹脂や水溶性樹脂等の親水性樹脂の原料として用いることができる。
【0074】
本発明の製造方法により得られたアクリル酸を、吸水性樹脂や水溶性樹脂等の親水性樹脂を製造するための原料として用いた場合、重合反応を制御しやすく、得られた親水性樹脂の品質が安定し、吸水性能、無機材料の分散性能等の各種性能が改善される。
親水性樹脂としては、吸水性樹脂であることが好ましい。
【0075】
吸水性樹脂を製造する場合には、例えば、本発明の製造方法により得られたアクリル酸又はその塩(アクリル酸を部分中和して得た塩)を単量体成分の主成分(好ましくは70モル%以上、より好ましくは90モル%以上)とし、更に0.001〜5モル%(アクリル酸に対する値)程度の架橋剤、0.001〜2モル%(単量体成分に対する値)程度のラジカル重合開始剤を用いて、架橋重合させた後、乾燥・粉砕することにより、吸水性樹脂を得ることができる。
【0076】
ここで、吸水性樹脂とは、架橋構造を有する水膨潤性水不溶性のポリアクリル酸であって、自重の3倍以上、好ましくは10〜1,000倍の純水又は生理食塩水を吸水することにより、水溶性成分(水可溶分)が好ましくは25質量%以下、より好ましくは10質量%以下である水不溶性ヒドロゲルを生成するポリアクリル酸を意味する。
このような吸水性樹脂の具体例や物性測定法は、例えば、米国特許第6,107,358号、米国特許第6,174,978号、米国特許第6,241,928号等に記載されている。
また、生産性向上の観点から好ましい製造方法は、例えば、米国特許第6,867,269号、米国特許第6,906,159号、米国特許第7,091,253号、国際公開第2001/038402号、国際公開第2006/034806号等に記載されている。
【0077】
アクリル酸を出発原料として、中和、重合、乾燥等により、吸水性樹脂を製造する一連の工程は、例えば以下の通りである。
本発明の製造方法により得られるアクリル酸の一部は、ラインを介して、吸水性樹脂の製造プロセスに供給される。吸収性樹脂の製造プロセスにおいては、アクリル酸を中和工程、重合工程、乾燥工程に導入して、所望の処理を施すことにより、吸水性樹脂を製造する。各種物性の改善を目的として所望の処理を施してもよく、例えば、重合中又は重合後に架橋工程を介在させてもよい。
【0078】
中和工程は、任意の工程であり、例えば、所定量の塩基性物質の粉末又は水溶液と、アクリル酸やポリアクリル酸(塩)とを混合する方法が例示されるが、従来公知の方法を採用すればよく、特に限定されるものではない。なお、中和工程は、重合前又は重合後のいずれで行なってもよく、また、重合前後の両方で行なってもよい。
アクリル酸やポリアクリル酸(塩)の中和に用いられる塩基性物質としては、例えば、炭酸(水素)塩、アルカリ金属の水酸化物、アンモニア、有機アミン等、従来公知の塩基性物質を適宜用いればよい。
また、ポリアクリル酸の中和率は、特に限定されるものではなく、任意の中和率(例えば、30〜100モル%の範囲内における任意の値)となるように調整すればよい。
【0079】
重合工程における重合方法は、特に限定されるものではなく、ラジカル重合開始剤による重合、放射線重合、電子線や活性エネルギー線の照射による重合、光増感剤による紫外線重合等、従来公知の重合方法を用いればよい。また、重合開始剤、重合条件等の各種条件については、任意に選択することができる。もちろん、必要に応じて、架橋剤や他の単量体、更には水溶性連鎖移動剤や親水性高分子等、従来公知の添加剤を添加してもよい。
【0080】
重合後のアクリル酸塩系ポリマー(すなわち、吸水性樹脂)は、乾燥工程に付される。乾燥方法としては、特に限定されるものではなく、熱風乾燥機、流動層乾燥機、ナウター式乾燥機等、従来公知の乾燥手段を用いて、所望の乾燥温度、好ましくは70〜230℃で、適宜乾燥させればよい。乾燥工程を経て得られた吸水性樹脂は、そのまま用いてもよく、更に所望の形状に造粒・粉砕、表面架橋をしてから用いてもよく、還元剤、香料、バインダー等の従来公知の添加剤を添加する等、用途に応じた後処理を施してから用いてもよい。
【実施例】
【0081】
以下に実施例を挙げて本発明をより具体的に説明するが、本発明は、下記実施例によって限定されるものではなく、適宜変更して実施することも可能であり、それらはいずれも本発明の技術的範囲に包含される。
【0082】
(調製例1)
(3−ヒドロキシプロピオン酸(3HP)を含む組成物の取得方法)
Klebsiella pneumoniae ATCC25955株のゲノムDNAをテンプテレ−トとしてグリセロールデヒドラターゼ遺伝子(GD遺伝子)及びグリセロールデヒドラターゼ再活性化因子(GDR遺伝子)を含む領域を、下記の2つのプライマーを用いてPCRで増幅し、増幅断片の末端を制限酵素NdeI、BglIIで切断し、電気泳動によって切断断片を回収した。なお、GD遺伝子及びGDR遺伝子配列を増幅する以下のプライマーはGenBank Accession number:NC_009648記載のDNA配列を元に設計した。
フォワードプライマー:
5’−GCGCGCCATATGTTAATTCGCCTGACCGGCC−3’
リバースプライマー:
5’−GCGCGCAGATCTTCAGTTTCTCTCACTTAACG−3’
【0083】
pACYCDuet−1プラスミド(タカラバイオ社)をテンプレートにして下記の2つのプライマーでベクター配列を増幅し、pACYCDuet−1プラスミドのT7プロモーターの後ろにNdeIサイト及びBgl IIサイトを持ったDNA断片を増幅した。
フォワードプライマー:
5’−GAAGGAGATATACATATGGCGCGC−3’
リバースプライマー:
5’−CCGATATCCAATTGAGATCTGCGCGC−3’
【0084】
増幅断片を制限酵素BglIIとNdeIで切断し、電気泳動によって切断断片を分離して回収した。この2つのDNA断片をライゲーションし、大腸菌TOP10コンピテントセル(インビトロジェン社)に導入し、クロラムフェニコール含有プレートに広げて培養したところ、クロラムフェニコール耐性大腸菌を得ることができた。クロラムフェニコール耐性大腸菌からプラスミドDNAを抽出し、制限酵素により分子量の確認を行ったところ、目的とするGD遺伝子及びGDR遺伝子がpACYCDuet−1プラスミドに挿入されていることが確認できた。構築した組換えプラスミドをGD−GDR/pACYCDuet−1と命名し、以降の実験に用いた。
【0085】
大腸菌K−12 W3110株のゲノムDNAをテンプテレ−トとしてγ−glutamyl−γ−aminobutyraldehyde dehydrogenase遺伝子(aldH遺伝子)を下記の2つのプライマーを用いてPCR法で増幅し、増幅断片の末端を制限酵素NdeI、BglIIで切断し、電気泳動によって切断断片を回収した。なお、以下プライマーはGenBank Accession number:AB200319記載のDNA配列を元に設計した。
フォワードプライマー:
5’−GGGGGGCCATATGAATTTTCATCATCTGGCTTACTG−3’
リバースプライマー:
5’−CCCCAGATCTTCAGGCCTCCAGGCTTATCCAGATG−3’
【0086】
pUC18プラスミドをテンプレートにして下記の2つのプライマーでベクター配列を増幅し、pUC18プラスミドのlacプロモーターの後ろにNdeIサイトを持ち、lacZ遺伝子の終始コドンの位置にBamHIサイトを持ったDNA断片を増幅した。
フォワードプライマー:
5’−CCCCCCCATATGTGTTTCCTGTGTGAAATTGTTATCCGCTCACAATTCCACACAATATACGAGCC−3’
リバースプライマー:
5’−CCCCGGATCCTTAGTTAAGCCAGCCCCGACACCCGCCAACACC−3’
【0087】
増幅断片を制限酵素BamHIとNdeIで切断し、電気泳動によって切断断片を分離して回収した。この2つのDNA断片をライゲーションし、大腸菌TOP10コンピテントセルに導入し、アンピシリン含有プレートに広げて培養した。得られた形質転換体からプラスミドを抽出し、制限酵素処理により分子量の確認をしたところ、目的どおり、aldHがpUC18プラスミドに挿入されていることを確認した。構築した組換えプラスミドをaldH/pUC18と命名し、以降の実験で使用した。
【0088】
構築したGD−GDR/pACYCDuet−1及びaldH/pUC18をEscherichia coli BL21(DE3) competent cell(Merck社製)のプロトコールに従って、ヒートショック法により導入し、E.coli(GD−GDR/pACYCDuet−1、aldH/pUC18)を作出した。
E.coli(GD−GDR/pACYCDuet−1、aldH/pUC18)を、アンピシリン100ppm、クロラムフェニコール50ppm添加LB液体培地5mL(LB培地1Lあたりの組成:トリプトン10g、酵母エキス5g、NaCl10g)で37℃、16時間、振盪培養し、前培養液を得た。次に前培養液5mLを、アンピシリン100ppm、クロラムフェニコール50ppm、添加NS液体培地1Lに植菌し、37℃、攪拌速度725rpm、通気量1L/minで通気攪拌培養を行った。なお、NS液体培地の組成は、グリセリン40g/L、硫酸アンモニウム10g/L、リン酸二水素カリウム2g/L、リン酸水素二カリウム6g/L、硫酸マグネシウム7水和物1g/L、酵母エキス40g/Lである。また、培養には、バイオット社製ジャーファーメンター:BMJ−02NP2を使用し、培養中はアンモニア水を用いて培養液中のpHを7にコントロールした。培養8時間後に1M IPTG溶液を1mL、8mMアデノシルコバラミン溶液を1mL添加し、培養途中にグリセリンが枯渇しないように適時グリセリンを添加しながら100時間培養を行った。得られた培養液を遠心分離にかけ、培養液上清を回収した。
【0089】
以下記載の高速液体クロマトグラフィーを用いた分析方法で培養液上清中の生成物の確認を行ったところ、生成物である3−ヒドロキシプロピオン酸のピークを7.9分の位置に確認することができ、培養液中の3−ヒドロキシプロピオン酸の濃度は2質量%であった。
高速液体クロマトグラフィーでの分析条件:
使用カラム: YMC−pACK FA(YMC社製)
流量:1mL/min
インジェクション量:10μL
溶離液:メタノール/アセトニトリル/H2O=40/5/55(V/V/V)
内部標準:2−Hydroxy−2−methyl−n−butyric acid
検出:UV400nm
【0090】
培養上清100μLに内部標準液200μLを加えた。ヒドロキシカルボン酸ラベル化試薬(YMC社製)の試薬A液200μL、試薬B液200μLを加え、よく混合した後、60℃、20分間処理した。ヒドロキシカルボン酸ラベル化試薬(YMC社製)の試薬C液200μLを添加し、よく混合した。60℃、15分間処理後、室温まで冷えたら0.45mmフィルターに通し、LC分析サンプルとして供した。
菌体を除去した2質量%の3−ヒドロキシプロピオン酸含有培養液からWO2002/090312号記載の方法で、原料組成物として12質量%の3HP水溶液を得た。
【0091】
(実施例1)
(3HPの第一の脱水工程)
特開2009−190915号公報の方法に従い、球状のZSM−5型ゼオライト(プロトン型)を合成した。内径10mmのステンレス製反応管に、ZSM−5型ゼオライトを充填し、その上にステンレス製の1.5mmのディクソンパッキンを蒸発層として積層した。反応管を電気炉にて300℃に加熱し、上記調製例1で得られた3HP水溶液を、毎時16.7gの速度で反応管の上部に供給した。同時に、毎時3Lの速度で窒素ガスを流した。
反応管の下部から抜き出した反応ガスを、冷却捕集し反応液を得た。得られた反応液を液体クロマトグラフィーで分析した。反応後1時間目から2時間目の間の反応成績は、3HPの転化率は100%、アクリル酸の収率は95モル%であった。反応後7時間目から8時間目の間の反応成績は、3HPの転化率は95%、アクリル酸の収率は91モル%であり、経時的な触媒活性低下が観察された。
反応後、ディクソンパッキン及び触媒を抜き出してみると、褐色の着色が見られた。
【0092】
(触媒の再生工程)
反応器から抜き出したディクソンパッキン及び触媒を、磁性皿に薄く広げ、焼成炉内に設置した。その後、焼成炉内に、0.5L/分の流量で空気を流通させ、1時間かけて500℃まで昇温し1時間保持して、触媒上の炭素状物質を酸化分解して、再生を行った。冷却後、取り出したディクソンパッキンは元の銀色に、触媒は元の白色に変化していた。
【0093】
(3HPの第二の脱水工程)
触媒再生工程で得られた触媒を、再度反応器に充填し、第一の脱水工程と同様の条件で、3HPの脱水反応を実施した。反応後1時間目から2時間目の間の反応成績は、3HPの転化率は100%、アクリル酸の収率は94モル%であった。反応後7時間目から8時間目の間の反応成績は、3HPの転化率は96%、アクリル酸の収率は92モル%であった。触媒を再生することによって、第一の脱水工程とほぼ同様の反応結果を第二の脱水工程でも得ることができた。
上記第一の脱水工程後の触媒(サンプル1−1)、触媒再生後の触媒(サンプル1−2)、第二の脱水工程後の触媒(サンプル1−3)を、示差熱−熱重量測定装置にて分析を行った。分析条件は、試料30mgを10℃/分で800℃まで昇温し、流通空気量は50mL/分とした。その結果、重量減少率は、サンプル1−1で5.0%、サンプル1−2で0.5%、サンプル1−3で5.1%であった。触媒再生工程によって、炭素状物質が減少し、触媒活性が回復したことが示唆される。
【0094】
(比較例1)
実施例1の3HPの第一の脱水工程を、16時間継続して行った。反応後15時間目から16時間目の間の反応成績は、3HPの転化率は82%、アクリル酸の収率は72モル%であった。
第一の脱水工程後に、触媒再生工程を経て、第二の脱水工程を行って合計16時間反応した実施例1に比べると、3HPの転化率、アクリル酸の収率とも大きく低下した。
上記第一の脱水工程後の触媒(比較サンプル1)を、実施例1と同様に示差熱−熱重量測定装置にて分析を行った。その結果、重量減少率は、比較サンプル1で13.2%であった。比較例1では、大量の炭素状物質が触媒上に付着していることが示唆された。
【0095】
(実施例2)
触媒を市販のγ−アルミナペレット(サンゴバン社製)に変えた以外は、実施例1と同様に反応、触媒再生を行った。
(3HPの第一の脱水工程)
反応後1時間目から2時間目の間の反応成績は、3HPの転化率は91%、アクリル酸の収率は86モル%であった。反応後7時間目から8時間目の間の反応成績は、3HPの転化率は79%、アクリル酸の収率は76モル%であり、経時的な触媒活性低下が観察された。
(触媒の再生工程)
第一の脱水工程終了後、反応器の温度を450℃に上げ、窒素を5L/時間の流量で1時間流通した。その後、1.5L/時間の流量で酸化剤含有ガス(酸素2容量%、残部は窒素)を流通させたところ、触媒層の温度の上昇が観察された。次に、酸化剤の含有量を多くしたガス(酸素5容量%、残部は窒素)を流通させ、最後に更に酸化剤の含有量を多くしたガス(酸素8容量%、残部は窒素)を、炭素状物質の酸化分解による発熱が収まるまでの約24時間流通させた。
(3HPの第二の脱水工程)
触媒の再生工程終了後、引き続き、第一の脱水工程と同様の条件で、3HPの脱水反応を実施した。反応後1時間目から2時間目の間の反応成績は、3HPの転化率は91%、アクリル酸の収率は86モル%であった。反応後7時間目から8時間目の間の反応成績は、3HPの転化率は78%、アクリル酸の収率は76モル%であった。触媒を再生することによって、第一の脱水工程とほぼ同様の反応結果を第二の脱水工程でも得ることができた。
【0096】
(実施例3)
調製例1で得られた12質量%の3HP水溶液の濃縮を行った。薄膜蒸発器を用いて水の除去を行った。得られた濃縮液を、水分計、液体クロマトグラフィー及びNMRで分析したところ、水が34質量%、3HPが61質量%、3HPのオリゴマーが5質量%含まれていた。
(3HPの第一の脱水工程)
上記濃縮液を原料組成物として用い、実施例1で使用したZSM−5を触媒として脱水反応を実施した。上記濃縮液を、毎時3gの速度で反応管の上部に供給した。同時に、毎時22Lの速度で窒素ガスを流した。反応後1時間目から2時間目の間の反応成績は、3HP及びオリゴマーの転化率は97%、アクリル酸の収率(対3HP及びオリゴマー基準)は93モル%であった。反応後7時間目から8時間目の間の反応成績は、3HP及びオリゴマーの転化率は91%、アクリル酸の収率は87モル%であり、経時的な触媒活性低下が観察された。
(触媒の再生工程)
第一の脱水工程終了後、反応器の温度を450℃に上げ、窒素を5L/時間の流量で1時間流通した。その後、1.5L/時間の流量で酸化剤含有ガス(酸素2容量%、残部は窒素)を流通させたところ、触媒層の温度の上昇が観察された。次に、酸化剤の含有量を多くしたガス(酸素5容量%、残部は窒素)を流通させ、最後に更に酸化剤の含有量を多くしたガス(酸素8容量%、残部は窒素)を、炭素状物質の酸化分解による発熱が収まるまでの約30時間流通させた。
(3HPの第二の脱水工程)
第一の脱水工程と同様の条件で、3HPの脱水反応を実施した。反応後1時間目から2時間目の間の反応成績は、3HP及びオリゴマーの転化率は96%、アクリル酸の収率は92モル%であった。反応後7時間目から8時間目の間の反応成績は、3HP及びオリゴマーの転化率は90%、アクリル酸の収率は87モル%であった。触媒を再生することによって、第一の脱水工程とほぼ同様の反応結果を第二の脱水工程でも得ることができた。
【0097】
上記実施例3における第一の脱水工程後の触媒(サンプル3−1)、触媒再生後の触媒(サンプル3−2)を、実施例1と同様に示差熱−熱重量測定装置にて分析を行った。その結果、重量減少率は、サンプル3−1で6.1%、サンプル3−2で0.9%であった。
【0098】
(実施例4)
調製例1で得られた12質量%の3HP水溶液の濃縮を行った。ロータリーエバポレーターを用いて水の除去を行った。得られた濃縮液を、水分計、液体クロマトグラフィー及びNMRで分析したところ、水が34質量%、3HPが30質量%、3HPのオリゴマーが36質量%含まれていた。
(3HPの第一の脱水工程)
上記濃縮液を原料組成物として用い、実施例1で使用したZSM−5を触媒として脱水反応を実施した。上記濃縮液を、毎時3gの速度で反応管の上部に供給した。同時に、毎時22Lの速度で窒素ガスを流した。反応後1時間目から2時間目の間の反応成績は、3HP及びオリゴマーの転化率は89%、アクリル酸の収率(3HP及びオリゴマー基準)は83モル%であった。反応後7時間目から8時間目の間の反応成績は、3HP及びオリゴマーの転化率は78%、アクリル酸の収率は73モル%であり、経時的な触媒活性低下が観察された。
(触媒の再生工程)
第一の脱水工程終了後、反応器の温度を450℃に上げ、窒素を5L/時間の流量で1時間流通した。その後、1.5L/時間の流量で酸化剤含有ガス(酸素2容量%、残部は窒素)を流通させたところ、触媒層の温度の上昇が観察された。次に、酸化剤の含有量を多くしたガス(酸素5容量%、残部は窒素)を流通させ、最後に更に酸化剤の含有量を多くしたガス(酸素8容量%、残部は窒素)を、炭素状物質の酸化分解による発熱が収まるまでの約38時間流通させた。
(3HPの第二の脱水工程)
第一の脱水工程と同様の条件で、3HPの脱水反応を実施した。反応後1時間目から2時間目の間の反応成績は、3HPの転化率は82%、アクリル酸の収率は76モル%であった。反応後7時間目から8時間目の間の反応成績は、3HP及びオリゴマーの転化率は72%、アクリル酸の収率は66モル%であった。
【0099】
上記触媒再生後の触媒(サンプル4)を、実施例1と同様に示差熱−熱重量測定装置にて分析を行った。その結果、重量減少率は、サンプル4で2.7%であった。
【0100】
(実施例5)
(触媒の調製)
硝酸カリウム(2.5g)とリン酸第2アンモニウム(1.6g)を100gの水に溶解した溶液に、酸化ケイ素(30g)を加え、湯浴上で均一に加熱混合した後、加熱乾固した。次いで、空気中120℃で20時間仮焼してから10〜24メッシュに破砕し、更に空気中500℃で2時間焼成することにより、酸素を除く組成がK1Si20P0.5からなる触媒を得た。
(3HPの第一の脱水工程)
内径10mmのステンレス管に、蒸発層としてステンレス製の1.5mmディクソンパッキンを充填し、電気炉内に設置し蒸発器とした。また内径10mmのステンレス管に、上記の触媒を充填し、電気炉内に設置し反応器とした。蒸発器の出口と反応器の入口をステンレス管で連結し、周囲を電気ヒーターで加熱できるようにした。
また、蒸発器内の温度を275℃とし、反応器内の温度を300℃とし、実施例1と同じ原料組成物を毎時33.5gの速度で蒸発器の上部に供給した。同時に、毎時6Lの速度で窒素ガスを流した。蒸発器の出口ガスはそのまま反応器へ供給し、24時間継続して反応を実施した。1〜2時間目及び23〜24時間目の、反応器の出口ガスを冷却捕集し、得られた液を分析した。結果を表1に示す。
(触媒の再生工程)
第一の脱水工程終了後、反応器の温度を450℃に上げ、窒素を5L/時間の流量で1時間流通した。その後、1.5L/時間の流量で酸化剤含有ガス(酸素2容量%、残部は窒素)を流通させたところ、触媒層の温度の上昇が観察された。次に、酸化剤の含有量を多くしたガス(酸素5容量%、残部は窒素)を流通させ、最後に空気を炭素状物質の酸化分解による発熱が収まるまでの約30時間流通させた。
(3HPの第二の脱水工程)
第一の脱水工程と同様の条件で、3HPの脱水反応を実施した。その後、触媒再生−3HP脱水反応を繰り返し実施した。これらの結果を表1に示す。
脱水反応を4回繰り返しても、触媒活性に変化はなかった。
【0101】
【表1】
【0102】
(実施例6)吸水性樹脂の製造例
実施例1で得られた精製アクリル酸に重合禁止剤を60質量ppm添加した。別途、鉄を0.2質量ppm含有する苛性ソーダから得られたNaOH水溶液に対して、上記の重合禁止剤添加アクリル酸を冷却下(液温35℃)で添加することにより、75モル%中和を行った。得られた、中和率75モル%、濃度35質量%のアクリル酸ナトリウム水溶液に、内部架橋剤としてポリエチレングリコールジアクリレート0.05モル%(アクリル酸ナトリウム水溶液に対する値)を溶解させることにより、単量体成分を得た。この単量体成分350gを容積1Lの円筒容器に入れ、2L/minの割合で窒素を吹き込んで、20分間脱気した。次いで、過硫酸ナトリウム0.12g/モル(単量体成分に対する値)及びL−アスコルビン酸0.005g/モル(単量体成分に対する値)の水溶液をスターラー攪拌下で添加して、重合を開始させた。重合開始後に攪拌を停止し、静置水溶液重合を行った。単量体成分の温度が約15分(重合ピーク時間)後にピーク重合温度108℃を示した後、30分間重合を進行させた。その後、重合物を円筒容器から取り出し、含水ゲル状架橋重合体を得た。
得られた含水ゲル状架橋重合体は、45℃でミートチョッパー(孔径:8mm)により細分化した後、170℃の熱風乾燥機で、20分間加熱乾燥させた。更に、乾燥重合体(固形分:約95%)をロールミルで粉砕し、JIS標準篩で粒径600〜300μmに分級することにより、ポリアクリル酸系吸水性樹脂(中和率:75%)を得た。
本発明の製造方法により得られたアクリル酸の重合性は、プロピレンを原料とするアクリル酸の製造方法により得られたアクリル酸の重合性と同等であり、得られた吸水性樹脂は、臭気がなく、物性も同等であった。
【0103】
このように、3−ヒドロキシカルボン酸又はそのエステルの脱水用触媒上の炭素状物質に、酸化剤を接触させて炭素状物質を除去する、本発明の脱水用触媒の再生方法を用いることによって、脱水用触媒の活性を効率的に回復させることができ、また、当該再生方法を含む(メタ)アクリル酸又はそのエステルの製造方法により、触媒上の炭素状物質による反応管の閉塞を抑制し、3−ヒドロキシカルボン酸又はそのエステルを効率良く(メタ)アクリル酸又はそのエステルに転化することができ、(メタ)アクリル酸又はそのエステルを長期間にわたり安定して製造することができるという作用機序は、すべて同様であるものと考えられる。
したがって、上記実施例の結果から、本発明の技術的範囲全般において、また、本明細書において開示した種々の形態において本発明が適用でき、有利な作用効果を発揮することができるといえる。
【特許請求の範囲】
【請求項1】
3−ヒドロキシカルボン酸又はそのエステルの脱水用触媒上の炭素状物質を除去することによって触媒を再生する方法であって、
該再生方法は、脱水用触媒に酸化剤を接触させて炭素状物質を除去することを特徴とする3−ヒドロキシカルボン酸又はそのエステルの脱水用触媒の再生方法。
【請求項2】
前記酸化剤は、ガス状酸化剤であることを特徴とする請求項1に記載の3−ヒドロキシカルボン酸又はそのエステルの脱水用触媒の再生方法。
【請求項3】
前記再生方法は、固定床反応器内に充填された3−ヒドロキシカルボン酸又はそのエステルの脱水用触媒を再生する方法であり、該固定床反応器内にガス状酸化剤を流通させて、炭素状物質を除去することを特徴とする請求項2に記載の3−ヒドロキシカルボン酸又はそのエステルの脱水用触媒の再生方法。
【請求項4】
請求項1〜3のいずれかに記載の3−ヒドロキシカルボン酸又はそのエステルの脱水用触媒の再生方法を含むことを特徴とする(メタ)アクリル酸又はそのエステルの製造方法。
【請求項5】
前記製造方法は、第一の脱水工程、触媒再生工程及び第二の脱水工程を含み、
該第一の脱水工程は、3−ヒドロキシカルボン酸又はそのエステルと脱水用触媒との気相接触反応により3−ヒドロキシカルボン酸又はそのエステルを脱水する工程であり、
該触媒再生工程は、前記再生方法により脱水用触媒を再生する工程であり、
該第二の脱水工程は、3−ヒドロキシカルボン酸又はそのエステルと触媒再生工程後の脱水用触媒との気相接触反応により3−ヒドロキシカルボン酸又はそのエステルを脱水する工程であることを特徴とする請求項4に記載の(メタ)アクリル酸又はそのエステルの製造方法。
【請求項1】
3−ヒドロキシカルボン酸又はそのエステルの脱水用触媒上の炭素状物質を除去することによって触媒を再生する方法であって、
該再生方法は、脱水用触媒に酸化剤を接触させて炭素状物質を除去することを特徴とする3−ヒドロキシカルボン酸又はそのエステルの脱水用触媒の再生方法。
【請求項2】
前記酸化剤は、ガス状酸化剤であることを特徴とする請求項1に記載の3−ヒドロキシカルボン酸又はそのエステルの脱水用触媒の再生方法。
【請求項3】
前記再生方法は、固定床反応器内に充填された3−ヒドロキシカルボン酸又はそのエステルの脱水用触媒を再生する方法であり、該固定床反応器内にガス状酸化剤を流通させて、炭素状物質を除去することを特徴とする請求項2に記載の3−ヒドロキシカルボン酸又はそのエステルの脱水用触媒の再生方法。
【請求項4】
請求項1〜3のいずれかに記載の3−ヒドロキシカルボン酸又はそのエステルの脱水用触媒の再生方法を含むことを特徴とする(メタ)アクリル酸又はそのエステルの製造方法。
【請求項5】
前記製造方法は、第一の脱水工程、触媒再生工程及び第二の脱水工程を含み、
該第一の脱水工程は、3−ヒドロキシカルボン酸又はそのエステルと脱水用触媒との気相接触反応により3−ヒドロキシカルボン酸又はそのエステルを脱水する工程であり、
該触媒再生工程は、前記再生方法により脱水用触媒を再生する工程であり、
該第二の脱水工程は、3−ヒドロキシカルボン酸又はそのエステルと触媒再生工程後の脱水用触媒との気相接触反応により3−ヒドロキシカルボン酸又はそのエステルを脱水する工程であることを特徴とする請求項4に記載の(メタ)アクリル酸又はそのエステルの製造方法。
【図1】

【公開番号】特開2013−46899(P2013−46899A)
【公開日】平成25年3月7日(2013.3.7)
【国際特許分類】
【出願番号】特願2012−144634(P2012−144634)
【出願日】平成24年6月27日(2012.6.27)
【出願人】(000004628)株式会社日本触媒 (2,292)
【Fターム(参考)】
【公開日】平成25年3月7日(2013.3.7)
【国際特許分類】
【出願日】平成24年6月27日(2012.6.27)
【出願人】(000004628)株式会社日本触媒 (2,292)
【Fターム(参考)】
[ Back to top ]