
3−メチルアミノ−1−(チエン−2−イル)プロパン−1−オールの製造方法
【課題】式(1)で表される3-メチルアミノ-1-(チエン-2-イル)プロパン-1-オールを製造するための酵素的および非酵素的方法、さらに、これらの方法を実施するための酵素、これらの酵素をコードする核酸配列、これらの核酸配列を含む発現カセット、ベクターならびに組換え宿主生物の提供。

【解決手段】a)チオフェンを、ルイス酸の存在下で、ハロゲン化β-ハロプロピオニルまたはハロゲン化アクリロイルと反応させ、その際、同時に、または反応が生じた後であるが反応生成物を単離する前にハロゲン化水素を通過させて、3-ハロ-1-(チエン-2-イル)プロパン-1-オンを得、そして、b)工程a)で得られたプロパノンを還元し、次いで、適宜に反応生成物を単離することなく、メチルアミンと反応させる方法。
【解決手段】a)チオフェンを、ルイス酸の存在下で、ハロゲン化β-ハロプロピオニルまたはハロゲン化アクリロイルと反応させ、その際、同時に、または反応が生じた後であるが反応生成物を単離する前にハロゲン化水素を通過させて、3-ハロ-1-(チエン-2-イル)プロパン-1-オンを得、そして、b)工程a)で得られたプロパノンを還元し、次いで、適宜に反応生成物を単離することなく、メチルアミンと反応させる方法。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、式I:
【化1】
【0002】
で表される3-メチルアミノ-1-(チエン-2-イル)プロパン-1-オールを調製するための方法に関し、特にS-エナンチオマーI-Sを調製するための方法に関する。
【背景技術】
【0003】
式I-S:
【化2】
【0004】
で表されるアミノプロパノールIのSエナンチオマーは、式II:
【化3】
【0005】
(式中、Bは、n価の負に荷電した無機酸または有機酸のラジカルであり、HnBは、医薬的に許容される酸である)で表される抗うつ剤デュロキセチンを合成するための重要な前駆体である。
【0006】
デュロキセチンまたはその対応する塩基を調製するための先行技術の方法は、複雑であり、キラルな試薬またはキラルな出発化合物の使用を必要とする。
【0007】
従って、EP-B-0273658には、デュロキセチンの対応する塩基を調製するための方法が記載されており、この方法は、Mannich反応で2-アセチルチオフェンをホルムアルデヒドおよびジメチルアミンと反応させ、得られたMannich塩基のケト基を還元してラセミ体(S)-3-N,N-ジメチルアミノ-1-(チエン-2-イル)プロパン-1-オールを得、そのアルコール官能基をフッ化ナフチルでエーテル化し、最終的にそのジメチルアミノ基をメチルアミノ官能基に変換することによるものである。ナフチルエーテルの所望のエナンチオマーは、キラルな出発材料を使用することによって、または最終生成物の工程でのラセミ化合物の分割によって、例えば光学活性酸との塩またはキラルな固定相でのクロマトグラフィーによって得られる。
【0008】
US-5,362,886には、類似する方法が記載されており、その方法では、ケト基の還元後に得られるラセミ体のプロパノールにS-マンデル酸を加える。これに関連して得られるアルコールのSエナンチオマーは後続の反応工程で使用される。
【0009】
さらに、EP-A-0457559には、EP-B-0273658に記載される方法と類似する方法が記載されている。この場合には、Mannich塩基のケト基を、不斉還元系であるLAH-lcb(水素化アルミニウムリチウム-[(2R,2S)-(-)-4-ジメチルアミノ-1,2-ジフェニル-3-メチル-2-ブタノール])により還元すると、Sエナンチオマーの形のアルコールが得られる。費用は別にして、この方法の難点は、還元系LAH-lcbが不安定であって、数分間しか安定でない点である。
【0010】
Journal of Labelled Compounds and Radiopharmaceuticals, volume XXXVI, No. 3, pp. 213〜223において、W. J. WheelerおよびF. Kuoもまた、デュロキセチンの調製方法を記載している。この方法では、チオフェン-2-カルボニルクロリドを、Stilleカップリングにおいて、DMPU(ジメチルプロピレンウレア)中、触媒量のベンジル(クロロ)-ビス(トリフェニルホスフィン)パラジウム(II)の存在下で、ビニルトリ-n-ブチルスタンナンと反応させて、式II:
【化4】
【0011】
で表される1-(チエン-2-イル)プロペノンを得、これを続いて塩化水素で処理することによって式III.1:
【化5】
【0012】
で表される3-クロロ-1-(チエン-2-イル)プロパン-1-オンに変換する。続いて、この方法で得られるクロロプロパノンIII.1をキラルなオキサザボロリジン(oxazaborolidine)およびBH3を用いて還元すると、式IV.1-S:
【化6】
【0013】
で表される(S)-3-クロロ-1-(チエン-2-イル)プロパン-1-オールが得られる。
【0014】
この方法で得られるアルコールIV.1-Sは、ヨウ化ナトリウム、次いで、メチルアミンと連続して反応させると、(S)-3-メチルアミノ-1-(チエン-2-イル)プロパン-1-オールI-Sに変換される。続いて水素化ナトリウム、1-フルオロナフタレンおよび塩化水素と連続して反応させることにより、デュロキセチンを塩酸塩の形態で得る。これに関連して、不都合な点は、第一に、クロロプロパノン中間体III.1を調製するために複数の工程および高価な試薬が必要である点である。第二に、クロロプロパノール IV.1-Sは、クロロプロパノンIII.1がアミノアルコールに変換されるときに単離される。しかし、本発明者らの行った研究によって、このクロロプロパノールは不安定であり、強い発熱反応できわめて容易に分解され、アミノアルコールに関して収量の低下を招くだけでなく、産業的規模でこの反応を管理することをより困難にすることが分かっている。
【0015】
W. J. Wheelerらによって記載される、デュロキセチン合成中に中間体として生成する3-クロロ-1-(チエン-2-イル)プロパン-1-オンの調製方法は、文献から公知である。しかし、先行技術より公知である方法の欠点は、クロロプロパノンIII.1が低収率で形成されるか、あるいは取り扱いの難しい試薬を用いる必要があるか、のいずれかである。従って、CR Acad. Sci., Ser. C, 1979, 288(1), 49-52において、A. Etienneらは、溶媒としてのニトロメタン中でルイス酸触媒としての三塩化アルミニウムの存在下に、塩化3-クロロプロピオニルとチオフェンとのFriedel-Crafts反応によって、クロロプロパノンIII.1を調製する方法を記載する。このクロロプロパノンIII.1は、わずか7%の収率でしか得られない。LiuらによってChirality, 12, 26-29(2000)に記載されている対応する反応もまた、ルイス酸触媒としての四塩化スズと、溶媒としてのベンゼンの存在下で、満足のいく収率をもたらさない。Meth-Cohnらは、Acta Chem. Scand. B 20(6), 1577-1587(1966)において、三塩化鉄または三塩化アルミニウムの存在下でのチオフェンに対するFriedel-Craftsアシル化を記載しており、クロロプロパノンIII.1は中程度から良好な収率で生成される。この方法の欠点は、溶媒として二硫化炭素を使用する必要がある点である。
【0016】
Kamal, G. B. R. Khanna, R. RamuおよびT. Krishnajiらは、Tetrahedron Lett. 44, 2003, 4783-4787に、デュロキセチン前駆体である(S)-3-ヒドロキシ-3-(チエン-2-イル)プロパンニトリルを調製する方法を記載しており、この方法では、塩化クロロアセチルでチオフェンをアセチル化し、そのケトンを水素化ホウ素ナトリウムで還元してラセミ体アルコールを得、その塩素ラジカルをシアニドで置換し、ラセミ体ニトリルアルコールをシュードモナス・セパシア(Pseudomonas cepacia)のリパーゼ(ケイ藻土に固定化したもの;Rエナンチオマーのエステル化を選択的に触媒する)の存在下で酢酸ビニルと反応させて、所望のSエナンチオマー(エステル化されないままで残る)が純粋な形態で単離できるようにする。この方法の欠点は、多数の反応工程を必要とする一方で、エステル化されたR成分がデュロキセチンを生じるようにそれ以上反応しないため、半量のニトリルアルコールが無駄になる点である。
【発明の開示】
【発明が解決しようとする課題】
【0017】
従って、本発明の目的は、上記の先行技術の欠点を克服した3-メチルアミノ-1-(チエン-2-イル)プロパン-1-オールIの調製方法を提供することである。さらに、この方法は、全体的に良い収率で化合物Iを供給し、特にまた、エナンチオ選択的にSエナンチオマーI-Sの調製を可能にすべきである。
【課題を解決するための手段】
【0018】
本発明者らは、上記の目的が以下の方法で達成されることを見出した。最初に、チオフェンを、Friedel-Crafts反応でルイス酸の存在下に、ハロゲン化β-ハロプロピオニルまたはハロゲン化アクリロイルと反応させるが、その際、反応生成物を単離する前にハロゲン化水素を通過させる。その後、得られた式III:
【化7】
【0019】
Hal=ハロゲン
で表される3-ハロ-1-(チエン-2-イル)-プロパン-1-オンのケト基を還元し、式IV:
【化8】
【0020】
で表される還元生成物をメチルアミンと反応させる。
【0021】
かくして、本発明は、式I:
【化9】
【0022】
で表される3-メチルアミノ-1-(チエン-2-イル)プロパン-1-オールの調製方法に関し、この方法では、
a)チオフェンを、ルイス酸の存在下で、ハロゲン化β-ハロプロピオニルまたはハロゲン化アクリロイルと反応させ、その際、同時に、または反応が生じた後であるが反応生成物を単離する前にハロゲン化水素を通過させて、3-ハロ-1-(チエン-2-イル)プロパン-1-オンを得、そして
b)工程a)で得られたプロパノンを還元し、次いで、適宜に反応生成物を単離することなく、メチルアミンと反応させる。
【0023】
本発明による方法は、全体として良い回収率で目的化合物Iを提供する。さらに、このハロプロパノン中間体IIIの調製は、高価な有機スズ試薬の使用を必要としない。取り扱いが困難なハロプロパノールIVを単離する必要もない。さらに、この方法は、(S)-3-ハロ-1-(チエン-2-イル)プロパン-1-オールI-Sの形成に関して選択性を示すキラル触媒またはキラル還元剤の存在下にて、ハロプロペノン中間体IIIを還元することによる簡便な方法で、エナンチオ選択的にSエナンチオマーI-Sを調製することを可能とする。
【図面の簡単な説明】
【0024】
【図1】本発明により単離されたLu10288デヒドロゲナーゼの活性染色ゲルを示す;レーン1:分子量標準、下から:47kDa、74kDa、121kDaおよび205kDa;レーン2:空き;レーン3:ホモジェネート上清;レーン4:有用なQセファロースフラクション;レーン5:有用なQセファロースフラクション(3倍量);レーン6:有用なSuperdexフラクション;レーン7:有用なMono-Qフラクション;レーン8:有用なMono-Pフラクション。
【図2】図2Aおよび2Bは、ラクトバチラス・ブレビス(Lactobacillus brevis)デヒドロゲナーゼを用いて還元することによって(S)-3-メチルアミノ-1-(チエン-2-イル)プロパン-1-オールI-Sを調製するために用いた異なる混合物の典型的な反応経過を示す。
【図3A】本発明により単離されたLu10288デヒドロゲナーゼのブロットバンドのN末端配列決定の結果を示す。
【図3B−1】本発明により精製されたLu10288デヒドロゲナーゼの異なるタンパク質分解断片の配列決定データを示す;配列決定ステップにつき複数のシグナルが得られた場合に、主要な配列を確立することができる限り、これは二重下線によって示される。読み取ることができなかった位置は「/」で示される;同定がやや不確かである位置については、そのアミノ酸残基が丸かっこで示される。いずれが明確に優先されるのか特定することができない場合には、1つの位置に2つのアミノ酸が示される。「?」は不明または同定不能なアミノ酸を示す。
【図3B−2】図3B−1の続き。
【図3B−3】図3B−2の続き。
【図3B−4】図3B−3の続き。
【図3B−5】図3B−4の続き。
【図3B−6】図3B−5の続き。
【発明を実施するための形態】
【0025】
本発明において、「エナンチオ選択性」とは、以下の式:
ee(%)=[Sエナンチオマー−Rエナンチオマー/(Sエナンチオマー+Rエナンチオマー)]×100
に従って、公知の方式で算出されるSエナンチオマーのエナンチオマー過剰率ee(%)が、少なくとも50%、好ましくは少なくとも80%、特に少なくとも90%、とりわけ少なくとも95%であることを意味する。
【0026】
アシル化の間、または好ましくはアシル化が起こった後であるが反応生成物を単離する前に、ハロゲン化水素を通過させることの結果として、実質的に少しの1-チエン-2-イル-プロペノンII(この化合物は先行技術の方法では副生成物として常に生成し、目的のアシル化生成物IIIの収率を減少させる)も、工程a)の最終生成物として得られない。
【0027】
1つの電子対空位を有する共有結合の金属ハロゲン化物および半金属ハロゲン化物は、ルイス酸として使用することに適している。一般に、これらは、チタン、スズ、アルミニウム、バナジウム、鉄またはホウ素のハロゲン化合物から選択される。塩化物が好ましいが、アルミニウムの場合には、二塩化モノアルキルアルミニウムおよび塩化ジアルキルアルミニウムも好適である。ホウ素の場合には、フッ化物もまた好ましい。好適なルイス酸の例は、四塩化チタン、三塩化ホウ素、三フッ化ホウ素、四塩化スズ、五塩化バナジウム、三塩化鉄、三塩化アルミニウム、二塩化アルキルアルミニウムおよび塩化ジアルキルアルミニウムである。三塩化アルミニウムを用いるのが特に好ましい。
【0028】
好適なハロゲン化β-ハロプロピオニルは、塩化3-クロロプロピオニルおよび臭化3-ブロモプロピオニルまたは塩化3-ブロモプロピオニルである。塩化3-クロロプロピオニルを用いるのが好ましい。
【0029】
ハロゲン化アクリロイルとしては、塩化アクリロイルを用いるのが好ましい。
【0030】
ハロゲン化β-ハロプロピオニルは、アシル化剤として工程a)に用いるのが好ましい。
【0031】
塩化水素および臭化水素が好適なハロゲン化水素である。ハロゲン化水素を使用するにあたり、そのハロゲン原子が用いるハロゲン化β-ハロプロピオニルのβ-ハロゲンラジカルに一致しているものが好ましい。従って、塩化3-クロロプロピオニルを用いる場合には、塩化水素を用いることが好ましい。
【0032】
Friedel-Craftsアシル化反応に通常用いられる溶媒はどれも、工程a)の反応で溶媒として用いるのに適している。原則的に、好適な溶媒は、用いる試薬(特にルイス酸)の反応性を低下させないか、または所定の反応条件下でルイス酸との競合反応に加わらない非プロトン性溶媒である。これら溶媒の例は、それ自体がアシル化される芳香族化合物(すなわち、チオフェン)、チオフェンよりもアシル化することが著しく困難な芳香族炭化水素、例えば、ベンゼン、ニトロベンゼンおよびハロゲン化芳香族炭化水素(例:クロロベンゼンおよびジクロロベンゼン)、さらに、ハロゲン化脂肪族炭化水素、特にハロアルカン、例えば、クロロメタン、ジクロロメタン、クロロホルム、四塩化炭素、クロロエタン、ジクロロエタンおよびトリクロロエタンである。上記溶媒の混合物もまた好適である。ニトロベンゼンまたはハロゲン化炭化水素のような溶媒を使用することも好ましく、この場合にはFriedel-Craftsアシル化を本質的に均一に進行させることができる。特に、ハロゲン化芳香族または脂肪族炭化水素を用いるのが好ましく、ハロアルカンが特に好ましいものである。とりわけジクロロエタンまたはクロロベンゼンが用いられる。
【0033】
ルイス酸は、理論上得られるアシル化チオフェンの量に基づいて少なくとも等モル量で用いなければならない。なぜなら、ルイス酸がケトンとの複合体を形成し、この複合体はアシル化の間に形成されて、Friedel-Crafts反応の慣用条件下で安定であるからである。ルイス酸は好ましくは、より小さな割合で用いられるアシル化成分(チオフェンまたはハロゲン化β-ハロプロピオニル) 1モルに対して、1:1〜1:2、好ましくは1:1〜1:1.5、特に1:1.1〜1:1.5のモル比率で用いられる。
【0034】
チオフェンおよびハロゲン化β-ハロプロピオニルは、好ましくは1:0.5〜1:2、好ましくは1:0.7〜1:1.5、特に1:0.8〜1:1.2のモル比率で用いられる。
【0035】
Friedel-Craftsアシル化に関連して一緒に加えられる試薬の順序は、二番目に重要である。従って、例えば、初めにルイス酸を溶媒に導入し、最初にハロゲン化β-ハロプロピオニルを、次いでチオフェンを加えることができる。あるいはまた、初めにチオフェンとルイス酸を導入し、それらにハロゲン化β-ハロプロピオニルを加えることも可能である。
【0036】
一般に、ハロゲン化β-ハロプロピオニルを加える場合には、冷却することが有利である。それはルイス酸と酸ハロゲン化物との反応が一般に、強く発熱するためである。反応温度は、特に溶媒に応じて、選択される。一般には、10℃〜40℃の範囲である。ハロ炭化水素、特にハロアルカンを用いる場合には、反応温度を最高でも50℃とすることが好適である。それは、そうしなければこの溶媒自体が反応するためである。反応温度は、好ましくは-20℃〜40℃、特に好ましくは0〜30℃である。
【0037】
原則的に、反応圧は限定的ではない。一般的に、反応は標準圧力下で行うが、しかし、また正の圧力または負の圧力下でも行うことができる。例えば、溶媒が非常に揮発性であるか、または標準条件下で液体でない場合、例えばクロロメタンの場合によくあることだが、正の圧力が用いられる。
【0038】
ハロゲン化水素は好ましくは、アシル化が起こったごに通過させる。先行技術の慣用方法(例えば、ガスクロマトグラフィー、薄層クロマトグラフィーまたはNMR分光法)は、アシル化反応の完了を達成するために用いることができる。ハロゲン化水素の通過にかける時間は、特に、バッチの大きさによって決まり、個々の場合において当業者が決定できる。一般に、ハロゲン化水素を、少なくとも、わずかな1-チエン-2-イルプロペノンも検出することができなくなるまで通過させる。
【0039】
アシル化生成物の後処理のために、反応混合物は一般に、形成されたケトン-ルイス酸複合体を分解するために、初めに加水分解によって処理する。水または希薄な鉱酸水溶液(例えば、希塩酸)を加水分解に用いる。さらなる精製および単離は、例えば、Organikum [有機化学の実際的な手段], VEB Deutscher Verlag der Wissenschaften, 第17版, 1988, 第323頁以降に記載されているような、公知の方法を用いて行う。
【0040】
本発明による方法は、工程a)において、高い収率で3-ハロ-(チエン-2-イル)-1-プロパノンIIIを生じる。アシル化反応の過程で形成されうる1-チエン-2-イルプロペノンIIは実質的に完全に3-ハロ-(チエン-2-イル)-1-プロパノンIIIに変換され、その結果反応混合物は、3-ハロ-(チエン-2-イル)-1-プロパノンIIIの収量に基づいて、せいぜい1モル%、特に好ましくは、せいぜい0.5モル%のプロペノンIIしか含まないようになる。
【0041】
工程b)におけるプロパノンIIIの還元は、例えば、金属水素化物もしくは半金属水素化物を用いるか、または好適な遷移金属触媒の存在下で水素を用いることによって、達成される。
【0042】
好適な金属水素化物または半金属水素化物はいずれも中性で、それぞれ金属および半金属の複合水素化物である。ケトンのアルコールへの還元に有用であることが判明している金属水素化物または半金属水素化物を用いると有利である。上記水素化物は好ましくは、ホウ素またはアルミニウムの水素化物である。
【0043】
好適な中性の水素化物の例は、ボラン(BH3;B2H6)、アラン(AlH3)、シラン(SiH4)、モノボランおよびジアルキルボラン、例えば、ビス(3-メチルブタ-2-イル)ボラン(ジシアミルボラン)または9-ボラビシクロ[3.3.1]ノナン(9-BBN)、モノアルキルアルミニウムおよびジアルキルアルミニウム化合物、例えば、ジイソブチルアルミニウム水素化物(DIBAL-H)、ならびにトリアルキルシラン、例えば、トリメチルシランまたはトリエチルシランである。
【0044】
好適な複合水素化物の例は、複合ホウ素水素化物、例えば、水素化ホウ素ナトリウム(NaBH4)、水素化ホウ素リチウム(LiBH4)もしくは水素化ホウ素カルシウム(Ca[BH4]2)、複合アルミニウム水素化物、例えば、水素化リチウムアルミニウム(LiAlH4)、複合アルキルホウ素水素化物もしくはアルコキシホウ素水素化物、例えば、水素化トリエチルホウ素リチウムもしくは水素化トリイソプロポキシホウ素カリウム(KB[OCH(CH3)2]3H)、または複合アルキルアルミニウム水素化物もしくはアルコキシアルミニウム水素化物、例えば、水素化ジエチルアルミニウムナトリウム、水素化ビス(2-メトキシエトキシ)アルミニウムリチウム(LiAl[OC2H4OCH3]2H2)、水素化ビス(2-メトキシエトキシ)アルミニウムナトリウム(NaAl[OC2H4OCH3]2H2;「red Al」)もしくは水素化トリス(tert-ブトキシ)アルミニウムリチウム(LiAl[OC(CH3)3]3H)などである。
【0045】
金属水素化物または半金属水素化物を用いた還元に好適な溶媒は、特に、用いる還元剤に左右されるが、反応条件下で還元される基をもともと含むべきではない。従って、上記の水素化物を用いた還元は、好ましくは、所定の反応条件下で還元され得る官能基を有しない非プロトン性溶媒中で行う。これらの溶媒の例は、芳香族ならびに脂肪族炭化水素、例えば、C5-C8-アルカンおよびC5-C8-シクロアルカン(例:ペンタン、ヘキサン、ヘプタン、シクロペンタン、シクロヘキサンおよびシクロオクタン)、芳香族化合物(例:ベンゼン、トルエン、ニトロベンゼン、クロロベンゼンおよびジクロロベンゼン)、さらに、4個〜8個の炭素原子を有する開放鎖および環状のエーテル(例:ジエチルエーテル、メチルtert-ブチルエーテル、テトラヒドロフランまたはジオキサン)、さらにまた、塩素化炭化水素、特にハロアルカン(例:クロロメタン、ジクロロメタン、クロロホルム、四塩化炭素、ジクロロエタンまたはトリクロロエタン)である。上記の溶媒の混合物も好適である。上記の芳香族炭化水素、エーテルまたはハロ炭化水素を用いることが好ましい。
【0046】
上記のいくつかの複合水素化物、特に反応性の低い水素化物(例えば、水素化ホウ素ナトリウム)を用いた還元はまた、プロトン性溶媒(C1-C3-アルコールなど、例えば、メタノール、エタノール、プロパノールもしくはイソプロパノール)の存在下で、または水溶液中でさえも行うことができる。この場合に、少なくとも1種の上記の非プロトン性溶媒と少なくとも1種のアルコールとからなる混合溶媒を用いることが好ましい。水素化物は、塩基性プロトン性溶液中でより安定であるために、プロトン性溶媒を用いる場合には、好適な塩基の存在下で反応を行うことが好ましい。好適な塩基の例は、水酸化アルカリ金属、例えば、水酸化ナトリウムもしくは水酸化カリウム、または炭酸アルカリ金属、例えば、炭酸ナトリウムもしくは炭酸カリウムである。水酸化ナトリウムを用いることが好ましい。
【0047】
多数の上記の金属水素化物または半金属水素化物を用いた場合に、還元はしばしば発熱を伴って進行するので、結果として反応熱を取り除きながら、すなわち冷却しながら、反応を行うことが好適である。この反応温度は、好ましくは-50〜40℃、特に好ましくは-30〜30℃、特に-20〜20℃である。この還元は、例えば、Organikum [有機化学の実際的な手段], VEB Deutscher Verlag der Wissenschaften, 第17版, 1988, 第492頁以降に記載されているような、先行技術の慣用方法を用いて実施できる。これらの方法においては、概してプロパノンIIIを初めに導入し、そして還元剤を少しずつ加える。しかし、プロパノンIIIと還元剤を少しずつ同時に加えることもできる。
【0048】
プロパノンIIIはまた、例えば、アルミニウムアルコキシド用いて還元することもできる。アルミニウムアルコキシドを用いたケトンの還元は、慣習的にメーヤワイン・ポンドルフ・ヴァーレイ還元とも称される。この還元において、ケトンは対応するアルコールに変換され、そしてアルコキシドは対応するアルデヒド(第一級アルコールから形成されたアルコキシドの場合)またはケトン(第二級アルコールから形成されたアルコキシドの場合)に同時に酸化される。この反応は、例えば、Organikum [有機化学の実際的な手段], VEB Deutscher Verlag der Wissenschaften, 第17版, 1988, 第486頁以降に記載されているような、公知の方法を用いて実施できる。
【0049】
さらに、プロパノンIIIはまた、接触水素化によって、すなわち、好適な遷移金属触媒の存在下にて、水素とプロパノンIIIとを反応させることによっても還元できる。この好適な遷移金属には、VIII族、VI族およびI族の金属、特に白金、ルテニウム、銅、クロムおよびニッケルが含まれる。この触媒は当然のことながら、これらがチオフェン基の水素化を触媒しないように選択すべきである。触媒は不均一系触媒または均一系触媒のいずれかとして用いられる。好適な方法は、原則的に公知であり、例えば、Transition Metals in Organic Synthesis. M. Beller, C. Bolm, Wiley-VCH, Weinheim, 1998, 第2巻, 第1頁以降(均一系触媒)または第81頁以降(不均一系触媒)に記載されている。
【0050】
工程b)における還元は、好ましくは、金属水素化物または半金属水素化物を用いて、特に好ましくは上記の複合金属水素化物または半金属水素化物の1種を用いて行われる。特に水素化ホウ素ナトリウムが用いられる。
【0051】
上記の非不斉還元剤を用いる場合には、プロキラルな3-ハロ-1-(チエン-2-イル)プロパン-1-オン IIIが主に、ラセミ体アルコール IVに還元される。これに対応して、メチルアミンを用いた後続の反応は、ラセミ体の3-メチルアミノ-1-(チエン-2-イル)プロパン-1-オールIをもたらす。
【0052】
好ましい実施形態においては、本発明による方法を用いて、式I-S:
【化10】
【0053】
で表される(S)-3-メチルアミノ-1-(チエン-2-イル)プロパン-1-オールを調製するか、またはこの化合物と共にそのRエナンチオマーI-Rを含む混合物(エナンチオマーI-Sが優勢である)を調製する。工程b)の還元は、(S)-3-メチルアミノ-1-(チエン-2-イル)プロパン-1-オールI-Sの形成に関して選択性を示すキラル還元剤またはキラル触媒の存在下で行われる。
【0054】
この方法を行う場合に、プロキラルなハロプロパノンIIIは、工程b)において、式IV-S:
【化11】
【0055】
で表されるS-ハロプロパノールまたはSおよびRエナンチオマーの混合物(Sエナンチオマーが優勢である)にエナンチオ選択的に還元される。この目的のために、不斉金属水素化物または不斉半金属水素化物を、例えば、工程b)の還元剤として用いるか、そうでなければこの還元を、不斉誘導を媒介する化合物の存在下で行う。
【0056】
不斉金属水素化物または不斉半金属水素化物は好ましくは、不斉アルミニウム水素化物、不斉ホウ素水素化物または不斉ケイ素水素化物である。好適な不斉ホウ素水素化物は、例えば、E. J. Corey, C. J. Helal, Angew. Chem. 1998, 110, 2092-2118またはM. M. Midland, L. A. Morell, Methoden Org. Chem. [有機化学の方法], Houben-Weyl, 第4版, volume E 21d, pp. 4082-4098, 1995(これらはその全体が本明細書中に参考として援用される)に記載されている。好適な不斉ケイ素水素化物は、特に、H. Brunner, Methoden Org. Chem. [有機化学の方法], Houben-Weyl, 第4版, volume E21d, pp. 4074-4081, 1995(これはその全体が本明細書中に参考として援用される)に記載されている。
【0057】
本発明において、不斉誘導を媒介する化合物とは、実際の還元剤に影響を及ぼすことによって(例えば、結合を形成するか、もしくは還元剤と複合体を形成することによって)、または水素原子もしくは別の還元構成成分を還元剤から取ることによって、プロパノンIIIの還元をエナンチオ選択的にする化合物として理解される。一般に、不斉誘導を媒介する化合物は、それら自体還元性ではない。
【0058】
一方で、これらの化合物は、不斉水素化触媒を含む。不斉水素化において、水素は、不斉触媒が生じうるエナンチオマーの1つのエナンチオ選択的な形成を促す間に、実際の還元剤として作用する。水素化は不均一相または均一相のいずれかで行うことができる。不均一相での不斉水素化の好適な触媒は、例えば、A. Baiker, H. U. BlaserらによってHandbook of Heterogeneous Catalysis, 第5巻(G. Ertl, H. KnoerzingerおよびJ.Weitkamp編集), Wiley-VCH, Weinheim, 1997, 2422-2436(これはその全体が本明細書中に参考として援用される)に記載されている。
【0059】
しかし、均一相での水素化の触媒は、非常に重要である。VIII族金属(例えば、白金またはルテニウム、特にルテニウム)と少なくとも1つのリン含有配位子とを含む触媒が、特に好適である。好適な触媒は、例えば、R. Noyori, T. Ohkuma, Angew. Chem. 2001, 113, 40-75(これはその全体が本明細書中に参考として援用される)に記載されている。ルテニウム(II)ジアミン複合体(ルテニウムが二座配位キラルジホスファン配位子にさらに結合している)が特に好適である。ハロプロパノンIIIをS-ハロプロパノール IV-Sに還元するのに好適であるジホスファン配位子の例は、以下の式Aの(R)-BINAP、式Bの(R,R)-DIOPおよび式Cの(R,R)-CHIRAPHOSである:
【化12】
【0060】
Ar=フェニル(BINAP) Ph=フェニル
4-メチルフェニル(TolBINAP)
3,5-ジメチルフェニル(XylBINAP)
【0061】
さらに、特に好ましい触媒は、ジアミン配位子を含む。好適なジアミン配位子の例は、それらのエナンチオマー形態における1,2-エチレンジアミン、1,2-ジフェニル-1,2-エチレンジアミンおよび1,2-シクロヘキサンジアミンである。
【0062】
概して、水素化は、上記の条件下にて実施される。
【0063】
以下の式D:
【化13】
【0064】
(式中、RはC1-C4-アルキル、例えば、メチル、エチル、プロピル、イソプロピル、n-ブチル、sec-ブチル、イソブチルまたはtert-ブチルであり、特にメチルである)で表されるオキサザボリリジン(oxazaborylidine)は、特に好ましい不斉誘導媒介化合物である。
【0065】
この試薬を用いて、高度のエナンチオ選択性でプロキラルケトンを第二級アルコールに変換することができる(E. J. Corey, Pure Appl. Chem. 62, 1209, 1990;E. J. Corey, J. O. Link, J. Am. Chem. Soc. 114, 1906, 1992)。ボラン(例えば、BH3、ジアルキルボラン、ジアルコキシボランまたはジアリールオキシボラン)が実際の還元剤として用いられる。クロロプロパノンIII.1とオキサザボリリジンD(R=メチル)およびBH3とを反応させてS-クロロプロパノール IV.1-Sを得る反応は、W. J. WheelerおよびF. KuoらによってJournal of Labelled Compounds and Radiopharmaceuticals, volume XXXVI, No. 3, pp. 213-223に既に記載されている。
【0066】
還元において、概して、オキサザボリリジンは触媒量で用いられる。ボランは、ハロプロパノンIIIに基づいて、少なくとも等モル量で用いられるが、過剰量で用いることが好ましい。この反応は一般に、好適な溶媒中で実施する。好適な溶媒とは、所定の反応条件下にて還元されうる基を有しない非プロトン性溶媒である。これらの溶媒には、特に上記のハロアルカン、芳香族化合物および環状または非環状エーテルが含まれる。エーテルを用いるのが好ましく、テトラヒドロフランを用いるのが特に好ましい。反応温度は、好ましくは-80〜20℃、特に好ましくは-30〜10℃である。この反応は概して、初めにオキサザボリリジンを溶媒に導入し、最初にボラン、次いでハロプロパノンIII、またはこれとは逆に、最初にハロプロパノンIII、次いでボランを所望の反応温度で加えることによって行われる。ただし、前者の添加手順が好ましい。反応の継続時間は、特に、バッチの大きさによって決まり、個々の場合において当業者によって決定されうる。
【0067】
意外にも、プロパノンIIIからS-ハロプロパノール IV-Sへのエナンチオ選択的な還元は、酵素によって触媒される場合によく進行し、特にデヒドロゲナーゼが存在する場合に都合よく行われることが見出された。従って、本発明による方法の別の好ましい実施形態では、デヒドロゲナーゼが還元剤として用いられる。
【0068】
本発明による方法で還元が完了した後、用いた還元方法に応じて、触媒または過剰量の還元剤を、適宜に、不活性化させるか、または除去する。金属水素化物または半金属水素化物を用いた場合には、この処理は一般的に加水分解(例えば、水性またはアルコール性溶液を用いる)によって行う。均一な水素化触媒を用いた場合、またはアルミニウムアルコキシドで還元した場合には、加水分解後処理もしばしば用いられる。還元が不均一相での接触水素化の形態で行われた場合、または還元剤として酵素を用いて行われた場合には、触媒または酵素を物理的な分離(例えば、デカントまたは濾過)によって取り除くことができる。しかし、均一還元系が用いられた場合、特に、金属水素化物または半金属水素化物を用いて還元を行った場合には、初めに不活性化しないことが好ましい。
【0069】
還元後に得られたハロプロパノール IV を単離することなく、その代わりにメチルアミンと直接的に反応させて、3-メチルアミノ-1-(チエン-2-イル)プロパン-1-オールIを得ることが有利であることが分かった。このために、反応混合物(適宜に不活性化してもよいし、不均一還元系から分離してもよい)をメチルアミンで処理し、好ましくは、昇温(例えば、30〜80℃、特に50〜70℃)にて反応させる。メチルアミンは気体または水溶液のいずれかとして用いることができるが、水溶液としての使用が好ましい。メチルアミンは、用いるハロプロパノンIIIの量に基づいて、好ましくは1〜100モル当量、特に好ましくは5〜10モル当量で用いる。この反応は1〜250バールの圧力下、特に系自体が発生する固有の圧力下にて行うことが好ましい。
【0070】
メチルアミンとの反応が完了した後、反応混合物を慣用方法で後処理する。そのために、触媒または還元剤を既に述べたように不活性化して分離し(このような処理がメチルアミンを加える前に行われなかった場合)、その後溶媒を除去し、純粋なメチルアミノプロパノールIを、例えば、結晶化、温浸、抽出またはクロマトグラフィーによって残渣から単離する。
【0071】
本発明による方法を用いて、良い収率で3-メチルアミノ-1-(チエン-2-イル)プロパン-1-オールIを得ることが可能である。特に、高価で、取り扱いが困難な試薬または溶媒を本方法の工程a)において必要とせず、またハロプロパノンIIIが非常に高い収率で生成される。工程b)では、不安定なハロプロパノール IVの単離、ならびにそれに伴う収率の低下および/またはハロプロパノール IVの困難な取り扱いが、回避される。さらに、本発明による方法を用いて、メチルアミノプロパノール I-SのSエナンチオマーを選択的に得ることができる。このSエナンチオマーは、工程b)における還元がキラル還元剤を用いて行われる場合に、デュロキセチンへのさらなる変換のために必須である。
【0072】
さらに、本発明はまた、3-ハロ-1-(チエン-2-イル)プロパン-1-オンIIIをエナンチオ選択的に還元することを含む、式I-Sの(S)-3-メチルアミノ-1-(チエン-2-イル)プロパン-1-オールの調製方法に関し、前記方法では、この還元をデヒドロゲナーゼの存在下にて行うことを含む。この方法の好ましい実施形態では、これに関連して得られる(S)-3-ハロ-1-(チエン-2-イル)プロパン-1-オールIV-Sを、単離することなく、メチルアミンと反応させる。好ましいデヒドロゲナーゼに関して、さらにこの方法の実施に関しては、以下の観察を特に参照されたい。
【0073】
生化学的な実施形態:
好適なデヒドロゲナーゼ(EC 1.1.X.X)は、特に、デヒドロゲナーゼ(E.C. 1.1.1.x)、特にアルコールデヒドロゲナーゼ(E.C.1.1.1.1またはE.C.1.1.1.2)である。これらは、ハロプロパノンIIIのS-ハロプロパノール IV-Sへの選択的な還元をもたらす。このデヒドロゲナーゼは好ましくは、微生物、特に好ましくは細菌または真菌(特に酵母)から得られる。これは、それぞれ菌株コレクションに寄託されたものか、または土壌試料、バイオマス試料などの天然供給源の分離株から得られる。デヒドロゲナーゼは特に酵母または乳酸菌に由来するものである。
【0074】
デヒドロゲナーゼは、精製されたもしくは部分的に精製された形態、または微生物自体の形態で用いることができる。デヒドロゲナーゼを微生物から単離および精製するための方法は、当業者に周知である(例えば、K. DrauzおよびH. WaldmannのEnzyme Catalysis in Organic Synthesis 2002, Vol.III, 991-1032, Wiley-VCH, WeinheimにおけるK. Nakamura & T. Matsudaらによる「Reduction of Ketons」)。デヒドロゲナーゼを産生させる組換え法も同じように知られている(例えば、W. Hummel, K. Abokitse, K. Drauz, C. Rollmannおよび H. Groegerらによる Adv. Synth. Catal. 2003, 345, Nos. 1+2, pp. 153-159)。
【0075】
好適な細菌の例は、シュードモナス(Pseudomonas)属、ブルクホリデリア(Burkholderia)属、アグロバクテリウム(Agrobacterium)属、ロドコッカス(Rhodococcus)属、ラクトバチラス(Lactobacillus)属、またはラクトコッカス(Lactococcus)属のものである。好適な酵母の例は、ゲオトリカム(Geotrichum)属、ピチア(Pichia)属、カンジダ(Candida)属、ハンセヌラ(Hansenula)属、またはサッカロマイセス(Saccharomyces)属のものである。
【0076】
酵母および乳酸菌に由来するデヒドロゲナーゼを用いることが特に好ましい。これらのデヒドロゲナーゼのうち、ゲオトリカム(Geotrichum)属もしくはカンジダ(Candida)属の酵母から得られるか、またはラクトバチラス(Lactobacillus)属、もしくはラクトコッカス(Lactococcus)属の乳酸菌から得られるデヒドロゲナーゼが好ましい。
【0077】
ラクトバチラス(Lactobacillus)属の菌種の例は、L.ブレビス(brevis)、L.ケフィア(kefir)、L.プランタルム(plantarum)、L.カセイ(casei)、L.アシドフィルス(acidophilus)、L.デルブリュッキィ(delbrueckii)およびL.サンフランシスコ(sanfrancisco)である。
【0078】
カンジダ(Candida)属の菌種の例は、C.マグノリア(magnoliae)、C.ルゴサ(rugosa)、C.ユチリス(utilis)、C.ボイジニィ(boidinii)、C.パラプシローシス(parapsilosis)およびC.アンタルティカ(antarctica)である。
【0079】
ゲオトリカム(Geotrichum)属の菌種の例は、G.カンディダム(candidum)、G.クラバタム(clavatum)およびG.フェルメンタンス(fermentans)である。
【0080】
カンジダ・マグノリア(Candida magnoliae)、ゲオトリカム・カンジドウム(Geotrichum candidum)またはラクトバチラス・ブレビス(Lactobacillus brevis)に由来するデヒドロゲナーゼを用いることが特に好ましい。
【0081】
デヒドロゲナーゼによる還元は一般に、好適な補酵素(補因子とも称される)の存在下にて行われる。NADHおよび/またはNADPHは一般に、ケトンを還元するための補酵素として用いられる。さらに、デヒドロゲナーゼを細胞系(もともと補因子を含むか、細胞系に選択的レドックス媒介物が加えられている)として用いることが可能である(K. DrauzおよびH. WaldmannらのEnzyme Catalysis Organic Synthesis 2002, Vol.III, 991-1032, Wiley-VCH, Weinheimにおける、A. Schmidt, F. Hollmann および B. Buehlerらの「Oxidation of Alcohols」)。
【0082】
デヒドロゲナーゼによる還元はまた、一般に好適な補基質の存在下にて行われる。この補基質は概して、還元過程の間に酸化される補酵素の還元剤として作用し、その結果この補酵素を再生させる。好適な補基質の例は、糖、特にヘキソース、例えば、グルコース、マンノースまたはフルクトース、および/または被酸化性アルコール、特にエタノール、プロパノール、またはイソプロパノールであり、またギ酸もそうである。第二のデヒドロゲナーゼ(例えば、グルコースを補基質として用いる場合には、グルコースデヒドロゲナーゼ)が補基質を酸化する目的で、またこれに関連して補酵素を再生させるために加えられる。このデヒドロゲナーゼは、遊離もしくは固定化酵素として、または遊離もしくは固定化細胞の形態で用いることができる。それは、(組換え)デヒドロゲナーゼ株において別々にまたは共発現によって調製することができる。
【0083】
本発明により用いられるデヒドロゲナーゼは、遊離または固定化形態で用いることができる。固定化酵素とは、不活性な支持体に固定された酵素を意味するものとして理解される。好適な支持体材料およびそれらに固定される酵素は、EP-A-1149849、EP-A-1069183およびDE-OS 100193773、またそれらに引用されている参考文献から公知である。これに関して、これら文献における開示内容は、その全体が本明細書中に参考として援用される。好適な支持体材料としては、例えば、粘土、粘土鉱物(例:カオリナイト、ケイ藻土)、パーライト、二酸化ケイ素、酸化アルミニウム、炭酸ナトリウム、炭酸カルシウム、セルロース粉末、アニオン交換材料、合成ポリマー(例:ポリスチレン、アクリル樹脂、フェノールホルムアルデヒド樹脂、ポリウレタン、およびポリエチレンやポリプロピレンなどのポリオレフィン)が挙げられる。この支持体材料は支持体に担持された酵素を調製するために用いられ、一般に微細な粒子状をしており、多孔形態が好ましい。支持体材料の粒径は一般に、5mmを超えず、特に2mmを超えない(粒度曲線)。類似の方法において、全細胞体触媒としてデヒドロゲナーゼを用いる場合には、遊離形態または固定化形態を選択することが可能である。支持体材料の例は、アルギン酸Caおよびカラギーナンである。酵素および細胞はグルタルアルデヒドで直接的に架橋することもできる(CLEAを得るための架橋)。
【0084】
対応するおよびさらなる固定化方法は、例えば、K. DrauzおよびH. WaldmannらのEnzyme Catalysis in Organic Synthesis 2002, Vol.III, 991-1032, Wiley-VCH, Weinheimにおける、J. LalondeおよびA. Margolinらによる「Immobilization of Enzymes」に記載されている。
【0085】
還元は、水性または非水性の反応媒体中または2相系もしくは(マイクロ)エマルションにて行うことができる。水性反応媒体は、好ましくは緩衝溶液であり、概して、pH5〜8、好ましくはpH6〜8である。水以外に、水性溶媒は少なくとも1つのアルコール(例えば、エタノールまたはイソプロパノール)をさらに含むことができる。
【0086】
「非水性反応媒体」とは、反応媒体の全重量に基づいて、1重量%未満、好ましくは0.5重量%未満の水を含む反応媒体を意味するものとして理解される。この反応は好ましくは、有機溶媒中で行われる。好適な溶媒の例は、脂肪族炭化水素、好ましくは5個〜8個の炭素原子有する脂肪族炭化水素(例:ペンタン、シクロペンタン、ヘキサン、シクロヘキサン、ヘプタン、オクタンもしくはシクロオクタン)、ハロゲン化脂肪族炭化水素、好ましくは1個〜2個の炭素原子を有するハロゲン化脂肪族炭化水素(例:ジクロロメタン、クロロホルム、四塩化炭素、ジクロロエタンもしくはテトラクロロエタン)、芳香族炭化水素(例:ベンゼン、トルエン、キシレン、クロロベンゼンまたはジクロロベンゼン)、脂肪族非環状エーテルおよび脂肪族環状エーテル、好ましくは4個〜8個の炭素原子を有するもの(例:ジエチルエーテル、メチルtert-ブチルエーテル、エチルtert-ブチルエーテル、ジプロピルエーテル、ジイソプロピルエーテル、ジブチルエーテルもしくはテトラヒドロフラン)、またはエステル(例:酢酸エチルもしくは酢酸n-ブチル)、またはケトン(例:メチルイソブチルケトンもしくはジオキサン)、あるいはそれらの混合物である。上記のエーテル、特にテトラヒドロフランを用いることが特に好ましい。
【0087】
例えば、デヒドロゲナーゼによる還元は、水性-有機反応溶媒、特に水性反応溶媒中にて行われる。
【0088】
ハロプロパノンIIIは、好ましくは、酵素による還元において、0.1g/l〜500g/l、特に好ましくは、1g/l〜50g/lの濃度で用いられ、その後、連続的にまたは断続的に供給することができる。
【0089】
一般に、酵素による還元は、用いるデヒドロゲナーゼが不活性化される温度よりも低い反応温度、好ましくは少なくとも-10℃である反応温度で行われる。例えば0〜100℃、好ましくは15〜60℃、特に20〜40℃の範囲、例えば約30℃とすることが特に好ましい。
【0090】
反応を実施するために、例えば、初めに、ハロプロパノンIIIをデヒドロゲナーゼ、溶媒、適宜に補酵素、適宜に補酵素を再生するための補助デヒドロゲナーゼ、および/または他の補基質と一緒に導入し、その混合物を、例えば撹拌または振とうによって、混合することが可能である。しかし、また、反応器(例えば、カラム)にデヒドロゲナーゼを固定化し、ハロプロパノンIIIと適宜に補酵素および/または補基質を含有する混合物をこの反応器に通過させることも可能である。このために、目的の変換が達成されるまで、循環方式にて混合物を反応器に通すことが可能である。これに関連して、ハロプロパノンIIIのケト基はOH基に還元され、それに伴ってハロプロパノール IV-S のSエナンチオマーが本質的に選択的に形成される。一般に、還元は、混合物中に存在するハロプロパノンIIIに基づいて、変換率が少なくとも70%、特に好ましくは少なくとも85%、特に少なくとも95%に達するまで行われる。これに関連して、反応の進行(すなわち、ケトンの連続的な還元)はガスクロマトグラフィーのような慣用方法によって追跡することができる。
【0091】
本発明による方法に特に好ましく用いられるデヒドロゲナーゼは、以下の特性の少なくとも1つを有するアルコールデヒドロゲナーゼである:
N末端領域に、a) 配列番号1に示される、少なくとも5、6、7または8個、好ましくは少なくとも10個(例えば、10、11、12、13、14または15個)連続したアミノ酸残基の構成的なアミノ酸配列を含み、配列番号1に示されるアミノ酸位置12に対応する位置が、好ましくはさらにバリンを表す;またはb) 配列番号2に示される、少なくとも5、6、7または8個、好ましくは少なくとも10個(例えば、10、11、12、13、14または15個)連続したアミノ酸残基の構成的なアミノ酸配列を含む、アミノ酸配列を有するアルコールデヒドロゲナーゼおよびそれらの機能的等価物。
【0092】
3-クロロ-1-(チエン-2-イル)プロパン-1-オンを(S)-3-クロロ-1-(チエン-2-イル)プロパン-1-オールに還元することが可能である、アルコールデヒドロゲナーゼ。
【0093】
少なくとも85%eeのエナンチオマー純度にて(NADHおよび/またはNADPHの存在下;30℃かつpH6.0にて)還元を触媒する、アルコールデヒドロゲナーゼ。
【0094】
配列番号3を含む核酸配列によってコードされるか、または配列番号4に示されるアミノ酸配列もしくは少なくとも図3に示される構成的な配列を含み、好ましくは、ラクトバチラス・ブレビス(Lactobacillus brevis)から得ることができるアルコールデヒドロゲナーゼ、およびそれらに由来する機能的に等価なアルコールデヒドロゲナーゼ。
【0095】
配列番号5を含む核酸によってコードされるか、または配列番号6を含むアミノ酸配列を有し、好ましくは、カンジダ・マグノリア(Candida magnoliae)(ATCC 12573)から得ることができるアルコールデヒドロゲナーゼ、およびそれらに由来する機能的に等価であるアルコールデヒドロゲナーゼ。
【0096】
本発明はまた、上に列挙した特性の少なくとも1つを有する(S)-3-クロロ-1-(チエン-2-イル)-プロパン-1-オールデヒドロゲナーゼに関する。
【0097】
デヒドロゲナーゼが安定である温度範囲は、好ましくは10〜80℃である。その活性が最適である温度は、好ましくは20〜60℃、特に好ましくは25〜40℃の範囲である。安定であるpH範囲は好ましくはpH4〜10である。脱水素に最適なpHは好ましくはpH5〜8の範囲である。
【0098】
さらに、本発明によるデヒドロゲナーゼは好ましくは、NAD+またはNADP+の存在下にて、(S)-3-クロロ-1-(チエン-2-イル)プロパン-1-オールを脱水素して3-クロロ-1-(チエン-2-イル)プロパン-1-オンを生成することが可能なものである。デヒドロゲナーゼの分子量は30±2キロダルトンの範囲にあることが好ましい。
【0099】
本発明はまた、本発明によるデヒドロゲナーゼのコード配列または構成的なコード配列を含む核酸配列に関する。この非限定的な例は、配列番号3または配列番号6に示される配列である。
【0100】
本発明はさらに、少なくとも1つの調節核酸配列と機能的に連結された、本発明による核酸配列を含む発現カセットに関する。
【0101】
本発明はさらに、本発明による発現カセットを少なくとも1つ含む組換えベクターに関する。
【0102】
さらに、本発明は、本発明によるベクターを少なくとも1つ用いて形質転換した原核細胞または真核細胞の宿主に関する。
【0103】
最後に、本発明は、(S)-3-ハロ-1-(チエン-2-イル)プロパン-1-オールを調製するための、特にデュロキセチンを調製するための、本発明によるデヒドロゲナーゼまたはこのデヒドロゲナーゼを産生する微生物の使用に関する。
【0104】
本発明によるデヒドロゲナーゼのさらなる改変:
本発明はまた、(S)-3-クロロ-1-(チエン-2-イル)プロパン-1-オールデヒドロゲナーゼ活性を有する明確に開示された酵素の「機能的等価物」および本発明の方法におけるそれら等価物の使用を含む。
【0105】
本発明に関して、明確に開示された酵素の「機能的等価物」または類似体は、これらの酵素とは異なるが、依然として所望の生物学的活性(例えば、基質特異性)を保有するポリペプチドである。従って、「機能的等価物」とは、例えば、3-クロロ-1-(チエン-2-イル)プロパン-1-オンを対応するSアルコールに還元し、以下に列挙される配列番号1、2、4もしくは6または図3に示されるアミノ酸配列の1つを含む酵素の少なくとも20%、好ましくは50%、さらに好ましくは75%、特に好ましくは90%の活性を有する酵素を意味するものとして理解される。機能的等価物はまた、好ましくはpH4〜10の間で安定であり、有利には、最適pHをpH5〜8に、最適温度を20℃〜80℃の範囲に有する。
【0106】
本発明によると、「機能的等価物」とはまた、特に、上記のアミノ酸配列の少なくとも1つの配列位置に、特に挙げたアミノ酸以外のアミノ酸を有するが、それでも上記の生物学的活性の1つを保有する変異体を意味するものとしても理解される。したがって、「機能的等価物」とは、1個または複数のアミノ酸付加、アミノ酸置換、アミノ酸欠失および/またはアミノ酸転移によって得ることができる変異体を含む。かかる変化は、本発明に従う特性プロファイルを有する変異体をもたらす限り、どのような配列位置にも出現しうる。また、機能的等価物が存在するのは、特に、変異型および非変異型のポリペプチドの反応パターンが質的に一致する場合、すなわち、同一の基質が例えば異なる速度で変換される場合である。
【0107】
好適なアミノ酸置換の例を以下の表に列挙する:
【0108】
上記趣旨での「機能的等価物」とはまた、記載したポリペプチドの「前駆体」ならびにそのポリペプチドの「機能的誘導体」および「塩」でもある。
【0109】
これに関連して、「前駆体」とは、ポリペプチドの天然または合成の前駆体であり、この前駆体は所望の生物学的活性を保持しても保持しなくてもよい。
【0110】
語句「塩」とは、本発明によるタンパク質分子のカルボキシル基の塩とアミノ基の酸付加塩との両方を意味するものとして理解される。カルボキシル基の塩は、それ自体公知の方法で調製することができ、無機塩(例えば、ナトリウム、カルシウム、アンモニウム、鉄および亜鉛の塩)、ならびに有機塩基(例えば、トリエタノールアミン、アルギニン、リジン、ピペリジンなどのアミン)との塩を含む。酸付加塩、例えば鉱酸(例:塩酸または硫酸)との塩ならびに有機酸(例:酢酸およびシュウ酸)との塩などは、本発明の一部を構成する。
【0111】
本発明によるポリペプチドの「機能的誘導体」は同様に、公知の手法を用いて、機能的アミノ酸の側鎖基またはそれらのN末端もしくはC末端に、調製することができる。これらの誘導体は、例えば、カルボン酸基の脂肪族エステル;カルボン酸基のアミド(このアミドは、アンモニアまたは第一級もしくは第二級アミンと反応させることによって得ることができる);遊離アミノ基のNアシル誘導体(この誘導体は、アシル基と反応させることによって調製される);または、遊離ヒドロキシル基のO-アシル誘導体(この誘導体はアシル基と反応させることによって調製される)を含む。
【0112】
タンパク質のグリコシル化が起こると考えられる場合、本発明による「機能的等価物」とは、上記のタイプのタンパク質の脱グリコシル化またはグリコシル化形態、ならびにグリコシル化パターンを変えることによって得られる改変形態を含む。
【0113】
「機能的等価物」とは当然に、他の生物体および天然の変異体から得ることができるポリペプチドもまた含む。例えば、相同配列領域の部分は配列比較によって確立することができ、また同等の酵素は本発明の具体的なのガイドラインを用いて同定することができる。
【0114】
「機能的等価物」とはまた、本発明によるポリペプチドの断片(好ましくは、個々のドメインまたは配列モチーフ)も含み、かかる断片は、例えば、所望の生物学的機能を有する。
【0115】
さらに、「機能的等価物」とは、上記のポリペプチド配列またはそれらに由来する機能的等価物の1つと、それらと機能的に異なりかつN末端またはC末端で機能的に(すなわち、融合タンパク質成分の相互の機能が顕著に損なわれることなく)連結されている少なくとも1つのさらなる異種配列と、を含む融合タンパク質である。このような異種配列の非限定的な例は、シグナルぺプチドまたは酵素である。
【0116】
さらに本発明に含まれる「機能的等価物」とは、具体的に開示されたタンパク質の相同体である。これらの相同体は、Proc. Natl. Acad, Sci. (USA) 85(8), 1988, 2444-2448に記載されるPearsonおよびLipmanのアルゴリズムを用いて算出した場合に、具体的に開示されたアミノ酸配列の1つに対して少なくとも60%、好ましくは少なくとも75%、特に少なくとも85%、例えば、90%、95%、97%または99%の相同性を有する。本発明による相同ポリペプチドの相同性パーセントとは、特に、本明細書中に具体的に記載される1つのアミノ酸配列の全長にわたるアミノ酸残基の同一性パーセントを意味する。
【0117】
本発明によるタンパク質またはポリペプチドの相同体は、変異誘発によって(例えば、タンパク質の点突然変異またはトランケーションによって)作製することが可能である。
【0118】
本発明のタンパク質の相同体は、トランケーション変異体のような変異体のコンビナトリアル・ライブラリーのスクリーニングによって同定することができる。例えば、タンパク質変異体の多様なライブラリーは、核酸レベルでのコンビナトリアル変異誘発によって、例えば合成オリゴヌクレオチドの混合物を酵素的にライゲートすることによって、作製することができる。縮重オリゴヌクレオチド配列から潜在的相同体のライブラリーを調製するために用いることができる方法は多数存在する。縮重遺伝子配列を自動DNA合成装置で化学的に合成して、この合成遺伝子を好適な発現ベクターにライゲートすることができる。縮重遺伝子セットを用いると、所望の潜在的タンパク質配列のセットをコードするすべての配列を混合物として調製することが可能となる。縮重オリゴヌクレオチドを合成するための方法は、当業者に公知である(例えば、Narang, S.A.(1983)Tetrahedron 39:3;Itakuraら(1984)Annu. Rev. Biochem. 53:323;Itakuraら,(1984)Science198:1056;Ikeら(1983)Nucleic Acids Res. 11:477)。
【0119】
点突然変異またはトランケーションによって作製されたコンビナトリアル・ライブラリーの遺伝子産物をスクリーニングするための手法、および所定の性質を有する遺伝子産物についてcDNAライブラリーをスクリーニングするための手法は、先行技術において公知である。これらの手法は、本発明の相同体のコンビナトリアル変異誘発によって生じた遺伝子ライブラリーを迅速にスクリーニングするために適合させることができる。ハイスループット分析にかけられる大きな遺伝子ライブラリーをスクリーニングするために最も頻繁に用いられる手法は、遺伝子ライブラリーを複製可能な発現ベクターにクローニングし、得られたベクターライブラリーを用いて好適な細胞を形質転換し、所望の活性の検出が遺伝子(その産物が検出された)をコードするベクターの単離を容易にする条件下にて、コンビナトリアル遺伝子を発現させることを含む。リカーシブ・アンサンブル変異誘発(recursive ensemble mutagenesis)(REM)は、ライブラリー中の機能的変異体の頻度を高める手法であり、相同体を同定するためにスクリーニング試験と組み合わせて用いることができる(Arkin およびYourvan(1992)PNAS 89:7811-7815;Delgraveら(1993)Protein Engineering 6(3):327-331)。
【0120】
本発明によるコード核酸配列のさらなる実施形態:
本発明は、特に、本発明のデヒドロゲナーゼ活性を有する酵素をコードする核酸配列(一本鎖および二本鎖DNAおよびRNA配列、例えばcDNAおよびmRNA)に関する。例えば、配列番号1、2、4、6もしくは図3に示されるアミノ酸配列またはそれらの特徴的な構成配列をコードする核酸配列、あるいは配列番号3もしくは5に示される核酸配列またはそれらの特徴的な構成配列を含む核酸配列が好ましいものである。
【0121】
本明細書中で言及するすべての核酸配列は、それ自体公知の方法で、ヌクレオチド構成単位から化学合成によって、例えば、二重鎖の個々の重複した相補性核酸構成単位の断片縮合によって作製することができる。オリゴヌクレオチドは、例えば、公知の方法にて、ホスホアミダイト法を用いて化学的に合成できる(Voet, Voet, 第2版, Wiley Press New York, pages 896-897)。合成オリゴヌクレオチドの結合およびDNAポリメラーゼKlenow 断片によるギャップのフィルインならびにライゲーション反応、さらに一般的なクローニング方法は、Sambrookら(1989), Molecular Cloning:A laboratory manual, Cold Spring Harbor Laboratory Pressに記載されている。
本発明はまた、上記のポリペプチドおよびそれらの機能的等価物(例えば、人工的なヌクレオチド類似体を用いて入手可能)の1つをコードする核酸配列(一本鎖および二本鎖DNAおよびRNA配列、例えば、cDNAおよびmRNA)に関する。
【0122】
本発明は、本発明のポリペプチドもしくはタンパク質または生物学的に活性なそれらのセグメントをコードする単離された核酸分子および核酸断片の両方に関し、かかる核酸断片は、例えば本発明によるコード核酸を同定または増幅するためのハイブリダイゼーションプローブまたはプライマーとして用いることができる。
【0123】
本発明による核酸分子はさらに、コード遺伝子領域の3’末端および/または5’末端に由来する非翻訳配列を含むことができる。
【0124】
本発明はまた、具体的に記載されたヌクレオチド配列に相補的である核酸分子またはそれらのセグメントを含む。
【0125】
本発明のヌクレオチド配列は、別の細胞型および生物体に含まれる相同配列を同定および/またはクローニングするために用いることができるプローブおよびプライマーの作製を可能にする。これらのプローブまたはプライマーは一般に、「ストリンジェント」な条件(下記参照)下にて、本発明の核酸配列のセンス鎖または対応するアンチセンス鎖中の少なくとも約12個、好ましくは少なくとも約25個(例えば、約40個、50個または75個)連続したヌクレオチドとハイブリダイズするヌクレオチド配列領域を含む。
【0126】
「単離された」核酸分子とは、核酸の天然源に存在する他の核酸分子から分離されており、さらに、組換え技術によって調製された場合には、他の細胞性材料もしくは培養培地を実質的に含まず、また化学的に合成された場合には、化学的前駆体もしくは他の化学薬品を実質的に含まないものである。
【0127】
本発明による核酸分子は、標準的な分子生物学的手法および本発明により提供される配列情報を用いて単離することができる。例えば、cDNAは、具体的に開示された完全長配列またはそれらのセグメントをハイブリダイゼーションプローブとして用いて、例えば、Sambrook, J., Fritsch, E.F. および Maniatis, T. らによるMolecular Cloning:A Laboratory Manual. 第2版, Cold Spring Harbor Laboratory, Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY, 1989に記載されるような、標準的なハイブリダイゼーション手法を用いて、好適なcDNA ライブラリーから単離することができる。さらに、ポリメラーゼ連鎖反応を用いて、開示された配列またはそれらのセグメントの1つを含む核酸分子を、この配列に基づいて構築されたオリゴヌクレオチドプライマーを使用して、単離することができる。この方法で増幅した核酸は、好適なベクターにクローン化し、そしてDNA配列解析によって特徴づけることができる。本発明によるオリゴヌクレオチドはまた、標準的な合成方法によって、例えば、自動DNA合成装置を用いて調製できる。原則的に、本発明による核酸配列は任意の生物体において同定して、該生物体から単離することができる。有利には、本発明による核酸配列またはそれらの相同体は、真菌、酵母または細菌から単離できる。言及できる細菌はグラム陰性細菌およびグラム陽性細菌である。本発明による核酸は好ましくは、グラム陰性細菌、有利にはαプロテオ細菌、βプロテオ細菌またはγプロテオ細菌、好ましくはエンテロバクテリアセア(Enterobacteriaceae)科、シュードモナダセア(Pseudomonadaceae)科またはリゾビアセア(Rhizobiaceae)科の細菌、特に好ましくはアグロバクテリウム(Agrobacterium)属、シュードモナス(Pseudomonas)またはブルクホリデリア(Burkholderia)属の細菌から、当業者に公知の方法を用いて単離される。
【0128】
本発明による核酸配列は、例えばゲノムライブラリーまたはcDNAライブラリーを用いた、通常のハイブリダイゼーション法またはPCR手法により、例えば他の真菌または細菌から単離できる。これらのDNA配列は本発明による配列と標準条件下でハイブリダイズする。保存領域(例えば、活性中心)に由来する短いオリゴヌクレオチドをハイブリダイゼーションに用いることが有利であり、これらのオリゴヌクレオチドは、当業者に公知の方法で本発明によるデヒドロゲナーゼと比較することによって、同定することが可能である。しかし、本発明による核酸のより長い断片または完全長配列をハイブリダイゼーションに用いることも可能である。これらの標準条件は、用いた核酸(オリゴヌクレオチド、長い断片もしくは完全長配列)に応じて、またはどの核酸型(DNAもしくはRNA)をハイブリダイゼーションに用いるかによって、様々に変化する。こうして、DNA:DNA ハイブリッドの融解温度は、例えば、同じ長さのDNA:RNA ハイブリッドの融解温度よりもおよそ10℃低い。
【0129】
核酸に応じて、標準条件とは、例えば、0.1〜5×SSC(1×SSC=0.15 M NaCl, 15mM クエン酸ナトリウム, pH7.2)の濃度を含有する水性緩衝溶液中において、またはさらに50%ホルムアミドの存在下にて、42〜58℃の温度(例えば、5×SSC、50%ホルムアミド中において42℃)であると理解される。有利には、DNA:DNA ハイブリッドのハイブリダイゼーション条件は、0.1×SSC、約20℃〜45℃、好ましくは約30℃〜45℃の温度である。DNA:RNAハイブリッドについては、ハイブリダイゼーション条件は有利には、0.1×SSC、約30℃〜55℃、好ましくは約45℃〜55℃の温度である。ハイブリダイゼーションのために特定される温度は融解温度の値であり、この値は、例として、ホルムアミドの非存在下で約100ヌクレオチド長および50% G+C含有量を有する核酸について計算されたものである。DNA ハイブリダイゼーションの実験条件は、遺伝学に関する専門書、例えば、Sambrookらによる「Molecular Cloning」, Cold Spring Harbor Laboratory, 1989に記載されており、当業者に公知の式を用いて、例えば核酸の長さ、ハイブリッドの性質またはG+C含有量に応じて、算出することができる。当業者は以下の文献からハイブリダイゼーションに関するさらなる情報を得ることができる:Ausubelら(編), 1985, Current Protocols in Molecular Biology, John Wiley & Sons, New York;HamesおよびHiggins(編), 1985, Nucleics Hybridization:A Practical Approach, IRL Press at Oxford University Press, Oxford;Brown(編), 1991, Essential Molecular Biology:A Practical Approach, IRL Press at Oxford University Press, Oxford。
【0130】
本発明はまた、具体的に開示されたまたは誘導することができる核酸配列の誘導体に関する。
【0131】
従って、本発明によるさらなる核酸配列は、例えば、配列番号3または5から誘導され、1個または複数のヌクレオチドの付加、置換、挿入または欠失によってそれらとは異なるが、それでも所望の特性プロファイルを有するポリペプチドをコードしている。
【0132】
本発明はまた、サイレント変異と称されるものを含むか、または具体的に挙げた配列と比較した場合に、特定の起源生物または宿主生物のコドン使用頻度に従って改変されている核酸配列、ならびにそれらの天然の変異体(例えば、スプライス変異体もしくは対立遺伝子変異体)を含む。
【0133】
本発明は同様に、保存的ヌクレオチド置換(例えば、問題のアミノ酸を、同一の電荷、大きさ、極性および/または可溶性を有するアミノ酸で置換する)によって得ることができる配列に関する。
【0134】
本発明はまた、具体的に開示された核酸から配列多形の結果として得られる分子に関する。これらの遺伝子多形は、天然の変異の結果として集団内の個体間に存在しうる。これらの天然の変異は通常、遺伝子のヌクレオチド配列に1〜5%の分散を生じさせる。
【0135】
「本発明による核酸配列の誘導体」とは、例えば、全長配列領域にわたって、推定アミノ酸レベルで少なくとも40%の相同性、好ましくは少なくとも60%の相同性、特に好ましくは少なくとも80、85、90、93、95または98%の相同性を有する対立遺伝子変異体を意味するものと理解される(アミノ酸レベルでの相同性に関しては、ポリペプチドに関する上記観察を参照のこと)。相同性は配列の構成的領域にわたって高いことが有利でありうる。
【0136】
さらに、「誘導体」とは、本発明による核酸配列の相同体、例えば、真菌または細菌の相同体、トランケート型配列、またはコードDNA配列および非コードDNA配列の一本鎖DNAもしくはRNAとしても理解される。従って、これらは、例えば、特定した全DNA領域にわたって、DNAレベルで、少なくとも40%、好ましくは少なくとも60%、さらに好ましくは少なくとも70%、特に好ましくは少なくとも80%の相同性を有する。
【0137】
さらに、「誘導体」とは、例えば、プロモーターとの融合体としても理解される。所与のヌクレオチド配列の上流に配置されるプロモーターは、プロモーターの機能または活性を損なうことなく、1個または複数のヌクレオチドの交換、挿入、逆位および/または欠失によって変化させることができる。さらに、プロモーターの活性はそれらの配列を変えることによって増加させることができるし、あるいはプロモーターをより活性のあるプロモーター(他の種に属する生物体に由来するものを含む)と完全に置き換えることができる。
【0138】
「誘導体」とはまた、遺伝子発現および/またはタンパク質発現が変化、好ましくは増加するように、開始コドンの上流-1〜-1000塩基または停止コドンの下流0〜1000塩基の領域においてヌクレオチド配列が変化している変異体としても理解される。
【0139】
本発明はまた、「ストリンジェントな条件」下にて、上記のコード配列とハイブリダイズする核酸配列をさらに含む。この特性は、ポリヌクレオチドまたはオリゴヌクレオチドがストリンジェントな条件下で実質的に相補的な配列と結合するが、こうした条件下で非相補的なパートナーとの間には非特異的結合を形成しない能力として理解される。そのために、これらの配列は70〜100%、好ましくは90〜100%相補的でなければならない。互いに結合することができる相補的配列の特性は、例えば、ノーザンブロット法もしくはサザンブロット法、またはPCRもしくはRT-PCRにおけるプライマー結合に関連して利用される。通常30塩基対またはそれ以上の長さのオリゴヌクレオチドがこの目的のために用いられる。ノーザンブロット法では、例えば、ストリンジェントな条件とは、非特異的にハイブリダイズしたcDNAプローブまたはオリゴヌクレオチドを溶出するために、50〜70℃、好ましくは60〜65℃の温度の洗浄液、例えば0.1%SDSを含有する0.1×SSC緩衝液(20×SSC:3M NaCl、0.3Mクエン酸Na、pH7.0)を用いることと理解される。これに関連して、上述したように、互いに結合し続ける核酸だけが高い相補性を有するものである。ストリンジェントな条件を確立することは、当業者に公知であり、例えば、AusubelらによるCurrent Protocols in Molecular Biology, John Wiley & Sons, N.Y.(1989), 6.3.1-6.3.6に記載されている。
【0140】
これらのポリヌクレオチドは、ゲノムライブラリーまたはcDNAライブラリーをスクリーニングすることによって見出し、適切な場合には、好適なプライマーを用いてPCRによってそれらから増幅し、次いで、例えば、好適なプローブを用いて単離することができる。さらに、本発明によるポリヌクレオチドは化学的に合成することもできる。
【0141】
本発明による構築物の実施形態
さらに、本発明はまた、調節核酸配列の遺伝子制御下に、本発明によるポリペプチドをコードする核酸配列を含む発現構築物に関し、さらに、これらの発現構築物の少なくとも1つを含むベクターに関する。
【0142】
本発明によるこれらの構築物は、好ましくは、所与のコード配列の5’側上流にプロモーターおよび3’側下流にターミネーター配列を含み、また適切な場合には、コード配列にそれぞれ作用可能に結合されるさらなる調節エレメントを含む。
【0143】
「作用可能な結合」とは、プロモーター、コード配列、ターミネーター、適切な場合には、さらなる調節エレメントの逐次的な配置の結果として、それぞれの調節エレメントが、必要に応じて、コード配列の発現に関連してその機能を果たすことができるようになることと理解される。作用可能に結合される配列の例は、ターゲッティング配列およびエンハンサー、ポリアデニル化シグナルなどである。他の調節エレメントとしては、選択マーカー、増幅シグナル、複製起点などが挙げられる。好適な調節配列は、例えば、GoeddelによるGene Expression Technology:Methods in Enzymology 185, Academic Press, San Diego, CA(1990)に記載されている。
【0144】
本発明による核酸構築物とは、特に、本発明によるデヒドロゲナーゼの遺伝子が、その遺伝子発現を調節する、例えば増加させるために、1個または複数の調節シグナルに作用可能にまたは機能的に結合されているものとして理解される。
【0145】
これらの調節配列に加えて、これらの配列の天然の調節要素(実際の構造遺伝子の上流にある)がなおも存在していてもよく、適切な場合には、この天然の調節要素がオフに切り替えられて、遺伝子の発現が増加されるように遺伝的に改変されている。しかし、核酸構築物はより単純な構成を有していてもよく、すなわち、さらなる調節シグナルがコード配列の上流に挿入されておらず、天然のプロモーターがその調節要素と共に除かれていない。その代わりに、もはやあらゆる調節が行われず、かつ遺伝子発現が増加されるように、天然の調節配列が突然変異される。
【0146】
好ましい核酸構築物はまた、有利には、1個または複数の上記のエンハンサー配列を含み、このエンハンサー配列はプロモーターに機能的に結合されて、核酸配列の発現を増加させることが可能である。また、DNA配列の3’末端に追加の有利な配列(例えば、さらなる調節エレメントまたはターミネーター)を挿入すことも可能である。本発明による核酸は1以上のコピー数で構築物中に存在しうる。この構築物はまた、適宜この構築物を選択するために、さらなるマーカー(例えば、抗生物質耐性遺伝子または栄養要求性補足遺伝子)を含むことができる。
【0147】
本発明による方法に有利な調節配列は、例えば、cos、tac、trp、tet、trp-tet、lpp、lac、lpp-lac、acIq、T7、T5、T3、gal、trc、ara、rhaP(rhaPBAD)SP6、λ-PRまたはλ-PLプロモーターのようなプロモーターに存在し、これらのプロモーターはグラム陰性細菌に有利に用いられる。他の有利な調節配列は、例えば、グラム陽性プロモーターのamyおよびSPO2、ならびに酵母または真菌プロモーターのADC1、MFα、AC、P-60、CYC1、GAPDH、TEF、rp28およびADHに存在する。ピルビン酸デカルボキシラーゼおよびメタノールオキシダーゼのプロモーター(例えば、ハンセヌラ(Hansenula)属由来)もまた、これに関連して有利である。また、人工プロモーターを調節に使用することも可能である。
【0148】
宿主生物内での発現のために、核酸構築物は宿主内で遺伝子を最適に発現することができるベクター(例えば、プラスミドまたはファージ)に有利に挿入される。プラスミドやファージに加えて、ベクターとは、当業者に公知の他のいずれかのベクター、すなわち、例えば、ウイルス(例:SV40、CMV、バキュロウイルスおよびアデノウイルス)、トランスポゾン、ISエレメント、ファスミド、コスミドおよび線状または環状DNAとして理解される。これらのベクターは宿主生物内で自律的に複製するかまたは染色体上で複製することができる。これらのベクターは、本発明のさらなる実施形態を構成する。好適なプラスミドの例は、大腸菌のpLG338、pACYC184、pBR322、pUC18、pUC19、pKC30、pRep4、pHS1、pKK223-3、pDHE19.2、pHS2、pPLc236、pMBL24、pLG200、pUR290、pIN-III113-B1、lgt11およびpBdCI、ストレプトミセス(Streptomyces)のpIJ101、pIJ364、pIJ702およびpIJ361、バシルス(Bacillus )属のpUB110、pC194およびpBD214、コリネバクテリウム(Corynebacterium)属のpSA77およびpAJ667、真菌のpALS1、pIL2およびpBB116、酵母の2αM、pAG-1、YEp6、YEp13およびpEMBLYe23、ならびに植物のpLGV23、pGHlac+、pBIN19、pAK2004 およびpDH51である。これらのプラスミドは可能性のあるプラスミドの小さなセレクションを構成する。他のプラスミドが当業者に周知であり、それらに関する情報は、例えば、Cloning Vectors (Pouwels P. H.ら編 Elsevier, Amsterdam-New York-Oxford, 1985, ISBN 0 444 904018)から得ることができる。
【0149】
存在する他の遺伝子の発現のために、核酸構築物はまた、発現を増加させるために3’末端および/または5’末端の調節配列を含むことも有利であり、これらの配列は所定の宿主生物および1以上の遺伝子に応じて最適な発現のために選択される。
【0150】
これらの調節配列は遺伝子の選択的発現ならびにタンパク質の発現を可能にすべきである。これは例えば、宿主生物に依存して、当該遺伝子のみが誘導後に発現または過剰発現されるか、あるいは、当該遺伝子がただちに発現および/または過剰発現されることを意味しうる。
【0151】
これに関連して、調節配列または調節因子は好ましくは、ポジティブな影響を及ぼし、それによって導入された遺伝子の発現を増加させることができる。従って、調節エレメントは、プロモーターおよび/またはエンハンサーのような強力な転写シグナルを用いることによって転写レベルで有利に増強させることができる。しかし、これに加えて、例えばmRNAの安定性を向上させることによって、翻訳を高めることも可能である。
【0152】
ベクターの別の実施形態においては、本発明の核酸構築物または本発明の核酸を含むベクターを線状DNAの形態で微生物に挿入し、非相同組換えまたは相同組換えによって宿主生物のゲノムに組み込むことも可能である。この線状DNAは線状化したベクター(例えば、プラスミド)からなるものであってもよいし、本発明の核酸構築物もしくは核酸からのみなるものであってもよい。
【0153】
生物体による異種遺伝子の最適な発現を可能とするために、その生物体に用いられている特有のコドン使用頻度に従って核酸配列を改変することは有利である。このコドン使用頻度は、対象の生物体に由来する他の既知遺伝子のコンピュータ解析によって容易に決定することができる。
【0154】
本発明の発現カセットは、好適なプロモーターを好適なコードヌクレオチド配列およびターミネーターシグナルまたはポリアデニル化シグナルに融合することによって作製される。このために、例えば、T. Maniatis, E.F. FritschおよびJ. SambrookらによるMolecular Cloning:A Laboratory Manual, Cold Spring Harbor Laboratory, Cold Spring Harbor, NY(1989)、T.J. Silhavy, M.L. BermanおよびL.W. EnquistによるExperiments with Genen Fusions, Cold Spring Harbor Laboratory, Cold Spring Harbor, NY(1984)、ならびにAusubel, F.M.らによるCurrent Protocols in Molecular Biology, Greene Publishing Assoc. and Wiley Interscience(1987)に記載されているような、通常の組換えおよびクローニング技術が用いられる。
【0155】
好適な宿主生物内での発現を達成するためには、組換え核酸構築物または遺伝子構築物を、宿主内での遺伝子の最適な発現を可能にする宿主特異的ベクターに挿入することが有利である。ベクターは当業者に周知であり、それらに関する情報は、例えば「Cloning Vectors」(Pouwels P. H.ら, Eds., Elsevier, Amsterdam-New York-Oxford, 1985)から得ることができる。
【0156】
本発明で用いることができる宿主生物
本発明によるベクターまたは構築物は、組換え微生物を調製するために用いることが可能である。これらの組換え微生物は、例えば、本発明による少なくとも1つのベクターで形質転換され、本発明によるポリペプチドの産生のために用いられる。有利には、本発明による上記組換え構築物を好適な宿主系に導入して発現させる。これに関連して、当業者に良く知られているクローニング方法およびトランスフェクション方法、例えば、共沈、プロトプラスト融合、エレクトロポレーション、レトロウイルスによるトランスフェクションなどが、所与の発現系にて核酸を発現させるために好ましく使用される。好適な系は、例えば、Current Protocols in Molecular Biology, F. Ausubelら編, Wiley Interscience, New York 1997,またはSambrookら Molecular Cloning:A Laboratory Manual, 第2版, Cold Spring Harbor Laboratory, Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY, 1989に記載されている。
【0157】
本発明により、相同的に組換えされた微生物を作製することもまた可能である。このために、本発明による遺伝子の少なくとも1つのセグメントを含むベクター、または本発明による配列を改変する(例えば、機能的に破壊する(「ノック・アウト」ベクター)ために少なくとも1つのアミノ酸の欠失、付加または置換が適宜導入されているコード配列の少なくとも1つのセグメントを含むベクターを作製することが必要である。導入される配列は、例えば、関連する微生物に由来する相同体であってもよいし、哺乳動物、酵母もしくは昆虫源に由来するものであってもよい。あるいはまた、相同組換えに用いられるベクターは、相同組換えに関連する内在性遺伝子が変異されるか、さもなければ改変されるが、機能的タンパク質をなおもコードするように構成することができる(例えば、下流調節領域が改変され、その結果として内在性タンパク質の発現を変化させることができる)。本発明による遺伝子の改変されたセグメントは、相同組換えベクター内にある。相同組換えに好適なベクターの構築は、例えば、Thomas, K.R.およびCapecchi, M.R.(1987)Cell 51:503に記載されている。
【0158】
原則的に、どのような原核生物または真核生物も、本発明による核酸または核酸構築物の組換え宿主生物として用いるのに適している。有利には、微生物、例えば細菌、真菌または酵母が宿主生物として用いられる。グラム陽性細菌またはグラム陰性細菌、好ましくはエンテロバクテリアセア(Enterobacteriaceae)科、シュードモナダセア(Pseudomonadaceae)科、リゾビアセア(Rhizobiaceae)科、ストレプトマイセタセア(Streptomycetaceae)科もしくはノカルディアセア(Nocardiaceae)科の細菌、特に好ましくはエスケリキア(Escherichia)属、シュードモナス(Pseudomonas)属、ストレプトマイセス(Streptomyces)属、ノカルディア(Nocardia)属、ブルクホリデリア(Burkholderia)属、サルモネラ(Salmonella)属、アグロバクテリウム(Agrobacterium)属またはロドコッカス(Rhodococcus)属の細菌が有利に用いられる。エスケリキア・コリ(Escherichia coli)の属および種が特に好ましい。これに加えて、他の有利な細菌はα-プロトバクテリア、β-プロトバクテリアおよびγ-プロトバクテリアの群に見出すことができる。
【0159】
これに関連して、宿主生物、または本発明による宿主生物は、本発明によるデヒドロゲナーゼ活性を有する酵素をコードする、本発明に記載の核酸配列、核酸構築物またはベクターを少なくとも1つ含むことが好ましい。
【0160】
本発明の方法に用いられる生物は、宿主生物に応じて、当業者に公知の方法で増殖させるかまたは培養する。微生物は一般的に、炭素源(通常は糖の形態)、窒素源(通常は有機窒素源の形態、例えば酵母エキス)、または塩(例えば、硫酸アンモニウム)、微量元素(例えば、鉄塩、マンガン塩およびマグネシウム塩)、適宜にビタミン類を含有する液体培地中で0℃〜100℃、好ましくは10℃〜60℃の温度にて酸素を供給しながら増殖させる。これに関連して、液体栄養培地のpHは一定の値(培養の間中調整される)に維持してもしなくてもよい。この培養はバッチ式、半バッチ式、または連続的に行うことができる。栄養分は発酵の開始時にはじめに導入されるか、または続いて半連続的にまたは連続的に供給できる。ケトンはこの培養物に直接的に加えることができるが、培養後に加えることが有利である。酵素は、実施例に記載の方法を用いて生物体から単離するか、そうでなければ、粗抽出物として反応に用いることができる。
【0161】
組換えによる本発明のポリペプチドの調製
本発明はさらに、本発明によるポリペプチドまたはそれらの機能的、生物学的に活性な断片を組換えによって調製するための方法に関する。この方法は、ポリペプチド産生微生物を培養し、ポリペプチドの発現を誘導し、適切な場合には、これらのポリペプチドを培養物から単離することを含む。所望であれば、このポリペプチドをこの方法により工業的規模で製造することが可能である。
【0162】
組換え微生物は公知の方法で培養および発酵させることができる。例えば、細菌をTB培地またはLB培地中で20〜40℃の温度、pH6〜9にて増殖させる。好適な培養条件は、例えば、T. Maniatis, E.F. FritschおよびJ. SambrookらによるMolecular Cloning:A Laboratory Manual, Cold Spring Harbor Laboratory, Cold Spring Harbor, NY(1989)に詳細に記載されている。
【0163】
ポリペプチドが培地中に分泌されないときには、細胞を破砕して、その溶解液から公知のタンパク質単離方法により産物を取得する。細胞は、所望により、高周波超音波、高圧(例えば、フレンチプレス細胞破砕機)、浸透圧、界面活性剤、溶解酵素もしくは有機溶媒の作用、ホモジェナイザーの使用または上記方法のいくつかの組合せによって、破砕することができる。
【0164】
ポリペプチドを精製するには、公知のクロマトグラフィーによる方法、例えば、分子ふるいクロマトグラフィー(ゲル濾過)、例えば、Qセファロースクロマトグラフィー、イオン交換クロマトグラフィーおよび疎水性クロマトグラフィー、さらに他の慣用方法、例えば、限外濾過、結晶化、塩析、透析および未変性ゲル電気泳動を用いることができる。好適な方法は、例えば、Cooper, F. G., Biochemische Arbeitsmethoden [生化学的操作方法], Verlag Walter de Gruyter, Berlin, New YorkまたはScopes, R., Protein Purification, Springer Verlag, New York, Heidelberg, Berlinに記載されている。
【0165】
組換えタンパク質を単離するためには、特定のヌクレオチド配列によってcDNAを伸長させ、それによって、例えば精製を簡便にするために用いられる、改変されたポリペプチドまたは融合タンパク質をコードするベクター系またはオリゴヌクレオチドを用いることが有利でありうる。このような好適な改変の例には、アンカーとして作用するタグと称されるものがあり、例えば、ヘキサヒスチジンアンカーとして知られる改変、または抗体によって抗原として認識され得るエピトープ(例えば、Harlow, E.およびLane, D., 1988, Antibodies:A Laboratory Manual. Cold Spring Harbor(N.Y.)Pressに記載されている)がある。これらのアンカーは、タンパク質を固相支持体(例えば、クロマトグラフィーカラムに充填されるポリマーマトリックスもしくはマイクロタイタープレートまたは別の支持体)に結合させるために用いることができる。
【0166】
同時に、これらのアンカーをタンパク質の認識に用いることも可能である。タンパク質の認識のためには、通常の標識、例えば、蛍光色素、基質との反応後に検出可能な反応生成物を形成する酵素標識または放射性標識を、単独でまたはアンカーと組み合わせて、タンパク質を誘導体化するために用いることが可能である。
【0167】
本発明による酵素的還元方法を実施するための他の実施形態
本発明の方法においては、デヒドロゲナーゼを遊離の酵素または固定化酵素として用いることができる。
【0168】
本発明の方法は0℃〜95℃、好ましくは10℃〜85℃、特に好ましくは15℃〜75℃の温度にて実施することが有利である。
【0169】
本発明の方法においては、pHをpH4〜12、好ましくはpH4.5〜9、特に好ましくはpH5〜8に維持する。
【0170】
本発明の方法において、エナンチオマー的に純粋なまたはキラルな生成物、例えば3-クロロ-1-(チエン-2-イル)-(S)-プロパン-1-オールは、エナンチオマー冨化を示すエナンチオマーであるとして理解されるべきである。本方法では、少なくとも70%ee、好ましくは少なくとも80%ee、さらに好ましくは少なくとも90%ee、特に好ましくは少なくとも98%eeのエナンチオマー純度を達成することが好ましい。
【0171】
本発明の方法のために、本発明による核酸、核酸構築物またはベクターを含む増殖中の細胞を用いることが可能である。また、休止細胞または破砕細胞を用いることも可能である。破砕細胞とは、例えば、溶媒で処理されたことによって透過性にされた細胞、または酵素で処理されたことによって、機械的に処理されたことによって(例えば、フレンチプレスもしくは超音波処理)もしくは別の方法によって破壊された細胞として理解される。このようにして得られた粗製抽出物は本発明による方法に適している。また、精製酵素または部分精製酵素を本方法に用いることもできる。本反応に有利に用いることができる固定化した微生物または酵素も同様に好適である。
【0172】
本発明による方法は、バッチ式、半バッチ式または連続的に行うことができる。
【0173】
本方法は、例えば、Biotechnology, 第3巻, 第2版, Rehmら編,(1993)、特に第II章に記載されるようなバイオリアクター内で有利に行うことができる。
【0174】
以下の例は、本発明を限定することなく例証することを目的とする。これに関して添付の図面を参照されたい。
【実施例】
【0175】
A. 化学的実施例
実施例1:3-クロロ-1-(チエン-2-イル)プロパン-1-オンIII.1の調製
33kgのチオフェンを150kgのジクロロエタンにまず導入し、その後62.7kgの三塩化アルミニウムを加えた。この反応混合物を10℃まで冷却し、55kgの塩化3-クロロプロピオニルを加えた。次いでこの混合物を室温にて12時間撹拌した。GCおよびNMRで調べると、反応混合物は、生成された3-クロロ-1-(チエン-2-イル)プロパン-1-オンおよび3-クロロ-1-(チエン-2-イル)プロペノンの合計量に基づいて、約3〜4モル%の3-クロロ-1-(チエン-2-イル)プロペノンをまだ含むことがわかった。その後、少しのプロペノンも検出することができなくなるまで塩化水素を室温にて通過させた(およそ30分間)。続いて、100kgの脱イオン水を加えることによってこの反応混合物を加水分解した。有機相を分離し、100kgの脱イオン水で洗浄した。真空にて溶媒を取り除いた後、65.7kg(理論値の96%)の表題化合物を油状物として得た。
【0176】
実施例2:水素化ホウ素ナトリウムで還元することによる3-メチルアミノ-1-(チエン-2-イル)プロパン-1-オールIの調製
実施例1から得られたプロパノンを初めに、0℃にて、400mlのトルエンと200gのメタノールの混合溶媒中に導入した。30%濃度の水酸化ナトリウム水溶液2gを加えた後、21.4gの水素化ホウ素ナトリウムを2.5時間かけて少しずつ加えた。0℃で40分間撹拌した後、この反応混合物を12.1gの40%濃度のメチルアミン水溶液で処理した。この混合物を、その固有の圧力下に60℃で6時間撹拌した。この混合物を室温まで冷却した後、溶媒を除去し、残留物をトルエンで温浸し、トルエンから濾過した。この濾液を乾燥させると、175.8gの表題化合物が淡黄色の固形物として得られた。
【0177】
B. 生化学的実施例
実施例3:補因子再生のためのグルコースデヒドロゲナーゼの調製
補因子を再生するために、販売元(例えば、Julich Fine Chemicals 注文番号22.10もしくは19.10)または本発明者ら自身の供給源から得られるグルコースデヒドロゲナーゼを用いることが可能であった。本発明者ら自身の供給源とは、枯草菌(Bacillus subtilis)(Genbank登録番号M12276)(Lu11293)に由来するグルコースデヒドロゲナーゼ遺伝子を含む大腸菌XL10 Gold pUC19クローンであった。
【0178】
以下の培地を大腸菌Lu11293の発酵のために調製した:
【0179】
2本の1Lエルレンマイヤーフラスコにそれぞれ150mlの培地を入れて滅菌し、5mlの滅菌塩溶液を補充した。LBアンピシリン寒天プレートから接種した後、予備培養物を37℃、200rpmで12時間インキュベートして、発酵培地に加えた。発酵は37℃、内圧0.1バール、pH7.0(20%リン酸および25%NaOHを用いて調整)、空気補給速度7.5L/分、および300rpmで開始した(空気を10〜20L/分および500〜1500rpmで流入させてpO2を20〜50%に調整した)。2時間後、0.1mM IPTGを誘導のために加え、発酵を全13時間後に終わらせた。細胞(1.3kg)を回収して、洗浄した後、使用するまで-20℃にて保存した(混合物中に2〜20g/L)。
【0180】
実施例4:3-クロロ-1-(チエン-2-イル)プロパン-1-オンを還元するためのデヒドロゲナーゼのスクリーニング
a)種々の細菌株および真菌株(主に種々の菌株コレクションに由来する)を20mlのLB培地、MRS培地(Difco)またはGYP培地(酵母に使用)(1%w/v D-グルコース、それぞれ0.5%w/v 酵母抽出物およびポリペプトン、pH6.0)中で30℃または37℃にて24〜48時間増殖させ、3mM Tris-HCl、pH7.5中で洗浄し、そして2mlの緩衝液に再懸濁させた。アリコートを振動粉砕器中の1容のガラスビーズ(直径0.3〜0.5mm)で破砕した(10分の氷上での冷却時間をはさんで3×5分)。濁りを遠心分離(4℃、14000rpmにて5分間)によって分離した後、透明な粗抽出物を得た。その後、細胞懸濁液および粗抽出物を、実施例3から3-クロロ-1-(チエン-2-イル)プロパン-1-オンを還元するためにそれらの活性について試験した。
【0181】
アッセイには以下を用いた:
150μlの細胞/粗抽出物
50μlの1Mグルコース溶液
50μlのグルコースデヒドロゲナーゼ(12〜15U/μl;20〜200mg MBM/ml)(実施例3から)
50μlの2mM NADH
50μlの2mM NADPH
10μlの0.5M 3-クロロ-1-(チエン-2-イル)プロパン-1-オン(メタノールに溶解したもの)
190μlの50mM MES, pH6.0またはTris-HCl, pH7.5
【0182】
インキュベーションは30℃で行った。このサンプル(200μl)は1時間、2時間または24時間後に3μlの濃HClを用いて停止させた。遠心分離後、上清を3-クロロ-1-(チエン-2-イル)プロパン-1-オンおよび3-クロロ-1-(チエン-2-イル)プロパン-1-オールについてHPLCで分析した(Chromolith Speed ROD, RP-18e 50-4.6mm、流速1.5ml/分、0.0〜1.0分:35%アセトニトリル+65%10mM KH2PO4緩衝液、pH2.5;1.1〜1.3分:80%アセトニトリル+20%10mM KH2PO4緩衝液、pH2.5;1.3〜2.0分:35%アセトニトリル+65%10mM KH2PO4緩衝液、pH2,5;230nm(アルコール)および260nm(ケトン)にて検出、保持時間 Rt=1.250分(3-クロロ-1-(チエン-2-イル)プロパン-1-オール)およびRt=1.500分(3-クロロ-1-(チエン-2-イル)プロパン-1-オン);HCl除去から生じうる副生成物(1-チエン-2-イルプロペン-1-オンおよび1-チエン-2-イルプロペン-1-オール)、Rt=0.971分およびRt=1.165分)。
【0183】
選択した菌株は、エナンチオマー過剰率(ee)を決定するために繰り返し試験したが、その際、サンプルをメチルtert-ブチルエーテル(MTBE)またはメチルイソブチルケトン(MIBK)を用いて抽出し、キラルGC分析またはHPLC分析によって特徴づけた。そのために以下の方法を用いた:GC:Hydrodex-β-6-TBDM、25m、90℃ 10分 5℃/分 180℃ 10分、ラン時間:38分、入口:ヒーター:200℃、圧力:106.8 kPa、全流量:102ml/分、スプリット比:125:1、スプリット流量:99.8ml/分、検出器:200℃またはHPLC:Chiracel OD-H、250*4.6mm(Daicel)、40℃、流速1.0ml/分、0.0〜1.0分:97.5% n-ヘキサン+2.5%イソプロパノール;230nm(アルコール)および260nm(ケトン)にて検出、Rt 9.50(3-クロロ-1-(チエン-2-イル)プロパン-1-オン)、Rt 16.60分(R-3-クロロ-1-(チエン-2-イル)プロパン-1-オール)およびRt 18.30分(S-3-クロロ-1-(チエン-2-イル)プロパン-1-オール)。
【0184】
b)種々のラクトバチラス(Lactobacillus)菌株を新たに単離した
ラクトバチラス・ブレビス(Lactobacillus brevis)株であるLu10288、10290および10291を以下のように単離した。
【0185】
b1)用いた培地
Kleymann培地:
以下の物質を915ml中に溶解した:
10g/l トリプトン(Difco-Becton Dickinson)
7g/l 酵母エキス(Difco-Becton Dickinson)
2g/l 牛肉エキス(Difco-Becton Dickinson)
5g/l フルクトース
2g/l マルトース
3.6g/l 50%グルコン酸
1.9g/l クエン酸*H2O
5g/l酢酸
1g/l Tween 80
0.2g/l MgSO4*7H2O
0.05g/l MnSO4
0.01g/l FeSO4
0.4g/l L-システイン
1.25ml NH4OH(25%)
寒天プレートについて:2% Bacto Agar(Difco-Becton Dickinson)を加える
前記溶液のpHをpH5.4に調整した。
前記溶液を121℃にて15分間オートクレーブ滅菌した。
オートクレーブ滅菌した後、50mlの滅菌グルコース溶液(5 gのグルコースをH2Oで50mlにしたもの)および40mlのエタノールを撹拌しながら加え、寒天プレートに注いだ。
【0186】
KMB培地:
10g/l トリプトン(Difco-Becton Dickinson)
7g/l 酵母エキス(Difco-Becton Dickinson)
2g/l KH2PO4
1g/l Tween 80
0.5g/l MgSO4*7H2O
1g/l MnSO4
20g/l CaCO3
10g/lグルコース
1.5%寒天(寒天プレート調製用)
前記溶液を121℃にて15分間オートクレーブ滅菌した。
【0187】
グルコースおよびCaCO3(Riedel de Haen)を別々に溶解してオートクレーブ滅菌し、プレートに注ぐ前に寒天と混合した。
【0188】
b2)微生物の単離:
北ドイツ地方(LUFA-Oldenburg)産の5〜10gのトウモロコシ貯蔵飼料を50mlエルレンマイヤーフラスコ中で20mlの塩水と共に37℃にて一晩、嫌気的にインキュベートした。得られた液体フラクションを1:100の割合で50mlのKleymann培地に接種し、穏やかに撹拌しながら室温で24時間嫌気的にインキュベートした。その後、この細胞懸濁液を上記のKleymann選択寒天プレートに播種した。このプレートを37℃で48〜72時間嫌気的にインキュベートし、得られたコロニーを、KMB培地上に繰り返しストリークすることによって単離した。
【0189】
この菌株は、20gのフルクトース/Lを含有する液体KMB培地中(24時間、37℃、50rpm)で酸発酵パターン(pHの測定、および培養上清のHPLC分析によるグルコース、フルクトース、乳酸、酢酸およびエタノールの濃度の測定)を決定することによって特徴づけた。酢酸および乳酸を形成した異種発酵菌株を単離し、そして16sRNAを分析することによって体系的に特徴づけた。
【0190】
c)スクリーニング結果
表1、2および3は、菌株の例および/または変換率およびee値を示す。
【表1】
【0191】
【表2】
【0192】
【表3】
【0193】
実施例5:ラクトバチラス・ブレビス(Lactobacillus brevis)由来のデヒドロゲナーゼの精製
ラクトバチラス・ブレビス(Lactobacillus brevis)Lu10288の発酵のために以下の培地を調製した:
【0194】
MRS寒天プレートから2つの予備培養物を接種し、この予備培養物を37℃にて17時間、200rpmでインキュベートして、発酵培地に加えた。この発酵は37℃、内圧0.1バール、pH5.8、空気補給速度1L/分、および100rpmで開始し(pO2の調節なし)、23時間後にOD600が14.8になった時点で終結させた。
洗浄した細胞試料の活性を、MES, pH6.0中の休止細胞を用いて実施例4a)と同様に測定した。
【0195】
Lu10288デヒドロゲナーゼは以下のように精製した:
均質化:
100gのLu10288(ラクトバチラス・ブレビス(Lactobacillus brevis))の湿ったバイオマスを、100mlのMES緩衝液、1mM MgCl2、pH7.1を用いて、5×20gに小分けして再懸濁させ、10に小分けして20分間、4000rpmにて氷で冷却しながら、ガラスビーズ(直径0.1mm〜0.2mm、50mlの懸濁細胞に対して50mlのガラスビーズ)を用いてボールミル内でホモジェナイズした。ガラスビーズをG2ガラス吸引フィルターを通して分離し、20mlの緩衝液で洗浄した。次いで、回収したホモジェネート(610ml)をGSAローター内で20分間、12000rpmにて清澄化した。
【0196】
a) Q-セファロースイオン交換クロマトグラフィー:
直径5cm、容積400mlのQ-セファロースファーストフロー(Pharmacia)カラムを20mM MES緩衝液、1mM MgCl2、 pH6.8中で平衡化した。610mlのホモジェネートを11錠のComplete(プロテアーゼ阻害剤混合物、Roche, Complete, EDTAフリー;カタログ番号:1873580)で処理し、Q-セファロースカラムに10ml/分の速度でロードした。280 nmにて検出した。次いで、このカラムを同じ緩衝液で3倍のカラム容量を用いて洗浄した。溶出のために、20mM MES、1mM MgCl2、1M NaCl、pH6.8の直線勾配(100分間で0%NaClから100%NaClまで)を適用した。10mlフラクションを回収して試験した。フラクション42〜62は、HPLC試験において3-クロロ-1-(チエン-2-イル)プロパン-1-オンとの活性を示した。
【0197】
b) Superdex分子ふるいクロマトグラフィー:
直径2.6cm、容積240mlのSuperdex 200分子ふるいカラム(Pharmacia)を20mM MES、1mM MgCl2、緩衝液1Lにつき1錠のComplete(EDTAフリー)、pH7.1中で平衡化した。Q-セファロースから得られた有用なフラクションを合わせて(240ml)、124gの硫酸アンモニウムを徐々に加えて80%飽和に調整した。pHは7.1に維持した。この溶液を4℃にて10分間撹拌し、次いで12000rpmにて20分間遠心分離した。得られたペレットを5mlの平衡緩衝液に再懸濁し、この懸濁液を1時間、4℃にて透析した(10 kDa排除体積)。透析した溶液を9ml部ずつ2分し、分子ふるいカラムに4ml/分の流速でロードした。4mlフラクションを回収して試験した。フラクション48〜56は両方の基質に対して再び活性を示した。
【0198】
c) Mono-Qイオン交換クロマトグラフィー:
容積20mlの分離用Mono-Q HRカラム(Pharmacia)を20mM MES、1mM MgCl2、pH7.1中で平衡化した。Superdexから得られた有用なフラクションを合わせたもの70mlを4ml/分の流速でロードした。カラムを洗浄した後、100分間で100%までの20mM MES、1mM MgCl2、0.5M NaCl、pH7.1の直線勾配を用いて展開した。4mlフラクションを回収した。活性のあるフラクション(フラクション36〜41)を合わせた。
【0199】
d) Mono-Pイオン交換クロマトグラフィー:
Mono-Pカラム(Pharmacia, 4ml)を20mM MOPS、1mM MgCl2、pH7.1中で平衡化した。21mlの有用なMono-Qフラクションを0.75ml/分の流速でロードした。基底値になるまで洗浄した後、100分間で100%までの20mM MOPS、1mM MgCl2、0.5M NaCl、pH7.1の直線勾配を適用した。フラクション(0.75ml)を回収した。活性のあるフラクション(34〜39)を合わせた。
【0200】
ゲルでの活性染色:
試料を同容量のNovex「未変性Tris-グリシン」試料緩衝液(Novex)で希釈した。Anamedのtris-グリシンゲル(SDSなし、厚さ1mm、10個の試料用ウェル)をランニング・チャンバーに設置した。Invitrogenの「Tris-グリシン未変性」ランニング緩衝液(Invitrogen)(10×)をランニング緩衝液として希釈後に用いた。試料ウェルにロードし、ゲルを200 V、約50 mAにて開始した。電気泳動を約1.5時間継続した。ランニング・チャンバーは氷水中に置いた。ゲルを取り外した後、ガラス皿に置き、50mM MES、8mM MgCl2、pH6.2中で洗浄し、そしてインキュベートした。
【0201】
次いで、0.35mM NADPおよび0.35mM NAD、19.6mgのNB-テトラゾリウムならびに2.1mgのフェナジンエトサルフェート(PES)をこの溶液中のゲルに加えた。続いて基質(3-クロロ-1-(チエン-2-イル)プロパン-1-オール)を加えて最終濃度を1mMとした。このゲルは染色されるまで暗所に保存した。典型的なゲルを図1に示す。
【0202】
【0203】
本方法で精製したデヒドロゲナーゼのN末端を、SDS-PAGEおよびゲル・ブロッティング後にEdman配列決定により決定した(配列番号1)。
【0204】
実施例6:カンジダ・マグノリア(Candida magnoliae)由来のデヒドロゲナーゼの精製
カンジダ・マグノリア(Candida magnoliae)Lu8742の発酵のために、2つの予備培養物を150mlの培地(8g/Lの酵母エキス(65%)、5g/Lのペプトン、3.5g/Lのグルコース、5g/LのKH2PO4、2g/LのMgSO4*7H2Oを含有、pH6.0)中で28℃、200rpmで15時間増殖させ、10.7g/Lのグルコースおよび4mlのテゴシポン(tegosipon)を含有する同様の培地13.2Lへの接種に用いた。発酵を28℃、内圧0.1バール、pH6.0、空気補給速度5L/分、および500rpmで開始し(pO2を>20%に調整)、26時間後にOD600が15.1となった時点で停止させた。
【0205】
均質化:
回収した細胞(378gの湿ったバイオマス)を、10錠のRoche Complete(EDTAフリー)プロテアーゼ阻害剤(およそ9×濃度)を含有する1Lの50mM MES+1mM MgCl2、pH6.5中に懸濁し、Z04ミクロフルイダイザー中1000バールで2回破壊した。遠心分離(20分/10000g)に続いて、澄んだ上清(1.3Lのホモジェネート)の半分を下記のとおりに精製した。これを行うとき、試料フラクションは、50mM MES、pH6.0、0.2mM NaDH/NaDPH、100mMグルコース、50μlのGDH/mlおよび10mMの3-クロロ-1-(チエン-2-イル)プロパン-1-オン中30℃にて、好適な希釈で、活性について試験した。
【0206】
Q-セファロースイオン交換クロマトグラフィー:
直径5cm、容積400mlのQ-セファロースファーストフローカラム(Pharmacia)を20mM MES緩衝液、1mM MgCl2、pH6.8中で平衡化した。650mlのホモジェネートを11錠のComplete(プロテアーゼ阻害剤混合物)で処理し、7.5ml/分の速度にてQ-セファロースカラムにロードした。280nmにて検出した。次にカラムを700mlの同一緩衝液を用いて洗浄した。溶出のために、20mM MES、1mM MgCl2、0.75M NaCl、pH6.8の直線勾配(100分間で0%NaClから100%NaCl)を適用し、その後カラムを200mlの20mM MES、1mM MgCl2、0.75M NaCl、pH6.8で洗浄した。10mlフラクションを回収して試験した。フラクション56〜96はHPLC試験において3-クロロ-1-(チエン-2-イル)プロパン-1-オンとの活性を示した。
【0207】
硫酸アンモニウム沈降:
41mlの有用なQ-セファロースピークフラクションを(NH4)2SO4(pH6.2)で90%飽和にし、4℃にて30分間撹拌した後、10000gにて10分間遠心分離した。得られたペレットを10mlの20mM MES、1mM MgCl2、1Lにつき1錠のRoche Complete(EDTAフリー)、pH6.2中に懸濁し、この懸濁液をPierce Slide-A-Lyzer(10 kDa排除体積)にて20mM MES、1mM MgCl2、1Lにつき1錠のRoche Complete(EDTAフリー)に対して30分間透析した。
【0208】
Superdex分子ふるいクロマトグラフィー:
直径2.6cm、容積240mlのSuperdex 200分子ふるいカラム(Pharmacia)を20mM MES、1mM MgCl2、1Lの緩衝液につき1錠のComplete(EDTAフリー)、pH6.2中で平衡化した。硫酸アンモニウム沈降から得られた透析物(2×7ml)を4ml/分の流速で分子ふるいカラムにロードした。4mlフラクションを回収して試験した。フラクション21〜24は両基質について再び活性を示した。
【0209】
Mono-Qイオン交換クロマトグラフィー:
容積1mlのMono-Q HR5/5カラム(Pharmacia製)を20mM MES、1mM MgCl2、pH6.8中で平衡化した。Superdexカラムからの有用なフラクションを合わせたもの10mlを1ml/分の流速でロードした。カラムを洗浄した後、100分間で100%までの20mM MES、1mM MgCl2、0.75M NaCl、pH6.8の直線勾配を用いて展開し、続いて20mM MES、1mM MgCl2、pH6.8を用いて10分間洗浄した。1mlフラクションを回収した(検出 226nm)。活性を有するフラクション(フラクション24〜27)を合わせた。
【0210】
単離の結果を以下の表に要約した。
【0211】
このようにして精製したデヒドロゲナーゼのN末端を、SDS-PAGEおよびゲル・ブロッティング後にEdman配列決定により決定した(配列番号2)。
【0212】
実施例7:ラクトバチラス・ブレビス(Lactobacillus brevis)デヒドロゲナーゼを用いた還元による(S)-3-メチルアミノ-1-(チエン-2-イル)プロパン-1-オールI-Sの調製
ラクトバチラス・ブレビス(Lactobacillus brevis)Lu10288を実施例4に記載されるように増殖させて回収した。休止細胞(10〜100g/Lのバイオマス)を0.2〜2mM NAD(P)+、実施例1からの1〜100g/Lのプロパノン(バッチ式または流加式)および18g/Lのグルコースで処理して、30℃にて2〜8時間インキュベートした。この反応を0.5M NaOHで滴定することによってpH6.0〜7.0を維持し、HPLC分析によって追跡した。図2AおよびBはそれぞれ、混合物1および2に対応する典型的な反応経過を示す。
【0213】
【0214】
続いて、バイオマスを遠心分離および/または濾過によって除去した。MTBE抽出物のNMR分析は60〜70%の3-クロロ-1-(チエン-2-イル)プロパン-1-オールの含有量を与えた。未反応の3-クロロ-1-(チエン-2-イル)プロパン-1-オンおよび副生成物である1-チエン-2-イルプロペン-1-オンの含有量は、それぞれ10%未満であった。1-チエン-2-イル-プロペン-1-オールは痕跡量で検出できたにすぎなかった。
【0215】
次いで、1.1gの40%メチルアミン水溶液を無細胞水性混合物に加えた。後者を次いで、60℃で6時間、固有の圧力下にて撹拌した。混合物を室温まで冷却した後、溶媒を除去した。その後残留物をトルエンで温浸し、トルエンから濾過した。別法として、アミノ化反応の後に細胞分離を行うことも可能であった。濾液を乾燥させた後、202mgの表題化合物を淡黄色の固形物として得た。Sエナンチオマーを95%eeのエナンチオマー過剰率で得た。ee値は、回転量を測定することよって(c=1、溶媒:メタノール)、またShift NMR(Shift試薬:2,2,2-トリフルオロ-1-(9-アンスリル)エタノール(+)(TFAE);溶媒:CDCl3;500 MHz)によって決定した。
【0216】
実施例8:ラクトバチラス・ブレビス(Lactobacillus brevis)Lu10288由来のデヒドロゲナーゼのクローニング
a) 先行プロトプラスト形成に続くLU10288由来の染色体DNAの調製:
(1) 必要な溶液
溶液1: 0.41Mスクロース
0.01M MgSO4*7H2O
5ml/L M12培地1:2
10ml/L 10%KH2PO4 pH6.7
2.5mg/mlリゾチーム(使用直前に加える)
溶液1は以下のように調製する:
14.03gのスクロース、0.25gのMgSO4*7H2O、5mlのM12(10×濃度)および1mlの10% KH2PO4、pH6.7を一緒にして100mlとなし、濾過滅菌する。
プロテイナーゼ: Qiagen製、20mg/ml ストック溶液
RNase : Qiagen製、100mg/ml ストック溶液
TE緩衝液: 10mM Tris*Cl pH8、1mM EDTA pH8
【0217】
(2) 培養および破砕:
− 250mlのバッフル付きエルレンマイヤーフラスコに入れた100mlのMRS培地(Difco製)中で37℃、200rpmにて一晩培養する;
− 細胞を遠心分離する:4000rpm、10分間、4℃;
− 上清を捨て、ペレットを5mlの溶液1(+リゾチーム)にとり、よく懸濁させる。インキュベーター(37℃)内で1〜4時間インキュベートする;
− プロトプラストを慎重に遠心分離する:3000rpm、10分間;
− 上清を捨て、ペレットを10mlの溶液1(リゾチームなし)で洗浄する:3000rpm、4℃、8分間;
− 上清を捨て、ペレットを10mlの0.01M Tris-HCl、pH8.0で洗浄する:3000rpm、4℃、8分間;
− 上清を捨て、ペレットを4mlのTE緩衝液に懸濁する;
− 0.5mlの10%SDSおよび0.5mlの5M NaClを加え、慎重に混合する;
− 1 mgのプロテイナーゼK(Qiagenのプロテイナーゼ:20mg/ml、すなわち50μl)を加えて、インキュベーター内で37℃にて一晩インキュベートする;
− この混合物をTE緩衝液を用いて20mlにする。
【0218】
(3) 抽出:
− フェノールを1:1、すなわち20mlのフェノール+20mlの混合物となるように加える。慎重に混合し、4℃、4000rpmで5分間遠心分離する;
− 上相を取り、それを新しいFalcon(20ml)に移す;
− 20mlのフェノール:クロロホルム:イソアミルアルコール(25:24:1)を加える。慎重に混合し、4℃、4000rpmで5分間遠心分離する;
− 上相を取り、新しいFalcon(約18ml)に移す;
− 18mlのクロロホルム:イソアミルアルコール(24:1、すなわち18mlのクロロホルム+333μlのイソアミルアルコール)を加える。慎重に混合して、4℃、4000rpmで5分間遠心分離する。この工程を上相が透明になるまで繰り返す;
− 上相(18ml)を2倍容量のエタノール(36ml)を用いて沈殿させる。1/50の3M 酢酸ナトリウム(約360μl)を加える。-20℃で少なくとも30分間沈殿させたままにする。その後、4℃、12000rpmで30分間遠心分離する;
− 上清を捨て、ペレットを1〜2mlのTE緩衝液にとる。TE緩衝液1mlに対して20μgのRNaseを加える。
インキュベーション:水浴中37℃にて1時間。
【0219】
(4) 透析:
RNase処理の後、混合物を透析バッグに移す。3回の透析を行うが、それぞれ1.5LのTE緩衝液中、4℃にて1時間行う。最後の透析工程を一晩かけて行うことも可能である。
− 透析したDNAをFalconに移し、数本のEppendorffチューブ(500μl)に分注する;
− 2倍容量のエタノール(1000μl)および1/3容量の2M LiCl(166μl)を加える;
− -20℃にて少なくとも30分間沈殿させる。その後、4℃、12000rpmで30分間遠心分離する;
− 上清を慎重に流出させる;
− ペレットを20mlの70%エタノールで洗浄し、4℃、12000rpmで15分間遠心分離する;
− 上清を慎重に流し出し、ペレットを風乾させ、適量の10mM Tris*HCl、pH8.0(ペレットの大きさによって100μl以上)にDNAを取る。
再解離を改善するために、DNAをEppendorf振とう器中で55〜60℃、低振動数(400rpm)で1〜2時間インキュベートした。
【0220】
b) ペプチドのトリプシン消化およびEdmann配列決定の後、さらなるアミノ酸配列を実施例5に記載のタンパク質精製より得た。配列解析の結果を図3Bにまとめた。
切断点のために、配列FVVDGGYTAQ(V8-RP Fr.7を参照)はおそらくC末端を表す。核酸配列(プライマーMke338および339)は、Lu10288染色体DNA上で行われるPCR増幅によりデヒドロゲナーゼ遺伝子をクローニングするために用いたものであるが(プロトコルは上記参照)、ラクトバチラス・ブレビス(Lactobacillus brevis)のコドン使用頻度を考慮して、上記配列およびN末端アミノ酸配列(配列番号1)から推定した。
【0221】
【0222】
PCRは、Pfuポリメラーゼ(Stratagene) および以下の温度プログラムを用いた標準的なStratageneプロトコルに従って行った:95℃にて5分;95℃にて45秒、52℃にて45秒、および72℃にて1分20秒を30サイクル;72℃にて10分;使用するまで10℃。PCR産物(0.8 kb)をアガロースゲル電気泳動(1.2% E-Gel、Invitrogen)およびカラムクロマトグラフィー(Mini-Elute、Qiagen)によって単離し、次いでNdeI/HindIIIを用いて消化して、同様に消化したpDHE19.2ベクター(pJOE誘導体、DE19848129)にクローン化した。ライゲーション混合物を大腸菌XL1 Blue(Stratagene)に形質転換させた。対応するクローンの配列決定は、得られたプラスミドpDHE10288adh1の挿入物として、配列番号3に示した核酸配列をもたらし、この配列は配列番号4のアミノ酸配列に対応した。実施例5および8で同定されるペプチドはすべて、この配列中に再び見出される。
【0223】
【0224】
実施例9:ラクトバチラス・ブレビス(Lactobacillus brevis)Lu10288由来の組換えデヒドロゲナーゼの活性の測定
プラスミドpDHE10288adh1を大腸菌TG10 pAgro4 pHSG575に再形質転換した(TG10:大腸菌TG1のRhaA- 誘導体(Stratagene);pAgro4:Takeshita, S;Sato, M;Toba, M;Masahashi, W;Hashimoto-Gotoh, T(1987)Gene 61, 63-74;pHSG575:T. Tomoyasuら(2001), Mol. Microbiol. 40(2), 397-413)。
【0225】
それぞれの場合に6個の形質転換体を、100mlのエルレンマイヤー(バッフル付き)フラスコに入れた20mlのLBAmp/Spec/Cm(100μg/LのAmp;50mg/LのSpec;10μg/LのCm)、0.1mM IPTG、0.5g/Lのラムノース中で37℃にて18時間増殖させ、次いで10分間、5000gにて遠心分離し、10mM Tris/HCl, pH7.0を用いて1回洗浄し、2mlの同一緩衝液に再懸濁させた。100μlの細胞懸濁液を900μlの50mM MES、pH6(50μl/mlのグルコースDH(実施例1)、100mMグルコース、100mM NaCl、1mM NADH、1mM NADPHおよび10mM 3-クロロ-1-(チエン-2-イル)プロパン-1-オンを含有する)中で振とうしながら20分間インキュベートした。この混合物を実施例4と同様に分析した。平均0.13mMの3-クロロ-1-(チエン-2-イル)プロパン-1-オールが生成され、これは培養懸濁液1Lあたり6.6Uの活性に相当した。粗抽出物(振動式ミルにて0.7mlのガラスビーズ(d=0.5mm)を用いた細胞破砕(3×5分、その合間に氷上で冷却した)によって得られたもの)を含有する同様のアッセイ試料においても、0.21mMの3-クロロ-1-(チエン-2-イル)プロパン-1-オール(これは10.7U/Lの活性に相当する)が測定された。培養物にラムノースを加えなかったコントロール実験においては、わずかな3-クロロ-1-(チエン-2-イル)プロパン-1-オールも検出することはできなかった。
【0226】
実施例10:カンジダ・マグノリア(Candida magnoliae)Lu8472由来のデヒドロゲナーゼのクローニング
以下のアミノ酸配列は、Edmann配列決定によってN末端配列を再度決定した後に、実施例6に記載されるタンパク質精製より得られたものである:
(S,GまたはT)(TまたはP)TSNALVTGGSRGIGAA
【0227】
以下のさらなるアミノ酸配列は、ペプチドのトリプシン消化およびEdmann配列決定の後に得られたものである:
IGVNSINPG
【0228】
核酸配列(プライマーMke366, 367および374)は、Lu8472染色体DNA上で行われるPCR増幅によってデヒドロゲナーゼ遺伝子をクローニングするために以下のように使用したものであるが(3倍濃縮のリチカーゼ溶液を用いたゲノムDNAキット、Qiagen、Hilden)、カンジダ・マグノリア(Candida magnoliae)のコドン使用頻度を考慮して、これらのペプチドから推定した。
【0229】
【0230】
プライマーMke366およびMke367を1:1の比率で混合した。PCRは、Pfu Turboポリメラーゼ(Stratagene)および以下の温度勾配プログラムを用いた標準的なStratageneプロトコルに従って行った:95℃にて1分;95℃にて1分、X℃1にて45秒および72℃にて2分を30サイクル;72℃にて10分;使用するまで10℃。PCR産物(約0.5 kb)をアガロースゲル電気泳動(1.2%E-Gel、Invitrogen)およびカラムクロマトグラフィー(GFX-Kit、Amersham Pharmacia)によって単離し、次いで配列決定した(配列決定プライマー:Mke366およびMke374)。得られた配列を配列番号5に示す。それから推定されるアミノ酸配列(配列番号6)は、カンジダ・マグノリア(Candida magnoliae)のカルボニル還元酵素(WO200140450)と53%の同一性を示す。タンパク質精製の後に決定したペプチド配列には、ごくわずかな相違が存在する。この相違は、配列決定の誤りによるものか、またはカンジダ・マグノリア(Candida magnoliae)Lu8742に複数のイソ酵素が存在することにより生じたと考えられる。
【0231】
112個のアッセイ試料を異なるアニーリング温度、すなわち25℃から45℃までの温度(それぞれの場合に約2℃の増分)で実験した。すべてのアッセイ試料において、類似した濃度の0.5kbバンドが主産物として形成された。
【0232】
【技術分野】
【0001】
本発明は、式I:
【化1】
【0002】
で表される3-メチルアミノ-1-(チエン-2-イル)プロパン-1-オールを調製するための方法に関し、特にS-エナンチオマーI-Sを調製するための方法に関する。
【背景技術】
【0003】
式I-S:
【化2】
【0004】
で表されるアミノプロパノールIのSエナンチオマーは、式II:
【化3】
【0005】
(式中、Bは、n価の負に荷電した無機酸または有機酸のラジカルであり、HnBは、医薬的に許容される酸である)で表される抗うつ剤デュロキセチンを合成するための重要な前駆体である。
【0006】
デュロキセチンまたはその対応する塩基を調製するための先行技術の方法は、複雑であり、キラルな試薬またはキラルな出発化合物の使用を必要とする。
【0007】
従って、EP-B-0273658には、デュロキセチンの対応する塩基を調製するための方法が記載されており、この方法は、Mannich反応で2-アセチルチオフェンをホルムアルデヒドおよびジメチルアミンと反応させ、得られたMannich塩基のケト基を還元してラセミ体(S)-3-N,N-ジメチルアミノ-1-(チエン-2-イル)プロパン-1-オールを得、そのアルコール官能基をフッ化ナフチルでエーテル化し、最終的にそのジメチルアミノ基をメチルアミノ官能基に変換することによるものである。ナフチルエーテルの所望のエナンチオマーは、キラルな出発材料を使用することによって、または最終生成物の工程でのラセミ化合物の分割によって、例えば光学活性酸との塩またはキラルな固定相でのクロマトグラフィーによって得られる。
【0008】
US-5,362,886には、類似する方法が記載されており、その方法では、ケト基の還元後に得られるラセミ体のプロパノールにS-マンデル酸を加える。これに関連して得られるアルコールのSエナンチオマーは後続の反応工程で使用される。
【0009】
さらに、EP-A-0457559には、EP-B-0273658に記載される方法と類似する方法が記載されている。この場合には、Mannich塩基のケト基を、不斉還元系であるLAH-lcb(水素化アルミニウムリチウム-[(2R,2S)-(-)-4-ジメチルアミノ-1,2-ジフェニル-3-メチル-2-ブタノール])により還元すると、Sエナンチオマーの形のアルコールが得られる。費用は別にして、この方法の難点は、還元系LAH-lcbが不安定であって、数分間しか安定でない点である。
【0010】
Journal of Labelled Compounds and Radiopharmaceuticals, volume XXXVI, No. 3, pp. 213〜223において、W. J. WheelerおよびF. Kuoもまた、デュロキセチンの調製方法を記載している。この方法では、チオフェン-2-カルボニルクロリドを、Stilleカップリングにおいて、DMPU(ジメチルプロピレンウレア)中、触媒量のベンジル(クロロ)-ビス(トリフェニルホスフィン)パラジウム(II)の存在下で、ビニルトリ-n-ブチルスタンナンと反応させて、式II:
【化4】
【0011】
で表される1-(チエン-2-イル)プロペノンを得、これを続いて塩化水素で処理することによって式III.1:
【化5】
【0012】
で表される3-クロロ-1-(チエン-2-イル)プロパン-1-オンに変換する。続いて、この方法で得られるクロロプロパノンIII.1をキラルなオキサザボロリジン(oxazaborolidine)およびBH3を用いて還元すると、式IV.1-S:
【化6】
【0013】
で表される(S)-3-クロロ-1-(チエン-2-イル)プロパン-1-オールが得られる。
【0014】
この方法で得られるアルコールIV.1-Sは、ヨウ化ナトリウム、次いで、メチルアミンと連続して反応させると、(S)-3-メチルアミノ-1-(チエン-2-イル)プロパン-1-オールI-Sに変換される。続いて水素化ナトリウム、1-フルオロナフタレンおよび塩化水素と連続して反応させることにより、デュロキセチンを塩酸塩の形態で得る。これに関連して、不都合な点は、第一に、クロロプロパノン中間体III.1を調製するために複数の工程および高価な試薬が必要である点である。第二に、クロロプロパノール IV.1-Sは、クロロプロパノンIII.1がアミノアルコールに変換されるときに単離される。しかし、本発明者らの行った研究によって、このクロロプロパノールは不安定であり、強い発熱反応できわめて容易に分解され、アミノアルコールに関して収量の低下を招くだけでなく、産業的規模でこの反応を管理することをより困難にすることが分かっている。
【0015】
W. J. Wheelerらによって記載される、デュロキセチン合成中に中間体として生成する3-クロロ-1-(チエン-2-イル)プロパン-1-オンの調製方法は、文献から公知である。しかし、先行技術より公知である方法の欠点は、クロロプロパノンIII.1が低収率で形成されるか、あるいは取り扱いの難しい試薬を用いる必要があるか、のいずれかである。従って、CR Acad. Sci., Ser. C, 1979, 288(1), 49-52において、A. Etienneらは、溶媒としてのニトロメタン中でルイス酸触媒としての三塩化アルミニウムの存在下に、塩化3-クロロプロピオニルとチオフェンとのFriedel-Crafts反応によって、クロロプロパノンIII.1を調製する方法を記載する。このクロロプロパノンIII.1は、わずか7%の収率でしか得られない。LiuらによってChirality, 12, 26-29(2000)に記載されている対応する反応もまた、ルイス酸触媒としての四塩化スズと、溶媒としてのベンゼンの存在下で、満足のいく収率をもたらさない。Meth-Cohnらは、Acta Chem. Scand. B 20(6), 1577-1587(1966)において、三塩化鉄または三塩化アルミニウムの存在下でのチオフェンに対するFriedel-Craftsアシル化を記載しており、クロロプロパノンIII.1は中程度から良好な収率で生成される。この方法の欠点は、溶媒として二硫化炭素を使用する必要がある点である。
【0016】
Kamal, G. B. R. Khanna, R. RamuおよびT. Krishnajiらは、Tetrahedron Lett. 44, 2003, 4783-4787に、デュロキセチン前駆体である(S)-3-ヒドロキシ-3-(チエン-2-イル)プロパンニトリルを調製する方法を記載しており、この方法では、塩化クロロアセチルでチオフェンをアセチル化し、そのケトンを水素化ホウ素ナトリウムで還元してラセミ体アルコールを得、その塩素ラジカルをシアニドで置換し、ラセミ体ニトリルアルコールをシュードモナス・セパシア(Pseudomonas cepacia)のリパーゼ(ケイ藻土に固定化したもの;Rエナンチオマーのエステル化を選択的に触媒する)の存在下で酢酸ビニルと反応させて、所望のSエナンチオマー(エステル化されないままで残る)が純粋な形態で単離できるようにする。この方法の欠点は、多数の反応工程を必要とする一方で、エステル化されたR成分がデュロキセチンを生じるようにそれ以上反応しないため、半量のニトリルアルコールが無駄になる点である。
【発明の開示】
【発明が解決しようとする課題】
【0017】
従って、本発明の目的は、上記の先行技術の欠点を克服した3-メチルアミノ-1-(チエン-2-イル)プロパン-1-オールIの調製方法を提供することである。さらに、この方法は、全体的に良い収率で化合物Iを供給し、特にまた、エナンチオ選択的にSエナンチオマーI-Sの調製を可能にすべきである。
【課題を解決するための手段】
【0018】
本発明者らは、上記の目的が以下の方法で達成されることを見出した。最初に、チオフェンを、Friedel-Crafts反応でルイス酸の存在下に、ハロゲン化β-ハロプロピオニルまたはハロゲン化アクリロイルと反応させるが、その際、反応生成物を単離する前にハロゲン化水素を通過させる。その後、得られた式III:
【化7】
【0019】
Hal=ハロゲン
で表される3-ハロ-1-(チエン-2-イル)-プロパン-1-オンのケト基を還元し、式IV:
【化8】
【0020】
で表される還元生成物をメチルアミンと反応させる。
【0021】
かくして、本発明は、式I:
【化9】
【0022】
で表される3-メチルアミノ-1-(チエン-2-イル)プロパン-1-オールの調製方法に関し、この方法では、
a)チオフェンを、ルイス酸の存在下で、ハロゲン化β-ハロプロピオニルまたはハロゲン化アクリロイルと反応させ、その際、同時に、または反応が生じた後であるが反応生成物を単離する前にハロゲン化水素を通過させて、3-ハロ-1-(チエン-2-イル)プロパン-1-オンを得、そして
b)工程a)で得られたプロパノンを還元し、次いで、適宜に反応生成物を単離することなく、メチルアミンと反応させる。
【0023】
本発明による方法は、全体として良い回収率で目的化合物Iを提供する。さらに、このハロプロパノン中間体IIIの調製は、高価な有機スズ試薬の使用を必要としない。取り扱いが困難なハロプロパノールIVを単離する必要もない。さらに、この方法は、(S)-3-ハロ-1-(チエン-2-イル)プロパン-1-オールI-Sの形成に関して選択性を示すキラル触媒またはキラル還元剤の存在下にて、ハロプロペノン中間体IIIを還元することによる簡便な方法で、エナンチオ選択的にSエナンチオマーI-Sを調製することを可能とする。
【図面の簡単な説明】
【0024】
【図1】本発明により単離されたLu10288デヒドロゲナーゼの活性染色ゲルを示す;レーン1:分子量標準、下から:47kDa、74kDa、121kDaおよび205kDa;レーン2:空き;レーン3:ホモジェネート上清;レーン4:有用なQセファロースフラクション;レーン5:有用なQセファロースフラクション(3倍量);レーン6:有用なSuperdexフラクション;レーン7:有用なMono-Qフラクション;レーン8:有用なMono-Pフラクション。
【図2】図2Aおよび2Bは、ラクトバチラス・ブレビス(Lactobacillus brevis)デヒドロゲナーゼを用いて還元することによって(S)-3-メチルアミノ-1-(チエン-2-イル)プロパン-1-オールI-Sを調製するために用いた異なる混合物の典型的な反応経過を示す。
【図3A】本発明により単離されたLu10288デヒドロゲナーゼのブロットバンドのN末端配列決定の結果を示す。
【図3B−1】本発明により精製されたLu10288デヒドロゲナーゼの異なるタンパク質分解断片の配列決定データを示す;配列決定ステップにつき複数のシグナルが得られた場合に、主要な配列を確立することができる限り、これは二重下線によって示される。読み取ることができなかった位置は「/」で示される;同定がやや不確かである位置については、そのアミノ酸残基が丸かっこで示される。いずれが明確に優先されるのか特定することができない場合には、1つの位置に2つのアミノ酸が示される。「?」は不明または同定不能なアミノ酸を示す。
【図3B−2】図3B−1の続き。
【図3B−3】図3B−2の続き。
【図3B−4】図3B−3の続き。
【図3B−5】図3B−4の続き。
【図3B−6】図3B−5の続き。
【発明を実施するための形態】
【0025】
本発明において、「エナンチオ選択性」とは、以下の式:
ee(%)=[Sエナンチオマー−Rエナンチオマー/(Sエナンチオマー+Rエナンチオマー)]×100
に従って、公知の方式で算出されるSエナンチオマーのエナンチオマー過剰率ee(%)が、少なくとも50%、好ましくは少なくとも80%、特に少なくとも90%、とりわけ少なくとも95%であることを意味する。
【0026】
アシル化の間、または好ましくはアシル化が起こった後であるが反応生成物を単離する前に、ハロゲン化水素を通過させることの結果として、実質的に少しの1-チエン-2-イル-プロペノンII(この化合物は先行技術の方法では副生成物として常に生成し、目的のアシル化生成物IIIの収率を減少させる)も、工程a)の最終生成物として得られない。
【0027】
1つの電子対空位を有する共有結合の金属ハロゲン化物および半金属ハロゲン化物は、ルイス酸として使用することに適している。一般に、これらは、チタン、スズ、アルミニウム、バナジウム、鉄またはホウ素のハロゲン化合物から選択される。塩化物が好ましいが、アルミニウムの場合には、二塩化モノアルキルアルミニウムおよび塩化ジアルキルアルミニウムも好適である。ホウ素の場合には、フッ化物もまた好ましい。好適なルイス酸の例は、四塩化チタン、三塩化ホウ素、三フッ化ホウ素、四塩化スズ、五塩化バナジウム、三塩化鉄、三塩化アルミニウム、二塩化アルキルアルミニウムおよび塩化ジアルキルアルミニウムである。三塩化アルミニウムを用いるのが特に好ましい。
【0028】
好適なハロゲン化β-ハロプロピオニルは、塩化3-クロロプロピオニルおよび臭化3-ブロモプロピオニルまたは塩化3-ブロモプロピオニルである。塩化3-クロロプロピオニルを用いるのが好ましい。
【0029】
ハロゲン化アクリロイルとしては、塩化アクリロイルを用いるのが好ましい。
【0030】
ハロゲン化β-ハロプロピオニルは、アシル化剤として工程a)に用いるのが好ましい。
【0031】
塩化水素および臭化水素が好適なハロゲン化水素である。ハロゲン化水素を使用するにあたり、そのハロゲン原子が用いるハロゲン化β-ハロプロピオニルのβ-ハロゲンラジカルに一致しているものが好ましい。従って、塩化3-クロロプロピオニルを用いる場合には、塩化水素を用いることが好ましい。
【0032】
Friedel-Craftsアシル化反応に通常用いられる溶媒はどれも、工程a)の反応で溶媒として用いるのに適している。原則的に、好適な溶媒は、用いる試薬(特にルイス酸)の反応性を低下させないか、または所定の反応条件下でルイス酸との競合反応に加わらない非プロトン性溶媒である。これら溶媒の例は、それ自体がアシル化される芳香族化合物(すなわち、チオフェン)、チオフェンよりもアシル化することが著しく困難な芳香族炭化水素、例えば、ベンゼン、ニトロベンゼンおよびハロゲン化芳香族炭化水素(例:クロロベンゼンおよびジクロロベンゼン)、さらに、ハロゲン化脂肪族炭化水素、特にハロアルカン、例えば、クロロメタン、ジクロロメタン、クロロホルム、四塩化炭素、クロロエタン、ジクロロエタンおよびトリクロロエタンである。上記溶媒の混合物もまた好適である。ニトロベンゼンまたはハロゲン化炭化水素のような溶媒を使用することも好ましく、この場合にはFriedel-Craftsアシル化を本質的に均一に進行させることができる。特に、ハロゲン化芳香族または脂肪族炭化水素を用いるのが好ましく、ハロアルカンが特に好ましいものである。とりわけジクロロエタンまたはクロロベンゼンが用いられる。
【0033】
ルイス酸は、理論上得られるアシル化チオフェンの量に基づいて少なくとも等モル量で用いなければならない。なぜなら、ルイス酸がケトンとの複合体を形成し、この複合体はアシル化の間に形成されて、Friedel-Crafts反応の慣用条件下で安定であるからである。ルイス酸は好ましくは、より小さな割合で用いられるアシル化成分(チオフェンまたはハロゲン化β-ハロプロピオニル) 1モルに対して、1:1〜1:2、好ましくは1:1〜1:1.5、特に1:1.1〜1:1.5のモル比率で用いられる。
【0034】
チオフェンおよびハロゲン化β-ハロプロピオニルは、好ましくは1:0.5〜1:2、好ましくは1:0.7〜1:1.5、特に1:0.8〜1:1.2のモル比率で用いられる。
【0035】
Friedel-Craftsアシル化に関連して一緒に加えられる試薬の順序は、二番目に重要である。従って、例えば、初めにルイス酸を溶媒に導入し、最初にハロゲン化β-ハロプロピオニルを、次いでチオフェンを加えることができる。あるいはまた、初めにチオフェンとルイス酸を導入し、それらにハロゲン化β-ハロプロピオニルを加えることも可能である。
【0036】
一般に、ハロゲン化β-ハロプロピオニルを加える場合には、冷却することが有利である。それはルイス酸と酸ハロゲン化物との反応が一般に、強く発熱するためである。反応温度は、特に溶媒に応じて、選択される。一般には、10℃〜40℃の範囲である。ハロ炭化水素、特にハロアルカンを用いる場合には、反応温度を最高でも50℃とすることが好適である。それは、そうしなければこの溶媒自体が反応するためである。反応温度は、好ましくは-20℃〜40℃、特に好ましくは0〜30℃である。
【0037】
原則的に、反応圧は限定的ではない。一般的に、反応は標準圧力下で行うが、しかし、また正の圧力または負の圧力下でも行うことができる。例えば、溶媒が非常に揮発性であるか、または標準条件下で液体でない場合、例えばクロロメタンの場合によくあることだが、正の圧力が用いられる。
【0038】
ハロゲン化水素は好ましくは、アシル化が起こったごに通過させる。先行技術の慣用方法(例えば、ガスクロマトグラフィー、薄層クロマトグラフィーまたはNMR分光法)は、アシル化反応の完了を達成するために用いることができる。ハロゲン化水素の通過にかける時間は、特に、バッチの大きさによって決まり、個々の場合において当業者が決定できる。一般に、ハロゲン化水素を、少なくとも、わずかな1-チエン-2-イルプロペノンも検出することができなくなるまで通過させる。
【0039】
アシル化生成物の後処理のために、反応混合物は一般に、形成されたケトン-ルイス酸複合体を分解するために、初めに加水分解によって処理する。水または希薄な鉱酸水溶液(例えば、希塩酸)を加水分解に用いる。さらなる精製および単離は、例えば、Organikum [有機化学の実際的な手段], VEB Deutscher Verlag der Wissenschaften, 第17版, 1988, 第323頁以降に記載されているような、公知の方法を用いて行う。
【0040】
本発明による方法は、工程a)において、高い収率で3-ハロ-(チエン-2-イル)-1-プロパノンIIIを生じる。アシル化反応の過程で形成されうる1-チエン-2-イルプロペノンIIは実質的に完全に3-ハロ-(チエン-2-イル)-1-プロパノンIIIに変換され、その結果反応混合物は、3-ハロ-(チエン-2-イル)-1-プロパノンIIIの収量に基づいて、せいぜい1モル%、特に好ましくは、せいぜい0.5モル%のプロペノンIIしか含まないようになる。
【0041】
工程b)におけるプロパノンIIIの還元は、例えば、金属水素化物もしくは半金属水素化物を用いるか、または好適な遷移金属触媒の存在下で水素を用いることによって、達成される。
【0042】
好適な金属水素化物または半金属水素化物はいずれも中性で、それぞれ金属および半金属の複合水素化物である。ケトンのアルコールへの還元に有用であることが判明している金属水素化物または半金属水素化物を用いると有利である。上記水素化物は好ましくは、ホウ素またはアルミニウムの水素化物である。
【0043】
好適な中性の水素化物の例は、ボラン(BH3;B2H6)、アラン(AlH3)、シラン(SiH4)、モノボランおよびジアルキルボラン、例えば、ビス(3-メチルブタ-2-イル)ボラン(ジシアミルボラン)または9-ボラビシクロ[3.3.1]ノナン(9-BBN)、モノアルキルアルミニウムおよびジアルキルアルミニウム化合物、例えば、ジイソブチルアルミニウム水素化物(DIBAL-H)、ならびにトリアルキルシラン、例えば、トリメチルシランまたはトリエチルシランである。
【0044】
好適な複合水素化物の例は、複合ホウ素水素化物、例えば、水素化ホウ素ナトリウム(NaBH4)、水素化ホウ素リチウム(LiBH4)もしくは水素化ホウ素カルシウム(Ca[BH4]2)、複合アルミニウム水素化物、例えば、水素化リチウムアルミニウム(LiAlH4)、複合アルキルホウ素水素化物もしくはアルコキシホウ素水素化物、例えば、水素化トリエチルホウ素リチウムもしくは水素化トリイソプロポキシホウ素カリウム(KB[OCH(CH3)2]3H)、または複合アルキルアルミニウム水素化物もしくはアルコキシアルミニウム水素化物、例えば、水素化ジエチルアルミニウムナトリウム、水素化ビス(2-メトキシエトキシ)アルミニウムリチウム(LiAl[OC2H4OCH3]2H2)、水素化ビス(2-メトキシエトキシ)アルミニウムナトリウム(NaAl[OC2H4OCH3]2H2;「red Al」)もしくは水素化トリス(tert-ブトキシ)アルミニウムリチウム(LiAl[OC(CH3)3]3H)などである。
【0045】
金属水素化物または半金属水素化物を用いた還元に好適な溶媒は、特に、用いる還元剤に左右されるが、反応条件下で還元される基をもともと含むべきではない。従って、上記の水素化物を用いた還元は、好ましくは、所定の反応条件下で還元され得る官能基を有しない非プロトン性溶媒中で行う。これらの溶媒の例は、芳香族ならびに脂肪族炭化水素、例えば、C5-C8-アルカンおよびC5-C8-シクロアルカン(例:ペンタン、ヘキサン、ヘプタン、シクロペンタン、シクロヘキサンおよびシクロオクタン)、芳香族化合物(例:ベンゼン、トルエン、ニトロベンゼン、クロロベンゼンおよびジクロロベンゼン)、さらに、4個〜8個の炭素原子を有する開放鎖および環状のエーテル(例:ジエチルエーテル、メチルtert-ブチルエーテル、テトラヒドロフランまたはジオキサン)、さらにまた、塩素化炭化水素、特にハロアルカン(例:クロロメタン、ジクロロメタン、クロロホルム、四塩化炭素、ジクロロエタンまたはトリクロロエタン)である。上記の溶媒の混合物も好適である。上記の芳香族炭化水素、エーテルまたはハロ炭化水素を用いることが好ましい。
【0046】
上記のいくつかの複合水素化物、特に反応性の低い水素化物(例えば、水素化ホウ素ナトリウム)を用いた還元はまた、プロトン性溶媒(C1-C3-アルコールなど、例えば、メタノール、エタノール、プロパノールもしくはイソプロパノール)の存在下で、または水溶液中でさえも行うことができる。この場合に、少なくとも1種の上記の非プロトン性溶媒と少なくとも1種のアルコールとからなる混合溶媒を用いることが好ましい。水素化物は、塩基性プロトン性溶液中でより安定であるために、プロトン性溶媒を用いる場合には、好適な塩基の存在下で反応を行うことが好ましい。好適な塩基の例は、水酸化アルカリ金属、例えば、水酸化ナトリウムもしくは水酸化カリウム、または炭酸アルカリ金属、例えば、炭酸ナトリウムもしくは炭酸カリウムである。水酸化ナトリウムを用いることが好ましい。
【0047】
多数の上記の金属水素化物または半金属水素化物を用いた場合に、還元はしばしば発熱を伴って進行するので、結果として反応熱を取り除きながら、すなわち冷却しながら、反応を行うことが好適である。この反応温度は、好ましくは-50〜40℃、特に好ましくは-30〜30℃、特に-20〜20℃である。この還元は、例えば、Organikum [有機化学の実際的な手段], VEB Deutscher Verlag der Wissenschaften, 第17版, 1988, 第492頁以降に記載されているような、先行技術の慣用方法を用いて実施できる。これらの方法においては、概してプロパノンIIIを初めに導入し、そして還元剤を少しずつ加える。しかし、プロパノンIIIと還元剤を少しずつ同時に加えることもできる。
【0048】
プロパノンIIIはまた、例えば、アルミニウムアルコキシド用いて還元することもできる。アルミニウムアルコキシドを用いたケトンの還元は、慣習的にメーヤワイン・ポンドルフ・ヴァーレイ還元とも称される。この還元において、ケトンは対応するアルコールに変換され、そしてアルコキシドは対応するアルデヒド(第一級アルコールから形成されたアルコキシドの場合)またはケトン(第二級アルコールから形成されたアルコキシドの場合)に同時に酸化される。この反応は、例えば、Organikum [有機化学の実際的な手段], VEB Deutscher Verlag der Wissenschaften, 第17版, 1988, 第486頁以降に記載されているような、公知の方法を用いて実施できる。
【0049】
さらに、プロパノンIIIはまた、接触水素化によって、すなわち、好適な遷移金属触媒の存在下にて、水素とプロパノンIIIとを反応させることによっても還元できる。この好適な遷移金属には、VIII族、VI族およびI族の金属、特に白金、ルテニウム、銅、クロムおよびニッケルが含まれる。この触媒は当然のことながら、これらがチオフェン基の水素化を触媒しないように選択すべきである。触媒は不均一系触媒または均一系触媒のいずれかとして用いられる。好適な方法は、原則的に公知であり、例えば、Transition Metals in Organic Synthesis. M. Beller, C. Bolm, Wiley-VCH, Weinheim, 1998, 第2巻, 第1頁以降(均一系触媒)または第81頁以降(不均一系触媒)に記載されている。
【0050】
工程b)における還元は、好ましくは、金属水素化物または半金属水素化物を用いて、特に好ましくは上記の複合金属水素化物または半金属水素化物の1種を用いて行われる。特に水素化ホウ素ナトリウムが用いられる。
【0051】
上記の非不斉還元剤を用いる場合には、プロキラルな3-ハロ-1-(チエン-2-イル)プロパン-1-オン IIIが主に、ラセミ体アルコール IVに還元される。これに対応して、メチルアミンを用いた後続の反応は、ラセミ体の3-メチルアミノ-1-(チエン-2-イル)プロパン-1-オールIをもたらす。
【0052】
好ましい実施形態においては、本発明による方法を用いて、式I-S:
【化10】
【0053】
で表される(S)-3-メチルアミノ-1-(チエン-2-イル)プロパン-1-オールを調製するか、またはこの化合物と共にそのRエナンチオマーI-Rを含む混合物(エナンチオマーI-Sが優勢である)を調製する。工程b)の還元は、(S)-3-メチルアミノ-1-(チエン-2-イル)プロパン-1-オールI-Sの形成に関して選択性を示すキラル還元剤またはキラル触媒の存在下で行われる。
【0054】
この方法を行う場合に、プロキラルなハロプロパノンIIIは、工程b)において、式IV-S:
【化11】
【0055】
で表されるS-ハロプロパノールまたはSおよびRエナンチオマーの混合物(Sエナンチオマーが優勢である)にエナンチオ選択的に還元される。この目的のために、不斉金属水素化物または不斉半金属水素化物を、例えば、工程b)の還元剤として用いるか、そうでなければこの還元を、不斉誘導を媒介する化合物の存在下で行う。
【0056】
不斉金属水素化物または不斉半金属水素化物は好ましくは、不斉アルミニウム水素化物、不斉ホウ素水素化物または不斉ケイ素水素化物である。好適な不斉ホウ素水素化物は、例えば、E. J. Corey, C. J. Helal, Angew. Chem. 1998, 110, 2092-2118またはM. M. Midland, L. A. Morell, Methoden Org. Chem. [有機化学の方法], Houben-Weyl, 第4版, volume E 21d, pp. 4082-4098, 1995(これらはその全体が本明細書中に参考として援用される)に記載されている。好適な不斉ケイ素水素化物は、特に、H. Brunner, Methoden Org. Chem. [有機化学の方法], Houben-Weyl, 第4版, volume E21d, pp. 4074-4081, 1995(これはその全体が本明細書中に参考として援用される)に記載されている。
【0057】
本発明において、不斉誘導を媒介する化合物とは、実際の還元剤に影響を及ぼすことによって(例えば、結合を形成するか、もしくは還元剤と複合体を形成することによって)、または水素原子もしくは別の還元構成成分を還元剤から取ることによって、プロパノンIIIの還元をエナンチオ選択的にする化合物として理解される。一般に、不斉誘導を媒介する化合物は、それら自体還元性ではない。
【0058】
一方で、これらの化合物は、不斉水素化触媒を含む。不斉水素化において、水素は、不斉触媒が生じうるエナンチオマーの1つのエナンチオ選択的な形成を促す間に、実際の還元剤として作用する。水素化は不均一相または均一相のいずれかで行うことができる。不均一相での不斉水素化の好適な触媒は、例えば、A. Baiker, H. U. BlaserらによってHandbook of Heterogeneous Catalysis, 第5巻(G. Ertl, H. KnoerzingerおよびJ.Weitkamp編集), Wiley-VCH, Weinheim, 1997, 2422-2436(これはその全体が本明細書中に参考として援用される)に記載されている。
【0059】
しかし、均一相での水素化の触媒は、非常に重要である。VIII族金属(例えば、白金またはルテニウム、特にルテニウム)と少なくとも1つのリン含有配位子とを含む触媒が、特に好適である。好適な触媒は、例えば、R. Noyori, T. Ohkuma, Angew. Chem. 2001, 113, 40-75(これはその全体が本明細書中に参考として援用される)に記載されている。ルテニウム(II)ジアミン複合体(ルテニウムが二座配位キラルジホスファン配位子にさらに結合している)が特に好適である。ハロプロパノンIIIをS-ハロプロパノール IV-Sに還元するのに好適であるジホスファン配位子の例は、以下の式Aの(R)-BINAP、式Bの(R,R)-DIOPおよび式Cの(R,R)-CHIRAPHOSである:
【化12】
【0060】
Ar=フェニル(BINAP) Ph=フェニル
4-メチルフェニル(TolBINAP)
3,5-ジメチルフェニル(XylBINAP)
【0061】
さらに、特に好ましい触媒は、ジアミン配位子を含む。好適なジアミン配位子の例は、それらのエナンチオマー形態における1,2-エチレンジアミン、1,2-ジフェニル-1,2-エチレンジアミンおよび1,2-シクロヘキサンジアミンである。
【0062】
概して、水素化は、上記の条件下にて実施される。
【0063】
以下の式D:
【化13】
【0064】
(式中、RはC1-C4-アルキル、例えば、メチル、エチル、プロピル、イソプロピル、n-ブチル、sec-ブチル、イソブチルまたはtert-ブチルであり、特にメチルである)で表されるオキサザボリリジン(oxazaborylidine)は、特に好ましい不斉誘導媒介化合物である。
【0065】
この試薬を用いて、高度のエナンチオ選択性でプロキラルケトンを第二級アルコールに変換することができる(E. J. Corey, Pure Appl. Chem. 62, 1209, 1990;E. J. Corey, J. O. Link, J. Am. Chem. Soc. 114, 1906, 1992)。ボラン(例えば、BH3、ジアルキルボラン、ジアルコキシボランまたはジアリールオキシボラン)が実際の還元剤として用いられる。クロロプロパノンIII.1とオキサザボリリジンD(R=メチル)およびBH3とを反応させてS-クロロプロパノール IV.1-Sを得る反応は、W. J. WheelerおよびF. KuoらによってJournal of Labelled Compounds and Radiopharmaceuticals, volume XXXVI, No. 3, pp. 213-223に既に記載されている。
【0066】
還元において、概して、オキサザボリリジンは触媒量で用いられる。ボランは、ハロプロパノンIIIに基づいて、少なくとも等モル量で用いられるが、過剰量で用いることが好ましい。この反応は一般に、好適な溶媒中で実施する。好適な溶媒とは、所定の反応条件下にて還元されうる基を有しない非プロトン性溶媒である。これらの溶媒には、特に上記のハロアルカン、芳香族化合物および環状または非環状エーテルが含まれる。エーテルを用いるのが好ましく、テトラヒドロフランを用いるのが特に好ましい。反応温度は、好ましくは-80〜20℃、特に好ましくは-30〜10℃である。この反応は概して、初めにオキサザボリリジンを溶媒に導入し、最初にボラン、次いでハロプロパノンIII、またはこれとは逆に、最初にハロプロパノンIII、次いでボランを所望の反応温度で加えることによって行われる。ただし、前者の添加手順が好ましい。反応の継続時間は、特に、バッチの大きさによって決まり、個々の場合において当業者によって決定されうる。
【0067】
意外にも、プロパノンIIIからS-ハロプロパノール IV-Sへのエナンチオ選択的な還元は、酵素によって触媒される場合によく進行し、特にデヒドロゲナーゼが存在する場合に都合よく行われることが見出された。従って、本発明による方法の別の好ましい実施形態では、デヒドロゲナーゼが還元剤として用いられる。
【0068】
本発明による方法で還元が完了した後、用いた還元方法に応じて、触媒または過剰量の還元剤を、適宜に、不活性化させるか、または除去する。金属水素化物または半金属水素化物を用いた場合には、この処理は一般的に加水分解(例えば、水性またはアルコール性溶液を用いる)によって行う。均一な水素化触媒を用いた場合、またはアルミニウムアルコキシドで還元した場合には、加水分解後処理もしばしば用いられる。還元が不均一相での接触水素化の形態で行われた場合、または還元剤として酵素を用いて行われた場合には、触媒または酵素を物理的な分離(例えば、デカントまたは濾過)によって取り除くことができる。しかし、均一還元系が用いられた場合、特に、金属水素化物または半金属水素化物を用いて還元を行った場合には、初めに不活性化しないことが好ましい。
【0069】
還元後に得られたハロプロパノール IV を単離することなく、その代わりにメチルアミンと直接的に反応させて、3-メチルアミノ-1-(チエン-2-イル)プロパン-1-オールIを得ることが有利であることが分かった。このために、反応混合物(適宜に不活性化してもよいし、不均一還元系から分離してもよい)をメチルアミンで処理し、好ましくは、昇温(例えば、30〜80℃、特に50〜70℃)にて反応させる。メチルアミンは気体または水溶液のいずれかとして用いることができるが、水溶液としての使用が好ましい。メチルアミンは、用いるハロプロパノンIIIの量に基づいて、好ましくは1〜100モル当量、特に好ましくは5〜10モル当量で用いる。この反応は1〜250バールの圧力下、特に系自体が発生する固有の圧力下にて行うことが好ましい。
【0070】
メチルアミンとの反応が完了した後、反応混合物を慣用方法で後処理する。そのために、触媒または還元剤を既に述べたように不活性化して分離し(このような処理がメチルアミンを加える前に行われなかった場合)、その後溶媒を除去し、純粋なメチルアミノプロパノールIを、例えば、結晶化、温浸、抽出またはクロマトグラフィーによって残渣から単離する。
【0071】
本発明による方法を用いて、良い収率で3-メチルアミノ-1-(チエン-2-イル)プロパン-1-オールIを得ることが可能である。特に、高価で、取り扱いが困難な試薬または溶媒を本方法の工程a)において必要とせず、またハロプロパノンIIIが非常に高い収率で生成される。工程b)では、不安定なハロプロパノール IVの単離、ならびにそれに伴う収率の低下および/またはハロプロパノール IVの困難な取り扱いが、回避される。さらに、本発明による方法を用いて、メチルアミノプロパノール I-SのSエナンチオマーを選択的に得ることができる。このSエナンチオマーは、工程b)における還元がキラル還元剤を用いて行われる場合に、デュロキセチンへのさらなる変換のために必須である。
【0072】
さらに、本発明はまた、3-ハロ-1-(チエン-2-イル)プロパン-1-オンIIIをエナンチオ選択的に還元することを含む、式I-Sの(S)-3-メチルアミノ-1-(チエン-2-イル)プロパン-1-オールの調製方法に関し、前記方法では、この還元をデヒドロゲナーゼの存在下にて行うことを含む。この方法の好ましい実施形態では、これに関連して得られる(S)-3-ハロ-1-(チエン-2-イル)プロパン-1-オールIV-Sを、単離することなく、メチルアミンと反応させる。好ましいデヒドロゲナーゼに関して、さらにこの方法の実施に関しては、以下の観察を特に参照されたい。
【0073】
生化学的な実施形態:
好適なデヒドロゲナーゼ(EC 1.1.X.X)は、特に、デヒドロゲナーゼ(E.C. 1.1.1.x)、特にアルコールデヒドロゲナーゼ(E.C.1.1.1.1またはE.C.1.1.1.2)である。これらは、ハロプロパノンIIIのS-ハロプロパノール IV-Sへの選択的な還元をもたらす。このデヒドロゲナーゼは好ましくは、微生物、特に好ましくは細菌または真菌(特に酵母)から得られる。これは、それぞれ菌株コレクションに寄託されたものか、または土壌試料、バイオマス試料などの天然供給源の分離株から得られる。デヒドロゲナーゼは特に酵母または乳酸菌に由来するものである。
【0074】
デヒドロゲナーゼは、精製されたもしくは部分的に精製された形態、または微生物自体の形態で用いることができる。デヒドロゲナーゼを微生物から単離および精製するための方法は、当業者に周知である(例えば、K. DrauzおよびH. WaldmannのEnzyme Catalysis in Organic Synthesis 2002, Vol.III, 991-1032, Wiley-VCH, WeinheimにおけるK. Nakamura & T. Matsudaらによる「Reduction of Ketons」)。デヒドロゲナーゼを産生させる組換え法も同じように知られている(例えば、W. Hummel, K. Abokitse, K. Drauz, C. Rollmannおよび H. Groegerらによる Adv. Synth. Catal. 2003, 345, Nos. 1+2, pp. 153-159)。
【0075】
好適な細菌の例は、シュードモナス(Pseudomonas)属、ブルクホリデリア(Burkholderia)属、アグロバクテリウム(Agrobacterium)属、ロドコッカス(Rhodococcus)属、ラクトバチラス(Lactobacillus)属、またはラクトコッカス(Lactococcus)属のものである。好適な酵母の例は、ゲオトリカム(Geotrichum)属、ピチア(Pichia)属、カンジダ(Candida)属、ハンセヌラ(Hansenula)属、またはサッカロマイセス(Saccharomyces)属のものである。
【0076】
酵母および乳酸菌に由来するデヒドロゲナーゼを用いることが特に好ましい。これらのデヒドロゲナーゼのうち、ゲオトリカム(Geotrichum)属もしくはカンジダ(Candida)属の酵母から得られるか、またはラクトバチラス(Lactobacillus)属、もしくはラクトコッカス(Lactococcus)属の乳酸菌から得られるデヒドロゲナーゼが好ましい。
【0077】
ラクトバチラス(Lactobacillus)属の菌種の例は、L.ブレビス(brevis)、L.ケフィア(kefir)、L.プランタルム(plantarum)、L.カセイ(casei)、L.アシドフィルス(acidophilus)、L.デルブリュッキィ(delbrueckii)およびL.サンフランシスコ(sanfrancisco)である。
【0078】
カンジダ(Candida)属の菌種の例は、C.マグノリア(magnoliae)、C.ルゴサ(rugosa)、C.ユチリス(utilis)、C.ボイジニィ(boidinii)、C.パラプシローシス(parapsilosis)およびC.アンタルティカ(antarctica)である。
【0079】
ゲオトリカム(Geotrichum)属の菌種の例は、G.カンディダム(candidum)、G.クラバタム(clavatum)およびG.フェルメンタンス(fermentans)である。
【0080】
カンジダ・マグノリア(Candida magnoliae)、ゲオトリカム・カンジドウム(Geotrichum candidum)またはラクトバチラス・ブレビス(Lactobacillus brevis)に由来するデヒドロゲナーゼを用いることが特に好ましい。
【0081】
デヒドロゲナーゼによる還元は一般に、好適な補酵素(補因子とも称される)の存在下にて行われる。NADHおよび/またはNADPHは一般に、ケトンを還元するための補酵素として用いられる。さらに、デヒドロゲナーゼを細胞系(もともと補因子を含むか、細胞系に選択的レドックス媒介物が加えられている)として用いることが可能である(K. DrauzおよびH. WaldmannらのEnzyme Catalysis Organic Synthesis 2002, Vol.III, 991-1032, Wiley-VCH, Weinheimにおける、A. Schmidt, F. Hollmann および B. Buehlerらの「Oxidation of Alcohols」)。
【0082】
デヒドロゲナーゼによる還元はまた、一般に好適な補基質の存在下にて行われる。この補基質は概して、還元過程の間に酸化される補酵素の還元剤として作用し、その結果この補酵素を再生させる。好適な補基質の例は、糖、特にヘキソース、例えば、グルコース、マンノースまたはフルクトース、および/または被酸化性アルコール、特にエタノール、プロパノール、またはイソプロパノールであり、またギ酸もそうである。第二のデヒドロゲナーゼ(例えば、グルコースを補基質として用いる場合には、グルコースデヒドロゲナーゼ)が補基質を酸化する目的で、またこれに関連して補酵素を再生させるために加えられる。このデヒドロゲナーゼは、遊離もしくは固定化酵素として、または遊離もしくは固定化細胞の形態で用いることができる。それは、(組換え)デヒドロゲナーゼ株において別々にまたは共発現によって調製することができる。
【0083】
本発明により用いられるデヒドロゲナーゼは、遊離または固定化形態で用いることができる。固定化酵素とは、不活性な支持体に固定された酵素を意味するものとして理解される。好適な支持体材料およびそれらに固定される酵素は、EP-A-1149849、EP-A-1069183およびDE-OS 100193773、またそれらに引用されている参考文献から公知である。これに関して、これら文献における開示内容は、その全体が本明細書中に参考として援用される。好適な支持体材料としては、例えば、粘土、粘土鉱物(例:カオリナイト、ケイ藻土)、パーライト、二酸化ケイ素、酸化アルミニウム、炭酸ナトリウム、炭酸カルシウム、セルロース粉末、アニオン交換材料、合成ポリマー(例:ポリスチレン、アクリル樹脂、フェノールホルムアルデヒド樹脂、ポリウレタン、およびポリエチレンやポリプロピレンなどのポリオレフィン)が挙げられる。この支持体材料は支持体に担持された酵素を調製するために用いられ、一般に微細な粒子状をしており、多孔形態が好ましい。支持体材料の粒径は一般に、5mmを超えず、特に2mmを超えない(粒度曲線)。類似の方法において、全細胞体触媒としてデヒドロゲナーゼを用いる場合には、遊離形態または固定化形態を選択することが可能である。支持体材料の例は、アルギン酸Caおよびカラギーナンである。酵素および細胞はグルタルアルデヒドで直接的に架橋することもできる(CLEAを得るための架橋)。
【0084】
対応するおよびさらなる固定化方法は、例えば、K. DrauzおよびH. WaldmannらのEnzyme Catalysis in Organic Synthesis 2002, Vol.III, 991-1032, Wiley-VCH, Weinheimにおける、J. LalondeおよびA. Margolinらによる「Immobilization of Enzymes」に記載されている。
【0085】
還元は、水性または非水性の反応媒体中または2相系もしくは(マイクロ)エマルションにて行うことができる。水性反応媒体は、好ましくは緩衝溶液であり、概して、pH5〜8、好ましくはpH6〜8である。水以外に、水性溶媒は少なくとも1つのアルコール(例えば、エタノールまたはイソプロパノール)をさらに含むことができる。
【0086】
「非水性反応媒体」とは、反応媒体の全重量に基づいて、1重量%未満、好ましくは0.5重量%未満の水を含む反応媒体を意味するものとして理解される。この反応は好ましくは、有機溶媒中で行われる。好適な溶媒の例は、脂肪族炭化水素、好ましくは5個〜8個の炭素原子有する脂肪族炭化水素(例:ペンタン、シクロペンタン、ヘキサン、シクロヘキサン、ヘプタン、オクタンもしくはシクロオクタン)、ハロゲン化脂肪族炭化水素、好ましくは1個〜2個の炭素原子を有するハロゲン化脂肪族炭化水素(例:ジクロロメタン、クロロホルム、四塩化炭素、ジクロロエタンもしくはテトラクロロエタン)、芳香族炭化水素(例:ベンゼン、トルエン、キシレン、クロロベンゼンまたはジクロロベンゼン)、脂肪族非環状エーテルおよび脂肪族環状エーテル、好ましくは4個〜8個の炭素原子を有するもの(例:ジエチルエーテル、メチルtert-ブチルエーテル、エチルtert-ブチルエーテル、ジプロピルエーテル、ジイソプロピルエーテル、ジブチルエーテルもしくはテトラヒドロフラン)、またはエステル(例:酢酸エチルもしくは酢酸n-ブチル)、またはケトン(例:メチルイソブチルケトンもしくはジオキサン)、あるいはそれらの混合物である。上記のエーテル、特にテトラヒドロフランを用いることが特に好ましい。
【0087】
例えば、デヒドロゲナーゼによる還元は、水性-有機反応溶媒、特に水性反応溶媒中にて行われる。
【0088】
ハロプロパノンIIIは、好ましくは、酵素による還元において、0.1g/l〜500g/l、特に好ましくは、1g/l〜50g/lの濃度で用いられ、その後、連続的にまたは断続的に供給することができる。
【0089】
一般に、酵素による還元は、用いるデヒドロゲナーゼが不活性化される温度よりも低い反応温度、好ましくは少なくとも-10℃である反応温度で行われる。例えば0〜100℃、好ましくは15〜60℃、特に20〜40℃の範囲、例えば約30℃とすることが特に好ましい。
【0090】
反応を実施するために、例えば、初めに、ハロプロパノンIIIをデヒドロゲナーゼ、溶媒、適宜に補酵素、適宜に補酵素を再生するための補助デヒドロゲナーゼ、および/または他の補基質と一緒に導入し、その混合物を、例えば撹拌または振とうによって、混合することが可能である。しかし、また、反応器(例えば、カラム)にデヒドロゲナーゼを固定化し、ハロプロパノンIIIと適宜に補酵素および/または補基質を含有する混合物をこの反応器に通過させることも可能である。このために、目的の変換が達成されるまで、循環方式にて混合物を反応器に通すことが可能である。これに関連して、ハロプロパノンIIIのケト基はOH基に還元され、それに伴ってハロプロパノール IV-S のSエナンチオマーが本質的に選択的に形成される。一般に、還元は、混合物中に存在するハロプロパノンIIIに基づいて、変換率が少なくとも70%、特に好ましくは少なくとも85%、特に少なくとも95%に達するまで行われる。これに関連して、反応の進行(すなわち、ケトンの連続的な還元)はガスクロマトグラフィーのような慣用方法によって追跡することができる。
【0091】
本発明による方法に特に好ましく用いられるデヒドロゲナーゼは、以下の特性の少なくとも1つを有するアルコールデヒドロゲナーゼである:
N末端領域に、a) 配列番号1に示される、少なくとも5、6、7または8個、好ましくは少なくとも10個(例えば、10、11、12、13、14または15個)連続したアミノ酸残基の構成的なアミノ酸配列を含み、配列番号1に示されるアミノ酸位置12に対応する位置が、好ましくはさらにバリンを表す;またはb) 配列番号2に示される、少なくとも5、6、7または8個、好ましくは少なくとも10個(例えば、10、11、12、13、14または15個)連続したアミノ酸残基の構成的なアミノ酸配列を含む、アミノ酸配列を有するアルコールデヒドロゲナーゼおよびそれらの機能的等価物。
【0092】
3-クロロ-1-(チエン-2-イル)プロパン-1-オンを(S)-3-クロロ-1-(チエン-2-イル)プロパン-1-オールに還元することが可能である、アルコールデヒドロゲナーゼ。
【0093】
少なくとも85%eeのエナンチオマー純度にて(NADHおよび/またはNADPHの存在下;30℃かつpH6.0にて)還元を触媒する、アルコールデヒドロゲナーゼ。
【0094】
配列番号3を含む核酸配列によってコードされるか、または配列番号4に示されるアミノ酸配列もしくは少なくとも図3に示される構成的な配列を含み、好ましくは、ラクトバチラス・ブレビス(Lactobacillus brevis)から得ることができるアルコールデヒドロゲナーゼ、およびそれらに由来する機能的に等価なアルコールデヒドロゲナーゼ。
【0095】
配列番号5を含む核酸によってコードされるか、または配列番号6を含むアミノ酸配列を有し、好ましくは、カンジダ・マグノリア(Candida magnoliae)(ATCC 12573)から得ることができるアルコールデヒドロゲナーゼ、およびそれらに由来する機能的に等価であるアルコールデヒドロゲナーゼ。
【0096】
本発明はまた、上に列挙した特性の少なくとも1つを有する(S)-3-クロロ-1-(チエン-2-イル)-プロパン-1-オールデヒドロゲナーゼに関する。
【0097】
デヒドロゲナーゼが安定である温度範囲は、好ましくは10〜80℃である。その活性が最適である温度は、好ましくは20〜60℃、特に好ましくは25〜40℃の範囲である。安定であるpH範囲は好ましくはpH4〜10である。脱水素に最適なpHは好ましくはpH5〜8の範囲である。
【0098】
さらに、本発明によるデヒドロゲナーゼは好ましくは、NAD+またはNADP+の存在下にて、(S)-3-クロロ-1-(チエン-2-イル)プロパン-1-オールを脱水素して3-クロロ-1-(チエン-2-イル)プロパン-1-オンを生成することが可能なものである。デヒドロゲナーゼの分子量は30±2キロダルトンの範囲にあることが好ましい。
【0099】
本発明はまた、本発明によるデヒドロゲナーゼのコード配列または構成的なコード配列を含む核酸配列に関する。この非限定的な例は、配列番号3または配列番号6に示される配列である。
【0100】
本発明はさらに、少なくとも1つの調節核酸配列と機能的に連結された、本発明による核酸配列を含む発現カセットに関する。
【0101】
本発明はさらに、本発明による発現カセットを少なくとも1つ含む組換えベクターに関する。
【0102】
さらに、本発明は、本発明によるベクターを少なくとも1つ用いて形質転換した原核細胞または真核細胞の宿主に関する。
【0103】
最後に、本発明は、(S)-3-ハロ-1-(チエン-2-イル)プロパン-1-オールを調製するための、特にデュロキセチンを調製するための、本発明によるデヒドロゲナーゼまたはこのデヒドロゲナーゼを産生する微生物の使用に関する。
【0104】
本発明によるデヒドロゲナーゼのさらなる改変:
本発明はまた、(S)-3-クロロ-1-(チエン-2-イル)プロパン-1-オールデヒドロゲナーゼ活性を有する明確に開示された酵素の「機能的等価物」および本発明の方法におけるそれら等価物の使用を含む。
【0105】
本発明に関して、明確に開示された酵素の「機能的等価物」または類似体は、これらの酵素とは異なるが、依然として所望の生物学的活性(例えば、基質特異性)を保有するポリペプチドである。従って、「機能的等価物」とは、例えば、3-クロロ-1-(チエン-2-イル)プロパン-1-オンを対応するSアルコールに還元し、以下に列挙される配列番号1、2、4もしくは6または図3に示されるアミノ酸配列の1つを含む酵素の少なくとも20%、好ましくは50%、さらに好ましくは75%、特に好ましくは90%の活性を有する酵素を意味するものとして理解される。機能的等価物はまた、好ましくはpH4〜10の間で安定であり、有利には、最適pHをpH5〜8に、最適温度を20℃〜80℃の範囲に有する。
【0106】
本発明によると、「機能的等価物」とはまた、特に、上記のアミノ酸配列の少なくとも1つの配列位置に、特に挙げたアミノ酸以外のアミノ酸を有するが、それでも上記の生物学的活性の1つを保有する変異体を意味するものとしても理解される。したがって、「機能的等価物」とは、1個または複数のアミノ酸付加、アミノ酸置換、アミノ酸欠失および/またはアミノ酸転移によって得ることができる変異体を含む。かかる変化は、本発明に従う特性プロファイルを有する変異体をもたらす限り、どのような配列位置にも出現しうる。また、機能的等価物が存在するのは、特に、変異型および非変異型のポリペプチドの反応パターンが質的に一致する場合、すなわち、同一の基質が例えば異なる速度で変換される場合である。
【0107】
好適なアミノ酸置換の例を以下の表に列挙する:
【0108】
上記趣旨での「機能的等価物」とはまた、記載したポリペプチドの「前駆体」ならびにそのポリペプチドの「機能的誘導体」および「塩」でもある。
【0109】
これに関連して、「前駆体」とは、ポリペプチドの天然または合成の前駆体であり、この前駆体は所望の生物学的活性を保持しても保持しなくてもよい。
【0110】
語句「塩」とは、本発明によるタンパク質分子のカルボキシル基の塩とアミノ基の酸付加塩との両方を意味するものとして理解される。カルボキシル基の塩は、それ自体公知の方法で調製することができ、無機塩(例えば、ナトリウム、カルシウム、アンモニウム、鉄および亜鉛の塩)、ならびに有機塩基(例えば、トリエタノールアミン、アルギニン、リジン、ピペリジンなどのアミン)との塩を含む。酸付加塩、例えば鉱酸(例:塩酸または硫酸)との塩ならびに有機酸(例:酢酸およびシュウ酸)との塩などは、本発明の一部を構成する。
【0111】
本発明によるポリペプチドの「機能的誘導体」は同様に、公知の手法を用いて、機能的アミノ酸の側鎖基またはそれらのN末端もしくはC末端に、調製することができる。これらの誘導体は、例えば、カルボン酸基の脂肪族エステル;カルボン酸基のアミド(このアミドは、アンモニアまたは第一級もしくは第二級アミンと反応させることによって得ることができる);遊離アミノ基のNアシル誘導体(この誘導体は、アシル基と反応させることによって調製される);または、遊離ヒドロキシル基のO-アシル誘導体(この誘導体はアシル基と反応させることによって調製される)を含む。
【0112】
タンパク質のグリコシル化が起こると考えられる場合、本発明による「機能的等価物」とは、上記のタイプのタンパク質の脱グリコシル化またはグリコシル化形態、ならびにグリコシル化パターンを変えることによって得られる改変形態を含む。
【0113】
「機能的等価物」とは当然に、他の生物体および天然の変異体から得ることができるポリペプチドもまた含む。例えば、相同配列領域の部分は配列比較によって確立することができ、また同等の酵素は本発明の具体的なのガイドラインを用いて同定することができる。
【0114】
「機能的等価物」とはまた、本発明によるポリペプチドの断片(好ましくは、個々のドメインまたは配列モチーフ)も含み、かかる断片は、例えば、所望の生物学的機能を有する。
【0115】
さらに、「機能的等価物」とは、上記のポリペプチド配列またはそれらに由来する機能的等価物の1つと、それらと機能的に異なりかつN末端またはC末端で機能的に(すなわち、融合タンパク質成分の相互の機能が顕著に損なわれることなく)連結されている少なくとも1つのさらなる異種配列と、を含む融合タンパク質である。このような異種配列の非限定的な例は、シグナルぺプチドまたは酵素である。
【0116】
さらに本発明に含まれる「機能的等価物」とは、具体的に開示されたタンパク質の相同体である。これらの相同体は、Proc. Natl. Acad, Sci. (USA) 85(8), 1988, 2444-2448に記載されるPearsonおよびLipmanのアルゴリズムを用いて算出した場合に、具体的に開示されたアミノ酸配列の1つに対して少なくとも60%、好ましくは少なくとも75%、特に少なくとも85%、例えば、90%、95%、97%または99%の相同性を有する。本発明による相同ポリペプチドの相同性パーセントとは、特に、本明細書中に具体的に記載される1つのアミノ酸配列の全長にわたるアミノ酸残基の同一性パーセントを意味する。
【0117】
本発明によるタンパク質またはポリペプチドの相同体は、変異誘発によって(例えば、タンパク質の点突然変異またはトランケーションによって)作製することが可能である。
【0118】
本発明のタンパク質の相同体は、トランケーション変異体のような変異体のコンビナトリアル・ライブラリーのスクリーニングによって同定することができる。例えば、タンパク質変異体の多様なライブラリーは、核酸レベルでのコンビナトリアル変異誘発によって、例えば合成オリゴヌクレオチドの混合物を酵素的にライゲートすることによって、作製することができる。縮重オリゴヌクレオチド配列から潜在的相同体のライブラリーを調製するために用いることができる方法は多数存在する。縮重遺伝子配列を自動DNA合成装置で化学的に合成して、この合成遺伝子を好適な発現ベクターにライゲートすることができる。縮重遺伝子セットを用いると、所望の潜在的タンパク質配列のセットをコードするすべての配列を混合物として調製することが可能となる。縮重オリゴヌクレオチドを合成するための方法は、当業者に公知である(例えば、Narang, S.A.(1983)Tetrahedron 39:3;Itakuraら(1984)Annu. Rev. Biochem. 53:323;Itakuraら,(1984)Science198:1056;Ikeら(1983)Nucleic Acids Res. 11:477)。
【0119】
点突然変異またはトランケーションによって作製されたコンビナトリアル・ライブラリーの遺伝子産物をスクリーニングするための手法、および所定の性質を有する遺伝子産物についてcDNAライブラリーをスクリーニングするための手法は、先行技術において公知である。これらの手法は、本発明の相同体のコンビナトリアル変異誘発によって生じた遺伝子ライブラリーを迅速にスクリーニングするために適合させることができる。ハイスループット分析にかけられる大きな遺伝子ライブラリーをスクリーニングするために最も頻繁に用いられる手法は、遺伝子ライブラリーを複製可能な発現ベクターにクローニングし、得られたベクターライブラリーを用いて好適な細胞を形質転換し、所望の活性の検出が遺伝子(その産物が検出された)をコードするベクターの単離を容易にする条件下にて、コンビナトリアル遺伝子を発現させることを含む。リカーシブ・アンサンブル変異誘発(recursive ensemble mutagenesis)(REM)は、ライブラリー中の機能的変異体の頻度を高める手法であり、相同体を同定するためにスクリーニング試験と組み合わせて用いることができる(Arkin およびYourvan(1992)PNAS 89:7811-7815;Delgraveら(1993)Protein Engineering 6(3):327-331)。
【0120】
本発明によるコード核酸配列のさらなる実施形態:
本発明は、特に、本発明のデヒドロゲナーゼ活性を有する酵素をコードする核酸配列(一本鎖および二本鎖DNAおよびRNA配列、例えばcDNAおよびmRNA)に関する。例えば、配列番号1、2、4、6もしくは図3に示されるアミノ酸配列またはそれらの特徴的な構成配列をコードする核酸配列、あるいは配列番号3もしくは5に示される核酸配列またはそれらの特徴的な構成配列を含む核酸配列が好ましいものである。
【0121】
本明細書中で言及するすべての核酸配列は、それ自体公知の方法で、ヌクレオチド構成単位から化学合成によって、例えば、二重鎖の個々の重複した相補性核酸構成単位の断片縮合によって作製することができる。オリゴヌクレオチドは、例えば、公知の方法にて、ホスホアミダイト法を用いて化学的に合成できる(Voet, Voet, 第2版, Wiley Press New York, pages 896-897)。合成オリゴヌクレオチドの結合およびDNAポリメラーゼKlenow 断片によるギャップのフィルインならびにライゲーション反応、さらに一般的なクローニング方法は、Sambrookら(1989), Molecular Cloning:A laboratory manual, Cold Spring Harbor Laboratory Pressに記載されている。
本発明はまた、上記のポリペプチドおよびそれらの機能的等価物(例えば、人工的なヌクレオチド類似体を用いて入手可能)の1つをコードする核酸配列(一本鎖および二本鎖DNAおよびRNA配列、例えば、cDNAおよびmRNA)に関する。
【0122】
本発明は、本発明のポリペプチドもしくはタンパク質または生物学的に活性なそれらのセグメントをコードする単離された核酸分子および核酸断片の両方に関し、かかる核酸断片は、例えば本発明によるコード核酸を同定または増幅するためのハイブリダイゼーションプローブまたはプライマーとして用いることができる。
【0123】
本発明による核酸分子はさらに、コード遺伝子領域の3’末端および/または5’末端に由来する非翻訳配列を含むことができる。
【0124】
本発明はまた、具体的に記載されたヌクレオチド配列に相補的である核酸分子またはそれらのセグメントを含む。
【0125】
本発明のヌクレオチド配列は、別の細胞型および生物体に含まれる相同配列を同定および/またはクローニングするために用いることができるプローブおよびプライマーの作製を可能にする。これらのプローブまたはプライマーは一般に、「ストリンジェント」な条件(下記参照)下にて、本発明の核酸配列のセンス鎖または対応するアンチセンス鎖中の少なくとも約12個、好ましくは少なくとも約25個(例えば、約40個、50個または75個)連続したヌクレオチドとハイブリダイズするヌクレオチド配列領域を含む。
【0126】
「単離された」核酸分子とは、核酸の天然源に存在する他の核酸分子から分離されており、さらに、組換え技術によって調製された場合には、他の細胞性材料もしくは培養培地を実質的に含まず、また化学的に合成された場合には、化学的前駆体もしくは他の化学薬品を実質的に含まないものである。
【0127】
本発明による核酸分子は、標準的な分子生物学的手法および本発明により提供される配列情報を用いて単離することができる。例えば、cDNAは、具体的に開示された完全長配列またはそれらのセグメントをハイブリダイゼーションプローブとして用いて、例えば、Sambrook, J., Fritsch, E.F. および Maniatis, T. らによるMolecular Cloning:A Laboratory Manual. 第2版, Cold Spring Harbor Laboratory, Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY, 1989に記載されるような、標準的なハイブリダイゼーション手法を用いて、好適なcDNA ライブラリーから単離することができる。さらに、ポリメラーゼ連鎖反応を用いて、開示された配列またはそれらのセグメントの1つを含む核酸分子を、この配列に基づいて構築されたオリゴヌクレオチドプライマーを使用して、単離することができる。この方法で増幅した核酸は、好適なベクターにクローン化し、そしてDNA配列解析によって特徴づけることができる。本発明によるオリゴヌクレオチドはまた、標準的な合成方法によって、例えば、自動DNA合成装置を用いて調製できる。原則的に、本発明による核酸配列は任意の生物体において同定して、該生物体から単離することができる。有利には、本発明による核酸配列またはそれらの相同体は、真菌、酵母または細菌から単離できる。言及できる細菌はグラム陰性細菌およびグラム陽性細菌である。本発明による核酸は好ましくは、グラム陰性細菌、有利にはαプロテオ細菌、βプロテオ細菌またはγプロテオ細菌、好ましくはエンテロバクテリアセア(Enterobacteriaceae)科、シュードモナダセア(Pseudomonadaceae)科またはリゾビアセア(Rhizobiaceae)科の細菌、特に好ましくはアグロバクテリウム(Agrobacterium)属、シュードモナス(Pseudomonas)またはブルクホリデリア(Burkholderia)属の細菌から、当業者に公知の方法を用いて単離される。
【0128】
本発明による核酸配列は、例えばゲノムライブラリーまたはcDNAライブラリーを用いた、通常のハイブリダイゼーション法またはPCR手法により、例えば他の真菌または細菌から単離できる。これらのDNA配列は本発明による配列と標準条件下でハイブリダイズする。保存領域(例えば、活性中心)に由来する短いオリゴヌクレオチドをハイブリダイゼーションに用いることが有利であり、これらのオリゴヌクレオチドは、当業者に公知の方法で本発明によるデヒドロゲナーゼと比較することによって、同定することが可能である。しかし、本発明による核酸のより長い断片または完全長配列をハイブリダイゼーションに用いることも可能である。これらの標準条件は、用いた核酸(オリゴヌクレオチド、長い断片もしくは完全長配列)に応じて、またはどの核酸型(DNAもしくはRNA)をハイブリダイゼーションに用いるかによって、様々に変化する。こうして、DNA:DNA ハイブリッドの融解温度は、例えば、同じ長さのDNA:RNA ハイブリッドの融解温度よりもおよそ10℃低い。
【0129】
核酸に応じて、標準条件とは、例えば、0.1〜5×SSC(1×SSC=0.15 M NaCl, 15mM クエン酸ナトリウム, pH7.2)の濃度を含有する水性緩衝溶液中において、またはさらに50%ホルムアミドの存在下にて、42〜58℃の温度(例えば、5×SSC、50%ホルムアミド中において42℃)であると理解される。有利には、DNA:DNA ハイブリッドのハイブリダイゼーション条件は、0.1×SSC、約20℃〜45℃、好ましくは約30℃〜45℃の温度である。DNA:RNAハイブリッドについては、ハイブリダイゼーション条件は有利には、0.1×SSC、約30℃〜55℃、好ましくは約45℃〜55℃の温度である。ハイブリダイゼーションのために特定される温度は融解温度の値であり、この値は、例として、ホルムアミドの非存在下で約100ヌクレオチド長および50% G+C含有量を有する核酸について計算されたものである。DNA ハイブリダイゼーションの実験条件は、遺伝学に関する専門書、例えば、Sambrookらによる「Molecular Cloning」, Cold Spring Harbor Laboratory, 1989に記載されており、当業者に公知の式を用いて、例えば核酸の長さ、ハイブリッドの性質またはG+C含有量に応じて、算出することができる。当業者は以下の文献からハイブリダイゼーションに関するさらなる情報を得ることができる:Ausubelら(編), 1985, Current Protocols in Molecular Biology, John Wiley & Sons, New York;HamesおよびHiggins(編), 1985, Nucleics Hybridization:A Practical Approach, IRL Press at Oxford University Press, Oxford;Brown(編), 1991, Essential Molecular Biology:A Practical Approach, IRL Press at Oxford University Press, Oxford。
【0130】
本発明はまた、具体的に開示されたまたは誘導することができる核酸配列の誘導体に関する。
【0131】
従って、本発明によるさらなる核酸配列は、例えば、配列番号3または5から誘導され、1個または複数のヌクレオチドの付加、置換、挿入または欠失によってそれらとは異なるが、それでも所望の特性プロファイルを有するポリペプチドをコードしている。
【0132】
本発明はまた、サイレント変異と称されるものを含むか、または具体的に挙げた配列と比較した場合に、特定の起源生物または宿主生物のコドン使用頻度に従って改変されている核酸配列、ならびにそれらの天然の変異体(例えば、スプライス変異体もしくは対立遺伝子変異体)を含む。
【0133】
本発明は同様に、保存的ヌクレオチド置換(例えば、問題のアミノ酸を、同一の電荷、大きさ、極性および/または可溶性を有するアミノ酸で置換する)によって得ることができる配列に関する。
【0134】
本発明はまた、具体的に開示された核酸から配列多形の結果として得られる分子に関する。これらの遺伝子多形は、天然の変異の結果として集団内の個体間に存在しうる。これらの天然の変異は通常、遺伝子のヌクレオチド配列に1〜5%の分散を生じさせる。
【0135】
「本発明による核酸配列の誘導体」とは、例えば、全長配列領域にわたって、推定アミノ酸レベルで少なくとも40%の相同性、好ましくは少なくとも60%の相同性、特に好ましくは少なくとも80、85、90、93、95または98%の相同性を有する対立遺伝子変異体を意味するものと理解される(アミノ酸レベルでの相同性に関しては、ポリペプチドに関する上記観察を参照のこと)。相同性は配列の構成的領域にわたって高いことが有利でありうる。
【0136】
さらに、「誘導体」とは、本発明による核酸配列の相同体、例えば、真菌または細菌の相同体、トランケート型配列、またはコードDNA配列および非コードDNA配列の一本鎖DNAもしくはRNAとしても理解される。従って、これらは、例えば、特定した全DNA領域にわたって、DNAレベルで、少なくとも40%、好ましくは少なくとも60%、さらに好ましくは少なくとも70%、特に好ましくは少なくとも80%の相同性を有する。
【0137】
さらに、「誘導体」とは、例えば、プロモーターとの融合体としても理解される。所与のヌクレオチド配列の上流に配置されるプロモーターは、プロモーターの機能または活性を損なうことなく、1個または複数のヌクレオチドの交換、挿入、逆位および/または欠失によって変化させることができる。さらに、プロモーターの活性はそれらの配列を変えることによって増加させることができるし、あるいはプロモーターをより活性のあるプロモーター(他の種に属する生物体に由来するものを含む)と完全に置き換えることができる。
【0138】
「誘導体」とはまた、遺伝子発現および/またはタンパク質発現が変化、好ましくは増加するように、開始コドンの上流-1〜-1000塩基または停止コドンの下流0〜1000塩基の領域においてヌクレオチド配列が変化している変異体としても理解される。
【0139】
本発明はまた、「ストリンジェントな条件」下にて、上記のコード配列とハイブリダイズする核酸配列をさらに含む。この特性は、ポリヌクレオチドまたはオリゴヌクレオチドがストリンジェントな条件下で実質的に相補的な配列と結合するが、こうした条件下で非相補的なパートナーとの間には非特異的結合を形成しない能力として理解される。そのために、これらの配列は70〜100%、好ましくは90〜100%相補的でなければならない。互いに結合することができる相補的配列の特性は、例えば、ノーザンブロット法もしくはサザンブロット法、またはPCRもしくはRT-PCRにおけるプライマー結合に関連して利用される。通常30塩基対またはそれ以上の長さのオリゴヌクレオチドがこの目的のために用いられる。ノーザンブロット法では、例えば、ストリンジェントな条件とは、非特異的にハイブリダイズしたcDNAプローブまたはオリゴヌクレオチドを溶出するために、50〜70℃、好ましくは60〜65℃の温度の洗浄液、例えば0.1%SDSを含有する0.1×SSC緩衝液(20×SSC:3M NaCl、0.3Mクエン酸Na、pH7.0)を用いることと理解される。これに関連して、上述したように、互いに結合し続ける核酸だけが高い相補性を有するものである。ストリンジェントな条件を確立することは、当業者に公知であり、例えば、AusubelらによるCurrent Protocols in Molecular Biology, John Wiley & Sons, N.Y.(1989), 6.3.1-6.3.6に記載されている。
【0140】
これらのポリヌクレオチドは、ゲノムライブラリーまたはcDNAライブラリーをスクリーニングすることによって見出し、適切な場合には、好適なプライマーを用いてPCRによってそれらから増幅し、次いで、例えば、好適なプローブを用いて単離することができる。さらに、本発明によるポリヌクレオチドは化学的に合成することもできる。
【0141】
本発明による構築物の実施形態
さらに、本発明はまた、調節核酸配列の遺伝子制御下に、本発明によるポリペプチドをコードする核酸配列を含む発現構築物に関し、さらに、これらの発現構築物の少なくとも1つを含むベクターに関する。
【0142】
本発明によるこれらの構築物は、好ましくは、所与のコード配列の5’側上流にプロモーターおよび3’側下流にターミネーター配列を含み、また適切な場合には、コード配列にそれぞれ作用可能に結合されるさらなる調節エレメントを含む。
【0143】
「作用可能な結合」とは、プロモーター、コード配列、ターミネーター、適切な場合には、さらなる調節エレメントの逐次的な配置の結果として、それぞれの調節エレメントが、必要に応じて、コード配列の発現に関連してその機能を果たすことができるようになることと理解される。作用可能に結合される配列の例は、ターゲッティング配列およびエンハンサー、ポリアデニル化シグナルなどである。他の調節エレメントとしては、選択マーカー、増幅シグナル、複製起点などが挙げられる。好適な調節配列は、例えば、GoeddelによるGene Expression Technology:Methods in Enzymology 185, Academic Press, San Diego, CA(1990)に記載されている。
【0144】
本発明による核酸構築物とは、特に、本発明によるデヒドロゲナーゼの遺伝子が、その遺伝子発現を調節する、例えば増加させるために、1個または複数の調節シグナルに作用可能にまたは機能的に結合されているものとして理解される。
【0145】
これらの調節配列に加えて、これらの配列の天然の調節要素(実際の構造遺伝子の上流にある)がなおも存在していてもよく、適切な場合には、この天然の調節要素がオフに切り替えられて、遺伝子の発現が増加されるように遺伝的に改変されている。しかし、核酸構築物はより単純な構成を有していてもよく、すなわち、さらなる調節シグナルがコード配列の上流に挿入されておらず、天然のプロモーターがその調節要素と共に除かれていない。その代わりに、もはやあらゆる調節が行われず、かつ遺伝子発現が増加されるように、天然の調節配列が突然変異される。
【0146】
好ましい核酸構築物はまた、有利には、1個または複数の上記のエンハンサー配列を含み、このエンハンサー配列はプロモーターに機能的に結合されて、核酸配列の発現を増加させることが可能である。また、DNA配列の3’末端に追加の有利な配列(例えば、さらなる調節エレメントまたはターミネーター)を挿入すことも可能である。本発明による核酸は1以上のコピー数で構築物中に存在しうる。この構築物はまた、適宜この構築物を選択するために、さらなるマーカー(例えば、抗生物質耐性遺伝子または栄養要求性補足遺伝子)を含むことができる。
【0147】
本発明による方法に有利な調節配列は、例えば、cos、tac、trp、tet、trp-tet、lpp、lac、lpp-lac、acIq、T7、T5、T3、gal、trc、ara、rhaP(rhaPBAD)SP6、λ-PRまたはλ-PLプロモーターのようなプロモーターに存在し、これらのプロモーターはグラム陰性細菌に有利に用いられる。他の有利な調節配列は、例えば、グラム陽性プロモーターのamyおよびSPO2、ならびに酵母または真菌プロモーターのADC1、MFα、AC、P-60、CYC1、GAPDH、TEF、rp28およびADHに存在する。ピルビン酸デカルボキシラーゼおよびメタノールオキシダーゼのプロモーター(例えば、ハンセヌラ(Hansenula)属由来)もまた、これに関連して有利である。また、人工プロモーターを調節に使用することも可能である。
【0148】
宿主生物内での発現のために、核酸構築物は宿主内で遺伝子を最適に発現することができるベクター(例えば、プラスミドまたはファージ)に有利に挿入される。プラスミドやファージに加えて、ベクターとは、当業者に公知の他のいずれかのベクター、すなわち、例えば、ウイルス(例:SV40、CMV、バキュロウイルスおよびアデノウイルス)、トランスポゾン、ISエレメント、ファスミド、コスミドおよび線状または環状DNAとして理解される。これらのベクターは宿主生物内で自律的に複製するかまたは染色体上で複製することができる。これらのベクターは、本発明のさらなる実施形態を構成する。好適なプラスミドの例は、大腸菌のpLG338、pACYC184、pBR322、pUC18、pUC19、pKC30、pRep4、pHS1、pKK223-3、pDHE19.2、pHS2、pPLc236、pMBL24、pLG200、pUR290、pIN-III113-B1、lgt11およびpBdCI、ストレプトミセス(Streptomyces)のpIJ101、pIJ364、pIJ702およびpIJ361、バシルス(Bacillus )属のpUB110、pC194およびpBD214、コリネバクテリウム(Corynebacterium)属のpSA77およびpAJ667、真菌のpALS1、pIL2およびpBB116、酵母の2αM、pAG-1、YEp6、YEp13およびpEMBLYe23、ならびに植物のpLGV23、pGHlac+、pBIN19、pAK2004 およびpDH51である。これらのプラスミドは可能性のあるプラスミドの小さなセレクションを構成する。他のプラスミドが当業者に周知であり、それらに関する情報は、例えば、Cloning Vectors (Pouwels P. H.ら編 Elsevier, Amsterdam-New York-Oxford, 1985, ISBN 0 444 904018)から得ることができる。
【0149】
存在する他の遺伝子の発現のために、核酸構築物はまた、発現を増加させるために3’末端および/または5’末端の調節配列を含むことも有利であり、これらの配列は所定の宿主生物および1以上の遺伝子に応じて最適な発現のために選択される。
【0150】
これらの調節配列は遺伝子の選択的発現ならびにタンパク質の発現を可能にすべきである。これは例えば、宿主生物に依存して、当該遺伝子のみが誘導後に発現または過剰発現されるか、あるいは、当該遺伝子がただちに発現および/または過剰発現されることを意味しうる。
【0151】
これに関連して、調節配列または調節因子は好ましくは、ポジティブな影響を及ぼし、それによって導入された遺伝子の発現を増加させることができる。従って、調節エレメントは、プロモーターおよび/またはエンハンサーのような強力な転写シグナルを用いることによって転写レベルで有利に増強させることができる。しかし、これに加えて、例えばmRNAの安定性を向上させることによって、翻訳を高めることも可能である。
【0152】
ベクターの別の実施形態においては、本発明の核酸構築物または本発明の核酸を含むベクターを線状DNAの形態で微生物に挿入し、非相同組換えまたは相同組換えによって宿主生物のゲノムに組み込むことも可能である。この線状DNAは線状化したベクター(例えば、プラスミド)からなるものであってもよいし、本発明の核酸構築物もしくは核酸からのみなるものであってもよい。
【0153】
生物体による異種遺伝子の最適な発現を可能とするために、その生物体に用いられている特有のコドン使用頻度に従って核酸配列を改変することは有利である。このコドン使用頻度は、対象の生物体に由来する他の既知遺伝子のコンピュータ解析によって容易に決定することができる。
【0154】
本発明の発現カセットは、好適なプロモーターを好適なコードヌクレオチド配列およびターミネーターシグナルまたはポリアデニル化シグナルに融合することによって作製される。このために、例えば、T. Maniatis, E.F. FritschおよびJ. SambrookらによるMolecular Cloning:A Laboratory Manual, Cold Spring Harbor Laboratory, Cold Spring Harbor, NY(1989)、T.J. Silhavy, M.L. BermanおよびL.W. EnquistによるExperiments with Genen Fusions, Cold Spring Harbor Laboratory, Cold Spring Harbor, NY(1984)、ならびにAusubel, F.M.らによるCurrent Protocols in Molecular Biology, Greene Publishing Assoc. and Wiley Interscience(1987)に記載されているような、通常の組換えおよびクローニング技術が用いられる。
【0155】
好適な宿主生物内での発現を達成するためには、組換え核酸構築物または遺伝子構築物を、宿主内での遺伝子の最適な発現を可能にする宿主特異的ベクターに挿入することが有利である。ベクターは当業者に周知であり、それらに関する情報は、例えば「Cloning Vectors」(Pouwels P. H.ら, Eds., Elsevier, Amsterdam-New York-Oxford, 1985)から得ることができる。
【0156】
本発明で用いることができる宿主生物
本発明によるベクターまたは構築物は、組換え微生物を調製するために用いることが可能である。これらの組換え微生物は、例えば、本発明による少なくとも1つのベクターで形質転換され、本発明によるポリペプチドの産生のために用いられる。有利には、本発明による上記組換え構築物を好適な宿主系に導入して発現させる。これに関連して、当業者に良く知られているクローニング方法およびトランスフェクション方法、例えば、共沈、プロトプラスト融合、エレクトロポレーション、レトロウイルスによるトランスフェクションなどが、所与の発現系にて核酸を発現させるために好ましく使用される。好適な系は、例えば、Current Protocols in Molecular Biology, F. Ausubelら編, Wiley Interscience, New York 1997,またはSambrookら Molecular Cloning:A Laboratory Manual, 第2版, Cold Spring Harbor Laboratory, Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY, 1989に記載されている。
【0157】
本発明により、相同的に組換えされた微生物を作製することもまた可能である。このために、本発明による遺伝子の少なくとも1つのセグメントを含むベクター、または本発明による配列を改変する(例えば、機能的に破壊する(「ノック・アウト」ベクター)ために少なくとも1つのアミノ酸の欠失、付加または置換が適宜導入されているコード配列の少なくとも1つのセグメントを含むベクターを作製することが必要である。導入される配列は、例えば、関連する微生物に由来する相同体であってもよいし、哺乳動物、酵母もしくは昆虫源に由来するものであってもよい。あるいはまた、相同組換えに用いられるベクターは、相同組換えに関連する内在性遺伝子が変異されるか、さもなければ改変されるが、機能的タンパク質をなおもコードするように構成することができる(例えば、下流調節領域が改変され、その結果として内在性タンパク質の発現を変化させることができる)。本発明による遺伝子の改変されたセグメントは、相同組換えベクター内にある。相同組換えに好適なベクターの構築は、例えば、Thomas, K.R.およびCapecchi, M.R.(1987)Cell 51:503に記載されている。
【0158】
原則的に、どのような原核生物または真核生物も、本発明による核酸または核酸構築物の組換え宿主生物として用いるのに適している。有利には、微生物、例えば細菌、真菌または酵母が宿主生物として用いられる。グラム陽性細菌またはグラム陰性細菌、好ましくはエンテロバクテリアセア(Enterobacteriaceae)科、シュードモナダセア(Pseudomonadaceae)科、リゾビアセア(Rhizobiaceae)科、ストレプトマイセタセア(Streptomycetaceae)科もしくはノカルディアセア(Nocardiaceae)科の細菌、特に好ましくはエスケリキア(Escherichia)属、シュードモナス(Pseudomonas)属、ストレプトマイセス(Streptomyces)属、ノカルディア(Nocardia)属、ブルクホリデリア(Burkholderia)属、サルモネラ(Salmonella)属、アグロバクテリウム(Agrobacterium)属またはロドコッカス(Rhodococcus)属の細菌が有利に用いられる。エスケリキア・コリ(Escherichia coli)の属および種が特に好ましい。これに加えて、他の有利な細菌はα-プロトバクテリア、β-プロトバクテリアおよびγ-プロトバクテリアの群に見出すことができる。
【0159】
これに関連して、宿主生物、または本発明による宿主生物は、本発明によるデヒドロゲナーゼ活性を有する酵素をコードする、本発明に記載の核酸配列、核酸構築物またはベクターを少なくとも1つ含むことが好ましい。
【0160】
本発明の方法に用いられる生物は、宿主生物に応じて、当業者に公知の方法で増殖させるかまたは培養する。微生物は一般的に、炭素源(通常は糖の形態)、窒素源(通常は有機窒素源の形態、例えば酵母エキス)、または塩(例えば、硫酸アンモニウム)、微量元素(例えば、鉄塩、マンガン塩およびマグネシウム塩)、適宜にビタミン類を含有する液体培地中で0℃〜100℃、好ましくは10℃〜60℃の温度にて酸素を供給しながら増殖させる。これに関連して、液体栄養培地のpHは一定の値(培養の間中調整される)に維持してもしなくてもよい。この培養はバッチ式、半バッチ式、または連続的に行うことができる。栄養分は発酵の開始時にはじめに導入されるか、または続いて半連続的にまたは連続的に供給できる。ケトンはこの培養物に直接的に加えることができるが、培養後に加えることが有利である。酵素は、実施例に記載の方法を用いて生物体から単離するか、そうでなければ、粗抽出物として反応に用いることができる。
【0161】
組換えによる本発明のポリペプチドの調製
本発明はさらに、本発明によるポリペプチドまたはそれらの機能的、生物学的に活性な断片を組換えによって調製するための方法に関する。この方法は、ポリペプチド産生微生物を培養し、ポリペプチドの発現を誘導し、適切な場合には、これらのポリペプチドを培養物から単離することを含む。所望であれば、このポリペプチドをこの方法により工業的規模で製造することが可能である。
【0162】
組換え微生物は公知の方法で培養および発酵させることができる。例えば、細菌をTB培地またはLB培地中で20〜40℃の温度、pH6〜9にて増殖させる。好適な培養条件は、例えば、T. Maniatis, E.F. FritschおよびJ. SambrookらによるMolecular Cloning:A Laboratory Manual, Cold Spring Harbor Laboratory, Cold Spring Harbor, NY(1989)に詳細に記載されている。
【0163】
ポリペプチドが培地中に分泌されないときには、細胞を破砕して、その溶解液から公知のタンパク質単離方法により産物を取得する。細胞は、所望により、高周波超音波、高圧(例えば、フレンチプレス細胞破砕機)、浸透圧、界面活性剤、溶解酵素もしくは有機溶媒の作用、ホモジェナイザーの使用または上記方法のいくつかの組合せによって、破砕することができる。
【0164】
ポリペプチドを精製するには、公知のクロマトグラフィーによる方法、例えば、分子ふるいクロマトグラフィー(ゲル濾過)、例えば、Qセファロースクロマトグラフィー、イオン交換クロマトグラフィーおよび疎水性クロマトグラフィー、さらに他の慣用方法、例えば、限外濾過、結晶化、塩析、透析および未変性ゲル電気泳動を用いることができる。好適な方法は、例えば、Cooper, F. G., Biochemische Arbeitsmethoden [生化学的操作方法], Verlag Walter de Gruyter, Berlin, New YorkまたはScopes, R., Protein Purification, Springer Verlag, New York, Heidelberg, Berlinに記載されている。
【0165】
組換えタンパク質を単離するためには、特定のヌクレオチド配列によってcDNAを伸長させ、それによって、例えば精製を簡便にするために用いられる、改変されたポリペプチドまたは融合タンパク質をコードするベクター系またはオリゴヌクレオチドを用いることが有利でありうる。このような好適な改変の例には、アンカーとして作用するタグと称されるものがあり、例えば、ヘキサヒスチジンアンカーとして知られる改変、または抗体によって抗原として認識され得るエピトープ(例えば、Harlow, E.およびLane, D., 1988, Antibodies:A Laboratory Manual. Cold Spring Harbor(N.Y.)Pressに記載されている)がある。これらのアンカーは、タンパク質を固相支持体(例えば、クロマトグラフィーカラムに充填されるポリマーマトリックスもしくはマイクロタイタープレートまたは別の支持体)に結合させるために用いることができる。
【0166】
同時に、これらのアンカーをタンパク質の認識に用いることも可能である。タンパク質の認識のためには、通常の標識、例えば、蛍光色素、基質との反応後に検出可能な反応生成物を形成する酵素標識または放射性標識を、単独でまたはアンカーと組み合わせて、タンパク質を誘導体化するために用いることが可能である。
【0167】
本発明による酵素的還元方法を実施するための他の実施形態
本発明の方法においては、デヒドロゲナーゼを遊離の酵素または固定化酵素として用いることができる。
【0168】
本発明の方法は0℃〜95℃、好ましくは10℃〜85℃、特に好ましくは15℃〜75℃の温度にて実施することが有利である。
【0169】
本発明の方法においては、pHをpH4〜12、好ましくはpH4.5〜9、特に好ましくはpH5〜8に維持する。
【0170】
本発明の方法において、エナンチオマー的に純粋なまたはキラルな生成物、例えば3-クロロ-1-(チエン-2-イル)-(S)-プロパン-1-オールは、エナンチオマー冨化を示すエナンチオマーであるとして理解されるべきである。本方法では、少なくとも70%ee、好ましくは少なくとも80%ee、さらに好ましくは少なくとも90%ee、特に好ましくは少なくとも98%eeのエナンチオマー純度を達成することが好ましい。
【0171】
本発明の方法のために、本発明による核酸、核酸構築物またはベクターを含む増殖中の細胞を用いることが可能である。また、休止細胞または破砕細胞を用いることも可能である。破砕細胞とは、例えば、溶媒で処理されたことによって透過性にされた細胞、または酵素で処理されたことによって、機械的に処理されたことによって(例えば、フレンチプレスもしくは超音波処理)もしくは別の方法によって破壊された細胞として理解される。このようにして得られた粗製抽出物は本発明による方法に適している。また、精製酵素または部分精製酵素を本方法に用いることもできる。本反応に有利に用いることができる固定化した微生物または酵素も同様に好適である。
【0172】
本発明による方法は、バッチ式、半バッチ式または連続的に行うことができる。
【0173】
本方法は、例えば、Biotechnology, 第3巻, 第2版, Rehmら編,(1993)、特に第II章に記載されるようなバイオリアクター内で有利に行うことができる。
【0174】
以下の例は、本発明を限定することなく例証することを目的とする。これに関して添付の図面を参照されたい。
【実施例】
【0175】
A. 化学的実施例
実施例1:3-クロロ-1-(チエン-2-イル)プロパン-1-オンIII.1の調製
33kgのチオフェンを150kgのジクロロエタンにまず導入し、その後62.7kgの三塩化アルミニウムを加えた。この反応混合物を10℃まで冷却し、55kgの塩化3-クロロプロピオニルを加えた。次いでこの混合物を室温にて12時間撹拌した。GCおよびNMRで調べると、反応混合物は、生成された3-クロロ-1-(チエン-2-イル)プロパン-1-オンおよび3-クロロ-1-(チエン-2-イル)プロペノンの合計量に基づいて、約3〜4モル%の3-クロロ-1-(チエン-2-イル)プロペノンをまだ含むことがわかった。その後、少しのプロペノンも検出することができなくなるまで塩化水素を室温にて通過させた(およそ30分間)。続いて、100kgの脱イオン水を加えることによってこの反応混合物を加水分解した。有機相を分離し、100kgの脱イオン水で洗浄した。真空にて溶媒を取り除いた後、65.7kg(理論値の96%)の表題化合物を油状物として得た。
【0176】
実施例2:水素化ホウ素ナトリウムで還元することによる3-メチルアミノ-1-(チエン-2-イル)プロパン-1-オールIの調製
実施例1から得られたプロパノンを初めに、0℃にて、400mlのトルエンと200gのメタノールの混合溶媒中に導入した。30%濃度の水酸化ナトリウム水溶液2gを加えた後、21.4gの水素化ホウ素ナトリウムを2.5時間かけて少しずつ加えた。0℃で40分間撹拌した後、この反応混合物を12.1gの40%濃度のメチルアミン水溶液で処理した。この混合物を、その固有の圧力下に60℃で6時間撹拌した。この混合物を室温まで冷却した後、溶媒を除去し、残留物をトルエンで温浸し、トルエンから濾過した。この濾液を乾燥させると、175.8gの表題化合物が淡黄色の固形物として得られた。
【0177】
B. 生化学的実施例
実施例3:補因子再生のためのグルコースデヒドロゲナーゼの調製
補因子を再生するために、販売元(例えば、Julich Fine Chemicals 注文番号22.10もしくは19.10)または本発明者ら自身の供給源から得られるグルコースデヒドロゲナーゼを用いることが可能であった。本発明者ら自身の供給源とは、枯草菌(Bacillus subtilis)(Genbank登録番号M12276)(Lu11293)に由来するグルコースデヒドロゲナーゼ遺伝子を含む大腸菌XL10 Gold pUC19クローンであった。
【0178】
以下の培地を大腸菌Lu11293の発酵のために調製した:
【0179】
2本の1Lエルレンマイヤーフラスコにそれぞれ150mlの培地を入れて滅菌し、5mlの滅菌塩溶液を補充した。LBアンピシリン寒天プレートから接種した後、予備培養物を37℃、200rpmで12時間インキュベートして、発酵培地に加えた。発酵は37℃、内圧0.1バール、pH7.0(20%リン酸および25%NaOHを用いて調整)、空気補給速度7.5L/分、および300rpmで開始した(空気を10〜20L/分および500〜1500rpmで流入させてpO2を20〜50%に調整した)。2時間後、0.1mM IPTGを誘導のために加え、発酵を全13時間後に終わらせた。細胞(1.3kg)を回収して、洗浄した後、使用するまで-20℃にて保存した(混合物中に2〜20g/L)。
【0180】
実施例4:3-クロロ-1-(チエン-2-イル)プロパン-1-オンを還元するためのデヒドロゲナーゼのスクリーニング
a)種々の細菌株および真菌株(主に種々の菌株コレクションに由来する)を20mlのLB培地、MRS培地(Difco)またはGYP培地(酵母に使用)(1%w/v D-グルコース、それぞれ0.5%w/v 酵母抽出物およびポリペプトン、pH6.0)中で30℃または37℃にて24〜48時間増殖させ、3mM Tris-HCl、pH7.5中で洗浄し、そして2mlの緩衝液に再懸濁させた。アリコートを振動粉砕器中の1容のガラスビーズ(直径0.3〜0.5mm)で破砕した(10分の氷上での冷却時間をはさんで3×5分)。濁りを遠心分離(4℃、14000rpmにて5分間)によって分離した後、透明な粗抽出物を得た。その後、細胞懸濁液および粗抽出物を、実施例3から3-クロロ-1-(チエン-2-イル)プロパン-1-オンを還元するためにそれらの活性について試験した。
【0181】
アッセイには以下を用いた:
150μlの細胞/粗抽出物
50μlの1Mグルコース溶液
50μlのグルコースデヒドロゲナーゼ(12〜15U/μl;20〜200mg MBM/ml)(実施例3から)
50μlの2mM NADH
50μlの2mM NADPH
10μlの0.5M 3-クロロ-1-(チエン-2-イル)プロパン-1-オン(メタノールに溶解したもの)
190μlの50mM MES, pH6.0またはTris-HCl, pH7.5
【0182】
インキュベーションは30℃で行った。このサンプル(200μl)は1時間、2時間または24時間後に3μlの濃HClを用いて停止させた。遠心分離後、上清を3-クロロ-1-(チエン-2-イル)プロパン-1-オンおよび3-クロロ-1-(チエン-2-イル)プロパン-1-オールについてHPLCで分析した(Chromolith Speed ROD, RP-18e 50-4.6mm、流速1.5ml/分、0.0〜1.0分:35%アセトニトリル+65%10mM KH2PO4緩衝液、pH2.5;1.1〜1.3分:80%アセトニトリル+20%10mM KH2PO4緩衝液、pH2.5;1.3〜2.0分:35%アセトニトリル+65%10mM KH2PO4緩衝液、pH2,5;230nm(アルコール)および260nm(ケトン)にて検出、保持時間 Rt=1.250分(3-クロロ-1-(チエン-2-イル)プロパン-1-オール)およびRt=1.500分(3-クロロ-1-(チエン-2-イル)プロパン-1-オン);HCl除去から生じうる副生成物(1-チエン-2-イルプロペン-1-オンおよび1-チエン-2-イルプロペン-1-オール)、Rt=0.971分およびRt=1.165分)。
【0183】
選択した菌株は、エナンチオマー過剰率(ee)を決定するために繰り返し試験したが、その際、サンプルをメチルtert-ブチルエーテル(MTBE)またはメチルイソブチルケトン(MIBK)を用いて抽出し、キラルGC分析またはHPLC分析によって特徴づけた。そのために以下の方法を用いた:GC:Hydrodex-β-6-TBDM、25m、90℃ 10分 5℃/分 180℃ 10分、ラン時間:38分、入口:ヒーター:200℃、圧力:106.8 kPa、全流量:102ml/分、スプリット比:125:1、スプリット流量:99.8ml/分、検出器:200℃またはHPLC:Chiracel OD-H、250*4.6mm(Daicel)、40℃、流速1.0ml/分、0.0〜1.0分:97.5% n-ヘキサン+2.5%イソプロパノール;230nm(アルコール)および260nm(ケトン)にて検出、Rt 9.50(3-クロロ-1-(チエン-2-イル)プロパン-1-オン)、Rt 16.60分(R-3-クロロ-1-(チエン-2-イル)プロパン-1-オール)およびRt 18.30分(S-3-クロロ-1-(チエン-2-イル)プロパン-1-オール)。
【0184】
b)種々のラクトバチラス(Lactobacillus)菌株を新たに単離した
ラクトバチラス・ブレビス(Lactobacillus brevis)株であるLu10288、10290および10291を以下のように単離した。
【0185】
b1)用いた培地
Kleymann培地:
以下の物質を915ml中に溶解した:
10g/l トリプトン(Difco-Becton Dickinson)
7g/l 酵母エキス(Difco-Becton Dickinson)
2g/l 牛肉エキス(Difco-Becton Dickinson)
5g/l フルクトース
2g/l マルトース
3.6g/l 50%グルコン酸
1.9g/l クエン酸*H2O
5g/l酢酸
1g/l Tween 80
0.2g/l MgSO4*7H2O
0.05g/l MnSO4
0.01g/l FeSO4
0.4g/l L-システイン
1.25ml NH4OH(25%)
寒天プレートについて:2% Bacto Agar(Difco-Becton Dickinson)を加える
前記溶液のpHをpH5.4に調整した。
前記溶液を121℃にて15分間オートクレーブ滅菌した。
オートクレーブ滅菌した後、50mlの滅菌グルコース溶液(5 gのグルコースをH2Oで50mlにしたもの)および40mlのエタノールを撹拌しながら加え、寒天プレートに注いだ。
【0186】
KMB培地:
10g/l トリプトン(Difco-Becton Dickinson)
7g/l 酵母エキス(Difco-Becton Dickinson)
2g/l KH2PO4
1g/l Tween 80
0.5g/l MgSO4*7H2O
1g/l MnSO4
20g/l CaCO3
10g/lグルコース
1.5%寒天(寒天プレート調製用)
前記溶液を121℃にて15分間オートクレーブ滅菌した。
【0187】
グルコースおよびCaCO3(Riedel de Haen)を別々に溶解してオートクレーブ滅菌し、プレートに注ぐ前に寒天と混合した。
【0188】
b2)微生物の単離:
北ドイツ地方(LUFA-Oldenburg)産の5〜10gのトウモロコシ貯蔵飼料を50mlエルレンマイヤーフラスコ中で20mlの塩水と共に37℃にて一晩、嫌気的にインキュベートした。得られた液体フラクションを1:100の割合で50mlのKleymann培地に接種し、穏やかに撹拌しながら室温で24時間嫌気的にインキュベートした。その後、この細胞懸濁液を上記のKleymann選択寒天プレートに播種した。このプレートを37℃で48〜72時間嫌気的にインキュベートし、得られたコロニーを、KMB培地上に繰り返しストリークすることによって単離した。
【0189】
この菌株は、20gのフルクトース/Lを含有する液体KMB培地中(24時間、37℃、50rpm)で酸発酵パターン(pHの測定、および培養上清のHPLC分析によるグルコース、フルクトース、乳酸、酢酸およびエタノールの濃度の測定)を決定することによって特徴づけた。酢酸および乳酸を形成した異種発酵菌株を単離し、そして16sRNAを分析することによって体系的に特徴づけた。
【0190】
c)スクリーニング結果
表1、2および3は、菌株の例および/または変換率およびee値を示す。
【表1】
【0191】
【表2】
【0192】
【表3】
【0193】
実施例5:ラクトバチラス・ブレビス(Lactobacillus brevis)由来のデヒドロゲナーゼの精製
ラクトバチラス・ブレビス(Lactobacillus brevis)Lu10288の発酵のために以下の培地を調製した:
【0194】
MRS寒天プレートから2つの予備培養物を接種し、この予備培養物を37℃にて17時間、200rpmでインキュベートして、発酵培地に加えた。この発酵は37℃、内圧0.1バール、pH5.8、空気補給速度1L/分、および100rpmで開始し(pO2の調節なし)、23時間後にOD600が14.8になった時点で終結させた。
洗浄した細胞試料の活性を、MES, pH6.0中の休止細胞を用いて実施例4a)と同様に測定した。
【0195】
Lu10288デヒドロゲナーゼは以下のように精製した:
均質化:
100gのLu10288(ラクトバチラス・ブレビス(Lactobacillus brevis))の湿ったバイオマスを、100mlのMES緩衝液、1mM MgCl2、pH7.1を用いて、5×20gに小分けして再懸濁させ、10に小分けして20分間、4000rpmにて氷で冷却しながら、ガラスビーズ(直径0.1mm〜0.2mm、50mlの懸濁細胞に対して50mlのガラスビーズ)を用いてボールミル内でホモジェナイズした。ガラスビーズをG2ガラス吸引フィルターを通して分離し、20mlの緩衝液で洗浄した。次いで、回収したホモジェネート(610ml)をGSAローター内で20分間、12000rpmにて清澄化した。
【0196】
a) Q-セファロースイオン交換クロマトグラフィー:
直径5cm、容積400mlのQ-セファロースファーストフロー(Pharmacia)カラムを20mM MES緩衝液、1mM MgCl2、 pH6.8中で平衡化した。610mlのホモジェネートを11錠のComplete(プロテアーゼ阻害剤混合物、Roche, Complete, EDTAフリー;カタログ番号:1873580)で処理し、Q-セファロースカラムに10ml/分の速度でロードした。280 nmにて検出した。次いで、このカラムを同じ緩衝液で3倍のカラム容量を用いて洗浄した。溶出のために、20mM MES、1mM MgCl2、1M NaCl、pH6.8の直線勾配(100分間で0%NaClから100%NaClまで)を適用した。10mlフラクションを回収して試験した。フラクション42〜62は、HPLC試験において3-クロロ-1-(チエン-2-イル)プロパン-1-オンとの活性を示した。
【0197】
b) Superdex分子ふるいクロマトグラフィー:
直径2.6cm、容積240mlのSuperdex 200分子ふるいカラム(Pharmacia)を20mM MES、1mM MgCl2、緩衝液1Lにつき1錠のComplete(EDTAフリー)、pH7.1中で平衡化した。Q-セファロースから得られた有用なフラクションを合わせて(240ml)、124gの硫酸アンモニウムを徐々に加えて80%飽和に調整した。pHは7.1に維持した。この溶液を4℃にて10分間撹拌し、次いで12000rpmにて20分間遠心分離した。得られたペレットを5mlの平衡緩衝液に再懸濁し、この懸濁液を1時間、4℃にて透析した(10 kDa排除体積)。透析した溶液を9ml部ずつ2分し、分子ふるいカラムに4ml/分の流速でロードした。4mlフラクションを回収して試験した。フラクション48〜56は両方の基質に対して再び活性を示した。
【0198】
c) Mono-Qイオン交換クロマトグラフィー:
容積20mlの分離用Mono-Q HRカラム(Pharmacia)を20mM MES、1mM MgCl2、pH7.1中で平衡化した。Superdexから得られた有用なフラクションを合わせたもの70mlを4ml/分の流速でロードした。カラムを洗浄した後、100分間で100%までの20mM MES、1mM MgCl2、0.5M NaCl、pH7.1の直線勾配を用いて展開した。4mlフラクションを回収した。活性のあるフラクション(フラクション36〜41)を合わせた。
【0199】
d) Mono-Pイオン交換クロマトグラフィー:
Mono-Pカラム(Pharmacia, 4ml)を20mM MOPS、1mM MgCl2、pH7.1中で平衡化した。21mlの有用なMono-Qフラクションを0.75ml/分の流速でロードした。基底値になるまで洗浄した後、100分間で100%までの20mM MOPS、1mM MgCl2、0.5M NaCl、pH7.1の直線勾配を適用した。フラクション(0.75ml)を回収した。活性のあるフラクション(34〜39)を合わせた。
【0200】
ゲルでの活性染色:
試料を同容量のNovex「未変性Tris-グリシン」試料緩衝液(Novex)で希釈した。Anamedのtris-グリシンゲル(SDSなし、厚さ1mm、10個の試料用ウェル)をランニング・チャンバーに設置した。Invitrogenの「Tris-グリシン未変性」ランニング緩衝液(Invitrogen)(10×)をランニング緩衝液として希釈後に用いた。試料ウェルにロードし、ゲルを200 V、約50 mAにて開始した。電気泳動を約1.5時間継続した。ランニング・チャンバーは氷水中に置いた。ゲルを取り外した後、ガラス皿に置き、50mM MES、8mM MgCl2、pH6.2中で洗浄し、そしてインキュベートした。
【0201】
次いで、0.35mM NADPおよび0.35mM NAD、19.6mgのNB-テトラゾリウムならびに2.1mgのフェナジンエトサルフェート(PES)をこの溶液中のゲルに加えた。続いて基質(3-クロロ-1-(チエン-2-イル)プロパン-1-オール)を加えて最終濃度を1mMとした。このゲルは染色されるまで暗所に保存した。典型的なゲルを図1に示す。
【0202】
【0203】
本方法で精製したデヒドロゲナーゼのN末端を、SDS-PAGEおよびゲル・ブロッティング後にEdman配列決定により決定した(配列番号1)。
【0204】
実施例6:カンジダ・マグノリア(Candida magnoliae)由来のデヒドロゲナーゼの精製
カンジダ・マグノリア(Candida magnoliae)Lu8742の発酵のために、2つの予備培養物を150mlの培地(8g/Lの酵母エキス(65%)、5g/Lのペプトン、3.5g/Lのグルコース、5g/LのKH2PO4、2g/LのMgSO4*7H2Oを含有、pH6.0)中で28℃、200rpmで15時間増殖させ、10.7g/Lのグルコースおよび4mlのテゴシポン(tegosipon)を含有する同様の培地13.2Lへの接種に用いた。発酵を28℃、内圧0.1バール、pH6.0、空気補給速度5L/分、および500rpmで開始し(pO2を>20%に調整)、26時間後にOD600が15.1となった時点で停止させた。
【0205】
均質化:
回収した細胞(378gの湿ったバイオマス)を、10錠のRoche Complete(EDTAフリー)プロテアーゼ阻害剤(およそ9×濃度)を含有する1Lの50mM MES+1mM MgCl2、pH6.5中に懸濁し、Z04ミクロフルイダイザー中1000バールで2回破壊した。遠心分離(20分/10000g)に続いて、澄んだ上清(1.3Lのホモジェネート)の半分を下記のとおりに精製した。これを行うとき、試料フラクションは、50mM MES、pH6.0、0.2mM NaDH/NaDPH、100mMグルコース、50μlのGDH/mlおよび10mMの3-クロロ-1-(チエン-2-イル)プロパン-1-オン中30℃にて、好適な希釈で、活性について試験した。
【0206】
Q-セファロースイオン交換クロマトグラフィー:
直径5cm、容積400mlのQ-セファロースファーストフローカラム(Pharmacia)を20mM MES緩衝液、1mM MgCl2、pH6.8中で平衡化した。650mlのホモジェネートを11錠のComplete(プロテアーゼ阻害剤混合物)で処理し、7.5ml/分の速度にてQ-セファロースカラムにロードした。280nmにて検出した。次にカラムを700mlの同一緩衝液を用いて洗浄した。溶出のために、20mM MES、1mM MgCl2、0.75M NaCl、pH6.8の直線勾配(100分間で0%NaClから100%NaCl)を適用し、その後カラムを200mlの20mM MES、1mM MgCl2、0.75M NaCl、pH6.8で洗浄した。10mlフラクションを回収して試験した。フラクション56〜96はHPLC試験において3-クロロ-1-(チエン-2-イル)プロパン-1-オンとの活性を示した。
【0207】
硫酸アンモニウム沈降:
41mlの有用なQ-セファロースピークフラクションを(NH4)2SO4(pH6.2)で90%飽和にし、4℃にて30分間撹拌した後、10000gにて10分間遠心分離した。得られたペレットを10mlの20mM MES、1mM MgCl2、1Lにつき1錠のRoche Complete(EDTAフリー)、pH6.2中に懸濁し、この懸濁液をPierce Slide-A-Lyzer(10 kDa排除体積)にて20mM MES、1mM MgCl2、1Lにつき1錠のRoche Complete(EDTAフリー)に対して30分間透析した。
【0208】
Superdex分子ふるいクロマトグラフィー:
直径2.6cm、容積240mlのSuperdex 200分子ふるいカラム(Pharmacia)を20mM MES、1mM MgCl2、1Lの緩衝液につき1錠のComplete(EDTAフリー)、pH6.2中で平衡化した。硫酸アンモニウム沈降から得られた透析物(2×7ml)を4ml/分の流速で分子ふるいカラムにロードした。4mlフラクションを回収して試験した。フラクション21〜24は両基質について再び活性を示した。
【0209】
Mono-Qイオン交換クロマトグラフィー:
容積1mlのMono-Q HR5/5カラム(Pharmacia製)を20mM MES、1mM MgCl2、pH6.8中で平衡化した。Superdexカラムからの有用なフラクションを合わせたもの10mlを1ml/分の流速でロードした。カラムを洗浄した後、100分間で100%までの20mM MES、1mM MgCl2、0.75M NaCl、pH6.8の直線勾配を用いて展開し、続いて20mM MES、1mM MgCl2、pH6.8を用いて10分間洗浄した。1mlフラクションを回収した(検出 226nm)。活性を有するフラクション(フラクション24〜27)を合わせた。
【0210】
単離の結果を以下の表に要約した。
【0211】
このようにして精製したデヒドロゲナーゼのN末端を、SDS-PAGEおよびゲル・ブロッティング後にEdman配列決定により決定した(配列番号2)。
【0212】
実施例7:ラクトバチラス・ブレビス(Lactobacillus brevis)デヒドロゲナーゼを用いた還元による(S)-3-メチルアミノ-1-(チエン-2-イル)プロパン-1-オールI-Sの調製
ラクトバチラス・ブレビス(Lactobacillus brevis)Lu10288を実施例4に記載されるように増殖させて回収した。休止細胞(10〜100g/Lのバイオマス)を0.2〜2mM NAD(P)+、実施例1からの1〜100g/Lのプロパノン(バッチ式または流加式)および18g/Lのグルコースで処理して、30℃にて2〜8時間インキュベートした。この反応を0.5M NaOHで滴定することによってpH6.0〜7.0を維持し、HPLC分析によって追跡した。図2AおよびBはそれぞれ、混合物1および2に対応する典型的な反応経過を示す。
【0213】
【0214】
続いて、バイオマスを遠心分離および/または濾過によって除去した。MTBE抽出物のNMR分析は60〜70%の3-クロロ-1-(チエン-2-イル)プロパン-1-オールの含有量を与えた。未反応の3-クロロ-1-(チエン-2-イル)プロパン-1-オンおよび副生成物である1-チエン-2-イルプロペン-1-オンの含有量は、それぞれ10%未満であった。1-チエン-2-イル-プロペン-1-オールは痕跡量で検出できたにすぎなかった。
【0215】
次いで、1.1gの40%メチルアミン水溶液を無細胞水性混合物に加えた。後者を次いで、60℃で6時間、固有の圧力下にて撹拌した。混合物を室温まで冷却した後、溶媒を除去した。その後残留物をトルエンで温浸し、トルエンから濾過した。別法として、アミノ化反応の後に細胞分離を行うことも可能であった。濾液を乾燥させた後、202mgの表題化合物を淡黄色の固形物として得た。Sエナンチオマーを95%eeのエナンチオマー過剰率で得た。ee値は、回転量を測定することよって(c=1、溶媒:メタノール)、またShift NMR(Shift試薬:2,2,2-トリフルオロ-1-(9-アンスリル)エタノール(+)(TFAE);溶媒:CDCl3;500 MHz)によって決定した。
【0216】
実施例8:ラクトバチラス・ブレビス(Lactobacillus brevis)Lu10288由来のデヒドロゲナーゼのクローニング
a) 先行プロトプラスト形成に続くLU10288由来の染色体DNAの調製:
(1) 必要な溶液
溶液1: 0.41Mスクロース
0.01M MgSO4*7H2O
5ml/L M12培地1:2
10ml/L 10%KH2PO4 pH6.7
2.5mg/mlリゾチーム(使用直前に加える)
溶液1は以下のように調製する:
14.03gのスクロース、0.25gのMgSO4*7H2O、5mlのM12(10×濃度)および1mlの10% KH2PO4、pH6.7を一緒にして100mlとなし、濾過滅菌する。
プロテイナーゼ: Qiagen製、20mg/ml ストック溶液
RNase : Qiagen製、100mg/ml ストック溶液
TE緩衝液: 10mM Tris*Cl pH8、1mM EDTA pH8
【0217】
(2) 培養および破砕:
− 250mlのバッフル付きエルレンマイヤーフラスコに入れた100mlのMRS培地(Difco製)中で37℃、200rpmにて一晩培養する;
− 細胞を遠心分離する:4000rpm、10分間、4℃;
− 上清を捨て、ペレットを5mlの溶液1(+リゾチーム)にとり、よく懸濁させる。インキュベーター(37℃)内で1〜4時間インキュベートする;
− プロトプラストを慎重に遠心分離する:3000rpm、10分間;
− 上清を捨て、ペレットを10mlの溶液1(リゾチームなし)で洗浄する:3000rpm、4℃、8分間;
− 上清を捨て、ペレットを10mlの0.01M Tris-HCl、pH8.0で洗浄する:3000rpm、4℃、8分間;
− 上清を捨て、ペレットを4mlのTE緩衝液に懸濁する;
− 0.5mlの10%SDSおよび0.5mlの5M NaClを加え、慎重に混合する;
− 1 mgのプロテイナーゼK(Qiagenのプロテイナーゼ:20mg/ml、すなわち50μl)を加えて、インキュベーター内で37℃にて一晩インキュベートする;
− この混合物をTE緩衝液を用いて20mlにする。
【0218】
(3) 抽出:
− フェノールを1:1、すなわち20mlのフェノール+20mlの混合物となるように加える。慎重に混合し、4℃、4000rpmで5分間遠心分離する;
− 上相を取り、それを新しいFalcon(20ml)に移す;
− 20mlのフェノール:クロロホルム:イソアミルアルコール(25:24:1)を加える。慎重に混合し、4℃、4000rpmで5分間遠心分離する;
− 上相を取り、新しいFalcon(約18ml)に移す;
− 18mlのクロロホルム:イソアミルアルコール(24:1、すなわち18mlのクロロホルム+333μlのイソアミルアルコール)を加える。慎重に混合して、4℃、4000rpmで5分間遠心分離する。この工程を上相が透明になるまで繰り返す;
− 上相(18ml)を2倍容量のエタノール(36ml)を用いて沈殿させる。1/50の3M 酢酸ナトリウム(約360μl)を加える。-20℃で少なくとも30分間沈殿させたままにする。その後、4℃、12000rpmで30分間遠心分離する;
− 上清を捨て、ペレットを1〜2mlのTE緩衝液にとる。TE緩衝液1mlに対して20μgのRNaseを加える。
インキュベーション:水浴中37℃にて1時間。
【0219】
(4) 透析:
RNase処理の後、混合物を透析バッグに移す。3回の透析を行うが、それぞれ1.5LのTE緩衝液中、4℃にて1時間行う。最後の透析工程を一晩かけて行うことも可能である。
− 透析したDNAをFalconに移し、数本のEppendorffチューブ(500μl)に分注する;
− 2倍容量のエタノール(1000μl)および1/3容量の2M LiCl(166μl)を加える;
− -20℃にて少なくとも30分間沈殿させる。その後、4℃、12000rpmで30分間遠心分離する;
− 上清を慎重に流出させる;
− ペレットを20mlの70%エタノールで洗浄し、4℃、12000rpmで15分間遠心分離する;
− 上清を慎重に流し出し、ペレットを風乾させ、適量の10mM Tris*HCl、pH8.0(ペレットの大きさによって100μl以上)にDNAを取る。
再解離を改善するために、DNAをEppendorf振とう器中で55〜60℃、低振動数(400rpm)で1〜2時間インキュベートした。
【0220】
b) ペプチドのトリプシン消化およびEdmann配列決定の後、さらなるアミノ酸配列を実施例5に記載のタンパク質精製より得た。配列解析の結果を図3Bにまとめた。
切断点のために、配列FVVDGGYTAQ(V8-RP Fr.7を参照)はおそらくC末端を表す。核酸配列(プライマーMke338および339)は、Lu10288染色体DNA上で行われるPCR増幅によりデヒドロゲナーゼ遺伝子をクローニングするために用いたものであるが(プロトコルは上記参照)、ラクトバチラス・ブレビス(Lactobacillus brevis)のコドン使用頻度を考慮して、上記配列およびN末端アミノ酸配列(配列番号1)から推定した。
【0221】
【0222】
PCRは、Pfuポリメラーゼ(Stratagene) および以下の温度プログラムを用いた標準的なStratageneプロトコルに従って行った:95℃にて5分;95℃にて45秒、52℃にて45秒、および72℃にて1分20秒を30サイクル;72℃にて10分;使用するまで10℃。PCR産物(0.8 kb)をアガロースゲル電気泳動(1.2% E-Gel、Invitrogen)およびカラムクロマトグラフィー(Mini-Elute、Qiagen)によって単離し、次いでNdeI/HindIIIを用いて消化して、同様に消化したpDHE19.2ベクター(pJOE誘導体、DE19848129)にクローン化した。ライゲーション混合物を大腸菌XL1 Blue(Stratagene)に形質転換させた。対応するクローンの配列決定は、得られたプラスミドpDHE10288adh1の挿入物として、配列番号3に示した核酸配列をもたらし、この配列は配列番号4のアミノ酸配列に対応した。実施例5および8で同定されるペプチドはすべて、この配列中に再び見出される。
【0223】
【0224】
実施例9:ラクトバチラス・ブレビス(Lactobacillus brevis)Lu10288由来の組換えデヒドロゲナーゼの活性の測定
プラスミドpDHE10288adh1を大腸菌TG10 pAgro4 pHSG575に再形質転換した(TG10:大腸菌TG1のRhaA- 誘導体(Stratagene);pAgro4:Takeshita, S;Sato, M;Toba, M;Masahashi, W;Hashimoto-Gotoh, T(1987)Gene 61, 63-74;pHSG575:T. Tomoyasuら(2001), Mol. Microbiol. 40(2), 397-413)。
【0225】
それぞれの場合に6個の形質転換体を、100mlのエルレンマイヤー(バッフル付き)フラスコに入れた20mlのLBAmp/Spec/Cm(100μg/LのAmp;50mg/LのSpec;10μg/LのCm)、0.1mM IPTG、0.5g/Lのラムノース中で37℃にて18時間増殖させ、次いで10分間、5000gにて遠心分離し、10mM Tris/HCl, pH7.0を用いて1回洗浄し、2mlの同一緩衝液に再懸濁させた。100μlの細胞懸濁液を900μlの50mM MES、pH6(50μl/mlのグルコースDH(実施例1)、100mMグルコース、100mM NaCl、1mM NADH、1mM NADPHおよび10mM 3-クロロ-1-(チエン-2-イル)プロパン-1-オンを含有する)中で振とうしながら20分間インキュベートした。この混合物を実施例4と同様に分析した。平均0.13mMの3-クロロ-1-(チエン-2-イル)プロパン-1-オールが生成され、これは培養懸濁液1Lあたり6.6Uの活性に相当した。粗抽出物(振動式ミルにて0.7mlのガラスビーズ(d=0.5mm)を用いた細胞破砕(3×5分、その合間に氷上で冷却した)によって得られたもの)を含有する同様のアッセイ試料においても、0.21mMの3-クロロ-1-(チエン-2-イル)プロパン-1-オール(これは10.7U/Lの活性に相当する)が測定された。培養物にラムノースを加えなかったコントロール実験においては、わずかな3-クロロ-1-(チエン-2-イル)プロパン-1-オールも検出することはできなかった。
【0226】
実施例10:カンジダ・マグノリア(Candida magnoliae)Lu8472由来のデヒドロゲナーゼのクローニング
以下のアミノ酸配列は、Edmann配列決定によってN末端配列を再度決定した後に、実施例6に記載されるタンパク質精製より得られたものである:
(S,GまたはT)(TまたはP)TSNALVTGGSRGIGAA
【0227】
以下のさらなるアミノ酸配列は、ペプチドのトリプシン消化およびEdmann配列決定の後に得られたものである:
IGVNSINPG
【0228】
核酸配列(プライマーMke366, 367および374)は、Lu8472染色体DNA上で行われるPCR増幅によってデヒドロゲナーゼ遺伝子をクローニングするために以下のように使用したものであるが(3倍濃縮のリチカーゼ溶液を用いたゲノムDNAキット、Qiagen、Hilden)、カンジダ・マグノリア(Candida magnoliae)のコドン使用頻度を考慮して、これらのペプチドから推定した。
【0229】
【0230】
プライマーMke366およびMke367を1:1の比率で混合した。PCRは、Pfu Turboポリメラーゼ(Stratagene)および以下の温度勾配プログラムを用いた標準的なStratageneプロトコルに従って行った:95℃にて1分;95℃にて1分、X℃1にて45秒および72℃にて2分を30サイクル;72℃にて10分;使用するまで10℃。PCR産物(約0.5 kb)をアガロースゲル電気泳動(1.2%E-Gel、Invitrogen)およびカラムクロマトグラフィー(GFX-Kit、Amersham Pharmacia)によって単離し、次いで配列決定した(配列決定プライマー:Mke366およびMke374)。得られた配列を配列番号5に示す。それから推定されるアミノ酸配列(配列番号6)は、カンジダ・マグノリア(Candida magnoliae)のカルボニル還元酵素(WO200140450)と53%の同一性を示す。タンパク質精製の後に決定したペプチド配列には、ごくわずかな相違が存在する。この相違は、配列決定の誤りによるものか、またはカンジダ・マグノリア(Candida magnoliae)Lu8742に複数のイソ酵素が存在することにより生じたと考えられる。
【0231】
112個のアッセイ試料を異なるアニーリング温度、すなわち25℃から45℃までの温度(それぞれの場合に約2℃の増分)で実験した。すべてのアッセイ試料において、類似した濃度の0.5kbバンドが主産物として形成された。
【0232】
【特許請求の範囲】
【請求項1】
式I:
【化1】
で表される3-メチルアミノ-1-(チエン-2-イル)プロパン-1-オールを調製するための方法であって、
a)チオフェンを、ルイス酸の存在下で、ハロゲン化β-ハロプロピオニルまたはハロゲン化アクリロイルと反応させ、その際、同時に、または反応が生じた後であるが反応生成物を単離する前にハロゲン化水素を通過させて、3-ハロ-1-(チエン-2-イル)プロパン-1-オンを得、そして
b)工程a)で得られたプロパノンを還元し、次いで、適宜に反応生成物を単離することなく、メチルアミンと反応させる、
ことを含んでなる、前記方法。
【請求項2】
工程a)に用いられるルイス酸が三塩化アルミニウムである、請求項1に記載の方法。
【請求項3】
工程a)における反応が、溶媒であるハロゲン化炭化水素中で実施される、請求項1または2に記載の方法。
【請求項4】
工程b)の還元が、還元剤として、金属水素化物もしくは半金属水素化物を用いて、または遷移金属触媒の存在下で水素を用いて行われる、請求項1〜3のいずれか1項に記載の方法。
【請求項5】
工程b)の還元を、(S)-3-メチルアミノ-1-(チエン-2-イル)プロパン-1-オールの形成に関して選択性を示すキラル還元剤またはキラル触媒の存在下で行うことを特徴とする、式I-S:
【化2】
で表される(S)-3-メチルアミノ-1-(チエン-2-イル)プロパン-1-オールを、適当な場合にはそのRエナンチオマーI-Rとの混合物として、ただし該混合物中には式I-Sのプロパノールを優勢に存在させて、調製するための請求項1〜4のいずれか1項に記載の方法。
【請求項6】
工程b)に用いられる前記還元剤が、不斉金属水素化物もしくは不斉半金属水素化物、または不斉遷移金属触媒の存在下での水素であるか、あるいは前記還元が、不斉誘導を媒介する化合物の存在下で行われる、請求項5に記載の方法。
【請求項7】
工程b)の還元が、デヒドロゲナーゼ(E.C. 1.1.x.x.)の存在下で行われる、請求項5に記載の方法。
【請求項8】
還元が、デヒドロゲナーゼ(E.C. 1.1.1.x)の存在下、特にアルコールデヒドロゲナーゼ(E.C.1.1.1.1またはE.C.1.1.1.2)の存在下で行われる、請求項7に記載の方法。
【請求項9】
前記デヒドロゲナーゼが、ゲオトリカム(Geotrichum)属、ピチア(Pichia)属、カンジダ(Candida)属、ハンセヌラ(Hansenula)属またはサッカロマイセス(Saccharomyces)属の酵母、およびシュードモナス(Pseudomonas)属、ブルクホリデリア(Burkholderia)属、アグロバクテリウム(Agrobacterium)属、ロドコッカス(Rhodococcus)属またはラクトバチラス(Lactobacillus)属の細菌に由来するデヒドロゲナーゼのなかから選択される、請求項7または8に記載の方法。
【請求項10】
前記デヒドロゲナーゼが、ゲオトリカム・カンジドウム(Geotrichum candidum)、カンジダ・マグノリア(Candida magnoliae)およびラクトバチラス・ブレビス(Lactobacillus brevis)に由来するデヒドロゲナーゼのなかから選択される、請求項9に記載の方法。
【請求項11】
N末端領域に、
a)配列番号1に示される少なくとも10個連続したアミノ酸残基の構成的なアミノ酸配列であって、配列番号1に示されるアミノ酸位置12に対応する位置が好ましくはさらにバリンを表す構成的なアミノ酸配列;または
b)配列番号2に示される少なくとも10個連続したアミノ酸残基の構成的なアミノ酸配列;
を含むアミノ酸配列を有するアルコールデヒドロゲナーゼ、ならびにそれに由来する機能的に等価であるアルコールデヒドロゲナーゼ。
【請求項12】
3-クロロ-1-(チエン-2-イル)プロパン-1-オンを(S)-3-クロロ-1-(チエン-2-イル)プロパン-1-オールに還元することが可能である、請求項11に記載のアルコールデヒドロゲナーゼ。
【請求項13】
少なくとも85% eeのエナンチオマー純度で(NADHおよび/またはNADPHの存在下;30℃、pH6.0にて)前記還元を触媒する、請求項12に記載のアルコールデヒドロゲナーゼ。
【請求項14】
配列番号3を含む核酸配列によってコードされるか、または配列番号4に示されるアミノ酸配列もしくは少なくとも図3に示される構成的な配列を含んでなり、好ましくはラクトバチラス・ブレビス(Lactobacillus brevis)から得ることができる、請求項11〜13のいずれか1項に記載のアルコールデヒドロゲナーゼ、ならびにそれに由来する機能的に等価なアルコールデヒドロゲナーゼ。
【請求項15】
配列番号5を含む核酸配列によってコードされるか、または配列番号6を含むアミノ酸配列を有し、好ましくはカンジダ・マグノリア(Candida magnoliae)(ATCC 12573)から得ることができる、請求項11〜13のいずれか1項に記載のアルコールデヒドロゲナーゼ、ならびにそれに由来する機能的に等価なアルコールデヒドロゲナーゼ。
【請求項16】
請求項11〜15のいずれか1項に記載のデヒドロゲナーゼのコード配列、特に配列番号3および5に示される配列、を含む核酸配列、ならびにそれに由来する誘導体。
【請求項17】
少なくとも1つの調節核酸配列と機能的に連結された請求項15に記載の核酸配列を含む発現カセット。
【請求項18】
請求項16に記載の少なくとも1つの発現カセットを含む、組換えベクター。
【請求項19】
請求項17に記載の少なくとも1つのベクターを用いて形質転換された原核細胞または真核細胞の宿主。
【請求項20】
(S)-3-ハロ-1-(チエン-2-イル)プロパン-1-オールの調製における、請求項11〜14のいずれか1項に記載のデヒドロゲナーゼ、またはこのデヒドロゲナーゼを産生する天然もしくは組換え微生物の使用。
【請求項21】
用いられる前記デヒドロゲナーゼが、請求項11〜14のいずれか1項に記載のアルコールデヒドロゲナーゼまたは、このデヒドロゲナーゼを産生する天然もしくは組換え微生物である、請求項7〜10のいずれか1項に記載の方法。
【請求項22】
式I-Sの(S)-3-メチルアミノ-1-(チエン-2-イル)プロパン-1-オールを調製するための方法であって、3-ハロ-1-(チエン-2-イル)プロパン-1-オンをエナンチオ選択的に還元し、この還元をデヒドロゲナーゼの存在下で行う、前記方法。
【請求項23】
還元において得られる(S)-3-ハロ-1-(チエン-2-イル)プロパン-1-オールを、単離することなくメチルアミンと反応させる、請求項21に記載の方法。
【請求項24】
前記デヒドロゲナーゼが、ゲオトリカム(Geotrichum)属、ピチア(Pichia)属、カンジダ(Candida)属、ハンセヌラ(Hansenula)属またはサッカロマイセス(Saccharomyces)属の酵母、およびシュードモナス(Pseudomonas)属、ブルクホリデリア(Burkholderia)属、アグロバクテリウム(Agrobacterium)属、ロドコッカス(Rhodococcus)属またはラクトバチラス(Lactobacillus)属の細菌に由来するデヒドロゲナーゼのなかから選択される、請求項21または22に記載の方法。
【請求項25】
前記デヒドロゲナーゼが、ゲオトリカム・カンジドウム(Geotrichum candidum)、カンジダ・マグノリア(Candida magnoliae)またはラクトバチラス・ブレビス(Lactobacillus brevis)に由来するデヒドロゲナーゼのなかから選択される、請求項23に記載の方法。
【請求項26】
前記デヒドロゲナーゼが、請求項11〜15のいずれか1項に記載のアルコールデヒドロゲナーゼのなかから選択される、請求項21に記載の方法。
【請求項1】
式I:
【化1】
で表される3-メチルアミノ-1-(チエン-2-イル)プロパン-1-オールを調製するための方法であって、
a)チオフェンを、ルイス酸の存在下で、ハロゲン化β-ハロプロピオニルまたはハロゲン化アクリロイルと反応させ、その際、同時に、または反応が生じた後であるが反応生成物を単離する前にハロゲン化水素を通過させて、3-ハロ-1-(チエン-2-イル)プロパン-1-オンを得、そして
b)工程a)で得られたプロパノンを還元し、次いで、適宜に反応生成物を単離することなく、メチルアミンと反応させる、
ことを含んでなる、前記方法。
【請求項2】
工程a)に用いられるルイス酸が三塩化アルミニウムである、請求項1に記載の方法。
【請求項3】
工程a)における反応が、溶媒であるハロゲン化炭化水素中で実施される、請求項1または2に記載の方法。
【請求項4】
工程b)の還元が、還元剤として、金属水素化物もしくは半金属水素化物を用いて、または遷移金属触媒の存在下で水素を用いて行われる、請求項1〜3のいずれか1項に記載の方法。
【請求項5】
工程b)の還元を、(S)-3-メチルアミノ-1-(チエン-2-イル)プロパン-1-オールの形成に関して選択性を示すキラル還元剤またはキラル触媒の存在下で行うことを特徴とする、式I-S:
【化2】
で表される(S)-3-メチルアミノ-1-(チエン-2-イル)プロパン-1-オールを、適当な場合にはそのRエナンチオマーI-Rとの混合物として、ただし該混合物中には式I-Sのプロパノールを優勢に存在させて、調製するための請求項1〜4のいずれか1項に記載の方法。
【請求項6】
工程b)に用いられる前記還元剤が、不斉金属水素化物もしくは不斉半金属水素化物、または不斉遷移金属触媒の存在下での水素であるか、あるいは前記還元が、不斉誘導を媒介する化合物の存在下で行われる、請求項5に記載の方法。
【請求項7】
工程b)の還元が、デヒドロゲナーゼ(E.C. 1.1.x.x.)の存在下で行われる、請求項5に記載の方法。
【請求項8】
還元が、デヒドロゲナーゼ(E.C. 1.1.1.x)の存在下、特にアルコールデヒドロゲナーゼ(E.C.1.1.1.1またはE.C.1.1.1.2)の存在下で行われる、請求項7に記載の方法。
【請求項9】
前記デヒドロゲナーゼが、ゲオトリカム(Geotrichum)属、ピチア(Pichia)属、カンジダ(Candida)属、ハンセヌラ(Hansenula)属またはサッカロマイセス(Saccharomyces)属の酵母、およびシュードモナス(Pseudomonas)属、ブルクホリデリア(Burkholderia)属、アグロバクテリウム(Agrobacterium)属、ロドコッカス(Rhodococcus)属またはラクトバチラス(Lactobacillus)属の細菌に由来するデヒドロゲナーゼのなかから選択される、請求項7または8に記載の方法。
【請求項10】
前記デヒドロゲナーゼが、ゲオトリカム・カンジドウム(Geotrichum candidum)、カンジダ・マグノリア(Candida magnoliae)およびラクトバチラス・ブレビス(Lactobacillus brevis)に由来するデヒドロゲナーゼのなかから選択される、請求項9に記載の方法。
【請求項11】
N末端領域に、
a)配列番号1に示される少なくとも10個連続したアミノ酸残基の構成的なアミノ酸配列であって、配列番号1に示されるアミノ酸位置12に対応する位置が好ましくはさらにバリンを表す構成的なアミノ酸配列;または
b)配列番号2に示される少なくとも10個連続したアミノ酸残基の構成的なアミノ酸配列;
を含むアミノ酸配列を有するアルコールデヒドロゲナーゼ、ならびにそれに由来する機能的に等価であるアルコールデヒドロゲナーゼ。
【請求項12】
3-クロロ-1-(チエン-2-イル)プロパン-1-オンを(S)-3-クロロ-1-(チエン-2-イル)プロパン-1-オールに還元することが可能である、請求項11に記載のアルコールデヒドロゲナーゼ。
【請求項13】
少なくとも85% eeのエナンチオマー純度で(NADHおよび/またはNADPHの存在下;30℃、pH6.0にて)前記還元を触媒する、請求項12に記載のアルコールデヒドロゲナーゼ。
【請求項14】
配列番号3を含む核酸配列によってコードされるか、または配列番号4に示されるアミノ酸配列もしくは少なくとも図3に示される構成的な配列を含んでなり、好ましくはラクトバチラス・ブレビス(Lactobacillus brevis)から得ることができる、請求項11〜13のいずれか1項に記載のアルコールデヒドロゲナーゼ、ならびにそれに由来する機能的に等価なアルコールデヒドロゲナーゼ。
【請求項15】
配列番号5を含む核酸配列によってコードされるか、または配列番号6を含むアミノ酸配列を有し、好ましくはカンジダ・マグノリア(Candida magnoliae)(ATCC 12573)から得ることができる、請求項11〜13のいずれか1項に記載のアルコールデヒドロゲナーゼ、ならびにそれに由来する機能的に等価なアルコールデヒドロゲナーゼ。
【請求項16】
請求項11〜15のいずれか1項に記載のデヒドロゲナーゼのコード配列、特に配列番号3および5に示される配列、を含む核酸配列、ならびにそれに由来する誘導体。
【請求項17】
少なくとも1つの調節核酸配列と機能的に連結された請求項15に記載の核酸配列を含む発現カセット。
【請求項18】
請求項16に記載の少なくとも1つの発現カセットを含む、組換えベクター。
【請求項19】
請求項17に記載の少なくとも1つのベクターを用いて形質転換された原核細胞または真核細胞の宿主。
【請求項20】
(S)-3-ハロ-1-(チエン-2-イル)プロパン-1-オールの調製における、請求項11〜14のいずれか1項に記載のデヒドロゲナーゼ、またはこのデヒドロゲナーゼを産生する天然もしくは組換え微生物の使用。
【請求項21】
用いられる前記デヒドロゲナーゼが、請求項11〜14のいずれか1項に記載のアルコールデヒドロゲナーゼまたは、このデヒドロゲナーゼを産生する天然もしくは組換え微生物である、請求項7〜10のいずれか1項に記載の方法。
【請求項22】
式I-Sの(S)-3-メチルアミノ-1-(チエン-2-イル)プロパン-1-オールを調製するための方法であって、3-ハロ-1-(チエン-2-イル)プロパン-1-オンをエナンチオ選択的に還元し、この還元をデヒドロゲナーゼの存在下で行う、前記方法。
【請求項23】
還元において得られる(S)-3-ハロ-1-(チエン-2-イル)プロパン-1-オールを、単離することなくメチルアミンと反応させる、請求項21に記載の方法。
【請求項24】
前記デヒドロゲナーゼが、ゲオトリカム(Geotrichum)属、ピチア(Pichia)属、カンジダ(Candida)属、ハンセヌラ(Hansenula)属またはサッカロマイセス(Saccharomyces)属の酵母、およびシュードモナス(Pseudomonas)属、ブルクホリデリア(Burkholderia)属、アグロバクテリウム(Agrobacterium)属、ロドコッカス(Rhodococcus)属またはラクトバチラス(Lactobacillus)属の細菌に由来するデヒドロゲナーゼのなかから選択される、請求項21または22に記載の方法。
【請求項25】
前記デヒドロゲナーゼが、ゲオトリカム・カンジドウム(Geotrichum candidum)、カンジダ・マグノリア(Candida magnoliae)またはラクトバチラス・ブレビス(Lactobacillus brevis)に由来するデヒドロゲナーゼのなかから選択される、請求項23に記載の方法。
【請求項26】
前記デヒドロゲナーゼが、請求項11〜15のいずれか1項に記載のアルコールデヒドロゲナーゼのなかから選択される、請求項21に記載の方法。
【図1】

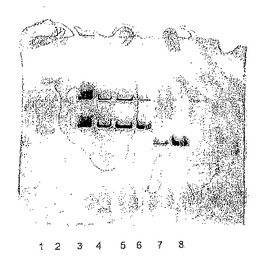
【図2】
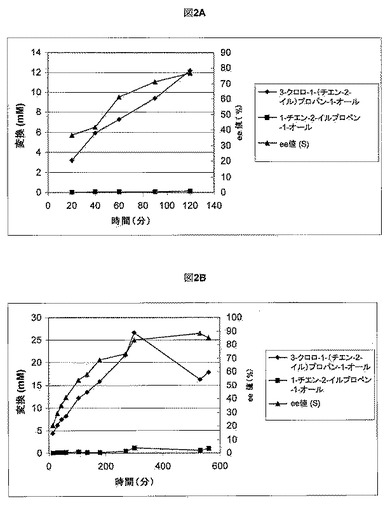
【図3A】
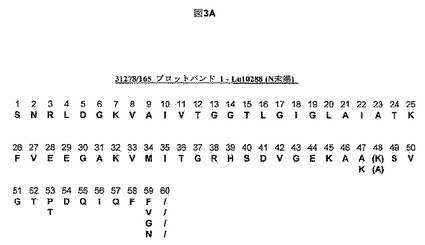
【図3B−1】

【図3B−2】

【図3B−3】

【図3B−4】

【図3B−5】

【図3B−6】

【図2】
【図3A】
【図3B−1】
【図3B−2】
【図3B−3】
【図3B−4】
【図3B−5】
【図3B−6】
【公開番号】特開2011−142908(P2011−142908A)
【公開日】平成23年7月28日(2011.7.28)
【国際特許分類】
【出願番号】特願2011−5361(P2011−5361)
【出願日】平成23年1月14日(2011.1.14)
【分割の表示】特願2006−530058(P2006−530058)の分割
【原出願日】平成16年9月30日(2004.9.30)
【出願人】(508020155)ビーエーエスエフ ソシエタス・ヨーロピア (2,842)
【氏名又は名称原語表記】BASF SE
【住所又は居所原語表記】D−67056 Ludwigshafen, Germany
【Fターム(参考)】
【公開日】平成23年7月28日(2011.7.28)
【国際特許分類】
【出願日】平成23年1月14日(2011.1.14)
【分割の表示】特願2006−530058(P2006−530058)の分割
【原出願日】平成16年9月30日(2004.9.30)
【出願人】(508020155)ビーエーエスエフ ソシエタス・ヨーロピア (2,842)
【氏名又は名称原語表記】BASF SE
【住所又は居所原語表記】D−67056 Ludwigshafen, Germany
【Fターム(参考)】
[ Back to top ]