
3−(2−ヒドロキシ−5−メチルフェニル)−N,N−ジイソプロピル−3−フェニルプロピルアミンの誘導体およびその使用方法
本発明は、トルテロジン、5−ヒドロキシメチルトルテロジン、フェソテロジンの新規誘導体、およびそれらの医薬的に許容される塩に関する。本発明は、本発明の化合物を含む組成物、およびムスカリン受容体遮断薬によって有利に治療される疾患および疾病を治療する方法におけるかかる組成物の用途についても提供する。

【発明の詳細な説明】
【技術分野】
【0001】
(関連出願の相互参照)
本願は、2008年4月9日出願の米国特許仮出願第61/043,729号の利益を主張するものであり、この出願の全教示内容は、参照することで本明細書に組み入れられる。
【0002】
本発明は、トルテロジン、5−ヒドロキシメチルトルテロジン、およびフェソテロジンの新規誘導体、ならびに、それらの医薬的に許容される塩、溶媒和物、および水和物に関する。本発明は、本発明の化合物を含む組成物、およびムスカリン受容体遮断薬によって有利に治療される疾患および疾病を治療する方法におけるかかる組成物の用途についても提供する。
【0003】
トルテロジンは、3−(2−ヒドロキシ−5−メチルフェニル)−N,N−ジイソプロピル−3−フェニルプロピルアミンであり、L−酒石酸との1:1塩としてデトロール(登録商標)の名称で販売されている。これは、強力かつ競合的なムスカリン受容体遮断薬であり、例えば、過活動膀胱をもつ成人の治療に有用である。
【0004】
デトロールは、頻尿、切迫性、切迫性尿失禁、またはこれらの症状の任意の組み合わせの症状をもつ過活動膀胱の患者の治療に承認されている。トルテロジンは、高齢者の記憶および認識における化合物の作用について臨床試験中でもある。
【0005】
経口投与後、トルテロジンは肝臓で代謝され、主要な薬理学的活性代謝産物である5−ヒドロキシメチルトルテロジンを産生する。トルテロジンと同様の抗ムスカリン活性を示す5−ヒドロキシメチル代謝産物は、治療効果に著しく寄与する。フェソテロジンは、5−ヒドロキシメチル代謝産物のプロドラッグであり、フマル酸塩(2−メチルプロピオン酸2−[3−(N,N−ジイソプロピルアミノ)−1(R)−フェニルプロピル]−4−(ヒドロキシメチル)フェニルエステルフマル酸塩、およびイソ酪酸2−[3−(ジイソプロピルアミノ)−1(R)−フェニルプロピル]−4−(ヒドロキシメチル)フェニルエステルフマル酸塩)としてトヴィアズ(登録商標)の名称で販売されている。フェソテロジンは、切迫性尿失禁および過活動膀胱の治療において欧州連合で承認されており、抗ムスカリンとして米国で臨床試験中である。
【0006】
トルテロジン、5−ヒドロキシメチルトルテロジン、およびフェソテロジンの有益な活性にも関わらず、上述の疾患および疾病を治療するための新規化合物への継続的な必要性がある。
【図面の簡単な説明】
【0007】
【図1】トルテロジンと比較した、ヒト肝ミクロソームにおける本発明の種々の化合物の代謝経過を示す。(定義)
【0008】
「改善する」および「治療する」という用語は、区別なく使用され、治療および予防的治療の両方を含む。両方の用語は、疾患(例えば、本明細書で使用する疾患または疾病)の発生または進行を減少する、抑制する、弱毒化する、軽減する、阻む、または安定化することを意味する。
【0009】
「疾患」とは、細胞、組織、または器官の正常機能を損なうまたは妨げるあらゆる症状または疾病のことを意味する。
【0010】
合成に用いられる化学物質の由来に応じて、天然同位体存在度におけるいくつかのばらつきが合成化合物で起こることが分かるであろう。従って、トルテロジン、5−ヒドロキシメチルトルテロジン、またはフェソテロジンの製剤は、少量の重水素化された同位体置換体を本質的に含む。天然に豊富で安定な水素および炭素同位体の濃度は、このばらつきに関わらず、少濃度であり、本発明の化合物の安定同位体置換の程度と比較して微々たるものである。例えば、Wada,Eら、Seikagaku,1994,66:15 Gannes,LZら、Comp Biochem Physiol Mol Integr Physiol,1998,119:725を参照されたい。本発明の化合物において、特定の位置が重水素を有するものとして指定される場合、その位置における重水素の存在度は、重水素の天然存在度である0.015%よりも実質的に多いことを理解されたい。「D」または「重水素」として指定される位置は、一般に、重水素の天然存在度(即ち、少なくとも45%重水素取り込み)よりも少なくとも3000倍の存在度で、その位置に重水素を有する。
【0011】
本明細書で使用する「同位体濃縮係数」という用語は、特定同位体の同位体存在度と天然存在度の間の比率を意味する。
【0012】
他の実施形態において、本発明の化合物は、指定された各重水素原子に対して、少なくとも3500(指定された各重水素原子で52.5%の重水素取り込み)、少なくとも4000(60%重水素取り込み)、少なくとも4500(67.5%重水素取り込み)、少なくとも5000(75%重水素取り込み)、少なくとも5500(82.5%重水素取り込み)、少なくとも6000(90%重水素取り込み)、少なくとも6333.3(95%重水素取り込み)、少なくとも6466.7(97%重水素取り込み)、少なくとも6600(99%重水素取り込み)、または少なくとも6633.3(99.5%重水素取り込み)の同位体濃縮係数を有する。
【0013】
本発明の化合物において、特定の同位体として特に指定されない任意の原子は、その原子の任意の安定同位体を表わすことを意味する。特に明記しない限り、位置が「H」または「水素」と具体的に指定される場合、その位置は、その天然存在度の同位体組成で水素を有すると理解される。
【0014】
「同位体置換体」という用語は、その同位体組成においてのみ本発明の特定化合物とは異なる種のことを言う。
【0015】
「化合物」という用語は、本発明の化合物のことを言う場合、分子の構成原子間において同位体変化があり得る以外は同一の化学構造を有する分子の集合体のことを言う。従って、示された重水素原子を含む特定の化学構造によって表わされる化合物は、その構造における1つ以上の指定された重水素の位置に水素原子を有するより少量の同位体置換体を含むことが当業者には明らかであろう。本発明の化合物におけるかかる同位体置換体の相対量は、化合物を作るのに用いる重水素化試薬の同位体純度や、化合物の調製に用いる種々の合成工程における重水素の取り込み効率を含む多くの要因による。しかしながら、上述のように、かかる同位体置換体のin totoの相対量は、化合物の55%未満である。他の実施形態において、かかる同位体置換体のin totoの相対量は、化合物の47.5%未満、40%未満、32.5%未満、25%未満、17.5%未満、10%未満、5%未満、3%未満、1%未満、または0.5%未満である。
【0016】
本発明は、本発明の化合物の塩、溶媒和物、または水和物についても提供する。
【0017】
本発明の化合物の塩は、酸と、アミノ官能基などの化合物の塩基性基との間で形成されるか、あるいは、塩基と、カルボキシル官能基などの化合物の酸性基との間で形成される。別の実施形態によれば、化合物は、医薬的に許容される酸付加塩である。
【0018】
本明細書で使用する「医薬的に許容される」という用語は、適切な医学的判断の範囲内で、過度の毒性、刺激、アレルギー反応等がなくヒトや他の哺乳類の組織に接触させて用いるのに適切であり、なおかつ、妥当な便益/リスク比に相応した成分のことを言う。「医薬的に許容される塩」とは、受容体への投与時に、本発明の化合物を直接的または間接的に提供することができる任意の非毒性塩のことを意味する。「医薬的に許容される対イオン」とは、受容体への投与時に塩から放出された際に、毒性のない塩のイオン部分のことである。
【0019】
医薬的に許容される塩を形成するのに一般的に用いられる酸としては、二硫化水素、塩酸、臭化水素酸、ヨウ化水素酸、硫酸、およびリン酸などの無機酸のみならず、パラ−トルエンスルホン酸、サリチル酸、酒石酸、二酒石酸、アスコルビン酸、マレイン酸、ベシル酸、フマル酸、グルコン酸、グルクロン酸、ギ酸、グルタミン酸、メタンスルホン酸、エタンスルホン酸、ベンゼンスルホン酸、乳酸、シュウ酸、パラ−ブロモフェニルスルホン酸、炭酸、コハク酸、クエン酸、安息香酸、および酢酸などの有機酸、ならびに関連の無機酸および有機酸が挙げられる。従って、かかる医薬的に許容される塩としては、硫酸塩、ピロ硫酸塩、重硫酸塩、亜硫酸塩、重亜硫酸塩、リン酸塩、一水素リン酸塩、二水素リン酸塩、メタリン酸塩、ピロリン酸塩、塩化物、臭化物、ヨウ化物、酢酸塩、プロピオン酸塩、デカン酸塩、カプリル酸塩、アクリル酸塩、ぎ酸塩、イソ酪酸塩、カプリン酸塩、ヘプタン酸塩、プロピオル酸塩、シュウ酸塩、マロン酸塩、コハク酸塩、スベリン酸塩、セバシン酸塩、フマル酸塩、マレイン酸塩、ブチン−1,4−ジオエート、ヘキシン−1,6−ジオエート、安息香酸塩、クロロ安息香酸塩、メチル安息香酸塩、ジニトロ安息香酸塩、ヒドロキシ安息香酸塩、メトキシ安息香酸塩、フタル酸塩、テレフタル酸塩、スルホン酸塩、キシレンスルホン酸塩、フェニル酢酸塩、フェニルプロピオン酸塩、フェニル酪酸塩、クエン酸塩、乳酸塩、β−ヒドロキシ酪酸塩、グリコール酸塩、マレイン酸塩、酒石酸塩、メタンスルホン酸塩、プロパンスルホン酸塩、ナフタレン−1−スルホン酸塩、ナフタレン−2−スルホン酸塩、マンデル酸塩、および他の塩が挙げられる。一実施形態において、医薬的に許容される酸付加塩としては、塩酸および臭化水素酸などの鉱酸で形成されたもの、特に、マレイン酸などの有機酸で形成されたものが挙げられる。
【0020】
本明細書で使用する「水和物」という用語は、非共有結合性の分子間力によって結合した化学量論量または非化学量論量の水をさらに含む化合物を意味する。
【0021】
本明細書で使用する「溶媒和物」という用語は、非共有結合性の分子間力によって結合した化学量論量または非化学量論量の溶媒(水、アセトン、エタノール、メタノール、ジクロロメタン、2−プロパノールなど)をさらに含む化合物を意味する。
【0022】
本発明の化合物(例えば、式I、式IIの化合物など)は、不斉炭素原子を含む。このように、本発明の化合物は、個々の鏡像異性体(例えば、(S)または(R)の1つ)としても存在し、2つの鏡像異性体の混合物としても存在することができる。(R)化合物の方が活性がより大きく、好適である。従って、本発明の化合物は、両方のラセミ混合物だけでなく、別の可能な立体異性体を実質的に含まない個々それぞれの立体異性体も含む。本明細書で使用する「他の立体異性体を実質的に含まない」という用語は、他の立体異性体を25%未満、好ましくは他の立体異性体を10%未満、より好ましくは他の立体異性体を5%未満、最も好ましくは他の立体異性体を2%未満、または他の立体異性体を「X」%未満(ここで、Xは0以上100以下の数)含むことを意味する。所与の化合物に対する個々の鏡像異性体を得るまたは合成する方法は、当該技術分野では周知であり、最終化合物または出発材料あるいは中間体に実用可能なものとして適用することができる。
【0023】
本明細書で使用する「安定化合物」という用語は、製造するのに十分な安定性を有し、本明細書で詳述した目的に有用な十分な期間にわたって化合物の完全性を維持する化合物のことを言う(例えば、治療薬に反応する疾患または疾病を治療するための治療薬、治療化合物の製造に使用する中間体、単離可能または保存可能な中間体化合物への製剤)。
【0024】
「D」とは、重水素のことを言う。
【0025】
「立体異性体」とは、鏡像異性体およびジアステレオマーのことを言う。
【0026】
「Tert」、「t」および「t−」は、それぞれ、三級のことを言う。
【0027】
「US」は、アメリカ合衆国のことを言う。
【0028】
「FDA」は、食品医薬品局のことを言う。
【0029】
「E.U.」は、欧州連合のことを言う。
【0030】
本明細書を通じて、「各R」への言及としては、該当する場合には独立して、全ての「R」基(例えば、R1、R2、R3、R4、およびR5)を含む。特に明記しない限り、変数が概括的なことを言う場合、特定の変数の具体的な実施形態を全て含むことを意味する。
(治療化合物)
【0031】
本発明は、式Iの化合物:
【化1】
式(I)、
または、その医薬的に許容される塩、溶媒和物、もしくは水和物を提供し、
式中:
R1は、CD3、CHD2、CH2D、CH3、CD2OH、CHDOH、またはCH2OHであり;
R2およびR3は、それぞれ独立して、0〜7個の重水素原子をもつイソプロピル基であり;
R4は、HまたはC(O)−C1−C6アルキルであり;
R1、R2、またはR3の少なくとも1つは重水素原子を含む。
【0032】
ある実施形態において、R1は、CH3、CD3、CD2OH、またはCH2OHから選択される。
【0033】
ある実施形態において、R2およびR3は同一である。
【0034】
他の実施形態において、R2およびR3は、−CH(CD3)2、−CD(CD3)2、−CH(CH3)2、および−CD(CH3)2から独立して選択される。
【0035】
ある実施形態において、R4はHである。
【0036】
他の実施形態において、R4はC(O)−CH(CH3)2である。
【0037】
さらに別の実施形態において、化合物は、表1(下記)に記載の化合物のいずれか1つから選択される:
表1:式Iの例示実施形態
【化2】
【0038】
さらに別の実施形態において、本発明は、式IIの化合物:
【化3】
式(II)、
またはその医薬的に許容される塩を提供し、
式中:
R2およびR3は、それぞれ独立して、0〜7個の重水素原子をもつイソプロピル基である。
【0039】
式IIの一実施形態において、R2およびR3の各々は、−CD(CD3)2および−CH(CH3)2から独立して選択される。より具体的な態様において、式IIの化合物は、化合物100および化合物112から選択される。
【0040】
さらに別の実施形態において、本発明は、式IIIの化合物:
【化4】
式(III)、
またはその医薬的に許容される塩を提供し、
式中:
R2およびR3は、それぞれ独立して、0〜7個の重水素原子をもつイソプロピル基であり;
Yは両方とも水素または重水素であり、
Yが両方とも水素である場合、R2またはR3の少なくとも1つは重水素原子を含む。
【0041】
式IIIの一実施形態において、R2およびR3の各々は、−CD(CD3)2および−CH(CH3)2から独立して選択される。より具体的な態様において、式IIIの化合物は、化合物116、化合物117、または化合物118から選択される。
【0042】
別の実施形態において、上述の実施形態のいずれかにおいて重水素として指定されない任意の原子は、その天然同位体存在度で存在する。
【0043】
式I、式IIなどの化合物の合成は、合成化学の分野における当業者によって容易に達成することができる。関連手順および中間体は、例えば、米国特許第7,119,212号、同第7,005,449号、同第5,382,600号、同第5,922,914号、同第5,686,464号、および同第5,559,269号;PCT出願刊行物国際公開第2005/006143号、同第2005/012227号、同第2004/078700号、同第2001/049649号、同第2003/014060号、および同第2006/074479号;Andersson,PGら、J Org Chem1998,63(22):8067;ならびに欧州特許公報第EP0957073号に開示されている。
【0044】
かかる方法は、重水素化され、必要に応じて、対応する他の同位体を含有する試薬、および/または、本明細書に記載の化合物を合成するための中間体を利用して、あるいは、同位体原子を化学構造に導入するため当該技術分野で周知の標準的な合成プロトコルを使用することで行うことができる。
(例示合成)
【0045】
式I、式IIなどの化合物を合成するための便利な方法についてスキーム1で説明する。
【0046】
スキーム1.式Iの化合物への一般経路。
【化5】
X=CD3、CD2H、CDH2、CH3、CD2OBn、CDHOBn、またはCH2OBn;Bn=ベンジル。
【0047】
スキーム1に示すように、式Iの化合物の調製は、Mg/CuBr−ジメチルスルフィドを用いて、適当に重水素化された臭化アリール11を公知の4(R)−フェニル−3−[3−フェニル−2(E)−プロペノイル]オキサゾリジン−2−オン(10)に位置選択的付加して、付加物12を生成することから始まる。THF/水中のLiOH/H2O2で12を加水分解して、カルボン酸13を得る。ベンゼン中のSOCl2/ピリジンと13を反応させて、酸塩化物14を生成し、これを適当に重水素化されたアミン20(市販のジイソプロピルアミン−d14[98原子%D]など)で処理し、対応するアミド15を得る。エーテル中のLiAlH4と15を還元させて、三級アミン16得、メタノール中のPd/CによりH2と水素化させて脱ベンジル化し、式Iの化合物(式中、R4はHである)を得る。17のフェノール性ヒドロキシル基をEt3Nの存在下で塩化アシル21によりアシル化し、式Iの化合物(式中、R4は、−C(O)−C1−C6アルキル)を得る。参照文献:Andersson,PGら、「Asymmetric total synthesis of (+)−tolterodine, a new muscarinic receptor antagonist, via copper−assisted asymmetric conjugate addition of aryl Grignard reagents to 3−phenyl−prop−2−enoyl−oxazolidinones」J Org Chem,1998,63(22):8067における一般手順;および欧州特許公報第EP957073号。
【0048】
適当に重水素化された臭化アリール11を調製するための例示的方法について以下のスキーム2Aおよび2Bに示す。
【0049】
スキーム2A.中間体11への一般経路。
【化6】
Y=CD3、CD2H、CDH2、CH3;
X=CD3、CD2H、CDH2、CH3、CD2OBn、CDHOBn、またはCH2OBn
【0050】
スキーム2Aに示すように(Aki,ら、J Phys Chem A 2002,106(14):3436−3444の一般手順に従う)、所望の臭化アリール30をMgおよびO2経由でフェノール31に変換した後、Br2で臭素化し、ブロモフェノール32を得る。ブロモフェノール32を塩化ベンジル経由で直接ベンジル保護して化合物11を生成してもよいし、あるいは、DDQで酸化させた後、NaBH4またはNaBD4(98原子%D)を用いて、適当に重水素化されたアルコール33に還元してもよい。次いで、ジオール33を塩化ベンジルでベンジル保護し、化合物11を得てもよい。
【0051】
スキーム2B:中間体11への代理経路。
【化7】
【0052】
スキーム2Bに示すように、市販の3−ブロモ−4−ヒドロキシ安息香酸を臭化ベンジルおよび炭酸カリウムで処理し、エステル45を得る。該エステルをLiAlH4またはLiAlD4(99原子%D)で還元させ、アルコール46を得る。これを臭化ベンジルおよび炭酸カリウムで処理し、化合物11(式中、XはCD2OBnまたはCH2OBnである)を得る。
【0053】
適当に重水素化されたアミン20の調製方法を以下のスキーム3Aおよび3Bに示す。
【0054】
スキーム3A.
【化8】
【0055】
スキーム3B.
【化9】
各Zは、独立して、CH3、CH2D、CHD2、またはCD3である。
【0056】
スキーム3Aに示すように、適当に重水素化されたアミン40を適当に重水素化された2−クロロプロパン41(または任意の他の適当に重水素化された2−ハロプロパン)でアルキル化して、所望のアミン20を得る(例:Zhang,C,ら、Zhongguo Yaowu Huaxue Zazhi 2004,14(3):161−164の同様の手順を参照されたい)。
【0057】
あるいは、スキーム3Bに示すように、アミン40を適当に重水素化された2−ケトプロパン42で縮合させ、イミン43を生成し、これを、適当に重水素化されたラネーニッケルおよび水素(または重水素)ガスで還元し、アミン20を生成する。例えば、Bunnelle,WH,ら、Synthesis,1997,4:439−442;および、Norton,DGら、J Org Chem 1954,19:1054−66を参照されたい。
【0058】
式I、式IIなどの化合物およびこれらの合成前駆体を合成するためのさらなる方法は、本明細書のスキームに明示的に示していない経路内のものを含み、当業者である化学者の通常の技術の範囲内である。適用可能な化合物の合成に有用な合成化学変換および保護基方法論(保護および脱保護)は、当該技術分野で周知であり、例えば、Larock R,Comprehensive Organic Transformations,VCH Publishers(1989);Greene TWら、Protective Groups in Organic Synthesis,3rd Ed.,John Wiley and Sons(1999);Fieser Lら、Fieser and Fieser’s Reagents for Organic Synthesis,John Wiley and Sons(1994);およびPaquette L,ed.,Encyclopedia of Reagents for Organic Synthesis,John Wiley and Sons(1995)、ならびにその後の版に記載されているものが挙げられる。
【0059】
本発明により想定された置換基および変数の組合せは、安定化合物の形成をもたらすもののみである。
(組成物)
【0060】
本発明は、有効量の、式I、式IIなど(例えば、本明細書の式のいずれかを含む)の化合物、または前記化合物の医薬的に許容される塩、溶媒和物、または水和物と;医薬的に許容される担体とを含むピロゲンフリー組成物についても提供する。一実施形態において、組成物はピロゲンフリーである。別の実施形態において、組成物は、医薬用途(「医薬組成物」)用に調合され、ここで、担体は医薬的に許容される担体である。担体(単数または複数)は、製剤の他の成分と相溶性があるという意味において「許容される」ものでなければならず、医薬的に許容される担体の場合、薬剤に通常使用される量でその受容体に無害なものでなければならない。
【0061】
本発明の医薬組成物に用いることができる医薬的に許容される担体、アジュバント、および賦形剤としては、限定的ではないが、イオン交換体、アルミナ、ステアリン酸アルミニウム、レシチン、血清タンパク質(例えば、ヒト血清アルブミン)、緩衝物質(例えば、リン酸塩)、グリシン、ソルビン酸、ソルビン酸カリウム、植物性飽和脂肪酸の部分グリセリド混合物、水、塩、または電解質(例えば、硫酸プロタミン)、リン酸水素二ナトリウム、リン酸水素カリウム、塩化ナトリウム、亜鉛塩、コロイド状シリカ、三ケイ酸マグネシウム、ポリビニルピロリドン、セルロースベースの物質、ポリエチレングリコール、カルボキシメチルセルロースナトリウム、ポリアクリレート、ワックス、ポリエチレン−ポリオキシプロピレン−ブロックポリマー、ポリエチレングリコール、およびラノリンが挙げられる。
【0062】
必要に応じて、医薬組成物における本発明の化合物の溶解度およびバイオアベイラビリティは、当該技術分野で周知の方法により高めることができる。1つの方法としては、製剤に脂質賦形剤を用いることが挙げられる。「Oral Lipid−Based Formulations:Enhancing the Bioavailability of Poorly Water−Soluble Drugs(Drugs and the Pharmaceutical Sciences)」、David J.Hauss,ed.Informa Healthcare,2007;および「Role of Lipid Excipients in Modifying Oral and Parenteral Drug Delivery:Basic Principles and Biological Examples」、Kishor M.Wasan,ed.Wiley−Interscience,2006を参照されたい。
【0063】
バイオアベイラビリティを高める別の公知の方法としては、LUTROL(商標)およびPLURONIC(商標)(BASF Corporation)などのポロクサマー、または、エチレンオキシドと酸化プロピレンのブロック共重合体と任意に調合された本発明の化合物を非晶形で用いることである。米国特許第7,014,866号;米国特許公報第20060094744号、および同第20060079502号を参照されたい。
【0064】
本発明の医薬組成物としては、経口、直腸、鼻腔、局所(口腔および舌下を含む)、膣内または非経口(皮下、筋肉内、静脈内、および皮内を含む)投与に適するものが挙げられる。ある実施形態において、本明細書の式の化合物は、(例えば、経皮貼り剤またはイオン導入技術を用いて)経皮投与される。他の製剤は、単位投与剤形(例えば、錠剤および徐放カプセル剤)や、リポソームで簡便に提供することができ、薬剤技術分野で周知のあらゆる方法で調製することができる。例えば、Remington’s Pharmaceutical Sciences,Mack Publishing Company,Philadelphia,PA(17th ed.1985)を参照されたい。
【0065】
かかる調製方法としては、1つ以上の副成分を構成する担体などの、投与される成分分子と会合させる工程が挙げられる。一般に、組成物は、有効成分を液体担体、リポソームまたは微細な固体担体、もしくは両方と均一かつ密接に会合させることで調製され、必要に応じて、生成物を成形する。
【0066】
ある実施形態において、化合物は経口投与される。経口投与に適当な本発明の組成物は、各々が所定量の有効成分を含むカプセル剤、サッシェ剤、または錠剤などの個別単位として;散剤または顆粒剤として;水性液体または非水液体中の溶液または懸濁液として;水中油乳濁液として;油中水乳濁液として;リポソーム中に包まれたものとして;あるいは、ボーラス剤等として提供することができる。軟質ゼラチンカプセル剤は、かかる懸濁液を含めるのに有用な場合があり、これは、化合物の吸収率を有利に増加させ得る。
【0067】
経口用途用の錠剤の場合、一般的に使用される担体としては、ラクトースおよびコーンスターチが挙げられる。ステアリン酸マグネシウムなどの滑沢剤も一般に添加される。カプセル形態の経口投与において、有用な希釈剤としては、ラクトースおよび乾燥コーンスターチが挙げられる。水性懸濁液を経口投与する場合、有効成分は乳化剤および懸濁剤と組合せられる。所望の場合、特定の甘味料および/または香料および/または着色剤を添加してもよい。
【0068】
経口投与に適当な組成物としては、芳香化基剤、通常、スクロースおよびアカシアまたはトラガカント中に成分を含むトローチ剤;不活性基剤、例えば、ゼラチンおよびグリセリン、またはスクロースおよびアカシア中に有効成分を含む香錠が挙げられる。
【0069】
非経口投与に適当な組成物としては、抗酸化剤、緩衝剤、静菌剤、および、製剤を目的の受容体の血液と等張させる溶質を含んでいてもよい水性および非水性の無菌注射液;懸濁剤および増粘剤を含んでいてもよい水性および非水性の無菌懸濁液が挙げられる。製剤は、単回投与用または複数投与用入れ物、例えば、密封アンプルおよびバイアルで提供してもよく、無菌液体担体、例えば、注射用水を使用直前に添加することのみを必要とするフリーズドライ(冷凍乾燥)状態で保存してもよい。即時注射液および懸濁液は、無菌散剤、顆粒剤、および錠剤から調製してもよい。
【0070】
かかる注射液は、例えば、滅菌注射用水性または油性懸濁剤の形態であってもよい。この懸濁液は、当該技術分野で公知の技術に従い、適切な分散剤または湿潤剤(例えば、ツイーン80など)および懸濁剤を用いて調合してもよい。滅菌注射用製剤はまた、例えば、1,3−ブタンジオール中の溶液として、非毒性の非経口的に許容される希釈剤または溶媒中の滅菌注射溶液または懸濁液であってもよい。使用できる許容可能な賦形剤や溶媒の中には、マンニトール、水、リンガー溶液、および等張性塩化ナトリウム溶液がある。さらに、滅菌固定油は、溶媒または懸濁媒体として従来使用されている。この目的では、合成のモノグリセリドまたはジグリセリドを含む任意の無刺激固定油を用いてもよい。オレイン酸およびそのグリセリド誘導体などの脂肪酸は、特に、そのポリオキシエチル化形態ではオリーブ油またはヒマシ油などの天然の医薬的に許容される油であるため、注射用製剤において有用である。これらの油状溶液または懸濁液は、長鎖アルコール希釈剤または分散剤を含有してもよい。
【0071】
本発明の医薬組成物は、直腸内投与用に坐剤形態で投与してもよい。これらの組成物は、本発明の化合物と、室温で固体であるが直腸温度では液体であるため、直腸内で溶解して有効成分を放出する適当な非刺激性賦形剤とを混合することにより調製することができる。かかる物質としては、限定的ではないが、ココアバター、蜜蝋、およびポリエチレングリコールが挙げられる。
【0072】
本発明の医薬組成物は、鼻エアロゾルまたは鼻吸入により投与してもよい。かかる組成物は、医薬製剤分野で公知の技術により調製され、なおかつ、ベンジルアルコールまたは他の適切な保存剤、バイオアベイラビリティを高めるための吸収促進剤、フルオロカーボン、および/または当該技術分野で公知の他の可溶化剤または分散剤を用いて、塩水中の溶液として調製することができる。例えば、Alexza Molecular Delivery Corporationに譲渡されているRabinowitz JD and Zaffaroni AC、米国特許第6,803,031号を参照されたい。
【0073】
本発明の医薬組成物の局所投与は、所望の治療が局所塗布により容易に近づける領域または器官に関与する場合に特に有用である。皮膚への局所塗布では、医薬組成物は、担体に懸濁または溶解させた有効成分を含有する適当な軟膏で調合すべきである。本発明の化合物の局所投与用の担体としては、限定的ではないが、鉱油、液化石油、白色石油、プロピレングリコール、ポリオキシエチレンポリオキシプロピレン化合物、乳化ワックス、および水が挙げられる。あるいは、医薬組成物は、担体に懸濁または溶解させた活性化合物を含有する適当なローションまたはクリームと調合することができる。適当な担体としては、限定的ではないが、鉱油、ソルビタンモノステアレート、ポリソルベート60、セチルエステルワックス、セテアリールアルコール、2−オクチルドデカノール、ベンジルアルコール、および水が挙げられる。本発明の医薬組成物は、直腸坐剤製剤または適当な浣腸製剤により、腸管低部に局所塗布してもよい。局所経皮貼り剤およびイオン導入投与も、本発明に含まれる。例えば、式Iの化合物は、例えば、PCT国際公開公報第2005107812号に記載のトルテロジンの当該技術分野で周知の調製方法と同様の方法で局所または経皮用組成物に調合してもよい。
【0074】
対象治療の適用は、対象部位に投与するように局所であってもよい。注射や、カテーテル、トロカール、投射物、プルロニックゲル、ステント、薬物徐放性ポリマー、または内部アクセスをもたらす他のデバイスを用いて対象部位に対象組成物をもたらす種々の技術を用いることができる。例えば、式I、式IIなどの化合物は、例えば、国際公開第2007011131号;同第2007022255号;同第2006021425号;同第2005105036号;同第2004105735号;同第2001034139号;および同第2000027364号に記載のトルテロジンの当該技術分野で周知の調製方法と同様の方法で徐放用に調合してもよい。
【0075】
従って、さらに別の実施形態によれば、本発明の化合物は、プロテーゼ、人工弁、代用血管、ステント、またはカテーテルなどの移植医療機器を被覆するための組成物に組み込むことができる。適当な被覆および被覆された移植機器の一般的調製は、当該技術分野で周知であり、米国特許第6,099,562号;同第5,886,026号;および同第5,304,121号に例示されている。被覆は、通常、ヒドロゲルポリマー、ポリメチルジシロキサン、ポリカプロラクトン、ポリエチレングリコール、ポリ乳酸、エチレン酢酸ビニル、およびその混合物などの生体適合性ポリマー材料である。被覆は、必要に応じて、フルオロシリコーン、ポリサッカリド、ポリエチレングリコール、リン脂質、またはその組合せの適当な上塗りによりさらに被覆し、組成物に徐放特性を付与してもよい。侵襲機器に対する被覆は、本明細書で使用される用語としての医薬的に許容される担体、アジュバント、または賦形剤の定義内に含まれるものである。
【0076】
別の実施形態によれば、本発明は、前記機器を上述の被覆組成物に接触させる工程を含む移植医療機器の被覆方法を提供する。当業者であれば、哺乳類への移植前に機器の被覆を行うことはすぐに分かるであろう。
【0077】
別の実施形態によれば、本発明は、前記薬物放出機器を本発明の化合物または組成物に接触させる工程を含む、移植型の薬物放出機器の含浸方法を提供する。移植型の薬物放出機器としては、限定的ではないが、生分解性のポリマーカプセル剤または薬剤(bullets)、非分解性、拡散性のポリマーカプセル剤、および生分解性のポリマーカシェ剤が挙げられる。
【0078】
別の実施形態によれば、本発明は、治療効果がある化合物または本発明の前記化合物を含む組成物で被覆した移植医療機器を提供する。
【0079】
別の実施形態によれば、本発明は、前記機器から放出され、かつ、治療効果がある化合物または本発明の化合物を含む組成物に含浸したもしくはを含む移植型の薬物放出機器を提供する。
【0080】
器官または組織が手術中に接触可能であるか、または、患者から除去される場合、かかる器官または組織を本発明の組成物を含む培地中に浴してもよく、本発明の組成物を器官上に塗布してもよく、あるいは、本発明の組成物を任意の他の簡便な方法で塗布してもよい。
【0081】
別の実施形態において、本発明の組成物は、第2の治療薬をさらに含む。第2の治療薬としては、トルテロジンと同じ作用機序をもつ化合物と一緒に投与した際に、有利な特性を有することが知られている、あるいは、有利な特性を示す任意の化合物または治療薬から選択することができる。かかる薬剤としては、トルテロジンと併用して有用であると示されるものが挙げられ、限定的ではないが、αアドレナリン受容体遮断薬(例えば、PCT国際特許出願国際公開第2001/021167号および同第2007/010509号に記載のもの);ビシファジン化合物(例えば、PCT国際特許出願国際公開第2006/102029号に記載のもの);スタチン(例えば、PCT国際特許出願国際公開第2006/008437号に記載のもの);デヒドロエピアンドロステロン(DHEA)同族体(例えば、PCT国際特許出願国際公開第2006/007312号に記載のもの;α2δサブユニットカルシウムチャネル調節因子(例えば、GABA類似体や、PCT国際特許出願国際公開第2004/084879号に記載の他のもの);選択的セロトニン再取り込み阻害薬(SSRI)または選択的ノルエピネフリン再取り込み阻害薬(例えば、フルオキセチン、パロキセチン、およびPCT国際特許出願国際公開第2004/019892および同第2001/062236号に記載の他のもの);アンドロゲン、エストロゲン、またはエストロゲン作用薬(例えば、PCT国際特許出願国際公開第2004/043429号、同第2003/039553号、および同第2003/039523号に記載のもの);EGF受容体拮抗薬(例えば、PCT国際特許出願国際公開第2003/039524号に記載のもの);5HT1a受容体修飾因子(例えば、PCT国際特許出願国際公開第2003/026564号に記載のもの);チエノピランカルボキサミド誘導体(例えば、PCT国際特許出願国際公開第2001/009140号に記載のもの);5a還元酵素阻害薬(例えば、PCT国際特許出願国際公開第2001/021167号に記載のもの)、ならびに、米国特許公報第2006−205682号および同第2006−160887号、PCT国際特許出願国際公開第2006/079625号、同第2006/015970号、同第2005/092342号、同第2005/092341号、および同第2001/062236号、日本国特許公報第2003−055261号および同第2005−015394号に記載の第2の薬剤が挙げられる。
【0082】
一実施形態において、第2の治療薬は、不安定膀胱もしくは過活動膀胱、失禁、感染、下部尿路疾患、記憶障害もしくは認知障害、心不全、肺炎(肺胞性肺炎を含む)、良性前立腺肥大症、前立腺肥大症、呼吸器疾患、喘息、女性性機能障害、または膀胱炎から選択される疾患または疾病の治療または予防に有用な薬剤である。
【0083】
一実施形態において、第2の治療薬はタムスロシンである。
【0084】
別の実施形態において、本発明は、本発明の化合物と、上述の第2の治療薬のいずれかの1つ以上との別々の剤形を提供し、ここで、化合物と第2の治療薬とは互いに会合している。本明細書で使用する「互いに会合している」という用語は、別々の剤形を一緒に販売し、投与すること(連続的または同時に互いに24時間未満内)を意図することが容易に明らかとなるように、別々の剤形が一緒に包装されている、あるいは、互いに取り付けられていることを意味する。
【0085】
本発明の医薬組成物では、本発明の化合物は、有効量で存在する。本明細書で使用する「有効量」という用語は、適切な投与計画で投与する場合、治療している疾患の重篤度、期間、または進行を軽減または改善する、治療している疾患の促進を予防する、治療している疾患を退行させる、あるいは、別の治療の予防または治療効果(単数または複数)を高めるまたは向上させるのに十分な量のことを言う。
【0086】
動物とヒトに対する用量の相互関係(体表面1平方メートル当たりのミリグラムに基づく)は、Freireichら、(1966)Cancer Chemother.Rep 50:219に記載されている。体表面積は、患者の身長および体重からおおよそ決定することができる。例えば、Scientific Tables,Geigy Pharmaceuticals,Ardsley,N.Y.,1970,537を参照されたい。
【0087】
一実施形態において、本発明の化合物の有効量は、成人患者に対して、0.1mg/日〜50mg/日の範囲であり得る。別の実施形態において、本発明の化合物の有効量は、成人患者に対して、1mg/日〜10mg/日の範囲であり得る。さらに別の実施形態において、有効量は、成人患者に対して、2〜6mg/日の範囲であり得る。さらに別の実施形態において、有効量は、成人患者に対して、約4mg/日である。
【0088】
有効量は、当業者により認識されるように、治療する疾患、疾患の重篤度、投与経路、患者の大きさ、性別、年齢、および一般的健康状態、賦形剤の使用、他の治療処置に伴う、例えば、他の薬剤の使用との併用の可能性、および治療医の判断に応じて変化させてもよい。例えば、有効量を選択するためのガイダンスは、トルテロジンの処方情報を参照することで決めることができる。
【0089】
第2の治療薬を含む医薬組成物において、第2の治療薬の有効量は、その薬剤のみを用いる単剤療法レジメンで通常使用される投与量の約20%〜100%の間である。有効量は、標準の単剤治療量の約70%〜100%の間であるのが好ましい。これら第2の治療薬の標準の単剤治療量は、当該技術分野で周知である。例えば、Wellsら、eds.,Pharmacotherapy Handbook,2nd Edition,Appleton and Lange,Stamford,Conn.(2000);PDR Pharmacopoeia,Tarascon Pocket Pharmacopoeia2000,Deluxe Edition,Tarascon Publishing,Loma Linda,Calif.(2000)を参照されたい。これら参照文献の各々は、参照により本明細書に完全に引用したものとする。
【0090】
上記で参照した第2の治療薬のいくつかは、本発明の化合物と相乗的に作用すると予想される。これが起こる場合には、第2の治療薬および/または本発明の化合物の有効薬量を単剤療法で必要な量より減らすことができる。これは、第2の治療薬または本発明の化合物いずれかの毒性の副作用の軽減、効果の相乗的な改善、投与または使用し易さの改善、および/または、化合物の調製または製剤の総費用の削減という利点を有する。
(治療方法)
【0091】
別の実施形態において、本発明は、本明細書に記載の式I、式IIなどの1つ以上の化合物を細胞に接触させる工程を含む、細胞におけるコリン作動性ムスカリン受容体の活性を調節する方法を提供する。
【0092】
別の実施形態によれば、本発明は、本発明の化合物または組成物の有効量をそれを必要とする対象に投与する工程を含む、トルテロジンにより有利に治療される疾患に罹患しているまたは罹患しやすい対象の治療方法を提供する。かかる疾患は、当該技術分野では周知であり、限定的ではないが、以下の特許および/または公開公報:米国特許公報第2006−205682号および同第2006−160887号、PCT国際特許出願国際公開第2006/079625号、同第2006/015970号、同第2005/092342号、同第2005/092341号、同第2001/062236号、同第2003/039553号、同第2003/039524号、および同第2003/026564号、ならびに日本国特許公報第2003−055261号および同第2005−015394号に開示されている。
【0093】
別の実施形態において、本発明の方法は、過活動膀胱、失禁、下部尿路感染症、喘息、良性前立腺肥大症、および記憶および認知障害から選択される疾患または疾病に罹患しているもしくは罹患しやすい対象の治療に用いる。ある実施形態において、本発明の方法は、過活動膀胱および失禁から選択される疾患または疾病に罹患しているもしくは罹患しやすい対象の治療に用いる。本明細書に記載の方法としては、対象が特定の治療の必要があると見なされているものも含む。かかる治療が必要である対象の同定は、対象または医療専門家の判断であってもよく、主観的(例えば、意見)または客観的(例えば、試験または診断法により測定可能)であってもよい。
【0094】
別の実施形態において、上記の治療方法のいずれかは、患者に1つ以上の第2の治療薬を同時投与する工程をさらに含む。第2の治療薬の選択は、トルテロジンとの同時投与に有用であると知られている任意の第2の治療薬から行ってよい。本発明の化合物と併用可能してもよいかかる薬剤、疾患、および疾病の例としては、(1)αアドレナリン受容体遮断薬(例えば、タムスロシン、および、PCT国際特許出願国際公開第2001/021167号および同第2007/010509号に記載のもの);ビシファジン化合物(PCT国際特許出願国際公開第2006/102029号に記載のもの);α2δサブユニットカルシウムチャネル調節因子(GABA類似体、および、PCT国際特許出願国際公開第2004/084879号に記載の他のもの);選択的セロトニン再取り込み阻害薬(SSRI)または選択的ノルエピネフリン再取り込み阻害薬(フルオキセチン、パロキセチン、および、PCT国際特許出願国際公開第2004/019892号および同第2001/062236号に記載の他のもの);5HT1a受容体修飾因子(PCT国際特許出願国際公開第2003/026564号に記載のもの);チエノピランカルボキサミド誘導体(PCT国際特許出願国際公開第2001/009140号に記載のもの);または、5a還元酵素阻害薬(PCT国際特許出願国際公開第2001/021167号に記載のもの)、各々は、下部尿路疾患(失禁、不安定膀胱もしくは過活動膀胱、および他の排尿障害を含む)の治療に用いる;(2)デヒドロエピアンドロステロン(DHEA)同族体(炎症の治療に用いるPCT国際特許出願国際公開第2006/007312号に記載のもの);(3)スタチン(呼吸器疾患の治療に用いるPCT国際特許出願国際公開第2006/008437号に記載のもの);(4)アンドロゲン、エストロゲン、またはエストロゲン作用薬(女性性機能障害の治療に用いるPCT国際特許出願国際公開第2004/043429号、同第2003/039553号、および同第2003039523号に記載のもの);(5)EGF受容体拮抗薬(PCT国際特許出願国際公開第2003/039524号に記載のもの);または、チエノピランカルボキサミド誘導体(良性前立腺肥大症または前立腺肥大症の治療に用いるPCT国際特許出願国際公開第2001/009140号に記載のもの);のみならず、米国特許公報第2006−205682号、同第2006−160887号、PCT国際特許出願国際公開第2006/079625号、同第2006/015970号、同第2005/092342号、同第2005/092341号、同第2001/062236号、日本国特許公報2003−055261号、および同第2005−015394号に記載のものが挙げられる。
【0095】
特に、本発明の併用療法としては、式Iの化合物および第2の治療薬を投与することによって以下の疾患:過活動膀胱および失禁の治療が挙げられる。
【0096】
一実施形態によれば、式Iの化合物は、下部尿路疾患を治療するためにタムスロシンと同時投与される。
【0097】
本明細書で使用する「同時投与する」という用語は、単一剤形(例えば、本発明の化合物と、上述の第2の治療薬とを含む本発明の組成物)の一部として、あるいは、別個の複数剤形として、第2の治療薬を本発明の化合物とともに投与することを意味する。あるいは、本発明の化合物の投与前、投与に続いて、または投与後に、追加の薬剤を投与してもよい。かかる併用療法による治療では、本発明の化合物と第2の治療薬(単数または複数)の両方を従来の方法で投与する。本発明の化合物と第2の治療薬との両方を含む本発明の組成物を対象に投与することは、治療期間中に別時で、同一治療薬、任意の他の第2の治療薬、または本発明の任意の化合物を前記対象に個別投与することを妨げるものではない。
【0098】
これらの第2の治療薬の有効量は、当業者に公知であり、投与ガイダンスは、本明細書に引用した特許および特許出願公開のみならず、Wellsら、eds.,Pharmacotherapy Handbook,2nd Edition,Appleton and Lange,Stamford,Conn.(2000);PDR Pharmacopoeia,Tarascon Pocket Pharmacopoeia 2000,Deluxe Edition,Tarascon Publishing,Loma Linda,Calif.(2000)、および他の医療テキストに見ることができる。しかしながら、第2の治療薬の最適な有効量の範囲を判断するのは当業者の十分な範囲内である。
【0099】
本発明の一実施形態において、第2の治療薬を対象に投与する場合、本発明の化合物の有効量は、第2の治療薬を投与しない場合におけるその有効量未満である。別の実施形態において、第2の治療薬の有効量は、本発明の化合物を投与しない場合におけるその有効量未満である。これにより、高用量のいずれの薬剤に伴う望ましくない副作用を最小限にすることができる。他の潜在的利点(限定的ではないが、投与計画の向上、および/または、薬剤費の削減を含む)は、当業者に明らかになるであろう。
【0100】
さらに別の態様において、本発明は、対象の上述の疾患、疾病、または症状を治療または予防するため、単一組成物または別々の剤形として、薬剤の製造において式I、式IIなどの化合物のみを使用、または、1つ以上の上述の第2の治療薬とともに式I、式IIなどの化合物を使用することを提供する。本発明の別の態様は、対象の本明細書に記載の疾患、疾病、または症状を治療または予防に使用する式I、式IIなどの化合物である。
【0101】
他の態様において、本明細書の方法としては、治療投与に対する対象の反応を監視する工程をさらに含むものが挙げられる。かかる監視としては、治療計画のマーカーまたは指標として、対象の組織、体液、試験片、細胞、タンパク質、化学マーカー、遺伝物質などの周期的サンプリングを含んでもよい。他の方法において、対象は、かかる治療に対する適性の関連マーカーまたは指標を評価することによって、かかる治療が必要であるかプレスクリーニングまたは同定される。
(診断方法およびキット)
【0102】
本発明の化合物および組成物は、溶液または生体試料(例、血漿)中のトルテロジン、5−ヒドロキシメチルトルテロジン、またはフェソテロジンの濃度を判断する方法、トルテロジン、5−ヒドロキシメチルトルテロジン、またはフェソテロジンの代謝を調べる方法、および他の分析研究における試薬としても有用である。
【0103】
一実施形態によれば、本発明は、溶液または生体試料中のトルテロジン、5−ヒドロキシメチルトルテロジン、またはフェソテロジンの濃度を判断する方法を提供し、該方法は:
a)公知濃度の式Iの化合物を生体試料の溶液に加える工程と;
b)式Iの化合物からトルテロジン、5−ヒドロキシメチルトルテロジン、またはフェソテロジンを区別する測定装置に溶液または生体試料をかける工程と;
c)式Iの化合物の検出量と、生体試料または溶液に加えられた式Iの化合物の公知濃度とを相関させる測定装置を較正する工程と;
d)前記較正された測定装置で生体試料中のトルテロジン、5−ヒドロキシメチルトルテロジン、またはフェソテロジンの量を測定する工程と;
e)式Iの化合物に対して得られた検出量および濃度間の相関を用いて、試料の溶液中のトルテロジン、5−ヒドロキシメチルトルテロジン、またはフェソテロジンの濃度を判断する工程とを含む。
【0104】
対応する式Iの化合物からトルテロジン、5−ヒドロキシメチルトルテロジン、またはフェソテロジンを区別することができる測定装置としては、同位体存在度においてのみ互いに異なる2つの化合物を区別することができる任意の測定装置が挙げられる。測定装置の例としては、質量スペクトロメータ、NMRスペクトロメータ、またはIRスペクトロメータが挙げられる。
【0105】
別の実施形態において、本発明は、式Iの化合物をある時間、代謝酵素源と接触させる工程と、前記時間の後、式Iの化合物の量と式Iの化合物の代謝産物を比較する工程とを含む、式Iの化合物の代謝安定性を評価する方法を提供する。
【0106】
関連実施形態において、本発明は、式Iの化合物を投与した後の患者内の式Iの化合物の代謝安定性を評価する方法を提供する。本方法は、式Iの化合物を対象に投与した一定期間の後、患者から血清、尿、または糞便試料を採取する工程と;式Iの化合物の量と血清、尿、または糞便試料における式Iの化合物の代謝産物を比較する工程とを含む。
【0107】
本発明は、過活動膀胱および失禁の治療に用いるキットも提供する。これらのキットは、a)式Iの化合物、またはその塩、水和物、もしくは溶媒和物を含む、入れ物に入った医薬組成物と;b)過活動膀胱および失禁を治療するための医薬組成物の使用方法を記載した説明書と、を含む。
【0108】
入れ物は、前記医薬組成物を保持できる任意の容器あるいは他の密閉されたまたは密閉可能な装置であってよい。例としては、ボトル、アンプル、分割型ボトル、またはマルチチャンバ式ホルダボトル(各区分またはチャンバは単回投与の前記組成物を含む)、分割型ホイルパケット(各区分は単回投与の前記組成物を含む)、または単回投与の前記組成物を分注するディスペンサが挙げられる。入れ物は、医薬的に許容される材料、例えば、紙または段ボール箱、ガラスまたはプラスチックボトルあるいは瓶(jar)、ジッパー付バッグ(例えば、異なる入れ物に配置するための「詰め替え」用錠剤を保持)、または治療スケジュールに従ってパックから押し出す個別用量を備えたブリスターパックからなる当該技術分野で周知の任意の従来の形状または型であってよい。用いる入れ物は、関連する正確な剤形に左右される。例えば、従来の段ボール箱は、液体懸濁液を保持するのには一般に使用されないであろう。単一剤形を販売するため単一包装に複数の入れ物を一緒に使用することも可能である。例えば、錠剤は、ボトルに収容し、次いで、ボックス内に収容してもよい。一実施形態において、入れ物はブリスターパックである。
【0109】
キットは、医師、薬剤師、または対象用の情報および/または説明書を収容するタイプの記憶補助をさらに含んでもよい。かかる記憶補助としては、特定された錠剤またはカプセル剤を服用すべきかの投与計画日数に対応する投与量を含有する各チャンバまたは区分上に印字された数字、各チャンバまたは区分上に印字された週の曜日、または同一種類の情報を含むカードが挙げられる。単回投与用ディスペンサでは、記憶補助としては、さらに、分注された日用量の数を示す機械的計数器や、液晶読み出し信号、および/または、例えば、日用量を最後に摂取した日付を読み出し、および/または、次に用量をいつ摂取するのかを気付かせる可聴リマインダ信号を連結させたバッテリー駆動のマイクロチップメモリが挙げられる。かかるキットに有用な他の記憶補助は、カード上に印字されたカレンダのみならず、容易に明らかとなる他の変形例がある。
【0110】
本発明のキットは、単回投与の医薬組成物を投与するまたは測り分ける機器をさらに含んでいてもよい。かかる機器としては、前記組成物が吸入可能組成物である場合は吸入器;前記組成物が注射用組成物である場合は注射器および針;前記組成物が口腔液体組成物である場合は容量マーキング付または無しの注射器、匙、ポンプ、または容器;あるいは、キット内に存在する組成物の投与製剤に適切な任意の他の計測機器または送達機器が挙げられる。
(実施例)
実施例1.種々の中間体11の合成。
【0111】
A.1−(ベンジルオキシ)−2−ブロモ−4−メチルベンゼン(11a、X=CH3)。
中間体11aは、Akiら、J Phys Chem,2002,106(14):3436−3444の一般手順に従って調製した。
スキーム4a。
【化10】
【0112】
1−(ベンジルオキシ)−2−ブロモ−4−メチルベンゼン(11a、X=CH3)。
アセトン(60mL)中の2−ブロモ−4−メチルフェノール32a(5g、26.7mmol)の溶液にK2CO3(11g、80mmol)を加え、続いて、臭化ベンジル(3.8mL、32mmol)を加えた。混合物を4時間環流加熱した後、室温に冷却した。固体を濾過で除去し、濾液を真空下(in vacuo)で濃縮した。カラムクロマトグラフィー(5% EtOAc/ヘプタン)で残渣を精製し、白色の半固体として11aを6.7g(収率90%)得た。
【0113】
B.1−(ベンジルオキシ)−2−ブロモ−4−(メチル−d3)ベンゼン(11b、X=CH3)。
スキーム4b。
【化11】
【0114】
工程1.1−(ベンジルオキシ)−4−ブロモベンゼン(53)。
アセトン(60mL)中の4−ブロモフェノール(52)(5g、29mmol)の溶液にK2CO3(12.4g、90mmol)を加え、続いて、臭化ベンジル(3.8mL、32mmol)を加えた。混合物を4時間環流加熱した後、室温に冷却した。固体を濾過で除去し、濾液を真空下で濃縮した。カラムクロマトグラフィー(5% EtOAc/ヘプタン)で残渣を精製し、白色固体として53を6.9g(収率90%)得た。
【0115】
工程2.1−(ベンジルオキシ)−4−(メチル−d3)ベンゼン(54)。
THF(無水、40mL)中の1−(ベンジルオキシ)−4−ブロモベンゼン(53)(5g、19mmol)の溶液を−78℃に冷却した後、n−BuLi(2.5Mヘキサン中に9.1mL、22.8mmol)を滴下した。反応混合物を30分間攪拌した後、CD3I[ケンブリッジアイソトープ、99原子%D](1.4mL、22.8mmol)を加えた。混合物を1時間攪拌した後、水性NH4Cl(飽和)を加えて急冷した。混合物をEtOAc(100mL)で抽出し、有機層をブラインで洗浄し、Na2SO4で乾燥させ、濾過し、真空下で濃縮し、カラムクロマトグラフィー(5% EtOAc/ヘプタン)で精製し、黄色油として54を3.05g(収率80%)得た。
【0116】
工程3.1−(ベンジルオキシ)−2−ブロモ−4−(メチル−d3)ベンゼン(11b、X=CD3)。
アセトニトリル(50mL)中の54(3.0g、15mmol)の溶液にN−ブロモスクシンイミド(3.2g、18mmol)を加え、続いて、NH4OAc(3.5g、45mmol)を加えた。混合物を3時間攪拌した後、水(30mL)を加え、混合物をEtOAc(150mL)で抽出した。有機層をブラインで洗浄し、Na2SO4で乾燥させ、濾過し、真空下で濃縮し、カラムクロマトグラフィー(5% EtOAc/ヘプタン)で精製し、黄色油として11bを3.6g(収率85%)得た。
【0117】
C.1−(ベンジルオキシ)−4−(ベンジルオキシメチル)−2−ブロモベンゼン(11c、X=CH2OBn)。
中間体11cは、Akiら、J Phys Chem,2002,106(14):3436−3444の一般手順に従って調製した。
スキーム4c。
【化12】
【0118】
工程1.4−(ベンジルオキシ)−3−ブロモ安息香酸ベンジル(45)。
アセトン(200mL)中の3−ブロモ−4−ヒドロキシ安息香酸44(10g、46mmol)の溶液にK2CO3(20g、145mmol)および臭化ベンジル(12mL、101mmol)を加えた。混合物を5時間環流加熱した後、室温に冷却した。固体を濾過で除去し、濾液を真空下で濃縮した。残渣をEtOAc(150mL)中に溶解させ、溶液をブラインで洗浄し、Na2SO4で乾燥させ、濾過し、真空下で濃縮し、シロップ状の残渣を得た。残渣を5% EtOAc/ヘプタンから再結晶化させ、白色固体として45を16.4g(収率90%)得た。
【0119】
工程2.(4−(ベンジルオキシ)−3−ブロモフェニル)メタノール(46a)。
4−(ベンジルオキシ)−3−ブロモ安息香酸ベンジル(45)(15g、37.8mmol)をドライTHF(150mL)中に溶解させ、0℃に冷却した。LiAlH4(2.9g、76mmol)を3分割してゆっくりと加えた。混合物を室温で12時間攪拌した後、H2O(3mL)、15% NaOH(3mL)、およびH2O(7.6mL)を加えて急冷した。沈殿物を濾過で除去し、EtOAcで洗浄した。組み合わせた濾液を濃縮し、無色油を得た。これをカラムクロマトグラフィー(5%〜10% EtOAc/ヘプタン)で精製し、無色油として46aを9.4g(収率84%)得た。
【0120】
工程3.1−(ベンジルオキシ)−4−(ベンジルオキシメチル)−2−ブロモベンゼン(11c、X=CH2OBn)。
アセトン(100mL)中の(4−(ベンジルオキシ)−3−ブロモフェニル)メタノール(46a)(9g、30.7mmol)の溶液にK2CO3(12.7g、92mmol)および臭化ベンジル(4mL、33.8mmol)を加えた。混合物を5時間攪拌した後、室温に冷却した。固体を濾過で除去し、濾液を真空下で残渣に濃縮し、これをカラムクロマトグラフィー(5% EtOAc/ヘプタン)で精製し、無色油として11cを10.6g(収率90%)得た。
【0121】
D.1−(ベンジルオキシ)−4−(ベンジルオキシ(メチル−d2))−2−ブロモベンゼン(11d、X=CH2OBn)。
スキーム4d。
【化13】
【0122】
工程1.(4−(ベンジルオキシ)−3−ブロモフェニル)−1,1−d2−メタノール(46b)。
ドライTHF(150mL)中の4−(ベンジルオキシ)−3−ブロモ安息香酸ベンジル(45)(15g、37.8mmol)の0℃の溶液にLiAlD4(ケンブリッジアイソトープ、99原子%D)(3.1g、75.6mmol)を3分割してゆっくりと加えた。混合物を室温で12時間攪拌した後、H2O(3mL)、15% NaOH(3mL)、およびH2O(7.6mL)を加えて急冷した。沈殿物を濾過し、EtOAcで洗浄した。濾液を真空下で濃縮し、無色油を得た。これをカラムクロマトグラフィー(5%〜10% EtOAc/ヘプタン)で精製し、無色油として46bを9.0g(収率81%)得た。
【0123】
工程2.1−(ベンジルオキシ)−4−(ベンジルオキシ(メチル−d2))−2−ブロモベンゼン(11d、X=CH2OBn)。
アセトン(100mL)中の(4−(ベンジルオキシ)−3−ブロモフェニル)−1,1−d2−メタノール(46b)(9.0g、30.5mmol)の溶液にK2CO3(12.7g、92mmol)および臭化ベンジル(4mL、33.8mmol)を加えた。混合物を5時間攪拌し、室温に冷却した。固体を濾過で除去した。濾液を真空下で濃縮し、カラムクロマトグラフィー(5% EtOAc/ヘプタン)で残渣を精製し、無色油として11dを10.4g(収率88%)得た。
実施例2.(R)−2−(3−(ジ(イソプロピル−d7)アミノ)−1−フェニルプロピル)−4−メチルフェノール(化合物106)の合成。
【0124】
適当に重水素化された試薬を用いて、スキーム1で概説したように化合物106を調製した。
【化14】
【0125】
工程1.(R)−3−((R)−3−(2−(ベンジルオキシ)−5−メチルフェニル)−3−フェニルプロパノイル)−4−フェニルオキサゾリジン−2−オン(12a、X=CH3)。
THF(無水、20mL)中の11a(5g、18mmol、実施例1aを参照されたい)の溶液にMg(648mg、27mmol)を加えた。小片のヨウ素を加え、混合物を80℃で1時間加熱した後、−50℃に冷却した。CuBr−Me2S(3.5g、9mmol)を加えた。混合物を−30℃で1時間攪拌した後、THF(無水、40mL)中の(R)−3−シンナモイル−4−フェニルオキサゾリジン−2−オン(10)(6.3g、21.6mmol、Andersson,PGら、J Org Chem,1998,63(22):8067およびNicolas,Eら、J Org Chem,1993,58(3):766−770に記載のように調製した)の溶液を加えた。混合物を室温にゆっくりと温めた後、さらに6時間攪拌し、水性NH4Cl(飽和)で急冷し、EtOAc(200mL)で抽出した。有機層をブラインで洗浄し、Na2SO4で乾燥させ、濾過し、真空下で濃縮した。カラムクロマトグラフィー(20% EtOAc/ヘプタン)で残渣を精製し、白色固体として12aを7.1g(収率80%)得た。
【0126】
工程2.(R)−3−(2−(ベンジルオキシ)−5−メチルフェニル)−3−フェニルプロパン酸(13a、X=CH3)。
THF(30mL)中の12a(4g、8.1mmol)の溶液に水(10mL)を加えた。混合物を0℃に冷却し、30% H2O2溶液(7.2mL、64mmol)を加え、続いて、LiOH(388mg、16.2mmol)を加えた。混合物を室温で4時間攪拌し、4N HClでpH=2に酸性化した後、EtOAc(50mL×3)で抽出した。有機層をNa2SO4で乾燥させ、濾過し、真空下で濃縮し、カラムクロマトグラフィー(20〜50% EtOAc/ヘプタン)で残渣を精製し、白色固体として13aを2.3g(収率83%)得た。
【0127】
工程3.(R)−3−(2−(ベンジルオキシ)−5−メチルフェニル)−3−フェニルプロパノイルクロリド(14a、X=CH3)。
トルエン(10mL)中の13a(1.1g、2.9mmol)の溶液に塩化チオニル(1.1mL、15mL)を加え、続いて、ドライピリジンを1滴加えた。混合物を1時間環流加熱した。塩化チオニルを真空下で除去した。ドライエーテル(20mL)を加え、得られた固体を濾過で除去した。濾液を真空下で濃縮し、淡黄色固体として粗14aを1.1g得た。
【0128】
工程4.(R)−3−(2−(ベンジルオキシ)−5−メチルフェニル)−N,N−ジ(イソプロピル−d7)−3−フェニルプロパンアミド(15a、X=CH3;R2=R3=CD(CD3)2)。
ドライエーテル(20mL)中の粗14a(1.1g)の溶液にジイソプロピルアミン−d14[CDNアイソトープ、98原子%D](517mg、4.5mmol)を加え、続いて、Et3N(1.25mL、9mmol)を加えた。混合物を4時間攪拌し、2N HCl(15mL)を加えて急冷した後、EtOAc(50mL)で抽出した。有機層を水性NaHCO3(飽和)、ブラインで洗浄し、Na2SO4で乾燥させ、濾過し、真空下で濃縮し、残渣をカラムクロマトグラフィー(30% EtOAc/ヘプタン)で精製し、無色油として15aを1.1g(収率86%)得た。
【0129】
工程5.(R)−3−(2−(ベンジルオキシ)−5−メチルフェニル)−N,N−ジ(イソプロピル−d7)−3−フェニルプロパン−1−アミン(16a、X=CH3;R2=R3=CD(CD3)2)。
THF(無水、15mL)中の15a(1.1g、2.48mmol)の0℃の溶液にLiAlH4(190mg、5.0mmol)を加えた。混合物を室温で14時間攪拌した後、H2O(0.2mL)、15% NaOH溶液(0.2mL)、およびH2O(0.5mL)を加えて急冷した。沈殿物を濾過し、EtOAcで洗浄した。濾液を真空下で濃縮し、無色油として16aを860mg(収率80%)得た。
【0130】
工程6.(R)−2−(3−(ジ(イソプロピル−d7)アミノ)−1−フェニルプロピル)−4−メチルフェノールL−タートレート(そのL−酒石酸塩として化合物106)。
メタノール(20mL)中の16a(850mg、1.97mmol)の溶液に10% Pd/C(50%水で2.1g、1.0mmol)を加えた。混合物を水素下(2atm)で10時間攪拌し、セリットパッドを通して濾過し、濾液を真空下で濃縮した。カラムクロマトグラフィー(5% MeOH/CH2Cl2中の2% Et3N)で残渣を精製し、その遊離塩基として化合物106を570mg(収率85%)得た。遊離塩基(570mg、1.68mmol)を10mLのエタノール(無水)中に溶解させ、続いて、L−酒石酸(277mg、1.85mmol)を加えた。混合物を1時間環流加熱した後、冷蔵庫で10時間保持した。得られた塩を濾過し、冷却エタノールで洗浄し、L−酒石酸塩として化合物106を820mg得た。1H−NMR(300MHz、DMSO−d6):δ 2.17(s、3H)、2.28〜2.33(m、2H)、2.67〜2.75(m、2H)、3.96(s、2H)、4.30(t、J=7.8、1H)、6.67(d、J=7.9、1H)、6.80(dd、J1=8.2、J2=1.8、1H)、7.02(d、J=2.0、1H)、7.13〜7.19(m、1H)、7.25〜7.33(m、4H)。13C−NMR(75MHz、DMSO−d6):δ 21.02、41.50、45.67、72.14、115.73、126.66、128.00、128.12、128.50、128.58、128.87、130.30、144.74、152.98、174.69。HPLC(方法:Waters Atlantis T3 2.1×50mm 3μm C18−RPカラム−傾斜法5〜95% ACN+0.1%ギ酸で14分(1.0mL/分)、95% ACNで4分保持した;波長:254nm):保持時間:4.88分;99+%純度。キラルHPLC(方法:250mm×4.6mmキラルODカラム−定組成法95%ヘキサン/5%イソプロパノールで40分間(0.50mL/分);波長:210nm):保持時間:8.84分(主鏡像異性体);11.59分(副鏡像異性体);99.4% ee純度。MS(M+H):340.2。元素分析(C26H23D14NO7):計算値:C=63.79、H=7.62、N=2.86。実測値:C=63.79、H=7.55、N=2.86。
実施例3.(R)−2−(3−(ジイソプロピル)アミノ)−1−フェニルプロピル)−4−(メチル−d3)フェノール(化合物112)の合成。
【0131】
適当に重水素化された試薬を用いて、スキーム1で概説したように化合物112を調製した。
【化15】
【0132】
工程1.(R)−3−((R)−3−(2−(ベンジルオキシ)−5−(メチル−d3)フェニル)−3−フェニルプロパノイル)−4−フェニルオキサゾリジン−2−オン(12b、X=CD3)。
THF(無水、20mL)中の1−((2−ブロモ−4−(メチル−d3)−フェノキシ)メチル)ベンゼン(11b)(3.5g、12.5mmol、実施例1bを参照されたい)の溶液にMg(450mg、18.8mmol)を加えた。小片のヨウ素を加え、混合物を80℃で1時間加熱した後、−50℃に冷却した。CuBr−Me2S(1.3g、6.3mmol)を加えた。混合物を−30℃で1時間攪拌した後、THF(無水、40mL)中の(R)−3−シンナモイル−4−フェニルオキサゾリジン−2−オン(10)(5.5g、18.75mmol、実施例2、工程1を参照されたい)の溶液を加えた。混合物を室温にゆっくりと温め、さらに6時間攪拌し、水性NH4Cl(飽和)を加えて急冷し、EtOAc(200mL)で抽出した。有機層をブラインで洗浄し、Na2SO4で乾燥させ、濾過し、濾液を真空下で濃縮した。カラムクロマトグラフィー(20% EtOAc/ヘプタン)で残渣を精製し、白色固体として12bを5.2g(収率83%)得た。
【0133】
工程2.(R)−3−(2−(ベンジルオキシ)−5−(メチル−d3)−フェニル)−3−フェニルプロパン酸(13b、X=CD3)。
THF(30mL)中の12b(4g、8.1mmol)の溶液に水(10mL)を加えた。混合物を0℃に冷却し、30% H2O2溶液(7.2mL、64mmol)を加え、続いて、LiOH(388mg、16.2mmol)を加えた。混合物を室温で4時間攪拌し、4N HClでpH=2に酸性化し、EtOAc(50mL×3)で抽出した。有機抽出物を組み合わせ、Na2SO4で乾燥させ、濾過し、濾液を真空下で濃縮した。カラムクロマトグラフィー(20〜50% EtOAc/ヘプタン)で残渣を精製し、白色固体として13bを2.3g(収率83%)得た。
【0134】
工程3.(R)−3−(2−(ベンジルオキシ)−5−(メチル−d3)−フェニル)−3−フェニルプロパノイルクロリド(14b、X=CD3)。
トルエン(10mL)中の13b(1.2g、3.0mmol)に塩化チオニル(1.1mL、15mL)を加え、続いて、ドライピリジンを1滴加えた。混合物を1時間環流加熱した。過剰の塩化チオニルを真空下で除去した。ドライエーテル(20mL)を加え、得られた固体を濾過で除去した。濾液を真空下で濃縮し、淡黄色固体として粗14bを1.1g得た。
【0135】
工程4.(R)−3−(2−(ベンジルオキシ)−5−(メチル−d3)−フェニル)−N,N−ジイソプロピル−3−フェニルプロパンアミド(15b、X=CD3、R2=R3=CH(CH3)2)。
ドライエーテル(20mL)中の粗14b(1.1g)の溶液にジイソプロピルアミン(2.1mL、15mmol)を加えた。反応混合物を4時間攪拌し、2N HCl(15mL)を加えて急冷し、EtOAc(50mL)で抽出した。有機層を水性NaHCO3(飽和)、ブラインで洗浄し、Na2SO4で乾燥させ、濾過し、濾液を真空下で濃縮した。カラムクロマトグラフィー(30% EtOAc/ヘプタン)で残渣を精製し、無色油として15bを1.1g(収率89%)得た。
【0136】
工程5.(R)−3−(2−(ベンジルオキシ)−5−(メチル−d3)−フェニル)−N,N−ジイソプロピル−3−フェニルプロパン−1−アミン(16b、X=CD3、R2=R3=CH(CH3)2)。
THF(無水、15mL)中の15b(1.1g、2.6mmol)の0℃の溶液にLiAlH4(210mg、5.2mmol)を加えた。混合物を室温で14時間攪拌した後、H2O(0.21mL)、15% NaOH溶液(0.21mL)、およびH2O(0.52mL)を加えて急冷した。沈殿物を濾過し、EtOAcで洗浄した。濾液を濃縮し、無色油として16bを880mg(収率80%)得た。
【0137】
工程6.2−((R)−3−(ジイソプロピルアミノ)−1−フェニルプロピル)−4−メチルフェノールL−タートレート(そのL−酒石酸塩として化合物112)。
メタノール(20mL)中の16b(870mg、2.08mmol)の溶液に10% Pd/C(50%水で2.1g、1.0mmol)を加えた。混合物を水素下(2atm)で10時間攪拌した後、セリットパッドを通して濾過し、真空下で濃縮した。カラムクロマトグラフィー(5% MeOH/CH2Cl2中の2% Et3N)で残渣を精製し、その遊離塩基として化合物112を560mg(収率81%)得た。遊離塩基(560mg、1.7mmol)を10mLのエタノール(無水)中に溶解させ、続いて、L−酒石酸(280mg、1.87mmol)を加えた。混合物を1時間環流加熱した後、冷蔵庫で10時間保持した。得られた塩を濾過し、冷却エタノールで洗浄し、L−酒石酸塩として化合物112を800mg得た。1H−NMR(300MHz、DMSO−d6):δ 1.08(d、J=6.6、12H)、2.15〜2.17(m、0.6H)、2.26〜2.32(m、2H)、2.65〜2.73(m、2H)、3.37〜3.45(m、2H)、3.98(s、2H)、4.30(t、J=7.8、1H)、6.66(d、J=8.0、1H)、6.80(dd、J1=8.2、J2=2.2、1H))、7.01(d、J=2.1、1H)、7.13〜7.19(m、1H)、7.24〜7.33(m、4H)。13C−NMR(75MHz、DMSO−d6):δ 18.91、33.71、41.49、45.60、52.73、72.18、115.72、126.63、127.88、128.09、128.50、128.58、128.86、130.37、144.82、152.98、174.64。HPLC(方法:Waters Atlantis T3 2.1×50mm 3μm C18−RPカラム−傾斜法5−95% ACN+0.1%ギ酸で14分(1.0mL/分)、95% ACNで4分保持した;波長:254nm):保持時間:4.87分;99+%純度。キラルHPLC(方法:250mm×4.6mmキラルODカラム−定組成法95%ヘキサン/5%イソプロパノールで40分間(0.50mL/分);波長:210nm):保持時間:8.88分(主鏡像異性体);11.62分(副鏡像異性体);99.8% ee純度。MS(M+H):329.1。元素分析(C26H34D3NO7):計算値:C=62.25、H=7.79、N=2.93。実測値:C=65.11、H=7.76、N=2.91。
実施例4.(R)−2−(3−(ジ(イソプロピル−d7))アミノ)−1−フェニルプロピル)−4−(メチル−d3)フェノール(化合物100)の合成。
【0138】
適当に重水素化された試薬を用いて、スキーム1で概説したように化合物100を調製した。
【化16】
【0139】
工程1.(R)−3−(2−(ベンジルオキシ)−5−(メチル−d3)−フェニル)−N,N−ジ(イソプロピル−d7)−3−フェニルプロパンアミド(15e、X=CD3、R2=R3=CD(CD3)2)。
ドライエーテル(20mL)中の粗14b(1.1g)(実施例3、工程3を参照)の溶液にジイソプロピルアミン−d14[CDNアイソトープ、99原子%D](517mg、4.5mmol)を加え、続いて、Et3N(1.25mL、9mmol)を加えた。混合物を4時間攪拌し、2N HCl(15mL)を加えて急冷した後、EtOAc(50mL)で抽出した。有機層を水性NaHCO3(飽和)、ブラインで洗浄し、Na2SO4で乾燥させ、濾過し、真空下で濃縮した。カラムクロマトグラフィー(30% EtOAc/ヘプタン)で残渣を精製し、無色油として15eを1.06g(収率83%)得た。
【0140】
工程2.(R)−3−(2−(ベンジルオキシ)−5−(メチル−d3)−フェニル)−N,N−ジ(イソプロピル−d7)−3−フェニルプロパン−1−アミン(16e、X=CD3、R2=R3=CD(CD3)2)。
15e(1.0g、2.47mmol)の溶液をTHF(無水、15mL)中に溶解させ、0℃に冷却し、続いて、LiAlH4(190mg、5.0mmol)を加えた。混合物を室温で14時間攪拌した後、H2O(0.2mL)、15% NaOH溶液(0.2mL)、およびH2O(0.5mL)を加えて急冷した。沈殿物を濾過し、EtOAcで洗浄した。濾液を真空下で濃縮し、無色油として16eを850mg(収率79%)得た。
【0141】
工程3.(R)−2−(3−(ジ(イソプロピル−d7))アミノ)−1−フェニルプロピル)−4−(メチル−d3)フェノール(化合物100)。
メタノール(20mL)中の16e(850mg、1.97mmol)の溶液に10% Pd/C(50%水で2.1g、1.0mmol)を加えた。混合物を水素下(2atm)で10時間攪拌した後、セリットパッドを通して濾過した。濾液を真空下で濃縮し、カラムクロマトグラフィー(5% MeOH/CH2Cl2中の2% Et3N)で残渣を精製し、遊離塩基として化合物100を550mg(収率80%)得た。遊離塩基(550mg、1.61mmol)を10mLのエタノール(無水)中に溶解させ、続いて、L−酒石酸(266mg、1.77mmol)を加えた。混合物を1時間環流加熱した後、冷蔵庫で10時間保持した。得られた塩を濾過し、冷却エタノールで洗浄し、L−酒石酸塩として化合物100を790mg得た。1H−NMR(300MHz、DMSO−d6):δ 1.04〜1.07(m、0.7H)、2.16〜2.18(m、0.7H)、2.26〜2.28(m、2H)、2.63〜2.75(m、2H)、3.43(微量)、3.95(s、2H)、4.30(t、J=7.8、1H)、6.67(d、J=7.9、1H)、6.81(dd、J1=8.0、J2=2.0、1H))、7.02(d、J=1.8、1H)、7.14〜7.19(m、1H)、7.25〜7.33(m、4H)。13C−NMR(75MHz、DMSO−d6):δ 41.48、45.60、72.02、115.72、126.65、128.00、128.11、128.50,128.58、128.87、130.31、144.78、152.97、174.61。HPLC(方法:Waters Atlantis T3 2.1×50mm 3μm C18−RPカラム−傾斜法5−95% ACN+0.1%ギ酸で14分(1.0mL/分)、95% ACNで4分保持した;波長:254nm):保持時間:4.79分;99+%純度。キラルHPLC(方法:250mm×4.6mmキラルODカラム−定組成法95%ヘキサン/5%イソプロパノールで40分間(0.50mL/分);波長:210nm):保持時間:8.87分(主鏡像異性体);11.61分(副鏡像異性体);99.6% ee純度。MS(M+H):343.2。元素分析(C26H20DI7NO7):計算値:C=63.40、H=7.57、N=2.84。実測値:C=63.54、H=7.73、N=2.77。
実施例5.(R)−2−(3−(ジ(イソプロピル−d7))アミノ)−1−フェニルプロピル)−4−(ヒドロキシメチル)フェノール(化合物109)の合成。
【0142】
適当に重水素化された試薬を用いて、スキーム1で概説したように化合物109を調製した。
【化17】
【0143】
工程1.(R)−3−((R)−3−(2−(ベンジルオキシ)−5−(ベンジルオキシメチル)フェニル)−3−フェニルプロパノイル)−4−フェニルオキサゾリジン−2−オン(12c、X=CH2OBn)。
THF(無水、20mL)中の11c(5g、13mmol、実施例1cを参照)の溶液にMg(480mg、20mmol)を加えた。小片のヨウ素を加え、混合物を80℃で1時間加熱した後、−50℃に冷却した。CuBr−Me2S(1.4g、7mmol)を加えた。混合物を−30℃で1時間攪拌した後、THF(無水、40mL)中の(R)−3−シンナモイル−4−フェニルオキサゾリジン−2−オン(10)(5.5g、19mmol、実施例2、工程1を参照されたい)の溶液を加えた。混合物を室温にゆっくりと温め、さらに6時間攪拌した後、水性NH4Cl(飽和)を加えて急冷し、EtOAc(200mL)で抽出した。有機層をブラインで洗浄し、Na2SO4で乾燥させ、濾過し、真空下で濃縮した。カラムクロマトグラフィー(20% EtOAc/ヘプタン)で残渣を精製し、白色固体として12cを6.0g(収率77%)得た。
【0144】
工程2.(R)−3−(2−(ベンジルオキシ)−5−(ベンジルオキシメチル)フェニル)−3−フェニルプロパン酸(13c、X=CH2OBn)。
THF(30mL)中の12c(4.8g、8.1mmol)の溶液に水(10mL)を加えた。混合物を0℃に冷却し、30% H2O2溶液(7.2mL、64mmol)を加え、続いて、LiOH(388mg、16.2mmol)を加えた。混合物を室温で4時間攪拌し、4N HClでpH=2に酸性化し、EtOAc(50mL×3)で抽出した。有機抽出物を組み合わせ、Na2SO4で乾燥させ、濾過し、濾液を真空下で濃縮した。カラムクロマトグラフィー(20〜50% EtOAc/ヘプタン)で残渣を精製し、白色固体として13cを2.95g(収率80%)得た。
【0145】
工程3.(R)−3−(2−(ベンジルオキシ)−5−(ベンジルオキシメチル)フェニル)−3−フェニルプロパノイルクロリド(14c、X=CH2OBn)。
トルエン(10mL)中の13c(1.3g、2.9mmol)に塩化チオニル(1.1mL、15mL)を加え、続いて、ドライピリジンを1滴加えた。混合物を1時間環流加熱した。塩化チオニルを真空下で除去した。ドライエーテル(20mL)を加えた。得られた固体を濾過で除去した。溶液を真空下で濃縮し、淡黄色固体として粗14cを1.3g得た。
【0146】
工程4.(R)−3−(2−(ベンジルオキシ)−5−(ベンジルオキシメチル)フェニル)−N,N−ジ(イソプロピル−d7)−3−フェニルプロパンアミド(15c、X=CH2OBn、R2=R3=CD(CD3)2)。
ドライエーテル(20mL)中の粗14c(1.3g)の溶液にジイソプロピルアミン−d14[CDNアイソトープ、98原子%D](500mg、4.35mmol)を加え、続いて、Et3N(1.25mL、9mmol)を加えた。混合物を4時間攪拌し、2N HCl(15mL)を加えて急冷し、EtOAc(50mL)で抽出した。有機層を水性NaHCO3(飽和)、ブラインで洗浄し、Na2SO4で乾燥させ、濾過し、濾液を真空下で濃縮した。カラムクロマトグラフィー(30% EtOAc/ヘプタン)で残渣を精製し、無色油として15cを1.2g(収率75%)得た。
【0147】
工程5.(R)−3−(2−(ベンジルオキシ)−5−(ベンジルオキシメチル)フェニル)−N,N−ジ(イソプロピル−d7)−3−フェニルプロパン−1−アミン(16c、X=CH2OBn、R2=R3=CD(CD3)2)。
THF(無水、15mL)中の15c(1.2g、2.2mmol)の溶液を0℃に冷却し、続いて、LiAlH4(167mg、4.4mmol)を加えた。混合物を室温で14時間攪拌した後、H2O(0.18mL)、15% NaOH溶液(0.18mL)、およびH2O(0.44mL)を加えて急冷した。沈殿物を濾過し、EtOAcで洗浄した。濾液を真空下で濃縮し、残渣をカラムクロマトグラフィー(50% EtOAc/ヘプタン中の5% Et3N)で精製し、無色油として16cを920mg(収率78%)得た。
【0148】
工程6.(R)−2−(3−(ジ(イソプロピル−d7))アミノ)−1−フェニルプロピル)−4−(ヒドロキシメチル)フェノール(化合物109)。
ジクロロメタン(20mL)中の16c(910mg、1.7mmol)の−78℃の溶液にBCl3(1Mジクロロメタン中の5.9mL、5.9mmol)を滴下した。混合物を3時間攪拌した後、H2Oを加えて急冷し、室温に温めた。混合物を水性NaHCO3(飽和)でpH=9にし、ジクロロメタンで抽出した。有機層をNa2SO4で乾燥させ、濾過し、濾液を真空下で濃縮した。カラムクロマトグラフィー(5%メタノール/ジクロロメタン中の5% Et3N)で残渣を精製し、黄色油として化合物109を440mg(収率72%)得た。
実施例6.(R)−2−(3−(ジ(イソプロピル−d7))アミノ)−1−フェニルプロピル)−4−(ヒドロキシメチル)フェニルイソブチレート(化合物117)の合成。
【0149】
化合物117は、スキーム1に概説したように、化合物109から調製した。
【化18】
【0150】
(R)−2−(3−(ジ(イソプロピル−d7))アミノ)−1−フェニルプロピル)−4−(ヒドロキシメチル)フェニルイソブチレート(化合物117)。
ジクロロメタン(10mL)中の化合物109(440mg、1.24mmol)の−30℃の溶液にジイソプロピルエチルアミン(0.23mL、1.36mmol)を加え、続いて、塩化イソブチリル(0.13mL、1.24mmol)を加えた。反応混合物を−30℃で3時間攪拌した後、水を加えて急冷し、ジクロロメタンで抽出した。有機層をNa2SO4で乾燥させ、濾過し、濾液を真空下で濃縮した。カラムクロマトグラフィー(20% EtOAc/ヘプタン中の5% Et3N、その後、5%メタノール/ジクロロメタン中の5% Et3N)で残渣を精製し、黄色油として化合物117を370mg(収率70%)得た。1H−NMR(300MHz、CDCl3):δ 0.92(m、0.3H)、1.30(d、J=11.1、3H)、1.33(d、J=11.1、3H)、2.07〜2.18(m、2H)、2.22〜2.41(m、2H)、2.76〜2.85(m、1H)、4.12(t、J=7.4、1H)、4.63(s、2H)、6.97(d、J=8.2、1H)、7.00〜7.31(m、6H)、7.34(d、J=1.5、1H)。13C−NMR(75MHz、CDCl3):δ 18.97、19.10、34.20、36.90、41.77、43.83、64.90、122.60、125.60、126.14、126.89、127.92、128.33、136.95、138.64、143.87、147.94、175.32。HPLC(方法:20mm C18−RPカラム−傾斜法2−95% ACN+0.1%ギ酸で3.3分、95% ACNで1.7分保持した;波長:210nm):保持時間:2.89分;90%純度。MS(M+H):426.3。
実施例7.(R)−2−(3−(ジイソプロピルアミノ)−1−フェニルプロピル)−4−(ヒドロキシル(メチル−d2))フェノール(化合物113)の合成。
【0151】
適当に重水素化された試薬を用いて、スキーム1で概説したように化合物113を調製した。
【化19】
【0152】
工程1.(R)−3−((R)−3−(2−(ベンジルオキシ)−5−(ベンジルオキシ(メチル−d2))フェニル)−3−フェニルプロパノイル)−4−フェニルオキサゾリジン−2−オン(12d、X=CD2OBn)。
THF(無水、20mL)中の11d(5g、13mmol、実施例1dを参照)の溶液にMg(480mg、20mmol)を加えた。小片のヨウ素を加え、混合物を80℃で1時間加熱した後、−50℃に冷却した。CuBr−Me2S(1.4g、7mmol)を加えた。混合物を−30℃で1時間攪拌した後、THF(無水、40mL)中の(R)−3−シンナモイル−4−フェニルオキサゾリジン−2−オン(10)(5.5g、19mmol、実施例2、工程1を参照されたい)の溶液を加えた。混合物を室温にゆっくりと温め、さらに6時間攪拌し、水性NH4Cl(飽和)を加えて急冷し、EtOAc(200mL)で抽出した。有機層をブラインで洗浄し、Na2SO4で乾燥させ、濾過し、濾液を真空下で濃縮した。カラムクロマトグラフィー(20% EtOAc/ヘプタン)で残渣を精製し、白色固体として12dを5.8g(収率74%)得た。
【0153】
工程2.(R)−3−(2−(ベンジルオキシ)−5−(ベンジルオキシ(メチル−d2))フェニル)−3−フェニルプロパン酸(13d、X=CD2OBn)。
THF(30mL)中の12d(5.0g、8.3mmol)の溶液に水(10mL)を加えた。混合物を0℃に冷却し、30% H2O2溶液(7.2mL、64mmol)を加え、続いて、LiOH(388mg、16.2mmol)を加えた。混合物を室温で4時間攪拌し、4N HClでpH=2に酸性化し、EtOAc(50mL×3)で抽出した。有機抽出物を組み合わせ、Na2SO4で乾燥させ、濾過し、濾液を真空下で濃縮した。カラムクロマトグラフィー(20〜50% EtOAc/ヘプタン)で残渣を精製し、白色固体として13dを2.82g(収率75%)得た。
【0154】
工程3.(R)−3−(2−(ベンジルオキシ)−5−(ベンジルオキシ(メチル−d2))フェニル)−3−フェニルプロパノイルクロリド(14d、X=CD2OBn)。
トルエン(10mL)中の13d(1.4g、3.1mmol)に塩化チオニル(1.1mL、15mmol)を加え、続いて、ドライピリジンを1滴加えた。混合物を1時間環流加熱した。過剰の塩化チオニルを真空下で除去した。ドライエーテル(20mL)を加えた。得られた固体を濾過で除去した。溶液を濃縮し、淡黄色固体として粗14dを1.3g得た。
【0155】
工程4.(R)−3−(2−(ベンジルオキシ)−5−(ベンジルオキシ(メチル−d2))フェニル)−N,N−ジイソプロピル−3−フェニルプロパンアミド(15d、X=CD2OBn)。
ドライエーテル(20mL)中の14dの溶液にジイソプロピルアミン(2.1mL、15mmol)を加えた。混合物を4時間攪拌し、2N HCl(15mL)を加えて急冷し、EtOAc(50mL)で抽出した。有機層をNaHCO3(飽和)、ブラインで洗浄し、Na2SO4で乾燥させ、濾過し、濾液を真空下で濃縮した。カラムクロマトグラフィー(30% EtOAc/ヘプタン)で残渣を精製し、無色油として15dを1.25g(収率75%)得た。
【0156】
工程5.(R)−3−(2−(ベンジルオキシ)−5−(ベンジルオキシメチル−d2)フェニル)−N,N−ジイソプロピル−3−フェニルプロパン−1−アミン(16d、X=CD2OBn)。
THF(無水、15mL)中の15d(1.2g、2.2mmol)の0℃の溶液にLiAlH4(170mg、4.4mmol)を加えた。混合物を室温で14時間攪拌した後、H2O(0.18mL)、15% NaOH溶液(0.18mL)、およびH2O(0.44mL)で急冷した。沈殿物を濾過し、EtOAcで洗浄した。有機層を真空下で濃縮し、残渣をカラムクロマトグラフィー(50%EtOAc/ヘプタン中の5% Et3N)で精製し、無色油として16dを920mg(収率80%)得た。
【0157】
工程6.(R)−2−(3−(ジイソプロピルアミノ)−1−フェニルプロピル)−4−(ヒドロキシル(メチル−d2))フェノール(化合物113)。
ジクロロメタン(20mL)中の16d(920mg、1.76mmol)の−78℃の溶液にBCl3(1Mジクロロメタン中の6.3mL、6.3mmol)を滴下した。混合物を3時間攪拌した後、H2Oで急冷し、室温に温めた。混合物をNaHCO3(飽和)でpH=9にし、ジクロロメタンで抽出した。有機層をNa2SO4で乾燥させ、濾過し、濾液を真空下で濃縮した。カラムクロマトグラフィー(5%メタノール/ジクロロメタン中の5% Et3N)で残渣を精製し、黄色油として化合物113を425mg(収率70%)得た。
実施例8.(R)−2−(3−(ジイソプロピルアミノ)−1−フェニルプロピル)−4−(ヒドロキシル(メチル−d2))フェニルイソブチレート(化合物116)の合成。
【0158】
化合物116は、スキーム1に概説したように、化合物113から調製した。
【化20】
【0159】
(R)−2−(3−(ジイソプロピルアミノ)−1−フェニルプロピル)−4−(ヒドロキシル(メチル−d2))フェニルイソブチレート(化合物116)。
ジクロロメタン(10mL)中の化合物113(420mg、1.22mmol)の溶液を−30℃に冷却した後、ジイソプロピルエチルアミン(0.23mL、1.35mmol)を加え、続いて、塩化イソブチリル(0.13mL、1.23mmol)を加えた。混合物を−30℃で3時間攪拌した後、水で急冷し、ジクロロメタンで抽出し、Na2SO4で乾燥させ、濾過し、濾液を真空下で濃縮した。カラムクロマトグラフィー(20% EtOAc/ヘプタン中の5% Et3N、その後、5%メタノール/ジクロロメタン中の5% Et3N)で残渣を精製し、黄色油として化合物116を360mg(収率70%)得た。1H−NMR(300MHz、CDCl3):δ 0.91(d、J=6.4、12H)、1.30(d、J=11.1、3H)、1.32(d、J=11.1、3H)、2.08〜2.17(m、2H)、2.30〜2.37(m、2H)、2.76〜2.85(m、1H)、2.91〜3.00(m、2H)、4.11(t、J=7.6、1H)、6.96(d、J=8.2、1H)、7.12〜7.32(m、6H)、7.34(d、J=2.3、1H)。13C−NMR(75MHz、CDCl3):δ 18.98、19.10、20.59、34.20、36.93、41.77、43.82、48.71、122.62、125.65、126.14、126.95、127.92、128.34、136.98、138.48、143.86、147.98、175.32。HPLC(方法:20mm C18−RPカラム−傾斜法2−95% ACN+0.1%ギ酸で3.3分、95% ACNで1.7分保持した;波長:210nm):保持時間:2.85分;90%純度。MS(M+H):414.3。
実施例9.(R)−2−(3−(ジ(イソプロピル−d7)アミノ)−1−フェニルプロピル)−4−(ヒドロキシル(メチル−d2))フェノール(化合物103)。
【0160】
適当に重水素化された試薬を用いて、スキーム1で概説したように化合物103を調製した。
【化21】
【0161】
工程1.(R)−3−(2−(ベンジルオキシ)−5−(ベンジルオキシ(メチル−d2))フェニル)−N,N−ジ(イソプロピル−d7)−3−フェニルプロパンアミド(15f、X=CD2OBn、R2=R3=CD(CD3)2)。
ドライエーテル(20mL)中の粗14d(1.3g)(実施例7、工程3を参照)の溶液にジイソプロピルアミン−d14[CDNアイソトープ、98原子%D](500mg、4.35mmol)を加え、続いて、Et3N(1.25mL、9mmol)を加えた。混合物を4時間攪拌し、2N HCl(15mL)で急冷し、EtOAc(50mL)で抽出した。有機層を水性NaHCO3(飽和)、ブラインで洗浄し、Na2SO4で乾燥させ、濾過し、濾液を真空下で濃縮した。カラムクロマトグラフィー(30% EtOAc/ヘプタン)で残渣を精製し、無色油として15fを1.26g(収率74%)得た。
【0162】
工程2.(R)−3−(2−(ベンジルオキシ)−5−(ベンジルオキシ(メチル−d2))フェニル)−N,N−ジ(イソプロピル−d7)−3−フェニルプロパン−1−アミン(16f、X=CD2OBn、R2=R3=CD(CD3)2)。
THF(無水、15mL)中の15f(1.2g、2.18mmol)の0℃の溶液にLiAlH4(170mg、4.4mmol)を加えた。混合物を室温で14時間攪拌した後、H2O(0.18mL)、15% NaOH溶液(0.18mL)、およびH2O(0.44mL)で急冷した。沈殿物を濾過し、EtOAcで洗浄した。濾液を真空下で濃縮し、カラムクロマトグラフィー(50% EtOAc/ヘプタン中の5% Et3N)で残渣を精製し、無色油として16fを960mg(収率82%)得た。
【0163】
工程3.(R)−2−(3−(ジ(イソプロピル−d7)−アミノ)−1−フェニルプロピル)−4−(ヒドロキシル(メチル−d2))フェノール(化合物103)。
ジクロロメタン(20mL)中の16f(960mg、1.79mmol)の−78℃の溶液にBCl3(1Mジクロロメタン中の6.3mL、6.3mmol)を滴下した。混合物を3時間攪拌した後、H2Oで急冷し、室温に温めた。混合物を水性NaHCO3(飽和)でpH=9にし、ジクロロメタンで抽出した。有機層をNa2SO4で乾燥させ、濾過し、濾液を真空下で濃縮した。カラムクロマトグラフィー(5%メタノール/ジクロロメタン中の5% Et3N)で残渣を精製し、黄色油として化合物103を460mg(収率73%)得た。
実施例10.(R)−2−(3−(ジ(イソプロピル−d7)アミノ)−1−フェニルプロピル)−4−(ヒドロキシル(メチル−d2))フェニルイソブチレート(化合物118)の合成。
【0164】
化合物118は、スキーム1に概説したように、化合物103から調製した。
【化22】
(R)−2−(3−(ジ(イソプロピル−d7)アミノ)−1−フェニルプロピル)−4−(ヒドロキシル(メチル−d2))フェニルイソブチレート(化合物118)。
ジクロロメタン(10mL)中の化合物103(460mg、1.29mmol)の−30℃の溶液にジイソプロピルエチルアミン(0.25mL、1.42mmol)を加え、続いて、塩化イソブチリル(0.14mL、1.29mmol)を加えた。混合物を−30℃で3時間攪拌した後、水で急冷し、ジクロロメタンで抽出し、Na2SO4で乾燥させ、濾過し、濾液を真空下で濃縮した。カラムクロマトグラフィー(20% EtOAc/ヘプタン中の5% Et3N、その後、5%メタノール/ジクロロメタン中の5% Et3N)で残渣を精製し、黄色油として化合物118を385mg(収率70%)得た。1H−NMR(300MHz、CDCl3):δ 0.90(微量)、1.30(d、J=11.1、3H)、1.33(d、J=11.1、3H)、2.13〜2.17(m、2H)、2.30〜2.36(m、2H)、2.76〜2.85(m、1H)、4.11(t、J=7.6、1H)、6.97(d、J=8.2、1H)、7.13〜7.33(m、6H)、7.34(d、J=1.5、1H)。13C−NMR(75MHz、CDCl3):δ 18.98、19.10、34.20、36.87、41.76、43.83、122.62、125.67、126.15、126.96、127.91、128.34、136.98、138.48、143.85、147.99、175.32。HPLC(方法:20mm C18−RPカラム−傾斜法2−95% ACN+0.1%ギ酸で3.3分、95% ACNで1.7分保持した;波長:210nm):保持時間:2.87分;90%純度。MS(M+H):428.4。
実施例11.ヒト肝ミクロソームにおける代謝安定性の評価。
【0165】
材料:XenoTech,LLC(Lenexa,KS))からヒト肝ミクロソーム(20mg/mL)を入手した。Sigma−Aldrichからβ−ニコチンアミドアデニンジヌクレオチドリン酸、還元型(NADPH)、塩化マグネシウム(MgCl2)、およびジメチル・スルホキシド(DMSO)を購入した。
【0166】
化合物100、化合物106、化合物112、およびトルテロジンのストック溶液(7.5mM)をDMSO中で別々に調製した。7.5mMのストック溶液をアセトニトリル(ACN)中で50μMに希釈した。20mg/mLヒト肝ミクロソームを3mMのMgCl2を含有するpH7.4の0.1Mリン酸カリウム緩衝液中で0.625mg/mLに希釈した。希釈したミクロソームを、2通りで、96ウェルディープウェルポリプロピレンプレートのウェルに加えた。試験化合物溶液50μMの1つの10μLをミクロソームに加え、混合物を10分間予熱した。予熱したNADPH溶液を加えることで反応を開始させた。最終反応体積は、0.5mLであり、0.5mg/mLのヒト肝ミクロソーム、1μMの試験化合物、および、pH7.4の、0.1Mリン酸カリウム緩衝液中の2mM NADPH、ならびに3mMのMgCl2を含有した。反応混合物を37℃でインキュベートし、50μLの一定分量を0分、5分、10分、20分、および30分で取り出し、内部標準によって50μLの氷冷ACNを含んだ浅いウェルの96ウェルプレートに加え、反応を止めた。100μLの水をプレートのウェルに加えた後、プレートを4℃で20分間保存し、遠心分離してペレット状の沈降タンパク質にした。上清を別の96ウェルプレートに移し、Applied Bio−systemsのAPI4000質量分析計を用いてLC−MS/MSによって親残余の量を分析した。7−エトキシクマリン(1μM)を陽性対照として用いた。実験を2回繰返して、結果を確認した。
【0167】
データ分析:試験化合物のin vitro半減期(t1/2s)は、以下の式を用いて、%親残余(ln)対インキュベーション時間の関係による線形回帰の傾きから算出した。in vitro t1/2=0.693/k、ここで、k=−[%親残余(ln)対インキュベーション時間の線形回帰の傾き]。データ分析は、マイクロソフトのエクセルソフトウェアを用いて行った。
【0168】
結果を図1および以下の表2に示す。
【0169】
表2.ヒト肝ミクロソームにおける本発明の化合物の半減期の算出値。
【化23】
*%差=[(重水素化種)−(非重水素化種)](100)/(非重水素化種)
【0170】
化合物100および112は、トルテロジンと比較して、ヒト肝ミクロソームにおける半減期が実質的に増加したことを示す。驚いたことに、14個の重水素原子を含む化合物106は、トルテロジンと比較した場合、安定性にほとんど変化を示さなかった。一方、3個の重水素原子しか含まない化合物112は、安定性が増すことを示した。代謝の時間経過はこれらの結果で一致している(図1)。
【0171】
理論に束縛されるものではないが、本出願人らは、フェニル部分上の5−メチル基の重水素化によって、そのフェニル部分上に2−ヒドロキシルを含む本発明の化合物の安定性増加がもたらされると考えている。
【0172】
さらに説明することなく、当業者は、前述の説明および例示実施例を用いて、本発明の化合物を作り、利用し、請求項に記載の方法を実行することができると考えられる。上述の議論および実施例は、特定の好適な実施形態の詳細な説明を単に述べたにすぎないこと理解すべきである。当業者には、本発明の精神および範囲から逸脱せずにさまざまな変更物および同等物の作成が可能であることが明らかであろう。
【技術分野】
【0001】
(関連出願の相互参照)
本願は、2008年4月9日出願の米国特許仮出願第61/043,729号の利益を主張するものであり、この出願の全教示内容は、参照することで本明細書に組み入れられる。
【0002】
本発明は、トルテロジン、5−ヒドロキシメチルトルテロジン、およびフェソテロジンの新規誘導体、ならびに、それらの医薬的に許容される塩、溶媒和物、および水和物に関する。本発明は、本発明の化合物を含む組成物、およびムスカリン受容体遮断薬によって有利に治療される疾患および疾病を治療する方法におけるかかる組成物の用途についても提供する。
【0003】
トルテロジンは、3−(2−ヒドロキシ−5−メチルフェニル)−N,N−ジイソプロピル−3−フェニルプロピルアミンであり、L−酒石酸との1:1塩としてデトロール(登録商標)の名称で販売されている。これは、強力かつ競合的なムスカリン受容体遮断薬であり、例えば、過活動膀胱をもつ成人の治療に有用である。
【0004】
デトロールは、頻尿、切迫性、切迫性尿失禁、またはこれらの症状の任意の組み合わせの症状をもつ過活動膀胱の患者の治療に承認されている。トルテロジンは、高齢者の記憶および認識における化合物の作用について臨床試験中でもある。
【0005】
経口投与後、トルテロジンは肝臓で代謝され、主要な薬理学的活性代謝産物である5−ヒドロキシメチルトルテロジンを産生する。トルテロジンと同様の抗ムスカリン活性を示す5−ヒドロキシメチル代謝産物は、治療効果に著しく寄与する。フェソテロジンは、5−ヒドロキシメチル代謝産物のプロドラッグであり、フマル酸塩(2−メチルプロピオン酸2−[3−(N,N−ジイソプロピルアミノ)−1(R)−フェニルプロピル]−4−(ヒドロキシメチル)フェニルエステルフマル酸塩、およびイソ酪酸2−[3−(ジイソプロピルアミノ)−1(R)−フェニルプロピル]−4−(ヒドロキシメチル)フェニルエステルフマル酸塩)としてトヴィアズ(登録商標)の名称で販売されている。フェソテロジンは、切迫性尿失禁および過活動膀胱の治療において欧州連合で承認されており、抗ムスカリンとして米国で臨床試験中である。
【0006】
トルテロジン、5−ヒドロキシメチルトルテロジン、およびフェソテロジンの有益な活性にも関わらず、上述の疾患および疾病を治療するための新規化合物への継続的な必要性がある。
【図面の簡単な説明】
【0007】
【図1】トルテロジンと比較した、ヒト肝ミクロソームにおける本発明の種々の化合物の代謝経過を示す。(定義)
【0008】
「改善する」および「治療する」という用語は、区別なく使用され、治療および予防的治療の両方を含む。両方の用語は、疾患(例えば、本明細書で使用する疾患または疾病)の発生または進行を減少する、抑制する、弱毒化する、軽減する、阻む、または安定化することを意味する。
【0009】
「疾患」とは、細胞、組織、または器官の正常機能を損なうまたは妨げるあらゆる症状または疾病のことを意味する。
【0010】
合成に用いられる化学物質の由来に応じて、天然同位体存在度におけるいくつかのばらつきが合成化合物で起こることが分かるであろう。従って、トルテロジン、5−ヒドロキシメチルトルテロジン、またはフェソテロジンの製剤は、少量の重水素化された同位体置換体を本質的に含む。天然に豊富で安定な水素および炭素同位体の濃度は、このばらつきに関わらず、少濃度であり、本発明の化合物の安定同位体置換の程度と比較して微々たるものである。例えば、Wada,Eら、Seikagaku,1994,66:15 Gannes,LZら、Comp Biochem Physiol Mol Integr Physiol,1998,119:725を参照されたい。本発明の化合物において、特定の位置が重水素を有するものとして指定される場合、その位置における重水素の存在度は、重水素の天然存在度である0.015%よりも実質的に多いことを理解されたい。「D」または「重水素」として指定される位置は、一般に、重水素の天然存在度(即ち、少なくとも45%重水素取り込み)よりも少なくとも3000倍の存在度で、その位置に重水素を有する。
【0011】
本明細書で使用する「同位体濃縮係数」という用語は、特定同位体の同位体存在度と天然存在度の間の比率を意味する。
【0012】
他の実施形態において、本発明の化合物は、指定された各重水素原子に対して、少なくとも3500(指定された各重水素原子で52.5%の重水素取り込み)、少なくとも4000(60%重水素取り込み)、少なくとも4500(67.5%重水素取り込み)、少なくとも5000(75%重水素取り込み)、少なくとも5500(82.5%重水素取り込み)、少なくとも6000(90%重水素取り込み)、少なくとも6333.3(95%重水素取り込み)、少なくとも6466.7(97%重水素取り込み)、少なくとも6600(99%重水素取り込み)、または少なくとも6633.3(99.5%重水素取り込み)の同位体濃縮係数を有する。
【0013】
本発明の化合物において、特定の同位体として特に指定されない任意の原子は、その原子の任意の安定同位体を表わすことを意味する。特に明記しない限り、位置が「H」または「水素」と具体的に指定される場合、その位置は、その天然存在度の同位体組成で水素を有すると理解される。
【0014】
「同位体置換体」という用語は、その同位体組成においてのみ本発明の特定化合物とは異なる種のことを言う。
【0015】
「化合物」という用語は、本発明の化合物のことを言う場合、分子の構成原子間において同位体変化があり得る以外は同一の化学構造を有する分子の集合体のことを言う。従って、示された重水素原子を含む特定の化学構造によって表わされる化合物は、その構造における1つ以上の指定された重水素の位置に水素原子を有するより少量の同位体置換体を含むことが当業者には明らかであろう。本発明の化合物におけるかかる同位体置換体の相対量は、化合物を作るのに用いる重水素化試薬の同位体純度や、化合物の調製に用いる種々の合成工程における重水素の取り込み効率を含む多くの要因による。しかしながら、上述のように、かかる同位体置換体のin totoの相対量は、化合物の55%未満である。他の実施形態において、かかる同位体置換体のin totoの相対量は、化合物の47.5%未満、40%未満、32.5%未満、25%未満、17.5%未満、10%未満、5%未満、3%未満、1%未満、または0.5%未満である。
【0016】
本発明は、本発明の化合物の塩、溶媒和物、または水和物についても提供する。
【0017】
本発明の化合物の塩は、酸と、アミノ官能基などの化合物の塩基性基との間で形成されるか、あるいは、塩基と、カルボキシル官能基などの化合物の酸性基との間で形成される。別の実施形態によれば、化合物は、医薬的に許容される酸付加塩である。
【0018】
本明細書で使用する「医薬的に許容される」という用語は、適切な医学的判断の範囲内で、過度の毒性、刺激、アレルギー反応等がなくヒトや他の哺乳類の組織に接触させて用いるのに適切であり、なおかつ、妥当な便益/リスク比に相応した成分のことを言う。「医薬的に許容される塩」とは、受容体への投与時に、本発明の化合物を直接的または間接的に提供することができる任意の非毒性塩のことを意味する。「医薬的に許容される対イオン」とは、受容体への投与時に塩から放出された際に、毒性のない塩のイオン部分のことである。
【0019】
医薬的に許容される塩を形成するのに一般的に用いられる酸としては、二硫化水素、塩酸、臭化水素酸、ヨウ化水素酸、硫酸、およびリン酸などの無機酸のみならず、パラ−トルエンスルホン酸、サリチル酸、酒石酸、二酒石酸、アスコルビン酸、マレイン酸、ベシル酸、フマル酸、グルコン酸、グルクロン酸、ギ酸、グルタミン酸、メタンスルホン酸、エタンスルホン酸、ベンゼンスルホン酸、乳酸、シュウ酸、パラ−ブロモフェニルスルホン酸、炭酸、コハク酸、クエン酸、安息香酸、および酢酸などの有機酸、ならびに関連の無機酸および有機酸が挙げられる。従って、かかる医薬的に許容される塩としては、硫酸塩、ピロ硫酸塩、重硫酸塩、亜硫酸塩、重亜硫酸塩、リン酸塩、一水素リン酸塩、二水素リン酸塩、メタリン酸塩、ピロリン酸塩、塩化物、臭化物、ヨウ化物、酢酸塩、プロピオン酸塩、デカン酸塩、カプリル酸塩、アクリル酸塩、ぎ酸塩、イソ酪酸塩、カプリン酸塩、ヘプタン酸塩、プロピオル酸塩、シュウ酸塩、マロン酸塩、コハク酸塩、スベリン酸塩、セバシン酸塩、フマル酸塩、マレイン酸塩、ブチン−1,4−ジオエート、ヘキシン−1,6−ジオエート、安息香酸塩、クロロ安息香酸塩、メチル安息香酸塩、ジニトロ安息香酸塩、ヒドロキシ安息香酸塩、メトキシ安息香酸塩、フタル酸塩、テレフタル酸塩、スルホン酸塩、キシレンスルホン酸塩、フェニル酢酸塩、フェニルプロピオン酸塩、フェニル酪酸塩、クエン酸塩、乳酸塩、β−ヒドロキシ酪酸塩、グリコール酸塩、マレイン酸塩、酒石酸塩、メタンスルホン酸塩、プロパンスルホン酸塩、ナフタレン−1−スルホン酸塩、ナフタレン−2−スルホン酸塩、マンデル酸塩、および他の塩が挙げられる。一実施形態において、医薬的に許容される酸付加塩としては、塩酸および臭化水素酸などの鉱酸で形成されたもの、特に、マレイン酸などの有機酸で形成されたものが挙げられる。
【0020】
本明細書で使用する「水和物」という用語は、非共有結合性の分子間力によって結合した化学量論量または非化学量論量の水をさらに含む化合物を意味する。
【0021】
本明細書で使用する「溶媒和物」という用語は、非共有結合性の分子間力によって結合した化学量論量または非化学量論量の溶媒(水、アセトン、エタノール、メタノール、ジクロロメタン、2−プロパノールなど)をさらに含む化合物を意味する。
【0022】
本発明の化合物(例えば、式I、式IIの化合物など)は、不斉炭素原子を含む。このように、本発明の化合物は、個々の鏡像異性体(例えば、(S)または(R)の1つ)としても存在し、2つの鏡像異性体の混合物としても存在することができる。(R)化合物の方が活性がより大きく、好適である。従って、本発明の化合物は、両方のラセミ混合物だけでなく、別の可能な立体異性体を実質的に含まない個々それぞれの立体異性体も含む。本明細書で使用する「他の立体異性体を実質的に含まない」という用語は、他の立体異性体を25%未満、好ましくは他の立体異性体を10%未満、より好ましくは他の立体異性体を5%未満、最も好ましくは他の立体異性体を2%未満、または他の立体異性体を「X」%未満(ここで、Xは0以上100以下の数)含むことを意味する。所与の化合物に対する個々の鏡像異性体を得るまたは合成する方法は、当該技術分野では周知であり、最終化合物または出発材料あるいは中間体に実用可能なものとして適用することができる。
【0023】
本明細書で使用する「安定化合物」という用語は、製造するのに十分な安定性を有し、本明細書で詳述した目的に有用な十分な期間にわたって化合物の完全性を維持する化合物のことを言う(例えば、治療薬に反応する疾患または疾病を治療するための治療薬、治療化合物の製造に使用する中間体、単離可能または保存可能な中間体化合物への製剤)。
【0024】
「D」とは、重水素のことを言う。
【0025】
「立体異性体」とは、鏡像異性体およびジアステレオマーのことを言う。
【0026】
「Tert」、「t」および「t−」は、それぞれ、三級のことを言う。
【0027】
「US」は、アメリカ合衆国のことを言う。
【0028】
「FDA」は、食品医薬品局のことを言う。
【0029】
「E.U.」は、欧州連合のことを言う。
【0030】
本明細書を通じて、「各R」への言及としては、該当する場合には独立して、全ての「R」基(例えば、R1、R2、R3、R4、およびR5)を含む。特に明記しない限り、変数が概括的なことを言う場合、特定の変数の具体的な実施形態を全て含むことを意味する。
(治療化合物)
【0031】
本発明は、式Iの化合物:
【化1】
式(I)、
または、その医薬的に許容される塩、溶媒和物、もしくは水和物を提供し、
式中:
R1は、CD3、CHD2、CH2D、CH3、CD2OH、CHDOH、またはCH2OHであり;
R2およびR3は、それぞれ独立して、0〜7個の重水素原子をもつイソプロピル基であり;
R4は、HまたはC(O)−C1−C6アルキルであり;
R1、R2、またはR3の少なくとも1つは重水素原子を含む。
【0032】
ある実施形態において、R1は、CH3、CD3、CD2OH、またはCH2OHから選択される。
【0033】
ある実施形態において、R2およびR3は同一である。
【0034】
他の実施形態において、R2およびR3は、−CH(CD3)2、−CD(CD3)2、−CH(CH3)2、および−CD(CH3)2から独立して選択される。
【0035】
ある実施形態において、R4はHである。
【0036】
他の実施形態において、R4はC(O)−CH(CH3)2である。
【0037】
さらに別の実施形態において、化合物は、表1(下記)に記載の化合物のいずれか1つから選択される:
表1:式Iの例示実施形態
【化2】
【0038】
さらに別の実施形態において、本発明は、式IIの化合物:
【化3】
式(II)、
またはその医薬的に許容される塩を提供し、
式中:
R2およびR3は、それぞれ独立して、0〜7個の重水素原子をもつイソプロピル基である。
【0039】
式IIの一実施形態において、R2およびR3の各々は、−CD(CD3)2および−CH(CH3)2から独立して選択される。より具体的な態様において、式IIの化合物は、化合物100および化合物112から選択される。
【0040】
さらに別の実施形態において、本発明は、式IIIの化合物:
【化4】
式(III)、
またはその医薬的に許容される塩を提供し、
式中:
R2およびR3は、それぞれ独立して、0〜7個の重水素原子をもつイソプロピル基であり;
Yは両方とも水素または重水素であり、
Yが両方とも水素である場合、R2またはR3の少なくとも1つは重水素原子を含む。
【0041】
式IIIの一実施形態において、R2およびR3の各々は、−CD(CD3)2および−CH(CH3)2から独立して選択される。より具体的な態様において、式IIIの化合物は、化合物116、化合物117、または化合物118から選択される。
【0042】
別の実施形態において、上述の実施形態のいずれかにおいて重水素として指定されない任意の原子は、その天然同位体存在度で存在する。
【0043】
式I、式IIなどの化合物の合成は、合成化学の分野における当業者によって容易に達成することができる。関連手順および中間体は、例えば、米国特許第7,119,212号、同第7,005,449号、同第5,382,600号、同第5,922,914号、同第5,686,464号、および同第5,559,269号;PCT出願刊行物国際公開第2005/006143号、同第2005/012227号、同第2004/078700号、同第2001/049649号、同第2003/014060号、および同第2006/074479号;Andersson,PGら、J Org Chem1998,63(22):8067;ならびに欧州特許公報第EP0957073号に開示されている。
【0044】
かかる方法は、重水素化され、必要に応じて、対応する他の同位体を含有する試薬、および/または、本明細書に記載の化合物を合成するための中間体を利用して、あるいは、同位体原子を化学構造に導入するため当該技術分野で周知の標準的な合成プロトコルを使用することで行うことができる。
(例示合成)
【0045】
式I、式IIなどの化合物を合成するための便利な方法についてスキーム1で説明する。
【0046】
スキーム1.式Iの化合物への一般経路。
【化5】
X=CD3、CD2H、CDH2、CH3、CD2OBn、CDHOBn、またはCH2OBn;Bn=ベンジル。
【0047】
スキーム1に示すように、式Iの化合物の調製は、Mg/CuBr−ジメチルスルフィドを用いて、適当に重水素化された臭化アリール11を公知の4(R)−フェニル−3−[3−フェニル−2(E)−プロペノイル]オキサゾリジン−2−オン(10)に位置選択的付加して、付加物12を生成することから始まる。THF/水中のLiOH/H2O2で12を加水分解して、カルボン酸13を得る。ベンゼン中のSOCl2/ピリジンと13を反応させて、酸塩化物14を生成し、これを適当に重水素化されたアミン20(市販のジイソプロピルアミン−d14[98原子%D]など)で処理し、対応するアミド15を得る。エーテル中のLiAlH4と15を還元させて、三級アミン16得、メタノール中のPd/CによりH2と水素化させて脱ベンジル化し、式Iの化合物(式中、R4はHである)を得る。17のフェノール性ヒドロキシル基をEt3Nの存在下で塩化アシル21によりアシル化し、式Iの化合物(式中、R4は、−C(O)−C1−C6アルキル)を得る。参照文献:Andersson,PGら、「Asymmetric total synthesis of (+)−tolterodine, a new muscarinic receptor antagonist, via copper−assisted asymmetric conjugate addition of aryl Grignard reagents to 3−phenyl−prop−2−enoyl−oxazolidinones」J Org Chem,1998,63(22):8067における一般手順;および欧州特許公報第EP957073号。
【0048】
適当に重水素化された臭化アリール11を調製するための例示的方法について以下のスキーム2Aおよび2Bに示す。
【0049】
スキーム2A.中間体11への一般経路。
【化6】
Y=CD3、CD2H、CDH2、CH3;
X=CD3、CD2H、CDH2、CH3、CD2OBn、CDHOBn、またはCH2OBn
【0050】
スキーム2Aに示すように(Aki,ら、J Phys Chem A 2002,106(14):3436−3444の一般手順に従う)、所望の臭化アリール30をMgおよびO2経由でフェノール31に変換した後、Br2で臭素化し、ブロモフェノール32を得る。ブロモフェノール32を塩化ベンジル経由で直接ベンジル保護して化合物11を生成してもよいし、あるいは、DDQで酸化させた後、NaBH4またはNaBD4(98原子%D)を用いて、適当に重水素化されたアルコール33に還元してもよい。次いで、ジオール33を塩化ベンジルでベンジル保護し、化合物11を得てもよい。
【0051】
スキーム2B:中間体11への代理経路。
【化7】
【0052】
スキーム2Bに示すように、市販の3−ブロモ−4−ヒドロキシ安息香酸を臭化ベンジルおよび炭酸カリウムで処理し、エステル45を得る。該エステルをLiAlH4またはLiAlD4(99原子%D)で還元させ、アルコール46を得る。これを臭化ベンジルおよび炭酸カリウムで処理し、化合物11(式中、XはCD2OBnまたはCH2OBnである)を得る。
【0053】
適当に重水素化されたアミン20の調製方法を以下のスキーム3Aおよび3Bに示す。
【0054】
スキーム3A.
【化8】
【0055】
スキーム3B.
【化9】
各Zは、独立して、CH3、CH2D、CHD2、またはCD3である。
【0056】
スキーム3Aに示すように、適当に重水素化されたアミン40を適当に重水素化された2−クロロプロパン41(または任意の他の適当に重水素化された2−ハロプロパン)でアルキル化して、所望のアミン20を得る(例:Zhang,C,ら、Zhongguo Yaowu Huaxue Zazhi 2004,14(3):161−164の同様の手順を参照されたい)。
【0057】
あるいは、スキーム3Bに示すように、アミン40を適当に重水素化された2−ケトプロパン42で縮合させ、イミン43を生成し、これを、適当に重水素化されたラネーニッケルおよび水素(または重水素)ガスで還元し、アミン20を生成する。例えば、Bunnelle,WH,ら、Synthesis,1997,4:439−442;および、Norton,DGら、J Org Chem 1954,19:1054−66を参照されたい。
【0058】
式I、式IIなどの化合物およびこれらの合成前駆体を合成するためのさらなる方法は、本明細書のスキームに明示的に示していない経路内のものを含み、当業者である化学者の通常の技術の範囲内である。適用可能な化合物の合成に有用な合成化学変換および保護基方法論(保護および脱保護)は、当該技術分野で周知であり、例えば、Larock R,Comprehensive Organic Transformations,VCH Publishers(1989);Greene TWら、Protective Groups in Organic Synthesis,3rd Ed.,John Wiley and Sons(1999);Fieser Lら、Fieser and Fieser’s Reagents for Organic Synthesis,John Wiley and Sons(1994);およびPaquette L,ed.,Encyclopedia of Reagents for Organic Synthesis,John Wiley and Sons(1995)、ならびにその後の版に記載されているものが挙げられる。
【0059】
本発明により想定された置換基および変数の組合せは、安定化合物の形成をもたらすもののみである。
(組成物)
【0060】
本発明は、有効量の、式I、式IIなど(例えば、本明細書の式のいずれかを含む)の化合物、または前記化合物の医薬的に許容される塩、溶媒和物、または水和物と;医薬的に許容される担体とを含むピロゲンフリー組成物についても提供する。一実施形態において、組成物はピロゲンフリーである。別の実施形態において、組成物は、医薬用途(「医薬組成物」)用に調合され、ここで、担体は医薬的に許容される担体である。担体(単数または複数)は、製剤の他の成分と相溶性があるという意味において「許容される」ものでなければならず、医薬的に許容される担体の場合、薬剤に通常使用される量でその受容体に無害なものでなければならない。
【0061】
本発明の医薬組成物に用いることができる医薬的に許容される担体、アジュバント、および賦形剤としては、限定的ではないが、イオン交換体、アルミナ、ステアリン酸アルミニウム、レシチン、血清タンパク質(例えば、ヒト血清アルブミン)、緩衝物質(例えば、リン酸塩)、グリシン、ソルビン酸、ソルビン酸カリウム、植物性飽和脂肪酸の部分グリセリド混合物、水、塩、または電解質(例えば、硫酸プロタミン)、リン酸水素二ナトリウム、リン酸水素カリウム、塩化ナトリウム、亜鉛塩、コロイド状シリカ、三ケイ酸マグネシウム、ポリビニルピロリドン、セルロースベースの物質、ポリエチレングリコール、カルボキシメチルセルロースナトリウム、ポリアクリレート、ワックス、ポリエチレン−ポリオキシプロピレン−ブロックポリマー、ポリエチレングリコール、およびラノリンが挙げられる。
【0062】
必要に応じて、医薬組成物における本発明の化合物の溶解度およびバイオアベイラビリティは、当該技術分野で周知の方法により高めることができる。1つの方法としては、製剤に脂質賦形剤を用いることが挙げられる。「Oral Lipid−Based Formulations:Enhancing the Bioavailability of Poorly Water−Soluble Drugs(Drugs and the Pharmaceutical Sciences)」、David J.Hauss,ed.Informa Healthcare,2007;および「Role of Lipid Excipients in Modifying Oral and Parenteral Drug Delivery:Basic Principles and Biological Examples」、Kishor M.Wasan,ed.Wiley−Interscience,2006を参照されたい。
【0063】
バイオアベイラビリティを高める別の公知の方法としては、LUTROL(商標)およびPLURONIC(商標)(BASF Corporation)などのポロクサマー、または、エチレンオキシドと酸化プロピレンのブロック共重合体と任意に調合された本発明の化合物を非晶形で用いることである。米国特許第7,014,866号;米国特許公報第20060094744号、および同第20060079502号を参照されたい。
【0064】
本発明の医薬組成物としては、経口、直腸、鼻腔、局所(口腔および舌下を含む)、膣内または非経口(皮下、筋肉内、静脈内、および皮内を含む)投与に適するものが挙げられる。ある実施形態において、本明細書の式の化合物は、(例えば、経皮貼り剤またはイオン導入技術を用いて)経皮投与される。他の製剤は、単位投与剤形(例えば、錠剤および徐放カプセル剤)や、リポソームで簡便に提供することができ、薬剤技術分野で周知のあらゆる方法で調製することができる。例えば、Remington’s Pharmaceutical Sciences,Mack Publishing Company,Philadelphia,PA(17th ed.1985)を参照されたい。
【0065】
かかる調製方法としては、1つ以上の副成分を構成する担体などの、投与される成分分子と会合させる工程が挙げられる。一般に、組成物は、有効成分を液体担体、リポソームまたは微細な固体担体、もしくは両方と均一かつ密接に会合させることで調製され、必要に応じて、生成物を成形する。
【0066】
ある実施形態において、化合物は経口投与される。経口投与に適当な本発明の組成物は、各々が所定量の有効成分を含むカプセル剤、サッシェ剤、または錠剤などの個別単位として;散剤または顆粒剤として;水性液体または非水液体中の溶液または懸濁液として;水中油乳濁液として;油中水乳濁液として;リポソーム中に包まれたものとして;あるいは、ボーラス剤等として提供することができる。軟質ゼラチンカプセル剤は、かかる懸濁液を含めるのに有用な場合があり、これは、化合物の吸収率を有利に増加させ得る。
【0067】
経口用途用の錠剤の場合、一般的に使用される担体としては、ラクトースおよびコーンスターチが挙げられる。ステアリン酸マグネシウムなどの滑沢剤も一般に添加される。カプセル形態の経口投与において、有用な希釈剤としては、ラクトースおよび乾燥コーンスターチが挙げられる。水性懸濁液を経口投与する場合、有効成分は乳化剤および懸濁剤と組合せられる。所望の場合、特定の甘味料および/または香料および/または着色剤を添加してもよい。
【0068】
経口投与に適当な組成物としては、芳香化基剤、通常、スクロースおよびアカシアまたはトラガカント中に成分を含むトローチ剤;不活性基剤、例えば、ゼラチンおよびグリセリン、またはスクロースおよびアカシア中に有効成分を含む香錠が挙げられる。
【0069】
非経口投与に適当な組成物としては、抗酸化剤、緩衝剤、静菌剤、および、製剤を目的の受容体の血液と等張させる溶質を含んでいてもよい水性および非水性の無菌注射液;懸濁剤および増粘剤を含んでいてもよい水性および非水性の無菌懸濁液が挙げられる。製剤は、単回投与用または複数投与用入れ物、例えば、密封アンプルおよびバイアルで提供してもよく、無菌液体担体、例えば、注射用水を使用直前に添加することのみを必要とするフリーズドライ(冷凍乾燥)状態で保存してもよい。即時注射液および懸濁液は、無菌散剤、顆粒剤、および錠剤から調製してもよい。
【0070】
かかる注射液は、例えば、滅菌注射用水性または油性懸濁剤の形態であってもよい。この懸濁液は、当該技術分野で公知の技術に従い、適切な分散剤または湿潤剤(例えば、ツイーン80など)および懸濁剤を用いて調合してもよい。滅菌注射用製剤はまた、例えば、1,3−ブタンジオール中の溶液として、非毒性の非経口的に許容される希釈剤または溶媒中の滅菌注射溶液または懸濁液であってもよい。使用できる許容可能な賦形剤や溶媒の中には、マンニトール、水、リンガー溶液、および等張性塩化ナトリウム溶液がある。さらに、滅菌固定油は、溶媒または懸濁媒体として従来使用されている。この目的では、合成のモノグリセリドまたはジグリセリドを含む任意の無刺激固定油を用いてもよい。オレイン酸およびそのグリセリド誘導体などの脂肪酸は、特に、そのポリオキシエチル化形態ではオリーブ油またはヒマシ油などの天然の医薬的に許容される油であるため、注射用製剤において有用である。これらの油状溶液または懸濁液は、長鎖アルコール希釈剤または分散剤を含有してもよい。
【0071】
本発明の医薬組成物は、直腸内投与用に坐剤形態で投与してもよい。これらの組成物は、本発明の化合物と、室温で固体であるが直腸温度では液体であるため、直腸内で溶解して有効成分を放出する適当な非刺激性賦形剤とを混合することにより調製することができる。かかる物質としては、限定的ではないが、ココアバター、蜜蝋、およびポリエチレングリコールが挙げられる。
【0072】
本発明の医薬組成物は、鼻エアロゾルまたは鼻吸入により投与してもよい。かかる組成物は、医薬製剤分野で公知の技術により調製され、なおかつ、ベンジルアルコールまたは他の適切な保存剤、バイオアベイラビリティを高めるための吸収促進剤、フルオロカーボン、および/または当該技術分野で公知の他の可溶化剤または分散剤を用いて、塩水中の溶液として調製することができる。例えば、Alexza Molecular Delivery Corporationに譲渡されているRabinowitz JD and Zaffaroni AC、米国特許第6,803,031号を参照されたい。
【0073】
本発明の医薬組成物の局所投与は、所望の治療が局所塗布により容易に近づける領域または器官に関与する場合に特に有用である。皮膚への局所塗布では、医薬組成物は、担体に懸濁または溶解させた有効成分を含有する適当な軟膏で調合すべきである。本発明の化合物の局所投与用の担体としては、限定的ではないが、鉱油、液化石油、白色石油、プロピレングリコール、ポリオキシエチレンポリオキシプロピレン化合物、乳化ワックス、および水が挙げられる。あるいは、医薬組成物は、担体に懸濁または溶解させた活性化合物を含有する適当なローションまたはクリームと調合することができる。適当な担体としては、限定的ではないが、鉱油、ソルビタンモノステアレート、ポリソルベート60、セチルエステルワックス、セテアリールアルコール、2−オクチルドデカノール、ベンジルアルコール、および水が挙げられる。本発明の医薬組成物は、直腸坐剤製剤または適当な浣腸製剤により、腸管低部に局所塗布してもよい。局所経皮貼り剤およびイオン導入投与も、本発明に含まれる。例えば、式Iの化合物は、例えば、PCT国際公開公報第2005107812号に記載のトルテロジンの当該技術分野で周知の調製方法と同様の方法で局所または経皮用組成物に調合してもよい。
【0074】
対象治療の適用は、対象部位に投与するように局所であってもよい。注射や、カテーテル、トロカール、投射物、プルロニックゲル、ステント、薬物徐放性ポリマー、または内部アクセスをもたらす他のデバイスを用いて対象部位に対象組成物をもたらす種々の技術を用いることができる。例えば、式I、式IIなどの化合物は、例えば、国際公開第2007011131号;同第2007022255号;同第2006021425号;同第2005105036号;同第2004105735号;同第2001034139号;および同第2000027364号に記載のトルテロジンの当該技術分野で周知の調製方法と同様の方法で徐放用に調合してもよい。
【0075】
従って、さらに別の実施形態によれば、本発明の化合物は、プロテーゼ、人工弁、代用血管、ステント、またはカテーテルなどの移植医療機器を被覆するための組成物に組み込むことができる。適当な被覆および被覆された移植機器の一般的調製は、当該技術分野で周知であり、米国特許第6,099,562号;同第5,886,026号;および同第5,304,121号に例示されている。被覆は、通常、ヒドロゲルポリマー、ポリメチルジシロキサン、ポリカプロラクトン、ポリエチレングリコール、ポリ乳酸、エチレン酢酸ビニル、およびその混合物などの生体適合性ポリマー材料である。被覆は、必要に応じて、フルオロシリコーン、ポリサッカリド、ポリエチレングリコール、リン脂質、またはその組合せの適当な上塗りによりさらに被覆し、組成物に徐放特性を付与してもよい。侵襲機器に対する被覆は、本明細書で使用される用語としての医薬的に許容される担体、アジュバント、または賦形剤の定義内に含まれるものである。
【0076】
別の実施形態によれば、本発明は、前記機器を上述の被覆組成物に接触させる工程を含む移植医療機器の被覆方法を提供する。当業者であれば、哺乳類への移植前に機器の被覆を行うことはすぐに分かるであろう。
【0077】
別の実施形態によれば、本発明は、前記薬物放出機器を本発明の化合物または組成物に接触させる工程を含む、移植型の薬物放出機器の含浸方法を提供する。移植型の薬物放出機器としては、限定的ではないが、生分解性のポリマーカプセル剤または薬剤(bullets)、非分解性、拡散性のポリマーカプセル剤、および生分解性のポリマーカシェ剤が挙げられる。
【0078】
別の実施形態によれば、本発明は、治療効果がある化合物または本発明の前記化合物を含む組成物で被覆した移植医療機器を提供する。
【0079】
別の実施形態によれば、本発明は、前記機器から放出され、かつ、治療効果がある化合物または本発明の化合物を含む組成物に含浸したもしくはを含む移植型の薬物放出機器を提供する。
【0080】
器官または組織が手術中に接触可能であるか、または、患者から除去される場合、かかる器官または組織を本発明の組成物を含む培地中に浴してもよく、本発明の組成物を器官上に塗布してもよく、あるいは、本発明の組成物を任意の他の簡便な方法で塗布してもよい。
【0081】
別の実施形態において、本発明の組成物は、第2の治療薬をさらに含む。第2の治療薬としては、トルテロジンと同じ作用機序をもつ化合物と一緒に投与した際に、有利な特性を有することが知られている、あるいは、有利な特性を示す任意の化合物または治療薬から選択することができる。かかる薬剤としては、トルテロジンと併用して有用であると示されるものが挙げられ、限定的ではないが、αアドレナリン受容体遮断薬(例えば、PCT国際特許出願国際公開第2001/021167号および同第2007/010509号に記載のもの);ビシファジン化合物(例えば、PCT国際特許出願国際公開第2006/102029号に記載のもの);スタチン(例えば、PCT国際特許出願国際公開第2006/008437号に記載のもの);デヒドロエピアンドロステロン(DHEA)同族体(例えば、PCT国際特許出願国際公開第2006/007312号に記載のもの;α2δサブユニットカルシウムチャネル調節因子(例えば、GABA類似体や、PCT国際特許出願国際公開第2004/084879号に記載の他のもの);選択的セロトニン再取り込み阻害薬(SSRI)または選択的ノルエピネフリン再取り込み阻害薬(例えば、フルオキセチン、パロキセチン、およびPCT国際特許出願国際公開第2004/019892および同第2001/062236号に記載の他のもの);アンドロゲン、エストロゲン、またはエストロゲン作用薬(例えば、PCT国際特許出願国際公開第2004/043429号、同第2003/039553号、および同第2003/039523号に記載のもの);EGF受容体拮抗薬(例えば、PCT国際特許出願国際公開第2003/039524号に記載のもの);5HT1a受容体修飾因子(例えば、PCT国際特許出願国際公開第2003/026564号に記載のもの);チエノピランカルボキサミド誘導体(例えば、PCT国際特許出願国際公開第2001/009140号に記載のもの);5a還元酵素阻害薬(例えば、PCT国際特許出願国際公開第2001/021167号に記載のもの)、ならびに、米国特許公報第2006−205682号および同第2006−160887号、PCT国際特許出願国際公開第2006/079625号、同第2006/015970号、同第2005/092342号、同第2005/092341号、および同第2001/062236号、日本国特許公報第2003−055261号および同第2005−015394号に記載の第2の薬剤が挙げられる。
【0082】
一実施形態において、第2の治療薬は、不安定膀胱もしくは過活動膀胱、失禁、感染、下部尿路疾患、記憶障害もしくは認知障害、心不全、肺炎(肺胞性肺炎を含む)、良性前立腺肥大症、前立腺肥大症、呼吸器疾患、喘息、女性性機能障害、または膀胱炎から選択される疾患または疾病の治療または予防に有用な薬剤である。
【0083】
一実施形態において、第2の治療薬はタムスロシンである。
【0084】
別の実施形態において、本発明は、本発明の化合物と、上述の第2の治療薬のいずれかの1つ以上との別々の剤形を提供し、ここで、化合物と第2の治療薬とは互いに会合している。本明細書で使用する「互いに会合している」という用語は、別々の剤形を一緒に販売し、投与すること(連続的または同時に互いに24時間未満内)を意図することが容易に明らかとなるように、別々の剤形が一緒に包装されている、あるいは、互いに取り付けられていることを意味する。
【0085】
本発明の医薬組成物では、本発明の化合物は、有効量で存在する。本明細書で使用する「有効量」という用語は、適切な投与計画で投与する場合、治療している疾患の重篤度、期間、または進行を軽減または改善する、治療している疾患の促進を予防する、治療している疾患を退行させる、あるいは、別の治療の予防または治療効果(単数または複数)を高めるまたは向上させるのに十分な量のことを言う。
【0086】
動物とヒトに対する用量の相互関係(体表面1平方メートル当たりのミリグラムに基づく)は、Freireichら、(1966)Cancer Chemother.Rep 50:219に記載されている。体表面積は、患者の身長および体重からおおよそ決定することができる。例えば、Scientific Tables,Geigy Pharmaceuticals,Ardsley,N.Y.,1970,537を参照されたい。
【0087】
一実施形態において、本発明の化合物の有効量は、成人患者に対して、0.1mg/日〜50mg/日の範囲であり得る。別の実施形態において、本発明の化合物の有効量は、成人患者に対して、1mg/日〜10mg/日の範囲であり得る。さらに別の実施形態において、有効量は、成人患者に対して、2〜6mg/日の範囲であり得る。さらに別の実施形態において、有効量は、成人患者に対して、約4mg/日である。
【0088】
有効量は、当業者により認識されるように、治療する疾患、疾患の重篤度、投与経路、患者の大きさ、性別、年齢、および一般的健康状態、賦形剤の使用、他の治療処置に伴う、例えば、他の薬剤の使用との併用の可能性、および治療医の判断に応じて変化させてもよい。例えば、有効量を選択するためのガイダンスは、トルテロジンの処方情報を参照することで決めることができる。
【0089】
第2の治療薬を含む医薬組成物において、第2の治療薬の有効量は、その薬剤のみを用いる単剤療法レジメンで通常使用される投与量の約20%〜100%の間である。有効量は、標準の単剤治療量の約70%〜100%の間であるのが好ましい。これら第2の治療薬の標準の単剤治療量は、当該技術分野で周知である。例えば、Wellsら、eds.,Pharmacotherapy Handbook,2nd Edition,Appleton and Lange,Stamford,Conn.(2000);PDR Pharmacopoeia,Tarascon Pocket Pharmacopoeia2000,Deluxe Edition,Tarascon Publishing,Loma Linda,Calif.(2000)を参照されたい。これら参照文献の各々は、参照により本明細書に完全に引用したものとする。
【0090】
上記で参照した第2の治療薬のいくつかは、本発明の化合物と相乗的に作用すると予想される。これが起こる場合には、第2の治療薬および/または本発明の化合物の有効薬量を単剤療法で必要な量より減らすことができる。これは、第2の治療薬または本発明の化合物いずれかの毒性の副作用の軽減、効果の相乗的な改善、投与または使用し易さの改善、および/または、化合物の調製または製剤の総費用の削減という利点を有する。
(治療方法)
【0091】
別の実施形態において、本発明は、本明細書に記載の式I、式IIなどの1つ以上の化合物を細胞に接触させる工程を含む、細胞におけるコリン作動性ムスカリン受容体の活性を調節する方法を提供する。
【0092】
別の実施形態によれば、本発明は、本発明の化合物または組成物の有効量をそれを必要とする対象に投与する工程を含む、トルテロジンにより有利に治療される疾患に罹患しているまたは罹患しやすい対象の治療方法を提供する。かかる疾患は、当該技術分野では周知であり、限定的ではないが、以下の特許および/または公開公報:米国特許公報第2006−205682号および同第2006−160887号、PCT国際特許出願国際公開第2006/079625号、同第2006/015970号、同第2005/092342号、同第2005/092341号、同第2001/062236号、同第2003/039553号、同第2003/039524号、および同第2003/026564号、ならびに日本国特許公報第2003−055261号および同第2005−015394号に開示されている。
【0093】
別の実施形態において、本発明の方法は、過活動膀胱、失禁、下部尿路感染症、喘息、良性前立腺肥大症、および記憶および認知障害から選択される疾患または疾病に罹患しているもしくは罹患しやすい対象の治療に用いる。ある実施形態において、本発明の方法は、過活動膀胱および失禁から選択される疾患または疾病に罹患しているもしくは罹患しやすい対象の治療に用いる。本明細書に記載の方法としては、対象が特定の治療の必要があると見なされているものも含む。かかる治療が必要である対象の同定は、対象または医療専門家の判断であってもよく、主観的(例えば、意見)または客観的(例えば、試験または診断法により測定可能)であってもよい。
【0094】
別の実施形態において、上記の治療方法のいずれかは、患者に1つ以上の第2の治療薬を同時投与する工程をさらに含む。第2の治療薬の選択は、トルテロジンとの同時投与に有用であると知られている任意の第2の治療薬から行ってよい。本発明の化合物と併用可能してもよいかかる薬剤、疾患、および疾病の例としては、(1)αアドレナリン受容体遮断薬(例えば、タムスロシン、および、PCT国際特許出願国際公開第2001/021167号および同第2007/010509号に記載のもの);ビシファジン化合物(PCT国際特許出願国際公開第2006/102029号に記載のもの);α2δサブユニットカルシウムチャネル調節因子(GABA類似体、および、PCT国際特許出願国際公開第2004/084879号に記載の他のもの);選択的セロトニン再取り込み阻害薬(SSRI)または選択的ノルエピネフリン再取り込み阻害薬(フルオキセチン、パロキセチン、および、PCT国際特許出願国際公開第2004/019892号および同第2001/062236号に記載の他のもの);5HT1a受容体修飾因子(PCT国際特許出願国際公開第2003/026564号に記載のもの);チエノピランカルボキサミド誘導体(PCT国際特許出願国際公開第2001/009140号に記載のもの);または、5a還元酵素阻害薬(PCT国際特許出願国際公開第2001/021167号に記載のもの)、各々は、下部尿路疾患(失禁、不安定膀胱もしくは過活動膀胱、および他の排尿障害を含む)の治療に用いる;(2)デヒドロエピアンドロステロン(DHEA)同族体(炎症の治療に用いるPCT国際特許出願国際公開第2006/007312号に記載のもの);(3)スタチン(呼吸器疾患の治療に用いるPCT国際特許出願国際公開第2006/008437号に記載のもの);(4)アンドロゲン、エストロゲン、またはエストロゲン作用薬(女性性機能障害の治療に用いるPCT国際特許出願国際公開第2004/043429号、同第2003/039553号、および同第2003039523号に記載のもの);(5)EGF受容体拮抗薬(PCT国際特許出願国際公開第2003/039524号に記載のもの);または、チエノピランカルボキサミド誘導体(良性前立腺肥大症または前立腺肥大症の治療に用いるPCT国際特許出願国際公開第2001/009140号に記載のもの);のみならず、米国特許公報第2006−205682号、同第2006−160887号、PCT国際特許出願国際公開第2006/079625号、同第2006/015970号、同第2005/092342号、同第2005/092341号、同第2001/062236号、日本国特許公報2003−055261号、および同第2005−015394号に記載のものが挙げられる。
【0095】
特に、本発明の併用療法としては、式Iの化合物および第2の治療薬を投与することによって以下の疾患:過活動膀胱および失禁の治療が挙げられる。
【0096】
一実施形態によれば、式Iの化合物は、下部尿路疾患を治療するためにタムスロシンと同時投与される。
【0097】
本明細書で使用する「同時投与する」という用語は、単一剤形(例えば、本発明の化合物と、上述の第2の治療薬とを含む本発明の組成物)の一部として、あるいは、別個の複数剤形として、第2の治療薬を本発明の化合物とともに投与することを意味する。あるいは、本発明の化合物の投与前、投与に続いて、または投与後に、追加の薬剤を投与してもよい。かかる併用療法による治療では、本発明の化合物と第2の治療薬(単数または複数)の両方を従来の方法で投与する。本発明の化合物と第2の治療薬との両方を含む本発明の組成物を対象に投与することは、治療期間中に別時で、同一治療薬、任意の他の第2の治療薬、または本発明の任意の化合物を前記対象に個別投与することを妨げるものではない。
【0098】
これらの第2の治療薬の有効量は、当業者に公知であり、投与ガイダンスは、本明細書に引用した特許および特許出願公開のみならず、Wellsら、eds.,Pharmacotherapy Handbook,2nd Edition,Appleton and Lange,Stamford,Conn.(2000);PDR Pharmacopoeia,Tarascon Pocket Pharmacopoeia 2000,Deluxe Edition,Tarascon Publishing,Loma Linda,Calif.(2000)、および他の医療テキストに見ることができる。しかしながら、第2の治療薬の最適な有効量の範囲を判断するのは当業者の十分な範囲内である。
【0099】
本発明の一実施形態において、第2の治療薬を対象に投与する場合、本発明の化合物の有効量は、第2の治療薬を投与しない場合におけるその有効量未満である。別の実施形態において、第2の治療薬の有効量は、本発明の化合物を投与しない場合におけるその有効量未満である。これにより、高用量のいずれの薬剤に伴う望ましくない副作用を最小限にすることができる。他の潜在的利点(限定的ではないが、投与計画の向上、および/または、薬剤費の削減を含む)は、当業者に明らかになるであろう。
【0100】
さらに別の態様において、本発明は、対象の上述の疾患、疾病、または症状を治療または予防するため、単一組成物または別々の剤形として、薬剤の製造において式I、式IIなどの化合物のみを使用、または、1つ以上の上述の第2の治療薬とともに式I、式IIなどの化合物を使用することを提供する。本発明の別の態様は、対象の本明細書に記載の疾患、疾病、または症状を治療または予防に使用する式I、式IIなどの化合物である。
【0101】
他の態様において、本明細書の方法としては、治療投与に対する対象の反応を監視する工程をさらに含むものが挙げられる。かかる監視としては、治療計画のマーカーまたは指標として、対象の組織、体液、試験片、細胞、タンパク質、化学マーカー、遺伝物質などの周期的サンプリングを含んでもよい。他の方法において、対象は、かかる治療に対する適性の関連マーカーまたは指標を評価することによって、かかる治療が必要であるかプレスクリーニングまたは同定される。
(診断方法およびキット)
【0102】
本発明の化合物および組成物は、溶液または生体試料(例、血漿)中のトルテロジン、5−ヒドロキシメチルトルテロジン、またはフェソテロジンの濃度を判断する方法、トルテロジン、5−ヒドロキシメチルトルテロジン、またはフェソテロジンの代謝を調べる方法、および他の分析研究における試薬としても有用である。
【0103】
一実施形態によれば、本発明は、溶液または生体試料中のトルテロジン、5−ヒドロキシメチルトルテロジン、またはフェソテロジンの濃度を判断する方法を提供し、該方法は:
a)公知濃度の式Iの化合物を生体試料の溶液に加える工程と;
b)式Iの化合物からトルテロジン、5−ヒドロキシメチルトルテロジン、またはフェソテロジンを区別する測定装置に溶液または生体試料をかける工程と;
c)式Iの化合物の検出量と、生体試料または溶液に加えられた式Iの化合物の公知濃度とを相関させる測定装置を較正する工程と;
d)前記較正された測定装置で生体試料中のトルテロジン、5−ヒドロキシメチルトルテロジン、またはフェソテロジンの量を測定する工程と;
e)式Iの化合物に対して得られた検出量および濃度間の相関を用いて、試料の溶液中のトルテロジン、5−ヒドロキシメチルトルテロジン、またはフェソテロジンの濃度を判断する工程とを含む。
【0104】
対応する式Iの化合物からトルテロジン、5−ヒドロキシメチルトルテロジン、またはフェソテロジンを区別することができる測定装置としては、同位体存在度においてのみ互いに異なる2つの化合物を区別することができる任意の測定装置が挙げられる。測定装置の例としては、質量スペクトロメータ、NMRスペクトロメータ、またはIRスペクトロメータが挙げられる。
【0105】
別の実施形態において、本発明は、式Iの化合物をある時間、代謝酵素源と接触させる工程と、前記時間の後、式Iの化合物の量と式Iの化合物の代謝産物を比較する工程とを含む、式Iの化合物の代謝安定性を評価する方法を提供する。
【0106】
関連実施形態において、本発明は、式Iの化合物を投与した後の患者内の式Iの化合物の代謝安定性を評価する方法を提供する。本方法は、式Iの化合物を対象に投与した一定期間の後、患者から血清、尿、または糞便試料を採取する工程と;式Iの化合物の量と血清、尿、または糞便試料における式Iの化合物の代謝産物を比較する工程とを含む。
【0107】
本発明は、過活動膀胱および失禁の治療に用いるキットも提供する。これらのキットは、a)式Iの化合物、またはその塩、水和物、もしくは溶媒和物を含む、入れ物に入った医薬組成物と;b)過活動膀胱および失禁を治療するための医薬組成物の使用方法を記載した説明書と、を含む。
【0108】
入れ物は、前記医薬組成物を保持できる任意の容器あるいは他の密閉されたまたは密閉可能な装置であってよい。例としては、ボトル、アンプル、分割型ボトル、またはマルチチャンバ式ホルダボトル(各区分またはチャンバは単回投与の前記組成物を含む)、分割型ホイルパケット(各区分は単回投与の前記組成物を含む)、または単回投与の前記組成物を分注するディスペンサが挙げられる。入れ物は、医薬的に許容される材料、例えば、紙または段ボール箱、ガラスまたはプラスチックボトルあるいは瓶(jar)、ジッパー付バッグ(例えば、異なる入れ物に配置するための「詰め替え」用錠剤を保持)、または治療スケジュールに従ってパックから押し出す個別用量を備えたブリスターパックからなる当該技術分野で周知の任意の従来の形状または型であってよい。用いる入れ物は、関連する正確な剤形に左右される。例えば、従来の段ボール箱は、液体懸濁液を保持するのには一般に使用されないであろう。単一剤形を販売するため単一包装に複数の入れ物を一緒に使用することも可能である。例えば、錠剤は、ボトルに収容し、次いで、ボックス内に収容してもよい。一実施形態において、入れ物はブリスターパックである。
【0109】
キットは、医師、薬剤師、または対象用の情報および/または説明書を収容するタイプの記憶補助をさらに含んでもよい。かかる記憶補助としては、特定された錠剤またはカプセル剤を服用すべきかの投与計画日数に対応する投与量を含有する各チャンバまたは区分上に印字された数字、各チャンバまたは区分上に印字された週の曜日、または同一種類の情報を含むカードが挙げられる。単回投与用ディスペンサでは、記憶補助としては、さらに、分注された日用量の数を示す機械的計数器や、液晶読み出し信号、および/または、例えば、日用量を最後に摂取した日付を読み出し、および/または、次に用量をいつ摂取するのかを気付かせる可聴リマインダ信号を連結させたバッテリー駆動のマイクロチップメモリが挙げられる。かかるキットに有用な他の記憶補助は、カード上に印字されたカレンダのみならず、容易に明らかとなる他の変形例がある。
【0110】
本発明のキットは、単回投与の医薬組成物を投与するまたは測り分ける機器をさらに含んでいてもよい。かかる機器としては、前記組成物が吸入可能組成物である場合は吸入器;前記組成物が注射用組成物である場合は注射器および針;前記組成物が口腔液体組成物である場合は容量マーキング付または無しの注射器、匙、ポンプ、または容器;あるいは、キット内に存在する組成物の投与製剤に適切な任意の他の計測機器または送達機器が挙げられる。
(実施例)
実施例1.種々の中間体11の合成。
【0111】
A.1−(ベンジルオキシ)−2−ブロモ−4−メチルベンゼン(11a、X=CH3)。
中間体11aは、Akiら、J Phys Chem,2002,106(14):3436−3444の一般手順に従って調製した。
スキーム4a。
【化10】
【0112】
1−(ベンジルオキシ)−2−ブロモ−4−メチルベンゼン(11a、X=CH3)。
アセトン(60mL)中の2−ブロモ−4−メチルフェノール32a(5g、26.7mmol)の溶液にK2CO3(11g、80mmol)を加え、続いて、臭化ベンジル(3.8mL、32mmol)を加えた。混合物を4時間環流加熱した後、室温に冷却した。固体を濾過で除去し、濾液を真空下(in vacuo)で濃縮した。カラムクロマトグラフィー(5% EtOAc/ヘプタン)で残渣を精製し、白色の半固体として11aを6.7g(収率90%)得た。
【0113】
B.1−(ベンジルオキシ)−2−ブロモ−4−(メチル−d3)ベンゼン(11b、X=CH3)。
スキーム4b。
【化11】
【0114】
工程1.1−(ベンジルオキシ)−4−ブロモベンゼン(53)。
アセトン(60mL)中の4−ブロモフェノール(52)(5g、29mmol)の溶液にK2CO3(12.4g、90mmol)を加え、続いて、臭化ベンジル(3.8mL、32mmol)を加えた。混合物を4時間環流加熱した後、室温に冷却した。固体を濾過で除去し、濾液を真空下で濃縮した。カラムクロマトグラフィー(5% EtOAc/ヘプタン)で残渣を精製し、白色固体として53を6.9g(収率90%)得た。
【0115】
工程2.1−(ベンジルオキシ)−4−(メチル−d3)ベンゼン(54)。
THF(無水、40mL)中の1−(ベンジルオキシ)−4−ブロモベンゼン(53)(5g、19mmol)の溶液を−78℃に冷却した後、n−BuLi(2.5Mヘキサン中に9.1mL、22.8mmol)を滴下した。反応混合物を30分間攪拌した後、CD3I[ケンブリッジアイソトープ、99原子%D](1.4mL、22.8mmol)を加えた。混合物を1時間攪拌した後、水性NH4Cl(飽和)を加えて急冷した。混合物をEtOAc(100mL)で抽出し、有機層をブラインで洗浄し、Na2SO4で乾燥させ、濾過し、真空下で濃縮し、カラムクロマトグラフィー(5% EtOAc/ヘプタン)で精製し、黄色油として54を3.05g(収率80%)得た。
【0116】
工程3.1−(ベンジルオキシ)−2−ブロモ−4−(メチル−d3)ベンゼン(11b、X=CD3)。
アセトニトリル(50mL)中の54(3.0g、15mmol)の溶液にN−ブロモスクシンイミド(3.2g、18mmol)を加え、続いて、NH4OAc(3.5g、45mmol)を加えた。混合物を3時間攪拌した後、水(30mL)を加え、混合物をEtOAc(150mL)で抽出した。有機層をブラインで洗浄し、Na2SO4で乾燥させ、濾過し、真空下で濃縮し、カラムクロマトグラフィー(5% EtOAc/ヘプタン)で精製し、黄色油として11bを3.6g(収率85%)得た。
【0117】
C.1−(ベンジルオキシ)−4−(ベンジルオキシメチル)−2−ブロモベンゼン(11c、X=CH2OBn)。
中間体11cは、Akiら、J Phys Chem,2002,106(14):3436−3444の一般手順に従って調製した。
スキーム4c。
【化12】
【0118】
工程1.4−(ベンジルオキシ)−3−ブロモ安息香酸ベンジル(45)。
アセトン(200mL)中の3−ブロモ−4−ヒドロキシ安息香酸44(10g、46mmol)の溶液にK2CO3(20g、145mmol)および臭化ベンジル(12mL、101mmol)を加えた。混合物を5時間環流加熱した後、室温に冷却した。固体を濾過で除去し、濾液を真空下で濃縮した。残渣をEtOAc(150mL)中に溶解させ、溶液をブラインで洗浄し、Na2SO4で乾燥させ、濾過し、真空下で濃縮し、シロップ状の残渣を得た。残渣を5% EtOAc/ヘプタンから再結晶化させ、白色固体として45を16.4g(収率90%)得た。
【0119】
工程2.(4−(ベンジルオキシ)−3−ブロモフェニル)メタノール(46a)。
4−(ベンジルオキシ)−3−ブロモ安息香酸ベンジル(45)(15g、37.8mmol)をドライTHF(150mL)中に溶解させ、0℃に冷却した。LiAlH4(2.9g、76mmol)を3分割してゆっくりと加えた。混合物を室温で12時間攪拌した後、H2O(3mL)、15% NaOH(3mL)、およびH2O(7.6mL)を加えて急冷した。沈殿物を濾過で除去し、EtOAcで洗浄した。組み合わせた濾液を濃縮し、無色油を得た。これをカラムクロマトグラフィー(5%〜10% EtOAc/ヘプタン)で精製し、無色油として46aを9.4g(収率84%)得た。
【0120】
工程3.1−(ベンジルオキシ)−4−(ベンジルオキシメチル)−2−ブロモベンゼン(11c、X=CH2OBn)。
アセトン(100mL)中の(4−(ベンジルオキシ)−3−ブロモフェニル)メタノール(46a)(9g、30.7mmol)の溶液にK2CO3(12.7g、92mmol)および臭化ベンジル(4mL、33.8mmol)を加えた。混合物を5時間攪拌した後、室温に冷却した。固体を濾過で除去し、濾液を真空下で残渣に濃縮し、これをカラムクロマトグラフィー(5% EtOAc/ヘプタン)で精製し、無色油として11cを10.6g(収率90%)得た。
【0121】
D.1−(ベンジルオキシ)−4−(ベンジルオキシ(メチル−d2))−2−ブロモベンゼン(11d、X=CH2OBn)。
スキーム4d。
【化13】
【0122】
工程1.(4−(ベンジルオキシ)−3−ブロモフェニル)−1,1−d2−メタノール(46b)。
ドライTHF(150mL)中の4−(ベンジルオキシ)−3−ブロモ安息香酸ベンジル(45)(15g、37.8mmol)の0℃の溶液にLiAlD4(ケンブリッジアイソトープ、99原子%D)(3.1g、75.6mmol)を3分割してゆっくりと加えた。混合物を室温で12時間攪拌した後、H2O(3mL)、15% NaOH(3mL)、およびH2O(7.6mL)を加えて急冷した。沈殿物を濾過し、EtOAcで洗浄した。濾液を真空下で濃縮し、無色油を得た。これをカラムクロマトグラフィー(5%〜10% EtOAc/ヘプタン)で精製し、無色油として46bを9.0g(収率81%)得た。
【0123】
工程2.1−(ベンジルオキシ)−4−(ベンジルオキシ(メチル−d2))−2−ブロモベンゼン(11d、X=CH2OBn)。
アセトン(100mL)中の(4−(ベンジルオキシ)−3−ブロモフェニル)−1,1−d2−メタノール(46b)(9.0g、30.5mmol)の溶液にK2CO3(12.7g、92mmol)および臭化ベンジル(4mL、33.8mmol)を加えた。混合物を5時間攪拌し、室温に冷却した。固体を濾過で除去した。濾液を真空下で濃縮し、カラムクロマトグラフィー(5% EtOAc/ヘプタン)で残渣を精製し、無色油として11dを10.4g(収率88%)得た。
実施例2.(R)−2−(3−(ジ(イソプロピル−d7)アミノ)−1−フェニルプロピル)−4−メチルフェノール(化合物106)の合成。
【0124】
適当に重水素化された試薬を用いて、スキーム1で概説したように化合物106を調製した。
【化14】
【0125】
工程1.(R)−3−((R)−3−(2−(ベンジルオキシ)−5−メチルフェニル)−3−フェニルプロパノイル)−4−フェニルオキサゾリジン−2−オン(12a、X=CH3)。
THF(無水、20mL)中の11a(5g、18mmol、実施例1aを参照されたい)の溶液にMg(648mg、27mmol)を加えた。小片のヨウ素を加え、混合物を80℃で1時間加熱した後、−50℃に冷却した。CuBr−Me2S(3.5g、9mmol)を加えた。混合物を−30℃で1時間攪拌した後、THF(無水、40mL)中の(R)−3−シンナモイル−4−フェニルオキサゾリジン−2−オン(10)(6.3g、21.6mmol、Andersson,PGら、J Org Chem,1998,63(22):8067およびNicolas,Eら、J Org Chem,1993,58(3):766−770に記載のように調製した)の溶液を加えた。混合物を室温にゆっくりと温めた後、さらに6時間攪拌し、水性NH4Cl(飽和)で急冷し、EtOAc(200mL)で抽出した。有機層をブラインで洗浄し、Na2SO4で乾燥させ、濾過し、真空下で濃縮した。カラムクロマトグラフィー(20% EtOAc/ヘプタン)で残渣を精製し、白色固体として12aを7.1g(収率80%)得た。
【0126】
工程2.(R)−3−(2−(ベンジルオキシ)−5−メチルフェニル)−3−フェニルプロパン酸(13a、X=CH3)。
THF(30mL)中の12a(4g、8.1mmol)の溶液に水(10mL)を加えた。混合物を0℃に冷却し、30% H2O2溶液(7.2mL、64mmol)を加え、続いて、LiOH(388mg、16.2mmol)を加えた。混合物を室温で4時間攪拌し、4N HClでpH=2に酸性化した後、EtOAc(50mL×3)で抽出した。有機層をNa2SO4で乾燥させ、濾過し、真空下で濃縮し、カラムクロマトグラフィー(20〜50% EtOAc/ヘプタン)で残渣を精製し、白色固体として13aを2.3g(収率83%)得た。
【0127】
工程3.(R)−3−(2−(ベンジルオキシ)−5−メチルフェニル)−3−フェニルプロパノイルクロリド(14a、X=CH3)。
トルエン(10mL)中の13a(1.1g、2.9mmol)の溶液に塩化チオニル(1.1mL、15mL)を加え、続いて、ドライピリジンを1滴加えた。混合物を1時間環流加熱した。塩化チオニルを真空下で除去した。ドライエーテル(20mL)を加え、得られた固体を濾過で除去した。濾液を真空下で濃縮し、淡黄色固体として粗14aを1.1g得た。
【0128】
工程4.(R)−3−(2−(ベンジルオキシ)−5−メチルフェニル)−N,N−ジ(イソプロピル−d7)−3−フェニルプロパンアミド(15a、X=CH3;R2=R3=CD(CD3)2)。
ドライエーテル(20mL)中の粗14a(1.1g)の溶液にジイソプロピルアミン−d14[CDNアイソトープ、98原子%D](517mg、4.5mmol)を加え、続いて、Et3N(1.25mL、9mmol)を加えた。混合物を4時間攪拌し、2N HCl(15mL)を加えて急冷した後、EtOAc(50mL)で抽出した。有機層を水性NaHCO3(飽和)、ブラインで洗浄し、Na2SO4で乾燥させ、濾過し、真空下で濃縮し、残渣をカラムクロマトグラフィー(30% EtOAc/ヘプタン)で精製し、無色油として15aを1.1g(収率86%)得た。
【0129】
工程5.(R)−3−(2−(ベンジルオキシ)−5−メチルフェニル)−N,N−ジ(イソプロピル−d7)−3−フェニルプロパン−1−アミン(16a、X=CH3;R2=R3=CD(CD3)2)。
THF(無水、15mL)中の15a(1.1g、2.48mmol)の0℃の溶液にLiAlH4(190mg、5.0mmol)を加えた。混合物を室温で14時間攪拌した後、H2O(0.2mL)、15% NaOH溶液(0.2mL)、およびH2O(0.5mL)を加えて急冷した。沈殿物を濾過し、EtOAcで洗浄した。濾液を真空下で濃縮し、無色油として16aを860mg(収率80%)得た。
【0130】
工程6.(R)−2−(3−(ジ(イソプロピル−d7)アミノ)−1−フェニルプロピル)−4−メチルフェノールL−タートレート(そのL−酒石酸塩として化合物106)。
メタノール(20mL)中の16a(850mg、1.97mmol)の溶液に10% Pd/C(50%水で2.1g、1.0mmol)を加えた。混合物を水素下(2atm)で10時間攪拌し、セリットパッドを通して濾過し、濾液を真空下で濃縮した。カラムクロマトグラフィー(5% MeOH/CH2Cl2中の2% Et3N)で残渣を精製し、その遊離塩基として化合物106を570mg(収率85%)得た。遊離塩基(570mg、1.68mmol)を10mLのエタノール(無水)中に溶解させ、続いて、L−酒石酸(277mg、1.85mmol)を加えた。混合物を1時間環流加熱した後、冷蔵庫で10時間保持した。得られた塩を濾過し、冷却エタノールで洗浄し、L−酒石酸塩として化合物106を820mg得た。1H−NMR(300MHz、DMSO−d6):δ 2.17(s、3H)、2.28〜2.33(m、2H)、2.67〜2.75(m、2H)、3.96(s、2H)、4.30(t、J=7.8、1H)、6.67(d、J=7.9、1H)、6.80(dd、J1=8.2、J2=1.8、1H)、7.02(d、J=2.0、1H)、7.13〜7.19(m、1H)、7.25〜7.33(m、4H)。13C−NMR(75MHz、DMSO−d6):δ 21.02、41.50、45.67、72.14、115.73、126.66、128.00、128.12、128.50、128.58、128.87、130.30、144.74、152.98、174.69。HPLC(方法:Waters Atlantis T3 2.1×50mm 3μm C18−RPカラム−傾斜法5〜95% ACN+0.1%ギ酸で14分(1.0mL/分)、95% ACNで4分保持した;波長:254nm):保持時間:4.88分;99+%純度。キラルHPLC(方法:250mm×4.6mmキラルODカラム−定組成法95%ヘキサン/5%イソプロパノールで40分間(0.50mL/分);波長:210nm):保持時間:8.84分(主鏡像異性体);11.59分(副鏡像異性体);99.4% ee純度。MS(M+H):340.2。元素分析(C26H23D14NO7):計算値:C=63.79、H=7.62、N=2.86。実測値:C=63.79、H=7.55、N=2.86。
実施例3.(R)−2−(3−(ジイソプロピル)アミノ)−1−フェニルプロピル)−4−(メチル−d3)フェノール(化合物112)の合成。
【0131】
適当に重水素化された試薬を用いて、スキーム1で概説したように化合物112を調製した。
【化15】
【0132】
工程1.(R)−3−((R)−3−(2−(ベンジルオキシ)−5−(メチル−d3)フェニル)−3−フェニルプロパノイル)−4−フェニルオキサゾリジン−2−オン(12b、X=CD3)。
THF(無水、20mL)中の1−((2−ブロモ−4−(メチル−d3)−フェノキシ)メチル)ベンゼン(11b)(3.5g、12.5mmol、実施例1bを参照されたい)の溶液にMg(450mg、18.8mmol)を加えた。小片のヨウ素を加え、混合物を80℃で1時間加熱した後、−50℃に冷却した。CuBr−Me2S(1.3g、6.3mmol)を加えた。混合物を−30℃で1時間攪拌した後、THF(無水、40mL)中の(R)−3−シンナモイル−4−フェニルオキサゾリジン−2−オン(10)(5.5g、18.75mmol、実施例2、工程1を参照されたい)の溶液を加えた。混合物を室温にゆっくりと温め、さらに6時間攪拌し、水性NH4Cl(飽和)を加えて急冷し、EtOAc(200mL)で抽出した。有機層をブラインで洗浄し、Na2SO4で乾燥させ、濾過し、濾液を真空下で濃縮した。カラムクロマトグラフィー(20% EtOAc/ヘプタン)で残渣を精製し、白色固体として12bを5.2g(収率83%)得た。
【0133】
工程2.(R)−3−(2−(ベンジルオキシ)−5−(メチル−d3)−フェニル)−3−フェニルプロパン酸(13b、X=CD3)。
THF(30mL)中の12b(4g、8.1mmol)の溶液に水(10mL)を加えた。混合物を0℃に冷却し、30% H2O2溶液(7.2mL、64mmol)を加え、続いて、LiOH(388mg、16.2mmol)を加えた。混合物を室温で4時間攪拌し、4N HClでpH=2に酸性化し、EtOAc(50mL×3)で抽出した。有機抽出物を組み合わせ、Na2SO4で乾燥させ、濾過し、濾液を真空下で濃縮した。カラムクロマトグラフィー(20〜50% EtOAc/ヘプタン)で残渣を精製し、白色固体として13bを2.3g(収率83%)得た。
【0134】
工程3.(R)−3−(2−(ベンジルオキシ)−5−(メチル−d3)−フェニル)−3−フェニルプロパノイルクロリド(14b、X=CD3)。
トルエン(10mL)中の13b(1.2g、3.0mmol)に塩化チオニル(1.1mL、15mL)を加え、続いて、ドライピリジンを1滴加えた。混合物を1時間環流加熱した。過剰の塩化チオニルを真空下で除去した。ドライエーテル(20mL)を加え、得られた固体を濾過で除去した。濾液を真空下で濃縮し、淡黄色固体として粗14bを1.1g得た。
【0135】
工程4.(R)−3−(2−(ベンジルオキシ)−5−(メチル−d3)−フェニル)−N,N−ジイソプロピル−3−フェニルプロパンアミド(15b、X=CD3、R2=R3=CH(CH3)2)。
ドライエーテル(20mL)中の粗14b(1.1g)の溶液にジイソプロピルアミン(2.1mL、15mmol)を加えた。反応混合物を4時間攪拌し、2N HCl(15mL)を加えて急冷し、EtOAc(50mL)で抽出した。有機層を水性NaHCO3(飽和)、ブラインで洗浄し、Na2SO4で乾燥させ、濾過し、濾液を真空下で濃縮した。カラムクロマトグラフィー(30% EtOAc/ヘプタン)で残渣を精製し、無色油として15bを1.1g(収率89%)得た。
【0136】
工程5.(R)−3−(2−(ベンジルオキシ)−5−(メチル−d3)−フェニル)−N,N−ジイソプロピル−3−フェニルプロパン−1−アミン(16b、X=CD3、R2=R3=CH(CH3)2)。
THF(無水、15mL)中の15b(1.1g、2.6mmol)の0℃の溶液にLiAlH4(210mg、5.2mmol)を加えた。混合物を室温で14時間攪拌した後、H2O(0.21mL)、15% NaOH溶液(0.21mL)、およびH2O(0.52mL)を加えて急冷した。沈殿物を濾過し、EtOAcで洗浄した。濾液を濃縮し、無色油として16bを880mg(収率80%)得た。
【0137】
工程6.2−((R)−3−(ジイソプロピルアミノ)−1−フェニルプロピル)−4−メチルフェノールL−タートレート(そのL−酒石酸塩として化合物112)。
メタノール(20mL)中の16b(870mg、2.08mmol)の溶液に10% Pd/C(50%水で2.1g、1.0mmol)を加えた。混合物を水素下(2atm)で10時間攪拌した後、セリットパッドを通して濾過し、真空下で濃縮した。カラムクロマトグラフィー(5% MeOH/CH2Cl2中の2% Et3N)で残渣を精製し、その遊離塩基として化合物112を560mg(収率81%)得た。遊離塩基(560mg、1.7mmol)を10mLのエタノール(無水)中に溶解させ、続いて、L−酒石酸(280mg、1.87mmol)を加えた。混合物を1時間環流加熱した後、冷蔵庫で10時間保持した。得られた塩を濾過し、冷却エタノールで洗浄し、L−酒石酸塩として化合物112を800mg得た。1H−NMR(300MHz、DMSO−d6):δ 1.08(d、J=6.6、12H)、2.15〜2.17(m、0.6H)、2.26〜2.32(m、2H)、2.65〜2.73(m、2H)、3.37〜3.45(m、2H)、3.98(s、2H)、4.30(t、J=7.8、1H)、6.66(d、J=8.0、1H)、6.80(dd、J1=8.2、J2=2.2、1H))、7.01(d、J=2.1、1H)、7.13〜7.19(m、1H)、7.24〜7.33(m、4H)。13C−NMR(75MHz、DMSO−d6):δ 18.91、33.71、41.49、45.60、52.73、72.18、115.72、126.63、127.88、128.09、128.50、128.58、128.86、130.37、144.82、152.98、174.64。HPLC(方法:Waters Atlantis T3 2.1×50mm 3μm C18−RPカラム−傾斜法5−95% ACN+0.1%ギ酸で14分(1.0mL/分)、95% ACNで4分保持した;波長:254nm):保持時間:4.87分;99+%純度。キラルHPLC(方法:250mm×4.6mmキラルODカラム−定組成法95%ヘキサン/5%イソプロパノールで40分間(0.50mL/分);波長:210nm):保持時間:8.88分(主鏡像異性体);11.62分(副鏡像異性体);99.8% ee純度。MS(M+H):329.1。元素分析(C26H34D3NO7):計算値:C=62.25、H=7.79、N=2.93。実測値:C=65.11、H=7.76、N=2.91。
実施例4.(R)−2−(3−(ジ(イソプロピル−d7))アミノ)−1−フェニルプロピル)−4−(メチル−d3)フェノール(化合物100)の合成。
【0138】
適当に重水素化された試薬を用いて、スキーム1で概説したように化合物100を調製した。
【化16】
【0139】
工程1.(R)−3−(2−(ベンジルオキシ)−5−(メチル−d3)−フェニル)−N,N−ジ(イソプロピル−d7)−3−フェニルプロパンアミド(15e、X=CD3、R2=R3=CD(CD3)2)。
ドライエーテル(20mL)中の粗14b(1.1g)(実施例3、工程3を参照)の溶液にジイソプロピルアミン−d14[CDNアイソトープ、99原子%D](517mg、4.5mmol)を加え、続いて、Et3N(1.25mL、9mmol)を加えた。混合物を4時間攪拌し、2N HCl(15mL)を加えて急冷した後、EtOAc(50mL)で抽出した。有機層を水性NaHCO3(飽和)、ブラインで洗浄し、Na2SO4で乾燥させ、濾過し、真空下で濃縮した。カラムクロマトグラフィー(30% EtOAc/ヘプタン)で残渣を精製し、無色油として15eを1.06g(収率83%)得た。
【0140】
工程2.(R)−3−(2−(ベンジルオキシ)−5−(メチル−d3)−フェニル)−N,N−ジ(イソプロピル−d7)−3−フェニルプロパン−1−アミン(16e、X=CD3、R2=R3=CD(CD3)2)。
15e(1.0g、2.47mmol)の溶液をTHF(無水、15mL)中に溶解させ、0℃に冷却し、続いて、LiAlH4(190mg、5.0mmol)を加えた。混合物を室温で14時間攪拌した後、H2O(0.2mL)、15% NaOH溶液(0.2mL)、およびH2O(0.5mL)を加えて急冷した。沈殿物を濾過し、EtOAcで洗浄した。濾液を真空下で濃縮し、無色油として16eを850mg(収率79%)得た。
【0141】
工程3.(R)−2−(3−(ジ(イソプロピル−d7))アミノ)−1−フェニルプロピル)−4−(メチル−d3)フェノール(化合物100)。
メタノール(20mL)中の16e(850mg、1.97mmol)の溶液に10% Pd/C(50%水で2.1g、1.0mmol)を加えた。混合物を水素下(2atm)で10時間攪拌した後、セリットパッドを通して濾過した。濾液を真空下で濃縮し、カラムクロマトグラフィー(5% MeOH/CH2Cl2中の2% Et3N)で残渣を精製し、遊離塩基として化合物100を550mg(収率80%)得た。遊離塩基(550mg、1.61mmol)を10mLのエタノール(無水)中に溶解させ、続いて、L−酒石酸(266mg、1.77mmol)を加えた。混合物を1時間環流加熱した後、冷蔵庫で10時間保持した。得られた塩を濾過し、冷却エタノールで洗浄し、L−酒石酸塩として化合物100を790mg得た。1H−NMR(300MHz、DMSO−d6):δ 1.04〜1.07(m、0.7H)、2.16〜2.18(m、0.7H)、2.26〜2.28(m、2H)、2.63〜2.75(m、2H)、3.43(微量)、3.95(s、2H)、4.30(t、J=7.8、1H)、6.67(d、J=7.9、1H)、6.81(dd、J1=8.0、J2=2.0、1H))、7.02(d、J=1.8、1H)、7.14〜7.19(m、1H)、7.25〜7.33(m、4H)。13C−NMR(75MHz、DMSO−d6):δ 41.48、45.60、72.02、115.72、126.65、128.00、128.11、128.50,128.58、128.87、130.31、144.78、152.97、174.61。HPLC(方法:Waters Atlantis T3 2.1×50mm 3μm C18−RPカラム−傾斜法5−95% ACN+0.1%ギ酸で14分(1.0mL/分)、95% ACNで4分保持した;波長:254nm):保持時間:4.79分;99+%純度。キラルHPLC(方法:250mm×4.6mmキラルODカラム−定組成法95%ヘキサン/5%イソプロパノールで40分間(0.50mL/分);波長:210nm):保持時間:8.87分(主鏡像異性体);11.61分(副鏡像異性体);99.6% ee純度。MS(M+H):343.2。元素分析(C26H20DI7NO7):計算値:C=63.40、H=7.57、N=2.84。実測値:C=63.54、H=7.73、N=2.77。
実施例5.(R)−2−(3−(ジ(イソプロピル−d7))アミノ)−1−フェニルプロピル)−4−(ヒドロキシメチル)フェノール(化合物109)の合成。
【0142】
適当に重水素化された試薬を用いて、スキーム1で概説したように化合物109を調製した。
【化17】
【0143】
工程1.(R)−3−((R)−3−(2−(ベンジルオキシ)−5−(ベンジルオキシメチル)フェニル)−3−フェニルプロパノイル)−4−フェニルオキサゾリジン−2−オン(12c、X=CH2OBn)。
THF(無水、20mL)中の11c(5g、13mmol、実施例1cを参照)の溶液にMg(480mg、20mmol)を加えた。小片のヨウ素を加え、混合物を80℃で1時間加熱した後、−50℃に冷却した。CuBr−Me2S(1.4g、7mmol)を加えた。混合物を−30℃で1時間攪拌した後、THF(無水、40mL)中の(R)−3−シンナモイル−4−フェニルオキサゾリジン−2−オン(10)(5.5g、19mmol、実施例2、工程1を参照されたい)の溶液を加えた。混合物を室温にゆっくりと温め、さらに6時間攪拌した後、水性NH4Cl(飽和)を加えて急冷し、EtOAc(200mL)で抽出した。有機層をブラインで洗浄し、Na2SO4で乾燥させ、濾過し、真空下で濃縮した。カラムクロマトグラフィー(20% EtOAc/ヘプタン)で残渣を精製し、白色固体として12cを6.0g(収率77%)得た。
【0144】
工程2.(R)−3−(2−(ベンジルオキシ)−5−(ベンジルオキシメチル)フェニル)−3−フェニルプロパン酸(13c、X=CH2OBn)。
THF(30mL)中の12c(4.8g、8.1mmol)の溶液に水(10mL)を加えた。混合物を0℃に冷却し、30% H2O2溶液(7.2mL、64mmol)を加え、続いて、LiOH(388mg、16.2mmol)を加えた。混合物を室温で4時間攪拌し、4N HClでpH=2に酸性化し、EtOAc(50mL×3)で抽出した。有機抽出物を組み合わせ、Na2SO4で乾燥させ、濾過し、濾液を真空下で濃縮した。カラムクロマトグラフィー(20〜50% EtOAc/ヘプタン)で残渣を精製し、白色固体として13cを2.95g(収率80%)得た。
【0145】
工程3.(R)−3−(2−(ベンジルオキシ)−5−(ベンジルオキシメチル)フェニル)−3−フェニルプロパノイルクロリド(14c、X=CH2OBn)。
トルエン(10mL)中の13c(1.3g、2.9mmol)に塩化チオニル(1.1mL、15mL)を加え、続いて、ドライピリジンを1滴加えた。混合物を1時間環流加熱した。塩化チオニルを真空下で除去した。ドライエーテル(20mL)を加えた。得られた固体を濾過で除去した。溶液を真空下で濃縮し、淡黄色固体として粗14cを1.3g得た。
【0146】
工程4.(R)−3−(2−(ベンジルオキシ)−5−(ベンジルオキシメチル)フェニル)−N,N−ジ(イソプロピル−d7)−3−フェニルプロパンアミド(15c、X=CH2OBn、R2=R3=CD(CD3)2)。
ドライエーテル(20mL)中の粗14c(1.3g)の溶液にジイソプロピルアミン−d14[CDNアイソトープ、98原子%D](500mg、4.35mmol)を加え、続いて、Et3N(1.25mL、9mmol)を加えた。混合物を4時間攪拌し、2N HCl(15mL)を加えて急冷し、EtOAc(50mL)で抽出した。有機層を水性NaHCO3(飽和)、ブラインで洗浄し、Na2SO4で乾燥させ、濾過し、濾液を真空下で濃縮した。カラムクロマトグラフィー(30% EtOAc/ヘプタン)で残渣を精製し、無色油として15cを1.2g(収率75%)得た。
【0147】
工程5.(R)−3−(2−(ベンジルオキシ)−5−(ベンジルオキシメチル)フェニル)−N,N−ジ(イソプロピル−d7)−3−フェニルプロパン−1−アミン(16c、X=CH2OBn、R2=R3=CD(CD3)2)。
THF(無水、15mL)中の15c(1.2g、2.2mmol)の溶液を0℃に冷却し、続いて、LiAlH4(167mg、4.4mmol)を加えた。混合物を室温で14時間攪拌した後、H2O(0.18mL)、15% NaOH溶液(0.18mL)、およびH2O(0.44mL)を加えて急冷した。沈殿物を濾過し、EtOAcで洗浄した。濾液を真空下で濃縮し、残渣をカラムクロマトグラフィー(50% EtOAc/ヘプタン中の5% Et3N)で精製し、無色油として16cを920mg(収率78%)得た。
【0148】
工程6.(R)−2−(3−(ジ(イソプロピル−d7))アミノ)−1−フェニルプロピル)−4−(ヒドロキシメチル)フェノール(化合物109)。
ジクロロメタン(20mL)中の16c(910mg、1.7mmol)の−78℃の溶液にBCl3(1Mジクロロメタン中の5.9mL、5.9mmol)を滴下した。混合物を3時間攪拌した後、H2Oを加えて急冷し、室温に温めた。混合物を水性NaHCO3(飽和)でpH=9にし、ジクロロメタンで抽出した。有機層をNa2SO4で乾燥させ、濾過し、濾液を真空下で濃縮した。カラムクロマトグラフィー(5%メタノール/ジクロロメタン中の5% Et3N)で残渣を精製し、黄色油として化合物109を440mg(収率72%)得た。
実施例6.(R)−2−(3−(ジ(イソプロピル−d7))アミノ)−1−フェニルプロピル)−4−(ヒドロキシメチル)フェニルイソブチレート(化合物117)の合成。
【0149】
化合物117は、スキーム1に概説したように、化合物109から調製した。
【化18】
【0150】
(R)−2−(3−(ジ(イソプロピル−d7))アミノ)−1−フェニルプロピル)−4−(ヒドロキシメチル)フェニルイソブチレート(化合物117)。
ジクロロメタン(10mL)中の化合物109(440mg、1.24mmol)の−30℃の溶液にジイソプロピルエチルアミン(0.23mL、1.36mmol)を加え、続いて、塩化イソブチリル(0.13mL、1.24mmol)を加えた。反応混合物を−30℃で3時間攪拌した後、水を加えて急冷し、ジクロロメタンで抽出した。有機層をNa2SO4で乾燥させ、濾過し、濾液を真空下で濃縮した。カラムクロマトグラフィー(20% EtOAc/ヘプタン中の5% Et3N、その後、5%メタノール/ジクロロメタン中の5% Et3N)で残渣を精製し、黄色油として化合物117を370mg(収率70%)得た。1H−NMR(300MHz、CDCl3):δ 0.92(m、0.3H)、1.30(d、J=11.1、3H)、1.33(d、J=11.1、3H)、2.07〜2.18(m、2H)、2.22〜2.41(m、2H)、2.76〜2.85(m、1H)、4.12(t、J=7.4、1H)、4.63(s、2H)、6.97(d、J=8.2、1H)、7.00〜7.31(m、6H)、7.34(d、J=1.5、1H)。13C−NMR(75MHz、CDCl3):δ 18.97、19.10、34.20、36.90、41.77、43.83、64.90、122.60、125.60、126.14、126.89、127.92、128.33、136.95、138.64、143.87、147.94、175.32。HPLC(方法:20mm C18−RPカラム−傾斜法2−95% ACN+0.1%ギ酸で3.3分、95% ACNで1.7分保持した;波長:210nm):保持時間:2.89分;90%純度。MS(M+H):426.3。
実施例7.(R)−2−(3−(ジイソプロピルアミノ)−1−フェニルプロピル)−4−(ヒドロキシル(メチル−d2))フェノール(化合物113)の合成。
【0151】
適当に重水素化された試薬を用いて、スキーム1で概説したように化合物113を調製した。
【化19】
【0152】
工程1.(R)−3−((R)−3−(2−(ベンジルオキシ)−5−(ベンジルオキシ(メチル−d2))フェニル)−3−フェニルプロパノイル)−4−フェニルオキサゾリジン−2−オン(12d、X=CD2OBn)。
THF(無水、20mL)中の11d(5g、13mmol、実施例1dを参照)の溶液にMg(480mg、20mmol)を加えた。小片のヨウ素を加え、混合物を80℃で1時間加熱した後、−50℃に冷却した。CuBr−Me2S(1.4g、7mmol)を加えた。混合物を−30℃で1時間攪拌した後、THF(無水、40mL)中の(R)−3−シンナモイル−4−フェニルオキサゾリジン−2−オン(10)(5.5g、19mmol、実施例2、工程1を参照されたい)の溶液を加えた。混合物を室温にゆっくりと温め、さらに6時間攪拌し、水性NH4Cl(飽和)を加えて急冷し、EtOAc(200mL)で抽出した。有機層をブラインで洗浄し、Na2SO4で乾燥させ、濾過し、濾液を真空下で濃縮した。カラムクロマトグラフィー(20% EtOAc/ヘプタン)で残渣を精製し、白色固体として12dを5.8g(収率74%)得た。
【0153】
工程2.(R)−3−(2−(ベンジルオキシ)−5−(ベンジルオキシ(メチル−d2))フェニル)−3−フェニルプロパン酸(13d、X=CD2OBn)。
THF(30mL)中の12d(5.0g、8.3mmol)の溶液に水(10mL)を加えた。混合物を0℃に冷却し、30% H2O2溶液(7.2mL、64mmol)を加え、続いて、LiOH(388mg、16.2mmol)を加えた。混合物を室温で4時間攪拌し、4N HClでpH=2に酸性化し、EtOAc(50mL×3)で抽出した。有機抽出物を組み合わせ、Na2SO4で乾燥させ、濾過し、濾液を真空下で濃縮した。カラムクロマトグラフィー(20〜50% EtOAc/ヘプタン)で残渣を精製し、白色固体として13dを2.82g(収率75%)得た。
【0154】
工程3.(R)−3−(2−(ベンジルオキシ)−5−(ベンジルオキシ(メチル−d2))フェニル)−3−フェニルプロパノイルクロリド(14d、X=CD2OBn)。
トルエン(10mL)中の13d(1.4g、3.1mmol)に塩化チオニル(1.1mL、15mmol)を加え、続いて、ドライピリジンを1滴加えた。混合物を1時間環流加熱した。過剰の塩化チオニルを真空下で除去した。ドライエーテル(20mL)を加えた。得られた固体を濾過で除去した。溶液を濃縮し、淡黄色固体として粗14dを1.3g得た。
【0155】
工程4.(R)−3−(2−(ベンジルオキシ)−5−(ベンジルオキシ(メチル−d2))フェニル)−N,N−ジイソプロピル−3−フェニルプロパンアミド(15d、X=CD2OBn)。
ドライエーテル(20mL)中の14dの溶液にジイソプロピルアミン(2.1mL、15mmol)を加えた。混合物を4時間攪拌し、2N HCl(15mL)を加えて急冷し、EtOAc(50mL)で抽出した。有機層をNaHCO3(飽和)、ブラインで洗浄し、Na2SO4で乾燥させ、濾過し、濾液を真空下で濃縮した。カラムクロマトグラフィー(30% EtOAc/ヘプタン)で残渣を精製し、無色油として15dを1.25g(収率75%)得た。
【0156】
工程5.(R)−3−(2−(ベンジルオキシ)−5−(ベンジルオキシメチル−d2)フェニル)−N,N−ジイソプロピル−3−フェニルプロパン−1−アミン(16d、X=CD2OBn)。
THF(無水、15mL)中の15d(1.2g、2.2mmol)の0℃の溶液にLiAlH4(170mg、4.4mmol)を加えた。混合物を室温で14時間攪拌した後、H2O(0.18mL)、15% NaOH溶液(0.18mL)、およびH2O(0.44mL)で急冷した。沈殿物を濾過し、EtOAcで洗浄した。有機層を真空下で濃縮し、残渣をカラムクロマトグラフィー(50%EtOAc/ヘプタン中の5% Et3N)で精製し、無色油として16dを920mg(収率80%)得た。
【0157】
工程6.(R)−2−(3−(ジイソプロピルアミノ)−1−フェニルプロピル)−4−(ヒドロキシル(メチル−d2))フェノール(化合物113)。
ジクロロメタン(20mL)中の16d(920mg、1.76mmol)の−78℃の溶液にBCl3(1Mジクロロメタン中の6.3mL、6.3mmol)を滴下した。混合物を3時間攪拌した後、H2Oで急冷し、室温に温めた。混合物をNaHCO3(飽和)でpH=9にし、ジクロロメタンで抽出した。有機層をNa2SO4で乾燥させ、濾過し、濾液を真空下で濃縮した。カラムクロマトグラフィー(5%メタノール/ジクロロメタン中の5% Et3N)で残渣を精製し、黄色油として化合物113を425mg(収率70%)得た。
実施例8.(R)−2−(3−(ジイソプロピルアミノ)−1−フェニルプロピル)−4−(ヒドロキシル(メチル−d2))フェニルイソブチレート(化合物116)の合成。
【0158】
化合物116は、スキーム1に概説したように、化合物113から調製した。
【化20】
【0159】
(R)−2−(3−(ジイソプロピルアミノ)−1−フェニルプロピル)−4−(ヒドロキシル(メチル−d2))フェニルイソブチレート(化合物116)。
ジクロロメタン(10mL)中の化合物113(420mg、1.22mmol)の溶液を−30℃に冷却した後、ジイソプロピルエチルアミン(0.23mL、1.35mmol)を加え、続いて、塩化イソブチリル(0.13mL、1.23mmol)を加えた。混合物を−30℃で3時間攪拌した後、水で急冷し、ジクロロメタンで抽出し、Na2SO4で乾燥させ、濾過し、濾液を真空下で濃縮した。カラムクロマトグラフィー(20% EtOAc/ヘプタン中の5% Et3N、その後、5%メタノール/ジクロロメタン中の5% Et3N)で残渣を精製し、黄色油として化合物116を360mg(収率70%)得た。1H−NMR(300MHz、CDCl3):δ 0.91(d、J=6.4、12H)、1.30(d、J=11.1、3H)、1.32(d、J=11.1、3H)、2.08〜2.17(m、2H)、2.30〜2.37(m、2H)、2.76〜2.85(m、1H)、2.91〜3.00(m、2H)、4.11(t、J=7.6、1H)、6.96(d、J=8.2、1H)、7.12〜7.32(m、6H)、7.34(d、J=2.3、1H)。13C−NMR(75MHz、CDCl3):δ 18.98、19.10、20.59、34.20、36.93、41.77、43.82、48.71、122.62、125.65、126.14、126.95、127.92、128.34、136.98、138.48、143.86、147.98、175.32。HPLC(方法:20mm C18−RPカラム−傾斜法2−95% ACN+0.1%ギ酸で3.3分、95% ACNで1.7分保持した;波長:210nm):保持時間:2.85分;90%純度。MS(M+H):414.3。
実施例9.(R)−2−(3−(ジ(イソプロピル−d7)アミノ)−1−フェニルプロピル)−4−(ヒドロキシル(メチル−d2))フェノール(化合物103)。
【0160】
適当に重水素化された試薬を用いて、スキーム1で概説したように化合物103を調製した。
【化21】
【0161】
工程1.(R)−3−(2−(ベンジルオキシ)−5−(ベンジルオキシ(メチル−d2))フェニル)−N,N−ジ(イソプロピル−d7)−3−フェニルプロパンアミド(15f、X=CD2OBn、R2=R3=CD(CD3)2)。
ドライエーテル(20mL)中の粗14d(1.3g)(実施例7、工程3を参照)の溶液にジイソプロピルアミン−d14[CDNアイソトープ、98原子%D](500mg、4.35mmol)を加え、続いて、Et3N(1.25mL、9mmol)を加えた。混合物を4時間攪拌し、2N HCl(15mL)で急冷し、EtOAc(50mL)で抽出した。有機層を水性NaHCO3(飽和)、ブラインで洗浄し、Na2SO4で乾燥させ、濾過し、濾液を真空下で濃縮した。カラムクロマトグラフィー(30% EtOAc/ヘプタン)で残渣を精製し、無色油として15fを1.26g(収率74%)得た。
【0162】
工程2.(R)−3−(2−(ベンジルオキシ)−5−(ベンジルオキシ(メチル−d2))フェニル)−N,N−ジ(イソプロピル−d7)−3−フェニルプロパン−1−アミン(16f、X=CD2OBn、R2=R3=CD(CD3)2)。
THF(無水、15mL)中の15f(1.2g、2.18mmol)の0℃の溶液にLiAlH4(170mg、4.4mmol)を加えた。混合物を室温で14時間攪拌した後、H2O(0.18mL)、15% NaOH溶液(0.18mL)、およびH2O(0.44mL)で急冷した。沈殿物を濾過し、EtOAcで洗浄した。濾液を真空下で濃縮し、カラムクロマトグラフィー(50% EtOAc/ヘプタン中の5% Et3N)で残渣を精製し、無色油として16fを960mg(収率82%)得た。
【0163】
工程3.(R)−2−(3−(ジ(イソプロピル−d7)−アミノ)−1−フェニルプロピル)−4−(ヒドロキシル(メチル−d2))フェノール(化合物103)。
ジクロロメタン(20mL)中の16f(960mg、1.79mmol)の−78℃の溶液にBCl3(1Mジクロロメタン中の6.3mL、6.3mmol)を滴下した。混合物を3時間攪拌した後、H2Oで急冷し、室温に温めた。混合物を水性NaHCO3(飽和)でpH=9にし、ジクロロメタンで抽出した。有機層をNa2SO4で乾燥させ、濾過し、濾液を真空下で濃縮した。カラムクロマトグラフィー(5%メタノール/ジクロロメタン中の5% Et3N)で残渣を精製し、黄色油として化合物103を460mg(収率73%)得た。
実施例10.(R)−2−(3−(ジ(イソプロピル−d7)アミノ)−1−フェニルプロピル)−4−(ヒドロキシル(メチル−d2))フェニルイソブチレート(化合物118)の合成。
【0164】
化合物118は、スキーム1に概説したように、化合物103から調製した。
【化22】
(R)−2−(3−(ジ(イソプロピル−d7)アミノ)−1−フェニルプロピル)−4−(ヒドロキシル(メチル−d2))フェニルイソブチレート(化合物118)。
ジクロロメタン(10mL)中の化合物103(460mg、1.29mmol)の−30℃の溶液にジイソプロピルエチルアミン(0.25mL、1.42mmol)を加え、続いて、塩化イソブチリル(0.14mL、1.29mmol)を加えた。混合物を−30℃で3時間攪拌した後、水で急冷し、ジクロロメタンで抽出し、Na2SO4で乾燥させ、濾過し、濾液を真空下で濃縮した。カラムクロマトグラフィー(20% EtOAc/ヘプタン中の5% Et3N、その後、5%メタノール/ジクロロメタン中の5% Et3N)で残渣を精製し、黄色油として化合物118を385mg(収率70%)得た。1H−NMR(300MHz、CDCl3):δ 0.90(微量)、1.30(d、J=11.1、3H)、1.33(d、J=11.1、3H)、2.13〜2.17(m、2H)、2.30〜2.36(m、2H)、2.76〜2.85(m、1H)、4.11(t、J=7.6、1H)、6.97(d、J=8.2、1H)、7.13〜7.33(m、6H)、7.34(d、J=1.5、1H)。13C−NMR(75MHz、CDCl3):δ 18.98、19.10、34.20、36.87、41.76、43.83、122.62、125.67、126.15、126.96、127.91、128.34、136.98、138.48、143.85、147.99、175.32。HPLC(方法:20mm C18−RPカラム−傾斜法2−95% ACN+0.1%ギ酸で3.3分、95% ACNで1.7分保持した;波長:210nm):保持時間:2.87分;90%純度。MS(M+H):428.4。
実施例11.ヒト肝ミクロソームにおける代謝安定性の評価。
【0165】
材料:XenoTech,LLC(Lenexa,KS))からヒト肝ミクロソーム(20mg/mL)を入手した。Sigma−Aldrichからβ−ニコチンアミドアデニンジヌクレオチドリン酸、還元型(NADPH)、塩化マグネシウム(MgCl2)、およびジメチル・スルホキシド(DMSO)を購入した。
【0166】
化合物100、化合物106、化合物112、およびトルテロジンのストック溶液(7.5mM)をDMSO中で別々に調製した。7.5mMのストック溶液をアセトニトリル(ACN)中で50μMに希釈した。20mg/mLヒト肝ミクロソームを3mMのMgCl2を含有するpH7.4の0.1Mリン酸カリウム緩衝液中で0.625mg/mLに希釈した。希釈したミクロソームを、2通りで、96ウェルディープウェルポリプロピレンプレートのウェルに加えた。試験化合物溶液50μMの1つの10μLをミクロソームに加え、混合物を10分間予熱した。予熱したNADPH溶液を加えることで反応を開始させた。最終反応体積は、0.5mLであり、0.5mg/mLのヒト肝ミクロソーム、1μMの試験化合物、および、pH7.4の、0.1Mリン酸カリウム緩衝液中の2mM NADPH、ならびに3mMのMgCl2を含有した。反応混合物を37℃でインキュベートし、50μLの一定分量を0分、5分、10分、20分、および30分で取り出し、内部標準によって50μLの氷冷ACNを含んだ浅いウェルの96ウェルプレートに加え、反応を止めた。100μLの水をプレートのウェルに加えた後、プレートを4℃で20分間保存し、遠心分離してペレット状の沈降タンパク質にした。上清を別の96ウェルプレートに移し、Applied Bio−systemsのAPI4000質量分析計を用いてLC−MS/MSによって親残余の量を分析した。7−エトキシクマリン(1μM)を陽性対照として用いた。実験を2回繰返して、結果を確認した。
【0167】
データ分析:試験化合物のin vitro半減期(t1/2s)は、以下の式を用いて、%親残余(ln)対インキュベーション時間の関係による線形回帰の傾きから算出した。in vitro t1/2=0.693/k、ここで、k=−[%親残余(ln)対インキュベーション時間の線形回帰の傾き]。データ分析は、マイクロソフトのエクセルソフトウェアを用いて行った。
【0168】
結果を図1および以下の表2に示す。
【0169】
表2.ヒト肝ミクロソームにおける本発明の化合物の半減期の算出値。
【化23】
*%差=[(重水素化種)−(非重水素化種)](100)/(非重水素化種)
【0170】
化合物100および112は、トルテロジンと比較して、ヒト肝ミクロソームにおける半減期が実質的に増加したことを示す。驚いたことに、14個の重水素原子を含む化合物106は、トルテロジンと比較した場合、安定性にほとんど変化を示さなかった。一方、3個の重水素原子しか含まない化合物112は、安定性が増すことを示した。代謝の時間経過はこれらの結果で一致している(図1)。
【0171】
理論に束縛されるものではないが、本出願人らは、フェニル部分上の5−メチル基の重水素化によって、そのフェニル部分上に2−ヒドロキシルを含む本発明の化合物の安定性増加がもたらされると考えている。
【0172】
さらに説明することなく、当業者は、前述の説明および例示実施例を用いて、本発明の化合物を作り、利用し、請求項に記載の方法を実行することができると考えられる。上述の議論および実施例は、特定の好適な実施形態の詳細な説明を単に述べたにすぎないこと理解すべきである。当業者には、本発明の精神および範囲から逸脱せずにさまざまな変更物および同等物の作成が可能であることが明らかであろう。
【特許請求の範囲】
【請求項1】
式IIの化合物:
【化1】
式(II)、またはその医薬的に許容される塩であって、
式中:
R2およびR3は、それぞれ独立して、0〜7個の重水素原子をもつイソプロピル基である。
【請求項2】
R2およびR3の各々が、独立して、−CD(CD3)2および−CH(CH3)2から選択される請求項1に記載の化合物。
【請求項3】
化合物100または化合物112から選択される請求項2に記載の化合物。
【請求項4】
重水素として指定されない任意の原子が、その天然同位体存在度で存在する請求項1〜3のいずれか1項に記載の化合物。
【請求項5】
有効量の請求項1に記載の化合物と、医薬的に許容される担体とを含むピロゲンフリー医薬組成物。
【請求項6】
不安定膀胱もしくは過活動膀胱、失禁、感染、下部尿路疾患、記憶障害もしくは認知障害、心不全、肺炎、良性前立腺肥大症、前立腺肥大症、呼吸器疾患、喘息、女性性機能障害、または膀胱炎から選択される疾患または疾病に罹患しているもしくは罹患しやすい患者の治療に有用な第2の治療薬を有効量でさらに含む請求項5に記載の組成物。
【請求項7】
前記第2の治療薬が、αアドレナリン受容体遮断薬;ビシファジン化合物;スタチン;デヒドロエピアンドロステロン(DHEA)同族体;α2δサブユニットカルシウムチャネル調節因子;選択的セロトニン再取り込み阻害薬(SSRI)または選択的ノルエピネフリン再取り込み阻害薬;アンドロゲン;エストロゲン;エストロゲン作用薬;EGF受容体拮抗薬;5HT1a受容体修飾因子;チエノピランカルボキサミド誘導体;および5a還元酵素阻害薬から選択される請求項6に記載の組成物。
【請求項8】
前記第2の治療薬がタムスロシンである請求項7に記載の組成物。
【請求項9】
不安定膀胱もしくは過活動膀胱、失禁、感染、下部尿路疾患、記憶障害もしくは認知障害、心不全、肺炎、良性前立腺肥大症、前立腺肥大症、呼吸器疾患、喘息、女性性機能障害、または膀胱炎から選択される疾患または疾病に罹患しているもしくは罹患しやすい患者の治療方法であって、
請求項5に記載の組成物をそれを必要とする前記患者に投与する工程を含む方法。
【請求項10】
前記疾患または疾病が、過活動膀胱および失禁から選択される請求項9に記載の方法。
【請求項11】
第2の治療薬が:
a.αアドレナリン受容体遮断薬、ビシファジン化合物、α2δサブユニットカルシウムチャネル調節因子、選択的セロトニン再取り込み阻害薬(SSRI)、選択的ノルエピネフリン再取り込み阻害薬、5HT1a受容体修飾因子、チエノピランカルボキサミド誘導体、および5a還元酵素阻害薬から選択され;前記患者は、下部尿路疾患に苦しんでいるあるいは罹患しやすく;
b.デヒドロエピアンドロステロン(DHEA)同族体;前記患者は、炎症に苦しんでいるあるいは罹患しやすく;
c.スタチン、前記患者は、呼吸器疾患に苦しんでいるあるいは罹患しやすく;
d.アンドロゲン、エストロゲン、またはエストロゲン作用薬から選択され;前記患者は、女性性機能障害に苦しんでいるあるいは罹患しやすく;または
e.EGF受容体拮抗薬、またはチエノピランカルボキサミド誘導体から選択され;前記患者は、良性前立腺肥大症または前立腺肥大症に罹患しているもしくは罹患しやすい、
である前記第2の治療薬をそれを必要とする前記患者に同時投与する工程をさらに含む請求項9または請求項10に記載の方法。
【請求項12】
前記第2の治療薬がタムスロシンであり、前記患者が良性前立腺肥大症または前立腺肥大症に罹患しているもしくは罹患しやすい請求項11に記載の方法。
【請求項1】
式IIの化合物:
【化1】
式(II)、またはその医薬的に許容される塩であって、
式中:
R2およびR3は、それぞれ独立して、0〜7個の重水素原子をもつイソプロピル基である。
【請求項2】
R2およびR3の各々が、独立して、−CD(CD3)2および−CH(CH3)2から選択される請求項1に記載の化合物。
【請求項3】
化合物100または化合物112から選択される請求項2に記載の化合物。
【請求項4】
重水素として指定されない任意の原子が、その天然同位体存在度で存在する請求項1〜3のいずれか1項に記載の化合物。
【請求項5】
有効量の請求項1に記載の化合物と、医薬的に許容される担体とを含むピロゲンフリー医薬組成物。
【請求項6】
不安定膀胱もしくは過活動膀胱、失禁、感染、下部尿路疾患、記憶障害もしくは認知障害、心不全、肺炎、良性前立腺肥大症、前立腺肥大症、呼吸器疾患、喘息、女性性機能障害、または膀胱炎から選択される疾患または疾病に罹患しているもしくは罹患しやすい患者の治療に有用な第2の治療薬を有効量でさらに含む請求項5に記載の組成物。
【請求項7】
前記第2の治療薬が、αアドレナリン受容体遮断薬;ビシファジン化合物;スタチン;デヒドロエピアンドロステロン(DHEA)同族体;α2δサブユニットカルシウムチャネル調節因子;選択的セロトニン再取り込み阻害薬(SSRI)または選択的ノルエピネフリン再取り込み阻害薬;アンドロゲン;エストロゲン;エストロゲン作用薬;EGF受容体拮抗薬;5HT1a受容体修飾因子;チエノピランカルボキサミド誘導体;および5a還元酵素阻害薬から選択される請求項6に記載の組成物。
【請求項8】
前記第2の治療薬がタムスロシンである請求項7に記載の組成物。
【請求項9】
不安定膀胱もしくは過活動膀胱、失禁、感染、下部尿路疾患、記憶障害もしくは認知障害、心不全、肺炎、良性前立腺肥大症、前立腺肥大症、呼吸器疾患、喘息、女性性機能障害、または膀胱炎から選択される疾患または疾病に罹患しているもしくは罹患しやすい患者の治療方法であって、
請求項5に記載の組成物をそれを必要とする前記患者に投与する工程を含む方法。
【請求項10】
前記疾患または疾病が、過活動膀胱および失禁から選択される請求項9に記載の方法。
【請求項11】
第2の治療薬が:
a.αアドレナリン受容体遮断薬、ビシファジン化合物、α2δサブユニットカルシウムチャネル調節因子、選択的セロトニン再取り込み阻害薬(SSRI)、選択的ノルエピネフリン再取り込み阻害薬、5HT1a受容体修飾因子、チエノピランカルボキサミド誘導体、および5a還元酵素阻害薬から選択され;前記患者は、下部尿路疾患に苦しんでいるあるいは罹患しやすく;
b.デヒドロエピアンドロステロン(DHEA)同族体;前記患者は、炎症に苦しんでいるあるいは罹患しやすく;
c.スタチン、前記患者は、呼吸器疾患に苦しんでいるあるいは罹患しやすく;
d.アンドロゲン、エストロゲン、またはエストロゲン作用薬から選択され;前記患者は、女性性機能障害に苦しんでいるあるいは罹患しやすく;または
e.EGF受容体拮抗薬、またはチエノピランカルボキサミド誘導体から選択され;前記患者は、良性前立腺肥大症または前立腺肥大症に罹患しているもしくは罹患しやすい、
である前記第2の治療薬をそれを必要とする前記患者に同時投与する工程をさらに含む請求項9または請求項10に記載の方法。
【請求項12】
前記第2の治療薬がタムスロシンであり、前記患者が良性前立腺肥大症または前立腺肥大症に罹患しているもしくは罹患しやすい請求項11に記載の方法。
【図1】

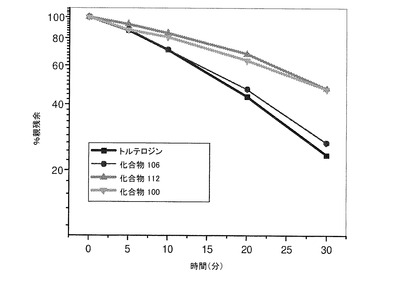
【公表番号】特表2011−518783(P2011−518783A)
【公表日】平成23年6月30日(2011.6.30)
【国際特許分類】
【出願番号】特願2011−504185(P2011−504185)
【出願日】平成21年4月9日(2009.4.9)
【国際出願番号】PCT/US2009/040126
【国際公開番号】WO2009/126844
【国際公開日】平成21年10月15日(2009.10.15)
【出願人】(509049012)コンサート ファーマシューティカルズ インコーポレイテッド (24)
【Fターム(参考)】
【公表日】平成23年6月30日(2011.6.30)
【国際特許分類】
【出願日】平成21年4月9日(2009.4.9)
【国際出願番号】PCT/US2009/040126
【国際公開番号】WO2009/126844
【国際公開日】平成21年10月15日(2009.10.15)
【出願人】(509049012)コンサート ファーマシューティカルズ インコーポレイテッド (24)
【Fターム(参考)】
[ Back to top ]