
3,3A,6,6A−テトラヒドロ−2H−シクロペンタン[b]フラン−2−オンの合成方法
本発明は3,3a,6,6a−テトラヒドロ−2H−シクロペンタン[b]フラン−2−オンの合成方法に関する。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、プロスタグランジンの合成の中間体として有用な、分子である3,3a,6,6a−テトラヒドロ−2H−シクロペンタン[b]フラン−2−オンの合成方法に関する。
【背景技術】
【0002】
プロスタグランジンは、多様な用途を有する重要な一連の分子群である。多くのプロスタグランジンの合成が知られており、3,3a,6,6a−テトラヒドロ−2H−シクロペンタン[b]フラン−2−オンは、そうしたいくつかの合成で知られた中間体である。3,3a,6,6a−テトラヒドロ−2H−シクロペンタン[b]フラン−2−オン(ラクトン)は、多くの方法により製造されてきた。ラセミ体は、ジクロロケテンとシクロペンタジオンとを反応させ、次いで、亜鉛で脱塩素化し、次に、Bayer-Willige酸化により製造できる(Grieco, P. A., J. Org. Chem. 1972年, 37巻, 2363-4頁)。この製法には、ジクロロケテン工程で黒色タールが形成されるという問題があり(Corey, E. J.; Snider, B. B., J. Org. Chem. 1974年, 39巻, 256-8頁;Covington, E. W.等, Tetrahedron Lett. 1983年, 3125-3128頁;Carnell, A. J.等、J. Chem. Soc., Chem. Commun. 1990年, 20巻, 1438-9頁; Bertolasi, V.等, Tetrahedron: Asymmetry (2001年), 12巻 (10号), 1479-1483頁)に記載されたような解決方法を必要としている。
【0003】
3,3a,6,6a−テトラヒドロ−2H−シクロペンタン[b]フラン−2−オンを、鏡像異性体が濃縮された形態で製造する他の方法は、シクロペンタジエン酢酸の無対称的ヒドロホウ素化反応を必要とする(Partridge, J. J.等, J. Am. Chem. Soc.1973年, 95巻, 7171-2頁;Partridge, J. J.等, Org. Syn. Coll. VII巻, 339-345頁)。この方法は、分解が避けられる利点を有するが、しかし、大規模な製造工程ではその利点を制限するいくつもの操作上の問題を有している。
【0004】
三番目の方法は、3−アシルオキシ−5−ヒドロキシシクロペンテンのClaisienの転位である。3S,5RモノアセテートのオルトエステルClaisien転位が文献に記載されている(Laumen, K.等, J. Chem. Soc. Chem. Comm. 1986年, 1298-1299頁; Laumen, K.等, Tetrahedron Lett. 1984年, 25巻, 5875-5878頁; Nara, M.等, Tetrahedron, 1980年, 36巻, 3161-3170頁; Takano, S.等, J. Chem. Soc. , Chem. Commun. 1976年, 6巻, 189-190頁)が、高温度(160℃)の使用を必要とし生産規模を達成するのが困難である。
【0005】
したがって、3,3a,6,6a−テトラヒドロ−2H−シクロペンタン[b]フラン−2−オンの、経済的で、鏡像異性体が濃縮された生成物として得られ、および、大規模の製造に適した、簡単で経済的な製造方法が必要とされている。
【発明の開示】
【課題を解決するための手段】
【0006】
本発明は、
a)式Iの3−アシルオキシ−5−ヒドロキシシクロペンテンと、
【化1】

式IIaのアミドアセタールまたは式IIbのケテンアミノアセタール
【化2】
(式中、
R1およびR′1は、C1〜C4アルキル、または
R1およびR′1は、一緒になって3〜7員環を形成し、
R2は、C1〜C4アルキル、
Acは、C1〜C4アルカノイルである)、
とをアルコールR2OH濃度を容量で3%以下に維持しながら、>90℃の沸点の適切な溶媒中で、90〜140℃において、
反応させて次の式IIIのアシルヒドロキシシクロペンテンアセトアミドを得る、
【化3】
【0007】
b)アルカリもしくはアルカリ土類の水酸化物、炭酸塩、重炭酸塩または第4級アンモニウム水酸化物溶液を加えて、均質なまたは2相性の混合物を得、そして
c)pKa<2の強酸を加えて、式IVのラクトンを得る、
【化4】
工程を含む3,3a,6,6a−テトラヒドロ−2H−シクロペンタン[b]フラン−2−オン(式IV)を製造する方法を提供する。
【0008】
すなわち本発明は、3,3a,6,6a−テトラヒドロ−2H−シクロペンタン[b]フラン−2−オンを、製造するための、以下の工程:
a)式Iの3−アシルオキシ−5−ヒドロキシシクロペンテンを、
【化5】
式IIaのアミドアセタールまたは式IIbのケテンアミノアセタールと、
【化6】
(式中、
R1およびR′1は、C1〜C4アルキル、または
R1およびR′1は、一緒になって3〜7員環を形成し、
R2は、C1〜C4アルキル、
Acは、C1〜C4アルカノイルである)、
アルコールR2OH濃度を容量で3%以下に維持しながら、>90℃の沸点の適切な溶媒で、90〜140℃において、反応させ、
式IIIのアシルヒドロキシシクロペンテンアセトアミドを得る工程、
【化7】
【0009】
b)アルカリもしくはアルカリ土類の水酸化物、炭酸塩、重炭酸塩または第4級アンモニウム水酸化物溶液を加えて、均質なまたは2相性の混合物を得る工程;
c)pKa<2の強酸を加えて、式IVの標題のラクトンを得る工程、
【化8】
を含む方法を提供するものである。
【0010】
工程aにおけるClaisienの転位に作用する、式IIaのアミドアセタール、または、式IIbのケテンアミノアセタールの使用は、そうした条件下での転位が、通常の製造機器で容易に達成できる、わずか90〜120℃の温度しか必要としないという、驚くべき結果をもたらす。
【0011】
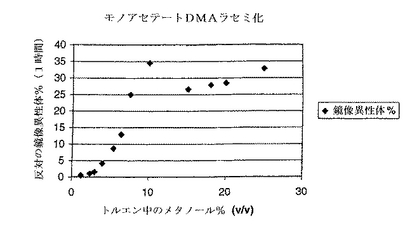
さらに、反応混合液中のアルコール(R2OH)濃度を3%より少なく維持すると、ラセミ化が最小限になるという予期せぬ結果が得られた。図1に示すように、3〜5%より高いアルコール濃度は、出発物質の式Iのアシルオキシヒドロキシシクロペンテンの大量のラセミ化をもたらす。図は、120℃の槽温度中でメタノールの種々の量において、トルエン中でジメチルアセトアミドジメチルアセタール(DMA)との工程aを行った時の、1時間の反応時間後に生じたラセミ化を示している。
【0012】
図1で示されているように、反応溶液中のアルコール濃度を3%未満、および好ましくは2%未満に維持することが重要である。このことは、出発物質中のアルコールの低濃度を確認すること、反応溶液からアルコール生成物を蒸留すること、場合によっては、アセタールまたはケテンアセタールを少量加えること、そして、場合によっては、反応器の頭部のスペースを窒素、アルゴン等のような不活性気体でパージすることによって、達成できる。
【0013】
工程aで、トルエン以外に適切な溶媒は、キシレン、メシチレン、アニソール、クロロベンゼン、ブロモベンゼン、o−ジクロロベンゼン、エチルベンゼン、インダン、テトラリン、デカリン、ヘプタン、オクタン、イソオクタン、および高級アルカン、メチルシクロヘキサン、ジメチルシクロヘキサン、エチルシクロヘキサン、ブロモブタンまたは他の高沸点ハロアルカン、エチレングリコールジメチルエーテル、エチレングリコールジエチルエーテル、高級エチレングリコールエーテル、ジエチレングリコールエーテル、プロピルエーテル、ブチルエーテルおよび同様の高沸点エーテル、ジメチルテトラヒドロフラン、1,4−ジオキサン、2,2−ジメチル−1,3−ジオキソラン、アセトアルデヒドジエチルアセタールおよび同様の高沸点アセタール、トリエチルアミンおよび高沸点第三級アミン、ジメチルアニリン、ピリジン、ルチジン、コリジン、n−メチルピペリジン、キノリン、キナルジン、レピジン、イソキノリン、プロピオンニトリル、ベンゾニトリルおよび同様の高沸点ニトリル、ならびに、通常圧で沸点が>90℃であるそれらの同類の溶媒である。アミドアセタールの非限定的な例には、ジメチルアセトアミドジメチルアセタール、ジメチルアセトアミドジエチルアセタール、ジエチルアセトアミドジメチルアセタール、および、N−1,1−ジメトキシエチルピロリジンが挙げられる。ケテンアミノアセタールの非限定的な例には、1−メトキシ−1−ジメチルアミノエチレン、および、1−メトキシ−1−ジエチルアミノエチレン、1−エトキシ−1−ジエチルアミノエチレン、ならびに、1−メトキシ−1−ピロリジニルエチレンが挙げられる。式IIIのアミドは、クロマトグラフィーにより単離および精製できるが、通常は、粗生成物として工程bで用いることができる。
【0014】
工程bにおいて、適切な塩基は、水酸化ナトリウム、水酸化カリウム、炭酸リチウム、炭酸セシウム、水酸化テトラブチルアンモニウム等の水性溶液である。二相性の条件下で、相転移触媒、例えば、水酸化ベンジルトリデシルアンモニウム等が、場合によって、使用できる。アミド加水分解の中間生成物は、通常は単離しないで、アルカリ性加水分解混合物の酸性化により直接に式IVのラクトンに変換される。
【0015】
最後の工程cで、酸性化のためのpKa<2の適切な酸には、塩酸、硫酸、臭化水素酸、リン酸、フルオロ硼酸、過塩素酸、トルエンスルホン酸、メタンスルホン酸、トリフルオロ酢酸等の水性溶液が含まれる。標題の化合物は、抽出、クロマトグラフィーおよび結晶化のような従来法により単離できる。
【0016】
さらなる努力なしに、当業者は、前記の記載を用いて、本発明を完全に実施できる。以下の詳細な実施例は、いかに種々の化合物を製造し、そして、いかに本発明の種々の方法を実施するかを記載しており、これは単に説明のためであり、いかなる意味でも前記の開示を制限するものではない。当業者は、この方法の、反応物ならびに反応条件および技法の両方に関して、適切な変更を容易に認識するだろう。
【0017】
実施例1: 1S,5R−2−オキサビシクロ[3.3.0]オクタ−6−エン−3−オン
【化9】
【0018】
工程a: (1R−シス)5−(アセチルオキシ)−N,N−ジメチル−2−シクロペンテン−1−アセトアミド
3S,5R 3−アセトキシ−5−ヒドロキシシクロペンテン(アルドリッチ・ケミカルCo.、17.55g、123ミリモル、98.8%ee)、ジメチルアセトアミドジメチルアセタール(アルドリッチ・ケミカルCo.、24.57g、182ミリモル)、およびトルエン351mLの混合物を、130℃に保っている油浴中で、メタノールを短絡路コンデンサーで蒸留しながら、加熱した。混合物からの最初のメタノールの蒸留の後、ゆっくりと窒素を通し始め、当初の容量を維持するために必要な追加のトルエンを加えた。加熱は6〜8時間継続し、トルエンを蒸留によって除去し、暗赤褐色の油状の粗生成物を得た。〔(5−(アセチルオキシ)−N,N−ジメチル−2−シクロペンテン−1−アセトアミドは、Ema, T.等、J.Org.Chem., 1996年, 61巻, 8610頁に記載されている。〕
1H NMR (CDCl3) δ 2.01 (s, 3H), 2.31−2.37 (m, 2H), 2.55 (dd, 1H, J = 16.0, 7.0Hz), 2.75 (dd, 1H, J = 16, 6.6Hz), 2.96 (s, 3H), 3.02 (s, 3H), 3.38 (m, 1H), 5.46 (m, 1H), 5.73 (m, 2H)。
13C NMR(CDCl3)δ21.42,32.30,35.77,37.59,39.57,44.74,75.42,128.46,133.41,170.83,172.03
【0019】
工程b: (1R−シス)5−ヒドロキシ−2−シクロペンテン−1−酢酸、カリウム塩
工程aからのアミド粗生成物をMTBE50mLに溶解した。水酸化カリウム(16.2g、246ミリモル)を水160mL中に溶解した溶液を加え、混合物を65℃の湯浴で1時間攪拌しながら加熱した。混合物を冷却し、相を分離した。水相をMTBE(50mL)で洗浄し、標題の化合物の水溶液を得た。
【0020】
工程c: 1S,5R−2−オキサビシクロ[3.3.0]オクタ−6−エン−3−オン
工程bのアルカリ性溶液を、濃塩酸を加えてpH1.0〜1.5に酸性化し、1時間攪拌した。混合物を塩化メチレン(3×50mL)で抽出し、有機抽出相を50〜70mLに濃縮し、シリカゲル(10g、230〜400メッシュ)を通して濾過した。このシリカゲルは、追加の塩化メチレン(75mL)で洗浄した。濾液を合体して、30℃(湯浴)で減圧下に濃縮し(100mL)、放置して結晶化するラクトンを得た。
1H NMR (CDCl3) δ 2.17 (d, 1H, J = 18Hz), 2.39−2.55 (m, 4H), 3.26 (m, 1H), 4.87(t, 1H, J = 5.6Hz), 5.33 (m, 1H), 5.52 (m, 1H)。
13C NMR(CDCl3)δ33.71,39.85,45.83,83.41,129.9,131.7,177.1
【0021】
実施例2: ジメチルアセトアミドジメチルアセタールのゆっくりした添加
工程a: (1R−シス)5−(アセチルオキシ)−N,N−ジメチル−2−シクロペンテン−1−アセトアミド
3S,5R 3−アセトキシ−5−ヒドロキシシクロペンテン30.0gをトルエン240mLに溶解し、少量の不溶性物質を除去するために、溶液をマグネソール(magnesol)に通して濾過した。濾過した溶液を100℃(内部温度)に加熱し、ジメチルアセトアミドジメチルアセタール64.6mL(28%(v/v)メタノール)をトルエン64.5mLに溶解した溶液を、ゆっくりした蒸留速度を維持しながら、6時間かけて小分けして加えた。反応混合物を、添加の完了後さらに4時間、加熱した。この混合物を次に減圧下で濃縮し、暗色油状の生成物を得た。
【0022】
工程b: (1R−シス)5−ヒドロキシ−2−シクロペンテン−1−酢酸、カリウム塩
この油状生成物をMTBE85mLに溶解し、水酸化カリウム27.7gおよび水109mLを加えた。二相性混合物を還流下で1時間加熱した。相を分離して、水相をMTBE85mLで抽出し、標題の化合物の水溶液を得た。
【0023】
工程c: 1S,5R−2−オキサビシクロ[3.3.0]オクタ−6−エン−3−オン
工程bの水相に濃塩酸を50mL加え(最終pH1.0)、次いで混合物を室温で1時間攪拌した。混合物を塩化メチレン(3×120mL)で抽出した。合体した塩化メチレン溶液をシリカ(17g、230〜400メッシュ)を通して濾過した。濾液を蒸発させ23.91g(総収率、91.2%)得た。
【図面の簡単な説明】
【0024】
【図1】反応混合液中のアルコール濃度と生成物中の反対鏡像異性体%の関係を示す。
【技術分野】
【0001】
本発明は、プロスタグランジンの合成の中間体として有用な、分子である3,3a,6,6a−テトラヒドロ−2H−シクロペンタン[b]フラン−2−オンの合成方法に関する。
【背景技術】
【0002】
プロスタグランジンは、多様な用途を有する重要な一連の分子群である。多くのプロスタグランジンの合成が知られており、3,3a,6,6a−テトラヒドロ−2H−シクロペンタン[b]フラン−2−オンは、そうしたいくつかの合成で知られた中間体である。3,3a,6,6a−テトラヒドロ−2H−シクロペンタン[b]フラン−2−オン(ラクトン)は、多くの方法により製造されてきた。ラセミ体は、ジクロロケテンとシクロペンタジオンとを反応させ、次いで、亜鉛で脱塩素化し、次に、Bayer-Willige酸化により製造できる(Grieco, P. A., J. Org. Chem. 1972年, 37巻, 2363-4頁)。この製法には、ジクロロケテン工程で黒色タールが形成されるという問題があり(Corey, E. J.; Snider, B. B., J. Org. Chem. 1974年, 39巻, 256-8頁;Covington, E. W.等, Tetrahedron Lett. 1983年, 3125-3128頁;Carnell, A. J.等、J. Chem. Soc., Chem. Commun. 1990年, 20巻, 1438-9頁; Bertolasi, V.等, Tetrahedron: Asymmetry (2001年), 12巻 (10号), 1479-1483頁)に記載されたような解決方法を必要としている。
【0003】
3,3a,6,6a−テトラヒドロ−2H−シクロペンタン[b]フラン−2−オンを、鏡像異性体が濃縮された形態で製造する他の方法は、シクロペンタジエン酢酸の無対称的ヒドロホウ素化反応を必要とする(Partridge, J. J.等, J. Am. Chem. Soc.1973年, 95巻, 7171-2頁;Partridge, J. J.等, Org. Syn. Coll. VII巻, 339-345頁)。この方法は、分解が避けられる利点を有するが、しかし、大規模な製造工程ではその利点を制限するいくつもの操作上の問題を有している。
【0004】
三番目の方法は、3−アシルオキシ−5−ヒドロキシシクロペンテンのClaisienの転位である。3S,5RモノアセテートのオルトエステルClaisien転位が文献に記載されている(Laumen, K.等, J. Chem. Soc. Chem. Comm. 1986年, 1298-1299頁; Laumen, K.等, Tetrahedron Lett. 1984年, 25巻, 5875-5878頁; Nara, M.等, Tetrahedron, 1980年, 36巻, 3161-3170頁; Takano, S.等, J. Chem. Soc. , Chem. Commun. 1976年, 6巻, 189-190頁)が、高温度(160℃)の使用を必要とし生産規模を達成するのが困難である。
【0005】
したがって、3,3a,6,6a−テトラヒドロ−2H−シクロペンタン[b]フラン−2−オンの、経済的で、鏡像異性体が濃縮された生成物として得られ、および、大規模の製造に適した、簡単で経済的な製造方法が必要とされている。
【発明の開示】
【課題を解決するための手段】
【0006】
本発明は、
a)式Iの3−アシルオキシ−5−ヒドロキシシクロペンテンと、
【化1】
式IIaのアミドアセタールまたは式IIbのケテンアミノアセタール
【化2】
(式中、
R1およびR′1は、C1〜C4アルキル、または
R1およびR′1は、一緒になって3〜7員環を形成し、
R2は、C1〜C4アルキル、
Acは、C1〜C4アルカノイルである)、
とをアルコールR2OH濃度を容量で3%以下に維持しながら、>90℃の沸点の適切な溶媒中で、90〜140℃において、
反応させて次の式IIIのアシルヒドロキシシクロペンテンアセトアミドを得る、
【化3】
【0007】
b)アルカリもしくはアルカリ土類の水酸化物、炭酸塩、重炭酸塩または第4級アンモニウム水酸化物溶液を加えて、均質なまたは2相性の混合物を得、そして
c)pKa<2の強酸を加えて、式IVのラクトンを得る、
【化4】
工程を含む3,3a,6,6a−テトラヒドロ−2H−シクロペンタン[b]フラン−2−オン(式IV)を製造する方法を提供する。
【0008】
すなわち本発明は、3,3a,6,6a−テトラヒドロ−2H−シクロペンタン[b]フラン−2−オンを、製造するための、以下の工程:
a)式Iの3−アシルオキシ−5−ヒドロキシシクロペンテンを、
【化5】
式IIaのアミドアセタールまたは式IIbのケテンアミノアセタールと、
【化6】
(式中、
R1およびR′1は、C1〜C4アルキル、または
R1およびR′1は、一緒になって3〜7員環を形成し、
R2は、C1〜C4アルキル、
Acは、C1〜C4アルカノイルである)、
アルコールR2OH濃度を容量で3%以下に維持しながら、>90℃の沸点の適切な溶媒で、90〜140℃において、反応させ、
式IIIのアシルヒドロキシシクロペンテンアセトアミドを得る工程、
【化7】
【0009】
b)アルカリもしくはアルカリ土類の水酸化物、炭酸塩、重炭酸塩または第4級アンモニウム水酸化物溶液を加えて、均質なまたは2相性の混合物を得る工程;
c)pKa<2の強酸を加えて、式IVの標題のラクトンを得る工程、
【化8】
を含む方法を提供するものである。
【0010】
工程aにおけるClaisienの転位に作用する、式IIaのアミドアセタール、または、式IIbのケテンアミノアセタールの使用は、そうした条件下での転位が、通常の製造機器で容易に達成できる、わずか90〜120℃の温度しか必要としないという、驚くべき結果をもたらす。
【0011】
さらに、反応混合液中のアルコール(R2OH)濃度を3%より少なく維持すると、ラセミ化が最小限になるという予期せぬ結果が得られた。図1に示すように、3〜5%より高いアルコール濃度は、出発物質の式Iのアシルオキシヒドロキシシクロペンテンの大量のラセミ化をもたらす。図は、120℃の槽温度中でメタノールの種々の量において、トルエン中でジメチルアセトアミドジメチルアセタール(DMA)との工程aを行った時の、1時間の反応時間後に生じたラセミ化を示している。
【0012】
図1で示されているように、反応溶液中のアルコール濃度を3%未満、および好ましくは2%未満に維持することが重要である。このことは、出発物質中のアルコールの低濃度を確認すること、反応溶液からアルコール生成物を蒸留すること、場合によっては、アセタールまたはケテンアセタールを少量加えること、そして、場合によっては、反応器の頭部のスペースを窒素、アルゴン等のような不活性気体でパージすることによって、達成できる。
【0013】
工程aで、トルエン以外に適切な溶媒は、キシレン、メシチレン、アニソール、クロロベンゼン、ブロモベンゼン、o−ジクロロベンゼン、エチルベンゼン、インダン、テトラリン、デカリン、ヘプタン、オクタン、イソオクタン、および高級アルカン、メチルシクロヘキサン、ジメチルシクロヘキサン、エチルシクロヘキサン、ブロモブタンまたは他の高沸点ハロアルカン、エチレングリコールジメチルエーテル、エチレングリコールジエチルエーテル、高級エチレングリコールエーテル、ジエチレングリコールエーテル、プロピルエーテル、ブチルエーテルおよび同様の高沸点エーテル、ジメチルテトラヒドロフラン、1,4−ジオキサン、2,2−ジメチル−1,3−ジオキソラン、アセトアルデヒドジエチルアセタールおよび同様の高沸点アセタール、トリエチルアミンおよび高沸点第三級アミン、ジメチルアニリン、ピリジン、ルチジン、コリジン、n−メチルピペリジン、キノリン、キナルジン、レピジン、イソキノリン、プロピオンニトリル、ベンゾニトリルおよび同様の高沸点ニトリル、ならびに、通常圧で沸点が>90℃であるそれらの同類の溶媒である。アミドアセタールの非限定的な例には、ジメチルアセトアミドジメチルアセタール、ジメチルアセトアミドジエチルアセタール、ジエチルアセトアミドジメチルアセタール、および、N−1,1−ジメトキシエチルピロリジンが挙げられる。ケテンアミノアセタールの非限定的な例には、1−メトキシ−1−ジメチルアミノエチレン、および、1−メトキシ−1−ジエチルアミノエチレン、1−エトキシ−1−ジエチルアミノエチレン、ならびに、1−メトキシ−1−ピロリジニルエチレンが挙げられる。式IIIのアミドは、クロマトグラフィーにより単離および精製できるが、通常は、粗生成物として工程bで用いることができる。
【0014】
工程bにおいて、適切な塩基は、水酸化ナトリウム、水酸化カリウム、炭酸リチウム、炭酸セシウム、水酸化テトラブチルアンモニウム等の水性溶液である。二相性の条件下で、相転移触媒、例えば、水酸化ベンジルトリデシルアンモニウム等が、場合によって、使用できる。アミド加水分解の中間生成物は、通常は単離しないで、アルカリ性加水分解混合物の酸性化により直接に式IVのラクトンに変換される。
【0015】
最後の工程cで、酸性化のためのpKa<2の適切な酸には、塩酸、硫酸、臭化水素酸、リン酸、フルオロ硼酸、過塩素酸、トルエンスルホン酸、メタンスルホン酸、トリフルオロ酢酸等の水性溶液が含まれる。標題の化合物は、抽出、クロマトグラフィーおよび結晶化のような従来法により単離できる。
【0016】
さらなる努力なしに、当業者は、前記の記載を用いて、本発明を完全に実施できる。以下の詳細な実施例は、いかに種々の化合物を製造し、そして、いかに本発明の種々の方法を実施するかを記載しており、これは単に説明のためであり、いかなる意味でも前記の開示を制限するものではない。当業者は、この方法の、反応物ならびに反応条件および技法の両方に関して、適切な変更を容易に認識するだろう。
【0017】
実施例1: 1S,5R−2−オキサビシクロ[3.3.0]オクタ−6−エン−3−オン
【化9】
【0018】
工程a: (1R−シス)5−(アセチルオキシ)−N,N−ジメチル−2−シクロペンテン−1−アセトアミド
3S,5R 3−アセトキシ−5−ヒドロキシシクロペンテン(アルドリッチ・ケミカルCo.、17.55g、123ミリモル、98.8%ee)、ジメチルアセトアミドジメチルアセタール(アルドリッチ・ケミカルCo.、24.57g、182ミリモル)、およびトルエン351mLの混合物を、130℃に保っている油浴中で、メタノールを短絡路コンデンサーで蒸留しながら、加熱した。混合物からの最初のメタノールの蒸留の後、ゆっくりと窒素を通し始め、当初の容量を維持するために必要な追加のトルエンを加えた。加熱は6〜8時間継続し、トルエンを蒸留によって除去し、暗赤褐色の油状の粗生成物を得た。〔(5−(アセチルオキシ)−N,N−ジメチル−2−シクロペンテン−1−アセトアミドは、Ema, T.等、J.Org.Chem., 1996年, 61巻, 8610頁に記載されている。〕
1H NMR (CDCl3) δ 2.01 (s, 3H), 2.31−2.37 (m, 2H), 2.55 (dd, 1H, J = 16.0, 7.0Hz), 2.75 (dd, 1H, J = 16, 6.6Hz), 2.96 (s, 3H), 3.02 (s, 3H), 3.38 (m, 1H), 5.46 (m, 1H), 5.73 (m, 2H)。
13C NMR(CDCl3)δ21.42,32.30,35.77,37.59,39.57,44.74,75.42,128.46,133.41,170.83,172.03
【0019】
工程b: (1R−シス)5−ヒドロキシ−2−シクロペンテン−1−酢酸、カリウム塩
工程aからのアミド粗生成物をMTBE50mLに溶解した。水酸化カリウム(16.2g、246ミリモル)を水160mL中に溶解した溶液を加え、混合物を65℃の湯浴で1時間攪拌しながら加熱した。混合物を冷却し、相を分離した。水相をMTBE(50mL)で洗浄し、標題の化合物の水溶液を得た。
【0020】
工程c: 1S,5R−2−オキサビシクロ[3.3.0]オクタ−6−エン−3−オン
工程bのアルカリ性溶液を、濃塩酸を加えてpH1.0〜1.5に酸性化し、1時間攪拌した。混合物を塩化メチレン(3×50mL)で抽出し、有機抽出相を50〜70mLに濃縮し、シリカゲル(10g、230〜400メッシュ)を通して濾過した。このシリカゲルは、追加の塩化メチレン(75mL)で洗浄した。濾液を合体して、30℃(湯浴)で減圧下に濃縮し(100mL)、放置して結晶化するラクトンを得た。
1H NMR (CDCl3) δ 2.17 (d, 1H, J = 18Hz), 2.39−2.55 (m, 4H), 3.26 (m, 1H), 4.87(t, 1H, J = 5.6Hz), 5.33 (m, 1H), 5.52 (m, 1H)。
13C NMR(CDCl3)δ33.71,39.85,45.83,83.41,129.9,131.7,177.1
【0021】
実施例2: ジメチルアセトアミドジメチルアセタールのゆっくりした添加
工程a: (1R−シス)5−(アセチルオキシ)−N,N−ジメチル−2−シクロペンテン−1−アセトアミド
3S,5R 3−アセトキシ−5−ヒドロキシシクロペンテン30.0gをトルエン240mLに溶解し、少量の不溶性物質を除去するために、溶液をマグネソール(magnesol)に通して濾過した。濾過した溶液を100℃(内部温度)に加熱し、ジメチルアセトアミドジメチルアセタール64.6mL(28%(v/v)メタノール)をトルエン64.5mLに溶解した溶液を、ゆっくりした蒸留速度を維持しながら、6時間かけて小分けして加えた。反応混合物を、添加の完了後さらに4時間、加熱した。この混合物を次に減圧下で濃縮し、暗色油状の生成物を得た。
【0022】
工程b: (1R−シス)5−ヒドロキシ−2−シクロペンテン−1−酢酸、カリウム塩
この油状生成物をMTBE85mLに溶解し、水酸化カリウム27.7gおよび水109mLを加えた。二相性混合物を還流下で1時間加熱した。相を分離して、水相をMTBE85mLで抽出し、標題の化合物の水溶液を得た。
【0023】
工程c: 1S,5R−2−オキサビシクロ[3.3.0]オクタ−6−エン−3−オン
工程bの水相に濃塩酸を50mL加え(最終pH1.0)、次いで混合物を室温で1時間攪拌した。混合物を塩化メチレン(3×120mL)で抽出した。合体した塩化メチレン溶液をシリカ(17g、230〜400メッシュ)を通して濾過した。濾液を蒸発させ23.91g(総収率、91.2%)得た。
【図面の簡単な説明】
【0024】
【図1】反応混合液中のアルコール濃度と生成物中の反対鏡像異性体%の関係を示す。
【特許請求の範囲】
【請求項1】
式I
【化1】
の3−アシルオキシ−5−ヒドロキシシクロペンテンと、式IIaのアミドアセタールまたは式IIbのケテンアミノアセタール
【化2】
(式中、
R1およびR′1は、C1〜C4アルキル、または
R1およびR′1は、一緒になって3〜7員環を形成し、
R2は、C1〜C4アルキル、
Acは、C1〜C4アルカノイルである)、
とを、アルコールR2OH濃度を容量で3%未満に維持しながら、90℃<の沸点を有する適切な溶媒中で、90〜140℃において反応させて、次の式III
【化3】
のアシルヒドロキシシクロペンテンアセトアミドを得ることを含む、
式IIIの5−(アシルオキシ)−N、N−ジアルキル−2−シクロペンテン−1−アセトアミドの製造方法。
【請求項2】
アルカリもしくはアルカリ土類の水酸化物、炭酸塩、重炭酸塩または第4級アンモニウム水酸化物溶液を加えて、均質なまたは2相性の混合物を得る工程;および、
pKa<2の強酸を加えて、式IV
【化4】
の標題のラクトンを得る工程、
を更に含む、(4R,5S)−3,3a,6,6a−テトラヒドロ−2H−シクロペンタン[b]フラン−2−オンを製造するための請求項1の方法。
【請求項3】
式I
【化5】
の3−アシルオキシ−5−ヒドロキシシクロペンテンと、式IIaのアミドアセタールまたは式IIbのケテンアミノアセタール
【化6】
(式中、
R1およびR′1は、C1〜C4アルキル、または
R1およびR′1は、一緒になって3〜7員環を形成し、
R2は、C1〜C4アルキル、
Acは、C1〜C4アルカノイルである)、
とをアルコールR2OH濃度を容量で3%未満に維持しながら、90℃<の沸点の適切な溶媒中で、90〜140℃において反応させ、式III
【化7】
のアシルヒドロキシシクロペンテンアセトアミドを得る
ことを含む、方法により製造される生成物。
【請求項4】
さらに、アルカリもしくはアルカリ土類の水酸化物、炭酸塩、重炭酸塩または第4級アンモニウム水酸化物溶液を加えて、均質なまたは2相性の混合物を得る工程;および、
pKa<2の強酸を加えて、式IV
【化8】
のラクトンを得る工程、
をさらに含む方法により製造する、請求項3の生成物。
【請求項1】
式I
【化1】
の3−アシルオキシ−5−ヒドロキシシクロペンテンと、式IIaのアミドアセタールまたは式IIbのケテンアミノアセタール
【化2】
(式中、
R1およびR′1は、C1〜C4アルキル、または
R1およびR′1は、一緒になって3〜7員環を形成し、
R2は、C1〜C4アルキル、
Acは、C1〜C4アルカノイルである)、
とを、アルコールR2OH濃度を容量で3%未満に維持しながら、90℃<の沸点を有する適切な溶媒中で、90〜140℃において反応させて、次の式III
【化3】
のアシルヒドロキシシクロペンテンアセトアミドを得ることを含む、
式IIIの5−(アシルオキシ)−N、N−ジアルキル−2−シクロペンテン−1−アセトアミドの製造方法。
【請求項2】
アルカリもしくはアルカリ土類の水酸化物、炭酸塩、重炭酸塩または第4級アンモニウム水酸化物溶液を加えて、均質なまたは2相性の混合物を得る工程;および、
pKa<2の強酸を加えて、式IV
【化4】
の標題のラクトンを得る工程、
を更に含む、(4R,5S)−3,3a,6,6a−テトラヒドロ−2H−シクロペンタン[b]フラン−2−オンを製造するための請求項1の方法。
【請求項3】
式I
【化5】
の3−アシルオキシ−5−ヒドロキシシクロペンテンと、式IIaのアミドアセタールまたは式IIbのケテンアミノアセタール
【化6】
(式中、
R1およびR′1は、C1〜C4アルキル、または
R1およびR′1は、一緒になって3〜7員環を形成し、
R2は、C1〜C4アルキル、
Acは、C1〜C4アルカノイルである)、
とをアルコールR2OH濃度を容量で3%未満に維持しながら、90℃<の沸点の適切な溶媒中で、90〜140℃において反応させ、式III
【化7】
のアシルヒドロキシシクロペンテンアセトアミドを得る
ことを含む、方法により製造される生成物。
【請求項4】
さらに、アルカリもしくはアルカリ土類の水酸化物、炭酸塩、重炭酸塩または第4級アンモニウム水酸化物溶液を加えて、均質なまたは2相性の混合物を得る工程;および、
pKa<2の強酸を加えて、式IV
【化8】
のラクトンを得る工程、
をさらに含む方法により製造する、請求項3の生成物。
【図1】

【公表番号】特表2006−511576(P2006−511576A)
【公表日】平成18年4月6日(2006.4.6)
【国際特許分類】
【出願番号】特願2004−561853(P2004−561853)
【出願日】平成15年12月10日(2003.12.10)
【国際出願番号】PCT/IB2003/005978
【国際公開番号】WO2004/056749
【国際公開日】平成16年7月8日(2004.7.8)
【出願人】(504396379)ファルマシア・アンド・アップジョン・カンパニー・エルエルシー (130)
【Fターム(参考)】
【公表日】平成18年4月6日(2006.4.6)
【国際特許分類】
【出願日】平成15年12月10日(2003.12.10)
【国際出願番号】PCT/IB2003/005978
【国際公開番号】WO2004/056749
【国際公開日】平成16年7月8日(2004.7.8)
【出願人】(504396379)ファルマシア・アンド・アップジョン・カンパニー・エルエルシー (130)
【Fターム(参考)】
[ Back to top ]