
3,4−ジアミノピリジン誘導体
本発明は、3,4-ジアミノピリジン誘導体、それらの製造方法および3,4-ジアミノピリジン誘導体の使用に関する。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、3,4-ジアミノピリジン誘導体、それらの製造方法および3,4-ジアミノピリジン誘導体の使用に関する。
【背景技術】
【0002】
ドナー置換ピリジンは、多くの合成的に重要な変換、例えば、アルコール、アミンまたはエノラートのアシル化における求核触媒として顕著な役割を担う1、2。
【0003】
最近、(4-ジメチルアミノ)ピリジン1(DMAP)または(4-ピロリジノ)ピリジン2(PPY)(スキーム1参照)の置換誘導体を適切に使用するラセミ化合物の速度論的分割実験において、大きな進歩が得られた[2〜8]。これらの発展にも関わらず、当該分野はやや不安定のままである。ここで種々の解決法が特定の合成問題、例えば第2級アルコールのラセミ化合物の速度論的分割に利用可能であるが、他の明らかに類似した課題、例えば第1級または第3級アルコールのラセミ化合物の速度論的分割は、実質的に解消されていないようである。
【0004】
【化1】

【0005】
スキーム1:(4-ジメチルアミノ)ピリジン1および(4-ピロリジノ)ピリジン2の構造式
【0006】
この状況において、単独の合成戦略内の幅広い範囲の構造的変動を可能にするというモジュール触媒の概念は、非常に望ましいように見える。最近、このタイプの概念が、ペプチド構造に基づき、およびイミダゾールを活性中心として使用する触媒について、Millerらによって研究された2d、9。ペプチドはまた、KawabataらおよびCampbellらによって開発されたPPY誘導体中の変動可能な構造的要素である3、4。これらの場合の全てにおいて、該ペプチド構造は、触媒反応サイクルの速度決定段階における基質と側鎖との間に構築されるさらなる触媒による反応過程に影響を及ぼす。しかしながら、求核触媒反応中心の電子的特性は、通常、ペプチド構造の変動によって変化しない。
【0007】
一方、1および2の3-置換誘導体のモジュール構造は、ピリジン環の求核性並びにその側鎖非対称性の両方を変化させる潜在能力を有する4〜8。これまで行われた実験は、立体的に区別する方法を調査することに限定されていた。
【0008】
既に、アシル化反応におけるドナー置換ピリジンの潜在的触媒能力に関する以前の研究は、触媒活性についての定性基準としてのアシルピリジニウムカチオン4Acの相対的安定性を示した10、11。
【0009】
【化2】
【0010】
スキーム2:アシルピリジニウムカチオンから置換ピリジンへのアシル転位反応(1)
【発明の概要】
【発明が解決しようとする課題】
【0011】
したがって、本発明の目的は、単独の合成戦略内の触媒として使用された場合、幅広い構造的変動を可能にする化合物を提供することにある。さらなる本発明の目的は、幅広い範囲の構造的変動で製造するのに簡単である化合物を提供することにある。特に、アシル化反応用並びにウレタンおよびポリウレタン製造用の触媒として使用し得る化合物を知ることが望ましいであろう。
【課題を解決するための手段】
【0012】
上記目的は、独立請求項の主題によって達成される。好ましい実施態様は、従属請求項から導かれ得る。
本発明の発明者らは、驚くべきことに、3,4-ジアミノピリジン誘導体が製造容易であり、および触媒として有利な特性を有することを見出した。
【0013】
本発明の第一の局面は、以下の式I:
【0014】
【化3】
【0015】
〔式中、R1およびR2は、それぞれ互いに独立して電子ドナーであり、ここでR2はHであってもよく、および
R3、R4、R5およびR6は、それぞれ独立して、H、置換または非置換の直鎖または分枝状のアルキル、アルケニル、アルキニル、アルコキシおよび置換または非置換のアリールの中から選択され、ここで基R3および/またはR4は、基R5および/またはR6と一緒になって環を形成し得る。〕
で示される3,4-ジアミノピリジン誘導体に関する。
【0016】
特に、本発明の3,4-ジアミノピリジン誘導体は、ピリジン環の電子特性の広い変動性に起因して、種々の目的に使用し得る触媒として適当である。ピリジン環の3位および4位における2つの窒素原子の間の架橋エチレン単位は、ピリジン環に対して有利な電子押出効果を発揮し得る。該エチレン架橋は一以上の置換基を有し得る。ピリジン環の4位における少なくとも窒素原子は、完全に置換される。すなわち、これは、もはや水素原子を有しない。
【図面の簡単な説明】
【0017】
【図1】図1は、上記スキーム1の等式(1)によるピリジン1、2および5と比較した、3,4-ジアミノピリジン6〜21についての反応のエンタルピーを示す。
【図2】図2は、化合物34の単結晶X線構造を示す。
【発明を実施するための形態】
【0018】
好ましい本発明の実施態様によれば、本発明者らは、電子ドナーR1およびR2が、それぞれ独立して、置換または非置換の直鎖または分枝状のアルキル、アルケニル、アルキニル、アシル、アルコキシおよび置換または非置換のアリール、より好ましくは置換または非置換のフェニルおよびベンジルの中から選択され、ここでR2はHであってもよい、式Iで示される3,4-ジアミノピリジン誘導体を開示する。
【0019】
本目的のために、電子ドナーは、それぞれの置換部位にて電子密度を高める置換基である。ここで、基R1およびR2は、ピリジン環の3位および4位における窒素原子上で電子密度を高める。
【0020】
本発明の目的のために、上記置換基アルキル、アルケニル、アルキニル、アシルおよびアルコキシは、いずれの場合も、互いに独立して以下のように定義される。
【0021】
用語「アルキル」は、飽和炭化水素基を意味する。該アルキル基は、好ましくは置換または非置換の直鎖または分枝状のC1-C20-アルキル、より好ましくはC1-C10-アルキル、さらにより好ましくはC1-C8-アルキル、さらにより好ましくはC2-C6-アルキルおよび最も好ましくはC3-C5-アルキルである。このようなアルキル置換基の例は、メチル、エチル、n-プロピル、イソプロピル、n-ブチル、sec-ブチルおよびt-ブチルである。
【0022】
用語「アルケニル」は、単独のまたは多重の不飽和基、すなわち、一以上の二重結合を有するアルキル基を意味する。該アルケニル基は、好ましくは置換または非置換の直鎖または分枝状のC2-C20-アルケニル、より好ましくはC2-C10-アルケニル、さらにより好ましくはC2-C8-アルケニル、さらにより好ましくはC3-C6-アルケニルおよび最も好ましくはC3-C5-アルケニルである。
【0023】
用語「アルキニル」は、三重結合による単独のまたは多重の不飽和である基、すなわち、一以上の三重結合を有するアルキル基を意味する。該基は、一以上の二重結合をさらに有し得る。該アルキニル基は、好ましくは置換または非置換の直鎖または分枝状のC2-C20-アルキニル、より好ましくはC2-C10-アルキニル、さらにより好ましくはC2-C8-アルキニル、さらにより好ましくはC3-C6-アルキニルおよび最も好ましくはC3-C5-アルキニルである。
【0024】
用語「アシル」は、-C(O)-R基〔式中、Rは、上記のようなアルキル、アルケニル、アルキニルまたはアリール基である〕を意味する。アシルは、好ましくは置換または非置換の直鎖または分枝状のC1-C20-アシル、より好ましくはC1-C10-アシル、さらにより好ましくはC1-C8-アシル、さらにより好ましくはC2-C6-アシルおよび最も好ましくはC3-C5-アシル、またはベンゾイルである。特に好ましいアシル基はアセチル基である。
【0025】
用語「アルコキシ」は、酸素原子を介して結合したアルキル基を意味する。該アルコキシ基は、好ましくは置換または非置換の直鎖または分枝状のC1-C20-アルコキシ、より好ましくはC1-C10-アルコキシ、さらにより好ましくはC1-C8-アルコキシ、さらにより好ましくはC2-C6-アルコキシおよび最も好ましくはC3-C5-アルコキシである。このようなアルキル置換基の例は、メトキシ、エトキシ、n-プロポキシ、イソプロポキシ、n-ブトキシ、sec-ブトキシおよびt-ブトキシである。
【0026】
さらなる本発明の実施態様において、基R3、R4、R5およびR6 の少なくとも一つはHではない。ここで、基R3、R4、R5およびR6の少なくとも一つがフェニルであることが好ましい。より好ましくは、2つの基R3およびR6が各々Hであり、および2つの基R4およびR5が一緒になって、以下の化合物11において例示されるような6員環を形成するように、1,4-ブタンジイルであることである。しかしながら、この6員環はまた、不飽和環または置換環であることが好ましい。
【0027】
別の好ましい本発明の実施態様にしたがって、本発明者らは、式Iで示される3,4-ジアミノピリジン誘導体であって、式Ia:
【0028】
【化4】
【0029】
〔式中、R1およびR2は、それぞれ互いに独立して電子ドナーであり、ここでR2はHであってもよく、および
R3およびR6は、それぞれ独立して、H、置換または非置換の直鎖または分枝状のアルキル、アルケニル、アルキニル、アルコキシ、および置換または非置換のアリールの中から選択され、
R4およびR5は、それぞれ独立して、置換または非置換の直鎖または分枝状のアルキル、アルケニル、アルキニル、アルコキシ、および置換または非置換のアリールの中から選択され、および
基R3および/またはR4は、基R5および/またはR6と一緒になって環を形成し得る。〕
で示される3,4-ジアミノピリジン誘導体を与えるように、2つの基R4およびR5が互いに対してシス位に配置される3,4-ジアミノピリジン誘導体を開示する。
【0030】
この実施態様において、2つの基R4およびR5は、ピリジン環の原子、ピリジン環の3位および4位における2つの窒素原子および架橋エチレン単位の炭素原子8および9により構成される平面に対してシス位に配置される。炭素原子8および9において複数の置換基が存在する場合、すなわち、基R3およびR4またはR5およびR6の少なくとも2つがHではない場合、基R4およびR5が環を形成する場合、該環がシス位に存在することが特に好ましい。環形成がない場合、基R3、R4、R5およびR6の中の2つのより嵩高い置換基が炭素原子8および9に関してシス位に存在することが好ましい。
【0031】
特に好ましい3,4-ジアミノピリジン誘導体において、R1およびR2はそれぞれエチルであり、R3およびR5はそれぞれHであり、およびR4およびR6は一緒になって、6員環を形成するように、1,4-ブタンジイルである。この化合物は、本明細書中で11として示されている。
【0032】
別の好ましい本発明の実施態様において、R1およびR2は、それぞれ互いに独立して置換または非置換の直鎖または分枝状のC1-C10-アルキル、好ましくは置換または非置換の直鎖または分枝状のC2-C8-アルキルおよび最も好ましくは置換または非置換の直鎖または分枝状のC3-C6-アルキルである。
【0033】
本発明の第二の局面は、式Iで示される3,4-ジアミノピリジン誘導体の製造方法であって、
・3,4-ジアミノピリジンを1,2-ジカルボニル化合物と反応させる工程、
・得られたジイミンを対応するジアミンに還元する工程、および
・該ピリジン環の4位における窒素上の少なくとも水素原子を置換する工程
を含む方法に関する。
【0034】
上記1,2-ジカルボニル化合物は、好ましくは、以下の式II:
【0035】
【化5】
【0036】
で示される化合物である。
第二の局面の好ましい実施態様において、ピリジン環の3位および4位における窒素原子の両方が置換される。
【0037】
ピリジン環の3位および/または4位における窒素原子上のアルキル置換が、窒素原子のアシル化およびその後の得られたアミドのアミンへの還元を含む二段階合成によって達成されることが好ましい。さらなる置換基R3およびR6が導入される場合、これは、好ましくは、アニオン性アルキル基によるジイミンの還元の間に達成される。
【0038】
本発明の第三の局面は、化学反応における触媒としての、上記式Iで示される3,4-ジアミノピリジンの使用に関する。この化学反応は、好ましくはアシル化反応、特にアルコールまたはアミンのアシル化反応、またはウレタンおよび/またはポリウレタン合成である。本発明の3,4-ジアミノピリジンは、有利には、イソシアネートとアルコールとの反応を触媒し得る。したがって、本発明の3,4-ジアミノピリジンは、ウレタンおよび/またはポリウレタンの合成に有用である。
【0039】
触媒の通例の使用において、本発明の3,4-ジアミノピリジンは、触媒される反応に必要な試薬および溶媒に添加される。これらは、アシル化反応またはウレタンおよび/またはポリウレタンの製造に必要な試薬であり得る。
また、本発明の化合物は、薬剤または殺草剤の製造に有利に使用し得る。
【実施例】
【0040】
以下、本発明を、実施例によって、および添付の図面を参照して、より詳細に記載する。
3,4-ジアミノピリジン部分に基づく触媒の開発は、対応するアシル中間体の安定性の理論推定値によって援助される。該理論推定値は、ホモデスミック(homodesmic)アシル転移反応(1)についての反応のエンタルピーによって定量的に表現し得る(スキーム2参照)。
【0041】
3,4-ジアミノピリジンについての多くの反応のエンタルピーを、その触媒潜在能力が以前の研究から既に知られている他のピリジン誘導体についての値と共に、表1に示す。これらとしては、ピリジン(4)、DMAP(1)、PPY(2)および三環式DMAP誘導体5が挙げられ、これらは全て本発明ではない。表1に示す結果を、図1にグラフ形式で示す。
【0042】
【表1】
【0043】
表1.スキーム1の等式(1)によるアセチル転移反応についての、298.15 Kでの反応のエンタルピーΔHrxn(298)。これらは、B3LYP/6-311+G(d,p)// B3LYP/6-31G(d)レベル(単位kJ mol-1)に関して算出された。a 元素e上の電荷の単位;b 電荷および距離パラメーターは、常に、最も好適な配座異性体に基づく。
【0044】
ここで考えられる構造的に最も簡単な3,4-ジアミノピリジンは、テトラヒドロピリド[3,4-b]ピラジン骨格に基づく6である。対応するアセチル中間体6Acの安定性は、より高い触媒活性の5の安定性に近い。1つ(7および21における)または2つ(8におけるような)のアセチル基による6中の2つのメチル基の置換は、ピリジン環中の電子密度を低減させ、したがって、対応するカチオン7Acおよび8Acの安定性も極めて顕著に低減させる。6Acおよび21Acの間の安定性の差(19.5 kJ mol-1)および6Acおよび7Acの間の安定性の差(37.9 kJ mol-1)は、4位中の窒素が、3位中の窒素よりも置換パターンの変化に対して顕著により感受性になることを示す。8中2つの置換基(6に対して56.3 kJ mol-1)の複合効果は、2つの個々の置換基(57.4 kJ mol-1)の合計と実質的に同一である。エチル基による6中の2つのメチル基の置換は、アセチル中間体の安定性を10 kJ mol-1高める。シス配置(9におけるような)またはトランス配置(10におけるような)のいずれかの系6上への飽和アルキル環の縮合も同様に、アセチル中間体の安定性を約15 kJ mol-1高める。三環系9および10に由来するアセチルピリジニウムカチオンの安定性は、二環系6について記載されたものと同様の様式でN置換基に依存する。したがって、2つのエチル基の導入は、最も安定性のアセチル中間体11Acおよび15Acを導く。最後に、6のエチレン架橋(例えば19におけるような)中へのアリール置換基の導入も、対応するカチオン性中間体の安定性を高める。これは、おそらく3,4-ジアミノ窒素原子への誘導電子移動に起因する。図1中の化合物のアセチル中間体についての安定性値は、実質的に90 kJ mol-1の範囲に渡る。化合物1、2および5の場合において、それらの安定性値は、触媒活性に関連することが見出された。理論に束縛されることを意図しないが、3,4-ジアミノピリジン18および11(これはスケールの2つの末端に印を付ける)の安定性値は、30の因子によるそれらの触媒活性に関して異なると考えられる。さらに、表1中の電荷および距離データは、より大きい熱力学安定性がより短いC-N結合長さおよびより小さいアセチル基電荷に対応することを示す。
【0045】
図1に示される化合物の幾つかを、3,4-ジアミノピリジン22から、三〜五段階の順序で、効率的に製造することができる(スキーム3参照)。22とグリオキサール、1,2-シクロヘキサジオンまたはベンジルとの縮合は、良好な収率のジイミン23〜25を導く[12]。次いで、ピリドピラジン23(R=H)は、テトラヒドロ誘導体26に還元され13、そして、Eschweiler-Clark条件下、アルキル化されて化合物6を形成することができた14。より長いアルキル鎖の導入は、ビス-アシル化およびその後の対応するジアミンへの還元を含む二段階の順序によって、より効率的に達成される15。このように、26から出発して、ジエチル化化合物20を48%の収率で製造できた。類似の手順は、ジアミン27から、2つの工程で37%の収率で得られるシクロヘキシル縮合系11の合成を可能にする。また、シス縮合ジアミン27も、24から、NaBH4/BH3/H2NCH2CH2OHを使用する選択的還元16によって74%の収率で、または-40℃でのLiAlH3を使用する還元によって90%の収率で得ることができる。化合物26について使用されるより簡単な還元方法の使用は、この場合、27のシス/トランス混合物を導く。
【0046】
【化6】
【0047】
スキーム3:3,4-ジアミノピリジン部分に基づく触媒の合成。
a) グリオキサール、1,2-シクロヘキサジエノンまたはベンジル、EtOH、70℃、5時間、95〜98%。
b) 粉末NaBH4、EtOH、40℃、24時間、50%。
c) 200当量のCH2O(水中37%)、100当量のHCO2H、110℃、48時間、57%。
d) (CH3CO)2O、ピリジン、100℃、24時間、80%。
e) 4.2当量のLiAlH4、2.6当量のAlCl3、MTBE、0℃、1時間、次いで、還流、8時間、60%。
f) LiAlH4、THF、-40℃〜室温、32時間、90%。
g) (CH3CO)2O、25 mol%のPPY、ピリジン、100℃、48時間、68%。
h) 4.2当量のLiAlH4、2.6当量のAlCl3、MTBE、0℃、1時間、次いで、還流、8時間、55%。
i) 1.1当量の(CH3CO)2O、0.2当量の2、NEt3、DCM、室温、3時間、82%。
j) 2.2当量のLiAlH4、1.3当量のAlCl3、MTBE、0℃、1時間、次いで、還流、8時間、65%。
k) 1) 1当量のn-BuLi、THF、-78℃、1時間、次いで、0℃、1時間;2) 1.1当量のCH3COCl、THF、-78℃〜室温、1時間、16%。
l) 粉末NaBH4、EtOH、40℃、48時間、74%。
m) 88当量のCH2O(水中37%)、200当量のHCO2H、110℃、48時間、90%。
n) 1) 1当量のn-BuLi、MTBE、-78℃、1時間、次いで、0℃、1時間;2) 1.1当量のCH3COCl、MTBE、-78〜0℃、1時間、36%。
o) 2.2当量のLiAlH4、1.3当量のAlCl3、THF、0℃、1時間、次いで、還流、18時間、53%。
【0048】
27の反応についてのより穏やかなアシル化方法の使用は、C4アミノ基の顕著により高い反応性を示し、これは、唯一の反応生成物としてのアミド28の優先的形成を説明する。したがって、従来通り、対応するアミンを形成するためのこのアミドの還元およびC3上の残留アミノ官能基のアシル化は、異なる強度の2つのドナー置換基を含有する触媒を製造するのを可能にする。C3およびC4上の2つのアミノ窒素原子を連結する架橋へのアリール置換基の影響を、化合物34の合成によって調査した。対応するピリドピラジン25の還元は、26について使用される条件下、容易に起こり得るが13、その後のEschweiler-Clarkアルキル化は、モノアルキル化段階にて停止して化合物32を与える。32のアセチル化は、驚くべきことに、先に使用した全ての条件下で困難であることがわかった。しかしながら、これは、最終的に、最初にn-BuLiを使用して脱プロトン化し、アミドアニオンとAcClとを反応させ、次いで、還元することによって行うことができ、その結果、34を形成することができた。
【0049】
スキーム3に示される全ての化合物の組成を、高分解能質量分析によって示す。ピリジン環の構造は、全ての化合物について、1H-NMRスペクトルの適切な領域(6.5〜8 ppm)における3つのシグナル(1つのシングレット、2つのダブレット)の存在から直接的に導くことができる。これは、化合物11、14および27〜30におけるシクロヘキサン縮合の立体化学的配置およびN-アルキル化およびN-アシル化反応の位置化学的制御の問題を残す。構造11(スキーム4参照)中の隣接プロトンHaおよびHeについての1H-NMRシグナルは、3JHH=2.8 Hzの結合定数を有し、および軸方向-エクアトリアル配置またはエクアトリアル-エクアトリアル配置のいずれかと調和し得る正のNOEシグナルを示す17。
【0050】
しかしながら、2つのプロトンのエクアトリアル-エクアトリアル配向は、除外することができる。なぜなら、3,4-ジアミノピリジン単位は、シクロヘキサン環に二軸縮合によって結合することがほとんどできないからである。
【0051】
【化7】
【0052】
スキーム4:矢印は、観察されたNOE強化を示す。
【0053】
28を形成するための27のアセチル化の位置選択性は、直接的に確認できなかった。なぜなら、28の分光データは、この点に関して情報を与えるものではなかったからである。しかしながら、28〜29の還元後、ピリジン環のC5位におけるエチル基とプロトンとの間の正のNOEシグナルを観察することができる。これは、スキーム3に示される構造を支持する。同様に、32を形成するための31のN-メチル化の位置化学は、メチル基についてのシグナル(2.85 ppm)と、ピリジン環のC1でのプロトンについてのシグナル(7.82 ppm)と、ベンジルのプロトンの一つのシグナル(4.44 ppm)との間のNOE強化の観察によって確率することができた。二重アルキル化生成物34におけるこの位置化学の保持は、この化合物(図2参照、イソヘキサン/ジクロロメタンから結晶化されたもの)の単結晶X線構造分析によって、疑いなく確認することができた。
【0054】
〔触媒特性〕
ピリジン1、2、5、6、8、11、20および34の触媒特性を、スキーム5に示す2つのアシル化反応AおよびBにおいて調査した。
【0055】
【化8】
【0056】
スキーム5:アシル化反応AおよびB
【0057】
第3級アルコール35は、以前の研究において、その低い反応性のためにモデル基質として使用された10。補助塩基としてのトリエチルアミンの存在下での無水酢酸(36)による35のアセチル化(反応A)は、少なくともDMAP(1)の反応性を有する触媒を10%の濃度(35に対して)で使用する限り、定量的に進行する。該反応の半減期τ1/2は、1H-NMRスペクトルにおける出発材料および生成物のシグナルの積分によって決定することができる。ここで、該半減期を触媒活性の指標として使用する。反応Bにおいて、同一のアルコールを、従来通り、アシル化試薬としてのイソ酪酸無水物(38)と反応させる。この反応は、36の反応よりも顕著にゆっくりと進行するため、該反応のために、より高い反応温度(40℃)を選択した。この選択は、2つの反応のほぼ同じ半減期を導く。
【0058】
DMAP触媒アシル化反応に関する最近の研究に基づいて18,19、非極性溶媒中の両方の反応について、スキーム6に示される機構を推測することができる。したがって、無水物および触媒を、まず、(通常迅速)の第一段階において反応させ、アシルピリジニウムカチオンとカルボキシレートアニオンとの錯体を形成する。この錯体は、通常、本明細書中で使用される条件下、1H-NMRスペクトル中に検出することができない。したがって、対応する平衡定数Kは、反応AおよびBの両方について、非常に小さいと推測することができる。
【0059】
【化9】
【0060】
スキーム6:反応AおよびBによるアシル化機構
【0061】
次いで、速度決定工程において、該アシルピリジニウムカチオンを、アルコール35と反応させてエステル生成物を形成し、および触媒を不活性化する。該触媒は、補助塩基NEt3の作用によって再生することができる。ここで測定される半減期τ1/2(表2)は、該工程の両方に対する触媒の影響を反映する。したがって、Kに対する置換基効果が、kcatに対する効果よりも大きい場合にのみ、アセチルピリジニウムカチオンについてのτ1/2と算出された安定性値(表1)との明らかな相関関係が予想され得る。
【0062】
【表2】
【0063】
表2.モデル反応AおよびBについての反応半減期τ1/2(単位:分)。括弧内の結果は、補助塩基としてHuenig塩基(EtN(i-Pr)2)を使用して得られた。a 参考文献20参照;b 参考文献23参照
【0064】
表2中の化合物1、2および5についての反応性データは、先行文献中の同一条件下での同一反応についての反応性データから幾分外れている10。これは、部分的に、該反応(実験部参照)に続くNMR測定における小さな差に起因する。さらに、比較的活性な触媒(例えば5)についての結果は、並外れて純粋な試料を使用する場合のみ、再現することができる。他方、厳密に嫌気的な条件下での反応の実施は必要ではない。触媒6、11および20についての表2中の結果は、ジアミノピリジンがモノ置換ピリジン(例えばDMAPまたはPPY)よりも顕著に反応性であることを示す。最高の結果は、上記炭素環式化合物5に対して同様の活性を有する化合物11について得られた。20および11についての結果の比較は、架橋部分のドナー特性の強い影響を示すが、N-メチル置換またはN-エチル置換(20対6)の間の差は、小さい効果を有する傾向にある。これらの結果は、表1および図1における安定性値と完全に一致するが、関連化合物19の安定性値によって測定された34の触媒活性は、驚くべきことに低い。該触媒活性は、アクセプター置換基の導入によって顕著に低減される。これは、30の低活性(11と比較して)によって、並びにN-アシル化化合物8を使用する場合の顕著に遅い反応によって示される。これは、対応するアセチル中間体の低安定性値の観点から予期されるべきことであったが、それにもかかわらず、本発明者らは、図1中の安定性スケールが、全ての場合において、表2中の実験的に決定された反応性データに一致しているわけではないことを認識しなければならない。基質の選択に対する反応性データの依存性は、反応AおよびBについての結果の比較によって明らかになる。最も活性な化合物11およびDMAPについての反応性比は、反応Aについて7.0であり、および反応Bについて4.8である。同様の結果は、ピリジン誘導体5について得られ、これは、7.9および4.5の反応性比を示す。補助塩基の選択は、触媒5および11について調査されたさらなる実験的変数を表す。ここで、驚くべきことに、両方の触媒についての触媒効率は、NEt3に比べてより強い補助塩基EtN(iPr)2(Huenig塩基)を使用する場合、比較可能なように低減することが見出された。
【0065】
アルキル置換3,4-ジアミノピリジンは、アルコールのアシル化についての触媒の新規クラスを表す。これらの化合物の触媒効率は、窒素置換基の変動によって、単独の合成戦略内で幅広い範囲に渡って変動し得る。これらの触媒の最も良好なものの触媒活性は、これまで既知のDMAPの炭素環式誘導体の最も良好なものに匹敵する。後者の化合物とは対照的に、最も活性な3,4-ジアミノピリジン触媒は、柔軟な3-または4-段階合成によって、簡単な方法で得ることができる。当該方法は、さらなる構造的変動のために、容易に改変することができる。
【0066】
幾何学の最適化は、原子価殻分割型基底関数6-31G(d)と組み合わせたBecke3LYPハイブリッド汎関数を使用して行った。298.15 Kの温度での熱化学的エンタルピー補正は、同じレベルでの振動数計算によって行う。一点計算をBecke3LYP/6-311+G(d,p)レベルについて行う。Becke3LYP/6-31G(d)レベルでの熱化学的補正によって得られたエネルギーの組合せは、本明細書中に記載されたH298値を与える。全ての計算は、Gaussian 03を使用して行う21。
【0067】
水分を除外して行う全ての反応は、乾燥溶媒および乾燥オーブンおよびヘアドライヤーによって乾燥した反応装置中、窒素雰囲気下で行う。THFは、窒素雰囲気下、水素化ナトリウムから蒸留する。メチルtert-ブチルエーテル(MTBE)、ピリジン、トリエチルアミンおよび重クロロホルムは、窒素雰囲気下、水素化カルシウムから蒸留する。無水酢酸およびイソ酪酸無水物は、減圧下、P4O10から乾燥K2CO3上に蒸留し、濾過し、および減圧下、分画する。両方の無水物は、窒素雰囲気下、4Å分子篩上で貯蔵する。エタノール、ジクロロメタン、酢酸エチルおよびメタノールは、ロータリーエバポレーターで蒸留する。n-BuLiは、ジフェニル酢酸で滴定し、モル濃度を決定する。全ての他の試薬は、本明細書中で他に示さない限り、さらなる精製をせずに、最も高い可能性のある品質で使用する。薄層クロマトグラフィーは、Merck KGaAからの蛍光染料でマークしたTLCプレート(シリカゲル60 F254、層厚さ:0.2 mm)およびFlukaプレート(塩基性酸化アルミニウムF254、Brockman活性:1、pH:9.5、層厚さ:0.2 mm)を用いて行う。フラッシュクロマトグラフィーは、Merck KGaAシリカゲル60(粒度:0.040-0.063 mm)およびFluka塩基性酸化アルミニウム(塩基性酸化アルミニウムF254、Brockman活性:1、pH:9.5、粒度:0.05-0.15 mm)を使用して、シリカゲルについて1.5 barの圧力にて、および塩基性酸化アルミニウムについて0.5 barの圧力にて行う。1H-および13C-NMRスペクトルは、Varian Mercury 200、Varian 300、Varian INOVA 400およびVarian 600機器を用いて記録する。NOEスペクトルは、CDCl3中、27℃にて記録する。化学シフトは、溶媒ピークに対するppmで報告する22。以下の略語を使用して1H-NMRスペクトルにおけるシグナルの多重度を特徴付ける:s=シングレット、d=ダブレット、t=トリプレット、q=カルテット、quin=クインテット、sex=セクステット、sep=セプテット、m=マルチプレット、b=ブロードおよび使用した略語の組合せ。全ての1Hおよび13Cシグナルは、COSY、NOESY、HSQC、HMBCおよびDEPT実験を用いて割り当てる。反応速度測定についての1H-NMRスペクトルは、プログラムVNMR 4.3 Rev. G0194を使用して評価する。関連ピークの積分は、社内で書かれたサブプログラムを使用して自動的に評価する。サブルーチンは、MAGICAL(商標) IIプログラミングを使用して記載する。IRスペクトルは、試料媒体としてKBrペレットを用いて、Perkin-Elmer 1420赤外分光計およびATR技術を使用するPerkin-Elmer FT-IRスペクトルBX分光計を使用して記録する。全てのシグナルは、vs=非常に強い、s=強い、m=中程度およびw=弱いとして報告する。質量スペクトルは、イオン源として電子衝撃イオン化(EI、70 eV)または化学イオン化(CI、イソブタン)を使用して、Finnigan MAT 95によって記録する。ESI-MSスペクトルは、Thermo Finnigan LTQ FT機器を使用して記録する。ガスクロマトグラムは、25 m CS-Supreme-5キャピラリーカラムを備えたVarian 3400 GCによって、検出器としてFinnigan MAT 95質量分析計を使用して記録する。電子が豊富なピリジン11、5、6、20および32の場合、溶媒は、フラッシュクロマトグラフィー後、窒素流をロータリーエバポレーター中に通じることによって留去する。
【0068】
〔反応性実験AおよびBについての一般的手順〕
重クロロホルム、トリエチルアミンおよびHuenig塩基を、使用前に、窒素雰囲気下、水素化カルシウムから新たに蒸留する。全ての反応速度測定を、反応Aについて23℃および反応Bについて40℃の一定の温度にて、Varian Mercury 200分光計を用いて記録する。以下の重クロロホルム中の標準溶液を、3つの乾燥した5 ml容のフラスコ中で作製した。
A:1.2 Mの無水酢酸溶液
B:0.6 Mのエチニルシクロヘキサノール溶液および1.8 MのトリエチルアミンまたはHuenig塩基溶液
C:0.06 Mの触媒溶液
【0069】
(反応Aについての試料調製および反応速度測定)
いずれの場合も、200 μlの上記標準溶液を、エッペンドルフ型ピペットを用いてNMRチューブ中に取る。該反応混合物を混合し、そして直ぐにNMR分光計中に置く。該反応を、所定の時間間隔にて100%変換までNMRスペクトルを記録することによって監視する。
【0070】
(反応Bについての試料調製および反応速度測定)
溶媒の蒸発を防止するために、NMRチューブを、バーナーを用いてフレームシールすること以外は、反応Aについて記載された手順。
【0071】
(反応Aについての半減期の決定)
アセチル基のプロトンに割り当てられ得る全てのシグナルを、スペクトルの記録後、自動的に積分する。ここで、無水酢酸、エステルおよび酢酸トリエチルアンモニウムを、以下の積分限界内で積分する:±8、±2および±6 Hz。該変換を、等式2にしたがって算出する。該変換を、等式4を用いて時間の関数として自己矛盾なく適合させる。該半減期を、関数4を使用して50%変換にて算出する。
【0072】
(反応Bについての半減期の決定)
イソ酪酸基のダブレットおよびジオキサンのシグナルを、NMRスペクトルの記録後、自動的に積分する。ここで、ジオキサンおよびイソ酪酸無水物を、同じ積分限界±2 Hzの範囲内で積分する。該変換を、等式3にしたがって算出する。該変換を、等式4を用いて時間の関数として自己矛盾なく適合させる。該半減期を、関数4を使用して50%変換にて算出する。
【0073】
【数1】
【0074】
〔方法〕
ピリド[3,4-b]ピラジン(23):
2.00 g(18.33 mmol)の3,4-ジアミノピリジンを、30 mlのエタノール中の2.82 mlのグリオキサール(水中40重量%、ρ=1.265 g/ml、62.48 mmol)の溶液に添加する。該反応混合物を、70℃の油浴温度にて5時間維持し、次いで、室温に冷却後、該溶媒をロータリーエバポレーターによって留去する。粗生成物を、フラッシュクロマトグラフィー(EtOAc/イソヘキサン、9:1)によって精製する。これにより2.35 g(17.96 mmol、98%)の白色粉末を得る。
Rf=0.27 (イソヘキサン/EtOAc, 1:9). 1H NMR (200 MHz, CDCl3):δ=7.94 (dd, 3J=5.8 Hz, 4J=0.6 Hz, 1H, H-7), 8.83 (d, 3J=5.8 Hz, 1H, H-8), 8.96 (d, 3J=1.6 Hz, 1H, H-3), 9.02 (d, 3J=1.6 Hz, 1H, H-2), 9.56 (d, 4J=0.6 Hz, 1H, H-5) ppm. 13C NMR (100 MHz, CDCl3):δ=121.6 (CH, C-7), 138.0 (Cq), 145.3 (Cq), 147.3 (C-H, C-8), 146.5 (C-H, C-3), 149.2 (CH, C-2), 154.8 (C-H, C-5) ppm. GC-MS (EI):保持時間5.45分, m/e (%):132 (8), 131 (M+, 100), 104 (25), 77 (13), 50 (10) IR (KBr):□=3435 (vs), 3092 (w), 3023 (m), 1969 (w), 1758 (w), 1631 (w), 1598 (vs), 1562 (m), 1536 (w), 1488 (s), 1436 (vs), 1416 (m), 1381 (m), 1351 (w), 1290 (w), 1279 (m), 1212 (m), 1201 (m), 1148 (w), 1033 (s), 1014 (s), 972 (w), 959 (vs), 931 (m), 881 (vs), 838 (m), 824 (s), 773 (w), 651 (s), 623 (m), 546 (w), 524 (w), 457 (s) cm-1.
【0075】
1,2,3,4-テトラヒドロピリド[3,4-b]ピラジン(26):
2.90 g(0.076 mol)のNaBH4粉末を、100 mlの乾燥エタノール中のキノキサリン23(2.90 g、0.022 mol)の溶液に添加する。該反応溶液を、40℃の油浴温度に加熱し、次いで、この温度にて24時間維持する。その後、該反応溶液を室温に冷却し、そして3 mlの水を添加する。該無機固体を吸引によって濾去し、そして20 mlのDCMで2回洗浄する。合わせた有機相を硫酸ナトリウムによって乾燥し、次いで、該溶媒をロータリーエバポレーターによって留去する。得られる粗生成物を、塩基性酸化アルミニウムによるフラッシュクロマトグラフィー(EtOAc/MeOH、10:1)によって精製する。これにより白色発泡体を得る。
Rf=0.15 (塩基性酸化アルミニウム、20:1、DCM:MeOH). 1H NMR:(200 MHz, CDCl3):δ=3.37 (d, 3J=5.8 Hz, 1H, H-3), 3.38 (d, 3J=5.8 Hz, 1H, H-2), 6.29 (d, 3J=5.4 Hz, 1H, H-8), 7.67 (d, 3J=5.4 Hz, 1H, H-7), 7.67 (s, 1H, H-5). 13C NMR:(100 MHz, CDCl3):δ=40.1 (CH2), 41.0 (CH2), 107.8 (C5), 129.6 (Cq), 135.3 (C-H, C7), 140.1 (Cq), 141.1 (C-H, C-5). GC-MS 保持時間8.09分 (EI) m/e (%):136 (8), 135 (M+ 79), 134 (M+-H+,100), 133 (10), 132 (7), 131 (2), 120 (4), 107 (7), 105 (4), 94 (2), 93 (7), 80 (2), 79 (3), 78 (4), 67 (4), 66 (2), 53 (2), 52 (2), 52 (3), 51 (2). IR (KBr):□=3349 (m), 2859 (m), 1593 (s), 1534 (s), 1474 (s), 1344 (s), 1311 (s), 1281 (s), 1256 (w) 1228 (m), 1182 (m), 1102 (m), 1051 (w), 1038 (m), 893 (m), 862 (m), 824 (s), 772 (m) cm-1. HRMS (EI) (%):C7H8N3 (M-H+)の計算値:134.0718、実測値:134.0709.
【0076】
1-(4-アセチル-3,4-ジヒドロ-2H-ピリド[3,4-b]ピラジン-1-イル)エタノン(8):
23 ml(24.71 g、24.20 mol)の無水酢酸を、60 mlのピリジン中のテトラヒドロキノキサリン26(1.49 g、0.011 mol)の溶液に0℃の温度にて添加する。その後、該反応混合物を100℃にて48時間加熱する。室温に冷却後、該溶媒を減圧下留去し、そして該黄色固体をフラッシュクロマトグラフィー(EtOAc:MeOH、10:3)によって精製する。これにより1.93 g(8.80 mmol、80%)の淡黄色固体を得る。
1H NMR (400 MHz, CDCl3):δ=2.25 (s, 3H, CH3), 2.31 (s, 3H, CH3), 3.90 (ddd, 2J=12 Hz, 3J=4 Hz, 3J=4 Hz, 2H, CH2), 3.94 (ddd, 2J=12 Hz, 3J=4 Hz, 3J=4 Hz, 2H, CH2), 7.67 (s, 1H, H-5), 7.87 (bs, 1H, H-5), 8.32 (d, 3J=4 Hz, 1H, H-7), 8.46 (m, 1H, H-8). 13C NMR (100 MHz, CDCl3):δ=22.3, 22.8 (CH3), 42.1, 46.5 (CH2), 117.4 (C5), 128.2 (Cq), 139.0 (Cq), 145.7 (C-7), 146.9 (C-8), 168.6 (C=O). GC-MS(EI) 保持時間9.66分, m/e (%):220 (14), 219 (M+, 81), 178 (11), 177 (M+-AcO,100), 176 (10), 162 (3), 159 (2), 136 (7), 135 (78), 134 (M+-2AcO,99), 133 (8), 132 (9), 120 (4), 119 (2), 107 (5), 105 (2), 93 (3), 80 (2), 79 (4), 78 (4), 52 (2), 51 (2), 43 (AcO+,20);IR (neat):□=2960 (w), 1684 (s) 1654 (vs) 1582 (m) 1494 (s), 1407 (vs), 1330 (m), 1320 (vs), 1277 (m), 1259 (s), 1248 (m), 1217 (s), 1248 (s), 1217 (s), 1179 (m), 1150 (w), 1118 (m), 1064 (m), 1033 (s), 969 (s), 886 (w), 857 (m), 846 (s), 799 (s), 764 (w), 749 (w), 704 (w) cm-1. HRMS (EI): C11H13N3O2 [M+]の計算値:219.1008;実測値:219.0995.
【0077】
1,4-ジエチル-1,2,3,4-テトラヒドロピリド[3,4-b]ピラジン(20):
1.56 g(11.69 mmol)のAlCl3を、60 mlのMTBE中に室温にて懸濁させる。45分間の撹拌後、該反応混合物を0℃に冷却し、およびLiAlH4(1.32 g、34.65 mmol)を少しずつ添加する。添加が完了した後、該混合物を別の15分間攪拌し、化合物8(1.00 g、4.56 mmol)を添加し、そして該混合物を、0℃にて別の1時間撹拌する。その後、該反応混合物を、8時間還流し、そして室温に冷却後、氷水中に注ぐ。沈澱した無機固体を吸引によって濾去し、そして30 mlのDCMで2回洗浄する。該水相をpH 12にし、そして40 mlのDCMで3回抽出する。合わせた抽出物をNa2SO4によって乾燥し、次いで、該溶媒をロータリーエバポレーターによって留去する。粗生成物を、シリカゲルによるフラッシュクロマトグラフィー(EtOAc/MeOH/NEt3、10:0.5:1)によって精製する。これにより、0.52 g(60%)の冷凍庫中で固化する無色油を得る。
Rf=0.47 (EtOAc/MeOH/NEt3、10:0.5:1). 1H NMR:(400 MHz, CDCl3):δ=1.15 (q, 3J=14 Hz, 6H, CH3), 3.22 (m, 3J=6 Hz, 2H, H-3), 3.23 (m, 2J=14 Hz, 4H, CH2), 3.42 (m, 3J=6 Hz, 2H, H-2), 6.34 (d, 3J=5.2 Hz, 1H, H-8), 7.69 (s, 1H, H-5), 7.75 (d, 3J=5.2 Hz, 1H, H-7). 13C NMR:(100 MHz, CDCl3):δ=10.1, 10.4 (CH3), 44.6, 46.5 (CH2, C-2, C-3), 44.9, 45.0 (CH2), 104.0 (C-8), 130.5, 130.1 (Cq), 131.8 (C-5), 140.7 (C-7). GC-MS(EI):保持時間8.63分, m/e (%):192 (10), 191 (M+,100) , 190 (4), 177 (8), 176 (95), 175 (5), 162 (11), 161 (8), 160 (5), 148 (13), 147 (10), 146 (8), 135 (2), 134 (9), 133 (6), 131 (8), 121 (2), 119 (4), 118 (3), 107 (3), 104 (2), 92 (2), 91 (2), 80 (8), 77 (4) IR (KBr):v=3436 (s), 3112 (w), 3019 (w), 2964 (w), 1661 (s), 1686 (vs), 1583 (m), 1497 (m), 1409 (s), 1363 (w), 1332 (m), 1311 (s), 1260 (m), 1249 (w), 1232 (m), 1220 (m), 1180 (m), 1150 (w), 1119 (m), 1064 (w), 1035 (m), 985 (m), 970 (m), 886 (s), 858 (m), 847 (m), 802 (m), 765 (w), 740 (w), 704 (w), 647 (w), 614 (w), 592 (w), 577(m), 570 (m), 507 (w) cm-1. HRMS (EI):C11H17N3 [M+]の計算値:191.1422;実測値:191.1430.
【0078】
1,4-ジメチル-1,2,3,4-テトラヒドロピリド[3,4-b]ピラジン(6):
35 mlのギ酸を、氷中で冷却しながらテトラヒドロキノキサリン26(1.26 g、9.32 mmol)に添加する。その後、12 mlのホルムアルデヒド溶液(水中<37%)を添加し、そして該混合物を110℃の油浴温度にて48時間加熱する。その後、室温まで冷却し、そして約150 mlの20%濃度の水酸化ナトリウム溶液を、氷中で冷却しながら添加する。該pHは、この添加の間に12を超えるべきではない。母液を、液体/液体抽出装置中で、250 mlのDCMを用いて、終夜抽出する。該有機相を、Na2SO4によって乾燥し、次いで、該溶媒をロータリーエバポレーターによって留去する。粗生成物を、シリカゲル(EtOAc/MeOH/NEt3、10:1:1)および塩基性酸化アルミニウム(EtOAc/MeOH、10:1)によるフラッシュクロマトグラフィーによって精製する。これにより0.849 g(5.32 mmol、57%)の黄色固体を得る。
Rf=0.56 (塩基性アルミナ、EtOAc/MeOH、10:1). 1H NMR:(400 MHz, CDCl3):δ=2.77 (s, 3H, CH3), 2.82 (s, 3H, CH3), 3.12 (m, 2H, CH2-B), 3.37 (m, 2H, CH2-C), 6.21 (d, 3J=5.6 Hz, 1H, H-7), 7.56 (s, 1H, H-5), 7.74 (d, 3J=5.6 Hz, 1H, H-8) ppm. 13C NMR:δ=(75 MHz, CDCl3):38.0 (N-1-CH3), 39.1 (N-4-CH3), 48.8 (C-3), 49.6 (C-2), 104.0 (C-7), 131.2 (C-5), 132.3 (C-4a), 141.6 (C-7), 142.2 (C-8a) ppm;GC-MS(EI):保持時間8.05分, m/e (%):164 (8), 163 (M+,100), 162 (21), 161 (5), 149 (4), 148 (40) 147 (9), 146 (8), 134 (8), 133 (11), 132 (6), 121 (4), 120 (2), 119 (6), 107 (3), 105 (2), 93 (2), 92 (4), 81 (6), 80 (4), 79 (2), 78 (4), 66 (3), 51 (2), 42 (4). IR (neat):□=3378 (w), 3035 (w), 2979 (w), 2873 (m), 2826 (s), 2792 (w), 1581 (s), 1519 (s), 1466 (s), 1454 (s), 1435 (m), 1416 (m), 1380 (w), 1335 (vs), 1290 (s), 1250 (w), 1235 (vs), 1214 (m), 1172 (s), 1114 (s), 1099 (s), 1069 (s), 1030 (m), 936 (w), 911 (w), 883 (s), 815 (s), 800 (s), 783 (s), 746 (m), 709 (m), 622 (m) cm-1. HRMS (EI):C9H13N3 [M+]の計算値:163.1109;実測値:163.1089.
【0079】
6,7,8,9-テトラヒドロピリド[3,4-b]キノキサリン(24):
1,2-シクロヘキサンジオン(1.52 g、13.93 mmol)を、50 mlのエタノール中の1.56 g(13.93 mmol)の3,4-ジアミノピリジンの懸濁液に添加する。その後、該反応混合物を70℃の油浴温度にて5時間加熱し、そして室温に冷却し、次いで、該溶媒をロータリーエバポレーターによって留去する。得られる生成物を、溶離剤として酢酸エチルを使用するフラッシュクロマトグラフィーによって精製する。これにより1.84 g(71%)の白色固体を得る。該固体は、冷蔵庫中に貯蔵し、そしてさらなる1〜2日後に処理すべきである。該固体を開口したフラスコ中で日光中に放置することによって、灰緑色への変色が生じる。
Rf=0.25 (EtOAc/ヘキサン、20:1). 1H NMR:(200 MHz, CDCl3):δ=2.05 (m, 4H, H-7,8), 3.18 (m, 4H, H-6,9), 7.79 (d, 3J=5.8 Hz, 1H), 8.72 (d, 3J =5.8 Hz, 1H), 9.38 (s, 1H) ppm. 13C NMR (75 MHz, CDCl3):δ=22.8 (CH2, C-7,8), 33.7 (CH2, C-6,9), 121.2 (CH), 136.9 (Cq), 144.1 (Cq), 146.8 (CH), 153.9 (CH), 156.8 (Cq) 159.9 (Cq) ppm. GC-MS(EI):保持時間8.44分, m/e (%) 186 (11), 185 (M+,100), 184 (39), 183 (3), 182 (3), 171 (2), 170 (21), 169 (4), 158 (2), 157 (5), 156 (5) 131 (2), 104 (2), 103 (4), 78 (3), 76 (4), 67 (2), 64 (2), 51 (2), 50 (6);IR (KBr):□=3435 (vs), 2947 (s), 2864 (m), 1594 (s), 1557 (w), 1461 (w), 1421 (m), 1385 (s), 1365 (w), 1330 (w), 1297 (m), 1211 (m), 1140 (w), 979 (m), 949 (w), 901 (m), 849 (m), 679 (w), 629 (w), 570 (w), 412 (w) cm-1. HRMS (EI):C11H11N3の計算値:185.0953 [M+]、実測値:185.0935
【0080】
5,5a,6,7,8,9a,10-オクタヒドロピリド[3,4-b]キノキサリン(27):
6.13 g(33.09 mmol)の24および100 mlのTHFを、250 mlのSchlenkフラスコ中に置く。該溶液を-40℃に冷却し、そして3.09 g(81.42 mmol、2.5当量)のLiAlH4を少量ずつ添加する。該反応混合物を室温まで温め、そして室温にて32時間撹拌する。その後、氷水中に注ぎ、次いで、該水相をpH 12にする。該無機固体を分離除去し、そして40 mlのDCMで2回洗浄する。母液を、液体/液体抽出装置中で、250 mlのDCMを用いて8時間抽出する。合わせた有機相をNa2SO4によって乾燥し、次いで、該溶媒をロータリーエバポレーターによって留去する。得られる粗生成物のフラッシュクロマトグラフィー(EtOAc/MeOH/NEt3、10:1:1)によって、5.63 g(29.78 mmol、90%)の白色固体を得る。
Rf=0.36 (EtOAc/MeOH/NEt3、10:1:1). 1H NMR:(300 MHz, CDCl3):δ=1.32-1.40 (m, 3H), 1.56-1.75 (m, 5H), 3.41 (m, 1H), 3.46 (s, 1H, N-H), 3.51 (m, 1H), 4.20 (s, 1H, N-H), 6.29 (d, 1H, 3J=5.4 Hz, H-3), 7.65 (d, 1H, 3J=5.4 Hz, H-4), 7.68 (s, 1H, H-1) ppm. 13C NMR (75 MHz, CDCl3):δ=22.0, 22.2 (C-7,8), 30.4, 30.8 (CH2, C-6,9), 49.2 (CH, C-5a), 50.1 (CH, C-9a), 107.7 (C-4), 128.8 (Cq, C-10a), 134.8 (C-3), 139.2 (Cq, C-4a) 140.7 (C-4) ppm. GC-MS (EI):保持時間10.06分, m/e (%) 190 (13), 189 (M+, 99), 188 (9), 187 (3), 185 (10), 184 (4), 170 (4), 160 (10), 159 (3), 158 (5), 148 (2) 147 (16), 146 (100), 145 (2), 134 (13), 133 (17), 132 (15), 120 (10), 119 (3), 105 (2) 94 (3), 93 (2), 78 (3), 66 (2), 40 (2);IR (neat):□=3220 (s), 2927 (s), 2852 (s), 2354 (m), 1725 (w), 1595 (vs), 1523 (vs), 1456 (w), 1443 (m), 1403 (w), 1362 (s), 1294 (s), 1269 (s), 1240 (m), 1206 (s), 1175 (s), 1087 (m), 1052 (m), 1003 (m), 938 (m), 884 (m) 810 (vs) 725 (m) cm-1. HRMS (EI):C11H15N3の計算値:189.1266 [M+]、実測値:189.1259.
【0081】
別の方法として、該反応は、試薬としてNaBH4/BH3/H2NCH2CH2OHを使用するOpatzらの方法でも行うことができる16。これにより74%の収率を得る。
【0082】
1-(10-アセチル-6,7,8,9,9a,10-ヘキサヒドロ-5aH-ピリド[3,4-b]キノキサリン-5-イル)エタノン(14):
27(1.20 g、6.34 mmol)、40 mlのピリジン、30 ml(32.36 g、317 mmol、ρ=1.082 g/ml)の無水酢酸および0.234 g(25 mol%)のPPYを、氷中で冷却しながら250 mlの丸底フラスコ中に導入する。該反応混合物を100℃の油浴温度に加熱し、そしてこの温度にて48時間維持する。該反応は、塩基性酸化アルミニウム(EtOAC/MeOH、10:1)によるTLCによって追跡することができる。室温に冷却後、該溶媒を減圧下留去し、そして得られる粗生成物を、塩基性酸化アルミニウムによるフラッシュクロマトグラフィー(EtOAc/MeOH、10:1)によって精製する。これにより暗黄色固体(1.17 g、4.31 mmol、68%)を得る。
Rf=0.45 (EtOAc/MeOH、10:1). 1H NMR:(400 MHz, CDCl3):δ=1.34 (m, 4H), 1.60 (d, 4H), 2.23 (s, 3H, H3C-CO-N), 2.27 (s, 3H, H3C-CO-N), 4.74 (m, 1H), 4.85 (m, 1H), 7.25 (d, 3J=5.6 Hz, 1H, H-4), 8.41 (d, 3J=5.6 Hz, H-3), 8.49 (s, 1H, H-1) ppm. 13C NMR (75 MHz, CDCl3):δ=21.7, 21.9 (C-7,8), 23.1, 23.6 (H3C-CO-N), 28.4, 28.5 (C-6,9), 55.2, 56.0 (CH, C-5a,9a), 119.6 (C-4), 130.8 (Cq, C-10a), 141.6 (Cq, C-4a), 147.1, 147.2 (C-1, C-3) 169.1, 169.2 (C=O) ppm. GC-MS(EI):保持時間10.45分, m/e (%) 274 (13), 273 (M+, 58), 232 (15), 231 (100), 230 (42), 216 (5), 214 (5), 213 (5), 203 (3), 202 (4), 190 (13), 189 (84), 188 (45), 173 (5), 172 (6), 160 (7), 159 (5), 158 (5), 147 (8), 146 (40) 134 (6), 133 (13), 132 (15), 120 (7), 43 (9);IR (neat):□=2940 (m), 1660 (vs), 1587 (m), 1559 (w), 1498 (s), 1448 (w), 1427 (w), 1388 (m), 1353 (w), 1337 (m), 1289 (s), 1270 (s), 1248 (s), 1248 (m), 1220 (w), 1183 (w), 1102 (w), 1036 (m), 978 (m), 857 (w), 839 (w) 777 (w) cm-1. HRMS (EI):C11H15N3の計算値:273.1477 [M+]、実測値:273.1482.
【0083】
5,10-ジエチル-5,5a,6,7,8,9,9a,10-オクタヒドロピリド[3,4-b]キノキサリン(11):
30 mlのMTBE中の0.77 g(5.80 mmol)のAlCl3の懸濁液を、室温にて45分間攪拌し、その後、0℃に冷却し、次いで、1.32 g(9.35 mmol)のLiAlH4を少量ずつ添加する。添加が完了した後、該混合物を別の15分間攪拌し、化合物14(0.64 g、2.23 mmol)を添加し、そして該混合物を0℃にて1時間撹拌する。その後、8時間還流し、次いで室温に冷却し、そして氷水中に注ぐ。無機沈澱を吸引によって濾去し、そして30 mlのDCMで2回洗浄する。該水相をpH 12にし、そして40 mlのDCMで3回抽出する。合わせた有機相をNa2SO4によって乾燥し、次いで、該溶媒をロータリーエバポレーターによって留去する。得られる粗生成物を、シリカゲル(EtOAc/MeOH/NEt3、10:2:1)および塩基性酸化アルミニウム(EtOAc/MeOH、10:1)によるフラッシュクロマトグラフィーによって精製する。これにより0.30 g(1.22 mmol、55%)の無色固体を得る。
Rf=0.32 (塩基性アルミナ、EtOAc/MeOH、10:1). 1H NMR:(400 MHz, CDCl3):δ=1.11, 1.13 (t, 3J=7.2 Hz, H3C-CH2-N-5, 3J=7.2 Hz, H3C-CH2-N-10, 6-H), 1.37 (m, 2H, H-7,8), 1.56 (m, 3Jaa=6.8 Hz, 3Jee=3.2 Hz, 4H, H-6, H-9, H-7, H-8), 1.76 (m, 3Jae=3.2 Hz, 1H, H-9), 1.89 (m, 3Jae=3.2 Hz, 1H), 3.18 (dq, 3J=7.2 Hz, 2J=14.4 Hz, 2-H, H3C-CHAB-N-5, H3C-CHAB-N-10), 3.23 (ddd, 3Jae=2.8 Hz, 3Jae=3.2 Hz, 3Jaa=6.8 Hz, 1H, H-5a), 3.34 (ddd, 3Jae=2.8 Hz, 3Jae=3.2 Hz, 3Jae=3.2 Hz, H-1, H-9a), 3.43 (dq, 3J=7.2 Hz, 2J=14.4 Hz, H3C-CHAB-N-5, H3C-CHAB-N-10, 2H, H-1), 6.33 (d, 3J=5.6 Hz, 1H, H-4), 7.67 (s, 1H, H-1), 7.72 (d, 1H, H-3) ppm. 13C NMR (100 MHz, CDCl3):δ=10.8 (H3C-CH2-N-5), 11.8 (H3C-CH2-N-10), 21.7 (C-7), 22.9 (C-8), 27.3 (C-6), 27.8 (C-9), 40.4 (H3C-CH2-N-5), 41.7 (H3C-CH2-N-10), 52.8 (C-9a), 56.4 (C-5a), 104.3 (C-4), 130.6 (C-10a, C-1), 139.4 (C-3), 141.2 (C-4a) ppm. GC-MS(EI):保持時間10.58分, m/e (%) 246 (18), 245 (M+, 100), 231 (9), 230 (57), 217 (7), 216 (44), 207 (6), 186 (7), 174 (15), 162 (6), 160 (16), 158 (6), 148 (12), 146 (6), 132 (8);IR (neat):□=3436 (s), 2970 (m), 2860 (vs), 2860 (s), 1628 (w), 1576 (vs), 1513 (vs), 1473 (w), 1447 (m), 1348 (s), 1317 (w), 1267 (s), 1211 (s), 1167 (w), 1125 (w), 1107 (w), 1077 (w), 1059 (w), 1040 (w), 920 (w), 798 (s) 777 (m), 746 (m) cm-1. HRMS (EI):C15H23N3の計算値:245.1892 [M+]、実測値:245.1889.
【0084】
1-(6,7,8,9,9a,10-ヘキサヒドロ-5aH-ピリド[3,4-b]キノキサリン-5-イル)エタノン(28):
27 (1.0 g、5.28 mmol)、20 mlのDCM、2.20 ml(15.84 mmol)のNEt3および5 mol%のPPY(39 mg、0.264 mmol)を、100 mlのSchlenkフラスコ中に置く。その後、0.54 ml(5.81 mmol)の無水酢酸を添加し、そして該反応混合物を30分間攪拌する。その後、該反応を2 mlのMeOHの添加により停止させ、そしてさらに10分間撹拌する。該溶媒をロータリーエバポレーターによって留去し、そして得られる粗生成物をフラッシュクロマトグラフィー(EtOAc/MeOH/NEt3、10:1:1)によって精製する。これにより1 g(4.31 mmol、82%)の28を得る。得られる生成物は、不純物としてPPYを含有し、そしてさらなる精製をせずにさらに反応させる。
Rf=0.44 (EtOAc/NEt3/MeOH、10:1:1). 1H NMR:(300 MHz, CDCl3):δ=1.44 (m, 5H), 1.76 (m, 3H), 2.29 (s, 3H, H3C-CO-N), 3.59 (m, 1H), 4.07 (m, 1H), 7.08 (bs, 1H, H-4), 7.85 (d, 3J= 5.4 Hz, 1H, H-3), 7.98 (s, H-1, H-1). 13C NMR (75 MHz, CDCl3):δ=18.5 (CH2), 23.5 (H3C-CO-N), 24.8, 25.4 (CH2), 31.08 (CH2), 48.9 (C-9a), 118.1 (Cq, C-4a), 127.9 (Cq, C-10a), 132.2 (C-4), 136.6 (C-1), 138.0 (C-3), 169.0 (C=O) ppm. HRMS (EI): C13H17N3Oの計算値:231.1372 [M+]、実測値:231.1368.
【0085】
5-エチル-5,5a,6,7,8,9a,10-オクタヒドロピリド[3,4-b]キノキサリン(29):
0.619 g(4.64 mmol)のAlCl3を30 mlのTHF中に懸濁させ、そして該混合物を室温にて45分間攪拌する。その後、該混合物を0℃に冷却し、そして0.300 g(7.91 mmol)のLiAlH4を少量ずつ添加する。別の15分間の撹拌後、化合物28(0.832 g、3.59 mmol)を添加し、そして該混合物を0℃にて1時間撹拌する。その後、該反応混合物を8時間還流し、そして冷却後、氷水中に注ぐ。該無機沈澱を濾去し、そして30 mlのDCMで2回洗浄する。母液をpH 12にし、そして40 mlのDCMで3回抽出する。合わせた有機相をNa2SO4によって乾燥し、次いで、該溶媒をロータリーエバポレーターによって留去する。該粗生成物を、シリカゲルによるフラッシュクロマトグラフィー(EtOAc/NEt3、10:1)によって精製する。これにより0.430 g(1.98 mmol、55%)の白色発泡体を得る。
Rf=0.24 (EtOAc/NEt3、10:1). 1H NMR:(300 MHz, CDCl3):δ=1.06 (t, 3J= 7.2 Hz, 3H, H3C-CH2-N-5), 1.25 (m, 2H), 1.54 (m, 5H), 1.78 (m, 1H), 3.20 (m, 4H), 3.79 (s, 1H, N-H), 6.21 (d, 3J= 5.7 Hz, 1H, H-4), 7.57 (s, H-1, H-1), 7.65 (d, 3J= 5.7 Hz, 1H, H-3). 13C NMR (75 MHz, CDCl3):δ=12.1 (H3C-CH2-N-5), 19.3 (C-7), 24.8 (C-8), 26.8 (C-6), 30.9 (C-9), 42.8 (H3C-CH2-N-5), 47.8 (C-5a), 58.4 (C-9a), 103.9 (C-4), 129.8 (Cq, C-10a), 133.4 (C-1), 138.4 (Cq, C-10a), 140.9 (C-3) ppm. GC-MS(EI):保持時間10.58分, m/e (%) 218 (17), 217 (M+, 100), 216 (7), 215 (7), 213 (5), 203 (5), 202 (51), 186 (17), 185 (5), 174 (12), 162 (6), 161 (8), 160 (9), 158 (7), 148 (5);148 (5), 146 (16), 145 (7), 134 (8), 133 (9), 132 (25), 120 (14), 78 (4);IR (neat):□=3212 (m), 3093 (w), 2971 (w), 2928 (s), 2851 (s), 1589 (s), 1560 (m), 1505 (vs), 1470 (w), 1442 (m), 1419 (m), 1363 (s), 1280 (vs), 1244 (s), 1210 (m), 1194 (s), 1177 (m), 1108 (m), 1072 (m), 1039 (m), 1007 (w) 968 (w), 897 (w), 886 (w), 795 (s), 740 (m) cm-1. HRMS (EI):C13H19N3の計算値:217.1579 [M+]、実測値:215.1573.
【0086】
1-(5-エチル-5a,6,7,8,9,9a-ヘキサヒドロ-5H-ピリド[3,4-b]キノキサリン-10-イル)エタノン(30):
29(0.250 g、1.15 mmol)および10 mlのTHFを、100 mlのSchlenkフラスコ中に置く。該溶液を-78℃に冷却し、0.51 mlのn-BuLi(1.27 mmol、1.1当量;ヘキサン中2.5 M)を添加し、そして該反応溶液を、冷却することなく30分間撹拌する。30分後、該反応溶液を、-78℃に再度冷却し、および0.09 ml(0.109 g、1.4 mmol、ρ=1.1051 g/ml)の塩化アセチルを添加し、冷却槽を除去し、そして該混合物を、冷却することなく1時間撹拌する。その後、該反応を4 mlによって停止させ、および20 mlのDCMで3回抽出する。合わせた有機相をNa2SO4によって乾燥し、次いで、該溶媒をロータリーエバポレーターによって留去する。該粗生成物をシリカゲル(EtOAc/MeOH、10:2)によって精製し、48 mg(0.18 mmol、16%)の無色固体を得る。
Rf=0.16 (EtOAc/NEt3、10:1). 1H NMR:(300 MHz, CDCl3):δ=1.19 (t, 3J= 7.2 Hz, 3H, H3C-CH2-N-5), 1.24 (m, 2H), 1.45 (m, 2H), 1.58 (m, 2H), 1.74 (m, 1H), 2.18 (m, 1H), 3.34 (m, 2J=14.4 Hz, 3J=7.2 Hz, 1H, H3C-CHAB-N-5), 3.53 (m, 1H, C-H), 3.57 (m, 2J=15.2 Hz, 3J=7.2 Hz, 1H, H3C-CHAB-N-5), 4.90 (bs, 1H, C-H), 6.62 (d, 3J=5.6 Hz, 1H, H-4), 8.12 (s, 1H, H-1), 8.08 (d, 3J=5.6 Hz, 1H, H-3). 13C NMR (100 MHz, CDCl3):δ=12.4 (H3C-CH2-N-5), 18.9 (C-7), 22.8 (H3C-CO-N-10), 24.7 (C-8), 25.8 (C-6), 28.5 (C-9), 40.0 (H3C-CH2-N-5), 48.7 (C-9a), 53.7 (C-5a), (C-4), 106.4 (C-4), 120.1 (Cq, C-10a), 145.1, 145.2 (C-1, Cq, C-4a), 147.1 (C-3), 167.1 (C=O) ppm. GC-MS(EI):保持時間10.39分, m/e (%) 260 (11), 259 (M+, 100), 230 (8), 218 (9), 217 (79), 216 (40), 203 (4), 202 (23), 203 (3), 202 (23), 201 (4), 189 (6), 188 (31), 187 (4), 186 (3), 174 (11), 162 (4), 161 (6), 160 (5), 158 (4), 148 (5), 146 (6), 136 (3), 132 (4), 131 (12), 120 (6), 43 (3);IR (neat):□=3398 (s), 2933 (s), 2863 (m), 1731 (m), 1637 (vs), 1595 (vs), 1512 (s), 1546 (w), 1449 (m), 1397 (s), 1365 (s), 1334 (m), 1285 (s), 1239 (m), 1198 (m), 1168 (vs), 1123 (w), 1067 (w), 1068 (w), 1016 (w), 802 (vs), 718 (w), 668 (m). HRMS (EI):C15H21N3Oの計算値:259.1685 [M+]、実測値:259.1687.
【0087】
2,3-ジフェニルピリド[3,4-b]キノキサリン(25)(文献からの変法):
5.51 g(0.026 mmol)のベンジルを、50 mlのエタノール中の2.86 g(0.026 mol)の3,4-ジアミノピリジンの懸濁液に添加し、そして該反応混合物を70℃の油浴温度にて6時間加熱する。室温に冷却後、沈澱した黄色固体を濾去する(氷水槽を用いる冷却が有用であり得る)。エタノールからの再結晶化により、6.91 g(0.024 mol、94%)の淡黄色固体を得る。
1H NMR:(400 MHz, CDCl3):δ=7.32-7.42 (m, 6H, 3,4,5-フェニル-H), 7.54-7.51(m, 4H, 2,6-フェニル-H), 7.98 (dd, 3J=6 Hz, 4J=0.8 Hz, 1H, 7-H), 8.82 (d, 3J =6 Hz, 1H, 8-H), 9.59 (d, 4J=0.8 Hz, 1H, 5-H) ppm. 13C NMR (100 MHz, CDCl3):δ=121.3 (C-7), 128.4, 129.4-129.9 (フェニル-C), 136.3 (Cq, C-4a), 143.5 (Cq, C-8a), 147.3 (C-8), 154.5 (C-5), 155.3, 157.9 (Cq, フェニル-C) ppm. GC-MS(EI):保持時間12.34分, m/e (%) 285 (3), 284 (M++H, 22), 283 (M+, 100), 282 (46), 206 (2), 181 (3), 180 (25), 179 (17), 154 (3), 153 (5), 152 (3) 142 (1), 141 (8), 140 (4), 127 (2), 104 (3), 103 (14), 102 (3), 78 (2), 77 (5), 76 (4), 51 (2), 50 (9);IR (neat):□=3061 (w), 1589 (m), 1577 (w), 1538 (w), 1492 (w), 1443 (m), 1419 (w), 1379 (s), 1346 (w), 1326 (m), 1315 (m), 1288 (w), 1211 (m), 1244 (w), 1227 (m), 1212 (w), 1180 (w), 1075 (m), 1058 (m), 1020 (m), 1001 (w), 976 (s), 921 (w), 892 (m), 892 (m), 830 (m), 819 (w), 811 (m), 763 (s), 735 (m), 708 (vs), 629 (s), 615 (m) cm-1.
【0088】
2,3-ジフェニル-1,2,3,4-テトラヒドロピリド[3,4-b]ピラジン(31):
25(6.25 g、22.05 mmol)および200 mlのエタノールを、500 mlのフラスコ中に置く。6.25 g(165 mmol)のNaBH4粉末を、該溶液に添加し、そして該反応混合物を40℃にて48時間撹拌する。その後、該反応混合物を室温に冷却し、そして該反応を10 mlの冷水によって停止する。さらに10分間撹拌した後、該反応混合物を、100 mlのDCMを用いて2回抽出し、そして合わせた有機相をNa2SO4によって乾燥する。該粗生成物を、シリカゲルによるフラッシュクロマトグラフィー(EtOAc/NEt3、10:1)によって精製する。これにより淡黄色固体を得る。
Rf=0.23 (EtOAc/NEt3, 10:1);1H NMR:(300 MHz, CDCl3):δ=3.99 (s, 1H, N-H), 4.59 (s, 2H, 2,3-H), 4.62 (s, 1H, N-H), 6.39 (d, 3J =5.4 Hz, 1H, 8-H), 6.80 (dd, 4J=1.2 Hz, 3J=9.3 Hz, 2H, 2,6-フェニル-H), 6.84 (dd, 4J=1.2 Hz, 3J=9.3 Hz, 2H, 2,6-フェニル-H), 7.12 (m, 6H, 3,4,5-フェニル-H) ppm. 13C NMR (75 MHz, CDCl3):δ=58.6, 60.1 (C-2,3), 108.0 (C-8), 127.9-128.4 (フェニル-C), 129.8 (C-4a), 139.8 (Cq, フェニル-C), 140.0 (Cq, C-8a), 134.9 (C-5), 141.3 (C-7) ppm;MS(EI):m/e (%) 288 (22), 287 (M+, 100), 286 (24), 285 (7), 284 (6), 283 (8), 282 (5), 211 (13), 210 (78), 209 (5), 208 (13), 197 (4), 196 (26), 181 (13), 179 (3), 127 (4), 104 (7), 92 (3), 91 (32), 77 (5);IR (neat):□=3215 (w), 2830 (w), 1596 (s), 1519 (s), 1493 (w), 1466 (w), 1452 (s), 1360 (m), 1290 (s), 1250 (m), 1231 (m), 1174 (s), 1120 (m), 1072 (m), 1050 (w), 1029 (w), 1005 (w), 989 (w), 950 (w), 905 (w), 844 (w), 810 (m), 770 (m), 723 (m), 675 (vs), 668 (w), 645 (w), 618 (m) cm-1. HRMS (EI):C19H17N3の計算値:287.1422 [M+]、実測値:287.1402.
【0089】
4-メチル-2,3-ジフェニル-1,2,3,4-テトラヒドロピリド[3,4-b]ピラジン(32):
100 mlの丸底フラスコ中、0.600 g(2.08 mmol)の31を、氷中で冷却しながら15.5 ml(417.6 mmol、ρ=1.22 g/ml)のギ酸中に溶解させ、そして5.1 mlのホルムアルデヒド 溶液(183.6 mmol、ρ=1.22 g/ml、水中<37%)を添加する。その後、該氷槽を除去し、次いで、該混合物を120℃の油浴温度にて48時間加熱する。0℃に冷却した後、50%濃度のNaOHを、氷中で冷却しながら母液のpHが12に上昇するまで添加する。その後、該混合物を50 mlのDCMで3回抽出し、そして合わせた有機相をNa2SO4によって乾燥する。該溶媒をロータリーエバポレーターで留去した後、粗生成物を、シリカゲルによるフラッシュクロマトグラフィー(CHCl3/イソヘキサン/NEt3、10:2:1)によって精製する。これにより0.564 g(1.87 mmol、90%)の黄色固体を得る。
Rf=0.40 (CHCl3/イソヘキサン/NEt3、10:2:1) 1H NMR:(400 MHz, CDCl3):δ=2.85 (s, 3H, CH3), 4.42 (s, 1H, N-H), 4.44 (d, 3J =3.6 Hz, 1H, H-3), 4.95 (d, 3J =3.6 Hz, 1H, H-2), 6.50 (d, 3J=5.2 Hz, 1H, H-8), 6.64 (dd, 4J=2.8 Hz, 3J=9.2 Hz, 2H, 2,6-フェニル-H), 6.90 (m, 2H, 2,6-フェニル-H) 7.05 (m, 2H, フェニル-H), 7.16 (m, 4H, フェニル-H), 7.82 (s, 1H, H-5), 7.84 (d, 3J=5.2 Hz ppm. 13C NMR (75 MHz, CDCl3):δ=37.2 (CH3), 57.8 (C-3), 67.9 (C-2), 107.6 (C-8), 127.4-128.5 (フェニル-C), 131.3 (C-5), 131.6 (Cq, C-4a) 137.4, 138.8 (Cq, フェニル-C), 139.9 (C-7, C-8a) ppm. MS(EI):m/e (%) 302 (22), 301 (M+, 100), 300 (5), 286 (5), 224 (10), 222 (4), 211 (13), 210 (87), 209 (3), 208 (6), 195 (11), 181 (6), 179 (3), 178 (3), 178 (3), 150 (7), 132 (3), 127 (3), 120 (3), 104 (4);92 (3), 91 (31), 85 (4), 83 (6), 78 (4), 77 (5);IR (neat):□=3200 (w), 3158 (w), 3029 (w), 2948 (w), 2881 (w), 2824 (w), 1578 (s), 1516 (vs), 1491 (m), 1452 (m), 1421 (m), 1374 (w), 1353 (w), 1326 (w), 1294 (w), 1256 (m), 1235 (m), 1199 (w), 1157 (m), 1131 (m), 1103 (w), 1073 (m), 1054 (m), 1031 (m), 1013 (m), 969 (w), 922 (w), 842 (w), 810 (s), 766 (s), 755 (m), 733 (m), 641 (vs) cm-1. HRMS (EI):C20H19N3の計算値:301.1574 [M+]、実測値:301.1559.
【0090】
4-(メチル-2,3-ジフェニル-1,2,3,4-テトラヒドロピリド[3,4-b]ピラジン-1-イル)エタノン(33):
2.90 g(9.62 mmol)の32および100 mlのMTBEを250 mlのSchlenkフラスコ中に置く。該溶液を-78℃に冷却し、そして4.62 mlのn-BuLi(11.50 mmol、1.1当量;ヘキサン中2.5 M)を10分間に渡って添加する。該反応混合物を、冷却せずに30分間攪拌する。その後、-78℃に冷却し、そして1.0 ml(0.96 g、12.55 mmol)の塩化アセチルを添加する。該反応混合物を室温になるまで放置し、そしてさらに30分間攪拌する。その後、該反応を、10 mlの水を添加することによって停止する。母液をDCMで3回抽出し、そして合わせた有機相をNa2SO4によって乾燥する。該溶媒をロータリーエバポレーターで留去した後、該粗生成物は、フラッシュクロマトグラフィー(CHCl3/イソヘキサン/NEt3、10:1:1)または酢酸エチルからの再結晶化のいずれかによって精製することができる。これにより1.2 g(3.49 mmol、36%)の白色固体を得る。
1H NMR:(600 MHz, CDCl3):δ=2.40 (s, 3H, H3C-CO-N), 2.86 (s, 3H, H3C), 4.99 (m, 1H, H-3), 5.35 (d, 3J =4.2 Hz, 1H, H-2), 6.53 (d, 3J=7.8 Hz, 2H, 2,6-フェニル-H), 6.80 (bs, 2H, 2,6-フェニル-H), 7.08 (t, 2H, 3J=7.2 Hz, フェニル-H), 7.17 (t, 3J=6.0 Hz, 2H, フェニル-H), 7.22 (t, 3J=6.0 Hz, 1H, フェニル-H), 7.25 (t, 3J=6.0 Hz, 1H, フェニル-H), 8.03 (d, 3J=6.6 Hz, 1H, H-8), 8.17 (s, 1H, H-5), 8.41 (d, 3J=6.6 Hz, 1H, H-7) ppm. 13C NMR (150 MHz, CDCl3):δ=24.60 (H3C-CO-N), 36.9 (H3C), 61.8 (C-3), 63.8 (C-2), 116.9 (C-8), 124.6 (C-5), 128.4-128.9 (フェニル-C), 134.7 (Cq, C-8a), 138.7 (Cq, C-4a), 171.0 (C=O) ppm;GC-MS(EI):保持時間1.22分, m/e (%) 345 (3), 344 (27), 343 (M+, 100), 315 (6), 302 (7), 301 (41), 300 (84), 224 (6), 223 (4), 222 (5), 211 (4), 210 (27), 208 (9), 195 (6), 181 (9), 179 (4), 178 (4), 146 (4), 118 (15), 104 (5);92 (9), 91 (94), 78 (4), 77 (4), 43 (6);IR (neat):□=2428 (m), 2039 (w), 1688 (s), 1613 (w), 1534 (s), 1492 (m), 1454 (w), 1388 (m), 1364 (m), 1345 (m), 1317 (w), 1292 (w), 1292 (w), 1272 (w), 1227 (vs), 1209 (vs), 1122 (w), 1103 (w), 1078 (m), 1028 (m), 998 (m), 822 (s), 799 (m), 768 (m), 704 (m), 691 (m), 638 (w) cm-1. HRMS (EI):C22H23N3Oの計算値:343.1685 [M+]、実測値:343.1699.
【0091】
1-エチル-4-メチル-2,3-ジフェニル-1,2,3,4-テトラヒドロピリド[3,4-b]ピラジン(34):
0.575 g(4.320 mmol)のAlCl3を、20 mlのTHF中に室温にて懸濁させ、その後、該混合物をこの温度にて45分間攪拌する。その後、該混合物を0℃に冷却し、そして0.277 g(7.290 mmol)のLiAlH4を少量ずつ添加する。添加が完了した後、該反応混合物をさらに15分間攪拌し、そして33 (1.00 g、3.32 mmol)を添加する。該混合物を0℃にて1時間撹拌し、その後、12時間還流する。次いで、該反応混合物を室温に冷却し、そして氷水中に注ぐ。該無機沈澱を濾去し、そして母液をDCMで抽出する。合わせた有機相をNa2SO4によって乾燥し、そして得られる粗生成物をフラッシュクロマトグラフィー(EtOAc/MeOH/NEt3、10/1/0.5)によって精製する。これにより0.580 g(1.76 mmol、53%)の黄色固体を得る。
Rf=0.57 (EtOAc/MeOH/NEt3、10/1/0.5). 1H NMR:(400 MHz, CDCl3):δ=1.05 (t, 3J =7.2 Hz, 3H, H3C-CH2-N), 2.71 (s, 3H, H3C), 3.17 (qd, 2J =14.8 Hz, 3J =7.2 Hz, 1H, CH3-CHAB-N), 3.38 (qd, 2J =14.8 Hz, 3J =7.2 Hz, 1H, CH3-CHAB-N), 4.40 (d, 3J=3.6 Hz, 1H, H-3), 4.55 (d, 3J=3.6 Hz, 1H, H-2), 6.52 (d, 1H, 3J=5.6 Hz, H-8), 6.73 (m, 4H, フェニル-H), 7.07 (t, 3J=7.2 Hz, 2H, フェニル-H), 7.11 (t, 3J=8 Hz, 2H, フェニル-H), 7.16 (m, 2H, フェニル-H), 7.92 (s, 1H, H-5), 7.96 (d, 3J=5.6 Hz, 1H, H-7) ppm. 13C NMR (100 MHz, CDCl3):δ=11.1 (H3C-CH2-N), 37.1 (H3C-N), 43.3 (H3C-CH2-N), 65.7 (C-2), 66.0 (C-3), 104.3 (C-8), 127.7-129.3 (フェニル-C), 133.0 (C-5), 133.3 (Cq, C-8a), 137.8, 138.6 (Cq, フェニル-C), 140.8 (Cq, C-8a), 141.0 (C-7) ppm. GC-HR-ESI-MS:保持時間0.53〜1.43分, C44H47N6の計算値:659.3862 [2M++H]、実測値:659.3823、C22H24N3の計算値:330.1970 [M++H]、実測値:330.1940;IR (neat):□=2965 (w), 2818 (w), 1578 (s), 1518 (vs), 1492 (m), 1452 (s), 1430 (w), 1371 (m), 1356 (m), 1308 (m), 1286 (m), 1266 (s), 1241 (s), 1215 (vs), 1165 (m), 1119 (m), 1067 (s), 1023 (s), 884 (w), 867 (w), 828 (w), 810 (s), 764 (s), 749 (m), 705 (vs), 660 (w), 615 (w) cm-1.
【0092】
〔参考文献〕
1) 以下のレビューを参照のこと:(a) G. Hoefle, W. Steglich, H. Vorbrueggen, Angew. Chem. 1978, 90, 602 - 615;Angew. Chem. Int. Ed. Engl. 1978, 17, 569 - 583;(b) E. F. V. Scriven, Chem. Soc. Rev. 1983, 12, 129 - 161;(c) A. Hassner, in Encyclopedia of Reagents for Organic Synthesis, Wiley, Chichester, 1995, 2022 - 2024;(d) U. Ragnarsson, L. Grehn, Acc. Chem. Res. 1998, 31, 494 - 501;(e) A. C. Spivey, A. Maddaford, A. Redgrave, Org. Prep. Proced. Int. 2000, 32, 331-365. (f) D. J. Berry, C. V. Digiovanna, S. S. Metrick, R. Murugan, Arkivoc 2001, 201 - 226;(g) A. C. Spivey, S. Arseniyadis, Angew. Chem. 2004, 116, 5552 - 5557;Angew. Chem. Int. Ed. Engl. 2004, 43, 5436 - 5441.
2) (a) E. Vedejs, M. Jure, Angew. Chem. 2005, 117, 4040 - 4069;Angew. Chem. Int. Ed. Engl. 2005, 44, 3971 - 4001. (b) P. I. Dalko, L. Moisan, Angew. Chem. 2004, 116, 5248 - 5286;Angew. Chem. Int. Ed. Engl. 2004, 43, 5138 - 5178. (c) G. Fu, Acc. Chem. Res. 2004, 37, 542 - 547. (d) S. France, D. J. Guerin, S. J. Miller, T. Lectka, Chem. Rev. 2003, 103, 2985 - 3012. (e) A. C. Spivey, A. Maddaford, A. Redgrave, Org. Prep. Proced. Int. 2000, 32, 331 - 365. (f) G. Fu, Acc. Chem. Res. 2000, 33, 412 - 420.
3) (a) T. Kawabata, R. Stragies, T. Fukaya, K. Fuji, Chirality 2003, 15, 71. (b) T. Kawabata, R. Stragies, T. Fukaya, Y. Nagaoka, H. Schedel, K. Fuji, Tetrahedron Lett. 2003, 44, 1545.
4) (a) G. Priem, B. Pelotier, S. J. F. Macdonald, M. S. Anson, I. B. Campbell, J. Org. Chem. 2003, 68, 3844. (b) B. Pelotier, G. Priem, S. J. F. Macdonald, M. S. Anson, R. J. Upton, I. B. Campbell, Tetrahedron Lett. 2005, 46, 9005.
5) (a) A. C. Spivey, T. Fekner, S. E. Spey, H. Adams, J. Org. Chem. 1999, 64, 9430、およびその引用文献. (b) A. C. Spivey, T. Fekner, S. E. Spey, J. Org. Chem. 2000, 65, 3154. (c) A. C. Spivey, A. Maddafort, T. Fekner, A. J. Redgrave, C. S. Frampton, J. Chem. Soc. Perkin Trans. 1. 2000, 3460. (d) A. C. Spivey, A. Maddafort, T. Fekner, D. P. Leese, A. J. Redgrave, C. S. Frampton, J. Chem. Soc. Perkin Trans. 1, 2001, 1785. (e) C. Malardier-Jugroot, A. C. Spivey, M. A. Whitehead, J. Mol. Struct. (THEOCHEM) 2003, 623, 263. (f) A. C. Spivey, D. P. Leese, F. Zhu, S. G. Davey, R. L. Jarvest, Tetrahedron 2004, 60, 4513. (g) A. C. Spivey, S. Arseniyadis, T. Fekner, A. Maddaford, D. P. Lees, Tetrahedron 2006, 62, 295.
6) (a) C. O. Dalaigh, S. J. Hynes, D. J. Maher, S. J. Connon, Org. Biomol. Chem. 2005, 3, 981. (b) C. O. Dalaigh, S. J. Hynes, J. E. O'Brien, T. McCabe, D. J. Maher, G. W. Watson, S. J. Connon, Org. Biomol. Chem. 2006, 4, 2785.
7) (a) S. A. Shaw, P. Aleman, E. Vedejs, J. Am. Chem. Soc. 2003, 125, 13368-13369. (b) S. A. Shaw, P. Aleman, J. Christy, J. W. Kampf, P. Va, E. Vedejs, J. Am. Chem. Soc. 2006, 128, 925-934.
8) S. Yamada, T. Misono, Y. Iwai, Tet. Lett. 2005, 46, 2239.
9) (a) S. J. Miller, Acc. Chem. Res. 2004, 37, 601-610、およびその引用文献. (b) M. B. Fierman, D. J. O’Leary, W. E. Steinmetz, S. J. Miller, J. Am. Chem. Soc. 2004, 126, 6967-6971. (c) J. W. Evans, M. B. Fierman, S. J. Miller, J. A. Ellman, J. Am. Chem. Soc. 2004, 126, 8134-8135. (d) B. R. Sculimbrene, Y. Xu, S. J. Miller, J. Am. Chem. Soc. 2004, 126, 13182-13183. (e) A. J. Morgan, Y. K. Wang, M. F. Roberts, S. J. Miller, J. Am. Chem. Soc. 2004, 126, 15370-15371. (f) C. A. Lewis, B. R. Sculimbrene, Y. Xu, S. J. Miller, Org. Lett. 2005, 3021. (g) Y. Xu, B. R. Sculimbrene, S. J. Miller, J. Org. Chem. 2006, 71, 4919-4928.
10) M. R. Heinrich, H. S. Klisa, H. Mayr, W. Steglich, H. Zipse, Angew. Chem. 2003, 115, 4975 - 4977;Angew. Chem. Int. Ed. 2003, 42, 4826 - 4828.
11) I. Held, A. Villinger, H. Zipse, Synthesis 2005, 1425 - 1426.
12) W. W. K. R. Mederski, D. Kux, M. Knoth, M. J. Schwarzkopf-Hofmann, Heterocycles, 2003, 4, 925-931.
13) J. Armand, L. Boulares, C. Bellec, J. Pinson, Can J. Chem. 1988, 66, 1500-1505.
14) M. L. Moore, Org. Reaction. 1949, 5, 301-330.
15) C. Sotirou-Leventis, Z. Mao, A.-M. M. Rawashdeh, J. Org. Chem. 2000, 65, 6017-6023.
16) C. Kison, N. Meyer, T. Opatz, Angew. Chem. Int. Ed. 2005, 44, 5662-5664. 該文献中に引用されたBH3・THF溶液の寿命に対する反応収率の依存性は、化合物24の合成について確認することができた。
17) Timothy D. W. Claridge, High-Resolution NMR Techniques in Organic Chemistry, Tetrahedron Organic Chemistry Series, 1999, 19, 298-299.
18) S. Xu, I. Held, B. Kempf, H. Mayr, W. Steglich, H. Zipse, Chem. Eur. J. 2005, 11, 4751 - 4757.
19) C. B. Fischer, S. Xu, H. Zipse, Chem. Eur. J. 2006, 12, 5779 - 5784.
20) この場合の反応速度は、背景反応の反応速度とわずかに異なる。このことは、本明細書中で使用される方法による半減期の正確な決定を格別に困難にする。表2中に示すデータは、25日間、17時間および30分間に渡る反応Aについての、並びに31日間、17時間および15分間に渡る反応Bについての、変換測定から得られる。両方の場合において、50%の変換は、これらの時間内で達成されない。
21) Gaussian 03, Revision B.03, M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, J. A. Montgomery, Jr., T. Vreven, K. N. Kudin, J. C. Burant, J. M. Millam, S. S. Iyengar, J. Tomasi, V. Barone, B. Mennucci, M. Cossi, G. Scalmani, N. Rega, G. A. Petersson, H. Nakatsuji, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, M. Klene, X. Li, J. E. Knox, H. P. Hratchian, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, P. Y. Ayala, K. Morokuma, G. A. Voth, P. Salvador, J. J. Dannenberg, V. G. Zakrzewski, S. Dapprich, A. D. Daniels, M. C. Strain, O. Farkas, D. K. Malick, A. D. Rabuck, K. Raghavachari, J. B. Foresman, J. V. Ortiz, Q. Cui, A. G. Baboul, S. Clifford, J. Cioslowski, B. B. Stefanov, G. Liu, A. Liashenko, P. Piskorz, I. Komaromi, R. L. Martin, D. J. Fox, T. Keith, M. A. Al-Laham, C. Y. Peng, A. Nanayakkara, M. Challacombe, P. M. W. Gill, B. Johnson, W. Chen, M. W. Wong, C. Gonzalez,およびJ. A. Pople, Gaussian, Inc., Wallingford CT, 2004.
22) H. E. Gottlieb, V. Kotyar, A. Nudelman, J. Org. Chem. 1997, 21 7512-7515.
23) この反応における変換は59%にて停止する。該反応混合物についてのESI-MS研究は、この時間にて該触媒がもはや完全のままではないことを示す。
【技術分野】
【0001】
本発明は、3,4-ジアミノピリジン誘導体、それらの製造方法および3,4-ジアミノピリジン誘導体の使用に関する。
【背景技術】
【0002】
ドナー置換ピリジンは、多くの合成的に重要な変換、例えば、アルコール、アミンまたはエノラートのアシル化における求核触媒として顕著な役割を担う1、2。
【0003】
最近、(4-ジメチルアミノ)ピリジン1(DMAP)または(4-ピロリジノ)ピリジン2(PPY)(スキーム1参照)の置換誘導体を適切に使用するラセミ化合物の速度論的分割実験において、大きな進歩が得られた[2〜8]。これらの発展にも関わらず、当該分野はやや不安定のままである。ここで種々の解決法が特定の合成問題、例えば第2級アルコールのラセミ化合物の速度論的分割に利用可能であるが、他の明らかに類似した課題、例えば第1級または第3級アルコールのラセミ化合物の速度論的分割は、実質的に解消されていないようである。
【0004】
【化1】
【0005】
スキーム1:(4-ジメチルアミノ)ピリジン1および(4-ピロリジノ)ピリジン2の構造式
【0006】
この状況において、単独の合成戦略内の幅広い範囲の構造的変動を可能にするというモジュール触媒の概念は、非常に望ましいように見える。最近、このタイプの概念が、ペプチド構造に基づき、およびイミダゾールを活性中心として使用する触媒について、Millerらによって研究された2d、9。ペプチドはまた、KawabataらおよびCampbellらによって開発されたPPY誘導体中の変動可能な構造的要素である3、4。これらの場合の全てにおいて、該ペプチド構造は、触媒反応サイクルの速度決定段階における基質と側鎖との間に構築されるさらなる触媒による反応過程に影響を及ぼす。しかしながら、求核触媒反応中心の電子的特性は、通常、ペプチド構造の変動によって変化しない。
【0007】
一方、1および2の3-置換誘導体のモジュール構造は、ピリジン環の求核性並びにその側鎖非対称性の両方を変化させる潜在能力を有する4〜8。これまで行われた実験は、立体的に区別する方法を調査することに限定されていた。
【0008】
既に、アシル化反応におけるドナー置換ピリジンの潜在的触媒能力に関する以前の研究は、触媒活性についての定性基準としてのアシルピリジニウムカチオン4Acの相対的安定性を示した10、11。
【0009】
【化2】
【0010】
スキーム2:アシルピリジニウムカチオンから置換ピリジンへのアシル転位反応(1)
【発明の概要】
【発明が解決しようとする課題】
【0011】
したがって、本発明の目的は、単独の合成戦略内の触媒として使用された場合、幅広い構造的変動を可能にする化合物を提供することにある。さらなる本発明の目的は、幅広い範囲の構造的変動で製造するのに簡単である化合物を提供することにある。特に、アシル化反応用並びにウレタンおよびポリウレタン製造用の触媒として使用し得る化合物を知ることが望ましいであろう。
【課題を解決するための手段】
【0012】
上記目的は、独立請求項の主題によって達成される。好ましい実施態様は、従属請求項から導かれ得る。
本発明の発明者らは、驚くべきことに、3,4-ジアミノピリジン誘導体が製造容易であり、および触媒として有利な特性を有することを見出した。
【0013】
本発明の第一の局面は、以下の式I:
【0014】
【化3】
【0015】
〔式中、R1およびR2は、それぞれ互いに独立して電子ドナーであり、ここでR2はHであってもよく、および
R3、R4、R5およびR6は、それぞれ独立して、H、置換または非置換の直鎖または分枝状のアルキル、アルケニル、アルキニル、アルコキシおよび置換または非置換のアリールの中から選択され、ここで基R3および/またはR4は、基R5および/またはR6と一緒になって環を形成し得る。〕
で示される3,4-ジアミノピリジン誘導体に関する。
【0016】
特に、本発明の3,4-ジアミノピリジン誘導体は、ピリジン環の電子特性の広い変動性に起因して、種々の目的に使用し得る触媒として適当である。ピリジン環の3位および4位における2つの窒素原子の間の架橋エチレン単位は、ピリジン環に対して有利な電子押出効果を発揮し得る。該エチレン架橋は一以上の置換基を有し得る。ピリジン環の4位における少なくとも窒素原子は、完全に置換される。すなわち、これは、もはや水素原子を有しない。
【図面の簡単な説明】
【0017】
【図1】図1は、上記スキーム1の等式(1)によるピリジン1、2および5と比較した、3,4-ジアミノピリジン6〜21についての反応のエンタルピーを示す。
【図2】図2は、化合物34の単結晶X線構造を示す。
【発明を実施するための形態】
【0018】
好ましい本発明の実施態様によれば、本発明者らは、電子ドナーR1およびR2が、それぞれ独立して、置換または非置換の直鎖または分枝状のアルキル、アルケニル、アルキニル、アシル、アルコキシおよび置換または非置換のアリール、より好ましくは置換または非置換のフェニルおよびベンジルの中から選択され、ここでR2はHであってもよい、式Iで示される3,4-ジアミノピリジン誘導体を開示する。
【0019】
本目的のために、電子ドナーは、それぞれの置換部位にて電子密度を高める置換基である。ここで、基R1およびR2は、ピリジン環の3位および4位における窒素原子上で電子密度を高める。
【0020】
本発明の目的のために、上記置換基アルキル、アルケニル、アルキニル、アシルおよびアルコキシは、いずれの場合も、互いに独立して以下のように定義される。
【0021】
用語「アルキル」は、飽和炭化水素基を意味する。該アルキル基は、好ましくは置換または非置換の直鎖または分枝状のC1-C20-アルキル、より好ましくはC1-C10-アルキル、さらにより好ましくはC1-C8-アルキル、さらにより好ましくはC2-C6-アルキルおよび最も好ましくはC3-C5-アルキルである。このようなアルキル置換基の例は、メチル、エチル、n-プロピル、イソプロピル、n-ブチル、sec-ブチルおよびt-ブチルである。
【0022】
用語「アルケニル」は、単独のまたは多重の不飽和基、すなわち、一以上の二重結合を有するアルキル基を意味する。該アルケニル基は、好ましくは置換または非置換の直鎖または分枝状のC2-C20-アルケニル、より好ましくはC2-C10-アルケニル、さらにより好ましくはC2-C8-アルケニル、さらにより好ましくはC3-C6-アルケニルおよび最も好ましくはC3-C5-アルケニルである。
【0023】
用語「アルキニル」は、三重結合による単独のまたは多重の不飽和である基、すなわち、一以上の三重結合を有するアルキル基を意味する。該基は、一以上の二重結合をさらに有し得る。該アルキニル基は、好ましくは置換または非置換の直鎖または分枝状のC2-C20-アルキニル、より好ましくはC2-C10-アルキニル、さらにより好ましくはC2-C8-アルキニル、さらにより好ましくはC3-C6-アルキニルおよび最も好ましくはC3-C5-アルキニルである。
【0024】
用語「アシル」は、-C(O)-R基〔式中、Rは、上記のようなアルキル、アルケニル、アルキニルまたはアリール基である〕を意味する。アシルは、好ましくは置換または非置換の直鎖または分枝状のC1-C20-アシル、より好ましくはC1-C10-アシル、さらにより好ましくはC1-C8-アシル、さらにより好ましくはC2-C6-アシルおよび最も好ましくはC3-C5-アシル、またはベンゾイルである。特に好ましいアシル基はアセチル基である。
【0025】
用語「アルコキシ」は、酸素原子を介して結合したアルキル基を意味する。該アルコキシ基は、好ましくは置換または非置換の直鎖または分枝状のC1-C20-アルコキシ、より好ましくはC1-C10-アルコキシ、さらにより好ましくはC1-C8-アルコキシ、さらにより好ましくはC2-C6-アルコキシおよび最も好ましくはC3-C5-アルコキシである。このようなアルキル置換基の例は、メトキシ、エトキシ、n-プロポキシ、イソプロポキシ、n-ブトキシ、sec-ブトキシおよびt-ブトキシである。
【0026】
さらなる本発明の実施態様において、基R3、R4、R5およびR6 の少なくとも一つはHではない。ここで、基R3、R4、R5およびR6の少なくとも一つがフェニルであることが好ましい。より好ましくは、2つの基R3およびR6が各々Hであり、および2つの基R4およびR5が一緒になって、以下の化合物11において例示されるような6員環を形成するように、1,4-ブタンジイルであることである。しかしながら、この6員環はまた、不飽和環または置換環であることが好ましい。
【0027】
別の好ましい本発明の実施態様にしたがって、本発明者らは、式Iで示される3,4-ジアミノピリジン誘導体であって、式Ia:
【0028】
【化4】
【0029】
〔式中、R1およびR2は、それぞれ互いに独立して電子ドナーであり、ここでR2はHであってもよく、および
R3およびR6は、それぞれ独立して、H、置換または非置換の直鎖または分枝状のアルキル、アルケニル、アルキニル、アルコキシ、および置換または非置換のアリールの中から選択され、
R4およびR5は、それぞれ独立して、置換または非置換の直鎖または分枝状のアルキル、アルケニル、アルキニル、アルコキシ、および置換または非置換のアリールの中から選択され、および
基R3および/またはR4は、基R5および/またはR6と一緒になって環を形成し得る。〕
で示される3,4-ジアミノピリジン誘導体を与えるように、2つの基R4およびR5が互いに対してシス位に配置される3,4-ジアミノピリジン誘導体を開示する。
【0030】
この実施態様において、2つの基R4およびR5は、ピリジン環の原子、ピリジン環の3位および4位における2つの窒素原子および架橋エチレン単位の炭素原子8および9により構成される平面に対してシス位に配置される。炭素原子8および9において複数の置換基が存在する場合、すなわち、基R3およびR4またはR5およびR6の少なくとも2つがHではない場合、基R4およびR5が環を形成する場合、該環がシス位に存在することが特に好ましい。環形成がない場合、基R3、R4、R5およびR6の中の2つのより嵩高い置換基が炭素原子8および9に関してシス位に存在することが好ましい。
【0031】
特に好ましい3,4-ジアミノピリジン誘導体において、R1およびR2はそれぞれエチルであり、R3およびR5はそれぞれHであり、およびR4およびR6は一緒になって、6員環を形成するように、1,4-ブタンジイルである。この化合物は、本明細書中で11として示されている。
【0032】
別の好ましい本発明の実施態様において、R1およびR2は、それぞれ互いに独立して置換または非置換の直鎖または分枝状のC1-C10-アルキル、好ましくは置換または非置換の直鎖または分枝状のC2-C8-アルキルおよび最も好ましくは置換または非置換の直鎖または分枝状のC3-C6-アルキルである。
【0033】
本発明の第二の局面は、式Iで示される3,4-ジアミノピリジン誘導体の製造方法であって、
・3,4-ジアミノピリジンを1,2-ジカルボニル化合物と反応させる工程、
・得られたジイミンを対応するジアミンに還元する工程、および
・該ピリジン環の4位における窒素上の少なくとも水素原子を置換する工程
を含む方法に関する。
【0034】
上記1,2-ジカルボニル化合物は、好ましくは、以下の式II:
【0035】
【化5】
【0036】
で示される化合物である。
第二の局面の好ましい実施態様において、ピリジン環の3位および4位における窒素原子の両方が置換される。
【0037】
ピリジン環の3位および/または4位における窒素原子上のアルキル置換が、窒素原子のアシル化およびその後の得られたアミドのアミンへの還元を含む二段階合成によって達成されることが好ましい。さらなる置換基R3およびR6が導入される場合、これは、好ましくは、アニオン性アルキル基によるジイミンの還元の間に達成される。
【0038】
本発明の第三の局面は、化学反応における触媒としての、上記式Iで示される3,4-ジアミノピリジンの使用に関する。この化学反応は、好ましくはアシル化反応、特にアルコールまたはアミンのアシル化反応、またはウレタンおよび/またはポリウレタン合成である。本発明の3,4-ジアミノピリジンは、有利には、イソシアネートとアルコールとの反応を触媒し得る。したがって、本発明の3,4-ジアミノピリジンは、ウレタンおよび/またはポリウレタンの合成に有用である。
【0039】
触媒の通例の使用において、本発明の3,4-ジアミノピリジンは、触媒される反応に必要な試薬および溶媒に添加される。これらは、アシル化反応またはウレタンおよび/またはポリウレタンの製造に必要な試薬であり得る。
また、本発明の化合物は、薬剤または殺草剤の製造に有利に使用し得る。
【実施例】
【0040】
以下、本発明を、実施例によって、および添付の図面を参照して、より詳細に記載する。
3,4-ジアミノピリジン部分に基づく触媒の開発は、対応するアシル中間体の安定性の理論推定値によって援助される。該理論推定値は、ホモデスミック(homodesmic)アシル転移反応(1)についての反応のエンタルピーによって定量的に表現し得る(スキーム2参照)。
【0041】
3,4-ジアミノピリジンについての多くの反応のエンタルピーを、その触媒潜在能力が以前の研究から既に知られている他のピリジン誘導体についての値と共に、表1に示す。これらとしては、ピリジン(4)、DMAP(1)、PPY(2)および三環式DMAP誘導体5が挙げられ、これらは全て本発明ではない。表1に示す結果を、図1にグラフ形式で示す。
【0042】
【表1】
【0043】
表1.スキーム1の等式(1)によるアセチル転移反応についての、298.15 Kでの反応のエンタルピーΔHrxn(298)。これらは、B3LYP/6-311+G(d,p)// B3LYP/6-31G(d)レベル(単位kJ mol-1)に関して算出された。a 元素e上の電荷の単位;b 電荷および距離パラメーターは、常に、最も好適な配座異性体に基づく。
【0044】
ここで考えられる構造的に最も簡単な3,4-ジアミノピリジンは、テトラヒドロピリド[3,4-b]ピラジン骨格に基づく6である。対応するアセチル中間体6Acの安定性は、より高い触媒活性の5の安定性に近い。1つ(7および21における)または2つ(8におけるような)のアセチル基による6中の2つのメチル基の置換は、ピリジン環中の電子密度を低減させ、したがって、対応するカチオン7Acおよび8Acの安定性も極めて顕著に低減させる。6Acおよび21Acの間の安定性の差(19.5 kJ mol-1)および6Acおよび7Acの間の安定性の差(37.9 kJ mol-1)は、4位中の窒素が、3位中の窒素よりも置換パターンの変化に対して顕著により感受性になることを示す。8中2つの置換基(6に対して56.3 kJ mol-1)の複合効果は、2つの個々の置換基(57.4 kJ mol-1)の合計と実質的に同一である。エチル基による6中の2つのメチル基の置換は、アセチル中間体の安定性を10 kJ mol-1高める。シス配置(9におけるような)またはトランス配置(10におけるような)のいずれかの系6上への飽和アルキル環の縮合も同様に、アセチル中間体の安定性を約15 kJ mol-1高める。三環系9および10に由来するアセチルピリジニウムカチオンの安定性は、二環系6について記載されたものと同様の様式でN置換基に依存する。したがって、2つのエチル基の導入は、最も安定性のアセチル中間体11Acおよび15Acを導く。最後に、6のエチレン架橋(例えば19におけるような)中へのアリール置換基の導入も、対応するカチオン性中間体の安定性を高める。これは、おそらく3,4-ジアミノ窒素原子への誘導電子移動に起因する。図1中の化合物のアセチル中間体についての安定性値は、実質的に90 kJ mol-1の範囲に渡る。化合物1、2および5の場合において、それらの安定性値は、触媒活性に関連することが見出された。理論に束縛されることを意図しないが、3,4-ジアミノピリジン18および11(これはスケールの2つの末端に印を付ける)の安定性値は、30の因子によるそれらの触媒活性に関して異なると考えられる。さらに、表1中の電荷および距離データは、より大きい熱力学安定性がより短いC-N結合長さおよびより小さいアセチル基電荷に対応することを示す。
【0045】
図1に示される化合物の幾つかを、3,4-ジアミノピリジン22から、三〜五段階の順序で、効率的に製造することができる(スキーム3参照)。22とグリオキサール、1,2-シクロヘキサジオンまたはベンジルとの縮合は、良好な収率のジイミン23〜25を導く[12]。次いで、ピリドピラジン23(R=H)は、テトラヒドロ誘導体26に還元され13、そして、Eschweiler-Clark条件下、アルキル化されて化合物6を形成することができた14。より長いアルキル鎖の導入は、ビス-アシル化およびその後の対応するジアミンへの還元を含む二段階の順序によって、より効率的に達成される15。このように、26から出発して、ジエチル化化合物20を48%の収率で製造できた。類似の手順は、ジアミン27から、2つの工程で37%の収率で得られるシクロヘキシル縮合系11の合成を可能にする。また、シス縮合ジアミン27も、24から、NaBH4/BH3/H2NCH2CH2OHを使用する選択的還元16によって74%の収率で、または-40℃でのLiAlH3を使用する還元によって90%の収率で得ることができる。化合物26について使用されるより簡単な還元方法の使用は、この場合、27のシス/トランス混合物を導く。
【0046】
【化6】
【0047】
スキーム3:3,4-ジアミノピリジン部分に基づく触媒の合成。
a) グリオキサール、1,2-シクロヘキサジエノンまたはベンジル、EtOH、70℃、5時間、95〜98%。
b) 粉末NaBH4、EtOH、40℃、24時間、50%。
c) 200当量のCH2O(水中37%)、100当量のHCO2H、110℃、48時間、57%。
d) (CH3CO)2O、ピリジン、100℃、24時間、80%。
e) 4.2当量のLiAlH4、2.6当量のAlCl3、MTBE、0℃、1時間、次いで、還流、8時間、60%。
f) LiAlH4、THF、-40℃〜室温、32時間、90%。
g) (CH3CO)2O、25 mol%のPPY、ピリジン、100℃、48時間、68%。
h) 4.2当量のLiAlH4、2.6当量のAlCl3、MTBE、0℃、1時間、次いで、還流、8時間、55%。
i) 1.1当量の(CH3CO)2O、0.2当量の2、NEt3、DCM、室温、3時間、82%。
j) 2.2当量のLiAlH4、1.3当量のAlCl3、MTBE、0℃、1時間、次いで、還流、8時間、65%。
k) 1) 1当量のn-BuLi、THF、-78℃、1時間、次いで、0℃、1時間;2) 1.1当量のCH3COCl、THF、-78℃〜室温、1時間、16%。
l) 粉末NaBH4、EtOH、40℃、48時間、74%。
m) 88当量のCH2O(水中37%)、200当量のHCO2H、110℃、48時間、90%。
n) 1) 1当量のn-BuLi、MTBE、-78℃、1時間、次いで、0℃、1時間;2) 1.1当量のCH3COCl、MTBE、-78〜0℃、1時間、36%。
o) 2.2当量のLiAlH4、1.3当量のAlCl3、THF、0℃、1時間、次いで、還流、18時間、53%。
【0048】
27の反応についてのより穏やかなアシル化方法の使用は、C4アミノ基の顕著により高い反応性を示し、これは、唯一の反応生成物としてのアミド28の優先的形成を説明する。したがって、従来通り、対応するアミンを形成するためのこのアミドの還元およびC3上の残留アミノ官能基のアシル化は、異なる強度の2つのドナー置換基を含有する触媒を製造するのを可能にする。C3およびC4上の2つのアミノ窒素原子を連結する架橋へのアリール置換基の影響を、化合物34の合成によって調査した。対応するピリドピラジン25の還元は、26について使用される条件下、容易に起こり得るが13、その後のEschweiler-Clarkアルキル化は、モノアルキル化段階にて停止して化合物32を与える。32のアセチル化は、驚くべきことに、先に使用した全ての条件下で困難であることがわかった。しかしながら、これは、最終的に、最初にn-BuLiを使用して脱プロトン化し、アミドアニオンとAcClとを反応させ、次いで、還元することによって行うことができ、その結果、34を形成することができた。
【0049】
スキーム3に示される全ての化合物の組成を、高分解能質量分析によって示す。ピリジン環の構造は、全ての化合物について、1H-NMRスペクトルの適切な領域(6.5〜8 ppm)における3つのシグナル(1つのシングレット、2つのダブレット)の存在から直接的に導くことができる。これは、化合物11、14および27〜30におけるシクロヘキサン縮合の立体化学的配置およびN-アルキル化およびN-アシル化反応の位置化学的制御の問題を残す。構造11(スキーム4参照)中の隣接プロトンHaおよびHeについての1H-NMRシグナルは、3JHH=2.8 Hzの結合定数を有し、および軸方向-エクアトリアル配置またはエクアトリアル-エクアトリアル配置のいずれかと調和し得る正のNOEシグナルを示す17。
【0050】
しかしながら、2つのプロトンのエクアトリアル-エクアトリアル配向は、除外することができる。なぜなら、3,4-ジアミノピリジン単位は、シクロヘキサン環に二軸縮合によって結合することがほとんどできないからである。
【0051】
【化7】
【0052】
スキーム4:矢印は、観察されたNOE強化を示す。
【0053】
28を形成するための27のアセチル化の位置選択性は、直接的に確認できなかった。なぜなら、28の分光データは、この点に関して情報を与えるものではなかったからである。しかしながら、28〜29の還元後、ピリジン環のC5位におけるエチル基とプロトンとの間の正のNOEシグナルを観察することができる。これは、スキーム3に示される構造を支持する。同様に、32を形成するための31のN-メチル化の位置化学は、メチル基についてのシグナル(2.85 ppm)と、ピリジン環のC1でのプロトンについてのシグナル(7.82 ppm)と、ベンジルのプロトンの一つのシグナル(4.44 ppm)との間のNOE強化の観察によって確率することができた。二重アルキル化生成物34におけるこの位置化学の保持は、この化合物(図2参照、イソヘキサン/ジクロロメタンから結晶化されたもの)の単結晶X線構造分析によって、疑いなく確認することができた。
【0054】
〔触媒特性〕
ピリジン1、2、5、6、8、11、20および34の触媒特性を、スキーム5に示す2つのアシル化反応AおよびBにおいて調査した。
【0055】
【化8】
【0056】
スキーム5:アシル化反応AおよびB
【0057】
第3級アルコール35は、以前の研究において、その低い反応性のためにモデル基質として使用された10。補助塩基としてのトリエチルアミンの存在下での無水酢酸(36)による35のアセチル化(反応A)は、少なくともDMAP(1)の反応性を有する触媒を10%の濃度(35に対して)で使用する限り、定量的に進行する。該反応の半減期τ1/2は、1H-NMRスペクトルにおける出発材料および生成物のシグナルの積分によって決定することができる。ここで、該半減期を触媒活性の指標として使用する。反応Bにおいて、同一のアルコールを、従来通り、アシル化試薬としてのイソ酪酸無水物(38)と反応させる。この反応は、36の反応よりも顕著にゆっくりと進行するため、該反応のために、より高い反応温度(40℃)を選択した。この選択は、2つの反応のほぼ同じ半減期を導く。
【0058】
DMAP触媒アシル化反応に関する最近の研究に基づいて18,19、非極性溶媒中の両方の反応について、スキーム6に示される機構を推測することができる。したがって、無水物および触媒を、まず、(通常迅速)の第一段階において反応させ、アシルピリジニウムカチオンとカルボキシレートアニオンとの錯体を形成する。この錯体は、通常、本明細書中で使用される条件下、1H-NMRスペクトル中に検出することができない。したがって、対応する平衡定数Kは、反応AおよびBの両方について、非常に小さいと推測することができる。
【0059】
【化9】
【0060】
スキーム6:反応AおよびBによるアシル化機構
【0061】
次いで、速度決定工程において、該アシルピリジニウムカチオンを、アルコール35と反応させてエステル生成物を形成し、および触媒を不活性化する。該触媒は、補助塩基NEt3の作用によって再生することができる。ここで測定される半減期τ1/2(表2)は、該工程の両方に対する触媒の影響を反映する。したがって、Kに対する置換基効果が、kcatに対する効果よりも大きい場合にのみ、アセチルピリジニウムカチオンについてのτ1/2と算出された安定性値(表1)との明らかな相関関係が予想され得る。
【0062】
【表2】
【0063】
表2.モデル反応AおよびBについての反応半減期τ1/2(単位:分)。括弧内の結果は、補助塩基としてHuenig塩基(EtN(i-Pr)2)を使用して得られた。a 参考文献20参照;b 参考文献23参照
【0064】
表2中の化合物1、2および5についての反応性データは、先行文献中の同一条件下での同一反応についての反応性データから幾分外れている10。これは、部分的に、該反応(実験部参照)に続くNMR測定における小さな差に起因する。さらに、比較的活性な触媒(例えば5)についての結果は、並外れて純粋な試料を使用する場合のみ、再現することができる。他方、厳密に嫌気的な条件下での反応の実施は必要ではない。触媒6、11および20についての表2中の結果は、ジアミノピリジンがモノ置換ピリジン(例えばDMAPまたはPPY)よりも顕著に反応性であることを示す。最高の結果は、上記炭素環式化合物5に対して同様の活性を有する化合物11について得られた。20および11についての結果の比較は、架橋部分のドナー特性の強い影響を示すが、N-メチル置換またはN-エチル置換(20対6)の間の差は、小さい効果を有する傾向にある。これらの結果は、表1および図1における安定性値と完全に一致するが、関連化合物19の安定性値によって測定された34の触媒活性は、驚くべきことに低い。該触媒活性は、アクセプター置換基の導入によって顕著に低減される。これは、30の低活性(11と比較して)によって、並びにN-アシル化化合物8を使用する場合の顕著に遅い反応によって示される。これは、対応するアセチル中間体の低安定性値の観点から予期されるべきことであったが、それにもかかわらず、本発明者らは、図1中の安定性スケールが、全ての場合において、表2中の実験的に決定された反応性データに一致しているわけではないことを認識しなければならない。基質の選択に対する反応性データの依存性は、反応AおよびBについての結果の比較によって明らかになる。最も活性な化合物11およびDMAPについての反応性比は、反応Aについて7.0であり、および反応Bについて4.8である。同様の結果は、ピリジン誘導体5について得られ、これは、7.9および4.5の反応性比を示す。補助塩基の選択は、触媒5および11について調査されたさらなる実験的変数を表す。ここで、驚くべきことに、両方の触媒についての触媒効率は、NEt3に比べてより強い補助塩基EtN(iPr)2(Huenig塩基)を使用する場合、比較可能なように低減することが見出された。
【0065】
アルキル置換3,4-ジアミノピリジンは、アルコールのアシル化についての触媒の新規クラスを表す。これらの化合物の触媒効率は、窒素置換基の変動によって、単独の合成戦略内で幅広い範囲に渡って変動し得る。これらの触媒の最も良好なものの触媒活性は、これまで既知のDMAPの炭素環式誘導体の最も良好なものに匹敵する。後者の化合物とは対照的に、最も活性な3,4-ジアミノピリジン触媒は、柔軟な3-または4-段階合成によって、簡単な方法で得ることができる。当該方法は、さらなる構造的変動のために、容易に改変することができる。
【0066】
幾何学の最適化は、原子価殻分割型基底関数6-31G(d)と組み合わせたBecke3LYPハイブリッド汎関数を使用して行った。298.15 Kの温度での熱化学的エンタルピー補正は、同じレベルでの振動数計算によって行う。一点計算をBecke3LYP/6-311+G(d,p)レベルについて行う。Becke3LYP/6-31G(d)レベルでの熱化学的補正によって得られたエネルギーの組合せは、本明細書中に記載されたH298値を与える。全ての計算は、Gaussian 03を使用して行う21。
【0067】
水分を除外して行う全ての反応は、乾燥溶媒および乾燥オーブンおよびヘアドライヤーによって乾燥した反応装置中、窒素雰囲気下で行う。THFは、窒素雰囲気下、水素化ナトリウムから蒸留する。メチルtert-ブチルエーテル(MTBE)、ピリジン、トリエチルアミンおよび重クロロホルムは、窒素雰囲気下、水素化カルシウムから蒸留する。無水酢酸およびイソ酪酸無水物は、減圧下、P4O10から乾燥K2CO3上に蒸留し、濾過し、および減圧下、分画する。両方の無水物は、窒素雰囲気下、4Å分子篩上で貯蔵する。エタノール、ジクロロメタン、酢酸エチルおよびメタノールは、ロータリーエバポレーターで蒸留する。n-BuLiは、ジフェニル酢酸で滴定し、モル濃度を決定する。全ての他の試薬は、本明細書中で他に示さない限り、さらなる精製をせずに、最も高い可能性のある品質で使用する。薄層クロマトグラフィーは、Merck KGaAからの蛍光染料でマークしたTLCプレート(シリカゲル60 F254、層厚さ:0.2 mm)およびFlukaプレート(塩基性酸化アルミニウムF254、Brockman活性:1、pH:9.5、層厚さ:0.2 mm)を用いて行う。フラッシュクロマトグラフィーは、Merck KGaAシリカゲル60(粒度:0.040-0.063 mm)およびFluka塩基性酸化アルミニウム(塩基性酸化アルミニウムF254、Brockman活性:1、pH:9.5、粒度:0.05-0.15 mm)を使用して、シリカゲルについて1.5 barの圧力にて、および塩基性酸化アルミニウムについて0.5 barの圧力にて行う。1H-および13C-NMRスペクトルは、Varian Mercury 200、Varian 300、Varian INOVA 400およびVarian 600機器を用いて記録する。NOEスペクトルは、CDCl3中、27℃にて記録する。化学シフトは、溶媒ピークに対するppmで報告する22。以下の略語を使用して1H-NMRスペクトルにおけるシグナルの多重度を特徴付ける:s=シングレット、d=ダブレット、t=トリプレット、q=カルテット、quin=クインテット、sex=セクステット、sep=セプテット、m=マルチプレット、b=ブロードおよび使用した略語の組合せ。全ての1Hおよび13Cシグナルは、COSY、NOESY、HSQC、HMBCおよびDEPT実験を用いて割り当てる。反応速度測定についての1H-NMRスペクトルは、プログラムVNMR 4.3 Rev. G0194を使用して評価する。関連ピークの積分は、社内で書かれたサブプログラムを使用して自動的に評価する。サブルーチンは、MAGICAL(商標) IIプログラミングを使用して記載する。IRスペクトルは、試料媒体としてKBrペレットを用いて、Perkin-Elmer 1420赤外分光計およびATR技術を使用するPerkin-Elmer FT-IRスペクトルBX分光計を使用して記録する。全てのシグナルは、vs=非常に強い、s=強い、m=中程度およびw=弱いとして報告する。質量スペクトルは、イオン源として電子衝撃イオン化(EI、70 eV)または化学イオン化(CI、イソブタン)を使用して、Finnigan MAT 95によって記録する。ESI-MSスペクトルは、Thermo Finnigan LTQ FT機器を使用して記録する。ガスクロマトグラムは、25 m CS-Supreme-5キャピラリーカラムを備えたVarian 3400 GCによって、検出器としてFinnigan MAT 95質量分析計を使用して記録する。電子が豊富なピリジン11、5、6、20および32の場合、溶媒は、フラッシュクロマトグラフィー後、窒素流をロータリーエバポレーター中に通じることによって留去する。
【0068】
〔反応性実験AおよびBについての一般的手順〕
重クロロホルム、トリエチルアミンおよびHuenig塩基を、使用前に、窒素雰囲気下、水素化カルシウムから新たに蒸留する。全ての反応速度測定を、反応Aについて23℃および反応Bについて40℃の一定の温度にて、Varian Mercury 200分光計を用いて記録する。以下の重クロロホルム中の標準溶液を、3つの乾燥した5 ml容のフラスコ中で作製した。
A:1.2 Mの無水酢酸溶液
B:0.6 Mのエチニルシクロヘキサノール溶液および1.8 MのトリエチルアミンまたはHuenig塩基溶液
C:0.06 Mの触媒溶液
【0069】
(反応Aについての試料調製および反応速度測定)
いずれの場合も、200 μlの上記標準溶液を、エッペンドルフ型ピペットを用いてNMRチューブ中に取る。該反応混合物を混合し、そして直ぐにNMR分光計中に置く。該反応を、所定の時間間隔にて100%変換までNMRスペクトルを記録することによって監視する。
【0070】
(反応Bについての試料調製および反応速度測定)
溶媒の蒸発を防止するために、NMRチューブを、バーナーを用いてフレームシールすること以外は、反応Aについて記載された手順。
【0071】
(反応Aについての半減期の決定)
アセチル基のプロトンに割り当てられ得る全てのシグナルを、スペクトルの記録後、自動的に積分する。ここで、無水酢酸、エステルおよび酢酸トリエチルアンモニウムを、以下の積分限界内で積分する:±8、±2および±6 Hz。該変換を、等式2にしたがって算出する。該変換を、等式4を用いて時間の関数として自己矛盾なく適合させる。該半減期を、関数4を使用して50%変換にて算出する。
【0072】
(反応Bについての半減期の決定)
イソ酪酸基のダブレットおよびジオキサンのシグナルを、NMRスペクトルの記録後、自動的に積分する。ここで、ジオキサンおよびイソ酪酸無水物を、同じ積分限界±2 Hzの範囲内で積分する。該変換を、等式3にしたがって算出する。該変換を、等式4を用いて時間の関数として自己矛盾なく適合させる。該半減期を、関数4を使用して50%変換にて算出する。
【0073】
【数1】
【0074】
〔方法〕
ピリド[3,4-b]ピラジン(23):
2.00 g(18.33 mmol)の3,4-ジアミノピリジンを、30 mlのエタノール中の2.82 mlのグリオキサール(水中40重量%、ρ=1.265 g/ml、62.48 mmol)の溶液に添加する。該反応混合物を、70℃の油浴温度にて5時間維持し、次いで、室温に冷却後、該溶媒をロータリーエバポレーターによって留去する。粗生成物を、フラッシュクロマトグラフィー(EtOAc/イソヘキサン、9:1)によって精製する。これにより2.35 g(17.96 mmol、98%)の白色粉末を得る。
Rf=0.27 (イソヘキサン/EtOAc, 1:9). 1H NMR (200 MHz, CDCl3):δ=7.94 (dd, 3J=5.8 Hz, 4J=0.6 Hz, 1H, H-7), 8.83 (d, 3J=5.8 Hz, 1H, H-8), 8.96 (d, 3J=1.6 Hz, 1H, H-3), 9.02 (d, 3J=1.6 Hz, 1H, H-2), 9.56 (d, 4J=0.6 Hz, 1H, H-5) ppm. 13C NMR (100 MHz, CDCl3):δ=121.6 (CH, C-7), 138.0 (Cq), 145.3 (Cq), 147.3 (C-H, C-8), 146.5 (C-H, C-3), 149.2 (CH, C-2), 154.8 (C-H, C-5) ppm. GC-MS (EI):保持時間5.45分, m/e (%):132 (8), 131 (M+, 100), 104 (25), 77 (13), 50 (10) IR (KBr):□=3435 (vs), 3092 (w), 3023 (m), 1969 (w), 1758 (w), 1631 (w), 1598 (vs), 1562 (m), 1536 (w), 1488 (s), 1436 (vs), 1416 (m), 1381 (m), 1351 (w), 1290 (w), 1279 (m), 1212 (m), 1201 (m), 1148 (w), 1033 (s), 1014 (s), 972 (w), 959 (vs), 931 (m), 881 (vs), 838 (m), 824 (s), 773 (w), 651 (s), 623 (m), 546 (w), 524 (w), 457 (s) cm-1.
【0075】
1,2,3,4-テトラヒドロピリド[3,4-b]ピラジン(26):
2.90 g(0.076 mol)のNaBH4粉末を、100 mlの乾燥エタノール中のキノキサリン23(2.90 g、0.022 mol)の溶液に添加する。該反応溶液を、40℃の油浴温度に加熱し、次いで、この温度にて24時間維持する。その後、該反応溶液を室温に冷却し、そして3 mlの水を添加する。該無機固体を吸引によって濾去し、そして20 mlのDCMで2回洗浄する。合わせた有機相を硫酸ナトリウムによって乾燥し、次いで、該溶媒をロータリーエバポレーターによって留去する。得られる粗生成物を、塩基性酸化アルミニウムによるフラッシュクロマトグラフィー(EtOAc/MeOH、10:1)によって精製する。これにより白色発泡体を得る。
Rf=0.15 (塩基性酸化アルミニウム、20:1、DCM:MeOH). 1H NMR:(200 MHz, CDCl3):δ=3.37 (d, 3J=5.8 Hz, 1H, H-3), 3.38 (d, 3J=5.8 Hz, 1H, H-2), 6.29 (d, 3J=5.4 Hz, 1H, H-8), 7.67 (d, 3J=5.4 Hz, 1H, H-7), 7.67 (s, 1H, H-5). 13C NMR:(100 MHz, CDCl3):δ=40.1 (CH2), 41.0 (CH2), 107.8 (C5), 129.6 (Cq), 135.3 (C-H, C7), 140.1 (Cq), 141.1 (C-H, C-5). GC-MS 保持時間8.09分 (EI) m/e (%):136 (8), 135 (M+ 79), 134 (M+-H+,100), 133 (10), 132 (7), 131 (2), 120 (4), 107 (7), 105 (4), 94 (2), 93 (7), 80 (2), 79 (3), 78 (4), 67 (4), 66 (2), 53 (2), 52 (2), 52 (3), 51 (2). IR (KBr):□=3349 (m), 2859 (m), 1593 (s), 1534 (s), 1474 (s), 1344 (s), 1311 (s), 1281 (s), 1256 (w) 1228 (m), 1182 (m), 1102 (m), 1051 (w), 1038 (m), 893 (m), 862 (m), 824 (s), 772 (m) cm-1. HRMS (EI) (%):C7H8N3 (M-H+)の計算値:134.0718、実測値:134.0709.
【0076】
1-(4-アセチル-3,4-ジヒドロ-2H-ピリド[3,4-b]ピラジン-1-イル)エタノン(8):
23 ml(24.71 g、24.20 mol)の無水酢酸を、60 mlのピリジン中のテトラヒドロキノキサリン26(1.49 g、0.011 mol)の溶液に0℃の温度にて添加する。その後、該反応混合物を100℃にて48時間加熱する。室温に冷却後、該溶媒を減圧下留去し、そして該黄色固体をフラッシュクロマトグラフィー(EtOAc:MeOH、10:3)によって精製する。これにより1.93 g(8.80 mmol、80%)の淡黄色固体を得る。
1H NMR (400 MHz, CDCl3):δ=2.25 (s, 3H, CH3), 2.31 (s, 3H, CH3), 3.90 (ddd, 2J=12 Hz, 3J=4 Hz, 3J=4 Hz, 2H, CH2), 3.94 (ddd, 2J=12 Hz, 3J=4 Hz, 3J=4 Hz, 2H, CH2), 7.67 (s, 1H, H-5), 7.87 (bs, 1H, H-5), 8.32 (d, 3J=4 Hz, 1H, H-7), 8.46 (m, 1H, H-8). 13C NMR (100 MHz, CDCl3):δ=22.3, 22.8 (CH3), 42.1, 46.5 (CH2), 117.4 (C5), 128.2 (Cq), 139.0 (Cq), 145.7 (C-7), 146.9 (C-8), 168.6 (C=O). GC-MS(EI) 保持時間9.66分, m/e (%):220 (14), 219 (M+, 81), 178 (11), 177 (M+-AcO,100), 176 (10), 162 (3), 159 (2), 136 (7), 135 (78), 134 (M+-2AcO,99), 133 (8), 132 (9), 120 (4), 119 (2), 107 (5), 105 (2), 93 (3), 80 (2), 79 (4), 78 (4), 52 (2), 51 (2), 43 (AcO+,20);IR (neat):□=2960 (w), 1684 (s) 1654 (vs) 1582 (m) 1494 (s), 1407 (vs), 1330 (m), 1320 (vs), 1277 (m), 1259 (s), 1248 (m), 1217 (s), 1248 (s), 1217 (s), 1179 (m), 1150 (w), 1118 (m), 1064 (m), 1033 (s), 969 (s), 886 (w), 857 (m), 846 (s), 799 (s), 764 (w), 749 (w), 704 (w) cm-1. HRMS (EI): C11H13N3O2 [M+]の計算値:219.1008;実測値:219.0995.
【0077】
1,4-ジエチル-1,2,3,4-テトラヒドロピリド[3,4-b]ピラジン(20):
1.56 g(11.69 mmol)のAlCl3を、60 mlのMTBE中に室温にて懸濁させる。45分間の撹拌後、該反応混合物を0℃に冷却し、およびLiAlH4(1.32 g、34.65 mmol)を少しずつ添加する。添加が完了した後、該混合物を別の15分間攪拌し、化合物8(1.00 g、4.56 mmol)を添加し、そして該混合物を、0℃にて別の1時間撹拌する。その後、該反応混合物を、8時間還流し、そして室温に冷却後、氷水中に注ぐ。沈澱した無機固体を吸引によって濾去し、そして30 mlのDCMで2回洗浄する。該水相をpH 12にし、そして40 mlのDCMで3回抽出する。合わせた抽出物をNa2SO4によって乾燥し、次いで、該溶媒をロータリーエバポレーターによって留去する。粗生成物を、シリカゲルによるフラッシュクロマトグラフィー(EtOAc/MeOH/NEt3、10:0.5:1)によって精製する。これにより、0.52 g(60%)の冷凍庫中で固化する無色油を得る。
Rf=0.47 (EtOAc/MeOH/NEt3、10:0.5:1). 1H NMR:(400 MHz, CDCl3):δ=1.15 (q, 3J=14 Hz, 6H, CH3), 3.22 (m, 3J=6 Hz, 2H, H-3), 3.23 (m, 2J=14 Hz, 4H, CH2), 3.42 (m, 3J=6 Hz, 2H, H-2), 6.34 (d, 3J=5.2 Hz, 1H, H-8), 7.69 (s, 1H, H-5), 7.75 (d, 3J=5.2 Hz, 1H, H-7). 13C NMR:(100 MHz, CDCl3):δ=10.1, 10.4 (CH3), 44.6, 46.5 (CH2, C-2, C-3), 44.9, 45.0 (CH2), 104.0 (C-8), 130.5, 130.1 (Cq), 131.8 (C-5), 140.7 (C-7). GC-MS(EI):保持時間8.63分, m/e (%):192 (10), 191 (M+,100) , 190 (4), 177 (8), 176 (95), 175 (5), 162 (11), 161 (8), 160 (5), 148 (13), 147 (10), 146 (8), 135 (2), 134 (9), 133 (6), 131 (8), 121 (2), 119 (4), 118 (3), 107 (3), 104 (2), 92 (2), 91 (2), 80 (8), 77 (4) IR (KBr):v=3436 (s), 3112 (w), 3019 (w), 2964 (w), 1661 (s), 1686 (vs), 1583 (m), 1497 (m), 1409 (s), 1363 (w), 1332 (m), 1311 (s), 1260 (m), 1249 (w), 1232 (m), 1220 (m), 1180 (m), 1150 (w), 1119 (m), 1064 (w), 1035 (m), 985 (m), 970 (m), 886 (s), 858 (m), 847 (m), 802 (m), 765 (w), 740 (w), 704 (w), 647 (w), 614 (w), 592 (w), 577(m), 570 (m), 507 (w) cm-1. HRMS (EI):C11H17N3 [M+]の計算値:191.1422;実測値:191.1430.
【0078】
1,4-ジメチル-1,2,3,4-テトラヒドロピリド[3,4-b]ピラジン(6):
35 mlのギ酸を、氷中で冷却しながらテトラヒドロキノキサリン26(1.26 g、9.32 mmol)に添加する。その後、12 mlのホルムアルデヒド溶液(水中<37%)を添加し、そして該混合物を110℃の油浴温度にて48時間加熱する。その後、室温まで冷却し、そして約150 mlの20%濃度の水酸化ナトリウム溶液を、氷中で冷却しながら添加する。該pHは、この添加の間に12を超えるべきではない。母液を、液体/液体抽出装置中で、250 mlのDCMを用いて、終夜抽出する。該有機相を、Na2SO4によって乾燥し、次いで、該溶媒をロータリーエバポレーターによって留去する。粗生成物を、シリカゲル(EtOAc/MeOH/NEt3、10:1:1)および塩基性酸化アルミニウム(EtOAc/MeOH、10:1)によるフラッシュクロマトグラフィーによって精製する。これにより0.849 g(5.32 mmol、57%)の黄色固体を得る。
Rf=0.56 (塩基性アルミナ、EtOAc/MeOH、10:1). 1H NMR:(400 MHz, CDCl3):δ=2.77 (s, 3H, CH3), 2.82 (s, 3H, CH3), 3.12 (m, 2H, CH2-B), 3.37 (m, 2H, CH2-C), 6.21 (d, 3J=5.6 Hz, 1H, H-7), 7.56 (s, 1H, H-5), 7.74 (d, 3J=5.6 Hz, 1H, H-8) ppm. 13C NMR:δ=(75 MHz, CDCl3):38.0 (N-1-CH3), 39.1 (N-4-CH3), 48.8 (C-3), 49.6 (C-2), 104.0 (C-7), 131.2 (C-5), 132.3 (C-4a), 141.6 (C-7), 142.2 (C-8a) ppm;GC-MS(EI):保持時間8.05分, m/e (%):164 (8), 163 (M+,100), 162 (21), 161 (5), 149 (4), 148 (40) 147 (9), 146 (8), 134 (8), 133 (11), 132 (6), 121 (4), 120 (2), 119 (6), 107 (3), 105 (2), 93 (2), 92 (4), 81 (6), 80 (4), 79 (2), 78 (4), 66 (3), 51 (2), 42 (4). IR (neat):□=3378 (w), 3035 (w), 2979 (w), 2873 (m), 2826 (s), 2792 (w), 1581 (s), 1519 (s), 1466 (s), 1454 (s), 1435 (m), 1416 (m), 1380 (w), 1335 (vs), 1290 (s), 1250 (w), 1235 (vs), 1214 (m), 1172 (s), 1114 (s), 1099 (s), 1069 (s), 1030 (m), 936 (w), 911 (w), 883 (s), 815 (s), 800 (s), 783 (s), 746 (m), 709 (m), 622 (m) cm-1. HRMS (EI):C9H13N3 [M+]の計算値:163.1109;実測値:163.1089.
【0079】
6,7,8,9-テトラヒドロピリド[3,4-b]キノキサリン(24):
1,2-シクロヘキサンジオン(1.52 g、13.93 mmol)を、50 mlのエタノール中の1.56 g(13.93 mmol)の3,4-ジアミノピリジンの懸濁液に添加する。その後、該反応混合物を70℃の油浴温度にて5時間加熱し、そして室温に冷却し、次いで、該溶媒をロータリーエバポレーターによって留去する。得られる生成物を、溶離剤として酢酸エチルを使用するフラッシュクロマトグラフィーによって精製する。これにより1.84 g(71%)の白色固体を得る。該固体は、冷蔵庫中に貯蔵し、そしてさらなる1〜2日後に処理すべきである。該固体を開口したフラスコ中で日光中に放置することによって、灰緑色への変色が生じる。
Rf=0.25 (EtOAc/ヘキサン、20:1). 1H NMR:(200 MHz, CDCl3):δ=2.05 (m, 4H, H-7,8), 3.18 (m, 4H, H-6,9), 7.79 (d, 3J=5.8 Hz, 1H), 8.72 (d, 3J =5.8 Hz, 1H), 9.38 (s, 1H) ppm. 13C NMR (75 MHz, CDCl3):δ=22.8 (CH2, C-7,8), 33.7 (CH2, C-6,9), 121.2 (CH), 136.9 (Cq), 144.1 (Cq), 146.8 (CH), 153.9 (CH), 156.8 (Cq) 159.9 (Cq) ppm. GC-MS(EI):保持時間8.44分, m/e (%) 186 (11), 185 (M+,100), 184 (39), 183 (3), 182 (3), 171 (2), 170 (21), 169 (4), 158 (2), 157 (5), 156 (5) 131 (2), 104 (2), 103 (4), 78 (3), 76 (4), 67 (2), 64 (2), 51 (2), 50 (6);IR (KBr):□=3435 (vs), 2947 (s), 2864 (m), 1594 (s), 1557 (w), 1461 (w), 1421 (m), 1385 (s), 1365 (w), 1330 (w), 1297 (m), 1211 (m), 1140 (w), 979 (m), 949 (w), 901 (m), 849 (m), 679 (w), 629 (w), 570 (w), 412 (w) cm-1. HRMS (EI):C11H11N3の計算値:185.0953 [M+]、実測値:185.0935
【0080】
5,5a,6,7,8,9a,10-オクタヒドロピリド[3,4-b]キノキサリン(27):
6.13 g(33.09 mmol)の24および100 mlのTHFを、250 mlのSchlenkフラスコ中に置く。該溶液を-40℃に冷却し、そして3.09 g(81.42 mmol、2.5当量)のLiAlH4を少量ずつ添加する。該反応混合物を室温まで温め、そして室温にて32時間撹拌する。その後、氷水中に注ぎ、次いで、該水相をpH 12にする。該無機固体を分離除去し、そして40 mlのDCMで2回洗浄する。母液を、液体/液体抽出装置中で、250 mlのDCMを用いて8時間抽出する。合わせた有機相をNa2SO4によって乾燥し、次いで、該溶媒をロータリーエバポレーターによって留去する。得られる粗生成物のフラッシュクロマトグラフィー(EtOAc/MeOH/NEt3、10:1:1)によって、5.63 g(29.78 mmol、90%)の白色固体を得る。
Rf=0.36 (EtOAc/MeOH/NEt3、10:1:1). 1H NMR:(300 MHz, CDCl3):δ=1.32-1.40 (m, 3H), 1.56-1.75 (m, 5H), 3.41 (m, 1H), 3.46 (s, 1H, N-H), 3.51 (m, 1H), 4.20 (s, 1H, N-H), 6.29 (d, 1H, 3J=5.4 Hz, H-3), 7.65 (d, 1H, 3J=5.4 Hz, H-4), 7.68 (s, 1H, H-1) ppm. 13C NMR (75 MHz, CDCl3):δ=22.0, 22.2 (C-7,8), 30.4, 30.8 (CH2, C-6,9), 49.2 (CH, C-5a), 50.1 (CH, C-9a), 107.7 (C-4), 128.8 (Cq, C-10a), 134.8 (C-3), 139.2 (Cq, C-4a) 140.7 (C-4) ppm. GC-MS (EI):保持時間10.06分, m/e (%) 190 (13), 189 (M+, 99), 188 (9), 187 (3), 185 (10), 184 (4), 170 (4), 160 (10), 159 (3), 158 (5), 148 (2) 147 (16), 146 (100), 145 (2), 134 (13), 133 (17), 132 (15), 120 (10), 119 (3), 105 (2) 94 (3), 93 (2), 78 (3), 66 (2), 40 (2);IR (neat):□=3220 (s), 2927 (s), 2852 (s), 2354 (m), 1725 (w), 1595 (vs), 1523 (vs), 1456 (w), 1443 (m), 1403 (w), 1362 (s), 1294 (s), 1269 (s), 1240 (m), 1206 (s), 1175 (s), 1087 (m), 1052 (m), 1003 (m), 938 (m), 884 (m) 810 (vs) 725 (m) cm-1. HRMS (EI):C11H15N3の計算値:189.1266 [M+]、実測値:189.1259.
【0081】
別の方法として、該反応は、試薬としてNaBH4/BH3/H2NCH2CH2OHを使用するOpatzらの方法でも行うことができる16。これにより74%の収率を得る。
【0082】
1-(10-アセチル-6,7,8,9,9a,10-ヘキサヒドロ-5aH-ピリド[3,4-b]キノキサリン-5-イル)エタノン(14):
27(1.20 g、6.34 mmol)、40 mlのピリジン、30 ml(32.36 g、317 mmol、ρ=1.082 g/ml)の無水酢酸および0.234 g(25 mol%)のPPYを、氷中で冷却しながら250 mlの丸底フラスコ中に導入する。該反応混合物を100℃の油浴温度に加熱し、そしてこの温度にて48時間維持する。該反応は、塩基性酸化アルミニウム(EtOAC/MeOH、10:1)によるTLCによって追跡することができる。室温に冷却後、該溶媒を減圧下留去し、そして得られる粗生成物を、塩基性酸化アルミニウムによるフラッシュクロマトグラフィー(EtOAc/MeOH、10:1)によって精製する。これにより暗黄色固体(1.17 g、4.31 mmol、68%)を得る。
Rf=0.45 (EtOAc/MeOH、10:1). 1H NMR:(400 MHz, CDCl3):δ=1.34 (m, 4H), 1.60 (d, 4H), 2.23 (s, 3H, H3C-CO-N), 2.27 (s, 3H, H3C-CO-N), 4.74 (m, 1H), 4.85 (m, 1H), 7.25 (d, 3J=5.6 Hz, 1H, H-4), 8.41 (d, 3J=5.6 Hz, H-3), 8.49 (s, 1H, H-1) ppm. 13C NMR (75 MHz, CDCl3):δ=21.7, 21.9 (C-7,8), 23.1, 23.6 (H3C-CO-N), 28.4, 28.5 (C-6,9), 55.2, 56.0 (CH, C-5a,9a), 119.6 (C-4), 130.8 (Cq, C-10a), 141.6 (Cq, C-4a), 147.1, 147.2 (C-1, C-3) 169.1, 169.2 (C=O) ppm. GC-MS(EI):保持時間10.45分, m/e (%) 274 (13), 273 (M+, 58), 232 (15), 231 (100), 230 (42), 216 (5), 214 (5), 213 (5), 203 (3), 202 (4), 190 (13), 189 (84), 188 (45), 173 (5), 172 (6), 160 (7), 159 (5), 158 (5), 147 (8), 146 (40) 134 (6), 133 (13), 132 (15), 120 (7), 43 (9);IR (neat):□=2940 (m), 1660 (vs), 1587 (m), 1559 (w), 1498 (s), 1448 (w), 1427 (w), 1388 (m), 1353 (w), 1337 (m), 1289 (s), 1270 (s), 1248 (s), 1248 (m), 1220 (w), 1183 (w), 1102 (w), 1036 (m), 978 (m), 857 (w), 839 (w) 777 (w) cm-1. HRMS (EI):C11H15N3の計算値:273.1477 [M+]、実測値:273.1482.
【0083】
5,10-ジエチル-5,5a,6,7,8,9,9a,10-オクタヒドロピリド[3,4-b]キノキサリン(11):
30 mlのMTBE中の0.77 g(5.80 mmol)のAlCl3の懸濁液を、室温にて45分間攪拌し、その後、0℃に冷却し、次いで、1.32 g(9.35 mmol)のLiAlH4を少量ずつ添加する。添加が完了した後、該混合物を別の15分間攪拌し、化合物14(0.64 g、2.23 mmol)を添加し、そして該混合物を0℃にて1時間撹拌する。その後、8時間還流し、次いで室温に冷却し、そして氷水中に注ぐ。無機沈澱を吸引によって濾去し、そして30 mlのDCMで2回洗浄する。該水相をpH 12にし、そして40 mlのDCMで3回抽出する。合わせた有機相をNa2SO4によって乾燥し、次いで、該溶媒をロータリーエバポレーターによって留去する。得られる粗生成物を、シリカゲル(EtOAc/MeOH/NEt3、10:2:1)および塩基性酸化アルミニウム(EtOAc/MeOH、10:1)によるフラッシュクロマトグラフィーによって精製する。これにより0.30 g(1.22 mmol、55%)の無色固体を得る。
Rf=0.32 (塩基性アルミナ、EtOAc/MeOH、10:1). 1H NMR:(400 MHz, CDCl3):δ=1.11, 1.13 (t, 3J=7.2 Hz, H3C-CH2-N-5, 3J=7.2 Hz, H3C-CH2-N-10, 6-H), 1.37 (m, 2H, H-7,8), 1.56 (m, 3Jaa=6.8 Hz, 3Jee=3.2 Hz, 4H, H-6, H-9, H-7, H-8), 1.76 (m, 3Jae=3.2 Hz, 1H, H-9), 1.89 (m, 3Jae=3.2 Hz, 1H), 3.18 (dq, 3J=7.2 Hz, 2J=14.4 Hz, 2-H, H3C-CHAB-N-5, H3C-CHAB-N-10), 3.23 (ddd, 3Jae=2.8 Hz, 3Jae=3.2 Hz, 3Jaa=6.8 Hz, 1H, H-5a), 3.34 (ddd, 3Jae=2.8 Hz, 3Jae=3.2 Hz, 3Jae=3.2 Hz, H-1, H-9a), 3.43 (dq, 3J=7.2 Hz, 2J=14.4 Hz, H3C-CHAB-N-5, H3C-CHAB-N-10, 2H, H-1), 6.33 (d, 3J=5.6 Hz, 1H, H-4), 7.67 (s, 1H, H-1), 7.72 (d, 1H, H-3) ppm. 13C NMR (100 MHz, CDCl3):δ=10.8 (H3C-CH2-N-5), 11.8 (H3C-CH2-N-10), 21.7 (C-7), 22.9 (C-8), 27.3 (C-6), 27.8 (C-9), 40.4 (H3C-CH2-N-5), 41.7 (H3C-CH2-N-10), 52.8 (C-9a), 56.4 (C-5a), 104.3 (C-4), 130.6 (C-10a, C-1), 139.4 (C-3), 141.2 (C-4a) ppm. GC-MS(EI):保持時間10.58分, m/e (%) 246 (18), 245 (M+, 100), 231 (9), 230 (57), 217 (7), 216 (44), 207 (6), 186 (7), 174 (15), 162 (6), 160 (16), 158 (6), 148 (12), 146 (6), 132 (8);IR (neat):□=3436 (s), 2970 (m), 2860 (vs), 2860 (s), 1628 (w), 1576 (vs), 1513 (vs), 1473 (w), 1447 (m), 1348 (s), 1317 (w), 1267 (s), 1211 (s), 1167 (w), 1125 (w), 1107 (w), 1077 (w), 1059 (w), 1040 (w), 920 (w), 798 (s) 777 (m), 746 (m) cm-1. HRMS (EI):C15H23N3の計算値:245.1892 [M+]、実測値:245.1889.
【0084】
1-(6,7,8,9,9a,10-ヘキサヒドロ-5aH-ピリド[3,4-b]キノキサリン-5-イル)エタノン(28):
27 (1.0 g、5.28 mmol)、20 mlのDCM、2.20 ml(15.84 mmol)のNEt3および5 mol%のPPY(39 mg、0.264 mmol)を、100 mlのSchlenkフラスコ中に置く。その後、0.54 ml(5.81 mmol)の無水酢酸を添加し、そして該反応混合物を30分間攪拌する。その後、該反応を2 mlのMeOHの添加により停止させ、そしてさらに10分間撹拌する。該溶媒をロータリーエバポレーターによって留去し、そして得られる粗生成物をフラッシュクロマトグラフィー(EtOAc/MeOH/NEt3、10:1:1)によって精製する。これにより1 g(4.31 mmol、82%)の28を得る。得られる生成物は、不純物としてPPYを含有し、そしてさらなる精製をせずにさらに反応させる。
Rf=0.44 (EtOAc/NEt3/MeOH、10:1:1). 1H NMR:(300 MHz, CDCl3):δ=1.44 (m, 5H), 1.76 (m, 3H), 2.29 (s, 3H, H3C-CO-N), 3.59 (m, 1H), 4.07 (m, 1H), 7.08 (bs, 1H, H-4), 7.85 (d, 3J= 5.4 Hz, 1H, H-3), 7.98 (s, H-1, H-1). 13C NMR (75 MHz, CDCl3):δ=18.5 (CH2), 23.5 (H3C-CO-N), 24.8, 25.4 (CH2), 31.08 (CH2), 48.9 (C-9a), 118.1 (Cq, C-4a), 127.9 (Cq, C-10a), 132.2 (C-4), 136.6 (C-1), 138.0 (C-3), 169.0 (C=O) ppm. HRMS (EI): C13H17N3Oの計算値:231.1372 [M+]、実測値:231.1368.
【0085】
5-エチル-5,5a,6,7,8,9a,10-オクタヒドロピリド[3,4-b]キノキサリン(29):
0.619 g(4.64 mmol)のAlCl3を30 mlのTHF中に懸濁させ、そして該混合物を室温にて45分間攪拌する。その後、該混合物を0℃に冷却し、そして0.300 g(7.91 mmol)のLiAlH4を少量ずつ添加する。別の15分間の撹拌後、化合物28(0.832 g、3.59 mmol)を添加し、そして該混合物を0℃にて1時間撹拌する。その後、該反応混合物を8時間還流し、そして冷却後、氷水中に注ぐ。該無機沈澱を濾去し、そして30 mlのDCMで2回洗浄する。母液をpH 12にし、そして40 mlのDCMで3回抽出する。合わせた有機相をNa2SO4によって乾燥し、次いで、該溶媒をロータリーエバポレーターによって留去する。該粗生成物を、シリカゲルによるフラッシュクロマトグラフィー(EtOAc/NEt3、10:1)によって精製する。これにより0.430 g(1.98 mmol、55%)の白色発泡体を得る。
Rf=0.24 (EtOAc/NEt3、10:1). 1H NMR:(300 MHz, CDCl3):δ=1.06 (t, 3J= 7.2 Hz, 3H, H3C-CH2-N-5), 1.25 (m, 2H), 1.54 (m, 5H), 1.78 (m, 1H), 3.20 (m, 4H), 3.79 (s, 1H, N-H), 6.21 (d, 3J= 5.7 Hz, 1H, H-4), 7.57 (s, H-1, H-1), 7.65 (d, 3J= 5.7 Hz, 1H, H-3). 13C NMR (75 MHz, CDCl3):δ=12.1 (H3C-CH2-N-5), 19.3 (C-7), 24.8 (C-8), 26.8 (C-6), 30.9 (C-9), 42.8 (H3C-CH2-N-5), 47.8 (C-5a), 58.4 (C-9a), 103.9 (C-4), 129.8 (Cq, C-10a), 133.4 (C-1), 138.4 (Cq, C-10a), 140.9 (C-3) ppm. GC-MS(EI):保持時間10.58分, m/e (%) 218 (17), 217 (M+, 100), 216 (7), 215 (7), 213 (5), 203 (5), 202 (51), 186 (17), 185 (5), 174 (12), 162 (6), 161 (8), 160 (9), 158 (7), 148 (5);148 (5), 146 (16), 145 (7), 134 (8), 133 (9), 132 (25), 120 (14), 78 (4);IR (neat):□=3212 (m), 3093 (w), 2971 (w), 2928 (s), 2851 (s), 1589 (s), 1560 (m), 1505 (vs), 1470 (w), 1442 (m), 1419 (m), 1363 (s), 1280 (vs), 1244 (s), 1210 (m), 1194 (s), 1177 (m), 1108 (m), 1072 (m), 1039 (m), 1007 (w) 968 (w), 897 (w), 886 (w), 795 (s), 740 (m) cm-1. HRMS (EI):C13H19N3の計算値:217.1579 [M+]、実測値:215.1573.
【0086】
1-(5-エチル-5a,6,7,8,9,9a-ヘキサヒドロ-5H-ピリド[3,4-b]キノキサリン-10-イル)エタノン(30):
29(0.250 g、1.15 mmol)および10 mlのTHFを、100 mlのSchlenkフラスコ中に置く。該溶液を-78℃に冷却し、0.51 mlのn-BuLi(1.27 mmol、1.1当量;ヘキサン中2.5 M)を添加し、そして該反応溶液を、冷却することなく30分間撹拌する。30分後、該反応溶液を、-78℃に再度冷却し、および0.09 ml(0.109 g、1.4 mmol、ρ=1.1051 g/ml)の塩化アセチルを添加し、冷却槽を除去し、そして該混合物を、冷却することなく1時間撹拌する。その後、該反応を4 mlによって停止させ、および20 mlのDCMで3回抽出する。合わせた有機相をNa2SO4によって乾燥し、次いで、該溶媒をロータリーエバポレーターによって留去する。該粗生成物をシリカゲル(EtOAc/MeOH、10:2)によって精製し、48 mg(0.18 mmol、16%)の無色固体を得る。
Rf=0.16 (EtOAc/NEt3、10:1). 1H NMR:(300 MHz, CDCl3):δ=1.19 (t, 3J= 7.2 Hz, 3H, H3C-CH2-N-5), 1.24 (m, 2H), 1.45 (m, 2H), 1.58 (m, 2H), 1.74 (m, 1H), 2.18 (m, 1H), 3.34 (m, 2J=14.4 Hz, 3J=7.2 Hz, 1H, H3C-CHAB-N-5), 3.53 (m, 1H, C-H), 3.57 (m, 2J=15.2 Hz, 3J=7.2 Hz, 1H, H3C-CHAB-N-5), 4.90 (bs, 1H, C-H), 6.62 (d, 3J=5.6 Hz, 1H, H-4), 8.12 (s, 1H, H-1), 8.08 (d, 3J=5.6 Hz, 1H, H-3). 13C NMR (100 MHz, CDCl3):δ=12.4 (H3C-CH2-N-5), 18.9 (C-7), 22.8 (H3C-CO-N-10), 24.7 (C-8), 25.8 (C-6), 28.5 (C-9), 40.0 (H3C-CH2-N-5), 48.7 (C-9a), 53.7 (C-5a), (C-4), 106.4 (C-4), 120.1 (Cq, C-10a), 145.1, 145.2 (C-1, Cq, C-4a), 147.1 (C-3), 167.1 (C=O) ppm. GC-MS(EI):保持時間10.39分, m/e (%) 260 (11), 259 (M+, 100), 230 (8), 218 (9), 217 (79), 216 (40), 203 (4), 202 (23), 203 (3), 202 (23), 201 (4), 189 (6), 188 (31), 187 (4), 186 (3), 174 (11), 162 (4), 161 (6), 160 (5), 158 (4), 148 (5), 146 (6), 136 (3), 132 (4), 131 (12), 120 (6), 43 (3);IR (neat):□=3398 (s), 2933 (s), 2863 (m), 1731 (m), 1637 (vs), 1595 (vs), 1512 (s), 1546 (w), 1449 (m), 1397 (s), 1365 (s), 1334 (m), 1285 (s), 1239 (m), 1198 (m), 1168 (vs), 1123 (w), 1067 (w), 1068 (w), 1016 (w), 802 (vs), 718 (w), 668 (m). HRMS (EI):C15H21N3Oの計算値:259.1685 [M+]、実測値:259.1687.
【0087】
2,3-ジフェニルピリド[3,4-b]キノキサリン(25)(文献からの変法):
5.51 g(0.026 mmol)のベンジルを、50 mlのエタノール中の2.86 g(0.026 mol)の3,4-ジアミノピリジンの懸濁液に添加し、そして該反応混合物を70℃の油浴温度にて6時間加熱する。室温に冷却後、沈澱した黄色固体を濾去する(氷水槽を用いる冷却が有用であり得る)。エタノールからの再結晶化により、6.91 g(0.024 mol、94%)の淡黄色固体を得る。
1H NMR:(400 MHz, CDCl3):δ=7.32-7.42 (m, 6H, 3,4,5-フェニル-H), 7.54-7.51(m, 4H, 2,6-フェニル-H), 7.98 (dd, 3J=6 Hz, 4J=0.8 Hz, 1H, 7-H), 8.82 (d, 3J =6 Hz, 1H, 8-H), 9.59 (d, 4J=0.8 Hz, 1H, 5-H) ppm. 13C NMR (100 MHz, CDCl3):δ=121.3 (C-7), 128.4, 129.4-129.9 (フェニル-C), 136.3 (Cq, C-4a), 143.5 (Cq, C-8a), 147.3 (C-8), 154.5 (C-5), 155.3, 157.9 (Cq, フェニル-C) ppm. GC-MS(EI):保持時間12.34分, m/e (%) 285 (3), 284 (M++H, 22), 283 (M+, 100), 282 (46), 206 (2), 181 (3), 180 (25), 179 (17), 154 (3), 153 (5), 152 (3) 142 (1), 141 (8), 140 (4), 127 (2), 104 (3), 103 (14), 102 (3), 78 (2), 77 (5), 76 (4), 51 (2), 50 (9);IR (neat):□=3061 (w), 1589 (m), 1577 (w), 1538 (w), 1492 (w), 1443 (m), 1419 (w), 1379 (s), 1346 (w), 1326 (m), 1315 (m), 1288 (w), 1211 (m), 1244 (w), 1227 (m), 1212 (w), 1180 (w), 1075 (m), 1058 (m), 1020 (m), 1001 (w), 976 (s), 921 (w), 892 (m), 892 (m), 830 (m), 819 (w), 811 (m), 763 (s), 735 (m), 708 (vs), 629 (s), 615 (m) cm-1.
【0088】
2,3-ジフェニル-1,2,3,4-テトラヒドロピリド[3,4-b]ピラジン(31):
25(6.25 g、22.05 mmol)および200 mlのエタノールを、500 mlのフラスコ中に置く。6.25 g(165 mmol)のNaBH4粉末を、該溶液に添加し、そして該反応混合物を40℃にて48時間撹拌する。その後、該反応混合物を室温に冷却し、そして該反応を10 mlの冷水によって停止する。さらに10分間撹拌した後、該反応混合物を、100 mlのDCMを用いて2回抽出し、そして合わせた有機相をNa2SO4によって乾燥する。該粗生成物を、シリカゲルによるフラッシュクロマトグラフィー(EtOAc/NEt3、10:1)によって精製する。これにより淡黄色固体を得る。
Rf=0.23 (EtOAc/NEt3, 10:1);1H NMR:(300 MHz, CDCl3):δ=3.99 (s, 1H, N-H), 4.59 (s, 2H, 2,3-H), 4.62 (s, 1H, N-H), 6.39 (d, 3J =5.4 Hz, 1H, 8-H), 6.80 (dd, 4J=1.2 Hz, 3J=9.3 Hz, 2H, 2,6-フェニル-H), 6.84 (dd, 4J=1.2 Hz, 3J=9.3 Hz, 2H, 2,6-フェニル-H), 7.12 (m, 6H, 3,4,5-フェニル-H) ppm. 13C NMR (75 MHz, CDCl3):δ=58.6, 60.1 (C-2,3), 108.0 (C-8), 127.9-128.4 (フェニル-C), 129.8 (C-4a), 139.8 (Cq, フェニル-C), 140.0 (Cq, C-8a), 134.9 (C-5), 141.3 (C-7) ppm;MS(EI):m/e (%) 288 (22), 287 (M+, 100), 286 (24), 285 (7), 284 (6), 283 (8), 282 (5), 211 (13), 210 (78), 209 (5), 208 (13), 197 (4), 196 (26), 181 (13), 179 (3), 127 (4), 104 (7), 92 (3), 91 (32), 77 (5);IR (neat):□=3215 (w), 2830 (w), 1596 (s), 1519 (s), 1493 (w), 1466 (w), 1452 (s), 1360 (m), 1290 (s), 1250 (m), 1231 (m), 1174 (s), 1120 (m), 1072 (m), 1050 (w), 1029 (w), 1005 (w), 989 (w), 950 (w), 905 (w), 844 (w), 810 (m), 770 (m), 723 (m), 675 (vs), 668 (w), 645 (w), 618 (m) cm-1. HRMS (EI):C19H17N3の計算値:287.1422 [M+]、実測値:287.1402.
【0089】
4-メチル-2,3-ジフェニル-1,2,3,4-テトラヒドロピリド[3,4-b]ピラジン(32):
100 mlの丸底フラスコ中、0.600 g(2.08 mmol)の31を、氷中で冷却しながら15.5 ml(417.6 mmol、ρ=1.22 g/ml)のギ酸中に溶解させ、そして5.1 mlのホルムアルデヒド 溶液(183.6 mmol、ρ=1.22 g/ml、水中<37%)を添加する。その後、該氷槽を除去し、次いで、該混合物を120℃の油浴温度にて48時間加熱する。0℃に冷却した後、50%濃度のNaOHを、氷中で冷却しながら母液のpHが12に上昇するまで添加する。その後、該混合物を50 mlのDCMで3回抽出し、そして合わせた有機相をNa2SO4によって乾燥する。該溶媒をロータリーエバポレーターで留去した後、粗生成物を、シリカゲルによるフラッシュクロマトグラフィー(CHCl3/イソヘキサン/NEt3、10:2:1)によって精製する。これにより0.564 g(1.87 mmol、90%)の黄色固体を得る。
Rf=0.40 (CHCl3/イソヘキサン/NEt3、10:2:1) 1H NMR:(400 MHz, CDCl3):δ=2.85 (s, 3H, CH3), 4.42 (s, 1H, N-H), 4.44 (d, 3J =3.6 Hz, 1H, H-3), 4.95 (d, 3J =3.6 Hz, 1H, H-2), 6.50 (d, 3J=5.2 Hz, 1H, H-8), 6.64 (dd, 4J=2.8 Hz, 3J=9.2 Hz, 2H, 2,6-フェニル-H), 6.90 (m, 2H, 2,6-フェニル-H) 7.05 (m, 2H, フェニル-H), 7.16 (m, 4H, フェニル-H), 7.82 (s, 1H, H-5), 7.84 (d, 3J=5.2 Hz ppm. 13C NMR (75 MHz, CDCl3):δ=37.2 (CH3), 57.8 (C-3), 67.9 (C-2), 107.6 (C-8), 127.4-128.5 (フェニル-C), 131.3 (C-5), 131.6 (Cq, C-4a) 137.4, 138.8 (Cq, フェニル-C), 139.9 (C-7, C-8a) ppm. MS(EI):m/e (%) 302 (22), 301 (M+, 100), 300 (5), 286 (5), 224 (10), 222 (4), 211 (13), 210 (87), 209 (3), 208 (6), 195 (11), 181 (6), 179 (3), 178 (3), 178 (3), 150 (7), 132 (3), 127 (3), 120 (3), 104 (4);92 (3), 91 (31), 85 (4), 83 (6), 78 (4), 77 (5);IR (neat):□=3200 (w), 3158 (w), 3029 (w), 2948 (w), 2881 (w), 2824 (w), 1578 (s), 1516 (vs), 1491 (m), 1452 (m), 1421 (m), 1374 (w), 1353 (w), 1326 (w), 1294 (w), 1256 (m), 1235 (m), 1199 (w), 1157 (m), 1131 (m), 1103 (w), 1073 (m), 1054 (m), 1031 (m), 1013 (m), 969 (w), 922 (w), 842 (w), 810 (s), 766 (s), 755 (m), 733 (m), 641 (vs) cm-1. HRMS (EI):C20H19N3の計算値:301.1574 [M+]、実測値:301.1559.
【0090】
4-(メチル-2,3-ジフェニル-1,2,3,4-テトラヒドロピリド[3,4-b]ピラジン-1-イル)エタノン(33):
2.90 g(9.62 mmol)の32および100 mlのMTBEを250 mlのSchlenkフラスコ中に置く。該溶液を-78℃に冷却し、そして4.62 mlのn-BuLi(11.50 mmol、1.1当量;ヘキサン中2.5 M)を10分間に渡って添加する。該反応混合物を、冷却せずに30分間攪拌する。その後、-78℃に冷却し、そして1.0 ml(0.96 g、12.55 mmol)の塩化アセチルを添加する。該反応混合物を室温になるまで放置し、そしてさらに30分間攪拌する。その後、該反応を、10 mlの水を添加することによって停止する。母液をDCMで3回抽出し、そして合わせた有機相をNa2SO4によって乾燥する。該溶媒をロータリーエバポレーターで留去した後、該粗生成物は、フラッシュクロマトグラフィー(CHCl3/イソヘキサン/NEt3、10:1:1)または酢酸エチルからの再結晶化のいずれかによって精製することができる。これにより1.2 g(3.49 mmol、36%)の白色固体を得る。
1H NMR:(600 MHz, CDCl3):δ=2.40 (s, 3H, H3C-CO-N), 2.86 (s, 3H, H3C), 4.99 (m, 1H, H-3), 5.35 (d, 3J =4.2 Hz, 1H, H-2), 6.53 (d, 3J=7.8 Hz, 2H, 2,6-フェニル-H), 6.80 (bs, 2H, 2,6-フェニル-H), 7.08 (t, 2H, 3J=7.2 Hz, フェニル-H), 7.17 (t, 3J=6.0 Hz, 2H, フェニル-H), 7.22 (t, 3J=6.0 Hz, 1H, フェニル-H), 7.25 (t, 3J=6.0 Hz, 1H, フェニル-H), 8.03 (d, 3J=6.6 Hz, 1H, H-8), 8.17 (s, 1H, H-5), 8.41 (d, 3J=6.6 Hz, 1H, H-7) ppm. 13C NMR (150 MHz, CDCl3):δ=24.60 (H3C-CO-N), 36.9 (H3C), 61.8 (C-3), 63.8 (C-2), 116.9 (C-8), 124.6 (C-5), 128.4-128.9 (フェニル-C), 134.7 (Cq, C-8a), 138.7 (Cq, C-4a), 171.0 (C=O) ppm;GC-MS(EI):保持時間1.22分, m/e (%) 345 (3), 344 (27), 343 (M+, 100), 315 (6), 302 (7), 301 (41), 300 (84), 224 (6), 223 (4), 222 (5), 211 (4), 210 (27), 208 (9), 195 (6), 181 (9), 179 (4), 178 (4), 146 (4), 118 (15), 104 (5);92 (9), 91 (94), 78 (4), 77 (4), 43 (6);IR (neat):□=2428 (m), 2039 (w), 1688 (s), 1613 (w), 1534 (s), 1492 (m), 1454 (w), 1388 (m), 1364 (m), 1345 (m), 1317 (w), 1292 (w), 1292 (w), 1272 (w), 1227 (vs), 1209 (vs), 1122 (w), 1103 (w), 1078 (m), 1028 (m), 998 (m), 822 (s), 799 (m), 768 (m), 704 (m), 691 (m), 638 (w) cm-1. HRMS (EI):C22H23N3Oの計算値:343.1685 [M+]、実測値:343.1699.
【0091】
1-エチル-4-メチル-2,3-ジフェニル-1,2,3,4-テトラヒドロピリド[3,4-b]ピラジン(34):
0.575 g(4.320 mmol)のAlCl3を、20 mlのTHF中に室温にて懸濁させ、その後、該混合物をこの温度にて45分間攪拌する。その後、該混合物を0℃に冷却し、そして0.277 g(7.290 mmol)のLiAlH4を少量ずつ添加する。添加が完了した後、該反応混合物をさらに15分間攪拌し、そして33 (1.00 g、3.32 mmol)を添加する。該混合物を0℃にて1時間撹拌し、その後、12時間還流する。次いで、該反応混合物を室温に冷却し、そして氷水中に注ぐ。該無機沈澱を濾去し、そして母液をDCMで抽出する。合わせた有機相をNa2SO4によって乾燥し、そして得られる粗生成物をフラッシュクロマトグラフィー(EtOAc/MeOH/NEt3、10/1/0.5)によって精製する。これにより0.580 g(1.76 mmol、53%)の黄色固体を得る。
Rf=0.57 (EtOAc/MeOH/NEt3、10/1/0.5). 1H NMR:(400 MHz, CDCl3):δ=1.05 (t, 3J =7.2 Hz, 3H, H3C-CH2-N), 2.71 (s, 3H, H3C), 3.17 (qd, 2J =14.8 Hz, 3J =7.2 Hz, 1H, CH3-CHAB-N), 3.38 (qd, 2J =14.8 Hz, 3J =7.2 Hz, 1H, CH3-CHAB-N), 4.40 (d, 3J=3.6 Hz, 1H, H-3), 4.55 (d, 3J=3.6 Hz, 1H, H-2), 6.52 (d, 1H, 3J=5.6 Hz, H-8), 6.73 (m, 4H, フェニル-H), 7.07 (t, 3J=7.2 Hz, 2H, フェニル-H), 7.11 (t, 3J=8 Hz, 2H, フェニル-H), 7.16 (m, 2H, フェニル-H), 7.92 (s, 1H, H-5), 7.96 (d, 3J=5.6 Hz, 1H, H-7) ppm. 13C NMR (100 MHz, CDCl3):δ=11.1 (H3C-CH2-N), 37.1 (H3C-N), 43.3 (H3C-CH2-N), 65.7 (C-2), 66.0 (C-3), 104.3 (C-8), 127.7-129.3 (フェニル-C), 133.0 (C-5), 133.3 (Cq, C-8a), 137.8, 138.6 (Cq, フェニル-C), 140.8 (Cq, C-8a), 141.0 (C-7) ppm. GC-HR-ESI-MS:保持時間0.53〜1.43分, C44H47N6の計算値:659.3862 [2M++H]、実測値:659.3823、C22H24N3の計算値:330.1970 [M++H]、実測値:330.1940;IR (neat):□=2965 (w), 2818 (w), 1578 (s), 1518 (vs), 1492 (m), 1452 (s), 1430 (w), 1371 (m), 1356 (m), 1308 (m), 1286 (m), 1266 (s), 1241 (s), 1215 (vs), 1165 (m), 1119 (m), 1067 (s), 1023 (s), 884 (w), 867 (w), 828 (w), 810 (s), 764 (s), 749 (m), 705 (vs), 660 (w), 615 (w) cm-1.
【0092】
〔参考文献〕
1) 以下のレビューを参照のこと:(a) G. Hoefle, W. Steglich, H. Vorbrueggen, Angew. Chem. 1978, 90, 602 - 615;Angew. Chem. Int. Ed. Engl. 1978, 17, 569 - 583;(b) E. F. V. Scriven, Chem. Soc. Rev. 1983, 12, 129 - 161;(c) A. Hassner, in Encyclopedia of Reagents for Organic Synthesis, Wiley, Chichester, 1995, 2022 - 2024;(d) U. Ragnarsson, L. Grehn, Acc. Chem. Res. 1998, 31, 494 - 501;(e) A. C. Spivey, A. Maddaford, A. Redgrave, Org. Prep. Proced. Int. 2000, 32, 331-365. (f) D. J. Berry, C. V. Digiovanna, S. S. Metrick, R. Murugan, Arkivoc 2001, 201 - 226;(g) A. C. Spivey, S. Arseniyadis, Angew. Chem. 2004, 116, 5552 - 5557;Angew. Chem. Int. Ed. Engl. 2004, 43, 5436 - 5441.
2) (a) E. Vedejs, M. Jure, Angew. Chem. 2005, 117, 4040 - 4069;Angew. Chem. Int. Ed. Engl. 2005, 44, 3971 - 4001. (b) P. I. Dalko, L. Moisan, Angew. Chem. 2004, 116, 5248 - 5286;Angew. Chem. Int. Ed. Engl. 2004, 43, 5138 - 5178. (c) G. Fu, Acc. Chem. Res. 2004, 37, 542 - 547. (d) S. France, D. J. Guerin, S. J. Miller, T. Lectka, Chem. Rev. 2003, 103, 2985 - 3012. (e) A. C. Spivey, A. Maddaford, A. Redgrave, Org. Prep. Proced. Int. 2000, 32, 331 - 365. (f) G. Fu, Acc. Chem. Res. 2000, 33, 412 - 420.
3) (a) T. Kawabata, R. Stragies, T. Fukaya, K. Fuji, Chirality 2003, 15, 71. (b) T. Kawabata, R. Stragies, T. Fukaya, Y. Nagaoka, H. Schedel, K. Fuji, Tetrahedron Lett. 2003, 44, 1545.
4) (a) G. Priem, B. Pelotier, S. J. F. Macdonald, M. S. Anson, I. B. Campbell, J. Org. Chem. 2003, 68, 3844. (b) B. Pelotier, G. Priem, S. J. F. Macdonald, M. S. Anson, R. J. Upton, I. B. Campbell, Tetrahedron Lett. 2005, 46, 9005.
5) (a) A. C. Spivey, T. Fekner, S. E. Spey, H. Adams, J. Org. Chem. 1999, 64, 9430、およびその引用文献. (b) A. C. Spivey, T. Fekner, S. E. Spey, J. Org. Chem. 2000, 65, 3154. (c) A. C. Spivey, A. Maddafort, T. Fekner, A. J. Redgrave, C. S. Frampton, J. Chem. Soc. Perkin Trans. 1. 2000, 3460. (d) A. C. Spivey, A. Maddafort, T. Fekner, D. P. Leese, A. J. Redgrave, C. S. Frampton, J. Chem. Soc. Perkin Trans. 1, 2001, 1785. (e) C. Malardier-Jugroot, A. C. Spivey, M. A. Whitehead, J. Mol. Struct. (THEOCHEM) 2003, 623, 263. (f) A. C. Spivey, D. P. Leese, F. Zhu, S. G. Davey, R. L. Jarvest, Tetrahedron 2004, 60, 4513. (g) A. C. Spivey, S. Arseniyadis, T. Fekner, A. Maddaford, D. P. Lees, Tetrahedron 2006, 62, 295.
6) (a) C. O. Dalaigh, S. J. Hynes, D. J. Maher, S. J. Connon, Org. Biomol. Chem. 2005, 3, 981. (b) C. O. Dalaigh, S. J. Hynes, J. E. O'Brien, T. McCabe, D. J. Maher, G. W. Watson, S. J. Connon, Org. Biomol. Chem. 2006, 4, 2785.
7) (a) S. A. Shaw, P. Aleman, E. Vedejs, J. Am. Chem. Soc. 2003, 125, 13368-13369. (b) S. A. Shaw, P. Aleman, J. Christy, J. W. Kampf, P. Va, E. Vedejs, J. Am. Chem. Soc. 2006, 128, 925-934.
8) S. Yamada, T. Misono, Y. Iwai, Tet. Lett. 2005, 46, 2239.
9) (a) S. J. Miller, Acc. Chem. Res. 2004, 37, 601-610、およびその引用文献. (b) M. B. Fierman, D. J. O’Leary, W. E. Steinmetz, S. J. Miller, J. Am. Chem. Soc. 2004, 126, 6967-6971. (c) J. W. Evans, M. B. Fierman, S. J. Miller, J. A. Ellman, J. Am. Chem. Soc. 2004, 126, 8134-8135. (d) B. R. Sculimbrene, Y. Xu, S. J. Miller, J. Am. Chem. Soc. 2004, 126, 13182-13183. (e) A. J. Morgan, Y. K. Wang, M. F. Roberts, S. J. Miller, J. Am. Chem. Soc. 2004, 126, 15370-15371. (f) C. A. Lewis, B. R. Sculimbrene, Y. Xu, S. J. Miller, Org. Lett. 2005, 3021. (g) Y. Xu, B. R. Sculimbrene, S. J. Miller, J. Org. Chem. 2006, 71, 4919-4928.
10) M. R. Heinrich, H. S. Klisa, H. Mayr, W. Steglich, H. Zipse, Angew. Chem. 2003, 115, 4975 - 4977;Angew. Chem. Int. Ed. 2003, 42, 4826 - 4828.
11) I. Held, A. Villinger, H. Zipse, Synthesis 2005, 1425 - 1426.
12) W. W. K. R. Mederski, D. Kux, M. Knoth, M. J. Schwarzkopf-Hofmann, Heterocycles, 2003, 4, 925-931.
13) J. Armand, L. Boulares, C. Bellec, J. Pinson, Can J. Chem. 1988, 66, 1500-1505.
14) M. L. Moore, Org. Reaction. 1949, 5, 301-330.
15) C. Sotirou-Leventis, Z. Mao, A.-M. M. Rawashdeh, J. Org. Chem. 2000, 65, 6017-6023.
16) C. Kison, N. Meyer, T. Opatz, Angew. Chem. Int. Ed. 2005, 44, 5662-5664. 該文献中に引用されたBH3・THF溶液の寿命に対する反応収率の依存性は、化合物24の合成について確認することができた。
17) Timothy D. W. Claridge, High-Resolution NMR Techniques in Organic Chemistry, Tetrahedron Organic Chemistry Series, 1999, 19, 298-299.
18) S. Xu, I. Held, B. Kempf, H. Mayr, W. Steglich, H. Zipse, Chem. Eur. J. 2005, 11, 4751 - 4757.
19) C. B. Fischer, S. Xu, H. Zipse, Chem. Eur. J. 2006, 12, 5779 - 5784.
20) この場合の反応速度は、背景反応の反応速度とわずかに異なる。このことは、本明細書中で使用される方法による半減期の正確な決定を格別に困難にする。表2中に示すデータは、25日間、17時間および30分間に渡る反応Aについての、並びに31日間、17時間および15分間に渡る反応Bについての、変換測定から得られる。両方の場合において、50%の変換は、これらの時間内で達成されない。
21) Gaussian 03, Revision B.03, M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, J. A. Montgomery, Jr., T. Vreven, K. N. Kudin, J. C. Burant, J. M. Millam, S. S. Iyengar, J. Tomasi, V. Barone, B. Mennucci, M. Cossi, G. Scalmani, N. Rega, G. A. Petersson, H. Nakatsuji, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, M. Klene, X. Li, J. E. Knox, H. P. Hratchian, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, P. Y. Ayala, K. Morokuma, G. A. Voth, P. Salvador, J. J. Dannenberg, V. G. Zakrzewski, S. Dapprich, A. D. Daniels, M. C. Strain, O. Farkas, D. K. Malick, A. D. Rabuck, K. Raghavachari, J. B. Foresman, J. V. Ortiz, Q. Cui, A. G. Baboul, S. Clifford, J. Cioslowski, B. B. Stefanov, G. Liu, A. Liashenko, P. Piskorz, I. Komaromi, R. L. Martin, D. J. Fox, T. Keith, M. A. Al-Laham, C. Y. Peng, A. Nanayakkara, M. Challacombe, P. M. W. Gill, B. Johnson, W. Chen, M. W. Wong, C. Gonzalez,およびJ. A. Pople, Gaussian, Inc., Wallingford CT, 2004.
22) H. E. Gottlieb, V. Kotyar, A. Nudelman, J. Org. Chem. 1997, 21 7512-7515.
23) この反応における変換は59%にて停止する。該反応混合物についてのESI-MS研究は、この時間にて該触媒がもはや完全のままではないことを示す。
【特許請求の範囲】
【請求項1】
以下の式I:
【化1】
〔式中、R1およびR2は、それぞれ互いに独立して電子ドナーであり、ここでR2はHであってもよく、および
R3、R4、R5およびR6は、それぞれ独立して、H、置換または非置換の直鎖または分枝状のアルキル、アルケニル、アルキニル、アルコキシおよび置換または非置換のアリールの中から選択され、ここで基R3および/またはR4は、基R5および/またはR6と一緒になって環を形成し得る。〕
で示される3,4-ジアミノピリジン誘導体。
【請求項2】
R1およびR2は、それぞれ独立して、置換または非置換の直鎖または分枝状のアルキル、アルケニル、アルキニル、アシル、アルコキシおよび置換または非置換のアリール、より好ましくは置換または非置換の直鎖または分枝状のC1-C20-アルキル、C2-C20-アルケニル、C2-C20-アルキニル、C1-C20-アシル、C1-C20-アルコキシ、置換または非置換のフェニルおよびベンジルの中から選択され、ここでR2はHであってもよい、請求項1に記載の3,4-ジアミノピリジン誘導体。
【請求項3】
R3、R4、R5およびR6は、それぞれ独立して、H、置換または非置換の直鎖または分枝状のC1-C20-アルキル、C2-C20-アルケニル、C2-C20-アルキニルおよび置換または非置換のフェニルおよびベンジルの中から選択される、請求項1または2に記載の3,4-ジアミノピリジン誘導体。
【請求項4】
基R3、R4、R5およびR6の少なくとも一つはHではなく、好ましくは、基R3、R4、R5およびR6の少なくとも一つはフェニルであり、または2つの基R3およびR5は各々Hであり、および2つの基R4およびR6は一緒になって、6員環を形成するように、1,4-ブタンジイルである、請求項1〜3のいずれかに記載の3,4-ジアミノピリジン誘導体。
【請求項5】
2つの基R4およびR5は、式Ia:
【化2】
〔式中、R1およびR2は、それぞれ互いに独立して電子ドナーであり、ここでR2はHであってもよく、
R3およびR6は、それぞれ独立して、H、置換または非置換の直鎖または分枝状のアルキル、アルケニル、アルキニル、アルコキシおよび置換または非置換のアリールの中から選択され、
R4およびR5は、それぞれ独立して、置換または非置換の直鎖または分枝状のアルキル、アルケニル、アルキニル、アルコキシおよび置換または非置換のアリールの中から選択され、および
基R3および/またはR4は、基R5および/またはR6と一緒になって環を形成し得る。〕
で示される3,4-ジアミノピリジン誘導体を与えるように、互いにシス位に配置されている、請求項1〜4のいずれかに記載の3,4-ジアミノピリジン誘導体。
【請求項6】
R1およびR2は、それぞれ互いに独立して置換または非置換の直鎖または分枝状のC1-C10-アルキル、好ましくは置換または非置換の直鎖または分枝状のC2-C8-アルキル、および最も好ましくは置換または非置換の直鎖または分枝状のC3-C6-アルキルである、請求項1〜5のいずれかに記載の3,4-ジアミノピリジン誘導体。
【請求項7】
R1およびR2は、それぞれエチルであり、R3およびR6は、それぞれHであり、およびR4およびR5は一緒になって、6員環を形成するように、1,4-ブタンジイルである、請求項1〜6のいずれかに記載の3,4-ジアミノピリジン誘導体。
【請求項8】
式Iで示される3,4-ジアミノピリジン誘導体の製造方法であって、
・3,4-ジアミノピリジンを1,2-ジカルボニル化合物と反応させる工程、
・得られたジイミンを対応するジアミンに還元する工程、および
・少なくとも、該ピリジン環の4位における窒素上の水素原子を置換する工程
を含む、方法。
【請求項9】
該ピリジン環の3位および4位における両方の窒素原子を置換する、請求項0に記載の方法。
【請求項10】
アルキル置換が、窒素原子のアシル化およびその後の得られたアミドのアミンへの還元を含む二段階合成によって達成される、請求項0または9に記載の方法。
【請求項11】
化学反応における触媒としての、請求項1〜0のいずれかに記載の3,4-ジアミノピリジン誘導体の使用。
【請求項12】
化学反応が、アシル化反応、特にアルコールまたはアミンのアシル化反応、またはウレタンおよび/またはポリウレタン合成である、請求項11に記載の使用。
【請求項13】
アシル化に必要な試薬に加えて、請求項1〜0のいずれかに記載の3,4-ジアミノピリジン誘導体を触媒として添加することを含む、アシル化アルコールまたはアシル化アミンの製造方法。
【請求項14】
ウレタンおよび/またはポリウレタンの製造に必要な試薬に加えて、請求項1〜0のいずれかに記載の3,4-ジアミノピリジン誘導体を触媒として添加することを含む、ポリウレタンの製造方法。
【請求項15】
薬剤または殺草剤を製造するための、請求項1〜7のいずれかに記載の3,4-ジアミノピリジン誘導体の使用。
【請求項16】
請求項1〜7のいずれかに記載の3,4-ジアミノピリジン誘導体を含む、医薬組成物または殺草剤組成物。
【請求項1】
以下の式I:
【化1】
〔式中、R1およびR2は、それぞれ互いに独立して電子ドナーであり、ここでR2はHであってもよく、および
R3、R4、R5およびR6は、それぞれ独立して、H、置換または非置換の直鎖または分枝状のアルキル、アルケニル、アルキニル、アルコキシおよび置換または非置換のアリールの中から選択され、ここで基R3および/またはR4は、基R5および/またはR6と一緒になって環を形成し得る。〕
で示される3,4-ジアミノピリジン誘導体。
【請求項2】
R1およびR2は、それぞれ独立して、置換または非置換の直鎖または分枝状のアルキル、アルケニル、アルキニル、アシル、アルコキシおよび置換または非置換のアリール、より好ましくは置換または非置換の直鎖または分枝状のC1-C20-アルキル、C2-C20-アルケニル、C2-C20-アルキニル、C1-C20-アシル、C1-C20-アルコキシ、置換または非置換のフェニルおよびベンジルの中から選択され、ここでR2はHであってもよい、請求項1に記載の3,4-ジアミノピリジン誘導体。
【請求項3】
R3、R4、R5およびR6は、それぞれ独立して、H、置換または非置換の直鎖または分枝状のC1-C20-アルキル、C2-C20-アルケニル、C2-C20-アルキニルおよび置換または非置換のフェニルおよびベンジルの中から選択される、請求項1または2に記載の3,4-ジアミノピリジン誘導体。
【請求項4】
基R3、R4、R5およびR6の少なくとも一つはHではなく、好ましくは、基R3、R4、R5およびR6の少なくとも一つはフェニルであり、または2つの基R3およびR5は各々Hであり、および2つの基R4およびR6は一緒になって、6員環を形成するように、1,4-ブタンジイルである、請求項1〜3のいずれかに記載の3,4-ジアミノピリジン誘導体。
【請求項5】
2つの基R4およびR5は、式Ia:
【化2】
〔式中、R1およびR2は、それぞれ互いに独立して電子ドナーであり、ここでR2はHであってもよく、
R3およびR6は、それぞれ独立して、H、置換または非置換の直鎖または分枝状のアルキル、アルケニル、アルキニル、アルコキシおよび置換または非置換のアリールの中から選択され、
R4およびR5は、それぞれ独立して、置換または非置換の直鎖または分枝状のアルキル、アルケニル、アルキニル、アルコキシおよび置換または非置換のアリールの中から選択され、および
基R3および/またはR4は、基R5および/またはR6と一緒になって環を形成し得る。〕
で示される3,4-ジアミノピリジン誘導体を与えるように、互いにシス位に配置されている、請求項1〜4のいずれかに記載の3,4-ジアミノピリジン誘導体。
【請求項6】
R1およびR2は、それぞれ互いに独立して置換または非置換の直鎖または分枝状のC1-C10-アルキル、好ましくは置換または非置換の直鎖または分枝状のC2-C8-アルキル、および最も好ましくは置換または非置換の直鎖または分枝状のC3-C6-アルキルである、請求項1〜5のいずれかに記載の3,4-ジアミノピリジン誘導体。
【請求項7】
R1およびR2は、それぞれエチルであり、R3およびR6は、それぞれHであり、およびR4およびR5は一緒になって、6員環を形成するように、1,4-ブタンジイルである、請求項1〜6のいずれかに記載の3,4-ジアミノピリジン誘導体。
【請求項8】
式Iで示される3,4-ジアミノピリジン誘導体の製造方法であって、
・3,4-ジアミノピリジンを1,2-ジカルボニル化合物と反応させる工程、
・得られたジイミンを対応するジアミンに還元する工程、および
・少なくとも、該ピリジン環の4位における窒素上の水素原子を置換する工程
を含む、方法。
【請求項9】
該ピリジン環の3位および4位における両方の窒素原子を置換する、請求項0に記載の方法。
【請求項10】
アルキル置換が、窒素原子のアシル化およびその後の得られたアミドのアミンへの還元を含む二段階合成によって達成される、請求項0または9に記載の方法。
【請求項11】
化学反応における触媒としての、請求項1〜0のいずれかに記載の3,4-ジアミノピリジン誘導体の使用。
【請求項12】
化学反応が、アシル化反応、特にアルコールまたはアミンのアシル化反応、またはウレタンおよび/またはポリウレタン合成である、請求項11に記載の使用。
【請求項13】
アシル化に必要な試薬に加えて、請求項1〜0のいずれかに記載の3,4-ジアミノピリジン誘導体を触媒として添加することを含む、アシル化アルコールまたはアシル化アミンの製造方法。
【請求項14】
ウレタンおよび/またはポリウレタンの製造に必要な試薬に加えて、請求項1〜0のいずれかに記載の3,4-ジアミノピリジン誘導体を触媒として添加することを含む、ポリウレタンの製造方法。
【請求項15】
薬剤または殺草剤を製造するための、請求項1〜7のいずれかに記載の3,4-ジアミノピリジン誘導体の使用。
【請求項16】
請求項1〜7のいずれかに記載の3,4-ジアミノピリジン誘導体を含む、医薬組成物または殺草剤組成物。
【図1】

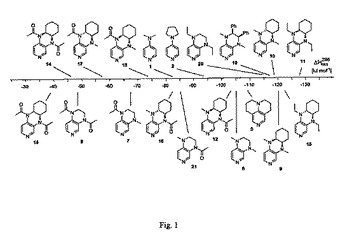
【図2】
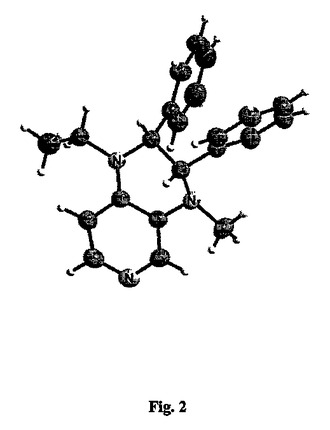
【図2】
【公表番号】特表2010−511646(P2010−511646A)
【公表日】平成22年4月15日(2010.4.15)
【国際特許分類】
【出願番号】特願2009−539650(P2009−539650)
【出願日】平成19年12月1日(2007.12.1)
【国際出願番号】PCT/EP2007/010460
【国際公開番号】WO2008/067965
【国際公開日】平成20年6月12日(2008.6.12)
【出願人】(504037346)バイエル・マテリアルサイエンス・アクチェンゲゼルシャフト (728)
【氏名又は名称原語表記】Bayer MaterialScience AG
【出願人】(509161152)ルートヴィッヒ−マクシミリアンス−ウニヴェルジテート・ミュンヘン (1)
【氏名又は名称原語表記】LUDWIG−MAXIMILIANS−UNIVERSITAET MUENCHEN
【Fターム(参考)】
【公表日】平成22年4月15日(2010.4.15)
【国際特許分類】
【出願日】平成19年12月1日(2007.12.1)
【国際出願番号】PCT/EP2007/010460
【国際公開番号】WO2008/067965
【国際公開日】平成20年6月12日(2008.6.12)
【出願人】(504037346)バイエル・マテリアルサイエンス・アクチェンゲゼルシャフト (728)
【氏名又は名称原語表記】Bayer MaterialScience AG
【出願人】(509161152)ルートヴィッヒ−マクシミリアンス−ウニヴェルジテート・ミュンヘン (1)
【氏名又は名称原語表記】LUDWIG−MAXIMILIANS−UNIVERSITAET MUENCHEN
【Fターム(参考)】
[ Back to top ]