
4−硫酸化ヒアルロン酸
【課題】ヒアルビウロン酸の4位が硫酸化されたヒアルビウロン酸オキサゾリン誘導体を設計し、羊睾丸由来ヒアルロニダーゼを作用せしめることにより4−硫酸化ヒアルロン酸を生成すること。
【解決手段】(1)一般式(I)で表される4−硫酸化ヒアルロン酸、(2)一般式(II)で表されるオキサゾリン誘導体にヒアルロン酸分解酵素を作用せしめることにより製造される4−硫酸化ヒアルロン酸、(3)ヒアルロン酸分解酵素が、ほ乳類由来のヒアルロニダーゼであることを特徴とする(2)記載の4−硫酸化ヒアルロン酸、(4)一般式(II)で表されるオキサゾリン誘導体にヒアルロン酸分解酵素を作用せしめるにあたり、pHを6.0〜7.0に調整して製造されることを特徴とする(2)又は(3)記載の4−硫酸化ヒアルロン酸を構成とする。
【解決手段】(1)一般式(I)で表される4−硫酸化ヒアルロン酸、(2)一般式(II)で表されるオキサゾリン誘導体にヒアルロン酸分解酵素を作用せしめることにより製造される4−硫酸化ヒアルロン酸、(3)ヒアルロン酸分解酵素が、ほ乳類由来のヒアルロニダーゼであることを特徴とする(2)記載の4−硫酸化ヒアルロン酸、(4)一般式(II)で表されるオキサゾリン誘導体にヒアルロン酸分解酵素を作用せしめるにあたり、pHを6.0〜7.0に調整して製造されることを特徴とする(2)又は(3)記載の4−硫酸化ヒアルロン酸を構成とする。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、オキサゾリン誘導体にヒアルロン酸分解酵素を作用せしめることにより製造される4−硫酸化ヒアルロン酸に関する。更に詳しくは、ヒアルロン酸分解酵素であるほ乳類由来のヒアルロニダーゼを触媒とし、オキサゾリン誘導体をモノマー基質として酵素的に重合させて製造される4−硫酸化ヒアルロン酸に関する。
【背景技術】
【0002】
ヒアルロン酸は、D−グルクロン酸とN−アセチルグルコサミンの二糖が直鎖状に交互に結合した枝分かれのない高分子多糖である。その機械的性質の改善や医学用材料に適した性質への改善のために誘導体合成が行われてきた。そのひとつが硫酸化である。ヒアルロン酸は、生体内において硫酸化を受けていないので、硫酸化することにより抗凝血活性付与が検討されてきた。この場合には、ヒアルロン酸中の多くの水酸基が硫酸化される程活性が高くなることが知られている。その他に、硫酸化ヒアルロン酸には抗炎症作用や腎疾患の予防または治療効果が知られている。また、反復単位(n)数が約6〜10で、硫酸化率が20〜50%のオリゴ硫酸化ヒアルロン酸には、皮膚の角化抑制効果が見いだされた。
【0003】
硫酸化ヒアルロン酸は、ヒアルロン酸の水酸基を化学修飾により硫酸化することによって合成される。しかし、化学修飾による硫酸化では、硫酸基の導入度及び導入位置を制御することは非常に困難である。従って、未だ4−硫酸化ヒアルロン酸のように、特定の位置だけが全て硫酸化された硫酸化ヒアルロン酸は合成されていない。
【特許文献1】特開平8−277224号公報
【特許文献2】特開平8−301771号公報
【特許文献3】特開平11−279042号公報
【非特許文献1】International Congress Series(2000),1196(NewFrontiers in Medical Sciences):Redefining Hyaluronan),203−212
【発明の開示】
【発明が解決しようとする課題】
【0004】
特定の位置が硫酸化されたヒアルロン酸は、構造と活性との相関を明らかにし、生理活性の高い分子を設計する上で有意義である。そのような硫酸化ヒアルロン酸は、未だに合成されていない。
本発明者は、鋭意研究を重ねた結果、ヒアルロン酸分解酵素を用いた酵素化学的手法により、4位だけをすべて硫酸化した4−硫酸化ヒアルロン酸をはじめて合成した。
更に詳しくは、本発明者はヒアルビウロン酸の4位が硫酸化されたヒアルビウロン酸オキサゾリン誘導体を設計し、羊睾丸由来ヒアルロニダーゼを作用せしめることにより4−硫酸化ヒアルロン酸が生成することを見い出し、本発明を完成するに至った。
【課題を解決するための手段】
【0005】
即ち、本発明は、(1)下記の一般式(I)で表される4−硫酸化ヒアルロン酸、
【化4】

(2)下記の一般式(II)で表されるオキサゾリン誘導体にヒアルロン酸分解酵素を作用せしめることにより製造される4−硫酸化ヒアルロン酸、
【化5】
(3)ヒアルロン酸分解酵素が、ほ乳類由来のヒアルロニダーゼであることを特徴とする請求項2記載の4−硫酸化ヒアルロン酸、
(4)下記の一般式(II)で表されるオキサゾリン誘導体にヒアルロン酸分解酵素を作用せしめるにあたり、pHを6.0〜7.0に調整して製造されることを特徴とする(2)又は(3)記載の4−硫酸化ヒアルロン酸である。
【化6】
【発明の効果】
【0006】
本発明の4−硫酸化ヒアルロン酸は、構造が明確な硫酸化ヒアルロン酸であり、4−硫酸化ヒアルロン酸の抗凝血活性などの活性と構造との相関検討により、高活性の硫酸化ヒアルロン酸を設計するために有用である。
【発明を実施するための最良の形態】
【0007】
以下、さらに詳しく本発明を説明する。
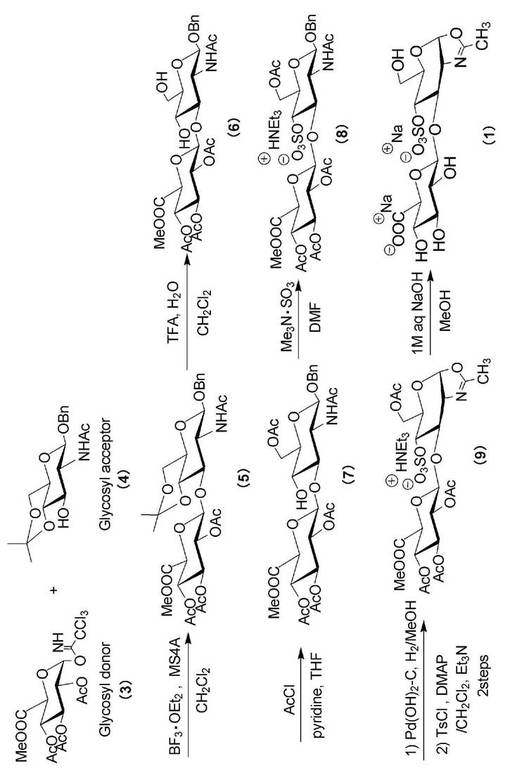
本発明で使用する基質モノマーの合成手法を、図1に示す合成スキーム中のオキサゾリン誘導体(1)の合成を例として、その手順の一例を以下に示す。
すなわち、メチル(2,3,4−トリ−O−アセチル−β−D−グルコピラノシルトリクロロアセトイミデート)ウロネート(3)を糖供与体、ベンジル (2−アセトアミド−2−デオキシ−4,6−O−イソプロピリデン−β−D−グルコピラノシド(4)を糖受容体として用い、モレキュラーシーブス(MS4A)存在下、ジクロロメタン中、プロモーターとしてBF3・OEt2を用い、0℃でグリコシル化を行い、ベンジル(メチル2,3,4−トリ−O−アセチル−β−D−グルコピラノシルウロネート)−(1→3) −2−アセトアミド−2−デオキシ−4,6−O−イソプロピリデン−β−D−グルコピラノシド(5)を合成する。得られた(5)の脱保護反応は、ジクロロメタンに溶解・攪拌し、次いで水、トリフルオロ酢酸を添加して0℃で反応を行う。反応終了後、TFA存在下で反応溶液の温度を上昇させないように、速やかに飽和NaHCO3水溶液で分液処理を行い、シリカゲルカラムで精製を行ない、ベンジル(メチル2,3,4−トリ−O−アセチル−β−D−グルコピラノシルウロネート)−(1→3)−2−アセトアミド−2−デオキシ−β−D−グルコピラノシド(6)を合成する。(6)をTHFとピリジン=1:1に溶解し、−78℃で反応させることにより、ベンジル(メチル2,3,4−トリ−O−アセチル−β−D−グルコピラノシルウロネート)−(1→3)−6−o−アセチル−2−アセトアミド−2−デオキシ−β−D−グルコピラノシド(7)を得る。
次に、4位の遊離水酸基に硫酸基を導入した後、アノマー位のベンジル基を脱保護してオキサゾリン化を行い、トリエチルアンモニウム ベンジル(メチル2,3,4,−トリ−O−アセチル−β−D−グルコピラノシルウロネート)−(1→3)−6−O−アセチル−4ーO−スルフォネート−2−アセトアミド−2−デオキシ−β−D−グルコピラノシド(8)を得る。(8)の精製をシリカゲルカラム及びゲルろ過により行い、2−メチル−4,5−ジヒドロ−[トリエチルアンモニウム−6−O−アセチル−4−O−スルフォネート−1,2−ジ−デオキシ−3−O−(2,3,4−トリ−O−アセチル−β−D−グルコピラノシルウロネート)−α−D−グルコピラノソ]−[2,1−d]−1,3−オキサゾ−ル(9)を得る。最後に、0℃でアルカリ分解により脱保護し、目的のソディウム(2−メチル−4,5−ジヒドロ−[4−O−スルフォ−1,2−ジデオキシ−3−O−(ソディウムβ−D−グルコピラノシルウロネート)−α−D−グルコピラノソ]−[2,1−d]−1,3−オキサゾール(1)を得ることができる。
【0008】
基質モノマーは、フリーの酸、ナトリウム、カリウム等の金属塩、アンモニウム塩、トリエチルアミン塩等の形であれば特に限定されない。生成する4−硫酸化ヒアルロン酸は、基質モノマーのグルクロン酸の塩の形態に依存し、フリーな酸あるいはナトリウム、カリウム塩等の金属塩、アンモニウム塩、トリエチルアミン塩等の形態が含まれる。
【0009】
かくして得られたオキサゾリン誘導体は重合触媒としてのヒアルロン酸分解酵素の基質モノマーとして好適に使用される。酵素反応時の基質モノマー濃度は実用面から0.1%以上、好ましくは1%以上で用いられる。
反応pHは酵素の反応性と基質モノマーの安定性を考慮し、5〜10望ましくは6.5〜7.5が好適に採用される。反応温度は5℃〜60℃で、通常は20℃〜40℃が用いられる。
【0010】
使用するヒアルロン酸分解酵素としては、ほ乳類由来のヒアルロニダーゼが好ましく、具体的にはエンド−β−N−アセチルヘキソサミダーゼ(EC3.2.1.35)に分類されるウシ睾丸由来又は羊睾丸由来ヒアルロニダーゼなどが好適であり、該酵素を適当な担体に固定化した固定化酵素の形態で使用することも可能である。バッチ反応あるいは連続反応形式いずれも採用される。
【0011】
反応は水溶媒あるいは水溶媒にメタノール、エタノール、n−プロパノール等のアルコール類、グリセリン、ポリエチレングリコール等のポリオール類、ジメチルスルフォキシド、ジメチルフォルムアミド、酢酸エチル、ジオキサン、反応に悪影響を及ぼさない各種無機塩類又はpH緩衝剤等を適宜添加した条件下でも進行する。
【0012】
上記条件下でバッチ反応を開始した場合、条件によって一概に規定できないが、数時間〜数日で反応は完了する。反応終了後、反応液を遠心分離、限外濾過、精密濾過、各種吸着カラム、溶媒沈殿及びクロマト分離等の公知精製手段を組み合わせることで高純度の4−硫酸化ヒアルロン酸を単離精製することが出来る。
得られる4−硫酸化ヒアルロン酸の分子量は、基質モノマーの酵素触媒による重合反応時の条件を制御することにより種々の分子量の4−硫酸化ヒアルロン酸を得ることが可能である。
【実施例】
【0013】
以下に本発明の詳細な内容について実施例で説明するが、本発明は以下の実施例に限定されるものではない。
【0014】
実施例1
ベンジル(メチル2,3,4−トリ−O−アセチル−β−D−グルコピラノシルウロネート)−(1→3)−2−アセトアミド−2−デオキシ−4,6−O−イソプロピリデン−β−D−グルコピラノシド(5)の合成
遮光二口フラスコ中で、メチル(2,3,4−トリ−O−アセチル−α−D−グルコピラノシル トリクロロアセトイミデート)ウロネート(3)(667mg,1.39mmol)とベンジル2−アセトアミド−2−デオキシ−4,6−O−イソプロピリデン−β−D−グルコピラノシド(4)(350mg,0.99mmol)を脱水ジクロロメタン(10ml)に溶解させ、モレキュラーシーブス4A(MS4A;1.2g)を加え、0℃で脱水ジクロロメタン(0.7ml)に溶解したBF3・OEt2(175μl,1.39mmol)の溶液を加えた。反応混合物をアルゴン雰囲気下0℃で4時間攪拌した。反応をEt3N(0.45ml)の添加で停止した。その混合物を珪藻土(Celite)でろ過し、飽和NaHCO3水溶液に注ぎ、CHCl3で抽出した。有機層を飽和NaCl水溶液で洗浄し、MgSO4で乾燥し、ろ過、濃縮した。その残渣をシリカゲルカラムクロマトグラフィー(2:1から1:2までn−ヘキサン−酢酸エチル)によって、精製し、白い固体として、ベンジル(メチル(2,3,4−トリ−O−アセチル−β−D−グルコピラノシルウロネート)−(1→3)−2−アセトアミド−2−デオキシ−4,6−O−イソプロピリデン−β−D−グルコピラノシド(476mg,0.69mmol,収率71%)を得た。
分析データは、以下の通りである。
1H−NMR (400MHz, CDCl3,TMS) :δ(ppm); 7.33−7.29 (m, 7H, aromatic), 5.78 (d, JNH,2 = 7.03 Hz, 1H, NH),5.24−5.15 (m, 3H, H−3', H−1, H−4’), 4.95 (t, J1’,2’ = J2’,3’ = 8.53 Hz, 1H, H−2’), 4.84 (dd, 2H,CH2Ph), 4.55−4.48(m, 3H, H−3, CH2Ph),3.95−3.91 (m, 2H, H−5', H−6a), 3.81−3.71 (m, 7H, H−6b, COOCH3, H−4), 3.40−3.34 (m, 1H, H−5),3.09−3.03 (dd, 1H, H−2), 2.08−1.99 (m, 14H, アセトアミドのCH3 , COCH3), 1.87 (s, 3H, isopropylidenのCH3), 1.65 (s, 3H, isopropylidenのCH3 ).
【0015】
実施例2
ベンジル(メチル2,3,4−トリ−O−アセチル−β−D−グルコピラノシルウロネート)−(1→3)−2−アセトアミド−2−デオキシ−β−D−グルコピラノシド(6)の合成
ナスフラスコ中で、ベンジル(メチル(2,3,4−トリ−O−アセチル−β−D−グルコピラノシルウロネート)−(1→3)−2−アセトアミド−2−デオキシ−4,6−O−イソプロピリデン−β−D−グルコピラノシド(124mg,0.18mmol)をジクロロメタン(10ml)と水(0.05ml)に溶解した。
その混合物に0℃でTFA(0.5ml,5%v/v)を加え、その後乾燥雰囲気下1時間攪拌した。反応溶液を飽和NaHCO3水溶液に入れ、CHCl3で抽出した。有機層を飽和塩化ナトリウム溶液で洗浄し、硫酸マグネシウムで乾燥し、ろ過、濃縮した。残基をシリカゲルクロマトグラフィー(1:2から1:3までクロロホルム−酢酸エチル,その後酢酸エチル)で精製し、白色固体として、ベンジル(メチル2,3,4−トリ−O−アセチル−β−D−グルコピラノシルウロネート)−(1→3)−2−アセトアミド−2−デオキシ−β−D−グルコピラノシド(96mg,0.14mmol,82%)を得た。
分析データは、以下の通りである。
Rf 0.23 (EtOAc); [α] D 27 −46°(c 1.0, CHCl3);mp 207.1℃;
1H−NMR (400MHz, CDCl3,TMS) :δ(ppm); 7.79 (d, JNH,2= 8.53 Hz,1H,NH), 7.34−7.27 (m,5H,aromatic), 5.33 (t, J2’,3’ = J3’,4’ = 9.53 Hz,1H,H−3’), 4.96−4.92(m,2H,H−4', H−1'), 4.81−4.77 (m, 2H, H−2',CH2Ph), 4.65 (t, J5,6−OH = 5.52 Hz, 1H, 6−OH), 4.54−4.51(m, 2H, CH2Ph, 4−OH),4.43−4.37 (m, 2H, H−5',H−1), 3.72−3.68 (m, 1H, H−6a), 3.63 (m, 5H, COOCH3, H−2, H−3), 3.54−3.49 (m, 1H,H−6b), 3.23−3.21 (m, 1H,H−4), 3.23−3.16 (m,1H, H−5), 1.99−1.83 (m, 12H,acetamido, COCH3)
13C−NMR (100MHz, CDCl3):δ(ppm);169.70−167.50 (COCH3,COOCH3), 138.15,128.31−127.41 (aromatic), 100.25 (C−1), 99.42 (C−1'), 82.88 (C−3), 76.73 (C−5),71.60 (C−3’), 70.97 (C−5’), 70.74 (C−2'), 69.62 (CH2Ph), 69.30 (C−4'), 68.61 (C−4),60.96 (C−6), 53.98 (C−2), 52.79 (OCH3)
High resolusion FAB Mass
計算値:[M+H]+ =628.2236 m/z (C28H38NO15)
実測値:628.2242 m/z
【0016】
実施例3
ベンジル(メチル 2,3,4,−トリ−O−アセチル−β−D−グルコピラノシルウロネート)−(1→3)−6−O−アセチル−2−アセトアミド−2−デオキシ−β−D−グルコピラノシド(7)の合成
ナスフラスコ中でベンジル(メチル 2,3,4−トリ−O−アセチル−β−D−グルコピラノシルウロネート)−(1→3)−2−アセトアミド−2−デオキシ−β−D−グルコピラノシド(93mg,0.14mmol)を脱水ピリジン(0.95ml)と脱水THF(s0.95ml)に溶解し、−78℃で脱水THF(0.5ml)に溶解したAcCl(10.5μl,0.14mmol)の溶液を加えた。その反応混合物を−78℃でアルゴン雰囲気下8時間攪拌した。反応は−78℃でメタノール(2ml)を添加することによって止め、その後溶液を濃縮して、飽和NaHCO3水溶液,1N塩酸水溶液に入れ、クロロホルムで抽出した。
有機層を飽和NaCl水溶液で洗浄し、硫酸マグネシウムで乾燥し、ろ過、濃縮した。残渣は、シリカゲルカラムクロマトグラフィー(1:1から1:3までn−ヘキサン−酢酸エチル,その後酢酸エチル)によって精製し、ベンジル(メチル2,3,4,−トリ−O−アセチル−β−D−グルコピラノシルウロネート)−(1→3)−6−O−アセチル−2−アセトアミド−2−デオキシ−β−D−グルコピラノシド(46mg,0.07mmol,収率47%)を白色の固体として得た。
分析データは、以下の通りである。
Rf 0.51 (酢酸エチル);
1H−NMR (400MHz, CDCl3,TMS) :δ(ppm); 6.06 (d, JNH,2 =7.52 Hz, 1H, NH), 5.29−5.16 (m, 2H, H−3’, H−4’), 5.00−4.96 (m, 1H, H−2’),4.91−4.83 (m, 2H, H−1, CH2Ph),4.66 (d, J1’,2’ =7.52 Hz, 1H, H−1’), 4.55−4.47 (m, 2H, CH2PH, H−6a), 4.36−4.28 (m, 2H, H−3, H−6b), 4.12 (d, J4’,5’ = 10.0 Hz, 1H, H−5’), 3.99 (s, 1H, OH),3.79−3.69 (m, 2H, COOCH3,H−5), 3.55 (d, J = 6.04 Hz, 1H, H−4), 3.23−3.16 (m, 1H, H−2), 2.09−1.94(m, 15H, CH3 ofacetamido, COCH3).
13C−NMR (100MHz, CDCl3):δ(ppm); 170.21−168.37 (COCH3),166.14 (COOCH3),127.73−127.25 (aromatic), 99.49(C−1'), 97.86(C−1), 82.34(C−3), 72.74 (C−5),71.30 (C−3’), 70.99 (C−2, CH2Ph),68.57 (C−4), 67.86 (C−4'), 62.70 (C−6), 56.10 (C−2), 52.29 (COOCH3) 19.91−19.48 (COCH3).
High resolusion FAB Mass
計算値:[M+H]+ =670.2342 m/z (C30H40NO16)
実測値:670.2343 m/z
【0017】
実施例4
トリエチルアンモニウム ベンジル(メチル 2,3,4,−トリ−O−アセチル−β−D−グルコピラノシルウロネート)−(1→3)−6−O−アセチル−4−O−スルフォネート−2−アセトアミド−2−デオキシ−β−D−グルコピラノシド(8)の合成
ナスフラスコ中で、ベンジル(メチル 2,3,4,−トリ−O−アセチル−β−D−グルコピラノシルウロネート)−(1→3)−6−O−アセチル−2−アセトアミド−2−デオキシ−β−D−グルコピラノシド(295mg,0.44mmol)をDMF(10.5ml)に溶解させ、SO3・NMe3(366mg,2.64mmol)を50℃でアルゴン雰囲気下加えた。50℃で10時間攪拌後、反応混合物を乾固した。残渣を0.5%v/vトリエチルアミン含有シリカゲルクロマトグラフィー(溶出溶媒:1:2 n−ヘキサン−酢酸エチルその後酢酸エチル、その後クロロホルム,20:1クロロホルム:メタノール)によって精製し、白色非結晶粉末としてトリエチルアンモニウム ベンジル(メチル2,3,4,−トリ−O−アセチル−β−D−グルコピラノシルウロネート)−(1→3)−6−O−アセチル−4−O−スルフォネート−2−アセトアミド−2−デオキシ−β−D−グルコピラノシド(370mg,0.43mmol,98%)を得た。
分析データは、以下の通りである。
Rf 0.16 (10:1 クロロホルム−メタノール);
1H−NMR (400MHz, CDCl3,TMS) :δ(ppm); 9.89 (s, 1H, (CH2CH3)3HN), 6.24 (d, JNH,2 = 8.52Hz,1H, NH), 5.32−5.27 (m, 1H,H−3), 5.14−5.10 (m, 2H, H−1', H−4’), 5.02−4.98 (m, 1H, H−2’), 4.85−4.78 (m, 2H,CH2PH, H−1),4.58−4.55 (m, 2H, CH2PH,H−4), 4.34−4.28 (m, 2H, H−6a, H−6b), 4.23−4.21 (m, 1H, H−5), 4.15−4.06 (m, 2H,H−5’, H−2), 3.70 (s, 3H, COOCH3),3.17−3.12 (m, 13H, (CH3CH2)3NH+),2.06−1.96 (m, 15H, CH3of acetamido, COCH3),1.28−1.24 (m, 21H, (CH3CH2)3NH+).
13C−NMR (100MHz, CDCl3):δ(ppm); 170.68, 170.23, 169.73 (CH3CO),167.49 (COOCH3),137.48−127.61 (aromatic), 99.05 (C−1), 97.72 (C−1'), 75.90 (C−3), 75.02 (C−5),73.02 (C−4), 72.13 (C−5'), 71.99 (C−3'), 70.51 (C−2'), 70.45 (CH2Ph), 69.69 (C−4'), 64.24 (C−6),52.61 (COOCH3),51.62 (C−2), 46.53 (HNCH2CH3), 8.66 (HNCH2CH3).
High resolusion FAB Mass
計算値:[M+H]+ =851.3114 m/z (C36H55N2O19S)
実測値 851.3132m/z
【0018】
実施例5
2−メチル−4,5−ジヒドロ−[トリエチルアンモニウム−6−O−アセチル−4−O−スルフォネート−1,2−ジ−デオキシ−3−O−(メチル2,3,4−トリ−O−アセチル−β−D−グルコピラノシルウロネート)−α−D−グルコピラノソ]−[2,1−d]−1,3−オキサゾ−ル(9)の合成
ナスフラスコ中で、トリエチルアンモニウム ベンジル(メチル2,3,4,−トリ−O−アセチル−β−D−グルコピラノシルウロネート)−(1→3)−6−O−アセチル−4−O−スルフォネート−2−アセトアミド−2−デオキシ−β−D−グルコピラノシド(443mg,0.52mmol)をメタノール(40mL)とトリエチルアミン(0.4ml)に溶解し、活性炭(265mg)上の20%水酸化パラジウムを加えた。
混合物を室温で水素雰囲気下1時間はげしく攪拌し、Celiteでろ過後、濃縮により乾固した。残渣に対して、脱水ジクロロメタン(27ml)にN,N−ジメチルアミノピリジン(DMAP,38mg,0.31mmol),トリエチルアミン(72μl,0.51mmol),TsCl(118mg,0.62mmol)加えた。反応混合物を室温アルゴン雰囲気下3時間攪拌し、その後、追加のDMAP(38mg,0.31mmol),トリエチルアミン(72μl,0.51mmol),TsCl(118mg,0.62mmol)を加えた。室温、アルゴン雰囲気下で一晩攪拌し、残渣をシリカゲルクロマトグラフィー(クロロホルム,その後0.5%v/vトリエチルアミンを含む20:1クロロホルム−メタノール)で精製した。さらに、セファデックス LH−20カラムクロマトグラフィー(1%v/vトリエタノールアミンを含むメタノール)で精製し、2−メチル−4,5−ジヒドロ−[トリエチルアンモニウム−6−O−アセチル−4−O−スルフォネート−1,2−ジ−デオキシ−3−O−(2,3,4−トリ−O−アセチル−β−D−グルコピラノシルウロネート)−α−D−グルコピラノソ]−[2,1−d]−1,3−オキサゾール(216mg,0.29mmol,56%)を白色非結晶粉末として得た。
分析データは、以下の通りである。
Rf 0.29 (10:1 クロロホルム−メタノール); [α] D 24 +11°;
1H−NMR (400MHz, CDCl3,TMS): δ(ppm); 9.89 (s, 1H, (CH2CH3)3HN), 5.56 (d, J1,2 = 7.00 Hz, 1H, H−1), 5.30−5.26 (m, 1H, H−3’), 5.19−514(m, 2H, H−1’ H−4’), 5.02−4.92 (m, 2H, H−2’), 4.68 (d, J = 1.76 Hz, H−4),4.41 (dd, J = 2.00 Hz, 1H, H−6a), 4.25 (d, J = 10.0 Hz, 1H, H−5’),4.16−4.08 (m, 2H, H−6a, H−2), 3.73 (s, 3H, COOCH3), 3.48−3.43 (m, 1H, H−5), 3.18−3.16 (m, 6H, (CH3CH2)3NH+), 1.37−1.25 (m, 9H, , (CH3CH2)3NH+).
13C−NMR (100MHz, CDCl3):δ(ppm); 171.41−169.81 (COCH3of acetamido), 167.76 (COOCH3),166.89 (COCH3 ofoxazoline), 101.08 (C−1’), 99.57 (C−1), 78.74 (C−3), 72.78 (C−3’), 72.56 (C−4),71.58 (C−2’), 69.86 (C−4’), 68.09 (C−5), 65.70 (C−6), 64.41 (C−2), 64.11 (C−5’),53.00 (COOCH3),21.24−20.92 (COCH3of acetamido), 14.32 (COCH3of oxazoline)
High resolusion FAB Mass
計算値:[M+H]+ =743.2539 (C29H47N2O18S)
実験値 743.2545.
【0019】
実施例6
ソディウム(2−メチル−4,5−ジヒドロ−[4−O−スルフォ−1,2−ジデオキシ−3−O−(ソディウムβ−D−グルコピラノシルウロネート)−α−D−グルコピラノソ]−[2,1−d]−1,3−オキサゾール(1)の合成
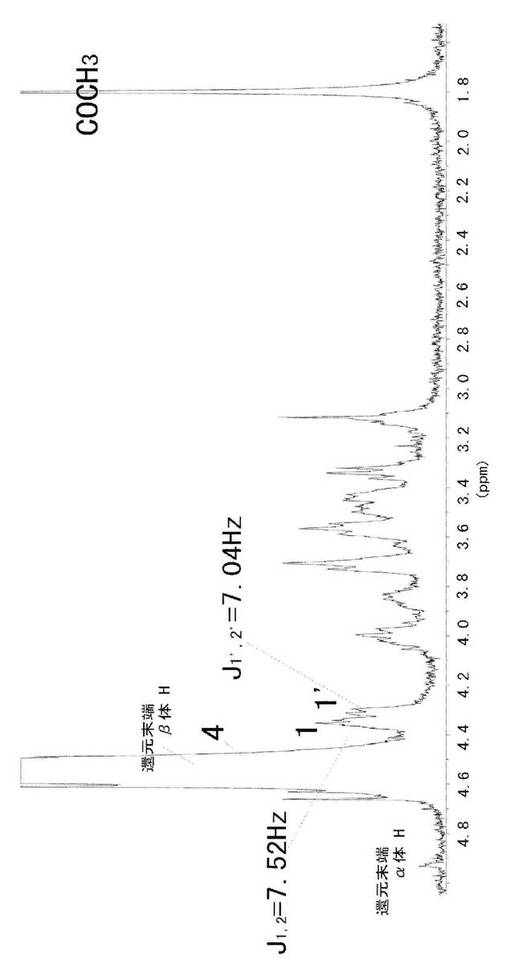
二口フラスコ中で、2−メチル−4,5−ジヒドロ−[トリエチルアンモニウム−6−O−アセチル−4−O−スルフォネート−1,2−ジ−デオキシ−3−O−(メチル 2,3,4−トリ−O−アセチル−β−D−グルコピラノシルウロネート)−α−D−グルコピラノソ]−[2,1−d]−1,3−オキサゾ−ル(92.1mg,0.12mmol)をメタノール(4.0ml)に溶解し、0℃で水酸化ナトリウム水溶液(1.0M,379μl,0.37mmol)を加え、1時間乾燥雰囲気下で攪拌し、その後室温で1時間攪拌した。その反応混合物にDowex50W−X8(H+form)を加えて、中和(pH10.4)した。その混合物を綿でろ過し、凍結乾燥して、黄色固体として、ソディウム(2−メチル−4,5−ジヒドロ−[4−O−スルフォ−1,2−ジデオキシ−3−O−(ソディウムβ−D−グルコピラノシルウロネート)−α−D−グルコピラノソ]−[2,1−d]−1,3−オキサゾール(62.3mg,純度98%)を得た。その1H−NMRの結果を図2に示した。1位プロトンに由来するピークが6.09ppmにJ1,2 = 7.52 Hzのダブレットピークとして観察され、これにより(1)がオキサゾリン環構造を取っていることが確認された。
分析データは、以下の通りである。
Rf 0.03 (2:1 クロロホルム−メタノール);
1H−NMR (400MHz, CDCl3,TMS):δ(ppm);6.09 (d, J1,2 =7.52 Hz, 1H, H−1), 4.69 (d, J = 8.04 Hz, 1H, H−4), 4.62 (d, J =7.52 Hz, 1H, H−1’), 4.40 (d, J = 2.52 Hz, 1H, H−3), 4.42 (d, J =7.00 Hz, 1H, H−2), 3.84−3.81 (dd, 1H, H−5’), 3.74−3.69 (m, 2H, H−4’, H−3’).352−3.48 (m, 2H, H−6a, H−6b), 3.45−3.41 (m, 1H, H−2’), 3.33−3.29 (m, 1H, H−5),2.22 (s, J = 2.22 Hz, 6H, acetone), 2.08 (s, 3H, CH3 of oxazoline).
13C−NMR (100MHz, CDCl3):δ(ppm); 176.33 (COONa), 169.29 (CH3C of oxazoline), 102.54 (C−1’), 100.06 (C−1), 77.26(C−5), 76.15 (C−3), 75.99 (C−4’), 73.38 (C−5), 72.96 (C−4), 72.46 (C−3'), 70.97(C−2'), 63.26 (C−2), 61.96 (C−6), 13.46 (CH3C of oxazoline)
High resolusion FAB Mass
計算値:[M+Na]+ =526.0214m/z(C14H19NO14SNa3)
実測値 526.0225m/z
【0020】
実施例7
酵素触媒重合による4−硫酸化ヒアルロン酸(2)の合成
基質モノマーであるソディウム(2−メチル−4,5−ジヒドロ−[4−O−スルフォ−1,2−ジデオキシ−3−O−(ソディウムβ−D−グルコピラノシルウロネート)−α−D−グルコピラノソ]−[2,1−d]−1,3−オキサゾール(10.5mg,20.8μmol)をリン酸緩衝液(50mM,pH6.5,210μl)に溶解し、羊精巣由来ヒアルロニダーゼ(Sigma社製,Lot No.122K1378,2660units/mg,以降、H−OTHと記す)を1.05mg加え、30℃で反応を行った。4時間反応後、反応懸濁液を90℃の湯浴中にて5分間加温することにより酵素を失活させた。その残渣を以下のサイズ排除クロマトグラフィー(SEC)条件で分析した。その結果、収率は62%であった。
SEC条件
検出器 :示差検出計
カラム :Shodex Ohpak SB−803HQ(8.0×300mm)
カラム温度:40℃
移動相 :0.1M 硝酸ナトリウム
流量 :0.5ml/min
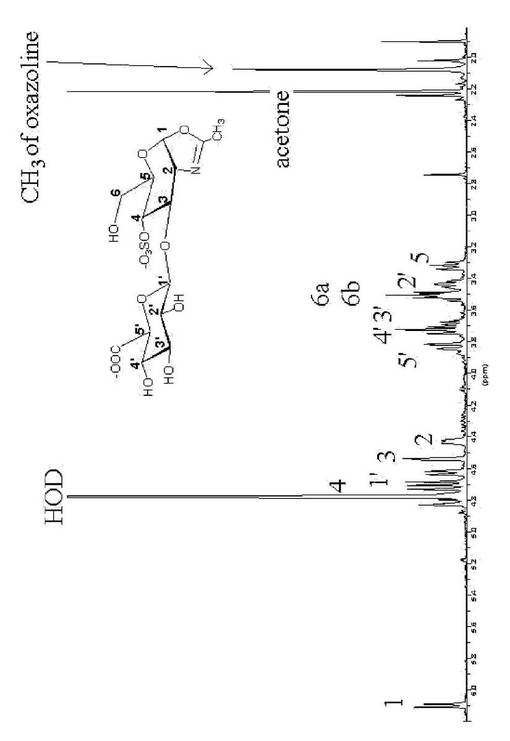
その重合生成物を0.1M硝酸ナトリウム溶液を移動層とし、Shodex OhpakSB−803HQカラムを用いたSECによって分離した。集合させたフラクションをSpectra/Por CE 透析膜(分子量分画500)を用いて蒸留水に対して透析により脱塩し、凍結乾燥して4−硫酸化ヒアルロン酸(5mg,収率47%)を得た。4−硫酸化ヒアルロン酸を重水に溶解し1H−NMRによる測定を行ったところ、内部グリコシド結合に由来するピークが見られた。その結果を図3に示す。
【0021】
実施例8
酵素触媒重合挙動の追跡
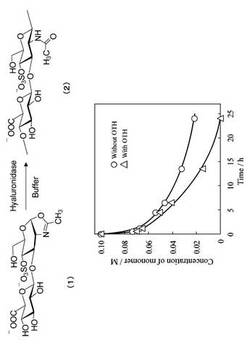
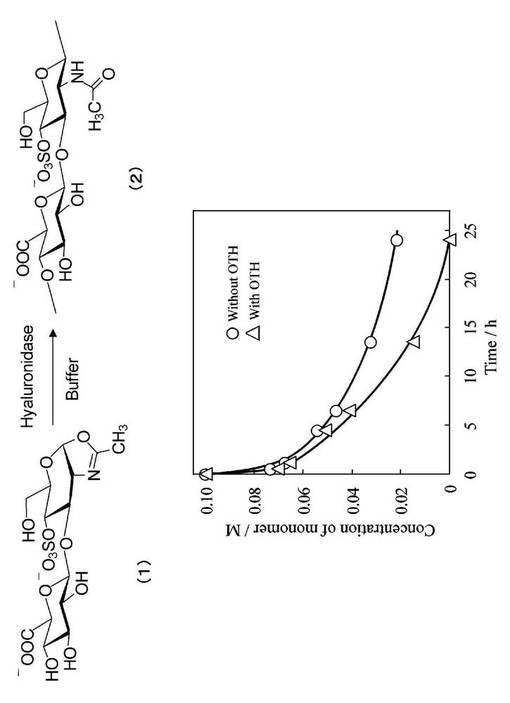
基質モノマーの消費をモニタリングする方法は以下の通りである。化合物(1)(10.5mg,20.8μmol)をリン酸緩衝D2O溶液(50mM,pD7.0,100μl)に溶解した。その溶液を50μlずつにわけた。対照液に対しては、リン酸緩衝D2O溶液(50mM,pD7.0,50μl)だけを加えた。もう一方の溶液にはリン酸緩衝D2O溶液(50)mM,pD7.0,50μl)に溶解したH−OTH(0.5mg)を加えた。これら2つの試料をNMR試験チューブ中30℃に置き、経時的に反応液を採取し、その化合物(1)の濃度を1H NMRスペクトロメトリーによるH−1プロトンとメチルプロトンのシグナルの積分値から計算した。追跡結果を図4に示した。図4より、酵素添加系において、モノマー消費が促進されたことがわかった。即ちこのモノマーがヒアルロニダーゼにより認識され、オキサゾリンの開環反応が促進されることが認識された。この反応液を以下の条件で、サイズ排除クロマトグラフィー(SEC)測定によりで分析した。
SEC条件
検出器 :示差検出計
カラム :Shodex Ohpak SB−803HQ(8.0×300mm)
カラム温度:40℃
移動相 :0.1M 硝酸ナトリウム
流量 :0.5ml/min
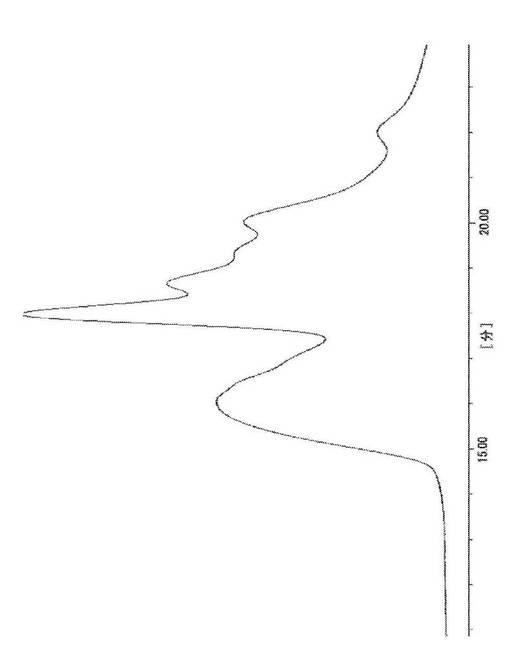
その結果、18分近傍の二糖加水分解物のピークに加え、これよりも高分子量側の16分近傍に重合物由来のピークが検出された。その結果を図5に示す。これにより、目的物(2)の生成していることが示唆された。
【0022】
実施例9
4−硫酸化ヒアルロン酸合成における反応pHの影響
重合反応において、触媒酵素として、OTH(2660unit/mg)を基質モノマーに対して、10%w/w濃度添加し、50mMリン酸緩衝液中、モノマー濃度0.1Mで、表に示す反応時間30℃でpHを変えて反応した。結果を表1に示した。モノマー(1)は、pH6.0から7.0で重合生成物が得られたのに対して、pH7.5以上の条件において急激な重合活性の低下が見られた。
【0023】
【表1】
【0024】
実施例10
4−硫酸化ヒアルロン酸合成における酵素濃度の影響
重合反応において、触媒酵素であるH−OTHの添加量と反応時間を表2に記載の条件に変更した以外は、実施例9と同様の反応及び分析を行った。その結果を表2に示す。酵素濃度は10%w/wのときに最も良好な収率(62%)で重合生成物が得られた。このときの分子量は2860であった。
【0025】
【表2】
【図面の簡単な説明】
【0026】
【図1】基質モノマーの合成スキームを示す。
【図2】基質モノマーの1H NMRスペクトルを示す。
【図3】ポリマーの1H NMRスペクトルを示す。
【図4】反応液モノマー基質の経時変化を示す。
【図5】サイズ排除クロマトグラフィー(SEC)による測定結果を示す。
【技術分野】
【0001】
本発明は、オキサゾリン誘導体にヒアルロン酸分解酵素を作用せしめることにより製造される4−硫酸化ヒアルロン酸に関する。更に詳しくは、ヒアルロン酸分解酵素であるほ乳類由来のヒアルロニダーゼを触媒とし、オキサゾリン誘導体をモノマー基質として酵素的に重合させて製造される4−硫酸化ヒアルロン酸に関する。
【背景技術】
【0002】
ヒアルロン酸は、D−グルクロン酸とN−アセチルグルコサミンの二糖が直鎖状に交互に結合した枝分かれのない高分子多糖である。その機械的性質の改善や医学用材料に適した性質への改善のために誘導体合成が行われてきた。そのひとつが硫酸化である。ヒアルロン酸は、生体内において硫酸化を受けていないので、硫酸化することにより抗凝血活性付与が検討されてきた。この場合には、ヒアルロン酸中の多くの水酸基が硫酸化される程活性が高くなることが知られている。その他に、硫酸化ヒアルロン酸には抗炎症作用や腎疾患の予防または治療効果が知られている。また、反復単位(n)数が約6〜10で、硫酸化率が20〜50%のオリゴ硫酸化ヒアルロン酸には、皮膚の角化抑制効果が見いだされた。
【0003】
硫酸化ヒアルロン酸は、ヒアルロン酸の水酸基を化学修飾により硫酸化することによって合成される。しかし、化学修飾による硫酸化では、硫酸基の導入度及び導入位置を制御することは非常に困難である。従って、未だ4−硫酸化ヒアルロン酸のように、特定の位置だけが全て硫酸化された硫酸化ヒアルロン酸は合成されていない。
【特許文献1】特開平8−277224号公報
【特許文献2】特開平8−301771号公報
【特許文献3】特開平11−279042号公報
【非特許文献1】International Congress Series(2000),1196(NewFrontiers in Medical Sciences):Redefining Hyaluronan),203−212
【発明の開示】
【発明が解決しようとする課題】
【0004】
特定の位置が硫酸化されたヒアルロン酸は、構造と活性との相関を明らかにし、生理活性の高い分子を設計する上で有意義である。そのような硫酸化ヒアルロン酸は、未だに合成されていない。
本発明者は、鋭意研究を重ねた結果、ヒアルロン酸分解酵素を用いた酵素化学的手法により、4位だけをすべて硫酸化した4−硫酸化ヒアルロン酸をはじめて合成した。
更に詳しくは、本発明者はヒアルビウロン酸の4位が硫酸化されたヒアルビウロン酸オキサゾリン誘導体を設計し、羊睾丸由来ヒアルロニダーゼを作用せしめることにより4−硫酸化ヒアルロン酸が生成することを見い出し、本発明を完成するに至った。
【課題を解決するための手段】
【0005】
即ち、本発明は、(1)下記の一般式(I)で表される4−硫酸化ヒアルロン酸、
【化4】
(2)下記の一般式(II)で表されるオキサゾリン誘導体にヒアルロン酸分解酵素を作用せしめることにより製造される4−硫酸化ヒアルロン酸、
【化5】
(3)ヒアルロン酸分解酵素が、ほ乳類由来のヒアルロニダーゼであることを特徴とする請求項2記載の4−硫酸化ヒアルロン酸、
(4)下記の一般式(II)で表されるオキサゾリン誘導体にヒアルロン酸分解酵素を作用せしめるにあたり、pHを6.0〜7.0に調整して製造されることを特徴とする(2)又は(3)記載の4−硫酸化ヒアルロン酸である。
【化6】
【発明の効果】
【0006】
本発明の4−硫酸化ヒアルロン酸は、構造が明確な硫酸化ヒアルロン酸であり、4−硫酸化ヒアルロン酸の抗凝血活性などの活性と構造との相関検討により、高活性の硫酸化ヒアルロン酸を設計するために有用である。
【発明を実施するための最良の形態】
【0007】
以下、さらに詳しく本発明を説明する。
本発明で使用する基質モノマーの合成手法を、図1に示す合成スキーム中のオキサゾリン誘導体(1)の合成を例として、その手順の一例を以下に示す。
すなわち、メチル(2,3,4−トリ−O−アセチル−β−D−グルコピラノシルトリクロロアセトイミデート)ウロネート(3)を糖供与体、ベンジル (2−アセトアミド−2−デオキシ−4,6−O−イソプロピリデン−β−D−グルコピラノシド(4)を糖受容体として用い、モレキュラーシーブス(MS4A)存在下、ジクロロメタン中、プロモーターとしてBF3・OEt2を用い、0℃でグリコシル化を行い、ベンジル(メチル2,3,4−トリ−O−アセチル−β−D−グルコピラノシルウロネート)−(1→3) −2−アセトアミド−2−デオキシ−4,6−O−イソプロピリデン−β−D−グルコピラノシド(5)を合成する。得られた(5)の脱保護反応は、ジクロロメタンに溶解・攪拌し、次いで水、トリフルオロ酢酸を添加して0℃で反応を行う。反応終了後、TFA存在下で反応溶液の温度を上昇させないように、速やかに飽和NaHCO3水溶液で分液処理を行い、シリカゲルカラムで精製を行ない、ベンジル(メチル2,3,4−トリ−O−アセチル−β−D−グルコピラノシルウロネート)−(1→3)−2−アセトアミド−2−デオキシ−β−D−グルコピラノシド(6)を合成する。(6)をTHFとピリジン=1:1に溶解し、−78℃で反応させることにより、ベンジル(メチル2,3,4−トリ−O−アセチル−β−D−グルコピラノシルウロネート)−(1→3)−6−o−アセチル−2−アセトアミド−2−デオキシ−β−D−グルコピラノシド(7)を得る。
次に、4位の遊離水酸基に硫酸基を導入した後、アノマー位のベンジル基を脱保護してオキサゾリン化を行い、トリエチルアンモニウム ベンジル(メチル2,3,4,−トリ−O−アセチル−β−D−グルコピラノシルウロネート)−(1→3)−6−O−アセチル−4ーO−スルフォネート−2−アセトアミド−2−デオキシ−β−D−グルコピラノシド(8)を得る。(8)の精製をシリカゲルカラム及びゲルろ過により行い、2−メチル−4,5−ジヒドロ−[トリエチルアンモニウム−6−O−アセチル−4−O−スルフォネート−1,2−ジ−デオキシ−3−O−(2,3,4−トリ−O−アセチル−β−D−グルコピラノシルウロネート)−α−D−グルコピラノソ]−[2,1−d]−1,3−オキサゾ−ル(9)を得る。最後に、0℃でアルカリ分解により脱保護し、目的のソディウム(2−メチル−4,5−ジヒドロ−[4−O−スルフォ−1,2−ジデオキシ−3−O−(ソディウムβ−D−グルコピラノシルウロネート)−α−D−グルコピラノソ]−[2,1−d]−1,3−オキサゾール(1)を得ることができる。
【0008】
基質モノマーは、フリーの酸、ナトリウム、カリウム等の金属塩、アンモニウム塩、トリエチルアミン塩等の形であれば特に限定されない。生成する4−硫酸化ヒアルロン酸は、基質モノマーのグルクロン酸の塩の形態に依存し、フリーな酸あるいはナトリウム、カリウム塩等の金属塩、アンモニウム塩、トリエチルアミン塩等の形態が含まれる。
【0009】
かくして得られたオキサゾリン誘導体は重合触媒としてのヒアルロン酸分解酵素の基質モノマーとして好適に使用される。酵素反応時の基質モノマー濃度は実用面から0.1%以上、好ましくは1%以上で用いられる。
反応pHは酵素の反応性と基質モノマーの安定性を考慮し、5〜10望ましくは6.5〜7.5が好適に採用される。反応温度は5℃〜60℃で、通常は20℃〜40℃が用いられる。
【0010】
使用するヒアルロン酸分解酵素としては、ほ乳類由来のヒアルロニダーゼが好ましく、具体的にはエンド−β−N−アセチルヘキソサミダーゼ(EC3.2.1.35)に分類されるウシ睾丸由来又は羊睾丸由来ヒアルロニダーゼなどが好適であり、該酵素を適当な担体に固定化した固定化酵素の形態で使用することも可能である。バッチ反応あるいは連続反応形式いずれも採用される。
【0011】
反応は水溶媒あるいは水溶媒にメタノール、エタノール、n−プロパノール等のアルコール類、グリセリン、ポリエチレングリコール等のポリオール類、ジメチルスルフォキシド、ジメチルフォルムアミド、酢酸エチル、ジオキサン、反応に悪影響を及ぼさない各種無機塩類又はpH緩衝剤等を適宜添加した条件下でも進行する。
【0012】
上記条件下でバッチ反応を開始した場合、条件によって一概に規定できないが、数時間〜数日で反応は完了する。反応終了後、反応液を遠心分離、限外濾過、精密濾過、各種吸着カラム、溶媒沈殿及びクロマト分離等の公知精製手段を組み合わせることで高純度の4−硫酸化ヒアルロン酸を単離精製することが出来る。
得られる4−硫酸化ヒアルロン酸の分子量は、基質モノマーの酵素触媒による重合反応時の条件を制御することにより種々の分子量の4−硫酸化ヒアルロン酸を得ることが可能である。
【実施例】
【0013】
以下に本発明の詳細な内容について実施例で説明するが、本発明は以下の実施例に限定されるものではない。
【0014】
実施例1
ベンジル(メチル2,3,4−トリ−O−アセチル−β−D−グルコピラノシルウロネート)−(1→3)−2−アセトアミド−2−デオキシ−4,6−O−イソプロピリデン−β−D−グルコピラノシド(5)の合成
遮光二口フラスコ中で、メチル(2,3,4−トリ−O−アセチル−α−D−グルコピラノシル トリクロロアセトイミデート)ウロネート(3)(667mg,1.39mmol)とベンジル2−アセトアミド−2−デオキシ−4,6−O−イソプロピリデン−β−D−グルコピラノシド(4)(350mg,0.99mmol)を脱水ジクロロメタン(10ml)に溶解させ、モレキュラーシーブス4A(MS4A;1.2g)を加え、0℃で脱水ジクロロメタン(0.7ml)に溶解したBF3・OEt2(175μl,1.39mmol)の溶液を加えた。反応混合物をアルゴン雰囲気下0℃で4時間攪拌した。反応をEt3N(0.45ml)の添加で停止した。その混合物を珪藻土(Celite)でろ過し、飽和NaHCO3水溶液に注ぎ、CHCl3で抽出した。有機層を飽和NaCl水溶液で洗浄し、MgSO4で乾燥し、ろ過、濃縮した。その残渣をシリカゲルカラムクロマトグラフィー(2:1から1:2までn−ヘキサン−酢酸エチル)によって、精製し、白い固体として、ベンジル(メチル(2,3,4−トリ−O−アセチル−β−D−グルコピラノシルウロネート)−(1→3)−2−アセトアミド−2−デオキシ−4,6−O−イソプロピリデン−β−D−グルコピラノシド(476mg,0.69mmol,収率71%)を得た。
分析データは、以下の通りである。
1H−NMR (400MHz, CDCl3,TMS) :δ(ppm); 7.33−7.29 (m, 7H, aromatic), 5.78 (d, JNH,2 = 7.03 Hz, 1H, NH),5.24−5.15 (m, 3H, H−3', H−1, H−4’), 4.95 (t, J1’,2’ = J2’,3’ = 8.53 Hz, 1H, H−2’), 4.84 (dd, 2H,CH2Ph), 4.55−4.48(m, 3H, H−3, CH2Ph),3.95−3.91 (m, 2H, H−5', H−6a), 3.81−3.71 (m, 7H, H−6b, COOCH3, H−4), 3.40−3.34 (m, 1H, H−5),3.09−3.03 (dd, 1H, H−2), 2.08−1.99 (m, 14H, アセトアミドのCH3 , COCH3), 1.87 (s, 3H, isopropylidenのCH3), 1.65 (s, 3H, isopropylidenのCH3 ).
【0015】
実施例2
ベンジル(メチル2,3,4−トリ−O−アセチル−β−D−グルコピラノシルウロネート)−(1→3)−2−アセトアミド−2−デオキシ−β−D−グルコピラノシド(6)の合成
ナスフラスコ中で、ベンジル(メチル(2,3,4−トリ−O−アセチル−β−D−グルコピラノシルウロネート)−(1→3)−2−アセトアミド−2−デオキシ−4,6−O−イソプロピリデン−β−D−グルコピラノシド(124mg,0.18mmol)をジクロロメタン(10ml)と水(0.05ml)に溶解した。
その混合物に0℃でTFA(0.5ml,5%v/v)を加え、その後乾燥雰囲気下1時間攪拌した。反応溶液を飽和NaHCO3水溶液に入れ、CHCl3で抽出した。有機層を飽和塩化ナトリウム溶液で洗浄し、硫酸マグネシウムで乾燥し、ろ過、濃縮した。残基をシリカゲルクロマトグラフィー(1:2から1:3までクロロホルム−酢酸エチル,その後酢酸エチル)で精製し、白色固体として、ベンジル(メチル2,3,4−トリ−O−アセチル−β−D−グルコピラノシルウロネート)−(1→3)−2−アセトアミド−2−デオキシ−β−D−グルコピラノシド(96mg,0.14mmol,82%)を得た。
分析データは、以下の通りである。
Rf 0.23 (EtOAc); [α] D 27 −46°(c 1.0, CHCl3);mp 207.1℃;
1H−NMR (400MHz, CDCl3,TMS) :δ(ppm); 7.79 (d, JNH,2= 8.53 Hz,1H,NH), 7.34−7.27 (m,5H,aromatic), 5.33 (t, J2’,3’ = J3’,4’ = 9.53 Hz,1H,H−3’), 4.96−4.92(m,2H,H−4', H−1'), 4.81−4.77 (m, 2H, H−2',CH2Ph), 4.65 (t, J5,6−OH = 5.52 Hz, 1H, 6−OH), 4.54−4.51(m, 2H, CH2Ph, 4−OH),4.43−4.37 (m, 2H, H−5',H−1), 3.72−3.68 (m, 1H, H−6a), 3.63 (m, 5H, COOCH3, H−2, H−3), 3.54−3.49 (m, 1H,H−6b), 3.23−3.21 (m, 1H,H−4), 3.23−3.16 (m,1H, H−5), 1.99−1.83 (m, 12H,acetamido, COCH3)
13C−NMR (100MHz, CDCl3):δ(ppm);169.70−167.50 (COCH3,COOCH3), 138.15,128.31−127.41 (aromatic), 100.25 (C−1), 99.42 (C−1'), 82.88 (C−3), 76.73 (C−5),71.60 (C−3’), 70.97 (C−5’), 70.74 (C−2'), 69.62 (CH2Ph), 69.30 (C−4'), 68.61 (C−4),60.96 (C−6), 53.98 (C−2), 52.79 (OCH3)
High resolusion FAB Mass
計算値:[M+H]+ =628.2236 m/z (C28H38NO15)
実測値:628.2242 m/z
【0016】
実施例3
ベンジル(メチル 2,3,4,−トリ−O−アセチル−β−D−グルコピラノシルウロネート)−(1→3)−6−O−アセチル−2−アセトアミド−2−デオキシ−β−D−グルコピラノシド(7)の合成
ナスフラスコ中でベンジル(メチル 2,3,4−トリ−O−アセチル−β−D−グルコピラノシルウロネート)−(1→3)−2−アセトアミド−2−デオキシ−β−D−グルコピラノシド(93mg,0.14mmol)を脱水ピリジン(0.95ml)と脱水THF(s0.95ml)に溶解し、−78℃で脱水THF(0.5ml)に溶解したAcCl(10.5μl,0.14mmol)の溶液を加えた。その反応混合物を−78℃でアルゴン雰囲気下8時間攪拌した。反応は−78℃でメタノール(2ml)を添加することによって止め、その後溶液を濃縮して、飽和NaHCO3水溶液,1N塩酸水溶液に入れ、クロロホルムで抽出した。
有機層を飽和NaCl水溶液で洗浄し、硫酸マグネシウムで乾燥し、ろ過、濃縮した。残渣は、シリカゲルカラムクロマトグラフィー(1:1から1:3までn−ヘキサン−酢酸エチル,その後酢酸エチル)によって精製し、ベンジル(メチル2,3,4,−トリ−O−アセチル−β−D−グルコピラノシルウロネート)−(1→3)−6−O−アセチル−2−アセトアミド−2−デオキシ−β−D−グルコピラノシド(46mg,0.07mmol,収率47%)を白色の固体として得た。
分析データは、以下の通りである。
Rf 0.51 (酢酸エチル);
1H−NMR (400MHz, CDCl3,TMS) :δ(ppm); 6.06 (d, JNH,2 =7.52 Hz, 1H, NH), 5.29−5.16 (m, 2H, H−3’, H−4’), 5.00−4.96 (m, 1H, H−2’),4.91−4.83 (m, 2H, H−1, CH2Ph),4.66 (d, J1’,2’ =7.52 Hz, 1H, H−1’), 4.55−4.47 (m, 2H, CH2PH, H−6a), 4.36−4.28 (m, 2H, H−3, H−6b), 4.12 (d, J4’,5’ = 10.0 Hz, 1H, H−5’), 3.99 (s, 1H, OH),3.79−3.69 (m, 2H, COOCH3,H−5), 3.55 (d, J = 6.04 Hz, 1H, H−4), 3.23−3.16 (m, 1H, H−2), 2.09−1.94(m, 15H, CH3 ofacetamido, COCH3).
13C−NMR (100MHz, CDCl3):δ(ppm); 170.21−168.37 (COCH3),166.14 (COOCH3),127.73−127.25 (aromatic), 99.49(C−1'), 97.86(C−1), 82.34(C−3), 72.74 (C−5),71.30 (C−3’), 70.99 (C−2, CH2Ph),68.57 (C−4), 67.86 (C−4'), 62.70 (C−6), 56.10 (C−2), 52.29 (COOCH3) 19.91−19.48 (COCH3).
High resolusion FAB Mass
計算値:[M+H]+ =670.2342 m/z (C30H40NO16)
実測値:670.2343 m/z
【0017】
実施例4
トリエチルアンモニウム ベンジル(メチル 2,3,4,−トリ−O−アセチル−β−D−グルコピラノシルウロネート)−(1→3)−6−O−アセチル−4−O−スルフォネート−2−アセトアミド−2−デオキシ−β−D−グルコピラノシド(8)の合成
ナスフラスコ中で、ベンジル(メチル 2,3,4,−トリ−O−アセチル−β−D−グルコピラノシルウロネート)−(1→3)−6−O−アセチル−2−アセトアミド−2−デオキシ−β−D−グルコピラノシド(295mg,0.44mmol)をDMF(10.5ml)に溶解させ、SO3・NMe3(366mg,2.64mmol)を50℃でアルゴン雰囲気下加えた。50℃で10時間攪拌後、反応混合物を乾固した。残渣を0.5%v/vトリエチルアミン含有シリカゲルクロマトグラフィー(溶出溶媒:1:2 n−ヘキサン−酢酸エチルその後酢酸エチル、その後クロロホルム,20:1クロロホルム:メタノール)によって精製し、白色非結晶粉末としてトリエチルアンモニウム ベンジル(メチル2,3,4,−トリ−O−アセチル−β−D−グルコピラノシルウロネート)−(1→3)−6−O−アセチル−4−O−スルフォネート−2−アセトアミド−2−デオキシ−β−D−グルコピラノシド(370mg,0.43mmol,98%)を得た。
分析データは、以下の通りである。
Rf 0.16 (10:1 クロロホルム−メタノール);
1H−NMR (400MHz, CDCl3,TMS) :δ(ppm); 9.89 (s, 1H, (CH2CH3)3HN), 6.24 (d, JNH,2 = 8.52Hz,1H, NH), 5.32−5.27 (m, 1H,H−3), 5.14−5.10 (m, 2H, H−1', H−4’), 5.02−4.98 (m, 1H, H−2’), 4.85−4.78 (m, 2H,CH2PH, H−1),4.58−4.55 (m, 2H, CH2PH,H−4), 4.34−4.28 (m, 2H, H−6a, H−6b), 4.23−4.21 (m, 1H, H−5), 4.15−4.06 (m, 2H,H−5’, H−2), 3.70 (s, 3H, COOCH3),3.17−3.12 (m, 13H, (CH3CH2)3NH+),2.06−1.96 (m, 15H, CH3of acetamido, COCH3),1.28−1.24 (m, 21H, (CH3CH2)3NH+).
13C−NMR (100MHz, CDCl3):δ(ppm); 170.68, 170.23, 169.73 (CH3CO),167.49 (COOCH3),137.48−127.61 (aromatic), 99.05 (C−1), 97.72 (C−1'), 75.90 (C−3), 75.02 (C−5),73.02 (C−4), 72.13 (C−5'), 71.99 (C−3'), 70.51 (C−2'), 70.45 (CH2Ph), 69.69 (C−4'), 64.24 (C−6),52.61 (COOCH3),51.62 (C−2), 46.53 (HNCH2CH3), 8.66 (HNCH2CH3).
High resolusion FAB Mass
計算値:[M+H]+ =851.3114 m/z (C36H55N2O19S)
実測値 851.3132m/z
【0018】
実施例5
2−メチル−4,5−ジヒドロ−[トリエチルアンモニウム−6−O−アセチル−4−O−スルフォネート−1,2−ジ−デオキシ−3−O−(メチル2,3,4−トリ−O−アセチル−β−D−グルコピラノシルウロネート)−α−D−グルコピラノソ]−[2,1−d]−1,3−オキサゾ−ル(9)の合成
ナスフラスコ中で、トリエチルアンモニウム ベンジル(メチル2,3,4,−トリ−O−アセチル−β−D−グルコピラノシルウロネート)−(1→3)−6−O−アセチル−4−O−スルフォネート−2−アセトアミド−2−デオキシ−β−D−グルコピラノシド(443mg,0.52mmol)をメタノール(40mL)とトリエチルアミン(0.4ml)に溶解し、活性炭(265mg)上の20%水酸化パラジウムを加えた。
混合物を室温で水素雰囲気下1時間はげしく攪拌し、Celiteでろ過後、濃縮により乾固した。残渣に対して、脱水ジクロロメタン(27ml)にN,N−ジメチルアミノピリジン(DMAP,38mg,0.31mmol),トリエチルアミン(72μl,0.51mmol),TsCl(118mg,0.62mmol)加えた。反応混合物を室温アルゴン雰囲気下3時間攪拌し、その後、追加のDMAP(38mg,0.31mmol),トリエチルアミン(72μl,0.51mmol),TsCl(118mg,0.62mmol)を加えた。室温、アルゴン雰囲気下で一晩攪拌し、残渣をシリカゲルクロマトグラフィー(クロロホルム,その後0.5%v/vトリエチルアミンを含む20:1クロロホルム−メタノール)で精製した。さらに、セファデックス LH−20カラムクロマトグラフィー(1%v/vトリエタノールアミンを含むメタノール)で精製し、2−メチル−4,5−ジヒドロ−[トリエチルアンモニウム−6−O−アセチル−4−O−スルフォネート−1,2−ジ−デオキシ−3−O−(2,3,4−トリ−O−アセチル−β−D−グルコピラノシルウロネート)−α−D−グルコピラノソ]−[2,1−d]−1,3−オキサゾール(216mg,0.29mmol,56%)を白色非結晶粉末として得た。
分析データは、以下の通りである。
Rf 0.29 (10:1 クロロホルム−メタノール); [α] D 24 +11°;
1H−NMR (400MHz, CDCl3,TMS): δ(ppm); 9.89 (s, 1H, (CH2CH3)3HN), 5.56 (d, J1,2 = 7.00 Hz, 1H, H−1), 5.30−5.26 (m, 1H, H−3’), 5.19−514(m, 2H, H−1’ H−4’), 5.02−4.92 (m, 2H, H−2’), 4.68 (d, J = 1.76 Hz, H−4),4.41 (dd, J = 2.00 Hz, 1H, H−6a), 4.25 (d, J = 10.0 Hz, 1H, H−5’),4.16−4.08 (m, 2H, H−6a, H−2), 3.73 (s, 3H, COOCH3), 3.48−3.43 (m, 1H, H−5), 3.18−3.16 (m, 6H, (CH3CH2)3NH+), 1.37−1.25 (m, 9H, , (CH3CH2)3NH+).
13C−NMR (100MHz, CDCl3):δ(ppm); 171.41−169.81 (COCH3of acetamido), 167.76 (COOCH3),166.89 (COCH3 ofoxazoline), 101.08 (C−1’), 99.57 (C−1), 78.74 (C−3), 72.78 (C−3’), 72.56 (C−4),71.58 (C−2’), 69.86 (C−4’), 68.09 (C−5), 65.70 (C−6), 64.41 (C−2), 64.11 (C−5’),53.00 (COOCH3),21.24−20.92 (COCH3of acetamido), 14.32 (COCH3of oxazoline)
High resolusion FAB Mass
計算値:[M+H]+ =743.2539 (C29H47N2O18S)
実験値 743.2545.
【0019】
実施例6
ソディウム(2−メチル−4,5−ジヒドロ−[4−O−スルフォ−1,2−ジデオキシ−3−O−(ソディウムβ−D−グルコピラノシルウロネート)−α−D−グルコピラノソ]−[2,1−d]−1,3−オキサゾール(1)の合成
二口フラスコ中で、2−メチル−4,5−ジヒドロ−[トリエチルアンモニウム−6−O−アセチル−4−O−スルフォネート−1,2−ジ−デオキシ−3−O−(メチル 2,3,4−トリ−O−アセチル−β−D−グルコピラノシルウロネート)−α−D−グルコピラノソ]−[2,1−d]−1,3−オキサゾ−ル(92.1mg,0.12mmol)をメタノール(4.0ml)に溶解し、0℃で水酸化ナトリウム水溶液(1.0M,379μl,0.37mmol)を加え、1時間乾燥雰囲気下で攪拌し、その後室温で1時間攪拌した。その反応混合物にDowex50W−X8(H+form)を加えて、中和(pH10.4)した。その混合物を綿でろ過し、凍結乾燥して、黄色固体として、ソディウム(2−メチル−4,5−ジヒドロ−[4−O−スルフォ−1,2−ジデオキシ−3−O−(ソディウムβ−D−グルコピラノシルウロネート)−α−D−グルコピラノソ]−[2,1−d]−1,3−オキサゾール(62.3mg,純度98%)を得た。その1H−NMRの結果を図2に示した。1位プロトンに由来するピークが6.09ppmにJ1,2 = 7.52 Hzのダブレットピークとして観察され、これにより(1)がオキサゾリン環構造を取っていることが確認された。
分析データは、以下の通りである。
Rf 0.03 (2:1 クロロホルム−メタノール);
1H−NMR (400MHz, CDCl3,TMS):δ(ppm);6.09 (d, J1,2 =7.52 Hz, 1H, H−1), 4.69 (d, J = 8.04 Hz, 1H, H−4), 4.62 (d, J =7.52 Hz, 1H, H−1’), 4.40 (d, J = 2.52 Hz, 1H, H−3), 4.42 (d, J =7.00 Hz, 1H, H−2), 3.84−3.81 (dd, 1H, H−5’), 3.74−3.69 (m, 2H, H−4’, H−3’).352−3.48 (m, 2H, H−6a, H−6b), 3.45−3.41 (m, 1H, H−2’), 3.33−3.29 (m, 1H, H−5),2.22 (s, J = 2.22 Hz, 6H, acetone), 2.08 (s, 3H, CH3 of oxazoline).
13C−NMR (100MHz, CDCl3):δ(ppm); 176.33 (COONa), 169.29 (CH3C of oxazoline), 102.54 (C−1’), 100.06 (C−1), 77.26(C−5), 76.15 (C−3), 75.99 (C−4’), 73.38 (C−5), 72.96 (C−4), 72.46 (C−3'), 70.97(C−2'), 63.26 (C−2), 61.96 (C−6), 13.46 (CH3C of oxazoline)
High resolusion FAB Mass
計算値:[M+Na]+ =526.0214m/z(C14H19NO14SNa3)
実測値 526.0225m/z
【0020】
実施例7
酵素触媒重合による4−硫酸化ヒアルロン酸(2)の合成
基質モノマーであるソディウム(2−メチル−4,5−ジヒドロ−[4−O−スルフォ−1,2−ジデオキシ−3−O−(ソディウムβ−D−グルコピラノシルウロネート)−α−D−グルコピラノソ]−[2,1−d]−1,3−オキサゾール(10.5mg,20.8μmol)をリン酸緩衝液(50mM,pH6.5,210μl)に溶解し、羊精巣由来ヒアルロニダーゼ(Sigma社製,Lot No.122K1378,2660units/mg,以降、H−OTHと記す)を1.05mg加え、30℃で反応を行った。4時間反応後、反応懸濁液を90℃の湯浴中にて5分間加温することにより酵素を失活させた。その残渣を以下のサイズ排除クロマトグラフィー(SEC)条件で分析した。その結果、収率は62%であった。
SEC条件
検出器 :示差検出計
カラム :Shodex Ohpak SB−803HQ(8.0×300mm)
カラム温度:40℃
移動相 :0.1M 硝酸ナトリウム
流量 :0.5ml/min
その重合生成物を0.1M硝酸ナトリウム溶液を移動層とし、Shodex OhpakSB−803HQカラムを用いたSECによって分離した。集合させたフラクションをSpectra/Por CE 透析膜(分子量分画500)を用いて蒸留水に対して透析により脱塩し、凍結乾燥して4−硫酸化ヒアルロン酸(5mg,収率47%)を得た。4−硫酸化ヒアルロン酸を重水に溶解し1H−NMRによる測定を行ったところ、内部グリコシド結合に由来するピークが見られた。その結果を図3に示す。
【0021】
実施例8
酵素触媒重合挙動の追跡
基質モノマーの消費をモニタリングする方法は以下の通りである。化合物(1)(10.5mg,20.8μmol)をリン酸緩衝D2O溶液(50mM,pD7.0,100μl)に溶解した。その溶液を50μlずつにわけた。対照液に対しては、リン酸緩衝D2O溶液(50mM,pD7.0,50μl)だけを加えた。もう一方の溶液にはリン酸緩衝D2O溶液(50)mM,pD7.0,50μl)に溶解したH−OTH(0.5mg)を加えた。これら2つの試料をNMR試験チューブ中30℃に置き、経時的に反応液を採取し、その化合物(1)の濃度を1H NMRスペクトロメトリーによるH−1プロトンとメチルプロトンのシグナルの積分値から計算した。追跡結果を図4に示した。図4より、酵素添加系において、モノマー消費が促進されたことがわかった。即ちこのモノマーがヒアルロニダーゼにより認識され、オキサゾリンの開環反応が促進されることが認識された。この反応液を以下の条件で、サイズ排除クロマトグラフィー(SEC)測定によりで分析した。
SEC条件
検出器 :示差検出計
カラム :Shodex Ohpak SB−803HQ(8.0×300mm)
カラム温度:40℃
移動相 :0.1M 硝酸ナトリウム
流量 :0.5ml/min
その結果、18分近傍の二糖加水分解物のピークに加え、これよりも高分子量側の16分近傍に重合物由来のピークが検出された。その結果を図5に示す。これにより、目的物(2)の生成していることが示唆された。
【0022】
実施例9
4−硫酸化ヒアルロン酸合成における反応pHの影響
重合反応において、触媒酵素として、OTH(2660unit/mg)を基質モノマーに対して、10%w/w濃度添加し、50mMリン酸緩衝液中、モノマー濃度0.1Mで、表に示す反応時間30℃でpHを変えて反応した。結果を表1に示した。モノマー(1)は、pH6.0から7.0で重合生成物が得られたのに対して、pH7.5以上の条件において急激な重合活性の低下が見られた。
【0023】
【表1】
【0024】
実施例10
4−硫酸化ヒアルロン酸合成における酵素濃度の影響
重合反応において、触媒酵素であるH−OTHの添加量と反応時間を表2に記載の条件に変更した以外は、実施例9と同様の反応及び分析を行った。その結果を表2に示す。酵素濃度は10%w/wのときに最も良好な収率(62%)で重合生成物が得られた。このときの分子量は2860であった。
【0025】
【表2】
【図面の簡単な説明】
【0026】
【図1】基質モノマーの合成スキームを示す。
【図2】基質モノマーの1H NMRスペクトルを示す。
【図3】ポリマーの1H NMRスペクトルを示す。
【図4】反応液モノマー基質の経時変化を示す。
【図5】サイズ排除クロマトグラフィー(SEC)による測定結果を示す。
【特許請求の範囲】
【請求項1】
下記の一般式(I)で表される4−硫酸化ヒアルロン酸。
【化1】
【請求項2】
下記の一般式(II)で表されるオキサゾリン誘導体にヒアルロン酸分解酵素を作用せしめることにより製造される4−硫酸化ヒアルロン酸。
【化2】
【請求項3】
ヒアルロン酸分解酵素が、ほ乳類由来のヒアルロニダーゼであることを特徴とする請求項2記載の4−硫酸化ヒアルロン酸。
【請求項4】
下記の一般式(II)で表されるオキサゾリン誘導体にヒアルロン酸分解酵素を作用せしめるにあたり、pHを6.0〜7.0に調整して製造されることを特徴とする請求項2又は3記載の4−硫酸化ヒアルロン酸。
【化3】
【請求項1】
下記の一般式(I)で表される4−硫酸化ヒアルロン酸。
【化1】
【請求項2】
下記の一般式(II)で表されるオキサゾリン誘導体にヒアルロン酸分解酵素を作用せしめることにより製造される4−硫酸化ヒアルロン酸。
【化2】
【請求項3】
ヒアルロン酸分解酵素が、ほ乳類由来のヒアルロニダーゼであることを特徴とする請求項2記載の4−硫酸化ヒアルロン酸。
【請求項4】
下記の一般式(II)で表されるオキサゾリン誘導体にヒアルロン酸分解酵素を作用せしめるにあたり、pHを6.0〜7.0に調整して製造されることを特徴とする請求項2又は3記載の4−硫酸化ヒアルロン酸。
【化3】
【図1】

【図2】
【図3】
【図4】
【図5】
【図2】
【図3】
【図4】
【図5】
【公開番号】特開2008−174642(P2008−174642A)
【公開日】平成20年7月31日(2008.7.31)
【国際特許分類】
【出願番号】特願2007−9616(P2007−9616)
【出願日】平成19年1月18日(2007.1.18)
【出願人】(000003296)電気化学工業株式会社 (1,539)
【Fターム(参考)】
【公開日】平成20年7月31日(2008.7.31)
【国際特許分類】
【出願日】平成19年1月18日(2007.1.18)
【出願人】(000003296)電気化学工業株式会社 (1,539)
【Fターム(参考)】
[ Back to top ]