
4−[3−(4−シクロプロパンカルボニル−ピペラジン−1−カルボニル)−4−フルオロ−ベンジル]−2H−フタラジン−1−オンの多形体
結晶形態Aとしての4-[3-(4-シクロプロパンカルボニル-ピペラジン-1-カルボニル)-4-フルオロ-ベンジル]-2H-フタラジン-1-オン。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、特定のフタラジノン誘導体の結晶形態および改良型合成方法、前記合成における中間体および医薬組成物、ならびに前記結晶形態の使用に関する。
【背景技術】
【0002】
哺乳動物の酵素PARP(113kDaのマルチドメインタンパク質)は、DNA一本鎖または二本鎖の切断部を認識し、それに迅速に結合する能力により、DNA損傷のシグナル伝達に関係しているとされている(非特許文献1)。
【0003】
いくつかの所見から、遺伝子増幅、細胞分裂、分化、アポトーシス、DNA塩基除去修復ならびにテロメア長および染色体安定性に及ぼす影響をはじめとする様々なDNA関連機能にPARPが関与しているという結論が得られている(非特許文献2)。
【0004】
PARPがDNA修復および他のプロセスをモジュレートする機序についての研究から、細胞核内でのポリ(ADP-リボース)鎖の形成におけるその重要性が明らかにされた(非特許文献3)。DNAに結合した活性型のPARPは、NADを利用して、種々の核標的タンパク質(トポイソメラーゼ、ヒストンおよびPARPそのものを含む)上でポリ(ADP-リボース)を合成する(非特許文献4)。
【0005】
ポリ(ADP-リボシル)化は、悪性形質転換とも関係していた。例えば、PARP活性は、SV40形質転換線維芽細胞の単離核においてより高く、一方、白血病細胞および結腸癌細胞のいずれもが、対応する正常な白血球および結腸粘膜よりも高い酵素活性を示す(非特許文献5;非特許文献6;および非特許文献7)。
【0006】
DNA修復におけるポリ(ADP-リボシル)化の機能的役割を解明するために、数種類の低分子量のPARP阻害剤が用いられた。アルキル化剤で処理した細胞では、PARPの阻害により、DNA鎖の切断と細胞の死滅が顕著に増加する(非特許文献8;非特許文献9)。
【0007】
その後、かかる阻害剤は、致死に至り得る損傷の修復を抑制することにより、放射線応答の効果を高めることが明らかになった(非特許文献10;非特許文献11)。PARP阻害剤は、低酸素性腫瘍細胞を放射線感受性にするのに効果的であると報告されている(特許文献1;特許文献2および特許文献3)。
【0008】
さらに、PARPノックアウト(PARP-/-)動物は、アルキル化剤およびγ線照射に応答してゲノムの不安定性を示す(非特許文献12;非特許文献13)。
【0009】
PARPの役割は、特定の血管疾患、敗血症性ショック、虚血性障害および神経毒性においても実証されている (非特許文献14;非特許文献15)。DNAの鎖切断が生じる酸素ラジカルDNA損傷(これは後にPARPによって認識される)は、PARP阻害剤の研究により明らかなように、かかる疾患状態の主な寄与因子である(非特許文献16;非特許文献17)。さらに最近では、PARPが出血性ショックの発病においてある役割を果たすことが実証されている(非特許文献18)。
【0010】
また、哺乳動物細胞への効率的なレトロウイルス感染がPARP活性の阻害により遮断されることも実証された。かかる組換えレトロウイルスベクター感染の阻害は、様々な異なる細胞型において起こることが明らかにされた(非特許文献19)。かくして、抗ウイルス療法および癌治療で使用するために、PARP阻害剤が開発された(特許文献4)。
【0011】
さらに、PARP阻害はヒト線維芽細胞における老化特性の発症を遅延させるということが推察されている(非特許文献20)。このことは、PARPがテロメア機能の制御において果たす役割と関連している可能性がある(非特許文献2)。
【0012】
特許文献5には、多数のフタラジノン誘導体、PARP阻害におけるそれらの活性、および放射線療法もしくは化学療法の補助剤(adjunct)としての、または単独型の薬剤としての癌治療におけるそれらの重要な用途が開示されている。
【0013】
特許文献6には、塩基除去修復(BER)阻害剤としてのPARP阻害剤、具体的にはフタラジノン誘導体の使用が開示されている。また相同組換え(HR)依存的DNA DSB修復活性に欠損のある癌(特に、BRCA1および/またはBRCA2欠損表現型を有する癌)の治療用製剤の製造におけるこれらの阻害剤の使用が開示されている。
【0014】
特許文献5に記載されている4-[3-(4-シクロプロパンカルボニル-ピペラジン-1-カルボニル)-4-フルオロ-ベンジル]-2H-フタラジン-1-オン(化合物A):
【化1】

【0015】
は、特に注目されている。
【0016】
特許文献5では、化合物Aは、多数のライブラリー化合物の1つとして、4-[4-フルオロ-3-(ピペラジン-1-カルボニル)-ベンジル]-2H-フタラジン-1-オン(化合物B):
【化2】
【0017】
から、ジクロロメタン中の(B)の溶液に塩化シクロプロパンカルボニル:
【化3】
【0018】
を添加し、続いてヒューニッヒ塩基(N,N-ジイソプロピルエチルアミン)を添加することにより合成された。この反応は室温で撹拌しながら16時間行われ、得られた化合物は分取HPLCにより精製される。
【0019】
ピペラジン誘導体(B)は、4-[2-フルオロ-5-(4-オキソ-3,4-ジヒドロ-フタラジン-1-イルメチル)-ベンゾイル]-ピペラジン-1-カルボン酸tert-ブチルエステル(化合物C):
【化4】
【0020】
を1時間、6M HClおよびエタノールを使用することにより脱保護し、次にpH9までアンモニアで塩基性化し、ジクロロメタンへ抽出することにより調製された。
【0021】
Boc保護ピペラジン誘導体(C)は、2-フルオロ-5-(4-オキソ-3,4-ジヒドロ-フタラジン-1-イルメチル)-安息香酸(化合物D):
【化5】
【0022】
から、ピペラジン-1-カルボン酸tert-ブチルエステル:
【化6】
【0023】
、2-(1H-ベンゾトリアゾール-1-イル)-1,1,3,3-テトラメチルウロニウムヘキサフルオロホスフェート(HBTU)、およびN,N,-ジイソプロピルエチルアミンをジメチルアセトアミド中に添加し、続いて18時間撹拌することにより調製された。
【先行技術文献】
【特許文献】
【0024】
【特許文献1】米国特許第5032617号
【特許文献2】米国特許第5215738号
【特許文献3】米国特許第5041653号
【特許文献4】WO 91/18591
【特許文献5】WO 2004/080976
【特許文献6】WO 2005/053662
【非特許文献】
【0025】
【非特許文献1】D'Amoursら, Biochem. J., 342, 249-268 (1999)
【非特許文献2】d'Adda di Fagagnaら, Nature Gen., 23(1), 76-80 (1999)
【非特許文献3】Althaus, F.R.およびRichter, C., ADP-Ribosylation of Proteins: Enzymology and Biological Significance, Springer-Verlag, Berlin (1987)
【非特許文献4】Rhunら, Biochem. Biophys. Res. Commun., 245, 1-10 (1998)
【非特許文献5】Miwaら, Arch. Biochem. Biophys., 181, 313-321 (1977)
【非特許文献6】Burzioら, Proc. Soc. Exp. Bioi. Med., 149, 933-938 (1975)
【非特許文献7】Hiraiら, Cancer Res., 43, 3441-3446 (1983)
【非特許文献8】Durkaczら, Nature, 283, 593-596 (1980)
【非特許文献9】Berger, N.A., Radiation Research, 101, 4-14 (1985)
【非特許文献10】Ben-Hurら, British Journal of Cancer, 49 (Suppl. VI), 34-42 (1984)
【非特許文献11】Schlickerら, Int. J. Radiat. Bioi., 75, 91-100 (1999)
【非特許文献12】Wangら, Genes Dev., 9, 509-520 (1995)
【非特許文献13】Menissier de Murciaら, Proc. Natl. Acad. Sci. USA, 94, 7303-7307 (1997)
【非特許文献14】Cantoniら, Biochim. Biophys. Acta, 1014, 1-7 (1989)
【非特許文献15】Szaboら, J. Clin. Invest., 100, 723-735 (1997)
【非特許文献16】Cosiら, J. Neurosci. Res., 39, 38-46 (1994)
【非特許文献17】Saidら, Proc. Natl. Acad. Sci. U.S.A., 93, 4688-4692 (1996)
【非特許文献18】Liaudetら, Proc. Natl. Acad. Sci. U.S.A., 97(3), 10203-10208 (2000)
【非特許文献19】Gakenら, J. Virology, 70(6), 3992-4000 (1996)
【非特許文献20】RattanおよびClark, Biochem. Biophys. Res. Comm., 201(2), 665-672 (1994)
【発明の概要】
【発明が解決しようとする課題】
【0026】
化合物Aの特定の形態は、例えば、その溶解度および/またはその安定性および/またはそのバイオアベイラビリティおよび/またはその不純物プロファイルおよび/またはその濾過特性および/またはその乾燥特性および/またはその吸湿性欠如に関して有利な特性を有している可能性がある。また/あるいは、前記形態は取り扱い易く、かつ/または微粉末化し易く、かつ/または錠剤へ製剤化し易い可能性がある。さらに、化合物Aを大容量スケールで合成するのに適した改良型合成方法も望まれている。
【課題を解決するための手段】
【0027】
したがって、本発明の第1の態様は、実質的に結晶形態の、具体的には形態Aの4-[3-(4-シクロプロパンカルボニル-ピペラジン-1-カルボニル)-4-フルオロ-ベンジル]-2H-フタラジン-1-オン(化合物A)を提供する。
【図面の簡単な説明】
【0028】
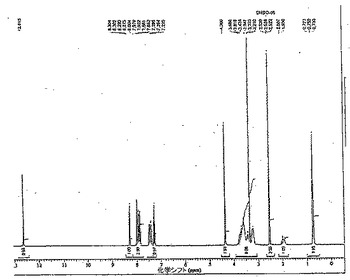
【図1】水処理後の化合物AのNMRを示す(実施例1)。
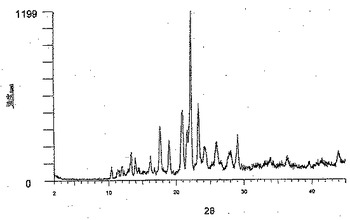
【図2】水処理後の形態Aとしての化合物Aの粉末X線回折パターンを示す (実施例1)。
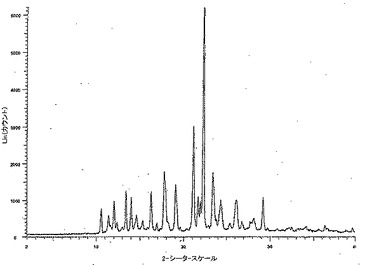
【図3】形態Aとしての化合物Aの代表的な粉末X線回折パターンを示す。
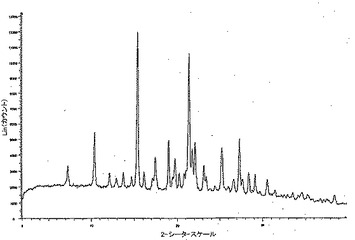
【図4】溶媒和された形態としての化合物Aの代表的な粉末X線回折パターンを示す。
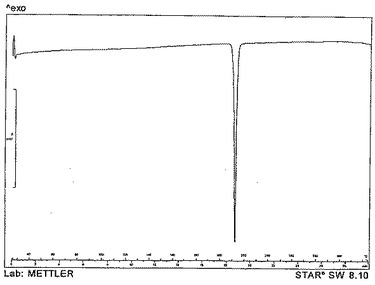
【図5】25℃〜325℃まで毎分10℃ずつ加熱することにより得られた、形態Aとしての化合物Aの代表的なDSCトレースを示す。
【発明を実施するための形態】
【0029】
上記で用いた「実質的に結晶形態」とは、化合物Aの少なくとも50重量%、好ましくは少なくとも70重量%、80重量%または90重量%が結晶形態であることを意味する。一部の実施形態では、少なくとも95重量%、99重量%または99.5重量%以上が結晶形態であってもよい。
【0030】
結晶形態Aとしての化合物Aは、次の特定のピークを有するX線回折パターン(λ=1.5418Å)を有する。
【表1】
【0031】
結晶形態Aとしての化合物Aは、次の追加ピークのX線回折パターン(λ=1.5418Å)も有する。
【表2】
【0032】
結晶形態Aとしての化合物Aは、上記リストの10のピークから選択される3つ以上のピークの任意の組み合わせを特徴とすることもできる。
【0033】
形態Aとしての化合物Aの代表的な粉末X線回折パターンを図3に示す。
【0034】
理論に制約されることを望むものではないが、化合物Aは、溶媒分子がその結晶格子内の位置を占めることができる構造を容易に形成し得る。かかる溶媒和物は、実際は必ずしも化学量論的ではないが、1種の純粋な溶媒和物から成っていてもよく(例えば、化合物Aメタノレートおよび化合物Aテトラヒドロフラネート)、あるいは可能性として、1種以上の溶媒成分(例えば、メタノールおよびジエチルエーテル)から成っていてもよい。溶媒分子は、典型的には、化合物A分子によって形成されているポケット内に存在する。ある状況では、これらのポケットの容量は一連の溶媒を包含するのに十分な程度に柔軟性があるため、物質の全体構造としての変化はほとんどなく、それ故、XRPD反射での変化はごく小さい。
【0035】
溶媒和物(同じ全体構造を共有するものを含む)は、純粋な溶媒としての、または他の溶媒と組み合わせた場合のジクロロメタン、酢酸エチル、メタノール、エタノール、イソプロパノール、2-ブタノン、t-ブチルメチルエーテル、トルエン、テトラヒドロフラン、水、シクロヘキサン、シクロプロピルメチルケトン、1,2-ジクロロエタン、エチルトリフルオロアセテート、フルオロベンゼンヘキサフルオロ-イソ-プロパノール、メチルノナフルオロブチルエーテル、2-メチル-1-プロパノール、ニトロメタン、プロピオニトリル、トリクロロエチレン、α,α,α-トリフルオロトルエン、ヘプタン、ジオキサン、アセトニトリルから溶液熟成(solution maturation)および結晶化実験により生じる。最も一般的な溶媒和物構造のX線回折パターンを図4に示すが、典型的には下記の位置で極めて強いピークを有する。
【表3】
【0036】
図に示したピークの相対強度は、試験の際の試料の方向、使用した器具の種類および設定により変わる可能性があるため、本明細書に記載のXRDトレースの強度は例示であり、絶対的な比較に用いることを意図するものではないことは理解されよう。
【0037】
化合物Aの形態Aは、溶媒を実質的に含まない。本明細書で使用する「溶媒を実質的に含まない」という用語は、極く僅かな量の任意の溶媒を有する形態、例えば総量で0.5重量%以下の任意の溶媒を含む形態を意味する。任意の溶媒(水を含む)の総量は、0.25重量%、0.1重量%、0.05重量%または0.025重量%以下であってもよい。
【0038】
また、化合物Aの形態Aは、示差走査熱量測定(DSC)を使用して特徴を決定することもできる。化合物Aの形態Aは、25℃〜325℃まで毎分10℃ずつ加熱した時、210.1℃±1℃で融解し始める。形態Aとしての化合物Aの代表的なDSCトレースを図5に示す。
【0039】
本発明の第2の態様は、結晶形態Aとしての4-[3-(4-シクロプロパンカルボニル-ピペラジン-1-カルボニル)-4-フルオロ-ベンジル]-2H-フタラジン-1-オン(化合物A)を得る方法であって、化合物Aを溶媒中で結晶化し、次に、置換剤を用いて前記結晶形態から溶媒を置換することを含む前記方法を提供する。置換剤は、水、またはC1-2アルコールと水の混合物であってもよい。
【0040】
第1の実施形態では、本方法は、
(i) 4-[3-(4-シクロプロパンカルボニル-ピペラジン-1-カルボニル)-4-フルオロ-ベンジル]-2H-フタラジン-1-オン(化合物A)を溶媒から結晶化するステップと;
(ii) 最初の溶媒がエタノールでない場合、結晶性化合物Aをエタノールで処理するステップと;
(iii) 結晶性化合物Aを水で処理し、封入されているエタノールを除去するステップと;
(iv) 得られた生成物を乾燥させるステップ
を含む。
【0041】
最初の結晶化で用いられる溶媒は、例えば、ジクロロメタンまたはアセトニトリルであってもよい。
【0042】
形態Aを得る方法は、一般に、溶媒置換を包含し得る。化合物Aは、溶媒を封入可能な結晶格子内チャンネルが形成されるように結晶化されるため、溶媒を除去することが困難になることが明らかとなっている。
【0043】
第1の実施形態の方法は、化合物Aの結晶化で使用される溶媒がジクロロメタンである場合に特に用いることができる。溶媒のジクロロメタンを溶媒のエタノールと交換するステップは、エタノールの存在下、大気圧で化合物Aの溶液を蒸留することにより行なうことができる。この交換は、ヘッド温度がエタノールの沸点(例えば、少なくとも73℃)に近づいた時に完了する。特にこの交換は、大部分のDCMを蒸留し、次いで、大量のエタノールを加えることにより行なうことができる。次いで、蒸留物のバッチを等量のエタノールで置き換えながら、蒸留を継続する。
【0044】
化合物Aのエタノール溶媒からの結晶化は、15℃以下に(好ましくは10℃未満、さらに好ましくは約8℃に)溶液を冷却することにより行うことができる。次いで、化合物Aの結晶を濾過により溶液から取り出すことができる。
【0045】
結晶性化合物Aは、水に結晶性物質を懸濁し、十分な時間(例えば、少なくとも3時間、好ましくは約4時間)加熱還流することにより、水で処理して封入されたエタノールを除去することができる。結晶性化合物Aは、濾過により水中懸濁液から取り出すことができる。
【0046】
上記ステップで得られた生成物は、容易に乾燥させることができる。例えば、少なくとも60℃、好ましくは約70℃の温度の加熱器で生成物を加熱することにより行われる。
【0047】
別のかかる実施形態では、本方法は、
(i) 溶媒を含有する結晶形態としての化合物Aを得るステップと;
(ii) 結晶形態の化合物Aの合成で使用される最初の溶媒が水とC1-2アルコール(すなわち、メタノール、エタノール)の混合物でない場合、結晶形態の化合物Aを水とC1-2アルコールの混合物で処理するステップと;
(ii) 得られた生成物を乾燥させるステップ
を含む。
【0048】
得られた生成物は、さらに水とC1-2アルコールの混合物で処理し、乾燥させ、さらに結晶形態Aの化合物Aを単離するすることができる。
【0049】
水とC1-2アルコールの混合物は、容量で2:1〜1:2の範囲にあるのが好ましく、さらには容量で1.5:1〜1:1.5の範囲にあるのが好ましい。特に好ましい混合物は、1部の水に対し1.2部のC1-2アルコールである。別の特に好ましい混合物は、2部の水に対し1部のC1-2アルコールである。C1-2アルコールはエタノールが好ましい。
【0050】
結晶形態としての化合物Aは、上記のように、化合物Aを溶媒から結晶化することにより得ることができる。
【0051】
ステップ(ii)の溶媒処理は、水とC1-2アルコールの混合物中に化合物Aを懸濁し、撹拌しながら加熱還流することにより行うことができる。続いてこれを55〜65℃に冷却し、例えばセライトパッドを通して濾過することができる。濾過パッドを水とC1-2アルコールの混合物で洗浄した後、周囲圧力(通常1気圧)以上で蒸留することができる。蒸留を止めて懸濁液を得て、これを室温に置いた後、濾過することができる。得られた濾過ケーキは、水で洗浄することができる。
【0052】
上記のステップで得られた生成物は、容易に乾燥することができる。例えば、少なくとも50℃、好ましくは約60℃の温度の加熱器で生成物を加熱することによって行われる。
【0053】
さらなる処理は、上記の方法と類似の方法で進めることができる。
【0054】
第3の実施形態では、本方法は、
(i) 溶媒としての水とC1-2アルコールの混合物中に化合物Aを懸濁するステップと;
(ii) 懸濁液を加熱還流するステップと;
(iii) 溶液を冷却し、形態Aとしての化合物Aを種結晶として加える(seeding)ステップと;
(iv) 得られた生成物を乾燥させるステップ
を含む。
【0055】
得られた生成物は、さらに水とC1-2アルコールの混合物で処理し、乾燥して、さらに結晶形態Aの化合物Aを単離することができる。
【0056】
水とC1-2アルコールの混合物は、容量で2:1〜1:5の範囲にあるのが好ましく、さらには容量で1:2〜1:4の範囲にあるのが好ましい。特に好ましい混合物は、1部の水に対し3部のC1-2アルコールである。C1-2アルコールはエタノールが好ましい。
【0057】
ステップ(iii)は、溶液を65〜75℃(例えば70℃)に冷却し、例えば、セライトパッドを通して濾過することを含む。濾過パッドを水とC1-2アルコールの混合物で洗浄した後、(周囲圧力以上で)蒸留することができる。種結晶の投与(seeding)は、得られた濾液が40〜50℃(例えば45℃)に冷却された後に始まり得る。得られた懸濁液は、2〜3時間(例えば2.5時間)で周囲温度(例えば20℃)まで冷却し、結晶化されるのに十分な時間、前記温度で維持することができる。この時間は12〜24時間であってもよく、約16時間であってもよい。この時間が終わったら、さらに水を加えることができる。その量は、存在する全溶媒(水およびC1-2アルコール)の容量とほぼ等しく、ゆっくりと(例えば、4〜6時間、例えば5時間かけて)加えることができる。水の添加後、その懸濁液は、例えば2時間、周囲温度で維持することができる。
【0058】
次いで、その懸濁液を濾過し、得られた濾過ケーキをC1-2アルコールと水の混合物(1:3〜1:2、例えば1:2.3の割合)で洗浄することができる。
【0059】
上記のステップで得られた生成物は、容易に乾燥することができる。例えば、40℃〜60℃の温度の真空下の加熱器で生成物を加熱することによって行われる。
【0060】
本発明の第3の態様は、化合物Bから化合物Aを合成する方法であって、
(i) 制御下で、適切な有機溶媒(例えば、DCM(ジクロロメタン))に溶解したトリエチルアミンおよび塩化シクロプロパンカルボニルの予混合溶液を、同一有機溶媒中の化合物Bに、溶液の温度が20℃未満となるように制御しながら添加するステップ含む前記方法を提供する。
【0061】
一部の実施形態では、本方法は、
(ii) (i)で得られた溶液を、溶液の温度を20℃以下に維持しながら反応が完了するまでかき混ぜる(例えば、撹拌する)ステップを含む。
【0062】
ステップ(i)の添加は、滴下法で実施することができる。
【0063】
この方法はWO 2004/080976に記載されている方法よりもより一層制御されているため、酸塩化物の付加がより位置選択的なものとなる。制御のほとんどない先行技術の方法では、酸塩化物の付加が所望のピペリジン窒素だけでなく、フタラジノンの窒素および/または酸素に起こる可能性がある。
【0064】
上記の方法は窒素雰囲気下で行なうのが好ましい。
【0065】
段階(ii)の溶液の温度は10〜15℃の間に維持されるのがさらに好ましい。
【0066】
上記反応の生成物は、少なくとも1回の水洗浄ステップにより後処理されるのが好ましい。より好ましくは、後処理は、最初と最終の水洗浄ステップと、希酸(例えば5%クエン酸溶液)を使用した後、希塩基(例えば5%炭酸ナトリウム溶液)を使用する中間洗浄ステップを含む。
【0067】
本発明の第4の態様は、化合物Dからの化合物Aを合成する方法であって、アミドカップリング剤および塩基、例えばアミン(具体的にはジイソプロピルエチルアミンなどの第三級アミンなど)の存在下で、化合物Dを1-(シクロプロピルカルボニル)ピペラジンまたはその鉱酸塩と反応させることを含む前記方法を提供する。
【0068】
鉱酸塩は、例えば塩酸塩であり得る。
【0069】
1-(シクロプロピルカルボニル)ピペラジンまたはその鉱酸塩の化合物Dへの添加は、任意の好適な溶媒(例えばアセトニトリル)中で行なうことができる。アミドカップリング剤は、好ましくは、2-(1H-ベンゾトリアゾール-1-イル)-1,1,3,3-テトラメチルウロニウムヘキサフルオロホスフェート(HBTU)である。好ましくは、それを1-(シクロプロピルカルボニル)ピペラジンまたはその鉱酸塩、ジイソプロピルエチルアミンおよび化合物Dの溶液に一定時間(例えば30分)かけて添加する。得られる溶液の温度は、25℃以下(または20℃以下、例えば18℃)で維持することができる。この添加後、得られた溶液を一定時間静置することができる。好ましい温度体制は、この溶液を室温で2時間維持することである。
【0070】
生じた化合物Aは、一定時間(例えば1時間)、10℃未満(または5℃未満、例えば3℃)に冷却し、その後濾過することにより、溶液から取り出すことができる。生じた化合物Aは、例えば冷却アセトニトリルで洗浄することができる。
【0071】
WO 2004/080976には、次の化合物Dへの経路が開示されている。
【化7】
【0072】
亜リン酸ジメチルをナトリウムメトキシドのメタノール溶液に0℃で滴下添加した。次いで、温度を5℃未満に維持しながら、2-カルボキシベンズアルデヒド(H)をメタノールスラリーとしてその反応混合物に分割添加した。得られた淡黄色溶液を20℃に1時間かけて加温した。メタンスルホン酸をその反応物に滴下添加し、得られた白色懸濁液を減圧蒸発させた。その白色残渣を水でクエンチし、クロロホルムで抽出した。この有機抽出物を合わせて水で洗浄し、MgSO4を用いて脱水し、減圧蒸発させて、白色固体として(3-オキソ-1,3-ジヒドロ-イソベンゾフラン-1-イル)ホスホン酸ジメチルエステル(G)(収率:95%)を得た。次いで、これをさらに精製せずに次の段階に使用した。
【0073】
(3-オキソ-1,3-ジヒドロ-イソベンゾフラン-1-イル)ホスホン酸ジメチルエステル(G)のテトラヒドロフラン溶液と2-フルオロ-5-ホルミルベンゾニトリル(F)のテトラヒドロフラン溶液の混合物に、温度を15℃未満に維持しながらトリエチルアミンを25分かけて滴下添加した。その反応混合物を1時間かけて徐々に20℃に加温し、減圧濃縮した。その白色残渣を水に30分間スラリー化し、濾過し、水、へキサンおよびエーテルで洗浄し、乾燥し、E異性体とZ異性体の50:50混合物として2-フルオロ-5-(3-オキソ-3H-イソベンゾフラン-1-イリデンメチル)ベンゾニトリル(E)(収率:96%)を得た。
【0074】
2-フルオロ-5-(3-オキソ-3H-イソベンゾフラン-1-イリデンメチル)ベンゾニトリル(E)の水中懸濁液に水酸化ナトリウム水溶液を添加し、その反応混合物を窒素下で90℃に30分間加熱した。その反応混合物を多少冷却して70℃とし、ヒドラジン水和物を添加し、70℃で18時間撹拌した。その反応物を室温に冷却し、2M HClを用いてpH4に酸性化した。その混合物を10分間撹拌し、濾過した。得られた固体を水、へキサン、エーテル、酢酸エチルで洗浄し、乾燥させ、淡いピンク色粉末の化合物D(収率:77%)を得た。
【0075】
また、化合物Dの改良型合成方法も望まれている。
【0076】
したがって、本発明の第5の態様は、化合物Dの合成方法であって、
(a) ジエチル(3-オキソ-1,3-ジヒドロ-2-ベンゾフラン-1-イル)ホスホネート(G')を2-カルボキシベンズアルデヒド(H)から合成するステップと;
(b) 2-フルオロ-5-[(E/Z)-(3-オキソ-2-ベンゾフラン-1(3H)-イリデン)メチル]ベンゾニトリル(E)をジエチル(3-オキソ-1,3-ジヒドロ-2-ベンゾフラン-1-イル)ホスホネートから合成するステップ
を含む方法を提供する。
【0077】
化合物G'は合成中に単離されないのが好ましい。この方法は、アルコール溶液中で不安定である亜リン酸ジメチルのナトリウム塩(Pelchowiczら, J.Chem.Soc, 4348-4350 (1961))の使用を避ける。ステップ(a)は、亜リン酸ジエチルのナトリウム塩が安定する2-メチルテトラヒドロフラン中で生じるのが好ましい。この塩は、2-メチルテトラヒドロフランに溶解したナトリウムt-アミレートの冷却溶液に亜リン酸ジエチルを添加することによりin situで形成することができる。亜リン酸ジエチルのナトリウム塩と反応させた後、メタンスルホン酸との反応を行うことができる。
【0078】
ステップ(b)は、2-メチルテトラヒドロフラン中でトリエチルアミンの添加により行うことができる。
【0079】
化合物Dの合成方法は、さらに、
(c) 化合物Eからヒドラジン水和物との反応により2-フルオロ-5-[(4-オキソ-3,4-ジヒドロフタラジン-1-イル)メチル]ベンゾニトリル(ED):
【化8】
【0080】
を合成するステップと;
(d) 化合物EDから水酸化ナトリウムとの反応により化合物Dを合成するステップ
を含んでいてもよい。
【0081】
ステップ(c)は、テトラヒドロフラン中の1.1〜1.3当量のヒドラジン水和物を使用し、続いて、過剰のヒドラジン水和物を酢酸を用いて中和させることにより実施することができる。
【0082】
本発明の第6の態様は、化合物ED:
【化9】
【0083】
および化合物Dの合成におけるその使用を提供する。
【0084】
本発明のさらなる態様は、1-(シクロプロピルカルボニル)ピペラジンの鉱酸塩、および、ピペラジンを酢酸と反応させ、その後、塩化シクロプロパンカルボニルを添加することによりそれを合成する方法を提供する。
【0085】
本発明の第7の態様は、第1の態様の化合物と製薬上許容可能な担体または希釈剤を含む医薬組成物を提供する。
【0086】
本発明の第8の態様は、ヒトまたは動物の身体の治療方法で使用するための第1の態様の化合物を提供する。
【0087】
本発明の第9の態様は、PARP活性の阻害剤の製造における本発明の第1の態様に定義した化合物の使用を提供する。
【0088】
本発明のさらなる態様は、血管疾患;敗血症性ショック;虚血性障害;神経毒性;出血性(haemorraghic)ショック;ウィルス感染;またはPARP活性を阻害することより寛解される疾患の治療用薬剤の製造における本発明の第1の態様に定義した化合物の使用を提供する。
【0089】
本発明の別のさらなる態様は、癌治療において補助剤として使用するための薬剤または電離放射線もしくは化学療法剤による治療で腫瘍細胞を増強するための薬剤の製造における本発明の第1の態様で定義した化合物の使用を提供する。
【0090】
本発明のさらなる別の態様は、PARPの阻害により寛解される疾患の治療であって、治療の必要な患者に、治療上有効な量の第1の態様に定義した化合物を好ましくは医薬組成物の剤形で投与することを含む治療、ならびに、癌の治療であって、治療の必要な患者に、治療上有効な量の第1の態様に定義した化合物を好ましくは医薬組成物の剤形で、電離放射線または化学療法剤と組み合わせて同時に、または連続して投与することを含む治療を提供する。
【0091】
本発明のさらなる態様では、本化合物は、相同組換え(HR)依存的DNA DSB修復活性に欠損のある癌の治療用薬剤の製造において使用可能であるか、または患者に治療上有効な量の化合物を投与することを含む、HR依存的DNA DSB修復活性に欠損のある癌の患者の治療において使用可能である。
【0092】
HR依存的DNA DSB修復経路は、連続DNAヘリックスを再形成する機序と同じ機序によって、DNAの二本鎖切断(DSB)を修復する(K.K. KhannaおよびS.P. Jackson, Nat. Genet. 27(3): 247-254 (2001))。HR依存的DNA DSB修復経路の成分としては、ATM(NM_000051)、RAD51(NM_002875)、RAD51L1(NM_002877)、RAD51C(NM_002876)、RAD51L3(NM_002878)、DMC1(NM_007068)、XRCC2(NM_005431)、XRCC3(NM_005432)、RAD52(NM_002879)、RAD54L(NM_003579)、RAD54B(NM_012415)、BRCA1(NM_007295)、BRCA2(NM_000059)、RAD50(NM_005732)、MRE11A(NM_005590)およびNBS1(NM_002485)が挙げられるが、これらに限定されるものではない。HR依存的DNA DSB修復経路に関与する他のタンパク質群としては、EMSYなどの制御因子が挙げられる(Hughes-Daviesら, Cell, 115, pp523-535)。またHR成分は、Woodら, Science, 291, 1284-1289 (2001)にも記載されている。
【0093】
HR依存的DNA DSB修復を欠損する癌は、この経路によるDNA DSB修復能力が正常細胞に比べて低下または抑制されている1個または複数の癌細胞を含み得るか、かかる癌細胞からなり得る。すなわち、HR依存的DNA DSB修復経路の活性が1個または複数の癌細胞において低下または消失している可能性がある。
【0094】
HR依存的DNA DSB修復経路の1種または複数の成分の活性は、HR依存的DNA DSB修復を欠損する癌に罹患した個体の1個または複数の癌細胞では消失する場合がある。HR依存的DNA DSB修復経路の成分は、当分野では特徴が十分に明らかとなっており(例えば、Woodら, Science, 291, 1284-1289 (2001)を参照)、上記成分などが挙げられる。
【0095】
ある好ましい実施形態では、癌細胞はBRCA1および/またはBRCA2欠損表現型を有している可能性がある。すなわち、癌細胞では、BRCA1および/またはBRCA2活性が低下または消失している。この表現型をもつ癌細胞はBRCA1および/またはBRCA2に欠損があり、すなわち、癌細胞では、例えば、コード核酸における変異もしくは多型により、または制御因子をコードする遺伝子(例えば、BRCA2制御因子をコードするEMSY遺伝子)の増幅、変異もしくは多型により、BRCA1および/またはBRCA2の発現および/または活性が低下または消失している可能性がある(Hughes-Daviesら, Cell, 115, 523-535)。
【0096】
BRCA1およびBRCA2は既知の腫瘍抑制因子であり、その野生型対立遺伝子はヘテロ接合性の保因者の腫瘍においてしばしば失われている(Jasin M., Oncogene, 21(58), 8981-93 (2002);Tuttら, Trends Mol Med., 8(12), 571-6, (2002))。BRCA1および/またはBRCA2突然変異と乳癌との関連性は、当分野では特徴が十分に明らかとなっている(Radice, P.J., Exp Clin Cancer Res., 21(3 Suppl), 9-12 (2002))。BRCA2結合因子をコードするEMSY遺伝子の増幅もまた、乳癌および卵巣癌と関連することが知られている。
【0097】
BRCA1および/またはBRCA2に変異のある保因者はまた、卵巣癌、前立腺癌および膵臓癌のリスクが高くなる。
【0098】
ある好ましい実施形態では、個体は、BRCA1および/またはBRCA2あるいはその制御因子における1つまたは複数の変異(例えば、突然変異および多型)に関しヘテロ接合である。BRCA1およびBRCA2における変異の検出は当分野では周知であり、例えば、EP 699 754、EP 705 903、Neuhausen, S.L.およびOstrander, E.A., Genet. Test, 1, 75-83 (1992);Chappnis, P.O.およびFoulkes, W.D., Cancer Treat Res, 107, 29-59 (2002);Janatova M.ら, Neoplasma, 50(4), 246-50 (2003); Jancarkova, N., Ceska Gynekol., 68(1), 11-6 (2003))に記載されている。BRCA2結合因子EMSYの増幅の測定は、Hughes-Daviesら, Cell, 115, 523-535)に記載されている。
【0099】
癌と関連している突然変異および多型は、変異核酸配列の存在を検出することにより核酸レベルで、または変異(すなわち、突然変異または対立遺伝子変異)ポリペプチドの存在を検出することによりタンパク質レベルで検出することができる。
【0100】
使用
本発明は、活性化合物、具体的にはPARP活性を阻害する活性がある化合物の形態Aとしての化合物Aを提供する。
【0101】
本明細書で使用される「活性(のある)」という用語は、PARP活性を阻害することができる化合物に関係する。本化合物が提供するPARP阻害を評価するのに簡便に用いることができる1つのアッセイを、下記の実施例で説明する。
【0102】
本発明はさらに、細胞におけるPARP活性を阻害する方法であって、その細胞を、好ましくは製薬上許容可能な組成物の形態の有効量の活性化合物と接触させることを含む方法を提供する。かかる方法は、in vitroまたはin vivoで行うことができる。
【0103】
例えば、細胞の試料をin vitroで増殖させ、活性化合物をその細胞と接触させ、その細胞に対する化合物の効果を観察することができる。「効果」の例として、一定時間に達成されたDNA修復の量を求めることができる。活性化合物が細胞に対して影響を与えることが認められる場合には、同じ細胞種の細胞を有する患者の治療方法において、その化合物の効力に関する予後マーカーまたは診断マーカーとしてそれを用いることができる。
【0104】
症状の治療に関して本明細書で使用される「治療」という用語は、一般に、ヒトに関するものか動物に関するものか(例:獣医用途)を問わず、例えば症状進行の阻害といった何らかの望ましい治療効果が得られる治療および療法に関係するものであり、その効果としては進行速度の低下、進行速度の停止、症状の改善および症状の治癒などが挙げられる。予防手段としての治療(すなわち予防)も含まれる。
【0105】
本明細書で使用される「補助剤(adjunct)」という用語は、公知の治療手段と組み合わせた活性化合物の使用を意味する。かかる手段には、各種癌の治療で用いられる薬剤の細胞傷害性療法および/または電離放射などがある。特に、当該活性化合物は、多くの癌化学療法の作用を増強することが知られており、そのようなものとしては、トポイソメラーゼのクラスの毒(例えば、トポテカン、イリノテカン、ルビテカン)、癌の治療に使用されている大部分の既知のアルキル化剤(例えば、DTIC、テモゾラミド)および白金系薬剤(例えば、カルボプラチン、シスプラチン)が挙げられる。
【0106】
活性化合物は、PARPを阻害するために細胞培養添加剤としても用いられ、例えばin vitroで公知の化学療法剤または電離放射線治療に対して細胞を増感させることもできる。
【0107】
活性化合物はin vitroアッセイの一部としても用いられ、例えば候補宿主において当該化合物による治療が有効であるかを決定することもできる。
【0108】
投与
本活性化合物または本活性化合物を含む医薬組成物は、全身投与/末梢投与、つまり所望の作用部位に関わらず、都合のよい投与経路によって被験者に投与することができ、それらの経路としては経口(例:経口摂取による);局所(例えば経皮、経鼻、眼内、口腔および舌下など);肺(例:例えばエアロゾルを用いた、例えば口もしくは鼻を介した吸入または通気療法);直腸;膣;例えば皮下、皮内、筋肉、静脈、動脈、心臓内、硬膜内、脊髄内、嚢内、被膜下、眼窩内、腹腔内、気管内、表皮下、関節内、クモ膜下および胸骨内などの注射による非経口;例えば皮下または筋肉でのデポー剤埋込物によるものなどがあるが、これらに限定されるものではない。
【0109】
被験体は、真核生物、動物、脊椎動物、哺乳動物、齧歯類(例:モルモット、ハムスター、ラット、マウス)、ネズミ類(例:マウス)、犬類(例:イヌ)、ネコ類(例:ネコ)、ウマ類(例:ウマ)、霊長類、類人猿(例:サルまたは類人猿)、サル類(例:マーモセット、ヒヒ)、類人猿(例:ゴリラ、チンパンジー、オランウータン、テナガザル)、またはヒトであってもよい。
【0110】
製剤
本活性化合物を単独で投与することは可能であるが、それを1種または複数の製薬上許容可能な担体、アジュバント、賦形剤、希釈剤、充填剤、緩衝剤、安定剤、保存剤、潤滑剤、または他の当業者には公知の物質ならびに適宜他の治療薬もしくは予防薬とともに、上記で定義した活性化合物を含む医薬組成物(例えば、製剤)として提供することが好ましい。
【0111】
そこで本発明はさらに、上記で定義した医薬組成物、ならびに本明細書に記載した1種または複数の製薬上許容可能な担体、賦形剤、緩衝剤、アジュバント、安定剤または他の物質ともに、上記で定義した活性化合物を、活性化合物が結晶形態Aとして得られるように混合することを含む、医薬組成物の製造方法を提供する。
【0112】
本明細書で使用される「製薬上許容可能な」という用語は、妥当な医学的判断の範囲内で、過度の毒性、刺激、アレルギー反応、または他の問題もしくは合併症を起こさず、妥当な利益/リスク比が得られて、被験者(例:ヒト)の組織との接触における使用に適した化合物、物質、組成物および/または剤形に関係するものである。各担体、賦形剤なども、製剤の他の成分と適合性であるという意味において「許容可能な」ものでなければならない。
【0113】
適切な担体、希釈剤、賦形剤などは標準的な薬学のテキストに記載されている。例えば、「Handbook of Pharmaceutical Additives」, 第2版(M. AshおよびI. Ash編), 2001 (Synapse Information Resources, Inc., Endicott, New York, USA)、「Remington's Pharmaceutical Sciences」, 第20版, pub. Lippincott, Williams and Wilkinsons, 2000;および「Handbook of Pharmaceutical Excipients」, 第2版, 1994を参照されたい。
【0114】
製剤は、好ましくは単位剤形で提供することができ、製薬分野で公知の任意の方法によって製造することができる。かかる方法には、1種または複数の副成分を構成する担体と本活性化合物を組み合わせるステップが含まれる。一般に、製剤は、液体担体もしくは微粉砕固体担体またはその両方を本活性化合物と均一かつ十分に組み合わせ、次に必要に応じて生成物を成形することにより製造される。
【0115】
製剤は、懸濁液、錠剤、粒剤、散剤、カプセル剤、カシェ剤(cachet)、丸剤またはペースト剤の形態とすることができる。
【0116】
経口投与(例えば、経口摂取)に好適な製剤は、カプセル剤、カシェ剤または錠剤などの、それぞれが所定量の活性化合物を含む個別の単位として;粉剤または粒剤として;水系もしくは非水系液体中の懸濁液として;あるいはペースト剤として提供することができる。
【0117】
錠剤は、適宜に1種または複数の副成分とともに、例えば圧縮または成形などの従来の手段によって製造することができる。圧縮錠は、適宜1種または複数の結合剤(例:ポビドン、ゼラチン、アラビアゴム、ソルビトール、トラガカント、ヒドロキシプロピルメチルセルロース);充填剤または希釈剤(例:乳糖、微結晶セルロース、リン酸水素カルシウム);潤滑剤(例:ステアリン酸マグネシウム、タルク、シリカ);崩壊剤(例:デンプングリコール酸ナトリウム、架橋ポビドン、架橋カルボキシメチルセルロースナトリウム);界面活性剤または分散剤または湿展剤(例:ラウリル硫酸ナトリウム);ならびに保存剤(例:p-ヒドロキシ安息香酸メチル、p-ヒドロキシ安息香酸プロピル、ソルビン酸)と混合されていてもよい粉末または顆粒などの自由流動形態の活性化合物を好適な機械で圧縮することで製造することができる。成形錠は、不活性液体希釈剤で湿らせた粉末化合物の混合物を適切な機械で成形することにより製造することができる。錠剤には適宜にコーティングまたは刻み目を施すことができ、例えばヒドロキシプロピルメチルセルロースを各種割合で用いて活性化合物を持続放出または制御放出して、所望の放出プロファイルを得るように製剤することができる。錠剤は、適宜腸溶コーティングを施して、胃以外の消化管部分で放出されるようにすることができる。
【0118】
カプセル剤は、懸濁液に溶解した活性化合物を含むことができる。
【0119】
局所投与(例えば、経皮投与、経鼻投与、眼内投与、口腔内投与および舌下投与)に適した製剤は、ペースト剤として製剤化することができる。
【0120】
眼部への局所投与に適した製剤には、好適な担体(特に活性化合物用の水性溶媒)中に活性化合物を懸濁させた点眼剤も含まれる。
【0121】
担体が固体である経鼻投与に適した製剤には、鼻での吸気を行うことで、すなわち鼻の近くに保持された粉剤容器から鼻道を通って急速な吸入によって投与される、粒径が例えば約20〜約500ミクロンの範囲の粗粉剤などがある。
【0122】
吸入による投与に好適な製剤には、ジクロロジフルオロメタン、トリクロロフルオロメタン、ジクロロテトラフルオロエタン、二酸化炭素、または他の好適なガスなどの適切な噴射剤を用いた、加圧パックからのエアロゾル噴霧剤として提供されるものなどがある。
【0123】
用量
活性化合物および活性化合物を含む組成物の適切な用量は患者ごとに変わり得ることは明らかであろう。最適用量の決定には、一般的に、本発明の治療のあらゆるリスクまたは有害な副作用に対して治療効果のレベルのバランスをとることが必要である。選択される用量レベルは種々の要因に依存し、こうした要因としては、特定化合物の活性、投与経路、投与時間、化合物の排出速度、治療の持続期間、併用される他の薬剤、化合物および/または物質、ならびに患者の年齢、性別、体重、症状、全身的な健康状態、および以前の病歴が挙げられるが、これらに限定されるものではない。化合物の量および投与経路は、最終的には医師の裁量に委ねられる。しかし一般に用量は、実質的に有害または有毒な副作用を起こすことなく、所望の効果を達成する作用部位での局所濃度を与えるようなものとする。
【0124】
in vivo投与は、治療過程全体にわたり連続的に、断続的に(例えば適当な時間間隔で分割用量として)または1回用量で行なうことができる。最も効果的な投与手段と用量を決定する方法は当業者によく知られており、治療に使用する製剤、治療目的、治療される標的細胞、および治療される被験体に応じて変わる。単回投与または複数回投与は、治療に当たっている医師により選択される用量レベルと投与パターンで行うことができる。
【0125】
一般に、本活性化合物の適当な用量は、1日当たり、約10mg〜約600mg/被験体のm2体面積重量(body area weight)の範囲である。
【実施例】
【0126】
実施例1:化合物Aの合成
【化10】
【0127】
出発物質(D)は、WO 2004/080976に開示されている方法により合成した。
【0128】
方法
分取HPLC
試料は、Waters 600 LCポンプ、Waters Xterra C18カラム(5μm 19mm×50mm)およびMicromass ZQ質量分析計を備え、陽イオンエレクトロスプレーイオン化モードで操作する、Waters社の質量分析計直結型精製システムを用いて精製した。移動相A(水中0.1%ギ酸)および移動相B(アセトニトリル中0.1%ギ酸)は、勾配をつけて用いた(7分間で5%Bから100%へ、3分間保持、流速20ml/分)。
【0129】
分析HPLC-MS
分析HPLCは、Spectra System P4000ポンプおよびJones Genesis C18カラム(4μm、50mm×4.6mm)を用いて行った。移動相A(水中0.1%ギ酸)および移動相B(アセトニトリル)は、勾配をつけて用いた(5%Bを1分間、5分間後に98%Bに到達、3分間保持、流速2ml/分)。検出は、254nm UVおよび210〜600nmの範囲のPDAでTSP UV 6000LP検出器を用いて行った。質量分析計は、陽イオンエレクトロスプレーモードで操作するFinnigan LCQであった。
【0130】
NMR
1H NMRスペクトルは、Bruker DPX 300分光計を300MHzで用いて記録した。化学シフトは、テトラメチルシラン内部標準と比較するδスケールで、100万分の1(ppm)で表した。特に断らない限り、すべての試料はDMSO-d6中に溶解させた。
【0131】
(a) 4-[2-フルオロ-5-(4-オキソ-3,4-ジヒドロ-フタラジン-1-イルメチル)-ベンゾイル]-ピペラジン-1-カルボン酸tert-ブチルエステル(C)
窒素下で室温にあるジメチルアセトアミド(DMA)(3561ml)中の出発物質D(850g)の撹拌溶液に、HBTU(2-(1H-ベンゾトリアゾール-1-イル)-1,1,3,3-テトラメチルウロニウムヘキサフルオロホスフェート)(1402g)を一度に加えた。次いで、温度を15〜25℃に維持しながらヒューニッヒ塩基(iPr2NEt、1096ml)を加え、続いて、温度を15〜25℃に維持しながら1-Bocピペラジン(637g)のDMA(1428ml)溶液を加えた。
【0132】
この溶液を室温で2時間撹拌し、完了についてサンプリングした(HPLC)。完了後、温度を15〜25℃に維持ながら前記溶液を激しく撹拌した水(17085ml)に加え、固体を濾過し、水(2×7131ml)、ヘキサン(2×7131ml)およびメチルtert-ブチルエーテル(MTBE)(2×3561ml)で洗浄した。次いで、固体を一晩乾燥させた後、水分および化学的純度を調べるためサンプリングした。
【0133】
その後、この反応を繰り返した(表を参照)。
【表4】
【0134】
(b) 4-[4-フルオロ-3-(ピペラジン-1-カルボニル)-ベンジル]-2H-フタラジン-1-オン(B)
工業用変性アルコール(IMS)(2200ml)および濃HCl(4400ml)の撹拌溶液に、窒素下で室温にて化合物C(2780.2g)を分割添加した。起泡は添加速度により調節した。次いで、この溶液を15〜25℃で30分間撹拌し、完了についてサンプリングした(HPLC)。
【0135】
完了後、溶液を蒸発させてすべてのIMSを除去し、CH2Cl2(2×3500ml)で水性部を抽出した後、濃縮アンモニアを使用してpHを>8に調節した。得られたスラリーを次に水(10000ml)で希釈し、CH2Cl2(4×3500ml)で抽出し、水(2×2000ml)で洗浄し、MgSO4(250g)により乾燥させ、蒸発させた。粗生成物を次にCH2Cl2(3500ml)でスラリー化し、MTBE(5000ml)に加えた。得られた懸濁液を濾過し、50℃で一晩乾燥させたところ、純度94.12%の物質611.0g(58.5%収率)が得られた。
【0136】
(c) 4-[3-(4-シクロプロパンカルボニル-ピペラジン-1-カルボニル)-4-フルオロ-ベンジル]-2H-フタラジン-1-オン(A)
窒素下のCH2Cl2(15480ml)に溶解した化合物B(1290g)の撹拌懸濁液に、温度を20℃未満に維持しながら、CH2Cl2(1290ml)に溶解のトリエチルアミン(470ml)および塩化シクロプロパンカルボニル(306ml)の予混合溶液を滴下添加した。この溶液を次に10〜15℃で15分間撹拌し、完了についてサンプリングした。この反応混合物は出発物質Bを1.18%しか含んでいないことが確認されたため、反応は完了していると考え、次にこのバッチを後処理した。
【0137】
反応混合物を水(7595ml)、5%クエン酸溶液(7595ml)、5%炭酸ナトリウム溶液(7595ml)および水(7595ml)で洗浄した。次いで、有機層を硫酸マグネシウム(500g)で乾燥させた。
【0138】
次いで、生成物を含有するCH2Cl2層を単離し、セライトにより濾過し、25L容器に充填した。次にCH2Cl2(8445ml)を大気圧で蒸留し、エタノール(10000ml)を加えた。次いで、蒸留は、除去された蒸留物4000ml毎にエタノール(4000ml)と置き換えて、ヘッド温度が73.7℃に達するまで継続した。その後、反応容量を、ヘッド温度が78.9℃に達する時間まで減少し(7730mlまで)、その溶液を一晩8℃に冷却させた。次いで、固体を濾別し、エタノール(1290ml)で洗浄し、一晩70℃で乾燥させた。
【0139】
収量=1377.3g(90%)。HPLC純度(99.34%[area%])。GCによると、4.93%のエタノールと0.45%のCH2Cl2が含有されていた。
【0140】
(d) 化合物Aの水処理
水(13770ml)に溶解した化合物A(実施例1の方法で生産したもの)(1377.0g)の懸濁液を4時間加熱還流し、室温に冷却し、濾過した。固体を水(2754ml)で洗浄し、70℃で一晩乾燥させた。
【0141】
収量=1274.8g(92.6%)。HPLC純度(99.49%[area%])。GCによると、0.01%のエタノールと0.01%のCH2Cl2が含有されていた。
【0142】
水処理後の化合物Aの1H NMRスペクトル(DMSO-d6)を図1に示す。
【0143】
水処理後の化合物Aの粉末X線回折パターンを図2に示すが、これによると化合物が形態Aであることが明らかである。
【0144】
実施例2:1-(シクロプロピルカルボニル)ピペラジンを使用する化合物Aの代替合成
【化11】
【0145】
方法(実施例3および4に関しても参照)
NMR
1H NMRスペクトルは、Bruker DPX 400分光計を400 MHzで用いて記録した。化学シフトは、テトラメチルシラン内部標準と比較するδスケールで100万分の1(ppm)で表した。特に断らない限り、すべての試料はDMSO-d6に溶解させた。
【0146】
質量スペクトル
質量スペクトルは、構造の確認にタンデム型質量分析(MS/MS)を使用して、Agilent XCTイオントラップ質量分析計に記録した。この装置は、陽イオンエレクトロスプレーモードで操作した。
【0147】
(a) 4-[3-(4-シクロプロパンカルボニル-ピペラジン-1-カルボニル)-4-フルオロ-ベンジル]-2H-フタラジン-1-オン(化合物A)
2-フルオロ-5-[(4-オキソ-3,4-ジヒドロフタラジン-1-イル)メチル]安息香酸(D)(15.23g、51.07mmol)をアセトニトリル(96ml)中に窒素下で撹拌しながら懸濁した。ジイソプロピルエチルアミン(19.6ml、112.3mmol)を加えた後、1-シクロプロピルカルボニルピペラジン(I)(9.45g、61.28mmol)およびアセトニトリル(1ml)を加えた。反応混合物を18℃に冷却した。O-ベンゾトリアゾール-1-イル-テトラメチルウロニウムヘキサフルオロホスフェート(25.18g、66.39mmol)を30分かけて加え、反応混合物を室温で2時間撹拌した。反応混合物を3℃に冷却し、この温度を1時間維持した後、濾過した。濾過ケーキを冷却(3℃)アセトニトリル(20ml)で洗浄し、40℃以下で真空乾燥したところ、浅黄色の固体(20.21g)として表題化合物が得られた。
【0148】
質量スペクトル:MH+ 435
1H NMR (400MHz, DMSO-d6) δ: 0.70 (m, 4H), 1.88 (br s, 1H), 3.20 (br s, 2H), 3.56 (m, 6H), 4.31 (s, 2H), 7.17 (t, 1H), 7.34 (dd, 1H), 7.41 (m, 1H), 7.77 (dt, 1H), 7.83 (dt, 1H), 7.92 (d, 1H), 8.25 (dd, 1H), 12.53 (s, 1H)。
【0149】
実施例3:1-(シクロプロピルカルボニル)ピペラジン塩酸塩を使用する化合物Aの代替合成
【化12】
【0150】
(a) 1-(シクロプロピルカルボニル)ピペラジン塩酸塩(I')
窒素下で撹拌しながら、酢酸(700ml)をピペラジン(50.00g、0.581mol)で少量ずつ15分間かけて処理した。反応混合物を40℃まで加温し、完全に溶解されるまでこの温度を維持した。塩化シクロプロパンカルボニル(59.2ml、0.638mol)を15分間かけて加えた。反応混合物を一晩室温で撹拌した。この反応混合物を濾過し、〜430mlの蒸留物が回収されるまで濾液を減圧蒸留した。反応混合物にトルエン(550ml)を加え、さらに400mlの蒸留物が回収されるまで減圧蒸留を続けた。さらに充填用トルエン(550ml)を加え、350mlの蒸留物が回収されるまで減圧蒸留を続けた。得られたスラリーをトルエン(200ml)で希釈し、一晩撹拌した。スラリーを回収するためさらにトルエン(500ml)を加えた。スラリーを濾過し、トルエン(100ml)で洗浄し、40℃で真空乾燥させたところ、表題化合物が白色固体(86.78g)として得られた。
【0151】
質量スペクトル:MH+ 155
1H NMR (400MHz, D2O) δ: 0.92 (m, 4H), 1.98 (m, 1H), 3.29 (m, 2H), 3.38 (m, 2H), 3.84 (m, 2H), 4.08 (m, 2H)。
【0152】
(b) 化合物A
2-フルオロ-5-[(4-オキソ-3,4-ジヒドロフタラジン-1-イル)メチル]安息香酸(D)(0.95g、3.19mmol)をアセトニトリル(4ml)中に窒素下で撹拌しながら懸濁させた。2-(1H-ベンゾトリアゾール-1-イル)-1,1,3,3-テトラメチルウロニウムヘキサフルオロホスフェート(HBTU)(1.45g、3.83mmol)を加え、続いて1-シクロプロピルカルボニルピペラジン塩酸塩(I')(0.73g、3.83mmol)を加えた。ジイソプロピルエチルアミン(1.39ml、7.98mmol)を3分かけて加え、反応混合物を室温で一晩撹拌した。この反応混合物を5℃に冷却し、1時間この温度で維持した後、濾過した。濾過ケーキを冷却(3℃)アセトニトリル(2ml)で洗浄し、次に40℃以下で真空乾燥させ、浅黄色固形物(0.93g)として表題化合物を得た。
【0153】
(c) 水性メタノールからの化合物Aの再結晶
ステップ(b)で得た化合物A(9.40g、21.64mmol)を水(100ml)とメタノール(120ml)の混合物に懸濁させた。この懸濁液を撹拌しながら加熱還流した。次いで、得られた混濁溶液を60℃に冷却し、harborliteパッドを通して濾過した。濾過パッドを水(5ml)とメタノール(5ml)の混合物で洗浄した。115mlの蒸留物が回収されるまで、濾液を大気圧で蒸留した。次いで、蒸留を止め、生じた懸濁液を室温まで冷却させた。得られた懸濁液を〜18時間撹拌した後、濾過した。濾過ケーキを水(20ml)で洗浄した後、60℃以下で真空乾燥させ、白色固体(8.67g)として形態Aの表題化合物を得た。
【0154】
質量スペクトル:MH+ 435
1H NMR (400MHz, DMSO-d6) δ: 0.70 (m, 4H), 1.88 (br s, 1H), 3.20 (br s, 2H), 3.56 (m, 6H), 4.31 (s, 2H), 7.17 (t, 1H), 7.34 (dd, 1H), 7.41 (m, 1H), 7.77 (dt, 1H), 7.83 (dt, 1H), 7.92 (d, 1H), 8.25 (dd, 1H), 12.53 (s, 1H)。
【0155】
(d) 水性エタノールからの化合物Aの再結晶
ステップ(b)で得た化合物A(9.40g、21.64mmol)を水(100ml)とエタノール(50ml)の混合物中に懸濁させた。この懸濁液を撹拌しながら加熱還流した。次いで、得られた混濁溶液を60℃に冷却し、harborliteパッドを通して濾過した。濾過パッドを水(5ml)とエタノール(5ml)の混合物で洗浄した。53mlの蒸留物を回収されるまで濾液を大気圧で蒸留した。次いで、蒸留を止め、生じた懸濁液を室温まで冷却させた。得られた懸濁液を〜18時間撹拌した後、濾過した。濾過ケーキを水(20ml)で洗浄した後、60℃以下で真空乾燥させ、白色固体((8.74g)として形態Aの表題化合物を得た。
【0156】
質量スペクトル:MH+435
1H NMR (400MHz, DMSO-d6) δ: 0.70 (m, 4H), 1.88 (br s, 1H), 3.20 (br s, 2H), 3.56 (m, 6H), 4.31 (s, 2H), 7.17 (t, 1H), 7.34 (dd, 1H), 7.41 (m, 1H), 7.77 (dt, 1H), 7.83 (dt, 1H), 7.92 (d, 1H), 8.25 (dd, 1H), 12.53 (s, 1H)。
【0157】
実施例4:化合物Dの代替合成
【化13】
【0158】
(a) 2-フルオロ-5-[(E/Z)-(3-オキソ-2-ベンゾフラン-1(3H)-イリデン)メチル]ベンゾニトリル(E)
ナトリウムt-アミレート(99.00g、0.854mol)および2-メチルテトラヒドロフラン(960ml)を窒素雰囲気下で2℃に冷却した。温度を5℃未満に維持しながら、亜リン酸ジエチル(110ml、0.855mol)を滴下添加した。2-メチルテトラヒドロフラン(40ml)をラインウォッシュ(line wash)として加えた。この反応系を1時間40分、2℃で撹拌した。2-カルボキシベンズアルデヒド(H)(80g、0.533mol)の2-メチルテトラヒドロフラン(200ml)溶液を、添加中温度を7℃未満に維持しながら加えた。2-メチルテトラヒドロフラン(40ml)のラインウォッシュを加えた。反応混合物を20℃まで加温し、20分間20℃で維持した。メタンスルホン酸(66ml、1.01mol)を1時間10分かけて加え、続いて2-メチルテトラヒドロフラン(40ml)を加えた。この反応混合物を一晩20℃で撹拌した。メタンスルホン酸(7ml、0.101mol)を、続いて2-メチルテトラヒドロフラン(7ml)を加え、この反応物をさらに4時間20℃で撹拌した。室温で水(400ml)を加え、得られた2相性混合物を室温で20分間撹拌した。水性の下層を除去し、有機層に水(400ml)に溶解した炭酸水素カリウム(53.50g、0.534mol)の溶液を加えた。この2相性混合物を20分間室温で撹拌し、次いで、水性の下層溶液を除去した。有機画分が得られた(ジエチル(3-オキソ1,3-ジヒドロ-2-ベンゾフラン-1-イル)ホスホネートの溶液)。この有機画分に2-フルオロ-5-ホルミルベンゾニトリル(64g、0.429mol)を加え、混合物を20℃で撹拌した。トリエチルアミン(66ml、0.473mol)を滴下添加し、続いて2-メチルテトラヒドロフラン(7ml)を加えた。反応混合物を一晩20℃で撹拌し、次いで5℃に冷却し、濾過し、工業用変性アルコール(480ml)で洗浄し、次いで、40℃以下で真空乾燥させ、表題化合物(91.2g)を得た。
【0159】
質量スペクトル:MH+266
1H NMR (400MHz, DMSO-d6) δ: 6.89 (s, 1H, 主要異性体), 6.94 (s, 1H, 副次異性体), 7.40 (dd, 1H, 副次異性体), 7.58 (t, 1H, 両異性体), 7.70 (t, 1H, 両異性体), 7.89 (t, 1H, 両異性体), 7.95 (d, 1H, 両異性体), 8.05 (d, 1H, 両異性体), 8.15 (m, 2H, 主要異性体)。
【0160】
(b) 2-フルオロ-5-[(4-オキソ-3,4-ジヒドロフタラジン-1-イル)メチル]ベンゾニトリル(ED)
2-フルオロ-5-[(E/Z)-(3-オキソ-2-ベンゾフラン-1(3H)-イリデン)メチル]ベンゾニトリル(E)(20g、75.40mmol)およびテトラヒドロフラン(200ml)を30分間窒素雰囲気下の室温で撹拌した。ヒドラジン一水和物(4.40ml、90.53mmol)を加え、続いてテトラヒドロフラン(4ml)のラインウォッシュを加えた。反応混合物を1時間45分室温で撹拌した。酢酸(1.10ml、19.20mmol)を加え、反応混合物を60℃に加温した。反応混合物を一晩60℃で維持した。反応混合物を50℃まで冷却し、水(200ml)を滴下添加した。添加の間、温度を45℃に維持した。反応混合物を20℃に冷却し、濾過し、水(30ml)とテトラヒドロフラン(30ml)の混合物で洗浄し、次に40℃以下で真空乾燥し、表題化合物(18.7g)を得た。
【0161】
質量スペクトル:MH+280
1H NMR (400MHz, DMSO-d6) δ: 4.38 (s, 2H), 7.46 (t, 1H), 7.72 (m, 1H), 7.85 (dt, 1H), 7.92 (m, 2H), 7.99 (d, 1H), 8.27 (dd, 1H), 12.57 (s, 1H)。
【0162】
(c) 2-フルオロ-5-[(4-オキソ-3,4-ジヒドロフタラジン-1-イル)メチル]安息香酸(D)
2-フルオロ-5-[(4-オキソ-3,4-ジヒドロフタラジン-1-イル)メチル]ベンゾニトリル(ED)(9.60g, 34.37mmol)および水(40ml)を20℃で撹拌した。2Mの水酸化ナトリウム(36ml、72.00mmol)を加え、反応混合物を90℃に加温し、一晩この温度で維持した。反応混合物を室温まで冷却し、濾過した。濾過パッドを水(10ml)で洗浄し、合わせた濾液を60℃で40分間かけて2MのHCl(56ml、112.00mmol)に加えた。得られた懸濁液を50℃まで冷却し、濾過し、水(57ml)で洗浄し、60℃以下で真空乾燥させ、白色固体(9.72g)として表題化合物を得た。
【0163】
質量スペクトル:MH+299
1H NMR (400MHz, DMSO-d6) δ: 4.36 (s, 2H), 7.24 (dd, 1H), 7.59 (m, 1H), 7.84 (dt, 2H), 7.90 (dt, 1H), 7.98 (d, 1H), 8.27 (dd, 1H), 12.59 (s, 1H), 13.22 (br s, 1H)。
【0164】
実施例5:水性エタノールからの化合物Aの再結晶
4-(3-{[4-(シクロプロピルカルボニル)ピペラジン-1-イル]カルボニル}-4-フルオロベンジル)フタラジン-1(2H)-オン(化合物A)(20.00g、44.66mmol)を、水(50ml)とエタノール(150ml)の混合物に懸濁した。この懸濁液を撹拌しながら加熱還流した。次に得られた溶液を70℃まで冷却し、濾過した。濾過パッドを水(8ml)とエタノール(22ml)の混合物で洗浄した。
【0165】
濾液を撹拌しながら45℃まで冷却した。混合物に種結晶を与えるために形態Aの4-(3-{[4-(シクロプロピルカルボニル)ピペラジン-1-イル]カルボニル}-4-フルオロベンジル)フタラジン-1(2H)-オン(化合物A)(0.08g)を加えた。得られた懸濁液を2.5時間かけて20℃まで冷却し、さらに16時間この温度で撹拌し、結晶化させた。温度を20℃に維持しながら、5時間かけて水(200ml)を加えた。添加終了後、懸濁液を2時間20℃で維持した。
【0166】
懸濁液を濾過し、濾過ケーキをエタノール(24ml)と水(56ml)の混合物で洗浄した。単離した固体を取り出し、40〜60℃で真空乾燥し、白色固体(18.1g)として表題化合物(形態A)を得た。
【0167】
図3〜5を得る方法
粉末X線回折− 図3(形態Aとしての化合物A)
粉末X線回折は、Bruker D5000回折装置により記録した(X線波長1.5418Å Cu線源、電圧40kV、フィラメント放出40mA)。試料は、2〜40°(2θ)の範囲で、0.02°のステップ幅および4秒のタイムカウントでスキャンした。
【0168】
粉末X線回折−図4(溶媒和形態としての化合物A)
溶媒和物ファミリーの粉末X線回折は、湾曲型の位置敏感型検出器(範囲120°(2θ))を取り付けたInel XRG-3000回折装置により記録した(X線波長1.5418Å Cu線源、電圧40kV、フィラメント放出30mA)。試料は、2.5〜40°(2θ)の範囲で、0.03°のステップ幅および一般に300秒の総回収時間でスキャンした。
【0169】
示差走査熱量測定法(DSC)−図5
DSCは、TSO801ROロボットシステムを備えたMettler DSC820Eを使用して記録した。通常、5mg未満の物質(穴の開いた蓋が取り付けられている40μLアルミニウム皿に入れたもの)を毎分10℃の一定の加熱速度で25℃〜325℃の温度範囲で加熱した。窒素パージガスを100ml/分の流速で使用した。
【0170】
実施例6
阻害作用
本活性化合物の阻害作用を評価するため、以下のアッセイを用いてIC50値を求めた。
【0171】
HeLa細胞核抽出物から単離された哺乳動物のPARPを、96ウェルFlashPlates(商標)(NEN、UK)中でZバッファー(25mM Hepes(Sigma);12.5mM MgCl2(Sigma);50mM KCl(Sigma);1mM DTT(Sigma);10%グリセロール(Sigma);0.001%NP-40(Sigma);pH7.4)とともにインキュベートし、様々な濃度の前記阻害剤を添加した。すべての化合物をDMSOで希釈し、最終アッセイ濃度を10〜0.01μMとした。DMSOの最終濃度は1%/ウェルであった。ウェル当たりの全アッセイ容量は40μlであった。
【0172】
30℃で10分間インキュベーションした後、NAD(5μM)、3H-NADおよび30mer二重鎖DNAオリゴを含有する反応混合物10μlを添加して反応を開始した。酵素活性(%)を計算するため、指定の正および負反応ウェルを化合物ウェル(未知)と組み合わせた。次いで、プレートを2分間振盪し、30℃で45分間インキュベートした。
【0173】
インキュベーション後、30%酢酸50μlを各ウェルに添加することにより反応をクエンチした。次いで、プレートを室温で1時間振盪した。
【0174】
プレートをTopCount NXT(商標)(Packard、UK)に移し、シンチレーションをカウントした。記録された値は、各ウェルを30秒カウントした後のカウント/分(cpm)である。
【0175】
次いで、各化合物の酵素活性(%)を以下の式によって計算する:
阻害率(%)=100−[100×(未知のcpm−平均負cpm)/(平均正cpm−平均負cpm)]
【0176】
IC50値(酵素活性の50%が阻害される濃度)を計算した。IC50値は異なる濃度の範囲、通常は10μM〜0.001μMにわたって求められる。かかるIC50値は、化合物の効力が増加したことを確認するための比較値として用いる。
【0177】
化合物Aは、IC50が約5nMであった。
【0178】
増強ファクター(Potentiation Factor)
活性化合物の増強ファクター(PF50)は、対照細胞増殖のIC50を細胞増殖+PARP阻害剤のIC50で割った比として計算される。対照と化合物で処理された細胞の両方の増殖阻害曲線は、アルキル化剤メタンスルホン酸メチル(MMS)の存在下のものである。試験化合物は0.2マイクロモルの固定濃度で使用した。MMSの濃度は0〜10μg/mlであった。
【0179】
細胞増殖をスルホローダミンB(SRB)アッセイによって評価した(Skehan, P.ら, (1990) New colorimetric cytotoxicity assay for anticancer-drug screening. J. Natl. Cancer Inst. 82, 1107-1112)。2,000個のHeLa細胞を平底96ウェルマイクロタイタープレートの各ウェルに100μlの容量で播種し、37℃で6時間インキュベートした。細胞を培地単独で置きかえるか、最終濃度が0.5、1もしくは5μMのPARP阻害剤含有培地で置き換えた。細胞をさらに1時間増殖させ、その後、一連の濃度(一般に0、1、2、3、5、7および10μg/ml)のMMSを未処理細胞またはPARP阻害剤処理細胞に添加した。PARP阻害剤のみで処理した細胞を使用して、PARP阻害剤による増殖阻害を評価した。
【0180】
細胞をさらに16時間静置し、その後培地を交換し、細胞を37℃でさらに72時間増殖させた。次いで培地を取り出し、細胞を氷冷10%(w/v)トリクロロ酢酸100μlで固定した。プレートを4℃で20分間インキュベートし、次いで水で4回洗浄した。次いで、各ウェルの細胞を0.4%(w/v)SRBの1%酢酸溶液100μlで20分間染色した後、1%酢酸で4回洗浄した。次いで、プレートを室温で2時間乾燥させた。10mM Tris Base 100μlを各ウェルに添加して、染色細胞からの色素を溶解させた。プレートを軽く振盪し、室温で30分間静置した後、Microquantマイクロタイタープレートリーダーを用いて564nMの光学濃度を測定した。
【0181】
化合物Aは、200nMにおけるPF50は少なくとも2.0であった。
【技術分野】
【0001】
本発明は、特定のフタラジノン誘導体の結晶形態および改良型合成方法、前記合成における中間体および医薬組成物、ならびに前記結晶形態の使用に関する。
【背景技術】
【0002】
哺乳動物の酵素PARP(113kDaのマルチドメインタンパク質)は、DNA一本鎖または二本鎖の切断部を認識し、それに迅速に結合する能力により、DNA損傷のシグナル伝達に関係しているとされている(非特許文献1)。
【0003】
いくつかの所見から、遺伝子増幅、細胞分裂、分化、アポトーシス、DNA塩基除去修復ならびにテロメア長および染色体安定性に及ぼす影響をはじめとする様々なDNA関連機能にPARPが関与しているという結論が得られている(非特許文献2)。
【0004】
PARPがDNA修復および他のプロセスをモジュレートする機序についての研究から、細胞核内でのポリ(ADP-リボース)鎖の形成におけるその重要性が明らかにされた(非特許文献3)。DNAに結合した活性型のPARPは、NADを利用して、種々の核標的タンパク質(トポイソメラーゼ、ヒストンおよびPARPそのものを含む)上でポリ(ADP-リボース)を合成する(非特許文献4)。
【0005】
ポリ(ADP-リボシル)化は、悪性形質転換とも関係していた。例えば、PARP活性は、SV40形質転換線維芽細胞の単離核においてより高く、一方、白血病細胞および結腸癌細胞のいずれもが、対応する正常な白血球および結腸粘膜よりも高い酵素活性を示す(非特許文献5;非特許文献6;および非特許文献7)。
【0006】
DNA修復におけるポリ(ADP-リボシル)化の機能的役割を解明するために、数種類の低分子量のPARP阻害剤が用いられた。アルキル化剤で処理した細胞では、PARPの阻害により、DNA鎖の切断と細胞の死滅が顕著に増加する(非特許文献8;非特許文献9)。
【0007】
その後、かかる阻害剤は、致死に至り得る損傷の修復を抑制することにより、放射線応答の効果を高めることが明らかになった(非特許文献10;非特許文献11)。PARP阻害剤は、低酸素性腫瘍細胞を放射線感受性にするのに効果的であると報告されている(特許文献1;特許文献2および特許文献3)。
【0008】
さらに、PARPノックアウト(PARP-/-)動物は、アルキル化剤およびγ線照射に応答してゲノムの不安定性を示す(非特許文献12;非特許文献13)。
【0009】
PARPの役割は、特定の血管疾患、敗血症性ショック、虚血性障害および神経毒性においても実証されている (非特許文献14;非特許文献15)。DNAの鎖切断が生じる酸素ラジカルDNA損傷(これは後にPARPによって認識される)は、PARP阻害剤の研究により明らかなように、かかる疾患状態の主な寄与因子である(非特許文献16;非特許文献17)。さらに最近では、PARPが出血性ショックの発病においてある役割を果たすことが実証されている(非特許文献18)。
【0010】
また、哺乳動物細胞への効率的なレトロウイルス感染がPARP活性の阻害により遮断されることも実証された。かかる組換えレトロウイルスベクター感染の阻害は、様々な異なる細胞型において起こることが明らかにされた(非特許文献19)。かくして、抗ウイルス療法および癌治療で使用するために、PARP阻害剤が開発された(特許文献4)。
【0011】
さらに、PARP阻害はヒト線維芽細胞における老化特性の発症を遅延させるということが推察されている(非特許文献20)。このことは、PARPがテロメア機能の制御において果たす役割と関連している可能性がある(非特許文献2)。
【0012】
特許文献5には、多数のフタラジノン誘導体、PARP阻害におけるそれらの活性、および放射線療法もしくは化学療法の補助剤(adjunct)としての、または単独型の薬剤としての癌治療におけるそれらの重要な用途が開示されている。
【0013】
特許文献6には、塩基除去修復(BER)阻害剤としてのPARP阻害剤、具体的にはフタラジノン誘導体の使用が開示されている。また相同組換え(HR)依存的DNA DSB修復活性に欠損のある癌(特に、BRCA1および/またはBRCA2欠損表現型を有する癌)の治療用製剤の製造におけるこれらの阻害剤の使用が開示されている。
【0014】
特許文献5に記載されている4-[3-(4-シクロプロパンカルボニル-ピペラジン-1-カルボニル)-4-フルオロ-ベンジル]-2H-フタラジン-1-オン(化合物A):
【化1】
【0015】
は、特に注目されている。
【0016】
特許文献5では、化合物Aは、多数のライブラリー化合物の1つとして、4-[4-フルオロ-3-(ピペラジン-1-カルボニル)-ベンジル]-2H-フタラジン-1-オン(化合物B):
【化2】
【0017】
から、ジクロロメタン中の(B)の溶液に塩化シクロプロパンカルボニル:
【化3】
【0018】
を添加し、続いてヒューニッヒ塩基(N,N-ジイソプロピルエチルアミン)を添加することにより合成された。この反応は室温で撹拌しながら16時間行われ、得られた化合物は分取HPLCにより精製される。
【0019】
ピペラジン誘導体(B)は、4-[2-フルオロ-5-(4-オキソ-3,4-ジヒドロ-フタラジン-1-イルメチル)-ベンゾイル]-ピペラジン-1-カルボン酸tert-ブチルエステル(化合物C):
【化4】
【0020】
を1時間、6M HClおよびエタノールを使用することにより脱保護し、次にpH9までアンモニアで塩基性化し、ジクロロメタンへ抽出することにより調製された。
【0021】
Boc保護ピペラジン誘導体(C)は、2-フルオロ-5-(4-オキソ-3,4-ジヒドロ-フタラジン-1-イルメチル)-安息香酸(化合物D):
【化5】
【0022】
から、ピペラジン-1-カルボン酸tert-ブチルエステル:
【化6】
【0023】
、2-(1H-ベンゾトリアゾール-1-イル)-1,1,3,3-テトラメチルウロニウムヘキサフルオロホスフェート(HBTU)、およびN,N,-ジイソプロピルエチルアミンをジメチルアセトアミド中に添加し、続いて18時間撹拌することにより調製された。
【先行技術文献】
【特許文献】
【0024】
【特許文献1】米国特許第5032617号
【特許文献2】米国特許第5215738号
【特許文献3】米国特許第5041653号
【特許文献4】WO 91/18591
【特許文献5】WO 2004/080976
【特許文献6】WO 2005/053662
【非特許文献】
【0025】
【非特許文献1】D'Amoursら, Biochem. J., 342, 249-268 (1999)
【非特許文献2】d'Adda di Fagagnaら, Nature Gen., 23(1), 76-80 (1999)
【非特許文献3】Althaus, F.R.およびRichter, C., ADP-Ribosylation of Proteins: Enzymology and Biological Significance, Springer-Verlag, Berlin (1987)
【非特許文献4】Rhunら, Biochem. Biophys. Res. Commun., 245, 1-10 (1998)
【非特許文献5】Miwaら, Arch. Biochem. Biophys., 181, 313-321 (1977)
【非特許文献6】Burzioら, Proc. Soc. Exp. Bioi. Med., 149, 933-938 (1975)
【非特許文献7】Hiraiら, Cancer Res., 43, 3441-3446 (1983)
【非特許文献8】Durkaczら, Nature, 283, 593-596 (1980)
【非特許文献9】Berger, N.A., Radiation Research, 101, 4-14 (1985)
【非特許文献10】Ben-Hurら, British Journal of Cancer, 49 (Suppl. VI), 34-42 (1984)
【非特許文献11】Schlickerら, Int. J. Radiat. Bioi., 75, 91-100 (1999)
【非特許文献12】Wangら, Genes Dev., 9, 509-520 (1995)
【非特許文献13】Menissier de Murciaら, Proc. Natl. Acad. Sci. USA, 94, 7303-7307 (1997)
【非特許文献14】Cantoniら, Biochim. Biophys. Acta, 1014, 1-7 (1989)
【非特許文献15】Szaboら, J. Clin. Invest., 100, 723-735 (1997)
【非特許文献16】Cosiら, J. Neurosci. Res., 39, 38-46 (1994)
【非特許文献17】Saidら, Proc. Natl. Acad. Sci. U.S.A., 93, 4688-4692 (1996)
【非特許文献18】Liaudetら, Proc. Natl. Acad. Sci. U.S.A., 97(3), 10203-10208 (2000)
【非特許文献19】Gakenら, J. Virology, 70(6), 3992-4000 (1996)
【非特許文献20】RattanおよびClark, Biochem. Biophys. Res. Comm., 201(2), 665-672 (1994)
【発明の概要】
【発明が解決しようとする課題】
【0026】
化合物Aの特定の形態は、例えば、その溶解度および/またはその安定性および/またはそのバイオアベイラビリティおよび/またはその不純物プロファイルおよび/またはその濾過特性および/またはその乾燥特性および/またはその吸湿性欠如に関して有利な特性を有している可能性がある。また/あるいは、前記形態は取り扱い易く、かつ/または微粉末化し易く、かつ/または錠剤へ製剤化し易い可能性がある。さらに、化合物Aを大容量スケールで合成するのに適した改良型合成方法も望まれている。
【課題を解決するための手段】
【0027】
したがって、本発明の第1の態様は、実質的に結晶形態の、具体的には形態Aの4-[3-(4-シクロプロパンカルボニル-ピペラジン-1-カルボニル)-4-フルオロ-ベンジル]-2H-フタラジン-1-オン(化合物A)を提供する。
【図面の簡単な説明】
【0028】
【図1】水処理後の化合物AのNMRを示す(実施例1)。
【図2】水処理後の形態Aとしての化合物Aの粉末X線回折パターンを示す (実施例1)。
【図3】形態Aとしての化合物Aの代表的な粉末X線回折パターンを示す。
【図4】溶媒和された形態としての化合物Aの代表的な粉末X線回折パターンを示す。
【図5】25℃〜325℃まで毎分10℃ずつ加熱することにより得られた、形態Aとしての化合物Aの代表的なDSCトレースを示す。
【発明を実施するための形態】
【0029】
上記で用いた「実質的に結晶形態」とは、化合物Aの少なくとも50重量%、好ましくは少なくとも70重量%、80重量%または90重量%が結晶形態であることを意味する。一部の実施形態では、少なくとも95重量%、99重量%または99.5重量%以上が結晶形態であってもよい。
【0030】
結晶形態Aとしての化合物Aは、次の特定のピークを有するX線回折パターン(λ=1.5418Å)を有する。
【表1】
【0031】
結晶形態Aとしての化合物Aは、次の追加ピークのX線回折パターン(λ=1.5418Å)も有する。
【表2】
【0032】
結晶形態Aとしての化合物Aは、上記リストの10のピークから選択される3つ以上のピークの任意の組み合わせを特徴とすることもできる。
【0033】
形態Aとしての化合物Aの代表的な粉末X線回折パターンを図3に示す。
【0034】
理論に制約されることを望むものではないが、化合物Aは、溶媒分子がその結晶格子内の位置を占めることができる構造を容易に形成し得る。かかる溶媒和物は、実際は必ずしも化学量論的ではないが、1種の純粋な溶媒和物から成っていてもよく(例えば、化合物Aメタノレートおよび化合物Aテトラヒドロフラネート)、あるいは可能性として、1種以上の溶媒成分(例えば、メタノールおよびジエチルエーテル)から成っていてもよい。溶媒分子は、典型的には、化合物A分子によって形成されているポケット内に存在する。ある状況では、これらのポケットの容量は一連の溶媒を包含するのに十分な程度に柔軟性があるため、物質の全体構造としての変化はほとんどなく、それ故、XRPD反射での変化はごく小さい。
【0035】
溶媒和物(同じ全体構造を共有するものを含む)は、純粋な溶媒としての、または他の溶媒と組み合わせた場合のジクロロメタン、酢酸エチル、メタノール、エタノール、イソプロパノール、2-ブタノン、t-ブチルメチルエーテル、トルエン、テトラヒドロフラン、水、シクロヘキサン、シクロプロピルメチルケトン、1,2-ジクロロエタン、エチルトリフルオロアセテート、フルオロベンゼンヘキサフルオロ-イソ-プロパノール、メチルノナフルオロブチルエーテル、2-メチル-1-プロパノール、ニトロメタン、プロピオニトリル、トリクロロエチレン、α,α,α-トリフルオロトルエン、ヘプタン、ジオキサン、アセトニトリルから溶液熟成(solution maturation)および結晶化実験により生じる。最も一般的な溶媒和物構造のX線回折パターンを図4に示すが、典型的には下記の位置で極めて強いピークを有する。
【表3】
【0036】
図に示したピークの相対強度は、試験の際の試料の方向、使用した器具の種類および設定により変わる可能性があるため、本明細書に記載のXRDトレースの強度は例示であり、絶対的な比較に用いることを意図するものではないことは理解されよう。
【0037】
化合物Aの形態Aは、溶媒を実質的に含まない。本明細書で使用する「溶媒を実質的に含まない」という用語は、極く僅かな量の任意の溶媒を有する形態、例えば総量で0.5重量%以下の任意の溶媒を含む形態を意味する。任意の溶媒(水を含む)の総量は、0.25重量%、0.1重量%、0.05重量%または0.025重量%以下であってもよい。
【0038】
また、化合物Aの形態Aは、示差走査熱量測定(DSC)を使用して特徴を決定することもできる。化合物Aの形態Aは、25℃〜325℃まで毎分10℃ずつ加熱した時、210.1℃±1℃で融解し始める。形態Aとしての化合物Aの代表的なDSCトレースを図5に示す。
【0039】
本発明の第2の態様は、結晶形態Aとしての4-[3-(4-シクロプロパンカルボニル-ピペラジン-1-カルボニル)-4-フルオロ-ベンジル]-2H-フタラジン-1-オン(化合物A)を得る方法であって、化合物Aを溶媒中で結晶化し、次に、置換剤を用いて前記結晶形態から溶媒を置換することを含む前記方法を提供する。置換剤は、水、またはC1-2アルコールと水の混合物であってもよい。
【0040】
第1の実施形態では、本方法は、
(i) 4-[3-(4-シクロプロパンカルボニル-ピペラジン-1-カルボニル)-4-フルオロ-ベンジル]-2H-フタラジン-1-オン(化合物A)を溶媒から結晶化するステップと;
(ii) 最初の溶媒がエタノールでない場合、結晶性化合物Aをエタノールで処理するステップと;
(iii) 結晶性化合物Aを水で処理し、封入されているエタノールを除去するステップと;
(iv) 得られた生成物を乾燥させるステップ
を含む。
【0041】
最初の結晶化で用いられる溶媒は、例えば、ジクロロメタンまたはアセトニトリルであってもよい。
【0042】
形態Aを得る方法は、一般に、溶媒置換を包含し得る。化合物Aは、溶媒を封入可能な結晶格子内チャンネルが形成されるように結晶化されるため、溶媒を除去することが困難になることが明らかとなっている。
【0043】
第1の実施形態の方法は、化合物Aの結晶化で使用される溶媒がジクロロメタンである場合に特に用いることができる。溶媒のジクロロメタンを溶媒のエタノールと交換するステップは、エタノールの存在下、大気圧で化合物Aの溶液を蒸留することにより行なうことができる。この交換は、ヘッド温度がエタノールの沸点(例えば、少なくとも73℃)に近づいた時に完了する。特にこの交換は、大部分のDCMを蒸留し、次いで、大量のエタノールを加えることにより行なうことができる。次いで、蒸留物のバッチを等量のエタノールで置き換えながら、蒸留を継続する。
【0044】
化合物Aのエタノール溶媒からの結晶化は、15℃以下に(好ましくは10℃未満、さらに好ましくは約8℃に)溶液を冷却することにより行うことができる。次いで、化合物Aの結晶を濾過により溶液から取り出すことができる。
【0045】
結晶性化合物Aは、水に結晶性物質を懸濁し、十分な時間(例えば、少なくとも3時間、好ましくは約4時間)加熱還流することにより、水で処理して封入されたエタノールを除去することができる。結晶性化合物Aは、濾過により水中懸濁液から取り出すことができる。
【0046】
上記ステップで得られた生成物は、容易に乾燥させることができる。例えば、少なくとも60℃、好ましくは約70℃の温度の加熱器で生成物を加熱することにより行われる。
【0047】
別のかかる実施形態では、本方法は、
(i) 溶媒を含有する結晶形態としての化合物Aを得るステップと;
(ii) 結晶形態の化合物Aの合成で使用される最初の溶媒が水とC1-2アルコール(すなわち、メタノール、エタノール)の混合物でない場合、結晶形態の化合物Aを水とC1-2アルコールの混合物で処理するステップと;
(ii) 得られた生成物を乾燥させるステップ
を含む。
【0048】
得られた生成物は、さらに水とC1-2アルコールの混合物で処理し、乾燥させ、さらに結晶形態Aの化合物Aを単離するすることができる。
【0049】
水とC1-2アルコールの混合物は、容量で2:1〜1:2の範囲にあるのが好ましく、さらには容量で1.5:1〜1:1.5の範囲にあるのが好ましい。特に好ましい混合物は、1部の水に対し1.2部のC1-2アルコールである。別の特に好ましい混合物は、2部の水に対し1部のC1-2アルコールである。C1-2アルコールはエタノールが好ましい。
【0050】
結晶形態としての化合物Aは、上記のように、化合物Aを溶媒から結晶化することにより得ることができる。
【0051】
ステップ(ii)の溶媒処理は、水とC1-2アルコールの混合物中に化合物Aを懸濁し、撹拌しながら加熱還流することにより行うことができる。続いてこれを55〜65℃に冷却し、例えばセライトパッドを通して濾過することができる。濾過パッドを水とC1-2アルコールの混合物で洗浄した後、周囲圧力(通常1気圧)以上で蒸留することができる。蒸留を止めて懸濁液を得て、これを室温に置いた後、濾過することができる。得られた濾過ケーキは、水で洗浄することができる。
【0052】
上記のステップで得られた生成物は、容易に乾燥することができる。例えば、少なくとも50℃、好ましくは約60℃の温度の加熱器で生成物を加熱することによって行われる。
【0053】
さらなる処理は、上記の方法と類似の方法で進めることができる。
【0054】
第3の実施形態では、本方法は、
(i) 溶媒としての水とC1-2アルコールの混合物中に化合物Aを懸濁するステップと;
(ii) 懸濁液を加熱還流するステップと;
(iii) 溶液を冷却し、形態Aとしての化合物Aを種結晶として加える(seeding)ステップと;
(iv) 得られた生成物を乾燥させるステップ
を含む。
【0055】
得られた生成物は、さらに水とC1-2アルコールの混合物で処理し、乾燥して、さらに結晶形態Aの化合物Aを単離することができる。
【0056】
水とC1-2アルコールの混合物は、容量で2:1〜1:5の範囲にあるのが好ましく、さらには容量で1:2〜1:4の範囲にあるのが好ましい。特に好ましい混合物は、1部の水に対し3部のC1-2アルコールである。C1-2アルコールはエタノールが好ましい。
【0057】
ステップ(iii)は、溶液を65〜75℃(例えば70℃)に冷却し、例えば、セライトパッドを通して濾過することを含む。濾過パッドを水とC1-2アルコールの混合物で洗浄した後、(周囲圧力以上で)蒸留することができる。種結晶の投与(seeding)は、得られた濾液が40〜50℃(例えば45℃)に冷却された後に始まり得る。得られた懸濁液は、2〜3時間(例えば2.5時間)で周囲温度(例えば20℃)まで冷却し、結晶化されるのに十分な時間、前記温度で維持することができる。この時間は12〜24時間であってもよく、約16時間であってもよい。この時間が終わったら、さらに水を加えることができる。その量は、存在する全溶媒(水およびC1-2アルコール)の容量とほぼ等しく、ゆっくりと(例えば、4〜6時間、例えば5時間かけて)加えることができる。水の添加後、その懸濁液は、例えば2時間、周囲温度で維持することができる。
【0058】
次いで、その懸濁液を濾過し、得られた濾過ケーキをC1-2アルコールと水の混合物(1:3〜1:2、例えば1:2.3の割合)で洗浄することができる。
【0059】
上記のステップで得られた生成物は、容易に乾燥することができる。例えば、40℃〜60℃の温度の真空下の加熱器で生成物を加熱することによって行われる。
【0060】
本発明の第3の態様は、化合物Bから化合物Aを合成する方法であって、
(i) 制御下で、適切な有機溶媒(例えば、DCM(ジクロロメタン))に溶解したトリエチルアミンおよび塩化シクロプロパンカルボニルの予混合溶液を、同一有機溶媒中の化合物Bに、溶液の温度が20℃未満となるように制御しながら添加するステップ含む前記方法を提供する。
【0061】
一部の実施形態では、本方法は、
(ii) (i)で得られた溶液を、溶液の温度を20℃以下に維持しながら反応が完了するまでかき混ぜる(例えば、撹拌する)ステップを含む。
【0062】
ステップ(i)の添加は、滴下法で実施することができる。
【0063】
この方法はWO 2004/080976に記載されている方法よりもより一層制御されているため、酸塩化物の付加がより位置選択的なものとなる。制御のほとんどない先行技術の方法では、酸塩化物の付加が所望のピペリジン窒素だけでなく、フタラジノンの窒素および/または酸素に起こる可能性がある。
【0064】
上記の方法は窒素雰囲気下で行なうのが好ましい。
【0065】
段階(ii)の溶液の温度は10〜15℃の間に維持されるのがさらに好ましい。
【0066】
上記反応の生成物は、少なくとも1回の水洗浄ステップにより後処理されるのが好ましい。より好ましくは、後処理は、最初と最終の水洗浄ステップと、希酸(例えば5%クエン酸溶液)を使用した後、希塩基(例えば5%炭酸ナトリウム溶液)を使用する中間洗浄ステップを含む。
【0067】
本発明の第4の態様は、化合物Dからの化合物Aを合成する方法であって、アミドカップリング剤および塩基、例えばアミン(具体的にはジイソプロピルエチルアミンなどの第三級アミンなど)の存在下で、化合物Dを1-(シクロプロピルカルボニル)ピペラジンまたはその鉱酸塩と反応させることを含む前記方法を提供する。
【0068】
鉱酸塩は、例えば塩酸塩であり得る。
【0069】
1-(シクロプロピルカルボニル)ピペラジンまたはその鉱酸塩の化合物Dへの添加は、任意の好適な溶媒(例えばアセトニトリル)中で行なうことができる。アミドカップリング剤は、好ましくは、2-(1H-ベンゾトリアゾール-1-イル)-1,1,3,3-テトラメチルウロニウムヘキサフルオロホスフェート(HBTU)である。好ましくは、それを1-(シクロプロピルカルボニル)ピペラジンまたはその鉱酸塩、ジイソプロピルエチルアミンおよび化合物Dの溶液に一定時間(例えば30分)かけて添加する。得られる溶液の温度は、25℃以下(または20℃以下、例えば18℃)で維持することができる。この添加後、得られた溶液を一定時間静置することができる。好ましい温度体制は、この溶液を室温で2時間維持することである。
【0070】
生じた化合物Aは、一定時間(例えば1時間)、10℃未満(または5℃未満、例えば3℃)に冷却し、その後濾過することにより、溶液から取り出すことができる。生じた化合物Aは、例えば冷却アセトニトリルで洗浄することができる。
【0071】
WO 2004/080976には、次の化合物Dへの経路が開示されている。
【化7】
【0072】
亜リン酸ジメチルをナトリウムメトキシドのメタノール溶液に0℃で滴下添加した。次いで、温度を5℃未満に維持しながら、2-カルボキシベンズアルデヒド(H)をメタノールスラリーとしてその反応混合物に分割添加した。得られた淡黄色溶液を20℃に1時間かけて加温した。メタンスルホン酸をその反応物に滴下添加し、得られた白色懸濁液を減圧蒸発させた。その白色残渣を水でクエンチし、クロロホルムで抽出した。この有機抽出物を合わせて水で洗浄し、MgSO4を用いて脱水し、減圧蒸発させて、白色固体として(3-オキソ-1,3-ジヒドロ-イソベンゾフラン-1-イル)ホスホン酸ジメチルエステル(G)(収率:95%)を得た。次いで、これをさらに精製せずに次の段階に使用した。
【0073】
(3-オキソ-1,3-ジヒドロ-イソベンゾフラン-1-イル)ホスホン酸ジメチルエステル(G)のテトラヒドロフラン溶液と2-フルオロ-5-ホルミルベンゾニトリル(F)のテトラヒドロフラン溶液の混合物に、温度を15℃未満に維持しながらトリエチルアミンを25分かけて滴下添加した。その反応混合物を1時間かけて徐々に20℃に加温し、減圧濃縮した。その白色残渣を水に30分間スラリー化し、濾過し、水、へキサンおよびエーテルで洗浄し、乾燥し、E異性体とZ異性体の50:50混合物として2-フルオロ-5-(3-オキソ-3H-イソベンゾフラン-1-イリデンメチル)ベンゾニトリル(E)(収率:96%)を得た。
【0074】
2-フルオロ-5-(3-オキソ-3H-イソベンゾフラン-1-イリデンメチル)ベンゾニトリル(E)の水中懸濁液に水酸化ナトリウム水溶液を添加し、その反応混合物を窒素下で90℃に30分間加熱した。その反応混合物を多少冷却して70℃とし、ヒドラジン水和物を添加し、70℃で18時間撹拌した。その反応物を室温に冷却し、2M HClを用いてpH4に酸性化した。その混合物を10分間撹拌し、濾過した。得られた固体を水、へキサン、エーテル、酢酸エチルで洗浄し、乾燥させ、淡いピンク色粉末の化合物D(収率:77%)を得た。
【0075】
また、化合物Dの改良型合成方法も望まれている。
【0076】
したがって、本発明の第5の態様は、化合物Dの合成方法であって、
(a) ジエチル(3-オキソ-1,3-ジヒドロ-2-ベンゾフラン-1-イル)ホスホネート(G')を2-カルボキシベンズアルデヒド(H)から合成するステップと;
(b) 2-フルオロ-5-[(E/Z)-(3-オキソ-2-ベンゾフラン-1(3H)-イリデン)メチル]ベンゾニトリル(E)をジエチル(3-オキソ-1,3-ジヒドロ-2-ベンゾフラン-1-イル)ホスホネートから合成するステップ
を含む方法を提供する。
【0077】
化合物G'は合成中に単離されないのが好ましい。この方法は、アルコール溶液中で不安定である亜リン酸ジメチルのナトリウム塩(Pelchowiczら, J.Chem.Soc, 4348-4350 (1961))の使用を避ける。ステップ(a)は、亜リン酸ジエチルのナトリウム塩が安定する2-メチルテトラヒドロフラン中で生じるのが好ましい。この塩は、2-メチルテトラヒドロフランに溶解したナトリウムt-アミレートの冷却溶液に亜リン酸ジエチルを添加することによりin situで形成することができる。亜リン酸ジエチルのナトリウム塩と反応させた後、メタンスルホン酸との反応を行うことができる。
【0078】
ステップ(b)は、2-メチルテトラヒドロフラン中でトリエチルアミンの添加により行うことができる。
【0079】
化合物Dの合成方法は、さらに、
(c) 化合物Eからヒドラジン水和物との反応により2-フルオロ-5-[(4-オキソ-3,4-ジヒドロフタラジン-1-イル)メチル]ベンゾニトリル(ED):
【化8】
【0080】
を合成するステップと;
(d) 化合物EDから水酸化ナトリウムとの反応により化合物Dを合成するステップ
を含んでいてもよい。
【0081】
ステップ(c)は、テトラヒドロフラン中の1.1〜1.3当量のヒドラジン水和物を使用し、続いて、過剰のヒドラジン水和物を酢酸を用いて中和させることにより実施することができる。
【0082】
本発明の第6の態様は、化合物ED:
【化9】
【0083】
および化合物Dの合成におけるその使用を提供する。
【0084】
本発明のさらなる態様は、1-(シクロプロピルカルボニル)ピペラジンの鉱酸塩、および、ピペラジンを酢酸と反応させ、その後、塩化シクロプロパンカルボニルを添加することによりそれを合成する方法を提供する。
【0085】
本発明の第7の態様は、第1の態様の化合物と製薬上許容可能な担体または希釈剤を含む医薬組成物を提供する。
【0086】
本発明の第8の態様は、ヒトまたは動物の身体の治療方法で使用するための第1の態様の化合物を提供する。
【0087】
本発明の第9の態様は、PARP活性の阻害剤の製造における本発明の第1の態様に定義した化合物の使用を提供する。
【0088】
本発明のさらなる態様は、血管疾患;敗血症性ショック;虚血性障害;神経毒性;出血性(haemorraghic)ショック;ウィルス感染;またはPARP活性を阻害することより寛解される疾患の治療用薬剤の製造における本発明の第1の態様に定義した化合物の使用を提供する。
【0089】
本発明の別のさらなる態様は、癌治療において補助剤として使用するための薬剤または電離放射線もしくは化学療法剤による治療で腫瘍細胞を増強するための薬剤の製造における本発明の第1の態様で定義した化合物の使用を提供する。
【0090】
本発明のさらなる別の態様は、PARPの阻害により寛解される疾患の治療であって、治療の必要な患者に、治療上有効な量の第1の態様に定義した化合物を好ましくは医薬組成物の剤形で投与することを含む治療、ならびに、癌の治療であって、治療の必要な患者に、治療上有効な量の第1の態様に定義した化合物を好ましくは医薬組成物の剤形で、電離放射線または化学療法剤と組み合わせて同時に、または連続して投与することを含む治療を提供する。
【0091】
本発明のさらなる態様では、本化合物は、相同組換え(HR)依存的DNA DSB修復活性に欠損のある癌の治療用薬剤の製造において使用可能であるか、または患者に治療上有効な量の化合物を投与することを含む、HR依存的DNA DSB修復活性に欠損のある癌の患者の治療において使用可能である。
【0092】
HR依存的DNA DSB修復経路は、連続DNAヘリックスを再形成する機序と同じ機序によって、DNAの二本鎖切断(DSB)を修復する(K.K. KhannaおよびS.P. Jackson, Nat. Genet. 27(3): 247-254 (2001))。HR依存的DNA DSB修復経路の成分としては、ATM(NM_000051)、RAD51(NM_002875)、RAD51L1(NM_002877)、RAD51C(NM_002876)、RAD51L3(NM_002878)、DMC1(NM_007068)、XRCC2(NM_005431)、XRCC3(NM_005432)、RAD52(NM_002879)、RAD54L(NM_003579)、RAD54B(NM_012415)、BRCA1(NM_007295)、BRCA2(NM_000059)、RAD50(NM_005732)、MRE11A(NM_005590)およびNBS1(NM_002485)が挙げられるが、これらに限定されるものではない。HR依存的DNA DSB修復経路に関与する他のタンパク質群としては、EMSYなどの制御因子が挙げられる(Hughes-Daviesら, Cell, 115, pp523-535)。またHR成分は、Woodら, Science, 291, 1284-1289 (2001)にも記載されている。
【0093】
HR依存的DNA DSB修復を欠損する癌は、この経路によるDNA DSB修復能力が正常細胞に比べて低下または抑制されている1個または複数の癌細胞を含み得るか、かかる癌細胞からなり得る。すなわち、HR依存的DNA DSB修復経路の活性が1個または複数の癌細胞において低下または消失している可能性がある。
【0094】
HR依存的DNA DSB修復経路の1種または複数の成分の活性は、HR依存的DNA DSB修復を欠損する癌に罹患した個体の1個または複数の癌細胞では消失する場合がある。HR依存的DNA DSB修復経路の成分は、当分野では特徴が十分に明らかとなっており(例えば、Woodら, Science, 291, 1284-1289 (2001)を参照)、上記成分などが挙げられる。
【0095】
ある好ましい実施形態では、癌細胞はBRCA1および/またはBRCA2欠損表現型を有している可能性がある。すなわち、癌細胞では、BRCA1および/またはBRCA2活性が低下または消失している。この表現型をもつ癌細胞はBRCA1および/またはBRCA2に欠損があり、すなわち、癌細胞では、例えば、コード核酸における変異もしくは多型により、または制御因子をコードする遺伝子(例えば、BRCA2制御因子をコードするEMSY遺伝子)の増幅、変異もしくは多型により、BRCA1および/またはBRCA2の発現および/または活性が低下または消失している可能性がある(Hughes-Daviesら, Cell, 115, 523-535)。
【0096】
BRCA1およびBRCA2は既知の腫瘍抑制因子であり、その野生型対立遺伝子はヘテロ接合性の保因者の腫瘍においてしばしば失われている(Jasin M., Oncogene, 21(58), 8981-93 (2002);Tuttら, Trends Mol Med., 8(12), 571-6, (2002))。BRCA1および/またはBRCA2突然変異と乳癌との関連性は、当分野では特徴が十分に明らかとなっている(Radice, P.J., Exp Clin Cancer Res., 21(3 Suppl), 9-12 (2002))。BRCA2結合因子をコードするEMSY遺伝子の増幅もまた、乳癌および卵巣癌と関連することが知られている。
【0097】
BRCA1および/またはBRCA2に変異のある保因者はまた、卵巣癌、前立腺癌および膵臓癌のリスクが高くなる。
【0098】
ある好ましい実施形態では、個体は、BRCA1および/またはBRCA2あるいはその制御因子における1つまたは複数の変異(例えば、突然変異および多型)に関しヘテロ接合である。BRCA1およびBRCA2における変異の検出は当分野では周知であり、例えば、EP 699 754、EP 705 903、Neuhausen, S.L.およびOstrander, E.A., Genet. Test, 1, 75-83 (1992);Chappnis, P.O.およびFoulkes, W.D., Cancer Treat Res, 107, 29-59 (2002);Janatova M.ら, Neoplasma, 50(4), 246-50 (2003); Jancarkova, N., Ceska Gynekol., 68(1), 11-6 (2003))に記載されている。BRCA2結合因子EMSYの増幅の測定は、Hughes-Daviesら, Cell, 115, 523-535)に記載されている。
【0099】
癌と関連している突然変異および多型は、変異核酸配列の存在を検出することにより核酸レベルで、または変異(すなわち、突然変異または対立遺伝子変異)ポリペプチドの存在を検出することによりタンパク質レベルで検出することができる。
【0100】
使用
本発明は、活性化合物、具体的にはPARP活性を阻害する活性がある化合物の形態Aとしての化合物Aを提供する。
【0101】
本明細書で使用される「活性(のある)」という用語は、PARP活性を阻害することができる化合物に関係する。本化合物が提供するPARP阻害を評価するのに簡便に用いることができる1つのアッセイを、下記の実施例で説明する。
【0102】
本発明はさらに、細胞におけるPARP活性を阻害する方法であって、その細胞を、好ましくは製薬上許容可能な組成物の形態の有効量の活性化合物と接触させることを含む方法を提供する。かかる方法は、in vitroまたはin vivoで行うことができる。
【0103】
例えば、細胞の試料をin vitroで増殖させ、活性化合物をその細胞と接触させ、その細胞に対する化合物の効果を観察することができる。「効果」の例として、一定時間に達成されたDNA修復の量を求めることができる。活性化合物が細胞に対して影響を与えることが認められる場合には、同じ細胞種の細胞を有する患者の治療方法において、その化合物の効力に関する予後マーカーまたは診断マーカーとしてそれを用いることができる。
【0104】
症状の治療に関して本明細書で使用される「治療」という用語は、一般に、ヒトに関するものか動物に関するものか(例:獣医用途)を問わず、例えば症状進行の阻害といった何らかの望ましい治療効果が得られる治療および療法に関係するものであり、その効果としては進行速度の低下、進行速度の停止、症状の改善および症状の治癒などが挙げられる。予防手段としての治療(すなわち予防)も含まれる。
【0105】
本明細書で使用される「補助剤(adjunct)」という用語は、公知の治療手段と組み合わせた活性化合物の使用を意味する。かかる手段には、各種癌の治療で用いられる薬剤の細胞傷害性療法および/または電離放射などがある。特に、当該活性化合物は、多くの癌化学療法の作用を増強することが知られており、そのようなものとしては、トポイソメラーゼのクラスの毒(例えば、トポテカン、イリノテカン、ルビテカン)、癌の治療に使用されている大部分の既知のアルキル化剤(例えば、DTIC、テモゾラミド)および白金系薬剤(例えば、カルボプラチン、シスプラチン)が挙げられる。
【0106】
活性化合物は、PARPを阻害するために細胞培養添加剤としても用いられ、例えばin vitroで公知の化学療法剤または電離放射線治療に対して細胞を増感させることもできる。
【0107】
活性化合物はin vitroアッセイの一部としても用いられ、例えば候補宿主において当該化合物による治療が有効であるかを決定することもできる。
【0108】
投与
本活性化合物または本活性化合物を含む医薬組成物は、全身投与/末梢投与、つまり所望の作用部位に関わらず、都合のよい投与経路によって被験者に投与することができ、それらの経路としては経口(例:経口摂取による);局所(例えば経皮、経鼻、眼内、口腔および舌下など);肺(例:例えばエアロゾルを用いた、例えば口もしくは鼻を介した吸入または通気療法);直腸;膣;例えば皮下、皮内、筋肉、静脈、動脈、心臓内、硬膜内、脊髄内、嚢内、被膜下、眼窩内、腹腔内、気管内、表皮下、関節内、クモ膜下および胸骨内などの注射による非経口;例えば皮下または筋肉でのデポー剤埋込物によるものなどがあるが、これらに限定されるものではない。
【0109】
被験体は、真核生物、動物、脊椎動物、哺乳動物、齧歯類(例:モルモット、ハムスター、ラット、マウス)、ネズミ類(例:マウス)、犬類(例:イヌ)、ネコ類(例:ネコ)、ウマ類(例:ウマ)、霊長類、類人猿(例:サルまたは類人猿)、サル類(例:マーモセット、ヒヒ)、類人猿(例:ゴリラ、チンパンジー、オランウータン、テナガザル)、またはヒトであってもよい。
【0110】
製剤
本活性化合物を単独で投与することは可能であるが、それを1種または複数の製薬上許容可能な担体、アジュバント、賦形剤、希釈剤、充填剤、緩衝剤、安定剤、保存剤、潤滑剤、または他の当業者には公知の物質ならびに適宜他の治療薬もしくは予防薬とともに、上記で定義した活性化合物を含む医薬組成物(例えば、製剤)として提供することが好ましい。
【0111】
そこで本発明はさらに、上記で定義した医薬組成物、ならびに本明細書に記載した1種または複数の製薬上許容可能な担体、賦形剤、緩衝剤、アジュバント、安定剤または他の物質ともに、上記で定義した活性化合物を、活性化合物が結晶形態Aとして得られるように混合することを含む、医薬組成物の製造方法を提供する。
【0112】
本明細書で使用される「製薬上許容可能な」という用語は、妥当な医学的判断の範囲内で、過度の毒性、刺激、アレルギー反応、または他の問題もしくは合併症を起こさず、妥当な利益/リスク比が得られて、被験者(例:ヒト)の組織との接触における使用に適した化合物、物質、組成物および/または剤形に関係するものである。各担体、賦形剤なども、製剤の他の成分と適合性であるという意味において「許容可能な」ものでなければならない。
【0113】
適切な担体、希釈剤、賦形剤などは標準的な薬学のテキストに記載されている。例えば、「Handbook of Pharmaceutical Additives」, 第2版(M. AshおよびI. Ash編), 2001 (Synapse Information Resources, Inc., Endicott, New York, USA)、「Remington's Pharmaceutical Sciences」, 第20版, pub. Lippincott, Williams and Wilkinsons, 2000;および「Handbook of Pharmaceutical Excipients」, 第2版, 1994を参照されたい。
【0114】
製剤は、好ましくは単位剤形で提供することができ、製薬分野で公知の任意の方法によって製造することができる。かかる方法には、1種または複数の副成分を構成する担体と本活性化合物を組み合わせるステップが含まれる。一般に、製剤は、液体担体もしくは微粉砕固体担体またはその両方を本活性化合物と均一かつ十分に組み合わせ、次に必要に応じて生成物を成形することにより製造される。
【0115】
製剤は、懸濁液、錠剤、粒剤、散剤、カプセル剤、カシェ剤(cachet)、丸剤またはペースト剤の形態とすることができる。
【0116】
経口投与(例えば、経口摂取)に好適な製剤は、カプセル剤、カシェ剤または錠剤などの、それぞれが所定量の活性化合物を含む個別の単位として;粉剤または粒剤として;水系もしくは非水系液体中の懸濁液として;あるいはペースト剤として提供することができる。
【0117】
錠剤は、適宜に1種または複数の副成分とともに、例えば圧縮または成形などの従来の手段によって製造することができる。圧縮錠は、適宜1種または複数の結合剤(例:ポビドン、ゼラチン、アラビアゴム、ソルビトール、トラガカント、ヒドロキシプロピルメチルセルロース);充填剤または希釈剤(例:乳糖、微結晶セルロース、リン酸水素カルシウム);潤滑剤(例:ステアリン酸マグネシウム、タルク、シリカ);崩壊剤(例:デンプングリコール酸ナトリウム、架橋ポビドン、架橋カルボキシメチルセルロースナトリウム);界面活性剤または分散剤または湿展剤(例:ラウリル硫酸ナトリウム);ならびに保存剤(例:p-ヒドロキシ安息香酸メチル、p-ヒドロキシ安息香酸プロピル、ソルビン酸)と混合されていてもよい粉末または顆粒などの自由流動形態の活性化合物を好適な機械で圧縮することで製造することができる。成形錠は、不活性液体希釈剤で湿らせた粉末化合物の混合物を適切な機械で成形することにより製造することができる。錠剤には適宜にコーティングまたは刻み目を施すことができ、例えばヒドロキシプロピルメチルセルロースを各種割合で用いて活性化合物を持続放出または制御放出して、所望の放出プロファイルを得るように製剤することができる。錠剤は、適宜腸溶コーティングを施して、胃以外の消化管部分で放出されるようにすることができる。
【0118】
カプセル剤は、懸濁液に溶解した活性化合物を含むことができる。
【0119】
局所投与(例えば、経皮投与、経鼻投与、眼内投与、口腔内投与および舌下投与)に適した製剤は、ペースト剤として製剤化することができる。
【0120】
眼部への局所投与に適した製剤には、好適な担体(特に活性化合物用の水性溶媒)中に活性化合物を懸濁させた点眼剤も含まれる。
【0121】
担体が固体である経鼻投与に適した製剤には、鼻での吸気を行うことで、すなわち鼻の近くに保持された粉剤容器から鼻道を通って急速な吸入によって投与される、粒径が例えば約20〜約500ミクロンの範囲の粗粉剤などがある。
【0122】
吸入による投与に好適な製剤には、ジクロロジフルオロメタン、トリクロロフルオロメタン、ジクロロテトラフルオロエタン、二酸化炭素、または他の好適なガスなどの適切な噴射剤を用いた、加圧パックからのエアロゾル噴霧剤として提供されるものなどがある。
【0123】
用量
活性化合物および活性化合物を含む組成物の適切な用量は患者ごとに変わり得ることは明らかであろう。最適用量の決定には、一般的に、本発明の治療のあらゆるリスクまたは有害な副作用に対して治療効果のレベルのバランスをとることが必要である。選択される用量レベルは種々の要因に依存し、こうした要因としては、特定化合物の活性、投与経路、投与時間、化合物の排出速度、治療の持続期間、併用される他の薬剤、化合物および/または物質、ならびに患者の年齢、性別、体重、症状、全身的な健康状態、および以前の病歴が挙げられるが、これらに限定されるものではない。化合物の量および投与経路は、最終的には医師の裁量に委ねられる。しかし一般に用量は、実質的に有害または有毒な副作用を起こすことなく、所望の効果を達成する作用部位での局所濃度を与えるようなものとする。
【0124】
in vivo投与は、治療過程全体にわたり連続的に、断続的に(例えば適当な時間間隔で分割用量として)または1回用量で行なうことができる。最も効果的な投与手段と用量を決定する方法は当業者によく知られており、治療に使用する製剤、治療目的、治療される標的細胞、および治療される被験体に応じて変わる。単回投与または複数回投与は、治療に当たっている医師により選択される用量レベルと投与パターンで行うことができる。
【0125】
一般に、本活性化合物の適当な用量は、1日当たり、約10mg〜約600mg/被験体のm2体面積重量(body area weight)の範囲である。
【実施例】
【0126】
実施例1:化合物Aの合成
【化10】
【0127】
出発物質(D)は、WO 2004/080976に開示されている方法により合成した。
【0128】
方法
分取HPLC
試料は、Waters 600 LCポンプ、Waters Xterra C18カラム(5μm 19mm×50mm)およびMicromass ZQ質量分析計を備え、陽イオンエレクトロスプレーイオン化モードで操作する、Waters社の質量分析計直結型精製システムを用いて精製した。移動相A(水中0.1%ギ酸)および移動相B(アセトニトリル中0.1%ギ酸)は、勾配をつけて用いた(7分間で5%Bから100%へ、3分間保持、流速20ml/分)。
【0129】
分析HPLC-MS
分析HPLCは、Spectra System P4000ポンプおよびJones Genesis C18カラム(4μm、50mm×4.6mm)を用いて行った。移動相A(水中0.1%ギ酸)および移動相B(アセトニトリル)は、勾配をつけて用いた(5%Bを1分間、5分間後に98%Bに到達、3分間保持、流速2ml/分)。検出は、254nm UVおよび210〜600nmの範囲のPDAでTSP UV 6000LP検出器を用いて行った。質量分析計は、陽イオンエレクトロスプレーモードで操作するFinnigan LCQであった。
【0130】
NMR
1H NMRスペクトルは、Bruker DPX 300分光計を300MHzで用いて記録した。化学シフトは、テトラメチルシラン内部標準と比較するδスケールで、100万分の1(ppm)で表した。特に断らない限り、すべての試料はDMSO-d6中に溶解させた。
【0131】
(a) 4-[2-フルオロ-5-(4-オキソ-3,4-ジヒドロ-フタラジン-1-イルメチル)-ベンゾイル]-ピペラジン-1-カルボン酸tert-ブチルエステル(C)
窒素下で室温にあるジメチルアセトアミド(DMA)(3561ml)中の出発物質D(850g)の撹拌溶液に、HBTU(2-(1H-ベンゾトリアゾール-1-イル)-1,1,3,3-テトラメチルウロニウムヘキサフルオロホスフェート)(1402g)を一度に加えた。次いで、温度を15〜25℃に維持しながらヒューニッヒ塩基(iPr2NEt、1096ml)を加え、続いて、温度を15〜25℃に維持しながら1-Bocピペラジン(637g)のDMA(1428ml)溶液を加えた。
【0132】
この溶液を室温で2時間撹拌し、完了についてサンプリングした(HPLC)。完了後、温度を15〜25℃に維持ながら前記溶液を激しく撹拌した水(17085ml)に加え、固体を濾過し、水(2×7131ml)、ヘキサン(2×7131ml)およびメチルtert-ブチルエーテル(MTBE)(2×3561ml)で洗浄した。次いで、固体を一晩乾燥させた後、水分および化学的純度を調べるためサンプリングした。
【0133】
その後、この反応を繰り返した(表を参照)。
【表4】
【0134】
(b) 4-[4-フルオロ-3-(ピペラジン-1-カルボニル)-ベンジル]-2H-フタラジン-1-オン(B)
工業用変性アルコール(IMS)(2200ml)および濃HCl(4400ml)の撹拌溶液に、窒素下で室温にて化合物C(2780.2g)を分割添加した。起泡は添加速度により調節した。次いで、この溶液を15〜25℃で30分間撹拌し、完了についてサンプリングした(HPLC)。
【0135】
完了後、溶液を蒸発させてすべてのIMSを除去し、CH2Cl2(2×3500ml)で水性部を抽出した後、濃縮アンモニアを使用してpHを>8に調節した。得られたスラリーを次に水(10000ml)で希釈し、CH2Cl2(4×3500ml)で抽出し、水(2×2000ml)で洗浄し、MgSO4(250g)により乾燥させ、蒸発させた。粗生成物を次にCH2Cl2(3500ml)でスラリー化し、MTBE(5000ml)に加えた。得られた懸濁液を濾過し、50℃で一晩乾燥させたところ、純度94.12%の物質611.0g(58.5%収率)が得られた。
【0136】
(c) 4-[3-(4-シクロプロパンカルボニル-ピペラジン-1-カルボニル)-4-フルオロ-ベンジル]-2H-フタラジン-1-オン(A)
窒素下のCH2Cl2(15480ml)に溶解した化合物B(1290g)の撹拌懸濁液に、温度を20℃未満に維持しながら、CH2Cl2(1290ml)に溶解のトリエチルアミン(470ml)および塩化シクロプロパンカルボニル(306ml)の予混合溶液を滴下添加した。この溶液を次に10〜15℃で15分間撹拌し、完了についてサンプリングした。この反応混合物は出発物質Bを1.18%しか含んでいないことが確認されたため、反応は完了していると考え、次にこのバッチを後処理した。
【0137】
反応混合物を水(7595ml)、5%クエン酸溶液(7595ml)、5%炭酸ナトリウム溶液(7595ml)および水(7595ml)で洗浄した。次いで、有機層を硫酸マグネシウム(500g)で乾燥させた。
【0138】
次いで、生成物を含有するCH2Cl2層を単離し、セライトにより濾過し、25L容器に充填した。次にCH2Cl2(8445ml)を大気圧で蒸留し、エタノール(10000ml)を加えた。次いで、蒸留は、除去された蒸留物4000ml毎にエタノール(4000ml)と置き換えて、ヘッド温度が73.7℃に達するまで継続した。その後、反応容量を、ヘッド温度が78.9℃に達する時間まで減少し(7730mlまで)、その溶液を一晩8℃に冷却させた。次いで、固体を濾別し、エタノール(1290ml)で洗浄し、一晩70℃で乾燥させた。
【0139】
収量=1377.3g(90%)。HPLC純度(99.34%[area%])。GCによると、4.93%のエタノールと0.45%のCH2Cl2が含有されていた。
【0140】
(d) 化合物Aの水処理
水(13770ml)に溶解した化合物A(実施例1の方法で生産したもの)(1377.0g)の懸濁液を4時間加熱還流し、室温に冷却し、濾過した。固体を水(2754ml)で洗浄し、70℃で一晩乾燥させた。
【0141】
収量=1274.8g(92.6%)。HPLC純度(99.49%[area%])。GCによると、0.01%のエタノールと0.01%のCH2Cl2が含有されていた。
【0142】
水処理後の化合物Aの1H NMRスペクトル(DMSO-d6)を図1に示す。
【0143】
水処理後の化合物Aの粉末X線回折パターンを図2に示すが、これによると化合物が形態Aであることが明らかである。
【0144】
実施例2:1-(シクロプロピルカルボニル)ピペラジンを使用する化合物Aの代替合成
【化11】
【0145】
方法(実施例3および4に関しても参照)
NMR
1H NMRスペクトルは、Bruker DPX 400分光計を400 MHzで用いて記録した。化学シフトは、テトラメチルシラン内部標準と比較するδスケールで100万分の1(ppm)で表した。特に断らない限り、すべての試料はDMSO-d6に溶解させた。
【0146】
質量スペクトル
質量スペクトルは、構造の確認にタンデム型質量分析(MS/MS)を使用して、Agilent XCTイオントラップ質量分析計に記録した。この装置は、陽イオンエレクトロスプレーモードで操作した。
【0147】
(a) 4-[3-(4-シクロプロパンカルボニル-ピペラジン-1-カルボニル)-4-フルオロ-ベンジル]-2H-フタラジン-1-オン(化合物A)
2-フルオロ-5-[(4-オキソ-3,4-ジヒドロフタラジン-1-イル)メチル]安息香酸(D)(15.23g、51.07mmol)をアセトニトリル(96ml)中に窒素下で撹拌しながら懸濁した。ジイソプロピルエチルアミン(19.6ml、112.3mmol)を加えた後、1-シクロプロピルカルボニルピペラジン(I)(9.45g、61.28mmol)およびアセトニトリル(1ml)を加えた。反応混合物を18℃に冷却した。O-ベンゾトリアゾール-1-イル-テトラメチルウロニウムヘキサフルオロホスフェート(25.18g、66.39mmol)を30分かけて加え、反応混合物を室温で2時間撹拌した。反応混合物を3℃に冷却し、この温度を1時間維持した後、濾過した。濾過ケーキを冷却(3℃)アセトニトリル(20ml)で洗浄し、40℃以下で真空乾燥したところ、浅黄色の固体(20.21g)として表題化合物が得られた。
【0148】
質量スペクトル:MH+ 435
1H NMR (400MHz, DMSO-d6) δ: 0.70 (m, 4H), 1.88 (br s, 1H), 3.20 (br s, 2H), 3.56 (m, 6H), 4.31 (s, 2H), 7.17 (t, 1H), 7.34 (dd, 1H), 7.41 (m, 1H), 7.77 (dt, 1H), 7.83 (dt, 1H), 7.92 (d, 1H), 8.25 (dd, 1H), 12.53 (s, 1H)。
【0149】
実施例3:1-(シクロプロピルカルボニル)ピペラジン塩酸塩を使用する化合物Aの代替合成
【化12】
【0150】
(a) 1-(シクロプロピルカルボニル)ピペラジン塩酸塩(I')
窒素下で撹拌しながら、酢酸(700ml)をピペラジン(50.00g、0.581mol)で少量ずつ15分間かけて処理した。反応混合物を40℃まで加温し、完全に溶解されるまでこの温度を維持した。塩化シクロプロパンカルボニル(59.2ml、0.638mol)を15分間かけて加えた。反応混合物を一晩室温で撹拌した。この反応混合物を濾過し、〜430mlの蒸留物が回収されるまで濾液を減圧蒸留した。反応混合物にトルエン(550ml)を加え、さらに400mlの蒸留物が回収されるまで減圧蒸留を続けた。さらに充填用トルエン(550ml)を加え、350mlの蒸留物が回収されるまで減圧蒸留を続けた。得られたスラリーをトルエン(200ml)で希釈し、一晩撹拌した。スラリーを回収するためさらにトルエン(500ml)を加えた。スラリーを濾過し、トルエン(100ml)で洗浄し、40℃で真空乾燥させたところ、表題化合物が白色固体(86.78g)として得られた。
【0151】
質量スペクトル:MH+ 155
1H NMR (400MHz, D2O) δ: 0.92 (m, 4H), 1.98 (m, 1H), 3.29 (m, 2H), 3.38 (m, 2H), 3.84 (m, 2H), 4.08 (m, 2H)。
【0152】
(b) 化合物A
2-フルオロ-5-[(4-オキソ-3,4-ジヒドロフタラジン-1-イル)メチル]安息香酸(D)(0.95g、3.19mmol)をアセトニトリル(4ml)中に窒素下で撹拌しながら懸濁させた。2-(1H-ベンゾトリアゾール-1-イル)-1,1,3,3-テトラメチルウロニウムヘキサフルオロホスフェート(HBTU)(1.45g、3.83mmol)を加え、続いて1-シクロプロピルカルボニルピペラジン塩酸塩(I')(0.73g、3.83mmol)を加えた。ジイソプロピルエチルアミン(1.39ml、7.98mmol)を3分かけて加え、反応混合物を室温で一晩撹拌した。この反応混合物を5℃に冷却し、1時間この温度で維持した後、濾過した。濾過ケーキを冷却(3℃)アセトニトリル(2ml)で洗浄し、次に40℃以下で真空乾燥させ、浅黄色固形物(0.93g)として表題化合物を得た。
【0153】
(c) 水性メタノールからの化合物Aの再結晶
ステップ(b)で得た化合物A(9.40g、21.64mmol)を水(100ml)とメタノール(120ml)の混合物に懸濁させた。この懸濁液を撹拌しながら加熱還流した。次いで、得られた混濁溶液を60℃に冷却し、harborliteパッドを通して濾過した。濾過パッドを水(5ml)とメタノール(5ml)の混合物で洗浄した。115mlの蒸留物が回収されるまで、濾液を大気圧で蒸留した。次いで、蒸留を止め、生じた懸濁液を室温まで冷却させた。得られた懸濁液を〜18時間撹拌した後、濾過した。濾過ケーキを水(20ml)で洗浄した後、60℃以下で真空乾燥させ、白色固体(8.67g)として形態Aの表題化合物を得た。
【0154】
質量スペクトル:MH+ 435
1H NMR (400MHz, DMSO-d6) δ: 0.70 (m, 4H), 1.88 (br s, 1H), 3.20 (br s, 2H), 3.56 (m, 6H), 4.31 (s, 2H), 7.17 (t, 1H), 7.34 (dd, 1H), 7.41 (m, 1H), 7.77 (dt, 1H), 7.83 (dt, 1H), 7.92 (d, 1H), 8.25 (dd, 1H), 12.53 (s, 1H)。
【0155】
(d) 水性エタノールからの化合物Aの再結晶
ステップ(b)で得た化合物A(9.40g、21.64mmol)を水(100ml)とエタノール(50ml)の混合物中に懸濁させた。この懸濁液を撹拌しながら加熱還流した。次いで、得られた混濁溶液を60℃に冷却し、harborliteパッドを通して濾過した。濾過パッドを水(5ml)とエタノール(5ml)の混合物で洗浄した。53mlの蒸留物を回収されるまで濾液を大気圧で蒸留した。次いで、蒸留を止め、生じた懸濁液を室温まで冷却させた。得られた懸濁液を〜18時間撹拌した後、濾過した。濾過ケーキを水(20ml)で洗浄した後、60℃以下で真空乾燥させ、白色固体((8.74g)として形態Aの表題化合物を得た。
【0156】
質量スペクトル:MH+435
1H NMR (400MHz, DMSO-d6) δ: 0.70 (m, 4H), 1.88 (br s, 1H), 3.20 (br s, 2H), 3.56 (m, 6H), 4.31 (s, 2H), 7.17 (t, 1H), 7.34 (dd, 1H), 7.41 (m, 1H), 7.77 (dt, 1H), 7.83 (dt, 1H), 7.92 (d, 1H), 8.25 (dd, 1H), 12.53 (s, 1H)。
【0157】
実施例4:化合物Dの代替合成
【化13】
【0158】
(a) 2-フルオロ-5-[(E/Z)-(3-オキソ-2-ベンゾフラン-1(3H)-イリデン)メチル]ベンゾニトリル(E)
ナトリウムt-アミレート(99.00g、0.854mol)および2-メチルテトラヒドロフラン(960ml)を窒素雰囲気下で2℃に冷却した。温度を5℃未満に維持しながら、亜リン酸ジエチル(110ml、0.855mol)を滴下添加した。2-メチルテトラヒドロフラン(40ml)をラインウォッシュ(line wash)として加えた。この反応系を1時間40分、2℃で撹拌した。2-カルボキシベンズアルデヒド(H)(80g、0.533mol)の2-メチルテトラヒドロフラン(200ml)溶液を、添加中温度を7℃未満に維持しながら加えた。2-メチルテトラヒドロフラン(40ml)のラインウォッシュを加えた。反応混合物を20℃まで加温し、20分間20℃で維持した。メタンスルホン酸(66ml、1.01mol)を1時間10分かけて加え、続いて2-メチルテトラヒドロフラン(40ml)を加えた。この反応混合物を一晩20℃で撹拌した。メタンスルホン酸(7ml、0.101mol)を、続いて2-メチルテトラヒドロフラン(7ml)を加え、この反応物をさらに4時間20℃で撹拌した。室温で水(400ml)を加え、得られた2相性混合物を室温で20分間撹拌した。水性の下層を除去し、有機層に水(400ml)に溶解した炭酸水素カリウム(53.50g、0.534mol)の溶液を加えた。この2相性混合物を20分間室温で撹拌し、次いで、水性の下層溶液を除去した。有機画分が得られた(ジエチル(3-オキソ1,3-ジヒドロ-2-ベンゾフラン-1-イル)ホスホネートの溶液)。この有機画分に2-フルオロ-5-ホルミルベンゾニトリル(64g、0.429mol)を加え、混合物を20℃で撹拌した。トリエチルアミン(66ml、0.473mol)を滴下添加し、続いて2-メチルテトラヒドロフラン(7ml)を加えた。反応混合物を一晩20℃で撹拌し、次いで5℃に冷却し、濾過し、工業用変性アルコール(480ml)で洗浄し、次いで、40℃以下で真空乾燥させ、表題化合物(91.2g)を得た。
【0159】
質量スペクトル:MH+266
1H NMR (400MHz, DMSO-d6) δ: 6.89 (s, 1H, 主要異性体), 6.94 (s, 1H, 副次異性体), 7.40 (dd, 1H, 副次異性体), 7.58 (t, 1H, 両異性体), 7.70 (t, 1H, 両異性体), 7.89 (t, 1H, 両異性体), 7.95 (d, 1H, 両異性体), 8.05 (d, 1H, 両異性体), 8.15 (m, 2H, 主要異性体)。
【0160】
(b) 2-フルオロ-5-[(4-オキソ-3,4-ジヒドロフタラジン-1-イル)メチル]ベンゾニトリル(ED)
2-フルオロ-5-[(E/Z)-(3-オキソ-2-ベンゾフラン-1(3H)-イリデン)メチル]ベンゾニトリル(E)(20g、75.40mmol)およびテトラヒドロフラン(200ml)を30分間窒素雰囲気下の室温で撹拌した。ヒドラジン一水和物(4.40ml、90.53mmol)を加え、続いてテトラヒドロフラン(4ml)のラインウォッシュを加えた。反応混合物を1時間45分室温で撹拌した。酢酸(1.10ml、19.20mmol)を加え、反応混合物を60℃に加温した。反応混合物を一晩60℃で維持した。反応混合物を50℃まで冷却し、水(200ml)を滴下添加した。添加の間、温度を45℃に維持した。反応混合物を20℃に冷却し、濾過し、水(30ml)とテトラヒドロフラン(30ml)の混合物で洗浄し、次に40℃以下で真空乾燥し、表題化合物(18.7g)を得た。
【0161】
質量スペクトル:MH+280
1H NMR (400MHz, DMSO-d6) δ: 4.38 (s, 2H), 7.46 (t, 1H), 7.72 (m, 1H), 7.85 (dt, 1H), 7.92 (m, 2H), 7.99 (d, 1H), 8.27 (dd, 1H), 12.57 (s, 1H)。
【0162】
(c) 2-フルオロ-5-[(4-オキソ-3,4-ジヒドロフタラジン-1-イル)メチル]安息香酸(D)
2-フルオロ-5-[(4-オキソ-3,4-ジヒドロフタラジン-1-イル)メチル]ベンゾニトリル(ED)(9.60g, 34.37mmol)および水(40ml)を20℃で撹拌した。2Mの水酸化ナトリウム(36ml、72.00mmol)を加え、反応混合物を90℃に加温し、一晩この温度で維持した。反応混合物を室温まで冷却し、濾過した。濾過パッドを水(10ml)で洗浄し、合わせた濾液を60℃で40分間かけて2MのHCl(56ml、112.00mmol)に加えた。得られた懸濁液を50℃まで冷却し、濾過し、水(57ml)で洗浄し、60℃以下で真空乾燥させ、白色固体(9.72g)として表題化合物を得た。
【0163】
質量スペクトル:MH+299
1H NMR (400MHz, DMSO-d6) δ: 4.36 (s, 2H), 7.24 (dd, 1H), 7.59 (m, 1H), 7.84 (dt, 2H), 7.90 (dt, 1H), 7.98 (d, 1H), 8.27 (dd, 1H), 12.59 (s, 1H), 13.22 (br s, 1H)。
【0164】
実施例5:水性エタノールからの化合物Aの再結晶
4-(3-{[4-(シクロプロピルカルボニル)ピペラジン-1-イル]カルボニル}-4-フルオロベンジル)フタラジン-1(2H)-オン(化合物A)(20.00g、44.66mmol)を、水(50ml)とエタノール(150ml)の混合物に懸濁した。この懸濁液を撹拌しながら加熱還流した。次に得られた溶液を70℃まで冷却し、濾過した。濾過パッドを水(8ml)とエタノール(22ml)の混合物で洗浄した。
【0165】
濾液を撹拌しながら45℃まで冷却した。混合物に種結晶を与えるために形態Aの4-(3-{[4-(シクロプロピルカルボニル)ピペラジン-1-イル]カルボニル}-4-フルオロベンジル)フタラジン-1(2H)-オン(化合物A)(0.08g)を加えた。得られた懸濁液を2.5時間かけて20℃まで冷却し、さらに16時間この温度で撹拌し、結晶化させた。温度を20℃に維持しながら、5時間かけて水(200ml)を加えた。添加終了後、懸濁液を2時間20℃で維持した。
【0166】
懸濁液を濾過し、濾過ケーキをエタノール(24ml)と水(56ml)の混合物で洗浄した。単離した固体を取り出し、40〜60℃で真空乾燥し、白色固体(18.1g)として表題化合物(形態A)を得た。
【0167】
図3〜5を得る方法
粉末X線回折− 図3(形態Aとしての化合物A)
粉末X線回折は、Bruker D5000回折装置により記録した(X線波長1.5418Å Cu線源、電圧40kV、フィラメント放出40mA)。試料は、2〜40°(2θ)の範囲で、0.02°のステップ幅および4秒のタイムカウントでスキャンした。
【0168】
粉末X線回折−図4(溶媒和形態としての化合物A)
溶媒和物ファミリーの粉末X線回折は、湾曲型の位置敏感型検出器(範囲120°(2θ))を取り付けたInel XRG-3000回折装置により記録した(X線波長1.5418Å Cu線源、電圧40kV、フィラメント放出30mA)。試料は、2.5〜40°(2θ)の範囲で、0.03°のステップ幅および一般に300秒の総回収時間でスキャンした。
【0169】
示差走査熱量測定法(DSC)−図5
DSCは、TSO801ROロボットシステムを備えたMettler DSC820Eを使用して記録した。通常、5mg未満の物質(穴の開いた蓋が取り付けられている40μLアルミニウム皿に入れたもの)を毎分10℃の一定の加熱速度で25℃〜325℃の温度範囲で加熱した。窒素パージガスを100ml/分の流速で使用した。
【0170】
実施例6
阻害作用
本活性化合物の阻害作用を評価するため、以下のアッセイを用いてIC50値を求めた。
【0171】
HeLa細胞核抽出物から単離された哺乳動物のPARPを、96ウェルFlashPlates(商標)(NEN、UK)中でZバッファー(25mM Hepes(Sigma);12.5mM MgCl2(Sigma);50mM KCl(Sigma);1mM DTT(Sigma);10%グリセロール(Sigma);0.001%NP-40(Sigma);pH7.4)とともにインキュベートし、様々な濃度の前記阻害剤を添加した。すべての化合物をDMSOで希釈し、最終アッセイ濃度を10〜0.01μMとした。DMSOの最終濃度は1%/ウェルであった。ウェル当たりの全アッセイ容量は40μlであった。
【0172】
30℃で10分間インキュベーションした後、NAD(5μM)、3H-NADおよび30mer二重鎖DNAオリゴを含有する反応混合物10μlを添加して反応を開始した。酵素活性(%)を計算するため、指定の正および負反応ウェルを化合物ウェル(未知)と組み合わせた。次いで、プレートを2分間振盪し、30℃で45分間インキュベートした。
【0173】
インキュベーション後、30%酢酸50μlを各ウェルに添加することにより反応をクエンチした。次いで、プレートを室温で1時間振盪した。
【0174】
プレートをTopCount NXT(商標)(Packard、UK)に移し、シンチレーションをカウントした。記録された値は、各ウェルを30秒カウントした後のカウント/分(cpm)である。
【0175】
次いで、各化合物の酵素活性(%)を以下の式によって計算する:
阻害率(%)=100−[100×(未知のcpm−平均負cpm)/(平均正cpm−平均負cpm)]
【0176】
IC50値(酵素活性の50%が阻害される濃度)を計算した。IC50値は異なる濃度の範囲、通常は10μM〜0.001μMにわたって求められる。かかるIC50値は、化合物の効力が増加したことを確認するための比較値として用いる。
【0177】
化合物Aは、IC50が約5nMであった。
【0178】
増強ファクター(Potentiation Factor)
活性化合物の増強ファクター(PF50)は、対照細胞増殖のIC50を細胞増殖+PARP阻害剤のIC50で割った比として計算される。対照と化合物で処理された細胞の両方の増殖阻害曲線は、アルキル化剤メタンスルホン酸メチル(MMS)の存在下のものである。試験化合物は0.2マイクロモルの固定濃度で使用した。MMSの濃度は0〜10μg/mlであった。
【0179】
細胞増殖をスルホローダミンB(SRB)アッセイによって評価した(Skehan, P.ら, (1990) New colorimetric cytotoxicity assay for anticancer-drug screening. J. Natl. Cancer Inst. 82, 1107-1112)。2,000個のHeLa細胞を平底96ウェルマイクロタイタープレートの各ウェルに100μlの容量で播種し、37℃で6時間インキュベートした。細胞を培地単独で置きかえるか、最終濃度が0.5、1もしくは5μMのPARP阻害剤含有培地で置き換えた。細胞をさらに1時間増殖させ、その後、一連の濃度(一般に0、1、2、3、5、7および10μg/ml)のMMSを未処理細胞またはPARP阻害剤処理細胞に添加した。PARP阻害剤のみで処理した細胞を使用して、PARP阻害剤による増殖阻害を評価した。
【0180】
細胞をさらに16時間静置し、その後培地を交換し、細胞を37℃でさらに72時間増殖させた。次いで培地を取り出し、細胞を氷冷10%(w/v)トリクロロ酢酸100μlで固定した。プレートを4℃で20分間インキュベートし、次いで水で4回洗浄した。次いで、各ウェルの細胞を0.4%(w/v)SRBの1%酢酸溶液100μlで20分間染色した後、1%酢酸で4回洗浄した。次いで、プレートを室温で2時間乾燥させた。10mM Tris Base 100μlを各ウェルに添加して、染色細胞からの色素を溶解させた。プレートを軽く振盪し、室温で30分間静置した後、Microquantマイクロタイタープレートリーダーを用いて564nMの光学濃度を測定した。
【0181】
化合物Aは、200nMにおけるPF50は少なくとも2.0であった。
【特許請求の範囲】
【請求項1】
結晶形態Aとしての4-[3-(4-シクロプロパンカルボニル-ピペラジン-1-カルボニル)-4-フルオロ-ベンジル]-2H-フタラジン-1-オン。
【請求項2】
粉末X線回折(XRD)において以下の特徴的ピークを有する、請求項1に記載の化合物。
【表1】
【請求項3】
粉末X線回折(XRD)において以下の特徴的ピークを有する、請求項1に記載の化合物。
【表2】
【請求項4】
示差走査熱量測定(DSC)において毎分10℃で25℃〜325℃まで加熱した時、210.1℃±1℃で融解し始める、請求項1〜3のいずれか1項に記載の化合物。
【請求項5】
結晶形態Aとしての4-[3-(4-シクロプロパンカルボニル-ピペラジン-1-カルボニル)-4-フルオロ-ベンジル]-2H-フタラジン-1-オン(化合物A)を得る方法であって、
(i) 4-[3-(4-シクロプロパンカルボニル-ピペラジン-1-カルボニル)-4-フルオロ-ベンジル]-2H-フタラジン-1-オンを溶媒から結晶化するステップと;
(ii) 最初の溶媒がエタノールでない場合、結晶性化合物Aをエタノールで処理するステップと;
(iii) 結晶性化合物Aを水で処理して封入されているエタノールを除去するステップと;
(iv) 得られた生成物を乾燥させるステップ
を含む前記方法。
【請求項6】
結晶形態Aとしての4-[3-(4-シクロプロパンカルボニル-ピペラジン-1-カルボニル)-4-フルオロ-ベンジル]-2H-フタラジン-1-オン(化合物A)を得る方法であって、
(i) 4-[3-(4-シクロプロパンカルボニル-ピペラジン-1-カルボニル)-4-フルオロ-ベンジル]-2H-フタラジン-1-オンを溶媒から結晶化するステップと;
(ii) 結晶形態の化合物Aの合成で使用される最初の溶媒が水とC1-2アルコールの混合物でない場合、水とC1-2アルコールの混合物で化合物を加熱するステップと;
(iii) 周囲圧力で混合物を蒸留するステップと;
(iv) 得られた生成物を乾燥させるステップ
を含む前記方法。
【請求項7】
結晶形態Aとしての4-[3-(4-シクロプロパンカルボニル-ピペラジン-1-カルボニル)-4-フルオロ-ベンジル]-2H-フタラジン-1-オン(化合物A)を得る方法であって、
(i) 溶媒としての水とC1-2アルコールの混合物に化合物Aを懸濁するステップと;
(ii) 懸濁液を加熱還流するステップと;
(iii) 溶液を冷却し、形態Aとしての化合物Aを種結晶として加える(seeding)ステップと;
(iv) 得られた生成物を乾燥させるステップ
を含む前記方法。
【請求項8】
4-[3-(4-シクロプロパンカルボニル-ピペラジン-1-カルボニル)-4-フルオロ-ベンジル]-2H-フタラジン-1-オンを4-[4-フルオロ-3-(ピペラジン-1-カルボニル)-ベンジル]-2H-フタラジン-1-オンから合成する方法であって、
(i) 制御下で、有機溶媒に溶解したトリエチルアミンおよび塩化シクロプロパンカルボニルの予混合溶液を、同一有機溶媒中の4-[4-フルオロ-3-(ピペラジン-1-カルボニル)-ベンジル]-2H-フタラジン-1-オンに、溶液の温度が20℃未満となるように制御しながら添加するステップ含む前記方法。
【請求項9】
4-[3-(4-シクロプロパンカルボニル-ピペラジン-1-カルボニル)-4-フルオロ-ベンジル]-2H-フタラジン-1-オンを2-フルオロ-5-(4-オキソ-3,4-ジヒドロ-フタラジン-1-イルメチル)-安息香酸から合成する方法であって、アミドカップリング剤の存在下で、2-フルオロ-5-(4-オキソ-3,4-ジヒドロ-フタラジン-1-イルメチル)-安息香酸を1-(シクロプロピルカルボニル)ピペラジンまたはその鉱酸塩と反応させることを含む前記方法。
【請求項10】
2-フルオロ-5-(4-オキソ-3,4-ジヒドロ-フタラジン-1-イルメチル)-安息香酸を合成する方法であって、
(a) ジエチル(3-オキソ-1,3-ジヒドロ-2-ベンゾフラン-1-イル)ホスホネートを2-カルボキシベンズアルデヒドから合成するステップと;
(b) 2-フルオロ-5-[(E/Z)-(3-オキソ-2-ベンゾフラン-1(3H)-イリデン)メチル]ベンゾニトリルをジエチル(3-オキソ-1,3-ジヒドロ-2-ベンゾフラン-1-イル)ホスホネートから合成するステップ
を含む前記方法。
【請求項11】
(c) 化合物Eからヒドラジン水和物との反応により2-フルオロ-5-[(4-オキソ-3,4-ジヒドロフタラジン-1-イル)メチル]ベンゾニトリル(ED):
【化1】
を合成するステップと;
(d) 化合物EDから水酸化ナトリウムとの反応により化合物Dを合成するステップ
をさらに含む、請求項10に記載の方法。
【請求項12】
2-フルオロ-5-[(4-オキソ-3,4-ジヒドロフタラジン-1-イル)メチル]ベンゾニトリル(ED)。
【化2】
【請求項13】
請求項1〜4のいずれか1項に記載の化合物と製薬上許容可能な担体または希釈剤を含む、医薬組成物。
【請求項14】
ヒトまたは動物の身体の治療方法に使用するための請求項1〜4のいずれか1項に記載の化合物。
【請求項15】
ヒトまたは動物の身体の治療におけるPARPの阻害方法に使用するための請求項1〜4のいずれか1項に記載の化合物。
【請求項16】
PARP活性の阻害剤の製造における請求項1〜4のいずれか1項に記載の化合物の使用。
【請求項17】
血管疾患;敗血症性ショック;虚血性障害;神経毒性;出血性(haemorraghic)ショック;ウィルス感染;またはPARPの活性を阻害することより寛解される疾患の治療用薬剤の製造における、請求項1〜4のいずれか1項に記載の化合物の使用。
【請求項18】
癌治療において補助剤(adjunct)として使用するための、あるいは電離放射線または化学療法剤による治療のための腫瘍細胞を増強するための、薬剤製造における、請求項1〜4のいずれか1項に記載の化合物の使用。
【請求項19】
個体における、相同組換え(HR)依存的DNA DSB修復経路に欠損のある癌の治療に使用するための薬剤の製造における、請求項1〜4のいずれか1項に記載の化合物の使用。
【請求項20】
前記癌が、HRによるDNA DSB修復能力が正常細胞に比べて低下または抑制された1個または複数の癌細胞を含む、請求項19に記載の使用。
【請求項21】
前記癌細胞がBRCA1またはBRCA2欠損表現型を有する、請求項20に記載の使用。
【請求項22】
前記癌細胞がBRCA1またはBRCA2に欠損がある、請求項21に記載の使用。
【請求項23】
前記個体が、HR依存的DNA DSB修復経路の成分をコードする遺伝子中の突然変異に関してヘテロ接合である、請求項19〜22のいずれか1項に記載の使用。
【請求項24】
前記個体がBRCA1および/またはBRCA2中の突然変異に関してヘテロ接合である、請求項23に記載の使用。
【請求項25】
前記癌が乳癌、卵巣癌、膵臓癌または前立腺癌である、請求項19〜24のいずれか1項に記載の使用。
【請求項26】
前記治療が電離放射線または化学療法剤の投与をさらに含む、請求項19〜25のいずれか1項に記載の使用。
【請求項1】
結晶形態Aとしての4-[3-(4-シクロプロパンカルボニル-ピペラジン-1-カルボニル)-4-フルオロ-ベンジル]-2H-フタラジン-1-オン。
【請求項2】
粉末X線回折(XRD)において以下の特徴的ピークを有する、請求項1に記載の化合物。
【表1】
【請求項3】
粉末X線回折(XRD)において以下の特徴的ピークを有する、請求項1に記載の化合物。
【表2】
【請求項4】
示差走査熱量測定(DSC)において毎分10℃で25℃〜325℃まで加熱した時、210.1℃±1℃で融解し始める、請求項1〜3のいずれか1項に記載の化合物。
【請求項5】
結晶形態Aとしての4-[3-(4-シクロプロパンカルボニル-ピペラジン-1-カルボニル)-4-フルオロ-ベンジル]-2H-フタラジン-1-オン(化合物A)を得る方法であって、
(i) 4-[3-(4-シクロプロパンカルボニル-ピペラジン-1-カルボニル)-4-フルオロ-ベンジル]-2H-フタラジン-1-オンを溶媒から結晶化するステップと;
(ii) 最初の溶媒がエタノールでない場合、結晶性化合物Aをエタノールで処理するステップと;
(iii) 結晶性化合物Aを水で処理して封入されているエタノールを除去するステップと;
(iv) 得られた生成物を乾燥させるステップ
を含む前記方法。
【請求項6】
結晶形態Aとしての4-[3-(4-シクロプロパンカルボニル-ピペラジン-1-カルボニル)-4-フルオロ-ベンジル]-2H-フタラジン-1-オン(化合物A)を得る方法であって、
(i) 4-[3-(4-シクロプロパンカルボニル-ピペラジン-1-カルボニル)-4-フルオロ-ベンジル]-2H-フタラジン-1-オンを溶媒から結晶化するステップと;
(ii) 結晶形態の化合物Aの合成で使用される最初の溶媒が水とC1-2アルコールの混合物でない場合、水とC1-2アルコールの混合物で化合物を加熱するステップと;
(iii) 周囲圧力で混合物を蒸留するステップと;
(iv) 得られた生成物を乾燥させるステップ
を含む前記方法。
【請求項7】
結晶形態Aとしての4-[3-(4-シクロプロパンカルボニル-ピペラジン-1-カルボニル)-4-フルオロ-ベンジル]-2H-フタラジン-1-オン(化合物A)を得る方法であって、
(i) 溶媒としての水とC1-2アルコールの混合物に化合物Aを懸濁するステップと;
(ii) 懸濁液を加熱還流するステップと;
(iii) 溶液を冷却し、形態Aとしての化合物Aを種結晶として加える(seeding)ステップと;
(iv) 得られた生成物を乾燥させるステップ
を含む前記方法。
【請求項8】
4-[3-(4-シクロプロパンカルボニル-ピペラジン-1-カルボニル)-4-フルオロ-ベンジル]-2H-フタラジン-1-オンを4-[4-フルオロ-3-(ピペラジン-1-カルボニル)-ベンジル]-2H-フタラジン-1-オンから合成する方法であって、
(i) 制御下で、有機溶媒に溶解したトリエチルアミンおよび塩化シクロプロパンカルボニルの予混合溶液を、同一有機溶媒中の4-[4-フルオロ-3-(ピペラジン-1-カルボニル)-ベンジル]-2H-フタラジン-1-オンに、溶液の温度が20℃未満となるように制御しながら添加するステップ含む前記方法。
【請求項9】
4-[3-(4-シクロプロパンカルボニル-ピペラジン-1-カルボニル)-4-フルオロ-ベンジル]-2H-フタラジン-1-オンを2-フルオロ-5-(4-オキソ-3,4-ジヒドロ-フタラジン-1-イルメチル)-安息香酸から合成する方法であって、アミドカップリング剤の存在下で、2-フルオロ-5-(4-オキソ-3,4-ジヒドロ-フタラジン-1-イルメチル)-安息香酸を1-(シクロプロピルカルボニル)ピペラジンまたはその鉱酸塩と反応させることを含む前記方法。
【請求項10】
2-フルオロ-5-(4-オキソ-3,4-ジヒドロ-フタラジン-1-イルメチル)-安息香酸を合成する方法であって、
(a) ジエチル(3-オキソ-1,3-ジヒドロ-2-ベンゾフラン-1-イル)ホスホネートを2-カルボキシベンズアルデヒドから合成するステップと;
(b) 2-フルオロ-5-[(E/Z)-(3-オキソ-2-ベンゾフラン-1(3H)-イリデン)メチル]ベンゾニトリルをジエチル(3-オキソ-1,3-ジヒドロ-2-ベンゾフラン-1-イル)ホスホネートから合成するステップ
を含む前記方法。
【請求項11】
(c) 化合物Eからヒドラジン水和物との反応により2-フルオロ-5-[(4-オキソ-3,4-ジヒドロフタラジン-1-イル)メチル]ベンゾニトリル(ED):
【化1】
を合成するステップと;
(d) 化合物EDから水酸化ナトリウムとの反応により化合物Dを合成するステップ
をさらに含む、請求項10に記載の方法。
【請求項12】
2-フルオロ-5-[(4-オキソ-3,4-ジヒドロフタラジン-1-イル)メチル]ベンゾニトリル(ED)。
【化2】
【請求項13】
請求項1〜4のいずれか1項に記載の化合物と製薬上許容可能な担体または希釈剤を含む、医薬組成物。
【請求項14】
ヒトまたは動物の身体の治療方法に使用するための請求項1〜4のいずれか1項に記載の化合物。
【請求項15】
ヒトまたは動物の身体の治療におけるPARPの阻害方法に使用するための請求項1〜4のいずれか1項に記載の化合物。
【請求項16】
PARP活性の阻害剤の製造における請求項1〜4のいずれか1項に記載の化合物の使用。
【請求項17】
血管疾患;敗血症性ショック;虚血性障害;神経毒性;出血性(haemorraghic)ショック;ウィルス感染;またはPARPの活性を阻害することより寛解される疾患の治療用薬剤の製造における、請求項1〜4のいずれか1項に記載の化合物の使用。
【請求項18】
癌治療において補助剤(adjunct)として使用するための、あるいは電離放射線または化学療法剤による治療のための腫瘍細胞を増強するための、薬剤製造における、請求項1〜4のいずれか1項に記載の化合物の使用。
【請求項19】
個体における、相同組換え(HR)依存的DNA DSB修復経路に欠損のある癌の治療に使用するための薬剤の製造における、請求項1〜4のいずれか1項に記載の化合物の使用。
【請求項20】
前記癌が、HRによるDNA DSB修復能力が正常細胞に比べて低下または抑制された1個または複数の癌細胞を含む、請求項19に記載の使用。
【請求項21】
前記癌細胞がBRCA1またはBRCA2欠損表現型を有する、請求項20に記載の使用。
【請求項22】
前記癌細胞がBRCA1またはBRCA2に欠損がある、請求項21に記載の使用。
【請求項23】
前記個体が、HR依存的DNA DSB修復経路の成分をコードする遺伝子中の突然変異に関してヘテロ接合である、請求項19〜22のいずれか1項に記載の使用。
【請求項24】
前記個体がBRCA1および/またはBRCA2中の突然変異に関してヘテロ接合である、請求項23に記載の使用。
【請求項25】
前記癌が乳癌、卵巣癌、膵臓癌または前立腺癌である、請求項19〜24のいずれか1項に記載の使用。
【請求項26】
前記治療が電離放射線または化学療法剤の投与をさらに含む、請求項19〜25のいずれか1項に記載の使用。
【図1】

【図2】
【図3】
【図4】
【図5】
【図2】
【図3】
【図4】
【図5】
【公表番号】特表2010−506894(P2010−506894A)
【公表日】平成22年3月4日(2010.3.4)
【国際特許分類】
【出願番号】特願2009−532883(P2009−532883)
【出願日】平成19年10月15日(2007.10.15)
【国際出願番号】PCT/GB2007/003888
【国際公開番号】WO2008/047082
【国際公開日】平成20年4月24日(2008.4.24)
【出願人】(503160629)クドス ファーマシューティカルズ リミテッド (23)
【Fターム(参考)】
【公表日】平成22年3月4日(2010.3.4)
【国際特許分類】
【出願日】平成19年10月15日(2007.10.15)
【国際出願番号】PCT/GB2007/003888
【国際公開番号】WO2008/047082
【国際公開日】平成20年4月24日(2008.4.24)
【出願人】(503160629)クドス ファーマシューティカルズ リミテッド (23)
【Fターム(参考)】
[ Back to top ]