
4−[4−(2−アダマンチルカルバモイル)−5−tert−ブチル−ピラゾル−1−イル]安息香酸の新規な結晶形態
4−[4−(2−アダマンチルカルバモイル)−5−tert−ブチル−ピラゾル−1−イル]安息香酸の新規な結晶形態;また、11βHSD1の阻害におけるそれらの使用、それらの調製方法およびそれらを含む医薬組成物も記載されている。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、4−[4−(2−アダマンチルカルバモイル)−5−tert−ブチル−ピラゾル−1−イル]安息香酸(本薬剤)の新規な結晶形態に関する。本薬剤は、ヒト11−β−ヒドロキシステロイド脱水素酵素1型(11βHSD1)阻害活性を有し、したがってメタボリック症候群などの病態の治療において価値があり、ヒトなどの温血動物の治療方法において有用である。また本発明は、本薬剤の結晶形態の製造工程、それらを含有する医薬組成物、およびヒトなどの温血動物において11βHSD1を阻害する医薬の製造におけるこれらの使用にも関する。
【0002】
本薬剤は下記の式(I)で示される。
【0003】
【化1】

【背景技術】
【0004】
グルココルチコイド(ヒトのコルチゾール、げっ歯類のコルチコステロン)は対抗制御的ホルモンである。すなわち、グルココルチコイドはインスリンの作用に対抗する(Dallman MF, Strack AM, Akana SF et al. 1993; Front Neuroendocrinol 14, 303-347)。グルココルチコイドは、糖新生に関与する肝酵素の発現を調節するとともに、脂肪組織からグリセロールを遊離し(脂肪分解の増加)筋組織からアミノ酸を遊離する(タンパク質合成の減少とタンパク質分解の増加)ことによって、基質の供給を高める。またグルココルチコイドは、トリグリセリドを蓄えることのできる成熟脂肪細胞への前駆脂肪細胞の分化において重要である(Bujalska IJ et al. 1999; Endocrinology 140, 3188-3196)。「ストレス」によって誘発されたグルココルチコイドが中心性肥満(それ自体が2型糖尿病、高血圧症および心疾患の強力な危険因子である)と関連している病態においては、このことは重大な意味を持ち得る(Bjorntorp P & Rosmond R 2000; Int. J. Obesity 24, S80-S85)。
【0005】
グルココルチコイド活性が、コルチゾールの分泌だけで制御されているのではなく、組織レベルにおいては、11−βヒドロキシステロイド脱水素酵素である11βHSD1(コルチゾンを活性化する)と11βHSD2(コルチゾールを不活性化する)による活性コルチゾールと不活性コルチゾンの細胞内相互変換によって制御されている、ということは現在十分に証明されている(Sandeep TC & Walker BR 2001 Trends in Endocrinol & Metab. 12, 446-453)。このメカニズムがヒトにおいて重要であり得るということは、カルベノキソロン(11βHSD1および2の両方を阻害する抗潰瘍薬)治療を用いてまず示された(Walker BR et al. 1995; J. Clin. Endocrinol. Metab. 80, 3155-3159)。この治療はインスリン感受性の増加につながり、それによって、活性グルココルチコイドの組織レベルを低下させることによって11βHSD1がインスリンの作用を調節している可能性がある、ということが示されている(Walker BR et al. 1995; J. Clin. Endocrinol. Metab. 80, 3155-3159)。
【0006】
臨床的には、クッシング症候群にはコルチゾールの過剰が関与しており、さらにその過剰には、耐糖能低下、中心性肥満(この貯蔵所における前駆脂肪細胞の分化の刺激に起因)、脂質異常症および高血圧症が関与している。クッシング症候群には、メタボリック症候群との明確な類似点が数多く見られる。メタボリック症候群には、通常、過剰の血中コルチゾール濃度は関与していないが(Jessop DS et al. 2001; J. Clin. Endocrinol. Metab. 86, 4109-4114)、組織中の11βHSD1活性が異常に高いと、同様の影響があることが予想される。肥満したヒトでは、血漿コルチゾール濃度は痩型対照群と同じかまたはそれよりも低いにもかかわらず、皮下脂肪中の11βHSD1活性が大きく増進されることが示された(Rask E et al. 2001; J. Clin. Endocrinol. Metab. 1418-1421)。さらに、メタボリック症候群に関与している中心性脂肪は、皮下脂肪よりもはるかに高いレベルの11βHSD1活性を示す(Bujalska IJ et al. 1997; Lancet 349, 1210-1213)。このように、グルココルチコイド、11βHSD1およびメタボリック症候群の間には関連性があるように思われる。
【0007】
11βHSD1ノックアウトマウスでは、グルココルチコイドに誘導される糖新生酵素の活性化が絶食に応じて減衰し、血漿グルコース濃度がストレスまたは肥満に応じて低下することが示されるが(Kotelevtsev Y et al. 1997; Proc. Natl. Acad. Sci USA 94, 14924-14929)、このことは、11βHSD1を阻害することが、2型糖尿病において血漿グルコースおよび肝糖産生を低下させる際に有用であることを示している。さらに、これらのマウスは、低トリグリセリド、高HDLコレステロールおよび高アポリポタンパク質AI濃度を示す、抗アテローム発生性リポタンパク質プロフィールを発現する(Morton NM et al. 2001; J. Biol. Chem. 276, 41293-41300)。この表現型は、脂肪異化酵素およびPPARαの肝での発現の増加によるものである。このこともまた、11βHSD1の阻害がメタボリック症候群の脂質異常症の治療において有用であることを示している。
【0008】
11βHSD1を過剰発現しているトランスジェニックマウスにおける最近の研究が、メタボリック症候群と11βHSD1との間の関連性を最も説得力をもって実証している(Masuzaki H et al. 2001; Science 294, 2166-2170)。脂肪特異的プロモーターの制御下で発現した場合、11βHSD1トランスジェニックマウスは、コルチコステロンの脂肪中濃度が高く、中心性肥満、インスリン抵抗性糖尿病、高脂血症および過食症を発症する。最も重要なことは、これらのマウスの脂肪中の11βHSD1活性の上昇レベルが、肥満した被験者と同等であることである。肝性11βHSD1活性および血漿コルチコステロン濃度は正常であったが、肝門脈コルチコステロン濃度は3倍に上昇しており、これが肝臓における代謝作用の原因であると考えられる。
【0009】
全体的に見ると、肥満したヒトと同等のレベルの11βHSD1を脂肪のみに過剰発現させるだけで、マウスでメタボリック症候群を完全に再現できることは、もはや明らかである。
【0010】
11βHSD1は組織内に広く分布しており、グルココルチコイド受容体の分布と重なっている。したがって、11βHSD1を阻害することによって、数多くの生理学的/病理学的役割におけるグルココルチコイドの作用が妨害される可能性がある。11βHSD1はヒト骨格筋に存在しており、タンパク質の代謝回転およびグルコース代謝に対するインスリンの同化作用をグルココルチコイドが妨害することは、十分に立証されている(Whorwood CB et al. 2001; J. Clin. Endocrinol. Metab. 86, 2296-2308)。したがって、骨格筋は11βHSD1に基づく治療の重要な標的になるはずである。
【0011】
グルココルチコイドはインスリン分泌も低下させるが、これによってグルココルチコイドに誘導されるインスリン抵抗性の作用が激化する可能性がある。膵島は11βHSD1を発現し、カルベノキソロンはインスリン放出に対する11−デヒドロコルチコステロンの作用を阻害することができる(Davani B et al. 2000; J. Biol. Chem. 275, 34841-34844)。このように、糖尿病の治療においては、11βHSD1阻害剤はインスリン抵抗性に対して組織レベルで作用するだけでなく、インスリンの分泌自体を増加させることができる。
【0012】
骨格の発育および骨の機能もまた、グルココルチコイドの作用によって調節されている。11βHSD1はヒト破骨細胞および骨芽細胞中に存在しており、健常ボランティアをカルベノキソロンで処置すると、骨吸収マーカーは減少したが、骨形成マーカーには変化は見られなかった(Cooper MS et al 2000; Bone 27, 375-381)。骨中の11βHSD1活性の阻害は、骨粗しょう症治療における防御機構として使用することができる。
【0013】
グルココルチコイドは緑内障などの眼病に関与している可能性もある。11βHSD1は、ヒトにおいて眼圧に影響を与えることが示されており、11βHSD1を阻害することによって、緑内障に伴う眼圧の上昇を緩和させることが期待される(Rauz S et al. 2001; Investigative Opthalmology & Visual Science 42, 2037-2042)。
【0014】
げっ歯類およびヒトの両方において、11βHSD1とメタボリック症候群との間には確実な関連性があると思われる。2型肥満糖尿病患者において11βHSD1を特異的に阻害する薬が、肝糖新生を減少させることによって血糖を下げ、中心性肥満を軽減し、アテローム発生性リポタンパク質表現型を改善し、血圧を下げ、かつインスリン抵抗性を低減するということを示唆する証拠がある。筋組織でのインスリンの作用は増強され、膵島ベータ細胞からのインスリン分泌も増加し得る。
【0015】
現在、メタボリック症候群の定義として主に2つのものが認識されている。
1)米国高脂血症治療ガイドライン(The Adult Treatment Panel(ATP III 2001 JMA))の定義では、患者が次の症状のうち3つ以上を有する場合、メタボリック症候群であるとしている。
男性の場合、腹囲40インチ(102cm)以上、女性の場合、35インチ(88cm)以上;
血清トリグリセリド濃度150mg/dl(1.69mmol/l)以上;
男性の場合、HDLコレステロール濃度40mg/dl(1.04mmol/l)未満、女性の場合、50mg/dl(1.29mmol/l)未満;
血圧135/80mmHg以上;および/または血糖(血清グルコース)110mg/dl(6.1mmol/l)以上。
2)WHO協議会は、因果関係を意味するものではなく、適当な時期にさらに改善することもできる実践的な定義として提案されている、次の定義を推奨している。
患者が、次の症状:耐糖能低下、耐糖能異常(IGT)もしくは糖尿病、および/またはインスリン抵抗性のうちの少なくとも1つを有し、かつ、次のうちの2つ以上を有する。
動脈圧の上昇;
血漿トリグリセリドの上昇;
中心性肥満;
微量アルブミン尿症。
【発明の概要】
【0016】
本発明者らは、本薬剤またはその薬学的に許容される塩は、有効な11βHSD1阻害剤であり、したがってメタボリック症候群に関連する病態の治療において価値があることを見出した。また本発明者らは、本発明の化合物は改良された特性を有し、それによって、医薬品として使用するためのより良い候補となることを見出した。
【0017】
本発明者らは、本薬剤のさらなる結晶形態を見出した。これらの形態は、形態2、形態3および形態4と称される。
【図面の簡単な説明】
【0018】
【図1】図1は、形態2のX線粉末回折パターンである。
【図2】図2は、形態2のDSC温度記録図である。
【図3】図3は、形態3のX線粉末回折パターンである。
【図4】図4は、形態3のDSC温度記録図である。
【図5】図5は、形態4のX線粉末回折パターンである。
【図6】図6は、形態4のDSC温度記録図である。
【図7】図7は、形態4のXRDパターンおよび面間隔dスペクトルである。
【発明を実施するための形態】
【0019】
したがって本発明の一態様は、およそ2−シータ=18.0の少なくとも1つの特異的ピークを有する測定したX線回折パターンを有する、4−[4−(2−アダマンチルカルバモイル)−5−tert−ブチル−ピラゾル−1−イル]安息香酸の結晶形態(形態2)に関する。
【0020】
2−シータ(θ)値はCuKa照射を用いて測定した。
本発明によれば、およそ2−シータ=18.0°および17.7°に少なくとも2つの特異的ピークを持つX線粉末回折パターンを有する本薬剤の結晶形態、形態2、が提供される。
【0021】
本発明によれば、およそ2−シータ=18.0、17.7および18.4°に特異的ピークを持つX線粉末回折パターンを有する本薬剤の結晶形態、形態2、が提供される。
本発明によれば、およそ2−シータ=18.0、17.7、18.4、8.9および20.5°に特異的ピークを持つX線粉末回折パターンを有する本薬剤の結晶形態、形態2、が提供される。
【0022】
本発明によれば、およそ2−シータ=18.0、17.7、18.4、8.9、20.5、10.4、21.9、13.4、27.6および16.7°に特異的ピークを持つX線粉末回折パターンを有する本薬剤の結晶形態、形態2、が提供される。
【0023】
本発明によれば、CuKa照射を用いた、図1に示されるX線粉末回折パターンと実質的に同一のX線粉末回折パターンを有する本薬剤の結晶形態、形態2、が提供される。
本発明によれば、2−シータ=18.0°プラスマイナス0.5°2−シータに少なくとも1つの特異的ピークを持つX線粉末回折パターンを有する本薬剤の結晶形態、形態2、が提供される。
【0024】
本発明によれば、2−シータ=18.0°および17.7°に少なくとも2つの特異的ピークを持つX線粉末回折パターンを有する本薬剤の結晶形態、形態2、が提供される。ここで、前記値はプラスマイナス0.5°2−シータであってよい。
【0025】
本発明によれば、2−シータ=18.0、17.7および18.4°に特異的ピークを持つX線粉末回折パターンを有する本薬剤の結晶形態、形態2、が提供される。ここで、前記値はプラスマイナス0.5°2−シータであってよい。
【0026】
本発明によれば、2−シータ=18.0、17.7、18.4、8.9および20.5°に特異的ピークを持つX線粉末回折パターンを有する本薬剤の結晶形態、形態2、が提供される。ここで、前記値はプラスマイナス0.5°2−シータであってよい。
【0027】
本発明によれば、2−シータ=18.0、17.7、18.4、8.9、20.5、10.4、21.9、13.4、27.6および16.7°に特異的ピークを持つX線粉末回折パターンを有する本薬剤の結晶形態、形態2、が提供される。ここで、前記値はプラスマイナス0.5°2−シータであってよい。
【0028】
本発明によれば、2−シータ=18.0°に少なくとも1つの特異的ピークを持つX線粉末回折パターンを有する本薬剤の結晶形態、形態2、が提供される。
本発明によれば、2−シータ=18.0および17.7°に少なくとも2つの特異的ピークを持つX線粉末回折パターンを有する本薬剤の結晶形態、形態2、が提供される。
【0029】
本発明によれば、2−シータ=18.0、17.7および18.4°に特異的ピークを持つX線粉末回折パターンを有する本薬剤の結晶形態、形態2、が提供される。
本発明によれば、2−シータ=18.0、17.7、18.4、8.9および20.5°に特異的ピークを持つX線粉末回折パターンを有する本薬剤の結晶形態、形態2、が提供される。
【0030】
本発明によれば、2−シータ=18.0、17.7、18.4、8.9、20.5、10.4、21.9、13.4、27.6および16.7°に特異的ピークを持つX線粉末回折パターンを有する本薬剤の結晶形態、形態2、が提供される。
【0031】
本発明によれば、CuKa照射を用いた、図1に示されるX線粉末回折パターンを有する本薬剤の結晶形態、形態2、が提供される。
【0032】
【表1】
【0033】
DSC分析から、形態2は309.9℃で融解を開始する高融点の固体であることがわかる。DSC温度記録図を図2に示す。
本発明によれば、およそ2−シータ=18.7°にピークを持つX線粉末回折パターンを有する、本薬剤の結晶形態、形態3が提供される。
【0034】
2−シータ(θ)値はCuKa照射を用いて測定した。
本発明によれば、およそ2−シータ=18.7°および11.7°に少なくとも2つの特異的ピークを持つX線粉末回折パターンを有する本薬剤の結晶形態、形態3、が提供される。
【0035】
本発明によれば、およそ2−シータ=18.7、11.7および19.2°に特異的ピークを持つX線粉末回折パターンを有する本薬剤の結晶形態、形態3、が提供される。
本発明によれば、およそ2−シータ=18.7、11.7、19.2、7.8、14.1、14.9および9.4°に特異的ピークを持つX線粉末回折パターンを有する本薬剤の結晶形態、形態3、が提供される。
【0036】
本発明によれば、およそ2−シータ=18.7、11.7、19.2、7.8、14.1、14.9、9.4、15.6、16.1および9.6°に特異的ピークを持つX線粉末回折パターンを有する本薬剤の結晶形態、形態3、が提供される。
【0037】
本発明によれば、CuKa照射を用いた、図3に示されるX線粉末回折パターンと実質的に同一のX線粉末回折パターンを有する本薬剤の結晶形態、形態3、が提供される。
本発明によれば、2−シータ=18.7°プラスマイナス0.5°2−シータに少なくとも1つの特異的ピークを持つX線粉末回折パターンを有する本薬剤の結晶形態、形態3、が提供される。
【0038】
本発明によれば、2−シータ=18.7°および11.7°に少なくとも2つの特異的ピークを持つX線粉末回折パターンを有する本薬剤の結晶形態、形態3、が提供される。ここで、前記値はプラスマイナス0.5°2−シータであってよい。
【0039】
本発明によれば、2−シータ=18.7、11.7および19.2°に特異的ピークを持つX線粉末回折パターンを有する本薬剤の結晶形態、形態3、が提供される。ここで、前記値はプラスマイナス0.5°2−シータであってよい。
【0040】
本発明によれば、2−シータ=18.7、11.7、19.2、7.8、14.1、14.9および9.4°に特異的ピークを持つX線粉末回折パターンを有する本薬剤の結晶形態、形態3、が提供される。ここで、前記値はプラスマイナス0.5°2−シータであってよい。
【0041】
本発明によれば、2−シータ=18.7、11.7、19.2、7.8、14.1、14.9、9.4、15.6、16.1および9.6°に特異的ピークを持つX線粉末回折パターンを有する本薬剤の結晶形態、形態3、が提供される。ここで、前記値はプラスマイナス0.5°2‐シータであってよい。
【0042】
本発明によれば、2−シータ=18.7°に少なくとも1つの特異的ピークを持つX線粉末回折パターンを有する本薬剤の結晶形態、形態3、が提供される。
本発明によれば、2−シータ=18.7および11.7°に少なくとも2つの特異的ピークを持つX線粉末回折パターンを有する本薬剤の結晶形態、形態3、が提供される。
【0043】
本発明によれば、2−シータ=18.7、11.7および19.2°に特異的ピークを持つX線粉末回折パターンを有する本薬剤の結晶形態、形態3、が提供される。
本発明によれば、2−シータ=18.7、11.7、19.2、7.8、14.1、14.9、9.4°に特異的ピークを持つX線粉末回折パターンを有する本薬剤の結晶形態、形態3、が提供される。
【0044】
本発明によれば、2−シータ=18.7、11.7、19.2、7.8、14.1、14.9、9.4、15.6、16.1および9.6°に特異的ピークを持つX線粉末回折パターンを有する本薬剤の結晶形態、形態3、が提供される。
【0045】
本発明によれば、CuKa照射を用いた、図3に示されるX線粉末回折パターンを有する本薬剤の結晶形態、形態3、が提供される。
【0046】
【表2】
【0047】
DSC分析から、形態3は309.3℃で融解を開始することがわかる。DSC温度記録図を図4に示す。
本発明によれば、およそ2−シータ=16.2にピークを持つX線回折パターンを有する、本薬剤の結晶形態、形態4が提供される。
【0048】
2−シータ(θ)値はCuKa照射を用いて測定された。
本発明によれば、およそ2−シータ=16.2°および20.6°に少なくとも2つの特異的ピークを持つX線粉末回折パターンを有する本薬剤の結晶形態、形態4、が提供される。
【0049】
本発明によれば、およそ2−シータ=16.2、20.6および17.7°に特異的ピークを持つX線粉末回折パターンを有する本薬剤の結晶形態、形態4、が提供される。
本発明によれば、およそ2−シータ=16.2、20.6、17.7、10.8および15.5°に特異的ピークを持つX線粉末回折パターンを有する本薬剤の結晶形態、形態4、が提供される。
【0050】
本発明によれば、およそ2−シータ=16.2、20.6、17.7、10.8、15.5、20.9、26.1、11.6、26.7および18.1°に特異的ピークを持つX線粉末回折パターンを有する本薬剤の結晶形態、形態4、が提供される。
【0051】
本発明によれば、CuKa照射を用いた、図5に示されるX線粉末回折パターンと実質的に同一のX線粉末回折パターンを有する本薬剤の結晶形態、形態4、が提供される。
本発明によれば、2−シータ=16.2°プラスマイナス0.5°2−シータに少なくとも1つの特異的ピークを持つX線粉末回折パターンを有する本薬剤の結晶形態、形態4、が提供される。
【0052】
本発明によれば、2−シータ=16.2°および20.6°に少なくとも2つの特異的ピークを持つX線粉末回折パターンを有する本薬剤の結晶形態、形態4、が提供される。ここで、前記値はプラスマイナス0.5°2−シータであってよい。
【0053】
本発明によれば、2−シータ=16.2、20.6および17.7°に特異的ピークを持つX線粉末回折パターンを有する本薬剤の結晶形態、形態4、が提供される。ここで、前記値はプラスマイナス0.5°2−シータであってよい。
【0054】
本発明によれば、2−シータ=16.2、20.6、17.7、10.8および15.5°に特異的ピークを持つX線粉末回折パターンを有する本薬剤の結晶形態、形態4、が提供される。ここで、前記値はプラスマイナス0.5°2−シータであってよい。
【0055】
本発明によれば、2−シータ=16.2、20.6、17.7、10.8、15.5、20.9、26.1、11.6、26.7および18.1°に特異的ピークを持つX線粉末回折パターンを有する本薬剤の結晶形態、形態4、が提供される。ここで、前記値はプラスマイナス0.5°2‐シータであってよい。
【0056】
本発明によれば、2−シータ=16.2°に少なくとも1つの特異的ピークを持つX線粉末回折パターンを有する本薬剤の結晶形態、形態4、が提供される。
本発明によれば、2−シータ=16.2および20.6°に少なくとも2つの特異的ピークを持つX線粉末回折パターンを有する本薬剤の結晶形態、形態4、が提供される。
【0057】
本発明によれば、2−シータ=16.2、20.6および17.7°に特異的ピークを持つX線粉末回折パターンを有する本薬剤の結晶形態、形態4、が提供される。
本発明によれば、2−シータ=16.2、20.6、17.7、10.8および15.5°に特異的ピークを持つX線粉末回折パターンを有する本薬剤の結晶形態、形態4、が提供される。
【0058】
本発明によれば、2−シータ=16.2、20.6、17.7、10.8、15.5、20.9、26.1、11.6および26.7°に特異的ピークを持つX線粉末回折パターンを有する本薬剤の結晶形態、形態4、が提供される。
【0059】
本発明によれば、CuKa照射を用いた、図5に示されるX線粉末回折パターンを有する本薬剤の結晶形態、形態4、が提供される。
【0060】
【表3】
【0061】
形態4のDSC分析から、初期事象は254.0℃に開始し、ピークは262.0℃で、その後312.0℃で融解を開始することがわかる。したがって形態4の融解開始は約312.0℃である。DSC温度記録図を図6に示す。
【0062】
形態4の別のより純粋なサンプルから、図7に示すXRDパターンおよび面間隔dスペクトルが得られた。2‐シータ値の位置および面間隔dをそれぞれ表Dおよび表Eに示す。
【0063】
【表4】
【0064】
【表5】
【0065】
表Cおよび表D中の2−シータ値の僅かなばらつきは、後述のX線粉末ディフラクトグラムにおける回折角の測定誤差が原因と考えられる。
したがって、測定誤差を考慮に入れた本発明の別の態様においては、2−シータ=16.1および20.5°に少なくとも2つの特異的ピークを持つX線粉末回折パターンを有する本薬剤の結晶形態、形態4、が提供される。
【0066】
本発明によれば、2−シータ=16.1、20.5および17.6°に特異的ピークを持つX線粉末回折パターンを有する本薬剤の結晶形態、形態4、が提供される。
本発明によれば、2−シータ=16.1、20.5、17.6、10.8および15.4°に特異的ピークを持つX線粉末回折パターンを有する本薬剤の結晶形態、形態4、が提供される。
【0067】
本発明によれば、2−シータ=16.1、20.5、17.6、10.8、15.4、20.9および26.1°に特異的ピークを持つX線粉末回折パターンを有する本薬剤の結晶形態、形態4、が提供される。
【0068】
本発明は形態2、3および4の結晶形態に関する、と述べられている場合、結晶化度は、都合良くは約60%より大きく、より都合良くは約80%より大きく、好ましくは約90%より大きく、より好ましくは約95%より大きい。最も好ましくは、結晶化度は約98%よりも大きい。
【0069】
別の態様においては、本発明は結晶形態2としての本薬剤に関する。
別の態様においては、本発明は結晶形態3としての本薬剤に関する。
別の態様においては、本発明は結晶形態4としての本薬剤に関する。
【0070】
別の態様においては、本発明は結晶形態1を実質的に含まない本薬剤の結晶形態2に関する。
別の態様においては、本発明は結晶形態1を実質的に含まない本薬剤の結晶形態3に関する。
【0071】
別の態様においては、本発明は結晶形態1を実質的に含まない本薬剤の結晶形態4に関する。
形態1を実質的に含まない結晶形態とは、形態1を30%未満しか有さない結晶形態を意味する。別の態様においては、「実質的に含まない」とは形態1を20%未満しか有さないことを意味する。別の態様においては、「実質的に含まない」とは形態1を10%未満しか有さないことを意味する。さらに別の態様においては、「実質的に含まない」とは形態1を5%未満しか有さないことを意味する。さらに別の態様においては、「実質的に含まない」とは形態1を1%未満しか有さないことを意味する。
【0072】
形態2、3および4は、図1、2および3に示したX線粉末回折パターンと実質的に同一のX線粉末回折パターンを提供し、表A、BおよびCに示した最も顕著な10のピーク(角度2−シータ値)を実質的に有している。X線粉末回折パターンの2−シータ値は、機器によって、または試料によってわずかに異なることがあるので、引用された値が絶対的なものと解釈すべきではない、ということが理解されるであろう。
【0073】
測定条件(使用する装置または機器など)によっては、1つまたは複数の測定誤差を有するX線粉末回折パターンが得られることがある、ということは公知である。特に、X線粉末回折パターンの強度は測定条件によって変動し得るということは、一般的に知られている。したがって、本発明の形態2、3および4は図1、2および3に示したX線粉末回折パターンと同一のX線粉末回折パターンを提供する結晶に限定されるわけではなく、図1、2および3に示したX線粉末回折パターンと実質的に同一のX線粉末回折パターンを提供するいずれの結晶も本発明の範囲内に含まれることを理解すべきである。X線粉末回折の当業者であれば、X線粉末回折パターンの実質的な同一性を判断することが可能である。
【0074】
X線粉末回折の当業者であれば、ピークの相対強度が、例えば、試料の分析にも影響し得る、30ミクロンを超える大きさの粒子および非ユニタリーアスペクト比に影響されることがある、ということは理解されるであろう。該当業者は、反射の位置が、回折計に置かれた試料の正確な高さ、および回折計のゼロ較正によって影響されることがある、ということも理解するであろう。試料の表面平面性もわずかに影響する場合がある。ゆえに、示された回折パターンデータが絶対的な値であると解釈すべきではない。(Jenkins, R & Snyder, R.L. ‘Introduction to X-Ray Powder Diffractometry’ John Wiley & Sons 1996; Bunn, C.W. (1948), Chemical Crystallography, Clarendon Press, London; Klug, H. P. & Alexander, L. E. (1974), X-Ray Diffraction Procedures)。
【0075】
一般に、X線粉末回折における回折角の測定誤差は、約5%以下、特にプラスマイナス0.5°2−シータである。典型的にはプラスマイナス0.2°2−シータである。図1、2、3および4のX線粉末回折パターンについて検討する場合や表A、B、CおよびDを読む場合には、こうした程度の測定誤差を考慮に入れておくべきである。さらに、実験条件および試料調製(優先配向)によって強度が変動する可能性があることを理解すべきである。
【0076】
使用した技術の詳細
X線粉末回折
【0077】
【表6】
【0078】
分析機器:シーメンスD5000
X線粉末回折スペクトルは、結晶材料の試料をシーメンスのシリコン単結晶(SSC)ウェハマウントに載せ、該試料を、顕微鏡用スライドで広げて薄層にすることによって決定した。試料を毎分30回転で回転させ(計数統計を改善するため)、1.5406オングストロームの波長で、40kVおよび40mAで作動させた銅製ロングファインフォーカス管(long−fine focus tube)によって発生させたX線を照射した。平行X線源はV20に設定された自動可変発散スリットを通過し、反射した放射線は2mmの散乱線除去スリット(antiscatter slit)と0.2mmの検出器スリットを通って方向づけられた。試料を、シータ−シータモードで2度から40度の2−シータ範囲にわたって、0.02度の2−シータ増加分あたり1秒間暴露した(連続スキャンモード)。稼働時間は31分41秒であった。機器には検出器としてシンチレーション計数管が備えられていた。制御およびデータ収集には、Diffract+ソフトウェアで作動する、デルOptiplex 686 NT 4.0 Workstationを用いた。X線粉末回折の当業者であれば、ピークの相対強度が、例えば、試料の分析にも影響し得る、30ミクロンを超える大きさの粒子および非ユニタリーアスペクト比に影響されることがある、ということは理解するであろう。該当業者は、反射の位置が、回折計に置かれた試料の正確な高さ、および回折計のゼロ較正によって影響されることがある、ということも理解するであろう。試料の表面平面性もわずかに影響する場合がある。ゆえに、示された回折パターンデータが絶対的な値であると解釈すべきではない。
【0079】
示差走査熱量測定
分析機器:ティー・エイ・インスツルメントQ1000 DSC
典型的には、蓋を取り付けた40μlのアルミ製の鍋に入った5mg未満の材料を、25℃〜325℃の温度範囲にわたって、1分間当たり10℃の一定の加熱速度で加熱した。窒素を使用したパージガスを用いた(流速100ml/分)。
【0080】
形態2、3および4は形態1からの競合スラリー化(competitive slurring)または播種によって調製することができる。
形態2はアセトニトリル中での競合スラリー化によって都合よく調製することができる。特にこれは45〜55℃の温度範囲、例えば約50℃で行われる。
【0081】
形態3はメタノール中での競合スラリー化によって都合よく調製することができる。特にこれは15〜30℃の温度範囲、例えば周囲温度近辺で行われる。
形態4は酢酸エチル中での競合スラリー化によって都合よく調製することができる。特にこれは15〜30℃の温度範囲、例えば周囲温度近辺で行われる。また、形態4は高温のアセトンまたはアセトニトリル中での競合スラリー化によって調製することができる。
【0082】
上述したように、本薬剤は11βHSD1阻害活性を有する。この特性は、以下のアッセイによって評価することができる。
アッセイ
11βHSD1の酸化還元酵素(oxo−reductase)活性による、コルチゾンのその活性型ステロイドであるコルチゾールへの変換は、競合均一時間分解蛍光アッセイ(HTRF)(セイエス ビオ アンテルナスィオナル(CisBio International)、研究開発、 管理および欧州事務所、インビトロテクノロジー(In Vitro Technologies)―HTRF(登録商標)/Bioassays BP 84175, 30204 バニョル/セーズ Cedex、フランス。Cortisol bulk HTRF kit: Cat No. 62CORPEC)によって測定することができる。
【0083】
本明細書に記載の化合物の評価は、N−末端に6−Hisタグを有する全長ヒト11βHSD1酵素(*1)を発現したバキュロウイルスを使用して行った。該酵素は、銅キレートカラムを用いて、洗剤で可溶化した細胞溶解物から精製した。11βHSD1の阻害剤はコルチゾンのコルチゾールへの変換を低減させるが、そのことは、上記アッセイにおいてシグナルの増加によって同定される。
【0084】
被験化合物を、10mMになるようにジメチルスルホキシド(DMSO)中に溶解させ、さらに1%DMSO含有アッセイ緩衝液で最終アッセイ濃度である10倍まで希釈した。次に、希釈化合物を黒384ウェルプレート(マトリックス(Matrix)、ハドソン、ニューハンプシャー州、米国)にまいた。
【0085】
アッセイは、コルチゾン(シグマ、プール、ドーセット州、英国、160nM)、グルコース−6−リン酸(ロシュ・ダイアグノスティックス、1mM)、NADPH(シグマ、プール、ドーセット州、英国、100μM)、グルコース−6−リン酸脱水素酵素(ロシュ・ダイアグノスティックス、12.5μg/ml)、EDTA(シグマ、プール、ドーセット州、英国、1mM)、アッセイ緩衝液(K2HPO4/KH2PO4、100mM)pH7.5、組換え11βHSD1および試験化合物からなる全量20μl中で(実行可能なアッセイウィンドウを得るために適切な希釈溶液を用いて−好適な希釈溶液の例としては、ストック酵素の1000倍希釈溶液がある)行った。アッセイプレートを37℃で25分間インキュベートし、その後、10μlの0.5mMのグリセルレチン酸、および抱合型コルチゾール(XL665またはD2)を添加して反応を停止した。次に、10μlの抗−コルチゾールクリプテートを添加し、プレートを密封して、室温で6時間インキュベートした。665nmおよび620nmでの蛍光を測定し、665nm:620nmの比をEnvisionプレートリーダーを用いて算出した。
【0086】
次に、これらのデータを使って、各化合物のIC50値(Origin 7.5、マイクロカルソフトウェア(Microcal software)、ノーサンプトン、マサチューセッツ州、米国)および/または30μMの化合物における阻害率(%)を算出した。
*1 The Journal of Biological Chemistry, Vol. 26, No 25, pp16653-16658
次の結果が得られた:参考例1 IC50 0.008μM
本発明の化合物の経口バイオアベイラビリティは、次のように試験することができる。
【0087】
PK試験におけるバイオアベイラビリティの決定
化合物を、25%HPBCDを含有するpH5.5ソレンソン緩衝液(sorrensons buffer)製剤に入れ、2mg/kg(2ml/kg)を静脈投与、5mg/kg(5ml/kg)を経口投与する。両方の経路について、血液サンプル(200μl)を、投与前、投与後0.25、0.5、1、2、3、4、5、6、8および24時間に採取し、遠心分離により血漿を調製する。血漿サンプルを下記のように分析する。PKパラメーター(クリアランス、分布容積、バイオアベイラビリティ、吸収率など)を、適切なPKソフトウェア(WinNon-Lin)を使用して標準PK法により算出する。
【0088】
血漿サンプルの生物分析
以下に説明するのは、DMPK解析に使用されるすべてのPK試験種に対して研究対象化合物を単一化合物投与またはカセット投与した後に、血漿サンプルを手作業で調製するための指針である。オープンアクセス(LC−MS/MS)またはマニュアル手法(LC−MS)による分析について説明する。
【0089】
内容
1.材料
2.一般的抽出方法
3.一般的プレート配置による実施例サンプルリスト
4.オープンアクセスのバッチサブミッションとシステムチェック
5.バッチパスの許容基準
1.材料
溶媒:メタノール、アセトニトリルおよびDMSO
水:精製またはHPLCグレード
1ml浅型96ウェルプレートまたはエッペンドルフチューブ
2ml深型96ウェルプレートおよび蓋
ブランク(対照)血漿
2.一般的抽出方法
化合物(1種または複数)を、塩係数も考慮に入れながら、DMSOを用いて可溶化し1mg/mlとする。このDMSOストック(1種または複数)を用いて、すべての検量線および品質管理(QC)サンプルを作成することができる。
2.i 単一化合物分析
2.i.a 検量線およびQCサンプルの作成:
1.標準溶液を以下のように調製する。
【0090】
【表7】
【0091】
2.50μlのブランク血漿を1mlの96ウェルプレート(浅型ウェル)の1つのウェルに移す。
3.5μlの各標準溶液を該プレートの他のウェルに移す。
【0092】
4.50μlのブランク血漿をこれらの各ウェルに加える。
5.QCサンプルを生成するために、100ng/ml、1,000ng/ml、および10,000ng/mlの各標準溶液について、各5μlを該プレートに加える(各濃度につきQCは3回)。
【0093】
6.50μlのブランク血漿をこれらのウェルのそれぞれに加える。
7.50μlの各PKサンプルを1mlの96ウェルプレートに移す。
8.5μlのメタノール(−化合物)を各PKサンプルに加える。
【0094】
9.すべての投与製剤がボルテックス混合で十分に混和しているか確認する。
10.予測濃度の静脈内(IV)および経口(PO)投与用の製剤を10μg/mlとなるようにメタノールで希釈する(例えば、予測濃度が2mg/mlとなるように調製された製剤は、1:200に希釈して10μg/mlの溶液とする)。
【0095】
11.血漿を50μlずつ(×6)プレートに加える。そのうちの3個のウェルに希釈IV製剤5μlを加え、残りの3個のウェルにはPO製剤を同様に加える。
12.研究関連内部標準(1μg/ml)を含有する100μlのアセトニトリルを、すべての検量線、QC、PKおよび製剤サンプルに加えることによって、タンパク質を沈殿させる。
【0096】
13.プレートをボルテックス混合後、4、000gで10分間遠心分離する。
14.100μlの上清を2mのl96ウェルプレートの各ウェル(下記のプレートマップ参照)に移す。ペレットを乱さないように注意する。
【0097】
15.50:50のメタノール:水、約1.5mlを後者のウェルに加える。
16.トリプルクワッド(triple quad)システムでの分析用に、400μlの水(HPLCグレード)を各サンプルに加え、静かに混合する。
【0098】
17.2mlプレートに、100,000ng/mlの各標準溶液のストック100μlを加え、次いで水900μlを加える。次に、他の一つのウェルに内部標準のサンプルを加える(プレートマップ参照)。これらは化合物調整のために行う(プレートマップ上に溶液調整と表示)。
【0099】
18.プラットフォームシステムでの分析用に、100μlの水(HPLCグレード)を各サンプルに加え、静かに混合する。
19.調製した化合物溶液を用いて、全化合物を手作業で5,000ng/mlに調整する(50,000ng/ml標準溶液100μlを900μlの水に添加する)。
2.ii カセット投与分析
2.iia 検量線およびQCサンプルの作成:
注記:カセット投与では、1mg/mlのストックを希釈するために必要なメタノールの量は、存在する化合物の数にしたがって調整する。
【0100】
1.バイアルに、必要とされる各1mg/mlのストック100μlを入れる。
2.必要量のメタノールを加え、全量1mlとする。
3.単一化合物の分析の場合と同様、以降の工程(上記工程2〜16)をすべて行う。
2.iii PKサンプルが定量上限(ULOQ)を超える場合。
【0101】
1.上記(工程1〜6)と同様にさらなる検量線およびQCサンプルを作成する。
2.50μl未満(例えば25μl)のULOQを超えるPKサンプルを移す。
3.これらのサンプルに十分なコントロール血漿を加え、最終血漿量を50μlとする。行った希釈を記録しておく。
【0102】
4.残りの全てのPKサンプルをそれぞれ50μl移す。
5.上記と同様(工程8〜16)、すべての製剤サンプルを調製し、すべてのサンプルを抽出する。
注記:検量線作成に使用する上限濃度は見直してもよいが、HPLCカラムまたはMS機器が飽和しないように注意しなければいけない。これはPKサンプルの希釈が推奨されているためである。
2.iv 感度が低い(定量下限(LLOQ)が高い)場合。
注記:血漿濃度の大部分が定量下限以下であるか、LLOQが10ng/mlより高い場合を高LLOQとする。これらのいずれかに当てはまる場合には、下記の方法を適用する。
【0103】
本発明のさらなる態様によれば、薬学的に許容される希釈剤または担体といっしょに、上記で定義したような、本薬剤、またはその薬学的に許容される塩を含む医薬組成物が提供される。
【0104】
本発明の組成物は、経口使用(例えば、錠剤、ロゼンジ剤、硬もしくは軟カプセル剤、水性もしくは油性の懸濁剤、乳剤、分散性粉剤もしくは顆粒剤、シロップ剤またはエリキシル剤として)、局所使用(例えば、クリーム剤、軟膏剤、ゲル剤、水性もしくは油性の液剤または懸濁剤として)、吸入投与(例えば、微粉または液体エアゾールとして)、通気投与(例えば、微粉として)、または非経口投与(例えば、静脈内、皮下、筋肉内、もしくは筋肉内投与用の無菌性の水性もしくは油性液剤として、または直腸投与用坐薬として)に適した形態であってよい。一般に、経口使用に適した形態の組成物が好ましい。
【0105】
本発明の組成物は、当技術分野において周知である、従来の医薬用賦形剤を使用する従来の手順により得ることができる。したがって、経口使用のための組成物は、例えば、1種または複数の着色剤、甘味剤、賦香剤、および/または防腐剤を含有していてもよい。
【0106】
錠剤用の好適な薬学的に許容される賦形剤としては、例えば、乳糖、炭酸ナトリウム、リン酸カルシウムまたは炭酸カルシウムなどの不活性希釈剤、コーンスターチまたはアルゲン酸などの造粒剤および崩壊剤、澱粉などの結合剤、ステアリン酸マグネシウム、ステアリン酸またはタルクなどの滑沢剤、p−ヒドロキシ安息香酸エチルまたはp−ヒドロキシ安息香酸プロピルなどの防腐剤、およびアスコルビン酸などの抗酸化剤が挙げられる。錠剤は、消化管内での崩壊およびそれに続く活性成分の吸収を加減するため、または錠剤の安定性および/もしくは外見を改良するために、コーティングされていなくてもされていてもよいが、いずれの場合においても、当技術分野において周知の従来のコーティング剤および手順を用いる。
【0107】
経口使用のための組成物は、活性成分が、炭酸カルシウム、リン酸カルシウムもしくはカオリンなどの不活性な固形希釈剤と混合されている、硬ゼラチンカプセル剤の形態、または活性成分が、水、またはピーナッツ油、流動パラフィンもしくはオリーブ油などの油と混合されている、軟ゼラチンカプセル剤の形態であってよい。
【0108】
水性懸濁剤は通常、微粉化した形態の活性成分ともに、1種または複数の、カルボキシメチルセルロースナトリウム、メチルセルロース、ヒドロキシプロピルメチルセルロース、アルギン酸ナトリウム、ポリビニル−ピロリドン、トラガカントゴムおよびアラビアゴムなどの懸濁剤、レシチン、またはアルキレンオキシドと脂肪酸との縮合生成物(例えば、ステアリン酸ポリオキシエチレン)、もしくはエチレンオキシドと長鎖脂肪族アルコールとの縮合生成物(例えば、ヘプタデカエチレンオキシセタノール)、もしくはエチレンオキシドと脂肪酸およびヘキシトールに由来する部分エステルとの縮合生成物(例えば、ポリオキシエチレンソルビトールモノオレアート)、もしくはエチレンオキシドと長鎖脂肪族アルコールとの縮合生成物(例えば、ヘプタデカエチレンオキシセタノール)、もしくはエチレンオキシドと脂肪酸およびヘキシトールに由来する部分エステルとの縮合生成物(例えば、ポリオキシエチレンソルビトールモノオレアート)、もしくはエチレンオキシドと脂肪酸およびヘキシトール無水物に由来する部分エステルとの縮合生成物(例えば、ポリエチレンソルビタンモノオレアート)などの分散剤または湿潤剤を含有する。また水性懸濁剤は、1種または複数の防腐剤(例えば、p−ヒドロキシ安息香酸エチルまたはp−ヒドロキシ安息香酸プロピル)、抗酸化剤(例えば、アスコルビン酸)、着色剤、賦香剤、および/または甘味剤(例えば、ショ糖、サッカリン、またはアスパルテーム)を含有していてもよい。
【0109】
油性懸濁剤は、活性成分を植物油(例えば、ラッカセイ油、オリーブ油、ゴマ油もしくはココナツ油)または鉱油(例えば、流動パラフィン)に懸濁させることによって製剤化することができる。また油性懸濁剤は、ミツロウ、固形パラフィンまたはセチルアルコールなどの増粘剤を含有していてもよい。口当たりのよい経口剤とするために、上述したような甘味剤、および賦香剤を添加してもよい。これらの組成物は、アスコルビン酸などの抗酸化剤を添加することによって保存することができる。
【0110】
水の添加による水性懸濁剤の調製に好適な分散性粉剤および顆粒剤は通常、活性成分とともに、分散剤または湿潤剤、懸濁剤、および1種または複数の防腐剤を含有する。好適な分散剤または湿潤剤、および懸濁剤は、すでに上述したものが代表的である。甘味剤、賦香剤および着色剤などの賦形剤がさらに存在してもよい。
【0111】
本発明の医薬組成物は、水中油型乳剤の形態であってもよい。油相は、オリーブ油やラッカセイ油などの植物油、流動パラフィンなどの鉱油、またはこれらのいずれかの混合物であってよい。好適な乳化剤は、例えば、アラビアゴムやトラガカントゴムなどの天然ゴム、大豆、レシチンなどの天然リン脂質、脂肪酸およびヘキシトール無水物に由来するエステルまたは部分エステル(例えば、ソルビタンモノオレアート)、ならびに該部分エステルとエチレンオキシドとの縮合生成物(例えば、ポリオキシエチレンソルビタンモノオレアート)であってよい。乳剤は、甘味剤、賦香剤および防腐剤を含有していてもよい。
【0112】
シロップ剤およびエリキシル剤は、グリセロール、プロピレングリコール、ソルビトール、アスパルテームまたはショ糖などの甘味剤とともに製剤化することができ、粘滑剤、防腐剤、賦香剤および/または着色剤を含有していてもよい。
【0113】
医薬組成物は、滅菌注射用の水性または油性懸濁液の形態であってもよく、上述した適当な分散剤または湿潤剤、および懸濁剤のうちの1種または複数を用いて、公知の手順に従って製剤化することができる。滅菌注射用の製剤は、無毒性で非経口的に許容される希釈剤もしくは溶剤中の滅菌注射用の溶液または懸濁液(例えば、1,3−ブタンジオール中の溶液)であってもよい。
【0114】
吸入投与用の組成物は、超微粒子状の固体小滴または液体小滴のいずれかを含有するエアゾールとして活性成分を調合するように調整された、従来の加圧エアゾールの形態であってよい。揮発性のフッ素化炭化水素や炭化水素などの従来のエアゾール噴射剤を使用することができ、エアゾール装置は、定量の活性成分を調合するように調整されると都合が良い。
【0115】
製剤に関するさらなる情報については、Comprehensive Medicinal Chemistry (Corwin Hansch; Chairman of Editorial Board), Pergamon Press 1990の第5巻、25.2章を参照されたい。
【0116】
単一剤形を製造するために1種または複数の賦形剤と組み合わされる活性成分の量は、被治療者および具体的な投与経路によって必然的に異なってくる。例えば、ヒトに経口投与するための製剤は、例えば、組成物全体の約5から約98重量%の間で異なり得る適切かつ使いやすい量の賦形剤と混合された、0.5mg〜2gの活性成分を通常含有する。投与単位剤形は、約1mg〜約500mgの活性成分を通常含有する。投与経路および投与法に関するさらなる情報については、Comprehensive Medicinal Chemistry (Corwin Hansch; Chairman of Editorial Board), Pergamon Press 1990の第5巻、25.3章を参照されたい。
【0117】
本発明者らは、本薬剤、またはその薬学的に許容される塩は、有効な11βHSD1阻害剤であり、したがってメタボリック症候群に関連する病態の治療において価値があることを見出した。
【0118】
当然のことながら、本明細書で「メタボリック症候群」という語を使用する場合は、1)および/もしくは2)の定義、またはこの症候群について認められているその他のあらゆる定義のメタボリック症候群に関する。当技術分野で使用される「メタボリック症候群」の同義語としては、リーベン症候群、インスリン抵抗性症候群および症候群Xが挙げられる。当然のことながら、本明細書で「メタボリック症候群」という語を使用する場合は、リーベン症候群、インスリン抵抗性症候群および症候群Xのことも指している。
【0119】
本発明のさらなる態様によれば、ヒトなどの温血動物の予防的または治療的処置方法における使用のための、上記に定義したような本薬剤、またはその薬学的に許容される塩が提供される。
【0120】
したがって、本発明のこの態様によれば、医薬として使用するための本薬剤、またはその薬学的に許容される塩がある。
本発明の別の特徴によれば、ヒトなどの温血動物での11βHSD1阻害作用の発現に使用される医薬の製造における、本薬剤、またはその薬学的に許容される塩の使用が提供される。
【0121】
11βHSD1阻害作用の発現、または11βHSD1阻害作用を発現させることに言及する場合、好適には、メタボリック症候群の治療を指す。また、11βHSD1阻害作用の発現に言及する場合は、糖尿病、肥満、高脂血症、高血糖症、高インスリン血症または高血圧症の治療を指し、特に2型糖尿病および肥満の治療を指す。あるいは、11βHSD1阻害作用の発現に言及する場合は、緑内障、骨粗しょう症、結核、痴呆、認知障害または鬱病の治療を指す。
【0122】
また、11βHSD1阻害作用の発現に言及する場合は、例えば、言語流暢性、言語記憶もしくは論理的記憶の改善による個人の認知能力の改善といった認知障害の治療、または軽度の認知障害の治療を指す。例えば、国際公開第03/086410号およびそれに含まれる参考文献、ならびにProceedings of National Academy of Sciences (PNAS), 2001, 98(8), 4717-4721を参照のこと。
【0123】
また、11βHSD1阻害作用の発現に言及する場合は、アテローム性動脈硬化症の治療、その発症の遅延、および/またはそのリスクの低減を指す。例えば、J. Experimental Medicine, 2005, 202(4), 517-527を参照のこと。
【0124】
また、11βHSD1阻害作用の発現に言及する場合は、アルツハイマー病および/または神経変性障害の治療を指す。
本発明のこの態様のさらなる特徴によれば、11βHSD1阻害作用を発現する治療を必要としているヒトなどの温血動物において11βHSD1阻害作用を発現する方法であって、該動物に、有効な量の式(1)の化合物、またはその薬学的に許容される塩を投与することを含む方法が提供される。
【0125】
治療薬におけるそれらの使用に加えて、本薬剤、またはその薬学的に許容される塩は、新しい治療薬の探索の一環として、ネコ、イヌ、ウサギ、サル、ラットおよびマウスなどの実験動物における11βHSD1阻害剤の効果を評価するための、インビトロおよびインビボ試験系の開発および標準化における薬理学的手段としても有用である。
【0126】
本明細書に記載の11βHSD1の阻害は、単一療法として適用してもよいか、または、本発明の主題に加えて、1種または複数の他の物質および/もしくは治療を伴ってもよい。そのような共同治療(conjoint treatment)は、治療の個々の成分を、同時に、順次、または別々に投与することにより行うことができる。同時の治療は、単一の錠剤または別々の錠剤で行ってよい。例えば、11βHSD1阻害剤、特に本発明の11βHSD1阻害剤と同時投与できる薬剤としては、治療別に大きく分けた以下のものが挙げられる。
1)インスリンおよびインスリン類似体;
2)スルホニル尿素薬などのインスリン分泌促進薬(例えば、グリベンクラミド、グリピジド)、食後血糖調節薬(例えば、レパグリニド、ナテグリニド)、グルカゴン様ペプチド1作動薬(GLP1作動薬)(例えば、エクセナチド、リラグルチド)およびジペプチジルペプチダーゼIV阻害剤(DPP−IV阻害剤);
3)PPARγ作動薬などのインスリン増感剤(例えば、ピオグリタゾンおよびロシグリタゾン);
4)肝糖産生を抑制する薬剤(例えば、メトホルミン);
5)腸からのグルコースの吸収を抑えるように設計された薬剤(例えば、アカルボース);
6)持続性高血糖症の合併症を治療するように設計された薬剤;例えば、アルドース還元酵素阻害剤
7)リン酸化チロシンホスファターゼ阻害剤、グルコース−6−ホスファターゼ阻害剤、グルカゴン受容体拮抗剤、グルコキナーゼ活性化剤、グリコーゲンホスホリラーゼ阻害剤、フルクトース1,6ビスホスファターゼ阻害剤、グルタミン:フルクトース−6−リン酸アミドトランスフェラーゼ阻害剤などのその他の抗糖尿病剤;
8)抗肥満剤(例えば、シブトラミンおよびオルリスタット);
9)HMG−CoA還元酵素阻害剤(プラバスタチンなどのスタチン類);PPARα作動薬(ゲムフィブロジルなどのフィブラート類);胆汁酸金属イオン封鎖剤(コレスチラミン);コレステロール吸収阻害剤(植物スタノール、合成阻害剤);回腸胆汁酸吸収阻害剤(IBATi)、コレステロールエステル転送タンパク質阻害剤、ならびにニコチン酸および類似体(ナイアシンおよび徐放性製剤)などの抗脂質異常症薬;
10)β遮断剤(例えば、アテノロール、インデラル);ACE阻害剤(例えば、リシノプリル);カルシウム拮抗剤(例えば、ニフェジピン);アンジオテンシン受容体拮抗剤(例えば、カンデサルタン)、α拮抗剤および利尿剤(例えば、フロセミド、ベンズチアジド)などの抗高血圧剤;
11)抗血栓剤、繊維素溶解活性化剤および抗血小板剤;トロンビン拮抗剤;因子Xa阻害剤;因子VIIa阻害剤;抗血小板剤(例えば、アスピリン、クロピドグレル);抗凝固剤(ヘパリンおよび低分子量類似体、ヒルジン)ならびにワルファリンなどの止血調節剤;
12)非ステロイド系抗炎症薬(例えば、アスピリン)およびステロイド系抗炎症薬(例えば、コルチゾン)などの抗炎症剤;および
13)腎臓によるグルコースの再吸収を防ぐ薬剤(SGLT阻害剤)。
【実施例】
【0127】
次に、本発明を下記の実施例により説明するが、特に断りがない限り、
(i)温度は摂氏(℃)で表し、操作は、室温または周囲温度、すなわち18〜25℃の温度範囲で、かつアルゴンなどの不活性ガス雰囲気下で行った。
(ii)溶媒の留去は、ロータリーエバポレーターを用いて、減圧下(600〜4000Pa;4.5〜30mmHg)、60℃以下の浴温で行った。
(iii)クロマトグラフィーは、シリカゲルのフラッシュクロマトグラフィーを意味する。
(iv)通常、反応の経過はTLCにより追跡し、反応時間は例示のためにのみ示す。
(v)収率は例示のためにのみ示し、必ずしも入念な過程開発によって得ることができるものではない。さらに物質が必要な場合には、調製を繰り返した。
(vi)NMRデータ(1H)は、示されている場合、特に断りがない限りは、溶媒として重水素化ジメチルスルホキシド(DMSO−d6)を用いて(特に断りがない限り)300または400MHzで測定し、テトラメチルシラン(TMS)に対する百万分率(ppm)で示した、主要な診断プロトンのデルタ値の形態である。ピークの多重度は次のように示す。s、シングレット;d、ダブレット;dd、ダブル−ダブレット;dt、ダブル−トリプレット;dm、ダブル−マルチプレット;t、トリプレット;m、マルチプレット;br、ブロード。
(vii)化学記号は通常の意味を有し、SI単位系および記号を用いる。
(viii)溶媒の割合は体積:体積(v/v)で表す。
(ix)質量スペクトル(MS)は、直接暴露プローブを用いて、化学イオン化(CI)モードにおいて70電子ボルトの電子エネルギーで実施した。イオン化は、示されている場合、電子衝撃(EI)、高速原子衝撃(FAB)または電子スプレー(ESP)により行った。m/zの値を示し、通常は、親質量を示すイオンのみを報告する。
(x)相対量(rel vol)とは、重要中間体の量と比較した相対的な量である。相対量は通常溶媒の量を指すのに用いられる。例えば重要中間体が100gで1000mlの溶媒を使用した場合、これは10相対量の溶媒と呼ばれる。
(xi)下記の略語を、以下において、または上記の工程の箇所で使用することがある。
【0128】
Et2O ジエチルエーテル
DMF ジメチルホルムアミド
DCM ジクロロメタン
THF テトラヒドロフラン
DMSO ジメチルスルホキシド
EtOAc 酢酸エチル
MTBE メチルtert−ブチルエーテル
DSC 示差走査熱量測定
参考例1
4−[4−(2−アダマンチルカルバモイル)−5−tert−ブチル−ピラゾル−1−イル]安息香酸
【0129】
【化2】
【0130】
2Mの水酸化ナトリウム水溶液(51.7mL、103.32mmol)をメタノール(100mL)中の4−[4−(2−アダマンチルカルバモイル)−5−tert−ブチル−ピラゾル−1−イル]安息香酸メチル(中間体#1)(4.5g、10.33mmol)に加えた。混合物を70℃で1時間攪拌し、次いで、周囲温度まで冷却し、減圧下で濃縮し、水(100mL)で希釈した。反応混合物を2MのHClでpH3に調節した。反応混合物をEtOAc(500mL)で抽出し、水(2×100mL)および飽和ブライン(50mL)で連続して洗浄した。有機層をMgSO4で乾燥させ、濾過し、エバポレートし、淡黄色の固体を得た。固体をEtOAc(20mL)で洗浄し、濾過により収集し、真空下で乾燥させ、4−[4−(2−アダマンチルカルバモイル)−5−tert−ブチル−ピラゾル−1−イル]安息香酸(3.89g、89%)を、クリーム色の結晶性固体として得た。
1H NMR(400.13MHz、DMSO−d6)δ1.19(9H、s)、1.49(2H、d)、1.70−1.96(10H、m)、2.09(2H、d)、3.98−4.01(1H、m)、7.49−7.53(2H、m)、7.61(1H、s)、8.06−8.09(2H、m)、8.20(1H、d)、13.30(1H、s)
m/z(ESI+)(M+H)+=422
m.p.308.8℃(開始)。
【0131】
参考例1は次のように調製することもできる。
水酸化ナトリウム水溶液(2M)(2.5当量)を、20℃のメタノール(10容量)中の4−[4−(2−アダマンチルカルバモイル)−5−tert−ブチル−ピラゾル−1−イル]安息香酸メチル(中間体#1)(1.0当量)の攪拌懸濁液に、5分間かけて少しずつ加えた(発熱20〜27℃)。得られた懸濁液を1時間、70℃(ジャケット温度)に加熱した(バッチ還流約60〜65℃)(LCMSにより完了)。オレンジ色の反応混合物を20℃まで冷却し(溶液にはわずかに混濁が残った)、セライトを通して濾過し少量の固体を除去した。次いで、濾液をフランジフラスコに注ぎ込み、水(25容量)を加えた。次いで、混合物を2MのHCl(約800〜850ml)でpH3に調節した(非常に濃くなる)。次いで水層を濾過し、淡黄色の固体を水で洗浄し、一晩吸引乾燥し、アセトニトリルおよび最後に1:1のアセトニトリル/ジエチルエーテルで洗浄し、真空下、50℃で72時間(週末)乾燥させて、4−[4−(2−アダマンチルカルバモイル)−5−tert−ブチル−ピラゾル−1−イル]安息香酸(80%)を固体として得た。
【0132】
中間体#2:4−ヒドラジニル安息香酸メチル塩酸塩
【0133】
【化3】
【0134】
ジオキサン中の4Mの塩化水素(100mL、399.60mmol)をMeOH(200mL)中の4−ヒドラジノ安息香酸(15.2g、99.90mmol)に加えた。得られた懸濁液を90℃で5時間攪拌した。20℃まで冷却した後、沈殿物を濾過により収集し、Et2O(100mL)で洗浄し、真空下で乾燥させ、2−(4−(メトキシカルボニル)フェニル)ヒドラジニウムクロリド(16.50g、82%)をクリーム色の結晶性固体として得た。
m/z(ESI−)(M−H)−=165;HPLCtR=1.12分
1H NMR(400.13MHz、DMSO−d6)δ3.81(3H、s)、6.99−7.02(2H、m)、7.86−7.90(2H、m)、8.98(1H、s)、10.47(3H、s)。
【0135】
中間体#2は次のように調製することもできる。
メタノール性塩酸溶液(4M)(4当量、新たに調製)を、窒素下で、メタノール(12.6容量)中の4−ヒドラジノ安息香酸懸濁液(1当量)に加えた。混合物を還流下で3時間攪拌し、次いで15℃未満まで冷却した。濾過により固体を収集し、MTBE(6.5容量)で洗浄し、風乾し、生成物を固体として得た。
TLC DCM:MeOH、9:1、生成物Rf0.87
mp 233.8〜234.6℃。
【0136】
中間体#3:N−(2−アダマンチル)−4,4−ジメチル−3−オキソ−ペンタンアミド
【0137】
【化4】
【0138】
THF中のリチウムビス(トリメチルシリル)アミド溶液の1M溶液(22.84ml、22.84mmol)をTHF(25mL)に加え、窒素下で−78℃まで冷却した。THF(25mL)中の3,3−ジメチル−2−ブタノン(2.287g、22.84mmol)の溶液を5分間にわたって滴下した。得られた溶液を、窒素下、−78℃で15分間攪拌した。THF(20mL)中の2−イソシアナトアダマンタン(R.Reck & C.Jochims Chem. Ber. 115(1982) p864の方法によって2−アダマンチルアミン塩酸塩から調製)(3.68g、20.76mmol)の溶液を5分間にわたって加えた。得られた溶液を−78℃で1時間攪拌した後、1時間かけて20℃まで昇温させた。反応混合物を飽和NH4Cl(150mL)に注ぎ込み、EtOAc(2×100mL)で抽出した。有機層を水(50mL)およびブライン(50mL)で洗浄し、MgSO4で乾燥させ、濾過し、エバポレートし、黄色の油を得た。粗生成物をフラッシュシリカクロマトグラフィー(溶出勾配:イソヘキサン中の0〜50%EtOAc)によって精製した。精製画分を蒸発乾固し、N−(2−アダマンチル)−4,4−ジメチル−3−オキソ−ペンタンアミド(4.64g、81%)を白色固体として得た。
1H NMR(400.13MHz、DMSO−d6)δ1.08−1.09(9H、m)、1.50(2H、d)、1.66−1.89(10H、m)、1.95−2.00(2H、m)、3.53(1.4H、s)、3.80−3.94(1H、m)、5.30(0.3H、s)、7.77−7.87(1H、m)、14.43(0.3H、s)(ケト型とエノール型の2:1混合物)
m/z(ESI+)(M+H)+=278。
【0139】
中間体#3は次のように調製することもできる。
水酸化ナトリウム水溶液(3M)(5容量)を水(5容量)中の2−アダマンチルアミン塩酸塩(1当量)の攪拌懸濁液に加えた。DCM(5容量)を得られた濃厚な懸濁液に加え、相を分離した。水層をDCM(4×5容量)で抽出し、合わせた有機層を濃縮し、遊離アミンを白色固体として得た。
【0140】
エチルピバロイルアセテート(1当量)を、窒素下で、キシレン(6.5容量)中の遊離アミンの懸濁液に加え、混合物を還流下で6.5時間攪拌した。バッチを室温まで冷却し、濃縮乾固した。残留物を、トルエン(3×1容量)、次いでヘキサン(3×1容量)でパージした。得られた固体を50℃で5分間ヘキサン中で攪拌し、その後室温まで冷却した。白色固体を濾過し、ヘキサン(2容量)で洗浄し風乾させた。
TLC ヘキサン:EtOAc、1:1、生成物Rf0.66
mp 124.5〜125.1℃。
【0141】
中間体#4:(2)−N−(2−アダマンチル)−2−(ジメチルアミノメチリデン)−4,4−ジメチル−3−オキソ−ペンタンアミド
【0142】
【化5】
【0143】
N,N−ジメチルホルムアミドジメチルアセタール(3.02mL、22.71mmol)を、窒素下で、1,4−ジオキサン(50mL)中のN−(2−アダマンチル)−4,4−ジメチル−3−オキソ−ペンタンアミド(中間体#3)(5.25g、18.93mmol)の攪拌懸濁液に加えた。得られた混合物を100℃で2時間攪拌した。反応混合物を蒸発乾固し、得られた淡いクリーム色の固体を真空下で乾燥させ、(2)−N−(2−アダマンチル)−2−(ジメチルアミノメチリデン)−4,4−ジメチル−3−オキソ−ペンタンアミド(5.83g、93%)を得た。
1H NMR(400.13MHz、DMSO−d6)δ1.13(9H、s)、1.47(2H、d)、1.69−1.83(10H、m)、2.03(2H、d)、2.92(6H、s)、3.90(1H、d)、7.24(1H、s)、7.94(1H、d)
m/z(ESI+)(M+H)+=333。
【0144】
中間体#4は次のように調製することもできる。
N,N−ジメチルホルムアミドジメチルアセタール(1.2当量)を、窒素下で、1,4−ジオキサン(9.6容量)中のN−(2−アダマンチル)−4,4−ジメチル−3−オキソ−ペンタンアミド(中間体#3)(1当量)の溶液に加えた。混合物を還流下で5時間加熱し、その後室温まで冷却した。溶媒を真空下で除去し、淡黄色の固体を次の工程でそのまま使用した。
TLC ヘキサン:EtOAc、1:1、生成物Rf0.94(不純物:Rf0.06+0.66)
mp 143.6〜147.6℃。
【0145】
中間体#1:4−[4−(2−アダマンチルカルバモイル)−5−tert−ブチル−ピラゾル−1−イル]安息香酸メチル
【0146】
【化6】
【0147】
4−ヒドラジニル安息香酸メチル塩酸塩(中間体#2)(3.04g、15.00mmol)を、エタノール(100mL)中の(2)−N−(2−アダマンチル)−2−(ジメチルアミノメチリデン)−4,4−ジメチル−3−オキソ−ペンタンアミド(中間体#4)(4.99g、15mmol)に一度に加えた。酢酸を5滴添加し、得られた溶液を80℃で2時間攪拌した。反応混合物を濃縮し、EtOAc(500mL)で希釈し、水(200mL)および飽和ブライン(200mL)で連続して洗浄した。有機層をMgSO4で乾燥させ、濾過し、エバポレートし、粗生成物を得た。
【0148】
該粗生成物をフラッシュシリカクロマトグラフィー(溶出勾配:イソヘキサン中の0〜50%EtOAc)によって精製した。精製画分を蒸発乾固し、4−[4−(2−アダマンチルカルバモイル)−5−tert−ブチル−ピラゾル−1−イル]安息香酸メチル(4.66g、71.3%)を黄色の固体として得た。
1H NMR(400.13MHz、DMSO−d6)δ1.19(9H、s)、1.50(2H、d)、1.69−1.95(10H、m)、2.09(2H、d)、3.91(3H、s)、3.99(1H、d)、7.53−7.56(2H、m)、7.62(1H、s)、8.09−8.12(2H、m)、8.20(1H、d)
m/z(ESI+)(M+H)+=436。
【0149】
中間体#1は次のように調製することもできる。
2−(4−(メトキシカルボニル)フェニル)ヒドラジニウムクロリド(中間体#2)(1当量)、次いで酢酸(0.023当量)を、窒素下で、メタノール(200容量)中の(2Z)−N−(2−アダマンチル)−2−(ジメチルアミノ−メチリデン)−4,4−ジメチル−3−オキソ−ペンタンアミド(中間体#4)(1当量)の溶液に加えた。混合物を還流下で1.5時間攪拌し、冷却し、3.5容量未満まで濃縮し、得られた懸濁液を酢酸エチル(96容量)で希釈した。該懸濁液を水(34.4容量)で洗浄して溶液を得、その溶液をブライン(34.4容量)で洗浄し、乾燥(MgSO4)し、濃縮乾固した。粗生成物をMTBE(9容量)中でスラリー状にし、15分間攪拌した。淡黄色の固体を濾過し、MTBE(11.4容量)で洗浄し、真空下、60℃で乾燥させた。
TLC DCM:MeOH、9:1、生成物Rf0.86(微量の不純物Rf0.68)
mp 193.6〜194.5℃。
【0150】
4−[4−(2−アダマンチルカルバモイル)−5−tert−ブチル−ピラゾル−1−イル]安息香酸は次のように調製することもできる。
塩酸(34.5%w/w、15.88g、5.48g@100.0%、0.1504モル、1.0モル当量)および水(70ml、1.4相対量)をメタノール(1250ml、12.5相対量)中の4−ヒドラジノ安息香酸(23.35g、22.88g@100.0%、0.1504モル、1.0モル当量)の懸濁液に加えた。得られた懸濁液を20〜25℃で30分間攪拌し、メタノール(250ml、5.0相対量)中の、20〜25℃のN−(2−アダマンチル)−2−(ジメチルアミノメチリデン)−4,4−ジメチル−3−オキソ−ペンタンアミド(中間体#4)(53.47g、50.0g@100.0%、4モル中0.150、1.0モル当量)の溶液を20分間かけて加え、次いで塩酸(34.5%w/w、2.38g、0.82g@100.0%、0.02264モル、0.15モル当量)および水(70.0ml、1.4相対量)を加えた。反応物の温度を62〜65℃まで上げ、90.0分間維持した。後処理として、残り6.0相対量(約300.0ml)となるまでメタノール溶液を大気中で濃縮した。得られた懸濁液を20〜25℃まで冷却し1.0時間攪拌した。生成物を濾過し、酢酸エチル(200.0ml、4.0相対量)で洗浄し、30分間吸引乾燥した。生成物を50℃で8時間、真空下(100mbar)で乾燥させ、未精製の4−[4−(2−アダマンチルカルバモイル)−5−tert−ブチル−ピラゾル−1−イル]安息香酸(47.0g、73.0%)を得た。
1H NMR(400.13MHz、DMSO−d6)δ1.19(9H、s)、1.49(2H、d)、1.70−1.96(10H、m)、2.09(2H、d)、3.98−4.01(1H、m)、7.49−7.53(2H、m)、7.61(1H、s)、8.06−8.09(2H、m)、8.20(1H、d)、13.30(1H、s)
m/z(ESI+)(M+H)+=422
m.p.308.8℃(開始)。
クロマトグラフィー条件:[HPLC]
Zorbax SB-Aq、150×4.6mm、5μ。使用する移動相は有機溶媒としてアセトニトリルを用いるギ酸緩衝液、流量1.0mL/分、注入量は20μL、実行時間は18分で、UV検出器の波長は220、320nmを使用する。
保持時間[HPLC]
4−[4−(2−アダマンチルカルバモイル)−5−tert−ブチル−ピラゾル−1−イル]安息香酸:(相対保持時間:0.77分)
(2)−N−(2−アダマンチル)−2−(ジメチルアミノメチリデン)−4,4−ジメチル−3−オキソ−ペンタンアミド(保持時間:14.2分)。
【0151】
10.0%w/w水酸化ナトリウム水溶液(109.1g、10.91g@100.0%、0.2727モル、1.15モル当量)を、未精製4−[4−(2−アダマンチルカルバモイル)−5−tert−ブチル−ピラゾル−1−イル]安息香酸(110.0g、100.0g@100.0%、0.2372モル、1.0モル当量)の水(1000.0ml、10.0相対量)中の懸濁液に加え、15.0分間攪拌した。不溶解生成物を濾過した。得られた透明な水溶液(濾液)にトルエン(600.0ml、6.0相対量)を加え、30.0分間攪拌した。相分離のために反応物を1.0時間静置した。水層を分離し、セライト床を通して濾過した。水層にメタノール(300.0ml、3.0相対量)を加えた。水層のpHを、希塩酸(3.8%w/w、261.6g、9.94@100.0%、0.2727モル、1.15モル当量)で2.25〜2.75に徐々に調節した。得られた懸濁液を1.5〜2.0時間攪拌し、生成物を濾過し、25.0%v/vメタノール水溶液[500.0ml、5.0相対量(メタノール125mlと水475mlの混合物)]で洗浄した。生成物を50〜60℃で16.0時間、真空下(100mbar)で乾燥させ、4−[4−(2−アダマンチルカルバモイル)−5−tert−ブチル−ピラゾル−1−イル]安息香酸(85.0g、85.0%)(形態1)を得た。
【0152】
中間体#3
N−(2−アダマンチル)−4,4−ジメチル−3−オキソ−ペンタンアミド
【0153】
【化7】
【0154】
中間体#3は次のように調製することもできる。トルエン(400.0ml、4.0相対量)を、2−アダマンタンアミン塩酸塩(100.0g、98.0g@100%、0.5221モル、1.00モル当量)の水(500.0ml、5.0相対量)中の溶液に加えた。10.0%w/wの水酸化ナトリウム水溶液(261.02g、26.1g@100.0%、0.6526モル、1.25モル当量)を上記溶液に加え、15分間攪拌した。有機層を水層から分離し、5.0%w/w塩化ナトリウム溶液(300.0ml、3.0相対量)で洗浄した。エチルピバロイルアセテート(115.9g、0.6526モル、1.25モル当量)を有機層に一度に加えた。反応物を加熱還流し、温度を110℃に維持しながら、トルエンを3〜3.5時間共沸して回収した(550.0ml、5.5相対量)。反応物を70〜80℃まで冷却し、n−ヘプタン(1000.0ml、10.0相対量)を15分間かけて加えた。反応物をさらに25℃まで冷却し、得られた懸濁液を1.0時間攪拌した。懸濁した固体を濾過により回収し、n−ヘプタン(400.0ml、4.0相対量)で洗浄し、生成物を50〜55℃で6.0時間、真空下(100mbar)で乾燥させ、N−(2−アダマンチル)−4,4−ジメチル−3−オキソ−ペンタンアミド(113.0g、75.8%)を白色結晶性固体として得た。
1H NMR(400.13MHz、DMSO−d6)δ1.08−1.09(9H、m)、1.50(2H、d)、1.66−1.89(10H、m)、1.95−2.00(2H、m)、3.53(1.4H、s)、3.80−3.94(1H、m)、5.30(0.3H、s)、7.77−7.87(1H、m)、14.43(0.3H、s)(ケト型とエノール型の2:1混合物)
m/z(ESI+)(M+H)+=278。
クロマトグラフィー条件:−(GC)
HP−5MSカラム、キャリヤーガスとしてヘリウム、流量1.0mL/分、溶媒遅延最大1.5分、オーブン温度は、初期温度50℃で2分間維持、その後20℃/分で280℃まで上昇、注入量は1.0μLとする。
保持時間(GC)
2−アダマンタンアミン塩酸塩(保持時間:8.1分)
N−(2−アダマンチル)−4,4−ジメチル−3−オキソ−ペンタンアミド(相対保持時間:1.617分)。
【0155】
中間体#4
(2)−N−(2−アダマンチル)−2−(ジメチルアミノメチリデン)−4,4−ジメチル−3−オキソ−ペンタンアミド
【0156】
【化8】
【0157】
中間体#4は次のように調製することもできる。N,N−ジメチルホルムアミドジメチルアセタール(69.25g、63.02g@100.0%、0.5288モル、1.5モル当量)を、N−(2−アダマンチル)−4,4−ジメチル−3−オキソ−ペンタンアミド(中間体3)(100g、97.8g@100.0%、0.3525モル、1.0モル当量)のn−ヘプタン(800.0ml、8.20相対量)およびトルエン(350.0ml、3.58相対量)中の懸濁液に加えた。反応物の温度を90〜95℃まで上げ5.0時間維持した後、80℃まで冷却し、n−ヘプタン(400.0ml、4.09相対量)を加えた。反応物温度をさらに25℃まで下げ、得られた懸濁液を濾過により回収し、n−ヘプタン(400.0ml、4.09相対量)で洗浄し、生成物を30分間吸引乾燥した。生成物を周囲温度(20〜25℃)で3.0時間、真空下(100mbar)で乾燥させ、(2)−N−(2−アダマンチル)−2−(ジメチルアミノメチリデン)−4,4−ジメチル−3−オキソ−ペンタンアミド(100g、79.7%)を得た。
1H NMR(400.13MHz、DMSO−d6)δ1.13(9H、s)、1.47(2H、d)、1.69−1.83(10H、m)、2.03(2H、d)、2.92(6H、s)、3.90(1H、d)、7.24(1H、s)、7.94(1H、d)。
m/z(ESI+)(M+H)+=333。
クロマトグラフィー条件:(HPLC)−
Sunfire C18、150×4.6mm、5μ。使用する移動相は有機溶媒としてメタノールを用いるリン酸水素二ナトリウム緩衝液、流量1.0mL/分、注入量は20μL、実行時間は20分で、示差屈折率検出器を使用する。
保持時間(HPLC):
N−(2−アダマンチル)−4,4−ジメチル−3−オキソ−ペンタンアミド(保持時間:11.0分)
(2)−N−(2−アダマンチル)−2−(ジメチルアミノメチリデン)−4,4−ジメチル−3−オキソ−ペンタンアミド(相対保持時間:1.18分)。
【0158】
参考例2
(2)−N−(2−アダマンチル)−2−(ジメチルアミノメチリデン)−4,4−ジメチル−3−オキソ−ペンタンアミドの合成
【0159】
【化9】
【0160】
2−アダマンタンアミン塩酸塩(25.0g、0.13モル)の水(75.0ml、3.0相対量)中の懸濁液に、トルエン(100.0ml、4.0相対量)を加えた。10.0%w/w水酸化ナトリウム水溶液(1.25モル当量)を上記溶液に入れ、10〜15分間攪拌した。有機層を分離し、水層をトルエン(75.0ml、3.0相対量)で再抽出し、分離した有機層と合わせた。合わせた有機層を5.0%w/w塩化ナトリウム溶液(75ml、3.0相対量)で洗浄し分離した。エチルピバロイルアセテート(26.01g、0.15モル)を有機層に加え、反応物を110〜112℃で加熱還流した。溶媒(4〜5相対量)を4〜5時間かけて共沸して回収した。反応物を40〜45℃まで冷却し、35〜40℃でn−ヘプタン(200.0ml、8.0相対量)を加え、次いで30〜35℃でDMF−DMA(26.45g、0.20モル)およびトリエチルアミン(13.48g、0.13モル)を加えた。反応物の温度を90〜93℃まで上げ2〜3時間維持した。副生成物として生成したメタノールを反応の間共沸して回収した。反応物を20〜25℃まで冷却し、その温度で1.0時間攪拌した。沈殿生成物を濾過し、床をn−ヘプタン(100.0ml、4.0相対量)で洗浄し、生成物を35〜40℃で3〜4時間、真空下(50〜100mbar)で乾燥させ、(2)−N−(2−アダマンチル)−2−(ジメチルアミノメチリデン)−4,4−ジメチル−3−オキソ−ペンタンアミドを得た(収率、86%)。室温では不安定であったため、生成物を窒素雰囲気下で充填し、10℃未満で貯蔵した。
【0161】
別の方法として、(2)−N−(2−アダマンチル)−2−(ジメチルアミノメチリデン)−4,4−ジメチル−3−オキソ−ペンタンアミドは次のように調製することができる。
【0162】
2−アダマンタンアミン塩酸塩(25.0g、0.13モル)の水(75.0ml、3.0相対量)中の懸濁液に、トルエン(100.0ml、4.0相対量)を加えた。10.0%w/w水酸化ナトリウム水溶液(1.25モル当量)を上記溶液に入れ、10〜15分間攪拌した。有機層を分離し、水層をトルエン(75.0ml、3.0相対量)で再抽出し、分離した有機層と合わせた。合わせた有機層を5.0%w/w塩化ナトリウム溶液(75ml、3.0相対量)で洗浄し分離した。エチルピバロイルアセテート(26.01g、0.15モル)を有機層に加え、反応物を110〜112℃で加熱還流した。溶媒(4〜5相対量)を4〜5時間かけて共沸して回収した。反応物を40〜45℃まで冷却し、35〜40℃でn−ヘプタン(200.0ml、8.0相対量)を加え、次いで同じ温度でDMF−DMA(26.45g、0.20モル)を加えた。反応物の温度を85〜90℃まで上げ4〜5時間維持した。副生成物として生成したメタノールを反応の間共沸して回収した。反応物を20〜25℃まで冷却し、その温度で1.0時間攪拌した。沈殿生成物を濾過し、床をn−ヘプタン(100.0ml、4.0相対量)で洗浄し、生成物を35〜40℃で3〜4時間、真空下(50〜100mbar)で乾燥させ、(2)−N−(2−アダマンチル)−2−(ジメチルアミノメチリデン)−4,4−ジメチル−3−オキソ−ペンタンアミドを得た(収率、72%)。室温では不安定であったため、生成物を窒素雰囲気下で充填し、10℃未満で貯蔵した。
クロマトグラフィー条件:−
Sunfire C18、150×4.6mm、5μ。使用する移動相は有機溶媒としてメタノールを用いるリン酸水素二ナトリウム緩衝液、流量1.0mL/分、注入量は20μL、実行時間は20分で、示差屈折率検出器を使用する。
保持時間:
N−(2−アダマンチル)−4,4−ジメチル−3−オキソ−ペンタンアミド 保持時間:11.0分
(2)−N−(2−アダマンチル)−2−(ジメチルアミノメチリデン)−4,4−ジメチル−3−オキソ−ペンタンアミド 相対保持時間:1.18分
1H NMR(400.13MHz、DMSO−d6)δ1.13(9H、s)、1.47(2H、d)、1.69−1.83(10H、m)、2.03(2H、d)、2.92(6H、s)、3.90(1H、d)、7.24(1H、s)、7.94(1H、d)
m/z(ESI+)(M+H)+=333。
【0163】
必要であれば該N−(2−アダマンチル)−4,4−ジメチル−3オキソ−ペンタンアミド中間体は単離することができる:
クロマトグラフィー条件:−
HP−5MSカラム、キャリヤーガスとしてヘリウム、流量1.0mL/分、溶媒遅延最大1.5分、オーブン温度は、初期温度50℃で2分間維持、その後20℃/分で280℃まで上昇、注入量は1.0μLとする。
保持時間:
2−アダマンタンアミン塩酸塩 保持時間8.1分
N−(2−アダマンチル)−4,4−ジメチル−3−オキソ−ペンタンアミド 相対保持時間:1.617分
1H NMR(400.13MHz、DMSO−d6)δ1.08−1.09(9H、m)、1.50(2H、d)、1.66−1.89(10H、m)、1.95−2.00(2H、m)、3.53(1.4H、s)、3.80−3.94(1H、m)、5.30(0.3H、s)、7.77−7.87(1H、m)、14.43(0.3H、s)(ケト型とエノール型の2:1混合物)
m/z(ESI+)(M+H)+=278。
【0164】
4−[4−(2−アダマンチルカルバモイル)−5−tert−ブチル−ピラゾル−1−イル]安息香酸(形態−1)の合成
【0165】
【化10】
【0166】
4−ヒドラジノ安息香酸塩酸塩(14.11g、0.075モル)、および(2)−N−(2−アダマンチル)−2−(ジメチルアミノメチリデン)−4,4−ジメチル−3−オキソ−ペンタンアミド(25.0g、0.075モル)をジャケット付き反応器に入れ、次いでイソプロピルアルコール(315ml、12.6相対量)および水(35ml、1.4相対量)を入れた。反応物を20〜25℃で約45〜60分間攪拌した。内容物を78〜80℃で加熱還流し、その温度で90分間維持した。反応物を50〜55℃まで冷却し、次いで同じ温度で水(150ml、6相対量)を加えた。内容物をさらに周囲温度(20〜25℃)まで冷却し、同じ温度で1.0時間攪拌した。沈殿生成物を濾過し、次いでイソプロピルアルコールと水(1:1)の混合物(250ml、10.0相対量)で洗浄し、4−[4−(2−アダマンチルカルバモイル)−5−tert−ブチル−ピラゾル−1−イル]安息香酸を得た。生成物を50〜55℃で4〜5時間、真空下で乾燥させ、さらに精製することなく用いた(収率:80%)。
1H NMR(400.13MHz、DMSO−d6)δ1.19(9H、s)、1.49(2H、d)、1.70−1.96(10H、m)、2.09(2H、d)、3.98−4.01(1H、m)、7.49−7.53(2H、m)、7.61(1H、s)、8.06−8.09(2H、m)、8.20(1H、d)、13.30(1H、s)
m/z(ESI+)(M+H)+=422
m.p.308.8℃(開始)。
クロマトグラフィー条件:−
Zorbax SB-Aq、150×4.6mm、5μ。使用する移動相は有機溶媒としてアセトニトリルを用いるギ酸緩衝液、流量1.0mL/分、注入量は20μL、実行時間は18分で、UV検出器の波長は220、320nmを使用する。
保持時間:
[((2)−N−(2−アダマンチル)−2−(ジメチルアミノメチリデン)−4,4−ジメチル−3−オキソ−ペンタンアミド 保持時間14.2分
4−[4−(2−アダマンチルカルバモイル)−5−tert−ブチル−ピラゾル−1−イル]安息香酸 相対保持時間0.77分(10.0分)
中間体 相対保持時間0.79(11.2分)。
【0167】
実施例1
4−[4−(2−アダマンチルカルバモイル)−5−tert−ブチル−ピラゾル−1−イル]安息香酸(形態2)
上記(参考例1−形態1)で調製した4−[4−(2−アダマンチルカルバモイル)−5−tert−ブチル−ピラゾル−1−イル]安息香酸約50mgを磁気攪拌子入りのバイアルに入れ、アセトニトリル約2mlを加えた。次いでバイアルを蓋できつく密封した。次いでスラリーを、磁気攪拌能力を有する加熱攪拌ブロック中、50℃で攪拌させた。3日後、試料をプレートより取り出し、蓋を外し、スラリーを周囲条件で乾燥させてから、XRPDおよびDSCで分析した。この形態(形態2)は、XRPDによって結晶性であることが確認され、先の形態とは異なっていることがわかった。この物質の融点は310.9℃(開始)であった。この物質は、CuKa照射を用いて測定される2シータのピークを18.0および17.7°に有していた。後にDSCを用いて融点を測定すると309.9℃であった。
【0168】
実施例2
4−[4−(2−アダマンチルカルバモイル)−5−tert−ブチル−ピラゾル−1−イル]安息香酸(形態3)
4−[4−(2−アダマンチルカルバモイル)−5−tert−ブチル−ピラゾル−1−イル]安息香酸(形態1)約20mgを磁気攪拌子入りのバイアルに入れ、メタノール約2mlを加え、次いでバイアルを蓋できつく密封し、マグネチックスターラープレート上で攪拌させた。3日後、試料をプレートより取り出し、蓋を外し、スラリーを周囲条件で乾燥させてから、XRPDおよびDSCで分析した。この形態(形態3)は、XRPDによって結晶性であることが確認され、先に見られた形態とは異なっていることがわかった。この物質の融点は309.4℃(開始)であった。この物質は、CuKa照射を用いて測定される2シータのピークを18.7および11.7°に有していた。後にDSCを用いて融点を測定すると309.3℃であった。
【0169】
実施例3
4−[4−(2−アダマンチルカルバモイル)−5−tert−ブチル−ピラゾル−1−イル]安息香酸(形態4)
形態1の4−[4−(2−アダマンチルカルバモイル)−5−tert−ブチル−ピラゾル−1−イル]安息香酸約20mgおよび形態3の物質20mgを磁気攪拌子入りのバイアルに入れ、酢酸エチル約2mlを加え、次いでバイアルを蓋できつく密封し、マグネチックスターラープレート上で攪拌させた。3日後、試料をプレートより取り出し、蓋を外し、スラリーを周囲条件で乾燥させてから、XRPDおよびDSCで分析した。この形態(形態4)は、XRPDによって結晶性であることが確認され、先に見られた形態とは異なっていることがわかった。この物質(形態4)の融点は309.1℃(開始)であった。この物質は、CuKa照射を用いて測定される2シータのピークを16.2および20.6°に有していた。後にDSCを用いて融点を測定すると312.0℃であった。
【0170】
別の方法として、高温においては溶媒としてアセトニトリルを用いてもよい。アセトニトリル(800ml、8.0相対量)を、乾燥純粋生成物((4−[4−(2−アダマンチルカルバモイル)−5−tert−ブチル−ピラゾル−1−イル]安息香酸、形態1(80.0g)に加え、スラリーを75〜78℃まで加熱した。反応物を72.0時間75〜78℃に維持し、次いで20〜25℃まで冷却し、20〜25℃で1.0時間攪拌した。生成物を濾過し吸引乾燥した。次いでアセトニトリル(240.0ml、3.0相対量)で洗浄し、30.0分間吸引乾燥した。次いで生成物を50℃で16.0時間、真空下(100mbar)で乾燥させ、4−[4−(2−アダマンチルカルバモイル)−5−tert−ブチル−ピラゾル−1−イル]安息香酸、形態−4(72.0g、90.0%)を得た。形態−4の物質(50mg、1%w/w、種)を、(4−[4−(2−アダマンチルカルバモイル)−5−tert−ブチル−ピラゾル−1−イル]安息香酸(形態1)5.0gのアセトニトリル50.0ml中の懸濁液に加え、12〜18時間、75〜78℃まで加熱した。反応物を20〜25℃まで冷却し、20〜25℃で1.0時間攪拌した。生成物を濾過し吸引乾燥した。次いでアセトニトリル(15ml)で洗浄し、5〜10.0分間吸引乾燥した。次いで生成物を50℃で16.0時間、真空下(100mbar)で乾燥させ、4−[4−(2−アダマンチルカルバモイル)−5−tert−ブチル−ピラゾル−1−イル]安息香酸、形態−4(4.5g、89〜90.0%)を得た。上記のように調製された4−[4−(2−アダマンチルカルバモイル)−5−tert−ブチル−ピラゾル−1−イル]安息香酸(形態1)をアセトニトリル(7容量)中に懸濁させ、(形態4)を5g播種し、3日間還流でスラリー状にした(ジャケット温度85℃)。サンプルを採取しDSCでチェックした(ピークを2つ示す)。このサンプルをさらに3日間(週末)還流で攪拌し、20℃まで冷却し、濾過し、アセトニトリル、次いでジエチルエーテルで十分に洗浄し、吸引乾燥し、50℃で48時間真空下で乾燥させて、淡黄色の固体(形態4)を得た(90%)。
【0171】
別の方法としては:
テトラヒドロフラン(9.0相対量)および水(0.5相対量)を4−[4−(2−アダマンチルカルバモイル)−5−tert−ブチル−ピラゾル−1−イル]安息香酸、形態1(20.0g、0.047モル)に加え、混合物を15分間攪拌した後、濾紙を通して濾過した。残留物をテトラヒドロフラン(1.0相対量)で洗浄し、合わせた濾液を反応器に移し、反応温度を58〜62℃まで上げた。反応を55〜65℃に維持しながらアセトニトリル(20.0相対量)を加えた。反応温度を68±2℃まで上げ、22時間その温度を維持し、その後20〜25℃まで冷却し2時間攪拌した。生成物を濾過し、床をアセトニトリル(5.0相対量)で洗浄した。ウエットケーキを45〜50℃で4時間、真空下(50〜100mbar)で乾燥させ、多形4を得て(収率80%)、XRPDで確認した。
【0172】
別の方法としては:
テトラヒドロフラン(10.0相対量)を4−[4−(2−アダマンチルカルバモイル)−5−tert−ブチル−ピラゾル−1−イル]安息香酸、形態1(5.0g、0.012モル)に加え、温度を58〜62℃まで上げた。反応を55〜65℃に維持しながらアセトニトリル(20.0相対量)を加えた。反応温度は20時間68±2℃で維持した。内容物を20〜25℃まで冷却し2時間攪拌した。生成物を濾過し、ウエットケーキをアセトニトリル(5.0相対量)で洗浄した後、45〜50℃で4時間、真空オーブン(50〜100mbar)内で乾燥させ、多形4を得て(収率90%)、XRPDおよび固体NMRで確認した。
【0173】
別の方法としては:
N,N−ジメチルホルムアミド(5.0相対量)およびアセトニトリル(5.0相対量)を4−[4−(2−アダマンチルカルバモイル)−5−tert−ブチル−ピラゾル−1−イル]安息香酸、形態1(5.0g、0.012モル)に加え、反応温度を60〜65℃まで上げた。温度を55〜65℃に維持しながらアセトニトリル(15.0相対量)を加えた。反応温度を75〜78℃まで上げ、20時間その温度を維持した。内容物を20〜25℃まで冷却し2時間攪拌した。生成物を濾過し、床をアセトニトリル(5.0相対量)で洗浄した後、45〜50℃で4時間、真空オーブン(50〜100mbar)内で乾燥させ、多形4を得て(収率88%)、XRPDで確認した。
【0174】
別の方法としては:
酢酸(10.0相対量)を4−[4−(2−アダマンチルカルバモイル)−5−tert−ブチル−ピラゾル−1−イル]安息香酸、形態1(5.0g、0.012モル)に加え、温度を75〜78℃まで上げた。温度を70〜78℃に維持しながらアセトニトリル(20.0相対量)を加えた。混合物を75〜78℃で攪拌し、22時間その温度を維持した。内容物を20〜25℃まで冷却し2時間攪拌した。生成物を濾過し、床をアセトニトリル(5.0相対量)で洗浄した後、45〜50℃で4時間、真空オーブン(50〜100mbar)内で乾燥させ、多形4を得て(収率66%)、XRPDで確認した。
【0175】
別の方法としては:
2−メチル−THF(10.0相対量)を4−[4−(2−アダマンチルカルバモイル)−5−tert−ブチル−ピラゾル−1−イル]安息香酸、形態1(5.0g、0.012モル)に加え、温度を70〜75℃まで上げた。温度を70〜75℃に維持しながらアセトニトリル(20.0相対量)を加えた後、75〜78℃で23時間攪拌させた。内容物を20〜25℃まで冷却し2時間攪拌した。生成物を濾過し、床をアセトニトリル(5.0相対量)で洗浄した後、45〜50℃で4時間、真空オーブン(50〜100mbar)内で乾燥させ、多形4を得て(収率93%)、XRPDで確認した。
【0176】
別の方法としては:
4−[4−(2−アダマンチルカルバモイル)−5−tert−ブチル−ピラゾル−1−イル]安息香酸(形態1−参考例2のように調製)(20.0g、0.047モル)、次いでテトラヒドロフラン(9.0相対量)および水(0.5相対量)を好適なジャケット付き反応器に加えた。内容物を15分間攪拌し、濾紙を通して濾過し、テトラヒドロフラン(1.0相対量)で洗浄した。合わせた濾液を反応器に移し、反応物の温度を58〜62℃まで上げた。温度を55〜65℃に維持しながらアセトニトリル(20.0相対量)を加えた。反応物の温度を68±2℃に上げ、22時間その温度を維持した。内容物を20〜25℃まで冷却し2時間攪拌した。生成物を濾過し、床をアセトニトリル(5.0相対量)で洗浄した。ウエットケーキを45〜50℃で4時間、真空下(50〜100mbar)で乾燥させ、多形形態4を得た(80%)。
【技術分野】
【0001】
本発明は、4−[4−(2−アダマンチルカルバモイル)−5−tert−ブチル−ピラゾル−1−イル]安息香酸(本薬剤)の新規な結晶形態に関する。本薬剤は、ヒト11−β−ヒドロキシステロイド脱水素酵素1型(11βHSD1)阻害活性を有し、したがってメタボリック症候群などの病態の治療において価値があり、ヒトなどの温血動物の治療方法において有用である。また本発明は、本薬剤の結晶形態の製造工程、それらを含有する医薬組成物、およびヒトなどの温血動物において11βHSD1を阻害する医薬の製造におけるこれらの使用にも関する。
【0002】
本薬剤は下記の式(I)で示される。
【0003】
【化1】
【背景技術】
【0004】
グルココルチコイド(ヒトのコルチゾール、げっ歯類のコルチコステロン)は対抗制御的ホルモンである。すなわち、グルココルチコイドはインスリンの作用に対抗する(Dallman MF, Strack AM, Akana SF et al. 1993; Front Neuroendocrinol 14, 303-347)。グルココルチコイドは、糖新生に関与する肝酵素の発現を調節するとともに、脂肪組織からグリセロールを遊離し(脂肪分解の増加)筋組織からアミノ酸を遊離する(タンパク質合成の減少とタンパク質分解の増加)ことによって、基質の供給を高める。またグルココルチコイドは、トリグリセリドを蓄えることのできる成熟脂肪細胞への前駆脂肪細胞の分化において重要である(Bujalska IJ et al. 1999; Endocrinology 140, 3188-3196)。「ストレス」によって誘発されたグルココルチコイドが中心性肥満(それ自体が2型糖尿病、高血圧症および心疾患の強力な危険因子である)と関連している病態においては、このことは重大な意味を持ち得る(Bjorntorp P & Rosmond R 2000; Int. J. Obesity 24, S80-S85)。
【0005】
グルココルチコイド活性が、コルチゾールの分泌だけで制御されているのではなく、組織レベルにおいては、11−βヒドロキシステロイド脱水素酵素である11βHSD1(コルチゾンを活性化する)と11βHSD2(コルチゾールを不活性化する)による活性コルチゾールと不活性コルチゾンの細胞内相互変換によって制御されている、ということは現在十分に証明されている(Sandeep TC & Walker BR 2001 Trends in Endocrinol & Metab. 12, 446-453)。このメカニズムがヒトにおいて重要であり得るということは、カルベノキソロン(11βHSD1および2の両方を阻害する抗潰瘍薬)治療を用いてまず示された(Walker BR et al. 1995; J. Clin. Endocrinol. Metab. 80, 3155-3159)。この治療はインスリン感受性の増加につながり、それによって、活性グルココルチコイドの組織レベルを低下させることによって11βHSD1がインスリンの作用を調節している可能性がある、ということが示されている(Walker BR et al. 1995; J. Clin. Endocrinol. Metab. 80, 3155-3159)。
【0006】
臨床的には、クッシング症候群にはコルチゾールの過剰が関与しており、さらにその過剰には、耐糖能低下、中心性肥満(この貯蔵所における前駆脂肪細胞の分化の刺激に起因)、脂質異常症および高血圧症が関与している。クッシング症候群には、メタボリック症候群との明確な類似点が数多く見られる。メタボリック症候群には、通常、過剰の血中コルチゾール濃度は関与していないが(Jessop DS et al. 2001; J. Clin. Endocrinol. Metab. 86, 4109-4114)、組織中の11βHSD1活性が異常に高いと、同様の影響があることが予想される。肥満したヒトでは、血漿コルチゾール濃度は痩型対照群と同じかまたはそれよりも低いにもかかわらず、皮下脂肪中の11βHSD1活性が大きく増進されることが示された(Rask E et al. 2001; J. Clin. Endocrinol. Metab. 1418-1421)。さらに、メタボリック症候群に関与している中心性脂肪は、皮下脂肪よりもはるかに高いレベルの11βHSD1活性を示す(Bujalska IJ et al. 1997; Lancet 349, 1210-1213)。このように、グルココルチコイド、11βHSD1およびメタボリック症候群の間には関連性があるように思われる。
【0007】
11βHSD1ノックアウトマウスでは、グルココルチコイドに誘導される糖新生酵素の活性化が絶食に応じて減衰し、血漿グルコース濃度がストレスまたは肥満に応じて低下することが示されるが(Kotelevtsev Y et al. 1997; Proc. Natl. Acad. Sci USA 94, 14924-14929)、このことは、11βHSD1を阻害することが、2型糖尿病において血漿グルコースおよび肝糖産生を低下させる際に有用であることを示している。さらに、これらのマウスは、低トリグリセリド、高HDLコレステロールおよび高アポリポタンパク質AI濃度を示す、抗アテローム発生性リポタンパク質プロフィールを発現する(Morton NM et al. 2001; J. Biol. Chem. 276, 41293-41300)。この表現型は、脂肪異化酵素およびPPARαの肝での発現の増加によるものである。このこともまた、11βHSD1の阻害がメタボリック症候群の脂質異常症の治療において有用であることを示している。
【0008】
11βHSD1を過剰発現しているトランスジェニックマウスにおける最近の研究が、メタボリック症候群と11βHSD1との間の関連性を最も説得力をもって実証している(Masuzaki H et al. 2001; Science 294, 2166-2170)。脂肪特異的プロモーターの制御下で発現した場合、11βHSD1トランスジェニックマウスは、コルチコステロンの脂肪中濃度が高く、中心性肥満、インスリン抵抗性糖尿病、高脂血症および過食症を発症する。最も重要なことは、これらのマウスの脂肪中の11βHSD1活性の上昇レベルが、肥満した被験者と同等であることである。肝性11βHSD1活性および血漿コルチコステロン濃度は正常であったが、肝門脈コルチコステロン濃度は3倍に上昇しており、これが肝臓における代謝作用の原因であると考えられる。
【0009】
全体的に見ると、肥満したヒトと同等のレベルの11βHSD1を脂肪のみに過剰発現させるだけで、マウスでメタボリック症候群を完全に再現できることは、もはや明らかである。
【0010】
11βHSD1は組織内に広く分布しており、グルココルチコイド受容体の分布と重なっている。したがって、11βHSD1を阻害することによって、数多くの生理学的/病理学的役割におけるグルココルチコイドの作用が妨害される可能性がある。11βHSD1はヒト骨格筋に存在しており、タンパク質の代謝回転およびグルコース代謝に対するインスリンの同化作用をグルココルチコイドが妨害することは、十分に立証されている(Whorwood CB et al. 2001; J. Clin. Endocrinol. Metab. 86, 2296-2308)。したがって、骨格筋は11βHSD1に基づく治療の重要な標的になるはずである。
【0011】
グルココルチコイドはインスリン分泌も低下させるが、これによってグルココルチコイドに誘導されるインスリン抵抗性の作用が激化する可能性がある。膵島は11βHSD1を発現し、カルベノキソロンはインスリン放出に対する11−デヒドロコルチコステロンの作用を阻害することができる(Davani B et al. 2000; J. Biol. Chem. 275, 34841-34844)。このように、糖尿病の治療においては、11βHSD1阻害剤はインスリン抵抗性に対して組織レベルで作用するだけでなく、インスリンの分泌自体を増加させることができる。
【0012】
骨格の発育および骨の機能もまた、グルココルチコイドの作用によって調節されている。11βHSD1はヒト破骨細胞および骨芽細胞中に存在しており、健常ボランティアをカルベノキソロンで処置すると、骨吸収マーカーは減少したが、骨形成マーカーには変化は見られなかった(Cooper MS et al 2000; Bone 27, 375-381)。骨中の11βHSD1活性の阻害は、骨粗しょう症治療における防御機構として使用することができる。
【0013】
グルココルチコイドは緑内障などの眼病に関与している可能性もある。11βHSD1は、ヒトにおいて眼圧に影響を与えることが示されており、11βHSD1を阻害することによって、緑内障に伴う眼圧の上昇を緩和させることが期待される(Rauz S et al. 2001; Investigative Opthalmology & Visual Science 42, 2037-2042)。
【0014】
げっ歯類およびヒトの両方において、11βHSD1とメタボリック症候群との間には確実な関連性があると思われる。2型肥満糖尿病患者において11βHSD1を特異的に阻害する薬が、肝糖新生を減少させることによって血糖を下げ、中心性肥満を軽減し、アテローム発生性リポタンパク質表現型を改善し、血圧を下げ、かつインスリン抵抗性を低減するということを示唆する証拠がある。筋組織でのインスリンの作用は増強され、膵島ベータ細胞からのインスリン分泌も増加し得る。
【0015】
現在、メタボリック症候群の定義として主に2つのものが認識されている。
1)米国高脂血症治療ガイドライン(The Adult Treatment Panel(ATP III 2001 JMA))の定義では、患者が次の症状のうち3つ以上を有する場合、メタボリック症候群であるとしている。
男性の場合、腹囲40インチ(102cm)以上、女性の場合、35インチ(88cm)以上;
血清トリグリセリド濃度150mg/dl(1.69mmol/l)以上;
男性の場合、HDLコレステロール濃度40mg/dl(1.04mmol/l)未満、女性の場合、50mg/dl(1.29mmol/l)未満;
血圧135/80mmHg以上;および/または血糖(血清グルコース)110mg/dl(6.1mmol/l)以上。
2)WHO協議会は、因果関係を意味するものではなく、適当な時期にさらに改善することもできる実践的な定義として提案されている、次の定義を推奨している。
患者が、次の症状:耐糖能低下、耐糖能異常(IGT)もしくは糖尿病、および/またはインスリン抵抗性のうちの少なくとも1つを有し、かつ、次のうちの2つ以上を有する。
動脈圧の上昇;
血漿トリグリセリドの上昇;
中心性肥満;
微量アルブミン尿症。
【発明の概要】
【0016】
本発明者らは、本薬剤またはその薬学的に許容される塩は、有効な11βHSD1阻害剤であり、したがってメタボリック症候群に関連する病態の治療において価値があることを見出した。また本発明者らは、本発明の化合物は改良された特性を有し、それによって、医薬品として使用するためのより良い候補となることを見出した。
【0017】
本発明者らは、本薬剤のさらなる結晶形態を見出した。これらの形態は、形態2、形態3および形態4と称される。
【図面の簡単な説明】
【0018】
【図1】図1は、形態2のX線粉末回折パターンである。
【図2】図2は、形態2のDSC温度記録図である。
【図3】図3は、形態3のX線粉末回折パターンである。
【図4】図4は、形態3のDSC温度記録図である。
【図5】図5は、形態4のX線粉末回折パターンである。
【図6】図6は、形態4のDSC温度記録図である。
【図7】図7は、形態4のXRDパターンおよび面間隔dスペクトルである。
【発明を実施するための形態】
【0019】
したがって本発明の一態様は、およそ2−シータ=18.0の少なくとも1つの特異的ピークを有する測定したX線回折パターンを有する、4−[4−(2−アダマンチルカルバモイル)−5−tert−ブチル−ピラゾル−1−イル]安息香酸の結晶形態(形態2)に関する。
【0020】
2−シータ(θ)値はCuKa照射を用いて測定した。
本発明によれば、およそ2−シータ=18.0°および17.7°に少なくとも2つの特異的ピークを持つX線粉末回折パターンを有する本薬剤の結晶形態、形態2、が提供される。
【0021】
本発明によれば、およそ2−シータ=18.0、17.7および18.4°に特異的ピークを持つX線粉末回折パターンを有する本薬剤の結晶形態、形態2、が提供される。
本発明によれば、およそ2−シータ=18.0、17.7、18.4、8.9および20.5°に特異的ピークを持つX線粉末回折パターンを有する本薬剤の結晶形態、形態2、が提供される。
【0022】
本発明によれば、およそ2−シータ=18.0、17.7、18.4、8.9、20.5、10.4、21.9、13.4、27.6および16.7°に特異的ピークを持つX線粉末回折パターンを有する本薬剤の結晶形態、形態2、が提供される。
【0023】
本発明によれば、CuKa照射を用いた、図1に示されるX線粉末回折パターンと実質的に同一のX線粉末回折パターンを有する本薬剤の結晶形態、形態2、が提供される。
本発明によれば、2−シータ=18.0°プラスマイナス0.5°2−シータに少なくとも1つの特異的ピークを持つX線粉末回折パターンを有する本薬剤の結晶形態、形態2、が提供される。
【0024】
本発明によれば、2−シータ=18.0°および17.7°に少なくとも2つの特異的ピークを持つX線粉末回折パターンを有する本薬剤の結晶形態、形態2、が提供される。ここで、前記値はプラスマイナス0.5°2−シータであってよい。
【0025】
本発明によれば、2−シータ=18.0、17.7および18.4°に特異的ピークを持つX線粉末回折パターンを有する本薬剤の結晶形態、形態2、が提供される。ここで、前記値はプラスマイナス0.5°2−シータであってよい。
【0026】
本発明によれば、2−シータ=18.0、17.7、18.4、8.9および20.5°に特異的ピークを持つX線粉末回折パターンを有する本薬剤の結晶形態、形態2、が提供される。ここで、前記値はプラスマイナス0.5°2−シータであってよい。
【0027】
本発明によれば、2−シータ=18.0、17.7、18.4、8.9、20.5、10.4、21.9、13.4、27.6および16.7°に特異的ピークを持つX線粉末回折パターンを有する本薬剤の結晶形態、形態2、が提供される。ここで、前記値はプラスマイナス0.5°2−シータであってよい。
【0028】
本発明によれば、2−シータ=18.0°に少なくとも1つの特異的ピークを持つX線粉末回折パターンを有する本薬剤の結晶形態、形態2、が提供される。
本発明によれば、2−シータ=18.0および17.7°に少なくとも2つの特異的ピークを持つX線粉末回折パターンを有する本薬剤の結晶形態、形態2、が提供される。
【0029】
本発明によれば、2−シータ=18.0、17.7および18.4°に特異的ピークを持つX線粉末回折パターンを有する本薬剤の結晶形態、形態2、が提供される。
本発明によれば、2−シータ=18.0、17.7、18.4、8.9および20.5°に特異的ピークを持つX線粉末回折パターンを有する本薬剤の結晶形態、形態2、が提供される。
【0030】
本発明によれば、2−シータ=18.0、17.7、18.4、8.9、20.5、10.4、21.9、13.4、27.6および16.7°に特異的ピークを持つX線粉末回折パターンを有する本薬剤の結晶形態、形態2、が提供される。
【0031】
本発明によれば、CuKa照射を用いた、図1に示されるX線粉末回折パターンを有する本薬剤の結晶形態、形態2、が提供される。
【0032】
【表1】
【0033】
DSC分析から、形態2は309.9℃で融解を開始する高融点の固体であることがわかる。DSC温度記録図を図2に示す。
本発明によれば、およそ2−シータ=18.7°にピークを持つX線粉末回折パターンを有する、本薬剤の結晶形態、形態3が提供される。
【0034】
2−シータ(θ)値はCuKa照射を用いて測定した。
本発明によれば、およそ2−シータ=18.7°および11.7°に少なくとも2つの特異的ピークを持つX線粉末回折パターンを有する本薬剤の結晶形態、形態3、が提供される。
【0035】
本発明によれば、およそ2−シータ=18.7、11.7および19.2°に特異的ピークを持つX線粉末回折パターンを有する本薬剤の結晶形態、形態3、が提供される。
本発明によれば、およそ2−シータ=18.7、11.7、19.2、7.8、14.1、14.9および9.4°に特異的ピークを持つX線粉末回折パターンを有する本薬剤の結晶形態、形態3、が提供される。
【0036】
本発明によれば、およそ2−シータ=18.7、11.7、19.2、7.8、14.1、14.9、9.4、15.6、16.1および9.6°に特異的ピークを持つX線粉末回折パターンを有する本薬剤の結晶形態、形態3、が提供される。
【0037】
本発明によれば、CuKa照射を用いた、図3に示されるX線粉末回折パターンと実質的に同一のX線粉末回折パターンを有する本薬剤の結晶形態、形態3、が提供される。
本発明によれば、2−シータ=18.7°プラスマイナス0.5°2−シータに少なくとも1つの特異的ピークを持つX線粉末回折パターンを有する本薬剤の結晶形態、形態3、が提供される。
【0038】
本発明によれば、2−シータ=18.7°および11.7°に少なくとも2つの特異的ピークを持つX線粉末回折パターンを有する本薬剤の結晶形態、形態3、が提供される。ここで、前記値はプラスマイナス0.5°2−シータであってよい。
【0039】
本発明によれば、2−シータ=18.7、11.7および19.2°に特異的ピークを持つX線粉末回折パターンを有する本薬剤の結晶形態、形態3、が提供される。ここで、前記値はプラスマイナス0.5°2−シータであってよい。
【0040】
本発明によれば、2−シータ=18.7、11.7、19.2、7.8、14.1、14.9および9.4°に特異的ピークを持つX線粉末回折パターンを有する本薬剤の結晶形態、形態3、が提供される。ここで、前記値はプラスマイナス0.5°2−シータであってよい。
【0041】
本発明によれば、2−シータ=18.7、11.7、19.2、7.8、14.1、14.9、9.4、15.6、16.1および9.6°に特異的ピークを持つX線粉末回折パターンを有する本薬剤の結晶形態、形態3、が提供される。ここで、前記値はプラスマイナス0.5°2‐シータであってよい。
【0042】
本発明によれば、2−シータ=18.7°に少なくとも1つの特異的ピークを持つX線粉末回折パターンを有する本薬剤の結晶形態、形態3、が提供される。
本発明によれば、2−シータ=18.7および11.7°に少なくとも2つの特異的ピークを持つX線粉末回折パターンを有する本薬剤の結晶形態、形態3、が提供される。
【0043】
本発明によれば、2−シータ=18.7、11.7および19.2°に特異的ピークを持つX線粉末回折パターンを有する本薬剤の結晶形態、形態3、が提供される。
本発明によれば、2−シータ=18.7、11.7、19.2、7.8、14.1、14.9、9.4°に特異的ピークを持つX線粉末回折パターンを有する本薬剤の結晶形態、形態3、が提供される。
【0044】
本発明によれば、2−シータ=18.7、11.7、19.2、7.8、14.1、14.9、9.4、15.6、16.1および9.6°に特異的ピークを持つX線粉末回折パターンを有する本薬剤の結晶形態、形態3、が提供される。
【0045】
本発明によれば、CuKa照射を用いた、図3に示されるX線粉末回折パターンを有する本薬剤の結晶形態、形態3、が提供される。
【0046】
【表2】
【0047】
DSC分析から、形態3は309.3℃で融解を開始することがわかる。DSC温度記録図を図4に示す。
本発明によれば、およそ2−シータ=16.2にピークを持つX線回折パターンを有する、本薬剤の結晶形態、形態4が提供される。
【0048】
2−シータ(θ)値はCuKa照射を用いて測定された。
本発明によれば、およそ2−シータ=16.2°および20.6°に少なくとも2つの特異的ピークを持つX線粉末回折パターンを有する本薬剤の結晶形態、形態4、が提供される。
【0049】
本発明によれば、およそ2−シータ=16.2、20.6および17.7°に特異的ピークを持つX線粉末回折パターンを有する本薬剤の結晶形態、形態4、が提供される。
本発明によれば、およそ2−シータ=16.2、20.6、17.7、10.8および15.5°に特異的ピークを持つX線粉末回折パターンを有する本薬剤の結晶形態、形態4、が提供される。
【0050】
本発明によれば、およそ2−シータ=16.2、20.6、17.7、10.8、15.5、20.9、26.1、11.6、26.7および18.1°に特異的ピークを持つX線粉末回折パターンを有する本薬剤の結晶形態、形態4、が提供される。
【0051】
本発明によれば、CuKa照射を用いた、図5に示されるX線粉末回折パターンと実質的に同一のX線粉末回折パターンを有する本薬剤の結晶形態、形態4、が提供される。
本発明によれば、2−シータ=16.2°プラスマイナス0.5°2−シータに少なくとも1つの特異的ピークを持つX線粉末回折パターンを有する本薬剤の結晶形態、形態4、が提供される。
【0052】
本発明によれば、2−シータ=16.2°および20.6°に少なくとも2つの特異的ピークを持つX線粉末回折パターンを有する本薬剤の結晶形態、形態4、が提供される。ここで、前記値はプラスマイナス0.5°2−シータであってよい。
【0053】
本発明によれば、2−シータ=16.2、20.6および17.7°に特異的ピークを持つX線粉末回折パターンを有する本薬剤の結晶形態、形態4、が提供される。ここで、前記値はプラスマイナス0.5°2−シータであってよい。
【0054】
本発明によれば、2−シータ=16.2、20.6、17.7、10.8および15.5°に特異的ピークを持つX線粉末回折パターンを有する本薬剤の結晶形態、形態4、が提供される。ここで、前記値はプラスマイナス0.5°2−シータであってよい。
【0055】
本発明によれば、2−シータ=16.2、20.6、17.7、10.8、15.5、20.9、26.1、11.6、26.7および18.1°に特異的ピークを持つX線粉末回折パターンを有する本薬剤の結晶形態、形態4、が提供される。ここで、前記値はプラスマイナス0.5°2‐シータであってよい。
【0056】
本発明によれば、2−シータ=16.2°に少なくとも1つの特異的ピークを持つX線粉末回折パターンを有する本薬剤の結晶形態、形態4、が提供される。
本発明によれば、2−シータ=16.2および20.6°に少なくとも2つの特異的ピークを持つX線粉末回折パターンを有する本薬剤の結晶形態、形態4、が提供される。
【0057】
本発明によれば、2−シータ=16.2、20.6および17.7°に特異的ピークを持つX線粉末回折パターンを有する本薬剤の結晶形態、形態4、が提供される。
本発明によれば、2−シータ=16.2、20.6、17.7、10.8および15.5°に特異的ピークを持つX線粉末回折パターンを有する本薬剤の結晶形態、形態4、が提供される。
【0058】
本発明によれば、2−シータ=16.2、20.6、17.7、10.8、15.5、20.9、26.1、11.6および26.7°に特異的ピークを持つX線粉末回折パターンを有する本薬剤の結晶形態、形態4、が提供される。
【0059】
本発明によれば、CuKa照射を用いた、図5に示されるX線粉末回折パターンを有する本薬剤の結晶形態、形態4、が提供される。
【0060】
【表3】
【0061】
形態4のDSC分析から、初期事象は254.0℃に開始し、ピークは262.0℃で、その後312.0℃で融解を開始することがわかる。したがって形態4の融解開始は約312.0℃である。DSC温度記録図を図6に示す。
【0062】
形態4の別のより純粋なサンプルから、図7に示すXRDパターンおよび面間隔dスペクトルが得られた。2‐シータ値の位置および面間隔dをそれぞれ表Dおよび表Eに示す。
【0063】
【表4】
【0064】
【表5】
【0065】
表Cおよび表D中の2−シータ値の僅かなばらつきは、後述のX線粉末ディフラクトグラムにおける回折角の測定誤差が原因と考えられる。
したがって、測定誤差を考慮に入れた本発明の別の態様においては、2−シータ=16.1および20.5°に少なくとも2つの特異的ピークを持つX線粉末回折パターンを有する本薬剤の結晶形態、形態4、が提供される。
【0066】
本発明によれば、2−シータ=16.1、20.5および17.6°に特異的ピークを持つX線粉末回折パターンを有する本薬剤の結晶形態、形態4、が提供される。
本発明によれば、2−シータ=16.1、20.5、17.6、10.8および15.4°に特異的ピークを持つX線粉末回折パターンを有する本薬剤の結晶形態、形態4、が提供される。
【0067】
本発明によれば、2−シータ=16.1、20.5、17.6、10.8、15.4、20.9および26.1°に特異的ピークを持つX線粉末回折パターンを有する本薬剤の結晶形態、形態4、が提供される。
【0068】
本発明は形態2、3および4の結晶形態に関する、と述べられている場合、結晶化度は、都合良くは約60%より大きく、より都合良くは約80%より大きく、好ましくは約90%より大きく、より好ましくは約95%より大きい。最も好ましくは、結晶化度は約98%よりも大きい。
【0069】
別の態様においては、本発明は結晶形態2としての本薬剤に関する。
別の態様においては、本発明は結晶形態3としての本薬剤に関する。
別の態様においては、本発明は結晶形態4としての本薬剤に関する。
【0070】
別の態様においては、本発明は結晶形態1を実質的に含まない本薬剤の結晶形態2に関する。
別の態様においては、本発明は結晶形態1を実質的に含まない本薬剤の結晶形態3に関する。
【0071】
別の態様においては、本発明は結晶形態1を実質的に含まない本薬剤の結晶形態4に関する。
形態1を実質的に含まない結晶形態とは、形態1を30%未満しか有さない結晶形態を意味する。別の態様においては、「実質的に含まない」とは形態1を20%未満しか有さないことを意味する。別の態様においては、「実質的に含まない」とは形態1を10%未満しか有さないことを意味する。さらに別の態様においては、「実質的に含まない」とは形態1を5%未満しか有さないことを意味する。さらに別の態様においては、「実質的に含まない」とは形態1を1%未満しか有さないことを意味する。
【0072】
形態2、3および4は、図1、2および3に示したX線粉末回折パターンと実質的に同一のX線粉末回折パターンを提供し、表A、BおよびCに示した最も顕著な10のピーク(角度2−シータ値)を実質的に有している。X線粉末回折パターンの2−シータ値は、機器によって、または試料によってわずかに異なることがあるので、引用された値が絶対的なものと解釈すべきではない、ということが理解されるであろう。
【0073】
測定条件(使用する装置または機器など)によっては、1つまたは複数の測定誤差を有するX線粉末回折パターンが得られることがある、ということは公知である。特に、X線粉末回折パターンの強度は測定条件によって変動し得るということは、一般的に知られている。したがって、本発明の形態2、3および4は図1、2および3に示したX線粉末回折パターンと同一のX線粉末回折パターンを提供する結晶に限定されるわけではなく、図1、2および3に示したX線粉末回折パターンと実質的に同一のX線粉末回折パターンを提供するいずれの結晶も本発明の範囲内に含まれることを理解すべきである。X線粉末回折の当業者であれば、X線粉末回折パターンの実質的な同一性を判断することが可能である。
【0074】
X線粉末回折の当業者であれば、ピークの相対強度が、例えば、試料の分析にも影響し得る、30ミクロンを超える大きさの粒子および非ユニタリーアスペクト比に影響されることがある、ということは理解されるであろう。該当業者は、反射の位置が、回折計に置かれた試料の正確な高さ、および回折計のゼロ較正によって影響されることがある、ということも理解するであろう。試料の表面平面性もわずかに影響する場合がある。ゆえに、示された回折パターンデータが絶対的な値であると解釈すべきではない。(Jenkins, R & Snyder, R.L. ‘Introduction to X-Ray Powder Diffractometry’ John Wiley & Sons 1996; Bunn, C.W. (1948), Chemical Crystallography, Clarendon Press, London; Klug, H. P. & Alexander, L. E. (1974), X-Ray Diffraction Procedures)。
【0075】
一般に、X線粉末回折における回折角の測定誤差は、約5%以下、特にプラスマイナス0.5°2−シータである。典型的にはプラスマイナス0.2°2−シータである。図1、2、3および4のX線粉末回折パターンについて検討する場合や表A、B、CおよびDを読む場合には、こうした程度の測定誤差を考慮に入れておくべきである。さらに、実験条件および試料調製(優先配向)によって強度が変動する可能性があることを理解すべきである。
【0076】
使用した技術の詳細
X線粉末回折
【0077】
【表6】
【0078】
分析機器:シーメンスD5000
X線粉末回折スペクトルは、結晶材料の試料をシーメンスのシリコン単結晶(SSC)ウェハマウントに載せ、該試料を、顕微鏡用スライドで広げて薄層にすることによって決定した。試料を毎分30回転で回転させ(計数統計を改善するため)、1.5406オングストロームの波長で、40kVおよび40mAで作動させた銅製ロングファインフォーカス管(long−fine focus tube)によって発生させたX線を照射した。平行X線源はV20に設定された自動可変発散スリットを通過し、反射した放射線は2mmの散乱線除去スリット(antiscatter slit)と0.2mmの検出器スリットを通って方向づけられた。試料を、シータ−シータモードで2度から40度の2−シータ範囲にわたって、0.02度の2−シータ増加分あたり1秒間暴露した(連続スキャンモード)。稼働時間は31分41秒であった。機器には検出器としてシンチレーション計数管が備えられていた。制御およびデータ収集には、Diffract+ソフトウェアで作動する、デルOptiplex 686 NT 4.0 Workstationを用いた。X線粉末回折の当業者であれば、ピークの相対強度が、例えば、試料の分析にも影響し得る、30ミクロンを超える大きさの粒子および非ユニタリーアスペクト比に影響されることがある、ということは理解するであろう。該当業者は、反射の位置が、回折計に置かれた試料の正確な高さ、および回折計のゼロ較正によって影響されることがある、ということも理解するであろう。試料の表面平面性もわずかに影響する場合がある。ゆえに、示された回折パターンデータが絶対的な値であると解釈すべきではない。
【0079】
示差走査熱量測定
分析機器:ティー・エイ・インスツルメントQ1000 DSC
典型的には、蓋を取り付けた40μlのアルミ製の鍋に入った5mg未満の材料を、25℃〜325℃の温度範囲にわたって、1分間当たり10℃の一定の加熱速度で加熱した。窒素を使用したパージガスを用いた(流速100ml/分)。
【0080】
形態2、3および4は形態1からの競合スラリー化(competitive slurring)または播種によって調製することができる。
形態2はアセトニトリル中での競合スラリー化によって都合よく調製することができる。特にこれは45〜55℃の温度範囲、例えば約50℃で行われる。
【0081】
形態3はメタノール中での競合スラリー化によって都合よく調製することができる。特にこれは15〜30℃の温度範囲、例えば周囲温度近辺で行われる。
形態4は酢酸エチル中での競合スラリー化によって都合よく調製することができる。特にこれは15〜30℃の温度範囲、例えば周囲温度近辺で行われる。また、形態4は高温のアセトンまたはアセトニトリル中での競合スラリー化によって調製することができる。
【0082】
上述したように、本薬剤は11βHSD1阻害活性を有する。この特性は、以下のアッセイによって評価することができる。
アッセイ
11βHSD1の酸化還元酵素(oxo−reductase)活性による、コルチゾンのその活性型ステロイドであるコルチゾールへの変換は、競合均一時間分解蛍光アッセイ(HTRF)(セイエス ビオ アンテルナスィオナル(CisBio International)、研究開発、 管理および欧州事務所、インビトロテクノロジー(In Vitro Technologies)―HTRF(登録商標)/Bioassays BP 84175, 30204 バニョル/セーズ Cedex、フランス。Cortisol bulk HTRF kit: Cat No. 62CORPEC)によって測定することができる。
【0083】
本明細書に記載の化合物の評価は、N−末端に6−Hisタグを有する全長ヒト11βHSD1酵素(*1)を発現したバキュロウイルスを使用して行った。該酵素は、銅キレートカラムを用いて、洗剤で可溶化した細胞溶解物から精製した。11βHSD1の阻害剤はコルチゾンのコルチゾールへの変換を低減させるが、そのことは、上記アッセイにおいてシグナルの増加によって同定される。
【0084】
被験化合物を、10mMになるようにジメチルスルホキシド(DMSO)中に溶解させ、さらに1%DMSO含有アッセイ緩衝液で最終アッセイ濃度である10倍まで希釈した。次に、希釈化合物を黒384ウェルプレート(マトリックス(Matrix)、ハドソン、ニューハンプシャー州、米国)にまいた。
【0085】
アッセイは、コルチゾン(シグマ、プール、ドーセット州、英国、160nM)、グルコース−6−リン酸(ロシュ・ダイアグノスティックス、1mM)、NADPH(シグマ、プール、ドーセット州、英国、100μM)、グルコース−6−リン酸脱水素酵素(ロシュ・ダイアグノスティックス、12.5μg/ml)、EDTA(シグマ、プール、ドーセット州、英国、1mM)、アッセイ緩衝液(K2HPO4/KH2PO4、100mM)pH7.5、組換え11βHSD1および試験化合物からなる全量20μl中で(実行可能なアッセイウィンドウを得るために適切な希釈溶液を用いて−好適な希釈溶液の例としては、ストック酵素の1000倍希釈溶液がある)行った。アッセイプレートを37℃で25分間インキュベートし、その後、10μlの0.5mMのグリセルレチン酸、および抱合型コルチゾール(XL665またはD2)を添加して反応を停止した。次に、10μlの抗−コルチゾールクリプテートを添加し、プレートを密封して、室温で6時間インキュベートした。665nmおよび620nmでの蛍光を測定し、665nm:620nmの比をEnvisionプレートリーダーを用いて算出した。
【0086】
次に、これらのデータを使って、各化合物のIC50値(Origin 7.5、マイクロカルソフトウェア(Microcal software)、ノーサンプトン、マサチューセッツ州、米国)および/または30μMの化合物における阻害率(%)を算出した。
*1 The Journal of Biological Chemistry, Vol. 26, No 25, pp16653-16658
次の結果が得られた:参考例1 IC50 0.008μM
本発明の化合物の経口バイオアベイラビリティは、次のように試験することができる。
【0087】
PK試験におけるバイオアベイラビリティの決定
化合物を、25%HPBCDを含有するpH5.5ソレンソン緩衝液(sorrensons buffer)製剤に入れ、2mg/kg(2ml/kg)を静脈投与、5mg/kg(5ml/kg)を経口投与する。両方の経路について、血液サンプル(200μl)を、投与前、投与後0.25、0.5、1、2、3、4、5、6、8および24時間に採取し、遠心分離により血漿を調製する。血漿サンプルを下記のように分析する。PKパラメーター(クリアランス、分布容積、バイオアベイラビリティ、吸収率など)を、適切なPKソフトウェア(WinNon-Lin)を使用して標準PK法により算出する。
【0088】
血漿サンプルの生物分析
以下に説明するのは、DMPK解析に使用されるすべてのPK試験種に対して研究対象化合物を単一化合物投与またはカセット投与した後に、血漿サンプルを手作業で調製するための指針である。オープンアクセス(LC−MS/MS)またはマニュアル手法(LC−MS)による分析について説明する。
【0089】
内容
1.材料
2.一般的抽出方法
3.一般的プレート配置による実施例サンプルリスト
4.オープンアクセスのバッチサブミッションとシステムチェック
5.バッチパスの許容基準
1.材料
溶媒:メタノール、アセトニトリルおよびDMSO
水:精製またはHPLCグレード
1ml浅型96ウェルプレートまたはエッペンドルフチューブ
2ml深型96ウェルプレートおよび蓋
ブランク(対照)血漿
2.一般的抽出方法
化合物(1種または複数)を、塩係数も考慮に入れながら、DMSOを用いて可溶化し1mg/mlとする。このDMSOストック(1種または複数)を用いて、すべての検量線および品質管理(QC)サンプルを作成することができる。
2.i 単一化合物分析
2.i.a 検量線およびQCサンプルの作成:
1.標準溶液を以下のように調製する。
【0090】
【表7】
【0091】
2.50μlのブランク血漿を1mlの96ウェルプレート(浅型ウェル)の1つのウェルに移す。
3.5μlの各標準溶液を該プレートの他のウェルに移す。
【0092】
4.50μlのブランク血漿をこれらの各ウェルに加える。
5.QCサンプルを生成するために、100ng/ml、1,000ng/ml、および10,000ng/mlの各標準溶液について、各5μlを該プレートに加える(各濃度につきQCは3回)。
【0093】
6.50μlのブランク血漿をこれらのウェルのそれぞれに加える。
7.50μlの各PKサンプルを1mlの96ウェルプレートに移す。
8.5μlのメタノール(−化合物)を各PKサンプルに加える。
【0094】
9.すべての投与製剤がボルテックス混合で十分に混和しているか確認する。
10.予測濃度の静脈内(IV)および経口(PO)投与用の製剤を10μg/mlとなるようにメタノールで希釈する(例えば、予測濃度が2mg/mlとなるように調製された製剤は、1:200に希釈して10μg/mlの溶液とする)。
【0095】
11.血漿を50μlずつ(×6)プレートに加える。そのうちの3個のウェルに希釈IV製剤5μlを加え、残りの3個のウェルにはPO製剤を同様に加える。
12.研究関連内部標準(1μg/ml)を含有する100μlのアセトニトリルを、すべての検量線、QC、PKおよび製剤サンプルに加えることによって、タンパク質を沈殿させる。
【0096】
13.プレートをボルテックス混合後、4、000gで10分間遠心分離する。
14.100μlの上清を2mのl96ウェルプレートの各ウェル(下記のプレートマップ参照)に移す。ペレットを乱さないように注意する。
【0097】
15.50:50のメタノール:水、約1.5mlを後者のウェルに加える。
16.トリプルクワッド(triple quad)システムでの分析用に、400μlの水(HPLCグレード)を各サンプルに加え、静かに混合する。
【0098】
17.2mlプレートに、100,000ng/mlの各標準溶液のストック100μlを加え、次いで水900μlを加える。次に、他の一つのウェルに内部標準のサンプルを加える(プレートマップ参照)。これらは化合物調整のために行う(プレートマップ上に溶液調整と表示)。
【0099】
18.プラットフォームシステムでの分析用に、100μlの水(HPLCグレード)を各サンプルに加え、静かに混合する。
19.調製した化合物溶液を用いて、全化合物を手作業で5,000ng/mlに調整する(50,000ng/ml標準溶液100μlを900μlの水に添加する)。
2.ii カセット投与分析
2.iia 検量線およびQCサンプルの作成:
注記:カセット投与では、1mg/mlのストックを希釈するために必要なメタノールの量は、存在する化合物の数にしたがって調整する。
【0100】
1.バイアルに、必要とされる各1mg/mlのストック100μlを入れる。
2.必要量のメタノールを加え、全量1mlとする。
3.単一化合物の分析の場合と同様、以降の工程(上記工程2〜16)をすべて行う。
2.iii PKサンプルが定量上限(ULOQ)を超える場合。
【0101】
1.上記(工程1〜6)と同様にさらなる検量線およびQCサンプルを作成する。
2.50μl未満(例えば25μl)のULOQを超えるPKサンプルを移す。
3.これらのサンプルに十分なコントロール血漿を加え、最終血漿量を50μlとする。行った希釈を記録しておく。
【0102】
4.残りの全てのPKサンプルをそれぞれ50μl移す。
5.上記と同様(工程8〜16)、すべての製剤サンプルを調製し、すべてのサンプルを抽出する。
注記:検量線作成に使用する上限濃度は見直してもよいが、HPLCカラムまたはMS機器が飽和しないように注意しなければいけない。これはPKサンプルの希釈が推奨されているためである。
2.iv 感度が低い(定量下限(LLOQ)が高い)場合。
注記:血漿濃度の大部分が定量下限以下であるか、LLOQが10ng/mlより高い場合を高LLOQとする。これらのいずれかに当てはまる場合には、下記の方法を適用する。
【0103】
本発明のさらなる態様によれば、薬学的に許容される希釈剤または担体といっしょに、上記で定義したような、本薬剤、またはその薬学的に許容される塩を含む医薬組成物が提供される。
【0104】
本発明の組成物は、経口使用(例えば、錠剤、ロゼンジ剤、硬もしくは軟カプセル剤、水性もしくは油性の懸濁剤、乳剤、分散性粉剤もしくは顆粒剤、シロップ剤またはエリキシル剤として)、局所使用(例えば、クリーム剤、軟膏剤、ゲル剤、水性もしくは油性の液剤または懸濁剤として)、吸入投与(例えば、微粉または液体エアゾールとして)、通気投与(例えば、微粉として)、または非経口投与(例えば、静脈内、皮下、筋肉内、もしくは筋肉内投与用の無菌性の水性もしくは油性液剤として、または直腸投与用坐薬として)に適した形態であってよい。一般に、経口使用に適した形態の組成物が好ましい。
【0105】
本発明の組成物は、当技術分野において周知である、従来の医薬用賦形剤を使用する従来の手順により得ることができる。したがって、経口使用のための組成物は、例えば、1種または複数の着色剤、甘味剤、賦香剤、および/または防腐剤を含有していてもよい。
【0106】
錠剤用の好適な薬学的に許容される賦形剤としては、例えば、乳糖、炭酸ナトリウム、リン酸カルシウムまたは炭酸カルシウムなどの不活性希釈剤、コーンスターチまたはアルゲン酸などの造粒剤および崩壊剤、澱粉などの結合剤、ステアリン酸マグネシウム、ステアリン酸またはタルクなどの滑沢剤、p−ヒドロキシ安息香酸エチルまたはp−ヒドロキシ安息香酸プロピルなどの防腐剤、およびアスコルビン酸などの抗酸化剤が挙げられる。錠剤は、消化管内での崩壊およびそれに続く活性成分の吸収を加減するため、または錠剤の安定性および/もしくは外見を改良するために、コーティングされていなくてもされていてもよいが、いずれの場合においても、当技術分野において周知の従来のコーティング剤および手順を用いる。
【0107】
経口使用のための組成物は、活性成分が、炭酸カルシウム、リン酸カルシウムもしくはカオリンなどの不活性な固形希釈剤と混合されている、硬ゼラチンカプセル剤の形態、または活性成分が、水、またはピーナッツ油、流動パラフィンもしくはオリーブ油などの油と混合されている、軟ゼラチンカプセル剤の形態であってよい。
【0108】
水性懸濁剤は通常、微粉化した形態の活性成分ともに、1種または複数の、カルボキシメチルセルロースナトリウム、メチルセルロース、ヒドロキシプロピルメチルセルロース、アルギン酸ナトリウム、ポリビニル−ピロリドン、トラガカントゴムおよびアラビアゴムなどの懸濁剤、レシチン、またはアルキレンオキシドと脂肪酸との縮合生成物(例えば、ステアリン酸ポリオキシエチレン)、もしくはエチレンオキシドと長鎖脂肪族アルコールとの縮合生成物(例えば、ヘプタデカエチレンオキシセタノール)、もしくはエチレンオキシドと脂肪酸およびヘキシトールに由来する部分エステルとの縮合生成物(例えば、ポリオキシエチレンソルビトールモノオレアート)、もしくはエチレンオキシドと長鎖脂肪族アルコールとの縮合生成物(例えば、ヘプタデカエチレンオキシセタノール)、もしくはエチレンオキシドと脂肪酸およびヘキシトールに由来する部分エステルとの縮合生成物(例えば、ポリオキシエチレンソルビトールモノオレアート)、もしくはエチレンオキシドと脂肪酸およびヘキシトール無水物に由来する部分エステルとの縮合生成物(例えば、ポリエチレンソルビタンモノオレアート)などの分散剤または湿潤剤を含有する。また水性懸濁剤は、1種または複数の防腐剤(例えば、p−ヒドロキシ安息香酸エチルまたはp−ヒドロキシ安息香酸プロピル)、抗酸化剤(例えば、アスコルビン酸)、着色剤、賦香剤、および/または甘味剤(例えば、ショ糖、サッカリン、またはアスパルテーム)を含有していてもよい。
【0109】
油性懸濁剤は、活性成分を植物油(例えば、ラッカセイ油、オリーブ油、ゴマ油もしくはココナツ油)または鉱油(例えば、流動パラフィン)に懸濁させることによって製剤化することができる。また油性懸濁剤は、ミツロウ、固形パラフィンまたはセチルアルコールなどの増粘剤を含有していてもよい。口当たりのよい経口剤とするために、上述したような甘味剤、および賦香剤を添加してもよい。これらの組成物は、アスコルビン酸などの抗酸化剤を添加することによって保存することができる。
【0110】
水の添加による水性懸濁剤の調製に好適な分散性粉剤および顆粒剤は通常、活性成分とともに、分散剤または湿潤剤、懸濁剤、および1種または複数の防腐剤を含有する。好適な分散剤または湿潤剤、および懸濁剤は、すでに上述したものが代表的である。甘味剤、賦香剤および着色剤などの賦形剤がさらに存在してもよい。
【0111】
本発明の医薬組成物は、水中油型乳剤の形態であってもよい。油相は、オリーブ油やラッカセイ油などの植物油、流動パラフィンなどの鉱油、またはこれらのいずれかの混合物であってよい。好適な乳化剤は、例えば、アラビアゴムやトラガカントゴムなどの天然ゴム、大豆、レシチンなどの天然リン脂質、脂肪酸およびヘキシトール無水物に由来するエステルまたは部分エステル(例えば、ソルビタンモノオレアート)、ならびに該部分エステルとエチレンオキシドとの縮合生成物(例えば、ポリオキシエチレンソルビタンモノオレアート)であってよい。乳剤は、甘味剤、賦香剤および防腐剤を含有していてもよい。
【0112】
シロップ剤およびエリキシル剤は、グリセロール、プロピレングリコール、ソルビトール、アスパルテームまたはショ糖などの甘味剤とともに製剤化することができ、粘滑剤、防腐剤、賦香剤および/または着色剤を含有していてもよい。
【0113】
医薬組成物は、滅菌注射用の水性または油性懸濁液の形態であってもよく、上述した適当な分散剤または湿潤剤、および懸濁剤のうちの1種または複数を用いて、公知の手順に従って製剤化することができる。滅菌注射用の製剤は、無毒性で非経口的に許容される希釈剤もしくは溶剤中の滅菌注射用の溶液または懸濁液(例えば、1,3−ブタンジオール中の溶液)であってもよい。
【0114】
吸入投与用の組成物は、超微粒子状の固体小滴または液体小滴のいずれかを含有するエアゾールとして活性成分を調合するように調整された、従来の加圧エアゾールの形態であってよい。揮発性のフッ素化炭化水素や炭化水素などの従来のエアゾール噴射剤を使用することができ、エアゾール装置は、定量の活性成分を調合するように調整されると都合が良い。
【0115】
製剤に関するさらなる情報については、Comprehensive Medicinal Chemistry (Corwin Hansch; Chairman of Editorial Board), Pergamon Press 1990の第5巻、25.2章を参照されたい。
【0116】
単一剤形を製造するために1種または複数の賦形剤と組み合わされる活性成分の量は、被治療者および具体的な投与経路によって必然的に異なってくる。例えば、ヒトに経口投与するための製剤は、例えば、組成物全体の約5から約98重量%の間で異なり得る適切かつ使いやすい量の賦形剤と混合された、0.5mg〜2gの活性成分を通常含有する。投与単位剤形は、約1mg〜約500mgの活性成分を通常含有する。投与経路および投与法に関するさらなる情報については、Comprehensive Medicinal Chemistry (Corwin Hansch; Chairman of Editorial Board), Pergamon Press 1990の第5巻、25.3章を参照されたい。
【0117】
本発明者らは、本薬剤、またはその薬学的に許容される塩は、有効な11βHSD1阻害剤であり、したがってメタボリック症候群に関連する病態の治療において価値があることを見出した。
【0118】
当然のことながら、本明細書で「メタボリック症候群」という語を使用する場合は、1)および/もしくは2)の定義、またはこの症候群について認められているその他のあらゆる定義のメタボリック症候群に関する。当技術分野で使用される「メタボリック症候群」の同義語としては、リーベン症候群、インスリン抵抗性症候群および症候群Xが挙げられる。当然のことながら、本明細書で「メタボリック症候群」という語を使用する場合は、リーベン症候群、インスリン抵抗性症候群および症候群Xのことも指している。
【0119】
本発明のさらなる態様によれば、ヒトなどの温血動物の予防的または治療的処置方法における使用のための、上記に定義したような本薬剤、またはその薬学的に許容される塩が提供される。
【0120】
したがって、本発明のこの態様によれば、医薬として使用するための本薬剤、またはその薬学的に許容される塩がある。
本発明の別の特徴によれば、ヒトなどの温血動物での11βHSD1阻害作用の発現に使用される医薬の製造における、本薬剤、またはその薬学的に許容される塩の使用が提供される。
【0121】
11βHSD1阻害作用の発現、または11βHSD1阻害作用を発現させることに言及する場合、好適には、メタボリック症候群の治療を指す。また、11βHSD1阻害作用の発現に言及する場合は、糖尿病、肥満、高脂血症、高血糖症、高インスリン血症または高血圧症の治療を指し、特に2型糖尿病および肥満の治療を指す。あるいは、11βHSD1阻害作用の発現に言及する場合は、緑内障、骨粗しょう症、結核、痴呆、認知障害または鬱病の治療を指す。
【0122】
また、11βHSD1阻害作用の発現に言及する場合は、例えば、言語流暢性、言語記憶もしくは論理的記憶の改善による個人の認知能力の改善といった認知障害の治療、または軽度の認知障害の治療を指す。例えば、国際公開第03/086410号およびそれに含まれる参考文献、ならびにProceedings of National Academy of Sciences (PNAS), 2001, 98(8), 4717-4721を参照のこと。
【0123】
また、11βHSD1阻害作用の発現に言及する場合は、アテローム性動脈硬化症の治療、その発症の遅延、および/またはそのリスクの低減を指す。例えば、J. Experimental Medicine, 2005, 202(4), 517-527を参照のこと。
【0124】
また、11βHSD1阻害作用の発現に言及する場合は、アルツハイマー病および/または神経変性障害の治療を指す。
本発明のこの態様のさらなる特徴によれば、11βHSD1阻害作用を発現する治療を必要としているヒトなどの温血動物において11βHSD1阻害作用を発現する方法であって、該動物に、有効な量の式(1)の化合物、またはその薬学的に許容される塩を投与することを含む方法が提供される。
【0125】
治療薬におけるそれらの使用に加えて、本薬剤、またはその薬学的に許容される塩は、新しい治療薬の探索の一環として、ネコ、イヌ、ウサギ、サル、ラットおよびマウスなどの実験動物における11βHSD1阻害剤の効果を評価するための、インビトロおよびインビボ試験系の開発および標準化における薬理学的手段としても有用である。
【0126】
本明細書に記載の11βHSD1の阻害は、単一療法として適用してもよいか、または、本発明の主題に加えて、1種または複数の他の物質および/もしくは治療を伴ってもよい。そのような共同治療(conjoint treatment)は、治療の個々の成分を、同時に、順次、または別々に投与することにより行うことができる。同時の治療は、単一の錠剤または別々の錠剤で行ってよい。例えば、11βHSD1阻害剤、特に本発明の11βHSD1阻害剤と同時投与できる薬剤としては、治療別に大きく分けた以下のものが挙げられる。
1)インスリンおよびインスリン類似体;
2)スルホニル尿素薬などのインスリン分泌促進薬(例えば、グリベンクラミド、グリピジド)、食後血糖調節薬(例えば、レパグリニド、ナテグリニド)、グルカゴン様ペプチド1作動薬(GLP1作動薬)(例えば、エクセナチド、リラグルチド)およびジペプチジルペプチダーゼIV阻害剤(DPP−IV阻害剤);
3)PPARγ作動薬などのインスリン増感剤(例えば、ピオグリタゾンおよびロシグリタゾン);
4)肝糖産生を抑制する薬剤(例えば、メトホルミン);
5)腸からのグルコースの吸収を抑えるように設計された薬剤(例えば、アカルボース);
6)持続性高血糖症の合併症を治療するように設計された薬剤;例えば、アルドース還元酵素阻害剤
7)リン酸化チロシンホスファターゼ阻害剤、グルコース−6−ホスファターゼ阻害剤、グルカゴン受容体拮抗剤、グルコキナーゼ活性化剤、グリコーゲンホスホリラーゼ阻害剤、フルクトース1,6ビスホスファターゼ阻害剤、グルタミン:フルクトース−6−リン酸アミドトランスフェラーゼ阻害剤などのその他の抗糖尿病剤;
8)抗肥満剤(例えば、シブトラミンおよびオルリスタット);
9)HMG−CoA還元酵素阻害剤(プラバスタチンなどのスタチン類);PPARα作動薬(ゲムフィブロジルなどのフィブラート類);胆汁酸金属イオン封鎖剤(コレスチラミン);コレステロール吸収阻害剤(植物スタノール、合成阻害剤);回腸胆汁酸吸収阻害剤(IBATi)、コレステロールエステル転送タンパク質阻害剤、ならびにニコチン酸および類似体(ナイアシンおよび徐放性製剤)などの抗脂質異常症薬;
10)β遮断剤(例えば、アテノロール、インデラル);ACE阻害剤(例えば、リシノプリル);カルシウム拮抗剤(例えば、ニフェジピン);アンジオテンシン受容体拮抗剤(例えば、カンデサルタン)、α拮抗剤および利尿剤(例えば、フロセミド、ベンズチアジド)などの抗高血圧剤;
11)抗血栓剤、繊維素溶解活性化剤および抗血小板剤;トロンビン拮抗剤;因子Xa阻害剤;因子VIIa阻害剤;抗血小板剤(例えば、アスピリン、クロピドグレル);抗凝固剤(ヘパリンおよび低分子量類似体、ヒルジン)ならびにワルファリンなどの止血調節剤;
12)非ステロイド系抗炎症薬(例えば、アスピリン)およびステロイド系抗炎症薬(例えば、コルチゾン)などの抗炎症剤;および
13)腎臓によるグルコースの再吸収を防ぐ薬剤(SGLT阻害剤)。
【実施例】
【0127】
次に、本発明を下記の実施例により説明するが、特に断りがない限り、
(i)温度は摂氏(℃)で表し、操作は、室温または周囲温度、すなわち18〜25℃の温度範囲で、かつアルゴンなどの不活性ガス雰囲気下で行った。
(ii)溶媒の留去は、ロータリーエバポレーターを用いて、減圧下(600〜4000Pa;4.5〜30mmHg)、60℃以下の浴温で行った。
(iii)クロマトグラフィーは、シリカゲルのフラッシュクロマトグラフィーを意味する。
(iv)通常、反応の経過はTLCにより追跡し、反応時間は例示のためにのみ示す。
(v)収率は例示のためにのみ示し、必ずしも入念な過程開発によって得ることができるものではない。さらに物質が必要な場合には、調製を繰り返した。
(vi)NMRデータ(1H)は、示されている場合、特に断りがない限りは、溶媒として重水素化ジメチルスルホキシド(DMSO−d6)を用いて(特に断りがない限り)300または400MHzで測定し、テトラメチルシラン(TMS)に対する百万分率(ppm)で示した、主要な診断プロトンのデルタ値の形態である。ピークの多重度は次のように示す。s、シングレット;d、ダブレット;dd、ダブル−ダブレット;dt、ダブル−トリプレット;dm、ダブル−マルチプレット;t、トリプレット;m、マルチプレット;br、ブロード。
(vii)化学記号は通常の意味を有し、SI単位系および記号を用いる。
(viii)溶媒の割合は体積:体積(v/v)で表す。
(ix)質量スペクトル(MS)は、直接暴露プローブを用いて、化学イオン化(CI)モードにおいて70電子ボルトの電子エネルギーで実施した。イオン化は、示されている場合、電子衝撃(EI)、高速原子衝撃(FAB)または電子スプレー(ESP)により行った。m/zの値を示し、通常は、親質量を示すイオンのみを報告する。
(x)相対量(rel vol)とは、重要中間体の量と比較した相対的な量である。相対量は通常溶媒の量を指すのに用いられる。例えば重要中間体が100gで1000mlの溶媒を使用した場合、これは10相対量の溶媒と呼ばれる。
(xi)下記の略語を、以下において、または上記の工程の箇所で使用することがある。
【0128】
Et2O ジエチルエーテル
DMF ジメチルホルムアミド
DCM ジクロロメタン
THF テトラヒドロフラン
DMSO ジメチルスルホキシド
EtOAc 酢酸エチル
MTBE メチルtert−ブチルエーテル
DSC 示差走査熱量測定
参考例1
4−[4−(2−アダマンチルカルバモイル)−5−tert−ブチル−ピラゾル−1−イル]安息香酸
【0129】
【化2】
【0130】
2Mの水酸化ナトリウム水溶液(51.7mL、103.32mmol)をメタノール(100mL)中の4−[4−(2−アダマンチルカルバモイル)−5−tert−ブチル−ピラゾル−1−イル]安息香酸メチル(中間体#1)(4.5g、10.33mmol)に加えた。混合物を70℃で1時間攪拌し、次いで、周囲温度まで冷却し、減圧下で濃縮し、水(100mL)で希釈した。反応混合物を2MのHClでpH3に調節した。反応混合物をEtOAc(500mL)で抽出し、水(2×100mL)および飽和ブライン(50mL)で連続して洗浄した。有機層をMgSO4で乾燥させ、濾過し、エバポレートし、淡黄色の固体を得た。固体をEtOAc(20mL)で洗浄し、濾過により収集し、真空下で乾燥させ、4−[4−(2−アダマンチルカルバモイル)−5−tert−ブチル−ピラゾル−1−イル]安息香酸(3.89g、89%)を、クリーム色の結晶性固体として得た。
1H NMR(400.13MHz、DMSO−d6)δ1.19(9H、s)、1.49(2H、d)、1.70−1.96(10H、m)、2.09(2H、d)、3.98−4.01(1H、m)、7.49−7.53(2H、m)、7.61(1H、s)、8.06−8.09(2H、m)、8.20(1H、d)、13.30(1H、s)
m/z(ESI+)(M+H)+=422
m.p.308.8℃(開始)。
【0131】
参考例1は次のように調製することもできる。
水酸化ナトリウム水溶液(2M)(2.5当量)を、20℃のメタノール(10容量)中の4−[4−(2−アダマンチルカルバモイル)−5−tert−ブチル−ピラゾル−1−イル]安息香酸メチル(中間体#1)(1.0当量)の攪拌懸濁液に、5分間かけて少しずつ加えた(発熱20〜27℃)。得られた懸濁液を1時間、70℃(ジャケット温度)に加熱した(バッチ還流約60〜65℃)(LCMSにより完了)。オレンジ色の反応混合物を20℃まで冷却し(溶液にはわずかに混濁が残った)、セライトを通して濾過し少量の固体を除去した。次いで、濾液をフランジフラスコに注ぎ込み、水(25容量)を加えた。次いで、混合物を2MのHCl(約800〜850ml)でpH3に調節した(非常に濃くなる)。次いで水層を濾過し、淡黄色の固体を水で洗浄し、一晩吸引乾燥し、アセトニトリルおよび最後に1:1のアセトニトリル/ジエチルエーテルで洗浄し、真空下、50℃で72時間(週末)乾燥させて、4−[4−(2−アダマンチルカルバモイル)−5−tert−ブチル−ピラゾル−1−イル]安息香酸(80%)を固体として得た。
【0132】
中間体#2:4−ヒドラジニル安息香酸メチル塩酸塩
【0133】
【化3】
【0134】
ジオキサン中の4Mの塩化水素(100mL、399.60mmol)をMeOH(200mL)中の4−ヒドラジノ安息香酸(15.2g、99.90mmol)に加えた。得られた懸濁液を90℃で5時間攪拌した。20℃まで冷却した後、沈殿物を濾過により収集し、Et2O(100mL)で洗浄し、真空下で乾燥させ、2−(4−(メトキシカルボニル)フェニル)ヒドラジニウムクロリド(16.50g、82%)をクリーム色の結晶性固体として得た。
m/z(ESI−)(M−H)−=165;HPLCtR=1.12分
1H NMR(400.13MHz、DMSO−d6)δ3.81(3H、s)、6.99−7.02(2H、m)、7.86−7.90(2H、m)、8.98(1H、s)、10.47(3H、s)。
【0135】
中間体#2は次のように調製することもできる。
メタノール性塩酸溶液(4M)(4当量、新たに調製)を、窒素下で、メタノール(12.6容量)中の4−ヒドラジノ安息香酸懸濁液(1当量)に加えた。混合物を還流下で3時間攪拌し、次いで15℃未満まで冷却した。濾過により固体を収集し、MTBE(6.5容量)で洗浄し、風乾し、生成物を固体として得た。
TLC DCM:MeOH、9:1、生成物Rf0.87
mp 233.8〜234.6℃。
【0136】
中間体#3:N−(2−アダマンチル)−4,4−ジメチル−3−オキソ−ペンタンアミド
【0137】
【化4】
【0138】
THF中のリチウムビス(トリメチルシリル)アミド溶液の1M溶液(22.84ml、22.84mmol)をTHF(25mL)に加え、窒素下で−78℃まで冷却した。THF(25mL)中の3,3−ジメチル−2−ブタノン(2.287g、22.84mmol)の溶液を5分間にわたって滴下した。得られた溶液を、窒素下、−78℃で15分間攪拌した。THF(20mL)中の2−イソシアナトアダマンタン(R.Reck & C.Jochims Chem. Ber. 115(1982) p864の方法によって2−アダマンチルアミン塩酸塩から調製)(3.68g、20.76mmol)の溶液を5分間にわたって加えた。得られた溶液を−78℃で1時間攪拌した後、1時間かけて20℃まで昇温させた。反応混合物を飽和NH4Cl(150mL)に注ぎ込み、EtOAc(2×100mL)で抽出した。有機層を水(50mL)およびブライン(50mL)で洗浄し、MgSO4で乾燥させ、濾過し、エバポレートし、黄色の油を得た。粗生成物をフラッシュシリカクロマトグラフィー(溶出勾配:イソヘキサン中の0〜50%EtOAc)によって精製した。精製画分を蒸発乾固し、N−(2−アダマンチル)−4,4−ジメチル−3−オキソ−ペンタンアミド(4.64g、81%)を白色固体として得た。
1H NMR(400.13MHz、DMSO−d6)δ1.08−1.09(9H、m)、1.50(2H、d)、1.66−1.89(10H、m)、1.95−2.00(2H、m)、3.53(1.4H、s)、3.80−3.94(1H、m)、5.30(0.3H、s)、7.77−7.87(1H、m)、14.43(0.3H、s)(ケト型とエノール型の2:1混合物)
m/z(ESI+)(M+H)+=278。
【0139】
中間体#3は次のように調製することもできる。
水酸化ナトリウム水溶液(3M)(5容量)を水(5容量)中の2−アダマンチルアミン塩酸塩(1当量)の攪拌懸濁液に加えた。DCM(5容量)を得られた濃厚な懸濁液に加え、相を分離した。水層をDCM(4×5容量)で抽出し、合わせた有機層を濃縮し、遊離アミンを白色固体として得た。
【0140】
エチルピバロイルアセテート(1当量)を、窒素下で、キシレン(6.5容量)中の遊離アミンの懸濁液に加え、混合物を還流下で6.5時間攪拌した。バッチを室温まで冷却し、濃縮乾固した。残留物を、トルエン(3×1容量)、次いでヘキサン(3×1容量)でパージした。得られた固体を50℃で5分間ヘキサン中で攪拌し、その後室温まで冷却した。白色固体を濾過し、ヘキサン(2容量)で洗浄し風乾させた。
TLC ヘキサン:EtOAc、1:1、生成物Rf0.66
mp 124.5〜125.1℃。
【0141】
中間体#4:(2)−N−(2−アダマンチル)−2−(ジメチルアミノメチリデン)−4,4−ジメチル−3−オキソ−ペンタンアミド
【0142】
【化5】
【0143】
N,N−ジメチルホルムアミドジメチルアセタール(3.02mL、22.71mmol)を、窒素下で、1,4−ジオキサン(50mL)中のN−(2−アダマンチル)−4,4−ジメチル−3−オキソ−ペンタンアミド(中間体#3)(5.25g、18.93mmol)の攪拌懸濁液に加えた。得られた混合物を100℃で2時間攪拌した。反応混合物を蒸発乾固し、得られた淡いクリーム色の固体を真空下で乾燥させ、(2)−N−(2−アダマンチル)−2−(ジメチルアミノメチリデン)−4,4−ジメチル−3−オキソ−ペンタンアミド(5.83g、93%)を得た。
1H NMR(400.13MHz、DMSO−d6)δ1.13(9H、s)、1.47(2H、d)、1.69−1.83(10H、m)、2.03(2H、d)、2.92(6H、s)、3.90(1H、d)、7.24(1H、s)、7.94(1H、d)
m/z(ESI+)(M+H)+=333。
【0144】
中間体#4は次のように調製することもできる。
N,N−ジメチルホルムアミドジメチルアセタール(1.2当量)を、窒素下で、1,4−ジオキサン(9.6容量)中のN−(2−アダマンチル)−4,4−ジメチル−3−オキソ−ペンタンアミド(中間体#3)(1当量)の溶液に加えた。混合物を還流下で5時間加熱し、その後室温まで冷却した。溶媒を真空下で除去し、淡黄色の固体を次の工程でそのまま使用した。
TLC ヘキサン:EtOAc、1:1、生成物Rf0.94(不純物:Rf0.06+0.66)
mp 143.6〜147.6℃。
【0145】
中間体#1:4−[4−(2−アダマンチルカルバモイル)−5−tert−ブチル−ピラゾル−1−イル]安息香酸メチル
【0146】
【化6】
【0147】
4−ヒドラジニル安息香酸メチル塩酸塩(中間体#2)(3.04g、15.00mmol)を、エタノール(100mL)中の(2)−N−(2−アダマンチル)−2−(ジメチルアミノメチリデン)−4,4−ジメチル−3−オキソ−ペンタンアミド(中間体#4)(4.99g、15mmol)に一度に加えた。酢酸を5滴添加し、得られた溶液を80℃で2時間攪拌した。反応混合物を濃縮し、EtOAc(500mL)で希釈し、水(200mL)および飽和ブライン(200mL)で連続して洗浄した。有機層をMgSO4で乾燥させ、濾過し、エバポレートし、粗生成物を得た。
【0148】
該粗生成物をフラッシュシリカクロマトグラフィー(溶出勾配:イソヘキサン中の0〜50%EtOAc)によって精製した。精製画分を蒸発乾固し、4−[4−(2−アダマンチルカルバモイル)−5−tert−ブチル−ピラゾル−1−イル]安息香酸メチル(4.66g、71.3%)を黄色の固体として得た。
1H NMR(400.13MHz、DMSO−d6)δ1.19(9H、s)、1.50(2H、d)、1.69−1.95(10H、m)、2.09(2H、d)、3.91(3H、s)、3.99(1H、d)、7.53−7.56(2H、m)、7.62(1H、s)、8.09−8.12(2H、m)、8.20(1H、d)
m/z(ESI+)(M+H)+=436。
【0149】
中間体#1は次のように調製することもできる。
2−(4−(メトキシカルボニル)フェニル)ヒドラジニウムクロリド(中間体#2)(1当量)、次いで酢酸(0.023当量)を、窒素下で、メタノール(200容量)中の(2Z)−N−(2−アダマンチル)−2−(ジメチルアミノ−メチリデン)−4,4−ジメチル−3−オキソ−ペンタンアミド(中間体#4)(1当量)の溶液に加えた。混合物を還流下で1.5時間攪拌し、冷却し、3.5容量未満まで濃縮し、得られた懸濁液を酢酸エチル(96容量)で希釈した。該懸濁液を水(34.4容量)で洗浄して溶液を得、その溶液をブライン(34.4容量)で洗浄し、乾燥(MgSO4)し、濃縮乾固した。粗生成物をMTBE(9容量)中でスラリー状にし、15分間攪拌した。淡黄色の固体を濾過し、MTBE(11.4容量)で洗浄し、真空下、60℃で乾燥させた。
TLC DCM:MeOH、9:1、生成物Rf0.86(微量の不純物Rf0.68)
mp 193.6〜194.5℃。
【0150】
4−[4−(2−アダマンチルカルバモイル)−5−tert−ブチル−ピラゾル−1−イル]安息香酸は次のように調製することもできる。
塩酸(34.5%w/w、15.88g、5.48g@100.0%、0.1504モル、1.0モル当量)および水(70ml、1.4相対量)をメタノール(1250ml、12.5相対量)中の4−ヒドラジノ安息香酸(23.35g、22.88g@100.0%、0.1504モル、1.0モル当量)の懸濁液に加えた。得られた懸濁液を20〜25℃で30分間攪拌し、メタノール(250ml、5.0相対量)中の、20〜25℃のN−(2−アダマンチル)−2−(ジメチルアミノメチリデン)−4,4−ジメチル−3−オキソ−ペンタンアミド(中間体#4)(53.47g、50.0g@100.0%、4モル中0.150、1.0モル当量)の溶液を20分間かけて加え、次いで塩酸(34.5%w/w、2.38g、0.82g@100.0%、0.02264モル、0.15モル当量)および水(70.0ml、1.4相対量)を加えた。反応物の温度を62〜65℃まで上げ、90.0分間維持した。後処理として、残り6.0相対量(約300.0ml)となるまでメタノール溶液を大気中で濃縮した。得られた懸濁液を20〜25℃まで冷却し1.0時間攪拌した。生成物を濾過し、酢酸エチル(200.0ml、4.0相対量)で洗浄し、30分間吸引乾燥した。生成物を50℃で8時間、真空下(100mbar)で乾燥させ、未精製の4−[4−(2−アダマンチルカルバモイル)−5−tert−ブチル−ピラゾル−1−イル]安息香酸(47.0g、73.0%)を得た。
1H NMR(400.13MHz、DMSO−d6)δ1.19(9H、s)、1.49(2H、d)、1.70−1.96(10H、m)、2.09(2H、d)、3.98−4.01(1H、m)、7.49−7.53(2H、m)、7.61(1H、s)、8.06−8.09(2H、m)、8.20(1H、d)、13.30(1H、s)
m/z(ESI+)(M+H)+=422
m.p.308.8℃(開始)。
クロマトグラフィー条件:[HPLC]
Zorbax SB-Aq、150×4.6mm、5μ。使用する移動相は有機溶媒としてアセトニトリルを用いるギ酸緩衝液、流量1.0mL/分、注入量は20μL、実行時間は18分で、UV検出器の波長は220、320nmを使用する。
保持時間[HPLC]
4−[4−(2−アダマンチルカルバモイル)−5−tert−ブチル−ピラゾル−1−イル]安息香酸:(相対保持時間:0.77分)
(2)−N−(2−アダマンチル)−2−(ジメチルアミノメチリデン)−4,4−ジメチル−3−オキソ−ペンタンアミド(保持時間:14.2分)。
【0151】
10.0%w/w水酸化ナトリウム水溶液(109.1g、10.91g@100.0%、0.2727モル、1.15モル当量)を、未精製4−[4−(2−アダマンチルカルバモイル)−5−tert−ブチル−ピラゾル−1−イル]安息香酸(110.0g、100.0g@100.0%、0.2372モル、1.0モル当量)の水(1000.0ml、10.0相対量)中の懸濁液に加え、15.0分間攪拌した。不溶解生成物を濾過した。得られた透明な水溶液(濾液)にトルエン(600.0ml、6.0相対量)を加え、30.0分間攪拌した。相分離のために反応物を1.0時間静置した。水層を分離し、セライト床を通して濾過した。水層にメタノール(300.0ml、3.0相対量)を加えた。水層のpHを、希塩酸(3.8%w/w、261.6g、9.94@100.0%、0.2727モル、1.15モル当量)で2.25〜2.75に徐々に調節した。得られた懸濁液を1.5〜2.0時間攪拌し、生成物を濾過し、25.0%v/vメタノール水溶液[500.0ml、5.0相対量(メタノール125mlと水475mlの混合物)]で洗浄した。生成物を50〜60℃で16.0時間、真空下(100mbar)で乾燥させ、4−[4−(2−アダマンチルカルバモイル)−5−tert−ブチル−ピラゾル−1−イル]安息香酸(85.0g、85.0%)(形態1)を得た。
【0152】
中間体#3
N−(2−アダマンチル)−4,4−ジメチル−3−オキソ−ペンタンアミド
【0153】
【化7】
【0154】
中間体#3は次のように調製することもできる。トルエン(400.0ml、4.0相対量)を、2−アダマンタンアミン塩酸塩(100.0g、98.0g@100%、0.5221モル、1.00モル当量)の水(500.0ml、5.0相対量)中の溶液に加えた。10.0%w/wの水酸化ナトリウム水溶液(261.02g、26.1g@100.0%、0.6526モル、1.25モル当量)を上記溶液に加え、15分間攪拌した。有機層を水層から分離し、5.0%w/w塩化ナトリウム溶液(300.0ml、3.0相対量)で洗浄した。エチルピバロイルアセテート(115.9g、0.6526モル、1.25モル当量)を有機層に一度に加えた。反応物を加熱還流し、温度を110℃に維持しながら、トルエンを3〜3.5時間共沸して回収した(550.0ml、5.5相対量)。反応物を70〜80℃まで冷却し、n−ヘプタン(1000.0ml、10.0相対量)を15分間かけて加えた。反応物をさらに25℃まで冷却し、得られた懸濁液を1.0時間攪拌した。懸濁した固体を濾過により回収し、n−ヘプタン(400.0ml、4.0相対量)で洗浄し、生成物を50〜55℃で6.0時間、真空下(100mbar)で乾燥させ、N−(2−アダマンチル)−4,4−ジメチル−3−オキソ−ペンタンアミド(113.0g、75.8%)を白色結晶性固体として得た。
1H NMR(400.13MHz、DMSO−d6)δ1.08−1.09(9H、m)、1.50(2H、d)、1.66−1.89(10H、m)、1.95−2.00(2H、m)、3.53(1.4H、s)、3.80−3.94(1H、m)、5.30(0.3H、s)、7.77−7.87(1H、m)、14.43(0.3H、s)(ケト型とエノール型の2:1混合物)
m/z(ESI+)(M+H)+=278。
クロマトグラフィー条件:−(GC)
HP−5MSカラム、キャリヤーガスとしてヘリウム、流量1.0mL/分、溶媒遅延最大1.5分、オーブン温度は、初期温度50℃で2分間維持、その後20℃/分で280℃まで上昇、注入量は1.0μLとする。
保持時間(GC)
2−アダマンタンアミン塩酸塩(保持時間:8.1分)
N−(2−アダマンチル)−4,4−ジメチル−3−オキソ−ペンタンアミド(相対保持時間:1.617分)。
【0155】
中間体#4
(2)−N−(2−アダマンチル)−2−(ジメチルアミノメチリデン)−4,4−ジメチル−3−オキソ−ペンタンアミド
【0156】
【化8】
【0157】
中間体#4は次のように調製することもできる。N,N−ジメチルホルムアミドジメチルアセタール(69.25g、63.02g@100.0%、0.5288モル、1.5モル当量)を、N−(2−アダマンチル)−4,4−ジメチル−3−オキソ−ペンタンアミド(中間体3)(100g、97.8g@100.0%、0.3525モル、1.0モル当量)のn−ヘプタン(800.0ml、8.20相対量)およびトルエン(350.0ml、3.58相対量)中の懸濁液に加えた。反応物の温度を90〜95℃まで上げ5.0時間維持した後、80℃まで冷却し、n−ヘプタン(400.0ml、4.09相対量)を加えた。反応物温度をさらに25℃まで下げ、得られた懸濁液を濾過により回収し、n−ヘプタン(400.0ml、4.09相対量)で洗浄し、生成物を30分間吸引乾燥した。生成物を周囲温度(20〜25℃)で3.0時間、真空下(100mbar)で乾燥させ、(2)−N−(2−アダマンチル)−2−(ジメチルアミノメチリデン)−4,4−ジメチル−3−オキソ−ペンタンアミド(100g、79.7%)を得た。
1H NMR(400.13MHz、DMSO−d6)δ1.13(9H、s)、1.47(2H、d)、1.69−1.83(10H、m)、2.03(2H、d)、2.92(6H、s)、3.90(1H、d)、7.24(1H、s)、7.94(1H、d)。
m/z(ESI+)(M+H)+=333。
クロマトグラフィー条件:(HPLC)−
Sunfire C18、150×4.6mm、5μ。使用する移動相は有機溶媒としてメタノールを用いるリン酸水素二ナトリウム緩衝液、流量1.0mL/分、注入量は20μL、実行時間は20分で、示差屈折率検出器を使用する。
保持時間(HPLC):
N−(2−アダマンチル)−4,4−ジメチル−3−オキソ−ペンタンアミド(保持時間:11.0分)
(2)−N−(2−アダマンチル)−2−(ジメチルアミノメチリデン)−4,4−ジメチル−3−オキソ−ペンタンアミド(相対保持時間:1.18分)。
【0158】
参考例2
(2)−N−(2−アダマンチル)−2−(ジメチルアミノメチリデン)−4,4−ジメチル−3−オキソ−ペンタンアミドの合成
【0159】
【化9】
【0160】
2−アダマンタンアミン塩酸塩(25.0g、0.13モル)の水(75.0ml、3.0相対量)中の懸濁液に、トルエン(100.0ml、4.0相対量)を加えた。10.0%w/w水酸化ナトリウム水溶液(1.25モル当量)を上記溶液に入れ、10〜15分間攪拌した。有機層を分離し、水層をトルエン(75.0ml、3.0相対量)で再抽出し、分離した有機層と合わせた。合わせた有機層を5.0%w/w塩化ナトリウム溶液(75ml、3.0相対量)で洗浄し分離した。エチルピバロイルアセテート(26.01g、0.15モル)を有機層に加え、反応物を110〜112℃で加熱還流した。溶媒(4〜5相対量)を4〜5時間かけて共沸して回収した。反応物を40〜45℃まで冷却し、35〜40℃でn−ヘプタン(200.0ml、8.0相対量)を加え、次いで30〜35℃でDMF−DMA(26.45g、0.20モル)およびトリエチルアミン(13.48g、0.13モル)を加えた。反応物の温度を90〜93℃まで上げ2〜3時間維持した。副生成物として生成したメタノールを反応の間共沸して回収した。反応物を20〜25℃まで冷却し、その温度で1.0時間攪拌した。沈殿生成物を濾過し、床をn−ヘプタン(100.0ml、4.0相対量)で洗浄し、生成物を35〜40℃で3〜4時間、真空下(50〜100mbar)で乾燥させ、(2)−N−(2−アダマンチル)−2−(ジメチルアミノメチリデン)−4,4−ジメチル−3−オキソ−ペンタンアミドを得た(収率、86%)。室温では不安定であったため、生成物を窒素雰囲気下で充填し、10℃未満で貯蔵した。
【0161】
別の方法として、(2)−N−(2−アダマンチル)−2−(ジメチルアミノメチリデン)−4,4−ジメチル−3−オキソ−ペンタンアミドは次のように調製することができる。
【0162】
2−アダマンタンアミン塩酸塩(25.0g、0.13モル)の水(75.0ml、3.0相対量)中の懸濁液に、トルエン(100.0ml、4.0相対量)を加えた。10.0%w/w水酸化ナトリウム水溶液(1.25モル当量)を上記溶液に入れ、10〜15分間攪拌した。有機層を分離し、水層をトルエン(75.0ml、3.0相対量)で再抽出し、分離した有機層と合わせた。合わせた有機層を5.0%w/w塩化ナトリウム溶液(75ml、3.0相対量)で洗浄し分離した。エチルピバロイルアセテート(26.01g、0.15モル)を有機層に加え、反応物を110〜112℃で加熱還流した。溶媒(4〜5相対量)を4〜5時間かけて共沸して回収した。反応物を40〜45℃まで冷却し、35〜40℃でn−ヘプタン(200.0ml、8.0相対量)を加え、次いで同じ温度でDMF−DMA(26.45g、0.20モル)を加えた。反応物の温度を85〜90℃まで上げ4〜5時間維持した。副生成物として生成したメタノールを反応の間共沸して回収した。反応物を20〜25℃まで冷却し、その温度で1.0時間攪拌した。沈殿生成物を濾過し、床をn−ヘプタン(100.0ml、4.0相対量)で洗浄し、生成物を35〜40℃で3〜4時間、真空下(50〜100mbar)で乾燥させ、(2)−N−(2−アダマンチル)−2−(ジメチルアミノメチリデン)−4,4−ジメチル−3−オキソ−ペンタンアミドを得た(収率、72%)。室温では不安定であったため、生成物を窒素雰囲気下で充填し、10℃未満で貯蔵した。
クロマトグラフィー条件:−
Sunfire C18、150×4.6mm、5μ。使用する移動相は有機溶媒としてメタノールを用いるリン酸水素二ナトリウム緩衝液、流量1.0mL/分、注入量は20μL、実行時間は20分で、示差屈折率検出器を使用する。
保持時間:
N−(2−アダマンチル)−4,4−ジメチル−3−オキソ−ペンタンアミド 保持時間:11.0分
(2)−N−(2−アダマンチル)−2−(ジメチルアミノメチリデン)−4,4−ジメチル−3−オキソ−ペンタンアミド 相対保持時間:1.18分
1H NMR(400.13MHz、DMSO−d6)δ1.13(9H、s)、1.47(2H、d)、1.69−1.83(10H、m)、2.03(2H、d)、2.92(6H、s)、3.90(1H、d)、7.24(1H、s)、7.94(1H、d)
m/z(ESI+)(M+H)+=333。
【0163】
必要であれば該N−(2−アダマンチル)−4,4−ジメチル−3オキソ−ペンタンアミド中間体は単離することができる:
クロマトグラフィー条件:−
HP−5MSカラム、キャリヤーガスとしてヘリウム、流量1.0mL/分、溶媒遅延最大1.5分、オーブン温度は、初期温度50℃で2分間維持、その後20℃/分で280℃まで上昇、注入量は1.0μLとする。
保持時間:
2−アダマンタンアミン塩酸塩 保持時間8.1分
N−(2−アダマンチル)−4,4−ジメチル−3−オキソ−ペンタンアミド 相対保持時間:1.617分
1H NMR(400.13MHz、DMSO−d6)δ1.08−1.09(9H、m)、1.50(2H、d)、1.66−1.89(10H、m)、1.95−2.00(2H、m)、3.53(1.4H、s)、3.80−3.94(1H、m)、5.30(0.3H、s)、7.77−7.87(1H、m)、14.43(0.3H、s)(ケト型とエノール型の2:1混合物)
m/z(ESI+)(M+H)+=278。
【0164】
4−[4−(2−アダマンチルカルバモイル)−5−tert−ブチル−ピラゾル−1−イル]安息香酸(形態−1)の合成
【0165】
【化10】
【0166】
4−ヒドラジノ安息香酸塩酸塩(14.11g、0.075モル)、および(2)−N−(2−アダマンチル)−2−(ジメチルアミノメチリデン)−4,4−ジメチル−3−オキソ−ペンタンアミド(25.0g、0.075モル)をジャケット付き反応器に入れ、次いでイソプロピルアルコール(315ml、12.6相対量)および水(35ml、1.4相対量)を入れた。反応物を20〜25℃で約45〜60分間攪拌した。内容物を78〜80℃で加熱還流し、その温度で90分間維持した。反応物を50〜55℃まで冷却し、次いで同じ温度で水(150ml、6相対量)を加えた。内容物をさらに周囲温度(20〜25℃)まで冷却し、同じ温度で1.0時間攪拌した。沈殿生成物を濾過し、次いでイソプロピルアルコールと水(1:1)の混合物(250ml、10.0相対量)で洗浄し、4−[4−(2−アダマンチルカルバモイル)−5−tert−ブチル−ピラゾル−1−イル]安息香酸を得た。生成物を50〜55℃で4〜5時間、真空下で乾燥させ、さらに精製することなく用いた(収率:80%)。
1H NMR(400.13MHz、DMSO−d6)δ1.19(9H、s)、1.49(2H、d)、1.70−1.96(10H、m)、2.09(2H、d)、3.98−4.01(1H、m)、7.49−7.53(2H、m)、7.61(1H、s)、8.06−8.09(2H、m)、8.20(1H、d)、13.30(1H、s)
m/z(ESI+)(M+H)+=422
m.p.308.8℃(開始)。
クロマトグラフィー条件:−
Zorbax SB-Aq、150×4.6mm、5μ。使用する移動相は有機溶媒としてアセトニトリルを用いるギ酸緩衝液、流量1.0mL/分、注入量は20μL、実行時間は18分で、UV検出器の波長は220、320nmを使用する。
保持時間:
[((2)−N−(2−アダマンチル)−2−(ジメチルアミノメチリデン)−4,4−ジメチル−3−オキソ−ペンタンアミド 保持時間14.2分
4−[4−(2−アダマンチルカルバモイル)−5−tert−ブチル−ピラゾル−1−イル]安息香酸 相対保持時間0.77分(10.0分)
中間体 相対保持時間0.79(11.2分)。
【0167】
実施例1
4−[4−(2−アダマンチルカルバモイル)−5−tert−ブチル−ピラゾル−1−イル]安息香酸(形態2)
上記(参考例1−形態1)で調製した4−[4−(2−アダマンチルカルバモイル)−5−tert−ブチル−ピラゾル−1−イル]安息香酸約50mgを磁気攪拌子入りのバイアルに入れ、アセトニトリル約2mlを加えた。次いでバイアルを蓋できつく密封した。次いでスラリーを、磁気攪拌能力を有する加熱攪拌ブロック中、50℃で攪拌させた。3日後、試料をプレートより取り出し、蓋を外し、スラリーを周囲条件で乾燥させてから、XRPDおよびDSCで分析した。この形態(形態2)は、XRPDによって結晶性であることが確認され、先の形態とは異なっていることがわかった。この物質の融点は310.9℃(開始)であった。この物質は、CuKa照射を用いて測定される2シータのピークを18.0および17.7°に有していた。後にDSCを用いて融点を測定すると309.9℃であった。
【0168】
実施例2
4−[4−(2−アダマンチルカルバモイル)−5−tert−ブチル−ピラゾル−1−イル]安息香酸(形態3)
4−[4−(2−アダマンチルカルバモイル)−5−tert−ブチル−ピラゾル−1−イル]安息香酸(形態1)約20mgを磁気攪拌子入りのバイアルに入れ、メタノール約2mlを加え、次いでバイアルを蓋できつく密封し、マグネチックスターラープレート上で攪拌させた。3日後、試料をプレートより取り出し、蓋を外し、スラリーを周囲条件で乾燥させてから、XRPDおよびDSCで分析した。この形態(形態3)は、XRPDによって結晶性であることが確認され、先に見られた形態とは異なっていることがわかった。この物質の融点は309.4℃(開始)であった。この物質は、CuKa照射を用いて測定される2シータのピークを18.7および11.7°に有していた。後にDSCを用いて融点を測定すると309.3℃であった。
【0169】
実施例3
4−[4−(2−アダマンチルカルバモイル)−5−tert−ブチル−ピラゾル−1−イル]安息香酸(形態4)
形態1の4−[4−(2−アダマンチルカルバモイル)−5−tert−ブチル−ピラゾル−1−イル]安息香酸約20mgおよび形態3の物質20mgを磁気攪拌子入りのバイアルに入れ、酢酸エチル約2mlを加え、次いでバイアルを蓋できつく密封し、マグネチックスターラープレート上で攪拌させた。3日後、試料をプレートより取り出し、蓋を外し、スラリーを周囲条件で乾燥させてから、XRPDおよびDSCで分析した。この形態(形態4)は、XRPDによって結晶性であることが確認され、先に見られた形態とは異なっていることがわかった。この物質(形態4)の融点は309.1℃(開始)であった。この物質は、CuKa照射を用いて測定される2シータのピークを16.2および20.6°に有していた。後にDSCを用いて融点を測定すると312.0℃であった。
【0170】
別の方法として、高温においては溶媒としてアセトニトリルを用いてもよい。アセトニトリル(800ml、8.0相対量)を、乾燥純粋生成物((4−[4−(2−アダマンチルカルバモイル)−5−tert−ブチル−ピラゾル−1−イル]安息香酸、形態1(80.0g)に加え、スラリーを75〜78℃まで加熱した。反応物を72.0時間75〜78℃に維持し、次いで20〜25℃まで冷却し、20〜25℃で1.0時間攪拌した。生成物を濾過し吸引乾燥した。次いでアセトニトリル(240.0ml、3.0相対量)で洗浄し、30.0分間吸引乾燥した。次いで生成物を50℃で16.0時間、真空下(100mbar)で乾燥させ、4−[4−(2−アダマンチルカルバモイル)−5−tert−ブチル−ピラゾル−1−イル]安息香酸、形態−4(72.0g、90.0%)を得た。形態−4の物質(50mg、1%w/w、種)を、(4−[4−(2−アダマンチルカルバモイル)−5−tert−ブチル−ピラゾル−1−イル]安息香酸(形態1)5.0gのアセトニトリル50.0ml中の懸濁液に加え、12〜18時間、75〜78℃まで加熱した。反応物を20〜25℃まで冷却し、20〜25℃で1.0時間攪拌した。生成物を濾過し吸引乾燥した。次いでアセトニトリル(15ml)で洗浄し、5〜10.0分間吸引乾燥した。次いで生成物を50℃で16.0時間、真空下(100mbar)で乾燥させ、4−[4−(2−アダマンチルカルバモイル)−5−tert−ブチル−ピラゾル−1−イル]安息香酸、形態−4(4.5g、89〜90.0%)を得た。上記のように調製された4−[4−(2−アダマンチルカルバモイル)−5−tert−ブチル−ピラゾル−1−イル]安息香酸(形態1)をアセトニトリル(7容量)中に懸濁させ、(形態4)を5g播種し、3日間還流でスラリー状にした(ジャケット温度85℃)。サンプルを採取しDSCでチェックした(ピークを2つ示す)。このサンプルをさらに3日間(週末)還流で攪拌し、20℃まで冷却し、濾過し、アセトニトリル、次いでジエチルエーテルで十分に洗浄し、吸引乾燥し、50℃で48時間真空下で乾燥させて、淡黄色の固体(形態4)を得た(90%)。
【0171】
別の方法としては:
テトラヒドロフラン(9.0相対量)および水(0.5相対量)を4−[4−(2−アダマンチルカルバモイル)−5−tert−ブチル−ピラゾル−1−イル]安息香酸、形態1(20.0g、0.047モル)に加え、混合物を15分間攪拌した後、濾紙を通して濾過した。残留物をテトラヒドロフラン(1.0相対量)で洗浄し、合わせた濾液を反応器に移し、反応温度を58〜62℃まで上げた。反応を55〜65℃に維持しながらアセトニトリル(20.0相対量)を加えた。反応温度を68±2℃まで上げ、22時間その温度を維持し、その後20〜25℃まで冷却し2時間攪拌した。生成物を濾過し、床をアセトニトリル(5.0相対量)で洗浄した。ウエットケーキを45〜50℃で4時間、真空下(50〜100mbar)で乾燥させ、多形4を得て(収率80%)、XRPDで確認した。
【0172】
別の方法としては:
テトラヒドロフラン(10.0相対量)を4−[4−(2−アダマンチルカルバモイル)−5−tert−ブチル−ピラゾル−1−イル]安息香酸、形態1(5.0g、0.012モル)に加え、温度を58〜62℃まで上げた。反応を55〜65℃に維持しながらアセトニトリル(20.0相対量)を加えた。反応温度は20時間68±2℃で維持した。内容物を20〜25℃まで冷却し2時間攪拌した。生成物を濾過し、ウエットケーキをアセトニトリル(5.0相対量)で洗浄した後、45〜50℃で4時間、真空オーブン(50〜100mbar)内で乾燥させ、多形4を得て(収率90%)、XRPDおよび固体NMRで確認した。
【0173】
別の方法としては:
N,N−ジメチルホルムアミド(5.0相対量)およびアセトニトリル(5.0相対量)を4−[4−(2−アダマンチルカルバモイル)−5−tert−ブチル−ピラゾル−1−イル]安息香酸、形態1(5.0g、0.012モル)に加え、反応温度を60〜65℃まで上げた。温度を55〜65℃に維持しながらアセトニトリル(15.0相対量)を加えた。反応温度を75〜78℃まで上げ、20時間その温度を維持した。内容物を20〜25℃まで冷却し2時間攪拌した。生成物を濾過し、床をアセトニトリル(5.0相対量)で洗浄した後、45〜50℃で4時間、真空オーブン(50〜100mbar)内で乾燥させ、多形4を得て(収率88%)、XRPDで確認した。
【0174】
別の方法としては:
酢酸(10.0相対量)を4−[4−(2−アダマンチルカルバモイル)−5−tert−ブチル−ピラゾル−1−イル]安息香酸、形態1(5.0g、0.012モル)に加え、温度を75〜78℃まで上げた。温度を70〜78℃に維持しながらアセトニトリル(20.0相対量)を加えた。混合物を75〜78℃で攪拌し、22時間その温度を維持した。内容物を20〜25℃まで冷却し2時間攪拌した。生成物を濾過し、床をアセトニトリル(5.0相対量)で洗浄した後、45〜50℃で4時間、真空オーブン(50〜100mbar)内で乾燥させ、多形4を得て(収率66%)、XRPDで確認した。
【0175】
別の方法としては:
2−メチル−THF(10.0相対量)を4−[4−(2−アダマンチルカルバモイル)−5−tert−ブチル−ピラゾル−1−イル]安息香酸、形態1(5.0g、0.012モル)に加え、温度を70〜75℃まで上げた。温度を70〜75℃に維持しながらアセトニトリル(20.0相対量)を加えた後、75〜78℃で23時間攪拌させた。内容物を20〜25℃まで冷却し2時間攪拌した。生成物を濾過し、床をアセトニトリル(5.0相対量)で洗浄した後、45〜50℃で4時間、真空オーブン(50〜100mbar)内で乾燥させ、多形4を得て(収率93%)、XRPDで確認した。
【0176】
別の方法としては:
4−[4−(2−アダマンチルカルバモイル)−5−tert−ブチル−ピラゾル−1−イル]安息香酸(形態1−参考例2のように調製)(20.0g、0.047モル)、次いでテトラヒドロフラン(9.0相対量)および水(0.5相対量)を好適なジャケット付き反応器に加えた。内容物を15分間攪拌し、濾紙を通して濾過し、テトラヒドロフラン(1.0相対量)で洗浄した。合わせた濾液を反応器に移し、反応物の温度を58〜62℃まで上げた。温度を55〜65℃に維持しながらアセトニトリル(20.0相対量)を加えた。反応物の温度を68±2℃に上げ、22時間その温度を維持した。内容物を20〜25℃まで冷却し2時間攪拌した。生成物を濾過し、床をアセトニトリル(5.0相対量)で洗浄した。ウエットケーキを45〜50℃で4時間、真空下(50〜100mbar)で乾燥させ、多形形態4を得た(80%)。
【特許請求の範囲】
【請求項1】
CuKa照射を用いて測定した次の2−シータ値:18.0および17.7°にピークを持つX線回折パターンを有する、4−[4−(2−アダマンチルカルバモイル)−5−tert−ブチル−ピラゾル−1−イル]安息香酸の結晶形態。
【請求項2】
CuKa照射を用いて測定した次の2−シータ値:18.0、17.7、18.4および8.9°にピークを持つX線粉末回折パターンを有する、請求項1に記載の結晶性化合物。
【請求項3】
CuKa照射を用いて測定した次の2−シータ値:18.0、17.7、18.4、8.9および20.5°にピークを持つX線粉末回折パターンを有する、請求項1に記載の化合物の結晶形態。
【請求項4】
CuKa照射を用いて測定した次の2−シータ値:18.0、17.7、18.4、8.9、20.5、10.4、21.9、13.4、27.6および16.7°にピークを持つX線粉末回折パターンを有する、請求項1に記載の化合物の結晶形態。
【請求項5】
CuKa照射を用いた、図1に示すものと実質的に同一のX線回折パターンを有する、請求項1に記載の結晶性化合物。
【請求項6】
約309.9℃(開始)の融点を有する、4−[4−(2−アダマンチルカルバモイル)−5−tert−ブチル−ピラゾル−1−イル]安息香酸の結晶形態。
【請求項7】
CuKa照射を用いて測定した次の2−シータ値:18.7および11.7°にピークを持つX線回折パターンを有する、4−[4−(2−アダマンチルカルバモイル)−5−tert−ブチル−ピラゾル−1−イル]安息香酸の結晶形態。
【請求項8】
CuKa照射を用いて測定した次の2−シータ値:18.7、11.7および19.2°にピークを持つX線粉末回折パターンを有する、請求項7に記載の結晶性化合物。
【請求項9】
CuKa照射を用いて測定した次の2−シータ値:18.7、11.7、19.2、7.8、14.1、14.9および9.4°にピークを持つX線粉末回折パターンを有する、請求項7に記載の化合物の結晶形態。
【請求項10】
CuKa照射を用いて測定した次の2−シータ値:18.7、11.7、19.2、7.8、14.1、14.9、9.4、15.6、16.1および9.6°にピークを持つX線粉末回折パターンを有する、請求項7に記載の化合物の結晶形態。
【請求項11】
CuKa照射を用いた、図3に示すものと実質的に同一のX線回折パターンを有する、請求項7に記載の結晶性化合物。
【請求項12】
約309.3℃(開始)の融点を有する、4−[4−(2−アダマンチルカルバモイル)−5−tert−ブチル−ピラゾル−1−イル]安息香酸の結晶形態。
【請求項13】
CuKa照射を用いて測定した次の2−シータ値:16.2および20.6°にピークを持つX線回折パターンを有する、4−[4−(2−アダマンチルカルバモイル)−5−tert−ブチル−ピラゾル−1−イル]安息香酸の結晶形態。
【請求項14】
CuKa照射を用いて測定した次の2−シータ値:16.2、20.6および17.7°にピークを持つX線粉末回折パターンを有する、請求項13に記載の結晶性化合物。
【請求項15】
CuKa照射を用いて測定した次の2−シータ値:16.2、20.6、17.7、10.8および15.5°にピークを持つX線粉末回折パターンを有する、請求項13に記載の化合物の結晶形態。
【請求項16】
CuKa照射を用いて測定した次の2−シータ値:16.2、20.6、17.7、10.8、15.5、20.9、26.1、11.6および26.7にピークを持つX線粉末回折パターンを有する、請求項13に記載の化合物の結晶形態。
【請求項17】
CuKa照射を用いた、図6に示すものと実質的に同一のX線回折パターンを有する、請求項13に記載の結晶性化合物。
【請求項18】
約312.0℃(開始)の融点を有する、4−[4−(2−アダマンチルカルバモイル)−5−tert−ブチル−ピラゾル−1−イル]安息香酸の結晶形態。
【請求項19】
請求項1、7および13のいずれか一項に記載の化合物を、薬学的に許容される希釈剤または担体と共に含む、医薬組成物。
【請求項20】
ヒトなどの温血動物の予防的または治療的処置方法における使用のための、請求項1、7および13のいずれか一項に記載の化合物。
【請求項21】
医薬として使用するための、請求項1、7および13のいずれか一項に記載の化合物。
【請求項22】
ヒトなどの温血動物での11βHSD1阻害作用の発現に使用される医薬の製造における、請求項1、7および13のいずれか一項に記載の化合物の使用。
【請求項23】
請求項1、7および13のいずれか一項に記載の化合物の有効量を、11βHSD1阻害作用を発現させるような治療を必要としている哺乳動物に投与することによって、11βHSD1阻害作用を発現させる方法。
【請求項24】
前記11βHSD1阻害作用が2型糖尿病を治療するためのものである、請求項23に記載の方法。
【請求項1】
CuKa照射を用いて測定した次の2−シータ値:18.0および17.7°にピークを持つX線回折パターンを有する、4−[4−(2−アダマンチルカルバモイル)−5−tert−ブチル−ピラゾル−1−イル]安息香酸の結晶形態。
【請求項2】
CuKa照射を用いて測定した次の2−シータ値:18.0、17.7、18.4および8.9°にピークを持つX線粉末回折パターンを有する、請求項1に記載の結晶性化合物。
【請求項3】
CuKa照射を用いて測定した次の2−シータ値:18.0、17.7、18.4、8.9および20.5°にピークを持つX線粉末回折パターンを有する、請求項1に記載の化合物の結晶形態。
【請求項4】
CuKa照射を用いて測定した次の2−シータ値:18.0、17.7、18.4、8.9、20.5、10.4、21.9、13.4、27.6および16.7°にピークを持つX線粉末回折パターンを有する、請求項1に記載の化合物の結晶形態。
【請求項5】
CuKa照射を用いた、図1に示すものと実質的に同一のX線回折パターンを有する、請求項1に記載の結晶性化合物。
【請求項6】
約309.9℃(開始)の融点を有する、4−[4−(2−アダマンチルカルバモイル)−5−tert−ブチル−ピラゾル−1−イル]安息香酸の結晶形態。
【請求項7】
CuKa照射を用いて測定した次の2−シータ値:18.7および11.7°にピークを持つX線回折パターンを有する、4−[4−(2−アダマンチルカルバモイル)−5−tert−ブチル−ピラゾル−1−イル]安息香酸の結晶形態。
【請求項8】
CuKa照射を用いて測定した次の2−シータ値:18.7、11.7および19.2°にピークを持つX線粉末回折パターンを有する、請求項7に記載の結晶性化合物。
【請求項9】
CuKa照射を用いて測定した次の2−シータ値:18.7、11.7、19.2、7.8、14.1、14.9および9.4°にピークを持つX線粉末回折パターンを有する、請求項7に記載の化合物の結晶形態。
【請求項10】
CuKa照射を用いて測定した次の2−シータ値:18.7、11.7、19.2、7.8、14.1、14.9、9.4、15.6、16.1および9.6°にピークを持つX線粉末回折パターンを有する、請求項7に記載の化合物の結晶形態。
【請求項11】
CuKa照射を用いた、図3に示すものと実質的に同一のX線回折パターンを有する、請求項7に記載の結晶性化合物。
【請求項12】
約309.3℃(開始)の融点を有する、4−[4−(2−アダマンチルカルバモイル)−5−tert−ブチル−ピラゾル−1−イル]安息香酸の結晶形態。
【請求項13】
CuKa照射を用いて測定した次の2−シータ値:16.2および20.6°にピークを持つX線回折パターンを有する、4−[4−(2−アダマンチルカルバモイル)−5−tert−ブチル−ピラゾル−1−イル]安息香酸の結晶形態。
【請求項14】
CuKa照射を用いて測定した次の2−シータ値:16.2、20.6および17.7°にピークを持つX線粉末回折パターンを有する、請求項13に記載の結晶性化合物。
【請求項15】
CuKa照射を用いて測定した次の2−シータ値:16.2、20.6、17.7、10.8および15.5°にピークを持つX線粉末回折パターンを有する、請求項13に記載の化合物の結晶形態。
【請求項16】
CuKa照射を用いて測定した次の2−シータ値:16.2、20.6、17.7、10.8、15.5、20.9、26.1、11.6および26.7にピークを持つX線粉末回折パターンを有する、請求項13に記載の化合物の結晶形態。
【請求項17】
CuKa照射を用いた、図6に示すものと実質的に同一のX線回折パターンを有する、請求項13に記載の結晶性化合物。
【請求項18】
約312.0℃(開始)の融点を有する、4−[4−(2−アダマンチルカルバモイル)−5−tert−ブチル−ピラゾル−1−イル]安息香酸の結晶形態。
【請求項19】
請求項1、7および13のいずれか一項に記載の化合物を、薬学的に許容される希釈剤または担体と共に含む、医薬組成物。
【請求項20】
ヒトなどの温血動物の予防的または治療的処置方法における使用のための、請求項1、7および13のいずれか一項に記載の化合物。
【請求項21】
医薬として使用するための、請求項1、7および13のいずれか一項に記載の化合物。
【請求項22】
ヒトなどの温血動物での11βHSD1阻害作用の発現に使用される医薬の製造における、請求項1、7および13のいずれか一項に記載の化合物の使用。
【請求項23】
請求項1、7および13のいずれか一項に記載の化合物の有効量を、11βHSD1阻害作用を発現させるような治療を必要としている哺乳動物に投与することによって、11βHSD1阻害作用を発現させる方法。
【請求項24】
前記11βHSD1阻害作用が2型糖尿病を治療するためのものである、請求項23に記載の方法。
【図1】

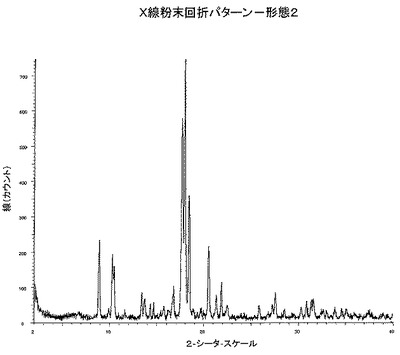
【図2】
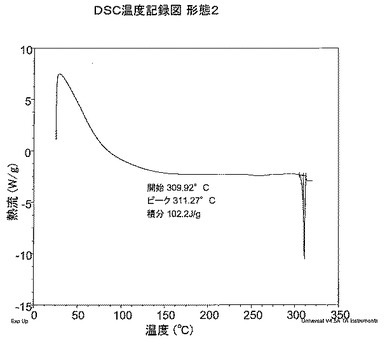
【図3】
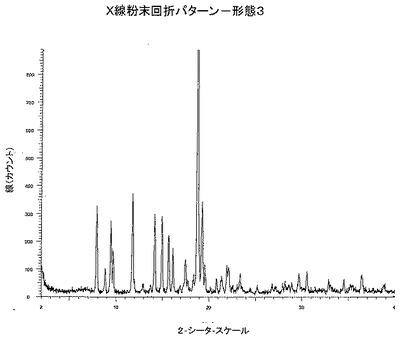
【図4】
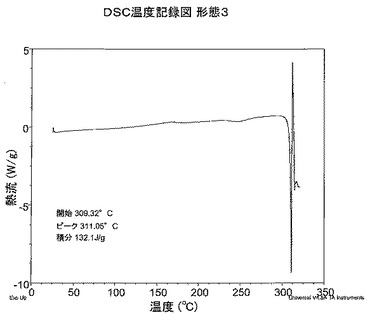
【図5】
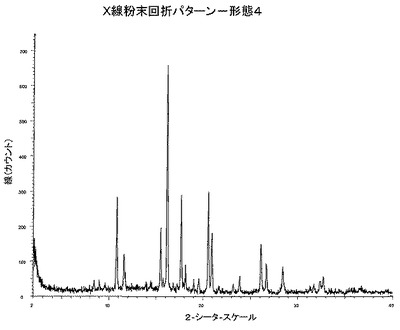
【図6】
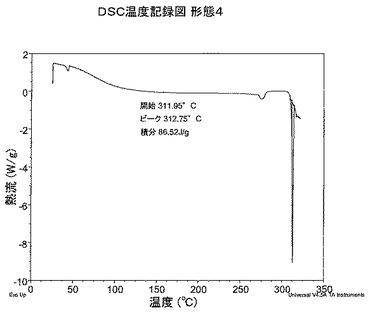
【図7】
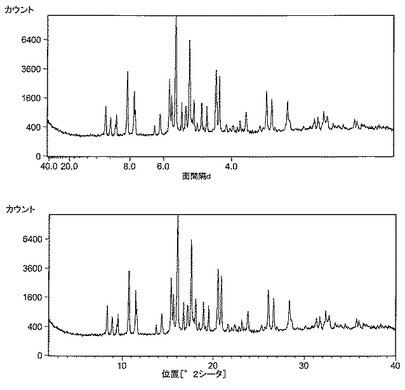
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【公表番号】特表2011−510966(P2011−510966A)
【公表日】平成23年4月7日(2011.4.7)
【国際特許分類】
【出願番号】特願2010−544790(P2010−544790)
【出願日】平成21年2月3日(2009.2.3)
【国際出願番号】PCT/GB2009/050096
【国際公開番号】WO2009/098501
【国際公開日】平成21年8月13日(2009.8.13)
【出願人】(300022641)アストラゼネカ アクチボラグ (581)
【Fターム(参考)】
【公表日】平成23年4月7日(2011.4.7)
【国際特許分類】
【出願日】平成21年2月3日(2009.2.3)
【国際出願番号】PCT/GB2009/050096
【国際公開番号】WO2009/098501
【国際公開日】平成21年8月13日(2009.8.13)
【出願人】(300022641)アストラゼネカ アクチボラグ (581)
【Fターム(参考)】
[ Back to top ]