
5−アミノサリチル酸の配糖体プロドラッグ
本発明の目的は、潰瘍性大腸炎治療剤として有用である5−アミノサリチル酸(5−ASA)を胃や小腸上部において殆ど吸収又は代謝されずに、効率よく疾患部位である大腸へ到達させることができかつ安全で長期投与可能な潰瘍性大腸炎治療剤を提供することにある。本発明は、次の一般式〔1〕で表されるD−ガラクトースを導入した5−ASAに関するものである。

本発明化合物は、効率よく作用部位である大腸まで到達することができ、腸内細菌叢によって分解され、大腸内で活性本体である5−ASAを生成することができる。
本発明化合物は、効率よく作用部位である大腸まで到達することができ、腸内細菌叢によって分解され、大腸内で活性本体である5−ASAを生成することができる。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、次の一般式〔1〕で表される5−アミノ−2−(β−D−ガラクトピラノシルオキシ)安息香酸(以下、「化合物〔1〕」という。)若しくは次の一般式〔2〕で表される5−アミノ−2−(α−D−ガラクトピラノシルオキシ)安息香酸(以下、「化合物〔2〕」という。)又はその医薬上許容される塩に関するものである。
【化1】
また、本発明は、化合物〔1〕若しくは化合物〔2〕又はその医薬上許容される塩を有効成分として含有する医薬組成物に関するものである。
更に、本発明は、化合物〔1〕、化合物〔2〕若しくは次の一般式〔3〕で表される5−アミノ−2−(β−D−グルコピラノシルオキシ)安息香酸(以下、「化合物〔3〕」という。)又はその医薬上許容される塩を有効成分として含有する潰瘍性大腸炎治療剤に関するものである。
【化2】
【背景技術】
【0002】
5−アミノサリチル酸(以下、「5−ASA」という。)は、フリーラジカル(DPPHL)還元作用、過酸化水素消去作用、次亜塩素酸イオン消去作用、過酸化脂質抑制作用やロイコトリエンB4生合成阻害作用を有することから、緩解と再燃を繰り返しながら生涯にわたる治療を必要とする慢性炎症性病変を本態とした難病である非特異性炎症性腸疾患(inflammatory bowel disease:IBD)と総称される潰瘍性大腸炎(ulcerative colitis:UD)及びクローン病(Crohn’s disease:CD)の治療薬として有用である(例えば、非特許文献1参照)。
しかしながら、5−ASAそのものを経口投与したのでは小腸上部で速やかにかつ完全に吸収され、炎症部位への局所作用によってその効果を示す5−ASAは疾患部位である大腸付近にほとんど到達しないことが知られている(例えば、非特許文献3参照)。
このような観点から、5−ASAを作用部位である大腸に到達させるため、5−ASAのドラッグデリバリーシステム(以下、「DDS」という。)及びプロドラッグ化の研究が行われている(例えば、非特許文献1,2,4参照)。
【0003】
5−ASAのDDS製剤としては、5−ASAをエチルセルロースの多孔性皮膜でコーティングすることによって、小腸から大腸にわたり徐々に5−ASAを放出するよう製剤化された5−ASA製剤(商品名:ペンタサ(登録商標)、日清キョーリン製薬(株)製)がある(例えば、非特許文献1,2参照)。しかしながら、健康成人に5−ASAとして1000mgを空腹時に単回経口投与した場合、血漿中未変化体濃度は5−ASAそのものを単回経口投与した場合と比較して最高で1/14(Cmax=1448.6±586.4ng/ml)に減少するものの、まだ相当量の5−ASAが血漿中に移行することが知られている(例えば、非特許文献5参照)。
【0004】
また、5−ASAのプロドラッグとしては、5−ASAのアミノ基がアゾ化された化合物サラゾスルファピリジン(以下、「SASP」という。)(商品名:サラゾピリン(登録商標)、ファイザー(株)製)がある(例えば、非特許文献3参照)。該化合物は、大腸内に存在するアゾ還元酵素を有する腸内細菌によって5−ASAに代謝される。潰瘍性大腸炎に対するSASPの有効性は認められているが、SASPが腸内細菌に分解される際に生成するスルファピリジン(SP)によって、薬剤過敏症、男性不妊症、吐き気、頭痛等の副作用が生じることが問題となっている(例えば、非特許文献3参照)。
【0005】
また、別のプロドラッグとしては、5−アミノサリチル酸メチルを高い水溶性を有するグルコース配糖体に誘導した化合物である、5−アミノ−2−(β−D−グルコピラノシルオキシ)安息香酸メチル及び2−アセトキシ−5−(β−D−グルコピラノシルアミノ)安息香酸メチルが知られている(例えば、非特許文献6,7参照)。該化合物の安全性は確認されているが、潰瘍性大腸炎に対する治療効果については全く検討されていない。
【0006】
更に、5−ASAではないが、潰瘍性大腸炎治療剤として有用なステロイド化合物のプロドラッグとして、デキサメタゾン、プレドニゾロンをグルコース等の配糖体に誘導した化合物が報告されている(例えば、特許文献1参照)。該化合物は大腸への特異的な薬剤付与を目的としているが、ラットに胃内投与した場合において、デキサメタゾンのグルコース誘導体は60%、プレドニゾロンのグルコース誘導体は15%以下しか盲腸に到達してないことが報告されている。
【0007】
以上のように、これまで潰瘍性大腸炎治療剤として有用である5−ASAを胃や小腸上部において殆ど吸収又は代謝されずに、効率よく疾患部位である大腸へ到達させることができる安全で長期投与可能な潰瘍性大腸炎治療剤はまだ知られていない。
【特許文献1】特公昭60−501105号公報
【非特許文献1】日薬理誌,104,pp.447−457(1994)
【非特許文献2】日薬理誌,104,pp.303−311(1994)
【非特許文献3】Scandinavian journal of gastroenterology,23,pp.107−112(1988)
【非特許文献4】Advanced Drug Delivery Reviews,7,pp.149−199(1991)
【非特許文献5】薬理と治療,22(Suppl.10),pp.S2467−S2495(1994)
【非特許文献6】Magyar Kemiai Folyoirat,97(4),pp.143−148(1991)
【非特許文献7】Archiv der Pharmazie An International Journal Pharmaceutical and Medicinal Chemistry,332(9),pp.321−326(1999)
【発明の開示】
【発明が解決しようとする課題】
【0008】
本発明の目的は、潰瘍性大腸炎治療剤として有用である5−ASAを胃や小腸上部において殆ど吸収又は代謝されずに、効率よく疾患部位である大腸へ到達させることができる安全で長期投与可能な潰瘍性大腸炎治療剤を提供することにある。
【課題を解決するための手段】
【0009】
本発明者らは、鋭意研究を重ねた結果、上記目的を達成しうる化合物を見出し、本発明を完成するに至った。
本発明として、例えば、化合物〔1〕若しくは化合物〔2〕又はその医薬上許容される塩を挙げることができる。
また、本発明として、化合物〔1〕若しくは化合物〔2〕又はそれらの医薬上許容される塩を有効成分として含有する医薬組成物を挙げることができ、更に、化合物〔1〕、化合物〔2〕若しくは化合物〔3〕(以下、便宜上あわせて「本発明化合物」という。)又はそれらの医薬上許容される塩を有効成分として含有する潰瘍性大腸炎治療剤を挙げることができる。
本発明化合物は、大腸内において腸内細菌叢により5−ASAに代謝されるので、全身性の副作用の低減を図ることができ、比較的多い投与量の本発明化合物を使用して比較的長い期間にわたって処方することが可能となった。
本明細書において使用する用語の定義は、以下の通りである。
「潰瘍性大腸炎」とは、主として粘膜を侵し、しばしば糜爛や潰瘍を形成する大腸の原因不明の糜爛性非特異性炎症である。
【0010】
以下、本発明について詳述する。
本発明化合物は、公知化合物又は容易に調製可能な中間体から、例えば、下記の方法に従って製造することができる。本発明化合物の製造において、原料が反応に影響を及ぼす置換基を有する場合には、原料をあらかじめ公知の方法により適当な保護基で保護した後に反応を行うのが一般的である。保護基は、反応後に、公知の方法により脱離することができる。
【0011】
製造法 1
【化3】
[式中、R1は、炭素数が1〜6の直鎖状又は分枝鎖状のアルキル、R2はD−グルコピラノシル又はD−ガラクトピラノシル(かかるR2の各水酸基はアセチル等の保護基で保護されていてもよい。)、Xはフッ素、塩素、臭素、ヨウ素等のハロゲンを表す。]
【0012】
工程1
本反応は、公知の化合物〔4〕のエステル化反応であり、公知の方法(非特許文献7参照)によって実施することができる。反応温度は、20〜200℃が適当である。反応溶媒は、一般的に、製造するカルボン酸エステルの種類によって異なるが、例えば、メタノール、エタノールなどのアルコール類を挙げることができる。酸としては、塩酸、硫酸などの無機酸を挙げることができる。反応時間は、使用する原料の種類、反応温度によって異なるが、通常、1〜72時間が適当である。
【0013】
工程2
本反応は、化合物〔5〕とグルコースやガラクトースのアノマー位がハロゲン化された化合物との縮合反応であって、それ自体公知の方法(非特許文献7参照)によって実施することができる。本反応は、触媒存在下において立体反転で進行する。本反応の触媒としては、例えば、酸化銀(I)、酸化水銀(II),AgOCOR3(R3は、炭素数が1〜6の直鎖状又は分枝鎖状のアルキルを表す。)を挙げることができる。反応溶媒は、一般的に反応に関与しなければ特に限定されないが、例えばキノリンを挙げることができる。反応温度は、0〜100℃が適当である。反応時間は、使用する原料の種類、反応温度等によって異なるが、通常、1〜72時間が適当である。さらに、必要であれば製造された化合物〔6〕のR2の各水酸基の保護基を公知の方法により脱離してもよい。
また、原料化合物である化合物〔9〕は、購入可能な化合物も存在するが、例えば、次の方法によっても製造することができる。本反応は、グルコースやガラクトースなどの糖のアノマー位をハロゲン化する反応であって、それ自体公知の方法によって実施することができる。ハロゲン化剤としては、一般的に、臭化水素−酢酸溶液、オキシ臭化リン、オキシ塩化リンなどが使用され、反応溶媒は、一般的に反応に関与しなければ特に限定されないが、例えば塩化メチレン、クロロホルム、1,2−ジクロロエタン等のハロゲン系溶媒を挙げることができる。反応温度は、0〜100℃が適当である。反応時間は、使用する原料の種類、反応温度によって異なるが、通常、1〜72時間が適当である。
【0014】
工程3
本反応は、化合物〔6〕を水素添加する反応であって、それ自体公知の方法(非特許文献7参照)により行うことができる。本反応は、例えば金属触媒存在下、適当な溶媒中、一般的に1〜10気圧の水素気圧下において、0〜100℃で実施することができる。金属触媒として、一般的にパラジウム炭素、パラジウム黒、二酸化白金、白金炭素などが使用され、反応溶媒は、反応に関与しなければ特に限定されないが、テトラヒドロフラン、1,4−ジオキサン、1,2−ジメトキシエタンなどのエーテル類、メタノール、エタノールなどのアルコール類、N,N−ジメチルホルムアミド、N,N−ジメチルアセトアミドなどのアミド類、ベンゼン、トルエン、キシレンなどの炭化水素類、又はこれらの混合溶媒を挙げることができる。反応時間は、使用する原料の種類、反応温度によって異なるが、通常、1〜48時間が適当である。
さらに、必要であれば製造された化合物〔7〕のR2の各水酸基の保護基を公知の方法により脱離してもよい。
【0015】
工程4
本反応は、化合物〔7〕のカルボン酸エステルを加水分解する反応であって、それ自体公知の方法により行うことができる。反応温度は、0〜100℃が適当である。反応溶媒は、一般的に反応に関与しなければ特に限定されないが、例えば、メタノール、エタノールなどのアルコール類を挙げることができる。塩基としては、水酸化ナトリウム、水酸化カリウムなどの無機塩基を挙げることができる。反応時間は、使用する原料の種類、反応温度によって異なるが、通常、1〜72時間が適当である。
また、化合物〔6〕の製造法の別法として下記のものを挙げることができる。
【0016】
製造法 2
【化4】
[式中、R1、R2は前記と同義である。X1は、フッ素、塩素、臭素、ヨウ素等のハロゲンを表す。]
【0017】
工程1
本反応は、公知の化合物〔10〕のエステル化反応であり、公知の方法(非特許文献7参照)によって実施することができる。反応温度は、20〜200℃が適当である。反応溶媒は、一般的に、製造するカルボン酸エステルの種類によって異なるが、例えば、メタノール、エタノールなどのアルコール類を挙げることができる。酸としては、塩酸、硫酸などの無機酸を挙げることができる。反応時間は、使用する原料の種類、反応温度によって異なるが、通常、1〜72時間が適当である。
【0018】
工程2
本反応は、化合物〔11〕とグルコース又はガラクトース誘導体〔12〕との縮合反応であって、それ自体公知の方法によって実施することができる。本反応の縮合剤としては、塩基を挙げることができる。本反応において、アノマー位の立体を制御できないので、シリカゲルクロマトグラフィー等により単一のジアステレオマーに分離精製する必要がある。この分離操作によりアノマー位の立体について両方の化合物(α体及びβ体)を得ることができる。本反応に使用する塩基としては、例えば、1,5−ジアザビシクロ[4.3.0]−5−ノネン、1,4−ジアザビシクロ[2.2.2]オクタン、1,8−ジアザビシクロ[5.4.0]−7−ウンデセンを挙げることができる。反応溶媒は、一般的に反応に関与しなければ特に限定されないが、例えばアセトニトリル、ジメチルスルホキシドを挙げることができる。反応温度は、0〜100℃が適当である。反応時間は、使用する原料の種類、反応温度等によって異なるが、通常、1〜72時間が適当である。さらに、必要であれば製造された化合物〔6〕のR2の各水酸基の保護基を公知の方法により脱離してもよい。
【0019】
このようにして製造される本発明化合物は、それ自体公知の手段、例えば、濃縮、液性変換、転溶、溶媒抽出、結晶化、再結晶、分留、クロマトグラフィーにより分離精製することができる。
本発明化合物は、そのまま医薬として用いることができるが、公知の方法により医薬上許容される塩の形にすることができる。
本発明化合物の「塩」としては、医薬上許容される塩、例えば、塩化水素、硫酸、硝酸、リン酸、フッ化水素酸、臭化水素酸等の無機酸の塩、酢酸、酒石酸、乳酸、クエン酸、フマル酸、マレイン酸、コハク酸、メタンスルホン酸、エタンスルホン酸、ベンゼンスルホン酸、トルエンスルホン酸、ナフタレンスルホン酸、カンファースルホン酸等の有機酸の塩、ナトリウム、カリウム、カルシウム等のアルカリ金属又はアルカリ土類金属の塩を挙げることができる。好ましい塩としては、塩酸塩を挙げることができる。
例えば、本発明化合物の塩酸塩は、本発明化合物を塩化水素のアルコール溶液又はジエチルエーテル溶液で処理し、析出結晶を濾取するか、結晶が析出しない場合は、溶液を濃縮して結晶を析出させた後、濾取することにより得ることができる。
【0020】
本発明化合物は、後記する試験例に示すように、殆ど血漿中へ移行しないという既存の5−ASA関連の医薬品にはない優れた特徴を有し、また経口投与により効率よく疾患部位である盲腸、近位結腸、遠位結腸および/または直腸に至る大腸全域に幅広く活性本体である5−ASAを到達させることができる。したがって、本発明化合物は、安全で長期投与可能な潰瘍性大腸炎治療剤として有用である。特に、化合物〔1〕は、その効果が顕著である。
本発明化合物は、後記する試験例に示すように、ラットのトリニトロベンゼンスルホン酸(TNBS)誘発大腸炎モデルにおける化合物〔1〕の有効性を検討した結果、ダメージスコア及び大腸湿重量を有意に抑制し、優れた潰瘍性大腸炎治療剤である。
【0021】
本発明化合物を医薬として投与する場合、本発明化合物又はその医薬上許容される塩をそのまま又は医薬的に許容される無毒性かつ不活性の担体中に、例えば、 0.1%〜99.5%、好ましくは 0.5%〜90%含有する医薬組成物として、人を含む哺乳動物に投与される。
医薬上許容される担体としては、固形、半固形、又は液状の希釈剤、充填剤、及びその他の処方用の助剤一種以上が用いられる。医薬組成物は、投与単位形態で投与することが望ましい。医薬組成物は、経口的又は非経口的(例えば、経直腸等)に投与することができる。これらの投与方法に適した剤型で投与されるのはもちろんである。例えば、経口投与が好ましい。
【0022】
本発明化合物又はその医薬上許容される塩の用量は、年齢、体重、疾病の性質、程度等の患者の状態、投与経路を考慮した上で調整することが望ましいが、通常は、成人に対して本発明化合物又はその医薬上許容される塩の有効成分量として、経口投与の場合、1日あたり、10mg〜10g/成人の範囲、好ましくは、1g〜4g/成人の範囲が適当である。場合によっては、これ以下でも足りるし、また逆にこれ以上の用量を必要とすることもある。通常、1日1回又は数回に分けて投与することができる。
【0023】
経口投与は固形又は液状の用量単位、例えば、末剤、散剤、錠剤、糖衣剤、カプセル剤、顆粒剤、液剤、シロップ剤、坐剤その他の剤型によって行うことができる。
末剤は、本発明化合物又はその医薬上許容される塩を適当な細かさにすることにより製造される。
散剤は、本発明化合物又はその医薬上許容される塩を適当な細かさと成し、次いで同様に細かくした医薬用担体、例えば、トウモロコシ澱粉、マンニトールのような可食性炭水化物、結晶セルロースその他と混合することにより製造される。必要に応じ、風味剤、保存剤、分散剤、着色剤、香料その他のものを混ぜてもよい。
カプセル剤は、まず上述のようにして粉末状となった末剤や散剤あるいは錠剤の項で述べるように顆粒化したものを、例えば、ゼラチンカプセルのようなカプセル外皮の中へ充填することにより製造される。滑沢剤や流動化剤、例えば、コロイド状のシリカ、タルク、ステアリン酸マグネシウム、ステアリン酸カルシウム、固形のポリエチレングリコールのようなものを粉末状態のものに混合し、然るのちに充填操作を行うこともできる。崩壊剤や可溶化剤、例えば、カルボキシメチルセルロース、カルボキシメチルセルロースカルシウム、低置換度ヒドロキシプロピルセルロース、ヒドロキシプロピルセルロース、クロスカルメロースナトリウム、カルボキシメチルスターチナトリウム、炭酸カルシウム、炭酸ナトリウム、無水リン酸水素カルシウムを添加すれば、カプセル剤が摂取されたときの医薬の有効性を改善することができる。
錠剤は、賦形剤を加えて粉末混合物を作り、顆粒化もしくはスラグ化し、次いで崩壊剤又は滑沢剤を加えたのち打錠することにより製造される。粉末混合物の製造には、適当に粉末化された物質を上述の希釈剤やベースと混合し、必要に応じ結合剤(例えば、カルボキシメチルセルロースナトリウム、メチルセルロース、ヒドロキシプロピルメチルセルロース、ゼラチン、ポリビニルピロリドン、ポリビニルアルコール)、溶解遅延化剤(例えば、パラフィン)、再吸収剤(例えば、四級塩)や吸着剤(例えば、ベントナイト、カオリン、リン酸ジカルシウム)を併用することができる。粉末混合物は、まず結合剤、例えば、シロップ、澱粉糊、アラビアゴム、セルロース溶液又は高分子物質溶液で湿らせ、攪拌混合し、これを乾燥、粉砕して顆粒とすることができる。このように粉末を顆粒化するかわりに、まず打錠機にかけたのち、得られる不完全な形態のスラグを破砕して顆粒にすることも可能である。このようにして作られる顆粒は、滑沢剤としてステアリン酸、ステアリン酸塩、タルク、ミネラルオイルその他を添加することにより、互いに付着することを防ぐことができる。このように滑沢化された混合物を次いで打錠する。こうして製造した素錠にフィルムコーティングや糖衣を施すことができる。
【0024】
また、本発明化合物又はその医薬上許容される塩は、上述のように顆粒化やスラグ化の工程を経ることなく、流動性の不活性担体と混合したのちに直接打錠をしてもよい。シェラックの密閉被膜からなる透明又は半透明の保護被覆、糖や高分子材料の被覆、及び、ワックスよりなる磨上被覆の如きも用いうる。他の経口投与剤型、例えば、溶液、シロップ、エリキシルもまたその一定量が薬物の一定量を含有するように用量単位形態にすることができる。シロップは、本発明化合物又はその医薬上許容される塩を適当な香味水溶液に溶解して製造され、またエリキシルは非毒性のアルコール性担体を用いることにより製造される。
必要であれば、経口投与のための用量単位処方は、マイクロカプセル化してもよい。該処方はまた被覆をしたり、高分子・ワックス等中に埋め込んだりすることにより作用時間の延長や持続放出をもたらすこともできる。
【0025】
非経口投与として坐剤等を用いることができる。直腸投与は、本発明化合物又はその医薬上許容される塩を低融点の、水に可溶又は不溶の固体、例えば、ポリエチレングリコール、カカオ脂、半合成の油脂(例えば、ウイテプゾール/登録商標)、高級エステル類(例えば、パルミチン酸ミリスチルエステル)及びそれらの混合物に溶解又は懸濁させて製造した坐剤等を用いることによって行うことができる。
【図面の簡単な説明】
【0026】
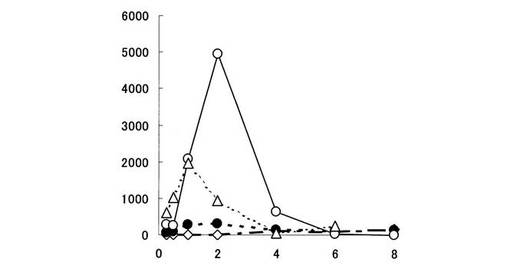
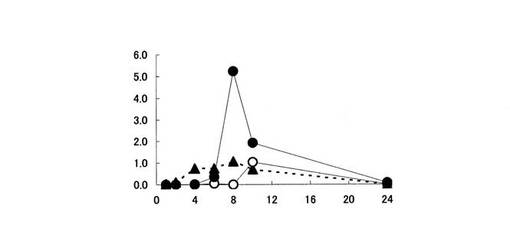
【図1】血漿中における5−ASAの濃度推移を示す。縦軸は、ラット血漿中に存在する5−ASAの濃度(ng/ml)を、横軸は時間(時間)をそれぞれ表す。黒丸印は化合物〔1〕を投与した場合、白菱形印は化合物〔2〕を投与した場合、白三角印は化合物〔3〕を投与した場合、白丸印はペンタサ(登録商標)を投与した場合の5−ASAの濃度推移をそれぞれ表す。
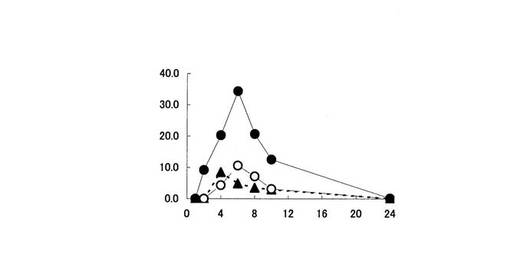
【図2】盲腸内容物中における5−ASA量の推移を示す。縦軸は、ラット盲腸内容物中に存在する5−ASA量(% of dose)を、横軸は時間(時間)をそれぞれ表す。黒丸印は化合物〔1〕を投与した場合、白丸印はペンタサ(登録商標)を投与した場合、黒三角印は5−ASAを投与した場合の5−ASA量の推移をそれぞれ表す。
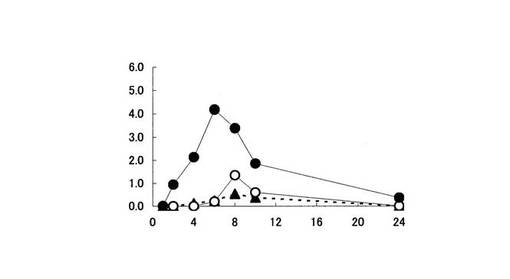
【図3】近位結腸内容物中における5−ASA量の推移を示す。縦軸は、ラット近位結腸内容物中に存在する5−ASA量(% of dose)を、横軸は時間(時間)をそれぞれ表す。黒丸は化合物〔1〕を投与した場合、白丸印はペンタサ(登録商標)を投与した場合、黒三角印は5−ASAを投与した場合の5−ASA量の推移をそれぞれ表す。
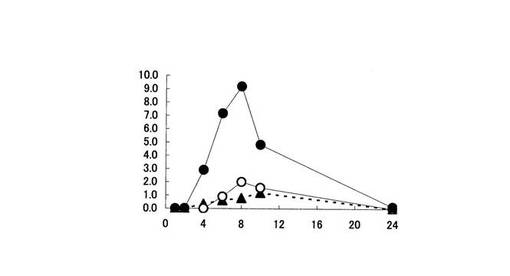
【図4】遠位結腸内容物中における5−ASA量の推移を示す。縦軸は、ラット遠位結腸内容物中に存在する5−ASA量(% of dose)を、横軸は時間(時間)をそれぞれ表す。黒丸印は化合物〔1〕を投与した場合、白丸印はペンタサ(登録商標)を投与した場合、黒三角印は5−ASAを投与した場合の5−ASA量の推移をそれぞれ表す。
【図5】直腸内容物中における5−ASA量の推移を示す。縦軸は、ラット直腸内容物中に存在する5−ASA量(% of dose)を、横軸は時間(時間)をそれぞれ表す。黒丸印は化合物〔1〕を投与した場合、白丸印はペンタサ(登録商標)を投与した場合、黒三角印は5−ASAを投与した場合の5−ASA量の推移をそれぞれ表す。
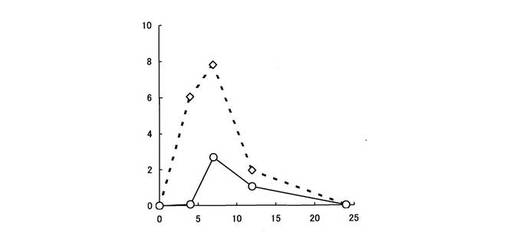
【図6】結腸組織中における5−ASAの濃度推移を示す。縦軸は、ラット結腸1g中に存在する5−ASAの濃度(μg/g)を、横軸は時間(時間)をそれぞれ表す。白菱形印は化合物〔2〕を投与した場合、白丸印はペンタサ(登録商標)を投与した場合の5−ASAの濃度推移をそれぞれ表す。
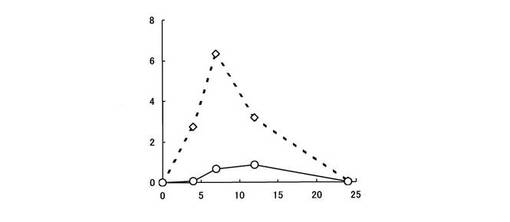
【図7】直腸組織中における5−ASAの濃度推移を示す。縦軸は、ラット直腸1g中に存在する5−ASAの濃度(μg/g)を、横軸は時間(時間)をそれぞれ表す。白菱形印は化合物〔2〕を投与した場合、白丸印はペンタサ(登録商標)を投与した場合の5−ASAの濃度推移をそれぞれ表す。
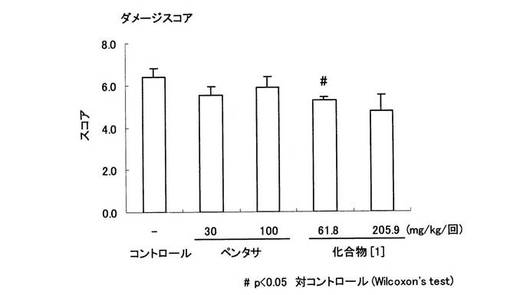
【図8】ラットTNBS誘起大腸炎に対するペンタサ(登録商標)及び化合物〔1〕の治療効果をダメージスコアによって示す。縦軸はダメージスコア、横軸は各被験薬物の投与用量(mg/kg/回)をそれぞれ表す。
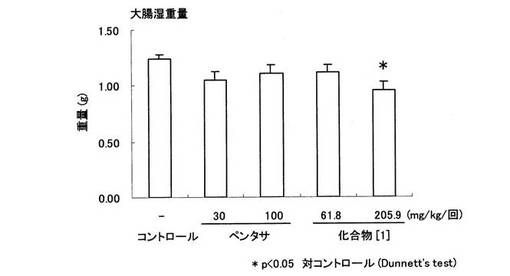
【図9】ラットTNBS誘起大腸炎に対するペンタサ(登録商標)及び化合物〔1〕の大腸炎発症に伴う組織湿重量の増加に対する作用を示す。縦軸は大腸湿重量(g)を示し、横軸は各被験薬物の投与用量(mg/kg/回)をそれぞれ表す。
【発明を実施するための最良の形態】
【0027】
以下に実施例、試験例及び製剤例を掲げて本発明を更に詳しく説明するが、本発明はこれらのみに限定されるものではない。
【0028】
実施例1 5−アミノ−2−(β−D−ガラクトピラノシルオキシ)安息香酸
工程1 5−ニトロサリチル酸メチル
5−ニトロサリチル酸30gの無水メタノール(500ml)溶液に、濃硫酸を滴下し、2日間加熱還流した。反応液を減圧濃縮し、酢酸エチル(500ml)で希釈し、水(500ml)を加え、氷冷下、飽和重曹水をゆっくり加えてアルカリ性(pH=9)にした。析出した黄色沈殿を濾過し、濾液の水層を酢酸エチルで抽出し、まとめた有機層を水、飽和食塩水で洗浄し、無水硫酸マグネシウムで乾燥後、濾過し、溶媒を濃縮し、5−ニトロサリチル酸メチル31.26gを得た。
【0029】
工程2−1 2’,3’,4’,6’−テトラ−O−アセチル−α−D−ガラクトピラノシルブロミド
1’,2’,3’,4’,6’−ペンタ−O−アセチル−β−D−ガラクトピラノース65gの塩化メチレン(500ml)溶液を氷冷し、30%臭化水素酢酸溶液(177.5g)を滴下した。反応混合物を室温において14時間攪拌した後、氷の入った飽和重曹水中へ投じた。有機層を飽和食塩水で洗浄し、無水硫酸マグネシウムで乾燥後、濾過し、溶媒を濃縮し、2’,3’,4’,6’−テトラ−O−アセチル−α−D−ガラクトピラノシルブロミド 68.7gを得た。
【0030】
工程2−2 5−ニトロ−2−(2’,3’,4’,6’−テトラ−O−アセチル−β−D−ガラクトピラノシルオキシ)安息香酸メチル
工程1で得られた5−ニトロサリチル酸メチル30.55gと工程2−1で得られた2’,3’,4’,6’−テトラ−O−アセチル−α−D−ガラクトピラノシルブロミド63.7gのキノリン(250ml)溶液に酸化銀(35.92g)を加え、遮光しながら室温において62 時間攪拌した。反応混合物を酢酸エチル(1000ml)で希釈した後、セライト濾過した。酢酸エチル層を2Nの塩酸(1000ml)で2回洗浄した後、水層を酢酸エチルで2回抽出した。まとめた有機層を飽和重曹水、水、飽和食塩水で洗浄後、硫酸ナトリウムで乾燥後、濾過し、溶媒を濃縮し、5−ニトロ−2−(2’,3’,4’,6’−テトラ−O−アセチル−β−D−ガラクトピラノシルオキシ)安息香酸メチル71.7gを得た。
【0031】
工程2−3 5−ニトロ−2−(β−D−ガラクトピラノシルオキシ)安息香酸メチル
工程2−2で得られた5−ニトロ−2−(2’,3’,4’,6’−テトラ−O−アセチル−β−D−ガラクトピラノシルオキシ)安息香酸メチル10.55gのメタノール溶液を60℃で攪拌し、ナトリウムメトキシドを加えた。30分攪拌した後、反応混合物をAmberlite IRC−50(5.0g)で中和した。濾過した後、有機層を濃縮し、5−ニトロ−2−(β−D−ガラクトピラノシルオキシ)安息香酸メチル4.90gを得た。
【0032】
工程3 5−アミノ−2−β−D−ガラクトピラノシルオキシ)安息香酸メチル
工程2−3で得られた5−ニトロ−2−(β−D−ガラクトピラノシルオキシ)安息香酸メチル4.90gのメタノール(100ml)溶液に10%パラジウム炭素(0.49g)を加え、水素1気圧、室温で接触還元反応を行った。20時間後、反応液を濾過し触媒を除き、有機層を濃縮し、5−アミノ−2−(β−D−ガラクトピラノシルオキシ)安息香酸メチル4.18gを得た。
【0033】
工程4 5−アミノ−2−(β−D−ガラクトピラノシルオキシ)安息香酸
工程2−4で得られた5−アミノ−2−(β−D−ガラクトピラノシルオキシ)安息香酸メチル4.18gの無水メタノール(120ml)懸濁液に、1N水酸化ナトリウム水溶液(12.7ml)を滴下し、加熱還流下において16時間攪拌した。反応液をそのまま減圧濃縮し、残渣を蒸留水で希釈した。その後、2N塩酸(6.4ml)を用いて中和した。その混合物を濃縮し、目的化合物3.41gを得た。
無色粉末物
MS(EI) m/z=338[M+Na]+
旋光度 [α]D20=−19.84(C=1.28,H2O)
元素分析値 (C13H17NO8として)
計算値(%) C:49.52 H:5.43 N:4.44
実測値(%) C:49.12 H:5.37 N:4.38
1H NMR (D2O)
3.74〜4.01(m,6H,H−2〜6),5.04(d,1H,J1,2=7.4Hz,H−1),7.30〜7.39(m,3H,Ph)
【0034】
実施例2 5−アミノ−2−(α−D−ガラクトピラノシルオキシ)安息香酸
工程1 2−フルオロ−5−ニトロ安息香酸メチル
2−フルオロ−5−ニトロ安息香酸12.0gの無水テトラヒドロフラン(60ml)及びジメチルホルムアミド(60μl)溶液を氷冷し、オキザリルクロライド(9.05g)を滴下した。滴下完了後、室温にて5時間撹拌した。反応液に、無水テトラヒドロフラン(30ml)及びメタノール(30ml)溶液を滴下し、室温下で終夜撹拌した。反応液を減圧濃縮し、酢酸エチル(240ml)で希釈し、5%炭酸水素ナトリウム水溶液、飽和食塩水で洗浄し、無水硫酸マグネシウムで乾燥後、濾過し、溶媒を濃縮した。濃縮残渣に、イソプロピルエーテル(24ml)を加えて溶解し、5℃に冷却して結晶を析出させた。析出晶を減圧濾過後、室温下で減圧乾燥し、2−フルオロ−5−ニトロ安息香酸メチル10.5gを得た。
【0035】
工程2 5−ニトロ−2−(2’,3’,4’,6’−テトラ−O−アセチル−α−D−ガラクトピラノシルオキシ)安息香酸メチル
工程1で得られた2−フルオロ−5−ニトロ安息香酸メチル7.13gと2’,3’,4’,6’−テトラ−O−アセチル−D−ガラクトピラノース12.50gのアセトニトリル(70ml)溶液にDBU4.95gを滴下し、室温にて2時間撹拌した。反応液を減圧濃縮し、酢酸エチル(300ml)で希釈し、1N塩酸(150ml)、5%炭酸水素ナトリウム水溶液(150ml)、飽和食塩水(150ml)で洗浄し、無水硫酸マグネシウムで乾燥後、濾過し、溶媒を濃縮した。濃縮物は、カラムクロマト(Wako gel(登録商標)C−200(和光純薬(株)製),n−へキサン:酢酸エチル=4〜1.5)で精製し、5−ニトロ−2−(2’,3’,4’,6’−テトラ−O−アセチル−α−D−ガラクトピラノシルオキシ)安息香酸メチル7.51g及び5−ニトロ−2−(2’,3’,4’,6’−テトラ−O−アセチル−β−D−ガラクトピラノシルオキシ)安息香酸メチル7.92gを得た。
【0036】
工程3−1 5−アミノ−2−(2’,3’,4’,6’−テトラ−O−アセチル−α−D−ガラクトピラノシルオキシ)安息香酸メチル
工程2−1で得られた5−ニトロ−2−(2’,3’,4’,6’−テトラ−O−アセチル−α−D−ガラクトピラノシルオキシ)安息香酸メチル7.00gのメタノール(210ml)溶液に10 %パラジウム炭素(0.70g)を加え、水素1気圧、室温で接触還元反応を行った。18時間後、反応液を濾過して触媒を除き、有機層を濃縮した。濃縮物は、カラムクロマト(Wako gel(登録商標)C−200(和光純薬(株)製),n−へキサン:酢酸エチル=3:1〜3:2)で精製し、5−アミノ−2−(2’,3’,4’,6’−テトラ−O−アセチル−α−D−ガラクトピラノシルオキシ)安息香酸メチル5.73gを得た。
【0037】
工程3−2 5−アミノ−2−(α−D−ガラクトピラノシルオキシ)安息香酸メチル
工程3−1で得られた5−アミノ−2−(2’,3’,4’,6’−テトラ−O−アセチル−α−D−ガラクトピラノシルオキシ)安息香酸メチル5.52gの無水テトラヒドロフラン−無水メタノール(1:1,110ml)に炭酸カリウム(307mg)を加え、室温にて15時間撹拌した。反応液を減圧濃縮し、濃縮物をカラムクロマト(Wako gel(登録商標)C−200(和光純薬(株)製), クロロホルム:メタノール=10:1〜5:1)で精製し、5−アミノ−2−(α−D−ガラクトピラノシルオキシ)安息香酸メチル2.71gを得た。
【0038】
工程4 5−アミノ−2−(α−D−ガラクトピラノシルオキシ)安息香酸
工程3−2で得られた5−アミノ−2−(α−D−ガラクトピラノシルオキシ)安息香酸メチル2.00gの水(40ml)懸濁液に、1N水酸化ナトリウム水溶液(6.07ml)を滴下し、50℃において2時間攪拌した。反応液を濾過して不溶物を除き、濾液に1N塩酸(6.07ml)を加えて中和した。反応液を減圧濃縮し、目的化合物1.34gを得た。
微黄色粉末物
MS(FAB) m/z=316[M+1]+
旋光度 [α]D20=79.37(C=1.28,H2O)
元素分析値 (C13H17NO8・0.8H2Oとして)
計算値(%) C:47.36 H:5.69 N:4.25
実測値(%) C:47.20 H:5.48 N:4.22
1H NMR (D2O)
3.70〜4.10(m,6H,H−2〜6),5.76(d,1H,J1,2=3.6Hz,H−1),7.37〜7.40(m,3H,Ph)
【0039】
参考例1 5−アミノ−2−(β−D−グルコピラノシルオキシ)安息香酸
工程1 5−ニトロサリチル酸メチル
実施例1の工程1と同様の方法を用いて合成した。
【0040】
工程2 5−ニトロ−2−(2’,3’,4’,6’−テトラ−O−アセチル−β−D−グルコピラノシルオキシ)安息香酸メチル
工程1で得られた5−ニトロサリチル酸メチル6.0gと2’,3’,4’,6’−テトラ−O−アセチル−α−D−グルコピラノシルブロミド18.8gとのキノリン(60ml)溶液に、酸化銀(10.5g)を加え室温で1時間激しく攪拌した。反応混合物を酢酸エチル(300ml)で希釈した後、セライト濾過した。酢酸エチル層を2N塩酸(2ml)で2回洗浄した後、水層を酢酸エチル(300ml)で2回抽出した。まとめた有機層を飽和重曹水、水、飽和食塩水で洗浄後、硫酸ナトリウムで乾燥後、濾過し、溶媒を濃縮し、5−ニトロ−2−(2’,3’,4’,6’−テトラ−O−アセチル−β−D−グルコピラノシルオキシ)安息香酸メチル 15.63gを得た。
【0041】
工程3−1 5−アミノ−2−(2’,3’,4’,6’−テトラ−O−アセチル−β−D−グルコピラノシルオキシ)安息香酸メチル
工程2で得られた5−ニトロ−2−(2’,3’,4’,6’−テトラ−O−アセチル−β−D−グルコピラノシルオキシ)安息香酸メチル12.0gのメタノール(400ml)懸濁液に10%パラジウム炭素(2.4g)を加え,水素3atm、30℃で接触還元反応を行った。3時間後、反応液をセライト濾過した。溶媒を濃縮し、5−アミノ−2−(2’,3’,4’,6’−テトラ−O−アセチル−β−D−グルコピラノシルオキシ)安息香酸メチル 11.2gを得た。
【0042】
工程3−2 5−アミノ−2−(β−D−グルコピラノシルオキシ)安息香酸メチル
工程3−1で得られた5−アミノ−2−(2’,3’,4’,6’−テトラ−O−アセチル−β−D−グルコピラノシルオキシ)安息香酸メチル0.68gの無水テトラヒドロフラン−メタノール(1:1,16ml)溶液に炭酸カリウム(37.8mg)を加え、室温で終夜攪拌した。反応液に4N塩化水素酢酸エチル溶液(0.14ml)を滴下し、そのまま溶媒を濃縮した。得られた粗成績体をシリカゲルカラムクロマトグラフィー(Wako gel(登録商標)C−200(和光純薬(株)製),塩化メチレン:メタノール=10:1〜8:1〜5:1)で精製し、5−アミノ−2−(β−D−グルコピラノシルオキシ)安息香酸メチル332mgを得た。
【0043】
工程4 5−アミノ−2−(β−D−グルコピラノシルオキシ)安息香酸
工程3−2で得られた5−アミノ−2−(β−D−グルコピラノシルオキシ)安息香酸メチル100mgのメタノール(3ml)懸濁液に、1N水酸化ナトリウム水溶液(0.3ml)を滴下し、50℃で攪拌した。5時間後室温に戻し溶媒を減圧留去した。得られた油状残渣を水1mlに溶解し氷冷下攪拌しながら、1N塩酸(0.3ml)を滴下した。この溶液を約3分の1の量まで濃縮し析出物を濾取して目的化合物93mgを得た。
元素分析値 (C13H17NO8・0.2H2Oとして)
計算値(%) C:48.97 H:5.50 N:4.39
実測値(%) C:48.80 H:5.35 N:4.31
【0044】
試験例1 血漿中5−ASA濃度の測定
7週齢のSD系雄性ラットに被験薬物として5−ASAを静脈内投与し、また化合物〔1〕、化合物〔2〕、化合物〔3〕及びペンタサ(登録商標)を5−ASAに換算して50mg/kg経口投与し、血漿中5−ASA濃度を高速液体クロマトグラフィー(HPLC)で測定した。なお、ペンタサ(登録商標)は、ペンタサ(登録商標)錠を粉砕して顆粒にしたものを使用した。
結果を表1に示す。
【表1】
上記結果から、ペンタサ(登録商標)を経口投与した場合、血漿中には5−ASAが比較的高濃度で検出された(図1参照)。本試験例の結果は、吸収部位である小腸上部において、投与されたペンタサの一部から5−ASAが放出されていることを示している。
一方、5−ASAの糖誘導体である化合物〔1〕、化合物〔2〕及び化合物〔3〕を経口投与した場合、血漿中5−ASA濃度は、ペンタサ(登録商標)に比べて低く推移した(図1参照)。また、化合物〔1〕及び化合物〔2〕を投与した場合、血漿中5−ASA濃度は、化合物〔3〕を投与した場合より低く推移し、5−ASAはほとんど検出されなかった(図1参照)。化合物〔1〕、化合物〔2〕、化合物〔3〕及びペンタサ(登録商標)を経口投与した場合の5−ASAの生物学的利用能(bioavailability)(表1参照)は、各々2%,0.8%,6%及び15%と算出された。すなわち、化合物〔1〕、化合物〔2〕及び化合物〔3〕は、ペンタサと比較して消化管からの吸収率が低いことが判明した。特に、化合物〔1〕及び化合物〔2〕は、生物学的利用能が著しく低いものであった。
この原因として、化合物〔1〕及び化合物〔2〕が化合物〔3〕に比べ胃や小腸において加水分解されにくく、胃や小腸上部において5−ASAを生成しないためと考えられる。
【0045】
試験例2 消化管内容物中の5−ASA濃度推移
7週齢のSD系雄性ラットに被験薬物として化合物〔1〕、ペンタサ(登録商標)及び5−ASAを5−ASAに換算して50mg/kg経口投与し、盲腸、近位結腸、遠位結腸および直腸内の内容物をホモジネートし、遠心分離後その上清を用いて、高速液体クロマトグラフィー(HPLC)により大腸各部位における5−ASA量を測定した。なお、ペンタサ(登録商標)は、ペンタサ(登録商標)錠を粉砕して顆粒にしたものを使用した。
結果を図2から図5に示す。
大腸各部位における5−ASA量は、化合物〔1〕を投与した時が最も高かった(図2,3,4,5参照)。
また、同様に被験薬物として化合物〔2〕及びペンタサ(登録商標)を5−ASAに換算して50mg/kg経口投与し、結腸及び直腸をホモジネートし、遠心分離後その上清を用いて、高速液体クロマトグラフィー(HPLC)により結腸組織中及び直腸組織中における5−ASA濃度を測定した。なお、ペンタサ(登録商標)は、ペンタサ(登録商標)錠を粉砕して顆粒にしたものを使用した。
結果を図6及び図7に示す。
化合物〔2〕についても、ペンタサ(登録商標)と比較して結腸組織中及び直腸組織中における5−ASA濃度が高かった(図6,7参照)。
試験例1において、化合物〔1〕及び化合物〔2〕は、胃や小腸において加水分解されにくく、胃や小腸上部において5−ASAを生成せず、消化管からの吸収率が低いことが確認されている。
試験例2の結果より、化合物〔1〕及び化合物〔2〕は、疾患部位である大腸に到達し、腸内細菌により5−ASAに代謝されることが明らかとなった。特に、化合物〔1〕は、大腸各部位において潰瘍性大腸炎の治療に有効な5−ASAが高濃度で検出された。
【0046】
試験例3 ラットのトリニトロベンゼンスルホン酸(以下、「TNBS」という。)誘発大腸炎モデルにおける化合物〔1〕の有効性の検討
24時間絶食した雌性SDラットにペントバルビタール麻酔下、TNBS/50%エタノール水溶液(30mg/0.25ml/ラット)を肛門から8cmの大腸内に経口投与用ゾンデを用いて投与することにより大腸炎を惹起した。TNBS投与3日後に大腸を摘出し、肛門から8cmの大腸の湿重量を測定すると共に、大腸炎発症の程度はWallaceら(Gastroenterology,96, p.29−36(1989))の方法に従いスコア化した。被験化合物として、ペンタサ(登録商標)は30,100mg/kg、化合物〔1〕は61.8,205.9mg/kg(5−ASA換算で30,100mg/kgに相当)の用量を1日2回(TNBS投与日は、TNBS投与4時間前1回のみ)経口投与した。なお、ペンタサ(登録商標)は、ペンタサ(登録商標)錠を粉砕して顆粒にしたものを使用した。
結果を図8及び図9に示す。
化合物〔1〕は、ダメージスコアを61.8mg/kg(5−ASAに換算して30mg/kgに相当)の用量において有意に抑制し、大腸湿重量を205.9mg/kg(5−ASAに換算して100mg/kgに相当)の用量において有意に抑制した(図8,9参照)。一方、ペンタサ(登録商標)はダメージスコアおよび大腸湿重量において共に明らかな作用を示さなかった。
【0047】
製剤例1 散剤(内服剤)
処方1剤700mg中
化合物〔1〕 500mg
トウモロコシ澱粉 127mg
結晶セルロース 35mg
ポリビニルアルコール 35mg
ステアリン酸マグネシウム 3mg
化合物〔1〕250g、トウモロコシ澱粉63.5g、結晶セルロース17.5gを流動層造粒乾燥機に投入し、ポリビニルアルコールの10%水溶液(175ml)を噴霧して造粒する。これにステアリン酸マグネシウム(0.4%(w/w))を加えて、700mg中に本化合物500mgを含む散剤を得る。
【0048】
製剤例2 錠剤(内服剤)
処方1剤600mg中
化合物〔1〕 400mg
トウモロコシ澱粉 153mg
結晶セルロース 42mg
ステアリン酸マグネシウム 5mg
化合物〔1〕400g、トウモロコシ澱粉153g、結晶セルロース42gの混合末を乾式造粒機で圧縮した後、顆粒状に解砕する。これにステアリン酸マグネシウム(0.8%(w/w))を加えて1錠重量600mg、錠径11mmに成形し、本化合物400mgを含む錠剤を得る。
【0049】
製剤例3 カプセル剤(内服剤)
処方1剤500mg中
化合物〔1〕 250mg
無水リン酸水素カルシウム
222.5mg
クロスカルメロースナトリウム 25mg
ステアリン酸マグネシウム 2.5mg
化合物〔1〕250g、無水リン酸水素カルシウム222.5g、クロスカルメロースナトリウム25gの混合末を乾式造粒機で圧縮した後、顆粒状に解砕する。これにステアリン酸マグネシウム(0.5%(w/w))を加えて500mgを0号硬カプセルに充填し、本化合物250mgを含むカプセル剤を得る。
【0050】
製剤例4 円柱状顆粒(内服剤)
処方1剤1000mg中
化合物〔1〕 750mg
トウモロコシ澱粉 170mg
結晶セルロース 50mg
ポリビニルアルコール 30mg
化合物〔1〕375g、トウモロコシ澱粉85g、結晶セルロース25gをニーダーに投入し、ポリビニルアルコールの12%水溶液(125ml)を加えて練合後、0.7mm径のスクリーンを装着した顆粒押出し成形機で押出する。これを乾燥して整粒し、1000mg中に本化合物750mgを含む顆粒剤を得る。
【0051】
製剤例5 球形コーティング顆粒(内服剤)
処方1剤1000mg中
ノンパレル 200mg
化合物〔1〕 500mg
トウモロコシ澱粉 170mg
低置換度ヒドロキシプロピルセルロース 40mg
ヒドロキシプロピルセルロース 50mg
ヒドロキシプロピルメチルセルロース 30mg
プロピレングリコール 6mg
酸化チタン 4mg
ノンパレル(24〜32メッシュ)200gを遠心流動造粒コーティング装置に投入し、ヒドロキシプロピルセルロースの8%水溶液(50%エタノール)を噴霧しながら、化合物〔1〕500g、トウモロコシ澱粉170g、低置換度ヒドロキシプロピルセルロース40gの混合末を徐々に添加して造粒し、乾燥して約900gの球状素顆粒を得る。
次に、この球状素顆粒400gを流動層造粒乾燥機に投入し、ヒドロキシプロピルメチルセルロース12.5g、プロピレングリコール2.5g、酸化チタン1.7gを含む水溶液(250ml)を噴霧して1000mg中に本化合物500mgを含むコーティング顆粒を得る。
【産業上の利用可能性】
【0052】
本発明化合物は、潰瘍性大腸炎の治療に有効な5−ASAを効率よく作用部位である大腸まで到達させることができ、また5−ASAを血漿中に移行させないという特徴を有する。即ち、全身性の副作用の低減を図ることができ、最大治療効果を得られるまで投与量を上げることも可能である。
【技術分野】
【0001】
本発明は、次の一般式〔1〕で表される5−アミノ−2−(β−D−ガラクトピラノシルオキシ)安息香酸(以下、「化合物〔1〕」という。)若しくは次の一般式〔2〕で表される5−アミノ−2−(α−D−ガラクトピラノシルオキシ)安息香酸(以下、「化合物〔2〕」という。)又はその医薬上許容される塩に関するものである。
【化1】
また、本発明は、化合物〔1〕若しくは化合物〔2〕又はその医薬上許容される塩を有効成分として含有する医薬組成物に関するものである。
更に、本発明は、化合物〔1〕、化合物〔2〕若しくは次の一般式〔3〕で表される5−アミノ−2−(β−D−グルコピラノシルオキシ)安息香酸(以下、「化合物〔3〕」という。)又はその医薬上許容される塩を有効成分として含有する潰瘍性大腸炎治療剤に関するものである。
【化2】
【背景技術】
【0002】
5−アミノサリチル酸(以下、「5−ASA」という。)は、フリーラジカル(DPPHL)還元作用、過酸化水素消去作用、次亜塩素酸イオン消去作用、過酸化脂質抑制作用やロイコトリエンB4生合成阻害作用を有することから、緩解と再燃を繰り返しながら生涯にわたる治療を必要とする慢性炎症性病変を本態とした難病である非特異性炎症性腸疾患(inflammatory bowel disease:IBD)と総称される潰瘍性大腸炎(ulcerative colitis:UD)及びクローン病(Crohn’s disease:CD)の治療薬として有用である(例えば、非特許文献1参照)。
しかしながら、5−ASAそのものを経口投与したのでは小腸上部で速やかにかつ完全に吸収され、炎症部位への局所作用によってその効果を示す5−ASAは疾患部位である大腸付近にほとんど到達しないことが知られている(例えば、非特許文献3参照)。
このような観点から、5−ASAを作用部位である大腸に到達させるため、5−ASAのドラッグデリバリーシステム(以下、「DDS」という。)及びプロドラッグ化の研究が行われている(例えば、非特許文献1,2,4参照)。
【0003】
5−ASAのDDS製剤としては、5−ASAをエチルセルロースの多孔性皮膜でコーティングすることによって、小腸から大腸にわたり徐々に5−ASAを放出するよう製剤化された5−ASA製剤(商品名:ペンタサ(登録商標)、日清キョーリン製薬(株)製)がある(例えば、非特許文献1,2参照)。しかしながら、健康成人に5−ASAとして1000mgを空腹時に単回経口投与した場合、血漿中未変化体濃度は5−ASAそのものを単回経口投与した場合と比較して最高で1/14(Cmax=1448.6±586.4ng/ml)に減少するものの、まだ相当量の5−ASAが血漿中に移行することが知られている(例えば、非特許文献5参照)。
【0004】
また、5−ASAのプロドラッグとしては、5−ASAのアミノ基がアゾ化された化合物サラゾスルファピリジン(以下、「SASP」という。)(商品名:サラゾピリン(登録商標)、ファイザー(株)製)がある(例えば、非特許文献3参照)。該化合物は、大腸内に存在するアゾ還元酵素を有する腸内細菌によって5−ASAに代謝される。潰瘍性大腸炎に対するSASPの有効性は認められているが、SASPが腸内細菌に分解される際に生成するスルファピリジン(SP)によって、薬剤過敏症、男性不妊症、吐き気、頭痛等の副作用が生じることが問題となっている(例えば、非特許文献3参照)。
【0005】
また、別のプロドラッグとしては、5−アミノサリチル酸メチルを高い水溶性を有するグルコース配糖体に誘導した化合物である、5−アミノ−2−(β−D−グルコピラノシルオキシ)安息香酸メチル及び2−アセトキシ−5−(β−D−グルコピラノシルアミノ)安息香酸メチルが知られている(例えば、非特許文献6,7参照)。該化合物の安全性は確認されているが、潰瘍性大腸炎に対する治療効果については全く検討されていない。
【0006】
更に、5−ASAではないが、潰瘍性大腸炎治療剤として有用なステロイド化合物のプロドラッグとして、デキサメタゾン、プレドニゾロンをグルコース等の配糖体に誘導した化合物が報告されている(例えば、特許文献1参照)。該化合物は大腸への特異的な薬剤付与を目的としているが、ラットに胃内投与した場合において、デキサメタゾンのグルコース誘導体は60%、プレドニゾロンのグルコース誘導体は15%以下しか盲腸に到達してないことが報告されている。
【0007】
以上のように、これまで潰瘍性大腸炎治療剤として有用である5−ASAを胃や小腸上部において殆ど吸収又は代謝されずに、効率よく疾患部位である大腸へ到達させることができる安全で長期投与可能な潰瘍性大腸炎治療剤はまだ知られていない。
【特許文献1】特公昭60−501105号公報
【非特許文献1】日薬理誌,104,pp.447−457(1994)
【非特許文献2】日薬理誌,104,pp.303−311(1994)
【非特許文献3】Scandinavian journal of gastroenterology,23,pp.107−112(1988)
【非特許文献4】Advanced Drug Delivery Reviews,7,pp.149−199(1991)
【非特許文献5】薬理と治療,22(Suppl.10),pp.S2467−S2495(1994)
【非特許文献6】Magyar Kemiai Folyoirat,97(4),pp.143−148(1991)
【非特許文献7】Archiv der Pharmazie An International Journal Pharmaceutical and Medicinal Chemistry,332(9),pp.321−326(1999)
【発明の開示】
【発明が解決しようとする課題】
【0008】
本発明の目的は、潰瘍性大腸炎治療剤として有用である5−ASAを胃や小腸上部において殆ど吸収又は代謝されずに、効率よく疾患部位である大腸へ到達させることができる安全で長期投与可能な潰瘍性大腸炎治療剤を提供することにある。
【課題を解決するための手段】
【0009】
本発明者らは、鋭意研究を重ねた結果、上記目的を達成しうる化合物を見出し、本発明を完成するに至った。
本発明として、例えば、化合物〔1〕若しくは化合物〔2〕又はその医薬上許容される塩を挙げることができる。
また、本発明として、化合物〔1〕若しくは化合物〔2〕又はそれらの医薬上許容される塩を有効成分として含有する医薬組成物を挙げることができ、更に、化合物〔1〕、化合物〔2〕若しくは化合物〔3〕(以下、便宜上あわせて「本発明化合物」という。)又はそれらの医薬上許容される塩を有効成分として含有する潰瘍性大腸炎治療剤を挙げることができる。
本発明化合物は、大腸内において腸内細菌叢により5−ASAに代謝されるので、全身性の副作用の低減を図ることができ、比較的多い投与量の本発明化合物を使用して比較的長い期間にわたって処方することが可能となった。
本明細書において使用する用語の定義は、以下の通りである。
「潰瘍性大腸炎」とは、主として粘膜を侵し、しばしば糜爛や潰瘍を形成する大腸の原因不明の糜爛性非特異性炎症である。
【0010】
以下、本発明について詳述する。
本発明化合物は、公知化合物又は容易に調製可能な中間体から、例えば、下記の方法に従って製造することができる。本発明化合物の製造において、原料が反応に影響を及ぼす置換基を有する場合には、原料をあらかじめ公知の方法により適当な保護基で保護した後に反応を行うのが一般的である。保護基は、反応後に、公知の方法により脱離することができる。
【0011】
製造法 1
【化3】
[式中、R1は、炭素数が1〜6の直鎖状又は分枝鎖状のアルキル、R2はD−グルコピラノシル又はD−ガラクトピラノシル(かかるR2の各水酸基はアセチル等の保護基で保護されていてもよい。)、Xはフッ素、塩素、臭素、ヨウ素等のハロゲンを表す。]
【0012】
工程1
本反応は、公知の化合物〔4〕のエステル化反応であり、公知の方法(非特許文献7参照)によって実施することができる。反応温度は、20〜200℃が適当である。反応溶媒は、一般的に、製造するカルボン酸エステルの種類によって異なるが、例えば、メタノール、エタノールなどのアルコール類を挙げることができる。酸としては、塩酸、硫酸などの無機酸を挙げることができる。反応時間は、使用する原料の種類、反応温度によって異なるが、通常、1〜72時間が適当である。
【0013】
工程2
本反応は、化合物〔5〕とグルコースやガラクトースのアノマー位がハロゲン化された化合物との縮合反応であって、それ自体公知の方法(非特許文献7参照)によって実施することができる。本反応は、触媒存在下において立体反転で進行する。本反応の触媒としては、例えば、酸化銀(I)、酸化水銀(II),AgOCOR3(R3は、炭素数が1〜6の直鎖状又は分枝鎖状のアルキルを表す。)を挙げることができる。反応溶媒は、一般的に反応に関与しなければ特に限定されないが、例えばキノリンを挙げることができる。反応温度は、0〜100℃が適当である。反応時間は、使用する原料の種類、反応温度等によって異なるが、通常、1〜72時間が適当である。さらに、必要であれば製造された化合物〔6〕のR2の各水酸基の保護基を公知の方法により脱離してもよい。
また、原料化合物である化合物〔9〕は、購入可能な化合物も存在するが、例えば、次の方法によっても製造することができる。本反応は、グルコースやガラクトースなどの糖のアノマー位をハロゲン化する反応であって、それ自体公知の方法によって実施することができる。ハロゲン化剤としては、一般的に、臭化水素−酢酸溶液、オキシ臭化リン、オキシ塩化リンなどが使用され、反応溶媒は、一般的に反応に関与しなければ特に限定されないが、例えば塩化メチレン、クロロホルム、1,2−ジクロロエタン等のハロゲン系溶媒を挙げることができる。反応温度は、0〜100℃が適当である。反応時間は、使用する原料の種類、反応温度によって異なるが、通常、1〜72時間が適当である。
【0014】
工程3
本反応は、化合物〔6〕を水素添加する反応であって、それ自体公知の方法(非特許文献7参照)により行うことができる。本反応は、例えば金属触媒存在下、適当な溶媒中、一般的に1〜10気圧の水素気圧下において、0〜100℃で実施することができる。金属触媒として、一般的にパラジウム炭素、パラジウム黒、二酸化白金、白金炭素などが使用され、反応溶媒は、反応に関与しなければ特に限定されないが、テトラヒドロフラン、1,4−ジオキサン、1,2−ジメトキシエタンなどのエーテル類、メタノール、エタノールなどのアルコール類、N,N−ジメチルホルムアミド、N,N−ジメチルアセトアミドなどのアミド類、ベンゼン、トルエン、キシレンなどの炭化水素類、又はこれらの混合溶媒を挙げることができる。反応時間は、使用する原料の種類、反応温度によって異なるが、通常、1〜48時間が適当である。
さらに、必要であれば製造された化合物〔7〕のR2の各水酸基の保護基を公知の方法により脱離してもよい。
【0015】
工程4
本反応は、化合物〔7〕のカルボン酸エステルを加水分解する反応であって、それ自体公知の方法により行うことができる。反応温度は、0〜100℃が適当である。反応溶媒は、一般的に反応に関与しなければ特に限定されないが、例えば、メタノール、エタノールなどのアルコール類を挙げることができる。塩基としては、水酸化ナトリウム、水酸化カリウムなどの無機塩基を挙げることができる。反応時間は、使用する原料の種類、反応温度によって異なるが、通常、1〜72時間が適当である。
また、化合物〔6〕の製造法の別法として下記のものを挙げることができる。
【0016】
製造法 2
【化4】
[式中、R1、R2は前記と同義である。X1は、フッ素、塩素、臭素、ヨウ素等のハロゲンを表す。]
【0017】
工程1
本反応は、公知の化合物〔10〕のエステル化反応であり、公知の方法(非特許文献7参照)によって実施することができる。反応温度は、20〜200℃が適当である。反応溶媒は、一般的に、製造するカルボン酸エステルの種類によって異なるが、例えば、メタノール、エタノールなどのアルコール類を挙げることができる。酸としては、塩酸、硫酸などの無機酸を挙げることができる。反応時間は、使用する原料の種類、反応温度によって異なるが、通常、1〜72時間が適当である。
【0018】
工程2
本反応は、化合物〔11〕とグルコース又はガラクトース誘導体〔12〕との縮合反応であって、それ自体公知の方法によって実施することができる。本反応の縮合剤としては、塩基を挙げることができる。本反応において、アノマー位の立体を制御できないので、シリカゲルクロマトグラフィー等により単一のジアステレオマーに分離精製する必要がある。この分離操作によりアノマー位の立体について両方の化合物(α体及びβ体)を得ることができる。本反応に使用する塩基としては、例えば、1,5−ジアザビシクロ[4.3.0]−5−ノネン、1,4−ジアザビシクロ[2.2.2]オクタン、1,8−ジアザビシクロ[5.4.0]−7−ウンデセンを挙げることができる。反応溶媒は、一般的に反応に関与しなければ特に限定されないが、例えばアセトニトリル、ジメチルスルホキシドを挙げることができる。反応温度は、0〜100℃が適当である。反応時間は、使用する原料の種類、反応温度等によって異なるが、通常、1〜72時間が適当である。さらに、必要であれば製造された化合物〔6〕のR2の各水酸基の保護基を公知の方法により脱離してもよい。
【0019】
このようにして製造される本発明化合物は、それ自体公知の手段、例えば、濃縮、液性変換、転溶、溶媒抽出、結晶化、再結晶、分留、クロマトグラフィーにより分離精製することができる。
本発明化合物は、そのまま医薬として用いることができるが、公知の方法により医薬上許容される塩の形にすることができる。
本発明化合物の「塩」としては、医薬上許容される塩、例えば、塩化水素、硫酸、硝酸、リン酸、フッ化水素酸、臭化水素酸等の無機酸の塩、酢酸、酒石酸、乳酸、クエン酸、フマル酸、マレイン酸、コハク酸、メタンスルホン酸、エタンスルホン酸、ベンゼンスルホン酸、トルエンスルホン酸、ナフタレンスルホン酸、カンファースルホン酸等の有機酸の塩、ナトリウム、カリウム、カルシウム等のアルカリ金属又はアルカリ土類金属の塩を挙げることができる。好ましい塩としては、塩酸塩を挙げることができる。
例えば、本発明化合物の塩酸塩は、本発明化合物を塩化水素のアルコール溶液又はジエチルエーテル溶液で処理し、析出結晶を濾取するか、結晶が析出しない場合は、溶液を濃縮して結晶を析出させた後、濾取することにより得ることができる。
【0020】
本発明化合物は、後記する試験例に示すように、殆ど血漿中へ移行しないという既存の5−ASA関連の医薬品にはない優れた特徴を有し、また経口投与により効率よく疾患部位である盲腸、近位結腸、遠位結腸および/または直腸に至る大腸全域に幅広く活性本体である5−ASAを到達させることができる。したがって、本発明化合物は、安全で長期投与可能な潰瘍性大腸炎治療剤として有用である。特に、化合物〔1〕は、その効果が顕著である。
本発明化合物は、後記する試験例に示すように、ラットのトリニトロベンゼンスルホン酸(TNBS)誘発大腸炎モデルにおける化合物〔1〕の有効性を検討した結果、ダメージスコア及び大腸湿重量を有意に抑制し、優れた潰瘍性大腸炎治療剤である。
【0021】
本発明化合物を医薬として投与する場合、本発明化合物又はその医薬上許容される塩をそのまま又は医薬的に許容される無毒性かつ不活性の担体中に、例えば、 0.1%〜99.5%、好ましくは 0.5%〜90%含有する医薬組成物として、人を含む哺乳動物に投与される。
医薬上許容される担体としては、固形、半固形、又は液状の希釈剤、充填剤、及びその他の処方用の助剤一種以上が用いられる。医薬組成物は、投与単位形態で投与することが望ましい。医薬組成物は、経口的又は非経口的(例えば、経直腸等)に投与することができる。これらの投与方法に適した剤型で投与されるのはもちろんである。例えば、経口投与が好ましい。
【0022】
本発明化合物又はその医薬上許容される塩の用量は、年齢、体重、疾病の性質、程度等の患者の状態、投与経路を考慮した上で調整することが望ましいが、通常は、成人に対して本発明化合物又はその医薬上許容される塩の有効成分量として、経口投与の場合、1日あたり、10mg〜10g/成人の範囲、好ましくは、1g〜4g/成人の範囲が適当である。場合によっては、これ以下でも足りるし、また逆にこれ以上の用量を必要とすることもある。通常、1日1回又は数回に分けて投与することができる。
【0023】
経口投与は固形又は液状の用量単位、例えば、末剤、散剤、錠剤、糖衣剤、カプセル剤、顆粒剤、液剤、シロップ剤、坐剤その他の剤型によって行うことができる。
末剤は、本発明化合物又はその医薬上許容される塩を適当な細かさにすることにより製造される。
散剤は、本発明化合物又はその医薬上許容される塩を適当な細かさと成し、次いで同様に細かくした医薬用担体、例えば、トウモロコシ澱粉、マンニトールのような可食性炭水化物、結晶セルロースその他と混合することにより製造される。必要に応じ、風味剤、保存剤、分散剤、着色剤、香料その他のものを混ぜてもよい。
カプセル剤は、まず上述のようにして粉末状となった末剤や散剤あるいは錠剤の項で述べるように顆粒化したものを、例えば、ゼラチンカプセルのようなカプセル外皮の中へ充填することにより製造される。滑沢剤や流動化剤、例えば、コロイド状のシリカ、タルク、ステアリン酸マグネシウム、ステアリン酸カルシウム、固形のポリエチレングリコールのようなものを粉末状態のものに混合し、然るのちに充填操作を行うこともできる。崩壊剤や可溶化剤、例えば、カルボキシメチルセルロース、カルボキシメチルセルロースカルシウム、低置換度ヒドロキシプロピルセルロース、ヒドロキシプロピルセルロース、クロスカルメロースナトリウム、カルボキシメチルスターチナトリウム、炭酸カルシウム、炭酸ナトリウム、無水リン酸水素カルシウムを添加すれば、カプセル剤が摂取されたときの医薬の有効性を改善することができる。
錠剤は、賦形剤を加えて粉末混合物を作り、顆粒化もしくはスラグ化し、次いで崩壊剤又は滑沢剤を加えたのち打錠することにより製造される。粉末混合物の製造には、適当に粉末化された物質を上述の希釈剤やベースと混合し、必要に応じ結合剤(例えば、カルボキシメチルセルロースナトリウム、メチルセルロース、ヒドロキシプロピルメチルセルロース、ゼラチン、ポリビニルピロリドン、ポリビニルアルコール)、溶解遅延化剤(例えば、パラフィン)、再吸収剤(例えば、四級塩)や吸着剤(例えば、ベントナイト、カオリン、リン酸ジカルシウム)を併用することができる。粉末混合物は、まず結合剤、例えば、シロップ、澱粉糊、アラビアゴム、セルロース溶液又は高分子物質溶液で湿らせ、攪拌混合し、これを乾燥、粉砕して顆粒とすることができる。このように粉末を顆粒化するかわりに、まず打錠機にかけたのち、得られる不完全な形態のスラグを破砕して顆粒にすることも可能である。このようにして作られる顆粒は、滑沢剤としてステアリン酸、ステアリン酸塩、タルク、ミネラルオイルその他を添加することにより、互いに付着することを防ぐことができる。このように滑沢化された混合物を次いで打錠する。こうして製造した素錠にフィルムコーティングや糖衣を施すことができる。
【0024】
また、本発明化合物又はその医薬上許容される塩は、上述のように顆粒化やスラグ化の工程を経ることなく、流動性の不活性担体と混合したのちに直接打錠をしてもよい。シェラックの密閉被膜からなる透明又は半透明の保護被覆、糖や高分子材料の被覆、及び、ワックスよりなる磨上被覆の如きも用いうる。他の経口投与剤型、例えば、溶液、シロップ、エリキシルもまたその一定量が薬物の一定量を含有するように用量単位形態にすることができる。シロップは、本発明化合物又はその医薬上許容される塩を適当な香味水溶液に溶解して製造され、またエリキシルは非毒性のアルコール性担体を用いることにより製造される。
必要であれば、経口投与のための用量単位処方は、マイクロカプセル化してもよい。該処方はまた被覆をしたり、高分子・ワックス等中に埋め込んだりすることにより作用時間の延長や持続放出をもたらすこともできる。
【0025】
非経口投与として坐剤等を用いることができる。直腸投与は、本発明化合物又はその医薬上許容される塩を低融点の、水に可溶又は不溶の固体、例えば、ポリエチレングリコール、カカオ脂、半合成の油脂(例えば、ウイテプゾール/登録商標)、高級エステル類(例えば、パルミチン酸ミリスチルエステル)及びそれらの混合物に溶解又は懸濁させて製造した坐剤等を用いることによって行うことができる。
【図面の簡単な説明】
【0026】
【図1】血漿中における5−ASAの濃度推移を示す。縦軸は、ラット血漿中に存在する5−ASAの濃度(ng/ml)を、横軸は時間(時間)をそれぞれ表す。黒丸印は化合物〔1〕を投与した場合、白菱形印は化合物〔2〕を投与した場合、白三角印は化合物〔3〕を投与した場合、白丸印はペンタサ(登録商標)を投与した場合の5−ASAの濃度推移をそれぞれ表す。
【図2】盲腸内容物中における5−ASA量の推移を示す。縦軸は、ラット盲腸内容物中に存在する5−ASA量(% of dose)を、横軸は時間(時間)をそれぞれ表す。黒丸印は化合物〔1〕を投与した場合、白丸印はペンタサ(登録商標)を投与した場合、黒三角印は5−ASAを投与した場合の5−ASA量の推移をそれぞれ表す。
【図3】近位結腸内容物中における5−ASA量の推移を示す。縦軸は、ラット近位結腸内容物中に存在する5−ASA量(% of dose)を、横軸は時間(時間)をそれぞれ表す。黒丸は化合物〔1〕を投与した場合、白丸印はペンタサ(登録商標)を投与した場合、黒三角印は5−ASAを投与した場合の5−ASA量の推移をそれぞれ表す。
【図4】遠位結腸内容物中における5−ASA量の推移を示す。縦軸は、ラット遠位結腸内容物中に存在する5−ASA量(% of dose)を、横軸は時間(時間)をそれぞれ表す。黒丸印は化合物〔1〕を投与した場合、白丸印はペンタサ(登録商標)を投与した場合、黒三角印は5−ASAを投与した場合の5−ASA量の推移をそれぞれ表す。
【図5】直腸内容物中における5−ASA量の推移を示す。縦軸は、ラット直腸内容物中に存在する5−ASA量(% of dose)を、横軸は時間(時間)をそれぞれ表す。黒丸印は化合物〔1〕を投与した場合、白丸印はペンタサ(登録商標)を投与した場合、黒三角印は5−ASAを投与した場合の5−ASA量の推移をそれぞれ表す。
【図6】結腸組織中における5−ASAの濃度推移を示す。縦軸は、ラット結腸1g中に存在する5−ASAの濃度(μg/g)を、横軸は時間(時間)をそれぞれ表す。白菱形印は化合物〔2〕を投与した場合、白丸印はペンタサ(登録商標)を投与した場合の5−ASAの濃度推移をそれぞれ表す。
【図7】直腸組織中における5−ASAの濃度推移を示す。縦軸は、ラット直腸1g中に存在する5−ASAの濃度(μg/g)を、横軸は時間(時間)をそれぞれ表す。白菱形印は化合物〔2〕を投与した場合、白丸印はペンタサ(登録商標)を投与した場合の5−ASAの濃度推移をそれぞれ表す。
【図8】ラットTNBS誘起大腸炎に対するペンタサ(登録商標)及び化合物〔1〕の治療効果をダメージスコアによって示す。縦軸はダメージスコア、横軸は各被験薬物の投与用量(mg/kg/回)をそれぞれ表す。
【図9】ラットTNBS誘起大腸炎に対するペンタサ(登録商標)及び化合物〔1〕の大腸炎発症に伴う組織湿重量の増加に対する作用を示す。縦軸は大腸湿重量(g)を示し、横軸は各被験薬物の投与用量(mg/kg/回)をそれぞれ表す。
【発明を実施するための最良の形態】
【0027】
以下に実施例、試験例及び製剤例を掲げて本発明を更に詳しく説明するが、本発明はこれらのみに限定されるものではない。
【0028】
実施例1 5−アミノ−2−(β−D−ガラクトピラノシルオキシ)安息香酸
工程1 5−ニトロサリチル酸メチル
5−ニトロサリチル酸30gの無水メタノール(500ml)溶液に、濃硫酸を滴下し、2日間加熱還流した。反応液を減圧濃縮し、酢酸エチル(500ml)で希釈し、水(500ml)を加え、氷冷下、飽和重曹水をゆっくり加えてアルカリ性(pH=9)にした。析出した黄色沈殿を濾過し、濾液の水層を酢酸エチルで抽出し、まとめた有機層を水、飽和食塩水で洗浄し、無水硫酸マグネシウムで乾燥後、濾過し、溶媒を濃縮し、5−ニトロサリチル酸メチル31.26gを得た。
【0029】
工程2−1 2’,3’,4’,6’−テトラ−O−アセチル−α−D−ガラクトピラノシルブロミド
1’,2’,3’,4’,6’−ペンタ−O−アセチル−β−D−ガラクトピラノース65gの塩化メチレン(500ml)溶液を氷冷し、30%臭化水素酢酸溶液(177.5g)を滴下した。反応混合物を室温において14時間攪拌した後、氷の入った飽和重曹水中へ投じた。有機層を飽和食塩水で洗浄し、無水硫酸マグネシウムで乾燥後、濾過し、溶媒を濃縮し、2’,3’,4’,6’−テトラ−O−アセチル−α−D−ガラクトピラノシルブロミド 68.7gを得た。
【0030】
工程2−2 5−ニトロ−2−(2’,3’,4’,6’−テトラ−O−アセチル−β−D−ガラクトピラノシルオキシ)安息香酸メチル
工程1で得られた5−ニトロサリチル酸メチル30.55gと工程2−1で得られた2’,3’,4’,6’−テトラ−O−アセチル−α−D−ガラクトピラノシルブロミド63.7gのキノリン(250ml)溶液に酸化銀(35.92g)を加え、遮光しながら室温において62 時間攪拌した。反応混合物を酢酸エチル(1000ml)で希釈した後、セライト濾過した。酢酸エチル層を2Nの塩酸(1000ml)で2回洗浄した後、水層を酢酸エチルで2回抽出した。まとめた有機層を飽和重曹水、水、飽和食塩水で洗浄後、硫酸ナトリウムで乾燥後、濾過し、溶媒を濃縮し、5−ニトロ−2−(2’,3’,4’,6’−テトラ−O−アセチル−β−D−ガラクトピラノシルオキシ)安息香酸メチル71.7gを得た。
【0031】
工程2−3 5−ニトロ−2−(β−D−ガラクトピラノシルオキシ)安息香酸メチル
工程2−2で得られた5−ニトロ−2−(2’,3’,4’,6’−テトラ−O−アセチル−β−D−ガラクトピラノシルオキシ)安息香酸メチル10.55gのメタノール溶液を60℃で攪拌し、ナトリウムメトキシドを加えた。30分攪拌した後、反応混合物をAmberlite IRC−50(5.0g)で中和した。濾過した後、有機層を濃縮し、5−ニトロ−2−(β−D−ガラクトピラノシルオキシ)安息香酸メチル4.90gを得た。
【0032】
工程3 5−アミノ−2−β−D−ガラクトピラノシルオキシ)安息香酸メチル
工程2−3で得られた5−ニトロ−2−(β−D−ガラクトピラノシルオキシ)安息香酸メチル4.90gのメタノール(100ml)溶液に10%パラジウム炭素(0.49g)を加え、水素1気圧、室温で接触還元反応を行った。20時間後、反応液を濾過し触媒を除き、有機層を濃縮し、5−アミノ−2−(β−D−ガラクトピラノシルオキシ)安息香酸メチル4.18gを得た。
【0033】
工程4 5−アミノ−2−(β−D−ガラクトピラノシルオキシ)安息香酸
工程2−4で得られた5−アミノ−2−(β−D−ガラクトピラノシルオキシ)安息香酸メチル4.18gの無水メタノール(120ml)懸濁液に、1N水酸化ナトリウム水溶液(12.7ml)を滴下し、加熱還流下において16時間攪拌した。反応液をそのまま減圧濃縮し、残渣を蒸留水で希釈した。その後、2N塩酸(6.4ml)を用いて中和した。その混合物を濃縮し、目的化合物3.41gを得た。
無色粉末物
MS(EI) m/z=338[M+Na]+
旋光度 [α]D20=−19.84(C=1.28,H2O)
元素分析値 (C13H17NO8として)
計算値(%) C:49.52 H:5.43 N:4.44
実測値(%) C:49.12 H:5.37 N:4.38
1H NMR (D2O)
3.74〜4.01(m,6H,H−2〜6),5.04(d,1H,J1,2=7.4Hz,H−1),7.30〜7.39(m,3H,Ph)
【0034】
実施例2 5−アミノ−2−(α−D−ガラクトピラノシルオキシ)安息香酸
工程1 2−フルオロ−5−ニトロ安息香酸メチル
2−フルオロ−5−ニトロ安息香酸12.0gの無水テトラヒドロフラン(60ml)及びジメチルホルムアミド(60μl)溶液を氷冷し、オキザリルクロライド(9.05g)を滴下した。滴下完了後、室温にて5時間撹拌した。反応液に、無水テトラヒドロフラン(30ml)及びメタノール(30ml)溶液を滴下し、室温下で終夜撹拌した。反応液を減圧濃縮し、酢酸エチル(240ml)で希釈し、5%炭酸水素ナトリウム水溶液、飽和食塩水で洗浄し、無水硫酸マグネシウムで乾燥後、濾過し、溶媒を濃縮した。濃縮残渣に、イソプロピルエーテル(24ml)を加えて溶解し、5℃に冷却して結晶を析出させた。析出晶を減圧濾過後、室温下で減圧乾燥し、2−フルオロ−5−ニトロ安息香酸メチル10.5gを得た。
【0035】
工程2 5−ニトロ−2−(2’,3’,4’,6’−テトラ−O−アセチル−α−D−ガラクトピラノシルオキシ)安息香酸メチル
工程1で得られた2−フルオロ−5−ニトロ安息香酸メチル7.13gと2’,3’,4’,6’−テトラ−O−アセチル−D−ガラクトピラノース12.50gのアセトニトリル(70ml)溶液にDBU4.95gを滴下し、室温にて2時間撹拌した。反応液を減圧濃縮し、酢酸エチル(300ml)で希釈し、1N塩酸(150ml)、5%炭酸水素ナトリウム水溶液(150ml)、飽和食塩水(150ml)で洗浄し、無水硫酸マグネシウムで乾燥後、濾過し、溶媒を濃縮した。濃縮物は、カラムクロマト(Wako gel(登録商標)C−200(和光純薬(株)製),n−へキサン:酢酸エチル=4〜1.5)で精製し、5−ニトロ−2−(2’,3’,4’,6’−テトラ−O−アセチル−α−D−ガラクトピラノシルオキシ)安息香酸メチル7.51g及び5−ニトロ−2−(2’,3’,4’,6’−テトラ−O−アセチル−β−D−ガラクトピラノシルオキシ)安息香酸メチル7.92gを得た。
【0036】
工程3−1 5−アミノ−2−(2’,3’,4’,6’−テトラ−O−アセチル−α−D−ガラクトピラノシルオキシ)安息香酸メチル
工程2−1で得られた5−ニトロ−2−(2’,3’,4’,6’−テトラ−O−アセチル−α−D−ガラクトピラノシルオキシ)安息香酸メチル7.00gのメタノール(210ml)溶液に10 %パラジウム炭素(0.70g)を加え、水素1気圧、室温で接触還元反応を行った。18時間後、反応液を濾過して触媒を除き、有機層を濃縮した。濃縮物は、カラムクロマト(Wako gel(登録商標)C−200(和光純薬(株)製),n−へキサン:酢酸エチル=3:1〜3:2)で精製し、5−アミノ−2−(2’,3’,4’,6’−テトラ−O−アセチル−α−D−ガラクトピラノシルオキシ)安息香酸メチル5.73gを得た。
【0037】
工程3−2 5−アミノ−2−(α−D−ガラクトピラノシルオキシ)安息香酸メチル
工程3−1で得られた5−アミノ−2−(2’,3’,4’,6’−テトラ−O−アセチル−α−D−ガラクトピラノシルオキシ)安息香酸メチル5.52gの無水テトラヒドロフラン−無水メタノール(1:1,110ml)に炭酸カリウム(307mg)を加え、室温にて15時間撹拌した。反応液を減圧濃縮し、濃縮物をカラムクロマト(Wako gel(登録商標)C−200(和光純薬(株)製), クロロホルム:メタノール=10:1〜5:1)で精製し、5−アミノ−2−(α−D−ガラクトピラノシルオキシ)安息香酸メチル2.71gを得た。
【0038】
工程4 5−アミノ−2−(α−D−ガラクトピラノシルオキシ)安息香酸
工程3−2で得られた5−アミノ−2−(α−D−ガラクトピラノシルオキシ)安息香酸メチル2.00gの水(40ml)懸濁液に、1N水酸化ナトリウム水溶液(6.07ml)を滴下し、50℃において2時間攪拌した。反応液を濾過して不溶物を除き、濾液に1N塩酸(6.07ml)を加えて中和した。反応液を減圧濃縮し、目的化合物1.34gを得た。
微黄色粉末物
MS(FAB) m/z=316[M+1]+
旋光度 [α]D20=79.37(C=1.28,H2O)
元素分析値 (C13H17NO8・0.8H2Oとして)
計算値(%) C:47.36 H:5.69 N:4.25
実測値(%) C:47.20 H:5.48 N:4.22
1H NMR (D2O)
3.70〜4.10(m,6H,H−2〜6),5.76(d,1H,J1,2=3.6Hz,H−1),7.37〜7.40(m,3H,Ph)
【0039】
参考例1 5−アミノ−2−(β−D−グルコピラノシルオキシ)安息香酸
工程1 5−ニトロサリチル酸メチル
実施例1の工程1と同様の方法を用いて合成した。
【0040】
工程2 5−ニトロ−2−(2’,3’,4’,6’−テトラ−O−アセチル−β−D−グルコピラノシルオキシ)安息香酸メチル
工程1で得られた5−ニトロサリチル酸メチル6.0gと2’,3’,4’,6’−テトラ−O−アセチル−α−D−グルコピラノシルブロミド18.8gとのキノリン(60ml)溶液に、酸化銀(10.5g)を加え室温で1時間激しく攪拌した。反応混合物を酢酸エチル(300ml)で希釈した後、セライト濾過した。酢酸エチル層を2N塩酸(2ml)で2回洗浄した後、水層を酢酸エチル(300ml)で2回抽出した。まとめた有機層を飽和重曹水、水、飽和食塩水で洗浄後、硫酸ナトリウムで乾燥後、濾過し、溶媒を濃縮し、5−ニトロ−2−(2’,3’,4’,6’−テトラ−O−アセチル−β−D−グルコピラノシルオキシ)安息香酸メチル 15.63gを得た。
【0041】
工程3−1 5−アミノ−2−(2’,3’,4’,6’−テトラ−O−アセチル−β−D−グルコピラノシルオキシ)安息香酸メチル
工程2で得られた5−ニトロ−2−(2’,3’,4’,6’−テトラ−O−アセチル−β−D−グルコピラノシルオキシ)安息香酸メチル12.0gのメタノール(400ml)懸濁液に10%パラジウム炭素(2.4g)を加え,水素3atm、30℃で接触還元反応を行った。3時間後、反応液をセライト濾過した。溶媒を濃縮し、5−アミノ−2−(2’,3’,4’,6’−テトラ−O−アセチル−β−D−グルコピラノシルオキシ)安息香酸メチル 11.2gを得た。
【0042】
工程3−2 5−アミノ−2−(β−D−グルコピラノシルオキシ)安息香酸メチル
工程3−1で得られた5−アミノ−2−(2’,3’,4’,6’−テトラ−O−アセチル−β−D−グルコピラノシルオキシ)安息香酸メチル0.68gの無水テトラヒドロフラン−メタノール(1:1,16ml)溶液に炭酸カリウム(37.8mg)を加え、室温で終夜攪拌した。反応液に4N塩化水素酢酸エチル溶液(0.14ml)を滴下し、そのまま溶媒を濃縮した。得られた粗成績体をシリカゲルカラムクロマトグラフィー(Wako gel(登録商標)C−200(和光純薬(株)製),塩化メチレン:メタノール=10:1〜8:1〜5:1)で精製し、5−アミノ−2−(β−D−グルコピラノシルオキシ)安息香酸メチル332mgを得た。
【0043】
工程4 5−アミノ−2−(β−D−グルコピラノシルオキシ)安息香酸
工程3−2で得られた5−アミノ−2−(β−D−グルコピラノシルオキシ)安息香酸メチル100mgのメタノール(3ml)懸濁液に、1N水酸化ナトリウム水溶液(0.3ml)を滴下し、50℃で攪拌した。5時間後室温に戻し溶媒を減圧留去した。得られた油状残渣を水1mlに溶解し氷冷下攪拌しながら、1N塩酸(0.3ml)を滴下した。この溶液を約3分の1の量まで濃縮し析出物を濾取して目的化合物93mgを得た。
元素分析値 (C13H17NO8・0.2H2Oとして)
計算値(%) C:48.97 H:5.50 N:4.39
実測値(%) C:48.80 H:5.35 N:4.31
【0044】
試験例1 血漿中5−ASA濃度の測定
7週齢のSD系雄性ラットに被験薬物として5−ASAを静脈内投与し、また化合物〔1〕、化合物〔2〕、化合物〔3〕及びペンタサ(登録商標)を5−ASAに換算して50mg/kg経口投与し、血漿中5−ASA濃度を高速液体クロマトグラフィー(HPLC)で測定した。なお、ペンタサ(登録商標)は、ペンタサ(登録商標)錠を粉砕して顆粒にしたものを使用した。
結果を表1に示す。
【表1】
上記結果から、ペンタサ(登録商標)を経口投与した場合、血漿中には5−ASAが比較的高濃度で検出された(図1参照)。本試験例の結果は、吸収部位である小腸上部において、投与されたペンタサの一部から5−ASAが放出されていることを示している。
一方、5−ASAの糖誘導体である化合物〔1〕、化合物〔2〕及び化合物〔3〕を経口投与した場合、血漿中5−ASA濃度は、ペンタサ(登録商標)に比べて低く推移した(図1参照)。また、化合物〔1〕及び化合物〔2〕を投与した場合、血漿中5−ASA濃度は、化合物〔3〕を投与した場合より低く推移し、5−ASAはほとんど検出されなかった(図1参照)。化合物〔1〕、化合物〔2〕、化合物〔3〕及びペンタサ(登録商標)を経口投与した場合の5−ASAの生物学的利用能(bioavailability)(表1参照)は、各々2%,0.8%,6%及び15%と算出された。すなわち、化合物〔1〕、化合物〔2〕及び化合物〔3〕は、ペンタサと比較して消化管からの吸収率が低いことが判明した。特に、化合物〔1〕及び化合物〔2〕は、生物学的利用能が著しく低いものであった。
この原因として、化合物〔1〕及び化合物〔2〕が化合物〔3〕に比べ胃や小腸において加水分解されにくく、胃や小腸上部において5−ASAを生成しないためと考えられる。
【0045】
試験例2 消化管内容物中の5−ASA濃度推移
7週齢のSD系雄性ラットに被験薬物として化合物〔1〕、ペンタサ(登録商標)及び5−ASAを5−ASAに換算して50mg/kg経口投与し、盲腸、近位結腸、遠位結腸および直腸内の内容物をホモジネートし、遠心分離後その上清を用いて、高速液体クロマトグラフィー(HPLC)により大腸各部位における5−ASA量を測定した。なお、ペンタサ(登録商標)は、ペンタサ(登録商標)錠を粉砕して顆粒にしたものを使用した。
結果を図2から図5に示す。
大腸各部位における5−ASA量は、化合物〔1〕を投与した時が最も高かった(図2,3,4,5参照)。
また、同様に被験薬物として化合物〔2〕及びペンタサ(登録商標)を5−ASAに換算して50mg/kg経口投与し、結腸及び直腸をホモジネートし、遠心分離後その上清を用いて、高速液体クロマトグラフィー(HPLC)により結腸組織中及び直腸組織中における5−ASA濃度を測定した。なお、ペンタサ(登録商標)は、ペンタサ(登録商標)錠を粉砕して顆粒にしたものを使用した。
結果を図6及び図7に示す。
化合物〔2〕についても、ペンタサ(登録商標)と比較して結腸組織中及び直腸組織中における5−ASA濃度が高かった(図6,7参照)。
試験例1において、化合物〔1〕及び化合物〔2〕は、胃や小腸において加水分解されにくく、胃や小腸上部において5−ASAを生成せず、消化管からの吸収率が低いことが確認されている。
試験例2の結果より、化合物〔1〕及び化合物〔2〕は、疾患部位である大腸に到達し、腸内細菌により5−ASAに代謝されることが明らかとなった。特に、化合物〔1〕は、大腸各部位において潰瘍性大腸炎の治療に有効な5−ASAが高濃度で検出された。
【0046】
試験例3 ラットのトリニトロベンゼンスルホン酸(以下、「TNBS」という。)誘発大腸炎モデルにおける化合物〔1〕の有効性の検討
24時間絶食した雌性SDラットにペントバルビタール麻酔下、TNBS/50%エタノール水溶液(30mg/0.25ml/ラット)を肛門から8cmの大腸内に経口投与用ゾンデを用いて投与することにより大腸炎を惹起した。TNBS投与3日後に大腸を摘出し、肛門から8cmの大腸の湿重量を測定すると共に、大腸炎発症の程度はWallaceら(Gastroenterology,96, p.29−36(1989))の方法に従いスコア化した。被験化合物として、ペンタサ(登録商標)は30,100mg/kg、化合物〔1〕は61.8,205.9mg/kg(5−ASA換算で30,100mg/kgに相当)の用量を1日2回(TNBS投与日は、TNBS投与4時間前1回のみ)経口投与した。なお、ペンタサ(登録商標)は、ペンタサ(登録商標)錠を粉砕して顆粒にしたものを使用した。
結果を図8及び図9に示す。
化合物〔1〕は、ダメージスコアを61.8mg/kg(5−ASAに換算して30mg/kgに相当)の用量において有意に抑制し、大腸湿重量を205.9mg/kg(5−ASAに換算して100mg/kgに相当)の用量において有意に抑制した(図8,9参照)。一方、ペンタサ(登録商標)はダメージスコアおよび大腸湿重量において共に明らかな作用を示さなかった。
【0047】
製剤例1 散剤(内服剤)
処方1剤700mg中
化合物〔1〕 500mg
トウモロコシ澱粉 127mg
結晶セルロース 35mg
ポリビニルアルコール 35mg
ステアリン酸マグネシウム 3mg
化合物〔1〕250g、トウモロコシ澱粉63.5g、結晶セルロース17.5gを流動層造粒乾燥機に投入し、ポリビニルアルコールの10%水溶液(175ml)を噴霧して造粒する。これにステアリン酸マグネシウム(0.4%(w/w))を加えて、700mg中に本化合物500mgを含む散剤を得る。
【0048】
製剤例2 錠剤(内服剤)
処方1剤600mg中
化合物〔1〕 400mg
トウモロコシ澱粉 153mg
結晶セルロース 42mg
ステアリン酸マグネシウム 5mg
化合物〔1〕400g、トウモロコシ澱粉153g、結晶セルロース42gの混合末を乾式造粒機で圧縮した後、顆粒状に解砕する。これにステアリン酸マグネシウム(0.8%(w/w))を加えて1錠重量600mg、錠径11mmに成形し、本化合物400mgを含む錠剤を得る。
【0049】
製剤例3 カプセル剤(内服剤)
処方1剤500mg中
化合物〔1〕 250mg
無水リン酸水素カルシウム
222.5mg
クロスカルメロースナトリウム 25mg
ステアリン酸マグネシウム 2.5mg
化合物〔1〕250g、無水リン酸水素カルシウム222.5g、クロスカルメロースナトリウム25gの混合末を乾式造粒機で圧縮した後、顆粒状に解砕する。これにステアリン酸マグネシウム(0.5%(w/w))を加えて500mgを0号硬カプセルに充填し、本化合物250mgを含むカプセル剤を得る。
【0050】
製剤例4 円柱状顆粒(内服剤)
処方1剤1000mg中
化合物〔1〕 750mg
トウモロコシ澱粉 170mg
結晶セルロース 50mg
ポリビニルアルコール 30mg
化合物〔1〕375g、トウモロコシ澱粉85g、結晶セルロース25gをニーダーに投入し、ポリビニルアルコールの12%水溶液(125ml)を加えて練合後、0.7mm径のスクリーンを装着した顆粒押出し成形機で押出する。これを乾燥して整粒し、1000mg中に本化合物750mgを含む顆粒剤を得る。
【0051】
製剤例5 球形コーティング顆粒(内服剤)
処方1剤1000mg中
ノンパレル 200mg
化合物〔1〕 500mg
トウモロコシ澱粉 170mg
低置換度ヒドロキシプロピルセルロース 40mg
ヒドロキシプロピルセルロース 50mg
ヒドロキシプロピルメチルセルロース 30mg
プロピレングリコール 6mg
酸化チタン 4mg
ノンパレル(24〜32メッシュ)200gを遠心流動造粒コーティング装置に投入し、ヒドロキシプロピルセルロースの8%水溶液(50%エタノール)を噴霧しながら、化合物〔1〕500g、トウモロコシ澱粉170g、低置換度ヒドロキシプロピルセルロース40gの混合末を徐々に添加して造粒し、乾燥して約900gの球状素顆粒を得る。
次に、この球状素顆粒400gを流動層造粒乾燥機に投入し、ヒドロキシプロピルメチルセルロース12.5g、プロピレングリコール2.5g、酸化チタン1.7gを含む水溶液(250ml)を噴霧して1000mg中に本化合物500mgを含むコーティング顆粒を得る。
【産業上の利用可能性】
【0052】
本発明化合物は、潰瘍性大腸炎の治療に有効な5−ASAを効率よく作用部位である大腸まで到達させることができ、また5−ASAを血漿中に移行させないという特徴を有する。即ち、全身性の副作用の低減を図ることができ、最大治療効果を得られるまで投与量を上げることも可能である。
【特許請求の範囲】
【請求項1】
5−アミノ−2−(β−D−ガラクトピラノシルオキシ)安息香酸若しくは5−アミノ−2−(α−D−ガラクトピラノシルオキシ)安息香酸又はその医薬上許容される塩。
【請求項2】
5−アミノ−2−(β−D−ガラクトピラノシルオキシ)安息香酸若しくは5−アミノ−2−(α−D−ガラクトピラノシルオキシ)安息香酸又はその医薬上許容される塩を有効成分として含有する医薬組成物。
【請求項3】
5−アミノ−2−(β−D−ガラクトピラノシルオキシ)安息香酸、5−アミノ−2−(α−D−ガラクトピラノシルオキシ)安息香酸若しくは5−アミノ−2−(β−D−グルコピラノシルオキシ)安息香酸又はその医薬上許容される塩を有効成分として含有する潰瘍性大腸炎治療剤。
【請求項1】
5−アミノ−2−(β−D−ガラクトピラノシルオキシ)安息香酸若しくは5−アミノ−2−(α−D−ガラクトピラノシルオキシ)安息香酸又はその医薬上許容される塩。
【請求項2】
5−アミノ−2−(β−D−ガラクトピラノシルオキシ)安息香酸若しくは5−アミノ−2−(α−D−ガラクトピラノシルオキシ)安息香酸又はその医薬上許容される塩を有効成分として含有する医薬組成物。
【請求項3】
5−アミノ−2−(β−D−ガラクトピラノシルオキシ)安息香酸、5−アミノ−2−(α−D−ガラクトピラノシルオキシ)安息香酸若しくは5−アミノ−2−(β−D−グルコピラノシルオキシ)安息香酸又はその医薬上許容される塩を有効成分として含有する潰瘍性大腸炎治療剤。
【図1】

【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【国際公開番号】WO2005/075492
【国際公開日】平成17年8月18日(2005.8.18)
【発行日】平成19年10月11日(2007.10.11)
【国際特許分類】
【出願番号】特願2005−517698(P2005−517698)
【国際出願番号】PCT/JP2005/001492
【国際出願日】平成17年2月2日(2005.2.2)
【出願人】(000004156)日本新薬株式会社 (46)
【Fターム(参考)】
【国際公開日】平成17年8月18日(2005.8.18)
【発行日】平成19年10月11日(2007.10.11)
【国際特許分類】
【国際出願番号】PCT/JP2005/001492
【国際出願日】平成17年2月2日(2005.2.2)
【出願人】(000004156)日本新薬株式会社 (46)
【Fターム(参考)】
[ Back to top ]