
5−尿素置換ナフタルイミド誘導体、製造方法、および癌治療用医薬組成物
新規なウレイル置換ナフタルイミド誘導体、薬学的に許容されるそれらの塩およびそれらの溶媒和物は、癌などの細胞増殖性疾患の治療のための医薬組成物の製造に有用である。本発明は既知の化合物の加水分解によるこのような誘導体の製造方法も提供する。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、新規な置換ナフタルイミド誘導体、それらの製造方法、ならびに、特に、様々な形態の癌の予防および/または治療における活性成分としてそれらを含む医薬組成物の形態の、抗腫瘍剤としてのそれらの医薬用途に関する。
【背景技術】
【0002】
アモナフィドを含めた様々な種類の置換ナフタルイミドは、抗腫瘍作用または他の有用な生物活性を有するとして当技術分野において知られている。特にWO2005/105753号は、癌などの細胞増殖性疾患の治療において活性を有する特異的な置換パターンを有するナフタルイミド誘導体を開示している。
【0003】
アモナフィドで見い出された活性のレベルは非常に興味深いものであったが、そしてそうあり続けているが、この材料は、重大な欠陥を有し、これは改善された特性を備える薬剤の継続的なニーズを示している。第1に、アモナフィドはある患者には毒性が強すぎることが見い出され、特に、薬剤の5日用量を摂取した患者が死に至ることもある実質的な骨髄毒性を生じる。さらに、アモナフィドは、マウスの白血病モデルにおいて中程度の活性しか有さないことが示された。また、アモナフィドは、結腸癌、肺癌および乳癌の、マウスにおけるヒトの移植腫瘍に活性を有さないことが示された。したがって、アモナフィドは顕著な生物活性を示すが、マウス腫瘍モデルにおいて実質的に広範囲の活性を有するものではない。Ajaniらは、Invest New Drugs (1988) 6:79-83において、アモナフィドは、インビトロで原発性ヒト充実性腫瘍において試験された場合に乏しい活性しか有さないことを示した。
【0004】
特定の形態の癌に対するアモナフィドなどの抗増殖剤の臨床活性が示されているが、腫瘍応答率、応答期間、骨髄毒性の減少および最終的に患者生存の改善が依然として求められている。当技術分野において、新規薬物と通常の抗腫瘍薬の適切な組合せを提供することによってヒトにおける抗増殖治療の有効性を改善することも必要とされている。
【発明の開示】
【発明が解決しようとする課題】
【0005】
アモナフィドなどの上記の欠点を考慮すると、当技術分野において、より有望な活性/副作用のバランスを示すナフタルイミド誘導体のニーズがある。
【課題を解決するための手段】
【0006】
本発明は、ナフタルイミジル部分の5位におけるウレイル基で置換されたナフタルイミド誘導体が、細胞増殖性障害の治療に有用であり、限定された数の反応ステップを通して効率的に製造することができ、先に知られた類似の誘導体の欠点のいくつかを示さないという最初の予想外の知見に基づくものである。本発明は、このようなウレイル置換されたナフタルイミド誘導体が、他の既知の置換ナフタルイミド誘導体の加水分解を通して高収率で容易に入手できるという予想外の知見にも基づいている。本発明は、このようなウレイル置換されたナフタルイミド誘導体が、十分な化学安定性を示し、例えば、ナノ粒子の形態の懸濁液としてまたは塩の形態の溶液として、薬剤に容易に製剤化することができるという予想外の知見にも基づいている。
【発明を実施するための最良の形態】
【0007】
定義
特に指定のない限り、置換基に関して本明細書で使用される用語「アルキル」は、1から4個の炭素原子を有する直鎖および分枝鎖の飽和非環式炭化水素の一価の基、例えば、メチル、エチル、プロピル、n−ブチル、1−メチルエチル(イソプロピル)、2−メチルプロピル(イソブチル)、および1,1−ジメチルエチル(ter−ブチル)などを意味する。
【0008】
特に指定のない限り、置換基のメンバーに関して本明細書で使用される用語「アルキレン」は、上記に定義されたアルキルに対応する二価の炭化水素基、限定されるものではないが、メチレン、ビス(メチレン)、トリス(メチレン)、テトラメチレンなどを意味する。
【0009】
特に指定のない限り、置換基に関して本明細書で使用される用語「アルコキシ」および「アルキルチオ」は、本明細書で上記に定義されたアルキル基が、単結合によって酸素原子または二価の硫黄原子に結合している置換基、限定されるものではないが、メトキシ、エトキシ、プロポキシ、ブトキシ、イソプロポキシ、sec−ブトキシ、tert−ブトキシ、チオメチル、チオエチル、チオプロピル、チオブチルなどを意味する。
【0010】
特に指定のない限り、置換原子に関して本明細書で使用される用語「ハロゲン」は、フッ素、塩素、臭素およびヨウ素からなる群から選択される任意の原子を意味する。
【0011】
特に指定のない限り、置換基に関して本明細書で使用される用語「ハロアルキル」は、1個または複数の水素原子が独立に1個または複数のハロゲン(好ましくはフッ素、塩素または臭素)で置換されているアルキル基(上記に定義されたようなもの)、限定されるものではないが、ジフルオロメチル、トリフルオロメチル、トリフルオロエチル、ジクロロメチルなどを意味する。
【0012】
特に指定のない限り、本明細書で使用される用語「溶媒和物」には、適切な無機溶媒(例えば、水から形成される水和物)または適切な有機溶媒、限定されるものではないが、アルコール、ケトン、エステルなどと、本発明のウレイル置換ナフタルイミド(イソキノリンジオン)誘導体とによって形成され得る任意の組合せが含まれる。
【0013】
特に指定のない限り、本明細書で使用される用語「抗転移性」は、新生物腫瘍組織からの細胞の転移を停止させ、それゆえこれらの細胞による新たな組織の定着を減少させる医薬成分の能力を意味する。
【0014】
本明細書で使用される用語「細胞増殖性障害」は、任意のタイプの癌または細胞増殖を伴う他の病態、限定されるものではないが、白血病、肺癌、結腸直腸癌、中枢神経系(CNS)癌、メラノーマ、卵巣癌、腎癌、前立腺癌、乳癌、神経膠腫、膀胱癌、骨癌、肉腫、頭頸部癌、肝癌、精巣癌、膵癌、胃癌、食道癌、骨髄癌、十二指腸癌、眼癌(網膜芽腫)およびリンパ腫などを意味する。
【0015】
第1の態様においては、本発明は、構造式(I)により表される置換ナフタルイミド誘導体:
【化1】

【0016】
[式中、
− R1はモノまたはジ−C1〜4アルキルアミノ−C1〜4アルキルであり;
− 置換基R3およびR4のそれぞれは、水素、ハロゲン、C1〜4アルキル、C1−7アルコキシ、C1〜4アルキルチオ、ニトロ、シアノ、アミノ、保護されたアミノおよびハロC1〜4アルキルからなる群から独立に選択され;
− mは置換基R3の数であって、0から3の範囲であり;
− nは置換基R4の数であって、0から2の範囲であり;
− R2はCONH2である。]、
ならびに/または薬学的に許容されるそれらの塩および/もしくはそれらの溶媒和物および/もしくはそれらの代謝産物のグループを提供する。
【0017】
構造式(I)により表される誘導体の代謝産物には、限定されるものではないが、次のもの:
− それらのモノ−N−オキシドおよびジ−N−オキシド;
− R2がCONHOHである誘導体;ならびに
− R3および/またはR4がヒドロキシルである誘導体
が含まれる。
【0018】
あるいは、本発明のナフタルイミド誘導体のモノ−およびジ−N−オキシドは、限定されるものではないが、過酸化水素(例えば酢酸の存在下で)またはクロロ過安息香酸などの過酸などの酸化剤で構造式(I)により表される誘導体を処理することによって直接合成することができる。
【0019】
上記に定義された新規な化合物は、共通して、限定されるものではないが、アモナフィドなどのアミノ置換ナフタルイミド(イソキノリンジオン)のアミノ基がウレイル基、またはその代謝形態においてウレイルN−オキシド基で置換されているという、構造的な特徴を有する。
【0020】
この第1の態様の好ましい実施形態においては、本発明は、
− n=0(R4が水素ではない場合)であり、かつ/または
− m=0(R3が水素ではない場合)であり、かつ/または
− m=2であり、両方の置換基R3は隣接しており、それらが結合している炭素原子と一緒になってフェニル基を形成し、かつ/または
− R1は1から3個の炭素原子を有するアルキレン基であり、ジメチルアミノもしくはジエチルアミノ基に連結しており、かつ/または
− R2はCONH2
である化合物、ならびに/または薬学的に許容されるそれらの塩および/もしくはそれらの溶媒和物および/もしくはそれらの代謝産物のサブグループに関する。
【0021】
この第1の態様の別の好ましい実施形態においては、本発明は、
− n=m=0(R3およびR4が水素ではない場合)であり、かつ/または
− R1は1もしくは2個の炭素原子を有するアルキレン基であり、ジメチルアミノもしくはジエチルアミノ基に連結しており、かつ/または
− R2はCONH2
である化合物、ならびに/または薬学的に許容されるそれらの塩および/もしくはそれらの溶媒和物および/もしくはそれらの代謝産物のサブグループに関する。
【0022】
この第1の態様のさらに別の好ましい実施形態においては、本発明は、mが1に等しい場合にR3がニトロではない化合物、それらの塩、溶媒和物または代謝産物のサブグループに関する。この第1の態様のさらに別の好ましい実施形態においては、本発明は、R3および/またはR4が、水素、ハロゲン、C1〜4アルキル、C1−7アルコキシ、C1〜4アルキルチオ、シアノ、アミノ、アシルアミノおよびハロC1〜4アルキルからなる群から選択される化合物、それらの塩、溶媒和物または代謝産物のサブグループに関する。
【0023】
別の好ましい実施形態においては、本発明は、N−{2−[2−(ジメチルアミノ)エチル]−1,3−ジオキソ−2,3−ジヒドロ−1H−ベンゾ[de]イソキノリン−5−イル}尿素、その塩または代謝産物に関する。
【0024】
第2の態様においては、本発明は、5−置換アモナフィドまたはアモナフィド誘導体(ここで、その5置換基は、それが加水分解によりウレイル基に変換できるように選択される)を加水分解することによる、構造式(I)により表されるウレイル置換ナフタルイミド(イソキノリンジオン)誘導体の製造方法を提供する。加水分解に適する5−置換アモナフィド誘導体には、限定されるものではないが、構造式(II)を有する化合物:
【化2】
【0025】
[式中、
− m、n、R1、R3およびR4のそれぞれは、構造式(I)について定義された通りであり、
− R’はC1〜4アルコキシアミドカルボニルまたはC1ハロアルキルアミドカルボニルである]
が含まれる。
【0026】
上記の構造式(II)を有するいくつかの化合物は、例えばWO2005/105753号から既に知られるが、例えばC1〜4アルコキシカルボニルイソシアネート(エトキシカルボニルイソシアネートなど)またはC1ハロアルキルカルボニルイソシアネート(トリクロロアセチルイソシアネートもしくはトリフルオロアセチルイソシアネートなど)とアモナフィドとの反応の生成物として、中程度の収率でしか入手可能ではなかった。したがって、本発明の別の態様は、より良い収率でこれらの中間体を入手することを可能にする反応条件を設計することである。この目的のための1つの方法は、C1〜4アルコキシカルボニルイソシアネートまたはC1ハロアルキルカルボニルイソシアネートとアモナフィドとの前記反応が、
− エーテル(例えばジエチルエーテル)、ケトン(例えば2−ブタノンまたはメチルエチルケトン)およびハロゲン化炭化水素(好ましくは最大2個の炭素原子および/または少なくとも1個の塩素原子を有するもの、例えばジクロロメタン)からなる群から選択される溶媒の存在、ならびに/あるいは
− 0℃未満の温度(例えば約−30℃から約−5℃の範囲の温度)、ならびに/あるいは
− 前記C1〜4アルコキシカルボニルイソシアネートまたはC1ハロアルキルカルボニルイソシアネートのモル過剰量、ならびに/あるいは
− その完了後に反応混合物に水を添加することにより反応をクエンチし、望ましくない環化副生成物の形成(C1〜4アルコキシカルボニルイソシアネートまたはC1ハロアルキルカルボニルイソシアネートのモル過剰量が使用される場合)を回避すること
を含む条件下で実施される方法である。
【0027】
上記反応条件の1つまたは複数が使用される場合、上記構造式(II)を有する化合物は、先行技術によるよりも同一またはより短い反応時間内で著しく向上した収率で得ることができる。当業者は、前述のプロセスの特徴のどの組合せが、R’、R1、R3およびR4の正確な性質などのパラメーターに応じて、可能な限り最短の反応時間内で最も適当な収率を提供し得るかを容易に決定することができる。
【0028】
限定されるものではないが、構造式(II)を有する化合物などの、その5−置換基がウレイル基に変換され得る5−置換アモナフィドまたはアモナフィド誘導体の加水分解は、酸性条件下または塩基性条件下のいずれかで実施し得る。当業者は、この種の加水分解は、限定されるものではないが、pH、温度、使用される酸または塩基の種類および反応混合物のための溶媒の種類などのパラメーターによって、副生成物としてアモナフィドを生成しやすく、このアモナフィドは、次いで、構造式(I)を有する所望の化合物から分離する必要があることを容易に理解されよう。アモナフィドの形成を最小化するための最適な条件の決定は、当業者の一般的知識に含まれる。本発明の1つの利点は、最終生成物中の残留アモナフィドの割合を3重量%未満に保つことは極めて容易であることがわかったことである。
【0029】
第3の態様においては、本発明は、
− 構造式(I)により表されるウレイル置換ナフタルイミド(イソキノリンジオン)誘導体、ならびに/または薬学的に許容されるその塩および/もしくはその溶媒和物および/もしくはその代謝産物の治療有効量;ならびに
− 1種または複数の薬学的に許容される担体
を含む医薬組成物を提供する。
【0030】
別の態様においては、本発明は、少なくとも1種の構造式(I)により表されるウレイル置換ナフタルイミド(イソキノリンジオン)誘導体、ならびに/または薬学的に許容されるその塩および/もしくはその溶媒和物および/もしくはその代謝産物と、1種または複数の抗新生物薬とを含有する組合せ製剤(好ましくは下記に詳述される相乗的組合せの形態における)を提供する。
【0031】
別の態様においては、本発明は、一般式(I)で表される置換ナフタルイミド(イソキノリンジオン)誘導体、ならびに/または薬学的に許容されるそれらの塩および/もしくはそれらの溶媒和物および/もしくはそれらの代謝産物が、アモナフィドの上述の欠点の多くを回避しながら、アモナフィドに比べて、特に腫瘍細胞に関して、著しくより高い生物活性を有するという予想外の知見に関する。特に、本発明に係るウレイル置換ナフタルイミド誘導体は、著しい抗転移効果を有する。転移は、細胞が、転移の主要な経路として血管またはリンパ管を使用して、新生物腫瘍組織から移動し、新組織を定着する生物学的プロセスを意味し、この生物学的プロセスは、転移プロセスとしても知られている。この知見に基づき、本発明は、ヒトの腫瘍の治療および/または予防方法を提供する。より具体的には、本発明は、細胞増殖性疾患を有する宿主の治療方法であって、前記宿主に、構造式(I)により表されるウレイル置換ナフタルイミド(イソキノリンジオン)誘導体、ならびに/または薬学的に許容されるそれらの塩および/もしくはそれらの溶媒和物および/もしくはそれらの代謝産物の有効量を接触させることを含む方法に関する。
【0032】
別の実施形態においては、本発明は、抗腫瘍剤としての、構造式(I)により表されるウレイル置換ナフタルイミド(イソキノリンジオン)誘導体、ならびに/または薬学的に許容されるそれらの塩および/もしくはそれらの溶媒和物および/もしくはそれらの代謝産物の使用を提供する。
【0033】
別の特定の実施形態においては、本発明は、上記構造式(I)を有し、薬学的に許容される塩の形態である、ウレイル置換ナフタルイミド(イソキノリンジオン)誘導体、ならびにそれらを活性成分として含む医薬組成物のグループに関する。後者には、構造式(I)を有する化合物が、塩形成薬剤と形成し得る任意の治療活性を有する非毒性塩が含まれる。このような付加塩は、本発明のウレイル置換ナフタルイミド(イソキノリンジオン)誘導体を適切な塩形成酸または塩基で処理することにより都合よく得ることができる。例えば、塩基性を有するウレイル置換ナフタルイミド(イソキノリンジオン)誘導体は、通常の手法に従い遊離塩基形態を適量の適切な酸で処理することによって対応する治療活性を有する非毒性酸塩の形態に変換し得る。このような適切な塩形成酸の例には、例えば、限定されるものではないが、塩の形成をもたらす無機酸、例えばハロゲン化水素酸塩(例えば塩酸塩および臭化水素酸塩)、硫酸塩、硝酸塩、リン酸塩、二リン酸塩、炭酸塩、重炭酸塩など;および、塩の形成をもたらす有機モノカルボン酸またはジカルボン酸、例えば酢酸塩、プロピオン酸塩、ヒドロキシ酢酸塩、2−ヒドロキシプロピオン酸塩、2−オキソプロピオン酸塩、乳酸塩、ピルビン酸塩、シュウ酸塩、マロン酸塩、コハク酸塩、マレイン酸塩、フマル酸塩、リンゴ酸塩、酒石酸塩、クエン酸塩、メタンスルホン酸塩、エタンスルホン酸塩、安息香酸塩、2−ヒドロキシ安息香酸塩、4−アミノ−2−ヒドロキシ安息香酸塩、ベンゼンスルホン酸塩、p−トルエン−スルホン酸塩、サリチル酸塩、p−アミノサリチル酸塩、パモ酸塩、重酒石酸塩、カンファースルホン酸塩、エデト酸塩、1,2−エタンジスルホン酸塩、フマル酸塩、グルコヘプタン酸塩、グルコン酸塩、グルタミン酸塩、ヘキシルレゾルシン酸塩、ヒドロキシナフトエ酸塩、ヒドロキシエタンスルホン酸塩、マンデル酸塩、メチル硫酸塩、パントテン酸塩、ステアリン酸塩など、ならびにエタン二酸、プロパン二酸、ブタン二酸、(Z)−2−ブテン二酸、(E)2−ブテン二酸、2−ヒドロキシブタン二酸、2,3−ジヒドロキシブタン二酸、2−ヒドロキシ−1,2,3−プロパン−トリカルボン酸、シクロヘキサン−スルファミン酸などから誘導される塩、が含まれる。
【0034】
酸性を有する構造式(I)を有するウレイル置換ナフタルイミド(イソキノリンジオン)誘導体は、同様の方法で対応する治療活性を有する非毒性塩基塩の形態に変換し得る。適切な塩形成塩基の例には、例えば、金属水酸化物のような無機塩基、限定されるものではないが、対応する金属塩をもたらす、カルシウム、リチウム、マグネシウム、カリウムおよびナトリウムなどのアルカリ金属およびアルカリ土類金属、または亜鉛の水酸化物など;窒素含有有機塩基、限定されるものではないが、アンモニア、アルキルアミン、ベンザチン、ヒドラバミン、アルギニン、リシン、N,N’−ジベンジルエチレンジアミン、クロロプロカイン、コリン、ジエタノールアミン、エチレンジアミン、N−メチルグルカミン、プロカインなどが含まれる。
【0035】
適切な塩形成酸または塩基で本発明のウレイル置換ナフタルイミド(イソキノリンジオン)誘導体(I)を処理するための反応条件は、それぞれ塩基性または酸性の異なる有機化合物を除いて、同一の酸または塩基が関与する標準条件と類似している。好ましくは、細胞増殖性疾患の治療のための医薬組成物におけるその使用または薬剤の製造におけるその使用を考慮して、薬学的に許容される塩は設計され、すなわち、塩形成酸または塩基は、本発明のウレイル置換ナフタルイミド(イソキノリンジオン)誘導体に、より大きな水溶性、より低い毒性、より大きな安定性および/またはより遅い溶出速度を付与するように選択される。
【0036】
本発明は、生物活性成分、すなわち活性成分(特に薬剤または診断薬)としての、あるいは薬剤または診断用キットの製造のための、構造式(I)により表されるウレイル置換ナフタルイミド(イソキノリンジオン)誘導体、または薬学的に許容されるそれらの塩もしくはそれらの溶媒和物および/もしくはそれらの代謝産物の使用をさらに提供する。特に、前記薬剤は、細胞増殖性障害からなる群から選択される病態の予防または治療用であり得る。
【0037】
本発明に係る化合物は、いくつかのタイプの癌に対して極めて活性である。したがって、それらの好ましい薬理学的特性のために、本発明に係る化合物は、細胞増殖に関係する疾患を患う患者の治療、特に癌の治療のための薬剤としての使用、または薬剤および組合せ製剤の製造における使用に特に適している。
【0038】
上記に記載された使用のいずれも、非医学的用途(例えば化粧用組成物における)、非治療用途、非診断用途、ヒト以外の用途(例えば獣医用組成物)、または専らインビトロでの使用、または動物から離れた細胞での使用に制限されてもよい。
【0039】
本発明は、さらに
(a)1種または複数の構造式(I)で表されるウレイル置換ナフタルイミド(イソキノリンジオン)誘導体、ならびに/または薬学的に許容されるそれらの塩および/もしくはそれらの溶媒和物および/もしくはそれらの代謝産物、ならびに
(b)1種または複数の薬学的に許容される担体
を含む医薬組成物に関する。
【0040】
別の実施形態においては、本発明は、1種または複数の構造式(I)で表されるウレイル置換ナフタルイミド(イソキノリンジオン)誘導体、ならびに/または薬学的に許容されるそれらの塩および/もしくはそれらの溶媒和物および/もしくはそれらの代謝産物と、抗新生物薬からなる群から選択されるのが好ましい1種または複数の生物活性薬物との組合せ製剤、好ましくは相乗的組合せを提供する。当技術分野で普通であるように、複合薬における相乗効果の評価は、ChouらによりAdv. Enzyme Reg. (1984) 22:27に記載された半有効原理を用いて個別の薬物の間の相互作用の定量化の分析によって行うことができる。簡単に説明すると、この原理は、2種の薬物の間の相互作用(相乗作用、相加作用、拮抗作用)は、次の方程式:
【数1】
【0041】
(式中、EDxは、所与の効果を生ずるために必要である、単独使用される第1もしくはそれぞれ第2の薬物(1a、2a)の用量、または第2もしくはそれぞれ第1の薬物(1c、2c)と組み合わせた用量である)
により定義される組合せ指数(以下CIという)を用いて定量化することができることを記載している。第1および第2の薬物は、それぞれCI<1、CI=1、またはCI>1に応じて、相乗作用または相加作用または拮抗作用を有する。本明細書で下記により詳しく説明されるように、この原理は、限定されるものではないが、細胞増殖に対する活性などのいくつかの所望の効果に適用され得る。
【0042】
本発明は、さらに、
(a)1種または複数の抗新生物薬、ならびに
(b)少なくとも1種の構造式(I)で表されるウレイル置換ナフタルイミド(イソキノリンジオン)誘導体、ならびに/または薬学的に許容されるそれらの塩および/もしくはそれらの溶媒和物および/もしくはそれらの代謝産物、ならびに
(c)場合によっては1種または複数の薬学的賦形剤または薬学的に許容される担体
を含有する、細胞増殖性障害の治療または予防における同時、別々または順次使用のための、組成物または細胞増殖に対する相乗効果を有する組合せ製剤に関する。
【0043】
本発明の相乗的な抗増殖医薬組成物または組合せ製剤に含まれるのに適する抗新生物薬は、好ましくは、アルカロイド、アルキル化剤(限定されるものではないが、アルキルスルホネート、アジリジン、エチレンイミン、メチルメラミン、ナイトロジェン・マスタードおよびニトロソ尿素を含む)、抗生物質、代謝拮抗物質(限定されるものではないが、葉酸類似体、プリン類似体およびピリミジン類似体を含む)、酵素、インターフェロンおよび白金錯体からなる群から選択される。より具体的な例には、アシビシン、アクラルビシン、アコダゾール、アクロニン、アドゼレシン、アルデスロイキン、アルトレタミン、アンボマイシン、アメタントロン、アミノグルテチミド、アムサクリン、アナストロゾール、アンスラマイシン、アスパラギナーゼ、アスペルリン、アザシチジン、アゼテパ、アゾトマイシン、バチマスタット、ベンゾデパ、ビカルタミド、ビサントレン、ビスナフィド、ビゼレシン、ブレオマイシン、ブレキナール、ブロピリミン、ブスルファン、カクチノマイシン、カルステロン、カラセミド、カルベチマー、カルボプラチン、カルムスチン、カルビシン、カルゼレシン、セデフィンゴール、クロラムブシル、シロレマイシン、シスプラチン、クラドリビン、クリスナトール、シクロホスファミド、シタラビン、ダカルバジン、ダクチノマイシン、ダウノルビシン、デシタビン、デキソルマプラチン、デザグアニン、ジアジクオン、ドセタキセル、ドキソルビシン、ドロロキシフェン、ドロモスタノロン、デュアゾマイシン、エダトレキサート、エフロミチン、エルサミトルシン、エンロプラチン、エンプロメート、エピプロピジン、エピルビシン、エルブロゾール、エソルビシン、エストラムスチン、エタニダゾール、エチオダイズ(ethiodized)化油I131、エトポシド、エトプリン、ファドロゾール、ファザラビン、フェンレチニド、フロキシウリジン、フルダラビン、フルオロウラシル、フルロシタビン、ホスキドン、ホストリエシン、ゲムシタビン、Gold 198、ヒドロキシ尿素、イダルビシン、イホスファミド、イルモホシン、インターフェロンα−2a、インターフェロンα−2b、インターフェロンα−n1、インターフェロンα−n3、インターフェロンβ−1a、インターフェロンγ−1b、イプロプラチン、イリノテカン、ランレオチド、レトロゾール、ロイプロリド、リアロゾール、ロメトレキソール、ロムスチン、ロソキサントロン、マソプロコール、マイタンシン、メクロレタミン、メゲストロール、メレンゲストロール、メルファラン、メノガリル、メルカプトプリン、メトトレキサート、メトプリン、メツレデパ、ミチンドミド、ミトカルシン、ミトクロミン、ミトギリン、ミトマルシン、マイトマイシン、ミトスペル、ミトタン、ミトキサントロン、ミコフェノール酸、ノコダゾール、ノガラ−マイシン、オルマプラチン、オキシスラン、パクリタキセル、ペガスパルガーゼ、ペリオマイシン、ペンタムスチン、ペプロマイシン、ペルホスファミド、ピポブロマン、ピポスルファン、ピロキサントロン、プリカマイシン、プロメスタン、ポルフィメル、プロフィロマイシン、プレドニムスチン、プロカルバジン、ピューロマイシン、ピラゾフリン、リボプリン、ログレチミド、サフィンゴール、セムスチン、シムトラゼン、スパルホセート、スパルソマイシン、スピロゲルマニウム、スピロムスチン、スピロプラチン、ストレプトニグリン、ストレプトゾシン、塩化ストロンチウム89、スロフェナル、タリソマイシン、タキサン、タキソイド、テコガラン、テガフール、テロキサントロン、テモポルフィン、テニポシド、テロキシロン、テストラクトン、チアミプリン、チオグアニン、チオテパ、チアゾフリン、チラパザミン、トポテカン、トレミフェン、トレストロン、トリシリビン、トリメトレキセート、トリプトレリン、ツブロゾール、ウラシルマスタード、ウレデパ、バプレオチド、ベルテポルフィン、ビンブラスチン、ビンクリスチン、ビンデシン、ビネピジン、ビングリシネート、ビンロイロシン、ビノレルビン、ビンロシジン、ビンゾリジン、ボロゾール、ゼニプラチン、ジノスタチン、ゾルビシンおよびその薬学的に許容される塩が含まれる。
【0044】
本発明の相乗的な抗増殖医薬組成物または組合せ製剤に含まれるのに適する他の抗新生物化合物には、20−エピ−1,25ジヒドロキシビタミンD3、5−エチニルウラシル、アビラテロン、アクラルビシン、アシルフルベン、アデシペノール、アドゼレシン、アルデスロイキン、ALL−TKアンタゴニスト、アルトレタミン、アンバムスチン、アミドックス、アミホスチン、アミノレブリン酸、アムルビシン、アムサクリン、アナグレリド、アナストロゾール、アンドログラホリド、血管新生阻害剤、アンタゴニストD、アンタゴニストG、アンタレリックス、抗背側形態形成タンパク質−1、限定はしないが、ベノルテロン、シオテロネル、シプロテロン、デルマジノン、オキセンドロン、トプテロン、ザノテロンのような抗アンドロゲン、限定はしないが、クロメテロン、デルマジノン、ナホキシジン、ニトロミフェン、ラロキシフェン、タモキシフェン、トレミフェン、トリオキシフェンのような抗エストロゲン、およびその薬学的に許容される塩、抗ネオプラストン、アンチセンスオリゴヌクレオチド、アフィジコリングリシネート、アポトーシス遺伝子調節因子、アポトーシス調節因子、アプリン酸、アラ−CDP−DL−PTBA、アルギニンデアミナーゼ、アスラクリン、アタメスタン、アトリムスチン、アキシナスタチン、アザセトロン、アザトキシン、アザチロシン、バッカチンIII誘導体、バラノール、バチマスタット、BCR/ABLアンタゴニスト、ベンゾクロリン、ベンゾイルスタウロスポリン、β−ラクタム誘導体、β−アレチン、βクラマイシンB、ベツリン酸、bFGF阻害剤、ビカルタミド、ビサントレン、ビサジリジニルスペルミン、ビスナフィド、ビストラテンA、ビゼレシン、ブレフレート、ブロピリミン、ブドチタン、ブチオニンスルホキシイミン、カルシポトリオール、カルホスチンC、カンプトテシン誘導体、カナリポックスIL−2、カペシタビン、カルボキサミド−アミノトリアゾール、カルボキシアミドトリアゾール、CaRest M3、CARN700、軟骨組織由来阻害剤、カルゼレシン、カゼインキナーゼ阻害剤、カスタノスペルミン、セクロピンB、セトロレリックス、クロリン、クロロキノキサリン、スルホンアミド、シカプロスト、シス−ポルフィリン、クロミフェンおよびその類似体、クロトリマゾール、コリスマイシンAおよびB、コンブレタスタチンおよびその類似体、コナゲニン、クラムベスシジン816、クリプトフィシンおよびその誘導体、クラシンA、シクロペンタントラキノン、シクロプラタム、シペマイシン、シタラビン、細胞溶解因子、シトスタチン、ダクリキシマブ、デヒドロジデムニンB、デスロレリン、デキシホスファミド、デキスラゾキサン、デキシベラパミル、ジデムニンB、ジドックス、ジエチルノルスペルミン、ジヒドロ−5−アザシチジン、ジヒドロタキソール、ジオキサマイシン、ジフェニルスピロムスチン、ドコサノール、ドラセトロン、ドキシフルリジン、ドロロキシフェン、ドロナビノール、デュオカルマイシンSA、エブセレン、エコムスチン、エデルホシン、エドレコロマブ、エレメン、エミテフル、エプリステリド、エストロゲンアゴニストおよびアンタゴニスト、エキセメスタン、フィルグラスチム、フィナステリド、フラボピリドール、フレゼラスチン、フルアステロン、フルオロダウノルニシン、ホルフェニメックス、ホルメスタン、ホテムスチン、ガドリニウムテキサフィリン、硝酸ガリウム、ガロシタビン、ガニレリックス、ゼラチナーゼ阻害剤、グルタチオン阻害剤、ヘプスルファム、ヘレグリン、ヘキサメチレンビスアセトアミド、ヒペリシン、イバンドロン酸、イドキシフェン、イドラマントン、イロマスタット、イミダゾアクリドン、イミキモド、免疫刺激性ペプチド、インスリン様増殖因子−1レセプター阻害剤、インターフェロンアゴニスト、イオベングアン、ヨードドキソルビシン、イポメアノール、イリノテカン、イロプラクト、イルソグラジン、イソベンガゾール、イソホモハリコンドリンB、イタセトロン、ジャスプラキノリド、カハラリドF、ラメラリン−N、レイナマイシン、レノグラスチム、レンチナン、レプトルスタチン、白血病阻害因子、リュープロレリン、レバミソール、リアロゾール、リソクリンアミド、ロバプラチン、ロムブリシン、ロニダミン、ロバスタチン、ロキソリビン、ルルトテカン、ルテチウムテキサフィリン、リソフィリン、マンノスタチンA、マリマスタット、マソプロコール、マスピン、マトリリシン阻害剤、マトリックスメタロプロテイナーゼ阻害剤、メルバロン、メテレリン、メチオニナーゼ、メトクロプラミド、MIF阻害剤、ミフェプリストン、ミルテホシン、ミリモスチム、ミトグアゾン、ミトラクトール、ミトナフィド、ミトトキシン繊維芽細胞増殖因子−サポリン、モファロテン、モルグラモスチム、ヒト絨毛膜ゴナドトロピンモノクローナル抗体、モピダモール、ミカペロキシドB、ミリアポロン、N−アセチルジナリン、N−置換ベンズアミド、ナファレリン、ナグレスチップ、ナロキソン、ペンタゾシン、ナパビン、ナフテルピン、ナルトグラスチム、ネダプラチン、ネモルビシン、ネリドロン酸、中性エンドペプチダーゼ、ニルタミド、ニサマイシン、酸化窒素調節因子、ニトロキシド抗酸化剤、ニトルリン、オクトレチド、オキセノン、オナプリストン、オンダンセトロン、オンダンセトロン、オラシン、オサテロン、オキサリプラチン、オキサウノマイシン、パラウアミン、パルミトイルリゾキシン、パミドロン酸、パナキシトリオール、パノミフェン、パラバクチン、パゼリプチン、ペルデシン、ペントサン、ペントスタチン、ペントロゾール、ペルフルブロン、ペリリルアルコール、フェナジノマイシン、フェニルアセテート、ホスファターゼ阻害剤、ピシバニル、ピロカルピン、ピラルビシン、ピリトレキシム、プラセチンAおよびB、プラスミノーゲン活性化阻害剤、プロピルビス−アクリドン、プロスタグランジンJ2、プロテアソーム阻害剤、タンパク質キナーゼC阻害剤、タンパク質チロシンホスファターゼ阻害剤、プリンヌクレオシドホスホリラーゼ阻害剤、プルプリン、ピラゾロアクリジン、ラルチトレキセド、ラモセトロン、rasファルネシルタンパク質トランスフェラーゼ阻害剤、ras阻害剤、ras−GAP阻害剤、レテリプチン、レニウム186エチドロネート、リゾキシン、レチンアミド、ロヒツキン、ロムルチド、ロキニメックス、ルビギノンB1、ルボキシル、サイントピン、サルコフィトールA、サルグラモスチム、シゾフィラン、ソブゾキサン、ナトリウムボロカプテート、フェニル酢酸ナトリウム、ソルベロール、ソマトメジン結合タンパク質、ソネルミン、スパルホシン酸、スピカマイシンD、スプレノペンチン、スポンギスタチン1、スクアラミン、幹細胞分裂阻害剤、スチピアミド、ストロメリシン阻害剤、スルフィノシン、スラジスタ、スラミン、スワインソニン、タリムスチン、タモキシフェン、タウロムスチン、タザロテン、テコガラン、テルラピリリウム、テロメラーゼ阻害剤、テモゾロミド、テトラクロロデカオキシド、テトラゾミン、タリブラスチン、チオコラリン、トロンボポエチン、チマルファシン、チモポエチンレセプターアゴニスト、チモトリナン、甲状腺刺激ホルモン、スズエチルエチオプルプリン、チタノセン、トプセンチン、トレチノイン、トリアセチルウリジン、トロピセトロン、ツロステリド、チロシンキナーゼ阻害剤、チルホスチン、ウベニメックス、尿生殖洞由来増殖阻害因子、ウロキナーゼレセプターアンタゴニスト、バリオリンB、ベラレソール、ベラミン、ベルジン、ベルテポルフィン、ビンキサルチン、ビタキシン、ザノテロン、ジラスコルブ、およびその薬学的に許容される塩が含まれる。
【0045】
細胞増殖に対する本発明の医薬組成物または組合せ製剤の相乗活性は、限定されるものではないが、腫瘍細胞系の培養物中の3H−チミジンの取り込みから生じる放射能の測定などの1種または複数の試験を用いて容易に判定することができる。例えば、限定されるものではないが、
− RPMI1788:ヒト末梢血白血球(PBL)白人腫瘍系、
− Jurkat:ヒト急性T細胞白血病、
− EL4:C57BI/6マウスリンパ腫、または
− THP−1:ヒト単球腫瘍系
などの様々な腫瘍細胞系が、試験化合物の抗腫瘍効果を評価するために選択され得る。
【0046】
選択された腫瘍細胞系に応じて、例えば、
− RPMI1788およびTHP−1に対して:RPMI−1640+10%FCS+1%NEAA+1%ピルビン酸ナトリウム+5×10−5メルカプト−エタノール+抗生物質(G−418 0.45μg/ml)、
− JurkatおよびEL4に対して:RPMI−1640+10%FCS+抗生物質(G−418 0.45μg/ml)
などの様々な培養培地が使用され得る。
【0047】
相乗作用測定試験の特定の実施形態においては、腫瘍細胞系を回収し、完全培地中の0.27×106細胞/mlの懸濁液を調製する。懸濁液(150μl)を、マイクロタイタープレートに3通りに添加する。試験濃度(50μl)における完全培地(対照)または試験化合物のいずれかを、マイクロタイタープレート中の細胞懸濁液に添加する。細胞を、5%CO2下37℃で約16時間インキュベートする。3H−チミジンを添加し、細胞をさらに8時間インキュベートする。細胞を回収し、放射能をβ−カウンターにおいて、カウント毎分(CPM)で測定する。3H−チミジン細胞含有量、したがって測定された放射能は、細胞系の増殖に比例している。相乗効果は、本明細書で先に開示された半有効解析法により評価する。
【0048】
本発明に係る、細胞増殖に対する相乗活性を有する医薬組成物または組合せ製剤は、製剤の正確な目的用途および期待効果に応じた広い含有量範囲にわたって、構造式(I)を有するウレイル置換ナフタルイミド(イソキノリンジオン)誘導体、ならびに/または薬学的に許容されるそれらの塩および/もしくはそれらの溶媒和物および/もしくはそれらの代謝産物を含有し得る。一般的に、組合せ製剤のウレイル置換ナフタルイミド(イソキノリンジオン)誘導体含有量は、約0.1から約99.9重量%、好ましくは1から99重量%、より好ましくは5から95重量%の範囲内である。
【0049】
本発明に係る医薬組成物および組合せ製剤は、経口または任意の他の適切な方法で投与し得る。経口投与が好ましく、製剤は、錠剤、水性分散剤、分散可能な散剤もしくは顆粒剤、乳剤、硬もしくは軟カプセル剤、シロップ剤、エリキシル剤またはゲル剤の形態を有し得る。投与剤形は、これらの医薬組成物の製造用の当技術分野で既知の任意の方法を用いて調製することができ、甘味料、香料、着色剤、保存料などを添加剤として含むことができる。担体材料および賦形剤は、本明細書で下記に詳述され、とりわけ、炭酸カルシウム、炭酸ナトリウム、乳糖、リン酸カルシウムまたはリン酸ナトリウム;造粒剤および崩壊剤、結合剤などを含むことができる。本発明の医薬組成物または組合せ製剤は、任意の不活性固体希釈剤または担体材料と混合されるゼラチンカプセル剤中に含まれ得、あるいは成分が水または油媒体と混合される軟ゼラチンカプセル剤の形態を有する。水性分散剤は、生物活性を有する組成物または組合せ製剤を、懸濁化剤、分散剤または湿潤剤と組み合わせて含み得る。油分散剤は、植物油などの懸濁化剤を含み得る。直腸投与も、例えば、坐剤またはゲル剤の形態で適用できる。注射(例えば、静脈内、筋肉内または腹腔内)も、治療すべき障害および患者の状態に応じて、例えば、注射溶剤または分散剤の形態で投与方法として適用できる。
【0050】
特に指定のない限り、医薬組成物および組合せ製剤に関係して本明細書で使用される用語「薬学的に許容される担体または賦形剤」は、任意の材料または物質を意味し、この任意の材料または物質と一緒に、活性成分(複数でもよい)、すなわち、本発明のウレイル置換ナフタルイミドおよび場合によっては抗新生物薬が、例えば、前記組成物を溶解すること、分散することまたは拡散することにより治療すべき位置へのその適用または散布を促進するために、かつ/あるいはその有効性を損なうことなくその貯蔵、輸送または取り扱いを促進するために製剤化され得る。薬学的に許容される担体は、固体または液体または圧縮されて液体を形成する気体であってよく、すなわち、本発明の組成物は、濃縮物、乳剤、溶剤、顆粒剤、微粉剤、噴霧剤、エアゾール剤、ペレット剤または散剤として適切に使用し得る。
【0051】
本発明の前記医薬組成物における使用のための適切な薬学的担体、およびその製剤化のための効率的な方法は、薬理学の当業者に周知である。本発明内でそれらの選択に特定の制限はないが、本発明のプテリジン誘導体の通例低いまたは非常に低い水溶性のために、予想される時間放出プロフィールを考慮して、それらを適切に製剤化する助けとなり得る適切な担体の組合せの選択に特別な注意が払われよう。適切な薬学的担体には、湿潤剤、分散剤、固着剤、接着剤、乳化剤または界面活性剤、増粘剤、錯化剤、ゲル化剤、溶媒、被覆剤、抗菌剤および抗真菌剤(例えば、フェノール、ソルビン酸、クロロブタノール)、等張剤(糖または塩化ナトリウムなど)などの添加剤が含まれるが、但し、これらは薬学的な実施と一致するものに限られ、すなわち、哺乳動物に永久的な損傷を生じない担体および添加物に限られる。本発明の医薬組成物は、例えば、選択された担体物質および(適切な場合)界面活性剤などのその他の添加剤と一緒に、1段階または多段階手順で、活性成分を均一に混合、溶解、噴霧乾燥、コーティングおよび/または粉砕することによって任意の既知の方法で調製し得る。本発明の医薬組成物は、例えば、通例約1から10μmの直径を有するミクロスフェアの形態でそれらを得るために、すなわち、生物活性成分(複数でもよい)の制御放出または持続放出用のマイクロカプセルの製造のために、微粉化によっても調製し得る。
【0052】
本発明の医薬組成物における使用のための適切な界面活性剤は、好ましくは、良好な乳化特性、分散特性および/または湿潤特性を有する非イオン性、陽イオン性および/または陰イオン性材料である。このような適切な陰イオン性界面活性剤には、水溶性石鹸および水溶性合成界面活性剤の両方が含まれる。適切な石鹸は、高級脂肪酸(C10−C22)のアルカリまたはアルカリ土類金属塩、非置換または置換アンモニウム塩、例えば、オレイン酸もしくはステアリン酸、またはココナッツ油もしくは獣脂油から得られる天然脂肪酸混合物のナトリウムまたはカリウム塩である。合成界面活性剤には、ポリアクリル酸のナトリウムまたはカルシウム塩;脂肪スルホネートおよびスルフェート;スルホン化ベンズイミダゾール誘導体およびアルキル−アリールスルホネートが含まれる。脂肪スルホネートおよびスルフェートは、通常、8から22個の炭素原子を有するアルキルまたはアシル基で置換されたアルカリまたはアルカリ土類金属塩、非置換アンモニウム塩またはアンモニウム塩の形態であり、例えば、リグノスルホン酸またはドデシルスルホン酸のナトリウムまたはカルシウム塩、あるいは天然脂肪酸から得られる脂肪アルコールスルフェートの混合物、硫酸またはスルホン酸エステル(ラウリル硫酸ナトリウムなど)のアルカリまたはアルカリ土類金属塩、および脂肪アルコール/エチレンオキシド付加物のスルホン酸である。適切なスルホン化ベンズイミダゾール誘導体は、好ましくは、8から22個の炭素原子を含有する。アルキルアリールスルホネートの例は、ドデシル−ベンゼンスルホン酸またはジブチル−ナフタレンスルホン酸またはナフタレンスルホン酸/ホルムアルデヒド縮合生成物のナトリウム、カルシウムまたはアルカノールアミン塩である。同様に、本発明の実施に適するのは、対応するホスフェート、例えば、リン酸エステルの塩、ならびにp−ノニル−フェノールとエチレンおよび/またはプロピレンオキシドの付加物、あるいはリン脂質である。本目的に適する適切なリン脂質には、限定されるものではないが、例えば、ホスファチジル−エタノールアミン、ホスファチジルセリン、ホスファチジルグリセリン、リゾレシチン、カルジオリピン、ジオクタニル−ホスファチジルコリン、ジパルミトイルホスファチジルコリンおよびそれらの混合物などのセファリンまたはレシチンタイプの天然(動物もしくは植物細胞に由来)または合成リン脂質が含まれる。
【0053】
適切な非イオン性界面活性剤には、分子中に少なくとも12個の炭素原子を含有するアルキルフェノール、脂肪アルコール、脂肪酸、脂肪族アミンまたはアミドのポリエトキシル化およびポリプロポキシル化誘導体、脂肪族および脂環式アルコール、飽和および不飽和脂肪酸およびアルキルフェノールのポリグリコールエーテル誘導体などのアルキルアレンスルホネートおよびジアルキルスルホスクシネートが含まれ、前記誘導体は、好ましくは、(脂肪族)炭化水素部分中に3から10個のグリコールエーテル基および8から20個の炭素原子、そしてアルキルフェノールのアルキル部分中に6から18個の炭素原子を含有する。さらに適切な非イオン性界面活性剤は、アルキル鎖中に1から10個の炭素原子を含有するポリエチレンオキシドとポリプロピレングリコール、エチレンジアミノポリプロピレングリコールの水溶性付加物であり、この付加物は、20から250個のエチレングリコールエーテル基および/または10から100個のプロピレングリコールエーテル基を含有する。このような化合物は、通常、プロピレングリコール単位当たり1から5個のエチレングリコール単位を含有する。非イオン性界面活性剤の代表例は、ノニルフェノール−ポリエトキシエタノール、ヒマシ油ポリグリコールエーテル、ポリプロピレン/ポリエチレンオキシド付加物、トリブチルフェノキシポリエトキシエタノール、ポリエチレングリコールおよびオクチルフェノキシポリエトキシエタノールである。ポリエチレンソルビタン(ポリオキシエチレンソルビタントリオレエートなど)、グリセロール、ソルビタン、ショ糖およびペンタエリトリトールの脂肪酸エステルも、適切な非イオン性界面活性剤である。
【0054】
本発明を実施するために適する陽イオン性界面活性剤には、限定されるものではないが、ハロ、フェニル、置換フェニルまたはヒドロキシで場合によっては置換されていてもよい4個の炭化水素基を有する第4級アンモニウム塩、好ましくはハロゲン化物;例えば、N−置換基として少なくとも1個のC8−C22アルキル基(例えば、セチル、ラウリル、パルミチル、ミリスチル、オレイルなど)と、さらなる置換基として非置換またはハロゲン化低級アルキル、ベンジルおよび/またはヒドロキシ−低級アルキル基を含有する第4級アンモニウム塩が含まれる。
【0055】
本目的に適した界面活性剤のより詳細な説明は、例えば、「McCutcheon's Detergents and Emulsifiers Annual」(MC Publishing Crop., Ridgewood, New Jersey, 1981)、「Tensid-Taschenbuch」、2nd ed. (Hanser Verlag, Vienna, 1981)および「Encyclopaedia of Surfactants」(Chemical Publishing Co., New York, 1981)に見い出し得る。
【0056】
構造形成剤、増粘剤またはゲル形成剤も、本発明の医薬組成物および組合せ製剤に含まれ得る。適切なこのような薬剤には、特に、限定されるものではないが、商標Aerosilとして市販されている製品などの高分散ケイ酸;ベントナイト;モンモリロナイトのテトラアルキルアンモニウム塩(例えば、商標Bentoneとして市販されている製品)(ここで、前記アルキル基のそれぞれは、1から20個の炭素原子を含有し得る);セトステアリルアルコールおよび変性ヒマシ油生成物(例えば、商標Antisettleとして市販されている製品)が含まれる。
【0057】
本発明の医薬組成物および組合せ製剤に同様に含まれ得るゲル化剤には、限定されるものではないが、カルボキシメチルセルロース、酢酸セルロースなどのセルロース誘導体;アラビアゴム、キサンタンガム、トラガカントガム、グアーガムなどの天然ゴム;ゼラチン;二酸化ケイ素;カルボマーなどの合成ポリマーおよび任意の適切な割合のそれらの混合物が含まれる。ゼラチンおよび変性セルロースは、ゲル化剤の好ましいクラスである。
【0058】
本発明の医薬組成物および組合せ製剤に同様に存在し得る他の任意選択の賦形剤には、限定されるものではないが、酸化マグネシウムなどの添加剤;アゾ染料;二酸化チタンなどの有機および無機顔料;UV吸収剤;安定剤;臭気マスキング剤;粘性増強剤;例えば、パルミチン酸アスコルビル、重亜硫酸ナトリウム、メタ重亜硫酸ナトリウムなどの抗酸化剤およびそれらの混合物;例えば、ソルビン酸カリウム、安息香酸ナトリウム、ソルビン酸、没食子酸プロピル、ベンジルアルコール、メチルパラベン、プロピルパラベンなどの保存料;エチレン−ジアミンテトラ酢酸などの金属イオン封鎖剤;天然バニリンなどの着香料;クエン酸および酢酸などの緩衝剤;ケイ酸塩、珪藻土、酸化マグネシウムまたは酸化アルミニウムなどの増量剤または充填剤;マグネシウム塩などの緻密化剤;ならびにそれらの混合物が含まれる。
【0059】
さらなる成分が、本発明の組成物および組合せ製剤における生物活性成分の作用の持続時間を制御するために含まれ得る。したがって、制御放出組成物は、例えば、ポリエステル、ポリアミノ酸、ポリビニル−ピロリドン、エチレン−ビニルアセテートコポリマー、メチルセルロース、カルボキシメチルセルロース、硫酸プロタミンなどの適切なポリマー担体を選択することによって得られる。薬物放出の速度および作用の持続時間も、活性成分を、粒子、例えば、ヒドロゲル、ポリ乳酸、ヒドロキシメチル−セルロース、ポリメチルメタクリレートおよびその他の上記に記載されたポリマーなどのポリマー物質のマイクロカプセルに組み込むことによって制御し得る。このような方法には、リポソーム、ミクロスフェア、マイクロエマルジョン、ナノ粒子、ナノカプセルなどのコロイド薬物送達系が含まれる。投与の経路に応じて、本発明の医薬組成物または組合せ製剤は、保護被覆も必要とし得る。
【0060】
注射用途に適した医薬剤形には、滅菌水溶液または分散液およびその即時調製用の滅菌粉末が含まれる。したがって、本目的のための一般的な担体には、生体適合性の水性緩衝液、エタノール、グリセロール、プロピレングリコール、ポリエチレングリコール、シクロデキストリンなどの錯化剤など、およびそれらの混合物が含まれる。
【0061】
構造式(I)を有するウレイル置換ナフタルイミド(イソキノリンジオン)誘導体、ならびに/または薬学的に許容されるそれらの塩および/もしくはそれらの溶媒和物および/もしくはそれらの代謝産物と、抗新生物薬とを含む組合せ製剤の場合においては、両方の成分は、治療すべき患者において必ずしも同時にそれらの相乗的治療効果を直接にもたらさないので、前記組合せ製剤は、2種の成分を別々に但し隣接させて含有する薬剤キットまたはパッケージの形態であってよい。後者においては、したがって、それぞれの成分は、他の成分の投与経路と異なる投与経路に適した様式で製剤化することができ、例えば、それらの一方は、経口または非経口製剤の形態であってよいが、他方は、静脈注射用アンプルまたはエアゾールの形態である。
【0062】
本発明はさらに、患者、好ましくは哺乳動物、より好ましくはヒトにおける細胞増殖性障害の予防または治療方法に関する。本発明の方法は、それを必要とする患者に、構造式(I)を有するウレイル置換ナフタルイミド(イソキノリンジオン)誘導体、ならびに/または薬学的に許容されるそれらの塩および/もしくはそれらの溶媒和物および/もしくはそれらの代謝産物の治療有効量を、場合によっては抗新生物薬の有効量と一緒に、または極めて詳細に上記に開示されたような上記を含む医薬組成物を投与することからなる。ウレイル置換ナフタルイミド(イソキノリンジオン)誘導体の有効量は、ヒトの体重1kg当たり1日当たり通常0.01mgから20mg、好ましくは0.1mgから5mgの範囲である。治療すべき病態および患者の状態に応じて、前記有効量は、1日当たり数回の副次的単位に分けることができ、2日以上の間隔で投与することができる。治療すべき患者は、前記病態を患っている任意の温血動物、好ましくはヒトであってよい。
【0063】
次の実施例は、その範囲を何ら制限することなく、ウレイル置換ナフタルイミドの調製、医薬製剤および生物学的評価を含む、本発明のいくつかの実施形態を例示することを意図している。
【実施例1】
【0064】
エチル({2−[2−(ジメチルアミノ)エチル]−1,3−ジオキソ−2,3−ジヒドロ−1H−ベンゾ[de]イソキノリン−5−イル}アミノ)カルボニルカルバメートの調製
1.086gのアモナフィドを窒素雰囲気下−20℃で80mLの2−ブタノンに溶解した。次いで、2mLの2−ブタノンに溶解した880mgのエトキシカルボニルイソシアネート(2モル当量)を、滴下ロートを用いて5分間かけて注意深く添加した。撹拌しつつ反応温度を−20℃に25分間維持した。次いで反応混合物を40分間45℃に温め、この時間の後250μLの水を添加した。この反応クエンチステップ後、形成された沈殿をろ紙で40℃でろ過した。乾燥後、1.162gの予想生成物(下記の構造式)を得た(収率:76%)。高速液体クロマトグラフィー(以下HPLCという)は95.6%を超える純度を示した。微量のアモナフィド(約2%)は依然存在した。
【化3】
【0065】
所望の生成物は、以下により特性決定した:
− プロトン核磁気共鳴(300MHz、CDCl3):WO2005/105753号の実施例4におけると同一のピークを示した、そして
− エレクトロスプレーオン化質量スペクトル:M+H+=399におけるピーク;および2M+H+=797における付加物の存在を示した。
【実施例2】
【0066】
N−{2−[2−(ジメチルアミノ)エチル]−1,3−ジオキソ−2,3−ジヒドロ−1H−ベンゾ[de]イソキノリン−5−イル}尿素の調製
実施例1の化合物の100mgをNaOH 0.1Mの100mL中に溶解した。反応混合物を温めて還流し1時間この温度を維持した。混合物をHPLCで分析し、主要生成物(収率76%)として期待された尿素(下記の構造式)の存在が示された。
【化4】
【0067】
この生成物は、次の技術により特性決定した:
− プロトン核磁気共鳴(RMN 1H, 300MHz, DMSO)では、以下に示されるピーク:9.40(NH-17, bs);8.53(H-2, d, J=1.8);8.48(H-4, d, J=1.8);8.26-8.32(H-6およびH-7, m);6.18(NH2-19, bs);4.14(H-14, t, J=6.6);2.51(H-13, m);および2.21(H-15およびH-16, s) ppmを示した;
− 13C NMR(75.4MHz, DMSO, TMS内部標準)では、以下に示されるピーク37.5(CH2, C-13);49.9(2×CH3, C-15およびC-16);57.0(CH, C14);119.0(CH, C-芳香族);122.2(C, C-芳香族);122.9(C, C-芳香族);123.5(C, C-芳香族);123.9(CH, C-芳香族);127.8(CH, C-芳香族);128.6(CH, C-芳香族);132.7(C, C-芳香族);133.8(CH, C-芳香族);140.3(C, C-芳香族);156.5(C, C-18);および163.8(C, C-12);164.0(C, C-11)ppmを示した;そして
− エレクトロスプレーオン化質量スペクトルでは、M+H+=327におけるピーク;および2M+H+=653における付加物を示した。
【実施例3】
【0068】
全体的な細胞増殖に対する影響
全体的な細胞増殖に対する実施例2の化合物の影響を迅速(すなわち5日以内)に測定するために試験を行った。本試験は、ミトコンドリア還元によって黄色生成物の3−(4,5−ジメチルチアゾール−2−イル)−2,5−ジフェニルテトラゾリウムブロミド(本明細書でMTTという)を青色生成物のホルマザン染料に変換し得る代謝活性を有する生細胞の数を測定する。分光光度計を用いて測定された本実験の最後に得られたホルマザンの量は、生細胞数と直接的に比例している。したがって、光学密度測定によって、対照条件(未処理細胞)および/または他の参照化合物と比較しての、被験化合物の影響の定量的測定が可能になる。
【0069】
表1に記載された6種のヒト癌細胞系を次のMTT試験に使用した。これらの細胞系は6種の癌の組織型を含み、前立腺癌、神経膠腫、膵臓癌、結腸癌、肺癌および乳癌である。使用される細胞型に応じて1ウェル当たり1,000から4,000個の細胞で1ウェル当たり細胞懸濁液の100μlの量で平底の96ウェルマイクロ−ウェル中で細胞を増殖させた。周知のMEM10%血清培養培地に、それぞれの細胞系を接種した。
【表1】
【0070】
詳細な実験手順は次の通りである。37℃でのインキュベーションの24時間後、被験化合物を次のモル濃度:10−9M、5.10−9M、10−8M、5.10−8M、10−7M、5.10−7M、10−6M、5.10−6Mおよび10−5Mで予め溶解した100μlの新しい培地で、培養培地を置換した。それぞれの実験を6回繰り返した。
【0071】
試験対象の化合物を含む(実験条件)または含まない(対照条件)37℃でのインキュベーションの72時間後、1mg/mlの濃度でRPMI(1640フェノールレッド非含有)に溶解した100μlのMTTと、培地を置換した。続いてマイクロウェルを37℃で3時間インキュベートし、400gで10分間遠心分離した。MTTを除去し、形成されたホルマザン結晶を100μlのDMSOに溶解した。マイクロウェルを5分間振とうし、570nm(最大ホルマザン吸収)および630nm(バックグラウンドノイズ)の波長において分光光度計を読み取った。
【0072】
それぞれの実験条件について、平均光学密度を計算し、また対照と比較した残存生細胞の百分率を計算した。
【0073】
下記の表2は実施例2の化合物についてのIC50値を示している。IC50は被験化合物が全体の腫瘍細胞増殖を50%だけ阻害したモル濃度の範囲を表している。
【表2】
【実施例4】
【0074】
細胞移動に対する効果
異なる細胞系、すなわちU−373 MG(神経膠腫)およびA549(肺癌)の細胞を移動実験の48時間前に培養フラスコに接種した。試験日に、表3の右欄に記載の通りに、制御した温度(37.0±0.1℃)で12時間または22時間緩衝培地を含有する密閉ファルコンディッシュ中で、実施例2の化合物を用いてまたは用いずに細胞を処理した。実施例2の化合物を3つの非細胞毒性の濃度(10−6M、10−7M、10−8M)において使用した。細胞の移動を位相差顕微鏡に備え付けたCCDカメラを用いて観測した。ノンパラメトリックMann−Whitney検定による移動の統計的分析を最も運動性の細胞の25%および50%についてそして細胞集団全体について実施した。下記の表3は被験化合物の抗移動作用を示している。
【表3】
【0075】
表3のデータは、実施例2の化合物は本研究で使用された非細胞毒性の濃度でU−373 MG癌細胞の移動レベルの減少を誘導したことを示している。特に、本化合物は細胞移動の統計的に有意な阻害を示している。
【実施例5】
【0076】
ナノ粒子懸濁液製剤
ナノ粒子懸濁液を実施例2の化合物の製剤用に使用する。この手法のために、ポリソルベート80(Tween 80)、Texapon K12(SDS)、PVA(ポリビニルアルコール)、Lutrol F68(Poloxamer 188)、Lutrol F127(Poloxamer 407)、ヒドロキシプロピル−β−シクロデキストリン、タウロコール酸ナトリウムおよび他のリン脂質(Lipoid S PC−3およびPhopholipon 90 H)を含む選択された賦形剤(特に張力活性剤)を使用する。
【0077】
賦形剤およびその量を選択後、実施例2の化合物を含有する懸濁液を、所望量の水にその予定量を単純に添加することによって調製する。次いで、懸濁液を、予備の粒径低減(微粉化)のために低温で、24000rpmでタラックス(turax)にかける。次いで、懸濁液を高圧でEmulsiFlexホモジナイザー(homogenisator)にかける。異なる圧力での3サイクルのホモジナイズを使用して期待される粒径を得ることができ、例えば、第1サイクルを7000psiで7分間実施し、第2サイクルを12000psiで8分間実施し、最後に最終サイクルを約21000〜24000psiで30分間実施する。次いで、粒径分布の測定をレーザー回折(各測定の間を20秒として5回の測定を行う)により行った。これらの5回の測定の平均は懸濁液の粒径分布を示す。
【実施例6】
【0078】
2,2,2−トリクロロ−N−[({2−[2−(ジメチルアミノ)エチル]−1,3−ジオキソ−2,3−ジヒドロ−1H−ベンゾ[de]イソキノリン−5−イル}アミノ)カルボニル]アセトアミドの調製
機械的撹拌機、還流冷却器、冷却氷浴、滴下ロートおよび温度調節器を備えた3つ首12L丸底フラスコに、窒素下でアモナフィド(90g)および3.6Lのメチルエチルケトン(MEK)を導入した。得られた懸濁液を−10℃に冷却し、600mLのMEK中の120gのトリクロロアセチルイソシアネート(0.64モル)の溶液を、温度を−5℃以下に維持しながら35分かけて滴下した。反応混合物を−10℃から−5℃の間で3時間撹拌し、次いで2.8mLの水をゆっくりと添加した。混合物を放置して室温に温め、得られた固体をろ過により単離し、100mLのMEKを用いてフィルター上で洗浄し、2日間風乾させて147gの所望の化合物を得た(収率:97%;純度:98.1%)。本化合物の特徴的なスペクトルはWO2005/105753号の実施例2に記載されたものと同じであった。
【実施例7】
【0079】
N−{2−[2−(ジメチルアミノ)エチル]−1,3−ジオキソ−2,3−ジヒドロ−1H−ベンゾ[de]イソキノリン−5−イル}尿素の代替調製法
機械的撹拌機、還流冷却器、および温度調節器を備えた3つ首22L丸底フラスコに、実施例6の化合物(250g)および水中の5%K2CO3溶液の7.5Lを導入した。(氷浴)を用いて混合物を10℃に冷却し、次いで7.5Lのメタノール(MeOH)を一度に添加した。温度は20℃に上がった。フラスコを氷浴から取り外し、出発原料の大部分が溶解するまで周囲温度で撹拌を続けた(約30〜45分)。混合物を急いでろ過して(清澄化して)少量の未反応材料および他の機械的不純物を取り除いた。混合物を室温で2時間撹拌し、4LのMeOHを一度に添加した。混合物を54〜56℃に3時間加熱した。反応の進行をHPLCでモニターして完了を確認した。反応混合物を冷却(氷浴)し8−10℃に2時間保った。得られた固体をろ過により単離し、フィルター上で洗浄(2×100mLの水)し、次いで2日間風乾させて156g(収率:90%)の所望の化合物(HPLC純度:99.47%)を得た。本化合物の特徴的なスペクトルは本明細書で上記の実施例2に記載されたものと同じであった。
【実施例8】
【0080】
乳酸ベースの溶液製剤
実施例2または実施例7により生成された化合物の液体溶液を次の通りに得た。
【0081】
第1に、2体積%の乳酸溶液を次の通り調製した。50mL容量フラスコに、クラスA−TDピペットを用いて、40mLの0.9%NaCl注射溶液、1.18mLの乳酸、85%ACS試薬を添加した。次いで、体積を0.9%NaCl注射溶液で50mLに調節し、全体を逆さまにして混合した。
【0082】
25mL容量フラスコに、実施例2および7の化合物の700mgを正確に計量した。この特定の量に、10mLの0.9%NaCl注射溶液および8.89mLの前述の2%乳酸水溶液を添加した。得られた溶液を10分間激しく撹拌し超音波処理した。溶液のpHは6.4〜6.6の間であった。次いで、前述の2%乳酸水溶液の少量(20μL)の注意深い添加によって、pHを5.75に調節した。0.9%NaCl注射溶液を使用して25mLの最終体積に調節した。その時点で、本発明の化合物の溶解が完了したことが目視検査で観察され、予め滅菌したシリンジフィルター(例えば、Milliporeフィルター−Durapore(PVDF)、0.22μm)に溶液を通すことによって溶液を滅菌し、こうして28mg/mLの溶液が得られた。
【0083】
この原液28mg/mLから、0.9%NaCl注射溶液を用いた希釈ステップを行うことによって、次の表に示された希釈溶液を10mL容量フラスコ中に得た。表4において、示された用量は、静脈内注射用の用量体積は1kg当たり5mLであるという仮定に対応する。
【表4】
【実施例9】
【0084】
N−{2−[2−(ジメチルアミノ)エチル]−1,3−ジオキソ−2,3−ジヒドロ−1H−ベンゾ[de]イソキノリン−5−イル}尿素の血液毒性
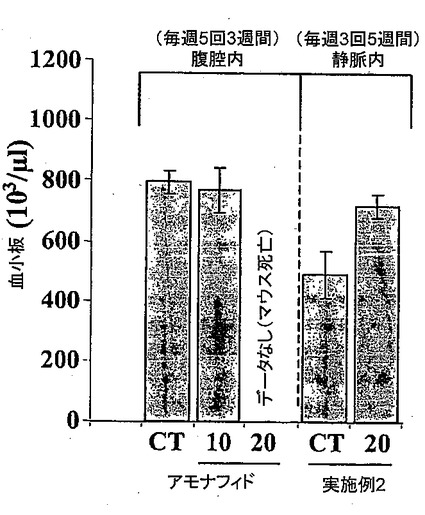
発明者らは、血小板、赤血球および白血球に対するアモナフィドの影響と比較した、血小板、赤血球および白血球に対する実施例2または実施例7により生成された化合物の化合物により誘発される潜在的な血液毒性を測定した。アモナフィドの影響をマウスへの腹腔内投与によって、10mg/kgおよび20mg/kgにおいて評価した。投与スケジュールは連続3週間で毎週5回であった。実施例2または実施例7により生成された化合物の影響はマウスへの静脈内投与によって20mg/kgで評価した。投与スケジュールは連続5週間で毎週3回(月曜日、水曜日および金曜日)であった。動物を最後の注射後3日に犠牲にした。1グループ当たり10匹のマウスが存在した。図1は、血小板に対する化合物により誘発された血液毒性についてのこのアッセイの結果を示している。図1は、10mg/kgの用量でのアモナフィドの15回の長期投与にマウスが耐えたが、20mg/kgの用量でのアモナフィドの15回の長期投与の全回を摂取する前に全ての動物が死亡したことを示している。対照的に、図1は、20mg/kgの用量での実施例2または実施例7により生成された化合物の15回の長期投与にマウスが耐えたことを示している。したがって、アモナフィドと異なり、実施例2または実施例7により生成された化合物は、これらの実験条件において治療用量で血液毒性を誘発しないことが見い出された。
【実施例10】
【0085】
N−{2−[2−(ジメチルアミノ)エチル]−1,3−ジオキソ−2,3−ジヒドロ−1H−ベンゾ[de]イソキノリン−5−イル}尿素とP−糖タンパク質の相互作用
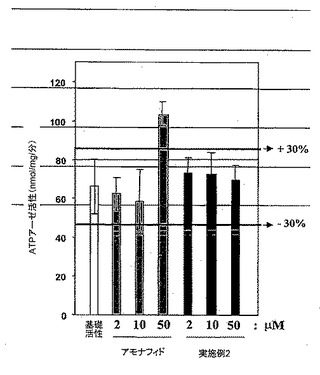
P−糖タンパク質(以下ではP−gpという)との薬物相互作用を試験するために、発明者らは濃縮P−gp膜小胞調製物(次のキットを使用した:SPI BIO Franceから市販されているP−gp薬物相互作用アッセイキット)からのATPアーゼ活性の調節の研究に基づくアッセイを使用した。P−gp ATPアーゼ活性を小胞懸濁液培地中のADP形成のモニターに基づく分光光度法によって測定した。基礎ATPアーゼ活性を、添加された薬物の不存在下で測定された活性と定義した。基礎的な活性の調節を、異なる濃度(それぞれ2、10、および50μM)でアモナフィドまたは実施例2もしくは実施例7により生成された化合物を添加することによって実施した。図2に示されたデータは、50μMでアッセイした場合にアモナフィドはATPアーゼ活性を有意に変更するが、本発明の化合物はATPアーゼ活性に殆ど影響を与えないことを示している。
【実施例11】
【0086】
ヒトの癌細胞における自食作用に関係した細胞死に対するN−{2−[2−(ジメチルアミノ)エチル]−1,3−ジオキソ−2,3−ジヒドロ−1H−ベンゾ[de]イソキノリン−5−イル}尿素の誘発作用
トポイソメラーゼII標的薬物の顕著な特徴はアポトーシスの誘発であり、これは、開裂可能な複合体の安定化および/またはトポイソメラーゼII鎖切断(strand-passage)活性の阻害の結果としての完全な染色体分離の達成の失敗による、DNA損傷レベルの細胞内増加の結果である。アモナフィドはトポイソメラーゼII阻害剤でありアポトーシスを誘発し、これは発明者らが、実施例2または実施例7により生成された化合物を含むヒトPC−3(表1を参照されたい)およびDU−145(ATCC番号:HTB−81)前立腺癌細胞において観測しなかった特徴である。
【0087】
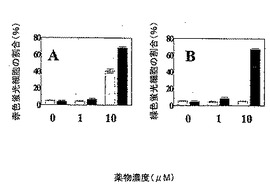
発明者らはアネキシンVおよびプロピジウムヨージドの両方についてPC−3およびDU−145陽性細胞の百分率を求めるためにフローサイトメトリー(Mijatovicら、Neoplasia 2006において見い出された手順による)を使用し、発明者らは実施例2または実施例7により生成された化合物の10μMでの処置を受けたPC−3またはDU−145細胞の最大で10%のみがアポトーシスプロセスを経験したことを観測した。
【0088】
発明者らは本化合物で処置したPC−3およびDU−145細胞において自食促進作用を観測した。発明者らは、それらを0(対照、未処置細胞)、1μMまたは10μMで処置した後に、PC−3(灰色棒)またはDU−145(黒色棒)細胞のアクリジン・オレンジ染色後に、酸性の小胞細胞小器官(赤色蛍光染色として表した)(Kanzawaら、Cell Death Differ 2004において見い出された手順による)を定量し、結果は図3Aに示されている。
【0089】
リソソームは細胞死をいくつかのレベルで制御することは周知である。内因性または外因性ストレス(化学療法を含む)への応答において、リソソーム膜の透過化(LMP)が起こり、異化加水分解酵素(catabolic hydrolase)の放出をもたらすことがあり、これはカスパーゼ依存的アポトーシス、カスパーゼ非依存性アポトーシス様細胞死または高レベルのLMPに続く壊死さえ媒介し得る。したがって、発明者らは、それらが実施例2または実施例7により生成された化合物の0(対照、未処置細胞)、1μMまたは10μMで処置された後に、PC−3(灰色棒)またはDU−145(黒色棒)の72時間の処置後に、酸性の小胞細胞小器官の「漏出」(緑色蛍光染色として表した)(Nylandsted, J.ら、Heat Shock Protein 70 Promotes Cell Survival by Inhibiting Lysosomal Membrane Permeabilization, J Exp Med. (2004) 16;200(4):425-35およびMijatovicら、Neoplasia 2006による)を定量し、結果は図3Bに示されている。発明者らはPC−3細胞の72時間の処置後にLMPを観測しなかったが、本発明の化合物の10μMで処置した場合にDU−145細胞に著しい薬物により誘発されたLMPプロセスが出現した。
【実施例12】
【0090】
DU−145ヒト前立腺癌細胞における老化に対するN−{2−[2−(ジメチルアミノ)エチル]−1,3−ジオキソ−2,3−ジヒドロ−1H−ベンゾ[de]イソキノリン−5−イル}尿素の誘発作用
実施例2または実施例7により生成された化合物が非アポトーシス細胞死を誘発したという特徴は、6日間本化合物の10μMで処置したヒトPC−3およびDU−145前立腺癌細胞において細胞イメージングを用いて形態学レベルでさらに観測された(細胞をFalconフラスコ(25cm2)中に接種し定量ビデオ顕微鏡で6日間分析した)。
【0091】
老化細胞はその形質膜の完全性を維持しているが、それらは永久の増殖停止を受け、その細胞の増殖能力(clonogenicity)を失うので、老化は「生細胞死」の1種であると考えることができる。老化は癌の進行に対する自然のバリアとして作用し得る。
【0092】
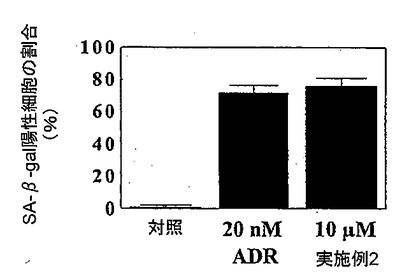
老化に一般的な特徴が、ヒトDU−145前立腺癌細胞において実施例2の化合物によって誘発された。老化細胞は扁平な細胞質および増加した粒度などの形態学的変化を一般的に示すことが知られている。老化に関係したβ−ガラクトシダーゼ活性の誘発は、老化をしている細胞に起こる特異的な事象であり、発明者らが、図4に明らかにしたように、現在の研究(Dimriら、A biomarker that identifies senescent human cells in culture and in aging skin in vivo, Proc Natl Acad Sci U S A. (1995) 92(20):9363-7において見い出された手順による)でもう一度観測した特徴である。ドキソルビシン(ADR)の中用量(nM範囲)は野性型ヒト癌細胞に老化を誘発することが知られている。したがって、発明者らは発明者らの実験において陽性対照としてドキソルビシンを使用した。図4に示されたように、本発明の化合物の10μMは、DU−145細胞中の20nMのドキソルビシンと類似の百分率の老化関連のβ−ガラクトシダーゼ陽性染色を誘発した。
【実施例13】
【0093】
N−{2−[2−(ジメチルアミノ)エチル]−1,3−ジオキソ−2,3−ジヒドロ−1H−ベンゾ[de]イソキノリン−5−イル}尿素により標的化される遺伝子の同定
生化学レベルにおいて、老化は、実施例2の化合物でインビトロで処置したPC−3細胞のゲノム解析を実施した場合に、現在の研究で本知見者らが観測した特徴である、代謝における変化を伴う。
【0094】
遺伝子レベルにおいて、発明者らは、本化合物で処置した場合にPC−3細胞におけるクロマチン構造および遺伝子発現パターンの改変も観測した。
【0095】
発明者らは、1μMもしくは10μMで1回、または1μMで1週間に5回(連続5日間に1日1回)のいずれかで本発明の化合物(N−{2−[2−(ジメチルアミノ)エチル]−1,3−ジオキソ−2,3−ジヒドロ−1H−ベンゾ[de]イソキノリン−5−イル}尿素)で処置したインビトロで増殖させたヒトPC−3癌細胞を使用するAffymetrix全ゲノムマイクロアレイを用いて遺伝子標的の評価の最初の実験を行った(全ゲノム解析はAffymetrixヒトゲノムU133セットプラス2.0(High Wycombe, United Kingdom)を使用してVIB MicroArray Facility(UZ Gasthuisberg, Catholic University of Leuven, Belgium)で実施した)。発明者らが得た最も顕著なデータは表5に報告され、インビトロで単一の高用量としてアッセイする場合(急性期インビトロ処置)、本化合物は、ヒストンの様々なタイプ(少なくともmRNAレベルで)の発現のレベルを顕著に増加させることによって核構成および生物発生を著しく改変したことを示す。本化合物により標的化された遺伝子の第2セットは、「アミノ酸代謝」と記した遺伝子のカテゴリーに属する(表5)。
【0096】
前立腺癌細胞における老化に関係した遺伝子の同定プロセスにおいて、先行技術は、DNA損傷により誘発された老化の状態における癌細胞中のets相同因子(EHF)の顕著な抑制を教示しており、EHFは老化および細胞周期停止を阻害することによってPC−3前立腺癌細胞における実質的な薬物耐性をもたらすことを示した。興味深いことに、本発明者らは、EHFも本発明の化合物の標的であることを見い出した。
【0097】
転写因子のE2Fファミリーは、細胞周期の進行に重要な役割を果たすことが知られている。E2F−1は、別のタンパク質DP−1とヘテロ二量体複合体となって、低リン酸化pRbに結合しているために通常不活性である。細胞がG1期からS期に進行する場合、pRbは過剰リン酸化になり結合E2F−1/DP−1ヘテロ二量体を放出し、次いで、これはTSおよびDHFRなどのDNAに含まれる遺伝子の転写を活性化する。機能性pRbの損失は遊離E2F−1レベルと次いでのTSおよびDHFRのレベル増加を生じ得る。ゲノムAffymetrix手法により明らかにされたように、発明者らは、連続5日間の1日1回の実施例2の化合物の1μMでのPC−3細胞の処置はE2F−1のmRNAレベルを2倍減少させることを見い出した。
【0098】
老化の初期の間、Rbはゲノム中の特異的位置におけるヘテロクロマチンの核生成を制御することがあり、次いでこれはヒストンメチルトランスフェラーゼの作用およびHP1タンパク質のリクルートによって拡がる。発明者らは、実施例2の化合物は、PC−3細胞中の少なくともmRNAレベルにおいてヒストンH1、H2およびH3の増加を通してPC−3細胞中のヘテロクロマチンのレベルを著しく増加させることを見い出した(表5)。対照的に、本化合物はH2AFYのmRNA発現のレベルを2.6倍減少させた。
【表5】
【図面の簡単な説明】
【0099】
【図1】アモナフィドと比較した、本発明の化合物による血小板に対する化合物により誘発された血液毒性を示す図である。
【図2】アモナフィドと比較した、本発明の化合物について分光光度計で測定されたP−gp ATPアーゼ活性を示す図である。
【図3】本発明の化合物の様々な濃度における、(A)酸性の小胞細胞小器官(赤色蛍光染色として示される)の定量により評価された、薬物により誘発された自食促進作用、および(B)アクリジン・オレンジ染色および緑色蛍光染色の定量に従って評価された、薬物により誘発されたリソソーム膜透過化(LMP)を示す図である。
【図4】ドキソルビシンと比較した、本発明の化合物により誘発されたDU−145ヒト前立腺癌細胞における老化に関連したβ−ガラクトシダーゼ活性を示す図である。
【技術分野】
【0001】
本発明は、新規な置換ナフタルイミド誘導体、それらの製造方法、ならびに、特に、様々な形態の癌の予防および/または治療における活性成分としてそれらを含む医薬組成物の形態の、抗腫瘍剤としてのそれらの医薬用途に関する。
【背景技術】
【0002】
アモナフィドを含めた様々な種類の置換ナフタルイミドは、抗腫瘍作用または他の有用な生物活性を有するとして当技術分野において知られている。特にWO2005/105753号は、癌などの細胞増殖性疾患の治療において活性を有する特異的な置換パターンを有するナフタルイミド誘導体を開示している。
【0003】
アモナフィドで見い出された活性のレベルは非常に興味深いものであったが、そしてそうあり続けているが、この材料は、重大な欠陥を有し、これは改善された特性を備える薬剤の継続的なニーズを示している。第1に、アモナフィドはある患者には毒性が強すぎることが見い出され、特に、薬剤の5日用量を摂取した患者が死に至ることもある実質的な骨髄毒性を生じる。さらに、アモナフィドは、マウスの白血病モデルにおいて中程度の活性しか有さないことが示された。また、アモナフィドは、結腸癌、肺癌および乳癌の、マウスにおけるヒトの移植腫瘍に活性を有さないことが示された。したがって、アモナフィドは顕著な生物活性を示すが、マウス腫瘍モデルにおいて実質的に広範囲の活性を有するものではない。Ajaniらは、Invest New Drugs (1988) 6:79-83において、アモナフィドは、インビトロで原発性ヒト充実性腫瘍において試験された場合に乏しい活性しか有さないことを示した。
【0004】
特定の形態の癌に対するアモナフィドなどの抗増殖剤の臨床活性が示されているが、腫瘍応答率、応答期間、骨髄毒性の減少および最終的に患者生存の改善が依然として求められている。当技術分野において、新規薬物と通常の抗腫瘍薬の適切な組合せを提供することによってヒトにおける抗増殖治療の有効性を改善することも必要とされている。
【発明の開示】
【発明が解決しようとする課題】
【0005】
アモナフィドなどの上記の欠点を考慮すると、当技術分野において、より有望な活性/副作用のバランスを示すナフタルイミド誘導体のニーズがある。
【課題を解決するための手段】
【0006】
本発明は、ナフタルイミジル部分の5位におけるウレイル基で置換されたナフタルイミド誘導体が、細胞増殖性障害の治療に有用であり、限定された数の反応ステップを通して効率的に製造することができ、先に知られた類似の誘導体の欠点のいくつかを示さないという最初の予想外の知見に基づくものである。本発明は、このようなウレイル置換されたナフタルイミド誘導体が、他の既知の置換ナフタルイミド誘導体の加水分解を通して高収率で容易に入手できるという予想外の知見にも基づいている。本発明は、このようなウレイル置換されたナフタルイミド誘導体が、十分な化学安定性を示し、例えば、ナノ粒子の形態の懸濁液としてまたは塩の形態の溶液として、薬剤に容易に製剤化することができるという予想外の知見にも基づいている。
【発明を実施するための最良の形態】
【0007】
定義
特に指定のない限り、置換基に関して本明細書で使用される用語「アルキル」は、1から4個の炭素原子を有する直鎖および分枝鎖の飽和非環式炭化水素の一価の基、例えば、メチル、エチル、プロピル、n−ブチル、1−メチルエチル(イソプロピル)、2−メチルプロピル(イソブチル)、および1,1−ジメチルエチル(ter−ブチル)などを意味する。
【0008】
特に指定のない限り、置換基のメンバーに関して本明細書で使用される用語「アルキレン」は、上記に定義されたアルキルに対応する二価の炭化水素基、限定されるものではないが、メチレン、ビス(メチレン)、トリス(メチレン)、テトラメチレンなどを意味する。
【0009】
特に指定のない限り、置換基に関して本明細書で使用される用語「アルコキシ」および「アルキルチオ」は、本明細書で上記に定義されたアルキル基が、単結合によって酸素原子または二価の硫黄原子に結合している置換基、限定されるものではないが、メトキシ、エトキシ、プロポキシ、ブトキシ、イソプロポキシ、sec−ブトキシ、tert−ブトキシ、チオメチル、チオエチル、チオプロピル、チオブチルなどを意味する。
【0010】
特に指定のない限り、置換原子に関して本明細書で使用される用語「ハロゲン」は、フッ素、塩素、臭素およびヨウ素からなる群から選択される任意の原子を意味する。
【0011】
特に指定のない限り、置換基に関して本明細書で使用される用語「ハロアルキル」は、1個または複数の水素原子が独立に1個または複数のハロゲン(好ましくはフッ素、塩素または臭素)で置換されているアルキル基(上記に定義されたようなもの)、限定されるものではないが、ジフルオロメチル、トリフルオロメチル、トリフルオロエチル、ジクロロメチルなどを意味する。
【0012】
特に指定のない限り、本明細書で使用される用語「溶媒和物」には、適切な無機溶媒(例えば、水から形成される水和物)または適切な有機溶媒、限定されるものではないが、アルコール、ケトン、エステルなどと、本発明のウレイル置換ナフタルイミド(イソキノリンジオン)誘導体とによって形成され得る任意の組合せが含まれる。
【0013】
特に指定のない限り、本明細書で使用される用語「抗転移性」は、新生物腫瘍組織からの細胞の転移を停止させ、それゆえこれらの細胞による新たな組織の定着を減少させる医薬成分の能力を意味する。
【0014】
本明細書で使用される用語「細胞増殖性障害」は、任意のタイプの癌または細胞増殖を伴う他の病態、限定されるものではないが、白血病、肺癌、結腸直腸癌、中枢神経系(CNS)癌、メラノーマ、卵巣癌、腎癌、前立腺癌、乳癌、神経膠腫、膀胱癌、骨癌、肉腫、頭頸部癌、肝癌、精巣癌、膵癌、胃癌、食道癌、骨髄癌、十二指腸癌、眼癌(網膜芽腫)およびリンパ腫などを意味する。
【0015】
第1の態様においては、本発明は、構造式(I)により表される置換ナフタルイミド誘導体:
【化1】
【0016】
[式中、
− R1はモノまたはジ−C1〜4アルキルアミノ−C1〜4アルキルであり;
− 置換基R3およびR4のそれぞれは、水素、ハロゲン、C1〜4アルキル、C1−7アルコキシ、C1〜4アルキルチオ、ニトロ、シアノ、アミノ、保護されたアミノおよびハロC1〜4アルキルからなる群から独立に選択され;
− mは置換基R3の数であって、0から3の範囲であり;
− nは置換基R4の数であって、0から2の範囲であり;
− R2はCONH2である。]、
ならびに/または薬学的に許容されるそれらの塩および/もしくはそれらの溶媒和物および/もしくはそれらの代謝産物のグループを提供する。
【0017】
構造式(I)により表される誘導体の代謝産物には、限定されるものではないが、次のもの:
− それらのモノ−N−オキシドおよびジ−N−オキシド;
− R2がCONHOHである誘導体;ならびに
− R3および/またはR4がヒドロキシルである誘導体
が含まれる。
【0018】
あるいは、本発明のナフタルイミド誘導体のモノ−およびジ−N−オキシドは、限定されるものではないが、過酸化水素(例えば酢酸の存在下で)またはクロロ過安息香酸などの過酸などの酸化剤で構造式(I)により表される誘導体を処理することによって直接合成することができる。
【0019】
上記に定義された新規な化合物は、共通して、限定されるものではないが、アモナフィドなどのアミノ置換ナフタルイミド(イソキノリンジオン)のアミノ基がウレイル基、またはその代謝形態においてウレイルN−オキシド基で置換されているという、構造的な特徴を有する。
【0020】
この第1の態様の好ましい実施形態においては、本発明は、
− n=0(R4が水素ではない場合)であり、かつ/または
− m=0(R3が水素ではない場合)であり、かつ/または
− m=2であり、両方の置換基R3は隣接しており、それらが結合している炭素原子と一緒になってフェニル基を形成し、かつ/または
− R1は1から3個の炭素原子を有するアルキレン基であり、ジメチルアミノもしくはジエチルアミノ基に連結しており、かつ/または
− R2はCONH2
である化合物、ならびに/または薬学的に許容されるそれらの塩および/もしくはそれらの溶媒和物および/もしくはそれらの代謝産物のサブグループに関する。
【0021】
この第1の態様の別の好ましい実施形態においては、本発明は、
− n=m=0(R3およびR4が水素ではない場合)であり、かつ/または
− R1は1もしくは2個の炭素原子を有するアルキレン基であり、ジメチルアミノもしくはジエチルアミノ基に連結しており、かつ/または
− R2はCONH2
である化合物、ならびに/または薬学的に許容されるそれらの塩および/もしくはそれらの溶媒和物および/もしくはそれらの代謝産物のサブグループに関する。
【0022】
この第1の態様のさらに別の好ましい実施形態においては、本発明は、mが1に等しい場合にR3がニトロではない化合物、それらの塩、溶媒和物または代謝産物のサブグループに関する。この第1の態様のさらに別の好ましい実施形態においては、本発明は、R3および/またはR4が、水素、ハロゲン、C1〜4アルキル、C1−7アルコキシ、C1〜4アルキルチオ、シアノ、アミノ、アシルアミノおよびハロC1〜4アルキルからなる群から選択される化合物、それらの塩、溶媒和物または代謝産物のサブグループに関する。
【0023】
別の好ましい実施形態においては、本発明は、N−{2−[2−(ジメチルアミノ)エチル]−1,3−ジオキソ−2,3−ジヒドロ−1H−ベンゾ[de]イソキノリン−5−イル}尿素、その塩または代謝産物に関する。
【0024】
第2の態様においては、本発明は、5−置換アモナフィドまたはアモナフィド誘導体(ここで、その5置換基は、それが加水分解によりウレイル基に変換できるように選択される)を加水分解することによる、構造式(I)により表されるウレイル置換ナフタルイミド(イソキノリンジオン)誘導体の製造方法を提供する。加水分解に適する5−置換アモナフィド誘導体には、限定されるものではないが、構造式(II)を有する化合物:
【化2】
【0025】
[式中、
− m、n、R1、R3およびR4のそれぞれは、構造式(I)について定義された通りであり、
− R’はC1〜4アルコキシアミドカルボニルまたはC1ハロアルキルアミドカルボニルである]
が含まれる。
【0026】
上記の構造式(II)を有するいくつかの化合物は、例えばWO2005/105753号から既に知られるが、例えばC1〜4アルコキシカルボニルイソシアネート(エトキシカルボニルイソシアネートなど)またはC1ハロアルキルカルボニルイソシアネート(トリクロロアセチルイソシアネートもしくはトリフルオロアセチルイソシアネートなど)とアモナフィドとの反応の生成物として、中程度の収率でしか入手可能ではなかった。したがって、本発明の別の態様は、より良い収率でこれらの中間体を入手することを可能にする反応条件を設計することである。この目的のための1つの方法は、C1〜4アルコキシカルボニルイソシアネートまたはC1ハロアルキルカルボニルイソシアネートとアモナフィドとの前記反応が、
− エーテル(例えばジエチルエーテル)、ケトン(例えば2−ブタノンまたはメチルエチルケトン)およびハロゲン化炭化水素(好ましくは最大2個の炭素原子および/または少なくとも1個の塩素原子を有するもの、例えばジクロロメタン)からなる群から選択される溶媒の存在、ならびに/あるいは
− 0℃未満の温度(例えば約−30℃から約−5℃の範囲の温度)、ならびに/あるいは
− 前記C1〜4アルコキシカルボニルイソシアネートまたはC1ハロアルキルカルボニルイソシアネートのモル過剰量、ならびに/あるいは
− その完了後に反応混合物に水を添加することにより反応をクエンチし、望ましくない環化副生成物の形成(C1〜4アルコキシカルボニルイソシアネートまたはC1ハロアルキルカルボニルイソシアネートのモル過剰量が使用される場合)を回避すること
を含む条件下で実施される方法である。
【0027】
上記反応条件の1つまたは複数が使用される場合、上記構造式(II)を有する化合物は、先行技術によるよりも同一またはより短い反応時間内で著しく向上した収率で得ることができる。当業者は、前述のプロセスの特徴のどの組合せが、R’、R1、R3およびR4の正確な性質などのパラメーターに応じて、可能な限り最短の反応時間内で最も適当な収率を提供し得るかを容易に決定することができる。
【0028】
限定されるものではないが、構造式(II)を有する化合物などの、その5−置換基がウレイル基に変換され得る5−置換アモナフィドまたはアモナフィド誘導体の加水分解は、酸性条件下または塩基性条件下のいずれかで実施し得る。当業者は、この種の加水分解は、限定されるものではないが、pH、温度、使用される酸または塩基の種類および反応混合物のための溶媒の種類などのパラメーターによって、副生成物としてアモナフィドを生成しやすく、このアモナフィドは、次いで、構造式(I)を有する所望の化合物から分離する必要があることを容易に理解されよう。アモナフィドの形成を最小化するための最適な条件の決定は、当業者の一般的知識に含まれる。本発明の1つの利点は、最終生成物中の残留アモナフィドの割合を3重量%未満に保つことは極めて容易であることがわかったことである。
【0029】
第3の態様においては、本発明は、
− 構造式(I)により表されるウレイル置換ナフタルイミド(イソキノリンジオン)誘導体、ならびに/または薬学的に許容されるその塩および/もしくはその溶媒和物および/もしくはその代謝産物の治療有効量;ならびに
− 1種または複数の薬学的に許容される担体
を含む医薬組成物を提供する。
【0030】
別の態様においては、本発明は、少なくとも1種の構造式(I)により表されるウレイル置換ナフタルイミド(イソキノリンジオン)誘導体、ならびに/または薬学的に許容されるその塩および/もしくはその溶媒和物および/もしくはその代謝産物と、1種または複数の抗新生物薬とを含有する組合せ製剤(好ましくは下記に詳述される相乗的組合せの形態における)を提供する。
【0031】
別の態様においては、本発明は、一般式(I)で表される置換ナフタルイミド(イソキノリンジオン)誘導体、ならびに/または薬学的に許容されるそれらの塩および/もしくはそれらの溶媒和物および/もしくはそれらの代謝産物が、アモナフィドの上述の欠点の多くを回避しながら、アモナフィドに比べて、特に腫瘍細胞に関して、著しくより高い生物活性を有するという予想外の知見に関する。特に、本発明に係るウレイル置換ナフタルイミド誘導体は、著しい抗転移効果を有する。転移は、細胞が、転移の主要な経路として血管またはリンパ管を使用して、新生物腫瘍組織から移動し、新組織を定着する生物学的プロセスを意味し、この生物学的プロセスは、転移プロセスとしても知られている。この知見に基づき、本発明は、ヒトの腫瘍の治療および/または予防方法を提供する。より具体的には、本発明は、細胞増殖性疾患を有する宿主の治療方法であって、前記宿主に、構造式(I)により表されるウレイル置換ナフタルイミド(イソキノリンジオン)誘導体、ならびに/または薬学的に許容されるそれらの塩および/もしくはそれらの溶媒和物および/もしくはそれらの代謝産物の有効量を接触させることを含む方法に関する。
【0032】
別の実施形態においては、本発明は、抗腫瘍剤としての、構造式(I)により表されるウレイル置換ナフタルイミド(イソキノリンジオン)誘導体、ならびに/または薬学的に許容されるそれらの塩および/もしくはそれらの溶媒和物および/もしくはそれらの代謝産物の使用を提供する。
【0033】
別の特定の実施形態においては、本発明は、上記構造式(I)を有し、薬学的に許容される塩の形態である、ウレイル置換ナフタルイミド(イソキノリンジオン)誘導体、ならびにそれらを活性成分として含む医薬組成物のグループに関する。後者には、構造式(I)を有する化合物が、塩形成薬剤と形成し得る任意の治療活性を有する非毒性塩が含まれる。このような付加塩は、本発明のウレイル置換ナフタルイミド(イソキノリンジオン)誘導体を適切な塩形成酸または塩基で処理することにより都合よく得ることができる。例えば、塩基性を有するウレイル置換ナフタルイミド(イソキノリンジオン)誘導体は、通常の手法に従い遊離塩基形態を適量の適切な酸で処理することによって対応する治療活性を有する非毒性酸塩の形態に変換し得る。このような適切な塩形成酸の例には、例えば、限定されるものではないが、塩の形成をもたらす無機酸、例えばハロゲン化水素酸塩(例えば塩酸塩および臭化水素酸塩)、硫酸塩、硝酸塩、リン酸塩、二リン酸塩、炭酸塩、重炭酸塩など;および、塩の形成をもたらす有機モノカルボン酸またはジカルボン酸、例えば酢酸塩、プロピオン酸塩、ヒドロキシ酢酸塩、2−ヒドロキシプロピオン酸塩、2−オキソプロピオン酸塩、乳酸塩、ピルビン酸塩、シュウ酸塩、マロン酸塩、コハク酸塩、マレイン酸塩、フマル酸塩、リンゴ酸塩、酒石酸塩、クエン酸塩、メタンスルホン酸塩、エタンスルホン酸塩、安息香酸塩、2−ヒドロキシ安息香酸塩、4−アミノ−2−ヒドロキシ安息香酸塩、ベンゼンスルホン酸塩、p−トルエン−スルホン酸塩、サリチル酸塩、p−アミノサリチル酸塩、パモ酸塩、重酒石酸塩、カンファースルホン酸塩、エデト酸塩、1,2−エタンジスルホン酸塩、フマル酸塩、グルコヘプタン酸塩、グルコン酸塩、グルタミン酸塩、ヘキシルレゾルシン酸塩、ヒドロキシナフトエ酸塩、ヒドロキシエタンスルホン酸塩、マンデル酸塩、メチル硫酸塩、パントテン酸塩、ステアリン酸塩など、ならびにエタン二酸、プロパン二酸、ブタン二酸、(Z)−2−ブテン二酸、(E)2−ブテン二酸、2−ヒドロキシブタン二酸、2,3−ジヒドロキシブタン二酸、2−ヒドロキシ−1,2,3−プロパン−トリカルボン酸、シクロヘキサン−スルファミン酸などから誘導される塩、が含まれる。
【0034】
酸性を有する構造式(I)を有するウレイル置換ナフタルイミド(イソキノリンジオン)誘導体は、同様の方法で対応する治療活性を有する非毒性塩基塩の形態に変換し得る。適切な塩形成塩基の例には、例えば、金属水酸化物のような無機塩基、限定されるものではないが、対応する金属塩をもたらす、カルシウム、リチウム、マグネシウム、カリウムおよびナトリウムなどのアルカリ金属およびアルカリ土類金属、または亜鉛の水酸化物など;窒素含有有機塩基、限定されるものではないが、アンモニア、アルキルアミン、ベンザチン、ヒドラバミン、アルギニン、リシン、N,N’−ジベンジルエチレンジアミン、クロロプロカイン、コリン、ジエタノールアミン、エチレンジアミン、N−メチルグルカミン、プロカインなどが含まれる。
【0035】
適切な塩形成酸または塩基で本発明のウレイル置換ナフタルイミド(イソキノリンジオン)誘導体(I)を処理するための反応条件は、それぞれ塩基性または酸性の異なる有機化合物を除いて、同一の酸または塩基が関与する標準条件と類似している。好ましくは、細胞増殖性疾患の治療のための医薬組成物におけるその使用または薬剤の製造におけるその使用を考慮して、薬学的に許容される塩は設計され、すなわち、塩形成酸または塩基は、本発明のウレイル置換ナフタルイミド(イソキノリンジオン)誘導体に、より大きな水溶性、より低い毒性、より大きな安定性および/またはより遅い溶出速度を付与するように選択される。
【0036】
本発明は、生物活性成分、すなわち活性成分(特に薬剤または診断薬)としての、あるいは薬剤または診断用キットの製造のための、構造式(I)により表されるウレイル置換ナフタルイミド(イソキノリンジオン)誘導体、または薬学的に許容されるそれらの塩もしくはそれらの溶媒和物および/もしくはそれらの代謝産物の使用をさらに提供する。特に、前記薬剤は、細胞増殖性障害からなる群から選択される病態の予防または治療用であり得る。
【0037】
本発明に係る化合物は、いくつかのタイプの癌に対して極めて活性である。したがって、それらの好ましい薬理学的特性のために、本発明に係る化合物は、細胞増殖に関係する疾患を患う患者の治療、特に癌の治療のための薬剤としての使用、または薬剤および組合せ製剤の製造における使用に特に適している。
【0038】
上記に記載された使用のいずれも、非医学的用途(例えば化粧用組成物における)、非治療用途、非診断用途、ヒト以外の用途(例えば獣医用組成物)、または専らインビトロでの使用、または動物から離れた細胞での使用に制限されてもよい。
【0039】
本発明は、さらに
(a)1種または複数の構造式(I)で表されるウレイル置換ナフタルイミド(イソキノリンジオン)誘導体、ならびに/または薬学的に許容されるそれらの塩および/もしくはそれらの溶媒和物および/もしくはそれらの代謝産物、ならびに
(b)1種または複数の薬学的に許容される担体
を含む医薬組成物に関する。
【0040】
別の実施形態においては、本発明は、1種または複数の構造式(I)で表されるウレイル置換ナフタルイミド(イソキノリンジオン)誘導体、ならびに/または薬学的に許容されるそれらの塩および/もしくはそれらの溶媒和物および/もしくはそれらの代謝産物と、抗新生物薬からなる群から選択されるのが好ましい1種または複数の生物活性薬物との組合せ製剤、好ましくは相乗的組合せを提供する。当技術分野で普通であるように、複合薬における相乗効果の評価は、ChouらによりAdv. Enzyme Reg. (1984) 22:27に記載された半有効原理を用いて個別の薬物の間の相互作用の定量化の分析によって行うことができる。簡単に説明すると、この原理は、2種の薬物の間の相互作用(相乗作用、相加作用、拮抗作用)は、次の方程式:
【数1】
【0041】
(式中、EDxは、所与の効果を生ずるために必要である、単独使用される第1もしくはそれぞれ第2の薬物(1a、2a)の用量、または第2もしくはそれぞれ第1の薬物(1c、2c)と組み合わせた用量である)
により定義される組合せ指数(以下CIという)を用いて定量化することができることを記載している。第1および第2の薬物は、それぞれCI<1、CI=1、またはCI>1に応じて、相乗作用または相加作用または拮抗作用を有する。本明細書で下記により詳しく説明されるように、この原理は、限定されるものではないが、細胞増殖に対する活性などのいくつかの所望の効果に適用され得る。
【0042】
本発明は、さらに、
(a)1種または複数の抗新生物薬、ならびに
(b)少なくとも1種の構造式(I)で表されるウレイル置換ナフタルイミド(イソキノリンジオン)誘導体、ならびに/または薬学的に許容されるそれらの塩および/もしくはそれらの溶媒和物および/もしくはそれらの代謝産物、ならびに
(c)場合によっては1種または複数の薬学的賦形剤または薬学的に許容される担体
を含有する、細胞増殖性障害の治療または予防における同時、別々または順次使用のための、組成物または細胞増殖に対する相乗効果を有する組合せ製剤に関する。
【0043】
本発明の相乗的な抗増殖医薬組成物または組合せ製剤に含まれるのに適する抗新生物薬は、好ましくは、アルカロイド、アルキル化剤(限定されるものではないが、アルキルスルホネート、アジリジン、エチレンイミン、メチルメラミン、ナイトロジェン・マスタードおよびニトロソ尿素を含む)、抗生物質、代謝拮抗物質(限定されるものではないが、葉酸類似体、プリン類似体およびピリミジン類似体を含む)、酵素、インターフェロンおよび白金錯体からなる群から選択される。より具体的な例には、アシビシン、アクラルビシン、アコダゾール、アクロニン、アドゼレシン、アルデスロイキン、アルトレタミン、アンボマイシン、アメタントロン、アミノグルテチミド、アムサクリン、アナストロゾール、アンスラマイシン、アスパラギナーゼ、アスペルリン、アザシチジン、アゼテパ、アゾトマイシン、バチマスタット、ベンゾデパ、ビカルタミド、ビサントレン、ビスナフィド、ビゼレシン、ブレオマイシン、ブレキナール、ブロピリミン、ブスルファン、カクチノマイシン、カルステロン、カラセミド、カルベチマー、カルボプラチン、カルムスチン、カルビシン、カルゼレシン、セデフィンゴール、クロラムブシル、シロレマイシン、シスプラチン、クラドリビン、クリスナトール、シクロホスファミド、シタラビン、ダカルバジン、ダクチノマイシン、ダウノルビシン、デシタビン、デキソルマプラチン、デザグアニン、ジアジクオン、ドセタキセル、ドキソルビシン、ドロロキシフェン、ドロモスタノロン、デュアゾマイシン、エダトレキサート、エフロミチン、エルサミトルシン、エンロプラチン、エンプロメート、エピプロピジン、エピルビシン、エルブロゾール、エソルビシン、エストラムスチン、エタニダゾール、エチオダイズ(ethiodized)化油I131、エトポシド、エトプリン、ファドロゾール、ファザラビン、フェンレチニド、フロキシウリジン、フルダラビン、フルオロウラシル、フルロシタビン、ホスキドン、ホストリエシン、ゲムシタビン、Gold 198、ヒドロキシ尿素、イダルビシン、イホスファミド、イルモホシン、インターフェロンα−2a、インターフェロンα−2b、インターフェロンα−n1、インターフェロンα−n3、インターフェロンβ−1a、インターフェロンγ−1b、イプロプラチン、イリノテカン、ランレオチド、レトロゾール、ロイプロリド、リアロゾール、ロメトレキソール、ロムスチン、ロソキサントロン、マソプロコール、マイタンシン、メクロレタミン、メゲストロール、メレンゲストロール、メルファラン、メノガリル、メルカプトプリン、メトトレキサート、メトプリン、メツレデパ、ミチンドミド、ミトカルシン、ミトクロミン、ミトギリン、ミトマルシン、マイトマイシン、ミトスペル、ミトタン、ミトキサントロン、ミコフェノール酸、ノコダゾール、ノガラ−マイシン、オルマプラチン、オキシスラン、パクリタキセル、ペガスパルガーゼ、ペリオマイシン、ペンタムスチン、ペプロマイシン、ペルホスファミド、ピポブロマン、ピポスルファン、ピロキサントロン、プリカマイシン、プロメスタン、ポルフィメル、プロフィロマイシン、プレドニムスチン、プロカルバジン、ピューロマイシン、ピラゾフリン、リボプリン、ログレチミド、サフィンゴール、セムスチン、シムトラゼン、スパルホセート、スパルソマイシン、スピロゲルマニウム、スピロムスチン、スピロプラチン、ストレプトニグリン、ストレプトゾシン、塩化ストロンチウム89、スロフェナル、タリソマイシン、タキサン、タキソイド、テコガラン、テガフール、テロキサントロン、テモポルフィン、テニポシド、テロキシロン、テストラクトン、チアミプリン、チオグアニン、チオテパ、チアゾフリン、チラパザミン、トポテカン、トレミフェン、トレストロン、トリシリビン、トリメトレキセート、トリプトレリン、ツブロゾール、ウラシルマスタード、ウレデパ、バプレオチド、ベルテポルフィン、ビンブラスチン、ビンクリスチン、ビンデシン、ビネピジン、ビングリシネート、ビンロイロシン、ビノレルビン、ビンロシジン、ビンゾリジン、ボロゾール、ゼニプラチン、ジノスタチン、ゾルビシンおよびその薬学的に許容される塩が含まれる。
【0044】
本発明の相乗的な抗増殖医薬組成物または組合せ製剤に含まれるのに適する他の抗新生物化合物には、20−エピ−1,25ジヒドロキシビタミンD3、5−エチニルウラシル、アビラテロン、アクラルビシン、アシルフルベン、アデシペノール、アドゼレシン、アルデスロイキン、ALL−TKアンタゴニスト、アルトレタミン、アンバムスチン、アミドックス、アミホスチン、アミノレブリン酸、アムルビシン、アムサクリン、アナグレリド、アナストロゾール、アンドログラホリド、血管新生阻害剤、アンタゴニストD、アンタゴニストG、アンタレリックス、抗背側形態形成タンパク質−1、限定はしないが、ベノルテロン、シオテロネル、シプロテロン、デルマジノン、オキセンドロン、トプテロン、ザノテロンのような抗アンドロゲン、限定はしないが、クロメテロン、デルマジノン、ナホキシジン、ニトロミフェン、ラロキシフェン、タモキシフェン、トレミフェン、トリオキシフェンのような抗エストロゲン、およびその薬学的に許容される塩、抗ネオプラストン、アンチセンスオリゴヌクレオチド、アフィジコリングリシネート、アポトーシス遺伝子調節因子、アポトーシス調節因子、アプリン酸、アラ−CDP−DL−PTBA、アルギニンデアミナーゼ、アスラクリン、アタメスタン、アトリムスチン、アキシナスタチン、アザセトロン、アザトキシン、アザチロシン、バッカチンIII誘導体、バラノール、バチマスタット、BCR/ABLアンタゴニスト、ベンゾクロリン、ベンゾイルスタウロスポリン、β−ラクタム誘導体、β−アレチン、βクラマイシンB、ベツリン酸、bFGF阻害剤、ビカルタミド、ビサントレン、ビサジリジニルスペルミン、ビスナフィド、ビストラテンA、ビゼレシン、ブレフレート、ブロピリミン、ブドチタン、ブチオニンスルホキシイミン、カルシポトリオール、カルホスチンC、カンプトテシン誘導体、カナリポックスIL−2、カペシタビン、カルボキサミド−アミノトリアゾール、カルボキシアミドトリアゾール、CaRest M3、CARN700、軟骨組織由来阻害剤、カルゼレシン、カゼインキナーゼ阻害剤、カスタノスペルミン、セクロピンB、セトロレリックス、クロリン、クロロキノキサリン、スルホンアミド、シカプロスト、シス−ポルフィリン、クロミフェンおよびその類似体、クロトリマゾール、コリスマイシンAおよびB、コンブレタスタチンおよびその類似体、コナゲニン、クラムベスシジン816、クリプトフィシンおよびその誘導体、クラシンA、シクロペンタントラキノン、シクロプラタム、シペマイシン、シタラビン、細胞溶解因子、シトスタチン、ダクリキシマブ、デヒドロジデムニンB、デスロレリン、デキシホスファミド、デキスラゾキサン、デキシベラパミル、ジデムニンB、ジドックス、ジエチルノルスペルミン、ジヒドロ−5−アザシチジン、ジヒドロタキソール、ジオキサマイシン、ジフェニルスピロムスチン、ドコサノール、ドラセトロン、ドキシフルリジン、ドロロキシフェン、ドロナビノール、デュオカルマイシンSA、エブセレン、エコムスチン、エデルホシン、エドレコロマブ、エレメン、エミテフル、エプリステリド、エストロゲンアゴニストおよびアンタゴニスト、エキセメスタン、フィルグラスチム、フィナステリド、フラボピリドール、フレゼラスチン、フルアステロン、フルオロダウノルニシン、ホルフェニメックス、ホルメスタン、ホテムスチン、ガドリニウムテキサフィリン、硝酸ガリウム、ガロシタビン、ガニレリックス、ゼラチナーゼ阻害剤、グルタチオン阻害剤、ヘプスルファム、ヘレグリン、ヘキサメチレンビスアセトアミド、ヒペリシン、イバンドロン酸、イドキシフェン、イドラマントン、イロマスタット、イミダゾアクリドン、イミキモド、免疫刺激性ペプチド、インスリン様増殖因子−1レセプター阻害剤、インターフェロンアゴニスト、イオベングアン、ヨードドキソルビシン、イポメアノール、イリノテカン、イロプラクト、イルソグラジン、イソベンガゾール、イソホモハリコンドリンB、イタセトロン、ジャスプラキノリド、カハラリドF、ラメラリン−N、レイナマイシン、レノグラスチム、レンチナン、レプトルスタチン、白血病阻害因子、リュープロレリン、レバミソール、リアロゾール、リソクリンアミド、ロバプラチン、ロムブリシン、ロニダミン、ロバスタチン、ロキソリビン、ルルトテカン、ルテチウムテキサフィリン、リソフィリン、マンノスタチンA、マリマスタット、マソプロコール、マスピン、マトリリシン阻害剤、マトリックスメタロプロテイナーゼ阻害剤、メルバロン、メテレリン、メチオニナーゼ、メトクロプラミド、MIF阻害剤、ミフェプリストン、ミルテホシン、ミリモスチム、ミトグアゾン、ミトラクトール、ミトナフィド、ミトトキシン繊維芽細胞増殖因子−サポリン、モファロテン、モルグラモスチム、ヒト絨毛膜ゴナドトロピンモノクローナル抗体、モピダモール、ミカペロキシドB、ミリアポロン、N−アセチルジナリン、N−置換ベンズアミド、ナファレリン、ナグレスチップ、ナロキソン、ペンタゾシン、ナパビン、ナフテルピン、ナルトグラスチム、ネダプラチン、ネモルビシン、ネリドロン酸、中性エンドペプチダーゼ、ニルタミド、ニサマイシン、酸化窒素調節因子、ニトロキシド抗酸化剤、ニトルリン、オクトレチド、オキセノン、オナプリストン、オンダンセトロン、オンダンセトロン、オラシン、オサテロン、オキサリプラチン、オキサウノマイシン、パラウアミン、パルミトイルリゾキシン、パミドロン酸、パナキシトリオール、パノミフェン、パラバクチン、パゼリプチン、ペルデシン、ペントサン、ペントスタチン、ペントロゾール、ペルフルブロン、ペリリルアルコール、フェナジノマイシン、フェニルアセテート、ホスファターゼ阻害剤、ピシバニル、ピロカルピン、ピラルビシン、ピリトレキシム、プラセチンAおよびB、プラスミノーゲン活性化阻害剤、プロピルビス−アクリドン、プロスタグランジンJ2、プロテアソーム阻害剤、タンパク質キナーゼC阻害剤、タンパク質チロシンホスファターゼ阻害剤、プリンヌクレオシドホスホリラーゼ阻害剤、プルプリン、ピラゾロアクリジン、ラルチトレキセド、ラモセトロン、rasファルネシルタンパク質トランスフェラーゼ阻害剤、ras阻害剤、ras−GAP阻害剤、レテリプチン、レニウム186エチドロネート、リゾキシン、レチンアミド、ロヒツキン、ロムルチド、ロキニメックス、ルビギノンB1、ルボキシル、サイントピン、サルコフィトールA、サルグラモスチム、シゾフィラン、ソブゾキサン、ナトリウムボロカプテート、フェニル酢酸ナトリウム、ソルベロール、ソマトメジン結合タンパク質、ソネルミン、スパルホシン酸、スピカマイシンD、スプレノペンチン、スポンギスタチン1、スクアラミン、幹細胞分裂阻害剤、スチピアミド、ストロメリシン阻害剤、スルフィノシン、スラジスタ、スラミン、スワインソニン、タリムスチン、タモキシフェン、タウロムスチン、タザロテン、テコガラン、テルラピリリウム、テロメラーゼ阻害剤、テモゾロミド、テトラクロロデカオキシド、テトラゾミン、タリブラスチン、チオコラリン、トロンボポエチン、チマルファシン、チモポエチンレセプターアゴニスト、チモトリナン、甲状腺刺激ホルモン、スズエチルエチオプルプリン、チタノセン、トプセンチン、トレチノイン、トリアセチルウリジン、トロピセトロン、ツロステリド、チロシンキナーゼ阻害剤、チルホスチン、ウベニメックス、尿生殖洞由来増殖阻害因子、ウロキナーゼレセプターアンタゴニスト、バリオリンB、ベラレソール、ベラミン、ベルジン、ベルテポルフィン、ビンキサルチン、ビタキシン、ザノテロン、ジラスコルブ、およびその薬学的に許容される塩が含まれる。
【0045】
細胞増殖に対する本発明の医薬組成物または組合せ製剤の相乗活性は、限定されるものではないが、腫瘍細胞系の培養物中の3H−チミジンの取り込みから生じる放射能の測定などの1種または複数の試験を用いて容易に判定することができる。例えば、限定されるものではないが、
− RPMI1788:ヒト末梢血白血球(PBL)白人腫瘍系、
− Jurkat:ヒト急性T細胞白血病、
− EL4:C57BI/6マウスリンパ腫、または
− THP−1:ヒト単球腫瘍系
などの様々な腫瘍細胞系が、試験化合物の抗腫瘍効果を評価するために選択され得る。
【0046】
選択された腫瘍細胞系に応じて、例えば、
− RPMI1788およびTHP−1に対して:RPMI−1640+10%FCS+1%NEAA+1%ピルビン酸ナトリウム+5×10−5メルカプト−エタノール+抗生物質(G−418 0.45μg/ml)、
− JurkatおよびEL4に対して:RPMI−1640+10%FCS+抗生物質(G−418 0.45μg/ml)
などの様々な培養培地が使用され得る。
【0047】
相乗作用測定試験の特定の実施形態においては、腫瘍細胞系を回収し、完全培地中の0.27×106細胞/mlの懸濁液を調製する。懸濁液(150μl)を、マイクロタイタープレートに3通りに添加する。試験濃度(50μl)における完全培地(対照)または試験化合物のいずれかを、マイクロタイタープレート中の細胞懸濁液に添加する。細胞を、5%CO2下37℃で約16時間インキュベートする。3H−チミジンを添加し、細胞をさらに8時間インキュベートする。細胞を回収し、放射能をβ−カウンターにおいて、カウント毎分(CPM)で測定する。3H−チミジン細胞含有量、したがって測定された放射能は、細胞系の増殖に比例している。相乗効果は、本明細書で先に開示された半有効解析法により評価する。
【0048】
本発明に係る、細胞増殖に対する相乗活性を有する医薬組成物または組合せ製剤は、製剤の正確な目的用途および期待効果に応じた広い含有量範囲にわたって、構造式(I)を有するウレイル置換ナフタルイミド(イソキノリンジオン)誘導体、ならびに/または薬学的に許容されるそれらの塩および/もしくはそれらの溶媒和物および/もしくはそれらの代謝産物を含有し得る。一般的に、組合せ製剤のウレイル置換ナフタルイミド(イソキノリンジオン)誘導体含有量は、約0.1から約99.9重量%、好ましくは1から99重量%、より好ましくは5から95重量%の範囲内である。
【0049】
本発明に係る医薬組成物および組合せ製剤は、経口または任意の他の適切な方法で投与し得る。経口投与が好ましく、製剤は、錠剤、水性分散剤、分散可能な散剤もしくは顆粒剤、乳剤、硬もしくは軟カプセル剤、シロップ剤、エリキシル剤またはゲル剤の形態を有し得る。投与剤形は、これらの医薬組成物の製造用の当技術分野で既知の任意の方法を用いて調製することができ、甘味料、香料、着色剤、保存料などを添加剤として含むことができる。担体材料および賦形剤は、本明細書で下記に詳述され、とりわけ、炭酸カルシウム、炭酸ナトリウム、乳糖、リン酸カルシウムまたはリン酸ナトリウム;造粒剤および崩壊剤、結合剤などを含むことができる。本発明の医薬組成物または組合せ製剤は、任意の不活性固体希釈剤または担体材料と混合されるゼラチンカプセル剤中に含まれ得、あるいは成分が水または油媒体と混合される軟ゼラチンカプセル剤の形態を有する。水性分散剤は、生物活性を有する組成物または組合せ製剤を、懸濁化剤、分散剤または湿潤剤と組み合わせて含み得る。油分散剤は、植物油などの懸濁化剤を含み得る。直腸投与も、例えば、坐剤またはゲル剤の形態で適用できる。注射(例えば、静脈内、筋肉内または腹腔内)も、治療すべき障害および患者の状態に応じて、例えば、注射溶剤または分散剤の形態で投与方法として適用できる。
【0050】
特に指定のない限り、医薬組成物および組合せ製剤に関係して本明細書で使用される用語「薬学的に許容される担体または賦形剤」は、任意の材料または物質を意味し、この任意の材料または物質と一緒に、活性成分(複数でもよい)、すなわち、本発明のウレイル置換ナフタルイミドおよび場合によっては抗新生物薬が、例えば、前記組成物を溶解すること、分散することまたは拡散することにより治療すべき位置へのその適用または散布を促進するために、かつ/あるいはその有効性を損なうことなくその貯蔵、輸送または取り扱いを促進するために製剤化され得る。薬学的に許容される担体は、固体または液体または圧縮されて液体を形成する気体であってよく、すなわち、本発明の組成物は、濃縮物、乳剤、溶剤、顆粒剤、微粉剤、噴霧剤、エアゾール剤、ペレット剤または散剤として適切に使用し得る。
【0051】
本発明の前記医薬組成物における使用のための適切な薬学的担体、およびその製剤化のための効率的な方法は、薬理学の当業者に周知である。本発明内でそれらの選択に特定の制限はないが、本発明のプテリジン誘導体の通例低いまたは非常に低い水溶性のために、予想される時間放出プロフィールを考慮して、それらを適切に製剤化する助けとなり得る適切な担体の組合せの選択に特別な注意が払われよう。適切な薬学的担体には、湿潤剤、分散剤、固着剤、接着剤、乳化剤または界面活性剤、増粘剤、錯化剤、ゲル化剤、溶媒、被覆剤、抗菌剤および抗真菌剤(例えば、フェノール、ソルビン酸、クロロブタノール)、等張剤(糖または塩化ナトリウムなど)などの添加剤が含まれるが、但し、これらは薬学的な実施と一致するものに限られ、すなわち、哺乳動物に永久的な損傷を生じない担体および添加物に限られる。本発明の医薬組成物は、例えば、選択された担体物質および(適切な場合)界面活性剤などのその他の添加剤と一緒に、1段階または多段階手順で、活性成分を均一に混合、溶解、噴霧乾燥、コーティングおよび/または粉砕することによって任意の既知の方法で調製し得る。本発明の医薬組成物は、例えば、通例約1から10μmの直径を有するミクロスフェアの形態でそれらを得るために、すなわち、生物活性成分(複数でもよい)の制御放出または持続放出用のマイクロカプセルの製造のために、微粉化によっても調製し得る。
【0052】
本発明の医薬組成物における使用のための適切な界面活性剤は、好ましくは、良好な乳化特性、分散特性および/または湿潤特性を有する非イオン性、陽イオン性および/または陰イオン性材料である。このような適切な陰イオン性界面活性剤には、水溶性石鹸および水溶性合成界面活性剤の両方が含まれる。適切な石鹸は、高級脂肪酸(C10−C22)のアルカリまたはアルカリ土類金属塩、非置換または置換アンモニウム塩、例えば、オレイン酸もしくはステアリン酸、またはココナッツ油もしくは獣脂油から得られる天然脂肪酸混合物のナトリウムまたはカリウム塩である。合成界面活性剤には、ポリアクリル酸のナトリウムまたはカルシウム塩;脂肪スルホネートおよびスルフェート;スルホン化ベンズイミダゾール誘導体およびアルキル−アリールスルホネートが含まれる。脂肪スルホネートおよびスルフェートは、通常、8から22個の炭素原子を有するアルキルまたはアシル基で置換されたアルカリまたはアルカリ土類金属塩、非置換アンモニウム塩またはアンモニウム塩の形態であり、例えば、リグノスルホン酸またはドデシルスルホン酸のナトリウムまたはカルシウム塩、あるいは天然脂肪酸から得られる脂肪アルコールスルフェートの混合物、硫酸またはスルホン酸エステル(ラウリル硫酸ナトリウムなど)のアルカリまたはアルカリ土類金属塩、および脂肪アルコール/エチレンオキシド付加物のスルホン酸である。適切なスルホン化ベンズイミダゾール誘導体は、好ましくは、8から22個の炭素原子を含有する。アルキルアリールスルホネートの例は、ドデシル−ベンゼンスルホン酸またはジブチル−ナフタレンスルホン酸またはナフタレンスルホン酸/ホルムアルデヒド縮合生成物のナトリウム、カルシウムまたはアルカノールアミン塩である。同様に、本発明の実施に適するのは、対応するホスフェート、例えば、リン酸エステルの塩、ならびにp−ノニル−フェノールとエチレンおよび/またはプロピレンオキシドの付加物、あるいはリン脂質である。本目的に適する適切なリン脂質には、限定されるものではないが、例えば、ホスファチジル−エタノールアミン、ホスファチジルセリン、ホスファチジルグリセリン、リゾレシチン、カルジオリピン、ジオクタニル−ホスファチジルコリン、ジパルミトイルホスファチジルコリンおよびそれらの混合物などのセファリンまたはレシチンタイプの天然(動物もしくは植物細胞に由来)または合成リン脂質が含まれる。
【0053】
適切な非イオン性界面活性剤には、分子中に少なくとも12個の炭素原子を含有するアルキルフェノール、脂肪アルコール、脂肪酸、脂肪族アミンまたはアミドのポリエトキシル化およびポリプロポキシル化誘導体、脂肪族および脂環式アルコール、飽和および不飽和脂肪酸およびアルキルフェノールのポリグリコールエーテル誘導体などのアルキルアレンスルホネートおよびジアルキルスルホスクシネートが含まれ、前記誘導体は、好ましくは、(脂肪族)炭化水素部分中に3から10個のグリコールエーテル基および8から20個の炭素原子、そしてアルキルフェノールのアルキル部分中に6から18個の炭素原子を含有する。さらに適切な非イオン性界面活性剤は、アルキル鎖中に1から10個の炭素原子を含有するポリエチレンオキシドとポリプロピレングリコール、エチレンジアミノポリプロピレングリコールの水溶性付加物であり、この付加物は、20から250個のエチレングリコールエーテル基および/または10から100個のプロピレングリコールエーテル基を含有する。このような化合物は、通常、プロピレングリコール単位当たり1から5個のエチレングリコール単位を含有する。非イオン性界面活性剤の代表例は、ノニルフェノール−ポリエトキシエタノール、ヒマシ油ポリグリコールエーテル、ポリプロピレン/ポリエチレンオキシド付加物、トリブチルフェノキシポリエトキシエタノール、ポリエチレングリコールおよびオクチルフェノキシポリエトキシエタノールである。ポリエチレンソルビタン(ポリオキシエチレンソルビタントリオレエートなど)、グリセロール、ソルビタン、ショ糖およびペンタエリトリトールの脂肪酸エステルも、適切な非イオン性界面活性剤である。
【0054】
本発明を実施するために適する陽イオン性界面活性剤には、限定されるものではないが、ハロ、フェニル、置換フェニルまたはヒドロキシで場合によっては置換されていてもよい4個の炭化水素基を有する第4級アンモニウム塩、好ましくはハロゲン化物;例えば、N−置換基として少なくとも1個のC8−C22アルキル基(例えば、セチル、ラウリル、パルミチル、ミリスチル、オレイルなど)と、さらなる置換基として非置換またはハロゲン化低級アルキル、ベンジルおよび/またはヒドロキシ−低級アルキル基を含有する第4級アンモニウム塩が含まれる。
【0055】
本目的に適した界面活性剤のより詳細な説明は、例えば、「McCutcheon's Detergents and Emulsifiers Annual」(MC Publishing Crop., Ridgewood, New Jersey, 1981)、「Tensid-Taschenbuch」、2nd ed. (Hanser Verlag, Vienna, 1981)および「Encyclopaedia of Surfactants」(Chemical Publishing Co., New York, 1981)に見い出し得る。
【0056】
構造形成剤、増粘剤またはゲル形成剤も、本発明の医薬組成物および組合せ製剤に含まれ得る。適切なこのような薬剤には、特に、限定されるものではないが、商標Aerosilとして市販されている製品などの高分散ケイ酸;ベントナイト;モンモリロナイトのテトラアルキルアンモニウム塩(例えば、商標Bentoneとして市販されている製品)(ここで、前記アルキル基のそれぞれは、1から20個の炭素原子を含有し得る);セトステアリルアルコールおよび変性ヒマシ油生成物(例えば、商標Antisettleとして市販されている製品)が含まれる。
【0057】
本発明の医薬組成物および組合せ製剤に同様に含まれ得るゲル化剤には、限定されるものではないが、カルボキシメチルセルロース、酢酸セルロースなどのセルロース誘導体;アラビアゴム、キサンタンガム、トラガカントガム、グアーガムなどの天然ゴム;ゼラチン;二酸化ケイ素;カルボマーなどの合成ポリマーおよび任意の適切な割合のそれらの混合物が含まれる。ゼラチンおよび変性セルロースは、ゲル化剤の好ましいクラスである。
【0058】
本発明の医薬組成物および組合せ製剤に同様に存在し得る他の任意選択の賦形剤には、限定されるものではないが、酸化マグネシウムなどの添加剤;アゾ染料;二酸化チタンなどの有機および無機顔料;UV吸収剤;安定剤;臭気マスキング剤;粘性増強剤;例えば、パルミチン酸アスコルビル、重亜硫酸ナトリウム、メタ重亜硫酸ナトリウムなどの抗酸化剤およびそれらの混合物;例えば、ソルビン酸カリウム、安息香酸ナトリウム、ソルビン酸、没食子酸プロピル、ベンジルアルコール、メチルパラベン、プロピルパラベンなどの保存料;エチレン−ジアミンテトラ酢酸などの金属イオン封鎖剤;天然バニリンなどの着香料;クエン酸および酢酸などの緩衝剤;ケイ酸塩、珪藻土、酸化マグネシウムまたは酸化アルミニウムなどの増量剤または充填剤;マグネシウム塩などの緻密化剤;ならびにそれらの混合物が含まれる。
【0059】
さらなる成分が、本発明の組成物および組合せ製剤における生物活性成分の作用の持続時間を制御するために含まれ得る。したがって、制御放出組成物は、例えば、ポリエステル、ポリアミノ酸、ポリビニル−ピロリドン、エチレン−ビニルアセテートコポリマー、メチルセルロース、カルボキシメチルセルロース、硫酸プロタミンなどの適切なポリマー担体を選択することによって得られる。薬物放出の速度および作用の持続時間も、活性成分を、粒子、例えば、ヒドロゲル、ポリ乳酸、ヒドロキシメチル−セルロース、ポリメチルメタクリレートおよびその他の上記に記載されたポリマーなどのポリマー物質のマイクロカプセルに組み込むことによって制御し得る。このような方法には、リポソーム、ミクロスフェア、マイクロエマルジョン、ナノ粒子、ナノカプセルなどのコロイド薬物送達系が含まれる。投与の経路に応じて、本発明の医薬組成物または組合せ製剤は、保護被覆も必要とし得る。
【0060】
注射用途に適した医薬剤形には、滅菌水溶液または分散液およびその即時調製用の滅菌粉末が含まれる。したがって、本目的のための一般的な担体には、生体適合性の水性緩衝液、エタノール、グリセロール、プロピレングリコール、ポリエチレングリコール、シクロデキストリンなどの錯化剤など、およびそれらの混合物が含まれる。
【0061】
構造式(I)を有するウレイル置換ナフタルイミド(イソキノリンジオン)誘導体、ならびに/または薬学的に許容されるそれらの塩および/もしくはそれらの溶媒和物および/もしくはそれらの代謝産物と、抗新生物薬とを含む組合せ製剤の場合においては、両方の成分は、治療すべき患者において必ずしも同時にそれらの相乗的治療効果を直接にもたらさないので、前記組合せ製剤は、2種の成分を別々に但し隣接させて含有する薬剤キットまたはパッケージの形態であってよい。後者においては、したがって、それぞれの成分は、他の成分の投与経路と異なる投与経路に適した様式で製剤化することができ、例えば、それらの一方は、経口または非経口製剤の形態であってよいが、他方は、静脈注射用アンプルまたはエアゾールの形態である。
【0062】
本発明はさらに、患者、好ましくは哺乳動物、より好ましくはヒトにおける細胞増殖性障害の予防または治療方法に関する。本発明の方法は、それを必要とする患者に、構造式(I)を有するウレイル置換ナフタルイミド(イソキノリンジオン)誘導体、ならびに/または薬学的に許容されるそれらの塩および/もしくはそれらの溶媒和物および/もしくはそれらの代謝産物の治療有効量を、場合によっては抗新生物薬の有効量と一緒に、または極めて詳細に上記に開示されたような上記を含む医薬組成物を投与することからなる。ウレイル置換ナフタルイミド(イソキノリンジオン)誘導体の有効量は、ヒトの体重1kg当たり1日当たり通常0.01mgから20mg、好ましくは0.1mgから5mgの範囲である。治療すべき病態および患者の状態に応じて、前記有効量は、1日当たり数回の副次的単位に分けることができ、2日以上の間隔で投与することができる。治療すべき患者は、前記病態を患っている任意の温血動物、好ましくはヒトであってよい。
【0063】
次の実施例は、その範囲を何ら制限することなく、ウレイル置換ナフタルイミドの調製、医薬製剤および生物学的評価を含む、本発明のいくつかの実施形態を例示することを意図している。
【実施例1】
【0064】
エチル({2−[2−(ジメチルアミノ)エチル]−1,3−ジオキソ−2,3−ジヒドロ−1H−ベンゾ[de]イソキノリン−5−イル}アミノ)カルボニルカルバメートの調製
1.086gのアモナフィドを窒素雰囲気下−20℃で80mLの2−ブタノンに溶解した。次いで、2mLの2−ブタノンに溶解した880mgのエトキシカルボニルイソシアネート(2モル当量)を、滴下ロートを用いて5分間かけて注意深く添加した。撹拌しつつ反応温度を−20℃に25分間維持した。次いで反応混合物を40分間45℃に温め、この時間の後250μLの水を添加した。この反応クエンチステップ後、形成された沈殿をろ紙で40℃でろ過した。乾燥後、1.162gの予想生成物(下記の構造式)を得た(収率:76%)。高速液体クロマトグラフィー(以下HPLCという)は95.6%を超える純度を示した。微量のアモナフィド(約2%)は依然存在した。
【化3】
【0065】
所望の生成物は、以下により特性決定した:
− プロトン核磁気共鳴(300MHz、CDCl3):WO2005/105753号の実施例4におけると同一のピークを示した、そして
− エレクトロスプレーオン化質量スペクトル:M+H+=399におけるピーク;および2M+H+=797における付加物の存在を示した。
【実施例2】
【0066】
N−{2−[2−(ジメチルアミノ)エチル]−1,3−ジオキソ−2,3−ジヒドロ−1H−ベンゾ[de]イソキノリン−5−イル}尿素の調製
実施例1の化合物の100mgをNaOH 0.1Mの100mL中に溶解した。反応混合物を温めて還流し1時間この温度を維持した。混合物をHPLCで分析し、主要生成物(収率76%)として期待された尿素(下記の構造式)の存在が示された。
【化4】
【0067】
この生成物は、次の技術により特性決定した:
− プロトン核磁気共鳴(RMN 1H, 300MHz, DMSO)では、以下に示されるピーク:9.40(NH-17, bs);8.53(H-2, d, J=1.8);8.48(H-4, d, J=1.8);8.26-8.32(H-6およびH-7, m);6.18(NH2-19, bs);4.14(H-14, t, J=6.6);2.51(H-13, m);および2.21(H-15およびH-16, s) ppmを示した;
− 13C NMR(75.4MHz, DMSO, TMS内部標準)では、以下に示されるピーク37.5(CH2, C-13);49.9(2×CH3, C-15およびC-16);57.0(CH, C14);119.0(CH, C-芳香族);122.2(C, C-芳香族);122.9(C, C-芳香族);123.5(C, C-芳香族);123.9(CH, C-芳香族);127.8(CH, C-芳香族);128.6(CH, C-芳香族);132.7(C, C-芳香族);133.8(CH, C-芳香族);140.3(C, C-芳香族);156.5(C, C-18);および163.8(C, C-12);164.0(C, C-11)ppmを示した;そして
− エレクトロスプレーオン化質量スペクトルでは、M+H+=327におけるピーク;および2M+H+=653における付加物を示した。
【実施例3】
【0068】
全体的な細胞増殖に対する影響
全体的な細胞増殖に対する実施例2の化合物の影響を迅速(すなわち5日以内)に測定するために試験を行った。本試験は、ミトコンドリア還元によって黄色生成物の3−(4,5−ジメチルチアゾール−2−イル)−2,5−ジフェニルテトラゾリウムブロミド(本明細書でMTTという)を青色生成物のホルマザン染料に変換し得る代謝活性を有する生細胞の数を測定する。分光光度計を用いて測定された本実験の最後に得られたホルマザンの量は、生細胞数と直接的に比例している。したがって、光学密度測定によって、対照条件(未処理細胞)および/または他の参照化合物と比較しての、被験化合物の影響の定量的測定が可能になる。
【0069】
表1に記載された6種のヒト癌細胞系を次のMTT試験に使用した。これらの細胞系は6種の癌の組織型を含み、前立腺癌、神経膠腫、膵臓癌、結腸癌、肺癌および乳癌である。使用される細胞型に応じて1ウェル当たり1,000から4,000個の細胞で1ウェル当たり細胞懸濁液の100μlの量で平底の96ウェルマイクロ−ウェル中で細胞を増殖させた。周知のMEM10%血清培養培地に、それぞれの細胞系を接種した。
【表1】
【0070】
詳細な実験手順は次の通りである。37℃でのインキュベーションの24時間後、被験化合物を次のモル濃度:10−9M、5.10−9M、10−8M、5.10−8M、10−7M、5.10−7M、10−6M、5.10−6Mおよび10−5Mで予め溶解した100μlの新しい培地で、培養培地を置換した。それぞれの実験を6回繰り返した。
【0071】
試験対象の化合物を含む(実験条件)または含まない(対照条件)37℃でのインキュベーションの72時間後、1mg/mlの濃度でRPMI(1640フェノールレッド非含有)に溶解した100μlのMTTと、培地を置換した。続いてマイクロウェルを37℃で3時間インキュベートし、400gで10分間遠心分離した。MTTを除去し、形成されたホルマザン結晶を100μlのDMSOに溶解した。マイクロウェルを5分間振とうし、570nm(最大ホルマザン吸収)および630nm(バックグラウンドノイズ)の波長において分光光度計を読み取った。
【0072】
それぞれの実験条件について、平均光学密度を計算し、また対照と比較した残存生細胞の百分率を計算した。
【0073】
下記の表2は実施例2の化合物についてのIC50値を示している。IC50は被験化合物が全体の腫瘍細胞増殖を50%だけ阻害したモル濃度の範囲を表している。
【表2】
【実施例4】
【0074】
細胞移動に対する効果
異なる細胞系、すなわちU−373 MG(神経膠腫)およびA549(肺癌)の細胞を移動実験の48時間前に培養フラスコに接種した。試験日に、表3の右欄に記載の通りに、制御した温度(37.0±0.1℃)で12時間または22時間緩衝培地を含有する密閉ファルコンディッシュ中で、実施例2の化合物を用いてまたは用いずに細胞を処理した。実施例2の化合物を3つの非細胞毒性の濃度(10−6M、10−7M、10−8M)において使用した。細胞の移動を位相差顕微鏡に備え付けたCCDカメラを用いて観測した。ノンパラメトリックMann−Whitney検定による移動の統計的分析を最も運動性の細胞の25%および50%についてそして細胞集団全体について実施した。下記の表3は被験化合物の抗移動作用を示している。
【表3】
【0075】
表3のデータは、実施例2の化合物は本研究で使用された非細胞毒性の濃度でU−373 MG癌細胞の移動レベルの減少を誘導したことを示している。特に、本化合物は細胞移動の統計的に有意な阻害を示している。
【実施例5】
【0076】
ナノ粒子懸濁液製剤
ナノ粒子懸濁液を実施例2の化合物の製剤用に使用する。この手法のために、ポリソルベート80(Tween 80)、Texapon K12(SDS)、PVA(ポリビニルアルコール)、Lutrol F68(Poloxamer 188)、Lutrol F127(Poloxamer 407)、ヒドロキシプロピル−β−シクロデキストリン、タウロコール酸ナトリウムおよび他のリン脂質(Lipoid S PC−3およびPhopholipon 90 H)を含む選択された賦形剤(特に張力活性剤)を使用する。
【0077】
賦形剤およびその量を選択後、実施例2の化合物を含有する懸濁液を、所望量の水にその予定量を単純に添加することによって調製する。次いで、懸濁液を、予備の粒径低減(微粉化)のために低温で、24000rpmでタラックス(turax)にかける。次いで、懸濁液を高圧でEmulsiFlexホモジナイザー(homogenisator)にかける。異なる圧力での3サイクルのホモジナイズを使用して期待される粒径を得ることができ、例えば、第1サイクルを7000psiで7分間実施し、第2サイクルを12000psiで8分間実施し、最後に最終サイクルを約21000〜24000psiで30分間実施する。次いで、粒径分布の測定をレーザー回折(各測定の間を20秒として5回の測定を行う)により行った。これらの5回の測定の平均は懸濁液の粒径分布を示す。
【実施例6】
【0078】
2,2,2−トリクロロ−N−[({2−[2−(ジメチルアミノ)エチル]−1,3−ジオキソ−2,3−ジヒドロ−1H−ベンゾ[de]イソキノリン−5−イル}アミノ)カルボニル]アセトアミドの調製
機械的撹拌機、還流冷却器、冷却氷浴、滴下ロートおよび温度調節器を備えた3つ首12L丸底フラスコに、窒素下でアモナフィド(90g)および3.6Lのメチルエチルケトン(MEK)を導入した。得られた懸濁液を−10℃に冷却し、600mLのMEK中の120gのトリクロロアセチルイソシアネート(0.64モル)の溶液を、温度を−5℃以下に維持しながら35分かけて滴下した。反応混合物を−10℃から−5℃の間で3時間撹拌し、次いで2.8mLの水をゆっくりと添加した。混合物を放置して室温に温め、得られた固体をろ過により単離し、100mLのMEKを用いてフィルター上で洗浄し、2日間風乾させて147gの所望の化合物を得た(収率:97%;純度:98.1%)。本化合物の特徴的なスペクトルはWO2005/105753号の実施例2に記載されたものと同じであった。
【実施例7】
【0079】
N−{2−[2−(ジメチルアミノ)エチル]−1,3−ジオキソ−2,3−ジヒドロ−1H−ベンゾ[de]イソキノリン−5−イル}尿素の代替調製法
機械的撹拌機、還流冷却器、および温度調節器を備えた3つ首22L丸底フラスコに、実施例6の化合物(250g)および水中の5%K2CO3溶液の7.5Lを導入した。(氷浴)を用いて混合物を10℃に冷却し、次いで7.5Lのメタノール(MeOH)を一度に添加した。温度は20℃に上がった。フラスコを氷浴から取り外し、出発原料の大部分が溶解するまで周囲温度で撹拌を続けた(約30〜45分)。混合物を急いでろ過して(清澄化して)少量の未反応材料および他の機械的不純物を取り除いた。混合物を室温で2時間撹拌し、4LのMeOHを一度に添加した。混合物を54〜56℃に3時間加熱した。反応の進行をHPLCでモニターして完了を確認した。反応混合物を冷却(氷浴)し8−10℃に2時間保った。得られた固体をろ過により単離し、フィルター上で洗浄(2×100mLの水)し、次いで2日間風乾させて156g(収率:90%)の所望の化合物(HPLC純度:99.47%)を得た。本化合物の特徴的なスペクトルは本明細書で上記の実施例2に記載されたものと同じであった。
【実施例8】
【0080】
乳酸ベースの溶液製剤
実施例2または実施例7により生成された化合物の液体溶液を次の通りに得た。
【0081】
第1に、2体積%の乳酸溶液を次の通り調製した。50mL容量フラスコに、クラスA−TDピペットを用いて、40mLの0.9%NaCl注射溶液、1.18mLの乳酸、85%ACS試薬を添加した。次いで、体積を0.9%NaCl注射溶液で50mLに調節し、全体を逆さまにして混合した。
【0082】
25mL容量フラスコに、実施例2および7の化合物の700mgを正確に計量した。この特定の量に、10mLの0.9%NaCl注射溶液および8.89mLの前述の2%乳酸水溶液を添加した。得られた溶液を10分間激しく撹拌し超音波処理した。溶液のpHは6.4〜6.6の間であった。次いで、前述の2%乳酸水溶液の少量(20μL)の注意深い添加によって、pHを5.75に調節した。0.9%NaCl注射溶液を使用して25mLの最終体積に調節した。その時点で、本発明の化合物の溶解が完了したことが目視検査で観察され、予め滅菌したシリンジフィルター(例えば、Milliporeフィルター−Durapore(PVDF)、0.22μm)に溶液を通すことによって溶液を滅菌し、こうして28mg/mLの溶液が得られた。
【0083】
この原液28mg/mLから、0.9%NaCl注射溶液を用いた希釈ステップを行うことによって、次の表に示された希釈溶液を10mL容量フラスコ中に得た。表4において、示された用量は、静脈内注射用の用量体積は1kg当たり5mLであるという仮定に対応する。
【表4】
【実施例9】
【0084】
N−{2−[2−(ジメチルアミノ)エチル]−1,3−ジオキソ−2,3−ジヒドロ−1H−ベンゾ[de]イソキノリン−5−イル}尿素の血液毒性
発明者らは、血小板、赤血球および白血球に対するアモナフィドの影響と比較した、血小板、赤血球および白血球に対する実施例2または実施例7により生成された化合物の化合物により誘発される潜在的な血液毒性を測定した。アモナフィドの影響をマウスへの腹腔内投与によって、10mg/kgおよび20mg/kgにおいて評価した。投与スケジュールは連続3週間で毎週5回であった。実施例2または実施例7により生成された化合物の影響はマウスへの静脈内投与によって20mg/kgで評価した。投与スケジュールは連続5週間で毎週3回(月曜日、水曜日および金曜日)であった。動物を最後の注射後3日に犠牲にした。1グループ当たり10匹のマウスが存在した。図1は、血小板に対する化合物により誘発された血液毒性についてのこのアッセイの結果を示している。図1は、10mg/kgの用量でのアモナフィドの15回の長期投与にマウスが耐えたが、20mg/kgの用量でのアモナフィドの15回の長期投与の全回を摂取する前に全ての動物が死亡したことを示している。対照的に、図1は、20mg/kgの用量での実施例2または実施例7により生成された化合物の15回の長期投与にマウスが耐えたことを示している。したがって、アモナフィドと異なり、実施例2または実施例7により生成された化合物は、これらの実験条件において治療用量で血液毒性を誘発しないことが見い出された。
【実施例10】
【0085】
N−{2−[2−(ジメチルアミノ)エチル]−1,3−ジオキソ−2,3−ジヒドロ−1H−ベンゾ[de]イソキノリン−5−イル}尿素とP−糖タンパク質の相互作用
P−糖タンパク質(以下ではP−gpという)との薬物相互作用を試験するために、発明者らは濃縮P−gp膜小胞調製物(次のキットを使用した:SPI BIO Franceから市販されているP−gp薬物相互作用アッセイキット)からのATPアーゼ活性の調節の研究に基づくアッセイを使用した。P−gp ATPアーゼ活性を小胞懸濁液培地中のADP形成のモニターに基づく分光光度法によって測定した。基礎ATPアーゼ活性を、添加された薬物の不存在下で測定された活性と定義した。基礎的な活性の調節を、異なる濃度(それぞれ2、10、および50μM)でアモナフィドまたは実施例2もしくは実施例7により生成された化合物を添加することによって実施した。図2に示されたデータは、50μMでアッセイした場合にアモナフィドはATPアーゼ活性を有意に変更するが、本発明の化合物はATPアーゼ活性に殆ど影響を与えないことを示している。
【実施例11】
【0086】
ヒトの癌細胞における自食作用に関係した細胞死に対するN−{2−[2−(ジメチルアミノ)エチル]−1,3−ジオキソ−2,3−ジヒドロ−1H−ベンゾ[de]イソキノリン−5−イル}尿素の誘発作用
トポイソメラーゼII標的薬物の顕著な特徴はアポトーシスの誘発であり、これは、開裂可能な複合体の安定化および/またはトポイソメラーゼII鎖切断(strand-passage)活性の阻害の結果としての完全な染色体分離の達成の失敗による、DNA損傷レベルの細胞内増加の結果である。アモナフィドはトポイソメラーゼII阻害剤でありアポトーシスを誘発し、これは発明者らが、実施例2または実施例7により生成された化合物を含むヒトPC−3(表1を参照されたい)およびDU−145(ATCC番号:HTB−81)前立腺癌細胞において観測しなかった特徴である。
【0087】
発明者らはアネキシンVおよびプロピジウムヨージドの両方についてPC−3およびDU−145陽性細胞の百分率を求めるためにフローサイトメトリー(Mijatovicら、Neoplasia 2006において見い出された手順による)を使用し、発明者らは実施例2または実施例7により生成された化合物の10μMでの処置を受けたPC−3またはDU−145細胞の最大で10%のみがアポトーシスプロセスを経験したことを観測した。
【0088】
発明者らは本化合物で処置したPC−3およびDU−145細胞において自食促進作用を観測した。発明者らは、それらを0(対照、未処置細胞)、1μMまたは10μMで処置した後に、PC−3(灰色棒)またはDU−145(黒色棒)細胞のアクリジン・オレンジ染色後に、酸性の小胞細胞小器官(赤色蛍光染色として表した)(Kanzawaら、Cell Death Differ 2004において見い出された手順による)を定量し、結果は図3Aに示されている。
【0089】
リソソームは細胞死をいくつかのレベルで制御することは周知である。内因性または外因性ストレス(化学療法を含む)への応答において、リソソーム膜の透過化(LMP)が起こり、異化加水分解酵素(catabolic hydrolase)の放出をもたらすことがあり、これはカスパーゼ依存的アポトーシス、カスパーゼ非依存性アポトーシス様細胞死または高レベルのLMPに続く壊死さえ媒介し得る。したがって、発明者らは、それらが実施例2または実施例7により生成された化合物の0(対照、未処置細胞)、1μMまたは10μMで処置された後に、PC−3(灰色棒)またはDU−145(黒色棒)の72時間の処置後に、酸性の小胞細胞小器官の「漏出」(緑色蛍光染色として表した)(Nylandsted, J.ら、Heat Shock Protein 70 Promotes Cell Survival by Inhibiting Lysosomal Membrane Permeabilization, J Exp Med. (2004) 16;200(4):425-35およびMijatovicら、Neoplasia 2006による)を定量し、結果は図3Bに示されている。発明者らはPC−3細胞の72時間の処置後にLMPを観測しなかったが、本発明の化合物の10μMで処置した場合にDU−145細胞に著しい薬物により誘発されたLMPプロセスが出現した。
【実施例12】
【0090】
DU−145ヒト前立腺癌細胞における老化に対するN−{2−[2−(ジメチルアミノ)エチル]−1,3−ジオキソ−2,3−ジヒドロ−1H−ベンゾ[de]イソキノリン−5−イル}尿素の誘発作用
実施例2または実施例7により生成された化合物が非アポトーシス細胞死を誘発したという特徴は、6日間本化合物の10μMで処置したヒトPC−3およびDU−145前立腺癌細胞において細胞イメージングを用いて形態学レベルでさらに観測された(細胞をFalconフラスコ(25cm2)中に接種し定量ビデオ顕微鏡で6日間分析した)。
【0091】
老化細胞はその形質膜の完全性を維持しているが、それらは永久の増殖停止を受け、その細胞の増殖能力(clonogenicity)を失うので、老化は「生細胞死」の1種であると考えることができる。老化は癌の進行に対する自然のバリアとして作用し得る。
【0092】
老化に一般的な特徴が、ヒトDU−145前立腺癌細胞において実施例2の化合物によって誘発された。老化細胞は扁平な細胞質および増加した粒度などの形態学的変化を一般的に示すことが知られている。老化に関係したβ−ガラクトシダーゼ活性の誘発は、老化をしている細胞に起こる特異的な事象であり、発明者らが、図4に明らかにしたように、現在の研究(Dimriら、A biomarker that identifies senescent human cells in culture and in aging skin in vivo, Proc Natl Acad Sci U S A. (1995) 92(20):9363-7において見い出された手順による)でもう一度観測した特徴である。ドキソルビシン(ADR)の中用量(nM範囲)は野性型ヒト癌細胞に老化を誘発することが知られている。したがって、発明者らは発明者らの実験において陽性対照としてドキソルビシンを使用した。図4に示されたように、本発明の化合物の10μMは、DU−145細胞中の20nMのドキソルビシンと類似の百分率の老化関連のβ−ガラクトシダーゼ陽性染色を誘発した。
【実施例13】
【0093】
N−{2−[2−(ジメチルアミノ)エチル]−1,3−ジオキソ−2,3−ジヒドロ−1H−ベンゾ[de]イソキノリン−5−イル}尿素により標的化される遺伝子の同定
生化学レベルにおいて、老化は、実施例2の化合物でインビトロで処置したPC−3細胞のゲノム解析を実施した場合に、現在の研究で本知見者らが観測した特徴である、代謝における変化を伴う。
【0094】
遺伝子レベルにおいて、発明者らは、本化合物で処置した場合にPC−3細胞におけるクロマチン構造および遺伝子発現パターンの改変も観測した。
【0095】
発明者らは、1μMもしくは10μMで1回、または1μMで1週間に5回(連続5日間に1日1回)のいずれかで本発明の化合物(N−{2−[2−(ジメチルアミノ)エチル]−1,3−ジオキソ−2,3−ジヒドロ−1H−ベンゾ[de]イソキノリン−5−イル}尿素)で処置したインビトロで増殖させたヒトPC−3癌細胞を使用するAffymetrix全ゲノムマイクロアレイを用いて遺伝子標的の評価の最初の実験を行った(全ゲノム解析はAffymetrixヒトゲノムU133セットプラス2.0(High Wycombe, United Kingdom)を使用してVIB MicroArray Facility(UZ Gasthuisberg, Catholic University of Leuven, Belgium)で実施した)。発明者らが得た最も顕著なデータは表5に報告され、インビトロで単一の高用量としてアッセイする場合(急性期インビトロ処置)、本化合物は、ヒストンの様々なタイプ(少なくともmRNAレベルで)の発現のレベルを顕著に増加させることによって核構成および生物発生を著しく改変したことを示す。本化合物により標的化された遺伝子の第2セットは、「アミノ酸代謝」と記した遺伝子のカテゴリーに属する(表5)。
【0096】
前立腺癌細胞における老化に関係した遺伝子の同定プロセスにおいて、先行技術は、DNA損傷により誘発された老化の状態における癌細胞中のets相同因子(EHF)の顕著な抑制を教示しており、EHFは老化および細胞周期停止を阻害することによってPC−3前立腺癌細胞における実質的な薬物耐性をもたらすことを示した。興味深いことに、本発明者らは、EHFも本発明の化合物の標的であることを見い出した。
【0097】
転写因子のE2Fファミリーは、細胞周期の進行に重要な役割を果たすことが知られている。E2F−1は、別のタンパク質DP−1とヘテロ二量体複合体となって、低リン酸化pRbに結合しているために通常不活性である。細胞がG1期からS期に進行する場合、pRbは過剰リン酸化になり結合E2F−1/DP−1ヘテロ二量体を放出し、次いで、これはTSおよびDHFRなどのDNAに含まれる遺伝子の転写を活性化する。機能性pRbの損失は遊離E2F−1レベルと次いでのTSおよびDHFRのレベル増加を生じ得る。ゲノムAffymetrix手法により明らかにされたように、発明者らは、連続5日間の1日1回の実施例2の化合物の1μMでのPC−3細胞の処置はE2F−1のmRNAレベルを2倍減少させることを見い出した。
【0098】
老化の初期の間、Rbはゲノム中の特異的位置におけるヘテロクロマチンの核生成を制御することがあり、次いでこれはヒストンメチルトランスフェラーゼの作用およびHP1タンパク質のリクルートによって拡がる。発明者らは、実施例2の化合物は、PC−3細胞中の少なくともmRNAレベルにおいてヒストンH1、H2およびH3の増加を通してPC−3細胞中のヘテロクロマチンのレベルを著しく増加させることを見い出した(表5)。対照的に、本化合物はH2AFYのmRNA発現のレベルを2.6倍減少させた。
【表5】
【図面の簡単な説明】
【0099】
【図1】アモナフィドと比較した、本発明の化合物による血小板に対する化合物により誘発された血液毒性を示す図である。
【図2】アモナフィドと比較した、本発明の化合物について分光光度計で測定されたP−gp ATPアーゼ活性を示す図である。
【図3】本発明の化合物の様々な濃度における、(A)酸性の小胞細胞小器官(赤色蛍光染色として示される)の定量により評価された、薬物により誘発された自食促進作用、および(B)アクリジン・オレンジ染色および緑色蛍光染色の定量に従って評価された、薬物により誘発されたリソソーム膜透過化(LMP)を示す図である。
【図4】ドキソルビシンと比較した、本発明の化合物により誘発されたDU−145ヒト前立腺癌細胞における老化に関連したβ−ガラクトシダーゼ活性を示す図である。
【特許請求の範囲】
【請求項1】
構造式(I)により表される置換ナフタルイミド誘導体:
【化1】
[式中、
R1はモノまたはジ−C1〜4アルキルアミノ−C1〜4アルキルであり;
R3およびR4のそれぞれは、水素、ハロゲン、C1〜4アルキル、C1〜4アルコキシ、C1〜4アルキルチオ、ニトロ、シアノ、アミノ、保護されたアミノおよびハロC1〜4アルキルからなる群から独立に選択され;
mは置換基R3の数であって、0から3の範囲であり;
nは置換基R4の数であって、0から2の範囲であり;
R2はCONH2である。]、
ならびに/または薬学的に許容されるその塩および/もしくはその溶媒和物および/もしくはその代謝産物。
【請求項2】
n=0であり、かつ/または
m=0であり、かつ/または
m=2であり、両方の置換基R3は隣接しており、それらが結合している炭素原子と一緒になってフェニル基を形成し、かつ/または
R1が1から3個の炭素原子を有するアルキレン基であり、ジメチルアミノもしくはジエチルアミノ基に連結しており、かつ/または
R2がCONH2である
構造式(I)により表される、請求項1に記載の置換ナフタルイミド誘導体ならびに/または薬学的に許容されるその塩および/もしくはその溶媒和物および/もしくはその代謝産物。
【請求項3】
mが1に等しい場合に、R3がニトロではない、請求項1に記載の置換ナフタルイミド誘導体。
【請求項4】
N−{2−[2−(ジメチルアミノ)エチル]−1,3−ジオキソ−2,3−ジヒドロ−1H−ベンゾ[de]イソキノリン−5−イル}尿素である、請求項1に記載の置換ナフタルイミド誘導体および/または薬学的に許容されるその塩および/またはその代謝産物。
【請求項5】
前記代謝産物が、
そのモノ−N−オキシドおよびジ−N−オキシド;
R2がCONHOHである誘導体;ならびに
R3および/またはR4がヒドロキシルである誘導体
からなる群から選択される、請求項1から4のいずれか一項に記載の置換ナフタルイミド誘導体。
【請求項6】
1種または複数の薬学的に許容される担体および請求項1から5のいずれか一項に記載の置換ナフタルイミド誘導体の治療有効量を含む医薬組成物。
【請求項7】
抗新生物薬をさらに含む、請求項6に記載の医薬組成物。
【請求項8】
前記置換ナフタルイミド誘導体が、N−{2−[2−(ジメチルアミノ)エチル]−1,3−ジオキソ−2,3−ジヒドロ−1H−ベンゾ[de]イソキノリン−5−イル}尿素および/または薬学的に許容されるその塩および/またはその代謝産物である、請求項6または7に記載の医薬組成物。
【請求項9】
構造式(II)を有する化合物:
【化2】
[式中、
m、n、R1、R3およびR4のそれぞれは請求項1で定義された通りであり、
R’はC1〜4アルコキシアミドカルボニルまたはC1ハロアルキルアミドカルボニルである。]
を加水分解するステップを含む、請求項1に記載の置換ナフタルイミド誘導体の製造方法。
【請求項10】
前記構造式(II)を有する化合物が、アモナフィドとC1〜4アルコキシカルボニルイソシアネートまたはC1ハロアルキルカルボニルイソシアネートとの反応の生成物である、請求項9に記載の方法。
【請求項11】
アモナフィドとC1〜4アルコキシカルボニルイソシアネートまたはC1ハロアルキルカルボニルイソシアネートとの前記反応が、エーテル、ケトンおよびハロゲン化炭化水素からなる群から選択される溶媒の存在下で、かつ/または0℃未満の温度で実施される、請求項10に記載の方法。
【請求項12】
アモナフィドとC1〜4アルコキシカルボニルイソシアネートまたはC1ハロアルキルカルボニルイソシアネートとの前記反応が、モル過剰の前記C1〜4アルコキシカルボニルイソシアネートまたはC1ハロアルキルカルボニルイソシアネートの存在下で実施され、かつ前記反応が完了後に反応混合物に水を添加することによりクエンチされる、請求項10または11に記載の方法。
【請求項13】
構造式(II)を有する化合物を加水分解するステップが塩基性条件下で実施される、請求項9から12のいずれか一項に記載の方法。
【請求項14】
薬剤としての、請求項1から5のいずれか一項に記載の置換ナフタルイミド誘導体、ならびに/または薬学的に許容されるその塩および/もしくはその溶媒和物および/もしくはその代謝産物の使用。
【請求項15】
前記薬剤が細胞増殖性障害の治療用である、請求項14に記載の使用。
【請求項16】
前記細胞増殖性障害が、白血病、肺癌、結腸直腸癌、中枢神経系(CNS)癌、メラノーマ、卵巣癌、腎癌、前立腺癌、乳癌、神経膠腫、膀胱癌、骨癌、肉腫、頭頸部癌、肝癌、精巣癌、膵癌、胃癌、食道癌、骨髄癌、十二指腸癌、眼癌(網膜芽腫)およびリンパ腫からなる群から選択される、請求項15に記載の使用。
【請求項17】
ヒトにおける細胞増殖性障害の治療方法であって、請求項1から5のいずれか一項に記載の置換ナフタルイミド誘導体、ならびに/または薬学的に許容されるその塩および/もしくはその溶媒和物および/もしくはその代謝産物の治療有効量の前記ヒトへの投与を含む方法。
【請求項18】
前記量が体重1kg当たり1日当たり0.01mgから20mgである、請求項17に記載の細胞増殖性障害の治療方法。
【請求項19】
抗新生物薬の有効量の投与をさらに含む、請求項17または18に記載の細胞増殖性障害の治療方法。
【請求項20】
前記投与が、静脈内投与、筋肉内投与、腹腔内投与、経口投与および直腸投与から選択される、請求項17に記載の細胞増殖性障害の治療方法。
【請求項1】
構造式(I)により表される置換ナフタルイミド誘導体:
【化1】
[式中、
R1はモノまたはジ−C1〜4アルキルアミノ−C1〜4アルキルであり;
R3およびR4のそれぞれは、水素、ハロゲン、C1〜4アルキル、C1〜4アルコキシ、C1〜4アルキルチオ、ニトロ、シアノ、アミノ、保護されたアミノおよびハロC1〜4アルキルからなる群から独立に選択され;
mは置換基R3の数であって、0から3の範囲であり;
nは置換基R4の数であって、0から2の範囲であり;
R2はCONH2である。]、
ならびに/または薬学的に許容されるその塩および/もしくはその溶媒和物および/もしくはその代謝産物。
【請求項2】
n=0であり、かつ/または
m=0であり、かつ/または
m=2であり、両方の置換基R3は隣接しており、それらが結合している炭素原子と一緒になってフェニル基を形成し、かつ/または
R1が1から3個の炭素原子を有するアルキレン基であり、ジメチルアミノもしくはジエチルアミノ基に連結しており、かつ/または
R2がCONH2である
構造式(I)により表される、請求項1に記載の置換ナフタルイミド誘導体ならびに/または薬学的に許容されるその塩および/もしくはその溶媒和物および/もしくはその代謝産物。
【請求項3】
mが1に等しい場合に、R3がニトロではない、請求項1に記載の置換ナフタルイミド誘導体。
【請求項4】
N−{2−[2−(ジメチルアミノ)エチル]−1,3−ジオキソ−2,3−ジヒドロ−1H−ベンゾ[de]イソキノリン−5−イル}尿素である、請求項1に記載の置換ナフタルイミド誘導体および/または薬学的に許容されるその塩および/またはその代謝産物。
【請求項5】
前記代謝産物が、
そのモノ−N−オキシドおよびジ−N−オキシド;
R2がCONHOHである誘導体;ならびに
R3および/またはR4がヒドロキシルである誘導体
からなる群から選択される、請求項1から4のいずれか一項に記載の置換ナフタルイミド誘導体。
【請求項6】
1種または複数の薬学的に許容される担体および請求項1から5のいずれか一項に記載の置換ナフタルイミド誘導体の治療有効量を含む医薬組成物。
【請求項7】
抗新生物薬をさらに含む、請求項6に記載の医薬組成物。
【請求項8】
前記置換ナフタルイミド誘導体が、N−{2−[2−(ジメチルアミノ)エチル]−1,3−ジオキソ−2,3−ジヒドロ−1H−ベンゾ[de]イソキノリン−5−イル}尿素および/または薬学的に許容されるその塩および/またはその代謝産物である、請求項6または7に記載の医薬組成物。
【請求項9】
構造式(II)を有する化合物:
【化2】
[式中、
m、n、R1、R3およびR4のそれぞれは請求項1で定義された通りであり、
R’はC1〜4アルコキシアミドカルボニルまたはC1ハロアルキルアミドカルボニルである。]
を加水分解するステップを含む、請求項1に記載の置換ナフタルイミド誘導体の製造方法。
【請求項10】
前記構造式(II)を有する化合物が、アモナフィドとC1〜4アルコキシカルボニルイソシアネートまたはC1ハロアルキルカルボニルイソシアネートとの反応の生成物である、請求項9に記載の方法。
【請求項11】
アモナフィドとC1〜4アルコキシカルボニルイソシアネートまたはC1ハロアルキルカルボニルイソシアネートとの前記反応が、エーテル、ケトンおよびハロゲン化炭化水素からなる群から選択される溶媒の存在下で、かつ/または0℃未満の温度で実施される、請求項10に記載の方法。
【請求項12】
アモナフィドとC1〜4アルコキシカルボニルイソシアネートまたはC1ハロアルキルカルボニルイソシアネートとの前記反応が、モル過剰の前記C1〜4アルコキシカルボニルイソシアネートまたはC1ハロアルキルカルボニルイソシアネートの存在下で実施され、かつ前記反応が完了後に反応混合物に水を添加することによりクエンチされる、請求項10または11に記載の方法。
【請求項13】
構造式(II)を有する化合物を加水分解するステップが塩基性条件下で実施される、請求項9から12のいずれか一項に記載の方法。
【請求項14】
薬剤としての、請求項1から5のいずれか一項に記載の置換ナフタルイミド誘導体、ならびに/または薬学的に許容されるその塩および/もしくはその溶媒和物および/もしくはその代謝産物の使用。
【請求項15】
前記薬剤が細胞増殖性障害の治療用である、請求項14に記載の使用。
【請求項16】
前記細胞増殖性障害が、白血病、肺癌、結腸直腸癌、中枢神経系(CNS)癌、メラノーマ、卵巣癌、腎癌、前立腺癌、乳癌、神経膠腫、膀胱癌、骨癌、肉腫、頭頸部癌、肝癌、精巣癌、膵癌、胃癌、食道癌、骨髄癌、十二指腸癌、眼癌(網膜芽腫)およびリンパ腫からなる群から選択される、請求項15に記載の使用。
【請求項17】
ヒトにおける細胞増殖性障害の治療方法であって、請求項1から5のいずれか一項に記載の置換ナフタルイミド誘導体、ならびに/または薬学的に許容されるその塩および/もしくはその溶媒和物および/もしくはその代謝産物の治療有効量の前記ヒトへの投与を含む方法。
【請求項18】
前記量が体重1kg当たり1日当たり0.01mgから20mgである、請求項17に記載の細胞増殖性障害の治療方法。
【請求項19】
抗新生物薬の有効量の投与をさらに含む、請求項17または18に記載の細胞増殖性障害の治療方法。
【請求項20】
前記投与が、静脈内投与、筋肉内投与、腹腔内投与、経口投与および直腸投与から選択される、請求項17に記載の細胞増殖性障害の治療方法。
【図1】

【図2】
【図3】
【図4】
【図2】
【図3】
【図4】
【公表番号】特表2009−536169(P2009−536169A)
【公表日】平成21年10月8日(2009.10.8)
【国際特許分類】
【出願番号】特願2009−508228(P2009−508228)
【出願日】平成19年5月7日(2007.5.7)
【国際出願番号】PCT/EP2007/003991
【国際公開番号】WO2007/128538
【国際公開日】平成19年11月15日(2007.11.15)
【出願人】(505129585)ユニバイオスクリーン エス.アー. (8)
【Fターム(参考)】
【公表日】平成21年10月8日(2009.10.8)
【国際特許分類】
【出願日】平成19年5月7日(2007.5.7)
【国際出願番号】PCT/EP2007/003991
【国際公開番号】WO2007/128538
【国際公開日】平成19年11月15日(2007.11.15)
【出願人】(505129585)ユニバイオスクリーン エス.アー. (8)
【Fターム(参考)】
[ Back to top ]