
5−FUおよび5−FUプロドラッグと併用してDPD阻害物質を投与するための方法
【課題】5−FUおよび5−FUプロドラッグと併用してDPD阻害物質を投与するための方法の提供。
【解決手段】DPD酵素の活性を実質的に失わせるDPD阻害物質を、その必要がある患者へと最初に投与し、その後に5−FUまたは5−FUプロドラッグを投与することを含む、5−FUおよび/または5−FUプロドラッグと組み合わせたDPD阻害物質の改善された投与および投薬の方法であって、5−FUまたは5−FUプロドラッグのレベルが患者におけるDPD阻害物質を上回る量である、方法を提供する。特定の実施形態では、5−FUまたは5−FUプロドラッグは、前記DPD阻害物質が投与されてから少なくとも約4時間後に投与される。
【解決手段】DPD酵素の活性を実質的に失わせるDPD阻害物質を、その必要がある患者へと最初に投与し、その後に5−FUまたは5−FUプロドラッグを投与することを含む、5−FUおよび/または5−FUプロドラッグと組み合わせたDPD阻害物質の改善された投与および投薬の方法であって、5−FUまたは5−FUプロドラッグのレベルが患者におけるDPD阻害物質を上回る量である、方法を提供する。特定の実施形態では、5−FUまたは5−FUプロドラッグは、前記DPD阻害物質が投与されてから少なくとも約4時間後に投与される。
【発明の詳細な説明】
【技術分野】
【0001】
(発明の背景)
(発明の分野)
本発明は、全体として癌治療に関し、より詳しくは、5−FUおよび/または5−FUプロドラッグと併用してDPD阻害物質を用いる癌治療に関する。
【背景技術】
【0002】
(関連技術の説明)
5−フルオロウラシル(5−FU)は、30年以上、癌患者のうち固形癌を処置するために臨床において使用されている(非特許文献1;非特許文献2;非特許文献3)。5−FUは、DNA合成およびRNA機能を妨害する、擬似ウリジンヌクレオチド(例えば、FUMP、FUDP、FUTP)およびデオキシウリジンヌクレオチド(例えば、FdUMP、FdUDP、FdUTP)へと代謝的変換により活性化されなければならない(非特許文献4において概説されている)。5−FUとその元の形であるウラシルとの違いは5位におけるわずか1個のフッ素による置換であることから、癌患者において容易に活性化される。しかし、ウラシルとのその構造類似性が徒となり、抗腫瘍活性を全く有しない分解生成物へと迅速且つ広範に変換されてしまう主な原因ともなっている。この代謝過程は不活性化と呼ばれる。5−FUは、酵素ジヒドロピリミジンデヒドロゲナーゼ(DPD:EC 1312,ウラシル還元酵素)によって容易に不活性化される(非特許文献4;非特許文献5)。したがって、癌を処置するための5−FUの抗腫瘍効果は、抗腫瘍ヌクレオチドへの代謝的変換(活性化)と、無用な代謝産物への代謝的変換(不活性化)との間の微妙な均衡に左右される。
【0003】
さらに、5−FUの代謝的不活性化によるいくつかの臨床的問題が生じている。第1に、DPDのレベルは個人差があり(非特許文献6;非特許文献7)、1日の流れの中でも個人差がある(非特許文献7;非特許文献8;非特許文献9)ため、所定の投与量から得られる全身の5−FUレベルのばらつきは非常に大きく、したがって、有効性および毒性は非常に予測しづらいものになっている。極端な場合、DPDを遺伝子的に欠損している患者に対し、「標準的な」治療的用量の5−FUによる処置は、重篤且つ時には致死的な毒性を示す(非特許文献10において概説されている)。第2に、胃腸におけるDPDレベルにばらつきがあるため(非特許文献11;非特許文献12;非特許文献13)、経口投与された5−FUの吸収は非常にばらつきがある(非特許文献14;非特許文献15;非特許文献16)、したがって、薬物血漿中レベルを予見することができず、望ましくない毒性または不適当な有効性をもたらし得る。第3に、高レベルのDPDを含有する腫瘍は、5−FU処置に対して反応する可能性が低い(非特許文献17;非特許文献18)。最後に、5−FUの分解生成物は、神経毒性(非特許文献19;非特許文献20)、心臓毒性(非特許文献21;非特許文献22)、手掌足底発赤知覚不全(手足症候群)(非特許文献23)、およびGI毒性(非特許文献24)を生じる可能性があり、抗腫瘍活性を妨害すると思われる(非特許文献24;非特許文献25)。
【0004】
DPDは、5−FUの分解(不活性化)の第1段階および律速段階である、遍在する酵素である。DPDの阻害は血漿中の5−FUの半減期を延長することが研究から示されている。不可逆的にDPDを不活性化するもの、ならびに、可逆的にDPDを阻害するもの等、いくつかのDPD阻害物質について研究されている。
【0005】
5−エチニルウラシルは、エニルウラシルとも呼ばれ、5−FUの代謝的不活性化を低減または失わせる不可逆性のDPD不活性化物質であるDPD阻害物質の一つの例である(概説については、非特許文献26;非特許文献27を参照されたい)。エニルウラシルと5−FUとの構造が類似しているため、エニルウラシルはDPDに対する基質となる。DPDがエニルウラシルを分解しようとすると、エニルウラシルは、DPDと不可逆的に結合する高度反応性化合物へと変換され、それによりこの酵素を不活性化する。したがって、少しでもエニルウラシルが存在すれば、DPDは破壊され、それ以上5−FUを不活性化することはできない。このような処置を患者に施しても、数日もすれば、DPD酵素のde novo合成の結果、活性なDPDのみが再び現れる。
【0006】
エニルウラシルは癌患者におけるフェーズI臨床試験において調べられている(非特許文献28;非特許文献29;非特許文献30において概説されている)。エニルウラシルは、毒性を生じることなく、強力にDPD活性を失わせた。0.74mg/m2(合計約1mg)の用量で、長時間にわたり、全DPD活性の90%以上を失わせた。実際、1回のエニルウラシル投与から24時間後に、DPDのレベルは投与前のレベルのわずか3%であった。5−FUの排出半減期は、エニルウラシルの一回の投与により、約10分から3.5時間に増大した。3.7mg/m2の用量のエニルウラシルは、5−FUの半減期を4.5時間に増大した。用量をさらに多くしたが、目立った効果は得られなかった。
【0007】
エニルウラシルはさらに、フェーズIIおよびフェーズIII臨床試験において経口投与されている(非特許文献28;非特許文献31に概説されている)。これらの治験では2つの投与計画が使用された。「5−dayスケジュール」では、エニルウラシルが、1日目から7日目まで1日当たり50mgの固定用量で投与された。エニルウラシルの投与後、2日目から6日目まで、約20mg/m2の用量で5−FUが投与された。「28−dayスケジュール」では、エニルウラシルおよび5−FUが、エニルウラシル10に対して5−FUを1とする固定された比で、b.i.d.(1日当たり2回)、28日間、共投与した。5−FUの用量は約1mg/m2とした。エニルウラシルは、5−FUが関連する手足症候群毒性を無効にし、5−FUが安全に経口投与されることを可能にし、結果として、5−FU血漿中レベルが十分予見可能となった。しかしながら、これらの投与計画抗腫瘍活性は残念なことに期待はずれであった。結腸直腸癌のための28−dayエニルウラシル投与計画を用いた2つの多施設フェーズIII試験において、エニルウラシルおよび5−FUを投与される患者には、エニルウラシルを使用しない標準的な5−FU投与計画によって処置された患者よりも低い抗腫瘍活性を示す傾向があった(非特許文献31)。
【先行技術文献】
【非特許文献】
【0008】
【非特許文献1】Ansfieldら,Cancer 39:34−40,1977
【非特許文献2】Gremら,Cancer Treat Rep 71:1249−1264,1987
【非特許文献3】Chabnerら,Cancer,Principles and Practice of Oncology,2nd Ed,pp 287−328 Philadelphia,PA:J B Lippincott Co,1985
【非特許文献4】Meyers,Pharmacol Rev,33:1−15,1981
【非特許文献5】Dasherら,Pharmac Ther 48:189−222,1990
【非特許文献6】Flemingら,Cancer Res 52:2899−2902,1992
【非特許文献7】Gremら,Cancer Chemother Pharmacol 40:117−125,1997
【非特許文献8】Harrisら,Cancer Res 50:197−201,1990
【非特許文献9】Petitら,Cancer Res 48:1676−1679,1988
【非特許文献10】Morrisonら,Oncol Nurs Forum 24:83−88,1997
【非特許文献11】Hoら,Anticancer Res 6:781−784,1986
【非特許文献12】Naguibら,Cancer Res 45:5405−5412,1985
【非特許文献13】Spectorら,Biochem Pharmacol 46:2243−2248,1993
【非特許文献14】Christophidisら,Clin Pharmacokinetics 3:330−336,1978
【非特許文献15】Cohenら,Cancer Chemother Rep 58:723−731,1974
【非特許文献16】Finchら,Br J Clin Pharmacol 7:613−617,1979
【非特許文献17】Etienneら,J Clin Oncol 13:1663−1670,1995
【非特許文献18】Fischelら,Clin Cancer Res 1:991−996,1995
【非特許文献19】Okedaら,Acta Neuropathol 81:66−73,1990
【非特許文献20】Koenigら,Arch Neurol 23:155−160,1970
【非特許文献21】Lancet 337:560,1991
【非特許文献22】Lemaireら,Br J Cancer 66:119−127,1992
【非特許文献23】Hohneker,Oncology 12:52−56,1998
【非特許文献24】Spectorら,Cancer Res 55:1239−1241,1995
【非特許文献25】Caoら,Pharmacol 59:953−960,2000
【非特許文献26】Spectorら,Drugs of The Future 19:565−571,1994
【非特許文献27】Paffら,Invest.New Drugs:18,365−371(2000)
【非特許文献28】Levinら,Invest New Drugs 18:383−90,2000
【非特許文献29】Bakerら,J Clin Oncol 18:915−926 2000
【非特許文献30】Schilskyら,J Clin Oncol 4:1450−7,1998
【非特許文献31】Schilskyら,J Clin Oncol:20:1519−26,2002
【発明の概要】
【発明が解決しようとする課題】
【0009】
したがって、当該分野では、5−FUおよび5−FUプロドラッグの抗腫瘍効果および治療指数を最大にし、投与の予見可能性を向上し、経口投与により5−FUおよび5−FUプロドラッグを効果的に投与するために、5−FUおよび5−FUプロドラッグが組み合わせて使用されるDPD阻害物質の最適な投与または投薬スケジュールを特定する必要性が重要視され、未だそれを満たすものはない。本発明はこれらの必要性を満たし、その他の関連する利点を提供する。
【課題を解決するための手段】
【0010】
(発明の要旨)
本発明は全体として、5−FUおよび5−FUプロドラッグと組み合わせたDPD阻害物質、例えば、エニルウラシルを投与するための改良された方法に関する。したがって、本発明の一つの態様にしたがって、DPD酵素の活性を実質的に失わせるDPD阻害物質を、患者へと最初に投与し、その後に5−FUまたは5−FUプロドラッグを投与することを含む、患者における癌の処置方法であって、5−FUまたは5−FUプロドラッグは、その投与時に、5−FUまたは5−FUプロドラッグがDPD阻害物質を実質的に上回る量で患者中に存在するような用量で投与される、方法を提供する。
【0011】
本発明の本態様にしたがう一つの実施形態において、5−FUまたは5−FUプロドラッグは、DPD阻害物質が投与されてから、少なくとも約4時間後、少なくとも約6時間後、少なくとも約8時間後、少なくとも約10時間後、少なくとも約12時間後、少なくとも約14時間後、少なくとも約24時間後、または少なくとも約36時間後に投与される。
【0012】
本発明の本態様にしたがう別の実施形態において、5−FUまたは5−FUプロドラッグは、DPD阻害物質が投与されてから、約4〜72時間後、4〜36時間後、4〜24時間後、4〜14時間後、6〜14時間後または8〜14時間後に投与される。
【0013】
本発明の本態様にしたがう別の実施形態において、5−FUまたは5−FUプロドラッグは、DPD阻害物質の投与後、DPD阻害物質の排出半減期の少なくとも約0.1〜4倍、排出半減期の1〜4倍、排出半減期の2〜4倍または排出半減期の3〜4倍が経過した時点で投与される。
【0014】
本発明の本態様にしたがう別の実施形態において、DPD阻害物質は、患者におけるDPD活性を患者におけるベースラインのDPD活性の約10%未満、約5%未満、約3%未満または約1%未満に低減させるに十分な用量で投与される。
【0015】
本発明の本態様にしたがう別の実施形態において、5−FUまたは5−FUプロドラッグは、その投与時に5−FUまたは5−FUプロドラッグが患者中にDPD阻害物質の少なくとも約2倍、少なくとも約3倍、少なくとも約5倍または少なくとも約100倍過剰で存在するような用量で投与される。
【0016】
本発明の本態様にしたがう別の実施形態において、DPD阻害物質は不可逆性DPD阻害物質である。
【0017】
本発明の本態様にしたがう別の実施形態において、DPD阻害物質は可逆性DPD阻害物質である。かかる実施形態において、特定の好ましい可逆性DPD阻害物質としては、過剰な阻害物質が身体から排除されるよりもゆっくりとDPDから脱離し、したがって、5−FUまたは5−FUプロドラッグを投与する際に実質的に過剰で存在しない、強固に結合している阻害物質を挙げることができることが理解されるであろう。その他の好ましい阻害物質としては、DPD活性を阻害するが、ウリジンホスホリラーゼ(UPase)、オロチン酸ホスホリボシルトランスフェラーゼ(OPRTase)およびチミジンホスホリラーゼ(TP)等のフルオロウラシルを活性化するその他の酵素を実質的に阻害しないものを挙げることができる。
【0018】
本発明の本態様にしたがう別の実施形態において、5−FUまたは5−FUプロドラッグは、以下の化合物およびそれらのリン酸エステル等の5’−エステルからなる群より選択される:5−フルオロウリジン、5−フルオロシチジン、5−フルオロ−2−デオキシウリジン、5−フルオロ−2−デオキシシチジン、および5−フルオロアラビノシルウラシル。
【0019】
本発明の本態様にしたがう別の実施形態において、5−FUまたは5−FUプロドラッグは、5’−デオキシ−4’,5−フルオロウリジン、5’−デオキシ−5−フルオロウリジン、1−(2−テトラヒドロフラニル)−5−フルオロウラシル、1−C1−8アルキルカルバモイル−5−フルオロウラシル誘導体、1−(2−テトラヒドロフリル)−5−フルオロウラシル、5’−デオキシ−5−フルオロ−N−[(ペンチルオキシ)カルボニル]−シチジン(カペシタビン)、またはインビボで5−FUに変換される化合物からなる群より選択される。本発明の本態様にしたがう別の実施形態において、DPD阻害物質はエニルウラシル、またはそのプロドラッグである。
【0020】
本発明の本態様にしたがう別の実施形態において、DPD阻害物質はエニルウラシルであり、0.8〜15、2〜15、5〜15または2.5〜5mg/m2の用量で投与される。
【0021】
本発明の本態様にしたがう別の実施形態において、DPD阻害物質はエニルウラシルであり、5−FUまたは5−FUプロドラッグは5−FUである。
【0022】
本発明の本態様にしたがう別の実施形態において、5−FUまたは5−FUプロドラッグは5−FUであり、0.5〜80、0.5〜40、10〜80、10〜60、10〜30または20〜60mg/m2の用量で投与される。
【0023】
本発明の別の実施形態において、エニルウラシルと、5−FUまたは5−FUプロドラッグは、1:3〜1:20、1:5〜1:15、または1:8〜1:12の比の用量で投与される。
【0024】
一つの好ましい実施形態において、エニルウラシルは、約15〜30mg/m2の用量での5−FUまたは5−FUプロドラッグの投与、または約5〜100mg/m2の用量でのカペシタビンの投与の少なくとも約10〜14時間前に、最初に約2.5〜5mg/m2の用量で投与される。場合により、さらなる用量の5−FUまたはカペシタビンを、エニルウラシルのさらなる投与を伴いまたは伴わずに、その後に投与することができる。
【0025】
本発明の本態様にしたがう別の実施形態において、DPD阻害物質はエニルウラシルであり、5−FUまたは5−FUプロドラッグはカペシタビンである。
【0026】
本発明の本態様にしたがう別の実施形態において、DPD阻害物質はエニルウラシルであり、5−FUまたは5−FUプロドラッグは5−FUまたはカペシタビンであり、エニルウラシルは約0.8〜15、2.5〜15、5〜15または2.5〜5mg/m2の用量で投与される。
【0027】
本発明の本態様にしたがう別の実施形態において、DPD阻害物質はエニルウラシルであり、5−FUまたは5−FUプロドラッグは5−FUであり、使用される投与スケジュールに応じて、エニルウラシルは約2.5〜5mg/m2の用量で投与され、5−FUは約0.5〜40mg/m2の用量で投与される。
【0028】
一つの例示的スケジュール、例えば、28日間(28−day)、1日当たり2回(b.i.d.)のスケジュールは、約0.5〜1.5mg/m2の用量の5−FUを使用する。別の例示的スケジュール、例えば、5日間(5−day)、1日当たり1回のスケジュールは、約10〜60mg/m2の用量の5−FUを使用する。より特定の実施形態において、5−FUの用量は約10〜30mg/m2である。別の実施形態において、5−FUの用量は約20〜60mg/m2である。
【0029】
別の例示的スケジュール、例えば、1週間当たり(weekly)1回のスケジュールは、約10〜80mg/m2の用量の5−FUを使用し得る。より特定の実施形態において、5−FUを約15〜40または10〜30mg/m2の用量で投与する。別の実施形態において、5−FUを約30〜80mg/m2の用量で投与する。
【0030】
本発明の本態様にしたがう別の実施形態において、DPD阻害物質はエニルウラシルであり、5−FUまたは5−FUプロドラッグはカペシタビンであり、エニルウラシルは約0.8〜15、約2〜15、約5〜15または約2.5〜5mg/m2の用量で投与され、カペシタビンは約0.8〜200mg/m2の用量で投与される。より特定の実施形態において、カペシタビンは、(例えば、特定の例示的な長期にわたる1日2回の日々の計画のために)約0.8〜10mg/m2または1.3〜4mg/m2の用量で投与される。
【0031】
本発明の本態様にしたがう別の実施形態において、DPD阻害物質は5位が置換されたウラシル類似体またはそのプロドラッグを含む。
【0032】
本発明の本態様にしたがう別の実施形態において、DPD阻害物質は、ハロゲン原子、C2−4アルケニル基、ハロゲンによって置換されたC2−4アルケニル基、C2−6アルキニル基、ハロゲンによって置換されたC2−6アルキニル基、シアノ基またはハロゲンによって置換されたC1−4アルキル基によって5位が置換されたウラシル類似体を含む。
【0033】
本発明の本態様にしたがう別の実施形態において、DPD阻害物質は、エニルウラシル、5−プロピニルウラシル、5−シアノウラシル、5−プロピニルウラシル、5−ブロモエチニルウラシル、5−(1−クロロビニル)ウラシル、5−ヨードウラシル、5−ブロモビニルウラシル、(E)−5−(2−ブロモビニル)ウラシル、5−ヘキサ−1−イニルウラシル、5−ビニルウラシル、5−トリフルオロウラシル、5−ブロモウラシルおよび5−(2−ブロモ−1−クロロビニル)ウラシルからなる群より選択されるウラシル類似体を含む。
【0034】
本発明の本態様にしたがう別の実施形態において、DPD阻害物質は、5−(フェニルセレネニル)ウラシル(PSU)、5−(フェニルチオ)ウラシル(PTU)、5−(フェニルセレネニル)バルビツール酸および5−(フェニルチオ)バルビツール酸からなる群より選択される。
【0035】
本発明の別の態様にしたがって、エニルウラシルを最初に投与し、その後に5−FUを投与することを含む、患者における癌の処置方法であって、5−FUは、その投与時に5−FUが患者におけるDPD阻害物質を実質的に上回る量で存在するような用量で投与される、方法を提供する。
【0036】
本発明の本態様にしたがう一つの実施形態において、5−FUは、エニルウラシルが投与されてから、少なくとも約4時間後、6時間後、8時間後、10時間後、12時間後、14時間後、24時間後または36時間後に投与される。一つの好ましい実施形態において、5−FUは、エニルウラシルが投与されてから、少なくとも約4時間後、少なくとも約8時間後または少なくとも約12時間後に投与される。
【0037】
本発明の本態様にしたがう別の実施形態において、5−FUは、エニルウラシルが投与されてから約4〜72時間後、4〜36時間後、4〜24時間後、4〜14時間後、6〜14時間後または8〜14時間後に投与される。一つの好ましい実施形態において、5−FUは、エニルウラシルが投与されてから約4〜14時間後に投与される。
【0038】
本発明の別の実施形態において、5−FUは、エニルウラシルの投与後、エニルウラシルの排出半減期の約0.1〜4倍、排出半減期の1〜4倍、排出半減期の2〜4倍または排出半減期の3〜4倍が経過した時点で投与される。
【0039】
本発明の本態様にしたがう別の実施形態において、エニルウラシルは、患者におけるDPD活性をベースラインのDPD活性の約10%未満、約5%未満、約3%未満または約1%未満に低減させるに十分な用量で投与する。
【0040】
本発明の本態様にしたがう別の実施形態において、5−FUは、その投与時に5−FUが患者中にエニルウラシルの少なくとも約2倍、少なくとも約3倍、少なくとも約5倍または少なくとも約100倍過剰で存在するような用量で投与される。
【0041】
本発明の本態様にしたがう別の実施形態において、エニルウラシルは約0.7〜15mg/m2の用量で投与される。本発明の別の実施形態において、エニルウラシルは約2.5〜5mg/m2の用量で投与される。本発明の別の実施形態において、エニルウラシルは約5〜15mg/m2の用量で投与される。
【0042】
本発明の本態様にしたがう別の実施形態において、エニルウラシルは約2.5〜5mg/m2の用量で投与され、5−FUは約0.5〜40mg/m2の用量で投与される。
【0043】
本発明の別の態様にしたがって、エニルウラシルを最初に投与し、その後に5−FUを投与することを含む、患者における癌の処置方法であって、エニルウラシルは約2.5〜5mg/m2の用量で投与され、5−FUは、エニルウラシルの投与後、エニルウラシルの排出半減期の約1〜4倍、2〜4倍または3〜4倍が経過した時点で投与される、方法を提供する。
【0044】
本発明の本態様にしたがう一つの実施形態において、5−FUは、エニルウラシルが投与されてから、少なくとも約4時間後、6時間後、8時間後、10時間後、12時間後または14時間後に投与される。
【0045】
本発明の本態様にしたがう一つの実施形態において、5−FUは、エニルウラシルが投与されてから、約4〜36時間後、4〜24時間後、4〜14時間後、6〜14時間後または8〜14時間後に投与される。
【0046】
本発明の本態様にしたがう別の実施形態において、5−FUは、その投与時に5−FUが患者中にエニルウラシルの少なくとも約2倍、少なくとも約3倍、少なくとも約5倍または少なくとも約100倍過剰で存在するような用量で投与される。
【0047】
本発明の本態様にしたがう別の実施形態において、5−FUは約0.5〜40mg/m2の用量で投与される。
【0048】
より特定の実施形態において、エニルウラシル(または別のDPD阻害物質)を最初に投与し、その後に、エニルウラシル(または別のDPD阻害物質)を場合により再度投与する前の望ましい時点で、5−FUまたは5−FUプロドラッグを複数回投与する。例えば、一つの好ましい実施形態において、エニルウラシルは最初、5−FUまたは5−FUプロドラッグの投与の少なくとも約10〜14時間前に、例えば、5−FUを約15〜30mg/m2の用量で投与されまたはカペシタビンは約5〜100mg/m2の用量で投与されるいずれかの初日の前の晩に、約2.5〜5mg/m2の用量で投与され、その後、同様の複数用量の5−FUまたはカペシタビンは投与される。例えば、一つの例示的実施形態において、エニルウラシルを最初に投与し、その後に、例えば、エニルウラシルを場合により再び投与する前に、5−FUまたはカペシタビンは、1週間に付き3日間は毎日複数回投与され、そして、このサイクルを繰り返す。
【0049】
本発明の別の態様にしたがって、最初にエニルウラシルを投与し、その後5−FUを投与することを含む、患者における癌の処置方法であって、エニルウラシルは約2.5〜5mg/m2の用量で投与され、5−FUは、エニルウラシルの投与後、エニルウラシルの排出半減期の1〜4倍、2〜4倍または3〜4倍が経過した時点で投与され、5−FUは約0.5〜40mg/m2の用量で投与される、方法を提供する。
【0050】
本発明の別の態様にしたがって、DPD阻害物質および5−FUまたは5−FUプロドラッグを含む医薬徐放製剤であって、患者へ製剤を投与した後、DPD阻害物質が放出されてから約0.5〜36時間後、4〜36時間後、4〜24時間後または4〜14時間後まで、5−FUまたは5−FUプロドラッグが実質的に放出されない、前記製剤を提供する。
【0051】
本発明の別の態様にしたがって、患者にDPD阻害物質を投与するためにDPD阻害物質および送達ビヒクルを含む製剤処方が提供される。別の実施形態において、製剤は、DPD阻害物質および5−FUまたは5−FUプロドラッグを含む。別の実施形態において、製剤は、送達ビヒクル、DPD阻害物質および5−FUまたは5−FUプロドラッグを含む。特定の実施形態において、送達ビヒクルはミクロスフィアである。関連する実施形態において、送達ビヒクルは、当技術分野において知られている製剤技術および送達技術を用いて癌細胞へのDPD阻害物質の優先的または選択的ターゲティングを可能にするミクロスフィアである。
【0052】
本発明のこれらおよびその他の態様は、以下の詳細な説明および添付の図面を参照した際に明らかになるであろう。本発明の各種態様をより具体的に示すために本明細書中で引用された特許およびその他の文献は、それらの全体を参照により本明細書に組み入れるものとする。
例えば、本願発明は以下の項目を提供する。
(項目1)
最初にDPD阻害物質を投与し、その後に5−FUまたは5−FUプロドラッグを投与することを含む、患者における癌の処置方法であって、該5−FUまたは5−FUプロドラッグは、その投与時に、該5−FUまたは5−FUプロドラッグが該DPD阻害物質を上回る量で該患者中に存在するような用量で投与される、方法。
(項目2)
前記5−FUまたは5−FUプロドラッグは、前記DPD阻害物質が投与されてから少なくとも約4時間後に投与される、項目1に記載の方法。
(項目3)
前記5−FUまたは5−FUプロドラッグは、前記DPD阻害物質が投与されてから少なくとも約12時間後に投与される、項目1に記載の方法。
(項目4)
前記5−FUまたは5−FUプロドラッグは、前記DPD阻害物質が投与されてから少なくとも約24時間後に投与される、項目1に記載の方法。
(項目5)
前記5−FUまたは5−FUプロドラッグは、前記DPD阻害物質の投与後、前記DPD阻害物質の排出半減期の少なくとも1〜4倍が経過した時点で投与される、項目1に記載の方法。
(項目6)
前記DPD阻害物質は、前記患者におけるDPD活性を該患者におけるベースラインのDPD活性の5%未満に低減させるに十分な用量で投与される、項目1に記載の方法。
(項目7)
前記5−FUまたは5−FUプロドラッグは、その投与時に該5−FUまたは5−FUプロドラッグが前記患者中に前記DPD阻害物質の少なくとも2倍過剰で存在するような用量で投与される、項目1に記載の方法。
(項目8)
前記5−FUまたは5−FUプロドラッグは、その投与時に該5−FUまたは5−FUプロドラッグが前記患者中に前記DPD阻害物質の少なくとも5倍過剰で存在するような用量で投与される、項目1に記載の方法。
(項目9)
前記DPD阻害物質が不可逆性DPD阻害物質である、項目1に記載の方法。
(項目10)
前記DPD阻害物質が可逆性DPD阻害物質である、項目1に記載の方法。
(項目11)
前記5−FUまたは5−FUプロドラッグが、5−フルオロウリジン、5−フルオロシチジン、5−フルオロ−2−デオキシウリジン、5−フルオロ−2−デオキシシチジン、5’−デオキシ−4’,5−フルオロウリジン、および5−フルオロアラビノシルウラシル、5’−デオキシ−5−フルオロウリジン、1−(2−テトラヒドロフラニル)−5−フルオロウラシル、1−C1−8アルキルカルバモイル−5−フルオロウラシル誘導体、1−(2−テトラヒドロフリル)−5−フルオロウラシル、5’−デオキシ−5−フルオロ−N−[(ペンチルオキシ)カルボニル]−シチジン(カペシタビン)、またはインビボで5−FUに変換される化合物からなる群およびそれらのリン酸エステルを含めた5’−エステルから選択される、項目1に記載の方法。
(項目12)
前記5−FUまたは5−FUプロドラッグが5−FUまたはカペシタビンである、項目1に記載の方法。
(項目13)
前記DPD阻害物質がエニルウラシルまたはそのプロドラッグである、項目1に記載の方法。
(項目14)
前記DPD阻害物質がエニルウラシルであり、前記5−FUまたは5−FUプロドラッグが5−FUまたはカペシタビンである、項目1に記載の方法。
(項目15)
前記DPD阻害物質がエニルウラシルであり、前記5−FUまたは5−FUプロドラッグが5−FUであり、前記エニルウラシルは約0.8〜15mg/m2の用量で投与される、項目1に記載の方法。
(項目16)
前記DPD阻害物質がエニルウラシルであり、前記5−FUまたは5−FUプロドラッグが5−FUであり、前記エニルウラシルは約2.5〜5mg/m2の用量で投与される、項目1に記載の方法。
(項目17)
前記DPD阻害物質がエニルウラシルであり、前記5−FUまたは5−FUプロドラッグが5−FUであり、前記エニルウラシルは約5〜15mg/m2の用量で投与される、項目1に記載の方法。
(項目18)
前記DPD阻害物質がエニルウラシルであり、前記5−FUまたは5−FUプロドラッグが5−FUであり、前記エニルウラシルは約2.5〜15mg/m2の用量で投与され、前記5−FUは約0.5〜80mg/m2の用量で投与される、項目1に記載の方法。
(項目19)
前記DPD阻害物質がエニルウラシルであり、前記5−FUまたは5−FUプロドラッグが5−FUであり、前記エニルウラシルは約2.5〜5mg/m2の用量で投与され、前記5−FUは約0.5〜40mg/m2の用量で投与される、項目1に記載の方法。
(項目20)
前記DPD阻害物質が5−置換ウラシル化合物またはそのプロドラッグを含む、項目1に記載の方法。
(項目21)
前記DPD阻害物質が、ハロゲン原子、C2−4アルケニル基、ハロゲンによって置換されたC2−4アルケニル基、C2−6アルキニル基、ハロゲンによって置換されたC2−6アルキニル基、シアノ基、C1−4アルキル基またはハロゲンによって置換されたC1−4アルキル基によって5位が置換されたウラシル化合物を含む、項目1に記載の方法。
(項目22)
前記DPD阻害物質が、エニルウラシル、5−プロピニルウラシル、5−シアノウラシル、5−プロピニルウラシル、5−ブロモエチニルウラシル、5−(1−クロロビニル)ウラシル、5−ヨードウラシル、5−ブロモビニルウラシル、(E)−5−(2−ブロモビニル)ウラシル、5−ヘキサ−1−イニルウラシル、5−ビニルウラシル、5−トリフルオロウラシル、5−ブロモウラシルおよび5−(2−ブロモ−1−クロロビニル)ウラシルからなる群より選択されるウラシル化合物を含む、項目1に記載の方法。
(項目23)
前記DPD阻害物質が、5−(フェニルセレネニル)ウラシル(PSU)、5−(フェニルチオ)ウラシル(PTU)、5−(フェニルセレネニル)バルビツール酸および5−(フェニルチオ)バルビツール酸からなる群より選択される、項目1に記載の方法。
(項目24)
最初にエニルウラシルを投与し、その後に5−FUを投与することを含む、患者における癌の処置方法であって、該5−FUは、その投与時に該5−FUが該DPD阻害物質を上回る量で該患者中に存在するような用量で投与される、方法。
(項目25)
前記5−FUは、前記エニルウラシルが投与されてから少なくとも約4時間後に投与される、項目24に記載の方法。
(項目26)
前記5−FUは、前記エニルウラシルが投与されてから少なくとも約12時間後に投与される、項目24に記載の方法。
(項目27)
前記5−FUは、前記エニルウラシルの投与後、エニルウラシルの排出半減期の少なくとも約1〜4倍が経過した時点で投与される、項目24に記載の方法。
(項目28)
前記エニルウラシルは、前記患者におけるDPD活性を前記患者におけるベースラインのDPD活性の5%未満に低減させるに十分な用量で投与される、項目24に記載の方法。
(項目29)
前記5−FUは、その投与時に前記5−FUが前記患者中に前記DPD阻害物質の少なくとも2倍過剰で存在するような用量で投与される、項目24に記載の方法。
(項目30)
前記5−FUは、その投与時に前記5−FUが前記患者中に前記DPD阻害物質の少なくとも5倍過剰で存在するような用量で投与される、項目24に記載の方法。
(項目31)
前記エニルウラシルは約2.5〜5mg/m2の用量で投与される、項目24に記載の方法。
(項目32)
前記エニルウラシルは約2.5〜15mg/m2の用量で投与され、前記5−FUは約0.5〜40mg/m2の用量で投与される、項目24に記載の方法。
(項目33)
最初にエニルウラシルを投与し、その後に5−FUを投与することを含む、患者における癌の処置方法であって、前記エニルウラシルは約2.5〜15mg/m2の用量で投与され、前記5−FUは、前記エニルウラシルの投与後、エニルウラシルの排出半減期の少なくとも1〜4倍が経過した時点で投与される、方法。
(項目34)
前記5−FUは、前記エニルウラシルが投与されてから少なくとも約4時間後に投与される、項目33に記載の方法。
(項目35)
前記5−FUは、前記エニルウラシルが投与されてから少なくとも約14時間後に投与される、項目33に記載の方法。
(項目36)
前記5−FUは、前記エニルウラシルが投与されてから少なくとも約18時間後に投与される、項目33に記載の方法。
(項目37)
前記5−FUは、その投与時に前記5−FUが前記患者中に前記DPD阻害物質の少なくとも2倍過剰で存在するような用量で投与される、項目33に記載の方法。
(項目38)
前記5−FUは、その投与時に前記5−FUが前記患者中に前記DPD阻害物質の少なくとも5倍過剰で存在するような用量で投与される、項目33に記載の方法。
(項目39)
前記5−FUは約0.5〜40mg/m2の用量で投与される、項目33に記載の方法。
(項目40)
最初にエニルウラシルを投与し、その後に5−FUを投与することを含む、患者における癌の処置方法であって、前記エニルウラシルは約2.5〜15mg/m2の用量で投与され、前記5−FUは、前記エニルウラシルを投与してから少なくとも約4時間後に投与され、前記5−FUは約0.5〜40mg/m2の用量で投与される、方法。
(項目41)
前記5−FUは、前記エニルウラシルが投与されてから少なくとも約14時間後に投与される、項目40に記載の方法。
(項目42)
前記5−FUは、前記エニルウラシルが投与されてから少なくとも約18時間後に投与される、項目40に記載の方法。
(項目43)
最初にエニルウラシルを投与し、その後にカペシタビンを投与することを含む、患者における癌の処置方法であって、該エニルウラシルは約2.5〜15mg/m2の用量で投与され、該5−FUは、該エニルウラシルの投与後、エニルウラシルの排出半減期の少なくとも1〜4倍が経過した時点で投与される、方法。
(項目44)
前記カペシタビンは、前記エニルウラシルが投与されてから少なくとも約4時間後に投与される、項目43に記載の方法。
(項目45)
前記カペシタビンは、前記エニルウラシルが投与されてから少なくとも約14時間後に投与される、項目43に記載の方法。
(項目46)
前記カペシタビンは、前記エニルウラシルが投与されてから少なくとも約24時間後に投与される、項目43に記載の方法。
(項目47)
前記カペシタビンは、その投与時に該カペシタビンが前記患者中に前記DPD阻害物質の少なくとも2倍過剰で存在するような用量で投与される、項目43に記載の方法。
(項目48)
前記カペシタビンは、その投与時に前記カペシタビンが前記患者中に前記DPD阻害物質の少なくとも5倍過剰で存在するような用量で投与される、項目43に記載の方法。
(項目49)
前記カペシタビンは約1.5〜4mg/m2の用量で投与される、項目43に記載の方法。
(項目50)
DPD阻害物質および5−FUまたは5−FUプロドラッグを含む経口医薬徐放製剤であって、前記製剤を患者へ投与した後に、該DPD阻害物質が放出されてから少なくとも約4時間後までの間は該5−FUまたは5−FUプロドラッグが実質的に放出されない、前記製剤。
(項目51)
前記DPD阻害物質が放出されてから少なくとも約12時間後までの間は前記5−FUまたは5−FUプロドラッグが実質的に放出されない、項目50に記載の製剤。
(項目52)
前記5−FUまたは5−FUプロドラッグが5−FUまたはカペシタビンである、項目50に記載の製剤。
(項目53)
前記DPD阻害物質がエニルウラシルである、項目50に記載の製剤。
(項目54)
DPD阻害物質および送達ビヒクルを含む経口医薬徐放製剤であって、該送達ビヒクルが、患者の癌細胞に対する前記DPD阻害物質の選択的標的化を容易にする、前記製剤。
(項目55)
前記送達ビヒクルがミクロスフィアである、項目54に記載の製剤。
(項目56)
最初にエニルウラシルを投与し、その後に5−FUを投与することを含む、患者における癌の処置方法であって、エニルウラシルは5−FUの投与の少なくとも約0.5時間前に投与され、該5−FUまたは5−FUプロドラッグは、その投与時に該5−FUまたは5−FUプロドラッグが該DPD阻害物質を上回る量で該患者中に存在するような用量で投与される、方法。
(項目57)
最初にエニルウラシルを投与し、その後に5−FUを投与することを含む、患者における癌の処置方法であって、1日あたり2回の長期投与スケジュール開始の少なくとも約12時間前に約2.5〜15mg/m2の用量でエニルウラシルは投与され、その後、各日における1日2回の最初の5−FU投与の前の約4〜6時間の間の時点で、約0.5〜2mg/m2の用量で該エニルウラシルは投与される、方法。
(項目58)
前記DPD阻害物質の用量と、前記5−FUまたは5−FUプロドラッグの用量とが、1:3〜1:20の比にある、項目1に記載の方法。
(項目59)
前記DPD阻害物質の用量と、前記5−FUまたは5−FUプロドラッグの用量とが、1:5〜1:15の比にある、項目1に記載の方法。
(項目60)
前記DPD阻害物質の用量と、前記5−FUまたは5−FUプロドラッグの用量とが、1:8〜1:12の比にある、項目1に記載の方法。
【図面の簡単な説明】
【0053】
【図1】図1は、エニルウラシルおよび5−FUの化学構造を示す。
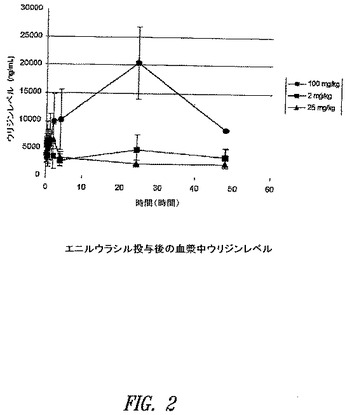
【図2】図2は、エニルウラシルがマウスにおける投与後に血漿中のウリジンレベル増大を引き起こすことを示す。
【図3】図3は、エニルウラシルおよび5−FUを含む錠剤形態の経口徐放製剤を例示する。
【発明を実施するための形態】
【0054】
(発明の詳細な説明)
上述の通り、本発明は全体として、少なくとも1つの5−FUまたは5−FUプロドラッグと組み合わせた少なくとも1つのDPD阻害物質を患者へと投与することを含む癌の処置方法に関し、また、そのような方法において有用な組成物および製剤に関する。本明細書中に記載の方法はすなわち、実例として、限定されるものではないが、乳癌、肺癌、結腸癌、膵臓癌、胃癌、膀胱癌、腎癌、頭頸部癌、食道癌、肝細胞癌、および全ての悪性白血病およびリンパ腫を含む、そこにおいて5−FUおよび/または5−FUプロドラッグが活性を有する本質的に任意のタイプの癌に対し適用可能である。さらに、本発明は5−FUおよび5−FUプロドラッグの抗腫瘍効果を向上させるため、従来5−FUに対する所望の応答性を示すとはいえなかったかもしれない癌のタイプが、本明細書中に記載の方法に従って投与される場合には、改善された応答性を示す可能性がある。
【0055】
エニルウラシル等のDPD阻害物質が5−FUおよび5−FUプロドラッグの代謝的活性化を阻害し得、それによりそれらの抗腫瘍活性を落とすことが、思いがけなく発見された。したがって、本発明の一般的な態様に従い、5−FUまたは5−FUプロドラッグが患者へと投与される時点で、5−FUまたは5−FUプロドラッグレベルがDPD阻害物質レベルを十分に上回るようにすることにより、それに対しDPD阻害物質が5−FUまたは5−FUプロドラッグの代謝的活性化を妨害し得る度合は都合よく最小化され、またこれらの薬物の抗腫瘍効果はそれにより向上する。したがって、特定の実施形態において、エニルウラシル等の不可逆性なDPD阻害物質はDPDを効果的に不活化する最低用量で投与される必要があり、また、好ましくは、5−FUを投与する前に血流中から過剰な阻害物質を除去するために十分な時間が経過する必要がある。さらに、DPDは5−FUまたは5−FUプロドラッグの投与前に患者から実質的に除去することができるため、所望のレベルの治療的活性を達成するために必要とされる5−FUまたは5−FUプロドラッグの用量を顕著に低減することができ、それにより本明細書中に記載の治療的な優位性に加えて経済的な優位性を提供できる。
【0056】
この開示に照らして、5−FUまたは5−FUプロドラッグのレベルが投与時に治療上効果的な量であり、かつ、5−FU代謝活性化の阻害を最小化するまたは無くすために患者中のDPD阻害物質レベルを十分に上回るようにさせながら本明細書中に記載の方法に使用することができる、多数の投与および服薬スケジュールが存在することが当業者の一部には理解されるであろう。このような全ての投与および服薬スケジュールが本発明の範囲内であると考えられる。
【0057】
本発明の1つの実例となる実施形態において、DPD阻害物質をまず、その患者中のDPD活性を実質的になくすために、その必要性を有する患者へと投与(すなわち、前投与)し、次いで5−FUまたは5−FUプロドラッグを投与する。「実質的になくす」とは、患者中のDPD活性のレベルが、DPD阻害物質投与前の患者におけるDPD活性のベースラインに対して少なくとも20%未満、10%未満、5%未満、3%未満または1%未満であることを意味する。患者のDPD活性のベースラインレベルは、例えば、患者から得たPBMC中で、公知の手法(例えば、Bakerら、J Clin Oncol 18:915−926 2000;Schilskyら、J Clin Oncol 4:1450−7,1998)を用いて、迅速に調べることができる。
【0058】
少なくとも1つのDPD阻害物質を最初に投与し、それにより実質的にその患者中のDPDの活性を失わせた後、次いで、DPD阻害物質が患者から排除され実質的になくなるのに十分な時間が経過した後に、5−FUまたは5−FUプロドラッグまたはそれらの組み合わせを、患者へと投与する。DPD阻害物質の投与と、5−FUまたは5−FUプロドラッグの投与との間の時間的な遅れは、5−FUまたは5−FUプロドラッグの投与条件により変化し得、それはその時点で患者中の残存するDPD阻害物質のレベルに対して実質的に過剰な量で患者中に存在する。1つの実例となる実施形態において、5−FUまたは5−FUプロドラッグを、その患者中に存在する5−FUまたは5−FUプロドラッグのレベルが少なくともその患者中に残存するDPD阻害物質よりも過剰に、例えば、5−FUまたは5−FUプロドラッグの投与時に、その患者中に残存するDPD阻害物質のレベルの少なくとも約2倍、少なくとも約3倍、少なくとも約5倍または少なくとも約100倍過剰で存在するような用量で投与する。当業者であれば、本明細書中に記載の実施形態に従って、DPD阻害物質に対する5−FUまたは5−FUプロドラッグの患者における過剰さの程度を計算および/または決定するために、多数の公知かつ適用可能な手法のうち任意のものを使用することができるであろう。このような例には、例えば、HPLC、LC−MS、ELISA等が挙げられる。上述の通り、5−FUまたは5−FUプロドラッグの投与時に、5−FUまたは5−FUプロドラッグが患者中のDPD阻害物質に対し十分に過剰なレベルで存在することを確実にすることにより、DPD阻害物質による5−FUまたは5−FUプロドラッグの代謝的活性化の妨害がそれにより最小化され、5−FUまたは5−FUプロドラッグの有効性がそれにより向上すると考えられる。
【0059】
本発明の別の実施形態において、5−FUまたは5−FUプロドラッグを、DPD阻害物質の投与後、DPD阻害物質の排出半減期の少なくとも0.1〜4倍、排出半減期の1〜4倍、排出半減期の2〜4倍、または排出半減期の3〜4倍が経過した時点で患者へと投与する。特定のDPD阻害物質に関する排出半減期が調べられており、調べられていないものについては、排出半減期は、よく知られ確立されたガスクロマトグラフィー/質量分析およびHPLC手法を用いて迅速に決定することができる(Bakerら、J Clin Oncol 18:915−926 2000;Schilskyら、J Clin Oncol 4:1450−7,1998参照)。ヒトにおけるエニルウラシルの排出半減期は約3.5時間であると報告されている(例えば、Bakerら、J Clin
Oncol 18:915−926 2000;Ochoaら、Ann Oncol 11:1313−22,2000)が、エニルウラシルおよびその他のDPD阻害物質の半減期は用量依存的であり、この用量依存性はDPD阻害物質、および5−FUまたは5−FUプロドラッグの間の適切な時間遅延を調べる際に考慮される必要がある。したがって、エニルウラシルをDPD阻害物質として利用する本発明のある種の実施形態については、エニルウラシルのレベルを5−FUまたは5−FUプロドラッグ投与前の除去によって十分に低減できるようにするために、5−FUまたは5−FUプロドラッグは、エニルウラシルの投与後、少なくとも約0.5時間、約2時間、約4時間、約6時間、約8時間、約10時間、約12時間、約14時間、約24時間または約36時間後に投与される。特定のその他の実施形態において、5−FUまたは5−FUプロドラッグは、エニルウラシル投与後約4〜72時間、4〜36時間、約4〜24時間、約4〜14時間、約6〜14時間または約8〜14時間後に投与される。もちろん、これらの範囲は事実上説明的なものであり、特定の投与計画が必要とされまたは所望される時には、5−FU投与時にエニルウラシルの存在が最小化されるかまたは存在しない状態となる限りにおいて変更し得る。
【0060】
本発明に従い使用されるDPD阻害物質は、可逆性または不可逆性のDPD酵素の阻害物質であり得る。DPD酵素の可逆性阻害物質の説明的な実例には、ウラシル、CDHPおよび3−シアノ−2,6−ジヒドロキリピリジン(CNDP)を含む。DPD酵素の可逆性阻害物質におけるその他の説明的な実例には、例えば、5−(フェニルセレネニル)ウラシル(PSU)、5−(フェニルチオ)ウラシル(PTU)、5−(フェニルセレネニル)バルビツール酸および5−(フェニルチオ)バルビツール酸等、米国特許第5,476,855号および国際公開第95/012400号中に記載のものを含み、それらの内容を、その全体を参照により本明細書に組み入れるものとする。特定の好ましい可逆性DPD阻害物質には、過剰な阻害物質が身体から排除されるよりもゆっくりとDPDから脱離する強固に結合した阻害物質、および/または、DPD活性を阻害するが、ウリジンホスホリラーゼ(UPase)、オロチン酸ホスホリボシルトランスフェラーゼ(OPRTase)およびチミジンホスホリラーゼ(TP)等のフルオロウラシルを活性化するその他の酵素を実質的に阻害しない阻害物質を含むことが理解されるであろう。
【0061】
本発明の特定の好ましい実施形態において、DPD阻害物質は不可逆的にDPD酵素を不活化するものである。これに関する説明的なDPD阻害物質としては、限定されるものではないが、5−置換ウラシル化合物、またはそのプロドラッグを含むDPD阻害物質、特に5位がハロゲン原子、所望によりハロゲンで置換された(例えば、2−ブロモビニル、1−クロロビニルまたは2−ブロモ−1−クロロビニル)C2−4 アルケニル基(例えば、ビニル)、所望によりハロゲン原子で置換されたC2−6 アルキニル基、シアノ基、またはハロゲンで置換されたC1−4 アルキル基(例えば、トリフルオロメチル)、で置換されたウラシル化合物を含むDPD阻害物質が挙げられる。
【0062】
本発明のより特定的な実施形態において、DPD阻害物質は、エニルウラシル、5−プロピニルウラシル、5−シアノウラシル、5−プロピニルウラシル、5−ブロモエチニルウラシル、5−(1−クロロビニル)ウラシル、5−ヨードウラシル、5−ブロモビニルウラシル、(E)−5−(2−ブロモビニル)ウラシル、5−ヘキサ−1−イニルウラシル、5−ビニルウラシル、5−トリフルオロウラシル、5−ブロモウラシル、および5−(2−ブロモ−l−クロロビニル)ウラシル、またはそれらのプロドラッグからなる群から選択される。
【0063】
別の実施形態において、DPD阻害物質は5−ブロモビニルウラシルのプロドラッグであり、説明的な1化合物は、化合物1−β−D−アラビノフラノシル−(E)−5−(2−ブロモビニル)ウラシル(BV−araUまたはソリブジンとも称する)により代表される。これに関する特定の説明的なプロドラッグ化合物としては、例えば、米国特許第4,386,076号が挙げられ、この開示を参照により本明細書に組み入れるものとする。
【0064】
本発明の好ましい1実施形態において、5−エチニル−2(1H)−ピリミジノン(4−酸素を失ったエニルウラシル)(Porterら、Biochem.Pharmacol 47:1165− 1171,1994)、エニルウラシルのヌクレオシドまたはデオキシヌクレオチドシド誘導体、インビボでエニルウラシルへと変換される化合物および/またはインビボで不活化剤へと変換されるDPD不活化剤誘導体等の、DPD阻害物質はエニルウラシルまたはエニルウラシルのプロドラッグである。一例として、このような化合物には、上記の5−置換ウラシル化合物に相当する核酸塩基、例えば、リボース、2’−デオキシリボース、2’,3’−ジデオキシリボース、アラビノースまたは、ハロゲンまたはエステル等の5’置換基等の2’−または3’−置換基をさらに含み得るその他の開裂可能な糖を含むヌクレオシド誘導体を含むヌクレオシド誘導体が挙げられる。このようなヌクレオシド誘導体のより特定的な例としては、1−(β−D−アラビノフラノシル)−5−プロプ−1−イニルウラシルおよび2’,3’−ジデオキシ−5−エチニル−3’−フルオロウリジンが挙げられる。
【0065】
本発明に従って使用される多数の5−FUプロドラッグもまた知られているだろう。5−FUのプロドラッグは5−フルオロウラシルへと代謝される化合物であり、例として、5−フルオロウリジン、5−フルオロシチジン、5−フルオロ−2−デオキシウリジン、5−フルオロ−2−デオキシシチジン、5−フルオロアラビノシルウラシル、およびそれらのリン酸エステル類を含む5’−エステル類を含む。その他の例示的な化合物には、5’−デオキシ−4’,5−フルオロウリジン、5’−デオキシ−5−フルオロウリジン1−(2−テトラヒドロフラニル)−5−フルオロウラシル、1−C1−8アルキルカルバモイル−5−フルオロウラシル誘導体、1−(2−テトラヒドロフリル)−5−フルオロウラシル、フトラフール(テガフール(Tegafur)、アジア諸国で広範に使用されている経口5−FUプロドラッグ)、および5’−デオキシ−5−フルオロ−N−[(ペンチルオキシ)カルボニル]−シチジン(カペシタビン、Roche Laboratories Inc.によりXeloda(登録商標)として販売)またはインビボで5−FUへと変換される化合物を含み得る。
【0066】
本発明の特に好ましい実施形態において、本明細書中に記載の方法は、5−FUと組み合わせたエニルウラシルを利用する。上述の通り、5−FUの抗腫瘍活性を最大化するため、患者におけるDPD活性を実質的になくし、さらに5−FUが投与されるときにエニルウラシルが過剰に存在しないようにさせる用量で、エニルウラシルを投与する。エニルウラシルは非常に強力な不可逆性のDPD不活化剤であるため、5−FUの前に投与されることが好ましい。エニルウラシルは迅速にDPDを不活化し、次いで、好ましい所定の時間で、5−FUの投与前に患者から排出されることにより、実質的になくなる。したがって、5−FUが患者へと投与される場合にDPD活性は実質的になくなり、エニルウラシルのレベルは都合よく低下するであろう。
【0067】
この開示に照らし、投与スケジュールが、5−FUまたは5−FUプロドラッグが、好ましくは5−FUまたは5−FUプロドラッグの投与時に患者中に残存するエニルウラシルのレベルを上回って存在するように選択される限り、本発明の方法は所望されるどのような遅延および用量特性を有する投与スケジュールをも含み得ることが理解されるであろう。エニルウラシルおよび5−FUの特定の組み合わせを利用する例示的な1実施形態において、本発明の方法は、5日間(5−day)、1日当たり1回、または1週間当たり(weekly)1回のスケジュールを含む。5−dayまたはweeklyのスケジュールに関し、エニルウラシルの用量範囲は、典型的には約0.8〜10mg/m2、好ましくは約2.5〜5mg/m2である。5−FU投与前に過剰分をなくすために十分な時間が経過するのであれば、より高いエニルウラシル用量が使用可能であろうことに留意されたい。
【0068】
簡便のため、ある種の実施形態においては、エニルウラシルの単純に固定化された用量が好ましい。本明細書中に記載するように、約2.5〜15mgの範囲の用量は通常、ほとんどの患者に好適であろう。特定の好ましい実施形態において、固定される用量は約2.5〜5mgの範囲内である。大きさの異なる患者における、24時間以上、およびある場合には3〜5日間以上DPDをなくすであろう例示的なエニルウラシルの固定用量を以下の表に示す。
【0069】
【表1】

1つの実例となる実施形態において、エニルウラシル投与および5−FU投与の間の時間的遅延は約4〜72時間、4〜36時間、4〜24時間または4〜14時間(または少なくともエニルウラシルの排出半減期の約1〜4倍)である。例えば、エニルウラシルを5−FU投与の前夜に投与してもよく、または、別の方法では朝に投与し、その後5−FUを夜に投与することができる。これらのスケジュールに関し、約20〜30mg/m2の例示的な5−FU用量を用いて(Levinら,Invest New Drugs 18:383−90,2000;Schilskyら,J Clin Oncol 4:1450−7,1998;Guoら,Cancer Chemother Pharmacol 52:79−85,2003)、例えば、5−FUは常にエニルウラシルに対し実質的に過剰である必要がある。
【0070】
別の実施形態において、長期投与スケジュール、例えば、28日間の投与スケジュールを使用することができる。例示的な28日間、b.i.dスケジュール(例えば、1日2回、28日間)は、約0.5〜2mg/m2、好ましくは約1mg/m2の5−FUの投与を含む(例えば、Bakerら,J Clin Oncol 18:915−926 2000)。エニルウラシルが低い5−FU用量を上回って存在しないことを保証するために、b.i.d.スケジュールの開始の少なくとも約12時間前、例えば、5−FUを投与する最初の日の前夜に、約2.5〜5mg/m2でエニルウラシルを投与することができる。次いで、各日における最初の5−FU投与の約2〜8時間前、好ましくは約4〜6時間前に約0.5〜2mg/m2、例えば1mg/m2の用量での1日1回のエニルウラシル投与を、28日間、行うことができる。この方法は、最初の投与に由来する5−FUレベルがエニルウラシルを十分に上回るようにさせ、2度目の投与に由来する5−FUレベルがエニルウラシルを十分に上回るようにさせる。続く処置の日々の間、例えば、2〜28日で、患者の血中エニルウラシルのレベル(例えば、1mg/m2で投与した場合)は4時間(最初の日々の5−FU投与)以内で約50%に減少し(0.5mg/m2に)、第2の5−FU投与により約94%(0.06mg/m2)まで減少する。エニルウラシルにおける最初の2.5〜5mg/m2および続いての1mg/m2の用量は、28日間の5−FU投与に関し、患者におけるDPD活性を効果的に失わせるための役割を果たすはずである。
【0071】
その他の実例となる実施形態において、エニルウラシル(またはその他のDPD阻害物質)を最初に投与し、次いで複数用量の5−FUまたは5−FUプロドラッグを、その後、エニルウラシルが所望により再度投与される前に、所望の時点で添加する。例えば、例示的な実施形態において、エニルウラシルを最初に投与し、次いで複数の5−FU用量を例示的な時点である約12時間、36時間、および54時間においても、エニルウラシルが所望により再度投与されてこのサイクルが繰り返される前に、投与する。
【0072】
特定の実施形態において、エニルウラシルおよび5−FUを、約1:5〜1:15または1:8〜1:12の割合で投与する。
【0073】
本発明の別の実施形態において、5−FU投与時に、エニルウラシルに対しモル数が過剰である本発明に従う限り、5−FUを、エニルウラシル投与後にかなり迅速に、例えば、0.5〜1時間で投与する。
【0074】
エニルウラシル投与における本発明の実施形態に記載の用量およびスケジュールは、先に用いられたフェーズIIおよびフェーズIIIヒト試験(Levinら,Invest
New Drugs 18:383−90,2000;Schilskyら,J Clin Oncol:20:1519−26,2002)に用いられたものとは異なる。これらの研究は高用量のエニルウラシルおよび、エニルウラシルと共に同時に、またはエニルウラシルの後わずか1時間後のいずれかに投与される5−FUを使用する。抗腫瘍活性のうちのあるものはフェーズIII試験において立証されていたが、全体の結果は所望されたものに及ばず、試験は成功しなかった。本発明の方法を用いることにより、過剰なエニルウラシルが存在しないようにさせることにより、またすなわち5−FUまたは5−FUプロドラッグが投与される時に5−FUの活性化を阻害し得ないことにより、向上された効果が達成されるであろうことが確信される。
【0075】
本発明は、単一の製剤中に共存してまたは本発明に従う別個の時点において投与される別個の製剤として存在する、少なくとも1つの薬学上許容可能な担体または賦形剤を含んでなり、さらにDPD阻害物質および/または5−FUまたは5−FUプロドラッグを含む、さらなる特徴を有する製剤処方を含む。一実施形態において、エニルウラシルおよび5−FU、約1:3〜1:20、1:5〜1:15、または1:8〜1:12の比率で1以上の製剤処方中に存在する。担体または賦形剤は製剤のその他の成分と適合性を有するという意味で「薬学上許容可能」であり、患者を害しない。製剤には、例えば、経口、直腸内、経鼻、局所(口内および舌下を含む)、経膣および腹腔内(皮下、筋肉内、静脈内および皮内)投与のために適合したものを含む。製剤は、簡便的に単位投与量形態で存在することができ、薬学的な技術においてよく知られた方法により調製することができる。このような方法には、活性成分と、1以上の副成分を構成する担体とをあわせる工程を含む。通常、製剤は均一に調製し、活性成分を液状担体または細粒の固体担体あるいはその両者と密にあわせる工程および、次いで必要であれば、この産物を成形する工程を含む。
【0076】
本発明の製剤を、原則的に任意の適用可能な手法を用いて調製し、および/または投与することができる。経口投与に適合した本発明の製剤は、例えば、予め決定した量の活性成分を;粉末または顆粒として;水系または非水系の溶液または懸濁液として;または水中油型液状エマルジョンまたは油中水型液状としてそれぞれが含むカプセル(capsule、cachet)または錠剤などの別個の単位であってもよい。活性成分は丸薬(bolus)、舐剤またはペーストであってもよい。経口投与は典型的には好ましい投与経路であるだろう。
【0077】
錠剤は、例えば、所望により1以上の副成分と共に圧縮または成型することにより製造してもよい。圧縮された錠剤は、粉末または顆粒等の流し込める形態の活性成分を、所望により結合剤(例えば、ポビドン、ゼラチン、ヒドロキシプロピルメチルセルロース)、滑剤、不活性希釈剤、保存剤、崩壊剤(例えば、デンプングリコール酸ナトリウム、架橋ポビドン、架橋カルボキシメチルセルロースナトリウム)、界面活性剤または分散剤と共に適切な機械において圧縮することにより調製できる。成型錠剤は、不活性希釈剤と共に湿らせた粉末化化合物を適切な機械において成型することにより調製できる。錠剤は所望により被覆したりまたは割線を入れたりしてもよく、活性成分の放出を制御するために、例えば、所望の放出プロファイルを提供する多様な割合のヒドロキシプロピルメチルセルロースをそこに用いて配合することができる。
【0078】
口内での局所投与のための製剤には、例えば、活性成分を風味付けした基礎原料、通常はスクロースおよびアカシアまたはトラガカント中に含むトローチ剤;活性成分をゼラチンおよびグリセリン、またはスクロースおよびアカシア等の不活性の基材中に含むトローチ(pastilles);および、活性成分を好適な液状担体中に含む洗口液を含む。直腸内投与のための製剤としては、例えば、例えば、カカオバターまたはサリチル酸塩等を含む好適な基材と共に座剤として調製してもよい。経膣投与のための製剤としては、例えば、活性成分に加えて技術的に知られたそのような担体を含むペッサリー、タンポン、クリーム、ジェル、ペースト、フォームまたはスプレー製剤が挙げられる。
【0079】
腹腔内投与のための製剤には、例えば、水系および非水系の、抗酸化剤、緩衝剤、静菌剤および所定の被投与者における血液と等張の処方を与える溶質を含み得る等張滅菌注射液;および、分散剤および増粘剤を含み得る水系および非水系の滅菌懸濁液が挙げられる。製剤は、例えば、アンプル、バイアル等の容器内に封入された単位投与または複数投与量であってもよく、例えば注射のために、使用前に直ちに滅菌液状担体を加えるだけでよいフリーズドライ(凍結乾燥)状態で保存することもできる。滅菌粉末、顆粒および前述の種類の錠剤から、即席の注射液を調製する。
【0080】
典型的には、1以上の活性な薬剤を含む液状の製剤を、好ましくはpH7〜11、通常は9.5〜10.5に緩衝化する。特定の単位投与量の製剤化には、活性成分の、本明細書中の上記に列挙されたような日々の投与量または単位、日々の部分用量、またはそれらの適切な一部を含み得る。
【0081】
本明細書中に記載のDPD阻害物質および5−FUプロドラッグを製造する方法は公知であり、従来の方法論を使用して実行することができる。例えば、上記に参照されるDPD阻害物質は、5−エチニルウラシルの調製に関してはHeterocycl.Chem.19(3)463−4(1982);5−(2−ブロモビニル)ウラシル、5−ブロモエチニルウラシルおよび5−(2−ブロモ−l− クロロビニル)ウラシルの調製に関してはJ.Chem.Soc.Perkin Trans.1(16),1665−70(1981);5−シアノ−ウラシルの調製に関してはNucleic Acid Chemistry,Vol.2,927−30(1978);5−ビニルウラシルの調製に関してはNucleic Acids Research,1(1)105−7(1974);5−トリフルオロメチルウラシルの調製に関してはZ.Chern 17(11)415−16(1977);5−(1−クロロビニル)ウラシルの調製に関してはNucleic Acids Research 3(10),2845(1976)に記載の方法により調製することができる。本発明の特定のその他の化合物は、2’,3’−ジデオキシ−5−エチニル−3’−フルオロウリジン等の3’−フルオロ−2’,3’−ジデオキシ5−アルキニルウリジン化合物の調製に関しては、欧州特許出願公開第356166号、および1−(b−D−アラビノフラノシル)−5−プロプ−I−イニルウラシル(1−(b−D−arabinofuranosyl)−5−prop−I−ynyluracil)等の5−アルキニルウラシルアラビノシドの調製に関しては欧州特許出願公開第272065号に記載の工程に従って調製することができる。これらおよびその他の合成手法は公知であり、本発明で使用するための化合物を製造するために利用することができる。
【0082】
一実施形態において、本発明は、製剤成分における所望の時間的放出を可能にする方法でDPD阻害物質および5−FUまたは5−FUプロドラッグを共に患者へと投与する組み合わせ経口製剤を提供する。2つの成分の差示的な時間的放出輸送は、公知の手法および材料を用いて達成できる。例えば、一実施形態において、例えば錠剤形態における経口製剤を、図3に説明的に示すような3つの異なる層から構成することができる。外側の層は、迅速に放出される構造中にエニルウラシルを含み得る。中間層は、本発明の5−FUまたは5−FUプロドラッグの放出を所望の程度まで遅らせる徐放性成分(例えば、徐放性緩衝剤)であることができ、そこにおいて、迅速に放出される構造中のコア層中に、5−FUまたは5−FUプロドラッグが局在する。DPD阻害物質および5−FUまたは5−FUプロドラッグを本明細書中に記載の適切な用量および割合で配合する。好ましい1実施形態において、DPD阻害物質はエニルウラシルであり、5−FUまたは5−FUプロドラッグは5−FUまたはカペシタビンである。
【0083】
別の実施形態において、別の配合は、5−FUまたは5−FUプロドラッグを含むミクロスフィア等の公知の送達ビヒクルを含むことができる。一実施形態において、例えば、5−FUまたは5−FUプロドラッグを、時間的放出成分(例えば、時間的放出崩壊緩衝材)のシェル内にカプセル化することができ、外側の層がDPD阻害物質の迅速な放出を提供する。好ましい1実施形態において、DPD阻害物質はエニルウラシルであり、5−FUまたは5−FUプロドラッグは5−FUまたはカペシタビンである。実例となる組み合わせ製剤におけるこれらおよびその他の実施例を、DPD阻害物質、および、5−FUまたは5−FUプロドラッグの送達の間における適切な時間遅延を1つの経口調製物中で可能にする公知の手法を用いて設計でき、製造することができる。
【0084】
別の実施形態において、DPD阻害物質は、癌細胞および腫瘍を選択的にまたは優先的に標的とすることができる。癌細胞および腫瘍におけるDPDの選択効果は、循環における半減期よりも長い化合物の半減期を可能にし、そのことが、活性化ヌクレオチドのより高い定常状態レベルおよび治療指数の向上を実現するであろう。一実施形態において、腫瘍に対するエニルウラシル等のDPD阻害物質の選択的標的化は、技術的によく知られた標的化されたミクロスフィアを用いて実現可能である。別の実施形態において、標的化は、化合物を肝細胞および肝臓癌細胞に対し標的化するアシアロ糖タンパク質(asiolglycoprotein)等の組織特異的な受容体を用いて達成される。このような標的化は、例えば、DPD阻害物質を化学的にまたはその他の方法で(例えば、エニルウラシルにおける3−6ガラクトース残基を置換することにより)修飾することにより、実現することができる。
【0085】
別の実施形態において、本明細書中に記載の方法は、ロイコボリンの投与をさらに含む。ロイコボリン、またはロイコボリンの活性な異性体であるイソロイコボリンは癌患者の治療に関し5−FUと併せてよく用いられる。それはまた、エニルウラシルおよび5−FUに関する上述の投与計画に付加してもよい。ロイコボリンは腫瘍を有するラットおよび組織培養においてエニルウラシルおよび5−FUの抗腫瘍効果を向上させることが示されており(Caoら、Cancer Res 90:1507−1510,1993;Fischelら、Biochem Pharmacol 53:1703−1709,1997)、また、エニルウラシルおよび5−FUを投与されている患者に対して投与されている(Schilskyら、J Clin Oncol 4:1450−7,1998;Guoら、Cancer Chemother Pharmacol 52:79−85,2003)。ロイコボリンは経口製剤において都合よく利用することもできる。
【0086】
本発明は、以下の非限定的な実施例を考慮してさらに理解することができる。
【実施例】
【0087】
(実施例1)
(過剰のエニルウラシルがエニルウラシルおよび5−FUの抗腫瘍活性を減少させる)
ラットにWard癌腫瘍を移植し、それらの腫瘍が先に記載されたように重量で3,000mgまで成長させた(Caoら,Cancer Res 90:1507−1510、1993)後に、以下の投与計画の1つを用いて処置した。3,000mgの腫瘤を有するラットに、0日目、7日目、および14日目に以下の処置を施した。
【0088】
【表2】
A群のラットには処置を行わなかった。B群のラットには1mg/kgのエニルウラシル(時間(t)=0)を腹腔内(ip)投与し、次いで5 mg/kgの5−FUをt=60において静脈内(iv)投与した。C群のラットには1mg/kgのエニルウラシル(t=0)をip投与し、次いで25mg/kgのエニルウラシルをt=55においてip投与し、かつ5mg/kgの5−FUをt=60においてiv投与した。動物には1週あたり1回、3週間の間投与を行った。エニルウラシルも、各週の処置における2日目および3日目にB群およびC群において1 mg/kgでラットへとip投与した。C群に対する処置5−FUが投与された時に高レベルのエニルウラシルが存在する場合の臨床試験を模した。
【0089】
A群における腫瘍は迅速に10,000mgへと成長し、ラットを犠牲にした。C群における腫瘍は平均して大きさがわずかに減少したのみであった一方、B群における腫瘍は迅速に消失した。これらの結果は、C群における過剰なエニルウラシルが5−FUの抗腫瘍活性を大きく減少させることを示し、それはエニルウラシルが5−FUの代謝的活性化を妨げることに起因すると思われる。
【0090】
(実施例2)
(エニルウラシルは活性なヌクレオチドへの代謝的活性化を抑制する)
HEK293細胞をまずエニルウラシル(10μM)で1時間処理した。4〜48時間の洗浄期間後に、細胞を[6−14C]−5−FU(66μM)で2時間、37℃にて処理した。対照は[6−14C]−5−FU(66μM)のみで2時間処理した、またはプレインキュベーションを行うことなくエニルウラシル(10μM)および[6−14C]−5−FU(66μM)を共投与したHEK293細胞であった。細胞溶解物中に存在する[6−14C]−5−FU異化産物/同化産物を定量するために、放射活性検出を伴う逆相HPLCを利用した。異なるセットの実験において、異なるエニルウラシル投与スケジュールにおける5−FUの細胞毒性を調べた。HEK293細胞を、エニルウラシル(5μM)と1時間プレインキュベーション後か、エニルウラシルを伴わないプレインキュベーション後に、またはプレインキュベーションなしで共投与されるエニルウラシル(5μM)と共に、一定濃度範囲の5−FUを用いて72時間、37℃にて処理した。MTS増殖アッセイおよび算出される5−FUに対するEC50値により、細胞毒性を分析した。
【0091】
エニルウラシルを用いた1時間の細胞の前処理、それに次ぐ、[6−14C]−5−FU処理に先立つ36時間または48時間の洗浄は、[6−14C]−5−FU処理単独、またはエニルウラシルおよび[6−14C]−5−FUの共投与の場合と比較して、[6−14C]−FUMP同化産物の生成の顕著な増加を実現する(p−値<0.05)。例えば、48時間の洗浄除去は、[6−14C]−5−FU処理のみに対して[6−14C]−FUMPの生成における70%の増加、また、エニルウラシルおよび[6−14C]−5−FUの共投与に対して[6−14C]−FUMPの生成における41%の増加を実現した。細胞毒性試験において、エニルウラシルによる1時間のプレインキュベーションは、5−FU処理単独(311.4nM±1.1)またはEUおよび5−FUのプレインキュベーションなしの共投与(307.8nM±1.03)と比較して減少したEC50値(264.0nM±1.7)(平均±SE)を伴って、5−FUの細胞毒性を増加させた(p<0.05)。
【0092】
これらの結果はさらに、5−FUを不活性な形態へと分解する異化経路を阻害する公知の役割に加えて、5−FUを活性な抗腫瘍ヌクレオチドへと変換する同化経路を阻害する際の、エニルウラシルの役割を裏付ける。
【0093】
(実施例3)
(エニルウラシルは血漿ウリジンレベルを増加させる)
エニルウラシル阻害がDPD阻害によるウラシルレベルの上昇をもたらすことが知られている一方、フルオロピリミジンを同化するその他の酵素への阻害効果がウリジン等のその他の同化産物のレベルを量的に変化させることが期待されるであろう。エニルウラシルを2mg/kg、25mg/kgおよび100mg/kgでマウスに投与した。0分、15分、30分、60分、2時間、24時間および48時間時点で血漿サンプルを取得した。LC−MSにより公知の手法を用いてウリジンレベルを測定し、アッセイの妥当性を検証するために標準物質を使用した。
【0094】
図2に示す、これらの実験から得られる結果は、エニルウラシルが投与後のウリジンレベルの上昇を引き起こすことを示す。この知見はウリジンホスホリラーゼ等の同化酵素に対する阻害効果を有するエニルウラシルと一致し、5−FUの活性なヌクレオチドへの同化的変換阻害におけるエニルウラシルの役割をさらに支持する。
【0095】
本発明の特定の実施形態が説明のために本明細書中に記載されるが、本発明の本発明の精神および範囲から逸脱することなくなされる多様な改変がなされ得、したがって、本発明は添付の請求項によるもの以外の限定を受けないことが、前述の記載から理解されるであろう。
【技術分野】
【0001】
(発明の背景)
(発明の分野)
本発明は、全体として癌治療に関し、より詳しくは、5−FUおよび/または5−FUプロドラッグと併用してDPD阻害物質を用いる癌治療に関する。
【背景技術】
【0002】
(関連技術の説明)
5−フルオロウラシル(5−FU)は、30年以上、癌患者のうち固形癌を処置するために臨床において使用されている(非特許文献1;非特許文献2;非特許文献3)。5−FUは、DNA合成およびRNA機能を妨害する、擬似ウリジンヌクレオチド(例えば、FUMP、FUDP、FUTP)およびデオキシウリジンヌクレオチド(例えば、FdUMP、FdUDP、FdUTP)へと代謝的変換により活性化されなければならない(非特許文献4において概説されている)。5−FUとその元の形であるウラシルとの違いは5位におけるわずか1個のフッ素による置換であることから、癌患者において容易に活性化される。しかし、ウラシルとのその構造類似性が徒となり、抗腫瘍活性を全く有しない分解生成物へと迅速且つ広範に変換されてしまう主な原因ともなっている。この代謝過程は不活性化と呼ばれる。5−FUは、酵素ジヒドロピリミジンデヒドロゲナーゼ(DPD:EC 1312,ウラシル還元酵素)によって容易に不活性化される(非特許文献4;非特許文献5)。したがって、癌を処置するための5−FUの抗腫瘍効果は、抗腫瘍ヌクレオチドへの代謝的変換(活性化)と、無用な代謝産物への代謝的変換(不活性化)との間の微妙な均衡に左右される。
【0003】
さらに、5−FUの代謝的不活性化によるいくつかの臨床的問題が生じている。第1に、DPDのレベルは個人差があり(非特許文献6;非特許文献7)、1日の流れの中でも個人差がある(非特許文献7;非特許文献8;非特許文献9)ため、所定の投与量から得られる全身の5−FUレベルのばらつきは非常に大きく、したがって、有効性および毒性は非常に予測しづらいものになっている。極端な場合、DPDを遺伝子的に欠損している患者に対し、「標準的な」治療的用量の5−FUによる処置は、重篤且つ時には致死的な毒性を示す(非特許文献10において概説されている)。第2に、胃腸におけるDPDレベルにばらつきがあるため(非特許文献11;非特許文献12;非特許文献13)、経口投与された5−FUの吸収は非常にばらつきがある(非特許文献14;非特許文献15;非特許文献16)、したがって、薬物血漿中レベルを予見することができず、望ましくない毒性または不適当な有効性をもたらし得る。第3に、高レベルのDPDを含有する腫瘍は、5−FU処置に対して反応する可能性が低い(非特許文献17;非特許文献18)。最後に、5−FUの分解生成物は、神経毒性(非特許文献19;非特許文献20)、心臓毒性(非特許文献21;非特許文献22)、手掌足底発赤知覚不全(手足症候群)(非特許文献23)、およびGI毒性(非特許文献24)を生じる可能性があり、抗腫瘍活性を妨害すると思われる(非特許文献24;非特許文献25)。
【0004】
DPDは、5−FUの分解(不活性化)の第1段階および律速段階である、遍在する酵素である。DPDの阻害は血漿中の5−FUの半減期を延長することが研究から示されている。不可逆的にDPDを不活性化するもの、ならびに、可逆的にDPDを阻害するもの等、いくつかのDPD阻害物質について研究されている。
【0005】
5−エチニルウラシルは、エニルウラシルとも呼ばれ、5−FUの代謝的不活性化を低減または失わせる不可逆性のDPD不活性化物質であるDPD阻害物質の一つの例である(概説については、非特許文献26;非特許文献27を参照されたい)。エニルウラシルと5−FUとの構造が類似しているため、エニルウラシルはDPDに対する基質となる。DPDがエニルウラシルを分解しようとすると、エニルウラシルは、DPDと不可逆的に結合する高度反応性化合物へと変換され、それによりこの酵素を不活性化する。したがって、少しでもエニルウラシルが存在すれば、DPDは破壊され、それ以上5−FUを不活性化することはできない。このような処置を患者に施しても、数日もすれば、DPD酵素のde novo合成の結果、活性なDPDのみが再び現れる。
【0006】
エニルウラシルは癌患者におけるフェーズI臨床試験において調べられている(非特許文献28;非特許文献29;非特許文献30において概説されている)。エニルウラシルは、毒性を生じることなく、強力にDPD活性を失わせた。0.74mg/m2(合計約1mg)の用量で、長時間にわたり、全DPD活性の90%以上を失わせた。実際、1回のエニルウラシル投与から24時間後に、DPDのレベルは投与前のレベルのわずか3%であった。5−FUの排出半減期は、エニルウラシルの一回の投与により、約10分から3.5時間に増大した。3.7mg/m2の用量のエニルウラシルは、5−FUの半減期を4.5時間に増大した。用量をさらに多くしたが、目立った効果は得られなかった。
【0007】
エニルウラシルはさらに、フェーズIIおよびフェーズIII臨床試験において経口投与されている(非特許文献28;非特許文献31に概説されている)。これらの治験では2つの投与計画が使用された。「5−dayスケジュール」では、エニルウラシルが、1日目から7日目まで1日当たり50mgの固定用量で投与された。エニルウラシルの投与後、2日目から6日目まで、約20mg/m2の用量で5−FUが投与された。「28−dayスケジュール」では、エニルウラシルおよび5−FUが、エニルウラシル10に対して5−FUを1とする固定された比で、b.i.d.(1日当たり2回)、28日間、共投与した。5−FUの用量は約1mg/m2とした。エニルウラシルは、5−FUが関連する手足症候群毒性を無効にし、5−FUが安全に経口投与されることを可能にし、結果として、5−FU血漿中レベルが十分予見可能となった。しかしながら、これらの投与計画抗腫瘍活性は残念なことに期待はずれであった。結腸直腸癌のための28−dayエニルウラシル投与計画を用いた2つの多施設フェーズIII試験において、エニルウラシルおよび5−FUを投与される患者には、エニルウラシルを使用しない標準的な5−FU投与計画によって処置された患者よりも低い抗腫瘍活性を示す傾向があった(非特許文献31)。
【先行技術文献】
【非特許文献】
【0008】
【非特許文献1】Ansfieldら,Cancer 39:34−40,1977
【非特許文献2】Gremら,Cancer Treat Rep 71:1249−1264,1987
【非特許文献3】Chabnerら,Cancer,Principles and Practice of Oncology,2nd Ed,pp 287−328 Philadelphia,PA:J B Lippincott Co,1985
【非特許文献4】Meyers,Pharmacol Rev,33:1−15,1981
【非特許文献5】Dasherら,Pharmac Ther 48:189−222,1990
【非特許文献6】Flemingら,Cancer Res 52:2899−2902,1992
【非特許文献7】Gremら,Cancer Chemother Pharmacol 40:117−125,1997
【非特許文献8】Harrisら,Cancer Res 50:197−201,1990
【非特許文献9】Petitら,Cancer Res 48:1676−1679,1988
【非特許文献10】Morrisonら,Oncol Nurs Forum 24:83−88,1997
【非特許文献11】Hoら,Anticancer Res 6:781−784,1986
【非特許文献12】Naguibら,Cancer Res 45:5405−5412,1985
【非特許文献13】Spectorら,Biochem Pharmacol 46:2243−2248,1993
【非特許文献14】Christophidisら,Clin Pharmacokinetics 3:330−336,1978
【非特許文献15】Cohenら,Cancer Chemother Rep 58:723−731,1974
【非特許文献16】Finchら,Br J Clin Pharmacol 7:613−617,1979
【非特許文献17】Etienneら,J Clin Oncol 13:1663−1670,1995
【非特許文献18】Fischelら,Clin Cancer Res 1:991−996,1995
【非特許文献19】Okedaら,Acta Neuropathol 81:66−73,1990
【非特許文献20】Koenigら,Arch Neurol 23:155−160,1970
【非特許文献21】Lancet 337:560,1991
【非特許文献22】Lemaireら,Br J Cancer 66:119−127,1992
【非特許文献23】Hohneker,Oncology 12:52−56,1998
【非特許文献24】Spectorら,Cancer Res 55:1239−1241,1995
【非特許文献25】Caoら,Pharmacol 59:953−960,2000
【非特許文献26】Spectorら,Drugs of The Future 19:565−571,1994
【非特許文献27】Paffら,Invest.New Drugs:18,365−371(2000)
【非特許文献28】Levinら,Invest New Drugs 18:383−90,2000
【非特許文献29】Bakerら,J Clin Oncol 18:915−926 2000
【非特許文献30】Schilskyら,J Clin Oncol 4:1450−7,1998
【非特許文献31】Schilskyら,J Clin Oncol:20:1519−26,2002
【発明の概要】
【発明が解決しようとする課題】
【0009】
したがって、当該分野では、5−FUおよび5−FUプロドラッグの抗腫瘍効果および治療指数を最大にし、投与の予見可能性を向上し、経口投与により5−FUおよび5−FUプロドラッグを効果的に投与するために、5−FUおよび5−FUプロドラッグが組み合わせて使用されるDPD阻害物質の最適な投与または投薬スケジュールを特定する必要性が重要視され、未だそれを満たすものはない。本発明はこれらの必要性を満たし、その他の関連する利点を提供する。
【課題を解決するための手段】
【0010】
(発明の要旨)
本発明は全体として、5−FUおよび5−FUプロドラッグと組み合わせたDPD阻害物質、例えば、エニルウラシルを投与するための改良された方法に関する。したがって、本発明の一つの態様にしたがって、DPD酵素の活性を実質的に失わせるDPD阻害物質を、患者へと最初に投与し、その後に5−FUまたは5−FUプロドラッグを投与することを含む、患者における癌の処置方法であって、5−FUまたは5−FUプロドラッグは、その投与時に、5−FUまたは5−FUプロドラッグがDPD阻害物質を実質的に上回る量で患者中に存在するような用量で投与される、方法を提供する。
【0011】
本発明の本態様にしたがう一つの実施形態において、5−FUまたは5−FUプロドラッグは、DPD阻害物質が投与されてから、少なくとも約4時間後、少なくとも約6時間後、少なくとも約8時間後、少なくとも約10時間後、少なくとも約12時間後、少なくとも約14時間後、少なくとも約24時間後、または少なくとも約36時間後に投与される。
【0012】
本発明の本態様にしたがう別の実施形態において、5−FUまたは5−FUプロドラッグは、DPD阻害物質が投与されてから、約4〜72時間後、4〜36時間後、4〜24時間後、4〜14時間後、6〜14時間後または8〜14時間後に投与される。
【0013】
本発明の本態様にしたがう別の実施形態において、5−FUまたは5−FUプロドラッグは、DPD阻害物質の投与後、DPD阻害物質の排出半減期の少なくとも約0.1〜4倍、排出半減期の1〜4倍、排出半減期の2〜4倍または排出半減期の3〜4倍が経過した時点で投与される。
【0014】
本発明の本態様にしたがう別の実施形態において、DPD阻害物質は、患者におけるDPD活性を患者におけるベースラインのDPD活性の約10%未満、約5%未満、約3%未満または約1%未満に低減させるに十分な用量で投与される。
【0015】
本発明の本態様にしたがう別の実施形態において、5−FUまたは5−FUプロドラッグは、その投与時に5−FUまたは5−FUプロドラッグが患者中にDPD阻害物質の少なくとも約2倍、少なくとも約3倍、少なくとも約5倍または少なくとも約100倍過剰で存在するような用量で投与される。
【0016】
本発明の本態様にしたがう別の実施形態において、DPD阻害物質は不可逆性DPD阻害物質である。
【0017】
本発明の本態様にしたがう別の実施形態において、DPD阻害物質は可逆性DPD阻害物質である。かかる実施形態において、特定の好ましい可逆性DPD阻害物質としては、過剰な阻害物質が身体から排除されるよりもゆっくりとDPDから脱離し、したがって、5−FUまたは5−FUプロドラッグを投与する際に実質的に過剰で存在しない、強固に結合している阻害物質を挙げることができることが理解されるであろう。その他の好ましい阻害物質としては、DPD活性を阻害するが、ウリジンホスホリラーゼ(UPase)、オロチン酸ホスホリボシルトランスフェラーゼ(OPRTase)およびチミジンホスホリラーゼ(TP)等のフルオロウラシルを活性化するその他の酵素を実質的に阻害しないものを挙げることができる。
【0018】
本発明の本態様にしたがう別の実施形態において、5−FUまたは5−FUプロドラッグは、以下の化合物およびそれらのリン酸エステル等の5’−エステルからなる群より選択される:5−フルオロウリジン、5−フルオロシチジン、5−フルオロ−2−デオキシウリジン、5−フルオロ−2−デオキシシチジン、および5−フルオロアラビノシルウラシル。
【0019】
本発明の本態様にしたがう別の実施形態において、5−FUまたは5−FUプロドラッグは、5’−デオキシ−4’,5−フルオロウリジン、5’−デオキシ−5−フルオロウリジン、1−(2−テトラヒドロフラニル)−5−フルオロウラシル、1−C1−8アルキルカルバモイル−5−フルオロウラシル誘導体、1−(2−テトラヒドロフリル)−5−フルオロウラシル、5’−デオキシ−5−フルオロ−N−[(ペンチルオキシ)カルボニル]−シチジン(カペシタビン)、またはインビボで5−FUに変換される化合物からなる群より選択される。本発明の本態様にしたがう別の実施形態において、DPD阻害物質はエニルウラシル、またはそのプロドラッグである。
【0020】
本発明の本態様にしたがう別の実施形態において、DPD阻害物質はエニルウラシルであり、0.8〜15、2〜15、5〜15または2.5〜5mg/m2の用量で投与される。
【0021】
本発明の本態様にしたがう別の実施形態において、DPD阻害物質はエニルウラシルであり、5−FUまたは5−FUプロドラッグは5−FUである。
【0022】
本発明の本態様にしたがう別の実施形態において、5−FUまたは5−FUプロドラッグは5−FUであり、0.5〜80、0.5〜40、10〜80、10〜60、10〜30または20〜60mg/m2の用量で投与される。
【0023】
本発明の別の実施形態において、エニルウラシルと、5−FUまたは5−FUプロドラッグは、1:3〜1:20、1:5〜1:15、または1:8〜1:12の比の用量で投与される。
【0024】
一つの好ましい実施形態において、エニルウラシルは、約15〜30mg/m2の用量での5−FUまたは5−FUプロドラッグの投与、または約5〜100mg/m2の用量でのカペシタビンの投与の少なくとも約10〜14時間前に、最初に約2.5〜5mg/m2の用量で投与される。場合により、さらなる用量の5−FUまたはカペシタビンを、エニルウラシルのさらなる投与を伴いまたは伴わずに、その後に投与することができる。
【0025】
本発明の本態様にしたがう別の実施形態において、DPD阻害物質はエニルウラシルであり、5−FUまたは5−FUプロドラッグはカペシタビンである。
【0026】
本発明の本態様にしたがう別の実施形態において、DPD阻害物質はエニルウラシルであり、5−FUまたは5−FUプロドラッグは5−FUまたはカペシタビンであり、エニルウラシルは約0.8〜15、2.5〜15、5〜15または2.5〜5mg/m2の用量で投与される。
【0027】
本発明の本態様にしたがう別の実施形態において、DPD阻害物質はエニルウラシルであり、5−FUまたは5−FUプロドラッグは5−FUであり、使用される投与スケジュールに応じて、エニルウラシルは約2.5〜5mg/m2の用量で投与され、5−FUは約0.5〜40mg/m2の用量で投与される。
【0028】
一つの例示的スケジュール、例えば、28日間(28−day)、1日当たり2回(b.i.d.)のスケジュールは、約0.5〜1.5mg/m2の用量の5−FUを使用する。別の例示的スケジュール、例えば、5日間(5−day)、1日当たり1回のスケジュールは、約10〜60mg/m2の用量の5−FUを使用する。より特定の実施形態において、5−FUの用量は約10〜30mg/m2である。別の実施形態において、5−FUの用量は約20〜60mg/m2である。
【0029】
別の例示的スケジュール、例えば、1週間当たり(weekly)1回のスケジュールは、約10〜80mg/m2の用量の5−FUを使用し得る。より特定の実施形態において、5−FUを約15〜40または10〜30mg/m2の用量で投与する。別の実施形態において、5−FUを約30〜80mg/m2の用量で投与する。
【0030】
本発明の本態様にしたがう別の実施形態において、DPD阻害物質はエニルウラシルであり、5−FUまたは5−FUプロドラッグはカペシタビンであり、エニルウラシルは約0.8〜15、約2〜15、約5〜15または約2.5〜5mg/m2の用量で投与され、カペシタビンは約0.8〜200mg/m2の用量で投与される。より特定の実施形態において、カペシタビンは、(例えば、特定の例示的な長期にわたる1日2回の日々の計画のために)約0.8〜10mg/m2または1.3〜4mg/m2の用量で投与される。
【0031】
本発明の本態様にしたがう別の実施形態において、DPD阻害物質は5位が置換されたウラシル類似体またはそのプロドラッグを含む。
【0032】
本発明の本態様にしたがう別の実施形態において、DPD阻害物質は、ハロゲン原子、C2−4アルケニル基、ハロゲンによって置換されたC2−4アルケニル基、C2−6アルキニル基、ハロゲンによって置換されたC2−6アルキニル基、シアノ基またはハロゲンによって置換されたC1−4アルキル基によって5位が置換されたウラシル類似体を含む。
【0033】
本発明の本態様にしたがう別の実施形態において、DPD阻害物質は、エニルウラシル、5−プロピニルウラシル、5−シアノウラシル、5−プロピニルウラシル、5−ブロモエチニルウラシル、5−(1−クロロビニル)ウラシル、5−ヨードウラシル、5−ブロモビニルウラシル、(E)−5−(2−ブロモビニル)ウラシル、5−ヘキサ−1−イニルウラシル、5−ビニルウラシル、5−トリフルオロウラシル、5−ブロモウラシルおよび5−(2−ブロモ−1−クロロビニル)ウラシルからなる群より選択されるウラシル類似体を含む。
【0034】
本発明の本態様にしたがう別の実施形態において、DPD阻害物質は、5−(フェニルセレネニル)ウラシル(PSU)、5−(フェニルチオ)ウラシル(PTU)、5−(フェニルセレネニル)バルビツール酸および5−(フェニルチオ)バルビツール酸からなる群より選択される。
【0035】
本発明の別の態様にしたがって、エニルウラシルを最初に投与し、その後に5−FUを投与することを含む、患者における癌の処置方法であって、5−FUは、その投与時に5−FUが患者におけるDPD阻害物質を実質的に上回る量で存在するような用量で投与される、方法を提供する。
【0036】
本発明の本態様にしたがう一つの実施形態において、5−FUは、エニルウラシルが投与されてから、少なくとも約4時間後、6時間後、8時間後、10時間後、12時間後、14時間後、24時間後または36時間後に投与される。一つの好ましい実施形態において、5−FUは、エニルウラシルが投与されてから、少なくとも約4時間後、少なくとも約8時間後または少なくとも約12時間後に投与される。
【0037】
本発明の本態様にしたがう別の実施形態において、5−FUは、エニルウラシルが投与されてから約4〜72時間後、4〜36時間後、4〜24時間後、4〜14時間後、6〜14時間後または8〜14時間後に投与される。一つの好ましい実施形態において、5−FUは、エニルウラシルが投与されてから約4〜14時間後に投与される。
【0038】
本発明の別の実施形態において、5−FUは、エニルウラシルの投与後、エニルウラシルの排出半減期の約0.1〜4倍、排出半減期の1〜4倍、排出半減期の2〜4倍または排出半減期の3〜4倍が経過した時点で投与される。
【0039】
本発明の本態様にしたがう別の実施形態において、エニルウラシルは、患者におけるDPD活性をベースラインのDPD活性の約10%未満、約5%未満、約3%未満または約1%未満に低減させるに十分な用量で投与する。
【0040】
本発明の本態様にしたがう別の実施形態において、5−FUは、その投与時に5−FUが患者中にエニルウラシルの少なくとも約2倍、少なくとも約3倍、少なくとも約5倍または少なくとも約100倍過剰で存在するような用量で投与される。
【0041】
本発明の本態様にしたがう別の実施形態において、エニルウラシルは約0.7〜15mg/m2の用量で投与される。本発明の別の実施形態において、エニルウラシルは約2.5〜5mg/m2の用量で投与される。本発明の別の実施形態において、エニルウラシルは約5〜15mg/m2の用量で投与される。
【0042】
本発明の本態様にしたがう別の実施形態において、エニルウラシルは約2.5〜5mg/m2の用量で投与され、5−FUは約0.5〜40mg/m2の用量で投与される。
【0043】
本発明の別の態様にしたがって、エニルウラシルを最初に投与し、その後に5−FUを投与することを含む、患者における癌の処置方法であって、エニルウラシルは約2.5〜5mg/m2の用量で投与され、5−FUは、エニルウラシルの投与後、エニルウラシルの排出半減期の約1〜4倍、2〜4倍または3〜4倍が経過した時点で投与される、方法を提供する。
【0044】
本発明の本態様にしたがう一つの実施形態において、5−FUは、エニルウラシルが投与されてから、少なくとも約4時間後、6時間後、8時間後、10時間後、12時間後または14時間後に投与される。
【0045】
本発明の本態様にしたがう一つの実施形態において、5−FUは、エニルウラシルが投与されてから、約4〜36時間後、4〜24時間後、4〜14時間後、6〜14時間後または8〜14時間後に投与される。
【0046】
本発明の本態様にしたがう別の実施形態において、5−FUは、その投与時に5−FUが患者中にエニルウラシルの少なくとも約2倍、少なくとも約3倍、少なくとも約5倍または少なくとも約100倍過剰で存在するような用量で投与される。
【0047】
本発明の本態様にしたがう別の実施形態において、5−FUは約0.5〜40mg/m2の用量で投与される。
【0048】
より特定の実施形態において、エニルウラシル(または別のDPD阻害物質)を最初に投与し、その後に、エニルウラシル(または別のDPD阻害物質)を場合により再度投与する前の望ましい時点で、5−FUまたは5−FUプロドラッグを複数回投与する。例えば、一つの好ましい実施形態において、エニルウラシルは最初、5−FUまたは5−FUプロドラッグの投与の少なくとも約10〜14時間前に、例えば、5−FUを約15〜30mg/m2の用量で投与されまたはカペシタビンは約5〜100mg/m2の用量で投与されるいずれかの初日の前の晩に、約2.5〜5mg/m2の用量で投与され、その後、同様の複数用量の5−FUまたはカペシタビンは投与される。例えば、一つの例示的実施形態において、エニルウラシルを最初に投与し、その後に、例えば、エニルウラシルを場合により再び投与する前に、5−FUまたはカペシタビンは、1週間に付き3日間は毎日複数回投与され、そして、このサイクルを繰り返す。
【0049】
本発明の別の態様にしたがって、最初にエニルウラシルを投与し、その後5−FUを投与することを含む、患者における癌の処置方法であって、エニルウラシルは約2.5〜5mg/m2の用量で投与され、5−FUは、エニルウラシルの投与後、エニルウラシルの排出半減期の1〜4倍、2〜4倍または3〜4倍が経過した時点で投与され、5−FUは約0.5〜40mg/m2の用量で投与される、方法を提供する。
【0050】
本発明の別の態様にしたがって、DPD阻害物質および5−FUまたは5−FUプロドラッグを含む医薬徐放製剤であって、患者へ製剤を投与した後、DPD阻害物質が放出されてから約0.5〜36時間後、4〜36時間後、4〜24時間後または4〜14時間後まで、5−FUまたは5−FUプロドラッグが実質的に放出されない、前記製剤を提供する。
【0051】
本発明の別の態様にしたがって、患者にDPD阻害物質を投与するためにDPD阻害物質および送達ビヒクルを含む製剤処方が提供される。別の実施形態において、製剤は、DPD阻害物質および5−FUまたは5−FUプロドラッグを含む。別の実施形態において、製剤は、送達ビヒクル、DPD阻害物質および5−FUまたは5−FUプロドラッグを含む。特定の実施形態において、送達ビヒクルはミクロスフィアである。関連する実施形態において、送達ビヒクルは、当技術分野において知られている製剤技術および送達技術を用いて癌細胞へのDPD阻害物質の優先的または選択的ターゲティングを可能にするミクロスフィアである。
【0052】
本発明のこれらおよびその他の態様は、以下の詳細な説明および添付の図面を参照した際に明らかになるであろう。本発明の各種態様をより具体的に示すために本明細書中で引用された特許およびその他の文献は、それらの全体を参照により本明細書に組み入れるものとする。
例えば、本願発明は以下の項目を提供する。
(項目1)
最初にDPD阻害物質を投与し、その後に5−FUまたは5−FUプロドラッグを投与することを含む、患者における癌の処置方法であって、該5−FUまたは5−FUプロドラッグは、その投与時に、該5−FUまたは5−FUプロドラッグが該DPD阻害物質を上回る量で該患者中に存在するような用量で投与される、方法。
(項目2)
前記5−FUまたは5−FUプロドラッグは、前記DPD阻害物質が投与されてから少なくとも約4時間後に投与される、項目1に記載の方法。
(項目3)
前記5−FUまたは5−FUプロドラッグは、前記DPD阻害物質が投与されてから少なくとも約12時間後に投与される、項目1に記載の方法。
(項目4)
前記5−FUまたは5−FUプロドラッグは、前記DPD阻害物質が投与されてから少なくとも約24時間後に投与される、項目1に記載の方法。
(項目5)
前記5−FUまたは5−FUプロドラッグは、前記DPD阻害物質の投与後、前記DPD阻害物質の排出半減期の少なくとも1〜4倍が経過した時点で投与される、項目1に記載の方法。
(項目6)
前記DPD阻害物質は、前記患者におけるDPD活性を該患者におけるベースラインのDPD活性の5%未満に低減させるに十分な用量で投与される、項目1に記載の方法。
(項目7)
前記5−FUまたは5−FUプロドラッグは、その投与時に該5−FUまたは5−FUプロドラッグが前記患者中に前記DPD阻害物質の少なくとも2倍過剰で存在するような用量で投与される、項目1に記載の方法。
(項目8)
前記5−FUまたは5−FUプロドラッグは、その投与時に該5−FUまたは5−FUプロドラッグが前記患者中に前記DPD阻害物質の少なくとも5倍過剰で存在するような用量で投与される、項目1に記載の方法。
(項目9)
前記DPD阻害物質が不可逆性DPD阻害物質である、項目1に記載の方法。
(項目10)
前記DPD阻害物質が可逆性DPD阻害物質である、項目1に記載の方法。
(項目11)
前記5−FUまたは5−FUプロドラッグが、5−フルオロウリジン、5−フルオロシチジン、5−フルオロ−2−デオキシウリジン、5−フルオロ−2−デオキシシチジン、5’−デオキシ−4’,5−フルオロウリジン、および5−フルオロアラビノシルウラシル、5’−デオキシ−5−フルオロウリジン、1−(2−テトラヒドロフラニル)−5−フルオロウラシル、1−C1−8アルキルカルバモイル−5−フルオロウラシル誘導体、1−(2−テトラヒドロフリル)−5−フルオロウラシル、5’−デオキシ−5−フルオロ−N−[(ペンチルオキシ)カルボニル]−シチジン(カペシタビン)、またはインビボで5−FUに変換される化合物からなる群およびそれらのリン酸エステルを含めた5’−エステルから選択される、項目1に記載の方法。
(項目12)
前記5−FUまたは5−FUプロドラッグが5−FUまたはカペシタビンである、項目1に記載の方法。
(項目13)
前記DPD阻害物質がエニルウラシルまたはそのプロドラッグである、項目1に記載の方法。
(項目14)
前記DPD阻害物質がエニルウラシルであり、前記5−FUまたは5−FUプロドラッグが5−FUまたはカペシタビンである、項目1に記載の方法。
(項目15)
前記DPD阻害物質がエニルウラシルであり、前記5−FUまたは5−FUプロドラッグが5−FUであり、前記エニルウラシルは約0.8〜15mg/m2の用量で投与される、項目1に記載の方法。
(項目16)
前記DPD阻害物質がエニルウラシルであり、前記5−FUまたは5−FUプロドラッグが5−FUであり、前記エニルウラシルは約2.5〜5mg/m2の用量で投与される、項目1に記載の方法。
(項目17)
前記DPD阻害物質がエニルウラシルであり、前記5−FUまたは5−FUプロドラッグが5−FUであり、前記エニルウラシルは約5〜15mg/m2の用量で投与される、項目1に記載の方法。
(項目18)
前記DPD阻害物質がエニルウラシルであり、前記5−FUまたは5−FUプロドラッグが5−FUであり、前記エニルウラシルは約2.5〜15mg/m2の用量で投与され、前記5−FUは約0.5〜80mg/m2の用量で投与される、項目1に記載の方法。
(項目19)
前記DPD阻害物質がエニルウラシルであり、前記5−FUまたは5−FUプロドラッグが5−FUであり、前記エニルウラシルは約2.5〜5mg/m2の用量で投与され、前記5−FUは約0.5〜40mg/m2の用量で投与される、項目1に記載の方法。
(項目20)
前記DPD阻害物質が5−置換ウラシル化合物またはそのプロドラッグを含む、項目1に記載の方法。
(項目21)
前記DPD阻害物質が、ハロゲン原子、C2−4アルケニル基、ハロゲンによって置換されたC2−4アルケニル基、C2−6アルキニル基、ハロゲンによって置換されたC2−6アルキニル基、シアノ基、C1−4アルキル基またはハロゲンによって置換されたC1−4アルキル基によって5位が置換されたウラシル化合物を含む、項目1に記載の方法。
(項目22)
前記DPD阻害物質が、エニルウラシル、5−プロピニルウラシル、5−シアノウラシル、5−プロピニルウラシル、5−ブロモエチニルウラシル、5−(1−クロロビニル)ウラシル、5−ヨードウラシル、5−ブロモビニルウラシル、(E)−5−(2−ブロモビニル)ウラシル、5−ヘキサ−1−イニルウラシル、5−ビニルウラシル、5−トリフルオロウラシル、5−ブロモウラシルおよび5−(2−ブロモ−1−クロロビニル)ウラシルからなる群より選択されるウラシル化合物を含む、項目1に記載の方法。
(項目23)
前記DPD阻害物質が、5−(フェニルセレネニル)ウラシル(PSU)、5−(フェニルチオ)ウラシル(PTU)、5−(フェニルセレネニル)バルビツール酸および5−(フェニルチオ)バルビツール酸からなる群より選択される、項目1に記載の方法。
(項目24)
最初にエニルウラシルを投与し、その後に5−FUを投与することを含む、患者における癌の処置方法であって、該5−FUは、その投与時に該5−FUが該DPD阻害物質を上回る量で該患者中に存在するような用量で投与される、方法。
(項目25)
前記5−FUは、前記エニルウラシルが投与されてから少なくとも約4時間後に投与される、項目24に記載の方法。
(項目26)
前記5−FUは、前記エニルウラシルが投与されてから少なくとも約12時間後に投与される、項目24に記載の方法。
(項目27)
前記5−FUは、前記エニルウラシルの投与後、エニルウラシルの排出半減期の少なくとも約1〜4倍が経過した時点で投与される、項目24に記載の方法。
(項目28)
前記エニルウラシルは、前記患者におけるDPD活性を前記患者におけるベースラインのDPD活性の5%未満に低減させるに十分な用量で投与される、項目24に記載の方法。
(項目29)
前記5−FUは、その投与時に前記5−FUが前記患者中に前記DPD阻害物質の少なくとも2倍過剰で存在するような用量で投与される、項目24に記載の方法。
(項目30)
前記5−FUは、その投与時に前記5−FUが前記患者中に前記DPD阻害物質の少なくとも5倍過剰で存在するような用量で投与される、項目24に記載の方法。
(項目31)
前記エニルウラシルは約2.5〜5mg/m2の用量で投与される、項目24に記載の方法。
(項目32)
前記エニルウラシルは約2.5〜15mg/m2の用量で投与され、前記5−FUは約0.5〜40mg/m2の用量で投与される、項目24に記載の方法。
(項目33)
最初にエニルウラシルを投与し、その後に5−FUを投与することを含む、患者における癌の処置方法であって、前記エニルウラシルは約2.5〜15mg/m2の用量で投与され、前記5−FUは、前記エニルウラシルの投与後、エニルウラシルの排出半減期の少なくとも1〜4倍が経過した時点で投与される、方法。
(項目34)
前記5−FUは、前記エニルウラシルが投与されてから少なくとも約4時間後に投与される、項目33に記載の方法。
(項目35)
前記5−FUは、前記エニルウラシルが投与されてから少なくとも約14時間後に投与される、項目33に記載の方法。
(項目36)
前記5−FUは、前記エニルウラシルが投与されてから少なくとも約18時間後に投与される、項目33に記載の方法。
(項目37)
前記5−FUは、その投与時に前記5−FUが前記患者中に前記DPD阻害物質の少なくとも2倍過剰で存在するような用量で投与される、項目33に記載の方法。
(項目38)
前記5−FUは、その投与時に前記5−FUが前記患者中に前記DPD阻害物質の少なくとも5倍過剰で存在するような用量で投与される、項目33に記載の方法。
(項目39)
前記5−FUは約0.5〜40mg/m2の用量で投与される、項目33に記載の方法。
(項目40)
最初にエニルウラシルを投与し、その後に5−FUを投与することを含む、患者における癌の処置方法であって、前記エニルウラシルは約2.5〜15mg/m2の用量で投与され、前記5−FUは、前記エニルウラシルを投与してから少なくとも約4時間後に投与され、前記5−FUは約0.5〜40mg/m2の用量で投与される、方法。
(項目41)
前記5−FUは、前記エニルウラシルが投与されてから少なくとも約14時間後に投与される、項目40に記載の方法。
(項目42)
前記5−FUは、前記エニルウラシルが投与されてから少なくとも約18時間後に投与される、項目40に記載の方法。
(項目43)
最初にエニルウラシルを投与し、その後にカペシタビンを投与することを含む、患者における癌の処置方法であって、該エニルウラシルは約2.5〜15mg/m2の用量で投与され、該5−FUは、該エニルウラシルの投与後、エニルウラシルの排出半減期の少なくとも1〜4倍が経過した時点で投与される、方法。
(項目44)
前記カペシタビンは、前記エニルウラシルが投与されてから少なくとも約4時間後に投与される、項目43に記載の方法。
(項目45)
前記カペシタビンは、前記エニルウラシルが投与されてから少なくとも約14時間後に投与される、項目43に記載の方法。
(項目46)
前記カペシタビンは、前記エニルウラシルが投与されてから少なくとも約24時間後に投与される、項目43に記載の方法。
(項目47)
前記カペシタビンは、その投与時に該カペシタビンが前記患者中に前記DPD阻害物質の少なくとも2倍過剰で存在するような用量で投与される、項目43に記載の方法。
(項目48)
前記カペシタビンは、その投与時に前記カペシタビンが前記患者中に前記DPD阻害物質の少なくとも5倍過剰で存在するような用量で投与される、項目43に記載の方法。
(項目49)
前記カペシタビンは約1.5〜4mg/m2の用量で投与される、項目43に記載の方法。
(項目50)
DPD阻害物質および5−FUまたは5−FUプロドラッグを含む経口医薬徐放製剤であって、前記製剤を患者へ投与した後に、該DPD阻害物質が放出されてから少なくとも約4時間後までの間は該5−FUまたは5−FUプロドラッグが実質的に放出されない、前記製剤。
(項目51)
前記DPD阻害物質が放出されてから少なくとも約12時間後までの間は前記5−FUまたは5−FUプロドラッグが実質的に放出されない、項目50に記載の製剤。
(項目52)
前記5−FUまたは5−FUプロドラッグが5−FUまたはカペシタビンである、項目50に記載の製剤。
(項目53)
前記DPD阻害物質がエニルウラシルである、項目50に記載の製剤。
(項目54)
DPD阻害物質および送達ビヒクルを含む経口医薬徐放製剤であって、該送達ビヒクルが、患者の癌細胞に対する前記DPD阻害物質の選択的標的化を容易にする、前記製剤。
(項目55)
前記送達ビヒクルがミクロスフィアである、項目54に記載の製剤。
(項目56)
最初にエニルウラシルを投与し、その後に5−FUを投与することを含む、患者における癌の処置方法であって、エニルウラシルは5−FUの投与の少なくとも約0.5時間前に投与され、該5−FUまたは5−FUプロドラッグは、その投与時に該5−FUまたは5−FUプロドラッグが該DPD阻害物質を上回る量で該患者中に存在するような用量で投与される、方法。
(項目57)
最初にエニルウラシルを投与し、その後に5−FUを投与することを含む、患者における癌の処置方法であって、1日あたり2回の長期投与スケジュール開始の少なくとも約12時間前に約2.5〜15mg/m2の用量でエニルウラシルは投与され、その後、各日における1日2回の最初の5−FU投与の前の約4〜6時間の間の時点で、約0.5〜2mg/m2の用量で該エニルウラシルは投与される、方法。
(項目58)
前記DPD阻害物質の用量と、前記5−FUまたは5−FUプロドラッグの用量とが、1:3〜1:20の比にある、項目1に記載の方法。
(項目59)
前記DPD阻害物質の用量と、前記5−FUまたは5−FUプロドラッグの用量とが、1:5〜1:15の比にある、項目1に記載の方法。
(項目60)
前記DPD阻害物質の用量と、前記5−FUまたは5−FUプロドラッグの用量とが、1:8〜1:12の比にある、項目1に記載の方法。
【図面の簡単な説明】
【0053】
【図1】図1は、エニルウラシルおよび5−FUの化学構造を示す。
【図2】図2は、エニルウラシルがマウスにおける投与後に血漿中のウリジンレベル増大を引き起こすことを示す。
【図3】図3は、エニルウラシルおよび5−FUを含む錠剤形態の経口徐放製剤を例示する。
【発明を実施するための形態】
【0054】
(発明の詳細な説明)
上述の通り、本発明は全体として、少なくとも1つの5−FUまたは5−FUプロドラッグと組み合わせた少なくとも1つのDPD阻害物質を患者へと投与することを含む癌の処置方法に関し、また、そのような方法において有用な組成物および製剤に関する。本明細書中に記載の方法はすなわち、実例として、限定されるものではないが、乳癌、肺癌、結腸癌、膵臓癌、胃癌、膀胱癌、腎癌、頭頸部癌、食道癌、肝細胞癌、および全ての悪性白血病およびリンパ腫を含む、そこにおいて5−FUおよび/または5−FUプロドラッグが活性を有する本質的に任意のタイプの癌に対し適用可能である。さらに、本発明は5−FUおよび5−FUプロドラッグの抗腫瘍効果を向上させるため、従来5−FUに対する所望の応答性を示すとはいえなかったかもしれない癌のタイプが、本明細書中に記載の方法に従って投与される場合には、改善された応答性を示す可能性がある。
【0055】
エニルウラシル等のDPD阻害物質が5−FUおよび5−FUプロドラッグの代謝的活性化を阻害し得、それによりそれらの抗腫瘍活性を落とすことが、思いがけなく発見された。したがって、本発明の一般的な態様に従い、5−FUまたは5−FUプロドラッグが患者へと投与される時点で、5−FUまたは5−FUプロドラッグレベルがDPD阻害物質レベルを十分に上回るようにすることにより、それに対しDPD阻害物質が5−FUまたは5−FUプロドラッグの代謝的活性化を妨害し得る度合は都合よく最小化され、またこれらの薬物の抗腫瘍効果はそれにより向上する。したがって、特定の実施形態において、エニルウラシル等の不可逆性なDPD阻害物質はDPDを効果的に不活化する最低用量で投与される必要があり、また、好ましくは、5−FUを投与する前に血流中から過剰な阻害物質を除去するために十分な時間が経過する必要がある。さらに、DPDは5−FUまたは5−FUプロドラッグの投与前に患者から実質的に除去することができるため、所望のレベルの治療的活性を達成するために必要とされる5−FUまたは5−FUプロドラッグの用量を顕著に低減することができ、それにより本明細書中に記載の治療的な優位性に加えて経済的な優位性を提供できる。
【0056】
この開示に照らして、5−FUまたは5−FUプロドラッグのレベルが投与時に治療上効果的な量であり、かつ、5−FU代謝活性化の阻害を最小化するまたは無くすために患者中のDPD阻害物質レベルを十分に上回るようにさせながら本明細書中に記載の方法に使用することができる、多数の投与および服薬スケジュールが存在することが当業者の一部には理解されるであろう。このような全ての投与および服薬スケジュールが本発明の範囲内であると考えられる。
【0057】
本発明の1つの実例となる実施形態において、DPD阻害物質をまず、その患者中のDPD活性を実質的になくすために、その必要性を有する患者へと投与(すなわち、前投与)し、次いで5−FUまたは5−FUプロドラッグを投与する。「実質的になくす」とは、患者中のDPD活性のレベルが、DPD阻害物質投与前の患者におけるDPD活性のベースラインに対して少なくとも20%未満、10%未満、5%未満、3%未満または1%未満であることを意味する。患者のDPD活性のベースラインレベルは、例えば、患者から得たPBMC中で、公知の手法(例えば、Bakerら、J Clin Oncol 18:915−926 2000;Schilskyら、J Clin Oncol 4:1450−7,1998)を用いて、迅速に調べることができる。
【0058】
少なくとも1つのDPD阻害物質を最初に投与し、それにより実質的にその患者中のDPDの活性を失わせた後、次いで、DPD阻害物質が患者から排除され実質的になくなるのに十分な時間が経過した後に、5−FUまたは5−FUプロドラッグまたはそれらの組み合わせを、患者へと投与する。DPD阻害物質の投与と、5−FUまたは5−FUプロドラッグの投与との間の時間的な遅れは、5−FUまたは5−FUプロドラッグの投与条件により変化し得、それはその時点で患者中の残存するDPD阻害物質のレベルに対して実質的に過剰な量で患者中に存在する。1つの実例となる実施形態において、5−FUまたは5−FUプロドラッグを、その患者中に存在する5−FUまたは5−FUプロドラッグのレベルが少なくともその患者中に残存するDPD阻害物質よりも過剰に、例えば、5−FUまたは5−FUプロドラッグの投与時に、その患者中に残存するDPD阻害物質のレベルの少なくとも約2倍、少なくとも約3倍、少なくとも約5倍または少なくとも約100倍過剰で存在するような用量で投与する。当業者であれば、本明細書中に記載の実施形態に従って、DPD阻害物質に対する5−FUまたは5−FUプロドラッグの患者における過剰さの程度を計算および/または決定するために、多数の公知かつ適用可能な手法のうち任意のものを使用することができるであろう。このような例には、例えば、HPLC、LC−MS、ELISA等が挙げられる。上述の通り、5−FUまたは5−FUプロドラッグの投与時に、5−FUまたは5−FUプロドラッグが患者中のDPD阻害物質に対し十分に過剰なレベルで存在することを確実にすることにより、DPD阻害物質による5−FUまたは5−FUプロドラッグの代謝的活性化の妨害がそれにより最小化され、5−FUまたは5−FUプロドラッグの有効性がそれにより向上すると考えられる。
【0059】
本発明の別の実施形態において、5−FUまたは5−FUプロドラッグを、DPD阻害物質の投与後、DPD阻害物質の排出半減期の少なくとも0.1〜4倍、排出半減期の1〜4倍、排出半減期の2〜4倍、または排出半減期の3〜4倍が経過した時点で患者へと投与する。特定のDPD阻害物質に関する排出半減期が調べられており、調べられていないものについては、排出半減期は、よく知られ確立されたガスクロマトグラフィー/質量分析およびHPLC手法を用いて迅速に決定することができる(Bakerら、J Clin Oncol 18:915−926 2000;Schilskyら、J Clin Oncol 4:1450−7,1998参照)。ヒトにおけるエニルウラシルの排出半減期は約3.5時間であると報告されている(例えば、Bakerら、J Clin
Oncol 18:915−926 2000;Ochoaら、Ann Oncol 11:1313−22,2000)が、エニルウラシルおよびその他のDPD阻害物質の半減期は用量依存的であり、この用量依存性はDPD阻害物質、および5−FUまたは5−FUプロドラッグの間の適切な時間遅延を調べる際に考慮される必要がある。したがって、エニルウラシルをDPD阻害物質として利用する本発明のある種の実施形態については、エニルウラシルのレベルを5−FUまたは5−FUプロドラッグ投与前の除去によって十分に低減できるようにするために、5−FUまたは5−FUプロドラッグは、エニルウラシルの投与後、少なくとも約0.5時間、約2時間、約4時間、約6時間、約8時間、約10時間、約12時間、約14時間、約24時間または約36時間後に投与される。特定のその他の実施形態において、5−FUまたは5−FUプロドラッグは、エニルウラシル投与後約4〜72時間、4〜36時間、約4〜24時間、約4〜14時間、約6〜14時間または約8〜14時間後に投与される。もちろん、これらの範囲は事実上説明的なものであり、特定の投与計画が必要とされまたは所望される時には、5−FU投与時にエニルウラシルの存在が最小化されるかまたは存在しない状態となる限りにおいて変更し得る。
【0060】
本発明に従い使用されるDPD阻害物質は、可逆性または不可逆性のDPD酵素の阻害物質であり得る。DPD酵素の可逆性阻害物質の説明的な実例には、ウラシル、CDHPおよび3−シアノ−2,6−ジヒドロキリピリジン(CNDP)を含む。DPD酵素の可逆性阻害物質におけるその他の説明的な実例には、例えば、5−(フェニルセレネニル)ウラシル(PSU)、5−(フェニルチオ)ウラシル(PTU)、5−(フェニルセレネニル)バルビツール酸および5−(フェニルチオ)バルビツール酸等、米国特許第5,476,855号および国際公開第95/012400号中に記載のものを含み、それらの内容を、その全体を参照により本明細書に組み入れるものとする。特定の好ましい可逆性DPD阻害物質には、過剰な阻害物質が身体から排除されるよりもゆっくりとDPDから脱離する強固に結合した阻害物質、および/または、DPD活性を阻害するが、ウリジンホスホリラーゼ(UPase)、オロチン酸ホスホリボシルトランスフェラーゼ(OPRTase)およびチミジンホスホリラーゼ(TP)等のフルオロウラシルを活性化するその他の酵素を実質的に阻害しない阻害物質を含むことが理解されるであろう。
【0061】
本発明の特定の好ましい実施形態において、DPD阻害物質は不可逆的にDPD酵素を不活化するものである。これに関する説明的なDPD阻害物質としては、限定されるものではないが、5−置換ウラシル化合物、またはそのプロドラッグを含むDPD阻害物質、特に5位がハロゲン原子、所望によりハロゲンで置換された(例えば、2−ブロモビニル、1−クロロビニルまたは2−ブロモ−1−クロロビニル)C2−4 アルケニル基(例えば、ビニル)、所望によりハロゲン原子で置換されたC2−6 アルキニル基、シアノ基、またはハロゲンで置換されたC1−4 アルキル基(例えば、トリフルオロメチル)、で置換されたウラシル化合物を含むDPD阻害物質が挙げられる。
【0062】
本発明のより特定的な実施形態において、DPD阻害物質は、エニルウラシル、5−プロピニルウラシル、5−シアノウラシル、5−プロピニルウラシル、5−ブロモエチニルウラシル、5−(1−クロロビニル)ウラシル、5−ヨードウラシル、5−ブロモビニルウラシル、(E)−5−(2−ブロモビニル)ウラシル、5−ヘキサ−1−イニルウラシル、5−ビニルウラシル、5−トリフルオロウラシル、5−ブロモウラシル、および5−(2−ブロモ−l−クロロビニル)ウラシル、またはそれらのプロドラッグからなる群から選択される。
【0063】
別の実施形態において、DPD阻害物質は5−ブロモビニルウラシルのプロドラッグであり、説明的な1化合物は、化合物1−β−D−アラビノフラノシル−(E)−5−(2−ブロモビニル)ウラシル(BV−araUまたはソリブジンとも称する)により代表される。これに関する特定の説明的なプロドラッグ化合物としては、例えば、米国特許第4,386,076号が挙げられ、この開示を参照により本明細書に組み入れるものとする。
【0064】
本発明の好ましい1実施形態において、5−エチニル−2(1H)−ピリミジノン(4−酸素を失ったエニルウラシル)(Porterら、Biochem.Pharmacol 47:1165− 1171,1994)、エニルウラシルのヌクレオシドまたはデオキシヌクレオチドシド誘導体、インビボでエニルウラシルへと変換される化合物および/またはインビボで不活化剤へと変換されるDPD不活化剤誘導体等の、DPD阻害物質はエニルウラシルまたはエニルウラシルのプロドラッグである。一例として、このような化合物には、上記の5−置換ウラシル化合物に相当する核酸塩基、例えば、リボース、2’−デオキシリボース、2’,3’−ジデオキシリボース、アラビノースまたは、ハロゲンまたはエステル等の5’置換基等の2’−または3’−置換基をさらに含み得るその他の開裂可能な糖を含むヌクレオシド誘導体を含むヌクレオシド誘導体が挙げられる。このようなヌクレオシド誘導体のより特定的な例としては、1−(β−D−アラビノフラノシル)−5−プロプ−1−イニルウラシルおよび2’,3’−ジデオキシ−5−エチニル−3’−フルオロウリジンが挙げられる。
【0065】
本発明に従って使用される多数の5−FUプロドラッグもまた知られているだろう。5−FUのプロドラッグは5−フルオロウラシルへと代謝される化合物であり、例として、5−フルオロウリジン、5−フルオロシチジン、5−フルオロ−2−デオキシウリジン、5−フルオロ−2−デオキシシチジン、5−フルオロアラビノシルウラシル、およびそれらのリン酸エステル類を含む5’−エステル類を含む。その他の例示的な化合物には、5’−デオキシ−4’,5−フルオロウリジン、5’−デオキシ−5−フルオロウリジン1−(2−テトラヒドロフラニル)−5−フルオロウラシル、1−C1−8アルキルカルバモイル−5−フルオロウラシル誘導体、1−(2−テトラヒドロフリル)−5−フルオロウラシル、フトラフール(テガフール(Tegafur)、アジア諸国で広範に使用されている経口5−FUプロドラッグ)、および5’−デオキシ−5−フルオロ−N−[(ペンチルオキシ)カルボニル]−シチジン(カペシタビン、Roche Laboratories Inc.によりXeloda(登録商標)として販売)またはインビボで5−FUへと変換される化合物を含み得る。
【0066】
本発明の特に好ましい実施形態において、本明細書中に記載の方法は、5−FUと組み合わせたエニルウラシルを利用する。上述の通り、5−FUの抗腫瘍活性を最大化するため、患者におけるDPD活性を実質的になくし、さらに5−FUが投与されるときにエニルウラシルが過剰に存在しないようにさせる用量で、エニルウラシルを投与する。エニルウラシルは非常に強力な不可逆性のDPD不活化剤であるため、5−FUの前に投与されることが好ましい。エニルウラシルは迅速にDPDを不活化し、次いで、好ましい所定の時間で、5−FUの投与前に患者から排出されることにより、実質的になくなる。したがって、5−FUが患者へと投与される場合にDPD活性は実質的になくなり、エニルウラシルのレベルは都合よく低下するであろう。
【0067】
この開示に照らし、投与スケジュールが、5−FUまたは5−FUプロドラッグが、好ましくは5−FUまたは5−FUプロドラッグの投与時に患者中に残存するエニルウラシルのレベルを上回って存在するように選択される限り、本発明の方法は所望されるどのような遅延および用量特性を有する投与スケジュールをも含み得ることが理解されるであろう。エニルウラシルおよび5−FUの特定の組み合わせを利用する例示的な1実施形態において、本発明の方法は、5日間(5−day)、1日当たり1回、または1週間当たり(weekly)1回のスケジュールを含む。5−dayまたはweeklyのスケジュールに関し、エニルウラシルの用量範囲は、典型的には約0.8〜10mg/m2、好ましくは約2.5〜5mg/m2である。5−FU投与前に過剰分をなくすために十分な時間が経過するのであれば、より高いエニルウラシル用量が使用可能であろうことに留意されたい。
【0068】
簡便のため、ある種の実施形態においては、エニルウラシルの単純に固定化された用量が好ましい。本明細書中に記載するように、約2.5〜15mgの範囲の用量は通常、ほとんどの患者に好適であろう。特定の好ましい実施形態において、固定される用量は約2.5〜5mgの範囲内である。大きさの異なる患者における、24時間以上、およびある場合には3〜5日間以上DPDをなくすであろう例示的なエニルウラシルの固定用量を以下の表に示す。
【0069】
【表1】
1つの実例となる実施形態において、エニルウラシル投与および5−FU投与の間の時間的遅延は約4〜72時間、4〜36時間、4〜24時間または4〜14時間(または少なくともエニルウラシルの排出半減期の約1〜4倍)である。例えば、エニルウラシルを5−FU投与の前夜に投与してもよく、または、別の方法では朝に投与し、その後5−FUを夜に投与することができる。これらのスケジュールに関し、約20〜30mg/m2の例示的な5−FU用量を用いて(Levinら,Invest New Drugs 18:383−90,2000;Schilskyら,J Clin Oncol 4:1450−7,1998;Guoら,Cancer Chemother Pharmacol 52:79−85,2003)、例えば、5−FUは常にエニルウラシルに対し実質的に過剰である必要がある。
【0070】
別の実施形態において、長期投与スケジュール、例えば、28日間の投与スケジュールを使用することができる。例示的な28日間、b.i.dスケジュール(例えば、1日2回、28日間)は、約0.5〜2mg/m2、好ましくは約1mg/m2の5−FUの投与を含む(例えば、Bakerら,J Clin Oncol 18:915−926 2000)。エニルウラシルが低い5−FU用量を上回って存在しないことを保証するために、b.i.d.スケジュールの開始の少なくとも約12時間前、例えば、5−FUを投与する最初の日の前夜に、約2.5〜5mg/m2でエニルウラシルを投与することができる。次いで、各日における最初の5−FU投与の約2〜8時間前、好ましくは約4〜6時間前に約0.5〜2mg/m2、例えば1mg/m2の用量での1日1回のエニルウラシル投与を、28日間、行うことができる。この方法は、最初の投与に由来する5−FUレベルがエニルウラシルを十分に上回るようにさせ、2度目の投与に由来する5−FUレベルがエニルウラシルを十分に上回るようにさせる。続く処置の日々の間、例えば、2〜28日で、患者の血中エニルウラシルのレベル(例えば、1mg/m2で投与した場合)は4時間(最初の日々の5−FU投与)以内で約50%に減少し(0.5mg/m2に)、第2の5−FU投与により約94%(0.06mg/m2)まで減少する。エニルウラシルにおける最初の2.5〜5mg/m2および続いての1mg/m2の用量は、28日間の5−FU投与に関し、患者におけるDPD活性を効果的に失わせるための役割を果たすはずである。
【0071】
その他の実例となる実施形態において、エニルウラシル(またはその他のDPD阻害物質)を最初に投与し、次いで複数用量の5−FUまたは5−FUプロドラッグを、その後、エニルウラシルが所望により再度投与される前に、所望の時点で添加する。例えば、例示的な実施形態において、エニルウラシルを最初に投与し、次いで複数の5−FU用量を例示的な時点である約12時間、36時間、および54時間においても、エニルウラシルが所望により再度投与されてこのサイクルが繰り返される前に、投与する。
【0072】
特定の実施形態において、エニルウラシルおよび5−FUを、約1:5〜1:15または1:8〜1:12の割合で投与する。
【0073】
本発明の別の実施形態において、5−FU投与時に、エニルウラシルに対しモル数が過剰である本発明に従う限り、5−FUを、エニルウラシル投与後にかなり迅速に、例えば、0.5〜1時間で投与する。
【0074】
エニルウラシル投与における本発明の実施形態に記載の用量およびスケジュールは、先に用いられたフェーズIIおよびフェーズIIIヒト試験(Levinら,Invest
New Drugs 18:383−90,2000;Schilskyら,J Clin Oncol:20:1519−26,2002)に用いられたものとは異なる。これらの研究は高用量のエニルウラシルおよび、エニルウラシルと共に同時に、またはエニルウラシルの後わずか1時間後のいずれかに投与される5−FUを使用する。抗腫瘍活性のうちのあるものはフェーズIII試験において立証されていたが、全体の結果は所望されたものに及ばず、試験は成功しなかった。本発明の方法を用いることにより、過剰なエニルウラシルが存在しないようにさせることにより、またすなわち5−FUまたは5−FUプロドラッグが投与される時に5−FUの活性化を阻害し得ないことにより、向上された効果が達成されるであろうことが確信される。
【0075】
本発明は、単一の製剤中に共存してまたは本発明に従う別個の時点において投与される別個の製剤として存在する、少なくとも1つの薬学上許容可能な担体または賦形剤を含んでなり、さらにDPD阻害物質および/または5−FUまたは5−FUプロドラッグを含む、さらなる特徴を有する製剤処方を含む。一実施形態において、エニルウラシルおよび5−FU、約1:3〜1:20、1:5〜1:15、または1:8〜1:12の比率で1以上の製剤処方中に存在する。担体または賦形剤は製剤のその他の成分と適合性を有するという意味で「薬学上許容可能」であり、患者を害しない。製剤には、例えば、経口、直腸内、経鼻、局所(口内および舌下を含む)、経膣および腹腔内(皮下、筋肉内、静脈内および皮内)投与のために適合したものを含む。製剤は、簡便的に単位投与量形態で存在することができ、薬学的な技術においてよく知られた方法により調製することができる。このような方法には、活性成分と、1以上の副成分を構成する担体とをあわせる工程を含む。通常、製剤は均一に調製し、活性成分を液状担体または細粒の固体担体あるいはその両者と密にあわせる工程および、次いで必要であれば、この産物を成形する工程を含む。
【0076】
本発明の製剤を、原則的に任意の適用可能な手法を用いて調製し、および/または投与することができる。経口投与に適合した本発明の製剤は、例えば、予め決定した量の活性成分を;粉末または顆粒として;水系または非水系の溶液または懸濁液として;または水中油型液状エマルジョンまたは油中水型液状としてそれぞれが含むカプセル(capsule、cachet)または錠剤などの別個の単位であってもよい。活性成分は丸薬(bolus)、舐剤またはペーストであってもよい。経口投与は典型的には好ましい投与経路であるだろう。
【0077】
錠剤は、例えば、所望により1以上の副成分と共に圧縮または成型することにより製造してもよい。圧縮された錠剤は、粉末または顆粒等の流し込める形態の活性成分を、所望により結合剤(例えば、ポビドン、ゼラチン、ヒドロキシプロピルメチルセルロース)、滑剤、不活性希釈剤、保存剤、崩壊剤(例えば、デンプングリコール酸ナトリウム、架橋ポビドン、架橋カルボキシメチルセルロースナトリウム)、界面活性剤または分散剤と共に適切な機械において圧縮することにより調製できる。成型錠剤は、不活性希釈剤と共に湿らせた粉末化化合物を適切な機械において成型することにより調製できる。錠剤は所望により被覆したりまたは割線を入れたりしてもよく、活性成分の放出を制御するために、例えば、所望の放出プロファイルを提供する多様な割合のヒドロキシプロピルメチルセルロースをそこに用いて配合することができる。
【0078】
口内での局所投与のための製剤には、例えば、活性成分を風味付けした基礎原料、通常はスクロースおよびアカシアまたはトラガカント中に含むトローチ剤;活性成分をゼラチンおよびグリセリン、またはスクロースおよびアカシア等の不活性の基材中に含むトローチ(pastilles);および、活性成分を好適な液状担体中に含む洗口液を含む。直腸内投与のための製剤としては、例えば、例えば、カカオバターまたはサリチル酸塩等を含む好適な基材と共に座剤として調製してもよい。経膣投与のための製剤としては、例えば、活性成分に加えて技術的に知られたそのような担体を含むペッサリー、タンポン、クリーム、ジェル、ペースト、フォームまたはスプレー製剤が挙げられる。
【0079】
腹腔内投与のための製剤には、例えば、水系および非水系の、抗酸化剤、緩衝剤、静菌剤および所定の被投与者における血液と等張の処方を与える溶質を含み得る等張滅菌注射液;および、分散剤および増粘剤を含み得る水系および非水系の滅菌懸濁液が挙げられる。製剤は、例えば、アンプル、バイアル等の容器内に封入された単位投与または複数投与量であってもよく、例えば注射のために、使用前に直ちに滅菌液状担体を加えるだけでよいフリーズドライ(凍結乾燥)状態で保存することもできる。滅菌粉末、顆粒および前述の種類の錠剤から、即席の注射液を調製する。
【0080】
典型的には、1以上の活性な薬剤を含む液状の製剤を、好ましくはpH7〜11、通常は9.5〜10.5に緩衝化する。特定の単位投与量の製剤化には、活性成分の、本明細書中の上記に列挙されたような日々の投与量または単位、日々の部分用量、またはそれらの適切な一部を含み得る。
【0081】
本明細書中に記載のDPD阻害物質および5−FUプロドラッグを製造する方法は公知であり、従来の方法論を使用して実行することができる。例えば、上記に参照されるDPD阻害物質は、5−エチニルウラシルの調製に関してはHeterocycl.Chem.19(3)463−4(1982);5−(2−ブロモビニル)ウラシル、5−ブロモエチニルウラシルおよび5−(2−ブロモ−l− クロロビニル)ウラシルの調製に関してはJ.Chem.Soc.Perkin Trans.1(16),1665−70(1981);5−シアノ−ウラシルの調製に関してはNucleic Acid Chemistry,Vol.2,927−30(1978);5−ビニルウラシルの調製に関してはNucleic Acids Research,1(1)105−7(1974);5−トリフルオロメチルウラシルの調製に関してはZ.Chern 17(11)415−16(1977);5−(1−クロロビニル)ウラシルの調製に関してはNucleic Acids Research 3(10),2845(1976)に記載の方法により調製することができる。本発明の特定のその他の化合物は、2’,3’−ジデオキシ−5−エチニル−3’−フルオロウリジン等の3’−フルオロ−2’,3’−ジデオキシ5−アルキニルウリジン化合物の調製に関しては、欧州特許出願公開第356166号、および1−(b−D−アラビノフラノシル)−5−プロプ−I−イニルウラシル(1−(b−D−arabinofuranosyl)−5−prop−I−ynyluracil)等の5−アルキニルウラシルアラビノシドの調製に関しては欧州特許出願公開第272065号に記載の工程に従って調製することができる。これらおよびその他の合成手法は公知であり、本発明で使用するための化合物を製造するために利用することができる。
【0082】
一実施形態において、本発明は、製剤成分における所望の時間的放出を可能にする方法でDPD阻害物質および5−FUまたは5−FUプロドラッグを共に患者へと投与する組み合わせ経口製剤を提供する。2つの成分の差示的な時間的放出輸送は、公知の手法および材料を用いて達成できる。例えば、一実施形態において、例えば錠剤形態における経口製剤を、図3に説明的に示すような3つの異なる層から構成することができる。外側の層は、迅速に放出される構造中にエニルウラシルを含み得る。中間層は、本発明の5−FUまたは5−FUプロドラッグの放出を所望の程度まで遅らせる徐放性成分(例えば、徐放性緩衝剤)であることができ、そこにおいて、迅速に放出される構造中のコア層中に、5−FUまたは5−FUプロドラッグが局在する。DPD阻害物質および5−FUまたは5−FUプロドラッグを本明細書中に記載の適切な用量および割合で配合する。好ましい1実施形態において、DPD阻害物質はエニルウラシルであり、5−FUまたは5−FUプロドラッグは5−FUまたはカペシタビンである。
【0083】
別の実施形態において、別の配合は、5−FUまたは5−FUプロドラッグを含むミクロスフィア等の公知の送達ビヒクルを含むことができる。一実施形態において、例えば、5−FUまたは5−FUプロドラッグを、時間的放出成分(例えば、時間的放出崩壊緩衝材)のシェル内にカプセル化することができ、外側の層がDPD阻害物質の迅速な放出を提供する。好ましい1実施形態において、DPD阻害物質はエニルウラシルであり、5−FUまたは5−FUプロドラッグは5−FUまたはカペシタビンである。実例となる組み合わせ製剤におけるこれらおよびその他の実施例を、DPD阻害物質、および、5−FUまたは5−FUプロドラッグの送達の間における適切な時間遅延を1つの経口調製物中で可能にする公知の手法を用いて設計でき、製造することができる。
【0084】
別の実施形態において、DPD阻害物質は、癌細胞および腫瘍を選択的にまたは優先的に標的とすることができる。癌細胞および腫瘍におけるDPDの選択効果は、循環における半減期よりも長い化合物の半減期を可能にし、そのことが、活性化ヌクレオチドのより高い定常状態レベルおよび治療指数の向上を実現するであろう。一実施形態において、腫瘍に対するエニルウラシル等のDPD阻害物質の選択的標的化は、技術的によく知られた標的化されたミクロスフィアを用いて実現可能である。別の実施形態において、標的化は、化合物を肝細胞および肝臓癌細胞に対し標的化するアシアロ糖タンパク質(asiolglycoprotein)等の組織特異的な受容体を用いて達成される。このような標的化は、例えば、DPD阻害物質を化学的にまたはその他の方法で(例えば、エニルウラシルにおける3−6ガラクトース残基を置換することにより)修飾することにより、実現することができる。
【0085】
別の実施形態において、本明細書中に記載の方法は、ロイコボリンの投与をさらに含む。ロイコボリン、またはロイコボリンの活性な異性体であるイソロイコボリンは癌患者の治療に関し5−FUと併せてよく用いられる。それはまた、エニルウラシルおよび5−FUに関する上述の投与計画に付加してもよい。ロイコボリンは腫瘍を有するラットおよび組織培養においてエニルウラシルおよび5−FUの抗腫瘍効果を向上させることが示されており(Caoら、Cancer Res 90:1507−1510,1993;Fischelら、Biochem Pharmacol 53:1703−1709,1997)、また、エニルウラシルおよび5−FUを投与されている患者に対して投与されている(Schilskyら、J Clin Oncol 4:1450−7,1998;Guoら、Cancer Chemother Pharmacol 52:79−85,2003)。ロイコボリンは経口製剤において都合よく利用することもできる。
【0086】
本発明は、以下の非限定的な実施例を考慮してさらに理解することができる。
【実施例】
【0087】
(実施例1)
(過剰のエニルウラシルがエニルウラシルおよび5−FUの抗腫瘍活性を減少させる)
ラットにWard癌腫瘍を移植し、それらの腫瘍が先に記載されたように重量で3,000mgまで成長させた(Caoら,Cancer Res 90:1507−1510、1993)後に、以下の投与計画の1つを用いて処置した。3,000mgの腫瘤を有するラットに、0日目、7日目、および14日目に以下の処置を施した。
【0088】
【表2】
A群のラットには処置を行わなかった。B群のラットには1mg/kgのエニルウラシル(時間(t)=0)を腹腔内(ip)投与し、次いで5 mg/kgの5−FUをt=60において静脈内(iv)投与した。C群のラットには1mg/kgのエニルウラシル(t=0)をip投与し、次いで25mg/kgのエニルウラシルをt=55においてip投与し、かつ5mg/kgの5−FUをt=60においてiv投与した。動物には1週あたり1回、3週間の間投与を行った。エニルウラシルも、各週の処置における2日目および3日目にB群およびC群において1 mg/kgでラットへとip投与した。C群に対する処置5−FUが投与された時に高レベルのエニルウラシルが存在する場合の臨床試験を模した。
【0089】
A群における腫瘍は迅速に10,000mgへと成長し、ラットを犠牲にした。C群における腫瘍は平均して大きさがわずかに減少したのみであった一方、B群における腫瘍は迅速に消失した。これらの結果は、C群における過剰なエニルウラシルが5−FUの抗腫瘍活性を大きく減少させることを示し、それはエニルウラシルが5−FUの代謝的活性化を妨げることに起因すると思われる。
【0090】
(実施例2)
(エニルウラシルは活性なヌクレオチドへの代謝的活性化を抑制する)
HEK293細胞をまずエニルウラシル(10μM)で1時間処理した。4〜48時間の洗浄期間後に、細胞を[6−14C]−5−FU(66μM)で2時間、37℃にて処理した。対照は[6−14C]−5−FU(66μM)のみで2時間処理した、またはプレインキュベーションを行うことなくエニルウラシル(10μM)および[6−14C]−5−FU(66μM)を共投与したHEK293細胞であった。細胞溶解物中に存在する[6−14C]−5−FU異化産物/同化産物を定量するために、放射活性検出を伴う逆相HPLCを利用した。異なるセットの実験において、異なるエニルウラシル投与スケジュールにおける5−FUの細胞毒性を調べた。HEK293細胞を、エニルウラシル(5μM)と1時間プレインキュベーション後か、エニルウラシルを伴わないプレインキュベーション後に、またはプレインキュベーションなしで共投与されるエニルウラシル(5μM)と共に、一定濃度範囲の5−FUを用いて72時間、37℃にて処理した。MTS増殖アッセイおよび算出される5−FUに対するEC50値により、細胞毒性を分析した。
【0091】
エニルウラシルを用いた1時間の細胞の前処理、それに次ぐ、[6−14C]−5−FU処理に先立つ36時間または48時間の洗浄は、[6−14C]−5−FU処理単独、またはエニルウラシルおよび[6−14C]−5−FUの共投与の場合と比較して、[6−14C]−FUMP同化産物の生成の顕著な増加を実現する(p−値<0.05)。例えば、48時間の洗浄除去は、[6−14C]−5−FU処理のみに対して[6−14C]−FUMPの生成における70%の増加、また、エニルウラシルおよび[6−14C]−5−FUの共投与に対して[6−14C]−FUMPの生成における41%の増加を実現した。細胞毒性試験において、エニルウラシルによる1時間のプレインキュベーションは、5−FU処理単独(311.4nM±1.1)またはEUおよび5−FUのプレインキュベーションなしの共投与(307.8nM±1.03)と比較して減少したEC50値(264.0nM±1.7)(平均±SE)を伴って、5−FUの細胞毒性を増加させた(p<0.05)。
【0092】
これらの結果はさらに、5−FUを不活性な形態へと分解する異化経路を阻害する公知の役割に加えて、5−FUを活性な抗腫瘍ヌクレオチドへと変換する同化経路を阻害する際の、エニルウラシルの役割を裏付ける。
【0093】
(実施例3)
(エニルウラシルは血漿ウリジンレベルを増加させる)
エニルウラシル阻害がDPD阻害によるウラシルレベルの上昇をもたらすことが知られている一方、フルオロピリミジンを同化するその他の酵素への阻害効果がウリジン等のその他の同化産物のレベルを量的に変化させることが期待されるであろう。エニルウラシルを2mg/kg、25mg/kgおよび100mg/kgでマウスに投与した。0分、15分、30分、60分、2時間、24時間および48時間時点で血漿サンプルを取得した。LC−MSにより公知の手法を用いてウリジンレベルを測定し、アッセイの妥当性を検証するために標準物質を使用した。
【0094】
図2に示す、これらの実験から得られる結果は、エニルウラシルが投与後のウリジンレベルの上昇を引き起こすことを示す。この知見はウリジンホスホリラーゼ等の同化酵素に対する阻害効果を有するエニルウラシルと一致し、5−FUの活性なヌクレオチドへの同化的変換阻害におけるエニルウラシルの役割をさらに支持する。
【0095】
本発明の特定の実施形態が説明のために本明細書中に記載されるが、本発明の本発明の精神および範囲から逸脱することなくなされる多様な改変がなされ得、したがって、本発明は添付の請求項によるもの以外の限定を受けないことが、前述の記載から理解されるであろう。
【特許請求の範囲】
【請求項1】
患者における癌を処置するための組合せ物であって、該組合せ物は、(i)DPD阻害物質を含む組成物および(ii)5−FUまたは5−FUプロドラッグを含む組成物を含み、該DPD阻害物質を含む該組成物は、最初に投与され、該5−FUまたは該5−FUプロドラッグを含む該組成物は、その少なくとも4時間後に投与され、該DPD阻害物質の用量と、該5−FUまたは5−FUプロドラッグの用量とが、1:3〜1:20の比にあり、該5−FUまたは5−FUプロドラッグを含む組成物は、その投与時に、該5−FUまたは5−FUプロドラッグが該DPD阻害物質を上回る量で該患者中に存在するような用量で投与され、該DPD阻害物質は、エニルウラシルであり、該エニルウラシルは2.5〜5mg/m2の用量で投与され、そして
該5−FUまたは5−FUプロドラッグは、5−フルオロウリジン、5−フルオロシチジン、5−フルオロ−2−デオキシウリジン、5−フルオロ−2−デオキシシチジン、5’−デオキシ−4’,5−フルオロウリジン、5−フルオロアラビノシルウラシル、5’−デオキシ−5−フルオロウリジン、1−(2−テトラヒドロフラニル)−5−フルオロウラシル、1−C1−8アルキルカルバモイル−5−フルオロウラシル、1−(2−テトラヒドロフリル)−5−フルオロウラシル、5’−デオキシ−5−フルオロ−N−[(ペンチルオキシ)カルボニル]−シチジン(カペシタビン)、およびリン酸エステルを含めたそれらの5’−エステルからなる群から選択されることを特徴とし、
該エニルウラシルの用量、および該5−FUまたは5−FUプロドラッグを投与する前の遅延は、該DPD阻害物質が該5−FUもしくは該5−FUプロドラッグから生成される5−FUの代謝活性化を妨害し、そして、5−FU抗腫瘍活性を弱める程度を最小化するように機能する、
組合せ物。
【請求項2】
前記5−FUまたは5−FUプロドラッグを含む前記組成物が、前記DPD阻害物質を含む前記組成物が投与されてから少なくとも12時間後または少なくとも14時間後に投与されることを特徴とする、請求項1に記載の組合せ物。
【請求項3】
前記5−FUまたは5−FUプロドラッグを含む前記組成物が、前記DPD阻害物質を含む前記組成物が投与されてから、該DPD阻害物質についての排出半減期の2〜4倍の時間で投与されることを特徴とする、請求項1に記載の組合せ物。
【請求項4】
前記5−FUまたは5−FUプロドラッグが5−FUまたはカペシタビンである、請求項1に記載の組合せ物。
【請求項5】
前記5−FUまたは5−FUプロドラッグが5−FUであり、該5−FUを含む前記組成物は10〜60mg/m2の用量で投与されることを特徴とする、請求項1に記載の組合せ物。
【請求項6】
前記5−FUまたは5−FUプロドラッグが5−FUであり、該5−FUを含む前記組成物は10〜30mg/m2の用量で投与されることを特徴とする、請求項1に記載の組合せ物。
【請求項7】
前記5−FUまたは5−FUプロドラッグが5−FUであり、該5−FUを含む前記組成物は20〜60mg/m2の用量で投与されることを特徴とする、請求項1に記載の組合せ物。
【請求項8】
患者における癌を処置するための組合せ物であって、該組合せ物は、エニルウラシルを含む組成物および5−FUを含む組成物を含み、該エニルウラシルを含む組成物は最初に投与され、該5−FUを含む該組成物は、その後に投与され、該エニルウラシルを含む該組成物は、2.5〜5mg/m2の用量で投与され、該5−FUを含む該組成物は、該エニルウラシルを含む該組成物が投与されてから少なくとも4時間後に投与され、そして該5−FUを含む該組成物は、その投与時に該5−FUが該患者中に該エニルウラシルの少なくとも5倍過剰で存在するような用量で投与されることを特徴とし、
該エニルウラシルの用量、および該5−FUを投与する前の遅延は、該エニルウラシルが該5−FUの代謝活性化を妨害し、そして、5−FU抗腫瘍活性を弱める程度を最小化するように機能する、
組合せ物。
【請求項9】
前記5−FUを含む前記組成物は、前記エニルウラシルを含む組成物が投与されてから少なくとも14時間後または少なくとも18時間後に投与されることを特徴とする、請求項8に記載の組合せ物。
【請求項10】
前記5−FUを含む前記組成物が、前記エニルウラシルを含む前記組成物が投与されてから、該エニルウラシルについての排出半減期の2〜4倍の時間で投与されることを特徴とする、請求項8に記載の組合せ物。
【請求項11】
前記5−FUまたは5−FUプロドラッグがカペシタビンである、請求項1に記載の組合せ物。
【請求項12】
前記カペシタビンを含む前記組成物は、前記エニルウラシルを含む組成物が投与されてから少なくとも14時間後または少なくとも18時間後に投与されることを特徴とする、請求項11に記載の組合せ物。
【請求項13】
前記5−FUを含む前記組成物が、前記エニルウラシルを含む前記組成物が投与されてから、該エニルウラシルについての排出半減期の2〜4倍の時間で投与されることを特徴とする、請求項11に記載の組合せ物。
【請求項14】
前記カペシタビンを含む前記組成物は0.8〜200mg/m2の用量で投与されることを特徴とする、請求項11に記載の組合せ物。
【請求項15】
前記カペシタビンを含む前記組成物は05〜100mg/m2の用量で投与されることを特徴とする、請求項11に記載の組合せ物。
【請求項16】
前記DPD阻害物質の用量と、前記5−FUまたは5−FUプロドラッグの用量とが、1:5〜1:15の比にある、請求項1に記載の組合せ物。
【請求項17】
前記DPD阻害物質の用量と、前記5−FUまたは5−FUプロドラッグの用量とが、1:8〜1:12の比にある、請求項1に記載の組合せ物。
【請求項18】
患者における癌を処置するための組合せ物であって、該組合せ物は、エニルウラシルを含む組成物および5−FUを含む組成物を含み、該エニルウラシルを含む組成物は最初に2.5〜5mg/m2の用量で投与され、そして該5−FUを含む組成物は、その後、該エニルウラシルの排出半減期の少なくとも3倍が経過してから投与され、該5−FUを含む該組成物の投与の時点での、該患者において存在するエニルウラシルの、5−FUに対する比は、1:5と1:15との間であることを特徴とし、
該エニルウラシルの用量、および該5−FUを投与する前の遅延は、該エニルウラシルが該5−FUの代謝活性化を妨害し、そして、5−FU抗腫瘍活性を弱める程度を最小化するように機能する、
組合せ物。
【請求項19】
前記5−FUを含む前記組成物の投与の時点での、前記患者において存在するエニルウラシルの、5−FUに対する比は、1:8と1:12との間である、請求項18に記載の組合せ物。
【請求項20】
前記エニルウラシルを含む前記組成物の投与と前記5−FUを含む組成物の投与との間での前記エニルウラシルの除去半減期の少なくとも3倍の遅延は、該5−FUを含む組成物が投与される時に過剰なエニルウラシルが前記患者に存在しないことを確実にするためのものである、請求項18に記載の組合せ物。
【請求項21】
患者における癌を処置するための組合せ物であって、該組合せ物は、エニルウラシルを含む組成物および5−FUを含む組成物を含み、該エニルウラシルを含む組成物は最初に2.5〜5mg/m2の用量で投与され、そして該5−FUを含む組成物は、その後、該エニルウラシルの排出半減期の少なくとも3倍が経過してから投与され、該5−FUを含む組成物は、該5−FUを含む該組成物の投与の時点で、該患者において該エニルウラシルの少なくとも5倍過剰で存在するような用量で投与されることを特徴とし、
該エニルウラシルの用量、および該5−FUを投与する前の遅延は、該エニルウラシルが該5−FUの代謝活性化を妨害し、そして、5−FU抗腫瘍活性を弱める程度を最小化するように機能する、
組合せ物。
【請求項1】
患者における癌を処置するための組合せ物であって、該組合せ物は、(i)DPD阻害物質を含む組成物および(ii)5−FUまたは5−FUプロドラッグを含む組成物を含み、該DPD阻害物質を含む該組成物は、最初に投与され、該5−FUまたは該5−FUプロドラッグを含む該組成物は、その少なくとも4時間後に投与され、該DPD阻害物質の用量と、該5−FUまたは5−FUプロドラッグの用量とが、1:3〜1:20の比にあり、該5−FUまたは5−FUプロドラッグを含む組成物は、その投与時に、該5−FUまたは5−FUプロドラッグが該DPD阻害物質を上回る量で該患者中に存在するような用量で投与され、該DPD阻害物質は、エニルウラシルであり、該エニルウラシルは2.5〜5mg/m2の用量で投与され、そして
該5−FUまたは5−FUプロドラッグは、5−フルオロウリジン、5−フルオロシチジン、5−フルオロ−2−デオキシウリジン、5−フルオロ−2−デオキシシチジン、5’−デオキシ−4’,5−フルオロウリジン、5−フルオロアラビノシルウラシル、5’−デオキシ−5−フルオロウリジン、1−(2−テトラヒドロフラニル)−5−フルオロウラシル、1−C1−8アルキルカルバモイル−5−フルオロウラシル、1−(2−テトラヒドロフリル)−5−フルオロウラシル、5’−デオキシ−5−フルオロ−N−[(ペンチルオキシ)カルボニル]−シチジン(カペシタビン)、およびリン酸エステルを含めたそれらの5’−エステルからなる群から選択されることを特徴とし、
該エニルウラシルの用量、および該5−FUまたは5−FUプロドラッグを投与する前の遅延は、該DPD阻害物質が該5−FUもしくは該5−FUプロドラッグから生成される5−FUの代謝活性化を妨害し、そして、5−FU抗腫瘍活性を弱める程度を最小化するように機能する、
組合せ物。
【請求項2】
前記5−FUまたは5−FUプロドラッグを含む前記組成物が、前記DPD阻害物質を含む前記組成物が投与されてから少なくとも12時間後または少なくとも14時間後に投与されることを特徴とする、請求項1に記載の組合せ物。
【請求項3】
前記5−FUまたは5−FUプロドラッグを含む前記組成物が、前記DPD阻害物質を含む前記組成物が投与されてから、該DPD阻害物質についての排出半減期の2〜4倍の時間で投与されることを特徴とする、請求項1に記載の組合せ物。
【請求項4】
前記5−FUまたは5−FUプロドラッグが5−FUまたはカペシタビンである、請求項1に記載の組合せ物。
【請求項5】
前記5−FUまたは5−FUプロドラッグが5−FUであり、該5−FUを含む前記組成物は10〜60mg/m2の用量で投与されることを特徴とする、請求項1に記載の組合せ物。
【請求項6】
前記5−FUまたは5−FUプロドラッグが5−FUであり、該5−FUを含む前記組成物は10〜30mg/m2の用量で投与されることを特徴とする、請求項1に記載の組合せ物。
【請求項7】
前記5−FUまたは5−FUプロドラッグが5−FUであり、該5−FUを含む前記組成物は20〜60mg/m2の用量で投与されることを特徴とする、請求項1に記載の組合せ物。
【請求項8】
患者における癌を処置するための組合せ物であって、該組合せ物は、エニルウラシルを含む組成物および5−FUを含む組成物を含み、該エニルウラシルを含む組成物は最初に投与され、該5−FUを含む該組成物は、その後に投与され、該エニルウラシルを含む該組成物は、2.5〜5mg/m2の用量で投与され、該5−FUを含む該組成物は、該エニルウラシルを含む該組成物が投与されてから少なくとも4時間後に投与され、そして該5−FUを含む該組成物は、その投与時に該5−FUが該患者中に該エニルウラシルの少なくとも5倍過剰で存在するような用量で投与されることを特徴とし、
該エニルウラシルの用量、および該5−FUを投与する前の遅延は、該エニルウラシルが該5−FUの代謝活性化を妨害し、そして、5−FU抗腫瘍活性を弱める程度を最小化するように機能する、
組合せ物。
【請求項9】
前記5−FUを含む前記組成物は、前記エニルウラシルを含む組成物が投与されてから少なくとも14時間後または少なくとも18時間後に投与されることを特徴とする、請求項8に記載の組合せ物。
【請求項10】
前記5−FUを含む前記組成物が、前記エニルウラシルを含む前記組成物が投与されてから、該エニルウラシルについての排出半減期の2〜4倍の時間で投与されることを特徴とする、請求項8に記載の組合せ物。
【請求項11】
前記5−FUまたは5−FUプロドラッグがカペシタビンである、請求項1に記載の組合せ物。
【請求項12】
前記カペシタビンを含む前記組成物は、前記エニルウラシルを含む組成物が投与されてから少なくとも14時間後または少なくとも18時間後に投与されることを特徴とする、請求項11に記載の組合せ物。
【請求項13】
前記5−FUを含む前記組成物が、前記エニルウラシルを含む前記組成物が投与されてから、該エニルウラシルについての排出半減期の2〜4倍の時間で投与されることを特徴とする、請求項11に記載の組合せ物。
【請求項14】
前記カペシタビンを含む前記組成物は0.8〜200mg/m2の用量で投与されることを特徴とする、請求項11に記載の組合せ物。
【請求項15】
前記カペシタビンを含む前記組成物は05〜100mg/m2の用量で投与されることを特徴とする、請求項11に記載の組合せ物。
【請求項16】
前記DPD阻害物質の用量と、前記5−FUまたは5−FUプロドラッグの用量とが、1:5〜1:15の比にある、請求項1に記載の組合せ物。
【請求項17】
前記DPD阻害物質の用量と、前記5−FUまたは5−FUプロドラッグの用量とが、1:8〜1:12の比にある、請求項1に記載の組合せ物。
【請求項18】
患者における癌を処置するための組合せ物であって、該組合せ物は、エニルウラシルを含む組成物および5−FUを含む組成物を含み、該エニルウラシルを含む組成物は最初に2.5〜5mg/m2の用量で投与され、そして該5−FUを含む組成物は、その後、該エニルウラシルの排出半減期の少なくとも3倍が経過してから投与され、該5−FUを含む該組成物の投与の時点での、該患者において存在するエニルウラシルの、5−FUに対する比は、1:5と1:15との間であることを特徴とし、
該エニルウラシルの用量、および該5−FUを投与する前の遅延は、該エニルウラシルが該5−FUの代謝活性化を妨害し、そして、5−FU抗腫瘍活性を弱める程度を最小化するように機能する、
組合せ物。
【請求項19】
前記5−FUを含む前記組成物の投与の時点での、前記患者において存在するエニルウラシルの、5−FUに対する比は、1:8と1:12との間である、請求項18に記載の組合せ物。
【請求項20】
前記エニルウラシルを含む前記組成物の投与と前記5−FUを含む組成物の投与との間での前記エニルウラシルの除去半減期の少なくとも3倍の遅延は、該5−FUを含む組成物が投与される時に過剰なエニルウラシルが前記患者に存在しないことを確実にするためのものである、請求項18に記載の組合せ物。
【請求項21】
患者における癌を処置するための組合せ物であって、該組合せ物は、エニルウラシルを含む組成物および5−FUを含む組成物を含み、該エニルウラシルを含む組成物は最初に2.5〜5mg/m2の用量で投与され、そして該5−FUを含む組成物は、その後、該エニルウラシルの排出半減期の少なくとも3倍が経過してから投与され、該5−FUを含む組成物は、該5−FUを含む該組成物の投与の時点で、該患者において該エニルウラシルの少なくとも5倍過剰で存在するような用量で投与されることを特徴とし、
該エニルウラシルの用量、および該5−FUを投与する前の遅延は、該エニルウラシルが該5−FUの代謝活性化を妨害し、そして、5−FU抗腫瘍活性を弱める程度を最小化するように機能する、
組合せ物。
【図1】

【図2】
【図3】

【図2】
【図3】
【公開番号】特開2012−229273(P2012−229273A)
【公開日】平成24年11月22日(2012.11.22)
【国際特許分類】
【外国語出願】
【出願番号】特願2012−186899(P2012−186899)
【出願日】平成24年8月27日(2012.8.27)
【分割の表示】特願2007−544558(P2007−544558)の分割
【原出願日】平成17年12月5日(2005.12.5)
【出願人】(507182575)アドヘレックス テクノロジーズ, インコーポレイテッド (2)
【Fターム(参考)】
【公開日】平成24年11月22日(2012.11.22)
【国際特許分類】
【出願番号】特願2012−186899(P2012−186899)
【出願日】平成24年8月27日(2012.8.27)
【分割の表示】特願2007−544558(P2007−544558)の分割
【原出願日】平成17年12月5日(2005.12.5)
【出願人】(507182575)アドヘレックス テクノロジーズ, インコーポレイテッド (2)
【Fターム(参考)】
[ Back to top ]