
5−FU又はそのプロドラッグと、DPD阻害剤との組合せに付随した神経毒性の処置
5−FU及び/又は5−FUプロドラッグと組み合わせたDPD阻害剤の改善された投与及び投薬の方法であって、患者内の神経組織と非神経組織の両方における酵素の活性を実質的に消失させるDPD阻害剤をそれを必要とする患者にまず投与すること、及びその後に5−FU又は5−FUプロドラッグを投与することを含み、5−FU又はプロドラッグから生成される5−FUのレベルが、患者におけるDPD阻害剤よりもかなり過剰である、方法が提供される。

【発明の詳細な説明】
【技術分野】
【0001】
関連出願への相互参照
本願は、2009年10月14日に出願された米国仮特許出願第61/251,449号の米国特許法第119条(e)項の優先権を主張し、この米国仮特許出願は、本明細書中に参考として援用される。
【0002】
発明の背景
発明の分野
本発明は、一般に癌治療に関し、より詳細には、DPD阻害剤と5−FU及び/又は5−FUプロドラッグを併用して癌治療に付随する神経毒性を防止又は最小限にする方法に関する。
【背景技術】
【0003】
先行技術の説明
5−フルオロウラシル(5−FU)は、癌患者における固形腫瘍の処置に30年以上臨床的に使用されてきた(Ansfield et al.,Cancer 39:34−40,1977;Grem et al.,Cancer Treat Rep 71:1249−1264,1987;Chabner et al.,Cancer,Principles and Practice of Oncology,2nd Ed,pp287−328 Philadelphia,PA:J B Lippincott Co,1985)。5−FUは、代謝変換によって、DNA合成及びRNA機能を阻害する偽ウリジンヌクレオチド(例えば、FUMP、FUDP、FUTP)及びデオキシウリジンヌクレオチド(例えば、FdUMP、FdUDP、FdUTP)に活性化されなければならない(Meyers,Pharmacol Rev,33:1−15,1981;Dasher et al.,Pharmac Ther 48:189−222,1990に概説されている)。5−FUは、その天然の相当物であるウラシルとは、5位のフッ素置換しか異ならないので、癌患者においては容易に活性化される。残念ながら、ウラシルとのその構造上の類似性は、抗腫瘍活性を持たない生成物に急速かつ大規模に変換される原因にもなっている。この代謝過程は、異化と称される。5−FUは、酵素ジヒドロピリミジンデヒドロゲナーゼ(DPD:EC1312、ウラシル還元酵素)によって急速に異化される(Meyers,Pharmacol Rev,33:1−15,1981;Dasher et al.,Pharmac Ther 48:189−222,1990)。したがって、癌治療用5−FUの抗腫瘍効果は、抗腫瘍ヌクレオチドへの代謝変換(活性化)と無用な代謝産物への代謝変換(異化)との繊細なバランスに依拠している。
【0004】
さらに、幾つかの臨床上の問題が5−FUの代謝異化に起因して生じる。第一に、DPDレベルは、個体間で異なり(Fleming et al.,Cancer Res 52:2899−2902,1992;Grem et al.,Cancer Chemother Pharmacol 40:117−125,1997)、個体内でも1日の間に変わるので(Grem et al.,Cancer Chemother Pharmacol 40:117−125,1997;Harris et al.,Cancer Res 50:197−201,1990;Petit et al.,Cancer Res 48:1676−1679,1988)、所与の用量から生じる5−FU又はプロドラッグ由来の5−FUの全身的レベルが大きく変化し、したがって有効性及び毒性を予測することが極めて難しい。極端な場合、遺伝的にDPDが欠乏した患者は、「標準」治療量の5−FUで処置すると、激しい、時には致死的な毒性を経験する(Morrison et al.,Oncol Nurs Forum 24:83−88,1997に概説されている)。第二に、胃腸のDPDレベルの変化は(Ho et al.,Anticancer Res 6:781−784,1986;Naguib et al.,Cancer Res 45:5405−5412,1985;Spector et al.,Biochem Pharmacol 46:2243−2248,1993)、経口投与された5−FUの吸収を大きく変化させ(Christophidis et al.,Clin Pharmacokinetics 3:330−336,1978;Cohen et al.,Cancer Chemother Rep 58:723−731,1974;Finch et al.,Br J Clin Pharmacol 7:613−617,1979)、薬物の血しょう中濃度を予測不可能にし、望ましくない毒性や不十分な効力をもたらし得る。第三に、高レベルのDPDを含む腫瘍は、5−FU治療に反応しない可能性がある(Etienne et al.,J Clin Oncol 13:1663−1670,1995;Fischel et al.,Clin Cancer Res 1:991−996,1995)。
【0005】
最後に、F−Balなどの5−FU分解産物は、神経毒性(Okeda et al.,Acta Neuropathol 81:66−73,1990;Koenig et al.Arch Neurol 23:155−160,1970;Davis ST,et al.Biochem Pharmacol 1994;48:233−6;Saif MW,et al.Anticancer Drugs 2001;12:525−31に概説されている)、心毒性(et al.,Lancet 337:560,1991;Lemaire et al.,Br J Cancer 66:119−127,1992)、手掌足底紅斑異感覚症候群(手足症候群)(Hohneker,Oncology 12:52−56,1998)、及びGI毒性(Spector et al.,Cancer Res 55:1239−1241,1995)を生じるおそれがあり、抗腫瘍活性を阻害するように思える(Spector et al.,Cancer Res 55:1239−1241,1995;Cao,et al.,Pharmacol 59:953−960,2000)。
【0006】
DPDは、5−FUの分解(異化)において第1かつ律速段階である広範に分布する酵素である。研究によれば、DPDを阻害すると、血しょう中の5−FUの半減期が大きく増加する。DPDを不可逆的に不活性化するもの、及びDPDを可逆的に阻害するものを含めて、幾つかのDPD阻害剤が研究されている。例えば、エニルウラシル(5−エチニルウラシル、776C85)は、強力な不可逆的DPD失活剤である。DPD及び異化経路中の一連の酵素は、最終的に、5−FUをα−フルオロ−β−アラニン(F−Bal)に変換するので(Spector et al.,Drugs of The Future 1994;19:565−71;Paff et al.,Invest New Drugs 2000;18:365−71に概説されている)、エニルウラシルは、5−FU排出経路を異化から腎排せつに変更し、それによって5−FU排出半減期を10〜20分から4.5〜6.5時間に延長する(Adjei et al.,J Clin Oncol 2002;20:1683−91;Ochoa et al.,Ann Oncol 2000;11:1313−22;Baker,Invest New Drugs 2000;18:373−81;Baker et al.,J Clin Oncol 1996;14:3085−96;Guo et al.,Cancer Chemother Pharmacol 2003;52:79−85;Schilsky et al.,J Clin Oncol 1998;16:1450−7)。
【0007】
胃腸管における5−FU分解を防止することによって、エニルウラシルは5−FUの経口投与も可能にする(Baker et al.,J Clin Oncol 1996;14:3085−96)。さらに、エニルウラシルは、5−FU関連神経毒性(Saif MW,et al.Anticancer Drugs 2001;12:525−31に概説されているDavis et al.,Biochem Pharmacol 1994;48:233−6)及び手足毒性症候群(Schilsky et al.,J Clin Oncol 2002;20:1519−26)の原因であると思われる、F−Balなどの5−FU異化産物の形成を防止する。さらに、F−Balなどの5−FU異化産物は、5−FUの抗腫瘍活性を低下させると思われる(Cao et al.,Biochem Pharmacol 2000;59:953−60;Spector T,et al.Cancer Res 1995;55:1239−41 Spector et al.,Drugs of The Future 1994;19:565−71;Paff et al.,Invest New Drugs 2000;18:365−71)。
【0008】
さらに、DPDは患者中に異なるレベルで存在するので、DPDがエニルウラシルによって不活性化されると、5−FUの極めて変わりやすい非線形薬物動態が、高度に予測可能な線形になる(Baker,Invest New Drugs 2000;18:373−81に概説されている)。実際、エニルウラシルは、腫瘍を有する実験動物において、5−FUの抗腫瘍効果をかなり改善し、治療指数を増加させた(Baccanari et al.,Proc Natl Acad Sci USA 1993;90:11064−812;Cao et al.,Cancer Res 1994;54:1507−10)。
【0009】
エニルウラシルは、癌患者において第一相臨床試験で試験された(Levin et al.,Invest New Drugs 18:383−90,2000;Baker et al.,J Clin Oncol 18:915−926 2000;Schilsky et al.,J Clin Oncol 4:1450−7,1998に概説されている)。これらの試験では、エニルウラシルは、毒性を生じずに、DPD活性を極めて強力に消失させた。例えば、用量0.74mg/m2(合計約1mg)は、末梢血細胞中のDPDの90%超を長期間除去した。5−FUの排出半減期は、1回のエニルウラシルによって約10分から3.5時間に増加した。用量3.7mg/m2のエニルウラシルは、5−FUの半減期を4.5〜6.5時間に延長した。用量をさらに高くしても、明白な利点は得られなかった。
【0010】
続いて、2回の多施設第三相試験が結腸直腸癌患者において5−FUの10倍過剰のエニルウラシルを含む複合丸剤を使用して実施された。患者に10mg/平方メートル体表面積(mg/m2)エニルウラシル及び1mg/m2 5−FUを12時間ごとに28日間投与した。薬物を1週間停止した後、このサイクルを繰り返した。服薬遵守が問題なかった北米の試験の結果は、有望な抗腫瘍活性、高い耐容性、及びごくわずかの手足症候群を示したが、投薬計画は、エニルウラシルのない5−FU/ロイコボリンの標準投薬計画よりも抗腫瘍性の利点が少ない傾向にあった(Schilsky et al.,J Clin Oncol 2002;20:1519−26)。これらの結果の説明は、当時は明らかでなかった。
【0011】
特許文献1は、5−FUが対象に投与された時点で過剰のエニルウラシルが存在すると、5−FUの抗腫瘍活性がかなり低下するという重要な知見を記載している。したがって、5−FUの抗腫瘍活性を最大にするために、5−FU投与時に5−FUがエニルウラシルよりもかなり過剰に存在するように、低用量のエニルウラシルを5−FUのかなり前に投与することを提案している。さもないと、5−FUの抗腫瘍効果が損なわれるおそれがある。これらの結果は、5−FU投与時の5−FUに対するエニルウラシル比が10:1であった第三相試験における予想よりも低い抗腫瘍活性を説明するものである。
【0012】
したがって、臨床試験は、5mg用量のエニルウラシル、続いて30〜160mg用量の5−FUを12〜24時間後に癌患者に投与して開始された。予想外に、この処置を受けた41名の患者の大多数は、軽度から重度の何らかの神経毒性を経験し、主な神経学的症候は、運動失調(不安定歩行)、神経障害、錯乱、めまい及び不明瞭発語であった。
【0013】
神経毒性を防止又は最小化するために、5−FU及び5−FUプロドラッグの抗腫瘍効果及び治療指数を最大にするために、投薬の予測可能性を改善するために、さらに、5−FU及び5−FUプロドラッグを経口投与によって効果的に投与できるようにするために、5−FU及び5−FUプロドラッグと併用されるDPD阻害剤の最適な投薬及び投与計画を確認する重要かつ未対処の要求が当該技術分野において残っていることは明らかである。本発明は、これらの要求を満たし、他の関連する利点を提供する。
【先行技術文献】
【特許文献】
【0014】
【特許文献1】国際公開第2006/060697号
【発明の概要】
【課題を解決するための手段】
【0015】
発明の要旨
したがって、本発明の一態様によれば、DPD阻害剤及び5−FU又は5−FUプロドラッグから選択される抗癌剤を含む組合せを用いて、癌患者の処置に付随する神経毒性を防止又は最小限にする方法であって、神経組織と非神経組織の両方におけるDPD活性を実質的に消失させるのに十分な用量でDPD阻害剤をまず投与する工程、及びその後に5−FU又は5−FUプロドラッグを投与する工程を含み、5−FU又は5−FUプロドラッグが、5−FU又は5−FUプロドラッグから生成される5−FUがDPD阻害剤よりもかなり過剰に患者中に存在するような用量で投与される、方法が提供される。
【0016】
本発明のこの態様の例示的一実施形態においては、DPD阻害剤は、約14〜40mg/m2又は約15〜40mg/m2又は約16〜40mg/m2の用量など、患者の神経組織と非神経組織の両方においてDPD活性を実質的に消失させるのに十分な用量で投与される。より具体的な実施形態においては、エニルウラシルの用量は約14〜30mg/m2又は約15〜30mg/m2又は約16〜30mg/m2である。更に別の具体的実施形態においては、エニルウラシルの用量は約14〜21mg/m2又は約15〜21mg/m2又は約16〜21mg/m2である。
【0017】
別の例示的一実施形態においては、5−FU又は5−FUプロドラッグは、DPD阻害剤が投与されてから約11〜16時間後に投与される。
【0018】
更に別の例示的一実施形態においては、5−FU又は5−FUプロドラッグは、DPD阻害剤が投与されてからDPD阻害剤の少なくとも約3〜5排出半減期が経過したときに投与される。
【0019】
別の例示的一実施形態においては、DPD阻害剤は、患者の神経及び非神経組織におけるDPD活性を患者におけるベースラインDPD活性の5%未満に低下させるのに十分な用量で投与される。
【0020】
更に別の例示的一実施形態においては、5−FU又は5−FUプロドラッグは、その投与時に5−FU又はプロドラッグから生成される5−FUがDPD阻害剤の少なくとも10倍過剰で患者中に存在するような用量で投与される。
【0021】
本発明に使用される例示的5−FUプロドラッグとしては、5−フルオロウリジン、5−フルオロシチジン、5−フルオロ−2−デオキシウリジン、5−フルオロ−2−デオキシシチジン、5’−デオキシ−4’,5−フルオロウリジン、及び5−フルオロアラビノシルウラシル、5’−デオキシ−5−フルオロウリジン、1−(2−テトラヒドロフラニル)−5−フルオロウラシル、1−C1〜8アルキルカルバモイル−5−フルオロウラシル誘導体、1−(2−テトラヒドロフリル)−5−フルオロウラシル、5’−デオキシ−5−フルオロ−N−[(ペンチルオキシ)カルボニル]−シチジン(カペシタビン)、又は5−FUにin vivoで変換される化合物からなる、リン酸エステルを含めた5’−エステルが挙げられるが、それだけに限定されない。
【0022】
好ましい一実施形態においては、本発明の方法に使用される抗癌剤は5−FU又はカペシタビンである。
【0023】
一般に、本発明の方法に有用であるDPD阻害剤としては、不可逆的DPD阻害剤が挙げられるが、それだけに限定されない。例えば、ある種の例示的DPD阻害剤は、5−置換ウラシル化合物又はそのプロドラッグを含む。より具体的な一実施形態においては、DPD阻害剤は、ハロゲン原子、C2〜4アルケニル基、ハロゲンで置換されたC2〜4アルケニル基、C2〜6アルキニル基、ハロゲンで置換されたC2〜6アルキニル基、シアノ基、C1〜4アルキル基、又はハロゲンで置換されたC1〜4アルキル基で、5位が置換されたウラシル化合物を含む。別の一具体的実施形態においては、DPD阻害剤は、エニルウラシル、5−プロパ−1−イニルウラシル、5−シアノウラシル、5−プロパ−1−イニルウラシル、5−ブロモエチニルウラシル、5−(1−クロロビニル)ウラシル、5−ヨードウラシル、5−(2−ブロモビニル)ウラシル、(E)−5−(2−ブロモビニル)ウラシル、5−ヘキサ−1−イニルウラシル、5−ビニルウラシル、5−トリフルオロウラシル、5−ブロモウラシル及び5−(2−ブロモ−1−クロロビニル)ウラシルからなる群から選択されるウラシル化合物を含む。
【0024】
本発明の好ましい一実施形態においては、DPD阻害剤は、エニルウラシル又はそのプロドラッグである。
【0025】
別の好ましい一実施形態においては、DPD阻害剤はエニルウラシルであり、抗癌剤は5−FUである。
【0026】
更に別の好ましい一実施形態においては、DPD阻害剤はエニルウラシルであり、抗癌剤はカペシタビンである。
【0027】
例示的一実施形態においては、DPD阻害剤はエニルウラシルであり、抗癌剤は5−FUであり、エニルウラシルは約16〜40mg/m2の用量又は本明細書に記載の別のDPD阻害剤用量若しくは範囲で投与され、5−FUはその約11〜16時間後に約15〜50mg/m2の用量で投与される。
【0028】
別の例示的一実施形態においては、DPD阻害剤はエニルウラシルであり、抗癌剤は5−FUプロドラッグであり、エニルウラシルは約16〜40mg/m2の用量又は本明細書に記載の別のDPD阻害剤用量若しくは範囲で投与され、5−FUプロドラッグはその約11〜16時間後に約40〜150mg/m2の用量で投与される。
【0029】
別の例示的一実施形態においては、DPD阻害剤はエニルウラシルであり、抗癌剤は5−FUであり、エニルウラシルは約16〜40mg/m2の用量又は本明細書に記載の別のDPD阻害剤用量若しくは範囲で投与され、エニルウラシルが投与されてからエニルウラシルの少なくとも約3〜5排出半減期が経過したときに5−FUが約15〜50mg/m2の用量で投与される。
【0030】
別の例示的一実施形態においては、DPD阻害剤はエニルウラシルであり、抗癌剤は5−FUプロドラッグであり、エニルウラシルは約16〜40mg/m2の用量又は本明細書に記載の別のDPD阻害剤用量若しくは範囲で投与され、エニルウラシルが投与されてからエニルウラシルの少なくとも約3〜5排出半減期が経過したときに5−FUプロドラッグが約40〜150mg/m2の用量で投与される。
【0031】
別の例示的一実施形態においては、DPD阻害剤はエニルウラシルであり、抗癌剤は5−FUであり、エニルウラシルは患者の神経及び非神経組織におけるDPD活性を患者におけるベースラインDPD活性の5%未満に低下させるのに十分な用量で投与され、5−FUはその約11〜16時間後に約15〜50mg/m2の用量で投与される。
【0032】
別の例示的一実施形態においては、DPD阻害剤はエニルウラシルであり、抗癌剤は5−FUプロドラッグであり、エニルウラシルは患者の神経及び非神経組織におけるDPD活性を患者におけるベースラインDPD活性の5%未満に低下させるのに十分な用量で投与され、5−FUプロドラッグはその約11〜16時間後に約40〜150mg/m2の用量で投与される。
【0033】
別の例示的一実施形態においては、DPD阻害剤はエニルウラシルであり、抗癌剤は5−FU又は5−FUプロドラッグであり、エニルウラシルは約16〜40mg/m2又は本明細書に記載の別のDPD阻害剤用量若しくは範囲の用量で投与され、5−FU又は5−FUプロドラッグはその約11〜16時間後に、5−FU又は5−FUプロドラッグから生成される5−FUがDPD阻害剤の少なくとも10倍過剰で患者中に存在するような用量で投与される。
【0034】
本発明の別の一態様によれば、DPD阻害剤及び5−FU又は5−FUプロドラッグを含む徐放性経口医薬製剤であって、製剤を患者に投与した後、5−FU又は5−FUプロドラッグは、DPD阻害剤が放出された後少なくとも約11〜16時間まで実質的に放出されず、その放出後に5−FU又はプロドラッグから生成される5−FUが、患者に残留するDPD阻害剤の少なくとも約10倍過剰で患者中に存在する、徐放性経口医薬製剤が提供される。
【0035】
本発明のこれら及び他の態様は、以下の詳細な説明及び添付の図を参照することによって明らかになるであろう。本発明の種々の態様をより具体的に記載するのに本明細書で引用する特許及び他の文書は、参照によりその全体が本明細書に援用される。
【図面の簡単な説明】
【0036】
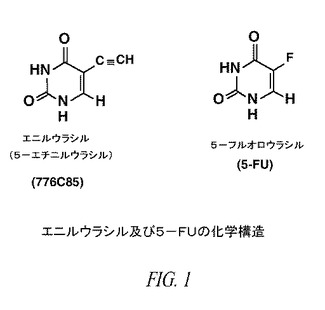
【図1】図1は、エニルウラシル及び5−FUの化学構造を示す。
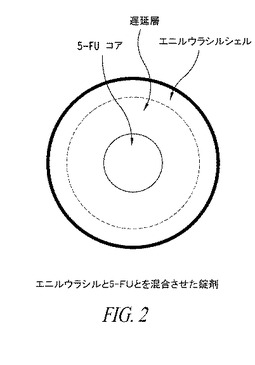
【図2】図2は、エニルウラシル及び5−FUを含む錠剤の形の例示的経口徐放製剤を示す。
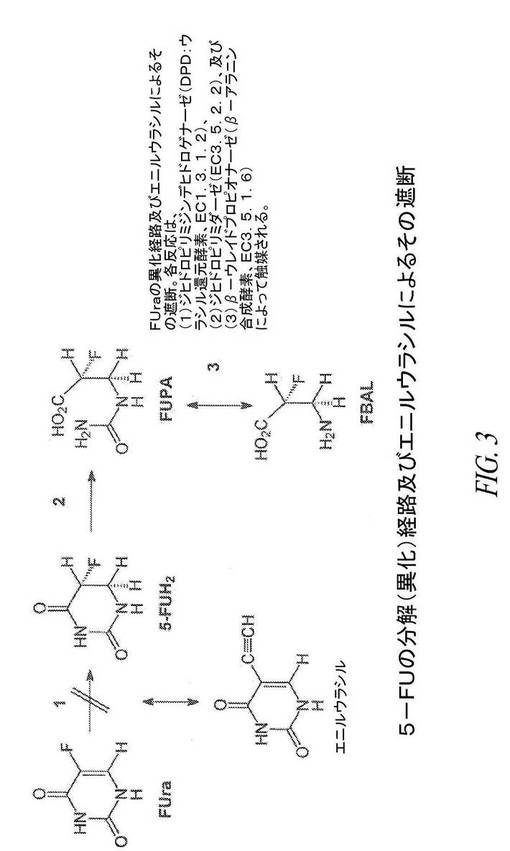
【図3】図3は、5−FU異化経路、及びエニルウラシルによるその遮断を示す。
【発明を実施するための形態】
【0037】
国際公開第2006/060697号に記載のように、5−FU及びプロドラッグから生成される5−FUよりも過剰な比のエニルウラシルなどのDPD阻害剤は、恐らくは代謝的活性化ステップの1つ以上を阻害することによって、その抗腫瘍活性を損ない得る。したがって、5−FU又はプロドラッグから生成される5−FUレベルが、5−FU又は5−FUプロドラッグが患者に投与された時点でDPD阻害剤レベルよりも十分過剰であるようにすることによって、DPD阻害剤が5−FU又は5−FUプロドラッグの抗腫瘍活性を阻害し得る程度が有利に最小化され、それによってこれらの薬剤の抗腫瘍効果が改善される。したがって、エニルウラシルなどの不可逆的DPD阻害剤は、DPDを効果的に不活性化する最低用量で投与されるべきであり、その投与時に5−FUがDPD阻害剤よりも過剰に存在するように、DPDに結合していない余分なDPD阻害剤がある程度除去されるように十分な時間が経過すべきである。
【0038】
この重要な知見に基づいて、臨床試験が開始され、患者におけるDPD活性を全身的に消失させるのに十分であると考えられる5mg用量のエニルウラシルが癌患者に投与された。次いで、5−FUが12〜24時間後に30〜160mgの用量で投与された。しかし、予想外に、この処置を受けた41名の患者の大多数が、軽度から重度の何らかの神経毒性を経験し、主な神経学的症候は、運動失調(不安定歩行)、神経障害、錯乱、めまい及び不明瞭発語であった。
【0039】
したがって、本発明は、神経組織と非神経組織の両方におけるDPDを十分に除去する投薬及びタイミングパラメータを適切に選択することによってこの神経毒性を防止又は最小化することができ、同時に5−FU又は5−FUプロドラッグが、その投与時に、残留DPD阻害剤よりも十分過剰であるようにすることによって、5−FU又はプロドラッグから生成される5−FUの抗腫瘍活性を阻害しない、方法に関する。
【0040】
本明細書に記載の方法は、限定的なものではない実例として、乳癌、肺癌、結腸癌、膵癌、胃癌、膀胱癌、腎臓癌、頭頚部癌、食道癌、肝細胞癌並びにすべての悪性白血病及びリンパ腫を含めて、5−FU及び/又は5−FUプロドラッグが活性である本質的にあらゆる癌タイプ(例えば、あらゆる5−FU反応性癌タイプ又は5−FUプロドラッグ反応性癌タイプ)の処置に適用可能である。さらに、本発明は5−FU及び5−FUプロドラッグの抗腫瘍効果を改善するので、5−FUに対して以前は望ましい反応性を示さないこともあった癌タイプが、本明細書に記載の方法によって投与されたときに改善された反応性を示すこともある。
【0041】
この開示に鑑みて、本明細書に記載の方法に使用することができ、患者の神経組織と非神経組織の両方におけるDPD活性を十分に阻害するのに十分なレベルでDPD阻害剤が確実に投与されるようにし、その投与時における5−FU又は5−FUプロドラッグのレベルが治療有効量であり、患者におけるDPD阻害剤レベルよりも十分過剰であり5−FU抗腫瘍活性の阻害を最小化又は解消するようにする、複数の投与及び投薬スケジュールが存在することが当業者には理解されるであろう。かかる投与及び投薬スケジュールはすべて本発明の範囲内とみなされる。
【0042】
本発明の例示的一実施形態においては、DPD阻害剤は、患者の神経組織と非神経組織の両方におけるDPD活性を実質的に消失させるのに十分な用量でそれを必要とする患者にまず投与され(すなわち、前投与され)、続いて5−FU又は5−FUプロドラッグが投与される。「実質的に消失させる」とは、患者の神経組織と非神経組織の両方におけるDPD活性レベルが、DPD阻害剤の投与前の患者におけるDPD活性のベースラインレベルの10%未満、好ましくは5%未満に低下することを意味する。患者のDPD活性のベースラインレベルは、患者から採取された生物試料において公知技術を使用して容易に決定することができる(例えば、Baker et al.,J Clin Oncol 18:915−926 2000;Schilsky et al.,J Clin Oncol 4:1450−7,1998)。しかし、循環血球などの非神経組織におけるDPD阻害の分析は、神経組織におけるDPD阻害の程度を過大評価するおそれがある。
【0043】
少なくとも1種類のDPD阻害剤をまず投与して、神経組織と非神経組織の両方において患者のDPD活性を実質的に消失させた後、次いで、DPD阻害剤が排出によって患者から完全にではないが実質的に除去されるのに十分な時間が経過した後に5−FU若しくは5−FUプロドラッグ又はその組合せを患者に投与する。この点について、ある実施形態においては、新規合成されるDPDを不活性化するために、低レベルのDPD阻害剤が5−FU投与まで及び/又は投与中の系内に残留することが望ましい場合もある。
【0044】
DPD阻害剤と5−FU又は5−FUプロドラッグの投与間の時間的遅延は変動し得る。ただし、5−FU又は5−FUプロドラッグの投与時には、それはその時点で患者に残留するDPD阻害剤レベルよりもかなり過剰に患者中に存在する。例示的一実施形態においては、5−FU又は5−FUプロドラッグは、5−FU又はプロドラッグから生成される5−FUのレベルが、患者に残留するDPD阻害剤よりも少なくともモル過剰で、例えば、5−FU又は5−FUプロドラッグが投与された時点で患者に残留するDPD阻害剤のレベルの少なくとも約2倍、少なくとも約3倍、少なくとも約5倍、少なくとも約10倍、少なくとも約25倍、少なくとも約50倍、又は少なくとも約100倍過剰で、患者中に存在するような用量で投与される。当業者は、本明細書に記載の実施形態に従ってDPD阻害剤に対して患者における5−FU又はプロドラッグから生成される5−FUの過剰レベルを計算及び/又は決定するのに幾つかの公知の利用可能な技術のいずれかを使用できることを認識するであろう。かかる技術としては、例えば、HPLC、LC−MS、ELISAなどが挙げられる。上述したように、5−FU又はプロドラッグから生成される5−FUが、5−FU又は5−FUプロドラッグが投与された時点で患者におけるDPD阻害剤のレベルよりも十分過剰に存在するようにすることによって、DPD阻害剤による5−FU又は5−FUプロドラッグの抗腫瘍性との干渉が最小化され、それによって5−FU又は5−FUプロドラッグの効力が改善される。
【0045】
本発明の更なる実施形態においては、5−FU又は5−FUプロドラッグは、DPD阻害剤が投与されてからDPD阻害剤の少なくとも約1、2、2.5、3、5、7、10、14又は21排出半減期が経過した後にのみ患者に投与される。あるDPD阻害剤の排出半減期は測定されており、測定されていない場合には、排出半減期は、周知の確立されたガスクロマトグラフィー/質量分析及びHPLC法によって容易に測定することができる(Baker et al.,J Clin Oncol 18:915−926 2000;Schilsky et al.,J Clin Oncol 4:1450−7,1998で言及されている)。ヒトにおけるエニルウラシルの排出半減期は約3.5時間と報告されているが(例えば、Baker et al.,J Clin Oncol 18:915−926 2000;Ochoa et al.,Ann Oncol 11:1313−22,2000)、DPD阻害剤の半減期は用量依存的であり得る。この用量依存性は、DPD阻害剤と5−FU又は5−FUプロドラッグの投与間の適切な時間的遅延を決定するときに考慮すべきである。
【0046】
エニルウラシルをDPD阻害剤として使用する本発明のある実施形態では、5−FU又は5−FUプロドラッグの投与前に排出によってエニルウラシルレベルが十分に低下するように、5−FU又は5−FUプロドラッグは、エニルウラシル投与から少なくとも約3時間、約6時間、約8時間、約11時間、約16時間、約20時間、約36時間、約48時間又は約72時間後に投与される。本発明のある関連実施形態においては、5−FU又は5−FUプロドラッグは、エニルウラシル投与から約11〜16時間、約8〜20時間、約6〜36時間、約3〜48時間又は約3〜72時間後に投与される。本発明の更に別の実施形態においては、5−FU又は5−FUプロドラッグは、その投与時に、患者におけるエニルウラシルと5−FUの比が約1:10、約1:5、約1:4又は約1:3未満になるまで投与されない。言うまでもなく、これらの範囲及び比は本質的に説明のためのものであって、必要に応じて、又は特定の投薬スケジュールに応じて、変わり得ることが理解されるであろう。ただし、5−FU又は5−FUプロドラッグが投与されるときにはエニルウラシルの存在は最小限であり、又は存在せず、さらに、DPD活性は、5−FU又は5−FUプロドラッグが投与される時点で神経組織と非神経組織の両方において所望の程度まで実質的に消失している。
【0047】
本発明によって使用されるDPD阻害剤は、好ましくは、DPDを不可逆的に不活性化するものである。したがって、エニルウラシルなどの阻害剤は、酵素を不活性化し、酵素に共有結合していない余分な阻害剤は、5−FU又は5−FUプロドラッグが投与される前にある程度除去される。例示的な不可逆的DPD阻害剤としては、5−置換ウラシル化合物、特にハロゲン原子、ハロゲンで場合によっては置換されたC2〜4アルケニル基(例えば、ビニル)(例えば、2−ブロモビニル、1−クロロビニル又は2−ブロモ−1−クロロビニル)、ハロゲン原子で場合によっては置換されたC2〜6アルキニル基、シアノ基、又はハロゲンで置換されたC1〜4アルキル基(例えば、トリフルオロメチル)で、5位が置換されたウラシル化合物を含むDPD阻害剤又はそのプロドラッグが挙げられるが、それだけに限定されない。
【0048】
本発明のより特別な実施形態においては、DPD阻害剤は、エニルウラシル、5−プロパ−1−イニルウラシル、5−シアノウラシル、5−プロピニルウラシル、5−ブロモエチニルウラシル、5−(1−クロロビニル)ウラシル、5−ヨードウラシル、5−(1−ブロモビニル)ウラシル、(E)−5−(2−ブロモビニル)ウラシル、5−ヘキサ−1−イニルウラシル、5−ビニルウラシル、5−トリフルオロウラシル、5−ブロモウラシル及び5−(2−ブロモ−1−クロロビニル)ウラシル、又はそのプロドラッグからなる群から選択される。
【0049】
別の例示的一実施形態においては、DPD阻害剤は5−ブロモビニルウラシルのプロドラッグであり、例示的一化合物は(BV−araU又はソリブジンとも称される)化合物1−β−D−アラビノフラノシル−(E)−5−(2−ブロモビニル)ウラシルである。これに関するある例示的プロドラッグ化合物は、例えば、米国特許第4,386,076号に記載されており、その開示を参照により本明細書に援用する。
【0050】
本発明の好ましい一実施形態においては、DPD阻害剤は、エニルウラシル若しくは5−エチニル−2(1H)−ピリミジノン(4−酸素のないエニルウラシル)などのエニルウラシルのプロドラッグ(Porter,et al.,Biochem.Pharmacol 47:1165−1171,1994)、エニルウラシルのヌクレオシド若しくはデオキシヌクレオシド誘導体、エニルウラシルにin vivoで変換される化合物、及び/又は失活剤にin vivoで変換されるDPD失活剤の誘導体である。例として、かかる化合物としては、上記5−置換ウラシル化合物に対応する核酸塩基を含むヌクレオシド誘導体、例えば、リボース、2’−デオキシリボース、2’,3’−ジデオキシリボース、アラビノース又は別の開裂可能な糖部分を含むヌクレオシド誘導体が挙げられ、ハロゲンなどの2’若しくは3’置換基又はエステルなどの5’置換基を更に含むことができる。かかるヌクレオシド誘導体のより特別な例としては、1−(β−D−アラビノフラノシル)−5−プロパ−1−イニルウラシル及び2’,3’−ジデオキシ−5−エチニル−3’−フルオロウリジンが挙げられる。
【0051】
やはり本発明によって使用することができる多数の5−FUプロドラッグが知られている。5−FUのプロドラッグは、in vivoで5−フルオロウラシルに代謝される化合物であり、実例として、5−フルオロウリジン、5−フルオロシチジン、5−フルオロ−2−デオキシウリジン、5−フルオロ−2−デオキシシチジン、5−フルオロアラビノシルウラシル、及びリン酸エステルを含めたその5’−エステルが挙げられる。別の例示的化合物としては、5’−デオキシ−4’,5−フルオロウリジン、5’−デオキシ−5−フルオロウリジン、1−(2−テトラヒドロフラニル)−5−フルオロウラシル、1−C1〜8アルキルカルバモイル−5−フルオロウラシル誘導体、1−(2−テトラヒドロフリル)−5−フルオロウラシル、Ftorafur(テガフール、アジア諸国で広く用いられる経口5−FUプロドラッグ)、及び5’−デオキシ−5−フルオロ−N−[(ペンチルオキシ)カルボニル]−シチジン(カペシタビン、Roche Laboratories Inc.によってXeloda(登録商標)として市販されている)、又は5−FUにin vivoで変換される化合物が挙げられる。
【0052】
この開示に鑑みて、本発明の方法は、所望のどんなタイプ、期間及び投薬特性の投与計画でも含み得ることが理解されるであろう。ただし、投与計画は、5−FUに付随する神経毒性が防止又は最小化されるように、さらに、5−FU又は5−FUプロドラッグが、5−FU又は5−FUプロドラッグが投与された時点で患者に残留するDPD阻害剤のレベルよりも十分過剰に存在するように、適切に選択される。
【0053】
本発明のある特に好ましい実施形態においては、本明細書に記載の方法は、5−FUと組み合わせたDPD阻害剤エニルウラシルの投与を利用する。例えば、例示的実施形態においては、毎週又は5日投薬スケジュールを含む投与計画を用いることができ、エニルウラシルは5−FUの前夜に投与され、5−FUは1日/週又は1回/日で5日間のみ投与される。
【0054】
本明細書に記載のように、神経組織と非神経組織の両方におけるDPD活性を実質的に阻害するのに十分な量が患者に投与されるのであれば、エニルウラシルの適切な用量は変わり得ることが理解されるであろう。ある実施形態においては、例えば、エニルウラシルは、好ましくは、約14〜40mg/m2又は約15〜40mg/m2又は約16〜40mg/m2の用量で投与される。より具体的な実施形態においては、エニルウラシルの用量は約14〜30mg/m2又は約15〜30mg/m2又は約16〜30mg/m2である。更に別の具体的実施形態においては、エニルウラシルの用量は約14〜21mg/m2又は約15〜21mg/m2又は約16〜21mg/m2又は約16〜25mg/m2又は約12〜35mg/m2である。本発明の更に別の実施形態においては、エニルウラシルは、約14〜50mg/m2、約15〜50mg/m2、約16〜50mg/m2、約20〜50mg/m2又は約30〜50mg/m2で投与することができる。
【0055】
言うまでもないが、好ましくは、本発明によって患者に投与するために選択されるDPD阻害剤投与量は、患者の神経組織と非神経組織の両方におけるDPD活性が実質的に消失するようにするのに少なくとも十分なものである。さらに、本明細書に記載の本発明の種々の実施形態に関連して、上記及び本明細書に記載のDPD阻害剤用量範囲のどれでも使用できることが理解されるであろう。
【0056】
本発明のある別の実施形態においては、5−FUはエニルウラシルから約11〜16時間後に投与され、それによって、エニルウラシルと5−FUの高い比に起因する抗腫瘍活性の低下を回避するために要求されるように、エニルウラシルと5−FUの比が確実に1.0をかなり下回るようになる。
【0057】
別の好ましい実施形態においては、エニルウラシルは約16〜40mg/m2又は本明細書に記載の別のDPD阻害剤用量若しくは範囲で投与され、5−FUは、約3〜5エニルウラシル排出半減期が経過した後にのみ投与される。
【0058】
更に別の好ましい実施形態においては、エニルウラシルは約16〜40mg/m2又は本明細書に記載の別のDPD阻害剤用量若しくは範囲で投与され、5−FUは、5−FU投与時にエニルウラシルと5−FUの比が≦1:10であるように十分な時間が経過した後にのみ投与される。
【0059】
14〜21mg/m2のエニルウラシル用量は以前に用いられたが(Schilsky et al.,J Clin Oncol 1998;16:1450−7;Baker et al.,J Clin Oncol 2000;18:915−26)、エニルウラシルは5−FU投与の1時間前又は5−FU投与と同時に投与された。それに対して、本発明は、独自に、5−FUの約11〜16時間前にエニルウラシルを投与し、ある好ましい実施形態においては、ヒト患者において認められた予想外の神経毒性を回避するために、神経組織と非神経組織の両方におけるDPDを不活性化するのに十分なDPD阻害剤用量を使用する。
【0060】
下記表1に示したエニルウラシル量を使用して16〜20mg/m2の範囲の用量を得ることができる。10.5時間(約3排出半減期)後に体内に残留するエニルウラシルの計算量も示す。したがって、毎週及び5日投薬スケジュールが約15〜50mg/m2の範囲の5−FU用量を使用する場合、エニルウラシルと5−FUの比は、5−FUが投与されるときには常に約1:6未満である。例えば、一般に使用される25mg/m2の5−FU用量がエニルウラシルから10.5時間後に投与されるときには、エニルウラシルと5−FUの比は約1:10以下である。
【0061】
【表1】
表1−表示BSAの患者に16〜20mg/m2を送達するエニルウラシルの用量、及び10.5時間(約3排出半減期)後に体内に残留するエニルウラシルの計算量。
【0062】
別の例では、1.9m2未満の患者にエニルウラシル30mgを投与した場合、及び≧1.9m2の患者にエニルウラシル45mgを投与した場合、これら2つの用量は、広範囲の体の大きさにわたって約16〜23mg/m2となる。さらに、エニルウラシルは無毒であり、最高50mg/日で7日間投与したときに安全であることが判明したので(Schilsky et al.,J Clin Oncol 4:1450−7,1998)、例示的投与スキームをより一層簡略にすることができる。例えば、すべての患者にエニルウラシル40mgを投与した場合、投薬範囲は約15〜31mg/m2を含む。さらに、すべての患者にエニルウラシル50mgを投与した場合、投薬範囲は約19〜39mg/m2である。
【0063】
したがって、別の実施形態においては、本発明によって使用されるエニルウラシル用量範囲は、有利には、約16〜23mg/m2、15〜31mg/m2及び19〜39mg/m2を含む。
【0064】
本発明の更に別の実施形態においては、エニルウラシルと5−FUの投与の時間間隔は、約11〜16時間、約8〜20時間、約6〜36時間、約3〜48時間又は約3〜72時間とすることができる。
【0065】
別の実施形態においては、少なくとも約3〜5、約2.5〜7、約2〜10、約1〜14又は1〜21エニルウラシル排出半減期が5−FU投与前に経過する。
【0066】
更なる実施形態においては、エニルウラシルは、5−FUが投与される前日に投与され、又は5−FUが投与される複数日前に投与される。
【0067】
更に別の実施形態においては、5−FUは、特に毎週及び5日投薬スケジュールで、約15〜40mg/m2、10〜50mg/m2、5〜60mg/m2又は5〜70mg/m2の用量で投与される。
【0068】
更なる実施形態においては、5−FUプロドラッグは、特に毎週及び5日投薬スケジュールで、約20〜60mg/m2、15〜80mg/m2、10〜100mg/m2又は5〜150mg/m2の用量で投与される。
【0069】
更なる実施形態においては、5−FUは、特に長期療法では、8、10、12、14又は16時間ごとに約0.8〜1.2mg/m2又は0.3〜1.8mg/m2の用量で投与される。
【0070】
本発明の更なる実施形態においては、本発明によって使用される5−FU投与計画は、毎週スケジュール、5日間スケジュール、毎日スケジュール、5−FUが指定日に複数回投与される毎日スケジュール、5−FUがエニルウラシル投与後1日を超えて投与され、エニルウラシルが5−FU療法中5−FU前に毎日、隔日又は3日ごとに投与される毎日スケジュール、5−FUがエニルウラシル投与から1日以上後に複数回投与され、エニルウラシルが5−FU療法中5−FU前に毎日、隔日又は3日ごとに投与される毎日スケジュールである。
【0071】
例示的一実施形態においては、エニルウラシルは、5−FU前夜に、約16〜40mg/m2の用量で、又は本明細書に記載の別のDPD阻害剤用量範囲で投与することができ、あるいは、夕方の5−FU投与に続いて朝に投与することができる。約20から30mg/m2の例示的5−FU用量をこれらのスケジュールで使用する場合(Levin et al.,Invest New Drugs 18:383−90,2000;Schilsky et al.,J Clin Oncol 4:1450−7,1998;Guo et al.,Cancer Chemother Pharmacol 52:79−85,2003)、例えば、5−FUはエニルウラシルよりも常に実質的に過剰であるべきである。
【0072】
別の例示的一実施形態においては、28日b.i.d.(28日間毎日2回)スケジュールを使用することができる。このタイプの投与計画は、5−FUが1mg/m2でしか投与されないので、異なる手法を必要とすることが理解されるであろう(例えば、Baker et al.,J Clin Oncol 2000;18:915−26を参照されたい)。したがって、エニルウラシルがこの低5−FU用量よりも過剰に存在しないように注意しなければならない。しかし、高用量のエニルウラシルはDPDを長期間不活性化された状態に維持するので、エニルウラシルは、例えば、2日ごと、場合によっては3日ごとに投与することができる。この戦略は、各エニルウラシル投与間にエニルウラシルと5−FUの比が各後続の5−FU投与につれて確実に低下するようにする。
【0073】
別の例示的一実施形態においては、エニルウラシル(又は別のDPD阻害剤)がまず投与され、次いで5−FU又は5−FUプロドラッグの複数回投与がその後所望の時点で投与され、その後エニルウラシルが場合によっては再度投与される。例えば、例示的一実施形態においては、エニルウラシルがまず投与され、次いで複数の5−FU用量がその約8、10、12、14又は16時間後の例示的時点において投与され、必要に応じて、その後エニルウラシルが場合によっては再度投与され、このサイクルが繰り返される。
【0074】
本発明は、更なる特徴として、単一製剤として一緒に、又は本発明に従って別々の時点で投与される個別の製剤として、少なくとも1種類の薬学的に許容される担体又は賦形剤を含み、DPD阻害剤及び/又は5−FU若しくは5−FUプロドラッグを更に含む、医薬製剤を含む。担体又は賦形剤は、製剤の他の成分と適合するという意味で「薬学的に許容され」、患者に無害である。製剤としては、例えば、経口、直腸、経鼻、(頬及び舌下を含めた)局所、膣及び(皮下、筋肉内、静脈内及び皮内を含めた)非経口投与に適合したものが挙げられる。製剤は、好都合には単位剤形として提供することができ、製薬技術分野で周知の任意の方法によって調製することができる。かかる方法は、活性成分を1種類以上の副成分を構成する担体と会合させる段階を含む。一般に、製剤は、活性成分を液状担体若しくは微粉担体又はその両方と均一かつ十分に会合させ、次いで必要に応じて生成物を成形することによって調製される。
【0075】
本発明による製剤は、本質的に任意の利用可能な技術を使用して調製及び/又は投与することができる。経口投与に適合した本発明の製剤は、例えば、各々が所定量の活性成分を含むカプセル剤、カシェ剤、錠剤などの個別単位として、散剤若しくは顆粒剤として、水性液体若しくは非水性液体中の溶液剤若しくは懸濁液剤として、又は水中油型乳剤若しくは油中水型乳剤として提供することができる。活性成分は、ボーラス、舐剤又はペーストとしても提供することができる。経口投与は、典型的に好ましい投与経路である。
【0076】
錠剤は、例えば、圧縮又は成形によって製造することができ、1種類以上の副成分を場合によっては含むことができる。圧縮錠剤は、結合剤(例えば、ポビドン、ゼラチン、ヒドロキシプロピルメチルセルロース)、滑沢剤、不活性希釈剤、防腐剤、崩壊剤(例えば、デンプングリコール酸ナトリウム、架橋ポビドン、架橋カルボキシメチルセルロースナトリウム)界面活性剤又は分散剤と場合によっては混合することができる、散剤、顆粒剤などの易流動性活性成分を適切な機械で圧縮して調製することができる。成形錠剤は、不活性希釈液で湿らせた粉末化合物の混合物を適切な機械で成形することによって製造することができる。錠剤は、場合によっては、被覆することができ、又は刻み目をつけることができ、例えば所望の放出プロファイルを与えるために様々な割合のヒドロキシプロピルメチルセルロースを使用して、その中の活性成分を制御放出するように処方することができる。
【0077】
口内局所投与用製剤としては、例えば、風味付けされた基剤、通常はスクロースとアラビアゴム又はトラガカント中に活性成分を含むロゼンジ、ゼラチンとグリセリン、スクロースとアラビアゴムなどの不活性基剤中に活性成分を含むトローチ剤、及び適切な液状担体中に活性成分を含む洗口剤が挙げられる。直腸投与用製剤は、例えば、例えばカカオ脂又はサリチラートを含む、適切な基材を含む坐剤として提供することができる。膣内投与用製剤は、例えば、活性成分に加えて、適切であることが当該技術分野で知られている担体を含有する、膣坐剤、タンポン、クリーム剤、ゲル剤、ペースト剤、泡剤又は噴霧剤として提供することができる。
【0078】
非経口投与用製剤としては、例えば、水性及び非水性の等張性無菌注射液が挙げられる。等張性無菌注射液は、抗酸化剤、緩衝剤、静菌剤、及び製剤を対象レシピエントの血液と等張にする溶質、並びに懸濁化剤及び増粘剤を含み得る水性及び非水性の無菌懸濁液を含むことができる。製剤は、単位用量又は複数用量の密閉容器、例えばアンプル及びバイアルとして提供することができ、使用直前に無菌液状担体、例えば注射用水の添加のみを必要とするフリーズドライ(凍結乾燥)状態で貯蔵することができる。即時注射液及び懸濁液は、上記種類の無菌散剤、顆粒剤及び錠剤から調製することができる。
【0079】
典型的には、1種類以上の活性薬剤を含む液剤は、好ましくはpH7から11、一般に9.5から10.5に緩衝される。ある単位投与製剤としては、活性成分の1日用量若しくは単位、上述した1日の分割用量(sub−dose)、又はその適切な一部分を含むものが挙げられる。
【0080】
本明細書に記載のDPD阻害剤及び5−FUプロドラッグを製造する方法は公知であり、従来の方法を使用して実施することができる。例えば、上記DPD阻害剤は、5−エチニルウラシルの調製の場合はHeterocycl.Chem.19(3)463−4(1982)、5−(2−ブロモビニル)ウラシル、5−ブロモエチニルウラシル及び5−(2−ブロモ−1−クロロビニル)ウラシルの調製の場合はJ.Chem.Soc.Perkin Trans.1(16),1665−70(1981)、5−シアノ−ウラシルの調製の場合はNucleic Acid Chemistry,Vol.2,927−30(1978)、5−ビニルウラシルの調製の場合はNucleic Acids Research,1(1)105−7(1974)、5−トリフルオロメチルウラシルの調製の場合はZ.Chern 17(11)415−16(1977)、5−(1−クロロビニル)ウラシルの調製の場合はNucleic Acids Research 3(10),2845(1976)に記載の方法によって調製することができる。本発明のある別の化合物は、2’,3’−ジデオキシ−5−エチニル−3’−フルオロウリジンなどの3’−フルオロ−2’,3’−ジデオキシ−5−アルキニルウリジン化合物の調製の場合は欧州特許明細書第356166号、及び1−(b−D−アラビノフラノシル)−5−プロパ−1−イニルウラシルなどの5−アルキニルウラシルアラビノシドの調製の場合は欧州特許明細書第272065号に記載のプロセスに従って調製することができる。本発明に使用される化合物を製造するためのこれら及び他の合成技術が公知であり、利用可能である。
【0081】
一実施形態においては、本発明は、製剤成分を患者にその所望の投与量範囲内で所望の時間放出することができるようにDPD阻害剤と5−FU又は5−FUプロドラッグが一緒に投与される混合経口製剤を提供する。2成分の差次的徐放性送達は、公知技術及び材料を使用して実施することができる。例えば、一実施形態においては、例えば錠剤の形の経口製剤は、図2に説明用に示したように、3つの異なる層で構成することができる。外層は、即放処方のエニルウラシルを含むことができる。中間層は、本発明によって5−FU又は5−FUプロドラッグの放出を所望の程度遅らせる徐放性成分(例えば、徐放性緩衝剤)とすることができる。5−FU又は5−FUプロドラッグは、即放処方のコア層に位置する。DPD阻害剤及び5−FU又は5−FUプロドラッグは、本明細書に記載の適切な用量及び比で処方される。好ましい一実施形態においては、DPD阻害剤はエニルウラシルであり、5−FU又は5−FUプロドラッグは5−FU又はカペシタビンである。
【0082】
別の一実施形態においては、別の製剤は、5−FU又は5−FUプロドラッグを含むミクロスフェアなどの公知の送達ビヒクルを含むことができる。一実施形態においては、例えば、5−FU又は5−FUプロドラッグは、徐放性成分(例えば、徐放性崩壊緩衝剤)のシェル、及びDPD阻害剤を即時に放出する外層で被包することができる。好ましい一実施形態においては、DPD阻害剤はエニルウラシルであり、5−FU又は5−FUプロドラッグは5−FU又はカペシタビンである。例示的混合製剤のこれら及び別の例は、単一経口製剤においてDPD阻害剤と5−FU又は5−FUプロドラッグの送達の間を適切に遅延することができる公知技術を使用して、設計及び製造することができる。
【0083】
別の一実施形態においては、本明細書に記載の方法は、さらに、ロイコボリンの投与を含む。ロイコボリン又はロイコボリンの活性異性体であるIsovorinは、一般に、癌患者の処置に5−FUと併用される。これは、エニルウラシルと5−FUの上記投与計画に追加することもできる。ロイコボリンは、腫瘍を有するラット及び組織培養においてエニルウラシル及び5−FUの抗腫瘍効果を改善することが判明し(Cao et al.,Cancer Res 90:1507−1510,1993;Fischel et al.,Biochem Pharmacol 53:1703−1709,1997)、エニルウラシル及び5−FUを投与された患者に投与されてきた(Schilsky et al.,J Clin Oncol 4:1450−7,1998;Guo et al.,Cancer Chemother Pharmacol 52:79−85,2003)。ロイコボリンは、経口製剤に都合よく利用することもできる。
【0084】
本発明は、以下の非限定的実施例を考察することによって更に理解することができる。
【実施例】
【0085】
(実施例1)
5−FUと組み合わせたエニルウラシルの投与に付随する予想外の神経毒性、及びそれを防止又は最小限にする方法
国際公開第2006/060697号に記載の重要な機構的知見に基づく臨床試験が開始された。より具体的には、癌患者に5mg用量のエニルウラシルが投与され、その12〜24時間後に30〜160mg用量の5−FUが投与された。しかし、予想外に、この処置を受けた41名の患者の大多数は、軽度から重度の何らかの神経毒性を経験し、主な神経学的症候は、運動失調(不安定歩行)、神経障害、錯乱、めまい及び不明瞭発語であった。
【0086】
F−Balは、5−FUの主要な分解(異化)産物である。図3に示した経路によれば、DPDは、5−FUをジヒドロフルオロウラシル(5−FUH2)に変換し、ジヒドロフルオロウラシルは、α−フルオロ−β−ウレイドプロピオン酸(FUPA)、次いでF−Balに変換される。エニルウラシルは、DPDを不活性化することによってこの経路を遮断する。
【0087】
5−FU自体は神経毒性を生じず、したがって臨床試験患者で認められた神経毒性の直接的原因ではなかったが、研究によれば、5−FUの分解産物の一つであるF−Balはマウス、サル、ネコ及びイヌにおいて神経毒性を生じ得ることが判明した(Saif et al.,Anticancer Drugs 2001;12:525−31)。さらに、イヌにおける研究は、F−Balが神経毒性を生じ得る更なる証拠を与えた(Davis et al.,Biochem Pharmacol 1994;48:233−6)。例えば、イヌへの5−FUの静脈内投与は、低血中濃度の5−FUしか得られず、発作、筋肉振戦及び運動失調を誘発した。しかし、イヌをエニルウラシルで前処理すると、高血中濃度の5−FUを神経毒性なしに得ることができた。したがって、神経組織における5−FUの異化を適切に遮断することによって、エニルウラシルは神経毒性を抑制した。
【0088】
F−Balは5−FUに付随する神経毒性の原因であると思われ、エニルウラシルはF−Balの形成を防止するので、エニルウラシルで処置された臨床試験患者における神経毒性の高罹患率は、特に、患者に使用された5mg用量のエニルウラシルがそのDPDを実質的に消失させるのに十分であると考えられ、したがって神経系における神経毒性5−FU異化産物の形成を防止するはずであったことを考えると、全く予想外であった。
【0089】
しかし、臨床試験データを分析し、さらに、科学文献に記載されたことを考慮すると、特定用量のエニルウラシルは、患者の循環血球などの非神経組織におけるDPDを不活性化するのには十分かもしれないが(Schilsky et al.,J Clin Oncol 1998;16:1450−7)、神経組織におけるDPDを十分に不活性化するのには必ずしも十分ではないことが現在では理解される。例えば、ラットにおいては、肝臓及びひ臓、腸管粘膜、肺などの他の非神経組織のDPDの50%を不活性化するのに必要な用量の約6倍のエニルウラシルが脳内のDPDの50%を不活性化するのに必要である(Spector et al.,Biochem Pharmacol 1993;46:2243−8)。神経組織中のDPD酵素に接近し、それを阻害するエニルウラシルの能力はどういうわけか妨害されるので、神経組織内のDPDを不活性化するには、非神経組織に必要な用量よりも高用量のエニルウラシルが必要である。したがって、臨床試験で使用されたエニルウラシルの用量は、ヒト神経組織におけるDPD活性を阻害するのには不十分であったと思われる。その結果、5−FUは、神経組織において、患者における神経毒性を生じるF−Balを含めた神経毒性異化産物に異化されたと思われる。
【0090】
さらに、5−FU異化が非神経組織において阻害されるときには、5−FUは神経組織により接近する可能性がある。その結果、エニルウラシルの用量が非神経組織中のDPDを阻害するのには十分であるが、神経組織中のDPDを十分に阻害するのには低すぎる場合、5−FUは神経組織において選択的にF−Balに変換されることになる。したがって、非神経組織中のDPDを十分に阻害するのには十分であるが、神経組織では不十分であるエニルウラシル用量は、5−FUによって誘導される神経毒性を引き起こすおそれがある。
【0091】
この理論は、患者における神経毒性の発生率が、より高用量のエニルウラシルを投与した患者において減少したという観察によって強く支持される。患者に5−FU投与前にエニルウラシル5mgを投与した臨床試験では、41名の患者の大多数が5−FUによって誘導される神経毒性を経験した。それに対して、5−FU投与前にエニルウラシル20mgを投与した患者では、神経毒性の発生率は17名のうち2名(12%)に減少した(Guo XD,et al.Cancer Chemother Pharmacol 2003;52:79−85;Saif et al.,Anticancer Drugs 2001;12:525−31)。これら2名の患者の体格が大きいことは特に注目すべきである。その体表面積(BSA)は2.1m2及び2.5m2であった。したがって、エニルウラシルの20mg用量は、これらの患者にそれぞれ9.5mg/m2及び8.0mg/m2のエニルウラシルを送達した。重要なことには、その末梢血細胞のDPDは完全に不活性化された。したがって、この分析に基づいて、神経組織のDPDを十分不活性化して神経毒性を防止するには少なくとも9.5mg/m2を超えるエニルウラシル用量が必要であると思われる。さらに、12時間ごとの約11.5mg/m2のエニルウラシル用量は、それでも重篤な神経毒性の全発生率が6%という結果になる(Schilsky et al.,J Clin Oncol 2002;20:1519−26)。
【0092】
したがって、臨床における神経毒性を回避するためには、エニルウラシルの用量が十分に高く、好ましくは約12mg/m2又は14mg/m2又は15mg/m2又は16mg/m2を超え、より好ましくは約12〜21mg/m2又は14〜21mg/m2又は15〜21mg/m2又は16〜21mg/m2又は16〜25mg/m2又は15〜40mg/m2又は16〜40mg/m2であり、非神経組織と神経組織の両方におけるDPDを不活性化することが重要である。さらに、5−FUの抗腫瘍活性を最大にするためには、5−FUは、その投与時に、特許におけるエニルウラシルと5−FUの比が好ましくは約1:10、1:5又は1:3以下であるような用量で投与すべきである。しかし、5−FU投与時にエニルウラシルレベルが完全には除去されていないことが重要であり得る。例えば、ある実施形態においては、エニルウラシルが除去された後に出現する新規合成されたDPDを不活性化するのに幾らかのエニルウラシルが存在することが好ましい(Spector T,et al.Biochem Pharmacol 1993;46:2243−8;Heslin MJ et al.Cancer Chemother Pharmacol 2003;52:399−404:Keith B,et al.Clin Cancer Res 2002;8:1045−50)。
【0093】
上記のことから、本発明の特定の実施形態を説明のために本明細書に記載したが、本発明の精神及び範囲から逸脱せずに種々の改変を成し得ることが理解されるであろう。したがって、本発明は、添付の特許請求の範囲による以外は限定されない。
【技術分野】
【0001】
関連出願への相互参照
本願は、2009年10月14日に出願された米国仮特許出願第61/251,449号の米国特許法第119条(e)項の優先権を主張し、この米国仮特許出願は、本明細書中に参考として援用される。
【0002】
発明の背景
発明の分野
本発明は、一般に癌治療に関し、より詳細には、DPD阻害剤と5−FU及び/又は5−FUプロドラッグを併用して癌治療に付随する神経毒性を防止又は最小限にする方法に関する。
【背景技術】
【0003】
先行技術の説明
5−フルオロウラシル(5−FU)は、癌患者における固形腫瘍の処置に30年以上臨床的に使用されてきた(Ansfield et al.,Cancer 39:34−40,1977;Grem et al.,Cancer Treat Rep 71:1249−1264,1987;Chabner et al.,Cancer,Principles and Practice of Oncology,2nd Ed,pp287−328 Philadelphia,PA:J B Lippincott Co,1985)。5−FUは、代謝変換によって、DNA合成及びRNA機能を阻害する偽ウリジンヌクレオチド(例えば、FUMP、FUDP、FUTP)及びデオキシウリジンヌクレオチド(例えば、FdUMP、FdUDP、FdUTP)に活性化されなければならない(Meyers,Pharmacol Rev,33:1−15,1981;Dasher et al.,Pharmac Ther 48:189−222,1990に概説されている)。5−FUは、その天然の相当物であるウラシルとは、5位のフッ素置換しか異ならないので、癌患者においては容易に活性化される。残念ながら、ウラシルとのその構造上の類似性は、抗腫瘍活性を持たない生成物に急速かつ大規模に変換される原因にもなっている。この代謝過程は、異化と称される。5−FUは、酵素ジヒドロピリミジンデヒドロゲナーゼ(DPD:EC1312、ウラシル還元酵素)によって急速に異化される(Meyers,Pharmacol Rev,33:1−15,1981;Dasher et al.,Pharmac Ther 48:189−222,1990)。したがって、癌治療用5−FUの抗腫瘍効果は、抗腫瘍ヌクレオチドへの代謝変換(活性化)と無用な代謝産物への代謝変換(異化)との繊細なバランスに依拠している。
【0004】
さらに、幾つかの臨床上の問題が5−FUの代謝異化に起因して生じる。第一に、DPDレベルは、個体間で異なり(Fleming et al.,Cancer Res 52:2899−2902,1992;Grem et al.,Cancer Chemother Pharmacol 40:117−125,1997)、個体内でも1日の間に変わるので(Grem et al.,Cancer Chemother Pharmacol 40:117−125,1997;Harris et al.,Cancer Res 50:197−201,1990;Petit et al.,Cancer Res 48:1676−1679,1988)、所与の用量から生じる5−FU又はプロドラッグ由来の5−FUの全身的レベルが大きく変化し、したがって有効性及び毒性を予測することが極めて難しい。極端な場合、遺伝的にDPDが欠乏した患者は、「標準」治療量の5−FUで処置すると、激しい、時には致死的な毒性を経験する(Morrison et al.,Oncol Nurs Forum 24:83−88,1997に概説されている)。第二に、胃腸のDPDレベルの変化は(Ho et al.,Anticancer Res 6:781−784,1986;Naguib et al.,Cancer Res 45:5405−5412,1985;Spector et al.,Biochem Pharmacol 46:2243−2248,1993)、経口投与された5−FUの吸収を大きく変化させ(Christophidis et al.,Clin Pharmacokinetics 3:330−336,1978;Cohen et al.,Cancer Chemother Rep 58:723−731,1974;Finch et al.,Br J Clin Pharmacol 7:613−617,1979)、薬物の血しょう中濃度を予測不可能にし、望ましくない毒性や不十分な効力をもたらし得る。第三に、高レベルのDPDを含む腫瘍は、5−FU治療に反応しない可能性がある(Etienne et al.,J Clin Oncol 13:1663−1670,1995;Fischel et al.,Clin Cancer Res 1:991−996,1995)。
【0005】
最後に、F−Balなどの5−FU分解産物は、神経毒性(Okeda et al.,Acta Neuropathol 81:66−73,1990;Koenig et al.Arch Neurol 23:155−160,1970;Davis ST,et al.Biochem Pharmacol 1994;48:233−6;Saif MW,et al.Anticancer Drugs 2001;12:525−31に概説されている)、心毒性(et al.,Lancet 337:560,1991;Lemaire et al.,Br J Cancer 66:119−127,1992)、手掌足底紅斑異感覚症候群(手足症候群)(Hohneker,Oncology 12:52−56,1998)、及びGI毒性(Spector et al.,Cancer Res 55:1239−1241,1995)を生じるおそれがあり、抗腫瘍活性を阻害するように思える(Spector et al.,Cancer Res 55:1239−1241,1995;Cao,et al.,Pharmacol 59:953−960,2000)。
【0006】
DPDは、5−FUの分解(異化)において第1かつ律速段階である広範に分布する酵素である。研究によれば、DPDを阻害すると、血しょう中の5−FUの半減期が大きく増加する。DPDを不可逆的に不活性化するもの、及びDPDを可逆的に阻害するものを含めて、幾つかのDPD阻害剤が研究されている。例えば、エニルウラシル(5−エチニルウラシル、776C85)は、強力な不可逆的DPD失活剤である。DPD及び異化経路中の一連の酵素は、最終的に、5−FUをα−フルオロ−β−アラニン(F−Bal)に変換するので(Spector et al.,Drugs of The Future 1994;19:565−71;Paff et al.,Invest New Drugs 2000;18:365−71に概説されている)、エニルウラシルは、5−FU排出経路を異化から腎排せつに変更し、それによって5−FU排出半減期を10〜20分から4.5〜6.5時間に延長する(Adjei et al.,J Clin Oncol 2002;20:1683−91;Ochoa et al.,Ann Oncol 2000;11:1313−22;Baker,Invest New Drugs 2000;18:373−81;Baker et al.,J Clin Oncol 1996;14:3085−96;Guo et al.,Cancer Chemother Pharmacol 2003;52:79−85;Schilsky et al.,J Clin Oncol 1998;16:1450−7)。
【0007】
胃腸管における5−FU分解を防止することによって、エニルウラシルは5−FUの経口投与も可能にする(Baker et al.,J Clin Oncol 1996;14:3085−96)。さらに、エニルウラシルは、5−FU関連神経毒性(Saif MW,et al.Anticancer Drugs 2001;12:525−31に概説されているDavis et al.,Biochem Pharmacol 1994;48:233−6)及び手足毒性症候群(Schilsky et al.,J Clin Oncol 2002;20:1519−26)の原因であると思われる、F−Balなどの5−FU異化産物の形成を防止する。さらに、F−Balなどの5−FU異化産物は、5−FUの抗腫瘍活性を低下させると思われる(Cao et al.,Biochem Pharmacol 2000;59:953−60;Spector T,et al.Cancer Res 1995;55:1239−41 Spector et al.,Drugs of The Future 1994;19:565−71;Paff et al.,Invest New Drugs 2000;18:365−71)。
【0008】
さらに、DPDは患者中に異なるレベルで存在するので、DPDがエニルウラシルによって不活性化されると、5−FUの極めて変わりやすい非線形薬物動態が、高度に予測可能な線形になる(Baker,Invest New Drugs 2000;18:373−81に概説されている)。実際、エニルウラシルは、腫瘍を有する実験動物において、5−FUの抗腫瘍効果をかなり改善し、治療指数を増加させた(Baccanari et al.,Proc Natl Acad Sci USA 1993;90:11064−812;Cao et al.,Cancer Res 1994;54:1507−10)。
【0009】
エニルウラシルは、癌患者において第一相臨床試験で試験された(Levin et al.,Invest New Drugs 18:383−90,2000;Baker et al.,J Clin Oncol 18:915−926 2000;Schilsky et al.,J Clin Oncol 4:1450−7,1998に概説されている)。これらの試験では、エニルウラシルは、毒性を生じずに、DPD活性を極めて強力に消失させた。例えば、用量0.74mg/m2(合計約1mg)は、末梢血細胞中のDPDの90%超を長期間除去した。5−FUの排出半減期は、1回のエニルウラシルによって約10分から3.5時間に増加した。用量3.7mg/m2のエニルウラシルは、5−FUの半減期を4.5〜6.5時間に延長した。用量をさらに高くしても、明白な利点は得られなかった。
【0010】
続いて、2回の多施設第三相試験が結腸直腸癌患者において5−FUの10倍過剰のエニルウラシルを含む複合丸剤を使用して実施された。患者に10mg/平方メートル体表面積(mg/m2)エニルウラシル及び1mg/m2 5−FUを12時間ごとに28日間投与した。薬物を1週間停止した後、このサイクルを繰り返した。服薬遵守が問題なかった北米の試験の結果は、有望な抗腫瘍活性、高い耐容性、及びごくわずかの手足症候群を示したが、投薬計画は、エニルウラシルのない5−FU/ロイコボリンの標準投薬計画よりも抗腫瘍性の利点が少ない傾向にあった(Schilsky et al.,J Clin Oncol 2002;20:1519−26)。これらの結果の説明は、当時は明らかでなかった。
【0011】
特許文献1は、5−FUが対象に投与された時点で過剰のエニルウラシルが存在すると、5−FUの抗腫瘍活性がかなり低下するという重要な知見を記載している。したがって、5−FUの抗腫瘍活性を最大にするために、5−FU投与時に5−FUがエニルウラシルよりもかなり過剰に存在するように、低用量のエニルウラシルを5−FUのかなり前に投与することを提案している。さもないと、5−FUの抗腫瘍効果が損なわれるおそれがある。これらの結果は、5−FU投与時の5−FUに対するエニルウラシル比が10:1であった第三相試験における予想よりも低い抗腫瘍活性を説明するものである。
【0012】
したがって、臨床試験は、5mg用量のエニルウラシル、続いて30〜160mg用量の5−FUを12〜24時間後に癌患者に投与して開始された。予想外に、この処置を受けた41名の患者の大多数は、軽度から重度の何らかの神経毒性を経験し、主な神経学的症候は、運動失調(不安定歩行)、神経障害、錯乱、めまい及び不明瞭発語であった。
【0013】
神経毒性を防止又は最小化するために、5−FU及び5−FUプロドラッグの抗腫瘍効果及び治療指数を最大にするために、投薬の予測可能性を改善するために、さらに、5−FU及び5−FUプロドラッグを経口投与によって効果的に投与できるようにするために、5−FU及び5−FUプロドラッグと併用されるDPD阻害剤の最適な投薬及び投与計画を確認する重要かつ未対処の要求が当該技術分野において残っていることは明らかである。本発明は、これらの要求を満たし、他の関連する利点を提供する。
【先行技術文献】
【特許文献】
【0014】
【特許文献1】国際公開第2006/060697号
【発明の概要】
【課題を解決するための手段】
【0015】
発明の要旨
したがって、本発明の一態様によれば、DPD阻害剤及び5−FU又は5−FUプロドラッグから選択される抗癌剤を含む組合せを用いて、癌患者の処置に付随する神経毒性を防止又は最小限にする方法であって、神経組織と非神経組織の両方におけるDPD活性を実質的に消失させるのに十分な用量でDPD阻害剤をまず投与する工程、及びその後に5−FU又は5−FUプロドラッグを投与する工程を含み、5−FU又は5−FUプロドラッグが、5−FU又は5−FUプロドラッグから生成される5−FUがDPD阻害剤よりもかなり過剰に患者中に存在するような用量で投与される、方法が提供される。
【0016】
本発明のこの態様の例示的一実施形態においては、DPD阻害剤は、約14〜40mg/m2又は約15〜40mg/m2又は約16〜40mg/m2の用量など、患者の神経組織と非神経組織の両方においてDPD活性を実質的に消失させるのに十分な用量で投与される。より具体的な実施形態においては、エニルウラシルの用量は約14〜30mg/m2又は約15〜30mg/m2又は約16〜30mg/m2である。更に別の具体的実施形態においては、エニルウラシルの用量は約14〜21mg/m2又は約15〜21mg/m2又は約16〜21mg/m2である。
【0017】
別の例示的一実施形態においては、5−FU又は5−FUプロドラッグは、DPD阻害剤が投与されてから約11〜16時間後に投与される。
【0018】
更に別の例示的一実施形態においては、5−FU又は5−FUプロドラッグは、DPD阻害剤が投与されてからDPD阻害剤の少なくとも約3〜5排出半減期が経過したときに投与される。
【0019】
別の例示的一実施形態においては、DPD阻害剤は、患者の神経及び非神経組織におけるDPD活性を患者におけるベースラインDPD活性の5%未満に低下させるのに十分な用量で投与される。
【0020】
更に別の例示的一実施形態においては、5−FU又は5−FUプロドラッグは、その投与時に5−FU又はプロドラッグから生成される5−FUがDPD阻害剤の少なくとも10倍過剰で患者中に存在するような用量で投与される。
【0021】
本発明に使用される例示的5−FUプロドラッグとしては、5−フルオロウリジン、5−フルオロシチジン、5−フルオロ−2−デオキシウリジン、5−フルオロ−2−デオキシシチジン、5’−デオキシ−4’,5−フルオロウリジン、及び5−フルオロアラビノシルウラシル、5’−デオキシ−5−フルオロウリジン、1−(2−テトラヒドロフラニル)−5−フルオロウラシル、1−C1〜8アルキルカルバモイル−5−フルオロウラシル誘導体、1−(2−テトラヒドロフリル)−5−フルオロウラシル、5’−デオキシ−5−フルオロ−N−[(ペンチルオキシ)カルボニル]−シチジン(カペシタビン)、又は5−FUにin vivoで変換される化合物からなる、リン酸エステルを含めた5’−エステルが挙げられるが、それだけに限定されない。
【0022】
好ましい一実施形態においては、本発明の方法に使用される抗癌剤は5−FU又はカペシタビンである。
【0023】
一般に、本発明の方法に有用であるDPD阻害剤としては、不可逆的DPD阻害剤が挙げられるが、それだけに限定されない。例えば、ある種の例示的DPD阻害剤は、5−置換ウラシル化合物又はそのプロドラッグを含む。より具体的な一実施形態においては、DPD阻害剤は、ハロゲン原子、C2〜4アルケニル基、ハロゲンで置換されたC2〜4アルケニル基、C2〜6アルキニル基、ハロゲンで置換されたC2〜6アルキニル基、シアノ基、C1〜4アルキル基、又はハロゲンで置換されたC1〜4アルキル基で、5位が置換されたウラシル化合物を含む。別の一具体的実施形態においては、DPD阻害剤は、エニルウラシル、5−プロパ−1−イニルウラシル、5−シアノウラシル、5−プロパ−1−イニルウラシル、5−ブロモエチニルウラシル、5−(1−クロロビニル)ウラシル、5−ヨードウラシル、5−(2−ブロモビニル)ウラシル、(E)−5−(2−ブロモビニル)ウラシル、5−ヘキサ−1−イニルウラシル、5−ビニルウラシル、5−トリフルオロウラシル、5−ブロモウラシル及び5−(2−ブロモ−1−クロロビニル)ウラシルからなる群から選択されるウラシル化合物を含む。
【0024】
本発明の好ましい一実施形態においては、DPD阻害剤は、エニルウラシル又はそのプロドラッグである。
【0025】
別の好ましい一実施形態においては、DPD阻害剤はエニルウラシルであり、抗癌剤は5−FUである。
【0026】
更に別の好ましい一実施形態においては、DPD阻害剤はエニルウラシルであり、抗癌剤はカペシタビンである。
【0027】
例示的一実施形態においては、DPD阻害剤はエニルウラシルであり、抗癌剤は5−FUであり、エニルウラシルは約16〜40mg/m2の用量又は本明細書に記載の別のDPD阻害剤用量若しくは範囲で投与され、5−FUはその約11〜16時間後に約15〜50mg/m2の用量で投与される。
【0028】
別の例示的一実施形態においては、DPD阻害剤はエニルウラシルであり、抗癌剤は5−FUプロドラッグであり、エニルウラシルは約16〜40mg/m2の用量又は本明細書に記載の別のDPD阻害剤用量若しくは範囲で投与され、5−FUプロドラッグはその約11〜16時間後に約40〜150mg/m2の用量で投与される。
【0029】
別の例示的一実施形態においては、DPD阻害剤はエニルウラシルであり、抗癌剤は5−FUであり、エニルウラシルは約16〜40mg/m2の用量又は本明細書に記載の別のDPD阻害剤用量若しくは範囲で投与され、エニルウラシルが投与されてからエニルウラシルの少なくとも約3〜5排出半減期が経過したときに5−FUが約15〜50mg/m2の用量で投与される。
【0030】
別の例示的一実施形態においては、DPD阻害剤はエニルウラシルであり、抗癌剤は5−FUプロドラッグであり、エニルウラシルは約16〜40mg/m2の用量又は本明細書に記載の別のDPD阻害剤用量若しくは範囲で投与され、エニルウラシルが投与されてからエニルウラシルの少なくとも約3〜5排出半減期が経過したときに5−FUプロドラッグが約40〜150mg/m2の用量で投与される。
【0031】
別の例示的一実施形態においては、DPD阻害剤はエニルウラシルであり、抗癌剤は5−FUであり、エニルウラシルは患者の神経及び非神経組織におけるDPD活性を患者におけるベースラインDPD活性の5%未満に低下させるのに十分な用量で投与され、5−FUはその約11〜16時間後に約15〜50mg/m2の用量で投与される。
【0032】
別の例示的一実施形態においては、DPD阻害剤はエニルウラシルであり、抗癌剤は5−FUプロドラッグであり、エニルウラシルは患者の神経及び非神経組織におけるDPD活性を患者におけるベースラインDPD活性の5%未満に低下させるのに十分な用量で投与され、5−FUプロドラッグはその約11〜16時間後に約40〜150mg/m2の用量で投与される。
【0033】
別の例示的一実施形態においては、DPD阻害剤はエニルウラシルであり、抗癌剤は5−FU又は5−FUプロドラッグであり、エニルウラシルは約16〜40mg/m2又は本明細書に記載の別のDPD阻害剤用量若しくは範囲の用量で投与され、5−FU又は5−FUプロドラッグはその約11〜16時間後に、5−FU又は5−FUプロドラッグから生成される5−FUがDPD阻害剤の少なくとも10倍過剰で患者中に存在するような用量で投与される。
【0034】
本発明の別の一態様によれば、DPD阻害剤及び5−FU又は5−FUプロドラッグを含む徐放性経口医薬製剤であって、製剤を患者に投与した後、5−FU又は5−FUプロドラッグは、DPD阻害剤が放出された後少なくとも約11〜16時間まで実質的に放出されず、その放出後に5−FU又はプロドラッグから生成される5−FUが、患者に残留するDPD阻害剤の少なくとも約10倍過剰で患者中に存在する、徐放性経口医薬製剤が提供される。
【0035】
本発明のこれら及び他の態様は、以下の詳細な説明及び添付の図を参照することによって明らかになるであろう。本発明の種々の態様をより具体的に記載するのに本明細書で引用する特許及び他の文書は、参照によりその全体が本明細書に援用される。
【図面の簡単な説明】
【0036】
【図1】図1は、エニルウラシル及び5−FUの化学構造を示す。
【図2】図2は、エニルウラシル及び5−FUを含む錠剤の形の例示的経口徐放製剤を示す。
【図3】図3は、5−FU異化経路、及びエニルウラシルによるその遮断を示す。
【発明を実施するための形態】
【0037】
国際公開第2006/060697号に記載のように、5−FU及びプロドラッグから生成される5−FUよりも過剰な比のエニルウラシルなどのDPD阻害剤は、恐らくは代謝的活性化ステップの1つ以上を阻害することによって、その抗腫瘍活性を損ない得る。したがって、5−FU又はプロドラッグから生成される5−FUレベルが、5−FU又は5−FUプロドラッグが患者に投与された時点でDPD阻害剤レベルよりも十分過剰であるようにすることによって、DPD阻害剤が5−FU又は5−FUプロドラッグの抗腫瘍活性を阻害し得る程度が有利に最小化され、それによってこれらの薬剤の抗腫瘍効果が改善される。したがって、エニルウラシルなどの不可逆的DPD阻害剤は、DPDを効果的に不活性化する最低用量で投与されるべきであり、その投与時に5−FUがDPD阻害剤よりも過剰に存在するように、DPDに結合していない余分なDPD阻害剤がある程度除去されるように十分な時間が経過すべきである。
【0038】
この重要な知見に基づいて、臨床試験が開始され、患者におけるDPD活性を全身的に消失させるのに十分であると考えられる5mg用量のエニルウラシルが癌患者に投与された。次いで、5−FUが12〜24時間後に30〜160mgの用量で投与された。しかし、予想外に、この処置を受けた41名の患者の大多数が、軽度から重度の何らかの神経毒性を経験し、主な神経学的症候は、運動失調(不安定歩行)、神経障害、錯乱、めまい及び不明瞭発語であった。
【0039】
したがって、本発明は、神経組織と非神経組織の両方におけるDPDを十分に除去する投薬及びタイミングパラメータを適切に選択することによってこの神経毒性を防止又は最小化することができ、同時に5−FU又は5−FUプロドラッグが、その投与時に、残留DPD阻害剤よりも十分過剰であるようにすることによって、5−FU又はプロドラッグから生成される5−FUの抗腫瘍活性を阻害しない、方法に関する。
【0040】
本明細書に記載の方法は、限定的なものではない実例として、乳癌、肺癌、結腸癌、膵癌、胃癌、膀胱癌、腎臓癌、頭頚部癌、食道癌、肝細胞癌並びにすべての悪性白血病及びリンパ腫を含めて、5−FU及び/又は5−FUプロドラッグが活性である本質的にあらゆる癌タイプ(例えば、あらゆる5−FU反応性癌タイプ又は5−FUプロドラッグ反応性癌タイプ)の処置に適用可能である。さらに、本発明は5−FU及び5−FUプロドラッグの抗腫瘍効果を改善するので、5−FUに対して以前は望ましい反応性を示さないこともあった癌タイプが、本明細書に記載の方法によって投与されたときに改善された反応性を示すこともある。
【0041】
この開示に鑑みて、本明細書に記載の方法に使用することができ、患者の神経組織と非神経組織の両方におけるDPD活性を十分に阻害するのに十分なレベルでDPD阻害剤が確実に投与されるようにし、その投与時における5−FU又は5−FUプロドラッグのレベルが治療有効量であり、患者におけるDPD阻害剤レベルよりも十分過剰であり5−FU抗腫瘍活性の阻害を最小化又は解消するようにする、複数の投与及び投薬スケジュールが存在することが当業者には理解されるであろう。かかる投与及び投薬スケジュールはすべて本発明の範囲内とみなされる。
【0042】
本発明の例示的一実施形態においては、DPD阻害剤は、患者の神経組織と非神経組織の両方におけるDPD活性を実質的に消失させるのに十分な用量でそれを必要とする患者にまず投与され(すなわち、前投与され)、続いて5−FU又は5−FUプロドラッグが投与される。「実質的に消失させる」とは、患者の神経組織と非神経組織の両方におけるDPD活性レベルが、DPD阻害剤の投与前の患者におけるDPD活性のベースラインレベルの10%未満、好ましくは5%未満に低下することを意味する。患者のDPD活性のベースラインレベルは、患者から採取された生物試料において公知技術を使用して容易に決定することができる(例えば、Baker et al.,J Clin Oncol 18:915−926 2000;Schilsky et al.,J Clin Oncol 4:1450−7,1998)。しかし、循環血球などの非神経組織におけるDPD阻害の分析は、神経組織におけるDPD阻害の程度を過大評価するおそれがある。
【0043】
少なくとも1種類のDPD阻害剤をまず投与して、神経組織と非神経組織の両方において患者のDPD活性を実質的に消失させた後、次いで、DPD阻害剤が排出によって患者から完全にではないが実質的に除去されるのに十分な時間が経過した後に5−FU若しくは5−FUプロドラッグ又はその組合せを患者に投与する。この点について、ある実施形態においては、新規合成されるDPDを不活性化するために、低レベルのDPD阻害剤が5−FU投与まで及び/又は投与中の系内に残留することが望ましい場合もある。
【0044】
DPD阻害剤と5−FU又は5−FUプロドラッグの投与間の時間的遅延は変動し得る。ただし、5−FU又は5−FUプロドラッグの投与時には、それはその時点で患者に残留するDPD阻害剤レベルよりもかなり過剰に患者中に存在する。例示的一実施形態においては、5−FU又は5−FUプロドラッグは、5−FU又はプロドラッグから生成される5−FUのレベルが、患者に残留するDPD阻害剤よりも少なくともモル過剰で、例えば、5−FU又は5−FUプロドラッグが投与された時点で患者に残留するDPD阻害剤のレベルの少なくとも約2倍、少なくとも約3倍、少なくとも約5倍、少なくとも約10倍、少なくとも約25倍、少なくとも約50倍、又は少なくとも約100倍過剰で、患者中に存在するような用量で投与される。当業者は、本明細書に記載の実施形態に従ってDPD阻害剤に対して患者における5−FU又はプロドラッグから生成される5−FUの過剰レベルを計算及び/又は決定するのに幾つかの公知の利用可能な技術のいずれかを使用できることを認識するであろう。かかる技術としては、例えば、HPLC、LC−MS、ELISAなどが挙げられる。上述したように、5−FU又はプロドラッグから生成される5−FUが、5−FU又は5−FUプロドラッグが投与された時点で患者におけるDPD阻害剤のレベルよりも十分過剰に存在するようにすることによって、DPD阻害剤による5−FU又は5−FUプロドラッグの抗腫瘍性との干渉が最小化され、それによって5−FU又は5−FUプロドラッグの効力が改善される。
【0045】
本発明の更なる実施形態においては、5−FU又は5−FUプロドラッグは、DPD阻害剤が投与されてからDPD阻害剤の少なくとも約1、2、2.5、3、5、7、10、14又は21排出半減期が経過した後にのみ患者に投与される。あるDPD阻害剤の排出半減期は測定されており、測定されていない場合には、排出半減期は、周知の確立されたガスクロマトグラフィー/質量分析及びHPLC法によって容易に測定することができる(Baker et al.,J Clin Oncol 18:915−926 2000;Schilsky et al.,J Clin Oncol 4:1450−7,1998で言及されている)。ヒトにおけるエニルウラシルの排出半減期は約3.5時間と報告されているが(例えば、Baker et al.,J Clin Oncol 18:915−926 2000;Ochoa et al.,Ann Oncol 11:1313−22,2000)、DPD阻害剤の半減期は用量依存的であり得る。この用量依存性は、DPD阻害剤と5−FU又は5−FUプロドラッグの投与間の適切な時間的遅延を決定するときに考慮すべきである。
【0046】
エニルウラシルをDPD阻害剤として使用する本発明のある実施形態では、5−FU又は5−FUプロドラッグの投与前に排出によってエニルウラシルレベルが十分に低下するように、5−FU又は5−FUプロドラッグは、エニルウラシル投与から少なくとも約3時間、約6時間、約8時間、約11時間、約16時間、約20時間、約36時間、約48時間又は約72時間後に投与される。本発明のある関連実施形態においては、5−FU又は5−FUプロドラッグは、エニルウラシル投与から約11〜16時間、約8〜20時間、約6〜36時間、約3〜48時間又は約3〜72時間後に投与される。本発明の更に別の実施形態においては、5−FU又は5−FUプロドラッグは、その投与時に、患者におけるエニルウラシルと5−FUの比が約1:10、約1:5、約1:4又は約1:3未満になるまで投与されない。言うまでもなく、これらの範囲及び比は本質的に説明のためのものであって、必要に応じて、又は特定の投薬スケジュールに応じて、変わり得ることが理解されるであろう。ただし、5−FU又は5−FUプロドラッグが投与されるときにはエニルウラシルの存在は最小限であり、又は存在せず、さらに、DPD活性は、5−FU又は5−FUプロドラッグが投与される時点で神経組織と非神経組織の両方において所望の程度まで実質的に消失している。
【0047】
本発明によって使用されるDPD阻害剤は、好ましくは、DPDを不可逆的に不活性化するものである。したがって、エニルウラシルなどの阻害剤は、酵素を不活性化し、酵素に共有結合していない余分な阻害剤は、5−FU又は5−FUプロドラッグが投与される前にある程度除去される。例示的な不可逆的DPD阻害剤としては、5−置換ウラシル化合物、特にハロゲン原子、ハロゲンで場合によっては置換されたC2〜4アルケニル基(例えば、ビニル)(例えば、2−ブロモビニル、1−クロロビニル又は2−ブロモ−1−クロロビニル)、ハロゲン原子で場合によっては置換されたC2〜6アルキニル基、シアノ基、又はハロゲンで置換されたC1〜4アルキル基(例えば、トリフルオロメチル)で、5位が置換されたウラシル化合物を含むDPD阻害剤又はそのプロドラッグが挙げられるが、それだけに限定されない。
【0048】
本発明のより特別な実施形態においては、DPD阻害剤は、エニルウラシル、5−プロパ−1−イニルウラシル、5−シアノウラシル、5−プロピニルウラシル、5−ブロモエチニルウラシル、5−(1−クロロビニル)ウラシル、5−ヨードウラシル、5−(1−ブロモビニル)ウラシル、(E)−5−(2−ブロモビニル)ウラシル、5−ヘキサ−1−イニルウラシル、5−ビニルウラシル、5−トリフルオロウラシル、5−ブロモウラシル及び5−(2−ブロモ−1−クロロビニル)ウラシル、又はそのプロドラッグからなる群から選択される。
【0049】
別の例示的一実施形態においては、DPD阻害剤は5−ブロモビニルウラシルのプロドラッグであり、例示的一化合物は(BV−araU又はソリブジンとも称される)化合物1−β−D−アラビノフラノシル−(E)−5−(2−ブロモビニル)ウラシルである。これに関するある例示的プロドラッグ化合物は、例えば、米国特許第4,386,076号に記載されており、その開示を参照により本明細書に援用する。
【0050】
本発明の好ましい一実施形態においては、DPD阻害剤は、エニルウラシル若しくは5−エチニル−2(1H)−ピリミジノン(4−酸素のないエニルウラシル)などのエニルウラシルのプロドラッグ(Porter,et al.,Biochem.Pharmacol 47:1165−1171,1994)、エニルウラシルのヌクレオシド若しくはデオキシヌクレオシド誘導体、エニルウラシルにin vivoで変換される化合物、及び/又は失活剤にin vivoで変換されるDPD失活剤の誘導体である。例として、かかる化合物としては、上記5−置換ウラシル化合物に対応する核酸塩基を含むヌクレオシド誘導体、例えば、リボース、2’−デオキシリボース、2’,3’−ジデオキシリボース、アラビノース又は別の開裂可能な糖部分を含むヌクレオシド誘導体が挙げられ、ハロゲンなどの2’若しくは3’置換基又はエステルなどの5’置換基を更に含むことができる。かかるヌクレオシド誘導体のより特別な例としては、1−(β−D−アラビノフラノシル)−5−プロパ−1−イニルウラシル及び2’,3’−ジデオキシ−5−エチニル−3’−フルオロウリジンが挙げられる。
【0051】
やはり本発明によって使用することができる多数の5−FUプロドラッグが知られている。5−FUのプロドラッグは、in vivoで5−フルオロウラシルに代謝される化合物であり、実例として、5−フルオロウリジン、5−フルオロシチジン、5−フルオロ−2−デオキシウリジン、5−フルオロ−2−デオキシシチジン、5−フルオロアラビノシルウラシル、及びリン酸エステルを含めたその5’−エステルが挙げられる。別の例示的化合物としては、5’−デオキシ−4’,5−フルオロウリジン、5’−デオキシ−5−フルオロウリジン、1−(2−テトラヒドロフラニル)−5−フルオロウラシル、1−C1〜8アルキルカルバモイル−5−フルオロウラシル誘導体、1−(2−テトラヒドロフリル)−5−フルオロウラシル、Ftorafur(テガフール、アジア諸国で広く用いられる経口5−FUプロドラッグ)、及び5’−デオキシ−5−フルオロ−N−[(ペンチルオキシ)カルボニル]−シチジン(カペシタビン、Roche Laboratories Inc.によってXeloda(登録商標)として市販されている)、又は5−FUにin vivoで変換される化合物が挙げられる。
【0052】
この開示に鑑みて、本発明の方法は、所望のどんなタイプ、期間及び投薬特性の投与計画でも含み得ることが理解されるであろう。ただし、投与計画は、5−FUに付随する神経毒性が防止又は最小化されるように、さらに、5−FU又は5−FUプロドラッグが、5−FU又は5−FUプロドラッグが投与された時点で患者に残留するDPD阻害剤のレベルよりも十分過剰に存在するように、適切に選択される。
【0053】
本発明のある特に好ましい実施形態においては、本明細書に記載の方法は、5−FUと組み合わせたDPD阻害剤エニルウラシルの投与を利用する。例えば、例示的実施形態においては、毎週又は5日投薬スケジュールを含む投与計画を用いることができ、エニルウラシルは5−FUの前夜に投与され、5−FUは1日/週又は1回/日で5日間のみ投与される。
【0054】
本明細書に記載のように、神経組織と非神経組織の両方におけるDPD活性を実質的に阻害するのに十分な量が患者に投与されるのであれば、エニルウラシルの適切な用量は変わり得ることが理解されるであろう。ある実施形態においては、例えば、エニルウラシルは、好ましくは、約14〜40mg/m2又は約15〜40mg/m2又は約16〜40mg/m2の用量で投与される。より具体的な実施形態においては、エニルウラシルの用量は約14〜30mg/m2又は約15〜30mg/m2又は約16〜30mg/m2である。更に別の具体的実施形態においては、エニルウラシルの用量は約14〜21mg/m2又は約15〜21mg/m2又は約16〜21mg/m2又は約16〜25mg/m2又は約12〜35mg/m2である。本発明の更に別の実施形態においては、エニルウラシルは、約14〜50mg/m2、約15〜50mg/m2、約16〜50mg/m2、約20〜50mg/m2又は約30〜50mg/m2で投与することができる。
【0055】
言うまでもないが、好ましくは、本発明によって患者に投与するために選択されるDPD阻害剤投与量は、患者の神経組織と非神経組織の両方におけるDPD活性が実質的に消失するようにするのに少なくとも十分なものである。さらに、本明細書に記載の本発明の種々の実施形態に関連して、上記及び本明細書に記載のDPD阻害剤用量範囲のどれでも使用できることが理解されるであろう。
【0056】
本発明のある別の実施形態においては、5−FUはエニルウラシルから約11〜16時間後に投与され、それによって、エニルウラシルと5−FUの高い比に起因する抗腫瘍活性の低下を回避するために要求されるように、エニルウラシルと5−FUの比が確実に1.0をかなり下回るようになる。
【0057】
別の好ましい実施形態においては、エニルウラシルは約16〜40mg/m2又は本明細書に記載の別のDPD阻害剤用量若しくは範囲で投与され、5−FUは、約3〜5エニルウラシル排出半減期が経過した後にのみ投与される。
【0058】
更に別の好ましい実施形態においては、エニルウラシルは約16〜40mg/m2又は本明細書に記載の別のDPD阻害剤用量若しくは範囲で投与され、5−FUは、5−FU投与時にエニルウラシルと5−FUの比が≦1:10であるように十分な時間が経過した後にのみ投与される。
【0059】
14〜21mg/m2のエニルウラシル用量は以前に用いられたが(Schilsky et al.,J Clin Oncol 1998;16:1450−7;Baker et al.,J Clin Oncol 2000;18:915−26)、エニルウラシルは5−FU投与の1時間前又は5−FU投与と同時に投与された。それに対して、本発明は、独自に、5−FUの約11〜16時間前にエニルウラシルを投与し、ある好ましい実施形態においては、ヒト患者において認められた予想外の神経毒性を回避するために、神経組織と非神経組織の両方におけるDPDを不活性化するのに十分なDPD阻害剤用量を使用する。
【0060】
下記表1に示したエニルウラシル量を使用して16〜20mg/m2の範囲の用量を得ることができる。10.5時間(約3排出半減期)後に体内に残留するエニルウラシルの計算量も示す。したがって、毎週及び5日投薬スケジュールが約15〜50mg/m2の範囲の5−FU用量を使用する場合、エニルウラシルと5−FUの比は、5−FUが投与されるときには常に約1:6未満である。例えば、一般に使用される25mg/m2の5−FU用量がエニルウラシルから10.5時間後に投与されるときには、エニルウラシルと5−FUの比は約1:10以下である。
【0061】
【表1】
表1−表示BSAの患者に16〜20mg/m2を送達するエニルウラシルの用量、及び10.5時間(約3排出半減期)後に体内に残留するエニルウラシルの計算量。
【0062】
別の例では、1.9m2未満の患者にエニルウラシル30mgを投与した場合、及び≧1.9m2の患者にエニルウラシル45mgを投与した場合、これら2つの用量は、広範囲の体の大きさにわたって約16〜23mg/m2となる。さらに、エニルウラシルは無毒であり、最高50mg/日で7日間投与したときに安全であることが判明したので(Schilsky et al.,J Clin Oncol 4:1450−7,1998)、例示的投与スキームをより一層簡略にすることができる。例えば、すべての患者にエニルウラシル40mgを投与した場合、投薬範囲は約15〜31mg/m2を含む。さらに、すべての患者にエニルウラシル50mgを投与した場合、投薬範囲は約19〜39mg/m2である。
【0063】
したがって、別の実施形態においては、本発明によって使用されるエニルウラシル用量範囲は、有利には、約16〜23mg/m2、15〜31mg/m2及び19〜39mg/m2を含む。
【0064】
本発明の更に別の実施形態においては、エニルウラシルと5−FUの投与の時間間隔は、約11〜16時間、約8〜20時間、約6〜36時間、約3〜48時間又は約3〜72時間とすることができる。
【0065】
別の実施形態においては、少なくとも約3〜5、約2.5〜7、約2〜10、約1〜14又は1〜21エニルウラシル排出半減期が5−FU投与前に経過する。
【0066】
更なる実施形態においては、エニルウラシルは、5−FUが投与される前日に投与され、又は5−FUが投与される複数日前に投与される。
【0067】
更に別の実施形態においては、5−FUは、特に毎週及び5日投薬スケジュールで、約15〜40mg/m2、10〜50mg/m2、5〜60mg/m2又は5〜70mg/m2の用量で投与される。
【0068】
更なる実施形態においては、5−FUプロドラッグは、特に毎週及び5日投薬スケジュールで、約20〜60mg/m2、15〜80mg/m2、10〜100mg/m2又は5〜150mg/m2の用量で投与される。
【0069】
更なる実施形態においては、5−FUは、特に長期療法では、8、10、12、14又は16時間ごとに約0.8〜1.2mg/m2又は0.3〜1.8mg/m2の用量で投与される。
【0070】
本発明の更なる実施形態においては、本発明によって使用される5−FU投与計画は、毎週スケジュール、5日間スケジュール、毎日スケジュール、5−FUが指定日に複数回投与される毎日スケジュール、5−FUがエニルウラシル投与後1日を超えて投与され、エニルウラシルが5−FU療法中5−FU前に毎日、隔日又は3日ごとに投与される毎日スケジュール、5−FUがエニルウラシル投与から1日以上後に複数回投与され、エニルウラシルが5−FU療法中5−FU前に毎日、隔日又は3日ごとに投与される毎日スケジュールである。
【0071】
例示的一実施形態においては、エニルウラシルは、5−FU前夜に、約16〜40mg/m2の用量で、又は本明細書に記載の別のDPD阻害剤用量範囲で投与することができ、あるいは、夕方の5−FU投与に続いて朝に投与することができる。約20から30mg/m2の例示的5−FU用量をこれらのスケジュールで使用する場合(Levin et al.,Invest New Drugs 18:383−90,2000;Schilsky et al.,J Clin Oncol 4:1450−7,1998;Guo et al.,Cancer Chemother Pharmacol 52:79−85,2003)、例えば、5−FUはエニルウラシルよりも常に実質的に過剰であるべきである。
【0072】
別の例示的一実施形態においては、28日b.i.d.(28日間毎日2回)スケジュールを使用することができる。このタイプの投与計画は、5−FUが1mg/m2でしか投与されないので、異なる手法を必要とすることが理解されるであろう(例えば、Baker et al.,J Clin Oncol 2000;18:915−26を参照されたい)。したがって、エニルウラシルがこの低5−FU用量よりも過剰に存在しないように注意しなければならない。しかし、高用量のエニルウラシルはDPDを長期間不活性化された状態に維持するので、エニルウラシルは、例えば、2日ごと、場合によっては3日ごとに投与することができる。この戦略は、各エニルウラシル投与間にエニルウラシルと5−FUの比が各後続の5−FU投与につれて確実に低下するようにする。
【0073】
別の例示的一実施形態においては、エニルウラシル(又は別のDPD阻害剤)がまず投与され、次いで5−FU又は5−FUプロドラッグの複数回投与がその後所望の時点で投与され、その後エニルウラシルが場合によっては再度投与される。例えば、例示的一実施形態においては、エニルウラシルがまず投与され、次いで複数の5−FU用量がその約8、10、12、14又は16時間後の例示的時点において投与され、必要に応じて、その後エニルウラシルが場合によっては再度投与され、このサイクルが繰り返される。
【0074】
本発明は、更なる特徴として、単一製剤として一緒に、又は本発明に従って別々の時点で投与される個別の製剤として、少なくとも1種類の薬学的に許容される担体又は賦形剤を含み、DPD阻害剤及び/又は5−FU若しくは5−FUプロドラッグを更に含む、医薬製剤を含む。担体又は賦形剤は、製剤の他の成分と適合するという意味で「薬学的に許容され」、患者に無害である。製剤としては、例えば、経口、直腸、経鼻、(頬及び舌下を含めた)局所、膣及び(皮下、筋肉内、静脈内及び皮内を含めた)非経口投与に適合したものが挙げられる。製剤は、好都合には単位剤形として提供することができ、製薬技術分野で周知の任意の方法によって調製することができる。かかる方法は、活性成分を1種類以上の副成分を構成する担体と会合させる段階を含む。一般に、製剤は、活性成分を液状担体若しくは微粉担体又はその両方と均一かつ十分に会合させ、次いで必要に応じて生成物を成形することによって調製される。
【0075】
本発明による製剤は、本質的に任意の利用可能な技術を使用して調製及び/又は投与することができる。経口投与に適合した本発明の製剤は、例えば、各々が所定量の活性成分を含むカプセル剤、カシェ剤、錠剤などの個別単位として、散剤若しくは顆粒剤として、水性液体若しくは非水性液体中の溶液剤若しくは懸濁液剤として、又は水中油型乳剤若しくは油中水型乳剤として提供することができる。活性成分は、ボーラス、舐剤又はペーストとしても提供することができる。経口投与は、典型的に好ましい投与経路である。
【0076】
錠剤は、例えば、圧縮又は成形によって製造することができ、1種類以上の副成分を場合によっては含むことができる。圧縮錠剤は、結合剤(例えば、ポビドン、ゼラチン、ヒドロキシプロピルメチルセルロース)、滑沢剤、不活性希釈剤、防腐剤、崩壊剤(例えば、デンプングリコール酸ナトリウム、架橋ポビドン、架橋カルボキシメチルセルロースナトリウム)界面活性剤又は分散剤と場合によっては混合することができる、散剤、顆粒剤などの易流動性活性成分を適切な機械で圧縮して調製することができる。成形錠剤は、不活性希釈液で湿らせた粉末化合物の混合物を適切な機械で成形することによって製造することができる。錠剤は、場合によっては、被覆することができ、又は刻み目をつけることができ、例えば所望の放出プロファイルを与えるために様々な割合のヒドロキシプロピルメチルセルロースを使用して、その中の活性成分を制御放出するように処方することができる。
【0077】
口内局所投与用製剤としては、例えば、風味付けされた基剤、通常はスクロースとアラビアゴム又はトラガカント中に活性成分を含むロゼンジ、ゼラチンとグリセリン、スクロースとアラビアゴムなどの不活性基剤中に活性成分を含むトローチ剤、及び適切な液状担体中に活性成分を含む洗口剤が挙げられる。直腸投与用製剤は、例えば、例えばカカオ脂又はサリチラートを含む、適切な基材を含む坐剤として提供することができる。膣内投与用製剤は、例えば、活性成分に加えて、適切であることが当該技術分野で知られている担体を含有する、膣坐剤、タンポン、クリーム剤、ゲル剤、ペースト剤、泡剤又は噴霧剤として提供することができる。
【0078】
非経口投与用製剤としては、例えば、水性及び非水性の等張性無菌注射液が挙げられる。等張性無菌注射液は、抗酸化剤、緩衝剤、静菌剤、及び製剤を対象レシピエントの血液と等張にする溶質、並びに懸濁化剤及び増粘剤を含み得る水性及び非水性の無菌懸濁液を含むことができる。製剤は、単位用量又は複数用量の密閉容器、例えばアンプル及びバイアルとして提供することができ、使用直前に無菌液状担体、例えば注射用水の添加のみを必要とするフリーズドライ(凍結乾燥)状態で貯蔵することができる。即時注射液及び懸濁液は、上記種類の無菌散剤、顆粒剤及び錠剤から調製することができる。
【0079】
典型的には、1種類以上の活性薬剤を含む液剤は、好ましくはpH7から11、一般に9.5から10.5に緩衝される。ある単位投与製剤としては、活性成分の1日用量若しくは単位、上述した1日の分割用量(sub−dose)、又はその適切な一部分を含むものが挙げられる。
【0080】
本明細書に記載のDPD阻害剤及び5−FUプロドラッグを製造する方法は公知であり、従来の方法を使用して実施することができる。例えば、上記DPD阻害剤は、5−エチニルウラシルの調製の場合はHeterocycl.Chem.19(3)463−4(1982)、5−(2−ブロモビニル)ウラシル、5−ブロモエチニルウラシル及び5−(2−ブロモ−1−クロロビニル)ウラシルの調製の場合はJ.Chem.Soc.Perkin Trans.1(16),1665−70(1981)、5−シアノ−ウラシルの調製の場合はNucleic Acid Chemistry,Vol.2,927−30(1978)、5−ビニルウラシルの調製の場合はNucleic Acids Research,1(1)105−7(1974)、5−トリフルオロメチルウラシルの調製の場合はZ.Chern 17(11)415−16(1977)、5−(1−クロロビニル)ウラシルの調製の場合はNucleic Acids Research 3(10),2845(1976)に記載の方法によって調製することができる。本発明のある別の化合物は、2’,3’−ジデオキシ−5−エチニル−3’−フルオロウリジンなどの3’−フルオロ−2’,3’−ジデオキシ−5−アルキニルウリジン化合物の調製の場合は欧州特許明細書第356166号、及び1−(b−D−アラビノフラノシル)−5−プロパ−1−イニルウラシルなどの5−アルキニルウラシルアラビノシドの調製の場合は欧州特許明細書第272065号に記載のプロセスに従って調製することができる。本発明に使用される化合物を製造するためのこれら及び他の合成技術が公知であり、利用可能である。
【0081】
一実施形態においては、本発明は、製剤成分を患者にその所望の投与量範囲内で所望の時間放出することができるようにDPD阻害剤と5−FU又は5−FUプロドラッグが一緒に投与される混合経口製剤を提供する。2成分の差次的徐放性送達は、公知技術及び材料を使用して実施することができる。例えば、一実施形態においては、例えば錠剤の形の経口製剤は、図2に説明用に示したように、3つの異なる層で構成することができる。外層は、即放処方のエニルウラシルを含むことができる。中間層は、本発明によって5−FU又は5−FUプロドラッグの放出を所望の程度遅らせる徐放性成分(例えば、徐放性緩衝剤)とすることができる。5−FU又は5−FUプロドラッグは、即放処方のコア層に位置する。DPD阻害剤及び5−FU又は5−FUプロドラッグは、本明細書に記載の適切な用量及び比で処方される。好ましい一実施形態においては、DPD阻害剤はエニルウラシルであり、5−FU又は5−FUプロドラッグは5−FU又はカペシタビンである。
【0082】
別の一実施形態においては、別の製剤は、5−FU又は5−FUプロドラッグを含むミクロスフェアなどの公知の送達ビヒクルを含むことができる。一実施形態においては、例えば、5−FU又は5−FUプロドラッグは、徐放性成分(例えば、徐放性崩壊緩衝剤)のシェル、及びDPD阻害剤を即時に放出する外層で被包することができる。好ましい一実施形態においては、DPD阻害剤はエニルウラシルであり、5−FU又は5−FUプロドラッグは5−FU又はカペシタビンである。例示的混合製剤のこれら及び別の例は、単一経口製剤においてDPD阻害剤と5−FU又は5−FUプロドラッグの送達の間を適切に遅延することができる公知技術を使用して、設計及び製造することができる。
【0083】
別の一実施形態においては、本明細書に記載の方法は、さらに、ロイコボリンの投与を含む。ロイコボリン又はロイコボリンの活性異性体であるIsovorinは、一般に、癌患者の処置に5−FUと併用される。これは、エニルウラシルと5−FUの上記投与計画に追加することもできる。ロイコボリンは、腫瘍を有するラット及び組織培養においてエニルウラシル及び5−FUの抗腫瘍効果を改善することが判明し(Cao et al.,Cancer Res 90:1507−1510,1993;Fischel et al.,Biochem Pharmacol 53:1703−1709,1997)、エニルウラシル及び5−FUを投与された患者に投与されてきた(Schilsky et al.,J Clin Oncol 4:1450−7,1998;Guo et al.,Cancer Chemother Pharmacol 52:79−85,2003)。ロイコボリンは、経口製剤に都合よく利用することもできる。
【0084】
本発明は、以下の非限定的実施例を考察することによって更に理解することができる。
【実施例】
【0085】
(実施例1)
5−FUと組み合わせたエニルウラシルの投与に付随する予想外の神経毒性、及びそれを防止又は最小限にする方法
国際公開第2006/060697号に記載の重要な機構的知見に基づく臨床試験が開始された。より具体的には、癌患者に5mg用量のエニルウラシルが投与され、その12〜24時間後に30〜160mg用量の5−FUが投与された。しかし、予想外に、この処置を受けた41名の患者の大多数は、軽度から重度の何らかの神経毒性を経験し、主な神経学的症候は、運動失調(不安定歩行)、神経障害、錯乱、めまい及び不明瞭発語であった。
【0086】
F−Balは、5−FUの主要な分解(異化)産物である。図3に示した経路によれば、DPDは、5−FUをジヒドロフルオロウラシル(5−FUH2)に変換し、ジヒドロフルオロウラシルは、α−フルオロ−β−ウレイドプロピオン酸(FUPA)、次いでF−Balに変換される。エニルウラシルは、DPDを不活性化することによってこの経路を遮断する。
【0087】
5−FU自体は神経毒性を生じず、したがって臨床試験患者で認められた神経毒性の直接的原因ではなかったが、研究によれば、5−FUの分解産物の一つであるF−Balはマウス、サル、ネコ及びイヌにおいて神経毒性を生じ得ることが判明した(Saif et al.,Anticancer Drugs 2001;12:525−31)。さらに、イヌにおける研究は、F−Balが神経毒性を生じ得る更なる証拠を与えた(Davis et al.,Biochem Pharmacol 1994;48:233−6)。例えば、イヌへの5−FUの静脈内投与は、低血中濃度の5−FUしか得られず、発作、筋肉振戦及び運動失調を誘発した。しかし、イヌをエニルウラシルで前処理すると、高血中濃度の5−FUを神経毒性なしに得ることができた。したがって、神経組織における5−FUの異化を適切に遮断することによって、エニルウラシルは神経毒性を抑制した。
【0088】
F−Balは5−FUに付随する神経毒性の原因であると思われ、エニルウラシルはF−Balの形成を防止するので、エニルウラシルで処置された臨床試験患者における神経毒性の高罹患率は、特に、患者に使用された5mg用量のエニルウラシルがそのDPDを実質的に消失させるのに十分であると考えられ、したがって神経系における神経毒性5−FU異化産物の形成を防止するはずであったことを考えると、全く予想外であった。
【0089】
しかし、臨床試験データを分析し、さらに、科学文献に記載されたことを考慮すると、特定用量のエニルウラシルは、患者の循環血球などの非神経組織におけるDPDを不活性化するのには十分かもしれないが(Schilsky et al.,J Clin Oncol 1998;16:1450−7)、神経組織におけるDPDを十分に不活性化するのには必ずしも十分ではないことが現在では理解される。例えば、ラットにおいては、肝臓及びひ臓、腸管粘膜、肺などの他の非神経組織のDPDの50%を不活性化するのに必要な用量の約6倍のエニルウラシルが脳内のDPDの50%を不活性化するのに必要である(Spector et al.,Biochem Pharmacol 1993;46:2243−8)。神経組織中のDPD酵素に接近し、それを阻害するエニルウラシルの能力はどういうわけか妨害されるので、神経組織内のDPDを不活性化するには、非神経組織に必要な用量よりも高用量のエニルウラシルが必要である。したがって、臨床試験で使用されたエニルウラシルの用量は、ヒト神経組織におけるDPD活性を阻害するのには不十分であったと思われる。その結果、5−FUは、神経組織において、患者における神経毒性を生じるF−Balを含めた神経毒性異化産物に異化されたと思われる。
【0090】
さらに、5−FU異化が非神経組織において阻害されるときには、5−FUは神経組織により接近する可能性がある。その結果、エニルウラシルの用量が非神経組織中のDPDを阻害するのには十分であるが、神経組織中のDPDを十分に阻害するのには低すぎる場合、5−FUは神経組織において選択的にF−Balに変換されることになる。したがって、非神経組織中のDPDを十分に阻害するのには十分であるが、神経組織では不十分であるエニルウラシル用量は、5−FUによって誘導される神経毒性を引き起こすおそれがある。
【0091】
この理論は、患者における神経毒性の発生率が、より高用量のエニルウラシルを投与した患者において減少したという観察によって強く支持される。患者に5−FU投与前にエニルウラシル5mgを投与した臨床試験では、41名の患者の大多数が5−FUによって誘導される神経毒性を経験した。それに対して、5−FU投与前にエニルウラシル20mgを投与した患者では、神経毒性の発生率は17名のうち2名(12%)に減少した(Guo XD,et al.Cancer Chemother Pharmacol 2003;52:79−85;Saif et al.,Anticancer Drugs 2001;12:525−31)。これら2名の患者の体格が大きいことは特に注目すべきである。その体表面積(BSA)は2.1m2及び2.5m2であった。したがって、エニルウラシルの20mg用量は、これらの患者にそれぞれ9.5mg/m2及び8.0mg/m2のエニルウラシルを送達した。重要なことには、その末梢血細胞のDPDは完全に不活性化された。したがって、この分析に基づいて、神経組織のDPDを十分不活性化して神経毒性を防止するには少なくとも9.5mg/m2を超えるエニルウラシル用量が必要であると思われる。さらに、12時間ごとの約11.5mg/m2のエニルウラシル用量は、それでも重篤な神経毒性の全発生率が6%という結果になる(Schilsky et al.,J Clin Oncol 2002;20:1519−26)。
【0092】
したがって、臨床における神経毒性を回避するためには、エニルウラシルの用量が十分に高く、好ましくは約12mg/m2又は14mg/m2又は15mg/m2又は16mg/m2を超え、より好ましくは約12〜21mg/m2又は14〜21mg/m2又は15〜21mg/m2又は16〜21mg/m2又は16〜25mg/m2又は15〜40mg/m2又は16〜40mg/m2であり、非神経組織と神経組織の両方におけるDPDを不活性化することが重要である。さらに、5−FUの抗腫瘍活性を最大にするためには、5−FUは、その投与時に、特許におけるエニルウラシルと5−FUの比が好ましくは約1:10、1:5又は1:3以下であるような用量で投与すべきである。しかし、5−FU投与時にエニルウラシルレベルが完全には除去されていないことが重要であり得る。例えば、ある実施形態においては、エニルウラシルが除去された後に出現する新規合成されたDPDを不活性化するのに幾らかのエニルウラシルが存在することが好ましい(Spector T,et al.Biochem Pharmacol 1993;46:2243−8;Heslin MJ et al.Cancer Chemother Pharmacol 2003;52:399−404:Keith B,et al.Clin Cancer Res 2002;8:1045−50)。
【0093】
上記のことから、本発明の特定の実施形態を説明のために本明細書に記載したが、本発明の精神及び範囲から逸脱せずに種々の改変を成し得ることが理解されるであろう。したがって、本発明は、添付の特許請求の範囲による以外は限定されない。
【特許請求の範囲】
【請求項1】
DPD阻害剤及び5−FU又は5−FUプロドラッグから選択される抗癌剤を含む組合せを用いて、癌患者の処置に付随する神経毒性を最小限にする方法であって、
神経組織と非神経組織の両方におけるDPD活性を実質的に消失させるのに十分な用量でDPD阻害剤をまず投与する工程、及び
その後に前記5−FU又は5−FUプロドラッグを投与する工程
を含み、前記5−FU又は5−FUプロドラッグが、前記5−FU又は前記5−FUプロドラッグから生成される5−FUが前記DPD阻害剤よりもかなり過剰に前記患者中に存在するような用量で投与される、方法。
【請求項2】
前記DPD阻害剤が約16〜40mg/m2の用量で投与される、請求項1に記載の方法。
【請求項3】
前記DPD阻害剤が約16〜25mg/m2の用量で投与される、請求項1に記載の方法。
【請求項4】
前記5−FU又は5−FUプロドラッグが、前記DPD阻害剤が投与されてから約11〜16時間後に投与される、請求項1に記載の方法。
【請求項5】
前記5−FU又は5−FUプロドラッグが、前記DPD阻害剤が投与されてから前記DPD阻害剤の少なくとも約3〜5排出半減期が経過したときに投与される、請求項1に記載の方法。
【請求項6】
前記DPD阻害剤が、前記患者における神経及び非神経組織において、DPD活性を前記患者におけるベースラインDPD活性の5%未満に低下させるのに十分な用量で投与される、請求項1に記載の方法。
【請求項7】
前記5−FU又は5−FUプロドラッグが、その投与時において前記5−FU又はプロドラッグから生成される5−FUが前記DPD阻害剤の少なくとも10倍過剰で前記患者中に存在するような用量で投与される、請求項1に記載の方法。
【請求項8】
前記5−FUプロドラッグが、5−フルオロウリジン、5−フルオロシチジン、5−フルオロ−2−デオキシウリジン、5−フルオロ−2−デオキシシチジン、5’−デオキシ−4’,5−フルオロウリジン、及び5−フルオロアラビノシルウラシル、5’−デオキシ−5−フルオロウリジン、1−(2−テトラヒドロフラニル)−5−フルオロウラシル、1−C1〜8アルキルカルバモイル−5−フルオロウラシル誘導体、1−(2−テトラヒドロフリル)−5−フルオロウラシル、5’−デオキシ−5−フルオロ−N−[(ペンチルオキシ)カルボニル]−シチジン(カペシタビン)、又は5−FUにin vivoで変換される化合物からなる群、及びリン酸エステルを含めたその5’−エステルから選択される、請求項1に記載の方法。
【請求項9】
前記抗癌剤が5−FUである、請求項1に記載の方法。
【請求項10】
前記抗癌剤がカペシタビンである、請求項1に記載の方法。
【請求項11】
前記DPD阻害剤が5−置換ウラシル化合物又はそのプロドラッグを含む、請求項1に記載の方法。
【請求項12】
前記DPD阻害剤が、ハロゲン原子、C2〜4アルケニル基、ハロゲンで置換されたC2〜4アルケニル基、C2〜6アルキニル基、ハロゲンで置換されたC2〜6アルキニル基、シアノ基、C1〜4アルキル基、又はハロゲンで置換されたC1〜4アルキル基で、5位が置換されたウラシル化合物を含む、請求項1に記載の方法。
【請求項13】
前記DPD阻害剤が、エニルウラシル、5−プロパ−1−イニルウラシル、5−シアノウラシル、5−プロパ−1−イニルウラシル、5−ブロモエチニルウラシル、5−(1−クロロビニル)ウラシル、5−ヨードウラシル、5−(2−ブロモビニル)ウラシル、(E)−5−(2−ブロモビニル)ウラシル、5−ヘキサ−1−イニルウラシル、5−ビニルウラシル、5−トリフルオロウラシル、5−ブロモウラシル及び5−(2−ブロモ−1−クロロビニル)ウラシルからなる群から選択されるウラシル化合物を含む、請求項1に記載の方法。
【請求項14】
前記DPD阻害剤がエニルウラシル又はそのプロドラッグである、請求項1に記載の方法。
【請求項15】
前記DPD阻害剤がエニルウラシルであり、前記抗癌剤が5−FUである、請求項1に記載の方法。
【請求項16】
前記DPD阻害剤がエニルウラシルであり、前記抗癌剤がカペシタビンである、請求項1に記載の方法。
【請求項17】
前記DPD阻害剤がエニルウラシルであり、前記抗癌剤が5−FUであり、前記エニルウラシルが約16〜40mg/m2の用量で投与され、前記5−FUがその約11〜16時間後に約15〜50mg/m2の用量で投与される、請求項1に記載の方法。
【請求項18】
前記DPD阻害剤がエニルウラシルであり、前記抗癌剤が5−FUプロドラッグであり、前記エニルウラシルが約16〜40mg/m2の用量で投与され、前記5−FUプロドラッグがその約11〜16時間後に約40〜150mg/m2の用量で投与される、請求項1に記載の方法。
【請求項19】
前記5−FUプロドラッグがカペシタビンである、請求項18に記載の方法。
【請求項20】
前記DPD阻害剤がエニルウラシルであり、前記抗癌剤が5−FUであり、前記エニルウラシルが約16〜40mg/m2の用量で投与され、前記エニルウラシルが投与されてから前記エニルウラシルの少なくとも約3〜5排出半減期が経過したときに前記5−FUが約15〜50mg/m2の用量で投与される、請求項1に記載の方法。
【請求項21】
前記DPD阻害剤がエニルウラシルであり、前記抗癌剤が5−FUプロドラッグであり、前記エニルウラシルが約16〜40mg/m2の用量で投与され、前記エニルウラシルが投与されてから前記エニルウラシルの少なくとも約3〜5排出半減期が経過したときに前記5−FUプロドラッグが約40〜150mg/m2の用量で投与される、請求項1に記載の方法。
【請求項22】
前記5−FUプロドラッグがカペシタビンである、請求項21に記載の方法。
【請求項23】
前記DPD阻害剤がエニルウラシルであり、前記抗癌剤が5−FUであり、前記エニルウラシルが前記患者における神経及び非神経組織においてDPD活性を前記患者におけるベースラインDPD活性の5%未満に低下させるのに十分な用量で投与され、前記5−FUがその約11〜16時間後に約15〜50mg/m2の用量で投与される、請求項1に記載の方法。
【請求項24】
前記DPD阻害剤がエニルウラシルであり、前記抗癌剤が5−FUプロドラッグであり、前記エニルウラシルが前記患者における神経及び非神経組織においてDPD活性を前記患者におけるベースラインDPD活性の5%未満に低下させるのに十分な用量で投与され、前記5−FUプロドラッグがその約11〜16時間後に約40〜150mg/m2の用量で投与される、請求項1に記載の方法。
【請求項25】
前記5−FUプロドラッグがカペシタビンである、請求項24に記載の方法。
【請求項26】
前記DPD阻害剤がエニルウラシルであり、前記抗癌剤が5−FU又は5−FUプロドラッグであり、前記エニルウラシルが約16〜40mg/m2の用量で投与され、前記5−FU又は5−FUプロドラッグがその約11〜16時間後に、前記5−FU又は前記5−FUプロドラッグから生成される5−FUが前記DPD阻害剤の少なくとも10倍過剰で前記患者中に存在するような用量で投与される、請求項1に記載の方法。
【請求項27】
DPD阻害剤及び5−FU又は5−FUプロドラッグを含む徐放性経口医薬製剤であって、前記製剤を患者に投与後、前記DPD阻害剤が放出された後少なくとも約11〜16時間まで前記5−FU又は5−FUプロドラッグが実質的に放出されず、その放出後に前記5−FU又はプロドラッグから生成される5−FUが、前記患者に残留する前記DPD阻害剤の少なくとも約10倍過剰で前記患者中に存在する、徐放性経口医薬製剤。
【請求項1】
DPD阻害剤及び5−FU又は5−FUプロドラッグから選択される抗癌剤を含む組合せを用いて、癌患者の処置に付随する神経毒性を最小限にする方法であって、
神経組織と非神経組織の両方におけるDPD活性を実質的に消失させるのに十分な用量でDPD阻害剤をまず投与する工程、及び
その後に前記5−FU又は5−FUプロドラッグを投与する工程
を含み、前記5−FU又は5−FUプロドラッグが、前記5−FU又は前記5−FUプロドラッグから生成される5−FUが前記DPD阻害剤よりもかなり過剰に前記患者中に存在するような用量で投与される、方法。
【請求項2】
前記DPD阻害剤が約16〜40mg/m2の用量で投与される、請求項1に記載の方法。
【請求項3】
前記DPD阻害剤が約16〜25mg/m2の用量で投与される、請求項1に記載の方法。
【請求項4】
前記5−FU又は5−FUプロドラッグが、前記DPD阻害剤が投与されてから約11〜16時間後に投与される、請求項1に記載の方法。
【請求項5】
前記5−FU又は5−FUプロドラッグが、前記DPD阻害剤が投与されてから前記DPD阻害剤の少なくとも約3〜5排出半減期が経過したときに投与される、請求項1に記載の方法。
【請求項6】
前記DPD阻害剤が、前記患者における神経及び非神経組織において、DPD活性を前記患者におけるベースラインDPD活性の5%未満に低下させるのに十分な用量で投与される、請求項1に記載の方法。
【請求項7】
前記5−FU又は5−FUプロドラッグが、その投与時において前記5−FU又はプロドラッグから生成される5−FUが前記DPD阻害剤の少なくとも10倍過剰で前記患者中に存在するような用量で投与される、請求項1に記載の方法。
【請求項8】
前記5−FUプロドラッグが、5−フルオロウリジン、5−フルオロシチジン、5−フルオロ−2−デオキシウリジン、5−フルオロ−2−デオキシシチジン、5’−デオキシ−4’,5−フルオロウリジン、及び5−フルオロアラビノシルウラシル、5’−デオキシ−5−フルオロウリジン、1−(2−テトラヒドロフラニル)−5−フルオロウラシル、1−C1〜8アルキルカルバモイル−5−フルオロウラシル誘導体、1−(2−テトラヒドロフリル)−5−フルオロウラシル、5’−デオキシ−5−フルオロ−N−[(ペンチルオキシ)カルボニル]−シチジン(カペシタビン)、又は5−FUにin vivoで変換される化合物からなる群、及びリン酸エステルを含めたその5’−エステルから選択される、請求項1に記載の方法。
【請求項9】
前記抗癌剤が5−FUである、請求項1に記載の方法。
【請求項10】
前記抗癌剤がカペシタビンである、請求項1に記載の方法。
【請求項11】
前記DPD阻害剤が5−置換ウラシル化合物又はそのプロドラッグを含む、請求項1に記載の方法。
【請求項12】
前記DPD阻害剤が、ハロゲン原子、C2〜4アルケニル基、ハロゲンで置換されたC2〜4アルケニル基、C2〜6アルキニル基、ハロゲンで置換されたC2〜6アルキニル基、シアノ基、C1〜4アルキル基、又はハロゲンで置換されたC1〜4アルキル基で、5位が置換されたウラシル化合物を含む、請求項1に記載の方法。
【請求項13】
前記DPD阻害剤が、エニルウラシル、5−プロパ−1−イニルウラシル、5−シアノウラシル、5−プロパ−1−イニルウラシル、5−ブロモエチニルウラシル、5−(1−クロロビニル)ウラシル、5−ヨードウラシル、5−(2−ブロモビニル)ウラシル、(E)−5−(2−ブロモビニル)ウラシル、5−ヘキサ−1−イニルウラシル、5−ビニルウラシル、5−トリフルオロウラシル、5−ブロモウラシル及び5−(2−ブロモ−1−クロロビニル)ウラシルからなる群から選択されるウラシル化合物を含む、請求項1に記載の方法。
【請求項14】
前記DPD阻害剤がエニルウラシル又はそのプロドラッグである、請求項1に記載の方法。
【請求項15】
前記DPD阻害剤がエニルウラシルであり、前記抗癌剤が5−FUである、請求項1に記載の方法。
【請求項16】
前記DPD阻害剤がエニルウラシルであり、前記抗癌剤がカペシタビンである、請求項1に記載の方法。
【請求項17】
前記DPD阻害剤がエニルウラシルであり、前記抗癌剤が5−FUであり、前記エニルウラシルが約16〜40mg/m2の用量で投与され、前記5−FUがその約11〜16時間後に約15〜50mg/m2の用量で投与される、請求項1に記載の方法。
【請求項18】
前記DPD阻害剤がエニルウラシルであり、前記抗癌剤が5−FUプロドラッグであり、前記エニルウラシルが約16〜40mg/m2の用量で投与され、前記5−FUプロドラッグがその約11〜16時間後に約40〜150mg/m2の用量で投与される、請求項1に記載の方法。
【請求項19】
前記5−FUプロドラッグがカペシタビンである、請求項18に記載の方法。
【請求項20】
前記DPD阻害剤がエニルウラシルであり、前記抗癌剤が5−FUであり、前記エニルウラシルが約16〜40mg/m2の用量で投与され、前記エニルウラシルが投与されてから前記エニルウラシルの少なくとも約3〜5排出半減期が経過したときに前記5−FUが約15〜50mg/m2の用量で投与される、請求項1に記載の方法。
【請求項21】
前記DPD阻害剤がエニルウラシルであり、前記抗癌剤が5−FUプロドラッグであり、前記エニルウラシルが約16〜40mg/m2の用量で投与され、前記エニルウラシルが投与されてから前記エニルウラシルの少なくとも約3〜5排出半減期が経過したときに前記5−FUプロドラッグが約40〜150mg/m2の用量で投与される、請求項1に記載の方法。
【請求項22】
前記5−FUプロドラッグがカペシタビンである、請求項21に記載の方法。
【請求項23】
前記DPD阻害剤がエニルウラシルであり、前記抗癌剤が5−FUであり、前記エニルウラシルが前記患者における神経及び非神経組織においてDPD活性を前記患者におけるベースラインDPD活性の5%未満に低下させるのに十分な用量で投与され、前記5−FUがその約11〜16時間後に約15〜50mg/m2の用量で投与される、請求項1に記載の方法。
【請求項24】
前記DPD阻害剤がエニルウラシルであり、前記抗癌剤が5−FUプロドラッグであり、前記エニルウラシルが前記患者における神経及び非神経組織においてDPD活性を前記患者におけるベースラインDPD活性の5%未満に低下させるのに十分な用量で投与され、前記5−FUプロドラッグがその約11〜16時間後に約40〜150mg/m2の用量で投与される、請求項1に記載の方法。
【請求項25】
前記5−FUプロドラッグがカペシタビンである、請求項24に記載の方法。
【請求項26】
前記DPD阻害剤がエニルウラシルであり、前記抗癌剤が5−FU又は5−FUプロドラッグであり、前記エニルウラシルが約16〜40mg/m2の用量で投与され、前記5−FU又は5−FUプロドラッグがその約11〜16時間後に、前記5−FU又は前記5−FUプロドラッグから生成される5−FUが前記DPD阻害剤の少なくとも10倍過剰で前記患者中に存在するような用量で投与される、請求項1に記載の方法。
【請求項27】
DPD阻害剤及び5−FU又は5−FUプロドラッグを含む徐放性経口医薬製剤であって、前記製剤を患者に投与後、前記DPD阻害剤が放出された後少なくとも約11〜16時間まで前記5−FU又は5−FUプロドラッグが実質的に放出されず、その放出後に前記5−FU又はプロドラッグから生成される5−FUが、前記患者に残留する前記DPD阻害剤の少なくとも約10倍過剰で前記患者中に存在する、徐放性経口医薬製剤。
【図1】

【図2】
【図3】
【図2】
【図3】
【公表番号】特表2013−508293(P2013−508293A)
【公表日】平成25年3月7日(2013.3.7)
【国際特許分類】
【出願番号】特願2012−534373(P2012−534373)
【出願日】平成22年10月14日(2010.10.14)
【国際出願番号】PCT/US2010/052734
【国際公開番号】WO2011/047195
【国際公開日】平成23年4月21日(2011.4.21)
【出願人】(512098625)アドヘレックス テクノロジーズ, インコーポレイテッド (1)
【Fターム(参考)】
【公表日】平成25年3月7日(2013.3.7)
【国際特許分類】
【出願日】平成22年10月14日(2010.10.14)
【国際出願番号】PCT/US2010/052734
【国際公開番号】WO2011/047195
【国際公開日】平成23年4月21日(2011.4.21)
【出願人】(512098625)アドヘレックス テクノロジーズ, インコーポレイテッド (1)
【Fターム(参考)】
[ Back to top ]