
5−HT6アンタゴニストとしてのスルホニルピラゾリンカルボキサミジン誘導体
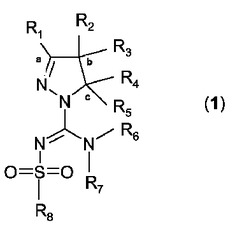
本発明は、5−HT6受容体のアンタゴニストとしてのスルホニルピラゾリンカルボキサミジン誘導体に、これらの化合物の製造方法に、そしてそれらの合成に有用な新規中間体に関する。本発明はまた、そのような化合物および組成物の使用、特にパーキンソン病、ハンチントン舞踏病、統合失調症、不安、鬱病、躁鬱病、精神病、癲癇、強迫神経症、気分障害、片頭痛、アルツハイマー病、加齢に伴う認識衰退、軽度認識障害、睡眠障害、摂食障害、食欲不振、過食症、無茶食い障害、パニック発作、静座不能、注意欠陥多動性障害、注意欠陥障害、コカイン、エタノール、ニコチンもしくはベンゾジアゼピンの乱用からの離脱、疼痛、脊椎外傷もしくは頭部損傷と関連する障害、水頭症、機能性腸疾患、過敏性腸症候群、肥満および2型糖尿病における治療効果を得るために患者にそれらを投与することにおけるそれらの使用にも関する。これらの化合物は一般式(1)
【化1】

[式中、記号は本明細書において示される意味を有する]
を有する。
【化1】
[式中、記号は本明細書において示される意味を有する]
を有する。
【発明の詳細な説明】
【索引】
【0001】
索引 頁
発明の名称 1
索引 1
技術分野 1
背景技術 1
開示 3
定義 8
略語 14
実施例1:分析方法 16
実施例2:合成の一般的態様 17
実施例3:ピラゾリン中間体の合成 19
実施例4:本発明の化合物の合成 41
実施例5:動物研究において使用する製剤 63
実施例6:薬理学的方法 63
実施例7:製薬学的製剤 65
文献目録 68
請求項 69
要約書 77
【技術分野】
【0002】
本発明は製薬および有機化学の分野に関し、そしてスルホニルピラゾリンカルボキサミジン誘導体、中間体、製剤および方法を提供する。
【背景技術】
【0003】
末梢および中枢神経系の重要な伝達物質、セロトニン(5−ヒドロキシトリプタミンもしくは5−HT)は、5−HT1、5−HT2、5−HT3、5−HT4、5−HT5、5−HT6および5−HT7と呼ばれる多数の受容体ファミリーを介して媒介される、広範囲の生理学的および病理学的機能を調節する。後者の3つの機能は他のものよりあまり理解されていないが、5−HTに媒介されるシグナル伝達を選択的に妨げる化合物は重要な新規薬剤標的であることは一般に認められている。
【0004】
ラット5−HT6受容体は、2つの異なるグループにより(非特許文献1;非特許文献2)、そして89%の配列同一性を共有するヒトのものはそのすぐ後にクローン化された(非特許文献3)。5−HT6受容体への最近の関心の多くは、いくつかの向精神薬がヒト5−HT6受容体での高親和性アンタゴニストであるからである(非特許文献3;非特許文献4)。これらの化合物には、アミトリプチリン(Ki=65nM)ならびに非定型抗精神病薬クロザピン(Ki=9.5nM)、オランザピン(Ki=10nM)およびクエチアピン(Ki=33nM)が包含される。しかしながら、これらの化合物のいずれも選択的ではない。報告される最初の選択的5−HT6受容体アンタゴニストは、Ro 04−6790およびRo 63−0563である。それらの有用性は、それらの中程度の親和性(それぞれ、Ki=50nMおよび12nM)および不十分な薬物動態により限定される(非特許文献5)。選択的5−HT6受容体アンタゴニストRo−04−6790およびSB−271046の最近の開発に伴い、認知機能のモデルにおけるこれらの化合物の活性に関するいくつかの報告がなされている。SB−271046は、モリス水迷路における能力を改善した(非特許文献6)。これらの結果は、5−HT6受容体配列に対
するアンチセンスオリゴヌクレオチドの長期脳室内投与がモリス水迷路における能力のいくつかの尺度の改善をもたらした結果と一致する(非特許文献7)。最近、ラットにおける食物摂取を減少する5−HT6アンタゴニストおよび5−HT6アンチセンスオリゴヌクレオチドの効果が報告されている(非特許文献8;非常特許文献9;非特許文献10)。肥満は、許容基準を上回る過剰体重をもたらす体脂肪含有量の増加を特徴とする症状である。肥満は西欧諸国における最も重大な栄養障害であり、そして全ての先進諸国における主要な健康問題である。この障害は心臓血管疾患、消化器系疾患、呼吸器系疾患、癌および2型糖尿病のような疾患の増加した発生率に起因する増加した死亡率をもたらす。
【0005】
5−HT6選択的リガンドは、パーキンソン病、ハンチントン舞踏病および/もしくは統合失調症、不安、鬱病、躁鬱病、精神病、癲癇、強迫神経症、気分障害、片頭痛、アルツハイマー病(認識記憶の増強)、加齢に伴う認識衰退、軽度認識障害、神経細胞成長障害を特徴とする神経変性疾患、睡眠障害、食欲不振および過食症のような摂食障害、無茶食い障害、パニック発作、静座不能、注意欠陥多動性障害(ADHD)、注意欠陥障害(ADD)、コカイン、エタノール、ニコチンおよびベンゾジアゼピンのような薬物乱用からの離脱、ならびに疼痛のような中枢神経系のある種の障害、ならびに脊椎外傷および/もしくは頭部損傷と関連する障害、例えば水頭症の処置もしくは予防において潜在的に有用であると同定されている。5−HT6選択的リガンドはまた、機能性腸疾患および過敏性腸症候群のようなある種の胃腸障害の処置においてそして肥満および2型糖尿病の処置もしくは予防において有用であり、体重のそして体重増加の減少をもたらすと予想される。体重のそして体重増加の減少(例えば、体重障害を処置すること)は、とりわけ食物摂取の減少によりもたらされる。
【0006】
本発明の目的は、ある種のCNS障害の処置に有用な化合物、既知の5−HT6アンタゴニストのいずれかと化学的に関係しない強力なそして選択的な5−HT6アンタゴニストを提供することであった。
【先行技術文献】
【非特許文献】
【0007】
【非特許文献1】Ruat,M.et al.(1993)Biochem.Biophys.Res.Commun.193:268−276
【非特許文献2】Sebben,M.et al.(1994)NeuroReport 5:2553−2557
【非特許文献3】Kohen,R.et al.(1996).J Neurochem 66(1):47−56
【非特許文献4】Roth,B.L.,et al.(1994).J Pharmacol Exp Ther 268(3):1403−10
【非特許文献5】Sleight,A.J.,et al.(1998).Br J Pharmacol 124(3):556−62
【非特許文献6】Rogers,D.C.,et al.(1999).Br J Pharmacol 127(suppl.).22P
【非特許文献7】Bentley,J.C.,et al.(1999b)Br J Pharmacol 126(7):1537−42
【非特許文献8】Bentley,J.C.et al.(1997)J.Psychopharmacol.Suppl.A64,255
【非特許文献9】Bentley,J.C.et al.(1999a)Br J Pharmacol.Suppl.126,P66
【非特許文献10】Woolley,M.L.et al.(2001)Neuropharmacology 41:210−219
【発明の開示】
【0008】
驚くべきことに、ある種のスルホニルピラゾリンカルボキサミジン誘導体は5−HT6受容体アンタゴニストであることが見出された。本発明は一般式(1):
【0009】
【化1】
【0010】
[式中:
−R1は水素、非置換のアルキル(C1〜4)基、1個もしくはそれ以上のハロゲン原子で置換されたアルキル(C1〜4)基、または場合により1個もしくはそれ以上のハロゲン原子で置換されていてもよいフェニル基を表し、
−R2およびR3は独立して水素、非置換のアルキル(C1〜4)基、1個もしくはそれ以上のハロゲン原子で置換されたアルキル(C1〜4)基、場合により1個もしくはそれ以上のハロゲン原子で置換されていてもよいアルキル(C1〜4)−O−アルキル(C1〜4)−フェニル基または場合により1個もしくはそれ以上のハロゲン原子で置換されていてもよいフェニル基を表すか、あるいは
R1およびR2は「a」および「b」の記号が付された炭素原子と一緒になってC5〜8−シクロアルキル環を形成するか、あるいは
R2およびR3は「b」の記号が付された炭素原子と一緒になってC3〜8−シクロアルキル環を形成するか、あるいは
R2およびR3は「b」の記号が付された炭素原子と一緒になって場合により置換されていてもよいC5〜8−ヘテロシクロアルキル環を形成するか、あるいは
−R4およびR5は独立して水素、非置換のアルキル(C1〜4)基、1個もしくはそれ以上のハロゲン原子で置換されたアルキル(C1〜4)基、場合により置換されていてもよい単環式芳香族基、場合により置換されていてもよい縮合した二環式芳香族基、場合により置換されていてもよい単環式ヘテロ芳香族基、場合により置換されていてもよい縮合した二環式ヘテロ芳香族基を表すか、あるいは
R3およびR4は「b」および「c」の記号が付された炭素原子と一緒になってC3〜8−シクロアルキル環を形成するか、あるいは
R3およびR4は「b」および「c」の記号が付された炭素原子と一緒になって場合により置換されていてもよいC5〜8−ヘテロシクロアルキル環を形成するか、あるいは
−R6およびR7は独立して水素原子、またはアルキル(C1〜4)基、または1個もしくはそれ以上のハロゲン原子で置換されたアルキル(C1〜4)基;あるいは(C1〜3)アルコキシ基、またはジアルキル(C1〜3)−アミノ−アルキル(C1〜3)基、または場合により置換されていてもよい単環式もしくは縮合した二環式芳香族もしくはヘテロ芳香族基、または場合により置換されていてもよいC5〜8−シクロアルキル基もしくは場合により置換されていてもよいC5〜8−ヘテロシクロアルキル基を表すか、あるいは
R6およびR7は、それらが結合している窒素原子と一緒になって、場合により置換されていてもよいC5〜8−ヘテロシクロアルキル基を形成し、
−R8は場合により置換されていてもよい単環式芳香族基、場合により置換されていてもよい縮合した二環式芳香族基、場合により置換されていてもよい単環式ヘテロ芳香族基、場合により置換されていてもよい縮合した二環式ヘテロ芳香族基、−CR9=CR10−アリール基(ここで、R9およびR10は独立して水素またはアルキル−(C1〜3)基を表す)、−C≡C−アリール基、場合により置換されていてもよいピペリジニル基、もしくは基−NR11R12(ここで、R11およびR12は独立して水素、アルキル−(C1〜3)基または場合により置換されていてもよいフェニルもしくはベンジル基を表す)を表す]
の化合物もしくは互変異性体、立体異性体、N−オキシド、同位体標識したアナログ、または前述のもののいずれかの薬理学的に許容しうる塩、水和物もしくは溶媒和物に関する。
【0011】
本発明は、式(1)を有する化合物のラセミ化合物、ジアステレオマーの混合物ならびに個々の立体異性体に関する。本発明はまた、式(1)を有する化合物のE異性体、Z異性体およびE/Z混合物にも関する。
【0012】
本発明は特に、
−R1が水素を表すかもしくはR1およびR2が「a」および「b」の記号が付された炭素原子と一緒になってシクロヘキシル環を形成し、
−R2およびR3が独立して水素もしくはアルキル(C1〜3)基を表すか、またはR2およびR3が「b」の記号が付された炭素原子と一緒になってシクロペンチルもしくはシクロヘキシル環を形成し、
−R4およびR5が独立して水素、アルキル(C1〜3)基を表すか、またはR3およびR4が「b」および「c」の記号が付された炭素原子と一緒になってC3〜8−シクロアルキル環を形成し、
−R6およびR7が独立して水素原子、またはアルキル(C1〜3)基、または1個
もしくはそれ以上のハロゲン原子で置換されたアルキル(C1〜4)基;またはメトキシ基、またはシクロヘキシル基、またはベンジル基、または4−ピペリジニル基を表し、
−R8が上記に示されるとおりの意味を有する
一般式(1)の化合物もしくは互変異性体、立体異性体、N−オキシド、同位体標識したアナログ、または前述のもののいずれかの薬理学的に許容しうる塩、水和物もしくは溶媒和物に関する。
【0013】
なおさらに特に、本発明は、R1、R4、R5およびR6が水素を表し、R2およびR3が独立してアルキル(C1〜3)基を表すか、またはR2およびR3が「b」の記号が付された炭素原子と一緒になってシクロペンチルもしくはシクロヘキシル環を形成し、R7がアルキル(C1〜3)基を表し、そしてR8が上記に示されるとおりの意味を有する一般式(1)の化合物もしくは互変異性体、立体異性体、N−オキシド、同位体標識したアナログ、または前述のもののいずれかの薬理学的に許容しうる塩、水和物もしくは溶媒和物に関する。
【0014】
別の態様において、本発明は、ピラゾリン環における2個の潜在的不斉炭素原子のいずれか一方もしくは両方が左旋性もしくは右旋性鏡像異性体である式(1)の化合物に関する。
【0015】
式(1)の本発明の化合物ならびにその薬理学的に許容しうる塩は、5−HT6受容体アンタゴニスト活性を有する。それらは、5−HT6受容体が関係するか、もしくはそれらの受容体の操作により処置できる障害を処置することにおいて有用である。例えば、パーキンソン病、ハンチントン舞踏病、統合失調症、不安、鬱病、躁鬱病、精神病、癲癇、強迫神経症、気分障害、片頭痛、アルツハイマー病、加齢に伴う認識衰退、軽度認識障害
、睡眠障害、摂食障害、食欲不振、過食症、無茶食い障害、パニック発作、静座不能、注意欠陥多動性障害、注意欠陥障害、コカイン、エタノール、ニコチンもしくはベンゾジアゼピンの乱用からの離脱、疼痛、脊椎外傷もしくは頭部損傷と関連する障害、水頭症、機能性腸疾患、過敏性腸症候群、肥満および2型糖尿病において。
【0016】
本発明の他の態様には:
例えば、5−HT6受容体をブロックすることにより処置できる障害もしくは症状を処置するための製薬学的組成物、該組成物は式(1)の化合物もしくはその製薬学的に許容しうる塩および製薬学的に許容しうる担体を含んでなる;
5−HT6受容体をブロックすることにより処置できる障害もしくは症状を処置する方法、該方法は式(1)の化合物もしくはその製薬学的に許容しうる塩をそのような処置を必要とする哺乳類に投与することを含んでなる;
例えば本明細書に記載される障害から選択される障害もしくは症状を処置するための製薬学的組成物;
本明細書に記載される障害から選択される障害もしくは症状を処置する方法、該方法は式(1)の化合物もしくはその製薬学的に許容しうる塩をそのような処置を必要とする哺乳類に投与することを含んでなる;
本明細書に記載される障害から選択される障害もしくは症状を処置するための製薬学的組成物、該組成物は式(1)の化合物もしくはその製薬学的に許容しうる塩および製薬学的に許容しうる担体を含んでなる;
本明細書に記載される障害から選択される障害もしくは症状を処置する方法、該方法は式(1)の化合物もしくはその製薬学的に許容しうる塩をそのような処置を必要とする患者に投与することを含んでなる;
式(1)の化合物の有効量をそれを必要とする患者に投与することを含んでなる5−HT6受容体に拮抗する方法
が包含されるがこれらに限定されるものではない。
【0017】
本発明はまた、薬剤の製造のための式(1)の化合物もしくは塩の使用も提供する。
【0018】
本発明はさらに、本発明の化合物もしくはその製薬学的に許容しうる塩、または本発明の化合物を含んでなる製薬学的組成物もしくは製剤が、記載される症状の1つもしくはそれ以上を処置するために、もう一つのもしくは複数の治療薬と同時にもしくは逐次もしくは組み合わせた製剤として投与される併用療法に関する。そのような他の治療薬(1つもしくは複数)は、本発明の化合物の投与の前に、それと同時に、もしくはその後に投与することができる。
【0019】
本発明はまた、本明細書に記載される障害から選択される障害もしくは症状を処置するための化合物、製薬学的組成物、キットおよび方法も提供し、該方法は式(1)の化合物もしくはその製薬学的に許容しうる塩をそのような処置を必要とする患者に投与することを含んでなる。
【0020】
本発明の化合物は、5−HT6受容体拮抗活性を保有する。本発明の化合物のこの活性は、例えば、本明細書に記述されるかもしくは当該技術分野において既知であるアッセイの1つもしくはそれ以上を用いて容易に示される。
【0021】
本発明はまた、本発明の化合物を製造する方法およびそれらの方法において使用される中間体も提供する。
【0022】
本明細書に記述される化合物および中間体の単離および精製は、所望に応じて、例えば濾過、抽出、結晶化、カラムクロマトグラフィー、薄層クロマトグラフィー、厚層クロマ
トグラフィー、分取低もしくは高圧液体クロマトグラフィー、またはこれらの方法の組み合わせのような任意の適当な分離もしくは精製方法によりもたらすことができる。適当な分離および単離方法の特定の実例は、製造および実施例から取ることができる。しかしながら、他の同等な分離もしくは単離方法もまたもちろん用いることができる。
【0023】
本発明の化合物は1個もしくはそれ以上の不斉中心を含有する可能性があり、従って、ラセミ化合物およびラセミ混合物、単一の鏡像異性体、ジアステレオマー混合物および個々のジアステレオマーとして存在することができる。
【0024】
様々な置換基の性質により、分子は追加の不斉中心を有し得る。各々のそのような不斉中心は、独立して2個の光学異性体をもたらす。混合物におけるそして純粋なもしくは部分的に精製された化合物としての、可能な光学異性体およびジアステレオマーの全ては本発明に属する。本発明は、これらの化合物の全てのそのような異性体を包含する。式(1)は、好ましい立体化学なしに化合物のクラスの構造を示す。これらのジアステレオマーの独立した合成もしくはそれらのクロマトグラフィー分離は、当該技術分野において既知であるようにそこに開示される方法論の適切な改変により成し遂げられることができる。それらの絶対立体化学は、必要に応じて、既知の絶対立体配置の不斉中心を含有する試薬で誘導体化される、結晶性生成物もしくは結晶性中間体のX線結晶学により決定されることができる。化合物のラセミ混合物は、ジアステレオマー混合物を生成せしめるための鏡像異性的に純粋な化合物への化合物のラセミ混合物のカップリング、続いて分別結晶もしくはクロマトグラフィーのような標準的な方法による個々のジアステレオマーの分離のような当該技術分野において周知である方法により個々の鏡像異性体に分離することができる。カップリングは、鏡像異性的に純粋な酸もしくは塩基、例えば(−)−ジ−p−トルオイル−D−酒石酸および/もしくは(+)−ジ−p−トルオイル−L−酒石酸を用いる塩の形成からなることが多い。次に、ジアステレオマー誘導体は、付加したキラル残基の切断により純粋な鏡像異性体に転化することができる。化合物のラセミ混合物はまた、キラル固定相を利用してクロマトグラフィー方法:当該技術分野において周知である方法により直接分離することもできる。あるいはまた、化合物の任意の鏡像異性体は、当該技術分野において周知である方法により既知の立体配置の光学的に純粋な出発物質もしくは試薬を用いて立体選択的合成により得ることができる。
【0025】
式(1)の化合物もしくはその製薬学的に許容しうる塩のシスおよびトランス異性体もまた本発明に属し、そしてこれはまた式(1)の化合物もしくはその製薬学的に許容しうる塩の互変異性体にも適用される。
【0026】
化合物の結晶形態のあるものは多形として存在する可能性があり:そのようなものとして本発明に属するものとする。さらに、化合物のあるものは水(すなわち、水和物)もしくは一般的な有機溶媒と溶媒和物を形成し得る。そのような溶媒和物もまた、本発明の範囲内に入る。
【0027】
PETもしくはSPECTにより検出可能であるように同位体標識された式(1)の化合物を包含する式(1)もしくはその製薬学的に許容しうる塩の同位体標識化合物もまた、本発明の範囲内に入る。同じことが、受容体結合もしくは代謝研究に適当な、[13C]−、[14C]−、[3H]−、[18F]−、[125I]−もしくは他の同位体濃縮原子で標識された式(1)の化合物に適用される。
【0028】
本発明の化合物はまた、神経学的機能、機能障害および疾患の生化学的研究における試薬もしくは基準として用いることもできる。
【0029】
[定義]
本明細書に開示される化合物の記述において使用する一般用語は、それらの通常の意味を有する。アルキルという用語は、本明細書において用いる場合、1価の飽和した分枝状もしくは直線状炭化水素鎖を意味する。他に記載されない限り、そのような鎖は1〜18個の炭素原子を含有することができる。そのようなアルキル基の代表は、メチル、エチル、プロピル、イソプロピル、ブチル、イソブチル、sec−ブチル、tert−ブチル、ペンチル、イソペンチル、ネオペンチル、tert−ペンチル、ヘキシル、イソヘキシル、ヘプチル、オクチル、ノニル、デシル、ウンデシル、ドデシル、トリデシル、テトラデシル、ペンタデシル、ヘキサデシル、ヘプタデシル、オクタデシルなどである。「低級」と限定される場合、アルキル基は1〜6個の炭素原子を含有する。同じ炭素含有量は、親用語「アルカン」に、そして「アルコキシ」のような派生用語に適用される。様々な炭化水素含有部分の炭素含有量は、該部分における炭素原子の最小および最大数を示す接頭辞により示され、すなわち、接頭辞Cx〜Cyは、包括的に整数「x」から整数「y」まで存在する炭素原子の数を定義する。例えば、「アルキル(C1〜3)」はメチル、エチル、n−プロピルもしくはイソプロピルを意味し、そして「アルキル(C1〜4)」は「メチル、エチル、n−プロピル、イソプロピル、n−ブチル、2−ブチル、イソブチルもしくは2−メチル−n−プロピル」を意味する。「アルケニル」という用語は、ビニル、アリル、ブテニルなどのような1個もしくはそれ以上の炭素−炭素二重結合を有する直線状もしくは分枝状炭化水素基を意味し、そして例えば(C2〜4)アルケニルを表す。「アルキニル」基において直線状もしくは分枝状炭化水素基は、エチニル、プロパルギル、1−ブチニル、2−ブチニルなどのような、1個もしくはそれ以上の炭素−炭素三重結合を有し、そして例えば(C2〜4)アルキニルを表す。他に記載されない限り、「アルケニル」および「アルキニル」鎖は1〜18個の炭素原子を含有することができる。「アシル」という用語は、アルキル(C1〜3)カルボニル、アリールカルボニルもしくはアリール−アルキル(C1〜3)カルボニルを意味する。
【0030】
「アリール」という用語は、フリル、チエニル、ピロリル、オキサゾリル、チアゾリル、イミダゾリル、イミダゾ[2,1−b][1,3]チアゾリル、ピラゾリル、イソオキサゾリル、イソチアゾリル、ピリジル、ピリダジニル、ピリミジニル、ピラジニル、1,3,5−トリアジニル、フェニル、インダゾリル、インドリル、インドリジニル、イソインドリル、ベンゾ[b]フラニル、1,2,3,4−テトラヒドロ−ナフチル、1,2,3,4−テトラヒドロイソキノリニル、インダニル、インデニル、ベンゾ[b]チエニル、2,3−ジヒドロ−1,4−ベンゾジオキシン−5−イル、ベンズイミダゾリル、ベンゾチアゾリル、ベンゾ[1,2,5]チア−ジアゾリル、プリニル、キノリニル、イソキノリニル、フタラジニル、キナゾリニル、キノキサリニル、1,8−ナフチリジニル、ナフチル、プテリジニルもしくはアズレニルが包含されるがこれらに限定されるものではない単環式もしくは縮合した二環式芳香族もしくはヘテロ芳香族基を包含する。「ハロ」もしくは「ハロゲン」はクロロ、フルオロ、ブロモもしくはヨードを意味し;「ヘテロアルキル、ヘテロ芳香族」などにおける場合に「ヘテロ」は、1個もしくはそれ以上のN、OもしくはS原子を含有することを意味する。「ヘテロアルキル」には任意の位置にヘテロ原子を有するアルキル基が包含され、従ってN−結合した、O−結合したもしくはS−結合したアルキル基が包含される。
【0031】
「置換された」という用語は、特定の基もしくは部分が1個もしくはそれ以上の置換基を有することを意味する。任意の基が多数の置換基を保有する可能性があり、そして様々な可能な置換基が提供される場合、これらの置換基は独立して選択され、そして同じものである必要はない。「非置換の」という用語は、特定の基が置換基を保有しないことを意味する。「場合により置換されていてもよい」は、基がC1〜8アルキル、C1〜8アルケニル、C1〜8アルキニル、アリール、フルオロ、クロロ、ブロモ、ヒドロキシル、C1〜8アルキルオキシ、C1〜8アルケニルオキシ、アリールオキシ、アシルオキシ、アミノ、C1〜8アルキルアミノ、ジアルキル(C1〜8)−アミノ、アリールアミノ、チ
オ、C1〜8アルキルチオ、アリールチオ、アルキルスルホニル、アリールスルホニル、アルキルスルフィニル、アリールスルフィニル、シアノ、オキソ、ニトロ、アシル、アミド、C1〜8アルキルアミド、ジアルキル(C1〜8)アミド、カルボキシルから選択される1個もしくはそれ以上の基でさらに置換されるかもしくはそうでない可能性があり、または2個の任意の置換基が、それらが結合している炭素原子と一緒になって、窒素、酸素もしくは硫黄から選択される0、1もしくは2個のヘテロ原子を含有する5もしくは6員の芳香もしくは非芳香環を形成することを意味する。任意の置換基は、それら自体が追加の任意の置換基を保有し得る。好ましい任意の置換基には、例えばメチル、エチルおよびトリフルオロメチルのようなC1〜3アルキル、フルオロ、クロロ、ブロモ、ヒドロキシル、例えばメトキシ、エトキシおよびトリフルオロメトキシのようなC1〜3アルキルオキシ、ならびにアミノが包含される。置換基に関連して、「独立して」という用語は、1個より多くのそのような置換基が可能である場合に、それらが同じものであるかもしくは相互に異なり得ることを意味する。
【0032】
「C3〜8シクロアルキル」は、シクロプロピル、シクロブチル、シクロペンチル、シクロヘキシル、シクロヘプチルもしくはシクロオクチルを意味し;「C5〜8ヘテロシクロアルキル」は、ピペリジニル、モルホリニル、アゼパニル、ピロリジニル、チオモルホリニル、ピペラジニル、テトラヒドロフリル、テトラヒドロピラニルが包含されるがこれらに限定されるものではないヘテロ原子含有環をさし;「C5〜10ビシクロアルキル基」は、ビシクロ[2.2.1]ヘプタニル、ビシクロ[3.3.0]オクタニルもしくはビシクロ[3.1.1]ヘプタニル基が包含されるがこれらに限定されるものではない炭素二環式環系をさし;「C6〜10トリシクロアルキル基」は、1−アダマンチル、ノルアダマンチルもしくは2−アダマンチル基が包含されるがこれらに限定されるものではない炭素三環式環系をさす。「C8〜11テトラシクロアルキル基」という略語は、キュビル、ホモキュビルもしくはビスホモキュビル基が包含されるがこれらに限定されるものではない炭素四環式環系をさす。
【0033】
「オキシ」、「チオ」および「カルボ」という用語は、別の基の一部として本明細書において用いる場合、それぞれ、例えばヒドロキシル、オキシアルキル、チオアルキル、カルボキシアルキルなどのような2つの基の間のリンカーとして働く、酸素原子、硫黄原子およびカルボニル(C=O)基をさす。「アミノ」という用語は、単独でもしくは別の基の一部として本明細書において用いる場合、末端であるかもしくは2つの他の基の間のリンカーであり得る窒素原子をさし、ここで、該基は第一級、第二級もしくは第三級(それぞれ、窒素原子に結合している2個の水素原子、窒素原子に結合している1個の水素原子、そして窒素原子に結合している水素原子がない)アミンであることができる。「スルフィニル」および「スルホニル」という用語は、別の基の一部として本明細書において用いる場合、それぞれ−SO−もしくは−SO2−基をさす。
【0034】
本明細書において用いる場合、「脱離基」(L)という用語は、置換(substitution)もしくは置換(displacement)反応中に離脱する荷電もしくは非荷電原子もしくは基を意味するものとする。該用語は、アミン、チオールもしくはアルコール求核試薬のような求核試薬により容易に置換可能な基をさす。そのような脱離基は、当該技術分野において周知である。例には、N−ヒドロキシスクシンイミド、N−ヒドロキシベンゾトリアゾール、ハロゲン化物(Br、Cl、I)、トリフラート、メシレート、トシレートなどが包含されるがこれらに限定されるものではない。
【0035】
上記の化合物のN−オキシドは、本発明に属する。第三級アミンは、N−オキシド代謝物を生じるかもしくはそうでない可能性がある。N−酸化が起こる程度は、微量からほぼ定量的転化まで様々である。N−酸化物は、それらの対応する第三級アミンより活性であるか、もしくは活性が低い可能性がある。N−酸化物は化学的手段によりそれらの対応す
る第三級アミンに容易に還元することができるが、人体においてこれは様々な程度に起こる。あるN−酸化物は、対応する第三級アミンへのほぼ定量的な還元的転化を受け、ある場合には転化はほんの微量の反応であるか、もしくは完全にないことさえある(Bickel,1969)。
【0036】
生物活性剤(すなわち、式(1)の化合物)を与えるようにインビボで代謝される任意の化合物は、本願の範囲および精神の範囲内のプロドラッグである。プロドラッグは、それ自体不活性であるが1種もしくはそれ以上の活性代謝物に転化される治療薬である。従って、本発明の処置の方法において、「投与すること」という用語は特に開示される化合物で、もしくは特に開示されないが患者への投与後にインビボで特定の化合物に転化する化合物で記述される様々な障害を処置することを包含するものとする。プロドラッグは、親薬剤分子の有用性へのいくつかの障害を克服するために用いられる薬剤分子の生可逆的(bioreversible)誘導体である。これらの障害には、溶解性、透過性、安定性、前全身的(presystemic)代謝およびターゲッティング制限が包含されるがこれらに限定されるものではない(Bundgaard,1985;King,1994;Stella,2004;Ettmayer,2004;Jaervinen,2005)。プロドラッグ、すなわち、任意の既知の経路によりヒトに投与した場合に式(1)を有する化合物に代謝される化合物は、本発明に属する。特に、これは第一級もしくは第二級アミノまたはヒドロキシ基を有する化合物に関する。そのような化合物は、有機酸と反応させて例えばアミジン、エナミン、マンニッヒ塩基、ヒドロキシル−メチレン誘導体、O−(アシルオキシメチレンカルバメート)誘導体、カルバメート、エステル、アミドもしくはエナミノン、しかしこれらに限定されるものではない、投与後に容易に取り除かれる付加的基が存在する式(1)を有する化合物を生成せしめることができる。
【0037】
「結晶形態」は、同じ化合物の様々な固体形態、例えば多形、溶媒和物および非晶質形態をさす。「多形」は、化合物が異なる結晶充填配置において結晶化することができる結晶構造であり、これらは全て同じ元素組成を有する。多形は、温度、過飽和のレベル、不純物の存在、溶媒の極性、冷却の速度のようないくつかの結晶化条件により影響される、高い頻度で起こる現象である。異なる多形は通常は異なるX線回折パターン、固体状態NMRスペクトル、赤外もしくはラマンスペクトル、融点、密度、硬度、結晶形、光学および電気特性、安定性ならびに溶解性を有する。再結晶溶媒、結晶化の速度、貯蔵温度および他の因子は、1つの結晶形態を顕著にさせ得る。「溶媒和物」は一般に溶媒の化学量論もしくは非化学量論量のいずれかを含有する結晶形態である。結晶化の過程中に、ある化合物は結晶性固体状態において溶媒分子の固定モル比を捕捉する傾向を有することが多く、このようにして溶媒和物を形成する。溶媒が水である場合、「水和物」が形成され得る。式(1)の化合物およびその製薬学的に許容しうる塩は、水和物もしくは溶媒和物の形態で存在することができ、そしてそのような水和物および溶媒和物もまた本発明に包含される。その例には、1/10水和物、1/4水和物、1/2水和物、1水和物、2塩酸塩1/2水和物、2塩酸塩2水和物、2塩酸塩3/2水和物などが包含される。「非晶質」形態は長距離秩序を有さない非結晶性物質であり、そして一般に特有の粉末X線回折パターンを与えない。結晶形態は一般に、Byrn(1995)およびMartin(1995)により記述されている。
【0038】
より簡潔な記述を提供するために、本明細書に示される量的表現のいくつかは「約」という用語で修飾されない。「約」という用語が明白に使用されようとなかろうと、本明細書に示されるあらゆる量は実際の既定値をさすものとし、そしてそれはまたそのような既定値の実験および/もしくは測定条件に起因する近似を包含する当該技術分野における通常の技量に基づいて合理的に推測されるそのような既定値への近似もさすものとすることが理解される。
【0039】
「選択的」および「選択性」という用語は、別の受容体(例えば、他の5−HT受容体サブタイプ)に対する実質的な交差反応性を示すことなしに特定の受容体(例えば5−HT6受容体)に対する反応性を示す化合物をさす。従って、例えば、本発明の選択的化合物は、他の5−HT受容体に対する実質的な交差反応性を示すことなしに5−HT6受容体に対する反応性を示すことができる。1つの態様において、本発明の化合物は5−HT6受容体に対する少なくとも約10倍の選択性、5−HT6受容体に対する少なくとも約50倍の選択性、5−HT6受容体に対する少なくとも約100倍の選択性、5−HT6受容体に対する少なくとも約250倍の選択性もしくは所望の標的に対する少なくとも約500倍の選択性を有する。
【0040】
本明細書の記述および請求項の全体にわたって、「含んでなる」という語および「含んでなること」および「含んでなる(comprises)」のような該語のバリエーションは、他の添加物、成分、整数もしくは段階を除くものではない。
【0041】
式(1)の化合物はそのままの(raw)化学物質として投与することが可能であり得るが、それらを「製薬学的組成物」として与えることが好ましい。さらなる態様に従って、本発明はその1つもしくはそれ以上の製薬学的に許容しうる担体および場合により1つもしくはそれ以上の他の治療成分と一緒に、式(1)の化合物またはその製薬学的に許容しうる塩もしくは溶媒和物を含んでなる製薬学的組成物を提供する。担体(1つもしくは複数)は、製剤の他の成分と適合しそしてそのレシピエントに有害でないという意味で「許容可能」でなければならない。
【0042】
「組成物」という用語は、本明細書において用いる場合、既定の量もしくは割合の特定の成分を含んでなる生成物ならびに特定量の特定の成分を合わせることから直接もしくは間接的に生じる任意の生成物を包含する。製薬学的組成物に関連して、この用語は1種もしくはそれ以上の有効成分、および不活性成分を含んでなる任意の担体を含んでなる生成物、ならびに任意の2種もしくはそれ以上の成分の組み合わせ、錯体形成もしくは凝集から、または1種もしくはそれ以上の成分の解離から、または1種もしくはそれ以上の成分の他のタイプの反応もしくは相互作用から直接もしくは間接的に生じる任意の生成物を包含する。一般に、製薬学的組成物は、有効成分を液状担体もしくは微粉化固形担体もしくは両方と均一にそして密接に会合させ、そして次に必要に応じて生成物を所望の製剤に成形することにより製造される。製薬学的組成物は、疾患の進行もしくは症状の際に所望の効果をもたらすために十分な活性目的化合物を含む。従って、本発明の製薬学的組成物は、本発明の化合物および製薬学的に許容しうる担体を混合することにより製造される任意の組成物を包含する。「製薬学的に許容しうる」は、担体、希釈剤もしくは賦形剤が製剤の他の成分と適合しそしてそのレシピエントに有害でない必要があることを意味する。
【0043】
本願の文脈内で、「組み合わせ製剤」という用語は、錠剤もしくは注入液のような1つの製剤において物理的に組み合わされた式(1)の化合物および1種もしくはそれ以上の他の薬剤を意味する真の組み合わせ、ならびに場合により成分化合物の投与の順守を容易にするためのさらなる手段、例えばラベルもしくは図面を有して、使用説明書と一緒に、別個の投与形態物において式(1)の化合物および1種もしくはそれ以上の他の薬剤を含んでなる「キットオブパーツ(kit−of−parts)」の両方を含んでなる。真の組み合わせで、定義による薬物療法は同時である。「キットオブパーツ」の中身は、同時にもしくは異なる時間間隔で投与することができる。同時もしくは逐次のいずれかである治療は、使用する他の薬剤の特徴、作用の発現および期間、血漿レベル、クリアランスなどのような特徴、ならびに疾患、その段階および個々の患者の特徴により決まる。
【0044】
5−HT6受容体に対する本発明の化合物の親和性は、上記のように決定された。式(1)の既定化合物について測定される結合親和性から、理論的最低有効用量を概算するこ
とができる。測定されるKi値の2倍に等しい化合物の濃度で、5−HT6受容体のほぼ100%が化合物により占められると思われる。患者のkg当たりの化合物のmgにその濃度を転化することにより、理想的な生物学的利用能を仮定して理論的最低有効用量がもたらされる。薬物動態、薬力学および他の考慮事項は、実際に投与される用量をより高いかもしくはより低い値に改変し得る。有効成分の典型的な毎日用量は広い範囲内で異なり、そして関連する適応症、投与の経路、患者の年齢、体重および性別のような様々な因子により決まり、そして医師により決定され得る。一般に、単一もしくは個々の用量における患者への総毎日用量投与は、例えば、毎日0.001〜10mg/kg体重そしてさらに通常は1日当たり0.01〜1,000mgの総有効成分の量であることができる。そのような投薬量は毎日1〜3回、もしくは効能に必要とされる限り何度も、そして少なくとも2ヶ月の期間にわたって、より典型的には少なくとも6ヶ月間、もしくは長期的に処置を必要とする患者に投与される。
【0045】
「治療的に有効な量」という用語は、本明細書において用いる場合、本発明の組成物を投与することにより処置できる症状を処置するための治療薬の量をさす。その量は、組織系、動物もしくはヒトにおいて検出可能な治療もしくは改善反応を示すために十分な量である。効果には、例えば本明細書に記載される症状を処置することを包含することができる。患者の正確な有効量は、患者のサイズおよび健康、処置する症状の性質および程度、処置する医師(研究者、獣医、医師もしくは他の臨床医)の推奨、ならびに投与のために選択される治療法もしくは治療法の組み合わせにより決まる。従って、前もって正確な有効量を特定することは有用でない。
【0046】
「製薬学的に許容しうる塩」という用語は、適切な医学的判断の範囲内で、過度の毒性、刺激、アレルギー反応などなしにヒトおよび下等動物の組織と接触して使用するのに適当でありそして妥当な利益/危険比に相応する塩をさす。製薬学的に許容しうる塩は、当該技術分野において周知である。それらは、本発明の化合物を最終的に単離しそして精製する場合にインサイチューで、または無機もしくは有機塩基および無機もしくは有機酸を包含する製薬学的に許容しうる無毒の塩基もしくは酸とそれらを反応させることにより別個に製造することができる(Berge,1977)。「遊離塩基」形態は、塩を塩基もしくは酸と接触させ、そして常法において親化合物を単離することにより再生することができる。化合物の親形態は、極性溶媒における溶解性のようなある種の物理的性質において様々な塩形態と異なるが、そのほかの点ではこれらの塩は本発明の目的のために化合物の親形態と同等である。
【0047】
「錯体」は、金属イオンと錯体形成した本発明、例えば式(1)の化合物の錯体をさし、ここで、少なくとも1個の金属イオンはキレートされるかもしくは封鎖される。錯体は、当該技術分野において周知である方法により製造される(Dwyer,1964)。
【0048】
「処置」という用語は、本明細書において用いる場合、哺乳類、例えばヒト症状もしくは疾患の任意の処置をさし、そして:(1)疾患もしくは症状を抑制すること、すなわち、その発症を止めること、(2)疾患もしくは症状を軽減すること、すなわち、症状を退行させること、または(3)疾患の症候を止めることが包含される。
【0049】
「抑制する」という用語には、進行、重症度もしくは結果として起こる症候を妨げること、防ぐこと、抑えること、軽減すこと、改善することおよび遅くすること、止めることもしくは回復させることを包含するその一般に認められている意味が包含される。そのようなものとして、本発明の方法には、必要に応じて、医学的治療および/もしくは予防投与の両方が包含される。
【0050】
本明細書において用いる場合、「医学的治療」という用語には、ヒトもしくは他の哺乳
類にインビボもしくはエクスビボで実施される予防、診断および治療処方計画が包含されるものとする。
【0051】
哺乳類には、ウシ、ヒツジおよびブタ動物のような経済的に重要な動物、特に肉を生産するもの、ならびに家畜、運動競技動物(sports animals)、動物園の動物およびヒトが包含され、後者が好ましい。「患者」という用語は、本明細書において用いる場合、処置、観察もしくは実験の対象である動物、好ましくは哺乳類、最も好ましくはヒトをさす。
【0052】
本明細書において用いる場合、「体重障害」という用語は、異常な(例えば過剰)体重をもたらす、エネルギー摂取とエネルギー消費との間の不均衡により引き起こされる障害をさす。そのような体重障害には肥満が包含される(Roth,1994;Sibley,1993;Sleigh,1995,1997)。「肥満」は、ヒトが少なくとも25.9の、身長の二乗当たりの体重(kg/m2)として計算される、ボディ・マス・インデックス(BMI)を有する症状をさす。通常、正常体重を有するヒトは、19.9〜25.9未満のBMIを有する。本明細書における肥満は、遺伝的であろうと環境的であろうと、任意の原因に起因し得る。肥満をもたらすかもしくは肥満の原因であり得る障害の例には、過食および過食症、多嚢胞卵巣疾患、頭蓋咽頭腫、プラダー・ウィリ症候群、フレーリッヒ症候群、II型糖尿病、GH欠損患者、正常バリアント低身長(normal
variant short stature)、ターナー症候群ならびに低下した代謝活性もしくは総除脂肪量の割合としての安静時エネルギー消費量の減少を示す他の病状、例えば急性リンパ芽球性白血病を有する子供が包含される。
【0053】
略語
ACE−クロリド クロロギ酸1−クロロエチル
ACN アセトニトリル
AcOH 酢酸
ADD 注意欠陥障害
ADHD 注意欠陥多動性障害
API 大気圧イオン化
BMI ボディ・マス・インデックス
n−BuOH n−ブタノール
t−BuOH t−ブタノール
(t)−BOC (第三級)−ブトキシカルボニル
CHO チャイニーズハムスター卵巣(細胞)
CNS 中枢神経系
CUR カーテンガス
DBU 1,8−ジアザビシクロ[5.4.0]ウンデク−7−エン
(1,2)−DCE (1,2)−ジクロロエタン
DCM ジクロロメタン
DF 偏向器電圧
DIPEA N,N−ジイソプロピルエチルアミン
DMAP 4−ジメチルアミノピリジン
DMC 2−クロロ−1,3−ジメチルイミダゾリニウムクロリド
DMF N,N’−ジメチルホルムアミド
DMSO ジメチルスルホキシド
EA 酢酸エチル
EP 入口電位(entrance potential)
EtOAc 酢酸エチル
EtOH エタノール
Et2O ジエチルエーテル
FCS ウシ胎仔血清
FP 集束電位(focusing potential)
g グラム
h 時間
5−HT 5−ヒドロキシトリプタミン、セロトニン
KOtBu カリウムtert−ブトキシド
MeI ヨウ化メチル
MeOH メタノール
mg ミリグラム
min 分
mlもしくはmL ミリリットル
m.p. 融点、c.q.融解範囲
MsCl メタンスルホニルクロリド(塩化メシル)
MTBE メチルtert−ブチルエーテル
NaHMDS ナトリウムヘキサメチルジシラザン
NEB ネブライザーガス
PA 石油エーテル(40〜60)
p−TsOH パラトルエンスルホン酸
Rf 保持係数(薄層クロマトグラフィー)
Rt 保持時間(LC/MS)
RT 室温
SCX 強陽イオン交換
TBAB 臭化テトラブチルアンモニウム
TEA トリエチルアミン
TEM 温度
TFA トリフルオロ酢酸
THF テトラヒドロフラン
【実施例1】
【0054】
分析方法
核磁気共鳴スペクトル(1H NMR)は、他に示されない限り、300KでBruker ARX400(1H:400MHz)もしくはVarian VXR200(1H:200MHz)装置を用いて示される溶媒において決定された。スペクトルは、Cambridge Isotope Laboratories Ltdから入手した重水素化クロロホルムもしくはDMSOにおいて決定された。化学シフト(δ)は、テトラメチルシラン(1H)からppmダウンフィールド単位(in ppm downfield)で示される。カップリング定数Jは、Hz単位で示される。NMRスペクトルにおけるピーク形状は、記号「q」(カルテット)、「dq」(ダブルカルテット)、「t」(トリプレット)、「dt」(ダブルトリプレット)、「d」(ダブレット)、「dd」(ダブルダブレット)、「ddd」(ダブルダブルダブレット)、「s」(シングレット)、「bs」(ブロードなシングレット)および「m」(マルチプレット)で示される。NHおよびOHシグナルは、サンプルを1滴のD2Oと混合した後に同定された。
【0055】
フラッシュクロマトグラフィーは、示される溶離剤およびシリカゲル(Merckシリカゲル60:0.040〜0.063mm)を用いる精製をさす。融点は、Buechi B−545融点装置上で記録された。湿気および/もしくは酸素に敏感な化合物を含む全ての反応は、無水窒素雰囲気下で実施された。反応は、示される溶離剤でシリカ被覆ガラスプレート(Merckプレコートシリカゲル60 F254)上で薄層クロマトグラフィー(TLC)を用いることによりモニターされた。スポットは、UV光(254nm)もしくはI2により視覚化された。
【0056】
液体クロマトグラフィー−質量分析(LC−MS):LC−MS系は、2台のPerkin Elmerシリーズ200マイクロポンプからなった。これらのポンプは、Gilson 215オートサンプラーに連結された、50μlのT字型ミキサーにより相互に連結された。方法は下記のとおりであった:
段階 総時間 流量(μl/分) A(%) B(%)
0 0 2000 95 5
1 1.8 2000 0 100
2 2.5 2000 0 100
3 2.7 2000 95 5
4 3.0 2000 95 5
A=0.025%HCOOHおよび10mmol NH4HCOO pH=±3を有する100%水
B=0.025%HCOOHを有する100%ACN
【0057】
オートサンプラーは2μlの注入ループを有し、そして3μm粒子を有するWaters Atlantis C18 30*4.6mmカラムに連結された。カラムは、40℃でPerkin Elmerシリーズ200カラムオーブンにおいてサーモスタットで調節された。カラムは、2.7μlフローセルを有するPerkin Elmerシリーズ200UV計に連結された。波長は254nmに設定された。UV計は、Sciex API 150EX質量分析計に連結された。質量分析計は以下のパラメーターを有した:
走査範囲:150〜900a.m.u.;極性:陽性;走査形態:プロフィール;分解能
Q1:UNIT;刻み幅:0.10a.m.u.;走査当たりの時間:0.500sec;NEB:10;CUR:10 IS:5200;TEM:325;DF:30;FP:225およびEP:10。光散乱検出器は、Sciex API 150に連結された。光散乱検出器は、50℃および3barのN2で作動するSedere Sedex 55であった。完全な系は、G3パワーマックにより制御された。
【実施例2】
【0058】
合成の一般的態様
ピラゾリン部分を含有する請求化合物および中間体の適当な合成は、市販されているかもしくは下記のとおり製造される4,5−ジヒドロ−1H−ピラゾールもしくは4,5−ジヒドロ−3H−ピラゾールビルディングブロックを用いて、WO 01/70700に以前に開示されたものと同様の経路をたどる。
【0059】
【化2】
【0060】
経路1は、例えば塩基の存在下でクロロギ酸メチルもしくはジ−tert−ブチルジカーボネートとスルホンアミドとの反応により製造することができる、一般式(I)のカルバミン酸スルホニルを用いる。次に、一般式(II)のピラゾリンとそれらの反応生成物をPCl3、POCl3/DMAPもしくは2−クロロ−1,3−ジメチルイミダゾリニウムクロリド(DMC)のようなハロゲン化剤を用いて一般式(III)のクロロイミン中間体に転化し、続いてアミンと反応させて一般式(IV)のスルホニルピラゾリンカルボキサミジン誘導体を得ることができる。
【0061】
【化3】
【0062】
経路2は、KOHの存在下でCS2との反応、続いてヨウ化メチルのようなハロゲン化アルキルとの反応によりスルホンアミドから製造することができる、一般式(V)のN−(ビス−アルキルスルファニル−メチレン)−スルホンアミド構造を用いる。次に、2個のS−アルキル官能基は、好ましくは一般式(VI)の構造を得るためにピラゾリンビルディングブロックから出発して、アミンで置換して、一般式(IV)のスルホニルピラゾリンカルボキサミジン誘導体で終わることができる。
【0063】
【化4】
【0064】
経路3はヨウ化メチルのようなハロゲン化アルキルとチオ尿素ビルディングブロックとの反応により都合よく製造される一般式(IX)のアルキル−イソチオ尿素フラグメントもしくはその適当な塩形態を用い、それを塩基の存在下でピラゾリンと反応させて一般式(X)のピラゾリンカルボキサミジン誘導体を得ることができる。後者を塩基の存在下でハロゲン化スルホニル(X=Br、Cl、F、好ましくはCl)と反応させて一般式(IV)のスルホニルピラゾリンカルボキサミジン誘導体を得ることができる。
【0065】
特定の合成方法の選択は、使用する試薬と官能基との適合性、保護基、触媒、活性化およびカップリング試薬を使用する可能性ならびに製造される最終化合物に存在する最終的な構造的特徴のような当業者に既知である因子により決まる。
【0066】
製薬学的に許容しうる塩は、例えば本発明の化合物を適当な酸、例えば無機酸もしくは有機酸と混合することにより、当該技術分野において周知である標準的な方法を用いて得ることができる。
【実施例3】
【0067】
ピラゾリン中間体の合成
【0068】
【化5】
【0069】
3−エチル−4,5−ジヒドロ−1H−ピラゾール
ヒドラジン水和物(24.55mL)をMeOH(50mL)に溶解し、そして氷浴において冷却した。この溶液に、温度が10℃未満に保たれるような速度でエチルビニルケトン(50mL)を加えた。氷浴を取り除き、そして混合物を室温で2時間攪拌し、その後でMeOHを減圧下で蒸発させた。生成物は真空蒸留(70℃、20mbar)により得られ、7.22gの無色の液体を生成せしめた。1H NMR(400MHz,CDCl3)δ1.15(t,J=8Hz,3H),2.34(q,J=8Hz,2H),2.59(t,J=10Hz,2H),3.10(br s,1H),3,34(t,J=10Hz,2H)。
【0070】
【化6】
【0071】
3−メチル−4,5−ジヒドロ−1H−ピラゾール
ヒドラジン水和物(29.2mL)をMeOH(50mL)に溶解した。この溶液に、温度が50℃未満に保たれるような速度でメチルビニルケトン(50mL)を加えた。混合物を50℃で2時間攪拌し、その後でMeOHを減圧下で蒸発させた。生成物は真空蒸留(68〜82℃、20mbar)により得られ、11.8gの無色の液体を生成せしめた。1H NMR(400MHz,DMSO−d6)δ1.88(s,3H),2.47(t,J=10Hz,2H),3.15(t,J=10Hz,2H),6.10(br s,1H)。
【0072】
【化7】
【0073】
4−エチル−4,5−ジヒドロ−1H−ピラゾール
ヒドラジン水和物(58mL)をMeOH(300mL)に溶解し、そして氷浴において冷却した。この混合物に、温度が10℃未満に保たれるような速度でMeOH(100mL)中の2−エチルアクロレイン(100g)の溶液を加えた。氷浴を取り除き、そして混合物を室温で一晩攪拌し、その後でMeOHを減圧下で蒸発させた。生成物が真空蒸留(70〜80℃、20mbar)により得られ、54.9gの無色の液体を生成せしめた。1H NMR(400MHz,CDCl3)δ0.98(t,J=8Hz,3H),1.42〜1.70(m,2H),2.89〜3.02(m,2H),3.43〜3.54(m,1H),6.78(br s,1H),NH 不可視。
【0074】
【化8】
【0075】
4−メチル−4,5−ジヒドロ−1H−ピラゾール
ヒドラジン水和物(16.65mL)をCH3CN(50mL)に溶解し、そして氷浴において冷却した。この混合物に、CH3CN(50mL)中の2−メチルアクロレイン(24.02g)の溶液を加えた。氷浴を取り除き、そして混合物を室温で一晩攪拌し、その後でCH3CNを減圧下で蒸発させた。生成物が真空蒸留(102〜108℃、250mbar)により得られ、7.0gの無色の液体を生成せしめた。1H NMR(400MHz,CDCl3)δ1.18(d,J=7Hz,3H),2.90(t,J=9H
z,1H),3.00〜3.12(m,1H),3.51(t,J=9Hz,1H),5.48(br s,1H),6.73(br s,1H)。
【0076】
【化9】
【0077】
5−エチル−4,5−ジヒドロ−1H−ピラゾール
ヒドラジン水和物(12.1mL)をMeOH(50mL)に溶解し、そして氷浴において冷却した。この混合物に、温度が10℃未満に保たれるような速度でMeOH(50mL)中の2−ペンテナール(24.4mL)の溶液を加えた。氷浴を取り除き、そして混合物を室温で2.5時間攪拌し、続いて減圧下で蒸発させた。生成物が真空蒸留(68〜72℃、25mbar)により得られ、8.25gの無色の液体を生成せしめた。1H NMR(400MHz,CDCl3)δ0.92(d,J=7.5Hz,3H),1.42〜1.61(m,2H),2.36(ddd,J=17,8および2Hz,1H),2.76(ddd,J=17,10および2Hz,1H),3.51〜3.62(m,1H),5.35(br s,1H),6.76(br s,1H)。
【0078】
【化10】
【0079】
4,4−ジメチル−4,5−ジヒドロ−3H−ピラゾール
2,2−ジメチル−1,3−プロパンジアミン(20.0g)をH2O(80mL)およびMeOH(20mL)に溶解し、そして氷浴において冷却した。同時に、H2O2(30%、120mL)およびNaClO(10%、350mL)を滴下して加えた。反応混合物を室温で一晩攪拌し、DCMで抽出し、有機層をNa2SO4上で乾燥させ、そして溶媒を減圧下で蒸発させた。真空蒸留(102〜105℃、250mbar)により11.4gの無色の非晶質油性化合物を生成せしめた。1H NMR(200MHz,CDCl3)δ1.05(s,6H),4.13(s,4H)。
【0080】
あるいはまた、この化合物は下記のとおり合成された:
【0081】
【化11】
【0082】
4,4−ジメチル−4,5−ジヒドロ−3H−ピラゾール
2,2−ジメチル−1,3−プロパンジアミン(8.97g)をH2O(45mL)に溶解し、そして氷浴において冷却した。同時に、温度を25℃未満に保ちながら、H2O2(30%、54mL)およびNaClO(10%、157mL)を滴下して加えた。次に、反応混合物を室温で1時間攪拌し、そしてDCM(2x45mL)で抽出した。合わせた有機層を水性亜硫酸ナトリウム(20%、25mL)で抽出し、水(2x25mL)で洗浄し、Na2SO4上で乾燥させ、そして減圧下で蒸発させて(50℃で>200mbar)8.89gの無色の液体(いくらかの残留DCMを含有する)を生成せしめた。1H NMR(200MHz,CDCl3)δ1.05(s,6H),4.13(s,4H)。
【0083】
【化12】
【0084】
2,2−ジエチル−マロノニトリル
マロノニトリル(15.2g)をTBAB(3.0g、4mol%)およびヨウ化エチル(36.8mL、2当量)と混合した。室温で30分間攪拌した後に、混合物を氷浴において冷却し、KOtBu(51.6g、2当量)を少しずつ加え、氷浴を取り除き、そして混合物を室温で30分間攪拌した。DCM/H2Oで抽出し、Na2SO4上で乾燥させ、そして減圧下で蒸発させて40グラムの粗物質を生成せしめ、それをDCMで溶出するフラッシュクロマトグラフィーにより精製した。これにより20.4グラムの橙色の油を生成せしめ、それは静置すると凝固した。1H NMR(400MHz,CDCl3)δ1.29(t,J=7.5Hz,6H),2.00(q,J=7.5Hz,4H)。
【0085】
2,2−ジエチル−プロパン−1,3−ジアミン
乾式Et2O(100mL)中のLiAlH4(4.66g)の懸濁液を氷浴において冷却し、そして温度が20℃未満に保たれるような速度でEt2O(50mL)中の2,2−ジエチル−マロノニトリル(5.0g)の溶液を滴下して加えた。混合物を室温で一晩攪拌し、氷浴において冷却し、そしてH2O(5mL)、2M水性NaOH(10mL)そして再びH2O(5mL)を加えることによりクエンチした。懸濁液を濾過し、フィルターケーキをEt2Oで洗浄し、そして合わせた濾液を減圧下で蒸発乾固させて5.0gの透明な薄黄色の液体を生成せしめた。1H NMR(400MHz,CDCl3)δ0.80(t,J=8Hz,6H),1.08(br s,4H),1.22(q,J=8Hz,4H),2.52(s,4H)。
【0086】
4,4−ジエチル−4,5−ジヒドロ−3H−ピラゾール
2,2−ジエチル−プロパン−1,3−ジアミン(5.0g)をH2O(40mL)およびMeOH(10mL)の混合物に溶解し、そして氷浴において冷却した。同時に、H2O2(24.2mLの30%溶液、6当量)およびNaClO(54.9mLの10%溶液、2.4当量)を滴下して加え、氷浴を取り除き、そして混合物を室温で2時間攪拌した。DCMで抽出し、Na2SO4上で乾燥させ、そして減圧下で蒸発させて77%の予想生成物および23%のジアミン出発物質を含有する3.51gの透明な黄色の液体を生成せしめた。この物質をさらに精製せずに次の段階において使用した。1H NMR(400MHz,CDCl3)δ0.78(t,J=7.5Hz,6H),1.36(q,J=7.5Hz,4H),4.14(s,4H)。
【0087】
【化13】
【0088】
シクロペンタン−1,1−ジカルボニトリル
マロノニトリル(15.0g)を乾式DMF(200mL)に溶解し、そして氷浴において冷却した。次に、DBU(75mL、2.2当量)および1,4−ジブロモブタン(29.6mL、1.1当量)を滴下して加えた。氷浴を取り除き、追加の100mLの乾式DMFを加え、そして混合物を80℃で2時間攪拌した。周囲温度まで冷却した後に、DCMを加え、そして混合物を5%水性NaHCO3で5回洗浄した。有機相をNa2SO4上で乾燥させ、そして減圧下で蒸発させて40gの黒色の油性物質を生成せしめた。これをPA:EA 9:1(Rf=0.35、I2で視覚化される)で溶出するフラッシュクロマトグラフィーにより精製して23.4gの無色の液体を生成せしめた。1H NMR(400MHz,CDCl3)δ1.94〜2.03(m,4H),2.41(t,J=7Hz,4H)。
【0089】
C−(1−アミノメチル−シクロペンチル)−メチルアミン
乾式Et2O(100mL)中のLiAlH4(4.74g)の懸濁液を氷浴において冷却し、そして温度が20℃未満に保たれるような速度でEt2O(50mL)中のシクロペンタン−1,1−ジカルボニトリル(5.0g)の溶液を滴下して加えた。混合物を室温で一晩攪拌し、氷浴において冷却し、そしてH2O(5mL)、2M水性NaOH(10mL)そして再びH2O(5mL)を加えることによりクエンチした。懸濁液を濾過し、フィルターケーキをEt2Oで洗浄し、そして合わせた濾液を減圧下で蒸発乾固させて4.95gの透明な無色の液体を生成せしめた。1H NMR(400MHz,CDCl3)δ1.24(br s,4H),1.22〜1.40(m,4H),1.55〜1.64(m,4H),2.62(s,4H)。
【0090】
2,3−ジアザ−スピロ[4,4]ノン−2−エン
C−(1−アミノメチル−シクロペンチル)−メチルアミン(4.87g)をH2O(40mL)およびMeOH(10mL)の混合物に溶解し、そして氷浴において冷却した。同時に、H2O2(23.9mLの30%溶液、6当量)およびNaClO(54.3mLの10%溶液、2.4当量)を滴下して加え、氷浴を取り除き、そして混合物を室温で2時間攪拌した。DCMで抽出し、Na2SO4上で乾燥させ、そして減圧下で蒸発させて90%の予想生成物および10%のジアミン出発物質を含有する3.74gの透明な薄黄色の液体を生成せしめた。この物質をさらに精製せずに次の段階において使用した。1H NMR(400MHz,CDCl3)δ1.48〜1.57(m,4H),1.62〜1.69(m,4H),4.26(s,4H)。
【0091】
【化14】
【0092】
シクロヘキサン−1,1−ジカルボニトリル
マロノニトリル(15.0g)を乾式DMF(200mL)に溶解した。次に、DBU(75mL)および1,5−ジブロモペンタン(34mL)を0℃(氷浴)で加えた。氷浴を取り除き、そして反応物を80℃で2時間攪拌した。冷却した後に、反応物をDCMに注ぎ込んだ。有機層を5%NaHCO3で数回洗浄し、有機層をNa2SO4上で乾燥させ、そして溶媒を減圧下で蒸発させた。粗生成物をPA:EtOAc(9:1)で溶出するフラッシュクロマトグラフィーにより精製して25.7gの白色の結晶を生成せしめた。1H NMR(400MHz,CDCl3)δ1.48〜1.61(m,2H),1.68〜1.84(m,4H),2.13(t,J=6Hz,4H)。
【0093】
C−(1−アミノメチル−シクロヘキシル)−メチルアミン
シクロヘキサン−1,1−ジカルボニトリル(20.0g)を乾式Et2O(70mL)に溶解した。氷浴において冷却した乾式Et2O(250mL)中のLiAlH4(17.0g)の懸濁液にこの混合物を滴下して加えた。混合物を室温で一晩攪拌し、氷浴において冷却し、そしてH2O(17.0mL)、2M水性NaOH(34.0mL)そして再びH2O(17mL)を加えることによりクエンチした。懸濁液を濾過し、フィルターケーキをEt2Oで洗浄し、そして合わせた濾液を蒸発乾固させて20.8gの透明な無色の液体を生成せしめた。1H NMR(400MHz,CDCl3)δ1.05〜1.55(m,14H),2.61(s,4H)。
【0094】
2,3−ジアザ−スピロ[4,5]デク−2−エン
C−(1−アミノメチル−シクロヘキシル)−メチルアミン(10.0g)をH2O(40mL)およびMeOH(10mL)の混合物に溶解し、そして氷浴において冷却した。同時に、H2O2(44.3mLの30%溶液、6当量)およびNaClO(125.5mLの10%溶液、2.4当量)を滴下して加え、氷浴を取り除き、そして混合物を室温で45分間攪拌した。DCMで抽出し、Na2SO4上で乾燥させ、そして減圧下で蒸発させて、8.7gの透明な薄黄色の液体を生成せしめた。1H NMR(400MHz,CDCl3)δ1.24〜1.53(m,10H),4.17(s,4H)。
【0095】
【化15】
【0096】
2−(2−メチル−[1,3]ジオキソラン−2−イル)−酪酸エチルエステル
メチル−2−エチルアセトアセテート(100mL)をトルエン(250mL)に溶解した。エチレングリコール(46.9mL、1.35当量)および触媒量のp−TsOH・H2Oを加え、そして混合物をディーン・スターク条件下で一晩還流させた。周囲温度まで冷却した後に、混合物を5%水性NaHCO3および飽和水性NaClで洗浄し、有機相をNa2SO4上で乾燥させ、そして減圧下で蒸発させた。残留物を反復真空蒸留(118〜128℃、15mbar)により精製し、85.5gの生成物を生成せしめた。1H NMR(400MHz,CDCl3)δ0.90(t,J=7Hz,3H),1.28(t,J=7Hz,3H),1.40(s,3H),1.59〜1.83(m,2H
),2.56(dd,J=11.5および4Hz,1H),3.90〜4.06(m,4H),4.18(m,2H)。
【0097】
2−(2−メチル−[1,3]ジオキソラン−2−イル)−ブタン−1−オール
2−(2−メチル−[1,3]ジオキソラン−2−イル)−酪酸エチルエステル(85.5g)を乾式Et2O(50mL)に溶解した。氷浴において冷却した乾式Et2O(200mL)中のLiAlH4(16.1g)の懸濁液にこの混合物を滴下して加えた。混合物を4時間還流させ、氷浴において冷却し、そしてH2O(16.1mL)、2M水性NaOH(32.2mL)そして再びH2O(16.1mL)を加えることによりクエンチした。懸濁液を濾過し、フィルターケーキをEt2Oで洗浄し、そして合わせた濾液を蒸発乾固させた。残留物(49g)を真空蒸留(112〜125℃、15mbar)により精製し、43.5gの透明な無色の液体を生成せしめた。1H NMR(400MHz,CDCl3)δ0.98(t,J=7.5Hz,3H),1.10〜1.24(m,1H),1.31(s,1H),1.50〜1.75(m,2H),3.12(br s,1H),3.59〜3.76(m,2H),3.94〜4.02(m,4H)。
【0098】
3−ヒドロキシメチル−ペンタン−2−オン
2−(2−メチル−[1,3]ジオキソラン−2−イル)−ブタン−1−オール(43.5g)をH2O(100mL)およびEtOH(10mL)の混合物に溶解し、そして濃水性HCl(1mL)を加えた。混合物を2時間還流させ、周囲温度に冷却し、2M水性NaOHで中和し、硫酸アンモニウムで飽和させ、そしてEt2Oで2回抽出した。合わせた有機層をNa2SO4上で乾燥させ、そして蒸発乾固させた。黄色がかった残留物(25.7g)を真空蒸留により精製して20.7gの透明な無色の油を生成せしめた。1H NMR(400MHz,CDCl3)δ0.95(t,J=7.5Hz,3H),1.49〜1.76(m,2H),2.21(s,3H),2.64(m,1H),3.68〜3.84(m,3H)。
【0099】
酢酸2−エチル−3−オキソ−ブチルエステル
3−ヒドロキシメチル−ペンタン−2−オン(20.7g)をCHCl3(150mL)に溶解し、そして氷浴において冷却した。無水酢酸(80mL)、続いてDMAP(2.18g)を加え、そして混合物を室温で一晩攪拌した。氷浴において冷却した後に、MeOH(120mL)を滴下して加え、そして混合物を飽和水性NaHCO3溶液に注ぎ込んだ。DCMで2回抽出した後に、合わせた有機相をNa2SO4上で乾燥させ、そして減圧下で蒸発させて28.0gの薄黄色の液体を生成せしめた。1H NMR(400MHz,CDCl3)δ0.93(t,J=8Hz,3H),1.46〜1.75(m,2H),2.03(s,3H),2.20(s,3H),2.77(quint.,J=6.5Hz,1H),4.20(d,J=7Hz,3H)。
【0100】
4−エチル−3−メチル−4,5−ジヒドロ−1H−ピラゾール
酢酸2−エチル−3−オキソ−ブチルエステル(23.0g)を乾式THF(75mL)に溶解し、そしてDBU(23.9mL)を加えた。混合物を室温で15分間攪拌して中間体3−メチレン−ペンタン−2−オンを生成せしめた。MeOH(75mL)を加え、続いてヒドラジン水和物(7.75mL)を滴下して加えた。得られる混合物を室温で一晩攪拌し、そして減圧下で蒸発させた。残留物を真空蒸留(94〜106℃、15mbar)により精製し、7.9gの透明な無色の液体を生成せしめた。1H NMR(400MHz,CDCl3)δ0.95(t,J=7.5Hz,3H),1.33〜1.83(m,2H),1.92(s,3H),2.78(m,1H),3.01(t,J=9.5Hz,1H),3.51(t,J=9,5Hz,1H)。
【0101】
【化16】
【0102】
2−ジメチルアミノメチル−シクロヘキサノン
シクロヘキサノン(259mL)にホルムアルデヒド(37.2mLの37%水溶液)およびジメチルアミン塩酸塩(40.8g)を加えた。攪拌した混合物をゆっくりと加熱し、そして1時間還流させた。周囲温度まで冷却した後にH2Oを加え、そして混合物をEt2Oで2回抽出した。50%水性NaOH(27.5mL)の添加により水層を塩基性にし、そして次にDCMで2回抽出した。合わせた有機相をNa2SO4上で乾燥させ、そして減圧下で蒸発させて66.6gの薄黄色の液体を生成せしめた。1H NMR(400MHz,CDCl3)δ1.34〜1.47(m,1H),1.60〜1.78(m,2H),1.81〜1.92(m,1H),1.98〜2.09(m,1H),2.16〜2.55(m,5H),2.21(s,6H),2.69(dd,J=13および6Hz,1H)。
【0103】
3,3a,4,5,6,7−ヘキサヒドロ−2H−インダゾール
ヒドラジン水和物(28.0mL)をn−BuOH(200mL)に溶解し、そして氷浴において冷却した。n−BuOH(50mL)中の2−ジメチルアミノメチル−シクロヘキサノン(64.0g)の溶液を滴下して加え、混合物をゆっくりと温め、そして20時間還流させた。溶媒を減圧下で蒸発させた。残留物を真空蒸留(64〜67℃、28Pa)により精製し、24.2gの透明な無色の液体を生成せしめた。この物質をさらに精製せずに次の段階において使用した。
【0104】
【化17】
【0105】
4−エチル−5−メチル−4,5−ジヒドロ−1H−ピラゾール
ヒドラジン水和物(12.4mL)をMeOH(100mL)に溶解し、そして氷浴において冷却した。この混合物に、温度が10℃未満に保たれるような速度でMeOH(50mL)中の2−エチル−ブト−2−エナール(25g)の溶液を加えた。氷浴を取り除き、そして混合物を室温で2日間攪拌した。溶媒を減圧下で蒸発させた。真空蒸留(90〜100℃、20mbar)により約1:2の比率でジアステレオマー混合物としての所望の生成物とヒドラゾンを含有する16.9gの薄黄色の液体を生成せしめた。この物質をさらに精製せずに次の段階において使用した。
【0106】
【化18】
【0107】
5−エチル−4−メチル−4,5−ジヒドロ−1H−ピラゾール
N2雰囲気下で、ヒドラジン水和物(63.9mL、10当量)をMeOH(100mL)に溶解し、そして氷浴において冷却した。この混合物に、温度が10℃未満に保たれるような速度でMeOH(50mL)中の2−メチル−ペント−2−エナール(15.0mL)の溶液を加えた。氷浴を取り除き、そして混合物を室温で一晩攪拌した。溶媒を減圧下で蒸発させた。真空蒸留(40〜45℃、15mbar)により約1:1の比率でジアステレオマー混合物としての所望の生成物とヒドラゾンを含有する9.5gの薄黄色の液体を生成せしめた。この物質をさらに精製せずに次の段階において使用した。
【0108】
【化19】
【0109】
4−ヒドロキシ−3,3−ジメチル−ブタン−2−オン
25mLの3−メチル−ブタン−2−オンに7.01gのパラホルムアルデヒドおよび36.0mLのトリフルオロ酢酸を加えた。混合物を7時間還流させた。冷却した後に、300mLのH2Oおよび100g(5当量)のNaHCO3を加えた。懸濁液を濾過し、そして有機層を分離した。フィルターケーキをDCMで2回洗浄し、合わせた濾液をNa2SO4上で乾燥させ、そして溶媒を減圧下で蒸発させて23.7gの橙色の液体を生成せしめた。1H NMR(400MHz,CDCl3)δ1.17(s,6H),2.17(s,3H),2.38(t,J=7Hz,1H),3.65(d,J=7Hz,2H)。
【0110】
メタンスルホン酸2,2−ジメチル−3−オキソ−ブチルエステル
23.7gの4−ヒドロキシ−3,3−ジメチル−ブタン−2−オンを150mLのDCMに溶解した。49.5mL(3当量)のピリジンおよび17.5mL(1.1当量)の塩化メシルを加え、そして混合物を室温で20時間攪拌した。懸濁液を濾過し、そしてフィルターケーキをDCMで2回洗浄した。濾液を1M HClで洗浄し、そして水層をDCMで2回抽出した。合わせた濾液をNa2SO4上で乾燥させ、そして溶媒を減圧下で蒸発させて41.2gの褐色の液体を生成せしめた。1H NMR(400MHz,CDCl3)δ1.24(s,6H),2.20(s,3H),3.03(s,3H),4.21(s,2H)。
【0111】
3,4,4−トリメチル−4,5−ジヒドロ−1H−ピラゾール
39.2gのメタンスルホン酸2,2−ジメチル−3−オキソ−ブチルエステルを200mLのMeOHに溶解し、そして氷浴において冷却した。21.6mL(2.2当量)のヒドラジン水和物を滴下して加え、そして反応混合物を室温で1時間攪拌した。反応混合物を濃縮し、5%NaHCO3を加え、そしてDCMで3回抽出した。合わせた有機層をNa2SO4上で乾燥させ、そして溶媒を減圧下で蒸発させて19.5gの橙色の液体を生成せしめた。この液体の10gの真空蒸留により6.4gの薄黄色の液体を生成せしめた(76〜78℃、20mbar)。1H NMR(400MHz,CDCl3)δ1.14(s,6H),1.86(s,3H),3.14(s,2H),4.00(br s,1H)。
【0112】
【化20】
【0113】
3−ジメチルアミノ−1−フェニル−プロパン−1−オン塩酸塩
40mLのEtOH中の0.5mLの濃水性HClの溶液に、アセトフェノン(30.0g)、パラホルムアルデヒド(10.0g)およびジメチルアミン塩酸塩(26.5g)を加え、そして混合物を3時間還流させた。混合物を室温に冷却し、そして沈殿物を濾過し、アセトンで洗浄し、そして真空中で乾燥させて37.2gの白色の結晶性物質を得た。1H NMR(200MHz,DMSO−d6)δ2.84(s,6H),3.38〜3.55(m,2H),3.57〜3.74(m,2H),7.48〜7.73(m,3H),7.97〜8.10(m,2H)。
【0114】
3−フェニル−4,5−ジヒドロ−1H−ピラゾール
N2雰囲気下で、3−ジメチルアミノ−1−フェニル−プロパン−1−オン塩酸塩(37.2g)を温かいMeOH(75mL)に溶解し、そして50℃で攪拌したMeOH(30mL)中のヒドラジン水和物(23mL)および50%水性NaOH(12mL)の溶液にゆっくりと加えた。混合物を2時間還流させ、そして減圧下で蒸発させた。氷水を残留物に加え、そして5分間攪拌した後に生じた固体を濾過して分離した。残留物をEt2Oに溶解し、Na2SO4上で乾燥させ、そして減圧下で蒸発乾固させて80%純粋である19.7gの黄色の油を生成せしめ、それをさらに精製せずに次の段階において使用した。
【0115】
【化21】
【0116】
1−(4−クロロ−フェニル)−3−ジメチルアミノ−プロパン−1−オン塩酸塩
EtOH(80mL)に、p−クロロアセトフェノン(77.3g、0.50mol)、ジメチルアミン塩酸塩(52.7g、0.65mol)、パラホルムアルデヒド(19.8g、0.66mol)および濃水性HCl(1mL)を加え、そして混合物を5時間還流させた。混合物を40℃に冷却し、アセトン(400mL)を加え、そして攪拌下で混合物を20℃までさらに冷却した。沈殿物を濾過し、アセトンおよびPAで洗浄し、そして空気乾燥させて69.5gの生成物を生成せしめ、それをさらに精製せずに次の段階において使用した。
【0117】
3−(4−クロロ−フェニル)−4,5−ジヒドロ−1H−ピラゾール
N2雰囲気下で、1−(4−クロロ−フェニル)−3−ジメチルアミノ−プロパン−1−オン塩酸塩(37.2g)を温かいMeOH(75mL)に溶解し、そして50℃で攪拌したMeOH(30mL)中のヒドラジン水和物(23mL)および50%水性NaOH(12mL)の溶液にゆっくりと加えた。混合物を2時間還流させ、そして減圧下で蒸
発させた。水を残留物に加え、続いてDCMで抽出した。有機相を水で2回洗浄し、乾燥させ、そして減圧下で蒸発させ、25.0gの黄色の固体、m.p.90〜100℃を生成せしめた。
【0118】
【化22】
【0119】
4−フェニル−4,5−ジヒドロ−1H−ピラゾール
N2雰囲気下で、ジメチルアミン塩酸塩(7.27g)およびホルマリン(37%)(6.63mL)をフェニルアセトアルデヒド(10mL)に加え、そして室温で一晩攪拌した。反応混合物をジエチルエーテルで1回抽出し、有機層をMgSO4上で乾燥させ、そして中間体2−フェニル−プロペナールを含有する溶液をMeOHに溶解した。ヒドラジン水和物(7.87mL)を加え、そして反応混合物を50℃で2時間攪拌した(Et2Oを蒸発させた)。混合物を減圧下で濃縮した。残留物をDCMに溶解し、そしてMgSO4上で乾燥させ、続いて減圧下で蒸発させ、3.12gの黄色の油を生成せしめ、それをさらに精製せずに次の段階において使用した。
【0120】
【化23】
【0121】
3−フェニル−ブト−3−エン−2−オン
1−フェニル−プロパン−2−オン(40.8g)を200mLのMeOHに溶解した。ホルマリン(37%)(79mL)、ピペリジン(4ml)およびHOAc(4ml)を加え、そして反応物を60℃で3時間攪拌した。反応混合物を減圧下で蒸発乾固させた。残留物をジエチルエーテルに溶解し、そして水で抽出した。有機層を1M HClで洗浄し、MgSO4上で乾燥させ、そして減圧下で蒸発させて36.3gの黄色の液体を生成せしめた。1H NMR(400MHz,CDCl3)δ2.41(s,3H),5.87(s,1H),6.18(s,1H),7.24〜7.40(m,5H)。
【0122】
3−メチル−4−フェニル−4,5−ジヒドロ−1H−ピラゾール
ヒドラジン水和物(12.06mL)をMeOH(200mL)中の3−フェニル−ブト−3−エン−2−オン(36.3g)に加えた。反応物を還流温度で一晩攪拌した。溶媒を減圧下で蒸発させた。残留物をジエチルエーテルに溶解し、そして水で洗浄した。有機相をNa2SO4上で乾燥させ、そして減圧下で蒸発させた。粗物質をDCM:MeOH=98:2で溶出するフラッシュカラムクロマトグラフィーにより精製して65%の所望の生成物を含有する19.7gの橙色の油を生成せしめ、それをさらに精製せずに次の段階において使用した。
【0123】
【化24】
【0124】
5−フェニル−4,5−ジヒドロ−1H−ピラゾール
N2雰囲気下で、ヒドラジン水和物(9.2mL)をt−BuOH(20mL)中の桂皮アルデヒド(10.0g)の溶液に加えた。混合物を一晩還流させ、続いて減圧下で濃縮した。水を残留物に加え、そして水相をDCMで2回抽出した。合わせた有機層を水で洗浄し、Na2SO4上で乾燥させ、そして減圧下で濃縮した。これにより85%の所望の生成物を含有する10.46gの黄色の油を生成せしめ、それをさらに精製せずに次の段階において使用した。1H NMR(200MHz,CDCl3)δ2.61〜2.80(m,1H),3.04〜3.23(m,1H),4.72(dd,J=8および10Hz,1H),5.60〜6.10(br s,1H),6.77〜6.87(m,1H),7.18〜7.47(m,5H)。
【0125】
【化25】
【0126】
5−フラン−2−イル−4,5−ジヒドロ−1H−ピラゾール
N2雰囲気下で、ヒドラジン水和物(4.0mL)をt−BuOH(25mL)中の3−(2−フリル)アクロレイン(5.0g)の溶液に加えた。混合物を2日間還流させ、続いて減圧下で蒸発させた。残留物をDCMに溶解し、そして5%水性NaHCO3で2回抽出した。有機相をNa2SO4上で乾燥させ、そして減圧下で蒸発させた。これにより45%の予想生成物および閉環を受けなかった55%のヒドラゾン中間体を含有する5.3gの黄色の油を生成せしめた。n−BuOHにおいてさらに24時間還流させて(処理(workup)後に)58%の予想生成物および42%のヒドラゾンを含有する5.6gの褐色の油を生成せしめた。この物質をさらに精製せずに次の段階において使用した。1H NMR(400MHz,CDCl3)における特有のピラゾリンシグナル:δ2.87〜3.08(m,2H),4.72〜4.81(m,1H),6.87(br s,1H)。
【0127】
【化26】
【0128】
3−(3,4−ジヒドロ−2H−ピラゾール−3−イル)−ピリジン
N2雰囲気下で、ヒドラジン水和物(3.65mL、2当量)をt−BuOH(25mL)中の3−(3−ピリジル)アクロレイン(5.0g)の溶液に加えた。混合物を3日間還流させ、続いて減圧下で蒸発させた。残留物をDCMに溶解し、そして5%水性NaHCO3で洗浄した。有機相をNa2SO4上で乾燥させ、そして減圧下で蒸発させた。
これにより74%の予想生成物および閉環を受けなかった26%のヒドラゾン中間体を含有する5.0gの赤色の油を生成せしめた。この物質をさらに精製せずに次の段階において使用した。1H NMR(400MHz,CDCl3)における特有のピラゾリンシグナル:δ2.63〜2.75(m,1H),3.13〜3.25(m,1H),4.72〜4.82(m,1H),6.85(br s,1H)。
【0129】
【化27】
【0130】
3−フラン−3−イル−プロペナール
6.08gの(トリフェニルホスホルアニリデン)アセトアルデヒドを10mLの乾式DMFに懸濁した。1.67mL(1当量)の3−フルアルデヒドを加え、そして混合物を80℃で一晩攪拌した。混合物をEAに溶解し、そして5%水性NaHCO3で4回洗浄し、有機相をNa2SO4上で乾燥させ、濾過し、そして真空中で濃縮した。残留物をPAに懸濁し、濾過し、そして真空中で濃縮して68%の所望の生成物を含有する1.47gの薄褐色の油を生成せしめた。この物質をさらに精製せずに次の段階において使用した。1H NMR(400MHz,CDCl3)における特有のシグナル:δ6.45(dd,J=8および16Hz,1H),9.63(d,J=8Hz,1H)。
【0131】
5−フラン−3−イル−4,5−ジヒドロ−1H−ピラゾール
5.84ml(10当量)のヒドラジン水和物を20mLのジエチルエーテルに加えた。エマルジョンを氷/NaCl浴で−10℃に冷却した。20mLのジエチルエーテル中1.47gの3−フラン−3−イル−プロペナールの溶液を滴下して加えた。混合物を一晩(氷浴で)攪拌し、そして室温にゆっくりと到達させた。5%水性NaHCO3を加え、そして混合物をEAで3回抽出した。合わせた有機層をNa2SO4上で乾燥させ、濾過し、そして真空中で濃縮した。残留物をSCXイオン交換カラム上で捕捉し、MeOHで洗浄し、そしてMeOH中1MのNH3で溶出して蒸発後に85%の所望の生成物を含有する950mgの橙色の油を生成せしめた。この物質をさらに精製せずに次の段階において使用した。1H NMR(400MHz,CDCl3)δ2.62〜2.72(m,1H),2.97〜3.07(m,1H),4.62〜4.71(m,1H),5.57〜5.74(br s,1H),6.36(br s,1H),6.87(br s,1H),7.35〜7.41(m,2H)。
【0132】
【化28】
【0133】
2−ピリジン−2−イル−プロペナール
6.08gの(トリフェニルホスホルアニリデン)アセトアルデヒドを10mLの乾式DMFに懸濁した。1.90mL(1当量)のピリジン−2−カルバルデヒドを加え、そして混合物を室温で一晩攪拌した。混合物をEAに溶解し、そして5%水性NaHCO3
で4回洗浄し、有機相をNa2SO4上で乾燥させ、濾過し、そして真空中で濃縮した。残留物をPAに懸濁し、濾過し、そして真空中で濃縮して80%の所望の生成物を含有する1.50gの暗黄色の油を生成せしめた。この物質をさらに精製せずに次の段階において使用した。1H NMR(400MHz,CDCl3)δ7.09(dd,J=8および16Hz,1H),7.30〜7.36(m,1H),7.49〜7.59(m,2H),7.77(dt,J=8.8および2Hz,1H),8.67〜8.74(m,1H),9.81(d,J=8Hz,1H)。
【0134】
2−(3,4−ジヒドロ−2H−ピラゾール−3−イル)−ピリジン
4.56ml(10当量)のヒドラジン水和物を20mLのジエチルエーテルに加えた。エマルジョンを氷/NaCl浴で−10℃に冷却した。20mLのジエチルエーテル中1.25gの3−ピリジン−2−イル−プロペナールの溶液を滴下して加えた。混合物を一晩(氷浴で)攪拌し、そして室温にゆっくりと到達させた。5%水性NaHCO3を加え、そしてEAで5回抽出した。合わせた有機層をNa2SO4上で乾燥させ、濾過し、そして真空中で濃縮した。残留物をSCXイオン交換カラム上で捕捉し、MeOHで洗浄し、そしてMeOH中1MのNH3で溶出して90%の所望の生成物を含有する1.28gの褐色の油を生成せしめた。この物質をさらに精製せずに次の段階において使用した。1H NMR(400MHz,CDCl3)δ2.84〜2.94(m,1H),3.19〜3.29(m,1H),4.82〜4.90(m,1H),6.83(br s,1H),7.17〜7.23(m,1H),7.35〜7.40(m,1H),7.69(dt,J=7.5,7.5および2Hz,1H),8.53〜8.58(m,1H)。
【0135】
【化29】
【0136】
3−ピリジン−4−イル−プロペナール
6.08gの(トリフェニルホスホルアニリデン)アセトアルデヒドを10mLの乾式DMFに懸濁した。1.93mL(1当量)のピリジン−4−カルバルデヒドを加え、そして混合物を室温で一晩攪拌した。混合物をEAに溶解し、そして5%水性NaHCO3で4回洗浄し、有機相をNa2SO4上で乾燥させ、濾過し、そして真空中で濃縮した。残留物をPAに懸濁し、濾過し、そして真空中で濃縮して80%の所望の生成物を含有する1.17gの黄色の油を生成せしめた。この物質をさらに精製せずに次の段階において使用した。1H NMR(400MHz,CDCl3)δ6.85(dd,J=8および16Hz,1H),7.39〜7.47(m,3H),8.70〜8.74(m,2H),9.78(d,J=8Hz,1H)。
【0137】
4−(3,4−ジヒドロ−2H−ピラゾール−3−イル)−ピリジン
4.27ml(10当量)のヒドラジン水和物を20mLのジエチルエーテルに加えた。エマルジョンを氷/NaCl浴で−10℃に冷却した。20mLのジエチルエーテル中1.17gの3−ピリジン−4−イル−プロペナールの溶液を滴下して加えた。混合物を一晩(氷浴で)攪拌し、そして室温にゆっくりと到達させた。5%水性NaHCO3を加え、そしてEAで5回抽出した。合わせた有機層をNa2SO4上で乾燥させ、濾過し、そして真空中で濃縮した。残留物をSCXイオン交換カラム上で捕捉し、MeOHで洗浄し、そしてMeOH中1MのNH3で溶出して90%の所望の生成物を含有する1.23gの褐色の油を生成せしめた。この物質をさらに精製せずに次の段階において使用した。
1H NMR(400MHz,CDCl3)δ2.61〜2.71(m,1H),3.15〜3.25(m,1H),4.68〜4.76(m,1H),6.82(br s,1H),7.25〜7.30(m,2H),8.55〜8.60(m,2H)。
【0138】
【化30】
【0139】
3−チオフェン−3−イル−プロペナール
10.0gの(トリフェニルホスホルアニリデン)アセトアルデヒドを10mLの乾式DMFに懸濁した。2.88mL(1当量)のチオフェン−3−カルバルデヒドを加え、そして混合物を80℃で一晩攪拌した。混合物をEAに溶解し、そして5%水性NaHCO3で4回洗浄し、有機相をNa2SO4上で乾燥させ、濾過し、そして真空中で濃縮した。残留物をPAに懸濁し、濾過し、そして真空中で濃縮して54%の所望の生成物を含有する4.16gの橙色の油を生成せしめた。この物質をさらに精製せずに次の段階において使用した。1H NMR(400MHz,CDCl3)における特有のシグナル:δ6.54(dd,J=8および16Hz,1H),9.66(d,J=8Hz,1H)。
【0140】
5−チオフェン−3−イル−4,5−ジヒドロ−1H−ピラゾール
14.6ml(10当量)のヒドラジン水和物を50mLのジエチルエーテルに加えた。エマルジョンを氷/NaCl浴で−10℃に冷却した。25mLのジエチルエーテル中4.16gの3−チオフェン−3−イル−プロペナールの溶液を滴下して加えた。混合物を(氷浴で)一晩攪拌し、そして室温にゆっくりと到達させた。5%水性NaHCO3を加え、そしてEAで3回抽出した。合わせた有機層をNa2SO4上で乾燥させ、濾過し、そして真空中で濃縮して70%の所望の生成物を含有する4.12gの橙色の油を生成せしめた。この物質をさらに精製せずに次の段階において使用した。1H NMR(400MHz,CDCl3)における特有のピラゾリンシグナル:δ2.78〜2.88(m,1H),3.03〜3.13(m,1H),4.77〜4.86(m,1H),6.86(br s,1H)。
【0141】
【化31】
【0142】
3−チオフェン−2−イル−プロペナール
10.0gの(トリフェニルホスホルアニリデン)アセトアルデヒドを10mLの乾式DMFに懸濁した。3.07mL(1当量)のチオフェン−2−カルバルデヒドを加え、そして混合物を80℃で一晩攪拌した。混合物をEAに溶解し、そして5%水性NaHCO3で4回洗浄し、有機相をNa2SO4上で乾燥させ、濾過し、そして真空中で濃縮した。残留物をPAに懸濁し、濾過し、そして真空中で濃縮して50%の所望の生成物を含有する4.27gの橙色の油を生成せしめた。この物質をさらに精製せずに次の段階において使用した。1H NMR(400MHz,CDCl3)における特有のシグナル:δ
6.52(dd,J=8および16Hz,1H),9.63(d,J=8Hz,1H)。
【0143】
5−チオフェン−2−イル−4,5−ジヒドロ−1H−ピラゾール
15.0ml(10当量)のヒドラジン水和物を50mLのジエチルエーテルに加えた。エマルジョンを氷/NaCl浴で−10℃に冷却した。25mLのジエチルエーテル中4.27gの3−チオフェン−2−イル−プロペナールの溶液を滴下して加えた。混合物を(氷浴で)一晩攪拌し、そして室温にゆっくりと到達させた。5%水性NaHCO3を加え、そしてEAで3回抽出した。合わせた有機層をNa2SO4上で乾燥させ、濾過し、そして真空中で濃縮して70%の所望の生成物を含有する5.58gの橙色の油を生成せしめた。この物質をさらに精製せずに次の段階において使用した。1H NMR(400MHz,CDCl3)における特有のピラゾリンシグナル:δ2.77〜2.86(m,1H),3.08〜3.18(m,1H),4.95〜5.03(m,1H),6.88(br s,1H)。
【0144】
【化32】
【0145】
3−イソプロピル−5−フェニル−4,5−ジヒドロ−1H−ピラゾール
0.38mLの3−メチル−2−ブタノンを10mLのDCMに溶解した。0.36mL(1当量)のベンズアルデヒドを加え、続いて1.50mLのトリフルオロメタンスルホン酸無水物を滴下して加えた。混合物を室温で1時間攪拌した。次に、10mLのMeOHおよび0.87mL(5当量)のヒドラジン水和物を加えた。混合物を室温で30分間攪拌し、そして真空中で濃縮した。残留物をDCMに溶解し、5%水性NaHCO3で抽出し、そして有機相をNa2SO4上で乾燥させ、濾過し、そして真空中で濃縮して約50%の所望の生成物を含有する520mgの褐色の油を生成せしめ、それをさらに精製せずに次の段階において使用した。
【0146】
【化33】
【0147】
4−メチル−5−フェニル−4,5−ジヒドロ−1H−ピラゾール
5.22ml(1当量)のヒドラジン水和物を100mLのジエチルエーテルに加えた。エマルジョンを氷浴で冷却した。15.0mLの2−メチル−3−フェニル−プロペナールを滴下して加え、そして混合物を室温で一晩攪拌した。H2Oを加え、有機層を分離し、そして水層をジエチルエーテルで抽出した。合わせた有機層をNa2SO4上で乾燥させ、濾過し、そして真空中で濃縮した。真空蒸留により5.9gの所望の生成物(ジアステレオマー対の混合物)を透明な液体として生成せしめた(76〜82℃、0.2〜0.3mbar)。第一のジアステレオマー対の1H NMR(400MHz,CDCl3):δ0.71(d,J=7Hz,3H),3.20〜3.31(m,1H),4.77(d,J=10Hz,1H),6.73(br s,1H),7.23〜7.42(m,
5H),8.55〜8.60(m,2H)。第二のジアステレオマー対の1H NMR(400MHz,CDCl3):δ1.24(d,J=7Hz,3H),2.90〜3.11(m,1H),4.22(d,J=11Hz,1H),6.71(br s,1H),7.23〜7.42(m,5H),8.55〜8.60(m,2H)。
【0148】
【化34】
【0149】
2−ベンジリデン−ブチルアルデヒド
30.0mLのベンズアルデヒドを150mLのEtOHに溶解し、そして氷浴で冷却した。5.01mLの45%KOH(0.2当量)を加え、続いて16.5mLのブチルアルデヒドを滴下して加えた。混合物を室温で3日間攪拌し、1M HClで酸性化し、そしてエーテルで抽出した。有機層をNa2SO4上で乾燥させ、濾過し、そして真空中で濃縮した。真空蒸留により70%の所望の生成物を含有する20.4gの黄色の液体を生成せしめた(78〜82℃、0.6mbar)。この物質をさらに精製せずに次の段階において使用した。1H NMR(400MHz,CDCl3)における特有のシグナル:δ1.15(d,J=7.5Hz,3H),2.57(q,J=7,5Hz,1H),7.22(s,1H),9.56(s,1H)。
【0150】
4−エチル−5−フェニル−4,5−ジヒドロ−1H−ピラゾール
62ml(10当量)のヒドラジン水和物を150mLのジエチルエーテルに加えた。エマルジョンを氷/NaCl浴で−10℃に冷却した。100mLのエーテル中20.4gの2−ベンジリデン−ブチルアルデヒドの溶液を−10℃で滴下して加え、そして−10℃で3時間攪拌した。混合物を(氷浴で)一晩攪拌し、そしてゆっくりと室温に到達させた。H2Oを加え、有機層を分離し、そして水層をジエチルエーテルで2回抽出した。合わせた有機層をNa2SO4上で乾燥させ、濾過し、そして真空中で濃縮した。真空蒸留により94%の所望生成物(ジアステレオマー対の混合物)を含有する6.1gの透明な液体を生成せしめた(102〜106℃、0.6mbar)。第一のジアステレオマー対の特有のシグナル:1H NMR(400MHz,CDCl3)δ0.83(t,J=6.5Hz,3H),3.03〜3.13(m,1H),4.74〜4.81(m,1H),6.83(br s,1H)。第二のジアステレオマー対の特有のシグナル:1H NMR(400MHz,CDCl3)δ1.00(d,J=7.5Hz,3H),2.84〜2.93(m,1H),4.28〜4.34(m,1H),6.76(br s,1H)。
【0151】
【化35】
【0152】
(1−メチル−1,2,5,6−テトラヒドロ−ピリジン−3−イル)−メタノール
15.0gの1−メチル−1,2,5,6−テトラヒドロ−ピリジン−3−カルボン酸メチルエステル臭化水素酸塩をEAに溶解し、そして2M NaOHで抽出した。有機層を分離し、そして水層を再びEAで抽出した。合わせた有機層をNa2SO4上で乾燥させ、濾過し、そして濃縮して8.27gの遊離塩基を黄色の油として生成せしめた(84%)。6.5gのLiAlH4(3.2当量)を100mLの乾式THFに懸濁し、そして氷浴で冷却した。これに50mLの乾式THF中8.27gの1−メチル−1,2,5,6−テトラヒドロ−ピリジン−3−カルボン酸メチルエステル(遊離塩基)の溶液を滴下して加えた。混合物を室温で3時間攪拌した。混合物を氷浴で冷却し、そして6.5mLのH2O、13mLの2M NaOHおよび6.5mLのH2Oを滴下して加えた。残留物を濾過し、エーテルで洗浄し、そして濾液を真空中で濃縮して6.7gの無色の油を生成せしめた。1H NMR(400MHz,CDCl3)δ2.18〜2.26(m,2H),2.36(s,3H),2.49(t,J=6Hz,2H),2.92〜2.97(m,2H),4.00(br s,2H),5.68(br s,1H)。
【0153】
1−メチル−1,2,5,6−テトラヒドロ−ピリジン−3−カルバルデヒド
2.88mLの塩化オキサリル(2.4当量)を20mLのDCMに溶解した。混合物を−78℃に冷却し、そして10mLのDCM中3.37mLのDMSO(2.0当量)の溶液を滴下して加えた。混合物を−78℃で15分間攪拌した。温度を−65℃未満に保ちながら10mLのDCM中3.0gの(1−メチル−1,2,5,6−テトラヒドロ−ピリジン−3−イル)−メタノールの溶液を滴下して加えた。混合物を−78℃で15分間攪拌した。9.81mLのトリエチルアミン(3.0当量)を滴下して加え、そして次に混合物を室温まで温めておいた。混合物を攪拌可能にしておくために50mLのDCMを加えた。混合物を室温で1時間攪拌した。H2Oを加え、有機層を分離し、そして水層を再びDCMで抽出した。合わせた有機層をNa2SO4上で乾燥させ、濾過し、そして濃縮して3.24gの橙色の油(85%純粋)を生成せしめ、それをさらに精製せずに次の段階において使用した。1H NMR(400MHz,CDCl3)δ2.43(s,3H),2.48〜2.60(m,4H),3.11〜3.15(m,2H),6.85(m,1H),9.43(s,1H)。
【0154】
5−メチル−3a,4,5,6,7,7a−ヘキサヒドロ−1H−ピラゾロ[4,3−c]ピリジン
3.2gの1−メチル−1,2,5,6−テトラヒドロ−ピリジン−3−カルバルデヒドを10mLのn−BuOHに溶解した。2当量のヒドラジン水和物を加え、混合物を24時間還流させ、そして次に真空中で濃縮した。残留物をDCMに溶解し、そして2M NaOHで抽出し、そして有機相をNa2SO4上で乾燥させ、濾過し、そして真空中で濃縮して1.78gの褐色の油を生成せしめ、それをさらに精製せずに次の段階において使用した。
【0155】
【化36】
【0156】
ベンジル−ビス−(2−クロロ−エチル)−アミン
ビス−(2−クロロ−エチル)−アミン塩酸塩を150mLのアセトニトリルに懸濁した。34.8gのK2CO3(3当量)および10.0mLの臭化ベンジル(1当量)を加えた。混合物を一晩還流させた。シリカ上で濃縮し、そしてフラッシュカラムクロマトグラフィー(溶離剤PA:エーテル=95.5)で精製して4.11gの無色の油を生成せしめた。1H NMR(400MHz,CDCl3)δ2.93(t,J=7Hz,4H),3.50(t,J=7Hz,4H),3.74(s,2H),7.22〜7.37(m,5H)。
【0157】
1−ベンジル−ピペリド(piperide)−4,4−ジカルボニトリル
0.57gのマロニトリル(Malonitrile)を20mLのDMFに溶解した。1.31gのK2CO3(1.1当量)を加え、そして混合物を65℃で2時間攪拌した。10mLのDMF中2.0gのベンジル−ビス−(2−クロロ−エチル)−アミン(1当量)の溶液を65℃で滴下して加え、そして混合物を65℃でさらに3時間攪拌した。冷却した後に、混合物をEAで希釈し、そして5%水性NaHCO3で抽出した。有機相をNa2SO4上で乾燥させ、濾過し、そして真空中で濃縮して85%の予想生成物および15%のベンジル−ビス−(2−クロロ−エチル)−アミンを含有する2.02gの橙色の油を生成せしめた。この物質をさらに精製せずに次の段階において使用した。1H NMR(400MHz,CDCl3)δ2.24(t,J=5.5Hz,4H),2.50〜2.75(br s,4H),3.55(s,2H),7.22〜7.42(m,5H)。
【0158】
C−(4−アミノメチル−1−ベンジル−ピペリジン−4−イル)−メチルアミン
1.50gのLiAlH4(3当量)を100mLの乾式ジエチルエーテルに懸濁し、そして氷浴で冷却した。これに50mLの乾式THF中2.99gの1−ベンジル−ピペリド−4,4−ジカルボニトリルの溶液を滴下して加えた。混合物を室温で一晩攪拌した。混合物を氷浴で冷却し、そして1.5mLのH2O、3mLの2M NaOHおよび1.5mLのH2Oを滴下して加えた。残留物を濾過し、THFで洗浄し、そして濾液を真空中で濃縮して60%の所望の生成物を含有する2.63gの黄色の油を生成せしめ、それをさらに精製せずに次の段階において使用した。1H NMR(400MHz,CDCl3)δ1.00〜1.60(br s,2H),1.46(t,J=5.5Hz,4H),2.40(t,J=5.5Hz,4H),2.65(s,4H),3.50(s,2
H),7.20〜7.35(m,5H)。
【0159】
8−ベンジル−2,3,8−トリアザ−スピロ[4,5]デク−2−エン
2.48gのC−(4−アミノメチル−1−ベンジル−ピペリジン−4−イル)−メチルアミンを40mLのH2Oおよび10mLのMeOHに懸濁し、そして氷浴で冷却した。同時に、6.7mLの30%H2O2(6当量)および15.2mLの10%NaClO(2.4当量)を滴下して加えた。混合物を室温で1時間攪拌した。混合物をDCMで2回抽出し、合わせた有機層をNa2SO4上で乾燥させ、濾過し、そして濃縮して2.20gの黄色の油を生成せしめ、それをさらに精製せずに次の段階において使用した。経路2に従って、化合物155をこのピラゾリンビルディングブロックで製造した。これから、ベンジル脱保護(1,2−DCE中のACE−クロリド、続いてMeOH)により化合物156を生成せしめ、それを還元的アルキル化(1,2−DCE中NaBH(OAc)3の存在下で(CH2O)n)によりメチル化して化合物157を生成せしめた。
【0160】
【化37】
【0161】
メチルマロニトリル(Methylmalonitrile)
マロニトリル(10.00g;151.32mmol;2.0当量)にヨードメタン(4.71ml;75.66mmol;1.0当量)および臭化テトラブチルアンモニウム(0.98g;3.03mmol;0.04当量)を加えた。混合物を室温で30分間攪拌し、次に氷浴で冷却し、そしてカリウムtert−ブトキシド(8.49g;75.66mmol;1.0当量)をゆっくりと加えた(添加は混合物が凝固する前に開始した)。混合物を室温で2時間攪拌した。水を加え、続いてDCMで2回抽出した。Na2SO4上で乾燥させ、濾過し、そして溶媒を除いて10gの褐色の液体を生成せしめ、それを溶離剤DCM:PA=1:1、3:1およびDCMでフラッシュカラムクロマトグラフィーにより精製して3.25gの透明な液体(静置すると凝固する)を生成せしめた。1H
NMR(400MHz,CDCl3)δ1.79(d,J=7.5Hz,3H),3.79(q,J=7.5Hz,1H)。
【0162】
2−ベンジルオキシメチル−2−メチル−マロノニトリル
メチルマロニトリル(3.22g;39.40mmol;1.0当量)をTHF(35ml)に溶解した。ベンジルクロロメチルエーテル(7.54g;43.35mmol;1.1当量)およびヨウ化ナトリウム(0.20g;1.33mmol;0.03当量)を加えた。黄色の懸濁液を氷浴で冷却し、そして水素化ナトリウム(1.89g;47.29mmol;1.2当量)を少しずつ加えた。さらに多くの白色の沈殿物が形成された。混合物を室温で30分間攪拌し、エーテルで希釈し、そして5%水性NaHCO3で抽出した。有機相をNa2SO4上で乾燥させ、濾過し、そして真空中で濃縮して9.6g
の黄色の液体/油を生成せしめた。フラッシュカラムクロマトグラフィー(溶離剤EA:PA=1:4)により精製して6.2gの黄色の油を生成せしめた。1H NMR(400MHz,CDCl3)δ1.78(s,3H),3.72(s,1H),4.71(s,2H),7.31−7.43(m,5H)。
【0163】
2−ベンジルオキシメチル−2−メチル−プロパン−1,3−ジアミン
LiAlH4(3.22g;84.84mmol;3.0当量)を30mLの乾式ジエチルエーテルに懸濁し、そして氷浴で冷却した。20mLの乾式ジエチルエーテル中の2−ベンジルオキシメチル−2−メチル−マロノニトリル(5.72g;28.28mmol;1.0当量)の溶液を滴下して加えた。懸濁液を室温で4時間攪拌し、そして次に氷浴で冷却した。これに3.22mLのH2O、6.44mLの2M NaOHおよび3.22mLのH2Oを加えた。沈殿物を濾過して分離し、そしてエーテルで洗浄した。濾液を減圧下で濃縮して5.47g(84%)の無色の油を生成せしめた。1H NMR(400MHz,CDCl3)δ0.84(s,3H),2.59〜2.69(m,4H),3.29(s,2H),4.49(s,2H),7.24〜7.39(m,5H)。
【0164】
4−ベンジルオキシメチル−4−メチル−4,5−ジヒドロ−3H−ピラゾール
5.92gの2−ベンジルオキシメチル−2−メチル−プロパン−1,3−ジアミンを水(40ml)およびNaOH(10ml)に溶解し、そして氷浴で冷却した。同時に、30%H2O2(16.1mL)および10%NaClO(36.5mL)を滴下して加えた。得られる白色のエマルジョンを室温で一晩攪拌した。混合物をDCMで抽出し、有機相をNa2SO4上で乾燥させ、濾過し、そして真空中で濃縮して70%の所望の生成物を含有する5.69gの淡黄色の油を生成せしめ、それをさらに精製せずに次の段階において使用した。1H NMR(400MHz,CDCl3)δ1.05(s,3H),3.22(s,2H),4.11〜4.20(m,2H),4.29〜4.38(m,2H),4.48(s,2H),7.24〜7.39(m,5H)。
【0165】
【化38】
【0166】
テトラヒドロ−ピラン−4,4−ジカルボニトリル
マロノニトリル(5.0g)をDMSO(5mL)に溶解した。次に、ビス(2−ブロモエチル)エーテル(9.49mL)およびTBAB(1.22g)を加え、続いてKOtBu(8.49g)を少しずつ加えた。混合物を室温で4時間攪拌し、DCMに溶解し、そして5%水性NaHCO3で3回抽出した。有機相をNa2SO4上で乾燥させ、そして減圧下で蒸発させた。粗物質をPA:Et2O 65:35(Rf=0.24、KMnO4で視覚化した)で溶出するフラッシュクロマトグラフィーにより精製して2.49g(24%)の固体を生成せしめた。1H NMR(400MHz,CDCl3)δ2.24(t,4H),3.87(t,4H)。
【0167】
C−(4−アミノメチル−テトラヒドロ−ピラン−4−イル)−メチルアミン
テトラヒドロ−ピラン−4,4−ジカルボニトリル(1.52g)を乾式THF(25mL)に溶解し、そして−10℃に冷却した。この溶液に、BH3・THF(56mLのTHF中1M溶液、5当量)を滴下して加え、混合物を室温まで温めておき、そして次に60℃で6時間攪拌した。混合物を氷浴において冷却し、そしてHCl(24.2mLの6M水溶液、13当量)を加えた。混合物を室温まで温めておき、そして2時間攪拌した。混合物を2M水性NaOHで中和し、そしてDCMで3回抽出した。水層を蒸発乾固させ、残留物をCHCl3と攪拌し、固体を濾過して分離し、そして有機相を減圧下で蒸発させて1.0g(62%)の黄色の油を生成せしめた。1H NMR(400MHz,CDCl3)δ1.46(t,4H),2.74(s,4H),3.67(t,4H)。
【0168】
8−オキサ−2,3−ジアザ−スピロ[4,5]デク−2−エン
C−(4−アミノメチル−テトラヒドロ−ピラン−4−イル)−メチルアミン(1.0g)をH2O(10mL)およびMeOH(2.5mL)の混合物に溶解し、そして氷浴において冷却した。同時に、H2O2(4.8mLの30%溶液、6当量)およびNaClO(12.4mLの10%溶液、2.4当量)を滴下して加え、氷浴を取り除き、そして混合物を室温で一晩攪拌した。DCMで抽出し、Na2SO4上で乾燥させ、そして減圧下で蒸発させて85%の予想生成物および15%のジアミン出発物質を含有する380mgの透明な薄黄色の液体を生成せしめた。この物質をさらに精製せずに次の段階において使用した。1H NMR(400MHz,CDCl3)δ1.49(t,4H),3.65(t,4H),4.28(s,4H)。
【0169】
【化39】
【0170】
2,2−ビス−(2,2,2−トリフルオロ−エチル)−マロノニトリル
マロノニトリル(20.15mmol)および1−ヨード−3,3,3−トリフルオロプロパン(42.65mmol)を30mlの乾式THFに溶解し、そして混合物を氷/塩浴で冷却した。温度を5℃未満に保ちながら、1.61gのNaH(40.3mmol)を少しずつ加えた。反応混合物を室温で2時間攪拌し、そして減圧下で蒸発させた。粗物質をDCMで溶出するフラッシュクロマトグラフィーにより精製し、0.76グラムの油を生成せしめた。1H NMR(400MHz,CDCl3)δ2.62〜2.49(m,4H),2.31〜2.24(m,4H)。
【0171】
2,2−ビス−(2,2,2−トリフルオロ−エチル)−プロパン−1,3−ジアミン
340mgのLiAlH4(8.95mmol)を15mlの乾式Et2Oに懸濁し、そして氷浴において冷却した。温度が20℃未満に保たれるような速度でEt2O中760mgの2,2−ビス−(2,2,2−トリフルオロ−エチル)−マロノニトリルの溶液を滴下して加えた。混合物を室温で一晩攪拌し、氷浴において冷却し、そしてH2O(0.35ml)、2M水性NaOH(0.70ml)そして再びH2O(0.35ml)を加えることによりクエンチした。懸濁液を濾過し、フィルターケーキをEt2Oで洗浄し、そして合わせた濾液を減圧下で蒸発乾固させて0.72gの油を生成せしめた。この物質をさらに精製せずに次の段階において使用した。
【0172】
4,4−ビス−(2,2,2−トリフルオロ−エチル)−4,5−ジヒドロ−3H−ピラゾール
2,2−ビス−(2,2,2−トリフルオロ−エチル)−プロパン−1,3−ジアミン(720mg)をH2O(3ml)およびMeOH(0.75ml)の混合物に溶解し、そして氷浴において冷却した。同時に、H2O2(1.7mLの30%溶液、6当量)およびNaClO(3.85mLの10%溶液、2.4当量)を滴下して加え、氷浴を取り
除き、そして混合物を室温で一晩攪拌した。混合物をDCMで抽出し、有機相をMgSO4上で乾燥させ、そして減圧下で蒸発させて、50%の予想生成物および50%のジアミン出発物質を含有する0.82gの油を生成せしめた。この物質をさらに精製せずに次の段階において使用した。1H NMR(400MHz,CDCl3)δ4.25(s,2H),2.24〜1.85(m,4H),1.85〜1.43(m,4H)。
【実施例4】
【0173】
特定の化合物の合成
【0174】
【化40】
【0175】
(2−クロロ−ベンゼンスルホニル)−カルバミン酸メチルエステル
25.0gの2−クロロ−ベンゼンスルホンアミドに75mLのアセトニトリルおよび45.2mL(2.5当量)のトリエチルアミンを加えた。混合物を氷浴で冷却し、そして15.1mLのクロロギ酸メチルを滴下してゆっくりと加えた。混合物を室温で一晩攪拌し、そして真空中で濃縮した。水を加え、そして水層をエーテルで2回洗浄した。2M
HClでの水層の酸性化は、白色の沈殿物の形成をもたらした。懸濁液を濾過し、残留物をH2Oで洗浄し、そして真空中で乾燥させて19.1gの白色の固体を生成せしめた。1H NMR(400MHz,DMSO−d6)δ3.58(s,3H),7.52〜7.61(m,1H),7.62〜7.72(m,2H),8.10(dd,J=8および1.5Hz,1H),12.42(br s,1H)。
【0176】
【化41】
【0177】
2−クロロ−N−4−エチル−4,5−ジヒドロ−ピラゾール−1−カルボニル)ベンゼンスルホンアミド
8.5gの4−エチル−4,5−ジヒドロ−1H−ピラゾールを75mLのトルエンに溶解した。19.0gの(2−クロロ−ベンゼンスルホニル)−カルバミン酸メチルエステルを加え、そして混合物を4時間還流させた。冷却した後に、沈殿物が形成された。懸濁液を濾過し、残留物をPAで洗浄し、そして真空中で乾燥させて20.3gの白色の結晶を生成せしめた。1H NMR(200MHz,DMSO−d6)δ0.90(t,J=7.5Hz,3H),1.30〜1.70(m,2H),3.00〜3.40(m,1
H),3.25(t,J=10.5Hz,1H),3.74(t,J=10.5Hz,1H),7.08(s,1H),7.40〜7.73(m,3H),8.03〜8.16(m,1H),10.00(br s,1H)。
【0178】
【化42】
【0179】
2−クロロ−N−[ジエチルアミノ−4−エチル−4,5−ジヒドロ−ピラゾール−1−イル]−メチレン]−ベンゼン−スルホンアミド(化合物1)
2.0gの2−クロロ−N−4−エチル−4,5−ジヒドロ−ピラゾール−1−カルボニル)ベンゼンスルホンアミドを10mLのDCEに溶解した。1.07gの2−クロロ−1,3−ジメチルイミダゾリニウムクロリド(DMC)および1.75mLのTEAを加え、そして混合物を1.5時間還流させてクロロイミン中間体をインサイチューで生成せしめた。次に、5mL(過剰)のジエチルアミンを加え、そして混合物を室温で一晩攪拌した。混合物を真空中で濃縮し、そしてH2Oを加えた。DCMで抽出し(2回)、合わせた有機層をNa2SO4上で乾燥させ、蒸発乾固させ、そしてフラッシュクロマトグラフィー(エーテル、Rf=0.35)で精製し、320mgの黄色の油を生成せしめた。1H NMR(400MHz,CDCl3)δ0.95(t,J=7.5Hz,3H),1.16(t,J=7Hz,6H),1.44〜1.66(m,2H),3.00〜3.10(m,1H),3.48(q,J=7Hz,4H),3.70(dd,J=11および7Hz,1H),4.11(t,J=11Hz,1H),6.97(d,J=2Hz,1H),7.30〜7.41(m,2H),7.46(dd,J=7.5および2Hz,1H),8.16(dd,J=7.5および2Hz,1H)。
【0180】
【化43】
【0181】
N−(ビス−メチルスルファニル−メチレン)−2−クロロ−ベンゼンスルホンアミド
41.6gの2−クロロ−ベンゼンスルホンアミドに300mLのDMFおよび22mLの二硫化炭素を加えた。混合物を氷浴で冷却した。温度が10℃未満に保たれるような速度で100mLのH2O中29gのKOH(15.0mL)の溶液を滴下して加えた。混合物を5℃で30分間攪拌した。次に、温度が10℃未満に保たれるような速度で32mLのMeIを滴下して加えた。次に、混合物を室温まで温めておき、そしてさらに30分間攪拌した。H2Oを加え、そして沈殿物が生じた。これを濾過して分離し、そしてH2Oで洗浄した。残留物をEtOHで研和し、濾過して分離し、そして真空中で乾燥させ
て42.6gの白色の結晶を生成せしめた。1H NMR(200MHz,CDCl3)δ2.57(s,6H),7.32〜7.60(m,3H),8.11〜8.27(br
d,J=7.5Hz,1H)。
【0182】
【化44】
【0183】
2−クロロ−N−[(4−エチル−4,5−ジヒドロ−ピラゾール−1−イル)−メチル−スルファニル−メチレン]ベンゼンスルホン−アミド
500mgの4−エチル−4,5−ジヒドロ−1H−ピラゾールを10mLのピリジンに溶解した。1.51gのN−(ビス−メチルスルファニル−メチレン)−2−クロロ−ベンゼンスルホンアミドを加え、そして混合物を一晩還流させた。混合物を真空中で濃縮し、そしてH2Oを加え、続いてDCMで2回抽出した。合わせた有機層をNa2SO4上で乾燥させ、そして真空中で濃縮した。粗生成物をフラッシュクロマトグラフィー(勾配DCM:アセトン=100:0〜95:5)で精製して1.30gの黄色の油を生成せしめた。1H NMR(400MHz,CDCl3)δ1.02(t,J=7.5Hz,3H),1.55〜1.77(m,2H),2.28(s,3H),3.27〜3.39(m,1H),4.13(dd,J=11.5および6.5Hz,1H),4.58(t,J=11.5Hz,1H),7.16(d,J=2Hz,1H),7.39(dt,J=7.5および2Hz,1H),7.46(dt,J=7.5および2Hz,1H),7.52(dd,J=7.5および2Hz,1H),8.17(dd,J=7.5および2Hz,1H)。
【0184】
【化45】
【0185】
2−クロロ−N−[エチルアミノ−(4−エチル−4,5−ジヒドロ−ピラゾール−1−イル)−メチレン]−ベンゼンスルホン−アミド(化合物2)
1.30gの2−クロロ−N−[(4−エチル−4,5−ジヒドロ−ピラゾール−1−イル)−メチル−スルファニル−メチレン]−ベンゼンスルホン−アミドを10mLのMeOHに溶解した。H2O中のエチルアミンの70%溶液5mL(過剰)を加え、そして混合物を室温で1時間攪拌した。混合物を真空中で濃縮し、そして粗生成物をフラッシュクロマトグラフィー(エーテル、Rf=0.33)により精製して1.09gの無色の油
を生成せしめた。1H NMR(400MHz,CDCl3)δ0.95(t,J=7.5Hz,3H),1.16(t、J=7Hz,3H),1.44〜1.69(m,2H),3.03〜3.18(m,1H),3.44〜3.58(m,2H),3.71(br
dd,J=11および7.5Hz,1H),4.12(br t,J=11Hz,1H),6.88(br s,1H),6.94(d,J=2Hz,1H),7.35(dt,J=7.5および2Hz,1H),7.40(dt,J=7.5および2Hz,1H),7.48(dd,J=7.5および2Hz,1H),8.18(dd,J=7.5および2Hz,1H)。
【0186】
同様にして、「経路2」と記した以下の表における化合物は製造されている。
【0187】
【化46】
【0188】
1−エチル−2−メチル−イソチオ尿素ヨウ化水素酸塩
20.5gのエチル−チオ尿素を100mLのEtOHに溶解した。混合物を氷浴で冷却し、そして13.5mL(1.1当量)のMeIを滴下して加えた。混合物を室温で1時間攪拌し、そして真空中で濃縮して48.3gの薄黄色の油を生成せしめた。1H NMR(400MHz,DMSO−d6)δ1.17(t,J=7.5Hz,3H),2.61(s,3H),3.34(q、J=7.5Hz,2H),9.10(br s,2H)。
【0189】
【化47】
【0190】
4−N−ジエチル−4,5−ジヒドロ−ピラゾール−1−カルボキサミジン塩酸塩
19.36gの4−エチル−4,5−ジヒドロ−1H−ピラゾールを100mLのトルエンに溶解した。48.5gの1−エチル−2−メチル−イソチオ尿素ヨウ化水素酸塩および33.8mLのDiPEAを加え、そして混合物を48時間還流させた。混合物を濃縮し、2M NaOHを加え、続いてDCMで抽出した(3回)。合わせた有機層をNa2SO4上で乾燥させ、そして溶媒を真空中で蒸発させてNMRによれば75%の所望の生成物を含有する32.7g(99%)の赤色の油を生成せしめた。油をEtOHに溶解し、そして194mLのEtOH中1M HClを滴下して加えた。混合物を室温で30分間攪拌し、そして真空中で濃縮した。CH3CN:MTBE=1:1からの結晶化により11.52g(29%)の所望の生成物がベージュ色の固体として得られた。1H NMR(400MHz,DMSO−d6)δ0.96(t,J=7.5Hz,3H),1.16(t,J=7Hz,3H),1.46〜1.72(m,2H),3.32(q,J=7Hz,2H),3.35〜3.45(m,1H),3.55(dd,J=10.5およ
び7Hz,1H),3.96(t,J=10.5Hz,1H),7.34(d,J=2Hz,1H),8.00(br s,2H)。
【0191】
【化48】
【0192】
ベンゾ[1,2,5]チアジアゾール−4−スルホン酸エチルアミノ−(4−エチル−4,5−ジヒドロ−ピラゾール−1−イル)−メチレンアミド(化合物78)
300mgの4,N−ジエチル−4,5−ジヒドロ−ピラゾール−1−カルボキサミジン塩酸塩を10mLのDCMに懸濁した。0.53mLのDiPEAおよび310mgのベンゾ[1,2,5]チアジアゾール−4−スルホニルクロリドを加え、そして混合物を室温で一晩攪拌した。混合物を5%NaHCO3および2M NaOHで洗浄し、有機層をNa2SO4上で乾燥させ、そして溶媒を真空中で蒸発させて410mgの赤色/褐色の油を生成せしめた。粗生成物をフラッシュクロマトグラフィー(DCM:アセトン=98:2、Rf=0.18)により精製して350mg(65%)の橙色の油を生成せしめた。1H NMR(400MHz,CDCl3)δ0.93(t,J=7.5Hz,3H),1.16(br t,J=7Hz,3H),1.41〜1.66(m,2H),3.01〜3.16(m,1H),3.39〜3.55(m,2H),3.59〜3.74(m,1H),3.95〜4.15(m,1H),6.94(br s,1H),6.95(br s,1H),7.68(dd,J=9および7Hz,1H),8.15(br d,J=9Hz,1H),8.31(br d,J=7Hz,1H)。
【0193】
【化49】
【0194】
1−エチル−2−メチル−イソチオ尿素ヨウ化水素酸塩
20.0gのエチル−チオ尿素を100mLのEtOHに懸濁し、そして30g(1.1当量)のMeIを滴下して加え、その間に混合物は透明な黄色の溶液になった。次に、混合物を室温で1時間攪拌し、そして真空中で濃縮して48.1gの黄色の油を生成せしめた。1H NMR(400MHz,DMSO−d6)δ1.17(t,J=7.5Hz,3H),2.61(s,3H),3.34(q,J=7.5Hz,2H),9.10(br s,2H)。
【0195】
【化50】
【0196】
N−エチル−4,4−ジメチル−4,5−ジヒドロ−ピラゾール−1−カルボキサミジン塩酸塩
12.0gの4,4−ジメチル−4,5−ジヒドロ−3H−ピラゾールを100mLのピリジンに溶解した。50mLのピリジン中30.0gの1−エチル−2−メチル−イソチオ尿素ヨウ化水素酸塩の溶液を加え、そして混合物を20時間還流させた。混合物を室温まで冷却し、そして減圧下で濃縮し、そして残留物をDCM(120mL)に溶解した。有機相を2N NaOH(2x120mL)で抽出し、水(120mL)で洗浄し、Na2SO4上で乾燥させ、そして減圧下で蒸発させて16.3g(79%)の橙色の油を生成せしめた。油(10.0g)をEtOAc(50mL)に溶解し、そして60℃に加熱した。熱源の除去後に、イソプロパノール(20mL)中のHClの5〜6N溶液を4分の期間にわたって投与した。室温まで冷却した後に、EtOAc(50mL)を4分の期間にわたって加え、そして混合物を20℃で90分間攪拌した。生じた結晶を濾過により集め、そしてEtOAc(20mL)で洗浄し、続いて穏やかな加熱で減圧下で乾燥させて、6.52g(54%)の所望の生成物を黄色の固体として生成せしめた。1H NMR(400MHz,DMSO−d6)δ1.13(t,J=7Hz,3H),1.24(s,6H),3.27〜3.34(m,2H),3.64(s,2H),7.26(s,1H),8.03(br s,2H),8.13(br s,1H)。
【0197】
【化51】
【0198】
3−クロロ−N−[(4,4−ジメチル−4,5−ジヒドロ−ピラゾール−1−イル)−エチルアミノ−メチレン]−ベンゼンスルホンアミド(化合物33)
6.39gのN−エチル−4,4−ジメチル−4,5−ジヒドロ−ピラゾール−1−カルボキサミジン塩酸塩を65mLのDCMに懸濁した。12.0mLのDiPEAおよび3.96mLの3−クロロ−ベンゼンスルホニルクロリドを加え、そして懸濁液を室温で20時間攪拌し、暗褐色の混濁溶液をもたらした。混合物を2M NaOH(2x125mL)および1M HCl(2x125mL)で抽出し、水(100mL)で洗浄し、そして有機層をNa2SO4上で乾燥させ、続いて減圧下で蒸発させて7.70gの褐色の油を生成せしめた。油(1.0g)を還流下でMTBE(3mL)に溶解し、そして溶液を室温までゆっくりと冷却し、結晶化を開始した。懸濁液を室温で10分間攪拌し、ヘキサン(6mL)を1分の期間にわたって加えた。得られる懸濁液を室温で20分そして0℃で50分間攪拌し、そして生成物を濾過により集め、そしてヘキサン(1mL)で洗浄した。40℃で減圧下で乾燥させて0.85gの薄褐色の固体、m.p.62〜67℃を生成せしめた。
【0199】
同様にして、「経路3」と記した以下の表における化合物は製造されている。
【0200】
【表1】
【0201】
【表2】
【0202】
【表3】
【0203】
【表4】
【0204】
【表5】
【0205】
【表6】
【0206】
【表7】
【0207】
【表8】
【0208】
【表9】
【0209】
【表10】
【0210】
【表11】
【0211】
【表12】
【0212】
【表13】
【0213】
【表14】
【0214】
【表15】
【0215】
S*=合成経路;Rf(x)=Rf値、括弧の間:TLC移動相:
(a)=ジエチルエーテル;(b)=MeOH:TEA=97:3;(c)=DCM:アセトン=99:1;(d)=DCM:MeOH=99:1;(e)=DCM:アセトン=98:2;(f)=DCM:アセトン=95:5;(g)=DCM:MeOH=98:2;(h)=EA:PA=1:2;(i)=EA:PA=1:3;(j)=EA:PA=1:1;(k)=EA:PA=1:4;(l)=DCM;(m)=DCM:MeOH=97:3;(n)=DCM:MeOH=95:5;(o)=EA;(p)=EA:MeOH:NH4OH=94.5:5:0.5;(q)=DCM:EA=3:1;(r)=DCM:ジエチルエーテル=1:4;(s)=ジエチルエーテル:PA=7:3;(t)=ジエチルエーテル:PA=8:2;(u)=EA:PA=3:1;(v)=DCM:MeOH:NH4OH=78:20:2;(w)=DCM:MeOH:NH4OH=94.5:5:
0.5;(x)=Et2O:EA=8:2;(y)=Et2O:EA=9:1;(z)=EA:PA=5:95;Rt=LC−MS分析における保持時間(分単位)。
【0216】
本発明の化合物は新規である。以下の表に示されるように、それらは5−HT6受容体に対する高い親和性(pKi)を有し、そして強力なアンタゴニスト(pA2)である。文献に開示される構造的に最も密接に関係する化合物は、WO 02/030881に開示されるスルホニルピロリジン誘導体のいくつかである:
【0217】
【化52】
【0218】
WO 02/030881において、実施例18(R=p−CH3)、25(R=p−Cl)、26(R=H)および27(R=o−NO2)について薬理学的データは示されなかったが、それらは疼痛および片頭痛を包含する多数の症候および症状の治療において有用なガバペンチン結合部位のモジュレーターであると請求される。これらの化合物は、本発明の化合物に関する合成研究中に一連の化合物がピラゾリン環(本発明の全ての化合物に存在する)と異なる環系で合成されそしてこれらの全てが5−HT6アンタゴニストとして不活性であることが見出されたので、5−HT6受容体に対する親和性を有すると思われない。WO 02/030881に開示されるものに最も近いのは:
【0219】
【化53】
【0220】
であった。この化合物は、ピラゾリン環を有する化合物(例えば、化合物28は7.7のpA2値を有する)と顕著に異なり不活性である(pA2<5.0)ことが見出された。上記に示されるピロリジン環を有する化合物のほかに、全く同じ構造を有するが異なる環を有する化合物もまた合成され(上記に開示されるものと同様の経路を用いて)、そして試験された。特に:フェニル、2−ピリジル、2−ピラジニル、2−フラニル、5−イソオキサゾリル、2−キノリルおよび1−イソキノリル環(上記に示される化合物における1−ピロリジン環の代わりに)を有する化合物は全て不活性である(pA2<5.0)ことが見出され、本発明の化合物のピラゾリン環は5−HT6受容体との相互作用にとって重要であることを示す。
【0221】
その合成が上に記述される特定の化合物は、より詳細に本発明をさらに説明するものであり、従って、決して本発明の範囲を限定すると見なされない。本発明の他の態様は、本明細書および本明細書に開示される本発明の実施の考察から当業者に明らかである。従って、本明細書および実施例は実例としてのみ考えられるものとする。
【実施例5】
【0222】
動物研究において使用する製剤
経口(p.o.)投与用:ガラス管中の一般式(1)の固体化合物の所望の量(0.5〜5mg)に、いくらかのガラスビーズを加え、そして2分間ボルテックスすることにより固体を粉砕した。水中1%のメチルセルロースおよび2%(v/v)のポロキサマー188(Lutrol F68)の溶液1mlの添加後に、10分間ボルテックスすることにより化合物を懸濁した。数滴の水性NaOH(0.1N)でpHを7に調整した。超音波浴を用いることにより懸濁液中の残留粒子をさらに懸濁した。
【0223】
腹腔内(i.p.)投与用:ガラス管中の一般式(1)の固体化合物の所望の量(0.5〜15mg)に、いくらかのガラスビーズを加え、そして2分間ボルテックスすることにより固体を粉砕した。水中1%のメチルセルロースおよび5%のマンニトールの溶液1mlの添加後に、10分間ボルテックスすることにより化合物を懸濁した。最後にpHを7に調整した。
【実施例6】
【0224】
薬理学的方法
ヒト5−HT6受容体に対するインビトロ親和性
[3H]−N−メチル−リセルグ酸ジエチルアミド([3H]−LSD)をリガンドとして用いる結合研究によりヒト5−HT6受容体でトランスフェクションしたCHO細胞の膜調製物においてヒト5−HT6受容体に対する親和性を測定した。膜調製物は、Euroscreen(ブリュッセル)により供給される細胞から調製した。CHO/Gα16/mtAEQ/h5HT6−A1細胞を1%の透析したFCS、2mMのL−グルタミン、ジェネティシン500μg/mlおよびゼオシン200μg/mlを補足したCHO−S−SFM II培地(Gibco BRL)中Tフラスコにおいて培養した。0.25%のトリプシン(1ml/T175フラスコ)を用いて細胞を採取し、遠心分離し、そして次にCHO−S−SFM II培地に懸濁し、そして−80℃で凍結させた。融解後に細胞を4℃で1500gで3分の間遠心分離した。ペレットから、2サイクルの均質化(Potter−Elvehjem 10ストローク、600rpm)および遠心分離(40,000g15分間、4℃)により細胞膜を調製した。定常状態条件を達成するようにそして特異的結合を最適化するようにアッセイを定めた。5−HT6受容体に関して、5x105の細胞からの膜を5.0nMの[3H]−LSDと37℃で30分間インキュベーションした。非特異的結合は10−5Mのセロトニンを用いて決定された。0.5%のポリエチレンイミンで前処理されているガラス繊維フィルター(GF/B)を通した真空濾過によりアッセイを止めた。全および結合放射活性を液体シンチレーション計数により決定した。80%より大きい特異的結合がこれらのアッセイの各々において得られた。化合物を4log濃度範囲で試験し;全ての決定は三重反復として行われた。IC50値は、Hill式曲線適合を用いて非線形回帰分析により決定された。阻害定数(Ki値)は、Cheng−Preushoff式:
Ki=IC50:(1+L/Kd)
[式中、Lはアッセイにおける放射性リガンド([3H]−LSD)濃度を、そしてKdは受容体に対する放射性リガンドの親和性を表す]
から計算された。結果は、pKi値、少なくとも3回の別個の実験の平均±SDとして表される。
【0225】
ヒト5−HT6受容体へのインビトロ機能活性((拮抗)作動作用((ant)agonism))
CHO−ヒト−5HT6−エクオリンアッセイは、Euroscreen、ブリュッセルから購入した(Euroscreen,技術書類一式、ヒト組換えセロトニン5−HT6−A1受容体、DNAクローンおよびCHO AequoScreenTM組換え細胞系、カタログn0:ES−316−A、2003年2月)。ヒト−5HT6−エクオリン
細胞は、ミトコンドリア標的化アポ−エクオリンを発現する。活性エクオリンを再構成するためには、細胞にコエレンテラジンを与えなければならない。ヒト5−HT6受容体へのアゴニストの結合後に細胞内カルシウム濃度は増加し、そしてアポ−エクオリン/コエレンテラジン複合体へのカルシウムの結合はコエレンテラジンの酸化反応を引き起こし、それはアポ−エクオリン、コエレンテラミド、CO2および光(λmax469nm)の生成をもたらす。この発光反応は、アゴニスト濃度により決まる。発光は、MicroBeta Jet(Perkin Elmer)を用いて測定される。化合物のアゴニスト作用は、pEC50として表される。化合物のアンタゴニスト作用は、10−8Mのα−メチルセロトニン誘起発光の阻害として決定され、そしてpA2はCheng−Preushoff式に従って計算された。化合物を5log濃度範囲で試験し、そして3回の独立した実験を二重反復で行った。
【実施例7】
【0226】
製薬学的製剤
臨床使用のために、式(1)の化合物は、該化合物、さらに特に本明細書に開示される特定の化合物を含有するので本発明の重要なそして新規の態様である製薬学的組成物に調合される。用いることができる製薬学的組成物のタイプには、錠剤、チュアブル錠、カプセル剤(マイクロカプセル剤を包含する)、液剤、非経口液剤、軟膏(クリームおよびゲル)、座薬、懸濁剤および本明細書に開示される他のタイプが包含されるがこれらに限定されるものではなく、もしくは本明細書および当該技術分野における一般的知識から当業者に明らかである。有効成分は例えば、シクロデキストリン、それらのエーテルもしくはそれらのエステルにおける包接錯体の形態であることもできる。組成物は経口、静脈内、皮下、気管、気管支、鼻腔内、肺、経皮、口腔、直腸、非経口もしくは他の投与方法に用いられる。製薬学的製剤は、製薬学的に許容しうる添加剤、希釈剤および/もしくは担体と混合した式(1)の少なくとも1つの化合物を含有する。有効成分の総量は、好適には製剤の約0.1%(w/w)〜約95%(w/w)、好適には0.5%〜50%(w/w)、そして好ましくは1%〜25%(w/w)の範囲である。
【0227】
本発明の化合物は、製薬学的に通常の液体もしくは固体充填剤および増量剤、溶媒、乳化剤、潤滑剤、香料、着色料および/もしくは緩衝物質のような、液体もしくは固体、粉末成分のような補助物質を用いて通常の方法によって投与に適当な形態にすることができる。頻繁に用いられる補助物質には、炭酸マグネシウム、二酸化チタン、ラクトース、サッカロース、ソルビトール、マンニトールおよび他の糖もしくは糖アルコール、タルク、乳タンパク質、ゼラチン、澱粉、アミロペクチン、セルロースおよびその誘導体、動物および植物油、例えば魚肝油、ヒマワリ油、ラッカセイ油もしくはゴマ油、ポリエチレングリコールおよび溶媒、例えば滅菌水および1価もしくは多価アルコール、例えばグリセロール、ならびに崩壊剤および潤滑剤、例えばステアリン酸マグネシウム、ステアリン酸カルシウム、ステアリルフマル酸ナトリウムおよびポリエチレングリコールワックスが包含される。次に、混合物を顆粒剤に加工するかもしくは錠剤に圧縮することができる。錠剤は、以下の成分を用いて製造される。
【0228】
【表16】
【0229】
成分を混ぜ合わせ、そして圧縮して各々230mgの重さの錠剤を生成せしめる。
【0230】
有効成分は、製剤を生成せしめるために混合する前に、他の非有効成分と別個に予混合することができる。有効成分はまた、製剤を生成せしめるために非有効成分と混合する前に、相互に混合することもできる。
【0231】
ソフトゼラチンカプセル剤は、本発明の有効成分、植物油、脂肪、もしくはソフトゼラチンカプセル剤用の他の適当な賦形剤の混合物を含有するカプセルで製造することができる。ハードゼラチンカプセル剤は、有効成分の顆粒を含有することができる。ハードゼラチンカプセル剤はまた、ラクトース、サッカロース、ソルビトール、マンニトール、ジャガイモ澱粉、コーンスターチ、アミロペクチン、セルロース誘導体もしくはゼラチンのような固形粉末成分と一緒に有効成分を含有することもできる。ハードゼラチンカプセル剤は、以下の成分を用いて製造することができる:
【0232】
【表17】
【0233】
上記の成分を混合し、そして120mgの量においてハードゼラチンカプセルに詰める。
【0234】
直腸投与用の投与単位は、(i)中性脂肪ベースと混合した有効成分を含有する座薬の形態で;(ii)植物油、パラフィン油もしくはゼラチン直腸カプセル剤用の他の適当な賦形剤との混合物において有効成分を含有するゼラチン直腸カプセル剤の形態で;(iii)既製のミクロ浣腸(micro enema)の形態で;または(iv)投与の直前に適当な溶媒において再構成される乾燥ミクロ浣腸製剤の形態で製造することができる。各々1mgの有効成分を含有する座薬を下記のとおり製造することができる。
【0235】
【表18】
【0236】
有効成分を適切なサイズのメッシュふるいに通し、そして必要な最小加熱を用いて前もって融解した飽和脂肪酸グリセリドに懸濁する。次に、混合物を標準2g容量の座薬型に注ぎ込み、そして冷却させる。
【0237】
液状製剤はシロップ剤、エリキシル剤、濃縮ドロップもしくは懸濁剤、例えば、有効成分ならびに例えば糖もしくは糖アルコールおよびエタノール、水、グリセロール、プロピレングリコールおよびポリエチレングリコールの混合物からなる残りを含有する液剤もしくは懸濁剤の形態で製造することができる。静脈内製剤は、下記のとおり製造することができる:
【0238】
【表19】
【0239】
化合物をArlatone GTM、EtOHおよび水に溶解し、そして次に溶液をさらなる水でゆっくりと希釈する。
【0240】
所望に応じて、そのような液状製剤は着色剤、香料、防腐剤、サッカリンおよびカルボキシメチルセルロースもしくは他の増粘剤を含有することができる。液状製剤はまた、使用前に適当な溶媒で再構成される乾燥粉末の形態で製造することもできる。非経口投与用の液剤は、製薬学的に許容しうる溶媒における本発明の製剤の液剤として製造することができる。これらの液剤はまた、安定化成分、防腐剤および/もしくは緩衝成分を含有することもできる。非経口投与用の液剤はまた、使用前に適当な溶媒で再構成される乾燥製剤として製造することもできる。
【0241】
また本発明に従って提供されるのは、医学的治療における使用のための、製剤および本発明の製薬学的組成物の成分の1つもしくはそれ以上を詰めた1つもしくはそれ以上の容器を含んでなる「キットオブパーツ」である。そのような容器(1つもしくは複数)に付随するのは様々な資料、例えば使用説明書、または製薬学的製品の製造、使用もしくは販売を規制する政府機関により規定される形態の通知であることができ、この通知はヒトもしくは動物投与のための製造、使用もしくは販売の該機関による承認を示す。5−HT6受容体の拮抗作用が必要とされるかもしくは所望される症状を処置することに使用する薬剤の製造における本発明の製剤の使用、および5−HT6受容体の拮抗作用が必要とされるかもしくは所望される症状を患っているかもしくは起こしやすい患者への式(1)の少なくとも1つの化合物の治療的に有効な総量の投与を含んでなる医学的処置の方法。
【0242】
例としてそして限定するものではなく、全身使用もしくは局所適用のための好ましい活性化合物を含んでなるいくつかの製薬学的組成物が示される。本発明の他の化合物もしくはその組み合わせを該化合物の代わりに(もしくはそれに加えて)用いることができる。有効成分の濃度は、本明細書に説明されるように広範囲にわたって変えることができる。含まれ得る成分の量およびタイプは、当該技術分野において周知である。
【0243】
文献目録
Bentley,J.C.et al.(1997)J.Psychopharmacol.Suppl.A64,255
Bentley,J.C.et al.(1999a)Br J Pharmacol.Suppl.126,P66
Bentley,J.C.,et al.(1999b).Br J Pharmacol 126(7):1537−42
Berge,S.M.:“Pharmaceutical salts”,J.Pharmaceutical Science,66,1−19(1977).
Bickel,M.H.,:“The pharmacology and Biochemistry of N−oxides”,Pharmacological Reviews,21(4),325−355,1969.
Bundgaard,H.(editor),“Design of Prodrugs”,Elsevier,1985.
Byrn et al.,Pharmaceutical Research,12(7),945−954,1995.
Dwyer & Meilor,:“Chelating agents and Metal Chelates”,Academic Press,chapter 7,1964.
Ettmayer,P.et al.,“Lessons learned from
marketed and investigational prodrugs”,J.Med.Chem.,47,2393−2404,2004.
Jaervinen,T.et al.,“Design and Pharmaceutical applications of prodrugs”,pages 733−796 in:S.C.Gad(editor):“Drug Discovery Handbook”,John Wiley & Sons Inc.,New Jersey,U.S.A.,2005.
King,F.D.,(editor),page 215 in:“Medicinal Chemistry:Principles and Practice”,1994,ISBN 0−85186−494−5.
Kohen,R.,et al.(1996).J Neurochem 66(1):47−56
Martin,E.W.(Editor),“Remington:The Science and Practice of Pharmacy”,Mack Publishing Company,19th Edition,Easton,Pa,Vol
2.,Chapter 83,1447−1462,1995.
Rogers,D.C.,et al.(1999).Br J Pharamcol
127(suppl.).22P
Roth,B.L.,et al.(1994).J Pharmacol Exp Ther 268(3):1403−10
Ruat,M.et al.(1993)Biochem.Biophys.Res.Commun.193:268−276
Sebben,M.et al.(1994)NeuroReport 5:2553
−2557
Sibley,D.R.et al.,Mol.Pharmacol.,1993,43,320−327
Sleight,A.J.,et al.,Neurotransmission,1995,11,1−5
Sleight,A.J.,et al.,Serotonin ID Research Alert,1997,2(3),115−8).
Sleight,A.J.,et al.(1998).Br J Pharmacol 124(3):556−62
Stella,J.,“Prodrugs as therapeutics”,Expert Opin.Ther.Patents,14(3),277−280,2004.
Woolley M.L.et al.(2001)Neuropharmacology 41:210−219
WO 01/070700およびWO 02/030881
【索引】
【0001】
索引 頁
発明の名称 1
索引 1
技術分野 1
背景技術 1
開示 3
定義 8
略語 14
実施例1:分析方法 16
実施例2:合成の一般的態様 17
実施例3:ピラゾリン中間体の合成 19
実施例4:本発明の化合物の合成 41
実施例5:動物研究において使用する製剤 63
実施例6:薬理学的方法 63
実施例7:製薬学的製剤 65
文献目録 68
請求項 69
要約書 77
【技術分野】
【0002】
本発明は製薬および有機化学の分野に関し、そしてスルホニルピラゾリンカルボキサミジン誘導体、中間体、製剤および方法を提供する。
【背景技術】
【0003】
末梢および中枢神経系の重要な伝達物質、セロトニン(5−ヒドロキシトリプタミンもしくは5−HT)は、5−HT1、5−HT2、5−HT3、5−HT4、5−HT5、5−HT6および5−HT7と呼ばれる多数の受容体ファミリーを介して媒介される、広範囲の生理学的および病理学的機能を調節する。後者の3つの機能は他のものよりあまり理解されていないが、5−HTに媒介されるシグナル伝達を選択的に妨げる化合物は重要な新規薬剤標的であることは一般に認められている。
【0004】
ラット5−HT6受容体は、2つの異なるグループにより(非特許文献1;非特許文献2)、そして89%の配列同一性を共有するヒトのものはそのすぐ後にクローン化された(非特許文献3)。5−HT6受容体への最近の関心の多くは、いくつかの向精神薬がヒト5−HT6受容体での高親和性アンタゴニストであるからである(非特許文献3;非特許文献4)。これらの化合物には、アミトリプチリン(Ki=65nM)ならびに非定型抗精神病薬クロザピン(Ki=9.5nM)、オランザピン(Ki=10nM)およびクエチアピン(Ki=33nM)が包含される。しかしながら、これらの化合物のいずれも選択的ではない。報告される最初の選択的5−HT6受容体アンタゴニストは、Ro 04−6790およびRo 63−0563である。それらの有用性は、それらの中程度の親和性(それぞれ、Ki=50nMおよび12nM)および不十分な薬物動態により限定される(非特許文献5)。選択的5−HT6受容体アンタゴニストRo−04−6790およびSB−271046の最近の開発に伴い、認知機能のモデルにおけるこれらの化合物の活性に関するいくつかの報告がなされている。SB−271046は、モリス水迷路における能力を改善した(非特許文献6)。これらの結果は、5−HT6受容体配列に対
するアンチセンスオリゴヌクレオチドの長期脳室内投与がモリス水迷路における能力のいくつかの尺度の改善をもたらした結果と一致する(非特許文献7)。最近、ラットにおける食物摂取を減少する5−HT6アンタゴニストおよび5−HT6アンチセンスオリゴヌクレオチドの効果が報告されている(非特許文献8;非常特許文献9;非特許文献10)。肥満は、許容基準を上回る過剰体重をもたらす体脂肪含有量の増加を特徴とする症状である。肥満は西欧諸国における最も重大な栄養障害であり、そして全ての先進諸国における主要な健康問題である。この障害は心臓血管疾患、消化器系疾患、呼吸器系疾患、癌および2型糖尿病のような疾患の増加した発生率に起因する増加した死亡率をもたらす。
【0005】
5−HT6選択的リガンドは、パーキンソン病、ハンチントン舞踏病および/もしくは統合失調症、不安、鬱病、躁鬱病、精神病、癲癇、強迫神経症、気分障害、片頭痛、アルツハイマー病(認識記憶の増強)、加齢に伴う認識衰退、軽度認識障害、神経細胞成長障害を特徴とする神経変性疾患、睡眠障害、食欲不振および過食症のような摂食障害、無茶食い障害、パニック発作、静座不能、注意欠陥多動性障害(ADHD)、注意欠陥障害(ADD)、コカイン、エタノール、ニコチンおよびベンゾジアゼピンのような薬物乱用からの離脱、ならびに疼痛のような中枢神経系のある種の障害、ならびに脊椎外傷および/もしくは頭部損傷と関連する障害、例えば水頭症の処置もしくは予防において潜在的に有用であると同定されている。5−HT6選択的リガンドはまた、機能性腸疾患および過敏性腸症候群のようなある種の胃腸障害の処置においてそして肥満および2型糖尿病の処置もしくは予防において有用であり、体重のそして体重増加の減少をもたらすと予想される。体重のそして体重増加の減少(例えば、体重障害を処置すること)は、とりわけ食物摂取の減少によりもたらされる。
【0006】
本発明の目的は、ある種のCNS障害の処置に有用な化合物、既知の5−HT6アンタゴニストのいずれかと化学的に関係しない強力なそして選択的な5−HT6アンタゴニストを提供することであった。
【先行技術文献】
【非特許文献】
【0007】
【非特許文献1】Ruat,M.et al.(1993)Biochem.Biophys.Res.Commun.193:268−276
【非特許文献2】Sebben,M.et al.(1994)NeuroReport 5:2553−2557
【非特許文献3】Kohen,R.et al.(1996).J Neurochem 66(1):47−56
【非特許文献4】Roth,B.L.,et al.(1994).J Pharmacol Exp Ther 268(3):1403−10
【非特許文献5】Sleight,A.J.,et al.(1998).Br J Pharmacol 124(3):556−62
【非特許文献6】Rogers,D.C.,et al.(1999).Br J Pharmacol 127(suppl.).22P
【非特許文献7】Bentley,J.C.,et al.(1999b)Br J Pharmacol 126(7):1537−42
【非特許文献8】Bentley,J.C.et al.(1997)J.Psychopharmacol.Suppl.A64,255
【非特許文献9】Bentley,J.C.et al.(1999a)Br J Pharmacol.Suppl.126,P66
【非特許文献10】Woolley,M.L.et al.(2001)Neuropharmacology 41:210−219
【発明の開示】
【0008】
驚くべきことに、ある種のスルホニルピラゾリンカルボキサミジン誘導体は5−HT6受容体アンタゴニストであることが見出された。本発明は一般式(1):
【0009】
【化1】
【0010】
[式中:
−R1は水素、非置換のアルキル(C1〜4)基、1個もしくはそれ以上のハロゲン原子で置換されたアルキル(C1〜4)基、または場合により1個もしくはそれ以上のハロゲン原子で置換されていてもよいフェニル基を表し、
−R2およびR3は独立して水素、非置換のアルキル(C1〜4)基、1個もしくはそれ以上のハロゲン原子で置換されたアルキル(C1〜4)基、場合により1個もしくはそれ以上のハロゲン原子で置換されていてもよいアルキル(C1〜4)−O−アルキル(C1〜4)−フェニル基または場合により1個もしくはそれ以上のハロゲン原子で置換されていてもよいフェニル基を表すか、あるいは
R1およびR2は「a」および「b」の記号が付された炭素原子と一緒になってC5〜8−シクロアルキル環を形成するか、あるいは
R2およびR3は「b」の記号が付された炭素原子と一緒になってC3〜8−シクロアルキル環を形成するか、あるいは
R2およびR3は「b」の記号が付された炭素原子と一緒になって場合により置換されていてもよいC5〜8−ヘテロシクロアルキル環を形成するか、あるいは
−R4およびR5は独立して水素、非置換のアルキル(C1〜4)基、1個もしくはそれ以上のハロゲン原子で置換されたアルキル(C1〜4)基、場合により置換されていてもよい単環式芳香族基、場合により置換されていてもよい縮合した二環式芳香族基、場合により置換されていてもよい単環式ヘテロ芳香族基、場合により置換されていてもよい縮合した二環式ヘテロ芳香族基を表すか、あるいは
R3およびR4は「b」および「c」の記号が付された炭素原子と一緒になってC3〜8−シクロアルキル環を形成するか、あるいは
R3およびR4は「b」および「c」の記号が付された炭素原子と一緒になって場合により置換されていてもよいC5〜8−ヘテロシクロアルキル環を形成するか、あるいは
−R6およびR7は独立して水素原子、またはアルキル(C1〜4)基、または1個もしくはそれ以上のハロゲン原子で置換されたアルキル(C1〜4)基;あるいは(C1〜3)アルコキシ基、またはジアルキル(C1〜3)−アミノ−アルキル(C1〜3)基、または場合により置換されていてもよい単環式もしくは縮合した二環式芳香族もしくはヘテロ芳香族基、または場合により置換されていてもよいC5〜8−シクロアルキル基もしくは場合により置換されていてもよいC5〜8−ヘテロシクロアルキル基を表すか、あるいは
R6およびR7は、それらが結合している窒素原子と一緒になって、場合により置換されていてもよいC5〜8−ヘテロシクロアルキル基を形成し、
−R8は場合により置換されていてもよい単環式芳香族基、場合により置換されていてもよい縮合した二環式芳香族基、場合により置換されていてもよい単環式ヘテロ芳香族基、場合により置換されていてもよい縮合した二環式ヘテロ芳香族基、−CR9=CR10−アリール基(ここで、R9およびR10は独立して水素またはアルキル−(C1〜3)基を表す)、−C≡C−アリール基、場合により置換されていてもよいピペリジニル基、もしくは基−NR11R12(ここで、R11およびR12は独立して水素、アルキル−(C1〜3)基または場合により置換されていてもよいフェニルもしくはベンジル基を表す)を表す]
の化合物もしくは互変異性体、立体異性体、N−オキシド、同位体標識したアナログ、または前述のもののいずれかの薬理学的に許容しうる塩、水和物もしくは溶媒和物に関する。
【0011】
本発明は、式(1)を有する化合物のラセミ化合物、ジアステレオマーの混合物ならびに個々の立体異性体に関する。本発明はまた、式(1)を有する化合物のE異性体、Z異性体およびE/Z混合物にも関する。
【0012】
本発明は特に、
−R1が水素を表すかもしくはR1およびR2が「a」および「b」の記号が付された炭素原子と一緒になってシクロヘキシル環を形成し、
−R2およびR3が独立して水素もしくはアルキル(C1〜3)基を表すか、またはR2およびR3が「b」の記号が付された炭素原子と一緒になってシクロペンチルもしくはシクロヘキシル環を形成し、
−R4およびR5が独立して水素、アルキル(C1〜3)基を表すか、またはR3およびR4が「b」および「c」の記号が付された炭素原子と一緒になってC3〜8−シクロアルキル環を形成し、
−R6およびR7が独立して水素原子、またはアルキル(C1〜3)基、または1個
もしくはそれ以上のハロゲン原子で置換されたアルキル(C1〜4)基;またはメトキシ基、またはシクロヘキシル基、またはベンジル基、または4−ピペリジニル基を表し、
−R8が上記に示されるとおりの意味を有する
一般式(1)の化合物もしくは互変異性体、立体異性体、N−オキシド、同位体標識したアナログ、または前述のもののいずれかの薬理学的に許容しうる塩、水和物もしくは溶媒和物に関する。
【0013】
なおさらに特に、本発明は、R1、R4、R5およびR6が水素を表し、R2およびR3が独立してアルキル(C1〜3)基を表すか、またはR2およびR3が「b」の記号が付された炭素原子と一緒になってシクロペンチルもしくはシクロヘキシル環を形成し、R7がアルキル(C1〜3)基を表し、そしてR8が上記に示されるとおりの意味を有する一般式(1)の化合物もしくは互変異性体、立体異性体、N−オキシド、同位体標識したアナログ、または前述のもののいずれかの薬理学的に許容しうる塩、水和物もしくは溶媒和物に関する。
【0014】
別の態様において、本発明は、ピラゾリン環における2個の潜在的不斉炭素原子のいずれか一方もしくは両方が左旋性もしくは右旋性鏡像異性体である式(1)の化合物に関する。
【0015】
式(1)の本発明の化合物ならびにその薬理学的に許容しうる塩は、5−HT6受容体アンタゴニスト活性を有する。それらは、5−HT6受容体が関係するか、もしくはそれらの受容体の操作により処置できる障害を処置することにおいて有用である。例えば、パーキンソン病、ハンチントン舞踏病、統合失調症、不安、鬱病、躁鬱病、精神病、癲癇、強迫神経症、気分障害、片頭痛、アルツハイマー病、加齢に伴う認識衰退、軽度認識障害
、睡眠障害、摂食障害、食欲不振、過食症、無茶食い障害、パニック発作、静座不能、注意欠陥多動性障害、注意欠陥障害、コカイン、エタノール、ニコチンもしくはベンゾジアゼピンの乱用からの離脱、疼痛、脊椎外傷もしくは頭部損傷と関連する障害、水頭症、機能性腸疾患、過敏性腸症候群、肥満および2型糖尿病において。
【0016】
本発明の他の態様には:
例えば、5−HT6受容体をブロックすることにより処置できる障害もしくは症状を処置するための製薬学的組成物、該組成物は式(1)の化合物もしくはその製薬学的に許容しうる塩および製薬学的に許容しうる担体を含んでなる;
5−HT6受容体をブロックすることにより処置できる障害もしくは症状を処置する方法、該方法は式(1)の化合物もしくはその製薬学的に許容しうる塩をそのような処置を必要とする哺乳類に投与することを含んでなる;
例えば本明細書に記載される障害から選択される障害もしくは症状を処置するための製薬学的組成物;
本明細書に記載される障害から選択される障害もしくは症状を処置する方法、該方法は式(1)の化合物もしくはその製薬学的に許容しうる塩をそのような処置を必要とする哺乳類に投与することを含んでなる;
本明細書に記載される障害から選択される障害もしくは症状を処置するための製薬学的組成物、該組成物は式(1)の化合物もしくはその製薬学的に許容しうる塩および製薬学的に許容しうる担体を含んでなる;
本明細書に記載される障害から選択される障害もしくは症状を処置する方法、該方法は式(1)の化合物もしくはその製薬学的に許容しうる塩をそのような処置を必要とする患者に投与することを含んでなる;
式(1)の化合物の有効量をそれを必要とする患者に投与することを含んでなる5−HT6受容体に拮抗する方法
が包含されるがこれらに限定されるものではない。
【0017】
本発明はまた、薬剤の製造のための式(1)の化合物もしくは塩の使用も提供する。
【0018】
本発明はさらに、本発明の化合物もしくはその製薬学的に許容しうる塩、または本発明の化合物を含んでなる製薬学的組成物もしくは製剤が、記載される症状の1つもしくはそれ以上を処置するために、もう一つのもしくは複数の治療薬と同時にもしくは逐次もしくは組み合わせた製剤として投与される併用療法に関する。そのような他の治療薬(1つもしくは複数)は、本発明の化合物の投与の前に、それと同時に、もしくはその後に投与することができる。
【0019】
本発明はまた、本明細書に記載される障害から選択される障害もしくは症状を処置するための化合物、製薬学的組成物、キットおよび方法も提供し、該方法は式(1)の化合物もしくはその製薬学的に許容しうる塩をそのような処置を必要とする患者に投与することを含んでなる。
【0020】
本発明の化合物は、5−HT6受容体拮抗活性を保有する。本発明の化合物のこの活性は、例えば、本明細書に記述されるかもしくは当該技術分野において既知であるアッセイの1つもしくはそれ以上を用いて容易に示される。
【0021】
本発明はまた、本発明の化合物を製造する方法およびそれらの方法において使用される中間体も提供する。
【0022】
本明細書に記述される化合物および中間体の単離および精製は、所望に応じて、例えば濾過、抽出、結晶化、カラムクロマトグラフィー、薄層クロマトグラフィー、厚層クロマ
トグラフィー、分取低もしくは高圧液体クロマトグラフィー、またはこれらの方法の組み合わせのような任意の適当な分離もしくは精製方法によりもたらすことができる。適当な分離および単離方法の特定の実例は、製造および実施例から取ることができる。しかしながら、他の同等な分離もしくは単離方法もまたもちろん用いることができる。
【0023】
本発明の化合物は1個もしくはそれ以上の不斉中心を含有する可能性があり、従って、ラセミ化合物およびラセミ混合物、単一の鏡像異性体、ジアステレオマー混合物および個々のジアステレオマーとして存在することができる。
【0024】
様々な置換基の性質により、分子は追加の不斉中心を有し得る。各々のそのような不斉中心は、独立して2個の光学異性体をもたらす。混合物におけるそして純粋なもしくは部分的に精製された化合物としての、可能な光学異性体およびジアステレオマーの全ては本発明に属する。本発明は、これらの化合物の全てのそのような異性体を包含する。式(1)は、好ましい立体化学なしに化合物のクラスの構造を示す。これらのジアステレオマーの独立した合成もしくはそれらのクロマトグラフィー分離は、当該技術分野において既知であるようにそこに開示される方法論の適切な改変により成し遂げられることができる。それらの絶対立体化学は、必要に応じて、既知の絶対立体配置の不斉中心を含有する試薬で誘導体化される、結晶性生成物もしくは結晶性中間体のX線結晶学により決定されることができる。化合物のラセミ混合物は、ジアステレオマー混合物を生成せしめるための鏡像異性的に純粋な化合物への化合物のラセミ混合物のカップリング、続いて分別結晶もしくはクロマトグラフィーのような標準的な方法による個々のジアステレオマーの分離のような当該技術分野において周知である方法により個々の鏡像異性体に分離することができる。カップリングは、鏡像異性的に純粋な酸もしくは塩基、例えば(−)−ジ−p−トルオイル−D−酒石酸および/もしくは(+)−ジ−p−トルオイル−L−酒石酸を用いる塩の形成からなることが多い。次に、ジアステレオマー誘導体は、付加したキラル残基の切断により純粋な鏡像異性体に転化することができる。化合物のラセミ混合物はまた、キラル固定相を利用してクロマトグラフィー方法:当該技術分野において周知である方法により直接分離することもできる。あるいはまた、化合物の任意の鏡像異性体は、当該技術分野において周知である方法により既知の立体配置の光学的に純粋な出発物質もしくは試薬を用いて立体選択的合成により得ることができる。
【0025】
式(1)の化合物もしくはその製薬学的に許容しうる塩のシスおよびトランス異性体もまた本発明に属し、そしてこれはまた式(1)の化合物もしくはその製薬学的に許容しうる塩の互変異性体にも適用される。
【0026】
化合物の結晶形態のあるものは多形として存在する可能性があり:そのようなものとして本発明に属するものとする。さらに、化合物のあるものは水(すなわち、水和物)もしくは一般的な有機溶媒と溶媒和物を形成し得る。そのような溶媒和物もまた、本発明の範囲内に入る。
【0027】
PETもしくはSPECTにより検出可能であるように同位体標識された式(1)の化合物を包含する式(1)もしくはその製薬学的に許容しうる塩の同位体標識化合物もまた、本発明の範囲内に入る。同じことが、受容体結合もしくは代謝研究に適当な、[13C]−、[14C]−、[3H]−、[18F]−、[125I]−もしくは他の同位体濃縮原子で標識された式(1)の化合物に適用される。
【0028】
本発明の化合物はまた、神経学的機能、機能障害および疾患の生化学的研究における試薬もしくは基準として用いることもできる。
【0029】
[定義]
本明細書に開示される化合物の記述において使用する一般用語は、それらの通常の意味を有する。アルキルという用語は、本明細書において用いる場合、1価の飽和した分枝状もしくは直線状炭化水素鎖を意味する。他に記載されない限り、そのような鎖は1〜18個の炭素原子を含有することができる。そのようなアルキル基の代表は、メチル、エチル、プロピル、イソプロピル、ブチル、イソブチル、sec−ブチル、tert−ブチル、ペンチル、イソペンチル、ネオペンチル、tert−ペンチル、ヘキシル、イソヘキシル、ヘプチル、オクチル、ノニル、デシル、ウンデシル、ドデシル、トリデシル、テトラデシル、ペンタデシル、ヘキサデシル、ヘプタデシル、オクタデシルなどである。「低級」と限定される場合、アルキル基は1〜6個の炭素原子を含有する。同じ炭素含有量は、親用語「アルカン」に、そして「アルコキシ」のような派生用語に適用される。様々な炭化水素含有部分の炭素含有量は、該部分における炭素原子の最小および最大数を示す接頭辞により示され、すなわち、接頭辞Cx〜Cyは、包括的に整数「x」から整数「y」まで存在する炭素原子の数を定義する。例えば、「アルキル(C1〜3)」はメチル、エチル、n−プロピルもしくはイソプロピルを意味し、そして「アルキル(C1〜4)」は「メチル、エチル、n−プロピル、イソプロピル、n−ブチル、2−ブチル、イソブチルもしくは2−メチル−n−プロピル」を意味する。「アルケニル」という用語は、ビニル、アリル、ブテニルなどのような1個もしくはそれ以上の炭素−炭素二重結合を有する直線状もしくは分枝状炭化水素基を意味し、そして例えば(C2〜4)アルケニルを表す。「アルキニル」基において直線状もしくは分枝状炭化水素基は、エチニル、プロパルギル、1−ブチニル、2−ブチニルなどのような、1個もしくはそれ以上の炭素−炭素三重結合を有し、そして例えば(C2〜4)アルキニルを表す。他に記載されない限り、「アルケニル」および「アルキニル」鎖は1〜18個の炭素原子を含有することができる。「アシル」という用語は、アルキル(C1〜3)カルボニル、アリールカルボニルもしくはアリール−アルキル(C1〜3)カルボニルを意味する。
【0030】
「アリール」という用語は、フリル、チエニル、ピロリル、オキサゾリル、チアゾリル、イミダゾリル、イミダゾ[2,1−b][1,3]チアゾリル、ピラゾリル、イソオキサゾリル、イソチアゾリル、ピリジル、ピリダジニル、ピリミジニル、ピラジニル、1,3,5−トリアジニル、フェニル、インダゾリル、インドリル、インドリジニル、イソインドリル、ベンゾ[b]フラニル、1,2,3,4−テトラヒドロ−ナフチル、1,2,3,4−テトラヒドロイソキノリニル、インダニル、インデニル、ベンゾ[b]チエニル、2,3−ジヒドロ−1,4−ベンゾジオキシン−5−イル、ベンズイミダゾリル、ベンゾチアゾリル、ベンゾ[1,2,5]チア−ジアゾリル、プリニル、キノリニル、イソキノリニル、フタラジニル、キナゾリニル、キノキサリニル、1,8−ナフチリジニル、ナフチル、プテリジニルもしくはアズレニルが包含されるがこれらに限定されるものではない単環式もしくは縮合した二環式芳香族もしくはヘテロ芳香族基を包含する。「ハロ」もしくは「ハロゲン」はクロロ、フルオロ、ブロモもしくはヨードを意味し;「ヘテロアルキル、ヘテロ芳香族」などにおける場合に「ヘテロ」は、1個もしくはそれ以上のN、OもしくはS原子を含有することを意味する。「ヘテロアルキル」には任意の位置にヘテロ原子を有するアルキル基が包含され、従ってN−結合した、O−結合したもしくはS−結合したアルキル基が包含される。
【0031】
「置換された」という用語は、特定の基もしくは部分が1個もしくはそれ以上の置換基を有することを意味する。任意の基が多数の置換基を保有する可能性があり、そして様々な可能な置換基が提供される場合、これらの置換基は独立して選択され、そして同じものである必要はない。「非置換の」という用語は、特定の基が置換基を保有しないことを意味する。「場合により置換されていてもよい」は、基がC1〜8アルキル、C1〜8アルケニル、C1〜8アルキニル、アリール、フルオロ、クロロ、ブロモ、ヒドロキシル、C1〜8アルキルオキシ、C1〜8アルケニルオキシ、アリールオキシ、アシルオキシ、アミノ、C1〜8アルキルアミノ、ジアルキル(C1〜8)−アミノ、アリールアミノ、チ
オ、C1〜8アルキルチオ、アリールチオ、アルキルスルホニル、アリールスルホニル、アルキルスルフィニル、アリールスルフィニル、シアノ、オキソ、ニトロ、アシル、アミド、C1〜8アルキルアミド、ジアルキル(C1〜8)アミド、カルボキシルから選択される1個もしくはそれ以上の基でさらに置換されるかもしくはそうでない可能性があり、または2個の任意の置換基が、それらが結合している炭素原子と一緒になって、窒素、酸素もしくは硫黄から選択される0、1もしくは2個のヘテロ原子を含有する5もしくは6員の芳香もしくは非芳香環を形成することを意味する。任意の置換基は、それら自体が追加の任意の置換基を保有し得る。好ましい任意の置換基には、例えばメチル、エチルおよびトリフルオロメチルのようなC1〜3アルキル、フルオロ、クロロ、ブロモ、ヒドロキシル、例えばメトキシ、エトキシおよびトリフルオロメトキシのようなC1〜3アルキルオキシ、ならびにアミノが包含される。置換基に関連して、「独立して」という用語は、1個より多くのそのような置換基が可能である場合に、それらが同じものであるかもしくは相互に異なり得ることを意味する。
【0032】
「C3〜8シクロアルキル」は、シクロプロピル、シクロブチル、シクロペンチル、シクロヘキシル、シクロヘプチルもしくはシクロオクチルを意味し;「C5〜8ヘテロシクロアルキル」は、ピペリジニル、モルホリニル、アゼパニル、ピロリジニル、チオモルホリニル、ピペラジニル、テトラヒドロフリル、テトラヒドロピラニルが包含されるがこれらに限定されるものではないヘテロ原子含有環をさし;「C5〜10ビシクロアルキル基」は、ビシクロ[2.2.1]ヘプタニル、ビシクロ[3.3.0]オクタニルもしくはビシクロ[3.1.1]ヘプタニル基が包含されるがこれらに限定されるものではない炭素二環式環系をさし;「C6〜10トリシクロアルキル基」は、1−アダマンチル、ノルアダマンチルもしくは2−アダマンチル基が包含されるがこれらに限定されるものではない炭素三環式環系をさす。「C8〜11テトラシクロアルキル基」という略語は、キュビル、ホモキュビルもしくはビスホモキュビル基が包含されるがこれらに限定されるものではない炭素四環式環系をさす。
【0033】
「オキシ」、「チオ」および「カルボ」という用語は、別の基の一部として本明細書において用いる場合、それぞれ、例えばヒドロキシル、オキシアルキル、チオアルキル、カルボキシアルキルなどのような2つの基の間のリンカーとして働く、酸素原子、硫黄原子およびカルボニル(C=O)基をさす。「アミノ」という用語は、単独でもしくは別の基の一部として本明細書において用いる場合、末端であるかもしくは2つの他の基の間のリンカーであり得る窒素原子をさし、ここで、該基は第一級、第二級もしくは第三級(それぞれ、窒素原子に結合している2個の水素原子、窒素原子に結合している1個の水素原子、そして窒素原子に結合している水素原子がない)アミンであることができる。「スルフィニル」および「スルホニル」という用語は、別の基の一部として本明細書において用いる場合、それぞれ−SO−もしくは−SO2−基をさす。
【0034】
本明細書において用いる場合、「脱離基」(L)という用語は、置換(substitution)もしくは置換(displacement)反応中に離脱する荷電もしくは非荷電原子もしくは基を意味するものとする。該用語は、アミン、チオールもしくはアルコール求核試薬のような求核試薬により容易に置換可能な基をさす。そのような脱離基は、当該技術分野において周知である。例には、N−ヒドロキシスクシンイミド、N−ヒドロキシベンゾトリアゾール、ハロゲン化物(Br、Cl、I)、トリフラート、メシレート、トシレートなどが包含されるがこれらに限定されるものではない。
【0035】
上記の化合物のN−オキシドは、本発明に属する。第三級アミンは、N−オキシド代謝物を生じるかもしくはそうでない可能性がある。N−酸化が起こる程度は、微量からほぼ定量的転化まで様々である。N−酸化物は、それらの対応する第三級アミンより活性であるか、もしくは活性が低い可能性がある。N−酸化物は化学的手段によりそれらの対応す
る第三級アミンに容易に還元することができるが、人体においてこれは様々な程度に起こる。あるN−酸化物は、対応する第三級アミンへのほぼ定量的な還元的転化を受け、ある場合には転化はほんの微量の反応であるか、もしくは完全にないことさえある(Bickel,1969)。
【0036】
生物活性剤(すなわち、式(1)の化合物)を与えるようにインビボで代謝される任意の化合物は、本願の範囲および精神の範囲内のプロドラッグである。プロドラッグは、それ自体不活性であるが1種もしくはそれ以上の活性代謝物に転化される治療薬である。従って、本発明の処置の方法において、「投与すること」という用語は特に開示される化合物で、もしくは特に開示されないが患者への投与後にインビボで特定の化合物に転化する化合物で記述される様々な障害を処置することを包含するものとする。プロドラッグは、親薬剤分子の有用性へのいくつかの障害を克服するために用いられる薬剤分子の生可逆的(bioreversible)誘導体である。これらの障害には、溶解性、透過性、安定性、前全身的(presystemic)代謝およびターゲッティング制限が包含されるがこれらに限定されるものではない(Bundgaard,1985;King,1994;Stella,2004;Ettmayer,2004;Jaervinen,2005)。プロドラッグ、すなわち、任意の既知の経路によりヒトに投与した場合に式(1)を有する化合物に代謝される化合物は、本発明に属する。特に、これは第一級もしくは第二級アミノまたはヒドロキシ基を有する化合物に関する。そのような化合物は、有機酸と反応させて例えばアミジン、エナミン、マンニッヒ塩基、ヒドロキシル−メチレン誘導体、O−(アシルオキシメチレンカルバメート)誘導体、カルバメート、エステル、アミドもしくはエナミノン、しかしこれらに限定されるものではない、投与後に容易に取り除かれる付加的基が存在する式(1)を有する化合物を生成せしめることができる。
【0037】
「結晶形態」は、同じ化合物の様々な固体形態、例えば多形、溶媒和物および非晶質形態をさす。「多形」は、化合物が異なる結晶充填配置において結晶化することができる結晶構造であり、これらは全て同じ元素組成を有する。多形は、温度、過飽和のレベル、不純物の存在、溶媒の極性、冷却の速度のようないくつかの結晶化条件により影響される、高い頻度で起こる現象である。異なる多形は通常は異なるX線回折パターン、固体状態NMRスペクトル、赤外もしくはラマンスペクトル、融点、密度、硬度、結晶形、光学および電気特性、安定性ならびに溶解性を有する。再結晶溶媒、結晶化の速度、貯蔵温度および他の因子は、1つの結晶形態を顕著にさせ得る。「溶媒和物」は一般に溶媒の化学量論もしくは非化学量論量のいずれかを含有する結晶形態である。結晶化の過程中に、ある化合物は結晶性固体状態において溶媒分子の固定モル比を捕捉する傾向を有することが多く、このようにして溶媒和物を形成する。溶媒が水である場合、「水和物」が形成され得る。式(1)の化合物およびその製薬学的に許容しうる塩は、水和物もしくは溶媒和物の形態で存在することができ、そしてそのような水和物および溶媒和物もまた本発明に包含される。その例には、1/10水和物、1/4水和物、1/2水和物、1水和物、2塩酸塩1/2水和物、2塩酸塩2水和物、2塩酸塩3/2水和物などが包含される。「非晶質」形態は長距離秩序を有さない非結晶性物質であり、そして一般に特有の粉末X線回折パターンを与えない。結晶形態は一般に、Byrn(1995)およびMartin(1995)により記述されている。
【0038】
より簡潔な記述を提供するために、本明細書に示される量的表現のいくつかは「約」という用語で修飾されない。「約」という用語が明白に使用されようとなかろうと、本明細書に示されるあらゆる量は実際の既定値をさすものとし、そしてそれはまたそのような既定値の実験および/もしくは測定条件に起因する近似を包含する当該技術分野における通常の技量に基づいて合理的に推測されるそのような既定値への近似もさすものとすることが理解される。
【0039】
「選択的」および「選択性」という用語は、別の受容体(例えば、他の5−HT受容体サブタイプ)に対する実質的な交差反応性を示すことなしに特定の受容体(例えば5−HT6受容体)に対する反応性を示す化合物をさす。従って、例えば、本発明の選択的化合物は、他の5−HT受容体に対する実質的な交差反応性を示すことなしに5−HT6受容体に対する反応性を示すことができる。1つの態様において、本発明の化合物は5−HT6受容体に対する少なくとも約10倍の選択性、5−HT6受容体に対する少なくとも約50倍の選択性、5−HT6受容体に対する少なくとも約100倍の選択性、5−HT6受容体に対する少なくとも約250倍の選択性もしくは所望の標的に対する少なくとも約500倍の選択性を有する。
【0040】
本明細書の記述および請求項の全体にわたって、「含んでなる」という語および「含んでなること」および「含んでなる(comprises)」のような該語のバリエーションは、他の添加物、成分、整数もしくは段階を除くものではない。
【0041】
式(1)の化合物はそのままの(raw)化学物質として投与することが可能であり得るが、それらを「製薬学的組成物」として与えることが好ましい。さらなる態様に従って、本発明はその1つもしくはそれ以上の製薬学的に許容しうる担体および場合により1つもしくはそれ以上の他の治療成分と一緒に、式(1)の化合物またはその製薬学的に許容しうる塩もしくは溶媒和物を含んでなる製薬学的組成物を提供する。担体(1つもしくは複数)は、製剤の他の成分と適合しそしてそのレシピエントに有害でないという意味で「許容可能」でなければならない。
【0042】
「組成物」という用語は、本明細書において用いる場合、既定の量もしくは割合の特定の成分を含んでなる生成物ならびに特定量の特定の成分を合わせることから直接もしくは間接的に生じる任意の生成物を包含する。製薬学的組成物に関連して、この用語は1種もしくはそれ以上の有効成分、および不活性成分を含んでなる任意の担体を含んでなる生成物、ならびに任意の2種もしくはそれ以上の成分の組み合わせ、錯体形成もしくは凝集から、または1種もしくはそれ以上の成分の解離から、または1種もしくはそれ以上の成分の他のタイプの反応もしくは相互作用から直接もしくは間接的に生じる任意の生成物を包含する。一般に、製薬学的組成物は、有効成分を液状担体もしくは微粉化固形担体もしくは両方と均一にそして密接に会合させ、そして次に必要に応じて生成物を所望の製剤に成形することにより製造される。製薬学的組成物は、疾患の進行もしくは症状の際に所望の効果をもたらすために十分な活性目的化合物を含む。従って、本発明の製薬学的組成物は、本発明の化合物および製薬学的に許容しうる担体を混合することにより製造される任意の組成物を包含する。「製薬学的に許容しうる」は、担体、希釈剤もしくは賦形剤が製剤の他の成分と適合しそしてそのレシピエントに有害でない必要があることを意味する。
【0043】
本願の文脈内で、「組み合わせ製剤」という用語は、錠剤もしくは注入液のような1つの製剤において物理的に組み合わされた式(1)の化合物および1種もしくはそれ以上の他の薬剤を意味する真の組み合わせ、ならびに場合により成分化合物の投与の順守を容易にするためのさらなる手段、例えばラベルもしくは図面を有して、使用説明書と一緒に、別個の投与形態物において式(1)の化合物および1種もしくはそれ以上の他の薬剤を含んでなる「キットオブパーツ(kit−of−parts)」の両方を含んでなる。真の組み合わせで、定義による薬物療法は同時である。「キットオブパーツ」の中身は、同時にもしくは異なる時間間隔で投与することができる。同時もしくは逐次のいずれかである治療は、使用する他の薬剤の特徴、作用の発現および期間、血漿レベル、クリアランスなどのような特徴、ならびに疾患、その段階および個々の患者の特徴により決まる。
【0044】
5−HT6受容体に対する本発明の化合物の親和性は、上記のように決定された。式(1)の既定化合物について測定される結合親和性から、理論的最低有効用量を概算するこ
とができる。測定されるKi値の2倍に等しい化合物の濃度で、5−HT6受容体のほぼ100%が化合物により占められると思われる。患者のkg当たりの化合物のmgにその濃度を転化することにより、理想的な生物学的利用能を仮定して理論的最低有効用量がもたらされる。薬物動態、薬力学および他の考慮事項は、実際に投与される用量をより高いかもしくはより低い値に改変し得る。有効成分の典型的な毎日用量は広い範囲内で異なり、そして関連する適応症、投与の経路、患者の年齢、体重および性別のような様々な因子により決まり、そして医師により決定され得る。一般に、単一もしくは個々の用量における患者への総毎日用量投与は、例えば、毎日0.001〜10mg/kg体重そしてさらに通常は1日当たり0.01〜1,000mgの総有効成分の量であることができる。そのような投薬量は毎日1〜3回、もしくは効能に必要とされる限り何度も、そして少なくとも2ヶ月の期間にわたって、より典型的には少なくとも6ヶ月間、もしくは長期的に処置を必要とする患者に投与される。
【0045】
「治療的に有効な量」という用語は、本明細書において用いる場合、本発明の組成物を投与することにより処置できる症状を処置するための治療薬の量をさす。その量は、組織系、動物もしくはヒトにおいて検出可能な治療もしくは改善反応を示すために十分な量である。効果には、例えば本明細書に記載される症状を処置することを包含することができる。患者の正確な有効量は、患者のサイズおよび健康、処置する症状の性質および程度、処置する医師(研究者、獣医、医師もしくは他の臨床医)の推奨、ならびに投与のために選択される治療法もしくは治療法の組み合わせにより決まる。従って、前もって正確な有効量を特定することは有用でない。
【0046】
「製薬学的に許容しうる塩」という用語は、適切な医学的判断の範囲内で、過度の毒性、刺激、アレルギー反応などなしにヒトおよび下等動物の組織と接触して使用するのに適当でありそして妥当な利益/危険比に相応する塩をさす。製薬学的に許容しうる塩は、当該技術分野において周知である。それらは、本発明の化合物を最終的に単離しそして精製する場合にインサイチューで、または無機もしくは有機塩基および無機もしくは有機酸を包含する製薬学的に許容しうる無毒の塩基もしくは酸とそれらを反応させることにより別個に製造することができる(Berge,1977)。「遊離塩基」形態は、塩を塩基もしくは酸と接触させ、そして常法において親化合物を単離することにより再生することができる。化合物の親形態は、極性溶媒における溶解性のようなある種の物理的性質において様々な塩形態と異なるが、そのほかの点ではこれらの塩は本発明の目的のために化合物の親形態と同等である。
【0047】
「錯体」は、金属イオンと錯体形成した本発明、例えば式(1)の化合物の錯体をさし、ここで、少なくとも1個の金属イオンはキレートされるかもしくは封鎖される。錯体は、当該技術分野において周知である方法により製造される(Dwyer,1964)。
【0048】
「処置」という用語は、本明細書において用いる場合、哺乳類、例えばヒト症状もしくは疾患の任意の処置をさし、そして:(1)疾患もしくは症状を抑制すること、すなわち、その発症を止めること、(2)疾患もしくは症状を軽減すること、すなわち、症状を退行させること、または(3)疾患の症候を止めることが包含される。
【0049】
「抑制する」という用語には、進行、重症度もしくは結果として起こる症候を妨げること、防ぐこと、抑えること、軽減すこと、改善することおよび遅くすること、止めることもしくは回復させることを包含するその一般に認められている意味が包含される。そのようなものとして、本発明の方法には、必要に応じて、医学的治療および/もしくは予防投与の両方が包含される。
【0050】
本明細書において用いる場合、「医学的治療」という用語には、ヒトもしくは他の哺乳
類にインビボもしくはエクスビボで実施される予防、診断および治療処方計画が包含されるものとする。
【0051】
哺乳類には、ウシ、ヒツジおよびブタ動物のような経済的に重要な動物、特に肉を生産するもの、ならびに家畜、運動競技動物(sports animals)、動物園の動物およびヒトが包含され、後者が好ましい。「患者」という用語は、本明細書において用いる場合、処置、観察もしくは実験の対象である動物、好ましくは哺乳類、最も好ましくはヒトをさす。
【0052】
本明細書において用いる場合、「体重障害」という用語は、異常な(例えば過剰)体重をもたらす、エネルギー摂取とエネルギー消費との間の不均衡により引き起こされる障害をさす。そのような体重障害には肥満が包含される(Roth,1994;Sibley,1993;Sleigh,1995,1997)。「肥満」は、ヒトが少なくとも25.9の、身長の二乗当たりの体重(kg/m2)として計算される、ボディ・マス・インデックス(BMI)を有する症状をさす。通常、正常体重を有するヒトは、19.9〜25.9未満のBMIを有する。本明細書における肥満は、遺伝的であろうと環境的であろうと、任意の原因に起因し得る。肥満をもたらすかもしくは肥満の原因であり得る障害の例には、過食および過食症、多嚢胞卵巣疾患、頭蓋咽頭腫、プラダー・ウィリ症候群、フレーリッヒ症候群、II型糖尿病、GH欠損患者、正常バリアント低身長(normal
variant short stature)、ターナー症候群ならびに低下した代謝活性もしくは総除脂肪量の割合としての安静時エネルギー消費量の減少を示す他の病状、例えば急性リンパ芽球性白血病を有する子供が包含される。
【0053】
略語
ACE−クロリド クロロギ酸1−クロロエチル
ACN アセトニトリル
AcOH 酢酸
ADD 注意欠陥障害
ADHD 注意欠陥多動性障害
API 大気圧イオン化
BMI ボディ・マス・インデックス
n−BuOH n−ブタノール
t−BuOH t−ブタノール
(t)−BOC (第三級)−ブトキシカルボニル
CHO チャイニーズハムスター卵巣(細胞)
CNS 中枢神経系
CUR カーテンガス
DBU 1,8−ジアザビシクロ[5.4.0]ウンデク−7−エン
(1,2)−DCE (1,2)−ジクロロエタン
DCM ジクロロメタン
DF 偏向器電圧
DIPEA N,N−ジイソプロピルエチルアミン
DMAP 4−ジメチルアミノピリジン
DMC 2−クロロ−1,3−ジメチルイミダゾリニウムクロリド
DMF N,N’−ジメチルホルムアミド
DMSO ジメチルスルホキシド
EA 酢酸エチル
EP 入口電位(entrance potential)
EtOAc 酢酸エチル
EtOH エタノール
Et2O ジエチルエーテル
FCS ウシ胎仔血清
FP 集束電位(focusing potential)
g グラム
h 時間
5−HT 5−ヒドロキシトリプタミン、セロトニン
KOtBu カリウムtert−ブトキシド
MeI ヨウ化メチル
MeOH メタノール
mg ミリグラム
min 分
mlもしくはmL ミリリットル
m.p. 融点、c.q.融解範囲
MsCl メタンスルホニルクロリド(塩化メシル)
MTBE メチルtert−ブチルエーテル
NaHMDS ナトリウムヘキサメチルジシラザン
NEB ネブライザーガス
PA 石油エーテル(40〜60)
p−TsOH パラトルエンスルホン酸
Rf 保持係数(薄層クロマトグラフィー)
Rt 保持時間(LC/MS)
RT 室温
SCX 強陽イオン交換
TBAB 臭化テトラブチルアンモニウム
TEA トリエチルアミン
TEM 温度
TFA トリフルオロ酢酸
THF テトラヒドロフラン
【実施例1】
【0054】
分析方法
核磁気共鳴スペクトル(1H NMR)は、他に示されない限り、300KでBruker ARX400(1H:400MHz)もしくはVarian VXR200(1H:200MHz)装置を用いて示される溶媒において決定された。スペクトルは、Cambridge Isotope Laboratories Ltdから入手した重水素化クロロホルムもしくはDMSOにおいて決定された。化学シフト(δ)は、テトラメチルシラン(1H)からppmダウンフィールド単位(in ppm downfield)で示される。カップリング定数Jは、Hz単位で示される。NMRスペクトルにおけるピーク形状は、記号「q」(カルテット)、「dq」(ダブルカルテット)、「t」(トリプレット)、「dt」(ダブルトリプレット)、「d」(ダブレット)、「dd」(ダブルダブレット)、「ddd」(ダブルダブルダブレット)、「s」(シングレット)、「bs」(ブロードなシングレット)および「m」(マルチプレット)で示される。NHおよびOHシグナルは、サンプルを1滴のD2Oと混合した後に同定された。
【0055】
フラッシュクロマトグラフィーは、示される溶離剤およびシリカゲル(Merckシリカゲル60:0.040〜0.063mm)を用いる精製をさす。融点は、Buechi B−545融点装置上で記録された。湿気および/もしくは酸素に敏感な化合物を含む全ての反応は、無水窒素雰囲気下で実施された。反応は、示される溶離剤でシリカ被覆ガラスプレート(Merckプレコートシリカゲル60 F254)上で薄層クロマトグラフィー(TLC)を用いることによりモニターされた。スポットは、UV光(254nm)もしくはI2により視覚化された。
【0056】
液体クロマトグラフィー−質量分析(LC−MS):LC−MS系は、2台のPerkin Elmerシリーズ200マイクロポンプからなった。これらのポンプは、Gilson 215オートサンプラーに連結された、50μlのT字型ミキサーにより相互に連結された。方法は下記のとおりであった:
段階 総時間 流量(μl/分) A(%) B(%)
0 0 2000 95 5
1 1.8 2000 0 100
2 2.5 2000 0 100
3 2.7 2000 95 5
4 3.0 2000 95 5
A=0.025%HCOOHおよび10mmol NH4HCOO pH=±3を有する100%水
B=0.025%HCOOHを有する100%ACN
【0057】
オートサンプラーは2μlの注入ループを有し、そして3μm粒子を有するWaters Atlantis C18 30*4.6mmカラムに連結された。カラムは、40℃でPerkin Elmerシリーズ200カラムオーブンにおいてサーモスタットで調節された。カラムは、2.7μlフローセルを有するPerkin Elmerシリーズ200UV計に連結された。波長は254nmに設定された。UV計は、Sciex API 150EX質量分析計に連結された。質量分析計は以下のパラメーターを有した:
走査範囲:150〜900a.m.u.;極性:陽性;走査形態:プロフィール;分解能
Q1:UNIT;刻み幅:0.10a.m.u.;走査当たりの時間:0.500sec;NEB:10;CUR:10 IS:5200;TEM:325;DF:30;FP:225およびEP:10。光散乱検出器は、Sciex API 150に連結された。光散乱検出器は、50℃および3barのN2で作動するSedere Sedex 55であった。完全な系は、G3パワーマックにより制御された。
【実施例2】
【0058】
合成の一般的態様
ピラゾリン部分を含有する請求化合物および中間体の適当な合成は、市販されているかもしくは下記のとおり製造される4,5−ジヒドロ−1H−ピラゾールもしくは4,5−ジヒドロ−3H−ピラゾールビルディングブロックを用いて、WO 01/70700に以前に開示されたものと同様の経路をたどる。
【0059】
【化2】
【0060】
経路1は、例えば塩基の存在下でクロロギ酸メチルもしくはジ−tert−ブチルジカーボネートとスルホンアミドとの反応により製造することができる、一般式(I)のカルバミン酸スルホニルを用いる。次に、一般式(II)のピラゾリンとそれらの反応生成物をPCl3、POCl3/DMAPもしくは2−クロロ−1,3−ジメチルイミダゾリニウムクロリド(DMC)のようなハロゲン化剤を用いて一般式(III)のクロロイミン中間体に転化し、続いてアミンと反応させて一般式(IV)のスルホニルピラゾリンカルボキサミジン誘導体を得ることができる。
【0061】
【化3】
【0062】
経路2は、KOHの存在下でCS2との反応、続いてヨウ化メチルのようなハロゲン化アルキルとの反応によりスルホンアミドから製造することができる、一般式(V)のN−(ビス−アルキルスルファニル−メチレン)−スルホンアミド構造を用いる。次に、2個のS−アルキル官能基は、好ましくは一般式(VI)の構造を得るためにピラゾリンビルディングブロックから出発して、アミンで置換して、一般式(IV)のスルホニルピラゾリンカルボキサミジン誘導体で終わることができる。
【0063】
【化4】
【0064】
経路3はヨウ化メチルのようなハロゲン化アルキルとチオ尿素ビルディングブロックとの反応により都合よく製造される一般式(IX)のアルキル−イソチオ尿素フラグメントもしくはその適当な塩形態を用い、それを塩基の存在下でピラゾリンと反応させて一般式(X)のピラゾリンカルボキサミジン誘導体を得ることができる。後者を塩基の存在下でハロゲン化スルホニル(X=Br、Cl、F、好ましくはCl)と反応させて一般式(IV)のスルホニルピラゾリンカルボキサミジン誘導体を得ることができる。
【0065】
特定の合成方法の選択は、使用する試薬と官能基との適合性、保護基、触媒、活性化およびカップリング試薬を使用する可能性ならびに製造される最終化合物に存在する最終的な構造的特徴のような当業者に既知である因子により決まる。
【0066】
製薬学的に許容しうる塩は、例えば本発明の化合物を適当な酸、例えば無機酸もしくは有機酸と混合することにより、当該技術分野において周知である標準的な方法を用いて得ることができる。
【実施例3】
【0067】
ピラゾリン中間体の合成
【0068】
【化5】
【0069】
3−エチル−4,5−ジヒドロ−1H−ピラゾール
ヒドラジン水和物(24.55mL)をMeOH(50mL)に溶解し、そして氷浴において冷却した。この溶液に、温度が10℃未満に保たれるような速度でエチルビニルケトン(50mL)を加えた。氷浴を取り除き、そして混合物を室温で2時間攪拌し、その後でMeOHを減圧下で蒸発させた。生成物は真空蒸留(70℃、20mbar)により得られ、7.22gの無色の液体を生成せしめた。1H NMR(400MHz,CDCl3)δ1.15(t,J=8Hz,3H),2.34(q,J=8Hz,2H),2.59(t,J=10Hz,2H),3.10(br s,1H),3,34(t,J=10Hz,2H)。
【0070】
【化6】
【0071】
3−メチル−4,5−ジヒドロ−1H−ピラゾール
ヒドラジン水和物(29.2mL)をMeOH(50mL)に溶解した。この溶液に、温度が50℃未満に保たれるような速度でメチルビニルケトン(50mL)を加えた。混合物を50℃で2時間攪拌し、その後でMeOHを減圧下で蒸発させた。生成物は真空蒸留(68〜82℃、20mbar)により得られ、11.8gの無色の液体を生成せしめた。1H NMR(400MHz,DMSO−d6)δ1.88(s,3H),2.47(t,J=10Hz,2H),3.15(t,J=10Hz,2H),6.10(br s,1H)。
【0072】
【化7】
【0073】
4−エチル−4,5−ジヒドロ−1H−ピラゾール
ヒドラジン水和物(58mL)をMeOH(300mL)に溶解し、そして氷浴において冷却した。この混合物に、温度が10℃未満に保たれるような速度でMeOH(100mL)中の2−エチルアクロレイン(100g)の溶液を加えた。氷浴を取り除き、そして混合物を室温で一晩攪拌し、その後でMeOHを減圧下で蒸発させた。生成物が真空蒸留(70〜80℃、20mbar)により得られ、54.9gの無色の液体を生成せしめた。1H NMR(400MHz,CDCl3)δ0.98(t,J=8Hz,3H),1.42〜1.70(m,2H),2.89〜3.02(m,2H),3.43〜3.54(m,1H),6.78(br s,1H),NH 不可視。
【0074】
【化8】
【0075】
4−メチル−4,5−ジヒドロ−1H−ピラゾール
ヒドラジン水和物(16.65mL)をCH3CN(50mL)に溶解し、そして氷浴において冷却した。この混合物に、CH3CN(50mL)中の2−メチルアクロレイン(24.02g)の溶液を加えた。氷浴を取り除き、そして混合物を室温で一晩攪拌し、その後でCH3CNを減圧下で蒸発させた。生成物が真空蒸留(102〜108℃、250mbar)により得られ、7.0gの無色の液体を生成せしめた。1H NMR(400MHz,CDCl3)δ1.18(d,J=7Hz,3H),2.90(t,J=9H
z,1H),3.00〜3.12(m,1H),3.51(t,J=9Hz,1H),5.48(br s,1H),6.73(br s,1H)。
【0076】
【化9】
【0077】
5−エチル−4,5−ジヒドロ−1H−ピラゾール
ヒドラジン水和物(12.1mL)をMeOH(50mL)に溶解し、そして氷浴において冷却した。この混合物に、温度が10℃未満に保たれるような速度でMeOH(50mL)中の2−ペンテナール(24.4mL)の溶液を加えた。氷浴を取り除き、そして混合物を室温で2.5時間攪拌し、続いて減圧下で蒸発させた。生成物が真空蒸留(68〜72℃、25mbar)により得られ、8.25gの無色の液体を生成せしめた。1H NMR(400MHz,CDCl3)δ0.92(d,J=7.5Hz,3H),1.42〜1.61(m,2H),2.36(ddd,J=17,8および2Hz,1H),2.76(ddd,J=17,10および2Hz,1H),3.51〜3.62(m,1H),5.35(br s,1H),6.76(br s,1H)。
【0078】
【化10】
【0079】
4,4−ジメチル−4,5−ジヒドロ−3H−ピラゾール
2,2−ジメチル−1,3−プロパンジアミン(20.0g)をH2O(80mL)およびMeOH(20mL)に溶解し、そして氷浴において冷却した。同時に、H2O2(30%、120mL)およびNaClO(10%、350mL)を滴下して加えた。反応混合物を室温で一晩攪拌し、DCMで抽出し、有機層をNa2SO4上で乾燥させ、そして溶媒を減圧下で蒸発させた。真空蒸留(102〜105℃、250mbar)により11.4gの無色の非晶質油性化合物を生成せしめた。1H NMR(200MHz,CDCl3)δ1.05(s,6H),4.13(s,4H)。
【0080】
あるいはまた、この化合物は下記のとおり合成された:
【0081】
【化11】
【0082】
4,4−ジメチル−4,5−ジヒドロ−3H−ピラゾール
2,2−ジメチル−1,3−プロパンジアミン(8.97g)をH2O(45mL)に溶解し、そして氷浴において冷却した。同時に、温度を25℃未満に保ちながら、H2O2(30%、54mL)およびNaClO(10%、157mL)を滴下して加えた。次に、反応混合物を室温で1時間攪拌し、そしてDCM(2x45mL)で抽出した。合わせた有機層を水性亜硫酸ナトリウム(20%、25mL)で抽出し、水(2x25mL)で洗浄し、Na2SO4上で乾燥させ、そして減圧下で蒸発させて(50℃で>200mbar)8.89gの無色の液体(いくらかの残留DCMを含有する)を生成せしめた。1H NMR(200MHz,CDCl3)δ1.05(s,6H),4.13(s,4H)。
【0083】
【化12】
【0084】
2,2−ジエチル−マロノニトリル
マロノニトリル(15.2g)をTBAB(3.0g、4mol%)およびヨウ化エチル(36.8mL、2当量)と混合した。室温で30分間攪拌した後に、混合物を氷浴において冷却し、KOtBu(51.6g、2当量)を少しずつ加え、氷浴を取り除き、そして混合物を室温で30分間攪拌した。DCM/H2Oで抽出し、Na2SO4上で乾燥させ、そして減圧下で蒸発させて40グラムの粗物質を生成せしめ、それをDCMで溶出するフラッシュクロマトグラフィーにより精製した。これにより20.4グラムの橙色の油を生成せしめ、それは静置すると凝固した。1H NMR(400MHz,CDCl3)δ1.29(t,J=7.5Hz,6H),2.00(q,J=7.5Hz,4H)。
【0085】
2,2−ジエチル−プロパン−1,3−ジアミン
乾式Et2O(100mL)中のLiAlH4(4.66g)の懸濁液を氷浴において冷却し、そして温度が20℃未満に保たれるような速度でEt2O(50mL)中の2,2−ジエチル−マロノニトリル(5.0g)の溶液を滴下して加えた。混合物を室温で一晩攪拌し、氷浴において冷却し、そしてH2O(5mL)、2M水性NaOH(10mL)そして再びH2O(5mL)を加えることによりクエンチした。懸濁液を濾過し、フィルターケーキをEt2Oで洗浄し、そして合わせた濾液を減圧下で蒸発乾固させて5.0gの透明な薄黄色の液体を生成せしめた。1H NMR(400MHz,CDCl3)δ0.80(t,J=8Hz,6H),1.08(br s,4H),1.22(q,J=8Hz,4H),2.52(s,4H)。
【0086】
4,4−ジエチル−4,5−ジヒドロ−3H−ピラゾール
2,2−ジエチル−プロパン−1,3−ジアミン(5.0g)をH2O(40mL)およびMeOH(10mL)の混合物に溶解し、そして氷浴において冷却した。同時に、H2O2(24.2mLの30%溶液、6当量)およびNaClO(54.9mLの10%溶液、2.4当量)を滴下して加え、氷浴を取り除き、そして混合物を室温で2時間攪拌した。DCMで抽出し、Na2SO4上で乾燥させ、そして減圧下で蒸発させて77%の予想生成物および23%のジアミン出発物質を含有する3.51gの透明な黄色の液体を生成せしめた。この物質をさらに精製せずに次の段階において使用した。1H NMR(400MHz,CDCl3)δ0.78(t,J=7.5Hz,6H),1.36(q,J=7.5Hz,4H),4.14(s,4H)。
【0087】
【化13】
【0088】
シクロペンタン−1,1−ジカルボニトリル
マロノニトリル(15.0g)を乾式DMF(200mL)に溶解し、そして氷浴において冷却した。次に、DBU(75mL、2.2当量)および1,4−ジブロモブタン(29.6mL、1.1当量)を滴下して加えた。氷浴を取り除き、追加の100mLの乾式DMFを加え、そして混合物を80℃で2時間攪拌した。周囲温度まで冷却した後に、DCMを加え、そして混合物を5%水性NaHCO3で5回洗浄した。有機相をNa2SO4上で乾燥させ、そして減圧下で蒸発させて40gの黒色の油性物質を生成せしめた。これをPA:EA 9:1(Rf=0.35、I2で視覚化される)で溶出するフラッシュクロマトグラフィーにより精製して23.4gの無色の液体を生成せしめた。1H NMR(400MHz,CDCl3)δ1.94〜2.03(m,4H),2.41(t,J=7Hz,4H)。
【0089】
C−(1−アミノメチル−シクロペンチル)−メチルアミン
乾式Et2O(100mL)中のLiAlH4(4.74g)の懸濁液を氷浴において冷却し、そして温度が20℃未満に保たれるような速度でEt2O(50mL)中のシクロペンタン−1,1−ジカルボニトリル(5.0g)の溶液を滴下して加えた。混合物を室温で一晩攪拌し、氷浴において冷却し、そしてH2O(5mL)、2M水性NaOH(10mL)そして再びH2O(5mL)を加えることによりクエンチした。懸濁液を濾過し、フィルターケーキをEt2Oで洗浄し、そして合わせた濾液を減圧下で蒸発乾固させて4.95gの透明な無色の液体を生成せしめた。1H NMR(400MHz,CDCl3)δ1.24(br s,4H),1.22〜1.40(m,4H),1.55〜1.64(m,4H),2.62(s,4H)。
【0090】
2,3−ジアザ−スピロ[4,4]ノン−2−エン
C−(1−アミノメチル−シクロペンチル)−メチルアミン(4.87g)をH2O(40mL)およびMeOH(10mL)の混合物に溶解し、そして氷浴において冷却した。同時に、H2O2(23.9mLの30%溶液、6当量)およびNaClO(54.3mLの10%溶液、2.4当量)を滴下して加え、氷浴を取り除き、そして混合物を室温で2時間攪拌した。DCMで抽出し、Na2SO4上で乾燥させ、そして減圧下で蒸発させて90%の予想生成物および10%のジアミン出発物質を含有する3.74gの透明な薄黄色の液体を生成せしめた。この物質をさらに精製せずに次の段階において使用した。1H NMR(400MHz,CDCl3)δ1.48〜1.57(m,4H),1.62〜1.69(m,4H),4.26(s,4H)。
【0091】
【化14】
【0092】
シクロヘキサン−1,1−ジカルボニトリル
マロノニトリル(15.0g)を乾式DMF(200mL)に溶解した。次に、DBU(75mL)および1,5−ジブロモペンタン(34mL)を0℃(氷浴)で加えた。氷浴を取り除き、そして反応物を80℃で2時間攪拌した。冷却した後に、反応物をDCMに注ぎ込んだ。有機層を5%NaHCO3で数回洗浄し、有機層をNa2SO4上で乾燥させ、そして溶媒を減圧下で蒸発させた。粗生成物をPA:EtOAc(9:1)で溶出するフラッシュクロマトグラフィーにより精製して25.7gの白色の結晶を生成せしめた。1H NMR(400MHz,CDCl3)δ1.48〜1.61(m,2H),1.68〜1.84(m,4H),2.13(t,J=6Hz,4H)。
【0093】
C−(1−アミノメチル−シクロヘキシル)−メチルアミン
シクロヘキサン−1,1−ジカルボニトリル(20.0g)を乾式Et2O(70mL)に溶解した。氷浴において冷却した乾式Et2O(250mL)中のLiAlH4(17.0g)の懸濁液にこの混合物を滴下して加えた。混合物を室温で一晩攪拌し、氷浴において冷却し、そしてH2O(17.0mL)、2M水性NaOH(34.0mL)そして再びH2O(17mL)を加えることによりクエンチした。懸濁液を濾過し、フィルターケーキをEt2Oで洗浄し、そして合わせた濾液を蒸発乾固させて20.8gの透明な無色の液体を生成せしめた。1H NMR(400MHz,CDCl3)δ1.05〜1.55(m,14H),2.61(s,4H)。
【0094】
2,3−ジアザ−スピロ[4,5]デク−2−エン
C−(1−アミノメチル−シクロヘキシル)−メチルアミン(10.0g)をH2O(40mL)およびMeOH(10mL)の混合物に溶解し、そして氷浴において冷却した。同時に、H2O2(44.3mLの30%溶液、6当量)およびNaClO(125.5mLの10%溶液、2.4当量)を滴下して加え、氷浴を取り除き、そして混合物を室温で45分間攪拌した。DCMで抽出し、Na2SO4上で乾燥させ、そして減圧下で蒸発させて、8.7gの透明な薄黄色の液体を生成せしめた。1H NMR(400MHz,CDCl3)δ1.24〜1.53(m,10H),4.17(s,4H)。
【0095】
【化15】
【0096】
2−(2−メチル−[1,3]ジオキソラン−2−イル)−酪酸エチルエステル
メチル−2−エチルアセトアセテート(100mL)をトルエン(250mL)に溶解した。エチレングリコール(46.9mL、1.35当量)および触媒量のp−TsOH・H2Oを加え、そして混合物をディーン・スターク条件下で一晩還流させた。周囲温度まで冷却した後に、混合物を5%水性NaHCO3および飽和水性NaClで洗浄し、有機相をNa2SO4上で乾燥させ、そして減圧下で蒸発させた。残留物を反復真空蒸留(118〜128℃、15mbar)により精製し、85.5gの生成物を生成せしめた。1H NMR(400MHz,CDCl3)δ0.90(t,J=7Hz,3H),1.28(t,J=7Hz,3H),1.40(s,3H),1.59〜1.83(m,2H
),2.56(dd,J=11.5および4Hz,1H),3.90〜4.06(m,4H),4.18(m,2H)。
【0097】
2−(2−メチル−[1,3]ジオキソラン−2−イル)−ブタン−1−オール
2−(2−メチル−[1,3]ジオキソラン−2−イル)−酪酸エチルエステル(85.5g)を乾式Et2O(50mL)に溶解した。氷浴において冷却した乾式Et2O(200mL)中のLiAlH4(16.1g)の懸濁液にこの混合物を滴下して加えた。混合物を4時間還流させ、氷浴において冷却し、そしてH2O(16.1mL)、2M水性NaOH(32.2mL)そして再びH2O(16.1mL)を加えることによりクエンチした。懸濁液を濾過し、フィルターケーキをEt2Oで洗浄し、そして合わせた濾液を蒸発乾固させた。残留物(49g)を真空蒸留(112〜125℃、15mbar)により精製し、43.5gの透明な無色の液体を生成せしめた。1H NMR(400MHz,CDCl3)δ0.98(t,J=7.5Hz,3H),1.10〜1.24(m,1H),1.31(s,1H),1.50〜1.75(m,2H),3.12(br s,1H),3.59〜3.76(m,2H),3.94〜4.02(m,4H)。
【0098】
3−ヒドロキシメチル−ペンタン−2−オン
2−(2−メチル−[1,3]ジオキソラン−2−イル)−ブタン−1−オール(43.5g)をH2O(100mL)およびEtOH(10mL)の混合物に溶解し、そして濃水性HCl(1mL)を加えた。混合物を2時間還流させ、周囲温度に冷却し、2M水性NaOHで中和し、硫酸アンモニウムで飽和させ、そしてEt2Oで2回抽出した。合わせた有機層をNa2SO4上で乾燥させ、そして蒸発乾固させた。黄色がかった残留物(25.7g)を真空蒸留により精製して20.7gの透明な無色の油を生成せしめた。1H NMR(400MHz,CDCl3)δ0.95(t,J=7.5Hz,3H),1.49〜1.76(m,2H),2.21(s,3H),2.64(m,1H),3.68〜3.84(m,3H)。
【0099】
酢酸2−エチル−3−オキソ−ブチルエステル
3−ヒドロキシメチル−ペンタン−2−オン(20.7g)をCHCl3(150mL)に溶解し、そして氷浴において冷却した。無水酢酸(80mL)、続いてDMAP(2.18g)を加え、そして混合物を室温で一晩攪拌した。氷浴において冷却した後に、MeOH(120mL)を滴下して加え、そして混合物を飽和水性NaHCO3溶液に注ぎ込んだ。DCMで2回抽出した後に、合わせた有機相をNa2SO4上で乾燥させ、そして減圧下で蒸発させて28.0gの薄黄色の液体を生成せしめた。1H NMR(400MHz,CDCl3)δ0.93(t,J=8Hz,3H),1.46〜1.75(m,2H),2.03(s,3H),2.20(s,3H),2.77(quint.,J=6.5Hz,1H),4.20(d,J=7Hz,3H)。
【0100】
4−エチル−3−メチル−4,5−ジヒドロ−1H−ピラゾール
酢酸2−エチル−3−オキソ−ブチルエステル(23.0g)を乾式THF(75mL)に溶解し、そしてDBU(23.9mL)を加えた。混合物を室温で15分間攪拌して中間体3−メチレン−ペンタン−2−オンを生成せしめた。MeOH(75mL)を加え、続いてヒドラジン水和物(7.75mL)を滴下して加えた。得られる混合物を室温で一晩攪拌し、そして減圧下で蒸発させた。残留物を真空蒸留(94〜106℃、15mbar)により精製し、7.9gの透明な無色の液体を生成せしめた。1H NMR(400MHz,CDCl3)δ0.95(t,J=7.5Hz,3H),1.33〜1.83(m,2H),1.92(s,3H),2.78(m,1H),3.01(t,J=9.5Hz,1H),3.51(t,J=9,5Hz,1H)。
【0101】
【化16】
【0102】
2−ジメチルアミノメチル−シクロヘキサノン
シクロヘキサノン(259mL)にホルムアルデヒド(37.2mLの37%水溶液)およびジメチルアミン塩酸塩(40.8g)を加えた。攪拌した混合物をゆっくりと加熱し、そして1時間還流させた。周囲温度まで冷却した後にH2Oを加え、そして混合物をEt2Oで2回抽出した。50%水性NaOH(27.5mL)の添加により水層を塩基性にし、そして次にDCMで2回抽出した。合わせた有機相をNa2SO4上で乾燥させ、そして減圧下で蒸発させて66.6gの薄黄色の液体を生成せしめた。1H NMR(400MHz,CDCl3)δ1.34〜1.47(m,1H),1.60〜1.78(m,2H),1.81〜1.92(m,1H),1.98〜2.09(m,1H),2.16〜2.55(m,5H),2.21(s,6H),2.69(dd,J=13および6Hz,1H)。
【0103】
3,3a,4,5,6,7−ヘキサヒドロ−2H−インダゾール
ヒドラジン水和物(28.0mL)をn−BuOH(200mL)に溶解し、そして氷浴において冷却した。n−BuOH(50mL)中の2−ジメチルアミノメチル−シクロヘキサノン(64.0g)の溶液を滴下して加え、混合物をゆっくりと温め、そして20時間還流させた。溶媒を減圧下で蒸発させた。残留物を真空蒸留(64〜67℃、28Pa)により精製し、24.2gの透明な無色の液体を生成せしめた。この物質をさらに精製せずに次の段階において使用した。
【0104】
【化17】
【0105】
4−エチル−5−メチル−4,5−ジヒドロ−1H−ピラゾール
ヒドラジン水和物(12.4mL)をMeOH(100mL)に溶解し、そして氷浴において冷却した。この混合物に、温度が10℃未満に保たれるような速度でMeOH(50mL)中の2−エチル−ブト−2−エナール(25g)の溶液を加えた。氷浴を取り除き、そして混合物を室温で2日間攪拌した。溶媒を減圧下で蒸発させた。真空蒸留(90〜100℃、20mbar)により約1:2の比率でジアステレオマー混合物としての所望の生成物とヒドラゾンを含有する16.9gの薄黄色の液体を生成せしめた。この物質をさらに精製せずに次の段階において使用した。
【0106】
【化18】
【0107】
5−エチル−4−メチル−4,5−ジヒドロ−1H−ピラゾール
N2雰囲気下で、ヒドラジン水和物(63.9mL、10当量)をMeOH(100mL)に溶解し、そして氷浴において冷却した。この混合物に、温度が10℃未満に保たれるような速度でMeOH(50mL)中の2−メチル−ペント−2−エナール(15.0mL)の溶液を加えた。氷浴を取り除き、そして混合物を室温で一晩攪拌した。溶媒を減圧下で蒸発させた。真空蒸留(40〜45℃、15mbar)により約1:1の比率でジアステレオマー混合物としての所望の生成物とヒドラゾンを含有する9.5gの薄黄色の液体を生成せしめた。この物質をさらに精製せずに次の段階において使用した。
【0108】
【化19】
【0109】
4−ヒドロキシ−3,3−ジメチル−ブタン−2−オン
25mLの3−メチル−ブタン−2−オンに7.01gのパラホルムアルデヒドおよび36.0mLのトリフルオロ酢酸を加えた。混合物を7時間還流させた。冷却した後に、300mLのH2Oおよび100g(5当量)のNaHCO3を加えた。懸濁液を濾過し、そして有機層を分離した。フィルターケーキをDCMで2回洗浄し、合わせた濾液をNa2SO4上で乾燥させ、そして溶媒を減圧下で蒸発させて23.7gの橙色の液体を生成せしめた。1H NMR(400MHz,CDCl3)δ1.17(s,6H),2.17(s,3H),2.38(t,J=7Hz,1H),3.65(d,J=7Hz,2H)。
【0110】
メタンスルホン酸2,2−ジメチル−3−オキソ−ブチルエステル
23.7gの4−ヒドロキシ−3,3−ジメチル−ブタン−2−オンを150mLのDCMに溶解した。49.5mL(3当量)のピリジンおよび17.5mL(1.1当量)の塩化メシルを加え、そして混合物を室温で20時間攪拌した。懸濁液を濾過し、そしてフィルターケーキをDCMで2回洗浄した。濾液を1M HClで洗浄し、そして水層をDCMで2回抽出した。合わせた濾液をNa2SO4上で乾燥させ、そして溶媒を減圧下で蒸発させて41.2gの褐色の液体を生成せしめた。1H NMR(400MHz,CDCl3)δ1.24(s,6H),2.20(s,3H),3.03(s,3H),4.21(s,2H)。
【0111】
3,4,4−トリメチル−4,5−ジヒドロ−1H−ピラゾール
39.2gのメタンスルホン酸2,2−ジメチル−3−オキソ−ブチルエステルを200mLのMeOHに溶解し、そして氷浴において冷却した。21.6mL(2.2当量)のヒドラジン水和物を滴下して加え、そして反応混合物を室温で1時間攪拌した。反応混合物を濃縮し、5%NaHCO3を加え、そしてDCMで3回抽出した。合わせた有機層をNa2SO4上で乾燥させ、そして溶媒を減圧下で蒸発させて19.5gの橙色の液体を生成せしめた。この液体の10gの真空蒸留により6.4gの薄黄色の液体を生成せしめた(76〜78℃、20mbar)。1H NMR(400MHz,CDCl3)δ1.14(s,6H),1.86(s,3H),3.14(s,2H),4.00(br s,1H)。
【0112】
【化20】
【0113】
3−ジメチルアミノ−1−フェニル−プロパン−1−オン塩酸塩
40mLのEtOH中の0.5mLの濃水性HClの溶液に、アセトフェノン(30.0g)、パラホルムアルデヒド(10.0g)およびジメチルアミン塩酸塩(26.5g)を加え、そして混合物を3時間還流させた。混合物を室温に冷却し、そして沈殿物を濾過し、アセトンで洗浄し、そして真空中で乾燥させて37.2gの白色の結晶性物質を得た。1H NMR(200MHz,DMSO−d6)δ2.84(s,6H),3.38〜3.55(m,2H),3.57〜3.74(m,2H),7.48〜7.73(m,3H),7.97〜8.10(m,2H)。
【0114】
3−フェニル−4,5−ジヒドロ−1H−ピラゾール
N2雰囲気下で、3−ジメチルアミノ−1−フェニル−プロパン−1−オン塩酸塩(37.2g)を温かいMeOH(75mL)に溶解し、そして50℃で攪拌したMeOH(30mL)中のヒドラジン水和物(23mL)および50%水性NaOH(12mL)の溶液にゆっくりと加えた。混合物を2時間還流させ、そして減圧下で蒸発させた。氷水を残留物に加え、そして5分間攪拌した後に生じた固体を濾過して分離した。残留物をEt2Oに溶解し、Na2SO4上で乾燥させ、そして減圧下で蒸発乾固させて80%純粋である19.7gの黄色の油を生成せしめ、それをさらに精製せずに次の段階において使用した。
【0115】
【化21】
【0116】
1−(4−クロロ−フェニル)−3−ジメチルアミノ−プロパン−1−オン塩酸塩
EtOH(80mL)に、p−クロロアセトフェノン(77.3g、0.50mol)、ジメチルアミン塩酸塩(52.7g、0.65mol)、パラホルムアルデヒド(19.8g、0.66mol)および濃水性HCl(1mL)を加え、そして混合物を5時間還流させた。混合物を40℃に冷却し、アセトン(400mL)を加え、そして攪拌下で混合物を20℃までさらに冷却した。沈殿物を濾過し、アセトンおよびPAで洗浄し、そして空気乾燥させて69.5gの生成物を生成せしめ、それをさらに精製せずに次の段階において使用した。
【0117】
3−(4−クロロ−フェニル)−4,5−ジヒドロ−1H−ピラゾール
N2雰囲気下で、1−(4−クロロ−フェニル)−3−ジメチルアミノ−プロパン−1−オン塩酸塩(37.2g)を温かいMeOH(75mL)に溶解し、そして50℃で攪拌したMeOH(30mL)中のヒドラジン水和物(23mL)および50%水性NaOH(12mL)の溶液にゆっくりと加えた。混合物を2時間還流させ、そして減圧下で蒸
発させた。水を残留物に加え、続いてDCMで抽出した。有機相を水で2回洗浄し、乾燥させ、そして減圧下で蒸発させ、25.0gの黄色の固体、m.p.90〜100℃を生成せしめた。
【0118】
【化22】
【0119】
4−フェニル−4,5−ジヒドロ−1H−ピラゾール
N2雰囲気下で、ジメチルアミン塩酸塩(7.27g)およびホルマリン(37%)(6.63mL)をフェニルアセトアルデヒド(10mL)に加え、そして室温で一晩攪拌した。反応混合物をジエチルエーテルで1回抽出し、有機層をMgSO4上で乾燥させ、そして中間体2−フェニル−プロペナールを含有する溶液をMeOHに溶解した。ヒドラジン水和物(7.87mL)を加え、そして反応混合物を50℃で2時間攪拌した(Et2Oを蒸発させた)。混合物を減圧下で濃縮した。残留物をDCMに溶解し、そしてMgSO4上で乾燥させ、続いて減圧下で蒸発させ、3.12gの黄色の油を生成せしめ、それをさらに精製せずに次の段階において使用した。
【0120】
【化23】
【0121】
3−フェニル−ブト−3−エン−2−オン
1−フェニル−プロパン−2−オン(40.8g)を200mLのMeOHに溶解した。ホルマリン(37%)(79mL)、ピペリジン(4ml)およびHOAc(4ml)を加え、そして反応物を60℃で3時間攪拌した。反応混合物を減圧下で蒸発乾固させた。残留物をジエチルエーテルに溶解し、そして水で抽出した。有機層を1M HClで洗浄し、MgSO4上で乾燥させ、そして減圧下で蒸発させて36.3gの黄色の液体を生成せしめた。1H NMR(400MHz,CDCl3)δ2.41(s,3H),5.87(s,1H),6.18(s,1H),7.24〜7.40(m,5H)。
【0122】
3−メチル−4−フェニル−4,5−ジヒドロ−1H−ピラゾール
ヒドラジン水和物(12.06mL)をMeOH(200mL)中の3−フェニル−ブト−3−エン−2−オン(36.3g)に加えた。反応物を還流温度で一晩攪拌した。溶媒を減圧下で蒸発させた。残留物をジエチルエーテルに溶解し、そして水で洗浄した。有機相をNa2SO4上で乾燥させ、そして減圧下で蒸発させた。粗物質をDCM:MeOH=98:2で溶出するフラッシュカラムクロマトグラフィーにより精製して65%の所望の生成物を含有する19.7gの橙色の油を生成せしめ、それをさらに精製せずに次の段階において使用した。
【0123】
【化24】
【0124】
5−フェニル−4,5−ジヒドロ−1H−ピラゾール
N2雰囲気下で、ヒドラジン水和物(9.2mL)をt−BuOH(20mL)中の桂皮アルデヒド(10.0g)の溶液に加えた。混合物を一晩還流させ、続いて減圧下で濃縮した。水を残留物に加え、そして水相をDCMで2回抽出した。合わせた有機層を水で洗浄し、Na2SO4上で乾燥させ、そして減圧下で濃縮した。これにより85%の所望の生成物を含有する10.46gの黄色の油を生成せしめ、それをさらに精製せずに次の段階において使用した。1H NMR(200MHz,CDCl3)δ2.61〜2.80(m,1H),3.04〜3.23(m,1H),4.72(dd,J=8および10Hz,1H),5.60〜6.10(br s,1H),6.77〜6.87(m,1H),7.18〜7.47(m,5H)。
【0125】
【化25】
【0126】
5−フラン−2−イル−4,5−ジヒドロ−1H−ピラゾール
N2雰囲気下で、ヒドラジン水和物(4.0mL)をt−BuOH(25mL)中の3−(2−フリル)アクロレイン(5.0g)の溶液に加えた。混合物を2日間還流させ、続いて減圧下で蒸発させた。残留物をDCMに溶解し、そして5%水性NaHCO3で2回抽出した。有機相をNa2SO4上で乾燥させ、そして減圧下で蒸発させた。これにより45%の予想生成物および閉環を受けなかった55%のヒドラゾン中間体を含有する5.3gの黄色の油を生成せしめた。n−BuOHにおいてさらに24時間還流させて(処理(workup)後に)58%の予想生成物および42%のヒドラゾンを含有する5.6gの褐色の油を生成せしめた。この物質をさらに精製せずに次の段階において使用した。1H NMR(400MHz,CDCl3)における特有のピラゾリンシグナル:δ2.87〜3.08(m,2H),4.72〜4.81(m,1H),6.87(br s,1H)。
【0127】
【化26】
【0128】
3−(3,4−ジヒドロ−2H−ピラゾール−3−イル)−ピリジン
N2雰囲気下で、ヒドラジン水和物(3.65mL、2当量)をt−BuOH(25mL)中の3−(3−ピリジル)アクロレイン(5.0g)の溶液に加えた。混合物を3日間還流させ、続いて減圧下で蒸発させた。残留物をDCMに溶解し、そして5%水性NaHCO3で洗浄した。有機相をNa2SO4上で乾燥させ、そして減圧下で蒸発させた。
これにより74%の予想生成物および閉環を受けなかった26%のヒドラゾン中間体を含有する5.0gの赤色の油を生成せしめた。この物質をさらに精製せずに次の段階において使用した。1H NMR(400MHz,CDCl3)における特有のピラゾリンシグナル:δ2.63〜2.75(m,1H),3.13〜3.25(m,1H),4.72〜4.82(m,1H),6.85(br s,1H)。
【0129】
【化27】
【0130】
3−フラン−3−イル−プロペナール
6.08gの(トリフェニルホスホルアニリデン)アセトアルデヒドを10mLの乾式DMFに懸濁した。1.67mL(1当量)の3−フルアルデヒドを加え、そして混合物を80℃で一晩攪拌した。混合物をEAに溶解し、そして5%水性NaHCO3で4回洗浄し、有機相をNa2SO4上で乾燥させ、濾過し、そして真空中で濃縮した。残留物をPAに懸濁し、濾過し、そして真空中で濃縮して68%の所望の生成物を含有する1.47gの薄褐色の油を生成せしめた。この物質をさらに精製せずに次の段階において使用した。1H NMR(400MHz,CDCl3)における特有のシグナル:δ6.45(dd,J=8および16Hz,1H),9.63(d,J=8Hz,1H)。
【0131】
5−フラン−3−イル−4,5−ジヒドロ−1H−ピラゾール
5.84ml(10当量)のヒドラジン水和物を20mLのジエチルエーテルに加えた。エマルジョンを氷/NaCl浴で−10℃に冷却した。20mLのジエチルエーテル中1.47gの3−フラン−3−イル−プロペナールの溶液を滴下して加えた。混合物を一晩(氷浴で)攪拌し、そして室温にゆっくりと到達させた。5%水性NaHCO3を加え、そして混合物をEAで3回抽出した。合わせた有機層をNa2SO4上で乾燥させ、濾過し、そして真空中で濃縮した。残留物をSCXイオン交換カラム上で捕捉し、MeOHで洗浄し、そしてMeOH中1MのNH3で溶出して蒸発後に85%の所望の生成物を含有する950mgの橙色の油を生成せしめた。この物質をさらに精製せずに次の段階において使用した。1H NMR(400MHz,CDCl3)δ2.62〜2.72(m,1H),2.97〜3.07(m,1H),4.62〜4.71(m,1H),5.57〜5.74(br s,1H),6.36(br s,1H),6.87(br s,1H),7.35〜7.41(m,2H)。
【0132】
【化28】
【0133】
2−ピリジン−2−イル−プロペナール
6.08gの(トリフェニルホスホルアニリデン)アセトアルデヒドを10mLの乾式DMFに懸濁した。1.90mL(1当量)のピリジン−2−カルバルデヒドを加え、そして混合物を室温で一晩攪拌した。混合物をEAに溶解し、そして5%水性NaHCO3
で4回洗浄し、有機相をNa2SO4上で乾燥させ、濾過し、そして真空中で濃縮した。残留物をPAに懸濁し、濾過し、そして真空中で濃縮して80%の所望の生成物を含有する1.50gの暗黄色の油を生成せしめた。この物質をさらに精製せずに次の段階において使用した。1H NMR(400MHz,CDCl3)δ7.09(dd,J=8および16Hz,1H),7.30〜7.36(m,1H),7.49〜7.59(m,2H),7.77(dt,J=8.8および2Hz,1H),8.67〜8.74(m,1H),9.81(d,J=8Hz,1H)。
【0134】
2−(3,4−ジヒドロ−2H−ピラゾール−3−イル)−ピリジン
4.56ml(10当量)のヒドラジン水和物を20mLのジエチルエーテルに加えた。エマルジョンを氷/NaCl浴で−10℃に冷却した。20mLのジエチルエーテル中1.25gの3−ピリジン−2−イル−プロペナールの溶液を滴下して加えた。混合物を一晩(氷浴で)攪拌し、そして室温にゆっくりと到達させた。5%水性NaHCO3を加え、そしてEAで5回抽出した。合わせた有機層をNa2SO4上で乾燥させ、濾過し、そして真空中で濃縮した。残留物をSCXイオン交換カラム上で捕捉し、MeOHで洗浄し、そしてMeOH中1MのNH3で溶出して90%の所望の生成物を含有する1.28gの褐色の油を生成せしめた。この物質をさらに精製せずに次の段階において使用した。1H NMR(400MHz,CDCl3)δ2.84〜2.94(m,1H),3.19〜3.29(m,1H),4.82〜4.90(m,1H),6.83(br s,1H),7.17〜7.23(m,1H),7.35〜7.40(m,1H),7.69(dt,J=7.5,7.5および2Hz,1H),8.53〜8.58(m,1H)。
【0135】
【化29】
【0136】
3−ピリジン−4−イル−プロペナール
6.08gの(トリフェニルホスホルアニリデン)アセトアルデヒドを10mLの乾式DMFに懸濁した。1.93mL(1当量)のピリジン−4−カルバルデヒドを加え、そして混合物を室温で一晩攪拌した。混合物をEAに溶解し、そして5%水性NaHCO3で4回洗浄し、有機相をNa2SO4上で乾燥させ、濾過し、そして真空中で濃縮した。残留物をPAに懸濁し、濾過し、そして真空中で濃縮して80%の所望の生成物を含有する1.17gの黄色の油を生成せしめた。この物質をさらに精製せずに次の段階において使用した。1H NMR(400MHz,CDCl3)δ6.85(dd,J=8および16Hz,1H),7.39〜7.47(m,3H),8.70〜8.74(m,2H),9.78(d,J=8Hz,1H)。
【0137】
4−(3,4−ジヒドロ−2H−ピラゾール−3−イル)−ピリジン
4.27ml(10当量)のヒドラジン水和物を20mLのジエチルエーテルに加えた。エマルジョンを氷/NaCl浴で−10℃に冷却した。20mLのジエチルエーテル中1.17gの3−ピリジン−4−イル−プロペナールの溶液を滴下して加えた。混合物を一晩(氷浴で)攪拌し、そして室温にゆっくりと到達させた。5%水性NaHCO3を加え、そしてEAで5回抽出した。合わせた有機層をNa2SO4上で乾燥させ、濾過し、そして真空中で濃縮した。残留物をSCXイオン交換カラム上で捕捉し、MeOHで洗浄し、そしてMeOH中1MのNH3で溶出して90%の所望の生成物を含有する1.23gの褐色の油を生成せしめた。この物質をさらに精製せずに次の段階において使用した。
1H NMR(400MHz,CDCl3)δ2.61〜2.71(m,1H),3.15〜3.25(m,1H),4.68〜4.76(m,1H),6.82(br s,1H),7.25〜7.30(m,2H),8.55〜8.60(m,2H)。
【0138】
【化30】
【0139】
3−チオフェン−3−イル−プロペナール
10.0gの(トリフェニルホスホルアニリデン)アセトアルデヒドを10mLの乾式DMFに懸濁した。2.88mL(1当量)のチオフェン−3−カルバルデヒドを加え、そして混合物を80℃で一晩攪拌した。混合物をEAに溶解し、そして5%水性NaHCO3で4回洗浄し、有機相をNa2SO4上で乾燥させ、濾過し、そして真空中で濃縮した。残留物をPAに懸濁し、濾過し、そして真空中で濃縮して54%の所望の生成物を含有する4.16gの橙色の油を生成せしめた。この物質をさらに精製せずに次の段階において使用した。1H NMR(400MHz,CDCl3)における特有のシグナル:δ6.54(dd,J=8および16Hz,1H),9.66(d,J=8Hz,1H)。
【0140】
5−チオフェン−3−イル−4,5−ジヒドロ−1H−ピラゾール
14.6ml(10当量)のヒドラジン水和物を50mLのジエチルエーテルに加えた。エマルジョンを氷/NaCl浴で−10℃に冷却した。25mLのジエチルエーテル中4.16gの3−チオフェン−3−イル−プロペナールの溶液を滴下して加えた。混合物を(氷浴で)一晩攪拌し、そして室温にゆっくりと到達させた。5%水性NaHCO3を加え、そしてEAで3回抽出した。合わせた有機層をNa2SO4上で乾燥させ、濾過し、そして真空中で濃縮して70%の所望の生成物を含有する4.12gの橙色の油を生成せしめた。この物質をさらに精製せずに次の段階において使用した。1H NMR(400MHz,CDCl3)における特有のピラゾリンシグナル:δ2.78〜2.88(m,1H),3.03〜3.13(m,1H),4.77〜4.86(m,1H),6.86(br s,1H)。
【0141】
【化31】
【0142】
3−チオフェン−2−イル−プロペナール
10.0gの(トリフェニルホスホルアニリデン)アセトアルデヒドを10mLの乾式DMFに懸濁した。3.07mL(1当量)のチオフェン−2−カルバルデヒドを加え、そして混合物を80℃で一晩攪拌した。混合物をEAに溶解し、そして5%水性NaHCO3で4回洗浄し、有機相をNa2SO4上で乾燥させ、濾過し、そして真空中で濃縮した。残留物をPAに懸濁し、濾過し、そして真空中で濃縮して50%の所望の生成物を含有する4.27gの橙色の油を生成せしめた。この物質をさらに精製せずに次の段階において使用した。1H NMR(400MHz,CDCl3)における特有のシグナル:δ
6.52(dd,J=8および16Hz,1H),9.63(d,J=8Hz,1H)。
【0143】
5−チオフェン−2−イル−4,5−ジヒドロ−1H−ピラゾール
15.0ml(10当量)のヒドラジン水和物を50mLのジエチルエーテルに加えた。エマルジョンを氷/NaCl浴で−10℃に冷却した。25mLのジエチルエーテル中4.27gの3−チオフェン−2−イル−プロペナールの溶液を滴下して加えた。混合物を(氷浴で)一晩攪拌し、そして室温にゆっくりと到達させた。5%水性NaHCO3を加え、そしてEAで3回抽出した。合わせた有機層をNa2SO4上で乾燥させ、濾過し、そして真空中で濃縮して70%の所望の生成物を含有する5.58gの橙色の油を生成せしめた。この物質をさらに精製せずに次の段階において使用した。1H NMR(400MHz,CDCl3)における特有のピラゾリンシグナル:δ2.77〜2.86(m,1H),3.08〜3.18(m,1H),4.95〜5.03(m,1H),6.88(br s,1H)。
【0144】
【化32】
【0145】
3−イソプロピル−5−フェニル−4,5−ジヒドロ−1H−ピラゾール
0.38mLの3−メチル−2−ブタノンを10mLのDCMに溶解した。0.36mL(1当量)のベンズアルデヒドを加え、続いて1.50mLのトリフルオロメタンスルホン酸無水物を滴下して加えた。混合物を室温で1時間攪拌した。次に、10mLのMeOHおよび0.87mL(5当量)のヒドラジン水和物を加えた。混合物を室温で30分間攪拌し、そして真空中で濃縮した。残留物をDCMに溶解し、5%水性NaHCO3で抽出し、そして有機相をNa2SO4上で乾燥させ、濾過し、そして真空中で濃縮して約50%の所望の生成物を含有する520mgの褐色の油を生成せしめ、それをさらに精製せずに次の段階において使用した。
【0146】
【化33】
【0147】
4−メチル−5−フェニル−4,5−ジヒドロ−1H−ピラゾール
5.22ml(1当量)のヒドラジン水和物を100mLのジエチルエーテルに加えた。エマルジョンを氷浴で冷却した。15.0mLの2−メチル−3−フェニル−プロペナールを滴下して加え、そして混合物を室温で一晩攪拌した。H2Oを加え、有機層を分離し、そして水層をジエチルエーテルで抽出した。合わせた有機層をNa2SO4上で乾燥させ、濾過し、そして真空中で濃縮した。真空蒸留により5.9gの所望の生成物(ジアステレオマー対の混合物)を透明な液体として生成せしめた(76〜82℃、0.2〜0.3mbar)。第一のジアステレオマー対の1H NMR(400MHz,CDCl3):δ0.71(d,J=7Hz,3H),3.20〜3.31(m,1H),4.77(d,J=10Hz,1H),6.73(br s,1H),7.23〜7.42(m,
5H),8.55〜8.60(m,2H)。第二のジアステレオマー対の1H NMR(400MHz,CDCl3):δ1.24(d,J=7Hz,3H),2.90〜3.11(m,1H),4.22(d,J=11Hz,1H),6.71(br s,1H),7.23〜7.42(m,5H),8.55〜8.60(m,2H)。
【0148】
【化34】
【0149】
2−ベンジリデン−ブチルアルデヒド
30.0mLのベンズアルデヒドを150mLのEtOHに溶解し、そして氷浴で冷却した。5.01mLの45%KOH(0.2当量)を加え、続いて16.5mLのブチルアルデヒドを滴下して加えた。混合物を室温で3日間攪拌し、1M HClで酸性化し、そしてエーテルで抽出した。有機層をNa2SO4上で乾燥させ、濾過し、そして真空中で濃縮した。真空蒸留により70%の所望の生成物を含有する20.4gの黄色の液体を生成せしめた(78〜82℃、0.6mbar)。この物質をさらに精製せずに次の段階において使用した。1H NMR(400MHz,CDCl3)における特有のシグナル:δ1.15(d,J=7.5Hz,3H),2.57(q,J=7,5Hz,1H),7.22(s,1H),9.56(s,1H)。
【0150】
4−エチル−5−フェニル−4,5−ジヒドロ−1H−ピラゾール
62ml(10当量)のヒドラジン水和物を150mLのジエチルエーテルに加えた。エマルジョンを氷/NaCl浴で−10℃に冷却した。100mLのエーテル中20.4gの2−ベンジリデン−ブチルアルデヒドの溶液を−10℃で滴下して加え、そして−10℃で3時間攪拌した。混合物を(氷浴で)一晩攪拌し、そしてゆっくりと室温に到達させた。H2Oを加え、有機層を分離し、そして水層をジエチルエーテルで2回抽出した。合わせた有機層をNa2SO4上で乾燥させ、濾過し、そして真空中で濃縮した。真空蒸留により94%の所望生成物(ジアステレオマー対の混合物)を含有する6.1gの透明な液体を生成せしめた(102〜106℃、0.6mbar)。第一のジアステレオマー対の特有のシグナル:1H NMR(400MHz,CDCl3)δ0.83(t,J=6.5Hz,3H),3.03〜3.13(m,1H),4.74〜4.81(m,1H),6.83(br s,1H)。第二のジアステレオマー対の特有のシグナル:1H NMR(400MHz,CDCl3)δ1.00(d,J=7.5Hz,3H),2.84〜2.93(m,1H),4.28〜4.34(m,1H),6.76(br s,1H)。
【0151】
【化35】
【0152】
(1−メチル−1,2,5,6−テトラヒドロ−ピリジン−3−イル)−メタノール
15.0gの1−メチル−1,2,5,6−テトラヒドロ−ピリジン−3−カルボン酸メチルエステル臭化水素酸塩をEAに溶解し、そして2M NaOHで抽出した。有機層を分離し、そして水層を再びEAで抽出した。合わせた有機層をNa2SO4上で乾燥させ、濾過し、そして濃縮して8.27gの遊離塩基を黄色の油として生成せしめた(84%)。6.5gのLiAlH4(3.2当量)を100mLの乾式THFに懸濁し、そして氷浴で冷却した。これに50mLの乾式THF中8.27gの1−メチル−1,2,5,6−テトラヒドロ−ピリジン−3−カルボン酸メチルエステル(遊離塩基)の溶液を滴下して加えた。混合物を室温で3時間攪拌した。混合物を氷浴で冷却し、そして6.5mLのH2O、13mLの2M NaOHおよび6.5mLのH2Oを滴下して加えた。残留物を濾過し、エーテルで洗浄し、そして濾液を真空中で濃縮して6.7gの無色の油を生成せしめた。1H NMR(400MHz,CDCl3)δ2.18〜2.26(m,2H),2.36(s,3H),2.49(t,J=6Hz,2H),2.92〜2.97(m,2H),4.00(br s,2H),5.68(br s,1H)。
【0153】
1−メチル−1,2,5,6−テトラヒドロ−ピリジン−3−カルバルデヒド
2.88mLの塩化オキサリル(2.4当量)を20mLのDCMに溶解した。混合物を−78℃に冷却し、そして10mLのDCM中3.37mLのDMSO(2.0当量)の溶液を滴下して加えた。混合物を−78℃で15分間攪拌した。温度を−65℃未満に保ちながら10mLのDCM中3.0gの(1−メチル−1,2,5,6−テトラヒドロ−ピリジン−3−イル)−メタノールの溶液を滴下して加えた。混合物を−78℃で15分間攪拌した。9.81mLのトリエチルアミン(3.0当量)を滴下して加え、そして次に混合物を室温まで温めておいた。混合物を攪拌可能にしておくために50mLのDCMを加えた。混合物を室温で1時間攪拌した。H2Oを加え、有機層を分離し、そして水層を再びDCMで抽出した。合わせた有機層をNa2SO4上で乾燥させ、濾過し、そして濃縮して3.24gの橙色の油(85%純粋)を生成せしめ、それをさらに精製せずに次の段階において使用した。1H NMR(400MHz,CDCl3)δ2.43(s,3H),2.48〜2.60(m,4H),3.11〜3.15(m,2H),6.85(m,1H),9.43(s,1H)。
【0154】
5−メチル−3a,4,5,6,7,7a−ヘキサヒドロ−1H−ピラゾロ[4,3−c]ピリジン
3.2gの1−メチル−1,2,5,6−テトラヒドロ−ピリジン−3−カルバルデヒドを10mLのn−BuOHに溶解した。2当量のヒドラジン水和物を加え、混合物を24時間還流させ、そして次に真空中で濃縮した。残留物をDCMに溶解し、そして2M NaOHで抽出し、そして有機相をNa2SO4上で乾燥させ、濾過し、そして真空中で濃縮して1.78gの褐色の油を生成せしめ、それをさらに精製せずに次の段階において使用した。
【0155】
【化36】
【0156】
ベンジル−ビス−(2−クロロ−エチル)−アミン
ビス−(2−クロロ−エチル)−アミン塩酸塩を150mLのアセトニトリルに懸濁した。34.8gのK2CO3(3当量)および10.0mLの臭化ベンジル(1当量)を加えた。混合物を一晩還流させた。シリカ上で濃縮し、そしてフラッシュカラムクロマトグラフィー(溶離剤PA:エーテル=95.5)で精製して4.11gの無色の油を生成せしめた。1H NMR(400MHz,CDCl3)δ2.93(t,J=7Hz,4H),3.50(t,J=7Hz,4H),3.74(s,2H),7.22〜7.37(m,5H)。
【0157】
1−ベンジル−ピペリド(piperide)−4,4−ジカルボニトリル
0.57gのマロニトリル(Malonitrile)を20mLのDMFに溶解した。1.31gのK2CO3(1.1当量)を加え、そして混合物を65℃で2時間攪拌した。10mLのDMF中2.0gのベンジル−ビス−(2−クロロ−エチル)−アミン(1当量)の溶液を65℃で滴下して加え、そして混合物を65℃でさらに3時間攪拌した。冷却した後に、混合物をEAで希釈し、そして5%水性NaHCO3で抽出した。有機相をNa2SO4上で乾燥させ、濾過し、そして真空中で濃縮して85%の予想生成物および15%のベンジル−ビス−(2−クロロ−エチル)−アミンを含有する2.02gの橙色の油を生成せしめた。この物質をさらに精製せずに次の段階において使用した。1H NMR(400MHz,CDCl3)δ2.24(t,J=5.5Hz,4H),2.50〜2.75(br s,4H),3.55(s,2H),7.22〜7.42(m,5H)。
【0158】
C−(4−アミノメチル−1−ベンジル−ピペリジン−4−イル)−メチルアミン
1.50gのLiAlH4(3当量)を100mLの乾式ジエチルエーテルに懸濁し、そして氷浴で冷却した。これに50mLの乾式THF中2.99gの1−ベンジル−ピペリド−4,4−ジカルボニトリルの溶液を滴下して加えた。混合物を室温で一晩攪拌した。混合物を氷浴で冷却し、そして1.5mLのH2O、3mLの2M NaOHおよび1.5mLのH2Oを滴下して加えた。残留物を濾過し、THFで洗浄し、そして濾液を真空中で濃縮して60%の所望の生成物を含有する2.63gの黄色の油を生成せしめ、それをさらに精製せずに次の段階において使用した。1H NMR(400MHz,CDCl3)δ1.00〜1.60(br s,2H),1.46(t,J=5.5Hz,4H),2.40(t,J=5.5Hz,4H),2.65(s,4H),3.50(s,2
H),7.20〜7.35(m,5H)。
【0159】
8−ベンジル−2,3,8−トリアザ−スピロ[4,5]デク−2−エン
2.48gのC−(4−アミノメチル−1−ベンジル−ピペリジン−4−イル)−メチルアミンを40mLのH2Oおよび10mLのMeOHに懸濁し、そして氷浴で冷却した。同時に、6.7mLの30%H2O2(6当量)および15.2mLの10%NaClO(2.4当量)を滴下して加えた。混合物を室温で1時間攪拌した。混合物をDCMで2回抽出し、合わせた有機層をNa2SO4上で乾燥させ、濾過し、そして濃縮して2.20gの黄色の油を生成せしめ、それをさらに精製せずに次の段階において使用した。経路2に従って、化合物155をこのピラゾリンビルディングブロックで製造した。これから、ベンジル脱保護(1,2−DCE中のACE−クロリド、続いてMeOH)により化合物156を生成せしめ、それを還元的アルキル化(1,2−DCE中NaBH(OAc)3の存在下で(CH2O)n)によりメチル化して化合物157を生成せしめた。
【0160】
【化37】
【0161】
メチルマロニトリル(Methylmalonitrile)
マロニトリル(10.00g;151.32mmol;2.0当量)にヨードメタン(4.71ml;75.66mmol;1.0当量)および臭化テトラブチルアンモニウム(0.98g;3.03mmol;0.04当量)を加えた。混合物を室温で30分間攪拌し、次に氷浴で冷却し、そしてカリウムtert−ブトキシド(8.49g;75.66mmol;1.0当量)をゆっくりと加えた(添加は混合物が凝固する前に開始した)。混合物を室温で2時間攪拌した。水を加え、続いてDCMで2回抽出した。Na2SO4上で乾燥させ、濾過し、そして溶媒を除いて10gの褐色の液体を生成せしめ、それを溶離剤DCM:PA=1:1、3:1およびDCMでフラッシュカラムクロマトグラフィーにより精製して3.25gの透明な液体(静置すると凝固する)を生成せしめた。1H
NMR(400MHz,CDCl3)δ1.79(d,J=7.5Hz,3H),3.79(q,J=7.5Hz,1H)。
【0162】
2−ベンジルオキシメチル−2−メチル−マロノニトリル
メチルマロニトリル(3.22g;39.40mmol;1.0当量)をTHF(35ml)に溶解した。ベンジルクロロメチルエーテル(7.54g;43.35mmol;1.1当量)およびヨウ化ナトリウム(0.20g;1.33mmol;0.03当量)を加えた。黄色の懸濁液を氷浴で冷却し、そして水素化ナトリウム(1.89g;47.29mmol;1.2当量)を少しずつ加えた。さらに多くの白色の沈殿物が形成された。混合物を室温で30分間攪拌し、エーテルで希釈し、そして5%水性NaHCO3で抽出した。有機相をNa2SO4上で乾燥させ、濾過し、そして真空中で濃縮して9.6g
の黄色の液体/油を生成せしめた。フラッシュカラムクロマトグラフィー(溶離剤EA:PA=1:4)により精製して6.2gの黄色の油を生成せしめた。1H NMR(400MHz,CDCl3)δ1.78(s,3H),3.72(s,1H),4.71(s,2H),7.31−7.43(m,5H)。
【0163】
2−ベンジルオキシメチル−2−メチル−プロパン−1,3−ジアミン
LiAlH4(3.22g;84.84mmol;3.0当量)を30mLの乾式ジエチルエーテルに懸濁し、そして氷浴で冷却した。20mLの乾式ジエチルエーテル中の2−ベンジルオキシメチル−2−メチル−マロノニトリル(5.72g;28.28mmol;1.0当量)の溶液を滴下して加えた。懸濁液を室温で4時間攪拌し、そして次に氷浴で冷却した。これに3.22mLのH2O、6.44mLの2M NaOHおよび3.22mLのH2Oを加えた。沈殿物を濾過して分離し、そしてエーテルで洗浄した。濾液を減圧下で濃縮して5.47g(84%)の無色の油を生成せしめた。1H NMR(400MHz,CDCl3)δ0.84(s,3H),2.59〜2.69(m,4H),3.29(s,2H),4.49(s,2H),7.24〜7.39(m,5H)。
【0164】
4−ベンジルオキシメチル−4−メチル−4,5−ジヒドロ−3H−ピラゾール
5.92gの2−ベンジルオキシメチル−2−メチル−プロパン−1,3−ジアミンを水(40ml)およびNaOH(10ml)に溶解し、そして氷浴で冷却した。同時に、30%H2O2(16.1mL)および10%NaClO(36.5mL)を滴下して加えた。得られる白色のエマルジョンを室温で一晩攪拌した。混合物をDCMで抽出し、有機相をNa2SO4上で乾燥させ、濾過し、そして真空中で濃縮して70%の所望の生成物を含有する5.69gの淡黄色の油を生成せしめ、それをさらに精製せずに次の段階において使用した。1H NMR(400MHz,CDCl3)δ1.05(s,3H),3.22(s,2H),4.11〜4.20(m,2H),4.29〜4.38(m,2H),4.48(s,2H),7.24〜7.39(m,5H)。
【0165】
【化38】
【0166】
テトラヒドロ−ピラン−4,4−ジカルボニトリル
マロノニトリル(5.0g)をDMSO(5mL)に溶解した。次に、ビス(2−ブロモエチル)エーテル(9.49mL)およびTBAB(1.22g)を加え、続いてKOtBu(8.49g)を少しずつ加えた。混合物を室温で4時間攪拌し、DCMに溶解し、そして5%水性NaHCO3で3回抽出した。有機相をNa2SO4上で乾燥させ、そして減圧下で蒸発させた。粗物質をPA:Et2O 65:35(Rf=0.24、KMnO4で視覚化した)で溶出するフラッシュクロマトグラフィーにより精製して2.49g(24%)の固体を生成せしめた。1H NMR(400MHz,CDCl3)δ2.24(t,4H),3.87(t,4H)。
【0167】
C−(4−アミノメチル−テトラヒドロ−ピラン−4−イル)−メチルアミン
テトラヒドロ−ピラン−4,4−ジカルボニトリル(1.52g)を乾式THF(25mL)に溶解し、そして−10℃に冷却した。この溶液に、BH3・THF(56mLのTHF中1M溶液、5当量)を滴下して加え、混合物を室温まで温めておき、そして次に60℃で6時間攪拌した。混合物を氷浴において冷却し、そしてHCl(24.2mLの6M水溶液、13当量)を加えた。混合物を室温まで温めておき、そして2時間攪拌した。混合物を2M水性NaOHで中和し、そしてDCMで3回抽出した。水層を蒸発乾固させ、残留物をCHCl3と攪拌し、固体を濾過して分離し、そして有機相を減圧下で蒸発させて1.0g(62%)の黄色の油を生成せしめた。1H NMR(400MHz,CDCl3)δ1.46(t,4H),2.74(s,4H),3.67(t,4H)。
【0168】
8−オキサ−2,3−ジアザ−スピロ[4,5]デク−2−エン
C−(4−アミノメチル−テトラヒドロ−ピラン−4−イル)−メチルアミン(1.0g)をH2O(10mL)およびMeOH(2.5mL)の混合物に溶解し、そして氷浴において冷却した。同時に、H2O2(4.8mLの30%溶液、6当量)およびNaClO(12.4mLの10%溶液、2.4当量)を滴下して加え、氷浴を取り除き、そして混合物を室温で一晩攪拌した。DCMで抽出し、Na2SO4上で乾燥させ、そして減圧下で蒸発させて85%の予想生成物および15%のジアミン出発物質を含有する380mgの透明な薄黄色の液体を生成せしめた。この物質をさらに精製せずに次の段階において使用した。1H NMR(400MHz,CDCl3)δ1.49(t,4H),3.65(t,4H),4.28(s,4H)。
【0169】
【化39】
【0170】
2,2−ビス−(2,2,2−トリフルオロ−エチル)−マロノニトリル
マロノニトリル(20.15mmol)および1−ヨード−3,3,3−トリフルオロプロパン(42.65mmol)を30mlの乾式THFに溶解し、そして混合物を氷/塩浴で冷却した。温度を5℃未満に保ちながら、1.61gのNaH(40.3mmol)を少しずつ加えた。反応混合物を室温で2時間攪拌し、そして減圧下で蒸発させた。粗物質をDCMで溶出するフラッシュクロマトグラフィーにより精製し、0.76グラムの油を生成せしめた。1H NMR(400MHz,CDCl3)δ2.62〜2.49(m,4H),2.31〜2.24(m,4H)。
【0171】
2,2−ビス−(2,2,2−トリフルオロ−エチル)−プロパン−1,3−ジアミン
340mgのLiAlH4(8.95mmol)を15mlの乾式Et2Oに懸濁し、そして氷浴において冷却した。温度が20℃未満に保たれるような速度でEt2O中760mgの2,2−ビス−(2,2,2−トリフルオロ−エチル)−マロノニトリルの溶液を滴下して加えた。混合物を室温で一晩攪拌し、氷浴において冷却し、そしてH2O(0.35ml)、2M水性NaOH(0.70ml)そして再びH2O(0.35ml)を加えることによりクエンチした。懸濁液を濾過し、フィルターケーキをEt2Oで洗浄し、そして合わせた濾液を減圧下で蒸発乾固させて0.72gの油を生成せしめた。この物質をさらに精製せずに次の段階において使用した。
【0172】
4,4−ビス−(2,2,2−トリフルオロ−エチル)−4,5−ジヒドロ−3H−ピラゾール
2,2−ビス−(2,2,2−トリフルオロ−エチル)−プロパン−1,3−ジアミン(720mg)をH2O(3ml)およびMeOH(0.75ml)の混合物に溶解し、そして氷浴において冷却した。同時に、H2O2(1.7mLの30%溶液、6当量)およびNaClO(3.85mLの10%溶液、2.4当量)を滴下して加え、氷浴を取り
除き、そして混合物を室温で一晩攪拌した。混合物をDCMで抽出し、有機相をMgSO4上で乾燥させ、そして減圧下で蒸発させて、50%の予想生成物および50%のジアミン出発物質を含有する0.82gの油を生成せしめた。この物質をさらに精製せずに次の段階において使用した。1H NMR(400MHz,CDCl3)δ4.25(s,2H),2.24〜1.85(m,4H),1.85〜1.43(m,4H)。
【実施例4】
【0173】
特定の化合物の合成
【0174】
【化40】
【0175】
(2−クロロ−ベンゼンスルホニル)−カルバミン酸メチルエステル
25.0gの2−クロロ−ベンゼンスルホンアミドに75mLのアセトニトリルおよび45.2mL(2.5当量)のトリエチルアミンを加えた。混合物を氷浴で冷却し、そして15.1mLのクロロギ酸メチルを滴下してゆっくりと加えた。混合物を室温で一晩攪拌し、そして真空中で濃縮した。水を加え、そして水層をエーテルで2回洗浄した。2M
HClでの水層の酸性化は、白色の沈殿物の形成をもたらした。懸濁液を濾過し、残留物をH2Oで洗浄し、そして真空中で乾燥させて19.1gの白色の固体を生成せしめた。1H NMR(400MHz,DMSO−d6)δ3.58(s,3H),7.52〜7.61(m,1H),7.62〜7.72(m,2H),8.10(dd,J=8および1.5Hz,1H),12.42(br s,1H)。
【0176】
【化41】
【0177】
2−クロロ−N−4−エチル−4,5−ジヒドロ−ピラゾール−1−カルボニル)ベンゼンスルホンアミド
8.5gの4−エチル−4,5−ジヒドロ−1H−ピラゾールを75mLのトルエンに溶解した。19.0gの(2−クロロ−ベンゼンスルホニル)−カルバミン酸メチルエステルを加え、そして混合物を4時間還流させた。冷却した後に、沈殿物が形成された。懸濁液を濾過し、残留物をPAで洗浄し、そして真空中で乾燥させて20.3gの白色の結晶を生成せしめた。1H NMR(200MHz,DMSO−d6)δ0.90(t,J=7.5Hz,3H),1.30〜1.70(m,2H),3.00〜3.40(m,1
H),3.25(t,J=10.5Hz,1H),3.74(t,J=10.5Hz,1H),7.08(s,1H),7.40〜7.73(m,3H),8.03〜8.16(m,1H),10.00(br s,1H)。
【0178】
【化42】
【0179】
2−クロロ−N−[ジエチルアミノ−4−エチル−4,5−ジヒドロ−ピラゾール−1−イル]−メチレン]−ベンゼン−スルホンアミド(化合物1)
2.0gの2−クロロ−N−4−エチル−4,5−ジヒドロ−ピラゾール−1−カルボニル)ベンゼンスルホンアミドを10mLのDCEに溶解した。1.07gの2−クロロ−1,3−ジメチルイミダゾリニウムクロリド(DMC)および1.75mLのTEAを加え、そして混合物を1.5時間還流させてクロロイミン中間体をインサイチューで生成せしめた。次に、5mL(過剰)のジエチルアミンを加え、そして混合物を室温で一晩攪拌した。混合物を真空中で濃縮し、そしてH2Oを加えた。DCMで抽出し(2回)、合わせた有機層をNa2SO4上で乾燥させ、蒸発乾固させ、そしてフラッシュクロマトグラフィー(エーテル、Rf=0.35)で精製し、320mgの黄色の油を生成せしめた。1H NMR(400MHz,CDCl3)δ0.95(t,J=7.5Hz,3H),1.16(t,J=7Hz,6H),1.44〜1.66(m,2H),3.00〜3.10(m,1H),3.48(q,J=7Hz,4H),3.70(dd,J=11および7Hz,1H),4.11(t,J=11Hz,1H),6.97(d,J=2Hz,1H),7.30〜7.41(m,2H),7.46(dd,J=7.5および2Hz,1H),8.16(dd,J=7.5および2Hz,1H)。
【0180】
【化43】
【0181】
N−(ビス−メチルスルファニル−メチレン)−2−クロロ−ベンゼンスルホンアミド
41.6gの2−クロロ−ベンゼンスルホンアミドに300mLのDMFおよび22mLの二硫化炭素を加えた。混合物を氷浴で冷却した。温度が10℃未満に保たれるような速度で100mLのH2O中29gのKOH(15.0mL)の溶液を滴下して加えた。混合物を5℃で30分間攪拌した。次に、温度が10℃未満に保たれるような速度で32mLのMeIを滴下して加えた。次に、混合物を室温まで温めておき、そしてさらに30分間攪拌した。H2Oを加え、そして沈殿物が生じた。これを濾過して分離し、そしてH2Oで洗浄した。残留物をEtOHで研和し、濾過して分離し、そして真空中で乾燥させ
て42.6gの白色の結晶を生成せしめた。1H NMR(200MHz,CDCl3)δ2.57(s,6H),7.32〜7.60(m,3H),8.11〜8.27(br
d,J=7.5Hz,1H)。
【0182】
【化44】
【0183】
2−クロロ−N−[(4−エチル−4,5−ジヒドロ−ピラゾール−1−イル)−メチル−スルファニル−メチレン]ベンゼンスルホン−アミド
500mgの4−エチル−4,5−ジヒドロ−1H−ピラゾールを10mLのピリジンに溶解した。1.51gのN−(ビス−メチルスルファニル−メチレン)−2−クロロ−ベンゼンスルホンアミドを加え、そして混合物を一晩還流させた。混合物を真空中で濃縮し、そしてH2Oを加え、続いてDCMで2回抽出した。合わせた有機層をNa2SO4上で乾燥させ、そして真空中で濃縮した。粗生成物をフラッシュクロマトグラフィー(勾配DCM:アセトン=100:0〜95:5)で精製して1.30gの黄色の油を生成せしめた。1H NMR(400MHz,CDCl3)δ1.02(t,J=7.5Hz,3H),1.55〜1.77(m,2H),2.28(s,3H),3.27〜3.39(m,1H),4.13(dd,J=11.5および6.5Hz,1H),4.58(t,J=11.5Hz,1H),7.16(d,J=2Hz,1H),7.39(dt,J=7.5および2Hz,1H),7.46(dt,J=7.5および2Hz,1H),7.52(dd,J=7.5および2Hz,1H),8.17(dd,J=7.5および2Hz,1H)。
【0184】
【化45】
【0185】
2−クロロ−N−[エチルアミノ−(4−エチル−4,5−ジヒドロ−ピラゾール−1−イル)−メチレン]−ベンゼンスルホン−アミド(化合物2)
1.30gの2−クロロ−N−[(4−エチル−4,5−ジヒドロ−ピラゾール−1−イル)−メチル−スルファニル−メチレン]−ベンゼンスルホン−アミドを10mLのMeOHに溶解した。H2O中のエチルアミンの70%溶液5mL(過剰)を加え、そして混合物を室温で1時間攪拌した。混合物を真空中で濃縮し、そして粗生成物をフラッシュクロマトグラフィー(エーテル、Rf=0.33)により精製して1.09gの無色の油
を生成せしめた。1H NMR(400MHz,CDCl3)δ0.95(t,J=7.5Hz,3H),1.16(t、J=7Hz,3H),1.44〜1.69(m,2H),3.03〜3.18(m,1H),3.44〜3.58(m,2H),3.71(br
dd,J=11および7.5Hz,1H),4.12(br t,J=11Hz,1H),6.88(br s,1H),6.94(d,J=2Hz,1H),7.35(dt,J=7.5および2Hz,1H),7.40(dt,J=7.5および2Hz,1H),7.48(dd,J=7.5および2Hz,1H),8.18(dd,J=7.5および2Hz,1H)。
【0186】
同様にして、「経路2」と記した以下の表における化合物は製造されている。
【0187】
【化46】
【0188】
1−エチル−2−メチル−イソチオ尿素ヨウ化水素酸塩
20.5gのエチル−チオ尿素を100mLのEtOHに溶解した。混合物を氷浴で冷却し、そして13.5mL(1.1当量)のMeIを滴下して加えた。混合物を室温で1時間攪拌し、そして真空中で濃縮して48.3gの薄黄色の油を生成せしめた。1H NMR(400MHz,DMSO−d6)δ1.17(t,J=7.5Hz,3H),2.61(s,3H),3.34(q、J=7.5Hz,2H),9.10(br s,2H)。
【0189】
【化47】
【0190】
4−N−ジエチル−4,5−ジヒドロ−ピラゾール−1−カルボキサミジン塩酸塩
19.36gの4−エチル−4,5−ジヒドロ−1H−ピラゾールを100mLのトルエンに溶解した。48.5gの1−エチル−2−メチル−イソチオ尿素ヨウ化水素酸塩および33.8mLのDiPEAを加え、そして混合物を48時間還流させた。混合物を濃縮し、2M NaOHを加え、続いてDCMで抽出した(3回)。合わせた有機層をNa2SO4上で乾燥させ、そして溶媒を真空中で蒸発させてNMRによれば75%の所望の生成物を含有する32.7g(99%)の赤色の油を生成せしめた。油をEtOHに溶解し、そして194mLのEtOH中1M HClを滴下して加えた。混合物を室温で30分間攪拌し、そして真空中で濃縮した。CH3CN:MTBE=1:1からの結晶化により11.52g(29%)の所望の生成物がベージュ色の固体として得られた。1H NMR(400MHz,DMSO−d6)δ0.96(t,J=7.5Hz,3H),1.16(t,J=7Hz,3H),1.46〜1.72(m,2H),3.32(q,J=7Hz,2H),3.35〜3.45(m,1H),3.55(dd,J=10.5およ
び7Hz,1H),3.96(t,J=10.5Hz,1H),7.34(d,J=2Hz,1H),8.00(br s,2H)。
【0191】
【化48】
【0192】
ベンゾ[1,2,5]チアジアゾール−4−スルホン酸エチルアミノ−(4−エチル−4,5−ジヒドロ−ピラゾール−1−イル)−メチレンアミド(化合物78)
300mgの4,N−ジエチル−4,5−ジヒドロ−ピラゾール−1−カルボキサミジン塩酸塩を10mLのDCMに懸濁した。0.53mLのDiPEAおよび310mgのベンゾ[1,2,5]チアジアゾール−4−スルホニルクロリドを加え、そして混合物を室温で一晩攪拌した。混合物を5%NaHCO3および2M NaOHで洗浄し、有機層をNa2SO4上で乾燥させ、そして溶媒を真空中で蒸発させて410mgの赤色/褐色の油を生成せしめた。粗生成物をフラッシュクロマトグラフィー(DCM:アセトン=98:2、Rf=0.18)により精製して350mg(65%)の橙色の油を生成せしめた。1H NMR(400MHz,CDCl3)δ0.93(t,J=7.5Hz,3H),1.16(br t,J=7Hz,3H),1.41〜1.66(m,2H),3.01〜3.16(m,1H),3.39〜3.55(m,2H),3.59〜3.74(m,1H),3.95〜4.15(m,1H),6.94(br s,1H),6.95(br s,1H),7.68(dd,J=9および7Hz,1H),8.15(br d,J=9Hz,1H),8.31(br d,J=7Hz,1H)。
【0193】
【化49】
【0194】
1−エチル−2−メチル−イソチオ尿素ヨウ化水素酸塩
20.0gのエチル−チオ尿素を100mLのEtOHに懸濁し、そして30g(1.1当量)のMeIを滴下して加え、その間に混合物は透明な黄色の溶液になった。次に、混合物を室温で1時間攪拌し、そして真空中で濃縮して48.1gの黄色の油を生成せしめた。1H NMR(400MHz,DMSO−d6)δ1.17(t,J=7.5Hz,3H),2.61(s,3H),3.34(q,J=7.5Hz,2H),9.10(br s,2H)。
【0195】
【化50】
【0196】
N−エチル−4,4−ジメチル−4,5−ジヒドロ−ピラゾール−1−カルボキサミジン塩酸塩
12.0gの4,4−ジメチル−4,5−ジヒドロ−3H−ピラゾールを100mLのピリジンに溶解した。50mLのピリジン中30.0gの1−エチル−2−メチル−イソチオ尿素ヨウ化水素酸塩の溶液を加え、そして混合物を20時間還流させた。混合物を室温まで冷却し、そして減圧下で濃縮し、そして残留物をDCM(120mL)に溶解した。有機相を2N NaOH(2x120mL)で抽出し、水(120mL)で洗浄し、Na2SO4上で乾燥させ、そして減圧下で蒸発させて16.3g(79%)の橙色の油を生成せしめた。油(10.0g)をEtOAc(50mL)に溶解し、そして60℃に加熱した。熱源の除去後に、イソプロパノール(20mL)中のHClの5〜6N溶液を4分の期間にわたって投与した。室温まで冷却した後に、EtOAc(50mL)を4分の期間にわたって加え、そして混合物を20℃で90分間攪拌した。生じた結晶を濾過により集め、そしてEtOAc(20mL)で洗浄し、続いて穏やかな加熱で減圧下で乾燥させて、6.52g(54%)の所望の生成物を黄色の固体として生成せしめた。1H NMR(400MHz,DMSO−d6)δ1.13(t,J=7Hz,3H),1.24(s,6H),3.27〜3.34(m,2H),3.64(s,2H),7.26(s,1H),8.03(br s,2H),8.13(br s,1H)。
【0197】
【化51】
【0198】
3−クロロ−N−[(4,4−ジメチル−4,5−ジヒドロ−ピラゾール−1−イル)−エチルアミノ−メチレン]−ベンゼンスルホンアミド(化合物33)
6.39gのN−エチル−4,4−ジメチル−4,5−ジヒドロ−ピラゾール−1−カルボキサミジン塩酸塩を65mLのDCMに懸濁した。12.0mLのDiPEAおよび3.96mLの3−クロロ−ベンゼンスルホニルクロリドを加え、そして懸濁液を室温で20時間攪拌し、暗褐色の混濁溶液をもたらした。混合物を2M NaOH(2x125mL)および1M HCl(2x125mL)で抽出し、水(100mL)で洗浄し、そして有機層をNa2SO4上で乾燥させ、続いて減圧下で蒸発させて7.70gの褐色の油を生成せしめた。油(1.0g)を還流下でMTBE(3mL)に溶解し、そして溶液を室温までゆっくりと冷却し、結晶化を開始した。懸濁液を室温で10分間攪拌し、ヘキサン(6mL)を1分の期間にわたって加えた。得られる懸濁液を室温で20分そして0℃で50分間攪拌し、そして生成物を濾過により集め、そしてヘキサン(1mL)で洗浄した。40℃で減圧下で乾燥させて0.85gの薄褐色の固体、m.p.62〜67℃を生成せしめた。
【0199】
同様にして、「経路3」と記した以下の表における化合物は製造されている。
【0200】
【表1】
【0201】
【表2】
【0202】
【表3】
【0203】
【表4】
【0204】
【表5】
【0205】
【表6】
【0206】
【表7】
【0207】
【表8】
【0208】
【表9】
【0209】
【表10】
【0210】
【表11】
【0211】
【表12】
【0212】
【表13】
【0213】
【表14】
【0214】
【表15】
【0215】
S*=合成経路;Rf(x)=Rf値、括弧の間:TLC移動相:
(a)=ジエチルエーテル;(b)=MeOH:TEA=97:3;(c)=DCM:アセトン=99:1;(d)=DCM:MeOH=99:1;(e)=DCM:アセトン=98:2;(f)=DCM:アセトン=95:5;(g)=DCM:MeOH=98:2;(h)=EA:PA=1:2;(i)=EA:PA=1:3;(j)=EA:PA=1:1;(k)=EA:PA=1:4;(l)=DCM;(m)=DCM:MeOH=97:3;(n)=DCM:MeOH=95:5;(o)=EA;(p)=EA:MeOH:NH4OH=94.5:5:0.5;(q)=DCM:EA=3:1;(r)=DCM:ジエチルエーテル=1:4;(s)=ジエチルエーテル:PA=7:3;(t)=ジエチルエーテル:PA=8:2;(u)=EA:PA=3:1;(v)=DCM:MeOH:NH4OH=78:20:2;(w)=DCM:MeOH:NH4OH=94.5:5:
0.5;(x)=Et2O:EA=8:2;(y)=Et2O:EA=9:1;(z)=EA:PA=5:95;Rt=LC−MS分析における保持時間(分単位)。
【0216】
本発明の化合物は新規である。以下の表に示されるように、それらは5−HT6受容体に対する高い親和性(pKi)を有し、そして強力なアンタゴニスト(pA2)である。文献に開示される構造的に最も密接に関係する化合物は、WO 02/030881に開示されるスルホニルピロリジン誘導体のいくつかである:
【0217】
【化52】
【0218】
WO 02/030881において、実施例18(R=p−CH3)、25(R=p−Cl)、26(R=H)および27(R=o−NO2)について薬理学的データは示されなかったが、それらは疼痛および片頭痛を包含する多数の症候および症状の治療において有用なガバペンチン結合部位のモジュレーターであると請求される。これらの化合物は、本発明の化合物に関する合成研究中に一連の化合物がピラゾリン環(本発明の全ての化合物に存在する)と異なる環系で合成されそしてこれらの全てが5−HT6アンタゴニストとして不活性であることが見出されたので、5−HT6受容体に対する親和性を有すると思われない。WO 02/030881に開示されるものに最も近いのは:
【0219】
【化53】
【0220】
であった。この化合物は、ピラゾリン環を有する化合物(例えば、化合物28は7.7のpA2値を有する)と顕著に異なり不活性である(pA2<5.0)ことが見出された。上記に示されるピロリジン環を有する化合物のほかに、全く同じ構造を有するが異なる環を有する化合物もまた合成され(上記に開示されるものと同様の経路を用いて)、そして試験された。特に:フェニル、2−ピリジル、2−ピラジニル、2−フラニル、5−イソオキサゾリル、2−キノリルおよび1−イソキノリル環(上記に示される化合物における1−ピロリジン環の代わりに)を有する化合物は全て不活性である(pA2<5.0)ことが見出され、本発明の化合物のピラゾリン環は5−HT6受容体との相互作用にとって重要であることを示す。
【0221】
その合成が上に記述される特定の化合物は、より詳細に本発明をさらに説明するものであり、従って、決して本発明の範囲を限定すると見なされない。本発明の他の態様は、本明細書および本明細書に開示される本発明の実施の考察から当業者に明らかである。従って、本明細書および実施例は実例としてのみ考えられるものとする。
【実施例5】
【0222】
動物研究において使用する製剤
経口(p.o.)投与用:ガラス管中の一般式(1)の固体化合物の所望の量(0.5〜5mg)に、いくらかのガラスビーズを加え、そして2分間ボルテックスすることにより固体を粉砕した。水中1%のメチルセルロースおよび2%(v/v)のポロキサマー188(Lutrol F68)の溶液1mlの添加後に、10分間ボルテックスすることにより化合物を懸濁した。数滴の水性NaOH(0.1N)でpHを7に調整した。超音波浴を用いることにより懸濁液中の残留粒子をさらに懸濁した。
【0223】
腹腔内(i.p.)投与用:ガラス管中の一般式(1)の固体化合物の所望の量(0.5〜15mg)に、いくらかのガラスビーズを加え、そして2分間ボルテックスすることにより固体を粉砕した。水中1%のメチルセルロースおよび5%のマンニトールの溶液1mlの添加後に、10分間ボルテックスすることにより化合物を懸濁した。最後にpHを7に調整した。
【実施例6】
【0224】
薬理学的方法
ヒト5−HT6受容体に対するインビトロ親和性
[3H]−N−メチル−リセルグ酸ジエチルアミド([3H]−LSD)をリガンドとして用いる結合研究によりヒト5−HT6受容体でトランスフェクションしたCHO細胞の膜調製物においてヒト5−HT6受容体に対する親和性を測定した。膜調製物は、Euroscreen(ブリュッセル)により供給される細胞から調製した。CHO/Gα16/mtAEQ/h5HT6−A1細胞を1%の透析したFCS、2mMのL−グルタミン、ジェネティシン500μg/mlおよびゼオシン200μg/mlを補足したCHO−S−SFM II培地(Gibco BRL)中Tフラスコにおいて培養した。0.25%のトリプシン(1ml/T175フラスコ)を用いて細胞を採取し、遠心分離し、そして次にCHO−S−SFM II培地に懸濁し、そして−80℃で凍結させた。融解後に細胞を4℃で1500gで3分の間遠心分離した。ペレットから、2サイクルの均質化(Potter−Elvehjem 10ストローク、600rpm)および遠心分離(40,000g15分間、4℃)により細胞膜を調製した。定常状態条件を達成するようにそして特異的結合を最適化するようにアッセイを定めた。5−HT6受容体に関して、5x105の細胞からの膜を5.0nMの[3H]−LSDと37℃で30分間インキュベーションした。非特異的結合は10−5Mのセロトニンを用いて決定された。0.5%のポリエチレンイミンで前処理されているガラス繊維フィルター(GF/B)を通した真空濾過によりアッセイを止めた。全および結合放射活性を液体シンチレーション計数により決定した。80%より大きい特異的結合がこれらのアッセイの各々において得られた。化合物を4log濃度範囲で試験し;全ての決定は三重反復として行われた。IC50値は、Hill式曲線適合を用いて非線形回帰分析により決定された。阻害定数(Ki値)は、Cheng−Preushoff式:
Ki=IC50:(1+L/Kd)
[式中、Lはアッセイにおける放射性リガンド([3H]−LSD)濃度を、そしてKdは受容体に対する放射性リガンドの親和性を表す]
から計算された。結果は、pKi値、少なくとも3回の別個の実験の平均±SDとして表される。
【0225】
ヒト5−HT6受容体へのインビトロ機能活性((拮抗)作動作用((ant)agonism))
CHO−ヒト−5HT6−エクオリンアッセイは、Euroscreen、ブリュッセルから購入した(Euroscreen,技術書類一式、ヒト組換えセロトニン5−HT6−A1受容体、DNAクローンおよびCHO AequoScreenTM組換え細胞系、カタログn0:ES−316−A、2003年2月)。ヒト−5HT6−エクオリン
細胞は、ミトコンドリア標的化アポ−エクオリンを発現する。活性エクオリンを再構成するためには、細胞にコエレンテラジンを与えなければならない。ヒト5−HT6受容体へのアゴニストの結合後に細胞内カルシウム濃度は増加し、そしてアポ−エクオリン/コエレンテラジン複合体へのカルシウムの結合はコエレンテラジンの酸化反応を引き起こし、それはアポ−エクオリン、コエレンテラミド、CO2および光(λmax469nm)の生成をもたらす。この発光反応は、アゴニスト濃度により決まる。発光は、MicroBeta Jet(Perkin Elmer)を用いて測定される。化合物のアゴニスト作用は、pEC50として表される。化合物のアンタゴニスト作用は、10−8Mのα−メチルセロトニン誘起発光の阻害として決定され、そしてpA2はCheng−Preushoff式に従って計算された。化合物を5log濃度範囲で試験し、そして3回の独立した実験を二重反復で行った。
【実施例7】
【0226】
製薬学的製剤
臨床使用のために、式(1)の化合物は、該化合物、さらに特に本明細書に開示される特定の化合物を含有するので本発明の重要なそして新規の態様である製薬学的組成物に調合される。用いることができる製薬学的組成物のタイプには、錠剤、チュアブル錠、カプセル剤(マイクロカプセル剤を包含する)、液剤、非経口液剤、軟膏(クリームおよびゲル)、座薬、懸濁剤および本明細書に開示される他のタイプが包含されるがこれらに限定されるものではなく、もしくは本明細書および当該技術分野における一般的知識から当業者に明らかである。有効成分は例えば、シクロデキストリン、それらのエーテルもしくはそれらのエステルにおける包接錯体の形態であることもできる。組成物は経口、静脈内、皮下、気管、気管支、鼻腔内、肺、経皮、口腔、直腸、非経口もしくは他の投与方法に用いられる。製薬学的製剤は、製薬学的に許容しうる添加剤、希釈剤および/もしくは担体と混合した式(1)の少なくとも1つの化合物を含有する。有効成分の総量は、好適には製剤の約0.1%(w/w)〜約95%(w/w)、好適には0.5%〜50%(w/w)、そして好ましくは1%〜25%(w/w)の範囲である。
【0227】
本発明の化合物は、製薬学的に通常の液体もしくは固体充填剤および増量剤、溶媒、乳化剤、潤滑剤、香料、着色料および/もしくは緩衝物質のような、液体もしくは固体、粉末成分のような補助物質を用いて通常の方法によって投与に適当な形態にすることができる。頻繁に用いられる補助物質には、炭酸マグネシウム、二酸化チタン、ラクトース、サッカロース、ソルビトール、マンニトールおよび他の糖もしくは糖アルコール、タルク、乳タンパク質、ゼラチン、澱粉、アミロペクチン、セルロースおよびその誘導体、動物および植物油、例えば魚肝油、ヒマワリ油、ラッカセイ油もしくはゴマ油、ポリエチレングリコールおよび溶媒、例えば滅菌水および1価もしくは多価アルコール、例えばグリセロール、ならびに崩壊剤および潤滑剤、例えばステアリン酸マグネシウム、ステアリン酸カルシウム、ステアリルフマル酸ナトリウムおよびポリエチレングリコールワックスが包含される。次に、混合物を顆粒剤に加工するかもしくは錠剤に圧縮することができる。錠剤は、以下の成分を用いて製造される。
【0228】
【表16】
【0229】
成分を混ぜ合わせ、そして圧縮して各々230mgの重さの錠剤を生成せしめる。
【0230】
有効成分は、製剤を生成せしめるために混合する前に、他の非有効成分と別個に予混合することができる。有効成分はまた、製剤を生成せしめるために非有効成分と混合する前に、相互に混合することもできる。
【0231】
ソフトゼラチンカプセル剤は、本発明の有効成分、植物油、脂肪、もしくはソフトゼラチンカプセル剤用の他の適当な賦形剤の混合物を含有するカプセルで製造することができる。ハードゼラチンカプセル剤は、有効成分の顆粒を含有することができる。ハードゼラチンカプセル剤はまた、ラクトース、サッカロース、ソルビトール、マンニトール、ジャガイモ澱粉、コーンスターチ、アミロペクチン、セルロース誘導体もしくはゼラチンのような固形粉末成分と一緒に有効成分を含有することもできる。ハードゼラチンカプセル剤は、以下の成分を用いて製造することができる:
【0232】
【表17】
【0233】
上記の成分を混合し、そして120mgの量においてハードゼラチンカプセルに詰める。
【0234】
直腸投与用の投与単位は、(i)中性脂肪ベースと混合した有効成分を含有する座薬の形態で;(ii)植物油、パラフィン油もしくはゼラチン直腸カプセル剤用の他の適当な賦形剤との混合物において有効成分を含有するゼラチン直腸カプセル剤の形態で;(iii)既製のミクロ浣腸(micro enema)の形態で;または(iv)投与の直前に適当な溶媒において再構成される乾燥ミクロ浣腸製剤の形態で製造することができる。各々1mgの有効成分を含有する座薬を下記のとおり製造することができる。
【0235】
【表18】
【0236】
有効成分を適切なサイズのメッシュふるいに通し、そして必要な最小加熱を用いて前もって融解した飽和脂肪酸グリセリドに懸濁する。次に、混合物を標準2g容量の座薬型に注ぎ込み、そして冷却させる。
【0237】
液状製剤はシロップ剤、エリキシル剤、濃縮ドロップもしくは懸濁剤、例えば、有効成分ならびに例えば糖もしくは糖アルコールおよびエタノール、水、グリセロール、プロピレングリコールおよびポリエチレングリコールの混合物からなる残りを含有する液剤もしくは懸濁剤の形態で製造することができる。静脈内製剤は、下記のとおり製造することができる:
【0238】
【表19】
【0239】
化合物をArlatone GTM、EtOHおよび水に溶解し、そして次に溶液をさらなる水でゆっくりと希釈する。
【0240】
所望に応じて、そのような液状製剤は着色剤、香料、防腐剤、サッカリンおよびカルボキシメチルセルロースもしくは他の増粘剤を含有することができる。液状製剤はまた、使用前に適当な溶媒で再構成される乾燥粉末の形態で製造することもできる。非経口投与用の液剤は、製薬学的に許容しうる溶媒における本発明の製剤の液剤として製造することができる。これらの液剤はまた、安定化成分、防腐剤および/もしくは緩衝成分を含有することもできる。非経口投与用の液剤はまた、使用前に適当な溶媒で再構成される乾燥製剤として製造することもできる。
【0241】
また本発明に従って提供されるのは、医学的治療における使用のための、製剤および本発明の製薬学的組成物の成分の1つもしくはそれ以上を詰めた1つもしくはそれ以上の容器を含んでなる「キットオブパーツ」である。そのような容器(1つもしくは複数)に付随するのは様々な資料、例えば使用説明書、または製薬学的製品の製造、使用もしくは販売を規制する政府機関により規定される形態の通知であることができ、この通知はヒトもしくは動物投与のための製造、使用もしくは販売の該機関による承認を示す。5−HT6受容体の拮抗作用が必要とされるかもしくは所望される症状を処置することに使用する薬剤の製造における本発明の製剤の使用、および5−HT6受容体の拮抗作用が必要とされるかもしくは所望される症状を患っているかもしくは起こしやすい患者への式(1)の少なくとも1つの化合物の治療的に有効な総量の投与を含んでなる医学的処置の方法。
【0242】
例としてそして限定するものではなく、全身使用もしくは局所適用のための好ましい活性化合物を含んでなるいくつかの製薬学的組成物が示される。本発明の他の化合物もしくはその組み合わせを該化合物の代わりに(もしくはそれに加えて)用いることができる。有効成分の濃度は、本明細書に説明されるように広範囲にわたって変えることができる。含まれ得る成分の量およびタイプは、当該技術分野において周知である。
【0243】
文献目録
Bentley,J.C.et al.(1997)J.Psychopharmacol.Suppl.A64,255
Bentley,J.C.et al.(1999a)Br J Pharmacol.Suppl.126,P66
Bentley,J.C.,et al.(1999b).Br J Pharmacol 126(7):1537−42
Berge,S.M.:“Pharmaceutical salts”,J.Pharmaceutical Science,66,1−19(1977).
Bickel,M.H.,:“The pharmacology and Biochemistry of N−oxides”,Pharmacological Reviews,21(4),325−355,1969.
Bundgaard,H.(editor),“Design of Prodrugs”,Elsevier,1985.
Byrn et al.,Pharmaceutical Research,12(7),945−954,1995.
Dwyer & Meilor,:“Chelating agents and Metal Chelates”,Academic Press,chapter 7,1964.
Ettmayer,P.et al.,“Lessons learned from
marketed and investigational prodrugs”,J.Med.Chem.,47,2393−2404,2004.
Jaervinen,T.et al.,“Design and Pharmaceutical applications of prodrugs”,pages 733−796 in:S.C.Gad(editor):“Drug Discovery Handbook”,John Wiley & Sons Inc.,New Jersey,U.S.A.,2005.
King,F.D.,(editor),page 215 in:“Medicinal Chemistry:Principles and Practice”,1994,ISBN 0−85186−494−5.
Kohen,R.,et al.(1996).J Neurochem 66(1):47−56
Martin,E.W.(Editor),“Remington:The Science and Practice of Pharmacy”,Mack Publishing Company,19th Edition,Easton,Pa,Vol
2.,Chapter 83,1447−1462,1995.
Rogers,D.C.,et al.(1999).Br J Pharamcol
127(suppl.).22P
Roth,B.L.,et al.(1994).J Pharmacol Exp Ther 268(3):1403−10
Ruat,M.et al.(1993)Biochem.Biophys.Res.Commun.193:268−276
Sebben,M.et al.(1994)NeuroReport 5:2553
−2557
Sibley,D.R.et al.,Mol.Pharmacol.,1993,43,320−327
Sleight,A.J.,et al.,Neurotransmission,1995,11,1−5
Sleight,A.J.,et al.,Serotonin ID Research Alert,1997,2(3),115−8).
Sleight,A.J.,et al.(1998).Br J Pharmacol 124(3):556−62
Stella,J.,“Prodrugs as therapeutics”,Expert Opin.Ther.Patents,14(3),277−280,2004.
Woolley M.L.et al.(2001)Neuropharmacology 41:210−219
WO 01/070700およびWO 02/030881
【特許請求の範囲】
【請求項1】
式(I):
【化1】
[式中:
−R1は水素、非置換のアルキル(C1〜4)基、1個もしくはそれ以上のハロゲン原子で置換されたアルキル(C1〜4)基、または場合により1個もしくはそれ以上のハロゲン原子で置換されていてもよいフェニル基を表し、
−R2およびR3は独立して水素、非置換のアルキル(C1〜4)基、1個もしくはそれ以上のハロゲン原子で置換されたアルキル(C1〜4)基、場合により1個もしくはそれ以上のハロゲン原子で置換されていてもよいアルキル(C1〜4)−O−アルキル(C1〜4)−フェニル基または場合により1個もしくはそれ以上のハロゲン原子で置換されていてもよいフェニル基を表すか、あるいは
R1およびR2は「a」および「b」の記号が付された炭素原子と一緒になってC5〜8−シクロアルキル環を形成するか、あるいは
R2およびR3は「b」の記号が付された炭素原子と一緒になってC3〜8−シクロアルキル環を形成するか、あるいは
R2およびR3は「b」の記号が付された炭素原子と一緒になって場合により置換されていてもよいC5〜8−ヘテロシクロアルキル環を形成するか、あるいは
−R4およびR5は独立して水素、非置換のアルキル(C1〜4)基、1個もしくはそれ以上のハロゲン原子で置換されたアルキル(C1〜4)基、場合により置換されていてもよい単環式芳香族基、場合により置換されていてもよい縮合した二環式芳香族基、場合により置換されていてもよい単環式ヘテロ芳香族基、場合により置換されていてもよい縮合した二環式ヘテロ芳香族基を表すか、あるいは
R3およびR4は「b」および「c」の記号が付された炭素原子と一緒になってC3〜8−シクロアルキル環を形成するか、あるいは
R3およびR4は「b」および「c」の記号が付された炭素原子と一緒になって場合により置換されていてもよいC5〜8−ヘテロシクロアルキル環を形成するか、あるいは
−R6およびR7は独立して水素原子、またはアルキル(C1〜4)基、または1個もしくはそれ以上のハロゲン原子で置換されたアルキル(C1〜4)基;あるいは(C1〜3)アルコキシ基、またはジアルキル(C1〜3)−アミノ−アルキル(C1〜3)基、または場合により置換されていてもよい単環式もしくは縮合した二環式芳香族もしくはヘテロ芳香族基、または場合により置換されていてもよいC5〜8−シクロアルキル基もしくは場合により置換されていてもよいC5〜8−ヘテロシクロアルキル基を表すか、あるいは
R6およびR7は、それらが結合している窒素原子と一緒になって、場合により置換されていてもよいC5〜8−ヘテロシクロアルキル基を形成し、
−R8は場合により置換されていてもよい単環式芳香族基、場合により置換されていてもよい縮合した二環式芳香族基、場合により置換されていてもよい単環式ヘテロ芳香族基
、場合により置換されていてもよい縮合した二環式ヘテロ芳香族基、−CR9=CR10−アリール基(ここで、R9およびR10は独立して水素もしくはアルキル−(C1〜3)基を表す)、−C≡C−アリール基、場合により置換されていてもよいピペリジニル基、もしくは基−NR11R12(ここで、R11およびR12は独立して水素、アルキル−(C1〜3)基または場合により置換されていてもよいフェニルもしくはベンジル基を表す)を表す]
の化合物もしくは互変異性体、立体異性体、N−オキシド、同位体標識したアナログ、または前述のもののいずれかの薬理学的に許容しうる塩、水和物もしくは溶媒和物。
【請求項2】
−R1が水素を表すかもしくはR1およびR2が「a」および「b」の記号が付された炭素原子と一緒になってシクロヘキシル環を形成し、
−R2およびR3が独立して水素もしくはアルキル(C1〜3)基を表すか、またはR2およびR3が「b」の記号が付された炭素原子と一緒になってシクロペンチルもしくはシクロヘキシル環を形成し、
−R4およびR5が独立して水素、アルキル(C1〜3)基を表すか、またはR3およびR4が「b」および「c」の記号が付された炭素原子と一緒になってC3〜8−シクロアルキル環を形成し、
−R6およびR7が独立して水素原子、またはアルキル(C1〜3)基、または1個
もしくはそれ以上のハロゲン原子で置換されたアルキル(C1〜4)基、またはメトキシ基、またはシクロヘキシル基、またはベンジル基または4−ピペリジニル基を表し、
−R8が請求項1に示されるとおりの意味を有する
請求項1に記載の式(1)の化合物、もしくは互変異性体、立体異性体、N−オキシド、同位体標識したアナログ、または前述のもののいずれかの薬理学的に許容しうる塩、水和物もしくは溶媒和物。
【請求項3】
R1、R4、R5およびR6が水素を表し、R2およびR3が独立してアルキル(C1〜3)基を表すか、またはR2およびR3が「b」の記号が付された炭素原子と一緒になってシクロペンチルもしくはシクロヘキシル環を形成し、R7がアルキル(C1〜3)基を表し、そしてR8が請求項1に示されるとおりの意味を有する請求項1に記載の式(1)の化合物、もしくは互変異性体、立体異性体、N−オキシド、同位体標識したアナログ、または前述のもののいずれかの薬理学的に許容しうる塩、水和物もしくは溶媒和物。
【請求項4】
式:
【化2】
のものから選択される化合物。
【請求項5】
該化合物が光学活性鏡像異性体である、請求項1〜4のいずれか1項に記載の化合物、もしくは互変異性体、立体異性体、N−オキシド、同位体標識したアナログ、または前述のもののいずれかの薬理学的に許容しうる塩、水和物もしくは溶媒和物。
【請求項6】
ピラゾリン環における2個の潜在的不斉炭素原子のいずれか一方もしくは両方が左旋性鏡像異性体である、請求項1〜4のいずれか1項に記載の化合物、もしくは互変異性体、立体異性体、N−オキシド、同位体標識したアナログ、または前述のもののいずれかの薬理学的に許容しうる塩、水和物もしくは溶媒和物。
【請求項7】
ピラゾリン環における2個の潜在的不斉炭素原子のいずれか一方もしくは両方が右旋性鏡像異性体である、請求項1〜4のいずれか1項に記載の化合物、もしくは互変異性体、立体異性体、N−オキシド、同位体標識したアナログ、または前述のもののいずれかの薬理学的に許容しうる塩、水和物もしくは溶媒和物。
【請求項8】
請求項1〜7のいずれか1項に記載の化合物またはその薬理学的に許容しうる塩、水和物もしくは溶媒和物を含んでなる薬剤。
【請求項9】
製薬学的に許容しうる担体および/もしくは少なくとも1種の製薬学的に許容しうる補助物質、有効成分として、請求項1〜7のいずれか1項の少なくとも1種の化合物またはその塩、水和物もしくは溶媒和物の薬理学的活性量を含んでなる製薬学的組成物。
【請求項10】
錠剤、丸剤、ロゼンジ、糖衣錠、トローチ、ハードもしくはソフトカプセル剤、散剤、カシェ剤、顆粒剤、座薬、液剤、水性もしくは油性懸濁剤、乳剤、ローション、シロップ剤、軟膏、ゲル、パスタ剤、クリーム、フォーム、吸入剤、スプレー、エアロゾルもしくは経皮パッチの形態の請求項8の薬剤もしくは請求項9の組成物。
【請求項11】
パーキンソン病、ハンチントン舞踏病、統合失調症、不安、鬱病、躁鬱病、精神病、癲癇、強迫神経症、気分障害、片頭痛、アルツハイマー病、加齢に伴う認識衰退、軽度認識障害、睡眠障害、摂食障害、食欲不振、過食症、無茶食い障害、パニック発作、静座不能、注意欠陥多動性障害、注意欠陥障害、コカイン、エタノール、ニコチンもしくはベンゾジアゼピンの乱用からの離脱、疼痛、脊椎外傷もしくは頭部損傷と関連する障害、水頭症、機能性腸疾患、過敏性腸症候群、肥満および2型糖尿病を処置するための請求項8の薬剤もしくは請求項9の組成物。
【請求項12】
請求項1に記載の化合物を含んでなる5−HT6受容体アンタゴニスト。
【請求項13】
少なくとも1種の追加の治療薬をさらに含んでなる請求項9に記載の製薬学的組成物。
【請求項14】
請求項1〜4の1項の化合物を投与に適当な形態にすることを特徴とする、請求項9もしくは請求項13に記載の製薬学的組成物の製造方法。
【請求項15】
パーキンソン病、ハンチントン舞踏病、統合失調症、不安、鬱病、躁鬱病、精神病、癲癇、強迫神経症、気分障害、片頭痛、アルツハイマー病、加齢に伴う認識衰退、軽度認識障害、睡眠障害、摂食障害、食欲不振、過食症、無茶食い障害、パニック発作、静座不能、注意欠陥多動性障害、注意欠陥障害、コカイン、エタノール、ニコチンもしくはベンゾジアゼピンの乱用からの離脱、疼痛、脊椎外傷もしくは頭部損傷と関連する障害、水頭症、機能性腸疾患、過敏性腸症候群、肥満および2型糖尿病の治療における同時、別個もしくは逐次使用のための、(i)一般式(1)の化合物、またはその薬理学的に許容しうる塩もしくは水和物および(ii)他の薬剤を含んでなる組み合わせ製剤。
【請求項16】
一般式(1)の化合物の合成において有用である、一般式(1x):
【化3】
[式中、XはハロゲンもしくはS−アルキル(C1〜3)のいずれかを表し、そして他の記号は請求項1に示されるとおりの意味を有する]
の化合物ならびにその互変異性体、立体異性体およびN−オキシド、ならびに式(1x)の該化合物ならびにその互変異性体、立体異性体およびN−オキシドの薬理学的に許容しうる塩、水和物および溶媒和物。
【請求項17】
一般式(1)の化合物の合成において有用である、一般式(1z):
【化4】
[式中、記号は請求項1に示されるとおりの意味を有する]
の化合物ならびにその互変異性体、立体異性体およびN−オキシド、ならびに式(1z)の該化合物ならびにその互変異性体、立体異性体およびN−オキシドの薬理学的に許容しうる塩、水和物および溶媒和物。
【請求項18】
(i)式(IX)の化合物をハロゲン化アルキル、例えばヨウ化メチルと反応させることにより得られ得る式(X)の化合物を塩基の存在下でピラゾリンと反応させて式(1z)の化合物を生成せしめる段階、
(ii)式(1z)の化合物を、ジイソプロピルエチルアミンのような塩基の存在下で、ジクロロメタンのような非プロトン性溶媒中で、式R8−SO2−X(式中、XはBr、ClもしくはFである)のハロゲン化スルホニルと反応させる段階
を含んでなる、R7が水素である請求項1に記載の式(1)の化合物、すなわち式(1’)(式中、全ての記号は請求項1に示されるとおりの意味を有する)を有する化合物の製造方法。
【化5】
【請求項19】
X=Cl、そしてハロゲン化アルキルがヨウ化メチルである請求項18に記載の方法。
【請求項20】
5−HT6受容体により媒介されるか、もしくはこれらの受容体の調節によって処置することができる障害もしくは症状の処置もしくは予防用の薬剤の製造のための請求項1〜7のいずれかに記載の化合物の使用。
【請求項21】
個体における、5−HT6受容体により媒介されるか、もしくはこれらの受容体の調節によって処置することができる障害もしくは症状の処置もしくは予防のための請求項1〜7のいずれかに記載の化合物の有効量の使用。
【請求項22】
請求項1に記載の化合物もしくはその薬理学的に許容しうる塩の有効用量をそのような処置を必要とする患者に投与することを含んでなる5−HT6受容体に拮抗する方法。
【請求項23】
パーキンソン病、ハンチントン舞踏病、統合失調症、不安、鬱病、躁鬱病、精神病、癲癇、強迫神経症、気分障害、片頭痛、アルツハイマー病、加齢に伴う認識衰退、軽度認識障害、睡眠障害、摂食障害、食欲不振、過食症、無茶食い障害、パニック発作、静座不能、注意欠陥多動性障害、注意欠陥障害、コカイン、エタノール、ニコチンもしくはベンゾジアゼピンの乱用からの離脱、疼痛、脊椎外傷もしくは頭部損傷と関連する障害、水頭症
、機能性腸疾患、過敏性腸症候群、肥満および2型糖尿病の処置もしくは予防用の製薬学的組成物の製造のための請求項1〜7のいずれかに記載の化合物の使用。
【請求項24】
請求項1に記載の式(1)の化合物の有効量を以下の疾患の1種もしくはそれ以上を有するヒトに投与することを含んでなる、パーキンソン病、ハンチントン舞踏病、統合失調症、不安、鬱病、躁鬱病、精神病、癲癇、強迫神経症、気分障害、片頭痛、アルツハイマー病、加齢に伴う認識衰退、軽度認識障害、睡眠障害、摂食障害、食欲不振、過食症、無茶食い障害、パニック発作、静座不能、注意欠陥多動性障害、注意欠陥障害、コカイン、エタノール、ニコチンもしくはベンゾジアゼピンの乱用からの離脱、疼痛、脊椎外傷もしくは頭部損傷と関連する障害、水頭症、機能性腸疾患、過敏性腸症候群、肥満および2型糖尿病の処置方法。
【請求項25】
請求項1〜7のいずれか1項に記載の化合物、またはその薬理学的に許容しうる塩、水和物もしくは溶媒和物、または請求項9に記載の組成物を含んでなり、そしてパーキンソン病、ハンチントン舞踏病、統合失調症、不安、鬱病、躁鬱病、精神病、癲癇、強迫神経症、気分障害、片頭痛、アルツハイマー病、加齢に伴う認識衰退、軽度認識障害、睡眠障害、摂食障害、食欲不振、過食症、無茶食い障害、パニック発作、静座不能、注意欠陥多動性障害、注意欠陥障害、コカイン、エタノール、ニコチンもしくはベンゾジアゼピンの乱用からの離脱、疼痛、脊椎外傷もしくは頭部損傷と関連する障害、水頭症、機能性腸疾患、過敏性腸症候群、肥満および2型糖尿病の処置におけるその使用説明書をそれに伴う販売用パッケージ。
【請求項1】
式(I):
【化1】
[式中:
−R1は水素、非置換のアルキル(C1〜4)基、1個もしくはそれ以上のハロゲン原子で置換されたアルキル(C1〜4)基、または場合により1個もしくはそれ以上のハロゲン原子で置換されていてもよいフェニル基を表し、
−R2およびR3は独立して水素、非置換のアルキル(C1〜4)基、1個もしくはそれ以上のハロゲン原子で置換されたアルキル(C1〜4)基、場合により1個もしくはそれ以上のハロゲン原子で置換されていてもよいアルキル(C1〜4)−O−アルキル(C1〜4)−フェニル基または場合により1個もしくはそれ以上のハロゲン原子で置換されていてもよいフェニル基を表すか、あるいは
R1およびR2は「a」および「b」の記号が付された炭素原子と一緒になってC5〜8−シクロアルキル環を形成するか、あるいは
R2およびR3は「b」の記号が付された炭素原子と一緒になってC3〜8−シクロアルキル環を形成するか、あるいは
R2およびR3は「b」の記号が付された炭素原子と一緒になって場合により置換されていてもよいC5〜8−ヘテロシクロアルキル環を形成するか、あるいは
−R4およびR5は独立して水素、非置換のアルキル(C1〜4)基、1個もしくはそれ以上のハロゲン原子で置換されたアルキル(C1〜4)基、場合により置換されていてもよい単環式芳香族基、場合により置換されていてもよい縮合した二環式芳香族基、場合により置換されていてもよい単環式ヘテロ芳香族基、場合により置換されていてもよい縮合した二環式ヘテロ芳香族基を表すか、あるいは
R3およびR4は「b」および「c」の記号が付された炭素原子と一緒になってC3〜8−シクロアルキル環を形成するか、あるいは
R3およびR4は「b」および「c」の記号が付された炭素原子と一緒になって場合により置換されていてもよいC5〜8−ヘテロシクロアルキル環を形成するか、あるいは
−R6およびR7は独立して水素原子、またはアルキル(C1〜4)基、または1個もしくはそれ以上のハロゲン原子で置換されたアルキル(C1〜4)基;あるいは(C1〜3)アルコキシ基、またはジアルキル(C1〜3)−アミノ−アルキル(C1〜3)基、または場合により置換されていてもよい単環式もしくは縮合した二環式芳香族もしくはヘテロ芳香族基、または場合により置換されていてもよいC5〜8−シクロアルキル基もしくは場合により置換されていてもよいC5〜8−ヘテロシクロアルキル基を表すか、あるいは
R6およびR7は、それらが結合している窒素原子と一緒になって、場合により置換されていてもよいC5〜8−ヘテロシクロアルキル基を形成し、
−R8は場合により置換されていてもよい単環式芳香族基、場合により置換されていてもよい縮合した二環式芳香族基、場合により置換されていてもよい単環式ヘテロ芳香族基
、場合により置換されていてもよい縮合した二環式ヘテロ芳香族基、−CR9=CR10−アリール基(ここで、R9およびR10は独立して水素もしくはアルキル−(C1〜3)基を表す)、−C≡C−アリール基、場合により置換されていてもよいピペリジニル基、もしくは基−NR11R12(ここで、R11およびR12は独立して水素、アルキル−(C1〜3)基または場合により置換されていてもよいフェニルもしくはベンジル基を表す)を表す]
の化合物もしくは互変異性体、立体異性体、N−オキシド、同位体標識したアナログ、または前述のもののいずれかの薬理学的に許容しうる塩、水和物もしくは溶媒和物。
【請求項2】
−R1が水素を表すかもしくはR1およびR2が「a」および「b」の記号が付された炭素原子と一緒になってシクロヘキシル環を形成し、
−R2およびR3が独立して水素もしくはアルキル(C1〜3)基を表すか、またはR2およびR3が「b」の記号が付された炭素原子と一緒になってシクロペンチルもしくはシクロヘキシル環を形成し、
−R4およびR5が独立して水素、アルキル(C1〜3)基を表すか、またはR3およびR4が「b」および「c」の記号が付された炭素原子と一緒になってC3〜8−シクロアルキル環を形成し、
−R6およびR7が独立して水素原子、またはアルキル(C1〜3)基、または1個
もしくはそれ以上のハロゲン原子で置換されたアルキル(C1〜4)基、またはメトキシ基、またはシクロヘキシル基、またはベンジル基または4−ピペリジニル基を表し、
−R8が請求項1に示されるとおりの意味を有する
請求項1に記載の式(1)の化合物、もしくは互変異性体、立体異性体、N−オキシド、同位体標識したアナログ、または前述のもののいずれかの薬理学的に許容しうる塩、水和物もしくは溶媒和物。
【請求項3】
R1、R4、R5およびR6が水素を表し、R2およびR3が独立してアルキル(C1〜3)基を表すか、またはR2およびR3が「b」の記号が付された炭素原子と一緒になってシクロペンチルもしくはシクロヘキシル環を形成し、R7がアルキル(C1〜3)基を表し、そしてR8が請求項1に示されるとおりの意味を有する請求項1に記載の式(1)の化合物、もしくは互変異性体、立体異性体、N−オキシド、同位体標識したアナログ、または前述のもののいずれかの薬理学的に許容しうる塩、水和物もしくは溶媒和物。
【請求項4】
式:
【化2】
のものから選択される化合物。
【請求項5】
該化合物が光学活性鏡像異性体である、請求項1〜4のいずれか1項に記載の化合物、もしくは互変異性体、立体異性体、N−オキシド、同位体標識したアナログ、または前述のもののいずれかの薬理学的に許容しうる塩、水和物もしくは溶媒和物。
【請求項6】
ピラゾリン環における2個の潜在的不斉炭素原子のいずれか一方もしくは両方が左旋性鏡像異性体である、請求項1〜4のいずれか1項に記載の化合物、もしくは互変異性体、立体異性体、N−オキシド、同位体標識したアナログ、または前述のもののいずれかの薬理学的に許容しうる塩、水和物もしくは溶媒和物。
【請求項7】
ピラゾリン環における2個の潜在的不斉炭素原子のいずれか一方もしくは両方が右旋性鏡像異性体である、請求項1〜4のいずれか1項に記載の化合物、もしくは互変異性体、立体異性体、N−オキシド、同位体標識したアナログ、または前述のもののいずれかの薬理学的に許容しうる塩、水和物もしくは溶媒和物。
【請求項8】
請求項1〜7のいずれか1項に記載の化合物またはその薬理学的に許容しうる塩、水和物もしくは溶媒和物を含んでなる薬剤。
【請求項9】
製薬学的に許容しうる担体および/もしくは少なくとも1種の製薬学的に許容しうる補助物質、有効成分として、請求項1〜7のいずれか1項の少なくとも1種の化合物またはその塩、水和物もしくは溶媒和物の薬理学的活性量を含んでなる製薬学的組成物。
【請求項10】
錠剤、丸剤、ロゼンジ、糖衣錠、トローチ、ハードもしくはソフトカプセル剤、散剤、カシェ剤、顆粒剤、座薬、液剤、水性もしくは油性懸濁剤、乳剤、ローション、シロップ剤、軟膏、ゲル、パスタ剤、クリーム、フォーム、吸入剤、スプレー、エアロゾルもしくは経皮パッチの形態の請求項8の薬剤もしくは請求項9の組成物。
【請求項11】
パーキンソン病、ハンチントン舞踏病、統合失調症、不安、鬱病、躁鬱病、精神病、癲癇、強迫神経症、気分障害、片頭痛、アルツハイマー病、加齢に伴う認識衰退、軽度認識障害、睡眠障害、摂食障害、食欲不振、過食症、無茶食い障害、パニック発作、静座不能、注意欠陥多動性障害、注意欠陥障害、コカイン、エタノール、ニコチンもしくはベンゾジアゼピンの乱用からの離脱、疼痛、脊椎外傷もしくは頭部損傷と関連する障害、水頭症、機能性腸疾患、過敏性腸症候群、肥満および2型糖尿病を処置するための請求項8の薬剤もしくは請求項9の組成物。
【請求項12】
請求項1に記載の化合物を含んでなる5−HT6受容体アンタゴニスト。
【請求項13】
少なくとも1種の追加の治療薬をさらに含んでなる請求項9に記載の製薬学的組成物。
【請求項14】
請求項1〜4の1項の化合物を投与に適当な形態にすることを特徴とする、請求項9もしくは請求項13に記載の製薬学的組成物の製造方法。
【請求項15】
パーキンソン病、ハンチントン舞踏病、統合失調症、不安、鬱病、躁鬱病、精神病、癲癇、強迫神経症、気分障害、片頭痛、アルツハイマー病、加齢に伴う認識衰退、軽度認識障害、睡眠障害、摂食障害、食欲不振、過食症、無茶食い障害、パニック発作、静座不能、注意欠陥多動性障害、注意欠陥障害、コカイン、エタノール、ニコチンもしくはベンゾジアゼピンの乱用からの離脱、疼痛、脊椎外傷もしくは頭部損傷と関連する障害、水頭症、機能性腸疾患、過敏性腸症候群、肥満および2型糖尿病の治療における同時、別個もしくは逐次使用のための、(i)一般式(1)の化合物、またはその薬理学的に許容しうる塩もしくは水和物および(ii)他の薬剤を含んでなる組み合わせ製剤。
【請求項16】
一般式(1)の化合物の合成において有用である、一般式(1x):
【化3】
[式中、XはハロゲンもしくはS−アルキル(C1〜3)のいずれかを表し、そして他の記号は請求項1に示されるとおりの意味を有する]
の化合物ならびにその互変異性体、立体異性体およびN−オキシド、ならびに式(1x)の該化合物ならびにその互変異性体、立体異性体およびN−オキシドの薬理学的に許容しうる塩、水和物および溶媒和物。
【請求項17】
一般式(1)の化合物の合成において有用である、一般式(1z):
【化4】
[式中、記号は請求項1に示されるとおりの意味を有する]
の化合物ならびにその互変異性体、立体異性体およびN−オキシド、ならびに式(1z)の該化合物ならびにその互変異性体、立体異性体およびN−オキシドの薬理学的に許容しうる塩、水和物および溶媒和物。
【請求項18】
(i)式(IX)の化合物をハロゲン化アルキル、例えばヨウ化メチルと反応させることにより得られ得る式(X)の化合物を塩基の存在下でピラゾリンと反応させて式(1z)の化合物を生成せしめる段階、
(ii)式(1z)の化合物を、ジイソプロピルエチルアミンのような塩基の存在下で、ジクロロメタンのような非プロトン性溶媒中で、式R8−SO2−X(式中、XはBr、ClもしくはFである)のハロゲン化スルホニルと反応させる段階
を含んでなる、R7が水素である請求項1に記載の式(1)の化合物、すなわち式(1’)(式中、全ての記号は請求項1に示されるとおりの意味を有する)を有する化合物の製造方法。
【化5】
【請求項19】
X=Cl、そしてハロゲン化アルキルがヨウ化メチルである請求項18に記載の方法。
【請求項20】
5−HT6受容体により媒介されるか、もしくはこれらの受容体の調節によって処置することができる障害もしくは症状の処置もしくは予防用の薬剤の製造のための請求項1〜7のいずれかに記載の化合物の使用。
【請求項21】
個体における、5−HT6受容体により媒介されるか、もしくはこれらの受容体の調節によって処置することができる障害もしくは症状の処置もしくは予防のための請求項1〜7のいずれかに記載の化合物の有効量の使用。
【請求項22】
請求項1に記載の化合物もしくはその薬理学的に許容しうる塩の有効用量をそのような処置を必要とする患者に投与することを含んでなる5−HT6受容体に拮抗する方法。
【請求項23】
パーキンソン病、ハンチントン舞踏病、統合失調症、不安、鬱病、躁鬱病、精神病、癲癇、強迫神経症、気分障害、片頭痛、アルツハイマー病、加齢に伴う認識衰退、軽度認識障害、睡眠障害、摂食障害、食欲不振、過食症、無茶食い障害、パニック発作、静座不能、注意欠陥多動性障害、注意欠陥障害、コカイン、エタノール、ニコチンもしくはベンゾジアゼピンの乱用からの離脱、疼痛、脊椎外傷もしくは頭部損傷と関連する障害、水頭症
、機能性腸疾患、過敏性腸症候群、肥満および2型糖尿病の処置もしくは予防用の製薬学的組成物の製造のための請求項1〜7のいずれかに記載の化合物の使用。
【請求項24】
請求項1に記載の式(1)の化合物の有効量を以下の疾患の1種もしくはそれ以上を有するヒトに投与することを含んでなる、パーキンソン病、ハンチントン舞踏病、統合失調症、不安、鬱病、躁鬱病、精神病、癲癇、強迫神経症、気分障害、片頭痛、アルツハイマー病、加齢に伴う認識衰退、軽度認識障害、睡眠障害、摂食障害、食欲不振、過食症、無茶食い障害、パニック発作、静座不能、注意欠陥多動性障害、注意欠陥障害、コカイン、エタノール、ニコチンもしくはベンゾジアゼピンの乱用からの離脱、疼痛、脊椎外傷もしくは頭部損傷と関連する障害、水頭症、機能性腸疾患、過敏性腸症候群、肥満および2型糖尿病の処置方法。
【請求項25】
請求項1〜7のいずれか1項に記載の化合物、またはその薬理学的に許容しうる塩、水和物もしくは溶媒和物、または請求項9に記載の組成物を含んでなり、そしてパーキンソン病、ハンチントン舞踏病、統合失調症、不安、鬱病、躁鬱病、精神病、癲癇、強迫神経症、気分障害、片頭痛、アルツハイマー病、加齢に伴う認識衰退、軽度認識障害、睡眠障害、摂食障害、食欲不振、過食症、無茶食い障害、パニック発作、静座不能、注意欠陥多動性障害、注意欠陥障害、コカイン、エタノール、ニコチンもしくはベンゾジアゼピンの乱用からの離脱、疼痛、脊椎外傷もしくは頭部損傷と関連する障害、水頭症、機能性腸疾患、過敏性腸症候群、肥満および2型糖尿病の処置におけるその使用説明書をそれに伴う販売用パッケージ。
【公表番号】特表2010−504302(P2010−504302A)
【公表日】平成22年2月12日(2010.2.12)
【国際特許分類】
【出願番号】特願2009−528720(P2009−528720)
【出願日】平成19年9月20日(2007.9.20)
【国際出願番号】PCT/EP2007/059944
【国際公開番号】WO2008/034863
【国際公開日】平成20年3月27日(2008.3.27)
【出願人】(501439149)ソルベイ・フアーマシユーチカルズ・ベー・ブイ (71)
【Fターム(参考)】
【公表日】平成22年2月12日(2010.2.12)
【国際特許分類】
【出願日】平成19年9月20日(2007.9.20)
【国際出願番号】PCT/EP2007/059944
【国際公開番号】WO2008/034863
【国際公開日】平成20年3月27日(2008.3.27)
【出願人】(501439149)ソルベイ・フアーマシユーチカルズ・ベー・ブイ (71)
【Fターム(参考)】
[ Back to top ]